Exhibit 99.2
CARISMA BUSINESS
Overview
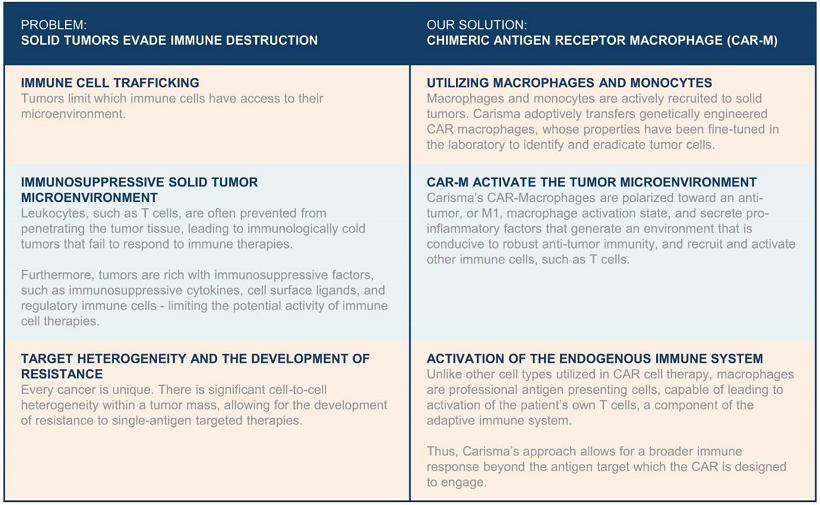
Carisma Therapeutics Inc., or Carisma, is a clinical stage cell therapy company focused on utilizing Carisma’s proprietary macrophage and monocyte cell engineering platform to develop transformative immunotherapies to treat cancer and other serious diseases. Carisma has created a comprehensive cell therapy platform to enable the therapeutic use of engineered macrophages and monocytes, which belong to a subgroup of white blood cells called myeloid cells. Macrophages and monocytes are part of the innate immune system and can detect and degrade harmful substances through a process referred to as phagocytosis, in which the harmful substance is engulfed and destroyed and in turn leads to the activation of a broad immune response.
To harness the powerful immunologic functions of macrophages against cancer, Carisma has developed a proprietary Chimeric Antigen Receptor Macrophage, or CAR-M, platform technology. Chimeric antigen receptors, or CARs, are synthetically engineered receptors that are designed to bestow immune cells with the ability to target specific antigens on the surface of cancer cells. By introducing CARs into macrophage and monocyte cells, Carisma aims to redirect their potent innate immune functions against cancer. Carisma’s CAR-M platform technology incorporates proprietary tumor targeting constructs, vectors to deliver CARs to macrophages and monocytes and novel manufacturing processes. Carisma’s CAR-M therapeutics are designed to infiltrate the solid tumor microenvironment, kill cancer cells via targeted phagocytosis, and activate other immune cells, such as T-cells, to initiate a robust anti-tumor immune response.
Carisma’s lead product candidate CT-0508, the first CAR-M to be evaluated in a human clinical trial, is an ex vivo autologous cell therapy product candidate, wherein immune cells from blood drawn from a patient are engineered outside of the body and reinfused into the same patient. CT-0508 is intended to treat solid tumors that overexpress HER2, a protein that is overexpressed on the surface of a variety of solid tumors, including breast cancer, gastric cancer, esophageal cancer, salivary gland cancer, and numerous others. Carisma has completed enrollment of the first group of patients in a Phase 1 clinical trial of CT-0508, with nine patients having been successfully dosed. In November 2022, Carisma presented preliminary clinical results from the first group of patients. CT-0508 was successfully manufactured using macrophages obtained from heavily pre-treated, advanced solid tumor patients and has shown high CAR expression, viability, and purity. In addition, CT-0508 has been generally well-tolerated after infusion with no dose-limiting toxicities reported to date from the nine patients enrolled in the first group. While the results from this early clinical trial data are both preliminary and limited, Carisma believes the results indicate that CT-0508 can be detected within the tumor microenvironment, or TME, lead to remodeling and activation of the TME, and potentially induce anti-tumor adaptive immunity. Carisma anticipates providing multiple clinical data updates over the next 18 months. In the combination setting, Carisma has observed the synergistic potential of CT-0508 with a PD1 blocking T-cell checkpoint inhibitor in pre-clinical models, enabling a combination trial with pembrolizumab. Carisma submitted a clinical protocol amendment to the United States Food and Drug Administration, or FDA, in September 2022 to allow Carisma to treat patients with the co-administration of CT-0508 and pembrolizumab, and opened the study for enrollment in December 2022. The FDA has granted “Fast Track” status to CT-0508 for the treatment of patients with HER2 overexpressing solid tumors and Carisma plans to prioritize development for this indication.
Beyond CT-0508, Carisma has a broad pipeline of cell therapy assets in various stages of pre-clinical development. In addition to the development of ex vivo CAR-M cell therapies, Carisma is also developing in vivo CAR-M gene therapies, wherein immune cells are directly engineered within the patient’s body. To advance its in vivo CAR-M therapeutics, Carisma established a strategic collaboration with ModernaTX, Inc., or Moderna, focused on the development and potential commercialization of up to 12 product candidates, of which four have already been nominated. In collaboration with Moderna, Carisma has established an approach that uses Moderna’s myeloid cell specific lipid nanoparticle/mRNA, or LNP/mRNA, technology, together with Carisma’s CAR-M platform technology, to create novel in vivo oncology gene therapies. Carisma believes this approach has the potential to enable a series of off-the-shelf product candidates to target a patient’s own myeloid cells against cancer cells directly within their body. As part of the agreement with Moderna, Carisma received a $45.0 million up-front cash payment and an investment by Moderna in the form of a $35.0 million convertible note, in addition to future research funding and the opportunity for milestone payments and royalties.
Through its robust internal discovery engine, Carisma is building upon its platform to enhance and expand the utility of macrophage cell and gene therapies, leading to the creation of multiple product candidates with the potential to treat cancer and other serious diseases. By replacing the targeting domain of the CAR, Carisma can reprogram the target antigen specificity of the CAR-M cell product and develop candidates against a range of cancer indications and therapeutic areas beyond oncology. As a result, Carisma believes the flexibility of its macrophage and monocyte cell engineering platform will allow Carisma to generate new product candidates suitable for clinical development in a cost-efficient manner to expand its pipeline. In addition to acting as a first line of defense in the innate immune system, macrophages are found in all tissues in the body where they serve key regulatory functions such as wound healing, termination of immune responses, and tissue regeneration. Using its macrophage and monocyte ex vivo and in vivo engineering platform, Carisma is pursuing early research and development of multiple assets for the potential treatment of diseases beyond oncology, including liver fibrosis, neurodegeneration, and other immunologic and inflammatory diseases.
By investing in early platform research and accessing key enabling technologies, Carisma is enhancing and expanding its platform capabilities and reinforcing its leadership position in the engineered macrophage field. Carisma has developed proprietary CAR-M platform enhancements directed toward key product parameters that are important for efficacy, safety, and patient access to its CAR-M therapies. Carisma plans to apply these technology enhancements to future CAR-M product candidates.
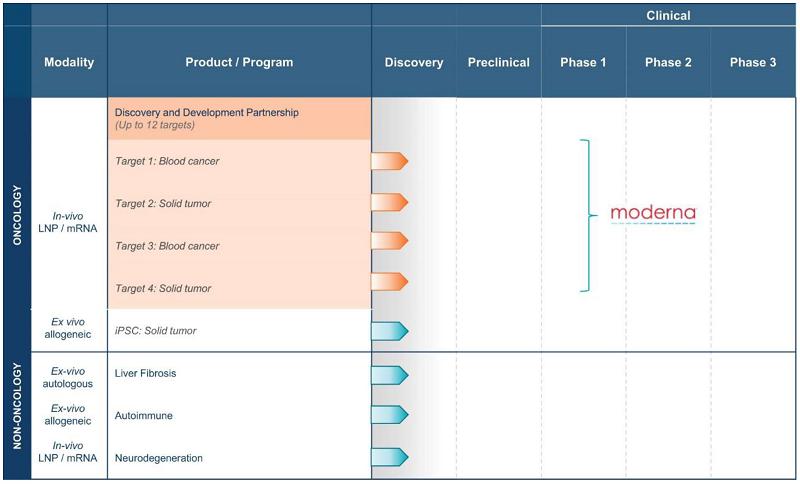
Carisma’s Pipeline Programs
Using its proprietary CAR-M platform technology, Carisma is developing a broad pipeline of product candidates, with a strong initial focus in oncology. Carisma’s ex vivo autologous CAR-M product candidates are summarized in the table below.

Carisma is also advancing discovery-stage candidates across a range of therapeutic areas, as summarized in the following table.
Carisma’s lead product candidate, CT-0508, is an ex vivo autologous cell therapy product candidate intended to treat solid tumors that overexpress HER2, a protein that is overexpressed on the surface of a variety of solid tumors including breast cancer, gastric cancer, esophageal cancer, salivary gland cancer and numerous others. CT-0508 is produced by engineering a patient’s own monocyte-derived macrophages from blood drawn from the patient with a chimeric adenoviral vector, Ad5f35, containing an anti-HER2 CAR.
Carisma has completed enrollment of the first group of patients in a Phase 1 clinical trial, with nine patients successfully dosed. The second group is currently open for enrollment, with nine additional patients to be dosed. In November 2022, Carisma presented preliminary clinical results from the first group of patients. CT-0508 was successfully manufactured using macrophages obtained from heavily pre-treated, advanced solid tumor patients and has shown high CAR expression, viability, and purity. In addition, CT-0508 has been generally well-tolerated after infusion with no dose-limiting toxicities reported to date from the nine patients enrolled in the first group. While the results from this early clinical trial data are both preliminary and limited, Carisma believes the results indicate that CT-0508 can be detected within the TME, lead to remodeling and activation of the TME, and potentially induce anti-tumor adaptive immunity. Carisma anticipates providing multiple clinical data updates over the next 18 months. In the combination setting, Carisma has observed the synergistic potential of CT-0508 with a PD1 blocking T-cell checkpoint inhibitor in pre-clinical models, enabling a combination trial with pembrolizumab. Carisma submitted a clinical protocol amendment to the FDA in September 2022 to allow it to treat patients with the co-administration of CT-0508 and pembrolizumab, and opened the study for enrollment in December 2022. The FDA has granted “Fast Track” status to CT-0508 for the treatment of patients with HER2 overexpressing solid tumors and Carisma plans to prioritize development for this indication.
Carisma is currently in the pre-clinical stage for another product candidate, CT-0525, which is also intended to treat solid tumors that overexpress HER2. By leveraging its discovery engine and preliminary clinical data from its Phase 1 clinical trial of CT-0508, Carisma is building upon its CAR-M platform to generate next-generation therapeutics that may increase potential efficacy and patient access. Notably, Carisma has developed a novel approach to CAR-M therapy to accelerate the manufacturing process, increase the cell yield, and improve upon the potential anti-tumor effect by engineering patients’ monocytes directly, without ex vivo differentiation into macrophages, as Carisma currently does for CT-0508. Carisma refers to this CAR-Monocyte approach as CAR-Mono. By increasing the cell yield, the CAR-Mono approach enables a larger potential dose, which may improve tumor control. The CAR-Mono approach reduces manufacturing time and leverages an automated, closed-system manufacturing process. CT-0525 is Carisma’s first CAR-Mono product candidate and is currently in the pre-clinical process development stage. Carisma expects to submit an IND to the FDA for CT-0525 in the second half of 2023 and initiate clinical development shortly thereafter.
Carisma is also expanding its pipeline to include multiple tumor targets, encompassing diverse solid tumor indications with significant unmet medical needs, including the following product candidates:
| · | CT-1119: CT-1119 is a mesothelin targeted CAR-M that Carisma plans to evaluate in patients with advanced mesothelin-positive solid tumors, including lung cancer, mesothelioma, pancreatic cancer, ovarian cancer, and others. Carisma anticipates nominating a lead next generation CAR construct for CT-1119 in the first half of 2024 and expects to submit an IND to the FDA in 2025. In November 2022, Carisma presented preliminary and limited data demonstrating that CT-1119 can mediate phagocytosis, tumor cell killing, and pro-inflammatory cytokine release and control tumor growth in pre-clinical lung cancer models. |
| · | CT-0729: CT-0729 is a prostate-specific membrane antigen, or PSMA, targeted CAR-M that Carisma plans to evaluate in patients with advanced, PSMA positive metastatic castrate resistant prostate cancer. CT-0729 is in the discovery stage. |
Carisma’s pipeline programs CT-1119 and CT-0729 may be developed using the next generation CAR-Mono approach.
Carisma’s current CAR-M cell therapy pipeline is informing the discovery and pre-clinical development of off-the-shelf engineered macrophage therapeutics. Carisma is developing in vivo reprogrammed LNP/mRNA CAR-M therapies for cancer through its collaboration with Moderna. In addition, Carisma is establishing an ex vivo allogeneic, induced pluripotent stem cell, or iPSC, derived macrophage and monocyte platform with the potential to develop iPSC-derived CAR-M and other macrophage therapies for indications in oncology and beyond, including indications such as liver fibrosis, neurodegeneration and auto-immunity.
Carisma’s Team
Carisma was founded in 2016 by leading cell therapy experts from the University of Pennsylvania. Dr. Saar Gill is a co-inventor of the CAR-M technology and a co-founder of Carisma. He is an Associate Professor of Medicine in the Division of Hematology-Oncology at the University of Pennsylvania. Dr. Michael Klichinsky, Pharm.D., Ph.D., is a co-inventor of the CAR-M technology, a scientific co-founder of Carisma, and Carisma’s current Chief Scientific Officer. Dr. Carl June, a co-inventor of the CAR-M technology, is the Richard W. Vague Professor in Immunotherapy in the Department of Pathology and Laboratory Medicine at the University of Pennsylvania. He also is currently Director of the Center for Cellular Immunotherapies at the Perelman School of Medicine and Director of the Parker Institute for Cancer Immunotherapy at the University of Pennsylvania, and Scientific Advisor to Carisma.
Carisma’s executive team has decades of experience in business operations, discovery, development, and manufacturing of advanced therapeutics for the treatment of serious diseases. Steven Kelly, Carisma’s Chief Executive Officer, brings over 35 years of experience in the biopharmaceutical industry at all phases of the business across multiple therapeutic categories. Carisma’s Chief Technology and Development Officer, Daniel Cushing, Ph.D., brings over 30 years of experience in the biopharmaceutical industry and is responsible for product development at Carisma. Richard Morris, Carisma’s Chief Financial Officer, has more than 25 years of experience in building and growing successful biotechnology organizations, with a focus on capital fundraising (including initial public offerings), financial strategy and operations execution, and business development efforts. Carisma’s Chief Business Officer, Tom Wilton, has over 25 years of biopharmaceutical industry experience, including corporate strategy, business development, research and development operations, and marketing.
Carisma’s Strategy
Carisma’s vision is to become a leading cell therapy company, developing and ultimately commercializing macrophage-based cell therapies that positively transform the treatment of cancer and other serious diseases. To achieve its vision, Carisma has developed its macrophage engineering platform, a pipeline of assets spanning numerous indications with unmet medical needs, a robust discovery engine, broad CAR-M intellectual property, robust manufacturing capabilities, and a dedicated executive team with extensive experience in cell therapy and drug development, manufacturing and commercialization and leading scientific expertise in the field. The key components of Carisma’s strategy are:
| · | Advance Carisma’s lead product candidate, CT-0508, through clinical development for the treatment of HER2 overexpressing solid tumors. CT-0508 is an ex vivo gene-modified autologous CAR-M cell therapy product candidate intended to treat solid tumors that overexpress HER2. Carisma has completed enrollment of the first group of patients in a Phase 1 clinical trial, with nine patients successfully dosed. The second group is currently open for enrollment, with nine additional patients to be dosed. In November 2022, Carisma presented preliminary clinical results from the first group of patients. CT-0508 was successfully manufactured using macrophages obtained from heavily pre-treated, advanced solid tumor patients and has shown high CAR expression, viability, and purity. In addition, CT-0508 has been generally well-tolerated after infusion with no dose-limiting toxicities reported to date from the nine patients enrolled in the first group. While the results from this early clinical trial data are both preliminary and limited, Carisma believes the results indicate that CT-0508 can be detected within the TME, leads to remodeling and activation of the TME, and potentially induces anti-tumor adaptive immunity. Additionally, the FDA granted “Fast Track” status to CT-0508 for the treatment of patients with HER2 overexpressing solid tumors and Carisma plans to prioritize development for this indication. Carisma has initiated a sub-study to evaluate the combination of CT-0508 with pembrolizumab, a PD1 blocking T cell checkpoint inhibitor, for patients with advanced HER2 overexpressing solid tumors. |
| · | Invest in Carisma’s CAR-Mono platform technology to further extend its leadership position in macrophage and monocyte based cellular therapy. As part of its ongoing platform enhancement effort, Carisma has developed its CAR-Mono approach, which significantly reduces manufacturing time and leverages an automated, closed-system manufacturing process. Carisma is currently in the pre-clinical process development stage for CT-0525, Carisma’s first anti-HER2 CAR-Mono product candidate, and expects to submit an IND in the second half of 2023. |
| · | Advance Carisma’s pre-clinical CAR-M oncology pipeline candidates to clinical development stage. Beyond its initial HER2 target, Carisma is expanding its pipeline into multiple tumor targets and constructs. CT-1119 is a mesothelin targeted CAR-M that Carisma plans to evaluate in patients with advanced mesothelin-positive solid tumors, with an IND expected to be submitted in 2025. In November 2022, Carisma presented preliminary and limited data demonstrating that CT-1119 can mediate phagocytosis, tumor cell killing, and pro-inflammatory cytokine release and control tumor growth in pre-clinical lung cancer models. Carisma anticipates nominating a next generation CAR construct for CT-1119 in the first half of 2024. Additionally, CT-0729 is a PSMA targeted CAR-M intended for use against metastatic castrate resistant prostate cancer and is currently in the discovery stage. Carisma is also developing product candidates targeting other cancer antigens. |
| · | Build next-generation technologies to expand the scope and capabilities of Carisma’s platform. Beyond its CAR-M and CAR-Mono technologies, Carisma is pursuing multiple platform enhancements for its CAR constructs, editing technologies and therapeutic delivery vehicles. Further, Carisma is actively developing a gene edited iPSC-derived macrophage platform and leveraging delivery technologies for its mRNA-based in vivo CAR-M platform for oncology. |
| · | Harness the potential of Carisma’s platform to develop novel product candidates to address therapeutic areas beyond oncology. While Carisma has initially been an oncology focused company, Carisma believes the breadth of the myeloid engineering platform enables significant opportunities outside of oncology. Based on early data related to Carisma’s novel therapeutic approach, Carisma believes its platform has significant potential across multiple therapeutic areas, including fibrosis, neurodegeneration, autoimmunity, and chronic inflammation, which are currently in the discovery stage. |
| · | Selectively enter into strategic partnerships and collaborations to maximize the potential of Carisma’s platform. Given the breadth of opportunities enabled by Carisma’s platform, Carisma may opportunistically enter into strategic collaborations intended to advance and accelerate its development programs, expand into new therapeutic areas and enhance the capabilities of its platform. Carisma currently has a broad strategic collaboration with Moderna focused on the development of in vivo CAR-M therapeutics for up to 12 oncology targets, of which four have already been nominated. |
Background
Cellular Immunotherapy
Cellular immunotherapy is a type of immuno-oncology approach whereby human immune cells are utilized to recognize and destroy cancer cells in a targeted manner. To date, cellular immunotherapy has focused on the transfer of T-cells or natural killer, or NK, cells. For example, T-cells with intrinsic tumor reactivity, such as tumor infiltrating lymphocytes, have been utilized, as well as T-cells genetically engineered with tumor targeting T-cell receptors, or TCRs, or CARs, have been tested in a variety of hematologic malignancies and solid tumors. The only FDA approved genetically modified cellular immunotherapies for cancer are CAR T-cell therapies for B cell hematologic malignancies expressing CD19 or multiple myeloma expressing B-cell maturation antigen, or BCMA.
Despite the incredible promise shown by cell therapies for hematologic malignancies, the success has not been replicated in the solid tumor setting. There are numerous challenges impacting T and NK cell immunotherapy in patients with solid tumors, such as the inability of cells to appropriately access the tumor microenvironment, overcome immunosuppression in the tumor microenvironment and overcome target antigen heterogeneity. Importantly, there have been challenges in targeting solid tumors with CAR T-cells without inducing toxicities against normal tissues or inducing severe systemic cytokine release syndrome, or CRS. To date, no CAR therapies for the treatment of solid tumors have received marketing approval.
Macrophages and Monocytes and the Tumor Microenvironment
Macrophages play a vital role in the innate immune system, the body’s first line of defense against foreign pathogens. Macrophages are highly plastic innate cells that mediate a multitude of protective and homeostatic functions, including elimination of pathogens through phagocytosis, clearance of cellular debris, induction or regulation of inflammation, antigen presentation, and tissue remodeling. Macrophages can arise from circulating bone marrow-derived monocytes or embryonic precursors and are found in all tissues in the human body. Depending on the environmental cues, macrophages can actively adopt distinct activation states, or phenotypes, to either initiate or terminate immune responses. While macrophage activation states are complex, they can be categorized into two general subsets:
| · | Classically activated (M1): M1 macrophages are pro-inflammatory and are associated with anti-tumoral functions. They initiate or enhance immune responses by recruiting T-cells, upregulating antigen processing machinery and co-stimulatory ligands, and secreting pro-inflammatory factors cytokines and chemokines, and ultimately promote T-cell responses. |
| · | Alternatively activated (M2): M2 macrophages are immunosuppressive and are associated with pro-tumoral functions. They accelerate tumor invasion and metastasis and promote angiogenesis (or formation of new blood vessels) by secreting inhibitory cytokines and upregulating immunosuppressive cell surface molecules, and ultimately inhibit T-cell responses. |
Macrophages are typically the most abundant immune cell in the TME of most cancers, where they generally adopt an M2 phenotype and are therefore associated with poor prognostic outcomes and increased intratumoral immunosuppression. For example, numerous studies have shown that patients with more M2 macrophages in their tumors have reduced responses to immune checkpoint inhibitors such as pembrolizumab.
Given the generally negative role of M2 macrophages in the TME, there have been numerous therapeutic approaches focused on inhibiting tumor associated macrophage, or TAM, infiltration or survival. Other approaches have sought to convert TAMs from an M2 to an M1 phenotype. While numerous studies have shown that TAM infiltration is typically associated with poor prognostic outcome, macrophages have been shown to have potent anti-tumor capabilities if appropriately activated and targeted.
The Opportunity for Engineered Macrophages in Treating Cancer
Carisma believes macrophage and monocyte cell therapies hold promise in addressing the limitations of other cell types and transforming the cell therapy treatment paradigm for solid tumors. The inherent biology of macrophages and monocytes offers several potential advantages that directly apply to current barriers for cell therapy efficacy in the solid tumor context.
Macrophages and monocytes are actively recruited into solid tumors, while other immune cells such as T-cells are often actively excluded. Macrophages are professional phagocytic cells capable of directly killing tumor cells through this unique mechanism. In addition to direct killing, macrophages can secrete pro-inflammatory factors that convert the immunosuppressive TME into an environment that promotes immunity. Importantly, macrophages and monocytes are professional antigen presenting cells, meaning they can directly present tumor-derived antigens to T-cells leading to anti-tumor T-cell responses, a phenomenon known as epitope spreading. Epitope spreading enables activity against tumor cells which either lack or lose expression of the initial antigen targeted by the CAR - a key challenge for cell therapies - and ultimately enables macrophages and monocytes to overcome target antigen heterogeneity within the patient’s cancer.
