Table of Contents
Exhibit 99.1

929 North Front Street
Wilmington, North Carolina 28401
[ ], 2010
Dear PPD Shareholder:
On October 28, 2009, we announced a plan to spin off our compound partnering business (the “Compound Partnering Business”) into a separate publicly traded U.S. company, Furiex Pharmaceuticals, Inc. (“Furiex”). Pharmaceutical Product Development, Inc. (“PPD”) will continue to own its core drug discovery and development services, or contract research organization, business (the “CRO Business”).
We expect to complete this spin-off on [ ], 2010. We will accomplish the spin-off through a pro rata dividend of the common stock of Furiex to PPD’s shareholders. At the time of the spin-off, you will receive one share of Furiex common stock for every shares of PPD common stock that you hold at 5:00 p.m., Eastern Time, on [ ], 2010, the record date for this dividend. However, if you sell your shares of PPD common stock prior to the ex-dividend date you also will be selling your right to receive shares of Furiex common stock. We will not issue any fractional shares of Furiex, so if you otherwise would have been entitled to a fractional share of Furiex in the spin-off, you would receive the net cash value of such fractional share instead. We have applied to have the common stock of Furiex listed on the Nasdaq Global Market under the symbol “FURX”. Shares of PPD will continue to be listed on the Nasdaq Global Select Market under the symbol “PPDI”.
Our Board of Directors has determined that a strategic separation of the Compound Partnering Business from the CRO Business is in the best interests of each of those businesses and our shareholders. We believe that the separation will enhance value for our shareholders by allowing them to realize the full potential of each company independently. We also expect that the separation of the businesses will allow each business to deploy and raise capital in accordance with the unique needs of its business. Specifically, we believe that the separation of the business will increase our ability to attract and retain employees by providing equity compensation tied directly to our company’s performance, and our research and development efforts in particular. In addition, we anticipate that the separation will permit the management of each business to better focus on its distinct type of operation and to more effectively pursue the strategies necessary to improve its business performance. Finally, we believe that the separation of the two businesses will simplify the profile of each company, allowing investors to more easily evaluate each company.
Enclosed please find an Information Statement that describes the spin-off and the business of Furiex, which we are providing to all PPD shareholders in accordance with U.S. securities laws. The Information Statement describes in detail the distribution of Furiex common stock to holders of PPD common stock and contains important business and financial information about Furiex. We encourage you to read this information carefully. Please note that shareholder approval is not required for this spin-off, so we are not asking you for a proxy. You will not need to take any action to receive Furiex shares and you will not be required to pay anything for the Furiex shares or surrender any of your PPD shares.
Table of Contents
If you have any questions regarding the spin-off, please contact our investor relations department by calling (910) 558-7585 or sending a letter to: Investor Relations, Pharmaceutical Product Development, Inc., 929 North Front Street, Wilmington, North Carolina 28401.
| Sincerely, |
| Fred N. Eshelman |
| Executive Chairman |
This Information Statement is first being mailed to shareholders on or about [ ], 2010. This Information Statement is furnished for informational purposes only.
Table of Contents

3900 Paramount Parkway
Suite 150
Morrisville, North Carolina 27560
[ ], 2010
Dear Future Furiex Shareholder:
It is my great pleasure to welcome you as a shareholder of Furiex Pharmeceuticals, Inc. (“Furiex”) and introduce you to our company. We are a drug development collaboration company that will pursue the Compound Partnering Business of our parent company, Pharmaceutical Product Development, Inc. (“PPD”).
We seek to collaborate with pharmaceutical and biotechnology companies to increase the value of their early stage drug candidates by applying our novel approach to drug development that we believe expedites research and development decision-making and can shorten drug development timelines. We share risk with our collaborators by financing the cost of development to the point of mutually agreed upon pre-determined clinical milestones. In exchange, Furiex shares the potential rewards with our collaborators, receiving milestone payments and royalties based on the continued development and commercialization success of the drug candidate. Most of the large pharmaceutical companies with which we collaborate have the option to continue late stage clinical development and commercialization of the drug candidate after it has reached the specified pre-determined milestones. If our collaborator is unable or unwilling to perform late stage development and commercialization, then we have the option to seek out a new development and commercialization partner. Furiex’s team is staffed with the same key PPD team members who demonstrated proven success in the drug development collaboration business while at PPD, as well as highly-qualified new members. Our strategy is to invest in programs that have well defined clinical endpoints, a relatively straightforward path to regulatory approval and a large potential addressable market.
Our current pipeline is comprised of several compounds in various stages of development and commercialization, including:
| • | Rights to royalties and sales-based milestones from the collaboration with ALZA Corporation, a Janssen-Cilag affiliate, on Priligy®, the first approved treatment in the world for premature ejaculation. To date, Priligy has been approved for marketing in Austria, Finland, Germany, Italy, Mexico, New Zealand, Portugal, South Korea, Spain and Sweden. |
| • | Rights to potential future regulatory milestones and, if approved, royalties and sales-based milestones from Takeda Pharmaceutical Company Limited on alogliptin and alogliptin-containing products. Alogliptin is a dipeptidyl peptidase IV, or DPP4, inhibitor for the treatment of type 2 diabetes. In June and September 2009, the U.S. Food and Drug Administration, or FDA, issued complete response letters to the alogliptin monotherapy and the fixed-dosed combination of alogliptin and Actos® New Drug Applications, requesting an additional cardiovascular safety trial with the alogliptin monotherapy to satisfy the FDA’s December 2008 guidance on anti-diabetic therapies. That trial is presently ongoing. |
| • | A fluoroquinolone antibiotic compound we licensed from Janssen Pharmaceutica, N.V., an affiliate of Johnson & Johnson, in November 2009 for the treatment of complicated skin and skin structure infections, such as abscesses that occur deep in the skin layers, and respiratory infections. This antibiotic has a broad spectrum of activity and is able to treat methicillin-resistant staphylococcus aureus, or MRSA, infections. Based on pre-clinical in vitro data, we believe this product may show superior bacteriocidal activity, compared to currently marketed antibiotics, for a number of difficult-to-treat pathogens. We are developing both oral and intravenous, or IV, formulations of this compound to allow doctors and patients to select the appropriate method of delivery under the |
Table of Contents
circumstances. We have initiated work with the goal of beginning a Phase I study with the IV formulation in the first half of 2010. We are planning to initiate Phase II clinical trials using the oral formulation for complicated skin infections and using both formulations for respiratory infections in 2010. |
| • | A compound that is a mu opioid receptor agonist and delta opioid receptor antagonist, which we call mu delta, we licensed from Janssen Pharmaceutica, N.V. in November 2009 for the treatment of diarrhea-predominant irritable bowel syndrome. We have begun work with the goal of initiating Phase II clinical trials during the second quarter of 2010. |
| • | A novel statin compound we refer to as PPD 10558 licensed from Ranbaxy Laboratories, Ltd. for the treatment of dyslipidemia, which is an excessive level of blood lipids such as cholesterol. Preclinical data suggests this statin will avoid some of the adverse interactions seen when currently marketed statins are taken with some other drugs by patients with high blood lipid levels. In addition, one of the most common side effects of statin usage is a condition of pain and/or muscle weakness, known as statin-associated myopathy, which is reported to occur in up to 10% of statin-treated patients. Preclinical and Phase I human studies suggest that PPD 10558 has similar cholesterol-lowering properties as a leading marketed statin, and also has pharmacologic properties which suggest that it may have a lower risk of myopathy than currently marketed statins. PPD 10558 may therefore be a useful treatment option for statin-intolerant patients. PPD completed a high-dose comparator study in healthy volunteers that indicated the drug was well-tolerated and suggested it compares favorably to currently marketed statins with respect to lowering lipid levels. |
| • | A variety of therapeutic candidates developed by Eli Lilly and Company that we obtained through our acquisition of Magen BioSciences, Inc. in April 2009, including vitamin D receptor modulators. We have tested two of these compounds for various safety and efficacy parameters for topical treatment of dermatology indications in the anti-proliferative and anti-inflammatory areas. We have elected, however, not to pursue the development of these compounds. We also are currently investigating other compounds under material transfer agreements to identify potential drug development candidates for dermatological indications, and are considering other strategic alternatives for our dermatology business. |
As you know, the Board of Directors of PPD has approved a plan to spin off Furiex into a separate publicly traded company. We expect to complete the spin-off on [ ], 2010. We have applied to have our common stock listed on the Nasdaq Global Market under the symbol “FURX”.
With our existing product rights, promising clinical pipeline, experienced management team and strong balance sheet, we believe that we will begin our future as an independent public company from a position of considerable strength. This spin-off should enable us to operate our business with even greater focus and agility. As a Furiex shareholder, you can share in our progress as we strive to continue strengthening and growing our business.
On behalf of the Furiex team, I invite you to learn more about Furiex and our opportunity as a soon-to-be independent publicly traded company by reading the attached Information Statement.
| Sincerely, |
June S. Almenoff, President and |
Chief Medical Officer |
Table of Contents
Information contained herein is subject to completion or amendment. A Registration Statement on Form 10 relating to these securities has been filed with the Securities and Exchange Commission.
Preliminary and Subject to Completion, dated April 29, 2010
Information Statement
Furiex Pharmaceuticals, Inc
(par value $0.001 per share)
We are furnishing this Information Statement to the shareholders of Pharmaceutical Product Development, Inc., or PPD, in connection with PPD’s distribution to holders of its common stock of all outstanding shares of common stock of Furiex Pharmaceuticals, Inc., or Furiex. At this time, Furiex is a wholly owned subsidiary of PPD. After the spin-off is completed, Furiex will be a separate publicly traded company and will own and operate the compound partnering business (the “Compound Partnering Business”) currently owned and operated by PPD. PPD will continue to own its drug discovery and development services, or contract research organization, business (the “CRO Business”).
If you are a holder of record of PPD common stock at 5:00 p.m., Eastern Time, on [ ], 2010, which will be the record date for the distribution, you will be entitled to receive one share of our common stock for every shares of PPD common stock that you hold on the record date. However, if you sell your shares of PPD common stock prior to the ex-dividend date, you also will be selling your right to receive shares of Furiex common stock. Our common stock will be issued in book-entry form only, which means no physical stock certificates will be issued. No fractional shares of Furiex common stock will be issued. If you otherwise would have been entitled to a fractional share of Furiex common stock in the distribution, you will receive the net cash value of such fractional share instead. Immediately after the distribution is completed on the distribution date, we will be an independent publicly traded company. We expect the distribution to occur on [ ], 2010.
No shareholder vote is required for the spin-off to occur. We are not asking you for a proxy, and you are requested not to send us a proxy. No action is necessary for you to receive the shares of our common stock to which you are entitled in the spin-off. This means that you donot need to:
| • | pay any consideration to Furiex or to PPD; or |
| • | surrender or exchange any shares of PPD common stock to receive your shares of our common stock. |
Currently, there is no public trading market for the common stock of Furiex, although we expect that a “when-issued” trading market will develop on or shortly after the record date for the distribution. We have applied to have our common stock traded on the Nasdaq Global Market under the symbol “FURX”.
As you review this Information Statement, you should carefully consider the matters described in “Risk Factors” beginning on page 11.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this Information Statement is truthful or complete. Any representation to the contrary is a criminal offense.
This Information Statement does not constitute an offer to sell or the solicitation of an offer to buy any securities.
If you have inquiries related to the distribution, you should contact PPD’s transfer agent, American Stock Transfer & Trust Company, at 59 Maiden Lane, Plaza Level, New York, New York 10038 or (800) 937-5449.
Table of Contents
| ii | ||
| 1 | ||
| 11 | ||
| 29 | ||
| 30 | ||
| 38 | ||
| 38 | ||
| 39 | ||
Management’s Discussion and Analysis of Financial Condition and Results of Operations | 55 | |
| 68 | ||
| 71 | ||
| 75 | ||
| 81 | ||
Security Ownership of Certain Beneficial Owners and Management | 90 | |
| 91 | ||
| 91 | ||
| 94 | ||
| 94 | ||
| 95 | ||
| 95 | ||
| 96 |
i
Table of Contents
Furiex is furnishing this Information Statement to you solely to provide you with information regarding both the spin-off and our company. It is not, and should not be construed as, an inducement or encouragement to buy or sell any securities of Furiex or PPD.
You should rely only on the information contained in this Information Statement. We have not authorized any other person to provide you with information different from that contained in this Information Statement. The information contained in this Information Statement is believed by us to be accurate as of its date. Therefore, you should assume that the information contained in this Information Statement is accurate only as of the date on the front cover of this Information Statement or other dates stated in this Information Statement, regardless of the time of delivery of this Information Statement. Our business, financial condition, results of operations and prospects may have changed since that date, and neither we nor PPD will update the information except in the normal course of our respective public disclosure obligations and practices or as specifically indicated in this Information Statement.
We own or have rights to trademarks, trade names, copyrights and other intellectual property used in our business. All other company names, tradenames and trademarks included in this Information Statement are trademarks, registered trademarks or trade names of their respective owners.
As used in this Information Statement, the terms “we,” “us,” “our,” and the “Company” mean Furiex and its subsidiaries (unless the context indicates a different meaning).
We describe in this Information Statement the Compound Partnering Business transferred to us by PPD in connection with the spin-off as though the Compound Partnering Business were our business for all historical periods described. However, Furiex is a newly-formed entity. Some of the actions necessary to transfer assets and liabilities of PPD to us have not occurred but will occur before the effectiveness of the spin-off. References in this Information Statement to the historical assets, liabilities, products, business or activities of our business are intended to refer to the historical assets, liabilities, products, business or activities of the Compound Partnering Business as those were conducted as part of PPD prior to the transfer and assumption by Furiex and the spin-off.
ii
Table of Contents
The following is a summary of some of the information contained in this Information Statement. We urge you to read this entire document carefully, including the risk factors, our historical combined financial statements and the notes to those financial statements, and our unaudited pro forma condensed combined financial statements.
Our Company
Furiex is a drug development collaboration company that will pursue the Compound Partnering Business of our parent company, PPD. We seek to collaborate with pharmaceutical or biotechnology companies to increase the value of their early stage drug candidates by applying our novel approach to drug development that we believe expedites research and development decision-making and can shorten drug development timelines.
In order to obtain regulatory approval from the FDA to market a drug, certain data about the safety and efficacy of the drug is required by the FDA. To obtain such data, drug developers frequently choose to run studies sequentially. For example, they may run one study, wait to see the results, and then they run the next study. Developers prefer this approach primarily to limit upfront expenditures since the success of any given study is not known and the decision may be made not to move forward due to negative data. This sequential approach slows down the development process. In addition, development is slowed due to time taken at each decision point at the end of each study.
Furiex approaches drug development by minimizing the time it takes to bring products to the market. Furiex’s novel approach manages drug development with parallel processing and efficient decision making. We use our drug development experience to predict possible outcomes of a study and take risks based on those predictions. By assuming in advance success at each critical decision point, as opposed to waiting for results to demonstrate success in order to make a decision, time is reduced. In addition, we seek to mitigate risks by contingency planning for potential problems. As a result, we can accelerate the development process by bridging steps across the developmental program as well as between studies, as was evidenced with alogliptin where it took just five years from the start of our collaboration to the filing of the NDA. Additionally, we focus our efforts on the essential studies necessary for regulatory approval and we do not include extraneous studies. This helps to shorten developmental timelines while still achieving success.
Our approach to drug development poses risks to us. If we miscalculate the assumed safety and efficacy of any drug as well as any assumed decision point in its development, the time to develop could be significantly extended, which would add to the expense of development. In addition, if our assumptions or calculations about any drug are incorrect or we encounter unforeseen difficulties with any drug, we might have invested significant amounts without any return on that investment. Further, the value of the drug candidate might be impaired, and possibly significantly so, which could result in material losses to us if the selected indication or the drug itself is abandoned. For example, we recorded an impairment charge of $10.4 million related to one of our dermatology compounds, MAG-131, due to efficacy data which led us to determine that the asset was impaired.
We share risk with our collaborators by financing the cost of development to the point of mutually agreed upon pre-determined clinical milestones. In exchange, Furiex shares the potential rewards with our collaborators, receiving milestone payments and royalties based on the continued development and commercialization success of the drug candidate. Most of the large pharmaceutical companies with which we collaborate have the option to continue late stage clinical development and commercialization of the drug candidate after it has reached the specified pre-determined milestones. If our collaborator is unable or unwilling to perform late stage development and commercialization, then we have the option to seek out a new development and commercialization partner. Furiex’s team is staffed with the same key PPD team members who demonstrated proven success in the drug
1
Table of Contents
development collaboration business while at PPD, as well as highly-qualified new members. Our strategy is to invest in programs that have well defined clinical endpoints, a relatively straightforward path to regulatory approval and a large potential addressable market.
Our current pipeline is comprised of several compounds in various stages of development and commercialization, including:
| • | Rights to royalties and sales-based milestones from the collaboration with ALZA Corporation, a Janssen-Cilag affiliate, on Priligy®, the first approved treatment in the world for premature ejaculation. To date, Priligy has been approved for marketing in Austria, Finland, Germany, Italy, Mexico, New Zealand, Portugal, South Korea, Spain and Sweden. |
| • | Rights to potential future regulatory milestones and, if approved, royalties and sales-based milestones from Takeda Pharmaceutical Company Limited on alogliptin and alogliptin-containing products. Alogliptin is a dipeptidyl peptidase IV, or DPP4, inhibitor for the treatment of type 2 diabetes. In June and September 2009, the U.S. Food and Drug Administration, or FDA, issued complete response letters to the alogliptin monotherapy New Drug Application, or NDA, and the fixed-dosed combination of alogliptin and Actos® NDA, requesting an additional cardiovascular safety trial with the alogliptin monotherapy to satisfy the FDA’s December 2008 guidance on anti-diabetic therapies. That trial is presently ongoing. |
| • | A fluoroquinolone antibiotic compound we licensed from Janssen Pharmaceutica, N.V., an affiliate of Johnson & Johnson, in November 2009 for the treatment of complicated skin and skin structure infections, such as abscesses that occur deep in the skin layers, and respiratory infections. This antibiotic has a broad spectrum of activity and is able to treat methicillin-resistant staphylococcus aureus, or MRSA, infections. Based on pre-clinical in vitro data, we believe this product may show superior bacteriocidal activity, compared to currently marketed antibiotics, for a number of difficult-to-treat pathogens. We are developing both oral and intravenous, or IV, formulations of this compound to allow doctors and patients to select the appropriate method of delivery under the circumstances. We have initiated work with the goal of beginning a Phase I study with the IV formulation in the first half of 2010. We are planning to initiate Phase II clinical trials using the oral formulation for complicated skin infections and using both formulations for respiratory infections in 2010. |
| • | A compound that is a mu opioid receptor agonist and delta opioid receptor antagonist, which we call mu delta, we licensed from Janssen Pharmaceutica, N.V. in November 2009 for the treatment of diarrhea-predominant irritable bowel syndrome. We have begun work with the goal of initiating Phase II clinical trials during the second quarter of 2010. |
| • | A novel statin compound we refer to as PPD 10558 licensed from Ranbaxy Laboratories, Ltd. for the treatment of dyslipidemia, which is an excessive level of blood lipids such as cholesterol. Preclinical data suggests this statin will avoid some of the adverse interactions seen when currently marketed statins are taken with some other drugs by patients with high blood lipid levels. In addition, one of the most common side effects of statin usage is a condition of pain and/or muscle weakness, known as statin-associated myopathy, which is reported to occur in up to 10% of statin-treated patients. Preclinical and Phase I human studies suggest that PPD 10558 has similar cholesterol-lowering properties as a leading marketed statin, and also has pharmacologic properties which suggest that it may have a lower risk of myopathy than currently marketed statins. PPD 10558 may therefore be a useful treatment option for statin-intolerant patients. PPD completed a high-dose comparator study in healthy volunteers that indicated the drug was well-tolerated and suggested it compares favorably to currently marketed statins with respect to lowering lipid levels. |
2
Table of Contents
| • | A variety of therapeutic candidates developed by Eli Lilly and Company that we obtained through our acquisition of Magen BioSciences, Inc. in April 2009, including vitamin D receptor modulators. We have tested two of these compounds for various safety and efficacy parameters for topical treatment of dermatology indications in the anti-proliferative and anti-inflammatory areas. We have elected, however, not to pursue the development of these compounds. We also are currently investigating other compounds under material transfer agreements to identify potential drug development candidates for dermatological indications, and are considering other strategic alternatives for our dermatology business. |
At the closing of the spin-off, PPD will capitalize our company with cash and cash equivalents amounting to $100.0 million, and we will assume current accounts payable and other accrued expenses payable to third parties and incurred in the ordinary course of our Compound Partnering Business, which as of December 31, 2009, totaled approximately $4.6 million. We expect that this cash contribution will fund Furiex’s operations and working capital requirements for at least 12 months after the closing of the spin-off, based on current operating plans. In addition to this cash contribution, we expect to receive future payments from our existing collaborations that will provide additional support for our operations and working capital requirements. We might, however, run into unexpected delays or other problems with our drug candidates, which could increase our development costs and deplete our cash resources more rapidly than we anticipate. Further, we might not receive anticipated payments from our collaborators because of failure to meet milestones or other reasons, which would reduce our income. If we are unable to raise additional capital, whether we face any of these development issues or not, we might have to curtail or cease development of one or more of our drug candidates.
We were incorporated in Delaware on October 21, 2009 as PPD Therapeutics, Inc. and changed our name to Furiex Pharmaceuticals, Inc. on February 22, 2010. Our principal executive office is located at 3900 Paramount Parkway, Suite 150, Morrisville, North Carolina 27560 and our telephone number at that location is (919) 380-2000. Our internet address is www.furiex.com. Our website and the information contained on that site, or connected to that site, are not incorporated into this Information Statement or our registration statement on Form 10.
Reasons for the Spin-Off
On October 28, 2009, PPD announced a plan to spin off its Compound Partnering Business into a separate publicly traded company. PPD and we believe that the separation of the Compound Partnering Business from the CRO Business will enhance value for holders of PPD and Furiex stock by creating significant opportunities and benefits, including:
| • | Market Recognition: allowing investors to recognize and realize the full potential value of each company independently; |
| • | Employee Incentives: increasing our ability to attract and retain employees by providing equity compensation tied directly to our company’s performance, and our research and development efforts in particular; |
| • | Improved Capital Flexibility: allowing each company to deploy capital and access additional financing, if appropriate, in accordance with its unique needs and business model; and |
| • | Business Focus: allowing each company to focus its efforts on and allocate its resources towards its own business opportunities and challenges. |
As a newly independent company, however, we might not realize any of these benefits for any number of reasons, including lack of market acceptance of the value of our business model or drug pipeline, an inability to attract and retain qualified employees, and the difficulty in managing our business independently, which we have never done.
3
Table of Contents
Summary of the Spin-Off
The following is a brief summary of the terms of the spin-off. Please see “The Spin-Off” beginning on page 30 for a more detailed description of the matters described below.
Distributing company | Pharmaceutical Product Development, Inc. |
Distributed company | Furiex Pharmaceuticals, Inc. |
Distribution ratio | Each holder of PPD common stock will receive one share of our common stock for every shares of PPD common stock held on the record date. |
Securities to be distributed | Approximately million shares of our common stock. The shares of our common stock to be distributed will constitute all of the outstanding shares of our common stock immediately after the spin-off. |
Fractional shares | We will not distribute any fractional shares of our common stock. Instead, the distribution agent will aggregate fractional shares into whole shares, sell the whole shares in the open market at prevailing market prices and distribute the aggregate net cash proceeds of the sales pro rata to each holder who otherwise would have been entitled to receive a fractional share in the distribution. Recipients of cash in lieu of fractional shares will not be entitled to any interest on the amounts of payment made in lieu of fractional shares. The receipt of cash in lieu of fractional shares generally will be taxable to the recipient shareholders as described in “The Spin-Off—Material U.S. Federal Income Tax Consequences of the Distribution” beginning on page 32. |
Distribution agent, transfer agent and registrar for Furiex shares | American Stock Transfer & Trust Company, LLC 59 Maiden Lane, Plaza Level New York, New York 10038 (800) 937-5449 |
Record Date | 5:00 p.m. Eastern Time on [ ], 2010 |
Distribution Date | [ ], 2010 |
Stock exchange listing | Currently there is no public market for our common stock. We have applied to have our common stock listed on the Nasdaq Global Market under the symbol “FURX”. |
U.S. federal income tax consequences | PPD has requested a private letter ruling from the Internal Revenue Service and is expecting an independent tax opinion confirming that the distribution will qualify as a tax-free reorganization for U.S. federal income tax purposes under Sections 368(a)(1)(D) and 355 of the Internal Revenue Code of 1986, as amended, or the Code. Assuming that the distribution is tax-free, for U.S. federal income tax purposes, no taxable gain or loss will be recognized by a shareholder that is subject to U.S. federal income tax, and no amount will be included in the taxable income of a shareholder that is subject to U.S. federal income tax, upon the receipt of our common stock pursuant to |
4
Table of Contents
the distribution. A shareholder that is subject to U.S. federal income tax generally will recognize taxable gain or loss with respect to any cash received in lieu of a fractional share. See “Risk Factors—Risks Relating to the Spin-off—If the distribution or internal transactions undertaken in anticipation of the separation are determined to be taxable for U.S. federal income tax purposes, we, our shareholders that are subject to U.S. federal income tax and PPD could incur significant U.S. federal income tax liabilities” on page 14 and “The Spin-Off—Material U.S. Federal Income Tax Consequences of the Distribution” beginning on page 32. |
Purposes of the Distribution | PPD’s Board and management have determined that the spin-off will enhance long-term shareholder value by providing the benefits set forth below under the caption “The Spin-Off—Reasons for the Spin-Off” beginning on page 30. |
Conditions to the distribution | The distribution of our common stock is subject to the satisfaction of the following conditions, among other conditions described in this information statement: |
| • | the Securities and Exchange Commission, or SEC, shall have declared effective our registration statement on Form 10, of which this Information Statement is a part, under the Securities Exchange Act of 1934, as amended, or Exchange Act, and no stop order relating to the registration statement is in effect; |
| • | all permits, registrations and consents required under the securities or blue sky laws of states or other political subdivisions of the United States or of other foreign jurisdictions in connection with the distribution shall have been received; |
| • | the listing of our common stock on the Nasdaq Global Market shall have been approved, subject to official notice of issuance; |
| • | the receipt of a favorable private letter ruling from the Internal Revenue Service and an independent tax opinion to the effect that the pro rata dividend distribution of our shares to PPD shareholders will be treated as a tax-free distribution to PPD and its shareholders; |
| • | all material government approvals and other consents necessary to consummate the distribution shall have been received; and |
| • | no order, injunction or decree issued by any court of competent jurisdiction or other legal restraint or prohibition preventing consummation of the distribution or any of the transactions related thereto, including the transfers of the assets and liabilities contemplated by the Separation and Distribution Agreement, shall be in effect. |
The fulfillment of these conditions does not create any obligation on PPD to effect the distribution, and the PPD Board has reserved the
5
Table of Contents
right, in its sole discretion, to amend, modify or abandon the distribution and related transactions at any time prior to the distribution date. PPD has the right not to complete the distribution if, at any time, the PPD Board determines, in its sole discretion, that the distribution is not in the best interests of PPD or its shareholders or that market conditions are such that it is not advisable to separate the Compound Partnering Business from PPD. |
Risks relating to ownership of our common stock and the distribution | Our business is subject to both general and specific risks and uncertainties relating to our business, our relationship with PPD and our being a separate, publicly traded company. Our business is also subject to risks relating to the separation. You should read carefully “Risk Factors” beginning on page 11. |
Agreements with PPD | Before and as a condition to the distribution, we will enter into a Separation and Distribution Agreement, a Master Development Services Agreement, a Transition Services Agreement and other agreements with PPD to effect the separation and distribution and provide a framework for our relationship with PPD after the separation. These agreements will govern the relationships among us and PPD after the completion of the separation plan and provide for the allocation among us and PPD of PPD’s assets, liabilities and obligations (including employee benefits and tax-related assets and liabilities) attributable to periods prior to our separation from PPD. For a discussion of these arrangements, see “Related Person Transactions” beginning on page 88. |
Questions and Answers about the Spin-off
How will the spin-off work? | The spin-off will be accomplished through a series of transactions by which PPD will contribute to us its Compound Partnering Business, which we refer to as the contribution, and PPD will distribute to its shareholders all of the outstanding shares of our common stock on a pro rata basis, which we refer to as the distribution. When we refer to the occurrence of the spin-off, we are referring to the date the spin-off is finalized and our stock is distributed to you. For additional information on the transactions in the spin-off, see “The Spin-Off—Manner of Effecting the Spin-Off” beginning on page 30. |
What will our relationship with PPD be after the spin-off? | PPD and Furiex each will be independent, publicly traded companies. However, we will enter into agreements with PPD or one of its wholly owned subsidiaries that will ease the transition of PPD and Furiex into two independent, publicly traded companies following the spin-off. These agreements will also allocate responsibility for obligations arising before and after the spin-off, including, among others, obligations relating to taxes. We will also enter into an agreement appointing PPD as the preferred provider of CRO services to us. For additional information on our relationship with PPD after the spin-off, see “Our Relationship with PPD after the Spin-Off” beginning on page 66. |
6
Table of Contents
When will the spin-off be completed? | PPD expects to complete the spin-off by distributing shares of our common stock on [ ], 2010, to holders of record of PPD common stock on the record date. As discussed under “The Spin-Off—Trading of PPD Common Stock After the Record Date and Prior to the Ex-Dividend Date,” if you sell your shares of PPD common stock in the “regular way” market after the record date and prior to the ex-dividend date, you also will be selling your right to receive shares of our common stock in connection with the spin-off. For additional information on the spin-off, see “The Spin-Off—Results of the Spin-Off” beginning on page 31. |
What do I have to do to participate in the distribution? | Nothing. You are not required to take any action to receive shares of our common stock in the spin-off. No vote of PPD shareholders is required or will be taken to authorize the spin-off. If you own shares of PPD common stock as of the close of business on the record date and do not sell those shares in the “regular way” market prior to the ex-dividend date, a book-entry account statement reflecting your ownership of shares of our common stock will be mailed to you, or your brokerage account will be credited for the shares, on or about [ ], 2010. Do not mail in PPD common stock certificates in connection with the spin-off. |
How many shares of your common stock will I receive? | PPD will distribute one share of our common stock for every shares of PPD common stock you own of record as of the close of business on the record date and do not sell in the “regular way” market prior to the ex-dividend date. Cash will be distributed in lieu of fractional shares, as described below. Based on approximately million shares of PPD common stock that we expect to be outstanding on the record date, PPD will distribute a total of approximately million shares of our common stock. The number of shares of our common stock that PPD will distribute to its shareholders will be reduced to the extent that cash payments are to be made in lieu of the issuance of fractional shares of our common stock and to the extent that shares of our common stock are held back and sold on the market to satisfy backup withholding taxes and non-U.S. holder dividend withholding taxes and brokerage and other costs, and will be increased to the extent, if any, that PPD options are exercised prior to the record date. For additional information on the distribution, see “The Spin-Off—Results of the Spin-Off” beginning on page 31. |
How will PPD distribute fractional shares of Furiex common stock? | PPD will not distribute any fractional shares of our common stock to its shareholders. Instead, the distribution agent will aggregate fractional shares into whole shares, sell the whole shares in the open market at prevailing market prices and distribute the aggregate net cash proceeds of the sales pro rata to each holder who otherwise would have been entitled to receive a fractional share in the distribution. Recipients of cash in lieu of fractional shares will not be entitled to any interest on the amounts of payment made in lieu of fractional shares. The receipt of cash in lieu of fractional shares |
7
Table of Contents
generally will be taxable to the recipient shareholders as described in “The Spin-Off—Material U.S. Federal Income Tax Consequences of the Distribution” beginning on page 32. |
Can PPD decide to cancel the distribution of Furiex common stock even if all the conditions have been met? | Yes. PPD has the right to terminate the distribution, and the spin-off, even if all of the conditions set forth in the Separation and Distribution Agreement are satisfied, if at any time the Board of PPD determines that the distribution is not in the best interest of PPD and its shareholders or that market conditions are such that it is not advisable to separate the Compound Partnering Business from PPD. |
Will I receive physical certificates representing shares of Furiex common stock following the separation? | No. We will not issue physical stock certificates in the distribution, even if requested. Instead, PPD, with the assistance of American Stock Transfer & Trust Company, the distribution agent, will electronically issue shares of our common stock to you or to your bank or brokerage firm on your behalf by way of direct registration in book-entry form. The book-entry system allows registered shareholders to hold their shares without physical stock certificates. A benefit of issuing stock electronically in book-entry form is that there will be none of the physical handling and safekeeping responsibilities that are inherent in owning physical stock certificates. For additional information, see “The Spin-Off—Manner of Effecting the Spin-Off” beginning on page 30. |
Do you intend to pay dividends on your common stock? | We currently do not intend to pay dividends on our common stock. The declaration and amount of dividends will be determined by our Board and will depend on our financial condition, earnings, capital requirements, legal requirements, regulatory constraints, contractual restrictions, and any other factors that our Board believes are relevant. See “Dividend Policy” on page 38 for additional information on our dividend policy following the spin-off. |
What if I want to sell my PPD common stock or Furiex common stock? | You should consult your financial advisors, such as your stockbroker, bank or tax advisor. Neither PPD nor Furiex makes any recommendation as to the purchase, retention or sale of shares of PPD common stock or the Furiex common stock to be distributed. |
If you decide to sell any shares before the ex-dividend date, you should make sure your stockbroker, bank or other nominee understands whether you want to sell your PPD common stock or the Furiex common stock you will receive in the distribution or both.
Where will I be able to trade shares of Furiex common stock? | There is no current trading market for our common stock. We plan to have our common stock authorized for listing on the Nasdaq Global Market under the symbol “FURX”. We expect that a limited market, commonly known as a “when-issued” trading market, for our common stock will begin on or shortly after [ ], 2010. The term “when-issued” means that shares can be traded prior to the time shares are actually available or issued. We expect that on the distribution date or the first trading day after the distribution date, “when-issued” trading in our common stock will end and “regular |
8
Table of Contents
way” trading will begin. “Regular way” trading refers to trading after a security has been issued and typically involves a transaction that settles on the third full business day following the date of a trade. Shares of our common stock generally will be freely tradable following the spin-off. For additional information regarding the trading of our common stock, see “The Spin-Off—Market for Our Common Stock; Trading of Our Common Stock in Connection with the Spin-Off” beginning on page 35. |
Will the number of PPD shares I own change as a result of the spin-off? | No. The number of shares of PPD common stock you own will not change as a result of the spin-off. |
What will happen to the listing of PPD common stock? | Nothing. It is expected that after the distribution of Furiex common stock, PPD common stock will continue to be traded on the Nasdaq Global Select Market under the symbol “PPDI”. |
Are there any risks to owning Furiex common stock? | Yes. Our business is subject to both general and specific risks relating to our operations, anticipated net losses, and our operating as a stand-alone company. Our business is also subject to risks relating to the separation. These and other risks are described in “Risk Factors” beginning on page 11. We encourage you to read that section carefully. |
Who do I contact for information regarding Furiex and the spin-off? | Before the spin-off, you should direct inquiries relating to the spin-off to: |
Investor Relations
Pharmaceutical Product Development, Inc.
929 North Front Street
Wilmington, North Carolina 28401
(910) 558-7585
After the spin-off, you should direct inquiries relating to our common stock to:
Investor Relations
Furiex Pharmaceuticals, Inc.
3900 Paramount Parkway, Suite 150
Morrisville, North Carolina 27560
(919) 380-2000
After the spin-off, the transfer agent and registrar for our common stock will be:
American Stock Transfer & Trust Company, LLC
59 Maiden Lane, Plaza Level
New York, New York 10038
(800) 937-5449
9
Table of Contents
Summary Historical Combined Financial Information
The tables below set forth selected historical financial data of the combined financial statements of Furiex. This information has been prepared from Furiex’s audited combined financial statements as of December 31, 2009 and 2008 and for each of the three years in the period ended December 31, 2009. Financial information as of December 31, 2007, 2006 and 2005 and for each of the two years in the period ended December 31, 2006 has been prepared from unaudited combined financial statements not included herein. During these periods, Furiex was an integrated business of PPD. The historical financial information is not likely to be indicative of Furiex’s future performance or its future financial position or results of operations, and it does not provide or reflect data as if Furiex had actually operated as a separate, stand-alone entity during the periods covered. Per share data has not been presented as no common shares were outstanding during the periods presented and such information would not be meaningful.
The selected historical financial data should be read in conjunction with the combined financial statements and related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations”, included elsewhere in this Information Statement.
Combined Statements of Operations Data (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2005 | 2006 | 2007 | 2008 | 2009 (1) | ||||||||||||||||
Net revenue | $ | 25,371 | $ | 15,857 | $ | 560 | $ | 18,419 | $ | 6,312 | ||||||||||
Operating expenses | 28,029 | 3,604 | 23,316 | 11,645 | 38,131 | |||||||||||||||
Income (loss) from operations(2) | (2,658 | ) | 12,253 | (22,756 | ) | 6,774 | (31,819 | ) | ||||||||||||
Other income (expense), net | (2 | ) | (329 | ) | 19 | 14 | (2 | ) | ||||||||||||
Income (loss) from continuing operations | (2,660 | ) | 11,924 | (22,737 | ) | 6,788 | (31,821 | ) | ||||||||||||
Discontinued operations(3) | 2,164 | (4,066 | ) | (185 | ) | (976 | ) | 22,890 | ||||||||||||
Net income (loss) | $ | (496 | ) | $ | 7,858 | $ | (22,922 | ) | $ | 5,812 | $ | (8,931 | ) | |||||||
Combined Balance Sheet Data (in thousands):
| As of December 31, | ||||||||||
| 2005 | 2006 | 2007 | 2008 | 2009 | ||||||
Total assets | 69,166 | 63,581 | 63,265 | 61,138 | 55,877 | |||||
PPD net investment(4) | 62,314 | 58,895 | 56,870 | 55,524 | 49,270 | |||||
| (1) | We acquired Magen BioSciences, Inc. in 2009. Results of operations for the acquisition are included in our combined results of operations as of and since the effective date of the acquisition. For further details, see Note 2 in the notes to the combined financial statements. |
| (2) | Impairments of intangible assets are included in income (loss) from operations. For 2009, the impairment of intangible asset was related to in-process research and development for the MAG-131 compound obtained through the acquisition of Magen Biosciences. For 2008, the impairment of intangible asset related to the remaining unamortized value of our royalty interest in SinuNase and other Accentia antifungal products. For further details, see Note 5 in the notes to the combined financial statements. |
| (3) | In 2009, we completed dispositions of Piedmont Research Center, LLC and PPD Biomarker Discovery Sciences, LLC. Results of operations for these dispositions are included in discontinued operations, net of provision for income tax of $0. For further details, see Note 2 in the notes to the to the combined financial statements. |
| (4) | The financial statements of the company represent a combination of various components of PPD comprising the Discovery Sciences segment. Because a direct ownership relationship did not exist among all the components comprising the company, PPD’s net investment in the company is shown in lieu of shareholder’s equity in the combined financial statements. The net investment account represents the cumulative investments in, distributions from and earnings (loss) of the company. |
10
Table of Contents
This Information Statement includes forward-looking statements. All statements other than statements of historical facts are forward-looking statements, including any projections of milestones, royalties or other financial items, any statements of the plans and objectives of management for future operations, any statements concerning proposed new products or licensing or collaborative arrangements, any statements regarding future economic conditions or performance, and any statement of assumptions underlying any of the foregoing. In some cases, forward-looking statements can be identified by the use of terminology such as “believes”, “might”, “will”, “expects”, “plans”, “anticipates”, “estimates”, “potential” or “continue”, or the negative thereof or other comparable terminology. Although we believe that the expectations reflected in the forward-looking statements contained in this Information Statement are reasonable, there can be no assurance that such expectations or any of the forward-looking statements will prove to be correct, and actual results could differ materially from those projected or assumed in the forward-looking statements. Our future financial condition and results of operations, as well as any forward-looking statements, are subject to inherent risks and uncertainties, including the risk factors set forth below, and for the reasons described elsewhere in this Information Statement, any of which could significantly adversely impact our business and cause a partial or complete loss of your Furiex stock’s value. All forward-looking statements and reasons why results might differ included in this Information Statement are made as of the date hereof, and we assume no obligation to update these forward-looking statements or reasons why actual results might differ.
Risks Relating to the Spin-Off
We might not realize the potential benefits from the spin-off; PPD shareholders might not realize the potential benefits of the spin-off.
We expect the spin-off to provide us with market recognition of our potential as an independent company and the attendant benefits of being able to incentivize our employees with compensation tied directly to our performance, deploy capital and access financing to meet our specific needs and focus our efforts on our own business opportunities and challenges. However, we might not realize the potential benefits that we expect from our spin-off from PPD. Further, PPD shareholders might not realize the intended benefits of the spin-off. We have described those anticipated benefits in greater detail elsewhere in this Information Statement. See “The Spin-Off—General”. There is a risk, however, that by separating from PPD we might be more susceptible to market fluctuations and other adverse events than we would have been were we still a part of PPD. In addition, we will incur significant costs, which might exceed our estimates, and we will incur some negative effects from our separation from PPD, including the loss of cash flow and revenue derived from PPD’s CRO Business.
Our historical and pro forma financial information is not necessarily indicative of our future financial position, future results of operations or future cash flows and does not reflect what our financial position, results of operations or cash flows would have been as a stand-alone company during the periods presented.
Our historical financial information included in this Information Statement does not necessarily reflect what our financial position, results of operations or cash flows would have been as a stand-alone company during the periods presented. In addition, it is not necessarily indicative of our future financial position, future results of operations or future cash flows. This is primarily a result of the following factors:
| • | Prior to our separation, our business was operated by PPD as part of its broader corporate organization and we did not operate as a stand-alone company; |
| • | Most general administrative functions were performed by PPD for the combined entity, so although our historical combined financial statements reflect allocations of costs for services shared with PPD, these allocations may differ from the costs we will incur for these services as an independent company; |
11
Table of Contents
| • | After the completion of our separation, the cost of capital for our business might be higher than PPD’s cost of capital prior to our separation; and |
| • | Prior to the separation, our financial statements include revenues and expenses of services that will not be continued by us subsequent to the separation. |
The unaudited pro forma condensed combined balance sheet as of December 31, 2009 assumes the funding by PPD of $100.0 million for a capital contribution based on the anticipated post-separation capital structure as well as the assumption by us of accounts payable to third parties in the ordinary course of our Compound Partnering Business. The pro forma financial position of Furiex as of December 31, 2009 and its results of operations for the years ended December 31, 2008 and 2009 also will differ from the financial position and results of operations of Furiex after the spin-off due to ongoing losses that are expected to continue after December 31, 2009. Please refer to “Management’s Discussion and Analysis of Financial Condition and Results of Operations”, “Unaudited Pro Forma Condensed Combined Financial Statements” and our historical combined financial statements and the notes to those statements included elsewhere in this Information Statement.
Our separation from PPD might present significant challenges.
There is a significant degree of difficulty and management distraction inherent in the process of our separating from PPD. These difficulties include:
| • | the challenge of effecting the separation while carrying on the ongoing operations of each business; |
| • | the potential difficulty in retaining key officers and personnel of each company; and |
| • | separating corporate infrastructure, including but not limited to systems, insurance, accounting, legal, finance, tax and human resources, for each of the two companies. |
Our separation from PPD might not be completed as successfully and cost-effectively as we anticipate. This could have an adverse effect on our business, financial condition and results of operations.
Our internal systems and resources might not be adequately prepared to meet the financial reporting and other requirements to which we will be subject following the separation.
Our financial results previously were included within the consolidated results of PPD. However, we were not directly subject to the reporting and other requirements of the Securities Exchange Act of 1934, as amended, which we refer to as the Exchange Act. As a result of the separation, we will be directly subject to the reporting and other obligations under the Exchange Act immediately after the separation. In addition, we expect to be subject to the requirements of Section 404 of the Sarbanes-Oxley Act of 2002 beginning with our financial statements for the year ending December 31, 2011, which will require annual management assessments of the effectiveness of our internal control over financial reporting and a report by our independent registered public accounting firm addressing the effectiveness of our internal control over financial reporting. These reporting and other obligations will place significant demands on our management and administrative and operational resources, including accounting resources.
To comply with these requirements, we might need to acquire or upgrade our systems, including information technology, implement additional financial and management controls, reporting systems and procedures and hire additional legal, accounting and finance staff. If we are unable to establish our financial and management controls, reporting systems, information technology and procedures in a timely and effective fashion, our ability to comply with our financial reporting requirements and other rules that apply to reporting companies could be impaired. In addition, if we are unable to conclude that our internal control over financial reporting is effective (or if the auditors are unable to express an opinion on the effectiveness of our internal controls), we could lose investor confidence in the accuracy and completeness of our financial reports.
12
Table of Contents
Our management will be responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) under the Exchange Act. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States. Any failure to achieve and maintain effective internal controls could have an adverse effect on our business, financial position and results of operations.
As a stand-alone company, we will not receive any of the revenue or cash flows derived from PPD’s CRO Business.
For fiscal 2009, PPD earned $1.4 billion or approximately 99.6% of its revenue from continuing operations from services revenues derived from PPD’s CRO Business. After the completion of the separation, we will not receive any such revenue. PPD will initially contribute to us cash and cash equivalents of $100.0 million, and we will assume current accounts payable and other accrued expenses payable to third parties and incurred in the ordinary course of our Compound Partnering Business, which as of December 31, 2009, totaled approximately $4.6 million. We expect that this cash contribution will fund Furiex’s operations and working capital requirements for at least 12 months after the closing of the spin-off, based on current operating plans. In addition to this cash contribution, we expect to receive future payments from our existing collaborations that will provide additional support for our operations and working capital requirements. Despite these resources and potential future revenue, we cannot assure you that such funds will meet our working capital and operational needs or that our working capital requirements will not increase beyond our current expectations. We might need to obtain additional financing from banks or other lenders, or through public offerings or private placements of debt or equity securities, strategic relationships or other arrangements to fully execute our business strategy.
Concerns about our prospects as a stand-alone company could affect our ability to retain employees.
The spin-off represents a substantial organizational and operational change and our employees might have concerns about our prospects as a stand-alone company, including our ability to successfully operate the new entity and our ability to maintain our independence after the spin-off. If we are not successful in assuring our employees of our prospects as an independent company, our employees might seek other employment, which could materially adversely affect our business.
We have no history operating as an independent company upon which you can evaluate us.
We do not have an operating history as a stand-alone entity. While our Compound Partnering Business has constituted a part of the historic operations of PPD since 1998, we have not operated as a stand-alone company without the CRO Business. Following the spin-off, as an independent company, our ability to satisfy our obligations and achieve profitability will be solely dependent upon the future performance of our Compound Partnering Business, and we will not be able to rely upon the capital resources and cash flows of the CRO Business remaining with PPD.
We might have received better terms from unaffiliated third parties than the terms we receive in our agreements with PPD.
The agreements we will enter into with PPD in connection with the spin-off, including the Master Development Services Agreement, the sublease, the Employee Matters Agreement and the Transition Services Agreement, were negotiated while we were still part of PPD. The terms of these agreements relate to, among other things, drug development services to be provided to us by PPD, the subleasing of our offices, employee benefit matters and the provision of transition services to us by PPD. The Master Development Services Agreement requires us to use PPD for specified drug development services for three years contingent on PPD’s expertise and capabilities to provide the needed services. While we believe the terms and conditions of these agreements with PPD are reasonable and acceptable to us, they might not reflect the same terms and conditions that we could have obtained had we sought competitive bids from and negotiated with unaffiliated parties. See “Our Relationship with PPD after the Spin-Off”.
