UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-K/A
Amendment No. 1
| |
þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2013
OR
| |
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES ACT OF 1934 |
For the transition period from ____________ to ____________
Commission File Number: 001-35994
HEAT BIOLOGICS, INC.
(Name of small business issuer in its charter)
| |
Delaware | 26-2844103 |
(State or other jurisdiction of incorporation or organization) | (IRS Employer Identification Number) |
| |
100 Europa Drive, Suite 420 | |
Chapel Hill, NC | 27517 |
(Address of principal executive offices) | (Zip Code) |
(919) 240-7133
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| |
| |
Title of Class | Name of each exchange on which registered |
Common Stock, $0.0002 par value per share | NASDAQ |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No þ
Indicate by check mark whether the issuer: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of issuer’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every interactive data file required to be submitted and posted pursuant to Rule 405 of Regulation S-T (section 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes þ No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated file, a non-accelerated file, or a smaller reporting company. See the definitions of “large accelerated filer, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| | | | | |
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |
| Non-accelerated filer | ¨ | Smaller reporting company | þ | |
| (Do not check if a smaller reporting company) | |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No þ
The aggregate market value of the registrant’s common stock held by non-affiliates of the registrant as of March 27, 2014, was approximately $29,384,995 based on $6.64, the price at which the registrant’s common stock was last sold on that date. The registrant has provided this information as of March 27, 2014 because its common stock was not publicly traded as of the last business day of its most recently completed second fiscal quarter.
As of March 31, 2014, the issuer had 6,452,341 shares of common stock outstanding.
Documents incorporated by reference: None.
EXPLANATORY NOTE
Heat Biologics, Inc. (the “Company”) is filing this Amendment No. 1 on Form 10-K/A (this “Amendment”) to amend its Annual Report on Form 10-K for the year ended December 31, 2013, filed with the Securities and Exchange Commission (the “SEC”) on March 31, 2014 (the “Original 10-K”).
This Amendment is being filed for the sole purpose of correcting a typographical error in the signature date on the Report of Independent Registered Public Accounting Firm in Part II, Item 8, which was dated March 31, 2013 instead of March 31, 2014. As required by the SEC, this Amendment includes new certifications pursuant to Sections 302 and 906 of the Sarbanes-Oxley Act of 2002, filed as Exhibits 31.1, 31.2, 32.1 and 32.2, hereto.
Except as described above, the Company has not modified or updated the Original 10-K or the financial statements included therein or modified any disclosures contained in the Original 10-K. Accordingly, this Amendment, with the exception of the foregoing, does not reflect events occurring after the date of filing of the Original 10-K, or modify or update any disclosures affected by subsequent events. Consequently, all other information not affected by the correction described above is unchanged and reflects the disclosures and other information made at the date of the filing of the Original 10-K and should be read in conjunction with our filings with the SEC subsequent to the filing of the Original 10-K, including amendments to those filings, if any.
HEAT BIOLOGICS, INC.
FORM 10-K
TABLE OF CONTENTS
PART I
Forward-Looking Statements
Most of the matters discussed within this report include forward-looking statements on our current expectations and projections about future events. In some cases you can identify forward-looking statements by terminology such as “may,” “should,” “potential,” “continue,” “expects,” “anticipates,” “intends,” “plans,” “believes,” “estimates,” and similar expressions. These statements are based on our current beliefs, expectations, and assumptions and are subject to a number of risks and uncertainties, many of which are difficult to predict and generally beyond our control, that could cause actual results to differ materially from those expressed, projected or implied in or by the forward-looking statements. Such risks and uncertainties include the risks noted under “Item 1A Risk Factors.” We do not undertake any obligation to update any forward-looking statements. Unless the context requires otherwise, references to “we,” “us,” “our,” and “Heat Biologics,” refer to Heat Biologics, Inc. and its subsidiaries.
Item 1.
Business
Overview
We are a development stage biopharmaceutical company engaged in the development of novel allogeneic, “off-the-shelf” cellular therapeutic vaccines to combat a wide range of cancers and infectious diseases. Our proprietaryImPACT™ImmunePanAntigenCytotoxicTherapy is being designed to deliver live, genetically-modified, irradiated human cells which are reprogrammed to “pump out” a broad spectrum of cancer-associated antigens together with a potent immune adjuvant called “gp96” to educate and activate a cancer patient’s immune system to recognize and kill cancerous cells. We intend for ourImPACT cells to secrete an antigen-adjuvant complex that generates anti-cancer immune responses in patients by mobilizing and activating cytotoxic “killer” T cells that target multiple cancer antigens, thus harnessing a patient’s own immune system to fight cancer.
Unlike autologous or “personalized” therapeutic vaccine approaches which require extraction and processing of cancer or blood from each individual patient, ourImPACT therapeutic vaccine uses a master cell line containing a host of known and unknown tumor associated antigens to mass-produce a single vaccine product applicable to all patients with a particular cancer type. We believe our off-the-shelf, allogeneic immunotherapy offers logistical, manufacturing and cost of goods benefits compared to autologous patient-specific approaches.
Our most advanced product candidates are HS-110 and HS-410.
HS-110
We have submitted a Phase 2 protocol to our open IND in non-small cell lung cancer (NSCLC) patients with our therapeutic vaccine candidate HS-110 (viagenpumatucel-L). HS-110 is a biologic product which consists of a lung cancer cell line that has been genetically modified using ourImPACT technology platform to secrete a wide range of lung cancer associated antigens bound to a gp96 adjuvant and is designed to activate a T-cell mediated pan-antigen immune response against the patient’s cancer. The Phase 2 trial will evaluate HS-110 in combination with low dose cyclophosphamide followed by sequential chemotherapy versus chemotherapy alone in third-line NSCLC patients. The trial will enroll 123 patients at approximately 20-30 investigative centers over 24 months. We anticipate recruitment to begin in the third quarter of 2014.
The inventor of theImPACT technology that we license recently reported results from a Phase 1 open-label, single center clinical trial of HS-110 in patients with advanced NSCLC. We believe the results provide clinical evidence that HS-110 is capable of generating anti-cancer immune responses. Eighteen patients were vaccinated, and 15 of the 18 vaccinated patients completed the first course of three planned courses of therapy. Two patients completed all three planned courses of therapy (defined as three, six week treatment cycles).
1
HS-110 showed no overt toxicity. There were no serious adverse events (SAEs) that were considered by the trial investigator to be treatment-related. Most of the adverse events (AEs) were reported as mild or moderate (grade 1 or 2) with the most frequent being skin induration and rash that were transitory and usually resolved in 1 to 2 weeks. HS-110 provides evidence of a CD8-CTL IFN-γ immune response in patients with advanced NSCLC. In 11 of the 15 patients (73%) that completed the first course of therapy with HS-110, there was a twofold or greater increase in CD8 cells secreting interferon gamma (CD8-CTL IFN-γ). These patients also exhibited an estimated median survival of 16.5 months (95% CI:7.1-20.0). In contrast, 4 patients were immune non-responders and survived 2.1, 2.3, 6.7, and 6.7 months, or a median survival of 4.5 months, which is consistent with the expected survival times in this patient population. The protocol required that we look for such responses, but, as is typical in immunotherapy, no partial or complete tumor responses were observed. The median one-year overall survival rate of patients in the study was 44% (95% CI:21.6-65.1), comparing favorably to a 5.5% rate based on published data from a 43-patient advanced lung cancer population. One of the late-stage lung cancer patients survived over four years since starting the therapy and another patient survived over three years since starting the therapy. These findings were consistent with multiple pre-clinical published studies onImPACT therapy.
HS-410
We have initiated dosing in a Phase 1/2 bladder cancer trial with HS-410. HS-410 is a biologic product which consists of a bladder cancer cell line which has been genetically modified using ourImPACT technology platform to secrete a wide range of bladder cancer antigens bound to a gp96 adjuvant and is designed to activate a T-cell mediated pan-antigen immune response against the patient’s bladder cancer. To date, we have dosed 1 patient in our 93-patient, Phase 1/2 trial to examine safety, tolerability, immune response and preliminary clinical activity of HS-410 in patients with high risk, superficial bladder cancer who have completed surgical resection and 3-6 weekly intravesical bacillus Calmette-Guérin (BCG) immunotherapy installations. We anticipate including approximately 10-15 clinical sites with an enrollment period of 18-24 months. Patient recruitment began in December 2013.
Additional Indications
We continue to evaluate other indications for ourImPACT therapeutic vaccines and have developed a cell line for ovarian cancer and one for triple negative breast cancer. Our decision to further pursue either of these two product candidates or any additional product candidates other than our two lead product candidates will be based in part upon available funding and partnering opportunities. To date, in excess of $14,000,000 of funding has been awarded to the primary inventor of the technology we license by the National Institutes of Health (NIH) and through other research and clinical grants, which has been used to further develop ourImPACT technology platform that we license. We have little control over the direction of the NIH grant funds that have been received by the primary inventor of the technology we license and since payment is made to the inventors as opposed to us we do not recognize any revenue from such grant funds nor do they fund any expenses that we incur. Although earmarked for further development of the technology that we license, any funds awarded to the primary inventor are used in his discretion and we have little control over his use of the funds. The NIH is also currently fully funding the primary inventor’s study of an HS-HIV product candidate in non-human primates with a therapeutic and prophylactic vaccine for the treatment and prevention of HIV utilizing theImPACT approach.
The table below summarizes our current product candidates and their stages of development:
| | | |
Product Candidate | Indication | Phase of Development | Upcoming Milestone(s) |
HS-110 | Non-Small Cell Lung Cancer (NSCLC) | Open commercial IND | 2014 - Initiate Phase 2 |
HS-410 | Bladder Cancer Adjuvant | Enrolling patients | 2015 - Report Phase 1 data on immune response and safety |
2
ImPACT Therapy—Novel Pan-Antigen Immune Activation
OurImPACT therapy is a novel technology platform designed to educate and stimulate the immune system to combat specific disease targets, such as cancer cells.ImPACT utilizes live attenuated, human-derived, genetically-modified cells to generate an array of tumor associated antigens and secrete an essential immunostimulatory protein called “gp96-Ig”. The secreted proteins are designed to generate an immune response against cancer cells by mobilizing and activating a patient’s own killer T cells to target a broad array of different tumor antigens with the goal of eliminating cancer cells. In contrast with other vaccine technologies that target only one antigen,ImPACT’s pan-antigen approach which may enable the body to induce and maintain an immune response against a broad array of tumor-specific proteins, by potentially providing a more robust and sustained immune response and limiting cancer cells’ ability to evade the immune system. We believe the clinical and pre-clinical results suggest thatImPACT generates anti-tumor immune responses capable of targeting and destroying tumors. We believe our novel, off-the-shelf, live cell therapy has the potential to be used to not only combat a wide range of cancers, but also against various infectious diseases, such as hepatitis C, malaria and HIV, for which non-human primate studies, which we believe are encouraging, have been completed. We have leveraged our existing infrastructure by developing additional product candidates in areas where we can use our proprietary technology. Our success will depend on the clinical and regulatory success of our product candidates and our ability to retain, on commercially reasonable terms, financial and managerial resources, which are currently limited. To date, we have not received regulatory approval for any of our product candidates or derived any revenues from their sales. Moreover, there can be no assurance that we will ever receive regulatory approval for any of our product candidates or derive any revenues from their sales. We should have sufficient capital to operate the company for at least 12 months.
Our Product Candidates and Clinical Development Programs
Our development program involves testing ourImPACT -based product candidates against a number of disease targets, including non-small cell lung cancer and bladder cancer. We have submitted our Phase 2 clinical trial protocol for HS-110, our lead drug candidate, against non-small cell lung cancer (NSCLC) to FDA and intend to initiate the trial in the third quarter of 2014. Our Phase 2 trial will expand upon the Phase 1 results obtained by the primary inventor as described below. In the fourth quarter of 2013, we initiated a Phase 1/2 clinical trial against bladder cancer using our HS-410 drug candidate. We plan to utilize this vaccine to delay or prevent the recurrence of bladder cancer in post-resected bladder cancer patients.
Our History
We were incorporated under the laws of the State of Delaware on June 10, 2008. Our principal offices are located at 100 Europa Drive, Suite 300, Chapel Hill, NC 27517. Our website address iswww.heatbio.com. The information contained in, and that can be accessed through, our website is not incorporated into and is not a part of this report.
We make available on our website our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q and Current Reports on Form 8-K as soon as reasonably practicable after those reports are filed with the SEC. The following Corporate Governance documents are also posted on our website: Code of Conduct, Code of Ethics for Financial Management and the Charters for the Audit Committee, Compensation Committee and Nominating Committee of the Board of Directors. Our phone number is (919) 240-7133 and our facsimile number is (919) 305-8566. Our filings may also be read and copied at the SEC's Public Reference Room at 100 F Street NE, Room 1580 Washington, DC 20549. Information on the operation of the Public Reference Room may be obtained by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of that website is www.sec.gov.
References to Heat Biologics also include references to our subsidiaries Heat Biologics I, Inc. (of which we own a 92.5% interest), Heat Biologics III, Inc., Heat Biologics IV, Inc. and Heat Biologics GmbH unless otherwise indicated. In June 2012, we divested our 92.5% interest in Heat Biologics II, Inc., which resulted in Heat Biologics II, Inc. being classified as discontinued operations in our consolidated financial statements for the years ended December 31, 2012. On May 30, 2012, we formed two wholly-owned subsidiaries, Heat Biologics III, Inc. and Heat Biologics IV, Inc. We assigned our proprietary rights related to the development and application of ourImPACT Therapy for the treatment of non-small lung cancer to Heat Biologics III, Inc. and our proprietary rights related to the development and application of ourImPACT Therapy to the treatment of bladder cancer to Heat Biologics IV, Inc.
3
Strategy
Our objective is to become a leading biopharmaceutical company specializing in the development and commercialization of allogeneic, off-the-shelf therapeutic vaccines. We are focused on discovering, developing and applying our core platformImPACT technology towards a number of disease indications. The key elements of our strategy are:
·
Develop and obtain regulatory approval for our ImPACT-based products. We plan to initiate a Phase 2 clinical trial in NSCLC in Q3-2014 and are currently conducting a Phase 1/2 clinical trial in bladder cancer, which we initiated in Q4-2013. After NSCLC and bladder cancers, depending upon funding and partnering opportunities, we plan to initiate additional clinical trials and in some cases expand current clinical trials against these and other disease targets utilizing ourImPACT technology platform.
·
Maximize commercial opportunity for our ImPACT technology. Our product candidates target large markets with significant unmet medical needs. For each of our product candidates, we seek to retain all manufacturing, marketing and distribution rights which should give us the ability to maximize the economic potential of any future U.S. or international commercialization efforts. We believe that we should be well positioned to successfully commercialize our product candidates independently or through U.S. and international corporate partnerships.
·
Enhance our partnering efforts.We are continually exploring partnerships for licensing and other collaborative relationships and remain opportunistic in seeking strategic partnerships.
·
Further expand our broad patent portfolio. We have made a significant investment in the development of our patent portfolio to protect our technologies and programs, and we intend to continue to do so. We have obtained exclusive rights to five different patent families directed to therapeutic compositions and methods related to our vaccine platform and preclinical development programs for cancer. These families comprise six PCT applications, ten issued patents, two allowed patent applications, and forty- eight pending patent applications. These patents and applications cover the United States, Europe, and Japan as well as several other countries having commercially significant markets.
·
Manage our business with efficiency and discipline. We believe we have efficiently utilized our capital and human resources to develop and acquire our product candidates and programs, and create a broad intellectual property portfolio. We operate cross-functionally and are led by an experienced management team with backgrounds in developing and commercializing product candidates. We use project management techniques to assist us in making disciplined strategic program decisions and to attempt to limit the risk profile of our product pipeline.
·
Obtain additional grant funding. To more fully develop ourImPACT technology platform and its application to a variety of human diseases, we plan to continue to seek and access external sources of grant funding on our own behalf and in conjunction with our academic and other partners to support the development of our pipeline programs. While we intend to work with our academic partners to secure additional grant funding, these partners have no obligation to work with us to secure such funding. We also intend to continue to evaluate opportunities and, as appropriate, acquire or license technologies that meet our business objectives.
·
Continue to both leverage and fortify our intellectual property portfolio. We believe that we have a strong intellectual property position relating to the development and commercialization of ourImPACT technology platform. We plan to continue to leverage this portfolio to create value. In addition to fortifying our existing intellectual property position, we intend to file new patent applications, in-license new intellectual property and take other steps to strengthen, leverage, and expand our intellectual property position.
Disease Targets and Markets
The Oncology Market
The American Cancer Society estimates that 1.66 million people in the U.S. will be diagnosed with cancer in 2013. The lifetime probability of being diagnosed with an invasive cancer is 45% for men and 38% for women. It is projected that 580,350 Americans will die from cancer in 2013.
4
Despite continuous advances made in the field of cancer research every year, there remains a significant unmet medical need as the overall five-year survival rate for cancer patients diagnosed between 2001 and 2007 is an average of 67%. According to the Center of Disease Control, in 2011, cancer was the second leading cause of mortality in the U.S. (23.2%) behind heart disease (24.1%). The American Cancer Society estimates that one in four deaths in the U.S. is due to cancer.
The main treatments for cancer are surgery, radiotherapy and chemotherapy. There are often, however, significant debilitating effects resulting from these treatments or lingering morbidity associated with these approaches to treatment of cancer. Our goal is to develop compounds that can lengthen survival times and improve the quality of life of cancer patients and survivors.
Although there are a large number of patients, treatment and management of cancer is performed by a relatively concentrated pool of medical professionals. We plan to reach this prescriber base using a relatively small commercial infrastructure that we intend to develop in the future by either hiring internally, partnering or contracting with one or more third-party entities with an established sales force. These development plans are dependent on our raising additional capital and receiving grant funding, the success of HS-110, and HS-410 and any technologies we might develop in the future and successful negotiation of commercial relationships, none of which we have completed to date. We believe, however, assuming the efficacy and safety of HS-110 and HS-410 and any other technology we might acquire, that our experienced management team will raise the capital and establish the commercial relationships necessary for success.
Limitations of Current Cancer Therapies
We believe current cancer treatment alternatives suffer from a number of limitations that impair their effectiveness in improving patient survival and overall quality of life including:
·
Toxicity. Chemotherapeutic agents are highly toxic to the human body and very often cause a variety of significant and debilitating side effects, including, but not limited to, nausea and vomiting, bleeding, anemia and mucositis. Some targeted therapeutics have fewer systemic toxicities, but still typically have off-target effects such as gastrointestinal inflammation, severe skin reactions and breathing difficulties. These side effects limit a patient's ability to tolerate treatment and as such can deprive the patient of the potential benefit of additional treatments or treatment combinations that might otherwise destroy or prevent the growth of cancer cells. Once they become aware of the limited efficacy, limited increased survival and potentially significant toxicity of existing treatment alternatives, many patients diagnosed with terminal cancer choose to limit or forego therapy in order to avoid further compromising their quality of life. Patients with advanced stage cancer also often cannot tolerate cancer therapy, and certain therapies can hasten death as the patient's health further deteriorates from the therapy applied.
·
Mechanism of action. While many current therapeutic approaches can be effective against specific targeted cells, the efficacy of these therapies in treating cancer over the long term generally is limited by the abundance and diversity of the cancer and tumor cells, which are believed to enable the targeted cells to adapt and become resistant to the current therapeutic approach over time.
·
Short-term approach. Other than tumor removal in a surgical procedure, curing the cancer is often not the intent or a potential outcome of many current cancer therapies. Rather, increased survival time is the primary focus of many currently marketed and development-stage cancer therapeutics. In this regard, many cancer therapies show only a modest impact on the overall survival of the patients and only affect the length of time that passes after treatment begins and before the patient’s disease worsens or the patient dies.
·
Immune system suppression. A weakened immune system not only inhibits the body's natural ability to fight cancer, but also causes patients to become more susceptible to infections and other diseases. Current approaches to cancer treatment generally involve introduction of an agent, such as a chemical, an antibody or radiation, which causes cell apoptosis (programmed cell death) or inhibits the proliferation of all cells, including immune cells, which has the unintended consequence of indirectly suppressing the immune system.
5
Immunotherapy Overview
OurImPACT technology is a form of immunotherapy. Immunotherapy involves administration of a therapeutic agent that enlists or boosts a subject’s immune system in order to fight disease.
Commonly recognized successful examples of immunotherapy includeprophylactic vaccines, such as, childhood immunizations against infectious diseases such as measles, mumps, and rubella. In these cases, usually weakened (attenuated) or inactivated viruses are injected into the body to educate certain immune system cells to recognize and remember small pieces of viral or bacterial proteins (antigens). If and when an individual is subsequently exposed to this same pathogen, the immune system will recognize these antigens immediately and mount a potent immune response to neutralize and eliminate the pathogenic threat.
Therapeutic vaccines, such asImPACT -based product candidates, operate in a fashion similar toprophylactic vaccines except thattherapeutic vaccines are administered after a particular disease is already present. In each case, the human immune system is educated and harnessed to recognize and fight the disease of interest. Cancer can be considered a failure of the immune system to effectively recognize and eliminate inappropriately dividing and multiplying (malignant) cells. Under ordinary circumstances the human immune system continuously monitors and eliminates inappropriately dividing cells. However, for reasons that are not entirely understood, under cancerous conditions the immune system fails to recognize malignant cells and such cells are permitted to inappropriately multiply, grow and metastasize to form tumors which eventually become life threatening. Our therapeutic vaccines are designed to assist the immune system in identifying and eliminating malignant cells. Our approach involves the introduction of cellular antigens that are characteristic of malignant cells with the goal of generating an immune response against the particular form of cancer. In our approach, in addition to introducing a number of cancer-specific antigens, we also introduce a protein known as gp96 which stimulates and primes the immune system to further recognize cancer antigens and generates a potent and broad pan-antigen immune response against cancerous cells.
Immunotherapy Approaches
Immunotherapy is designed to stimulate and enhance the body’s natural mechanism for killing cancer cells and virus-infected cells. Generally, immunotherapeutic approaches to treat disease can be separated into two distinct classes, passive and active, based on their mechanism of action.
Passive Immunotherapy: Passive immunotherapies generally consist of monoclonal antibodies directed at a single disease-specific enzyme or protein on the surface of the targeted cells with the goal of either killing the targeted cells or preventing them from dividing. Rather than stimulate or otherwise use the body’s immune system to initiate the attack on the disease, the attack is made by the therapy which is producedex vivo, or outside of the body. These therapies also are not usually personalized for the patient.
Active Immunotherapy: Active immunotherapies generally consist of therapies intended to trigger or stimulate the body’s own immune system to fight disease. Active immunotherapies have no direct therapeutic action but rather contain antigens specifically designed to activate the patient’s own immune system to find and kill the targeted cells that carry the same antigen. Active immunotherapies depend on the patient’s immune system to seek out and destroy targeted cells or tumors. Most active immunotherapies utilize off-the-shelf antigens, known as “defined” antigens, rather than individualized, patient specific antigens, and are often paired with adjuvants, which are agents that generally activate the immune system cells to increase immune response.
Shortcomings of Immunotherapies: Both passive and active immunotherapy approaches have shortcomings, which include:
·
Most active immunotherapies use normal, non-mutated, self-antigens which are typically poor at stimulating immune responses, even from healthy immune systems. In fact, the human immune system generally does not generate immune responses against self-antigens. Most passive and active immunotherapies also target one or only a few antigens, which increases the probability that infected cells will escape detection by the immune system and immunotherapy.
·
Most active immunotherapies employ defined antigens that are not effective against multiple types of cancer.
6
·
Most immunotherapies produce toxic effects resulting in damage to healthy tissues if the target antigen is absorbed by normal cells in addition to the targeted cancer or virus-infected cells.
·
Many patients may not be able to mount effective immune responses with immunotherapy due to tumor or virus induced immunosuppression of accessory cells such as CD4+ helper T cells, which are necessary for the immunotherapies to be effective but may be functionally impaired by the patient’s disease.
·
It can be difficult to commercialize immunotherapies based on cells derived from individual patients in a cost-effective manner as a result of the added complexity, limited patient material for production of multiple doses, and the need to store and ship the individual doses.
·
Immunotherapies that rely on defined, off-the-shelf antigens or a single targeted antigen may have limited effectiveness because even within the same type of cancer, the genetic makeup and distinct antigens of a tumor can vary significantly from patient to patient.
These shortcomings were highlighted by the findings of a study recently published inNature Medicine (Finak and Park (2008), Stromal gene expression predicts clinical outcome in breast cancer, Nature Medicine, 14, 518 – 527) where the whole genomes of 50 patients’ breast cancer tumors were sequenced alongside matching DNA from the same patients’ healthy cells to identify the genetic alterations present in the cancerous cells. The study found that the genomic pattern of each of the tumors varied significantly. Of the approximately 1,700 gene mutations found in total, most were specific and unique to the individual patients’ cancerous tumors, and that only three of the genetic mutations occurred in 10% or more of the patients.
Although many of the immunotherapies currently in clinical development have shown promising results, we believe that specific proprietary elements of the ImPACT platform, especially the specific targeting of tumor antigens to patient CD8+ T cells, combined with an appropriate clinical strategy (focused on non-immunogenic tumors) position Heat favorably to competitive compounds.
Our Solution: ImPACT Therapy
We believe ourImPACT Therapy has a number of advantages over existing therapies. These advantages, elaborated below, may enable us to develop commercial products that extend the survival of, and improve the quality of life for, cancer patients:
·
It is designed to fight cancer by activating the immune system against a wide variety of cancer antigens (both known and unknown).
·
It is intended to continually secrete a wide variety of cancer-associated antigens, thus initiating a broad and sustained pan-antigen cytotoxic T cell attack against the targeted cancer. We believe this broad-based attack increases the probability of destroying the targeted cancer.
·
It is designed to stimulate a natural immune response against specific cancer cells. We believe this may limit serious adverse events related to treatment.
·
We believe that the novel mechanism of action, good tolerability and favorable safety profile will enable ourImPACT product candidates to have potential benefits across multiple disease stages and tumor types and in combination with other therapies. We believe ourImPACT technology can be targeted to additional specific tumor types by modifying cells from the cancer type of interest.
·
OurImPACT Therapy represents a first-in-class adjuvant that functions as both an immune activator and an antigen-delivery vehicle.ImPACT is the only adjuvant technology platform currently known to us in clinical development that is specifi c to CD8+ cytotoxic T cell immune response, which is especially important for developing therapeutics in oncology as well as a number of other infectious disease indications.
·
We believe many patients who are too ill to tolerate chemotherapy due to the associated toxicities may be able to benefit from ourImPACT product candidates.
7
ImPACT TECHNOLOGY PLATFORM
ImPACT Background
OurImPACT technology represents an allogenic or “off-the-shelf” method to deliver cancer antigens accompanied by heat shock proteins, or HSPs, to illicit an immune response. HSPs are used as a signaling mechanism by the immune system to identify mutated proteins (“antigens”), including those from tumor cells. Although always present within certain cells, HSPs are normally only released when cells die by necrosis or unnatural cell death (rather than apoptosis or natural programmed cell death) and upon release are recognized by the host’s immune system. When a cell dies an unnatural death through “necrosis”, such as when it is infected and killed by a flu virus or other pathogen, the cell releases its contents into circulation setting off a molecular warning to the immune system thereby generating a rapid and potent immune response. Because HSPs very rarely leave cells, the immune system has evolved to recognize HSPs that have been released from dying cells as the sentries of a molecular alarm system. Upon detection of HSPs, the immune system then directs an immune response against any foreign (pathogenic) proteins bound to the HSP at the time the cell that released it died.
HSP’s have several functions including:
·
Protecting tissues from pathogens by activating the immune system.
·
Acting as a chaperone to:
o
Facilitate proper protein folding within the endoplasmic reticulum.
o
Enable proper function of toll-like receptors and the innate immune system.
o
Carry irreparable proteins to intracellular garbage disposals to be degraded into peptides (short chains of amino acids – that are protein fragments).
·
Loading peptides onto another class of proteins known as MHC I molecules. MHC I molecules move to the cellular surface where they are monitored by the immune system.
HSP gp96 is one of the most abundantly expressed proteins in the human body and is expressed by all cells. It is normally retained within cells in a compartment called the endoplasmic reticulum (ER), where it facilitates the folding of newly synthesized proteins so that they may perform their various tasks properly. Gp96 is particularly important in the process of detecting antigens as it is present in all cell types and, it is able to recognize all antigens. It also induces the immune system to activate CD8+ (“killer”) T cells which then seek out and destroy the cells that are marked by antigens. Gp96 is normally only contained inside the ER of cells, however when a cell dies an abnormal death through necrosis it breaks open and releases gp96 into the surrounding tissue microenvironment.ImPACT works by modifying the chemical structure of gp96 so that a cell can continuously secrete it into the extracellular space accompanied by the unique peptide that it is folding at the time without causing necrosis. This allows the immune system to seek out and destroy cells characterized with antigens before the body would otherwise have detected them.
ImPACT Technology Overview
A limitation of utilizing gp96 as a cancer immunotherapy is that it is normally retained within cells by a small region called a “KDEL sequence” that acts like a “leash”, preventing gp96 from leaving the ER. Therefore, in order to utilize gp96 as a therapeutic, gp96 must either be purified from individual cells or engineered to be secreted from cells.
8
To overcome this limitation, a team of scientists led by Eckhard Podack, MD, Ph.D., the Chairman of our Scientific Advisory Board and the inventor of our technology, deleted this KDEL sequence and replaced it with another sequence that causes the new fusion protein, called gp96-Ig, to be secreted from cells continuously. Multiple tumor cell lines were then made to express gp96-Ig, and as expected, secreted it continuously into the extracellular space in a complex with tumor proteins. Dr. Podack demonstrated in the laboratory that gp96-Ig vaccination effectively cross-presented tumor specific antigens to immune cells, led to expansion of Cytotoxic T Lymphocytes (CTL) and the subsequent rejection of injected tumor cells. Importantly, these studies demonstrated that the secreted protein gp96-Ig maintained the critical characteristics of the native gp96 protein required to generate anti-tumor immune responses. Thus,in vitro proof-of-principle was established that the innovation, gp96-Ig, not only retained the desired properties of the native gp96 protein, but enhanced those functions and led to tumor-killing immune responses.
Our ImPACT technology platform:
·
Effectively cross-presents tumor antigens and leads to cytotoxic killer T cell activation
Published studies in mice showed that killer T cell activation was approximately 10 million times greater withImPACT secreted gp96-Ig than with a corresponding gp96 protein injection. The modified cell secretes gp96 in a sustained release for several days after injection. This creates a sustained immune response. These data suggest that gp96-chaperoned peptides may represent the most efficient, robust pathway for presenting a cell’s antigens to the immune system and activating killer T cell.
·
Binds and presents all potential tumor antigens to the immune system simultaneously
A single type of tumor (or virus) might have multiple strains derived from numerous tumor cells. These different strains have different antigens, all of which are capable of initiating an immune response. By creating a vaccine from a native tumor-cell line, we believe thatImPACT’s technology can develop a therapy that shares many common features with patients’ tumors of the same origin. We believe this “blanket” approach will provide each patient with a higher likelihood of a positive response to the therapy.
·
Features killer T cell activation that is independent of CD4+ T cell help
Animal studies have confirmed that our technology initiates a mechanism called cross-presentation that is critical to inducing tumor rejection. Importantly, it does this independently and successfully without additional CD4+ T cell (also known as a helper T cell) recruitment, which is typically required in a normal immune system response. This is particularly important in cancer and HIV because helper T cell activity is frequently impaired in these disease states.
·
May cause few side effects
We believe our technology allows the body to recognize cancer as a foreign entity and uses the body’s natural immune mechanism to recognize and fight it. In doing so, we believe our product candidates will generate fewer side effects than conventional chemotherapy and that patients will be able to maintain a higher quality of life.
The distinguishing characteristics ofImPACT are:
(i)
While most other immunotherapy approaches target only a single antigen,Heat’s patented approach uses modified heat shock proteins to stimulate an immune response against multiple antigens contained within cancer cells (both known and unknown). Cancer cells express different antigens that can be used to initiate an immune response. EachImPACT vaccine is created from a native tumor-cell line that we believe expresses the widest array of antigens common to a particular type of cancer. We believe this “pan-antigen” approach provides each patient with a higher likelihood of a response to the therapy.
9
(ii)
Heat’s product candidates are made from “off-the-shelf” (allogeneic) cells and may therefore beless expensive to manufacture than patient-specific (autologous) vaccines. Heat’s vaccines are mass-produced from a single source while other immunotherapy approaches require physicians to extract a patient’s blood and/or cells, send them to a facility where a personalized vaccine is created, and then have them shipped back to the physician for injection into the patient.
(iii)
While competing companies are developing therapies that are both “off-the-shelf” and which target multiple antigens,Heat’sImPACT technology is the onlyknown “off-the-shelf” (allogeneic)vaccine to us that directly induces “cross-presentation” to the CD8+ (“killer”) T cells, which are the cytotoxic arm of the immune system. Stimulating these CD8 (killer) T cells through “cross-presentation” has recently been shown to be critical to the induction of effective anti-tumor immunity. We believe our product candidates are able to leverage gp96 to serve as their own powerful immune stimulant (adjuvant) while other companies’ technologies rely on the use of a secondary adjuvant like GMCSF or Alum.
Our Product Candidates and Clinical Development Programs
We have initiated development programs to target ourImPACT technology platform against a range of diseases, including non-small cell lung cancer and bladder cancer. We have submitted a Phase 2 protocol to our open IND with our first therapeutic vaccine, HS-110, against NSCLC in March 2014, and we initiated a Phase 1/2 clinical trial for bladder cancer in Q4-2013. Our lead scientist has also completed a study in primates for the development of a therapeutic and prophylactic vaccine for the treatment and prevention of HIV. This study continues to be fully funded by the NIH. The HIV trials were initiated by the primary inventor and to date have been funded by grants awarded to the primary inventor, which can be used in the discretion of the inventor. We have no funding obligation for such trials and the primary inventor is responsible for future development and research; nonetheless any research conducted by the primary inventor contributes to our body of research and we may choose to progress any such research to further clinical trials and incorporate such research into our future development plans.
Summary of HS-110 Clinical Trial
Phase 1 HS-110 Clinical Trial
Background
A Phase 1 clinical trial with HS-110 in patients with very late stage IIIB/IV NSCLC was undertaken by the inventor of the technology which we license at the Sylvester Comprehensive Cancer Center with a total of 18 patients dosed, 15 of which completed the first course of three planned courses of therapy and were evaluated. Two of these 15 patients completed all three planned courses. The primary purpose of this trial was to evaluate safety of HS-110, while the secondary objectives were to study gp96-Ig specific immune responses and to monitor clinical progress. The patients were divided into 3 arms. Due to statistical and safety considerations and early termination of the study, the patients in the trial were not evenly divided among the three arms. Arm 1, which consisted of 11 patients, received 40 million cells every two weeks for 18 weeks, arm 2, which consisted of 4 patients, received 20 million cells every week for 18 weeks and arm 3, which consisted of 3 patients, received 10 million cells twice a week for 18 weeks. Three of the patients, who were late stage lung cancer patients, died before their immune response could be evaluated and were not included in the evaluation set at the end of the trial.
The Phase 1 trial was conducted under an investigator-sponsored IND and was fully funded by the NIH. The main criteria for inclusion were: (i) patients with histologically confirmed NSCLC stage IIIB, stage IV, or recurrent disease; (ii) at least one site of bi-dimensionally measurable disease; (iii) treated brain metastasis must be stable by CT scan or MRI for at least 8 weeks; (iv) patient must have received and failed at least two lines of therapy (one of them erlotinib); (v) age≥ 18 years; ECOG performance status 0-2; life expectancy≥ 3 months; and (vi) signed informed consent.
The median age was 67 years (range 38-86). HS-110 showed no overt toxicity. There were no serious adverse events (SAEs) that were considered by the trial investigator to be treatment-related. Most of the adverse events (AEs) were reported as mild or moderate (grade 1 or 2) with the most frequent being skin induration and rash that were transitory and usually resolved in 1 to 2 weeks.
10
HS-110 provides evidence of a CD8-CTL IFN-γ immune response in patients with advanced NSCLC. In 11 of the 15 patients (73%) that completed the first course of therapy with HS-110, there was a twofold or greater increase in CD8 cells secreting interferon gamma (CD8-CTL IFN-γ). These patients also exhibited an estimated median survival of 16.5 months (95% CI:7.1-20.0). In contrast, 4 patients were immune non-responders and survived 2.1, 2.3, 6.7, and 6.7 months, or a median survival of 4.5 months, which is consistent with the expected survival times in this patient population. The protocol required that we look for such responses, but, as is typical in immunotherapy, no partial or complete tumor responses were observed. The median one-year overall survival rate of patients in the study was 44% (95% CI:21.6-65.1). For comparative purposes, while there was a wide range of survival times, the one-year overall survival rate in a published one-year, 43-patient, advanced lung cancer population was 5.5%. One of the late-stage lung cancer patients survived over four years since starting the therapy and another patient survived over three years since starting the therapy. These findings were consistent with multiple pre-clinical published studies onImPACT therapy.
HS-110 Safety
We believe HS-110 showed no overt toxicity. There were no serious adverse events (SAEs) that were considered by the trial investigator to be treatment-related. Most of the adverse events (AEs) were reported as mild or moderate (grade 1 or 2) with the most frequent being skin induration and rash that were transitory and usually resolved in 1 to 2 weeks. The single grade 3 AE was in the “Body as a Whole” category (fatigue) and was rated as “possibly” related. There were no immune-related events with the vaccine or the vaccinations.
Skin reactions at the vaccination site were minimal and of short duration and there was no evidence of the generation of any autoimmune phenomena. In lieu of a dose escalation design, the design of the Phase I trial involved increasing the frequency of vaccination, while still retaining the total dose of vaccine cells administered. A more frequent vaccination schedule caused increased tumor rejection in preclinical models.
