Exhibit 99.6
PROTOCOL An Open Label Escalating Study to Determine Maximum Necessary Dose (MND) of Homatropine Methylbromide needed for Palliation of Hot Flashes in Menopausal Women. March 14, 2013 Version 6.1 Eaton Scientific Systems, Inc. |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
| Sponsor: | Eaton Scientific Systems Ltd. |
Michael Borkowski, President
9595 Wilshire Blvd Suite 900
Beverly Hills, CA 90212
310.281.6923
| Study Title: | An Open Label Escalating Study to Determine Maximum Necessary (MND) Dose of Homatropine Methylbromide needed for Palliation of Hot Flashes in Menopausal Women |
| Principal Investigator: | Dr. Michael Guice |
| Study Monitor: | David Stark, DC, QME, CCRA |
6980 Eagle Ridge Road
Penngrove, CA 94951
(707) 795 3005
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
TABLE OF CONTENTS
| 5 |
| 5 |
| 6 |
| 6 |
| 7 |
| 10 |
| 11 |
| 11 |
| 11 |
| 12 |
| 13 |
| 13 |
| 14 |
| 14 |
| 15 |
| 15 |
| 16 |
| 16 |
| 16 |
| 16 |
| 16 |
| 16 |
| 17 |
| 18 |
| 19 |
| 19 |
| 19 |
| 19 |
| 20 |
| 21 |
| 22 |
| 22 |
| 22 |
| 22 |
| 22 |
| 22 |
| 23 |
| 25 |
| 25 |
| 27 |
| 27 |
| 28 |
| 28 |
| 28 |
| 28 |
| 29 |
| 30 |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
| 30 |
| 31 |
| 31 |
| 31 |
| 32 |
| 32 |
| 32 |
| 33 |
| 33 |
| 33 |
| 33 |
| 33 |
| 33 |
| 33 |
| 35 |
| 35 |
| 35 |
| 35 |
| 36 |
| 36 |
| 37 |
| 38 |
| 46 |
| 48 |
| 49 |
| 51 |
| 52 |
| 53 |
| 54 |
| 57 |
| 58 |
| 59 |
| 60 |
| 62 |
| 63 |
| 64 |
| 65 |
| 66 |
| 67 |
| 69 |
| 70 |
| 72 |
| 73 |
| 73 |
| 73 |
| 73 |
| 73 |
| 1. | Study A. is a single-center, prospective, open label, dose escalating study to determine the maximum dose of homatropine methylbromide needed to alleviate or palliate Hot Flashes in subjects who are experiencing Hot Flashes and/or night sweats. |
| 2. | Homatropine methylbromide has been used as a drug in humans for over 50 years. Currently, it is used to discourage overdose in the popular narcotic cough syrup Hycodan® and also to decrease motility in the gut. |
| 3. | Due to the long established use of homatropine methylbromide in humans both orally and intraocularly, the usual bench toxicity and animal studies will not be performed for this IND application. Instead, a single-center, prospective, open label dose escalating study to determine maximum needed dose will be used. Safety for use in human subjects has already been determined in previous studies. |
| 4. | Up to 22 subjects will be recruited from the general population in the Principal Investigator’s practice and surrounding community. Menopause Rating Scale (MRS), Greene Climacteric Rating Scale (GRS), Bother Scale questionnaires and patient diaries will be used to determine eligibility and to collect data during the study. |
| 5. | Study A. will be run in 2 stages: (1) a baseline run-in stage where subjects take no drug and record selected baseline climacteric symptoms for a duration of 7 days, and (2) a dose escalation stage where subjects take the study drug starting at the lowest dose and can escalate the drug to the next highest does if the drug is not working based on improvement of symptoms, minimization of side effect and tolerability duration 14 days. After maximum dose needed is decided then Study B can begin. |
| 6. | At screening, subjects who meet inclusion criteria will be asked to sign an informed consent form prior to any clinical trial procedures. |
| 7. | Stage 1 will consist of a 1-week no drug run-in, where baseline data will be collected on selected climacteric symptoms, menopausal rating scale, bother scale questionnaire and using Subject Diary Pages. All enrolled subjects who remain on Study A will participate in this stage. |
| 8. | After completion of Stage 1, up to 22 eligible subjects will be enrolled into Stage 2 starting at the lowest dose, and escalating the dose as needed see Section 1.4.1 for listed doses. |
| 9. | This is a dose escalating study. Subjects will follow the protocol dose escalation schedule, and will have the opportunity to increase the dose during the study based on symptoms/efficacy. |
| 10. | Depending on improvement of menopausal symptoms, each subject will have the opportunity to increase the dose of the study drug if there is no relief of menopausal symptoms. If the subject experiences any adverse reactions, the subject will contact the PI or study nurse they will give the subject instructions. Typically, if the subject has an adverse reaction, the PI or study nurse will lower the study drug dose to the dose listed before the adverse reactions occurred and instruct the subject to stay at the does until the end of the study. However if the subject continues to experience adverse reactions the subject can move to the next lower dose again until the adverse reactions have subsided. |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
| 11. | Data evaluations of symptoms and side effects will be made at each dose level. Each subject is compared to themselves at baseline run-in and dose of drug independent of the other subjects. After Stage 2 has resulted in a determination of the maximum needed dose (MND), Study B can be started (separate protocol and Trial). |
| 12. | Blood samples will be obtained during the screening visit (and again at the last visit) for CBC, lipid panel, hepatic profile, and serum hormone levels including FSH, Estradiol, and Total and Free testosterone. Each patient will record their symptoms and adverse events during the dose escalation part of the study to evaluate their symptoms and a review of side effects will be made. At that time the subject will either be asked to stay at the current dose of the drug or they will increase the dose. There will be approximately 3 office visits for stage 2. In the event of an adverse event, the subject should call the office immediately and speak with the study nurse or PI. The subject will be instructed as to the course of action to take. |
| 13. | Blood tests for hormone levels will be conducted at the first visit (baseline), and at the end of the study. |
| 14. | Subjects will be asked to complete self-reporting questionnaires (MRS, GCS, and Bother scale questionnaires) at baseline, during the study and at the end of Study A Stage 1 and 2. In addition subjects will be asked to complete subject diaries. |
| 15. | Subjects will be asked to log the number of episodes of Hot Flashes and/or night sweats and other subjective symptoms on a daily basis, using the Subject Diary Pages. |
| 16. | After completion of Study A an analysis will be completed to determine maximum needed dose to improve Hot Flashes or night sweats. |
Hot Flashes and night sweats are common and salient symptoms experienced by menopausal woman that typically occur during the transition time from peri-menopause to menopause. They can continue to occur to up to 5 years post-menopause (75% of women experience Hot Flashes, and of those, 25% experience them for more than 5 years)1. The “hot flush” is a result of sudden or acute drop in estrogen levels. This sudden drop can be due to natural events (menopause), or as a result of surgical (oophorectomy) or medical (hormone therapy, chemotherapy) removal of ovarian function. It is estimated that 80% of women undergoing natural or iatrogenic menopause experience Hot Flashes. Over time, the frequency and intensity of Hot Flashes do diminish, but they are still present in up to 50% of women for up to 5 years.
A hot flush is subjectively described as a sensation of intense warmth lasting as little as 30 seconds or as long as 5 minutes. It can be accompanied by tachycardia or palpitations, headache, faintness, or vertigo, and typically ends in profuse sweating and a cold sensation. At night, the frequency and severity of Hot Flashes increase, affecting a woman’s sleep and ultimately her overall quality of life.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Estrogen therapy has been the mainstay of treatment for menopausal symptoms over the years, but concerns about the risks of hormone replacement therapy have made the search for alternative therapies critical for many women.
Mild symptoms may be improved by avoidance of triggering substances or situations. Caffeine, alcohol, spicy foods, and hot beverages may trigger Hot Flashes. Exercise, lowering stress levels, and smoking cessation are thought to help relieve the symptoms. Dressing in layers, wearing breathable clothing such as cotton and natural linen, and using fans, may also aid in comfort.
Studies investigating the benefits of dietary soy and vitamin E have mixed results as to the benefits on Hot Flashes2. There is up to a 40% placebo effect in most studies on Hot Flashes, and randomized, blinded studies involving phytoestrogens derived from soy or red leaf clover do not show additional benefit3. There is a concern that phytoestrogens are not risk-free as they stimulate cellular activity in breast cysts and can contribute to postmenopausal bleeding.
Black cohosh is an herbal supplement, which has been shown to have benefit for Hot Flashes without altering FSH levels or endometrial thickness3. Side effects such as nausea, headaches, dizziness and liver toxicity have been reported3. A black cohosh/St John’s Wort combination reduced the Menopause rating scale by 50% and the Hamilton Depression Scale by 41%4.
Many other herbal remedies have been proposed, such as dong quai, evening primrose oil, chaste tree, wild yam and ginseng, but placebo-controlled studies have not supported an efficacy3.
There are no prescription medications that are as effective as estrogen for the treatment of Hot Flashes, but many have a positive impact in some women. Unfortunately, all have side effects that balance their clinical use. Venlafaxine, paroxetine, and fluoxetine have all shown an approximate 60% reduction in Hot Flashes but with side effects common to the SSRIs. Veralipride reduces Hot Flashes but caused weight gain and galactorrhea.
Clonidine has a 50% reduction in symptoms, but causes dry mouth, sedation and hypotension. Gabapentin has a 45% reduction in frequency, and 54% decrease in severity but can cause somnolence, fatigue, tremors, nausea, edema and ataxia. High dose progestins or megace may help, but can cause PMS symptoms, depression and fluid retention. Bellergal was used in the past, but has addictive potential5.
The Women’s Health Initiative (WHI) was the largest placebo controlled study of hormone replacement therapy to date, and showed an increase in thromboembolism, stroke, and breast cancer in the estrogen-progestin group, leaving many women with the uncomfortable feeling that they were compromising their long-term health by using hormone therapy. Most women trying to find symptomatic relief from debilitating symptoms would welcome any low-risk alternative therapy.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
In certain severe cases, a physician prescribes medication. Progestins such as megestrol acetate (Megace) have been prescribed. The hormone estrogen is the most effective treatment for Hot Flashes. It can help not only in this aspect, but also in lubricating the vagina and urinary tract, improving sexual function, and decreasing and preventing the incidence of urinary tract infections.
However, drawbacks of standard estrogen replacement therapy include the potential for increased risk of breast cancer, cardiovascular disease, general discomfort, and ineffectiveness. Recent studies have linked hormone replacement therapy to an increase risk of breast cancer. Furthermore, not all women can or want to take HRT, depending on their medical history and family history. Documenting the effectiveness of alternative treatments and the development of new, non-hormonal treatments with low incidence of side effects and lower costs are desirable1-5. At present, other than SSRI’s which are marginally effective, there are no pharmaceutical or over the counter products that offer effective palliation of Hot Flashes.
Male hormones (androgens) have been studied as a supplement for estrogen replacement. It is known that with oral administration of exogenous estrogen, sex hormone binding globulin (SHBG) increases; thereby binding the free and bioavailable testosterone and estrogen. This then lowers the amount of bioavailable estrogen and androgen the body can use, and can exacerbate symptoms of menopause. The addition of androgen therapy can better control the symptoms and also allow better symptom control with lower doses of estrogen treatment.
The Food and Drug Administration has approved hormone replacement therapy as treatments of Hot Flashes. In addition to estrogen replacement therapy, addition of androgens has been shown to have a potentiating effect with estrogen resulting in greater relief of Hot Flashes21. Side effects of androgen therapy are hirsuitism, acne, aggression, and erythrocytosis. Progestin therapy has also been shown to reduce Hot Flashes22. Medroxyprogesterone acetate given at 150 mg intramuscular injections every three months has been shown in one study to improve symptoms by 90-100%.
Other medications, which may reduce Hot Flashes, include Clonidine (0.05-0.4 mg/day), B-blockers, and bellergal. Clonidine is an a-alpha adrenergic receptor agonist who is often used for Hot Flashes but it has several side effects including dry mouth, insomnia, headache, nausea, fatigue, and depression23. Propranolol is a peripherally and centrally acting b-blocker with reports of a 70% decrease in the number of Hot Flashes episodes24. Finally, bellergal is a combination of Phenobarbital, belladonna, and ergotamine tartrate that reports a 60% reduction of Hot Flashes25. It has, however, sedative effects and may interact with other medications.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Alternative medicines are also available and remain a very popular option. These therapies, however, are not regulated nor approved by the FDA. The choices include vitamins and behavioral therapy including yoga, biofeedback, and meditation. Herbal remedies are also being used which include dong quai, ginseng, black cohosh, and phytoestrogens. Native Americans have used black cohosh for years for premenstrual syndrome, dysmenorrhea, and menopausal symptoms. Numerous German studies have shown its effectiveness in treating Hot Flashes26. Dong quai appears to be less effective. A placebo-controlled trial showed it was ineffective in treating Hot Flashes27. Phytoestrogens essentially have a weak estrogenic action and are found in many foods, including whole grains, flaxseed, rye, millet, legumes, fruits, and vegetables. The isoflavones are a particular group of phytoestrogen found in soybean, clover, and alfalfa. Finally, other known alternatives are evening primrose oil, acupuncture and acupressure.
1.2.2.1 Hot Flush Therapy Recently Approved By the FDA
Estrasorb Clinical Studies: Effects on Vasomotor Symptoms
In a 12-week randomized, placebo-controlled clinical trial, a total of 200 postmenopausal women (average age 52 + 6 years, 79% Caucasian in the Estrasorb treatment group; average age 51.8 ± 6 years, 72% Caucasian in the placebo treatment group) were assigned to receive Estrasorb (3.45 grams containing 2.5 mg of estradiol per gram) or placebo for 12 weeks duration. Estrasorb was shown to be statistically better than placebo at Weeks 4 and 12 for relief of both the frequency and severity of moderate to severe vasomotor symptoms (p-value <0.001 for Weeks 4 and 12).
Frequency results are shown in Table 1.2.A Severity results are shown in Table 1.2.B.
