Table of Contents
Filed Pursuant to Rule 424(b)(5)
| Registration No. 333-204830 | |
| PROSPECTUS SUPPLEMENT | ||
| (To Prospectus dated June 19, 2015) |
2,646,091 Shares
 |
Common Stock
______________
We are offering 2,646,091 shares of our common stock, par value $0.001 per share, directly to investors in this offering at a price of $6.875 per share. In a concurrent private placement, we are selling to each such investor warrants to purchase the number of shares of our common stock purchased by such investor in this offering. The warrants and the shares of our common stock issuable upon the exercise of the warrants are not being registered under the Securities Act of 1933, as amended (the “Securities Act”), are not being offered pursuant to this prospectus supplement and the accompanying prospectus and are being offered pursuant to the exemption provided in Section 4(a)(2) under the Securities Act and Rule 506(b) promulgated thereunder.
Our common stock is listed on The NASDAQ Capital Market under the symbol “PLXP.” The consolidated closing bid price and the last reported sale price of our common stock on June 8,2017were $6.75 per share and $6.95 per share, respectively.
The warrants will be exercisable six months and one day after the date of issuance of the shares of our common stock offered hereunder, and will remain exercisable until the 10th anniversary of the date of issuance. The exercise price for the warrants initially will be $7.50, subject to certain adjustments.
The warrants are not registered and are not and will not be listed for trading on The NASDAQ Capital Market, or any other securities exchange or nationally recognized trading system. There is no market through which the warrants may be sold, and purchasers may not be able to resell the warrants purchased in the private placement conducted concurrently with this offering. This may affect the pricing of the warrants in the secondary market, the transparency and availability of trading prices, and the liquidity of the warrants.
By means of this prospectus supplement, we are offering $18,191,869 of securities pursuant to General Instruction I.B.6 of Form S-3. As of April 19, 2017, the aggregate market value of our outstanding common stock held by non-affiliates, or the public float, was approximately $56.0 million based on 5,229,661 shares of common stock held by non-affiliates and a per share price of $10.70, which was the closing price of our common stock on April 19, 2017. Pursuant to General Instruction I.B.6 of Form S-3, in no event will we sell our securities in a primary public offering with a value in excess of one-third of our public float in any 12-month period so long as our public float remains below $75,000,000. We have not offered any securities pursuant to General Instruction I.B.6 of Form S-3 during the 12-calendar months prior to and including the date of this prospectus supplement.
We are an “emerging growth company” as that term is used in the Jumpstart Our Business Startups Act of 2012 and, as such, have elected to comply with certain reduced public company reporting requirements for this prospectus supplement and future filings.
Investing in our common stock involves risks. See “Risk Factors” beginning on page S-11 of this prospectus supplement, on page6 of the accompanying prospectus and in the documents incorporated by reference into this prospectus supplement.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus supplement or the accompanying prospectus. Any representation to the contrary is a criminal offense.
We have engaged Raymond James & Associates, Inc. and Janney Montgomery Scott LLC as our placement agents in connection with this offering. The placement agents have no obligation to buy any of the securities from us or to arrange for the purchase or sale of any specific number or dollar amount of securities. We have agreed to pay the placement agent fees set forth in the table below, which assumes that we sell all of the common shares we are offering pursuant to this prospectus supplement and all of the warrants we are offering in the concurrent private placement. See “Plan of Distribution” beginning on page S-93 of this prospectus supplement for more information regarding these arrangements.
Per Share | Total | |||||||
Public offering price | $ | 6.875 | $ | 18,191,869 | ||||
Placement agent fees (1) | $ | 0.4125 | $ | 1,091,512 | ||||
Proceeds to us, before expenses (2) | $ | 6.4625 | $ | 17,100,357 | ||||
__________________
| (1) | For additional information about the expenses for which we have agreed to reimburse the placement agents in connection with this offering, see “Plan of Distribution” beginning on page S-93 of this prospectus supplement. |
| (2) | The amount of the offering proceeds to us presented in this table does not give effect to any exercise of the warrants being issued in the concurrent private placement. |
Certain of our directors and management team have agreed to purchase an aggregate of $525,000 of our shares of common stock and warrants in this offering and the concurrent private placement.
The placement agents expect to deliver the shares of common stock on or about June 14, 2017, subject to the satisfaction of certain conditions.
Lead Placement Agent | Co-Lead Placement Agent JANNEY MONTGOMERY SCOTT |
| |
The date of this prospectus supplement is June 9, 2017 | |
PROSPECTUSSUPPLEMENT
S-iii | |
S-iv | |
S-1 | |
S-9 | |
S-11 | |
S-38 | |
S-39 | |
S-39 | |
S-39 | |
S-41 | |
S-42 | |
S-87 | |
| PRIVATE PLACEMENT TRANSACTION | S-92 |
| PLAN OF DISTRIBUTION | S-93 |
S-100 | |
S-100 | |
S-100 | |
S-100 |
PROSPECTUS
1 | |
2 | |
6 | |
7 | |
8 | |
9 | |
10 | |
12 | |
15 | |
18 | |
20 | |
22 | |
23 | |
25 | |
25 | |
25 | |
25 |
ABOUTTHIS PROSPECTUS SUPPLEMENT
This prospectus supplement and the accompanying prospectus are part of a registration statement that we filed with the U.S. Securities and Exchange Commission, or SEC, utilizing a “shelf” registration process. This document is in two parts. The first part is this prospectus supplement, which describes the terms of the offering of the securities offered hereby and also adds to and updates the information contained in the accompanying prospectus and the documents incorporated by reference into this prospectus supplement and the accompanying prospectus. The second part is the accompanying prospectus, which provides more general information, some of which may not apply to this offering and some of which may have been supplemented or superseded by information in this prospectus supplement or documents incorporated or deemed to be incorporated by reference in this prospectus supplement that we filed with the SEC subsequent to the date of the prospectus. To the extent that there is any conflict between the information contained in this prospectus supplement, on the one hand, and the information contained in the accompanying prospectus or any document incorporated by reference herein or therein, on the other hand, you should rely on the information in this prospectus supplement.
You should rely only on the information contained or incorporated by reference in this prospectus supplement, the accompanying prospectus and any free writing prospectuses we may provide to you in connection with this offering. We have not, and the placement agents have not, authorized any other person to provide you with any information that is different. If anyone provides you with different or inconsistent information, you should not rely on it. We are not making an offer to sell these securities in any jurisdiction where the offer or sale is not permitted. The information contained in this prospectus supplement, the accompanying prospectus, the documents incorporated by reference herein and any free writing prospectuses we may provide to you in connection with this offering is accurate only as of their respective dates. Our business, financial condition, results of operations and prospects may have changed since those dates. You should not consider this prospectus supplement or the accompanying prospectus to be an offer or solicitation relating to the securities in any jurisdiction in which such an offer or solicitation relating to the securities is not authorized. Persons outside the United States who come into possession of this prospectus supplement must inform themselves about, and observe any restrictions relating to, the offering of the securities and the distribution of this prospectus supplement outside the United States. Furthermore, you should not consider this prospectus supplement or the accompanying prospectus to be an offer or solicitation relating to the securities if the person making the offer or solicitation is not qualified to do so, or if it is unlawful for you to receive such an offer or solicitation.
Aspertec™ and PLxGuard™ are trademarks of PLx. All other trademarks used in this prospectus supplement are the property of their respective owners. Use or display by us of other parties’ trademarks, trade names or service marks is not intended to and does not imply a relationship with, or endorsement or sponsorship by us of, the trademark, trade name or service mark owner.
The industry and market data contained or incorporated by reference in this prospectus supplement are based either on our management’s own estimates or on independent industry publications, reports by market research firms or other published independent sources. Although we believe these sources are reliable, we have not independently verified the information and cannot guarantee its accuracy and completeness, as industry and market data are subject to change and cannot always be verified with complete certainty due to limits on the availability and reliability of raw data, the voluntary nature of the data gathering process and other limitations and uncertainties inherent in any statistical survey of market shares. Accordingly, you should be aware that the industry and market data contained or incorporated by reference in this prospectus supplement, and estimates and beliefs based on such data, may not be reliable. Unless otherwise indicated, all information contained or incorporated by reference in this prospectus supplement concerning our industry in general or any segment thereof, including information regarding our general expectations and market opportunity, is based on management’s estimates using internal data, data from industry related publications, consumer research and marketing studies and other externally obtained data.
CAUTIONARYSTATEMENT CONCERNING FORWARD-LOOKING STATEMENTS
This prospectus supplement, the accompanying prospectus and the documents we have filed with the SEC that are incorporated by reference into this prospectus supplement and the accompanying prospectus contain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. These statements relate to future events or to our future operating or financial performance and involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. Forward-looking statements may include, but are not limited to, statements about:
● | our expected uses of the net proceeds to us from this offering; |
● | our ability to bring both Aspertec 81 mg and Aspertec 325 mg to market-readiness; |
● | our ability to maintain regulatory approval of Aspertec 325 mg or obtain and maintain regulatory approval of Aspertec 81 mg and any future product candidates; |
● | the benefits of the use of Aspertec 325 mg and Aspertec 81 mg; |
● | the projected dollar amounts of future sales of established and novel gastrointestinal(GI)-safer technologies for non-steroidal anti-inflammatory drugs (NSAIDs) and other analgesics; |
● | our ability to successfully commercialize our Aspertec products, or any future product candidates; |
● | the rate and degree of market acceptance of our Aspertec products or any future product candidates; |
● | our expectations regarding government and third-party payor coverage and reimbursement; |
● | our ability to scale up manufacturing of our Aspertec products to commercial scale; |
● | our ability to successfully build a specialty sales force and commercial infrastructure or collaborate with a firm that has these capabilities; |
● | our ability to compete with companies currently producing GI-safer technologies for NSAIDs and other analgesics; |
● | our reliance on third parties to conduct our clinical studies; |
● | our reliance on third-party contract manufacturers to manufacture and supply our product candidates for us; |
● | our reliance on our collaboration partners’ performance over which we do not have control; |
● | our ability to retain and recruit key personnel, including development of a sales and marketing function; |
● | our ability to obtain and maintain intellectual property protection for our Aspertec products or any future product candidates; |
● | the actual receipt and timing of any milestone payments or royalties from our collaborators; |
● | our estimates of our expenses, ongoing losses, future revenue, capital requirements and our needs for or ability to obtain additional financing; |
● | our expectations regarding the time during which we will be an emerging growth company under the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”); |
● | our ability to identify, develop, acquire and in-license new products and product candidates; |
● | our ability to successfully establish and successfully maintain appropriate collaborations and derive significant revenue from those collaborations; |
● | our financial performance; and |
● | developments and projections relating to our competitors or our industry. |
These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described in “Risk Factors.” Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this prospectus supplement and the accompanying prospectus may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements.
You should not rely upon forward-looking statements as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur. Moreover, except as required by law, neither we nor any other person assumes responsibility for the accuracy and completeness of the forward-looking statements. We undertake no obligation to update publicly any forward-looking statements for any reason after the date of this prospectus supplement to conform these statements to actual results or to changes in our expectations.
You should read this prospectus supplement and the accompanying prospectus and the documents that we reference in this prospectus supplement and the accompanying prospectus and have filed with the SEC as exhibits to the registration statement of which this prospectus supplement and the accompanying prospectus are a part with the understanding that our actual future results, levels of activity, performance and events and circumstances may be materially different from what we expect.
PROSPECTUSSUPPLEMENT SUMMARY
This summary highlights certain information about us, thisoffering,and information appearing elsewhere in this prospectus supplement or the accompanying prospectus and in the documents we incorporate by reference.This summary is not complete and does not contain all of the information that you should consider before investing in our securities.The following summary is qualified in its entirety by, and should be read in conjunction with, the more detailed information and financial statements and notes thereto appearing elsewhere in this prospectus supplement or incorporated herein by reference.Before you decide to invest in our securities, to fully understand this offering and its consequences to you, you should carefully read the entire prospectus supplement, including the matters set forth under the caption“Risk Factors” beginning on pageS-11 of this prospectus supplement and page6 of the accompanying prospectus, and the consolidated financial statements and related notes included or incorporated by reference in this prospectus supplement, the accompanying prospectus and the other documents incorporated by reference herein and therein.
On April19, 2017,Dipexium Acquisition Corp.,a Delaware corporation (“Merger Sub”) anda wholly-owned subsidiary of Dipexium Pharmaceuticals, Inc., a Delaware corporation (“Dipexium”),merged with and into PLx Pharma Inc., a privately-held Delaware corporation (“Old PLx”),pursuant to the terms of that certain Agreementand Plan of Merger and Reorganizationdated as ofDecember 22, 2016 (the“Merger Agreement”) by and among Dipexium, Merger Sub and Old PLx (the“Merger”).As part of the Merger, Dipexium was re-named PLx Pharma Inc.(“New PLx”)and Old PLx was re-namedPLx Opco Inc.Following completion of the Merger, Old PLx became a wholly-owned subsidiary ofNewPLx. Since the completion of the Merger, the business we have conducted has been primarily the business of Old PLx. Unless the context requires otherwise, references in this Prospectus Supplement to “we”, “us”, “our”, the “Company”, and “PLx” refer to New PLx and its subsidiaries.
References in this prospectussupplement to“pro forma” or“on a pro forma basis” shall mean that the results for the stated period or other information have been adjusted to reflect the consummation of the Merger.Unless otherwise provided, all per share amounts and other data with respect to our common stock in this prospectus supplement give effect to a 1-for-8 reverse stock split effected by Dipexium in April 2017. Unless otherwise indicated, discussions of historical results reflect the results of Old PLx prior to the completion of the Merger and do not include the results of Dipexium prior to the completion of the Merger.
Overview
We are a late-stage specialty pharmaceutical company initially focused on developing our clinically validated and patent-protected PLxGuard delivery system to provide safer and more effective aspirin products. Our PLxGuard delivery system works by releasing active pharmaceutical ingredients into the duodenum, the first part of the small intestine immediately below the stomach, rather than in the stomach itself. We believe this improves the absorption of many drugs currently on the market or in development, and reduces acute gastrointestinal (GI) side effects — including erosions, ulcers and bleeding — associated with aspirin and ibuprofen, and potentially other drugs.
Our U.S. Food and Drug Administration, or FDA, approved lead product, Aspertec 325 mg, is a novel formulation of aspirin that uses the PLxGuard delivery system that is intended to significantly reduce acute GI side effects while providing superior antiplatelet effectiveness for cardiovascular disease prevention as compared with the current standard of care, enteric coated aspirin. Aspertec 325 mg (formerly PL2200 Aspirin 325 mg) was originally approved under the drug name aspirin, and the proprietary name ‘Aspertec’ was granted subsequent to the FDA approval. A companion 81 mg dose of the same novel formulation — Aspertec 81 mg — is in late-stage development and will be the subject of a supplemental New Drug Application, or sNDA, leveraging the already approved status of Aspertec 325 mg.
Our commercialization strategy will target both the over-the-counter, or OTC, and prescription markets, taking advantage of the existing OTC distribution channels for aspirin while leveraging the FDA approval of Aspertec 325 mg and expected approval for Aspertec 81 mg for OTC and prescription use when recommended by physicians for cardiovascular disease treatment and prevention. Given our clinical demonstration of better antiplatelet efficacy (as compared with enteric coated aspirin) and better acute GI safety, we intend to use a physician-directed sales force to inform physicians — and, by extension, consumers — about our product’s clinical results in an effort to command both greater market share and a higher price for our superior aspirin product. Our product pipeline also includes other oral nonsteroidal anti-inflammatory drugs, or NSAIDs, using the PLxGuard delivery system that may be developed, including a clinical-stage, GI-safer ibuprofen — PL1200 Ibuprofen 200 mg — for pain and inflammation.
PLxGuardTM Delivery System
Our PLxGuard delivery system uses surface acting lipids, such as phospholipids and free fatty acids, to modify the physiochemical properties of various drugs to selectively release these drugs to targeted portions of the GI tract. Unlike tablet or capsule polymer coating technologies (e.g., enteric coating), which rely solely on drug release based on pH differences in the GI tract, the PLxGuard delivery system uses the differential in pH and bile acid contents between the stomach and duodenum to target Aspertec’s release. This approach is intended to more reliably release active pharmaceutical ingredients in the duodenum and decrease their exposure to the stomach, which is more susceptible to NSAID-induced bleeding and ulceration. The PLxGuard delivery system is a platform technology that we believe may be useful in improving the absorption of many acid labile, corrosive, and insoluble or impermeable drugs.
We believe our PLxGuard delivery system has the potential to improve many already-approved drugs and drugs in development because it may:
● | enhance the efficacy of the drug using our technology; |
● | improve the GI safety of the drug; |
● | provide new or extended patent protection for an already-approved or development-stage drug: and |
● | utilize the 505(b)(2) New Drug Application, or NDA, regulatory path, which may provide a faster and lower-cost FDA approval route when used with already-approved drugs. |
The PLxGuard delivery system has clinically proven these benefits with our novel formulations using aspirin and has clinical evidence supporting the potential for a GI-safer ibuprofen and preclinical evidence supporting the potential for a GI-safer oral diclofenac and intravenous indomethacin products. Other existing or new drugs in development that may benefit from the PLxGuard delivery system will be evaluated either by us or through collaboration agreements with other companies.
Product Pipeline
Our lead product, Aspertec 325 mg, has been approved by the FDA for OTC distribution and is the first-ever FDA-approved liquid-fill aspirin capsule. All the clinical trials necessary for product launch have been completed. In clinical trials in diabetic patients at risk for cardiovascular disease, Aspertec 325 mg demonstrated better antiplatelet efficacy compared with enteric coated aspirin, which is the current standard of care for cardiovascular disease prevention and treatment. Aspertec 325 mg delivers faster antiplatelet efficacy than enteric coated aspirin with a median time to 99% inhibition of serum Thromboxane B2 of two hours compared with 48 hours for enteric coated aspirin. Serum Thromboxane B2 is a clinically accepted marker for antiplatelet efficacy, which is sometimes referred to as aspirin response.
Aspertec 325 mg provides more reliable, predictable and sustained antiplatelet benefits than enteric coated aspirin with a 3 – 5 times greater chance of a complete aspirin antiplatelet effect than enteric coated aspirin. Aspertec 325 mg has demonstrated a statistically significant 65% reduction in the risk of acute ulcers compared with immediate release aspirin in healthy subjects with an age associated risk for cardiovascular disease. This acute GI safety benefit may also be important for acute coronary syndrome, or ACS, patients. Moreover, we believe ACS patients who are also diabetics and suffer from gastroparesis, or a lack of digestive stomach motility, could also benefit from Aspertec due to its more predictable absorption when compared to enteric coated aspirin. The acute GI safety benefit may also be used to differentiate Aspertec 325 mg from products intended for use in conditions associated with pain and inflammation, including other aspirin and NSAID products.
Aspertec 81 mg is our lower-dose companion product for Aspertec 325 mg (the two dose forms are often referred to in this prospectus supplement together as “Aspertec”). This product utilizes exactly the same formulation as the 325 mg product (except delivered in a capsule one quarter the size) and will be the subject of an sNDA, which we expect to submit to the FDA in the last quarter of 2017. We will rely on the clinical results for Aspertec 325 mg for the Aspertec 81 mg sNDA and do not anticipate any additional clinical trials will be required, effectively positioning this product as an end of Phase 3 status. We intend to begin selling both products by the end of 2018.
We believe our technology may be used with other selected NSAIDs, such as ibuprofen. We have used the PLxGuard delivery system to create a lipid-based formulation of ibuprofen, PL1200 Ibuprofen 200 mg, for the OTC market, and PL1100 Ibuprofen 400 mg, for prescription doses of ibuprofen. We have OTC and prescription (Rx) Investigational New Drug applications, or INDs, active with the FDA and have demonstrated bioequivalence with the OTC 200 mg dose ibuprofen to support a 505(b)(2) NDA in fasted-state clinical trials at three different doses, 200 mg, 400 mg and 800 mg. Using the PL1200 capsules at prescription doses, we demonstrated better GI safety in osteoarthritic patients with equivalent analgesic and anti-inflammatory efficacy, when compared with prescription ibuprofen in a six-week endoscopy pilot clinical trial. PL1200 and PL1100 Ibuprofen may be considered as being in Phase 1 in the FDA process and may qualify for the 505(b)(2) NDA path.
The following table summarizes information regarding our product candidates and development program:
 |
The Market
Cardiovascular/Aspirin Market
Aspirin, also known as acetylsalicylic acid, or ASA, is a foundational medicine which is widely used for the treatment and prevention of cardiovascular disease, or CVD—a collective term for chest pain, coronary artery disease, heart attack, heart failure, high blood pressure and stroke—which is the leading cause of death in the United States. Aspirin is widely used to treat patients exhibiting signs and symptoms of heart attack or stroke (collectively referred to as ACS), and is commonly prescribed or recommended by physicians, in addition to other drugs such as cholesterol or blood pressure medications, as the standard of care for treating ACS and preventing recurrent ACS for the duration of a patient’s life.
The 325 mg dose is generally prescribed:
● | in an acute setting for the treatment of ACS; |
● | following an interventional procedure (such as placement of a stent); and |
● | for preventing heart attack or stroke during the high-risk period following an event or intervention. |
Eventually, most patients move to the 81 mg dose for secondary prevention applications and for high-risk primary prevention, as they are typically directed to take aspirin every day for the rest of their life. The 81 mg aspirin dose is the most prevalent, representing approximately 70% to 80% of aspirin sales in the United States, followed by the 325 mg dose and then several other dose forms including powdered and effervescent aspirin products. In the United States, the primary use for aspirin is for the prevention and treatment of cardiovascular disease as evidenced by the predominance of the 81 mg dose.
Aspirin is also used for pain relief and fever reduction and is the most common NSAID available. The most widely used dose for pain is 325 mg. A GI-safer Aspertec 325 mg may be able to capture market share from other aspirin and NSAID products in the large pain and inflammation market.
The leading aspirin brand reported global annual sales in 2016 exceeding $1 billion. Branded OTC aspirin products include Bayer®, Ecotrin® and St. Joseph®, and there are numerous private label or store brands.Because of the known GI toxicity issues associated with immediate release or regular aspirin, enteric coated aspirin has evolved to become the leading aspirin dose form, representing over 90% of U.S. aspirin sales in 2013. This success is largely based on early clinical studies suggesting that enteric coated aspirin showed a reduction in superficial gastric lesions as compared with regular aspirin. However, when measured using contemporary clinical procedures, enteric coated aspirin has not continued to demonstrate such reductions. These deficiencies create a significant opportunity for the demonstrated efficacy of Aspertec.
Pain & Inflammation/Ibuprofen
The U.S. OTC analgesic market was over $4 billion in annual sales in 2016 (Consumer Healthcare Products Association). The leading OTC NSAID brands in the United States represented greater than $1 billion in annual sales in 2016 with one leading OTC ibuprofen brand generating more than $750 million in worldwide sales in 2016 (Evaluate). This class of OTC drugs is one of the most widely used drugs worldwide. Increasing concerns over liver toxicity associated with acetaminophen products, coupled with known ibuprofen analgesic superiority over acetaminophen, indicate there is a substantial global opportunity for a GI safer ibuprofen product. While we do not believe our technology will work with the entire NSAID class, it is possible that our technology may be successfully applied to other NSAIDs beyond aspirin and ibuprofen. For example, we have preclinical data suggesting that diclofenac — a leading NSAID for pain and inflammation outside the United States — is a viable product candidate.
Our Key Competitive Strengths
● | Our lead product,Aspertec 325 mg, has been approved by the FDA under the 505(b)(2) NDA process. We intend to leverage clinical studies and market research to launch Aspertec 325 mg on a commercial scale using a combined prescription and OTC strategy that takes advantage of the existing OTC distribution channels for aspirin while leveraging the FDA approval of Aspertec 325 mg for prescription and recommendation by physicians for cardiovascular disease treatment and prevention. We will be seeking approval of the companion Aspertec 81 mg dose via an sNDA, which we intend to file in the last quarter of 2017, that should benefit from the prior approval of Aspertec 325 mg. |
● | Our management team has extensive experience in development and commercialization of OTC products. Mr. Valentino, our Executive Chairman of the Board, brings over 30 years of experience in the healthcare industry. Mr. Valentino successfully built Adams Respiratory Therapeutics into a fully integrated specialty pharmaceutical company with more than $490 million in annual revenue and leading OTC brands such as Mucinex® and Delsym®. In December 2007, Adams Respiratory Therapeutics sold to Reckitt Benckiser for approximately $2.3 billion. Ms. Giordano, our President and Chief Executive Officer, has over 20 years in the life science industry and has successfully developed commercialization plans for several new product launches across various therapeutic areas, specifically building physician directed sales teams, including with one of the most successful products in the cardiology sector, Lipitor. |
● | Aspertec 325 mg has demonstrated clinically superior antiplatelet efficacy in diabetic patients when compared to enteric coated aspirin, the current standard of care for cardiovascular disease prevention. Aspertec 325 mg delivers faster and more reliable, predictable and sustained antiplatelet benefits than enteric coated aspirin as clinically demonstrated in diabetic patients at risk for cardiovascular disease with a median time to 99% inhibition of serum Thromboxane B2 (an accepted clinical marker of anti-platelet efficacy) of two hours compared with 48 hours and with a 3 – 5 times greater chance of a complete aspirin antiplatelet effect than enteric coated aspirin. |
● | Aspertec 325 mg has demonstrated clinically superior GI safety when compared to regular aspirin. Aspertec 325 mg has demonstrated a statistically significant 65% reduction in the risk of acute ulcers compared with regular aspirin in healthy subjects with an age associated risk for cardiovascular disease. Based on physician market research, we believe that our clinical data supporting superior acute GI safety as compared to regular aspirin is important for physicians who are having difficulty keeping high-risk patients on sustained aspirin use due to lack of tolerability. |
● | Our PLxGuard delivery system is a platform technology that may improve GI safety and efficacy when applied to already approved drugs. Our PLxGuard delivery system is a platform technology that we believe could improve the GI safety of selected NSAIDs that meet specific criteria. The U.S. analgesic market was over $4 billion in annual sales in 2016 (Consumer Healthcare Products Association). The leading NSAID brands in the United States represented greater than $1 billion annual sales in 2016 (Statista Inc.) with one leading OTC ibuprofen brand generating more than $750 million in worldwide sales in 2016 (Evaluate). We intend to explore new development opportunities and utilize the 505(b)(2) NDA regulatory pathway to decrease the time and costs associated with obtaining FDA approval for new products. |
● | Our PLxGuard delivery system may lower development risk and costs by leveraging the 505(b)(2) NDA pathway. A 505(b)(2) NDA is permitted to rely on the FDA’s prior conclusions regarding the safety and effectiveness of a previously approved drug, or to rely in part on data in the public domain. Reliance on the FDA's prior findings or data collected by others may expedite the development program for our product candidates by potentially decreasing the amount of clinical data that we would need to generate to submit an NDA. As the FDA has previously approved Aspertec 325 mg pursuant to a 505(b)(2) NDA, we believe that there is a strong likelihood that our future products would similarly qualify. The factors related to this qualification are expected to reduce the time and costs associated with clinical trials when compared to a traditional NDA for a new chemical entity. We also believe the strategy of targeting drugs with proven safety and efficacy provides a better prospect of clinical success of our proprietary development portfolio as compared to de novo drug development. We believe that the average time to market and cost of clinical trials for our products could be less than that required to develop a new drug. |
| ● | Our issued patents and patent applications in the United States and worldwide may protect us from generic competition and enable a higher price point than current aspirin products. We believe our patent portfolio provides strong protection of our delivery platform, drug formulations and manufacturing processes with 44 issued patents and additional pending applications with expirations ranging from December 19, 2021 through September 29, 2032. |
Our Strategy
Our goal is to become a leading specialty pharmaceutical company, commercializing both the Aspertec 325 mg and Aspertec 81 mg dose forms and developing additional branded products using our PLxGuard technology. The key elements of our strategy are to:
Successfully commercialize Aspertec 325 mg. Prior to launch, we will need to finalize labeling with the FDA, scale up our commercial production capabilities, and conduct three validation manufacturing runs required to satisfy FDA pre-launch requirements. We intend to hire, train and deploy initially a 45-person sales force growing to 125 persons to support a national launch to appropriate healthcare providers. This will provide a technical focus on Aspertec 325 mg and 81 mg products based upon our compelling clinical results to enhance the value of these products. We intend to begin selling both products by the end of 2018.
Obtain FDA approval for Aspertec 81 mg and prepare it for commercial launch. This will include, in addition to the efforts described above, submission of an sNDA seeking approval to launch Aspertec 81 mg simultaneously with the already-approved 325 mg strength. Once our Aspertec 81 mg product becomes available as the result of planned manufacturing activities, we expect to conduct a clinical crossover trial of Aspertec 81 mg comparing pharmacokinetic and pharmacodynamic endpoints with 81 mg enteric coated aspirin. The trial would be intended for commercial purposes and is not otherwise required in connection with any near-term regulatory filings or approvals. We intend to begin selling both products by the end of 2018.
Initiate a market launch strategy targeting physicians who desire reliable and predictable antiplatelet efficacy for their highest risk cardiovascular patients. Our market launch strategy will focus on targeting physicians who are seeking reliable and predictable antiplatelet efficacy for their highest risk cardiovascular patients. This strategy is designed to drive product sales professionally with a technical sales approach. We anticipate taking advantage of aspirin’s unique OTC and prescription approval by using a sales force to call on physicians to inform them of the clinically validated attributes of Aspertec. This physician sales effort will be designed to create interest in Aspertec among healthcare professionals and to influence their recommendations and prescriptions of Aspertec over other aspirin products.
Move beyond aspirin to leverage our PLxGuard delivery systemin new products. While we are currently focused on Aspertec, there is a pipeline of additional opportunities that can be exploited. We are developing novel formulations that combine already-approved selected NSAIDs that meet our specific criteria with our proprietary PLxGuard technology to make safer and more effective new products. In addition to Aspertec, we have clinical data for a GI-safer OTC ibuprofen product, PL1200 Ibuprofen 200 mg, and preclinical data for a GI-safer diclofenac and a GI-safer intravenous indomethacin. We believe that the PLxGuard technology may also prove a novel drug delivery platform for corrosive, acid labile and insoluble and impermeable drugs providing delivery along the GI tract. We believe that by focusing initially on commercializing Aspertec we can demonstrate the viability of our PLxGuard delivery system as a versatile platform technology.
Hire additional senior management and key personnel to support the formulation and execution of successful product launches. Though we will continue to evaluate other market launch options, including co-promotion in the United States with one or more existing firms, we currently expect to launch Aspertec in the United States using our own sales and marketing team. This will require additional hiring of both management and sales personnel.
Seek strategic partners to commercialize Aspertec and other products in global markets. We have licensed the rights to commercialize Aspertec in the People’s Republic of China (including Macao and Hong Kong), along with an option for commercialization in Cambodia, Indonesia, Laos, Malaysia, Myanmar, Papua New Guinea, Philippines, Singapore, Taiwan, Thailand and Vietnam. We will continue to seek partners in other global markets.
Recent Developments
On May 10, 2017, we announced that we had been awarded a $1.9 million grant by the National Cancer Institute (NCI) of the National Institutes of Health (NIH) in support of our novel formulation of aspirin for chemoprevention of colorectal cancer. Further research of our novel phosphatidylcholine (PC)-associated aspirin (Aspirin-PC) for the prevention of colorectal cancer and potentially other cancers is supported by two recently published studies in peer-reviewed journals of the American Association for Cancer Research (AACR).
On May 12, 2017, we entered into a non-binding term sheet with Silicon Valley Bank regarding a potential $15 million debt financing. The financing would (a) be contingent upon completion of this offering and a minimum threshold of available cash on hand upon completion of this offering, (b) provide for $7.5 million at closing with the remaining $7.5 million subject to the achievement of certain milestones, (c) have a maximum term of 42 months, with interest-only payments being made during the first 18 months, (d) be secured by a first-lien security interest on all of our assets, and (e) include a requirement that upon each $7.5 million tranche funding we grant Silicon Valley Bank a warrant exercisable for ten years from the date of issuance for the purchase of shares of our common stock equal to 5% of the amount committed by Silicon Valley Bank divided by an exercise price equal to the lower of the average price per share over the preceding 10 trading days or the price per share on the day prior to funding. The terms of the financing remain subject to negotiation and definitive documentation, and we will continue to evaluate our liquidity options going forward.
Risks Associated With Our Business
Our business is subject to numerous risks and uncertainties related to our status as a development-stage company, our financial condition and need for additional capital, the commercialization of Aspertec, development of other product candidates, our reliance on third parties, our intellectual property and government regulation. These risks include those highlighted in the section entitled “Risk Factors” immediately following this prospectus supplement summary, including the following:
● | We have not yet generated significant revenues, have a limited operating history, have incurred net losses in each year since our inception and anticipate that we will continue to incur significant losses for the foreseeable future, and if we are unable to achieve and sustain profitability, the market value of our common stock will likely decline. |
● | We will need substantial additional funding. If we are unable to raise capital when needed, we could be forced to delay, reduce or terminate our operations or commercialization efforts. |
● | We are substantially dependent on the success of our lead product candidate, Aspertec. If we are unable to successfully commercialize Aspertec or experience significant delays in doing so, our business could be materially harmed. |
● | Even though Aspertec 325 mg has already obtained regulatory approval, it may never achieve market acceptance necessary for commercial success and the market opportunity may be smaller than we estimate. |
● | Our ability to market Aspertec for long-term use may be hampered by lack of trial results demonstrating long-term GI-safety benefits. |
● | We currently have no sales and marketing staff or distribution organization. If we are unable to develop a sales and marketing and distribution capability on our own or through third parties, we will not be successful in commercializing our future products. |
● | We face substantial competition and our competitors may discover, develop or commercialize products faster or more successfully than us. |
● | Our business will be highly dependent on professional and public reputation and perception, which may change, leading to volatile levels of sales. |
● | We may not be able to protect our intellectual property rights throughout the world. |
● | The regulatory approval process is expensive, time consuming and uncertain and may prevent us from obtaining, or cause delays in obtaining, approvals for the commercialization of the 81 mg dose form of Aspertec or future product candidates, which will materially impair our ability to generate revenue. |
Corporate Information
Dipexium was originally organized as a limited liability company under Delaware law in January 2010. In March 2014, Dipexium converted from a Delaware limited liability company to a Delaware corporation. Dipexium’s common stock previously traded on The NASDAQ Capital Market under the symbol “DPRX.” In connection with the Merger, our common stock began trading on The NASDAQ Capital Market under the symbol “PLXP” on April 20, 2017.
Our principal executive offices are located at 8285 El Rio Street, Suite 130, Houston, Texas, 77054, and our telephone number is (713) 842-1249. Our website address iswww.plxpharma.com. The information contained on our website is not incorporated by reference into this prospectus supplement or the accompanying prospectus, and you should not consider any information contained on, or that can be accessed through, our website as part of this prospectus supplement or the accompanying prospectus or in deciding whether to purchase our common stock.
Implications of Being an Emerging Growth Company
As a company with less than $1.0 billion in revenue during our last fiscal year, we qualify as an “emerging growth company” as defined in the Jumpstart Our Business Startups Act, or JOBS Act, enacted in April 2012. An “emerging growth company” may take advantage of exemptions from some of the reporting requirements that are otherwise applicable to public companies. These exceptions include:
● | being permitted to present only two years of audited financial statements and only two years of related disclosure in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our Annual Report on Form 10-K for the year ended December 31, 2016; |
● | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as amended; |
● | reduced disclosure obligations regarding executive compensation in our periodic reports, proxy statements and registration statements; and |
● | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We may take advantage of these provisions until the earlier of (i) December 31, 2019, (ii) the last day of the fiscal year (a) in which we have total annual gross revenue of at least $1.0 billion or (b) in which we are deemed to be a large accelerated filer, which means the market value of our equity securities that is held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter, and (iii) the date on which we have issued more than $1.0 billion of non-convertible debt in any three-year period.
We have elected to take advantage of certain of the reduced disclosure obligations, and may elect to take advantage of other reduced reporting requirements in future filings. As a result, the information that we provide to our stockholders may be different from what you might receive from other public reporting companies in which you hold equity interests. In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. We have irrevocably elected not to avail ourselves of this exemption and, therefore, we will be subject to the same new or revised accounting standards, and at the same time, as other public companies that are not emerging growth companies.
THEOFFERING
Common stock offered by us in this offering | 2,646,091 shares |
Common stock outstanding beforethis offering | 6,037,824 shares |
Common stock to be outstanding immediately after this offering | 8,683,915 shares |
Use of proceeds | We intend to use these net proceeds to: advance Aspertec 325 mg to market-readiness; obtain supplemental regulatory approval of Aspertec 81 mg; fund the technology transfer and the commercial scale validation and manufacturing necessary to support both efforts; expand our management team; fund a PK/PD clinical study (for marketing purposes only) comparing Aspertec 81mg to enteric coated aspirin; and to fund working capital, capital expenditures and other general corporate purposes, which may include the acquisition or licensing of other products, businesses or technologies. See “Use of Proceeds” for additional information. |
Risk factors | See “Risk Factors” beginning on page S-11 of this prospectus supplement and page 6 of the accompanying prospectus, the documents incorporated by reference into this prospectus supplement and the accompanying prospectus, and the other information included in this prospectus supplement and the accompanying prospectus for a discussion of factors you should carefully consider before deciding to invest in our common stock. |
| Concurrent Private Placement | In a concurrent private placement, we are selling to the purchasers of shares of our common stock in this offering warrants to purchase the number of shares of our common stock purchased by such investors in this offering, or up to 2,646,091 warrants. We will receive gross proceeds from the concurrent private placement transaction only to the extent such warrants are exercised for cash. The warrants will be exercisable six months and one day after the issuance date at an exercise price of $7.50 per share and will expire on the 10th anniversary of the issuance date. The warrants and the shares of our common stock issuable upon the exercise of the warrants are not being offered pursuant to this prospectus supplement and the accompanying prospectus and are being offered pursuant to the exemption provided in Section 4(a)(2) under the Securities Act and Rule 506(b) promulgated thereunder. See "Private Placement Transaction." |
NASDAQ symbol | “PLXP” |
The number of shares of common stock to be outstanding after this offering is based on 1,391,218 shares of Dipexium common stock outstanding as of March 31, 2017, and excludes the following:
● | 266,668 shares of common stock issuable upon the exercise of stock options outstanding as of March 31, 2017, at a weighted average exercise price of $73.12 per share under Dipexium’s 2013 Equity Incentive Plan (the “2013 Plan”); and |
● | 978 shares of our common stock, subject to increase on an annual basis, reserved for future issuance under the 2013 Plan as of March 31, 2017. |
On a pro forma basis, the number of shares of our common stock to be outstanding after this offering is based on 6,025,349 shares of our common stock outstanding as of March 31, 2017, and excludes the following:
● | 691,386 shares of our common stock issuable upon the exercise of stock options outstanding as of March 31, 2017, at a weighted average exercise price of $12.44 per share under Old PLx’s 2015 Omnibus Incentive Plan (the “2015 Plan”); |
● | 450,580 shares of our common stock reserved for future issuance under the 2015 Plan as of March 31, 2017; |
| ● | 266,668 shares of common stock issuable upon the exercise of stock options outstanding as of March 31, 2017, at a weighted average exercise price of $73.12 per share under the 2013 Plan; |
| ● | 978 shares of common stock issuable upon the exercise of stock options outstanding as of March 31, 2017, at a weighted average exercise price of $73.12 per share under the 2013 Plan; and |
| ● | 2,646,091 shares of common stock issuable upon exercise of warrants issued to investors in the concurrent private placement. |
The shares of common stock issuable upon the exercise of the warrants issued to investors in the concurrent private placement and the exercise price of such warrants are subject to anti-dilution adjustments in certain circumstances. See “Dilution” for more information about these possible anti-dilution adjustments.
RISKFACTORS
Investing in our common stock involves a high degree of risk.Before deciding to invest in our common stock, you should carefully consider each of the following risk factors and all other information set forth in this prospectus supplement,theaccompanying prospectus, the documents incorporated herein and therein by reference and any related free writing prospectus.The following risks and the risks described elsewhere in this prospectussupplement and the accompanying prospectus (and any documents incorporated herein by reference), including in the section entitled“Management’s discussion and analysis of financial condition and results of operations,” could materially harm our business, financial condition, operating results, cash flow and prospects.If that occurs, the trading price of our common stock could decline, and you may lose all or part of your investment.
Please also read carefully the section entitled “Cautionary Statement Concerning Forward-Looking Statements” included in this prospectus supplement.
Risks Related to Our Business and Capital Requirements
We have not yet generated significant revenues, have a limited operating history, have incurred net losses in each year since our inception and anticipate that we will continue to incur significant losses for the foreseeable future, and if we are unable to achieve and sustain profitability, the market value of our common stock will likely decline.
We have not generated any revenue from the sale of products, have generated minimal revenue from licensing activities, and have incurred losses in each year since we commenced operations. Old PLx’s net loss for the year ended December 31, 2016 was $4.9 million. Similarly, Dipexium incurred a net loss of $21.3 million for the year ended December 31, 2016. As of March 31, 2017, we had an accumulated deficit of approximately $52.7 million on a pro forma basis.
We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future as we continue the development and commercialization of Aspertec and our other product candidates. Our expenses will also increase substantially if and when we:
● | discover and develop additional product candidates; |
● | establish a sales, marketing and distribution infrastructure to commercialize Aspertec and any other product candidates for which we may obtain marketing approval; |
● | establish a manufacturing and supply chain sufficient for commercial quantities of Aspertec and any other product candidates for which we may obtain marketing approval; |
● | maintain, expand and protect our intellectual property portfolio; |
● | hire additional clinical, scientific and commercial personnel; |
● | add operational, financial and management information systems and personnel, including personnel to support our product development and planned future commercialization efforts, as well as to support our obligations as a publicly reporting company; and |
● | acquire or in-license other product candidates and technologies. |
Even if we do generate revenues, we may never achieve profitability, and even if we do achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ equity and working capital. If we are unable to achieve and sustain profitability, the market value of our common stock will likely decline. Because of the numerous risks and uncertainties associated with developing biopharmaceutical products, we are unable to predict the extent of any future losses or when, if ever, we will become profitable.
We will need substantial additional funding. If we are unable to raise capital when needed, we could be forced to delay, reduce or terminate our operations or commercialization efforts.
As of March 31, 2017, on a pro forma basis, we had working capital of approximately $10.2 million and cash and cash equivalents of approximately $12.8 million. We anticipate that we will need to raise substantial additional financing in the future to fund our operations.
