UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
(Mark One)
x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended: December 31, 2013
or
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _______________ to _______________
Commission file number: 333-172647
Neurotrope, Inc.
(Exact name of registrant as specified in its charter)
Nevada | 46-3522381 |
| (State or other jurisdiction of | (IRS Employer Identification No.) |
| incorporation or organization) | |
10732 Hawk’s Vista Street | |
Plantation, FL | 33324 |
| (Address of principal executive offices) | (Postal Code) |
Registrant’s telephone number, including area code: 1-954-632-6630
Securities registered under Section 12(b) of the Act: None
Securities registered under Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Exchange Act. Yes x No ¨
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ¨ No ¨ (Note: The registrant is a voluntary filer of reports under Section 13 or 15(d) of the Securities Exchange Act of 1934; the registrant has filed during the preceding 12 months all reports it would have been required to file by Section 13 or 15(d) of the Securities Exchange Act of 1934 if the registrant had been subject to one of such Sections.)
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a smaller reporting company. See the definitions of the “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large Accelerated Filer ¨ | Accelerated Filer ¨ |
Non-Accelerated Filer ¨ | Smaller reporting company x |
| (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the voting and non-voting common equity of the registrant on June 30, 2013, the last business day of the registrant’s most recently completed second fiscal quarter, cannot be determined, because the registrant’s common stock, $0.001 par value per share (its only class of voting or non-voting common equity) did not begin to be quoted in the over-the-counter markets until on or about October 2, 2013.
As of April 10, 2014, there were 21,889,006shares of the registrant’s common stock, par value $0.001 per share, issued and outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
None.
TABLE OF CONTENTS
Item Number and Caption | | Page |
| | | |
| Cautionary Note Regarding Forward-Looking Statements | | 3 |
| Explanatory Note | | 3 |
| | | | |
PART I | | | |
| | | | |
| 1. | Business | | 5 |
| 1A. | Risk Factors | | 22 |
| 1B. | Unresolved Staff Comments | | 33 |
| 2. | Properties | | 33 |
| 3. | Legal Proceedings | | 33 |
| 4. | Mine Safety Disclosures | | 33 |
| | | | |
PART II | | | 34 |
| | | | |
| 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | | 34 |
| 6. | Selected Financial Data | | 36 |
| 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | | 36 |
| 7A | Quantitative and Qualitative Disclosures About Market Risk. | | 40 |
| 8. | Financial Statements and Supplementary Data | | 40 |
| 9. | Changes in and Disagreements with Accountants on Accounting, and Financial Disclosure | | 40 |
| 9A. | Controls and Procedures | | 41 |
| 9B. | Other Information | | 42 |
| | | | |
PART III | | | 43 |
| | | | |
| 10. | Directors, Executive Officers and Corporate Governance | | 43 |
| 11. | Executive Compensation | | 50 |
| 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | | 53 |
| 13. | Certain Relationships and Related Transactions, and Director Independence | | 55 |
| 14. | Principal Accountant Fees and Services | | 61 |
| | | | |
PART IV | | | 62 |
| | | | |
| 15. | Exhibits, Financial Statement Schedules | | 62 |
| | | | |
Financial Statements | | F-1 |
CAUTIONARY NOTE REGARDING FORWARD-LOOKING INFORMATION
This report contains forward-looking statements, including, without limitation, in the sections captioned “Business,” “Risk Factors,” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and elsewhere. Any and all statements contained in this report that are not statements of historical fact may be deemed forward-looking statements. Terms such as “may,” “might,” “would,” “should,” “could,” “project,” “estimate,” “pro-forma,” “predict,” “potential,” “strategy,” “anticipate,” “attempt,” “develop,” “plan,” “help,” “believe,” “continue,” “intend,” “expect,” “future,” and terms of similar import (including the negative of any of the foregoing) may be intended to identify forward-looking statements. However, not all forward-looking statements may contain one or more of these identifying terms. Forward-looking statements in this report may include, without limitation, statements regarding (i) the plans and objectives of management for future operations, including plans or objectives relating to the development of commercially viable pharmaceuticals, (ii) a projection of income (including income/loss), earnings (including earnings/loss) per share, capital expenditures, dividends, capital structure or other financial items, (iii) our future financial performance, including any such statement contained in a discussion and analysis of financial condition by management or in the results of operations included pursuant to the rules and regulations of the SEC, and (iv) the assumptions underlying or relating to any statement described in points (i), (ii) or (iii) above.
The forward-looking statements are not meant to predict or guarantee actual results, performance, events or circumstances and may not be realized because they are based upon our current projections, plans, objectives, beliefs, expectations, estimates and assumptions and are subject to a number of risks and uncertainties and other influences, many of which we have no control over. Actual results and the timing of certain events and circumstances may differ materially from those described by the forward-looking statements as a result of these risks and uncertainties. Factors that may influence or contribute to the inaccuracy of the forward-looking statements or cause actual results to differ materially from expected or desired results may include, without limitation, our inability to obtain adequate financing, the significant length of time associated with drug development and related insufficient cash flows and resulting illiquidity, our inability to expand our business, significant government regulation of pharmaceuticals and the healthcare industry, lack of product diversification, volatility in the price of our raw materials, existing or increased competition, results of arbitration and litigation, stock volatility and illiquidity, and our failure to implement our business plans or strategies. A description of some of the risks and uncertainties that could cause our actual results to differ materially from those described by the forward-looking statements in this report appears in the section captioned “Risk Factors” and elsewhere in this report.
Readers are cautioned not to place undue reliance on forward-looking statements because of the risks and uncertainties related to them and to the risk factors. We disclaim any obligation to update the forward-looking statements contained in this report to reflect any new information or future events or circumstances or otherwise.
EXPLANATORY NOTE
We were incorporated as BlueFlash Communications, Inc. in Florida on January 11, 2011. Prior to the Merger and Split-Off (each as defined below), our business was to provide software solutions to deliver geo-location targeted coupon advertising to mobile internet devices.
On August 9, 2013, we reincorporated in the State of Nevada by merging into a newly-formed special-purpose subsidiary, Neurotrope, Inc., which was the surviving corporation in the merger. As a result of this reincorporation merger, (i) we changed our name to Neurotrope, Inc., (ii) we changed our jurisdiction of incorporation from Florida to Nevada, (iii) we increased our authorized capital stock from 300,000,000 shares of common stock, par value $0.0001, to 300,000,000 shares of common stock, par value $0.0001, and 50,000,000 shares of “blank check” preferred stock, par value $0.0001, (iv) each share of BlueFlash Communications, Inc., common stock outstanding at the time of the reincorporation merger was automatically converted into 2.242 shares of Neurotrope, Inc., common stock, with the result that the 10,200,000 shares of common stock outstanding immediately prior to the reincorporation merger was converted into 22,868,400 shares of common stock outstanding immediately thereafter. All share and per share numbers in this Report relating to the common stock of Neurotrope, Inc., prior to this reincorporation merger have been adjusted to give effect to this conversion, unless otherwise stated.
In addition, in connection with the reincorporation, we changed our fiscal year from a fiscal year ending on January 31 of each year to one ending on December 31 of each year.
On August 23, 2013, our wholly owned subsidiary, Neurotrope Acquisition, Inc., a corporation formed in the State of Nevada on August 15, 2013 (“Acquisition Sub”) merged (the “Merger”) with and into Neurotrope BioScience, Inc., a corporation incorporated in the State of Delaware on October 31, 2012 (“Neurotrope BioScience”). Neurotrope BioScience was the surviving corporation in the Merger and became our wholly owned subsidiary. All of the outstanding Neurotrope BioScience common stock was converted into shares of our common stock, par value $0.0001 per share (the “Common Stock”), on a one-for-one basis.
In connection with the Merger and pursuant to the Split-Off Agreement (defined below), we transferred our pre-Merger business to our pre-Merger majority stockholder, in exchange for the surrender by her and cancellation of 20,178,000 shares of our Common Stock. See Item 13, “Split-Off”, below.
As a result of the Merger and Split-Off, we discontinued our pre-Merger business and acquired the business of Neurotrope BioScience, and will continue the existing business operations of Neurotrope BioScience as a publicly-traded company under the name Neurotrope, Inc.
Also on August 23, 2013, Neurotrope BioScience closed a private placement of 11,533,375 shares of its Series A convertible preferred stock, for a purchase price of $1.00 per share, for aggregate gross proceeds of $11,533,375 (before deducting placement agent fees and expenses of the offering estimated at approximately $1,500,000). Neurotrope BioScience had previously closed between February and May 2013 on private placements of 10,386,625 shares of its Series A convertible preferred stock, for a purchase price of $1.00 per share, for aggregate gross proceeds of $10,386,625 (before deducting placement agent fees and expenses of the offering). These private placement offerings (the “PPO”) were exempt from registration under Section 4(2) of the Securities Act of 1933, as amended (the “Securities Act”), in reliance upon the exemptions provided by Regulation D promulgated by the SEC thereunder. The PPO was sold to “accredited investors,” as defined in Regulation D. All of the outstanding Neurotrope BioScience Series A convertible preferred stock was converted into shares of our Series A convertible preferred stock (the “Series A Preferred Stock”) on a one-for-one basis in the Merger. Additional information concerning the PPO and the terms of the Series A Preferred Stock is presented below under Item 13, “The PPO.”
In accordance with “reverse merger” accounting treatment, our historical financial statements as of period ends, and for periods ended, prior to the Merger will be replaced with the historical financial statements of Neurotrope BioScience prior to the Merger in all future filings with the SEC.
DEFINITIONS
When used in this report, the terms, “we,” the “Company,” “our,” and “us” refers to Neurotrope, Inc., a Nevada corporation (formerly BlueFlash Communications, Inc., a Florida corporation), and its subsidiary.
PART I
Immediately following the Merger, the business of Neurotrope BioScience became our business. Neurotrope BioScience was originally formed to develop and market, principally, two product platforms: a drug candidate called bryostatin for the treatment of Alzheimer’s Disease and a diagnostic test for Alzheimer’s Disease, both of which are in the clinical testing stage.
History
As described above, we were incorporated in Florida as BlueFlash Communications, Inc. on January 11, 2011. Prior to the Merger and Split-Off (each as defined above), our business was to provide software solutions to deliver geo-location targeted coupon advertising to mobile internet devices. As a result of the Split-Off and the Merger, the Company discontinued its pre-Merger business and acquired the business of Neurotrope BioScience.
Our authorized capital stock currently consists of 300,000,000 shares of common stock, par value $0.0001, and 50,000,000 shares of “blank check” preferred stock, par value $0.0001, 24,325,000 of which has been designated as Series A Preferred Stock. Our common stock is quoted on the OTC Markets (OTCQB) under the symbol “NTRP.”
Our principal executive offices are located at 10732 Hawk’s Vista Street, Plantation, Florida 33324, USA. Our telephone number is 1-954-632-6630. Our website address iswww.neurotropebioscience.com.
Neurotrope BioScience was incorporated on October 31, 2012, under the laws of the State of Delaware. It is a clinical stage biopharmaceutical and diagnostics company. The Company has been principally focused on developing two product platforms, a drug candidate called bryostatin for the treatment of Alzheimer’s Disease (sometimes referred to herein as AD) and a diagnostic test for Alzheimer’s Disease, both of which are in the clinical testing stage.
We are considered to be a development stage company, as defined by Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) 915-10, in that we are devoting substantially all of our efforts to establishing a new business, where planned principal operations have commenced, but no revenues have been derived from these operations.
Neurotrope BioScience entered into a Technology License and Services Agreement with the Blanchette Rockefeller Neurosciences Institute (“BRNI”) and its affiliate NRV II, LLC, pursuant to which the company was granted an exclusive non-transferable license to certain patents and technologies required to develop our proposed products. (For additional information, see “Business—Intellectual Property—Technology License and Services Agreement.”) Neurotrope BioScience was formed for the primary purpose of commercializing certain technologies, which were initially developed by BRNI, for therapeutic or diagnostic applications for Alzheimer’s Disease or other neurodegenerative disorders. These technologies have been under development by BRNI since 1999 and have been financed through significant funding from a variety of non-investor sources (which include not-for-profit foundations, the National Institutes of Health (which is part of the U.S. Department of Health and Human Services) and contributions from individuals).
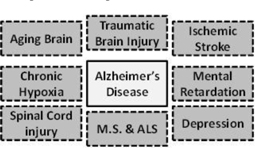
BRNI conducted one compassionate use protocolin familial Alzheimer’s Disease with the experimental drug bryostatin under a U.S. Food and Drug Administration (“FDA”) approved study protocol. This protocol has concluded, and now BRNI plans to modestly expand this clinical effort in Alzheimer’s Disease in the 2014 – 2015 timeframe to include additional patients. Also, we plan to begin controlled clinical trials with bryostatin in AD patients in late 2014. Bryostatin modulates the same enzyme target used by the diagnostic test for the detection of AD. We believe bryostatin may restore synaptic structures and functions damaged by AD, leading to improvements in cognition and memory. Beyond AD, several other neurodegenerative diseases, such as ischemic stroke, traumatic brain injury, Fragile X mental retardation, depression and aging in the brain, may be amenable to treatment with the same approach.
Since licensing the AD diagnostic technology from BRNI, we have conducted extensive analysis of the underlying technology and commercial sales potential of an AD diagnostic product. Based upon our analyses to date and our current resources, our Board of Directors may decide that allocation of future expenditures relating to the AD diagnostic product (beyond completion of our existing statement of work with BRNI for development of the diagnostic; see “Diagnostic Test for AD” below) may be better spent on development of bryostatin in the treatment of AD. As such, we are continuing to evaluate our original timeline for commercialization of the diagnostic product to insure the budget to develop bryostatin for the treatment of AD is fully funded over next 24 months. This on-going process could result in a delay of development of the diagnostic product.
In addition to bryostatin and the diagnostic test for Alzheimer’s Disease, we intend to pursue development of selected other technology platforms with applications related to the treatment of Alzheimer’s Disease and other neurodegenerative disorders based on our current licensed technology or technology available from third party licensors or collaborators.
Alzheimer’s Disease and the Potential Market for our Products
The Epidemic of Alzheimer’s Disease
According to the Alzheimer’s Association, it was estimated that 36 million people worldwide had AD in 2010 The prevalence of AD is independent of race, ethnicity, geography, life style and, to a large extent, genetics. The most common cause of developing AD is living a long life. In developing countries where the median age of death is less than 65 years old, AD is rarely recognized or diagnosed. In the U.S. in 2013, 5.2 million persons are estimated to have AD, and 96% of these people are older than 65 years of age.
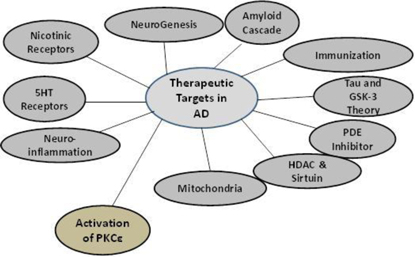
Researchers continue to explore a wide range of drug mechanisms in hopes of developing drugs to combat this disease.Figure 1 illustrates the range of mechanisms under consideration. Our approach, which involves the activation of the enzyme called protein kinase C epsilon (“PKCe”), represents a novel mechanism in the armamentarium of potential AD drug therapies.
Figure 1. Different Pharmacologic Targets being pursued for the Treatment of AD1
It has been shown that, during several years preceding the diagnosis of dementia associated with Alzheimer’s Disease, there is a gradual cognition decline, which at first may have rather benign characteristics. Entering the mild cognitive impairment (MCI) phase of the disease marks progression of AD to the point where there is a significant loss of synapses (the junctions between nerve cells) preventing effective neurotransmission (Figure 2). This precursor phase transitions into mild, moderate and, finally, severe stages of the disease that are characterized by greater systemic loss of neurons in the brain tissue.
1 Business Insights: Reference Code B100040-005, Publication Date May 2011, “Advances in Alzheimer’s Disease Drug Discovery”
Figure 2. Early Diagnosis of AD is Essential to Effective Treatment2
This progressive degeneration produces some abnormalities in the brain’s neurotransmitter systems. Multiple failures inacetylcholine and glutamate neurotransmitter systems (neurotransmitters) appear to underlie some of the symptomology of AD, and thus these systems have become targets for pharmacologic intervention.
The loss of neuronal function and neuronal cell death is also related to the abnormal processing of β amyloid (Aβ) peptide, ultimately leading to the formation of Aβ plaques (protein deposits) in the brain. As illustrated inFigure 2, this amyloid load in the brain usually becomes marked before the symptoms of the mild cognitive impairment (MCI) phase appear in AD patients.
The conventional amyloid cascade hypothesis holds that amyloid pathology leads to tau proteins (a protein found in nerve cells) being deposited within neurons in the form of insoluble tangles, excitotoxicity (overstimulation of nerve cells byneurotransmitters), inflammation and finally synaptic depletion and neuronal death. The majority of drug development efforts to date have focused on stopping the production of Aβ or its fragments, and the elimination of these peptides from either intracellular or extracellular locations has represented the preponderance of drug design efforts to halt the progression of AD. However, these efforts have been largely unsuccessful.
Neurotrope believes the current failures of therapies clearing formed amyloid come from an incorrect view of the process. In our view, amyloid is merely a signal of the final phase of the pathology, and its appearance is an indication that the cessation of neuronal function in neurons “stained” with plaque is inevitable.
In animal studies we found that PKCe activation in neurons targets the loss of synapses in the Alzheimer’s brain, and can delay or temporarily arrest other elements of the disease, i.e. the elevation of the toxic Aβ peptide, the loss of neurons, the appearance of plaques, and the loss of cognitive function.
Potential Market for Our Products
According to an article titled “Progress in Alzheimer’s Disease” published in The Journal of Neurology in 2012, there has been a dearth of new product introductions in the last 20 years either for the treatment of AD symptoms or its definitive diagnosis in patients who begin exhibiting the memory and cognitive disorders associated with the disease. According to the Alzheimer’s Association, all of the products introduced to date for the treatment of AD have yielded negative or marginal results with no long-term effect on the progression of AD and no improvement in the memory or cognitive performance of the patients receiving these therapies. With 36 million people worldwide estimated to have had AD in 2010, there is significant commercial potential for a new therapeutic that is effective in delaying the progression of the disease.
We believe the markets for drugs, therapies or diagnostics to treat and analyze AD exist exclusively in the developed world and principally comprise the North American, European and Japanese markets. The aggregate AD market is subdivided into four distinct segments, which are shown inFigure 3, as are the projected compounded annual growth rates (CAGRs) for these segments over the 2009-2014 timeframe.
2 Lancet Neurol. 2010;9,119. CR Jack et al, “ Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade”
Sales of the major drug therapies available only by prescription are reported inFigure 3, which includes, among others, the acetylcholinesterase inhibitors (Exelon®, Razadyne®, and Aricept®) and the glutamate antagonist Namenda®. These drugs are approved for the symptomatic treatment of the cognitive aspects of AD but have no meaningful effect on disease progression, giving only temporary improvement in cognitive decline. Despite their limited efficacy, this group of drugs had collective worldwide sales in 2011 in excess of approximately $6 billion, according to a BBC Research Report. The negative CAGR for this segment reflects the fact that this class of drugs faces generic competition over the timeframe considered.
Figure 3. Global Market for Alzheimer’s Disease ($ mm): 2009-20143
A much higher growth rate is projected for the use of biomarkers and diagnostics over the 2009-2014 timeframe. The use of techniques or tools to measure disease progression and clinical trial endpoints are, in our estimation, in high demand across the industry. We believe that there is currently no diagnostic test for AD that has achieved significant market penetration.
The “Therapeutics for Treatment of Symptoms” category cited inFigure 3 represents drugs from other classes that are being used to temporarily treat some of the symptoms of AD.4
Neurotrope’s Proposed Products
Challenges in Treating AD
One of the challenges in treating AD is that its symptoms become manifest only years after the disease process has actually commenced. Treatment strategies attempting to intervene once symptoms become apparent are focused on stimulating the neurotransmitter activity of still healthy neurons, or removing the amyloid plaque deposited in the brain. All drug development efforts to date that have targeted the removal of beta-amyloid or tau protein as their therapeutic mechanism of action have failed, and drugs approved for stimulating neurotransmitter activity offer short-lived, palliative results for AD patients. As such, these strategies have yielded negative or marginal results with no effect on the progression of AD and no improvement in the memory or cognitive performance of the patients receiving these therapies.
Dead or dying neurons cannot be returned to function, and many in the AD field currently believe that stemming the progression of the disease may only be possible with very early stage intervention. The FDA is encouraging the pharmaceutical industry to increase efforts to investigate such early stage interventional treatments by recommending that modified clinical endpoints, both functional and cognitive, be established to monitor the efficacy of drug prototypes being tested in early stage AD patients, according to an article published in The New England Journal of Medicine5.
3 BCC Research Report PHM062A Alzheimer’s Disease Therapeutics and Diagnostics: Global Markets, January 2010. Available at http://www.bccresearch.com/market-research/pharmaceuticals/alzheimers-disease-therapeutics-phm062a.html.
4 See footnote 1.
5 NEJM.org: The New England Journal of Medicine, March 15, 2013, page 1: Drug Development of Early Alzheimer’s Disease, N. Kozauer, M.D., and Russell Katz, M.D.
In contrast, we believe that our data from various preclinical animal models demonstrates that activation of PKCe in central nervous system neurons improves neuronal vitality and function in areas of the brain damaged by AD, resulting in the improvement of memory and cognition.
Synaptogenesis
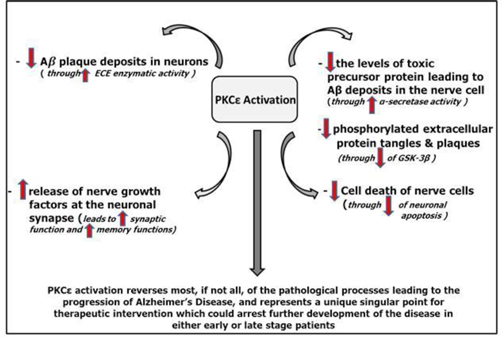
We believe that deficient activity or low concentrations of PKCε in aging subjects is one of the main causes of the neurodegeneration seen in AD. The schematic inFigure 4 illustrates only a portion of the changes mediated by PKCε, and how they may help reverse the neuronal damage and loss central to the pathogenic process in Alzheimer’s Disease.
Figure 4. PKCε Activation Involves 5 Different Mechanisms to Stop the Progression of Alzheimer’s Disease6
Activation of PKCe has been achieved with drug prototypes that mimic the activity of diacylglycerol and phosphatidylserine, which are the natural binding targets for this enzyme. In addition, a variety of in vitro and in vivo animal models have demonstrated that these drug prototypes are effective in restoring the structure and function of neuronal synapses. The first clinical application of the PKCe activators are focused on the treatment of Alzheimer’s Disease, but a number of other neurodegenerative diseases may be amenable to similar treatment. A list of these potential future drug targets is shown inFigure 5.
6 Based on unpublished BRNI research.
Figure 5. Therapeutic targets for neuroregeneration through PKCε activation
Treatment of AD by Stimulating Synaptic Regeneration and Prevention of Neuronal Death
BRNI’s research program in this area lies outside the conventional wisdom that has dominated research efforts in the industry. The pathology of AD likely has multiple layers in its development, and the accumulating presence of tau phosphorylated tangles and Aβ are causative factors in the poisoning of neurons and the resultant cognitive and memory disorders. However, once this process presents clinical manifestations of AD, restoring synaptic function thus far has not been effectively achieved by removing Aβ plaques with experimental drug interventions. Once neurons are poisoned with Aβ, the loss of function to the patient is irreversible.
BRNI’s and our approach is to restore general viability and hence synaptic function in still functioning neurons by stimulating the regeneration and growth of the dendritic branches in these neurons. (Dendrites are the branched projections of a neuron that act to propagate the electrochemical stimulation received from other neural cells.) This process can be visualized at the microscopic level in the neuronal cells of rats whose neurons have been damaged by ischemic shock (depriving oxygen) or traumatic injury to the brain. The morphology of the damaged neurons in these animal models looks strikingly different after they are treated with experimental drugs that activate PKCe. The new growth of dendritic trees on the damaged neurons creates a multiplicity of new synaptic connections, basically re-wiring the damaged neurons and restoring their function. Earlier therapeutic intervention with a PKCe activator produces better outcomes in tests measuring restored animal cognitive function.
PKCε Activation Stimulates the Formation of New Synaptic Connections
The new synaptic connections formed from the damaged neurons in rats can be demonstrated in various behavioral models for the animals that are used to measure memory functions. Treatment with bryostatin, for 12 weeks in genetically modified rodents pre-disposed to develop an AD-type of pathology showed that bryostatin promoted the growth of new synapses and preserved the existing synapses. In addition, these drugs also stopped the decrease of PKCeand the increase of soluble amyloid7.
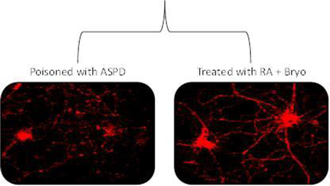
In cell tissue cultures, the difference in morphology between neurons damaged by the application of ASPD (a modified form of Aβ) as compared to neurons activated by the application of bryostatin + retinoic acid (a metabolite of vitamin A) is seen inFigure 6. Treatment with bryostatin, through PKCe activation, stimulates the regeneration of neurons and the formation of new synaptic connections.
7 Journal of Neuroscience 2011, 31 (2), 630, D. Alkon et al.
Figure 6. Synaptogenesis in Hippocampus Neurons8
The Central Role of PKCε in Maintaining Neuron Structure and Function
Upon activation, PKCe migrates from the intracellular fluid to the cell membrane, where it activates signal-regulating enzymes (specifically the MAP kinases Erk1/2 and NF-κβ), causing a series of changes leading to increased DNA transcription, synaptic maturation, a consequent increase in levels of growth factor proteins (such as nerve growth factor and brain-derived neurotrophic factor), an inhibition of programmed cell-death and a reduction ofβ amyloid.
This myriad of events is orchestrated by PKCe, and prompts a number of secondary events occurring in both the pre- and post-synaptic portions of the neuron. Cellular visualization of this effect shows an increase in the number of pre-synaptic vesicles in the neurons, an increase in pre-synaptic levels of PKCe and an increase in the number of mushroom spines associated with individual synaptic boutons (knoblike enlargements at the end of a nerve fiber, where it forms a synapse), which spines may be important in memory. Their genesis in these neurons is responsible for the formation of new synapses.
The central role of PKCe activation in these dynamic events does not contradict the amyloid hypotheses for AD, but offers an alternative target for therapeutic intervention which could prevent the formation of tangles and plaque.
Decreased amyloid formation from PKCe activation results from an increase in the rate of Aβ degradation by ECE (endothelin converting enzyme) and induction ofα secretase cleavage of amyloid precursor protein (the precursor molecule to Aβ) through phosphorylation of an enzyme known as Erk. In rodent models genetically predisposed to forming large amounts of amyloid deposits in their brains, PKCe activation was found to interrupt the ongoing formation of amyloid, suggesting that this approach may delay the progression of AD.
The key to BRNI’s innovation in this area has been in identifying highly potent drug prototypes that at low concentrations cause the specific and transient activation of PKCe, without interacting with the other isozyme variants of PKC whose inactivation would negate the synaptogenic properties of thee isoform.
Testing PKCe Activation in Humans
The basic drug mechanism invoking PKCe activation for neuronal regeneration has never been evaluated in man for any drug class or therapeutic application. We believe that the research in this field as described above is an ideal platform for testing this approach in human subjects.
We have licensed a body of biomedical research comprised of new methods and drug prototypes designed to stimulate neuronal regeneration. For additional information, see “Business—Intellectual Property—Technology License and Services Agreement.” We believe the commercial application of this technology has potential to impact Alzheimer’s Disease as well as traumatic brain injury, ischemic stroke, post-traumatic stress syndrome and learning disorders.
8 Based on unpublished BRNI research.
Drug Prototypes That Treat AD through Regeneration
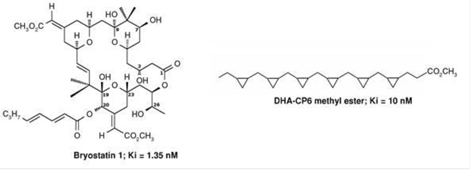
BRNI has developed a new chemical family of polyunsaturated fatty acid (PUFA) analogs, which appear to be effective in the activation of PKCe. Representative structures of bryostatin and a lead PUFA analog are shown inFigure 7.
Figure 7.Structures of Bryostatin 1 and a PUFA Analog Effective in the Activation of PKCe9
Ki values = effective concentration of the drug in achieving 50% activation of PKCε
These molecules activate PKCe by binding to two different and distinct active sites on the enzyme. The natural ligands that bind to these sites and act as activators for PKCe are diacylglycerol and phosphatidylserine. Bryostatin acts as a mimetic (mimic) for diacylglycerol by binding to the diacylglycerol site and, similarly, the PUFA analogs act as mimetics for phosphatidylserine by binding to the phosphatidylserine site. Both chemical families show a high level of specificity in activating PKCe.
Part of the hierarchal array of in vitro and in vivo tests useful in optimizing the potency of our potential drug prototypes is displayed inFigure 8.
9 Trends in Biochemical Sciences V. 34, #3, p.136. T.J. Nelson et al, “ Neuroprotective versus Tumorigenic protein kinase C activators”
Figure 8. Optimization of PKCe Activation Effects in Lead Drug Candidates: Array of in vitro and in vivo Test Models10
Bryostatin
The lead product in our armamentarium is bryostatin. Bryostatin is a natural product isolated from a marine organism. Bryostatin is a PKCα ande activator that was originally developed as a potential anticancer drug. According to Clinical Cancer Research, this drug candidate was previously evaluated in 63 clinical studies involving more than 1,200 patients at the National Cancer Institute (the “NCI”) for the treatment of various forms of cancer. While having failed these studies as an experimental anti-cancer therapy, much useful information on the safety, pharmacodynamics and toxicity of the drug was obtained from these in-human trials.
It was discovered that at a much lower dose than what was used in these anticancer trials, bryostatin is a potent activator of PKCe and may have efficacy in treating AD. As described above, activation of PKCe has now been shown to partially restore synaptic function in neurons damaged by AD in in vitro and in vivo animal models.