Adverse Events by Body System
| | |
Body System | Number of Events (N=219) | Severity Grade (# of events) |
Injection Site Reactions | 166 (75.8%) | Grade 1 (166) |
Respiratory System | 9 (4.1%) | Grade 2(5) |
Body as a Whole (general disorders including fever) | 8(3.7%) | Grade 1(4) Grade 2(3)a Grade 3(1)b |
Nervous System | 8(3.7%) | Grade 2(1) |
Musculoskeletal | 7(3.2%) | Grade 2(5) |
Digestive System | 7(3.2%) | Grade 1(7) |
Metabolic and Nutrition | 6(2.7%) | Grade 1(6) |
Skin and Appendages (non-injection site reactions) | 4(1.8%) | Grade 2(1) |
Cardiovascular System | 2(0.9%) | Grade 2(1) |
Urogenital System | 1(0.5%) | Grade 1(1) |
Endocrine System | 1(0.5%) | Grade 2(1) |
Hemic and Lymphatic | — | — |
———————
a
All grade 2 AEs except 4 were classified as non-related to treatment. The grade 2 treatment-related AEs were 1 musculoskeletal event (joint pain) rated as definitely related. 1 musculoskeletal event (knee weakness) rated as possibly related. 1 endocrine event (hot flashes) rated as unlikely related and 1 skin event (pruritus) rated as unlikely related.
b
The single grade 3 AE was in the body as a whole category (fatigue) and was rated as possibly related.
11
Injection Site Reactions
| |
Injection Site Reaction (ISR) | Number of Events (N = 166) |
Pain | 17 (10%) |
Induration | 58 (35%) |
Pruritus | 8 (5%) |
Hyperpigmentation/Discoloration | 3 (2%) |
Rash | 78 (47%) |
ISR non-specific | 2 (1%) |
Positive Immunological Response
In 11 of the 15 patients (73%) completing the first course of therapy with HS-110, there was a twofold or greater increase in CD8 cells secreting interferon gamma (CD8-CTL IFN-γ) following vaccination.

|
CD8 IFN-γ response. Samples from 15 patients collected for immune response at baseline and after at least one course of vaccination were available for analysis of the CD8 IFN-γ response. 20,000 purified patient CD8 T cells were stimulated with vaccine cells for 40h in ELI-spot plates and the frequency of IFN-γ secreting cells determined. + indicates first increase. Solid indicate immune response (IR+), dashed lines no response (IR− ). |
Since NSCLC is known to be highly immunosuppressive, we believe that by overcoming tumor-induced-suppression with frequent vaccinations as observed anecdotally in the Phase 1 study and the generation of an observed potent polyepitope specific CD8 CTL is encouraging and warrants further study.
12
Clinical Response
Seven of 15 patients completing the first course of therapy (39%; 95% CI: 17.3- 64.3%) achieved disease stabilization after the first course of vaccinations (6 weeks) and 8 patients had disease progression. While the protocol required that we look for such responses, as is typical in immunotherapy, no partial or complete tumor responses were noted in the study. Although clinicians and patients may perceive disease stabilization as beneficial, without a control arm the FDA does not consider it to be a clinical benefit for regulatory purposes. In order to obtain FDA approval, we will be required to show an improvement in progression-free survival (or, PFS) or overall survival (or, OS) when compared to a control arm in a randomized study. The Kaplan Meier estimate of median time to progression was 1.4 months (95% CI: 1.3-2.7), and the PFS rates at 1, 2 and 3 months were 88.9% (95% CI: 62.4- 97.1%), 38.9% (95% CI: 17.5-60.0%), and 11.1% (95% CI: 1.9-29.8%), respectively. Of note, two patients remained progression free for just over 7 months.
The typical median survival period for late-stage lung cancer is 4.5 months for patients who are not receiving any treatment. Two of the fifteen patients who completed the first course of therapy were followed for over 3 years and 4 years, respectively. The Kaplan-Meier estimate of median overall survival was 8.1 months (95% CI: 6.7- 18.2), and the 1, 2, and 3-year OS rates were 44.4% (95% CI: 21.6-65.1%), 19.0% (95% CI: 4.8- 40.3%), and 9.5% (95% CI: 0.8-32.1%), respectively. While these results may be encouraging, apparent differences in outcome between population-based survival estimates and treatment groups from a clinical study can arise from differences other than drug treatment. The reliability of such comparisons must also be considered in light of the unblinded nature of the study data at the time that the comparator was chosen. Moreover, the wide range of values in the 95% confidence intervals in our study suggests that the actual median survival times could lie anywhere in the reported intervals.
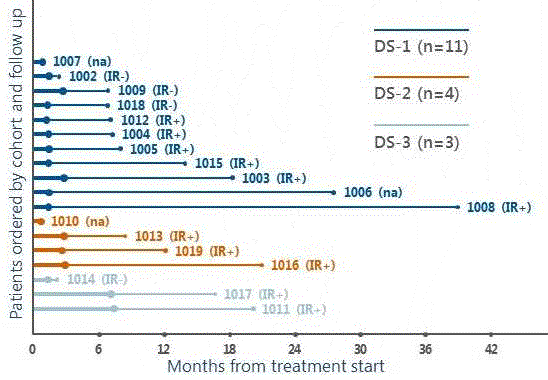
|
Time to progression (thick line) and additional follow up (thin line) by dose-schedule cohort. Patients are shown within cohort in order of increasing follow up (shortest at top). Filled diamonds indicate disease progression; open diamonds indicate stable disease at last assessment. Filled circles indicate death; open circles last follow up of surviving patients. IR+: more than twofold increase in CD8 from baseline. IR− : no CD8 immune response. na: not assessed for immune response. |
In 11 of the 15 patients (73%) completing the first course of therapy with HS-110, there was a twofold or greater increase in CD8 cells secreting interferon gamma (CD8-CTL IFN-y) following vaccination. In a non-prespecified analysis, the responders saw a threefold increase in median overall survival compared to the non-responders on the trial.
13

Summary
In summary, based on the results of this Phase 1 trial in 18 patients, we believe HS-110 showed no overt toxicity and appears to be capable of generating CD8-CTL IFN-γ immune responses in patients with advanced NSCLC. These results are encouraging and may be predictive of clinical benefit based on stabilization of disease, overall survival and the immune responder results.
ONCOLOGY INDICATIONS ofImPACT
Lung Cancer
Disease
Lung cancer is the leading cause of cancer-related death in the United States. According to the National Cancer Institute, in 2013, lung cancer is expected to account for 26% of all female cancer deaths and 28% of all male cancer deaths. An expected 228,190 people will be diagnosed with lung cancer in the United States in 2012. Of these lung cancers, roughly 85% will present as non-small cell lung cancer. Patients with advanced clinical stage IIIB/IV disease visible on chest radiography have a 5-year survival rate as low as 1-5%.
14
Clinical Development
The technology that we license was the subject of an investigator initiated Phase 1 clinical trial conducted at the Sylvester Cancer Center for the treatment of non-small cell lung cancer (“NSCLC” or “lung cancer”) to establish safety and proof of concept clinical efficacy.
After completion of the 18 patient Phase 1 trial, in which 15 patients completed the first course of three planned course of therapy and were evaluated, we successfully opened a new IND to conduct additional trials with HS-110 in patients with NSCLC. Our Phase 2 study, which has been submitted to the FDA, has been designed to investigate the combination of HS-110 with low dose cyclophosphamide followed by sequential chemotherapy versus chemotherapy alone in third-line NSCLC patients. The trial is structured as a multicenter randomized, study to evaluate the immune response, safety and efficacy endpoints of HS-110 when administered weekly for 12 weeks in combination with low-dose cyclophosphamide in an induction period followed by monotherapy HS-110 every three weeks during maintenance. Upon first progression, patients will be treated with a regimen from the list of allowable chemotherapies with continued administration of HS-110 for up to 2 years. Patients randomized to the comparator arm will be treated with one chemotherapy regimen until first progression and then switched to an alternate chemotherapy regimen until second progression. Blood samples will be taken to evaluate the immune response and their correlation to overall survival, and where considered appropriate by the investigator, patients will be invited to consent for pre- and post-treatment biopsies for exploratory biomarker analysis. The primary endpoint is overall survival; secondary endpoints follow objective responses and immune response. The trial will enroll 123 patients at approximately 20-30 investigative centers over 2 years. We anticipate recruitment to begin in Q3-2014.
In addition to our Phase 2 study, our chief scientist has received a grant award from the Marcus Foundation that fully funds a 36 patient Phase 1/2 investigator-sponsored Phase 1/2 study for use of HS-110 as a combination therapy with theophylline and oxygen. This study is anticipated to begin during Q2 2014 and is listed as identifier NCT01799161.
Bladder Cancer
Disease
In the United States, bladder cancer is the fourth most common type of cancer in men and the ninth most common cancer in women. According to the National Institutes of Cancer, 1 in 42 men and women will be diagnosed with bladder cancer during their lifetime, a total of more than half a million patients in the US. There are more than 60,000 cases of bladder cancer diagnosed each year in the United States, resulting in over 14,000 deaths per year. Available treatments are currently not effective, thus this remains an area of high unmet need.
Clinical Development
The Bladder Cancer Phase 1/2 Trial
We opened an IND in support of HS-410 for bladder cancer with no clinical hold. The first protocol submitted to the IND is a 93 patient, Phase 1/2 trial to examine safety, tolerability, immune response and preliminary clinical activity of HS-410 in patients with high risk, superficial bladder cancer who have completed surgical resection and 3-6 weekly intravesical bacillus Calmette-Guérin (BCG) immunotherapy instillations. We anticipate including approximately 10-15 clinical sites with an enrollment period of 18-24 months.
The Phase 1 portion will enroll two cohorts of 9 patients each to either a high or low dose group. Patients will receive weekly intradermal injections of HS-410 for 12 weeks followed by 3 monthly injections, and immune response will be evaluated at baseline, week 7, week 14 and week 29. The first 3 patients in each dose group will be enrolled at 2 week intervals to allow opportunity to assess safety and tolerability of HS-410. At the completion of the Phase 1 portion of the study, the dose resulting in the optimal safety and immune response will be advanced to Phase 2. In the Phase 2 portion, 75 patients will be enrolled in 2:1 fashion to HS-410 or placebo. Primary endpoint will examine time to 1st recurrence of bladder cancer. Other endpoints will include recurrence rate, progression rate and immune response.
15
Other Cancers
OurImPACT -technology is a broad based approach and can be used to combat a variety of cancers. We continue to evaluate other indications for ourImPACT therapeutic vaccines and have developed a cell line for ovarian cancer and one for triple negative breast cancer. Our decision to further pursue these or any additional product candidates other than our two lead product candidates will be based in part upon available funding and partnering opportunities.
Infectious Diseases
To date, over $4,000,000 in governmental and institutional funding has been provided to the inventor of the technology we license for HIV and hepatitis C virus (HCV) research using ourImPACT -technology. We do not intend to use any of our current funds to further any HIV or HCV research and instead plan to conduct additional research with respect to the use of ourImPACT-technology for the treatment of such diseases solely through additional governmental and institutional grants, if any, that may be received.
Manufacturing
We rely on third-party manufacturers to produce and store our product candidates for clinical use and currently do not own or operate manufacturing facilities.
We have retained Lonza Walkersville, Inc. a vendor, who has begun production of HS-110 to be used in Phase 2 and our potential Phase 3 clinical trials. We entered into an eight year Manufacturing Services Agreement, dated October 19, 2011, with the vendor (the “Manufacturing Agreement”). The Manufacturing Agreement provides that the vendor will manufacture products based on ourImPACT technology intended for use in pharmaceutical or medicinal end products, including, without limitation, products in a final packaged form and labeled for use in clinical trials or for commercial sale to end users in accordance with the terms and conditions of individual statements of work. The Manufacturing Agreement requires that we purchase certain minimum amounts each year from the vendor. The Manufacturing Agreement may be terminated by the parties upon mutual agreement, and by each party for a material breach by the other party that is not cured within the cure period, upon notice that a clinical trial for which product is being produced under the agreement is suspended or terminated or upon the other party’s insolvency, dissolution or liquidation.
The HS-110 used in the inventor’s Phase 1, and planned for use in our Phase 2 clinical trial and HS-410 used in our Phase 1/2clinical trial was and is currently manufactured under current good manufacturing practices, or cGMP. The vaccine is grown in large quantities and quality tested according to FDA guidelines. Following testing, the vaccine is irradiated, which is a commonly used attenuation process that eliminates the ability of the gp96-Ig-containing vaccine cell lines to replicate but allows them to continue secreting gp96-Ig for a period of several days. Quality tested, irradiated batches of the vaccine are then dispensed into individual doses and frozen in liquid nitrogen. These batches of frozen vaccine are stable for long periods of time, and are thawed immediately prior to administration to patients. Sufficient material to initiate the first few patients in the HS-110 Phase 2 study has already been produced, and preparations are underway to produce quantities required for trial completion and subsequent clinical trials. Sufficient material to complete the Phase I portion of the HS-410 Phase1/ 2 study has already been produced, and preparations are underway to produce quantities required for trial completion and subsequent clinical trials.
Competition
The pharmaceutical industry and biologics industry are each highly competitive and characterized by a number of established, large pharmaceutical companies and other companies, as well as smaller companies like ours. If our competitors market products that are less expensive, safer or more effective than any future products developed from our product candidates, or that reach the market before our approved product candidates, we may not achieve commercial success. Technological developments in our field of research and development occur at a rapid rate and we expect competition to intensify as advances in this field are made. We will be required to continue to devote substantial resources and efforts to our research and development activities.
16
As a biotech company with a cancer immunotherapy as its lead therapeutic, we compete with a broad range of companies. At the highest level, cancer immunotherapy can be seen as both a complement and a potential competitor to any oncology therapy, most notably chemotherapy, biologics and small molecule drugs. Not only do we compete with companies engaged in various cancer treatments including radiology and chemotherapy but we also compete with various companies that have developed or are trying to develop immunology vaccines for the treatment of cancer. Certain of our competitors have substantially greater capital resources, large customer bases, broader product lines, sales forces, greater marketing and management resources, larger research and development staffs and larger facilities than we do and have more established reputations as well as global distribution channels. Our most significant competitors, among others, are fully integrated pharmaceutical companies such as Eli Lilly (Alimta), Bristol-Myers Squibb (Erbitux) and Sanofi-Aventis (Eloxatin), and more established biotechnology companies such as Roche/Genentech (Avastin and Tarceva), and competing cancer immunotherapy companies such as Dendreon, New Link Genetics and others which have substantially greater financial, technical, sales, marketing, and human resources than we do. These companies might succeed in obtaining regulatory approval for competitive products more rapidly than we can for our products. In addition, competitors might develop technologies and products that are less expensive, safer or more effective than those being developed by us or that would render our technology obsolete. In addition, the pharmaceutical and biotechnology industry is characterized by rapid technological change. Because our research approach integrates many technologies, it may be difficult for us to remain current with the rapid changes in each technology. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Our competitors may render our technologies obsolete by advancing their existing technological approaches or developing new or different approaches.
We expect to compete with other pharmaceutical and biotechnology companies, and our competitors may:
·
develop and market products that are less expensive, more effective or safer than our future products;
·
commercialize competing products before we can launch any products developed from our product candidates;
·
operate larger research and development programs, possess greater manufacturing capabilities or have substantially greater financial resources than we do;
·
initiate or withstand substantial price competition more successfully than we can;
·
have greater success in recruiting skilled technical and scientific workers from the limited pool of available talent;
·
a more effectively negotiate third-party licenses and strategic relationships; and
·
take advantage of acquisition or other opportunities more readily than we can.
We expect to compete for market share against large pharmaceutical and biotechnology companies, smaller companies that are collaborating with larger pharmaceutical companies, new companies, academic institutions, government agencies and other public and private research organizations.
Many major pharmaceutical companies have at least one immunotherapy drug or therapeutic in development, either directly or in partnership with a smaller biotech firm. Some of our competitors that are developing competitive immunology drugs and therapeutics include Merck kGaA/Oncothyreon’s Stimuvax for the treatment of breast cancer and NSCLC; Transgene and its product TG4010 for the treatment of NSCLC lung cancer; GlaxoSmithKline and its product MAGE-A3 for the treatment of melanoma, NSCLC, multiple myeloma and squamous cell carcinoma; Oxford BioMedica and its product TroVax for the treatment of prostate, kidney and colorectal cancer); NewLink Genetics and its HyperAcute treatments for pancreatic cancer, lung cancer, melanoma and renal cell cancer; Celldex/Pfizer and their product CDX-110 for the treatment of malignant brain cancer; and Dendreon and its product Provenge for the treatment of prostate cancer.
17
The primary treatments for non-small cell lung cancer are surgery, radiation, chemotherapy and various combinations of each of these treatments. A large number of patients, particularly with advanced disease, are refractory to these treatments and are subsequently treated with a number of emerging biologic agents, including immunotherapy. Some examples of therapies commonly attempted with stage IIIB/IV NSCLC patients include: Alimta (pemetrexed), Avastin (bevacizumab), Tarceva (EGF inhibitor), Gemzar (gemcitabine), Erbitux (cetuximab), Carboplatin, Taxol, VP16 and Arlibercept. It is unlikely that biologic agents will compete with more traditional therapies in the short-term, but many oncologists believe that such therapies will eventually become the mainstay of lung cancer therapy. None of these agents have proven particularly effective for stage IIIB/IV NSCLC patients, with the most effective therapies only increasing survival by a few months. As a result, we do not consider these agents to be direct competitors to HS-110 because they are likely to be given either in sequence or in conjunction with some of the agents listed. Furthermore, many patients cannot tolerate many of the chemotherapeutics listed. Thus, we believe if HS-110 has a positive safety profile (without observation of local or systemic toxicities, none of which have been seen to date), it is likely that HS-110 would be preferred both by physicians and patients in this stage of disease.
As previously stated we compete with other forms of cancer treatment such as biologic therapies in addition to immunology therapies. There are several biologic therapies in clinical development against NSCLC that have been identified as potential competitors to HS-110. In particular, a cell-based vaccine therapy, Lucanix, is in development by NovaRx. Lucanix has recently completed Phase 3 clinical trials and failed to reach the primary endpoint, although these data have yet to be formally published.
.
Our strategy is to emphasize what we believe to be our competitive advantages which are that the therapy will have less side effects than most other chemotherapies, will be available at lower prices than other therapies and will work on almost all types of cancer and not just one specific type.
Although all chemotherapy drugs have severe side-effects such as overall damage to the immune system, not only to cancerous cells, leading to hair loss, nausea and vomiting, and considerable pain, etc., the side effects from immunotherapy are typically reduced because immunotherapy works with the body’s own immune response.
According to Schreiber et al, patient-specific vaccines are not more effective than off-the shelf vaccines in reducing tumors. Furthermore, patient-specific vaccines cost far more to produce than off the shelf (allogeneic) vaccines, where any donor tissue can be used. Over 95% of newly developed cancer immunotherapies cost over $20,000 per course of treatment and we expect that our treatment will be less expensive.
Grant Funding
To date, in excess of $14,000,000 in grants, have been awarded to the primary inventor of the technology we license to fund development ofImPACT technology and clinical trials upon which our clinical programs are based. We have little control over the direction of the NIH grant funds that have been received by the primary inventor of the technology we license and since payment is made to the inventors as opposed to us we do not recognize any revenue from such grant funds nor do they fund any expenses that we incur. Although earmarked for further development of the technology that we license, any funds awarded to the primary inventor are used in his discretion and we have little control over his use of the funds. Our strategy is to continue to apply for grants that will enable us to leverage our core technology platform. We have applied for grant funding from the NIH, DOD, CPRIT and other public and private foundations to be used for our research and development activities. We have applied for a grant in the amount of approximately $19,000,000 from CPRIT to be matched with $6,000,000 from us to be used to expand our bladder cancer clinical trial and commence other research and development activities. Although we have recently been granted a meeting with CPRIT to further discuss our grant application, there can no assurance that such grant funding will be awarded to us. Our primary inventor also applies for academic grants to enhance the core technology platform. Grant funds received by our primary inventor are not utilized by us. Rather, these funds support our primary inventor's academic interests and may benefit us to the extent that these grants enable him to enhance the technology platform or generate additional data to support our programs. Currently, our primary inventor's academic grants are supporting the HS-110 NSCLC combination study as well as the HIV study. All other clinical programs, including our Phase 2 NSCLC study and our Phase 1/2 bladder cancer study are supported by us.
18
Previous Grant awards for development ofImPACT
| | |
Grant Title | Granting Organization | Amount |
Regulation of Anti-Tumor Immunity | NIH | $6,187,904 |
Molecular Mechanism of Anti-Tumor and Anti-Bacterial Cytotoxicity | NIH | $897,295 |
Mechanisms of mucosal protection by HPV-SIV and gp96-Ig-SIV vaccines | NIH | $2,000,000 |
Systemic and mucosal HIV-immunity by HSP-gp96 vaccines | NIH | $451,410 |
Induction of mucosal SIV immunity in non-human primates by secreted HSP-gp96 | NIH | $2,124,733 |
Clinical Translation of Gene Therapy for Lung Cancer Award Recipient | Alliance for Cancer Gene Therapy | $1,000,000 |
Clinical Translation of Gene Therapy for Lung Cancer Award Recipient | State of Florida | $100,000 |
QTDP Grant | Dept. of Treasury | $244,479 |
Use of HS-110 as a Combination Therapy with Theophylline and Oxygen in Advanced Lung Cancer Patients | Marcus Foundation | $840,000 |
Intellectual Property
License Agreements and Intellectual Property
Our goal is to obtain, maintain and enforce patent protection for our products, formulations, processes, methods and other proprietary technologies, preserve our trade secrets and rights in our unique biological materials, and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. Our policy is to actively seek to obtain, where appropriate, the strongest intellectual property protection possible for our current product candidates (ImPACT therapy) and any future product candidates, proprietary information and proprietary technology through a combination of contractual arrangements and patents, both in the U.S. and abroad. However, even patent protection may not always afford us with complete protection against competitors who seek to circumvent our patents. See “Risk Factors - Risks Relating to Our Business” – “We have limited protection of our intellectual property.”
We will continue to depend upon the skills, knowledge and experience of our scientific and technical personnel, as well as that of our advisors, consultants and other contractors, none of which is patentable. To help protect our proprietary know-how, which is not patentable, and for inventions for which patents may be difficult to enforce, we currently rely and will in the future rely on trade secret protection and confidentiality agreements to protect our interests. To this end, we require all of our employees, consultants, advisors and other contractors to enter into confidentiality agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business.
19
License Agreements
In July 2008, we entered into an exclusive license agreement with the University of Miami (the “University”) for intellectual and tangible property rights relating to ourImPACT, technology. This license agreement was subsequently assigned to our subsidiary Heat Biologics I, Inc. which issued to the University shares of its common stock representing seven and one half percent (7.5%) of its common stock. The term of the license is the length of the last to expire patent, unless terminated earlier. The license agreement grants Heat Biologics I, Inc. exclusive, worldwide rights to make, use or sell licensed materials based upon the following patent-related rights:
·
U.S. patent applications: Serial number 60/075,358 (the “ ‘358 application”) entitled “Modified Heat Shock Protein-Antigenic Peptide Complex” and filed on February 20, 1998; Serial number 09/253,439 (the “ ‘439 application”) entitled “Modified Heat Shock Protein-Antigenic Peptide Complex ” and filed on February 19, 1999; serial number 11/878,460 (the “ ‘460 application”) entitled “Recombinant Cancer Cell Secreting Modified Heat Shock Protein-Antigenic Peptide Complex” and filed on July 24, 2007; and all U.S. patents and foreign patents and patent applications based on these U.S. applications; as well as all divisionals, continuations, and those claims in continuations-in-parts to the extent they are sufficiently described in the ‘358, ‘439, or ‘460 applications of the foregoing, and any re-examinations or reissues of the foregoing (the “GP96 Vaccine Technology Portfolio”).
As consideration for the rights granted in the license agreement, the licensee is obligated to pay the University upfront license fees, additional yearly and milestone payments and a royalty based on net sales of products covered by the patent-related rights set forth above. More specifically, the licensee is obligated to pay the University (i) all past and future patent costs associated with the licensed patent-related rights; (ii) a license issue fee of $150,000; (iii) annual payments of $10,000 in 2010, 2011 and 2012, and $20,000 each year thereafter; (iv) a milestone payment of $250,000 by the earlier of May 31, 2017 or approval of a BLA for the lung cancer vaccine or for a cancer vaccine other than lung cancer; and (v) royalties equal to a percent (in the low-to-mid single digits) of net sales of licensed products. The royalty rate is subject to reduction if additional license rights from third parties are required to commercialize licensed products. In the event of a sublicense to a third party, Heat Biologics I, Inc. is obligated to pay royalties to the University equal to a percentage of what it would have been required to pay to the University had it sold the products under sublicense itself. In exchange for additional consideration, the University agreed to postpone the payment due dates of this license agreement.
In February 2011, our subsidiary, Heat Biologics I, Inc., entered into four additional exclusive license agreements with the University. The terms of each of these additional licenses runs until all the patent-related rights licensed therein have expired, unless terminated earlier. In of these additional exclusive license agreements, Heat Biologics I, Inc. obtained exclusive, worldwide rights to make, use or sell products covered under the following patent-related rights:
·
U.S. patent application serial number 61/347,336 entitled “Cancer Treatment” and filed on May 21, 2010, all U.S. patents and foreign patents and patent applications based on these U.S. applications; as well as all divisionals, continuations, and those claims in continuations-in-parts (to the extent they are sufficiently described in the applications) of the foregoing, and any re-examinations or reissues of the foregoing (the “Cancer Treatment Portfolio”).
·
U.S. patent application serial number 61/033,425 entitled “Allogeneic Cancer –Based Immunotherapy” and filed on March 3, 2008 and PCT application number PCT/2009/001330 entitled “Allogeneic Cancer –Based Immunotherapy” filed on March 3, 2009, all U.S. patents and foreign patents and patent applications based on these U.S. applications as well as all divisionals, continuations, and those claims in continuations-in-parts (to the extent they are sufficiently described in the applications) of the foregoing, and any re-examinations or reissues of the foregoing (the “Allogeneic Cancer –Based Immunotherapy Portfolio”).
·
U.S. patent application serial number 61/033,425 entitled “Heat Shock Protein GP96 Vaccination and Methods of Using Same” filed on March 20, 2008 and PCT application number PCT/ 2009/001727 entitled “Heat Shock Protein GP96 Vaccination and Methods of Using Same “filed on March 19, 2009, all U.S. patents and foreign patents and patent applications based on these U.S. applications as well as all divisionals, continuations, and those claims in continuations-in-parts (to the extent they are sufficiently described in the applications) of the foregoing, and any re-examinations or reissues of the foregoing (the “Heat Shock Protein GP96 Vaccination Portfolio”).
20
·
U.S. patent application serial number 61/116.971 entitled “HIV/SIV Vaccine for the Generation of Mucosal and Systemic Immunity” filed November 28, 2008 and PCT application number PCT/ 2009/065500” entitled “HIV/SIV Vaccine for the Generation of Mucosal and Systemic Immunity” filed on November 23, 2009 all U.S. patents and foreign patents and patent applications based on these U.S. applications as well as all divisionals, continuations, and those claims in continuations-in-parts (to the extent they are sufficiently described in the applications) of the foregoing, and any re-examinations or reissues of the foregoing (the “HIV/SIV Vaccine Portfolio”).
As consideration for the rights granted in these additional four license agreements, the licensee is obligated to pay the University certain upfront license fees, past and future patent costs and royalties based on net sales of commercialized products covered by the patent-related rights set forth above. No annual or milestone payments are required under any of these four additional license agreements. The upfront license fees for the Cancer Treatment Portfolio and the HIV/SIV Vaccine Portfolio license agreements are $10,000 and $50,000, respectively. No upfront license fees were required under the license agreements for the Allogeneic Cancer–Based Immunotherapy and the Heat Shock Protein GP96 portfolios. Under each of these four additional license agreements, the royalties are equal to a percent (in the low-to-mid single digits) of net sales of products covered by the patent-related rights in the respective license agreements. These royalty rates are subject to reduction if additional license rights from third parties are required to commercialize licensed products. In the event of a sublicense to a third party, Heat Biologics I, Inc. is obligated to pay royalties to the University equal to a percentage of what it would have been required to pay to the University had it sold the products under sublicense itself. Each of these additional license agreements also provide that the licensee will not have to pay more than above royalty rates and sublicense fees if more than one license from the University is required to sell products covered by the licensed patent-related rights. In exchange for additional consideration (including the requirement that Heat Biologics I, Inc. pay additional milestone payments of $25,000 before initiation of any Phase 3 clinical trials for products covered by any of the license agreements, and an additional payment equal to 18% annual interest on the amounts due or a note convertible into an equivalent value of shares in Heat’s Preferred Stock), the University agreed to postpone the payment due dates for each of these four additional licenses.
All five of the above-described license agreements provide that the licensor has the right to terminate a subject license if the licensee has (i) not introduced, or at least use it best efforts to introduce, a licensed product in the commercial marketplace in the US, EU, or Japan by December 31, 2020; (ii) not otherwise exercise diligence to bring licensed products to market; or (iii) files, or has filed against it, a proceeding under the Bankruptcy Act, is adjudged insolvent, makes an assignment for the benefit of its creditors, or has an unreleased or unsatisfied writ of attachment or execution levied upon it.
In March 2014, our subsidiary, Heat Biologics I, Inc., entered into an additional exclusive license agreement with the University. The term of this license runs until all the patent-related rights licensed therein have expired, unless terminated earlier. In this exclusive license agreement, Heat Biologics I, Inc. obtained exclusive, worldwide rights to make, use or sell products covered under the University’s interest in the following patent-related rights:
·
U.S. Provisional Patent Application serial number 61/445,884 entitled “Combined Cell Based Gp96-IG-SIV/HIV; Recombinant Gp120 Protein Vaccination For Protection From SIV/HIV” and filed February 23, 2011 (the “884 application”); PCT Application Serial No. PCT/US2012/26256 entitled “Combined Cell Based Gp96-IG-SIV/HIV, Recombinant Gp120 Protein Vaccination For Protection From SIV/HIV” and filed February 23, 2012 (the “ ‘256 application”); and all U.S. patents and foreign patents and patent applications based on these applications; as well as all divisionals, continuations, and those claims in continuations-in-parts (to the extent they are sufficiently described in the ‘884 or ‘256 applications) of the foregoing, and any re-examinations or reissues of the foregoing (the “Combination HIV/SIV Vaccine Portfolio Portfolio”).
21
The patent rights in the Combination HIV/SIV Vaccine Portfolio are co-owned by the University and the National Institutes of Health (the “NIH”). Heat Biologics I, Inc. has only licensed the University’s rights therein. The NIH’s rights in this portfolio have not been licensed by Heat Biologics I, Inc. As consideration for the rights granted in this license agreement, the licensee is obligated to pay the University an upfront license fee, past patent costs, and royalties based on net sales on commercialized products covered by the patent-related rights set forth above. No annual payments are required under this license agreement. The licensee is obligated to make milestone payments under this license agreement as follows: $50,000 upon completion of a phase I clinical trial, $100,000 upon completion of a phase II trial, $100,000 upon completion of a phase III trial, and $100,000 upon acceptance of a BLA by the FDA or its foreign equivalent. Under this license agreement, the royalties are equal to a percent (low single digits) of net sales of products covered by the patent-related rights. This royalty rate is subject to reduction if additional license rights from third parties are required to commercialize licensed products. In the event of a sublicense to a third party, Heat Biologics I, Inc. is obligated to pay royalties to the University equal to a percentage of what it would have been required to pay to the University had it sold the products under sublicense itself. This license agreement also provides that the licensee will not have to pay more than the above sublicense fees or a royalty in the low-to-mid single digits if more than one license from the University is required to sell products covered by the licensed patent-related rights. The licensor has the right to terminate this license if the licensee has (i) not introduced, or at least use it best efforts to introduce, a licensed product in the commercial marketplace in the US, EU, or Japan by December 31, 2023; (ii) not otherwise exercise diligence to bring licensed products to market; or (iii) files, or has filed against it, a proceeding under the Bankruptcy Act, is adjudged insolvent, makes an assignment for the benefit of its creditors, or has an unreleased or unsatisfied writ of attachment or execution levied upon it.
Upon an uncured material breach of an obligation under any one of the above six license agreements by a party, the other party has the right to terminate that agreement upon 90 days notice or 30 days notice if the breach relates to payments due to the University. In the event of a termination, Heat Biologics I, Inc. will be obligated to pay all amounts that accrued prior to such termination. Each of the above license agreements also contains other customary clauses and terms as are common in similar agreements between industry and academia, including the licensee’s agreement to indemnify the University for liabilities arising out of the negligence of licensee, making the license grant subject to the Bayh-Dole act (35 U.S.C. 200 et seq.), the reservation of licensor of the right to use the licensed intellectual property rights for its internal, non-commercial purposes, limitations/disclaimers of various warranties and representations, reporting and record-keeping requirements, and licensee liability insurance requirements.
Under the above-described license agreements with the University, we have obtained exclusive rights to six different patent families. These families comprise six PCT applications, ten issued patents, two allowed patent applications, and forty-eight pending patent applications which cover the United States, Europe and Japan as well as several other countries having commercially significant markets. The six patent families associated with ourImPACT platform are:
“Recombinant cancer cell secreting modified heat shock protein-antigenic peptide complex.”
This family of patent filings relates to methods and compositions for enhancing an immune response. More particularly, the application describes the creation of a tumor cell therapy including a cancer cell that has been engineered to secrete a heat shock protein (gp96), and the use of such therapy to enhance an anti-tumor immune response. Within this family are one soon to be granted US patent, one pending US application, one granted Australian patent, one pending Australian patent application, three granted European patents (collectively validated in 28 countries), one pending Japanese application, and one granted Japanese patent. Not including any patent term adjustments or extensions (e.g., for patent office delays or extensions/exclusivity periods provided for new drug approvals in the US and some foreign countries), the term for patents in this family extends until 2019.
“Heat Shock Protein gp96 Vaccination and Methods of Using Same”
This family of patent filings also relates to methods and compositions for enhancing an immune response. It further describes: (a) how intraperitoneal gp96-Ig administration increases recruitment of innate immune cells into the administration site, mediates proliferation of dendritic cells (DCs) and CD8 cells, and activates natural killer (NK) cells; (b) that gp96-Ig-secreting cell vaccines are more effective when gp96-Ig is continuously released; (c) that frequent gp96 immunizations can overcome tumor-induced immune suppression and retards tumor growth; and (d) that B cell depletion can enhance gp96-Ig -mediated recruitment of NK cells and retention of DCs in the administration site. Within this family are one granted Australian patent, and one pending application each in the U.S., Canada, China, Europe, Israel, India, Japan, South Korea, and Hong Kong. Not including any patent term adjustments or extensions, the term for patents in this family extends until 2029.
22
“Allogenic Cancer Cell Based Immunotherapy”
This family of patent filings also relates to methods and compositions for enhancing an immune response. It further describes: (a) making vaccines cells allogeneic by expressing exogenous major histocompatibility complex (MHC) antigens; (b) B cell depletion to augment the effectiveness of the vaccines; and (c) the enhancement of anti-tumor immune responses using multiple immunizations less than two weeks apart. Within this family are one granted Australian patent, one granted U.S. patent, one granted European patent and one pending application each in the US, Canada, China, Europe, Israel, India, Japan, and South Korea. Not including any patent term adjustments or extensions, the term for patents in this family extends until 2029.
“Cancer Treatment”
This family of patent filings contains results from a Phase 1 clinical trial of human subjects with cancer. Within this family are one pending application each in the U.S., Australia, China, Europe, India, Israel, Japan, and South Korea. Filings in Canada and Hong Kong are intended to be made before the respective deadlines. Not including any patent term adjustments or extensions, the term for patents in this family extends until 2031.
“HIV/SIV Vaccines to Generate Mucosal and Systemic Immunity” This patent family relates to the use of host cells that have been engineered to secrete a heat shock protein (gp96) to treat various chronic viral infections including those caused by HIV. Within this family are one granted South African patent, two pending applications in the US, one allowed Australian application and one pending application Canada, China, Europe, India, the Philippines, Singapore, and Hong Kong. Not including any patent term adjustments or extensions, the term for patents in this family extends until 2029.
“Combined Cell Based Gp96-Ig-SIV/HIV, Recombinant Gp120 Protein Vaccination for Protection From SIV/HIV”
This patent family relates to combination therapies for treating chronic viral infections including HIV. The combination therapy uses host cells that have been engineered to secrete a heat shock protein (gp96) to induce antiviral T cell responses and soluble viral antigens to induce antiviral antibody responses. Within this family are one pending application each in the U.S., Australia, Canada, China, Europe, India, Japan, the Philippines, South Africa, and South Korea. Not including any patent term adjustments or extensions, the term for patents in this family extends until 2032.
In April 2013, we entered into an agreement with the University under which the University granted us an option to obtain an exclusive license to the following patent-related rights:
·
U.S. patent application serial number 12/303,036 entitled “Perforin-2 Proteins” filed December 2, 2008 and U.S. patent application serial number 61/637.455 entitled “Perforin-2 Defense Against Invasive and Multi-drug Resistant Bacteria” filed on April 21, 2012; all U.S. patents and foreign patents and patent applications based on these U.S. applications; as well as all divisionals, continuations, and those claims in continuations-in-parts (to the extent they are sufficiently described in the aforementioned applications) of the foregoing, and any re-examinations or reissues of the foregoing.
In consideration for the option, we are obligated to pay the University an option fee of $2,000 and to reimburse the University $3,000 for past patent costs. The term of the option is twelve months and is extendible so long as we continue to pay ongoing patent expenses. We are currently in the process of negotiating the terms of an exclusive license for this portfolio.