13
Table of Contents
The ownership by our executive officers and some of our directors of shares of common stock and/or options to purchase shares of common stock of PPD might create, or might create the appearance of, conflicts of interest.
Due to their current or former employment with or service to PPD, several of our executive officers and directors own shares of common stock of PPD and hold options to purchase shares of common stock of PPD. As of March 31, 2010, the following individuals owned the following amounts of common stock and options to purchase common stock of PPD (exercisable within 60 days of March 31, 2010): Fred N. Eshelman, our founding Chairman and a director, 7,669,601 shares and 776,665 options; Stuart Bondurant, a director, 7,160 shares and 35,399 options; Gail McIntyre, our Senior Vice President – Research, 3,220 shares and 121,032 options; and Paul S. Covington, our Senior Vice President – Clinical Operations, 881 shares and 58,334 options. If the options were exercised, Dr. Eshelman would own 7.1% and the other individuals would own less than 1.0% of PPD’s outstanding common stock as of that date. These individual holdings of common stock and/or options to purchase common stock of PPD may be significant compared to the individual’s total assets. This ownership by our directors and officers, after our separation, of common stock and/or options to purchase common stock of PPD creates, or, might create the appearance of, conflicts of interest when these directors and officers are faced with decisions that could have different implications for PPD than for us.
If the distribution or internal transactions undertaken in anticipation of the separation are determined to be taxable for U.S. federal income tax purposes, we, our shareholders that are subject to U.S. federal income tax and PPD could incur significant U.S. federal income tax liabilities.
PPD is seeking a private letter ruling from the Internal Revenue Service regarding the U.S. federal income tax consequences of the distribution of our common stock to the PPD shareholders substantially to the effect that the distribution, except for cash received in lieu of a fractional share of our common stock, will qualify as tax-free under Sections 368(a)(1)(D) and 355 of the Code. The private letter ruling is also expected to provide that any internal transactions undertaken in anticipation of the separation will qualify for favorable treatment under the Code. The private letter ruling relies or will rely on facts and assumptions, and representations and undertakings, from us and PPD regarding the past and future conduct of our respective businesses and other matters. Notwithstanding the private letter ruling, the Internal Revenue Service could determine on audit that the distribution or the internal transactions should be treated as taxable transactions if it determines that any of these facts, assumptions, representations or undertakings is not correct or has been violated, or that the distribution should be taxable for other reasons, including as a result of significant changes in stock or asset ownership after the distribution. If the distribution ultimately is determined to be taxable, the distribution could be treated as a taxable dividend or capital gain to you for U.S. federal income tax purposes, and you could incur significant U.S. federal income tax liabilities. In addition, PPD would recognize taxable gain in an amount equal to the excess, if any, of the fair market value of our common stock distributed to PPD shareholders on the distribution date over PPD’s tax basis in these common shares, if the distribution were held to be taxable.
In addition, under the terms of the Tax Sharing Agreement that we will enter into with PPD, in the event the distribution or the internal transactions were determined to be taxable and such determination was the result of actions taken after the distribution by us or PPD, the party responsible for such actions would be responsible for all resulting taxes imposed on us or PPD. If such determination is not the result of actions taken after the distribution by us or PPD, then PPD would be responsible for any resulting taxes imposed on us or PPD. These taxes could be significant.
We might not be able to engage in desirable strategic transactions and equity issuances following the separation because of restrictions relating to U.S. federal income tax requirements for tax-free distributions.
Our ability to engage in significant equity transactions could be limited or restricted after the distribution in order to preserve for U.S. federal income tax purposes the tax-free nature of the distribution by PPD. In addition, similar limitations and restrictions will apply to PPD. Even if the distribution otherwise qualifies for tax-free
14
Table of Contents
treatment under Sections 368(a)(1)(D) and 355 of the Code, it might result in corporate-level taxable gain to PPD under Section 355(e) of the Code if 50% or more, by vote or value, of our shareholder equity or PPD shareholder equity is acquired or issued as part of a plan or series of related transactions that includes the distribution. For this purpose, any acquisitions or issuances of PPD’s shareholder equity within two years before the distribution, and any acquisitions or issuances of our shareholder equity or PPD shareholder equity within two years after the distribution, generally are presumed to be part of such a plan, although we or PPD might be able to rebut that presumption. If an acquisition or issuance of our shareholder equity or PPD shareholder equity triggers the application of Section 355(e) of the Code, PPD would recognize taxable gain as described above, and could incur significant U.S. federal income tax liabilities as a result of the application of Section 355(e) of the Code.
Under the Tax Sharing Agreement, there are restrictions on our ability to take actions that could cause the distribution or internal transactions undertaken in anticipation of the separation to fail to qualify as tax-favored, which could include entering into, approving or allowing any transaction that results in a change in ownership of more than 50% of our common shares, a redemption of equity securities, a sale or other disposition of a substantial portion of our assets, an acquisition of a business or assets with shareholder equity to the extent one or more persons would acquire 50% or more of our shareholder equity, or engaging in certain internal transactions. These restrictions apply at any time after the distribution, unless we obtain a private letter ruling from the Internal Revenue Service or an opinion that such action will not cause the distribution or the internal transactions undertaken in anticipation of the separation to fail to qualify as tax-favored transactions, and such letter ruling or opinion, as the case may be, is acceptable to the parties. PPD is subject to similar restrictions under the Tax Sharing Agreement. Moreover, the Tax Sharing Agreement generally provides that a party thereto is responsible for any taxes imposed on any other party thereto as a result of the failure of the distribution or internal transactions to qualify as a tax-favored transaction under the Code if the failure is attributable to post-distribution actions taken by or in respect of the responsible party or its shareholders, regardless of whether the other parties consent to such actions or such party obtains a favorable letter ruling or opinion as described above. For example, we would be responsible for the acquisition of us by a third party at a time and in a manner that would cause such failure. These restrictions might prevent us from entering into transactions that might be advantageous to our shareholders, or might increase the cost of the transactions.
If PPD waives one or more conditions to the spin-off, it could adversely impact our operations, the tax treatment of the spin-off, the liquidity of our common stock or have other consequences.
PPD can waive certain of the conditions to the spin-off in it sole and absolute discretion. See “The Spin-Off—Distribution Conditions and Termination”. If PPD waived the condition that it receive a private letter ruling and an independent tax opinion to the effect that the distribution will qualify as a tax-free reorganization for U.S. federal income tax purposes, then the distribution might be taxable to you as PPD stockholders. Waivers of other conditions by PPD also could have material adverse consequences. However, PPD cannot waive the condition that the SEC declare our registration statement effective or that our common stock be approved for listing on Nasdaq. While PPD has the ability to waive the condition that there not be an order, injunction or decree that prohibits or prevents the consummation of the spin-off, PPD would not waive this condition because it would be illegal to do so. Further, we are not aware of any material federal or state regulatory requirements that must be complied with or any material permits, registrations or approvals that must be obtained, other than compliance with SEC rules and regulations and the declaration of effectiveness of our registration statement by the SEC, in connection with the distribution. In addition, we have received all necessary third party consents for the transfer of assets from PPD to Furiex as part of the spin-off.
15
Table of Contents
Risks Relating to Furiex’s Business
We anticipate that we will incur losses for the foreseeable future. We might never achieve or sustain profitability. If additional capital is not available, we might have to curtail or cease operations.
Our business has experienced significant net losses. We had net losses of $22.9 million in 2007, net income of $5.8 million in 2008 and a net loss of $8.9 million in 2009. The net income for 2008 included aggregate milestone payments of $18.0 million. We expect to continue to incur additional net losses over the next several years as we continue our research and development activities and incur significant preclinical and clinical development costs. Since we or our collaborators or licensees might not successfully develop additional products, obtain required regulatory approvals, manufacture products at an acceptable cost or with appropriate quality, or successfully market products with desired margins, our expenses might continue to exceed any revenues we receive. Our commitment of resources to the continued development of our products might require significant additional funds for development. Our operating expenses also might increase if we:
| • | move our earlier stage potential products into later stage clinical development, which is generally a more expensive stage of development; |
| • | select additional preclinical product candidates for preclinical development and then clinical development; |
| • | pursue clinical development of our potential products in new indications; |
| • | increase the number of patents we are prosecuting or otherwise expend additional resources on patent prosecution or defense; |
| • | invest in or acquire additional technologies, product candidates or businesses although we have no current agreements to do so; or |
| • | impair any of our investments in our product candidates. |
In the absence of substantial licensing, milestone and other revenues from third-party collaborators, royalties on sales of products licensed under our intellectual property rights, future revenues from our products in development or other sources of revenues, we will continue to incur operating losses and might require additional capital to fully execute our business strategy. The likelihood of reaching, and time required to reach, sustained profitability are highly uncertain.
Although we expect that we will have sufficient cash to fund our operations and working capital requirements for at least 12 months after the spin-off based on current operating plans, we might need to raise additional capital in the future to:
| • | acquire complementary businesses or technologies; |
| • | respond to competitive pressures; |
| • | fund our research and development programs; or |
| • | commercialize our product candidates. |
Our future capital needs depend on many factors, including:
| • | the scope, duration and expenditures associated with our research and development programs; |
| • | continued scientific progress in these programs; |
16
Table of Contents
| • | the outcome of potential licensing transactions, if any; |
| • | competing technological developments; |
| • | our proprietary patent position, if any, in our product candidates; |
| • | the regulatory approval process for our product candidates; and |
| • | the cost of attracting and retaining employees. |
We might seek to raise necessary funds through public or private equity offerings, debt financings or additional collaborations and licensing arrangements. We might not be able to obtain additional financing on terms favorable to us, if at all. General market conditions might make it very difficult for us to seek financing from the capital markets. We might have to relinquish rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us, in order to raise additional funds through collaborations or licensing arrangements. If adequate funds are not available, we might have to delay, reduce or eliminate one or more of our research or development programs and reduce overall overhead expenses or cease operations. These actions might reduce the market price of our common stock.
Our near-term success is largely dependent on the success of Priligy and our lead drug candidate, alogliptin, as well as our other drug candidates, and we cannot be certain that we will be able to obtain regulatory approval for or commercialize any of these drug candidates.
We have invested a significant portion of our time and financial resources in the development of Priligy, which began being marketed in 2009 in certain countries, and of our lead drug candidate, alogliptin for the treatment of type 2 diabetes. Priligy is further developed than our other product candidates and is being marketed in various foreign countries. As a result, we currently are relying on Priligy to generate revenue for us to supplement our cash. While Priligy is approved for marketing outside of the U.S., it has not been approved in the U.S. and the FDA issued a not approvable letter to our collaborative partner Janssen-Cilag in October 2005. Janssen-Cilag is investigating regulatory strategies for a potential refiling with the FDA. Our collaborative partner on alogliptin must perform a cardiovascular safety trial for alogliptin and does not expect results from that trial to be available for submission to the FDA for approximately two years after that trial began. We have also invested a significant amount of time and financial resources in the development of our other drug candidates. We anticipate that our success will depend largely on the receipt of regulatory approval and successful commercialization of these drug candidates. The future success of these drug candidates will depend on several factors, including the following:
| • | our ability to provide acceptable evidence of their safety and efficacy; |
| • | receipt of marketing approval from the FDA and any similar foreign regulatory authorities; |
| • | obtaining and maintaining commercial manufacturing arrangements with third-party manufacturers or establishing commercial-scale manufacturing capabilities; |
| • | collaborating with pharmaceutical companies or contract sales organizations to market and sell any approved drug; |
| • | acceptance of any approved drug in the medical community and by patients and third-party payors; and |
| • | successful completion of the alogliptin cardiovascular safety trial that generates safety data acceptable to the FDA. |
17
Table of Contents
Many of these factors are beyond our control. Accordingly, we cannot assure you that we will be able to continue generating revenues through the sale of Priligy or generate any revenue from the sale of alogliptin or other drug candidates.
We might obtain future financing through the issuance of debt or equity, which might have an adverse effect on our shareholders or otherwise adversely affect our business.
If we raise funds through the issuance of debt or equity, any debt securities or preferred stock issued will have rights, preferences and privileges senior to those of holders of our common stock in the event of liquidation. In such event, there is a possibility that once all senior claims are settled, there might be no assets remaining to pay out to the holders of our common stock. In addition, if we raise funds through the issuance of additional equity, whether through private placements or public offerings, such an issuance would dilute the ownership of our then current shareholders.
The terms of debt securities might also impose restrictions on our operations, which might include limiting our ability to incur additional indebtedness, to pay dividends on or repurchase our capital stock, or to make certain acquisitions or investments. In addition, we might be subject to covenants requiring us to satisfy certain financial tests and ratios, and our ability to satisfy such covenants may be affected by events outside of our control.
Our operating expenses and results and any revenue likely will fluctuate in future periods.
Our revenues and expenses are unpredictable and likely will fluctuate from quarter to quarter due to, among other things, the timing and the unpredictable nature of clinical trials and related expenses, including payments owed by us and to us under collaborative agreements for reimbursement of expenses, future milestone revenues under collaborative agreements, sales of Priligy and any future sales of other products. In addition, the recognition of clinical trial and other expenses that we otherwise would recognize over a period of time under applicable accounting principles might be accelerated in certain circumstances. In such a case, it might cause our expenses during that period to be higher than they otherwise would have been had the circumstances not occurred. For example, if we terminate a clinical trial for which we paid non-refundable upfront fees to a clinical research organization and in which we did not accrue all of the patient costs, the recognition of the expense associated with those fees that we were recognizing as we accrued patient costs would be accelerated and recognized in the period in which the termination occurred.
We are dependent on the performance of service providers.
We rely on service providers, such as contract manufacturers, clinical research organizations, medical institutions and clinical investigators, including physician sponsors, to conduct nearly all of our clinical trials, including recruiting and enrolling patients in the trials. Before the distribution, we will enter into a Master Development Services Agreement with PPD pursuant to which PPD will provide us clinical development activities on a preferred provider basis. If PPD or any of these other parties do not successfully carry out their contractual duties or meet expected deadlines, we might be delayed or may not obtain regulatory approval for or be able to commercialize our product candidates. If any of the third parties upon whom we rely to conduct our clinical trials do not comply with applicable laws, successfully carry out their obligations or meet expected deadlines, our clinical trials may be extended, delayed or terminated.
If the quality or accuracy of the clinical data obtained by third party contractors is compromised due to their failure to adhere to applicable laws or our clinical protocols, or for other reasons, we might not obtain regulatory approval for or successfully commercialize any of our product candidates. If our relationship with any of these organizations or individuals terminates, replacing any of these third parties could delay our clinical trials and could jeopardize our ability to obtain regulatory approvals and commercialize our product candidates on a timely basis, if at all.
18
Table of Contents
We might not successfully operate the Compound Partnering Business following the spin-off.
It takes many years for a drug development business like ours to generate revenue and income. Although we have experience operating our Compound Partnering Business within PPD’s Discovery Sciences segment since 1998, we might not be successful in operating this business as a stand-alone company following the spin-off. Generating revenue and income, consistently or at all, from our drug development business and compound partnering activities will depend on our ability to:
| • | develop products internally or obtain rights to them from others on favorable terms; |
| • | successfully complete non-clinical and clinical studies; |
| • | obtain clinical trial materials of sufficient quality or quantity; |
| • | obtain and maintain intellectual property rights to these products; |
| • | obtain and maintain regulatory approvals; |
| • | enter into agreements with third parties to continue the development and commercialization of drug candidates; and |
| • | enter into arrangements with third parties to manufacture products on our behalf and to provide sales and marketing functions. |
We must attract and retain key employees in order to succeed.
To be successful, we must attract and retain qualified clinical, scientific, management and other personnel and we face significant competition for experienced personnel. If we are unsuccessful in attracting and retaining qualified personnel, particularly at the management level, our business could be impaired. We rely on the services of our senior management, particularly our President and Chief Medical Officer, June Almenoff, our Senior Vice President – Research, Gail McIntyre, and our Senior Vice President – Clinical Operations, Paul Covington, as well as our Vice President – Legal Affairs, Nadine Chien, and our Vice President – Strategic Development, Sailash Patel, the loss of any of whom could adversely impact our operations. We do not carry key man insurance on any of these individuals or any of our other officers or employees. Any inability to hire additional qualified personnel might also require an increase in the workload for both existing and new personnel. We might not be successful in attracting new scientists or management, or in retaining or motivating our existing personnel. The shortage of experienced scientists and managers, or other factors, might lead to increased recruiting, relocation and compensation costs for these professionals, which might exceed our forecasts. These increased costs might make hiring new scientists and managers impracticable. If we are unable to attract and retain any of these personnel, our ability to execute our business plan will be adversely affected.
If our product identification efforts are not successful, we might not be able to effectively develop new products.
Our product candidates are in various stages of development and some are in an early development stage. Some or all of our product candidates may never be developed for any number of reasons, including failure to meet clinical trial tests and failure to receive regulatory approval. For example, we suspended our MAG-131 program due to efficacy data that was discovered in late 2009. To maintain our business, we need to have a sufficient pipeline of product candidates. Our success in identifying new product candidates depends upon our ability to identify and validate new targets through in-licensing or collaborative arrangements. In order to increase the possibilities of identifying compounds with a reasonable chance for success in clinical studies, part of our business strategy is to identify a higher number of potential targets than we expect to be able to progress through clinical development. If we are unsuccessful in our efforts to identify or obtain rights to new product candidates that lead to the required regulatory approvals and the successful commercialization of products, our business could be harmed.
19
Table of Contents
Our product development is dependent on our collaborators.
Our ability to succeed in our drug development business will depend on successfully executing existing and new arrangements we enter into for the development and commercialization of drug candidates. The third parties that we collaborate with might not perform their obligations as expected or they might breach or terminate their agreements with us or otherwise fail to conduct their collaborative activities successfully or in a timely manner. Further, parties collaborating with us might elect not to develop the product candidates or devote sufficient resources to the development, manufacture, regulatory strategy and approvals, marketing or sale of these product candidates. If the parties to our collaborative agreements do not fulfill their obligations, elect not to develop a candidate or fail to devote sufficient resources to it, our business could be materially and adversely affected.
Many of our drug candidates are in development and we might not be able to obtain regulatory approval for our product candidates.
The development and commercialization of pharmaceutical products are subject to extensive governmental regulation in the United States and foreign countries. Government approvals are required to develop, market and sell the potential drug candidates we develop alone or with others under our risk-sharing arrangements. Especially for the early-stage compounds we target for in-licensing, obtaining government approval to develop, market and sell drug candidates is time-consuming and expensive. Further, clinical trial results for a particular drug candidate might not satisfy requirements to obtain government approvals. For example, in late 2005, Janssen-Cilag, our collaborator on dapoxetine, received a “not approvable” letter from the FDA. In addition, governmental approvals might not be received in a timely manner, if at all, and we and our collaborative partners might not be able to meet other regulatory requirements for our products. For example, in late 2008, the FDA notified Takeda that it would not be able to complete its review of the alogliptin NDA before the Prescription Drug Use Fee Act date due to the lack of internal resources.In addition, requirements for government approval to market and sell drug candidates are subject to change. For example, the Division of Endocrinologic and Metabolic Drug Products in the Center for Drug Evaluation and Research decided that concerns about cardiovascular risk should be more thoroughly addressed during drug development programs, and, in December 2008, issued final guidance on the topic titled “Guidance for Industry—Diabetes Mellitus—Evaluating Cardiovascular Risk in New Antidiabetic Therapies to Treat Type 2 Diabetes”. As a result, in June 2009 and September 2009, the FDA issued complete responses to Takeda on its NDAs for the alogliptin monotherapy and the fixed-dose combination of alogliptin and Actos requesting an additional cardiovascular safety trial on alogliptin prior to further regulatory review. Finally, even if we are successful in obtaining all required approvals to market and sell a drug candidate, post-approval requirements and the failure to comply with other regulations could result in suspension or limitation of government approvals.
In connection with drug development activities outside the United States, we and our collaborators will be subject to foreign regulatory requirements governing the testing, approval, manufacture, labeling, marketing and sale of pharmaceutical products. These requirements vary from country to country. Even if approval has been obtained for a product in the United States, approvals in foreign countries must be obtained prior to marketing the product in those countries. The approval process in foreign countries may be more or less rigorous and the time required for approval may be longer or shorter than that required in the United States. Clinical studies conducted outside of any particular country may not be accepted by that country, and the approval of a pharmaceutical product in one country does not assure that the product will be approved in another country.
The failure to gain market acceptance of our product candidates among the medical community would adversely affect our revenue.
Even if approved, our product candidates might not gain market acceptance among physicians, patients, third-party payors and the medical community. We might not achieve market acceptance even if clinical trials demonstrate safety and efficacy and we obtain the necessary regulatory and reimbursement approvals. The degree of market acceptance of any product candidates that we develop will depend on a number of factors, including:
| • | establishment and demonstration of clinical efficacy and safety; |
20
Table of Contents
| • | cost-effectiveness of our product candidates versus competing products; |
| • | their potential advantage over alternative treatment methods; |
| • | pricing requirements in various markets; |
| • | reimbursement policies of government and third-party payors; and |
| • | marketing and distribution support for our product candidates, including the efforts of our collaborators where they have marketing and distribution responsibilities. |
Physicians will not recommend our products until clinical data or other factors demonstrate the safety and efficacy of our product as compared to conventional drug and other treatments. Even if we establish the clinical safety and efficacy of our product candidates, physicians might elect not to use our product for any number of other reasons, including whether the mode of administration of our products is effective for certain indications. The failure of our product candidates to achieve significant market acceptance would materially harm our business, financial condition and results of operations.
We face significant competition.
We face significant competition, including from entities that have substantially greater resources and more experience in the commercialization and marketing of pharmaceuticals than we have. Potential competitors in the United States and other countries include major pharmaceutical and biotechnology companies and specialized pharmaceutical companies. These entities have developed and are developing compounds that might compete with our products in development. These competitors might succeed in more rapidly developing and marketing technologies and products that are more effective than our product candidates or technologies or that would render any future commercialized products or technology obsolete or noncompetitive. Our product candidates and any future commercialized products might also face significant competition from both brand-name and generic manufacturers that could adversely affect any future sales of our products.
Any product that we or our collaborators succeed in developing and for which regulatory approval is obtained must then compete for market acceptance and market share. The relative speed with which we and our collaborators can develop products, complete the clinical testing and approval processes, and supply commercial quantities of the products to the market compared to competitive companies will affect market success. In addition, the amount of marketing and sales resources and the effectiveness of the marketing used with respect to a product will affect its marketing success. Other factors affecting the ability of our products to compete include their efficacy and safety, the manner and frequency of their administration, and the extent of any reimbursement coverage.
In addition, some CRO services providers and private equity funds are developing risk sharing models to finance the pharmaceutical industry’s pipeline. As these types of business models evolve, there will be increasing competition for compounds and funds to develop those compounds.
We must protect our patent and other intellectual property rights to succeed.
Our success is dependent in significant part on our ability to develop and protect patent and other intellectual property rights and operate without infringing the intellectual property rights of others.
Our pending patent applications might not result in the issuance of valid patents or the claim scope of our issued patents may not provide competitive advantages. Also, our patent protection might not prevent others from developing competitive products using related or other technology that does not infringe our patent rights. In addition, our patent for Priligy is for method of use and not composition of matter. Further, patent applications
21
Table of Contents
are confidential for a period of time after filing. We therefore might not know that a competitor has filed a patent application covering subject matter similar to subject matter in one of our patent applications or that we were the first to invent the innovation we seek to patent. This might lead to disputes including interference proceeding or litigation to determine rights to patentable subject matter. These disputes are often expensive and might result in our being unable to patent an innovation.
The scope, enforceability and effective term of patents can be highly uncertain and often involve complex legal and factual questions and proceedings. No consistent policy has emerged regarding the breadth of claims in pharmaceutical or biotechnology patents, so that even issued patents might later be modified or revoked by the relevant patent authorities or courts. These proceedings could be expensive, last several years and either prevent issuance of additional patents to us or result in a significant reduction in the scope or invalidation of our patents. Any limitation in claim scope could reduce our ability to negotiate future collaborative research and development agreements based on these patents. Moreover, the issuance of a patent in one country does not assure the issuance of a patent with similar claim scope in another country, and claim interpretation and infringement laws vary among countries, so we are unable to predict the extent of patent protection in any country.
In certain cases, we are reliant on our collaborator to file, negotiate and maintain patents covering a licensed product. Our collaborators may fail to adequately obtain and maintain such patents.
In addition to seeking the protection of patents and licenses, we also rely upon trade secrets, know-how and continuing technological innovation that we seek to protect, in part, by confidentiality agreements with employees, consultants, suppliers and licensees. If these agreements are not honored, we might not have adequate remedies for any breach. Additionally, our trade secrets might otherwise become known or patented by our competitors.
We might need to obtain patent licenses from others in order to manufacture or sell our potential products and we might not be able to obtain these licenses on terms acceptable to us or at all.
Other companies, universities and research institutions might obtain patents that could limit our ability to use, import, manufacture, market or sell our products or impair our competitive position. As a result, we might need to obtain licenses from others before we could continue using, importing, manufacturing, marketing, or selling our products. We might not be able to obtain required licenses on terms acceptable to us, if at all. If we do not obtain required licenses, we might encounter significant delays in product development while we redesign potentially infringing products or methods or we might not be able to market our products at all.
If our collaborations are not successful or are terminated by our collaborators, we might not effectively develop and market some of our product candidates.
We have agreements under which we rely on collaborators to manufacture our product candidates and essential components for those product candidates, design and conduct clinical trials, compile and analyze the data received from these trials, obtain regulatory approvals and, if approved, market these products. As a result, we may have limited or no control over the manufacturing, development and marketing of these potential products. In addition, the performance of our collaborators may not be sufficient or appropriate for regulatory review and approval for our product candidates. Further, we often rely on one manufacturer or other collaborator for such services, the loss of which could significantly delay the development of any of our drug candidates.
Our collaborators can terminate our collaborative agreements under certain conditions, and Janssen Pharmaceutica can do so on short notice under our development agreements for the fluoroquinolone and mu delta compounds. A collaborator may terminate its agreement with us or separately pursue alternative products, therapeutic approaches or technologies as a means of developing treatments for the diseases targeted by us, or our collaborative effort. Even if a collaborator continues to contribute to the arrangement, it may nevertheless decide not to actively pursue the development or commercialization of any resulting products. In these circumstances, our ability to further develop potential products could be severely limited. While we generally
22
Table of Contents
seek non-compete terms in our agreements with our collaborators for the products we are developing, the enforcement of a non-compete can be expensive and difficult to monitor and enforce and might be subject to being invalidated by a court or judge.
Continued funding and participation by collaborators will depend on the continued timely achievement of our research and development objectives, the retention of key personnel performing work under those agreements and on each collaborator’s own financial, competitive, marketing and strategic capabilities and priorities. These considerations include:
| • | the commitment of each collaborator’s management to the continued development of the licensed products or technology; |
| • | the relationships among the individuals responsible for the implementation and maintenance of the development efforts; and |
| • | the relative advantages of alternative products or technology being marketed or developed by each collaborator or by others, including their relative patent and proprietary technology positions, and their ability to manufacture potential products successfully. |
The willingness of our existing collaborators to continue development of our potential products and our ability to enter into new relationships depends upon, among other things, our patent position with respect to such products. If we are unable to successfully obtain and maintain patents, we might be unable to collect royalties on existing licensed products or enter into additional agreements.
In addition, our collaborators might independently develop products that are competitive with products that we have licensed to them. This could reduce our revenues or the likelihood of achieving revenues under our agreements with these collaborators.
We or our collaborators might not be able to attract a sufficient number of sites or enroll a sufficient number of patients in a timely manner in order to complete our clinical trials.
The rate of completion of clinical trials is significantly dependent upon the rate of patient enrollment. Patient enrollment is a function of many factors, including:
| • | the size of the patient population; |
| • | perceived risks and benefits of the drug under study; |
| • | availability of competing therapies, including those in clinical development; |
| • | availability of clinical drug supply; |
| • | participation of qualified clinical trial sites; |
| • | availability and willingness of potential participants to enroll in clinical trials; |
| • | design of the protocol; |
| • | proximity of and access by patients to clinical sites; |
| • | patient referral practices of physicians; |
| • | eligibility criteria for the study in question; and |
| • | efforts of the sponsor of and clinical sites involved in the trial to facilitate timely enrollment. |
We might have difficulty obtaining sufficient patient enrollment or clinician support to conduct our clinical trials as planned, and we might need to expend additional funds to obtain access to resources or delay or modify
23
Table of Contents
our plans significantly. These considerations might result in our being unable to successfully achieve our projected development timelines, or potentially even lead us to consider the termination of ongoing clinical trials or development of a product for a particular indication.
Changes in the U.S. and international healthcare industry, including reimbursement rates, could adversely affect the commercial value of our development product candidates.
The U.S. and international healthcare industry is subject to changing political, economic and regulatory influences that may significantly affect the purchasing practices and pricing of pharmaceuticals. The laws and regulations governing and issued by applicable regulatory agencies may change and additional government regulations might be enacted, which could prevent or delay regulatory approval of our product candidates. The U.S. Congress has also considered and may adopt healthcare reform or other legislation that could have the effect of putting downward pressure on the prices that pharmaceutical and biotechnology companies can charge for prescription drugs. Cost-containment measures, whether instituted by healthcare providers or imposed by government health administration regulators or new regulations, could result in greater selectivity in the purchase of drugs. As a result, third-party payors might challenge the price and cost effectiveness of our products. In addition, in many major markets outside the United States, including some where Priligy is approved, pricing approval is required before sales may commence. As a result, significant uncertainty exists as to the reimbursement status of approved healthcare products.
We might not be able to obtain or maintain our desired price for the products we develop. Any product we introduce might not be considered cost-effective relative to alternative therapies. As a result, adequate third-party reimbursement might not be available to enable us to obtain or maintain prices sufficient to realize an appropriate return on our investment in product development. Also, the trend towards managed healthcare in the United States and the concurrent growth of organizations such as health maintenance organizations, as well as legislative proposals to reform healthcare or reduce government insurance programs, might all result in lower prices, reduced reimbursement levels and diminished markets for our products. These factors will also affect the products that are marketed by our collaborators and licensees. We cannot predict the likelihood, nature or extent of adverse government regulation that might arise from legislation or administrative action, either in the United States or abroad. If we are not able to maintain regulatory compliance, we might not be permitted to market our future products and our business could suffer.
Manufacturing changes might result in delays in obtaining regulatory approval or marketing for our products.
If we make changes in the manufacturing process for any of our products, we might be required to demonstrate to the applicable regulatory agencies that the changes have not caused the resulting drug material to differ significantly from the drug material previously produced. Further, any significant manufacturing changes for the production of our product candidates could result in delays in development or regulatory approval or in the reduction or interruption of commercial sales of our product candidates. Our contract manufacturers’ inability to maintain manufacturing operations in compliance with applicable regulations within our planned time and cost parameters could materially harm our business, financial condition and results of operations.
We have made manufacturing changes and could make additional manufacturing changes for the production of our products currently in clinical development. These manufacturing changes or an inability to immediately show comparability between the older material and the newer material after making manufacturing changes could result in delays in development or regulatory approvals or in reduction or interruption of commercial sales and could impair our competitive position.
Our business might be harmed if we cannot obtain sufficient quantities of raw materials.
We depend on outside vendors for the supply of raw materials used to produce our product candidates for use in clinical trials. Once a supplier’s materials have been selected for use in the manufacturing process, the supplier in effect becomes a sole or limited source of that raw material due to regulatory compliance procedures.
24
Table of Contents
If the third-party suppliers were to cease production or otherwise fail to supply us with quality raw materials and we were unable to contract on acceptable terms for these services with alternative suppliers, our ability to produce our products and to conduct preclinical testing and clinical trials of product candidates would be adversely affected. This could impair our competitive position.
We must comply with extensive government regulations and laws.
We and our collaboration partners are subject to extensive regulation by federal government, state governments, and the foreign countries in which we conduct our business.
In particular, we are subject to extensive and rigorous government regulation as a developer of drug candidates. For example, the FDA regulates, among other things, the development, testing, research, manufacture, record-keeping, labeling, storage, approval, quality control, adverse event reporting, advertising, promotion, sale and distribution of pharmaceutical products. Our product candidates are subject to extensive regulation by foreign governments. The regulatory review and approval process, which includes preclinical studies and clinical trials of each product candidate, is lengthy, expensive and uncertain.
We must rely on our contract manufacturers and third-party suppliers for regulatory compliance and adhering to the FDA’s current Good Manufacturing Practices, or cGMP, requirements. If these manufacturers or suppliers fail to comply with applicable regulations, including FDA pre-or post-approval inspections and cGMP requirements, then the FDA could sanction us. These sanctions could include fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our products, delay, suspension or withdrawal of approvals, license revocation, product seizures or recalls, operational restrictions or criminal prosecutions, any of which could significantly and adversely affect our operating results.
If our operations are found to violate any applicable law or other governmental regulations, we might be subject to civil and criminal penalties, damages and fines. Similarly, if the hospitals, physicians or other providers or entities with which we do business are found non-compliant with applicable laws, they might be subject to sanctions, which could also have a negative impact on us. The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations, and additional legal or regulatory change. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and damage our reputation.
We expend a significant amount of resources on compliance efforts and such expenses are unpredictable and might adversely affect our operating results. Changing laws, regulations and standards might also create uncertainty and increase insurance costs. We are committed to compliance and maintaining high standards of corporate governance and public disclosure. As a result, we intend to invest all reasonably necessary resources to comply with evolving standards, and this investment might result in increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities.
Even if one of our in-licensed drug candidates successfully completes Phase II clinical trial testing, we will need to have a collaborator for final development and commercialization of that drug candidate or undertake those activities on our own.
We generally conduct our drug development business in two stages. During the first stage, we in-license a drug candidate from a collaborator and develop that candidate through Phase II clinical trials. If the drug candidate successfully completes Phase II testing, we enter a second stage during which we seek a collaborator, which might be the same collaborator as in the first stage, for the continued late stage development and ultimate commercialization of the drug candidate. If we cannot find a collaborator for final development and commercialization, we might not be able to complete the development and commercialization on our own due to the significant costs associated with these activities. As a result, we may not be able to recoup all or any part of our investment in the drug candidate.
25
Table of Contents
If we are unable to enter into agreements with third parties to market and sell our drug candidates or are unable to establish our own sales and marketing capabilities, we might be unable to generate product revenue.
We do not currently have the resources to sell, market or distribute any pharmaceutical products. In order to market any of our products that receive regulatory approval, we must make arrangements with third parties to perform these services, or build our sales, marketing, managerial and other non-technical capabilities. If we are unable to do so, we might not be able to generate product revenue and might not become profitable.
We might incur significant costs in order to comply with environmental regulations or to defend claims arising from accidents involving the use of hazardous materials.
We are subject to federal, state and local laws and regulations governing the use, discharge, handling and disposal of materials and wastes used in our operations. As a result, we might be required to incur significant costs to comply with these laws and regulations. We cannot eliminate the risk of accidental contamination or injury from these materials. In the event of such an accident, we could be held liable for any resulting damages and incur liabilities, which exceed our resources. In addition, we cannot predict the extent of the adverse effect on our business or the financial and other costs that might result from any new government requirements arising out of future legislative, administrative or judicial actions.
We might be subject to product liability claims, and our insurance coverage and indemnification rights might not be adequate to cover these claims.
We face an inherent business risk of exposure to product liability and other types of claims in the event that the use of products during research and development efforts or after commercialization results in death, personal injury or other adverse effects. This risk exists even with respect to any products that receive regulatory approval for commercial sale. While we will procure and maintain liability insurance with coverage up to $10 million per occurrence and in the aggregate and generally have indemnification rights under our collaboration agreements, our insurance might not be sufficient to satisfy any or all liabilities that may arise and our indemnification rights might not apply or be sufficient to cover such claims. Also, adequate insurance coverage might not be available in the future at acceptable cost, if at all.
Our operations might be affected by the occurrence of a natural disaster or other catastrophic event.
We depend on our collaboration partners, service providers, our own laboratories and other facilities for the continued operation of our business. Although we have contingency plans in effect for natural disasters or other catastrophic events, these events, including terrorist attacks, pandemic flu, hurricanes and ice storms, could still disrupt our operations or those of our collaboration partners, which could also affect us. Even though we carry business interruption insurance policies and typically have provisions in our contracts that protect us in certain events, we might suffer losses as a result of business interruptions that exceed the coverage available under our insurance policies or for which we do not have coverage. Any natural disaster or catastrophic event affecting us or our collaboration partners could have a significant negative impact on our operations and financial performance.
Risks Relating to Our Common Stock
There is no existing market for our common stock and a trading market that will provide you with adequate liquidity might not develop. In addition, once our common stock begins trading, the market price for our shares might fluctuate widely.
There is currently no public market for our common stock. It is anticipated that on or shortly after the record date for the distribution, trading of shares of our common stock will begin on a “when-issued” basis and will continue up to either the distribution date or the first trading date after the distribution date, after which “regular way” trading of our common stock will begin. However, an active trading market for our common stock might not develop as a result of the distribution or be sustained in the future.
26
Table of Contents
Market prices for securities of pharmaceutical companies have been highly volatile, and we expect such volatility to continue for the foreseeable future, so that investment in our securities involves substantial risk. Additionally, the stock market from time to time has experienced significant price and volume fluctuations unrelated to the operating performance of particular companies. The following are some of the factors that might have a significant effect on the market price of our common stock:
| • | developments or disputes as to patent or other proprietary rights; |
| • | approval or introduction of competing products and technologies; |
| • | results of clinical trials; |
| • | failures or unexpected delays in timelines for our potential products in development, including the obtaining of regulatory approvals; |
| • | delays in manufacturing or clinical trial plans; |
| • | fluctuations in our operating results; |
| • | market reaction to announcements by other biotechnology or pharmaceutical companies; |
| • | initiation, termination or modification of agreements with our collaborators or disputes or disagreements with collaborators; |
| • | loss of key personnel; |
| • | litigation or the threat of litigation; |
| • | public concern as to the safety of drugs developed by us; |
| • | sales of our common stock held by our directors and executive officers; and |
| • | comments and expectations of results made by securities analysts or investors. |
If any of these factors causes us to fail to meet the expectations of securities analysts or investors, or if adverse conditions prevail or are perceived to prevail with respect to our business, the price of our common stock would likely drop significantly. A significant drop in the price of a company’s common stock often leads to the filing of securities class action litigation against such a company. This type of litigation against us could result in substantial costs and a diversion of management’s attention and resources.
Substantial sales of common stock might occur in connection with this distribution, which could cause our stock price to decline.
The shares of our common stock that PPD intends to distribute to its shareholders generally may be sold immediately in the public market. Although we have no actual knowledge of any plan or intention on the part of any of our expected 5% or greater shareholders to sell our common stock following the distribution, we believe that many current PPD shareholders are particularly focused on the value of the CRO Business. Therefore, it is possible that some PPD shareholders, including possibly some of our large shareholders, will sell our common stock received in the distribution. In addition, PPD shareholders might sell our stock because our business profile or market capitalization as an independent company does not fit their investment objectives. The sales of significant amounts of our common stock or the perception in the market that this will occur might result in the lowering of the market price of our common stock.
Your percentage ownership in Furiex might be diluted in the future.
Your percentage ownership in Furiex might be diluted in the future because of equity awards that we expect will be granted to our directors, officers and employees, as well as any future equity financing. For a more detailed description of our equity compensation arrangements, see “Management—Executive Compensation”.
27
Table of Contents
Provisions in our amended and restated certificate of incorporation and bylaws and of Delaware law might prevent or delay an acquisition of our company, which could decrease the trading price of our common stock.
Our amended and restated certificate of incorporation, bylaws and Delaware law contain provisions that are intended to deter coercive takeover practices and inadequate takeover bids by making such practices or bids unacceptably expensive to the raider and to encourage prospective acquirors to negotiate with our Board rather than to attempt a hostile takeover. These provisions include, among others:
| • | no right of our shareholders to act by written consent; |
| • | procedures requiring advance notice of shareholder proposals or nominations for directors for election at shareholder meetings; |
| • | the right of our Board to issue preferred stock without shareholder approval; and |
| • | no shareholder rights to call a special shareholders meeting. |
Delaware law also imposes some restrictions on mergers and other business combinations between us and any holder of 15% or more of our outstanding common stock. For more information, see “Description of Capital Stock”.
We believe these provisions protect our shareholders from coercive or otherwise unfair takeover tactics by requiring potential acquirors to negotiate with our Board and by providing our Board with more time to assess any acquisition proposal. These provisions are not intended to make our company immune from takeovers. However, these provisions apply even if the offer might be considered beneficial by some shareholders and could delay or prevent an acquisition that our Board determines is not in the best interests of our company and our shareholders.
We do not expect to pay any dividends in the foreseeable future.
We do not expect to declare dividends in the foreseeable future. We currently intend to retain any potential future earnings to support our operations and to finance the growth and development of our business. We might not ever have sufficient surplus under Delaware law to be able to pay any dividends.
28
Table of Contents
This Information Statement includes forward-looking statements within the meaning of Section 27A of the Securities Act and Section 21E of the Exchange Act of 1934. All statements other than statements of historical facts are forward-looking statements for purposes of these provisions, including any projections of milestones, royalties or other financial items, any statements of the plans and objectives of management for future operations, any statements concerning proposed new products or licensing or collaborative arrangements, any statements regarding future economic conditions or performance, and any statement of assumptions underlying any of the foregoing. In some cases, forward-looking statements can be identified by the use of terminology such as “believes”, “might”, “will”, “expects”, “plans”, “anticipates”, “estimates”, “potential” or “continue”, or the negative thereof or other comparable terminology. Although we believe that the expectations reflected in the forward-looking statements contained in this Information Statement are reasonable, there can be no assurance that such expectations or any of the forward-looking statements will prove to be correct, and actual results could differ materially from those projected or assumed in the forward-looking statements. Our future financial condition and results of operations, as well as any forward-looking statements, are subject to inherent risks and uncertainties, including the risk factors set forth above, and for the reasons described elsewhere in this Information Statement. All forward-looking statements and reasons why results might differ included in this Information Statement are made as of the date hereof, and we assume no obligation to update these forward-looking statements or reasons why actual results might differ.
29
Table of Contents
Reasons for the Spin-Off
PPD began its business in 1985, initially focusing on the CRO Business. Building on its outsourcing relationship with pharmaceutical and biotechnology clients, PPD established its Discovery Sciences business in 1997. This business primarily focuses on the Compound Partnering Business, namely compound development and commercialization collaborations. PPD has developed a risk-sharing research and development model to help pharmaceutical and biotechnology clients develop compounds. Through collaborative arrangements based on this model, PPD assists its clients by sharing the risks and potential rewards of the development and commercialization of drugs at various stages of development.
PPD’s Board has explored strategic alternatives available to increase long-term shareholder value from the Compound Partnering Business and the CRO Business. While the two businesses were historically related, they have different business models, distinct cost, revenue and return-on-investment structures, and can be run independently with limited operational overlap. Specifically, the profitable CRO services business has different needs and constraints than those of a pharmaceutical research and development business. Some of the alternatives the PPD Board considered were selling or out-licensing some or all of its rights to compounds, selling a minority or majority interest in the Compound Partnering Business, or issuing a separate class of PPD stock to reflect the performance of the Compound Partnering Business. Ultimately, the PPD Board concluded a spin-off was in the best interests of PPD and its shareholders. The CRO Business will continue to operate under the PPD name and will be focused solely on its drug discovery and development service businesses and will no longer be coupled with the earnings dilution from the Compound Partnering Business. The Compound Partnering Business will have the opportunity to focus on developing and commercializing its existing and potential new drug candidates and to access external capital, if needed, without any constraints associated with operating in combination with the CRO Business. After consideration of these and other factors, PPD’s Board believed that maximizing value to shareholders might best be achieved by the separation and independent operation of the Compound Partnering Business and the CRO Business.
Accordingly, in October 2009, PPD’s Board decided to pursue the separation of the two businesses through the spin-off. On October 28, 2009, PPD announced that its Board had authorized in principle the distribution of our common stock to PPD’s shareholders in a spin-off of the Compound Partnering Business from PPD.
PPD and we believe that the spin-off of our Compound Partnering Business from PPD will provide several opportunities and benefits that are expected to enhance shareholder value, including the following:
| • | Market Recognition: allowing investors to recognize and realize the full potential value of each company independently; |
| • | Employee Incentives: increasing our ability to attract and retain employees by providing equity compensation tied directly to our company’s performance, and our research and development efforts in particular; |
| • | Improved Capital Flexibility: allowing each company to deploy capital and access additional financing, if appropriate, in accordance with its unique needs and business model; and |
| • | Business Focus: allowing each company to focus its efforts on and allocate its resources towards its own business opportunities and challenges. |
In determining whether to effect the separation, the PPD Board was mindful of the costs associated with the separation and the risks Furiex faces as a public company, which weighed against the separation. The Board determined, however, that for the reasons stated above, the separation provided the separated companies with opportunities and benefits that could enhance shareholder value.
Manner of Effecting the Spin-Off
The general terms and conditions of the spin-off will be set forth in the Separation and Distribution Agreement to be entered into by PPD and us. For a description of the expected terms of that agreement, see “Our Relationship with PPD after the Spin-Off—Separation and Distribution Agreement”.
30
Table of Contents
Overview. The spin-off will be accomplished through a series of transactions, pursuant to the terms and conditions of the Separation and Distribution Agreement, by which PPD will contribute to Furiex its Compound Partnering Business, including intellectual property, and distribute to its shareholders of record on the record date all of the outstanding shares of Furiex’s common stock. As discussed under “The Spin-Off—Trading of PPD Common Stock After the Record Date and Prior to the Ex-Dividend Date”, if a holder of record of PPD common stock sells those shares in the “regular way” market prior to the ex-dividend date, that shareholder also will be selling the right to receive shares of Furiex common stock in the distribution. The distribution will be made in book-entry form on the basis of one share of Furiex common stock for every shares of PPD common stock held on the record date, which is [ ], 2010. We will instruct American Stock Transfer & Trust Company, as distribution agent, to record the distribution on the distribution date to the holders of PPD common stock at the close of business on the record date (or their designated transferees), unless the shares of PPD common stock had been sold prior to the ex-dividend date. Each share of Furiex common stock that PPD distributes will be validly issued, fully paid and nonassessable and free of preemptive rights.
Fractional Shares. PPD will not distribute any fractional shares of our common stock to its shareholders. Instead, the distribution agent will aggregate fractional shares into whole shares, sell the whole shares in the open market at prevailing market prices and distribute the aggregate net cash proceeds of the sales pro rata (based on the fractional share such holder would otherwise be entitled to receive) to each holder who otherwise would have been entitled to receive a fractional share in the distribution. The distribution agent, in its sole discretion, without any influence by PPD or us, will determine when, how, through which broker-dealer and at what price to sell the whole shares. Any broker-dealer used by the distribution agent will not be an affiliate of either PPD or us. Recipients of cash in lieu of fractional shares will not be entitled to any interest on the amounts of payment made in lieu of fractional shares. If you physically hold PPD common stock certificates and are the registered holder, you will receive a check from the distribution agent in an amount equal to your pro rata share of the aggregate net cash proceeds of the sales. We estimate that it will take approximately four to six weeks from the distribution date for the distribution agent to complete the distributions of the aggregate net cash proceeds. If you hold your PPD common stock through a bank or brokerage firm, your bank or brokerage firm will receive on your behalf your pro rata share of the aggregate net cash proceeds of the sales, which should electronically credit your account for your share of such proceeds.