Table 1.2.A: Mean Number and Mean Change From Baseline in the Number of Moderate to Severe Vasomotor Symptoms Per Day (Intent-To-Treat Population) Treatment Group
| Baseline | Placebo | | Estrasorb |
| Observed value | (N = 100) | | (N = 100) |
Mean Number of Hot Flashes (SD) | | | |
| Week 4 | (N = 97) | | (N = 96) |
| Mean Number of Hot Flashes (SD) | 7.46 (6.42) | | 4.42 (5.60) |
| Mean Change from Baseline (SD) | – 5.97 (4.76) | | – 8.56 (6.19) |
| P-value vs. Placebo NA <0.001 | | | |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
| Week 12 | (N = 90) | | (N = 90) |
| Mean Number of Hot Flashes (SD) | 5.88 (6.17) | | 2.00 (3.64) |
| Mean Change from Baseline (SD) | – 7.20 (5.39) | | – 11.11 (6.84) |
| P-value vs. Placebo NA <0.001 | | | |
Table 1.2.B: Mean Change from Baseline in the Severity Score1 of Hot Flashes Per Day, Intent-to-Treat Population, Most Recent Value Carried Forward Treatment Group
| Baseline | Placebo | | Estrasorb |
| (observed value) | (N = 100) | | (N = 100) |
| Mean Severity Score per Day (SD) | 2.44 (0.37) | | 2.36 (0.36) |
| Week 4 | (N = 97) | | (N = 96) |
| Mean Severity Score per Day (SD) | 1.99 (0.81) | | 1.47 (1.03) |
| Mean Change from Baseline (SD) | - 0.45 (0.75) | | - 0.89 (1.04) |
| P-value versus Placebo NA <0.001 | | | |
| Week 12 | (N = 90) | | (N = 90) |
| Mean Severity Score per Day (SD) | 1.99 (0.98) | | 0.92 (1.00) |
| Mean Change from Baseline (SD) | -0.55 (0.91) | | -1.44 (1.04) |
| P-value versus Placebo NA <0.001 | | | |
1 = The severity score per day is determined by calculating the sum of recorded daily severity and dividing this number by the total number of Hot Flashes on that day.
The pathophysiology of how the decrease in estrogen affects the hypothalamic regulation of body temperature is largely unknown. Normally, there are peripheral temperature sensors as well as central temperature sensors in the anterior hypothalamus. The hypothalamus integrates these signals and initiates both conscious and unconscious responses. Anterior hypothalamic stimulation causes suppression of shivering and cutaneous vasodilation to lower body temperature. Posterior hypothalamic stimulation causes shivering to raise body temperature.24
There have been studies showing the presence of muscarinic receptors in the brain and more specifically the hypothalamus, Cortes et al showed that the highest densities of muscarinic cholinergic receptors were found in the striatum, olfactory tubercle and tuberal nuclei of the hypothalamus. Wess and his colleagues have found that, in normal mice, M3 receptors are abundant in the hypothalamus shown in their studies on eating and satiation. Whether or not these receptors are affected during menopause is unknown.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
According to the Greene Climacteric scale (a validated instrument used in this study), there are 21 common symptoms associated with a woman’s climacteric stage:
| o | heart beating quickly or strongly | | o | loss of interest in most things | | o | headaches |
| | | | | | | | |
| o | feeling tense or nervous | | o | feeling unhappy or depressed | | o | muscle and joint pains |
| | | | | | | | |
| o | difficulty in sleeping | | o | crying spells | | o | loss of feeling in hands and feet |
| | | | | | | | |
| o | excitability | | o | irritability | | o | breathing difficulties |
| | | | | | | | |
| o | attacks of panic | | o | feeling dizzy or faint | | o | Hot Flashes |
| | | | | | | | |
| o | difficulty in concentrating | | o | pressure or tightness in head or body | | o | sweating at night |
| | | | | | | | |
| o | feeling tired or lacking in energy | | o | parts of the body feel numb or tingling | | o | loss of interest in sex |
Homatropine methylbromide has been in use as a drug in humans for over 50 years 25. Indeed, it has been in use for so long that its value as a drug was “re-evaluated” as long ago as 1952 ibid. Homatropine methylbromide —the investigational product being studied in this protocol— should not be confused with the related chemical compound, homatropine hydrobromide (see Section 1.3.1.1).
One commercial use of homatropine methylbromide is as an ingredient in Hycodan® (a cough syrup). Homatropine was initially included to discourage over-dosage of the narcotic hydrocodone. The other ingredients in Hycodan® tablets are: calcium phosphate, dibasic, colloidal silicone dioxide, lactose, magnesium stearate, starch, and stearic acid. In Hycodan® suspension, the other ingredients are: caramel coloring, FD&C Red 40, liquid sugar, methylparaben, propylparaben, sorbitol solution and wild cherry imitation flavor.
Because homatropine has not been studied as an isolated ingredient of Hycodan®, it is difficult to identify side effects that can be wholly attributed to it. However, anecdotal reports of a common side effect that can be reasonably attributed to homatropine is dry mouth. Dry mouth is a common side effect with people who take Hycodan® Syrup. Dry mouth occurs because homatropine dries up the body. Weight gain was also reported, but this was likely due to the use of sugar to make the sorbital suspension.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
1.3.1.1 Homatropine hydrobromide
Another commercial use of homatropine is in the form of homatripine hydrobromide, which is used as an anticholinergic / mydriatric agent. It is
manufactured in Germany by Biehringer Ingelheim Pharma GmbH Pharma & Co. in Binger Str. 173, 55216 Ingelheim Am Rheim, Germany.
The molecular formula of Homatropine hydrobromide is C16 H21 NO3 HBR.
As this form of homatropine is an ophthalmic medication called "Isopto Homatropine", its known side effects are mostly related to the ophthalmic use such as: temporary drowsiness or blurred vision. However rare side effects to Isopto Homatropine have also been reported, such as:
”problem behavior,” dry mouth, dry skin, drowsiness, hallucination, fever, low energy, abnormal manner of walking, rash, temporary redness of the face and neck, slurred speech, fast heartbeat, swelling of the abdomen, and confusion.
Homatropine hydrobromide is an anti-muscarinic which acts on the eye muscles to produce dilatation of the pupil (mydriasis), and prevents the eye from accommodating for near vision (cyclopegia). An optometrist or ophthalmologist, to facilitate examination of the inside of a patient’s eye, typically uses this substance.
1.3.1.2 Anecdotal Use of Homatropine
Homatropine methylbromide was noted anecdotally in 2005 to have a positive effect on Hot Flashes. Homatropine methylbromide is ingredient of the prescription drug Hycodan; which is an FDA-approved, prescription anti-tussive manufactured by Endo Labs, containing hydrocodone bitartrate 5 mg and homatropine methylbromide 1.5 mg in each tablet or per 5cc of the suspension. Anecdotal reports of relief of Hot Flashes from peri-menopausal and post-menopausal women using Hycodan have been self-reported. The ingredient homatropine methylbromide was then isolated by a pharmacist as the ingredient responsible for the relief of symptoms.
Homatropine in oral form is currently not commercially available. The purpose of this clinical trial is to test the short-term efficacy and safety of a new Homatropine methylbromide oral suspension to ultimately improve Hot Flashes in perimenopausal and menopausal females and provide peer-reviewable data for the dietary supplement industry.
Oral absorption of Homatropine is poor and irregular. Total absorption is about 10—25%. Homatropine is thought to undergo hepatic metabolism by enzymatic hydrolysis and is excreted through the urine and feces. However, since the study drug is in the form of a suspension, it is yet to be determined how the body will react to the drug. However, general characteristics can be assumed by studying the family of drugs from which Homatropine originates.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Homatropine is a belladonna alkaloid that has an anticholinergic effect. Anticholinergic drugs work by affecting the cholinoreceptor stimulants, which are responsible for the nicotinic and muscurinic receptors. These receptors are responsible for the nerve, heart & smooth muscle, glands & endothelium, neuromuscular & skeletal muscle, and autonomic ganglion cells. Generally speaking, overstimulation of these receptors leads to diarrhea, painful/difficult urination and retention, miosis, bradycardia, bronchorrhea, emesis, lacrimation, and salivation.
Other muscarinic symptoms include nausea, vomiting, and diarrhea, as well as eye pain, blurred or dim vision, and conjuctival injection. Nicotinic stimulation causes muscle pain, tremors, weakness, hypertension, and fasciculation’s. The use of anticholinergic drugs is generally known to lead to antimuscurinic and antinicotinic actions. For example, Anti-Cholinergic drugs are routinely given to people with urinary incontinence to prevent frequent urination.
The anticholinergics are thought to act peripherally (not in the CNS, i.e. hypothalamus) to block the muscarinic receptors located on tissues, which receive parasympathetic postganglionic nerves. One exception is the sweat glands; which receive sympathetic-cholinergic nerves (the preganglionic nerves are sympathetic in origin (leave the thoraco-lumbar segments of the spinal cord) but the postganglionic nerves release the neurotransmitter acetylcholine (instead of norepinephrine which is released from other sympathetic postganglionic nerves).
The hypothalamus is one of several brain areas that regulate the discharge rate of parasympathetic and sympathetic nerves by descending nerve fibers that synapse with either the parasympathetic preganglionic or sympathetic preganglionic nerves. It appears that the hypothalamus is the major brain area that regulates the discharge rate of the autonomic nerves (which may increase or decrease).
In its experimental form, the test article (homatropine methylbromide oral suspension) is made of: Homatropine methylbromide (1.5mg / 5cc), red food coloring, Splenda, methylparaben, propylparaben, sorbitol solution and cherry imitation flavor.
Homatropine methylbromide is 8-Azoniabicyclo [3.2.1]octane,3-[(hydroxyphenylacetyl) oxy]-8,8-dimethyl-,bromide, endo-; a white crystal or fine white crystalline powder, with a molecular weight of (370.29).
There will be no placebo for this study.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
The test article will contain the specific amount of homatropine methylbromide necessary for the study (1.5mg/5cc). As the study is a dose escalating study, then the percentages and the concentration of homatropine methylbromide will remain the same, while only the amount taken will change.
For this study, homatropine methylbromide will be supplied as an oral suspension (“study drug”). Subjects will be asked to take the study drug at the following dose levels, four times per day. Subjects are to record their intake on a daily basis.
Homatropine methylbromide and its look-alike placebo will be provided for this clinical trial by Rox-San pharmacy. Homatropine and placebo will be provided in identical packaging. The placebo’s physical appearance will be the same as homatropine. The subjects will receive either homatropine or the placebo from the Principal Investigator, who will provide and store the respective substances.
All of the oral suspensions, bottling, labeling, etc. necessary for the study will be made and provided solely by Rox-San Pharmacy.
Rox-San Pharmacy
465 N. Roxbury Drive
Beverly Hills, Ca 90210
Tel: (310) 273 – 1644
Fax: (310) 276 - 4152
www.roxsan.com
The dose cohorts, and corresponding amounts of study drug, are defined as follows in Table 1.4.A. The beginning dosage in Dose is the same concentration of homatropine methylbromide as is found in Hycodan®, an FDA approved drug that has been in use for many years. Subsequent dose escalations are designed for those patients who may need a higher dose to experience relief of symptoms. Any risks of side effects and/or toxicity will be closely monitored.
Table 1.4.A – Dose
| No. | Dosage | Amount of Study Drug* |
| Dose 1 | 1 full teaspoon as needed every 4-6 hours | 5cc = 1.5 mg 4-6x/day |
| Dose 2 | 2 full teaspoons as needed every 4-6 hours | 10cc = 3.0 mg 4-6x/day |
| Dose 3 | 3 full teaspoons as needed every 4-6 hours | 15cc = 4.5 mg 4-6x/day |
| Dose 4 | 4 full teaspoons as needed every 4-6 hours | 20cc = 6.0 mg 4-6x/day |
*In active study drug formulation only. 1 tsp = 4.92881cc. Concentration of study drug 1.5mg/5cc.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Study A. Stage 1
Run-in phase no drug. Questionnaires will be filled out and menopausal symptoms recorded.
Study A. Stage 2 Dose Levels (study drug only):
Stage 2a - 1 full teaspoon as needed every 4-6 hours
Stage 2b - 2 full teaspoons as needed every 4-6 hours
Stage 2c - 3 full teaspoons as needed every 4-6 hours
Stage 2d - 4 full teaspoons as needed every 4-6 hours
| | Modifications: | If >1 dose-limiting toxicity (e.g. adverse event) (Dry mouth and thirst. Also decreased sweating, which could lead to an increase in body temperature (especially on a hot day). |
Unlikely, but still serious: Diarrhea, slow heartbeat, vomiting, and nausea.
Rare and serious: Eye pain, blurred or dim vision, painful or difficult urination and retention, muscle pain, tremors, weakness, and high blood pressure) occurs with any dose, dosage reverts to the most recent lower level prior to occurrence of symptoms.
Study A. Stage 1 - 1 week run-in (all subjects)
Study A. Stage 2 – Toxicity & MND Determination – Study Drug only start dose 1, if no relief of menopausal symptoms and no adverse events, increase to the next higher dose.
| 1. | Stage 2 Dose 1 – 14 days @ 1 full teaspoon as needed every 4-6 hours can escalate to dose 2. |
| 2. | Stage 2 Dose 2 – 14 days @ 2 full teaspoons as needed every 4-6 hours can escalate to dose 3. |
| 3. | Stage 2 Dose 3 – 14 days @ 3 full teaspoons as needed every 4-6 hours can escalate to dose 4. |
| 4. | Stage 2 Dose 4 – 14 days @ 4 full teaspoons as needed every 4-6 hours. |
The investigator will maintain accurate records of receipt of all test articles, including dates of receipt. In addition, accurate records will be kept regarding when each test article is dispensed to each individual subject in the study, as well as how much they use during the study.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
The research staff will review with the subject the use of the assigned test article during each visit to the trial center. Reasons for departure from the prescribed regimen must also be recorded. At the conclusion of the study, a Drug Dispensing Form (see Section 8), provided for this purpose, and will be signed by the investigator. At completion of the study, to satisfy regulatory requirements regarding drug accountability and destruction, the principal investigator will return all used, unused, empty, and partially used unit-dosing packages, with dispensing records to the sponsor for final accountability, inventory, and destruction.
Storage should be at 25°C (77°F); excursions permitted to 15°-30°C (59°-86°F).
The purpose of this study is to evaluate the safety, tolerability and maximum needed dose of homatropine methylbromide in menopausal patients experiencing symptoms of Hot Flashes and/or night sweats. This data will be used to support an Investigational New Drug (IND) application to the FDA.
1. Study A. Evaluate the safety, tolerability and maximum needed dose of homatropine in an escalating dosage regimen on an as needed basis.
1. Determine the maximum needed dose of homatropine.
A. Study A.:
| 1) | The primary intent-to-treat analysis will begin with evaluation of the homatropine dose at the one and two week visits. The analysis will also compare the incidence of adverse events for Homatropine in the overall safety population, specifically the events of Vasomotor Symptoms in Med DRA Preferred Terms that include sensations of heat, sweating, nausea, and vomiting. |
Single Center
The study will be conducted at one performance site, for better control of subject safety during this study.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Prospective
Data will be prospectively collected to support an Investigative New Drug (IND) application for this use of homatropine. This is a new use for an already FDA approved drug.
Randomized
Subjects will not be randomized in Study A.
Dose Escalating
Doses will be escalated in response to improvement in Hot Flashes/and or night sweats. All subjects will start with the first dose listed in 1.4.1. The doses will be taken once every 4-6 hours as needed. If there is no improvement in symptoms, the subjects can escalate to the next dose.