Carisma believes an approach which harnesses the direct effector functions of macrophages or monocytes, optimizes their activation status toward an inflammatory M1 phenotype, and redirects phagocytosis with molecular specificity would represent a major advance in cancer immunotherapy.
Carisma’s Novel Platform
CAR-M have the potential to address the key challenges involved in treating solid tumors:
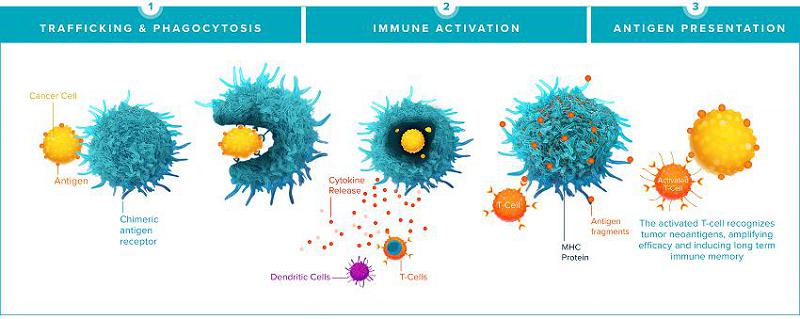
CAR-M have the ability to infiltrate solid tumors, phagocytose and destroy tumor cells directly, and present tumor-derived antigens leading to activation of the adaptive immune system. CAR-M mount anti-tumor immunity in numerous ways. First, CAR-M leverage the natural tumor-homing ability of macrophages and monocytes, the naturally most abundant immune cells in the TME, to traffic to both primary tumors and metastases, enabling engineered macrophages to act as a “Trojan horse,” tricking the tumor into recruiting engineered, anti-tumor CAR-M as if they were normal monocytes or macrophages. Once within the tumor, CAR-M directly kill antigen-expressing tumor cells through phagocytosis and secretion of cytotoxic factors. CAR-M secrete inflammatory cytokines and chemokines that promote a pro-inflammatory environment and lead to the recruitment of T-cells and other leukocytes. Finally, CAR-M serve as professional antigen-presenting cells for T-cells, inducing epitope spreading, systemic anti-tumor immunity, and immune memory against tumor antigens, expanding anti-tumor immunity to target negative tumor cells and potentially preventing antigen negative relapse.
Historically, macrophages have been challenging to genetically engineer due to their inherent resistance to most commonly used genetic manipulation methods. Furthermore, controlling the activation state of macrophages has been a long-standing challenge. Carisma believes that it has overcome these challenges with its proprietary platform that efficiently engineers macrophage-based cell therapies and enables control of their activation state.
Carisma’s proprietary platform enables the therapeutic use of engineered macrophages and monocytes for the treatment of cancer and other serious diseases and disorders. In its first application, solid tumors that overexpress HER2, the CAR-M platform is designed to identify and eradicate HER2 overexpressing tumor cells.
Currently, CAR-M are an individualized therapy that begin with the isolation of monocytes, the pre-cursor cell to macrophages, from blood drawn from a patient through a process called apheresis. The cells are purified, cultured, differentiated, and engineered with a CAR which bestows the macrophage with the ability to identify and eradicate cancer cells.
To enable its proprietary CAR-M therapy, Carisma had to overcome several key technical challenges, which are summarized by its platform capabilities:
| · | Gene Delivery: Carisma has identified Ad5f35, a chimeric adenoviral vector, as a highly efficient vector for introducing genes such as CARs into primary human macrophages and monocytes. Carisma has further developed additional proprietary technologies for ex vivo and in vivo macrophage engineering. |
| · | Activation State: Carisma demonstrated that Ad5f35 transduction leads to M1 polarization of human macrophages and monocytes and renders them resistant to conversion to M2 by immunosuppressive environments. |
| · | Tumor targeting: Carisma demonstrated that macrophage function can be harnessed against tumors in a targeted fashion via CARs. Carisma’s CARs enable antigen specific activation of macrophages and monocytes, antigen specific cancer cell phagocytosis and killing, and antigen specific release of pro-inflammatory cytokines and chemokines. |
| · | T-cell activation: By appropriately engineering and polarizing CAR-M, Carisma has demonstrated that they are able to recruit and activate T-cells - a key aspect to solid tumor immunotherapy. |
| · | Cell manufacturing processes: Carisma has developed manufacturing processes that enable the production of genetically engineered macrophages or monocytes for therapeutic use. |
In the case of Carisma’s lead product, CT-0508, a chimeric adenoviral vector, or Ad5f35, is used to deliver an anti-HER2 CAR which enables the macrophages to detect, phagocytose, kill, release inflammatory mediators, and initiate an immune reaction in response to HER2 overexpressing tumor cells. The resulting CAR-M, which are adenovirally transduced and locked into a pro-inflammatory M1 phenotype during the manufacturing process, are cryopreserved and shipped back to the patient for reinfusion. Reinfused CAR-M rapidly egress from peripheral blood and infiltrate tumor sites. Once in the tumor, CAR-M are activated by tumor-associated antigen engagement with the CAR, signaling via an intracellular signaling domain to phagocytose the tumor cell and release pro-inflammatory cytokines and chemokines that “warm up” the TME. They produce locally acting mediators that reprogram the TME, drawing in T-cells and NK cells, activating nearby antigen presenting cells, or APCs, such as dendritic cells, or DCs, and repolarizing immunosuppressive TAMs toward an M1 phenotype. In addition to direct phagocytosis of tumor cells, CAR-M present a patient’s unique array of tumor antigens to T-cells, leading to a broad adaptive immune response that has the potential to generate broad anti-tumor immunity.
Pre-clinical Data
Carisma evaluated its CAR-M platform in a variety of pre-clinical in vitro and in vivo model systems and published its foundational data in Nature Biotechnology in March 2020.
First, Carisma found that Ad5f35 led to the efficient transduction of human macrophages and could be utilized to produce human CAR-M. CAR-M mediated potent antigen-specific phagocytosis and tumor killing in a targeted fashion. CAR-M took on an activated M1 phenotype, expressed pro-inflammatory cytokines and chemokines, converted bystander M2 macrophages toward an M1 phenotype, recruited T-cells, and increased antigen presentation to activate T-cells. Enhanced anti-tumor T-cell responses mediated by CAR-M were noted in humanized murine models and the findings are summarized below.
Human CAR-M Anti-Tumoral Function In Vitro
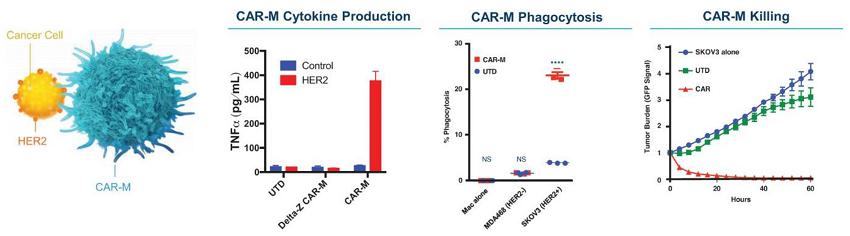
* SKOV3 = Human HER2+ ovarian cancer cell line
* UTD = Untransduced
* CAR = Anti-HER2 human CAR-M
* DeltaZ = CAR-M with a non-signaling control CAR
CAR-M were able to traffic to established tumors and co-localized with metastatic foci in the lung after intravenous administration without a pre-conditioning regimen. CAR-M treatment induced significant reduction in tumor burden and improved overall survival compared to mice treated with control macrophages in multiple mouse tumor models.
Transduction of macrophages with Ad5f35 led to the induction of a durable M1 phenotype. Despite the purported plasticity of macrophage phenotype, Ad5f35 transduced macrophages did not convert to M2 upon stimulation. CAR-M maintained a pro-inflammatory M1 state within the TME, while control macrophages were converted to M2. Additionally, CAR-M induced a pro-inflammatory signature in the surrounding TME. Given that solid tumors are rich in TAMs, Carisma evaluated the bidirectional interaction of CAR-M and M2 macrophages. While M2 macrophages failed to convert CAR-M from M1 to M2, CAR-M converted M2 macrophages to M1. Additionally, the presence of M2 macrophages did not impact the tumor killing capacity of CAR-M, highlighting their resistance to the immunosuppressive components of the TME.
Finally, CAR-M were shown to interact with cells of the adaptive immune system. CAR-M upregulated antigen presentation pathways and demonstrated heightened T-cell stimulation capacity as compared to control macrophages. Notably, CAR-M were able to present antigens to T-cells following phagocytosis. In addition, CAR-M were able to directly recruit various subtypes of T-cells.
To further its understanding of CAR-M, Carisma sought to model their function in fully immunocompetent mouse models which have an intact TME and immune system, enabling recapitulation of the complex immunological environment in human cancer patients. Toward that goal, Carisma developed a murine surrogate CAR-M to demonstrate the mechanism of action of CAR-M in vivo in mice which have a fully intact immune system. First, Carisma validated comparability between human and murine CAR-M. Carisma demonstrated that the same vector utilized in its clinical pipeline, Ad5f35, could be used to engineer primary murine macrophages, and confirmed that T-cells were viable, expressed CAR, and were similarly polarized to an M1 phenotype. Functional studies showed CAR-M mediated tumor killing of target cancer cells and enhanced the in vitro function of T-cells. Furthermore, murine CAR-M released pro-inflammatory cytokines similarly to human CAR-M.
Pre-clinical immunocompetent solid tumor models were established via subcutaneous, or SC, injection and engraftment of the murine colorectal cancer cell line, or CT26, engineered to express human HER2. In this model, intratumoral, or IT, CAR-M monotherapy significantly reduced tumor growth and prolonged overall survival compared to untransduced macrophages (macrophages not expressing a CAR). By rechallenging complete responders several months post tumor clearance with the same tumor cells lacking HER2 expression, Carisma was able to demonstrate that CAR-M therapy leads to epitope spreading and immune memory which confers protection against antigen-negative relapse.
CAR-M Control Tumor Progression, Improve Survival and Induce Long-Term Protection against Antigen-negative Relapse
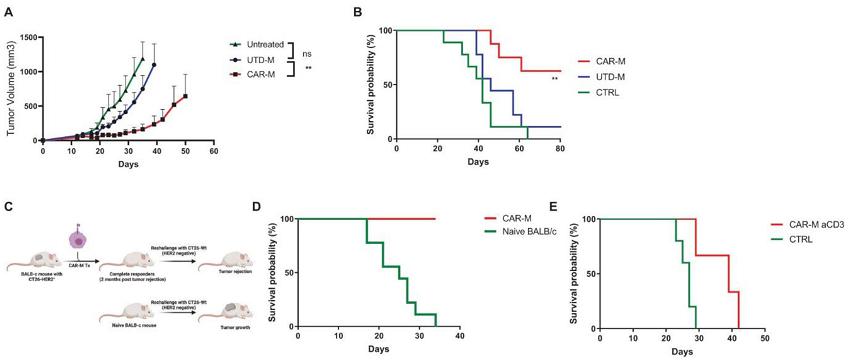
* UTD-M = Untransduced macrophage
CT26-HER2+ tumors were implanted subcutaneously in immunocompetent syngeneic mice. After 15 days, mice were treated with intratumoral CAR-M, UTD-M, or left untreated. CAR-M significantly reduced tumor progression.
CAR-M significantly increased long term survival compared to control groups.
Mice achieving complete responses, or CR, post CAR-M therapy were re-challenged with HER2-negative CT26-Wt tumors to model antigen negative relapse.
Naïve mice succumbed to disease within 35 days, while 100% of the mice from the CAR-M treatment group survived, indicating long-term tumor protection against antigen negative relapse.
T-cell depletion reversed CAR-M induced protection against antigen negative relapse, indicating that CAR-M treatment led to epitope spreading and anti-tumor T-cell memory.
Analysis of the tumor microenvironment of mice receiving CAR-M therapy demonstrated the ability of Carisma’s therapy to recruit additional immune cells, including T-cells, into the tumor. CAR-M led to immune activation in the TME associated with T-cell expansion, activation, and modulation of the overall T-cell repertoire of tumor infiltrating lymphocytes - suggesting the induction of a broad anti-tumor immune response.
CAR-M Reprogram the TME and Prime T-cells
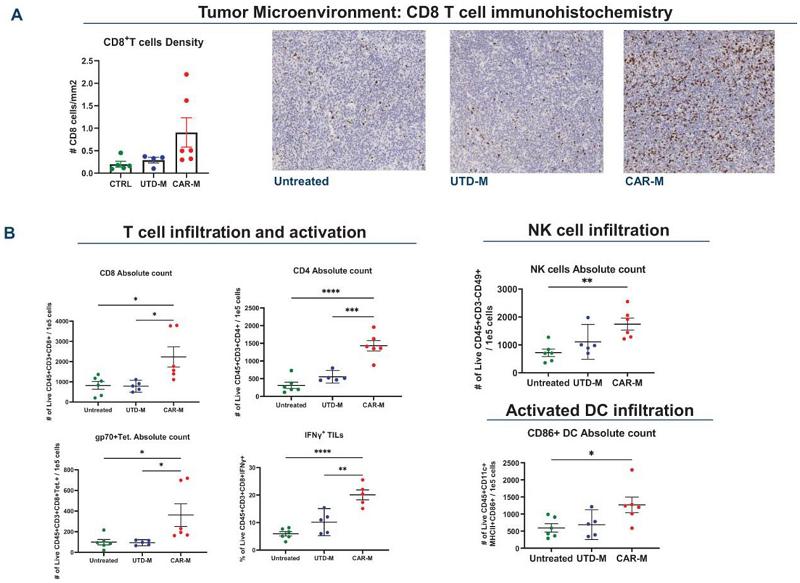
Immunohistochemistry assessment showed that CAR-M treatment increased tumor CD8+ T-cell infiltration in the CT26-HER2+ model indicating activation of the TME.
Flow cytometric analysis showed increased tumor infiltration of T-cells, NK cells, activated CD86+ DCs and tumor associated antigen specific CD8 T-cells (gp70 Tet+) in CAR-M treated mice, suggesting epitope spreading.
Combined, these results demonstrate that CAR-M have the potential to overcome some of the key challenges cell therapies encounter in the solid tumor setting and represent a novel immunotherapeutic platform that can be broadly applied to diverse tumor antigen targets.
Carisma is applying its CAR-M platform to a broad pipeline of product candidates, and Carisma intends to build a fully integrated immunotherapy company spanning autologous cell therapy, allogeneic cell therapy, and in vivo macrophage gene therapy. Carisma currently owns all rights to its product candidates and programs outside of its Moderna collaboration, which is limited to direct in vivo reprogrammed CAR-M in the field of oncology.
Macrophage and Monocyte Engineering Platform
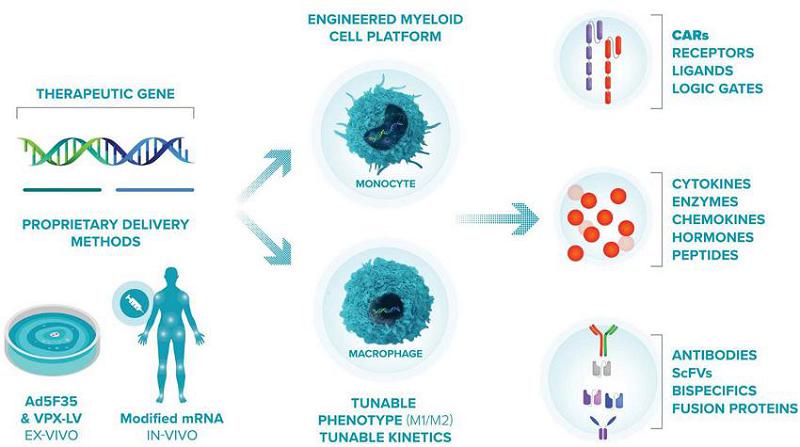
Gene Delivery
At the core of Carisma’s platform are its proprietary viral and non-viral approaches for delivering different payloads into macrophages and monocytes and engineering them into a variety of phenotypes depending on the disease which they are intended to treat.
For its initial solid tumor programs, Carisma’s adenoviral vector enables Carisma to generate an abundant supply of CAR-M cells and engineer the cells to be locked into an M1 phenotype. Carisma selected Ad5f35 after testing commonly utilized viral and non-viral approaches and demonstrating Ad5f35’s high efficiency in transducing primary human monocytes and macrophages. Ad5f35 can transduce macrophages with high efficiency, viability and reproducibility amongst donors. In addition to being highly efficient, transduction with Ad5f35 polarizes and locks macrophages into an M1 phenotype.
In addition to Ad5f35, Carisma’s platform includes two other proprietary methods for delivering genes into macrophages. The first is a modified lentiviral vector, or Vpx-LV, which carries viral protein X. Vpx-LV depletes SAMHD1 and permits lentiviral transduction of primary human monocytes, macrophages, and dendritic cells. Vpx-LV was developed by Dr. Nathaniel Landau at New York University, and Carisma holds a global exclusive license to develop this vector. Unlike Ad5f35, which induces a potent M1 phenotype upon transduction, Vpx-LV has minimal impact on macrophage phenotype and can be utilized as a flexible tool to generate M0 (non-activated), M1 (pro-inflammatory), or M2 (anti-inflammatory) polarized myeloid cell therapies with durable gene expression. Additionally, Carisma has developed a proprietary non-viral mRNA-based approach to transiently engineered macrophage and a companion method to induce a durable pro-inflammatory M1 phenotype. Carisma has successfully generated M1-primed non-viral CAR-M using a research manufacturing process consisting of mRNA transfection to deliver the CAR transgene followed by IFNβ priming to polarize the cells to an M1 anti-tumoral phenotype. Non-viral CAR-M demonstrated high viability, high CAR expression, M1 polarization and anti-tumoral function ex vivo similar to Ad5f35 engineered CAR-M. Additionally, in partnership with Moderna, Carisma is developing a myeloid tropic LNP/mRNA platform to program CAR-M directly in vivo.
Approach to Pipeline
Carisma’s proprietary technology and engineering capabilities enabled it to pioneer the CAR-M field and conduct the first in human CAR-M clinical trial, establishing its leading position in the engineered macrophage space. Carisma’s goal is to advance ex vivo autologous cell therapies and off-the-shelf therapies including allogeneic cell therapies and direct in vivo reprogramming approaches in oncology and other indications:
Expansion: Building Upon the Learnings of Autologous Cell Therapy
While the first iteration of its platform is the CAR macrophage, Carisma has expanded its capabilities to include multiple myeloid cell types (monocytes, macrophages, and dendritic cells), multiple gene delivery modalities (Ad5f35, Vpx-LV, and mRNA), various phenotypes (M1, M2, and subtypes thereof), and a broad variety of payloads including CARs, immune ligands, secreted or tethered cytokines, transcription factors, and other genes that enhance efficacy. Importantly, Carisma has expanded its platform to enable in vivo engineering of myeloid cells directly within the patient’s body. Additionally, Carisma has established a robust process to edit the genome of human myeloid cells by utilizing tools such as CRISPR/Cas9, enabling gene edited macrophages with inhibitory pathways such as SIRPα genetically removed from the cell product. Carisma’s engineered macrophage platform enables fine tuning the activation state of the engineered macrophage or monocyte. Finally, Carisma has established a novel Engineered Myeloid Microenvironment Converter, or EM-C, platform that utilizes proprietary synthetic cytokine switch receptors to generate engineered macrophages that respond to M2 cytokines with M1 responses (for oncology applications) or to generate engineered macrophages that respond to M1 cytokines with M2 responses, for auto-immune or chronic inflammatory diseases.
Carisma’s Pipeline of Product Candidates and Discovery Programs
Using its proprietary CAR-M platform, Carisma is developing a broad pipeline of product candidates, with an initial focus in oncology.
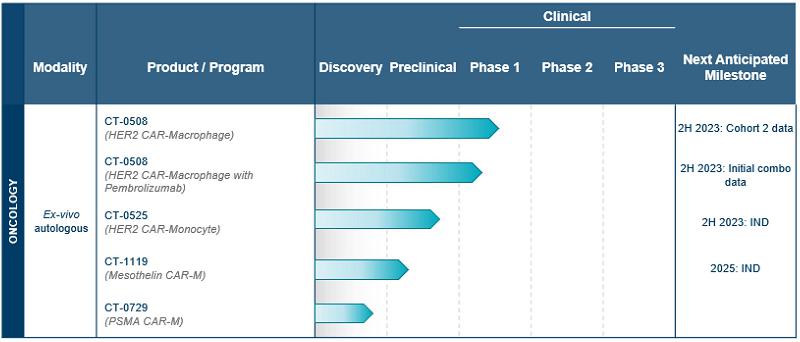
Lead Product Candidate: CT-0508
CT-0508 is a cell product comprised of autologous, peripheral blood monocyte-derived, pro inflammatory macrophages, transduced with a chimeric adenoviral vector, Ad5f35, containing an anti-HER2 CAR. The anti-HER2 CAR is a first-generation CAR composed of a fully human single-chain variable fragment, or scFv, derived from the monoclonal antibody trastuzumab, which is specific for human HER2. The anti-HER2 scFv is fused to a CAR backbone containing a cluster of differentiation CD8 hinge, CD8 transmembrane domain, and a CD3ζ intracellular domain. The CAR is cloned into an adenoviral vector backbone and transduced into monocyte-derived macrophages. Based on the pre-clinical data generated to date, CT-0508 CAR-M are able to specifically recognize HER2 overexpressing tumor cells, which triggers both direct killing of tumor cells and phagocytosis. Additionally, CAR engagement by HER2 on tumor cells results in the secretion of a broad array of pro-inflammatory cytokines and chemokines, which contribute to the recruitment and activation of additional immune cells to the TME, including effector T-cells and other antigen presenting cells. CT-0508 CAR-M are antigen presenting cells, and after phagocytosing tumor cells they process tumor-derived antigens and present them to T-cells, leading to T-cell immunity against tumor antigens. This additional activation of the adaptive immune system amplifies anti-tumor immune response and can lead to long term immune memory not only against HER2, the primary target, but other tumor specific neoantigens as well.
The Phase 1 clinical trial of CT-0508 is currently ongoing. As of March 7, 2022, seven clinical sites were open for screening and enrollment: (i) the University of Pennsylvania Abramson Cancer Center, (ii) the University of North Carolina Lineberger Comprehensive Cancer Center, (iii) the City of Hope National Medical Center, (iv) the MD Anderson Cancer Center, (v) the Sarah Cannon Cancer Research Institute, (vi) Oregon Health & Science University and (vii) Fred Hutchinson Cancer Center.
In November 2022, Carisma presented preliminary clinical results from the first group of patients. CT-0508 was successfully manufactured using macrophages obtained from heavily pre-treated, advanced solid tumor patients and has shown high CAR expression, viability, and purity. In addition, CT-0508 has been generally well-tolerated after infusion with no dose-limiting toxicities reported to date from the nine patients enrolled in the first group. While the results from this early clinical trial data are both preliminary and limited, Carisma believes the results indicate that CT-0508 can be detected within the TME, lead to remodeling and activation of the TME, and potentially induce anti-tumor adaptive immunity.