We may obtain additional financing through public or private equity offerings, debt financings (including related-party financings), a credit facility or strategic collaborations. Additional financing may not be available to us when we need it or it may not be available to us on favorable terms, if at all. Our failure to raise capital as and when needed could have a negative impact on our financial condition and our ability to pursue our business strategies. Our future financing requirements will depend on many factors, some of which are beyond our control, including:
● | our ability to enter into additional collaboration, licensing or other arrangements and the terms and timing of such arrangements; |
● | the type, number, costs and results of the product candidate development programs which we are pursuing or may choose to pursue in the future; |
● | the rate of progress and cost of our clinical trials, preclinical studies and other discovery and research and development activities; |
● | the timing of, and costs involved in, seeking and obtaining FDA and other regulatory approvals; |
● | the costs of preparing, filing, prosecuting, maintaining and enforcing any patent claims and other intellectual property rights, including litigation costs and the results of such litigation; |
● | the emergence of competing technologies and other adverse market developments; |
● | the resources we devote to marketing, and, if approved, commercializing our product candidates; |
● | the scope, progress, expansion, and costs of manufacturing our product candidates; |
● | our ability to enter into collaborative agreements to support the development of our product candidates and development efforts; |
● | the amount of funds we receive in this offering; and |
● | the costs associated with being a public company. |
Future capital requirements will also depend on the extent to which we acquire or invest in additional complementary businesses, products and technologies. We currently have no understandings, commitments or agreements relating to any of these types of transactions.
If we are unable to raise additional funds when needed, we may be required to sell or license to others technologies or clinical product candidates or programs that we would prefer to develop and commercialize ourselves. Without additional funding — or, alternatively, a partner willing to collaborate and fund development — we will be unable to continue development of PL1200 Ibuprofen or any other development-stage products in our pipeline.
We are substantially dependent on the success of our lead product candidate, Aspertec. If we are unable to successfully commercialize Aspertec or experience significant delays in doing so, our business could be materially harmed.
Our future success is substantially dependent on our ability to successfully commercialize Aspertec, which will depend on several factors, including the following:
● | establishing commercial manufacturing and supply arrangements; |
● | establishing a commercial infrastructure; |
● | identifying and successfully establishing one or more collaborations to commercialize Aspertec; |
● | acceptance of the product by patients, the medical community and third-party payors; |
● | obtaining market share while competing with more established companies; |
● | a continued acceptable safety and adverse event profile of the product; and |
● | qualifying for, identifying, registering, maintaining, enforcing and defending intellectual property rights and claims covering the product. |
Serious adverse events, undesirable side effects or other unexpected properties of Aspertec or any other product candidate may be identified after approval that could delay, prevent or cause the withdrawal of regulatory approval, limit the commercial potential, or result in significant negative consequences following marketing approval.
Serious adverse events or undesirable side effects caused by, or other unexpected properties of, Aspertec or our other product candidates could cause us, an institutional review board, or regulatory authorities to interrupt, delay or halt our manufacturing and distribution operations and could result in a more restrictive label, the imposition of distribution or use restrictions or the delay or denial of regulatory approval by the FDA or comparable foreign regulatory authorities. If Aspertec or any of our other product candidates are associated with serious adverse events or undesirable side effects or have properties that are unexpected, we may need to abandon their development or limit development to certain uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. Many compounds that initially showed promise in clinical or earlier stage testing have later been found to cause undesirable or unexpected side effects that prevented further development of the compound.
Undesirable side effects or other unexpected adverse events or properties of Aspertec or any of our other product candidates could arise or become known either during clinical development or, if approved, after the approved product has been marketed. If such an event occurs during development, our trials could be suspended or terminated and the FDA or comparable foreign regulatory authorities could order us to cease further development of, or deny approval of, our other product candidates. If such an event occurs with respect to Aspertec, a number of potentially significant negative consequences may result, including:
● | regulatory authorities may withdraw the approval of such product; |
● | regulatory authorities may require additional warnings on the label or impose distribution or use restrictions; |
● | regulatory authorities may require one or more post-market studies; |
● | we may be required to create a medication guide outlining the risks of such side effects for distribution to patients; |
● | we could be sued and held liable for harm caused to patients; and |
● | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product candidate, or could substantially increase commercialization costs and expenses, which could delay or prevent us from generating revenue from the sale of our products and harm our business and results of operations.
We may not be able to realize the anticipated benefits from combining the businesses of Dipexium and Old PLx.
The Merger involved the integration of two companies that previously have operated independently with principal offices in two distinct locations. Significant management attention and resources have been and will be required to integrate the two companies. The failure to successfully integrate and manage successfully the challenges presented by the integration process may result in the combined organization’s failure to achieve some or all of the anticipated benefits of the Merger.
Potential difficulties that may be encountered in the integration process include the following:
● | using the combined organization’s cash and other assets efficiently to develop the business of the combined organization; |
● | appropriately managing the liabilities of the combined organization; and |
● | potential unknown or currently unquantifiable liabilities associated with the Merger and the operations of the combined organization. |
Delays in the integration process could adversely affect our business, financial results, financial condition and stock price. Even if we are able to integrate the business operations successfully, there can be no assurance that this integration will result in the realization of the full benefits of synergies, innovation and operational efficiencies that may be possible from this integration and that these benefits will be achieved within a reasonable period of time.
Even though Aspertec 325 mg has already obtained regulatory approval, it may never achieve market acceptance by physicians, patients, and others in the medical community necessary for commercial success and the market opportunity may be smaller than we estimate.
Even if we are able to launch Aspertec commercially, it may not achieve market acceptance among physicians, patients, hospitals (including pharmacy directors) and third-party payors and, ultimately, may not be commercially successful. Market acceptance of any product candidate for which we receive approval depends on a number of factors, including:
● | the efficacy and safety of the product candidate as demonstrated in clinical trials; |
● | relative convenience and ease of administration; |
● | the clinical indications for which the product candidate is approved; |
● | the potential and perceived advantages and disadvantages of the product candidates, including cost and clinical benefit relative to alternative treatments; |
● | strength of competitive products; |
● | the effectiveness of our sales and marketing efforts; |
● | the strength of marketing and distribution support; |
● | the willingness of physicians to recommend or prescribe the product; |
● | the willingness of hospital pharmacy directors to purchase our products for their formularies; |
● | our ability to maintain regulatory approvals for Aspertec; |
● | acceptance by physicians, operators of hospitals and treatment facilities and parties responsible for reimbursement of the product; |
● | the availability of adequate coverage and reimbursement by third-party payors and government authorities; |
● | limitations or warnings, including distribution or use restrictions, contained in the product’s approved labeling or an approved risk evaluation and mitigation strategy; |
● | the approval of other new products for the same indications; |
● | the timing of market introduction of the approved product as well as competitive products; and |
● | adverse publicity about the product or favorable publicity about competitive products. |
For example, while we believe that the safety profile and certain efficacy data will allow us to differentiate Aspertec from other aspirin products in the market, we may not be able to make direct comparative claims regarding the safety or efficacy of Aspertec and other aspirin products in our promotional materials for Aspertec. Any failure by Aspertec or any other product candidate that obtains regulatory approval to achieve market acceptance or commercial success would adversely affect our business prospects.
Our ability to market Aspertec for long-term use may be hampered by lack of trial results demonstrating long-term GI-safety benefits.
While demonstrating a statistically significant reduction in mucosal damage at 42 days when evaluated using the same clinical endpoints used for early studies involving enteric coated aspirin, Aspertec 325 mg did not demonstrate a reduction in ulcer risk over the course of a 42-day trial when more contemporary clinical endpoints were used. This lack of demonstrated long-term GI benefits could hamper our ability to market Aspertec 325 mg for long-term use.
For many new product candidates, we will rely on third parties to conduct our preclinical studies and all of our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be unable to obtain regulatory approval for or commercialize any of our product candidates.
If we elect to pursue new products, we will rely on medical institutions, clinical investigators, contract laboratories and other third parties, such as contract research organizations, to conduct our preclinical studies and clinical trials on our product candidates in compliance with applicable regulatory requirements. These third parties are not our employees and, except for restrictions imposed by our contracts with such third parties, we have limited ability to control the amount or timing of resources that they devote to our programs. Although we rely on these third parties to conduct our preclinical studies and clinical trials, we remain responsible for ensuring that each of our preclinical studies and clinical trials is conducted in accordance with its investigational plan and protocol and the applicable legal, regulatory, and scientific standards, and our reliance on these third parties does not relieve us of our regulatory responsibilities. The FDA and regulatory authorities in other jurisdictions require us to comply with regulations and standards, commonly referred to as current good clinical practices, or cGCPs, for conducting, monitoring, recording and reporting the results of clinical trials, in order to ensure that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. If we or any of our third-party contractors fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. In addition, we are required to report certain financial interests of our third party investigators if these relationships exceed certain financial thresholds and meet other criteria. Our clinical trials must also generally be conducted with products produced under current good manufacturing practice, or cGMP, regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
Many of the third parties with whom we contract may also have relationships with other commercial entities, some of which may compete with us. If the third parties conducting our preclinical studies or our clinical trials do not perform their contractual duties or obligations or comply with regulatory requirements we may need to enter into new arrangements with alternative third parties. This could be costly, and our preclinical studies or clinical trials may need to be extended, delayed, terminated or repeated, and we may not be able to obtain regulatory approval in a timely fashion, or at all, for the applicable product candidate, or to commercialize such product candidate being tested in such studies or trials. If any of our relationships with these third parties terminate, we may not be able to enter into arrangements with alternative third party contractors or to do so on commercially reasonable terms. Though we carefully manage our relationships with our contract research organizations, there can be no assurance that we will not encounter similar challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
Clinical trials for future products may be delayed or prevented.
Clinical trials may be delayed or prevented for a broad range of reasons, including:
● | Difficulties obtaining regulatory approval to begin trials; |
● | Delays in reaching agreements on acceptable terms with contract manufacturers and contract research organizations; |
● | Insufficient or inadequate supply or quality of a product candidate or other materials necessary to conduct our clinical trials; |
● | Challenges recruiting and enrolling subjects to participate in clinical trials for a variety of reasons, including size and nature of subject population, proximity of subjects to clinical sites, eligibility criteria for the trial, nature of trial protocol, the availability of approved effective treatments for the relevant disease and competition from other clinical trial programs for similar indications; |
● | Difficulties maintaining contact with subjects after treatment, which results in incomplete data; |
● | Receipt by a competitor of marketing approval for a product targeting an indication that our product targets, such that we are not “first to market” with our product candidate; |
● | Governmental or regulatory delays and changes in regulatory requirements, policy and guidelines; |
● | Inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities; |
● | Unforeseen safety issues, including serious adverse events associated with a product candidate, or lack of effectiveness; and |
● | Lack of adequate funding to continue the clinical trial. |
One or more of these difficulties could result in delayed or cancelled trials and have a significant negative impact on our earnings.
We will rely on third-party contract manufacturing organizations to manufacture and supply Aspertec and other product candidates for us, as well as certain raw materials used in the production thereof. If one of our suppliers or manufacturers fails to perform adequately we may be required to incur significant delays and costs to find new suppliers or manufacturers.
We currently have limited experience in, and we do not own facilities for, manufacturing our product candidates, including Aspertec. We rely upon third-party manufacturing organizations to manufacture and supply our product candidates and certain raw materials used in the production thereof. Some of our key components for the production of Aspertec have a limited number of suppliers.
We will not control the manufacturing process of, and will be completely dependent on, our contract manufacturing partners for compliance with cGMP regulations for manufacture of our drug products. We will be relying on our contract manufacturers to successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or others. In addition, although we will have no day-to-day control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel, we are nonetheless responsible for ensuring that our drug products are manufactured in accordance with cGMPs. If the facilities that manufacture our drug products fail to maintain a cGMP compliance status acceptable to the FDA or a comparable foreign regulatory authority, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved. The FDA or a comparable foreign regulatory authority could also take enforcement action with regard to the facilities or the drug products.
We do not have commercial supply agreements with our suppliers. In the event that we and our suppliers cannot agree to the terms and conditions for them to provide clinical and commercial supply needs, we would not be able to manufacture our product candidates until a qualified alternative supplier is identified, which could also delay the development of, and impair our ability to commercialize, our product candidates.
Our third-party suppliers may not be able to meet our supply needs or timelines and this may negatively affect our business. The failure of third-party manufacturers or suppliers to perform adequately or the termination of our arrangements with any of them may adversely affect our business.
A key ingredient for our products is currently available from only a single provider.
One key ingredient is currently limited to a single provider, Lipoid GmbH, or Lipoid, who supplies cGMP lecithin and is a leader in supplying high quality lipids to the global pharmaceutical industry. Lipoid developed this particular cGMP lecithin with us over a several year period, and has informed us that we are currently the only buyer of the product. We do not have a long-term contract with Lipoid for the supply of commercial quantities of this product, and there can be no assurances that Lipoid will be able to supply sufficient commercial quantities in compliance with regulatory requirements at an acceptable cost.
We may be subject to costly product liability claims related to our products and product candidates and, if we are unable to obtain adequate insurance or are required to pay for liabilities resulting from a claim excluded from, or beyond the limits of, our insurance coverage, a material liability claim could adversely affect our financial condition.
We face the risk that the use of our product candidates may result in adverse side effects. Although we have product liability insurance, our insurance may be insufficient to reimburse us for any expenses or losses we may suffer, and we may be required to increase our product liability insurance coverage as we increase the size of our operations. We do not know whether we will be able to continue to obtain product liability coverage and obtain expanded coverage if we require it, on acceptable terms, if at all. We may not have sufficient resources to pay for any liabilities resulting from a claim excluded from, or beyond the limits of, our insurance coverage. To the extent that we are required to provide indemnities in favor of third parties, there is also a risk that these third parties could incur liability and bring a claim under such indemnities. An individual may bring a product liability claim against us alleging that one of our product candidates or products has caused an injury or is found to be unsuitable for consumer use. Any product liability claim brought against us, with or without merit, could result in:
● | the inability to commercialize Aspertec or future product candidates; |
● | decreased demand for Aspertec or future candidates; |
● | regulatory investigations that could require costly recalls or product modifications; |
● | loss of revenue; |
● | substantial costs of litigation; |
● | liabilities that substantially exceed our product liability insurance, which we would then be required to pay ourselves; |
● | an increase in our product liability insurance rates or the inability to maintain insurance coverage in the future on acceptable terms, if at all; |
● | the diversion of management’s attention from our business; and |
● | damage to our reputation and the reputation of our products. |
Product liability claims may subject us to the foregoing and other risks, which could have a material adverse effect on our business, results of operations, financial condition and prospects.
We currently have no sales and marketing staff or distribution organization. If we are unable to develop a sales and marketing and distribution capability on our own or through third parties, we will not be successful in commercializing our future products.
We currently have no sales, marketing or distribution organization or history. To achieve commercial success for any approved product candidate, we must either develop a sales, marketing and distribution organization or outsource these functions to third parties. If we rely on third parties for marketing and distributing our approved products, any revenue we receive will depend upon the efforts of third parties, which may not be successful and are only partially within our control, and our product revenue may be lower than if we directly marketed or sold our products. We have no historical operations in this area, and if such efforts were necessary, we may not be able to successfully commercialize our future products. If we are not successful in commercializing our future products, either on our own or through third parties, any future product revenue will be materially and adversely affected.
We face substantial competition and our competitors may discover, develop or commercialize products faster or more successfully than us.
The development and commercialization of new drug products is highly competitive. We face competition from major pharmaceutical companies and biotechnology companies worldwide with respect to Aspertec and other product candidates that we may seek to develop or commercialize in the future. There are a number of pharmaceutical and biotechnology companies that currently market and sell products or are pursuing the development of product candidates that compete directly or indirectly with Aspertec. Potential competitors also include academic institutions, government agencies and other public and private research organizations. Our competitors may succeed in developing, acquiring or licensing technologies and drug products that are more effective, safer or less costly than Aspertec or any other product candidates that we are currently developing or that we may develop, which could render our product candidates obsolete and noncompetitive.
Many of our competitors have materially greater name recognition and financial, manufacturing, marketing, research and drug development resources than we do. Additional mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated in our competitors. Large pharmaceutical companies in particular have extensive expertise in commercial sales, preclinical and clinical testing and in obtaining regulatory approvals for drugs. In addition, academic institutions, government agencies, and other public and private organizations conducting research may seek patent protection with respect to potentially competitive products or technologies. These organizations may also establish exclusive collaborative or licensing relationships with our competitors.
Finally, the success of any product that is successfully commercialized will depend in large part on our ability to prevent competitors from launching a generic version that would compete with such product. If such competitors are able to establish that our patents are invalid or that the generic version would not infringe upon our product, they may be able to launch a generic product prior to the expected expiration of our relevant patents, and any generic competition could have a material adverse effect on our business, results of operations, financial condition and prospects.
We may fail to innovate and be competitive.
We cannot state with certainty when or whether any of our products under development will be launched, whether we will be able to develop, license, or otherwise acquire compounds or products, or whether any products will be commercially successful. Failure to launch successful new products or new indications for existing products may cause our products to become obsolete, causing our revenues and operating results to suffer.
We expect to compete with a large number of multinational pharmaceutical companies, biotechnology companies, and generic pharmaceutical companies. To successfully expand our product offerings, we must continue to deliver to the market innovative, cost-effective products that meet important medical needs. Our product revenues can be adversely affected by the introduction by competitors of branded products that are perceived as superior by the marketplace, by generic or biosimilar versions of our branded products, and by generic or biosimilar versions of other products in the same therapeutic class as our branded products. Our revenues can also be adversely affected by treatment innovations that eliminate or minimize the need for treatment with drugs.
We may attempt to form collaborations in the future with respect to our products, but we may not be able to do so, which may cause us to alter our development and commercialization plans.
We may form strategic alliances, create joint ventures or collaborations or enter into licensing arrangements with third parties with respect to our programs that we believe will complement or augment our existing business. For example, we have entered into a licensing arrangement with Lee’s Pharmaceutical Holdings Limited for the commercialization of Aspertec in China and with an option for additional countries in Southeast Asia. We may attempt to find other strategic partners for other geographic jurisdictions and we may also attempt to find one or more strategic partners for the development or commercialization of one or more of our other product candidates. We face significant competition in seeking appropriate strategic partners, and the negotiation process to secure appropriate terms is time-consuming and complex. We may not be successful in our efforts to establish such a strategic partnership for any product candidates and programs on terms that are acceptable to us, or at all.
Any delays in identifying suitable collaborators and entering into agreements to develop or commercialize our product candidates could negatively impact the development or commercialization of our product candidates in geographic regions where we do not have development and commercialization infrastructure. Absent a collaboration partner, we would need to undertake development or commercialization activities at our own expense. If we elect to fund and undertake development or commercialization activities on our own, we may need to obtain additional expertise and additional capital, which may not be available to us on acceptable terms or at all. If we are unable to do so, we may not be able to develop our product candidates or bring them to market and our business may be materially and adversely affected.
We may be unable to realize the potential benefits of any collaboration.
Even if we are successful in entering into a collaboration with respect to the development or commercialization of one or more product candidates, there is no guarantee that the collaboration will be successful. Collaborations may pose a number of risks, including:
● | collaborators may not perform their obligations as expected; |
● | disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the course of development, might cause delays or termination of the development or commercialization of product candidates, and might result in legal proceedings, which would be time-consuming, distracting and expensive; |
● | collaborators may be impacted by changes in their strategic focus or available funding, or business combinations involving them, which could cause them to divert resources away from the collaboration; |
● | collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; |
● | the collaborations may not result in our achieving revenue to justify such transactions; and |
● | collaborations may be terminated and, if terminated, may result in a need for us to raise additional capital to pursue further development or commercialization of the applicable product candidate. |
As a result, a collaboration may not result in the successful development or commercialization of our product candidates.
We will need to grow our organization, and we may experience difficulties in managing growth.
As of June 1, 2017, we had nine employees. We will need to expand our managerial, operational, financial and other resources in order to manage our operations, continue our development activities, commercialize Aspertec or other product candidates and comply with our obligations as a publicly reporting company. Our management and personnel, systems and facilities currently in place may not be adequate to support this future growth. Our need to effectively execute our business strategy requires that we:
● | manage our internal discovery and development efforts effectively while carrying out our contractual obligations to licensors, contractors, government agencies, any future collaborators and other third parties; |
● | continue to improve our operational, financial and management controls, reporting systems and procedures; and |
● | identify, recruit, maintain, motivate and integrate additional employees. |
If we are unable to expand our managerial, operational, financial, and other resources to the extent required to manage our development and commercialization activities, our business will be materially adversely affected.
We are highly dependent on the services of our executive management team, and on our ability to attract and retain qualified personnel.
We may not be able to attract or retain qualified management and scientific and clinical personnel in the future due to the intense competition for qualified personnel among biotechnology, pharmaceutical and other businesses. We are highly dependent on the principal members of our management and scientific staff, particularly our Executive Chairman of the Board, Michael J. Valentino, and our President and Chief Executive Officer, Natasha Giordano. If we are not able to retain Mr. Valentino, Ms. Giordano, or our incoming Chief Financial Officer, Rita O’Connor, or are not able to attract, on acceptable terms, additional qualified personnel necessary for the continued development of our business, we may not be able to sustain our operations or grow. Although we have executed employment agreements with each member of our current executive management team, including Mr. Valentino, Ms. Giordano and Ms. O’Connor, we may not be able to retain their services as expected.
In addition, we have scientific and clinical advisors who assist us in formulating our product development and clinical strategies. These advisors are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us, or may have arrangements with other companies to assist in the development of products that may compete with ours.
If we are not able to attract, retain and motivate necessary personnel to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
Our business involves the use of hazardous materials and we and our third-party manufacturers must comply with environmental laws and regulations, which may be expensive and restrict how we do business.
Our third-party manufacturers’ activities and our own activities may involve the controlled storage, use and disposal of hazardous materials, including the components of our pharmaceutical product candidates, test samples and reagents, biological materials and other hazardous compounds. We and our manufacturers are subject to federal, state, local and foreign laws and regulations governing the use, generation, manufacture, storage, handling and disposal of these hazardous materials. We currently carry no insurance specifically covering environmental claims relating to the use of hazardous materials. Although we believe that our safety procedures for handling and disposing of these materials and waste products comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental injury or contamination from the use, storage, handling or disposal of hazardous materials. In the event of an accident, state or federal or other applicable authorities may curtail our use of these materials and/or interrupt our business operations. In addition, if an accident or environmental discharge occurs, or if we discover contamination caused by prior operations, including by prior owners and operators of properties we acquire, we could be liable for cleanup obligations, damages and fines. If such unexpected costs are substantial, this could significantly harm our financial condition and results of operations.
We or the third parties upon whom we depend may be adversely affected by natural disasters.
Changes to global climate, extreme weather and natural disasters could affect demand for our products and services, cause disruptions in manufacturing and distribution networks, alter the availability of goods and services within the supply chain, and affect the overall design and integrity of our operations.
Our corporate headquarters is located in Houston, Texas, which in the past has experienced hurricanes. Hurricanes or other natural disasters could severely disrupt our operations, and have a material adverse effect on our business, operations, financial condition and prospects.
If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as our information technology systems, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time.
If such an event were to affect our supply chain, it could have a material adverse effect on our business.
Our employees, independent contractors, principal investigators, consultants and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, principal investigators, consultants and vendors may engage in fraudulent or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or disclosure of unauthorized activities to us that violates:
● | FDA regulations, including those laws requiring the reporting of true, complete and accurate information to the FDA; |
● | manufacturing standards; |
● | federal and state healthcare fraud and abuse laws and regulations; or |
● | laws that require the true, complete and accurate reporting of financial information or data. |
Specifically, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Activities subject to these laws also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation.
It is not always possible to identify and deter misconduct by our employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
If we are not able to implement the requirements of Section 404 of the Sarbanes-Oxley Act of 2002 in a timely manner or with adequate compliance, we may be subject to sanctions by regulatory authorities.
Section 404 of the Sarbanes-Oxley Act of 2002 requires that we evaluate and determine the effectiveness of our internal controls over financial reporting and, beginning with our annual report for the year ending December 31, 2017, provide a management report on the internal control over financial reporting. If we have a material weakness in our internal controls over financial reporting, we may not detect errors on a timely basis and our financial statements may be materially misstated. We will be evaluating our internal controls systems to allow management to report on, and eventually our independent auditors to attest to, the effectiveness of the operation of our internal controls. We will be performing the system and process evaluation and testing (and any necessary remediation) required to comply with the management certification and eventual auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002. The aforementioned auditor attestation requirements will not apply to us until we are no longer an “emerging growth company.”
To date, we have not conducted a review of our internal controls for the purpose of providing the reports required by these rules. We cannot be certain as to the timing of completion of our evaluation, testing and remediation actions or the impact of the same on our operations. If we are not able to implement the requirements of Section 404 in a timely manner or with adequate compliance, we may be subject to sanctions or investigation by regulatory authorities, such as the SEC or NASDAQ. Any such action could adversely affect our financial results or investors’ confidence in us and could cause our stock price to fall. Moreover, if we are not able to comply with the requirements of Section 404 in a timely manner, or if we or our independent registered public accounting firm identifies deficiencies in our internal controls that are deemed to be material weaknesses, we could be subject to sanctions or investigations by NASDAQ, the SEC or other regulatory authorities, which would entail expenditure of additional financial and management resources and could materially adversely affect our stock price. Deficient internal controls could also cause us to fail to meet our reporting obligations or cause investors to lose confidence in our reported financial information, which could have a negative effect on our stock price.
Our ability to utilize Dipexium’s or Old PLx’s net operating loss and tax credit carryforwards in the future is subject to substantial limitations and may be further limited as a result of the Merger.
Under Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, if a corporation undergoes an “ownership change” (generally defined as a greater than 50 percent change (by value) in its equity ownership over a three-year period), the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes to offset its post-change income may be limited. Further, if the historic business of Dipexium is not treated as being continued by us for the two-year period beginning on the date of the merger (referred to as the “continuity of business requirement”), the pre-Merger net operating loss carryforward deductions become substantially reduced or unavailable for use by the surviving corporation in the transaction. It is expected that the Merger resulted in an “ownership change” of Dipexium. Accordingly, our ability to utilize Dipexium’s net operating loss and tax credit carryforwards will be substantially limited. These limitations, in turn, could result in increased future tax payments for the combined organization, which could have a material adverse effect on the business, financial condition or results of operations of the combined organization.
Further, after a change in ownership such as the Merger, a subsequent change in ownership within the following two-year period will effectively eliminate our ability to use net operating loss carryforwards arising prior to the Merger. We expect that this offering will likely result in such a change in ownership.
We now possess not only all of the assets but also all of the liabilities of both Dipexium and Old PLx. Discovery of previously undisclosed or unknown liabilities could have an adverse effect on the combined organization’s business, operating results and financial condition.
Acquisitions involve risks, including inaccurate assessment of undisclosed, contingent or other liabilities or problems. As a result of the Merger, we possess not only all of the assets, but also all of the liabilities of both Dipexium and Old PLx. Although Dipexium conducted a due diligence investigation of Old PLx and its known and potential liabilities and obligations, and Old PLx conducted a due diligence investigation of Dipexium and its known and potential liabilities and obligations, it is possible that undisclosed, contingent or other liabilities or problems may arise in the future, which could have an adverse effect on our business, operating results and financial condition.
Risks Related to Product Safety and Efficacy Issues
Our understanding of the safety and efficacy of Aspertec could change as larger portions of the population begin using Aspertec.
Aspertec, like all NSAIDs, poses specific risks, including stomach bleeding and, for aspirin, Reyes syndrome. As the product is used by additional patients, we may discover new risks associated with Aspertec which may result in changes to the distribution program and additional restrictions on the use of Aspertec which may decrease demand for the product. Regulatory authorities have been moving towards more active and transparent pharmacovigilance and are making greater amounts of stand-alone safety information and clinical trial data directly available to the public through websites and other means, e.g. periodic safety update report summaries, risk management plan summaries and various adverse event data. Safety information, without the appropriate context and expertise, may be misinterpreted and lead to misperception or legal action which may potentially cause our product sales or stock price to decline. Further, if serious safety, resistance or drug interaction issues arise with our marketed products, sales of these products could be limited or halted by us or by regulatory authorities and our results of operations would be adversely affected.
Adverse safety events involving our marketed products may have a negative impact on our business.
Discovery of safety issues with our products could create product liability and could cause additional regulatory scrutiny and requirements for additional labeling, withdrawal of products from the market, and the imposition of fines or criminal penalties. Adverse safety events may also damage physician and patient confidence in our products and our reputation. Any of these could result in liabilities, loss of revenue, material write-offs of inventory, material impairments of intangible assets, goodwill and fixed assets, material restructuring charges and other adverse impacts on our results of operations. The reporting of adverse safety events involving our products or products similar to ours and public rumors about such events may increase claims against us and may also cause our product sales or stock price to decline or experience periods of volatility. Restrictions on use or significant safety warnings that may be required to be included in the label of our products — such as the risk of developing an allergic reaction to soy, stomach bleeding or Reyes syndrome, in the label for Aspertec — may significantly reduce expected revenues for this product and require significant expense and management time.
Unexpected safety or efficacy concerns can arise with respect to marketed products, whether or not scientifically justified, leading to product recalls, withdrawals, or declining sales, as well as product liability, consumer fraud and/or other claims, including potential civil or criminal governmental actions.
Our business will be highly dependent on professional and public reputation and perception, which may change, leading to volatile sales.
Market perceptions of us are very important to our business, especially market perceptions of our company and brands and the safety and quality of our products. If we, our partners and suppliers, or our brands suffer from negative publicity, or if any of our products or similar products which other companies distribute are subject to market withdrawal or recall or are proven to be, or are claimed to be, ineffective or harmful to consumers, then this could have a material adverse effect on our business, financial condition, results of operations, cash flows, and/or share price. Also, because we are dependent on market perceptions, negative publicity associated with product quality, patient illness, or other adverse effects resulting from, or perceived to be resulting from, our products, or our partners’ and suppliers’ manufacturing facilities, could have a material adverse effect on our business, financial condition, results of operations, cash flows, or share price.
We must be able to adapt to changed circumstances and quickly update product labels, which could be costly or harm our reputation.
We may be required by regulatory authorities to change the labeling for any pharmaceutical product, including after a product has been marketed for several years. These changes are often the result of additional data from post-marketing studies, head-to-head trials, adverse events reports, studies that identify biomarkers (objective characteristics that can indicate a particular response to a product or therapy) or other studies or post-marketing experience that produce important additional information about a product. New information added to a product’s label can affect its risk-benefit profile, leading to potential recalls, withdrawals, or declining revenue, as well as product liability claims. Sometimes additional information from these studies identifies a portion of the patient population that may be non-responsive to a medicine or would be at higher risk of adverse reactions and labeling changes based on such studies may limit the patient population. The studies providing such additional information may be sponsored by us, but they could also be sponsored by competitors, insurance companies, government institutions, managed care organizations, scientists, investigators, or other interested parties. While additional safety and efficacy information from such studies can assist us and healthcare providers in identifying the best patient population for each product, it can also negatively impact our revenues due to inventory returns and a more limited patient population going forward. Additionally, certain study results, especially from head-to-head trials, could affect a product’s reimbursement status or priority with certain payors, which could also adversely affect revenues.
Risks Related to Intellectual Property
If we are unable to obtain and maintain sufficient intellectual property protection for Aspertec or our future product candidates, or if the scope of the intellectual property protection is not sufficiently broad, our competitors could develop and commercialize products similar or identical to ours, and our ability to successfully commercialize our product candidates may be adversely affected.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our technologies. If we do not adequately protect our intellectual property, competitors may be able to use our technologies and erode or negate any competitive advantage we may have, which could harm our business and ability to achieve profitability. In particular, our success depends in large part on our ability to obtain and maintain patent protection in the United States and other countries with respect to our product candidates. However, we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. We may also fail to identify patentable aspects of our research and development before it is too late to obtain patent protection.
Further, the patentability of inventions, and the validity, enforceability and scope of patents in the pharmaceutical field involve complex legal and scientific questions and can be uncertain. As a result, patent applications that we own or license may fail to result in issued patents in the United States or in other foreign countries for many reasons. For example, since patent applications in the United States and most other countries are confidential for a period of time after filing, we cannot be certain that we were the first to file any patent application related to our product candidates. Even if patents have issued, or do successfully issue, from patent applications, third parties may challenge the validity, enforceability or scope thereof, which may result in such patents being narrowed, invalidated or held unenforceable. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property or prevent others from designing around our claims. If the breadth or strength of protection provided by the patents and patent applications we hold, license or pursue with respect to our product candidates is threatened, it could threaten our ability to commercialize our product candidates. Further, if we encounter delays in our clinical trials, the period of time during which we could market any of our product candidates under patent protection, if approved, would be reduced. Changes to the patent laws in the United States and other jurisdictions could also diminish the value of our patents and patent applications or narrow the scope of our patent protection.
If we are unable to protect the confidentiality of our trade secrets, the value of our technology could be materially adversely affected and our business would be harmed.
In addition to the protection afforded by patents, we rely on confidential proprietary information — including trade secrets and know-how — to develop and maintain our competitive position. Any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding our competitive position in our market. We seek to protect our confidential proprietary information, in part, by confidentiality agreements and invention assignment agreements with our employees and confidentiality agreements with consultants, scientific advisors, contractors and collaborators. These agreements are designed to protect our proprietary information. However, we cannot be certain that such agreements have been entered into with all relevant parties, and we cannot be certain that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. For example, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. We also seek to preserve the integrity and confidentiality of our confidential proprietary information by maintaining physical security of our premises and physical and electronic security of our information technology systems, but it is possible that these security measures could be breached. If any of our confidential proprietary information were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position. If we are unable to prevent material disclosure of the intellectual property related to our technologies to third parties, we will not be able to establish or maintain a competitive advantage in our market, which could materially adversely affect our business, results of operations and financial condition.
We are parties to a lawsuit which, if adversely decided against us, could impact our rights to Locilex®.
In April 2010, Dipexium acquired the worldwide rights to develop pexiganan, the active pharmaceutical ingredient in Locilex®, from Genaera Liquidating Trust, which was put in place to liquidate the assets of Genaera Corporation. In June 2012, Dipexium, along with its two senior executives and several other unrelated defendants, were sued in the Federal District Court for the Eastern District of Pennsylvania by a former shareholder of Genaera Corporation and purported to be on behalf of other Genaera Corporation shareholders, alleging, in pertinent part, that Dipexium’s acquisition of the rights to pexiganan (the active ingredient in Locilex®, and which rights included the rights to the prior formulation of Locilex®) was for what was alleged to be inadequate consideration, and as a result, it was alleged that Dipexium and its senior executives aided and abetted a breach of fiduciary duty by Genaera Corporation and the Genaera Liquidating Trust to the former shareholders of Genaera Corporation. It was also alleged that Dipexium and its senior executives aided and abetted a breach of the duty of the trustee at common law and under a certain trust agreement which was alleged to exist and which was executed by Argyce LLC (or Argyce), as trustee. The agreement called for Argyce to create the Genaera Liquidating Trust pursuant to which Argyce apparently was appointed to liquidate the assets formerly held by Genaera Corporation. One of these assets was pexiganan, which Dipexium acquired via public auction conducted by Argyce on behalf of the Genaera Liquidating Trust.
The case against Dipexium and its senior executives was dismissed with prejudice by the Federal District Court, without leave to refile, on August 12, 2013 based on the argument that Plaintiff’s claims were time barred, and a subsequent motion to reconsider such dismissal was denied by the Federal District Court. Prior to the dismissal there was no request or action to seek class certification by the plaintiff though it was purportedly filed on behalf of other former Genaera Corporation shareholders. Plaintiff appealed the dismissal of the suit as well as the denial of the motion to reconsider to the Third Circuit Appellate Court, which granted Plaintiff’s appeal.
On October 17, 2014, the Third Circuit Appellate Court, in a 2-1 decision with a strong dissenting opinion, reversed the trial court’s dismissal of Plaintiff’s claims based on the expiration of the applicable statutes of limitation and remanded the case to the Federal District Court. In its opinion, the Third Circuit held that more information was necessary to determine when Plaintiff should have been on notice of his claims to determine the applicability of the discovery rule, which could serve to extend the time frame in which Plaintiff could bring his claims. Due to the strong dissent, all defendants filed the necessary documents requesting a petition for rehearing en banc, by the majority of the Third Circuit justices who are in active service. The Third Circuit denied the request for en banc hearing and remanded this case to District Court.
Upon remand to the Federal District Court, all defendants moved to dismiss the complaint for reasons other than being time barred. Dipexium and its executives moved for dismissal based on Plaintiff’s inability to make a case for aiding and abetting a breach of fiduciary duty because there was no underlying breach and such an aiding and abetting claim requires an element of knowing participation in the fiduciary breach which cannot be established by Plaintiff.
The District Court held a hearing on this in September 2015 and the District Court delivered an Order on November 10, 2015 pursuant to which the District Court granted the Motion to Dismiss filed by each and every defendant including the Company and its executives. In December 2015, Plaintiff appealed the Federal District Court’s decision to the Third Circuit Appellate Court and we anticipate a decision on whether to grant Plaintiff’s appeal by the Third Circuit Appellate Court in the first half of 2017. We will continue to vigorously defend Plaintiff’s claims on the factual record, which we believe will prove that we are not liable to the Plaintiff in any regard.
If we were to lose this case, we could be required to pay damages, which could have a material adverse effect on us, our business plans and results of operations.
If we are sued for infringing intellectual property rights of third parties, such litigation could be costly and time consuming and could prevent or delay us from developing or commercializing our product candidates.
There is a substantial amount of intellectual property litigation in the biotechnology and pharmaceutical industries, and we may become party to, or threatened with, litigation or other adversarial proceedings regarding intellectual property rights with respect to our technology or product candidates, including post-grant or inter-partes proceedings, interference or derivation proceedings before the U.S. Patent and Trademark Office, or USPTO. Third parties may assert infringement claims against us based on existing or future intellectual property rights. The outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. The pharmaceutical and biotechnology industries have produced a significant number of patents, and it may not always be clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that our product candidates, products or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid, and we may not be able to do this. Proving that a patent is invalid is difficult. For example, in the United States, proving invalidity requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and the time and attention of our management and scientific personnel could be diverted in pursuing these proceedings, which could have a material adverse effect on us. Even if we are successful in defending these claims, we may incur substantial costs and the time and attention of our management and scientific personnel could be diverted in pursuing these proceedings, which could have a material adverse effect on us.
If we are found to infringe a third party’s intellectual property rights, we could be forced, including by court order, to cease developing, manufacturing or commercializing the infringing product candidate or product. Alternatively, we may be required to obtain a license from such third party in order to use the infringing technology and continue developing, manufacturing or marketing the infringing product candidate. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees, if we are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could materially harm our business. We may also elect to enter into license agreements in order to settle patent infringement claims or to resolve disputes prior to litigation, and any such license agreements may require us to pay royalties and other fees that could be significant. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business.
We may be involved in lawsuits to protect or enforce our intellectual property rights which could be expensive, time consuming and unsuccessful.
Competitors may infringe or otherwise violate our patents, the patents of our licensors, or our other intellectual property rights. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. Any claims that we assert against perceived infringers could also provoke these parties to assert counterclaims against us alleging that we infringe their intellectual property rights. In addition, in an infringement proceeding, a court may decide that a patent of ours is not valid or is unenforceable, in whole or in part, or may refuse to stop the other party in such infringement proceeding from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated, held unenforceable or interpreted narrowly, and could put any of our patent applications at risk of not yielding an issued patent.
Post-grant or inter-parte proceedings, interference or derivation proceedings provoked by third parties or brought by the USPTO or any foreign patent authority may be necessary to determine the priority of inventions or other matters of inventorship with respect to our patents or patent applications. We may also become involved in other proceedings, such as re-examination or opposition proceedings, before the USPTO or its foreign counterparts relating to our intellectual property or the intellectual property rights of others. An unfavorable outcome in any such proceedings could require us to cease using the related technology or to attempt to license rights to it from the prevailing party, or could cause us to lose valuable intellectual property rights. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms, if any license is offered at all. Litigation or other proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may also become involved in disputes with others regarding the ownership of intellectual property rights. For example, we jointly develop intellectual property with certain parties, and disagreements may therefore arise as to the ownership of the intellectual property developed pursuant to these relationships. If we are unable to resolve these disputes, we could lose valuable intellectual property rights.
We may not be able to prevent misappropriation of our trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock. Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses, and could distract our technical and/or management personnel from their normal responsibilities. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. Uncertainties resulting from the initiation and continuation of intellectual property litigation or other proceedings could negatively affect our ability to compete in the marketplace.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on all of our product candidates throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection but where enforcement is not as strong, or where standards are different than they are in the United States. These products may compete with our products in jurisdictions where we do not have any issued patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing. Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions, including China, where we currently have granted a license for Aspertec 325 mg. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
If we breach any of the agreements under which we license the use, development and commercialization rights to our product candidates from third parties, we could lose license rights that are important to our business.
In addition to our own patents, an important patent family covering Aspertec is owned by the Board of Regents of The University of Texas System. Our development and commercialization of Aspertec is subject to our license agreement with The University of Texas System as is our license agreement with Lee’s Pharmaceutical Holdings Limited. Under our existing license agreements, we are subject to various obligations, including diligence obligations with respect to development and commercialization activities, payment obligations for achievement of certain milestones and royalties on product sales, as well as other material obligations. If we fail to comply with any of these obligations or otherwise breach our license agreements, The University of Texas System may have the right to terminate the applicable license in whole or in part. Specifically, Section 4.6 of our license agreement with The University of Texas System (as amended) provides that “Reasonable commercial diligence shall require that PLX . . . . [o]n or before September 8, 2013, Sell or offer for Sale a Licensed Product.” While we believe that we have exercised reasonable commercial diligence to actively attempt such commercialization, we have not yet successfully commercialized a licensed product. As such, the Board of Regents of The University of Texas System may have the option to terminate the license agreement, or to limit the exclusivity of the license in certain territories.
The loss of our license agreement with The University of Texas System could materially adversely affect our ability to proceed with the development or potential commercialization of Aspertec as currently planned, and could materially adversely affect our ability to proceed with any development or potential commercialization of PL1200 Ibuprofen and other NSAID programs.