Parallel Group Design
There is no parallel group design in Study A.
Study A. is a single-center, prospective, open label, dose escalating study to test safety and maximum needed dose of homatropine methylbromide in subjects who are experiencing Hot Flashes and night sweats in symptomatic menopausal women who are not currently receiving hormonal replacement therapy. A Continual Reassessment Method (CRM) design is used assuming the dose-limiting toxicity is a binary outcome (i.e., toxicity is not recorded along a continuum) and the toxicity risk is an increasing function of dose which is instrumental in the mapping of the dose-toxicity curve as an estimate of the risk associated with the actual dose. Moreover, with the CRM design, response and toxicity is defined so that they are disjointed, that is, defined so that a subject may not experience both a response and toxicity. Given this design and since this trial is not being designed to distinguish between toxicity with and without response, the subject has one of three outcomes: response, toxicity, or neither. Thus, a dose is acceptable only if both its response rate and its toxicity rate are acceptable.
The Continual Reassessment Method (CRM) method is a Bayesian dose-finding algorithm that first requires specification of a fixed target toxicity probability, qT*. A given dose is denoted by d, and the probability of response at that dose by qR(d), and the probability of toxicity at by d by qT(d). Based on prior clinical experience, the fixed standards qR* for the minimum probability of response and qT* for the maximum probability of toxicity that are appropriate for this particular trial are specified as qR* = x.xxx and qT* = x.xxx. As each successive dose cohort is treated, interim decisions are made in terms of the highest probability of response.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
A total of up to 22 patients with Hot Flashes and/or night sweats will be enrolled in Study A stages 1 and 2.
Study A Stage 1 - Before the run-in baseline period, all prospective subjects will be screened to assure they meet the study inclusion/exclusion criteria (i.e., three or more per day or approximately 21 per week Hot Flashes). These criteria will be confirmed from data collected on Subject Diary Pages (see Section 8). Subjects in this stage will be filling out subject diaries for 7 days.
Study A Stage 2 - Once found to be enrollment candidates, up to 22 subjects will be enrolled into the dose escalation stage. This stage of the trial will be conducted by treating all of the subjects, with starting with the first dose then escalating the dose if menopause symptoms do not improve see Section 1.4.1. Subjects in this stage will be taking the study drug (homatropine methylbromide oral suspension).
All patients who meet the inclusion/exclusion criteria will be enrolled into the study. The decision to escalate the drug will be based on still having symptoms and tolerating the dose.
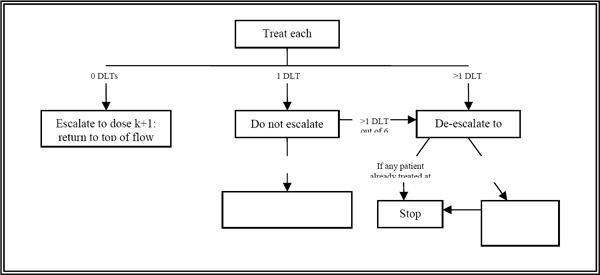
Figure 2.3. A below depicts a typical “3 + 3” dose escalation design in which the maximum needed dose (MND) is usually defined as the highest dose at which 0 or 1 DLTs are observed in six patients.
Figure 2.3.A: A standard “3 + 3” dose escalation design starting at dose k.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Typical tools used to assess the change from baseline in the frequency, duration, and interference of Hot Flashes measured on an ordinal scale (more than two categories) include adjacent-category logit models, proportional odds models, and the continuation ratio model. Due to limitations specific to the adjacent-category logit model and proportional odds model (i.e., inappropriate for repeated measures, assumes balanced data, and pre-specified fixed time points for repeated measures), the continuation ratio model seems bested suited for this particular study design. The continuation ratio model may be considered as a series of logistic regression models fitted to partitioned data. Partitioning the data allows the ordered (or hierarchical) outcomes to be viewed as a series of conditionally independent binary outcomes, each of which may be modeled via binary logistic regression. With the continuation ratio model it is assumed that the effect of the explanatory variables is the same across all partitions; then each partition is conditionally independent.
The continuation ratio model is particularly appropriate for the analysis of hierarchical data, where there is a natural baseline category (i.e., vasomotor symptoms) and where movement from one category to another denotes a shift from one state to another. Moreover, it is appropriate in situations where the response is really discrete, as opposed to course groupings of a liner scale.
Compute absolute and relative risk ratios (95% CI) of DLTs and Adverse Reactions in the Homatropine at the 1- and 2-week evaluations at Phase I and Phase II.
Covariate: Race, age, previous years since menopause, and type of menopause (i.e., natural vs. surgical). Any concomitant medications such as SSRI or other HTN medications etc.
A successful outcome is characterized by a composite endpoint composed of the clinically significant reduction in the frequency, and duration Hot Flashes as well as the interference with overall quality of life associated with Hot Flashes.
Prospective subjects will be recruited from the general population of the Los Angeles, California area by physician referral, advertising, and word-of-mouth.
The female subject will be included only if she:
The female subject will be included only if she:
| 1. | Naturally or surgically menopausal women and experiencing Hot Flashes and/or night sweats approximately 3-21/week |
| 2. | Has given written informed consent to participate in the study |
| 3. | Menopause. (Defined as FSH greater than 20, and/or absence of menses for at least 12 months prior to screening |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
| 4. | Reports episodes of Hot Flashes and/or night sweats affecting their quality of life, as determined by items 1 and 3 of the Menopause Rating Scale (see Section 8) and the Bother scale score of 7-21. |
| 5. | Is willing to report the number of episodes of Hot Flashes and /or night sweats on a daily basis |
| 6. | Must be able to successfully complete all study-related instruments, including all questionnaires |
| 7. | If, as agreed by the principal investigator, she meets all specific inclusion and exclusion study criteria |
| 8. | Has not been receiving oral, transdermal, or herbal supplements with estrogenic, androgenic, or progestagenic effects for a minimum of 30 days prior to entry into the study |
| 9. | Is not receiving hormone replacement therapy (HRT) for a minimum of 30 days prior to entry into the study |
| 10. | Is not receiving any medication to treat Hot Flashes for a minimum of 30 days prior to entry into the |
| a. | No herbal treatments or remedies |
| b. | SSRI therapy for more than 3 months due to anxiety or depression is allowed, with the Principal Investigator’s approval |
| c. | No other anticholinergic medications |
| d. | Other medications are acceptable for inclusion, with the Principal Investigator’s approval. No subjects will be asked to stop existing medications to be on this study |
Subjects will be excluded for any of the following reasons:
| 1. | Unwillingness or inability to comply with any aspect of the clinical trial protocol; and |
| 2. | Allergy to, or expresses problems with, ingredients in homatropine methylbromide; and |
| 3. | Primary glaucoma (or any family history of glaucoma), or subjects with narrow angle or close angle glaucoma; and |
| 4. | Hypersensitivity to belladonna alkaloids; and |
| 5. | Abnormal muscle weakness or myasthenia gravis; and |
| 6. | Those who have experienced or have thyrotoxicosis; and |
| 7. | Cardiovascular disease defined as: |
| a. | history of myocardial infarction, stroke, transient ischemic attack (TIA), carotid or other peripheral vascular disease, |
| b. | uncontrolled hypertension, |
| c. | a strong family history of heart attack before age 55, or |
| d. | instance of life-threatening arrhythmia within the past six months. |
| 8. | Insulin-dependent diabetes |
| 9. | Clinically significant hematological, renal or hepatic abnormalities; and |
| 10. | Active cancer, other than breast or endometrial; and |
| 11. | Pulmonary disease of any type; and |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
| 12. | Self-reported history of unrelated alcohol or controlled substance abuse within the past year; and |
| 13. | Current use of methadone, anti-coagulants, or other similar medications; and |
| 14. | Currently taking or using psychotropic drugs or trazodone: |
| a. | Subjects on SSRI’s for depression or anxiety will be allowed into the study as long as their dosage has been stable for at least three consecutive months prior to study entry; and |
| b. | This same SSRI dosage must be maintained throughout the study; and |
| c. | Subjects whose SSRI dosage is changed or discontinued during the study cannot be included in the study. |
| 15. | Severe vaginal or pelvic symptomatology; and |
| 16. | Any clinically significant abnormality from the screening physical examination or safety laboratory test results; and |
| 17. | Any medical condition, psychological condition, or social circumstance that would impair her ability to participate in the study, or who may increase the risk to herself or others by participating; and |
| 18. | ACTIVE Psychological or psychiatric therapy of depressive symptoms during the study (except for pre-existing SSRI therapy as noted); and |
| 19. | Use of any experimental (i.e., non-approved) drug within the past three months; and |
| 20. | The subject has a disease or condition that, in the principal investigator’s opinion, compromises the integrity of the clinical trial or the safety of the subject; and |
| 21. | Currently taking or using any anticholinergic medication for overactive bladder or any other condition; and |
| 22. | Has a personal history of breast or uterine cancer, or any medical condition which precludes hormone replacement therapy as a treatment option for climacteric symptoms. |
If a patient has been receiving any other medication for some period of time prior to the commencement of the study, as a matter of necessity for the treatment of a medical condition, then the medication may be permitted for the duration of the study, at the discretion of the investigator. It is the responsibility of the investigator to ensure that all changes in medication for a patient already on medication, or the commencement of medication during the study for a patient not initially on such medication at the study commencement, are recorded in full in the case report form in a manner corresponding to the entries in the patient’s medical records.
The exclusion criteria identified above are based upon general safety concerns identified with the condition, product, or disease state from recommendations made by the study physician, confounders identified by the biostatistician, or information identified in product ingredients’ research.
Subjects violating any of the inclusion/exclusion criteria will not be entered in the study.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Study subjects will be recruited from:
| | (a) referrals from women’s health and other medical offices by means of advertising brochures and flyers sent to medical providers and office staff (see Section 8), |
| | (b) the same flyer posted in the waiting and exam rooms of the principal investigator’s office, or |
| | (c) by the same advertising brochure (a.k.a. “Study Information Sheet”), which will be available throughout the principal investigator’s office, and |
| | (d) referrals by word-of-mouth. They will be screened initially by phone by the recruitment coordinator. Recruitment materials can be found in Section 8. |
Interested persons will either call the principal investigator’s office, or ask directly of the office staff about the study. Information will be provided based on a Recruitment Outline (Section 8), and the following procedures will be followed:
Interested callers will be asked for verbal consent to ask questions about their potential eligibility. A Screening & Eligibility Form (Section 8) will be started, and if the caller is determined to be potentially eligible, and expresses interest in being screened for the study, an appointment will be made and a research file started for that person.
If the caller is found to be not eligible, declines to answer questions, or decides for any other reason not to be screened for the study, they will be thanked for their time, and the Screening & Eligibility Form destroyed. A record of the phone call will be made on the Screening & Eligibility Log (Section 8).
The consent discussion will take place in a private setting, such as an examination room or private room in the investigator’s office.
The following documents may be used (depending on the site’s location and status), and are referred to in this procedure as “consent documents.”
| 1) | The study informed consent forms [Section 8] |
| i. | Separate forms for Stages 1-2 |
| 2) | The HIPAA Authorization form [Section 8] |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
| 3) | The California Experimental Subject’s Bill of Rights [Section 8] |
4.2.3 Consent Procedures:
| 1) | The investigator (or his/her authorized designee) will explain the study, its procedures, and requirements. |
| 2) | Potential subjects will be given an opportunity to read the study consent form, to have their questions answered to their satisfaction, and to bring the consent form home (if desired) to discuss study participation with significant others. |
| 3) | It will be carefully explained to subjects that they may withdraw from participation at any time, without prejudice or jeopardy to their standard medical care. |
| 4) | The subject may exercise her autonomy and choose to sign the consent form during the same visit, if so desired. This is not a requirement, but is often most convenient for the subject. |
The subject will be provided with a copy of the signed enrollment forms (in California, this would include a copy of the Experimental Subject’s Bill of Rights). Original enrollment forms shall be filed in a locked filing cabinet in the investigator’s office, as they will be the only study form that identifies the subject by name.
The subject will be asked to return to the Principal Investigator’s office for up to 4 study visits, according to the following visit schedule. (See Table 4.3.A for a complete representation of all study visits, and what will be done at each.)
| 1. | Stage 1 = One screening/enrollment visit (Visit 1) plus a baseline visit (Visit 2) to determine final eligibility. If eligible, subjects will then be enrolled into Stage 2 during the same visit. |
| a. | After the screening visit (Visit 1), vital signs will be taken, and eligible subjects will be given diaries and asked to fill them out daily. The subject will be instructed where to have blood drawn. |
| b. | At the baseline visit (Visit 2), subjects will be evaluated for compliance with the protocol, and baseline labs and examinations will be performed. Eligible subjects will then be enrolled into the study, and provided with the first does of the study drug. |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
| 2. | Stage 2 = One follow-up visit and an end of study visit (Visits 3,and 4) will be scheduled.. |
| a. | During Visits 3 subjects will be evaluated for potential adverse events, subject diaries will be collected and questionnaires will be filled out. Study drug will be dispensed. |
| b. | During visit 4 subject will be evaluated for potential adverse event, blood will be collected, subject diaries will be collected, questionnaires will be filled out and an end of study physical exam will be performed. |
| 3. | Subjects will complete self-reporting questionnaires at baseline (Visit 2), visit 3 and at the end of the study. These will consist of a Menopause Rating Scale (MRS, Section 8 and the Greene Climacteric Scale GCS, Section 8 and the bother scale) |
| | 4. | Subjects will complete daily study diary pages at home during the study, to collect information about menopausal symptoms, and possible relief of those symptoms and any side effect or adverse events. |
Table 4.3.A: Visit Schedule: Homatropine Dosing
| Procedures | Visit 1 Screening | Visit 2 Baseline | Visit 3 | Visit 4 |
| Study A | Stage 1 | Stage 2 | Stage 2 | Stage 2 |
| Basic Eligibility | X | X | | |
| Informed Consent | X | X | | |
| MRS Questionnaire | X | X | X | X |
| GCS Questionnaire | X | X | X | X |
| Bother Scale Questionnaire | X | X | X | X |
| Medical History & Eligibility Questions | X | | | |
| Dispense Study Drug | | X | X | |
| Blood Draw | X1 | | | X |
| Physical Exam | X | X | X | X |
| Review Of Rx And Non Rx Drugs | X | X | X | X |
| Turn In And Evaluate Diary Pages | | X2 | X | X |
| Hand Out Diary Pages | X | X | X | |
| Assess Subject Compliance | | X | X | X |
| Review Adverse Events | | | X | X |
| Collect Study Drug | | | X | X |
| Exit Exam | | | | X |
1The blood draw may happen on Visit 1 (screening) day or anytime before Visit 2 (baseline)
2The and of stage 1 and the beginning of stage 2 may happen on the same day. The diary pages collected on Visit 2 will be from stage 1.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
See Section 4.4 for a detailed description of all study activities.