Book Entry Statements. A book-entry account statement reflecting your ownership of shares of Furiex common stock will be mailed to you, or your brokerage account should be credited for the shares, on or about [ ], 2010. We will not issue physical stock certificates in the spin-off, even if requested.
Cash Contribution. At the closing of the spin-off, PPD will contribute to us cash and cash equivalents of $100.0 million, and we will assume current accounts payable and other accrued expenses payable to third parties and incurred in the ordinary course of our Compound Partnering Business, which as of December 31, 2009, totaled approximately $4.6 million. We expect that this cash contribution will fund Furiex’s operations and working capital requirements for at least 12 months after the closing of the spin-off, based on current operating plans. In addition to this cash contribution, we expect to receive future payments from our existing collaborations that will provide additional support for our operations and working capital requirements. We will assess our capital needs on a regular basis and might raise additional capital as deemed necessary.
Formation of a Holding Company Prior to the Spin-Off
In connection with and prior to the spin-off, PPD organized Furiex in October 2009 as a Delaware corporation for the purpose of transferring to Furiex the Compound Partnering Business and completing the spin-off.
Results of the Spin-Off
Following the spin-off, Furiex will be an independent, publicly traded company owning and operating what had previously been PPD’s Compound Partnering Business. We expect to have approximately million shares of our common stock issued and outstanding immediately following the spin-off based on the distribution
31
Table of Contents
ratio described above and the anticipated number of outstanding shares of PPD common stock on [ ], 2010, the record date. The actual number of shares to be distributed will be determined based on the number of shares of PPD common stock outstanding on the record date and will be reduced to the extent that cash payments are to be made in lieu of the issuance of fractional shares of our common stock and to the extent that shares of our common stock are held back and sold on the market to satisfy backup withholding taxes and non-U.S. holder dividend withholding taxes and brokerage and other costs, and may be increased if PPD option holders exercise any stock options prior to the record date.
Material U.S. Federal Income Tax Consequences of the Distribution
The following is a summary of the material U.S. federal income tax consequences of the distribution and is based on the Code, the Treasury regulations promulgated thereunder, and interpretations of the Code and Treasury regulations by the courts and the Internal Revenue Service, all as they exist as of the date of this Information Statement. This summary does not discuss all tax considerations that may be relevant to PPD shareholders in light of their particular circumstances, nor does it address the consequences to PPD shareholders subject to special treatment under the U.S. federal income tax laws, such as tax-exempt entities, nonresident alien individuals, non-U.S. entities, non-U.S. trusts and estates and beneficiaries thereof, persons who acquire PPD common stock pursuant to the exercise of employee stock options or otherwise as compensation, insurance companies and dealers in securities. In addition, this summary does not address the U.S. federal income tax consequences to PPD shareholders who do not hold their PPD common stock as a capital asset or any state, local or non-U.S. tax consequences of the transactions.
You should consult your tax advisor as to the specific tax consequences of the distribution to you, including the effect of any state, local or non-U.S. tax laws and of changes in applicable tax laws.
Material U.S. Federal Income Tax Consequences of the Distribution to PPD and Shareholders of PPD
PPD is seeking a private letter ruling from the Internal Revenue Service and an independent tax opinion substantially to the effect that, for U.S. federal income tax purposes, the distribution will qualify as tax-free to PPD and its shareholders under Sections 368(a)(1)(D) and 355 of the Code. The private letter ruling and tax opinion are expected to provide that:
| • | no gain or loss will be recognized by PPD for U.S. federal income tax purposes as a result of the distribution; |
| • | no gain or loss will be recognized by, or be included in the income of, a holder of shares of PPD common stock for U.S. federal income tax purposes solely as the result of the receipt of shares of our common stock in the distribution, except that gain or loss generally will be recognized with respect to any cash received in lieu of fractional shares; |
| • | for U.S. federal income tax purposes, the basis of the PPD common stock and our common stock in the hands of PPD shareholders immediately after the distribution, including any fractional share interest for which cash is received, will be the same as the basis of the PPD common stock immediately before the distribution, and will be allocated among the PPD common stock and our common stock, including any fractional share interest for which cash is received, in proportion to their relative fair market values on the date of the distribution; |
| • | the holding period for U.S. federal income tax purposes of shares of our common stock received by a PPD shareholder, including any fractional share interest for which cash is received, will include the holding period of the shareholder’s PPD common stock with respect to which our shares were issued, provided that such PPD shares are held as a capital asset on the date of the distribution; and |
32
Table of Contents
| • | a PPD shareholder who receives cash in lieu of a fractional share in the distribution will be treated as having sold such fractional share for cash and generally will recognize gain or loss for U.S. federal income tax purposes in an amount equal to the difference between the amount of cash received and the PPD shareholder’s adjusted tax basis in the fractional share. That gain or loss will be a capital gain or loss if the PPD common stock is held as a capital asset and a long-term capital gain or loss if the shareholder’s holding period for its PPD common stock exceeds one year. |
The private letter ruling and independent tax opinion are also expected to provide that certain internal transactions undertaken in anticipation of the separation will qualify for favorable tax treatment under the Code.
Material U.S. Federal Income Tax Consequences to PPD and Shareholders of PPD if the Distribution is Taxable
Although a private letter ruling is generally binding on the Internal Revenue Service, it will be based on assumptions and representations made by us and PPD that certain conditions that are necessary to obtain favorable tax treatment under the Code have been satisfied. Thus, the private letter ruling will not constitute an independent determination by the Internal Revenue Service that these conditions have been satisfied. If the factual representations and assumptions are incorrect in any material respect at the time of the distribution, the private letter ruling could be revoked retroactively or modified by the Internal Revenue Service. We are not aware of any facts or circumstances, however, that would cause these representations or assumptions to be untrue or incomplete in any material respect.
If, notwithstanding the conclusions expected to be provided in the private letter ruling and independent tax opinion, it is ultimately determined that the distribution does not qualify as tax-free for U.S. federal income tax purposes, then PPD would recognize gain in an amount equal to the excess of the fair market value of our common stock distributed to PPD shareholders on the distribution date over PPD’s tax basis in such shares.
In addition, if, notwithstanding the conclusions expected to be provided in the private letter ruling and the independent tax opinion, it is ultimately determined that the distribution does not qualify as tax-free for U.S. federal income tax purposes, then each shareholder that is subject to U.S. federal income tax and who receives our common stock in the distribution could be treated as receiving a taxable distribution in an amount equal to the fair market value of such shares. You could be taxed on the full value of the shares that you receive, without reduction for any portion of your basis in your PPD common stock, as a dividend for U.S. federal income tax purposes to the extent of your pro rata share of PPD’s current and accumulated earnings and profits, including earnings and profits resulting from PPD’s recognition of gain on the distribution. Under Treasury regulations, distributions are presumed to be taxable dividends for U.S. federal income tax purposes unless or to the extent we can demonstrate that the distributions are not from earnings and profits computed under U.S. federal income tax principles. If the value of our distributed stock is less than PPD’s accumulated earnings and profits through the end of the year of distribution, PPD’s earnings and profits for the year of the distribution will be as great as the value of our distributed stock, so the entire value of a taxable distribution generally would be taxable as a dividend. If the value of our distributed stock exceeds PPD’s accumulated earnings and profits through the end of the year of distribution, only a portion of the distribution would be taxable as a dividend. Under current law, assuming certain holding period and other requirements are met, individual citizens or residents of the United States are subject to U.S. federal income tax on dividends at a maximum rate of 15%. Amounts in excess of your pro rata share of PPD’s current and accumulated earnings and profits could be treated as a nontaxable return of capital to the extent of your basis in your PPD common stock and thereafter as capital gain, assuming you hold your PPD common stock as a capital asset. Under current law, individual citizens or residents of the United States are subject to U.S. federal income tax on long-term capital gains (that is, capital gains on assets held for more than one year) at a maximum rate of 15%. Certain PPD shareholders would be subject to additional special rules governing taxable distributions, such as those that relate to the dividends received deduction and extraordinary dividends. A shareholder’s income tax basis in our common stock received in a taxable distribution generally would equal the fair market value of our common stock on the distribution date, and the holding period for those shares would begin the day after the distribution date. The holding period for the shareholder’s PPD common stock would not be affected by the fact that the distribution was taxable.
33
Table of Contents
Even if the distribution otherwise qualifies for tax-free treatment under Sections 368(a)(1)(D) and 355 of the Code, it may result in corporate-level taxable gain to PPD under Section 355(e) of the Code if 50% or more, by vote or value, of our shareholder equity or PPD’s shareholder equity is acquired or issued as part of a plan or series of related transactions that includes the distribution. For this purpose, any acquisitions or issuances of PPD’s shareholder equity within two years before the distribution, and any acquisitions or issuances of our shareholder equity or PPD’s shareholder equity within two years after the distribution, generally are presumed to be part of such a plan, although we or PPD may be able to rebut that presumption. We are not aware of any such acquisitions or issuances of PPD’s shareholder equity within the two years before the distribution. If an acquisition or issuance of our shares or PPD’s shares triggers the application of Section 355(e) of the Code, PPD would recognize taxable gain as described above, and could incur significant U.S. federal income tax liabilities as a result of the application of Section 355(e) of the Code.
Material U.S. Federal Income Tax Consequences if the Internal Transactions are Taxable
If, notwithstanding the conclusions expected to be provided in the private letter ruling and tax opinion, it is ultimately determined that certain internal transactions undertaken in anticipation of the separation do not qualify for favorable tax treatment, we or PPD would incur significant tax liabilities.
Material Consequences under the Tax Sharing Agreement if the Distribution or the Internal Transactions are Taxable
In connection with the distribution, we and PPD will enter into a Tax Sharing Agreement pursuant to which we and PPD will agree to be responsible for certain tax liabilities and obligations following the distribution. Our indemnification obligations will include a covenant to indemnify PPD for any taxes and costs that they incur as a result of any action, misrepresentation or omission by us that causes the distribution or the internal transactions undertaken in anticipation of the separation to fail to qualify for favorable tax treatment under the Code. In addition, PPD will similarly agree to indemnify us for any taxes or costs that they cause us to incur as a result of their actions, misrepresentations or omissions that causes the distribution or the internal transactions to fail to qualify for favorable tax treatment under the Code. PPD will be responsible for any taxes resulting from the failure of the distribution or any internal transactions to qualify for favorable tax treatment under the Code, if the failure is not due to the actions, misrepresentations or omissions of us or PPD. In addition, even if we were not contractually required to indemnify PPD for tax liabilities if the distribution or the internal transactions were to fail to qualify for favorable tax treatment under the Code, we nonetheless might be legally liable under applicable U.S. federal income tax law for certain U.S. federal income tax liabilities incurred by PPD or U.S. affiliates of PPD if they were to fail to pay such tax liabilities.
Information Reporting by PPD Shareholders
Current U.S. Treasury regulations require each PPD shareholder that owns at least 5% of the total outstanding stock of PPD and receives stock in the distribution to attach to its United States federal income tax return for the year in which the distribution occurs a detailed statement containing certain information relating to the tax-free nature of the distribution. Upon request, PPD will provide shareholders of 5% or more of PPD’s outstanding stock who received Furiex common stock in the distribution with any pertinent information that is in PPD’s possession and is reasonably available, to the extent necessary to comply with that requirement.
The foregoing is a summary of material U.S. federal income tax consequences of the distribution under current law and is for general information only. The foregoing does not purport to address all U.S. federal income tax consequences or tax consequences that might arise under the tax laws of other jurisdictions or apply to particular categories of shareholders. Each PPD shareholder should consult his, her or its tax advisor as to the particular tax consequences of the distribution to such shareholder, including the application of U.S. federal, state, local and foreign tax laws, and the effect of possible changes in tax laws that might affect the tax consequences described above.
34
Table of Contents
Market for Our Common Stock; Trading of Our Common Stock in Connection with the Spin-Off
There is currently no trading market for our common stock. We have applied to have our common stock authorized for listing on the Nasdaq Global Market under the symbol “FURX”. We expect that a limited market, commonly known as a “when-issued” trading market, for our common stock will develop on or shortly after [ ], 2010, the record date of the distribution. The term “when-issued” means that our shares will trade even though PPD has not yet issued and distributed the Furiex shares. “When-issued” trading in our common stock will end and “regular way” trading will begin either on the distribution date or the first trading date after the distribution date. “Regular way” trading with respect to our common stock refers to trading after PPD has issued and distributed Furiex shares to PPD’s shareholders. Neither PPD nor we will set the initial trading price of our common stock; the public markets will establish our trading price.
We cannot predict the price at which our common stock will trade either in the “when-issued” trading market or in the “regular way” trading market after the spin-off. In fact, the combined trading prices of a share of our common stock, adjusted for the distribution ratio, and a share of PPD common stock after the spin-off might not equal or exceed the trading price of a share of PPD common stock immediately prior to the spin-off. The price at which our common stock trades is likely to fluctuate significantly, particularly until an orderly public market develops, if it ever does. Prices for our common stock will be determined in the public markets and might be influenced by many factors, many of which are beyond our control, including:
| • | lack of a trading history; |
| • | changes in expectations concerning our future financial performance and the future performance of the pharmaceutical industry in general, including financial estimates and recommendations by securities analysts; |
| • | the progress of planned and existing clinical trials involving our product candidates; |
| • | the receipt or denial of regulatory approval for any of our product candidates; |
| • | announcements regarding existing or new collaborations; |
| • | our financial results and differences between our actual financial and operating results and those expected by investors and analysts; |
| • | strategic moves by us or our competitors, such as acquisitions or restructurings; |
| • | changes in the regulatory environment governing our business; |
| • | our capital structure, including the amount of any indebtedness; |
| • | general economic, industry and market conditions; |
| • | the depth and liquidity of the market for our common stock; |
| • | investor and analyst perceptions of our business and us; and |
| • | the impact of the factors referred to in “Risk Factors”. |
We have appointed American Stock Transfer & Trust Company, LLC to serve as transfer agent and registrar for our common stock.
Shares of our common stock distributed to holders of PPD common stock in connection with the spin-off will be transferable under the Securities Act, except for shares received by persons who are our affiliates. Persons who are our affiliates after the spin-off generally include individuals or entities that control, are controlled by or
35
Table of Contents
are under common control with us and might include some of our officers, directors or principal shareholders. After we become a publicly traded company, securities held by our affiliates will be subject to the resale restrictions under the Securities Act. Our affiliates will be permitted to sell shares of our common stock only pursuant to an effective registration statement or an exemption from the registration requirements of the Securities Act, such as the exemption afforded by Rule 144 under the Securities Act.
Trading of PPD Common Stock After the Record Date and Prior to the Ex-Dividend Date
Beginning on the record date and through the day prior to the ex-dividend date, PPD common stock will trade “regular way” with the symbol “PPDI”. On the “regular way” market, from the record date through the date before the ex-dividend date set by Nasdaq, shares of PPD common stock will trade with an entitlement to shares of our common stock distributed in connection with the spin-off. Beginning on the ex-dividend date, shares of PPD common stock will trade without an entitlement to shares of our common stock distributed in connection with the spin-off. Therefore, if you own shares of PPD common stock at 5:00 p.m. Eastern Time on the record date and sell those shares on the regular way market prior to the ex-dividend date, you also will be selling your right to receive the shares of our common stock that would have been distributed to you in connection with the spin-off. If you hold those shares of PPD common stock held on the record date through the ex-dividend date, then PPD will distribute to you shares of our common stock with respect to your ownership of those shares of PPD common stock, even if you sell the shares of PPD common stock thereafter.
During the time period between the record date and the ex-dividend date, we anticipate that PPD common stock will also be available to trade on an “ex-distribution when-issued market “with the symbol “PPDIV”. On an “ex-distribution” market, shares of PPD common stock would trade without an entitlement to shares of our common stock distributed in connection with the spin-off. The “ex-distribution when-issued market” will cease to exist as of the ex-dividend date.
Distribution Conditions and Termination
We expect that the distribution will be effective, and the spin-off complete, on the distribution date, [ ], 2010, provided that, among other things:
| • | the SEC has declared effective our registration statement on Form 10, of which this Information Statement is a part, under the Exchange Act and no stop order relating to our Form 10 registration statement is in effect; |
| • | PPD and Furiex have received all permits, registrations and consents required under the securities or blue sky laws of states or other political subdivisions of the United States or of foreign jurisdictions in connection with the distribution; |
| • | PPD and Furiex have received all permits, registrations, clearances and consents from governmental authorities and third persons necessary to effect the spin-off and to permit the operation of our businesses thereafter; |
| • | the Nasdaq Global Market has approved our common stock for listing, subject to official notice of issuance; |
| • | the Internal Revenue Service has issued a favorable private letter ruling and we have received an independent tax opinion to the effect that the distribution will qualify as a tax-free reorganization for U.S. federal income tax purposes under Sections 368(a)(1)(D) and 355 of the Code; |
| • | no order, injunction or decree issued by any court of competent jurisdiction or other legal restraint or prohibition preventing consummation of the spin-off or any of the transactions related thereto, including the transfers of assets and liabilities contemplated by the Separation and Distribution Agreement, is in effect; |
36
Table of Contents
| • | no other events or developments shall have occurred that, in the judgment of the Board of PPD, in its sole and absolute discretion, would result in the spin-off having a material adverse effect on PPD or its shareholders; and |
| • | the PPD Board gives its final approval. |
The fulfillment of these conditions will not create any obligation on PPD’s part to effect the distribution, and the Board of PPD can amend, modify or abandon the distribution and the related transactions at any time prior to the distribution date. The Board of PPD may waive certain of these conditions in its sole and absolute discretion. However, we are not aware of any material federal or state regulatory requirements that must be complied with or any material permits, registrations or approvals that must be obtained, other than compliance with SEC rules and regulations and the declaration of effectiveness of our registration statement by the SEC, in connection with the distribution. In addition, we have received all necessary third party consents for the transfer of assets from PPD to Furiex as part of the spin-off. We believe that practical considerations would prevent PPD from waiving the requirement that we receive the private letter ruling and an independent tax opinion regarding the tax treatment of the spin-off. Further, PPD cannot waive the condition that the SEC declare our registration statement effective or that our common stock be approved for listing on Nasdaq. While PPD has the ability to waive the condition that there not be an order, injunction or decree that prohibits or prevents the consummation of the spin-off, PPD would not waive this condition because it would be illegal to do so.
Reason for Furnishing this Information Statement
This Information Statement is being furnished solely to provide information to shareholders of PPD who will receive shares of Furiex common stock in connection with our spin-off. It is not provided as an inducement or encouragement to buy or sell any of our securities. You should not assume that the information contained in this Information Statement is accurate as of any date other than the date set forth on the cover. Changes to the information contained in this Information Statement might occur after that date, and we undertake no obligation to update the information.
37
Table of Contents
The following table sets forth Furiex’s capitalization as of December 31, 2009 on a historical basis and on a pro forma basis to give effect to the pro forma adjustments included in our unaudited pro forma condensed combined balance sheet. The pro forma adjustments are based upon available information and assumptions that management believes are reasonable; however, such adjustments are subject to change based on the finalization of the terms of the spin-off and the transaction agreements. In addition, such adjustments are estimates and might not prove to be accurate or indicative of future adjustments.
You should read this table together with “Management’s Discussion and Analysis of Financial Condition and Results of Operations”, “Unaudited Pro Forma Condensed Combined Financial Statements” and our historical combined financial statements and the notes to those statements included elsewhere in this Information Statement.
| December 31, 2009 | |||||||
| Historical | Pro Forma | ||||||
| (In thousands, except share amounts) | |||||||
Cash and cash equivalents | $ | 6 | $ | 100,006 | (1) | ||
Shareholders’ equity: | |||||||
Preferred stock, $0.001 par value; none authorized, issued and outstanding; 10 million shares authorized pro forma; none issued and outstanding pro forma | — | — | |||||
Common stock, $0.001 par value; 40 million shares authorized; shares issued and outstanding; 40 million shares authorized pro forma; million shares issued and outstanding pro forma | (2 | ) | |||||
Additional paid-in capital | |||||||
Accumulated other comprehensive loss | |||||||
Pharmaceutical Product Development, Inc. net investment | 49,270 | 145,930 | |||||
Total shareholders’ equity | 49,270 | 145,930 | |||||
Total capitalization | $ | 49,270 | $ | 145,930 | |||
| (1) | Represents the contribution of $100.0 million in connection with the spin-off of the Compound Partnering Business. |
| (2) | Represents the distribution of million shares of our common stock to holders of PPD common stock based on the number of shares of PPD common stock outstanding at December 31, 2009. |
We do not currently anticipate paying any dividends for the foreseeable future. The declaration and payment of dividends are subject to the discretion of our Board. Any future determination to pay dividends will depend on our financial condition, earnings, capital requirements, legal requirements, regulatory constraints, contractual restrictions and other factors deemed relevant at the time by our Board.
38
Table of Contents
Furiex is a drug development collaboration company that will continue the Compound Partnering Business started by our parent company, PPD, in 1998.
The goal of compound partnering is to in-license from or form strategic alliances with pharmaceutical and biotechnology businesses to develop and commercialize therapeutics in which the risks and rewards are shared. We seek to collaborate with pharmaceutical and biotechnology companies to increase the value of their early stage drug candidates by applying our novel approach to drug development that we believe expedites research and development decision-making and can shorten drug development timelines. We share risk with our collaborators by financing the cost of development to the point of mutually agreed upon pre-determined clinical milestones. Furiex’s team is staffed with the same key PPD team members who demonstrated proven success in the drug development collaboration business while at PPD, as well as highly-qualified new members. Our strategy is to invest in drug candidates that have a relatively straightforward path to regulatory approval and a large potential addressable market. Every drug candidate we review is subjected to our rigorous due diligence process by our team of experts who possess experience in all aspects of the drug development process.
Once we in-license or form an alliance, we use our drug development experience and financial resources to advance the drug candidate through clinical development. Furiex applies a novel approach that shortens drug development timelines that we believe transforms research and development into revenues more rapidly than the typical development cycle for such collaborations. Specifically, Furiex sets the development strategy based on a product candidate’s best market position, designs and manages nonclinical and clinical studies, manages the drug manufacturing programs, and evaluates the efficacy and safety data necessary to obtain regulatory approvals for the drug candidate. Furiex will use service providers to execute the tasks needed to develop and commercialize its product candidates.
Most of the large pharmaceutical companies with which we collaborate have the option to continue late stage clinical development and commercialization of the drug candidate after it has reached the specified pre-determined milestones. If our collaborator is unable or unwilling to perform late stage development and commercialization, then we have the option to seek out a new development and commercialization partner.
In exchange for our drug development efforts and sharing the risk with our collaborator, we are entitled to receive milestone payments and royalties based on the continued development and commercialization success of the drug candidate.
Currently, we have rights in several compounds in various stages of development and commercialization, including:
| • | Rights to royalties and sales-based milestones from the collaboration with ALZA Corporation, a Janssen-Cilag affiliate, on Priligy, the first approved treatment in the world for premature ejaculation. To date, Priligy has been approved for marketing in Austria, Finland, Germany, Italy, Mexico, New Zealand, Portugal, South Korea, Spain and Sweden. |
| • | In June and September 2009, the FDA issued complete response letters to the NDAs covering the alogliptin monotherapy and the fixed-dose combination of alogliptin and Actos. A complete response letter indicates that the review cycle for an application is complete and that the application can not be approved in its present form, and informs sponsors of changes that must be made before an application can be approved, with no implication as to the ultimate approvability of the application. In the complete response letters, the FDA requested an additional cardiovascular safety trial with the alogliptin monotherapy be completed prior to approval of both NDAs. The cardiovascular trial requested by the FDA is ongoing. |
| • | A fluoroquinolone antibiotic compound we licensed from Janssen Pharmaceutica, N.V., an affiliate of Johnson & Johnson, in November 2009 for the treatment of complicated skin and skin structure |
39
Table of Contents
infections, such as abscesses that occur deep in the skin layers, and respiratory infections. This antibiotic has a broad spectrum of activity and is able to treat MRSA infections. Based on pre-clinical in vitro data, we believe this product may show superior bacteriocidal activity, compared to currently marketed antibiotics, for a number of difficult-to-treat pathogens. We are developing both oral and IV formulations of this compound to allow doctors and patients to select the appropriate method of delivery under the circumstances. We have initiated work with the goal of beginning a Phase I study with the IV formulation in the first half of 2010. We are planning to initiate Phase II clinical trials using the oral formulation for complicated skin infections and using both formulations for respiratory infections in 2010. |
| • | A compound that is a mu opioid receptor agonist and delta opioid receptor antagonist, which we call mu delta, we licensed from Janssen Pharmaceutica, N.V. in November 2009 for the treatment of diarrhea-predominant irritable bowel syndrome. We have begun work with the goal of initiating Phase II clinical trials during the second quarter of 2010. |
| • | A novel statin compound we refer to as PPD 10558 licensed from Ranbaxy Laboratories, Ltd. for the treatment of dyslipidemia, which is an excessive level of blood lipids such as cholesterol. Preclinical data suggests this statin will avoid some of the adverse interactions seen when currently marketed statins are taken with some other drugs by patients with high blood lipid levels. In addition, one of the most common side effects of statin usage is a condition of pain and/or muscle weakness, known as statin-associated myopathy, which is reported to occur in up to 10% of statin-treated patients. Preclinical and Phase I human studies suggest that PPD 10558 has similar cholesterol-lowering properties as a leading marketed statin, and also has pharmacologic properties which suggest that it may have a lower risk of myopathy than currently marketed statins. PPD 10558 may therefore be a useful treatment option for statin-intolerant patients. PPD completed a high-dose comparator study in healthy volunteers that indicated the drug was well-tolerated and suggested it compares favorably to currently marketed statins with respect to lowering lipid levels. |
| • | A variety of therapeutic candidates developed by Eli Lilly and Company that we obtained through our acquisition of Magen BioSciences, Inc. in April 2009, including vitamin D receptor modulators. We have tested two of these compounds for various safety and efficacy parameters for topical treatment of dermatology indications in the anti-proliferative and anti-inflammatory areas. The antiproliferative and anti-inflammatory dermatological conditions include psoriasis, atopic dermatitis, chronic dermatitis, acne, rosacea, actinic keratosis and skin cancer. We have elected, however, not to pursue the development of these compounds. We also are currently investigating other compounds under material transfer agreements to identify potential drug development candidates for dermatological indications. We also are considering other strategic alternatives for our dermatology business. |
40
Table of Contents
The following chart summarizes our compounds:
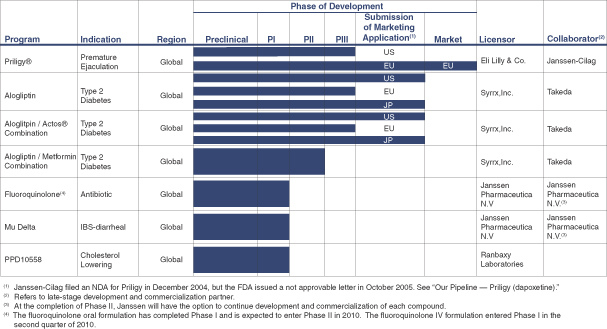
Drug Development Industry
Over the past two decades, technological advances have dramatically changed the drug discovery process. New and improved technologies have evolved such as ultra high-throughput screening, newin vitroandin vivopreclinical profiling techniques, biomarker research and genomics. These processes have produced many more molecules with potential viability as drug candidates. In addition, the U.S. biotechnology industry has grown rapidly and generated significant numbers of new drug candidates for development and regulatory approval. Consequently, the number of drug compounds in various stages of development has increased steadily over the past five years, with particularly strong growth in the number of compounds in early stages of development.
While research and development spending and the number of drug candidates are increasing, the time and cost required to develop a new drug candidate have also increased. According to the Tufts Center for the Study of Drug Development and PAREXEL International Corporation, the fully capitalized cost of developing and commercializing a new pharmaceutical product typically ranges from more than $900.0 million to over $1.0 billion. In addition, it generally takes approximately 12 years to develop a new prescription drug from preclinical studies through FDA approval to market in the United States.
Concurrently, pharmaceutical companies are also facing multiple upcoming patent expirations, with approximately $60 billion of at-risk sales projected in the U.S alone through 2011 according to “Combating Generics: Counter-Generics Strategy, Tactics and Execution,” a report published by Research and Markets. Biotechnology companies are also facing challenges, finding it difficult to attract the funding needed to sustain operations and advance clinical candidates, particularly in the wake of the global financial crisis.
As a result, many pharmaceutical and biotechnology companies do not have the necessary staff, capacity, or financial resources to independently develop the large numbers of molecules in their pipelines. In this challenging environment, Furiex offers an attractive partnership proposition, presenting a more cost-effective, innovative and rapid means of developing drug candidates, with incremental investment and risk-sharing to mitigate the costs and challenges of the drug development process.
41
Table of Contents
Our Solution
To address these industry issues, we have developed what we believe is a novel approach to drug development. Our approach to drug development involves applying proven solutions from our extensive global experience to reduce development timelines and expedite the decision-making cycle, planning for success and bridging steps in development by conducting earlier elements of a program while simultaneously planning for later phases.
In order to obtain regulatory approval from the FDA to market a drug, certain data about the safety and efficacy of the drug is required by the FDA. To obtain such data, drug developers frequently choose to run studies sequentially. For example, they may run one study, wait to see the results, and then they run the next study. Developers prefer this approach primarily to limit upfront expenditures since the success of any given study is not known and the decision may be made not to move forward due to negative data. This sequential approach slows down the development process. In addition, development is slowed due to time taken at each decision point at the end of each study.
We approach drug development by minimizing the time it takes to bring products to the market. Our novel approach manages drug development with parallel processing and efficient decision-making. We use our drug development experience to predict possible outcomes of a study and take risks based on those predictions. By assuming in advance success at each critical decision point, as opposed to waiting for results to demonstrate success in order to make a decision, time is reduced. In addition, we seek to mitigate risks by contingency planning for potential problems. As a result, we can accelerate the development process by bridging steps across the developmental program as well as between studies, as was evidenced with alogliptin where it took just five years from the start of our collaboration to the filing of the NDA. Additionally, we focus our efforts on the essential studies necessary for regulatory approval and we do not include extraneous studies. This helps to shorten developmental timelines while still achieving success.
Two key elements to our approach are our due diligence process and our planning for the success of each compound. Before we enter into a collaboration for a compound, we subject it to an intense due diligence review covering every step in the development process, from preclinical and clinical studies through marketing. We generally look for and enter into collaborations with respect to compounds that appear to have the following characteristics:
| • | large market potential; |
| • | a straightforward regulatory path; |
| • | a reasonable development time; |
| • | reasonable predictability of non-clinical models; |
| • | clinical evidence no later than Phase Ib; |
| • | a solid patent estate; |
| • | acceptable estimated cost of goods; and |
| • | attractive economic terms with the compound’s innovator and ultimate commercial collaborator. |
If a compound meets our diligence review standards, we then plan the entire development timeline upfront, using a set of assumptions and, in the event those assumptions do not materialize, a set of reasonable contingency plans. Part of the upfront planning involves initiating long-term studies, such as carcinogenicity studies, earlier than usual. We also use real-time data analysis tools to monitor the clinical study data of a drug candidate. By initiating long-term studies earlier and reviewing data in real time, we can significantly reduce the time needed after the conclusion of clinical studies to complete the necessary documentation for regulatory filing.
42
Table of Contents
We believe this approach works well because the core development team is empowered to make decisions, real-time technology tools facilitate rapid data review, development programs are designed to optimize market position, and timelines are driven by science and “must have” studies. The resulting ability to reduce development timelines in turn allows us to capitalize more quickly on our investment. We believe our success evolves from our development efficiency.
According to the Tufts University Center for the Study of Drug Development Outlook 2009, since 2002 the average time from the filing of an Investigational New Drug application, or IND, to the filing of an NDA is over eight years. By contrast, we advanced alogliptin as a treatment for type 2 diabetes (for the monotherapy program) from IND to NDA in only 39 months.
We believe our nearly 12 years of development experience has earned us a reputation in the pharmaceutical industry such that pharmaceutical companies approach us as a potential compound collaborator. We believe our most recent partnering project, which we entered into in November 2009 with Janssen Pharmaceutica, evolved from our reputation for success.
Our Strategy
Our strategy is to leverage our drug development experience to in-license, develop and commercialize novel early-stage drug candidates that address medical conditions with large markets and demand for new medicines. The key elements of our strategy include:
| • | Advance the clinical development of our current drug candidates. We plan to advance drug candidates based on the results of preclinical and clinical testing and assessment of market potential. Specifically, we intend to initiate the Phase II clinical trials for the fluoroquinolone antibiotic and the mu delta compound in 2010. |
| • | In-license or acquire additional early-stage compounds. The in-licensing of additional early-stage drug candidates in order to expand our portfolio and therefore increase long-term revenue opportunities is an objective of our company. We may consider acquisitions to accomplish this objective, although we have no current agreements for any such transactions. |
| • | Establish strategic commercialization collaborations.We plan to develop and commercialize our drug candidates through partnering or licensing arrangements like those with ALZA for Priligy and Takeda for alogliptin. This strategy allows us to focus on our core development strengths and leverage the resources and commercialization capabilities already in place at our collaborators. |
Our Pipeline
A major area of our business is the pursuit and maintenance of strategic collaborations and licensing activities to help us execute our strategy and share the costs and mitigate the risks of drug development. Our strategic collaborations are described in greater detail below.
Priligy (dapoxetine)
PPD entered into its first compound partnering arrangement in 1998 when it acquired an exclusive license, as part of a development collaboration with Eli Lilly and Company, to develop and commercialize the compound dapoxetine for genitourinary indications, including premature ejaculation. In December 2003, PPD acquired Lilly’s patents and remaining rights to develop and commercialize dapoxetine in the field of genitourinary disorders. PPD also agreed to pay Lilly a royalty of 5% on annual sales of dapoxetine, if any, in excess of $800.0 million. PPD developed the compound through Phase II proof-of-concept and, in January 2001, out-licensed it to ALZA, which is now part of Johnson & Johnson. ALZA is responsible for all clinical, regulatory, manufacturing,
43
Table of Contents
sales and marketing costs associated with the compound. As of December 31, 2009, we had received $35.9 million in combined development and regulatory milestone payments and sales-based royalties. If the related triggering events and product sales occur, we are entitled to future regulatory milestone payments of $ million, sales-based milestones of up to $50.0 million, and sales-based royalties ranging in the low double digit percentages depending on product sales volumes.
In December 2004, Janssen-Cilag, an affiliate of ALZA, submitted an NDA to the FDA for dapoxetine. Janssen-Cilag received a “not approvable” letter from the FDA in October 2005, but continued its global development program. In 2009, Austria, Finland, Germany, Italy, Mexico, New Zealand, Portugal, South Korea, Spain and Sweden approved the drug for marketing under the name Priligy. Janssen-Cilag continues to seek approval for Priligy in other countries and is investigating regulatory strategies for a potential refiling with the FDA.
No products are currently approved for the treatment of premature ejaculation in the U.S. We estimate that approximately 29 million men in the U.S. between the ages of 25 and 80 suffer from premature ejaculation. Our estimate is based on a U.S. male population between the ages of 25 and 80 (approximately 96 million males), as estimated for 2009 by the U.S. Census Bureau, based on data from the 2000 Census. We then multiplied the 96 million males by 30%, which is the percentage of the U.S. male population that the National Health and Social Life Survey (published in 1994) indicates suffers from premature ejaculation.
PPD will contribute to us all its rights and obligations with respect to dapoxetine. ALZA has worldwide rights to develop and commercialize dapoxetine. In addition to all the rights to receive milestone payments and royalties, Furiex will receive the following material rights and/or obligations:
| • | to prosecute patent applications and maintain granted patents, and allow ALZA to comment on such prosecution; |
| • | to notify ALZA of any actual, potential or suspected infringement of licensed patents by a third party of which Furiex becomes aware and of any claim of infringement of a third party’s proprietary rights of which Furiex receives notice with regard to the license grant; |
| • | to not publish any data regarding dapoxetine; |
| • | to not assert any patent or other intellectual property right owned by PPD against ALZA; |
| • | to indemnify ALZA for various claims and losses arising out of or under the agreement; |
| • | to defend licensed patents against infringers (in the event ALZA does not desire to defend) and be entitled to all recoveries, damages or awards if it defends. |
Alogliptin
In 2003, PPD made an investment in Syrrx, Inc., a privately held drug discovery company, and entered into a collaboration agreement to develop Syrrx’s orally active DPP4 inhibitors to treat type 2 diabetes and other major human diseases. In March 2005, Takeda Pharmaceutical Company Limited acquired Syrrx. In July 2005, Takeda acquired development and commercialization rights to these DPP4 inhibitors from PPD for an upfront payment, potential milestone payments and royalties associated with the future development and commercialization of specified DPP4 inhibitors and the right to serve as the sole provider of clinical and bioanalytical services to Takeda for Phase II and Phase III trials of DPP4 inhibitors conducted in the United States and Europe. PPD will retain the right to provide CRO services.
In December 2007, Takeda submitted an NDA for alogliptin to the FDA. In September 2008, Takeda submitted an NDA for alogliptin in Japan. In September 2008, Takeda also submitted an NDA for a fixed dose
44
Table of Contents
product containing alogliptin and Actos to the FDA. In June 2009, the FDA issued a complete response to Takeda on its alogliptin NDA and requested Takeda to conduct an additional cardiovascular safety trial that satisfies the FDA’s December 2008 guidance on anti-diabetes therapies. In September 2009, the FDA issued a complete response to Takeda on its NDA for the fixed dose combination of alogliptin and Actos, stating that further review would be dependent on the cardiovascular safety data that would be submitted in support of the alogliptin monotherapy NDA. This trial is presently ongoing. As of December 31, 2009, we had received $48.0 million in development and regulatory milestone payments. If the related triggering events and product sales occur, we are entitled to receive future regulatory milestones of up to $52.5 million, sales-based milestones of up to $33.0 million, and sales-based royalties ranging from the mid single digit to low double digit percentages depending on product sales volumes.
Takeda is awaiting issuance of final European Medicines Agency, or EMEA, guidance with respect to cardiovascular safety requirements for its Type 2 diabetes drugs. The EMEA issued draft guidance with respect to cardiovascular safety requirements for its Type 2 diabetes drugs on February 10, 2010. In the event Takeda files a marketing authorization application to the EMEA, we will have the right to receive a $10.0 million milestone payment from Takeda.
The American Diabetes Association estimates that there are approximately 24 million people in the U.S. with type 1 and type 2 diabetes. The World Health Organization estimates that more than 170 million people worldwide have type 1 and type 2 diabetes and that the number will double by 2030. 2008 worldwide sales of antidiabetic treatments were $27.3 billion.
In addition to all the rights to receive milestone payments and royalties, Furiex will receive the following material rights and/or obligations:
| • | to indemnify Takeda for various claims and losses arising out of or under the agreement; and |
| • | to not discover, develop, or commercialize any product directed to the DPP4 inhibitors. |
Janssen Pharmaceutica N.V. Collaboration
In November 2009, we entered into agreements with Janssen Pharmaceutica N.V., an affiliate of Johnson & Johnson, to develop and commercialize two Phase II-ready therapeutic compounds. The mu delta compound will be studied as a treatment for diarrhea-predominant irritable bowel syndrome, or IBS-d. The fluoroquinolone compound will be studied as a treatment for complicated skin and skin structure infections such as abscesses that occur deep in the skin layers and respiratory infections. This antibiotic has a broad spectrum of activity and is able to treat methicillin-resistant staphylococcus aureus, or MRSA and pneumonia infections. Under the two agreements, we in-licensed the two compounds and we will advance the compounds through Phase II development. At the completion of Phase II, Janssen Pharmaceutica will have the option to continue development and commercialization of each compound. In exchange, we may receive, for each compound, up to $90.0 million in regulatory milestone payments and up to $75.0 million in sales-based milestone payments, as well as royalties on sales of each compound if approved for marketing. In the event Janssen Pharmaceutica elects not to continue a program, we have the option to continue developing and commercializing the compound for that program and Janssen Pharmaceutica may receive, for each compound, up to $50.0 million in regulatory milestone payments and up to $75.0 million in sales-based milestone payments, as well as sales-based royalties ranging from the mid single to low double digit percentages depending on sales volumes, if approved for marketing. As of December 31, 2009, we had paid Janssen Pharmaceutica $7.0 million in up-front in-licensing payments.
We believe that the market for prescription treatments for IBS-d is underserved due to the limited number of available treatments and the adverse side effects associated with those treatments. The mu delta compound is a potential first-in-class locally active mu opioid receptor agonist and delta opioid receptor antagonist. Based on a market report in Nature Reviews Drug Discovery (February 2006), we estimate that IBS-d affects approximately
45
Table of Contents
29 million Americans age 15 and older. Our estimate is based on a U.S. population over the age of 15 (approximately 245 million people), as estimated for 2009 by the U.S. Census Bureau, based on data from the 2000 Census. We then multiplied the 245 million people by 12%, which is the percentage of the U.S. population that is reported to suffer from IBS-d, as reported in February 2006 by Nature Reviews Drug Discovery (Ashburn T., Gupta M. The IBS Market.).
The fluoroquinolone compound is a broad-spectrum antibiotic with activity against gram negative and gram positive bacteria, including MRSA. It is being developed as both oral and IV therapy to treat skin and respiratory infections. Bacterial infections are a major cause of morbidity and mortality, and antibiotic resistant infections have become a growing public health concern. More than 14 million ambulatory physician visits each year are related to skin and soft-tissue infections, and approximately 94,000 Americans developed serious MRSA infections in 2005, according to a recent study published in the Journal of the American Medical Association. We estimate that the worldwide market for antibiotics to treat MRSA is approximately $1.5 billion annually, based on 2007 sales of $951 million for Zyvox, $274 million for Cubicin, $122 million for Tygacil and $135 million for generic vancomycin (all sales as reported by Business Insights, Ltd., The Anti-Infectives Market Outlook to 2013, published in 2008), which are the products primarily used to treat MRSA.
PPD 10558
In early 2007, PPD exercised its option to acquire an exclusive worldwide license from Ranbaxy Laboratories, Ltd. to develop, manufacture and market its novel statin for the treatment of dyslipidemia. Dyslipidemia is an abnormal level of lipids, or fat, in the blood. Most dyslipidemias are hyperlipidemias, which are increased levels of lipids in the blood, such as high cholesterol. PPD will contribute this agreement and all related intellectual property to us. The preclinical toxicology, drug metabolism and pharmacokinetic data suggest that the statin has the potential to offer an improved safety profile over currently marketed statins as the compound will have less chance for drug-drug interactions and statin-induced myopathy, which is muscle weakness or pain. As of December 31, 2009, we had paid Ranbaxy $1.5 million in up-front and development milestone payments. If the related triggering events and product sales occur, Ranbaxy will be entitled to receive sales-based royalties ranging from the mid to high single digit percentages, as well as sales-based milestone payments of up to $38.0 million and development milestone payments of up to $5.0 million. Ranbaxy has retained co-marketing rights to the compound in India. We will be solely responsible and will bear all costs and expenses for the development, manufacture, marketing and commercialization of the compound and licensed products.
In 2007, PPD filed an IND with the FDA and completed a Phase I single dose study in humans. The compound was safe and well-tolerated at all doses in that trial. PPD also completed a first-in-patient study, and a drug-drug interaction study to evaluate the interaction between its statin and gemfibrozil, a fibrate commonly used to lower triglycerides. PPD completed a high-dose comparator study in healthy volunteers. The drug was well-tolerated and the results suggest the statin compound compares favorably to currently marketed statins with respect to improving lipid levels. We continue to evaluate various alternatives regarding the future development and commercialization of this compound.
The American Heart Association estimates that there are more than 35 million adults in the U.S. with total cholesterol greater than or equal to 240 milligrams per deciliter of blood, or mg/dL, and that there are more than 71 million adults in the U.S. with low-density lipoprotein, or LDL, equal to or greater than 130 mg/dL. The American Heart Association has determined that a total cholesterol level of 240 mg/dL and above presents high risk for heart disease and that an LDL level of between 130 and 159 mg/dL presents borderline high risk of heart disease. In 2007, worldwide sales of lipid regulators amounted to $35.3 billion.
Dermatology Business
In April 2009, PPD acquired Magen BioSciences, Inc., a biotechnology company founded in 2006 focused on dermatologic therapies. This company will be a wholly owned subsidiary of ours. As a result of this acquisition, we
46
Table of Contents
gained advanced screening and research capability for dermatological compounds, which enabled our Compound Partnering Business to expand into dermatological therapies. We have an exclusive license with Eli Lilly and Company to develop and commercialize numerous Vitamin D receptor modulators and an exclusive option to evaluate and negotiate a license for up to six programs in the anti-proliferative and anti-inflammatory areas for topical dermatological indication. In October 2009, we filed an IND on our then lead dermatology drug candidate, a Vitamin D receptor modulator we call MAG-131, and anticipated initiating clinical studies in psoriasis by early 2010. However, we suspended the MAG-131 program due to efficacy data that was discovered in late 2009. We recently completed the testing of another Vitamin D receptor modulator licensed from Lilly and determined not to pursue it for further development. As of December 31, 2009, we had not paid any development or regulatory milestones to Lilly. If the related triggering events and product sales occur, Lilly will be entitled to development and regulatory milestone payments of $21.4 million and sales-based royalties ranging from the mid single digit to low double digit percentages depending on product sales volumes. We also are currently investigating other compounds under material transfer agreements to identify potential drug development candidates for dermatological indications. We are targeting compounds with anti-inflammatory, anti-proliferative, sebum reduction, skin barrier restoration, anti-pruritic and other mechanisms, low oral/systemic bioavailability, rapid systemic metabolism/clearance, and good dermal penetration. We also are considering other strategic alternatives for our dermatology business.
Our Drug Development Capabilities
Our drug development capabilities embody nearly 12 years of experience as a research and development unit within PPD. This experience includes a deep understanding of the biological causes of human diseases and the factors that impact all aspects of successful drug development such as manufacturing, formulation, the cause of drug side effects, drug interactions and drug pharmacokinetics. We believe that our drug development capability and proven success rate will continue to provide a pipeline of unique compounds. Depending upon the availability of our development resources, our preclinical candidates might be added to our own internal clinical pipeline, or out-licensed to other companies for clinical development and commercialization.
Our Patents and Other Proprietary Rights
We own a patent on Priligy in the U.S. that expires in December 2012. We own patents covering the method of dosing of Priligy in Australia, Bulgaria, Canada, China, Czech Republic, Armenia, Azerbaijan, Belarus, Kazakhstan, Moldova, Russia, Tajikistan, Turkmenistan, Japan, Hong Kong, Philippines, Indonesia, South Korea, Mexico, New Zealand, Singapore, Taiwan, Ukraine, South Africa, Albania, Austria, Belgium, Switzerland, Cyprus, Germany, Denmark, Spain, Finland, France, Greece, Ireland, Italy, Lithuania, Luxembourg, Latvia, Monaco, Macedonia, Netherlands, Portugal, Romania, Sweden, Slovenia, and the United Kingdom that expire in August 2020, and in Malaysia that expires in August 2021.