The National Cancer Institute has entered into a material transfer agreement with BRNI to provide the bryostatin required for pre-clinical research as well as the Phase 2 clinical trials planned by the Company. The clinical material transfer agreement specifies that BRNI retains all of the bryostatin intellectual property. Our license agreement with BRNI (see “Business—Intellectual Property—Technology License and Services Agreement”) permits our access to new bryostatin clinical trial data and information held by the National Cancer Institute, as well as past clinical, safety and toxicity data compiled by the National Cancer Institute during the time this drug was being evaluated for its anticancer properties. See Item 1A, “Risk Factors—We are dependent upon the National Cancer Institute to supply bryostatin for our clinical trials.”
BRNI has received FDA approval to conduct an exploratory evaluation of bryostatin on a compassionate use basis in AD patients who have an inherited form of AD, frequently called familial Alzheimer’s Disease. Familial Alzheimer’s Disease results from one of four major mutations in the genome, and this mutation is passed on from generation to generation within a family that carries the defective gene. The tragic consequence of familial Alzheimer’s Disease is that it strikes its victims at an early age, often while they are in their twenties. The aggressive progression of familial Alzheimer’s Disease can render these patients in the terminal stages of Alzheimer’s Disease in their late 30s and early 40s.
10 Based on unpublished BRNI research.
We intend to further the clinical study in Familial Alzheimer’s Disease initiated in 2013. We also plan to initiate a clinical trial in up to 15 AD patients to assess the safety and tolerability of the drug candidate in 2014. This clinical trial will also provide us the opportunity to assess the efficacy of bryostatin on improving the cognition of these patients.
As of March 12, 2014, we entered into a statement of work (“SOW”) with BRNI to continue pre-clinical activities relating to the commercialization of the Company’s therapeutic product. The Company is obligated to pay BRNI a total of $465,000 (subject to a 20% cost overage, which may not be exceeded without our consent). Of this amount, the Company has paid $358,470, and the remainder of the total ($106,530, subject to a 20% cost overage, which may not be exceeded without our consent) is to be paid upon the completion by BRNI of certain activities relating to: transferring test materials; bio-analytical testing; contracting with a suitable contract research organization; completion of testing assays; and finalizing a clinical study protocol.
PUFA Analogs
Several other drug prototypes termed the PUFA analogs have been synthesized at BRNI and evaluated for their PKCe activating properties in models of AD. The PUFA analogs are not structurally related to bryostatin and activate PKCe at a different site. We believe the PUFA analogs represent a potential source for follow-on drug candidates. PKCe activators from the PUFA family of drug prototypes have demonstrated neuroregeneration efficacy roughly equivalent to that of bryostatin. If the PUFA analogs show adequate potency in preclinical models of AD, we would plan to advance a drug prototype from this chemical family into clinical testing in the 2016 timeframe.
Diagnostic Test for AD
If accurate biomarkers are established to allow clinicians to make an early diagnosis of AD and a determination of its severity, drug treatment of the disease could start earlier and perhaps delay its progression and end-stage consequences.
The definitive determination of AD in patients is currently achieved only after death upon autopsy; clinicians are unable to definitively diagnose the disease in living patients. BRNI’s research on the role of PKCe in neuroregeneration has allowed it to develop different peripheral biomarkers that have a high correlation level with the presence of AD. BRNI has developed three peripheral biomarkers whose expression is mediated by PKCe.
Using fibroblasts (a type of cell) obtained from a small skin biopsy, a series of in vitro tests are used to amplify the expression of these biomarkers, which we believe can be used to detect AD. The skin fibroblast displays the same basic pathophysiology indicative of AD as a neuron in the central nervous system. At present, there have been four studies of our diagnostic test involving a total of 174 patients, which have analyzed AD patients, patients with mixed dementia (AD plus other dementia), patients with non-AD dementia and normal (or “control”) patients.
Effective August 28, 2013, the Company signed an SOW with BRNI pursuant to its Technology License and Services Agreement, whereby the Company has contracted for the further development of its AD diagnostic product. The project is intended to validate each of three biomarkers in a heterogeneous patient population to determine sensitivity and selectivity parameters for each biomarker, or combination of biomarkers, to detect Alzheimer’s Disease. The three biomarkers to be evaluated are: the PKCe levels, the Erk1/2 ratios, and the fibroblast morphology test. Pursuant to the SOW, the Company is obligated to pay BRNI a total of $1,645,470 in twelve equal monthly installments of $137,123, payable on the first business day of each month commencing September 2013. These payments are for operating expenses associated with BRNI’s diagnostic laboratories. Operating expenses that are incurred in excess of this total amount are the responsibility of BRNI unless prior approval is obtained from the Company. The SOW may be extended if BRNI provides the Company with two months advanced notice that the SOW objectives are not met within the initial twelve month period.
Since licensing the AD diagnostic technology from BRNI, we have conducted extensive analysis of the underlying technology and commercial sales potential of an AD diagnostic product. Based upon our analyses to date and our current resources, our Board of Directors may decide that allocation of future expenditures relating to the AD diagnostic product (beyond completion of our existing statement of work with BRNI for development of the diagnostic; see “Diagnostic Test for AD” below) may be better spent on development of bryostatin in the treatment of AD. As such, we are continuing to evaluate our original timeline for commercialization of the diagnostic product to insure the budget to develop bryostatin for the treatment of AD is fully funded over next 24 months. This on-going process could result in a delay of development of the diagnostic product.
If we continue development of our diagnostic product, we will actively seek a marketing partner to assist both in the promotion of the diagnostic test and to provide laboratory support to process tests that exceed BRNI’s laboratory capacity.
Other Potential Products
We may acquire, by license or otherwise, other development stage products that are consistent with our product portfolio objectives and commercialization strategy.
Discontinued Research
We had planned to develop two other lines of research related to learning and memory disorders: (i) drug prototypes that activate or inhibit the enzyme carbonic anhydrase to modulate the attention status of animals, which may have had applications for attention deficit disorder (ADD) and post-traumatic stress disorder (PTSD), and (ii) generalizing the application of a blood-brain-barrier delivery system to a variety of drugs through a contract research service to be offered to other pharmaceutical companies seeking to improve the penetration of their drug prototypes into the brain.
We have decided, however, to focus our efforts on neurodegenerative diseases and the Alzheimer’s diagnostic, which are the most advanced programs in our portfolio, and therefore will not be pursuing either the drug candidate for activating carbonic anhydrase or the blood-brain-barrier delivery system.
Corporate Development Plan
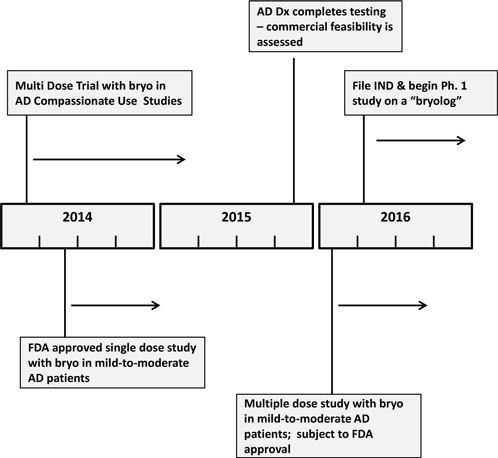
Our priorities over the next 24 months are focused on achieving the following milestone events:
| · | The first of these milestone events commences with our FDA single dose clinical trial with bryostatin in AD patients. The results of this study are expected to be reported by the fourth quarter of 2014. |
| · | BRNI received approval from the FDA to treat a second AD patient using a multiple dose protocol with bryostatin on a compassionate use basis. We expect the initial results from this protocol to be available by the end of the fourth quarter of 2015. |
| · | Assuming our Board decides to continue development of our in vitro diagnostic test system for the detection of Alzheimer’s Disease, we would plan to finish the testing phase of that development in 2015, whereupon we will evaluate the feasibility of its commercialization. |
Our operating plan for attaining these objectives is illustrated inFigure 9.
Figure 9. Operating Plan for the next 24 Months
We have engaged a contract research organization to help plan, execute and analyze our first clinical trial in AD. We plan to seek a “fast track” clinical development plan, and if the Phase 2 clinical development program for bryostatin is a success, we believe this product could possibly obtain accelerated FDA approval. However, there can be no assurance that the FDA will grant fast track approval. See “Governmental Regulation and Product Approval—United States Regulation—Fast Track Approval” below. If we do not receive fast track approval, then our operating plan for the development of bryostatin would be delayed.
Intellectual Property
Technology License and Services Agreement
Neurotrope BioScience entered into a Technology License and Services Agreement with BRNI and its affiliate NRV II, LLC (as amended, the “License Agreement”). Pursuant to the License Agreement, BRNI and NRV II, LLC granted to Neurotrope BioScience an exclusive, non-transferable, world-wide, royalty-bearing license to certain patents owned by BRNI or licensed to NRV II, LLC by BRNI as of or subsequent to October 31, 2012 to develop, use, manufacture, market and sell products or services for therapeutic or diagnostic applications for Alzheimer’s Disease or other cognitive dysfunctions. There are certain exceptions to the exclusivity of the license, including for BRNI and its affiliates to use the licensed intellectual property to engage in research and development and other non-commercial activities and to provide services to us or to perform other activities in connection with the License Agreement, and with respect to intellectual property acquired by BRNI or NRV II, LLC that would be subject to the License Agreement but is subject to a license existing as of the date of acquisition. Pursuant to the License Agreement, BRNI has the exclusive right (but not the obligation) to apply for, file, prosecute or maintain patents and patent applications for the technologies licensed to us. However, in order to maintain our rights to use the licensed technologies, we must reimburse BRNI for all of the attorney’s fees and other costs and expenses related to any of the foregoing.
We are required to pay to BRNI a royalty in the amount of 5% of our revenues in connection with the licensed technology depending upon the percentage of our ownership held by Neuroscience Research Ventures, Inc., which is an affiliate of BRNI (the amount of the royalty generally increases as the percentage ownership by Neuroscience Research Ventures, Inc. decreases). Within 30 days after our receipt of any amount of capital raised in a financing prior to a public offering, we are required to pay to BRNI 5% of such amount as an advance payment of future royalties. In addition, upon the February 28, 2013 closing on the sale of Series A Preferred Stock, we were required to pay BRNI a fee in the amount of approximately $1.6 million.
The License Agreement further requires us to pay BRNI (i) a fixed research fee equal to a pro-rata amount of $1 million in the year during which the we close on a $25 million round of financing, (ii) a fixed research fee of $1 million per year for each of the five calendar years following the completion of such financing and (iii) an annual fixed research fee in an amount to be negotiated and agreed upon no later than 90 days prior to the end of the fifth calendar year following the completion of such financing to be paid with respect to each remaining calendar year during the term of the License Agreement.
The term of the License Agreement continues until the later of the date (i) the last of the licensed patents expires, is abandoned or is declared unenforceable or invalid (in each case, determined in accordance with the License Agreement) and (ii) the last of the licensed technology enters the public domain. BRNI has the right to terminate the License Agreement after 30 days prior notice in certain circumstances, including if we were to materially breach any provisions of the License Agreement after a 60-day cure period for breaches that are capable of being cured, in the event of certain bankruptcy or insolvency proceedings or in the event of the termination of that certain Stockholders Agreement dated October 31, 2012, with respect to us. (See Item 13, “Certain Relationships and Related Transactions, and Director Independence—Common Stockholders Agreement: for more information about this agreement.)
Our Licensed Intellectual Property
We have licensed from BRNI an extensive intellectual property portfolio that includes issued patents, pending patents and provisional patent filings, in the U.S. and elsewhere, which, we believe, together cover the world’s key pharmaceutical markets. Composition of matter and method of use patents have been issued to BRNI that cover the use of the PUFA family of molecules for the same therapeutic applications.
We believe the License Agreement provides the Company rights to the patents and technologies required to develop its proposed products. The patents and technologies licensed to the Company pursuant to the License Agreement include, without limitation, the following:
| | · | drug prototypes composed of the bryostatin and PUFA chemical families; and |
| | | |
| | · | an in vitro diagnostic system to detect AD. |
A number of BRNI’s patents for treatment of neurological disorders have been under active prosecution for many years and have been the subject of multiple rejections for anticipation and/or obviousness based on prior art. There are no guarantees that BRNI’s pending patent applications will issue into commercially meaningful patents. The terms of the License Agreement do not permit Neurotrope to participate in the prosecution of these patents.If these patent applications are not approved or successfully prosecuted, then the Company will attempt to seek other means of protecting its proprietary position including, but not limited to, trade secrets, proprietary formulations and methods, etc.
A substantial amount of in-human data exists that was generated by the National Cancer Institute that involves the earlier evaluation of bryostatin as an anticancer agent. The National Cancer Institute also holds the existing inventory of the bryostatin drug product which is suitable for use in man. Our use of the substantial data package generated by the National Cancer Institute on bryostatin, as well as access to the clinical supply of this substance, is permitted under a material transfer agreements entered into and between the National Cancer Institute and BRNI.
There are no known patent conflicts or freedom to operate issues at this time which could encumber our ability to commercialize either the AD diagnostic system or the PKCe activators for the treatment of cognition and memory disorders. However, we cannot provide any assurance that such conflicts will not arise in the future. See the risk factors captioned “Our commercial success will depend, in part, on our ability, and the ability of our licensors, to obtain and maintain patent protection. Our licensors’ failure to obtain and maintain patent protection for our products may have a material adverse effect on our business.” and “Our licensed patented technologies may infringe on other patents, which may expose us to costly litigation.” under “Risk Factors—Risks Related to our Business.”
Governmental Regulation and Product Approval
The manufacturing and marketing of our potential products and our ongoing research and development activities are subject to extensive regulation by the FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries.
United States Regulation
Before any of our products can be marketed in the United States, they must receive approval from the FDA. To receive this approval, any drug we develop must undergo rigorous preclinical testing and clinical trials that demonstrate the product candidate’s safety and effectiveness for each indicated use. This extensive regulatory process controls, among other things, the development, testing, manufacture, safety, efficacy, record keeping, labeling, storage, approval, advertising, promotion, sale, and distribution of pharmaceutical products.
In general, before any new ethical pharmaceutical product can be marketed in the United States, the process typically required by the FDA includes:
| | · | preclinical laboratory and animal tests; |
| | · | submission of an investigational new drug application, or IND, which must become effective before human clinical trials may begin; |
| | · | adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use; |
| | · | pre-approval inspection of manufacturing facilities and selected clinical investigators; |
| | · | Submission of a New Drug Application, or NDA, to the FDA; and |
| | · | FDA approval of an NDA, or an NDA supplement (for subsequent indications or other modifications, including a change in location of the manufacturing facility). |
Preclinical Testing
In the United States, drug candidates are tested in animals until adequate proof of safety and efficacy is established. These preclinical studies generally evaluate the mechanism of action and pharmacology of the product and assess the potential safety and efficacy of the product. Tested compounds must be produced according to applicable current good manufacturing practice requirements and preclinical safety tests must be conducted in compliance with FDA and international regulations regarding good laboratory practices. The results of the preclinical tests, together with manufacturing information and analytical data, are generally submitted to the FDA as part of an IND, which must become effective before human clinical trials may commence. The IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA requests an extension or raises concerns about the conduct of the clinical trials as outlined in the application. If the FDA has any concerns, the sponsor of the application and the FDA must resolve the concerns before clinical trials can begin. Regulatory authorities may require additional preclinical data before allowing the clinical studies to commence or proceed from one phase to another, and could demand that the studies be discontinued or suspended at any time if there are significant safety issues. Furthermore, an independent institutional review board for each medical center proposing to participate in the conduct of the clinical trial, must review and approve the clinical protocol and patient informed consent form before commencement of the study at the respective medical center.
In March 2014 we were made aware that the NCI withdrew its IND supporting the use of bryostatin for cancer treatment in 2011 because it did not intend to pursue additional studies under the IND. BRNI's IND supporting the use of bryostatin for treatment of patients with Alzheimer's Disease cross-references the NCI IND for certain information, including manufacturing and controls information, nonclinical toxicology studies and clinical safety data. BRNI is currently in discussions with both the NCI and the FDA to confirm which, if any, NCI information should be resubmitted to the FDA under BRNI’s IND, and a near-term resolution of this issue is expected to be achieved in the coming months. We are dependent on both BRNI's management of its IND and its regulatory dialogue with the FDA. There can be no assurance that the FDA will consider BRNI's submission adequate to replace all cross references to the withdrawn NCI IND or that the resolution will be achieved in a timely fashion so as not to negatively impact the development schedule for our bryostatin therapeutic described herein.
Clinical Trials
Clinical trials for new drug candidates are typically conducted in three sequential phases that may overlap. In phase 1, the initial introduction of the drug candidate into healthy human volunteers, the emphasis is on testing for safety or adverse effects, dosage, tolerance, metabolism, distribution, excretion, and clinical pharmacology. Phase 2 involves studies in a limited patient population to determine the initial efficacy of the drug candidate for specific targeted indications, to determine dosage tolerance and optimal dosage and to identify possible adverse side effects and safety risks. Once a compound shows evidence of activity and is found to have an acceptable safety profile in phase 2 evaluations, pivotal phase 3 trials are undertaken to more fully evaluate clinical outcomes and to establish the overall risk/benefit profile of the drug, and to provide, if appropriate, an adequate basis for product labeling. During all clinical trials, physicians will monitor patients to determine effectiveness of the drug candidate and to observe and report any reactions or safety risks that may result from use of the drug candidate. The FDA, the trial sites internal review board or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk.
The data from the clinical trials, together with preclinical data and other supporting information that establishes a drug candidate’s profile, are submitted to the FDA in the form of a New Drug Application (“NDA”) or NDA supplement (for approval of a new indication if the product candidate is already approved for another indication). Under applicable laws and FDA regulations, each NDA submitted for FDA approval is usually given an internal administrative review within 45 to 60 days following submission of the NDA. If deemed complete, the FDA will “file” the NDA, thereby triggering substantive review of the application. The FDA can refuse to file any NDA that it deems incomplete or not properly reviewable. The FDA has established internal substantive review goals of six months for priority NDAs (for drugs addressing serious or life threatening conditions for which there is an unmet medical need) and ten months for regular NDAs. The FDA, however, is not legally required to complete its review within these periods, and these performance goals may change over time. Moreover, the outcome of the review, even if generally favorable, is not typically an actual approval, but an “action letter” that describes additional work that must be done before the NDA can be approved. The FDA’s review of a NDA may involve review and recommendations by an independent FDA advisory committee. The FDA may deny approval of an NDA or an NDA supplement if the applicable regulatory criteria are not satisfied, or it may require additional clinical data and/or an additional pivotal phase 3 clinical trial. Even if such data are submitted, the FDA may ultimately decide that the NDA or NDA supplement does not satisfy the criteria for approval.
Data Review and Approval
Substantial financial resources are necessary to fund the research, clinical trials and related activities necessary to satisfy FDA requirements or similar requirements of state, local and foreign regulatory agencies. It normally takes many years to satisfy these various regulatory requirements, assuming they are satisfied. Information generated in this process is susceptible to varying interpretations that could delay, limit, or prevent regulatory approval at any stage of the process. Accordingly, the actual time and expense required to bring a product to market may vary substantially. We cannot assure you that we will submit applications for required authorizations to manufacture and/or market potential products or that any such application will be reviewed and approved by the appropriate regulatory authorities in a timely manner, if at all. Data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. Success in early stage clinical trials does not ensure success in later stage clinical trials. Even if a product candidate receives regulatory approval, the approval may be significantly limited to specific disease states, patient populations and dosages, or have conditions placed on them that restrict the commercial applications, advertising, promotion or distribution of these products.
Once issued, the FDA may withdraw product approval if ongoing regulatory standards are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved products which have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs. The FDA may also request additional clinical trials after a product is approved. These so-called phase 4 studies may be made a condition to be satisfied after a drug receives approval. The results of phase 4 studies can confirm the effectiveness of a product candidate and can provide important safety information via the FDA’s voluntary adverse drug reaction reporting system. Any products manufactured or distributed by us pursuant to FDA approvals would be subject to continuing regulation by the FDA, including record-keeping requirements and reporting of adverse experiences with the drug. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with good manufacturing practices, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. We cannot be certain that we or our present or future suppliers will be able to comply with the good manufacturing practices regulations and other FDA regulatory requirements. If our present or future suppliers are not able to comply with these requirements, the FDA may halt our clinical trials, require us to recall a drug from distribution or withdraw approval of the NDA for that drug. Furthermore, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market.
The FDA closely regulates the marketing and promotion of drugs. Approval may be subject to post-marketing surveillance and other record keeping and reporting obligations, and involve ongoing requirements. Product approvals may be withdrawn if compliance with regulatory standards is not maintained or if problems occur following initial marketing. A company can make only those claims relating to safety and efficacy that are approved by the FDA. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available drugs for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturers’ communications on the subject of such off-label use.
Fast Track Approval
The Federal Food, Drug, and Cosmetic Act, as amended, and FDA regulations provide certain mechanisms for the accelerated “Fast Track” approval of potential products intended to treat serious or life-threatening illnesses which have demonstrated the potential to address unmet medical needs. The procedures permit early consultation and commitment from the FDA regarding the preclinical and clinical studies necessary to gain marketing approval. Provisions of this regulatory framework also permit, in certain cases, NDAs to be approved on the basis of valid indirect measurements of benefit of product effectiveness, thus accelerating the normal approval process. In the future, certain potential products employing our technology might qualify for this accelerated regulatory procedure. Even if the FDA agrees that these potential products qualify for accelerated approval procedures, the FDA may deny approval of our drugs or may require additional studies before approval. The FDA may also require us to perform post-approval, or phase 4, studies as a condition of such early approval. In addition, the FDA may impose restrictions on distribution and/or promotion in connection with any accelerated approval, and may withdraw approval if post-approval studies do not confirm the intended clinical benefit or safety of the potential product.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States. Orphan drug designation must be requested before submitting a NDA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process. If a product that has orphan drug designation subsequently receives FDA approval for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same disease, except in very limited circumstances, for seven years. These very limited circumstances are (i) an inability to supply the drug in sufficient quantities or (ii) a situation in which a new formulation of the drug has shown superior safety or efficacy. This exclusivity, however, also could block the approval of our product for seven years if a competitor obtains earlier approval of the same drug for the same indication.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products in foreign countries. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Under European Union regulatory systems, we may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure, which is available for medicines produced by biotechnology or which are highly innovative, provides for the grant of a single marketing authorization that is valid for all EU member states. This authorization is a marketing authorization application. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessment report, each member state must decide whether to recognize approval. This procedure is referred to as the mutual recognition procedure.
The policies of the FDA and foreign regulatory authorities may change and additional government regulations may be enacted which could prevent or delay regulatory approval of our products and could also increase the cost of regulatory compliance. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the United States or abroad.
Regulation of Diagnostic Tests
Our diagnostic test must be offered in a manner than complies with the regulatory framework developed by the FDA and the Centers for Medicare and Medicaid Services (CMS) with respect to diagnostic tests. See the risk factors below captioned“If we are unable to engage a lab certified under CLIA to process our diagnostic test at its facilities, the commercialization of our diagnostic test may be unsuccessful” and“If the FDA were to begin requiring approval or clearance of our tests, we could incur substantial costs and time delays associated with meeting requirements for pre-market clearance or approval.”
Other Government Regulation
Our research and development activities use biological and hazardous materials that are dangerous to human health and safety or the environment. We are subject to a variety of federal, state and local laws and regulations governing the use, generation, manufacture, storage, handling and disposal of these materials and wastes resulting from these materials. We are also subject to regulation by the Occupational Safety and Health Administration and federal and state environmental protection agencies and to regulation under the Toxic Substances Control Act.
In addition, once our products are marketed commercially, we will have to comply with the various laws relating to the Medicare, Medicaid and other federal healthcare programs. These federal laws include, by way of example, the following:
| | · | The anti-kickback statute (Section 1128B(b) of the Social Security Act) prohibits certain business practices and relationships that might affect the provision and cost of healthcare services reimbursable under Medicare, Medicaid and other federal healthcare programs, including the payment or receipt of remuneration for the referral of patients whose care will be paid by Medicare or other governmental programs; |
| | · | The physician self-referral prohibition (Ethics in Patient Referral Act of 1989, as amended, commonly referred to as the Stark Law, Section 1877 of the Social Security Act), which prohibits referrals by physicians of Medicare or Medicaid patients to providers of a broad range of designated healthcare services in which the physicians (or their immediate family members) have ownership interests or with which they have certain other financial arrangements; |
| | · | The anti-inducement law (Section 1128A(a)(5) of the Social Security Act), which prohibits providers from offering anything to a Medicare or Medicaid beneficiary to induce that beneficiary to use items or services covered by either program; |
| | · | The False Claims Act (31 U.S.C. § 3729 et seq.), which prohibits any person from knowingly presenting or causing to be presented false or fraudulent claims for payment to the federal government (including the Medicare and Medicaid programs); |
| | · | The Civil Monetary Penalties Law (Section 1128A of the Social Security Act), which authorizes the United States Department of Health and Human Services to impose civil penalties administratively for fraudulent or abusive acts. |
| | | |
| | · | The Physician Payment Sunshine Act (Section 1128G of the Social Security Act), which requires manufacturers of drugs, medical devices and biologicals that participate in U.S. federal health care programs to report certain payments and items of value given to physicians and teaching hospitals. |
Sanctions for violating these federal laws include criminal and civil penalties that range from punitive sanctions, damage assessments, money penalties, imprisonment, denial of Medicare and Medicaid payments, or exclusion from the Medicare and Medicaid programs, or both. These laws also impose an affirmative duty on those receiving Medicare or Medicaid funding to ensure that they do not employ or contract with persons excluded from the Medicare and other government programs.
Competition
We compete with many companies, research institutes, hospitals, governments and universities that are working to develop products and processes to treat or diagnose Alzheimer’s Disease. Many of these entities have substantially greater financial, technical, manufacturing, marketing, distribution and other resources than we do. However, there has been a dearth of new product introductions in the last 20 years either for the treatment of AD symptoms or its definitive diagnosis in patients who begin exhibiting the memory and cognitive disorders associated with the disease. All of the products introduced to date for the treatment of AD have yielded negative or marginal results with no effect on the progression of AD and no improvement in the memory or cognitive performance of the patients receiving these therapies. We believe that there is currently no diagnostic test for AD that has achieved significant market penetration, and thus the absolute determination of AD in patients is currently achieved only upon autopsy. We believe we are the only company currently pursuing PKCε activation as a mechanism to treat AD and neurodegenerative disease. Although we believe that we have no direct competitors working in this same field at the present time, we cannot provide assurance that our competitors will not discover compounds or processes that may be competitive with our products and introduce such products or processes before us.
Employees
As of the date of this report, the Company has six full-time employees, our Chief Executive Officer and Chief Financial Officer, a Vice President—Chief Medical Officer, a Vice President—Commercial Operations, a Vice President—Regulatory Affairs, a Manager of Clinical Operations and we have no part-time employees.
We plan to hire an additional approximately six full-time employees within the next twelve months whose principal responsibilities will be the support of our clinical development activities.
An investment in our securities involves a high degree of risk. We face a variety of risks that may affect our operations or financial results and many of those risks are driven by factors that we cannot control or predict. Before investing in our securities you should carefully consider the following risks, together with the financial and other information contained in this report. If any of the following risks actually occurs, our business, prospects, financial condition and results of operations could be materially adversely affected. In that case, the trading price of our common stock would likely decline and you may lose all or a part of your investment. Only those investors who can bear the risk of loss of their entire investment should invest in our securities.
Risks Related to Our Business and Financial Condition
Our ongoing viability as a company depends on our ability to successfully develop and commercialize bryostatin and our diagnostic test for the detection of Alzheimer’s Disease.
We are principally focused on developing two product platforms, a drug, bryostatin, for the treatment of Alzheimer’s Disease and a diagnostic test for the detection of Alzheimer’s Disease, both of which are still in the clinical testing stage and have not yet been fully developed. Our potential success is highly uncertain since both of our principal product candidates are in the development stage and are subject to regulatory approval. Our potential success depends upon our ability to complete development and successfully commercialize in a timely manner bryostatin for the treatment of Alzheimer’s Disease and the diagnostic test for Alzheimer’s Disease. We must develop bryostatin, test for efficacy in the targeted patient population and manufacture the finished dosage form of bryostatin on a commercial scale to meet regulatory standards and receive regulatory approvals. The development and commercialization process is both time-consuming and costly, and involves a high degree of business risk. Bryostatin is still at an early stage in its product development cycle, and our follow-on products are still at the concept stage. In order to make our diagnostic test system for Alzheimer’s Disease commercially available, we must make investments to upgrade BRNI’s laboratory facilities or contract with a third-party lab for the processing of the test. The results of clinical testing of our products are uncertain and we cannot assure that we will be able to obtain regulatory approvals of our products. If obtained, regulatory approvals may take longer or be more expensive than anticipated. Furthermore, even if regulatory approvals are obtained, our products may not perform as we expect and we may not be able to successfully and profitably produce and market our products. Delays in any part of the process or our inability to obtain regulatory approval of our products could adversely affect our future operating results by restricting (or even prohibiting) the introduction and sale of our products.
We are a development stage company and have a limited operating history upon which investors can evaluate our future prospects. For that reason, it is difficult to judge our prospects.
We are in the early development stage and we are subject to all of the risks inherent in the establishment of a new business enterprise. While development of our products was started in 1999 by BRNI, we incorporated our business on October 31, 2012 and on that same date entered into an exclusive license agreement for the continuing development and commercialization of our products, and therefore have a limited operating history. Our proposed products are currently in the research and development stage and we have not generated any revenues, nor do we expect our products to generate revenues for at least the next 24 to 36 months, if ever. As a result, the Company must be evaluated in light of the potential problems, delays, uncertainties and complications encountered in connection with a newly established pharmaceutical development business. The risks include, but are not limited to, the possibilities that any or all of our potential products will be found to be ineffective or, that the products once developed, although effective, are not economical to market; that our competitors hold proprietary rights that preclude us from marketing such products; that our competitors market a superior or equivalent product; or the failure to receive necessary regulatory clearances for our proposed products. To achieve profitable operations, we must successfully develop, obtain regulatory approval for, introduce and successfully market at a profit products that are currently in the research and development phase. Much of the clinical development work and testing for our product candidates remains to be completed. No assurance can be given that our research and development efforts will be successful, that required regulatory approvals will be obtained, that any of our products will be safe and effective, that any products, if developed and introduced, will be successfully marketed or achieve market acceptance or that products will be marketed at prices necessary to generate profits. Failure to successfully develop, obtain regulatory approvals for, or introduce and market our products would have material adverse effects on our business prospects, financial condition and results of operations.