The risks described elsewhere pertaining to our patents and other intellectual property rights also apply to the intellectual property rights that we license, and any failure by us or our licensors to obtain, maintain and enforce these rights could have a material adverse effect on our business. In some cases we do not have control over the prosecution, maintenance or enforcement of the patents that we license, and may not have sufficient ability to consult and input into the patent prosecution and maintenance process with respect to such patents, and our licensors may fail to take the steps that we believe are necessary or desirable in order to obtain, maintain and enforce the licensed patents.
Limitations on intellectual property rights may result in other threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business or permit us to maintain our competitive advantage. The following examples are illustrative:
● | others may be able to make compounds that are similar to Aspertec or our future product candidates but that are not covered by the claims of the patents that we own or license; |
● | we or our licensors or collaborators might not have been the first to make the inventions covered by an issued patent or pending patent application that we own or license; |
● | we or our licensors or collaborators might not have been the first to file patent applications covering an invention; |
● | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
● | pending patent applications that we own or license may not lead to issued patents; |
● | issued patents that we own or license may not provide us with any competitive advantages, or may be held invalid or unenforceable, as a result of legal challenges by our competitors; |
● | our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; and |
● | we may not develop or in-license additional proprietary technologies that are patentable. |
We may be subject to claims that our employees or consultants have wrongfully used or disclosed alleged trade secrets of former or other employers.
Some of our employees, consultants, advisors, and members of our Board of Directors, including our senior management, have been employed or retained by other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these individuals have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such individuals’ former or other employer. We are not aware of any material threatened or pending claims related to these matters, but in the future litigation may be necessary to defend against such claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
Risks Related to Government Regulation
The regulatory approval process is expensive, time consuming and uncertain and may prevent us from obtaining, or cause delays in obtaining, approvals for the commercialization of Aspertec 81 mg or future product candidates, which will materially impair our ability to generate revenue.
The design, development, research, testing, manufacturing, labeling, storage, recordkeeping, approval, selling, import, export, advertising, promotion, and distribution of drug products are subject to extensive and evolving regulation by federal, state and local governmental authorities in the United States, principally by the FDA, and foreign regulatory authorities, with regulations differing from country to country. Failure to obtain marketing approval for a product candidate will prevent us from commercializing the product candidate. While we are permitted to market Aspertec 325 mg, neither we nor any future partner are permitted to market any other product candidate in the United States until we receive regulatory approval of an NDA from the FDA.
We have not submitted an application or obtained marketing approval for doses of Aspertec other than the 325 mg dose, or for any other product candidate anywhere in the world. An NDA must include extensive preclinical and clinical data and supporting information to establish to the FDA’s satisfaction the product candidate’s safety and efficacy for each desired indication. The NDA must also include significant information regarding the chemistry, manufacturing and controls for the product candidate. Obtaining regulatory approval of an NDA can be a lengthy, expensive and uncertain process. In addition, failure to comply with FDA and other applicable U.S. and foreign regulatory requirements may subject us to administrative or judicially imposed sanctions, including:
● | warning or untitled letters; |
● | civil and criminal penalties; |
● | injunctions; |
● | withdrawal of approved products; |
● | product recalls; | |
| ● | seizure of products; |
● | total or partial suspension of production; and |
● | refusal to approve pending NDAs or supplements to approved NDAs. |
These actions could result in, among other things, substantial modifications to our business practices and operations; refunds of our products; the inability to obtain future approvals or marketing authorizations; and withdrawals or suspensions of current products from the market. Any of these events could disrupt our business and have a material adverse effect on our revenues, profitability and financial condition.
Prior to receiving approval to commercialize any future product candidates in the United States or abroad, we and any applicable collaboration partners must demonstrate with substantial evidence from well-controlled clinical trials, and to the satisfaction of the FDA and other regulatory authorities abroad, that such product candidates are safe and effective for their intended uses. Preclinical testing and clinical trials are long, expensive and uncertain processes. We may spend several years completing our testing for any particular product candidate, and failure can occur at any stage. Negative or inconclusive results or adverse medical events during a clinical trial could also cause the FDA or us to terminate a clinical trial or require that we repeat it or conduct additional clinical trials. Additionally, data obtained from preclinical studies and clinical trials can be interpreted in different ways and the FDA or other regulatory authorities may interpret the results of our studies and trials less favorably than we do. Even if we believe the preclinical or clinical data for a product candidate is promising, such data may not be sufficient to support approval by the FDA and other regulatory authorities. Administering any product candidates to humans may produce undesirable side effects, which could interrupt, delay or halt clinical trials of such product candidates and result in the FDA or other regulatory authorities denying approval of such product candidates for any or all targeted indications. The FDA or other regulatory authorities may determine that certain doses of Aspertec or any other product candidate that we develop are not effective, or are only moderately effective, or have undesirable or unintended side effects, toxicities, safety profile or other characteristics that preclude marketing approval or prevent or limit commercial use. In addition, any marketing approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially viable.
We are subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense and subject us to restrictions, withdrawal from the market, or penalties if we fail to comply with applicable regulatory requirements or if we experience unanticipated problems with our product candidates, when and if approved.
An approved product and its manufacturer are subject to continual review by the FDA and, as applicable, non-U.S. regulatory authorities. Any regulatory approval that we receive for our product candidates may be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for potentially costly post-marketing follow-up studies or surveillance to monitor the safety and efficacy of the product. In addition, if the FDA or non-U.S. regulatory authorities approve any of our product candidates, we will be subject to extensive and ongoing regulatory requirements by the FDA and other regulatory authorities with regard to labeling, packaging, adverse event reporting, storage, distribution, advertising, promotion, recordkeeping and submission of safety and other post-market information. Manufacturers of our products and manufacturers’ facilities are required to comply with cGMP regulations, which include requirements related to quality control and quality assurance as well as the corresponding maintenance of records and documentation. Further, regulatory authorities must approve these manufacturing facilities before they can be used to manufacture our products, and these facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP regulations. Accordingly, we and others with whom we work must continue to expend time, money and effort in all areas of regulatory compliance, including manufacturing, production and quality control.
We, and our direct and indirect suppliers, remain subject to the periodic inspection of our plants and facilities, review of production processes, and testing of our products to confirm that we are in compliance with all applicable regulations. For example, the FDA conducts ongoing inspections to determine whether our record keeping, production processes and controls, personnel and quality control are in compliance with the cGMP regulations, and other FDA regulations. Adverse findings during regulatory inspections may result in the implementation of Risk Evaluation and Mitigation Strategies programs, completion of government mandated post-marketing clinical studies, and government enforcement action relating to labeling, advertising, marketing and promotion, as well as regulations governing manufacturing controls noted above. The FDA has increased its enforcement activities related to the advertising and promotion of pharmaceutical, biological and medical device products. We will also be required to report certain adverse reactions and production problems, if any, to the FDA and to comply with requirements concerning advertising and promotion for our products. If we, any future collaboration partner or a regulatory authority discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory authority may impose restrictions on that product, the collaboration partner, the manufacturer or us, including requiring withdrawal of the product from the market or suspension of manufacturing.
The FDA closely regulates the post-approval marketing and promotion of drugs to ensure drugs are marketed only for the approved indications and in accordance with the provisions of the approved labeling and regulatory requirements. The FDA also imposes stringent restrictions on manufacturers’ communications regarding off-label use and if we do not restrict the promotion of our products only to their approved indications, we may be subject to enforcement action for off-label promotion. If we, our product candidates or the manufacturing facilities for our product candidates fail to comply with regulatory requirements of the FDA and/or other non-U.S. regulatory authorities, we could be subject to administrative or judicially imposed sanctions, including:
● | mandated modifications to promotional materials or the required provision of corrective information to healthcare practitioners; |
● | restrictions imposed on the product or its manufacturers or manufacturing processes; |
● | restrictions imposed on the labeling or marketing of the product; |
● | restrictions imposed on product distribution or use; |
● | requirements for post-marketing clinical trials; |
● | suspension of any ongoing clinical trials; |
● | suspension of or withdrawal of regulatory approval; |
● | voluntary or mandatory product recalls and publicity requirements; |
● | refusal to approve pending applications for marketing approval of new products or supplements to approved applications filed by us; |
● | restrictions on operations, including costly new manufacturing requirements; |
● | seizure or detention of our products; |
● | refusal to permit the import or export of our products; |
● | required entry into a consent decree, which can include imposition of various fines (including restitution or disgorgement of profits or revenue), reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; |
● | civil or criminal penalties; or |
● | injunctions. |
Widely publicized events concerning the safety risk of certain drug products have resulted in the withdrawal of drug products, revisions to drug labeling that further limit use of the drug products and the imposition by the FDA of risk evaluation and mitigation strategies, or REMS, to ensure that the benefits of the drug outweigh its risks. In addition, because of the serious public health risks of high profile adverse safety events with certain products, the FDA may require, as a condition of approval, costly REMS programs.
The regulatory requirements and policies may change and additional government regulations may be enacted with which we may also be required to comply. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or in other countries. If we or any future collaboration partner are not able to maintain regulatory compliance, we or such collaboration partner, as applicable, will not be permitted to market our future products and our business will suffer.
Failure to obtain regulatory approvals in foreign jurisdictions will prevent us from marketing our products internationally.
We may seek a distribution and marketing collaborator for Aspertec or other product candidates commercialized outside of the United States. In order to market our product candidates in the European Economic Area, or EEA (which comprises the 28 Member States of the EU, plus Norway, Iceland and Liechtenstein), and many other foreign jurisdictions, we or our collaboration partners must obtain separate regulatory approvals. We have had limited interactions with foreign regulatory authorities, and approval procedures vary among countries and can involve additional clinical testing. In addition, the time required to obtain approval from foreign regulatory authorities may differ from that required to obtain FDA approval. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one or more foreign regulatory authorities does not ensure approval by regulatory authorities in other foreign countries or by the FDA. However, a failure or delay in obtaining regulatory approval in one country may have a negative effect on our ability to obtain approval in other countries. The foreign regulatory approval process generally includes all of the risks associated with obtaining FDA approval. In addition, in many countries outside the United States, it is required that the product be approved for reimbursement before the product can be approved for sale in that country. We may or may not obtain foreign regulatory approvals on a timely basis, if at all. We may not be able to file for regulatory approvals and even if we file, we may not receive necessary approvals to commercialize our product candidates in any market.
Healthcare reform measures could hinder or prevent our product candidates’ commercial success.
In the United States, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare system that could affect our future revenue and profitability and the future revenue and profitability of our potential customers. Federal and state lawmakers regularly propose and, at times, enact legislation that results in significant changes to the healthcare system, some of which is intended to contain or reduce the costs of medical products and services. The Affordable Care Act contained a number of provisions, including those governing enrollment in federal healthcare programs, reimbursement changes and fraud and abuse measures that have impacted and will continue to impact existing government healthcare programs and will result in the development of new programs.
We currently benefit from regulations that mandate full reimbursement without cost sharing for aspirin when prescribed by a health care provider. Changes to these regulations could significantly reduce reimbursement rates in a manner that negatively affects our sales.
As a result of regulations enacted as part of the Affordable Care Act, we expect that Aspertec will qualify for 100% coverage when prescribed by physicians for the prevention of cardiovascular disease in patients with certain age-associated risks, requiring no out-of-pocket payments. While this will initially have the potential to expand the demand for Aspertec, changes to these regulations could have a significant adverse effect on reimbursement rates and, indirectly, on sales of Aspertec.
We are subject to healthcare laws, regulation and enforcement and our failure to comply with those laws could adversely affect our business, operations and financial condition.
Even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. We could be subject to healthcare fraud and abuse and patient privacy regulation by both the federal government and the states in which we conduct our business. The regulations that may affect our ability to operate include, without limitation:
● | the federal Anti-Kickback Statute, which prohibits, among other things, any person from knowingly and willfully offering, soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; |
● | the federal False Claims Act, which prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, false claims, or knowingly using false statements, to obtain payment from the federal government, and which may apply to entities that provide coding and billing advice to customers; |
● | federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
● | the federal physician sunshine requirements under the Affordable Care Act, which require manufacturers of drugs, devices, biologics, and medical supplies to report annually to the Centers for Medicare & Medicaid Services information related to payments and other transfers of value to physicians, other healthcare providers, and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members; and |
● | the federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act (collectively, “HIPAA”), which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information. |
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. In addition, recent healthcare reform legislation has strengthened these laws. For example, the recently enacted Affordable Care Act, among other things, amends the intent requirement of the Federal Anti-Kickback Statute and criminal healthcare fraud statutes. A person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it. In addition, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the Federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
These laws and regulations are broad in scope and they are subject to change and evolving interpretations, which could require us to incur substantial costs associated with compliance or to alter one or more of our sales or marketing practices. In addition, any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, the exclusion from participation in federal and state healthcare programs, imprisonment, or the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our financial results.
Failure to comply with domestic and international privacy and security laws can result in the imposition of significant civil and criminal penalties. The costs of compliance with these laws, including protecting electronically stored information from cyberattacks, and potential liability associated with failure to do so could adversely affect our business, financial condition and results of operations. We are subject to various domestic and international privacy and security regulations, including but not limited to HIPAA. HIPAA mandates, among other things, the adoption of uniform standards for the electronic exchange of information in common healthcare transactions, as well as standards relating to the privacy and security of individually identifiable health information, which require the adoption of administrative, physical and technical safeguards to protect such information. In addition, many states have enacted comparable laws addressing the privacy and security of health information, some of which are more stringent than HIPAA.
Our international operations will be subject to the Foreign Corrupt Practices Act.
As we pursue international licensing and sales arrangements outside the United States, we will be heavily regulated and expect to have significant interaction with foreign officials. Additionally, in many countries outside the United States, the health care providers who prescribe human pharmaceuticals are employed by the government and the purchasers of human pharmaceuticals are government entities; therefore, our interactions with these prescribers and purchasers would be subject to regulation under the Foreign Corrupt Practices Act, or FCPA, which prohibits any U.S. individual or business from paying, offering or authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business.
Compliance with these regulations may be costly, and may limit our ability to expand into certain markets. Further, we may inadvertently be found to be in violation of these and other regulations, which could result in material sanctions and penalties.
Risks Related to Our Common Stock, Warrants to Purchase Our Common Stock and this Offering
The price of our common stock may be volatile, and you may not be able to resell your shares at or above the public offering price.
The market price for the shares of our common stock may fluctuate significantly in response to a number of factors including:
● | ability to commercialize or delays in commercializing Aspertec; |
● | ability to commercialize or obtain regulatory approval for our product candidates, or delays in commercializing or obtaining regulatory approval; |
● | any need to suspend or discontinue clinical trials due to side effects or other safety risks, or any need to conduct studies on the long-term effects associated with the use of our product candidates; |
● | manufacturing issues related to Aspertec, our product candidates for clinical trials or future products for commercialization; |
● | commercial success and market acceptance of our product candidates following regulatory approval; |
● | undesirable side effects caused by product candidates after they have entered the market; |
● | ability to discover, develop and commercialize additional product candidates; |
● | announcements relating to collaborations that we may enter into with respect to the development or commercialization of our product candidates, or the timing of payments we may make or receive under these arrangements; |
● | success of our competitors in discovering, developing or commercializing products; |
● | strategic transactions undertaken by us; |
● | additions or departures of key personnel; |
● | product liability claims related to our clinical trials or product candidates; |
● | prevailing economic conditions; |
● | business disruptions caused by earthquakes or other natural disasters; |
● | disputes concerning our intellectual property or other proprietary rights; |
● | FDA or other U.S. or foreign regulatory actions affecting us or our industry; |
● | healthcare reform measures in the United States; |
● | sales of our common stock by our officers, directors or significant stockholders; |
● | future sales or issuances of equity or debt securities by us; |
● | fluctuations in our quarterly operating results; and |
● | the issuance of new or changed securities analysts’ reports or recommendations regarding us. |
In addition, the stock markets in general, and the markets for pharmaceutical stocks in particular, have experienced extreme volatility that has often been unrelated to the operating performance of the issuer. These broad market fluctuations may adversely affect the trading price or liquidity of our common stock. In the past, when the market price of a stock has been volatile, holders of that stock have sometimes instituted securities class action litigation against the issuer. If any of our stockholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the attention of our management would be diverted from the operation of our business.
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
Based on the beneficial ownership of our common stock as of June 1, 2017, after this offering, our officers and directors, together with holders of 5% or more of our outstanding common stock before this offering and their respective affiliates, will beneficially own approximately 10.0% of our common stock. Accordingly, these stockholders will continue to have significant influence over the outcome of corporate actions requiring stockholder approval, including the election of directors, any merger, consolidation or sale of all or substantially all of our assets or any other significant corporate transaction. The interests of these stockholders may not be the same as or may even conflict with your interests. For example, these large stockholders could delay or prevent a change of control of our company, even if such a change of control would benefit our other stockholders, which could deprive our stockholders of an opportunity to receive a premium for their common stock as part of a sale of our company or our assets and might affect the prevailing market price of our common stock. The significant concentration of stock ownership may adversely affect the trading price of our common stock due to investors’ perception that conflicts of interest may exist or arise.
We are an “emerging growth company” and we cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
In addition, Section 102 of the JOBS Act also provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933 (as amended, the “Securities Act”) for complying with new or revised accounting standards. As an “emerging growth company,” we can therefore delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. However, we are choosing to “opt out” of such extended transition period, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable.
Future sales of our common stock or securities convertible or exchangeable for our common stock may depress our stock price.
If our existing stockholders or holders of our options or warrants sell, or indicate an intention to sell, substantial amounts of our common stock in the public market, the trading price of our common stock could decline. The perception in the market that these sales may occur could also cause the trading price of our common stock to decline. Based on the number of shares of common stock outstanding as of June 1, 2017, upon the completion of this offering, we will have outstanding a total of 8,683,915 shares of common stock.
A total of 7,799,388 shares of common stock issued and outstanding are not subject to lock-up agreements with the placement agents and therefore may be eligible for sale in the public market upon closing of this offering. We expect that the lock-up agreements with the placement agents pertaining to this offering will expire 90 days from the date of this prospectus supplement. After the lock-up agreements expire, up to an additional 884,527 shares of common stock issued and outstanding will be eligible for sale in the public market, subject to volume limitations under Rule 144 under the Securities Act with respect to shares held by directors, executive officers and other affiliates. The placement agents may, however, in their sole discretion, permit our officers, directors and other stockholders and the holders of our outstanding options and warrants who are subject to the lock-up agreements to sell shares prior to the expiration of the lock-up agreements. Sales of these shares, or perceptions that they will be sold, could cause the trading price of our common stock to decline.
In addition, based on the number of shares subject to outstanding awards under the 2013 Plan and 2015 Plan, or available for issuance thereunder, as of March 31, 2017 on a pro forma basis, and including the initial reserves under the 2013 Plan and 2015 Plan, 1,409,611 shares of common stock that are either subject to outstanding options, outstanding but subject to vesting, or reserved for future issuance under the 2013 Plan and 2015 Plan will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules, the lock-up agreements and Rule 144 and Rule 701 under the Securities Act. We have also filed a registration statement permitting shares of common stock issued in the future pursuant to the 2013 Plan and 2015 Plan to be freely resold by plan participants in the public market, subject to the lock-up agreements, applicable vesting schedules and, for shares held by directors, executive officers and other affiliates, volume limitations under Rule 144. The 2015 Plan also contains a provision for the annual increase of the number of shares reserved for issuance under such plan, as described elsewhere in this prospectus supplement, which shares have also been registered. If the shares we may issue from time to time under the 2013 Plan and 2015 Plan are sold, or if it is perceived that they will be sold, by the award recipient in the public market, the trading price of our common stock could decline.
Investors in this offering will suffer immediate and substantial dilution of their investment.
If you purchase common stock in this offering, you will pay more for your shares than our adjusted net tangible book value per share. Based upon the public offering price of $6.875 per share, you will incur immediate and substantial dilution of $3.74 per share, representing the difference between the public offering price and our as adjusted net tangible book value per share.
We may be at risk of securities class action litigation.
We may be at risk of securities class action litigation. This risk is especially relevant for us due to our dependence on positive clinical trial outcomes and regulatory approvals of each of our product candidates. In the past, pharmaceutical companies have experienced significant stock price volatility, particularly when associated with binary events such as clinical trials and product approvals. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business and result in a decline in the market price of our common stock.
Raising additional capital may cause dilution to our existing stockholders or involve the issuance of securities with rights, preferences and privileges senior to those of holders of our common stock.
To raise capital, we may from time to time issue additional shares of common stock at a discount from the then-current trading price of our common stock. As a result, our common stockholders would experience immediate dilution upon the purchase of any shares of our common stock sold at such discount. In addition, as opportunities present themselves, we may enter into financing or similar arrangements in the future, including the issuance of debt securities, preferred stock or common stock. Whether or not we issue additional shares of common stock at a discount, any issuance of common stock will, and any issuance of other equity securities or of options, warrants or other rights to purchase common stock may, result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to decline. New investors could also gain rights, preferences and privileges senior to those of holders of our common stock, which could cause the price of our common stock to decline.
We will have broad discretion in the use of the net proceeds of this offering and may not use them effectively.
Our management will have broad discretion over the use of the net proceeds from this offering. Because of the number and variability of factors that will determine our use of such proceeds, you may not agree with how we allocate or spend the proceeds from this offering. We may pursue collaborations, clinical trials or discovery and development programs that do not result in an increase in the market value of our common stock and that may increase our losses. Our failure to allocate and spend the net proceeds from this offering effectively would have a material adverse effect on our financial condition and business. Until the net proceeds are used, they may be placed in investments that do not produce significant investment returns or that may lose value.
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of PLx more difficult and may prevent attempts by our stockholders to replace or remove management.
Provisions in our certificate of incorporation and bylaws may delay or prevent an acquisition or a change in management. In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits stockholders owning in excess of 15% of the outstanding combined company voting stock from merging or combining with the combined company. Although we believe these provisions collectively will provide for an opportunity to receive higher bids by requiring potential acquirors to negotiate with our board of directors, they would apply even if the offer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove then current management by making it more difficult for stockholders to replace members of the board of directors, which is responsible for appointing the members of management.
Provisions of our charter documents limit the liability of our officers and directors, which could limit the ability of stockholders (and outside parties) to bring claims against such officers and directors.
Our certificate of incorporation contains provisions that limit the liability of our current and former directors for monetary damages to the fullest extent permitted by Delaware law. Delaware law provides that directors of a corporation will not be personally liable for monetary damages for any breach of fiduciary duties as directors, except liability for:
● | any breach of the director’s duty of loyalty to the corporation or its stockholders; |
● | any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
● | unlawful payments of dividends or unlawful stock repurchases or redemptions; or |
● | any transaction from which the director derived an improper personal benefit. |
Such limitation of liability does not apply to liabilities arising under federal securities laws and does not affect the availability of equitable remedies, such as injunctive relief or rescission.
Our certificate of incorporation and our bylaws provide that we are required to indemnify our directors to the fullest extent permitted by Delaware law. Our bylaws also provide that, upon satisfaction of certain conditions, we shall advance expenses incurred by a director in advance of the final disposition of any action or proceeding, and permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in that capacity regardless of whether we would otherwise be permitted to indemnify him or her under the provisions of Delaware law. Our certificate of incorporation and bylaws provide our board of directors with discretion to indemnify our officers and employees when determined appropriate by the board. We have entered and expect to continue to enter into agreements to indemnify our directors and executive officers. With certain exceptions, these agreements provide for indemnification for related expenses including, among other things, attorneys’ fees, judgments, fines and settlement amounts incurred by any of these individuals in any action or proceeding. We believe that these bylaw provisions and indemnification agreements are necessary to attract and retain qualified persons as directors and officers. We also maintain customary directors’ and officers’ liability insurance.
The limitation of liability and indemnification provisions in our certificate of incorporation and bylaws may discourage stockholders from bringing a lawsuit against our directors for breach of their fiduciary duty. They may also reduce the likelihood of derivative litigation against our directors and officers, even though an action, if successful, might benefit us and other stockholders. Further, a stockholder’s investment may be adversely affected to the extent that we pay the costs of settlement and damage awards against directors and officers as required by these indemnification provisions. At present, there is no pending litigation or proceeding involving any of our directors, officers or employees for which indemnification is sought, and we are not aware of any threatened litigation that may result in claims for indemnification.
We do not anticipate paying any cash dividends on our capital stock in the foreseeable future; therefore capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
We have never declared or paid cash dividends on our capital stock. We do not anticipate paying any cash dividends on our capital stock in the foreseeable future. We currently intend to retain all available funds and any future earnings to fund the development and growth of our business. In addition, the terms of any future debt financing arrangement may contain terms prohibiting or limiting the amount of dividends that may be declared or paid on our common stock. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
If securities or industry analysts do not publish research, or publish inaccurate or unfavorable research, about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend, in part, on the research and reports that securities or industry analysts publish about us or our business. Securities and industry analysts do not currently, and may never, publish research on us. If no securities or industry analysts commence coverage of us, the trading price for our stock would likely be negatively impacted. In the event securities or industry analysts initiate coverage, if one or more of the analysts who cover us downgrade our stock or publish inaccurate or unfavorable research about our business, our stock price would likely decline. In addition, if our operating results fail to meet the forecast of analysts, our stock price would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, demand for our stock could decrease, which might cause our stock price and trading volume to decline.
You may not be able to resell your warrants purchased in the private placement conducted concurrently with this offering.
There is no established trading market for the warrants purchased in the private placement conducted concurrently with this offering, and we do not expect such a market to develop. In addition, we do not intend to apply for listing of the warrants on any securities exchange or other nationally recognized trading system, and you may not be able to resell your warrants. If your warrants cannot be resold, you will have to depend upon any appreciation in the value of our common stock over the exercise price of the warrants in order to realize a return on your investment in the warrants.
Investors will have no rights as a holder of common stock with respect to their warrants until they exercise their warrants and acquire our common stock.
Until you acquire shares of our common stock upon exercise of your warrants, you will have no rights with respect to the shares of our common stock underlying such warrants. Upon exercise of your warrants, you will be entitled to exercise the rights of a holder of common stock only as to matters for which the record date occurs after the exercise date.
USEOF PROCEEDS
We estimate that the net proceeds from our issuance and sale of 2,646,091 shares of our common stock in this offering will be approximately $16.8 million after deducting placement agent fees and estimated offering expenses payable by us.
We currently estimate that we will use the net proceeds from this offering, together with our cash and cash equivalents, as follows:
● | to advance Aspertec 325 mg to market-readiness; |
● | to obtain supplemental regulatory approval of Aspertec 81 mg; |
● | to fund the technology transfer and the commercial scale validation and manufacturing necessary to support both efforts; | |
| ● | to expand our management team; |
● | to fund a PK/PD clinical study (for marketing purposes only) comparing Aspertec 81mg to enteric coated aspirin; and |
● | to fund working capital, capital expenditures and other general corporate purposes, which may include the acquisition or licensing of other products, businesses or technologies. |
This expected use of the net proceeds from this offering and our existing cash and cash equivalents represents our intentions based upon our current plans and business conditions. The amounts and timing of our actual expenditures may vary significantly depending on numerous factors, including the progress of our development and commercialization efforts and the status of and results from clinical studies, as well as any collaborations that we may enter into with third parties and any unforeseen cash needs. As a result, our management will retain broad discretion over the allocation of the net proceeds from this offering. We have no current understandings, agreements or commitments for any material acquisitions or licenses of any products, businesses or technologies.
Pending our use of the net proceeds from this offering, we intend to invest the net proceeds in a variety of capital preservation investments, including short-term, investment grade, interest bearing instruments and U.S. government securities.
PRICERANGE OF COMMON STOCK
Our common stock is listed on The NASDAQ Capital Market and trades under the symbol “PLXP.” Prior to April 20, 2017, Dipexium’s common stock traded on The NASDAQ Capital Market under the symbol “DPRX”. The following table sets forth, for the quarterly periods indicated, the high and low sale price per share of Dipexium’s common stock for all periods prior to April 20, 2017 and our common stock since that date as reported on The NASDAQ Capital Market, and all such sale prices give effect to the 1-for-8 reverse stock split effected by Dipexium on April 19, 2017:
| Low | High | |||||
Year ended December 31, 2015 | ||||||
First Quarter | $ | 85.60 | $ | 121.12 | ||
Second Quarter | $ | 90.00 | $ | 120.00 | ||
Third Quarter | $ | 86.40 | $ | 136.80 | ||
Fourth Quarter | $ | 80.96 | $ | 117.28 | ||
Year ended December 31, 2016 | ||||||
First Quarter | $ | 48.32 | $ | 97.84 | ||
Second Quarter | $ | 68.32 | $ | 105.60 | ||
Third Quarter | $ | 76.00 | $ | 142.00 | ||
Fourth Quarter | $ | 9.20 | $ | 126.72 | ||
Year ending December 31, 2017 | ||||||
First Quarter | $ | 8.80 | $ | 12.80 | ||
Second Quarter (through June 8, 2017) | $ | 5.60 | $ | 11.20 |
On June 8, 2017, the consolidated closing bid price and the last reported sale price of our common stock on The NASDAQ Capital Market were $6.75 per share and $6.95 per share, respectively. As of June 1, 2017, we had approximately 125 holders of record of our common stock. The actual number of stockholders is greater than this number of record holders and includes stockholders who are beneficial owners but whose shares are held in street name by brokers and other nominees.
DIVIDENDPOLICY
We have never declared or paid, and do not anticipate declaring or paying, in the foreseeable future, any cash dividends on our capital stock. Future determination as to the declaration and payment of dividends, if any, will be at the discretion of our board of directors and will depend on then existing conditions, including our operating results, financial conditions, contractual restrictions, capital requirements, business prospects and other factors our board of directors may deem relevant.
The following table sets forth cash and cash equivalents and capitalization as of March 31, 2017:
| ● | for PLx on an actual basis as recast for the effects of the Merger; |
| ● | for Dipexium on an actual basis as adjusted for reverse merger acquisition accounting; and |
| ● | on an as adjusted, pro forma basis to reflect the sale by us of 2,646,091 shares of common stock in this offeringat an offering price of $6.875 per share after deducting the placement agent fees and estimated offering expenses payable by us, excluding the proceeds, if any, from the exercise of warrants issued in the concurrent private placement. |
You should read this table together with the section in the documents incorporated by reference in this prospectus supplement entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and related notes included elsewhere in this prospectus supplement, the accompanying prospectus and the documents incorporated herein and therein by reference.
As of March 31, 2017 (unaudited) | ||||||||||||
PLx actual as recast for the Merger | Dipexium actual as adjusted for reverse merger acquisition accounting | Pro-forma combined, as adjusted for this offering | ||||||||||
Cash and cash equivalents | $ | 425,785 | $ | 12,414,004 | $ | 29,615,146 | ||||||
Stockholders equity: | ||||||||||||
Common stock, $0.001 par value; 100,000,000shares authorized, 4,383,433 issued and outstanding,PLx actual as recast; 30,000,000 shares authorized,11,129,747 issued and outstanding, Dipexium actualas adjusted; 100,000,000 shares authorized, 8,683,915shares issued and outstanding, pro forma combined,as adjusted | $ | 4,383 | $ | 1,601 | $ | 8,630 | ||||||
Additional paid in capital | 49,849,711 | 17,509,417 | 84,131,839 | |||||||||
Accumulated deficit | (53,412,484 | ) | 682,500 | (52,729,984 | ) | |||||||
Total stockholders' equity | $ | (3,558,390 | ) | $ | 18,193,518 | $ | 31,410,485 | |||||
Total capitalization | $ | (3,558,390 | ) | $ | 18,193,518 | $ | 31,410,485 | |||||
The number of shares of common stock to be outstanding after this offering is based on 1,391,218 shares of Dipexium common stock outstanding as of March 31, 2017, and excludes the following:
| ● | 266,668 shares of common stock issuable upon the exercise of stock options outstanding as of March 31, 2017, at a weighted average exercise price of $73.12 per share under the 2013 Plan; and |
| ● | 978 shares of our common stock, subject to increase on an annual basis, reserved for future issuance under the 2013 Plan as of March 31, 2017. |
On a pro forma basis, the number of shares of our common stock to be outstanding after this offering is based on 6,025,349 shares of our common stock outstanding as of March 31, 2017, and excludes the following:
| ● | 691,386 shares of our common stock issuable upon the exercise of stock options outstanding as of March 31, 2017 at a weighted average exercise price of $12.44 per share under the 2015 Plan; |
| ● | 450,580 shares of our common stock reserved for future issuance under the 2015 Plan as of March 31, 2017; |
| ● | 266,668 shares of common stock issuable upon the exercise of stock options outstanding as of March 31, 2017, at a weighted average exercise price of $73.12 per share under the 2013 Plan; |
| ● | 978 shares of our common stock, subject to increase on an annual basis, reserved for future issuance under the 2013 Plan as of March 31, 2017; and |
| ● | 2,646,091 shares of common stock issuable upon exercise of warrants issued to investors in the concurrent private placement. |
Dilution is the amount by which the offering price paid by the purchasers of the shares of common stock sold in the offering exceeds the net tangible book value per share of our common stock after this offering. The historical net tangible book value of our common stock as of March 31, 2017 was approximately $10.5 million, or $1.74 per share on a pro forma basis.
After giving effect to our receipt of the net proceeds from our sale of 2,646,091 shares of our common stock at an offering price of $6.875 per share after deducting placement agent fees and estimated offering expenses payable by us, our as adjusted pro-forma net tangible book value as of March 31, 2017 would have been approximately $27.3 million, or $3.14 per share. This represents an immediate increase in as adjusted net tangible book value of $1.40 per share to our existing stockholders and an immediate dilution of $3.74 per share to investors purchasing common stock in this offering.
The following table illustrates this dilution on a per share basis to new investors:
Assumed public offering price per share | $ | 6.875 | ||||||
Historical pro-forma net tangible book value per share as of March 31, 2017 | $ | 1.74 | ||||||
Increase in as adjusted pro-forma net tangible book value per share attributable to new investors | $ | 1.40 | ||||||
As adjusted pro-forma net tangible book value per share after this offering | $ | 3.14 | ||||||
Dilution per share to investors participating in this offering | $ | 3.74 |
The number of shares of common stock to be outstanding after this offering is based on 1,391,218 shares of Dipexium common stock outstanding as of March 31, 2017, and excludes the following:
| ● | 266,668 shares of common stock issuable upon the exercise of stock options outstanding as of March 31, 2017, at a weighted average exercise price of $73.12 per share under the 2013 Plan; and |
| ● | 978 shares of our common stock, subject to increase on an annual basis, reserved for future issuance under the 2013 Plan as of March 31, 2017. |
On a pro forma basis, the number of shares of our common stock to be outstanding after this offering is based on 6,025,349 shares of our common stock outstanding as of March 31, 2017, and excludes the following:
| ● | 691,386 shares of our common stock issuable upon the exercise of stock options outstanding as of March 31, 2017 at a weighted average exercise price of $12.44 per share under the 2015 Plan; |
| ● | 450,580 shares of our common stock reserved for future issuance under the 2015 Plan as of March 31, 2017; |
| ● | 266,668 shares of common stock issuable upon the exercise of stock options outstanding as of March 31, 2017, at a weighted average exercise price of $73.12 per share under the 2013 Plan; |
| ● | 978 shares of our common stock, subject to increase on an annual basis, reserved for future issuance under the 2013 Plan as of March 31, 2017; and |
| ● | 2,646,091 shares of common stock issuable upon exercise of warrants issued to investors in the concurrent private placement. |
The shares of common stock issuable upon the exercise of the warrants issued to investors in the concurrent private placement and the exercise price of such warrants are subject to adjustments in certain circumstances. See “Private Placement Transaction—Description of Warrants” for more information regarding these adjustments.
TO THE EXTENT OUR OUTSTANDING OPTIONS AND WARRANTS ARE EXERCISED, YOU MAY EXPERIENCE FURTHER DILUTION. THE ABOVE ILLUSTRATION OF DILUTION PER SHARE TO INVESTORS PARTICIPATING IN THIS OFFERING ASSUMES NO EXERCISE OF OUTSTANDING OPTIONS OR OUTSTANDING WARRANTS TO PURCHASE SHARES OF OUR COMMON STOCK.
We are a late-stage specialty pharmaceutical company initially focused on developing our clinically validated and patent-protected PLxGuard delivery system to provide safer and more effective aspirin products. Our PLxGuard delivery system works by releasing active pharmaceutical ingredients into the duodenum, the first part of the small intestine immediately below the stomach, rather than in the stomach itself. We believe this improves the absorption of many drugs currently on the market or in development, and reduces acute GI side effects — including erosions, ulcers and bleeding — associated with aspirin and ibuprofen, and potentially other drugs.
Our FDA approved lead product, Aspertec 325 mg, is a novel formulation of aspirin that uses the PLxGuard delivery system to significantly reduce acute GI side effects while providing superior antiplatelet effectiveness for cardiovascular disease prevention as compared with the current standard of care, enteric coated aspirin. Aspertec 325 mg was originally approved under the drug name aspirin, and the proprietary name ‘Aspertec’ was granted subsequent to the NDA approval. A companion 81 mg dose of the same novel formulation — Aspertec 81 mg — is in late-stage development and will be the subject of a supplemental New Drug Application, or sNDA, leveraging the already approved status of Aspertec 325 mg.
Our commercialization strategy will target both the OTC and prescription markets, taking advantage of the existing OTC distribution channels for aspirin while leveraging the FDA approval of Aspertec 325 mg and expected approval for Aspertec 81 mg for OTC and prescription use when recommended by physicians for cardiovascular disease treatment and prevention. Given our clinical demonstration of better antiplatelet efficacy (as compared with enteric coated aspirin) and better acute GI safety, we intend to use a physician-directed sales force to inform physicians — and, by extension, consumers — about our product’s clinical results in an effort to command both greater market share and a higher price for our superior aspirin product. Our product pipeline also includes other oral NSAIDs using the PLxGuard delivery system that may be developed, including a clinical-stage, GI-safer ibuprofen — PL1200 Ibuprofen 200 mg — for pain and inflammation.
PLxGuardTM Delivery System
Our PLxGuard delivery system uses surface acting lipids, such as phospholipids and free fatty acids, to modify the physiochemical properties of various drugs to selectively release these drugs to targeted portions of the GI tract. Unlike tablet or capsule polymer coating technologies (e.g., enteric coating), which rely solely on drug release based on pH differences in the GI tract, the PLxGuardsystem uses the differential in pH and bile acid contents between the stomach and duodenum, to target Aspertec’s release. This approach is intended to more reliably release active pharmaceutical ingredients in the duodenum and decrease their exposure to the stomach, which is more susceptible to NSAID-induced bleeding and ulceration. The PLxGuard system is a platform technology that we believe may be useful in improving the absorption of many acid labile, corrosive, and insoluble or impermeable drugs.
We believe our PLxGuard delivery system has the potential to improve many already approved drugs and drugs in development, because it:
● | enhances the efficacy of the drug using our technology; |
● | improves the GI safety of the drug; |
● | provides new or extended patent protection for an already approved or in development drug: and |
● | can utilize the 505(b)(2) New Drug Application (NDA) regulatory path, which may provide a faster and lower-cost FDA approval route when used with already approved drugs. |
The PLxGuard delivery system has clinically proven these benefits with our novel formulations using aspirin and ibuprofen and has preclinical evidence supporting the potential for a GI-safer oral diclofenac and intravenous indomethacin products. Other existing or new drugs in development that may benefit from the PLxGuard delivery system will be evaluated either by us or through collaboration agreements with other companies.
Product Pipeline
Our lead product, Aspertec 325 mg, has been approved by the FDA for OTC distribution and is the first-ever FDA-approved liquid-fill aspirin capsule. All the clinical trials necessary for product launch have been completed. In clinical trials in diabetic patients at risk for cardiovascular disease, Aspertec 325 mg demonstrated better antiplatelet efficacy compared with enteric coated aspirin, which is the current standard of care for cardiovascular disease prevention and treatment. Aspertec 325 mg delivers faster antiplatelet efficacy than enteric coated aspirin with a median time to 99% inhibition of serum Thromboxane B2 of two hours compared with 48 hours for enteric coated aspirin. Serum Thromboxane B2 is a clinically accepted marker for antiplatelet efficacy, which is sometimes referred to as aspirin response.
Aspertec 325 mg provides more reliable, predictable and sustained antiplatelet benefits than enteric coated aspirin with a 3 – 5 times greater chance of a complete aspirin antiplatelet effect than enteric coated aspirin. Aspertec 325 mg has demonstrated a statistically significant 65% reduction in the risk of acute ulcers compared with immediate release aspirin in healthy subjects with an age-associated risk for cardiovascular disease. This acute GI safety benefit may also be important for acute coronary syndrome (ACS) patients. Moreover, we believe ACS patients who are also diabetics and suffer from gastroparesis, or a lack of digestive stomach motility, could also benefit from Aspertec due to its more predictable absorption when compared to enteric coated aspirin. The acute GI safety benefit may also be used to differentiate Aspertec 325 mg from products intended for use in conditions associated with pain and inflammation, including other aspirin and NSAID products.
Aspertec 81 mg is our lower-dose companion product for Aspertec 325 mg. This product utilizes exactly the same formulation as the 325 mg product (except delivered in a capsule one quarter the size) and will be the subject of an sNDA, which we expect to file with the FDA in the last quarter of 2017. We will rely on the clinical results for Aspertec 325 mg for the Aspertec 81 mg sNDA and do not anticipate any additional clinical trials will be required, effectively positioning this product as an end of Phase 3 status. We intend to begin selling both products by the end of 2018.
Early studies suggest that Aspirin could be beneficial in the prevention of different cancers by enhancing the efficacy of some current cancer therapies when co-administered with certain drugs. Aspertec has been or continues to be evaluated in this regard at The University of Texas M.D. Anderson Cancer Center, The University of Texas Health Science Center at Houston and Baylor College of Medicine. Recent publications in peer reviewed medical journals of some of the preclinical research using Aspertec suggest Aspertec may be more beneficial in this regard than current marketed aspirin for colorectal cancer prevention as published in Cancer Prevention Research (Cancer Prev Res January 13 2017), and when co-administered with Bevacizumab (Avastin®), Aspertec may provide better efficacy for ovarian cancer therapy than Bevacizumab alone or in combination with currently marketed aspirin as published in Molecular Cancer Therapeutics (Mol Cancer Ther December 1 2016 (15) (12) 2894-2904). The US Preventive Services Task Force (USPSTF) recently recommended that individuals ages 50 to 69 years old daily take low-dose aspirin for 10 years or longer to prevent colorectal cancer, with the provision they consult their physician given the GI risk of aspirin. More research would need to be conducted using Aspertec in order to confirm these early findings.