In addition to the licenses obtained from the University, we have entered into agreements with (i) the Regents of the University of Michigan (“U.Mich”); and (ii) the American Type Culture Collection (“ATCC”) for the evaluation of, acquisition of commercial rights to, certain biological materials.
23
In July 2011, we exercised an option agreement with U.Mich and entered into an exclusive license agreement with U.Mich to use, market, offer for sale, sell and/or sublicense materials and processes related to certain bladder cancer cell lines. The term of the license is perpetual, unless terminated earlier by us or by U.Mich. As consideration for the rights granted in the license agreement, we agreed to pay U.Mich up-front license fees and additional yearly and milestone payments. We also assumed under the license agreement responsibility for any infringement of third party rights caused by our use of the licensed materials. We paid an option fee of $2,000, a license issue fee of $10,000 and are obligated to pay an annual maintenance fee of $10,000 each year until the first commercial sale of a licensed product at which time the annual maintenance fee increases to $50,000. In addition, we are obligated to make milestone payments of $25,000, $50,000 and $75,000 upon completion of a Phase 1, Phase 2 and Phase 3 trial and $250,000 upon the first commercial sale of a licensed product and $350,000 upon annual net sales of $100,000,000 or more. The license agreements provide that the licensor has the right to terminate the license should we cease to carry on our business, fail to make a required payment or otherwise materially breach or default in our obligations under the license agreement following the giving of notice and an opportunity to cure any such breach. The license agreement provides that if we do not achieve the following milestones within the required period, U.Mich has the right to terminate the license agreement: completion of a Phase 1 clinical trial on or before January 1, 2015, a Phase 2 clinical trial on or before January 1, 2017, a Phase 3 clinical trial on or before January 1, 2019 and the first commercial sale of a product that includes the materials supplied by U.Mich on or before January 1, 2020. The license agreement also contains other customary clauses and terms as are common in similar agreements between industry and academia.
In April 2011 we entered into an evaluation and biological material license agreement with the ATCC to evaluate, use, market, offer for sale, sell and/or sublicense materials and processes related to various different cell lines. In October 2013 and March 2014, this agreement was amended to add additional cell lines in exchange for additional fees. The agreement with ATCC provides for an evaluation term of twelve months subject to two additional renewals, and a non-exclusive commercial use license upon termination of the evaluation period to utilize the products we obtain in the evaluation to develop, make, use and sell licensed products. The agreement with ATCC has a term of forty years. We paid an evaluation fee and two renewal evaluation fees totaling $15,000, and are obligated to pay a $50,000 fee upon initiation of the commercial license and a less than 1% royalty based on sales of licensed products. In addition, we are obligated to make milestone payments of $15,000, $30,000 and $60,000 upon initiation of a Phase 1, Phase 2, and Phase 3 trial, respectively; and $200,000 upon receipt of marketing authorization.
Government Regulation
FDA Approval Process
In the United States, pharmaceutical products are subject to extensive regulation by the U.S. Food and Drug Administration, or the FDA. The Federal Food, Drug, and Cosmetic Act, or the FDC Act, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Biological products used for the prevention, treatment, or cure of a disease or condition of a human being are subject to regulation under the FDC Act, except the section of the FDC Act which governs the approval of new drug applications, or NDAs. Biological products are approved for marketing under provisions of the Public Health Service Act, or PHSA, via a Biologics License Application, or BLA. However, the application process and requirements for approval of BLAs are very similar to those for NDAs, and biologics are associated with similar approval risks and costs as drugs. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs or BLAs, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
Pharmaceutical product development for a new product or certain changes to an approved product in the U.S. typically involves preclinical laboratory and animal tests, the submission to the FDA of an investigational new drug application, or IND, which must become effective before clinical testing may commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity, and novelty of the product or disease.
24
Preclinical tests include laboratory evaluation of product chemistry, formulation, and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements, including good laboratory practices. The results of preclinical testing are submitted to the FDA as part of an IND along with other information, including information about product chemistry, manufacturing and controls, and a proposed clinical trial protocol. Long term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has neither commented on nor questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin.
Clinical trials involve the administration of the investigational new drug or biologic to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted: (i) in compliance with federal regulations; (ii) in compliance with good clinical practice, or GCP, an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators, and monitors; as well as (iii) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND.
The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board, or IRB, for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
Clinical trials to support NDAs or BLAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase 1, the initial introduction of the drug or biologic into healthy human subjects or patients, the product is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses, and, if possible, early evidence on effectiveness. Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug or biologic for a particular indication, dosage tolerance, and optimum dosage, and to identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug or biologic and to provide adequate information for the labeling of the product. In most cases, the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy of the drug or biologic. A single Phase 3 trial with other confirmatory evidence may be sufficient in rare instances where the study is a large multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible.
After completion of the required clinical testing, an NDA or BLA is prepared and submitted to the FDA. FDA approval of the NDA or BLA is required before marketing of the product may begin in the U.S. The NDA or BLA must include the results of all preclinical, clinical, and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture, and controls. The cost of preparing and submitting an NDA or BLA is substantial. The submission of most NDAs and BLAs is additionally subject to a substantial application user fee, currently exceeding $1,958,000, and the manufacturer and/or sponsor under an approved new drug application are also subject to annual product and establishment user fees, currently exceeding $98,000 per product and $526,000 per establishment. These fees are typically increased annually.
25
The FDA has 60 days from its receipt of an NDA or BLA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of NDAs and BLAs. Most such applications for standard review drug or biologic products are reviewed within ten to twelve months; most applications for priority review drugs or biologics are reviewed in six to eight months. The FDA can extend these reviews by three months. Priority review can be applied to drugs that the FDA determines offer major advances in treatment, or provide a treatment where no adequate therapy exists. For biologics, priority review is further limited only for products intended to treat a serious or life-threatening disease relative to the currently approved products. The review process for both standard and priority review may be extended by the FDA for three additional months to consider certain late-submitted information, or information intended to clarify information already provided in the submission.
The FDA may also refer applications for novel drug or biologic products, or drug or biologic products that present difficult questions of safety or efficacy, to an advisory committee – typically a panel that includes clinicians and other experts – for review, evaluation, and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA or BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. FDA will not approve the product unless compliance with current good manufacturing practice, or cGMP, is satisfactory and the NDA or BLA contains data that provide substantial evidence that the drug or biologic is safe and effective in the indication studied.
After the FDA evaluates the NDA or BLA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA or BLA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included.
An approval letter authorizes commercial marketing of the drug or biologic with specific prescribing information for specific indications. As a condition of NDA or BLA approval, the FDA may require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the drug or biologic outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the product. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the product’s safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or BLA or NDA or BLA supplement before the change can be implemented. An NDA or BLA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA or BLA supplements as it does in reviewing NDAs or BLAs.
Post-Approval Requirements
Once an NDA or BLA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of drugs and biologics, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. Drugs and biologics may be marketed only for the approved indications and in accordance with the provisions of the approved labeling.
26
Adverse event reporting and submission of periodic reports is required following FDA approval of an NDA or BLA. The FDA also may require post-marketing testing, known as Phase 4 testing, REMS, and surveillance to monitor the effects of an approved product, or the FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control, drug manufacture, packaging, and labeling procedures must continue to conform to cGMPs after approval. Drug and biologic manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality-control to maintain compliance with cGMPs. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs or biologics intended to treat a rare disease or condition – generally a disease or condition that affects fewer than 200,000 individuals in the U.S. Orphan drug designation must be requested before submitting an NDA or BLA. After the FDA grants orphan drug designation, the generic identity of the drug or biologic and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first NDA or BLA applicant to receive FDA approval for a particular active ingredient to treat a particular disease with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the U.S. for that product, for that indication. During the seven-year exclusivity period, the FDA may not approve any other applications to market the same drug or biologic for the same disease, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. Orphan drug exclusivity does not prevent the FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA or BLA application user fee.
Fast Track Designation and Accelerated Approval
The FDA is required to facilitate the development, and expedite the review, of drugs or biologics that are intended for the treatment of a serious or life-threatening disease or condition for which there is no effective treatment and which demonstrate the potential to address unmet medical needs for the condition. Under the fast track program, the sponsor of a new drug or biologic candidate may request that the FDA designate the candidate for a specific indication as a fast track drug or biologic concurrent with, or after, the filing of the IND for the candidate. The FDA must determine if the drug or biologic candidate qualifies for fast track designation within 60 days of receipt of the sponsor’s request.
Under the fast track program and FDA’s accelerated approval regulations, the FDA may approve a drug or biologic for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions, or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A drug or biologic candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, will allow the FDA to withdraw the drug or biologic from the market on an expedited basis. All promotional materials for drug candidates approved under accelerated regulations are subject to prior review by the FDA.
In addition to other benefits such as the ability to use surrogate endpoints and engage in more frequent interactions with the FDA, the FDA may initiate review of sections of a fast track product’s NDA or BLA before the application is complete. This rolling review is available if the applicant provides, and the FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, the FDA’s time period goal for reviewing an application does not begin until the last section of the NDA or BLA is submitted. Additionally, the fast track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
27
Pediatric Information
Under the Pediatric Research Equity Act, or PREA, NDAs or BLAs or supplements to NDAs or BLAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant full or partial waivers, or deferrals, for submission of data. Unless otherwise required by regulation, PREA does not apply to any drug for an indication for which orphan designation has been granted.
Additional Controls for Biologics
To help reduce the increased risk of the introduction of adventitious agents, the PHSA emphasizes the importance of manufacturing controls for products whose attributes cannot be precisely defined. The PHSA also provides authority to the FDA to immediately suspend licenses in situations where there exists a danger to public health, to prepare or procure products in the event of shortages and critical public health needs, and to authorize the creation and enforcement of regulations to prevent the introduction or spread of communicable diseases in the United States and between states.
After a BLA is approved, the product may also be subject to official lot release as a condition of approval. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot. The FDA may also perform certain confirmatory tests on lots of some products, such as viral vaccines, before releasing the lots for distribution by the manufacturer. In addition, the FDA conducts laboratory research related to the regulatory standards on the safety, purity, potency, and effectiveness of biological products. As with drugs, after approval of biologics, manufacturers must address any safety issues that arise, are subject to recalls or a halt in manufacturing, and are subject to periodic inspection after approval.
Biosimilars
The Biologics Price Competition and Innovation Act of 2009, or BPCIA, creates an abbreviated approval pathway for biological products shown to be highly similar to or interchangeable with an FDA-licensed reference biological product. Biosimilarity sufficient to reference a prior FDA-approved product requires that there be no differences in conditions of use, route of administration, dosage form, and strength, and no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency. Biosimilarity must be shown through analytical studies, animal studies, and at least one clinical study, absent a waiver by the Secretary. A biosimilar product may be deemed interchangeable with a prior approved product if it meets the higher hurdle of demonstrating that it can be expected to produce the same clinical results as the reference product and, for products administered multiple times, the biologic and the reference biologic may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. No biosimilar or interchangeable products have been approved under the BPCIA to date. Complexities associated with the larger, and often more complex, structures of biological products, as well as the process by which such products are manufactured, pose significant hurdles to implementation which are still being evaluated by the FDA.
A reference biologic is granted twelve years of exclusivity from the time of first licensure of the reference product, and no application for a biosimilar can be submitted for four years from the date of licensure of the reference product. The first biologic product submitted under the abbreviated approval pathway that is determined to be interchangeable with the reference product has exclusivity against a finding of interchangeability for other biologics for the same condition of use for the lesser of (i) one year after first commercial marketing of the first interchangeable biosimilar, (ii) eighteen months after the first interchangeable biosimilar is approved if there is no patent challenge, (iii) eighteen months after resolution of a lawsuit over the patents of the reference biologic in favor of the first interchangeable biosimilar applicant, or (iv) 42 months after the first interchangeable biosimilar’s application has been approved if a patent lawsuit is ongoing within the 42-month period.
28
Cell and Tissue Based Biologics
Establishments that manufacture cell and tissue based products must comply with the FDA’s current good tissue practices, or cGTP, which are FDA regulations that govern the methods used in, and the facilities and controls used for, the manufacture of such products. The primary intent of the cGTP requirements is to ensure that cell and tissue based products are manufactured in a manner designed to prevent the introduction, transmission and spread of communicable disease. FDA regulations also include requirements for a unified registration and listing system, donor screening and testing, adverse reaction reporting, and labeling.
Cell and tissue based products may also be subject to the same approval standards, including demonstration of safety and efficacy, as other biologic and drug products if they meet certain criteria such as if the cells or tissues are more than minimally manipulated or if they are intended for a non-homologous use.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of FDA-regulated products, including drugs, are required to register and disclose certain clinical trial information. Information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to discuss the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
Non-U.S. Regulation
Before our products can be marketed outside of the U.S., they are subject to regulatory approval of the respective authorities in the country in which the product should be marketed. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary widely from country to country. No action can be taken to market any product in a country until an appropriate application has been approved by the regulatory authorities in that country. The current approval process varies from country to country, and the time spent in gaining approval varies from that required for FDA approval. In certain countries, the sales price of a product must also be approved. The pricing review period often begins after market approval is granted. Even if a product is approved by a regulatory authority, satisfactory prices might not be approved for such product.
In Europe, marketing authorizations may be submitted at a centralized, a decentralized or national level. The centralized procedure is mandatory for the approval of biotechnology products and provides for the grant of a single marketing authorization that is valid in all European Union member states. As of January 1995, a mutual recognition procedure is available at the request of the applicant for all medicinal products that are not subject to the centralized procedure. There can be no assurance that the chosen regulatory strategy will secure regulatory approvals on a timely basis or at all.
While we intend to market our products outside the United States in compliance with our respective license agreements, we have not made any applications with non-U.S. authorities and have no timeline for such applications or marketing.
Research and Development
We have built an internal and external research and development organization that includes expertise in discovery research, preclinical development, product formulation, analytical and medicinal chemistry, manufacturing, clinical development and regulatory and quality assurance. We engage third parties on a limited basis to conduct portions of our preclinical research; however, we are not substantially dependent upon any third parties for our preclinical research nor do any of these third parties conduct a major portion of our preclinical research. Research and development expenses were $2,737,688 and $902,938 during the years ended December 31, 2013 and 2012, respectively.
29
Employees
As of March 15, 2014, we had a total of 9 employees, of which 8 are full-time employees and 1 is part-time. We believe our relationships with our employees are satisfactory. None of our employees is represented by a labor union. We anticipate that we will need to identify, attract, train and retain other highly skilled personnel to pursue our development program. Hiring for such personnel is competitive, and there can be no assurance that we will be able to retain our key employees or attract, assimilate or retain the qualified personnel necessary for the development of our business.
Legal Proceedings
There are currently no pending legal proceedings against the Company or its subsidiaries.
Item 1A.
Risk Factors
Investing in our common stock involves a high degree of risk. In addition to the risks related to our business set forth in this Form 10-K and the other information included and incorporated by reference in this Form 10-K, you should carefully consider the risks described below before purchasing our common stock. Additional risks, uncertainties and other factors not presently known to us or that we currently deem immaterial may also impair our business operations.
Risks Relating to our Company
We have had limited operations to date.
We are a start-up entity and have had limited operations to date. As a start-up entity, we are subject to many of the risks common to such enterprises, including our ability to implement our business plan, market acceptance of our proposed business and products, under-capitalization, cash shortages, limitations with respect to personnel, financing and other resources, competition from better funded and experienced companies, and uncertainty of our ability to generate revenues. There is no assurance that our activities will be successful or will result in any revenues or profit, and the likelihood of our success must be considered in light of the stage of our development. Even if we generate revenue, there can be no assurance that we will be profitable. In addition, no assurance can be given that we will be able to consummate our business strategy and plans, or that financial, technological, market, or other limitations may force us to modify, alter, significantly delay, or significantly impede the implementation of such plans. We have insufficient results for investors to use to identify historical trends. Investors should consider our prospects in light of the risk, expenses and difficulties we will encounter as an early stage company. Our revenue and income potential is unproven and our business model is continually evolving. We are subject to the risks inherent to the operation of a new business enterprise, and cannot assure you that we will be able to successfully address these risks.
We have a limited operating history upon which to evaluate our ability to commercialize our products.
We are a development-stage company and our success is dependent upon our ability to obtain regulatory approval for and commercialize our products and we have not demonstrated an ability to perform the functions necessary for the approval or successful commercialization of any product candidates. The successful commercialization of any product candidates will require us to perform a variety of functions, including:
·
continuing to undertake pre-clinical development and initiate clinical trials;
·
participating in regulatory approval processes;
·
formulating and manufacturing products; and
·
conducting sales and marketing activities.
While various members of our management and staff have significant experience in conducting cancer trials, the Company, to date, has not successfully completed any clinical trials and has no experience conducting or enrolling patients in clinical trials. Until recently, our operations have been limited primarily to organizing and staffing the Company, acquiring, developing and securing our proprietary technology and undertaking pre-clinical trials of our product candidates. These operations provide a limited basis for you to assess our ability to commercialize our product candidates and the advisability of investing in our securities.
30
We currently have no product revenues and may not generate revenue at any time in the near future, if at all.
We currently have no products for sale and we cannot guarantee that we will ever have any drug products approved for sale. We and our product candidates are subject to extensive regulation by the U.S. Food and Drug Administration, or FDA, and comparable regulatory authorities in other countries governing, among other things, research, testing, clinical trials, manufacturing, labeling, promotion, marketing, adverse event reporting and recordkeeping of our product candidates. Until, and unless, we receive approval from the FDA and other regulatory authorities for our product candidates, we cannot commercialize our product candidates and will not have product revenues. For the foreseeable future, we will have to fund all of our operations and capital expenditures from cash on hand, grants, and, potentially, future offerings. We believe we have sufficient cash on hand to fund our current operating plans and capital expenditure requirements for at least 12 months. However, changes may occur that would consume our available capital before that time, including changes in and progress of our development activities, acquisitions of additional candidates and changes in regulation. Moreover, pre-clinical studies and clinical trials may not start or be completed as we forecast and may not achieve the desired results.
We may continue to generate operating losses and experience negative cash flows and it is uncertain whether we will achieve profitability.
For the years ended December 31, 2013 and December 31, 2012, we incurred a net loss of ($6,609,864) and ($2,471,147), respectively. We have also incurred a deficit accumulated during the development stage of ($12,346,630). We may continue to incur operating losses until such time, if ever, as we are able to achieve sufficient levels of revenue from operations. Our ability to achieve profitability will depend on the market acceptance of our product offerings and our capacity to develop, introduce and sell our products to our targeted markets. There can be no assurance that we will ever generate significant sales or achieve profitability. Accordingly, the extent of future losses and the time required to achieve profitability, if ever, cannot be predicted at this point.
Even if we succeed in developing and commercializing one or more product candidates, we expect to incur substantial losses for the foreseeable future and may never become profitable. We also expect to continue to incur significant operating and capital expenditures and anticipate that our expenses will increase substantially in the foreseeable future as we:
·
continue to undertake pre-clinical development and initiate clinical trials for product candidates;
·
seek regulatory approvals for product candidates;
·
implement additional internal systems and infrastructure; and
·
hire additional personnel.
We also expect to experience negative cash flows for the foreseeable future as we fund our operating losses and capital expenditures. As a result, we will need to generate significant revenues or raise additional financing in order to achieve and maintain profitability. We may not be able to generate these revenues or achieve profitability in the future. Our failure to achieve or maintain profitability would likely negatively impact the value of our securities and could prevent us from continuing as a going concern.
31
Risks Relating to our Business
If we do not obtain the necessary regulatory approvals in the U.S. and/or other countries we will not be able to sell our product candidates.
We cannot assure you that we will receive the approvals necessary to commercialize any of our product candidates or any product candidates we acquire or develop in the future. We will need FDA approval to commercialize our product candidates in the U.S. and approvals from the FDA-equivalent regulatory authorities in foreign jurisdictions to commercialize our product candidates in those jurisdictions. In order to obtain FDA approval of any product candidate, we must submit to the FDA a Biologics License Application, or BLA, demonstrating that the product candidate is safe, pure and potent, or effective for its intended use. This demonstration requires significant research including pre-clinical studies, as well as clinical trials. Satisfaction of the FDA’s regulatory requirements typically takes many years, depends upon the type, complexity and novelty of the product candidate and requires substantial resources for research, development and testing. We cannot predict whether our clinical trials will demonstrate the safety and efficacy of our product candidates or if the results of any clinical trials will be sufficient to advance to the next phase of development or for approval from the FDA. We also cannot predict whether our research and clinical approaches will result in drugs or therapeutics that the FDA considers safe and effective for the proposed indications. The FDA has substantial discretion in the drug approval process. The approval process may be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals may:
·
prevent or delay commercialization of, and our ability to derive product revenues from, our product candidates; and
·
diminish any competitive advantages that we may otherwise believe that we hold.
Even if we comply with all FDA requests, the FDA may ultimately reject one or more of our BLAs. We may never obtain regulatory clearance for any of our product candidates. Failure to obtain FDA approval of any of our product candidates will severely undermine our business by leaving us without a saleable product, and therefore without any source of revenues, until another product candidate can be developed. There is no guarantee that we will ever be able to develop or acquire another product candidate.
In addition, the FDA may require us to conduct additional pre-clinical and clinical testing or to perform post-marketing studies, as a condition to granting marketing approval of a product. Regulatory approval of oncology products often requires that patients in clinical trials be followed for long periods to assess their overall survival. The results generated after approval could result in loss of marketing approval, changes in product labeling, and/or new or increased concerns about the side effects or efficacy of a product. The FDA has significant post-market authority, including the explicit authority to require post-market studies and clinical trials, labeling changes based on new safety information, and compliance with FDA-approved risk evaluation and mitigation strategies. The FDA’s exercise of its authority has in some cases resulted, and in the future could result, in delays or increased costs during product development, clinical trials and regulatory review, increased costs to comply with additional post-approval regulatory requirements and potential restrictions on sales of approved products.
In foreign jurisdictions, we must also receive approval from the appropriate regulatory authorities before we can commercialize any vaccines. Foreign regulatory approval processes generally include all of the risks associated with the FDA approval procedures described above. There can be no assurance that we will receive the approvals necessary to commercialize our product candidate for sale outside the United States.
Our product candidates are in early stages of development.
Because our product candidates are in early stages of development they will require extensive pre-clinical and clinical testing. Only one product candidate is currently ready for Phase 2 clinical trials. We cannot predict with any certainty if or when we might submit a BLA for regulatory approval for any of our product candidates or whether any such BLA will be accepted for review by the FDA, or whether any BLA will be approved upon review.
32
Even if our clinical trials are completed as planned, we cannot be certain that their results will support our proposed indications. Success in pre-clinical testing and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the results of later clinical trials will replicate the results of prior clinical trials and pre-clinical testing. For example, the only clinical study completed to date with one of our product candidates by the inventor of the technology that we license showed evidence of an immune response in late-stage NSCLC patients exposed to HS-110. However, our future HS-110 trials will use doses and dosing regimens which have previously been tested in only 0 to 3 subjects, and will be conducted in patients with less advanced disease who may have different responses. In addition, immune response is not an acceptable regulatory endpoint for approval, and no actual clinical or tumor responses were observed in that study. Moreover, the HS-110 Phase 1 trial involved a small sample size, was not blinded and was sponsored by an individual who has a significant financial interest in the success of the product candidate. The clinical trial process may fail to demonstrate that our product candidates are safe and effective for their proposed uses. This failure could cause us to abandon a product candidate and may delay development of other product candidates. Any delay in, or termination of, our clinical trials will delay and possibly preclude the filing of any BLAs with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues.
Clinical trials are very expensive, time-consuming and difficult to design and implement.
As part of the regulatory process, we must conduct clinical trials for each product candidate to demonstrate safety and efficacy to the satisfaction of the FDA and other regulatory authorities. The number and design of the clinical trials that will be required varies depending upon product candidate, the condition being evaluated and the trial results themselves. Therefore, it is difficult to accurately estimate the cost of the clinical trials. Clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. The clinical trial process is also time consuming. We estimate that clinical trials of our product candidates will take at least several years to complete. Furthermore, failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed or prevented by several factors, including:
·
unforeseen safety issues;
·
failure to determine appropriate dosing;
·
greater than anticipated cost of our clinical trials;
·
failure to demonstrate effectiveness during clinical trials;
·
slower than expected rates of patient recruitment or difficulty obtaining investigators;
·
patient drop-out or discontinuation;
·
inability to monitor patients adequately during or after treatment;
·
third party contractors failing to comply with regulatory requirements or meet their contractual obligations to us in a timely manner;
·
insufficient or inadequate supply or quality of product candidates or other necessary materials to conduct our trials;
·
potential additional safety monitoring, or other conditions required by FDA or comparable foreign regulatory authorities regarding the scope or design of our clinical trials, or other studies requested by regulatory agencies;
·
problems engaging IRBs to oversee trials or in obtaining and maintaining IRB approval of studies;
·
imposition of clinical hold or suspension of our clinical trials by regulatory authorities; and
·
inability or unwillingness of medical investigators to follow our clinical protocols.
In addition, we or the FDA may suspend or terminate our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if the FDA finds deficiencies in our Investigational New Drug, or IND, submissions or the conduct of these trials. Therefore, we cannot predict with any certainty when, if ever, future clinical trials will commence or be completed. We submitted the protocol for our planned Phase 2 trial of HS-110 to the FDA in March 2014. There can be no assurance that the FDA will not have comments regarding the protocol.
33
There is uncertainty as to market acceptance of our technology and product candidates.
Even if the FDA approves one or more of our product candidates, the products may not gain broad market acceptance among physicians, healthcare payers, patients, and the medical community. We have conducted our own research into the markets for our product candidates; however we cannot guarantee market acceptance of our product candidates, if approved, and have somewhat limited information on which to estimate our anticipated level of sales. Our product candidates, if approved, will require patients, healthcare providers and doctors to adopt our technology. Our industry is susceptible to rapid technological developments and there can be no assurance that we will be able to match any new technological advances. If we are unable to match the technological changes in the needs of our customers the demand for our products will be reduced. Acceptance and use of any products we market will depend upon a number of factors including:
·
perceptions by members of the health care community, including physicians, about the safety and effectiveness of our products;
·
limitation on use or warnings required by FDA in our product labeling;
·
cost-effectiveness of our products relative to competing products;
·
convenience and ease of administration;
·
potential advantages of alternative treatment methods;
·
availability of reimbursement for our products from government or other healthcare payers; and
·
effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any.
Because we expect virtually all of our product revenues for the foreseeable future to be generated from sales of our current product candidates, if approved, the failure of these therapeutics to find market acceptance would substantially harm our business and would adversely affect our revenue.
Our development program depends upon third-party researchers who are outside our control.
We are dependent upon independent investigators and collaborators, such as universities and medical institutions, to conduct our pre-clinical and clinical trials under agreements with us. These collaborators are not our employees and we cannot control the amount or timing of resources that they devote to our programs. These investigators may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking such programs ourselves. If outside collaborators fail to devote sufficient time and resources to our development programs, or if their performance is substandard, the approval of our FDA applications, if any, and our introduction of new product candidates, if any, will be delayed if obtained at all. These collaborators may also have relationships with other commercial entities, some of whom may compete with us. If our collaborators assist our competitors at our expense, our competitive position would be harmed.
To date, in excess of $14,000,000 of funding has been awarded by the NIH to the primary inventor of the technology we license. We have little control over the direction of the NIH grant funds that have been received by the primary inventor of the technology we license and since payment is made to the inventor as opposed to us, we do not recognize any revenue from such grant funds nor do they fund any expenses that we incur.
Although earmarked for further development of the technology that we license, any funds awarded to the primary inventor are used in his discretion and we have little control over his use of the funds.
34
We will rely exclusively on third parties to formulate and manufacture our product candidates.
We have no experience in the formulation, development or manufacturing of biologics and do not intend to establish our own manufacturing facilities. We lack the resources and expertise to formulate or manufacture our own product candidates. The investigational products for our planned Phase 1 and Phase 2 clinical trials are manufactured by our contractors under current good manufacturing practices, or cGMPs and we have entered into agreements with commercial-scale manufacturers for the production and supply of investigational product for additional Phase 2 and Phase 3 clinical trials as well as commercialization. We must also develop and validate a potency assay prior to submission of a license application. Such assays have traditionally proven difficult to develop for cell-based products and must be established prior to initiating any Phase 3 clinical trials. If any of our current product candidates, or any product candidates we may develop or acquire in the future, receive FDA approval, we will rely on one or more third-party contractors for manufacturing. Our anticipated future reliance on a limited number of third-party manufacturers exposes us to the following risks:
We may be unable to identify manufacturers on acceptable terms or at all because the number of potential manufacturers with appropriate expertise and facilities is limited.
If we change manufacturers at any point during the development process or after approval we will be required to demonstrate comparability between the products made by the old and new manufacturers. If we are unable to do so, we may need to conduct additional clinical trials with product manufactured by the new manufacturer. For example, the manufacturer of the clinical trial material we intend to use for any future Phase 3 trials of HS-110 and of our commercial product, if approved, is a different manufacturer from the manufacturer of the inventor’s completed Phase 1 trial of HS-110 and the early portion of our planned initial Phase 2 trial of HS-110. Accordingly, it may be necessary to evaluate the comparability of the HS-101 produced by the two different manufacturers during the third stage of our planned Phase 2 trial of HS-110.
If we change the manufacturer of a product subsequent to the approval of the product, we will need to obtain approval from the FDA of the change in manufacturer. Any such approval would likely require significant testing and expense, and the new manufacturer may be subject to a cGMP inspection prior to approval.
Our third-party manufacturers might be unable to formulate and manufacture our product candidates in the volume and with the quality required to meet our clinical needs and commercial needs, if any.
Our contract manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store and distribute our product candidates.
Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA, and corresponding state agencies to ensure compliance with cGMPs and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards.
If any third-party manufacturer makes improvements in the manufacturing process for our products, we may not own, or may have to share, the intellectual property rights to the innovation.
Our contract manufacturers have in the past and may in the future encounter difficulties in achieving quality control and quality assurance and may experience shortages in qualified personnel. Our contract manufacturers are subject to inspections by the FDA and comparable agencies in other jurisdictions to assess compliance with applicable regulatory requirements. Any failure to follow cGMP or other regulatory requirements or delay, interruption or other issues that arise in the manufacture, packaging, or storage of our products as a result of a failure of the facilities or operations of third parties to comply with regulatory requirements or pass any regulatory authority inspection could significantly impair our ability to develop and commercialize our products, including leading to significant delays in the availability of products for our clinical studies or the termination or hold on a clinical study, or the delay or prevention of a filing or approval of marketing applications for our product candidates. Significant noncompliance could also result in the imposition of sanctions, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approvals for our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of products, operating restrictions and criminal prosecutions, any of which could damage our reputation. If we or our contract manufacturers are not able to maintain regulatory compliance, we may not be permitted to market our products and/or may be subject to product recalls, seizures, injunctions, or criminal prosecution.
Each of these risks could delay our clinical trials, the approval, if any, of our product candidates by the FDA or the commercialization of our product candidates or result in higher costs or deprive us of potential product revenues.
35
Even if we are able to obtain regulatory approval for our product candidates, we will continue to be subject to ongoing and extensive regulatory requirements, and our failure, or the failure of our contract manufacturers, to comply with these requirements could substantially harm our business.
If the FDA approves any of our product candidates, the labeling, manufacturing, packaging, adverse event reporting, storage, advertising, promotion and record-keeping for our products will be subject to ongoing FDA requirements and continued regulatory oversight and review. We may also be subject to additional FDA post-marketing obligations. If we are not able to maintain regulatory compliance, we may not be permitted to market our product candidates and/or may be subject to product recalls or seizures. The subsequent discovery of previously unknown problems with any marketed product, including adverse events of unanticipated severity or frequency, may result in restrictions on the marketing of the product, and could include withdrawal of the product from the market.
We have no experience selling, marketing or distributing products and have no internal capability to do so.
We currently have no sales, marketing or distribution capabilities. We do not anticipate having the resources in the foreseeable future to allocate to the sales and marketing of our proposed products, if approved. Our future success depends, in part, on our ability to enter into and maintain collaborative relationships for such capabilities, the collaborator’s strategic interest in the products under development and such collaborator’s ability to successfully market and sell any such products. We intend to pursue collaborative arrangements regarding the sales and marketing of our products, however, there can be no assurance that we will be able to establish or maintain such collaborative arrangements, or if able to do so, that our collaborators will have effective sales forces. To the extent that we decide not to, or are unable to, enter into collaborative arrangements with respect to the sales and marketing of our proposed products, significant capital expenditures, management resources and time will be required to establish and develop an in-house marketing and sales force with technical expertise. There can also be no assurance that we will be able to establish or maintain relationships with third party collaborators or develop in-house sales and distribution capabilities. To the extent that we depend on third parties for marketing and distribution, any revenues we receive will depend upon the efforts of such third parties, and there can be no assurance that such efforts will be successful. In addition, there can also be no assurance that we will be able to successfully market and sell our products in the United States or overseas on our own.
We may not be successful in establishing and maintaining strategic partnerships, which could adversely affect our ability to develop and commercialize products.
We may seek to enter into strategic partnerships in the future, including alliances with other biotechnology or pharmaceutical companies, to enhance and accelerate the development and commercialization of our products. We face significant competition in seeking appropriate strategic partners and the negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for any future product candidates and programs because our research and development pipeline may be insufficient, our product candidates and programs may be deemed to be at too early of a stage of development for collaborative effort and/or third parties may not view our product candidates and programs as having the requisite potential to demonstrate safety and efficacy or return on investment. Even if we are successful in our efforts to establish strategic partnerships, the terms that we agree upon may not be favorable to us and we may not be able to maintain such strategic partnerships if, for example, development or approval of a product candidate is delayed or sales of an approved product are disappointing.
If we ultimately determine that entering into strategic partnerships is in our best interest but either fail to enter into, are delayed in entering into or fail to maintain such strategic partnerships:
·
the development of certain of our current or future product candidates may be terminated or delayed;
·
our cash expenditures related to development of certain of our current or future product candidates may increase significantly and we may need to seek additional financing;
·
we may be required to hire additional employees or otherwise develop expertise, such as sales and marketing expertise, for which we have not budgeted;
·
we will bear all of the risk related to the development of any such product candidates;
·
the competitiveness of any product candidate that is commercialized could be reduced.
36
To the extent we elect to enter into licensing or collaboration agreements to partner our product candidates, our dependence on such relationships may adversely affect our business.
Our commercialization strategy for certain of our product candidates may depend on our ability to enter into agreements with collaborators to obtain assistance and funding for the development and potential commercialization of these product candidates. Supporting diligence activities conducted by potential collaborators and negotiating the financial and other terms of a collaboration agreement are long and complex processes with uncertain results. Even if we are successful in entering into one or more collaboration agreements, collaborations may involve greater uncertainty for us, as we have less control over certain aspects of our collaborative programs than we do over our proprietary development and commercialization programs. We may determine that continuing a collaboration under the terms provided is not in our best interest, and we may terminate the collaboration. Our collaborators could delay or terminate their agreements, and our product candidates subject to collaborative arrangements may never be successfully developed or commercialized.
Further, our future collaborators may develop alternative products or pursue alternative technologies either on their own or in collaboration with others, including our competitors, and the priorities or focus of our collaborators may shift such that our programs receive less attention or fewer resources than we would like, or they may be terminated altogether. Any such actions by our collaborators may adversely affect our business prospects and ability to earn revenues. In addition, we could have disputes with our future collaborators, such as the interpretation of terms in our agreements. Any such disagreements could lead to delays in the development or commercialization of any potential products or could result in time-consuming and expensive litigation or arbitration, which may not be resolved in our favor.
If we cannot compete successfully for market share against other drug companies, we may not achieve sufficient product revenues and our business will suffer.
The market for our product candidates is characterized by intense competition and rapid technological advances. If any of our product candidates receives FDA approval, it will compete with a number of existing and future drugs and therapies developed, manufactured and marketed by others. Existing or future competing products may provide greater therapeutic convenience or clinical or other benefits for a specific indication than our products, or may offer comparable performance at a lower cost. If our products fail to capture and maintain market share, we may not achieve sufficient product revenues and our business will suffer.
We will compete against fully integrated pharmaceutical companies and smaller companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors have oncology compounds already approved or in development. In addition, many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs or have substantially greater financial resources than we do, as well as significantly greater experience in:
·
developing drugs, biologics and other therapies;
·
undertaking pre-clinical testing and clinical trials;
·
obtaining FDA and other regulatory approvals of drugs, biologics and other therapies;
·
formulating and manufacturing drugs, biologics and other therapies; and
·
launching, marketing and selling drugs, biologics and other therapies.
We have limited protection of our intellectual property.
We intend to rely on a combination of common law copyright, patent, trademark, and trade secret laws and measures to protect our proprietary information. We have obtained exclusive rights to license the technology for which patent protection has been obtained; however such protection does not prevent unauthorized use of such technology. Trademark and copyright protections may be limited, and enforcement could be too costly to be effective. It may also be possible for unauthorized third parties to copy aspects of, or otherwise obtain and use, our proprietary information without authorization, including, but not limited to, product design, software, customer and prospective customer lists, trade secrets, copyrights, patents and other proprietary rights and materials. Other parties can use and register confusingly similar business, product and service names, as well as domain names, which could divert customers, resulting in a material adverse effect on our business, operating results and financial condition.
37
If we fail to successfully enforce our intellectual property rights, our competitive position could suffer, which could harm our operating results. Competitors may challenge the validity or scope of our patents or future patents we may obtain. In addition, our licensed patents may not provide us a meaningful competitive advantage. We may be required to spend significant resources to monitor and police our licensed intellectual property rights. We may not be able to detect infringement and our competitive position may be harmed. In addition, competitors may design around our technology or develop competing technologies. Intellectual property rights may also be unavailable or limited in some foreign countries, which could make it easier for competitors to capture market share.
The technology we license, our products or our development efforts may be found to infringe third-party intellectual property rights.