We currently rely on BRNI, and may also rely on other independent third-party contract research organizations, to perform clinical and non-clinical studies of our drug candidate and diagnostic test and to perform other research and development services.
Our license and services agreement with BRNI requires the Company to use BRNI to provide research and development services or other scientific assistance and support services, including clinical trials, under certain conditions. The BRNI license could limit the Company’s ability to make certain decisions without BRNI’s consent. See “Business—Intellectual Property—Technology License and Services Agreement.” Under certain conditions, we may, however, also rely on independent third-party contract research organizations to perform clinical and non-clinical studies of our drug candidate and diagnostic test (each such third-party contract research organization, along with BRNI, a “CRO”). Many important aspects of the services that may be performed for us by CROs would be out of our direct control. If there were to be any dispute or disruption in our relationship with such CROs, the development of our diagnostic test and drug candidate may be delayed. Moreover, in our regulatory submissions, we would expect to rely on the quality and validity of the clinical work performed by third-party CROs. If any of our CROs’ processes, methodologies or results were determined to be invalid or inadequate, our own clinical data and results and related regulatory approvals could be materially adversely impacted.
We have relied on the representations and materials provided by BRNI, including scientific, peer-reviewed and non-peer reviewed publications, abstracts, slides, internal documents, verbal communications, patents and related patent filings, with respect to the results of its research related to our proposed products.
BRNI began the development of the intellectual property that forms the basis for our proposed products in 1999. We have relied on the quality and validity of the research results obtained by BRNI with respect to this intellectual property, and we have conducted limited verification of the raw preclinical and clinical data produced by BRNI. If any of BRNI’s basic processes, methodologies or results were determined to be invalid or inadequate, our own clinical data and results and related regulatory approvals, could be materially adversely impacted.
If we do not obtain the necessary regulatory approvals in the U.S. and/or other countries, we will not be able to sell our drug candidates.
We cannot assure you that we will receive the approvals necessary to commercialize any of our drug candidates or any drug candidates we acquire or develop in the future. We will need approval from the FDA to commercialize our drug candidates in the U.S. and approvals from similar regulatory authorities in foreign jurisdictions to commercialize our drug candidates in those jurisdictions. In order to obtain FDA approval of bryostatin or any other drug candidate for the treatment of Alzheimer’s Disease, we must submit to the FDA a New Drug Application (“NDA”), demonstrating that the drug candidate is safe, pure and potent, or effective for its intended use. This demonstration requires significant research including completion of clinical trials. Satisfaction of the FDA’s regulatory requirements typically takes many years, depends upon the type, complexity and novelty of the drug candidate and requires substantial resources for research, development and testing. We cannot predict whether our clinical trials will demonstrate the safety and efficacy of our drug candidates or if the results of any clinical trials will be sufficient to advance to the next phase of development or for approval from FDA. We also cannot predict whether our research and clinical approaches will result in drugs or therapeutics that the FDA considers safe and effective for the proposed indications. The FDA has substantial discretion in the drug approval process. The approval process may be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals may: prevent or delay commercialization of, and our ability to derive revenues from, our drug candidates; and diminish any competitive advantages that we may otherwise believe that we hold. Even if we comply with all FDA requests, the FDA may ultimately reject one or more of our applications. We may never obtain regulatory clearance for any of our drug candidates. Failure to obtain FDA approval of our drug candidates will leave us without a saleable product and therefore without any source of revenues. In addition, the FDA may require us to conduct additional clinical testing or to perform post-marketing studies, as a condition to granting marketing approval of a drug product. If conditional marketing approval is obtained, the results generated after approval could result in loss of marketing approval, changes in product labeling, and/or new or increased concerns about the side effects or efficacy of a product. The FDA has significant post-market authority, including the explicit authority to require post-market studies and clinical trials, labeling changes based on new safety information and compliance with FDA-approved risk evaluation and mitigation strategies. The FDA’s exercise of its authority has in some cases resulted, and in the future could result, in delays or increased costs during product development, clinical trials and regulatory review, increased costs to comply with additional post-approval regulatory requirements and potential restrictions on sales of approved drugs. In foreign jurisdictions, the regulatory approval processes generally include the same or similar risks as those associated with the FDA approval procedures described above. We cannot assure you that we will receive the approvals necessary to commercialize our drug candidates for sale either within or outside the United States.
In March 2014 we were made aware that the NCI withdrew its IND supporting the use of bryostatin for cancer treatment in 2011 because it did not intend to pursue additional studies under the IND. BRNI's IND supporting the use of bryostatin for treatment of patients with Alzheimer's Disease cross-references the NCI IND for certain information, including manufacturing and controls information, nonclinical toxicology studies and clinical safety data. BRNI is currently in discussions with both the NCI and the FDA to confirm which, if any, NCI information should be resubmitted to the FDA under BRNI’s IND, and a near-term resolution of this issue is expected to be achieved in the coming months. We are dependent on both BRNI's management of its IND and its regulatory dialogue with the FDA. There can be no assurance that the FDA will consider BRNI's submission adequate to replace all cross references to the withdrawn NCI IND or that the resolution will be achieved in a timely fashion so as not to negatively impact the development schedule for our bryostatin therapeutic described herein.
If we are unable to engage a CLIA-certified lab to process our diagnostic test at its facilities, the commercialization of our diagnostic test may be unsuccessful.
Assuming our Board of Directors determines to continue development of our diagnostic test for Alzheimer’s Disease, we plan to offer the test in the United States in a laboratory that has been accredited under the Clinical Laboratory Improvement Amendments of 1988 (“CLIA”) to perform high complexity testing. We believe that if our test is solely available through a CLIA-certified lab, we may market the test as a laboratory developed test (“LDT”). Under current FDA enforcement policies and guidance, LDT’s generally do not require FDA premarket clearance or approval before commercialization, and we plan to market our test on that basis. If we are unable to contract with a CLIA-certified lab for the processing of our diagnostic test, we may not be able to market the test as a LTD, which could result in substantial delay in the commercialization of our diagnostic test.
If the FDA were to begin requiring approval or clearance of our tests, we could incur substantial costs and time delays associated with meeting requirements for pre-market clearance or approval.
Although the FDA maintains that it has authority to regulate the development and use of LDT’s, such as ours, as medical devices, it has not exercised its authority with respect to most LDT’s as a matter of enforcement discretion. The FDA does not generally extend its enforcement discretion to reagents or software provided by third parties and used to perform LDT’s, and therefore these products must typically comply with FDA medical device regulations, which are wide-ranging and govern, among other things: product design and development, product testing, product labeling, product storage, pre-market clearance or approval, advertising and promotion and product sales and distribution.
We believe that our diagnostic test, as utilized in a CLIA-certified laboratory testing, is an LDT. As a result, we believe that pursuant to the FDA’s current policies and guidance the FDA, in its discretion, will not require that we obtain regulatory clearances or approvals for our test. The container we provide for collection and transport of biopsy samples from a pathology laboratory to our clinical reference laboratory may be a medical device subject to FDA regulation but, we believe, will not receive pre-market review by the FDA because we expect the FDA to exercise its discretion not to enforce the requirement. While we believe that we are currently in material compliance with applicable laws and regulations, we cannot assure you that the FDA or other regulatory agencies would agree with our determination, and a determination that we have violated these laws, or a public announcement that we are being investigated for possible violations of these laws, could adversely affect our business prospects, financial condition and results of operations.
Moreover, FDA guidance and policy pertaining to diagnostic testing is continuing to evolve and is subject to ongoing review and revision. A significant change in any of the laws, regulations or policies may require us to change our business model in order to maintain regulatory compliance. At various times since 2006, the FDA has issued guidance documents or announced draft guidance regarding initiatives that may require varying levels of FDA oversight of our test. For example, in June 2010, the FDA announced a public meeting to discuss the agency’s oversight of LDT’s prompted by the increased complexity of LDT’s and their increasingly important role in clinical decision-making and disease management, particularly in the context of personalized medicine. The FDA indicated that it was considering a risk-based application of oversight to LDT’s and that, following public input and discussion, it might issue separate draft guidance on the regulation of LDT’s, which ultimately could require that we seek and obtain either pre-market clearance or approval of LDT’s, depending upon the risk-based approach FDA adopts. The public meeting was held in July 2010 and further public comments were submitted to the FDA through September 2010. The FDA has stated it is continuing to develop draft guidance in this area. Section 1143 of the Food and Drug Administration Safety and Innovation Act, signed by the U.S. President on July 9, 2012, requires the FDA to notify the U.S. Congress at least 60 days prior to issuing a draft or final guidance regulating LDT’s and provide details of the anticipated action.
We cannot provide any assurance that FDA regulation, including pre-market review, will not be required in the future for our tests, whether through additional guidance issued by the FDA, new enforcement policies adopted by the FDA or new legislation enacted by Congress. We believe it is possible that legislation will be enacted into law or guidance could be issued by the FDA which may result in increased regulatory burdens for us to offer our test.
The requirement of pre-market review could negatively affect our business until such review is completed and clearance to market or approval is obtained. The FDA could require that we stop selling our tests pending pre-market clearance or approval. If the FDA allows our test to remain on the market but there is uncertainty about our tests, if they are labeled investigational by the FDA or if labeling claims FDA allows us to make are very limited, orders or reimbursement may decline. The regulatory approval process may involve, among other things, successfully completing additional clinical trials and making a 510(k) submission, or filing a pre-market approval application with the FDA. If the FDA requires pre-market review, our tests may not be cleared or approved on a timely basis, if at all. We may also decide voluntarily to pursue FDA pre-market review of our tests if we determine that doing so would be appropriate.
If we were required to conduct additional clinical trials prior to continuing to offer our test, those trials could lead to delays or failure to obtain necessary regulatory approval, which could cause significant delays in commercializing any future products and harm our ability to generate revenue.
We have not generated any revenues since our inception and we do not expect to generate revenue for the foreseeable future. If we do not generate revenues and achieve profitability, we may have to curtail or cease our development plans and operations.
Our ability to generate revenues depends upon many factors, including our ability to complete development of our proposed products, our ability to obtain necessary regulatory approvals for our proposed products and our ability to successfully commercialize, market and sell our products. We have not generated any revenues since our inception on October 31, 2012. We expect to incur significant operating losses over the next several years. If we do not generate revenues, do not achieve profitability and do not have other sources of financing for our business, we may have to curtail or cease our development plans and operations, which could cause investors to lose the entire amount of their investment.
We will likely need additional financing to continue our operations. If we are unable to obtain additional financing on acceptable terms, we may have to curtail or cease our development plans and operations.
Since our inception on October 31, 2012, we have raised approximately $19,700,000 (net of offering expenses and advanced royalties paid to BRNI) from the sale of shares of our Series A Preferred Stock and, as of the date of this report, we had approximately $13.2 million of available cash. We expect that our operating expenses over the next several years will increase as we expand our product acquisition, facilities, infrastructure and research and development activities. Based upon our projected activities, we believe that our current available cash will be sufficient to support our current operating plan for approximately the next 24 months. However, if our operating plan changes or we incur significant unanticipated expenses, we may require additional capital before this timeframe. Additional funds may be raised through debt financing and/or the issuance of equity securities, there being no assurance that any type of financing on terms acceptable to us will be available or otherwise occur. Debt financing must be repaid regardless of whether we generate revenues or cash flows from operations and may be secured by substantially all of our assets. Any equity financing or debt financing that requires the issuance of warrants or other equity securities to the lender would cause the percentage ownership by our current shareholders to be diluted, which dilution may be substantial. Also, any additional equity securities issued may have rights, preferences or privileges senior to those of existing shareholders. If such financing is not available when required or is not available on acceptable terms, we may be required to reduce or eliminate certain product candidates and development activities related to bryostatin or the PUFA analogs and it may ultimately require us to suspend or cease operations, which could cause investors to lose the entire amount of their investment.
If our License Agreement were terminated, we may be required to cease operations.
Our rights to develop, commercialize and sell certain of our proposed products are, in part, dependent upon our License Agreement with BRNI and its affiliate NRV II, LLC. BRNI has the right to terminate this agreement after 30 days prior notice in certain circumstances, including if we were to materially breach any provisions of the agreement after a 60-day cure period for breaches that are capable of being cured, in the event of certain bankruptcy or insolvency proceedings or in the event of the termination of the Common Stockholders Agreement with respect to the Company. Additionally, this agreement provides that the license may not be assigned, including by means of a change of control of the Company, or sublicensed without the consent of BRNI. For additional information regarding the License Agreement, see “Business—Intellectual Property—Technology License and Services Agreement.” If the License Agreement were terminated, we would no longer have the rights to develop, commercialize and sell some of our proposed products, and we may be required to cease operations.
Our commercial success will depend, in part, on our ability, and the ability of our licensors, to obtain and maintain patent protection. Our licensors’ failure to obtain and maintain patent protection for our products may have a material adverse effect on our business.
Pursuant to our License Agreement, we have obtained rights to certain patents owned by BRNI or licensed to NRV II, LLC by BRNI as of or subsequent to October 31, 2012. For additional information regarding the License Agreement, see “Business—Intellectual Property—Technology License and Services Agreement.” In the future, we may seek rights from third parties to other patents or patent applications. Our success will depend, in part, on our ability and the ability of our licensors to maintain and/or obtain and enforce patent protection for our proposed products and to preserve our trade secrets, and to operate without infringing upon the proprietary rights of third parties. Patent positions in the field of biotechnology and pharmaceuticals are generally highly uncertain and involve complex legal and scientific questions. We cannot be certain that we or our licensors were the first inventors of inventions covered by our licensed patents or that we or they were the first to file. Accordingly, the patents licensed to us may not be valid or afford us protection against competitors with similar technology. The failure to maintain and/or obtain patent protection on the technologies underlying our proposed products may have material adverse effects on our competitive position and business prospects.
Our licensed patented technologies may infringe on other patents, which may expose us to costly litigation.
It is possible that our licensed patented technologies may infringe on patents or other rights owned by others. We may have to alter our products or processes, pay additional licensing fees, pay to defend an infringement action or challenge the validity of the patents in court or cease activities altogether because of patent rights of third parties, thereby causing additional unexpected costs and delays to us. Patent litigation is costly and time consuming, and we may not have sufficient resources to pay for such litigation. Pursuant to our License Agreement, BRNI has the exclusive right (but not the obligation) to apply for, file, prosecute or maintain patents and patent applications for our licensed technologies. However, in order to maintain our rights to use our licensed technologies, we must reimburse BRNI for all of the attorney’s fees and other costs and expenses related to any of the foregoing. For additional information regarding the License Agreement, see “Business—Intellectual Property—Technology License and Services Agreement.” If the patents licensed to us are determined to infringe a patent owned by a third party and we do not obtain a license under such third-party patents, or if we are found liable for infringement or are not able to have such third-party patents declared invalid, we may be liable for significant money damages, we may encounter significant delays in bringing products to market or we may be precluded from participating in the manufacture, use or sale of products or methods of treatment requiring such licenses.
We may not be able to protect our trade secrets and other unpatented proprietary technologies, which could give our competitors an advantage over us.
In addition to our reliance on patents and pending patents owned by BRNI, we rely upon trade secrets and other unpatented proprietary technologies. We may not be able to adequately protect our rights with regard to such unpatented proprietary technologies or competitors may independently develop substantially equivalent technologies. We seek to protect trade secrets and proprietary knowledge, in part through confidentiality agreements with our employees, consultants, advisors and collaborators. Nevertheless, these agreements may not effectively prevent disclosure of our confidential information and may not provide us with an adequate remedy in the event of unauthorized disclosure of such information and, as a result, our competitors could gain a competitive advantage over us.
We are dependent on Dr. James New, our Chief Executive Officer, for the successful execution of our business plan. The loss of Dr. New, or other key members of our management team he may recruit, could have a material adverse effect on our business prospects.
We are highly dependent on Dr. James New, our Chief Executive Officer. We currently have an employment agreement with Dr. New, the material terms of which are described under “Executive and Director Compensation—Executive Compensation.” We are also dependent on his network of contacts and experience to recruit key talent to the Company. We do not have key-man insurance on any of our officers. Loss of the services of Dr. New or other key members of our prospective management team could have a material adverse effect on our business prospects, financial condition and results of operations.
If we are unable to hire additional qualified personnel our business prospects may suffer.
Although we have not experienced any problems attracting and retaining key executives in the recent past and do not know that any key employee has plans to retire or leave our company, our success and achievement of our business plans depend upon our ability to recruit, hire, train and retain other highly qualified technical and managerial personnel. Competition for qualified employees among pharmaceutical and biotechnology companies is intense, and the loss of any of such persons, or an inability to attract, retain and motivate any additional highly skilled employees required for the implementation of our business plans and activities, could have a materially adverse effect on us. Our inability to attract and retain the necessary technical and managerial personnel and consultants and scientific and/or regulatory consultants and advisors could have a material adverse effect on our business prospects, financial condition and results of operations.
We may not be able to in-license or acquire new development-stage products or technologies.
Our product commercialization strategy relies, to some extent, on our ability to in-license or acquire product formulation techniques, new chemical entities, or related know-how in these fields that have proprietary protection. We may also seek to acquire, by license or otherwise, other development stage products that are consistent with our product portfolio objectives and commercialization strategy.The acquisition of products requires the identification of appropriate candidates, negotiation of terms of acquisition, financing for the acquisition and integration of the candidates into our portfolio. Failure to accomplish any of these tasks may diminish our growth rate and adversely alter our competitive position.
We are dependent upon the National Cancer Institute to supply bryostatin for our clinical trials.
BRNI has entered into a material transfer agreement with the National Cancer Institute, pursuant to which the National Cancer Institute has agreed to supply bryostatin to BRNI and one or more of its private sector collaborators and clinical trial investigators for the conduct of controlled studies and compassionate use protocols. This agreement does not provide for a sufficient amount of bryostatin to support the completion of all clinical trials required to ultimately obtain FDA approval of bryostatin for the treatment of Alzheimer’s Disease. Therefore, BRNI or we will have to enter into one or more subsequent agreements with the National Cancer Institute for the supply of additional amounts of bryostatin. If BRNI or we are unable to secure such additional agreements or if the National Cancer Institute otherwise discontinues for any reason supplying us with bryostatin, then we would have to either secure another source of bryostatin or discontinue our efforts to develop and commercialize bryostatin for the treatment of Alzheimer’s Disease. We are currently exploring alternative sources of bryostatin and believe it will be feasible to secure future bryostatin supplies from these sources on commercially reasonable terms, although there can be no assurance this will be the case.
We are dependent on BRNI to conduct clinical and non-clinical studies of our drug candidate and diagnostic test and to provide services for certain core aspects of our business, and we may engage other third parties to provide such services in the future. Any interruption or failure by these third parties to meet their obligations pursuant to various agreements with us could have a material adverse effect on our business, results of operations and financial condition.
We rely on BRNI to conduct clinical and non-clinical studies of our drug candidate and diagnostic test and provide us with other services, and we have engaged other third parties to provide such services when the cost, quality and timeliness of these services are superior to those existing at BRNI. Such third party contractors are subject to FDA requirements. Our business and financial viability are dependent on the regulatory compliance of these third parties, and on the strength, validity and terms of our various contracts with these third parties. Any interruption or failure by these third party contractors to meet their obligations pursuant to various agreements with us may be outside of our control and could have a material adverse effect on our business, financial condition and the results of operations.
We expect to rely on third parties to manufacture our proposed products and, as a result, we may not be able to control our product development.
We currently do not have an FDA approved manufacturing facility. We expect to rely on contract manufacturers to produce quantities of products and substances necessary for product commercialization. See also the risk factor above captioned “We are dependent upon the National Cancer Institute to supply bryostatin for our clinical trials.” Contract manufacturers that we may use must adhere to current good manufacturing practice regulations enforced by the FDA through its facilities inspection program. If the facilities of such manufacturers cannot pass a pre-approval plant inspection, the FDA pre-market approval of our products will not be granted. As a result:
| | · | there are a limited number of manufacturers that could produce the products for us and we may not be able to identify and enter into acceptable agreements with any manufacturers; |
| | · | the products may not be produced at costs or in quantities necessary to make them commercially viable; |
| | · | the quality of the products may not be acceptable to us and/or regulatory authorities; |
| | · | our manufacturing partners may go out of business or file for bankruptcy; |
| | · | our manufacturing partners may decide not to manufacture our products for us; |
| | · | our manufacturing partners could fail to manufacture to our specifications; |
| | · | there could be delays in the delivery of quantities needed; |
| | · | we could be unable fulfill our commercial needs in the event we obtain regulatory approvals and there is strong market demand; or |
| | · | ongoing inspections by the FDA or other regulatory authorities may result in suspensions, seizures, recalls, fines, injunctions, revocations and/or criminal prosecutions. |
If we are unable to engage contract manufacturers or suppliers to manufacture or have packaged our products or if we are unable to contract for a sufficient supply of required products and substances on acceptable terms, or if we encounter delays or difficulties in our relationships with these manufacturers, or with a regulatory agency, then the submission of products for regulatory approval and subsequent sales of such products would be delayed. Any such delay may have a materially adverse effect on our business prospects, financial condition and results of operations.
We may rely on third parties for marketing and sales and our revenue prospects may depend on their efforts.
We currently have no experience in sales, marketing or distribution. We do not anticipate having the resources in the foreseeable future to allocate to the sales and marketing of our proposed products. As a result, our future success may depend, in part, on our ability to enter into and maintain collaborative relationships with one or more third parties, the collaborator’s strategic interest in the products we have under development and such collaborator’s ability to successfully market and sell any such products. We intend to pursue collaborative arrangements regarding the sales and marketing of our products as appropriate. However, we may not be able to establish or maintain such collaborative arrangements or, if we are able to do so, they may not have effective sales forces. To the extent that we decide not to, or are unable to, enter into collaborative arrangements with respect to the sales and marketing of our proposed products, significant capital expenditures, management resources and time will be required to establish and develop an in-house marketing and sales force with technical expertise. To the extent that we depend on third parties for marketing and distribution, any revenues received by us will depend upon the efforts of such third parties, which may not be successful.
If our products are not accepted by the medical community or health insurance companies, our business prospects will suffer.
Commercial sales of our proposed products will substantially depend upon the products’ efficacy and on their acceptance by the medical community, providers of comprehensive healthcare insurance, healthcare benefit plan managers, the Centers for Medicare and Medicaid Services (which is the U.S. federal agency which administers Medicare, Medicaid and the State Children’s Health Insurance Program) and other organizations. Widespread acceptance of our products will require educating the medical community and third party payers of medical treatments as to the benefits and reliability of the products. Our proposed products may not be accepted, and, even if they are accepted, we are unable to estimate the length of time it would take to gain such acceptance.
The branded prescription segment of the pharmaceutical industry in which we operate is competitive, and we are particularly subject to the risks of such competition.
The branded prescription segment of the pharmaceutical industry in which we operate is competitive, in part, because the products that are sold require extensive sales and marketing resources invested in their commercialization. The increasing cost of prescription pharmaceuticals has caused providers of comprehensive healthcare insurance, healthcare benefit plan managers, the Centers for Medicare and Medicaid Services, as well as other organizations, collectively known as third party payers, to tightly control and dictate their drug formulary plans to control the costs associated with the use of prescription pharmaceutical products used by enrollees in these plans. Our ability to gain formulary access to drug plans supported by these third party payers is substantially dependent on the differentiated patient benefit that our proposed products can provide compared closely to similar products claiming the same benefits or advantages. We may not be able to differentiate our proposed products from those of our competitors, successfully develop or introduce new products that are less costly or offer better performance than those of our competitors, or offer purchasers of our proposed products payment and other commercial terms as favorable as those offered by our competitors. We expect that some of our proposed products will eventually face competition from a significant number of biotechnology or large pharmaceutical companies. Because certain of our competitors have substantially greater financial and other resources than we have, we are particularly subject to the risks inherent in competing with them. The effects of this competition could materially adversely affect our business prospects, financial condition and results of operations.
Our business will expose us to potential product liability risks, which could result in significant product liability exposure.
Our business will expose us to potential product liability risks that are inherent in the testing, designing, manufacturing and marketing of human diagnostic and therapeutic products. Product liability insurance in the pharmaceutical industry is generally expensive, and we may not be able to obtain or maintain product liability insurance in the future on acceptable terms or with adequate coverage against potential liabilities, if at all. A successful products liability claim brought against us could have a material adverse effect on our business prospects, financial condition and results of operations.
We do not currently have clinical trial liability insurance for our products and a successful clinical trial liability claim against us could have a material adverse effect on our financial condition even with such insurance coverage.
Our business will expose us to potential liability that results from risks associated with conducting clinical trials of our products. We do not currently have clinical trial liability insurance for our bryostatin drug product. Although we are in the process of procuring insurance coverage for our bryostatin product prior to initiation of our planned clinical trials, there is no guarantee that we will be able to procure such coverage on favorable rates if at all and, even if procured, there is no guarantee that we will procure adequate coverage to satisfy any liability we may incur. A successful clinical trial liability claim brought against us could have a material adverse effect on our business prospects, financial condition and results of operations even if we successfully obtain clinical trial insurance.
We do not currently have comprehensive insurance and a successful liability claim against us could have a material adverse effect on our financial condition.
Our business and actions can expose us to potential liability risks that are inherent in business. We do not currently maintain commercial general liability insurance. A successful liability claim brought against us could have a material adverse effect on our business prospects, financial condition and results of operations.
Reforms in the health care industry and the uncertainty associated with pharmaceutical pricing, reimbursement and related matters could adversely affect the marketing, pricing and demand for our products.
Public and private entities are seeking ways to reduce or contain increasing health care costs. All generic pharmaceutical manufacturers whose products are covered by the Medicaid program are required to rebate to each state a percentage of their “average manufacturer price” for the products in question. The extension of prescription drug coverage to all Medicare recipients was approved by Congress several years ago. Numerous other proposals to curb rising pharmaceutical prices have also been introduced or proposed in Congress and in some state legislatures. We cannot predict the nature of the measures that may be adopted or their effect on our competitive position. Our ability to market our products depends, in part, on reimbursement levels for them and related treatment established by health care providers, private health insurers and other organizations, including health maintenance organizations and managed care organizations. In the event that governmental authorities enact additional legislation or adopt regulations that affect third party coverage and reimbursement, demand for our products may be reduced, which may materially adversely affect our business prospects, financial condition and results of operations.
Consolidation in the pharmaceutical industry could materially affect our ability to operate as an independent entity.
The dearth of new product introductions in the branded prescription segment of the pharmaceutical market is a direct result of a long drought in basic research productivity in the industry. The pressure to grow revenues while containing the escalating costs of basic research and development has resulted in an increase in mergers and acquisitions in our industry. More consolidation in the pharmaceutical industry is expected over the next five years. We could become an acquisition target by a larger competitor and, as a consequence, suffer serious disruptions to our business model or even lose control of our ability to operate as an independent entity. Such events could have a material adverse effect on our product development efforts or the commercialization of our proposed products.
Risks Related to Our Common Stock
There currently is a limited public market for our Common Stock. Failure to develop or maintain an active trading market could negatively affect the value of our Common Stock and make it difficult or impossible for you to sell your shares.
There is currently a limited public market for shares of our Common Stock, and an active trading market may never develop or if developed, may not be maintained. Our Common Stock is quoted on the OTC Markets. The OTC Markets is a thinly traded market and lacks the liquidity of certain other public markets with which some investors may have more experience. We may not ever be able to satisfy the listing requirements for our Common Stock to be listed on a national securities exchange, which are often a more widely-traded and liquid market. Some, but not all, of the factors which may delay or prevent the listing of our Common Stock on a more widely-traded and liquid market include the following: our stockholders’ equity may be insufficient; the market value of our outstanding securities may be too low; our net income from operations may be too low; our Common Stock may not be sufficiently widely held; we may not be able to secure market makers for our Common Stock; and we may fail to meet the rules and requirements mandated by the several exchanges and markets to have our Common Stock listed. Should we fail to satisfy the initial listing standards of the national exchanges, or our Common Stock is otherwise rejected for listing and remains listed on the OTC Markets or suspended from the OTC Markets, the trading price of our Common Stock could suffer and the trading market for our Common Stock may be less liquid and our Common Stock price may be subject to increased volatility.
We cannot assure you that our Common Stock will become liquid or that it will be listed on a securities exchange.
Until our Common Stock is listed on a national securities exchange such as the New York Stock Exchange or the Nasdaq Stock Market, we expect our Common Stock to remain eligible for quotation on the OTC Markets, or on another over-the-counter quotation system, or in the “pink sheets.” In those venues, however, an investor may find it difficult to obtain accurate quotations as to the market value of our Common Stock. In addition, if we fail to meet the criteria set forth in SEC regulations, various requirements would be imposed by law on broker-dealers who sell our securities to persons other than established customers and accredited investors. Consequently, such regulations may deter broker-dealers from recommending or selling our Common Stock, which may further affect the liquidity of our Common Stock. This would also make it more difficult for us to raise capital.
Our Common Stock is subject to the “penny stock” rules of the SEC and the trading market in the securities is limited, which makes transactions in the stock cumbersome and may reduce the value of an investment in the stock.
The SEC has adopted Rule 15g-9 which establishes the definition of a “penny stock,” for the purposes relevant to us, as any equity security that has a market price of less than $5.00 per share or with an exercise price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, the rules require:
| | · | that a broker or dealer approve a person’s account for transactions in penny stocks; and |
| | · | the broker or dealer receive from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased. |
In order to approve a person’s account for transactions in penny stocks, the broker or dealer must:
| | · | obtain financial information and investment experience objectives of the person; and |
| | · | make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks. |
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the SEC relating to the penny stock market, which, in highlight form sets forth:
| | · | the basis on which the broker or dealer made the suitability determination; and |
| | · | that the broker or dealer received a signed, written agreement from the investor prior to the transaction. |
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of common stock and cause a decline in the market value of stock.
Disclosure also has to be made about the risks of investing in penny stocks in both public offerings and in secondary trading and about the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks. If we remain subject to the penny stock rules for any significant period, it could have an adverse effect on the market, if any, for our securities. If our securities are subject to the penny stock rules, investors will find it more difficult to dispose of our securities.
The price of our Common Stock may become volatile, which could lead to losses by investors and costly securities litigation.