We license the rights to the following patents related to our product candidates:
| • | PPD 10558. Licensed from Ranbaxy Laboratories. The license expires 10 years after the first commercial sale or expiration of the last to expire enforceable patent claim; |
| • | Mu Delta. Licensed from Janssen Pharmaceutica. The license expires upon the exercise of an option by Janssen to continue the development and commercialization of mu delta after completion of Phase II studies. If Janssen rejects the option, then license the continues until no further payments are owed to Janssen; |
| • | Fluoroquinolone. Licensed from Janssen Pharmaceutica. The license expires upon the exercise of an option by Janssen to continue the development and commercialization of fluoroquinolone after completion of Phase II studies. If Janssen rejects the option, then the license continues until no further payments are owed to Janssen; and |
47
Table of Contents
| • | Dermatology Compounds. Licensed from Eli Lilly. The license expires when no remaining royalty payments are owed to Eli Lilly on a country-by-country basis. |
We expend a significant amount of our resources on research and development efforts to develop innovative therapies. Obtaining, maintaining and protecting the intellectual property rights, including patent rights, developed through our research and development efforts, is essential for our business to succeed. To that end, we actively seek to implement patent strategies to maximize the effectiveness of our intellectual property positions. We have been issued numerous U.S. and foreign patents and have a variety of patent applications pending in the United States and various foreign countries covering, among other things, compositions of matter, drug formulations, methods of use and action, and manufacturing. We also have obtained licenses to other patents from pharmaceutical companies.
Pursuant to the terms of the Uruguay Round Agreements Act, patents issued from applications filed on or after June 8, 1995, have a term of 20 years from the date of filing, no matter how long it takes for the patent to issue. Because patent applications in the pharmaceutical industry often take a long time to issue, this method of patent term calculation can result in a shorter period of patent protection afforded to us compared to the prior method of term calculation, which was 17 years from the date of issue. Our issued U.S. patents expire between 2023 and 2029, excluding any potential patent term extension available under U.S. federal law. We actively seek full patent term adjustment following allowance of a patent. We also actively seek patent term extensions covering products following marketing approval. Under the Drug Price Competition and Patent Term Restoration Act of 1984 and the Generic Animal Drug and Patent Term Restoration Act of 1988, a patent that claims a product, use or method of manufacture covering drugs may be extended for up to five years to compensate the patent holder for a portion of the time required for FDA review. However, we might not be able to take advantage of the patent term extension provisions of this law.
While we file and prosecute patent applications to protect our inventions, our pending patent applications might not result in the issuance of patents or our issued patents might not provide competitive advantages. Also, our patent protection might not prevent others from developing competitive products using related or other technology.
In addition to seeking the protection of patents and licenses, we also rely upon trade secrets, know-how and continuing technological innovation, which we seek to protect, in part, by confidentiality agreements with employees, consultants, suppliers and licensees. If these agreements are not honored, we might not have adequate remedies for any breach. Additionally, our trade secrets might otherwise become known or patented by our competitors.
The scope, enforceability and effective term of issued patents can be highly uncertain and often involve complex legal and factual questions. No consistent policy has emerged regarding the breadth of claims in pharmaceutical patents, so that even issued patents might later be modified or revoked by the relevant patent authorities or courts. Moreover, the issuance of a patent in one country does not assure the issuance of a patent with similar claim scope in another country, and claim interpretation and infringement laws vary among countries, so we are unable to predict the extent of patent protection in any country. The patents we obtain and the unpatented proprietary technology we hold might not afford us significant commercial protection. Additional information regarding risks associated with our patents and other proprietary rights that affect our business is contained under the headings “We must protect our patents and other intellectual property rights to succeed” and “We might need to obtain patent licenses from others in order to manufacture or sell our potential products and we might not be able to obtain these licenses on terms acceptable to us or at all” under the heading “Risk Factors”.
Manufacturing and Supply
We currently rely on our collaborators and contract manufacturers to produce drug substances and drug products required for our clinical trials under current good manufacturing practices, with oversight by our internal managers. We plan to continue to rely upon contract manufacturers and collaboration partners to
48
Table of Contents
manufacture commercial quantities of our drug candidates if and when approved for marketing by the applicable regulatory agency. We generally rely on one manufacturer for the active pharmaceutical ingredient and another manufacturer for the formulated drug product for each of our drug candidate programs. At the early stage of clinical studies, we do not believe that we are substantially dependent on any supplier, nor that additional manufacturers would be beneficial due the possibility of changes in the method of manufacturing of the drug candidate. As a drug candidate moves to later stages of development and the drug formulation method is established, we then seek additional manufacturers for the drug.
Sales and Marketing
We currently have no marketing, sales or distribution capabilities. We plan to rely on third parties to market our products, like ALZA for Priligy. We generally plan to outlicense our commercial rights in a territory to a third party with marketing, sales and distribution capabilities in exchange for one or more of the following: up-front payments; research funding; development funding; milestone payments; and royalties on drug sales. In some instances, however, we might choose to develop our own staff for marketing, sales or distribution.
Government Regulation
The manufacturing, testing, labeling, approval and storage of our products are subject to rigorous regulation by numerous governmental authorities in the United States and other countries at the federal, state and local level, including the FDA. The process of obtaining approval for a new pharmaceutical product or for additional therapeutic indications within this regulatory framework requires expenditure of substantial resources and usually takes several years. Companies in the pharmaceutical and biotechnology industries, including us, have suffered significant setbacks in various stages of clinical trials, even in advanced clinical trials after promising results had been obtained in earlier trials.
The process for obtaining FDA approval of drug candidates customarily begins with the filing of an IND with the FDA for the use of a drug candidate to treat a particular indication. If the IND is accepted by the FDA, we would then start human clinical trials to determine, among other things, the proper dose, safety and efficacy of the drug candidate in the stated indication. The clinical trial process is customarily divided into three phases—Phase I, Phase II and Phase III. Each successive phase is generally larger and more time-consuming and expensive than the preceding phase. Throughout each phase we are subject to extensive regulation and oversight by the FDA. Even after a drug is approved and being marketed for commercial use, the FDA may require that we conduct additional trials, including Phase IV trials, to further study safety or efficacy.
As part of the regulatory approval process, we must demonstrate to the FDA the ability to manufacture a pharmaceutical product before we receive marketing approval. The manufacturing and quality control procedures we and our manufacturing collaborators must undertake must conform to rigorous standards in order to receive FDA approval and the validation of these procedures is a costly endeavor. Pharmaceutical manufacturers are subject to inspections by the FDA and local authorities as well as inspections by authorities of other countries. To supply pharmaceutical products for use in the United States, foreign manufacturers must comply with these FDA-approved guidelines. These foreign manufacturers are also subject to periodic inspection by the FDA or by corresponding regulatory agencies in these countries under reciprocal agreements with the FDA. Moreover, state, local and other authorities may also regulate pharmaceutical product manufacturing facilities. Before we are able to manufacture commercial products, we or our contract manufacturer, as the case may be, must meet FDA guidelines.
Both before and after marketing approval is obtained, a pharmaceutical product, its manufacturer and the holder of the Biologics License Application, or BLA or NDA for the pharmaceutical product are subject to comprehensive regulatory oversight. The FDA may deny approval to a BLA or NDA if applicable regulatory criteria are not satisfied. Moreover, even if regulatory approval is granted, such approval may be subject to
49
Table of Contents
limitations on the indicated uses for which we may market the pharmaceutical product. Further, marketing approvals may be withdrawn if compliance with regulatory standards is not maintained or if problems with the pharmaceutical product occur following approval. In addition, under a BLA or NDA, the manufacturer of the product continues to be subject to facility inspections and the applicant must assume responsibility for compliance with applicable pharmaceutical product and establishment standards. Violations of regulatory requirements at any stage may result in various adverse consequences, which may include, among other adverse actions, withdrawal of the previously approved pharmaceutical product or marketing approvals or the imposition of criminal penalties against the manufacturer or BLA or NDA holder.
For the development of pharmaceutical products outside the United States, we and our collaborators are subject to foreign regulatory requirements and the ability to market a drug is contingent upon receiving marketing authorizations from the appropriate regulatory authorities. The requirements governing the conduct of clinical trials and marketing authorization vary widely from country to country. In countries other than European Union countries, foreign marketing authorizations are applied for at a national level. Within the European Union, procedures are available to companies wishing to market a product in more than one European Union member state. Clinical trial applications must be filed with the relevant regulatory authority in each country in which we would want to conduct a clinical trial. Assuming approval and the success of any clinical trial, we would then need to seek marketing approval for the drug. The process for obtaining marketing approval of drug candidates in the European Union begins with the filing with the European Medicines Agency, or EMEA, of a Marketing Authorization Application, or MAA, for the use of a drug candidate to treat a particular indication. Similar processes and outcomes of such human clinical trials that are required by the FDA are also required by the EMEA including testing for dose, safety and efficacy in three phases. Similar to the FDA, we are subject to extensive regulation and oversight by the European regulators throughout each phase. Even after a drug is approved and being marketed for commercial use, the EMEA may require that we conduct additional trials, including Phase IV trials, to further study safety or efficacy. As a result, the EMEA regulatory approval process includes all of the risks associated with FDA approval set forth above.
If and when necessary, we will choose the appropriate route of European or other international regulatory filing to accomplish the most rapid regulatory approvals. Requirements relating to manufacturing, conduct of clinical trials and product licensing vary widely in different countries, and the chosen regulatory strategy might not secure regulatory approvals or approvals of our chosen product indications. In addition, if a particular product to be used outside of the United States is manufactured in the United States, FDA requirements and U.S. export provisions will apply.
Outside of the United States, many countries require us to obtain pricing approval in addition to regulatory approval prior to launching the product in the approving country. We or our licensees may encounter difficulties or unanticipated costs or price controls in our respective efforts to secure necessary governmental approvals. Failure to obtain pricing approval in a timely manner or approval of pricing which would support an adequate return on investment or generate a sufficient margin to justify the economic risk might delay or prohibit the commercial launch of the product in those countries.
The marketing and sale of approved pharmaceutical product is subject to strict regulation. Promotional materials and activities must comply with the approving agency’s regulations and other guidelines. Physicians may prescribe pharmaceutical or biologic products for uses that are not described in a product’s labeling or differ from those approved by the approving agency. While such “off-label” uses are common and regulatory agencies do not regulate physicians’ choice of treatments, many approving agencies restrict a company’s communications on the subject of “off-label” use. Companies cannot promote approved pharmaceutical or biologic products for off-label uses. If any advertising or promotional activities we undertake fail to comply with applicable regulations or guidelines regarding “off-label” use, we may be subject to warnings or enforcement action.
50
Table of Contents
Competition
The pharmaceutical industry is highly competitive. Many of our competitors are worldwide conglomerates with substantially greater resources than we to develop and commercialize their drugs and drug candidates. Potential competitors have developed and are developing compounds for treating the same indications as our product candidates. In addition, a number of academic and commercial organizations are actively pursuing similar technologies, and several companies have developed or may develop technologies that might compete with our compounds.
Priligy, indicated for premature ejaculation, competes with Cromadyn, a generic paroxetine sold by More Pharmaceuticals in Mexico. We are aware of three other compounds in development for premature ejaculation: PD502, a novel metered-dose aerosol formulation of lidocaine and prilocaine being developed by Plethora Solutions and Sciele Pharma; BVF-324, a repurposed drug with an undisclosed mechanism, being developed by Biovail; and alprostadil, a vasodilator being developed for erectile dysfunction and premature ejaculation by Warner Chilcott. All of these products are in late stage (Phase II/III) development.
If approved, alogliptin will compete in the type 2 diabetes space with two DPP4 inhibitors currently on the market, Bristol-Myers Squibb/AstraZeneca’s Onglyza® (saxagliptin) and Merck’s Januvia® (sitagliptin). Merck also markets Janumet®, a fixed-dose combination of sitagliptin and metformin. Novartis markets the DPP4 inhibitor Galvus® (vildagliptin) and Eucreas® (vildagliptin/metformin) in Europe. Other marketed oral anti-diabetic competitors include generic metformin, generic sulfonylureas, and thiazolidinediones, including GlaxoSmithKline’s Avandia® (rosiglitazone) and Takeda’s Actos (pioglitazone). Generic competitors to Avandia® and Actos® are expected to enter the market in 2012.
The diabetes pipeline is crowded, with, to our knowledge, approximately 50 compounds in Phase I development, approximately 70 in Phase II development, and approximately 30 in Phase III development or preregistration. In addition to DPP4 inhibitors, competitors are also developing GLP-1 agonists, SGLT-2 antagonists, PPAR agonists, and compounds with other mechanisms for treatment of diabetes. Other companies with DPP4 inhibitors in clinical development of which we are aware include Amgen/Servier, Arisaph Pharmaceuticals, Boehringer Ingelheim, Dong-A Pharmaceuticals (South Korea), Dainippon Sumitomo Pharma, Forest Laboratories/Phenomix, Glenmark Pharmaceuticals, Kyorin Pharmaceuticals, LG Life Sciences (South Korea), Mitsubishi Tanabe Pharma, and Sanwa Kagaku Kenkyusho (Japan). In addition, Bristol-Myers Squibb is developing a fixed-dose combination of saxagliptin and metformin, currently in Phase III development, and Merck is developing a fixed-dose combination of sitagliptin and pioglitazone (Actos), currently in Phase III development.
If approved, PPD10558, indicated for the treatment of dyslipidemia, would compete with a wide variety of lipid lowering drugs. Generic statins are expected to dominate the market by 2012 when Pfizer’s Lipitor® (atorvastatin) patent expires. PPD10558 may be differentiated from these products if it is proved to be safe in patients who cannot tolerate currently marketed statins. There are several competing statins and statin combination products in development, including AstraZeneca/Abbott’s Certriad® (rosuvastatin/fibrate), Merck’s MK-0524A (laropiprant/niacin/simvastatin), Sciele Pharma’s fenofibrate/pravastatin combination, Abbott/Solvay’s Zolip® (fenofibrate/simvastatin), and NicOx’s NCX-6560, a novel statin. Companies developing compounds with other mechanisms of action for use in hyperlipidemia of which we are aware include Aegerion Pharmaceuticals, Amarin Corporation, Bristol-Myers Squibb, Cortria Corporation, Dr. Reddys Laboratories, Esperion Therapeutics, Essentialis, Genfit, GlaxoSmithKline, Isis Pharmaceuticals/Genzyme, Japan Tobacco, Karo Bio, Kythera Biopharmaceuticals, The Medicines Company, Merck, Metabasis Therapeutics, Metabolex, Mitsubishi Tanabe Pharma, Sanofi-Aventis, and Surface Logix.
If approved, the fluoroquinolone antibiotic compound licensed from Janssen Pharmaceutica will compete with other fluoroquinolones currently on the market, including Johnson and Johnson’s Levaquin® (levofloxacin), Bayer/Merck’s Avelox® (moxifloxacin), Bayer/Merck’s Cipro® (ciprofloxacin), and Oscient’s Factive® (gemifloxacin). Generic versions of ciprofloxacin are currently available, and generic versions of moxifloxacin
51
Table of Contents
and levofloxacin will likely become available when the patents covering these products expire in 2011. If the fluoroquinolone from Janssen Pharmaceutica is found to be effective against MRSA infections, it would compete with Pfizer’s Zyrox® (linezolid), Cubist’s Cubicin® (daptomycin), Wyeth’s Tygacil® (tigecycline), Theravance’s Vibativ® (telavancin), and the generic drug vancomycin.
Companies developing compounds to treat MRSA infections in clinical trials include aRigen, Cerexa (now Forest Laboratories), Destiny Pharma, e-Therapeutics, FAB Pharma, NovaBay, Novexel (now AstraZeneca), Phico Therapeutics, Rib-X Pharmaceuticals, Theravance, and Wockhardt (India). The Rib-X and Wockhardt compounds are both fluoroquinolones. In addition, MerLion Pharmaceuticals is developing a fluoroquinolone in Phase II. Merck and Nabi Biopharmaceuticals are both developing vaccines against staphylococcus aureus.
If approved, the mu delta compound, also licensed from Janssen Pharmaceutica, will compete with Lotronex® (alosetron), marketed by Prometheus Laboratories, and over-the-counter treatments for diarrhea-predominant irritable bowel syndrome (IBS) such as loperamide (Imodium®). The pipeline for diarrhea-predominant IBS includes: asimadoline, which is being developed by Tioga Pharmaceuticals and is entering Phase III; crofelemer and rifaximin, currently in Phase II and III development, respectively, by Salix Pharmaceuticals; AGI-003, currently in Phase III development by AGI Therapeutics; ROSE-010, currently in Phase II development by Rose Pharma; dextofisopam, currently in Phase II development by Pharmos Corporation; and LX1031, currently in Phase II development by Lexicon Pharmaceuticals.
Competitors might succeed in more rapidly developing and marketing technologies and products that are more effective than our products or that would render our products or technology obsolete or noncompetitive. Our collaborators might also independently develop products that are competitive with products that we have licensed to them. Any product that we or our collaborators succeed in developing and for which regulatory approval is obtained must then compete for market acceptance and market share. The relative speed with which we and our collaborators can develop products, complete clinical testing and approval processes, and supply commercial quantities of the products to the market compared to competitive companies will affect market success. In addition, the amount of marketing and sales resources, and the effectiveness of the marketing used with respect to a product will affect its success. In addition, some CRO services providers and private equity funds are developing risk sharing models to finance the pharmaceutical industry’s pipeline. NovaQuest, a subsidiary of Quintiles Transnational, is active in this business. As these types of business models evolve, there will be increasing competition for compounds and funds that will affect our ability to add to our portfolio.
Other competitive factors affecting our business generally include:
| • | product efficacy and safety; |
| • | timing and scope of regulatory approval; |
| • | product availability, marketing and sales capabilities; |
| • | reimbursement coverage; |
| • | the amount of clinical benefit of our product candidates relative to their cost; |
| • | method of and frequency of administration of any of our product candidates which may be commercialized; |
| • | patent protection of our product candidates; |
| • | the capabilities of our collaborators; and |
| • | the ability to hire qualified personnel. |
52
Table of Contents
Employees
Following the spin-off, we estimate that we will have approximately 45 full-time employees. Of the total, the vast majority of employees will be engaged in research and development activities. Our success will depend in large part on our ability to attract and retain skilled and experienced employees. None of our employees is covered by a collective bargaining agreement. We consider our relations with our employees to be good.
Properties
The following table identifies the location and general character of each of our principal facilities, all of which we lease:
Location | Principal Uses | Approximate Floor Area (Sq. Ft.) | Lease Expiration Date | |||
Morrisville, NC | Headquarters | 4,650 | 2012 | |||
Waltham, MA | Dermatology Program | 13,000 | 2012 |
We have options to extend the terms of our Waltham, MA lease for up to five years and our Morrisville, NC sublease from PPD for up to one year. We own substantially all of the equipment used in our facilities.
Legal Proceedings
In the normal course of business, we might be a party to various claims and legal proceedings. As of this time, there are no outstanding claims that management believes will have a material effect upon our financial condition, results of operations or cash flows.
53
Table of Contents
Selected Financial Data
The tables below set forth selected historical financial data of the combined financial statements of Furiex. This information has been prepared from Furiex’s audited combined financial statements as of December 31, 2009 and 2008 and for each of the three years in the period ended December 31, 2009. Financial information as of December 31, 2007, 2006 and 2005 and for each of the two years in the period ended December 31, 2006 has been prepared from unaudited combined financial statements not included herein. During these periods, Furiex was an integrated business of PPD. The historical financial information is not likely to be indicative of Furiex’s future performance or its future financial position or results of operations, and it does not provide or reflect data as if Furiex had actually operated as a separate, stand-alone entity during the periods covered. Per share data has not been presented as no common shares were outstanding during the periods presented and such information would not be meaningful.
The selected historical financial data should be read in conjunction with the combined financial statements and related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations”, included elsewhere in this Information Statement.
Combined Statements of Operations Data (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2005 | 2006 | 2007 | 2008 | 2009 (1) | ||||||||||||||||
Net revenue | $ | 25,371 | $ | 15,857 | $ | 560 | $ | 18,419 | $ | 6,312 | ||||||||||
Operating expenses | 28,029 | 3,604 | 23,316 | 11,645 | 38,131 | |||||||||||||||
Income (loss) from operations(2) | (2,658 | ) | 12,253 | (22,756 | ) | 6,774 | (31,819 | ) | ||||||||||||
Other income (expense), net | (2 | ) | (329 | ) | 19 | 14 | (2 | ) | ||||||||||||
Income (loss) from continuing operations | (2,660 | ) | 11,924 | (22,737 | ) | 6,788 | (31,821 | ) | ||||||||||||
Discontinued operations(3) | 2,164 | (4,066 | ) | (185 | ) | (976 | ) | 22,890 | ||||||||||||
Net income (loss) | $ | (496 | ) | $ | 7,858 | $ | (22,922 | ) | $ | 5,812 | $ | (8,931 | ) | |||||||
Combined Balance Sheet Data (in thousands):
| As of December 31, | ||||||||||
| 2005 | 2006 | 2007 | 2008 | 2009 | ||||||
Total assets | 69,166 | 63,581 | 63,265 | 61,138 | 55,877 | |||||
PPD net investment(4) | 62,314 | 58,895 | 56,870 | 55,524 | 49,270 | |||||
| (1) | We acquired Magen BioSciences, Inc. in 2009. Results of operations for the acquisition are included in our combined results of operations as of and since the effective date of the acquisition. For further details, see Note 2 in the notes to the combined financial statements. |
| (2) | Impairments of intangible assets are included in income (loss) from operations. For 2009, the impairment of intangible asset was related to in-process research and development for the MAG-131 compound obtained through the acquisition of Magen Biosciences. For 2008, the impairment of intangible asset related to the remaining unamortized value of our royalty interest in SinuNase and other Accentia antifungal products. For further details, see Note 5 in the notes to the combined financial statements. |
| (3) | In 2009, we completed dispositions of Piedmont Research Center, LLC and PPD Biomarker Discovery Sciences, LLC. Results of operations for these dispositions are included in discontinued operations, net of provision for income tax of $0. For further details, see Note 2 in the notes to the to the combined financial statements. |
| (4) | The financial statements of the company represent a combination of various components of PPD comprising the Discovery Sciences segment. Because a direct ownership relationship did not exist among all the components comprising the company, PPD’s net investment in the company is shown in lieu of shareholder’s equity in the combined financial statements. The net investment account represents the cumulative investments in, distributions from and earnings (loss) of the company. |
54
Table of Contents
Management’s Discussion and Analysis of Financial Condition and Results of Operations
This Information Statement includes forward-looking statements. All statements other than statements of historical facts are forward-looking statements, including any projections of milestones, royalties or other financial items, any statements of the plans and objectives of management for future operations, any statements concerning proposed new products or licensing or collaborative arrangements, any statements regarding future economic conditions or performance, and any statement of assumptions underlying any of the foregoing. In some cases, forward-looking statements can be identified by the use of terminology such as “believes”, “might”, “will”, “expects”, “plans”, “anticipates”, “estimates”, “potential” or “continue”, or the negative thereof or other comparable terminology. Although we believe that the expectations reflected in the forward-looking statements contained in this Information Statement are reasonable, there can be no assurance that such expectations or any of the forward-looking statements will prove to be correct, and actual results could differ materially from those projected or assumed in the forward-looking statements. Our future financial condition and results of operations, as well as any forward-looking statements, are subject to inherent risks and uncertainties, including the risk factors set forth below under “Economic and Industry-Wide Factors” and above under “Risk Factors” beginning on page 11, and for the reasons described elsewhere in this Information Statement. All forward-looking statements and reasons why results might differ included in this Information Statement are made as of the date hereof, and we assume no obligation to update these forward-looking statements or reasons why actual results might differ.
Executive Overview
Furiex is a drug development collaboration company that will pursue the Compound Partnering Business of our parent company, PPD. We seek to collaborate with pharmaceutical and biotechnology companies to increase the value of their early stage drug candidates by applying our novel approach to drug development that we believe expedites research and development decision-making and can shorten drug development timelines. We share risk with our collaborators by financing the cost of development to the point of mutually agreed upon pre-determined clinical milestones. In exchange, we share the potential rewards with our collaborators, receiving milestone payments and royalties based on the continued development and commercialization success of the drug candidate. Most of our collaborators that are large pharmaceutical companies have the option to continue late stage clinical development and commercialization of the drug candidate after it has reached the specified pre-determined milestones. If our collaborator is unable or unwilling to perform late stage development and commercialization, then we have the option to seek out a new development and commercialization partner. Our team is staffed with the same key PPD team members who demonstrated proven success in the drug development collaboration business. Our strategy is to invest in programs that have well defined clinical endpoints, a relatively straightforward path to regulatory approval and a large potential addressable market.
Our current pipeline is comprised of several compounds in various stages of development and commercialization including, two compounds which have been out-licensed to global pharmaceutical companies, one of which is currently marketed in several European countries, one compound currently being considered for out-licensing and several compounds currently undergoing active research and development by us.
We operate in one reportable operating segment. See Note 14 “Segment and Related Information” in the notes to our combined financial statements for information regarding the operating segment. Historically, our revenues consisted primarily of milestone and royalty payments from collaborators from out-licensed compounds. Our 2009 revenues included $5.0 million in regulatory approval milestones and $0.9 million in royalties from the sale of Priligy by our collaborator, ALZA. Priligy was our first compound to receive regulatory approval for marketing. Our revenues in 2008 included milestones from the Takeda aloglipton programs related to regulatory filings in the United States and Japan.
We incurred research and development expenses of $24.3 million, $8.1 million, and $21.0 million for the years ended December 30, 2009, 2008, and 2007, respectively. Our research and development expenses include
55
Table of Contents
costs incurred for our current pre-clinical and clinical-stage drug candidates, including the Ranbaxy statin, MAG-131 and the two compounds in-licensed from Janssen Pharmacuetica N.V. These costs include clinical research services provided by PPD, pre-clinical testing and clinical drug manufacturing provided by third parties, the direct cost of Furiex personnel managing the programs and upfront payments to our collaborators. All research and development costs for our drug candidates and external collaborations are expensed as incurred.
The timing and amount of any future expenses, completion dates, and revenues related to our drug candidates is subject to significant uncertainty due to the early stage of our development programs. We do not know if we will be successful in developing or commercializing any of our drug candidates. The timing and amount of our development expenses will depend upon the costs associated with the potential future clinical trials of our drug candidates, and the related expansion of our research and development organization, regulatory requirements, advancement of our preclinical programs and manufacturing costs. There are numerous risks and uncertainties associated with the duration and cost of clinical trials, which vary significantly over the life of a project as a result of unanticipated events arising during clinical development. For example, if the FDA or another regulatory authority were to require us to conduct clinical trials beyond those which we currently anticipate to complete clinical development of a drug candidate, or if we experience significant delays in enrollment in any of our clinical trials, we would be required to expend significant additional financial resources and time on the completion of clinical development. The timing and amount of revenues, if any, is equally dependent upon the success of the clinical trials as well as the commercial success of these products in the marketplace, all of which are subject to a variety of risk factors and uncertainties.
As of December 31, 2009, we had four collaborations that involve potential future expenditures. The first is our collaboration with Janssen-Cilag for Priligy. In connection with this collaboration, we have an obligation to pay a royalty to Lilly of 5% on annual net sales of the compound in excess of $800.0 million. If the related triggering events and product sales occur, we are entitled to future regulatory milestone payments of $ million, sales-based milestones of up to $50.0 million, and sales-based royalties ranging in the low double digit percentages depending on product sales volumes. There are currently no ongoing costs of development for this compound as it has been licensed back to Janssen-Cilag. The second collaboration involving future expenditures is with Ranbaxy for the statin compound. If all criteria are met, the total amount of potential clinical and sales-based milestones that we are obligated to pay Ranbaxy would be $42.0 million. We also would be obligated to pay Ranbaxy sales-based royalties ranging from the mid to high single digit percentages. We will be solely responsible and will bear all costs and expenses for the development, manufacture, marketing and commercialization of the compound and licensed products. The current estimated cost of the next clinical trials phase of development is $10.0 million to $20.0 million which costs would accrue over the next one to two years. The third collaboration involving future expenditures is with Lilly for the dermatology compounds. If all criteria are met, the total potential clinical development milestones that we are obligated to pay would be $21.4 million per compound developed. We also would be obligated to pay Lilly sales-based royalties ranging from the low single digit to low double digit percentages. Current spend to screen compounds is on the order of $5.0 million to $10.0 million per year. The fourth collaboration involving future expenditures is with Janssen Pharmaceutica N.V. for the fluoroquinolone and mu delta compounds. The expenses associated with the development of our in-licensed compounds from Janssen Pharmaceutica are expected to be between $70.0 and $80.0 million over the next two to three years. At the completion of Phase II, Janssen will have the option to continue development and commercialization of each compound. In exchange, we may receive, for each compound, up to $90.0 million in regulatory milestone payments and up to $75 million in sales-based milestone payments, as well as royalties on sales of each compound if approved for marketing. At this point in time, potential sales of any of these compounds, as well as the time such sales might begin, are too uncertain to forecast with any degree of accuracy.
In addition to the four collaborations that involve potential future expenditures described above we are collaborating with Takeda on alogliptin. There are no expenditures on our part related to this collaboration,
however if the related triggering events and product sales occur, we are entitled to receive future regulatory
56
Table of Contents
milestones of up to $52.5 million, sales-based milestones of up to $33.0 million, and sales-based royalties ranging from the mid single digit to low double digit percentages depending on product sales volumes.
During the year ended December 31, 2009, we reported a loss from continuing operations of $31.8 million and a net loss of $8.9 million. During the year ended December 31, 2008, we reported income from continuing operations of $6.8 million and net income of $5.8 million. We expect to continue to incur significant net losses for the foreseeable future and that such losses will fluctuate from quarter to quarter and that such fluctuations may be substantial.
Our business is subject to various risks and uncertainties. See “Risk Factors” for additional information on these risks and uncertainties.
Basis of Accounting and Combination
The accompanying combined financial statements have been derived from the combined financial statements and accounting records of PPD, from the historical cost basis of the assets and liabilities of the various activities that reflect the combined results of operations, financial condition and cash flows of the Discovery Sciences segment of PPD. All the business components of the Discovery Sciences segment have been included in the historical statements because they were managed by common segment management, and because they reflect the historical performance of segment management. In 2009, PPD completed its disposition of Piedmont Research Center, LLC and PPD Biomarker Discovery Services, LLC. Due to the unique service offerings of these two subsidiaries, PPD determined these business units were no longer a long-term strategic fit and elected to sell them. These business units are recorded as discontinued operations in the statements of operations. Additionally, the Discovery Sciences segment includes pre-clinical consulting services that will not continue after the spin-off. All rights and obligations related to pre-clinical consulting services and the definitive purchase agreements related to Piedmont Research Center, LLC and PPD Biomarker Discovery Services, LLC will be retained by PPD.
Furiex has been allocated certain expenses from PPD such as executive oversight, risk management, accounting, tax, legal, investor relations, human resources, information technology, facilities and depreciation, but has not been allocated the underlying productive assets, such as certain information systems equipment, and furniture and facilities that will not be assigned to Furiex but from which Furiex has benefited. Such expenses have been reflected in the combined financial statements as expense allocations from PPD. The basis of these allocations includes full-time equivalent employees for the respective periods presented and square footage of occupied space. See Note 13 for further discussion of the allocations.
Management believes that the assumptions and allocations underlying the combined financial statements are reasonable. The financial information in these combined financial statements does not include all of the expenses that would have been incurred had Furiex been a separate, stand-alone publicly traded entity. The combined financial statements include assets, liabilities and operations for Piedmont Research Center, LLC, PPD Biomarker Discovery Science, LLC and preclinical consulting services that will not be included in Furiex at the time the spin-off is effected. As such, the financial information herein does not reflect the combined financial position, results of operations or cash flows of Furiex in the future or what the Company would have been had it been a separate, stand-alone entity during the periods presented.
The Separation of Furiex from PPD
On October 28, 2009, PPD announced its intention to separate its Compound Partnering Business into an independent, publicly traded company through a spin-off of 100% of our stock to PPD shareholders. Completion of the spin-off is expected in the middle of 2010, and is subject to various conditions, including final approval from PPD’s Board to complete the spin-off. Immediately following the distribution, PPD’s shareholders will own 100% of the equity in both companies. The separation will not require a vote by or approval of PPD shareholders.
57
Table of Contents
The Compound Partnering Business discussed herein represents the historical operating results and financial condition of Furiex. Any references to “we”, “us”, “Furiex” or the “Company” refer to the Compound Partnering Business as operated as a part of PPD prior to the spin-off.
Critical Accounting Policies and the Use of Estimates
The preparation of our combined financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the amounts reported in our combined financial statements and accompanying notes. Actual results could differ materially from those estimates. The items in our combined financial statements requiring significant estimates and judgments are as follows:
Revenue Recognition
We generate revenue in the form of upfront payments, development and regulatory milestone payments, royalties and sales-based milestone payments in connection with the out-licensing of compounds. All future milestones and royalties will depend on the future success of our collaborators in developing and commercializing the compound. Upfront payments are generally paid within a short period of time following the execution of an out-license and collaboration agreement. Milestone payments are typically one-time payments to us triggered by the collaborator’s achievement of specified development and regulatory events such as the commencement of Phase III trials or regulatory submission or approval. Royalties are payments received by us based on net product sales of a collaborator. Sales-based milestone payments are typically one-time payments to us triggered when the aggregate net sales of product by a collaborator for a specified period (for example, an annual period) reach an agreed upon threshold amount. We recognize these payments from our collaborators when the event which triggers the obligation of payment has occurred, there are no further obligations on our part in connection with the payment, and collection is reasonably assured.
We have also historically recorded revenue from service contracts, other than time-and-material contracts, on a proportional performance basis. To measure performance under these contracts on a given date, we compared effort expended to date to the estimated total effort to be expended to complete the contract using metrics such as the number of units to be delivered. Changes in the estimated total effort required to complete a contract without a corresponding proportional change to the contract value resulted in a cumulative adjustment to the amount of revenue recognized in the period the change in estimate was determined. For time-and-material contracts, we recognized revenue as hours were worked, multiplied by the applicable hourly rate. All service revenue contracts relate to service revenues that will not be included in the spin-off.
Goodwill
The fair value of goodwill could be impacted by future adverse changes such as future declines in our operating results, a decline in the valuation of pharmaceutical and biotechnology company stocks, including the valuation of our common stock, a further significant slowdown in the worldwide economy or the pharmaceutical and biotechnology industry, failure to meet the performance projections included in our forecasts of future operating results or the delay or abandonment of any of our in-process research and development programs.
We review goodwill for impairment annually on October 1 and whenever events or changes in circumstances indicate that the carrying amount of an asset might not be recoverable. This analysis utilizes a discounted cash flow method using the expected future inflows and outflows of our business and an appropriate discount rate. Based on our review as of October 1, 2009, our reporting unit’s fair value was in excess of our book value.
Intangible Assets
The value of our intangible assets could be impacted by future adverse changes such as changes in regulatory conditions, the introduction of competing products, changes in medical reimbursement rates, the
58
Table of Contents
discovery of manufacturing or safety concerns of products being marketed or developed, significant slowdowns in the worldwide economy or the pharmaceutical and biotechnology industry, or the delay or abandonment of any of our in-process research and development programs.
We evaluate intangible assets, which consist of royalty rights and acquired in-process research and development, at any time we believe indicators of impairment exist. We initially record these intangible assets at fair value and use fair value measurements to evaluate impairment. The fair value of our intangible assets is determined by estimating the costs to develop the acquired technology into commercially viable products, estimating the resulting net cash flows from the programs, and discounting the net cash flows to present value. Additionally, our estimates take into account the relevant market size and growth factors, expected trends in technology, and the nature and expected timing of new product introductions by us and our competitors. The resulting net cash flows from such programs are based on management’s estimates of cost of sales, operating expenses, and income taxes from such programs. The rates utilized to discount the net cash flows to their present value were commensurate with the stage of development of the program and uncertainties in the economic estimates used in the projections described above. During 2008, we reported an impairment of $1.6 million related to the royalty rights purchased from Accentia as a result of Accentia’s discontinuation of the sale of antifungal products and subsequent bankruptcy. We suspended the MAG-131 program due to efficacy data that was discovered in late 2009. As a result, we evaluated the asset for impairment using forecasts based on then currently available data, and determined that this asset was impaired. We recorded a $10.4 million impairment of our acquired in-process research and development asset as of December 31, 2009.
Share-Based Compensation
We measure share-based compensation cost at grant date, based on the fair value of the award, and recognize it as expense over the employee’s requisite service period. The fair value of each option award is estimated on the grant date using the Black-Scholes option-pricing model. The model requires the use of the following assumptions: an expected dividend yield; expected volatility; risk-free interest rate; and expected term. Some employees of Furiex have historically received awards from PPD. Based on our assumptions for the relevant factors, the weighted-average fair value of PPD options granted to our employees during the year ended December 31, 2009 was $8.80 per option. A change in these assumptions could have a significant impact on the weighted-average fair value of options. See Note 8 in the notes to our combined financial statements for details regarding the assumptions used in estimating fair value for the years ended December 31, 2007, 2008 and 2009 regarding equity awards granted to Furiex’s employees by PPD.
Tax Valuation Allowances
The use of estimates and judgments is necessary in determining the recoverability of certain deferred tax assets, which arise from net operating losses, tax credit carryforwards and temporary differences between the tax and financial statement recognition of revenue and expense. We reduce our deferred tax assets by a valuation allowance if, based on the weight of available evidence, it is more likely than not that some portion or all of the recorded deferred tax assets will not be realized in future periods.
In evaluating our ability to recover our deferred tax assets, in full or in part, we consider all available positive and negative evidence, including our past operating results, the existence of cumulative losses in the most recent fiscal years and our forecast of future taxable income on a jurisdiction-by-jurisdiction basis. In determining future taxable income, assumptions include the amount of state and federal pretax operating income, the reversal of temporary differences and the implementation of feasible and prudent tax planning strategies. These assumptions require significant judgment about the forecasts of future taxable income and are consistent with the plans and estimates we use to manage the underlying businesses. Based on our analysis of the above factors, we determined that a valuation allowance of $7.8 million was required as of December 31, 2009 for the full value of our current and long-term deferred tax assets based on our history of operating losses. Changes in our assumptions could result in an adjustment to the valuation allowance, up or down, in the future.
59
Table of Contents
Results of Operations
Year Ended December 31, 2008 versus Year Ended December 31, 2009
The following table sets forth amounts from our combined financial statements for the full years of 2008 and 2009.
| Year Ended December 31, | ||||||||
| (in thousands, except per share data) | 2008 | 2009 | ||||||
Net revenue: | ||||||||
Milestones | $ | 18,000 | $ | 5,000 | ||||
Royalties | — | 923 | ||||||
Service | 419 | 389 | ||||||
Total net revenue | 18,419 | 6,312 | ||||||
Direct expenses | 153 | 126 | ||||||
Research and development expenses | 8,053 | 24,334 | ||||||
Selling, general and administrative expenses | 1,738 | 3,127 | ||||||
Depreciation and amortization | 94 | 183 | ||||||
Impairment of intangible asset | 1,607 | 10,361 | ||||||
Income (loss) from operations | 6,774 | (31,819 | ) | |||||
Other income (expense), net | 14 | (2 | ) | |||||
Income (loss) from continuing operations | 6,788 | (31,821 | ) | |||||
Discontinued operations, net of taxes | (976 | ) | 22,890 | |||||
Net income (loss) | $ | 5,812 | $ | (8,931 | ) | |||
Net Revenue. Total net revenue decreased $12.1 million to $6.3 million in 2009. Milestones revenue decreased by $13.0 million based on the timing and progress of the various out-licensed products being developed by our collaborators. Milestones revenue for 2008 included a $15.0 million milestone we earned from Takeda as a result of the FDA’s acceptance of the alogliptin NDA and a $3.0 million milestone payment we earned from Takeda’s submission of the alogliptin NDA in Japan, while 2009 milestone revenue included a $5.0 million milestone based on regulatory approvals of the ALZA Priligy product. Royalty revenues are dependent on the approval and sale of products by our collaborators. We received our first royalties of $0.9 million from sales of Priligy as a result of the marketing approval in various countries outside the United States. Service revenues relate to consulting services provided to customers of PPD that will not be performed by us subsequent to the completion of the spin-off.
Expenses. Research and development, or R&D, expenses increased $16.3 million to $24.3 million in 2009. R&D expenses fluctuate significantly from period to period depending on the number and mix of acquired and in-licensed R&D programs. The increase in R&D expense was primarily due to development costs of $12.3 million associated with our dermatology compounds which we acquired as part of the Magen acquisition in April 2009 and $7.0 million of upfront payments and $2.2 million in development costs related to the two therapeutic compounds in-licensed as part of the agreement with Janssen Pharmaceutica N.V., offset by the $5.6 million reduction in the spending on our statin compound in-licensed from Ranbaxy.
60
Table of Contents
The following table sets forth amounts from our combined statements of operations for R&D expenses along with the dollar amount of the change from 2008 to 2009.
| Year Ended December 31, | $Inc (Dec) | |||||||||
| (in thousands) R&D by project: | 2008 | 2009 | ||||||||
Dermatology compounds | $ | — | $ | 12,346 | $ | 12,346 | ||||
Upfront payments to Janssen Pharmaceutica | — | 7,000 | 7,000 | |||||||
Mu delta compound | — | 572 | 572 | |||||||
Fluoroquinolone compound | — | 1,595 | 1,595 | |||||||
Statin compound | 6,848 | 1,288 | (5,560 | ) | ||||||
Other R&D | 1,205 | 1,533 | 328 | |||||||
Total R&D | $ | 8,053 | $ | 24,334 | $ | 16,281 | ||||
Costs related to the programs for compounds in-licensed from Janssen Pharmaceutica, if successful, are expected to be $70.0 to $80.0 million over the next two to three years until the programs are out-licensed to a collaborator. We continue to conduct limited development activities with respect to the Ranbaxy statin compound while we evaluate alternatives for future development and commercialization. We are currently screening our dermatology compounds and other compounds under material transfer agreements with other pharmaceutical companies to identify additional drug development candidates for dermatological indications, as well as seeking additional compounds to in-license. Consequently, the future spend levels for our dermatology compound development is highly dependent on any future determinations regarding those compounds. We plan to continue evaluating other compound partnering opportunities, which could result in significant additional R&D expense in future periods.
Selling, general and administrative, or SG&A, expenses increased $1.4 million to $3.1 million in 2009. The increase in SG&A expenses was the result of $1.1 million related to costs incurred related to the spin-off, and the additional SG&A expense related to the post-acquisition operations of Magen, which we acquired in April 2009.
Impairment. During 2009, we acquired in-process research and development of $10.4 million through the acquisition of Magen, which was related solely to the MAG-131 research program. At the time of acquisition, this program was in the pre-IND phase of research and we estimated that it would take approximately four to five years to complete research and development. We filed an IND in October 2009 but suspended the MAG-131 program due to efficacy data that was discovered in late 2009. As a result, we evaluated the asset for impairment. We reassessed the fair value of the program using a discounted cash flow model based on Level 3 inputs such as the estimated remaining costs to develop the acquired technology into commercially viable products, estimated net cash flows from the program, and a discount rate commensurate with the stage of development. Based on this analysis, we determined that the acquired in-process research and development asset was impaired and recorded a charge of $10.4 million as of December 31, 2009.
In 2008, Accentia announced its Phase III clinical trial for SinuNase failed to meet its goal in treating chronic sinusitis participants, discontinued the sales of anti-fungal products on which we received royalties and declared bankruptcy. As a result, we wrote off the $1.6 million of remaining unamortized value of our royalty interest in the anti-fungal products of Accentia.
Income Taxes. During 2008 and 2009, we did not record a tax provision related to our operating losses as we have provided full valuation allowances against our assets based on our history of operating losses. Additionally, with the exception of the pre-acquisition federal and state tax filings for Magen BioSciences, Inc. and certain separate state filings, our operations have been included in the consolidated federal and combined state tax returns of PPD and the resulting tax attributes have been fully utilized by PPD and are no longer available to us for future use. Subsequent to the completion of the spin-off from PPD, we will file federal and state returns separately from PPD and will be able to accumulate tax attributes of our income before tax for our future use. However, we do expect the need to provide a full valuation allowance against any deferred tax assets to continue until we are able to demonstrate a consistent pattern of profitability.
61
Table of Contents
Results of Operations. Income (loss) from operations decreased $38.6 million to a loss of $31.8 million in 2009. Income from operations in 2009 included the impairment charge of $10.4 million along with the additional R&D expense of $16.3 million mentioned above. Income from operations was also negatively impacted by the decrease of $13.0 million in milestone payments.
Results of operations from discontinued operations was $22.9 million in 2009. In May 2009, we completed our disposition of the assets of Piedmont Research Center, our preclinical oncology service business. In December 2009, we completed the disposition of our wholly owned subsidiary, PPD Biomarker Discovery Sciences, LLC, which owned and operated a biomarker discovery business. As a result, these business units have been shown as discontinued operations for 2008 and 2009. We recognized a net gain from the sale of businesses of $26.7 million related to these dispositions.
Net loss of $8.9 million in 2009 represents a $14.7 million decrease from net income of $5.8 million in 2008. Net loss for 2009 included the impairment charge of $10.4 million along with the additional R&D expense of $16.3 million, offset partially by the net gain from the sale of businesses of $26.7 million, as mentioned above.
Year Ended December 31, 2007 versus Year Ended December 31, 2008
The following table sets forth amounts from our combined financial statements for the full years of 2007 and 2008.
| Year Ended December 31, | ||||||||
| (in thousands, except per share data) | 2007 | 2008 | ||||||
Net revenue: | ||||||||
Milestones | $ | — | $ | 18,000 | ||||
Service | 560 | 419 | ||||||
Total net revenue | 560 | 18,419 | ||||||
Direct expenses | 185 | 153 | ||||||
Research and development expenses | 21,081 | 8,053 | ||||||
Selling, general and administrative expenses | 1,780 | 1,738 | ||||||
Depreciation and amortization | 270 | 94 | ||||||
Impairment of intangible asset | — | 1,607 | ||||||
Income (loss) from operations | (22,756 | ) | 6,774 | |||||
Other income (expense), net | 19 | 14 | ||||||
Income (loss) from continuing operations | (22,737 | ) | 6,788 | |||||
Discontinued operations, net of taxes | (185 | ) | (976 | ) | ||||
Net income (loss) | $ | (22,922 | ) | $ | 5,812 | |||
Net Revenue. Total net revenue increased $17.9 million to $18.4 million in 2008. Milestones revenue increased by $18.0 million based on the timing and progress of the various out-licensed products being developed by our collaborators. Milestones revenue for 2008 included a $15.0 million milestone we earned from Takeda as a result of the FDA’s acceptance of the alogliptin NDA and a $3.0 million milestone payment we earned from Takeda’s submission of the alogliptin NDA in Japan, while there were no milestones triggered by our out-licensed programs in 2007. Service revenues relate to consulting services provided to customers of PPD that will not be performed by us subsequent to the completion of the spin-off.
Expenses. R&D expenses decreased $13.0 million to $8.1 million in 2008. R&D expenses fluctuate significantly from period to period depending on the number, status and mix of our acquired and in-licensed R&D programs. The decrease in R&D expense during 2008 was due to the completion of the clinical trials associated with the statin compound we licensed from Ranbaxy and are developing as a potential treatment for dyslipidemia.
62
Table of Contents
The following table sets forth amounts from our combined statements of operations for R&D expenses along with the dollar amount of the change from 2007 to 2008.
| Year Ended December 31, | $Inc (Dec) | |||||||||
| (in thousands) R&D by project: | 2007 | 2008 | ||||||||
Statin compound | $ | 20,292 | $ | 6,848 | $ | (13,444 | ) | |||
Other R&D | 789 | 1,205 | 416 | |||||||
Total R&D | $ | 21,081 | $ | 8,053 | $ | (13,028 | ) | |||
We continue to conduct limited development activities with respect to the Ranbaxy statin compound while we evaluate alternatives for future development and commercialization. We plan to continue evaluating other compound partnering opportunities, which could result in significant additional R&D expense in future periods.