Third parties may in the future assert claims or initiate litigation related to their patent, copyright, trademark and other intellectual property rights in technology that is important to us. The asserted claims and/or litigation could include claims against us, our licensors or our suppliers alleging infringement of intellectual property rights with respect to our products or components of those products. Regardless of the merit of the claims, they could be time consuming, result in costly litigation and diversion of technical and management personnel, or require us to develop a non-infringing technology or enter into license agreements. We have not undertaken an exhaustive search to discover any third party intellectual patent rights which might be infringed by commercialization of the product candidates described herein. Although we are not currently aware of any such third party intellectual patent rights, it is possible that such rights currently exist or might be obtained in the future. In the event that a third party controls such rights and we are unable to obtain a license to such rights on commercially reasonable terms, we may not be able to sell or continue to develop our products, and may be liable for damages for such infringement. We cannot assure you that licenses will be available on acceptable terms, if at all. Furthermore, because of the potential for significant damage awards, which are not necessarily predictable, it is not unusual to find even arguably unmeritorious claims resulting in large settlements. If any infringement or other intellectual property claim made against us by any third party is successful, or if we fail to develop non-infringing technology or license the proprietary rights on commercially reasonable terms and conditions, our business, operating results and financial condition could be materially adversely affected.
If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we may have to:
·
obtain licenses, which may not be available on commercially reasonable terms, if at all;
·
abandon an infringing drug or therapy candidate;
·
redesign our products or processes to avoid infringement;
·
stop using the subject matter claimed in the patents held by others;
·
pay damages; or
·
defend litigation or administrative proceedings which may be costly whether we win or lose, and which could result in a substantial diversion of our financial and management resources.
We rely on licenses to use various technologies that are material to our business and if the agreements were to be terminated or if other rights which may be necessary or we deem advisable for commercializing our intended products cannot be obtained, itwould halt our ability to market our products and technology, as well as have an immediate material adverse effect on our business, operating results and financial condition.
We have licensing agreements with certain universities granting us the right to use certain critical intellectual property. The terms of the licensing agreements continues until the end of the life of the last patent to expire. If we breach the terms of these licensing agreements, including any failure to make minimum royalty payments required thereunder or failure to reach certain developmental milestones, using best efforts to introduce a licensed product in certain territories by certain dates, the licensor has the right to terminate the license. If we were to lose or otherwise be unable to maintain these licenses on acceptable terms, or find that it is necessary or appropriate to secure new licenses from other third parties, it would halt our ability to market our products and technology, which would have an immediate material adverse effect on our business, operating results and financial condition.
38
We may be unable to generate sufficient revenues to meet the minimum royalties or developmental milestones required under our license agreements.
For the years ended December 31, 2014, 2015, 2016, and 2017 our minimum royalty obligations under our licensing agreements, required to be paid with the passage of time, are $30,000, $30,000, $30,000, and $280,000, respectively, and thereafter through December 31, 2022, $30,000 per year. No assurance can be given that we will generate sufficient revenue or raise additional financing to make these minimum royalty payments. The license agreements also provide for certain developmental milestones. No assurance can be given that we will meet all of the required developmental milestones. Any failure to make the payments or reach the milestones required by the license agreements would permit the licensor to terminate the license. If we were to lose or otherwise be unable to maintain these licenses, it would halt our ability to market our products and technology, which would have an immediate material adverse effect on our business, operating results and financial condition.
Our ability to generate product revenues will be diminished if our therapies sell for inadequate prices or patients are unable to obtain adequate levels of reimbursement.
Our ability to commercialize our vaccines, alone or with collaborators, will depend in part on the extent to which reimbursement will be available from:
·
government and health administration authorities;
·
private health maintenance organizations and health insurers; and
·
other healthcare payers.
Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Healthcare payers, including Medicare, are challenging the prices charged for medical products and services. Cost control initiatives could decrease the price that we would receive for any products in the future, which would limit our revenue and profitability. Government and other healthcare payers increasingly attempt to contain healthcare costs by limiting both coverage and the level of reimbursement for drugs and therapeutics. We might need to conduct post-marketing studies in order to demonstrate the cost-effectiveness of any future products to such payers’ satisfaction. Such studies might require us to commit a significant amount of management time and financial and other resources. Our future products might not ultimately be considered cost-effective. Even if one of our product candidates is approved by the FDA, insurance coverage may not be available, and reimbursement levels may be inadequate, to cover such vaccines. If government and other healthcare payers do not provide adequate coverage and reimbursement levels for one of our products, once approved, market acceptance of such product could be reduced.
Legislative and regulatory changes affecting the health care industry could adversely affect our business.
Political, economic and regulatory influences are subjecting the health care industry to potential fundamental changes that could substantially affect our results of operations. U.S. and foreign governments, for example, continue to propose and pass legislation designed to reduce the cost of healthcare. In some foreign markets, the government controls the pricing and profitability of prescription pharmaceuticals. In the U.S., we expect that there will continue to be federal and state proposals to implement similar governmental controls. In addition, recent changes in the Medicare program and increasing emphasis on managed care in the United States will continue to put pressure on pharmaceutical product pricing. It is uncertain whether or when any legislative proposals will be adopted or what actions federal, state, or private payers for health care treatment and services may take in response to any health care reform proposal or legislation. We cannot predict the effect health care reforms may have on our business and we can offer no assurances that any of these reforms will not have a material adverse effect on our business. These actual and potential changes are causing the marketplace to put increased emphasis on the delivery of more cost-effective treatments. In addition, uncertainly remains regarding proposed significant reforms to the U.S. health care system.
We may not successfully effect our intended expansion.
Our success will depend upon the expansion of our operations and the effective management of our growth, which will place a significant strain on our management and on our administrative, operational and financial resources. To manage this growth, we must expand our facilities, augment our operational, financial and management systems and hire and train additional qualified personnel. If we are unable to manage our growth effectively, our business would be harmed.
39
We may be exposed to liability claims associated with the use of biological and hazardous materials and chemicals.
Our research and development activities may involve the controlled use of biological and hazardous materials and chemicals. Although we believe that our safety procedures for using, storing, handling and disposing of these materials comply with federal, state and local laws and regulations, we cannot completely eliminate the risk of accidental injury or contamination from these materials. In the event of such an accident, we could be held liable for any resulting damages and any liability could materially adversely affect our business, financial condition and results of operations. In addition, the federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous or radioactive materials and waste products may require us to incur substantial compliance costs that could materially adversely affect our business, financial condition and results of operations.
We rely on key executive officers and scientific and medical advisors, and their knowledge of our business and technical expertise would be difficult to replace.
We are highly dependent on our principal scientific, regulatory and medical advisors and our chief executive officer. Other than a $2,000,000 insurance policy on the life of Jeffrey Wolf, we do not have “key person” life insurance policies for any of our officers or advisors. The loss of the technical knowledge and management and industry expertise of any of our key personnel could result in delays in product development, loss of customers and sales and diversion of management resources, which could adversely affect our operating results.
If we are unable to hire additional qualified personnel, our ability to grow our business may be harmed.
We will need to hire additional qualified personnel with expertise in pre-clinical and clinical research, government regulation, formulation and manufacturing, sales and marketing and accounting and financing. In particular, over the next 12 months, we expect to hire additional new employees. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for such individuals is intense, and we cannot be certain that our search for such personnel will be successful. Attracting and retaining qualified personnel will be critical to our success.
Certain of our officers may have a conflict of interest.
One of our officers is currently working for the Company on a part-time basis. This part-time employee also works at other jobs and has discretion to decide what time he devotes to our activities, which may result in a lack of availability when needed due to responsibilities at other jobs. We expect that any part-time officers may join the Company on a full-time basis, but there can be no assurance given that any of our officers will be so employed.
We may incur substantial liabilities and may be required to limit commercialization of our products in response to product liability lawsuits.
The testing and marketing of drug and biological product candidates entail an inherent risk of product liability. Product liability claims might be brought against us by consumers, health care providers or others selling or otherwise coming into contact with our products. Clinical trial liability claims may be filed against us for damages suffered by clinical trial subjects or their families. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products which could impact our ability to continue as a going concern. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop, alone or with collaborators. In addition, regardless of merit or eventual outcome, product liability claims may result in:
·
decreased demand for any approved product candidates;
·
impairment of our business reputation;
·
withdrawal of clinical trial participants;
·
costs of related litigation;
·
distraction of management’s attention;
·
substantial monetary awards to patients or other claimants;
·
loss of revenues; and
·
the inability to successfully commercialize any approved drug candidates.
40
International expansion of our business exposes us to business, regulatory, political, operational, financial and economic risks associated with doing business outside of the United States.
Our business strategy incorporates international expansion, including establishing and maintaining clinician marketing and education capabilities outside of the United States and expanding our relationships with distributors and manufacturers. Doing business internationally involves a number of risks, including:
·
multiple, conflicting and changing laws and regulations such as tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals, permits and licenses;
·
failure by us or our distributors to obtain regulatory approvals for the sale or use of our product candidates in various countries;
·
difficulties in managing foreign operations;
·
complexities associated with managing multiple payor-reimbursement regimes or self-pay systems;·
·
limits on our ability to penetrate international markets if our product candidates cannot be processed by a manufacturer appropriately qualified in such markets;
·
financial risks, such as longer payment cycles, difficulty enforcing contracts and collecting accounts receivable and exposure to foreign currency exchange rate fluctuations;·
·
reduced protection for intellectual property rights;
·
natural disasters, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; and
·
failure to comply with the Foreign Corrupt Practices Act, including its books and records provisions and its anti-bribery provisions, by maintaining accurate information and control over sales and distributors’ activities.
Any of these risks, if encountered, could significantly harm our future international expansion and operations and, consequently, have a material adverse effect on our financial condition, results of operations and cash flows.
We may acquire other businesses or form joint ventures or make investments in other companies or technologies that could harm our operating results, dilute our stockholders’ ownership, increase our debt or cause us to incur significant expense.
As part of our business strategy, we may pursue acquisitions of businesses and assets. We also may pursue strategic alliances and joint ventures that leverage our core technology and industry experience to expand our offerings or distribution. We have no experience with acquiring other companies and limited experience with forming strategic alliances and joint ventures. We may not be able to find suitable partners or acquisition candidates, and we may not be able to complete such transactions on favorable terms, if at all. If we make any acquisitions, we may not be able to integrate these acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. Any future acquisitions also could result in significant write-offs or the incurrence of debt and contingent liabilities, any of which could have a material adverse effect on our financial condition, results of operations and cash flows. Integration of an acquired company also may disrupt ongoing operations and require management resources that would otherwise focus on developing our existing business. We may experience losses related to investments in other companies, which could have a material negative effect on our results of operations. We may not identify or complete these transactions in a timely manner, on a cost-effective basis, or at all, and we may not realize the anticipated benefits of any acquisition, technology license, strategic alliance or joint venture.
To finance any acquisitions or joint ventures, we may choose to issue shares of our common stock as consideration, which would dilute the ownership of our stockholders. If the price of our common stock is low or volatile, we may not be able to acquire other companies or fund a joint venture project using our stock as consideration. Alternatively, it may be necessary for us to raise additional funds for acquisitions through public or private financings. Additional funds may not be available on terms that are favorable to us, or at all.
41
Declining general economic or business conditions may have a negative impact on our business.
Continuing concerns over United States health care reform legislation and energy costs, geopolitical issues, the availability and cost of credit and government stimulus programs in the United States and other countries have contributed to increased volatility and diminished expectations for the global economy. These factors, combined with low business and consumer confidence and high unemployment, precipitated an economic slowdown and recession. If the economic climate does not improve or continues to deteriorate, our business, as well as the financial condition of our suppliers and our third-party payors, could be adversely affected, resulting in a negative impact on our business, financial condition and results of operations.
The U.S. government may have “march-in rights” to certain of our intellectual property.
Because federal grant monies were used in support of the research and development activities that resulted in certain of our issued pending U.S. patent applications, the federal government retains what are referred to as “march-in rights” to patents that are granted on these applications.
In particular, the National Institutes of Health, which administered grant monies to the primary inventor of the technology we license, technically retain the right to require us, under certain specific circumstances, to grant the U.S. government either a nonexclusive, partially exclusive or exclusive license to the patented invention in any field of use, upon terms that are reasonable for a particular situation. Circumstances that trigger march-in rights include, for example, failure to take, within a reasonable time, effective steps to achieve practical application of the invention in a field of use, failure to satisfy the health and safety needs of the public and failure to meet requirements of public use specified by federal regulations. The National Institutes of Health can elect to exercise these march-in rights on their own initiative or at the request of a third-party.
Risks Related to Our Common Stock
Certain of our officers and directors have sufficient voting power to make corporate governance decisions that could have a significant effect on us and the other stockholders.
As of March 31, 2014, our officers and directors together beneficially own approximately 34% of our outstanding common stock on a fully diluted basis. Mr. Wolf alone through his direct and indirect holdings beneficially owns approximately 20.6% of our outstanding common stock on a fully diluted basis. As a result, Mr. Wolf, alone will be able to exert a significant degree of influence over our management and affairs and over matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. In addition, this concentration of ownership may delay or prevent a change in our control and might affect the market price of our common stock, even when a change in control may be in the best interest of all stockholders. Furthermore, the interests of this concentration of ownership may not always coincide with our interests or the interests of other stockholders. Accordingly, these stockholders could cause us to enter into transactions or agreements that we would not otherwise consider.
The possible issuance of common stock subject to options and warrants may dilute the interest of stockholders.
In 2009, we adopted a 2009 Stock Option and Restricted Stock Plan under which we may grant awards to purchase 869,565 shares of our common stock, of which, 633,482 options were outstanding and 99,906 shares of restricted stock were outstanding as of December 31, 2013. In addition, as of December 31, 2013, we have 53,159 shares issuable upon exercise of warrants granted to third parties in connection with prior private placements of our equity securities and debt which excludes 125,000 shares of common stock issuable at $12.50 per share upon exercise of warrants issued to the underwriters in connection with our initial public offering. To the extent that outstanding stock options and warrants are exercised, or additional securities are issued, dilution to the interests of our stockholders may occur. Moreover, the terms upon which we will be able to obtain additional equity capital may be adversely affected since the holders of the outstanding options can be expected to exercise them at a time when we would, in all likelihood, be able to obtain any needed capital on terms more favorable to us than those provided in such outstanding options.
42
We have additional securities available for issuance, which, if issued, could adversely affect the rights of the holders of our common stock.
Our Third Amended and Restated Certificate of Incorporation authorizes the issuance of 50,000,000 shares of our common stock and 8,212,500 shares of Preferred Stock. In certain circumstances, the common stock and preferred stock, as well as the awards available for issuance under the 2009 Stock Option and Restricted Stock Plan, can be issued by our board of directors, without stockholder approval. Any future issuances of such stock would further dilute the percentage ownership of us held by holders of Preferred Stock and common stock. In addition, the issuance of Preferred Stock may be used as an “anti-takeover” device without further action on the part of our stockholders, and may adversely affect the holders of the common stock.
We have never paid dividends and have no plans to pay dividends in the future.
Holders of shares of our common stock are entitled to receive such dividends as may be declared by our board of directors. To date, we have paid no cash dividends on our shares of our preferred or common stock and we do not expect to pay cash dividends in the foreseeable future. We intend to retain future earnings, if any, to provide funds for operations of our business. Therefore, any return investors in our preferred or common stock may have will be in the form of appreciation, if any, in the market value of their shares of common stock.
We are an “emerging growth company,” and any decision on our part to comply with certain reduced disclosure requirements applicable to emerging growth companies could make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act enacted in April 2012, and, for as long as we continue to be an emerging growth company, we may choose to take advantage of exemptions from various reporting requirements applicable to other public companies including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, not being required to comply with any new requirements adopted by the Public Company Accounting Oversight Board, or the PCAOB, requiring mandatory audit firm rotation or a supplement to the auditor's report in which the auditor would be required to provide additional information about the audit and the financial statements of the issuer, not being required to comply with any new audit rules adopted by the PCAOB after April 5, 2012 unless the SEC determines otherwise, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We could remain an emerging growth company until the earliest of: (i) the last day of the fiscal year in which we have total annual gross revenues of $1 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of the date of our first sale of common equity securities pursuant to an effective registration statement; (iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer. We cannot predict if investors will find our common stock less attractive if we choose to rely on these exemptions. If some investors find our common stock less attractive as a result of any choices to reduce future disclosure, there may be a less active trading market for our common stock and our stock price may be more volatile. Further, as a result of these scaled regulatory requirements, our disclosure may be more limited than that of other public companies and you may not have the same protections afforded to shareholders of such companies.
Under Section 107(b) of the Jumpstart Our Business Startups Act, emerging growth companies can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
As a result of being a public company, we are subject to additional reporting and corporate governance requirements that will require additional management time, resources and expense.
As a public company we are obligated to file with the U.S. Securities and Exchange Commission annual and quarterly information and other reports that are specified in the U.S. Securities Exchange Act of 1934, as amended, or the Exchange Act. We are also subject to other reporting and corporate governance requirements under the Sarbanes-Oxley Act of 2002, as amended, and the rules and regulations promulgated thereunder, all of which impose significant compliance and reporting obligations upon us and require us to incur additional expense in order to fulfill such obligations.
43
We have identified material weaknesses in our internal controls, and we cannot provide assurances that these weaknesses will be effectively remediated or that additional material weaknesses will not occur in the future. If our internal control over financial reporting or our disclosure controls and procedures are not effective, we may not be able to accurately report our financial results, prevent fraud, or file our periodic reports in a timely manner, which may cause investors to lose confidence in our reported financial information and may lead to a decline in our stock price.
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting, as defined in Rule 13a-15(f) under the Exchange Act. Prior to the closing of our initial public offering in July 2013, we operated as a private company and the number and qualifications of our finance and accounting staff have not been consistent with those of a public company. We have identified material weaknesses in our internal controls with respect to our financial statement closing process of our condensed consolidated financial statements for the years ended December 31, 2013 and 2012. Our management discovered certain conditions that we deemed to be material weaknesses and significant deficiencies in our internal controls, as follows:
A lack of accounting and finance resources as well as effective oversight by those in charge of governance resulted in insufficient controls over timely financial statement preparation and review as well as the preparation and review around accounting for certain complex transactions.
The design of monitoring controls used to assess the design and operating effectiveness of our internal controls is inadequate. We also do not have an adequate internal process to report deficiencies in internal control to management on a timely basis.
We have begun to take actions that we believe will substantially remediate the material weaknesses identified. In response to the identification of our material weaknesses, we: (i) have retained a part-time Chief Financial Officer to segregate the duties of Chief Executive Officer and Chief Financial Officer; (ii) are in the process of establishing a review process for key aspects of our financial reporting process, including the accounting for complex transactions; and (iii) will seek to establish better operating controls and involve our board of directors in our internal controls process, which will involve establishing formal procedures to communicate deficiencies in internal controls on a timely basis, and encourage our board of directors to more actively participate in guiding management as it relates to internal controls matters. However, we cannot assure you that our internal control over financial reporting, as modified, will enable us to identify or avoid material weaknesses in the future. We will be required to expend time and resources to further improve our internal controls over financial reporting, including by expanding our finance and accounting staff.
Future sales of our common stock by our existing shareholders could cause our stock price to decline.
We currently have 6,452,341 shares of our common stock outstanding, all of which are currently eligible for sale in the public market, subject, in certain circumstances to the volume, manner of sale and other limitations under Rule 144 or 701 promulgated under the Securities Act. It is conceivable that shareholders may wish to sell some or all of their shares. If our shareholders sell substantial amounts of our common stock in the public market at the same time, the market price of our common stock could decrease significantly due to an imbalance in the supply and demand of our common stock. Even if they do not actually sell the stock, the perception in the public market that our shareholders might sell significant shares of our common stock could also depress the market price of our common stock.
A decline in the price of shares of our common stock might impede our ability to raise capital through the issuance of additional shares of our common stock or other equity securities, and may cause shareholders to lose part or all of their investment in our shares of common stock.
44
Our shares of common stock are from time to time thinly traded, so stockholders may be unable to sell at or near ask prices or at all if they need to sell shares to raise money or otherwise desire to liquidate their shares.
Our common stock has from time to time been “thinly-traded,” meaning that the number of persons interested in purchasing our common stock at or near ask prices at any given time may be relatively small or non-existent. This situation is attributable to a number of factors, including the fact that we are a small company that is relatively unknown to stock analysts, stock brokers, institutional investors and others in the investment community that generate or influence sales volume, and that even if we came to the attention of such persons, they tend to be risk-averse and would be reluctant to follow an unproven company such as ours or purchase or recommend the purchase of our shares until such time as we became more seasoned and viable. As a consequence, there may be periods of several days or more when trading activity in our shares is minimal or non-existent, as compared to a seasoned issuer which has a large and steady volume of trading activity that will generally support continuous sales without an adverse effect on share price. We cannot give stockholders any assurance that a broader or more active public trading market for our common shares will develop or be sustained, or that current trading levels will be sustained.
Our need for future financing may result in the issuance of additional securities which will cause investors to experience dilution.
Our cash requirements may vary from those now planned depending upon numerous factors, including the result of future research and development activities. We believe that the proceeds we received from the sale of the shares in our initial public offering and our private placement will provide us with sufficient working capital for at least the next twelve months. Thereafter, we expect to require additional funds in the future to conduct additional clinical trials. There are no other commitments by any person for future financing. Our securities may be offered to other investors at a price lower than the price per share offered to current shareholders, or upon terms which may be deemed more favorable than those offered to current shareholders. In addition, the issuance of securities in any future financing may dilute an investor's equity ownership. Moreover, we may issue derivative securities, including options and/or warrants, from time to time, to procure qualified personnel or for other business reasons. The issuance of any such derivative securities, which is at the discretion of our board of directors, may further dilute the equity ownership of our stockholders. No assurance can be given as to our ability to procure additional financing, if required, and on terms deemed favorable to us. To the extent additional capital is required and cannot be raised successfully, we may then have to limit our then current operations and/or may have to curtail certain, if not all, of our business objectives and plans.
Certain provisions of the General Corporation Law of the State of Delaware may have anti-takeover effects which may make an acquisition of our company by another company more difficult.
We are subject to the provisions of Section 203 of the General Corporation Law of the State of Delaware, which prohibits a Delaware corporation from engaging in any business combination, including mergers and asset sales, with an interested stockholder (generally, a 15% or greater stockholder) for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner. The operation of Section 203 may have anti-takeover effects, which could delay, defer or prevent a takeover attempt that a holder of our common stock might consider in its best interest.
45
Item 1B.
Unresolved Staff Comments
None.
Item 2.
Properties
Facilities
Our executive offices are located at 100 Europa Drive, Chapel Hill, North Carolina. We currently lease approximately 2,111 square feet of office space at such location for monthly rent of $4,046 on a month to month basis and intend to continue to do so until our new office space is available. On January 24, 2014 we entered into a five year lease for 5,303 square feet of office and laboratory space at 801 Capitola Drive, Chapel Hill, North Carolina 27517 for monthly rent of $10,341 exclusive of payments required for maintenance of common areas and utilities. We intend to move our executive offices to the Capitola Drive location at the end of April 2014. Based on our current operational plans, we believe that such facilities are adequate for our operations for the near future.
Item 3.
Legal Proceedings
None.
Item 4.
Mine Safety Disclosures
Not applicable.
46
PART II
Item 5.
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchase of Equity Securities
Our common stock has traded on the NASDAQ under the symbol “HTBX” since July 29, 2013. Prior to that time, there was no public market for our common stock. As a result, we have only set forth quarterly information with respect to high and low prices for our common stock for the two most recent fiscal quarters. The following table states the range of the high and low sales prices of our common stock for each of the last two calendar quarters during the year ended December 31, 2013. These quotations represent inter-dealer prices, without retail mark-up, markdown, or commission, and may not represent actual transactions. The last price of our common stock as reported on the NASDAQ on March 27, 2014 was $6.64 per share. As of March 27, 2014, there were approximately 31 stockholders of record of our common stock. This number does not include beneficial owners from whom shares are held by nominees in street name.
| | | | | | | | |
| | High | | | Low | |
YEAR ENDED DECEMBER 31, 2013 | | | | | | | | |
Fourth quarter | | $ | 15.29 | | | | 7.01 | |
Third quarter | | $ | 13.50 | | | | 9.01 | |
Dividend Policy
We have never paid any cash dividends on our common stock to date, and do not anticipate paying such cash dividends in the foreseeable future. Whether we declare and pay dividends is determined by our Board of Directors at their discretion, subject to certain limitations imposed under Delaware corporate law. The timing, amount and form of dividends, if any, will depend on, among other things, our results of operations, financial condition, cash requirements and other factors deemed relevant by our Board of Directors.
Equity Compensation Plan Information
Securities Authorized for Issuance Under Equity Compensation Plans
The following table contains information about our equity compensation plans as of December 31, 2013.
Equity Compensation Plan Information
| | | | | | | | |
Plan Category | | Number of
securities to be
issued upon
exercise of
outstanding
options (1) | | Weighted-average
exercise price of
outstanding
options | | Number of
securities
remaining
available for
future issuance
under equity
compensation
plans (excluding
securities reflected
in column (a)) | |
| | (a) | | (b) | | (c) | |
Equity compensation plans approved by security holders | | | | | | | |
2009 Equity Incentive Plan | | 633,482 | | $ | 3.36 | | 32,644 | |
| | | | | | | |
Equity compensation plans not approved by security holders | | — | | — | | — | |
Total | | 633,482 | | — | | 32,644 | |
———————
(1)
Does not include 99,906 shares of restricted stock which are fully vested and 103,583 shares of common stock issued upon option exercises. Does not include options exercisable for 32,251 shares of common stock that were issued subsequent to year end.
47
Recent Sales of Unregistered Securities
All sales of unregistered securities have been previously reported.
Purchase of Equity Securities
We have not purchased any of our registered securities during the period covered by this Annual Report on Form 10-K.
Use Of Proceed From Registered Securities
In connection with our initial public offering, we sold 2,700,000 (including the 200,000 over-allotment option shares) shares of our common stock at a price of $10.00 per share. Aggregate gross proceeds from the IPO, were $27 million and net proceeds received after underwriting commissions and offering expenses of $2.7 million were approximately $24.3 million.
As of December 31, 2013, we have used approximately $4.2 million of the net proceeds, in connection with our clinical trials, manufacturing and general and administrative expenses. Following year-end certain bonuses were paid to our executive officers and other employees in the amount of $183,125. There has been no material change in our planned use of the balance of the net proceeds from the offering as described in the prospectus filed with the SEC pursuant to Rule 424(b) under the Securities Act other than as previously reported.
Item 6.
Selected Financial Data
Not applicable because we are a smaller reporting company.
Item 7.
Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion of our financial condition and results of operations should be read in conjunction with the audited financial statements and notes thereto for the year ended December 31, 2013 found in this report. In addition to historical information, the following discussion contains forward-looking statements that involve risks, uncertainties and assumptions. Where possible, we have tried to identify these forward looking statements by using words such as “anticipate,” “believe,” “intends,” or similar expressions. Our actual results could differ materially from those anticipated by the forward-looking statements due to important factors and risks including, but not limited to, those set forth under “Risk Factors” in Part I, Item 1A of this Report.
OVERVIEW
We are a development stage biopharmaceutical company engaged in the development of novel allogeneic, “off-the-shelf” cellular therapeutic vaccines to combat a wide range of cancers and infectious diseases. Our proprietaryImPACT™ImmunePanAntigenCytotoxicTherapy is being designed to deliver live, genetically-modified, irradiated human cells which are reprogrammed to “pump out” a broad spectrum of cancer-associated antigens together with a potent immune adjuvant called “gp96” to educate and activate a cancer patient’s immune system to recognize and kill cancerous cells. We intend for ourImPACT cells to secrete an antigen-adjuvant complex that generates anti-cancer immune responses in patients by mobilizing and activating cytotoxic “killer” T cells that target multiple cancer antigens, thus harnessing a patient’s own immune system to fight cancer.
Unlike autologous or “personalized” therapeutic vaccine approaches which require extraction and processing of cancer or blood from each individual patient, ourImPACT therapeutic vaccine uses a master cell line containing a host of known and unknown tumor associated antigens to mass-produce a single vaccine product applicable to all patients with a particular cancer type. We believe our off-the-shelf, allogeneic immunotherapy offers logistical, manufacturing and cost benefits compared to autologous patient-specific approaches.
48
We commenced active operations in June 2008. Our operations to date have been primarily limited to organizing and staffing our company, business planning, raising capital, acquiring and developing our technology, identifying potential product candidates and undertaking preclinical and clinical studies of our most advanced product candidates. To date, we have not generated any revenues and have financed our operations with net proceeds from the private placement of our preferred stock and our initial public offering in which we received gross proceeds of $27 million. As of December 31, 2013, we had a deficit accumulated during the development stage of $12,346,630. We had net losses of $6,609,864 and $2,471,147 for the years ended December 31, 2013 and 2012, respectively. We expect to incur significant expenses and increasing operating losses for the foreseeable future. We expect our expenses to increase in connection with our ongoing activities, particularly as we continue the research and development and initiate and conduct clinical trials of, and seek marketing approval for, our product candidates. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant commercialization expenses related to product sales, marketing, manufacturing and distribution. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. Adequate additional financing may not be available to us on acceptable terms, or at all. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce or eliminate our research and development programs or any future commercialization efforts. We expect our existing cash will enable us to fund our current operating plan and capital expenditure requirements for at least 12 months. This is based on our current estimates, and we could use our available capital resources sooner than we currently expect. We will need to generate significant revenues to achieve profitability, and we may never do so.
CRITICAL ACCOUNTING POLICIES AND SIGNIFICANT JUDGMENTS AND ESTIMATES
We believe that several accounting policies are important to understanding our historical and future performance. We refer to these policies as "critical" because these specific areas generally require us to make judgments and estimates about matters that are uncertain at the time we make the estimate, and different estimates—which also would have been reasonable—could have been used, which would have resulted in different financial results.
Our management’s discussion and analysis of financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of our consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenue and expenses and related disclosure of contingent assets and liabilities. On an ongoing basis, we evaluate our estimates based on historical experience and make various assumptions, which management believes to be reasonable under the circumstances, which form the basis for judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
The Company has elected to follow the extended transition period guidance provided for in Securities Act Section 7(a)(2)(B) for complying with new or revised accounting standards. The Company will disclose the date on which adoption of such standards is required for non-emerging growth companies and the date on which the Company will adopt the recently issued accounting standards.
The notes to our audited consolidated financial statements contain a summary of our significant accounting policies. We consider the following accounting policies critical to the understanding of the results of our operations:
·
Revenue Recognition
·
Stock-based compensation, and
·
Research and development costs
Revenue Recognition
We recognize government grants when there is reasonable assurance that they will comply with the conditions attached to the grants and the grants will be received. The grants are recognized using an income approach and grant revenue is recognized as the related expenses are incurred.
49
Stock-Based Compensation
Calculating stock-based compensation expense requires the input of highly subjective assumptions. We apply the Black-Scholes-Merton option pricing model to determine the fair value of our stock options. Inherent in this model are assumptions related to expected stock-price volatility, option life, risk-free interest rate and dividend yield. We estimate the volatility of our common stock at the date of grant based on historical volatility. We estimate the expected life of our option using the contractual term of the option. The risk-free interest rate is based on the U.S. Treasury zero-coupon yield curve on the grant date for a maturity similar to the expected life of the options. The dividend rate is based on our historical rate, which we anticipate to remain at zero. The assumptions used in calculating the fair value of stock options represent our best estimates, however these estimates involve inherent uncertainties and the application of management judgment. As a result, if factors change and different assumptions are used, the stock-based compensation expense could be materially different in the future. In addition, we are required to estimate the expected forfeiture rate and only recognize expense for those stock options expected to vest over the service period.
Research and Development Costs
We expense research and development costs associated with developmental products not yet approved by the FDA to research and development expense as incurred. Research and development costs consist primarily of license fees (including upfront payments), milestone payments, manufacturing costs, salaries, stock-based compensation and related personnel costs, fees paid to consultants and outside service providers for laboratory development, legal expenses resulting from intellectual property prosecution and other expenses relating to the design, development, testing and enhancement of our product candidates.
RESULTS OF OPERATIONS
Year Ended December 31, 2013 and 2012
Revenues
Our revenues are entirely comprised of grant awards. There were no grant awards in 2013 and $3,110 in grant awards in 2012. We will continue our efforts to secure future grant funding to subsidize ongoing research and developments costs.
Operating Expenses
Total operating expenses for the year ended December 31, 2013 (the “2013 Period”) increased approximately 180% to approximately $6,564,641 compared to $2,345,787 for the year ended December 31, 2012 (the “2012 Period”). Operating expenses are primarily comprised of research and development, clinical and regulatory and general and administrative expenses. For the 2013 Period, research and development expenses were $2,737,688, clinical and regulatory expenses were $1,397,157 and general and administrative expenses were $2,429,796 as compared to research and development expenses of $902,938, clinical and regulatory expenses of $253,189 and general and administrative expenses of $1,189,660 for the 2012 Period. For the year ended December 31, 2013, research and development expenses represented approximately 42% of operating expenses, clinical trials and regulatory represented approximately 21% of operating expenses, and general and administrative expenses represented approximately 37% of operating expenses. For the year ended December 31, 2012, research and development expenses represented approximately 38% of operating expenses, clinical trials and regulatory represented approximately 11% of operating expenses, and general and administrative expenses represented approximately 51% of operating expenses.
Research and development expense.
Research and development expense for the 2013 Period increased 203% to $2,737,688 compared to $902,938 for the the 2012 Period. The $1,834,750 increase from the 2012 Period to the 2013 Period is primarily related to an increase of $1,530,000 in pre-manufacturing costs associated with preparing to produce vaccines for use in our clinical trials. Personnel costs, including outside consultants, increased by $187,000 primarily due to increased stock compensation expense. Patent costs also increased by $118,000 as we continued our efforts to expand our patent portfolio.
50
Clinical and regulatory expense.
Clinical and regulatory expense for the 2013 Period increased 452% to $1,397,157 compared to $253,189 for the 2012 Period. The $1,143,968 increase from the 2012 Period to the 2013 Period resulted from an increase of $698,000 in manufacturing and other clinical trial expenditures incurred in preparation for the initiation of the clinical trials. Consulting related to clinical trials increased by $293,000 from the 2012 Period to the 2013 Period. Personnel costs for our clinical and research staff also increased by $153,000 as we moved closer to launching clinical trials.
General and administrative expense.
General and administrative expense for the 2013 Period increased 104% to $2,429,796 compared to $1,189,660 for the 2012 Period. The $1,240,136 increase from the 2012 Period to the 2013 Period resulted from an increase of $743,000 in personnel costs, including consultants, of which $321,000 was non-cash stock based compensation. The remainder was primarily attributable to the hiring of a Director of Finance and Chief Financial Officer and related employee benefits associated with these positions. Insurance expense increased by $184,000 related primarily to directors and officers insurance that increased when the company went public. Marketing expense increased by $129,000 due to an increase in expenses such as the website enhancement and the initial public offering road show. Travel expense increased by $93,000 primarily related to fund-raising activities. The Company incurred $86,000 in additional costs associated with being a public company during the 2013 Period.
Interest expense
Interest expense decreased to $79,119 for the 2013 Period from $101,086 for the 2012 Period as the majority of the Company’s debt was extinguished in the 2013.
BALANCE SHEET AS OF DECEMBER 31, 2013 AND 2012
Prepaid expenses.
Prepaid expenses were $1,066,638 as of December 31, 2013 compared to $58,436 as of December 31, 2012. The increase of $1,008,202 was due primarily to prepayments for contract research, which increased by $643,000, as the Company prepared for clinical trials. Prepayments related to insurance, which increased due to the company becoming public in 2013, increased by $214,000 from the 2012 Period to the 2013 Period. The Company also had prepayments for software enhancements, brokers and other entities in the amount of $151,000 that did not exist at the end of the 2012 Period.
Accounts Payable.
Accounts payable was $651,917 as of December 31, 2013 compared to $505,471 as of December 31, 2012. This increase of $146,446 was primarily related to an increase of volume in payments as the company increased activity related to clinical trials.
LIQUIDITY AND CAPITAL RESOURCES
Sources of liquidity
To date, we have not generated any revenues. Since our inception in June, 2008, we have financed our operations principally through private placements and through our initial public offering, which we closed in July, 2013 and the closings of the partial exercises of the underwriter’s over-allotment option, which we closed in August 2013 and September 2013. The total gross proceeds raised from the offering and over-allotment option were $27 million, before underwriting discounts and commissions and other offering expenses payable by the Company for net proceeds of approximately $24.3 million. We believe that the proceeds we received from the sale of the shares in our private placement and our initial public offering will provide us with sufficient working capital to fund our current operating plans and capital expenditure requirements for at least 12 months. Thereafter, we expect to require additional funds in the future to conduct additional clinical trials. As of December 31, 2013, we had $21,864,157 in cash and cash equivalents and short term investments.
51
Cash flows
Operating activities. The use of cash in all periods resulted primarily from our net losses adjusted for non-cash charges and unfavorable changes in the components of working capital. The significant increase in cash used in operating activities for the 2013 Period compared to the 2012 Period is due to an increase in operating expenses as we increased manufacturing costs for both research, development and clinical and regulatory as we prepare for clinical trials, as well as an increase in general and administrative costs primarily associated with our initial public offering and costs associated with being a public company.
Investing activities. The use of cash in the 2013 Period was primarily due to the purchase of short term investments which were purchased with the cash obtained from the initial public offering in July 2013.
Financing activities. Cash provided by financing activities during the 2013 Period of approximately $28.4 million resulted primarily from the initial public offering and partial exercises of the over-allotment option which resulted in gross proceeds to us of approximately $24.3 million after underwriting discounts and commissions and other offering expenses paid by the Company.
OFF-BALANCE SHEET ARRANGEMENTS
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined under Securities and Exchange Commission rules.