The FDA has granted “Fast Track” status to CT-0508 for the treatment of patients with HER2 overexpressing solid tumors. The components of CT-0508 cells are shown below:
Key Components of First-Generation CAR Construct
Figure Legend: CT-0508 is an autologous monocyte derived macrophage cell product engineered with the adenoviral vector Ad5f35 to express an anti-HER2 CAR. The CAR is comprised of a single chain variable fragment derived from a humanized anti-HER2 antibody which provides specificity against the target antigen. The scFv is linked to a hinge domain derived from the human CD8 protein, which enables extension and flexibility from the cell membrane surface. The hinge is linked to a CD8 transmembrane, or TM, domain which spans the cell membrane, linking the extracellular portion of the CAR to the intracellular portion of the CAR, which is comprised of CD3ζ. CD3ζ signaling is activated when the CAR binds to the target antigen, leading to macrophage activation, phagocytosis, tumor cell killing, and release of pro-inflammatory factors such as cytokines and chemokines.
CT-0508 Therapy for HER2+ Solid Tumors
While therapies targeting solid tumors that overexpress HER2 have led to improved survival in breast and gastric or gastro-esophageal junction cancers, there remains an unmet need in patients with advanced HER2 positive, or HER2+, cancers and HER2 expressing cancers, including metastatic lung, ovarian, colon, bladder, and other cancers for which there are no HER2 targeted agents.
Approximately 20% of breast cancers overexpress HER2, a protein that is overexpressed on the surface of a variety of solid tumors. In addition to breast, gastric, and gastroesophageal junction cancers, HER2 is also overexpressed in a number of solid tumor indications including but not limited to bladder cancer, ovarian cancer, lung cancer and colon cancer.
| | HER2 Positivity Frequencies Across Tumor Types | |
| | Tumor type | | HER2
positivity (%) | |
| | Bladder cancer | | 8 - 70 | |
| | Breast cancer | | 11.0 - 25.0 | |
| | Cervical cancer | | 2.8 - 3.9 | |
| | Colorectal cancer | | 1.6 - 5.0 | |
| | Esophageal cancer | | 12.0 - 14.0 | |
| | Extrahepatic Cholangiocarcinoma | | 6.3 - 9.0 | |
| | Gallbladder cancer | | 9.8 - 12.8 | |
| | Gastric adenocarcinoma | | 7.0 - 34.0 | |
| | Ovarian cancer | | 26 | |
| | Salivary duct carcinoma | | 30 - 40 | |
| | Salivary mucoepidermoid carcinomas | | 17.6 | |
| | Testicular cancer | | 2.4 | |
| | Uterine cancer | | 3.0 | |
CT-0508 Clinical Study Design - Study 101
The ongoing Phase 1 clinical trial of CT-0508 is a single-arm, open-label study of systemic intravenous administration of CT-0508. This study is intended to evaluate safety, tolerability, cell trafficking, cell-manufacturing feasibility, and preliminary evidence of efficacy in approximately 18 subjects with locally advanced or metastatic solid tumors overexpressing HER2 who have failed available therapies.
A summary of the clinical trial design, dosing regimen, sample collection regimen, and primary and secondary objectives are shown below:
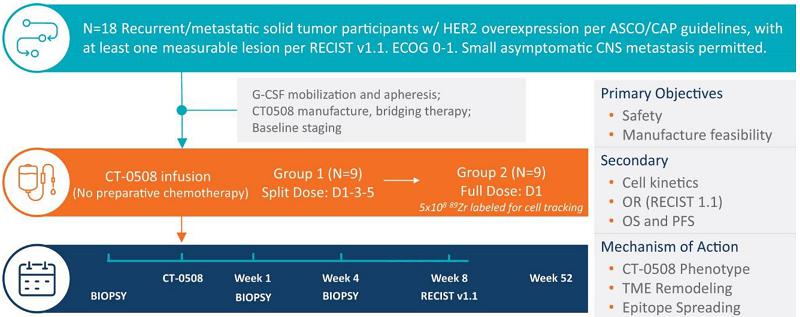
Filgrastim, or recombinant G-CSF, is administered to patients for four days prior to apheresis to mobilize monocytes into the peripheral blood, increasing the available circulating monocyte count prior to collection by apheresis. The CT-0508 cell product is then prepared, cryopreserved, and released following quality control testing. The first three participants in the study were hospitalized for eight days after the first infusion of CT-0508 (Day 1 to Day 8) as part of the pre-determined study design. There is no preparative chemotherapy prior to the cell product infusion. The first group of nine patients have been treated with a divided dose regimen consisting of:
| · | Day 1: Up to 0.5 × 109 cells; |
| · | Day 3: Up to 1.5 × 109 cells; and |
| · | Day 5: Up to 3.0 × 109 cells. |
Adverse event reporting begins at the start of mobilization and continues until any toxicities resolve or are deemed irreversible. Participants are continually reassessed for evidence of acute and/or cumulative toxicity. Approximately nine participants in the second group of patients will receive up to 5.0 × 109 total manufactured CT-0508 cells in a single infusion on Day 1.
HER2 has several advantages as a target antigen for CAR-M. In addition to being expressed in a variety of solid tumor types with significant unmet medical needs, HER2 is not shed or internalized and is only expressed at low levels in non-tumor tissues. As HER2 expression is typically maintained over the course of disease, CT-0508 may be developed for treatment of metastatic disease, for example, in the liver and lung, as well as primary tumors. Additionally, HER2 is typically not lost after patients with metastatic cancer progress on available HER2 targeted therapies, rendering HER2 refractory patients potentially eligible for CT-0508 therapy.
Participants enrolled in Study 101 undergo one pre-treatment and two on-treatment biopsies to assess CT-0508 trafficking, impact on the TME, induction of anti-tumor T-cell immunity, and other biomarkers. Blood samples are also collected over a period of 52 weeks for evaluation of pharmacokinetics and biomarkers associated with safety and efficacy.
Based upon clinical data, Carisma may seek Regenerative Medicine Advanced Therapy, or RMAT, and PRIority MEdicine, or PRIME, designations for CT-0508, which provide an expedited developmental and approval pathway, in the United States and the European Union, respectively.
CT-0508 Clinical Data - Study 101
Enrollment of the first group of nine patients in the Phase 1 clinical trial of CT-0508 has been completed and enrollment in the second group is currently ongoing. Carisma successfully generated CT-0508 product for all participants enrolled in the first group of the study with an average cell viability of 89%, an average purity of 85%, and an average CAR transduction of 81%.
Carisma has reported on the safety, clinical response, and correlative studies for the first nine patients dosed in the first group of the clinical trial. Of such patients, two had HER2 overexpression levels of 2+ by immunohistochemistry staining, or IHC, with additional fluorescent in-situ hybridization FISH confirmation; and seven had levels of HER2 3+ by IHC. HER2 grading was performed based on the ASCO/CAP guidelines. Patients in the trial had three median prior therapies with a range of two to 11. Patients had a median of two prior HER2 targeted therapies, with a range of zero to nine.
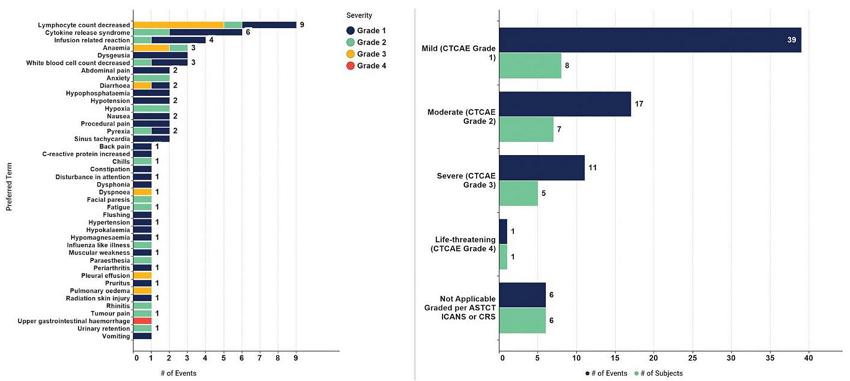
CT-0508 was well tolerated with no dose-limiting toxicities. The majority (55.5%) of subjects with treatment-emergent adverse events, or TEAEs, by maximum severity were Grades 1 and 2. One subject experienced an unrelated Grade 4 TEAE (related to progression of disease of the patient’s cancer). For subjects with TEAEs considered related to CT-0508, the majority (66.6%) were Grades 1 and 2, and none were Grades 4 or 5 severity. With respect to TEAEs of special interest, six patients experienced CRS, and all of these were Grades 1 and 2. There were no episodes of immune cell therapy associated neurotoxic syndrome, or ICANS, reported. No patients had severe CRS. No AEs, or SAEs, led to CT-0508 dose modification or discontinuation. No major organ toxicity was observed. The majority of AEs, regardless of relatedness, were Grades 1 and 2.
| | Overview of Subjects with Treatment-Emergent Adverse Events (Safety Population) | |
| | Category | | n = 9 (%) | |
| | Treatment-Emergent Adverse Event (TEAE) | | 9 (100.0) | |
| | TEAE, Related to CT-0508 | | 8 (88.9) | |
| | TEAE, Serious AE | | 4 (44.4) | |
| | TEAE, Serious AE Related to CT-0508 | | 2 (22.2) | |
| | TEAE, AEs of Special Interest | | 7 (77.8) | |
| | ◦ Grade 1 | | 3 (33.3) | |
| | ◦ Grade 2 | | 4 (44.4) | |
| | ◦ Grade 3 | | 0 (0.0) | |
| | ◦ Grade 4 | | 0 (0.0) | |
| | ◦ Grade 5 | | 0 (0.0) | |
| | Discontinued CT-0508 due to TEAE | | 0 (0.0) | |
Summary of AEs by Preferred Term and by Severity
The best overall response was stable disease per RECIST 1.1 criteria. A best overall response of stable disease was observed in four out of nine patients.
Transient and low-grade fever was observed in seven out of nine patients post CT-0508 infusion. All fevers resolved within 48 hours. In concordance with clinical observations, a transient increase in serum IL-6, a pro-inflammatory cytokine, was observed.
Carisma evaluated the pharmacokinetics, or PK, of CT-0508 in the peripheral blood and the tumor. Similar peripheral blood PK was observed for all nine participants with CT-0508 detectable only on infusion days for four to eight hours post-infusion, consistent with rapid migration of CAR-M from the blood to tissues following infusion. CT-0508 was detected within the TME of eight out of nine evaluable patients assessed to date using RNAscope™ technology as shown below. These data suggest that CT-0508 rapidly egresses from the peripheral blood and successfully traffic to the biopsied tumor mass.
CT-0508 Rapidly Migrates Out of the Blood and is Detected within the TME of 8/9 Participants Evaluated

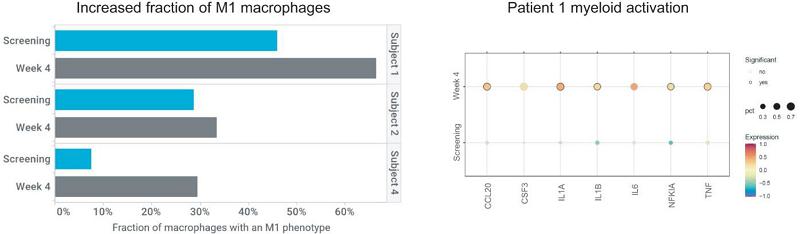
To evaluate the mechanism of action of CT-0508, single cell RNA sequencing, or scRNAseq, analysis was performed on fresh tumor biopsy to investigate changes within the TME following CT-0508 infusion. Analysis of screening (n=5), Day 8 post-infusion (n=5) and Week 4 post-infusion (n=3) biopsies revealed increases in CD8 T-cells, macrophages, and neutrophils on treatment consistent with inflammation and activation of an immune response.
These increases were also associated with reprogramming of the infiltrating macrophages toward an M1 phenotype by Week 4.
Single Cell RNAseq Analysis Demonstrates Remodeling of the Tumor Immune Landscape Following CT-0508 Infusion
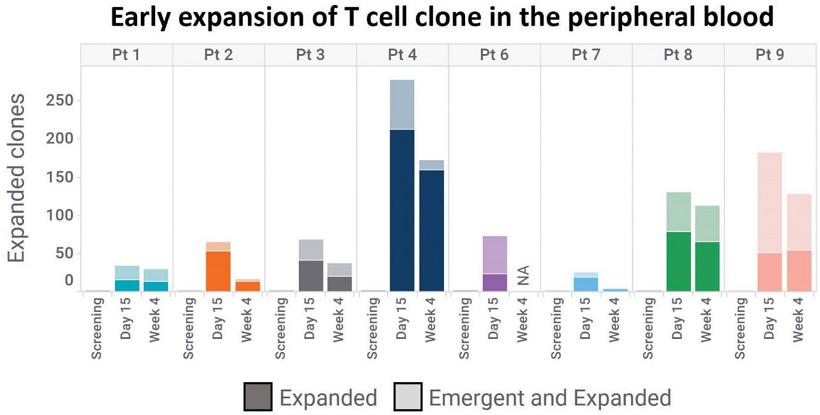
To evaluate whether CT-0508 was able to initiate anti-tumor adaptive immunity, TCR repertoire analysis was performed utilizing the Adaptive Biotechnologies™ TCR sequencing platform. The analysis was performed on peripheral blood and tumor tissue. Peripheral blood TCR repertoire analysis revealed an expansion of T-cell clones in the blood of participants post CT-0508 infusion, indicative of the initiation of an adaptive immune response.
Early Expansion of T-cells in the Periphery Following CT-0508 Infusion
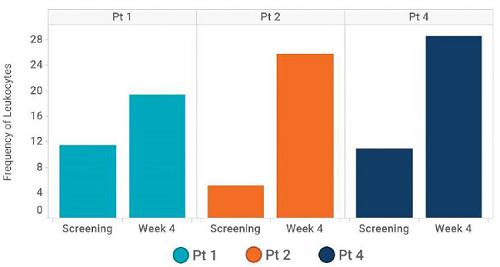
Based on scRNAseq analysis, the frequency of effector T-cells increased in all three participants with available screening and Week 4 biopsies (shown below). Furthermore, Carisma evaluated T-cell subtypes and found that Participant 1 demonstrated an increase in proliferating and effector memory CD8 T-cells, Participant 2 demonstrated an increase of all subsets except for activated CD8 T-cells, and Participant 4 demonstrated an increase in activated CD4, activated CD8 and effector memory CD8 T-cells.
Frequency of effector T-cells in TME
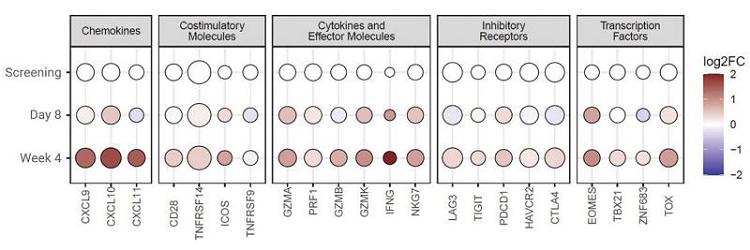
Differential gene expression analysis performed on biopsies collected at Week 4 post-CT-0508 infusion demonstrated an increased expression of genes associated with cytotoxic CD8 T-cell activation when compared to pre-treatment expression TME samples from six patients analyzed by RNA sequencing.
Increased T-Cell Activation in the TME
TCR repertoire analysis of the TME revealed that newly expanded peripheral clones accumulated over time within the TME, suggesting that these clones are tumor reactive. Expanding T-cell clonotypes from patient 1, 2 and 4 at Week 4 post-infusion clustered with cells expressing high levels of CD8α, perforin, granzyme B, Ki67, IFNγ, and PD1 demonstrated their activated and/or proliferating CD8 cytotoxic T lymphocyte phenotype.
Peripherally Expanded T-cell Clones Accumulate in the TME and Adopt a Cytotoxic Phenotype

While preliminary, the clinical data to date confirmed that CT-0508 is successfully manufactured from heavily pre-treated solid tumor patients, has been well tolerated, traffics to the tumor, activates the TME, and may initiate anti-tumor adaptive immunity.
Additional CT-0508 Studies
CT-0508 and Pembrolizumab combination sub study
This open-label sub study will assess the safety and feasibility of co-administering CT-0508 in combination with the PD-1 inhibitor, pembrolizumab. The target population for this sub study are subjects at least 18 years of age who meet inclusion criteria per the main protocol and have HER2 over-expressing solid tumors and meet the sub study specific eligibility criteria. Carisma expects to report clinical data for this sub study in the second half of 2023.
CT-0508 Intraperitoneal administration sub study
This sub study has been designed to assess the safety and feasibility of CT-0508 via regional administration into the peritoneal cavity. The target population for this sub study are subjects at least 18 years of age who meet inclusion criteria per the main protocol, that have HER2 over-expressing gynecological cancers including but not limited to ovarian, fallopian tube, primary peritoneal, and endometrial cancers, who have disease spread mainly within the peritoneal cavity that meet the sub study specific eligibility criteria. Subjects will be enrolled at select clinical sites participating in Study 101 that have the capability to enroll and adequately treat subjects with intraperitoneal administration of CT-0508. Carisma expects to report clinical data for this sub study in the second half of 2023.
CT-0508 Biodistribution sub study
This open-label sub study is designed to evaluate the whole body biodistribution of CT-0508 after intravenous administration using radiolabeled CT-0508 and longitudinal PET/CT imaging. This sub study includes 89Zr-oxine radiolabeling a fraction of the CT-0508 cell product, followed by administration on Day 1 and PET/CT imaging approximately on Day 1, 4, 8, 15, and 28 to assess trafficking and biodistribution of CT-0508. The target population for this sub study are subjects at least 18 years of age that meet inclusion criteria per the main protocol. Subjects will be enrolled at specific sites in Study 101 that have the capability to perform 89Zr-oxine labeling, administration, and routine PET/CT analysis.
Synergistic Potential of CAR-M Therapy with T-Cell Checkpoint Inhibitors
Blocking the immune checkpoint molecule programmed cell death 1, or PD-1, has revolutionized cancer treatment for patients with a multitude of solid tumor indications. Pembrolizumab is a potent humanized immunoglobulin G4, or IgG4, monoclonal antibody, or mAb, with high specificity of binding to the PD-1 receptor, inhibiting its interaction with programmed cell death ligand 1, or PD-L1, and programmed cell death ligand 2, or PD-L2. While pembrolizumab is currently indicated for the treatment of patients across several solid tumor indications, the majority of patients have either primary or secondary resistance to immune checkpoint blockade and may benefit from combinatorial therapy that could overcome immune cell exclusion, poor antigen presentation, low T-cell infiltration, high TAM infiltration, a lack of productive co-stimulation, low mutational burden, IT immunosuppression, and a low frequency of tumor reactive T-cell clones.
Based on the data generated during pre-clinical development, CT-0508 cell product is able to specifically recognize, cancer cells through the binding of the CAR to HER2 expressed on the surface of these cells. This interaction triggers activation of the CAR-macrophages and results in direct anti-tumor effect by killing and phagocytosis of the tumor cells. In addition, CT-0508 recruits T-cells, activates the TME, and as professional antigen presenting cells, can process and present tumor associated antigen and/or neoantigens expressed by the tumor cells, leading to T-cell immunity against these specific antigens. However, this indirect anti-tumor effect involves the engagement of T-cells that may be actively suppressed, or exhausted, within the tumor micro-environment by a variety of factors including secreted immune-modulatory factors and inhibitory ligands expressed on both immune and tumor cells. Additionally, several studies have demonstrated that patients with low mutational burden, low MHC expression, defective antigen presentation, low CD8+ T-cell infiltration, or minimal Th1 cytokine signatures tend to be unresponsive to PD-1 blockade. Therefore, based on the mechanism of action of CT-0508 and the limitations of PD-1 blockade, the combination of CAR-M therapy with PD-1 blockade therapy may be beneficial by enhancing antigen presentation (innate immunity) to initiate a robust anti-tumor T-cell response (adaptive immunity).
CAR-M and PD-1 blockade combination therapy: Pre-clinical Development
To model the combination of CT-0508 cell therapy with anti-PD-1 inhibitors, Carisma used a murine colorectal cancer cell line engineered to overexpress human HER2. Tumors were established in the flank of the immune competent mice and 14 days post tumor inoculation, mice were randomized and received either murine CAR-M alone (IV), murine PD-1 blockade alone (IP) or a combination of both treatments. Using a regimen where CAR-M was injected first when the tumor was well established, followed by the anti-PD-1 inhibitor a few days later, neither murine CAR-M (Anti-HER2 muCAR-Mac) nor murine anti-PD1 monotherapy had a significant effect on tumor growth and overall survival. However, when co-administered the combination of both therapies resulted in significant tumor growth delay associated with prolonged survival of the mice (all mice in the combination group survived until the end of the study). All mice treated with the combination of Anti-HER2 muCAR-Mac and anti-PD1 mAb completely cleared their tumors (below).
Assessment of Tumor Burden and overall survival in a Syngeneic Mouse Model of Colon Carcinoma in response to treatment with IV CAR-M and Anti-PD-1 Therapy
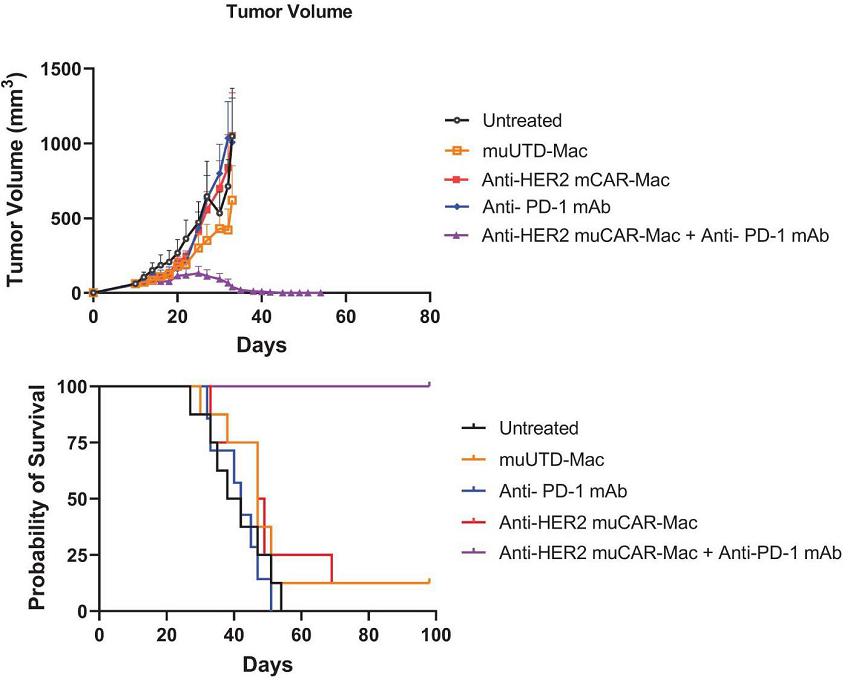
*Anti-HER2 muCAR-Mac are murine CAR-M and muUTD-Mac are untransduced murine CAR-M.
To determine the impact of Anti-HER2 muCAR-Mac and anti-PD-1 mAb combination therapy on tumor burden, tumor volumes were recorded during the treatment period (depicted in the left panel above) and mice were monitored for survival (depicted in the right panel above).
In addition to the IV CAR-M alone and in combination with anti-PD-1 therapy study described above, Carisma has performed studies with IT administered murine CAR-M. In this study, anti-PD-1 therapy was administered four times, at 3-day intervals starting 14 days post tumor inoculation (CAR-M therapy initiated on Day 15). IT murine CAR-M and anti-PD-1 therapy improved tumor control and significantly improved survival probability. Cumulative survival for all groups: 0% CR CTRL, 8.3% CR anti-PD-1, 38.9% CR CAR-M and 66.7% CR CAR-M and anti-PD-1:
IT CAR-M and Anti-PD-1 Combination Therapy Improves Survival in the CT26-HER2 Model

Analysis of immune cell populations in the TME showed that macrophages were more abundant in the CAR-M samples while other myeloid cells and DCs showed the greatest infiltration in the combination group. A significant increase in total tumor infiltrating T-cells, and in particular helper T-cells, was noted in the combination therapy.