We believe our technology may be used with other selected NSAIDs, such as ibuprofen. For the OTC market, we have used the PLxGuard delivery system to create a lipid-based formulation of ibuprofen, PL1200 Ibuprofen 200 mg, and PL1100 Ibuprofen 400 mg product for prescription doses of ibuprofen. We have OTC and prescription (Rx) Investigational New Drug applications, or INDs, active with the FDA and have demonstrated bioequivalence with the OTC 200 mg dose ibuprofen to support a 505(b)(2) NDA in fasted-state clinical trials at three different doses, 200 mg, 400 mg and 800 mg. Using the PL1200 capsules at prescription doses, we demonstrated better GI safety in osteoarthritic patients with equivalent analgesic and anti-inflammatory efficacy, when compared with prescription ibuprofen in a six-week endoscopy pilot clinical trial. PL1200 and PL1100 Ibuprofen may be considered as being in Phase 1 in the FDA process and may qualify for the 505(b)(2) NDA path.
The following table summarizes information regarding our product candidates and development program:
 |
We have several active INDs related to our aspirin and ibuprofen development programs, as shown below:
Product | IND(s) | Status | Filed On | Clinical Trials | ||||
Aspertec (OTC) | 074290 | Active | 12/20/2007 | PL-ASA-001 | ||||
Aspertec (Rx) | 077280 | Active | 12/20/2007 | PL-ASA-002 | ||||
PL-ASA-003 | ||||||||
PL-ASA-004 | ||||||||
PL-ASA-005 | ||||||||
| PL-ASA-006 | |||||||
PL1200 Ibuprofen, 200 mg (OTC) | 102678 | Active | 10/06/2009 | PL-IB-001 | ||||
PL1100 Ibuprofen, 400 mg (Rx) | 062824 | Active | 12/22/2003 | PL-IB-002 | ||||
PL-IB-003 | ||||||||
| PL-IB-004 |
Additionally, we have one withdrawn IND related to the naproxen development program, as shown below:
Product | IND(s) | Status | Filed On | Clinical Trials | ||||
PL3100 Naproxen, 250 mg (Rx) | 103964 | Withdrawn | 08/29/2009 | PL-NAP-001 | ||||
Development terminated | PL-NAP-002 |
The Market
Cardiovascular/Aspirin Market
Aspirin, also known as acetylsalicylic acid, or ASA, is a foundational medicine for the treatment and prevention of cardiovascular disease, or CVD — a collective term for chest pain, coronary artery disease, heart attack, heart failure, high blood pressure and stroke — which is the leading cause of death in the United States. Aspirin is widely used to treat patients exhibiting signs and symptoms of heart attack or stroke (collectively referred to as ACS), and is commonly prescribed or recommended by physicians, in addition to other drugs such as cholesterol or blood pressure medications, as the standard of care for treating ACS and preventing recurrent ACS for the duration of a patient’s life.
The 325 mg dose is generally prescribed:
● | in an acute setting for the treatment of ACS; |
● | following an interventional procedure (such as placement of a stent); and |
● | for preventing heart attack or stroke during the high-risk period following an event or intervention. |
Eventually, most patients move to the 81 mg dose for secondary prevention applications and for high-risk primary prevention, as they are typically directed to take aspirin every day for the rest of their life. The 81 mg aspirin dose is the most prevalent, representing approximately 70% to 80% of aspirin sales in the United States, followed by the 325 mg dose and then several other dose forms including powdered and effervescent aspirin products. In the United States, the primary use for aspirin is for the prevention and treatment of cardiovascular disease as evidenced by the predominance of the 81 mg dose. The Centers for Disease Control and Prevention recommends that approximately 34%, or 107 million people, of the U.S. population (U.S. Census Bureau July 2014 estimate) should be taking aspirin while fewer than half of those actually do so.
Aspirin is also used for pain relief and fever reduction and is the most common NSAID available. The most widely used dose for pain is 325 mg. A GI-safer Aspertec 325 mg may be able to capture market share from other aspirin and NSAID products in the large pain and inflammation market.
The leading aspirin brand reported global annual sales in 2016 exceeding $1 billion. Branded OTC U.S. aspirin products include Bayer®, Ecotrin® and St. Joseph®, and there are numerous private label or store brands. Because of the known GI toxicity issues associated with immediate release, or regular aspirin, enteric coated aspirin has evolved to become the leading aspirin dose form, representing over 90% of U.S. aspirin sales in 2013. This success is largely based on early clinical studies suggesting that enteric coated aspirin showed a reduction in superficial gastric lesions as compared with regular aspirin. However, when measured using contemporary clinical procedures, enteric coated aspirin has not continued to demonstrate such reductions. These deficiencies create a significant opportunity for the demonstrated efficacy of Aspertec.
Pain & Inflammation/Ibuprofen
The U.S. OTC analgesic market was over $4 billion in annual sales in 2016 (Consumer Healthcare Products Association). The leading OTC NSAID brands in the United States represented greater than $1 billion in annual sales in 2016 with one leading OTC ibuprofen brand generating greater than $750 million in worldwide in sales in 2016 (Evaluate). This class of OTC drugs is one of the most widely used drugs worldwide. Increasing concerns over liver toxicity associated with acetaminophen products coupled with known ibuprofen analgesic superiority over acetaminophen, indicate there is a substantial global opportunity for a GI safer ibuprofen product. While we do not believe our technology will work with the entire NSAIDs class, it is possible that our technology may be successfully applied to other NSAIDs beyond aspirin and ibuprofen. For example, we have preclinical data suggesting that diclofenac — a leading NSAID for pain and inflammation outside the United States — is a viable product candidate.
Our Key Competitive Strengths
● | Our lead product, Aspertec 325 mg, has been approved by the FDA under the 505(b)(2) NDA process. We intend to leverage clinical studies and market research to launch Aspertec on a commercial scale using a combined prescription and OTC strategy that takes advantage of the existing OTC distribution channels for aspirin while leveraging the FDA approval of Aspertec 325 mg for prescription and recommendation by physicians for cardiovascular disease treatment and prevention. We will be seeking approval of the companion Aspertec 81 mg dose via an sNDA that should benefit from the prior approval of Aspertec 325 mg. |
● | Our management team has extensive experience in development and commercialization of OTC products. Mr. Valentino, our Executive Chairman of the Board, brings over 30 years of experience in the healthcare industry. Mr. Valentino successfully built Adams Respiratory Therapeutics into a fully integrated specialty pharmaceutical company with more than $490 million in annual revenue and leading OTC brands such as Mucinex® and Delsym®. In December 2007, Adams Respiratory Therapeutics sold to Reckitt Benckiser for approximately $2.3 billion. Ms. Giordano, our President and Chief Executive Officer, has over 20 years in the life science industry and has successfully developed commercialization plans for several new product launches across various therapeutic areas, specifically building physician directed sales teams, including with one of the most successful products in the cardiology sector, Lipitor. |
● | Aspertec 325 mg has demonstrated clinically superior antiplatelet efficacy in diabetic patients when compared to enteric coated aspirin, the current standard of care for cardiovascular disease prevention. Aspertec 325 mg delivers faster and more reliable, predictable and sustained antiplatelet benefits than enteric coated aspirin as clinically demonstrated in diabetic patients at risk for cardiovascular disease with a median time to 99% inhibition of serum Thromboxane B2 (an accepted clinical maker of auto-platelet efficacy) of two hours compared with 48 hours and with a 3 – 5 times greater chance of a complete aspirin antiplatelet effect than enteric coated aspirin. |
● | Aspertec 325 mg has demonstrated clinically superior GI safety when compared to regular aspirin. Aspertec 325 mg has demonstrated a statistically significant 65% reduction in the risk of acute ulcers compared with regular aspirin in healthy subjects with an age associated risk for cardiovascular disease. Based on physician market research, we believe that our clinical data supporting superior acute GI safety as compared to regular aspirin is important for physicians who are having difficulty keeping high-risk patients on sustained aspirin use due to lack of tolerability. |
● | Our PLxGuard delivery system is a platform technology that may improve the GI safety and efficacy when applied to already approved drugs. Our PLxGuard delivery system is a platform technology that we believe could improve the GI safety of selected NSAIDs that meet specific criteria. The U.S. analgesic market was over $4 billion in annual sales in 2016 (Consumer Healthcare Products Association). The leading NSAID brands in the United States represented greater than $1 billion annual sales in 2016 (Statista Inc.) with one leading OTC ibuprofen brand generating more than $750 million in worldwide sales in 2016 (Evaluate). We intend to explore new development opportunities and utilize the 505(b)(2) NDA regulatory pathway to decrease the time and costs associated with obtaining FDA approval for new products. |
● | Our PLxGuard delivery system may lower development risk and costs by leveraging the 505(b)(2) NDA pathway. A 505(b)(2) NDA is permitted to rely on the FDA’s prior conclusions regarding the safety and effectiveness of a previously approved drug, or to rely in part on data in the public domain. Reliance on the FDA’s prior findings or data collected by others may expedite the development program for our product candidates by potentially decreasing the amount of clinical data that we would need to generate to submit an NDA. As the FDA has previously approved Aspertec 325 mg pursuant to a 505(b)(2) NDA, we believe that there is a strong likelihood that our future products would similarly qualify. The factors related to this qualification are expected to reduce the time and costs associated with clinical trials when compared to a traditional NDA for a new chemical entity. We also believe the strategy of targeting drugs with proven safety and efficacy provides a better prospect of clinical success of our proprietary development portfolio as compared to de novo drug development. We believe that the average time to market and cost of clinical trials for our products could be less than that required to develop a new drug. |
● | Our issued patents and patent applications in the United States and worldwide may protect us from generic competition and enable a higher price point than current aspirin products. We believe our patent portfolio provides strong protection of our delivery platform, drug formulations and manufacturing processes with 44 issued patents and additional pending applications with expirations ranging from December 19, 2021 through September 29, 2032. |
Our Strategy
Our goal is to become a leading specialty pharmaceutical company, commercializing both the Aspertec 325 mg and Aspertec 81 mg dose forms and developing additional branded products using our PLxGuard technology. The key elements of our strategy are to:
Successfully commercialize Aspertec 325 mg. Prior to launch, we will need to finalize labeling with the FDA, scale up our commercial production capabilities, and conduct three validation manufacturing runs required to satisfy FDA pre-launch requirements. We intend to hire, train and deploy a 45- to 125-person sales force to support a national launch to appropriate healthcare providers. This will provide a technical focus on Aspertec 325 mg and 81 mg products based upon our compelling clinical results to enhance the value of these products. We intend to begin selling both products by the end of 2018.
Obtain FDA approval for Aspertec 81 mg and prepare it for commercial launch. This will include, in addition to the efforts described above, submission of an sNDA seeking approval to launch Aspertec 81 mg simultaneously with the already-approved 325 mg strength. Once Aspertec 81 mg product becomes available as the result of planned manufacturing activities, PLx expects to conduct a clinical crossover trial of Aspertec 81 mg comparing pharmacokinetic and pharmacodynamic endpoints with 81 mg enteric coated aspirin. The trial would be intended for commercial purposes and is not otherwise required in connection with any near-term regulatory filings or approvals.
Initiate a physician-targeted market launch strategy. Our market launch strategy will focus on physician detailing by targeting physicians who are seeking reliable and predictable antiplatelet efficacy for their highest risk cardiovascular patients. This strategy is designed to drive product sales professionally with a technical sales approach. We anticipate taking advantage of aspirin’s unique OTC and prescription approval by using a sales force to call on physicians to inform them of the clinically validated attributes of Aspertec. This physician sales effort will be designed to create interest in Aspertec among healthcare professionals and to influence their recommendations and prescriptions of Aspertec over other aspirin products.
Move beyond aspirin to leverage our PLxGuard delivery system in new products. While we are currently focused on Aspertec, there is a pipeline of additional opportunities that can be exploited. We are developing novel formulations that combine already-approved selected NSAIDs that meet our specific criteria with our proprietary PLxGuard technology to make safer and more effective new products. In addition to Aspertec, we have clinical data for a GI-safer OTC ibuprofen product, PL1200 Ibuprofen 200 mg, and preclinical data for a GI-safer diclofenac and a GI-safer intravenous indomethacin. We believe that the PLxGuard technology may also prove a novel drug delivery platform for corrosive, acid labile and insoluble and impermeable drugs providing delivery along the GI tract. We believe that by focusing initially on commercializing Aspertec we can demonstrate the viability of our PLxGuard delivery system as a versatile platform technology.
Hire additional senior management and key personnel to support the formulation and execution of successful product launches. Though we will continue to evaluate other market launch options, including co-promotion in the United States with one or more existing firms, we currently expect to launch Aspertec in the United States using our own sales and marketing team. This will require additional hiring of both management and sales personnel.
Seek strategic partners to commercialize Aspertec and other products in global markets. We have licensed the rights to commercialize Aspertec in the People’s Republic of China (including Macao and Hong Kong), along with an option for commercialization in Cambodia, Indonesia, Laos, Malaysia, Myanmar, Papua New Guinea, Philippines, Singapore, Taiwan, Thailand and Vietnam. We will continue to seek partners in other global markets.
Use of Aspirin in Management of Cardiovascular Disease
Aspirin is critical to the management of cardiovascular disease. However, current aspirin products have an elevated risk of GI side effects, such as ulceration and dyspepsia that can lead to discontinuation. Discontinuation of aspirin leads to more than a three-fold increase in risk of recurrent major cardiac events. Enteric coated aspirin is the most commonly used aspirin formulation in the United States, with greater than 90% market share in 2013. This formulation does not decrease the risk of GI side effects and may have less heart health benefits than immediate release aspirin. We believe the variable and incomplete absorption of aspirin from the enteric coated dose form is responsible for “aspirin resistance” — incomplete antiplatelet activity that has been linked to an elevated risk of cardiovascular disease.
Aspirin remains a critical antiplatelet agent for the treatment and prevention of cardio and cerebrovascular disease. Over 50 million additional Americans who should be using aspirin daily for cardio disease prevention are not. The primary reason being they cannot tolerate aspirin’s GI toxicity. GI toxicity is a well-known side effect that is manifested as gastroduodenal ulceration, bleeding and dyspepsia. Among these side effects, ulceration and dyspepsia are most frequently reported with incidences of 5 – 15% and 20 – 40%, respectively.
After ischemic stroke or ACS, which is defined as stable or unstable angina or myocardial infarction, aspirin doses of 325 mg/day or less are used on a daily basis for the duration of a patient’s life. Compliance to such a regimen is critically important because discontinuation of aspirin leads to more than a three-fold increase in risk of a major cardiac event such myocardial infarction, stroke, or cardiovascular death.
Aspirin-induced GI mucosal injury, ulceration, and bleeding have been attributed to contact injury to the hydrophobic surface of the gastroduodenal tract, leading to an aberrant back-diffusion of acid into the mucosa that can trigger ulcerogenesis. Enteric coated, or EC, formulations of aspirin and co-therapy with antisecretory drugs have been developed to mitigate such injury. However, such approaches may attenuate antiplatelet activity.
Due to increasing concern over aspirin GI bleeding, ulceration, and dyspepsia, enteric coated aspirin has become the leading aspirin dose form in the United States with greater than 90% of aspirin sales. Despite its widespread adoption there have not been successful multiple large outcome studies supporting its use for secondary prevention. There is a growing body of evidence that suggests the absorption of aspirin from enteric coated aspirin products is highly variable and markedly reduced, resulting in reduced antiplatelet activity. This is in addition to the FDA’s opinion that the enteric coated aspirin results in “erratic absorption.” Moreover, there is no evidence of a decreased risk of clinically relevant ulceration or dyspepsia for enteric coated aspirin.
The basis for the use of aspirin for prevention of a cardiac event is largely derived from a meta-analysis consisting almost entirely of immediate-release formulations. The European Medicines Agency acknowledges that sufficient efficacy and safety data for a modified release formation (e.g. enteric coated aspirin) are absent, going so far as to state “…all valid clinical studies proving the efficacy and safety of ASA for the secondary prevention of cardiovascular events have been performed using immediate release formulations of ASA”. This is further supported by the results of a clinical trial in Japan of 14,000 high-risk primary prevention subjects published in theJournal of the American Medical Association in December 2014 that demonstrated once-daily, low-dose enteric coated aspirin compared to no aspirin did not significantly reduce the risk of the composite outcome of cardiovascular death, nonfatal stroke, and nonfatal myocardial infarction among Japanese patients 60 years or older with atherosclerotic risk factors. This study was stopped after five years when no statistical benefit was found for enteric coated aspirin.
Over time, with increasing recognition of the elevated risk of life threatening gastrointestinal bleeding and the elevated risk of secondary cardiovascular events upon aspirin discontinuation, enteric coated aspirin has become the prominent dose form because of the conceptually attractive mechanism of the selective release of ASA in the intestine while sparing the surface of the stomach from superficial injury. Although EC products have been shown to decrease the risk of superficial lesions, enteric coated aspirin has not been shown to decrease the risk of clinically relevant mucosal damage such as ulceration and bleeding. While commonly assumed to decrease the risk of dyspepsia, the rate of aspirin-induced incident dyspepsia appears no different among enteric coated aspirin and immediate release aspirin users.
In addition to the lack of evidence of improved GI safety or dyspepsia profile of enteric coated aspirin, at least one published study has shown that enteric coated aspirin is the primary cause of reduced antiplatelet activity or “aspirin resistance” (Grosser et al., “Drug Resistance and Pseudoresistance: An Unintended Consequence of Enteric Coating Aspirin”, Circulation , 2013;127:377 – 385). We have extended these findings and shown that aspirin resistance is mediated by decreased absorption of aspirin in diabetics. Except for non-compliance, we believe the lower bioavailability mediated by enteric coated aspirin is the major mechanism of aspirin resistance.
This provides a significant market opportunity for an OTC aspirin product that has predictable antiplatelet activity and lower risk of acute GI injury. To address the need for an aspirin product with improved safety and predictable absorption, Aspertec 325 mg has been developed and approved by the FDA in the United States. Aspertec is an aspirin product that selectively releases aspirin in the duodenum through its lipid-based formulation, a process that requires high pH and bile to emulsify the lipid matrix and release ASA. Unlike enteric coated aspirin whose release is only pH dependent, Aspertec has pH as well as bile sensitivity which (i) reduces the risk to the stomach of acute ulcers compared with current immediate release aspirin products, (ii) increases the rate and extent of absorption and antiplatelet activity compared to enteric coated aspirin, and (iii) reduces hyper-variable absorption when concomitantly administered with food.
Of the over 250 cardiovascular outcome studies done with aspirin, the seminal studies, which show a remarkable 21% to 51% decrease in mortality, have been done with immediate release aspirin products, with only one such study purportedly testing enteric coated aspirin. In that lone EC study, however, patients were instructed to chew the initial dose, which essentially voids the purpose of the enteric coating, making the dose, in effect, immediate release aspirin. The European Medicines Agency (EMA) — the EU’s equivalent of the FDA — concluded, as a result, that all valid clinical studies proving the efficacy and safety of aspirin for secondary prevention of cardiovascular events have been performed using immediate release formulations of aspirin.
Enteric coated aspirin was heavily marketed to cardiologists and consumers in the 1980s and 1990s as “safer.” The marketing was based on endoscopic findings that showed lower risk of gastric mucosal injury. In spite of the evidence to the contrary, most physicians that we have interacted with still equate signs and symptoms of dyspepsia to the presence of peptic ulcer disease. As such, physicians have erroneously presumed that enteric coated aspirin has a lower risk of dyspepsia. There have been no randomized prospective studies comparing enteric coated aspirin and immediate release aspirin induced gastrointestinal symptoms. Studies using enteric coated aspirin showed no difference in the rate of incidence of dyspepsia with enteric coated aspirin, and concluded that the choice to substitute enteric coated aspirin for regular aspirin for the prevention of dyspepsia was based on expert opinion, not on evidence available in medical literature.
Although there is some evidence that minor erosive damage is decreased with enteric coated aspirin, these products do not decrease the risk of clinically relevant ulceration or the risk of upper GI bleeding. In the early studies of enteric coated aspirin, ulcers were defined as superficial mucosal breaks without depth. In contrast, the contemporary definition of an ulcer as a mucosal break that penetrates the muscularis, is associated with life-threatening GI bleeding. Based on this definition, enteric coated aspirin has been associated with clinically relevant gastroduodenal ulceration.
Enteric coating markedly increases the inter-individual and intra-individual variability of the rate and extent of aspirin bioavailability due to “erratic absorption” of enteric coated aspirin. This variability is further exacerbated by concomitant administration of enteric coated aspirin with food, which is commonly employed to decrease the risk of dyspepsia.
Comparison of inter- and intra-individual variability of
immediate release aspirin (Alka Seltzer) with enteric coated aspirin (Ecotrin)
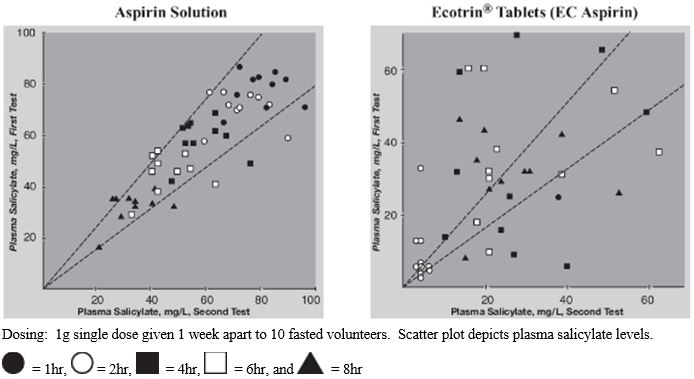 |
Source: Leonards, J.R. and G. Levy, Absorption and Metabolism of Aspirin Administered in Enteric-Coated Tablets.
JAMA, 1965. 193: p. 99 – 104.
Food further increases the variability of the rate
and extent of bioavailability of enteric coated aspirin
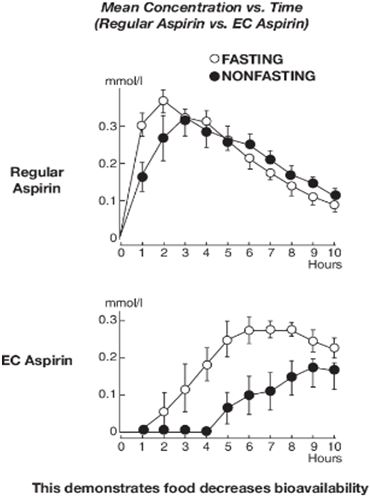 |
Source: Bogentoft, C., et al., Influence of food on the absorption of acetylsalicylic acid from enteric-coated dosage forms.European Journal of Clinical Pharmacology, 1978. 14(5): p. 351 – 355.
It is a generally accepted practice to take enteric coated aspirin with food in order to improve tolerability and increase compliance. However, this reduces the rate and extent of absorption, as illustrated below.
Individual Concentration vs. Time
(Regular Aspirin vs. EC Aspirin)
 |
Source: Bogentoft, C., et al., Influence of food on the absorption of acetylsalicylic acid from enteric-coated dosage forms.European Journal of Clinical Pharmacology, 1978. 14(5): p. 351 – 355.
Randomized clinical studies suggest that enteric coated aspirin may exhibit an attenuated antiplatelet activity. Such reduced antiplatelet efficacy may be due to lower bioavailability of aspirin from these delayed-release preparations. Enteric coated aspirin at a cardiovascular (CV) dose (81 – 325 mg) or an analgesic dose (325 – 1000 mg) has an acetylsalicylic acid Tmax that is approximately 3 hours later, a Cmax that is four times lower, and an area under the curve (AUC) that is two times lower than those for immediate-release aspirin formulations. Taking enteric coated aspirin with food may delay the rate of absorption and reduce the amount of aspirin absorption even more. In the Bogentoft study, which is represented above, three of the eight subjects (37.5%) did not register any meaningful aspirin absorption after taking enteric coated aspirin with food and thus were not receiving any antiplatelet benefit for cardiovascular disease prevention.
Aspertec
Aspertec 325 mg has been developed and approved by the FDA in the United States. Aspertec is an aspirin product that selectively releases aspirin in the duodenum through its lipid-based formulation, a process that requires high pH and bile to emulsify the lipid matrix and release ASA. Unlike enteric coated aspirin whose release is only pH dependent, Aspertec has pH as well as bile sensitivity which (i) reduces the risk to the stomach of acute ulcers compared with current immediate release aspirin products, (ii) increases the rate and extent of absorption and antiplatelet activity compared to enteric coated aspirin, and (iii) reduces hyper-variable absorption when concomitantly administered with food.
Aspirin (≤ 325 mg) is one of the most frequently used drugs in the world for CVD prevention. However, approximately 45% of patients with multiple CVD factors are not treated with antiplatelet agents including aspirin because the risk of GI bleeding may outweigh the vascular benefit.
The primary risk factors for GI bleeding induced by aspirin include the following:
● | A history of peptic ulcers, especially complicated ulcers, was associated with a three-fold increase in the risk; |
● | Advanced age and gender. The relative increase of the risk starts at age 60 years and rises in a nonlinear fashion with age. In patients aged 60 and over, the risk is slightly higher in males than in females; |
● | Presence of severe CVD; |
● | Use of high doses of aspirin or concomitant use of other NSAID, including cyclo-oxygenase-2 selective inhibitors; and |
● | Concomitant use of clopidogrel. Combination therapy with clopidogrel and aspirin is associated with a two-fold increased risk of upper GI complications compared to aspirin alone. |
Daily aspirin administration frequently induces gastric mucosal damage, with an incidence of 40 – 50% in healthy volunteers and in cardiovascular patients. In addition, it is ulcerogenic even at a very low dose of 10 mg per day, and increases the risk of GI bleeding. Advancing age and concomitant use of other NSAIDs and antiplatelet agents further increase the risk of life-threatening aspirin-induced GI bleeding. In secondary prevention of CVD, the addition of the antiplatelet agent clopidogrel to aspirin prophylaxis further decreases the rate of recurrent vascular events and mortality, but with elevated risk of GI bleeding. Moreover, the concomitant use of aspirin with other NSAIDs, cyclo-oxygenase-2 (COX-2) selective inhibitors, and with anticoagulant drugs also leads to synergistic increases in GI bleeding. Therefore, patients at risk for GI bleeding or ulceration are often co-prescribed gastric anti-secretory drugs, H2 receptor antagonists (H2RAs) or PPIs to decrease upper GI injury.
However, this is confounded by the FDA warning against the use of certain PPIs, such as omeprazole, with clopidogrel (Plavix). These drugs are associated with increased risk of:
● | Drug interactions (e.g. Plavix-clopidogrel); |
● | Osteoporosis related fractures of the hip, wrist or spine; |
● | Clostridium difficile associated diarrhea; and |
● | Hypomagnesemia. |
In the US, ACS patients are typically discharged from the hospital and prescribed, per practice guidelines:
● | Enteric coated aspirin; |
● | Adenosine diphosphate (ADP) receptor inhibitor such as clopidogrel (Plavix); |
● | Heparin; and |
● | Proton pump inhibitors such as omeprazole. |
The highest risk of a recurrent vascular event and GI damage is within 30 days of a revascularization procedure. As these procedures induce marked damage to the arterial endothelium, there is a high risk of focal thromboxane generation and thrombus formation leading re-occlusion. This increased risk of recurrent vascular events during this sub-acute period is associated with the risk of life threatening GI bleeding. The primary risk factors for such bleeding are:
● | dual antiplatelet agents (e.g. ADP inhibitors and aspirin); |
● | age; |
● | diabetes; |
● | anticoagulant agents; and |
● | severity of vascular disease. |
To decrease this sub-acute risk of GI bleeding induced by aspirin mucosal injury, enteric coated aspirin is the most predominately used chronic use aspirin product in the United States for the prevention of ACS, and specifically designated as the aspirin formulation of choice in clinical guidelines. As detailed herein, enteric coated aspirin has highly variable antiplatelet activity, and no evidence of lower risk of serious mucosal damage that leads to life threatening GI bleeding. As such, there is a clear need for an aspirin product that provides predictable antiplatelet activity and a lower risk of mucosal injury, which may improve cardiovascular outcome during this sub-chronic period.
The importance of GI risk upon initiation of aspirin therapy is supported by quantitative market research. A survey conducted by Weinman Schnee Morais, Inc. of 201 cardiologists — 40% of whom were interventional cardiologists — and 100 neurologists, yielded positive results. When the survey participants were shown a product profile of Aspertec:
● | About 80% said it is extremely/very important to them that Aspertec reduces the risk of acute gastric erosions and acute gastric ulcers. |
● | About 67 – 77% said that reducing these acute lesions could improve compliance and therefore lead to better outcomes for their patients. |
● | About 30 – 35% said that patients are most at risk to discontinue aspirin use due to tolerability issues in the first 10 days. |
Additional market research results are shown in the two figures below.
Physicians Indicate High Intent to Prescribe Aspertec
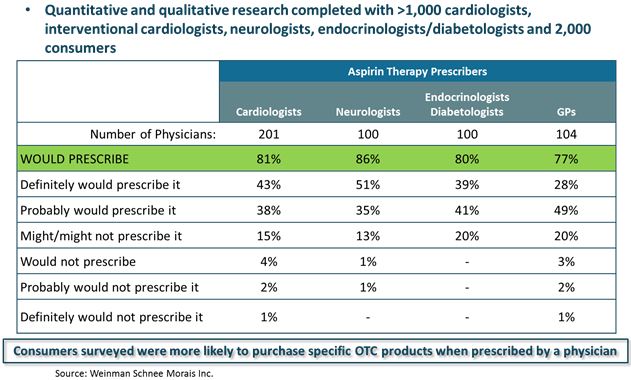 |
How Doctors Would Prescribe Aspertec
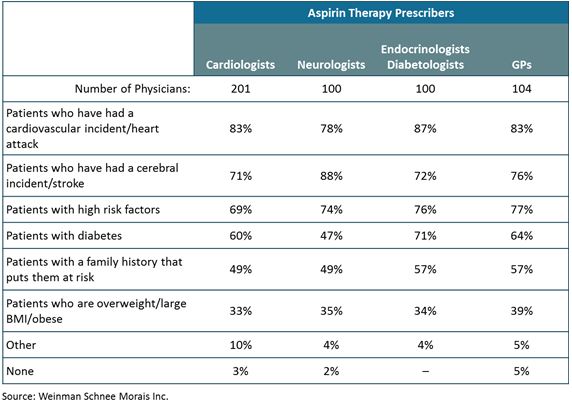 |
We believe that Aspertec has the potential to be better than enteric coated aspirin as the primary aspirin product for those with the greatest risk of cardiovascular disease, including the treatment and prevention of ACS and ischemic stroke. This is primarily because:
● | Aspertec has more predictable pharmacokinetics and antiplatelet activity than enteric coated aspirin. The FDA recognizes all enteric coated aspirin products have “erratic absorption” in the monograph Professional Labeling. Aspertec has clinical evidence of superior bioavailability and antiplatelet activity. |
● | Aspertec has a lower risk of acute GI ulceration than immediate release aspirin, which is the gold standard product for aspirin heart health benefits. Ulcers are a validated surrogate marker for risk of GI bleeding. As the risk of GI bleeding is highest within 30 days after ACS, Aspertec could become an important aspirin product for hospital use and discharge from the hospital after ACS including for patients using dual antiplatelet therapy. |
In two separate endoscopy trials involving healthy subjects with an age-associated risk for cardiovascular disease, Aspertec 325 mg demonstrated a statistically significant reduction of at least 65% in the risk of ulcers compared with 325 mg regular aspirin when taken once a day over a seven-day period. In an acute setting, Aspertec 325 mg is demonstrably GI safer than regular aspirin for patients in our target market. While Aspertec 325 mg demonstrated a statistically significant reduction in mucosal damage at 42 days similar to the endpoint used for enteric coated aspirin early studies, Aspertec 325 mg did not demonstrate a difference in the risk of getting an ulcer.
Aspertec 325 mg has also demonstrated increased effectiveness over enteric coated aspirin. We have successfully completed two pharmacokinetic and pharmacodynamic studies using diabetic patients with an increased risk for cardiovascular disease. The first study (PL-ASA-004) used a crossover study design where all the patients took both Aspertec 325 mg and a leading 325 mg enteric coated aspirin product once a day for three days. The second study (PL-ASA-006) compared both products again in all patients taking the products daily for ten days. Both studies demonstrated that Aspertec 325 mg was faster acting and provided more reliable, predictable and sustainable antiplatelet efficacy than the enteric coated aspirin 325 mg.
On February 14, 2017, we announced that the results of a 40-subject pharmacokinetic/ pharmacodynamic (PK/PD) trial of Aspertec had been published by the Journal of the American College of Cardiology (JACC). The publication, entitled ‘‘Enteric Coating and Aspirin Nonresponsiveness in Patients with Type 2 Diabetes Mellitus,’’ describes results of the study, which evaluated Aspertec 325 mg versus both plain and enteric coated aspirin over 72 hours.
Statistical significance is important and when used herein is denoted by p-values. The p-value is the probability that the reported result was achieved purely by chance (for example, a p-value <0.001 means that there is a less than a 0.1% chance that the observed change was purely due to chance). Generally, a p-value less than 0.05 is considered to be statistically significant.
FDA Interactions Related to Aspertec
Aspertec 325 mg was approved by the FDA on January 14, 2013. Upon approval, the FDA published the NDA approval letter as well as reviewers’ comments on the application. We noted in the published reviewer comments a mistake in a reviewer’s interpretation of the PL-ASA-002 endoscopy study results which suggested the trial was not successful. However, this trial was successful and the reviewer misinterpreted the results. This trial was not used as a basis for approval but was submitted to the FDA as part of the application and thus, it was commented on by the clinical reviewer. On October 3, 2014, we submitted a response to correct the reviewer’s misinterpretation of the PL-ASA-002 results.
On April 5, 2013, our regulatory legal counsel submitted a request on our behalf to the Office of Chief Counsel of the FDA to ask for their assistance in interacting with the Office of Drug Evaluation and Research regarding their decision to not approve a heart graphic and the words ‘safe’ and ‘reliable’ on the proposed label for Aspertec 325 mg. We brought to the attention of the Office of Chief Counsel that our NDA approval was subject to an unfair burden on limitations being put on our label which other OTC Monograph products that had no prior label approval were not subject to. On January 3, 2014, the FDA agreed with our position, allowed the heart graphic to be included on the label, and provided guidance on requesting language such as “safe” and “reliable.”
On December 7, 2015, our regulatory legal counsel submitted a request on our behalf to the Office of Chief Counsel (OCC) of the FDA to ask for their assistance in interacting with the Division of Medication Error Prevention and Analysis (DMEPA) regarding their decision to not approve the proposed brand name Aspertec. In response to this request, the OCC requested that we resubmit a proprietary name review request for Aspertec to DMEPA. We submitted this proprietary name review request to DMEPA on May 4, 2016, and on July 29, 2016, DMEPA concluded that the proprietary name Aspertec is acceptable for the product. We intend to finalize the label for Aspertec 325 mg via a labeling supplement to the NDA which will include the allowed proprietary name, the heart graphic, and other related formatting changes.
Aspertec Clinical Studies
OTC | ● | PL-ASA-001 — Pivotal PK study for 505(b)(2) NDA | ||
| Market Access Studies | ‒ | Study bridged Aspertec to the only aspirin formulation with valid CV outcome studies (immediate release aspirin) | ||
| ● | PL-ASA-003 — Food effect PK study for OTC, which showed: | |||
| ‒ | Food does not affect rate and extent of absorption | |||
Physician | Best | ● | PL-ASA-004 — PK/PD in Type II Diabetics without CV disease, which showed: | |
| to Support Product Launch | Activity | ‒ | PK and PD bioequivalent to IR release aspirin | |
| ● | PL-ASA-004 & PL-ASA-006 PK/PD in Type II Diabetics without CV disease, which showed: | |||
| ‒ | Aspirin resistance is due to enteric coated aspirin because it decreases aspirin absorption | |||
| ‒ | Aspertec is more reliable than enteric coated aspirin because of 1) faster onset, 2) greater chance of complete response — i.e. lower risk of aspirin resistance, 3) less variable absorption and antiplatelet activity | |||
Faster Onset | ● | PL-ASA-002 & PL-ASA-005 endoscopic studies in subjects with CV disease age associate risk, which showed: | ||
| Less Variability Safer than Immediate Release Aspirin | ‒ | Aspertec has a lower risk of sub-chronic gastroduodenal ulceration than immediate release aspirin | ||
Further detail regarding the Aspertec 325 mg clinical trials is provided below:
Protocol Number | Study Arms (Number of | Dosing Protocol | Study Center | Study Population | |||
PL-ASA-001 | – | 325 mg Aspertec (n=16) | Single 325 mg dose of study drug or single 650 mg dose of study drug | Houston, TX | Healthy volunteers at least 21 years of age. | ||
| – | 325 mg Immediate Release Aspirin Tablets (n=16) | ||||||
| – | 650 mg Aspertec (n=16) | ||||||
| – | 650 mg Immediate Release Aspirin Tablets (n=16) | ||||||
Study Initiation: 02/11/2008 | |||||||
Study Completion: 06/10/2008 |
| ||||||
PL-ASA-002 | – | 325 mg Aspertec (n=97) | 325 mg dose of study drug once daily for 7 days | Dallas, TX | Healthy volunteers between 50 and 75 years old.
Avg. age: 57.3 ± 6.20 | ||
| – | 325 mg Immediate-Release (IR) Aspirin Tablets (n=101) | ||||||
Study Initiation: 12/15/2008 | |||||||
Study Completion: 06/12/2009 |
| ||||||
PL-ASA-003 | – | 650 mg Aspertec in the fed state (n=20) | Single 650 mg dose of study drug | Houston, TX | Healthy volunteers between 21 and 65 years old, with a body mass index (BMI) between 20 and 32 kg/m2.
Avg. age: 36.8 ± 8.55 | ||
| – | 650 mg Aspertec in the fasted state (n=20) | ||||||
Study Initiation: 10/23/2010 | |||||||
Study Completion: 12/05/2010 |
| ||||||
PL-ASA-004 | – | 325 mg Aspertec (n=30) | 325 mg dose of study drug once daily for 3 days | Cincinnati, OH | Type II diabetes without previous history of vascular disease between 21 to 79 years old.
Avg. age: 52.9 ± 10.12 | ||
| – | 325 mg Enteric-Coated (EC) Aspirin Caplets (n=30) | ||||||
| – | 325 mg Immediate-Release (IR) Aspirin Tablets (n=30) | ||||||
Study Initiation: 02/20/2012 | |||||||
Study Completion: 06/16/2012 |
| ||||||
PL-ASA-005 | – | 325 mg Aspertec (n=110) | 325 mg dose of study drug once daily for six (6) weeks (42 days) | New York, NY | Healthy volunteers between 50 and 75 years of age, with a body mass index (BMI) between 20 and 32 kg/m2.
Avg. age: 57.3 ± 5.77 | ||
| – | 325 mg Immediate-Release (IR) Aspirin Tablets (n=120) | ||||||
Study Initiation: 09/04/2012 | |||||||
Study Completion: 06/24/2013 |
| ||||||
PL-ASA-006 | – | 325 mg Aspertec (n=57) | 325 mg dose of study drug once daily for 10 days | Cincinnati, OH | Type II diabetes without previous history of vascular disease between 20 to 79 years old. | ||
| – | 325 mg Enteric-Coated (EC) Aspirin (n=57) | ||||||
Study Initiation: 12/23/2013 | |||||||
Study Completion: 04/07/2014 |
| ||||||
We have engaged two separate firms, Weinman Schnee Morais, Inc. and Healogix, LLC, to conduct qualitative and quantitative physician market research regarding the significance of our findings, and in our most recent market study of 500 physicians (including cardiologists, neurologists and endocrinologists), more than 80% of the physicians surveyed expressed interest in prescribing or recommending Aspertec for their high-risk patients. This level of interest, when taken into account with both U.S. Department of Health and Human Services estimates of potential aspirin users and the increased efficacy and GI safety offered by our product, suggests a very important role for Aspertec to play in the prevention of cardiovascular disease in both the over-the-counter and prescription markets.
How the PLxGuard delivery system differs from enteric coating technology
The enteric coating of aspirin tablets utilizes methacrylic polymers that release the contents of the tablet at pH 5.5 or greater. As the release is solely dependent on pH, the release of any active pharmaceutical ingredient, or API, from coated tablets, granules, or capsules are dependent on the highly variable physiologic process of gastric emptying and the pH of intestinal fluids. These processes are markedly affected by the presence of food and disease specific factors.
Low-dose enteric coated aspirin products have lower bioavailability than regular aspirin
PK Parameters |
| Immediate-Release Aspirin |
| Enteric Coated Aspirin | ||||
(N=14 per group) |
| Cardiprin |
| Platin |
| Astrix |
| Cartia |
Cmax (ug/L) |
| 1979 ± 580 |
| 2721 ± 761 |
| 411 ± 133 |
| 186 ± 202 |
Tmax (h) |
| 0.48 ± 0.35 |
| 0.35 ± 0.10 |
| 3.73 ± 0.94 |
| 6.84 ± 2.71 |
AUC (mg/L*h) |
| 1.60 ± 0.41 |
| 1.54 ± 0.27 |
| 0.73 ± 0.17 |
| 0.56 ± 0.26 |
Source: Bochner, F., A.A. Somogyi, and K.M. Wilson, Bioinequivalence of four 100 mg oral aspirin formulations in healthy volunteers. Clin Pharmacokinet, 1991. 21(5): p. 394-9.
The mechanism by which aspirin bioavailability from enteric coated aspirin is reduced compared to plain aspirin in patients is unknown, but may be related to factors that affect tablet disintegration, as illustrated in Figure 5 below.
Enteric coated aspirin decreases absorption and increases variability
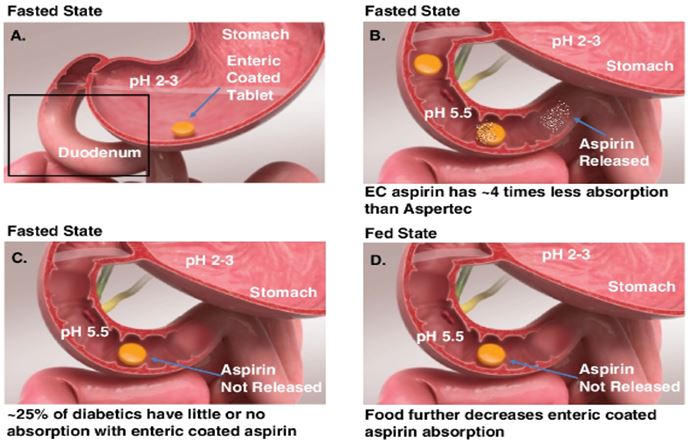 |
As illustrated above, enteric coated aspirin tablets are coated with a polymer that selectively releases at pH 5.5 or greater. As the normal fasted pH of the stomach is typically less than 3, the tablets remain intact in the stomach (Panel A). In the fasted state, gastric fluid is buffered by pancreatic bicarbonate in the duodenum (Panel B). The unfavorable pH for aspirin dissolution and unpredictable gastro-duodenal transit in the duodenum results in a lower extent and increased variability of absorption. An example of a partially dissolved capsule within a gastric ulcer is shown below.