Current and Future Financing Needs
We have incurred an accumulated deficit of $12,346,630 through December 31, 2013. We have incurred negative cash flows from operations since we started our business. We have spent, and expect to continue to spend, substantial amounts in connection with implementing our business strategy, including our planned product development efforts, our clinical trials, and our research and discovery efforts.
Based on our current plans, we believe that our cash will be sufficient to enable us to meet our planned operating needs for at least the next 12 months.
However, the actual amount of funds we will need to operate is subject to many factors, some of which are beyond our control. These factors include the following:
·
the progress of our research activities;
·
the number and scope of our research programs;
·
the progress of our preclinical and clinical development activities;
·
the progress of the development efforts of parties with whom we have entered into research and development agreements;
·
our ability to maintain current research and development licensing arrangements and to establish new research and development and licensing arrangements;
·
our ability to achieve our milestones under licensing arrangements;
·
the costs involved in prosecuting and enforcing patent claims and other intellectual property rights;
·
the costs and timing of regulatory approvals; and
·
profitability of our clinical laboratory diagnostic and microbiology services business.
We have based our estimate on assumptions that may prove to be wrong. We may need to obtain additional funds sooner or in greater amounts than we currently anticipate. Potential sources of financing include strategic relationships, public or private sales of our shares or debt and other sources. We may seek to access the public or private equity markets when conditions are favorable due to our long-term capital requirements. We do not have any committed sources of financing at this time, and it is uncertain whether additional funding will be available when we need it on terms that will be acceptable to us, or at all. If we raise funds by selling additional shares of common stock or other securities convertible into common stock, the ownership interest of our existing stockholders will be diluted. If we are not able to obtain financing when needed, we may be unable to carry out our business plan. As a result, we may have to significantly limit our operations and our business, financial condition and results of operations would be materially harmed.
52
License and Contractual Agreement Obligations
Below is a table of our contractual obligations for the years 2014 through 2018 as of December 31, 2013(in thousands).
| | | | | | | | | | | | | | | | | | | | | | | | |
| | Year ended December 31, | | | | |
| | 2014 | | | 2015 | | | 2016 | | | 2017 | | | 2018 | | | Total | |
License Agreements | | $ | 30,000 | | | $ | 30,000 | | | $ | 30,000 | | | $ | 280,000 | | | $ | 30,000 | | | $ | 400,000 | |
Lease Agreements(1) | | | 82,353 | | | | 183,137 | | | | 188,631 | | | | 194,290 | | | | 200,119 | | | | 848,530 | |
Total | | $ | 112,353 | | | $ | 213,137 | | | $ | 218,631 | | | $ | 474,290 | | | $ | 230,119 | | | $ | 1,248,530 | |
———————
(1)
We anticipate moving to new office space in April 2014. The numbers set forth above for lease agreements include lease payments for the new office space for a portion of 2014 and lease payments for new office space for the full years of 2015, 2016, 2017 and 2018.
Additional In-Licensed Programs
We may enter into additional license agreements relating to new product candidates.
Item 7A.
Quantitative and Qualitative Disclosures About Market Risk
Not applicable because we are a smaller reporting company.
Item 8.
Financial Statements and Supplemental Data
See pages F-1 through F-27.
Item 9.
Changes In and Discussions with Accountants on Accounting and Financial Disclosures
None
Item 9A.
Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Disclosure Controls and Procedures
Our management has adopted and maintains disclosure controls and procedures that are designed to provide reasonable assurance that information required to be disclosed in the reports filed under the Exchange Act, such as this Form 10-K, is collected, recorded, processed, summarized and reported within the time periods specified in the rules of the SEC. Our disclosure controls and procedures are also designed to ensure that such information is accumulated and communicated to management to allow timely decisions regarding required disclosure. As required under Exchange Act Rule 13a-15, our management, including the Chief Executive Officer and Chief Financial Officer, has conducted an evaluation of the effectiveness of disclosure controls and procedures as of the end of the period covered by this report. Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures are not effective at the reasonable assurance level due to the material weaknesses discussed in ITEM 1A. Risk Factors to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act, is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
53
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Exchange Act Rule 13a-15. Our internal control over financial reporting is designed to provide reasonable assurance to our management and Board of Directors regarding the preparation and fair presentation of published financial statements. Management conducted an assessment of our internal control over financial reporting based on the framework and criteria established by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (1992). Based on the assessment, management concluded that, as of December 31, 2013, our internal controls over financial reporting were not effective based on those criteria. Prior to the closing of our initial public offering in July 2013, we operated as a private company and the number and qualifications of our finance and accounting staff have not been consistent with those of a public company. We have identified material weaknesses in our internal controls with respect to our financial statement closing process of our consolidated financial statements for the year ended December 31, 2013. Our management discovered certain conditions that we deemed to be material weaknesses and significant deficiencies in our internal controls, as follows:
·
A lack of accounting and finance resources as well as effective oversight by those in charge of governance resulted in insufficient controls over timely financial statement preparation and review as well as the preparation and review around accounting for certain complex transactions.
·
The design of monitoring controls used to assess the design and operating effectiveness of our internal controls is inadequate. We also do not have an adequate internal process to report deficiencies in internal control to management on a timely basis.
We have begun to take actions that we believe will substantially remediate the material weaknesses identified. In response to the identification of our material weaknesses, we: (i) have retained a part-time Chief Financial Officer to segregate the duties of Chief Executive Officer and Chief Financial Officer; (ii) are in the process of establishing a review process for key aspects of our financial reporting process, including the accounting for complex transactions; and (iii) will seek to establish better operating controls and involve our board of directors in our internal controls process, which will involve establishing formal procedures to communicate deficiencies in internal controls on a timely basis, and encourage our board of directors to more actively participate in guiding management as it relates to internal controls matters. However, we cannot assure you that our internal control over financial reporting, as modified, will enable us to identify or avoid material weaknesses in the future. We will be required to expend time and resources to further improve our internal controls over financial reporting, including by expanding our finance and accounting staff.
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures and our internal control processes will prevent all error and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of error or fraud, if any, within our company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that the breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the control. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions, or the degree of compliance with the policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and may not be detected. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Changes in Internal Control over Financial Reporting
There has been no change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act) that occurred during our fiscal quarter ended December 31, 2013 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
54
This Annual Report on Form 10-K does not include an attestation report of the Company’s registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by the Company’s registered public accounting firm pursuant to rules of the SEC that permit the Company to provide only management’s report in this Annual Report on Form 10-K.
Item 9B.
Other Information
None.
55
PART III
Item 10.
Directors, Executive Officers and Corporate Governance
Below is certain information regarding our directors and executive officers.
| | | | | | |
Name | | Age | | Position | | Served as an Officer
or Director Since |
Jeffrey Wolf | | 50 | | Chairman, Chief Executive Officer and Director | | 2008 |
Matthew E. Czajkowski | | 64 | | Chief Financial Officer | | 2013 |
Melissa Price Ph.D. | | 40 | | Vice President of Clinical and Regulatory Affairs | | 2013 |
Anil K. Goyal Ph.D. | | 50 | | Vice President of Business Development | | 2013 |
Vadim Deyev, MD, Ph.D. | | 49 | | Director of Applied Research | | 2011 |
Taylor Schreiber | | 34 | | Vice President of Research and Development | | 2014 |
John Monahan, Ph.D. | | 67 | | Director | | 2009 |
Paul Belsky, MD | | 57 | | Director | | 2009 |
Michael Kharitonov, Ph.D. | | 50 | | Director | | 2009 |
Edward B. Smith | | 38 | | Director | | 2009 |
Louis C. Bock | | 49 | | Director | | 2013 |
All of the officers listed above are full-time employees of the Company other than Mr. Czajkowski, who works on a part-time basis.
Jeffrey Wolf,Chairman and Chief Executive Officer
Mr. Wolf founded Heat Biologics in August, 2008. Prior to founding Heat, from June 1997 to March 2011, Mr. Wolf has served as managing director at Seed-One Ventures, LLC a venture firm focused on launching and growing exceptional healthcare companies from the ground up. Since founding Seed-One, Mr. Wolf has founded and run several medical companies. Mr. Wolf’s start-ups include Avigen, a San Francisco-based gene therapy company where he was a co-founder and director; TyRx Pharma, a Princeton-based company focused on the development of bio-compatible polymers where he was a co-founder and Chairman; EluSys Therapeutics, a New Jersey company focused on the development of a novel technology to remove blood-borne pathogens where he was a co-founder, Chairman and Chief Executive Officer; and GenerationOne, a Miami-based company focused on mobile-based collaborative care, where he was the founder, Chairman and Chief Executive Officer. Mr. Wolf received his M.B.A. from Stanford Business School, his J.D. from New York University School of Law and his B.A. from the University of Chicago, where he graduated with honors in Economics.Mr. Wolf serves as a director of several Seed-One portfolio companies and serves as a director of Synthetic Biologics, Inc., a biotechnology company focused on the development of novel anti-infective biologic and drug candidates targeting specific pathogens that cause serious infections and other diseases.
We selected Mr. Wolf to serve on our Board as our chairman because he brings to the board extensive knowledge of the pharmaceutical and biotechnology industries. Having served in senior corporate positions in several biomedical companies, he has a vast knowledge of the industry and brings to the board significant executive leadership and operational experience. His business experience provides him with a broad understanding of the operational, financial and strategic issues facing public companies and his service on other public company boards provides him with extensive corporate governance knowledge.
Matthew Czajkowski,Chief Financial Officer
Mr. Czajkowski joined Heat Biologics in May 2013 as its Chief Financial Officer. Prior to joining Heat Biologics, Mr. Czajkowski worked from 2011-2012 as the Chief Executive Officer of NextRay, Inc., a company developing x-ray imaging technology. From 2007 -2010, he served as an independent advisor to various mid stage software and biotech companies where his responsibilities included fundraising. Prior thereto, from 2004-2006, he served as the Chief Financial and Administrative Officer of AAI Pharma Inc. and was part of the work out team for its Chapter 11 filing and from 2000-2004 served as the Chief Financial Officer of Pozen Inc., a publically traded biotechnology company. Prior to this, Mr. Czajkowski was at Goldman, Sachs & Co. where he founded and ran their Asia/Pacific mergers and acquisitions business. Mr. Czajkowski received his MBA from Harvard University in 1983 and his BA from Harvard University in 1977.
56
Melissa Price, Ph.D.,Vice President of Clinical and Regulatory Affairs
Dr. Price is responsible for coordinating the clinical development and operational efforts at Heat Biologics. Prior to joining Heat Biologics, Inc.,Dr. Price served in various positions at INC Research including Vice President of Global FSP Solutions at INC Research from February 2012 until October 2013 and Executive Director, Strategic Alliance Management from January 2010 until February 2012. From June 2009 until January 2010, Dr. Price served as the Senior Director, Drug Development Partnerships at Novaquest, a Quintiles Company. Prior thereto, from 2006 until 2009 she served in various positions at INC Research.Dr. Price received herPh.D. in Organic Chemistry from Yale University.
Anil Goyal, Ph.D., Vice President of Business Development
Dr Goyal joined Heat Biologics in December 2013 as Vice President of Business Development of the Company. Prior to joining Heat Biologics,Dr. Goyal served as President and Chief Executive Officer of Qualiber, Inc., a company which he co-founded, from April 2010 until December 2013 and Managing Director of OpenDoors Group, LLC, a company he founded, from August 2008 until December 2013. From January 2009 until January 2010, Dr. Goyal served as the Vice President of Business Development at Optherion, Inc. and from January 2003 until January 2008 he served as Vice President of Business Development of Serenex, Inc. Prior thereto, he served in various key management and development positions at Millennium Pharmaceuticals, Genome Therapeutics Corporation and Merck & Co.
Vadim V. Deyev, M.D., Ph.D.,Director of Applied Research
Dr. Deyev joined Heat Biologics in January 2009 as Director of Applied Research. Prior to joining Heat Biologics, Dr. Deyev worked from 2006-2008 as Associate Scientist of Microbiology and Immunology and Hybridoma and Fusion Protein Core Director at the University of Miami School of Medicine. Working with Dr. Eckhard Podack, Heat Biologics’ Scientific Advisor and Chairman of its Scientific Advisory Board, Dr. Deyev has made major contributions to the development of technologies later licensed by the Company. Since 2001, Dr. Deyev has authored numerous publications on immunology and oncology based upon his work with Dr. Podack at the University of Miami. Dr. Deyev joined the team at University of Miami in 1996 until present, after leading the Immunopharmacology Group at the Cancer Research Center in Moscow, Russia Dr. Deyev received his Ph.D. in Immunology/Oncology from Cancer Research Center in Moscow, Russia and his M.D. from Russian State Medical University.
Taylor H. Schreiber Ph.D.,Vice President of Research and Development
Dr. Schreiber joined Heat Biologics in March 2014 as Vice President of Research and Development. Dr. Schreiber is the co-inventor of significant elements of the Company’sImPACT Technology platform and has been intimately involved in the progression of gp96 heat shock protein immunotherapy both as a Ph.D. researcher and as a post-doctoral fellow in the laboratory of Eckhard Podack, M.D., Ph.D., the inventor of Heat’sImPACT Technology platform. Dr. Schreiber joins the Company after completing the M.D. / Ph.D. program at the University of Miami Miller School of Medicine, which he attended from 2004 until February 2014. In 2010, Dr. Schreiber received his Ph.D. degree from the Sheila and David Fuente Program in Cancer Biology at the University of Miami Miller School of Medicine after completing the four year Ph.D. program. Following his degree, Dr. Schreiber completed a post-doctoral fellowship with Dr. Eckhard Podack, M.D., Ph.D. studying the immunobiology of TNFRSF25 from 2010-2012. Dr. Schreiber received the best overall research award at the National Student Research Forum in 2008 and was nominated as a Future Leader in Cancer Research by the American Association for Cancer Research in 2011. Dr. Schreiber is an emerging expert in the field of tumor immunology and TNFRSF25 biology.
57
John Monahan, Ph.D.,Director
Dr. Monahan is currently the Chief Technology Officer of Synthetic Biologics, Inc., a biotechnology company focused on the development of synthetic DNA-based therapeutics and innovative disease-modifying medicines for serious illnesses. Dr. Monahan Co-Founded Avigen Inc. (NASDAQ:AVGN) in 1992, a company which has become a leader in its sector for the development of novel pharmaceutical products for the treatment of serious human diseases. Over a 12 year period as CEO of Avigen he raised over $235M in several private and public financings including its IPO. From 1989-1992, he was VP of R&D at Somatix Therapy Corp., Alameda, CA and from 1985-1989 he was Director of Molecular & Cell Biology at Triton Biosciences Inc., Alameda, CA. Prior to that from 1982-1985, he was Research Group Chief, Department of Molecular Genetics, Hoffmann-LaRoche, Inc. Nutley, NJ, and from 1975 to 1977 he was an Instructor at Baylor College of Medicine, Houston TX. He received his Ph.D. in Biochemistry in 1974 from McMaster University, Canada and his B.Sc. from University College Dublin, Ireland in 1969. Dr. Monahan is a board member of Tacere Therapeutics, CA. He is also a board member of a number of Irish biotech companies including Genable, Cellix, Luxcel, Identigen, Pharmatrin and GK Technologies.
We selected Dr. Monahan to serve on our Board because he brings to the board extensive knowledge of the pharmaceutical and biologics industry. Having served in senior corporate positions in many medical companies he has a vast knowledge of the industry.
Paul Belsky, M.D.,Director
Dr. Belsky has served on our Board since November 2009. Dr. Belsky is currently a medical and scientific advisor at Seed-One Ventures and has been a partner at Concorde Medical Group, LLC since June of 1998. Dr. Belsky served as a scientific advisor to Elusys Therapeutics, Sensatex, GenerationOne and TyRx Pharma. Dr. Belsky has extensive expertise in the clinical practice of internal medicine and cardiovascular diseases, and was formerly on the clinical academic faculty at Weill College of Medicine, Cornell University. He is a fellow of the American College of Cardiology and the American College of Chest Physicians, is a member of the American College of Physicians, and a Clinical Assistant Professor of Medicine at New York University School of Medicine. Dr. Belsky received his MD from the University of California at San Francisco, and his AB in Biology from Brown University, where he was elected Phi Beta Kappa.
We selected Dr. Belsky to serve on our Board because he brings to the board extensive knowledge of the medical industry. His medical background aids in the understanding of the detailed science behind our intellectual property.
Michael Kharitonov, Ph.D.,Director
Dr. Kharitonov has been the Chief Executive Officer of Voleon Capital Management, an investment management firm, since July 2007 until present. He is a high technology entrepreneur and computer scientist whose areas of expertise include advanced computer and communication technologies and quantitative finance. Dr. Kharitonov is a founder and CEO of Voleon Capital Management LLC. Dr. Kharitonov was a co-founder and former Chairman and CEO of Netli, Inc., a successful Silicon Valley startup that pioneered the development of Application Delivery Networks. Under Dr. Kharitonov’s leadership Netli raised over $20 million in venture financing from a number of Silicon Valley’s best known venture capital firms. In 2007 Netli was acquired by Akamai Technologies (NASDAQ: AKAM). Dr. Kharitonov also served as a Vice President of D. E. Shaw and Co., an international investment firm known as one of the most quantitatively advanced and computerized securities trading firms in the world. Dr. Kharitonov holds a Ph.D. degree from the Department of Computer Science at Stanford University. At Stanford he was awarded a Hertz Fellowship and was a winner of several scholarly awards. He also holds a B.A. in Computer Science and Mathematics with highest honors from University of California at Berkeley.
We selected Dr. Kharitonov to serve on our Board because he brings a strong start-up and finance background to the Company, and adds significant strategic, business and financial experience. His prior successful management experience and fundraisings provides him with a broad understanding issues faced by growing companies and of the financial markets and the financing opportunities available to us.
58
Edward B. Smith,Director
Since April 2005, Mr. Smith has been the Managing Partner of Brightline Capital Management, LLC (“BCM”), a New York-based investment firm founded in 2005. BCM is the investment manager of Brightline Ventures I, LLC, Brightline Ventures II, LLC, Brightline Ventures III, LLC and Brightline Capital Partners, LP. Prior to founding BCM, Mr. Smith worked at Gracie Capital from 2004-2005, GTCR Golder Rauner from 1999-2001 and Credit Suisse First Boston from 1997-1999. Mr. Smith holds a Bachelor of Arts in Social Studies from Harvard College and a Masters in Business Administration from Harvard Business School. He is currently a Director of Z Trim Holdings Inc. (OTC:ZTHO), a manufacturer of environmentally friendly agricultural functional ingredients.
We selected Mr. Smith to serve on our Board because he brings a strong business background to the Company, and adds significant strategic, business and financial experience. Mr. Smith’s business background provides him with a broad understanding of the issues facing us, the financial markets and the financing opportunities available to us. His service on other public company boards provides him with extensive corporate governance knowledge and insight into issues faced by companies similar to ours.
Louis C Bock,Director
Louis C. Bock was a Managing Director of Scale Venture Partners, a venture capital firm, until 2012. Mr. Bock joined Scale Venture Partners in September 1997 from Gilead Sciences, Inc., a biopharmaceutical company where he worked from September 1989 to September 1997. Prior to Gilead, he was a research associate at Genentech, Inc. from November 1987 to September 1989. He currently serves as a director of the following publicly traded companies: Horizon Therapeutics, Inc., for which he also serves as a member of the audit and compensation committees, and Zogenix, Inc., for which he also serves as a member of the audit committee. In addition, Mr. Bock serves on the board of directors of the following privately-held companies: Ascenta Therapeutics, Inc., for which he also serves as a member of the audit committee, and Sonexa Therapeutics, Inc., and also serves on the board of directors of Arizona Technology Enterprises, LLC, a non-profit organization. Mr. Bock is responsible for Scale Venture Partners’ investment in Somaxon Pharmaceuticals, Inc. In the past five years, Mr. Bock has also served as a member of the boards of directors of the following publicly traded companies: diaDexus Inc. and SGX Pharmaceuticals, Inc. Mr. Bock received his B.S. in Biology from California State University, Chico and an M.B.A. from California State University, San Francisco.
We selected Mr. Bock to serve on our Board because of his extensive clinical and leadership experience in the biotechnology and biopharmaceuticals industries, including experience in research, project management, business development and sales from his time at Gilead. His membership on other companies’ boards of directors, including positions on other audit and nominating/corporate governance committees provides him with extensive corporate governance knowledge and insight into issues faced by companies similar to ours.
Scientific Advisory Board
In addition to our Board, we also have a scientific advisory board comprised of six individuals. The Scientific Advisory Board is responsible for providing scientific advice and for assessing the scientific progress of our research and development efforts. We have entered into written agreements and confidentiality agreements with all of our members of our Scientific Advisory Board. The members of our Scientific Advisory Board are compensated for their services. Drs. Allison, Stebbing and Nemunaitis are each entitled to receive $1,500 per board meeting in addition to a reimbursement for travel and related. In addition, Drs. Allison, Stebbing and Von Hoff each received options to purchase 15,000 shares of our common stock, which options vest over a four year period. Dr. Von Hoff is entitled to receive $4,000 per onsite advisory board meeting, $2,000 per telephonic meeting and an hourly rate of $500 per hour for consultative discussions with management. Dr. Podack receives consulting fees equal to $3,125 per month subject to increase to $4,167 per month.
59
Eckhard Podack, M.D., Ph.D.,Scientific Advisor and Chairman, Scientific Advisory Board
Dr. Podack, the inventor of the Company’s technology, serves as Chairman of its Scientific Advisory Board. Dr. Podack received his medical degree from the Johan Wolfgang Goethe University in Frankfurt in 1968 and his Medical License in 1970. Following service in the German Army as Captain and Battalion Physician, he completed his Ph.D. in the field of Biochemistry at the Georg August University in Gottingen. From 1974-1984 he studied Immunology at the Scripps Clinic and Research Foundation in La Jolla CA where he received an Established Investigatorship from the American Heart Association. Dr. Podack is the discoverer of Perforin and well recognized as the “Father” of the field of core forming proteins. Dr. Podack is the Sylvester Distinguished Professor of Microbiology & Immunology and Medicine and Chairman of the Department of Microbiology at the University of Miami, Miller School of Medicine.
James Allison, Ph.D.,Scientific Advisor
Dr. Allison is a leader in the field of immunology, particularly in developing ways to help the immune system recognize and destroy cancer cells. His research is focused on the mechanisms that regulate the immunological response of T lymphocytes, especially strategies to manipulate those responses in clinically relevant areas, including autoimmunity, allergies, vaccinations, and tumor therapy. Dr. Allison is Chairman Department of Immunology at the MD Anderson Cancer Center and was formerly Chairman of the Immunology Program, Director of the Ludwig Center for Cancer Immunotherapy, Attending Immunologist, and David H. Koch Chair in Immunologic Studies at Memorial Sloan-Kettering Cancer Center in New York City. He is a member of the National Academy of Sciences and the Institute of Medicine as well as a fellow of the American Academy of Microbiology and the American Association for the Advancement of Science. He also is an investigator of the Howard Hughes Medical Institute.
Sol Barer, Ph.D.,Scientific Advisor
Dr. Barer is the former Chairman and Chief Executive Officer of Celgene Corp., a global biopharmaceutical company engaged in the discovery, development, and commercialization of novel therapies for the treatment of cancer and inflammatory diseases. Dr. Barer has spent the last 20 years with Celgene and its predecessor, Celanese Research Company, serving as President, COO, CEO, Senior Vice President of Science and Technology, and Vice President/General Manager of the Chiral Products Division. Dr. Barer received his B.S. from Brooklyn College and his Ph.D. in organic chemistry from Rutgers University.
John Nemunaitis, M.D.,Scientific Advisor
Dr. Nemunaitis is an oncologist and Executive Medical Director of the Mary Crowley Cancer Research Centers (MCCRC) and has been exploring novel targeted therapies for treating cancer patients for over 20 years. Dr. Nemunaitis received his B.A. and M.D. degrees from Case Western Reserve University. He completed his residency at Boston City Hospital and then performed his Hematology and Oncology fellowship at the University of Washington and the Fred Hutchinson Cancer Research Center in Seattle from 1988 to 1993. Dr. Nemunaitis came to Dallas in 1993 to establish the clinical research program for Texas Oncology Physicians Association (TOPA). He later established a not-for-profit translational research program (the MCCRC). He is a committee member of the Western Institutional Review Board (WIRB) and recently co-founded a molecular therapeutic/vaccine biotechnology company with GMP manufacturing capacity called Gradalis, Inc. Dr. Nemunaitis has authored over 250 peer-reviewed publications and 36 book chapters. He has instituted study establishment of over 350 trials, overseen FDA sponsored experimental treatment of nearly 4,000 cancer patients at MCCRC, and has carried out 14 government regulatory (FDA, RAC) presentations for biotechnology product development. He is also developer and holder of 8 new molecular and vaccine Investigational New Drug Applications (IND’s). His research focus is clinical in orientation and involves determination of molecular signals in order to optimize targeted therapy, development of RNAi based therapeutics, and cancer vaccine approaches.
60
Justin Stebbing, M.D., MA, FRCP, FRCPath, Ph.D.,Scientific Advisor and Clinical Advisor
Dr. Stebbing is a member of the Royal College of Physicians, American Board of Internal Medicine and a Fellow of the Royal College of Pathologists. Originally, Justin trained in medicine at Trinity College Oxford, obtaining a triple first class degree. After completion of junior doctor posts in Oxford, he undertook a residency (junior doctor) training at The Johns Hopkins Hospital in the US, before returning to London to continue his training in oncology at The Royal Marsden. Justin then undertook a PhD, funded by the Medical Research Council, investigating the interplay between the immune system and cancer. Specifically, the role of heat shock proteins in viral infections and tumorigenesis were examined helping in the development of vaccines that are currently in clinical trials. Dr. Stebbing has published over 300 peer-reviewed papers in journals such as the Lancet, New England Journal, Blood, PNAS, The Journal of Clinical Oncology and Annals of Internal Medicine, the majority as first or last author, as well as over 100 book chapters. His publications mainly focus on early and late stage trials of new drugs, mechanisms of disease, and prognostic indicators. He is on the scientific advisory board of a number of biotechnology companies and the editorial board of a number of world-leading journals such as the Journal of Clinical Oncology. He is now a senior lecturer at Imperial College, London.
Daniel D. Von Hoff, M.D.,Scientific Advisor
Daniel D. Von Hoff, M.D., is currently Physician in Chief and Director of Translational Research at TGen (Translational Genomics Research Institute) in Phoenix, Arizona. He is also Chief Scientific Officer for Scottsdale Healthcare’s Clinical Research Institute and Scientific Medical Officer for US Oncology. He holds an appointment as Clinical Professor of Medicine, University of Arizona, College of Medicine. Dr. Von Hoff’s major interest is in the development of new anti-cancer agents, both in the clinic and in the laboratory. He and his colleagues were involved in the beginning of the development of many of the agents that are now used routinely, including: mitoxantrone, fludarabine, paclitaxel, docetaxel, gemcitabine, irinotecan, nelarabine, capecitabine, lapatinib and others. At present, he and his colleagues are concentrating on the development of molecularly targeted therapies particularly for patients with advanced pancreatic cancer. Dr. Von Hoff has published more than 559 papers, 134 book chapters and over 1,000 abstracts.
Dr. Von Hoff served as an appointee to President Bush’s National Cancer Advisory Board from June 2004 to March 2010. Dr. Von Hoff is the past President of the American Association for Cancer Research (the world’s largest cancer research organization), a Fellow of the American College of Physicians, and a member and past board member of the American Society of Clinical Oncology. He is a founder of ILEX™ Oncology, Inc. (acquired by Genzyme after Ilex had 2 agents, alemtuzumab and clofarabine approved for patients with leukemia). He is founder and the Editor Emeritus of Investigational New Drugs – The Journal of New Anticancer Agents; and, Editor-in-Chief of Molecular Cancer Therapeutics. He is also proud to have been a mentor and teacher for multiple medical students, medical oncology fellows, graduate students, and post-doctoral fellows. He is a co-founder of the AACR/ASCO Methods in Clinical Cancer Research Workshop. Dr. Von Hoff currently serves as Physician in Chief for the Translational Genomics Research Institute (TGen) in Phoenix, Arizona and Chief Scientific Officer of Scottsdale Healthcare and US Oncology. Dr. Von Hoff received his MD degree from Columbia University.
Clinical Advisory Board
In addition to our Board and Scientific Advisory Board we also have a Clinical Advisory Board comprised of five individuals, Dr Stebbing who is also a member of our Scientific Advisory Board, Dr. Gary Action, Dr. Roger Cohen, Dr Llew Keltner and Dr. Mark Schoenberg. The clinical advisory board will work with our clinical team todesign and guide our clinical trials. We have entered into written agreements and confidentiality agreements with all of our members of our Clinical Advisory Board. The members of our Scientific Advisory Board are compensated for their services.
Justin Stebbing, M.D., Ph.D.,Scientific Advisor and Clinical Advisor
See above bio.
61
Gary Acton, M.D.,Clinical Advisor
Dr. Acton is a London-based clinician providing oncology drug development advice, predominantly to US and European biotechnology companies. Twenty years of pharmaceutical experience have left him with wide ranging clinical, commercial and corporate capabilities. He has expertise in all stages of drug development and through into the marketplace. This includes successful US NDA and European MAA approvals. Dr. Acton has been involved in drug development programs for most solid and hematological malignant indications. He has worked in North American, European, and Japanese pharmaceutical companies. Dr. Acton has served at Board level in both private and publicly traded entities. He originally studied medicine at Oxford and London Universities. Prior to moving into the pharmaceutical industry, Dr. Acton obtained a number of post-graduate qualifications while undergoing general medical and oncology training at a variety of London teaching hospitals.
Roger Cohen, M.D.,Clinical Advisor
Dr. Cohen is Professor of Medicine at the University of Pennsylvania and Associate Director for Clinical Research for the Abramson Cancer Center. He is a graduate of Harvard Medical School and completed internal medicine and hematology training at Mount Sinai Hospital (NY) followed by research fellowships at the Memorial Sloan-Kettering Cancer Center and National Institutes of Health and a medical oncology fellowship at the National Cancer Institute. He was a medical officer at the FDA Center for Biologics from 1989-1994 where he was Deputy Director, Division of Monoclonal Antibodies. Prior to his arrival at Penn, Dr. Cohen was Director of the Clinical Trials Office at the University of Virginia Cancer Center in Charlottesville and then Director of the Phase 1 Program at the Fox Chase Cancer Center where he also served as interim Medical Oncology Department Chair for more than 2 years. He is an active investigator on a number of first-in-humans clinical trials with research interests that focus on evaluation of novel therapies, including monoclonal antibodies, immune therapies, and small molecule cell-signaling pathway inhibitors. He primarily sees patients with lung and head and neck cancer.
Llew Keltner, M.D., Ph.D., Clinical Advisor
Dr. Keltner has been the Chief Executive Officer of AgonOx, a biotech company developing OX40 agonists for use in cancer therapy. Dr. Keltner was the President of Novici Biotech, a privately-held gene and protein optimization firm. He is also Chief Executive Officer of EPISTAT, an international healthcare technology transfer, corporate risk management, and healthcare strategy company that he founded in 1972. Dr. Keltner was Chief Executive Officer and President of Light Sciences Oncology, a privately-held biotechnology company developing a late stage, light-activated therapy for hepatocellular cancer and other solid tumors from 2001 to 2010. From 1997 to 2004, Dr. Keltner was Chief Executive Officer of Metastat, a development-stage biotech company focused on cancer metastasis. He is currently a member of the American Society of Clinical Oncology, American Medical Association, International Association of Tumor Marker Oncology, American Association of Clinical Chemistry, and Drug Information Association. Dr. Keltner received an M.S. in Epidemiology and Biostatistics, a Ph.D. in Biomedical Informatics, and an M.D. from Case Western Reserve University in Cleveland, Ohio. Dr. Keltner has also authored several research publications.
Mark Schoenberg, M.D., Clinical Advisor
Dr. Schoenberg is the Bernard L. Schwartz Distinguished Professor of Urologic Oncology at The Johns Hopkins University. His clinical practice is centered on the care of patients with invasive bladder cancer. It has been announced that in April 2014, Dr. Schoenberg will assume the position of Chairman of the Department of Urology at Montefiore Medical Center and Albert Einstein College of Medicine of Yeshiva University, Bronx, New York. Dr. Schoenberg’s research program has focused on the translational validation of urinary markers for the early detection of cancer, the development of regenerative medicine solutions to the challenges of lower urinary tract reconstruction, and minimally invasive therapies for urologic malignancies. He is the past chair of the Medical Advisory Board of the Bladder Cancer Advocacy Network, author ofThe Guide to Living with Bladder Cancer, co-editor ofThe Textbook of Bladder Cancer, a contributor toCampbell’s Urology, and former seminars editor of the journal Urologic Oncology. Dr. Schoenberg has served as principal investigator and co-investigator on numerous clinical trials and from 2005-2009 served as the national principal investigator for the Early Detection Research Network (EDRN/NCI) validation trial of microsatellite analysis for bladder cancer detection. He received his M.D. from the University of Texas Health Science Center and completed residency at the Hospital of the University of Pennsylvania and a basic research and clinical urologic oncology fellowship at Johns Hopkins.
62
Committees of the Board of Directors
Our common stock is listed on the NASDAQ Capital Market. Under the rules of NASDAQ, independent directors must comprise a majority of a listed company’s board of directors and all members our audit, compensation and nominating and corporate governance committees be independent. Audit committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Exchange Act. Under the rules of The NASDAQ Stock Market, a director will only qualify as an “independent director” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
In order to be considered to be independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors, or any other board committee: (1) accept, directly or indirectly, any consulting, advisory or other compensatory fee from the listed company or any of its subsidiaries or (2) be an affiliated person of the listed company or any of its subsidiaries.
Our Board undertook a review of its composition, the composition of its committees and the independence of each director. Based upon information requested from and provided by each director concerning his background, employment and affiliations, including family relationships, our Board has determined that Dr. Belsky, Dr. Kharitonov, Dr. Monahan, Mr. Smith and Mr. Bock, representing five of our six directors, do not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is “independent” as that term is defined under the rules of The NASDAQ Stock Market. In making this determination, our Board considered the relationships that each non-employee director has with us and all other facts and circumstances our Board deemed relevant in determining their independence, including the beneficial ownership of our capital stock by each non-employee director. We intend to comply with the other independence requirements for committees within the time periods specified above.
We currently have: (i) an audit committee comprised of Dr. Monahan, Mr. Smith, and Mr. Bock, each all of whom are deemed to be independent in accordance with the NASDAQ definition of independence as well as qualify as “audit committee financial experts” as that term is used in Section 407 of Regulation S-K; (ii) a compensation committee comprised of Dr. Belsky, Dr. Monahan and Dr. Kharitonov, each of whom is deemed to be independent in accordance with the NASDAQ definition of independence; and (iii) a nominating and corporate governance committee comprised of Dr. Belsky, Dr. Kharitonov and Mr. Smith.
Leadership Structure
Our Chief Executive Officer also serves as our Chairman of the Board. Our Board does not have a lead independent director. Our board of directors has determined its leadership structure was appropriate and effective for us given our stage of development.
2013 Director Compensation
Compensation of Directors
The following table sets forth information for the fiscal year ended December 31, 2013 regarding the compensation of our directors who at December 31, 2013 were not also named executive officers.
| | | | | | | | | | | | | | | | |
Name | | Fees Earned or Paid in Cash | | | Option Awards(1) | | | Other Compensation | | | Total | |
Paul Belsky, MD | | $ | 10,870 | | | $ | 39,423 | (2) | | $ | — | | | $ | 50,293 | |
Michael Kharitonov, Ph.D. | | $ | 10,870 | | | $ | 39,423 | (2) | | $ | — | | | $ | 50,293 | |
John Monahan, Ph.D. | | $ | 10,870 | | | $ | 39,423 | (2) | | $ | — | | | $ | 50,293 | |
Edward Smith | | $ | 10,870 | | | $ | 39,423 | (2) | | $ | — | | | $ | 50,293 | |
Louis Bock | | $ | 5,000 | | | $ | 238,162 | (3) | | $ | — | | | $ | 243,162 | |
————————
(1)
The amounts in the “Option Awards” column reflect the dollar amounts recognized as compensation expense for the financial statement reporting purposes for stock options for the fiscal year ended December 31, 2013 in accordance with SFAS 123(R). The fair value of the options was determined using the Black-Scholes-Merton model.
63
(2)
Represents 5,435 options granted on April 29, 2013 to each individual director with vesting of 1/16th on the grant on the last day of each calendar quarter following the vesting commencement date, subject to remaining on the Board of Directors.
(3)
Represents 21,740 options granted on September 19, 2013 with vesting of 1/16th of the grant on the last day of each calendar quarter following the vesting commencement date, subject to remaining on the Board of Directors.
As of December 31, 2013 the following table sets forth the number of aggregate outstanding option awards held by each of our directors who were not also named executive officers:
| | | | |
Name | | Aggregate Number of Option Awards | |
Paul Belsky, MD | | $ | 26,958 | |
Michael Kharitonov, Ph.D. | | $ | 34,567 | |
John Monahan, Ph.D. | | $ | 34,567 | |
Edward Smith | | $ | 26,958 | |
Louis Bock | | $ | 21,740 | |
Following our successful initial public offering and in light of the additional responsibilities being undertaken by our board members due to our transition to a public company, our Compensation Committee conducted an evaluation of the compensation of the members of our board of directors. In order to aid its decision- making,the Compensation Committee considered the compensation practices and the competitive market for directors at companies with which we compete for personnel and an independent compensation advisor was retained to conduct a study of our peer group compensation. Based substantially upon the results of the study, commencing January 2014, directors who are not employees receive an annual cash fee of $25,000 as well as a cash fee of $5,000 for each committee on which they serve and the Chairman of the Audit and Compensation Committees receive an additional $2,000. Upon election to the Board, each non-employee director receives a grant of stock options exercisable for 21,740 shares of common stock vesting over four years having an exercise price equal to the fair market value of the common stock on the date of the grant. Each nonemployee director also receives an annual option grant on the date of the Annual Meeting of Stockholders having a value of $25,000 on such date.