Profound TME Modulation in Response to IT CAR-M and CAR-M +Anti-PD-1 Combination Therapy
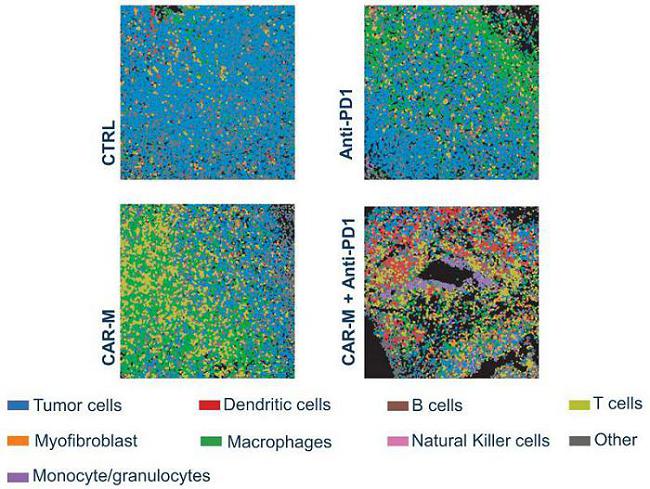
Analysis of the TCR repertoire demonstrated that IT administration of CAR-M in combination with a PD-1 blocking monoclonal antibody led to increased frequency of T-cells in the periphery and significantly modulated the TCR repertoire in the TME suggesting enhanced adaptive anti-tumor immunity.
Based on these data, CAR-M and pembrolizumab represent a potentially synergistic immunotherapeutic combination regimen that combines CAR-M to infiltrate the TME, degrade the tumor via phagocytosis, and recruit and prime T-cells and pembrolizumab to prevent or reverse T-cell exhaustion. Patients with HER2 overexpressing tumors, such as metastatic breast cancer, gastric cancer, ovarian cancer, esophageal cancer, and others are generally poor responders to pembrolizumab. Carisma has initiated a Phase 1 clinical study to evaluate CT-0508 in combination with pembrolizumab.
Additional Pipeline Candidates
Carisma’s additional pipeline candidates are CAR-M therapies that incorporate all of the core elements of its macrophage cell engineering platform, along with certain new platform enhancements that Carisma is currently developing. The CT-1119 product candidate targets the mesothelin tumor associated antigen that is found on lung cancer, mesothelioma, pancreatic cancer, ovarian cancer, and numerous other solid tumors. The CT-0729 product candidate targets the PSMA tumor associated antigen that is found on prostate cancer.
CT-1119 (Anti-Mesothelin CAR-M)
Mesothelin is a well validated tumor associated antigen. Mesothelin has been shown to be aberrantly expressed on the surface of tumor cells and plays an important role in promoting cancer invasion and proliferation. Mesothelin has been demonstrated to be expressed at high levels in mesothelioma, lung cancer, ovarian cancer, pancreatic cancer, and other solid tumors with limited expression in normal tissue, though recent data suggests inflammation may induce expression. There are no approved anti-mesothelin agents and no approved cell therapies targeting any of the solid tumor types that overexpress mesothelin. Mesothelin positive solid tumors represent a significant unmet medical need.
While there are no mesothelin targeted approved agents, numerous clinical trials have been conducted targeting mesothelin and safety has been established with a variety of modalities including monoclonal antibodies, antibody drug conjugates, and CAR-T-cells. Targeting mesothelin enables a similar strategy to Carisma’s CT-0508 HER2 Phase 1 trial in that it enables (i) a basket trial design that includes patients with diverse tumor types and (ii) separate arms for systemic and regional administration. There is a significant opportunity for regional administration of CT-1119, including intraperitoneal administration for mesothelin positive ovarian cancer with peritoneal metastasis and intrapleural administration for patients with malignant mesothelioma and lung tumors. There is also a significant opportunity for patients with mesothelin positive solid tumors with systemic metastasis.
To develop a mesothelin targeted CAR-M, Carisma has screened anti-mesothelin scFv’s using mRNA to identify humanized anti-mesothelin binders. Carisma obtained exclusive rights to a humanized anti-mesothelin scFv from the University of Pennsylvania. Carisma demonstrated that human CAR-M engineered with an Ad5f35 vector show high viability and efficiently express an anti-mesothelin (meso) CAR. Similar to CT-0508, CT-1119 adopts an M1 macrophage activation state.
CT-1119 effectively phagocytose mesothelin positive lung cancer (A549) and ovarian cancer (MesOV) cells as shown by two independent phagocytosis assays.
To evaluate the effector function of CT-1119, Carisma utilized mesothelin positive lung adenocarcinoma (A549) and ovarian cystadenocarcinoma (MesOV) cell lines. In vitro, CT-1119 shows robust killing of lung cancer cells expressing mesothelin and CAR engagement induces the release of the pro-inflammatory cytokine TNF-α following stimulation with mesothelin expressing but not wild type (Wt) cell lines:
CT-1119 Kill and Produce Cytokine in Response to Biologically Relevant Targets

In order to evaluate the direct anti-tumor activity of CT-1119 in a relevant animal model, Carisma engrafted immunodeficient NSG-S mice with A549 lung adenocarcinoma cells expressing mesothelin by intravenous administration, which creates a lung metastasis model. CT1119 demonstrated the ability to reduce tumor progression and reduced the number of metastatic tumor nodules.
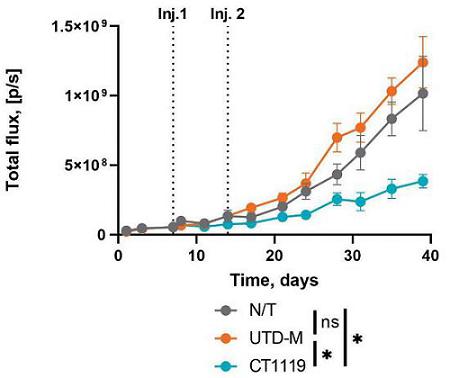
These findings demonstrate that CT-1119, an autologous human anti-mesothelin CAR-M, can effectively phagocytose and kill target tumor cells as well as initiate pro-inflammatory cytokine production in response to mesothelin. Carisma believes that CAR-M is a feasible approach for the treatment of mesothelin expressing solid tumors and is advancing the development of this program toward a clinical trial. CT-1119 is anticipated to be a CAR-Mono product encompassing a next generation CAR to enhance function. Carisma anticipates nominating a next generation CAR construct for CT-1119 in the first half of 2024. The IND is expected to be submitted in 2025.
CT-0729 (PSMA CAR-M)
Prostate-specific membrane antigen, or PSMA, is highly specific to prostate cancer cells. In vitro studies have been conducted demonstrating that Carisma can:
| · | Express anti-PSMA CARs on human macrophages |
| · | Mediate phagocytosis of PSMA overexpressing tumor cells |
| · | Induce killing of PSMA overexpressing tumor cells |
| · | Initiate cytokine release in a PSMA specific manner |
| · | Generate M1 polarized anti-PSMA CAR-M |
CT-0729 is in the discovery stage and a lead construct has not yet been nominated.
CAR-Mono: Pre-clinical Development
Currently, the CAR-M platform requires differentiation of circulating monocytes into macrophage ex vivo prior to transduction with Ad5f35 to express the CAR. Ex vivo differentiation takes approximately one week and is associated with the loss of a fraction of cells during the differentiation process. Carisma hypothesized that monocytes could be directly engineered to express CARs, shortening the ex vivo manufacturing process from approximately eight days to approximately one to two days. By bypassing ex vivo differentiation, CAR monocytes will be administered to patients, wherein they will traffic to and enter tumor tissue, differentiating into macrophages in vivo rather than ex vivo. CAR-Mono are a precursor to the CAR macrophage. Carisma further hypothesized that CAR-Mono may have improved tumor trafficking potential, given their smaller size and increased chemokine receptor expression.
To determine the feasibility of generating CAR monocytes, or CAR-Mono, Carisma conducted in vitro time course studies to assess cell viability and CAR expression compared to untransduced monocytes. Following transduction with Ad5f35, CAR expression and cell viability were tracked in vitro for 28 days. Viability was high (>90%) and CAR expression was high (>80%), and both stayed high for the entire 28 days of culture. Durable CAR expression is critical to enable the cells to (a) retain CAR expression while trafficking to the tumor, (b) retain CAR expression during differentiation into CAR macrophages, and (c) to enable sustained anti-tumor activity.
Carisma’s first CAR-Mono program is CT-0525, an autologous anti-HER2 CAR-Mono. CT-0525 is an advanced pre-clinical program, and Carisma plans to submit an IND for CT-0525 in the second half of 2023, followed by initiation of a Phase 1 clinical trial.
The M1 phenotype of Carisma’s CAR-M platform is an important aspect of the mechanism of action. As monocytes are the precursors to macrophages, differentiation and cell morphology was evaluated after two days and seven days of culture. After seven days in culture, CAR-Mono showed a progressively increasing M1 phenotype (high CD80 and CD86 expression) and expressed CAR. Compared to untransduced monocytes cultured for two or seven days in vitro, Ad5f35 transduced CAR-Mono upregulated M1 markers CD80 and CD86, confirming that Ad5f35 transduction similarly induces an M1 macrophage phenotype when added at the monocyte stage. Importantly, CAR-Mono-derived CAR-M had a similar morphology to CAR-M generated using the standard method - confirming morphologically that CAR-Mono differentiate into CAR-M.
To confirm that CAR-Mono differentiate into CAR-M and take on an M1 phenotype in vivo, NSG-S mice were engrafted with NCI-H2444 (Non-Small Cell Lung Cancer). NSG-S mice are highly immunodeficient mice that express human Interleukin (IL)-3, human GM-CSF, and human stem cell factor. These animals support enhanced engraftment of myeloid cells compared to NOD/SCID Il2rg-/- (NSG) mice, they are ideal for studies investigating the adoptive transfer of myeloid cells. Untransduced control or CAR-Mono were intratumorally injected (N=3 donors) and tumors were harvested seven days post injection. Human immune cells were enriched using flow sorting and processed for scRNA sequencing. By comparing the gene expression of in vivo and in vitro differentiated untransduced and CAR monocytes, Carisma’s data suggest that the monocytes have the potential to differentiate into macrophages and adopt an M1 like phenotype.
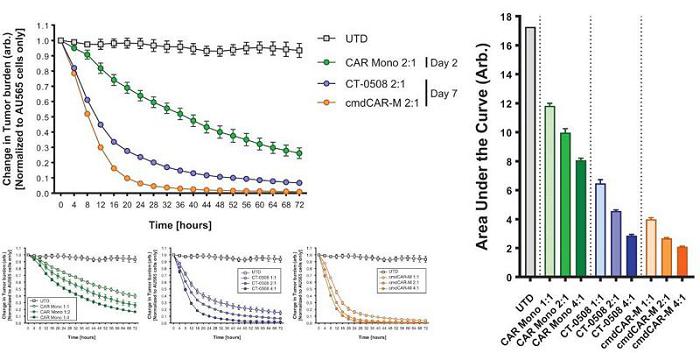
CAR-M are able to directly kill tumor cells via phagocytosis and release of cytotoxic mediators such as TNFα. Carisma evaluated the direct tumor killing capacity of CAR-Mono at Day 2 (monocyte phase) and Day 7 (macrophage phase). AU565, a HER2+ breast cancer cell line, was utilized as the target tumor cell.
Carisma’s data show that CAR-Mono mediated effective killing at Day 2 and that fully differentiated CAR-Mono-derived CAR-M (Day 7) also efficiently cleared tumor cells. When comparing CAR-Mono-derived CAR-M (Day 7) to CT-0508, Carisma found that CAR-Mono led to improved tumor killing and inflammatory cytokine production.
CAR-Mono-derived CAR-M Show Robust Tumor Killing Activity
CAR-Mono-derived CAR-M Cocultures Show Robust Proinflammatory Cytokine Production

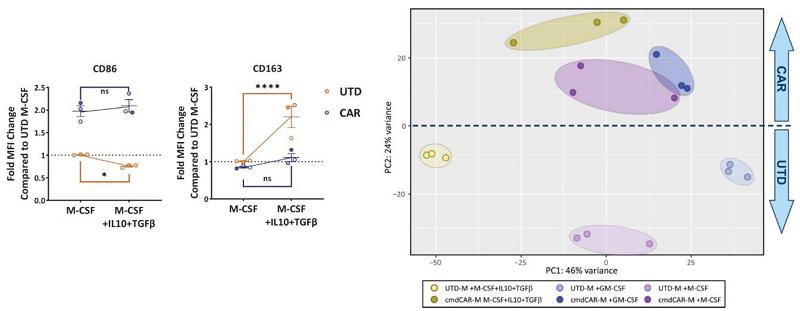
Carisma previously demonstrated that CT-0508 CAR-M are locked into an M1 phenotype by Ad5f35 transduction, and resist M2 conversion by immunosuppressive cytokines. Carisma evaluated whether CAR-Mono similarly resisted M2 environments by culturing the cells for seven days in the presence of M-CSF (differentiation factor) or M-CSF plus the immunosuppressive cytokines IL-10 and TGF-β during differentiation. CAR-Mono showed resistance to polarization and continued to express CD86 (M1) and not CD163 (M2) as demonstrated by flow cytometry and bulk RNA sequencing of untransduced and CAR-Mono-derived CAR-M. Additionally, untransduced monocytes but not CAR-Mono significantly upregulated CD163 in response to IL-10 and TGF-β.
CAR-Mono Are Protected Against M2 Polarization
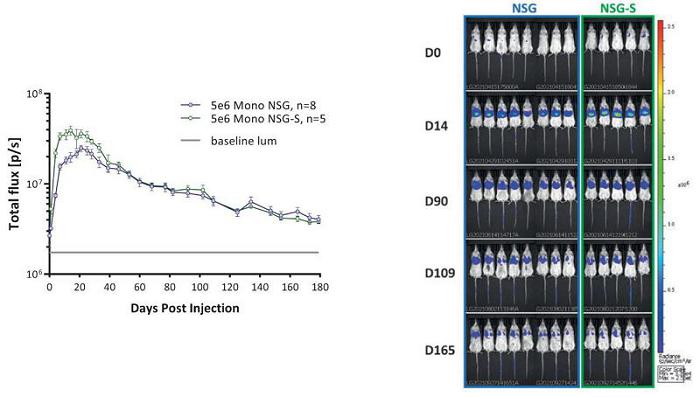
An important element to Carisma’s cell therapies is the long-term expression of its engineered CAR payloads by human myeloid cells. To evaluate persistence in vivo, monocytes were engineered with a modified Ad5f35 vector that induces the co-expression of CAR and luciferase under a single promoter by using a ribosomal skip site. This approach enables the ability to track luciferase using bioluminescent imaging and infer not only the viable persistence but also the CAR expression of human monocytes in mice. Ad5f35 engineered CAR-Luciferase Mono was injected intravenously into NSG or NSG-S mice and imaged for 180 days. While both NSG and NSG-S mice are immunodeficient, only NSG-S mice constitutively express human cytokines that promote myeloid cell survival (GM-CSF, IL3, and SCF). Carisma found that human CAR-Mono persisted for at least 180 days in vivo, independent of cytokine support.
CT-0525 Show Long Term Persistence In Vivo
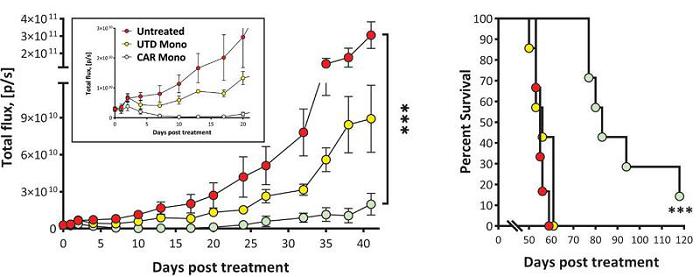
To determine whether CAR-Mono are able to control tumor growth in xenograft models, Carisma utilized a SKOV3 HER2+ ovarian cancer intraperitoneal carcinomatosis model. Anti-HER2 CAR-Mono significantly suppressed tumor growth and prolonged survival up to 120 days post treatment, while mice that received untransduced control monocytes or mice that were left untreated only survived for <60 days. Carisma is currently evaluating CAR-Mono in immunocompetent models.
CT-0525 Suppress Tumor Growth In Vivo
In summary, CAR-Mono were successfully generated with high efficiency and viability in a rapid, one-to-two day manufacturing process. CAR-Mono demonstrated stable CAR expression and viability in vitro, and persisted for at least six months in vivo. CAR-Mono differentiated into CAR-M efficiently and adopted an M1 macrophage phenotype, and resisted conversion to M2 in immunosuppressive environments. CAR-Mono were able to kill tumors cells in the monocyte phase and the macrophage phase. CAR-Mono controlled tumor growth in a xenograft mouse model of cancer. Based on the pre-clinical data to date, Carisma believes that CAR-Mono represents a potentially promising approach for cancer immunotherapy, while meaningfully expanding Carisma’s proprietary platform.
Next Generation Constructs
Carisma’s discovery team is developing a next generation CAR-M platform utilizing enhanced CAR constructs to increase potency and functionality of the engineered cells. This includes optimization of each element of the CAR itself - the binder (which gives the CAR specificity to a target antigen), the hinge (which connects the binder to the transmembrane domain and gives the CAR length and flexibility), the transmembrane domain (which spans the cell membrane), and the intracellular signaling domains (which are responsible for activation of immune cell function). It is well accepted in the immunology field that T-cells require multiple signals for activation - signal 1 deriving from the TCR, and signal 2 deriving from co-stimulatory receptors such as CD28 or 4-1BB. Thus, all approved CAR-T products are second generation CARs, incorporating CD3ζ as a primary signaling domain and either 4-1BB or CD28 as co-stimulatory domains. Third generation CARs, incorporating three signaling domains, have also been evaluated in T-cells. Unlike T-cells, macrophages do not require co-stimulation for activation and can be activated through a single signal, such as through an Fc receptor. However, multiple signaling pathways have the ability to enhance the macrophage response and may improve target-cell killing, cytokine/chemokine release, and macrophage activation.
Next Generation CAR-M Constructs
Carisma has been routinely developing and evaluating novel CAR-M constructs. Carisma’s well-established CAR-M assays enable a distinct opportunity to identify improved constructs in an efficient manner. Based on Carisma’s early findings, CD3ζ is a potent activator of macrophage function and induces phagocytosis, cytokine release, chemokine release, killing, and activation of pro-inflammatory genes. Given this finding, Carisma has been evaluating the addition of other innate immune receptors such as Toll-like receptors, CD40, MyD88, Dectin-1, to CD3ζ to improve upon CAR-M functionality.
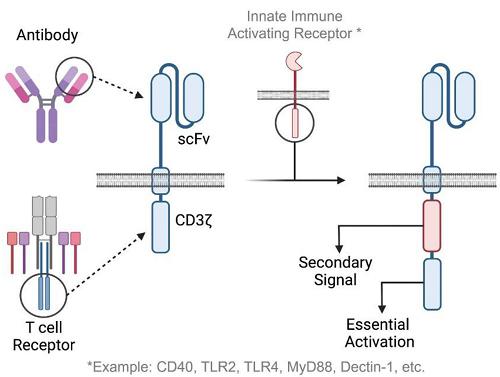
Next Generation Construct: Pre-clinical Development
Carisma’s preliminary data assessing tumor cell killing activity with CAR variants at a low “stress test” dose shows increased potency at low effector (macrophage) to target (tumor cell) ratios. CD40 is an activating receptor found on antigen presenting cells including macrophages that is activated after binding to CD40-Ligand, typically expressed on activated T-cells. CD40 signals through numerous second messengers leading to the activation of NF-ϰB and other transcription factors that induce a potent M1 phenotype and activate antigen presenting cells. MyD88 is expressed in macrophages and acts as an adaptor that plays a pivotal role in the signaling of Toll-like receptors. Carisma has found that addition of MyD88 and CD40 to CD3ζ CAR-M, in a specific sequence with a specific hinge domain, leads to a significant improvement to macrophage anti-tumor activity.
Addition of the MyD88/CD40 pathway significantly increases CAR-M potency
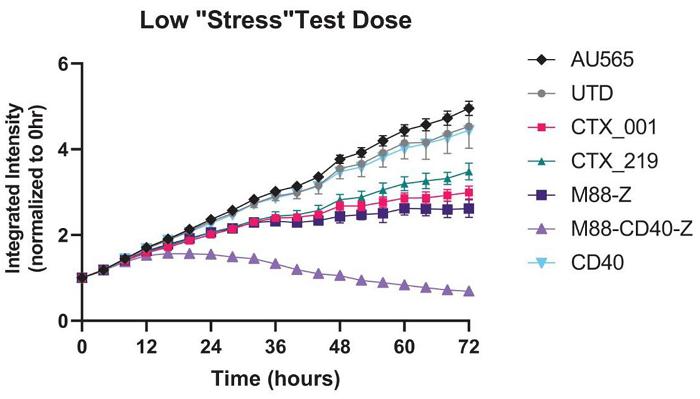
Further testing of the M88-CD40-CD3ζ CAR and other novel CAR-M are ongoing. Leading second/third generation CAR candidates may be incorporated into future autologous, allogeneic, or in vivo LNP programs.