Screening Procedures to Determine Eligibility
At Visit 1 (screening), subjects will sign an informed consent form prior to any clinical trial procedures. Study procedures will be conducted by the principal investigator (or designee). Subjects will:
| a) | Participate in a medical history, medication use, and screening assessment (See Screening & Eligibility Form, Section 8). |
| i. | Recording of basic demographic information. |
| ii. | Recording of medical history and any current drug treatment. |
| b) | Undergo a focused physical examination (see Section 8). |
| c) | Preliminary determination of eligibility based on inclusion and exclusion criteria. |
| d) | Patient questionnaires MRS, GCS and Bother scale questionnaire |
If Eligible To Be In the Study
If preliminary screening procedures result in a determination of eligibility, the following procedures will also be done at Visit 1.
Subjects will be provided with:
| e) | a 1-week supply of Stage 1 diary pages, to be completed daily; and |
| f) | an appointment for Visit 2 (1 week hence), instructions on how and when to have blood drawn, approximately 2 teaspoons, for screening laboratory tests, and detailed instructions on complying with study procedure |
If any screening or follow-up procedure during any study visit indicates that the subject is not eligible for the study while she is still in the Principal Investigator’s office, she will be thanked for her time, provided prorated compensation (see Section 5.5) i.e., $20 for completing the screening visit, and referred for continued women’s health care to her primary care physician. Particular emphasis will be made for the patient to follow-up with her personal physician if any physical abnormality is indicated during any screening or follow-up procedures.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Visit 2 will occur approximately 1 week after Visit 1 (+/- 2 days).
Procedures to Determine Compliance
After 1-week run-in period, prior to Visit 2, the subject will have blood drawn for screening laboratory tests, so that the results will be available at the time of the visit. At Visit 2, subjects will be evaluated with the 1-week run-in period by the following procedures:
| a) | Assessment of completion of diary pages; and |
| b) | Re-confirmation of consent |
Procedures to Determine Continued Eligibility
After determining that the subject remains a good candidate for the study, the following procedures will be performed as baseline measurements, and for subject safety:
| c) | Undergo a focused physical examination (review of systems). |
| d) | Assess & discuss blood results for baseline hormone panel, CBC, lipid panel, and hepatic profile; and |
| e) | Complete baseline questionnaire assessments related to eligibility (MRS, GCS and Bother scale); and |
| f) | After baseline blood result, office will contact subject and tell them how to proceed; and |
| g) | Review prescription and non-prescription drugs. |
Note: if the blood or other screening tests reveal a significant abnormality, a referral for appropriate follow-up medical care will be made, and the subject discontinued from the study. The discontinued subject or her insurance will pay for the costs for any follow-up as a result of the blood tests.
Enrollment in Dose Escalation (If Subject Remains Eligible for the Study)
If the additional screening procedures above indicate that the subject is still eligible to participate in the study, the following enrollment procedures will be done:
| h) | Review consent form with subject again, and write the first does in the space provided on the “Study Schedule and Dose Reminder” section of the consent form (see Section 8) with both subject and the Principal Investigator signing in the additional space provided |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
| i) | Dispense study medication (homatropine methylbromide oral suspension) |
| j) | Dispense a sufficient quantity of Stage 2 Subject Diary Pages, at least until the next study visit |
| k) | Make an appointment for next visit (Visit 3). Subject needs to be given directions and explained how to escalate the dose and how to record adverse events. |
Visit 3 will occur approximately 1 week after Visit 2 (+/- 1 day).
Procedures to Evaluate Subject Safety
| a) | Assess compliance with study procedures; and |
| b) | Assess adverse events; and |
| c) | Collect Subject Diary Pages and dispense additional ones as needed; and |
| d) | Collect study drug and dispense study drug; and |
| e) | Collect Questionnaires. |
If any parameter indicates that the subject has experienced, an adverse event or dose limiting toxicity (DLT), she will be contacted by telephone or in person and instructed to revert to the previous dose level and keep her next appointment. In the event that a subject experiences an adverse event they are to call the office immediately. This will be recorded on the Visit Form. If she chooses not to continue to participate at this time, she will be thanked for her time, and asked to bring all study materials back to the Principal Investigator’s office as soon as possible. There, she will be provided prorated compensation (see Section 5.5) if the subject participates in the study stage 2 and then drops out they will be given the screening amount plus the amount listed for stage 2, thanked for her participation, and referred for continued women’s health care to her primary care physician. This will be recorded on the Subject Withdrawal Form. Particular emphasis will be made for the patient to follow-up with her personal physician if any physical abnormality was indicated during any screening or follow-up procedures.
Visit 4 will occur approximately 1 week after Visit 3 (+/- 1 day).
(Same procedures as Visit 3)
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Additional Procedures
| a) | Collect any remaining study medication |
| b) | Complete Stage 2 questionnaire assessments (MRS, GCS and Bother scale) |
The following assessments and procedures will be conducted while the subject is participating in this clinical trial.
There are no randomization procedures for Study A Stage 1 or Stage 2.
| a) | Menopause Rating Scale |
The MRS is a validated questionnaire listing symptoms associated with menopause. Subjects will report on number, frequency and intensity of symptoms on this validated questionnaire. This will be administered at Visits 2, 3, and 4.
| b) | Greene Climacteric Scale |
The GCS is a validated questionnaire that looks at the total score and sub scores of the psychological, physical, and vasomotor symptoms during menopause. This will be administered at Visits 2, 3, and 4.
The menopause assessment scale is designed to indicate the severity of the symptoms you may be experiencing. These can be physical, psychological, sexual or a mixture of all three. This questionnaire will be used to document your level of “bother”. This will be administered at Visit’s 2, 3 and 4.
| | 1. | Medical History, Medications, and Screening Evaluation |
| | 2. | Hormone Panel: Tests for FSH, LH, Estradiol, Testosterone (Free and Total), DHEA, DHEAS, and SHBG. |
| § | CBC, Lipid Profile, and Hepatic Profile |
| | 4. | Abbreviated Physical Examination |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
| | 5. | Assessment of Compliance |
Compliance is defined as subjects who use 80% or more of the assigned product and complete 80% or more of the clinical trial forms and logs. This assessment will be recorded on the Study Visit Forms (see Section 8).
The primary safety variables will be adverse events encountered by the study population. Treatment group, event, and body system will tabulate the incidence of study-emergent adverse experiences.
An adverse event means any serious and unexpected adverse effect on health or safety or any life-threatening problem or death caused by, or associated with a test article, if that effect, problem, or death was not previously identified in nature, severity, or degree of incidence in the investigational plan or application (including a supplementary plan or application), or any other unanticipated serious problem associated with a test article that relates to the rights, safety, or welfare of subjects.
In addition, and adverse event could be any harm that comes to any subject as a result of participation in the study. The event could be physical harm unrelated to the test article, such as a traffic accident en route to a study appointment, or it could be non-physical harm, such as an inadvertent loss of privacy.
For this study, all serious and unanticipated or unexpected adverse device effects and adverse events shall be classified as either a “Clinical” or “Non-Clinical” adverse event, and reported by the investigator within 3 business days to the sponsor, monitor, and IRB by means of the Adverse Event Report form (see the form and further instructions in Section 8).
Important medical events that may not result in death, be life-threatening, or require hospitalization may be considered a serious adverse event when, based upon appropriate medical judgment, they may jeopardize the subject and may require medical or surgical intervention to prevent one of the outcomes mentioned above.
The sponsor will make medical treatment available at no cost to the subject in the event of a physical injury that is related to the study medications used in this study.
For more information on subject management related to adverse events, see Section 5.4.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
4.4.4.1 Adverse Event Severity Definitions
| · | Mild – The adverse event, taken as an isolated event, would cause no limitations of usual activities. |
| · | Moderate – The adverse event, taken as an isolated event, would cause some limitation of usual activities. |
| · | Severe – The adverse event, taken as an isolated event, would cause severe limitations or inability to carry out usual activities. |
4.4.4.2 Relationship to Clinical Trial Product Definitions
| · | Not Related – Unrelated to the clinical trial product consumption. |
| · | Remote – Possibility of relationship to clinical trial product consumption is remote, but can not be ruled out with certainty. |
| · | Possible – Possible relationship to the clinical trial product consumption. |
| · | Probable – Relationship to clinical trial product consumption is fairly certain. |
| · | Definite – Relationship to clinical trial product consumption is certain. |
Subjects will be permanently discontinued from the study prematurely if any of the following occurs. Data collected up to the time of discontinuation will be used in the final statistical analysis.
| · | An unexpected and serious adverse event occurs (unless the investigator and sponsor judge the adverse event to be clinically acceptable); |
| · | The patient requests to be withdrawn from the study (see Section 5.2 below); |
| · | A need for a concomitant medication prohibited by the protocol arises; |
| · | The principal investigator, study monitor, or medical monitor decides that it is in the patient’s best interest; |
| · | The patient is non-compliant with the protocol; or, |
| · | The investigator or sponsor decides to discontinue the study. |
If the subject is withdrawn due to an adverse event(s), the subject will be monitored until the adverse event has resolved or until the event is determined to be due to a stable or chronic condition or inter-current illness(s).
A Subject Withdrawal Form (see Section 8) will be completed to document the reason for subject discontinuation.
All study-related procedures will cease immediately upon discontinuation. In the event of a study-related injury, the sponsor will make arrangements for follow-up medical care at no cost to the subject. Insurance or other third-party payers would be billed as appropriate.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Adverse Events
Dry mouth and thirst. Also decreased sweating, which could lead to an increase in body temperature (especially on a hot day).
To repeat: Contact the Study Doctor if you have a fever. Homatropine is known to reduce sweat activity, which can make it harder for your body to cool itself.
Unlikely, but still serious:
Diarrhea, slow heartbeat, vomiting, and nausea.
Rare and serious:
Eye pain, blurred or dim vision, painful or difficult urination and retention, muscle pain, tremors, weakness, and high blood pressure.
In the event a subject withdraws from the clinical trial prematurely, every effort will be made to collect the assigned product container and used or unused assigned product.
A Subject Withdrawal Form will be completed to document the reason for subject withdrawal or discontinuation.
If a subject withdraws from the clinical trial, the subject’s identification number will not be reassigned.
This clinical trial may be prematurely terminated, if in the opinion of the sponsor, principal investigator, medical monitor, or study monitor there is sufficient reasonable cause. Circumstances that may warrant termination include, but are not limited to:
| · | Determination of unexpected, significant, or unacceptable risk to the subjects; |
| · | Plans to modify, suspend or discontinue the development of the study drug.. |
For a definition of what constitutes an adverse event, refer to Section 4.4.4.
In the event that a serious adverse event occurs (unless the investigator and sponsor judge the adverse event to be clinically acceptable), the subject will be immediately discontinued from the study.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
The sponsor will make medical treatment available at no cost to the subject in the event of a physical injury that is related to the test articles used in this study.
If the subject is withdrawn due to an adverse event(s), the subject will be monitored until the adverse event has resolved or until the event is determined to be due to a stable or chronic condition or inter-current illness(s).
A Subject Withdrawal Form will be completed to document the reason for subject discontinuation.
The IRB will be notified of all serious and unexpected adverse events using the Subject Withdrawal Form and forms specified by the IRB within a time frame specified by the IRB.
Subjects will be compensated according to the following prorated schedule, based upon what stage of the study procedures is completed. Financial remuneration is intended to compensate the subject for her time and travel expenses incurred during her participation in the study.
| Completion of | | | Amount: | |
| | | | | |
| Screening Procedures | (Visit 1) | | $ | 20 | |
| Study A Stage 1 | (Visit 2) | | $ | 50 | |
| Study A Stage 2 | (Visit 4) | | $ | 50 | |
| Parking Stage 1 and 2 | (Visit 2) | | $ | 30 | |
| Partial participation compensation | | Amount: | |
| | | | |
| Screening only | | $ | 50 | |
| Partial or full participation visit 1 and 2 | | $ | 50 | |
| Partial or full participation visit 4 | | $ | 50 | |
| Parking screening only | | $ | 15 | |
| Parking Partial or full any stage | | $ | 15 | |
The intent-to-treat population is defined as subjects who use at least one dose of assigned product.
The evaluable population is defined as subjects who:
| a) | use 85% or more of the assigned product and complete 85% or more of the clinical trial forms and logs. |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
| b) | Have reductions in hot flashes and/or night sweats that have been reduced to below a mild affliction as determined by the patient. |
The primary safety variables will be adverse events encountered by the study-population. Treatment group, event, and body system will tabulate the incidence of study-emergent adverse experiences. Because widespread usage of homatropine is already present, and no notable adverse effects have been noted, efficacy of efficacy of homatropine is deemed to be the primary concern.
The assessments of adverse events and research staff measurements will be compared within each individual subject not between subjects from reports and measurements at baseline, and at 3 weeks.
TBD
To be determined, based on recommendations made by the FDA during IND application.
Incidence of reports of adverse events will be summarized for subjects who consume at least one dose of their assigned product during the clinical trial. Incidence of reports of adverse events will be compared between the homatropine and placebo groups. Fisher’s exact tests will be used to compare homatropine versus placebo for numbers of subjects experiencing one or more adverse events and numbers of subjects experiencing one or more adverse events considered related to DS-FSE.
Potential confounders or effect modifiers include:
| · | baseline primary efficacy measurements |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
| · | social habits or lifestyle |
| · | number of prior treatments |
| · | use and method of birth control |
The co-morbid risk score will be used (see Table 6.3.A below). The co-morbid risk is comprised of 1 point for each of the major co-morbid factors plus points awarded for smoking, caffeine use, and alcohol consumption. The typical distribution in our clinical trials ranges from 0 to 6 or 7 points. Based on the specific distribution, 3 or 4 points is set as the classification of individuals at a higher co-morbid risk than the bulk of the distribution.
| Table 6.3.A: Co-Morbid Risk Scoring Example |
| Variable | Subject A | Subject B |
| Diabetes | 1 | 0 |
| Hypertension | 0 | 1 |
| Heart disease | 0 | 0 |
COPD1 | 0 | 1 |
| Ulcers | 0 | 0 |
| History of cancer | 0 | 0 |
Heavy smoker2 | 1 | 0 |
Heavy caffeine use3 | 1 | 0 |
Elevated alcohol use4 | 0 | 1 |
Total Risk Score5 | 3 | 3 |
1Chronic obstructive pulmonary disease. 2Smokes more than 1 pack per day (ppd). 3Consumes more than 3 cups of coffee or the caffeine equivalent per day. 4Consumes more than 4 standard alcoholic drinks per week. 5In this example both subjects have a risk score of 3 points. |
Adjustments will be made for potential confounders by one of two methods. This will include analyses of the differences of means and categorical analysis. Variables such as age, co-morbid risk score, and baseline primary efficacy measurements may be assessed against the primary end-point (a difference variable is created by subtracting the baseline value from the end-of-study value for each given parameter) by both the difference of means and by categorical analysis.