Item 11.
Executive Compensation
Set forth below is the compensation that was paid to all executive officers during the years ended December 31, 2013 and December 31, 2012 that exceeded $100,000.
Summary Compensation Table
| | | | | | | | | | | | | | | | | | | | | | |
Name and Principal Position | | Year | | Salary | | | Bonus | | | Options | | | Other(1) | | | Total | |
Jeffrey Wolf | | 2013 | | $ | 250,000 | | | $ | 125,000 | (3) | | $ | — | | | $ | 11,472 | | | $ | 386,472 | |
Chairman & CEO | | 2012 | | $ | 250,000 | | | $ | 58,333 | (2) | | $ | 11,492 | | | $ | 11,156 | | | $ | 330,981 | |
Matt Czajkowski (4) | | 2013 | | $ | 65,645 | | | $ | 13,125 | (3) | | $ | 277,970 | | | $ | — | | | $ | 356,740 | |
Chief Financial Officer | | 2012 | | | — | | | | — | | | | — | | | | — | | | | — | |
Melissa Price (5) | | 2013 | | $ | 52,500 | | | $ | 10,000 | (3) | | $ | 538,400 | | | $ | — | | | $ | 600,900 | |
Vice President of Clinical and Regulatory Affairs | | 2012 | | | — | | | | — | | | | — | | | | — | | | | — | |
Jennifer Harris (6) | | 2013 | | $ | 109,857 | | | $ | — | | | $ | 64,816 | | | $ | — | | | $ | 174,673 | |
Former Vice President of Clinical and Regulatory Affairs | | 2012 | | $ | 142,904 | | | $ | — | | | $ | — | | | $ | — | | | $ | 142,904 | |
————————
(1)
Represents payment for health insurance.
(2)
This bonus was accrued in 2012 and paid in 2013.
(3)
This bonus was accrued in 2013 but paid in 2014.
(4)
Mr. Czajkowski was appointed at the Company’s Chief Financial Officer in May 2013.
(5)
Ms. Price was appointed as the Company’s Vice President of Clinical and Regulatory Affairs in October 2013.
(6)
The Company and Jennifer Harris terminated their relationship on September 4, 2013.
64
Outstanding Equity Awards At Fiscal Year-End (December 31, 2013)
| | | | | | | | | | | | | |
Name and Principal Position | | Number of securities underlying unexercised options/ exercisable | | | Number of securities underlying unexercised options/un- exercisable | | | Option exercise price | | Option expiration date |
Jeffrey Wolf | | | 10,965 | (1) | | | — | | | $ | 2.30 | | 12/18/2019 |
Chairman of the Board, Chief Executive Officer | | | 108,696 | (1) | | | — | | | $ | 0.71 | | 4/7/2016 |
Matthew Czajkowski | | | 7,458 | (2) | | | 30,906 | | | $ | 8.81 | | 5/15/2023 |
Chief Financial Officer | | | | | | | | | | | | | |
Melissa Price | | | 3,125 | (3) | | | 46,875 | | | $ | 12.57 | | 10/1/2023 |
Vice President of Clinical and Regulatory Affairs | | | 9,509 | | | | 12,231 | | | $ | 0.64 | | 9/4/2013 |
Jennifer Harris (4) | | | 905 | | | | 7,771 | | | $ | 8.81 | | 9/4/2013 |
Former Vice President of Clinical and Regulatory Affairs | | | | | | | | | | | | | |
————————
(1)
All shares are fully vested as of December 31, 2013.
(2)
Mr. Czajkowski’s shares vest monthly over a 36 month period. These shares will be fully vested in May 2017.
(3)
Mrs. Price’s shares vest monthly over a 48 month period. These shares will be fully vested in October 2017.
(4)
The Company and Jennifer Harris terminated their relationship on September 4, 2013. Mrs. Harris exercised 9,509 shares in November 2013.
Employment Agreements
Following our successful initial public offering and in light of the additional responsibilities being undertaken by our management due to our transition to a public company, our Compensation Committee conducted an evaluation of the compensation of certain members of our management. In order to aid its decision-making,the Compensation Committee considered the compensation practices and the competitive market for executives at companies with which we compete for personnel and an independent compensation advisor was retained to conduct a study of our peer group compensation.
On December 18, 2009, we entered into an employment agreement with Jeffrey Wolf to act as our Chief Executive Officer, which was amended on November 22, 2011 and further amended on January 20, 2014. Mr. Wolf receives an annual base salary of $395,000 per year. He also may receive, at the sole discretion of the board, additional performance-based bonuses equal to up to 50% of this then outstanding base salary at the end of each year. Upon execution of the agreement, Mr. Wolf was issued options exercisable for 119,661 shares of our common stock. In addition, he is to receive certain options to purchase 2% of our fully diluted equity at an exercise price equal to the then current market price if our stock is traded on a nationally recognized exchange or NASDAQ and our market capitalization is at least $250 million for at least 5 days. In January 2014, in accordance with the terms of his amended employment agreement, Mr. Wolf was also granted options exercisable for 100,000 shares of common stock, vesting annuallypro rata over a two-year period of time, subject to approval of the shareholders of the 2014 Equity Incentive Plan. The decision to amend Mr. Wolf’s Employment Agreement to effect an upward adjustment in his compensation was substantially based on the Compensation Committee’s review of competitive market information, including the study conducted by the compensation advisor. The competitive market information and peer group study results indicated that the overall compensation of our CEO was below market, in fact it was below the 25th percentile of the peer group, and that following the upward adjustment it remains below but closer to the 25th percentile of the peer group.
If Mr. Wolf’s employment contract is terminated for death or disability (as defined in the agreement), he (or his estate in the event of death) will receive six months severance. If Mr. Wolf’s employment is terminated by us other than for cause, he will receive twelve months severance. In addition, if Mr. Wolf’s employment is terminated by us other than for cause all Restricted Shares, common stock and options to purchase common stock that would have vested shall immediately vest. Mr. Wolf will not be entitled to any additional severance in the event he is terminated for cause or voluntarily resigns. Under his employment agreement, Mr. Wolf has also agreed to non-competition provisions.
65
On May 15, 2013, we entered into an employment agreement with Matthew E. Czajkowski to act as our Chief Financial Officer, which was amended on January 20, 2014. Mr. Czajkowski receives an annual base salary of $135,000 per year for his provision of services to us for fifty-percent of his professional time. In addition, Mr. Czajkowski may receive, at the sole discretion of the board, additional performance-based bonuses equal to up to 50% of this then outstanding base salary at the end of each year. Upon execution of the agreement, Mr. Czajkowski was issued options exercisable for 38,364 shares of our common stock, which options are exercisable over a ten year period and vest monthly over three years at an exercise price of $8.81 per share. Upon reaching full-time employment status, he will be entitled to all benefits to which our other executive officers are entitled. If Mr. Czajkowski’s employment contract is terminated by the board of directors not for cause (as defined in the agreement) he (or his estate in the event of death) will receive three month’s severance. If Mr. Czajkowski’s employment contract is terminated for death or disability (as defined in the agreement), he (or his estate in the event of death) will be entitled to receive all unpaid compensation up to such date of termination and such number of options that would have vested upon the date of termination will immediately vest. Under his employment agreement, Mr. Czajkowski has also agreed to customary non-competition provisions.
Effective December 16, 2013, we appointed Anil K. Goyal, Ph.D. as our Vice President of Business Development. In connection with his appointment, Dr. Goyal entered into a four-year employment agreement with us (the “Goyal Employment Agreement”). Pursuant to the Goyal Employment Agreement, Dr. Goyal will be entitled to an annual base salary of $220,000 and will be eligible for discretionary performance bonus payments. Additionally, Dr. Goyal was granted an option to purchase 40,000 shares of our common stock with an exercise price equal to the Company’s per share market price on the date of issue. These options vestpro rata, on a monthly basis, over forty-eight months. Dr. Goyal is also eligible to receive, on the one year anniversary of his employment, an option to purchase 10,000 shares of our common stock if certain milestones, which are yet to be agreed to, are met by such date. The Goyal Employment Agreement also includes confidentiality obligations and inventions assignments by Dr. Goyal. If Dr. Goyal’s employment is terminated for any reason, he or his estate as the case may be, will be entitled to receive the accrued base salary, vacation pay, expense reimbursement and any other entitlements accrued by him to the extent not previously paid (the “Accrued Obligations”); provided, however, that if his employment is terminated (1) by us without Just Cause (as defined in the Goyal Employment Agreement) or (2) by Dr. Goyal for Good Reason (as defined in the Goyal Employment Agreement) then in addition to paying the Accrued Obligations: (x) we shall continue to pay his then current base salary for a period of four months; (y) he shall receive a pro-rated amount of the annual bonus which he would have received during the year without the occurrence of such termination; and (z) he will have the right to exercise any vested options and any options that would have vested in the next four months until the earlier of the expiration of the severance or the expiration of the term of the option.
Effective October 1, 2013, we appointed Melissa Price, Ph.D. as our Vice President of Clinical and Regulatory Affairs, which was amended on January 20, 2014. In connection with her appointment, Dr. Price entered into a four-year employment agreement with us (the “Price Employment Agreement”). Pursuant to the Price Employment Agreement, Dr. Price receives an annual base salary of $210,000 and will be eligible for discretionary performance bonus payments. Additionally, Dr. Price was granted an option to purchase 50,000 shares of our common stock with an exercise price equal to our per share market price on the date of issue. These options vest pro rata, on a monthly basis, over forty-eight months. Dr. Price is also eligible to receive, an option to purchase 10,000 shares of our common stock if at any time prior to December 31, 2014, certain agreed to milestones are attained. The Price Employment Agreement also includes confidentiality obligations and inventions assignments by Dr. Price. If Dr. Price’s employment is terminated for any reason, she or her estate as the case may be, will be entitled to receive the Accrued Obligations accrued by her to the extent not previously paid; provided, however, that if her employment is terminated (1) by us without Just Cause (as defined in the Price Employment Agreement) or by Dr. Price for Good Reason (as defined in the Price Employment Agreement) then in addition to paying the Accrued Obligations, (x) we shall continue to pay her then current base salary for a period of four months; (y) she shall receive a pro-rated amount of the annual bonus which she would have received during the year without the occurrence of such termination and (z) she will have the right to exercise any vested options and any options that would have vested in the next four months until the earlier of the expiration of the severance or the expiration of the term of the option.
Effective March 3, 2014, we appointed Taylor Schreiber, M.D., Ph.D., as our Vice President of Research and Development. In connection with his appointment, Dr. Schreiber entered into a four-year employment agreement with us. Pursuant to the employment agreement, Dr. Schreiber receives an annual base salary of $210,000 and will be eligible for discretionary performance bonus payments. Additionally, on the date that the Company’s shareholders approve a new stock incentive plan, we have agreed to grant Dr. Schreiber an option to purchase 50,000 shares of our common stock with an exercise price equal to our per share market price on the date of issue. These options will vest pro rata, on a monthly basis, over forty-eight months, with a certain percentage vesting immediately upon grant. Dr. Schreiber is also eligible to receive, on the one year anniversary of his employment, an option to purchase 10,000 shares of our common stock if certain milestones, which are yet to be agreed to, are met by such date. The employment agreement also includes confidentiality obligations and inventions assignments by Dr. Schreiber.
66
If Dr. Schreiber’s employment is terminated for any reason, he or his estate as the case may be, will be entitled to receive the Accrued Obligations accrued by him to the extent not previously paid (the “Accrued Obligations”); provided, however, that if his employment is terminated (1) by the Company without Just Cause (as defined in the Employment Agreement) or by Dr. Schreiber for Good Reason (as defined in the Employment Agreement) then in addition to paying the Accrued Obligations, (x) the Company shall continue to pay his then current base salary for a period of four months; (y) he shall receive a pro-rated amount of the annual bonus which he would have received during the year without the occurrence of such termination and (z) he will have the right to exercise any vested options until the earlier of the expiration of the severance or the expiration of the term of the option.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Securities Exchange Act of 1934 requires our executive officers, directors and persons who beneficially own more than 10 percent of a registered class of the Heat Biologics’ equity securities, to file with the SEC initial reports of ownership and reports of changes in ownership of our common stock. Such officers, directors and persons are required by SEC regulation to furnish us with copies of all Section 16(a) forms that they file with the SEC.
Based solely on a review of the copies of such forms that were received by us, or written representations from certain reporting persons that no Form 5s were required for those persons, we are not aware of any failures to file reports or report transactions in a timely manner during the year ended December 31, 2013 other than Sandra Silberman who did not file a Form 4 after receiving an option grant.
Code of Ethics
We have long maintained a Code of Conduct which is applicable to all of our directors, officers and employees. In addition, we have adopted a Code of Ethics for Financial Management which applies to our Chief Executive Officer, Chief Financial Officer, Treasurer and Controller. We undertake to provide a printed copy of these codes free of charge to any person who requests. Any such request should be sent to our principal executive offices attention: Corporate Secretary.
67
Item 12.
Security Ownership of Certain Beneficial Owners
The following table sets forth information, as of March 31, 2014, or as otherwise set forth below, with respect to the beneficial ownership of our common stock (i) all persons know to us to be the beneficial owners of more than 5% of the outstanding shares of our common stock, (ii) each of our directors and our executive officer named in the Summary Compensation Table, and (iii) all of our directors and our executive officer as a group. As of March 31, 2014 we had 6,452,341 shares of common stock outstanding.
Principal Stockholders Table
Unless otherwise indicated the mailing address of each of the stockholders below is c/o Heat Biologics, Inc., 100 Europa Drive, Chapel Hill, North Carolina 27517. Except as otherwise indicated, and subject to applicable community property laws, except to the extent authority is shared by both spouses under applicable law, the Company believes the persons named in the table have sole voting and investment power with respect to all shares of common stock held by them.
| | | | | | | | | | |
Name of Beneficial Owner | | | Number of Shares Beneficially Owned | | | Percentage Ownership | |
Executive Officers & Directors (1) | | | | | | | | |
Paul Belsky, M.D. (Director)(2) | | | | 59,183 | | | | * | | |
Louis Bock (Director) (3) | | | | 4,075 | | | | * | | |
Matthew E. Czajkowski (CFO)(4) | | | | 12,786 | | | | * | | |
Vadim Deyev, MD, Ph.D.(5) | | | | 10,870 | | | | * | | |
Anil Goyal, Ph.D.(6) | | | | 5,833 | | | | * | | |
Michael Kharitonov, Ph.D. (Director)(7) | | | | 68,527 | | | | 1.1 | % | |
John Monahan, Ph.D. (Director)(8) | | | | 19,778 | | | | * | | |
Melissa Price, Ph.D.(9) | | | | 8,333 | | | | * | | |
Taylor Schreiber (10) | | | | 21,740 | | | | * | | |
Edward Smith (Director)(11) | | | | 711,796 | | | | 11.0 | % | |
Jeffrey Wolf (Director, CEO, Treasurer & Secretary)(12) | | | | 1,353,387 | | | | 20.6 | % | |
| | | | | | | | * | | |
All Executive Officers & Directors, as a group (11 persons) | | | | 2,276,308 | | | | 34.0 | % | |
| | | | | | | | | | |
5% Stockholders(1) | | | | | | | | | | |
Brightline Ventures III, LLC(13) | | | | 697,303 | | | | 10.8 | % | |
Orion Holdings V, LLC (14) | | | | 695,653 | | | | 10.8 | % | |
Seed-One Holdings VI, LLC(14) | | | | 536,862 | | | | 8.3 | % | |
FW Heat Biologics, LLC(15) | | | | 453,673 | | | | 7.0 | % | |
Franklin Resources, Inc. (16) | | | | 657,800 | | | | 10.2 | % | |
————————
*less than 1%
(1)
Unless otherwise set forth below, the mailing address of Executive Officers, Directors and 5% or greater holders is c/o the Company, 100 Europa Drive, Chapel Hill, NC 27517.
(2)
Dr. Belsky has been issued options exercisable for 26,958 shares of common stock, of which 12,341 shares are vested as of March 31, 2014 and 2,152 shares will vest within 60 days of March 31, 2014 and are included in the number of shares beneficially owned by Dr. Belsky.
(3)
Mr. Bock has been issued options exercisable for 21,740 shares of common stock, of which 2,717 shares are vested as of March 31, 2014 and 1,358 will vest within 60 days of March 31, 2014 and are included in the number of shares beneficially owned by Mr. Bock and are included in the number of shares beneficially owned by Mr. Bock.
(4)
Mr. Czajkowski has been issued options exercisable for 38,364 shares of common stock, of which 10,655 shares are vested as of March 31, 2014 and 5,328 will vest within 60 days of March 31, 2014 and are included in the number of shares beneficially owned by Mr. Czajkowski.
(5)
Dr. Deyev has been issued options exercisable for 10,870 shares of common stock, of which 10,870 shares are vested as of March 31, 2014 and included in the number of shares beneficially owned by Mr. Deyev.
(6)
Dr. Goyal has been issued options exercisable for 40,000 shares of common stock, of which 1,667 shares are vested as of March 31, 2014 and 4,166 will vest within 60 days of March 31, 2014 and are included in the number of shares beneficially owned by Dr. Goyal.
68
(7)
Includes 49,960 shares of common stock held by Dr. Kharitonov. Dr. Kharitonov disclaims beneficial ownership of these shares except to the extent of any pecuniary interest (as defined in Rule 16a – 1(a)(2) promulgated under the Exchange Act) that he may have in the Sunrise Equity, LLC. Dr. Kharitonov has been issued options exercisable for 34,567 shares of common stock, of which 18,114 shares are vested as of March 31, 2014 and 2,492 will vest within 60 days of March 31, 2014 and are included in the number of shares beneficially owned by Dr. Kharitonov.
(8)
Dr. Monahan has been issued options exercisable for 34,567 shares of common stock, of which 18,114 shares are vested as of March 31, 2014 and 2,492 will vest within 60 days of March 31, 2014 and are included in the number of shares beneficially owned by Dr. Monahan. Includes 1,211 shares of common stock held by Dr. Monahan.
(9)
Dr. Price has been issued options exercisable for 50,000 shares of common stock, of which 6,250 shares are vested as of March 31, 2014 and 5,208 will vest within 60 days of March 31, 2014 and are included in the number of shares beneficially owned by Dr. Price.
(10)
Dr. Schreiber and an entity controlled by Dr. Schreiber have been issued an aggregate of 39,092 shares of common stock that are included in the number of shares beneficially owned by Dr. Schreiber. Does not include options exercisable for 50,000 shares of common stock that we have agreed to issue to Mr. Schreiber upon approval by our shareholders of our 2014 Stock Incentive Plan.
(11)
Mr. Smith has been issued options exercisable for 26,958 shares of common stock, of which 14,040 shares are vested as of March 31, 2014 and 2,152 will vest within 60 days of March 31, 2014 and are included in the number of shares beneficially owned by Mr. Smith. Includes 697,303 shares of common stock owned by Brightline Ventures III, LLC, of which Mr. Smith disclaims beneficial ownership except to the extent of any pecuniary interest.
(12)
Includes 695,653 shares of common stock held by Orion Holdings V, LLC and 536,862 shares of common stock held by Seed-One Holdings VI, LLC, entities for which Mr. Wolf serves as the managing member. Mr. Wolf is deemed to beneficially own the shares held by such entities as in his role as the managing member he has the control over the voting and disposition of any shares held by these entities. Does not include 86,957 shares of common stock beneficially owned by Mr. Wolf’s children’s trust which Mr. Wolf is not the trustee of Mr. Wolf disclaims beneficial ownership of these shares except to the extent of any pecuniary interest (as defined in Rule 16a – 1(a)(2) promulgated under the Exchange Act) that he may have in such entities. In addition, if our Company is traded on a recognized national exchange or NASDAQ while Mr. Wolf is employed by us and the market capitalization of our Company is in excess of $250 million for at least five consecutive trading days, then Mr. Wolf will be entitled to receive an additional stock option equal to 2% of the then outstanding shares of our common stock, at an exercise price equal to the then current market price as determined in good faith by the board. Mr. Wolf has been issued options exercisable for 219,661 shares of common stock, of which 119,661 shares are vested and exercisable within 60 days of March 15, 2014 and are included in the beneficial ownership of Mr. Wolf and the remaining 100,000 are subject to forfeiture if our 2014 Stock Incentive Plan is not approved by our shareholders.
(13)
Mr. Smith disclaims beneficial ownership of these shares except to the extent of any pecuniary interest (as defined in Rule 16a – 1(a)(2) promulgated under the Exchange Act) that he may have in such entities.
(14)
Mr. Wolf serves as the managing member of such entity. Mr. Wolf is deemed to beneficially own the shares held by such entity as in his role as the managing member he has the control over the voting and disposition of any shares held by this entity. Mr. Wolf disclaims beneficial ownership of these shares except to the extent of any pecuniary interest (as defined in Rule 16a – 1(a)(2) promulgated under the Exchange Act) that he may have in such entity.
(15)
Information obtained from a Form 3 filed by FW Heat Investors L.P on July 23, 2013. Includes 447,937 shares of common stock FW Heat Genpar, LLC is the sole general partner of FW Heat Investors L.P. GenPar's voting and disposition is decisions are further controlled by its sole member RMB Holdings, LLC ("Holdings"), Holdings' member Live Oak UAD 3/25/2010 (the "Trust") and the Trusts' trustees, Robert M. Bass and Anne T. Bass. The above conversion amount reflects the reverse stock split effected on May 29, 2013. Each of GenPar, Holdings, the Trust and each of the trustees disclaims his, her or its beneficial ownership except to the extent of his, her or its pecuniary interest. The mailing address ofFW Heat Investors L.P is 201 Main Street, Fort Worth, Texas 76102.
(16)
Information obtained from a Schedule 13G filed with the Securities and Exchange Commission on November 12, 2013. Charles B. Johnson and Rupert H. Johnson, Jr. (the “Principal Shareholders”) each own in excess of 10% of the outstanding common stock of Franklin Resources, Inc. (“FRI”) and are the principal stockholders of FRI. Franklin Advisor, Inc. a management subsidiary of FRI is also deemed to be a beneficial owner of the common stock owned by FRI. The address of Franklin Resources, Inc. isOne Franklin Parkway, San Mateo, California 94403-1906.
69
Item 13.
Certain Relationships and Related Transactions, and Director Independence
Pursuant to our charter, our Audit Committee shall review on an on-going basis for potential conflicts of interest, and approve if appropriate, all our “Related Party Transactions” as required by of NASDAQ Rule 4350(h). For purposes of the Audit Committee Charter, “Related Party Transactions” shall mean those transactions required to be disclosed pursuant to SEC Regulation S-K, Item 404.
The following is a summary of transactions since January 1, 2013 to which we have been a party in which the amount involved exceeded the lesser of $120,000 or one percent of the average of our total assets at the end of the most recent completed fiscal year and in which any of our executive officers, directors or beneficial holders of more than five percent of our capital stock had or will have a direct or indirect material interest, other than compensation arrangements which are described under the section of this Annual Report on Form 10-K entitled “Management—Non-Employee Director Compensation” and “Management — Executive Compensation.”
Pursuant to our funding agreement with the University of Miami, the University has been issued shares of Heat Biologics I, Inc. representing 7.5% of the outstanding shares of Heat Biologics I, Inc.
In March 2013, Dr. Belsky, Dr. Monahan and Mr. Wolf each purchased 2,622 shares of the Company’s Series B-1 Preferred Stock at a per share price of $2.67 in its private placement that consummated in March 2013 which converted into 1,160 shares of our common stock upon consummation of our initial public offering. In addition, each of Dr. Belsky, Dr. Monahan and Mr. Wolf were issued 51 shares of our common stock upon consummation of our initial public offering in in lieu of Series B-2 Preferred Stock that they had committed to purchase upon our receipt of certain grant funding and the shares underlying the warrants to be issued at such time.
Upon consummation of our initial public offering we issued to Michael Kharitonov 49,960 shares of our common stock upon the automatic conversion of shares of Series 1 Preferred Stock.
Upon consummation of our initial public offering we issued to Brightline Ventures III, LLC 697,303 shares of our common stock upon the automatic conversion of shares of Series A Preferred Stock.
Item 14.
Principal Accountant Fees and Services
Independent Registered Public Accounting Firm Fees and Services
The following table sets forth the aggregate fees including expenses billed to us for the years ended December 31, 2013 and 2012 by BDO USA, LLP.
| | | | | | | |
| | December 31,
2013 | | December 31,
2012 | |
Audit Fees and Expenses (1) | | $ | 180,349 | | $ | 112,730 | |
———————
(1)
Audit fees and expenses were for professional services rendered for the audit and reviews of the consolidated financial statements of the Company, professional services rendered for issuance of consents and assistance with review of documents filed with the SEC.
The Audit Committee has adopted procedures for pre-approving all audit and non-audit services provided by the independent registered public accounting firm, including the fees and terms of such services. These procedures include reviewing detailed back-up documentation for audit and permitted non-audit services. The documentation includes a description of, and a budgeted amount for, particular categories of non-audit services that are recurring in nature and therefore anticipated at the time that the budget is submitted. Audit Committee approval is required to exceed the pre-approved amount for a particular category of non-audit services and to engage the independent registered public accounting firm for any non-audit services not included in those pre-approved amounts. For both types of pre-approval, the Audit Committee considers whether such services are consistent with the rules on auditor independence promulgated by the SEC and the PCAOB. The Audit Committee also considers whether the independent registered public accounting firm is best positioned to provide the most effective and efficient service, based on such reasons as the auditor’s familiarity with our business, people, culture, accounting systems, risk profile, and whether the services enhance our ability to manage or control risks and improve audit quality. The Audit Committee may form and delegate pre-approval authority to subcommittees consisting of one or more members of the Audit Committee, and such subcommittees must report any pre-approval decisions to the Audit Committee at its next scheduled meeting. All of the services provided by the independent registered public accounting firm were pre-approved by the Audit Committee.
70
PART IV
Item 15.
Exhibits and Financial Statement Schedules
| | |
| (a)(1) | The following financial statements are included in this Annual Report on Form 10-K for the fiscal years ended December 31, 2013 and 2012. |
| | |
| 1. | Independent Registered Public Accounting Firm |
| | |
| 2. | Consolidated Balance Sheets as of December 31, 2013 and 2012 |
| | |
| 3. | Consolidated Statements of Operations for the years ended December 31, 2013 and 2012 |
| | |
| 4. | Consolidated Statements of changes in Stockholders’ Equity for the years ended December 31, 2013 and 2012 |
| | |
| 5. | Consolidated Statements of Cash Flows for the years ended December 31, 2013 and 2012 |
| | |
| 6. | Notes to Consolidated Financial Statements |
| | |
| (a)(2) | All financial statement schedules have been omitted as the required information is either inapplicable or included in the Consolidated Financial Statements or related notes. |
| | |
| (a)(3) | The following exhibits are either filed as part of this report or are incorporated herein by reference: |
| | |
Exhibit No. | | Description |
1.1 | | Form of Underwriting Agreement between Heat Biologics, Inc. and Aegis Capital Corp., as representative of the several underwriters (1) |
3.1 | | Certificate of Incorporation filed on June 10, 2008(2) |
3.2 | | Amended and Restated Bylaws, as currently in effect(2) |
3.3 | | Amended and Restated Certificate of Incorporation filed on October 16, 2009(2) |
3.4 | | Second Amended and Restated Certificate of Incorporation filed on December 16, 2011(2) |
3.5 | | Third Amended and Restated Certificate of Incorporation, as currently in effect(2) |
3.6 | | Certificate of Amendment to the Third Amended and Restated Certificate of Incorporation filed on May 29, 2013(1) |
4.1 | | 2009 Stock Incentive Plan(2)## |
4.2 | | First Amendment of the 2009 Stock Incentive Plan(2)## |
4.3 | | Second Amendment of the 2009 Stock Incentive Plan(2)## |
4.4 | | Third Amendment of the 2009 Stock Incentive Plan(2)## |
4.5 | | Fourth Amendment of the 2009 Stock Incentive Plan(2)## |
4.6 | | Warrant issued to Square 1 Bank(2) |
4.7 | | Warrant issued to North Carolina Biotechnology Center(1) |
4.8 | | Specimen Common Stock Certificate of Heat Biologics, Inc.(2) |
4.9 | | Form of Stock Purchase Agreement by and among Heat Biologics, Inc. and the Series B investors (Portions of the exhibit have been omitted pursuant to a request for confidential treatment. The omitted portions have been filed with the Commission)(2)## |
4.10 | | Form of Representative’s Warrant (1) |
4.11 | | Amendment to Stock Warrant with North Carolina Biotechnology Center(1) |
10.1 | | License Agreement (UMJ110) between the University of Miami and Heat Biologics, Inc. effective February 18, 2011 (2)## |
10.2 | | License Agreement (97-14) between the University of Miami and its School of Medicine and Heat Biologics, Inc. effective July 11, 2008(2) |
10.3 | | License Agreement (143) between the University of Miami and its School of Medicine and Heat Biologics I, Inc. effective February 11, 2011(2) |
10.4 | | License Agreement (D-107) between the University of Miami and its School of Medicine and Heat Biologics I, Inc. effective February 18, 2011(2) |
10.5 | | License Agreement (SS114A) between the University of Miami and its School of Medicine and Heat Biologics I, Inc. effective February 18, 2011 (2) |
10.6 | | Promissory Note with North Carolina Biotechnology Center dated December 14, 2011(2) |
10.7 | | Loan Agreement with North Carolina Biotechnology Center dated December 14, 2011(2) |
71
| | |
10.8 | | Common Stock Subscription Agreement between the University of Miami and Heat Biologics I, Inc. dated July 7, 2009(2) |
10.9 | | Employment Agreement with Jeffrey Wolf dated December 18, 2009(2)## |
10.10 | | Amendment to Employment Agreement with Jeffrey Wolf dated as of January 1, 2011(2)## |
10.11 | | Lease with Europa Center dated as of November 18, 2011(2) |
10.12 | | Non-Exclusive Evaluation and Biological Material License Agreement with American Type Culture Collection effective April 12, 2011(2) ## |
10.13 | | Manufacturing Services Agreement with Lonza Walkersville, Inc. dated as of October 20, 2011(2) |
10.14 | | Assignment and Assumption Agreement dated June 26, 2009(2) |
10.15 | | Termination Agreement UM97-114 dated June 26, 2009(2) |
10.16 | | Loan and Security Agreement with Square 1 Bank dated August 7, 2012(2) |
10.17 | | Employment Agreement with Jennifer Harris dated November 3, 2011 and amendment thereto dated May 1, 2013(1)## |
10.18 | | Amendment to License Agreement (UM97-14) dated April 29, 2009(2) |
10.19 | | First Amendment to Loan and Security Agreement with Square 1 Bank dated November 30, 2012(2) |
10.20 | | Second Amendment to License Agreement (UMSS-114) dated August 11, 2009(2) |
10.21 | | Exclusive License between Heat Biologics, Inc. and the University of Michigan dated July 22, 2011(2) |
10.22 | | 1st Lease Modification Agreement dated December 19, 2012(2) |
10.23 | | Form of Co Sale and First Refusal Agreement by and among Heat Biologics, Inc. and the Series B investors(2) |
10.24 | | Form of Voting Agreement by and among Heat Biologics, Inc. and the Series B investors(2) |
10.25 | | Form of Investor’s Rights Agreement by and among Heat Biologics, Inc. and the Series B investors(2) |
10.26 | | Second Amendment to Loan and Security Agreement with Square 1 Bank dated January 14, 2013(2) |
10.27 | | Third Amendment to Loan and Security Agreement with Square 1 Bank dated February 28, 2013(2) |
10.28 | | Fourth Amendment to Loan and Security Agreement with Square 1 Bank dated March 19, 2013(2) |
10.29 | | Option Contract for Exclusive License between Heat Biologics, Inc. and the University of Miami dated April 1, 2013(2) |
10.30 | | Fifth Amendment to the Loan and Security Agreement with Square 1 Bank dated April 18, 2013(2) |
10.31 | | Employment Agreement with Matthew Czajkowski dated May 15, 2013(1)## |
10.32 | | Form of Lock-up Agreement(1) |
10.33 | | Form of Agreement with Series B Preferred Stockholders to amend Stock Purchase Agreement(1) |
10.34 | | Employment Agreement, dated as of October 1, 2013, by and between Melissa Price and the Company(2)## |
10.34 | | Employment Agreement, dated as of December 16, 2013, by and between Anil K. Goyal and the Company(4)## |
10.35 | | Amendment to Employment Agreement, dated as of January20 , 2014 between the Company and Jeffrey Wolf(5)## |
10.36 | | Amendment to Employment Agreement, dated as of January20, 2014 between the Company and Melissa Price(5)## |
10.37 | | Amendment to Employment Agreement, dated as of January20, 2014 between the Company and Matthew Czajkowski(5)## |
10.38 | | Employment Agreement, dated as of March 3, 2014 between the Company and Taylor Schreiber (6)## |
10.39 | | Lease Agreement dated January 24, 2014 ǂ |
10.40 | | License Agreement (UMK-161) between the University of Miami and its School of Medicine and Heat Biologics I, Inc. effective March 4, 2014 ǂ *** |
21.1 | | List of Subsidiaries ǂ |
23.1 | | Consent of Independent Registered Public Accounting Firm (BDO USA, LLP) * |
31.1 | | Certification of Jeffrey Wolf, Chief Executive Officer, pursuant to Rule 13a-14(a)/15d-14(a) * |
31.2 | | Certification of Matthew Czajkowski, Chief Financial Officer pursuant to Rule 13a-14(a)/15d-14(a) * |
32.1 | | Certification of Jeffrey Wolf, Chief Executive Officer pursuant to Section 1350 of the Sarbanes-Oxley Act of 2002 * |
32.2 | | Certification Matthew Czajkowski, Chief Financial Officer pursuant to Section 1350 of the Sarbanes-Oxley Act of 2002 * |
101.INS | | XBRL Instance Document ** |
101.SCH | | XBRL Taxonomy Extension Schema Document ** |
101.CAL | | XBRL Taxonomy Extension Calculation Linkbase Document ** |
101.DEF | | XBRL Taxonomy Extension Definition Linkbase Document ** |
101.LAB | | XBRL Taxonomy Extension Label Linkbase Document ** |
101.PRE | | XBRL Taxonomy Extension Presentation Linkbase Document ** |
———————
(1)
Previously filed on Form S-1 with the Securities and Exchange Commission on May 6, 2013.
(2)
Previously filed on Form S-1 with the Securities and Exchange Commission on May 30, 2013.
(3)
Previously filed on Form 8-K with the Securities and Exchange Commission on October 1, 2013.
72
(4)
Previously filed on Form 8-K with the Securities and Exchange Commission on December 19, 2013.
(5)
Previously filed on Form 8-K with the Securities and Exchange Commission on January 21, 2014.
(6)
Previously filed on Form 8-K with the Securities and Exchange Commission on March 5, 2014.
*
Filed herewith.
##
Management contract or compensatory plan or arrangement required to be identified pursuant to Item 15(a)(3) of this report.
**
As provided in Rule 406T of Regulation S-T, this information is deemed furnished and not filed for purposes of Sections 11 and 12 of the Securities Act of 1933, as amended, and Section 18 of the Securities Exchange Act of 1934, as amended.
***
Confidential treatment has been requested as to certain portions of this exhibit pursuant to Rule 24b-2 of the Securities Exchange Act of 1934, as amended.
ǂ
Previously filed.
73
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned.
| | | |
| HEAT BIOLOGICS, INC. |
| |
| By: | /s/ Jeffrey Wolf | |
| Jeffrey Wolf |
| Chief Executive Officer and Director |
| (Principal Executive Officer) |
| Date: October 10, 2014 |
| | | |
| By: | /s/ Matthew Czajkowski | |
| Matthew Czajkowski |
| Chief Financial Officer |
| (Principal Financial and Principal Accounting Officer) |
| Date: October 10, 2014 |
74
INDEX TO FINANCIAL STATEMENTS
| | | | |
| | Page | |
| | | |
Report of Independent Registered Public Accounting Firm | | | F-2 | |
| | | | |
Consolidated Balance Sheets | | | F-3 | |
| | | | |
Consolidated Statements of Operations | | | F-4 | |
| | | | |
Consolidated Statement of Stockholders’ Equity (Deficit) | | | F-5 | |
| | | | |
Consolidated Statements of Cash Flows | | | F-7 | |
| | | | |
Notes to Consolidated Financial Statements | | | F-9 | |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
Heat Biologics, Inc.
(A Development Stage Company)
Chapel Hill, North Carolina
We have audited the accompanying consolidated balance sheets of Heat Biologics, Inc. (the “Company”) (a development stage company) as of December 31, 2013 and 2012 and the related consolidated statements of operations, stockholders’ equity (deficit), and cash flows for each of the two years in the period ended December 31, 2013, and for the period from June 30, 2008 (inception) to December 31, 2013. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Heat Biologics, Inc. at December 31, 2013 and 2012, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2013 and the period from June 10, 2008 (inception) to December 31, 2013, in conformity with accounting principles generally accepted in the United States of America.
/s/ BDO USA, LLP
BDO USA, LLP
Raleigh, North Carolina
March 31, 2014
F-2
HEAT BIOLOGICS, INC.