Impairment. In 2008, Accentia announced that its Phase III clinical trial for SinuNase failed to meet its goal in treating chronic sinusitis participants, discontinued the sales of anti-fungal products on which we received royalties and declared bankruptcy. As a result, we wrote off the $1.6 million of remaining unamortized value of our royalty interest in the anti-fungal products of Accentia.
Income Taxes. During 2007 and 2008, we did not record a tax provision related to our operating losses as we have provided full valuation allowances against our assets based on our history of operating losses. Additionally, our operations have historically been included in the consolidated federal and combined state tax returns of PPD and the resulting tax attributes have been fully utilized by PPD and are no longer available to us for future use. Subsequent to the completion of the spin-off from PPD, we will file federal and state returns separately from PPD and will be able to accumulate tax attributes of our income before tax for our future use. However, we do expect the need to provide a full valuation allowance against any deferred tax assets to continue until we are able to demonstrate a consistent pattern of profitability.
Income from operations increased $29.5 million to $6.8 million in 2008. Income from operations in 2008 included the $18.0 million in milestone payments from Takeda under our collaboration agreement and a $13.0 million decrease in R&D expenses, as discussed above.
Results of Operations. Results of operations from discontinued operations was a loss of $1.0 million in 2008. In May 2009, we completed our disposition of the assets of Piedmont Research Center, our preclinical oncology service business. In December 2009, we completed the disposition of our wholly owned subsidiary, PPD Biomarker Discovery Sciences, LLC, which owns and operates a biomarker discovery business. As a result, these business units have been shown as discontinued operations for 2007 and 2008.
Net income of $5.8 million in 2008 represents an increase of $28.7 million from a loss of $22.9 million in 2007. Net income for 2008 included the $18.0 million in milestone payments from Takeda under our collaboration agreement and a $13.0 million decrease in R&D expenses, as mentioned above.
Liquidity and Capital Resources
As of December 31, 2009, all cash and investments were held and managed by PPD. Accordingly, cash used to pay our expenses or cash collected from collaboration agreements, royalties or customer contracts by PPD on behalf of us are recorded as an increase or decrease in the PPD net investment.
Upon the spin-off, PPD has committed to provide us with a cash contribution of $100.0 million, and we will assume current accounts payable and other accrued expenses payable to third parties and incurred in the ordinary course of our Compound Partnering Business, which as of December 31, 2009, totaled approximately $4.6 million. We expect that this cash contribution will fund Furiex’s operations and working capital requirements for at least 12
63
Table of Contents
months after the closing of the spin-off, based on current operating plans. In addition to this cash contribution, we expect to receive future payments from our existing collaborations that will provide additional support for our operations and working capital requirements. Prior to the spin-off, our operations are being funded entirely by PPD.
Our future capital requirements will depend on numerous factors, including, among others: the cost and expense of continuing the research and development activities of our existing and potential new drug candidates; new collaborative agreements that we might enter into in the future; progress of product candidates in clinical trials; the ability of our licensees to obtain regulatory approval and successfully manufacture and market products licensed under our patents; the continued or additional support by our collaborators or other third parties of R&D efforts and clinical trials; investment in existing and new R&D programs; time required to gain regulatory approvals; our ability to obtain funding from third parties under collaborative arrangements; the demand for our potential products, if and when approved; potential acquisitions of technology, product candidates or businesses by us; and the costs of defending or prosecuting any patent opposition or litigation necessary to protect our proprietary technologies. In order to develop and obtain regulatory approval for our potential products we might need to raise substantial additional funds through equity or debt financings, collaborative arrangements, the use of sponsored research efforts or other means. Additional financing might not be available on acceptable terms, if at all, and such financing might only be available on terms dilutive to our shareholders.
In 2009, our operating activities used $24.1 million in cash as compared to providing $9.0 million for the previous year. The change in cash flow was due primarily to a decrease in net income of $14.7 million, an increase in other accrued expense of $2.8 million and the net gain on sale of business of $26.7 million, partially offset by the change in impairment of intangible assets of $8.8 million and the decrease in accounts payable of $2.1 million.
In 2009, our investing activities provided $25.8 million in cash. Cash was provided from the sale of businesses of $40.3 million, which was partially offset by cash used to make capital expenditures of $0.5 million and business acquisitions of $12.1 million.
In 2009, our financing activities used $1.7 million of cash from the net change in investment of PPD.
We have been involved in compound development and commercialization collaborations within PPD since 1998. We developed a risk-sharing research and development model to help pharmaceutical and biotechnology clients develop compounds. Through collaborative arrangements based on this model, we assist our clients by sharing the risks and potential rewards associated with the development and commercialization of drugs at various stages of development. As of December 31, 2009, our four main collaborations were with Janssen-Cilag, an affiliate of Johnson & Johnson, Takeda Pharmaceuticals Company Limited, Janssen Pharmaceutica N.V., an affiliate of Johnson & Johnson, and Eli Lilly and Company, and relate to, respectively, the product Priligy, the late stage candidate alogliptin, two Phase II-ready therapeutic compounds, and a series of early stage candidates including Vitamin D receptor modulators as well as up to six other programs for topical dermatological indications, respectively. They involve the potential future receipt of one or more of the following forms of revenue: payments upon the achievement of specified regulatory and sales-based milestones; and royalty payments if the compound is approved for sale. To date, Austria, Finland, Germany, Italy, Mexico, New Zealand, Portugal, South Korea, Spain and Sweden have approved Priligy for marketing. We received a $2.5 million milestone on each of the first two of these national approvals, for a total of $5.0 million, in the first quarter of 2009. We are entitled to royalties on net sales of Priligy and sales-based milestones if requisite sales levels are reached. We recorded the first royalties from the sales of Priligy in 2009. With regard to alogliptin, in June 2009, the FDA issued a complete response to Takeda on its alogliptin NDA, requesting Takeda conduct an additional cardiovascular safety trial that satisfies the FDA’s December 2008 guidance on anti-diabetes therapies. In September 2009, the FDA issued a complete response to Takeda on its NDA for the fixed dose combination of alogliptin and ACTOS NDA stating that further review would be dependent on the cardiovascular safety data that would be submitted in support of the alogliptin monotherapy NDA. The compounds related to Lilly and Janssen Pharmaceutica are still in discovery and development,
64
Table of Contents
respectively, and have not generated any regulatory milestone payments yet. Due to the risks associated with drug development and commercialization, including poor or unexpected preclinical and clinical trial results, obtaining regulatory approval to sell in any country and changing regulatory requirements, we might not receive any further milestone payments, royalties or other payments with respect to any of our drug development collaborations.
As of December 31, 2009, we had four collaborations that involve potential future expenditures. The first is our collaboration with Janssen-Cilag, for Priligy. In connection with this collaboration, we have an obligation to pay a royalty to Lilly of 5% on annual net sales of the compound in excess of $800.0 million.
The second collaboration involving future expenditures is with Ranbaxy. In February 2007, we exercised an option to license from Ranbaxy a statin compound that we are developing as a potential treatment for dyslipidemia, a metabolic disorder characterized by high cholesterol levels. Under the agreement, we have an exclusive license to make, use, sell, import and sublicense the compound and any licensed product anywhere in the world for any human use. We are solely responsible, and will bear all costs and expenses, for the development, manufacture, marketing and commercialization of the compound and licensed products. We are obligated to pay Ranbaxy milestone payments upon the occurrence of specified clinical development events. If a licensed product is approved for sale, we must also pay Ranbaxy royalties based on sales of the product, as well as commercial milestone payments based on the achievement of specified worldwide sales targets. If all criteria are met, the total amount of potential clinical and sales-based milestones that we are obligated to pay Ranbaxy would be $43.0 million. We also would be obligated to pay Ranbaxy sales-based royalties ranging from the mid to high single digit percentages. We completed a high dose comparator study in healthy volunteers. The drug was well-tolerated and a preliminary review of results suggests the statin compound compares favorably to currently marketed statins. We continue to conduct limited development activities with respect to the Ranbaxy statin compound while we evaluate alternatives for future development and commercialization.
The third collaboration involving future expenditures is with Lilly. In April 2009, we acquired Magen BioSciences, Inc., a biotechnology company founded in 2006 to discover dermatologic therapies. As a result, we expanded the Compound Partnering Business into dermatology and gained screening and research capabilities for dermatologic treatments. We have an exclusive license to develop and commercialize Vitamin D receptor modulator compounds for use as topical treatments in dermatological indications. We also have an option agreement with Lilly to screen compounds from six additional platforms for utility in dermatology and are investigating compounds from other potential collaborators under material transfer agreements. Through the acquisition of Magen, we acquired in-process research and development of $10.4 million. At acquisition, the acquired in-process research and development was related to the MAG-131 research program, which was in the pre-IND phase of research. We filed an IND for the MAG-131 compound in October 2009, but subsequently suspended the MAG-131 program due to efficacy data that was discovered in late 2009. We recently completed the testing of another Vitamin D receptor modulator licensed from Lilly and determined not to pursue it for further development. Under the license arrangements with Lilly, we are obligated to pay clinical development milestones as well as royalties based on the sales of the product. If all criteria are met, the total potential clinical development milestones that we are obligated to pay would be $21.4 million per compound developed. We also would be obligated to pay Lilly sales-based royalties ranging from the low single digit to low double digit percentages. We also are currently investigating other compounds under material transfer agreements to identify potential drug development candidates for dermatological indications, and are considering other strategic alternatives for our dermatology business.
The fourth collaboration involving future expenditures is with Janssen Pharmaceutica N.V. In November 2009, we entered into agreements with Janssen Pharmaceutica to develop and commercialize two Phase II-ready therapeutic compounds. We plan to study the mu delta compound as a treatment for diarrhea-predominant irritable bowel syndrome, or IBS-d and the fluoroquinolone compound as a treatment for community-acquired bacterial pneumonia and complicated skin and skin structure infections caused by gram negative or gram positive bacteria, including MRSA. Under the two agreements, we in-licensed the two compounds and we will advance the compounds through Phase II development. At the completion of Phase II, Janssen Pharmaceutica will have
65
Table of Contents
the option to continue development and commercialization of each compound. In exchange, we may receive, for each compound, up to $90.0 million in regulatory milestone payments and up to $75.0 million in sales-based milestone payments, as well as royalties on sales of each compound if approved for marketing. In the event Janssen Pharmaceutica elects not to continue a program, we have the option to continue developing and commercializing the compound for that program and we would be obligated to pay Janssen Pharmaceutica, for each compound, up to $50.0 million in regulatory milestone payments and up to $75.0 million in sales-based milestone payments, as well as sales-based royalties ranging from the mid single digit to low double digit percentages if approved for marketing.
We actively evaluate potential acquisitions and in-licensing that might require additional external financing, and we might seek funds from public or private issuances of equity or debt securities. While we believe we will have adequate sources of liquidity after the spin-off to fund our operations for at least 12 months after the completion of the spin-off, our sources of liquidity could be affected by: our dependence on future royalties and milestones from products under development risks related to the development, regulatory approval and commercialization of our product candidates; changes in the regulatory compliance requirements; reliance on key personnel; personal injury or other tort claims; international risks; environmental or intellectual property claims; or other factors described under “Risk Factors” and under the following subheadings in this “Managements’ Discussion and Analysis”: “Contractual Obligations”; “Critical Accounting Policies and Estimates”; “Potential Volatility of Operating Results”; and “Quantitative and Qualitative Disclosures about Market Risk”.
Contractual Obligations
As of December 31, 2009, future minimum payments on all our contractual obligations for years subsequent to December 31, 2009 were as follows:
| Payments Due by Period (in thousands) | |||||||||||||||
| 2010 | 2011 - 2012 | 2013 - 2014 | 2015 and thereafter | Total | |||||||||||
Operating leases | $ | 420 | $ | 846 | $ | 6 | $ | — | $ | 1,272 | |||||
As of December 31, 2009, we were contingently obligated under collaboration agreements that have not been included in the table above due to the inherent uncertainty in the amounts and timing of payments. For more information, see “Liquidity and Capital Resources.”
Off-balance Sheet Arrangements
We have no off-balance sheet arrangements except for operating leases entered into in the normal course of business.
Recent Accounting Pronouncements
In December 2007, the Financial Accounting Standards Board, or FASB, issued revised guidance on the accounting for business combinations. The revised guidance expands the definitions of a business and a business combination and requires that: the purchase price of an acquisition, including the issuance of equity securities to be determined on the acquisition date, be recorded at fair value at the acquisition date; all assets, liabilities, contingent consideration, contingencies and in-process research and development costs of an acquired business be recorded at fair value at the acquisition date; acquisition costs generally be expensed as incurred; restructuring costs generally be expensed in periods subsequent to the acquisition date; and changes be made in accounting for deferred tax asset valuation allowances and acquired income tax uncertainties after the measurement period to impact income tax expense. In April 2009, the FASB issued additional guidance on the accounting for business combinations which requires an acquirer to recognize at fair value an asset acquired or a liability assumed in a business combination that arises from a contingency if its acquisition-date fair value can be determined during the measurement period. If the acquisition-date fair value cannot be determined, the acquirer applies the recognition criteria in the accounting for contingencies guidance to determine whether the contingency should be
66
Table of Contents
recognized as of the acquisition date or thereafter. This revised guidance applies to business combinations for which the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2008. Except for the accounting for in-process research and development discussed in Note 5 and the disallowance of capitalization of acquisition costs, this pronouncement did not have a material impact on reporting of acquisitions in the financial statements.
In May 2009, the FASB issued a new accounting standard on the accounting for and disclosure of events that occur after the balance sheet date but before financial statements are issued or available to be issued, also known as “subsequent events.” The standard sets forth the period after the balance sheet date during which management of a reporting entity should evaluate events or transactions that occur for a potential recognition or disclosure in the financial statements, the circumstances under which an entity should recognize events or transactions occurring after the balance sheet date in its financial statements and the disclosures that an entity should make about events or transactions that occurred after the balance sheet date. This standard is effective for interim and annual periods ending after June 15, 2009. The adoption of this standard did not have a material impact on our financial statements.
Income Taxes
Except for the pre-acquisition federal and state tax filings for Magen BioSciences, Inc. and certain separate state filings, our operations have been included in the consolidated federal and combined state tax returns of PPD. As such, except for the pre-acquisition tax attributes of Magen and some losses from certain separate filing states, the tax attributes of our operations have been utilized or paid by PPD. Thus, the tax attributes which have been included in PPD’s combined returns have not been accounted for in the results of our operations.
Potential Volatility of Annual Operating Results
Our annual operating results have fluctuated in the past, and we expect that they will continue to fluctuate in the future. Factors that could cause these fluctuations to occur include:
| • | the timing of our milestone payments or other revenue, if any; |
| • | our dependence on a small number of compounds and collaborations; |
| • | success or failure of clinical trials and other aspects of developing and commercializing our product candidates; |
| • | our ability to properly manage our growth; |
| • | impairment of intangible assets; |
| • | the timing and amount of costs associated with R&D and compound partnering collaborations; |
| • | our ability to recruit and retain experienced personnel; |
| • | the timing and extent of new government regulations; and |
| • | intellectual property risks. |
Quantitative and Qualitative Disclosures about Market Risk
We expect to invest the cash and cash equivalents contributed to us by PPD consistent with PPD’s current investment policies. Therefore, we expect to maintain a non-trading investment portfolio of investment grade,
67
Table of Contents
highly liquid debt securities, which are designed to limit the amount of credit exposure to any one issue, issuer or type of instrument. We do not plan to use derivative financial instruments for speculative or trading purposes. We expect to carry our investments in debt securities at fair value, estimated as the amount at which an asset or liability could be bought or sold in a current transaction between willing parties. We expect to diversify our credit risk and invest in debt securities with high credit quality.
Our Relationship with PPD after the Spin-Off
General
Immediately prior to the spin-off, we will be a wholly owned subsidiary of PPD. After the spin-off, PPD will not have any ownership interest in our common stock, and we will be an independent, publicly traded company.
We will enter into agreements with PPD or one of its wholly owned subsidiaries prior to the spin-off to govern the terms of the spin-off and to define our ongoing relationship following the spin-off including PPD’s appointment as a preferred provider of CRO services to us under the Master Development Services Agreement, allocating responsibility for obligations arising before and after the spin-off, including obligations with respect to liabilities relating to PPD’s business and to Furiex’s business and obligations with respect to our employees, transition services and taxes. These agreements were negotiated while we were still part of PPD. While we believe the terms of these agreements are reasonable, they might not reflect terms that would have resulted from arm’s-length negotiations between unaffiliated third parties.
The following descriptions are summaries of the terms of the agreements. Any of these agreements that are material have been or will be filed as exhibits to the registration statement into which this Information Statement is incorporated and the summaries of such agreements are qualified in their entirety by reference to the full text of such agreements. We encourage you to read, in their entirety, each of the material agreements when they become available. The terms of these agreements have not yet been finalized; changes, some of which might be material, may be made prior to the spin-off.
Separation and Distribution Agreement
Prior to our separation from PPD, we will enter into the Separation and Distribution Agreement with PPD. The Separation and Distribution Agreement will set forth our agreements with PPD regarding the principal transactions necessary to separate us from PPD and other agreements that will govern our relationship with PPD after the completion of the separation, as described below.
The Separation and Distribution Agreement will identify assets to be transferred, liabilities to be assumed and contracts to be assigned to us as part of the separation of PPD into two independent companies, and will describe when and how these transfers, assumptions and assignments will occur. In particular, the Separation and Distribution Agreement will provide that, subject to the terms and conditions contained in the Separation and Distribution Agreement:
| • | All of the assets and liabilities associated or primarily used in connection with the Compound Partnering Business of PPD, including the related collaboration agreements, will be transferred to us, including all of PPD’s compound partnering intellectual property assets, and other assets and liabilities of the Compound Partnering Business; and |
| • | The CRO Business and related assets and liabilities will be retained by PPD. |
Except as expressly set forth in the Separation and Distribution Agreement or any ancillary agreement, all assets will be transferred to us on an “as is,” “where is” basis. So long as PPD is in compliance with the terms of the Separation and Distribution Agreement relating to the transfer, Furiex will bear the economic and legal risks that any conveyance will prove to be insufficient to vest in us good title, free and clear of any security interest,
68
Table of Contents
that any necessary consents or government approvals are not obtained and that any requirements of laws or judgments are not complied with. Information in this Information Statement with respect to the assets and liabilities of the parties following the separation is presented based on the allocation of such assets and liabilities pursuant to the Separation and Distribution Agreement, unless the context otherwise requires.
To the extent that any transfers contemplated by the Separation and Distribution Agreement have not been consummated on or prior to the date of the separation, the parties will agree to cooperate to affect such transfers as promptly as practicable following the date of the separation. In addition, each of the parties will agree to cooperate with each other and use reasonable commercial efforts to take or to cause to be taken all actions, and to do, or to cause to be done, all things reasonably necessary under applicable law or contractual obligations to consummate and make effective the transactions contemplated by the Separation and Distribution Agreement and the ancillary agreements.
The Separation and Distribution Agreement will also govern the rights and obligations of the parties regarding the proposed distribution. PPD will cause the distribution agent to electronically distribute to PPD shareholders that hold shares of PPD common stock as of the applicable record date all the issued and outstanding shares of our common stock. PPD will have the sole and absolute discretion to determine the terms of, and whether to proceed with, the distribution.
Except as otherwise provided in the Separation and Distribution Agreement or any ancillary agreement, each party will release and forever discharge the other party from all liabilities existing or arising from any acts or events occurring or failing to occur or alleged to have occurred or to have failed to occur or any conditions existing or alleged to have existed on or before the separation. The releases will not extend to obligations or liabilities under any agreements between the parties that remain in effect following the separation pursuant to the Separation and Distribution Agreement or any ancillary agreement. The Separation and Distribution Agreement provides that Furiex and PPD will indemnify the other for claims arising after the spin-off that relate to each entity’s business subsequent to the spin-off.
The Separation and Distribution Agreement will provide for “tail” insurance and the rights of the parties to report claims for occurrences prior to the separation and set forth procedures for the administration of insured claims. In addition, the Separation and Distribution Agreement will require PPD and us to maintain insurance policies for occurrences after the separation.
Other matters governed by the Separation and Distribution Agreement include, among others, access to financial and other records and information, legal privilege, confidentiality and resolution of disputes between the parties relating to the Separation and Distribution Agreement and the ancillary agreements and the agreements and transactions contemplated thereby.
Transition Services Agreement
Prior to our separation from PPD, we will enter into a Transition Services Agreement with PPD pursuant to which PPD will provide Furiex with a variety of administrative services for a period of time following the spin-off. Among the principal services PPD will provide to us are:
| • | accounting and finance support; |
| • | information technology services; and |
| • | human resources services. |
PPD will agree to make each service available to Furiex for cost plus 5%. The term of these transition services will be 12 months, with us having two six-month renewal options.
69
Table of Contents
Employee Matters Agreement
Prior to our separation from PPD, we will enter into an Employee Matters Agreement, which will govern the employee benefit obligations of PPD and us as they relate to current and former employees. The Employee Matters Agreement allocates liabilities and responsibilities relating to employee benefit matters, including 401(k) plan matters subject to ERISA in connection with the separation, as well as other employee benefit programs and for treatment of outstanding employee stock options.
Tax Sharing Agreement
Prior to our separation from PPD, we will enter into a Tax Sharing Agreement that generally will govern PPD’s and our respective rights, responsibilities and obligations after the separation with respect to taxes. Under the Tax Sharing Agreement, all tax liabilities and any tax refunds and credits (1) attributable to the Compound Partnering Business for any and all periods or portions thereof ending prior to or on the distribution date, (2) resulting or arising from the contribution of the Compound Partnering Business to us, and the distribution of our shares of common stock and the other separation transactions and (3) otherwise attributable to PPD, will be borne solely by or solely for the benefit of PPD. As a result, we generally expect to be liable only for tax liabilities attributable to, or incurred with respect to, the Compound Partnering Business after the distribution date. However, if we take any actions after PPD’s distribution of our shares that result in or cause the distribution to be taxable to PPD, we would be responsible under the Tax Sharing Agreement for any resulting taxes imposed on us or on PPD.
Master Development Services Agreement
For three years following the spin-off, PPD will provide Furiex with drug development services, specifically Phase I through IV clinical development services, pre-clinical services, post-approval services, program management, general drug development consulting, biostatistics and data management services, medical communications, regulatory consulting, laboratory services, including, but not limited to, analytical, bioanalytical, central laboratory and vaccine testing services, and such other drug development services as we require for our drug development candidates. Furiex will use PPD for its drug development needs as long as PPD has the expertise and capabilities to provide the required services. If PPD does not have the expertise and capabilities to provide the services Furiex requires, Furiex may seek to obtain those services from a third party provider. In addition, if PPD is in material breach of its obligations to provide services and does not cure such breach within 30 days after receipt of written notice (or if such breach cannot reasonably be so cured, PPD commences diligent efforts to cure during that period and continues them until the cure is completed within a reasonable time period), Furiex may terminate the applicable project agreement and use another party to provide the services. Furthermore, Furiex may terminate the Master Development Services agreement and the preferred provider relationship in the event that in any consecutive 12-month period, PPD on two or more occasions fails to remedy project specific material breaches within the applicable cure period, or PPD on two or more occasions fails to remedy a material breach under the agreement (other than the project specific breaches noted above) within the applicable cure period. PPD will also have customary termination rights if Furiex breaches the agreement and fails to cure within the applicable cure period.
Real Estate Agreements
Prior to the separation, we will enter into sublease and other agreements with PPD for office space to be utilized by us in our operations at our principal executive office in Morrisville, North Carolina and in other locations in the United States. Under these agreements, we will pay PPD base rent, additional rent associated with common area maintenance and other customary charges, and costs associated with PPD’s WAN connection and internet technology help desk support at the premises. We will also have to pay for any leasehold improvements we request. The sublease for our principal executive office has an initial term of two years with an option to renew for one additional year. The agreements covering our use of office space in other PPD locations have an initial term of one year with two six-month renewal options.
70
Table of Contents
Unaudited Pro Forma Condensed Combined Financial Statements
The unaudited pro forma combined financial statements presented below consist of the unaudited pro forma combined statement of operations for the year ended December 31, 2009 and the unaudited pro forma combined balance sheet as of December 31, 2009. The unaudited pro forma combined financial statements presented below should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our combined financial statements and corresponding notes included elsewhere in this information statement. The unaudited pro forma combined financial statements have been prepared giving effect to (1) the acquisition of Magen Biosciences, Inc., which occurred on April 2, 2009 and (2) the distribution and related transactions as if they occurred as of January 1, 2009.
The unaudited pro forma combined balance sheet and the unaudited pro forma combined statements of operations included in this information statement have been derived from the audited combined financial statements, and do not purport to represent what our financial position and results of operations actually would have been had the distribution and related transactions occurred on the date indicated, or to project our financial performance for any future period.
In connection with the proposed transaction to separate the company’s Compound Partnering Business from PPD’s core CRO Business, PPD will contribute substantially all of the assets and liabilities of its Compound Partnering Business to us, along with $100.0 million in cash and cash equivalents, all as a contribution to our capital. In addition, we will assume current accounts payable and other accrued expenses payable to third parties and incurred in the ordinary course of our Compound Partnering Business, which as of December 31, 2009, totaled approximately $4.6 million and has been included in the audited combined financial statements.
71
Table of Contents
FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
PRO FORMA COMBINED STATEMENT OF OPERATIONS (UNAUDITED)
(in thousands, except per share data)
| Historical (a) Company for the year ended 12/31/09 | Historical Magen for the period 1/1/09 – 4/2/09 | Subtotal Giving effect for the acquisition | Pro Forma Adjustments | As of December 31, 2009 Pro Forma | ||||||||||||||||
Net revenue: | ||||||||||||||||||||
Milestones | $ | 5,000 | $ | — | $ | 5,000 | $ | — | $ | 5,000 | ||||||||||
Royalties | 923 | — | 923 | — | 923 | |||||||||||||||
Service(b) | 389 | — | 389 | (389 | ) | — | ||||||||||||||
Total net revenue | 6,312 | — | 6,312 | (389 | ) | 5,923 | ||||||||||||||
Direct expenses(b) | 126 | — | 126 | (126 | ) | — | ||||||||||||||
Research and development expenses(c) | 24,334 | 1,110 | 25,444 | — | 25,444 | |||||||||||||||
Selling, general and administrative expenses (c) | 3,127 | 1,526 | 4,653 | — | 4,653 | |||||||||||||||
Depreciation and amortization | 183 | — | 183 | — | 183 | |||||||||||||||
Impairment of intangible asset(d) | 10,361 | — | 10,361 | — | 10,361 | |||||||||||||||
Total operating expenses | 38,131 | 2,636 | 40,767 | (126 | ) | 40,641 | ||||||||||||||
Loss from operations | (31,819 | ) | (2,636 | ) | (34,455 | ) | (263 | ) | (34,718 | ) | ||||||||||
Other income (expense), net(c) | (2 | ) | 1 | (1 | ) | — | (1 | ) | ||||||||||||
Loss from continuing operations | (31,821 | ) | (2,635 | ) | (34,456 | ) | (263 | ) | (34,719 | ) | ||||||||||
Basic and diluted net loss from continuing operations per share(e) | $ | — | ||||||||||||||||||
Basic and diluted weighted average shares outstanding(e) | — | |||||||||||||||||||
The accompanying notes are an integral part of these combined financial statements.
72
Table of Contents
FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
PRO FORMA COMBINED BALANCE SHEET (UNAUDITED)
(in thousands)
| As of December 31, 2009 Pro Forma | ||||||||||
| Historical | Adjustments | Pro Forma | ||||||||
| Assets | ||||||||||
Current assets: | ||||||||||
Cash and cash equivalents(f) | $ | 6 | $ | 100,000 | $ | 100,006 | ||||
Accounts receivable and unbilled services | 561 | — | 561 | |||||||
Prepaid expenses | 169 | — | 169 | |||||||
Other current assets(g) | 5,299 | (5,299 | ) | — | ||||||
Total current assets | 6,035 | 94,701 | 100,736 | |||||||
Property and equipment, net | 726 | — | 726 | |||||||
Goodwill | 49,116 | — | 49,116 | |||||||
Total assets | $ | 55,877 | $ | 94,701 | $ | 150,578 | ||||
| Liabilities and Pharmaceutical Product Development, Inc. Net Investment | ||||||||||
Current liabilities: | ||||||||||
Accounts payable | $ | 75 | $ | — | $ | 75 | ||||
Other accrued expenses(g) | 6,489 | (1,959 | ) | 4,530 | ||||||
Total current liabilities | 6,564 | (1,959 | ) | 4,605 | ||||||
Long-term deferred rent | 43 | — | 43 | |||||||
Total liabilities | 6,607 | (1,959 | ) | 4,648 | ||||||
Commitments and contingencies | ||||||||||
Shareholders’ Equity: | ||||||||||
Pharmaceutical Product Development, Inc. net investment(h) | 49,270 | (49,270 | ) | — | ||||||
Common stock, $0.001 par value, 100 shares authorized, issued and outstanding | — | — | — | |||||||
Additional paid-in capital | — | 145,930 | 145,930 | |||||||
Total liabilities and net investment | $ | 55,877 | $ | 94,701 | $ | 150,578 | ||||
The accompanying notes are an integral part of these combined financial statements.
73
Table of Contents
Notes to Unaudited Pro Forma Combined Financial Statements
Please note that, due to regulations governing the preparation of pro forma financial statements, the pro forma combined financial statements do not reflect certain estimated incremental expenses associated with being an independent, public company. We estimate these additional expenses, excluding stock option compensation for stock options which may be issued in the future and which the expense is not known at this time, are estimated to be approximately $3.4 million for the year ended December 31, 2009. The estimated incremental expenses associated with being an independent, public company include costs associated with corporate administrative service costs, executive compensation, internal audit, directors’ insurance, stock exchange listing fees, investor and public relations, legal and intellectual property costs. These unaudited pro forma combined financial statements reflect all other adjustments that, in the opinion of management, are necessary to present fairly the pro forma results of operations and financial position. The pro forma adjustments to the accompanying historical financial information for the fiscal year ended December 31, 2009 are described below:
| (a) | A separate column for Furiex Pharmaceuticals, Inc. has not been included because all balances of such entity have been included in this column. |
| (b) | Service revenue and related direct costs associated with services currently being provided to customers that will be provided by PPD to customers after the spin-off. |
| (c) | Reflects the results of operations assuming the Magen acquisition was consummated as of January 1, 2009. The amounts are based upon assumptions and estimates the Company believes are reasonable. |
| (d) | The impairment charge relates to the IPR&D acquired from Magen. |
| (e) | The actual number of our basic and diluted shares outstanding will not be known until closer to the actual distribution date. This information will be included prior to the effectiveness of the Form 10. |
| (f) | Reflects the contribution of $100.0 million in cash and cash equivalents to be received from PPD in connection with the distribution. |
| (g) | Includes (i) the $1.8 million withheld by PPD from the purchase price and placed in escrow to secure the sellers’ indemnification obligations and the associated liability related to the acquisition of Magen and (ii) the $3.4 million withheld by the purchaser of the assets of Piedmont Research Center and placed in escrow to secure PPD’s indemnification obligations. The funds in the escrow accounts and the associated rights and liabilities will remain with PPD after the spin-off. |
| (h) | The PPD net investment account represents the cumulative investments in, distribution from, and earnings (losses) of our company. PPD net investment at the date of the separation will be contributed to us as additional paid-in capital. As a result, these amounts have been reflected as equity (common stock and additional paid-in capital) as of December 31, 2009. |
74
Table of Contents
The following table sets forth information as of March 31, 2010 regarding individuals who serve as Furiex’s executive officers, including their positions.
Name | Age | Expected Position | ||
June S. Almenoff | 53 | President and Chief Medical Officer | ||
Gail McIntyre | 47 | Senior Vice President-Research | ||
Paul S. Covington | 53 | Senior Vice President-Clinical Operations | ||
Marshall Woodworth | 52 | Vice President—Finance and Treasurer |
June S. Almenoff is our President and Chief Medical Officer, which position she began on March 16, 2010. Dr. Almenoff had over 12 years of pharmaceutical industry experience at GlaxoSmithKline, or GSK, from 1997 to 2010. Most recently, she was Vice President in the Clinical Safety and Pharmacovigilance organization at GSK, where she served on the company’s senior governing medical boards and managed a diverse therapeutic portfolio supporting numerous regulatory approvals. Dr. Almenoff led the GSK teams that developed three pioneering systems for minimizing risk in early and late-stage drug development, which have been widely implemented by pharmaceutical companies and regulatory agencies. Dr. Almenoff worked in GSK’s licensing group, where she led the scientific diligence effort for the acquisition of Stiefel Laboratories and established a licensing program for a drug development unit. Dr. Almenoff received her B.A.cum laude from Smith College. She graduated from the M.D.-Ph.D. program at the Mt. Sinai School of Medicine and completed a residency in Internal Medicine and a Fellowship in Infectious Diseases at Stanford University Medical Center. Dr. Almenoff is a board-certified Fellow of the American College of Physicians with 10 years of clinical practice experience. Prior to joining GSK, Dr. Almenoff was on the faculty of Duke University Medical Center. She has more than 45 publications and one pending patent.
Gail McIntyre serves as our Senior Vice President—Research, which position she began on April 1, 2010. Prior to joining us, Dr. McIntyre was Senior Vice President, Research at PPD, a position she has held since October 2003. She has been with PPD for over 12 years. Dr. McIntyre has over 17 years in the drug discovery and development areas across numerous therapeutic areas (oncology, antiinfectives, antivirals, CNS, metabolic/endocrine, and dermatology) and various therapies (small drugs, treatment vaccines, immunomodulators, antibodies, immunoconjugates, and peptide mimetics). She has authored over 30 regulatory submissions and has successfully ushered compounds through the lead optimization phase to early drug development and IND as well as from early drug development through NDA. Dr. McIntyre received her B.A. (biology) cum laude from Merrimack College and was awarded her M.S. and Ph.D. degrees in Biochemistry and Biophysics from the University of North Carolina at Chapel Hill. She has worked in small private biotechnology companies and within a large pharmaceutical organization where she headed up the Preclinical Submissions Group within Regulatory Affairs. She is board certified in clinical pathology (hematology and clinical chemistry) by the American Society of Clinical Pathology and is a Diplomate of the American Board of Toxicology. She is a member of the American College of Toxicology, the American Society of Clinical Pathologists, the Drug Information Association, and the American Association for the Advancement of Science.
Paul S. Covington became our Senior Vice President-Clinical Operations on January 15, 2010. Dr. Covington was the Executive Vice President—Development with PPD from 2002 to 2008. From 2008 to 2010, Dr. Covington was an independent consultant. Dr. Covington joined PPD in September 1991 as a Medical Director. Dr. Covington is board certified in internal medicine and licensed in North Carolina and Alabama. Dr. Covington was in private practice in Clanton, Alabama from 1985 to 1990 where he served as Chief of Staff and Director of Critical Care and Cardiopulmonary Disease for the local hospital. From 1991 to 1992, he was Medical Director for the Birmingham site of Future Healthcare Research Centers.
Marshall Woodworth joined us on January 29, 2010 as our Vice President—Finance and Treasurer. Prior to that, Mr. Woodworth served as Vice President of Finance with Xerium Technologies, Inc. from March 2006 to
75
Table of Contents
May 2009. He was Division Finance Leader, Chemical and Industrial Divisions and then promoted to European Financial Leader while with Milliken & Company from April 2000 to March 2006. Mr. Woodworth received a B.S. degree in Geology from the University of Maryland, College Park and his M.B.A. in Finance from Indiana University.
Members of the Board of Directors
Our Board currently is comprised of five members. We do not have a classified Board. The terms of each director will expire upon the election and qualification of the directors to be elected at the 2011 annual meeting of shareholders.
The following individuals are serving on our Board:
Name | Age | |
Stuart Bondurant, M.D. | 80 | |
Peter B. Corr, Ph.D. | 61 | |
Wendy L. Dixon, Ph.D. | 54 | |
Fred N. Eshelman, Pharm.D. | 61 | |
Robert P. Ruscher | 49 |
Stuart Bondurant, M.D.is Emeritus Dean and Emeritus Professor of Medicine at the School of Medicine of the University of North Carolina at Chapel Hill. He was Interim Executive Vice President and Executive Dean for Health Sciences at Georgetown University in Washington, D.C. from 2004 to 2007. He was Dean of the School of Medicine at the University of North Carolina (Chapel Hill) from 1979 to 1994 and from 1996 to 1997. He served as Professor of Medicine at the University of North Carolina (Chapel Hill) from 1979 to 2004. He served as President and Dean of Albany Medical College from 1974 to 1979. Dr. Bondurant was Director of Epidemiologic Studies at the New York Academy of Medicine from January 1995 until March 1996. Dr. Bondurant has served as President of the American College of Physicians and the Association of American Physicians. He has also served as Interim President of the Institute of Medicine of the National Academy of Science and as Vice President of the American Heart Association and the American Society for Clinical Investigation. He served as the Chairman of the Board of Directors of the North Carolina Biotechnology Center from 1988 to 1992. Dr. Bondurant has served as a director of PPD since 1994. Dr. Bondurant also serves on the board of directors of PPD. Among other experience, qualifications, attributes and skills, Dr. Bondurant’s medical knowledge and experience and management skills support his service as a director of our company in light of our business and structure.
Peter B. Corr, Ph.D.is Co-Founder and General Partner of Celtic Therapeutics Management LLP. Dr. Corr retired from Pfizer Inc. in December 2006 where he was Senior Vice President for Science and Technology, a position he had held since 2002. In 2002 and 2003, he also headed worldwide pharmaceutical research and development for Pfizer. Previously, Dr. Corr served as Executive Vice President, Pfizer Global Research & Development; and President, Worldwide Development. Prior to Pfizer, Dr. Corr served as Senior Vice President, Discovery Research, at Monsanto/Searle and President of Pharmaceutical Research and Development at Warner Lambert/Parke Davis. Dr. Corr, who received his Ph.D. from Georgetown University School of Medicine, spent 18 years as a researcher in molecular biology and pharmacology at Washington University in St. Louis. When he left Washington University, Dr. Corr was Professor, Department of Medicine (Cardiology) and Professor, Department of Pharmacology and Molecular Biology. His research has been published in more than 160 scientific manuscripts. In addition to his work at Pfizer, Dr. Corr was Chairman of the Science & Regulatory Executive Committee of the Pharmaceutical Research and Manufacturers of America (PhRMA); Chairman of the PhRMA Foundation Board of Directors; and Chairman of the Hever Group, representing Chief Scientific Officers (CSO’s) across the European and U.S. based pharmaceutical industry. Among other experience, qualifications, attributes and skills, Dr. Corr’s executive experience at a leading pharmaceutical research and development company supports his service as a director of our company in light of our business and structure.
76
Table of Contents
Wendy L. Dixon, Ph.D.has a 30 year career in the pharmaceutical and biotechnology business, combining a technical background and experience in drug development and regulatory affairs with commercial responsibilities in building and leading organizations and launching and growing more than 20 pharmaceutical products including Tagamet, Fosamax, Singulair, Plavix, Abilify, Reyataz, and Baraclude. From 2001 to 2009 she was Chief Marketing Officer and President, Global Marketing for Bristol-Myers Squibb, and served on the CEO’s Executive Committee. From 1996 to 2001 she was Senior VP Marketing at Merck and prior to that she held executive management positions at West Pharmaceuticals, Osteotech, and Centocor and various positions at SmithKline and French (now GSK) in marketing, regulatory affairs, project management and as a biochemist. Dr. Dixon has served on the board of directors of DENTSPLY International (Nasdaq:XRAY), a publicly traded company, since July 2005. Among other experience, qualifications, attributes and skills, Dr. Dixon’s technical background in drug development and regulatory affairs supports her service as a director of our company in light of our business and structure.
Fredric N. Eshelman, Pharm.D.is our Founding Chairman, and served as Chief Executive Officer of PPD from July 1990 until July 2009, and as Vice Chairman of the Board of Directors of PPD from July 1993 until July 2009. Since July 2009, he has been PPD’s Executive Chairman. Dr. Eshelman founded PPD’s predecessor and served as its Chief Executive Officer until its sale to the company in 1989. Dr. Eshelman served as Senior Vice President, Development and as a director of Glaxo Inc., a subsidiary of Glaxo Holdings plc, from 1989 through 1990 before rejoining PPD. Among other experience, qualifications, attributes and skills, as PPD’s founder and a key participant in the development of our Compound Partnering Business, Dr. Eshelman’s unique experience and perspective on our business supports his service as a director of our company in light of our business and structure.
Robert P. Ruscherhas more than 20 years of experience working closely with emerging growth, technology-based companies (privately held/venture capital backed and publicly traded), as well as the investors and investment banks that help finance them. Mr. Ruscher began his business career as a certified public accountant working with San Francisco Bay area technology companies for three years, which was followed by seven years as a corporate attorney representing primarily life science companies, including those focused in the fields of biotechnology and specialty pharmaceuticals. In 1995 Mr. Ruscher joined Salix Pharmaceuticals (gastroenterology specialty pharmaceutical company) as Director, Business Development and advanced to positions of increasing responsibility, culminating in becoming President and Chief Executive Officer and a member of the Board of Directors in 1999. As President and Chief Executive Officer, Mr. Ruscher formulated and implemented Salix’s strategic change from a “virtual” model to a successful product-based sales and marketing focused specialty pharmaceutical company, including managing significant employee growth (7 to 125), overseeing a successful FDA New Drug Application approval process for Salix’s first product and receiving an FDA approvable letter for its second product. Mr. Ruscher served as President and Chief Executive Officer of Salix from 1999 to 2002, served as Executive Chairman of the Board from 2002 to 2003, and retired from Salix in 2005. Since that time he has been a private investor. Among other experience, qualifications, attributes and skills, Mr. Ruscher’s experience in public company management, business development and accounting supports his service as a director of our company in light of our business and structure.
Independence of Directors
All of our directors, other than Dr. Eshelman, qualify as independent directors as defined in Rule 4200 of the Nasdaq Marketplace rules for listed companies.
Each expected member of each of our Compensation, Nominating and Governance and Audit Committees is also expected to qualify as independent under Nasdaq’s Marketplace rules for listed companies.
Board Committees
Our Board has established the following three standing committees: Audit Committee; Compensation Committee; and Nominating and Governance Committee. Directors Bondurant, Dixon and Ruscher are the members of the Audit Committee and Mr. Ruscher qualifies as an “audit committee financial expert” as that term is defined in
77
Table of Contents
Item 407(d) of Regulation S-K promulgated by the SEC. Directors Corr, Dixon and Ruscher are the members of the Compensation Committee. Directors Bondurant, Corr and Ruscher are the members of the Nominating and Governance Committee.
Audit Committee
The primary purpose of our Audit Committee will be to oversee our accounting and financial reporting process and the audits of our combined financial statements, and on our compliance with legal and regulatory requirements.
The functions of our Audit Committee will include:
| • | Hiring the independent registered public accounting firm to conduct the annual audit of our financial statements and monitoring their independence and performance; |
| • | Reviewing and approving the planned scope of the annual audit and the results of the annual audit; |
| • | Pre-approving all audit services and permissible non-audit services provided by our independent registered public accounting firm; |
| • | Reviewing the accounting and reporting principles we apply in preparing our combined financial statements; |
| • | Reviewing our internal financial, operating and accounting controls with management and our independent registered public accounting firm; |
| • | Reviewing with management and our independent registered public accounting firm, as appropriate, our financial reports and our compliance with legal and regulatory requirements; and |
| • | Reviewing and approving any transaction that may present potential for conflict of interest, such as with our officers, directors or significant shareholders. |
Our Audit Committee will be responsible for reviewing and approving all related person transactions, including transactions with executive officers and directors, for potential conflicts of interests or other improprieties. Under SEC rules, related person transactions are those transactions to which we are or may be a party in which the amount involved exceeds $120,000, and in which any of our directors or executive officers or any other related person had or will have a direct or indirect material interest, excluding, among other things, compensation arrangements with respect to employment and Board membership. Our Audit Committee would approve a related person transaction if it determined that the transaction is in our best interests.
Our directors will be required to disclose to the committee or the full Board any potential conflict of interest, or personal interest in a transaction that our Board is considering. Our executive officers will be required to disclose any related person transaction to the Audit Committee. We also plan to poll our directors on an annual basis with respect to related person transactions and their service as an officer or director of other entities.
Any director involved in a related person transaction that is being reviewed or approved must recuse himself or herself from participation in any related deliberation or decision. Whenever possible, the transaction should be approved in advance and if not approved in advance, must be submitted for ratification as promptly as practical.
Compensation Committee
The primary purpose of our Compensation Committee will be to discharge our Board’s responsibilities relating to compensation of our executive officers and employees and to administer our equity compensation and other benefit plans. In carrying out these responsibilities, our Compensation Committee will review all components of executive officer and employee compensation for consistency with our Compensation Committee’s compensation philosophy, as in effect from time to time.
78
Table of Contents
The functions of our Compensation Committee will include:
| • | Designing and implementing competitive compensation policies to attract and retain key personnel; |
| • | Reviewing and formulating policy and determining the compensation of our executive officers and employees; |
| • | Reviewing and recommending to the Board the compensation of our directors; and |
| • | Administering our equity incentive plans and granting equity awards to our employees and directors under these plans. |
Our principal executive officer will not participate in the determination of her own compensation or the compensation of directors. However, it is intended that she would make recommendations to our Compensation Committee regarding the amount and composition of the compensation of our other officers and participate in the Compensation Committee’s deliberations about these other officers’ compensation.
The Compensation Committee will have the authority to engage compensation consultants or other advisors that it deems appropriate to assist it with its duties.
PPD’s Compensation Committee retained an independent compensation consultant, Frederic W. Cook & Co., Inc., to advise on various matters related to compensation of officers and directors and general compensation programs and matters, including in connection with planning for the spin-off. We expect to continue the engagement of the Cook firm to advise on these matters for us after the spin-off.
Our Compensation Committee would generally engage independent compensation consultants to provide:
| • | Assistance in selecting a peer group of companies for executive compensation comparison purposes; |
| • | Comparative market data on officer and board of director compensation practices and programs of peer companies and competitors; |
| • | Guidance on industry best practices and emerging trends and developments in officer and board director compensation; and |
| • | Advice on determining the total compensation of each of our officers and the material elements of total compensation, including (1) annual base salaries, (2) target cash bonus amounts, (3) stock option awards and (4) restricted stock awards. |
Nominating and Governance Committee
The primary purpose of our Nominating and Governance Committee will be to:
| • | Identify and evaluate individuals for possible Board members; |
| • | Select, and recommend to our Board, director nominees for each election of directors; |
| • | Develop and recommend to our Board criteria for selecting qualified director candidates; |
| • | Recommend any corporate governance guidelines it deems appropriate; and |
| • | Provide oversight in the evaluation of our Board and each committee of our Board. |
Our Nominating and Governance Committee will regularly assess the optimum size of our Board and its committees and the needs of our Board for various skills, background and business experience in determining whether it is advisable to consider additional candidates for nomination.
79
Table of Contents
In fulfilling its responsibilities to select, and recommend to our Board, director nominees for each election of directors, our Nominating and Governance Committee will consider the following factors:
| • | the appropriate size of our Board and its committees; |
| • | the perceived needs of our Board for particular skills, background and business experience; |
| • | the skills, background, reputation, and business experience of nominees compared to the skills, background, reputation, and business experience already possessed by other Board members; |
| • | nominees’ independence from management; and |
| • | applicable regulatory and listing requirements, including independence requirements and legal considerations. |
The goal of our Nominating and Governance Committee’s will be to assemble a Board that brings to Furiex a variety of perspectives and skills derived from high quality business and professional experience. Directors should possess high personal and professional ethics, integrity and values, and be committed to representing the best interests of our shareholders. They must also have an inquisitive and objective perspective and mature judgment. Director candidates, in the judgment of our Nominating and Governance Committee, must also have sufficient time available to perform all Board and committee responsibilities. Board members are expected to prepare for, attend and participate in all Board and applicable committee meetings.