Categories will be created for each of these potential confounders. As an example, age categories will be based on five-year intervals. The intervals for co-morbid risk, medications, baseline primary efficacy measurements, duration of reduced sexual function, and number of prior treatments will depend on the distribution of the data. A potential co-morbid distribution has a range from 0 to 10 points (we typically observe a range of 0 to 7 points as stated earlier). It would be most likely that these intervals would be approximately 0 to 2 (low co-morbid risk), 3 to 5 (moderate co-morbid risk), and 6 or more (high co-morbid risk). These categories form the basis of the confounder analysis.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
The primary efficacy measurements collected at baseline and follow-up visits are:
Typically, the responses are grouped together to represent mild symptoms. Categories using the more severe responses may be grouped together based on the purpose of the analysis. These categories are used to adjust for the end-point responses.
In order to maintain subject privacy, all subject records will identify subjects by their subject identification number only. The sponsor will grant monitor(s) and auditor(s) from the United States Department of Health and Human Services (DHHS), the United States Food and Drug Administration (FDA), and the IRB access to subject records to verify data and to audit the data collection process. Subject confidentiality will be maintained and will not be made publicly available to the extent permitted by the applicable laws and regulations.
The Principal Investigator will maintain all clinical trial records according to Good Clinical Practices (GCP). Records will be retained for at least three years after the clinical trial is completed.
This study must be conducted in compliance with Institutional Review Board/ Independent Ethics Committee (IRB/IEC), informed consent regulations and ICH GCP Guidelines. In addition, all local regulatory requirements will be adhered to, in particular, those that afford greater protection to the safety of the trial participants. This study will be conducted according to the current revision of the Declaration of Helsinki (Revised Edinburgh 2000), The Common Rule and The Belmont Report, and with local laws and regulations relevant to the use of new therapeutic agents in the local area.
Before initiating a trial, the investigator/institution should have written and dated approval/favorable opinion from the IRB/IEC for the trial protocol/amendment(s), written informed consent form, consent form updates, subject recruitment procedures and materials (e.g. advertisements) and written information to be provided to subjects.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
This protocol is subject to change after review by an IRB to ensure human subject protection in clinical research. Investigators will conduct the clinical trial in compliance with the IRB-approved protocol. Modifications to the protocol will not be made without written IRB approval except when the modification is needed to eliminate an immediate hazard(s) to subjects or if the change(s) involve only logistical or administrative aspects of the clinical trial. Any departures from the IRB-approved protocol will be fully documented in the source documentation.
The investigator, or a person designated by the investigator, will explain the benefits and risks of participation in the study to each subject, subject’s legally acceptable representative or impartial witness and obtain written informed consent prior to the subject entering the study (before initiation of non-routine tests and administration of study drug).
The principal investigator and research staff will be trained in the basic elements of human subject protection. Copies of important documents governing the protection of human subjects such as The Common Rule, The Belmont Report and the Declaration of Helsinki are available to clinical trial staff and subjects upon request from by visiting the following websites:
| · | The Common Rule is available at http://ohrp.osophs.dhhs.gov/humansubjects/guidance/45cfr46.htm |
| · | The Belmont Report is available at http://ohrp.osophs.dhhs.gov/humansubjects/guidance/belmont.htm |
| · | Declaration of Helsinki is available at http://www.wma.net/e/policy/17-c_e.html |
A periodic review of procedures and clinical trial execution by a non-involved third party (the study monitor) will be conducted.
This clinical trial will be conducted in accordance with Good Clinical Practices (GCP) and the appropriate regulatory requirements. The principal investigator is thoroughly familiar with the appropriate use of the clinical trial procedures as described in the protocol. Essential clinical documents will be maintained to demonstrate the validity of the clinical trial and the integrity of the data collected. Master files will be established at the beginning of the clinical trial, maintained for the duration of the clinical trial and retained according to the appropriate regulations.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
8.1 Recruitment & Enrollment Materials
| Appendix 8.1.1a | | Consent Form for Study A Stages 1 |
| Appendix 8.1.2 | | HIPAA Authorization Form |
| Appendix 8.1.3 | | California Experimental Subject’s Bill of Rights |
| Appendix 8.1.4a | | Study Schedule & Dose Reminder for Study A Stages 1 |
| Appendix 8.1.5 | | Study Recruitment Flyer |
| Appendix 8.1.6 | | Study Information Sheet |
| Appendix 8.1.7 | | Recruitment Outline |
| Appendix 8.1.8 | | Screening & Eligibility Form |
| Appendix 8.1.9 | | Screening & Enrollment Log |
8.2 Subject Management Forms
| Appendix 8.2.1 | | Focused Physical Examination Form |
| Appendix 8.2.2 | | Medical History & Medications Assessment |
| Appendix 8.2.3 | | Study Visit Forms (1 per visit) |
| Appendix 8.2.4a | | Data Safety & Evaluation Form (Part 1) |
| Appendix 8.2.4b | | Data Safety & Evaluation Form (Part 2) |
| Appendix 8.2.5 | | Subject Contact Log |
| Appendix 8.2.6 | | Subject Withdrawal Form |
| Appendix 8.2.7 | | Adverse Event Report Form |
| Appendix 8.2.8 | | Drug Usage Chart |
| Appendix 8.2.9 | | Randomization Log |
| Appendix 8.2.10 | | Dose Escalation Log (Stage 2) |
| Appendix 8.2.11 | | Study Drug Distribution Log |
8.3 Subject Questionnaires & Forms
| Appendix 8.3.1 | | Subject Diary Pages |
8.4 References
| Appendix 8.4.1 | | Literature References |
| Appendix 8.4.2 | | CTCAE v3 Toxicity Criteria |
8.5 Investigator’s Brochure
8.6 Standard Instruments (not attached to this protocol)
| Appendix 8.6.1 | | Menopause Rating Scale (MRS) |
| Appendix 8.6.2 | | Greene Climacteric Scale (GCS) |
| Appendix 8.6.3 | | Bother Scale |
(See next page)
CONSENT TO PARTICIPATE IN RESEARCH
STUDY A. STAGES 1-2
Project Title: Study A. Open Label Dose Escalating Study to Determine Maximum Necessary Dose (MND) of Homatropine Methylbromide needed for Palliation of Hot Flashes in Menopausal Women November
Principal Investigator (“Study Doctor”):
Location:
Telephone:
Being in a research study is different than being a patient. As a patient, your doctor is focused on your care. If you agree to be a research subject, your doctor will also be acting as the “Study Doctor,” and will have to follow the rules of the study. If there is a conflict between the study rules and your medical care, the Study Doctor’s first responsibility is for your safety and welfare. This consent form describes the research study and your participation. Please read this form carefully. If you have any questions, do not hesitate to ask the study doctor. The contents of this form should also be explained to you verbally by the study doctor or nurse. |
What is the Purpose of this Study?
The purpose of this study is to look at the effectiveness and safety of a drug called “homatropine,” to see if it will provide relief symptoms of menopause (such as Hot Flashes and night sweats), and perhaps improve the quality of life in women who are past the childbearing age.
You have been asked to participate in this study because you are going through menopause, and because you would not normally get treatment for your symptoms with traditional hormone replacement therapy because of one of the following:
| 1. | A history of cancer in your breast or uterus |
| 2. | Some other form of a hormone-dependent tumor |
| 3. | You have decided that hormone replacement therapy is not for you. |
Your participation in this study is entirely voluntary. You don’t have to take part if you don’t want to.
What is Homatropine?
Homatropine methylbromide (its full name) is an existing drug that has been approved by the FDA for many years for a variety of uses. Most commonly, it is used in a prescription cough syrup called “Hycodan®” to discourage overdose. For this study, homatropine will be provided in a pink liquid that you take with a teaspoon. Its most obvious side effect is to reduce sweating, which can lead to an increased body temperature.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Contact the Study Doctor if you have a fever. Homatropine is known to reduce sweat activity, which can make it harder for your body to cool itself.
Why is Homatropine Being Studied?
Currently, women who experience Hot Flashes are given drugs (for example, Premarin®) as part of what’s called “hormone replacement” therapy. However, when a woman has had cancer of the breast or uterus (or some other hormone-dependent tumor), hormone replacement therapy is not an option.
This study will look at homatropine as an alternative medication for improvement in Hot Flashes and/or night sweats.. The researchers hope that knowledge gained from this study might allow an alternative treatment for Hot Flashes or night sweats when hormone replacement therapy is not an option, or when women do not wish to take hormones.
This study is designed to work in 2 stages:
Study A. Stage 1 – Run-in period-for 1 week, you will monitor your menopause symptoms in a diary.
Study A. Stage 2 – Those who qualify and want to continue on the study will be assigned to stage 2 to take increasing doses of homatropine, to see how safe it is and what dose is ok.
How Long Will the Whole Study Last?
It is expected that up to 22 women will participate in this study as research subjects, over a period of 3 weeks. The time that each person will participate in each stage is as follows:
Stage 1 – 1 week
Stage 2 – 2 weeks
There will be 4 office visits over the study..
What Procedures Will Be Done?
If you agree to be in this study, some things will be done right after you sign this form. You may reschedule if that is more convenient for you. There would be 1-4 study visits at this office.
A Study Schedule & Dose Reminder is attached to this form for your review and convenience.
Study A. Stage 1 Screening Procedures (Visit 1)
This visit is expected to take about 60 minutes. (You could start right now if you want to.)
At this visit you will be asked many questions and take part in several tests to see if you qualify to be in the study. These activities will include:
| · | Questions about your medical history, medications, and symptoms |
| · | A brief physical examination |
| · | Filling out three questionnaires describing your menopausal symptoms: |
| o | The MRS, or “Menopause Rating Scale,” and |
| o | The GCS, or “Greene Climacteric Scale” |
Based on the results of these exams and tests, the Principal investigator will decide whether you are eligible to begin this study. The results will be discussed with you, and if there are any abnormal findings, appropriate referrals will be made for your medical care.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Whether or not you are eligible to be in the study, you will be given $20 as compensation for your time and expenses. The following shows compensation that will be given to you for participation in the study:
| Screening (Visit) 2 | | $ | 20 | |
| Study A Stage 1 (Visit 2) | | $ | 50 | |
| Study A Stage 2 (Visit 4) | | $ | 50 | |
| Parking both stages (Visit 2) | | $ | 30 | |
If you are eligible, and still want to participate, a weeks worth of diary pages will be provided for you to write down what symptoms you feel during this time. You will also be told when and where to have blood drawn for your final eligibility tests. The blood tests will involve:
| · | Collecting two small tubes of blood (about 2 teaspoons total) from a vein in your arm for these laboratory tests: |
| o | Lipid (blood fat) panel |
An appointment for Visit 2 baseline (1 week later) will be made, all your questions will be answered, and you will be given copies of all the study consent forms. You will also be given instructions about how much drug to take, and what to do with the forms.
Begin Study A. Stage 2 (Visit 2) baseline
This visit is expected to take about 30-60 minutes.
During visit 2 you will be asked more questions to make sure you can still be in the study. These activities will include:
| · | Another brief physical examination |
| · | Filling out 2 questionnaires describing your menopausal symptoms: |
| o | The MRS, or “Menopause Rating Scale,” and |
| o | The GCS, or “Greene Climacteric Scale” |
When you are done with the questionnaires, you will be given study supplies to last until your next visit.
| · | A 1-weeks supply of homatropine |
| · | More “study diary” pages on which to record your menopause symptoms at home. |
You will start the study drug at the lowest dose 4-6 times a day as needed. If you are not experiencing any adverse events and the study drug is not relieving your menopausal symptoms, you will be asked to take the next higher dose 4-6 times a day as needed. If you experience any adverse effects, call the study doctor immediately at XXX-XXX-XXXX. The study doctor will instruct you on what to do next.
An appointment for Visit 3 (a week later 1+/- day) will be made and all your questions will be answered.
Study A. Stage 2 (Visit 3)
This visit is expected to take about 30 minutes. The following procedures will be done:
| · | A brief physical examination |
| · | Turn in any diary pages completed at home since the last study visit and receive new ones as needed |
| · | Bring in your bottle of homatropine so the researchers can record how much of the study drug you have used so far. |
| · | Fill out subject questionnaires |
You will be reminded of study procedures and how much study drug to take. Any questions you have will be answered and you will receive an appointment for the next study visit (Visit 4) (end of study visit), approximately 1 week later..
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Study A. End of stage 2 visit (Visit 4)
This visit is expected to take about 30 minutes. The same procedures will be done as for visit 3.. In addition, blood will be drawn.
This visit is expected to take about 30 minutes. This will be the end of the study. You will not be given any more study supplies at this time.
Other procedures to be done include:
| · | Filling out two questionnaires describing your menopausal symptoms: |
| o | The MRS, or “Menopause Rating Scale,” and |
| o | The GCS, or “Greene Climacteric Scale” |
| o | Collection of two small tubes of blood (about 2 teaspoons total) from a vein in your arm to test for the same profiles as previous. |
After this visit, you will wait for one phone call from the Study Doctor or nurse. The phone call will be to notify you of the blood test results.
What are the Risks of this Study?
Risks of Homatropine - There is only a small expected risk from using homatropine. Known side effects include:
Likely:
Dry mouth and thirst. Also decreased sweating, which could lead to an increase in body temperature (especially on a hot day).
To repeat: Contact the Study Doctor if you have a fever. Homatropine is known to reduce sweat activity, which can make it harder for your body to cool itself.
Unlikely, but still serious:
Diarrhea, slow heartbeat, vomiting, and nausea.
Rare and serious:
Eye pain, blurred or dim vision, painful or difficult urination and retention, muscle pain, tremors, weakness, and high blood pressure.
Risks of Loss of Treatment –
If it turns out that homatropine does provide temporary relief of your symptoms associated with menopause, you should be aware that you will no longer have access to homatropine after the study is over and the likely recurrence of Hot Flashes without treatment if the study drug was successful.
Risks of Study Questionnaires –
These risks will be different for every woman, depending on how she feels about the personal questions that are asked.
If you are sensitive to personal questions about symptoms related to menopause, including physical symptoms, sexual performance, and other related issues, you should think carefully about whether you want to be in this study.
Personal questions always carry the risk of unknown psychological harms, including the remembrance of old things that may have happened in your past. The Study Doctor or nurse is always available to answer any questions you might have about the study questionnaires.
If you want, a referral can be given for someone to talk to about any feelings you might have (for example, a therapist or women’s health counselor). However, any expenses you might have as a result of feelings that come up because of this study will be the responsibility of you or your insurance.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Confidentiality –
Study questionnaires and other documents will be maintained according to the highest standards of research confidentiality. Only a code number will identify study documents and blood samples. The list linking the code number to your name will be kept confidential and available only to the Study Doctor or authorized staff. Of course, there may be a need to contact you by telephone if any study tests have significant safety findings.