(A development stage company)
Consolidated Balance Sheets
| | | | | | | |
| | December 31, 2013 | | December 31, 2012 | |
Assets | | | | | |
| | | | | |
Current Assets | | | | | |
Cash and cash equivalents | | $ | 4,566,992 | | $ | 5,030 | |
Short term investments | | | 17,297,165 | | | — | |
Related party receivable | | | 24,946 | | | 9,571 | |
Prepaid expenses and other current assets | | | 1,066,638 | | | 58,436 | |
Total Current Assets | | | 22,955,741 | | | 73,037 | |
| | | | | | | |
Property and Equipment, net | | | 53,753 | | | 10,782 | |
| | | | | | | |
Other Assets | | | | | | | |
Restricted cash | | | 1,252 | | | 26,214 | |
Debt issuance costs, net | | | — | | | 28,229 | |
Deposits | | | 9,320 | | | 9,320 | |
Total Other Assets | | | 10,572 | | | 63,763 | |
| | | | | | | |
Total Assets | | $ | 23,020,066 | | $ | 147,582 | |
| | | | | | | |
Liabilities and Stockholders' Equity (Deficit) | | | | | | | |
| | | | | | | |
Current Liabilities | | | | | | | |
Accounts payable | | $ | 651,917 | | $ | 505,471 | |
Accrued expenses and other payables | | | 503,050 | | | 129,208 | |
Accrued interest | | | 25,364 | | | 13,763 | |
Notes payable - current portion | | | — | | | 66,806 | |
Total Current Liabilities | | | 1,180,331 | | | 715,248 | |
| | | | | | | |
Long Term Liabilities | | | | | | | |
Notes payable - less current portion | | | — | | | 658,194 | |
Convertible notes payable | | | — | | | 197,099 | |
Stock warrants liability | | | 122,590 | | | 92,150 | |
Total Liabilities | | | 1,302,921 | | | 1,662,691 | |
| | | | | | | |
Stockholders' Equity (Deficit) | | | | | | | |
Series 1 preferred stock, $0.0001 par value; 112,500 shares authorized, 112,500 shares issued and outstanding at December 31, 2012 and 0 shares issued and outstanding at December 31, 2013 | | | — | | | 11 | |
Series A preferred stock, $0.0001 par value; 2,000,000 shares authorized, 1,863,128 shares issued and outstanding at December 31, 2012 and 0 shares issued and outstanding at December 31, 2013 | | | — | | | 186 | |
Common stock, $.0002 par value; 50,000,000 shares authorized, 6,375,426 and 2,144,542 shares issued and 6,375,426 and 1,858,971 shares outstanding at December 31, 2013 and 2012, respectively | | | 961 | | | 405 | |
Additional paid in capital | | | 34,337,591 | | | 4,495,832 | |
Deficit accumulated during the development stage | | | (12,346,630 | ) | | (5,935,282 | ) |
Total Stockholders' Equity (Deficit) | | | 21,991,922 | | | (1,438,848 | ) |
Non-Controlling Interest | | | (274,777 | ) | | (76,261 | ) |
Total Stockholders' Equity (Deficit) – Heat Biologics, Inc. | | | 21,717,145 | | | (1,515,109 | ) |
| | | | | | | |
Total Liabilities and Stockholders' Equity (Deficit) | | $ | 23,020,066 | | $ | 147,582 | |
See Notes to Consolidated Financial Statements
F-3
HEAT BIOLOGICS, INC.
(A development stage company)
Consolidated Statements of Operations
| | | | | | | | | | | |
| | Year ended,
December 31, | | | Period from
June 10, 2008
(inception) to
December 31, | |
| | 2013 | | 2012 | | | 2013 | |
Grant awards | | $ | — | | $ | 3,110 | | | $ | 585,589 | |
| | | | | | | | | | | |
Operating expenses: | | | | | | | | | | | |
Research and development | | | 2,737,688 | | | 902,938 | | | | 6,095,170 | |
Clinical and regulatory | | | 1,397,157 | | | 253,189 | | | | 1,825,000 | |
General and administrative | | | 2,429,796 | | | 1,189,660 | | | | 4,725,322 | |
Total operating expenses | | | 6,564,641 | | | 2,345,787 | | | | 12,645,492 | |
| | | | | | | | | | | |
Loss from operations | | | (6,564,641 | ) | | (2,342,677 | ) | | | (12,059,903 | ) |
| | | | | | | | | | | |
Interest income | | | 10,068 | | | 2 | | | | 10,755 | |
Other income (expense) | | | 23,828 | | | (7,257 | ) | | | 15,045 | |
Interest expense | | | (79,119 | ) | | (101,086 | ) | | | (298,580 | ) |
Total non-operating expenses | | | (45,223 | ) | | (108,341 | ) | | | (272,780 | ) |
| | | | | | | | | | | |
Loss from continuing operations | | | (6,609,864 | ) | | (2,451,018 | ) | | | (12,332,683 | ) |
| | | | | | | | | | | |
Loss from discontinued operations | | | — | | | (20,129 | ) | | | (288,724 | ) |
Net loss | | | (6,609,864 | ) | | (2,471,147 | ) | | | (12,621,407 | ) |
Net loss - non-controlling interest | | | (198,516 | ) | | (50,947 | ) | | | (274,777 | ) |
Beneficial conversion charge | | | (2,300,000 | ) | | — | | | | (2,300,000 | ) |
Preferred stock dividend | | | (361,668 | ) | | — | | | | (361,668 | ) |
Net loss attributable to common stockholders | | $ | (9,073,016 | ) | $ | (2,420,200 | ) | | $ | (15,008,298 | ) |
| | | | | | | | | | | |
Net loss per share attributable to common stockholders—basic and diluted | | $ | (2.42 | ) | $ | (1.32 | ) | | | | |
| | | | | | | | | | | |
Weighted-average number of common shares used in net loss per share attributable to common stockholders—basic and diluted | | | 3,747,357 | | | 1,831,769 | | | | | |
See Notes to Consolidated Financial Statements
F-4
HEAT BIOLOGICS INC.
(A development stage company)
Consolidated Statements of Stockholders’ Equity (Deficit)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Preferred Stock | | | Common | | | | | | Accumulated | | | Non-Controlling | | | Total | |
| | Series 1 | | | Series A | | | Series B | | | Stock | | | APIC | | | Deficit | | | Interest | | | Stockholders | |
Common Stock Issued: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
June 10, 2008, 1,395,559 shares | | $ | — | | | $ | — | | | $ | — | | | $ | 321 | | | $ | — | | | $ | — | | | $ | — | | | $ | 321 | |
July 11, 2008, 260,870 shares | | | — | | | | — | | | | — | | | | 60 | | | | — | | | | — | | | | — | | | | 60 | |
July 11, 2008, 184,048 shares | | | — | | | | — | | | | — | | | | 42 | | | | — | | | | — | | | | — | | | | 42 | |
Non-cash consideration for rent | | | — | | | | — | | | | — | | | | — | | | | 4,104 | | | | — | | | | — | | | | 4,104 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (281,971 | ) | | | — | | | | (281,971 | ) |
Balance December 31, 2008 | | | — | | | | — | | | | — | | | | 423 | | | | 4,104 | | | | (281,971 | ) | | | — | | | | (277,444 | ) |
Common Stock Issued: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
January 1, 2009, 60,871 shares | | | — | | | | — | | | | — | | | | 14 | | | | — | | | | — | | | | — | | | | 14 | |
April 20, 2009, 21,835 shares | | | — | | | | — | | | | — | | | | 5 | | | | — | | | | — | | | | — | | | | 5 | |
April 20, 2009, 98,626 shares | | | — | | | | — | | | | — | | | | 23 | | | | — | | | | — | | | | — | | | | 23 | |
Common Stock Cancelled: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
June 26, 2009, (282,672) shares | | | — | | | | — | | | | — | | | | (65 | ) | | | 65 | | | | — | | | | — | | | | — | |
Preferred Stock Issued: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
November 3, 2009, 112,500 shares at $2.22 per share | | | — | | | | 11 | | | | — | | | | — | | | | 249,989 | | | | — | | | | — | | | | 250,000 | |
Non-cash consideration for rent | | | — | | | | — | | | | — | | | | — | | | | 5,760 | | | | — | | | | — | | | | 5,760 | |
Stock based compensation | | | — | | | | — | | | | — | | | | — | | | | 13,364 | | | | — | | | | — | | | | 13,364 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (416,789 | ) | | | (6,650 | ) | | | (423,439 | ) |
Balance December 31, 2009 | | | — | | | | 11 | | | | — | | | | 400 | | | | 273,282 | | | | (698,760 | ) | | | (6,650 | ) | | | (431,717 | ) |
Non-cash consideration for rent | | | — | | | | — | | | | — | | | | — | | | | 5,760 | | | | — | | | | — | | | | 5,760 | |
Stock based compensation | | | — | | | | — | | | | — | | | | — | | | | 30,791 | | | | — | | | | — | | | | 30,791 | |
Stock issuance costs | | | — | | | | — | | | | — | | | | — | | | | (7,584 | ) | | | — | | | | — | | | | (7,584 | ) |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (711,438 | ) | | | (10,406 | ) | | | (721,844 | ) |
Balance December 31, 2010 | | | — | | | | 11 | | | | — | | | | 400 | | | | 302,249 | | | | (1,410,198 | ) | | | (17,056 | ) | | | (1,124,594 | ) |
Notes Payable Converted to Preferred Stock: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
September 30, 2011, 1,273,800 shares at $2.10 per share | | | — | | | | 127 | | | | — | | | | — | | | | 2,674,853 | | | | — | | | | — | | | | 2,674,980 | |
Preferred Stock Issued: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
December 20, 2011, 73,455 shares at $2.10 per share | | | — | | | | 7 | | | | — | | | | — | | | | 154,248 | | | | — | | | | — | | | | 154,255 | |
Preferred Series A Converted to Preferred Series 1, December 16, 2011, 112,500 shares at $2.22 per share | | | 11 | | | | (11 | ) | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | |
Stock based compensation | | | — | | | | — | | | | — | | | | — | | | | 91,984 | | | | — | | | | — | | | | 91,984 | |
Stock issuance costs | | | — | | | | — | | | | — | | | | — | | | | (17,581 | ) | | | — | | | | — | | | | (17,581 | ) |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (2,104,884 | ) | | | (8,258 | ) | | | (2,113,142 | ) |
Balance December 31, 2011 | | $ | 11 | | | $ | 134 | | | $ | — | | | $ | 400 | | | $ | 3,205,753 | | | $ | (3,515,082 | ) | | $ | (25,314 | ) | | $ | (334,098 | ) |
See Notes to Consolidated Financial Statements
F-5
HEAT BIOLOGICS INC.
(A development stage company)
Consolidated Statements of Stockholders’ Equity (Deficit) (Continued)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Preferred Stock | | | Common | | | | | | Accumulated | | | Non-Controlling | | | Total | |
| | Series 1 | | | Series A | | | Series B | | | Stock | | | APIC | | | Deficit | | | Interest | | | Stockholders | |
Balance December 31, 2011 | | $ | 11 | | | $ | 134 | | | $ | — | | | $ | 400 | | | $ | 3,205,753 | | | $ | (3,515,082 | ) | | $ | (25,314 | ) | | $ | (334,098 | ) |
Preferred Stock Issued: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
March 7, 2012, 47,619 shares at $2.10 per share | | | — | | | | 5 | | | | — | | | | — | | | | 99,995 | | | | — | | | | — | | | | 100,000 | |
April 3, 2012, 39,683 shares at $2.10 per share | | | — | | | | 4 | | | | — | | | | — | | | | 83,330 | | | | — | | | | — | | | | 83,334 | |
April 27, 2012, 428,571 shares at $2.10 per share | | | — | | | | 43 | | | | — | | | | — | | | | 899,957 | | | | — | | | | — | | | | 900,000 | |
Common Stock Issued: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
December 27, 2012, 1,087 shares | | | — | | | | — | | | | — | | | | — | | | | 825 | | | | — | | | | — | | | | 825 | |
December 31, 2012, 21,740 shares | | | — | | | | — | | | | — | | | | 5 | | | | 10,495 | | | | — | | | | — | | | | 10,500 | |
Vesting of restricted stock, 97,007 shares | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | |
Stock based compensation | | | — | | | | — | | | | — | | | | — | | | | 217,896 | | | | — | | | | — | | | | 217,896 | |
Stock issuance costs | | | — | | | | — | | | | — | | | | — | | | | (22,419 | ) | | | — | | | | — | | | | (22,419 | ) |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (2,420,200 | ) | | | (50,947 | ) | | | (2,471,147 | ) |
Balance at December 31, 2012 | | | 11 | | | | 186 | | | | — | | | | 405 | | | | 4,495,832 | | | | (5,935,282 | ) | | | (76,261 | ) | | | (1,515,109 | ) |
Preferred Stock Issued: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
March 25, 2013, 1,891,419 shares at $2.67 per share | | | — | | | | — | | | | 189 | | | | — | | | | 5,049,901 | | | | — | | | | — | | | | 5,050,090 | |
Common Stock Issued: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Preferred Series 1 Converted to Common Stock, July 29, 2013, 49,960 | | | (11 | ) | | | — | | | | — | | | | — | | | | 11 | | | | — | | | | — | | | | — | |
Preferred Series A Converted to Common Stock, July 29, 2013, 810,057 | | | — | | | | (186) | | | | — | | | | — | | | | 186 | | | | — | | | | — | | | | — | |
Preferred Series B Converted to Common Stock, July 29, 2013, 836,666 | | | — | | | | — | | | | (189) | | | | — | | | | 189 | | | | — | | | | — | | | | — | |
Common Stock Issued: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Initial public offering, 2,700,000 shares, net of underwriting discounts | | | — | | | | — | | | | — | | | | 540 | | | | 25,110,000 | | | | — | | | | — | | | | 25,110,540 | |
Exercise of stock options, 80,706 shares | | | — | | | | — | | | | — | | | | 16 | | | | 54,026 | | | | — | | | | — | | | | 54,042 | |
Vesting of restricted stock, 2,899 shares | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | |
Preferred Stock Dividend, July 29, 2013, 36,167 shares | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | |
Stock based compensation | | | — | | | | — | | | | — | | | | — | | | | 571,924 | | | | — | | | | — | | | | 571,924 | |
Stock issuance costs | | | — | | | | — | | | | — | | | | — | | | | (944,478 | ) | | | — | | | | — | | | | (944,478 | ) |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (6,411,348 | ) | | | (198,516 | ) | | | (6,609,864 | ) |
Balance at December 31, 2013 | | $ | — | | | $ | — | | | $ | — | | | $ | 961 | | | $ | 34,337,591 | | | $ | (12,346,630 | ) | | $ | (274,777 | ) | | $ | 21,717,145 | |
See Notes to Consolidated Financial Statements
F-6
HEAT BIOLOGICS, INC.
(A development stage company)
Consolidated Statements of Cash Flows
| | | | | | | | | | | | |
| | | | | | | | June 10, 2008 | |
| | For the year ended | | | (Inception) to | |
| | December 31, | | | December 31, | |
| | 2013 | | 2012 | | | 2013 | |
Cash Flows from Operating Activities | | | | | | | | |
Net loss | | $ | (6,609,864 | ) | | | (2,471,147 | ) | | | (12,621,407 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
Depreciation | | | 6,348 | | | | 2,587 | | | | 9,559 | |
Amortization of debt issuance costs | | | 28,229 | | | | 58,458 | | | | 126,742 | |
Amortization of bond premium | | | 50,781 | | | | — | | | | 50,781 | |
Re-measurement of fair value of stock warrants liability | | | 30,440 | | | | 3,540 | | | | 35,020 | |
Non-cash consideration for rent | | | — | | | | — | | | | 15,624 | |
Stock based compensation | | | 571,924 | | | | 217,896 | | | | 925,959 | |
Increase (decrease) in cash arising from changes in assets and liabilities: | | | | | | | | | | | | |
Related party receivable | | | (15,375 | ) | | | (9,571 | ) | | | (24,946 | ) |
Prepaid expenses and other current assets | | | (1,008,202 | ) | | | (52,843 | ) | | | (1,066,638 | ) |
Restricted cash | | | 24,962 | | | | (24,502 | ) | | | (1,252 | ) |
Deposits | | | — | | | | 200 | | | | (9,320 | ) |
Accounts payable | | | 146,446 | | | | 85,323 | | | | 651,917 | |
Accrued expenses and other payables | | | 373,842 | | | | 103,307 | | | | 503,050 | |
Accrued interest | | | 11,601 | | | | 13,077 | | | | 76,635 | |
Net Cash Used by Operating Activities | | | (6,388,868 | ) | | | (2,073,675 | ) | | | (11,328,276 | ) |
| | | | | | | | | | | | |
Cash Flows from Investing Activities | | | | | | | | | | | | |
Purchases of short term investments | | | (17,347,946 | ) | | | — | | | | (17,347,946 | ) |
Purchase of property and equipment | | | (49,319 | ) | | | (1,780 | ) | | | (63,312 | ) |
Net Cash Used in Investing Activities | | | (17,397,265 | ) | | | (1,780 | ) | | | (17,411,258 | ) |
| | | | | | | | | | | | |
Cash Flows from Financing Activities | | | | | | | | | | | | |
Proceeds from initial public offering, net of underwriting discounts | | | 25,110,000 | | | | — | | | | 25,110,000 | |
Related party payable | | | — | | | | (12,500 | ) | | | — | |
Borrowings on notes payable | | | 200,000 | | | | 950,000 | | | | 1,150,000 | |
Borrowings on line of credit | | | — | | | | — | | | | 273,427 | |
Payments on notes payable | | | (925,000 | ) | | | (225,000 | ) | | | (1,150,000 | ) |
Payments on line of credit | | | — | | | | — | | | | (273,427 | ) |
Issuance of convertible notes payable, net of issuance costs | | | — | | | | 197,099 | | | | 2,781,636 | |
Payments on convertible notes payable | | | (197,099 | ) | | | — | | | | (197,099 | ) |
Issuance of common stock | | | 540 | | | | 11,325 | | | | 12,330 | |
Exercise of stock options | | | 54,042 | | | | — | | | | 54,042 | |
Issuance of series A preferred stock | | | — | | | | 1,083,334 | | | | 1,487,589 | |
Issuance of series B-1 preferred stock | | | 5,050,090 | | | | — | | | | 5,050,090 | |
Stock issuance costs | | | (944,478 | ) | | | (22,419 | ) | | | (992,062 | ) |
Net Cash Provided by Financing Activities | | | 28,348,095 | | | | 1,981,839 | | | | 33,306,526 | |
| | | | | | | | | | | | |
Net Increase in Cash and Cash Equivalents | | | 4,561,962 | | | | (93,616 | ) | | | 4,566,992 | |
| | | | | | | | | | | | |
Cash and Cash Equivalents – Beginning of Period | | | 5,030 | | | | 98,646 | | | | — | |
| | | | | | | | | | | | |
Cash and Cash Equivalents – End of Period | | $ | 4,566,992 | | | $ | 5,030 | | | $ | 4,566,992 | |
See Notes to Consolidated Financial Statements
F-7
HEAT BIOLOGICS, INC.
(A development stage company)
Consolidated Statements of Cash Flows (Continued)
| | | | | | | | | | | | |
| | | | | | | | June 10, 2008 | |
| | The year ended | | | (Inception) to | |
| | December 31, | | | December 31, | |
| | 2013 | | 2012 | | | 2013 | |
| | | | | | | | |
Supplemental Disclosure for Cash Flow Information | | | | | | | | |
Interest paid | | $ | 60,922 | | | $ | 29,049 | | | $ | 135,606 | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
Supplemental Schedule of Noncash Investing and Financing Activities | | | | | | | | | | | | |
Beneficial conversion charge | | $ | 2,300,000 | | | $ | — | | | $ | 2,300,000 | |
| | | | | | | | | | | | |
Issuance of preferred stock warrants and debt issuance costs | | $ | — | | | $ | 31,680 | | | $ | 87,570 | |
| | | | | | | | | | | | |
Cancellation of common stock | | $ | — | | | $ | — | | | $ | 65 | |
| | | | | | | | | | | | |
Non-cash consideration for rent | | $ | — | | | $ | — | | | $ | 15,624 | |
| | | | | | | | | | | | |
Non-cash conversion of preferred stock into common stock | | $ | 386 | | | $ | — | | | $ | 386 | |
| | | | | | | | | | | | |
Preferred stock dividend | | $ | 361,668 | | | $ | — | | | $ | 361,668 | |
See Notes to Consolidated Financial Statements
F-8
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1.
Organization
Heat Biologics, Inc. (“Heat” or “the Company”), was incorporated in 2008 pursuant to the laws of the state of Delaware. Heat Biologics, Inc. is a development stage company focused on the development and commercialization ofImPact Therapy, a platform technology that offers a novel approach to treating cancer and other diseases by using live, modified cell lines to activate the immune system against specific defined targets. Heat is currently in Phase II clinical trials with its first drug for patients with advanced non-small cell lung cancer. During 2010 and part of 2011, Heat was headquartered in Miami Beach, Florida. In July 2011, Heat moved its headquarters to Chapel Hill, North Carolina.
Heat has owned 92.5% interests in two subsidiaries, Heat Biologics I, Inc. and Heat Biologics II, Inc. since their incorporation in the state of Delaware and commencement of operations on April 28, 2009. In April of 2012, the Board of Directors approved the sale of Heat’s entire 92.5% interest in Heat II. An independent appraisal report, issued on April 18, 2012, was concurrently approved by the Board as an accurate assessment of Heat II’s fair value of $0.0025 per share. On June 25, 2012 a stock purchase agreement was executed for the purchase of 3,700,000 shares of Heat II common stock by a related party. The operations of Heat II during fiscal year 2012 through June 25, 2012, and inception to date, are presented in the accompanying consolidated statements of operations as a loss from discontinued operations. At December 31, 2013 and 2012, there were no assets or liabilities on the consolidated balance sheets related to the discontinued operations of Heat II.
On May 30, 2012, Heat formed two-wholly owned subsidiaries, Heat Biologics III, Inc. (“Heat III”) and Heat Biologics, IV, Inc. (“Heat IV”). Heat also formed Heat Biologics GmbH (Heat GmbH), a wholly-owned limited liability company, organized in Germany on September 11, 2012.
Heat’s product candidates require clinical trials and approvals from regulatory agencies, as well as acceptance in the marketplace. Part of Heat’s strategy is to develop and commercialize some of its product candidates by continuing existing arrangements with academic and corporate collaborators and licensees and by entering into new collaborations.
2.
Summary of Significant Accounting Policies
Basis of Accounting
The accompanying consolidated financial statements have been prepared on the accrual basis of accounting in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”). Activities during the development stage include developing the business plan, raising capital, and developing the Company’s platform technology.
Principles of Consolidation
The consolidated financial statements include the accounts of Heat Biologics, Inc. and its subsidiaries, Heat Biologics I, Inc. (“Heat I”) and Heat Biologics II, Inc. (“Heat II”), Heat Biologics III, Inc. (“Heat III”), Heat Biologics IV, Inc. (“Heat IV”) and Heat Biologics GmbH. All significant intercompany accounts and transactions have been eliminated in consolidation. At December 31, 2013 and 2012, Heat held a 92.5% controlling interest in Heat I and accounts for its less than 100% interest in the consolidated financial statements in accordance with U.S. GAAP. Accordingly, the Company presents non-controlling interests as a component of stockholders’ equity (deficit) on its consolidated balance sheets and reports non-controlling interest net loss under the heading “net loss – non-controlling interest” in the consolidated statements of operations. In June 2012, the Company sold its entire 92.5% interest in Heat II. The operations of Heat II through June 25, 2012, and inception to date, are presented in the accompanying consolidated statements of operations as a loss from discontinued operations.
F-9
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Estimates are used for, but not limited to, useful lives of fixed assets, income taxes and stock-based compensation. Actual results may differ from those estimates.
Cash and Cash Equivalents and Restricted Cash
The Company considers all cash and other highly liquid investments with initial maturities from the date of purchase of three months or less to be cash and cash equivalents. The Company had a restricted cash balance of $1,252 and $26,214 at December 31, 2013 and 2012, respectively. The United States Patent and Trade Office (“USPTO”) requires the Company to maintain an account with a minimum of $1,000 to be used to pay fees associated with new trademarks of the Company and one of the Company’s lenders required a minimum $25,000 cash balance to be maintained with the lending bank during 2012.
Concentration of Credit Risk
At times, cash balances may exceed the Federal Deposit Insurance Corporation (“FDIC”) insurable limits. The Company has never experienced any losses related to these balances. All of the Company’s cash balances were fully insured at December 31, 2012. As of December 31, 2013, cash amounts in excess of $250,000 were not fully insured. The uninsured cash balance as of December 31, 2013 was $4,046,451. The Company does not believe it is exposed to significant credit risk on cash and cash equivalents.
Debt Issuance Costs, net
Debt issuance costs include the costs incurred to obtain financing, including the fair value of preferred stock warrants at the date of their issuance, and are amortized using the straight-line method, which approximates the effective interest method, over the life of the related debt. Debt issuance costs are included in the accompanying consolidated balance sheets net of amortization.
Property and Equipment
Property and equipment are stated at cost and are capitalized if the cost exceeds $500. Depreciation is calculated using the straight-line method and is based on estimated useful lives of 3 years for computer equipment and seven years for furniture and fixtures.
Stock Warrants Liability
In December 2011 and August 2012, the Company entered into a promissory note with each of two lenders and issued preferred stock warrants to each lender as consideration. The Company has accounted for these freestanding warrants as liabilities at their fair value on the accompanying consolidated balance sheets. The warrants are subject to re-measurement at each balance sheet date, and the change in fair value, if any, is recognized as other income (expense). The warrants converted from preferred stock warrants into warrants to purchase common stock upon the completion of the initial public offering in July 2013 and the number of shares were adjusted for the 1-for-2.3 reverse stock split. However, since the warrants still have an anti-dilution provision, they remain liabilities and are subject to re-measurement at each balance sheet date. The warrants are valued using a Monte Carlo simulation which is a generally accepted statistical method used to generate a defined number of stock price paths in order to develop a reasonable estimate of the range of the Company’s future expected stock prices and minimizes standard error.
F-10
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Significant assumptions used in the valuation of the stock warrants liability were as follows:
| | | | | | | | |
| | December 31, | |
| | 2013 | | | 2012 | |
| | | | | | |
Exercise price | | $ | 4.83 | | | $ | 4.83 | |
Risk-free interest rate | | | 2.75 | % | | | 1.78 | % |
Expected volatility | | | 71.6-71.9 | % | | | 75.6-76.3 | % |
Expected life (years) | | | 7.96-8.6 | | | | 10 | |
Expected dividend yield | | | 0 | % | | | 0 | % |
Beneficial Conversion Feature
When the Company issues an equity security that is convertible into common stock at a discount from the fair value of the common stock at the date the equity security counterparty is legally committed to purchase such a security (Commitment Date), a beneficial conversion charge is measured and recorded on the Commitment Date for the difference between the fair value of the Company’s common stock and the effective conversion price of the equity security. If the intrinsic value of the beneficial conversion feature is greater than the proceeds allocated to the equity security, the amount of the discount assigned to the beneficial conversion feature is limited to the amount of the proceeds allocated to the equity.
The amount allocated to the beneficial conversion feature is presented as an immediate charge to earnings available to common shareholders for convertible preferred stock instruments that are convertible by the shareholders at any time. In connection with the Company’s issuance of Series B-1 Preferred Stock during fiscal year 2013, the Company recorded a beneficial conversion charge of $2.3 million representing the difference between the effective conversion price of $6.14 and the fair value of the Company’s common stock as of the Commitment Date of $8.81.
Net Loss per Share
Basic net loss per share is computed by dividing net loss by the weighted average number of common shares outstanding during each year. Fully diluted net loss per share is computed using the weighted average number of common shares and dilutive securities outstanding during each year. Dilutive securities having an anti-dilutive effect on diluted loss per share are excluded from the calculation.
Fair Value of Financial Instruments
The carrying amount of certain of the Company’s financial instruments, including cash and cash equivalents, prepaid expenses and other current assets, deposits, accounts payable and accrued expenses and other payables approximate fair value due to their short maturities. The carrying value of the Company’s notes payable and convertible notes payable at December 31, 2012 approximated fair value because the interest rates under those obligations approximated market rates of interest available to the Company for similar instruments.
As a basis for determining the fair value of certain of the Company’s financial instruments, the Company utilizes a three-tier value hierarchy, which prioritizes the inputs used in measuring fair value as follows:
Level I – Observable inputs such as quoted prices in active markets for identical assets or liabilities.
Level II – Observable inputs, other than Level I prices, such as quoted prices for similar assets or liabilities, quoted prices in markets that are not active or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level III – Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
F-11
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
This hierarchy requires the Company to use observable market data, when available, and to minimize the use of unobservable inputs when determining fair value. The Company’s financial instruments that are measured at fair value on a recurring basis consist only of the stock warrants liability. The Company’s stock warrants liability was classified within Level III of the fair value hierarchy and as of December 31, 2013 and 2012.
The change in the fair value of the Level III warrants liability is summarized below:
| | | | |
Fair value at December 31, 2012 | | $ | 92,150 | |
Issuances | | | — | |
Change in fair value during the period | | | 30,440 | |
| | | | |
Fair value at December 31, 2013 | | $ | 122,590 | |
The change in the fair value of the Level III stock warrants liability is summarized below:
| | | | | | | | | | | | | |
| | December 31, 2013 | |
| | Identical Assets | | Observable Inputs | | Unobservable Inputs | | Total December 31, | |
Description | | (Level 1) | | (Level 2) | | (Level 3) | | 2013 | |
Liabilities measured at fair value | | | | | | | | | | | | | |
Stock Warrant Liability | | $ | — | | $ | — | | $ | (122,590 | ) | $ | (122,590 | ) |
| | | | | | | | | | | | | |
Total Liabilities measured at fair value | | $ | — | | $ | — | | $ | (122,590 | ) | $ | (122,590 | ) |
| | | | | | | | | | | | | |
| | December 31, 2012 | |
| | Identical Assets | | Observable Inputs | | Unobservable Inputs | | Total December 31, | |
Description | | (Level 1) | | (Level 2) | | (Level 3) | | 2012 | |
Liabilities measured at fair value | | | | | | | | | | | | | |
Stock Warrant Liability | | $ | — | | $ | — | | $ | (92,150 | ) | $ | (92,150 | ) |
| | | | | | | | | | | | | |
Total Liabilities measured at fair value | | $ | — | | $ | — | | $ | (92,150 | ) | $ | (92,150 | ) |
Marketing
Marketing costs are expensed as incurred. Marketing expense totaled $135,366 and $5,921 for the years ended December 31, 2013 and 2012, respectively. Marketing expenses from inception through December 31, 2013 totaled $183,274.
Income Tax
Income taxes are accounted for using the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to temporary differences between the financial statements carrying amounts of assets and liabilities and their respective tax bases, operating loss carryforwards, and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
In accordance with FASB ASC 740,Accounting for Income Taxes, the Company reflects in the financial statements the benefit of positions taken in a previously filed tax return or expected to be taken in a future tax return only when it is considered ‘more-likely-than-not’ that the position taken will be sustained by a taxing authority. As of December 31, 2013 and 2012, the Company had no unrecognized income tax benefits and correspondingly there is no impact on the Company’s effective income tax rate associated with these items. The Company’s policy for recording interest and penalties relating to uncertain income tax positions is to record them as a component of income tax expense in the accompanying consolidated statements of operations, As of December 31, 2013 and 2012, the Company had no such accruals.
F-12
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Stock-Based Compensation
The Company accounts for stock-based compensation arrangements with employees and non-employee directors using a fair value method which requires the recognition of compensation expense for costs related to all stock-based payments, including stock options. The fair value method requires the Company to estimate the fair value of stock-based payment awards on the date of grant using an option pricing model.
Stock-based compensation costs are based on the fair value of the underlying option calculated using the Black-Scholes-Merton option pricing model on the date of grant for stock options and recognized as expense on a straight-line basis over the requisite service period, which is the vesting period. Determining the appropriate fair value model and related assumptions requires judgment, including estimating stock price volatility, forfeiture rates and expected term. The expected volatility rates are estimated based on the actual volatility of comparable public companies over the expected term. The expected term for the years ended December 31, 2013 and 2012 represents the average time that options are expected to be outstanding based on the mid-point between the vesting date and the end of the contractual term of the award. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The Company has not paid dividends and does not anticipate paying a cash dividend in the foreseeable future and, accordingly, uses an expected dividend yield of zero. The risk-free interest rate is based on the rate of U.S. Treasury securities with maturities consistent with the estimated expected term of the awards. The measurement of nonemployee share-based compensation is subject to periodic adjustments as the underlying equity instruments vest and is recognized as an expense over the period over which services are received.
Net loss attributable to non-controlling interests
Net loss attributable to non-controlling interests is the result of the Company's consolidation of subsidiaries of which it does not own 100%. The Company's net loss attributable to non-controlling interests relates to the University's ownership in Heat I, and its ownership interest in Heat II before the divestiture of Heat II on June 25, 2012.
Revenue Recognition
The Company recognizes government grants when there is reasonable assurance that they will comply with the conditions attached to the grants and the grants will be received. The grants are recognized using an income approach and grant revenue is recognized as the related expenses are incurred.
Research and Development
Research and development costs are expensed as incurred. The Company has acquired exclusive licensing rights to intellectual property to further its research and development. These costs are expensed as incurred. The Company also incurs legal costs relating to the filing and application fees for patents which are owned by the universities with which the Company has license agreements. These costs are also expensed as research and development expense as incurred.
3.
Discontinued Operations
In April of 2012, the Company's board approved a plan to sell its 92.5% interest in Heat II to a related party entity. On June 25, 2012, the Company sold all of its interest in Heat II to the related party in exchange for $9,250 in cash and a receivable from the related party of $296,244. The receivable is due in full approximately seven years from the date of the transaction with interest accruing at a rate of 6% per annum. The Company performed a fair value analysis of the receivable from the related party and determined that due to the uncertainty surrounding the collectibility of the receivable, the fair value was $0. The Company's estimate of the fair value of the receivable is based upon several factors including the long-term maturity of the receivable, an analysis of the related party's ability and willingness to pay the receivable given the current financial position, and that fact that Heat II is likely years away from generating product revenues.
The $9,250 in cash was recorded as a reduction to the loss from discontinued operations in the consolidated statement of operations for the year ended December 31, 2012. The operations of Heat II through June 25, 2012, and inception to date, are presented in the accompanying consolidated statements of operations as a loss from discontinued operations.
F-13
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
4.
Investments
Investments - Investments in certain securities may be classified into three categories:
·
Held-to-maturity - Debt securities that the Company has the positive intent and ability to hold to maturity are reported at amortized cost.
·
Trading securities - Debt and equity securities that are bought and held principally for the purpose of selling in the near term are reported at fair value with unrealized gains and losses included in earnings.
·
Available-for-sale - Debt and equity securities not classified as either securities held-to-maturity or trading securities are reported at fair value with unrealized gains or losses excluded from earnings and reported as a separate component of stockholders’ equity.
The Company reassesses the appropriateness of the classification of its investments at the end of each reporting period. The Company has determined that its debt securities should be classified as held-to-maturity as of December 31, 2013. The Company held no investments at December 31, 2012. This classification was based upon management’s determination that it has the positive intent and ability to hold the securities until their maturity dates, as the underlying cash invested in these securities is not required for current operations. Investments consist of short-termFDIC insured certificates of deposit, commercial paper rated A1/P1 or above and corporate notes and bonds rated A and above carried at amortized cost using the effective interest method.
The following table summarizes information about short term investments at December 31, 2013:
| | | | | | | | | | | | |
| | Amortized
Cost | | | Gross
Unrealized
Losses | | | Estimated
Fair Value | |
Certificates of deposit, commercial paper | | $ | 17,297,165 | | | $ | 16,493 | | | $ | 17,280,672 | |
As of December 31, 2013, the estimated fair value of the investments was less than the amortized cost. Because management intends to hold the investments until their maturity dates, these unrealized losses were not recorded in the consolidated financial statements.
The maturities of held-to-maturity investments at December 31, 2013 were as follows:
| | | | | | | | |
| | Less than
1 Year | | | Total | |
Certificates of deposit, commercial paper | | $ | 17,297,165 | | | $ | 17,297,165 | |
5.
Property and Equipment
Property and equipment are recorded at cost and depreciated using the straight-line method, over estimated useful lives, ranging generally from five to seven years. Expenditures for maintenance and repairs are charged to expense as incurred.
Property and equipment consisted of the following:
| | | | | | | | |
| | December 31, | |
| | 2013 | | | 2012 | |
| | | | | | |
Furniture and fixtures | | $ | 10,780 | | | $ | 10,780 | |
Computers | | | 13,175 | | | | 3,213 | |
Lab equipment | | | 39,357 | | | | — | |
| | | | | | | | |
Total | | | 63,312 | | | | 13,993 | |
Accumulated depreciation | | | (9,559 | ) | | | (3,211 | ) |
| | | | | | | | |
Property and equipment, net | | $ | 53,753 | | | $ | 10,782 | |
F-14
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Depreciation expense totaled $6,348 and $2,587 for the years ended December 31, 2013 and 2012, respectively.
6.
Accrued Expenses
Accrued expenses consist of the following:
| | | | | | | | |
| | December 31, | |
| | 2013 | | | 2012 | |
| | | | | | |
Compensation and related benefits | | $ | 356,588 | | | $ | 105,927 | |
Accrued patent fees | | | 40,000 | | | | 20,000 | |
Miscellaneous expenses | | | 106,462 | | | | 3,281 | |
| | $ | 503,050 | | | $ | 129,208 | |
7.
Debt Issuance Costs
In December 2011, the Company recorded $55,890 of debt issuance costs related to the issuance of warrants to purchase Series A Preferred Stock to a lender. The warrants were issued in conjunction with a promissory note issued to the lender. In December 2011, the Company began amortizing the debt issuance costs over the three year term of the promissory note resulting in $883 of interest expense for the year ended December 31, 2011. The note payable associated with the preferred stock warrants was paid in full and terminated during 2012. The remaining balance of $55,007 was amortized and written off during 2012.
In August 2012, the Company recorded $31,680 of debt issuance costs related to the issuance of warrants to purchase Series A Preferred Stock to a lender. The warrants were issued in conjunction with a promissory note issued to the lender. At this time, the Company began amortizing the debt issuance costs over the four year term of the promissory note resulting in $3,451 of interest expense for the year ended December 31, 2012.