Other than the foregoing, there are no stated minimum criteria for director nominees, although our Nominating and Governance Committee may also consider such other factors as it may deem, from time to time, to be in the best interests of the company and our shareholders.
Our Nominating and Governance Committee will annually evaluate our Board members who are willing to continue in service against the criteria set forth above in determining whether to recommend these directors for re-election.
Candidates for nomination as director may come to the attention of our Nominating and Governance Committee from time to time through incumbent directors, management, shareholders or third parties. These candidates may be considered at meetings of our Nominating and Governance Committee at any point during the year. Such candidates will be evaluated against the criteria set forth above. If our Nominating and Governance Committee believes at any time that it is desirable that our Board consider additional candidates for nomination, the Committee may poll directors and management for suggestions or conduct research to identify possible candidates and may, if our Nominating and Governance Committee believes it is appropriate, engage a third-party search firm to assist in identifying qualified candidates.
Our Nominating and Governance Committee’s policy will be to evaluate any recommendation for director nominee proposed by a shareholder and our bylaws also permit shareholders to nominate directors for consideration at an annual meeting, subject to certain conditions. Any recommendation for director nominee must be submitted in writing to:
Furiex Pharmaceuticals, Inc.
Attention: Corporate Secretary
3900 Paramount Parkway, Suite 150
Morrisville, North Carolina 27560
Our bylaws will require that any director nomination made by a shareholder for consideration at an annual meeting must be received in writing not more than 90 days nor less than 60 days in advance of the meeting, and at a special meeting called for the purpose of the election of directors not later than the close of business on the tenth business day following the date on which notice of such meeting is first given to our shareholders.
80
Table of Contents
Each written notice containing a shareholder nomination of a director at an annual or special meeting must include:
| • | the name and address of the shareholder who intends to make the nomination and any shareholder associated with such shareholder, and the name and residence address of the person or persons to be nominated; |
| • | the class and number of shares that are beneficially owned by the shareholder and any associated shareholder; |
| • | a representation that the shareholder is a holder of record of stock entitled to vote at the meeting and intends to appear in person or by proxy at the meeting to nominate the person or persons specified in the notice; |
| • | a description of all arrangements or understandings between the shareholder and each nominee and any other person or persons (naming such person or persons) pursuant to which the nomination or nominations are to be made by the shareholder; |
| • | such other information regarding each nominee proposed by such shareholder as would be required to be disclosed in solicitations of proxies for election of directors, or as would otherwise be required, in each case pursuant to Regulation 14A under the Exchange Act including any information that would be required to be included in a proxy statement filed pursuant to Regulation 14A had the nominee been nominated by the board of directors; |
| • | the written consent of each nominee to be named in a proxy statement and to serve as director of the corporation if so elected; |
| • | whether and the extent to which any hedging or other transaction or series of transactions has been entered into by or on behalf of, or any other agreement, arrangement or understanding (including any short position or any borrowing or lending of shares) has been made, the effect or intent of which is to mitigate loss to or manage risk or benefit of share price changes for, or to increase or decrease the voting power of, such shareholder of any associated shareholder with respect to any share of our common stock; and |
| • | to the extent known by the shareholder giving the notice, the name and address of any other shareholder supporting the nominee for election or reelection as a director or the proposal of other business on the date of such shareholder’s notice. |
Compensation of Directors
The cash and equity compensation payable to non-employee directors (“Outside Directors”) is described below. Members of our Board who are also employees of Furiex are not entitled to any compensation with respect to their service as Board members.
Cash Compensation
Each Outside Director will receive a retainer of $25,000 per year.
The Chairperson of the Audit Committee will receive a retainer of $10,000 per year and the Chairperson of the Compensation Committee and the Nominating and Governance Committee each will receive a retainer of $7,500 per year for his or her service.
81
Table of Contents
We intend for each Outside Director to also receive cash compensation for attendance at meetings of our Board and committees of our Board in the amount of $2,000 for attendance in person and $1,000 for attendance by telephone.
All cash compensation payable to Outside Directors for their service on our Board and its committees and attendance on meetings will be paid on a quarterly basis in arrears.
We also intend to reimburse our directors for their travel expenses for Board and committee meetings.
Equity Compensation
For Outside Directors elected or appointed prior to the spin-off, each Outside Director will receive an equity award consisting of options and/or shares of deferred stock in the amount of $100,000. Outside Directors will receive annual grants of options and/or shares of deferred stock in the amount of $70,000. Each grant will be made out of the 2010 Stock Plan. Options granted to our Board members for annual service would vest immediately prior to the next election of directors, subject to the Board member’s continued service.
The initial equity grants to Outside Directors are expected to be made soon after the distribution date.
Consulting Agreement with Dr. Eshelman
We intend to enter into a consulting agreement with Dr. Eshelman immediately prior to the distribution. Pursuant to the consulting agreement, Dr. Eshelman will provide us senior strategic oversight services, assist our President and Chief Medical Officer with executive functions, executive decision-making and senior management integration, assist us with investor relations matters and provide other services as needed or requested by us. We believe these services will be important to our initial and long-term success, given Dr. Eshelman’s knowledge of our business as well as his extensive experience and expertise in drug development. For his services, we will grant Dr. Eshelman an incentive stock option to purchase shares of our common stock equal to % of our common stock outstanding immediately after the completion of the spin-off. The option will vest over three years, with one third of the option vesting on each of the first, second and third anniversaries of the grant. The option exercise price will be equal to the fair market value of our common stock on the date of grant. We also will reimburse Dr. Eshelman for his reasonable and necessary business expenses incurred in connection with providing the services. The agreement may be terminated by us or by Dr. Eshelman at any time and for any reason upon 30 days written notice. If Dr. Eshelman terminates his engagement for any reason or if his engagement terminates due to his death or disability, then unless our board approves otherwise, any unvested options will terminate. If we terminate Dr. Eshelman’s engagement at any time without cause (as defined in our stock plan), then all unvested options will immediately vest. If we terminate Dr. Eshelman’s engagement for cause, then all unexercised options, whether vested or not, will terminate.
Compensation Discussion and Analysis
We have separated our discussion of executive compensation into the following sections:
| • | the guiding philosophy and objectives for the executive compensation program we intend to implement after the spin-off; |
| • | treatment of outstanding equity awards in connection with the spin-off; and |
| • | historical compensation of our named executive officers prior to the spin-off under PPD’s executive compensation programs. |
82
Table of Contents
Compensation Program Objectives
We seek to establish and implement a compensation system for our executive officers that is performance-oriented and designed to meet the following objectives:
| • | enable the Company to attract, retain and motivate its executive officers; |
| • | provide for compensation that is both externally competitive and internally equitable; |
| • | reward for outstanding Company and individual performance; |
| • | provide long-term incentive compensation through equity grants; and |
| • | promote long-term shareholder value. |
To achieve these objectives, we seek to design our executive compensation program and set compensation levels that are comparable to those of other companies that compete with us for executive talent. We plan to use both objective and subjective criteria to evaluate company and individual performance and set compensation. This approach should allow the Compensation Committee to exercise discretion and not rely solely on rigid formulas and quantitative analyses. We also generally target cash compensation at the 50th percentile of our peer companies and supplement that with additional executive compensation in the form of equity grants. Through a discretionary annual cash incentive plan, the Compensation Committee will also seek to reward outstanding performance, incentivize future achievement and assist in attracting and retaining our executive officers. We expect to provide long-term compensation in the form of stock options that generally vest over a three-year period. We believe these awards will allow the executive officers to participate in the long-term success of the Company, align their interests with those of our shareholders, reward for past performance and incentivize future performance, and help retain talented executive management personnel.
We intend to rely on various sources of information to assist us in establishing and maintaining our compensation program, including an independent compensation consulting firm, surveys and the experience and knowledge of our senior management. The Compensation Committee will use information from these sources to assist it in evaluating the competitiveness of the company’s executive compensation programs, setting compensation levels for the executive officers and meeting the Compensation Committee’s stated compensation objectives.
We believe that the Company’s compensation programs for its executive officers are competitive and appropriately designed to attract and retain key employees, reward superior performance and promote long-term shareholder value. The Compensation Committee plans to continue to review the compensation payable to the Company’s executive officers, periodically evaluate the Company’s compensation practices with the assistance of an independent compensation consultant and make any changes it deems appropriate to the Company’s compensation structure to ensure that the programs are designed and implemented to achieve the Committee’s stated goals.
Executive Compensation Policy
The principal components of compensation for the Company’s executive officers are base salary, discretionary annual cash incentive awards and long-term incentive compensation in the form of equity grants. The company also provides severance benefits upon a change in control of the Company, a 401(k) retirement savings plan with matching contributions from the Company, a group health plan, group term life insurance, and short-term disability and long-term disability coverage. The Company does not maintain supplemental retirement programs for its executive officers because we believe that the existing compensation arrangements enable the Company’s executive officers to adequately plan for their retirement.
83
Table of Contents
The Company has entered into employment agreements with each of its executive officers. We believe that employment agreements with shorter terms and annual performance reviews promote better performance by Company executives. Thus, we follow a general policy that employment agreements for executives will have an initial term of one year, with a provision for successive one-year renewal terms unless either party gives notice to the other that the agreement will not be renewed.
Peer Group Compensation Data
We expect that an important component of our executive compensation analyses will be a comparison of our executive compensation to that paid by a group of peer companies. We believe it will be a useful tool because we compete with the peer companies for executive talent.
Tally Sheets
We also intend to review tally sheets setting forth and totaling all components of compensation for each of the Company’s executive officers, including base salary, annual cash incentive awards, equity awards and other perquisites. The overall purpose of these tally sheets is to bring together, in one place, all the elements of compensation of the executive officers. This will allow us to quantitatively analyze both the individual elements of compensation and the aggregate amounts of compensation. It also will facilitate our review of the compensation of each of the executive officers compared to the others, which we will do in order to evaluate the internal equity of our executive compensation program.
Compensation Program Elements
Base Salary
Furiex’s base salary is designed to recognize the duties and responsibilities of each executive office and the experience, knowledge, ability and skill of the executive officer that holds each such position. We believe that base salaries are an important component of the Company’s executive compensation program and are critical in attracting and retaining executive talent. The Compensation Committee plans to review base salaries of the Company’s executive officers on an annual basis. In setting annual base salaries, the Compensation Committee will take into consideration the Company’s overall financial and operating performance in the prior year, the Company-wide target for base salary increases for all employees, market and competitive salary information as noted above, inflation, changes in the scope of an executive officer’s job responsibilities, other components of compensation and other relevant factors. The Compensation Committee will also review the executive officer’s individual performance. For the principal executive officer, the Compensation Committee will evaluate performance and determine the salary adjustment. For the other executive officers, the Compensation Committee will receive an evaluation from the principal executive officer on that person’s performance and a recommendation for a salary adjustment.
Annual Incentive Cash Compensation
We expect that annual cash bonuses will be an important element of the Company’s executive compensation program. This component of compensation provides an incentive to the executive officers to achieve both Company and individual performance objectives and to be rewarded for those achievements. The Compensation Committee expects to establish bonus ranges and targets that are designed to ensure that targeted incentive and total cash compensation is competitive.
2010 Stock Plan
Our 2010 Stock Plan, or the Stock Plan, was approved by our Board and our sole shareholder, PPD, in February 2010. The types of equity awards that may be granted include stock options, stock appreciation rights, restricted stock and other stock-based or cash-based awards.
84
Table of Contents
An amount of shares equal to 18% of our outstanding common stock immediately after the spin-off will be initially authorized and reserved for issuance under the Stock Plan. The Stock Plan’s available share reserve is reduced by one share for each share issued upon exercise of a stock option or stock appreciation right, or pursuant to a “full value award” (e.g., restricted stock). Shares subject to awards which expire or are cancelled or forfeited will again become available for issuance under the Stock Plan. The shares available will not be reduced by awards settled in cash or by shares withheld to satisfy tax withholding obligations. Only the net number of shares issued upon the exercise of stock appreciation rights or options exercised by means of a net exercise or by tender of previously owned shares will be deducted from the shares available under the Stock Plan.
Awards may be granted under the Stock Plan to our employees, officers, directors and consultants or those of any present or future parent or subsidiary corporation or other affiliated entity. While we may grant incentive stock options only to employees, we may grant nonstatutory stock options, stock appreciation rights, restricted stock and other stock-based or cash-based awards to any eligible participant.
Appropriate adjustments will be made in the number of shares and reserved under the Stock Plan and in outstanding awards to prevent dilution or enlargement of participants’ rights in the event of a stock split or other change in our capital structure.
In the event of an acquisition as described in the Stock Plan, the acquiring or successor entity, unless otherwise provided by the board or a board designated committee, in its sole discretion, shall assume or continue all or any awards outstanding under the Stock Plan or substitute substantially equivalent awards. For stock rights that are so assumed or substituted, in the event of a termination of grantee’s employment or consulting relationship by the Company or its successor other than for cause or by grantee for good reason as described in the Stock Plan within 60 days prior to and 180 days after an acquisition, all stock rights held by such grantee will become vested and immediately and fully exercisable and all forfeiture restrictions will be waived. If the board, the board designated committee, or the successor board does not make appropriate provisions for the continuation of such stock rights by either assumption or substitution, unless otherwise provided by the board or committee in its sole discretion, stock rights will become vested and fully and immediately exercisable and all forfeiture restrictions will be waived and all stock rights not exercised at the time of the closing of an acquisition will terminate.
We intend to grant stock options to our executive officers and, as noted above, to our directors, shortly after the distribution date.
Policy on Timing of Equity Grants
We do not have any plan or practice to time equity grants in coordination with our public release or disclosure of material nonpublic information. We also do not time our release of material nonpublic information for purposes of affecting the value of compensation to employees, including our officers. Our equity grant policies provide that:
| • | Equity grants approved by the Compensation Committee will be granted effective as of the date of the meeting, unless another later date is specified by the Compensation Committee, at its discretion, including because the public announcement of material information is anticipated. |
| • | Equity grants approved by the Compensation Committee pursuant to a unanimous written consent will be effective as of the first business day of the week following the receipt by the company of the last signature required for such consent, unless another effective date is specified by the terms of such consent, which date shall be no earlier than the date the written consent shall become effective. |
Employment Agreements
June S. Almenoff, MD, Ph.D., President and Chief Medical Officer, entered into an employment agreement with the Company effective March 16, 2010. The employment agreement provides for automatic one-year
85
Table of Contents
renewals unless either party gives notice to the other at least 90 days prior to the end of the then current term that the agreement will not be renewed. In the event we were to terminate Dr. Almenoff’s employment without cause, she will receive severance pay equal to 12 months of base salary at the time of termination. Ms. Almenoff’s base salary will be $350,000.
Gail McIntyre, Senior Vice President-Research, entered into an employment agreement with the Company effective April 1, 2010. The employment agreement provides for automatic one-year renewals unless either party gives notice to the other at least 90 days prior to the end of the then current term that the agreement will not be renewed. In the event we were to terminate Dr. McIntyre’s employment without cause, she will be paid the salary for the remaining term of the agreement. Dr. McIntyre’s base salary will be $265,000.
Paul S. Covington, M.D., Senior Vice President-Clinical Operations, entered into an employment agreement with the Company effective January 15, 2010. The employment agreement provides for automatic one-year renewals unless either party gives notice to the other at least 90 days prior to the end of the then current term that the agreement will not be renewed. In the event we were to terminate Dr. Covington’s employment without cause, he will be paid the salary for the remaining term of the agreement. Dr. Covington’s base salary will be $285,000.
Marshall Woodworth, Vice President-Finance, entered into an employment agreement with the Company effective January 29, 2010. The employment agreement provides for automatic one-year renewals unless either party gives notice to the other at least 90 days prior to the end of the then current term that the agreement will not be renewed. In the event we were to terminate Mr. Woodworth’s employment without cause, he will be paid the salary for the remaining term of the agreement. Mr. Woodworth’s base salary will be $220,000.
Change in Control Severance Agreements
We have entered into severance agreements with each of our executive officers. Each agreement provides for a severance payment upon termination of the executive officer’s employment, other than for cause, within one year after a change in control of the company, including constructive termination of employment due to a substantial diminution in duties, reduction in base salary or required relocation. The amount of the severance payment equals a factor times the sum of (i) the executive officer’s base salary for the 12-month period prior to termination of employment plus (ii) the greater of (A) the executive officer’s target bonus under the Company’s incentive cash bonus plan in effect immediately prior to termination of employment or (B) the average of the cash bonuses received by the executive officer in the two successive 12-month periods immediately prior to termination of employment. Dr. Almenoff is entitled to a severance payment equal to two times the severance amount as calculated above. Dr. Covington, Dr. McIntyre and Mr. Woodworth are entitled to a severance payment equal to one times the severance amount calculated above. If triggered, the Company is obligated to make the severance payment to the executive officer in a lump sum within a specified time following the termination of employment with the Company.
In addition to the severance payment, the company is obligated to pay any accrued but unpaid bonus or deferred compensation owed, and all outstanding unvested stock options and unvested restricted stock granted at least six months prior to the executive officer’s termination of employment will vest upon termination. In addition, the executive officer shall continue to be treated under each stock award agreement as an employee of the Company until the first to occur of (i) the third anniversary of the termination of employment, or (ii) the expiration of the exercise period provided in the stock award agreement. The Company is also obligated to continue to pay for and provide the benefits that the executive officer was receiving immediately prior to termination of employment, including health, medical, dental, vision, wellness, accidental death and dismemberment, disability, and group term life insurance benefits. Dr. Almenoff is entitled to these benefits for two years following termination of her employment. Dr. Covington, Dr. McIntyre and Mr. Woodworth are entitled to these benefits for one year following termination of their employment. If triggered, the Company may discontinue providing these benefits to any executive officer on the date on which he or she becomes eligible for
86
Table of Contents
similar coverage under another employer’s plan. The company will also pay all legal expenses incurred by an executive officer in successfully enforcing the change in control severance agreement. The Company is not obligated to “gross up” an executive officer’s compensation for any excise, income or other tax due with respect to the severance benefits payable under these agreements.
Perquisites and Other Benefits
The Company plans to maintain broad-based benefits that are provided to all employees, including our executive officers. These benefits include a 401(k) retirement savings plan with matching contribution from the Company, a group health plan, group term life insurance, and short-term disability and long-term disability coverage.
Tax Considerations
Our Compensation Committee will consider the provisions of Section 162(m) of the Code and related Treasury regulations, which restrict deductibility for federal income tax purposes of executive compensation paid to our principal executive officer and each of the four other most highly compensated executive officers holding office at the end of any year to the extent such compensation exceeds $1,000,000 for any of such officers in any year and does not qualify for an exception under the statute or regulations. Our Compensation Committee’s policy will be to qualify our executives’ compensation for deductibility under applicable tax laws to the maximum extent possible, consistent with our compensation objectives. The members of our Compensation Committee will qualify as outside directors for purposes of exempting executive compensation from the limits on deductibility under Section 162(m). Our Compensation Committee will be solely responsible for granting stock options and other equity awards to officers.
Prohibition Against Certain Equity Transactions
Our Insider Trading Policy prohibits our officers and directors from engaging in “short” sales which could reasonably cause our officers and directors to have interests adverse to our shareholders. “Short” sales, which are sales of shares of common stock by a person that does not own the shares at the time of the sale, evidence an expectation that the value of the shares will decline in value. We prohibit our officers and directors from entering into “short” sales because such transactions signal to the market that the officer and director has no confidence in us or our short-term prospects and may reduce the officer’s or director’s incentive to improve our performance. In addition, Section 16(c) of the Exchange Act expressly prohibits officers and directors from engaging in short sales.
Treatment of Outstanding PPD Equity Awards in Connection with the Spin-Off
Each unvested option held by a PPD employee who becomes an employee of Furiex or its subsidiaries on the distribution date will automatically terminate on the distribution date. Each vested option held by such a person on the distribution date will remain outstanding and must be exercised prior to the earlier of the termination of the employee’s employment with Furiex or any of its parent or subsidiary companies for any reason (provided that the employee will be allowed to exercise during any post-termination period provided by the option) or the expiration date of the option. The per share exercise price of the option will be adjusted to be equal to the product of the pre-distribution option exercise price multiplied by a fraction, the numerator of which will be the per share price of PPD common stock on the day after the distribution and the denominator of which will be the per share price of PPD common stock on the day of the distribution. Following this adjustment to the per share exercise price, the number of shares subject to the option will be adjusted, if necessary, to ensure that the intrinsic value of the option is the same on the day after the distribution as it is on the day of the distribution.
87
Table of Contents
Historical PPD Compensation of Our Executive Officers Prior to the Spin-Off
The following tables do not include June S. Almenoff, Paul S. Covington or Marshall Woodworth because they joined us in 2010 and were not employed by PPD in any prior period except for Dr. Covington who received compensation from PPD in January 2009 prior to his resignation from PPD.
The following table sets forth information concerning the compensation earned by the officer of PPD who is expected to join us upon the spin-off and would be one of our named executive officers based on the compensation she received from PPD in 2009. This officer is Gail McIntyre, who is expected to be our Senior Vice President—Research. All of the information included in these tables reflects compensation earned by this individual, referred to as the named executive officer, for services with PPD. All references in the following tables to stock options, restricted stock, restricted stock units, and other stock awards relate to awards granted by PPD in regard to PPD common stock. The amounts and forms of compensation reported below do not necessarily reflect the compensation this person will receive following the spin-off, which could be higher or lower, because historical compensation was determined by PPD and future compensation levels will be determined based on the compensation policies, programs and procedures to be established by our Compensation Committee.
Summary Compensation Table
Name and Principal Position | Year | Salary ($) | Bonus ($) | Non-Equity Incentive Plan Compensation ($) | Stock Awards ($) | Option Awards (1) ($) | All Other Compensation (2) ($) | Total ($) | |||||||||||||
Gail McIntyre, Senior Vice President—Research | 2009 | $ | 241,657 | — | $ | 44,000 | — | $ | 130,584 | $ | 6,289 | $ | 416,997 | ||||||||
| (1) | The dollar value represents the aggregate grant date fair value of option awards granted in the fiscal year using the Black Scholes valuation methodology. Values are computed in accordance with Financial Accounting Standards Board Accounting Standards Codification Topic 718, Compensation— Stock Compensation. |
| (2) | All Other Compensation reflects $756 in a taxable benefit for premiums paid for PPD group life insurance and a PPD 401K match of $ 6,073. |
Grants of Plan-Based Awards During 2009
The following table lists each equity award granted by PPD during 2009 to our named executive officer.
Name | Grant Date | All Other Stock Awards: Number of shares of Stock or Units (#) | All Other Option Awards: Number of Securities Underlying Options (#) | Exercise or Base Price of Option Awards ($/Share) | Grant Date Fair Value of Stock and Option Award($)(1) | |||||||
Gail McIntyre | 02/18/09 | — | 20,000 | $ | 26.64 | $ | 130,584 | |||||
| (1) | The dollar value represents the aggregate grant date fair value of option awards granted in the fiscal year using the Black Scholes valuation methodology. Values are computed in accordance with Financial Accounting Standards Board Accounting Standards Codification Topic 718, Compensation— Stock Compensation. |
88
Table of Contents
Option Exercises and Stock Vested in 2009
The following table lists the number of shares of PPD’s common stock acquired upon exercise of options by our named executive officer during 2009 and the number of shares of restricted common stock that vested during 2009 held by our named executive officer.
| Option Awards | Stock Awards | |||||||
Name | Number of Shares Acquired on Exercise (#) | Value Realized on Exercise(1) ($) | Number of Share Acquired on Vesting (#) | Value Realized on Vesting (2) ($) | ||||
Gail McIntyre | — | — | — | — | ||||
Outstanding Equity Awards at December 31, 2009
The following table sets forth information regarding each unexercised option to purchase shares of PPD’s common stock of PPD held as of December 31, 2009 by our named executive officer.
| Option Awards | |||||||||||
Name | Number of Securities Underlying Unexercised Options(#) Exercisable | Number of Securities Underlying Unexercised Options(#) Unexercisable | Option Exercise Prices($) | Option Expiration Date | |||||||
Gail McIntyre | 60,000 | (1) | $ | 21.19 | 11/16/14 | ||||||
| 20,000 | (2) | $ | 34.62 | 02/22/16 | |||||||
| 10,000 | (3) | 5,000 | $ | 33.61 | 02/21/17 | ||||||
| 6,683 | (4) | 13,367 | $ | 44.78 | 02/20/18 | ||||||
| 3,000 | (5) | 6,000 | $ | 39.01 | 10/03/18 | ||||||
| 3,000 | (6) | 6,000 | $ | 20.64 | 11/20/18 | ||||||
| 0 | 20,000 | (7) | $ | 26.64 | 02/18/19 | ||||||
| (1) | These options were granted on November 16, 2004. |
| (2) | These options were granted on February 22, 2006. |
| (3) | These options were granted on February 21, 2007. |
| (4) | These options were granted on February 20, 2008. |
| (5) | These options were granted on October 3, 2008. |
| (6) | These options were granted on November 20, 2008. |
| (7) | These options were granted on February 18, 2009. |
89
Table of Contents
Potential Payments Upon Termination or Change in Control
The terms of our retention and severance for all of our executive officers are described under the heading “Change in Control and Severance Benefits” in the “Compensation Discussion and Analysis” section of this Information Statement. If the severance payments called for in the agreements for Dr. Almenoff, Dr. McIntyre, Dr. Covington and Mr. Woodworth were to be triggered on December 31, 2010, we would be obligated to make the following payments (assuming no bonuses are due the executive officer in 2010):
Name | Cash Payment | Benefits ($ per month)(1) and (# of months paid) | Number of Options that Would Vest (Market Value)(2) | ||||||||
June S. Almenoff | $ | 700,000 | $ | 1,446 | (24 mos. | ) | — | ||||
Gail McIntyre | $ | 265,000 | $ | 114 | (12 mos. | ) | — | ||||
Paul S. Covington | $ | 285,000 | $ | 1,446 | (12 mos. | ) | — | ||||
Marshall Woodworth | $ | 220,000 | $ | 1,442 | (12 mos. | ) | — | ||||
| (1) | The benefit amounts are the illustrative rates provided by the insurance company because coverage has not been finalized. |
| (2) | No options to purchase shares of our common stock have been granted to our executive officers. |
Security Ownership of Certain Beneficial Owners and Management
As of the date of this Information Statement, all of the outstanding shares of our common stock are owned by PPD. In connection with the spin-off, PPD will distribute to its shareholders all of the outstanding shares of our common stock and will immediately thereafter own none of our common stock. The following table provides information with respect to the expected beneficial ownership of our common stock immediately upon the spin-off by (1) each of our shareholders who we believe would be a beneficial owner of more than 5% of our outstanding common stock based on currently available information, (2) each member of our Board, (3) each executive officer and (4) all of our executive officers and directors as a group. We based the share amounts on each person’s beneficial ownership of PPD common stock as of , 2010, unless we indicate some other basis for the share amounts, and assuming a distribution ratio of one share of our common stock for every shares of PPD common stock. To the extent our directors and executive officers own PPD common stock at the time of the separation, they will participate in the distribution on the same terms as other holders of PPD common stock. Except as otherwise noted in the footnotes below, each person or entity identified below has sole voting and investment power with respect to such securities. As used in this Information Statement, “beneficial ownership” means that a person has, or may have within 60 days, the sole or shared power to vote or direct the voting of a security and/or the sole or shared investment power with respect to a security (i.e., the power to dispose or direct the disposition of a security). Unless otherwise specified, the address of each named individual in the table below is the address of Furiex.
Name of Beneficial Owner or Identity of Group(1) | Shares Beneficially Owned | Percent of Outstanding | ||
BlackRock, Inc. (2) | ||||
Stuart Bondurant | ||||
Capital World Investors (3) | ||||
Peter R. Corr | ||||
Wendy L. Dixon | ||||
Frederic N. Eshelman | ||||
Robert P. Ruscher | ||||
June S. Almenoff | ||||
Gail McIntyre | ||||
Paul S.Covington | ||||
Marshall Woodworth |
| * | Less than 1% |
90
Table of Contents
| (1) | Except as indicated in the footnotes to this table, the persons named in the table have sole voting and investment power with respect to all shares of common stock shown as beneficially owned by them, subject to community property laws where applicable. |
| (2) | Based on the amount of PPD common stock reported on Schedule 13G filed on January 29, 2010. The Schedule 13G filed by BlackRock, Inc. does not list any natural persons having voting or investment powers over the shares beneficially owned by BlackRock, Inc. The address for BlackRock, Inc. is 40 East 52ndStreet, New York, New York 10022. |
| (3) | Based on the amount of PPD common stock reported on Schedule 13G Amendment No. 2 filed on February 10, 2010. The Schedule 13G filed by Capital World Investors does not list any natural persons having voting power and/or investment powers over the shares reported as beneficially owned by Capital World Investors. The address of Capital World Investors is 333 South Hope Street, Los Angeles, California 90071. |
Following PPD’s distribution of our common stock to PPD’s shareholders, we will have a continuing relationship with PPD as a result of the agreements we are entering into in connection with the distribution, including the Separation and Distribution Agreement, the Master Development Services Agreement, the Transition Services Agreement, the Employee Matters Agreement, and the Tax Sharing Agreement. For a detailed discussion of each of these agreements, please see “Our Relationship with PPD after the Spin-Off”.
Some of our officers and directors own shares of PPD common stock or options to acquire additional shares of PPD common stock because of their prior employment relationship with PPD or their service on the Board of PPD. Such shares represent approximately 6.5%, and the options represent less than 1.0%, of PPD stock outstanding as of March 31, 2010. The options to acquire PPD common stock held by Furiex employees that are unvested will terminate on the distribution date. Any vested options held by Furiex employees after the distribution will terminate on the earlier of the expiration of the options existing term or termination of the optionee’s employment with Furiex for any reason, provided that the employee will be allowed to exercise during any post-termination period provided by the option. Ownership of PPD common stock and options to acquire PPD common stock could create or appear to create conflicts of interest for such officers and directors when faced with decisions that could have disparate implications for PPD and us.
General
The following is a summary of information concerning our capital stock. The summary below does not purport to be complete statements of the relevant provisions of our amended and restated certificate of incorporation or of our bylaws. The summary is qualified in its entirety by reference to these documents, which you must read for complete information on our capital stock. Our amended and restated certificate of incorporation and bylaws are included as exhibits to our registration statement on Form 10.
Authorized Capital Stock
Immediately following the distribution, our authorized capital stock will consist of up to 40 million shares of common stock, par value $0.001 per share, and 10 million shares of preferred stock, par value $0.001 per share.
Common Stock
As of , 2010, PPD had approximately [ ] shareholders of record each of which would become a shareholder of Furiex upon the distribution if such shareholder continued to hold its PPD shares of common stock through the ex-dividend date for the spin-off distribution. As of , 2010, PPD had
91
Table of Contents
issued and outstanding [ ] shares of common stock, which would result in our having [ ] shares of common stock outstanding based on a distribution ratio of one share of our common stock for every shares of PPD common stock held on the record date.
Holders of common stock are entitled to one vote for per share for the election of directors and all other matters submitted to a vote of our shareholders. Subject to the rights of any holders of preferred stock that may be issued in the future, the holders of common stock are entitled to share ratably in such dividends as may be declared by our Board out of funds legally available therefor. In the event of our dissolution, liquidation or winding up, holders of common stock are entitled to share ratably in all assets remaining after payment of all liabilities and liquidation preferences of any preferred stock. Holders of common stock have no preemptive, subscription, redemption, conversion rights or similar rights. Our amended and restated certificate of incorporation does not provide for cumulative voting rights with respect to the election of directors. All outstanding common stock issued in the distribution will be fully paid and nonassessable.
Preferred Stock
Under the terms of our amended and restated certificate of incorporation, our Board will be authorized, subject to limitations prescribed by the Delaware General Corporation Law, or the DGCL, and by our amended and restated certificate of incorporation, to issue preferred stock in one or more series without shareholder approval. Our Board will have the discretion, subject to limitations prescribed by the DGCL and by our amended and restated certificate of incorporation, to determine the rights, preferences, privileges and restrictions, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences, of each series of preferred stock.
Anti-takeover Effects of our Amended and Restated Certificate of Incorporation and Bylaws and Delaware Law
Provisions of our amended and restated certificate of incorporation and bylaws and of Delaware law could make the following more difficult:
| • | acquisition of us by means of a tender offer; |
| • | acquisition of us by means of a proxy contest or otherwise; or |
| • | removal of our incumbent officers and directors. |
These provisions, which are summarized below, are expected to discourage coercive takeover practices and inadequate takeover bids. These provisions are also designed to encourage persons seeking to acquire control of us to first negotiate with our Board. We believe that the benefits of increased protection give us the potential ability to negotiate with the proponent of an unsolicited proposal to acquire or restructure us and outweigh the disadvantages of discouraging those proposals because negotiation of the proposals could result in an improvement of their terms.
Exclusive Right to Fix Size of Board of Directors and to Fill Vacancies
Our amended and restated certificate of incorporation and bylaws provide that the number of directors in our Board shall initially be three and, thereafter, shall be exclusively fixed by our Board, which has set the number of directors at five. Newly created directorships resulting from any increase in our authorized number of directors and any vacancies in our Board resulting from death, resignation, retirement, disqualification or other cause (including removal from office by a vote of the shareholders) will be filled by a majority of our Board then in office.
92
Table of Contents
Elimination of Shareholder Action by Written Consent
Our amended and restated certificate of incorporation and bylaws expressly eliminate the right of our shareholders to act by written consent. Shareholder action must take place at the annual or special meeting of our shareholders.
Special Shareholder Meetings
Our amended and restated certificate of incorporation and bylaws provide that only our Board or such person or persons designated by our Board may call special meetings of our shareholders.
Requirements for Advance Notification of Shareholder Nominations and Proposals
Our bylaws have advance notice procedures with respect to shareholder proposals and nominations of candidates for election as directors, other than nominations made by or at the direction of our Board or a committee of our Board. The business to be conducted at a meeting will be limited to business properly brought before the meeting by or at the direction of our Board or a duly authorized committee thereof or by a shareholder of record who has given timely written notice to our secretary of that shareholder’s intention to bring such business before such meeting.
Delaware Anti-takeover Law
Upon the distribution, we will be governed by Section 203 of the DGCL. Section 203, subject to certain exceptions, prohibits a Delaware corporation from engaging in any business combination with any interested shareholder for a period of three years following the time that such shareholder became an interested shareholder, unless:
| • | prior to such time, the board of directors of the corporation approved either the business combination or the transaction that resulted in the shareholder becoming an interested shareholder; or |
| • | upon consummation of the transaction that resulted in the shareholder becoming an interested shareholder, the interested shareholder owned at least 85.0% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding specified shares; or |
| • | at or subsequent to such time, the business combination is approved by the board of directors and authorized at an annual or special meeting of shareholders, by the affirmative vote of at least 66 2/3% of the outstanding voting stock that is not owned by the interested shareholder. The shareholders cannot authorize the business combination by written consent. |
The application of Section 203 may limit the ability of shareholders to approve a transaction that they may deem to be in their best interests.
In general, Section 203 defines “business combination” to include:
| • | any merger or consolidation involving the corporation and the interested shareholder; or |
| • | any sale, lease, exchange, mortgage, pledge, transfer or other disposition of 10.0% or more of the assets of the corporation to or with the interested shareholder; or |
| • | subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any of its stock to the interested shareholder; or |
93
Table of Contents
| • | any transaction involving the corporation that has the effect of increasing the proportionate share of the stock of any class or series of the corporation beneficially owned by the interested shareholder; or |
| • | the receipt by the interested shareholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation. |
In general, Section 203 defines an “interested shareholder” as any person that is:
| • | the owner of 15% or more of the outstanding voting stock of the corporation; or |
| • | an affiliate or associate of the corporation who was the owner of 15% or more of the outstanding voting stock of the corporation at any time within three years immediately prior to the relevant date; or |
| • | the affiliates and associates of the above. |
Under specific circumstances, Section 203 makes it more difficult for an “interested shareholder” to effect various business combinations with a corporation for a three-year period, although the shareholders may, by adopting an amendment to the corporation’s certificate of incorporation or bylaws, elect not to be governed by this section, effective 12 months after adoption.
Our amended and restated certificate of incorporation and bylaws do not exclude us from the restrictions imposed under Section 203. We anticipate that the provisions of Section 203 might encourage companies interested in acquiring us to negotiate in advance with our Board since the shareholder approval requirement would be avoided if a majority of the directors then in office approve either the business combination or the transaction that resulted in the shareholder becoming an interested shareholder.
No Cumulative Voting
Our amended and restated certificate of incorporation and bylaws do not provide for cumulative voting in the election of directors.
Undesignated Preferred Stock
The authorization in our amended and restated certificate of incorporation of undesignated preferred stock makes it possible for our Board to issue shares of our preferred stock with voting or other rights or preferences that could impede the success of any attempt to change control of us. The provisions in our amended and restated certificate of incorporation authorizing such preferred stock may have the effect of deferring hostile takeovers or delaying changes of control of our management.
Transfer Agent and Registrar
The transfer agent for our common stock is American Stock Transfer & Trust Company, LLC. Their address is 59 Maiden Lane, Plaza Level, New York, New York 10038. Their telephone number is (800) 937-5449.
Shareholders of the Furiex are not entitled to appraisal or dissenters’ rights with respect to the spin-off under Delaware law or our amended and restated certificate of incorporation or bylaws.
Indemnification of Officers and Directors
Section 145 of the DGCL authorizes a court to award, or a corporation’s board to grant, indemnity to directors and officers in terms sufficiently broad to permit such indemnification under certain circumstances for liabilities (including reimbursement for expenses incurred) arising under the Securities Act of 1933. Our amended and restated certificate of incorporation and bylaws provide for indemnification of our officers and directors to the maximum extent permitted by Delaware law.
94
Table of Contents
Distribution of Information Statement
We will pay the costs of distributing this Information Statement. The distribution will be made by mail.
Where to Obtain More Information
We have filed with the SEC a registration statement on Form 10 under the Exchange Act for the common stock being issued to you in the distribution. This Information Statement, filed as an exhibit to the registration statement and incorporated therein by reference, omits certain information contained in the registration statement and the other exhibits and schedules thereto, to which reference is hereby made. Statements contained herein concerning the provisions of any documents filed as exhibits to the registration statement are not necessarily complete, and are qualified by reference to the copy of such document. The registration statement, including exhibits and schedules filed therewith, may be inspected and copied at the public reference facilities maintained by the SEC at 100 F Street, N.E., Washington, D.C. 20549. Copies of such materials may be obtained at prescribed rates by writing to the SEC. You may obtain information regarding the operation of the public reference room by calling 1-800-SEC-0330. The SEC also maintains a website (http://www.sec.gov) that contains reports, proxy and information statements and other information regarding registrants that file electronically with the SEC.
We are not currently subject to the informational requirements of the Exchange Act. Following the distribution, we will be subject to such informational requirements, and in accordance therewith, we will file reports, proxy and information statements and other information with the SEC. Such reports, proxy and information statements and other information can be inspected and copied at the address set forth above. We intend to furnish our shareholders with annual reports containing financial statements audited by our independent accountants and quarterly reports for the first three quarters of each fiscal year containing unaudited summary financial information.
We will maintain an Internet site at www.furiex.com, which we expect to be operational on or before the date that the Form 10 is declared effective. Our website and the information contained on that site, or connected to that site, are not incorporated into this Information Statement or the registration statement on Form 10.
95
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
The combined financial statements of Furiex Pharmaceuticals, Inc. represent the financial position, operating results, and cash flows of the component businesses of the Discovery Sciences Segment of Pharmaceutical Product Development, Inc. These financial statements will become the historical financial statements of Furiex Pharmaceuticals, Inc. subsequent to the completion of its spin-off from PPD.
FURIEX PHARMACEUTICALS, INC.
The financial statements of Furiex Pharmaceuticals, Inc. represent the stand-alone financial statements solely of the recently organized registrant that will succeed to the business being spun off from PPD. These financial statements have been included because they are required by Securities and Exchange Commission rules related to recently organized registrants. Subsequent to the spin-off, these stand-alone financial statements will no longer be required.
| F-28 | ||
| F-29 | ||
| F-30 | ||
| F-31 | ||
| F-32 | ||
96
Table of Contents
MAGEN BIOSCIENCES, INC.
(A DEVELOPMENT STAGE COMPANY)
The financial statements of Magen Biosciences, Inc. (a development stage company) represent the financial position, operating results, and cash flows prior to being acquired by Pharmaceutical Product Development, Inc. These financial statements have been included because they are required by Securities and Exchange Commission Regulation S-X, Rule 3-05,Financial Statements of Businesses Acquiredor to be Acquired.
| F-37 | ||
| F-38 | ||
| F-39 | ||
| F-41 | ||
| F-42 | ||
97
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
INDEX TO COMBINED FINANCIAL STATEMENTS
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of Pharmaceutical Product Development, Inc.
We have audited the accompanying combined balance sheets of the component businesses of the Discovery Sciences segment of Pharmaceutical Product Development, Inc. (collectively the “Combined Financial Statements of Furiex Pharmaceuticals, Inc.” or the “Company”) as of December 31, 2009 and 2008, and the related combined statements of operations, changes in Pharmaceutical Product Development, Inc. net investment, and cash flows for each of the three years in the period ended December 31, 2009. These financial statements are the responsibility of the management of Pharmaceutical Product Development, Inc. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such combined financial statements present fairly, in all material respects, the combined financial position of the Company at December 31, 2009 and 2008, and the combined results of its operations and its cash flows for each of the three years in the period ended December 31, 2009, in conformity with accounting principles generally accepted in the United States of America.
| /s/ DELOITTE & TOUCHE LLP |
Charlotte, North Carolina |
February 24, 2010 |
F-2
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
COMBINED STATEMENTS OF OPERATIONS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(in thousands)
| 2007 | 2008 | 2009 | ||||||||||
Net revenue: | ||||||||||||
Milestones | $ | — | $ | 18,000 | $ | 5,000 | ||||||
Royalties | — | — | 923 | |||||||||
Service | 560 | 419 | 389 | |||||||||
Total net revenue | 560 | 18,419 | 6,312 | |||||||||
Direct expenses | 185 | 153 | 126 | |||||||||
Research and development expenses | 21,081 | 8,053 | 24,334 | |||||||||
Selling, general and administrative expenses | 1,780 | 1,738 | 3,127 | |||||||||
Depreciation and amortization | 270 | 94 | 183 | |||||||||
Impairment of intangible assets | — | 1,607 | 10,361 | |||||||||
Total operating expenses | 23,316 | 11,645 | 38,131 | |||||||||
Income (loss) from operations | (22,756 | ) | 6,774 | (31,819 | ) | |||||||
Other income (expense), net | 19 | 14 | (2 | ) | ||||||||
Income (loss) from continuing operations | (22,737 | ) | 6,788 | (31,821 | ) | |||||||
Discontinued operations, net of income taxes | (185 | ) | (976 | ) | 22,890 | |||||||
Net income (loss) | $ | (22,922 | ) | $ | 5,812 | $ | (8,931 | ) | ||||
The accompanying notes are an integral part of these combined financial statements.
F-3
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
COMBINED BALANCE SHEETS
AS OF DECEMBER 31, 2008 AND 2009
(in thousands)
| 2008 | 2009 | |||||
| Assets | ||||||
Current assets: | ||||||
Cash and cash equivalents | $ | — | $ | 6 | ||
Accounts receivable and unbilled services | 118 | 561 | ||||
Prepaid expenses | 37 | 169 | ||||
Other current assets | — | 5,299 | ||||
Current assets of discontinued operations | 2,825 | — | ||||
Total current assets | 2,980 | 6,035 | ||||
Property and equipment, net | 1 | 726 | ||||
Goodwill | 45,129 | 49,116 | ||||
Long-term assets of discontinued operations | 13,028 | — | ||||
Total assets | $ | 61,138 | $ | 55,877 | ||
| Liabilities and Pharmaceutical Product Development, Inc. Net Investment | ||||||
Current liabilities: | ||||||
Accounts payable | $ | — | $ | 75 | ||
Other accrued expenses | 387 | 6,489 | ||||
Unearned income | 20 | — | ||||
Current liabilities of discontinued operations | 4,459 | — | ||||
Total current liabilities | 4,866 | 6,564 | ||||
Long-term deferred rent | — | 43 | ||||
Long-term liabilities of discontinued operations | 748 | — | ||||
Total liabilities | 5,614 | 6,607 | ||||
Commitments and contingencies (Notes 7 and 11) | ||||||
Pharmaceutical Product Development, Inc. net investment | 55,524 | 49,270 | ||||
Total liabilities and net investment | $ | 61,138 | $ | 55,877 | ||
The accompanying notes are an integral part of these combined financial statements.
F-4
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
COMBINED STATEMENTS OF CHANGES IN PHARMACEUTICAL PRODUCT
DEVELOPMENT, INC. NET INVESTMENT
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(in thousands)
Balance at December 31, 2006 | $ | 58,895 | ||
Net transfers from parent | 20,897 | |||
Net loss | (22,922 | ) | ||
Balance at December 31, 2007 | 56,870 | |||
Net transfers to parent | (7,158 | ) | ||
Net income | 5,812 | |||
Balance at December 31, 2008 | 55,524 | |||
Net transfers from parent | 2,677 | |||
Net loss | (8,931 | ) | ||
Balance at December 31, 2009 | $ | 49,270 | ||
The accompanying notes are an integral part of these combined financial statements.
F-5
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
COMBINED STATEMENTS OF CASH FLOWS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(in thousands)
| 2007 | 2008 | 2009 | ||||||||||
Cash flows from operating activities: | ||||||||||||
Net income (loss) | $ | (22,922 | ) | $ | 5,812 | $ | (8,931 | ) | ||||
Adjustments to reconcile net income (loss) to net cash provided by operating activities: | ||||||||||||
Depreciation and amortization | 1,972 | 1,413 | 1,271 | |||||||||
Impairment of intangible assets | — | 1,607 | 10,361 | |||||||||
Net gain on sale of businesses | — | — | (26,707 | ) | ||||||||
(Gain) loss on disposal of assets, net | (25 | ) | — | 7 | ||||||||
Changes in operating assets and liabilities, net of acquisitions: | ||||||||||||
Accounts receivable and unbilled services, net | (474 | ) | 810 | 377 | ||||||||
Prepaid expenses and other current assets | (51 | ) | 185 | 506 | ||||||||
Accrued income taxes | 36 | (58 | ) | — | ||||||||
Accounts payable | (128 | ) | 1,019 | (1,050 | ) | |||||||
Other accrued expenses | 1,503 | (1,739 | ) | 1,058 | ||||||||
Deferred rent | (95 | ) | (147 | ) | (675 | ) | ||||||
Unearned income | 717 | 144 | (364 | ) | ||||||||
Net cash provided by (used in) operating activities | (19,467 | ) | 9,046 | (24,147 | ) | |||||||
Cash flows from investing activities: | ||||||||||||
Purchases of property and equipment | (1,052 | ) | (1,816 | ) | (512 | ) | ||||||
Net proceeds from sale of businesses | — | — | 40,267 | |||||||||
Changes in restricted cash | — | — | (1,835 | ) | ||||||||
Net cash paid for acquisition | — | — | (12,092 | ) | ||||||||
Net cash provided by (used in) investing activities | (1,052 | ) | (1,816 | ) | 25,828 | |||||||
Cash flows from financing activities: | ||||||||||||
Net change in investment from Pharmaceutical Product Development, Inc. | 20,844 | (7,230 | ) | (1,675 | ) | |||||||
Repayment of capital lease obligations | (325 | ) | — | — | ||||||||
Net cash provided by (used in) financing activities | 20,519 | (7,230 | ) | (1,675 | ) | |||||||
Net increase in cash and cash equivalents | — | — | 6 | |||||||||
Cash and cash equivalents, beginning of the year | — | — | — | |||||||||
Cash and cash equivalents, end of the year | $ | — | $ | — | $ | 6 | ||||||
The accompanying notes are an integral part of these combined financial statements.