The trading price of our Common Stock is likely to be highly volatile and could fluctuate in response to factors such as:
| | · | actual or anticipated variations in our operating results; |
| | · | announcements of developments by us or our competitors; |
| | · | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments; |
| | · | adoption of new accounting standards affecting our Company’s industry; |
| | · | additions or departures of key personnel; |
| | · | sales of our Common Stock or other securities in the open market; |
| | · | changes in our industry; |
| | · | regulatory and economic developments, including our ability to obtain working capital financing; |
| | · | our ability to execute our business plan; and |
| | · | other events or factors, many of which are beyond our control. |
The stock market is subject to significant price and volume fluctuations. In the past, following periods of volatility in the market price of a company’s securities, securities class action litigation has often been initiated against the public company. Litigation initiated against us, whether or not successful, could result in substantial costs and diversion of our management’s attention and resources, which could harm our business and financial condition.
We do not anticipate dividends to be paid on our Common Stock, and investors may lose the entire amount of their investment.
Cash dividends have never been declared or paid on our Common Stock, and we do not anticipate such a declaration or payment for the foreseeable future. We expect to use future earnings, if any, to fund business growth. Therefore, stockholders will not receive any funds absent a sale of their shares. If we do not pay dividends, our Common Stock may be less valuable because a return on your investment will only occur if our stock price appreciates. We cannot assure stockholders of a positive return on their investment when they sell their shares, nor can we assure that stockholders will not lose the entire amount of their investment.
If securities analysts do not initiate coverage or continue to cover our Common Stock or publish unfavorable research or reports about our business, this may have a negative impact on the market price of our Common Stock.
The trading market for our Common Stock will depend on the research and reports that securities analysts publish about our business and the Company. It is often more difficult to obtain analyst coverage for companies whose securities are traded on the OTC Markets. We do not have any control over securities analysts. There is no guarantee that securities analysts will cover our Common Stock. If securities analysts do not cover our Common Stock, the lack of research coverage may adversely affect its market price. If we are covered by securities analysts, and our stock is the subject of an unfavorable report, our stock price and trading volume would likely decline. If one or more of these analysts ceases to cover the Company or fails to publish regular reports on the Company, we could lose visibility in the financial markets, which could cause our stock price or trading volume to decline.
State Blue Sky registration requirements could limit resale of the shares.
The holders of the shares of the Company, and persons who desire to purchase the shares in any trading market that might develop in the future, should be aware that there may be significant state law restrictions upon the ability of investors to resell the securities. Accordingly, investors should consider the secondary market for the Company’s securities to be a limited one. It is the intention of our management to seek coverage and publication of information regarding the Company in an accepted publication which permits a “manuals exemption.” This manuals exemption permits a security to be sold by stockholders in a particular state without being registered if the company issuing the security has a listing for that security in a securities manual recognized by that state. The listing entry must contain (i) the names of issuers, officers, and directors, (ii) an issuer’s balance sheet, and (iii) a profit and loss statement for either the fiscal year preceding the balance sheet or for the most recent fiscal year of operations. The principal accepted manuals are those published by Standard and Poor’s, and Mergent, Inc. Many states expressly recognize these manuals. A smaller number of states declare that they recognize securities manuals, but do not specify the recognized manuals. Among others, the following states do not have any provisions and, therefore, do not expressly recognize the manuals exemption: Alabama, Illinois, Kentucky, Louisiana, New York, Tennessee and Virginia.
You may experience dilution of your ownership interests because of the future issuance of additional shares of our Common Stock.
Any future issuance of our equity or equity-backed securities may dilute then-current stockholders’ ownership percentages and could also result in a decrease in the fair market value of our equity securities, because our assets would be owned by a larger pool of outstanding equity. As described above, we may need to raise additional capital through public or private offerings of our common or preferred stock or other securities that are convertible into or exercisable for our common or preferred stock. We may also issue such securities in connection with hiring or retaining employees and consultants (including stock options issued under our equity incentive plans), as payment to providers of goods and services, in connection with future acquisitions or for other business purposes. Our Board of Directors may at any time authorize the issuance of additional common or preferred stock without common stockholder approval, subject only to the total number of authorized common and preferred shares set forth in our articles of incorporation. The terms of equity securities issued by us in future transactions may be more favorable to new investors, and may include dividend and/or liquidation preferences, superior voting rights and the issuance of warrants or other derivative securities, which may have a further dilutive effect. Also, the future issuance of any such additional shares of common or preferred stock or other securities may create downward pressure on the trading price of the common stock. There can be no assurance that any such future issuances will not be at a price (or exercise prices) below the price at which shares of the common stock are then traded.
The conversion of our issued and outstanding shares of Series A preferred stock could have the effect of diluting the voting power of our common stock holders.
The conversion of our shares of Series A preferred stock could have the effect of changing or preventing a change of control of the Company and could dilute your interests. Our authorized capital stock includes 50,000,000 shares of preferred stock, of which 24,325,000 shares are designated as Series A Preferred Stock.
The effects of the conversion of shares of our Series A preferred stock upon the rights of our common shareholders might include, among other things, restricting dividends on our common stock, diluting the voting power of our common stock holders, reducing the market price of the common stock, or impairing the liquidation rights of the common stock.
We may obtain additional capital through the issuance of preferred stock, which may limit your rights as a holder of our Common Stock.
Without any stockholder vote or action, our Board of Directors may designate and approve for issuance shares of our preferred stock. The terms of any preferred stock may include priority claims to assets and dividends and special voting rights which could limit the rights of the holders of our Common Stock. The designation and issuance of preferred stock favorable to current management or stockholders could make any possible takeover of the Company or the removal of our management more difficult.
Being a public company is expensive and administratively burdensome.
As a public reporting company we are subject to the information and reporting requirements of the Securities Act of 1933, as amended (the “Securities Act”), the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and other federal securities laws, rules and regulations related thereto, including compliance with the Sarbanes-Oxley Act. Complying with these laws and regulations requires the time and attention of our Board of Directors and management, and increases our expenses. Among other things, we must:
| | · | maintain and evaluate a system of internal controls over financial reporting in compliance with the requirements of Section 404 of the Sarbanes-Oxley Act and the related rules and regulations of the SEC and the Public Company Accounting Oversight Board; |
| | · | maintain policies relating to disclosure controls and procedures; |
| | · | prepare and distribute periodic reports in compliance with our obligations under federal securities laws; |
| | · | institute a more comprehensive compliance function, including with respect to corporate governance; and |
| | · | involve, to a greater degree, our outside legal counsel and accountants in the above activities. |
The costs of preparing and filing annual and quarterly reports, proxy statements and other information with the SEC and furnishing audited reports to stockholders is expensive and much greater than that of a privately-held company, and compliance with these rules and regulations may require us to hire additional financial reporting, internal controls and other finance personnel, and will involve a material increase in regulatory, legal and accounting expenses and the attention of management. There can be no assurance that we will be able to comply with the applicable regulations in a timely manner, if at all. In addition, being a public company has made it more expensive for us to obtain director and officer liability insurance. In the future, we may be required to accept reduced coverage or incur substantially higher costs to obtain this coverage. These factors could also make it more difficult for us to attract and retain qualified executives and members of our Board of Directors, particularly directors willing to serve on an audit committee.
Any failure to maintain effective internal control over our financial reporting could materially adversely affect us.
Section 404 of the Sarbanes-Oxley Act of 2002 requires us to include in our annual reports on Form 10-K an assessment by management of the effectiveness of our internal control over financial reporting. (See Item 9A, “Controls and Procedures,” below.) In addition, at such time, if any, as we are no longer a “smaller reporting company,” our independent auditors will have to attest to and report on management’s assessment of the effectiveness of such internal control over financial reporting. While we intend to diligently and thoroughly document, review, test and improve our internal control over financial reporting in order to ensure compliance with Section 404, management has concluded that our internal control over financial reporting is currently not effective due to certain material weaknesses. Furthermore, even if management were to reach a conclusion that our internal control over financial reporting is effective, if our independent auditors are not satisfied with the adequacy of our internal control over financial reporting, or if the independent auditors interpret the requirements, rules or regulations differently than we do, then they may decline to attest to management’s assessment or may issue a report that is qualified. Any of these events could result in a loss of investor confidence in the reliability of our financial statements, which in turn could negatively impact the price of our Common Stock.
In particular, we must perform system and process evaluation and testing of our internal control over financial reporting to allow management and (if required in future) our independent registered public accounting firm to report on the effectiveness of our internal control over financial reporting, as required by Section 404. Our compliance with Section 404 may require that we incur substantial accounting expense and expend significant management efforts. We currently do not have an internal audit group, and we will need to retain the services of additional accounting and financial staff or consultants with appropriate public company experience and technical accounting knowledge to satisfy the ongoing requirements of Section 404.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
Our principal executive offices are currently located in the home of our Chief Executive Officer at 10732 Hawk’s Vista Street, Plantation, Florida 33324, USA. We incur no costs for the use of this office. We intend to relocate our headquarters to leased office space in the near future.
The 9,000 square foot BRNI lab that currently serves as the base of operations for the development of our diagnostic test is located on the campus of John Hopkins University in Rockville, Maryland.We do not lease space in this laboratory; rather, BRNI provides services to us from this lab pursuant to arrangements entered into between us and BRNI under the License Agreement, for which we compensate BRNI.
ITEM 3. LEGAL PROCEEDINGS
From time to time, we may become involved in various lawsuits and legal proceedings that arise in the ordinary course of business. Litigation is subject to inherent uncertainties, and an adverse result in these or other matters may arise from time to time that may harm our business, financial condition or results of operation. There are currently no pending legal proceedings that we believe will have individually or in the aggregate, a material adverse effect on our business, financial condition or operating results. As far as we are aware, no governmental authority is contemplating any proceeding to which we are a party or to which any of our properties is subject.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information and Holders
Our common stock is currently eligible for quotation and trades on the OTC Market under the symbol “NTRP.” The quotation of our common stock began on or about October 2, 2013. There has been very limited trading in our common stock to date.
As of April 10, 2014, we had 21,889,006 shares of our common stock issued and outstanding held by approximately 16 stockholders of record. To date, we have not paid dividends on our common stock.
We also have outstanding:
| · | 22,900,000 shares of Series A convertible preferred stock held by approximately 197 stockholders of record, convertible at any time into shares of common stock on a one-for-one basis, subject to adjustment in certain circumstances as provided therein; |
| o | 900,000 shares of our common stock, at an exercise price of $0.01 per share, subject to adjustment in certain circumstances as provided therein; and |
| o | 1,325,000 shares of our Series A convertible preferred stock, at an exercise price of $1.00 per share, subject to adjustment in certain circumstances as provided therein; and |
| · | options to purchase 6,579,952 shares of our common stock at a weighted average exercise price of $1.50 per share. |
The following table sets forth the high and low closing bid prices for our common stock for the fiscal quarter indicated as reported on OTC Markets. The quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not represent actual transactions. Our common stock is very thinly traded and, thus, pricing of our common stock on OTC Markets does not necessarily represent its fair market value.
| Period | | High | | Low | |
| | | | | | | | |
| Quarter ended December 31, 2013 (commencing October 2, 2013) | | $ | 2.30 | | $ | 1.20 | |
| Quarter ended March 31, 2014 | | $ | 2.69 | | $ | 1.44 | |
| Quarter ending June 30, 2014 (through April 11, 2014) | | $ | 1.67 | | $ | 1.21 | |
Dividends
We have never paid any cash dividends on our capital stock and do not anticipate paying any cash dividends on our Common Stock in the foreseeable future. We intend to retain future earnings to fund ongoing operations and future capital requirements. Any future determination to pay cash dividends will be at the discretion of our Board of Directors and will be dependent upon financial condition, results of operations, capital requirements and such other factors as the Board of Directors deems relevant. Other than provisions of the Nevada Revised Statutes requiring post-dividend solvency according to certain measures, there are no material restrictions limiting, or that are likely to limit, our ability to pay dividends on our common stock.
Recent Sales of Unregistered Securities
Except as previously disclosed in Current Reports on Form 8-K that we have filed, we have not sold any of our equity securities during the period covered by this Report that were not registered under the Securities Act.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
During the fourth quarter of the fiscal year covered by this report, no purchases were made by or on behalf of the Company or any “affiliated purchaser,” as defined in Rule 10b-18(a)(3) under the Exchange Act, of shares or other units of any class of the Company’s equity securities.
Securities Authorized for Issuance under Equity Compensation Plans
On August 22, 2013, our Board of Directors adopted, and on August 22, 2013, our stockholders approved, our 2013 Equity Incentive Plan, which reserves a total of 7,000,000 shares of our Common Stock for issuance pursuant to awards granted under the 2013 Plan.
The following table provides information as of December 31, 2013, with respect to the shares of common stock that may be issued under our existing equity compensation plans:
Equity Compensation Plan Information
| | | | | | | | | |
| | | | | | | | Number of securities | |
| | | Number of securities | | | | | remaining available for | |
| | | to be issued upon | | Weighted-average | | future issuance under equity | |
| | | exercise of | | exercise price of | | compensation plans | |
| | | outstanding options, | | outstanding options, | | (excluding securities | |
| Plan category | | warrants and rights | | warrants and rights | | reflected in column (a)) | |
| | | (a) | | | (b) | | (c) | |
| Equity compensation plans approved by security holders (1) | | 6,249,952 | | $ | 1.486 | | 750,048 | |
| Equity compensation plans not approved by security holders | | 0 | | | 0 | | 0 | |
| Total | | 6,249,952 | | | | | 750,048 | |
(1) 2013 Equity Incentive Plan
As described below, incentive awards authorized under the 2013 Plan include, but are not limited to, incentive stock options within the meaning of Section 422 of the internal Revenue Code of 1986, as amended (the “Code”). If an award granted under the 2013 Plan expires, terminates, is unexercised or is forfeited, or if any shares are surrendered to us in connection with the exercise of an incentive award, the shares subject to such award and the surrendered shares will become available for further awards under the 2013 Plan.
The number of shares of our Common Stock subject to the 2013 Plan, any number of shares subject to any numerical limit in the 2013 Plan, to the terms of any incentive award or to any combination of the foregoing, is expected to be adjusted in the event of any change in our outstanding our Common Stock by reason of any stock dividend, spin-off, split-up, stock split, reverse stock split, recapitalization, reclassification, merger, consolidation, liquidation, business combination or exchange of shares or similar transaction.
Administration
The Compensation Committee of the Board, or the Board in the absence of such a committee, will administer the 2013 Plan. Subject to the terms of the 2013 Plan, the Compensation Committee or the Board has complete authority and discretion to determine the terms of awards under the 2013 Plan.
Grants
The 2013 Plan authorizes the grant to participants of nonqualified stock options, incentive stock options, restricted stock awards, restricted stock units, performance grants intended to comply with Section 162(m) of the Internal Revenue Code (as amended, the “Code”) and stock appreciation rights, as described below:
| | · | Options granted under the 2013 Plan entitle the grantee, upon exercise, to purchase a specified number of shares from us at a specified exercise price per share. The exercise price for shares of our Common Stock covered by an option generally cannot be less than the fair market value of our Common Stock on the date of grant unless agreed to otherwise at the time of the grant. In addition, in the case of an incentive stock option granted to an employee who, at the time the incentive stock option is granted, owns stock representing more than 10% of the voting power of all classes of stock of the Company or any parent or subsidiary, the per share exercise price will be no less than 110% of the fair market value of our Common Stock on the date of grant. |
| | · | Restricted stock awards and restricted stock units may be awarded on terms and conditions established by the compensation committee, which may include performance conditions for restricted stock awards and the lapse of restrictions on the achievement of one or more performance goals for restricted stock units. |
| | · | The Compensation Committee may make performance grants, each of which will contain performance goals for the award, including the performance criteria, the target and maximum amounts payable, and other terms and conditions. |
| | · | The 2013 Plan authorizes the granting of stock awards. The compensation committee will establish the number of shares of our Common Stock to be awarded and the terms applicable to each award, including performance restrictions. |
| | · | Stock appreciation rights (“SARs”) entitle the participant to receive a distribution in an amount not to exceed the number of shares of our Common Stock subject to the portion of the SAR exercised multiplied by the difference between the market price of a share of our Common Stock on the date of exercise of the SAR and the market price of a share of our Common Stock on the date of grant of the SAR. |
Duration, Amendment, and Termination
The Board has the power to amend, suspend or terminate the 2013 Plan without stockholder approval or ratification at any time or from time to time. No change may be made that increases the total number of shares of our Common Stock reserved for issuance pursuant to incentive awards or reduces the minimum exercise price for options or exchange of options for other incentive awards, unless such change is authorized by our stockholders within one year. Unless sooner terminated, the 2013 Plan would terminate ten years after it is adopted.
ITEM 6. SELECTED FINANCIAL DATA
Not applicable to a smaller reporting company.
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
The following management’s discussion and analysis should be read in conjunction with the Company’s historical financial statements and the related notes thereto contained in this report. The management’s discussion and analysis contains forward-looking statements, such as statements of our plans, objectives, expectations and intentions. Any statements that are not statements of historical fact are forward-looking statements. When used, the words “believe,” “plan,” “intend,” “anticipate,” “target,” “estimate,” “expect” and the like, and/or future tense or conditional constructions (“will,” “may,” “could,” “should,” etc.), or similar expressions, identify certain of these forward-looking statements. These forward-looking statements are subject to risks and uncertainties, including those under “Risk Factors” in this report that could cause actual results or events to differ materially from those expressed or implied by the forward-looking statements. The Company’s actual results and the timing of events could differ materially from those anticipated in these forward-looking statements as a result of several factors. The Company does not undertake any obligation to update forward-looking statements to reflect events or circumstances occurring after the date of this report.
As the result of the Merger and the change in business and operations of the Company, from engaging in the business of providing software solutions to deliver geo-location targeted coupon advertising to mobile internet devices, to the business of developing two product platforms, including a drug candidate called bryostatin for the treatment of Alzheimer’s Disease (“AD”) and a diagnostic test for AD, a discussion of the past financial results of Neurotrope, Inc. (formerly BlueFlash Communications, Inc.) is not pertinent, and under applicable accounting principles the historical financial results of Neurotrope BioScience, the accounting acquirer, prior to the Merger are considered the historical financial results of the Company.
The following discussion highlights the Company’s results of operations and the principal factors that have affected our financial condition as well as our liquidity and capital resources for the periods described, and provides information that management believes is relevant for an assessment and understanding of the statements of financial condition and results of operations presented herein. The following discussion and analysis are based on the Company’s audited financial statements contained in this report, which we have prepared in accordance with United States generally accepted accounting principles. You should read the discussion and analysis together with such financial statements and the related notes thereto.
Basis of Presentation
References in this section to “Neurotrope,” “we,” “us,” “our,” “the Company” and “our Company” refer to Neurotrope, Inc. and its consolidated subsidiary Neurotrope BioScience, Inc. (“Neurotrope Bioscience”).
The audited financial statements for our fiscal years ended December 31, 2013 and 2012, include a summary of our significant accounting policies and should be read in conjunction with the discussion below. In the opinion of management, all material adjustments necessary to present fairly the results of operations for such periods have been included in these audited financial statements. All such adjustments are of a normal recurring nature.
Overview
Neurotrope Bioscience was founded as a Delaware corporation in October 2012. Activities since the Company’s inception through December 31, 2013, were devoted primarily to the development and commercialization of Alzheimer’s Disease (“AD”) diagnostics and related therapeutic products for large and growing markets using innovative licensed patented technology. This licensed technology has been under development since 1999 and has been financed by BRNI from a variety of non-investor sources including not-for-profit foundations, the National Institute of Health and individual philanthropists up until March 2013. From March 2013 forward, the licensed technology has been funded principally through collaboration with Neurotrope Bioscience (see the description of Neurotrope Bioscience financings below in “Financial Condition, Liquidity and Capital Resources – Sources and Uses of Liquidity”. For a detailed description of the License Agreement, refer to Item 1, “Business—Intellectual Property—Technology License and Services Agreement” above.
On August 23, 2013, a wholly owned subsidiary of Neurotrope, Inc. (formerly “BlueFlash Communications, Inc.”), Neurotrope Acquisition, Inc., a corporation formed in the State of Nevada on August 15, 2013 (“Acquisition Sub”), merged (the “Merger”) with and into Neurotrope Bioscience. Neurotrope Bioscience was the surviving corporation in the Merger and became the Company’s wholly owned subsidiary. All of the outstanding Neurotrope Bioscience common stock was converted into shares of Neurotrope, Inc. common stock on a one-for-one basis.
As a result of the Merger, Neurotrope, Inc., discontinued its pre-Merger business and acquired the business of Neurotrope Bioscience, and will continue the existing business operations of Neurotrope Bioscience as a publicly-traded company. A discussion of the pre-Merger financial results of Neurotrope, Inc., is not pertinent, and under applicable accounting principles the historical financial results of Neurotrope BioScience, the accounting acquirer in the Merger, prior to the Merger are considered the historical financial results of the Company.
As of December 31, 2013, the Company had devoted substantially all of its efforts to product development, raising capital and building infrastructure through utilizing its strategic alliances, specifically with BRNI. The Company did not, as of that date, realize any revenues from its planned principal operations. Accordingly, the Company is considered to be in the development stage.
Strategy
One of the central tenets developed by BRNI is that the neurodegeneration underlying these neurological diseases can be halted and reversed if treatment is initiated early enough. This process occurs by an improvement in nerve cell viability and synaptic function through activation of an enzyme called protein kinase C (PKC). This enzyme is actually a super family of isozymes (α, β, γ, δ, ε…) which have different activities in different tissues. The PKCepsilon (aka PKCε) variant has a very high concentration in the synapses of neurons, suggesting it plays a role in maintaining synaptic function. Deficient activity or low concentrations of PKCepsilon in aging subjects is thoughtto be once of main causes of the neurodegeneration seen in AD. Through a variety of the latest biomedical techniques and animal models developed to map and quantify neuroregeneration, BRNI has established a very central role for PKCepsilon in re-modeling or restoring synaptic function in both healthy and diseased neurons in the central nervous system.
The flagship product in the Neurotrope armamentarium is bryostatin. This drug has previously been evaluated in 1,200 patients at the National Cancer Institute (“NCI”) for the treatment of various forms of cancer. While bryostatin did not show sufficient anti-cancer effects to warrant commercialization of the compound, much useful information on the safety, pharmacodynamics, and toxicity of the drug was gleaned from these in-human trials.
It was discovered that at a much lower dose than that which was used in these anticancer trials, bryostatin is a potent activator of PKCε and may have efficacy in treating AD. Activation of PKCε has now been shown to partially restore synaptic function in neurons damaged by AD, ischemic stroke or traumatic brain injury in in vitro and in vivo animal models.
The NCI has allowed BRNI access to the valuable chemical, animal and human data from its cancer studies, to which Neurotrope in turn has access under the License Agreement to be used in our own research and regulatory programs.
The Company’s strategy is to efficiently utilize our licensed proprietary and patented technologies to further the development of those technologies toward commercializing a therapeutic and a diagnostic product for AD and potentially utilize these technologies to diagnose and treat other neurological diseases. We may also seek to acquire, by license or otherwise, other development stage products that are consistent with our product portfolio objectives and commercialization strategy.
Critical Accounting Policies, Estimates, and Judgments
Our financial statements are prepared in accordance with accounting principles that are generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. We continually evaluate our estimates and judgments, the most critical of which are those related to valuation of long-lived assets, our commitments to strategic alliance partners and the timing of the achievement of collaboration milestones. We base our estimates and judgments on historical experience and other factors that we believe to be reasonable under the circumstances. Materially different results can occur as circumstances change and additional information becomes known. Besides the estimates identified above that are considered critical, we make many other accounting estimates in preparing our financial statements and related disclosures. All estimates, whether or not deemed critical, affect reported amounts of assets, liabilities, revenues and expenses, as well as disclosures of contingent assets and liabilities. These estimates and judgments are also based on historical experience and other factors that are believed to be reasonable under the circumstances. Materially different results can occur as circumstances change and additional information becomes known, even for estimates and judgments that are not deemed critical.
Results of Operations
Neurotrope Bioscience began operations in October 2012. As a result, a comparison of the year ended December 31, 2013, to the period from inception to December 31, 2012, would not be meaningful.
Year ended December 31, 2013
Revenues
The Company has not generated any revenues for the year ended December 31, 2013.
Operating Expenses
Overview
Total operating expenses for the year ended December 31, 2013 were $7,590,782. Most significantly, during this period, the Company incurred related party research and development expenses for its ongoing development of its future diagnostic testing and pharmaceutical products in collaboration with BRNI, administrative expenses to manage the Company’s business and non-cash expenses associated with the issuance of stock-based compensation.
Research and Development Expenses
For the year ended December 31, 2013, the Company incurred $1,367,644 to BRNI for ongoing research and development principally relating to the development of the Company’s diagnostic and therapeutic products. Of these expenses, $867,386 relate to the continuing development of the Company’s AD diagnostic product, $194,630 relates to the development of the Company’s AD therapeutic product, $248,319 was paid for patent expenses associated with the two licensed technology platforms BRNI, and $51,200 was incurred to fund an ongoing compassionate use protocol with the Company’s AD therapeutic product under development.
General and Administrative Expenses
The Company incurred related party general and administrative expenses totaling $4,301,731 during the year ended December 31, 2013. Of this amount, approximately $2.85 million related to issuance of stock options as a non-cash expense, approximately $1.0 was paid to BRNI as a prepaid royalty against future royalties payable to BRNI upon commercialization of licensed technologies, $305,932 was incurred for salaries, taxes, insurance, travel expenses and accrued vacation of our Chief Executive Officer, $42,972 was incurred for financial and business development consulting expenses, $45,000 was paid to our Chairman for services provided to the Company and $14,332 was incurred for travel, insurance and other administrative expenses associated with Company operating activities.
The Company incurred $1,921,407 of other general and administrative expenses for the year ended December 31, 2013. $731,056 was incurred for legal expenses, principally relating to the License Agreement and for the closing of the Company’s merger and private placement financing, $451,348 was incurred for outside operations consulting services, $241,822 was incurred for issuance of stock options as a non-cash expense, $268,199 was incurred for professional fees associated with financial and accounting advisory services, $105,111 was incurred for salary, vacation and payroll expenses for our Chief Financial Officer, $43,303 was incurred for accounting services and $104,708 was incurred for office supplies, insurance, license fees, filing costs and other expenses.
Other Income
The Company earned $1,378 of interest income for the year ended December 31, 2013 on funds temporarily deposited in an interest bearing money market account.
Fiscal year ended December 31, 2012
Revenues
The Company did not generate revenues for the year ended December 31, 2012.
Operating Expenses
Overview
Total operating expenses for the year ended December 31, 2012 were $1,437,793. Most significantly, during this period, the Company incurred related party research and development expenses for its ongoing development of its future diagnostic testing and pharmaceutical product in collaboration with BRNI and administrative expenses to manage the Company’s business.
Research and Development Expenses
For the year ended December 31, 2012, the Company incurred expenses to BRNI totaling $983,491 for ongoing research and development incurred to fund the Company’s use of BRNI resources.
General and Administrative Expenses
The Company incurred related party general and administrative expenses totaling $257,145. Of this amount, $139,901 was incurred by BRNI for expenses associated with patent estate maintenance and licensing and administrative services provided to the Company, $41,667 was incurred for consulting fees paid to our current Chief Executive Officer (who was acting as a consultant rather than an employee at that time) and $27,552 was incurred for travel expenses.
The Company incurred $197,157 of general and administrative salaries for the year ended December 31, 2012. $193,594 was incurred for legal expenses, principally relating to the License Agreement and employment agreement expenses and $3,562 was incurred for professional fees associated with financial and accounting advisory services.
Financial Condition, Liquidity and Capital Resources
Since its inception, the Company has primarily devoted its efforts to negotiating the License Agreement and using BRNI’s resources to further the development of the Company’s diagnostic and therapeutic products toward commercialization while conducting business planning and recruiting executive management. Accordingly, the Company is considered to be in the development stage.
Cash and Working Capital
Since inception, the Company incurred negative cash flows from operations. As of December 31, 2013, the Company had an accumulated deficit of $9,029,366 and had working capital of $14,890,387 as compared to a working capital deficit of $1,437,793 as of December 31, 2012. The $16,328,180 increase in working capital was primarily attributable to the Company’s private placements of 23,000,000 shares of Series A convertible, redeemable preferred stock (“Series A”) sold at $1.00 per share in February, May, August and October of 2013, providing total net proceeds (after commissions and prepaid royalties) of approximately $19.7 million, offset by cash operating losses incurred by the Company for the year ended December 31, 2013.
Sources and Uses of Liquidity
Since inception, the Company has satisfied its operating cash requirements from the private placement of Series A sold principally to outside investors.
In February, 2013, through a private placement, the Company issued 9,073,300 shares of Series A at $1.00 per share, resulting in gross proceeds of $9,073,300. In connection with the closing of the private placement, the Company paid a placement agent $882,330. In May, 2013, the Company issued an additional 1,313,325 shares of Series A at $1.00 per share, resulting in gross proceeds of $1,313,325, on which the Company paid a placement agent $131,332. In August, 2013, through an additional private placement, the Company issued 11,533,375 of Series A at $1.00 per share, resulting in gross proceeds of $11,533,375. In connection with the closing of the August 2013 private placement, the Company paid a placement agent $1,103,338. Further, Neurotrope Bioscience became a wholly-owned subsidiary of a publicly traded company in the Merger, which management believes will provide additional alternatives to issue securities and raise capital in the future. In October, 2013, through an additional private placement, the Company issued 1,080,000 of Series A at $1.00 per share, resulting in gross proceeds of $1,080,000.In connection with the closing of the private placement, the Company paid a placement agent $108,000.
As of December 31, 2013, the Company had cash and cash equivalents totaling $15.2 million, which decreased to approximately $13.2 million as of the date of this report. With the proceeds from the private placements of Series A, management believes the Company has sufficient capital to fund the Company for at least the next 24 months of operations under its current product development plans. However, if our operating plan changes or we incur significant unanticipated expenses, we may require additional capital before this timeframe. During the next two years, we expect to spend approximately $10.5 million on development of both our AD diagnostic and therapeutic products. With these expenditures, we anticipate launching our commercialized AD diagnostic product and conducting significant clinical trials on our AD therapeutic. We expect to also incur approximately $6.0 million in general and administrative costs in that period to support our ongoing research and development expenses and our expenses associated with being a publicly traded company. Management believes the Company will generate revenues through commercialization of its diagnostic product within the next 36 months. Additional funds may be raised through debt financing and/or the issuance of equity securities; at this time no additional fundraising has been initiated, and no specific terms have been set. There can be no assurance that financing will be available when required in sufficient amounts, on acceptable terms or at all.