Total amortization expense for the debt issuance costs was $28,229 and $58,548 during fiscal year 2013 and 2012, respectively.
8.
Convertible Notes Payable
On October 20, 2011, the Company entered into a convertible note agreement with a vendor for an amount up to $950,000. The note accrues 12% simple interest per annum beginning on the day of the first advance. The note is convertible into common or Series A preferred stock at the latest valuation. The type of security converted will depend on whether common or Series A preferred stock is issued as part of a successful future equity raise of at least $7.5 million at the qualified offering price. Unless earlier converted into equity, the note will be payable upon demand after the eighth anniversary of the execution date of the vendor agreement which occurs in October 2019. The agreement allows the vendor to treat unpaid invoices as advances of principal under the promissory note. As of December 31, 2012, the outstanding balance on the note was $197,099. The note payable was terminated and paid off in July 2013.
Accrued interest on outstanding debt obligations was $0 and $13,763 at December 31, 2013 and 2012, respectively.
9.
Notes Payable
On December 14, 2011, the Company entered into a loan agreement with the North Carolina Biotechnology Center (the “Center”) for an amount up to $250,000 to be used by the Company to develop certain of its proprietary technology and processes as defined by the loan agreement during a one year period ended December 14, 2012. The principal of the loan, plus accrued interest, was due in full on December 14, 2014, with annual installments of 5% of the outstanding balance due on December 14, 2012 and 2013. The loan agreement accrues interest at 4.25% per annum beginning on the day of the first advance. As of December 31, 2011, the outstanding balance was $0 and no draw downs occurred during fiscal year 2011. During the year ended December 31, 2012, the Company drew down $225,000 of the loan and then repaid the principal balance, including accrued interest, in full in August 2012. The loan agreement was canceled upon the repayment.
F-15
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
In conjunction with this loan agreement, the Company issued warrants to purchase 29,762 shares of Series A Preferred Stock with an exercise price of $4.83 per share and an expiration date of December 13, 2021. Per the terms of the warrant agreement, the exercise price of $4.83 per share is subject to adjustment if at any time subsequent to the date of the warrant agreement, Preferred Series A shares are issued at a price less than $4.83 per share. The warrants converted from preferred stock warrants into warrants to purchase common stock upon the completion of the initial public offering in July 2013 and the number of shares were adjusted for the 1-for-2.3 reverse stock split. However, since the warrants still have an anti-dilution provision, they remain liabilities and are subject to re-measurement at each balance sheet date. The total number of warrants post the 1-for-2.3 reverse stock split is 12,940.
On August 7, 2012, the Company entered into a loan and security agreement (“the Loan and Security Agreement”) with a bank. The terms of the agreement provide for a $1,000,000 term loan (“Tranche A”) to be available to the Company as of the date of the Loan and Security Agreement. The Tranche A term loan may be increased to $2,775,000 upon the Company receiving grant funding totaling at least $16,000,000. The Tranche A term loan accrues interest monthly at an interest rate of 3% plus Prime or 6% per annum, whichever is greater. The Tranche A term loan principal balance, along with any accrued interest, is to be paid in thirty-six equal monthly installments beginning September 7, 2013 and ending August 7, 2016. As of December 31, 2012, the Company’s outstanding principal balance on the Tranche A term loan was $500,000.
Additionally, the Loan and Security Agreement provides for a term loan in an aggregate principal amount not to exceed $225,000 (“Term Loan B”). Payments of 5% of the outstanding principal balance, plus accrued interest are each due on August 2013 and 2014, with the remaining principal balance, plus all accrued interest, due December 14, 2014. The term loan accrues interest monthly at 4.25% per annum. Proceeds from the $225,000 Term Loan B were used to pay in full the principal balance of the loan with the Center as noted above. On August 27, 2013, the Company repaid the entire outstanding balance on the Tranche A term loan and the Term Loan B with the bank, in the amount of $725,000 and the loan agreement was terminated.
On January 10, 2013, the Company signed a Second Amendment to its Loan and Security Agreement which granted an extension of credit in the form of a Non-Formula Revolving Line (“the Non-Formula Line”) for an amount up to $200,000. This increase in credit was through a limited guaranty by an investor who secured the additional obligation by maintaining as collateral a money market account of a minimum of $200,000 with the bank. This guarantee was only for the amounts arising from the Line. It was the intention of both the investor and the Company that the Line was to be repaid within a reasonable time period after the successful raise of capital but no later than January 9, 2014, the maturity date of the Line. The payoff of the Line would release the investor of its obligation to the bank. The Company borrowed $200,000 on the Line in January 2013, and the entire balance was paid in April 2013.
In conjunction with the Loan and Security Agreement, the Company issued warrants to the bank to purchase 17,500 shares of Heat’s Series A Preferred Stock. The warrants were issued on August 7, 2012 with an initial exercise price of $4.83 per share and expire on August 7, 2022. The warrants converted from preferred stock warrants into warrants to purchase common stock upon the completion of the initial public offering in July 2013 and the number of shares were adjusted for the 1-for-2.3 reverse stock split. However, since the warrants still have an anti-dilution provision, they remain liabilities and are subject to re-measurement at each balance sheet date. The total number of warrants post the 1-for-2.3 reverse stock split is 7,609.
10.
License Agreements
On July 11, 2008, Heat entered into two agreements with The University of Miami (the “University”) to license, from the University, certain technology and processes in various stages of patent pursuit on an exclusive basis for use in its research and development and commercial activities (“License Agreement 03-31, 05-39” and “License Agreement 97-14”, or collectively “License Agreements”). Heat has the right to grant sublicenses under the License Agreements.
Heat is also responsible for all patent costs, past and future, associated with the preparation, filing, prosecution, issuance, and maintenance of United States patent applications. Heat is also required to make minimum royalty payments to the University under the terms of the License Agreements.
In connection with the License Agreements, Heat agreed to issue to the University 10% of all issued and outstanding common stock in each class and series on a fully-diluted basis together with rights to participate in future stock offerings.
F-16
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
In April 2009, Heat and the University agreed to amend the original License Agreements of July 11, 2008 to extend the terms of payments. For the additional consideration of $12,500 and additional stock of 2.5% of fully-dilutable shares issued and outstanding for each License Agreement, a revised extension date of August 11, 2009 was granted for all past due license fees and patent costs. Furthermore, the 10% original stock holdings were given assurance of anti-dilution protection until a “Qualified Investment” pursuant to this agreement. This anti-dilution protection has been distinguished with the subsequent agreement described below.
On June 26, 2009, Heat assigned all rights and obligations of License Agreement 03-31, 05-39 and License Agreement 97-14 to its subsidiaries, Heat II and Heat I, respectively. All previous stock ownership and rights of the University to participate in future stock offerings by Heat were mutually terminated. Heat I and Heat II agreed to issue the University 5% of each subsidiary’s issued and outstanding common stock in each class and series on a fully-diluted basis, together with fully-dilutable common shares equal to 2.5% of the total number of shares in each class and series issued outstanding. As a result, the University owns 7.5% of Heat I and Heat II’s issued and outstanding common stock. For each agreement, the Company agreed to make minimum royalty payments of $10,000 for three years beginning 2010 due on the anniversary date of the agreements. Beginning in 2013, and thereafter for the life of the agreements, the minimum royalty payments shall be $20,000 due on the same date. A milestone payment is due to the University from the Company no later than March 2022 of $400,000 for License Agreement 03-31, 05-39. Another milestone payment is due no later than May 2017 of $250,000 for License Agreement 97-14.
In August 2009, Heat II and the University entered into a second amendment (“Amendment 2”) to License Agreement 03-31, 05-39 to extend the foregoing payment due dates for all past due license fees and patent costs.
In August 2009, Heat I and the University entered into a second amendment (“Amendment 2”) to License Agreement 97-14 to extend the foregoing payment due dates for all past due license fees and patent costs.
In February 2010, Heat II and the University entered into a third amendment (“Amendment 4”) to License Agreement 03-31, 05-39 to grant back to the University a certain non-exclusive license. In all other respects, the original agreement remained the same.
On August 30, 2010, Heat entered into an option agreement with the University of Michigan (“University II”) to acquire the right to negotiate an exclusive license for certain materials which includes cancer bladder cells and all unmodified derivatives of these cells. An option fee of $2,000 was paid on September 8, 2010 to grant a period of nine months for this consideration. In July 2011, the Company exercised the option to acquire the license for $10,000.
In October 2010, Heat II and the University entered into a fourth amendment (“Amendment 5”) to License Agreement 03-31, 05-39 to grant to the licensor a non-exclusive license right for certain technology as research reagents and research tools.
On December 12, 2010, Heat II entered into another license agreement (“I-176”) with the University for one component of complimentary technology to the July 11, 2008 agreement. Heat II agreed to pay the University a license fee of $50,000 and a reimbursement of $15,797 for past patent fees. Heat II also agreed to make a minimum royalty payment of $10,000 during 2012.
On February 18, 2011, Heat I entered into a license agreement (“SS114A”) with the University to obtain additional technology related to License Agreement 97-14. Heat I agreed to reimburse the University for all past patent costs of $37,381. As partial consideration for the license, Heat II agreed to grant back certain exclusive rights to the University.
On February 18, 2011, Heat I entered into a license agreement (“143”) with the University to obtain additional technology related to License Agreement 97-14. In consideration for the license, Heat I agreed to pay the University a fee of $50,000 and reimburse them for past patent costs of $14,158.
On February 18, 2011, Heat I entered into a license agreement (“J110”) with the University to obtain additional technology related to License Agreement 97-14. In consideration for the license, Heat I agreed to pay the University a fee of $10,000 and reimburse them for past patent costs of $1,055.
F-17
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
On February 18, 2011, Heat I entered into a license agreement (“D-107”) with the University to obtain additional technology related to License Agreement 97-14.
On April 12, 2011, Heat entered into a non-exclusive evaluation and biological material license agreement with a not-for-profit corporation for evaluation and production of vaccines. In consideration for the evaluation and commercial use license, Heat agreed to pay the not-for-profit corporation a fee of $5,000 and $50,000, respectively. Heat has the option to renew the license once the original term has expired. Milestone payments are due upon certain events agreed upon by Heat and the not-for-profit corporation.
At December 31, 2011, Heat owed the University $160,000 in unpaid license fees. At December 19, 2012, Heat I owed the University $102,784 in unpaid license fees. Heat entered into a payment agreement on December 19, 2012 to extend the payment due of Heat I obligations until the earlier of the closing of a Series B financing round or June 1, 2013. As consideration for the extension of payment Heat I made an additional payment to the University equal to 18% annual interest of the outstanding balance on or before the due date or at the University’s option convert into shares of preferred stock according to the terms stipulated in the agreement.
Future minimum royalty payments as of December 31, 2013 are as follows:
| | | | |
Year ended December 31, | | | |
| | | |
2014 | | $ | 30,000 | |
2015 | | | 30,000 | |
2016 | | | 30,000 | |
2017 | | | 280,000 | |
2018 | | | 30,000 | |
Thereafter | | | 120,000 | |
| | | | |
Total | | $ | 520,000 | |
11.
Stock Warrants Liability
The summary of stock warrants liability activity for the years ended December 31, 2013 and 2012 is as follows:
| | | | | | | | | | | | | | | | |
| | Number of Warrants | | | Weighted Average Exercise Price | | | Weighted Average Remaining Contractual Life (in years) | | | Weighted Average Grant Date Fair Value | |
| | | | | | | | | | | | |
Outstanding at December 31, 2011 | | | 12,940 | | | $ | 4.83 | | | | 9.9 | | | $ | 1.91 | |
Granted | | | 7,609 | | | $ | 4.83 | | | | 9.9 | | | $ | 1.81 | |
Exercised | | | — | | | | — | | | | — | | | | — | |
Expired/cancelled | | | — | | | | — | | | | — | | | | — | |
Outstanding at December 31, 2012 | | | 20,549 | | | $ | 4.83 | | | | 8.9 | | | $ | 1.64 | |
Granted | | | — | | | | — | | | | — | | | | — | |
Exercised | | | — | | | | — | | | | — | | | | — | |
Expired/cancelled | | | — | | | | — | | | | — | | | | — | |
Outstanding at December 31, 2013 | | | 20,549 | | | $ | 4.83 | | | | 8.20 | | | $ | 5.95 | |
The aggregate intrinsic value of the stock warrants in the table above is $46,646 and $0 at December 31, 2013 and 2012, respectively. The aggregate intrinsic value is before applicable income taxes and is calculated based on the difference between the exercise price of the warrants and the estimated fair market value of the Company’s common stock as of the respective dates.
F-18
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
12.
Stockholders’ Equity (Deficit)
Authorized Capital
Heat has authorized 112,500 shares of Series 1 Preferred Stock (par value $0.0001) as of December 31, 2013 and 2012. Of the Series 1 Preferred Stock, 112,500 were issued and outstanding as of December 31, 2012. No Series 1 Preferred Stock was outstanding at December 31, 2013. Heat has authorized 2,000,000 shares of Series A Preferred Stock (par value $0.0001) as of December 31, 2013 and 2012. Of the Series A Preferred Stock, 1,863,128 shares were issued and outstanding as of December 31, 2012. No Series A Preferred Stock was outstanding at December 31, 2013. In 2013, Heat authorized 4,100,000 shares of Series B Preferred Stock (par value $0.0002). In March 2013, the Company sold an aggregate of 1,891,419 shares of the Company’s Series B-1 Preferred Stock for gross proceeds of approximately $5.0 million in our Series B Preferred Stock private placement. All shares of the Series B Preferred Stock, together with accrued dividends, automatically converted into shares of the Company’s common stock upon the consummation of the Company’s initial public offering on July 29, 2013.
Heat had 50,000,000 shares of common stock (par value $0.0002) authorized as of December 31, 2013 and 2012. Of the 50,000,000 common stock shares, 6,375,426 and 2,144,542 were issued and 6,375,426 and 1,858,971 were outstanding as December 31, 2013 and 2012, respectively.
Preferred Stock
Series 1, Series A, Series B-1, and Series B-2
Automatic Conversion
Each share of Preferred Stock automatically converts to common stock upon the earlier to occur of (i) on the date of consummation of a sale of common stock in a firm commitment underwritten public offering resulting in aggregate net cash proceeds to the Company (after deducting applicable underwriting discounts and commissions) of at least $15 million net proceeds; (ii) with respect to the Series A Preferred Stock, if 2/3 of the Series A Preferred Stock holders (including one of the larger investors so long as they hold 40% of the Series A Preferred Stock) vote in favor of a conversion then the Series A will automatically convert to common stock; (iii) with respect to the Series 1 Preferred Stock, if 2/3 of the Series 1 Preferred Stock holders vote in favor of a conversion then the Series 1 will automatically convert to common stock; and (iv) with respect to the Series B Preferred Stock if 2/3 of the Series B Preferred Stock holders vote in favor of a conversion then the Series B will automatically convert to common stock. As a result of the IPO, all outstanding shares of preferred stock were automatically converted to common stock.
Optional Conversion
The preferred stock is convertible into common stock at the option of the holder at any time. The conversion ratio for each share of the Series 1 Preferred Stock and the Series A Preferred Stock was its Original Issue Price ($2.35 and $2.10 for each share of the Series 1 Preferred Stock and Series A Preferred Stock, respectively) divided by its Conversion Price, as adjusted for stock splits, stock dividends, reorganizations, recapitalizations and the like, which Conversion Price initially was the Original Issue Price. The conversion ratio for each share of the Series B-1 Preferred Stock and the Series B-2 Preferred Stock was its Original Issue Price ($2.67 and $5.00 for each share of the Series B-1 Preferred Stock and Series B-2 Preferred Stock, respectively) plus accrued but unpaid dividends thereon divided by its conversion price, as adjusted for stock splits, stock dividends, reorganizations, recapitalizations and the like, which conversion price initially was the Original Issue Price. As a result of the 1-for-2.3 reverse stock split, the conversion ratio for the Preferred Stock was 0.4348.
F-19
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
In the event the Company at any time or from time to time after the Initial Series B Issuance Date shall issue additional shares of common stock without consideration or for consideration per share less than the Series 1 Conversion Price, Series A Conversion Price, Series B-1 Conversion Price, or Series B-2 Conversion Price, in effect on the date of and immediately prior to such issue, then the Series 1 Conversion Price, Series A Conversion Price, the Series B-1 Conversion Price, Series B-2 Conversion Price, shall be reduced, to a price determined by multiplying the Series 1 Conversion Price, Series A Conversion Price, the Series B-1 Conversion Price, or the Series B-2 Conversion Price in effect by a fraction, (A) the numerator of which shall be the number of shares of common stock outstanding immediately prior to such issuance, on a fully-diluted basis, plus the number of shares of common stock which the aggregate consideration received by the Company for the total number of Additional Shares of Common Stock so issued would purchase at the Series 1 Conversion Price, Series A Conversion Price, the Series B-1 Conversion Price, or the Series B-2 Conversion Price, as in effect immediately prior to such issuance, and (B) the denominator of which shall be the number of shares of common stock outstanding immediately prior to such issuance, on a fully-diluted basis, plus the number of such Additional Shares of common stock so issued. As a result of the IPO, all outstanding shares of preferred stock were automatically converted to common stock.
The preferred stock was determined to have characteristics more akin to equity than debt. Particularly, the preferred stock had no mandatory redemption provision nor was it redeemable at the option of the holder. As a result, the conversion option was determined to be clearly and closely related to the preferred stock and therefore did not need to be bifurcated and classified as a liability.
Dividends
The Series B Preferred Stock has a priority with respect to dividend distributions and distributions upon liquidation. The Series B Preferred Stock receive dividends when and as and if declared by the Board at a rate of 5% of their original issue price of such shares which is $6.14 per share for the Series B-1 Preferred Stock and $11.50 per share for the Series B-2 Preferred Stock. If the Company declares or pays a dividend upon the common stock, they must also pay to the holders of the Series A, 1 and B Preferred Stock the dividends that would have been declared with respect to common stock issuable upon conversion of the Series A, 1 and B Preferred Stock; provided, however that the Company cannot declare or pay a dividend unless and until all accrued dividends on the Series B Preferred Stock have been paid.
Liquidation
In the event of a liquidation, the holders of the Series B-1 and B-2 Preferred Stock are entitled to receive before any payment to any other Preferred Stockholder or common stock holder and pari passu with the holders of the Series 1 Preferred Stock an amount per share equal to the greater of $6.14 for the Series B-1 Preferred Stock and $11.50 for the Series B-2 Preferred Stock plus any dividends accrued and unpaid whether or not declared. After payment in full of the Series B Preferred Stockholders the holders of the Series A Preferred Stock are entitled to receive before any payment to the common stock holder and pari passu with the holders of the Series 1 Preferred Stock an amount per share equal to $4.83 plus any dividends declared but unpaid. In the event of a liquidation, the holders of the Series 1 Preferred Stock are entitled to receive before any payment to the common stock holder and pari passu with any distribution to the Series A Preferred Stock an amount per share equal to $5.41 plus any dividends declared but unpaid. After the payment in full of the amounts set forth above, the Company’s assets will be distributed ratably to all holders of common stock and Series B Preferred Stock on an as converted basis except that the Series B Preferred Stockholders shall not continue to share in such distribution after each has received 3 times its Original Issue Price.
Voting Rights
Each holder of Preferred Stock is entitled to vote on all matters stockholders are entitled to vote and to cast the number of votes as shall equal the whole number of shares of common stock into which their shares of Preferred Stock are convertible.
F-20
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Preferred Stock Dividend
In March 2013, the Company sold an aggregate of 1,891,419 shares of the Company’s Series B-1 Preferred Stock for gross proceeds of approximately $5.0 million in our Series B Preferred Stock private placement. All shares of the Series B Preferred Stock, together with accrued dividends, automatically converted into shares of the Company’s common stock upon the consummation of the Company’s initial public offering on July 29, 2013. In addition, the investors in the Series B-1 Preferred Stock were issued shares of the Company’s common stock having a value based upon the initial public offering price of $361,668 and the Company’s obligation to issue, and the investors, obligation to purchase, Series B-2 Preferred Stock and warrants upon fulfillment of certain conditions specified in the Company’s stock purchase agreement dated as of March 25, 2013 entered into in connection with such private placement (the “Stock Purchase Agreement”) terminated. The issuance of common stock to the Series B-1 Preferred stockholders totaling $361,668 has been accounted for as a preferred stock dividend, and as a result, has been included as an expense attributable to common stockholders in the Company’s condensed consolidated statements of operations.
Initial Public Offering
On July 29, 2013, the Company sold 2,500,000 shares of common stock at a public offering price of $10.00 per share upon the closing of the Company’s initial public offering (“IPO”) with gross proceeds of $25 million and net proceeds of $22.4 million. On August 15, 2013, the Company sold an additional 100,000 shares of common stock at a public offering price of $10.00 per share pursuant to the partial exercise of the over-allotment option granted to the underwriters resulting in additional gross proceeds to the Company of $1,000,000 and additional net proceeds of $930,000. On September 6, 2013, the Company sold an additional 100,000 shares of common stock at a public offering price of $10.00 per share pursuant to the partial exercise of the over-allotment option granted to the underwriters resulting in additional gross proceeds to the Company of $1,000,000 and additional net proceeds of $930,000. The total gross proceeds raised from the offering and over-allotment option were $27,000,000, before underwriting discounts, commissions and other offering expenses payable by the Company. The total net proceeds from the offering were approximately $24.3 million. Upon the closing of the IPO, all shares of the Company’s then-outstanding preferred stock automatically converted into an aggregate of 1,696,683 shares of common stock. In addition, upon the closing of the IPO, the Company issued an additional 36,167 shares of common stock to the Series B Preferred Stockholders as a Preferred Stock dividend. This transaction is discussed above under “Preferred Stock Dividend”. At that time, the Company’s obligation to issue, and the Series B Preferred Stockholders’ obligation to purchase Series B-2 Preferred Stock under the Stock Purchase Agreement terminated.
Restricted Stock
A summary of the Company's unvested restricted stock activity as of December 31, 2013 is as follows:
| | | | | | | | |
| | Shares | | | Weighted- Average Fair Value | |
Unvested at December 31, 2012 | | | 2,899 | | | $ | 2.23 | |
Vested | | | (2,899 | ) | | $ | 8.81 | |
Unvested at December 31, 2013 | | | — | | | $ | — | |
As of December 31, 2013, all restricted stock has vested and accordingly all stock-based compensation expense related to vested restricted stock has been recognized.
Common Stock Warrants
There are 20,549 warrants outstanding that are convertible into common stock that have an exercise price of $4.83 per share and expire 10 years from the date of issuance. These warrants were issued to lenders and were originally exercisable into Series A Preferred stock. The warrants converted from preferred stock warrants into warrants to purchase common stock upon the completion of the initial public offering in July 2013. However, since the warrants still have an anti-dilution provision, they remain liabilities and are subject to re-measurement at each balance sheet date. Refer to Footnote 11.
F-21
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
On March 10, 2011, the Company issued warrants to purchase 32,610 shares of common stock to non-employee placement agents in consideration for a private equity placement transaction. The warrants have an exercise price of $0.48 per share and expire 10 years from the issuance date. These warrants do not meet the criteria required to be classified as liability awards and therefore they are treated as equity awards.
In connection with our initial public offering, the Company issued warrants to the underwriters for 125,000 shares of common stock issuable at $12.50 per share upon exercise. The warrants have a ten-year life and expire on July 29, 2023. These warrants do not meet the criteria required to be classified as liability awards and therefore they are treated as equity awards.
The following table summarizes the activity of the Company’s common stock warrants, retro actively adjusted for the 1-for-2.3 reverse stock split.
| | | | |
| | Common Stock Warrants | |
Outstanding, January 1, 2012 | | | 45,550 | |
Granted | | | 7,609 | |
Exercised | | | — | |
Expired | | | — | |
Outstanding, December 31, 2012 | | | 53,159 | |
Granted to underwriters | | | 125,000 | |
Exercised | | | — | |
Expired | | | — | |
Outstanding, December 31, 2013 | | | 178,159 | |
Equity Compensation Plan
2009 Stock Incentive Plan
In 2009, the Company adopted the 2009 Stock Option Plan of Heat Biologics, Inc. (the “2009 Plan”), under which stock options to acquire 500,000 common shares could be granted to key employees, directors, and independent contractors. Under the 2009 Plan, both incentive and non-qualified stock options could be granted under terms and conditions established by the Board of Directors. The exercise price for incentive stock options was the fair market value of the related common stock on the date the stock option was granted. Stock options granted under the 2009 Plan generally have terms of 10 years and have various vesting schedules.
The Company amended the 2009 Stock Option Plan and all related addendum agreements in April 2011. This second amendment increased the number of shares available for issuance from 500,000 to 1,500,000. As of December 31, 2013 and 2012, there were 633,482 and 590,047 and stock options outstanding under the 2009 Plan, respectively.
The following table summarizes the components of the Company’s stock-based compensation included in net loss:
| | | | | | | | |
| | December 31, | |
| | 2013 | | | 2012 | |
| | | | | | |
Employee stock options | | $ | 131,178 | | | $ | 60,956 | |
Non-employee stock options | | | 415,212 | | | | 128,157 | |
Restricted stock awards | | | 25,534 | | | | 28,783 | |
| | $ | 571,924 | | | $ | 217,896 | |
F-22
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Stock Options
The fair value of each stock option is estimated on the date of grant using the Black-Scholes-Merton option pricing model with the following assumptions for stock options granted during the years ended December 31, 2013 and 2012:
| | | | | | | | |
| | December 31, | |
| | 2013 | | | 2012 | |
Dividend yield | | | 0.0% | | | | 0.0% | |
Expected volatility | | | 90-112% | | | | 80-90% | |
Risk-free interest rate | | | 1.39-2.26% | | | | 0.72-0.97% | |
Expected lives (years) | | | 5.75-6.5 | | | | 5-6.25 | |
The risk-free interest rate is based on U.S. Treasury interest rates at the time of the grant whose term is consistent with the expected life of the stock options. The Company used an average historical stock price volatility based on an analysis of reported data for a peer group of comparable companies that have issued stock options with substantially similar terms, as the Company did not have any trading history for its common stock. Expected term represents the period that the Company’s stock option grants are expected to be outstanding. The Company elected to utilize the “simplified” method to value stock option grants. Under this approach, the weighted-average expected life is presumed to be the average of the vesting term and the contractual term of the option.
Expected dividend yield was considered to be 0% in the option pricing formula since the Company had not paid any dividends and had no plans to do so in the future. The forfeiture rate was considered to be none insofar as the historical experience of the Company is very limited. As required by ASC 718, the Company will adjust the estimated forfeiture rate based upon actual experience.
The Company recognized $571,924 and $217,896 in stock-based compensation expense for the years ended December 31, 2013 and 2012, respectively for the Company’s stock option awards.
The following tables summarize the stock option activity for the years ended December 31, 2012 and 2013:
| | | | | | | | |
| | Shares | | | Weighted Average Exercise Price | |
Outstanding, January 1, 2012 | | | 471,905 | | | $ | 0.64 | |
Granted | | | 178,742 | | | $ | 0.76 | |
Exercised | | | (22,827 | ) | | $ | 0.51 | |
Expired/Cancelled | | | (37,773 | ) | | $ | 0.02 | |
Outstanding, December 31, 2012 | | | 590,047 | | | $ | 0.71 | |
Granted | | | 186,736 | | | $ | 10.07 | |
Exercised | | | (80,706 | ) | | $ | 0.67 | |
Expired/Cancelled | | | (62,595 | ) | | $ | 1.96 | |
Outstanding, December 31, 2013 | | | 633,482 | | | $ | 3.36 | |
The weighted average grant-date fair value of stock options granted during the years ended December 31, 2013 and 2012 was $8.51 and $1.31, respectively.
The total fair value of stock options that vested during the year ended December 31, 2013 was approximately $440,820.
F-23
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
The following table summarizes information about stock options outstanding at December 31, 2013:
| | | | | | | | | | | | | | | | | | | | | | | | |
Options Outstanding | | | Options Exercisable | | | Options Vested or Expected to Vest |
Balance as of 12/31/2013 | | | Weighted Average Remaining Contractual Life (Years) | | | Weighted Average Exercise Price | | | Balance as of 12/312013 | | | Weighted Average Remaining Contractual Life (Years) | | | Weighted Average Exercise Price | | | Balance as of 12/31/2013 | | | Weighted Average Remaining Contractual Life (Years) | | | Weighted Average Exercise Price |
633,482 | | | 7.27 | | | $3.36 | | | 426,423 | | | 6.31 | | | $1.23 | | | 426,423 | | | 6.31 | | | $1.23 |
As of December 31, 2013, the unrecognized stock-based compensation expense related to unvested stock options was approximately $1,604,200 that is expected to be recognized over a weighted average period of approximately 14 months.
A summary of the activity of the Company’s unvested stock options is as follows:
| | | | | | | | |
| | Shares | | | Weighted Average Exercise Price | |
Balance, January 1, 2012 | | | 248,439 | | | $ | 0.64 | |
Granted | | | 178,742 | | | $ | 0.76 | |
Vested | | | (257,510 | ) | | $ | 0.51 | |
Forfeited | | | (11,594 | ) | | $ | 0.02 | |
Outstanding, December 31, 2012 | | | 158,077 | | | $ | 0.71 | |
Granted | | | 186,736 | | | $ | 10.07 | |
Vested | | | (91,020) | | | $ | 0.67 | |
Forfeited | | | (46,734 | ) | | $ | 1.96 | |
Outstanding, December 31, 2013 | | | 207,059 | | | $ | 3.36 | |
13.
Income Tax
The components of income tax expense (benefit) attributable to continuing operations are as follows:
| | | | | | | | |
| | Year ended December 31, | |
| | 2013 | | | 2012 | |
| | | | | | |
Current expense: | | | | | | |
Federal | | $ | — | | | $ | — | |
State | | | — | | | | — | |
| | | | | | | | |
Deferred expense (benefit): | | | | | | | | |
Federal | | $ | — | | | $ | — | |
State | | | — | | | | — | |
| | | | | | | | |
Total | | $ | — | | | $ | — | |
F-24
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
The differences between the Company’s consolidated income tax expense attributable to continuing operations and the expense computed at the 34% United States statutory income tax rate were as follows:
| | | | | | | | |
| | Year ended December 31, | |
| | 2013 | | | 2012 | |
| | | | | | |
Federal income tax expense at statutory rate | | $ | (2,247,353 | ) | | $ | (840,190 | ) |
State and local income taxes, net of federal benefit | | | (186,475 | ) | | | (105,170 | ) |
Non-deductible expenses | | | 4,985 | | | | 54,991 | |
Prior-period true-up | | | 162,061 | | | | (152,306 | ) |
Research & development credit | | | (180,687 | ) | | | (57,293 | ) |
Change in tax rate | | | 32,774 | | | | — | |
Increase in valuation allowance | | | 2,414,695 | | | | 1,099,968 | |
| | $ | — | | | $ | — | |
The income tax effects of temporary differences from continuing operations that give rise to significant portions of deferred income tax assets (liabilities) are presented below:
| | | | | | | | |
| | December 31, | |
| | 2013 | | | 2012 | |
| | | | | | |
Deferred tax assets: | | | | | | |
Net operating loss carryforward | | $ | 4,167,785 | | | $ | 2,186,432 | |
Research & development credit | | | 424,739 | | | | 189,350 | |
Other | | | 247,966 | | | | 50,013 | |
Valuation allowance | | | (4,840,490 | ) | | | (2,425,795 | ) |
| | | | | | | | |
Deferred income taxes | | $ | — | | | $ | — | |
During 2013, the Company’s valuation allowance increased by $2,414,695. This increase was primarily due to the generation of additional net operating loss carryforwards and income tax credits.
The Company has approximately $20,999,761 of federal and state operating loss carryforwards which begin to expire in 2023.
In accordance with FASB ASC 740,Accounting for Income Taxes, the Company reflects in the financial statements the benefit of positions taken in a previously filed tax return or expected to be taken in a future tax return only when it is considered ‘more-likely-than-not’ that the position taken will be sustained by a taxing authority. As of December 31, 2013, the Company had no unrecognized income tax benefits and correspondingly there is no impact on the Company’s effective income tax rate associated with these items. The Company’s policy for recording interest and penalties relating to uncertain income tax positions is to record them as a component of income tax expense in the accompanying statements of income. As of December 31, 2013 and 2012, the Company had no such accruals.
The Company files income tax returns in the United States and various state jurisdictions. The Company is subject to examination by taxing authorities for the tax years ended December 31, 2008 through 2012.
14.
Commitments and Contingencies
In November 2011, the Company entered into a thirteen-month lease agreement for office space commencing on January 1, 2012. The monthly base rent is $3,870, which commenced February 1, 2012. On December 19, 2012, we entered into a lease modification agreement that extended the lease term and increased the monthly rent to $4,046. The Company will remain in the office space on a month to month basis until new office space is available.
F-25
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
In connection with the convertible note agreement entered into on October 20, 2011 with a vendor for an amount up to $950,000, the Company is required to use the vendor exclusively for the manufacture and supply of the material for the Company’s Phase III clinical trials and commercialization efforts. This note is no longer outstanding as of December 31, 2013.
15.
Related Party
The Chairman of the Company’s Scientific Advisory Board was paid $0, $18,750 and $140,625 in consulting fees for the years ended December 31, 2013 and 2012 and the period from inception through December 31, 2013, respectively.
A member of the Company’s Scientific Advisory Board was paid $0 for the years ended December 31, 2013 and 2012, respectively. The consulting fees paid since inception was $50,000.
A member of the Company’s management was paid $34,480, $30,910, and $70,910 in consulting fees for the years ended December 31, 2013 and 2012 and the period from inception through December 31, 2013, respectively.
The Company paid three members of the Clinical Advisory Board for consulting during 2013. These members received $45,000, $16,590 and $14,700 for their services during the year ended December 31, 2013. These members were not paid prior to 2013.
The Company compensates its board members. Board members received between $5,000 and $10,870 for services rendered during 2013. Board members were not compensated prior to the Company’s initial public offering in 2013.
The Company had a related party payable balance of $13,000 and $0 as of December 31, 2013 and 2012, respectively.
In June 2012, the Company sold its 92.5% ownership interest in Heat II to a related party in exchange for $9,250 in cash and a receivable of $296,224 to be paid in full in seven years from the date of the purchase. Interest accrues on the receivable at a rate of 6% per annum. At December 31, 2012, the Company also has a related party receivable from this entity for $9,571 related to invoices received by the Company pertaining to expenses of Heat II incurred subsequent to the sale of Heat II.
16.
Net Loss Per Share
Basic net loss per common share is computed by dividing net loss applicable to common stockholders by the weighted-average number of common shares outstanding during the periods. Fully diluted net loss per common share is computed using the weighted average number of common and dilutive common equivalent shares outstanding during the periods. Common equivalent shares consist of stock options that are computed using the treasury stock method.
For the years ended December 31, 2013 and 2012, all of the Company’s common stock options and warrants, preferred stock, and preferred stock warrants are anti-dilutive and therefore have been excluded from the diluted calculation.
The following table reconciles net loss to net loss applicable to common shareholders:
| | | | | | | |
| | For the year ended December 31, | |
| | 2013 | | 2012 | |
Net loss | | $ | (6,609,864 | ) | $ | (2,471,147 | ) |
Net loss: Non-controlling interest | | | (198,516 | ) | | (50,947 | ) |
Beneficial conversion charge | | | (2,300,000 | ) | | — | |
Preferred Stock Dividend | | | (361,668 | ) | | — | |
Net loss applicable to common stockholders | | $ | (9,073,016 | ) | $ | (2,420,200 | ) |
| | | | | | | |
Weighted-average number of common shares used in net loss per share applicable to common stockholders—basic and diluted | | | 3,747,357 | | | 1,831,769 | |
Net loss per share applicable to common stockholders—basic and diluted | | $ | (2.42 | ) | $ | (1.32 | ) |
F-26
HEAT BIOLOGICS, INC.
(A development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
The following potentially dilutive securities were excluded from the calculation of diluted net loss per share due to their anti-dilutive effect):
| | | | | | | | |
| | | For the year ended December 31, | |
| | | 2013 | | 2012 | |
Preferred stock (on an as converted basis) | | | | — | | | 860,017 | |
Preferred stock warrants | | | | — | | | 20,549 | |
Outstanding stock options | | | | 633,482 | | | 590,047 | |
Unvested restricted stock | | | | — | | | 2,899 | |
Common stock warrants | | | | 53,159 | | | 32,610 | |
Reverse Stock Split
In May 2013, the Company's board of directors and stockholders approved a 1-for-2.3 reverse stock split of the Company's common stock. The reverse stock split became effective on May 29, 2013. All share and per share amounts in the financial statements have been retroactively adjusted for all periods presented to give effect to the reverse stock split, including reclassifying an amount equal to the increase in par value to additional paid-in capital.
17.
Subsequent Events
In the first quarter of 2014, the Company entered into a lease agreement to lease a new office facility. The new lease commences on or around May 1, 2014 and includes escalating rent payments and a sixty-month term. Rent expense will be recorded on a straight-line basis over the lease term.
Future minimum lease payments are as follows:
| | | | |
2014 | | $ | 82,353 | |
2015 | | | 183,137 | |
2016 | | | 188,631 | |
2017 | | | 194,290 | |
2018 | | | 200,119 | |
| | | | |
Total | | $ | 848,530 | |
Total rent expense for the years ended December 31, 2013 and 2012 was $48,377 and $43,341, respectively.
In March 2014, the subsidiary, Heat Biologics I, Inc. entered into an additional exclusive license agreement with the University of Miami. No annual payments are required under this license agreement. The Company is obligated to make milestone payments under this license agreement as follows: $50,000 upon completion of a phase I clinical trial, $100,000 upon completion of a phase II trial, $100,000 upon completion of a phase III trial, and $100,000 upon acceptance of a BLA by the FDA or its foreign equivalent.
F-27