Novel Modalities
Carisma is applying learnings from its autologous CAR-M data, tools, and processes to establish off-the-shelf engineered macrophage therapeutics. To broaden the application of CAR myeloid cell therapy, Carisma is actively seeking to develop a gene edited iPSC-derived macrophage platform. The goal is to establish a process to generate allogeneic, iPSC-derived M1, M2, or CAR myeloid cells. One approach is using allogeneic iPSC myeloid cells manufactured using the following summary process:
iPSC-Derived Myeloid Cells: Summary

Initial in vitro studies show that iPSCs can be converted to monocytes or macrophages, skewed to an M1 or M2 phenotype with cytokine culture, and can be engineered with Carisma’s proprietary vectors to express tumor targeting CARs. In addition to offering an allogeneic, universal donor platform, iPSCs are expandable cells - unlike primary human monocytes or macrophages which are terminally differentiated. In the first 15 days of the process, Carisma noted >1,000x expansion. It took approximately 15 days to generate iPSC-derived monocytes and approximately 20 days to generate iPSC-derived macrophages. The process is currently being performed in a non-GMP research environment at research scale but has the potential to be developed into a commercial scale, GMP process in the future. The macrophages generated with this process express canonical macrophage markers, appear macrophage-like with Giemsa staining and microscopic evaluation, and importantly demonstrate cell-to-cell uniformity in morphology.
iCAR-M Production
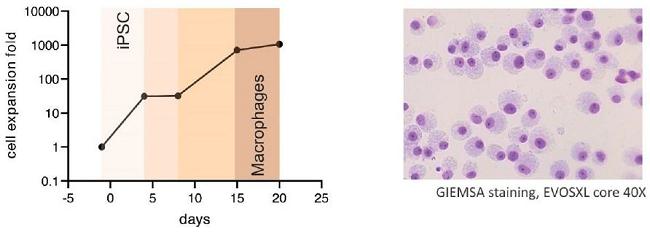
iCAR-M Anti-Tumor Activity
In order to confirm that iPSC-derived macrophages are effectively redirected against tumor associated antigens, Carisma introduced the anti-HER2 CAR into iPSC-derived macrophages using Ad5f35. Engineered iPSC-derived macrophages efficiently expressed CAR, upregulated M1 markers, and demonstrated an acceptable viability. To evaluate anti-tumor function, Carisma conducted an in vitro killing assay in which AU565 HER2+ breast cancer cells were cultured alone, together with iPSC-derived control macrophages, or iPSC-derived CAR-M. As shown below, while the control iPSC macrophages had minimal anti-tumor effect (orange), the iPSC-derived CAR-M cleared tumor cells over approximately 72 hours.
iPSC-Derived CAR-M Exert Potent Anti-Tumor Activity
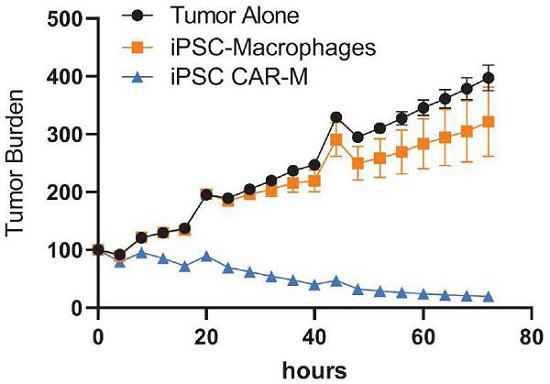
While iCAR-M appear to be able to exert direct anti-tumor functionality, there are inherent complexities that will be critical to inform the ultimate allogeneic macrophage strategy. CAR-M exert anti-tumor immunity through tumor infiltration, phagocytosis, cytokine/chemokine release, TME activation, T-cell recruitment, and antigen presentation. To enable an allogeneic off-the-shelf iCAR-M program, the cells will inherently be either MHC mismatched or MHC-edited (for example, MHC-I and MHC-II may be deleted using CRISPR/Cas9 editing to generate universally accepted macrophages), and thus may have limited direct antigen presentation potential. However, iCAR-M are expected to maintain the other mechanisms of action, as summarized below. Future studies will evaluate whether allogeneic CAR-M are capable of inducing epitope spreading, as indirect mechanisms of antigen presentation have not been ruled out.
Importantly, the iPSC-derived myeloid cell platform, combined with Carisma’s gene engineering capabilities, has the potential to be produced in multiple ways. First, iPSC-derived myeloid cells can be expanded, qualified, and banked prior to being used as a master cell bank source for the production of engineered myeloid cell therapies. In this example, iPSC-derived myeloid cells would be engineered with Carisma’s proprietary methods (Ad5f35, Vpx-LV, modified mRNA, or other) after differentiation into the desired myeloid cell subtype. Alternatively, Carisma is optimizing methods to introduce the CAR (or other genetic payload) at the iPSC stage using targeted integration into desired genomic loci, isolating iPSC clones with integrated genes and additional potential genetic edits, and differentiating these cells into monocytes or macrophages, and skewing them to an M1 phenotype using Carisma’s proprietary polarization processes. For this approach, care must be taken to ensure that the CAR (or other payload) is not epigenetically downregulated or lost during the myeloid differentiation process.
While the current focus of Carisma’s discovery efforts is to ultimately generate iCAR-M, the platform is expected to be readily adaptable to either develop (a) myeloid cells engineered with other anti-tumor payloads, or (b) myeloid cells engineered with payloads designed to ameliorate disease outside of oncology. Notably, Carisma has early non-oncology programs in liver fibrosis, neurodegeneration, and auto-immunity/chronic inflammation which can be combined with the iPSC-derived myeloid cell platform. Currently, Carisma’s pipeline is focused on autologous approaches and direct in vivo reprogramming, and the allogeneic iPSC-derived platform is at the pre-clinical discovery stage.
LNP/mRNA Platform (Moderna Collaboration)
In collaboration with Moderna, Carisma is developing an mRNA based in vivo CAR-M platform for oncology. This approach is highly differentiated in the cell therapy space - not only because it relies on myeloid cells as the engineered effectors, but also because it utilizes direct in vivo reprogramming of a patients’ own cells with a well-validated LNP/mRNA platform. By engineering a patients’ own cells directly within their body, ex vivo autologous or allogeneic cell manufacturing is entirely bypassed - significantly increasing the commercial potential of the therapy. Importantly, while this approach enables an off-the-shelf therapy, the engineered cells are autologous, as it is the patients’ own cells being engineered into CAR-M in vivo, or directly within their body. This strategic partnership enables Carisma to apply the learnings gleaned from autologous CAR-M development to expand its pipeline to up to 12 additional oncology candidates.
Studies with the myeloid tropic LNP have shown mRNA delivery is specific for myeloid cells (monocytes, macrophages, dendritic cells). Based on clinical data using other (non-CAR) payloads, Moderna has previously demonstrated that the LNP was well-tolerated after systemic administration and could furthermore be re-dosed. Preliminary data have demonstrated that the LNP is efficient in transfecting myeloid cells in vitro and in vivo. In addition, preliminary data confirms high CAR expression, viability, and CAR-M function. The platform summary for the in vivo CAR-M approach is shown below:
Myeloid Tropic LNP/mRNA Platform (Moderna Collaboration)
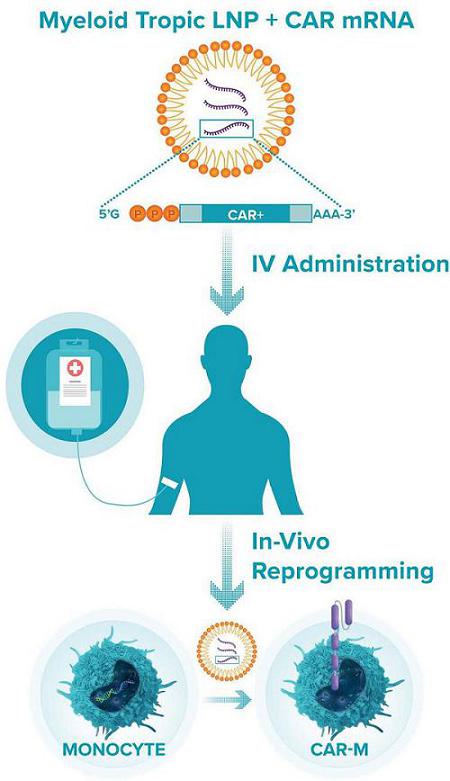
LNP/mRNA Pre-clinical Data (Moderna Collaboration): Pre-clinical Development
In vitro pre-clinical studies with the myeloid tropic LNP/mRNA platform have shown efficient transfection. Carisma has optimized conditions for ex vivo LNP/mRNA delivery to human and murine monocytes/macrophages, as well as primary murine myeloid cells to establish various relevant murine tumor models. Carisma’s goal was to establish a platform with high viability (>70%), high transfection efficiency (>70%), and significantly increased CAR-M killing activity compared to untransfected control macrophages. Key data for the anti-HER2 CAR constructs CTX_001 and CTX_219 are shown in the figures below:
LNP Transfection is Well Tolerated by Human Macrophages

LNP Engineering leads to Dose Dependent CAR Expression

In addition to CAR expression and viability, Carisma has shown that one day post transfection, CTX001 and CTX219 mediate potent killing activity against target AU565 cells.
LNP Engineered CAR-M Display Effective Killing Function
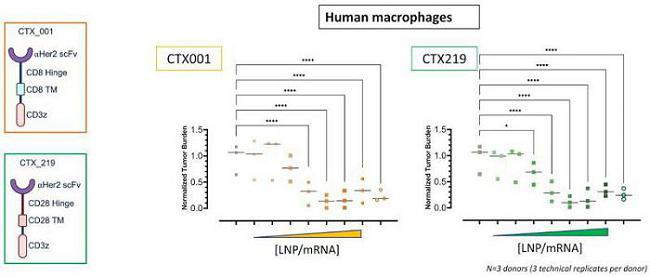
Statistical analysis was determined using Two-way ANOVA, * p<0.5 **** p<0.0001. The data were generated from N=3 donors (3 technical replicates per donor).
LNP Transfection is Well Tolerated by Human Monocytes
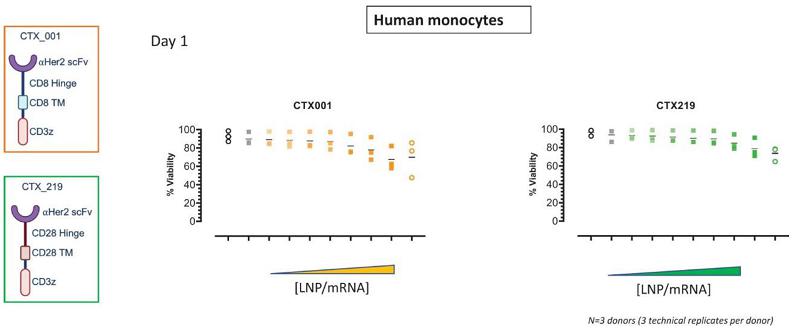
LNP Engineering leads to Dose Dependent CAR Expression in Monocytes
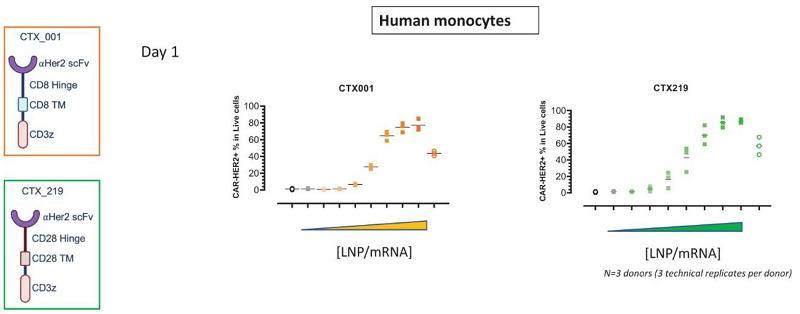
In addition to CAR expression and viability, Carisma has shown that monocytes engineered with LNP/mRNA encoding the CARs CTX001 and CTX219 mediate potent killing activity against target AU565 breast cancer cells. These findings demonstrate similarity between LNP engineered macrophage and monocyte effector function.
LNP Engineered CAR-Mono Display Effective Killing Function
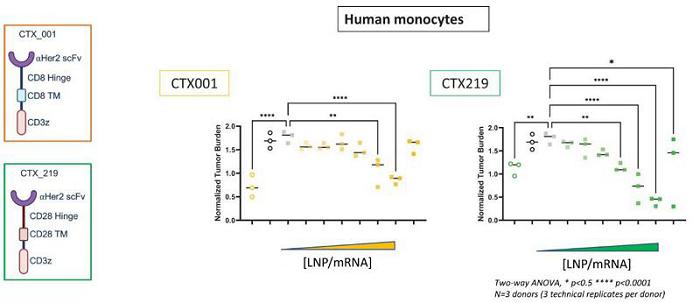
Statistical analysis was determined using Two-way ANOVA, * p<0.5 **** p<0.0001. The data were generated from N=3 donors (three technical replicates per donor).
To model direct reprogramming of TAMs to CAR-M, Carisma generated tumor spheroids comprised of HER2+ breast cancer cells and human TAMs. Carisma confirmed that the macrophages embedded within these spheroids adopted an M2, TAM-like phenotype. Carisma added LNPs (containing CAR mRNA and an M1 polarizing gene) directly to the tumor spheroids and found that the reprogrammed TAMs mediated tumor spheroid shrinking over a 72-hour period.
Directly Reprogramming Myeloid Cells within Tumors with LNP/mRNA

The first four indications have been nominated by Moderna, spanning both solid tumors and hematologic malignancies. Pre-clinical platform optimization, as well as target discovery and CAR development, are ongoing in parallel. Early in vivo proof of concept is expected in 2023.
EM-C: A novel platform for regulation of inflammation
Cytokines in tissue microenvironments regulate the balance between pro- and anti-inflammatory signaling. Dysregulated cytokine expression causes deleterious immunosuppression or inflammation, underpinning the pathophysiology of numerous diseases. As examples, anti-inflammatory cytokines in solid tumors suppress immune activation and safeguard the tumor, whereas pro-inflammatory cytokines in rheumatoid arthritis drive chronic inflammation. Rebalancing inflammation/immunosuppression by targeting aberrant cytokine signaling offers a generalizable approach to treating many diseases, but systemic cytokine blockade carries risks such as increased risk of serious infection. Cellular immunotherapies may offer a localized platform that could activate in response to cytokines then proportionately remodel the microenvironment’s inflammatory state as needed.
Macrophages are tissue infiltrating homeostatic regulators responsible for the initiation and resolution of inflammation. Carisma leveraged the ability of macrophages to regulate inflammation by generating macrophages that express genetically engineered, synthetic cytokine switch receptors (SR). Carisma termed this platform EM-C and evaluated its modular ability to convert immunosuppressive M2 signals into pro-inflammatory M1 responses for solid tumor microenvironment conversion, or vice versa for the conversion of pro-inflammatory M1 signals into immunosuppressive M2 responses for auto-immune or inflammatory diseases.
EM-Cs were generated by expressing SRs in primary human macrophages. First, Carisma generated SRs that convert immunosuppressive signals into inflammatory signals - focusing on IL-10. To convert IL-10 (a cytokine well known to polarize macrophages toward an M2 phenotype) in the TME into a pro-inflammatory signal, an IL-10 SR was designed by fusing the extracellular domain of the IL-10 receptor to an engineered portion of the IFNλ receptor. The in vitro response of IL-10 EM-Cs to IL-10 was monitored by the expression of M1 and M2 markers. While control untransduced or CAR (a control irrelevant construct) expressing macrophages responded to IL-10 through the downregulation of M1 markers and upregulation of M2 markers, IL10-SR EM-Cs upregulated M1 markers and downregulated M2 markers. These data and others demonstrate that Carisma’s IL10-SR EM-Cs can induce an inflammatory response when exposed to immunosuppressive conditions. Carisma has generated numerous additional EM-Cs that respond to other immunosuppressive factors, such as TGFβ.
Conversely, Carisma sought to develop EM-Cs that can convert inflammatory cytokines into anti-inflammatory responses. As myeloid cells are recruited to sites of inflammation. This approach would enable a myeloid cell therapy that accumulates at inflammatory sites and locally shuts down inflammation. This approach may have broad utility in autoimmunity, chronic inflammation, fibrosis, transplant, and graft versus host disease. Carisma generated EM-Cs targeting IFN-γ and demonstrated that while control untransduced or CAR (an irrelevant payload) macrophages respond to IFN-γ by upregulating M1 markers and downregulating M2 markers, IFN-γ SR EM-Cs responded to IFN-γ by downregulating M1 markers and upregulating M2 markers. These data demonstrate that Carisma’s proprietary EM-C platform can be broadly utilized to generate engineered macrophages with the ability to control the inflammatory nature of environments, and act as living converters, with broad potential applicability to oncology, autoimmunity, chronic inflammation, and other indications. This platform offers broad opportunity for strategic partnership.
Macrophages Expressing Cytokine Switch Receptors Can Invert
Pro- or Anti-Inflammatory Signals
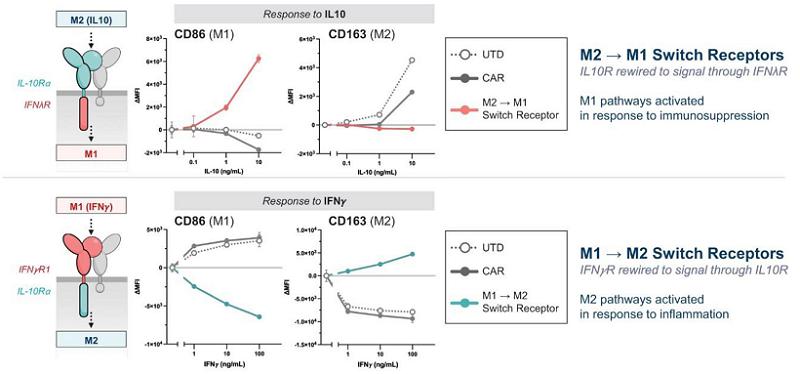
Indications Beyond Oncology
While Carisma is an oncology focused company, the macrophage engineering platform Carisma has established offers broad opportunity to develop cell therapies for indications beyond oncology. Carisma has numerous early-stage research programs designed to produce development candidates for liver fibrosis, neurodegenerative disease, and autoimmunity/chronic inflammation. Carisma’s new product candidates will incorporate all the core elements of its macrophage cell engineering platform, plus certain indication specific modifications uniquely designed to address the pathology of each indication. While autologous cell therapy may be utilized for proof of concept, these indications have the potential to be combined with Carisma’s allogeneic or off-the-shelf therapeutic approaches. Indications beyond oncology offer opportunities for strategic partnerships to accelerate the development of these programs.
Manufacturing and Delivery
Carisma does not own or operate, and currently has no plans to establish, any manufacturing facilities. Carisma currently relies on and expects to continue to rely on third-party contract development manufacturing organizations, or CDMOs, for the manufacturing and release testing of viral vectors and cell drug products. Carisma also currently relies on third parties for patient leukapheresis material logistics as well as to package, label, store, and distribute the cell drug products.
Carisma has established and will continue to establish arrangements with contract manufacturers to supply clinical materials and manufacturing capabilities for Carisma’s clinical trials. Carisma currently obtains its supplies from these manufacturers on a purchase order basis and does not have long-term supply arrangements in place. Should any of these manufacturers become unavailable to Carisma for any reason, Carisma believes that there are several potential replacements, although Carisma may incur some delay in identifying and qualifying such replacements.
Carisma also plans to continue to expand the scope and number of its collaborations to further develop its manufacturing capabilities and to minimize manufacturing risk. As Carisma scales to commercialization, it expects to increase its capacity with its current suppliers and evaluate other options to secure commercial scale capacity.
Manufacturing Process for CT-0508
A CDMO is used to produce cGMP lots of viral vector. The CT-0508 drug substance process begins by isolating the monocyte population from a single fresh patient leukopak mobilized by donor pretreatment with filgrastim. The resulting monocytes are cultured in the presence of a cytokine and other factors to induce differentiation into macrophages. The resulting macrophages are then transduced with the Ad5f35 vector encoding an anti-HER2 CAR. The resulting cells are then harvested as drug substance. Macrophages derived from a single leukopak from one patient comprise one batch of CT-0508. Final formulation is performed and transferred into freezing bags to generate drug product. The drug product is carefully frozen in a controlled process and then placed into secured storage and maintained at a temperature of ≤-135⁰ Celsius. Safety and specification tests are performed and if found acceptable the product is released and shipped to clinical trial sites. The current process from receipt of leukopak to drug product and cryopreservation is eight days.
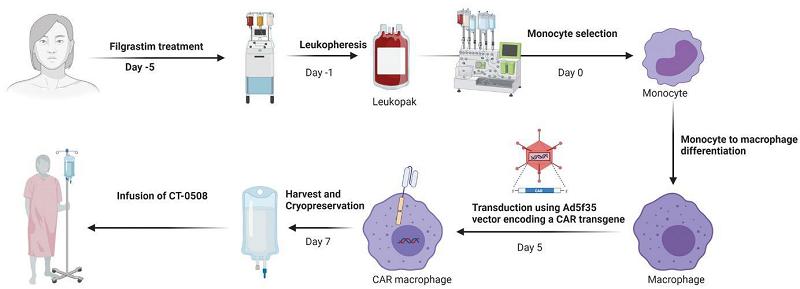
Carisma plans to continue to invest in process improvements to reduce the overall manufacturing process time and improve costs for the viral vector and cell drug product.
Intellectual Property
Carisma strives to protect and enhance its proprietary technology, inventions and improvements that Carisma believes are commercially important to the development of its business, including through seeking, maintaining and defending patent rights, whether developed internally or licensed from third parties. Carisma also intends to rely on trade secrets related to its proprietary technology platform and its know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen and maintain its proprietary position in the fields of cancer and other indications including those related to neurodegeneration, liver fibrosis and autoimmune disease, which may be important for the development of Carisma’s business. Carisma additionally may rely on regulatory protection afforded through data exclusivity, market exclusivity and patent term extensions, where available.
Carisma’s commercial success may depend, in part, on its ability to obtain and maintain patent and other proprietary protection for commercially important technology, inventions and know-how related to its business, defend, and enforce its patents, preserve the confidentiality of its trade secrets and operate without infringing the valid enforceable patents and proprietary rights of third parties. Carisma’s ability to stop third parties from making, using, selling, offering to sell or importing its products may depend on the extent to which it has rights under valid and enforceable licenses, patents or trade secrets that cover these activities. With respect to both Carisma’s owned and licensed intellectual property, Carisma cannot be sure that patents will be granted with respect to any of its pending patent applications or with respect to any patent applications filed by Carisma in the future, nor can it be sure that any of its existing patents or any patents that may be granted to Carisma in the future will be commercially useful in protecting its commercial products and methods of manufacturing such products, as well as being held valid if challenged.
Carisma currently controls over 15 granted patents and over 43 patent applications pending in several jurisdictions, including the United States, Europe, Australia, Brazil, Canada, China, Israel, Japan, Korea, Mexico, New Zealand, and Singapore. Intellectual property is a critical component of Carisma’s business plan for maximizing return on its investments. Carisma is actively developing intellectual property and will continue to maintain and defend United States and international patent rights for its products, technology, and development and improvement of its discovery platforms.
To maintain its competitive position in the market, Carisma has spent considerable effort securing intellectual property rights, including several patent rights related to its proprietary CAR technology and myeloid cell engineering technology.