Pursuant to a statement of work signed with BRNI on August 28, 2013, we are obligated to pay BRNI a total of $1,645,470 in twelve equal monthly installments of $137,123, payable on the first business day of each month, commencing September 2013. In addition, we have agreed to pay an estimated $877,300 in external costs to complete the first clinical trial of its AD diagnostic. As of December 31, 2013, up to approximately $2 million remained potentially payable under this SOW. In addition, on November 22, 2013, the Company paid BRNI $251,939 to conduct additional work on the development of the AD therapeutic product.
On March 12, 2014, we signed an SOW with BRNI to continue pre-clinical activities relating to the commercialization of the Company’s therapeutic product. The Company is obligated to pay BRNI a total of $465,000 (subject to a 20% cost overage, which may not be exceeded without our consent). Of this amount, the Company has paid $358,470, and the remainder of the total ($106,530) is to be paid upon the completion by BRNI of certain activities relating to: transferring test materials; bio-analytical testing; contracting with a suitable contract research organization; completion of testing assays; and finalizing a clinical study protocol.
Off-Balance Sheet Arrangements
The Company did not engage in any “off-balance sheet arrangements” (as that term is defined in Item 303(a)(4)(ii) of Regulation S-K) as of December 31, 2013.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
Not applicable to a smaller reporting company.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTAL DATA
Our audited consolidated financial statements as of, and for the year ended, December 31, 2013, and as of December 31, 2012, and for the period October 31, 2012 (Inception) through December 31, 2012, and for the period October 31, 2012 (Inception) through December 31, 2013, are included beginning on Page F-1 immediately following the signature page to this report. See Item 15 for a list of the financial statements included herein.
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
On August 23, 2013, Lake & Associates CPA’s LLC, was dismissed as our independent registered public accountant. On the same date, Friedman LLP was engaged as our new independent public accounting firm. The Board of Directors of the Company approved the dismissal of Lake & Associates CPA’s LLC, and approved the engagement of Friedman LLP as our independent auditor.
None of the reports of Lake & Associates CPA’s LLC on our financial statements for either of the past two years or subsequent interim period contained an adverse opinion or disclaimer of opinion, or was qualified or modified as to uncertainty, audit scope or accounting principles, except that our audited financial statements contained in our Annual Reports on Form 10-K for the fiscal years ended January 31, 2013, and January 31, 2012, filed with the SEC, included a going concern qualification in the report of Lake & Associates CPA’s LLC.
During the Company’s two most recent fiscal years ended January 31, 2013 and 2012, and the subsequent interim periods preceding their dismissal, there were no disagreements with Lake & Associates CPA’s LLC, whether or not resolved, on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedure, which, if not resolved to the satisfaction of Lake & Associates CPA’s LLC, would have caused them to make reference to the subject matter of the disagreement in connection with their report on the Company’s financial statements.
The Company provided Lake & Associates CPA’s LLC with a copy of the disclosures it is making in this report and requested that Lake & Associates CPA’s LLC, furnish it with a letter addressed to the SEC stating whether they agree with the above statements. Such letter was filed as an exhibit to Amendment No. 2 to our Current Report on Form 8-K, filed with the SEC on September 6, 2013.
On August 29, 2013, the Company was informed by letter from the SEC that effective August 13, 2013, the Public Company Accounting Oversight Board (“PCAOB”) revoked the registration of Lake & Associates CPA’s LLC and barred Jay Charles Lake, CPA, from being an associated person of a registered public accounting firm, on the basis of the PCAOB’s findings that Lake & Associates CPA’s LLC and Mr. Lake (the “Respondents”) violated PCAOB rules and auditing standards in auditing the 2009 financial statements of four issuer clients, that the firm violated PCAOB quality control standards, and that Mr. Lake directly and substantially contributed to the Firm’s violation of PCAOB quality control standards. Specifically, the PCAOB alleges that Respondents failed to, among other things: (a) plan the audits adequately; (b) perform sufficient audit procedures on material accounts, including revenue, accounts receivable, and inventory; and (c) prepare and maintain sufficient audit documentation. The full text of the PCAOB’s order can be found athttp://pcaobus.org/Enforcement/Decisions/Documents/ 08132013_Lake.pdf. The Company was unaware of this action by the PCAOB at the time that our Board of Directors approved the dismissal of Lake & Associates CPA’s LLC as our independent registered public accountant. No report of Lake & Associates CPA’s LLC is included in this report. The Company is not one of the issuers whose financial statements were referred to in the PCAOB’s order.
During the two most recent fiscal years and the interim periods preceding the engagement, and through the date of this report, neither the Company nor anyone on its behalf has previously consulted with Friedman LLP regarding either (a) the application of accounting principles to a specified transaction, either completed or proposed; or the type of audit opinion that might be rendered on the Company’s financial statements, and neither a written report was provided nor oral advice was provided to the Company that Friedman LLP concluded was an important factor considered by the Company in reaching a decision as to the accounting, auditing or financial reporting issue; or (b) any matter that was either the subject of a disagreement (as defined in paragraph 304(a)(1)(iv) of Regulation S-K and the related instructions thereto) or a reportable event (as described in paragraph 304(a)(1)(v)) of Regulation S-K).
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
We carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of our disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) as of December 31, 2013. Based upon that evaluation, our principal executive officer and principal financial officer concluded that, as of the end of the period covered in this report, because of certain weaknesses in internal control over financial reporting discussed below under “Management’s Report on Internal Control over Financial Reporting ,” our disclosure controls and procedureswerenot effective to ensurethat information required to be disclosed in reports filed by us under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported within the required time periods and is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure.
Our management, including our principal executive officer and principal financial officer, does not expect that our disclosure controls and procedures or our internal controls will prevent all errors or fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints and the benefits of controls must be considered relative to their costs. Due to the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. Management believes that the financial statements included in this report fairly present in all material respects our financial condition, results of operations and cash flows for the periods presented.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Securities Exchange Act of 1934 as a process designed by, or under the supervision of, the company’s principal executive and principal financial officers and effected by the Company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America and includes those policies and procedures that:
| | | |
| | · | Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the company; |
| | | |
| | · | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with accounting principles generally accepted in the United States of America and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and |
| | | |
| | · | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect all misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation. Because of the inherent limitations of internal control, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
As of December 31, 2013, our management, including our principal executive officer and principal financial officer, assessed the effectiveness of our internal control over financial reporting based on the criteria for effective internal control over financial reporting established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) and SEC guidance on conducting such assessments. Based on that evaluation, they concluded that, during the period covered by this report, such internal controls and procedures were not fully effective to detect the inappropriate application of US generally accepted accounting principles as more fully described below. This was due to deficiencies that existed in the design or operation of our internal controls over financial reporting that adversely affected our internal controls and that may be considered to be material weaknesses. The matters involving internal controls and procedures that our management considered to be material weaknesses under the standards of the Public Company Accounting Oversight Board were:
(1) lack of a functioning audit committee during 2013 resulting in ineffective oversight in the establishment and monitoring of required internal controls and procedures;
(2) inadequate segregation of duties consistent with control objectives; and
(3) ineffective controls over period end financial disclosure and reporting processes.
Management believes that the material weaknesses set forth above did not have an effect on our financial results.
Management’s Remediation Initiatives
Management believes that the appointment in the fourth quarter of 2014 of an audit committee consisting of three independent directors has remedied the internal control weaknesses that had previously existed relating to both the lack of a functioning audit committee and a lack of a majority of outside directors on our Board.
In an effort to remediate the other identified material weaknesses and other deficiencies and enhance our internal controls, we have initiated, or plan to initiate, the following series of measures:
We will create a position to segregate duties consistent with control objectives and will increase our personnel resources and technical accounting expertise within the accounting function when funds are available to us.
We anticipate that this initiative will be at least partially, if not fully, implemented by December 31, 2014.
Changes in Internal Control over Financial Reporting
There has been no change in our internal control over financial reporting identified in connection with the evaluation referred to above that occurred during our last completed fiscal quarter that has materially negatively affected, or is reasonably likely to materially affect, the registrant's internal control over financial reporting.
Attestation Report of the Registered Public Accounting Firm
Since we are neither an accelerated filer nor a large accelerated filer (as defined in Exchange Act Rule 12b-2), we are not required to provide an attestation report of our registered public accounting firm's on our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
None.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE
Directors and Executive Officers
Below are the names of and certain information regarding the Company’s current executive officers and directors who were appointed effective as of the closing of the Merger:
Name | | Age | | Position | | Date Named to Board of Directors or as Executive Officer | |
| | | | | | | | |
| John Abeles | | 69 | | Director and Chairman of the Board | | August 23, 2013 | |
| | | | | | | | |
| | Jim New | | 60 | | Director and Chief Executive Officer | | August 23, 2013 |
| | | | | | | | |
| | Paul E. Freiman | | 79 | | Director | | October 18, 2013 |
| | | | | | | | |
| James Gottlieb | | 66 | | Director | | December 10, 2013 | |
| | | | | | | | |
| | Jay M. Haft | | 78 | | Director | | August 23, 2013 |
| | | | | | | | |
| | William Singer | | 73 | | Director | | August 23, 2013 |
| | | | | | | | |
| | Robert Weinstein | | 54 | | Chief Financial Officer, Treasurer and Secretary | | August 23, 2013 |
| | | | | | | | | | | | | | |
Directors are elected to serve until the next annual meeting of stockholders and until their successors are elected and qualified. Directors are elected by a plurality of the votes cast at the annual meeting of stockholders and hold office until the expiration of the term for which he or she was elected and until a successor has been elected and qualified.
A majority of the authorized number of directors constitutes a quorum of the Board of Directors for the transaction of business. The directors must be present at the meeting to constitute a quorum. However, any action required or permitted to be taken by the Board of Directors may be taken without a meeting if all members of the Board of Directors individually or collectively consent in writing to the action.
The authorized number of directors to constitute our Board of Directors is seven. Pursuant to the terms of the Merger Agreement, Neurotrope BioScience and the Company agreed that the Company’s Board of Directors, as of the closing of the Merger, would consist of five members, of whom Neurotrope BioScience had the right to nominate four directors. In addition, (a) as described above, the holders of Series A Preferred Stock have the right to nominate one director, and (b) Hannah Rose Holdings LLC, a pre-reverse merger stockholder of the Company beneficially owning approximately 13.3% of our common stock, has the right to nominate one “independent” director, subject to the Company’s approval, not to be unreasonably withheld. Our Board of Directors is now comprised of six members: Messrs. Abeles, New, Singer and Gottlieb, who were nominated by Neurotrope BioScience, Mr. Haft, who was nominated by the holders of Series A Preferred Stock, and Mr. Freiman, who was nominated by Hannah Rose Holdings LLC. Executive officers are appointed by the Board of Directors and serve at its pleasure. The Company agreed in the Merger Agreement cause to be appointed as soon as practicable after the closing two additional “independent” directors, including the Hannah Rose Holdings nominee, for a total of seven directors. See Item 13, “Certain Relationships and Related Transactions, and Director Independence—Common Stockholders’ Agreement” and “—Voting Agreement” below.
The principal occupation and business experience during the past five years for our executive officers and directors is as follows:
Dr. John H. Abeles – Director and Chairman of the Board of Directors.Dr. Abeles is the President and founder of MedVest, Inc., which has provided consulting services to health care and high technology companies since 1980. His many board positions, both past and present, provide him with a unique perspective to counsel emerging companies on their strategic planning functions, capital raising activities, and operational effectiveness.
Dr. Abeles was born in 1945 in Rhodesia (now Zimbabwe). He practiced Medicine in London, before joining the Pharmaceutical Industry as a Senior Medical Executive, with Sterling Drug, Pfizer Inc. and Revlon Health Care. In 1975 he became a Wall Street health-care analyst with Kidder Peabody until 1980. Dr. Abeles was one of the first physicians to become an analyst at a Wall Street investment firm.
He is also the Managing Member of Dalyda Finance LLC, a family company engaging promising artistic and performing talent.
Since March 1992, Dr. Abeles has been the Founder, Sole Investor and General Partner of Northlea Partners LLLP, a private venture capital and private capital family office headquartered in Boca Raton, Florida.
Since 1998, he has served as the Chairman of UniMedica Inc., a web-enabled medical school education consulting and publishing firm, and as Assistant Professor of Clinical Pharmacology and Therapeutics at the International University of Health Sciences. He has served as a Director of Higuchi Bio-Science Institute, University of Kansas since 1997, and as a Director of the College of Chemistry Advisory Board at the University of California at Berkeley since 2001.
Dr. Abeles received his Medical degree as well as a degree in Pharmacology from the University of Birmingham, England. He is a Fellow of the Royal Society of Medicine, London.
Dr. Jim New – Chief Executive Officer and Director.Dr. New has served as Chief Executive Officer and a director of the Company since November 1, 2012 (from November 1, 2012, until February 28, 2013, as an independent contractor and since February 28, 2013, as an employee of the Company).
Dr. New’s industry experience spans the science and business elements of commercial pharmaceutical operations, as well as both the brand and generic pharmaceutical product development functions in the industry. Most recently he was CEO of AIKO Biotechnology (2011), and prior to this CEO of Lifecycle Pharma (2009). From 2002-2007 he was CEO and co-founder of Abrika Pharmaceuticals, which was acquired by Actavis. From 1999-2001 he worked for Novartis in Switzerland, first as Head of Worldwide Business Development, and later as Head of Mergers and Acquisitions. He was Senior Director of Licensing and Development at Pfizer from 1994-1999 and Director of Corporate Development at GlaxoSmithKline in 1993. From 1988 to 1992 he worked as a Director in the Licensing Department at Bristol-Myers Squibb, and from 1980 -1987 he worked as a medicinal chemist at BMS.
Dr. New received his undergraduate degree in Biology at SUNY Geneseo, a M.S. degree in Biochemistry at the University of Maine, his Ph.D. in Medicinal Chemistry at the University of Buffalo, and his MBA degree from Columbia Business School.
Paul E. Freiman – Director.Mr. Freiman has extensive pharmaceutical and biotechnology industry operating experience as a board member and Chief Executive Officer of private and publicly traded companies and is a pharmacist. He is currently an independent pharmaceutical and biotechnology industry consultant. He serves as Chairman of Chronix BioMedical, Inc., and is a member of the Board of Directors of NovaBay Pharmaceuticals, Inc. In the past, Mr. Freiman served on the boards of Otsuka America, Inc., and several biotechnology companies based in the United States and Singapore. Prior to his current consulting and board member roles, Mr. Freiman was a partner of Burrill Brasil Investimentos based in Rio de Janiero, Brazil. He also served as President and Chief Executive Officer of Neurobiological Technologies, Inc., and as a member of its Board of Directors.
Mr. Freiman also served as Chairman and Chief Executive Officer of Syntex Corporation, which was sold to The Roche Group during his tenure. He was involved with the marketing of Syntex’s lead product, Naprosyn®, and was responsible for moving the product to over-the-counter status, marketed as Aleve®.
Mr. Freiman served on the Board of the Pharmaceutical Research and Manufacturers Association of America (PhRMA) and was its Chairman. He served on a number of industry task forces both domestically and internationally. He also served as Chairman of the University of California (UCSF) Foundation, The United Way of Silicon Valley and a number of not-for-profit organizations over the years.
Mr. Freiman received a B.S. in pharmacy from Fordham University and an honorary doctorate from the Arnold & Marie Schwartz College of Pharmacy.
James Gottlieb – Director. Mr. Gottlieb has extensive senior level experience in a broad range of positions in Washington, D.C., in the U.S. House and Senate, and in the private and non-profit sectors. During his time in Washington, Mr. Gottlieb directed significant congressional policy and oversight matters regarding the Food and Drug Administration (“FDA”) and health-related aspects of the Veterans Administration, the largest civilian health provider in the nation.
Mr. Gottlieb is currently a partner with Capital Counsel LLC, where he leads the commerce team. His clients include a broad range of matters including healthcare, aviation, telecommunications, e-commerce, oversight and investigations and mergers & acquisitions. Prior to joining Capital Counsel LLC and based upon his in-depth experience in legislation, oversight and investigations (including with the FDA) and political roles on Capitol Hill, and foundation management, consulting and strategic advisory roles outside of government, Mr. Gottlieb formed Gottlieb Strategic Consulting, a government affairs firm.
Mr. Gottlieb served as the chief of staff for Representative Ted Weiss (D-NY) and later the Subcommittee on Human Resources & Government Relations of the House Committee on Government Operations (now Committee on Oversight & Government Reform), where he directed a wide range of oversight investigation and legislation in health, education, and veterans’ matters. Mr. Gottlieb served as Senator Jay Rockefeller’s chief counsel and staff director for the Senate Committee on Veterans Affairs. He also served as Senator Rockefeller’s Chief-of-Staff and was responsible for the Senator’s legislative community, economic development and political operations.
Mr. Gottlieb then served as chief management consultant to BRNI as a strategic policy and advocacy advisor and managed the restructuring of this multi-million dollar medical research facility for the study of brain disorders and continued to serve as legal counsel and policy advisor to Senator Rockefeller, BRNI’s Chairman. He served as a Board of Directors member of BRNI and serves as Treasurer of Friends of Jay Rockefeller.
Mr. Gottlieb received a B.A. in Business Administration from Michigan State University, a Master of Education from New York University and a law degree from New York Law School.
Jay M. Haft – Director.Mr. Haft served as Chairman of the Board of Directors at DUSA Pharmaceuticals from 2008 until December 2012, when the company merged win Sun Pharmaceutical Industries, Inc. Since 2005 to the present, Mr. Haft has been a partner and a member of the Investment Committee of Columbus Nova, a private investment arm of the Renova Group. From 1989 to 1994 he was a senior corporate partner of the law firm of Parker, Duryee, Rosoff and Haft, and was of counsel to the same firm from 1994 until 2002. He is currently of counsel to the law firm of Reed Smith LLP. Mr. Haft has served on approximately 30 corporate boards, including his tenure as Chairman of the Executive Committee of Emerson Radio Corporation. He is also active in the non-profit sector as well, particularly in the areas of education and art. Mr. Haft earned his Bachelor’s degree and graduated Phi Beta Kappa from Yale University and earned his law degree from Yale Law School.
William S. Singer – Director.Mr. Singer serves as President of BRNI and is also on its board of directors. He was a partner in the Chicago office of the law firm of Kirkland & Ellis LLP from 1980 until 2006 and has been of-counsel to that firm since that time, and concentrates his practice on corporate, real estate, and legislative matters. Since 2006, he also has been the sole proprietor of Singer Consulting LLC, which advises clients on federal legislation. He has been listed in Crain’s Who’s Who in Chicago Business 2000, 2001, 2002, 2003, and 2004 editions.
Mr. Singer has been prominently active in Chicago public service, serving as an Alderman for several years as a candidate for Mayoral office.
Robert Weinstein – Chief Financial Officer.Mr. Weinstein joined the Company in June 2013 as its acting Chief Financial Officer. He has extensive accounting and finance experience, spanning more than 25 years, as a public accountant, investment banker, healthcare private equity fund principal and chief financial officer.
From September 2011 to current, Mr. Weinstein has been an independent consultant for several healthcare companies in the pharmaceutical and biotechnology industries. From March 2010 to August 2011, he was the Chief Financial Officer of Green Energy Management Services Holdings, Inc., a publicly-traded energy consulting company. From August 2007 to February 2010, Mr. Weinstein served as Chief Financial Officer of Xcorporeal, Inc., a publicly-traded, development-stage medical device company which was sold in March 2010 to Fresenius Medical USA, the largest provider of dialysis equipment and services worldwide.
Mr. Weinstein received his MBA degree in finance and international business from the University of Chicago Graduate School of Business, is a Certified Public Accountant (inactive), and received his BS degree in accounting from the State University of New York at Albany.
Other Key Personnel and Advisor
Dr. Daniel Alkon – Chief Scientific Officer.Dr. Alkon has been the founding Scientific Director of BRNI since 1999. He received his undergraduate degree in chemistry in 1965 at the University of Pennsylvania. After earning his M.D. at Cornell University and finishing an internship in medicine at the Mt Sinai Hospital in New York, he joined the staff of the National Institutes of Health where during his 30 year career he became a medical Director in the U.S. Public Health Service at the NINDS and Chief of the Laboratory of Adaptive Systems. Dr. Alkon occupies the Toyota Chair in Neuroscience at BRNI. In this position he and his team conduct multidisciplinary research on the molecular and biophysical mechanisms of memory and memory dysfunction in psychiatric and neurological disorders, particularly Alzheimer’s disease. He is also a Professor at BRNI and a Professor of Neurology at West Virginia University. Dr. Alkon is not an employee of Neurotrope.
As an internationally recognized pioneer in research on brain-based neural networks and the molecular basis of memory, he has authored hundreds of scientific articles as well as several books including Memory Traces in the Brain by Cambridge University Press, and the popular book Memory’s Voice by Harper Collins.
Dr. Richard Scheyer – Vice President and Chief Medical Officer. Dr. Richard Scheyer has 30 years of professional medical experience, which includes over 16 years dedicated to clinical drug development. Dr. Scheyer was most recently President ofin vivo veritas, a firm specialized in translational medicine and Phase 1 / 2a pharmaceutical development. Prior to this position, Dr. Scheyer was Executive Director Translational Medicine / Clinical Science at Daiichi Sankyo Pharma Development, overseeing early clinical, biomarker, and pharmacogenomic strategy and execution for more than 60 development candidates across therapeutic areas. Dr. Scheyer’s also led the Clinical Advice function in Daiichi Medical Research, responsible for clinical studies across phases and disciplines, and Clinical Pharmacology in Daiichi Sankyo. Before joining Daiichi Sankyo, Dr. Scheyer was Director of Clinical Pharmacology, later Clinical Discovery and Human Pharmacology, at Hoechst Marion Roussel and Aventis Pharma, with special interest in early demonstration of clinical efficacy, and later served as Senior Director and Global Project Team Leader for Phase 2b / 3 programs at Aventis Pharma and Sanofi-Aventis. Dr. Scheyer led the development of teriflunomide (Aubagio) for multiple sclerosis from Phase 1 to Phase 3.
Dr. Scheyer has served on the Foundation for the NIH Biomarkers Consortium, and on Board of Directors of the Serious Adverse Event Consortium. Dr. Scheyer has published over 60 manuscripts and abstracts, with focus on clinical pharmacology and therapeutic activity in areas ranging from diabetes to oncology.
Dr. Scheyer received his B.S. in Physics from Stanford University, his M.D. from The State University of New York, Upstate Medical University, and completed residency training in Neurology and fellowship training in Epilepsy and Clinical Pharmacology at Yale University before joining the Yale faculty, serving as Associate Professor of Neurology.
Ira Weisberg – Vice President, Commercial Operations.Mr. Weisberg is an accomplished pharmaceuticals and biotech executive with over 35 years in business and corporate development experience managing companies, fund raising, product development, sales and distribution channels, compliance, regulatory, managed care, reimbursement systems, licensing, and other corporate and business development transactions in both the U.S. and international markets. Mr. Weisberg worked in both large pharmaceutical companies including American Home Products, Cooper Laboratories and emerging pharmaceutical and biotech companies including Interferon Sciences and LifeCycle Pharma. He has led or been involved with corporate partnering, mergers and acquisitions, new venture formation and financing within a number of therapeutic areas such as cancer and cardiovascular medicine. Mr. Weisberg was President and CEO of AlphaVax, an emerging cancer vaccine development company. He has also led product launches in the pharmaceutical, biopharmaceutical and biotechnology sectors, including managing the Critical Care business for Aventis Behring.
Mr. Weisberg earned his B.S. in Microbiology at St. John’s University.
David Crockford – Vice President, Regulatory Affairs.Mr. Crockford has more than 30 years of professional experience in the biotechnology and pharmaceutical industries. He led the development and obtained marketing approval of 18 drug products, 17 immunodiagnostic tests, and an intraoperative medical device. He co-founded and presided over one of the first publicly-held companies in the field of genetic engineering, pioneering the development and commercial application of monoclonal antibodies. Mr. Crockford has organized, presented and led discussions in many face-to-face meetings and teleconferences with a variety of Divisions and Centers (CBER, CDER and CDRH) at the FDA. He provided the regulatory strategy and direction and led the clinical development of a regenerative peptide for the treatment of patients with certain ailments including neurodegenerative diseases.
Mr. Crockford is the author of a number of articles and sole inventor/co-inventor of approximately twenty patents and applications disclosing compositions, methods of drug use and delivery. He earned a Bachelor of Arts degree in biology from Boston University’s College of Arts and Sciences and completed seminars in clinical chemistry, sponsored by Princeton University/Princeton Hospital, and reproductive medicine at Wayne State’s Mott Center for Human Growth and Development and UCLA Medical School.
Board of Directors
Our Board of Directors is authorized to consist of seven members and currently consists of six members, leaving one vacancy.
Management has been delegated the responsibility for meeting defined corporate objectives, implementing approved strategic and operating plans, carrying on the Company’s business in the ordinary course, managing cash flow, evaluating new business opportunities, recruiting staff and complying with applicable regulatory requirements. The Board of Directors exercises its supervision over management by reviewing and approving long-term strategic, business and capital plans, material contracts and business transactions, and all debt and equity financing transactions and stock issuances.
Director Independence
We are not currently subject to listing requirements of any national securities exchange or inter-dealer quotation system which has requirements that a majority of the board of directors be “independent” and, as a result, we are not at this time required to have our Board of Directors comprised of a majority of “independent directors.” Nevertheless, our Board of Directors has determined that each of Messrs. Freiman, Gottlieb and Haft is “independent” under the applicable standards of the SEC and the NYSE MKT stock market.
Director Attendance at Annual Meetings
Each of our directors is expected to be present at annual meetings of our stockholders absent exigent circumstances that prevent their attendance. Where a director is unable to attend an annual meeting in person but is able to do so by electronic conferencing, we will arrange for the director's participation by means where the director can hear, and be heard by, those present at the meeting. Last year, we did not hold an annual meeting of stockholders.
Board Committees and Meetings
Our Board of Directors has established three committees, each of which is compose solely of independent directors:
| · | The Audit Committee consists of Mr. Haft, as Chairman, Mr. Freiman and Mr. Gottlieb. |
| · | The Compensation Committee consists of Mr. Freiman, as Chairman, Mr. Gottlieb and Mr. Haft. |
| · | The Nominating and Corporate Governance Committee consists of Mr. Gottlieb, as Chairman, Mr. Freiman and Mr. Haft. |
Each of the Committees has a written charter adopted by the Board of Directors; a current copy of each such charter is available to security holders on our web site,www.neurotropebioscience.com.
The Board has at least four regularly scheduled meetings per year. In addition, the Board holds special meetings whenever requested by the Chairman of the Board, the Chief Executive Officer, the President or any director.
During 2013, the Board of Directors held two meetings, and took action by unanimous written consent in lieu of a meeting five times. During 2013, each director attended at least 75% of the aggregate of (i) the total number of meetings of the board of directors (held during the period for which he or she has been a director); and (ii) the total number of meetings held by all committees of the board on which he or she served (during the periods that he or she served).
Nominating and Corporate Governance Committee
The Nominating and Corporate Governance Committee assists the Board in (i) identifying qualified individuals to become directors, (ii) determining the composition of the Board and its committees, (iii) developing succession plans for executive officers, (iv) monitoring a process to assess Board effectiveness and (v) developing and implementing the Company’s corporate governance procedures and policies.
The Nominating and Corporate Governance Committee was established on December 11, 2013, and did not meet in 2013.
The Nominating and Corporate Governance Committee does not have a policy with regard to the consideration of any director candidates recommended by security holders. To date no security holders have made any such recommendations.
Pursuant to our by-laws, nominations of persons for election to the Board of Directors at an annual meeting or at any special meeting of stockholders for the purpose of electing directors may be made by or at the direction of the Board of Directors, by any nominating committee or person appointed for such purpose by the Board of Directors, or by any stockholder of record entitled to vote for the election of directors at the meeting who complies with the following notice procedures. Such nominations, other than those made by, or at the direction of, or under the authority of the Board of Directors, shall be made pursuant to timely notice in writing to the Secretary of the Company by a stockholder of record at such time. To be timely, a stockholder’s notice must be delivered to or mailed and received at the principal executive offices of the Company (a) in the case of an annual meeting, not less than 90 nor more than 120 days prior to the one-year anniversary of the date of the annual meeting of the previous year;provided, however, that if the annual meeting is called for a date that is not within 30 days before or after such anniversary date, notice by the stockholder in order to be timely must be so received not earlier than 120 days prior to such annual meeting and not later than the close of business on the tenth day following the day on which notice of the date of the annual meeting was mailed or public disclosure of the date of the annual meeting was made, whichever first occurs; and (b) in the case of a special meeting of stockholders for the purpose of electing directors, not earlier than 120 days prior to such special meeting and not later than the close of business on the tenth day following the day on which notice of the date of the special meeting was mailed or public disclosure of the date of the special meeting was made, whichever first occurs. Such stockholder’s notice to the Secretary must set forth (a) as to each person whom the stockholder proposes to nominate for election or re-election as a director, (i) the name, age, business address and residence address of the person, (ii) the principal occupation or employment of the person, (iii) the class and number of shares of capital stock of the Company, if any, which are beneficially owned by the person and (iv) any other information relating to the person that is required to be disclosed in solicitations for proxies for election of directors pursuant to Regulation 14A under the Exchange Act or other applicable law; and (b) as to the stockholder giving the notice (i) the name and record address of the stockholder and (ii) the class and number of shares of capital stock of the Company which are beneficially owned by the stockholder. The chairman of the meeting may, if the facts warrant, determine and declare to the meeting that a nomination was not made in accordance with the foregoing procedures, and the defective nomination will be disregarded.
Audit Committee
The Audit Committee (a) assists the Board of Directors in fulfilling its oversight of: (i) the quality and integrity of the Company’s financial statements; (ii) the Company’s compliance with legal and regulatory requirements relating to the Company’s financial statements and related disclosures; (iii) the qualifications and independence of the Company’s independent auditors; and (iv) the performance of the Company’s independent auditors; and (b) prepares any reports that the rules of the SEC require be included in the Company’s annual proxy statement..
The Audit Committee was established on December 11, 2013, and did not meet in 2013.