F-6
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
| 1. | Summary of Operations and Significant Accounting Policies: |
Organization and Business Description
In October 2009, the board of directors of Pharmaceutical Product Development, Inc. (“PPD”) authorized management of PPD to proceed with preparations to spin-off its compound partnering business from its core contract research organization, or CRO, business. In order to carry out the proposed spin-off of the compound partnering business, PPD formed a new wholly-owned subsidiary, Furiex Pharmaceuticals, Inc., a Delaware corporation (“Furiex” or the “Company”), into which it will transfer the Compound Partnering Business, including assets, employees, intellectual property rights and liabilities comprising that business, and approximately $100.0 million in the form of cash and cash equivalents. The Company will assume current accounts payable and other accrued expenses payable to third parties and incurred in the ordinary course of the Compound Partnering Business, which have been included as a component of the combined balance sheet as of December 31, 2009. PPD intends to effect the spin-off through a tax-free, pro-rata dividend distribution of all of the shares of the Company to PPD shareholders. PPD is seeking a private letter ruling from the Internal Revenue Service regarding the tax-free nature of the proposed spin-off. If the spin-off is completed, the Company will become an independent, publicly traded company. PPD will not have any ownership or other form of equity interest in the Company following the spin-off. Upon completion of the contemplated transaction, PPD will continue to own and operate its CRO business and the Company will own and operate its compound partnering business.
In connection with the spin-off, the Company and PPD expect to enter into a series of agreements, including a separation and distribution agreement, transition services agreement, sublease and license agreements, employee matters agreement, tax sharing agreement and a master development services agreement. Completion of the separation is subject to numerous conditions, including final approval by the PPD board of directors, receipt of a private letter ruling from the Internal Revenue Service and/or an independent opinion that the spin-off will be tax-free to PPD and its shareholders, and the filing and effectiveness of a Form 10 with the Securities and Exchange Commission. Approval by PPD’s shareholders is not required as a condition to the completion of the proposed spin-off.
Furiex is a drug development company that will continue the compound partnering business started by PPD in 1998. The goal of compound partnering is to in-license from, or form strategic alliances with, pharmaceutical and biotechnology companies to develop and commercialize therapeutics in which the risks and rewards are shared. The Company’s operations will be headquartered in Morrisville, North Carolina.
Basis of Accounting and Combination
The accompanying combined financial statements have been derived from the combined financial statements and accounting records of PPD from the historical cost basis of the assets and liabilities of the various activities that reflect the combined results of operations, financial condition and cash flows of the Discovery Sciences segment of PPD. All the business components of the Discovery Sciences segment have been included in the historical statements because they were managed by common segment management, and because they reflect the historical performance of segment management. In 2009, PPD completed its disposition of Piedmont Research Center, LLC and PPD Biomarker Discovery Services, LLC, both of which were included in the Discovery Sciences segment. Due to the unique service offerings of these two subsidiaries, PPD determined these business units were no longer a long-term strategic fit and elected to sell them. These business units are recorded
F-7
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
as discontinued operations in the financial statements of operations. Additionally, the Discovery Sciences segment of PPD includes pre-clinical consulting services that will not continue after the spin-off. All rights and obligations related to pre-clinical consulting services and the definitive purchase agreements related to Piedmont Research Center, LLC and PPD Biomarker Discovery Services, LLC will be retained by PPD.
The Company has been allocated certain expenses from PPD such as executive oversight, risk management, accounting, tax, legal, investor relations, human resources, information technology, facilities and depreciation, but has not been allocated the underlying productive assets, such as certain information systems equipment, and furniture and facilities that will not be assigned to the Company but from which the Company has benefited. Such expenses have been reflected in the combined financial statements as expense allocations from PPD. The basis of these allocations includes full-time equivalent employees for the respective periods presented and square footage of occupied space. See Note 13 for further discussion of the allocations.
Management believes that the assumptions and allocations underlying the combined financial statements are reasonable. The financial information in these combined financial statements does not include all of the expenses that would have been incurred had the Company been a separate, stand-alone publicly traded entity. The combined financial statements include assets, liabilities and operations, Piedmont Research Center, LLC, PPD Biomarker Discovery Science, LLC and pre-clinical consulting services that will not be included in the Company at the time the spin-off is effected. As such, the financial information herein does not reflect the combined financial position, results of operations or cash flows of the Company in the future or what they would have been had it been a separate, stand-alone entity during the periods presented.
Use of Estimates in Preparation of the Financial Statements
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires the Company’s management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Pharmaceutical Product Development, Inc.’s Net Investment
The accompanying combined financial statements have been derived from the combined financial statements and accounting records of PPD, from the historical cost basis of the assets and liabilities of the various activities that reflect the combined results of operations, financial condition and cash flows of the Discovery Sciences segment of PPD. Because a direct ownership relationship did not exist among all the components comprising the Company, PPD’s net investment in the Company is shown in lieu of shareholder’s equity in the combined financial statements. The net investment account represents the cumulative investments in, distributions from and earnings (loss) of the Company.
All cash and investments are held and managed by PPD. Accordingly, cash used to pay the Company’s expenses or cash collected from collaboration agreements, royalties or customer contracts by PPD on behalf of the Company is recorded as an increase or decrease in PPD’s net investment, with the exception of a small amount of petty cash that was part of an acquisition in 2009 that is included in the combined balance sheet for the year ended December 31, 2009.
F-8
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
Earnings Per Share
Because the Company did not historically exist as one company, but was comprised of several wholly owned subsidiaries of PPD, no earnings per share calculation is included in the combined financial statements.
Separation Costs
The Company has incurred and expects to incur legal, tax and other costs specifically associated with the planned separation, which are recorded as a component of selling, general and administrative expenses.
Revenue Recognition
The Company generates revenue in the form of upfront payments, development and regulatory milestone payments, royalties and sales-based milestone payments in connection with the out-licensing of compounds. The payment of future milestones and royalties will depend on the future success of the Company’s collaborators in developing and commercializing the compound. Upfront payments are generally paid within a short period of time following the execution of an out-license and collaboration agreement. Milestone payments are typically one-time payments to the Company triggered by the collaborator’s achievement of specified development and regulatory events such as the commencement of Phase III trials or regulatory submission or approval. Royalties are payments received by the Company based on net product sales of a collaborator. Sales-based milestone payments are typically one-time payments to the Company triggered when aggregate net sales of product by a collaborator for a specified period (for example, an annual period) reach an agreed upon threshold amount. The Company recognizes upfront payments, development and regulatory milestone payments, royalty payments, and sales-based milestone payments from our collaborators when the event which triggers the obligation of payment has occurred, there are no further obligations on our part in connection with the payment, and collection is reasonably assured.
The Company has also historically recorded revenue from service contracts, other than time-and-material contracts, on a proportional performance basis. To measure performance under these contracts on a given date, the Company compared effort expended to date to the estimated total effort to be expended to complete the contract using metrics such as the number of units to be delivered. Changes in the estimated total effort required to complete a contract without a corresponding proportional change to the contract value resulted in a cumulative adjustment to the amount of revenue recognized in the period the change in estimate was determined. For time-and-material contracts, the Company recognized revenue as hours were worked, multiplied by the applicable hourly rate. All service revenue contracts relate to service revenues that will not be included in the spin-off.
Concentration of Credit Risk
The Company’s collaborators, which are its sources of revenue, are primarily pharmaceutical and biotechnology companies. One collaborator accounted for the majority of the Company’s net revenue in 2008 and 2009. A concentration of credit risk with respect to revenue exists due to the small number of collaborators. A single collaborator accounted for 100% of the Company’s receivable balance at December 31, 2009.
F-9
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
Property and Equipment
The Company records property and equipment at cost less accumulated depreciation. The Company records depreciation using the straight-line method, based on the following estimated useful lives:
Furniture and equipment | 5-10 years | |
Computer equipment and software | 2-5 years |
The Company amortizes leasehold improvements over the shorter of the terms of the respective lives of the leases or the useful lives of the improvements. The Company depreciates property under capital leases over the term of the lease or the service life of the property, whichever is shorter.
Operating Leases
The Company records rent expense for operating leases on a straight-line basis over the term of the lease. The Company begins amortization on the date of initial possession, which is generally when the Company enters the space and begins to make improvements in preparation for its intended use. The Company accounts for the difference between rent expense and rent paid as deferred rent. The Company records a deferred rent liability at the inception of the lease term and amortizes the deferred rent over the term of the lease as a reduction to rent expense.
Goodwill
The excess of the purchase price of a business acquired over the fair value of net tangible assets and identifiable intangible assets at the date of the acquisition has been assigned to goodwill. The Company evaluates goodwill for impairment on an annual basis at October 1 or more frequently if events or changes in circumstances indicate that goodwill might be impaired. Any impairment could have a material adverse effect on the Company’s financial condition and results of operations.
Acquired In-Process Research and Development (IPR&D)
Acquired IPR&D represents the fair value assigned to research and development programs that the Company acquires that have not been completed at the date of acquisition and which have no future alternative use. The value assigned to acquired IPR&D is determined by estimating the costs to develop the acquired technology into commercially viable products, estimating the resulting net cash flows from the projects, and discounting the net cash flows to present value. Additionally, the Company’s estimates take into account the relevant market size and growth factors, expected trends in technology, and the nature and expected timing of new product introductions by the Company and its competitors. The resulting net cash flows from such programs are based on management’s estimates of cost of sales, operating expenses, and income taxes from such programs. The rates utilized to discount the net cash flows to their present value were commensurate with the stage of development of the program and uncertainties in the economic estimates used in the projections described above. Acquired IPR&D assets will be amortized, once the related project has been successfully developed and regulatory approval for a product launch obtained, over their estimated useful lives.
F-10
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
Realizability of Carrying Value of Long-Lived Assets
The Company reviews the recoverability of long-lived and finite-lived intangible assets when circumstances indicate that the carrying amount of assets might not be recoverable. This evaluation is based on various analyses, including undiscounted cash flow projections. In the event undiscounted cash flow projections indicate impairment, the Company would record an impairment based on the fair value of the assets at the date of the impairment.
Income Taxes
The provision for income taxes has been determined using the asset and liability approach of accounting for income taxes. Under this approach, deferred taxes represent the future tax consequences expected to occur when the reported amounts of assets and liabilities are recovered or paid. The provision for income taxes represents income taxes paid or payable for the year, plus the change in deferred taxes during the year. Deferred taxes result from differences between the financial reporting and tax basis of the Company’s assets and liabilities. Deferred tax assets and liabilities are measured using the currently enacted tax rates that apply to taxable income in effect for the years in which those tax attributes are expected to be recovered or paid, and are adjusted for changes in tax rates and tax laws when changes are enacted.
Valuation allowances are recorded to reduce deferred tax assets when it is more likely than not that a tax benefit will not be realized. The assessment of whether or not a valuation allowance is required often requires significant judgment, including the long-range forecast of future taxable income and the evaluation of tax planning initiatives. Adjustments to the deferred tax valuation allowances are made to earnings in the period when such assessments are made. Due to the historical losses from the Company’s operations, it has recorded a full valuation allowance on deferred tax assets.
The Company’s operations are conducted solely within the United States and have historically been included in PPD’s consolidated U.S. federal and state income tax returns, except in certain separate filing states. The income tax provision is calculated using the separate return basis, as if the Company had filed separate income tax returns under its existing structure for the periods presented. The operating losses and research and development credits generated by the Company were consolidated and utilized within the PPD tax return in all periods presented, except for certain separate state losses. Accordingly, the only net operating losses or research and development tax credit carryforwards reported in the accompanying combined financial statements are those attributable to the separate filing states or those which were acquired prior to the spin-off, and may be utilized by the Company subject to Section 382 and 383 limitations for the next 20 years.
Taxes payable to and from PPD are not reported in these financial statements and settlements of these balances are reflected as changes in PPD net investment.
Share-Based Compensation
Certain of the Company’s employees hold PPD stock options and participate in the PPD employee stock purchase plan. Share-based compensation expense for the Company is recognized based on PPD’s share-based compensation expense for the Company employees. The Company measures share-based compensation cost at grant date, based on the fair value of the award, and recognizes it as expense over the employee’s requisite service period.
F-11
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
Research and Development Costs
Research and development costs consist primarily of costs associated with preclinical studies and the clinical trials of the Company’s product candidates, development materials, patent costs, labor and related benefit charges associated with personnel performing research and development work, supplies associated with this work, consulting services, and an allocation of facility and information technology costs. The Company charges research and development costs to operations as incurred, and discloses them in the combined statements of operations. These costs include clinical research services provided by PPD, pre-clinical testing and clinical drug manufacturing provided by third parties, the direct cost of the Company’s personnel managing the programs and upfront and milestone payments to the Company’s collaborators. All research and development costs for the Company’s drug candidates and external collaborations are expensed as incurred.
Recent Accounting Pronouncements
In December 2007, the Financial Accounting Standards Board, or FASB, issued revised guidance on the accounting for business combinations. The revised guidance expands the definitions of a business and a business combination and requires that: the purchase price of an acquisition, including the issuance of equity securities to be determined on the acquisition date, be recorded at fair value at the acquisition date; all assets, liabilities, contingent consideration, contingencies and in-process research and development costs of an acquired business be recorded at fair value at the acquisition date; acquisition costs generally be expensed as incurred; restructuring costs generally be expensed in periods subsequent to the acquisition date; and changes be made in accounting for deferred tax asset valuation allowances and acquired income tax uncertainties after the measurement period to impact income tax expense. In April 2009, the FASB issued additional guidance on the accounting for business combinations which requires an acquirer to recognize at fair value an asset acquired or a liability assumed in a business combination that arises from a contingency if its acquisition-date fair value can be determined during the measurement period. If the acquisition-date fair value cannot be determined, the acquirer applies the recognition criteria in the accounting for contingencies guidance to determine whether the contingency should be recognized as of the acquisition date or thereafter. This revised guidance applies to business combinations for which the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2008. Except for the accounting for in-process research and development discussed in Note 5 and the disallowance of capitalization of acquisition costs, this pronouncement did not have a material impact on reporting of acquisitions in the financial statements.
In May 2009, the FASB issued a new accounting standard on the accounting for, and disclosure of, events that occur after the balance sheet date but before financial statements are issued or available to be issued, also known as “subsequent events.” The standard sets forth the period after the balance sheet date during which management of a reporting entity should evaluate events or transactions that occur for a potential recognition or disclosure in the financial statements, the circumstances under which an entity should recognize events or transactions occurring after the balance sheet date in its financial statements, and the disclosures that an entity should make about events or transactions that occurred after the balance sheet date. This standard is effective for interim and annual periods ending after June 15, 2009. The adoption of this standard did not have a material impact on the Company’s financial statements.
F-12
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
| 2. | Acquisitions and Dispositions: |
Acquisition
On April 2, 2009, the Company acquired 100 percent of the outstanding equity interests of Magen BioSciences, Inc., a biotechnology company focused on the development of dermatologic therapies, for total consideration of $14.9 million. Of this amount, the Company paid $13.1 million in connection with the closing and is obligated to pay the remaining $1.8 million, less any indemnification claims, in the second quarter of 2010, which is recorded as a component of other accrued expenses as of December 31, 2009. Additionally, the Company has $1.8 million in cash held in escrow to secure the seller’s indemnification obligations and the associated liability related to the acquisition of Magen. The funds in the escrow account and the associated rights and liabilities will remain with PPD after the spin-off. Through the acquisition, the Company expanded its compound partnering program into dermatology and gained screening capability for dermatologic compounds.
Acquisition costs related to Magen were $0.2 million and were included in selling, general and administrative expenses in the combined statements of operations. The Company does not expect to be able to deduct the goodwill related to the acquisition of Magen for tax purposes.
The Company accounted for this acquisition under the purchase method of accounting, using appropriate fair value techniques. Accordingly, the Company allocated the purchase price for this acquisition to the estimated fair value of assets acquired and liabilities assumed, which are set forth in the following table:
| Magen | |||
Assets acquired: | |||
Current assets, including cash of $939 | $ | 2,991 | |
Net property and equipment | 609 | ||
Goodwill | 3,987 | ||
Value of identifiable intangible assets: | 10,361 | ||
Total assets acquired | $ | 17,948 | |
Liabilities assumed: | |||
Current liabilities | 3,082 | ||
Total liabilities assumed | 3,082 | ||
Net assets acquired | $ | 14,866 | |
The unaudited pro forma results of operations for the Company assuming the Magen acquisition was consummated as of January 1 of each respective year, were as follows:
| December 31, | ||||||||
| 2008 | 2009 | |||||||
Net revenue | $ | 18,419 | $ | 6,312 | ||||
Loss from operations | (247 | ) | (34,455 | ) | ||||
Net loss | (1,079 | ) | (11,566 | ) | ||||
F-13
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
The above amounts are based upon assumptions and estimates the Company believes are reasonable. The pro forma financial information presented above is not necessarily indicative of either the results of operations that would have occurred had the acquisition taken place at the beginning of the period indicated or of future results of operations of the Company. Since the date of the acquisition, Magen has not generated any revenue and has generated a net loss of $8.9 million.
Dispositions
In May 2009, the Company completed its disposition of substantially all of the assets of its wholly owned subsidiary Piedmont Research Center, LLC for total consideration of $46.0 million. The purchaser has an indemnification holdback of $3.4 million which the Company has included as a component of other current assets. The Company should receive this in the second quarter of 2010. Piedmont Research Center provided preclinical research and evaluation of anticancer agents and therapies.
In December 2009, PPD completed its disposition of its wholly owned subsidiary PPD Biomarker Discovery Sciences, LLC for total consideration of $0.1 million and the right to receive a percentage of future revenues received by the purchaser from contracts that had been awarded as of the date of the disposition. Contingent consideration will be recognized when and if received in the future. The rights and obligations under this agreement will remain with PPD after the spin-off. PPD Biomarker Discovery Sciences provided biomarker discovery services and participant sample analysis.
Due to the unique service offerings of these subsidiaries, PPD determined these business units were no longer a long-term strategic fit and elected to sell them.
Included in the combined balance sheet as of December 31, 2008 were the following assets and liabilities.
Current assets of discontinued operations: | |||
Accounts receivable and unbilled services | $ | 2,738 | |
Prepaid expenses | 87 | ||
Total current assets of discontinued operations | $ | 2,825 | |
Long-term assets of discontinued operations: | |||
Property and equipment, net | $ | 4,541 | |
Goodwill | 8,487 | ||
Total long-term assets of discontinued operations | $ | 13,028 | |
Current liabilities of discontinued operations: | |||
Accounts payable | $ | 1,137 | |
Other accrued expenses | 1,339 | ||
Unearned Income | 1,983 | ||
Total current liabilities of discontinued operations | $ | 4,459 | |
Long-term liabilities of discontinued operations: | |||
Deferred rent | $ | 748 | |
Other long-term liabilities of discontinued operations | $ | 748 | |
F-14
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
The results of Piedmont Research Center and PPD Biomarker Discovery Services are reported as discontinued operations within the combined statements of operation as set forth in the following table:
| Twelve months Ended December 31, | ||||||||||||
| 2007 | 2008 | 2009 | ||||||||||
Net revenue | $ | 19,537 | $ | 18,517 | $ | 7,058 | ||||||
Loss from discontinued operations | (185 | ) | (976 | ) | (3,817 | ) | ||||||
Net gain on sale of businesses | — | — | 26,707 | |||||||||
| 3. | Accounts Receivable and Unbilled Services: |
Accounts receivable and unbilled services consisted of the following amounts on the dates set forth below:
| December 31, | ||||||
| 2008 | 2009 | |||||
Billed | $ | 70 | $ | 551 | ||
Unbilled | 48 | 10 | ||||
Total accounts receivable and unbilled services | $ | 118 | $ | 561 | ||
The Company did not record a provision for doubtful accounts as of December 31, 2008 and 2009 based on its assessment of collection risks.
| 4. | Property and Equipment: |
Property and equipment, stated at cost, consisted of the following amounts on the dates set forth below:
| December 31, | ||||||||
| 2008 | 2009 | |||||||
Leasehold improvements | — | 36 | ||||||
Fixed assets not placed in service | — | 29 | ||||||
Furniture and equipment | — | 890 | ||||||
Computer equipment and software | 17 | 305 | ||||||
Total property and equipment | 17 | 1,260 | ||||||
Less accumulated depreciation | (16 | ) | (534 | ) | ||||
Total property and equipment, net | $ | 1 | $ | 726 | ||||
F-15
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
| 5. | Goodwill and Intangible Assets: |
Changes in the carrying amount of goodwill for the twelve months ended December 31, 2008 and 2009 were as follows:
| Total | |||
Balance as of January 1, 2008 | $ | 45,129 | |
Changes | — | ||
Balance as of December 31, 2008 | 45,129 | ||
Goodwill recorded during the period for current year acquisition | 3,987 | ||
Balance as of December 31, 2009 | $ | 49,116 | |
During 2009, the Company acquired in-process research and development of $10.4 million through the acquisition of Magen, which was related solely to the MAG-131 research program. At the time of acquisition, this program was in the pre-Investigational New Drug application (“IND”) phase of research. The Company estimated that it would take approximately four to five years to complete research and development. The fair value of the in process research and development was determined using the discounted cash flow method. The discounted cash flow was determined based upon projected revenue, expenses and contributory assets related to the specific project and a discount rate based upon the overall weighted average cost of capital for the asset and the additional risk related to the uncertainty of the project. The Company also assessed the current status of development, nature and timing of efforts to complete such development, uncertainties, and other factors when estimating the fair value.
The Company filed an IND for MAG-131 in October 2009 but subsequently suspended the program for that compound due to efficacy data that was discovered in late 2009. As a result, the Company evaluated the asset for impairment. The Company reassessed the fair value of the program using a discounted cash flow model based on Level 3 inputs such as the estimated remaining costs to develop the acquired technology into commercially viable products, estimated net cash flows from the program, and a discount rate commensurate with the stage of development of the program. Based on this analysis, the Company determined that the acquired in-process research and development asset was impaired and recorded a charge of $10.4 million as of December 31, 2009. Because the intangible asset was an indefinite-lived asset, the Company had not amortized this asset during 2009.
In September 2004, the Company entered into a royalty agreement with Accentia Biopharmaceuticals, Inc. under which it paid $2.5 million to Accentia in exchange for the right to receive royalties on sales of specified antifungal products, primarily SinuNase. During 2008, Accentia reported that SinuNase did not meet its goal in treating chronic sinusitis patients in its Phase III clinical trial, discontinued the sale of all antifungal products, and filed for bankruptcy. As a result, the Company determined that the right under its agreement with Accentia to receive royalties on future sales of SinuNase was impaired, and recorded an impairment of $1.6 million for the remaining unamortized value of its royalty interest in SinuNase.
F-16
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
| 6. | Other Accrued Expenses: |
Other accrued expenses consisted of the following amounts on the dates set forth below:
| December 31, | ||||||
| 2008 | 2009 | |||||
Accrued salaries, wages, benefits and related costs | $ | 302 | $ | 633 | ||
Additional purchase price | — | 1,835 | ||||
Professional fees | — | 1,773 | ||||
Clinical supplies | — | 1,893 | ||||
Other | 85 | 355 | ||||
| $ | 387 | $ | 6,489 | |||
| 7. | Lease Obligations: |
On behalf of the Company, PPD is obligated under noncancellable operating leases which were acquired in the Magen acquisition. These leases expire at various dates through 2012 relating to a building and certain equipment. Rental expense, related to these leases, has been recorded in continuing operations in the amounts of $0, $0, and $0.3 million for the years ended December 31, 2007, 2008 and 2009, respectively.
As of December 31, 2009, future minimum payments for lease obligations for subsequent years were as follows:
2010 | $ | 420 | |
2011 | 435 | ||
2012 | 411 | ||
2013 | 4 | ||
2014 and thereafter | 2 | ||
| $ | 1,272 | ||
| 8. | Share-Based Compensation: |
Equity Compensation Plan
The Company has adopted an equity compensation plan (the “Plan”). The Plan provides for the grant of incentive stock options, non-qualified stock options, restricted stock and other types of equity awards to its directors, officers and employees and consultants. The plan will be administered by the board of directors of the Company, or a committee designated by its board of directors. Some employees of the Company have historically received awards from PPD. Accordingly, the following information regarding share-based compensation has been derived from the equity awards granted to Company employees by PPD.
The exercise price of each option granted is equal to the market price of PPD’s common stock on the date of grant, and the maximum exercise term of each option granted does not exceed ten years. Options are granted upon approval of the Compensation Committee of the board of directors of PPD. The majority of the options vest
F-17
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
ratably over a period of three years. The options expire on the earlier of ten years from the date of grant, or within specified time limits following termination of employment, retirement or death. Shares are issued from authorized, but unissued stock. PPD does not pay dividends on unexercised options.
For the years ended December 31, 2007, 2008 and 2009, stock-based compensation cost for the Company’s employees totaled $0.2 million, $0.4 million and $0.3 million, respectively, and is included in the accompanying combined financial statements.
For the years ended December 31, 2007, 2008 and 2009, the amount of cash received by PPD from the exercise of PPD stock options granted to the Company’s employees was $0.4 million, $0.7 million and $1.1 million, respectively.
A summary of option activity under PPD’s plan for the Company’s employees as of December 31, 2007, 2008 and 2009, and changes during the years, is presented below:
| Shares | Weighted- Average Exercise Price | Weighted- Average Remaining Contractual Life | Aggregate Intrinsic Value | |||||||||
Outstanding at January 1, 2007 | 189 | $ | 23.66 | |||||||||
Granted | 38 | 33.61 | ||||||||||
Exercised | (17 | ) | 21.65 | |||||||||
Forfeited | — | — | ||||||||||
Expired | — | — | ||||||||||
Outstanding at December 31, 2007 | 210 | $ | 25.60 | |||||||||
Exercisable at December 31, 2007 | 110 | $ | 21.47 | |||||||||
Outstanding at January 1, 2008 | 210 | $ | 25.60 | |||||||||
Granted | 78 | 37.77 | ||||||||||
Exercised | (47 | ) | 21.41 | |||||||||
Forfeited | (6 | ) | 33.34 | |||||||||
Expired | — | — | ||||||||||
Outstanding at December 31, 2008 | 235 | $ | 30.33 | |||||||||
Exercisable at December 31, 2008 | 120 | $ | 24.40 | |||||||||
Outstanding at January 1, 2009 | 235 | $ | 30.33 | |||||||||
Granted | 95 | 27.01 | ||||||||||
Exercised | (5 | ) | 16.46 | |||||||||
Forfeited | (60 | ) | 32.01 | |||||||||
Expired | (28 | ) | 28.82 | |||||||||
Outstanding at December 31, 2009 | 237 | $ | 29.02 | 7.9 years | $ | (1,325 | ) | |||||
Exercisable at December 31, 2009 | 154 | $ | 27.96 | 7.5 years | $ | (698 | ) | |||||
Vested or expected to vest at December 31, 2009 | 228 | $ | 29.03 | 7.9 years | $ | (1,276 | ) | |||||
F-18
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
The following table summarizes information about PPD’s stock options outstanding for the Company’s employees as of December 31, 2009:
| Options Outstanding | Options Exercisable | |||||||||||
Range of Exercise Prices | Number Outstanding at 12/31/09 | Weighted-Average Remaining Contractual Life | Weighted Average Exercise Price | Number Exercisable at 12/31/09 | Weighted- Average Exercise Price | |||||||
$15.43 – 22.00 | 100 | 4.7 years | $ | 21.20 | 86 | $ | 21.23 | |||||
$22.01 – 39.00 | 93 | 7.8 years | $ | 30.92 | 50 | $ | 34.25 | |||||
$39.01 – 44.78 | 44 | 8.4 years | $ | 42.97 | 18 | $ | 42.99 | |||||
| 237 | 7.9 years | $ | 29.02 | 154 | $ | 27.96 | ||||||
All options granted during the years ended December 31, 2007, 2008 and 2009 were granted with an exercise price equal to the fair value of PPD’s common stock on the grant date. The fair value of PPD’s common stock on the grant date is equal to the Nasdaq closing price of the stock on the date of grant. The weighted-average grant date fair value per share of options granted to the Company’s employees during the years ended December 31, 2007, 2008 and 2009 was $10.78, $9.33 and $7.55, respectively. The aggregate fair value of options granted to the Company’s employees during the years ended December 31, 2007, 2008 and 2009 was $0.4 million, $0.7 million and $0.7 million, respectively. The total intrinsic value (the amount by which the market value of PPD’s common stock exceeded the exercise price of the options on the date of exercise) of options exercised during the years ended December 31, 2007, 2008 and 2009 was approximately $0.3 million, $1.0 million and $22,000, respectively.
A summary of the status of unvested PPD options held by the Company’s employees as of December 31, 2009, and changes during the year then ended, is presented below:
Unvested Options | Shares | Weighted- Average Grant Date Fair Value | ||||
Unvested at January 1, 2009 | 115 | $ | 10.43 | |||
Granted | 82 | 8.80 | ||||
Vested | (54 | ) | 11.65 | |||
Forfeited | (60 | ) | 8.20 | |||
Unvested at December 31, 2009 | 83 | $ | 7.80 | |||
As of December 31, 2009, the total unrecognized compensation cost related to unvested PPD stock options held by the Company’s employees was approximately $0.5 million. The Company will amortize this cost over a weighted-average period of 1.6 years in accordance with the vesting periods of the options, up until the date of the spin-off at which time the unvested options will be forfeited. The total fair value of shares vested during the years ended December 31, 2007, 2008 and 2009 was $0.6 million, $0.7 million and $0.6 million, respectively.
F-19
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
The fair value of each grant is estimated on the grant date using the Black-Scholes option-pricing model. The following table indicates the assumptions used in estimating fair value for the years ended December 31, 2007, 2008 and 2009.
| 2007 | 2008 | 2009 | ||||
Expected term (years) | 4.00 | 3.75 | 3.50 | |||
Dividend yield (%) | 0.31-0.37 | 0.93-1.21 | 1.72-2.74 | |||
Risk-free interest rate (%) | 4.13-4.91 | 2.05-3.22 | 1.14-1.67 | |||
Expected volatility (%) | 30.67-33.00 | 29.88-30.94 | 36.58-39.27 |
The expected term represents an estimate of the period of time options are expected to remain outstanding and is based on historical exercise and termination data. The dividend yield is based on the most recent dividend payment over the market price of the PPD stock at the beginning of the period. The risk-free interest rate is based on the rate at the date of grant for a zero-coupon U.S. Treasury bond with a term that approximates the expected term of the option. Expected volatilities are based on the historical volatility of PPD’s stock price over the expected term of the options.
Employee Stock Purchase Plan
As of December 31, 2009, the Company’s employees participated in PPD’s employee stock purchase plan (the “PPD ESPP”). The PPD ESPP has two six-month offering periods (each an “Offering Period”) each year, beginning January 1 and July 1, respectively. Eligible employees can elect to make payroll deductions from 1% to 15% of their base pay during each payroll period of an Offering Period. None of the contributions made by eligible employees to purchase PPD’s common stock under the PPD ESPP are tax-deductible to the employees. The purchase price is 90% of the lesser of (a) the reported closing price of PPD’s common stock for the first day of the Offering Period or (b) the reported closing price of the common stock for the last day of the Offering Period.
Employees eligible to participate in the PPD ESPP include employees of the Company except employees who customarily work less than 20 hours per week or five months in a year. Because the eligible employee criteria determines both participation in and contributions to the PPD ESPP, it is not possible to determine the benefits and amounts that would be received by an eligible participant or group of participants in the future.
The fair value of each PPD ESPP share is estimated using the Black-Scholes option-pricing model. The following table indicates the assumptions used in estimating fair value for the years ended December 31, 2007, 2008 and 2009.
| 2007 | 2008 | 2009 | ||||
Expected term (years) | 0.50 | 0.50 | 0.50 | |||
Dividend yield (%) | 0.31-0.37 | 0.93-0.99 | 1.72-2.58 | |||
Risk-free interest rate (%) | 4.74-4.90 | 2.12-3.37 | 0.27-0.35 | |||
Expected volatility (%) | 21.20-28.44 | 31.47-31.99 | 31.32-36.68 |
The Company’s compensation costs for the PPD ESPP, as determined based on the fair value of the discount and option feature of the underlying PPD ESPP grant, were approximately $44,000, $0.1 million and $0.1 million for years ended December 31, 2007, 2008 and 2009, respectively. The weighted-average grant date
F-20
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
fair value per share during the years ended December 31, 2007, 2008 and 2009 was $5.80, $6.83 and $4.16, respectively. As of December 31, 2009, there was no unrecognized compensation cost related to PPD ESPP shares.
For the years ended December 31, 2007, 2008 and 2009, the value of stock issued to Company employees for PPD ESPP purchases was $0.2 million, $0.3 million and $0.3 million, respectively.
During the years ended December 31, 2007, 2008 and 2009, the Company’s employees contributed $0.2 million, $0.3 million and $0.3 million, respectively, to the PPD ESPP for the purchase of approximately 7,600, 8,200 and 13,300 shares, respectively. The aggregate fair value of shares purchased during the years ended December 31, 2007, 2008 and 2009 was $0.3 million, $0.3 million and $0.4 million, respectively. Contributions for the second Offering Period of 2009 were not used to purchase shares until January 2010.
| 9. | Income Taxes: |
Taxes computed at the statutory U.S. federal income tax rate of 35% are reconciled to the provision for income taxes as follows:
| Year Ended December 31, | ||||||||||||
| 2007 | 2008 | 2009 | ||||||||||
Effective tax rate | 0 | % | 0 | % | 0 | % | ||||||
Statutory rate of 35% | $ | (7,958 | ) | $ | 2,376 | $ | (11,062 | ) | ||||
State taxes, net of federal benefit | — | — | — | |||||||||
Nontaxable income net of nondeductible expenses | (294 | ) | (203 | ) | (22 | ) | ||||||
Change in valuation allowance | (3,832 | ) | (3,492 | ) | 3,776 | |||||||
Net operating loss offset by consolidated group | 12,084 | 1,319 | 7,308 | |||||||||
Provision for income taxes | $ | — | $ | — | $ | — | ||||||
Components of the current deferred tax assets were as follows:
| December 31, | ||||||||
| 2008 | 2009 | |||||||
Future benefit of carryforward losses | $ | — | $ | 434 | ||||
Accrued expenses | 39 | 1,205 | ||||||
Valuation allowance | (39 | ) | (1,641 | ) | ||||
Other | — | 2 | ||||||
Total current deferred tax asset | $ | — | $ | — | ||||
F-21
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
Components of the long-term deferred tax assets were as follows:
| December 31, | ||||||||
| 2008 | 2009 | |||||||
Other depreciation and amortization | $ | 1,280 | $ | 6,018 | ||||
Patent depreciation | 2,637 | — | ||||||
Deferred rent | — | 27 | ||||||
Stock options | 353 | 468 | ||||||
Deferred compensation | 22 | 26 | ||||||
Valuation allowance | (4,647 | ) | (9,099 | ) | ||||
Future benefit of carryforward losses | 355 | 2,446 | ||||||
Other | — | 114 | ||||||
Total long-term deferred tax asset | $ | — | $ | — | ||||
Federal operating loss carry forwards of approximately $8.0 million expire at various dates between 2026 and 2029. State operating loss carry forwards of approximately $8.0 million expire at various dates between 2021 and 2024.
| 10. | Employee Savings Plan: |
Savings plan
The employees of the Company participate in PPD’s 401(k) Retirement Savings Plan. The Company matches 50% of an employee’s savings up to 6% of pay and these contributions vest ratably over a four-year period. The Company’s contributions to the plan, net of forfeitures, were $0.2 million, $0.2 million and $0.1 million for the years ended December 31, 2007, 2008 and 2009, respectively.
Non-Qualified Deferred Compensation Plans
The employees of the Company participate in PPD’s non-qualified, unfunded deferred compensation plans that permit certain highly paid executive employees to defer current income for future financial and retirement needs. There are not any Company contributions to these plans and other than accruals for interest or dividend equivalents, all amounts credited to these plans are derived from elective deferrals of compensation otherwise payable to participants. Cash amounts deferred each quarter accrue interest based upon the three-month LIBOR rate plus 1.5%. The total liability related to this plan is $62,000 and $75,000 at December 31, 2008 and 2009, respectively, and is included as a component of other accrued expenses.
| 11. | Commitments and Contingencies: |
The Company (through PPD) currently maintains insurance for risks, among others, associated with the operation of its business and ownership of property. These policies provide coverage for a variety of potential losses, including loss or damage to property, bodily injury, general commercial liability, professional errors and omissions and medical malpractice. PPD’s retentions and deductibles associated with these insurance policies range from $0 to $5.0 million.
F-22
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
PPD is self-insured for health insurance for the majority of its employees, including the employees of the Company, but maintains stop-loss insurance on a “claims made” basis for expenses in excess of $0.28 million per member per year. PPD maintained a reserve on its consolidated balance sheets, to cover open claims and estimated claims incurred but not reported. The Company’s portion is less than $0.1 million and is included in PPD’s net investment on its combined balance sheets.
The Company has been involved in compound development and commercialization collaborations within PPD since 1998. The Company developed a risk-sharing research and development model to help pharmaceutical and biotechnology clients develop compounds. Through collaborative arrangements based on this model, the Company assists its clients by sharing the risks and potential rewards associated with the development and commercialization of drugs at various stages of development. As of December 31, 2009, the Company’s four main collaborations were with Janssen-Cilag, an affiliate of Johnson & Johnson, Takeda Pharmaceuticals Company Limited (“Takeda”), Janssen Pharmaceutica N.V., an affiliate of Johnson & Johnson and Eli Lilly and Company (“Lilly”), and relate to, respectively, the product Priligy, the late stage candidate alogliptin, two Phase II-ready therapeutic compounds, and a series of early stage candidates including Vitamin D receptor modulators as well as up to six other programs for topical dermatological indications, respectively. They involve the potential future receipt of one or more of the following forms of revenue: payments upon the achievement of specified regulatory and sales-based milestones; and royalty payments if the compound is approved for sale. To date, Austria, Finland, Germany, Italy, Mexico, New Zealand, Portugal, South Korea, Spain and Sweden have approved Priligy for marketing. The Company received a $2.5 million milestone on each of the first two of these national approvals, for a total of $5.0 million, in the first quarter of 2009. It is entitled to royalties on net sales of Priligy and sales-based milestones if requisite sales levels are reached. The Company recorded the first royalties from the sales of Priligy in 2009. With regard to alogliptin, in June 2009, the FDA issued a complete response to Takeda on its alogliptin New Drug Application or “NDA”, requesting Takeda conduct an additional cardiovascular safety trial that satisfies the FDA’s December 2008 guidance on anti-diabetes therapies. In September 2009, the FDA issued a complete response to Takeda on its NDA for the fixed dose combination of alogliptin and ACTOS stating that further review would be dependent on the cardiovascular safety data that would be submitted in support of the alogliptin monotherapy NDA. The compounds related to Lilly and Janssen Pharmaceutica are still in discovery and development, respectively, and have not generated any regulatory milestone payments yet. Due to the risks associated with drug development and commercialization, including poor or unexpected preclinical and clinical trial results, obtaining regulatory approval to sell in any country and changing regulatory requirements, the Company might not receive any further milestone payments, royalties or other payments with respect to any of the Company’s drug development collaborations.
As of December 31, 2009, the Company had four collaborations that involve potential future expenditures. The first is the Company’s collaboration with ALZA for Priligy. In connection with this collaboration, the Company has an obligation to pay a royalty to Lilly of 5% on annual net sales of the compound in excess of $800.0 million.
The second collaboration involving future expenditures is with Ranbaxy. In February 2007, the Company exercised an option to license from Ranbaxy a statin compound that the Company is developing as a potential treatment for dyslipidemia, a metabolic disorder characterized by high cholesterol levels. Under the agreement, the Company has an exclusive license to make, use, sell, import and sublicense the compound and any licensed product anywhere in the world for any human use. The Company is solely responsible, and will bear all costs and expenses, for the development, manufacture, marketing and commercialization of the compound and licensed products. It is obligated to pay Ranbaxy milestone payments upon the occurrence of specified clinical development events. If a
F-23
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
licensed product is approved for sale, the Company must also pay Ranbaxy royalties based on sales of the product, as well as commercial milestone payments based on the achievement of specified worldwide sales targets. If all criteria are met, the total amount of potential clinical and sales-based milestones that the Company is obligated to pay Ranbaxy would be $43.0 million. The Company also would be obligated to pay Ranbaxy sales-based royalties ranging from the mid to high single digit percentages. The Company completed a high dose comparator study in healthy volunteers. The drug was well-tolerated and a preliminary review of results suggests the statin compound compares favorably to currently marketed statins. The Company continues to conduct limited development activities with respect to the Ranbaxy statin compound while we evaluate alternatives for future development and commercialization.
The third collaboration involving future expenditures is with Lilly. In April 2009, the Company acquired Magen BioSciences, Inc., a biotechnology company founded in 2006 to discover dermatologic therapies. As a result, the Company expanded its compound development business into dermatology and gained advanced screening and research capabilities for dermatologic treatments. The Company has an exclusive license to develop and commercialize Vitamin D receptor modulator compounds for use as topical treatments in dermatological indications. It also has an option agreement with Lilly to screen compounds from six additional platforms for utility in dermatology and is investigating compounds from other potential collaborators under material transfer agreements. Through the acquisition of Magen, the Company acquired in-process research and development of $10.4 million. At acquisition, the acquired in-process research and development was related to the MAG-131 research program, which was in the pre-IND phase of research. The Company filed an IND for this compound in October 2009, but subsequently suspended the MAG-131 program due to efficacy data that was discovered in late 2009. The Company is currently screening additional Vitamin D receptor modulators from Lilly and compounds that regulate other targets to identify additional drug development candidates for other dermatological indications. Under the license arrangements with Lilly, the Company is obligated to pay clinical development milestones as well as royalties based on the sales of the product. If all criteria are met, the total potential clinical development milestones that the Company is obligated to pay would be $21.4 million per compound developed. The Company also would be obligated to pay Lilly sales-based royalties ranging from the mid single digit to low double digit percentages.
The fourth collaboration involving future expenditures is with Janssen Pharmaceutica N.V. In November 2009, the Company entered into agreements with Janssen Pharmaceutica to develop and commercialize two Phase II-ready therapeutic compounds. The Company plans to study the mu delta compound as a treatment for diarrhea-predominant irritable bowel syndrome, or IBSd and the fluoroquinolone compound as a treatment for community-acquired bacterial pneumonia and complicated skin and skin structure infections caused by gram negative or gram positive bacteria, including MRSA. Under the two agreements, the Company in-licensed the two compounds and will advance the compounds through Phase II development. At the completion of Phase II, Janssen Pharmaceutica will have the option to continue development and commercialization of each compound. In exchange, the Company may receive, for each compound, up to $90.0 million in regulatory milestone payments and up to $75.0 million in sales-based milestone payments, as well as royalties on sales of each compound if approved for marketing. In the event Janssen Pharmaceutica elects not to continue a program, the Company has the option to continue developing and commercializing the compound for that program and the Company would be obligated to pay Janssen Pharmaceutica, for each compound, up to $50.0 million in regulatory milestone payments and up to $75.0 million in sales-based milestone payments, as well as sales-based royalties ranging from the mid single digit to low double digit percentages if approved for marketing. During
F-24
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
2009, the Company expensed $7.0 million of upfront payments related to the two therapeutic compounds in-licensed as part of the agreement with Janssen Pharmaceutica.
| 12. | Fair Value of Financial Instruments: |
Accounts Receivable, Accounts Payable and Accrued Liabilities
The carrying amount approximates fair value because of the short maturity of these items.
| 13. | Net Investment of Pharmaceutical Product Development, Inc: |
The following table reflects a summary of the transfers to/from parent included in the combined statements of changes in Pharmaceutical Product Development, Inc. net investment:
| 2007 | 2008 | 2009 | ||||||
Corporate overhead allocations | 729 | 850 | 712 | |||||
Research and development services | 10,613 | 3,689 | 4,416 | |||||
Cash from parent for acquisitions | — | — | 21,503 | |||||
Transfer of proceeds from sale of businesses | — | — | (40,267 | ) | ||||
Transfers to (from) parent, net | 9,555 | (11,697 | ) | 16,313 | ||||
Total | 20,897 | (7,158 | ) | 2,677 |
Corporate Overhead Allocations
For each of the periods presented, the Company’s operations were fully integrated with PPD, including executive services, finance, treasury, corporate income tax, human resources, information technology, facilities, legal services and investor relations. The accompanying combined financial statements reflect the application of estimates and allocations of operating expenses. Management believes the methods used to allocate these operating expenses are reasonable. The allocation methods include relative time devoted by executive management to the Company’s business, and related benefit received by the Company for other services, and benefits such as costs associated with being a public company. Allocations of expense for these services of $0.2 million, $0.3 million, and $0.4 million associated with continuing operations and $0.5 million, $0.5 million, and $0.3 million associated with discontinued operations for the years ended December 31, 2007, 2008 and 2009, respectively, are reflected in the accompanying combined statements of operations.
Research and Development Services
PPD performed drug development work for the Company, and the expenses related to these services are included in research and development expenses in the accompanying combined set of financial statements. Such amounts were $10.6 million, $3.7 million and $4.4 million for the years ended December 31, 2007, 2008 and 2009, respectively.
Cash Transferred from Parent for Acquisitions
During 2009, the Company acquired Magen Biosciences, Inc. (See Note 2) and entered into the Janssen collaboration (see Note 11). These transactions were funded by a transfer of $21.5 million in cash and assets to the Company.
F-25
Table of Contents
COMBINED FINANCIAL STATEMENTS OF FURIEX PHARMACEUTICALS, INC.
(A COMPONENT OF PHARMACEUTICAL PRODUCT DEVELOPMENT, INC.)
NOTES TO COMBINED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2007, 2008 AND 2009
(dollars in tables in thousands)
Transfer of Proceeds from Sale of Business
As discussed in Note 2, the Company disposed of Piedmont Research Center, LLC and PPD Biomarker discovery Sciences, LLC during 2009. The cash proceeds of $40.3 million received from these transactions was transferred to PPD.
| 14. | Segment and Related Information: |
The Company business consists primarily of compound partnering activities. Accordingly, the Company operates in one reportable business segment.
The Company’s revenues are derived from royalties received from a U.S. company based on sales of a product sold in Europe. All of the Company’s long-lived assets are located in the United States.
F-26
Table of Contents
FURIEX PHARMACEUTICALS, INC.
INDEX TO FINANCIAL STATEMENTS
| Page | ||
| F-28 | ||
| F-29 | ||
| F-30 | ||
| F-31 | ||
| F-32 | ||
F-27
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of Pharmaceutical Product Development, Inc.