This consent form, and other forms you are asked to sign, will of course have your name on them. These forms will be kept in a locked filing cabinet in the study doctor’s office.
Because this is a research study, the records could be looked at by other people. Representatives of the sponsor, the study monitor, the U.S. Food and Drug Administration (FDA), and the IRB (see below) may review these records to make sure that the study is being conducted correctly. (This explained in detail on a separate, attached form.)
What are the Potential Benefits to Others and Me?
As this is just an initial dosing study, there are probably no anticipated benefits from participating.
The researchers hope that there may be some relief of menopause symptoms as a result of taking homatropine.
The data from this study might help other women in the future, if homatropine is found to be effective, and becomes available for the treatment of menopause symptoms.
Finances: Cost and Compensation
You will be compensated according to the following schedule for your time, and the costs of travel that come from being in this study. There should be no costs other than those for your transportation and parking in getting to the study doctor’s office. No doctors or anyone else who may refer you to this study are being paid any fee.
| o | $20 if you completed the screening tests, plus |
| o | $50 if you completed Stage 1, plus |
| o | $50 if you completed Stage 2 plus |
| o | $30 for parking stage 1 and 2 |
No matter what, the total compensation amount will be provided shortly after when you decide to end your participation in the study, or after the study is over. The total amount could be anywhere from $20 to $150.
Partial participation and compensation are listed below:
The following compensation will be paid:
| Screening only | | $ | 20 | |
| Partial or full participation visit 1 and 2 | | $ | 50 | |
| Partial or full participation visit 4 | | $ | 50 | |
| Parking screening only | | $ | 15 | |
| Parking Partial or full any stage | | $ | 15 | |
All study tests and procedures will be performed at no cost to you or your insurance. However, any referrals made for medical care due to test results or findings from this study will be the responsibility of you or your insurance company.
If you are injured as a direct result of being in this study, the sponsor will make follow-up medical care available at no cost to you or your insurance.
You should know that <<insert PI name>> is being compensated by the sponsor, Eaton Scientific Systems, Inc., for her role in this study as the Principal Investigator.
Questions or Concerns
Please feel free at any time to ask any questions you have or talk about any concerns. If you have any questions about the study, or your scheduled appointments or tests, call the Study Doctor, <<insert PI name>>, at:
<<insert PI phone>>
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
If you have any questions about the medical significance of your study tests, please contact your Primary Care Provider and/or Gynecologist.
If you have any questions about the study that you don’t feel comfortable discussing with those mentioned, you may call a company called “IRC.” They are the Institutional Review Board (IRB) that is responsible for protecting the safety of human research subjects involved in this study. They can be reached during normal business hours at (800) 472-3241.
Alternatives
You do not have to be in this study. You can say “no.”
Other alternate treatments for menopause symptoms available to you include:
| · | Some prescription medications other than Hormone Replacement Therapy are available, but all have side effects. |
| · | You can avoid foods, substances, or situations that trigger symptoms, and try to dress comfortably. |
| · | You can lower your stress levels, and quit smoking. |
| · | You can take certain dietary or herbal supplements. |
You should talk to your own health care provider if you are interested in any of these alternate treatments for symptoms of menopause.
Voluntary Participation and Rights
Participation should be voluntary. You have the right to stop being in this study at any time. You can refuse any procedure or can withdraw at any time without any harm to your regular medical care.
There are other forms that you will need to consider after this one. (You can check them off as you read them)
| □ | The study schedule and diary pages that will be used in the study are attached to this form. They should help inform you what you will be asked to do if you decide to be in the study. |
| □ | One extra form is to notify you how your private medical information will be handled during this study. |
| □ | Another form describes your rights as a research subject. |
If you decide to be in the study, you will be given copies of all these materials to keep, as well as a copy of all the forms that you sign for this study, including this consent form.
| If you decide to be in the study, please sign and date this form below. You should not sign until you have read all the materials and have received satisfactory answers to all of your questions. |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 through 2 |
Signatures and Permissions
| | Participant: I have been told about this study and have taken time to ask questions and think about my decision. I agree to participate. | Person obtains consent: I have talked with the participant about the study and answered all of her questions. I accept her consent. |
| Printed name | | |
Signature | | |
Date/ Time | | |
Subject Number _________
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
RESEARCH CONSENT FORM ADDENDUM
DISCLOSURE OF YOUR PERSONAL HEALTH INFORMATION
You are being invited to be in a research study. As part of that study lots of data will be generated. Some of it will be your personal health information.If you don’t authorize use or disclosure of your health information, you cannot participate in the study as it would not contribute to the outcome.
1. WHO can give out the information? <<Insert PI name>>
2. WHAT information may be disclosed?
| · | Study-related medical information, including test results |
3. TO WHOM and WHY will the information above will be disclosed?
| · | The sponsor, Eaton Scientific Systems, Inc. (for research and product development purposes) |
| · | Stark-SMO, Inc., (a contract research group helping the sponsor to conduct the study and analyze study data) |
| · | The U.S. Food and Drug Administration (FDA), to audit. |
| · | The Institutional Review Board (IRB) responsible for safety oversight of this research is entitled to inspect the above information. |
The Privacy Rule may no longer protect your personal health information if any of these groups re-disclose it to somebody else. (There could be other rules they must follow, however.)
4. REVOCATION: You may cancel this authorization at any time, by notifying the following person in writing:
<<Insert PI name & address>>
If you cancel this authorization, your health information collected during the study will only be used to make administrative or safety reports required by the study. You will also have to be withdrawn from the study.
5. EXPIRATION: This authorization will expire automatically at the end of the study.
6. REFUSAL: If you decline to sign this authorization, it will not affect regular, non-research treatment by your doctor, payment from your insurance, enrollment in any health plan, or eligibility for their benefits. However, you cannot participate in the study if you do not sign.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
7. ACCESS TO INFORMATION: You may inspect and get a copy of the information disclosed under this Authorization.
8. COMPENSATION: Eaton Scientific Systems, Inc. will compensate <<insert PI name>> a small amount of money for the cost of obtaining this Authorization, as part of their normal compensation for participating in this study.
Subject Signature Investigator/Coordinator Signature
| | Signature | Printed Name | Date |
| | I am authorizing use of my health information in the way it is described above. After we sign this, I will get a copy. |
Subject | | | |
| | We will allow this subject’s information to be used only as described above. After we sign this, we will keep the original. |
Investigator | | | |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
Any person requested to consent to participate as a subject in a research study involving a medical experiment, or is requested to consent on behalf of another, has the right to:
| | As an “experimental” or “research” subject you have the following rights: | |
| | | | |
| | 1. | Be told what the study is trying to find out. | |
| | | | |
| | 2. | Be told what will happen to you and whether any of the procedures, drugs, or devices is different from what would be used in standard practice. | |
| | | | |
| | 3. | Be told about the frequent and/or important risks, side effects, or discomforts of the research drugs, devices, or procedures. | |
| | | | |
| | 4. | Be told if you can expect any benefit from participating and, if so, what the benefit might be. | |
| | | | |
| | 5. | Be told the other choices you have and how they compare to being in the study. | |
| | | | |
| | 6. | Be allowed to ask any questions concerning the study both before agreeing to be involved and during the course of the study. | |
| | | | |
| | 7. | Be told what sort of medical treatment is available if any complications arise. | |
| | | | |
| | 8. | Refuse to participate at all or to change your mind about participation after the study is started. This decision will not affect your right to receive the care you would receive if you were not in the study. | |
| | | | |
| | 9. | Receive a copy of the signed and dated consent form. | |
| | | | |
| | 10. | Be free of pressure when considering whether you wish to agree to be in the study. | |
If you have any general questions about the study and your rights as a research subject, you should ask the researcher or research assistant. You may also contact the Institutional Review Board responsible for the protection of the rights of research subjects during this study. Their name is Independent Review Consulting (IRC), and they can be reached during normal business hours at (800) 472-3241.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
This schedule is provided as a place for you to write down your appointments. It also tells you what dose you should be taking of the study medication, and when. If you have any questions, or need to report problems with the study drug or your health, contact the study doctor at:
<<Insert PI name & phone>>
Study A Stage 1 Visit 1 (Screening)
Visit 1 (Screening) – This is the first day of the study for you.
| Date & Time | What Will Be Done | How Much Study Drug to Take Until Your Next Visit |
| | Consent discussion, screening examinations (including a physical exam), study questionnaires, medical history, and review of medications. (about 1 hour total) | None |
Reminder: Get additional blood tests done 2-3 working days before Visit 2 (baseline).
Visit 2 End of Study A Stage 1 and strart of Study A Stage 2, you will be taking homatropine oral suspension for 2 weeks, in escalating dose every 4-6 hours as needed. If you do not experience any adverse effects and the drug is not improving your menopausal systems, you can escalate the study drug to the next highest dose every 4-6 hours as needed. You will have 1 checkup visit – and one at the end of the study.
Visit 2 (Baseline Stage 2) – 1 week after Visit 1.
Reminder: Bring completed diary pages to each visit.
| Date & Time | What Will Be Done | How Much Study Drug to Take Until Your Next Visit |
| | Final eligibility; get new study drug; learn what dose to take; turn in diary pages and get new ones. (about 20-30 minutes total), brief physical exam, complete MRS, GCS and Bother scale questionnaire | Starting with dose 1 every 4-6 hours as needed. If no effect and no adverse events increase to the next higher dose every 4-6 hours as needed. |
| The dose for this Stage has been explained. | Subject’s Initials: | Doctor’s Initials: |
Reminder: Bring your study medication and completed diary pages to each visit.
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
Visit 3 – 1 week +/- day after Visit 2.
| Date & Time | What Will Be Done | How Much Study Drug to Take Until Your Next Visit |
| | Brief physical examination; review of symptoms and/or problems; check study drug level; turn in diary pages and get new ones as needed. (about 30 minutes total), dispense drug, complete MRS, GCS and Bother scale questionnaire | Continue study drug at current dose every 4-6 hours as needed. If no effect and no adverse events increase to the next highest does every 4-6 hours as needed. |
| The dose for this Stage has been explained. | Subject’s Initials: | Doctor’s Initials: |
Note: The study doctor or nurse will call you 1-2 days after this visit to notify you of your blood test results.
Reminder: Bring your study medication and completed diary pages to each visit.
Visit 4 (End of Study) – This is the end of study visit, to be scheduled 1 week +/- 1 day after Visit 3.
| Date & Time | What Will Be Done | How Much Study Drug to Take Until Your Next Visit |
| | Blood collection for lab tests; brief physical examination; review of symptoms and/or problems; check study drug level; turn in diary pages complete MRS, GCS and Bother scale questionnaire (about 30 minutes total) | |
| The dose for this Stage has been explained. | Subject’s Initials: | Doctor’s Initials: |
Note: The study doctor or nurse will call you 1-2 days after this visit to notify you of your blood test results.
-- END OF STUDY -- THANK YOU FOR YOUR PARTICIPATION --
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
Research Study of Menopause Symptoms
Would you like to volunteer to help find out if a Non-hormonal drug can relieve “Hot Flashes” and “night sweats” associated with menopause?
<<Insert PI name>> is looking for volunteers to participate in a research study of a study drug called, to see if it is safe and effective in treating Hot Flashes and night sweats, which are common symptoms of menopause. Dr. Michael Guice is participating as an investigator in this research study.
If you agree to be in this study, you will participate anywhere from 1 to 3 weeks,. The total time of your involvement could be up to 3 weeks with 4 office visits. Fixed compensation will be provided to help cover your expenses. Eligible women would have 2 or more episodes of Hot Flashes or night sweats per day, and enrollment will be based on inclusion and exclusion criteria being met.
For more information about the this study contact Dr. Michael Guice
<<Insert PI Contact Info>>
Menopause Study | Menopause Study | Menopause Study | Menopause Study | Menopause Study | Menopause Study | Menopause Study | Menopause Study | Menopause Study | Menopause Study | Menopause Study |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
(To be developed later and submitted to the IRB for review & approval before use)
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
INSTRUCTIONS:
When potential subjects call the PI’s office inquiring about participation in this study, the following information must be provided:
| 1. | Do not collect the caller’s personal information. Protect her confidentiality as best as possible. Use a “Screening & Eligibility Form” to record information about this call. |
| a) | If the caller declines to be in the study, destroy the Screening & Eligibility Form, and make a notation of the call on the Screening & Eligibility Log. |
| b) | If the caller makes an appointment for consent and screening, forward the partially-completed Screening & Eligibility Form to the PI. |
2. Give the PI’s name and address, the title of the study, and explain what it’s about.
3. Required discussion:
| a) | Ask for verbal consent to discuss private medical information. |
| b) | Every attempt is made to screen patients by phone prior to their travel to this office. |
| c) | The eligibility criteria are reviewed. |
| d) | The coordinator will then describe the study procedures and time commitment. |
| e) | The caller is clearly informed that this program is experimental and that an evaluation on site is necessary to make the final decision on whether a person qualifies for the study. |
| f) | The risks of homatropine will be discussed. |
| g) | The caller is informed that no subject will be accepted unless all eligibility criteria are met. |
4. Questions & answers
5. Ask if potential subject remains interested and is tentatively qualified
| | a) | If she is interested, make an appointment for screening and provide directions and instructions, then follow step 1b above. |
| | b) | If she is not interested, thank her for her time, and follow step 1a above. |
| | c) | If she is uncertain, do not collect information to re-contact the caller. Let her know how she can call back anytime for more information. |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
Subject Number | Screening & Eligibility Form – Page 1 of 3 |
Study Title: Study A. An Open Label Escalating Study to Determine Maximum Necessary Dose (MND) of Homatropine Methylbromide needed for Palliation of Hot Flashes in Menopausal Women,
Section ONE: Initial Telephone Contact Date: _____________
IMPORTANT! Any “NO” answer means the person is ineligible for the study. Thank them for their time, and ask if they would let others know about the study. DESTROY THIS FORM, and start a new one for the next caller.
| The interested person is… | Yes | No | ? |
Is aged 45-65. and Is menopausal. (FSH greater than 20, and/or no menses for at least): · 12 months before screening. | | | |
| Reports episodes Hot Flashes and/or night sweats approximately 21 times a week or greater, affecting their quality of life, and is willing to keep track of them on a daily basis | | | |
Has a personal history of breast or uterine cancer, or any medical condition which precludes hormone replacement therapy as a treatment option for climacteric symptoms, or · Has decided that hormone replacement therapy is not effective or desirable for her condition. | | | |
| Is willing and able to provide documentation of a normal pelvic exam and Pap smear within the past 12 months. | | | |
| Is not receiving hormone replacement therapy (HRT) or any medication to treat menopause symptoms for the past 4 weeks. | | | |
DOES NOT HAVE primary glaucoma (or any family history of glaucoma), or narrow angle or close angle glaucoma. | | | |
DOES NOT HAVE known allergy to homatropine or belladonna alkyloids | | | |
DOES NOT HAVE problems with alcohol or substance abuse of any kind | | | |
DOES NOT HAVE cardiac disease, insulin-dependent diabetes, other cancers, or disease of the kidney, blood, liver, or immune system and is not taking any anticholinergic medications. | | | |
If answers to all the above are “Yes,” discuss the other issues on the Recruitment Outline.