Partiallydissolved EC aspirin in a gastric ulcer crater
 |
Disease factors that affect GI motility, as in diabetic patients with gastroparesis, may further increase the incidence of erratic release and absorption (Panel C). Concomitant administration of enteric coated aspirin tablets with food, which can markedly affect gastric emptying, further decreases the rate and extent of absorption, and increases the variability of the absorptive process (Panel D).
Aspirin absorption is clearly dissolution-limited, as pre-dissolved solutions or sodium salts of acetylsalicylic acid result in a faster rate and extent of absorption than that observed for aspirin tablets. The release and dissolution of aspirin from EC dose forms is dependent on the normally stochastic process of gastric emptying. Thus, aspirin absorption from EC formulations is highly erratic, with marked inter- and intra-individual variability compared to solutions or immediate-release dose forms, including Aspertec. The unpredictable nature of ASA absorption from enteric coated aspirin is exacerbated by concomitant administration of food.
This intrinsic variability is confounded by co-morbidities found in diabetics. The incidence of gastroparesis in patients with diabetes may be as high as 30 – 40%. Because blood glucose is important in regulating gastric emptying, it is possible that patients treated with enteric coated aspirin have variable gastric emptying and a higher frequency of prolonged tablet retention in the stomach. With a greater residence time and susceptibility of ASA to hydrolyze in the GI tract, the amount of ASA available to be dissolved and absorbed in the upper GI tract, with its favorable pH for ASA dissolution, may be lower with enteric coated aspirin than with plain aspirin or Aspertec.
To address the need for an aspirin product with improved GI safety as well as predictable pharmacokinetics and pharmacodynamics, Aspertec 325 mg has been developed and approved in the United States. Aspertec is a product that selectively releases aspirin in the duodenum through its lipid-based formulation.
Aspertecprovides predictable absorption and lower variability
 |
As illustrated above, in Aspertec capsules, aspirin is suspended in a high solid content lipidic carrier. Release of aspirin from this matrix requires pH > 5.5, bile acid, and pancreatic enzymes. In the fasted state, as gastric fluid has a pH typically less than 3, and is generally devoid of any lipolytic enzymes and bile acids, the lipid matrix remains intact in the stomach (Panel A). In the fasted state, gastric fluid is buffered by pancreatic secretions including bicarbonate and lipase in the duodenum, which emulsify the oil matrix and release aspirin (Panel B). In spite of the unfavorable and unpredictable gastro-duodenal transit in the duodenum, aspirin is solvated and absorbed with minimal variability. As the emulsification process is generally independent of gastric emptying, patients with gastroparesis have predictable absorption (Panel C). Concomitant administration of Aspertec with food, which can markedly affect gastric emptying, does not affect aspirin absorption (Panel D).
Thein vivo site of release of aspirin from Aspertec capsules and from a leading immediate release aspirin tablets was estimated byin vitro dissolution in simulated gastric fluid, upper duodenal fluid, and lower duodenal fluid. The typical gastric, duodenal, and duodenojejunal residence times are approximately 30, 40, and 60 minutes, respectively. Over the typical residence times, the release and dissolution of aspirin from Aspertec was minimal (approximately 20%) in simulated gastric and upper duodenal fluid, in spite of high agitation. However, the release of aspirin was rapid in simulated lower duodenal fluid, resulting from the high pH and emulsification of Aspertec lipid by bile acids and pancreatic enzymes.
In contrast, the release and dissolution of aspirin from immediate-release aspirin tablets, under the same dissolution conditions as used for Aspertec, was rapid and non-selective with near complete release by 7.5 minutes in both simulated gastric and upper duodenal fluid. These data suggest that aspirin in the Aspertec matrix remains lipid-bound and therefore highly hydrophobic in the low pH environment of the stomach and upper duodenum. Coupled with observations of reduced gastric injury and increased hydrophobicity at gastroduodenal pH, these data further support the position that lipid-bound aspirin at low pH reduces the risk of gastric surface injury and that there is selective release of aspirin in lower duodenum.
The selective release in the duodenum results in reduced exposure of the aspirin to the gastric mucosa leading to lower gastric ulcerations in two clinical studies (PL-ASA-002 and PL-ASA-005). In an acute endoscopic study, PL-ASA-002, use of Aspertec has been shown to decrease the incidence of acute gastroduodenal ulceration in volunteers with an age-associated risk of GI bleeding.
Aspertec has a lower incidence of gastroduodenal ulceration in at-risk subjects
 |
Source: Cryer, B., et al., Low-dose aspirin-induced ulceration is attenuated by aspirin-phosphatidylcholine: a randomized clinical trial. Am J Gastroenterol, 2011. 106(2): p. 272-7.
In this PL-ASA-002 study, subjects with an age-associated risk of NSAID gastropathy were randomized to Aspertec 325 mg or immediate release aspirin. These subjects wereH. pylori negative and free from any gastroduodenal erosions or ulcers prior to enrollment. Subjects were treated with either Aspertec 325 mg or immediate release aspirin once daily for 7 days. Ulcers were defined as lesions of 3mm or greater with unequivocal depth as assessed by esophageal gastroduodenal endoscopy. The presence or absence of ulceration was independently evaluated by a blinded central review, and ulcer diagnoses were further subjected to rigorous blinded adjudication. In the intent to treat population, Aspertec 325 mg treatment had a 71% lower incidence of ulcers (95% confidence interval, 22.85 – 100%) than seen with immediate release aspirin at the day 7 endoscopy: Ulcers developed in 5 subjects (5.1%) in the Aspertec 325 mg group and in 18 subjects (17.6%) in the aspirin group. This risk reduction was largely driven by a decrease in gastric ulceration. Four gastric and two duodenal ulcers were found in the Aspertec 325 mg group, and twelve gastric and six duodenal ulcers were found in the immediate release aspirin group.
The decreased risk of acute ulceration (over 7 days of dosing) was confirmed in a similarly designed second endoscopic study (PL-ASA-005). In this study, all the major inclusion and exclusion criteria were identical. This study included two notable designed differences: (i) mucosal damage was measured by serial endoscopy at day 7 and day 42 after the same dose and schedule of Aspertec 325 mg and immediate release aspirin, and (ii) the diagnosis of ulceration was only confirmed by a blinded central reviewer. Aspertec 325 mg showed a 65% lower risk of ulceration than regular aspirin over 7 days and cumulatively over day 42. The decrease in cumulative risk reduction was driven by a marked decrease in gastric ulceration on day 7 findings because no difference in duodenal ulcerations were found at day 7 and no difference in either locations was found at day 42.
The risk of non-ulcerative erosive damage was measured by mean erosion number over a 42-day period in PL-ASA-005 study. Collectively, these data suggest that in subjects at risk for an age associated risk of GI bleeding (age 55 – 75), Aspertec 325 mg (i) has a lower risk of general erosive damage for at least a period of 42 days after aspirin initiation, and (ii) is particularly useful for decreasing the risk of life-threatening upper-GI mucosal injury for at least 7 days.
Combining the seven day endoscopy results for both PL-ASA-002 and PL-ASA-005 studies suggests a minimum reduction of 65% in the risk of upper GI ulcers.
Two studies demonstrate a 65% reduction in acute ulcer risk
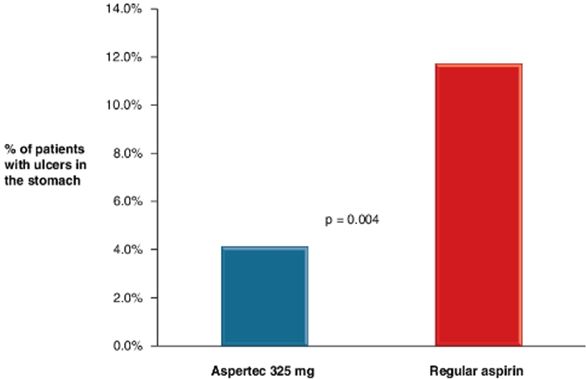 |
As the majority of aspirin-induced life-threatening upper-GI bleeding is due to mucosal lesions in the stomach and the greater risk of GI bleeding falls with 10 days after an ACS, Aspertec may be an alternative to enteric coated aspirin for use after an ACS when predictable and reliable antiplatelet activity is required.
As modified release aspirin products such as enteric coated aspirin can markedly affect pharmacokinetics (PK) and antiplatelet effects, the PK and onset of antiplatelet activity of Aspertec 325 mg were compared to that of enteric coated aspirin in patients with diabetes mellitus without cardiovascular disease in two crossover studies (PL-ASA-004 and PL-ASA-006). This population was selected because diabetes may be a univariant risk factor for aspirin-induced GI bleeding and diabetic hypercoagulability and obesity may be risk factors for aspirin non-responsiveness.
In these crossover studies, the rate and extent of aspirin were measured based on plasma levels of acetylsalicylic acid. The rate and extent of antiplatelet activity was measured by serum thromboxane generation, which is a direct and the most accurate method of assessing the Cycloxygenase I (COX-1) in platelets. The same diabetic patients were either initially dosed with enteric coated aspirin or Aspertec 325 mg, or vice versa, with a 2 week intervening washout period. All potential confounders, such as compliance, timing of food, concomitant drugs, drug interactions, and other factor were controlled.
Single dose pharmacokinetic/pharmacodynamic (PK/PD) study
The comparison of the rate and extent of single dose aspirin absorption in diabetics from Aspertec 325 mg and a leading 325 mg enteric coated aspirin product is illustrated below. Compared to enteric coated aspirin, Aspertec 325 mg had 4.0 to 5.5, and 3.3 to 5.6 fold greater AUC0-t and Cmax, respectively, of ASA with significantly greater incidence of patients with complete depletion of platelet COX-1 (99.0% Inhibition of thromboxane generation over baseline or ≤ 3.1ng/mL). This greater rate and extent of absorption and enzymatic inhibition was associated with markedly less variability.
A. Comparison of Single Dose Aspirin Absorption
 |
B. Incidence of Complete Aspirin Response
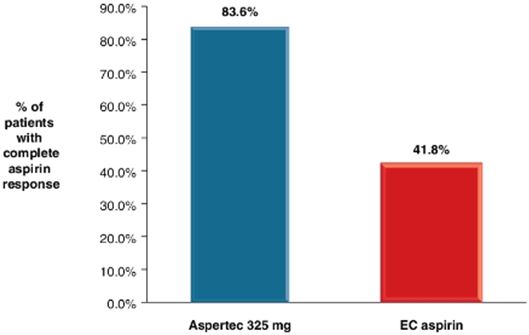 |
Note that in these crossover studies enteric coated aspirin treated patients have minimal absorption of aspirin and reduced responsiveness. After a single dose, the greater rate and extent of absorption of Aspertec 325 mg was associated with statistically significant greater incidence of a complete aspirin response in diabetic patients as compared with treatment with enteric coated aspirin. The incidence and 95% confidence interval of complete aspirin response of Aspertec 325 mg and enteric coated aspirin were, respectively, 83.6% (72.78, 91.94) and 41.8% (30.10, 55.92).Source: Reliable Inhibition of Thrombocyte Activity: Comparison of Aspertec Capsules, 325 MG and Enteric-Coated Aspirin (RITE Study), September 24, 2014
Interestingly, many of the same patients that had complete absorption and complete inhibition of TXB2 after Aspertec, had negligible absorption after enteric coated aspirin, with consequently lower COX-1 inhibition. The incidence of negligible absorption, defined as Cmax < 150ng/ml, after enteric coated aspirin was 40% (14/35) in PL-ASA-004, and 24.1% (13/54) in PL-ASA-006. These data strongly suggest aspirin resistance may be simply mediated by formulation dependent bioavailability.
In a suspected ACS patient in the United States, a loading of 325 mg aspirin is administered to enable rapid absorption and antiplatelet activity. Aspirin may be administered in the form of a chewed enteric coated aspirin, effervescent aspirin and in the case ofnil per os patients, via rectal suppository. After this loading dose, maintenance doses (81 to 325 mg) are administered to perpetuity, with ≥90% of the doses as EC coated tablets.
Aspirin’s antiplatelet effects are known to be cumulative and may require at least 24 hours to elicit a maximal response. As such, we compared the antiplatelet activity of Aspertec 325 mg with enteric coated aspirin, the rate and extent of COX-1 inhibition up to 4 days (in PL-ASA-004) and up to 11 days (in PL-ASA-006). In both studies, Aspertec had greater probability of eliciting an aspirin response than enteric coated aspirin, suggesting that when rapid and complete antiplatelet activity is required, as in ACS, Aspertec may be a better choice than enteric coated aspirin; Aspertec has a 3.4x to 5.0x greater chance of achieving a complete antiplatelet response than EC ASA in two replicate studies. Although the rate of a complete aspirin response is clearly slower, enteric coated aspirin treated patients do eventually elicit a complete response after multiple doses. The 75th percentile indicated that by 3 hours after the first administration of Aspertec 325 mg, at least 75% of the patients reached 99.0% inhibition, but it took over 96 hours (and 4 doses) before 75% of the patients reached 99.0% inhibition when administered enteric coated aspirin.
These complete aspirin responses were eventually achieved after perfect compliance to therapy and after administration of every dose in a carefully controlled fasted state. Even after multiple doses, the variability of COX-1 inhibition is 2 – 3 times higher with enteric coated aspirin than with Aspertec. In clinical practice, only approximately 50% of patients adhere to their aspirin regimen. As previously discussed, enteric coated aspirin has about 3 – 4 times lower bioavailability than Aspertec, which is further exacerbated by concomitant use of food, and 25 – 37% of fasted doses have intrinsically no bioavailability. With approximately 63% of cardiologists (out of 504 surveyed) recommending aspirin be taken with food, coupled with the high rate of non-compliance, the use of enteric coated aspirin with these clinical realities may not be the ideal choice of aspirin to constitutively suppress the generation of thromboxane.
For both the high risk clinical setting where the risks of cardiovascular and GI events are high, and in real clinical practice where compliance is generally poor, Aspertec may be a more attractive choice of an aspirin dose form than enteric coated aspirin to constitutively suppress platelet derived thromboxane generation.
Aspertec has a greater chance of achieving a complete aspirin response in two studies
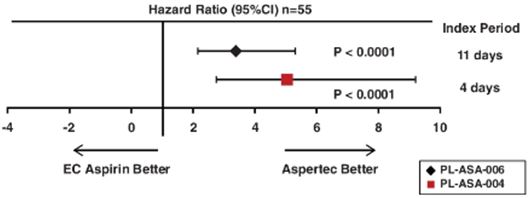 |
Hazard ratios, a measure of risk, for two studies reveal Aspertec has a 3.4x to 5.0x greater chance of achieving a complete antiplatelet response than enteric coated aspirin.Source: A Randomized, Actively Controlled, Crossover Pharmacodynamic Evaluation of Aspertec Versus Enteric-Coated and Immediate-Release Aspirin in Patients with Type II Diabetes, May 15, 2013; PL-ASA-004 CSR and Reliable Inhibition of Thrombocyte Activity: Comparison of Aspertec Capsules, 325 MG and Enteric-Coated Aspirin (RITE Study).
Overall, these studies show that Aspertec 325 mg depletes platelet COX-1 activity in diabetics at a rate and extent that is significantly greater than enteric coated aspirin and with less variability. Enteric coated aspirin is associated with a high risk of non-responsiveness, due to lower bioavailability. For patients with cardiovascular disease and at risk for acute GI damage, Aspertec may be an alternative with a lower risk for upper GI acute ulceration and more predicable PK and PD than enteric coated aspirin. Our findings, which show highly variable and incomplete antiplatelet activity associated with enteric coated aspirin as well as others’ studies demonstrating minimal or no evidence of improved GI safety, does not support the use of enteric coated aspirin in patients with diabetes mellitus.
On February 14, 2017, PLx announced that the results of a 40-subject pharmacokinetic/pharmacodynamics (PK/PD) trial of Aspertechad been published by theJournal of the American College of Cardiology(JACC). The publication, entitled ‘‘Enteric Coating and Aspirin Nonresponsiveness in Patients with Type 2 Diabetes Mellitus,’’ describes results of the study, which evaluated Aspertec 325 mg versus both plain and enteric coated aspirin over 72 hours.
FDA Approval for Aspertec 325 mg
Aspirin, the active ingredient in Aspertec 325 mg, was classified into the therapeutic class for Internal Analgesic, Antipyretic, Antirheumatic Drug Products for Over-the-Counter Human Use (IAAA). The Advance Notice for Proposed Rulemaking (ANPR) was published in 1977, and in 1988, the FDA published the tentative final monograph (TFM) for IAAA. The IAAA TFM proposes conditions under which OTC IAAA products may be marketed without prior review by the FDA, including labeling such as therapeutic indications, dosage instructions, and warnings about side effects and ways of preventing misuse. Although it has been updated and amended since its original publication, the IAAA monograph has not been finalized.
Aspertec 325 mg is different from the IAAA TFM aspirin products, as our product is the first ever liquid-filled capsule containing a lipidic suspension of aspirin. The lecithin excipient used in the drug product exceeds the approved amount listed in the FDA inactive ingredients guide (IIG) for an orally administered drug product. As a result, Aspertec 325 mg was considered a new drug which required NDA approval for marketing. We used the 505(b)(2) NDA path. Upon FDA approval of the 505(b)(2) NDA, Aspertec 325 mg was granted OTC labeling similar to that of monograph aspirin products. OTC aspirin, including Aspertec 325 mg, is indicated for the temporary relief of minor aches and pains associated with a cold, headache, backache, toothache, premenstrual and menstrual cramps, minor pain of arthritis, and to temporarily reduce fever.
All aspirin products available on the market today are being sold via the OTC monograph process. When Aspertec 325 mg is commercialized we believe it will be the only NDA-approved OTC aspirin product available.
Aspertec 325 mg FDA Approved OTC Indications
● | For the temporary relief of minor aches and pains associated with a cold, headache, backache, toothache, premenstrual & menstrual cramps, and minor pain of arthritis. |
● | Temporarily reduces fever. |
Although the IAAA TFM is still pending finalization, professional labeling for aspirin was finalized in 1998 to allow physicians to prescribe aspirin for cardiovascular and rheumatologic uses. With the approval of the Aspertec 325 mg (505)(b)(2) NDA, Aspertec is also eligible to be marketed to physicians for professional uses, as summarized below.
In the “Drug Facts” section that is a required part of labeling for all aspirin products, Aspertec will have an additional alert unique to Aspertec — “This product contains soy.”
Aspertec 325 mg Professional Labeling Indications
Professional labeling for Aspertec 325mg includes the following indications:
Vascular Indications (Ischemic Stroke, Transient Ischemic Attack (TIA), Acute Myocardial Infarction (MI), Prevention of Recurrent MI, Unstable Angina Pectoris, and Chronic Stable Angina Pectoris): Aspirin is indicated to: (1) reduce the combined risk of death and nonfatal stroke in patients who have had ischemic stroke or transient ischemia of the brain due to fibrin platelet emboli, (2) reduce the risk of vascular mortality in patients with a suspected acute MI, (3) reduce the combined risk of death and nonfatal MI in patients with a previous MI or unstable angina pectoris, and (4) reduce the combined risk of MI and sudden death in patients with chronic stable angina pectoris.
Revascularization Procedures (Coronary Artery Bypass Graft (CABG), Percutaneous Transluminal Coronary Angioplasty (PTCA), and Carotid Endarterectomy): Aspirin is indicated in patients who have undergone revascularization procedures (i.e., CABG, PTCA, or carotid endarterectomy) when there is a preexisting condition for which aspirin is already indicated.
Rheumatologic Disease Indications (Rheumatoid Arthritis, Juvenile Rheumatoid Arthritis, Spondyloarthropathies, Osteoarthritis, and the Arthritis and Pleurisy of Systemic Lupus Erythematosus (SLE)): Aspirin is indicated for the relief of the signs and symptoms of rheumatoid arthritis, juvenile rheumatoid arthritis, osteoarthritis, spondyloarthropathies, and arthritis and pleurisy associated with SLE.
We are currently completing the development of an 81 mg strength of Aspertec, which is expected to have similar indications.
Other Pipeline Opportunities Using PLxGuard Delivery System
We are developing novel formulations that combine already-approved nonsteroidal anti-inflammatory drugs (NSAIDs) with our patented PLxGuard technology to make safer and more effective new products. We are using the PLxGuard delivery system to create a lipid-based formulation of ibuprofen, PL1200 Ibuprofen Capsules, 200 mg. We have an IND active with the FDA and have demonstrated bioequivalence with OTC 200 mg dose ibuprofen to support a 505(b)(2) NDA in fasted-state clinical trials at three different doses, 200 mg, 400 mg and 800 mg. Using the PL1200 formulation at Rx doses, we demonstrated better GI safety in osteoarthritic patients with equivalent analgesic and anti-inflammatory efficacy, when compared with Rx ibuprofen in a six-week endoscopy pilot clinical trial, PL-IB-002 (Lanza et al.).
GI Safety Trial Comparing PL1200 Ibuprofen 200 mg with Regular Ibuprofen Using the PLxGuard Delivery System
 |
Source: Lanza et al., Clinical trial: comparison of ibuprofen-phosphatidylcholine and ibuprofen on the gastrointestinal safety and analgesic efficacy in osteoarthritic patients.Aliment Pharmacol Ther, 2008, 28(4):431-42).
ZavrylTM is a potential trademark for PL1200 Ibuprofen. This trademark has not been submitted to the FDA and there is no assurance it will be approved by the FDA.
While we do not believe our technology will work with the entire NSAIDs class, it is possible that our technology may be successfully applied to other NSAIDs beyond aspirin and ibuprofen.
For example, there is preclinical data suggesting diclofenac is a viable product candidate. Diclofenac is a leading NSAID for pain and inflammation outside the United States. In addition, the National Institutes of Health has provided funding to support preclinical efforts for a GI-safer intravenous indomethacin (PL4100). We believe PL4100 for Patent Ductus Arteriosus (PDA), a heart condition primarily affecting newborns, may be able to qualify for orphan drug designation, because PDA affects fewer than 200,000 individuals in the United States. (The number of registered births in the United States for 2013 was 3,932,181, while the estimated incidence of PDA in children born in the United States at term is between 0.02% and 0.006% of births. Assuming the higher percentage of 0.02%, this represents 78,644 infants per year). An evaluation and determination will occur later in this product’s development, should the product advance further.
We have attempted to expand the technology to other NSAIDs with limited success. A two week endoscopy trial with naproxen (PL3100 250 mg) failed to demonstrate better GI safety. From these efforts we have learned which NSAIDs are more likely to benefit from the technology. In our view, the PLxGuard technology may also prove a novel drug delivery platform for corrosive, acid labile and insoluble and impermeable drugs providing delivery along the GI tract for many non-NSAID drugs.
Commercialization Strategy
We plan to commercialize our branded Aspertec products in the United States using our own commercial infrastructure. As described above in the section entitled “Risk Factors,” we have little experience with commercialization, and have no existing marketing or distribution operation. We intend to use a specialty sales force of sales representatives targeting the highest prescribers of antiplatelet medication. This includes specialists whose practices consist primarily of patients with a high risk of cardiovascular disease including cardiologists, neurologists and endocrinologists (diabetologists). We intend to demonstrate to physicians the clinical differentiation of Aspertec to influence their aspirin prescribing and recommendations in favor of Aspertec. We estimate that there are approximately 33,000 physicians within these initial target specialties with the top three to five deciles generating 75% to 85% of prescribing volume within the target specialties. This narrows the initial target for the early sales effort to 12,000 to 15,000 physicians with an appropriately sized sales force. Pharmacists will also be targeted as they represent a trusted healthcare professional involved in patient care.
We will also evaluate co-promotion opportunities with companies that have an existing sales force targeting physician groups that prescribe or recommend aspirin. We will employ appropriate medical education, direct marketing, journal advertising, electronic health record communication and social media in addition to personal promotion. We believe that patients and consumers will be made aware of Aspertec by their physician initially. We also believe that direct to consumer advertising is an opportunity to be considered after the product has been established.
Our focus will be on secondary prevention patients and high risk primary prevention patients, those most at risk for cardiovascular disease. We anticipate selling Aspertec 81 mg and Aspertec 325 mg at the same retail price of $22.50 for a 30 count bottle representing a month’s supply. This potential price may change prior to the product launches. Applying this potential pricing to the target market in the table below demonstrates an implied market opportunity. It is estimated based upon this potential pricing and an expected mix of 81 mg and 325 mg sales that each 1% market share of this target high cardiovascular risk market may represent approximately $100 million in potential annual retail sales.
Aspertec Initially Targets 4 Sizeable U.S. Patient Populations;
Focus is on Highest Risk Populations that Can Benefit the Most from Aspertec.
Secondary Coronary Artery Disease (“CAD”) Prevention | Secondary Stroke Patients (1) | Diabetes with | High Risk Primary Prevention | |||||
Definition | Patients who have previously suffered from a CAD event including myocardial infarctions, CABG and Percutaneous Coronary Intervention | Patients who have previously suffered a stroke event including: | Patients who have been diagnosed | Patients over the age of 45 who also have >1 risk factor | ||||
– Cerebral Vascular Accident | ||||||||
– Transient Ischemic Attack | ||||||||
– Carotid Endarterectomy/ | ||||||||
Addressable Patient Population | ~17 million | ~8 million | ~7 million | ~15 million | ||||
Total Retail Market Size (3) | $4.7 billion | $2.2 billion | $1.9 billion | $4.1 billion |
Total Retail Market Potential: ~ $12.9 Billion
~1% of this Market = ~$125 Million of Retail Revenue
~1% of this Market = ~$100 Million of PLx Net Revenue
___________________
(1) Omits hemorrhagic and cardioembolic population
(2) Omits population with CVD and those accounted for within primary prevention
(3) Proposed retail pricing of $0.75 per dose taken daily
We are utilizing a similar commercial strategy that was successfully employed by our Executive Chairman, Michael Valentino, when he was Chief Executive Officer for Adams Respiratory Therapeutics for their Mucinex® product. A graphic representation of the Adams Respiratory Therapeutics commercial strategy and resulting sales is below.
|
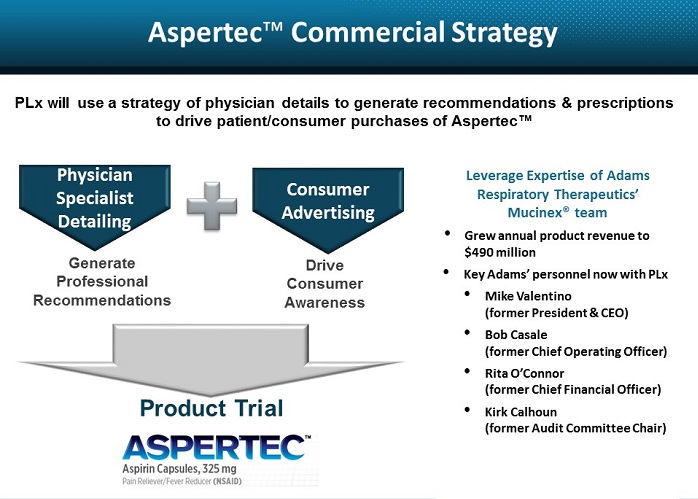
Manufacturing
We do not own or operate manufacturing facilities for the production of our product candidates, nor do we have plans to develop or own manufacturing operations in the foreseeable future. We currently rely on third-party contract manufacturers for all of our required ingredients and finished products for Aspertec and other product candidates. We do not have any long term contracts with any of these third parties. We do not have any contractual relationships for the manufacture of commercial supplies of Aspertec or other product candidates if they are approved. We have identified a preferred contract manufacturer and packager for commercial supplies of Aspertec and intend to enter into negotiations with them following the consummation of this offering. As commercial sales grow it is anticipated we will enter into agreements with one or more back-up manufacturers as appropriate for additional commercial production of Aspertec or other product candidates. We currently employ internal resources to manage our manufacturing contractors. The relevant manufacturers and potential manufacturers of Aspertec have orally advised us that they are compliant with both current Good Manufacturing Practices, or cGMP, and current Good Laboratory Practices, or cGLP. There can be no assurance that Aspertec or other product candidates, if approved, can be manufactured in sufficient commercial quantities, in compliance with regulatory requirements and at an acceptable cost.
We and our contract manufacturers are and will be subject to extensive government regulation in connection with the manufacture of any pharmaceutical product. We and our contract manufacturers must ensure that all of the processes, methods and equipment are compliant with cGMP and cGLP for drugs on an ongoing basis, as mandated by the FDA and other regulatory authorities, and conduct extensive audits of vendors, contract laboratories and suppliers. We believe that most of the ingredients we require to manufacture Aspertec are readily available from multiple suppliers and are commonly used in the pharmaceutical industry. One key ingredient is currently limited to a single provider, Lipoid, who supplies cGMP lecithin and is a leader in supplying high quality lipids to the global pharmaceutical industry. Lipoid is subject to a confidentiality agreement and developed this cGMP lecithin with PLx over a several year period and currently we are the only buyer of this product. Lipoid has represented the capability to provide this product from two different manufacturing sites in Germany, mitigating the risk of a shutdown at one site ceasing supply. We intend to negotiate a supply contract with Lipoid prior to commercially launching Aspertec. We do not at this time have any long term contract with Lipoid for the supply of commercial quantities of this product, and there can be no assurances that Lipoid will be able to supply sufficient commercial quantities in compliance with regulatory requirements at an acceptable cost.
Competition
The pharmaceutical industry is characterized by rapidly advancing technologies, intense competition and a strong emphasis of proprietary products. We face competition and potential competition from a number of sources, including pharmaceutical and biotechnology companies, generic drug companies, drug delivery companies and academic and research institutions. We believe the key competitive factors that will affect the development and commercial success of our product candidates include the therapeutic efficacy, safety and tolerability profiles and cost. Many of our competitors have substantially greater financial, technical and human resources than we do, as well as more experience in the development of product candidates, obtaining FDA and other regulatory approvals of products, and the commercialization of those products. Consequently, our competitors may develop products that may be more effective, better tolerated and less costly than our product candidates. Our competitors may also be more successful in manufacturing and marketing their products than we are. We will also face competition in recruiting and retaining qualified personnel and establishing clinical trial sites and patient enrollment in clinical trials.
Aspertec faces competition from many companies with aspirin products. These include OTC branded products from Bayer AG, Prestige Brands, Inc. (Ecotrin, Goody’s, BC Powder) and Bedrock Brands LLC (St. Joseph) and OTC private label or store brands (CVS, Walgreens) whose prices range between $0.02 and $0.09 per tablet, as well as prescription products from Aralez Pharmaceuticals (formerly Pozen, Inc.) (Yosprala) and Espero Pharmaceutical (Durlaza). OTC Aspirin is approved in the United States for multiple uses. In addition to cardiovascular disease prevention and treatment, aspirin may be used for the treatment of pain, inflammation and fever. There are a variety of aspirin and NSAID products in various stages of development in the United States and globally that represent potential competition when and if they become approved by the FDA and are commercialized. Companies and academic institutions involved include Takeda Pharmaceutical Company Limited (Takepron), Oxford Pharmascience Group Plc, Antibe Therapeutics Inc. and The City College of New York. Aspertec and other pain and inflammation product candidates such as PL1200 Ibuprofen will face competition from many firms. These include OTC and prescription products. Major competitors include Pfizer Inc. (Advil), Johnson & Johnson (MotrinIB, Tylenol), Bayer AG (Aleve) and private label or store brands (CVS, Walgreens).
The aspirin market is currently predominantly composed of generic products either branded (e.g. Bayer) or private label (e.g. CVS). Aspertec 325 mg is the only liquid-fill aspirin capsule product to be approved by the FDA. Aspertec went through a different regulatory approval process than the current OTC aspirin products being marketed in the US. Aspertec was approved under the 505(b)(2) NDA process and when launched is expected to be the only OTC available aspirin based product that successfully passed this rigorous process. We believe the clinical trials that demonstrated superior efficacy and safety will assist us in differentiating Aspertec from the competition and support the anticipated price. We anticipate that Aspertec’s attributes will fit well with wellness and loyalty programs that retailers have to stimulate patient compliance with chronic diseases (e.g. cardiovascular disease, diabetes and obesity) and represents the potential to have a higher margin and more profit for retailers than other OTC aspirin products, including their own store brands. Market research suggests patients and physicians are generally accepting of a $1.00 price per capsule for Aspertec for daily use, equating to $30.00 per month. Other product candidates will undergo clinical trials to provide differentiation as part of their product development and commercialization.
Intellectual Property
Our success depends, in part, upon our ability to protect our core novel technology. To establish and protect our proprietary rights we rely on a combination of patents, patent applications, trademarks, copyrights, trade secrets and know-how, license agreements, confidentiality procedures, non-disclosure agreements with third parties, employee disclosure and invention assignment agreements, and other contractual rights.
On January 8, 2003, we entered into a worldwide, exclusive license agreement with the Board of Regents of The University of Texas System which was subsequently amended multiple times and restated December 11, 2009 and subsequently amended April 15, 2011 and December 17, 2011. The patents in-licensed under this agreement constitute an important part of our intellectual property. This family of patents includes composition of matter, methods of manufacturing and methods of treatment that provides protection for Aspertec, PL1200 Ibuprofen and other NSAID product candidates in the United States and in a number of global markets. The following is a summary of the patents and expiration dates:
Methods and compositions employing formulations of lecithin oils and NSAIDs for protecting the gastrointestinal tract
Issued: Four U.S. patents expiring on March 1, 2022 or on March 23, 2022 and 23 additional foreign patents expiring December 19, 2021
Notice of Allowance: One U.S. patent expiring on December 19, 2021
Pending: Two U.S. patent applications (seeking expansion of claims) and two foreign patent applications (Brazil and Hong Kong)
Compositions and methods for treating and/or ameliorating cancer, the onset of cancers or the symptoms of cancers
Issued: United States, Australia, Canada, China, Hong Kong and Singapore expiring August 2, 2024
Sterile preparations of phospholipids and anti-inflammatory pharmaceuticals and methods of making and using same
Issued: Australia, Canada, India, Israel and Singapore expiring August 2, 2024
Purified phospholipid non-steroidal anti-inflammatory drug associated compositions and methods of preparing and using same
Issued: United States expiring June 3, 2026 and Australia and Mexico expiring October 12, 2025
We have developed our own patent applications entitledpH dependent carriers for targeted release of pharmaceuticals along the gastrointestinal tract, compositions therefrom and making and using same of which some have been issued and others, if issued with claims as filed, will provide patent protection for Aspertec, other NSAID products and other active pharmaceutical ingredients that will broaden the opportunity for new products to include many different drug classes. The following is a summary of the patents and expiration dates:
pH dependent carriers for targeted release of pharmaceuticals along the gastrointestinal tract, compositions therefrom and making and using same
Issued:Two U.S. patents with claims related to NSAIDs, including Aspertec and one Mexico patent with NSAID related claims similar to the U.S. issued patents, expiring September 29, 2032
Notice of Allowance: One U.S. and one Australia covering expanded claims for many different drug ingredients expiring September 29, 2032
Pending: Europe, Canada, China, Hong Kong, India, Japan, Mexico (expansion of claims) and South Korea with claims expected to expire September 29, 2032.
US patent no. 8,865187 “Compositions comprising lecithin oils and NSAIDs for protecting the gastrointestinal tract and providing enhanced therapeutic activity” and US patent no. 9,101637 “Methods of treating inflammation with compositions comprising lecithin oils and NSAIDs for protecting the gastrointestinal tract and providing enhanced therapeutic activity” and “pH dependent carriers for targeted release of pharmaceuticals along the gastrointestinal tract, compositions therefrom, and making and using same” are listed in the FDA Orange Book. As new patents are issued relative to FDA approved products such as Aspertec they will be added to the Orange Book and, as new products are approved by the FDA, the relevant patents will be added to the Orange Book. The Orange Book lists patents that protect each drug. Patent listings and use codes are provided by the drug application owner, and the FDA is obliged to list them. In order for a generic drug manufacturer to win approval of a drug under the Hatch-Waxman Act, the generic manufacturer must certify that they will not launch their generic until after the expiration of the Orange Book-listed patent, or that the patent is invalid, unenforceable, or that the generic product will not infringe the listed patent.
The term of individual patents depends upon the legal term for patents in the countries in which they are granted. In most countries, including the United States, the patent term is generally 20 years from the earliest claimed filing date of a non-provisional patent application in the applicable country. In the United States, a patent’s term may, in certain cases, be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in examining and granting a patent, or may be shortened if a patent is terminally disclaimed over a commonly owned patent or a patent naming a common inventor and having an earlier expiration date. The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, permits a patent term extension of up to five years beyond the expiration date of a U.S. patent as partial compensation for the length of time the drug is under regulatory review.
License Agreements
UT License Agreement
On January 8, 2003, we entered into a worldwide, exclusive license agreement with the Board of Regents of The University of Texas System which was amended and restated December 11, 2009, and subsequently amended April 15, 2011 and December 17, 2011. The patents in-licensed under this agreement constitute an important part of our intellectual property. This family of patents includes composition of matter, methods of manufacturing and methods of treatment that provides protection for Aspertec, PL1200 Ibuprofen and other NSAID product candidates in the United States and in a number of global markets. The Board of Regents of The University of Texas System granted us an exclusive license under its patents and know-how related to their NSAID-phospholipid technology to develop and commercialize NSAID products for use anywhere in the world. Certain of the technology was developed with government funding, and the exclusivity of our license is therefore subject to certain retained rights of the U.S. federal government. We are responsible for the development and commercialization of the licensed products under the agreement. The agreement is in effect as long as the patents are valid and we may terminate this agreement at our option with appropriate notice. Also, if we fail to actively attempt to commercialize licensed products for a specific period of time, the Board of Regents of The University of Texas System may have the option to terminate or limit the exclusivity of the license in certain territories. Specifically, Section 4.6 of our license agreement with The University of Texas System (as amended) provides that “Reasonable commercial diligence shall require that PLx . . . . [o]n or before September 8, 2013, Sell or offer for Sale a Licensed Product.” While we believe that we have exercised reasonable commercial diligence to actively attempt such commercialization, we have not yet successfully commercialized a licensed product. As such, the Board of Regents of The University of Texas System may have the option to terminate the license agreement, or to limit the exclusivity of the license in certain territories. The agreement provides for milestone payments by us related to the first product to obtain regulatory approval and the selling of, and payment for, a licensed product. The agreement provides for future potential milestone payments by us based upon the aggregate revenue from the sale of all licensed products in aggregate totaling $350,000. It is unlikely that any of these milestones will be triggered in the next twelve months. In addition to the milestone payments we will owe a royalty on the net sales of the licensed products. The amount of the royalty depends upon who is selling the product. Should we commercialize a product ourselves there is a running royalty obligation in the low single digit range based upon net sales. If a product is commercialized by another company under a sublicense agreement with PLx then the Board of Regents of The University of Texas System receives a share of consideration received by PLx that is in the low double digit range. There is a minimum annual royalty payment obligation. We prosecute and maintain the licensed patents at our expense. We are responsible for prosecution and control any action for infringement related to any product that does, or may, compete with one of our marketed licensed products and any claim within a licensed patent that covers or relates to such marketed licensed product.
International License Agreement with Lee’s
On March 12, 2012, Old PLx entered into a license agreement with Lee’s Pharmaceutical Holdings Limited, Zhaoke Pharmaceutical (HEFEI) Co. Ltd and Lee’s Pharmaceutical (Guangzhou) Limited (collectively, Lee’s) granting Lee’s exclusive rights to develop Aspertec in the People’s Republic of China including Hong Kong and Macao special Administrative Regions. This agreement was amended on December 31, 2013, and amended and restated effective June 19, 2015. This agreement is a sublicense of the Board of Regents of The University of Texas System patents providing Lee’s access to these and the PLx patents. As such, under the terms of the main license with the Board of Regents of The University of Texas System, any sublicense is automatically assigned to The University of Texas System if the main license agreement is terminated for any reason. Lee’s is responsible for developing and commercializing licensed products in their territories and has certain clinical and regulatory obligations. Lee’s will develop a 100 mg Aspertec and conduct bioequivalence, pharmacodynamics and endoscopy clinical studies appropriate for approval in China and other licensed territories. We prosecute and maintain the licensed patents and are reimbursed by Lee’s for this expense. Lee’s is responsible to prosecute and control any action for infringement related to any product that does, or may, compete with the marketed licensed products and any claim within a licensed patent that covers or relates to such marketed licensed product. PLx has received $400,000 in payments per the terms of the contract, which does not provide for any additional milestone payments in the future. Lee’s will pay a royalty in the low double digit range based upon net sales until the expiration of the last patent. At that time the royalty will reduce. There is a minimum annual royalty obligation. Lee’s may terminate the agreement with appropriate notice.
Government regulation
Government authorities in the United States, at the federal, state and local level, in the European Union and in other countries and jurisdictions extensively regulate, among other things, the research, development, testing, manufacture, including any manufacturing changes, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, import and export of pharmaceutical products such as those we are developing. The processes for obtaining regulatory approvals in the United States and in foreign countries and jurisdictions, along with subsequent compliance with applicable statutes and regulations, require the expenditure of substantial time and financial resources.
U.S. drug approval process
In the United States, the FDA regulates drugs under the federal Food, Drug, and Cosmetic Act, or FDCA, and implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA’s refusal to approve pending NDAs, withdrawal of an approval, imposition of a clinical hold, issuance of untitled or warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
● | completion of preclinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s current good laboratory practice, or cGLP, regulations; |
● | submission to the FDA of an IND which must become effective before human clinical trials may begin; |
● | approval by an independent institutional review board, or IRB, at each clinical site before each trial may be initiated; |
● | performance of adequate and well-controlled human clinical trials in accordance with current good clinical practices, or cGCP, to establish the safety and efficacy of the proposed drug or biological product for each indication; |
● | submission to the FDA of an NDA; |
● | satisfactory completion of an FDA advisory committee review, if applicable; |
● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with cGMP, and to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
● | FDA review and approval of the NDA. |
Preclinical studies and submission of an IND
Preclinical studies include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies to assess its potential safety and efficacy. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data and any available clinical data or literature, among other things, to the FDA as part of an IND. Some preclinical testing may continue even after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA’s allowing clinical trials to commence.