Exclusively Licensed Intellectual Property - UPenn
Carisma has exclusive rights to three patent families, and non-exclusive rights to related know-how by virtue of a license agreement with the University of Pennsylvania. These patent families are directed to, among other things, methods of efficiently expressing CARs in myeloid cells, including monocytes, macrophages, and dendritic cells and enhancing effector activity, as well as the modified cells and compositions including such modified cells for use in several indications including various oncology targets. The applications will have an expiration date of no earlier than 2036. This licensed patent portfolio includes:
| · | A patent family that includes nine issued U.S. patents and two pending U.S. patent applications relating to modified macrophages, monocytes and dendric cells comprising CARs. These U.S. patents are expected to expire in 2036, absent any term adjustments or extensions. Corresponding foreign applications have been filed and are pending in Australia (including one issued patent), Brazil (including one issued patent) Canada, China (including one issued patent), Europe, Israel (including one issued patent), India, Japan (including one issued patent), Korea, Mexico (including one issued patent), New Zealand, Russia (including one issued patent), Singapore, Thailand and South Africa. |
| · | A patent family that includes one pending U.S. patent application relating to modified macrophages, monocytes and dendric cells in protein aggregate-associated disorders. Patent applications in this family are expected to expire in 2039, absent any term adjustments or extensions. Corresponding foreign applications have been filed and are pending in Australia, Canada, China, Europe, Israel, Japan, Korea, New Zealand, and Singapore. |
| · | A patent family that includes one pending U.S. patent application relating to activation of antigen presenting cells. Patent applications in this family are expected to expire in 2040, absent any term adjustments or extensions. A corresponding foreign application has been filed in Europe. |
Exclusively Licensed Intellectual Property - NYU
Carisma has exclusive rights to one patent family, and non-exclusive rights to related know-how by virtue of a license agreement with New York University, or NYU. The rights granted under the NYU license are to all indications for human use. This licensed patent portfolio includes:
| · | A patent family that includes one U.S. patent relating to a chimeric HIV-1 vector with an SIV minimal Vpx packaging domain and method of making virions with enhanced infectivity for macrophages and dendric cells. The U.S. patent is expected to expire in 2033, absent any term adjustments or extensions. |
Carisma Owned Intellectual Property
Carisma currently owns five U.S. patent families. This owned patent portfolio includes:
| · | A patent family that includes one issued U.S. patent and two pending U.S. patent applications relating to macrophages, monocytes and dendric cells comprising novel CAR constructs. Patent applications in this family are expected to expire in 2041, absent any term adjustments or extensions. |
| · | A patent family that includes one pending Patent Cooperation Treaty, or PCT, application relating to mRNA transfection of macrophages, monocytes and dendric cells comprising CARs. Patent applications in this family are expected to expire in 2041, absent any term adjustments or extensions. |
| · | A patent family that includes one pending PCT application relating to modified immune cells for fibrosis and inflammation. Patent applications in this family are expected to expire in 2041, absent any term adjustments or extensions. |
| | | |
| | · | A patent family that includes one pending PCT application relating to in vivo delivery of CARs to a patient's own macrophages, monocytes, dendritic cells, or stem cells. Patent applications in this family are expected to expire in 2042, absent any term adjustments or extensions. |
| · | A patent family that includes one pending PCT application relating to self-polarizing immune cells. Patent applications in this family are expected to expire in 2042, absent any term adjustments or extensions. |
| · | Carisma will also seek to generate additional intellectual property that covers enhancements to all aspects of the platform, including novel CARs, combinations, gene editing and manufacturing improvements. Where appropriate, Carisma will also look to in-license relevant technology from third parties. |
Patent Term and Patent Term Extensions
The term of individual patents depends upon the legal term for patents in the countries in which they are obtained. In most countries, including the United States, the patent term is 20 years from the earliest filing date of a non-provisional patent application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the USPTO in examining and granting a patent or may be shortened if a patent is terminally disclaimed over an earlier filed patent. The term of a patent that covers a drug, biological product or medical device approved pursuant to a pre-market approval may also be eligible for patent term extension, or PTE, when FDA approval is granted, provided statutory and regulatory requirements are met. The length of the PTE is related to the length of time the drug is under regulatory review while the patent is in force. The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, permits a patent term extension of up to five years beyond the expiration date set for the patent. Patent extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent applicable to each regulatory review period may be granted an extension and only those claims regarding the approved drug are extended. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug.
Trademarks
Carisma’s trademark portfolio currently includes registered U.S. trademarks for Carisma in the United States, Europe, Great Britain and Japan. In order to supplement the protection of its brand, Carisma also has a registered internet domain name. Going forward, Carisma will consider additional trademarks to enhance its brand and support its products.
Trade Secrets
Carisma relies, in some circumstances, on trade secrets to protect its unpatented technology. However, trade secrets can be difficult to protect in certain circumstances. Carisma seeks to protect its trade secrets and proprietary technology and processes, including through confidentiality agreements with its employees, consultants, scientific advisors and contractors. Carisma also seeks to preserve the integrity and confidentiality of its data and trade secrets by maintaining physical security of its premises and physical and electronic security of its information technology systems. While Carisma has confidence in these individuals, organizations and systems, agreements or security measures may be breached. Carisma may not have adequate remedies for any breach and could lose its trade secrets through such a breach. In addition, Carisma’s trade secrets may otherwise become known or be independently discovered by competitors. To the extent that its consultants, contractors or collaborators use intellectual property owned by others in their work for Carisma, disputes may arise as to the rights in related or resulting trade secrets, know-how and inventions.
Moderna Collaboration Agreement
In January 2022, Carisma entered into a Collaboration and License Agreement with Moderna providing for a broad strategic partnership to discover, develop and commercialize in vivo engineered CAR-M therapeutics for up to 12 oncology programs. Carisma refers to this agreement as the Moderna Collaboration Agreement.
In collaboration with Moderna, Carisma has established an approach that uses Moderna’s LNP/mRNA technology, together with Carisma’s CAR-M platform technology, to create novel in vivo oncology medications.
Under the Moderna Collaboration Agreement, the parties initiate research programs during a research term, focused on the discovery and research of products directed to biological targets. Either party may nominate a target for inclusion in a research program, subject to certain exclusions. Carisma refers to a target included in a research program pursuant to designated procedures as a research target. Moderna may replace research targets pursuant to designated procedures. The first four research targets have been nominated and all programs are currently in the discovery phase at Carisma. Moderna funds the cost of Carisma’s activities in accordance with an agreed research budget.
Moderna has the right to designate up to 12 research targets as development targets during a specified development target nomination period upon payment of a development target designation milestone payment. Moderna can replace development targets with research targets during a specified period of time. If Moderna exercises its right to designate a development target, Moderna will have a worldwide, exclusive license under patents and know-how controlled by Carisma to develop and commercialize products directed to the applicable development target, subject to certain diligence obligations.
Commencing a specified time after the effective date of the Moderna Collaboration Agreement, Moderna will have the right to nominate targets relating to diseases outside the field of oncology for inclusion in research programs in specified circumstances. Such right is subject to the same exclusions as Moderna’s right to nominate other targets for inclusion in research programs.
During the term of the Moderna Collaboration Agreement, Carisma and its affiliates are subject to various exclusivity obligations under which Carisma is not permitted to research, develop or commercialize particular products outside of the collaboration, including products for use as in vivo therapies in the field of oncology, products directed to any target included in the collaboration, or products containing a polypeptide provided by Carisma to Moderna in connection with a research program that are directed to any development target.
Under the terms of the Moderna Collaboration Agreement, Carisma received a $45.0 million up-front cash payment. Assuming Moderna develops and commercializes 12 products, each directed to a different development target, Carisma is also eligible to receive up to between $247.0 million and $253.0 million per product in development target designation, development, regulatory and commercial milestone payments. In addition, Carisma is eligible to receive mid to high single digit tiered royalties on net sales of any products that are commercialized under the agreement, which may be subject to reductions. Moderna has also agreed to cover the cost of certain milestone payments and royalties Carisma owes to a licensor under one of its intellectual property in-license agreements that Carisma is sublicensing to Moderna under the Moderna Collaboration Agreement, which royalties Moderna may deduct in part from any royalties owed to Carisma.
Unless earlier terminated, the Moderna Collaboration Agreement will expire upon the expiration of all royalty obligations thereunder. The royalty period for each product developed under the Moderna Collaboration Agreement will expire on a country-by-country basis upon the later of (1) the expiration of the last-to-expire valid patent claim of specified patents, (2) the expiration of regulatory-based exclusivity for such product in such country or (3) ten years after the first commercial sale with respect to such product in such country. Moderna has the right to terminate the Moderna Collaboration Agreement for convenience in its entirety or with respect to a specific product or target on ninety days’ prior notice. Either Carisma or Moderna may terminate the Moderna Collaboration Agreement in its entirety if the other party is in material breach and such breach is not cured within the specified cure period, except in the case of Moderna’s breach of its diligence obligations, termination by Carisma is limited to the applicable target and product. In addition, either Carisma or Moderna may terminate the Moderna Collaboration Agreement in the event of specified insolvency events involving the other party. As an alternative to termination in the event of Carisma’s uncured material breach of certain sections of the agreement, Moderna has the option to continue the collaboration under the agreement with reduced payment obligations.
University of Pennsylvania License Agreement
In November 2017, Carisma entered into a license agreement with the Trustees of the University of Pennsylvania, or Penn, which was amended in February 2018, January 2019, March 2020 and June 2021. Carisma refers to this agreement as the Penn License Agreement. Pursuant to the Penn License Agreement, Penn granted Carisma (1) an exclusive, worldwide license, with specified rights to sublicense, under Penn’s interest in specified patents related to CAR macrophages, monocytes or dendritic cells, which Carisma refers to collectively as CAR-M, (2) an exclusive, worldwide license, with specified rights to sublicense, under Penn’s interest in specified patents related to CAR-M directed to mesothelin, and (3) a nonexclusive, worldwide license under Penn’s interest in specified know-how related to CAR-M, with limited rights to sublicense only in combination with specified products or patents. These licensed patents and know-how arose primarily from research conducted by Dr. Saar Gill and Dr. Michael Klichinsky at the University of Pennsylvania, co-founders of Carisma. The foregoing licenses are subject to rights retained by Penn for specified non-commercial uses and rights retained by the United States government. Under the Penn License agreement, Carisma is obligated to use commercially reasonable efforts to pursue development and commercialization of at least one CAR-M product in oncology and non-oncology fields.
Carisma is responsible for paying Penn an annual license maintenance fee in the low tens of thousands of dollars, payable until Carisma’s first payment of a royalty. Carisma is required to pay Penn up to $10.9 million per product in development and regulatory milestone payments, up to $30.0 million per product in commercial milestone payments, and up to an additional $1.7 million in development and regulatory milestone payments for the first CAR-M product directed to mesothelin. While the agreement remains in effect, Carisma is required to pay Penn low to mid-single digit percentage tiered royalties on annual net sales of licensed products, which may be subject to reductions. Penn is guaranteed a minimum royalty payment amount in the low hundreds of thousands of dollars for each year after the first commercial sale of a licensed product. Carisma must also pay Penn a percentage in the mid-single digits to low double digits of certain types of income Carisma receives from sublicensees. In addition, Carisma is required to pay Penn an annual alliance management fee in the low tens of thousands of dollars, ending after several years, unless Carisma provides funding to Penn for research and development activities that extend beyond a specified date, in which case Carisma will continue to owe the alliance management fee for each year in which Carisma continues to fund such activities. Carisma also paid Penn an upfront fee in the low hundreds of thousands of dollars for the license to the patents related to the mesothelin binder that is incorporated into the CAR design for Carisma’s mesothelin product candidate. Carisma is responsible for a pro rata share of costs relating to the prosecution and maintenance of the licensed patents.
The royalty period for each licensed product will expire on a product-by-product basis upon the later of (1) the expiration of the last-to-expire valid patent claim of the licensed patents covering such product in the country of sale or in the country of manufacture, or (2) the expiration of regulatory-based exclusivity for such product in the country of sale. The license agreement remains in effect until the later of (1) expiration or abandonment of the last licensed patent or (2) loss of regulatory exclusivity. Carisma may terminate the agreement for convenience upon thirty days’ prior notice. Penn may terminate the agreement for Carisma’s material breach, subject to a specified cure period, except for certain breaches for which Penn may terminate immediately. Penn may also terminate if Carisma becomes the subject of a specified insolvency event.
New York University License Agreement
In July 2020, Carisma entered into a license agreement with NYU. Carisma refers to this agreement as the NYU License Agreement. Pursuant to the NYU License Agreement, NYU granted Carisma (1) an exclusive, worldwide license, with specified rights to sublicense, under NYU’s interest in specified patents related to the Vpx-LV and (2) a nonexclusive, worldwide license, with specified rights to sublicense, under NYU’s interest in specified know-how related to the Vpx-LV, in each case to develop, manufacture, use and sell products developed using the Vpx-LV, which Carisma refers to as Licensed Products. The foregoing licenses are subject to rights retained by NYU to use, and to permit other non-commercial entities to use, the licensed patents and licensed know-how for educational and research purposes, as well as rights retained by the United States government. Under the NYU License Agreement, Carisma is obligated to use reasonable diligence to carry out a specified development plan and to obtain regulatory approval for Licensed Products in the U.S. and each of the other countries in which Carisma or its sublicensees intend to produce, use, and/or sell Licensed Products, as well as to begin the regular commercial production, use, and sale of the Licensed Products in good faith in accordance with the development plan and to continue diligently thereafter to commercialize the Licensed Products.
Carisma is required to pay NYU an annual license maintenance fee in the mid tens of thousands of dollars; up to $1,685,000 per Licensed Product in development and regulatory milestone payments; and low single digit percentage tiered royalties on annual net sales of Licensed Products on a country-by-country basis until the later of (1) 12 years after first commercial sale of a Licensed Product in such country or (2) expiration of the last to expire licensed patent. Carisma must also pay NYU a percentage in the low single digits to low double digits of certain types of income Carisma receives from sublicensees or assignees of the agreement. Carisma is also responsible for all costs relating to the prosecution, maintenance, and defense of the licensed patents.
The NYU License Agreement remains in effect until the expiration of all royalty terms in all countries. Either party may terminate the NYU License Agreement for the other party’s uncured material breach or insolvency or bankruptcy.
Competition
The biopharmaceutical industry, and in particular the cell therapy field, is characterized by intense investment and competition aimed at rapidly advancing new technologies, intense competition, and a strong emphasis on intellectual property and proprietary products. Carisma’s platform and therapeutic product candidates are expected to face substantial competition from multiple technologies, marketed products, and numerous other therapies being developed by other biopharmaceutical companies, academic research institutions, governmental agencies, and public and private research institutions. Many of Carisma’s potential competitors have substantially greater financial, technical, and other resources, such as larger research and development staff, established manufacturing capabilities and facilities, and experienced marketing organizations with well-established sales forces, and any product candidates that Carisma successfully develops and commercializes will compete with existing therapies and new therapies that may become available in the future. In addition, there is substantial patent infringement litigation in the biopharmaceutical industry, and, in the future, Carisma may bring or defend such litigation against its competitors.
The key competitive factors affecting the success of Carisma’s product candidates, if approved, are likely to be their efficacy, safety, convenience and price, the level of competition, and the availability of coverage and adequate reimbursement from third-party payors.
Unlike other cell therapy approaches, Carisma’s CAR-M platform is based on engineering macrophages and monocytes with proprietary vectors, constructs, and processes, enabling a differentiated platform from other cell therapy competitors that primarily focus on T or NK cells. While Carisma believes that its scientific expertise, novel technology, and intellectual property position offer competitive advantages, Carisma faces competition from multiple other cell therapy technologies and companies. Other companies developing engineered myeloid cell therapies include, among others, Myeloid Therapeutics, Shoreline Biosciences, Inceptor Bio, Thunder Bio, Resolution Therapeutics, CellOrigin, Sirpant Therapeutics, and others.
Due to the broad promise of cell therapies, and the potential of myeloid cell-based approaches to expand cell therapy efficacy into solid tumors, Carisma expects increasing competition from new and existing companies across several fronts, which include, among others:
| · | Myeloid cell therapies: Myeloid Therapeutics, Shoreline Biosciences, Inceptor Bio, Thunder Bio, CellOrigin, Deverra |
| · | Autologous T-cell therapies: Adaptimmune Therapeutics, Autolus Therapeutics, Bluebird, Bristol Myers Squibb, Kite/Gilead, Novartis, Gracell, TCR2, Poseida, Vor, TMunity, among others |
| · | Allogeneic T-cell therapies: Allogene, Atara, Century, Cellectis, Celyad, CRISPR, Fate, Gracell, Kite/Gilead, Legend, Poseida, Precision Bio, Sana, Vor, among others |
| · | NK and other cell therapies: Artiva, Celularity, Century, Editas, Fate, Fortress, ImmunityBio, Nkarta, NKGen, Takeda, Adicet, Gamida Cell, among others |
| · | Direct in vivo reprogrammed cell therapies: Umoja, Ensoma, Interius, Tidal/Sanofi, BioNTech |
In addition to competition from other cell therapy companies, any products that Carisma develops may also face competition from other types of therapies. Other companies developing non-cell therapies, including gene therapies, include Gilead, ALX Oncology, Trillium, Five-Prime, Immune-Onc, Pionyr, Infinity, NextCure, OncoResponse, Curis, Faron, Apexigen, Pfizer, Dren, and multiple biotechnology and pharmaceutical companies developing other directly competitive technologies such as small molecules, immune agonists, antibodies, bi/tri specific antibodies, antibody drug conjugates, and other solid tumor therapeutics.
Carisma also competes with third parties for retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, its programs. Carisma may pursue the in-license or acquisition of rights to complementary technologies and product candidates on an opportunistic basis. The acquisition and licensing of technologies and product candidates is a competitive area, and a number of more established companies also have similar strategies to in-license or acquire technologies and product candidates that Carisma may consider attractive. These established companies may have a competitive advantage over Carisma due to their size, cash resources and greater development and commercialization capabilities. In addition, companies that perceive Carisma to be a competitor may be unwilling to assign or license rights to Carisma. Carisma also may be unable to in-license or acquire the relevant technology or product candidate on terms that would allow it to make an appropriate return on its investment.
Carisma’s commercial opportunity could be reduced or eliminated if its competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that it may develop. Carisma’s competitors also may obtain FDA or other regulatory approval for their products more rapidly than it may obtain approval for Carisma’s, which could result in its competitors establishing a strong market position before it is able to enter the market. In addition, Carisma’s ability to compete may be affected in many cases by insurers or other third-party payors seeking to encourage the use of generic products.
Government Regulation
Government authorities in the United States, at the federal, state and local level, and in other countries and jurisdictions, including the European Union, extensively regulate, among other things, the research, development, testing, manufacture, pricing, reimbursement, sales, quality control, approval, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, post-approval monitoring and reporting, and import and export of pharmaceutical products, including biological products. The processes for obtaining marketing approvals in the United States and in foreign countries and jurisdictions, along with subsequent compliance with applicable statutes and regulations and other regulatory authorities, require the expenditure of substantial time and financial resources.
Licensure and Regulation of Biologics in the United States
In the United States, Carisma’s product candidates are regulated as biological products, or biologics, under the Public Health Service Act, or PHSA, and the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implementing regulations and guidance. A company, institution, or organization which takes responsibility for the initiation and management of a clinical development program for such products, and for their regulatory approval, is typically referred to as a sponsor. The failure of a sponsor to comply with the applicable U.S. requirements at any time during the product development process, including pre-clinical testing, clinical testing, the approval process, or post-approval process, may subject a sponsor to delays in the conduct of the study, regulatory review, and approval, and/or administrative or judicial sanctions.
A sponsor seeking approval to market and distribute a new biologic in the United States generally must satisfactorily complete each of the following steps:
| · | pre-clinical laboratory tests, animal studies, and formulation studies all performed in accordance with the FDA’s Good Laboratory Practices, or GLP, regulations and standards and other applicable regulations; |
| · | completion of the manufacture, under cGMP conditions, of the drug substance and drug product that the sponsor intends to use in human clinical trials along with required analytical and stability testing; |
| · | design of a clinical protocol and submission to the FDA of an Investigational New Drug application, or IND, for human clinical testing, which must become effective before human clinical trials may begin; |
| · | approval by an independent institutional review board, or IRB, representing each clinical site before each clinical trial may be initiated; |
| · | performance of adequate and well-controlled human clinical trials to establish the safety, potency, and purity of the product candidate for each proposed indication, in accordance with current good clinical practices, or GCP; |
| · | preparation and submission to the FDA of a biologics license application, or BLA, for a biologic product requesting marketing for one or more proposed indications, including submission of detailed information on the manufacture and composition of the product in clinical development and proposed labelling; |
| · | review of the product by an FDA advisory committee, where appropriate or if applicable; |
| · | satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities, including those of third parties, at which the product, or components thereof, are produced to assess compliance with cGMP requirements and to assure that the facilities, methods, and controls are adequate to preserve the product’s identity, strength, quality, and purity; |
| · | satisfactory completion of any FDA audits of the pre-clinical studies and clinical trial sites to assure compliance with GLP, as applicable, and GCP, and the integrity of clinical data in support of the BLA; |
| · | payment of user Prescription Drug User Fee Act, or PDUFA, securing FDA approval of the BLA and licensure of the new biologic product; and |
| · | compliance with any post-approval requirements, including the potential requirement to implement a risk evaluation and mitigation strategy, or REMS, and any post-approval studies or other post-marketing commitments required by the FDA. |
Pre-clinical Studies
Before testing any biologic product candidate in humans, the product candidate must undergo pre-clinical testing. Pre-clinical tests include laboratory evaluations of product chemistry, formulation and stability, as well as studies to evaluate the potential for efficacy and toxicity in animal studies. These studies are generally referred to as IND-enabling studies. The conduct of the pre-clinical tests and formulation of the compounds for testing must comply with federal regulations and requirements, including GLP regulations and standards and the United States Department of Agriculture’s Animal Welfare Act, if applicable. The results of the pre-clinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND application.
Investigational New Drug Application
An IND is an exemption from the FDCA that allows an unapproved product candidate to be shipped in interstate commerce for use in an investigational clinical trial and a request for FDA authorization to administer such investigational product to humans. The IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about the product or conduct of the proposed clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In that case, the IND sponsor and the FDA must resolve any outstanding FDA concerns before the clinical trials can begin or recommence. As a result, submission of the IND may result in the FDA not allowing the trials to commence or allowing the trial to commence on the terms originally specified by the sponsor in the IND.
If the FDA raises concerns or questions either during this initial 30-day period, or at any time during the IND process, it may choose to impose a partial or complete clinical hold. Clinical holds are imposed by the FDA whenever there is concern for patient safety and may be a result of new data, findings, or developments in clinical, pre-clinical, and/or chemistry, manufacturing, and controls. This order issued by the FDA would delay either a proposed clinical trial or cause suspension of an ongoing trial, until all outstanding concerns have been adequately addressed and the FDA has notified the company that investigations may proceed. This could cause significant delays or difficulties in completing Carisma’s planned clinical trial or future clinical trials in a timely manner.
Following the issuance of a clinical hold or partial clinical hold, a clinical investigation may only resume once the FDA has notified the sponsor that the investigation may proceed. The FDA will base that determination on information provided by the sponsor correcting the deficiencies previously cited or otherwise satisfying the FDA that the investigation can proceed or recommence. Occasionally, clinical holds are imposed due to manufacturing issues that may present safety issues for the clinical study subjects.
Expanded Access to an Investigational Drug for Treatment Use
Expanded access, sometimes called “compassionate use,” is the use of investigational products outside of clinical trials to treat patients with serious or immediately life-threatening diseases or conditions when there are no comparable or satisfactory alternative treatment options. The rules and regulations related to expanded access are intended to improve access to investigational products for patients who may benefit from investigational therapies. FDA regulations allow access to investigational products under an IND by the company or the treating physician for treatment purposes on a case-by-case basis for: individual patients (single-patient IND applications for treatment in emergency settings and non-emergency settings); intermediate-size patient populations; and larger populations for use of the investigational product under a treatment protocol or treatment IND application.
When considering an IND application for expanded access to an investigational product with the purpose of treating a patient or a group of patients, the sponsor and treating physicians or investigators will determine suitability when all of the following criteria apply: patient(s) have a serious or immediately life-threatening disease or condition, and there is no comparable or satisfactory alternative therapy to diagnose, monitor, or treat the disease or condition; the potential patient benefit justifies the potential risks of the treatment and the potential risks are not unreasonable in the context or condition to be treated; and the expanded use of the investigational drug for the requested treatment will not interfere initiation, conduct, or completion of clinical investigations that could support marketing approval of the product or otherwise compromise the potential development of the product.
There is no obligation for a sponsor to make its drug products available for expanded access; however, as required by the 21st Century Cures Act, or Cures Act, passed in 2016, if a sponsor has a policy regarding how it evaluates and responds to expanded access requests, sponsors are required to make such policies publicly available upon the earlier of initiation of a Phase 2 or Phase 3 clinical trial, or 15 days after the investigational drug or biologic receives designation as a breakthrough therapy, fast track product, or regenerative medicine advanced therapy.
In addition to and separate from expanded access, on May 30, 2018, the Right to Try Act was signed into law. The law, among other things, provides a federal framework for certain patients to access certain investigational products that have completed a Phase 1 clinical trial and that are undergoing investigation for FDA approval. Under certain circumstances, eligible patients can seek treatment without enrolling in clinical trials and without obtaining FDA permission under the FDA expanded access program. There is no obligation for a manufacturer to make its investigational products available to eligible patients as a result of the Right to Try Act.