| PLEASE ALSO ASK HOW THEY HEARD OF THIS STUDY | YES | NO |
| Did their physician refer them? | | |
OR, how else did they hear about this study? Study poster and/or brochure, or other (specify): | | |
NOTE: This is the only information that may be gathered before full informed consent is obtained.
| If the caller declines to participate, DESTROY THIS FORM | If the caller is eligible and wants to learn more, MAKE AN APPOINTMENT |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
SCREENING APPOINTMENT: INITIALS OF STAFF PERSON: _____________
| DO NOT COLLECT ANY NAMES OR CONTACT INFORMATION AT THIS POINT |
| PI: | Date/Time |
Subject Number | Screening & Eligibility Form – Page 2 of 3 |
Section TWO: The Consent Process Date: _____________
IMPORTANT: The first goal is to gain appropriate informed consent from the potential subject.
| Step 1: Explain the study verbally INITIALS OF STAFF PERSON: |
Step 2: Allow the potential subject to read all the Enrollment Forms o The correct Informed Consent Form, with Study Schedule o The HIPAA Authorization Form o The California Experimental Subject’s Bill of Rights NOTE: The potential subject may choose to bring the enrollment forms home to think about being in the study. She does not have to make a decision right now. |
Step 3: Obtain signatures on all the Enrollment Forms o Make copies of all forms for the subject o File originals in a locked filing cabinet in your office o Record the enrollment on the Enrollment Log o Assign a Study Number, and write it on all forms (DO NOT write the subject’s name!) |
Section THREE: Screening Procedures Date: _____________
If subject has signed all enrollment forms, proceed with screening procedures.
| REFER TO PROTOCOL FOR SPECIFIC ELIGIBILITY CRITERIA | YES | NO |
Has the subject met all of the inclusion and exclusion criteria? (See sections 3.2 and 3.3 of the protocol) | | |
Have you explained the screening & baseline procedures to the subject? (See section 4.3.1 of the protocol) | | |
| Check “Yes” or “No” for each of the following procedures: | YES | NO |
| Are the medical history and current meds supportive of eligibility? | | |
| Is the physical examination supportive of eligibility? | | |
| Does this subject meet the criteria for preliminary eligibility? | | |
| Do the Study A. Stage 1 diary pages verify ≥ 21 Hot Flashes and/or night sweats per week? | | |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
Decision of Principal Investigator: This subject ( o is ) ( o is not) eligible for this study, based on information evaluated up to this point. |
Signature of Principal Investigator | Date |
Subject Number | Screening & Eligibility Form – Page 3 of 3 |
IMPORTANT: If subject is NOT eligible, explain why, and what advice or referrals were given:
Section FOUR: Baseline Procedures
If subject is eligible for the study based on preliminary information, proceed with baseline procedures. (Check off as completed.)
| o | Provide a 1-week supply of Stage A Stage 1 diary pages, and instruct subject on their use |
| o | Make an appointment for Visit 2 ((1 +/- 1 day) week hence) Appt Date/Time: ____________ |
| o | Provide a study requisition for blood tests, and instruct subject when and where the blood samples need to be collected |
| o | Answer all subject’s questions, and confirm understanding of all instructions |
INITIALS OF STAFF PERSON: _____________
Date checked by Study Monitor: Signature:
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
INSTRUCTIONS: This is a confidential form; to be used ONLY by the Principal Investigator, or his/her authorized designee. Keep this form in a locked filing cabinet in the Principal Investigator’s office when not in use. Enter the specific information described below for all subjects who are screened; whether or not they are eligible.
| TO MAKE STUDY NUMBER: Date (YYMMDD) - Sequential Number Ex: 060925-01 |
If Eligible: Assign and enter the Subject ID #. Check status “Enrolled,” and circle what stages subject has been enrolled to complete (e.g. 1 and 2 only).
If Ineligible: Do NOT write any names or numbers. Check status “Ineligible,” and provide the reason the subject is ineligible.
(Study Monitor): FILL OUT the rest of the information, as study events occur
Enrollment Goal = Maximum of 22 ELIGIBLE Subjects (Do NOT Number Ineligible Subjects)
| Date of Screening | Subject Name and Study ID # | Status | Reason Ineligible | End Date & Reason | Staff Initials |
| | (Leave blank if ineligible) | o Enrolled Stages: 1 2 o Ineligible | | | |
| | (Leave blank if ineligible) | o Enrolled Stages: 1 2 o Ineligible | | | |
| | (Leave blank if ineligible) | o Enrolled Stages: 1 2 o Ineligible | | | |
| | (Leave blank if ineligible) | o Enrolled Stages: 1 2 o Ineligible | | | |
| | (Leave blank if ineligible) | o Enrolled Stages: 1 2 o Ineligible | | | |
| | (Leave blank if ineligible) | o Enrolled Stages: 1 2 o Ineligible | | | |
| | (Leave blank if ineligible) | o Enrolled Stages: 1 2 o Ineligible | | | |
| | (Leave blank if ineligible) | o Enrolled Stages: 1 2 o Ineligible | | | |
Date checked by Study Monitor: Signature:
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
FOCUSED PHYSICAL EXAMINATION
Subject Number: | oInitial Assessment oUpdate (Visit # _____) | Date: |
| Please check the appropriate boxes and, if abnormal, describe. | Age ____________ |
| | | | | |
| Height _______________ | (feet, inches) | Weight __________ | | (pounds – no footwear) |
| | | | | |
| Vital Signs: Temp _____________ | o°F o °C | B/P ___________ | | Pulse: ___________ |
| Normal | Abnormal | PHYSICAL EXAM - Describe any abnormality |
| | | | | | | | | | |
| o | o | Ears, nose and throat: | | |
| o | o | Ophthalmic: | | | | | |
| o | o | Respiratory: | | | | | |
| | | Smoker: oYes o No:o | # packs/week | |
| o | o | Cardiovascular | | | |
| o | o | Gastrointestinal | | | |
| o | o | Hepatic | | | | | | |
| o | o | Renal | | | | | | | |
| o | o | Urogenital | | | | | |
| o | o | Neurological | | | | |
| o | o | Endocrine | | | | | |
| o | o | Musculoskeletal | | | | |
| o | o | Skin | | | | | | | |
| o | o | Psychiatric | | | | | |
| o | o | Drug Allergies | | | | |
| This subject | o Is | o Is not | eligible to participate (or to continue to participate) in this study. |
| Principal | | |
| Investigator | | |
| Signature: | | | Date: ____/____/____ |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
Subject Number: | o Initial Assessment o Update (Visit # _____) | Date: |
MEDICAL HISTORY (Skip if update only)
Current or Previous Diseases? o No o Other (explain): oBreast CA dx date: ____________ o Endometrial CA dx date: ____________ |
Current or Previous Surgeries? o No o Other (explain): oMastectomy date: ____________ o Hystorectomy date: ____________ |
Current or Previous Injuries? o No o Yes (explain): |
| Date of Absence of Menses: Last FSH (if known): |
| Date of Last Pelvic Exam: Result: |
| Date of Last Pap Smear: Result: |
| Number of Hot Flush episodes per week: |
| Number of Night Sweat episodes per week: |
| Number of alcoholic beverages consumed per week: |
Any recreational drug use? o No o Other (explain): |
Ever participated on a research study before? o No o Other (explain): |
Any other significant history? |
MEDICATIONS and SUPPLEMENTS (Note any changes since last study visit)
| Herbal Remedies or Medications and Dose | | Indication | | Start Date | | Stop Date |
| | | | | | | |
| | | | | | | |
| | | | | | | |
| | | | | | | |
This subject o Is o Is not eligible to participate (or to continue to participate) in this study. |
| Principal Investigator Signature: ______________________________________ Date: ____/____/____ |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
INSTRUCTIONS: Circle the Visit Number below, perform visit procedures, and fill out the rest of this form.
| Procedures | Visit 1 Screening | Visit 2 Baseline | Visit 3 | Visit 4 |
| Study A | Stage 1 | Stage 2 | Stage 2 | Stage 2 |
| Final Eligibility | | X | | |
| Re-Consent | | X | | |
| Enroll in stage 2 | | X | | |
| MRS Questionnaire | | X | X | X |
| GCS Questionnaire | Use Screening & Eligibility form | X | X | X |
| Bother Scale Questionnaire | X | X | X |
| Dispense Study Drug | X | X | |
| Blood Draw | | | X |
| Physical Exam | X | X | X |
| Review Of Rx And Non Rx Drugs | X | X | X |
| Turn In And Evaluate Diary Pages | | X | X | X |
| Hand Out Diary Pages | | X | X | |
| Assess Subject Compliance & Study drug use | | | X | X |
| Review Adverse Events | | | X | X |
| Collect Study Drug | | | X | X |
| Exit Exam | | | | X |
| Results of Physical Exam & Update of Current Medications: | o OK | o N/A | o Other (explain below) |
Review of Diary Pages: # Complete: ____% Compliant: ____ | o OK | o N/A | o Other (explain below) |
■ Drug Usage: Volume: ____ (oz.)% Compliant: ____ (see Drug Usage Chart) |
| Review of Subject Compliance & Study Drug Use: | o OK | o N/A | o Other (explain below) |
Report of any Adverse Events: (OK only if none) | o OK | o N/A | o Other (explain below) |
| Completion of MRS, GCS and Bother Questionnaires: | o OK | o N/A | o Other (explain below) |
Results of Blood Tests for Toxicity: (see Data Safety Form) | o OK | o N/A | o Other (explain below) |
| | | | |
o Subject Notified of Results Date: ___________ By: (Initials) ________ |
| Special Procedures for This Study Visit: | o None | Staff Initials: |
o Enroll in Stage 2 Starting at dose 1 | o Dispense Study Drug and Amount Dispensed: | |
Explain any Unusual Results This Visit: o Check if continued on the back of this form
This subject o Is o Is not eligible to participate (or to continue to participate) in this study. |
| Principal Investigator Signature: ______________________________________ Date: ____/____/___ |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
Appendix 8.2.4a - Data Evaluation Form (Part 1)
INSTRUCTIONS: Use one of these forms for each subject; for the entire study. At each visit, compare the blood test results with the toxicity levels and definitions found in the CTCAE v3 Toxicity Criteria (see Reference Section 8.4.2). Record the highest toxicity level reached by any test parameter; then follow the visit instructions.
| Study A. Stage 2 | Subject Number: |
| Study Visit #2 (Baseline visit) | Visit Date: |
| (Blood tests results should be available at the time of the subject’s visit.) |
| | |
| |
| |
| Signature of Principal Investigator: | Date Reviewed by PI: Date checked by Study Monitor: |
-- END OF STUDY --
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
INSTRUCTIONS: Use one of these forms for each subject participating in Study A. Stage 2. At each visit, compare the blood test results with the toxicity levels and definitions found in the CTCAE v3 Toxicity Criteria (see Reference Section 8.4.2). Record the highest toxicity level reached by any test parameter; then follow the visit instructions.
| Dose Level: | Subject Number: |
| Study Visit #3 | Visit Date: |
| |
| | Dose Limiting Toxicity? o Yes o No |
| |
| |
| Signature of Principal Investigator: | Date Reviewed by PI: Date checked by Study Monitor: |
| Study Visit #4 end of study | Visit Date: |
(Blood tests results will be available after the subject’s visit - please process results immediately) |
| Highest toxicity level of any parameter: | Dose Limiting Toxicity? o Yes o No |
If any parameter has a toxicity level of 2 or more, explain: |
If any parameter has a toxicity level of 3 or more, notify the PI immediately, fill out an Adverse Event Report Form (Section 8.2.7), and fax it to the IRB and Study Monitor. |
| Signature of Principal Investigator: | Date Reviewed by PI: Date checked by Study Monitor: |
-- END OF STAGE 2 --
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
Instructions: If the subject calls or is contacted by the Principal Investigator’s office regarding this study, the date, time, and reason for the contact should be recorded, as well as who initiated the contact. Write a brief explanation of the event and its outcome, and follow the procedures for reporting any adverse events as needed.
DO NOT WRITE THE SUBJECT’S NAME, TELEPHONE NUMBER, OR ANY IDENTIFYING INFORMATION ON THIS FORM
Date / Time + Subject No. | Reason for Call / Outcome | Staff Initials |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
| Subject ID Number: | Study Stages Completed (circle): 1 2 3 |
Subject Withdrawal
Person reporting this withdrawal: | Today’s Date: |
During what part of the study did the subject decide to withdraw? |
Subject Complaints: ( ) None ( ) Describe: |
Other Subject Comments: |
NOTE: Inform subject that this is the end of the study (and THANK them!). Make sure that arrangements are made to return any unused study drug or placebo, and subject diary pages. |
Signature of person filling out this form: | Date signed: |
Signature of Principal Investigator (review on same date): | Date signed: |
Date checked by Study Monitor: Signature:
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
| Check one | Check one | Subject Identification Number: |
o ADVERSE EVENT (Non-Clinical) | o INITIAL REPORT | |
o SAFETY REPORT (Clinical) | o FOLLOW-UP REPORT |
| CONTACTS | Name | Phone | Fax | Email |
| Person submitting | | | | |
| Principal Investigator | | | | |
| Monitor | Stark-SMO | 415-888-3700 | 415-888-3600 | |
| IRB | | | | |
| EVENT | Adverse Event (Non-Clinical) | Safety Report (Clinical) |
| Date of occurrence | | |
| Date PI Notified | | |
| Date faxed to Monitor & IRB | | |
| Type of Event | o Loss of privacy o Lost specimen o Lost paperwork o Other | o Injury o Fainting o Other: |
| Describe Event Briefly (Key Words) | | |
For the Principal Investigator to fill out:
| Expectedness Seriousness | o Serious o Not serious | o Serious o Not serious |
| Related to Study Procedure? | o Definitely o Somewhat o Not at all | o Definitely o Somewhat o Not at all |
| Resolution | o Ongoing o Resolved | o Ongoing o Resolved |
Describe the event in your own words: (Attach an additional sheet of paper, if necessary) | How was the event, issue, or problem resolved? |
| | |
Does the consent form language address this problem? | o Yes o No |
| Should the consent form be modified or should a consent addendum be issued? |
| | o Yes o No |
What costs did the subject have? How were the costs of care or resolution covered? | |
| SIGNATURES | Signature | Print Name (legibly) | Date |
Person filling out form | | | |
Principal Investigator | | | |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
INSTRUCTIONS: Consult this form to determine how much study drug should be dispensed depending on the subject’s current menopausal symptoms and adverse events.. In addition, consult this form to determine how much study drug should be used by the time the subject presents for the return study visit Discrepancies should be noted on the Study Visit Form, and reported to the PI, IRB, and Study Monitor as a Protocol Violation.
| AMOUNT SHOWN IS THE AMOUNT THAT SHOULD BE REMAINING |
| Stage and Group | Stage 1 | Stage 2 Dose 1 | Stage 2 Dose 2 | Stage 2 Dose 3 | Stage 2 Dose 4 |
| Bottles | | 2 bottle | 4 bottles | 6 bottles | 8 bottles |
| Volume | | 28 oz. | 56 oz. | 84 oz. | 112 oz. |
| Visit 1 | | | | | — |
| Visit 2 | | Distribute | | | |
| Visit 3 | | Distribute | 28-56 oz. | 56-84 oz. | 84-112 oz. |
| Visit 4 | | End of study | | | |
 | Each bottle = 14 oz. when full ¾ bottle = approx. 11.5 oz. remaining ½ bottle = approx. 7 oz. remaining ¼ bottle = approx. 3.5 oz. remaining |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
INSTRUCTIONS: As subjects are eligible for Stage 2, write their Subject Number and the date in the next available row in the currently active section of the log. DO NOT ENROLL SUBJECTS INTO A SECTION UNTIL THE DATA HAS BEEN ANALYZED FROM THE PREVIOUS COHORT, AND THE STUDY MONITOR HAS GIVEN WRITTEN APPROVAL TO PROCEED. Up to six (6) subjects may be enrolled into each cohort; a minimum of 3 is required to proceed to the next cohort.