Clinical trials
Clinical trials involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with cGCP requirements, which include the requirement that all research subjects provide their informed consent (assent, if applicable) in writing for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an institutional review board, or IRB, at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their ClinicalTrials.gov website.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined:
● | Phase 1: The drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness. |
● | Phase 2: The drug is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
● | Phase 3: The drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product. In most cases, the FDA requires two adequate and well controlled Phase 3 clinical trials to demonstrate the efficacy of the drug. A single Phase 3 trial with other confirmatory evidence may be sufficient in rare instances where the study is a large multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the clinical trial patients are being exposed to an unacceptable health risk. The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Marketing approval
Assuming successful completion of the required clinical testing, the results of the preclinical and clinical studies, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. In most cases, the submission of an NDA is subject to a substantial application user fee, which is typically increased annually. Under the new Prescription Drug User Fee Act, or PDUFA, guidelines that are currently in effect, the FDA has a goal of ten months from the date of the FDA’s filing of a standard non-priority NDA to review and act on the submission.
The FDA also may require submission of a risk evaluation and mitigation strategy, or REMS, plan to mitigate any identified or suspected serious risks. The REMS plan could include medication guides, physician communication plans, assessment plans and elements to assure safe use, or ETASU, such as restricted distribution methods, patient registries or other risk minimization tools.
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality and purity. The FDA may refer an application for a novel drug to an advisory committee, which is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured, which is not under the control of the product sponsor. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with cGCP.
After the FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If, or when, those conditions have been met to the FDA’s satisfaction, the FDA will issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved indications for use for the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Post-approval requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies to determine compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend significant time, money and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
● | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
● | fines, untitled or warning letters or holds on post-approval clinical trials; |
● | refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product license approvals; |
● | product seizure or detention, or refusal to permit the import or export of products; or |
● | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved labeling, although doctors may prescribe drugs for off-label purposes.
The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act, or PDMA, which regulates the distribution of drugs and drug samples at the federal level, and sets minimum standards for the registration and regulation of drug distributors by the states. In addition, prescription drug manufacturers in the United States must comply with applicable provisions of the Drug Supply Chain Security Act and provide and receive product tracing information, maintain appropriate licenses, ensure they only work with other properly licensed entities, and have procedures in place to identify and properly handle suspect and illegitimate products.
Section 505(b)(2) NDAs
Most drug products obtain FDA marketing approval pursuant to an NDA or an abbreviated new drug application, or ANDA. A third alternative is a special type of NDA, commonly referred to as a Section 505(b)(2) NDA, which enables the applicant to rely, in part, on the FDA’s prior findings of safety and/or effectiveness for a similar product or published literature in support of its application.
Section 505(b)(2) NDAs often provide an alternate path to FDA approval for new or improved formulations or new uses of previously approved products. Section 505(b)(2) of the FDCA permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by, or for, the applicant and for which the applicant has not obtained a right of reference. If the Section 505(b)(2) applicant can establish that reliance on the FDA’s prior findings of safety and/or effectiveness is scientifically appropriate, it may eliminate the need to conduct certain preclinical or clinical studies of the new product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new product candidate for all, or some, of the indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.
Hatch-Waxman exclusivity
Marketing exclusivity provisions under the FDCA can delay the submission or the approval of certain applications for competing products. The FDCA provides a five-year period of non-patent exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity, or NCE, if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the NCE exclusivity period, the FDA may not accept for review an Abbreviated New Drug Application, or ANDA, or a Section 505(b)(2) NDA submitted by another company that references the previously approved drug. However, an ANDA or Section 505(b)(2) NDA may be submitted after four years if it contains a certification of patent invalidity or non-infringement (a Paragraph IV certification). If the ANDA or Section 505(b)(2) applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the ANDA applicant.
The FDCA also provides three years of marketing exclusivity for an NDA, Section 505(b)(2) NDA or supplement to an existing NDA or Section 505(b)(2) NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant, are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages, strengths or dosage forms of an existing drug. This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and, as a general matter, does not prohibit the FDA from approving ANDAs or Section 505(b)(2) NDAs for generic versions of the original, unmodified drug product. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Orphan drug designation and exclusivity
In the United States, the Orphan Drug Act provides incentives for the development of products intended to treat rare diseases or conditions. Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug or biological product available in the United States for this type of disease or condition will be recovered from sales of the product. If a sponsor demonstrates that a drug is intended to treat a rare disease or condition, the FDA may grant orphan designation for that product for the orphan disease indication. If a sponsor seeks orphan designation for a drug that contains an active moiety that has already been approved by the FDA for the same orphan indication, regardless of whether the approved product obtained orphan designation or orphan marketing exclusivity, the FDA will require the sponsor to provide a plausible hypothesis of the clinical superiority to the approved drug in order for the subsequent drug to be granted orphan designation. Clinical superiority can be shown on the basis of greater safety, greater efficacy, or making a major contribution to patient care. Orphan designation must be requested before submitting an NDA. After the FDA grants orphan product designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation, however, does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
Orphan drug designation provides manufacturers with research grants, tax credits, and eligibility for orphan drug exclusivity. If a product that has orphan drug designation subsequently receives the first FDA approval of the active moiety for that disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which for seven years prohibits the FDA from approving another product with the same active ingredient for the same indication, except in limited circumstances. If a drug’s orphan designation is predicated on a plausible hypothesis of clinical superiority, the FDA will require the sponsor to demonstrate clinical superiority in order for the product to be granted orphan drug exclusivity. If a drug designated as an orphan product receives marketing approval for an indication broader than the orphan indication for which it received the designation, it will not be entitled to orphan drug exclusivity. Orphan exclusivity will not bar approval of another product under certain circumstances, including if a subsequent product with the same active ingredient moiety for the same indication is shown to be clinically superior to the approved product on the basis of greater efficacy or safety, or providing a major contribution to patient care, or if the company with orphan drug exclusivity is not able to meet market demand. Further, the FDA may approve more than one product for the same orphan indication or disease as long as the products contain different active ingredients. Moreover, competitors may receive approval of different products for the indication for which the orphan product has exclusivity or obtain approval for the same product but for a different indication for which the orphan product has exclusivity.
In the European Union, the EMA’s Committee for Orphan Medicinal Products, or COMP, grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than 5 in 10,000 persons in the European Union community. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the European Union would be sufficient to justify the necessary investment in developing the drug.
In the European Union, orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity is granted following drug approval. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
NDA vs. OTC Monograph products
OTC drugs can be brought to market via two routes: the NDA approval process and the OTC monograph process. A drug product is eligible to be brought to market via the OTC monograph process if it is not a new drug, and the drug product meets the FDA’s established conditions for general recognition of safety and effectiveness (GRASE). The OTC drug monographs are a kind of “rule book” of conditions for each therapeutic category covering acceptable ingredients, uses (indications), doses, formulations, labeling, and testing.
The OTC Drug Review is a three-phase public rulemaking process established by the FDA to evaluate the safety and effectiveness of OTC drug products marketed in the United States prior to May 11, 1972. The three-phase rulemaking process can be summarized as follows:
● | 1. Advisory Review Panel — Advisory review panel appointed by the FDA analyzes data available on OTC drug active ingredients to determine if the active ingredients can be classified as GRASE, not GRASE, or insufficient data are available. Results of the advisory review panel’s analyses are published in the Federal Register as an Advance Notice for Proposed Rulemaking (ANPR). |
● | 2. Tentative Final Monograph — After the FDA reviews the advisory review panels’ findings, as well as additional data that may have become available and the public’s comments, the FDA publishes its conclusions in the Federal Register as a Proposed Rule also called a Tentative Final Monograph (TFM). |
● | 3. Final Monograph — After publication of the TFM, a period of time is allotted for interested parties to submit comments or data in response to the FDA’s proposal. The final regulations in the form of drug monographs provide a standard for GRASE OTC drug products. |
If a product deviates from the conditions under the TFM or final monograph and was not marketed before May 1972, then the drug product is considered a new drug and requires an NDA to be legally marketed. Aspirin was classified into the therapeutic class for Internal Analgesic, Antipyretic, and Antirheumatic Drug Products (IAAA). The ANPR was published in 1977, and in 1988, the FDA published the TFM for IAAA. The IAAA TFM proposes conditions under which OTC IAAA products may be marketed without prior review by the FDA, including labeling such as therapeutic indications, dosage instructions, and warnings about side effects and ways of preventing misuse. Although it has been updated and amended since its original publication, the IAAA monograph has not been finalized.
Differences between the NDA approval process and the OTC monograph process are listed below.
| ||
NDA Approval Process |
| OTC Monograph Process |
Pre-market approval — FDA reviews and approves formulation and labeling prior to marketing. |
| No pre-market approval — FDA sets forth specific conditions for GRASE, or in the case of a developing monograph, sets forth conditions that allow for continued marketing pending a final monograph. |
Confidential filing |
| Public process |
Drug-product specific |
| Active ingredient-specific and evaluated by OTC drug category |
May require a user fee |
| No user fees |
Potential for marketing exclusivity |
| No marketing exclusivity |
FDA reviews timelines |
| Manufacturers responsible for ensuring compliant product with no FDA-mandated review (either pre- or post-market) |
May require clinical studies, including studies on label comprehension and actual use |
| Generally does not require clinical studies. Label comprehension and actual use studies are not required for ingredients already covered by a final or tentative final monograph. |
Approved labeling is unique to the drug |
| Labeling is defined by the monograph. Once marketed, FDA can review the complete labeling at any time to determine whether it is truthful or misleading. |
Approved NDA is “license” to market |
| Final monograph is open to anyone |
Trade name reviewed prior to marketing |
| No review of trade name prior to marketing. Once marketed, FDA can review the trade name at any time. |
When Aspertec 325 mg is commercialized, we believe it will be the only NDA-approved OTC aspirin product available. Approval of the Aspertec 325mg NDA granted Aspertec 325mg labeling similar to that of monograph aspirin products.
Professional Labeling
Although the IAAA TFM has not been finalized for OTC use, final regulations for the professional labeling of aspirin were published in 1988. Professional labeling is labeling that provides specific information to health professionals for uses not included in OTC drug labeling. Professional labeling can be provided solely to healthcare professionals. Professional labeling may not be used on consumer products or on consumer-directed labeling. Under the IAAA regulations for professional labeling of aspirin, patients can only use aspirin for cardiovascular-related uses when directed to do so by a physician.
Professional labeling for aspirin includes the following indications:
● | Vascular Indications (Ischemic Stroke, Transient Ischemic Attack (TIA), Acute Myocardial Infarction (MI), Prevention of Recurrent MI, Unstable Angina Pectoris, and Chronic Stable Angina Pectoris): Aspirin is indicated to: (1) reduce the combined risk of death and nonfatal stroke in patients who have had ischemic stroke or transient ischemia of the brain due to fibrin platelet emboli, (2) reduce the risk of vascular mortality in patients with a suspected acute MI, (3) reduce the combined risk of death and nonfatal MI in patients with a previous MI or unstable angina pectoris, and (4) reduce the combined risk of MI and sudden death in patients with chronic stable angina pectoris. |
● | Revascularization Procedures (Coronary Artery Bypass Graft (CABG), Percutaneous Transluminal Coronary Angioplasty (PTCA), and Carotid Endarterectomy): Aspirin is indicated in patients who have undergone revascularization procedures (i.e., CABG, PTCA, or carotid endarterectomy) when there is a preexisting condition for which aspirin is already indicated. |
● | Rheumatologic Disease Indications (Rheumatoid Arthritis, Juvenile Rheumatoid Arthritis, Spondyloarthropathies, Osteoarthritis, and the Arthritis and Pleurisy of Systemic Lupus Erythematosus (SLE)): Aspirin is indicated for the relief of the signs and symptoms of rheumatoid arthritis, juvenile rheumatoid arthritis, osteoarthritis, spondyloarthropathies, and arthritis and pleurisy associated with SLE. |
FDA Oversight vs. FTC Oversight
Since 1971, the FDA and the Federal Trade Commission (FTC) have had a Memorandum of Understanding in place which dictates that the FDA has primary responsibility over OTC drug labeling, while the FTC has primary responsibility over OTC drug advertising.
| Rx Products |
| OTC Products | |
Labeling |
| FDA |
| FDA |
Advertising |
| FDA |
| FTC |
For an NDA-approved product, OTC labeling, that is, the label on the product bottle and the labeling on the box, must be submitted for review and approval prior to distribution. As indicated above, this is different from monograph products, which are not subjected to the FDA’s labeling review and approval processes prior to launching to market. Promotional labeling for OTC products (including brochures, leaflets and handouts) is under the purview of the FDA. Advertising for OTC products is under the purview of the FTC. Promotional advertisements (including in print and on radio and TV) are not required to be submitted to the FTC prior to distribution.
Under FTC regulations, claims in advertisements, including OTC medicine advertisements, must be truthful and cannot be misleading or unfair.
Advertisers must have substantiation that all objective express and implied claims in advertising are true before making the claims. The standard for substantiation of health claims is “competent and reliable scientific evidence.” For drug claims, competent and reliable scientific evidence generally has been interpreted as requiring at least one or two adequate and well-controlled human clinical studies of the product, or of an essentially equivalent product, that conform to acceptable designs and protocols and whose results, when considered in light of the entire body of relevant and reliable scientific evidence, are sufficient to substantiate that the representation is true.
Beyond FTC regulation of advertising, industry self-regulation plays an important role. The National Advertising Division (NAD) of the Council of Better Business Bureaus reviews advertising complaints by competitors. The NAD generally applies the same standard as the FTC. If the NAD determines that the substantiation does not support the claims or that it is otherwise false and misleading, it will recommend that the advertiser revise or discontinue the advertisement. If the advertiser does not agree to do so, the NAD will forward the case to the FTC or the FDA for review.
New legislation and regulations
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the testing, approval, manufacturing and marketing of products regulated by the FDA. In addition to new legislation, FDA regulations and policies are often revised or interpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether further legislative changes will be enacted or FDA regulations, guidance, policies or interpretations will be changed, or what the impact of such changes, if any, may be.
Foreign regulation
In order to market any product outside of the United States, we would need to comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of our products. The cost of establishing a regulatory compliance system for numerous varying jurisdictions can be very significant. Whether or not we obtain FDA approval for a product, we would need to obtain the necessary approvals by the comparable foreign regulatory authorities before we can commence clinical trials or marketing of the product in foreign countries and jurisdictions. Although many of the issues discussed above with respect to the United States apply similarly in the context of the European Union, the approval process varies between countries and jurisdictions and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others.
Pursuant to the European Clinical Trials Directive, a system for the approval of clinical trials in the European Union has been implemented through national legislation of the member states. Under this system, we must obtain approval from both the competent national authority of a European Union member state in which the clinical trial is to be conducted, and a favorable opinion from the competent ethics committee. Our clinical trial application must be accompanied by an investigational medicinal product dossier with supporting information prescribed by the European Clinical Trials Directive and corresponding national laws of the member states and further detailed in applicable guidance documents.
To obtain marketing approval of a drug under European Union regulatory systems, we may submit a Marketing Authorization Application, or MAA, either under a centralized or decentralized procedure. The centralized procedure provides for the grant of a single marketing authorization by the European Commission that is valid for all European Union member states. The centralized procedure is compulsory for specific products, including medicines produced by certain biotechnological processes, products designated as orphan medicinal products, advanced therapy products and products with a new active substance indicated for the treatment of certain diseases. For products with a new active substance indicated for the treatment of other diseases and products that are highly innovative or for which a centralized process is in the interest of patients, the centralized procedure may be optional. Under the centralized procedure, the Committee for Medicinal Products for Human Use, or the CHMP, established at the EMA is responsible for conducting the initial assessment of a drug. The CHMP also is responsible for several post-authorization and maintenance activities, such as the assessment of modifications or extensions to an existing marketing authorization. Under the centralized procedure in the European Union, the maximum timeframe for the evaluation of an MAA is 210 days, excluding clock stops, when additional information or written or oral explanation is requested by the CHMP but has not yet been provided. Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is of major interest from the point of view of public health and in particular from the viewpoint of therapeutic innovation. In this circumstance, the EMA ensures that the opinion of the CHMP is given within 150 days.
The decentralized procedure is available to applicants who wish to market a product in various European Union member states where such product has not previously received marketing approval in any European Union member state. The decentralized procedure provides for approval by one or more other, or concerned, member states of an assessment of an application performed by one member state designated by the applicant, known as the reference member state. Under this procedure, an applicant submits an application based on identical dossiers and related materials, including a draft summary of product characteristics, and draft labeling and package leaflet, to the reference member state and concerned member states. The reference member state prepares a draft assessment report and drafts of the related materials within 120 days after receipt of a valid application. Within 90 days of receiving the reference member state’s assessment report and related materials, each concerned member state must decide whether to approve the assessment report and related materials.
If a member state cannot approve the assessment report and related materials on the grounds of potential serious risk to public health, the disputed points are subject to a dispute resolution mechanism and may eventually be referred to the European Commission, whose decision is binding on all member states.
In the European Union, new chemical entities qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity. This data exclusivity, if granted, prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a generic (abbreviated) application for eight years, after which generic marketing authorization can be submitted, and the innovator’s data may be referenced, but not approved for two years. The overall ten-year period will be extended to a maximum of eleven years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. Even if a compound is considered to be a new chemical entity and the sponsor is able to gain the prescribed period of data exclusivity, another company nevertheless could also market another version of the drug if such company can complete a full MAA with a complete database of pharmaceutical test, preclinical tests and clinical trials and obtain marketing approval of its product.
Pharmaceutical coverage, pricing and reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any drug products for which we may obtain regulatory approval. Sales of any of our product candidates, if approved, will depend, in part, on the extent to which the costs of the products will be covered by third-party payors, including government health programs such as Medicare and Medicaid, commercial health insurers and managed care organizations. The process for determining whether a third-party payor will provide coverage for a drug product typically is separate from the process for setting the price of a drug product or for establishing the reimbursement rate that a payor will pay for the drug product once coverage is approved. Third-party payors may limit coverage to specific drug products on an approved list, also known as a formulary, which might not include all of the approved drugs for a particular indication.
In order to secure coverage and reimbursement for any product that might be approved for sale, we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product, in addition to the costs required to obtain FDA or other comparable regulatory approvals. Whether or not we conduct such studies, our product candidates may not be considered medically necessary or cost-effective. A third-party payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Third party reimbursement may not be sufficient to enable us to maintain price levels high enough to realize an appropriate return on our investment in product development.
The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of drugs have been a focus in this effort. Third-party payors are increasingly challenging the prices charged for medical products and services, examining the medical necessity and reviewing the cost-effectiveness of drug products and medical services and questioning safety and efficacy. If these third-party payors do not consider our products to be cost-effective compared to other available therapies, they may not cover our products after FDA approval or, if they do, the level of payment may not be sufficient to allow us to sell our products at a profit. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs to limit the growth of government-paid healthcare costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. Adoption of such controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals such as our drug product candidates and could adversely affect our net revenue and results.
Pricing and reimbursement schemes vary widely from country to country. Some countries provide that drug products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular product candidate to currently available therapies. For example, the European Union provides options for its member states to restrict the range of drug products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. European Union member states may approve a specific price for a drug product or they may instead adopt a system of direct or indirect controls on the profitability of the company placing the drug product on the market. Other member states allow companies to fix their own prices for drug products, but monitor and control company profits. The downward pressure on healthcare costs in general, particularly prescription drugs, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert competitive pressure that may reduce pricing within a country. Any country that has price controls or reimbursement limitations for drug products may not allow favorable reimbursement and pricing arrangements for any of our products.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, emphasis on managed care in the United States has increased and we expect will continue to increase the pressure on drug pricing. Coverage policies, third-party reimbursement rates and drug pricing regulation may change at any time. In particular, the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010, which we collectively refer to as the Affordable Care Act or ACA, contains provisions that have the potential to substantially change healthcare financing, including by impacting the profitability of drugs. For example, the Affordable Care Act revised the methodology by which rebates owed by manufacturers to the state and federal government for covered outpatient drugs under the Medicaid Drug Rebate Program, extended the Medicaid Drug Rebate Program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations and subjected manufacturers to new annual fees and taxes for certain branded prescription drugs. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Healthcare law and regulation
Healthcare providers, physicians and third-party payors play a primary role in the recommendation and prescribing of any product candidates for which we may obtain marketing approval. Our business operations and arrangements with investigators, healthcare professionals, consultants, third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations. These laws may constrain the business or financial arrangements and relationships through which we research, market, sell and distribute our products that obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations, include, but are not limited to, the following:
● | the federal healthcare Anti-Kickback Statute prohibits, among other things, persons or entities from knowingly and willfully soliciting, offering, receiving or paying any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid; |
● | the federal false claims laws and civil monetary penalties law impose penalties and provide for civil whistleblower or qui tam actions against individuals or entities for, among other things, knowingly presenting, or causing to be presented, to the federal government, claims for payment or approval that are false or fraudulent or making a false record or statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, among other things, imposes criminal liability for knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, also imposes certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information without written authorization; |
● | the federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; |
● | the federal transparency requirements under the Affordable Care Act require manufacturers of drugs, devices, biologics and medical supplies to report annually to the U.S. Department of Health and Human Services, or HHS, information related to payments and other transfers of value to physicians and teaching hospitals and certain physician ownership and investment interests; and |
● | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, that may apply to our business operations, including our sales or marketing arrangements, and claims involving healthcare items or services reimbursed by governmental third-party payors, and in some instances, also such claims reimbursed by non-governmental third-party payors, including private insurers. |
Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures. State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and/or administrative penalties, damages, fines, disgorgement, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other providers or entities with whom we expect to do business are found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
The Foreign Corrupt Practices Act
The Foreign Corrupt Practices Act, or FCPA, prohibits any U.S. individual or business from paying, offering or authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the United States to comply with accounting provisions requiring the company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations.
As we pursue international licensing and sales arrangements outside the United States, we will be heavily regulated and expect to have significant interaction with foreign officials. Additionally, in many countries outside the United States, the health care providers who prescribe human pharmaceuticals are employed by the government and the purchasers of human pharmaceuticals are government entities; therefore, our interactions with these prescribers and purchasers would be subject to regulation under the FCPA.
In addition to the U.S. application and enforcement of the FCPA, the various jurisdictions in which we operate and supply our products have laws and regulations aimed at preventing and penalizing corrupt and anticompetitive behavior. In recent years, several jurisdictions, including China, Brazil, and the United Kingdom, have enhanced their laws and regulations in this area, increased their enforcement activities, and/or increased the level of cross-border coordination and information sharing.
Employees
As of June 1, 2017, we had nine employees. None of our employees are represented by a labor union or covered by collective bargaining agreements. We consider our relationship with our employees to be good.
Facilities
Our principal facilities consist of office space in Houston, Texas. We occupy approximately 3,905 square feet of office and lab space, with rent of $4,652 per month, under a lease that expires December 31, 2017. We believe that this space or replacement office space will be available on commercially reasonable terms.
Compliance with Environmental Regulations
Our third-party manufacturers’ activities and our own activities may involve the controlled storage, use and disposal of hazardous materials, including the components of our pharmaceutical product candidates, test samples and reagents, biological materials and other hazardous compounds. We and our manufacturers are subject to federal, state, local and foreign laws and regulations governing the use, generation, manufacture, storage, handling and disposal of these hazardous materials. We currently carry no insurance specifically covering environmental claims relating to the use of hazardous materials. Although we believe that our safety procedures for handling and disposing of these materials and waste products comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental injury or contamination from the use, storage, handling or disposal of hazardous materials. In the event of an accident, state or federal or other applicable authorities may curtail our use of these materials and/or interrupt our business operations. In addition, if an accident or environmental discharge occurs, or if we discover contamination caused by prior operations, including by prior owners and operators of properties we acquire, we could be liable for cleanup obligations, damages and fines. If such unexpected costs are substantial, this could significantly harm our financial condition and results of operations.
Legal proceedings
In April 2010, Dipexium acquired the worldwide rights to develop pexiganan, the active pharmaceutical ingredient in Locilex®, from Genaera Liquidating Trust, which was put in place to liquidate the assets of Genaera Corporation. In June 2012, Dipexium, along with its two senior executives and several other unrelated defendants, were sued in the Federal District Court for the Eastern District of Pennsylvania by a former shareholder of Genaera Corporation and purported to be on behalf of other Genaera Corporation shareholders, alleging, in pertinent part, that Dipexium’s acquisition of the rights to pexiganan (the active ingredient in Locilex®, and which rights included the rights to the prior formulation of Locilex®) was for what was alleged to be inadequate consideration, and as a result, it was alleged that Dipexium and its senior executives aided and abetted a breach of fiduciary duty by Genaera Corporation and the Genaera Liquidating Trust to the former shareholders of Genaera Corporation. It was also alleged that Dipexium and its senior executives aided and abetted a breach of the duty of the trustee at common law and under a certain trust agreement which was alleged to exist and which was executed by Argyce LLC (or Argyce), as trustee. The agreement called for Argyce to create the Genaera Liquidating Trust pursuant to which Argyce apparently was appointed to liquidate the assets formerly held by Genaera Corporation. One of these assets was pexiganan, which Dipexium acquired via public auction conducted by Argyce on behalf of the Genaera Liquidating Trust.
The case against Dipexium and its senior executives was dismissed with prejudice by the Federal District Court, without leave to refile, on August 12, 2013 based on the argument that Plaintiff’s claims were time barred, and a subsequent motion to reconsider such dismissal was denied by the Federal District Court. Prior to the dismissal there was no request or action to seek class certification by the plaintiff though it was purportedly filed on behalf of other former Genaera Corporation shareholders. Plaintiff appealed the dismissal of the suit as well as the denial of the motion to reconsider to the Third Circuit Appellate Court, which granted Plaintiff’s appeal.
On October 17, 2014, the Third Circuit Appellate Court, in a 2-1 decision with a strong dissenting opinion, reversed the trial court’s dismissal of Plaintiff’s claims based on the expiration of the applicable statutes of limitation and remanded the case to the Federal District Court. In its opinion, the Third Circuit held that more information was necessary to determine when Plaintiff should have been on notice of his claims to determine the applicability of the discovery rule, which could serve to extend the time frame in which Plaintiff could bring his claims. Due to the strong dissent, all defendants filed the necessary documents requesting a petition for rehearing en banc, by the majority of the Third Circuit justices who are in active service. The Third Circuit denied the request for en banc hearing and remanded this case to District Court.
Upon remand to the Federal District Court, all defendants moved to dismiss the complaint for reasons other than being time barred. Dipexium and its executives moved for dismissal based on Plaintiff’s inability to make a case for aiding and abetting a breach of fiduciary duty because there was no underlying breach and such an aiding and abetting claim requires an element of knowing participation in the fiduciary breach which cannot be established by Plaintiff.
The District Court held a hearing on this in September 2015 and the District Court delivered an Order on November 10, 2015 pursuant to which the District Court granted the Motion to Dismiss filed by each and every defendant including the Company and its executives. In December 2015, Plaintiff appealed the Federal District Court’s decision to the Third Circuit Appellate Court and we anticipate a decision on whether to grant Plaintiff’s appeal by the Third Circuit Appellate Court in the first half of 2017. We will continue to vigorously defend Plaintiff’s claims on the factual record, which we believe will prove that we are not liable to the Plaintiff in any regard.
If we were to lose this case, we could be required to pay damages, which could have a material adverse effect on us, our business plans and results of operations.
CERTAINMATERIAL U.S. FEDERAL INCOME TAX CONSIDERATIONS
The following is a summary of certain material U.S. federal income tax considerations relating to the acquisition, ownership and disposition of shares of our common stock issued pursuant to this offering and the acquisition, ownership and disposition of the warrants issued to investors in the concurrent private placement. This summary deals only with shares of our common stock acquired by a stockholder in this offering, and with warrants acquired by an investor in the concurrent private placement, and that are held as a capital asset within the meaning of Section 1221 of the Internal Revenue Code of 1986, as amended, or the Code. This summary does not address the U.S. federal income tax considerations applicable to a stockholder or investor that is subject to special treatment under U.S. federal income tax laws, including: a dealer in securities or currencies; a financial institution; a regulated investment company; a real estate investment trust; a tax-exempt organization; an insurance company; a person holding our common stock or warrants as part of a hedging, integrated, conversion or straddle transaction or a person deemed to sell our common stock or warrants under the constructive sale provisions of the Code; a trader in securities that has elected the mark-to-market method of accounting; an entity that is treated as a partnership for U.S. federal income tax purposes; a person that received our common stock or warrants in connection with services provided to the Company or any of its affiliates; a U.S. person whose “functional currency” is not the U.S. dollar; a “controlled foreign corporation”; a “passive foreign investment company”; or a U.S. expatriate.
This summary is based upon provisions of the Code, and applicable Treasury regulations promulgated or proposed thereunder, rulings and judicial decisions, all as in effect as of the date hereof. Those authorities may be changed, perhaps with retroactive effect, or may be subject to differing interpretations, which could result in U.S. federal income tax consequences different from those discussed below. This summary does not address all aspects of U.S. federal income tax, does not address all tax considerations that may be relevant to stockholders in light of their personal circumstances and does not address any state, local, foreign, gift, estate or alternative minimum tax considerations.
For purposes of this discussion, a “U.S. holder” is a beneficial holder of our common stock or warrants that is: an individual citizen or resident of the United States for U.S. federal income tax purposes; a corporation (or any other entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any state thereof or the District of Columbia; an estate the income of which is subject to U.S. federal income taxation regardless of its source; or a trust if it (1) is subject to the primary supervision of a court within the United States and one or more U.S. persons (as defined in the Code) have the authority to control all substantial decisions of the trust or (2) has a valid election in effect under applicable U.S. Treasury regulations to be treated as a U.S. person.
For purposes of this discussion, a “non-U.S. holder” is a beneficial holder of our common stock or warrants that is for U.S. federal income tax purposes an individual, corporation, estate or trust and is not a U.S. holder.
If a partnership (or an entity or arrangement that is treated as a partnership for U.S. federal income tax purposes) holds our common stock or warrants, the tax treatment of a person treated as a partner in the partnership for U.S. federal income tax purposes generally will depend upon the status of the partner and the activities of the partnership. Partnerships and other entities that are treated as partnerships for U.S. federal income tax purposes and persons holding our common stock or warrants through a partnership or other entity treated as a partnership for U.S. federal income tax purposes are urged to consult their own tax advisors.
This summary is for general information only and is not intended to be tax advice. Holders of our common stock or warrants are urged to consult their own tax advisors concerning the tax considerations related to the acquisition, ownership and disposition of our common stock or warrants in light of their particular circumstances, as well as any tax considerations arising under the laws of any other jurisdiction, including any state, local and foreign income and other tax laws.
U.S. Holders
The following discussion is a summary of certain U.S. federal income tax considerations relevant to a U.S. holder of our common stock or warrants.
Distributions
Distributions with respect to our common stock, if any, generally will be includible in the gross income of a U.S. holder as ordinary dividend income to the extent of our current or accumulated earnings and profits, as determined for U.S. federal income tax purposes. Any portion of a distribution in excess of current and accumulated earnings and profits will be treated as a non-taxable return of capital, up to the U.S. holder’s adjusted tax basis in its shares of our common stock with respect to which the distribution was made. Any such distribution in excess of the U.S. holder’s adjusted tax basis in its shares will be treated as capital gain and as long-term capital gain if the U.S. holder’s holding period exceeds one year. If certain requirements are met (including certain holding period requirements), distributions constituting dividends paid to individual U.S. holders generally will qualify for a reduced tax rate on qualified dividend income.
Distributions constituting dividends for U.S. federal income tax purposes that are paid to U.S. holders that are corporations may qualify for the 70% dividends received deduction, or “DRD,” which is generally available to corporations that own less than 20% of the voting power or value of the outstanding stock of the distributing corporation. A U.S. holder that is a corporation holding 20% or more of the distributing corporation (by vote and value) may be eligible for an 80% DRD with respect to any such dividends. No assurance can be given that we will have sufficient earnings and profits (as determined for U.S. federal income tax purposes) to cause any distributions to be treated as dividends eligible for a DRD. In addition, a DRD is available only if certain other requirements (including certain holding period requirements) are satisfied, and a DRD may be subject to limitations in certain circumstances, which are not discussed herein.
Sale, Exchange, Redemption or Certain Other Taxable Dispositions of Our Common Stock
A U.S. holder of shares of our common stock generally will recognize gain or loss on the taxable sale, exchange, redemption (provided the redemption is treated as a sale or exchange), or other taxable disposition of such shares in an amount equal to the difference between such U.S. holder’s amount realized on such disposition and such U.S. holder’s adjusted tax basis in its shares of our common stock disposed of. A U.S. holder’s amount realized generally will equal the amount of cash and the fair market value of any property received in consideration for the shares of common stock disposed of. Such gain or loss will be capital gain or loss, and will be long-term capital gain or loss if the U.S. holder’s holding period for the shares of our common stock disposed of exceeds one year at the time of disposition. The deductibility of capital losses is subject to certain limitations. U.S. holders should consult their tax advisors regarding the treatment of capital gains and capital losses.
Exercise of the Warrants by U.S. Holders
The exercise of the warrants should not be a taxable event. Gain on the sale of a warrant would be long-term capital gain if the warrant is held for more than one year. Non-corporate U.S. holders (including individuals) may be eligible for preferential rates of U.S. federal income tax in respect of long-term capital gains. The U.S. holders’ tax basis in the common stock acquired on exercise of the warrants would include an allocable portion of the purchase price plus the exercise price. Distributions on the common stock acquired on the exercise of a warrant would be taxed as described in “—U.S. Holders —Distributions” above. A U.S. holder’s holding period with respect to the common stock acquired on the exercise of a warrant would begin at the time of such exercise. Gain on the sale of common stock acquired on exercise of the warrants would be taxable as described in “—U.S. Holders —Sale, Exchange, Redemption or Certain Other Taxable Dispositions of Our Common Stock” above.
Medicare Tax on Net Investment Income
An additional 3.8% Medicare tax will be imposed on certain net investment income of certain U.S. holders that are individuals, estates or trusts. Such tax applies to the lesser of (i) the U.S. holder’s net investment income for the relevant taxable year and (ii) the excess of the U.S. holder’s adjusted gross income (with certain adjustments) over a specified threshold amount. Net investment income generally includes dividends and net gains from the disposition of shares of stock in a corporation. U.S. holders that are individuals, estates or trusts should consult their tax advisors regarding the effect, if any, of the Medicare tax on their ownership and disposition of our common stock.
Information Reporting and Backup Withholding Tax
In general, information reporting will apply to payments of dividends on shares of our common stock and proceeds of a disposition of shares of our common stock to U.S. holders, other than certain exempt recipients such as corporations. Under U.S. federal income tax law, dividends and proceeds from the sale of shares of our common stock paid to a U.S. holder (other than an exempt recipient) may be subject to “backup” withholding at the then applicable rate. Backup withholding generally applies to a U.S. holder if the holder (i) fails to furnish to us or our paying agent a correct social security number or other taxpayer identification number, or TIN, or fails to furnish a certification of exempt status, (ii) has been notified by the IRS that it is subject to backup withholding as a result of the failure to properly report payments of interest or dividends or (iii) under certain circumstances, fails to provide a certified statement, signed under penalty of perjury, that the TIN provided is its correct number and that it is a U.S. person that is not subject to backup withholding. Backup withholding is not an additional tax. Any amounts withheld from payments to a U.S. holder under the backup withholding rules will be allowed as a credit against such holder’s U.S. federal income tax liability and may entitle the holder to a refund, provided that the required information is timely furnished to the IRS. Certain U.S. persons are exempt from backup withholding, including corporations, provided that their exemptions from backup withholding are properly established.
Non-U.S. Holders
The following is a summary of certain U.S. federal tax considerations applicable to a non-U.S. holder of our common stock or warrants.
Distributions
Distributions treated as dividends for U.S. federal income tax purposes (as described above under “— U.S. Holders — Distributions”), if any, that are paid to a non-U.S. holder with respect to shares of our common stock will be subject to U.S. federal withholding tax at a 30% rate (or a lower rate prescribed by an applicable income tax treaty) unless the dividends are effectively connected with the non-U.S. holder’s conduct of a trade or business in the United States (and, if required by an applicable income tax treaty, is attributable to a permanent establishment or fixed base maintained in the United States). To claim the exemption from withholding with respect to any such effectively connected income, the non-U.S. holder must furnish to us or our paying agent a properly executed IRS Form W-8ECI (or applicable successor form), certifying under penalties of perjury that a dividend paid on our common stock is not subject to withholding tax. The certification requirement also may require a non-U.S. holder to provide its U.S. taxpayer identification number.
If a non-U.S. holder is engaged in a trade or business in the United States and dividends with respect to our common stock are effectively connected with the conduct of such trade or business and, if required by an applicable income tax treaty, are attributable to a U.S. permanent establishment or fixed base, then the non-U.S. holder generally will be subject to U.S. federal income tax on such dividends on a net income basis in the same manner as if received by a U.S. holder (although the dividends will be exempt from the 30% U.S. federal withholding tax, provided the certification requirements are satisfied). In addition, if the non-U.S. holder is a corporation for U.S. federal income tax purposes, such holder may, under certain circumstances, be subject to an additional branch profits tax equal to 30% (or a lower rate prescribed by an applicable income tax treaty) of its effectively connected earnings and profits for the taxable year.
A non-U.S. holder who wishes to claim the benefit of an exemption or reduced rate of U.S. federal withholding tax under an applicable income tax treaty must furnish to us or our paying agent a valid IRS Form W-8BEN (or applicable successor form) certifying, under penalties of perjury, such non-U.S. holder’s qualification for the exemption or reduced rate. If a non-U.S. holder is eligible for an exemption or a reduced rate of U.S. federal withholding tax pursuant to an applicable income tax treaty, it may obtain a refund of any excess amounts withheld by filing an appropriate claim for refund with the IRS.
If a distribution exceeds our current and accumulated earnings and profits, the excess will be treated as a non-taxable return of capital, up to the non-U.S. holder’s adjusted tax basis in its shares of our common stock. Any remaining excess will be treated as capital gain, subject to the tax treatment described below in “— Sale, Exchange, Redemption or Certain Other Taxable Dispositions of Our Common Stock.” If we are not able to determine whether or not a distribution will exceed current and accumulated earnings and profits at the time a distribution is made, we may withhold tax on the entire amount of such distribution at the same rate as we would withhold on a dividend. However, a non-U.S. holder may obtain a refund of any excess withholding by filing an appropriate claim for refund with the IRS.
Any distribution described in this section would also be subject to the discussion below in “Foreign Account Tax Compliance Act.”
Sale, Exchange, Redemption or Certain Other Taxable Dispositions of Our Common Stock
Subject to the discussions below regarding backup withholding and the Foreign Account Tax Compliance Act, a non-U.S. holder generally will not be subject to U.S. federal income tax or withholding tax on gain realized upon a sale, exchange or other taxable disposition of shares of our common stock unless: (i) the gain is effectively connected with the conduct of a trade or business in the United States (and, if required by an applicable income tax treaty, is attributable to a U.S. permanent establishment or a fixed base) of the non-U.S. holder; (ii) the non-U.S. holder is a non-resident alien individual who is present in the United States for 183 days or more in the taxable year of that disposition, and certain other conditions are met; or (iii) we are or have been a “U.S. real property holding corporation”, or a USRPHC, for U.S. federal income tax purposes at any time within the shorter of the five-year period preceding the disposition and the non-U.S. holder’s holding period for our common stock.
If the first exception applies, the non-U.S. holder generally will be subject to U.S. federal income tax on a net basis with respect to such gain in the same manner as if such holder were a resident of the United States. In addition, if the non-U.S. holder is a corporation for U.S. federal income tax purposes, such gains may, under certain circumstances, also be subject to the branch profits tax at a rate of 30% (or at a lower rate prescribed by an applicable income tax treaty).
If the second exception applies, the non-U.S. holder generally will be subject to U.S. federal income tax at a rate of 30% tax on the gain from a disposition of our common stock, which may be offset by capital losses allocable to U.S. sources during the taxable year of disposition (even though the non-U.S. holder is not considered a resident of the United States).
With respect to the third exception above, we believe we currently are not, and we do not anticipate becoming, a USRPHC for U.S. federal income tax purposes. Because the determination of whether we are a USRPHC depends on the fair market value of our U.S. real property interests relative to the fair market value of our other trade or business assets and our foreign real property interests, there can be no assurances that we will not become a USRPHC in the future. Generally, a corporation is a USRPHC only if the fair market value of its U.S. real property interests (as defined in the Code) equals or exceeds 50% of the sum of the fair market value of its worldwide real property interests plus its other assets used or held for use in a trade or business. Even if we are or become a USRPHC, a non-U.S. holder would not be subject to U.S. federal income tax on a sale, exchange or other taxable disposition of our common stock by reason of our status as a USRPHC so long as (i) our common stock continues to be regularly traded on an established securities market (within the meaning of Section 897(c)(3) of the Code) during the calendar year in which such disposition occurs and (ii) such non-U.S. holder does not own and is not deemed to own (directly, indirectly, or constructively) more than 5% of our common stock at any time during the relevant period. If we are a USRPHC and the requirements of (i) or (ii) are not met, gain on the disposition of shares of our common stock generally will be taxed in the same manner as gain that is effectively connected with the conduct of a U.S. trade or business, except that the branch profits tax will not apply.
Exercise of the Warrants by Non-U.S. Holders
The exercise of the warrants should not be a taxable event. Gain on the sale of a warrant generally will not be subject to U.S. federal income tax or any withholding thereof unless one of the circumstances described under “—Non-U.S. Holders —Sale, Exchange, Redemption or Certain Other Taxable Dispositions of Our Common Stock” above applies. The non-U. S. holders’ tax basis in the common stock acquired on exercise of the warrants would include an allocable portion of the purchase price plus the exercise price. Distributions on the common stock acquired on the exercise of a warrant would be taxed as described in “—Non-U.S. Holders—Distributions” above. A non-U.S. holder’s holding period with respect to the common stock acquired on the exercise of a warrant would begin at the time of such exercise. Gain on the sale of common stock acquired on exercise of the warrants would be taxable as described in “—Non-U.S. Holders — Sale, Exchange, Redemption or Certain Other Taxable Dispositions of Our Common Stock” above.