Human Clinical Trials in Support of a BLA
Clinical trials involve the administration of the investigational product candidate to healthy volunteers or patients with the disease or condition to be treated under the supervision of a qualified principal investigator in accordance with GCP requirements. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, inclusion and exclusion criteria, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND.
A sponsor who wishes to conduct a clinical trial outside the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. When a foreign clinical trial is conducted under an IND, all FDA IND requirements must be met unless waived. When a foreign clinical trial is not conducted under an IND, the sponsor must ensure that the trial complies with certain regulatory requirements, including GCP requirements, of the FDA in order to use the trial as support for an IND or application for marketing approval. The GCP requirements encompass both ethical and data integrity standards for clinical trials. The FDA’s regulations are intended to help ensure the protection of human subjects enrolled in non-IND foreign clinical trials, as well as the quality and integrity of the resulting data. They further help ensure that non-IND foreign trials are conducted in a manner comparable to that required for clinical trials in the United States.
Further, each clinical trial must be reviewed and approved by an IRB either centrally or individually at each institution at which the clinical trial will be conducted. The IRB will consider, among other things, clinical trial design, patient informed consent, ethical factors, the safety of human subjects, and the possible liability of the institution. An IRB must operate in compliance with FDA regulations. The FDA, IRB, or the clinical trial sponsor may suspend or discontinue a clinical trial at any time for various reasons, including a finding that the clinical trial is not being conducted in accordance with FDA requirements or that the participants are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive GCP rules and the requirements for informed consent.
Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board, or DSMB. This group may recommend continuation of the trial as planned, changes in trial conduct, or cessation of the trial at designated check points based on certain available data from the trial to which only the DSMB has access.
Clinical trials typically are conducted in three sequential phases, but the phases may overlap or be combined. Additional studies may be required after approval.
| · | Phase 1 clinical trials are initially conducted in a limited population to test the product candidate for safety, including adverse effects, dose tolerance, absorption, metabolism, distribution, excretion, and pharmacodynamics in healthy humans or, on occasion, in patients, such as cancer patients. |
| · | Phase 2 clinical trials are generally conducted in a limited patient population to identify possible adverse effects and safety risks, evaluate the efficacy of the product candidate for specific targeted indications and determine dose tolerance and optimal dosage. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more costly Phase 3 clinical trials. |
| · | Phase 3 clinical trials proceed if the Phase 2 clinical trials demonstrate that a dose range of the product candidate is potentially effective and has an acceptable safety profile. Phase 3 clinical trials are undertaken within an expanded patient population to further evaluate dosage, provide substantial evidence of clinical efficacy, and further test for safety in an expanded and diverse patient population at multiple, geographically dispersed clinical trial sites. A well-controlled, statistically robust Phase 3 trial may be designed to deliver the data that regulatory authorities will use to decide whether or not to approve, and, if approved, how to appropriately label a biologic; such Phase 3 studies are referred to as “pivotal.” |
In some cases, the FDA may approve a BLA for a product but require the sponsor to conduct additional clinical trials to further assess the product’s safety and effectiveness after approval. Such post-approval trials are typically referred to as Phase 4 clinical trials. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication and to document a clinical benefit in the case of biologics approved under accelerated approval regulations. If the FDA approves a product while a company has ongoing clinical trials that were not necessary for approval, a company may be able to use the data from these clinical trials to meet all or part of any Phase 4 clinical trial requirement or to request a change in the product labeling. The failure to exercise due diligence with regard to conducting Phase 4 clinical trials could result in withdrawal of approval for products.
A clinical trial may combine the elements of more than one phase and the FDA often requires more than one Phase 3 trial to support marketing approval of a product candidate. A company’s designation of a clinical trial as being of a particular phase is not necessarily indicative that the study will be sufficient to satisfy the FDA requirements of that phase because this determination cannot be made until the protocol and data have been submitted to and reviewed by the FDA. Generally, pivotal trials are Phase 3 trials, but they may be Phase 2 trials if the design provides a well-controlled and reliable assessment of clinical benefit, particularly in an area of unmet medical need.
In December 2022, with the passage of Food and Drug Omnibus Reform Act, or FDORA, Congress required sponsors to develop and submit a diversity action plan for each Phase 3 clinical trial or any other “pivotal study” of a new drug or biological product. These plans are meant to encourage the enrollment of more diverse patient populations in late-stage clinical trials of FDA-regulated products. Specifically, actions plans must include the sponsor's goals for enrollment, the underlying rationale for those goals, and an explanation of how the sponsor intends to meet them. In addition to these requirements, the legislation directs the FDA to issue new guidance on diversity action plans.
Finally, sponsors of clinical trials are required to register and disclose certain clinical trial information on a public registry (clinicaltrials.gov) maintained by the U.S. National Institutes of Health, or NIH. In particular, information related to the product, patient population, phase of investigation, study sites and investigators and other aspects of the clinical trial is made public as part of the registration of the clinical trial. The failure to submit clinical trial information to clinicaltrials.gov, as required, is a prohibited act under the FDCA with violations subject to potential civil monetary penalties of up to $10,000 for each day the violation continues. Although the FDA has historically not enforced these reporting requirements due to the Department of Health and Human Services', or HHS, long delay in issuing final implementing regulations, those regulations have now been issued and the FDA has issued several Notices of Noncompliance to manufacturers since April 2021.
Special Regulations and Guidance Governing Gene Therapy Products
The FDA has defined a gene therapy product as one that mediates its effects by transcription and/or translation of transferred genetic material and/or by integrating into the host genome and which is administered as nucleic acids, viruses, or genetically engineered microorganisms. The products may be used to modify cells in vivo or transferred to cells ex vivo prior to administration to the recipient. Within the FDA, the Center for Biologics Evaluation and Research, or CBER, regulates gene therapy products. Within CBER, the review of gene therapy and related products is consolidated in the Office of Tissues and Advance Therapies, or OTAT, and the FDA has established the Cellular, Tissue and Gene Therapies Advisory Committee to advise CBER on its reviews. In September 2022, the FDA announced retitling of OTAT to the Office of Therapeutic Products, or OTP, and elevation of OTP to a "Super Office" to meet its growing cell and gene therapy workload and new commitments under the Prescription Drug User Fee Act agreement for fiscal years 2023-2027. CBER works closely with the Local Biosafety Board, a federal advisory committee, in reviewing proposed and ongoing gene therapy protocols and engaging in a public discussion of scientific, safety, ethical, and societal issues related to those protocols. The NIH and the Recombinant DNA Advisory Committee, or RAC, a federal advisory committee, also advise the FDA on gene therapy issues and other issues related to emerging technologies. The FDA and the NIH have published guidance documents with respect to the development and submission of gene therapy protocols.
The FDA has issued various guidance documents regarding gene therapies, including final guidance documents released in January 2020 relating to chemistry, manufacturing and controls information for gene therapy INDs, gene therapies for rare diseases and gene therapies for retinal disorders, as well as draft guidance in January 2021 for Human Gene Therapy for Neurodegenerative Diseases. Although the FDA has indicated that these and other guidance documents it previously issued are not legally binding, Carisma believes that its compliance with them is likely necessary to gain approval for any gene therapy product candidate Carisma may develop. The guidance documents provide additional factors that the FDA will consider at each of the above stages of development and relate to, among other things, the proper pre-clinical assessment of gene therapies; the chemistry, manufacturing, and control information that should be included in an IND application; the proper design of tests to measure product potency in support of an IND or BLA application; and measures to observe delayed adverse effects in subjects who have been exposed to investigational gene therapies when the risk of such effects is high. Further, the FDA usually recommends that sponsors observe subjects for potential gene therapy-related delayed adverse events for a 15-year period, including a minimum of five years of annual examinations followed by 10 years of annual queries, either in person or by questionnaire.
Further, to facilitate adverse event reporting and dissemination of additional information about gene therapy trials, the FDA and the NIH established the Genetic Modification Clinical Research Information System, or GeMCRIS. Investigators and sponsors of human gene transfer trials can utilize this web-based system to report serious adverse events and to provide annual reports.
Finally, for a gene therapy product, the FDA also will not approve the product if the manufacturer is not in compliance with good tissue practices, or GTP. These standards are found in FDA regulations and guidance that govern the methods used in, and the facilities and controls used for, the manufacture of human cells, tissues, and cellular and tissue-based products, or HCT/Ps, which are human cells or tissue intended for implantation, transplant, infusion, or transfer into a human recipient. The primary intent of the GTP requirements is to ensure thatT-cell and tissue-based products are manufactured in a manner designed to prevent the introduction, transmission, and spread of communicable disease. FDA regulations also require tissue establishments to register and list their HCT/Ps with the FDA and, when applicable, to evaluate donors through screening and testing.
Pediatric Studies
Under the Pediatric Research Equity Act of 2003, or PREA, a BLA or supplement thereto must contain data that are adequate to assess the safety and effectiveness of the product for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. Sponsors must also submit pediatric study plans prior to the assessment data. Those plans must contain an outline of the proposed pediatric study or studies the sponsor plans to conduct, including study objectives and design, any deferral or waiver requests, and other information required by regulation. The sponsor, the FDA, and the FDA’s internal review committee must then review the information submitted, consult with each other, and agree upon a final plan. The FDA or the sponsor may request an amendment to the plan at any time.
The FDA may, on its own initiative or at the request of the sponsor, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. A deferral may be granted for several reasons, including a finding that the product or therapeutic candidate is ready for approval for use in adults before pediatric trials are completed. The FDA is required to send a PREA Non-Compliance letter to sponsors who have failed to submit their pediatric assessments under PREA, have failed to seek or obtain a deferral or deferral extension or have failed to request approval for a required pediatric formulation. Unless otherwise required by regulation, the pediatric data requirements do not apply to products with orphan designation, although FDA has recently taken steps to limit what it considers abuse of this statutory exemption in PREA. The FDA also maintains a list of diseases that are exempt from PREA requirements due to low prevalence of disease in the pediatric population.
Compliance with cGMP Requirements
In connection with its review of a BLA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in full compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. The PHSA emphasizes the importance of manufacturing control for products like biologics whose attributes cannot be precisely defined.
Manufacturers and others involved in the manufacture and distribution of products must also register their establishments with the FDA and certain state agencies. Both domestic and foreign manufacturing establishments must register and provide additional information to the FDA upon their initial participation in the manufacturing process. Any product manufactured by or imported from a facility that has not registered, whether foreign or domestic, is deemed misbranded under the FDCA. The PREVENT Pandemics Act, which was enacted in December 2022, clarifies that foreign drug manufacturing establishments are subject to registration and listing requirements even if a drug or biologic undergoes further manufacture, preparation, propagation, compounding, or processing at a separate establishment outside of the United States prior to being imported or offered for import into the United States.
Establishments may be subject to periodic unannounced inspections by government authorities to ensure compliance with cGMPs and other laws. Inspections must follow a “risk-based schedule” that may result in certain establishments being inspected more frequently. Manufacturers may also have to provide, on request, electronic or physical records regarding their establishments. Delaying, denying, limiting, or refusing inspection by the FDA may lead to a product being deemed to be adulterated. Changes to the manufacturing process, specifications or container closure system for an approved product are strictly regulated and often require prior FDA approval before being implemented. The FDA’s regulations also require, among other things, the investigation and correction of any deviations from cGMP and the imposition of reporting and documentation requirements upon the sponsor and any third-party manufacturers involved in producing the approved product.
Submission and Filing of a BLA
The results of product candidate development, pre-clinical testing, and clinical trials, including negative or ambiguous results as well as positive findings, are submitted to the FDA as part of a BLA requesting license to market the product. The BLA must contain extensive manufacturing information and detailed information on the composition of the product and proposed labeling as well as payment of a user fee. Under federal law, the submission of most BLAs is subject to an application user fee, which for federal fiscal year 2023 is $3,242,026 for an application requiring clinical data. The sponsor of a licensed BLA is also subject to an annual program fee, which for federal fiscal year 2023 is $393,933. Certain exceptions and waivers are available for some of these fees, such as an exception from the application fee for products with orphan designation and a waiver for certain small businesses.
Following submission of a BLA, the FDA has 60 days to conduct a preliminary review of the application and it must inform the sponsor within that period of time whether the BLA is sufficiently complete to permit substantive review. In the event that FDA determines that an application does not satisfy this standard, it will issue a Refuse to File, or RTF, determination to the sponsor. The FDA may request additional information and studies, and the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing.
Once the submission has been accepted for filing, the FDA begins an in-depth review of the application. Under the goals and policies agreed to by the FDA under the PDUFA, the FDA has ten months in which to complete its initial review of a standard application and respond to the sponsor, and six months for a priority review of the application. The FDA does not always meet its PDUFA goal dates for standard and priority BLAs. The review process may often be significantly extended by FDA requests for additional information or clarification. The review process and the PDUFA goal date may be extended by three months if the FDA requests or if the sponsor otherwise provides additional information or clarification regarding information already provided in the submission within the last three months before the PDUFA goal date.
The FDA also may inspect the sponsor and one or more clinical trial sites to assure compliance with IND and GCP requirements and the integrity of the clinical data submitted to the FDA. With passage of FDORA, Congress clarified the FDA's authority to conduct inspections by expressly permitting inspection of facilities involved in the preparation, conduct, or analysis of clinical and non-clinical studies submitted to FDA as well as other persons holding study records or involved in the study process. To ensure cGMP and GCP compliance by its employees and third-party contractors, a sponsor may incur significant expenditure of time, money and effort in the areas of training, record keeping, production and quality control.
The FDA may also refer the application to an advisory committee for review, evaluation, and recommendation as to whether the application should be approved. In particular, the FDA may refer applications for novel biologic products or biologic products that present difficult questions of safety or efficacy to an advisory committee. Typically, an advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates, and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
The FDA’s Decision on a BLA
Under the PHSA, the FDA may approve a BLA if it determines that the product is safe, pure, and potent, and the facility where the product will be manufactured meets standards designed to ensure that it continues to be safe, pure, and potent. Specifically, the FDA must determine that the expected benefits of the proposed product outweigh its potential risks to patients. This “benefit-risk” assessment is informed by the extensive body of evidence about the proposed product in the BLA. On the basis of the FDA’s evaluation of the application and accompanying information, including the results of the inspection of the manufacturing facilities and any FDA audits of pre-clinical and clinical trial sites to assure compliance with GCPs, the FDA may issue a complete response letter, or CRL, or an approval letter.
If the application is not approved, the FDA will issue a CRL, which will contain the conditions that must be met in order to secure final approval of the application and will outline recommended actions the sponsor might take to obtain approval of the application. Sponsors that receive a CRL may submit to the FDA information that represents a complete response to the issues identified by the FDA, withdraw the application or request a hearing. The FDA will not approve an application until issues identified in the CRL have been addressed. If a CRL is issued, the sponsor will have one year to respond to the deficiencies identified by the FDA, at which time the FDA can deem the application withdrawn or, in its discretion, grant the sponsor an additional six-month extension to respond. For those seeking to challenge the FDA's CRL decision, the FDA has indicated that sponsors may request a formal hearing on the CRL or they may file a request for reconsideration or a request for a formal dispute resolution.
An approval letter, on the other hand, authorizes commercial marketing of the product with specific prescribing information for specific indications. The FDA may limit the approved indication(s) for use of the product. It may also require that contraindications, warnings, or precautions be included in the product labeling. In addition, the FDA may call for post-approval studies, including Phase 4 clinical trials, to further assess the product’s efficacy and/or safety after approval. The agency may also require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms, including REMS, to help ensure that the benefits of the product outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patent registries. The FDA may prevent or limit further marketing of a product based on the results of post-market studies or surveillance programs. After approval, many types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Expedited Review Programs
The FDA is authorized to expedite the review of applications in several ways. None of these expedited programs changes the standards for approval but each may help expedite the development or approval process governing product candidates.
| · | Fast Track designation. The sponsor of a product candidate may request the FDA to designate the product for a specific indication as a Fast Track product concurrent with or after the filing of the IND. Candidate products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product candidate and the specific indication for which it is being studied. In addition to other benefits, such as the ability to have greater interactions with the FDA, the FDA may initiate review of sections of a Fast Track application before the application is complete, a process known as rolling review. |
| · | Breakthrough therapy designation. To qualify for the breakthrough therapy program, product candidates must be intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence must indicate that such product candidates may demonstrate substantial improvement on one or more clinically significant endpoints over existing therapies. The FDA will seek to ensure the sponsor of a breakthrough therapy product candidate receives intensive guidance on an efficient drug development program, intensive involvement of senior managers and experienced staff on a proactive, collaborative and cross-disciplinary review and rolling review. |
| · | Priority review. A product candidate is eligible for priority review if it treats a serious condition and, if approved, it would be a significant improvement in the safety or effectiveness of the treatment, diagnosis or prevention compared to marketed products. FDA aims to complete its review of priority review applications within six months as opposed to 10 months for standard review. |
| · | Accelerated approval. Drug or biologic products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval. Accelerated approval means that a product candidate may be approved on the basis of adequate and well controlled clinical trials establishing that the product candidate has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity and prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a drug or biologic product candidate receiving accelerated approval perform adequate and well controlled post-marketing clinical trials. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials. With passage of the FDORA in December 2022, Congress modified certain provisions governing accelerated approval of drug and biologic products. Specifically, the new legislation authorized the FDA to: require a sponsor to have its confirmatory clinical trial underway before accelerated approval is awarded, require a sponsor of a product granted accelerated approval to submit progress reports on its post-approval studies to the FDA every six months (until the study is completed); and use expedited procedures to withdraw accelerated approval of an NDA or BLA after the confirmatory trial fails to verify the product's clinical benefit. Further, the FDORA requires the FDA to publish on its website “the rationale for why a post-approval study is not appropriate or necessary” whenever it decides not to require such a study upon granting accelerated approval. |
| · | Regenerative advanced therapy. With passage of the Cures Act, Congress authorized the FDA to accelerate review and approval of products designated as regenerative advanced therapies. A product is eligible for this designation if it is a regenerative medicine therapy that is intended to treat, modify, reverse or cure a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the product candidate has the potential to address unmet medical needs for such disease or condition. The benefits of a regenerative advanced therapy designation include early interactions with the FDA to expedite development and review, benefits available to breakthrough therapies, potential eligibility for priority review and accelerated approval based on surrogate or intermediate endpoints. |
Post-Approval Regulation
If regulatory approval for marketing of a product or new indication for an existing product is obtained, the sponsor will be required to comply with all regular post-approval regulatory requirements as well as any post-approval requirements that the FDA have imposed as part of the approval process. The sponsor will be required to report certain adverse reactions and production problems to the FDA, provide updated safety and efficacy information and comply with requirements concerning advertising and promotional labeling requirements. Manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including current good manufacturing process, or cGMP, regulations, which impose certain procedural and documentation requirements upon manufacturers. Accordingly, the sponsor and its third-party manufacturers must continue to expend time, money, and effort in the areas of production and quality control to maintain compliance with cGMP regulations and other regulatory requirements.
A product may also be subject to official lot release, meaning that the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official lot release, the manufacturer must submit samples of each lot, together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot, to the FDA. The FDA may in addition perform certain confirmatory tests on lots of some products before releasing the lots for distribution. Finally, the FDA will conduct laboratory research related to the safety, purity, potency, and effectiveness of pharmaceutical products.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
| · | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| · | fines, warning letters or holds on post-approval clinical trials; |
| · | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
| · | product seizure or detention, or refusal to permit the import or export of products; or |
| · | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Products may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. In September 2021, the FDA published final regulations which describe the types of evidence that the FDA will consider in determining the intended use of a biologic product.
It may be permissible, under very specific, narrow conditions, for a manufacturer to engage in nonpromotional, non-misleading communication regarding off-label information, such as distributing scientific or medical journal information. Moreover, with passage of the Pre-Approval Information Exchange Act in December 2022, sponsors of products that have not been approved may proactively communicate to payors certain information about products in development to help expedite patient access upon product approval. Previously, such communications were permitted under FDA guidance but the new legislation explicitly provides protection to sponsors who convey certain information about products in development to payors, including unapproved uses of approved products.
If a company is found to have promoted off-label uses, it may become subject to adverse public relations and administrative and judicial enforcement by the FDA, the Department of Justice, or the Office of the Inspector General of the Department of Health and Human Services, as well as state authorities. This could subject a company to a range of penalties that could have a significant commercial impact, including civil and criminal fines and agreements that materially restrict the manner in which a company promotes or distributes drug products.
Orphan Drug Designation and Exclusivity
Orphan drug designation in the United States is designed to encourage sponsors to develop products intended for rare diseases or conditions. In the United States, a rare disease or condition is statutorily defined as a condition that affects fewer than 200,000 individuals in the United States or that affects more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available the biologic for the disease or condition will be recovered from sales of the product in the United States.
Orphan drug designation qualifies a company for tax credits and market exclusivity for seven years following the date of the product’s marketing approval if granted by the FDA. An application for designation as an orphan product can be made any time prior to the filing of an application for approval to market the product. A product becomes an orphan when it receives orphan drug designation from the Office of Orphan Products Development at the FDA based on acceptable confidential requests made under the regulatory provisions. The product must then go through the review and approval process like any other product.
A sponsor may request orphan drug designation of a previously unapproved product or new orphan indication for an already marketed product. In addition, a sponsor of a product that is otherwise the same product as an already approved orphan drug may seek and obtain orphan drug designation for the subsequent product for the same rare disease or condition if it can present a plausible hypothesis that its product may be clinically superior to the first drug. More than one sponsor may receive orphan drug designation for the same product for the same rare disease or condition, but each sponsor seeking orphan drug designation must file a complete request for designation.
If a product with orphan designation receives the first FDA approval for the disease or condition for which it has such designation or for a select indication or use within the rare disease or condition for which it was designated, the product generally will receive orphan drug exclusivity. Orphan drug exclusivity means that the FDA may not approve another sponsor’s marketing application for the same product for the same indication for seven years, except in certain limited circumstances. If a product designated as an orphan drug ultimately receives marketing approval for an indication broader than what was designated in its orphan drug application, it may not be entitled to exclusivity.
The period of exclusivity begins on the date that the marketing application is approved by the FDA. Orphan drug exclusivity will not bar approval of another product under certain circumstances, including if a subsequent product with the same drug for the same condition is shown to be clinically superior to the approved product on the basis of greater efficacy or safety, or providing a major contribution to patient care, or if the company with orphan drug exclusivity is not able to meet market demand. Under Omnibus legislation enacted in December 2020, the requirement for a product to show clinical superiority applies to drugs and biologics that received orphan drug designation before enactment of the FDA Reauthorization Act of 2017, but have not yet been approved or licensed by FDA. In addition, the FDA may approve a second application for the same product for a different use or a second application for a clinically superior version of the product for the same use.
The FDA and Congress may further reevaluate the Orphan Drug Act and its regulations and policies. This may be particularly true in light of a decision from the Court of Appeals for the 11th Circuit in September 2021 finding that, for the purpose of determining the scope of exclusivity, the term “same disease or condition” means the designated “rare disease or condition” and could not be interpreted by the FDA to mean the “indication or use.” Although there have been legislative proposals to overrule this decision, they have not been enacted into law. On January 23, 2023, the FDA announced that, in matters beyond the scope of that court order, the FDA will continue to apply its existing regulations tying orphan-drug exclusivity to the uses or indications for which the orphan drug was approved.