Each person who served on the Audit Committee during fiscal 2013 is financially literate under the current listing standards of the NYSE MKT and has been determined by the Board of Directors to be independent. The Audit Committee has at this time not determined whether any director is an “audit committee financial expert” within the meaning of Item 407(d)(5) of Regulation S-K.
All directors comprising the Audit Committee have held various executive management positions in which they were responsible for receiving financial information relating to the entities to which they were executive managers. They had, or developed, an understanding of financial statements generally and how those statements are used to assess the financialposition of a company and its operating results. Each member of the Audit Committee also has a significant understanding of the business in which the Company is engaged in and has an appreciation for the relevant accounting principles for the business of the Company. See “Directors and Executive Officers” above for descriptions of the relevant education and experience of each member of the Audit Committee.
At no time since the commencement of the Company’s most recently completed fiscal year was a recommendation of the Audit Committee to nominate or compensate an external auditor not adopted by the Board of Directors.
The Audit Committee is responsible for the oversight of the Company’s financialreporting process on behalf of the Board of Directors and such other matters as specified in the Committee’s charter or as directed by the Board. Our Audit Committee is directly responsible for the appointment, compensation, retention and oversight of the work of any registered public accounting firm engaged by us for the purpose of preparing or issuing an audit report or performing other audit, review or attest services for us (or to nominate the independent registered public accounting firm for stockholder approval), and each such registered public accounting firm must report directly to the Audit Committee. Our Audit Committee must approve in advance all audit, review and attest services and all non-audit services (including, in each case, the engagement and terms thereof) to be performed by our independent auditors, in accordance with applicable laws, rules and regulations.
Compensation Committee
The Compensation Committee (i) assists the Board of Directors in discharging its responsibilities with respect to compensation of the Company’s executive officers and directors, (ii) evaluates the performance of the executive officers of the Company, and (iii) administers the Company’s stock and incentive compensation plans and recommends changes in such plans to the Board as needed.
The Compensation Committee was established on December 11, 2013, and did not meet in 2013..
Family Relationships
There are no family relationships among our Directors or executive officers.
Involvement in Certain Legal Proceedings
None of our directors or executive officers has been involved in any of the following events during the past ten years:
| | · | any bankruptcy petition filed by or against any business of which such person was a general partner or executive officer either at the time of the bankruptcy or within two years prior to that time, except that Mr. Weinstein was the Chief Financial Officer of Able Laboratories, Inc., until November 2004; that company filed for bankruptcy protection in July 2005; |
| | · | any conviction in a criminal proceeding or being subject to a pending criminal proceeding (excluding traffic violations and other minor offences); |
| | · | being subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining, barring, suspending or otherwise limiting his or her involvement in any type of business, securities or banking activities; or |
| | · | being found by a court ofcompetent jurisdiction (in a civil action), the Commission or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated. |
Section 16(a) Beneficial Ownership Reporting Compliance
Since we do not have any class of equity securities registeredpursuant to section 12 of the Exchange Act, this disclosure is not applicable
Code of Ethics
During the fiscal year ended December 31, 2013, we adopted a Code of Ethics and Business Conduct (“Code of Ethics”) for our employees, officers and directors (including our principal executive officer, principal financial officer and principal accounting officer) that complies with SEC regulations. The Code of Ethics is available free of charge on our website atwww.neurotropebioscience.com and is filed as an exhibit to this report.. We intend to timely disclose any amendments to, or waivers from, our code of ethics and business conduct that are required to be publicly disclosed pursuant to rules of the SEC and any securities exchange on which our shares may be listed by filing such amendment or waiver with the SEC.
Stockholder Communication with the Board
Stockholders may communicate with the Board of Directors, members of particular committees or individual directors, by sending a letter to such persons in care of our Chief Executive Officer at our principal executive offices. The Chief Executive Officer has the authority to disregard any inappropriate communications or to take other appropriate actions with respect to any inappropriate communications. If deemed an appropriate communication, the Chief Executive Officer will submit the correspondence to the Chairman of the Board or to any committee or specific director to whom the correspondence is directed. Procedures for sending communications to the Board of Directors can be found on our website atwww.neurotropebioscience.com. Please note that all such communications must be accompanied by a statement of the type and amount of our securities that the person holds; any special interest, meaning an interest that is not derived from the proponent's capacity as a shareholder, of the person in the subject matter of the communication; and the address, telephone number and e-mail address, if any, of the person submitting the communication.
ITEM 11. | EXECUTIVE COMPENSATION |
Summary Compensation Table
The following table sets forth information concerning the total compensation paid or accrued by us during the last two fiscal years ended December 31, 2013, to (i) all individuals that served as our principal executive officer or acted in a similar capacity for us at any time during the fiscal year ended December 31, 2013; (ii) all individuals that served as our principal financial officer or acted in a similar capacity for us at any time during the fiscal year ended December 31, 2013; (iii) two most highly compensated executive officers other than the principal executive officer or principal financial officer that were serving as executive officers at December 31, 2013, and who received total compensation for the fiscal year ended December 31, 2013, in excess of $100,000; and (iv) up to two additional individuals for whom disclosure would have been requires pursuant to clause (iii) above but for the fact that the individual was not serving as an executive officer at December 31, 2013 (collectively, the “named executive officers”). The Compensation Committee of the Board of Directors is responsible for determining executive compensation.
| | | Fiscal | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Year | | | | | | | | | | | | | | | Non-Equity | | | Non-Qualified | | | | | | | |
| Name & | | ended | | | | | | | | | | | | | | | Incentive Plan | | | Deferred | | | All Other | | | | |
| Principal | | December | | | | | | Bonus | | | Stock | | Option | | | Compensation | | | Compensation | | | Compensation | | | | |
| Position | | 31, | | Salary ($) | | | ($) | | | Awards($) | | Awards($) | | | ($) | | | Earnings ($) | | | ($) (4) | | | Total ($) | |
| Marissa | | 2013 | | | - | | | - | | | - | | | - | | | - | | | - | | | - | | | - | |
| Watson (1) | | 2012 | | | - | | | - | | | - | | | - | | | - | | | - | | | - | | | - | |
| Jim New (2) | | 2013 | | $ | 220,873 | | | - | | | - | | $ | 562,540 | | | - | | | - | | $ | 41,666 | | $ | 825,079 | |
| | | 2012 | | | 41,667 | | | - | | | - | | | - | | | - | | | - | | | - | | | 41,677 | |
| Robert | | 2013 | | $ | 65,531 | | | - | | | - | | $ | 529,040 | | | - | | | - | | $ | 80,094 | | $ | 674,665 | |
| Weinstein (3) | | 2012 | | | - | | | - | | | - | | | - | | | - | | | - | | | - | | | - | |
| (1) | On June 20, 2013, Ms. Watson resigned as our sole officer and director. |
| (2) | Reflects compensation received from Neurotrope BioScience, which was formed in 2012, through the date of the reverse Merger in August 2013. Salary was accrued in 2012 but paid in 2013. On August 23, 2013, Mr. New was appointed as our Chief Executive Officer and President. |
| (3) | Mr. Weinstein became our Chief Financial Officer on August 23, 2013. |
| (4) | Represents compensation earned as a consultant prior to becoming an employee. |
Outstanding Equity Awards at Fiscal Year-End
We have one compensation plan approved by our stockholders, the 2013 Plan. The following table provides information regarding 2013 Plan awards for each named executive officer outstanding as of December 31, 2013.
| | | Option awards | | Stock awards | |
| | | | | | | | | | | | | | Equity | |
| | | | | | | | | | | | | | incentive | |
| | | | | | | | | | | | | | plan | |
| | | | | | | | | | | | | Equity | awards: | |
| | | | | | | | | | | | | incentive | Market or | |
| | | | | Equity | | | | | | | | plan | payout | |
| | | | | incentive | | | | | | | | awards: | value of | |
| | | | | plan awards: | | | | | | | Market | Number of | unearned | |
| | | Number of | Number of | Number of | | | | | | Number of | value of | unearned | shares, | |
| | | securities | securities | securities | | | | | | shares or | shares of | shares, | units or | |
| | | underlying | underlying | underlying | | | | | | units of | units of | units or | other | |
| | | unexercised | unexercised | unexercised | | | Option | | | stock that | stock that | other rights | rights that | |
| | | options | options | unearned | | | exercise | Option | | have not | have not | that have | have not | |
| | | (#) | (#) | options | | | price | expiration | | vested | vested | not vested | vested | |
| Name | | exercisable | unexercisable | (#) | | | ($) | date | | (#) | ($) | (#) | ($) | |
| (a) | | (b) | (c) | (d) | | | (e) | (f) | | (g) | (h) | (i) | (j) | |
| Marissa Watson | | - | - | - | | | - | - | | - | - | - | - | |
| Jim New | | 707,000 | - | - | | $ | 1.75 | August 22, 2023(1) | | - | - | - | - | |
| Robert Weinstein | | - | 650,000 | - | | $ | 1.00 | October 1, 2023(2) | | - | - | - | - | |
| (1) | These options were fully vested on the date of grant (August 23, 2013). |
| (2) | These options vest 25% on each of the first four anniversaries of the date of grant (October 2, 2013). |
We have no plans in place and have never maintained any plans that provide for the payment of retirement benefits or benefits that will be paid primarily following retirement including, but not limited to, tax qualified deferred benefit plans, supplemental executive retirement plans, tax-qualified deferred contribution plans and nonqualified deferred contribution plans.
Except as indicated below, we have no contracts, agreements, plans or arrangements, whether written or unwritten, that provide for payments to the named executive officers listed above.
Employment Agreements
Jim New. Dr. Jim New has served as Chief Executive Officer and a director of Neurotrope BioScience since November 1, 2012 (from November 1, 2012, until February 28, 2013, as an independent contractor and from February 28, 2013, as an employee) and of Neurotrope since August 23, 2013. Neurotrope BioScience paid Dr. New $250,000 (on an annualized basis) for his services as Chief Executive Officer of the Company during the period when Dr. New was an independent contractor to Neurotrope (November 1, 2012, until February 28, 2013). Neurotrope BioScience entered into an employment agreement with Dr. New dated as of February 25, 2013. Pursuant to the terms of that employment agreement, upon the closing of the Merger, the Company, as successor to Neurotrope BioScience, became the counterparty to the employment agreement with Dr. New. Pursuant to his employment agreement, Dr. New currently serves as Chief Executive Officer of the Company. Dr. New’s employment agreement continues for a term of four years from February 28, 2013. Pursuant to Dr. New’s employment agreement, his initial salary is $250,000 per year, which will increase to $300,000 per year beginning on the later of (the date of increase is referred to as the “Salary Increase Date”) (i) February 28, 2015 (the second annual anniversary of the commencement of Dr. New’s employment term) and (ii) the Company’s closing on a B round of financing in an amount equal to $25 million. In addition, Dr. New is entitled to a guaranteed cash bonus equal to $50,000 per year during the periods prior to the Salary Increase Date and $100,000 per year during periods after the Salary Increase Date. The Company also is required to make available to Dr. New any benefits and perquisites that may from time to time be generally provided to its executive officers. Until the Company implements a group health insurance policy for its employees, the Company has agreed to reimburse Dr. New for the cost of family health insurance coverage up to $1,254 per month on a grossed-up tax basis. Neurotrope BioScience also paid Dr. New $10,000 as a reimbursement of legal fees incurred in connection with Dr. New’s employment agreement and his purchase of shares of common stock in Neurotrope BioScience. Dr. New is entitled to holidays, personal days and sick days in accordance with the Company’s standard policies and procedures in effect from time to time and four weeks of paid vacation per year. The Company may terminate Dr. New’s employment agreement at any time without cause (as defined in Dr. New’s employment agreement), and Dr. New may resign at any time for good reason (as defined in Dr. New’s employment agreement), upon not less than 30 days prior written notice, in which case the Company would be required to pay Dr. New severance in an amount equal to one year of salary plus payment for any accrued but unused vacation or leave time and one year of family health insurance. In the event the Company were to terminate Dr. New’s employment for cause or if Dr. New were to resign without good reason, then Dr. New would be entitled only to accrued but unpaid salary and accrued but unused vacation or leave time through the date of termination.
Robert Weinstein.The Company is party to an Employment Agreement dated as of October 1, 2013, with Robert Weinstein, pursuant to which he serves as the Company’s Chief Financial Officer and Executive Vice President. Under the terms of the agreement, the Company will pay Mr. Weinstein an annual base salary of not less than $240,000 per year for the period from the effective date to December 31, 2014; and $275,000 per year for the period January 1, 2015 to December 31, 2015. Such annual base salary may be reviewed annually and increased (but not decreased) at the discretion of the Board or a committee thereof, provided, however, that the salary will, at a minimum, be increased annually, beginning January 1, 2016, based upon the percentage increase in the Consumer Price Index for the immediately preceding year. The Company will pay Mr. Weinstein an annual incentive bonus of no less than $35,000 for the year ending December 31, 2013; an annual bonus of no less than $50,000 for the year ending December 31, 2014; and an annual bonus targeted at 50% of his annual base salary for all years beginning January 1, 2015, to be earned and payable based upon attainment of annual performance goals as determined by the Board or a committee thereof. Mr. Weinstein’s annual bonus opportunity may be periodically reviewed and increased at the discretion of the Board or a committee thereof. Mr. Weinstein will also be eligible to participate in all Company benefits generally available to the Company’s officers in accordance with the terms of those benefit plans and all retirement, life, disability, medical and dental plan benefits generally available to the Company’s officers in accordance with the terms of those plans.
Pursuant to the employment agreement, the Board granted an incentive stock option to Mr. Weinstein under the Company’s 2013 Equity Incentive Plan to purchase 650,000 shares of the Company’s common stock. The option vests with respect to 162,500 shares on each of the first, second, third and fourth anniversaries of October 1, 2013, subject to the executive’s continued employment with the Company on each such day. Mr. Weinstein will be entitled to additional options and/or equity-based awards as determined in the discretion of the Board or a committee thereof. All of his options and/or equity awards will cease vesting as of the date of termination of the employment agreement, provided that in the event of (i) Mr. Weinstein’s termination for Good Reason (defined in the Agreement) or (ii) termination of his employment by the Company without Cause (defined in the Agreement), his options and any other equity awards will be deemed to have vested as of the date of termination with respect to that number of shares that would have vested had his employment with the Company continued for a period of one year after the date of termination, and provided, further, that if Mr. Weinstein’s termination for Good Reason or the termination of his employment by the Company without Cause occurs within six months after the occurrence of a change of control of the Company, then all of his options and any other equity awards will be deemed to have vested as of the date of termination.
If Mr. Weinstein’s employment is terminated by the Company for a reason other than Cause or by him for Good Reason, and subject to Executive's compliance with other terms of the employment agreement, and certain other conditions, then the Company will pay him a severance amount equal to his annual base salary, payable in a single lump sum. In addition, if he elects health care continuation coverage under COBRA, the Company will pay for such health insurance coverage for a period of 18 months following the termination of his employment, as the same rate as it pays for health insurance coverage for its active employees (with Mr. Weinstein required to pay for any employee-paid portion of such coverage). If Mr. Weinstein’s employment is terminated by non-renewal or due to his death or disability, he will be entitled to any unpaid prorated Annual Bonus for the year in which his employment terminates.
The Company will reimburse Mr. Weinstein for reasonable attorney’s fees and expenses that he incurs in connection with the negotiation, preparation and execution of the employment agreement up to $10,000.
Subject to earlier termination by Mr. Weinstein’s death or disability, or by the Company for Cause, the term of the employment agreement is four years and will be extended automatically for successive one-year periods, unless either party gives written notice of termination to the other party at least 90 days prior to the end of the then-current term.
Director Compensation
The Company currently does not pay any cash compensation to members of its board of directors for their services as directors of the Company, except that it pays Dr. Abeles $9,000 per month in consideration of his service as Chairman of the board of directors. However, the Company reimburses its directors for all reasonable out-of-pocket expenses incurred in connection with their attendance at meetings of the board of directors. The Company will grant to each new director, at the time of such director's appointment, an option to purchase 250,000 common shares, to the Chairman of the Audit Committee, at the time of his or her appointment, an option to purchase an additional 50,000 common shares, and to each other member of the Chairman of the Audit Committee, at the time of his or her appointment, an option to purchase an additional 25,000 common shares, all such options with a term of ten years, exercisable at the fair market value per share of the common stock on the date of grant, which will vest 20% per year for each of five years after the date of grant (subject to acceleration of vesting upon a change of control of the Company).
The following table provides information concerning the compensation of our directors for the year ended December 31, 2013.
| | | | | | | | | | | | Nonqualified | | | | | | |
| | | Fees earned | | | | | | | Non-equity | | deferred | | | | | | |
| | | or paid in | | Stock | | | Option | | incentive plan | | compensation | | All other | | | | |
| | | cash | | awards | | | awards | | compensation | | earnings | | compensation | | | Total | |
| Name | | ($) | | ($) | | | ($) | | ($) | | ($) | | ($) | | | ($) | |
| (a) | | (b) | | (c) | | | (d) | | (e) | | (f) | | (g) | | | (h) | |
| John Abeles | $ | 45,000 | | - | | $ | 205,214 | | - | | - | | - | | $ | 250,214 | |
| Jim New | | - | | - | | | - | | - | | - | | - | | | - | |
| Paul E. Freiman | | - | | - | | | 313,096 | | - | | - | | - | | | 313,096 | |
| James Gottlieb | | - | | - | | | 322,317 | | - | | - | | - | | | 322,317 | |
| Jay M. Haft | | - | | - | | | 266,912 | | - | | - | | - | | | 266,912 | |
| William Singer | | - | | - | | | 246,256 | | - | | - | | - | | | 246,256 | |
ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The following table sets forth information with respect to the beneficial ownership of our Common Stock as of April 14, 2014 (the “Determination Date”), by (i) each stockholder known by us to be the beneficial owner of more than 5% of our Common Stock or Series A Preferred Stock (our only classes of voting securities), (ii) each of our directors and executive officers, and (iii) all of our directors and executive officers as a group. To the best of our knowledge, except as otherwise indicated, each of the persons named in the table has sole voting and investment power with respect to the shares of our Common Stock or Series A Preferred Stock beneficially owned by such person, except to the extent such power may be shared with a spouse. To our knowledge, none of the shares listed below are held under a voting trust or similar agreement, except as noted. Other than the Merger, to our knowledge, there is no arrangement, including any pledge by any person of securities of the Company or any of its parents, the operation of which may at a subsequent date result in a change in control of the Company.
Unless otherwise indicated in the following table, the address for each person named in the table is c/o Neurotrope, Inc.,10732 Hawk’s Vista Street, Plantation, Florida 33324, USA.
| | | | | Percent of Common | | Series A Preferred | | Percent of Series A | |
| Name and Address | | Common Stock | | Stock Beneficially | | Stock Beneficially | | Preferred Stock | |
| of Beneficial Owner | | Beneficially Owned | | Owned(1) | | Owned | | Beneficially Owned(1) | |
| | | | | | | | | | |
Neurosciences Research Ventures, Inc.(2)
364 Patteson Drive, #279
Morgantown, WV 26505 | | 10,236,250 | | 43.5 | % | 0 | | - | |
| | | | | | | | | | |
Hannah Rose Holdings, LLC(3)
101 Grovers Mill Road Suite #200
Lawrenceville, NJ 08648 | | 3,389,424 | | 14.6 | % | 1,375,000 | | 6.0 | % |
| | | | | | | | | | |
E. Jeffrey Peierls(4)
73 South Holman Way
Golden, CO 80401 | | 1,825,000 | | 7.7 | % | 1,825,000 | | 8.0 | % |
| | | | | | | | | | |
Charles Ramat(5)
111 Jim Stephenson Rd.
Swan Lake, NY 12783 | | 1,231,938 | | 5.3 | % | 1,000,000 | | 4.4 | % |
| | | | | | | | | | |
Dr. Jim New(6) | | 5,303,100 | | 23.5 | % | 0 | | - | |
| | | | | | | | | | |
Robert Weinstein(7) | | 0 | | - | | 0 | | - | |
| | | | | | | | | | |
Dr. John H. Abeles(8) | | 6,673,424 | | 28.2 | % | 750,000 | | 3.3 | % |
| | | | | | | | | | |
William Singer(9) | | 48,329 | | 0.2 | % | 0 | | - | |
| | | | | | | | | | |
Paul Freiman(10) | | 40,192 | | 0.2 | % | 0 | | - | |
| | | | | | | | | | |
James Gottlieb(11) | | 27,863 | | 0.1 | % | 0 | | - | |
| | | | | | | | | | |
Jay Haft(12) | | 119,315 | | 0.5 | % | 74,000 | | 0.3 | % |
| | | | | | | | | | |
| All directors and executive officers as a group (7 persons) | | 11,262,223 | | 45.8 | % | 824,000 | | 3.6 | % |
| (1) | Applicable percentage ownership is based on 21,889,006 shares of common stock outstanding and 22,851,000 shares of Series A Preferred Stock outstanding as of the Determination Date, together with securities exercisable or convertible into shares of common stock or Series A Preferred Stock, respectively, within 60 days of the Determination Date for each shareholder. Each share of Series A Preferred Stock is convertible at any time into one share of common stock, at the option of the holder. Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities. The shares issuable pursuant to the exercise or conversion of such securities are deemed outstanding for the purpose of computing the percentage of ownership of the security holder, but are not treated as outstanding for the purpose of computing the percentage of ownership of any other person. |
| (2) | Includes 1,662,500 shares underlying stock options held by Neurosciences Research Ventures, Inc. that are vested as of the Determination Date or will vest within 60 days thereafter. Does not include 451,250 Indemnification Escrow Shares that Neurosciences Research Ventures, Inc. may be entitled to receive under the Indemnification Escrow Agreement (as defined below), which Dr. Jim New, as Indemnification Representative, has the sole right to vote. (See Item 13, “Certain Relationships and Related Transactions, and Director Independence—Merger Agreement” below.) Neurosciences Research Ventures, Inc. is an affiliate of the Blanchette Rockefeller Neurosciences Institute. In addition to such shares, Neurosciences Research Ventures, Inc. may be deemed to be in a group with Dr. Daniel Alkon, Northlea Partners LLLP, Dr. Jim New, Hannah Rose Holdings, LLC and another stockholder, and thereby deemed to beneficially own any shares that are beneficially owned by such shareholders, as a result of their agreement to vote their shares in the Company in favor of certain matters pursuant to an Amended and Restated Stockholders Agreement and a Voting Agreement, each dated August 23, 2013 (see also “Certain Relationships and Related Transactions, and Director Independence”). |
| (3) | Hannah Rose Holdings, LLC is controlled by Matt Rosenblum. Hannah Rose Holdings, LLC and Mr. Rosenblum may be deemed to be in a group with Neurosciences Research Ventures, Inc., Dr. Daniel Alkon, Dr. Jim New, Northlea Partners LLLP and another stockholder, and thereby deemed to beneficially own any shares that are beneficially owned by such shareholders, as a result of their agreement to vote their shares in the Company in favor of certain matters pursuant to a Voting Agreement dated August 23, 2013. (See also “Certain Relationships and Related Transactions, and Director Independence”). |
| (4) | The shares of common stock and Series A Preferred Stock indicated as beneficially owned by E. Jeffrey Peierls include shares of Series A Preferred Stock that are owned by Brian E. Peierls, E. Jeffrey Peierls, The Peierls Bypass Trust, U.D.E.F. Peierls for Brian E. Peierls, U.D.E.F. Peierls for E. Jeffrey Peierls, U.D.J.N. Peierls for Brian Eliot Peierls, U.D.J.N. Peierls for E. Jeffrey Peierls, U.D.E.S. Peierls for E.F. Peierls et al., U.W.E.S. Peierls for Brian E. Peierls Accumulation, U.W.E.S. Peierls for E. Jeffrey Peierls Accumulation, U.W.J.N. Peierls for Brian E. Peierls, U.W.J.N. Peierls for E. Jeffrey Peierls, The Peierls Foundation, Inc., U.D. Ethel F. Peierls Charitable Lead Trust and U.W. Libby Peierls Marital Trust. E. Jeffrey Peierls has sole power to vote or direct the vote, and to dispose or direct the disposition, of such shares of common stock and Series A Preferred Stock. |
| (5) | Includes 231,938 shares underlying stock options beneficially owned by Mr. Ramat that are vested as of the Determination Date or will vest within 60 days thereafter, but does not include 372,466 shares underlying stock options that will not vest within 60 days after the Determination Date. The shares of common stock and Series A Preferred Stock indicated as beneficially owned by Charles Ramat are held by Mr. Ramat, Ramat Consulting and NTR21 Holdings, LLC. Mr. Ramat is an affiliate of each of Ramat Consulting and NTR21 Holdings, LLC, and has sole power to vote or direct the vote, and to dispose or direct the disposition, of such shares of common stock and Series A Preferred Stock. |
| (6) | Includes 707,000 shares underlying stock options held by Mr. New that are vested as of the Determination Date or will vest within 60 days thereafter. Includes 950,000 Indemnification Escrow Shares held in escrow under the Indemnification Escrow Agreement, which Mr. New, as Indemnification Representative, has the sole right to vote until such shares are released from escrow. Mr. New disclaims beneficial ownership of such shares except for the 191,900 shares in which he has a pecuniary interest. (See Item 13, “Certain Relationships and Related Transactions, and Director Independence—Merger Agreement” below.) In addition to the shares of common stock indicated as beneficially owned by Dr. Jim New, Dr. New may be deemed to be in a group with Neurosciences Research Ventures, Inc., Dr. Daniel Alkon, Northlea Partners LLLP, Hannah Rose Holdings, LLC and another stockholder, and thereby deemed to beneficially own any shares that are beneficially owned by such shareholders, as a result of their agreement to vote their shares in the Company in favor of certain matters pursuant to an Amended and Restated Stockholders Agreement and a Voting Agreement, each dated August 23, 2013 (see also “Certain Relationships and Related Transactions, and Director Independence”). |
| (7) | Does not include 650,000 shares underlying stock options held by Mr. Weinstein that will not vest within 60 days after the Determination Date. |
| (8) | Includes 995,774 shares underlying stock options held by Mr. Abeles that are vested as of the Determination Date or will vest within 60 days thereafter, but does not include 209,726 shares underlying stock options that will not vest within 60 days after the Determination Date. Does not include 259,350 Indemnification Escrow Shares that Mr. Abeles may be entitled to receive under the Indemnification Escrow Agreement, which Dr. Jim New, as Indemnification Representative, has the sole right to vote. The shares of common stock and Series A Preferred Stock indicated as beneficially owned by Dr. John H. Abeles are held by Northlea Partners LLLP. Dr. Abeles is an affiliate of Northlea Partners LLLP and has sole power to vote or direct the vote, and to dispose or direct the disposition, of such shares of common stock and Series A Preferred Stock. In addition to such shares, Dr. Abeles and Northlea Partners LLLP may be deemed to be in a group with Neurosciences Research Ventures, Inc., Dr. Daniel Alkon, Dr. Jim New, Hannah Rose Holdings, LLC and another stockholder, and thereby deemed to beneficially own any shares that are beneficially owned by such shareholders, as a result of their agreement to vote their shares in the Company in favor of certain matters pursuant to an Amended and Restated Stockholders Agreement and a Voting Agreement, each dated August 23, 2013 (see also “Certain Relationships and Related Transactions, and Director Independence”). |
| (9) | Includes 48,329 shares underlying stock options held by Mr. Singer that are vested as of the Determination Date or will vest within 60 days thereafter, but does not include 251,671 shares underlying stock options that will not vest within 60 days after the Determination Date. |
| (10) | Includes 40,192 shares underlying stock options held by Mr. Freiman that are vested as of the Determination Date or will vest within 60 days thereafter, but does not include 284,808 shares underlying stock options that will not vest within 60 days after the Determination Date. |
| (11) | Includes 27,863 shares underlying stock options held by Mr. Gottlieb that are vested as of the Determination Date or will vest within 60 days thereafter, but does not include 247,137 shares underlying stock options that will not vest within 60 days after the Determination Date. |
| (12) | Includes 45,315 shares underlying stock options held by Mr. Haft that are vested as of the Determination Date or will vest within 60 days thereafter, but does not include 254,685 shares underlying stock options that will not vest within 60 days after the Determination Date. |
ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
SEC rules require us to disclose any transaction or currently proposed transaction in which the Company is a participant and in which any related person has or will have a direct or indirect material interest involving the lesser of $120,000.00 or one percent (1%) of the average of the Company’s total assets as of the end of last two completed fiscal years. A related person is any executive officer, director, nominee for director, or holder of 5% or more of the Company’s common stock, or an immediate family member of any of those persons.
The descriptions set forth above under the captions “Executive Compensation—Employment Agreements” and —Director Compensation” are incorporated herein by reference.
Merger Agreement
On August 23, 2013 (the “Closing Date”), the Company, Acquisition Sub and Neurotrope BioScience entered into an Agreement and Plan of Merger and Reorganization (the “Merger Agreement”), which closed on the same date. Pursuant to the terms of the Merger Agreement, Acquisition Sub merged with and into Neurotrope BioScience, which was the surviving corporation and thus became a wholly-owned subsidiary of Neurotrope, Inc.
Pursuant to the Merger, we ceased to engage in the business of providing software solutions to deliver geo-location targeted coupon advertising to mobile internet devices and acquired the business of Neurotrope BioScience to engage in developing two product platforms, including a diagnostic test for Alzheimer’s Disease (“AD”) and a drug candidate called bryostatin for the treatment of Alzheimer’s Disease, both of which are in the clinical testing stage. See “Split-Off” below.
At the closing of the Merger, (a) each of the 19,000,000 shares of Neurotrope BioScience’s common stock issued and outstanding immediately prior to the closing of the Merger was converted into one share of our Common Stock, and (b) each of the 21,920,000 shares of Neurotrope BioScience’s Series A convertible preferred stock issued and outstanding immediately prior to the closing of the Merger was converted into one share of our Series A Preferred Stock. As a result, an aggregate of (a) 19,000,000 shares of our Common Stock were issued to the holders of Neurotrope BioScience’s common stock, and (b) 21,920,000 shares of our Series A Preferred Stock were issued to the holders of Neurotrope BioScience’s Series A convertible preferred stock. In addition, warrants issued to the placement agents and permitted sub-agents to purchase 900,000 shares of Neurotrope BioScience’s common stock were converted into Agent Warrants (as defined below) to purchase 900,000 shares of our Common Stock, and warrants issued to the placement agents and permitted sub-agents to purchase 1,217,000 shares of Neurotrope BioScience’s Series A convertible preferred stock were converted into Agent Warrants to purchase 1,217,000 shares of our Series A Preferred Stock. Neurotrope BioScience did not have any other stock purchase warrants or any stock options outstanding at the time of the Merger. The warrants to purchase 900,000 shares of our Common Stock and 1,217,000 shares of our Series A Preferred Stock issued on conversion of the warrants issued by Neurotrope BioScience as described above are referred to, collectively, as the “Agent Warrants.”