We have audited the accompanying balance sheet of Furiex Pharmaceuticals, Inc. (the “Company”) as of December 31, 2009, and the related statements of operations and cash flows for the period October 21, 2009 (date of inception) through December 31, 2009. These financial statements are the responsibility of the management of Pharmaceutical Product Development, Inc. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of Furiex Pharmaceuticals, Inc.’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, such financial statements present fairly, in all material respects, the financial position of Furiex Pharmaceuticals, Inc. at December 31, 2009 and the results of its operations and its cash flows for the period October 21, 2009 (date of inception) through December 31, 2009, in conformity with accounting principles generally accepted in the United States of America.
| /s/ DELOITTE & TOUCHE LLP |
| Charlotte, North Carolina |
| February 24, 2010 |
F-28
Table of Contents
STATEMENT OF OPERATIONS
FOR THE PERIOD OCTOBER 21, 2009 (DATE OF INCEPTION)
THROUGH DECEMBER 31, 2009
(in thousands)
| 2009 | ||||
Research and development expenses | $ | 9,306 | ||
General and administrative expenses | 82 | |||
Loss from operations | (9,388 | ) | ||
Net loss | $ | (9,388 | ) | |
The accompanying notes are an integral part of these financial statements.
F-29
Table of Contents
BALANCE SHEET
AS OF DECEMBER 31, 2009
(in thousands)
| 2009 | ||||
Current liabilities: | ||||
Related party payable | $ | 7,640 | ||
Other accrued expenses | 1,748 | |||
Total current liabilities | 9,388 | |||
Total liabilities | 9,388 | |||
Commitments and contingencies (Note 4) | ||||
Shareholder’s Equity: | ||||
Common stock, $0.001 par value, 100 shares authorized, issued and outstanding | — | |||
Accumulated deficit | (9,388 | ) | ||
Total liabilities and shareholder’s equity | $ | — | ||
The accompanying notes are an integral part of these financial statements.
F-30
Table of Contents
STATEMENT OF CASH FLOWS
FOR THE PERIOD OCTOBER 21, 2009 (DATE OF INCEPTION)
THROUGH DECEMBER 31, 2009
(in thousands)
| 2009 | ||||
Cash flows from operating activities: | ||||
Net loss | $ | (9,388 | ) | |
Changes in operating liabilities: | ||||
Other accrued expenses | 1,748 | |||
Net cash used by operating activities | (7,640 | ) | ||
Cash flows from financing activities: | ||||
Related party payable | 7,640 | |||
Net cash provided by financing activities | 7,640 | |||
Net increase in cash and cash equivalents | — | |||
Cash and cash equivalents, beginning of the period | — | |||
Cash and cash equivalents, end of the period | $ | — | ||
The accompanying notes are an integral part of these financial statements.
F-31
Table of Contents
NOTES TO FINANCIAL STATEMENTS
AS OF DECEMBER 31, 2009
(in thousands)
| 1. | Summary of Operations and Significant Accounting Policies: |
Organization and Business Description
In October 2009, the board of directors of Pharmaceutical Product Development, Inc. (“PPD”) authorized management of PPD to proceed with preparations to spin-off its compound partnering business from its core contract research organization, or CRO, business. In order to carry out the proposed spin-off of the Compound Partnering Business, PPD formed a new wholly-owned subsidiary, Furiex Pharmaceuticals, Inc., a Delaware corporation (“Furiex” or the “Company”), on October 21, 2009 into which it will transfer the Compound Partnering Business, including assets, employees, intellectual property rights and liabilities comprising that business, and approximately $100.0 million in the form of cash and cash equivalents. The Company will assume current accounts payable and other accrued expenses payable to third parties and incurred in the ordinary course of the Compound Partnering Business. PPD intends to effect the spin-off through a tax-free, pro-rata dividend distribution of all of the shares of the Company to PPD shareholders. The Company is seeking a private letter ruling from the Internal Revenue Service regarding the tax-free nature of the proposed spin-off. If the spin-off is completed, the Company will become an independent, publicly traded company. PPD will not have any ownership or other form of equity interest in the Company following the spin-off. Upon completion of the contemplated transaction, PPD will continue to own and operate its CRO business and the Company will own and operate the compound partnering business.
Furiex is a drug development company that will continue the Compound Partnering Business started by PPD in 1998. The goal of compound partnering is to in-license from, or form strategic alliances with, pharmaceutical and biotechnology businesses to develop and commercialize therapeutics in which the risks and rewards are shared. The Company’s operations will be located in Raleigh, North Carolina.
The Company is a component business of the Discovery Sciences segment of PPD, and its operations to date are comprised solely of the Janssen collaboration agreement as described below and the purchase of research and development services from PPD to support that collaboration. The financial information in these combined financial statements does not include all of the expenses that would have been incurred had the Company been a separate, stand-alone publicly traded entity or the impact of the proposed spin-out transaction. As such, the financial information herein does not reflect the financial position, results of operations or cash flows of the Company in the future or what they would have been had the Company been a separate, stand-alone entity during the periods presented.
Use of Estimates in Preparation of the Financial Statements
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires the Company’s management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Income Taxes
The provision for income taxes has been determined using the asset and liability approach of accounting for income taxes. Under this approach, deferred taxes represent the future tax consequences expected to occur when the reported amounts of assets and liabilities are recovered or paid. The provision for income taxes represents income taxes paid or payable for the year, plus the change in deferred taxes during the year. Deferred taxes result
F-32
Table of Contents
FURIEX PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS
AS OF DECEMBER 31, 2009
(in thousands)
from differences between the financial reporting and tax basis of our assets and liabilities. Deferred tax assets and liabilities are measured using the currently enacted tax rates that apply to taxable income in effect for the years in which those tax attributes are expected to be recovered or paid, and are adjusted for changes in tax rates and tax laws when changes are enacted.
Valuation allowances are recorded to reduce deferred tax assets when it is more likely than not that a tax benefit will not be realized. The assessment of whether or not a valuation allowance is required often requires significant judgment, including the long-range forecast of future taxable income and the evaluation of tax planning initiatives. Adjustments to the deferred tax valuation allowances are made to earnings in the period when such assessments are made. Due to the historical losses from the Company’s operations, it has recorded a full valuation allowance on deferred tax assets.
The Company’s operations are conducted solely within the United States and have historically been included in PPD’s consolidated US federal and state income tax returns. The income tax provision is calculated using the separate return basis, as if the Company had filed separate income tax returns. The operating losses and research and development credits generated by the Company were consolidated and utilized within the PPD return. Accordingly, no net operating losses or research and development tax credit carryforwards are reported in the accompanying financial statements.
Taxes payable to and from PPD and settlements of these balances are reflected as changes in the related party payable.
Research and Development Costs
Research and development costs consist primarily of costs associated with preclinical costs and the clinical trials of the Company’s product candidates, development materials, patent costs, labor and related benefit charges associated with personnel performing research and development work, supplies associated with this work, consulting services, and an allocation of facility and information technology costs. The Company charges research and development costs to operations as incurred, and discloses them in the statements of income.
In November 2009, the Company entered into a collaboration agreement with Janssen Pharmaceutica N.V. in which the Company paid $7.0 million in upfront payments which were expensed as incurred as the associated compounds have not completed the required work to establish the technological feasibility or receive regulatory approval for marketing.
| 2. | Other Accrued Expenses: |
Other accrued expenses consisted of the following amounts on the dates set forth below:
| December 31, 2009 | |||
Contract labor | $ | 626 | |
Professional fees | 72 | ||
Clinical supplies | 1,050 | ||
| $ | 1,748 | ||
F-33
Table of Contents
FURIEX PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS
AS OF DECEMBER 31, 2009
(in thousands)
| 3. | Income Taxes: |
Taxes computed at the statutory U.S. federal income tax rate of 35% are reconciled to the provision for income taxes as follows:
| December 31, 2009 | ||||
Effective tax rate | 0 | % | ||
Statutory rate of 35% | $ | (3,286 | ) | |
Nontaxable income net of nondeductible expenses | (43 | ) | ||
Change in valuation allowance | 2,482 | |||
Net operating loss by consolidated group | 847 | |||
Provision for income taxes | $ | — | ||
Components of the long-term deferred tax assets were as follows:
| December 31, 2009 | ||||
Other depreciation and amortization | $ | 2,482 | ||
Valuation allowance | (2,482 | ) | ||
Total long-term deferred tax asset | $ | — | ||
| 4. | Commitments and Contingencies: |
The Company has a collaboration involving future expenditures with Janssen Pharmaceutica N.V. (“Janssen”). In November 2009, the Company entered into agreements with Janssen Pharmaceutica to develop and commercialize two Phase II-ready therapeutic compounds. The Company plans to study the mu delta compound as a treatment for diarrhea-predominant irritable bowel syndrome, or IBSd and the fluoroquinolone compound as a treatment for community-acquired bacterial pneumonia and complicated skin and skin structure infections caused by gram negative or gram positive bacteria, including MRSA. Under the two agreements, the Company in-licensed the two compounds and the Company will advance the compounds through Phase II development. At the completion of Phase II, Janssen Pharmaceutica will have the option to continue development and commercialization of each compound. In exchange, the Company may receive, for each compound, up to $90.0 million in regulatory milestone payments and up to $75.0 million in sales-based milestone payments, as well as royalties on sales of each compound if approved for marketing. In the event Janssen Pharmaceutica elects not to continue a program, the Company has the option to continue developing and commercializing the compound for that program and Janssen Pharmaceutica may receive, for each compound, up to $50.0 million in regulatory milestone payments and up to $75.0 million in sales-based milestone payments, as well as royalties on sales of each compound if approved for marketing. During 2009, the Company expensed $7.0 million of upfront payments related to the two therapeutic compounds in-licensed as part of the agreement with Janssen Pharmaceutica.
In the normal course of business, the Company might be a party to various claims and legal proceedings. As of December 31, 2009, there are no outstanding claims that management believes will have a material effect upon the Company’s financial condition, results of operations or cash flows.
F-34
Table of Contents
FURIEX PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS
AS OF DECEMBER 31, 2009
(in thousands)
| 5. | Fair Value of Financial Instruments: |
Accrued Liabilities
The carrying amount approximates fair value because of the short maturity of these items.
| 6. | Related Party Transactions: |
The Company’s operations were fully integrated with PPD. PPD performed drug development work for the Company of $0.4 million and expenses related to these services are included in research and development expenses in the accompanying set of financial statements.
As of December 31, 2009, the related party payable was $7.6 million and has been included in current liabilities.
| 7. | Segment Information: |
The Company business consists solely of compound partnering activities. Accordingly, the Company operates in one reportable business segment.
| 8. | Subsequent Event: |
The Company has evaluated subsequent events through February 24, 2010, the date of issuance of the accompanying financial statements and no such events existed.
F-35
Table of Contents
MAGEN BIOSCIENCES, INC.
(A Development Stage Company)
TABLE OF CONTENTS
| Page | ||
| F-37 | ||
| F-38 | ||
| F-39-F-40 | ||
| F-41 | ||
| F-42-F-52 | ||
F-36
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of
Pharmaceutical Product Development, Inc.
We have audited the accompanying statements of operations, changes in stockholders’ equity, and cash flows of Magen BioSciences, Inc. (a development stage company) (the “Company”) for the year ended December 31, 2008, the period from January 1, 2009 through April 2, 2009, and the cumulative period January 25, 2006 (date of inception) through April 2, 2009. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with auditing standards generally accepted in the United States of America. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such financial statements present fairly, in all material respects, the results of the Company’s operations and its cash flows for the year ended December 31, 2008, the period from January 1, 2009 to April 2, 2009, and the cumulative period January 25, 2006 (date of inception) to April 2, 2009, in conformity with accounting principles generally accepted in the United States of America.
As discussed in Note 9 to the financial statements, the Company was purchased on April 2, 2009.
/s/ DELOITTE & TOUCHE LLP
Charlotte, North Carolina
February 24, 2010
F-37
Table of Contents
(A Development Stage Company)
FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM JANUARY 1, 2009
TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM JANUARY 25, 2006
(DATE OF INCEPTION) TO APRIL 2, 2009
| 2008 | Period From January 1, 2009 To April 2, 2009 | Cumulative Period From January 25, 2006 (Date of Inception) To April 2, 2009 | ||||||||||
OPERATING EXPENSES: | ||||||||||||
Research and development | $ | 4,493,176 | $ | 1,110,049 | $ | 9,654,083 | ||||||
General and administrative | 2,528,275 | 1,526,371 | 8,023,413 | |||||||||
Total operating expenses | 7,021,451 | 2,636,420 | 17,677,496 | |||||||||
LOSS FROM OPERATIONS | (7,021,451 | ) | (2,636,420 | ) | (17,677,496 | ) | ||||||
INTEREST INCOME | 130,847 | 1,057 | 649,354 | |||||||||
NET LOSS | $ | (6,890,604 | ) | $ | (2,635,363 | ) | $ | (17,028,142 | ) | |||
F-38
Table of Contents
(A Development Stage Company)
STATEMENT OF CHANGES IN STOCKHOLDERS’ EQUITY
FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM JANUARY 1, 2009
TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM JANUARY 25, 2006
(DATE OF INCEPTION) TO APRIL 2, 2009
| Common Stock | Additional Paid-In Capital | Other Comprehensive Income | Deficit Accumulated During Development Stage | Total Stockholders’ Deficit | ||||||||||||||||
| Shares | $0.001 Par Value | |||||||||||||||||||
OPENING BALANCE—January 25, 2006 | ||||||||||||||||||||
(date of inception): | ||||||||||||||||||||
Issuance of nonvested restricted stock to founders | 849,610 | $ | 850 | $ | — | $ | — | $ | — | $ | 850 | |||||||||
Issuance of common stock in exchange for license | 81,700 | 82 | 8,088 | 8,170 | ||||||||||||||||
Issuance of nonvested common stock to officer | 140,625 | 140 | 13,922 | 14,062 | ||||||||||||||||
Employee stock option compensation expense | 1,050 | 1,050 | ||||||||||||||||||
Nonemployee stock compensation expense | 931 | 931 | ||||||||||||||||||
Nonemployee stock option compensation expense | 3,841 | 3,841 | ||||||||||||||||||
Accretion of preferred stock to redemption value | (22,777 | ) | (22,777 | ) | ||||||||||||||||
Net loss | — | ��� | — | — | (1,714,270 | ) | (1,714,270 | ) | ||||||||||||
Total comprehensive loss | — | — | — | — | — | (1,714,270 | ) | |||||||||||||
BALANCE—December 31, 2006 | 1,071,935 | 1,072 | 5,055 | (1,714,270 | ) | (1,708,143 | ) | |||||||||||||
Vesting of nonvested common stock | 570,313 | 570 | 16,087 | 16,657 | ||||||||||||||||
Employee stock option compensation expense | 7,822 | 7,822 | ||||||||||||||||||
Nonemployee stock compensation expense | 1,865 | 1,865 | ||||||||||||||||||
Nonemployee stock option compensation expense | 5,904 | 5,904 | ||||||||||||||||||
Accretion of preferred stock to redemption value | (36,733 | ) | (5,482 | ) | (42,215 | ) | ||||||||||||||
Other comprehensive income | 1,406 | 1,406 | ||||||||||||||||||
Net loss | (5,787,905 | ) | (5,787,905 | ) | ||||||||||||||||
Total comprehensive loss | — | — | — | — | — | (5,786,499 | ) | |||||||||||||
BALANCE—December 31, 2007 | 1,642,248 | 1,642 | — | 1,406 | (7,507,657 | ) | (7,504,609 | ) | ||||||||||||
F-39
Table of Contents
MAGEN BIOSCIENCES, INC.
(A Development Stage Company)
STATEMENT OF CHANGES IN STOCKHOLDERS’ EQUITY
FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM JANUARY 1, 2009
TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM JANUARY 25, 2006
(DATE OF INCEPTION) TO APRIL 2, 2009
| Common Stock | Additional Paid-In Capital | Other Comprehensive Income | Deficit Accumulated During Development Stage | Total Stockholders’ Deficit | |||||||||||||||||
| Shares | $0.001 Par Value | ||||||||||||||||||||
BALANCE—December 31, 2007 | 1,642,248 | 1,642 | — | 1,406 | (7,507,657 | ) | (7,504,609 | ) | |||||||||||||
Vesting of nonvested common stock | 162,500 | 163 | 16,087 | 16,250 | |||||||||||||||||
Employee stock option compensation expense | 40,110 | 40,110 | |||||||||||||||||||
Non-employee stock compensation | 2,240 | 2,240 | |||||||||||||||||||
Accretion of preferred stock to redemption value | (59,886 | ) | (59,886 | ) | |||||||||||||||||
Other comprehensive income | (1,406 | ) | (1,406 | ) | |||||||||||||||||
Net loss | — | — | — | — | (6,890,604 | ) | (6,890,604 | ) | |||||||||||||
Total comprehensive loss | — | — | — | — | — | (6,892,010 | ) | ||||||||||||||
BALANCE—December 31, 2008 | 1,804,748 | 1,805 | (1,449 | ) | — | (14,398,261 | ) | (14,397,905 | ) | ||||||||||||
Vesting of nonvested common stock | 40,625 | 41 | 4,022 | 4,063 | |||||||||||||||||
Employee stock option compensation expense | 12,395 | 12,395 | |||||||||||||||||||
Non-employee stock compensation expense | 560 | 560 | |||||||||||||||||||
Accretion of preferred stock to redemption value | (15,535 | ) | (15,535 | ) | |||||||||||||||||
Other comprehensive income | — | ||||||||||||||||||||
Net loss | (2,635,363 | ) | (2,635,363 | ) | |||||||||||||||||
Total comprehensive loss | — | — | — | — | — | (2,635,363 | ) | ||||||||||||||
BALANCE—April 2, 2009 | 1,845,373 | $ | 1,846 | $ | (7 | ) | $ | — | $ | (17,033,624 | ) | $ | (17,031,785 | ) | |||||||
See notes to financial statements.
F-40
Table of Contents
(A Development Stage Company)
STATEMENTS OF CASH FLOWS
FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM JANUARY 1, 2009
TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM JANUARY 25, 2006
(DATE OF INCEPTION) TO APRIL 2, 2009
| 2008 | Period From January 1, 2009 To April 2, 2009 | Cumulative Period From January 25, 2006 (Date of Inception) To April 2, 2009 | ||||||||||
CASH FLOWS USED IN OPERATING ACTIVITIES: | ||||||||||||
Net loss | $ | (6,890,604 | ) | $ | (2,635,363 | ) | $ | (17,028,142 | ) | |||
Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
Depreciation | 199,797 | 52,015 | 368,842 | |||||||||
Stock-based compensation expense related to the issuance of common stock and stock options | 42,350 | 12,955 | 76,718 | |||||||||
Noncash research and development expense | — | — | 8,170 | |||||||||
Amortization of investments | (42,681 | ) | — | (42,681 | ) | |||||||
Loss on disposal of equipment | — | — | 774 | |||||||||
Changes in assets and liabilities | — | |||||||||||
Prepaid expenses and other current assets | 65,931 | 15,247 | (71,490 | ) | ||||||||
Accounts payable | (109,164 | ) | (199,158 | ) | (68,625 | ) | ||||||
Deferred rent | 30,430 | 3,622 | 34,052 | |||||||||
Accrued expenses | (248,950 | ) | 1,150,910 | 1,308,588 | ||||||||
Net cash used in operating activities | (6,952,891 | ) | (1,599,772 | ) | (15,413,794 | ) | ||||||
CASH FLOWS USED IN INVESTING ACTIVITIES: | ||||||||||||
Purchase of short-term investments | (4,761,763 | ) | — | (13,457,320 | ) | |||||||
Sales and maturities of short-term investments | 6,750,000 | — | 13,500,000 | |||||||||
Increase in restricted cash | — | — | (128,818 | ) | ||||||||
Payments to purchase property and equipment | (373,255 | ) | — | (978,832 | ) | |||||||
Net cash provided by (used in) investing activities | 1,614,982 | — | (1,064,970 | ) | ||||||||
CASH FLOWS PROVIDED BY FINANCING ACTIVITIES: | ||||||||||||
Proceeds from issuance of preferred stock—net of issuance costs of $100,673, $0, $214,360, respectively | 6,910,690 | — | 16,949,964 | |||||||||
Proceeds from loan | 350,000 | |||||||||||
Proceeds from nonvested issuance of common stock to officer and founders | — | — | 77,175 | |||||||||
Net cash provided by financing activities | 6,910,690 | 350,000 | 17,027,139 | |||||||||
NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS | 1,572,781 | (1,249,772 | ) | 898,375 | ||||||||
CASH AND CASH EQUIVALENTS—Beginning of period | 575,366 | 2,148,147 | — | |||||||||
CASH AND CASH EQUIVALENTS—End of period | $ | 2,148,147 | $ | 898,375 | $ | 898,375 | ||||||
NONCASH ACTIVITIES: | ||||||||||||
Accretion of Redeemable Convertible Preferred Stock to redemption value | $ | 59,886 | $ | 15,535 | $ | 140,413 | ||||||
See notes to financial statements.
F-41
Table of Contents
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
AS OF AND FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM
JANUARY 1, 2009 TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM
JANUARY 25, 2006 (DATE OF INCEPTION) TO APRIL 2, 2009
| 1. | NATURE OF BUSINESS AND BASIS OF PRESENTATION |
Nature of Business—Magen BioSciences, Inc. (a development stage company) (the “Company”) was a privately held biotechnology company incorporated in January 2006, to develop and commercialize innovative products focused on improving the health and appearance of human skin. The Company employed an in-licensing strategy to build a diversified product portfolio to address a number of skin disorders and the prevention of ultra-violet radiation related skin damage. The Company operated as a single segment in Cambridge, Massachusetts.
The Company was subject to a number of risks common to emerging development stage companies in the life sciences industry. Principal among those risks were the uncertainties of the drug development process, technological innovations, dependence on key individuals, development of the same or similar technological innovations by the Company’s competitors, protection of proprietary technology, compliance with government regulations and approval requirements, uncertainty of market acceptance of products, product liability, and the need to obtain additional financing necessary to fund future operations. Activities to date consisted primarily of research and development and, accordingly, the Company was classified as a development stage enterprise.
On April 2, 2009, Pharmaceutical Product Development, Inc. (PPD) acquired all of the ownership interests in the Company for $14.9 million. (see Note 9).
Stock Split—In connection with the Company’s second tranche of Series A-1 Redeemable Convertible Preferred Stock (“Series A-1 Preferred Stock”) financing completed in January 2008, the stockholders approved a stock split of the Company’s Series A-1 Preferred Stock on a 1.6 for 1 basis. Accordingly, all share amounts have been restated to reflect the 1.6 for 1 stock split as if it had occurred from inception.
| 2. | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
Use of Estimates—The preparation of the Company’s financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
Cash Equivalents—Cash equivalents represent highly liquid investments with remaining maturities of 90 days or less at the date of purchase. Cash equivalents as of December 31, 2008 and April 2, 2009 consisted of money market funds and investments with less than 90 days maturity when purchased.
Short-Term Investments—The Company had determined that all investments in corporate debt securities were short-term marketable securities classified as available-for-sale. Available-for-sale securities were carried at fair value, with the unrealized gains and losses reported in stockholders’ deficit in the accompanying balance sheets under the caption Other comprehensive income. The amortized cost of debt securities was adjusted for amortization of premiums and discounts to maturity. Such amortization was included in the Interest income line
F-42
Table of Contents
MAGEN BIOSCIENCES, INC.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
AS OF AND FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM
JANUARY 1, 2009 TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM
JANUARY 25, 2006 (DATE OF INCEPTION) TO APRIL 2, 2009
item on the accompanying statements of operations. The cost of securities sold was based on the specific identification method. Interest on securities classified as available-for-sale were included in the Interest income line item on the statements of operation. During the year ended December 31, 2008, the period ended April 2, 2009, and the period from January 25, 2006 (date of inception) the Company recognized $46,829, $0 and $200,829 in realized gains on securities. At December 31, 2008 and April 2, 2009 the Company had no unrealized gains and no short-term investments.
Concentrations of Credit Risk—Financial instruments that potentially subjected the Company to concentrations of credit risk consisted primarily of cash and cash equivalents and short-term investments. Substantially all of the Company’s cash and cash equivalents were held at one financial institution.
Depreciation of Property and Equipment—Property and equipment were recorded at cost. Expenditures for maintenance and repairs were charged to expense as incurred, whereas major betterments were capitalized as additions to property and equipment. Depreciation was provided using the straight-line method over the estimated useful life of three years for computer equipment, five years for laboratory equipment, five years for furniture and fixtures, and the shorter of three years or the term of the lease for leasehold improvements. Depreciation expense for the year ended December 31, 2008, the period from January 1, 2009 to April 2, 2009, and the cumulative period from January 25, 2006 (date of inception) to April 2, 2009 was $199,797, $52,015, and $368,842 respectively.
Research and Development Expenses—Research and development expenses were charged to operations as incurred or as third parties provide services. Clinical materials and supplies were charged to research and development expense as purchased. Patent costs were expensed as incurred and included in general and administrative expense. Up-front and milestone payments related to licensing of compounds which have not received regulatory approval for sale and are not currently being marketed were expensed as research and development expenses.
Stock-Based Compensation—Compensation expense related to stock based awards to employees and directors was recognized on a straight-line basis over the requisite service period for the entire award (straight-line attribution method), ensuring that the amount of compensation cost recognized at any date at least equaled the portion of the grant-date fair value of the award that was vested at that time. The measurement of stock-based compensation was subject to periodic adjustments as the underlying equity investments vested. The measurement date for employee awards was the date of grant. The measurement date for nonemployee awards was the date the performance of services was completed. Stock-based compensation is allocated to research and development or selling, general and administrative costs based the allocation of each individual employee’s current salary is allocated.
Income Taxes—The difference between the financial statement and tax basis of the assets and liabilities was determined for each period presented. Deferred income tax assets and liabilities were computed using the tax laws and rates that were expected to apply to periods in which such differences reversed. Valuation allowances were established, if necessary, to reduce the deferred tax asset to the amount that would more likely than not be realized.
| 3. | COMMITMENTS AND CONTINGENCIES |
License Agreements—The Company entered into a license agreement with an institution in May 2006. The license agreement provides the Company with exclusive worldwide rights to certain intellectual property in
F-43
Table of Contents
MAGEN BIOSCIENCES, INC.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
AS OF AND FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM
JANUARY 1, 2009 TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM
JANUARY 25, 2006 (DATE OF INCEPTION) TO APRIL 2, 2009
the defined field of use. The Company was required to use diligent commercial effort to develop and introduce the licensed product to the marketplace. In exchange for these rights, the Company issued 81,700 shares of common stock with a fair value at the date of issuance of $8,170, which was recorded in research and development expenses at the date of issuance. The Company was obligated to prosecute and maintain patent applications and patents and pay all patent related costs, to make payments ranging from $20,000 to $150,000 upon the achievement of certain milestones and to pay royalty payments on net sales. No development milestones were reached or sales made through April 2, 2009. As of April 1, 2009, this agreement was terminated.
The Company entered into an exclusive license agreement with a pharmaceutical company in November 2007 and February 2009. The license agreements provided the Company with exclusive worldwide rights to develop and commercialize products containing the licensed compounds for diagnosis and treatment of dermatological diseases and conditions in humans and animals.
In exchange for these exclusive worldwide licenses, the Company paid a nonrefundable $1,000,000 license fee payment when each agreement was entered into which was accounted for as research and development expense. In addition, the Company was obligated to make payments of up to an aggregate of $21,400,000 upon the achievement of development milestones under each agreement, if and when achieved. The agreements also provided for royalty payments as a percentage of net sales. There were no development milestones met or sales made through April 2, 2009 under either of these agreements.
Employment Agreements—The Company maintained an employment agreement with the Company’s chief executive officer that contained severance terms obligating the Company to pay severance plus medical and dental benefits for terms ranging from twelve to eighteen months in the event employment was terminated without cause or if the officer terminates for good reason, as defined in the agreement. No payments were due in the event of voluntary termination or termination for cause, as defined. During the period from January 1 2009 to April 2, 2009, the Company expensed in the aggregate $737,246 for bonuses to employees and for severance due to the chief executive officer under his employment agreement due to the acquisition of the Company on April 2, 2009 by Pharmaceutical Product Development, Inc. Additionally, immediately prior to the acquisition the Company terminated all of the employment agreements with employees (see Note 9).
Leases—In November 2007, the Company leased office space and obtained services (facilities management and office and laboratory services) under an operating lease that expires in November 2012. Future minimum lease payments under the operating lease as of April 2, 2009 are as follows:
2009 (remainder of 2009) | 268,052 | ||
2010 | 416,153 | ||
2011 | 430,718 | ||
2012 | 406,509 | ||
| $ | 1,521,432 | ||
F-44
Table of Contents
MAGEN BIOSCIENCES, INC.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
AS OF AND FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM
JANUARY 1, 2009 TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM
JANUARY 25, 2006 (DATE OF INCEPTION) TO APRIL 2, 2009
Rent expense, which was recorded on a straight-line basis, totaled $428,930, $103,617 and $849,715 for the year ended December 31, 2008, the period from January 1, 2009 to April 2, 2009, and the period from January 25, 2006 (date of inception) to April 2, 2009 respectively.
Loan Agreement—In March 2009, the Company borrowed $350,000 from PPD pursuant to the terms and subject to the conditions set forth in the Loan Agreement, by and between PPD and the Company. Upon acquisition of the Company by PPD the loan was offset against the purchase price paid by PPD. See Note 9.
| 4. | EMPLOYEE BENEFIT PLAN |
In July 2007, the Company established a simple individual retirement arrangements (IRA) retirement plan for eligible employees. Under a simple IRA plan, employees and employers make contributions to traditional IRAs set up for employees, subject to certain limits. The Company matches employee contributions dollar for dollar up to 1% of salary. Federal contribution limits per employee are $10,500 in 2008 and $11,500 in 2009. If the employee is age 50 or over, a “catch-up” contribution of $2,500 is allowed both in 2008 and 2009. During the year ended December 31, 2008, the period ended April 2, 2009 and the period from January 25,2006 (date of inception) to April 2, 2009, the Company made contributions totaling $15,335, $15,185 and $36,039, respectively.
F-45
Table of Contents
MAGEN BIOSCIENCES, INC.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
AS OF AND FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM
JANUARY 1, 2009 TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM
JANUARY 25, 2006 (DATE OF INCEPTION) TO APRIL 2, 2009
| 5. | REDEEMABLE CONVERTIBLE PREFERRED STOCK |
The Company had 14,180,000 shares of $0.001 par value preferred stock authorized as of December 31, 2008 and April 2, 2009, of which 1,840,000 shares were designated as Series A redeemable convertible preferred stock (“Series A Preferred Stock”) and 12,340,000 shares were designated as Series A-1 Preferred Stock. Because the Company’s shares of preferred stock were considered temporary equity, they have not been included in the accompanying Statement of Changes in Stockholder’s Equity. Changes in the balances of Redeemable Convertible Preferred Stock were as follows:
| Series A Redeemable Convertible Preferred Stock | Series A-1 Redeemable Convertible Preferred Stock | |||||||||
| Shares | $0.001 Par Value | Shares | $0.001 Par Value | |||||||
OPENING BALANCE—January 25, 2006 (date of inception): | ||||||||||
Issuance of Series A Redeemable Convertible Preferred Stock—net of issuance costs of $91,691 | 1,840,000 | $ | 1,748,309 | |||||||
Issuance of Series A-1 Redeemable Convertible referred Stock—net of issuance costs of $122,669 | 6,730,907 | $ | 8,290,965 | |||||||
Accretion of preferred stock to redemption value | 13,812 | 8,965 | ||||||||
BALANCE—December 31, 2006 | 1,840,000 | 1,762,121 | 6,730,907 | 8,299,930 | ||||||
Accretion of preferred stock to redemption value | 17,446 | 24,769 | ||||||||
BALANCE—December 31, 2007 | 1,840,000 | 1,779,567 | 6,730,907 | 8,324,699 | ||||||
Issuance of Series A-1 Preferred Stock—net of issuance costs of $100,673 | 5,609,092 | 6,910,690 | ||||||||
Accretion of preferred stock to redemption value | 16,671 | 43,216 | ||||||||
BALANCE—December 31, 2008 | 1,840,000 | 1,796,238 | 12,339,999 | 15,278,605 | ||||||
Accretion of preferred stock to redemption value | 4,168 | 11,367 | ||||||||
BALANCE—April 2, 2009 | 1,840,000 | $ | 1,800,406 | 12,339,999 | $ | 15,289,972 | ||||
Series A Preferred Stock—In February 2006, the Company issued 1,840,000 shares of Series A Preferred Stock at $1.00 per share for proceeds of $1,748,309 net of issuance costs of $91,691. The Series A issuance costs of $91,691 were being accreted to the earliest redemption date.
Series A-1 Preferred Stock—In August 2006, the Company received proceeds of $8,290,965, net of issuance costs of $122,669, from the issuance of 6,730,907 shares of Series A-1 Preferred Stock at $1.25 per share. The Series A-1 issuance costs of $122,669 were being accreted to the earliest redemption date.
In January 2008, the Company received proceeds of $6,910,690, net of issuance costs of $100,673, from the issuance of 5,609,092 shares of Series A-1 Preferred Stock at $1.25 per share. The Series A-1 issuance costs of $100,673 were being accreted to the earliest redemption date.
F-46
Table of Contents
MAGEN BIOSCIENCES, INC.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
AS OF AND FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM
JANUARY 1, 2009 TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM
JANUARY 25, 2006 (DATE OF INCEPTION) TO APRIL 2, 2009
The rights, preferences, and privileges of the Series A and Series A-1 Preferred Stock (collectively, the “Series A/A-1 Preferred Stock”) were as follows:
Voting Rights—The holder of each share of the Series A/A-1Preferred Stock had the right to one vote for each share of common stock into which the Preferred Stock could then be converted, and such holder would have full voting rights and powers equal to those of the holders of common stock and be entitled to vote on all matters. The holders of the common stock were entitled to one vote for each share held. Series A/A-1 Preferred Stockholders and holders of the common stock, voting separately, had the right to elect four and three members of the Board of Directors, respectively, and voting together had the right to elect two members of the Board of Directors.
Dividends—Dividends could be declared and paid on the capital stock of the Company when and if determined by the Board of Directors and, for the common stock, were subject to any preferential dividend rights of any then outstanding Series A/A-1 Preferred Stock. Dividends could not be declared, paid, or set aside on shares of common stock in any year unless the Series A Preferred Stockholders and the Series A-1 Preferred Stockholders received dividends of at least $0.08 and $0.10 per share, respectively, if and when declared by the Board of Directors. Dividends were not cumulative. No such dividends had been declared through April 2, 2009.
Liquidation—In the event of any liquidation, dissolution, or winding up of the Company, either voluntary or involuntary the holders of the Series A Preferred Stock and Series A-1 Preferred Stock were entitled to receive cash in the amount of $1.00 and $1.25 per share, respectively, subject to appropriate adjustment for any stock splits, stock dividends, combination, or any other similar recapitalization affecting such shares, plus any dividends declared but unpaid. The Series A/A-1 Preferred stockholders had preference over the holders of common stock. In the event that assets were insufficient to pay Series A/A-1 Preferred stockholders in the full amount to which they were entitled, they were to be distributed on a pro rata basis. After the payment of all preferential amounts, any remaining assets available for distribution would be distributed among the holders of the common stock then outstanding. The merger or consolidation of the Company would be deemed to be a liquidation event at the election of 55% of the holders of the preferred stock.
Redemption—First Redemption Shares (Series A/A-1 Preferred Stock issued and outstanding on or before October 31, 2006) and Second Redemption Shares (Series A-1 Preferred Stock issued and outstanding after October 31, 2006) were redeemable on August 19, 2011, August 20, 2012 and August 20, 2013 and on December 20, 2012, December 20, 2013 and December 19, 2014, respectively, at the then effective preference amount, which was equal to the original issuance prices of the Series A Preferred Stock and A-1 Preferred Stock of $1.00 and $1.25, respectively, plus any declared and unpaid dividends. The Series A Preferred Stock and A-1 Preferred Stock were redeemable as follows:
| First Redemption Shares | Second Redemption Shares | |||
Year Ended December 31: | ||||
2011 | 613,333 | |||
2012 | 613,333 | 5,141,666 | ||
2013 | 613,334 | 5,141,666 | ||
2014 | 5,141,667 |
F-47
Table of Contents
MAGEN BIOSCIENCES, INC.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
AS OF AND FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM
JANUARY 1, 2009 TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM
JANUARY 25, 2006 (DATE OF INCEPTION) TO APRIL 2, 2009
Conversion—Each share of Series A/A-1 Preferred Stock was convertible, at the option of the holder, at any time after issuance into a number of shares of common stock determined by dividing the original issue price by the conversion price, subject to certain antidilutive adjustments. The initial conversion price for the Series A Preferred Stock was $1.00 and for the Series A-1 Preferred Stock is $1.25. The initial conversion ratio for the Series A Preferred Stock was one-for-one and the initial conversion ratio for the Series A-1 Preferred Stock was one-for-one.
Each share of Preferred Stock would automatically convert into shares of common stock at the then effective conversion price upon the closing of an initial public offering for which the offering price was at least $5.00 per share resulting in at least $50,000,000 of net proceeds to the Company, or upon a vote of 55% or more of the Series A/A-1 Preferred Stockholders.
In the event that any Series A/A-1 Preferred Stock holder did not participate in a Qualified Financing, defined as an offering with a price per share less than the most recent equity round, each share of Series A/A-1 Preferred Stock held by such stockholder would automatically convert into shares of common stock at the respective Series A/A-1 Preferred Stock conversion price then in effect.
On April 2, 2009 all shares of preferred stock were purchased in connection with the sale of the Company to PPD (see Note 9).
| 6. | COMMON STOCK |
The Company had 19,200,000 shares of $0.001 par value common stock authorized as of December 31, 2008 and April 2, 2009 of which the Company sold 2,175,000 shares of unvested restricted common stock to founders and a nonemployee consultant at the fair value of $0.001 per share in January 2006, and sold 750,000 shares to an officer at the fair value of $0.10 per share in June 2006. Total proceeds for the issuance of stock were $77,175. All shares were subject to vesting and repurchasing provisions at the option of the Company as defined in the respective purchase agreements. The shares vest over four years from the date the grants, as provided in each agreement, with vesting commencement dates ranging from the effective date of the agreement to one year from the effective date of the agreement. Upon termination of services or employment, the Company may repurchase nonvested shares at their original purchase price. As of December 31, 2008, a total of 2,130,859 of the shares were vested and 794,141 were nonvested. As of April 2, 2009, a total of 2,273,438 of the shares were vested and 651,562 remained nonvested.
In May 2006, the Company issued 81,700 shares of common stock as partial exchange for a license agreement. The common stock was valued at $8,170, based on the fair value of the Company’s common stock which was recorded in research and development expenses (see Note 3).
Each share of common stock was entitled to one vote. The holders of common stock were also entitled to receive dividends whenever funds were legally available and when declared by the Board of Directors, subject to the prior rights of holders of all classes of stock outstanding.
F-48
Table of Contents
MAGEN BIOSCIENCES, INC.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
AS OF AND FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM
JANUARY 1, 2009 TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM
JANUARY 25, 2006 (DATE OF INCEPTION) TO APRIL 2, 2009
At April 2, 2009, the Company had the following shares of common stock reserved for future issuance:
Conversion of Series A preferred stock | 1,840,000 | |
Conversion of Series A-1 preferred stock | 12,339,999 | |
Granted stock options | 1,137,000 | |
Future stock grants | 348,583 | |
Total capital stock reserved for future issuance | 15,665,582 | |
On April 2, 2009 all shares of common stock were purchased in connection with the sale of the Company to PPD (see Note 9).
| 7. | SHARE-BASED PAYMENTS |
Under the Company’s 2006 equity incentive plan (the “Plan”), up to 1,485,583 shares of common stock could be granted to the Company’s employees, officers, directors, consultants, and advisors in the form of options, restricted stock awards, or other stock-based awards. As administrator of the Plan, the Board of Directors (the “Board”) had broad authority over its operation, including, but not limited to, the authority to determine (a) which of the eligible employees, consultants, and directors were granted options and awards; (b) when such options and awards were granted; (c) the amount and terms with respect to each grant, including the vesting period; and (d) whether the options granted were nonstatutory (nonqualified) stock options or incentive stock options under the Internal Revenue Code (the “Code”).
The exercise price per share relating to options granted under the Plan would not be less than the fair market value of the common stock on the date of grant. The vesting period for stock options was generally four years. Options granted under the Plan expired no more than 10 years from the date of grant.
F-49
Table of Contents
MAGEN BIOSCIENCES, INC.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
AS OF AND FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM
JANUARY 1, 2009 TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM
JANUARY 25, 2006 (DATE OF INCEPTION) TO APRIL 2, 2009
For the period January 1, 2009 through April 2, 2009 there were no new options granted, forfeited or other option activity. A summary of the stock option activity under the Plan from January 25, 2006 (date of inception) to December 31, 2008, is as follows:
| Number of Shares | Weighted- Average Exercise Price | Weighted- Average Remaining Contractual Life | Intrinsic Value | ||||||||
Options outstanding—January 25, 2006 | — | $ | — | ||||||||
(date of inception) | |||||||||||
Granted | 150,000 | 0.15 | |||||||||
Forfeited | (10,000 | ) | 0.10 | ||||||||
Options outstanding—January 1, 2007 | 140,000 | 0.15 | 9.55 | $ | 6,500 | ||||||
Granted | 212,000 | 0.17 | |||||||||
Options outstanding—January 1, 2008 | 352,000 | 0.15 | 8.83 | $ | 20,180 | ||||||
Granted | 785,000 | 0.24 | |||||||||
Options outstanding—December 31, 2008 | 1,137,000 | $ | 0.20 | 8.38 | $ | 20,180 | |||||
Granted | — | ||||||||||
Options outstanding—April 2, 2009 | 1,137,000 | $ | 0.20 | 8.13 | $ | 20,180 | |||||
Vested and expected to vest—April 2, 2009 | 430,465 | $ | 0.20 | 8.13 | $ | 20,180 | |||||
Available for grant—April 2, 2009 | 348,583 | $ | 0.21 | ||||||||
During the years ended December 31, 2007 and 2008, the Company granted options to employees with a total fair value of $27,753 and $115,866 and a weighted-average grant date fair value of $0.16 and $0.15 per share, respectively. Unrecognized compensation cost that was to be recognized as of April 2, 2009 is $105,426 which was expected to be recognized over 3 years.
The following table sets forth information regarding options outstanding at April 2, 2009:
Exercise Price | Options Outstanding | Number of Shares | Exercisable | ||||||
| Number of Shares | Weighted- Average Remaining Life (Years) | Weighted- Average Exercise Price | |||||||
$0.10 | 65,000 | 7.10 | 54,375 | $ | 0.10 | ||||
0.20 | 277,000 | 7.85 | 146,021 | 0.20 | |||||
0.24 | 795,000 | 9.00 | 230,069 | 0.24 | |||||
$0.10 – $0.24 | 1,137,000 | 8.13 | 430,465 | $ | 0.20 | ||||
The Company recorded total stock-based compensation expense of $40,110, $12,395 and $71,122 for the year ended December 31, 2008, the period from January 1, 2009 to April 2, 2009, and the period from January 25, 2006 (date of inception) to April 2, 2009, respectively, for options issued to employees and
F-50
Table of Contents
MAGEN BIOSCIENCES, INC.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
AS OF AND FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM
JANUARY 1, 2009 TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM
JANUARY 25, 2006 (DATE OF INCEPTION) TO APRIL 2, 2009
nonemployees. Stock-based compensation expense for options issued recorded as research and development expense was $26,072, $8,057 and $41,050 for the year ended December 31, 2008, the period from January 1, 2009 to April 2, 2009 and the period from January 25, 2006 (date of inception) to April 2, 2009, respectively, and expense recorded as general and administrative expense totaled $14,038, $4,338 and $30,072 for the year ended December 31, 2008, the period from January 1, 2009 to April 2, 2009 and the period from January 25, 2006 (date of inception) to April 2, 2009, respectively.
The fair value of each employee and director stock option grant was estimated at the date of grant using the Black-Scholes option-pricing model with the following assumptions for the year ended December 31, 2008:
| 2008 | ||
Expected volatility of underlying stock | 66% | |
Expected Life | 6.25 years | |
Weighted-average risk-free rate | 1.75% | |
Expected dividend yield | 0% |
Since the Company’s stock was not publicly traded, and the Company had a limited history, the expected volatility in 2008 for the Company’s common stock was determined based on the historical volatility of comparable publicly traded companies in a similar industry. The expected term of the options granted was calculated using the simplified method, which represents the average of the contractual term of the option and the weighted-average vesting period of the option. The risk-free interest rate was based on a treasury instrument, which term is consistent with the expected life of the stock options.
Included in the compensation expense above is expense related to nonemployee grants of $3,336, $916 and $13,997 for the years ended December 31, 2008, the period from January 1, 2009 to April 2, 2009 and the period from January 25, 2006 (date of inception) to April 2, 2009, respectively. During the years ended December 31, 2007 and 2008 the Company granted 65,000 and 10,000 stock options to nonemployees. The expense for the options was recognized as the options are vested. The weighted-average grant date fair value of nonemployee stock options granted for the years ended December 31, 2007 and 2008 was $0.24 and $0.15 per share, using the Black-Scholes option pricing model and the assumptions were the same as employee options listed above.
On April 2, 2009 all stock options were purchased in connection with the sale of the Company to PPD (see Note 9).
F-51
Table of Contents
MAGEN BIOSCIENCES, INC.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
AS OF AND FOR THE YEAR ENDED DECEMBER 31, 2008, THE PERIOD FROM
JANUARY 1, 2009 TO APRIL 2, 2009, AND FOR THE CUMULATIVE PERIOD FROM
JANUARY 25, 2006 (DATE OF INCEPTION) TO APRIL 2, 2009
| 8. | INCOME TAXES |
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s net deferred tax asset as of December 31, 2008 and April 2, 2009 were as follows:
| December 31, 2008 | April 2, 2009 | |||||||
Deferred tax asset (liability): | ||||||||
Net operating losses | $ | 3,040,000 | $ | 3,312,000 | ||||
Tax credits | 248,000 | 268,000 | ||||||
Accrued expenses | 195,000 | 390,000 | ||||||
Capitalized costs | 2,836,000 | 3,190,000 | ||||||
Depreciation | (87,000 | ) | (100,000 | ) | ||||
Other | (25,000 | ) | (24,000 | ) | ||||
Net deferred tax assets | 6,207,000 | 7,036,000 | ||||||
Deferred tax asset valuation allowance | (6,207,000 | ) | (7,036,000 | ) | ||||
Net deferred tax assets | $ | — | $ | — | ||||
The Company has recorded a full valuation allowance as of December 31, 2008 and April 2, 2009, as it was more likely than not that the benefits of the net operating loss carryforwards and other deferred tax assets would not be utilized.
At April 2, 2009, the Company had net operating losses being carried forward of approximately $17 million, which expire at various dates through 2029. The research and development tax credits total approximately $0.5 million and expire at various dates through 2029. The Company’s ability to use the losses and tax credits to offset future taxable income was subject to restrictions that could severely limit the Company’s future use of the loss carryforwards since certain ownership changes described in the code had occurred. The availability of the Company to fully utilize the net operating losses against future taxable income could have been limited. No income taxes were paid during the year ended December 31, 2008, for the period from January 1, 2009 to April 2, 2009 or for the period from January 25, 2006 (date of inception) to December 31, 2007.
| 9. | SUBSEQUENT EVENT |
On April 2, 2009, Pharmaceutical Product Development, Inc. (PPD) acquired all of the ownership interests in the Company for $14.9 million. Subsequent events have been updated through February 24, 2010.
* * * * * *
F-52