Information Reporting and Backup Withholding Tax
We must report annually to the IRS and to each non-U.S. holder the gross amount of the distributions on our common stock paid to such holder and the tax withheld, if any, with respect to such distributions, regardless of whether withholding was required. This information also may be made available under a specific treaty or agreement with the tax authorities in the country in which the non-U.S. holder resides or is established. A non-U.S. holder will generally be subject to backup withholding at the then applicable rate for dividends paid to such holder unless such holder furnishes a valid IRS Form W-8BEN (or such other applicable form and documentation as required by the Code or the Treasury regulations) certifying under penalties of perjury that it is a non-U.S. holder (and the payor does not have actual knowledge or reason to know that such holder is a U.S. person as defined under the Code), or otherwise establishes an exemption. Dividends paid to non-U.S. holders subject to U.S. federal withholding tax, as described above in “Distributions,” generally will be exempt from U.S. backup withholding.
Information reporting and, depending on the circumstances, backup withholding will apply to the payment of the proceeds of a sale or other disposition of shares of our common stock by a non-U.S. holder effected by or through the U.S. office of any broker, U.S. or foreign, unless the holder certifies that it is not a U.S. person (as defined under the Code) and satisfies certain other requirements, or otherwise establishes an exemption. Generally, information reporting and backup withholding will not apply to a payment of disposition proceeds to a non-U.S. holder where the transaction is effected outside the United States through a non-U.S. office of a broker. However, for information reporting purposes, dispositions effected through a non-U.S. office of a broker with substantial U.S. ownership or operations generally will be treated in a manner similar to dispositions effected through a U.S. office of a broker. Prospective investors should consult their own tax advisors regarding the application of the information reporting and backup withholding rules to them.
Copies of the information returns may be made available to the tax authorities in the country in which the non-U.S. holder resides or is incorporated under the provisions of an applicable treaty or agreement.
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules may be allowed as a credit against a non-U.S. holder’s U.S. federal income tax liability, if any, and may entitle such holder to a refund, provided that an appropriate claim is timely filed with the IRS.
Foreign Account Tax Compliance Act
Under the Foreign Account Tax Compliance Act, or FATCA, a 30% withholding tax will apply to dividends on, or gross proceeds from the sale or other disposition of, shares of our common stock paid to certain non-U.S. entities (including financial intermediaries) unless various information reporting and due diligence requirements, which are different from and in addition to the certification requirements described elsewhere in this discussion, have been satisfied (generally relating to ownership by U.S. persons of interests in or accounts with those entities). The withholding rules apply to payments of gross proceeds from dispositions of U.S. common stock beginning January 1, 2017.
Holders of our common stock should consult their tax advisors regarding the possible impact of FATCA on their investment in our common stock, including, without limitation, the process and deadlines for meeting the applicable requirements to prevent the imposition of the 30% withholding tax under FATCA.
In a concurrent private placement, or the Private Placement Transaction, we are selling to the investors in this offering warrants, or the Warrants, to purchase 2,646,091 shares of our common stock.
The Warrants and the shares of our common stock issuable upon exercise of the Warrants, or the Warrant Shares, are not being registered under the Securities Act pursuant to the registration statement of which this prospectus supplement and the accompanying prospectus form a part and are not being offered pursuant to this prospectus supplement and the accompanying prospectus. The Warrants and the Warrant Shares are being offered pursuant to an exemption from the registration requirements of the Securities Act provided in Section 4(a)(2) of the Securities Act and/or Regulation D promulgated thereunder. Accordingly, investors may only sell shares of common stock issued upon exercise of the Warrants pursuant to an effective registration statement under the Securities Act covering the resale of those shares, an exemption under Rule 144 under the Securities Act, or Rule 144, or another applicable exemption under the Securities Act.
Description of the Warrants
The Warrants are exercisable beginning on six months and one day following the date of issuance at an exercise price of $7.50 per share and will expire on the 10th anniversary of the original issue date of the Warrants. The exercise price and number of shares of our common stock issuable upon exercise of the Warrants will be subject to adjustment for combinations, stock splits and similar capital transactions, as described in the Warrants. In addition, in the event of a Fundamental Transaction (as defined below), the holder of the Warrant will have the right thereafter to receive, upon exercise of the Warrant, the same amount and kind of securities, cash or property as it would have been entitled to receive upon the occurrence of such Fundamental Transaction if it had been, immediately prior to such Fundamental Transaction, the holder of the number of Warrant Shares then issuable upon exercise in full of the Warrant, and the holder of the Warrant shall no longer have the right to receive Warrant Shares upon exercise of the Warrant. For purposes of each Warrant, a “Fundamental Transaction” is deemed to have occurred if, at any time while the Warrant is outstanding, (i) we effect (a) any merger of the Company with or into another person, (b) any sale, lease, license (other than in the normal course of business), assignment, transfer, conveyance or other disposition of all or substantially all of the Company’s assets in one or a series of related transactions, (c) any, direct or indirect, purchase offer, tender offer or exchange offer (whether by us or another person) pursuant to which holders of our common stock are permitted to sell, tender or exchange their shares for other securities, cash or property and has been accepted by the holders of 50% or more of our outstanding common stock, (iv) a reorganization or recapitalization of our common stock or any compulsory share exchange pursuant to which our common stock is effectively converted into or exchanged for other securities, cash or property, or (v) a stock or share purchase agreement or other business combination (including, without limitation, a reorganization, recapitalization, spinoff or scheme of arrangement) with another person or group of persons whereby such other person or group acquires more than 50% of the outstanding shares of our common stock (not including any shares of our common stock held by the other person or other persons making or party to, or associated or affiliated with the other persons making or party to, such stock or share purchase agreement or other business combination).
Raymond James & Associates, Inc. and Janney Montgomery Scott LLC, which we refer to as the placement agents, have agreed to act as the placement agents in connection with this offering, and Raymond James & Associates, Inc. has agreed to act as the representative of the placement agents, subject to the terms and conditions of a placement agent agreement, dated June 9, 2017. The placement agents may engage selected dealers to assist in the placement of the shares. The placement agents are not purchasing or selling any shares offered by this prospectus supplement and the accompanying prospectus, nor are they required to arrange the purchase or sale of any specific number or dollar amount of the shares, but have agreed to use best efforts to arrange for the sale of all of the shares offered hereby. We will enter into a securities purchase agreement directly with investors in connection with this offering and we may not sell the entire amount of shares offered pursuant to this prospectus supplement and the accompanying prospectus. The purchase price for each share has been determined based upon arm’s-length negotiations between the purchasers and us.
Investors purchasing the securities offered hereby will execute a securities purchase agreement with us, providing such investors with certain representations, warranties and covenants from us.
Commission and Expenses
We have agreed to pay the placement agents an aggregate cash placement fee equal to 6.0% of the gross proceeds in this offering. The following table shows the per share and total cash placement agent fees we will pay to the placement agents in connection with the sale of the shares offered pursuant to this prospectus supplement and the accompanying prospectus assuming the purchase of all of the shares offered hereby:
Per Share | $0.4625 |
|
|
Total | $1,091,512 |
Because there is no minimum offering amount required as a condition to closing in this offering, the actual total offering commissions, if any, are not presently determinable and may be substantially less than the maximum amount set forth above. We have also agreed to reimburse the representative for its out-of-pocket expenses in an aggregate amount equal to $100,000, with a separate limitation of $75,000 for legal expenses.
Our obligation to issue and sell shares to the purchasers is subject to the conditions set forth in the securities purchase agreement, which may be waived by us at our discretion. A purchaser’s obligation to purchase shares is subject to the conditions set forth in his or her securities purchase agreement as well, which may also be waived.
We currently anticipate that the sale of the shares will be completed on or about June 14, 2017. We estimate the total offering expenses of this offering that will be payable by us, excluding the placement agent fees, will be approximately $350,000 which includes legal and printing costs, various other fees and reimbursement of the placements agents’ expenses. At the closing, The Depository Trust Company will credit the shares of common stock to the respective accounts of the investors.
Certain of our directors and management team have agreed to purchase an aggregate of approximately $525,000 of our shares of common stock and warrants in this offering and the concurrent private placement.
Indemnification
We have agreed to indemnify the placement agents against liabilities under the Securities Act. We have also agreed to contribute to payments the placement agents may be required to make in respect of such liabilities.
Electronic Distribution
This prospectus supplement and the accompanying prospectus may be made available in electronic format on websites or through other online services maintained by the placement agents, or by an affiliate thereof. Other than this prospectus supplement and the accompanying prospectus in electronic format, the information on the placement agents’ websites and any information contained in any other website maintained by the placement agents is not part of this prospectus supplement and the accompanying prospectus or the registration statement of which this prospectus supplement and the accompanying prospectus form a part, has not been approved and/or endorsed by us or the placement agents, and should not be relied upon by investors.
The foregoing does not purport to be a complete statement of the terms and conditions of the placement agent agreement and securities purchase agreement. A copy of the placement agent agreement and the form of securities purchase agreement with the investors are included as exhibits to our current report on Form 8-K that will be filed with the SEC and incorporated by reference into the Registration Statement of which this prospectus supplement forms a part. See “Where Can You Find More Information” on page S-100.
Regulation M Restrictions
The placement agents may be deemed to be underwriters within the meaning of Section 2(a)(11) of the Securities Act, and any commissions received by them and any profit realized on the resale of the shares sold by them while acting as principals might be deemed to be underwriting discounts or commissions under the Securities Act. As an underwriter, each placement agent would be required to comply with the requirements of the Securities Act and the Exchange Act including, without limitation, Rule 415(a)(4) under the Securities Act and Rule 10b-5 and Regulation M under the Exchange Act. These rules and regulations may limit the timing of purchases and sales of shares by the placement agents acting as principals. Under these rules and regulations, the placement agents:
| ● | must not engage in any stabilization activity in connection with our securities; and |
| ● | must not bid for or purchase any of our securities or attempt to induce any person to purchase any of our securities, other than as permitted under the Exchange Act, until they have completed their participation in the distribution. |
Other
From time to time, the placement agents and their respective affiliates may in the future provide various investment banking, financial advisory and other services to us and our affiliates for which services they may receive customary fees. In the course of their business, the placement agents and their respective affiliates may actively trade our securities or loans for their own account or for the accounts of customers, and, accordingly, the placement agents and their respective affiliates may at any time hold long or short positions in such securities or loans. Except for services provided in connection with this offering and the Merger, the placement agents have not provided any investment banking or other financial services during the 180-day period preceding the date of this prospectus supplement and we do not expect to retain the placement agents to perform any investment banking or other financial services for at least 90 days after the date of this prospectus supplement.
Notice to Non-U.S. Investors
Investors are advised to contact their legal, financial or tax advisers to obtain an independent assessment of the financial and tax consequences of an investment in shares of common stock.
Belgium
The offering is exclusively conducted under applicable private placement exemptions under Belgian securities laws and therefore it has not been and will not be notified to, and this document or any other offering material relating to the shares of common stock has not been and will not be approved by, the Belgian Banking, Finance and Insurance Commission (“Commission bancaire, financière et des assurances/Commissie voor het Bank, Financie en Assurantiewezen”). Any representation to the contrary is unlawful.
Each placement agent has undertaken not to offer sell, resell, transfer or deliver directly or indirectly, any shares of common stock, or to take any steps relating/ancillary thereto, and not to distribute or publish this document or any other material relating to the shares of common stock or to the offering in a manner which would be construed as: (a) a public offering under the Belgian Royal Decree of 7 July 1999 on the public character of financial transactions; or (b) an offering of securities to the public under Directive 2003/71/EC which triggers an obligation to publish a prospectus in Belgium. Any action contrary to these restrictions will cause the recipient and us to be in violation of the Belgian securities laws.
France
Neither this prospectus supplement nor any other offering material relating to the shares of common stock has been submitted to the clearance procedures of the Autorité des marchés financiers in France. The shares of common stock have not been offered or sold and will not be offered or sold, directly or indirectly, to the public in France. Neither this prospectus supplement nor any other offering material relating to the shares of common stock has been or will be: (a) released, issued, distributed or caused to be released, issued or distributed to the public in France; or (b) used in connection with any offer for subscription or sale of the shares of common stock to the public in France. Such offers, sales and distributions will be made in France only: (i) to qualified investors (investisseurs qualifiés) and/or to a restricted circle of investors (cercle restreint d’investisseurs), in each case investing for their own account, all as defined in and in accordance with Articles L.411-2, D.411-1, D.411-2, D.734-1, D.744-1, D.754-1 and D.764-1 of the French Code monétaire et financier; (ii) to investment services providers authorised to engage in portfolio management on behalf of third parties; or (iii) in a transaction that, in accordance with article L.411-2-II-1°-or-2°-or 3° of the French Code monétaire et financier and article 211-2 of the General Regulations (Règlement Général) of the Autorité des marchés financiers, does not constitute a public offer (appel public à l’épargne). Such shares of common stock may be resold only in compliance with Articles L.411-1, L.411-2, L.412-1 and L.621-8 through L.621-8-3 of the French Code monétaire et financier.
United Kingdom / Germany / Norway / The Netherlands
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (each, a “Relevant Member State”) an offer to the public of any shares of common stock which are the subject of the offering contemplated by this prospectus supplement may not be made in that Relevant Member State other than the offers contemplated in this prospectus supplement in name(s) of Member State(s) where a prospectus will be approved or passported for the purposes of a non-exempt offer once this prospectus supplement has been approved by the competent authority in such Member State and published and passported in accordance with the Prospectus Directive as implemented in the name(s) of relevant Member State(s) except that an offer to the public in that Relevant Member State of any shares of common stock may be made at any time under the following exemptions under the Prospectus Directive, if they have been implemented in that Relevant Member State:
(a) to legal entities which are authorised or regulated to operate in the financial markets or, if not so authorised or regulated, whose corporate purpose is solely to invest in securities;
(b) to any legal entity which has two or more of (1) an average of at least 250 employees during the last financial year; (2) a total balance sheet of more than €43,000,000 and (3) an annual net turnover of more than €50,000,000, as shown in its last annual or consolidated accounts;
(c) by the representative to fewer than 100 natural or legal persons (other than qualified investors as defined in the Prospectus Directive); or
(d) in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of shares of common stock shall result in a requirement for the publication by the Company or any placement agent of a prospectus pursuant to Article 3 of the Prospectus Directive.
For the purposes of this provision, the expression an “offer to the public” in relation to any shares of common stock in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any shares of common stock to be offered so as to enable an investor to decide to purchase any shares of common stock, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State and the expression “Prospectus Directive” means Directive 2003/71/EC and includes any relevant implementing measure in each Relevant Member State.
This prospectus supplement and any other material in relation to the shares of common stock is only being distributed to, and is only directed at, persons in the United Kingdom that are qualified investors within the meaning of Article 2(1)(e) of the Prospective Directive (“qualified investors”) that also (i) have professional experience in matters relating to investments falling within Article 19(5) of the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005, as amended, or the Order, (ii) fall within Article 49(2)(a) to (d) of the Order or (iii) may otherwise lawfully have such material communicated to them (all such persons together being referred to as “relevant persons”). The shares of common stock are only available to, and any invitation, offer or agreement to purchase or otherwise acquire such shares of common stock will be engaged in only with, relevant persons. This prospectus supplement and its contents are confidential and should not be distributed, published or reproduced (in whole or in part) or disclosed by recipients to any other person in the United Kingdom. Any person in the United Kingdom that is not a relevant person should not act or rely on this prospectus supplement or any of its contents.
Israel
In the State of Israel, the shares of common stock offered hereby may not be offered to any person or entity other than the following:
(a) a fund for joint investments in trust (i.e., mutual fund), as such term is defined in the Law for Joint Investments in Trust, 5754-1994, or a management company of such a fund;
(b) a provident fund as defined in Section 47(a)(2) of the Income Tax Ordinance of the State of Israel, or a management company of such a fund;
(c) an insurer, as defined in the Law for Oversight of Insurance Transactions, 5741-1981;
(d) a banking entity or satellite entity, as such terms are defined in the Banking Law (Licensing), 5741-1981, other than a joint services company, acting for their own account or for the account of investors of the type listed in Section 15A(b) of the Securities Law 1968;
(e) a company that is licensed as a portfolio manager, as such term is defined in Section 8(b) of the Law for the Regulation of Investment Advisors and Portfolio Managers, 5755-1995, acting on its own account or for the account of investors of the type listed in Section 15A(b) of the Securities Law 1968;
(f) a company that is licensed as an investment advisor, as such term is defined in Section 7(c) of the Law for the Regulation of Investment Advisors and Portfolio Managers, 5755-1995, acting on its own account;
(g) a company that is a member of the Tel Aviv Stock Exchange, acting on its own account or for the account of investors of the type listed in Section 15A(b) of the Securities Law 1968;
(h) an underwriter fulfilling the conditions of Section 56(c) of the Securities Law, 5728-1968;
(i) a venture capital fund (defined as an entity primarily involved in investments in companies which, at the time of investment, (i) are primarily engaged in research and development or manufacture of new technological products or processes and (ii) involve above-average risk);
(j) an entity primarily engaged in capital markets activities in which all of the equity owners meet one or more of the above criteria; and
(k) an entity, other than an entity formed for the purpose of purchasing shares of common stock in this offering, in which the shareholders equity (including pursuant to foreign accounting rules, international accounting regulations and U.S. generally accepted accounting rules, as defined in the Securities Law Regulations (Preparation of Annual Financial Statements), 1993) is in excess of NIS 250 million.
Any offeree of the shares of common stock offered hereby in the State of Israel shall be required to submit written confirmation that it falls within the scope of one of the above criteria. This prospectus supplement will not be distributed or directed to investors in the State of Israel who do not fall within one of the above criteria.
Italy
The offering of the shares of common stock offered hereby in Italy has not been registered with the Commissione Nazionale per la Società e la Borsa (“CONSOB”) pursuant to Italian securities legislation and, accordingly, the shares of common stock offered hereby cannot be offered, sold or delivered in the Republic of Italy (“Italy”) nor may any copy of this prospectus supplement or any other document relating to the shares of common stock offered hereby be distributed in Italy other than to professional investors (operatori qualificati) as defined in Article 31, second paragraph, of CONSOB Regulation No. 11522 of 1 July, 1998 as subsequently amended. Any offer, sale or delivery of the shares of common stock offered hereby or distribution of copies of this prospectus supplement or any other document relating to the shares of common stock offered hereby in Italy must be made:
(a) by an investment firm, bank or intermediary permitted to conduct such activities in Italy in accordance with Legislative Decree No. 58 of 24 February 1998 and Legislative Decree No. 385 of 1 September 1993 (the “Banking Act”);
(b) in compliance with Article 129 of the Banking Act and the implementing guidelines of the Bank of Italy; and
(c) in compliance with any other applicable laws and regulations and other possible requirements or limitations which may be imposed by Italian authorities.
Sweden
This prospectus supplement has not been nor will it be registered with or approved by Finansinspektionen (the Swedish Financial Supervisory Authority). Accordingly, this prospectus supplement may not be made available, nor may the shares of common stock offered hereunder be marketed and offered for sale in Sweden, other than under circumstances which are deemed not to require a prospectus under the Financial Instruments Trading Act (1991: 980).
Switzerland
The shares of common stock being offered pursuant to this prospectus supplement will not be offered, directly or indirectly, to the public in Switzerland and this prospectus supplement does not constitute a public offering prospectus as that term is understood pursuant to art. 652a or art. 1156 of the Swiss Federal Code of Obligations. We have not applied for a listing of the shares of common stock being offered pursuant to this prospectus supplement on the SWX Swiss Exchange or on any other regulated securities market, and consequently, the information presented in this prospectus supplement does not necessarily comply with the information standards set out in the relevant listing rules. The shares of common stock being offered pursuant to this prospectus supplement have not been registered with the Swiss Federal Banking Commission as foreign investment funds, and the investor protection afforded to acquirers of investment fund certificates does not extend to acquirers of shares of common stock.
Canada
Notice to Canadian Residents
This document constitutes an “exempt offering document” as defined in and for the purposes of applicable Canadian securities laws. No prospectus has been filed with any securities commission or similar regulatory authority in Canada in connection with the offer and sale of the securities described herein (the “Securities”). No securities commission or similar regulatory authority in Canada has reviewed or in any way passed upon this document or on the merits of the Securities and any representation to the contrary is an offence.
Canadian investors are advised that this document has been prepared in reliance on section 3A.3 of National Instrument 33-105Underwriting Conflicts (“NI 33-105”).Pursuant to section 3A.3 of NI 33-105, this document is exempt from the requirement to provide investors with certain conflicts of interest disclosure pertaining to“connected issuer” and/or“related issuer” relationships as would otherwise be required pursuant to subsection 2.1(1) of NI 33-105.
Resale Restrictions
The offer and sale of the Securities in Canada is being made on a private placement basis only and is exempt from the requirement to prepare and file a prospectus under applicable Canadian securities laws. Any resale of Securities acquired by a Canadian investor in this offering must be made in accordance with applicable Canadian securities laws, which may vary depending on the relevant jurisdiction, and which may require resales to be made in accordance with Canadian prospectus requirements, a statutory exemption from the prospectus requirements, in a transaction exempt from the prospectus requirements or otherwise under a discretionary exemption from the prospectus requirements granted by the applicable local Canadian securities regulatory authority. These resale restrictions may under certain circumstances apply to resales of the Securities outside of Canada.
Representations of Purchasers
Each Canadian investor who purchases the Securities will be deemed to have represented to the issuer and to each dealer from whom a purchase confirmation is received, as applicable, that the investor (i) is purchasing as principal, or is deemed to be purchasing as principal in accordance with applicable Canadian securities laws, for investment only and not with a view to resale or redistribution; (ii) is an “accredited investor” as such term is defined in section 1.1 of National Instrument 45-106Prospectus Exemptions (“NI 45-106”) or, in Ontario, as such term is defined in section 73.3(1) of the Securities Act (Ontario); and (iii) is a “permitted client” as such term is defined in section 1.1 of National Instrument 31-103Registration Requirements, Exemptions and Ongoing Registrant Obligations.
Taxation and Eligibility for Investment
Any discussion of taxation and related matters contained in this document does not purport to be a comprehensive description of all of the tax considerations that may be relevant to a Canadian investor when deciding to purchase the Securities and, in particular, does not address any Canadian tax considerations. No representation or warranty is hereby made as to the tax consequences to a resident, or deemed resident, of Canada of an investment in the Securities or with respect to the eligibility of the Securities for investment by such investor under relevant Canadian federal and provincial legislation and regulations.
Rights of Action for Damages or Rescission
Securities legislation in certain of the Canadian jurisdictions provides certain purchasers of securities pursuant to an offering memorandum, including where the distribution involves an “eligible foreign security” as such term is defined in Ontario Securities Commission Rule 45-501Ontario Prospectus and Registration Exemptions and in Multilateral Instrument 45-107 Listing Representation and Statutory Rights of Action Disclosure Exemptions, as applicable, with a remedy for damages or rescission, or both, in addition to any other rights they may have at law, where the offering memorandum, or other offering document that constitutes an offering memorandum, and any amendment thereto, contains a “misrepresentation” as defined under applicable Canadian securities laws. These remedies, or notice with respect to these remedies, must be exercised or delivered, as the case may be, by the purchaser within the time limits prescribed under, and are subject to limitations and defences under, applicable Canadian securities legislation. In addition, these remedies are in addition to and without derogation from any other right or remedy available at law to the investor.
Language of Documents
Upon receipt of this document, each Canadian investor hereby confirms that it has expressly requested that all documents evidencing or relating in any way to the sale of the Securities described herein (including for greater certainty any purchase confirmation or any notice) be drawn up in the English language only.Par la réception de ce document, chaque investisseur canadien confirme par les présentes qu’il a expressément exigé que tous les documents faisant foi ou se rapportant de quelque manière que ce soit à la vente des valeurs mobilières décrites aux présentes (incluant, pour plus de certitude, toute confirmation d’achat ou tout avis) soient rédigés en anglais seulement.
LEGALMATTERS
Jackson Walker L.L.P., of Dallas, Texas, will pass upon the validity of the shares of common stock offered hereby. The placement agents are being represented by Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C, of New York, New York, in connection with the offering.
The consolidated financial statements of PLx Pharma Inc. (f/k/a Dipexium Pharmaceuticals, Inc.) at December 31, 2016 and 2015, and for each of the years in the two-year period ended December 31, 2016, incorporated by reference in this prospectus supplement have been audited by CohnReznick LLP, independent registered public accounting firm, as set forth in their report incorporated by reference herein, and are included in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
Old PLx’s historical consolidated financial statements as of and for the years ended December 31, 2016 and 2015 and for the three-month periods ended March 31, 2017 and 2016, as filed with our Current Report on Form 8-K dated May 5, 2017 and incorporated by reference in this prospectus supplement, have been audited by GBH CPAs PC, independent registered public accounting firm, as set forth in their report incorporated by reference herein, and are included in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
WHERECAN YOU FIND MORE INFORMATION
This prospectus supplement and the accompanying prospectus are part of the registration statement on Form S-3 we filed with the SEC under the Securities Act and do not contain all the information set forth in the registration statement. Whenever a reference is made in this prospectus supplement or the accompanying prospectus to any of our contracts, agreements or other documents, the reference may not be complete and you should refer to the exhibits that are a part of the registration statement or the exhibits to the reports or other documents incorporated by reference in this prospectus supplement and the accompanying prospectus for a copy of such contract, agreement or other document. Because we are subject to the information and reporting requirements of the Exchange Act, we file annual, quarterly and current reports, proxy statements and other information with the SEC. Our SEC filings are available to the public over the Internet at the SEC’s website at http://www.sec.gov. You may also read and copy any document we file at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for further information on the operation of the Public Reference Room.
INCORPORATIONBY REFERENCE
The SEC allows us to “incorporate by reference” information from other documents that we file with it, which means that we can disclose important information to you by referring you to those documents. The information incorporated by reference is considered to be part of this prospectus supplement and the accompanying prospectus. Information contained in this prospectus supplement and the accompanying prospectus and information that we file with the SEC in the future and incorporate by reference in this prospectus supplement and the accompanying prospectus will automatically update and supersede this information. We incorporate by reference the documents listed below and any future filings (other than Current Reports on Form 8-K furnished under Item 2.02 or Item 7.01 and exhibits filed on such form that are related to such items) we make with the SEC under Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act after the date of this prospectus supplement and before the sale of all the securities covered by this prospectus supplement:
● | our Annual Report on Form 10-K for the fiscal year ended December 31, 2016, filed on January 20, 2017; |
● | our Quarterly Report on Form 10-Q for the three months ended March 31, 2017, filed on April 17, 2017; |
● | our Current Reports on Form 8-K filed on April 20, 2017, May 2, 2017, and May 5, 2017; |
● | the description of our common stock contained in our Registration Statement on Form 8-A, filed with the SEC on March 12, 2014, including any amendments or reports filed for the purpose of updating that description. |
Pursuant to General Instruction B of Form 8-K, any information submitted under Item 2.02, results of Operations and Financial Condition, or Item 7.01, Regulation FD Disclosure, of Form 8-K is not deemed to be “filed” for the purpose of Section 18 of the Exchange Act, and we are not subject to the liabilities of Section 18 with respect to information submitted under Item 2.02 or Item 7.01 of Form 8-K. We are not incorporating by reference any information submitted under Item 2.02 or Item 7.01 of Form 8-K into any filing under the Securities Act or the Exchange Act or into this prospectus supplement, unless otherwise indicated on such Form 8-K.
You may request a copy of these filings, at no cost, by writing or calling us at the following address or telephone number:
8285 El Rio Street, Suite 130
Houston, Texas, 77054
(713) 842-1249
PROSPECTUS
DIPEXIUM PHARMACEUTICALS, INC.
$100,000,000.00
COMMON STOCK
DEBT SECURITIES
WARRANTS
RIGHTS
PURCHASE CONTRACTS
UNITS
This prospectus will allow us to issue, from time to time at prices and on terms to be determined at or prior to the time of the offering, up to $100,000,000.00 of any combination of the securities described in this prospectus, either individually or in units. We may also offer common stock upon conversion of or exchange for the debt securities; common stock or debt securities upon the exercise of warrants, rights or performance of purchase contracts; or any combination of these securities upon the performance of purchase contracts.
This prospectus describes the general terms of these securities and the general manner in which these securities will be offered. We will provide you with the specific terms of any offering in one or more supplements to this prospectus. The prospectus supplements will also describe the specific manner in which these securities will be offered and may also supplement, update or amend information contained in this document. You should read this prospectus and any prospectus supplement, as well as any documents incorporated by reference into this prospectus or any prospectus supplement, carefully before you invest.
Our securities may be sold directly by us to you, through agents designated from time to time or to or through underwriters or dealers. For additional information on the methods of sale, you should refer to the section entitled “Plan of Distribution” in this prospectus and in the applicable prospectus supplement. If any underwriters or agents are involved in the sale of our securities with respect to which this prospectus is being delivered, the names of such underwriters or agents and any applicable fees, commissions or discounts and over-allotment options will be set forth in a prospectus supplement. The price to the public of such securities and the net proceeds that we expect to receive from such sale will also be set forth in a prospectus supplement.
Our common stock is listed on the NASDAQ Capital Market, under the symbol “DPRX.” On June 8, 2015, the last reported sale price of our common stock on NASDAQ Capital Market was $14.99 per share.
Investing in our securities involves a high degree of risk. Before deciding whether to invest in our securities, you should consider carefully the risks that we have described on page 6 of this prospectus under the caption “Risk Factors.” We may include specific risk factors in supplements to this prospectus under the caption “Risk Factors.” This prospectus may not be used to sell our securities unless accompanied by a prospectus supplement.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this prospectus is June 19, 2015.
TABLE OF CONTENTS
| Page |
|
|
1 | |
|
|
2 | |
|
|
6 | |
|
|
7 | |
|
|
8 | |
|
|
9 | |
|
|
10 | |
|
|
12 | |
|
|
15 | |
|
|
18 | |
|
|
20 | |
|
|
22 | |
|
|
23 | |
|
|
25 | |
|
|
25 | |
|
|
25 | |
|
|
25 |
ABOUTTHIS PROSPECTUS
This prospectus is part of a registration statement that we filed with the Securities and Exchange Commission, or SEC, utilizing a “shelf” registration process. Under this shelf registration process, we may offer shares of our common stock, various series of debt securities and/or warrants, rights or purchase contracts to purchase any of such securities, either individually or in units, in one or more offerings, with a total value of up to $100,000,000.00. This prospectus provides you with a general description of the securities we may offer. Each time we offer a type or series of securities under this prospectus, we will provide a prospectus supplement that will contain specific information about the terms of that offering.
This prospectus does not contain all of the information included in the registration statement. For a more complete understanding of the offering of the securities, you should refer to the registration statement, including its exhibits. The prospectus supplement may also add, update or change information contained or incorporated by reference in this prospectus. However, no prospectus supplement will offer a security that is not registered and described in this prospectus at the time of its effectiveness. This prospectus, together with the applicable prospectus supplements and the documents incorporated by reference into this prospectus, includes all material information relating to the offering of securities under this prospectus. You should carefully read this prospectus, the applicable prospectus supplement, the information and documents incorporated herein by reference and the additional information under the heading “Where You Can Find More Information” before making an investment decision.
You should rely only on the information we have provided or incorporated by reference in this prospectus or any prospectus supplement. We have not authorized anyone to provide you with information different from that contained or incorporated by reference in this prospectus. No dealer, salesperson or other person is authorized to give any information or to represent anything not contained or incorporated by reference in this prospectus. You must not rely on any unauthorized information or representation. This prospectus is an offer to sell only the securities offered hereby, but only under circumstances and in jurisdictions where it is lawful to do so. You should assume that the information in this prospectus or any prospectus supplement is accurate only as of the date on the front of the document and that any information we have incorporated herein by reference is accurate only as of the date of the document incorporated by reference, regardless of the time of delivery of this prospectus or any sale of a security.
We further note that the representations, warranties and covenants made by us in any agreement that is filed as an exhibit to any document that is incorporated by reference in the accompanying prospectus were made solely for the benefit of the parties to such agreement, including, in some cases, for the purpose of allocating risk among the parties to such agreements, and should not be deemed to be a representation, warranty or covenant to you. Moreover, such representations, warranties or covenants were accurate only as of the date when made. Accordingly, such representations, warranties and covenants should not be relied on as accurately representing the current state of our affairs.
This prospectus may not be used to consummate sales of our securities, unless it is accompanied by a prospectus supplement. To the extent there are inconsistencies between any prospectus supplement, this prospectus and any documents incorporated by reference, the document with the most recent date will control.
We were organized originally as a limited liability company under the laws of the State of Delaware in January 2010. On March 12, 2014, we converted Dipexium Pharmaceuticals, LLC from a Delaware limited liability company to a Delaware corporation. See “Prospectus Summary — Corporate Conversion” for more information.
Unless the context otherwise requires, “Dipexium,” “the Company,” “we,” “us,” “our” and similar terms refer to Dipexium Pharmaceuticals, Inc.
PROSPECTUSSUMMARY
The following is a summary of what we believe to be the most important aspects of our business and the offering of our securities under this prospectus. We urge you to read this entire prospectus, including the more detailed consolidated financial statements, notes to the consolidated financial statements and other information incorporated by reference from our other filings with the SEC or included in any applicable prospectus supplement. Investing in our securities involves risks. Therefore, carefully consider the risk factors set forth in any prospectus supplements and in our most recent annual and quarterly filings with the SEC, as well as other information in this prospectus and any prospectus supplements and the documents incorporated by reference herein or therein, before purchasing our securities. Each of the risk factors could adversely affect our business, operating results and financial condition, as well as adversely affect the value of an investment in our securities.
Overview
We are a late stage pharmaceutical company focused on the development and commercialization of Locilex® (pexiganan cream 0.8%), a novel, first-in-class, broad spectrum, topical antibiotic. Locilex® is a chemically synthesized, 22-amino acid peptide isolated from the skin of the African Clawed Frog. Its novel mechanism of action kills microbial targets by disrupting the bacterial cell membrane; a process known as cell membrane permeability. Locilex® is initially being targeted for the treatment of mild infections of diabetic foot ulcers (or Mild DFI). In 2011, the market for diabetic foot infection therapeutics worldwide was approximately $1.46 billion. Our primary objective is to establish Locilex® as the standard of care for the treatment of patients with Mild DFI. Thereafter, our growth strategy includes potentially expanding the indications for Locilex® to include moderate infections of diabetic foot ulcers (or Moderate DFI) and certain other mild or moderate skin and skin structure infections in superficial wounds.
We believe that we have a clear clinical and regulatory pathway with the potential for near term United States Food and Drug Administration (or FDA) approval of Locilex®. We have reached agreement with the FDA through a special protocol assessment (or SPA) for our Phase 3 program. We intend to complete two pivotal Phase 3, double blind, placebo-controlled superiority studies. We began enrollment in these studies in the third quarter of 2014, and we anticipate completing enrollment in the second half of 2015. We also conducted two separate Phase 1 skin irritation and skin sensitization studies and we reported data on the first of these two Phase 1 studies in Q3 2014 and reported data on the second Phase 1 study in Q1 2015. If the data from our Phase 3 studies are sufficient to meet the primary endpoint, we expect to submit our new drug application (or NDA) for Locilex® to the FDA soon thereafter. We expect to receive a response from the FDA within six months of our NDA submission.
According to the Infectious Disease Society of America (or IDSA), diabetic foot infections (or DFI) may be classified by their clinical severity as mild, moderate, or severe. 47% of DFI patients first present to a treating physician in the mild stage of infection, 34% of DFI patients first present at the moderate stage and 18% of DFI patients first present at the severe stage. At the mild stage, patients can typically be treated on an outpatient basis and amputation risk is minimal (2% to 3% in Mild DFI). When not managed effectively, the potential for Mild DFI to progress to a limb- or life-threatening infection increases dramatically. Published research suggests amputation rates increase in Moderate DFI and severe infections of diabetic foot ulcers (or Severe DFI) to approximately 45% and 75%, respectively. Similarly, the hospitalization rate for Mild DFI patients is approximately 10%, increasing in Moderate DFI and Severe DFI to approximately 55% and 85%, respectively. Thus, DFIs are a major cause of patient morbidity, a substantial burden to the healthcare system, and a source of high financial costs.
Systemic antibiotics currently prescribe off-label to treat Mild DFI generate resistant pathogens which create infections that are more difficult to treat. Such antibiotics are also associated with toxic side effects in patients who typically have some degree of compromised liver and kidney function. We believe that a topical preparation like Locilex®, which is locally administered on the open wound and skin, offers significant advantages over systemic treatments. As a topical antibiotic, Locilex® affects only the area to which it is applied, and unlike most systemic antibiotics, it does not spread to the entire body through the bloodstream, which is host to a number of different bacteria other than bacteria causing the skin infection. Topical antibiotics are also delivered to the infected area at a higher concentration than those antibiotics that are delivered systemically. As a result, we believe that topical antibiotics are less likely to develop bacterial resistance, both because the higher antibiotic concentration more effectively kills the infection-causing bacteria before they develop resistance, and because other bacteria in thebloodstream are not exposed to the Locilex® topical cream. Currently, there are no products, including no antibiotics, specifically approved by the FDA for the treatment of Mild DFI, nor are there any topical antibiotics currently approved for any severity of DFI. As such, we believe that Locilex® has the potential to be the first topical antibiotic approved for the treatment of DFI, as well as the first product of any kind to be labeled specifically for the treatment of Mild DFI.
We believe that the key attributes of Locilex® are: (i) it has not generated resistant bacteria systemically; (ii) it has not generated cross resistance with other antibiotics; (iii) it has demonstrated activity against a broad spectrum of pathogens, including difficult to treat gram negative, and anaerobic bacteria; (iv) it has not been systemically absorbed; (v) it has not caused any significant safety or tolerability issues in over 500 patients treated; and (vi) it has demonstrated significant success treating multi-drug resistant bacteria in several laboratory tests and clinical trials performed to date. These attributes lead us to believe that Locilex® has the potential to be positioned as the standard of care to treat patients with Mild DFI. In addition, data generated to date support the potential use of Locilex® to treat a broad array of mild or moderate skin and skin structure infections in superficial wounds.
As reported in published research, Locilex® has previously demonstrated statistical non-inferiority to a systemic antibiotic in a large-scale, randomized, active-controlled double blind, multi-center clinical study in patients with Mild or Moderate DFI. We have conducted microbiology studies that highlight the sensitivity of resistant bacteria, including methicillin-resistant staphylococcus aureus (or MRSA), vancomycin-resistant enterococcus (or VRE), extended-spectrum b-lactamase (or ESBL) and multi-drug resistant (or MDR) bacteria, to pexiganan, the active pharmaceutical ingredient (or API) in Locilex®. Due to the increased global prevalence of resistant bacteria in all types of skin infections, Locilex® may provide an important therapeutic advance.
We have contracted with third party vendors with respect to all key elements of our clinical and regulatory program, including vendors to: (i) conduct the Phase 3 and Phase 1 clinical trials for Locilex®; (ii) manufacture the API; (iii) formulate the finished product; and (iv) label and package the product. We believe these key relationships will help drive our clinical and regulatory program for Locilex® in a timely and efficient manner.
Locilex® was originally sponsored by Magainin Pharmaceuticals, Inc. (or Magainin), which engaged in the FDA review process during 1998 and 1999, ultimately receiving a non-approvable letter based upon two manufacturing issues. We acquired the worldwide rights to pexiganan, the API in Locilex®, from a third party in April 2010. These rights included the prior formulation and all of the clinical and preclinical data generated by Magainin in its FDA review process. This includes data from over 1,000 evaluable patients, including 835 in large-scale, randomized, active-controlled, double blind, multi-center clinical studies as compared to a systemic quinolone standard of care as more fully described below. We believe we have corrected the manufacturing problems encountered by the prior sponsor.
Our Strategy
Our primary objective is to establish Locilex® as the standard of care to treat patients with Mild DFI. The key elements of our strategy are as follows:
● Complete the Phase 3 program for Locilex®. As a result of our SPA for Locilex®, we believe the clinical pathway for Locilex® is clear. Working with our key third party vendors, we commenced our Phase 3 program in the third quarter of 2014 and we anticipate completing enrollment in the second half of 2015.
● Obtain FDA approval of Locilex® for Mild DFI. If our Phase 3 trials meet their primary endpoints, we plan to submit our NDA for Locilex®.
● Commercially launch Locilex® in the U.S. We plan to utilize a small specialty sales force to launch Locilex®, if it is approved, in the U.S., initially targeting podiatrists and potentially expanding to other specialty healthcare providers.
● Expand Locilex®’s FDA-approved uses. If we are able to obtain FDA approval in Mild DFI, we will consider obtaining additional FDA approvals that will enable us to expand the Locilex® label to include patients with Moderate DFI and certain other mild or moderate skin and skin structure infections in superficial wounds.
● Commercialize Locilex in Europe. We received formal clinical and regulatory guidance from the European Medicines Agency (EMA) in May 2015. Based upon the EMA guidance, we anticipate filing a marketing authorization application (or MAA) in the European Union under the centralized process in 2016 at or shortly after our intended submission of the new drug application (or NDA) to the FDA in the United States provided that we receive favorable efficacy, safety and microbiology data upon completion of our Phase 3 program in the United States.
We will rely on our strong management team, board of directors and scientific advisors to execute our strategy. The individuals on our management team, board of directors and scientific advisors will contribute their significant business development, finance, and regulatory experience to the development and commercialization of Locilex®.
Corporate Conversion
We were organized originally as a limited liability company under the laws of the State of Delaware in January 2010. On March 12, 2014, we converted Dipexium Pharmaceuticals, LLC from a Delaware limited liability company to a Delaware corporation. As a result of the corporate conversion:
● The Class A Membership Interests of Dipexium Pharmaceuticals, LLC became shares of common stock of Dipexium Pharmaceuticals, Inc. pursuant to a conversion ratio of seven shares of common stock of Dipexium Pharmaceuticals, Inc. for each Class A membership interest of Dipexium Pharmaceuticals, LLC previously held. Accordingly, 767,911 Class A Membership Interests of Dipexium Pharmaceuticals, LLC issued and outstanding immediately prior to the corporate conversion were converted automatically into 5,375,377 shares of Dipexium Pharmaceuticals, Inc.
● All of the outstanding warrants to purchase Class A Membership Interests of Dipexium Pharmaceuticals, LLC became warrants to purchase shares of common stock of Dipexium Pharmaceuticals, Inc. in a ratio of seven shares of common stock of Dipexium Pharmaceuticals, Inc. for each Class A membership interest of Dipexium Pharmaceuticals, LLC underlying such warrants, with the effect that warrants to purchase 4,900 Class A Membership Interests of Dipexium Pharmaceuticals, LLC outstanding immediately prior to the corporate conversion automatically converted into warrants to purchase 34,300 shares of Dipexium Pharmaceuticals, Inc. upon consummation of the corporate conversion.