The Merger Agreement contained customary representations and warranties and pre- and post-closing covenants of each party and customary closing conditions. Breaches of the representations and warranties will be subject to customary indemnification provisions, subject to specified aggregate limits of liability.
The Merger Agreement provides that in the event that until the second anniversary of the Closing Date, the Company incurs damages (a) with respect to liabilities, obligations or indebtedness of the Split-Off Subsidiary (as defined below), whenever accruing, or of the Company accruing prior to the Merger and not previously disclosed, or (b) that a pre-Merger common stockholder of Neurotrope BioScience is entitled to under the indemnification provisions of the Merger Agreement, then the Company will issue to, in the case of clause (a) above, all of the pre-Merger common stockholders of Neurotrope BioScience, or, in the case of clause (b) above, such pre-Merger common stockholder of Neurotrope BioScience so entitled to indemnification, a number of shares of our stock that (in addition to the merger consideration shares to which such person was entitled) would result from dividing the whole dollar amount representing such damages by $1.00 (subject to equitable adjustment in the event of any stock split, stock dividend, reverse stock split or similar event). The limit on the aggregate number of shares of our stock issuable under this provision is 500,000 shares.
Pursuant to the Merger Agreement, the pre-Merger common stockholders of Neurotrope BioScience received initially only 95% of the Company Common Stock that they were entitled to receive; the remaining 5% of such shares, totaling 950,000 shares (the “Indemnification Escrow Shares”) were placed in escrow pursuant to an Indemnification Escrow Agreement among the Company, Dr. Jim New, as Indemnification Representative, and Gottbetter & Partners, LLP, as escrow agent. In the event that the Company should have a claim for indemnification under the Merger Agreement against any of the pre-Merger common stockholders of Neurotrope BioScience, it would have the right to sell some or all of the Indemnification Escrow Shares to satisfy such claim, and such right of sale would be the Company’s exclusive remedy for such claims against such persons except in the case of fraud or willful misconduct by them. If not so sold, the Indemnification Escrow Shares will be released from escrow and distributed to such holders on or about August 23, 2015. The Indemnification Representative has sole power to vote the Indemnification Escrow Shares while they are in escrow.
The Merger will be treated as a recapitalization of the Company for financial accounting purposes. Neurotrope BioScience will be considered the acquirer for accounting purposes, and the historical financial statements of Neurotrope, Inc., before the Merger will be replaced with the historical financial statements of Neurotrope BioScience before the Merger in all future filings with the SEC.
The parties have taken all actions necessary to ensure that the Merger is treated as a tax-free exchange under Section 351 of the Internal Revenue Code of 1986, as amended.
The issuance of shares of our Common Stock and Series A Preferred Stock to holders of Neurotrope BioScience’s capital stock in connection with the Merger was not registered under the Securities Act, in reliance upon the exemption from registration provided by Section 4(2) of the Securities Act, which exempts transactions by an issuer not involving any public offering, and Regulation D promulgated by the SEC under that section. These securities may not be offered or sold in the United States absent registration or an applicable exemption from the registration requirement, and are subject to further contractual restrictions on transfer as described below.
We also agreed not to register under the Securities Act the resale of the shares of our Common Stock received in the Merger by our officers, directors and key employees and holders of 10% or more of our Common Stock for a period of two years following the closing of the Merger.
The form of the Merger Agreement is filed as an exhibit to this report.All descriptions of the Merger Agreement herein are qualified in their entirety by reference to the text thereof filed as an exhibit hereto, which is incorporated herein by reference.
Split-Off
Upon the closing of the Merger, under the terms of a split-off agreement and a general release agreement, the Company transferred all of its pre-Merger operating assets and liabilities to its wholly-owned special-purpose subsidiary, Blue Flash Communications Corp., a Nevada corporation (“Split-Off Subsidiary”), formed on August 15, 2013. Thereafter, pursuant to the split-off agreement, the Company transferred all of the outstanding shares of capital stock of Split-Off Subsidiary to Marisa Watson, the pre-Merger majority stockholder of the Company, and the former sole officer and director of the Company (the “Split-Off”), in consideration of and in exchange for (i) the surrender and cancellation of an aggregate of 20,178,000 shares of our Common Stock held by Ms. Watson (which were cancelled and will resume the status of authorized but unissued shares of our Common Stock) and (ii) certain representations, covenants and indemnities. All descriptions of the split-off agreement and the general release agreement herein are qualified in their entirety by reference to the text thereof filed as exhibits hereto, which are incorporated herein by reference.
The PPO
Concurrently with the closing of the Merger and in contemplation of the Merger, Neurotrope BioScience held a closing of its private placement offering (the “PPO”) in which it sold 11,533,375 shares of its Series A preferred stock, at a price of $1.00 per share, for gross proceeds (before deducting commissions and expenses of the offering) of $11,533,375. This closing of the PPO and the closing of the Merger were conditioned upon each other.
Neurotrope BioScience had previously held closings of the PPO between February and May 2013 for 10,386,625 shares of its Series A convertible preferred stock, for a purchase price of $1.00 per share, for aggregate gross proceeds of $10,386,625 (before deducting placement agent fees and expenses of the offering). All of the outstanding shares of Neurotrope BioScience’s Series A convertible preferred stock were converted on a one-for-one basis into shares of Neurotrope, Inc.’s Series A convertible preferred stock in connection with the Merger.
Subsequent to the closing of the Merger, held a final closing for 1,080,000 additional shares of our Series A convertible preferred stock at $1.00 per share, resulting in gross proceeds of $1,080,000, for a total of $23,000,000 of gross proceeds raised between February and October 2013.
A description of the powers, designations, preferences, limitations, restrictions and relative rights of our Series A convertible preferred stock, and of certain other rights thereof and restrictions thereon under the Preferred Stockholders Agreement entered into by the Company with the holders of the Series A convertible preferred stock, is set forth in Item 2.01 of Amendment No. 1 to the Company’s Current Report on Form 8-K filed with the SEC on August 30, 2013, under the caption “Description of Securities—Preferred Stock,” which is incorporated herein by reference, and under the caption “Registration Rights” below.
Northlea Partners LLLP, which is controlled by our Chairman of the Board, Dr. John Abeles, purchased an aggregate of 750,000 shares of Series A Preferred Stock in the PPO for an aggregate purchase price of $750,000. Jay Haft, our director, purchased an aggregate of 25,000 shares of Series A Preferred Stock in the PPO for an aggregate purchase price of $25,000.
Neurotrope BioScience agreed to pay the placement agents in the PPO, EDI Financial, Inc. and Allied Beacon Financial, Inc., registered broker-dealers, a cash commission of 10% of the gross funds raised from investors in the PPO. In addition, the placement agents received (a) for the first $12,000,000 of aggregate gross PPO proceeds (including the prior closings), (i) warrants exercisable for a period of ten (10) years to purchase a number of shares of Neurotrope BioScience common stock equal to 7.5% of the number of shares of Neurotrope BioScience Series A convertible preferred stock sold to investors introduced by them, with a per share exercise price of $0.01, and (ii) warrants exercisable for a period of ten (10) years to purchase a number of shares of Neurotrope BioScience Series A convertible preferred stock equal to 2.5% of the number of shares of Neurotrope BioScience Series A convertible preferred stock sold to investors introduced by them, with a per share exercise price of $1.00; and (b) on aggregate gross PPO proceeds in excess of $12,000,000, warrants exercisable for a period of ten (10) years to purchase a number of shares of Neurotrope BioScience Series A convertible preferred stock equal to 10% of the number of shares of Neurotrope BioScience Series A convertible preferred stock sold to investors introduced by them, with an exercise price of $1.00 per share.
As a result of the foregoing, the placement agents and their permitted sub-agents were paid an aggregate commission of $2,225,000 and were issued warrants purchase an aggregate of 900,000 shares of Neurotrope BioScience common stock and warrants to purchase an aggregate of 1,325,000 shares of Neurotrope BioScience Series A convertible preferred stock, which were converted into Agent Warrants to purchase an aggregate of 900,000 shares of our Common Stock and an aggregate of 1,325,000 shares of our Series A Preferred Stock, as described more fully above. Neurotrope BioScience was also required to reimburse the placement agents $25,000 of legal expenses incurred in connection with the PPO. Neurotrope BioScience agreed to indemnify the placement agents and their sub-agents to the fullest extent permitted by law, against certain liabilities that may be incurred in connection with this PPO, including certain civil liabilities under the Securities Act, and, where such indemnification is not available, to contribute to the payments the placement agents and its sub-agents may be required to make in respect of such liabilities.
The PPO was exempt from registration under Section 4(2) of the Securities Act of 1933, as amended, in reliance upon the exemption provided by Rule 506(b) of Regulation D promulgated by the SEC thereunder. The PPO was sold to “accredited investors,” as defined in Regulation D.
A description of the voting powers, preferences and other rights of the Series A Convertible Preferred Stock is set forth in Amendment No. 1 to the Company’s Current Report on Form 8-K, filed with the Securities and Exchange Commission on August 30, 2013, under the caption “Description of Securities—Preferred Stock—Series A Preferred Stock,” which description is incorporated herein by reference. A copy of the Certificate of Designations, Preferences and Rights for the Series A Convertible Preferred Stock, as filed with the Nevada Secretary of State, is filed as an exhibit to the Company’s Quarterly Report on Form 10-Q for the quarter ended July 31, 2013.
The holders of the Agent Warrants have agreed that, until the Company raises at least $25,000,000 in a financing subsequent to the PPO, our Board of Directors (acting through at least a majority) will have the right to vote any shares of our common stock issued upon exercise of Agent Warrants.
All descriptions of the Agent Warrants herein are qualified in their entirety by reference to the text thereof filed as exhibits hereto, which are incorporated herein by reference.
Registration Rights
In connection with the PPO, we entered into a Preferred Stockholders Agreement, pursuant to which we have agreed to file a registration statement registering for resale the shares of our Common Stock underlying the shares of Series A Preferred Stock issued in the Merger in exchange for the shares of Neurotrope BioScience Series A convertible preferred stock sold in the PPO (the “Registrable Securities”) as follows: If at any time after the earlier of (i) five years after the closing of the PPO or (ii) 180 days after the effective date of the registration statement for an underwritten public offering of equity securities of the Company under the Securities Act with a price to the public of at least $5.00 per share and aggregate gross proceeds to the Company of at least $30,000,000 (a “QPO”), the Company receives a request from holders of at least 40% of the Registrable Securities then outstanding that the Company file a Form S-1 registration statement with the SEC with respect to Registrable Securities having an anticipated aggregate offering price, net of selling expenses, of at least $15,000,000, then the Company shall (x) within ten (10) days after the date such request is given, give notice thereof to all other holders of Registrable Securities; and (y) as soon as practicable, and in any event within sixty (60) days after the date such request is given, file a Form S-1 registration statement under the Securities Act covering all Registrable Securities that the initiating holders requested to be registered and any additional Registrable Securities requested to be included in such registration by any other holders, subject to certain limitations.
If at any time when it is eligible to use a Form S-3 registration statement, the Company receives a request from holders of at least thirty percent (30%) of the Registrable Securities then outstanding that the Company file a Form S-3 registration statement with respect to outstanding Registrable Securities of such holders having an anticipated aggregate offering price, net of selling expenses, of at least $1,000,000, then the Company shall (i) within ten (10) days after the date such request is given, give a notice to all other holders of Registrable Securities; and (ii) as soon as practicable, and in any event within forty-five (45) days after the date such request is given, file a Form S-3 registration statement under the Securities Act covering all Registrable Securities requested to be included in such registration, subject to certain limitations.
If the Company proposes to register (including, for this purpose, a registration effected by the Company for stockholders other than the holders of Series A Preferred Stock) any of its Common Stock under the Securities Act in connection with the public offering of such securities solely for cash (other than certain excluded registrations), the Company shall give each holder of Registrable Securities notice of such registration, and upon the request of each holder, shall, subject to certain limitations, cause to be registered all of the Registrable Securities that each such Holder has requested to be included in such registration.
We have agreed to maintain the effectiveness of any registration statement referred to above for at least 120 days after the date on which the registration statement is declared effective by the SEC, or, if earlier, until the distribution contemplated by the registration statement has been completed.
We will pay all expenses in connection with any registration obligation provided in the registration rights agreement, including, without limitation, all registration, filing, stock exchange fees, printing expenses, all fees and expenses of complying with applicable securities laws, and the fees and disbursements of our counsel and of our independent accountants. Each investor will be responsible for its own underwriting discounts and commissions, transfer taxes and the expenses of any attorney or other advisor such investor decides to employ.
Registration rights shall terminate upon the earliest of (i) the date when shares of registrable securities are eligible to be sold without restriction under Rule 144, (ii) the closing of a Deemed Liquidation Event, as such term is defined in the Company's certificate of incorporation and (iii) the fifth anniversary of the effective date of the company's first QPO after the Closing Date.
All descriptions of the Preferred Stockholders Agreement herein are qualified in their entirety by reference to the text thereof filed as an exhibit hereto, which is incorporated herein by reference.
The holders of the Agent Warrants have “piggyback” registration rights for the shares Common Stock underlying the Agent Warrants (including shares of Common Stock issuable upon conversion of the Series A Preferred Stock underlying the Agent Warrants) with respect to any registration statement filed by the Company that would permit the inclusion of such underlying shares, and subject to customary limitations.
Common Stockholders’ Agreement
The Company has entered into an Amended and Restated Stockholders Agreement (the "Common Stockholders Agreement"), dated August 23, 2013, with all of the holders of Neurotrope BioScience’s common stock prior to the Merger: Neurosciences Research Ventures, Inc. (“NRV”), Dr. Daniel Alkon (“Alkon”), Northlea Partners LLLP (“Northlea”) and Dr. Jim New (“New”), which amends and restates in its entirety the agreement between such stockholders and Neurotrope BioScience in effect prior to the Merger. Pursuant to the Common Stockholders Agreement, the parties agreed to certain corporate governance matters pertaining to the Company (including with respect to the composition of the Board of Directors) and to certain restrictions on the transfer of shares of Common Stock held by such parties. Each shareholder who is a party to the Common Stockholders Agreement agreed that:
| | · | the authorized number of directors of the Company will be seven, with six directors elected by the holders of a majority of the outstanding Common Stock, voting as a separate class (each, a “Common Director”) and one director elected by the holders of a majority of the outstanding Series A Preferred Stock, voting as a separate class on an as-converted basis; |
| | · | the following five persons shall be nominated and elected to the Board as Common Directors: |
| | o | two representatives designated by NRV (each, an “NRV Designee”), who are currently William S. Singer and James Gottlieb; |
| | o | two representatives designated by designated by the “Majority Abeles Stockholders” (as defined therein) (each, an “Abeles Designee”), who initially shall be Dr. John Abeles and Dr. Jim New; and |
| | o | one independent representative designated by the Board (the “Neurotrope Designee”), which designee has not been determined as of the date of this report; and |
| | · | subject to the provisions of applicable law, no NRV Designee, Abeles Designee or Neurotrope Designee shall be removed from the Board unless such removal is requested in writing by the party that designated such designee. |
In addition, each shareholder who is a party to the Common Stockholders Agreement granted the other parties (including the Company) certain rights of first refusal with respect to the sale or transfer of his or its shares of Common Stock in the Company. Each shareholder who is a party to the Common Stockholders Agreement also agreed to certain tag-along rights and drag-along obligations with respect to their shares of common stock in the Company. In addition, Northlea Partners LLLP, Dr. Abeles and Dr. Jim New agreed to vote together for certain specified matters that are approved by the NRV Designees (and against such matters that are not approved by the NRV Designees), including with respect to (i) licensing or sublicensing of intellectual property licensed from BRNI, (ii) certain fundamental changes or changes in ownership with respect to the Company and (iii) the liquidation, dissolution or winding up of the Company. The voting obligations described in the preceding sentence will terminate on the earlier of the effective date of a Company QPO and the closing of a sale by the Company of equity securities resulting in gross proceeds to the Company of at least $12,000,000.
Voting Agreement
The Company, NRV, Northlea, New, Alkon, Hannah Rose Holdings, LLC (“HRH”) and another stockholder of the Company entered into a Voting Agreement dated as of August 23, 2013, pursuant to which HRH and such other stockholder (which holds 488,079 shares of Common Stock) agreed to vote all of their Common Stock such that:
| | · | the authorized number of directors of the Company will be seven, with six directors elected by the holders of a majority of the outstanding Common Stock, voting as a separate class (each, a “Common Director”) and one director elected by the holders of a majority of the outstanding Series A Preferred Stock, voting as a separate class on an as-converted basis; |
| | · | the following five persons shall be nominated and elected to the Board as Common Directors: |
| | o | two NRV Designees, who are currently William S. Singer and James Gottlieb; |
| | o | two representatives designated by the holders of a majority in interest of the shares of Common Stock held by New and Northlea (each, an “Abeles Designee”), who initially shall be Dr. John Abeles and Dr. Jim New; and |
| | o | one independent Neurotrope Designee, which designee has not been determined as of the date of this report; and |
| | · | subject to the provisions of applicable law, no NRV Designee, Abeles Designee or Neurotrope Designee shall be removed from the Board unless such removal is requested in writing by the party that designated such designee. |
In addition, each of NRV, Northlea, New and Alkon agreed to vote all of their Common Stock in favor of electing as a Common Director after the Merger one independent representative designated by HRH (the “HRH Designee”) as soon as practicable after such representative has been identified by HRH. NRV, Abeles, New and Alkon agree to vote their Common Stock in favor of the election of the HRH Designee only at the time of such individual's initial appointment to the Parent's Board, and nothing obligates them to vote in favor of the election of any other individual as an HRH Designee or in favor of the continuing service of the HRH Designee once elected to the Board.
For information on numbers of shares of our stock held by NRV, Abeles, New, Alkon and HRH, see “Security Ownership of Certain Beneficial Owners and Management” above.
2013 Equity Incentive Plan
Before the Merger, our Board of Directors adopted, and our stockholders approved, our 2013 Plan, which provides for the issuance of incentive awards of up to 7,000,000 shares of our Common Stock to officers, key employees, consultants and directors. Following the closing of the Merger, options to purchase an aggregate of 5,154,404 shares of our Common Stock were issued to four founding stockholders of Neurotrope BioScience, our five directors and a consultant. See “Market Price of and Dividends on Common Equity and Related Stockholder Matters - Securities Authorized for Issuance under Equity Compensation Plans” below for more information about the 2013 Plan and the outstanding stock options.
Departure and Appointment of Directors and Officers
Our Board of Directors is authorized to consist of seven members and currently consists of six members, leaving one vacancy. On the Closing Date, Ronald Warren, our sole director before the Merger, resigned his position as a director, and John Abeles, Jim New, William Singer, Ralph Bean and Jay Haft were appointed to the Board of Directors. (Mr. Bean subsequently resigned and was replaced by James Gottlieb.)
Also on the Closing Date, Mr. Warren, our President, Secretary, Treasurer and sole officer before the Merger, resigned from these positions, and Jim New was appointed as our Chief Executive Officer and President, Robert Weinstein was appointed as our Chief Financial Officer and Treasurer, William Singer was appointed as our Secretary and Dan Alkon was appointed our Chief Scientific Officer by the Board.
See “Directors, Executive Officers, and Corporate Governance– Directors and Executive Officers” above for information about our directors and executive officers.
Lock-up Agreements and Other Restrictions
In connection with the Merger, each of our executive officers and directors named above and each person holding 10% or more of our Common Stock after giving effect to the Merger, the Split-Off and the PPO (the “Restricted Holders”), holding at that date in the aggregate 19,000,000 shares of our Common Stock, entered into agreements (the “Lock-Up and No Shorting Agreements”), whereby they are restricted for a period of 24 months after the Merger from certain sales or dispositions of our Common Stock held by them immediately after the Merger, except in certain limited circumstances (the “Lock-Up”).
Further, for a period of 24 months after the Merger, each Restricted Holder has agreed in the Lock-Up and No Shorting Agreements to be subject to restrictions on engaging in certain transactions, including effecting or agreeing to effect short sales, whether or not against the box, establishing any “put equivalent position” with respect to our Common Stock, borrowing or pre-borrowing any shares of our Common Stock, or granting other rights (including put or call options) with respect to our Common Stock or with respect to any security that includes, relates to or derives any significant part of its value from our Common Stock, or otherwise seeks to hedge his position in our Common Stock.
BRNI License
Our rights to develop, commercialize and sell our proposed products are dependent upon our Technology License and Services Agreement with BRNI. Neurosciences Research Ventures, Inc., which is an affiliate of BRNI, beneficially owns 43.6% of our outstanding common stock, and Dr. Dan Alkon, who is the Scientific Director of BRNI and the Chief Scientific Officer of the Company, beneficially owns 4.9% of our outstanding common stock. We are required to pay significant fees and royalties to BRNI pursuant to the Technology License and Services Agreement. For additional information, see Item 1, “Business—Intellectual Property—Technology License and Services Agreement” above.
The Company’s director, William S. Singer, also currently serves as President of BRNI and serves as a member of BRNI’s board of directors.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Audit Fees
The aggregate fees billed to us by our principal accountant for services rendered during the fiscal years ended December 31, 2013 and 2012, are set forth in the table below:
| | | Fiscal year | | Fiscal year | |
| | | ended | | ended | |
| | | December 31, | | December 31, | |
| Fee Category | | 2013 | | 2012 | |
Audit fees(1) | | $ | 78,000 | | $ | 25,700 | |
| Audit-related fees | | | - | | | - | |
| Tax fees | | | - | | | - | |
| All other fees | | | - | | | - | |
| Total fees | | $ | 78,000 | | $ | 25,700 | |
| (1) | Audit fees consists of fees incurred for professional services rendered for the audit of consolidated financial statements, for reviews of our interim consolidated financial statements included in our quarterly reports on Forms 10-Q and for services that are normally provided in connection with statutory or regulatory filings or engagements. |
Audit Committee’s Pre-Approval Practice
Our Audit Committee is directly responsible for the appointment, compensation, retention and oversight of the work of any registered public accounting firm engaged by us for the purpose of preparing or issuing an audit report or performing other audit, review or attest services for us, and each such registered public accounting firm must report directly to the Audit Committee. Our Audit Committee must approve in advance all audit, review and attest services and all permissible non-audit services (including, in each case, the engagement fees therefore and terms thereof) to be performed by our independent auditors, in accordance with applicable laws, rules and regulations.
Prior to the formation of our Audit Committee, our Board of Directors selected Friedman LLP as our independent registered public accountants for purposes of auditing our financial statements for the years ended December 31, 2013 and 2012. Friedman LLP was pre-approved by the Board to perform these audit services for us prior to its engagement.
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
Financial Statement Schedules
The consolidated financial statements of Neurotrope, Inc., and subsidiary are listed on the Index to Financial Statements on this annual report on Form 10-K beginning on page F-1.
All financial statement schedules are omitted because they are not applicable or the required information is shown in the financial statements or notes thereto.
Exhibits
The following exhibits are being filed with this Annual Report on Form 10-K:
In reviewing the agreements included or incorporated by reference as exhibits to this Annual Report on Form 10-K, please remember that they are included to provide you with information regarding their terms and are not intended to provide any other factual or disclosure information about the Company or the other parties to the agreements. The agreements may contain representations and warranties by each of the parties to the applicable agreement. These representations and warranties have been made solely for the benefit of the parties to the applicable agreement and:
· | should not in all instances be treated as categorical statements of fact, but rather as a way of allocating the risk to one of the parties if those statements prove to be inaccurate; |
· | have been qualified by disclosures that were made to the other party in connection with the negotiation of the applicable agreement, which disclosures are not necessarily reflected in the agreement; |
· | may apply standards of materiality in a way that is different from what may be viewed as material to you or other investors; and |
· | were made only as of the date of the applicable agreement or such other date or dates as may be specified in the agreement and are subject to more recent developments. |
Accordingly, these representations and warranties may not describe the actual state of affairs as of the date they were made or at any other time. Additional information about the Company may be found elsewhere in this Annual Report on Form 10-K and the Company’s other public filings, which are available without charge through the SEC’s website athttp://www.sec.gov.
Exhibit
Number | | Description |
| | | |
| | 2.1 | | Agreement and Plan of Merger, dated June 20, 2013, between BlueFlash Communications, Inc. and Neurotrope, Inc. (incorporated by reference from Exhibit 2.1 to the Registrant’s Current Report on Form 8-K filed with the SEC on August 8, 2013) |
| | | | |
| | 2.2 | | Amendment to Agreement and Plan of Merger, dated July 10, 2013, between BlueFlash Communications, Inc. and Neurotrope, Inc.(incorporated by reference from Exhibit 2.2 to the Registrant’s Current Report on Form 8-K filed with the SEC on August 8, 2013) |
| | | | |
| | 2.3* | | Agreement and Plan of Merger and Reorganization, dated as of August 23, 2013, by and among the Registrant, Acquisition Sub and Neurotrope BioScience, Inc. |
| | | | |
| | 3.1 | | Articles of Incorporation of the Registrant(incorporated by reference from Exhibit 3.1 to the Registrant’s Current Report on Form 8-K filed with the SEC on August 8, 2013) |
| | | | |
| | 3.2 | | Florida Articles of Merger of BlueFlash Communications, Inc. with and into Neurotrope, Inc., filed August 5, 2013 (incorporated by reference from Exhibit 3.3 to the Registrant’s Current Report on Form 8-K filed with the SEC on August 8, 2013) |
| | | | |
| | 3.3 | | Nevada Articles of Merger of BlueFlash Communications, Inc. with and into Neurotrope, Inc., filed August 5, 2013 (incorporated by reference from Exhibit 3.4 to the Registrant’s Current Report on Form 8-K filed with the SEC on August 8, 2013) |
| | | | |
Exhibit
Number | | Description |
| | | |
| 3.4* | | Certificate of Merger of Neurotrope BioScience, Inc., with and into Neurotrope Acquisition, Inc., filed August 23, 2013 |
| | | |
| 3.5 | | Certificate of Designations of Series A Convertible Preferred Stock of Neurotrope, Inc., filed with the Nevada Secretary of State on August 22, 2013(incorporated by reference from Exhibit 3.1 to the Registrant’s Quarterly Report on Form 10-Q for the quarter ended July 31, 2013) |
| | | |
| 3.6 | | Amended and Restated By-Laws of the Registrant(incorporated by reference from Exhibit 3.2 to the Registrant’s Current Report on Form 8-K filed with the SEC on August 8, 2013) |
| | | |
| 10.1* | | Split-Off Agreement, dated as of August 23, 2013, by and among the Registrant, Blue Flash Communications Corp. and Marissa Watson |
| | | |
| 10.2* | | General Release Agreement, dated as of August 23, 2013, by and among the Registrant, Blue Flash Communications Corp. and Marissa Watson |
| | | |
| 10.3* | | Form of Lock-Up and No Short Selling Agreement between the Registrant and the officers, directors and shareholders party thereto |
| | | |
| 10.4* | | Form of Subscription Agreement between Neurotrope BioScience, Inc., and the investors party thereto |
| | | |
| 10.5* | | Form of Agent Warrant for Common Stock of the Registrant |
| | | |
| 10.6* | | Form of Agent Warrant for Series A Preferred Stock of the Registrant |
| | | |
| 10.7* | | Placement Agency Agreement, dated June 25, 2013, between Neurotrope BioScience, Inc., and EDI Financial, Inc. |
| | | |
| 10.8* | | Amendment to Placement Agency Agreement, dated August 12, 2013, between Neurotrope BioScience, Inc., and EDI Financial, Inc. |
| | | |
| 10.9*† | | Employment Agreement, dated February 25, 2013, between the Registrant (as successor to Neurotrope BioScience) and Dr. James New |
| | | |
| 10.10*† | | Consulting Agreement, dated as of June 2, 2013, between the Registrant and Medical Cash Management Solutions, LLC (assigned to the Registrant) |
| | | |
| 10.11*† | | The Registrant’s 2013 Equity Incentive Plan |
| | | |
| 10.12*† | | Form of Option Agreement under 2013 Equity Incentive Plan |
| | | |
| 10.13* | | Technology License and Services Agreement, dated October 31, 2012, among the Registrant, BRNI and NRV II, LLC |
| | | |
| 10.14* | | Amendment #1 to Technology License and Services Agreement, dated August 21, 2013 among the Registrant, BRNI and NRV II, LLC |
| | | |
| 10.15* | | Common Stockholders Agreement, dated August 23, 2013, among the Registrant and the stockholders party thereto |
| | | |
| 10.16* | | Form of Preferred Stockholders Agreement, dated August 23, 2013, among the Registrant and the stockholders party thereto |
| | | |
| 10.17* | | Voting Agreement dated as of August 23, 2013, among the Registrant and the stockholders party thereto |
| | | |
| 10.18 | | Statement of Work Agreement dated August 20,2013, between Neurotrope BioScience and BRNI(incorporated by reference from identically numbered exhibit filed with the Amendment No. 1 to the Company’s Current Report on Form 8-K filed with the SEC on August 30, 2013) |
Exhibit
Number | | Description |
| | | |
| 14.1** | | Registrant’s Code of Business Conduct and Ethics |
| | | |
| 16.1 | | Letter from Lake & Associates CPA’s LLC to the Securities and Exchange Commission(incorporated by reference from identically numbered exhibit filed with the Amendment No. 2 to the Company’s Current Report on Form 8-K filed with the SEC on September 6, 2013) |
| | | |
| 21.1** | | Subsidiaries of the Registrant |
| | | |
| 31.1** | | Rule 13(a)-14(a)/15(d)-14(a) Certification of Principal Executive Officer |
| | | |
| 31.2** | | Rule 13(a)-14(a)/15(d)-14(a) Certification of Principal Financial And Accounting Officer |
| | | |
| 32.1** | | Section 1350 Certification of Principal Executive Officer(This certification is being furnished and shall not be deemed “filed” with the SEC for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of that section, and shall not be deemed to be incorporated by reference into any filing under the Securities Act or the Exchange Act, except to the extent that the registrant specifically incorporates it by reference.) |
| | | |
| 32.2** | | Section 1350 Certification of Principal Financial And Accounting Officer(This certification is being furnished and shall not be deemed “filed” with the SEC for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of that section, and shall not be deemed to be incorporated by reference into any filing under the Securities Act or the Exchange Act, except to the extent that the registrant specifically incorporates it by reference.) |
| | | |
| 101§ | | Interactive Data Files of Financial Statements and Notes. |
| | | |
| 101.ins§ | | Instant Document |
| | | |
| 101.sch§ | | XBRL Taxonomy Schema Document |
| | | |
| 101.cal§ | | XBRL Taxonomy Calculation Linkbase Document |
| | | |
| 101.def§ | | XBRL Taxonomy Definition Linkbase Document |
| | | |
| 101.lab§ | | XBRL Taxonomy Label Linkbase Document |
| | | |
| 101.pre§ | | XBRL Taxonomy Presentation Linkbase Document |
| | | |
__________________
| * Incorporated by reference from identically numbered exhibit filed with the Company’s Current Report on Form 8-K filed with the SEC on August 29, 2013. |
† Management contract or compensatory plan or arrangement.
| § Pursuant to Rule 406T of Regulation S-T, the XBRL related information in Exhibit 101 to this report shall not be deemed to be “filed” for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of that section, and shall not be deemed part of a registration statement, prospectus or other document filed under the Securities Act or the Exchange Act, except as shall be expressly set forth by specific reference in such filings. |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | NEUROTROPE, INC. | |
| | | | |
Dated: April 15, 2014 | By: | /s/ Jim New | |
| | | Jim New | |
| | | | Chief Executive Officer |
| | | | |
| | By: | /s/ Robert Weinstein | |
| | | Robert Weinstein | |
| | | | Chief Financial Officer |
| | | | | | |
In accordance with the Exchange Act, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| SIGNATURE | | TITLE | | DATE |
| | | | | |
| | | | |
/s/ John Abeles | | Chairman of the Board | | April 15, 2014 |
| John Abeles | | | | |
| | | | |
/s/ Jim New | | Chief Executive Officer,
Director | | April 15, 2014 |
| Jim New | | | | |
| | | | | |
| | | | |
/s/ Paul E. Freiman | | Director | | April 15, 2014 |
| Paul E. Freiman | | | | |
| | | | | |
| | | | |
/s/ Jay M. Haft | | Director | | April 15, 2014 |
| Jay M. Haft | | | | |
INDEX TO FINANCIAL STATEMENTS
| | | Page |
| | | |
| Report of Independent Registered Public Accounting Firm | | F-2 |
| | | |
| Consolidated Balance Sheets as of December 31, 2013 and 2012 | | F-3 |
| | | |
| Consolidated Statements of Operations for the year ended December 31, 2013, the period October 31, 2012 (inception) through December 31, 2012, and the period October 31, 2012 (inception) through December 31, 2013 | | F-4 |
| | | |
| Consolidated Statements of Changes in Shareholders’ Deficit for the year ended December 31, 2013, the period October 31, 2012 (inception) through December 31, 2012, and the period October 31, 2012 (inception) through December 31, 2013 | | F-5 |
| | | |
| Consolidated Statements of Cash Flows for the year ended December 31, 2013, the period October 31, 2012 (inception) through December 31, 2012, and the period October 31, 2012 (inception) through December 31, 2013 | | F-6 |
| | | |
| Notes to Consolidated Financial Statements | | F-8 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of Neurotrope, Inc.