Pediatric Exclusivity
Pediatric exclusivity is another type of non-patent exclusivity in the United States and for biologics, if granted, provides for the attachment of an additional six months of regulatory exclusivity to the term of any existing regulatory exclusivity, including orphan exclusivity. This six-month exclusivity may be granted if a BLA sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity that cover the product are extended by six months.
Biosimilars and Exclusivity
The 2010 Patient Protection and Affordable Care Act, which was signed into law in March 2010, included a subtitle called the BPCIA. The BPCIA established a regulatory scheme authorizing the FDA to approve biosimilars and interchangeable biosimilars. A biosimilar is a biological product that is highly similar to an existing FDA-licensed “reference product.” As of January 1, 2021, the FDA has approved 29 biosimilar products for use in the United States. To date, the FDA has approved a number of biosimilars, and the first interchangeable biosimilar product was approved on July 30, 2021, and a second product previously approved as a biosimilar was designated as interchangeable in October 2021. The FDA has also issued numerous guidance documents outlining its approach to reviewing and licensing biosimilars and interchangeable biosimilars under the PHSA, including a draft guidance issued in November 2020 that seeks to provide additional clarity to manufacturers of interchangeable biosimilars.
Under the BPCIA, a manufacturer may submit an application for licensure of a biologic product that is “biosimilar to” or “interchangeable with” a previously approved biological product or “reference product.” In order for the FDA to approve a biosimilar product, it must find that there are no clinically meaningful differences between the reference product and proposed biosimilar product in terms of safety, purity, and potency. For the FDA to approve a biosimilar product as interchangeable with a reference product, the agency must find that the biosimilar product can be expected to produce the same clinical results as the reference product, and (for products administered multiple times) that the biologic and the reference biologic may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. In December 2022, Congress clarified through the FDORA that the FDA may approve multiple first interchangeable biosimilar biological products so long as the products are all approved on the first day on which such a product is approved as interchangeable with the reference product.
Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date of approval of the reference product. The FDA may not approve a biosimilar product until 12 years from the date on which the reference product was approved. Even if a product is considered to be a reference product eligible for exclusivity, another company could market a competing version of that product if the FDA approves a full BLA for such product containing the sponsor’s own pre-clinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity, and potency of their product. The BPCIA also created certain exclusivity periods for biosimilars approved as interchangeable products. There have been recent government proposals to reduce the 12-year reference product exclusivity period, but none has been enacted to date. At the same time, since passage of the BPCIA, many states have passed laws or amendments to laws, which address pharmacy practices involving biosimilar products.
Federal and State Data Privacy and Security Laws
Under HIPAA, the U.S. Department of Health and Human Services has issued regulations to protect the privacy and security of protected health information used or disclosed by covered entities including certain healthcare providers, health plans, and healthcare clearinghouses. HIPAA also regulates standardization of data content, codes, and formats used in healthcare transactions and standardization of identifiers for health plans and providers. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and their regulations, including the omnibus final rule published on January 25, 2013, also imposes certain obligations on the business associates of covered entities that obtain protected health information in providing services to or on behalf of covered entities. In addition to federal privacy regulations, there are a number of state laws governing confidentiality and security of health information that are applicable to Carisma’s business. In addition to possible federal civil and criminal penalties for HIPAA violations, state attorneys general are authorized to file civil actions for damages or injunctions in federal courts to enforce HIPAA and seek attorney’s fees and costs associated with pursuing federal civil actions. Accordingly, state attorneys general (along with private plaintiffs) have brought civil actions seeking injunctions and damages resulting from alleged violations of HIPAA’s privacy and security rules. New laws and regulations governing privacy and security may be adopted in the future as well.
Additionally, California recently enacted legislation that has been dubbed the first “GDPR-like” law in the United States, the California Consumer Privacy Act, or CCPA. The CCPA creates new individual privacy rights for consumers (as that word is broadly defined in the law) and places increased privacy and security obligations on entities handling personal data of consumers or households. The CCPA went into effect on January 1, 2020 and requires covered companies to provide new disclosures to California consumers, provide such consumers new ways to opt-out of certain sales of personal information, and allow for a new cause of action for data breaches. The CCPA could impact Carisma’s business activities depending on how it is interpreted and exemplifies the vulnerability of Carisma’s business to not only cyber threats but also the evolving regulatory environment related to personal data and protected health information.
In November 2020, California voters passed a ballot initiative for the California Privacy Rights Act, or the CPRA, which went into effect on January 1, 2023, and significantly expanded the CCPA to incorporate additional GDPR-like provisions including requiring that the use, retention, and sharing of personal information of California residents be reasonably necessary and proportionate to the purposes of collection or processing, granting additional protections for sensitive personal information, and requiring greater disclosures related to notice to residents regarding retention of information. The CPRA also created a new enforcement agency - the California Privacy Protection Agency - whose sole responsibility is to enforce the CPRA, which will further increase compliance risk. The provisions in the CPRA may apply to some of our business activities. In addition, other states, including Virginia, Colorado, Utah, and Connecticut, already have passed state privacy laws. Virginia's privacy law also went into effect on January 1, 2023, and the laws in the other three states will go into effect later in the year. Other states will be considering these laws in the future, and Congress has also been debating passing a federal privacy law. These laws may impact our business activities, including our identification of research subjects, relationships with business partners and ultimately the marketing and distribution of our products.
Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available under such laws, it is possible that some of Carisma’s current or future business activities, including certain clinical research, sales, and marketing practices and the provision of certain items and services to Carisma’s customers, could be subject to challenge under one or more of such privacy and data security laws. The heightening compliance environment and the need to build and maintain robust and secure systems to comply with different privacy compliance and/or reporting requirements in multiple jurisdictions could increase the possibility that a healthcare company may fail to comply fully with one or more of these requirements. If Carisma’s operations are found to be in violation of any of the privacy or data security laws or regulations described above that are applicable to Carisma, or any other laws that apply to Carisma, Carisma may be subject to penalties, including potentially significant criminal, civil, and administrative penalties, damages, fines, imprisonment, contractual damages, reputational harm, diminished profits and future earnings, additional reporting requirements, and/or oversight if Carisma becomes subject to a consent decree or similar agreement to resolve allegations of non-compliance with these laws, and the curtailment or restructuring of Carisma’s operations, any of which could adversely affect Carisma’s ability to operate its business and its results of operations. To the extent that any of Carisma’s product candidates, once approved, are sold in a foreign country, Carisma may be subject to similar foreign laws.
Patent Term Restoration and Extension
In the United States, a patent claiming a new biologic product, its method of use or its method of manufacture may be eligible for a limited patent term extension under the Hatch-Waxman Act, which permits a patent extension of up to five years for patent term lost during product development and FDA regulatory review. Assuming grant of the patent for which the extension is sought, the restoration period for a patent covering a product is typically one-half the time between the effective date of the IND clearing clinical studies and the submission date of the BLA, plus the time between the submission date of the BLA and the ultimate approval date. Patent term restoration cannot be used to extend the remaining term of a patent past a total of 14 years from the product’s approval date in the United States. Only one patent applicable to an approved product is eligible for the extension, and the application for the extension must be submitted prior to the expiration of the patent for which extension is sought. A patent that covers multiple products for which approval is sought can only be extended in connection with one of the approvals. The USPTO reviews and approves the application for any patent term extension in consultation with the FDA.
FDA Approval of Companion Diagnostics
In August 2014, the FDA issued final guidance clarifying the requirements that will apply to approval of therapeutic products and in vitro companion diagnostics. According to the guidance, for novel drugs, a companion diagnostic device and its corresponding therapeutic should be approved or cleared contemporaneously by the FDA for the use indicated in the therapeutic product’s labeling. Approval or clearance of the companion diagnostic device will ensure that the device has been adequately evaluated and has adequate performance characteristics in the intended population. In July 2016, the FDA issued a draft guidance intended to assist sponsors of the drug therapeutic and in vitro companion diagnostic device on issues related to co-development of the products. The 2014 guidance also explains that a companion diagnostic device used to make treatment decisions in clinical trials of a biologic product candidate generally will be considered an investigational device, unless it is employed for an intended use for which the device is already approved or cleared. If used to make critical treatment decisions, such as patient selection, the diagnostic device generally will be considered a significant risk device under the FDA’s Investigational Device Exemption, or IDE, regulations. Thus, the sponsor of the diagnostic device will be required to comply with the IDE regulations. According to the guidance, if a diagnostic device and a product are to be studied together to support their respective approvals, both products can be studied in the same investigational study, if the study meets both the requirements of the IDE regulations and the IND regulations. The guidance provides that depending on the details of the study plan and subjects, a sponsor may seek to submit an IND alone, or both an IND and an IDE.
In April 2020, the FDA issued additional guidance which describes considerations for the development and labeling of companion diagnostic devices to support the indicated uses of multiple biological oncology products, when appropriate. The 2020 guidance expands on the policy statement in the 2014 guidance by recommending that companion diagnostic developers consider a number of factors when determining whether their test could be developed, or the labeling for approved companion diagnostics could be revised through a supplement, to support a broader labeling claim such as use with a specific group of oncology therapeutic products (rather than listing an individual therapeutic product(s)).
Under the FDCA, in vitro diagnostics, including companion diagnostics, are regulated as medical devices. In the United States, the FDCA and its implementing regulations, and other federal and state statutes and regulations govern, among other things, medical device design and development, pre-clinical and clinical testing, premarket clearance or approval, registration and listing, manufacturing, labeling, storage, advertising and promotion, sales and distribution, export and import, and post market surveillance. Unless an exemption applies, diagnostic tests require marketing clearance or approval from the FDA prior to commercial distribution.
The FDA previously has required in vitro companion diagnostics intended to select the patients who will respond to the product candidate to obtain pre-market approval, or PMA, simultaneously with approval of the therapeutic product candidate. The PMA process, including the gathering of clinical and pre-clinical data and the submission to and review by the FDA, can take several years or longer. It involves a rigorous premarket review during which the sponsor must prepare and provide the FDA with reasonable assurance of the device’s safety and effectiveness and information about the device and its components regarding, among other things, device design, manufacturing and labeling. PMA applications are subject to an application fee. For federal fiscal year 2023, the standard fee is $441,547 and the small business fee is $110,387.
Coverage, Pricing, and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any product candidates for which Carisma may seek regulatory approval by the FDA or other government authorities. In the United States and markets in other countries, patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Patients are unlikely to use any product candidates Carisma may develop unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of such product candidates. Even if any product candidates Carisma may develop are approved, sales of such product candidates will depend, in part, on the extent to which third-party payors, including government health programs in the United States such as Medicare and Medicaid, commercial health insurers, and managed care organizations, provide coverage, and establish adequate reimbursement levels for, such product candidates. The process for determining whether a payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors are increasingly challenging the prices charged, examining the medical necessity, and reviewing the cost-effectiveness of medical products and services and imposing controls to manage costs. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the approved products for a particular indication.
In order to secure coverage and reimbursement for any product that might be approved for sale, a company may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product, in addition to the costs required to obtain FDA or other comparable marketing approvals. Nonetheless, product candidates may not be considered medically necessary or cost effective. A decision by a third-party payor not to cover any product candidates Carisma may develop could reduce physician utilization of such product candidates once approved and have a material adverse effect on Carisma’s sales, results of operations and financial condition. Additionally, a payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage and reimbursement for the product, and the level of coverage and reimbursement can differ significantly from payor to payor. Third-party reimbursement and coverage may not be available to enable Carisma to maintain price levels sufficient to realize an appropriate return on Carisma’s investment in product development. In addition, any companion diagnostic tests require coverage and reimbursement separate and apart from the coverage and reimbursement for their companion pharmaceutical or biological products. Similar challenges to obtaining coverage and reimbursement, applicable to pharmaceutical or biological products, will apply to any companion diagnostics.
The containment of healthcare costs also has become a priority of federal, state and foreign governments and the prices of pharmaceuticals have been a focus in this effort. Governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement, and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit a company’s revenue generated from the sale of any approved products. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which a company or its collaborators receive marketing approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Outside the United States, ensuring adequate coverage and payment for any product candidates Carisma may develop will face challenges. Pricing of prescription pharmaceuticals is subject to governmental control in many countries. Pricing negotiations with governmental authorities can extend well beyond the receipt of regulatory marketing approval for a product and may require Carisma to conduct a clinical trial that compares the cost effectiveness of any product candidates Carisma may develop to other available therapies. The conduct of such a clinical trial could be expensive and result in delays in Carisma’s commercialization efforts.
In the European Union, pricing and reimbursement schemes vary widely from country to country. Some countries provide that products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular product candidate to currently available therapies (so called health technology assessments) in order to obtain reimbursement or pricing approval. For example, the European Union provides options for its member states to restrict the range of products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. European Union member states may approve a specific price for a product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the product on the market. Other member states allow companies to fix their own prices for products but monitor and control prescription volumes and issue guidance to physicians to limit prescriptions. Recently, many countries in the European Union have increased the amount of discounts required on pharmaceuticals and these efforts could continue as countries attempt to manage healthcare expenditures, especially in light of the severe fiscal and debt crises experienced by many countries in the European Union. The downward pressure on healthcare costs in general, particularly prescription products, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. Political, economic, and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various European Union member states, and parallel trade (arbitrage between low-priced and high-priced member states), can further reduce prices. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of Carisma’s products, if approved in those countries.
Healthcare Law and Regulation
Healthcare providers and third-party payors play a primary role in the recommendation and prescription of pharmaceutical products that are granted marketing approval. Arrangements with providers, consultants, third-party payors, and customers are subject to broadly applicable fraud and abuse, anti-kickback, false claims laws, reporting of payments to physicians and teaching physicians and patient privacy laws and regulations and other healthcare laws and regulations that may constrain Carisma’s business and/or financial arrangements. Restrictions under applicable federal and state healthcare laws and regulations, include the following:
| · | the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, paying, receiving, or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid; |
| · | the federal civil and criminal false claims laws, including the civil False Claims Act, and civil monetary penalties laws, which prohibit individuals or entities from, among other things, knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false, fictitious, or fraudulent or knowingly making, using, or causing to made or used a false record or statement to avoid, decrease, or conceal an obligation to pay money to the federal government; |
| · | the Foreign Corrupt Practices Act, which prohibits companies and their intermediaries from making, or offering or promising to make improper payments to non-U.S. officials for the purpose of obtaining or retaining business or otherwise seeking favorable treatment; and |
| · | the federal transparency requirements under the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the ACA, which require certain manufacturers of drugs, devices, biologics and medical supplies to report annually to the Centers for Medicare & Medicaid Services, or CMS, within the U.S. Department of Health and Human Services, information related to payments and other transfers of value made by that entity to physicians, other healthcare providers, and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. |
Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring pharmaceutical manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures. In addition, certain state and local laws require drug manufacturers to register pharmaceutical sales representatives in the jurisdiction. State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
If Carisma’s operations are found to be in violation of any of these laws or any other governmental regulations that may apply to Carisma, Carisma may be subject to significant civil, criminal, and administrative penalties, damages, fines, disgorgement, exclusion from government funded healthcare programs, such as Medicare and Medicaid, integrity oversight and reporting obligations, and the curtailment or restructuring of Carisma’s operations.
Healthcare Reform
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. There have been a number of federal and state proposals during the last few years regarding the pricing of pharmaceutical and biopharmaceutical products, limiting coverage and reimbursement for drugs and other medical products, government control and other changes to the healthcare system in the United States.
In March 2010, the United States Congress enacted the ACA, which, among other things, includes changes to the coverage and payment for products under government healthcare programs. Other legislative changes have been proposed and adopted in the United States since the ACA was enacted. For example, in August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2012 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of up to 2% per fiscal year, which will remain in effect through 2031 pursuant to the Coronavirus Aid, Relief and Economic Security Act or CARES Act. The American Taxpayer Relief Act of 2012, which was enacted in January 2013, among other things, further reduced Medicare payments to several providers, including hospitals, imaging centers, and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Under current legislation, the actual reductions in Medicare payments may vary up to 4%. Further, with passage of the Inflation Reduction Act, or the IRA, in August 2022, Congress extended the expansion of ACA premium tax credits through 2025. Those subsidies were originally extended through 2022 under the American Rescue Plan Act of 2021. These laws may result in additional reductions in Medicare and other healthcare funding and otherwise affect the prices Carisma may obtain for any of Carisma’s product candidates for which Carisma may obtain regulatory approval or the frequency with which any such product candidate is prescribed or used.
Since enactment of the ACA, there have been, and continue to be, numerous legal challenges and Congressional actions to repeal and replace provisions of the law. For example, with enactment of the Tax Cuts and Jobs Act of 2017, or the TCJA, which was signed by President Trump on December 22, 2017, Congress repealed the “individual mandate.” The repeal of this provision, which requires most Americans to carry a minimal level of health insurance, became effective in 2019 Further, on December 14, 2018, a U.S. District Court judge in the Northern District of Texas ruled that the individual mandate portion of the PPACA is an essential and inseverable feature of the ACA and therefore because the mandate was repealed as part of the Tax Act, the remaining provisions of the ACA are invalid as well. The U.S. Supreme Court heard this case on November 10, 2020 and, on June 17, 2021, dismissed this action after finding that the plaintiffs do not have standing to challenge the constitutionality of the ACA. Litigation and legislation over the ACA are likely to continue, with unpredictable and uncertain results.
Although the previous administration took executive actions to undermine or delay implementation of the ACA, those actions were rescinded with issuance of an Executive Order on January 28, 2021, by President Biden which directs federal agencies to reconsider rules and other policies that limit Americans’ access to health care, and consider actions that will protect and strengthen that access. Under this Executive Order, federal agencies are directed to re-examine: policies that undermine protections for people with pre-existing conditions, including complications related to COVID-19; demonstrations and waivers under Medicaid and the ACA that may reduce coverage or undermine the programs, including work requirements; policies that undermine the Health Insurance Marketplace or other markets for health insurance; policies that make it more difficult to enroll in Medicaid and the ACA; and policies that reduce affordability of coverage or financial assistance, including for dependents.
The prices of prescription pharmaceuticals have also been the subject of considerable discussion in the United States. There have been several recent U.S. congressional inquiries, as well as proposed and enacted state and federal legislation designed to, among other things, bring more transparency to pharmaceutical pricing, review the relationship between pricing and manufacturer patient programs, and reduce the costs of pharmaceuticals under Medicare and Medicaid. In 2020, President Trump issued several executive orders intended to lower the costs of prescription products and certain provisions in these orders have been incorporated into regulations. These regulations include an interim final rule implementing a most favored nation model for prices that would tie Medicare Part B payments for certain physician-administered pharmaceuticals to the lowest price paid in other economically advanced countries, effective January 1, 2021. That rule, however, has been subject to a nationwide preliminary injunction and, on December 29, 2021, CMS issued a final rule to rescind it. With issuance of this rule, CMS stated that it will explore all options to incorporate value into payments for Medicare Part B pharmaceuticals and improve beneficiaries’ access to evidence-based care.
In addition, in October 2020, HHS and the FDA published a final rule allowing states and other entities to develop a Section 804 Importation Program, or SIP, to import certain prescription products from Canada into the United States. The final rule is currently the subject of ongoing litigation, but at least six states (Vermont, Colorado, Florida, Maine, New Mexico, and New Hampshire) have passed laws allowing for the importation of products from Canada with the intent of developing SIPs for review and approval by the FDA. Further, on November 20, 2020, HHS finalized a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers, unless the price reduction is required by law. The final rule would eliminate the current safe harbor for Medicare drug rebates and create new safe harbors for beneficiary point-of-sale discounts and pharmacy benefit manager service fees. It originally was set to go into effect on January 1, 2022, but with passage of the IRA has been delayed by Congress to January 1, 2032.
More recently, on August 16, 2022, the IRA was signed into law by President Biden. The new legislation has implications for Medicare Part D, which is a program available to individuals who are entitled to Medicare Part A or enrolled in Medicare Part B to give them the option of paying a monthly premium for outpatient prescription drug coverage. Among other things, the IRA requires manufacturers of certain drugs to engage in price negotiations with Medicare (beginning in 2026), with prices that can be negotiated subject to a cap; imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation (first due in 2023); and replaces the Part D coverage gap discount program with a new discounting program (beginning in 2025). The IRA permits the Secretary of the HHS to implement many of these provisions through guidance, as opposed to regulation, for the initial years.
Specifically, with respect to price negotiations, Congress authorized Medicare to negotiate lower prices for certain costly single-source drug and biologic products that do not have competing generics or biosimilars and are reimbursed under Medicare Part B and Part D. CMS may negotiate prices for ten high-cost drugs paid for by Medicare Part D starting in 2026, followed by 15 Part D drugs in 2027, 15 Part B or Part D drugs in 2028, and 20 Part B or Part D drugs in 2029 and beyond. This provision applies to drug products that have been approved for at least 9 years and biologics that have been licensed for 13 years, but it does not apply to drugs and biologics that have been approved for a single rare disease or condition. Further, the legislation subjects drug manufacturers to civil monetary penalties and a potential excise tax for failing to comply with the legislation by offering a price that is not equal to or less than the negotiated “maximum fair price” under the law or for taking price increases that exceed inflation. The legislation also requires manufacturers to pay rebates for drugs in Medicare Part D whose price increases exceed inflation. The new law also caps Medicare out-of-pocket drug costs at an estimated $4,000 a year in 2024 and, thereafter beginning in 2025, at 2,000 a year.
On July 9, 2021, President Biden signed Executive Order 14063, which focuses on, among other things, the price of pharmaceuticals. The Order directs HHS to create a plan within 45 days to combat “excessive pricing of prescription pharmaceuticals and enhance domestic pharmaceutical supply chains, to reduce the prices paid by the federal government for such pharmaceuticals, and to address the recurrent problem of price gouging.” On September 9, 2021, HHS released its plan to reduce pharmaceutical prices. The key features of that plan are to: (a) make pharmaceutical prices more affordable and equitable for all consumers and throughout the health care system by supporting pharmaceutical price negotiations with manufacturers; (b) improve and promote competition throughout the prescription pharmaceutical industry by supporting market changes that strengthen supply chains, promote biosimilars, and increase transparency; and (c) foster scientific innovation to promote better healthcare and improve health by supporting public and private research and making sure that market incentives promote discovery of valuable and accessible new treatments.
At the state level, individual states are increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. A number of states, for example, require pharmaceutical manufacturers and other entities in the supply chain, including health carriers, pharmacy benefit managers, wholesale distributors, to disclose information about pricing of pharmaceuticals. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. These measures could reduce the ultimate demand for Carisma’s products, once approved, or put pressure on Carisma’s product pricing. Carisma expects that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for Carisma’s product candidates or additional pricing pressures.
Employees and Human Capital Resources
As of March 7, 2023, Carisma had 96 full-time employees, including a total of 34 employees with M.D. or Ph.D. degrees. Of these full-time employees, 87 are engaged in research and development activities. None of Carisma’s employees are represented by labor unions or covered by collective bargaining agreements. Carisma considers its relationship with its employees to be good.
Carisma’s human capital objectives are focused on attracting, developing, and retaining talent. Cash compensation plans and equity grants are designed to attract, retain and to motivate employees, directors, and select consultants to achieve our corporate objectives.
Facilities
Carisma’s principal facilities consist of office and laboratory space in Philadelphia, Pennsylvania. Carisma occupies approximately 4,369 square feet of office space under a lease that is expected to expire in January 2030 and approximately 3,600 square feet of laboratory space under a lease that expires in October 2023. Carisma believes that its facilities are sufficient to meet its current needs.
Legal Proceedings
Carisma is currently not a party to any material legal proceedings.