● The exercise price of all of the outstanding warrants was adjusted in the same ratio as the seven-for-one conversion ratio noted above such that all of our outstanding warrants to purchase Class A Membership Interests of Dipexium Pharmaceuticals, LLC which were exercisable at $60 per Class A membership interest were automatically adjusted such that the new exercise price for the outstanding warrants upon consummating the corporate conversion was $8.57 per share, subject to certain adjustments noted in each of the warrants.
In connection with the corporate conversion, Dipexium Pharmaceuticals, Inc. continued to hold all property of Dipexium Pharmaceuticals, LLC and assumed all of the debts and obligations of Dipexium Pharmaceuticals, LLC. Dipexium Pharmaceuticals, Inc. is governed by a Certificate of Incorporation filed with the Delaware Secretary of State and Bylaws. On the effective date of the corporate conversion, the members of the board of directors of Dipexium Pharmaceuticals, LLC became the members of the board of directors of Dipexium Pharmaceuticals, Inc. and the officers of Dipexium Pharmaceuticals, LLC became the officers of Dipexium Pharmaceuticals, Inc. The purpose of the corporate conversion was to reorganize our corporate structure so that our company would continue as a corporation rather than a limited liability company, and so that our existing investors would own our common stock rather than equity interests in a limited liability company. In order to consummate the corporate conversion, a certificate of conversion was filed with the Secretary of State of the State of Delaware on March 12, 2014.
Corporate Information
Dipexium is headquartered in New York, New York, and we were organized originally as a limited liability company under the laws of the State of Delaware in January 2010. Our principal offices are located at 61 Broadway, Suite 1905, New York, NY 10006, and our telephone number is (212) 269-2834. Our principal website is www.dipexiumpharmaceuticals.com. The information on or that can be accessed through our website is not part of this prospectus. Our common stock is listed on the NASDAQ Capital Market and trades under the symbol “DPRX.”
Offerings Under This Prospectus
Under this prospectus, we may offer shares of our common stock, various series of debt securities and/or warrants, rights or purchase contracts to purchase any of such securities, either individually or in units, with a total value of up to $100,000,000.00, from time to time at prices and on terms to be determined by market conditions at the time of the offering. This prospectus provides you with a general description of the securities we may offer. Each time we offer a type or series of securities under this prospectus, we will provide a prospectus supplement that will describe the specific amounts, prices and other important terms of the securities, including, to the extent applicable:
The prospectus supplement also may add, update or change information contained in this prospectus or in documents we have incorporated by reference into this prospectus. However, no prospectus supplement will fundamentally change the terms that are set forth in this prospectus or offer a security that is not registered and described in this prospectus at the time of its effectiveness.
We may sell the securities directly to investors or to or through agents, underwriters or dealers. We, and our agents or underwriters, reserve the right to accept or reject all or part of any proposed purchase of securities. If we offer securities through agents or underwriters, we will include in the applicable prospectus supplement:
● the names of those agents or underwriters;
● applicable fees, discounts and commissions to be paid to them;
● details regarding over-allotment options, if any; and
● the net proceeds to us.
This prospectus may not be used to consummate a sale of any securities unless it is accompanied by a prospectus supplement.
RISKFACTORS
Please carefully consider the risk factors described in our periodic reports filed with the SEC, which are incorporated by reference in this prospectus. Before making an investment decision, you should carefully consider these risks as well as other information we include or incorporate by reference in this prospectus or include in any applicable prospectus supplement. Additional risks and uncertainties not presently known to us or that we deem currently immaterial may also impair our business operations or adversely affect our results of operations or financial condition.
CAUTIONARYNOTE REGARDING FORWARD-LOOKING STATEMENTS
We are including the following cautionary statement in this prospectus to make applicable and take advantage of the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 for any forward-looking statements made by, or on behalf of us. With the exception of historical matters, the matters discussed in this prospectus are forward-looking statements (as defined in Section 21E of the Exchange Act) that involve risks and uncertainties that could cause actual results to differ materially from projected results. Accordingly, investors should not place undue reliance on forward-looking statements as a prediction of actual results. The forward-looking statements may include projections and estimates concerning the timing and success of specific projects and our future production, revenues, income and capital spending. When we use the words “believe,” “intend,” “expect,” “may,” “should,” “anticipate,” “could,” “estimate,” “plan,” “predict,” “project,” or their negatives, or other similar expressions, the statements which include those words are usually forward-looking statements. When we describe strategy that involves risks or uncertainties, we are making forward-looking statements. The forward-looking statements in this prospectus speak only as of the date of this prospectus; we disclaim any obligation to update these statements unless required by securities law, and we caution you not to rely on them unduly. We have based these forward-looking statements on our current expectations and assumptions about future events. While our management considers these expectations and assumptions to be reasonable, they are inherently subject to significant business, economic, competitive, regulatory and other risks, contingencies and uncertainties, most of which are difficult to predict and many of which are beyond our control. These risks, contingencies and uncertainties relate to, among other matters, the following:
● our limited operating history and our history of significant operating losses;
● risks and uncertainties associated with our research and development activities, including our clinical trials;
● our dependence on Locilex® as our only product;
● our ability to raise capital when needed;
● the timing of and our ability to achieve U.S. or international regulatory approvals for Locilex® or any other product candidates we may develop;
● our dependence on others to conduct clinical research of, and to manufacture and market, Locilex®;
● the terms of future licensing arrangements, and whether we can enter into such arrangements at all;
● risks associated with the timing and receipt of licensing and milestone revenues, if any;
● our ability to gain market acceptance for Locilex® or any other product candidates we may develop;
● our ability to maintain or protect the validity of our patents and other intellectual property, including in connection with pending or future litigation against us;
● our ability to secure registration for our current and future patent applications;
● our ability to extend our licensed composition of matter patent No. 5,912,231 under the Hatch-Waxman Act with the cooperation of Scripps;
● our estimates of the size of the prospective markets in which we may offer Locilex®;
● our expectations regarding minimizing our development risk;
● our ability to establish new relationships and maintain current relationships;
● our ability to attract and retain key personnel;
● developments relating to our competitors or our industry; and
● acceptance of our business model by investors.
RATIOOF EARNINGS TO FIXED CHARGES
Any time debt securities are offered pursuant to this prospectus, we will provide a table setting forth our ratio of earnings to fixed charges on a historical basis in the applicable prospectus supplement, if required.
USEOF PROCEEDS
We cannot assure you that we will receive any proceeds in connection with securities which may be offered pursuant to this prospectus. Unless otherwise indicated in the applicable prospectus supplement, we intend to use any net proceeds from the sale of securities under this prospectus for our operations and for other general corporate purposes, including, but not limited to, our internal research and development programs and the development of new programs, general working capital and possible future acquisitions. We have not determined the amounts we plan to spend on any of the areas listed above or the timing of these expenditures. As a result, our management will have broad discretion to allocate the net proceeds, if any, we receive in connection with securities offered pursuant to this prospectus for any purpose. Pending application of the net proceeds as described above, we may initially invest the net proceeds in short-term, investment-grade, interest-bearing securities or apply them to the reduction of short-term indebtedness.
PLANOF DISTRIBUTION
General Plan of Distribution
We may offer securities under this prospectus from time to time pursuant to underwritten public offerings, negotiated transactions, block trades or a combination of these methods. We may sell the securities (1) through underwriters or dealers, (2) through agents or (3) directly to one or more purchasers, or through a combination of such methods. We may distribute the securities from time to time in one or more transactions at:
● a fixed price or prices, which may be changed from time to time;
● market prices prevailing at the time of sale;
● prices related to the prevailing market prices; or
● negotiated prices.
We may directly solicit offers to purchase the securities being offered by this prospectus. We may also designate agents to solicit offers to purchase the securities from time to time. We will name in a prospectus supplement any underwriter or agent involved in the offer or sale of the securities.
If we utilize a dealer in the sale of the securities being offered by this prospectus, we will sell the securities to the dealer, as principal. The dealer may then resell the securities to the public at varying prices to be determined by the dealer at the time of resale.
If we utilize an underwriter in the sale of the securities being offered by this prospectus, we will execute an underwriting agreement with the underwriter at the time of sale, and we will provide the name of any underwriter in the prospectus supplement which the underwriter will use to make re-sales of the securities to the public. In connection with the sale of the securities, we, or the purchasers of the securities for whom the underwriter may act as agent, may compensate the underwriter in the form of underwriting discounts or commissions. The underwriter may sell the securities to or through dealers, and the underwriter may compensate those dealers in the form of discounts, concessions or commissions.
With respect to underwritten public offerings, negotiated transactions and block trades, we will provide in the applicable prospectus supplement information regarding any compensation we pay to underwriters, dealers or agents in connection with the offering of the securities, and any discounts, concessions or commissions allowed by underwriters to participating dealers. Underwriters, dealers and agents participating in the distribution of the securities may be deemed to be underwriters within the meaning of the Securities Act of 1933, as amended, or the Securities Act, and any discounts and commissions received by them and any profit realized by them on resale of the securities may be deemed to be underwriting discounts and commissions. We may enter into agreements to indemnify underwriters, dealers and agents against civil liabilities, including liabilities under the Securities Act, or to contribute to payments they may be required to make in respect thereof.
If so indicated in the applicable prospectus supplement, we will authorize underwriters or other persons acting as our agents to solicit offers by certain institutions to purchase securities from us pursuant to delayed delivery contracts providing for payment and delivery on the date stated in the prospectus supplement. Each contract will be for an amount not less than, and the aggregate amount of securities sold pursuant to such contracts shall not be less nor more than, the respective amounts stated in the prospectus supplement. Institutions with whom the contracts, when authorized, may be made include commercial and savings banks, insurance companies, pension funds, investment companies, educational and charitable institutions and other institutions, but shall in all cases be subject to our approval. Delayed delivery contracts will not be subject to any conditions except that:
● the purchase by an institution of the securities covered under that contract shall not at the time of delivery be prohibited under the laws of the jurisdiction to which that institution is subject; and
● if the securities are also being sold to underwriters acting as principals for their own account, the underwriters shall have purchased such securities not sold for delayed delivery. The underwriters and other persons acting as our agents will not have any responsibility in respect of the validity or performance of delayed delivery contracts.
Shares of our common stock sold pursuant to the registration statement of which this prospectus is a part will be authorized for quotation and trading on the NASDAQ Capital Market. The applicable prospectus supplement will contain information, where applicable, as to any other listing, if any, on the NASDAQ Capital Market or any securities market or other securities exchange of the securities covered by the prospectus supplement. We can make no assurance as to the liquidity of or the existence of trading markets for any of the securities.
In order to facilitate the offering of the securities, certain persons participating in the offering may engage in transactions that stabilize, maintain or otherwise affect the price of the securities. This may include over-allotments or short sales of the securities, which involve the sale by persons participating in the offering of more securities than we sold to them. In these circumstances, these persons would cover such over-allotments or short positions by making purchases in the open market or by exercising their over-allotment option. In addition, these persons may stabilize or maintain the price of the securities by bidding for or purchasing the applicable security in the open market or by imposing penalty bids, whereby selling concessions allowed to dealers participating in the offering may be reclaimed if the securities sold by them are repurchased in connection with stabilization transactions. The effect of these transactions may be to stabilize or maintain the market price of the securities at a level above that which might otherwise prevail in the open market. These transactions may be discontinued at any time.
In compliance with the guidelines of the Financial Industry Regulatory Authority, Inc., or FINRA, the maximum consideration or discount to be received by any FINRA member or independent broker dealer may not exceed 8% of the aggregate amount of the securities offered pursuant to this prospectus and any applicable prospectus supplement.
The underwriters, dealers and agents may engage in other transactions with us, or perform other services for us, in the ordinary course of their business.
DESCRIPTIONOF CAPITAL STOCK
Our certificate of incorporation, which was adopted on March 12, 2014, at the time of our corporate conversion, authorizes the issuance of 30,000,000 shares of common stock, $0.001 par value per share. As of June 1, 2015, there were 8,566,047 shares of our common stock issued and outstanding (on an as converted basis after giving effect to the conversion of each Class A membership interest of Dipexium Pharmaceuticals, LLC into seven shares of common stock of Dipexium Pharmaceuticals, Inc.) and held of record by approximately 107 stockholders. The following is a summary of our capital stock following consummation of our corporate conversion.
Common Stock
Holders of our common stock are entitled to one vote for each share held on all matters submitted to a vote of stockholders and are not entitled to cumulative voting rights.
Holders of our common stock are entitled to receive ratably such dividends, if any, as may be declared by our board of directors out of funds legally available therefor, subject to any preferential distribution rights of third parties (of which there are presently none). Upon our liquidation, dissolution or winding up, the holders of our common stock are entitled to receive ratably our net assets available after the payment of all debts and other liabilities.
Holders of our common stock have no preemptive, subscription, redemption or conversion rights. There are no redemption or sinking fund provisions applicable to the common stock. All of the outstanding shares of our common stock are fully-paid and nonassessable. The rights, preferences and privileges of holders of our common stock are subject to, and may be adversely affected by, the rights of the holders of any indebtedness of our company.
Preferred Stock
Our certificate of incorporation does not permit our board of directors to designate and issue shares of preferred stock.
Warrants
As of June 1, 2015, warrants for the issuance of 28,000 shares of our common stock were outstanding, all of which are exercisable at an exercise price of $8.57 per share, after taking into effect the conversion of each Class A membership interest of Dipexium Pharmaceuticals, LLC into seven shares of common stock of Dipexium Pharmaceuticals, Inc. All of the warrants are exercisable through various dates expiring between July 23, 2015 and November 15, 2018. The exercise price and the number of warrant shares purchasable upon the exercise of the warrants are subject to adjustment upon the occurrence of certain events, including stock dividends, stock splits, combinations and reclassifications of our capital stock. The warrants also contain a “cashless exercise” provision.
Registration Rights
Each of our investors in our previous private placements are party to an Investor Rights Agreement affording them certain “piggy back” registration rights with respect to their Class A membership interests in our company and Class A membership interests underlying warrants held by such investors. This is comprised of 402,248 shares of our common stock on a post-conversion basis. We refer to all of such Class A membership interests as “registrable securities.” If at any time when there is not an effective registration statement covering all of the registrable securities, we determine to prepare and file with the SEC a registration statement relating to an offering for our own account or the account of others under the Securities Act of any of our equity securities (other than on Form S-4 or Form S-8), we are required to send to each holder of registrable securities written notice of such determination and, if within seven business days after receipt of such notice, any such holder shall so request in writing (which request shall specify the registrable securities intended to be disposed of by the holder), we will cause the registration under the Securities Act of all registrable securities which we have been so requested to register by the holder, except in connection with this registration statement, for which each holder has waived any and all rights to have its shares included; provided, however, that, in connection with any underwritten public offering of our securities, we maintain the right to not register all or any portion of the registrable securities if it is determined that such registration would materially and adversely affect such underwritten public offering.
Transfer Agent and Registrar
The transfer agent and registrar of our common stock is VStock Transfer LLC.
Delaware Law and Certain Charter and By-Law Provisions
Delaware Anti-Takeover Law. Since our corporate conversion on March 12, 2014, we are subject to Section 203 of the Delaware General Corporation Law (or DGCL). Section 203 generally prohibits a public Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless:
● prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder;
● upon consummation of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding specified shares; or
● at or subsequent to the date of the transaction, the business combination is approved by the board of directors and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 662/3% of the outstanding voting stock which is not owned by the interested stockholder.
Section 203 defines a business combination to include:
● any merger or consolidation involving the corporation and the interested stockholder;
● any sale, lease, exchange, mortgage, pledge, transfer or other disposition of 10% or more of the assets of the corporation to or with the interested stockholder;
● subject to exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder;
● subject to exceptions, any transaction involving the corporation that has the effect of increasing the proportionate share of the stock of any class or series of the corporation beneficially owned by the interested stockholder; or
● the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation.
In general, Section 203 defines an “interested stockholder” as any person that is:
● the owner of 15% or more of the outstanding voting stock of the corporation;
● an affiliate or associate of the corporation who was the owner of 15% or more of the outstanding voting stock of the corporation at any time within three years immediately prior to the relevant date; or
● the affiliates and associates of the above.
Under specific circumstances, Section 203 makes it more difficult for an “interested stockholder” to effect various business combinations with a corporation for a three-year period, although the stockholders may, by adopting an amendment to the corporation’s certificate of incorporation or bylaws, elect not to be governed by this section, effective 12 months after adoption.
Our certificate of incorporation and bylaws do not exclude us from the restrictions of Section 203. We anticipate that the provisions of Section 203 might encourage companies interested in acquiring us to negotiate in advance with our board of directors since the stockholder approval requirement would be avoided if a majority of the directors then in office approve either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder.
Certificate of Incorporation and Bylaws. Our certificate of incorporation and bylaws contain provisions that could have the effect of discouraging potential acquisition proposals or tender offers or delaying or preventing a change of control of our company. These provisions are as follows:
● they do not permit our stockholders to act by written consent. As a result, any action by our stockholders must be taken at a duly called annual or special meeting of stockholders;
● they provide that special meetings of stockholders may be called only by the board of directors, our Chairman of the board of directors, one of our executive officers, or at the request in writing by stockholders of record owning at least sixty-six and two thirds (662/3%) percent of the issued and outstanding voting shares of common stock; and
● they do not include a provision for cumulative voting in the election of directors. Under cumulative voting, a minority stockholder holding a sufficient number of shares may be able to ensure the election of one or more directors. The absence of cumulative voting may have the effect of limiting the ability of minority stockholders to effect changes in our board of directors.
Elimination of Monetary Liability for Officers and Directors
Our certificate of incorporation incorporates certain provisions permitted under the DGCL relating to the liability of directors. The provisions eliminate a director’s liability for monetary damages for a breach of fiduciary duty, including gross negligence, except in circumstances involving certain wrongful acts, such as the breach of director’s duty of loyalty or acts or omissions, which involve intentional misconduct or a knowing violation of law. These provisions do not eliminate a director’s duty of care. Moreover, these provisions do not apply to claims against a director for certain violations of law, including knowing violations of federal securities law. Our certificate of incorporation also contains provisions to indemnify the directors, officers, employees or other agents to the fullest extent permitted by the DGCL. We believe that these provisions will assist us in attracting and retaining qualified individual to serve as directors.
Indemnification of Officers and Directors
Our certificate of incorporation also contains provisions to indemnify the directors, officers, employees or other agents to the fullest extent permitted by the DGCL. These provisions may have the practical effect in certain cases of eliminating the ability of shareholders to collect monetary damages from directors. We are also a party to indemnification agreements with each of our directors. We believe that these provisions will assist us in attracting or retaining qualified individuals to serve as our directors.
Disclosure of Commission Position on Indemnification for Securities Act Liabilities
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers and controlling persons pursuant to the foregoing provisions, we have been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable.
DESCRIPTIONOF DEBT SECURITIES
The following description, together with the additional information we include in any applicable prospectus supplements, summarizes the material terms and provisions of the debt securities that we may offer under this prospectus. While the terms we have summarized below will apply generally to any future debt securities we may offer pursuant to this prospectus, we will describe the particular terms of any debt securities that we may offer in more detail in the applicable prospectus supplement. If we so indicate in a prospectus supplement, the terms of any debt securities offered under such prospectus supplement may differ from the terms we describe below, and to the extent the terms set forth in a prospectus supplement differ from the terms described below, the terms set forth in the prospectus supplement shall control.
We may sell from time to time, in one or more offerings under this prospectus, debt securities, which may be senior or subordinated. We will issue any such senior debt securities under a senior indenture that we will enter into with a trustee to be named in the senior indenture. We will issue any such subordinated debt securities under a subordinated indenture, which we will enter into with a trustee to be named in the subordinated indenture. We use the term “indentures” to refer to either the senior indenture or the subordinated indenture, as applicable. The indentures will be qualified under the Trust Indenture Act of 1939, as in effect on the date of the indenture. We use the term “debenture trustee” to refer to either the trustee under the senior indenture or the trustee under the subordinated indenture, as applicable.
The following summaries of material provisions of the senior debt securities, the subordinated debt securities and the indentures are subject to, and qualified in their entirety by reference to, all the provisions of the indenture applicable to a particular series of debt securities.
General
Each indenture will provide that debt securities may be issued from time to time in one or more series and may be denominated and payable in foreign currencies or units based on or relating to foreign currencies. Neither indenture will limit the amount of debt securities that may be issued thereunder, and each indenture will provide that the specific terms of any series of debt securities shall be set forth in, or determined pursuant to, an authorizing resolution and/or a supplemental indenture, if any, relating to such series.
We will describe in each prospectus supplement the following terms relating to a series of debt securities:
● the title or designation;
● the aggregate principal amount and any limit on the amount that may be issued;
● the currency or units based on or relating to currencies in which debt securities of such series are denominated and the currency or units in which principal or interest or both will or may be payable;
● whether we will issue the series of debt securities in global form, the terms of any global securities and who the depositary will be;
● the maturity date and the date or dates on which principal will be payable;
● the interest rate, which may be fixed or variable, or the method for determining the rate and the date interest will begin to accrue, the date or dates interest will be payable and the record dates for interest payment dates or the method for determining such dates;
● whether or not the debt securities will be secured or unsecured, and the terms of any secured debt;
● the terms of the subordination of any series of subordinated debt;
● the place or places where payments will be payable;
● our right, if any, to defer payment of interest and the maximum length of any such deferral period;
● the date, if any, after which, and the price at which, we may, at our option, redeem the series of debt securities pursuant to any optional redemption provisions;
● the date, if any, on which, and the price at which we are obligated, pursuant to any mandatory sinking fund provisions or otherwise, to redeem, or at the holder’s option to purchase, the series of debt securities;
● whether the indenture will restrict our ability to pay dividends, or will require us to maintain any asset ratios or reserves;
● whether we will be restricted from incurring any additional indebtedness;
● a discussion on any material or special U.S. federal income tax considerations applicable to a series of debt securities;
● the denominations in which we will issue the series of debt securities, if other than denominations of $1,000 and any integral multiple thereof; and
● any other specific terms, preferences, rights or limitations of, or restrictions on, the debt securities.
We may issue debt securities that provide for an amount less than their stated principal amount to be due and payable upon declaration of acceleration of their maturity pursuant to the terms of the indenture. We will provide you with information on the federal income tax considerations and other special considerations applicable to any of these debt securities in the applicable prospectus supplement.
Conversion or Exchange Rights
We will set forth in the prospectus supplement the terms, if any, on which a series of debt securities may be convertible into or exchangeable for our common stock or our other securities. We will include provisions as to whether conversion or exchange is mandatory, at the option of the holder or at our option. We may include provisions pursuant to which the number of shares of our common stock or our other securities that the holders of the series of debt securities receive would be subject to adjustment.
Information Concerning the Debenture Trustee
The debenture trustee, other than during the occurrence and continuance of an event of default under the applicable indenture, undertakes to perform only those duties as are specifically set forth in the applicable indenture. Upon an event of default under an indenture, the debenture trustee under such indenture must use the same degree of care as a prudent person would exercise or use in the conduct of his or her own affairs. Subject to this provision, the debenture trustee is under no obligation to exercise any of the powers given it by the indentures at the request of any holder of debt securities unless it is offered reasonable security and indemnity against the costs, expenses and liabilities that it might incur.
Payment and Paying Agents
Unless we otherwise indicate in the applicable prospectus supplement, we will make payment of the interest on any debt securities on any interest payment date to the person in whose name the debt securities, or one or more predecessor securities, are registered at the close of business on the regular record date for the interest.
We will pay principal of and any premium and interest on the debt securities of a particular series at the office of the paying agents designated by us, except that unless we otherwise indicate in the applicable prospectus supplement, we will make interest payments by check which we will mail to the holder. Unless we otherwise indicate in a prospectus supplement, we will designate the corporate trust office of the debenture trustee in the City of New York as our sole paying agent for payments with respect to debt securities of each series. We will name in the applicable prospectus supplement any other paying agents that we initially designate for the debt securities of a particular series. We will maintain a paying agent in each place of payment for the debt securities of a particular series.
All money we pay to a paying agent or the debenture trustee for the payment of the principal of or any premium or interest on any debt securities which remains unclaimed at the end of two years after such principal, premium or interest has become due and payable will be repaid to us, and the holder of the security thereafter may look only to us for payment thereof.
Governing Law
The indentures and the debt securities will be governed by and construed in accordance with the laws of the State of New York, except to the extent that the Trust Indenture Act is applicable.
Subordination of Subordinated Debt Securities
Our obligations pursuant to any subordinated debt securities will be unsecured and will be subordinate and junior in priority of payment to certain of our other indebtedness to the extent described in a prospectus supplement. The subordinated indenture does not limit the amount of senior indebtedness we may incur. It also does not limit us from issuing any other secured or unsecured debt.
Transfer Agent and Registrar
The transfer agent and registrar for any warrants we offer will be set forth in the applicable prospectus supplement.
DESCRIPTIONOF WARRANTS
General
We may issue warrants to our stockholders to purchase shares of our common stock. We may offer warrants separately or together with one or more debt securities, common stock, rights or purchase contracts, or any combination of those securities in the form of units, as described in the applicable prospectus supplement. Each series of warrants will be issued under a separate warrant agreement to be entered into between us and a bank or trust company, as warrant agent. The warrant agent will act solely as our agent in connection with the certificates relating to the rights of the series of certificates and will not assume any obligation or relationship of agency or trust for or with any holders of rights certificates or beneficial owners of rights. The following description sets forth certain general terms and provisions of the rights to which any prospectus supplement may relate. The particular terms of the warrant to which any prospectus supplement may relate and the extent, if any, to which the general provisions may apply to the rights so offered will be described in the applicable prospectus supplement. To the extent that any particular terms of the warrant, warrant agreement or warrant certificates described in a prospectus supplement differ from any of the terms described below, then the terms described below will be deemed to have been superseded by that prospectus supplement. We encourage you to read the applicable warrant agreement and warrant certificate for additional information before you decide whether to purchase any of our rights.
We will provide in a prospectus supplement the following terms of the rights being issued:
● the date of determining the stockholders entitled to the rights distribution;
● the aggregate number of shares of common stock or other securities purchasable upon exercise of the rights;
● the exercise price;
● the aggregate number of rights issued;
● whether the rights are transferrable and the date, if any, on and after which the rights may be separately transferred;
● the date on which the right to exercise the rights will commence, and the date on which the right to exercise the rights will expire;
● the method by which holders of rights will be entitled to exercise;
● the conditions to the completion of the offering, if any;
● the withdrawal, termination and cancellation rights, if any;
● whether there are any backstop or standby purchaser or purchasers and the terms of their commitment, if any;
● whether stockholders are entitled to oversubscription rights, if any;
● any applicable U.S. federal income tax considerations; and
● any other terms of the rights, including terms, procedures and limitations relating to the distribution, exchange and exercise of the rights, as applicable.
Each warrant will entitle the holder of rights to purchase for cash the principal amount of shares of common stock or other securities at the exercise price provided in the applicable prospectus supplement. Warrants may be exercised at any time up to the close of business on the expiration date for the rights provided in the applicable prospectus supplement.
Holders may exercise warrants as described in the applicable prospectus supplement. Upon receipt of payment and the warrant certificate properly completed and duly executed at the corporate trust office of the rights agent or any other office indicated in the prospectus supplement, we will, as soon as practicable, forward the shares of common stock or other securities, as applicable, purchasable upon exercise of the rights. If less than all of the warrants issued in any rights offering are exercised, we may offer any unsubscribed securities directly to persons other than stockholders, to or through agents, underwriters or dealers or through a combination of such methods, including pursuant to standby arrangements, as described in the applicable prospectus supplement.
Warrant Agent
The warrant agent for any rights we offer will be set forth in the applicable prospectus supplement.
DESCRIPTIONOF RIGHTS
General
We may issue rights to our stockholders to purchase shares of our common stock or the other securities described in this prospectus. We may offer rights separately or together with one or more additional rights, debt securities, common stock, warrants or purchase contracts, or any combination of those securities in the form of units, as described in the applicable prospectus supplement. Each series of rights will be issued under a separate rights agreement to be entered into between us and a bank or trust company, as rights agent. The rights agent will act solely as our agent in connection with the certificates relating to the rights of the series of certificates and will not assume any obligation or relationship of agency or trust for or with any holders of rights certificates or beneficial owners of rights. The following description sets forth certain general terms and provisions of the rights to which any prospectus supplement may relate. The particular terms of the rights to which any prospectus supplement may relate and the extent, if any, to which the general provisions may apply to the rights so offered will be described in the applicable prospectus supplement. To the extent that any particular terms of the rights, rights agreement or rights certificates described in a prospectus supplement differ from any of the terms described below, then the terms described below will be deemed to have been superseded by that prospectus supplement. We encourage you to read the applicable rights agreement and rights certificate for additional information before you decide whether to purchase any of our rights.
We will provide in a prospectus supplement the following terms of the rights being issued:
● the date of determining the stockholders entitled to the rights distribution;
● the aggregate number of shares of common stock or other securities purchasable upon exercise of the rights;
● the exercise price;
● the aggregate number of rights issued;
● whether the rights are transferrable and the date, if any, on and after which the rights may be separately transferred;
● the date on which the right to exercise the rights will commence, and the date on which the right to exercise the rights will expire;
● the method by which holders of rights will be entitled to exercise;
● the conditions to the completion of the offering, if any;
● the withdrawal, termination and cancellation rights, if any;
● whether there are any backstop or standby purchaser or purchasers and the terms of their commitment, if any;
● whether stockholders are entitled to oversubscription rights, if any;
● any applicable U.S. federal income tax considerations; and
● any other terms of the rights, including terms, procedures and limitations relating to the distribution, exchange and exercise of the rights, as applicable.
Each right will entitle the holder of rights to purchase for cash the principal amount of shares of common stock or other securities at the exercise price provided in the applicable prospectus supplement. Rights may be exercised at any time up to the close of business on the expiration date for the rights provided in the applicable prospectus supplement.
Holders may exercise rights as described in the applicable prospectus supplement. Upon receipt of payment and the rights certificate properly completed and duly executed at the corporate trust office of the rights agent or any other office indicated in the prospectus supplement, we will, as soon as practicable, forward the shares of common stock or other securities, as applicable, purchasable upon exercise of the rights. If less than all of the rights issued in any rights offering are exercised, we may offer any unsubscribed securities directly to persons other than stockholders, to or through agents, underwriters or dealers or through a combination of such methods, including pursuant to standby arrangements, as described in the applicable prospectus supplement.
Rights Agent
The rights agent for any rights we offer will be set forth in the applicable prospectus supplement.
DESCRIPTIONOF PURCHASE CONTRACTS
We may issue purchase contracts, including contracts obligating holders to purchase from us, and for us to sell to holders, a specific or variable number of our debt securities, shares of common stock, warrants or rights, or securities of an entity unaffiliated with us, or any combination of the above, at a future date or dates. Alternatively, the purchase contracts may obligate us to purchase from holders, and obligate holders to sell to us, a specific or variable number of our debt securities, shares of common stock, warrants, rights or other property, or any combination of the above. The price of the securities or other property subject to the purchase contracts may be fixed at the time the purchase contracts are issued or may be determined by reference to a specific formula described in the purchase contracts. We may issue purchase contracts separately or as a part of units each consisting of a purchase contract and one or more of our other securities described in this prospectus or securities of third parties, including U.S. Treasury securities, securing the holder’s obligations under the purchase contract. The purchase contracts may require us to make periodic payments to holders or vice versa and the payments may be unsecured or pre-funded on some basis. The purchase contracts may require holders to secure the holder’s obligations in a manner specified in the applicable prospectus supplement.
The applicable prospectus supplement will describe the terms of any purchase contracts in respect of which this prospectus is being delivered, including, to the extent applicable, the following:
● whether the purchase contracts obligate the holder or us to purchase or sell, or both purchase and sell, the securities subject to purchase under the purchase contract, and the nature and amount of each of those securities, or the method of determining those amounts;
● whether the purchase contracts are to be prepaid;
● whether the purchase contracts are to be settled by delivery, or by reference or linkage to the value, performance or level of the securities subject to purchase under the purchase contract;
● any acceleration, cancellation, termination or other provisions relating to the settlement of the purchase contracts;
● any applicable U.S. federal income tax considerations; and
● whether the purchase contracts will be issued in fully registered or global form.
The preceding description sets forth certain general terms and provisions of the purchase contracts to which any prospectus supplement may relate. The particular terms of the purchase contracts to which any prospectus supplement may relate and the extent, if any, to which the general provisions may apply to the purchase contracts so offered will be described in the applicable prospectus supplement. To the extent that any particular terms of the purchase contracts described in a prospectus supplement differ from any of the terms described above, then the terms described above will be deemed to have been superseded by that prospectus supplement. We encourage you to read the applicable purchase contract for additional information before you decide whether to purchase any of our purchase contracts.
DESCRIPTIONOF UNITS
The following description, together with the additional information that we include in any applicable prospectus supplements summarizes the material terms and provisions of the units that we may offer under this prospectus. While the terms we have summarized below will apply generally to any units that we may offer under this prospectus, we will describe the particular terms of any series of units in more detail in the applicable prospectus supplement. The terms of any units offered under a prospectus supplement may differ from the terms described below.
We will incorporate by reference from reports that we file with the SEC, the form of unit agreement that describes the terms of the series of units we are offering, and any supplemental agreements, before the issuance of the related series of units. The following summaries of material terms and provisions of the units are subject to, and qualified in their entirety by reference to, all the provisions of the unit agreement and any supplemental agreements applicable to a particular series of units. We urge you to read the applicable prospectus supplements related to the particular series of units that we may offer under this prospectus, as well as any related free writing prospectuses and the complete unit agreement and any supplemental agreements that contain the terms of the units.
General
We may issue units consisting of common stock, one or more debt securities, warrants, rights or purchase contacts for the purchase of common stock and/or debt securities in one or more series, in any combination. Each unit will be issued so that the holder of the unit is also the holder of each security included in the unit. Thus, the holder of a unit will have the rights and obligations of a holder of each security included in the unit. The unit agreement under which a unit is issued may provide that the securities included in the unit may not be held or transferred separately, at any time or at any time before a specified date.
We will describe in the applicable prospectus supplement the terms of the series of units being offered, including:
● the designation and terms of the units and of the securities comprising the units, including whether and under what circumstances those securities may be held or transferred separately;
● any provisions of the governing unit agreement that differ from those described below; and
● any provisions for the issuance, payment, settlement, transfer or exchange of the units or of the securities comprising the units.
The provisions described in this section, as well as those set forth in any prospectus supplement or as described under “Description of Common Stock,” “Description of Debt Securities,” “Description of Warrants,” “Description of Rights” and “Description of Purchase Contracts” will apply to each unit, as applicable, and to any common stock, debt security, warrant, right or purchase contract included in each unit, as applicable.
Unit Agent
The name and address of the unit agent for any units we offer will be set forth in the applicable prospectus supplement.
Issuance in Series
We may issue units in such amounts and in such numerous distinct series as we determine.
Enforceability of Rights by Holders of Units
Each unit agent will act solely as our agent under the applicable unit agreement and will not assume any obligation or relationship of agency or trust with any holder of any unit. A single bank or trust company may act as unit agent for more than one series of units. A unit agent will have no duty or responsibility in case of any default by us under the applicable unit agreement or unit, including any duty or responsibility to initiate any proceedings at law or otherwise, or to make any demand upon us. Any holder of a unit may, without the consent of the related unit agent or the holder of any other unit, enforce by appropriate legal action its rights as holder under any security included in the unit.
Provisions of Delaware Law Governing Business Combinations
We are subject to the “business combination” provisions of Section 203 of the DGCL. In general, such provisions prohibit a publicly held Delaware corporation from engaging in any “business combination” transactions with any “interested stockholder” for a period of three years after the date on which the person became an “interested stockholder,” unless:
● prior to such date, the board of directors approved either the “business combination” or the transaction which resulted in the “interested stockholder” obtaining such status; or
● upon consummation of the transaction which resulted in the stockholder becoming an “interested stockholder,” the “interested stockholder” owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the voting stock outstanding (but not the outstanding voting stock owned by the “interested stockholder”) those shares owned by (a) persons who are directors and also officers and (b) employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or
● at or subsequent to such time the “business combination” is approved by the board of directors and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock which is not owned by the “interested stockholder.”
A “business combination” is defined to include mergers, asset sales and other transactions resulting in financial benefit to a stockholder. In general, an “interested stockholder” is a person who, together with affiliates and associates, owns 15% or more of a corporation’s voting stock or within three years did own 15% or more of a corporation’s voting stock. The statute could prohibit or delay mergers or other takeover or change in control attempts with respect to us and, accordingly, may discourage attempts to acquire us.
Limitations on Liability and Indemnification of Officers and Directors
Section 145 of the DGCL authorizes a court to award, or a corporation’s board of directors to grant, indemnity to directors and officers in terms sufficiently broad to permit such indemnification under certain circumstances for liabilities (including reimbursement for expenses incurred) arising under the Securities Act of 1933. Our amended and restated certificate of incorporation limits the liability of our officers and directors to the fullest extent permitted by the DGCL, and our amended and restated certificate of incorporation provides that we will indemnify our officers and directors to the fullest extent permitted by such law.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers or persons controlling the registrant pursuant to the foregoing provisions, the registrant has been information that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
LEGALMATTERS
Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C., New York, New York, will pass upon the validity of the issuance of the securities to be offered by this prospectus.
The balance sheets of Dipexium Pharmaceuticals, Inc. as of December 31, 2014 and 2013, and the related statements of operations, changes in members’ and shareholders’ equity and cash flows for each of the years in the two-year period ended December 31, 2014, have been audited by CohnReznick LLP, independent registered public accounting firm, as stated in their report which is incorporated by reference. Such financial statements have been incorporated by reference in reliance on the report of such firm given upon their authority as experts in accounting and auditing.
WHEREYOU CAN FIND MORE INFORMATION
We are subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, and file annual, quarterly and current reports, proxy statements and other information with the SEC. You may read and copy these reports, proxy statements and other information at the SEC’s public reference facilities at 100 F Street, N.E., Room 1580, Washington, D.C. 20549. You can request copies of these documents by writing to the SEC and paying a fee for the copying cost. Please call the SEC at 1-800-SEC-0330 for more information about the operation of the public reference facilities. SEC filings are also available at the SEC’s web site at http://www.sec.gov.
This prospectus is only part of a registration statement on Form S-3 that we have filed with the SEC under the Securities Act and therefore omits certain information contained in the registration statement. We have also filed exhibits and schedules with the registration statement that are excluded from this prospectus, and you should refer to the applicable exhibit or schedule for a complete description of any statement referring to any contract or other document. You may inspect a copy of the registration statement, including the exhibits and schedules, without charge, at the public reference room or obtain a copy from the SEC upon payment of the fees prescribed by the SEC.
We also maintain a website at www.dipexiumpharmaceuticals.com, through which you can access our SEC filings. The information set forth on, or accessible from, our website is not part of this prospectus.
INCORPORATIONOF INFORMATION BY REFERENCE
The SEC allows us to “incorporate by reference” information that we file with them. Incorporation by reference allows us to disclose important information to you by referring you to those other documents. The information incorporated by reference is an important part of this prospectus, and information that we file later with the SEC will automatically update and supersede this information. This prospectus omits certain information contained in the registration statement, as permitted by the SEC. You should refer to the registration statement and any prospectus supplement filed hereafter, including the exhibits, for further information about us and the securities we may offer pursuant to this prospectus. Statements in this prospectus regarding the provisions of certain documents filed with, or incorporated by reference in, the registration statement are not necessarily complete and each statement is qualified in all respects by that reference. Copies of all or any part of the registration statement, including the documents incorporated by reference or the exhibits, may be obtained upon payment of the prescribed rates at the offices of the SEC listed above in “Where You Can Find More Information.” The documents we are incorporating by reference are:
● our Annual Report on Form 10-K for the fiscal year ended December 31, 2014 filed on March 23, 2015;
● our Quarterly Report on Form 10-Q filed on May 13, 2015;
● our Current Reports on Form 8-K filed on March 24, 2015 and May 19, 2015;
● our Definitive Proxy Statement on Schedule 14A filed on April 1, 2015, but only to the extent that such information was incorporated by reference into our Annual Report on Form 10-K for the year ended December 31, 2014;
● the description of our common stock contained in our Registration Statement on Form 8-A, filed on March 12, 2014 pursuant to Section 12(b) of the Exchange Act, which incorporates by reference the description of the shares of our common stock contained in our Registration Statement on Form S-1 (File No. 333-193780) filed on February 6, 2014 and declared effective by the SEC on March 12, 2014, and any amendment or report filed with the SEC for purposes of updating such description; and
● all reports and other documents subsequently filed by us pursuant to Sections 13(a), 13(c), 14 and 15(d) of the Exchange Act after the date of this prospectus and prior to the termination or completion of the offering of securities under this prospectus shall be deemed to be incorporated by reference in this prospectus and to be a part hereof from the date of filing such reports and other documents;
Unless otherwise noted, the SEC file number for each of the documents listed above is 001-36351.
In addition, all reports and other documents filed by us pursuant to the Exchange Act after the date of the initial registration statement and prior to effectiveness of the registration statement shall be deemed to be incorporated by reference into this prospectus.
Any statement contained in this prospectus or in a document incorporated or deemed to be incorporated by reference into this prospectus will be deemed to be modified or superseded for purposes of this prospectus to the extent that a statement contained in this prospectus or any other subsequently filed document that is deemed to be incorporated by reference into this prospectus modifies or supersedes the statement. Any statement so modified or superseded will not be deemed, except as so modified or superseded, to constitute a part of this prospectus.
You may request, orally or in writing, a copy of any or all of the documents incorporated herein by reference. These documents will be provided to you at no cost, by contacting: Investor Relations, Dipexium Pharmaceuticals, Inc., 61 Broadway, Suite 1905, New York, New York 10006, or call (212) 269-2834.
You should rely only on information contained in, or incorporated by reference into, this prospectus and any prospectus supplement. We have not authorized anyone to provide you with information different from that contained in this prospectus or incorporated by reference in this prospectus. We are not making offers to sell the securities in any jurisdiction in which such an offer or solicitation is not authorized or in which the person making such offer or solicitation is not qualified to do so or to anyone to whom it is unlawful to make such offer or solicitation.
2,646,091 Shares
|
Common Stock
PROSPECTUS SUPPLEMENT
Lead Placement Agent | Co-Lead Placement Agent JANNEY MONTGOMERY SCOTT |
June 9, 2017