Write the subject’s name and phone number, to be used only in case of emergency.
WHEN A SECTION IS COMPLETE, FAX THIS FORM TO THE STUDY MONITOR AT 415-888-3600
KEEP THIS LOG IN A LOCKED FILE IN THE PRINCIPAL INVESTIGATOR’S OFFICE
Starting Dose #1 – 1 tsp. 4-6 x/day as needed for 14 days increase to next higher dose if no effect and no adverse events
| Subject Number | Date | Name | Phone | |
| | | | | |
| | | | | |
| | | | | |
Date checked by Study Monitor: Signature: OK to proceed? o Yes o No |
| Subject Number | Date | Name | Phone | |
| | | | | |
| | | | | |
| | | | | |
Date checked by Study Monitor: �� Signature: OK to proceed? o Yes o No |
Dose #2 – 2 tsp. 4-6 x/day as needed for 14 days increase to next higher dose if no effect and no adverse events
| Subject Number | Date | Name | Phone | |
| | | | | |
| | | | | |
| | | | | |
Date checked by Study Monitor: Signature: OK to proceed? o Yes o No |
| Subject Number | Date | Name | Phone | |
| | | | | |
| | | | | |
| | | | | |
Date checked by Study Monitor: Signature: OK to proceed? o Yes o No |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
| Appendix 8.2.10 Dose Escalation Log (Stage 2 only) | Page 2 of 2 |
Dose #3 – 3 tsp. 4-6 x/day as needed for 14 days increase to next higher dose if no effect and no adverse events
| Subject Number | Date | Name | Phone | |
| | | | | |
| | | | | |
| | | | | |
Date checked by Study Monitor: Signature: OK to proceed? o Yes o No |
| Subject Number | Date | Name | Phone | |
| | | | | |
| | | | | |
| | | | | |
Date checked by Study Monitor: Signature: OK to proceed? o Yes o No |
Dose #4 – 4 tsp. 4-6 x/day as needed for 14 days increase to next higher dose if no effect and no adverse events
| Subject Number | Date | Name | Phone | |
| | | | | |
| | | | | |
| | | | | |
Date checked by Study Monitor: Signature: OK to proceed? o Yes o No |
| Subject Number | Date | Name | Phone | |
| | | | | |
| | | | | |
| | | | | |
Date checked by Study Monitor: Signature: OK to proceed? o Yes o No |
-- END OF DOSE SCHEDULE --
FINAL (MND) DOSE DETERMINATION FOR Study B.
Maximum Needed Dose Level for Stage 2: Designated Dose Level for Study B: Signature: Date: |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
INSTRUCTIONS: Use one form per subject. Consult this form to determine what type of study drug should be distributed and when. As study drug is distributed, write the bottle code (or use the peel-off label) in the space provided.
Study Drug Distribution Table | Subject Number: |
| Study Visit | Type of Drug | Volume | Bottle Code(s) |
| 1 | No drug | | |
| 2 | Homatropine – Dose 1 (1 tsp 4-6/day as needed for 14 days) | (28 oz.) 2-14 oz bottle | |
| 2 | Homatropine – Group 2 (2 tsp 4-6/day as needed for 14 days) | (56 oz.) 4-14 oz bottles | |
| 2 | Homatropine – Group 3 (3 tsp 4-6/day as needed for 14 days)) | (84 oz.) 6-14 oz bottles | |
| 2 | Homatropine – Group 4 (4 tsp 4-6/day as needed for 14 days) | (112 oz.) 8-14 oz bottles | |
| | | | |
Write in the information below according to instructions provided after Stage 2:
Date checked by Study Monitor: Signature:
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
| Subject Number: Date: |
| | Dose: | Page 1 of 2 |
INSTRUCTIONS: Each day you are in the study, you are asked to write on this form how many Hot Flashes you had, how bad they were, and how long they lasted. Please write down whether you had night sweats, too. On days when you have Hot Flashes or night sweats, please write how annoying they were to you; and whether they interfered with your activities (and explain those activities). Thank you for your participation in this study.
If you have any questions about this form, call DO NOT WRITE YOUR NAME ON THIS FORM !
Write the date you began this page: (Write dates each day, too) | RATE YOUR HOT FLASHES HERE ( See below for more instructions ) | Did you also have night sweats? (Circle YES or NO) | If you had any Hot Flashes or night sweats, please write how annoying they were to you; and whether they interfered with your activities (and explain those activities). Use the back of this page if you need more room. |
HOW MANY? (Write a number) | HOW BAD? (Circle a number) | HOW LONG? (Write in minutes) |
| Day 1 | | 0 1 2 3 | | YES NO | |
| Day 2 | | 0 1 2 3 | | YES NO | |
| Day 3 | | 0 1 2 3 | | YES NO | |
| Day 4 | | 0 1 2 3 | | YES NO | |
| Day 5 | | 0 1 2 3 | | YES NO | |
| Day 6 | | 0 1 2 3 | | YES NO | |
| Day 7 | | 0 1 2 3 | | YES NO | |
| Day 8 | | 0 1 2 3 | | YES NO | |
| Day 9 | | 0 1 2 3 | | YES NO | |
| Day 10 | | 0 1 2 3 | | YES NO | |
| Day 11 | | 0 1 2 3 | | YES NO | |
| Day 12 | | 0 1 2 3 | | YES NO | |
| Day 13 | | 0 1 2 3 | | YES NO | |
| Day 14 | | 0 1 2 3 | | YES NO | |
| HOW MANY? | = | The number of Hot Flashes that you had for the entire 24-hour period. | ALWAYS FILL IN THE NEXT PAGE, AND BRING YOUR STUDY DIARY PAGES (AND STUDY DRUG SUPPLY) WITH YOU TO YOUR STUDY APPOINTMENTS. Checked by: Date: |
| HOW BAD? | 0 = | No feeling of heat or sweating for the entire 24-hour period. |
| | 1 = | Mild feeling of heat without any sweating most of the time. |
| | 2 = | Moderate feeling of heat with sweating most of the time, but able to do activities. |
| | 3 = | Severe feeling of heat with sweating most of the time, and activities interrupted. |
| HOW LONG? | = | Estimate the average amount of time (in minutes) that the Hot Flashes lasted |
| | | | |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
| Appendix 8.3.1 – Subject Diary Pages | Subject Number: Date: |
Page 2 of 2
INSTRUCTIONS: On the day of your study appointment, please answer the questions on this page, and bring your study diary pages and entire study drug supply (even empty bottles) to your study appointment.
Please answer the questions by circling YES or NO, and explain any YES answers. Thank you again for your participation in this study!
1. Since we last saw or contacted you, have you experienced any unusual symptoms, problems with or changes to your health? YES NO If “yes” please write the symptom(s): When did it start? How long did it last? Have you ever had this symptom before? If so, when? Was the symptom different this time? If so, how? Have you reported this symptom yet during this study? 2. Have you been using any medications or health supplements? YES NO If “yes” please fill in the section below: Checked by: Date: |
| What is the name of medication or health supplement? | What is the dose, or how often you take it? | For how long have you been using it? | For what reason or health problem are you taking it? | Is it new? |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
If you have any questions about this form, call DO NOT WRITE YOUR NAME ON THIS FORM !
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
| 1. | Belchetz, P.E. - Hormonal treatment of postmenopausal women. NEJM. 1994; 330:1062-71. |
| 2. | Krebs, E, et al Phytoestrogens for treatment of menopausal symptoms: A systematic review. Ob Gyn 104, 2004 824-836. |
| 3. | Fitzpatrick,L Alternatives to estrogen. Medical Clinics of N America 87 (2003)1091-1113, Tice, JA et al Phytoestrogen supplements for the treatment of Hot Flashes: The isoflavone clover extract study. JAMA July 9, 2003, 290. 207. |
| 4. | Uebelhack, R et al Balsck cohash and St. John’s Wort for climacteric complaints. Ob & Gyn 107, Feb 2006. 247-55. |
| 5. | Fitzpatrick, Vasomotor symptoms, Ob Gyn supplement Oct 2004 104, 106s-117s. |
| 6. | Doermann DJ. Sexual dysfunction. Gale Encyclopedia of Alternative Medicine. Gale Group; 2003. |
Available from URL: http://www.findarticles.com/cf_0/g2601/0012/2601001242/print.jhtml
| 7. | Kaplan SA. Female sexual dysfunction: Diagnosis and treatment in 2002. Patient Care; Feb. 2002. Gale Group; 2003. |
Available from URL: http://www.findarticles.com/cf_0/m3233/4_36/83736088/print.jhtml
| 8. | Spector HP, Carey MP. Incidence and prevalence of the sexual dysfunctions: A critical review of the empirical literature. Archives of Sexual Behavior 1990; 19(4):389-408. |
| 9. | Roberts AJ, O’Brien ME. Nutraceuticals: The Complete Encyclopedia of Supplements, Herbs, Vitamins, and Healing Foods. New York: The Berkley Publishing Group; 2001. |
| 10. | Challener C. Specialty supplements are the bright spot in US dietary supplement market. Chemical Market Reporter; July 2003. |
Available at URL: http://www.findarticles.com/cf_0/m0FVP/1_264/105553671/print.jhtml
| 11. | Allison DB, Fontaine KR, Heshka S, Mentore JL, Heymsfield SB. Alternative Treatments for Weight Loss: A Critical Review. Crit Rev Food Sci Nutr 2001;41(1):1-28 |
| 12. | Meinert, CL. Clinical Trials: Design, Conduct, and Analysis. New York: Oxford University Press; 1986. |
| 13. | Rosen R, et al. Female sexual function index. Journal of Marital and Sex Therapy 2000;26:191-208. |
| 14. | Larroy C. Comparing visual-analog and numeric scales for assessing menstrual pain. Behavioral Medicine; Wntr 2002. Gale Group; 2002. |
Available from URL: http://www.findarticles.com/cf_0/m0GDQ/4_27/90217535/print.jhtml.
| 15. | Altman, DG. Practical Statistics for Medical Research. London: Chapman and Hall; 1993;181-191, 232-250. |
| 16. | Bailar III, JC, Mosteller, F, editors. P Values. In: Medical Uses of Statistics. 2nd ed. Boston, Massachusetts: NEJM Books (New England Journal of Medicine); 1992; Chapter 10. |
| 17. | Basson, Rosemary, et al. 2000. Report of the International Consensus Development Conference on Female Sexual Dysfunction: Definitions and Classifications. The Journal of Urology. 163: 888-893. |
| 15. | CortesCortes R, Probst A, Palacios JM. Quantitative light microscopic autoradiographic localization of cholinergic muscarinic receptors in the human brain: forebrain. Neuroscience 1987 Jan;20(1):65-107. |
| 16. | Watts NB, Notelovitz M, Timmons MC, . Comparison of oral estrogens and estorgens plus androgen on bone mirela density, menopausal symptoms, and lipid-lowering profiles in surgical menopause. Obstet Gynecol. 1995; 85:529-537. |
| Homatropine Protocol v6.1 | Clinical Protocol – Stages 1 and 2 only |
| 17. | Morrison JC, martin DC, Blair RA. The use of medroxyprogesterone acetate for relief of climacteric symptoms. Am J Obstet Gynecol 1980; 138: 99-104. |
| 18. | Clayden JR, Bell JW, Pollard P. Menopausal flushin: double-blind trial of a non-hormonal medication. Br Med J 1974, 1: 409-412 |
| 19. | Wyon Y, Lindgrin R, Lundeberg T, HammarM. Effects of acupuncture on climacteric vasomotor symptoms, quality of life, and urinary excretion of neuropeptides among postmenopausal women. Menopause. 1995; 2:3-12 |
| 20. | Lebherz TB, French L. Non-hormonal treatment of the menopausal syndrome. A double-blind evaluation of an autonomic system stabilizer. Obstet Gynecol |
| 21. | Daiber W. Climacteric complaints: success withouth using hormones. Arztliche Praxis. 1983; 35:1946-1947. |
| 22. | Hirata JD, Swiersz LM, Zell B. Does dong quai have estrogenic effects in postmenopausal women? A double-blind, placebo-controlled trial. Fertility Sterility. 1997; 68:981-986. |
| 23. | Shoupe, D. Practical Strategies for Treating Hot Flashes. Women’s Health in Primary Care 2001; 4:170-182. |
| 24. | Freedman, RE Reduced termoregulatory null zone in postmentopausal women with Hot Flashes.Am J ob Gyn 181, July 1999. 66-70 |
(Standard reference – not provided) http://ctep.cancer.gov/forms/CTCAEv3.pdf
(Provided as a separate document)
| Menopause Rating Scale (MRS) |
| | |
| Information: | http://www.menopause-rating-scale.info/ |
| Document : | http://www.menopause-rating-scale.info/documents/MRS_English.pdf |
| | |
| Greene Climacteric Scale (GCS) |
| | |
| Information: | http://www.menopausematters.co.uk/greenescale.php |
| | |
| Appendix 8.6.3 | Bother Scale |
| | |
| Information: | |