We have audited the accompanying consolidated balance sheets of Neurotrope, Inc. (the “Company”) as of December 31, 2013 and 2012, and the related consolidated statements of operations, changes in shareholders’ deficiency, and cash flows for the year ended December 31, 2013, the period from October 31, 2012 (inception) to December 31, 2012 and the period from October 31, 2012 (inception) to December 31, 2013. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2013 and 2012, and the results of its operations and its cash flows for the year ended December 31, 2013, the period of October 31, 2012 (inception) to December 31, 2012 and the period of October 31, 2012 (inception) to December 31, 2013 in conformity with accounting principles generally accepted in the United States of America.
/s/ Friedman LLP
East Hanover, New Jersey
April 15, 2014
NEUROTROPE, INC.
(A Development Stage Company)
CONSOLIDATED BALANCE SHEETS
| | | | December 31, | | | December 31, | |
| | | | 2013 | | | 2012 | |
| ASSETS | | | | | | | |
| CURRENT ASSETS | | | | | | | |
| Cash and cash equivalents | | $ | 15,211,744 | | $ | - | |
| Prepaid expenses | | | 87,059 | | | - | |
| | | | | | | | |
| TOTAL CURRENT ASSETS | | | 15,298,803 | | | - | |
| | | | | | | | |
| TOTAL ASSETS | | $ | 15,298,803 | | $ | - | |
| | | | | | | | |
| LIABILITIES AND SHAREHOLDERS' DEFICIT | | | | | | | |
| CURRENT LIABILITIES | | | | | | | |
| Accounts payable and accrued expenses - related party | | $ | 251,779 | | $ | 1,240,636 | |
| Accounts payable and accrued expenses | | | 156,637 | | | 192,342 | |
| Advances from related party | | | - | | | 4,815 | |
| | | | | | | | |
| TOTAL CURRENT LIABILITIES | | | 408,416 | | | 1,437,793 | |
| | | | | | | | |
| Commitments and contingencies | | | | | | | |
| | | | | | | | |
| Convertible redeemable preferred stock, Series A, $.0001 par value, 50,000,000 | | | | | | | |
| shares authorized; 23,000,000 shares issued and outstanding (liquidation | | | | | | | |
| preference of $23,000,000 plus dividends accruable at 8% per annum of $1,023,616) | | | 19,943,572 | | | - | |
| | | | | | | | |
| SHAREHOLDERS' DEFICIT | | | | | | | |
| Common stock - 300,000,000 shares authorized, $.0001 par value; | | | | | | | |
| 21,690,406 issued and outstanding at December 31, 2012; | | | | | | | |
| 21,740,006 shares issued and outstanding at December 31, 2013 | | | 2,174 | | | 2,169 | |
| Additional paid-in capital | | | 3,974,007 | | | - | |
| Deficit accumulated during the development stage | | | (9,029,366) | | | (1,439,962) | |
| | | | | | | | |
| TOTAL SHAREHOLDERS' DEFICIT | | | (5,053,185) | | | (1,437,793) | |
| | | | | | | | |
| TOTAL LIABILITIES AND SHAREHOLDERS' DEFICIT | | $ | 15,298,803 | | $ | - | |
See accompanying notes to consolidated financial statements.
NEUROTROPE, INC.
(A Development Stage Company)
CONSOLIDATED STATEMENTS OF OPERATIONS
| | | | | | | | | | |
| | | | | | Period from | | Period from | |
| | | | | | October 31, | | October 31, | |
| | | | | | 2012 | | 2012 | |
| | | Year Ended | | (inception) to | | (inception) to | |
| | | December 31, | | December 31, | | December 31, | |
| | | 2013 | | 2012 | | 2013 | |
| OPERATING EXPENSES: | | | | | | | | | | |
| Research and development - related party | | $ | 1,367,644 | | $ | 983,491 | | $ | 2,351,135 | |
| General and administrative - related party | | | 4,301,731 | | | 257,145 | | | 4,558,876 | |
| General and administrative | | | 1,921,407 | | | 197,157 | | | 2,118,564 | |
| | | | | | | | | | | |
| TOTAL OPERATING EXPENSES | | | 7,590,782 | | | 1,437,793 | | | 9,028,575 | |
| | | | | | | | | | | |
| OTHER INCOME: | | | | | | | | | | |
| Interest income | | | 1,378 | | | - | | | 1,378 | |
| | | | | | | | | | | |
| Net loss before income taxes | | | (7,589,404) | | | (1,437,793) | | | (9,027,197) | |
| | | | | | | | | | | |
| Provision for income taxes | | | - | | | - | | | - | |
| | | | | | | | | | | |
| Net loss | | $ | (7,589,404) | | $ | (1,437,793) | | $ | (9,027,197) | |
| | | | | | | | | | | |
| PER SHARE DATA: | | | | | | | | | | |
| | | | | | | | | | | |
| Basic and diluted loss per common share | | $ | (0.35) | | $ | (0.07) | | | | |
| | | | | | | | | | | |
| Basic and diluted weighted average common shares outstanding | | | 21,702,415 | | | 21,690,406 | | | | |
See accompanying notes to consolidated financial statements.
NEUROTROPE, INC.
(A Development Stage Company)
CONSOLIDATED STATEMENT OF CHANGES IN SHAREHOLDERS' DEFICIT
| | | | | | | | | | | Deficit | | | | |
| | | | | | | | | | | Accumulated | | | | |
| | | | | | | | Additional | | During the | | | | |
| | | Common Stock | | Paid-In | | Development | | | | |
| | | Shares | | Amount | | Capital | | Stage | | | Total | |
| Balance, October 31, 2012 | | 2,690,400 | | $ | 269 | | $ | - | | $ | (269) | | $ | - | |
| Issuance of common stock | | 19,000,006 | | | 1,900 | | | - | | | (1,900) | | | - | |
| Net loss | | | | | | | | - | | | (1,437,793) | | | (1,437,793) | |
| Balance, December 31, 2012 | | 21,690,406 | | | 2,169 | | | - | | | (1,439,962) | | | (1,437,793) | |
| Issuance of common stock for consulting fees | | 49,600 | | | 5 | | | 44,635 | | | - | | | 44,640 | |
| Stock based compensation | | | | | | | | 3,122,944 | | | - | | | 3,122,944 | |
| Issuance of common stock warrants | | | | | | | | 806,428 | | | - | | | 806,428 | |
| Net loss | | | | | | | | - | | | (7,589,404) | | | (7,589,404) | |
| Balance, December 31, 2013 | | 21,740,006 | | $ | 2,174 | | $ | 3,974,007 | | $ | (9,029,366) | | $ | (5,053,185) | |
See accompanying notes to consolidated financial statements.
NEUROTROPE, INC.
(A Development Stage Company)
CONSOLIDATED STATEMENTS OF CASH FLOWS
| | | | | | | Period from | | Period from | |
| | | | | | | October 31, | | October 31, | |
| | | Year | | 2012 | | 2012 | |
| | | ended | | (inception) to | | (inception) to | |
| | | December 31, | | December 31, | | December 31, | |
| | | 2013 | | 2012 | | 2013 | |
| CASH FLOWS FROM OPERATING ACTIVITIES | | | | | | | | | | |
| Net loss | | $ | (7,589,404) | | $ | (1,437,793) | | $ | (9,027,197) | |
| Adjustments to reconcile net loss to net | | | | | | | | | | |
| cash used by operating activities | | | | | | | | | | |
| Stock based compensation | | | 3,122,944 | | | - | | | 3,122,944 | |
| Consulting services paid by issuance of common stock | | | 44,640 | | | - | | | 44,640 | |
| Change in assets and liabilities | | | | | | | | | | |
| Increase in prepaid expenses | | | (87,059) | | | - | | | (87,059) | |
| (Decrease) increase in accounts payable | | | | | | | | | | |
| and accrued expenses - related party | | | (988,857) | | | 1,240,636 | | | 251,779 | |
| (Decrease) increase in accounts payable and accrued expenses | | | (35,705) | | | 192,342 | | | 156,637 | |
| Total adjustments | | | 2,055,963 | | | 1,432,978 | | | 3,488,941 | |
| | | | | | | | | | | |
| Net Cash Used by Operating Activities | | | (5,533,441) | | | (4,815) | | | (5,538,256) | |
| | | | | | | | | | | |
| CASH FLOWS FROM FINANCING ACTIVITIES | | | | | | | | | | |
Issuance of preferred stock, preferred stock warrants and
common stock warrants net of transaction costs | | | 20,750,000 | | | - | | | 20,750,000 | |
| (Repayment of advances) advances from related party | | | (4,815) | | | 4,815 | | | - | |
| | | | | | | | | | | |
| Net Cash Provided by Financing Activities | | | 20,745,185 | | | 4,815 | | | 20,750,000 | |
| | | | | | | | | | | |
| NET INCREASE IN CASH | | | 15,211,744 | | | - | | | 15,211,744 | |
| | | | | | | | | | | |
| CASH AT BEGINNING OF YEAR | | | - | | | - | | | - | |
| | | | | | | | | | | |
| CASH AT END OF YEAR | | $ | 15,211,744 | | $ | - | | $ | 15,211,744 | |
See accompanying notes to consolidated financial statements.
Neurotrope, Inc.
A Development Stage Company
Notes to Consolidated Financial Statements
December 31, 2013 (audited)
Note 1 – Organization and Nature of Planned Business:
References in these notes to the consolidated financial statements to “Neurotrope, Inc.,” “we,” “us,” “our Company” refer to Neurotrope, Inc. and its consolidated subsidiary Neurotrope Biosciences, Inc. (“NBI”). NBI was incorporated in Delaware on October 31, 2012. NBI was formed to advance new diagnostic and therapeutic technologies in the field of neurodegenerative disease, primarily Alzheimer’s Disease. NBI plans to collaborate with the Blanchette Rockefeller Neurosciences Institute (“BRNI”), a related party, in this process. The exclusive rights to the licensed technology transferred to the Company on February 28, 2013 (see Note 4).
On August 23, 2013, a wholly owned subsidiary of Neurotrope, Inc. (formerly “BlueFlash Communications, Inc.”), Neurotrope Acquisition, Inc., a corporation formed in the State of Nevada on August 15, 2013 (“Acquisition Sub”) merged (the “Reverse Merger”) with and into NBI. NBI was the surviving corporation in the Reverse Merger and became the Company’s wholly owned subsidiary. All of the outstanding NBI common stock was converted into shares of Neurotrope, Inc. common stock on a one-for-one basis.
The Merger is being accounted for as a reverse-merger and recapitalization with NBI as the acquirer for financial reporting purposes and Neurotrope, Inc. as the acquired company. Consequently, the assets and liabilities and the operations that are reflected in the historical financial statements prior to the Merger are those of NBI and are recorded at the historical cost basis of NBI and the consolidated financial statements after completion of the Merger include the assets and liabilities of NBI and Neurotrope, Inc., and the historical operations of Neurotrope, Inc. and NBI from the closing date of the Merger. The stockholders’ equity section has been retroactively restated for all periods presented to reflect the accounting effect of the reverse merger transaction on the basis of the1:1 exchange ratio on the Merger date.
As a result of the Reverse Merger, Neurotrope, Inc. discontinued its pre-Reverse Merger business and acquired the business of NBI, and will continue the existing business operations of NBI as a publicly-traded company.
On February 28, 2013,NBI effected a stock split in which each of the 1,000 issued and outstanding shares of its common stock was converted into 19,000 shares of its common stock.
Note 2 – Contractual Commitments:
From January 1, 2013 through February 28, 2013, the Company compensated its President under an independent contractor agreement at the rate of $20,833 per month (see Note 4). On February 25, 2013, the Company executed a four-year employment agreement, effective March 1, 2013, with its President. This agreement provides for an annual salary of $250,000 and annual bonus of $50,000, which shall increase to an annual salary of $300,000 and an annual bonus of $100,000 effective as of the later of (a) February 28, 2015 or (b) the closing by the Company of a Series B Preferred Stock financing as described in Note 4 below.
Neurotrope, Inc.
A Development Stage Company
Notes to Condensed Consolidated Financial Statements
December 31, 2013 (audited)
Effective February 28, 2013, the Company executed an agreement with Ramat Consulting Corp. (“Ramat”), a related party, for consulting services, including business development and marketing consulting, for a five-year period, subject to annual renewals thereafter. Ramat's annual fee is $50,000, payable in monthly installments of $4,167, plus pre-approved travel and other reimbursable expenses. Ramat is also entitled to an option, with a term of ten years, to purchase300,000 shares of common stock of the Company at an exercise price of $1.00 per share. The option shall be deemed to have vested with respect to20% of the shares as of February 28, 2013, and the balance shall vest on a daily basis over the four-year period beginning on February 28, 2013. An entity related to Ramat purchased one million shares of Series A preferred stock on the effective date of this agreement.
Effective June 2, 2013, the Company executed a consulting agreement with Medical Cash Management Solutions, LLC (“MCMS”) for services as the Company's Chief Financial Officer, on an independent contractor basis, through November 30, 2013 at a fee of $20,000 per month plus reimbursable expenses. This agreement is subject to renewal terms as agreed to by the parties.
On October 1, 2013, the Company canceled its agreement with MCMS and executed a four-year employment agreement, effective October 1, 2013, with its Chief Financial Officer. This agreement provides for an annual salary of $240,000 which shall increase to an annual salary of $275,000 beginning January 1, 2015. In addition, the agreement provides for a $35,000 bonus for year ended December 31, 2013, a $50,000 bonus for year ended December 31, 2014 and a targeted bonus of50% of base salary for all subsequent years. In addition, on October 1, 2013 the Board granted a qualified stock option to the CFO under the Company’s 2013 Equity Incentive Plan to purchase650,000 shares of the Company’s common stock, with a term of ten years, exercisable at $1.00 per share. These options vest25% per year over four years, with accelerated vesting of 25% upon a termination without cause or for good reason.
Note 3 - Summary of Significant Accounting Policies:
Development Stage:
The Company is considered to be a development stage company, as defined by Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) 915-10, in that the Company is devoting substantially all of its efforts to establishing a new business where planned principal operations have commenced, but no revenues have been derived from these operations.
In addition, the Company’s licensed technology may not be ready for commercialization for several years, if at all. The Company expects to continue to incur losses at least until commercialization, because it anticipates that its expenditures on research and development and administrative activities will significantly exceed any potential revenues during the period. The Company cannot predict when, if ever, it will become profitable.
Use of Estimates:
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Neurotrope, Inc.
A Development Stage Company
Notes to Condensed Consolidated Financial Statements
December 31, 2013 (audited)
Cash and Cash Equivalents:
The Company considers all highly liquid temporary cash investments with an original maturity of three months or less when purchased to be cash equivalents. At December 31, 2013, the Company’s cash balances may exceed the current insured amounts under the Federal Deposit Insurance Corporation.
Research and Development Costs:
All research and development costs, including costs to maintain or expand the patent portfolio which do not meet the criteria for capitalization are expensed when incurred. FASB ASC Topic 730 requires companies involved in research and development activities to capitalize non-refundable advance payments for such services pursuant to contractual arrangements because the right to receive those services represents an economic benefit. Such capitalized advances will be expensed when the services occur and the economic benefit is realized. There were no capitalized research and development services at December 31, 2013 and 2012.
Income Taxes:
Income taxes are provided for the tax effects of transactions reported in the financial statements and consist of taxes currently due and deferred taxes. Deferred taxes are recognized for differences between the bases of assets and liabilities for financial statement and income tax purposes and the tax effects of net operating loss and other carry forwards. The deferred tax assets and liabilities represent the future tax consequences of those differences and carry forwards, which will either be taxable or deductible when the related assets, liabilities or carry forwards are recovered or settled. Deferred tax assets are reduced by a valuation allowance when, based on the weight of available evidence, it is more likely than not that some portion or all of the deferred tax assets will not be realized.
Uncertain Tax Positions:
The Company applies the provisions of FASB ASC 740-10,Accounting for Uncertain Tax Positions, which clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements and prescribes a recognition threshold and measurement process for financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. The standard also provides guidance on de-recognition, classification, interest and penalties, and accounting in interim periods, disclosure and transitions.
The Company has concluded that there are no significant uncertain tax positions requiring recognition in the accompanying financial statements. The tax period that is subject to examination by major tax jurisdictions is from October 31, 2012 (inception) through December 31, 2012. The Company has not yet filed tax returns for the year ended December 31, 2013.
In the event the Company was to receive an assessment for interest and/or penalties, it will be classified in the financial statements as selling, general and administrative expense when assessed.
Neurotrope, Inc.
A Development Stage Company
Notes to Condensed Consolidated Financial Statements
December 31, 2013 (audited)
Risks and Uncertainties:
The Company operates in an industry that is subject to rapid technological change, intense competition and significant government regulation, including but not limited to the United States FDA drug and device approval processes. The Company’s operations are subject to significant risk and uncertainties including financial, operational, technological, regulatory and other risks including the potential for business failure.
Stock Compensation:
The Company accounts for stock-based awards to employees in accordance with applicable accounting principles, which requires compensation expense related to share-based transactions, including employee stock options, to be measured and recognized in the financial statements based on a determination of the fair value of the stock options. The grant date fair value is determined using the Black-Scholes-Merton (“Black-Scholes”) pricing model. For all employee stock options, we recognize expense over the employee’s requisite service period (generally the vesting period of the equity grant). The Company’s option pricing model requires the input of highly subjective assumptions, including the expected stock price volatility, expected term, and forfeiture rate. Any changes in these highly subjective assumptions significantly impact stock-based compensation expense.
Options awarded to purchase shares of common stock issued to non-employees in exchange for services are accounted for as variable awards in accordance with applicable accounting principles. Such options are valued using the Black-Scholes option pricing model.
Earnings per Share:
Basic earnings per common share amounts are based on weighted average number of common shares outstanding. Diluted earnings per share amounts are based on the weighted average number of common shares outstanding, plus the incremental shares that would have been outstanding upon the assumed exercise of all potentially dilutive stock options, warrants and convertible stock, subject to anti-dilution limitations. All such potentially dilutive instruments were anti-dilutive as of December 31, 2013 and 2012. At December 31, 2013 and 2012, respectively, approximately31.5 million and0 shares underlying the convertible debentures, options and warrants were anti-dilutive.
Note 4 – Related Party Transactions:
The Company incurred consulting fees of $41,667 to its President during the period from January 1, 2013 to February 28, 2013 (see Note 3).
One director of the Company, until November 8, 2013, is also a director of BRNI. A second director of the Company is both the president and a director of BRNI. Both of these current and former directors are stockholders of a corporation, Neuroscience Research Ventures, Inc. (“NRVI”) that owns41.5% of the Company's outstanding common stock.
Effective October 31, 2012, the Company executed a Technology License and Services Agreement with BRNI, a related party, and two entities affiliated with BRNI (the “Agreement”). Under the terms of the Agreement, BRNI provides research services and grants the Company the exclusive and nontransferable license right to certain patent and intellectual property which became effective upon the Company's completion of a Series A Preferred Stock financing generating net proceeds to the Company of at least $8,000,000 on February 28, 2013. The Agreement terminates on the latter of the date (i) the last of the licensed patent expires, is abandoned, or is declared unenforceable or invalid or (ii) the last of the intellectual property enters the public domain.
The research services provided under the Agreement commenced on April 2, 2012. The Agreement requires the Company to reimburse BRNI for services rendered (the “Services Reimbursement”) on a pro-rated, thirty month basis, with respect to the period of time elapsed from April 2, 2012 through the date of the Series A Preferred Stock financing on February 28, 2013. BRNI invoiced the Company $1,198,696 for service reimbursements through December 31, 2012 and an additional $266,666 from January 1, 2013 to February 28, 2013.
Neurotrope, Inc.
A Development Stage Company
Notes to Condensed Consolidated Financial Statements
December 31, 2013 (audited)
The Agreement may require the Company to enter into scope of work agreements with BRNI. The Company shall not engage any other person other than BRNI to perform research or development services or other related scientific assistance without prior written consent of BRNI with such consent not unreasonably withheld. BRNI and Neurotrope may agree to have a third party provide services identical or similar to such services to Neurotrope in the case where BRNI is demonstrably unable to do so or such third party is demonstrably in a superior position to do so.
In addition to the fees under the services reimbursement and scope of work agreements, the Agreement requires the Company to pay BRNI a “Fixed Research Fee”, commencing the date that the Company completes a Series B Preferred Stock financing resulting in net proceeds of at least $25,000,000 (the “Series B Financing”). The fixed research fee is (i) a pro- rata amount of $1,000,000 in the year the Company completes such financing (ii) $1,000,000 per year for five calendar years subsequent to such financing and (iii) an annual fixed research fee in an amount to be negotiated and agreed upon no later than 90 days prior to the end of the fifth calendar year following the completion of such financing to be paid with respect to each remaining calendar year during the term of the Agreement. The Company had not completed this Series B Financing at December 31, 2013 (see Note 7) and, accordingly, no such fee was due as of that date.
The Agreement also requires the payment of royalties ranging between2% and5% of the Company’s revenues generated from the licensed patents and other intellectual property, dependent upon the percentage ownership that NRVI holds in the Company. Under the Agreement, the Company is required to prepay royalty fees at a rate of5% of all investor funds raised in the Series A or Series B Preferred Stock financings or any subsequent rounds of financing prior to a public offering, less commissions. On March 25, 2013, the Company prepaid $409,549 in royalties under the Agreement and paid the remainder due of $60,349 in July 2013 relating to the May 7, 2013 Series A preferred stock financing. On August 29, 2013, the Company paid $520,252 in prepaid royalties relating to the August 23, 2013 Series A preferred stock financing.On October 4, 2013, the Company paid $48,600in prepaid royalties relating to the Series A preferred stock financing. These royalties are expensed in “general and administrative expenses – related party” in the statement of operations.
On August 21, 2013, the Company and BRNI amended the Agreement to clarify certain provisions.
On August 22, 2013, the Board of Directors agreed to compensate its Chairman $9,000 per month for ongoing services to the Company relating to product development and negotiation of agreements with BRNI and outside contractors and consultants.
Effective August 28, 2013, the Company signed a statement of work (“SOW”) with BRNI pursuant to its licensing agreement (noted above) whereby the Company has contracted for the further development of its AD diagnostic product. Pursuant to the SOW, the Company is obligated to pay BRNI a total of $1,645,470 in12 equal monthly installments of $137,123, payable on the first business day of each month. These payments are for operating expenses associated with BRNI’s diagnostic laboratories. Operating expenses that are incurred in excess of this total amount are the responsibility of BRNI unless prior approval is obtained from the Company. The SOW may be extended if BRNI provides the Company with two months advanced notice that the SOW objectives are not met within the initial 12 month period. The Company will agree to continue funding the SOW at the same monthly rate for a period not to exceed an additional six (6) months to conclude the first anticipated clinical trial for the AD diagnostic product. In addition, the Company has agreed to pay an estimated $877,300 in external costs to complete the first clinical trial.
Neurotrope, Inc.
A Development Stage Company
Notes to Condensed Consolidated Financial Statements
December 31, 2013 (audited)
Effective November 13, 2013, the Company agreed to an SOW with BRNI pursuant to its licensing agreement (noted above) whereby the Company has contracted for the further development of its AD therapeutic product. Pursuant to the SOW, the Company paid BRNI $251,939 for related personnel and research services. The Company has expensed this entire amount in year ended December 31, 2013.
Note 5 – Income Taxes:
The Company incurred a net operating loss for income tax purposes of $9,027,197 for the period from October 31, 2012 (inception) through December 31, 2013. This amount is available for carry forward for use in offsetting taxable income of future years through 2032. The net operating loss carry forward resulted in a deferred tax asset of $3,069,247 at December 31, 2013, which is reduced to zero by an offsetting valuation allowance. As a result, there is no provision for income taxes.
Availability and utilization of the net operating loss carry forward may be limited due to changes in control.
Note 6 – Common Stock:
On February 28, 2013, NBI amended and restated its Certificate of Incorporation to authorize45,000,000 common shares at a par value of $0.01. On that date, the Company issued19,000,000 common shares. This recapitalization has been presented retroactively in the accompanying financial statements.
On June 20, 2013, BlueFlash Communications, Inc., a Florida corporation (“BlueFlash”), entered into an Agreement and Plan of Merger (the “Plan of Merger”) with Neurotrope, Inc., its wholly owned Nevada subsidiary, pursuant to which BlueFlash would merge with and into Neurotrope (the “Reincorporation Merger”). The Plan of Merger was amended by the Amendment to Agreement and Plan of Merger between the parties, dated July 10, 2013 (the “Amendment”). The purpose of the Merger was to re-domicile BlueFlash from Florida to Nevada, and to effect a name change and recapitalization as described below.
On August 5, 2013, Articles of Merger were filed with both the Secretary of State of the State of Nevada and the Secretary of State of the State of Florida, pursuant to which the Reincorporation Merger was effective as of August 9, 2013. Upon effectiveness of the Merger, Neurotrope, Inc.’s Articles of Incorporation and Neurotrope, Inc.’s Amended and Restated Bylaws became the Articles of Incorporation and Amended and Restated Bylaws of the registrant.
Neurotrope, Inc., has an authorized share capital of300,000,000 shares of common stock, par value $0.0001 per share (“Neurotrope Common Stock”) and50,000,000 shares of “blank check” preferred stock, par value $0.0001 per share. Prior to the Reincorporation Merger, Neurotrope, Inc. had100 shares of its Neurotrope Common Stock outstanding, held by BlueFlash, and therefore was a wholly-owned subsidiary of BlueFlash. Prior to the Merger, Neurotrope, Inc. had no assets, liabilities or business.
Pursuant to the Plan of Merger, among other things, (i) each share of common stock of BlueFlash, $0.0001 par value per share (“BlueFlash Common Stock”) was automatically converted into 2.242 shares of Neurotrope Common Stock.
Neurotrope, Inc.
A Development Stage Company
Notes to Condensed Consolidated Financial Statements
December 31, 2013 (audited)
On August 21, 2013, the NBI amended its articles of incorporation to increase its authorized shares of common stock from45,000,000 shares to57,000,000 shares.
On August 23, 2013, the Reverse Merger occurred as described in Note 1 above, and all of the outstanding Neurotrope BioScience common stock was converted into shares of Neurotrope, Inc. common stock on a one-for-one basis.
In connection with the Reverse Merger and pursuant to a Split-Off Agreement, Neurotrope, Inc. transferred its pre-Reverse Merger business to Marissa Watson, its pre-Reverse Merger majority stockholder, in exchange for the surrender by her and cancellation of20,178,000 shares of Neurotrope, Inc. common stock.
Note 7 – Preferred Stock:
Through a private placement, NBI issued9,073,300 preferred shares at $1 per share on that date, resulting in gross proceeds of $9,073,300. In connection with the February 28, 2013 closing of the private placement, the Company was required to pay the placement agent, Allied Beacon Partners, Inc., a cash fee equal to the sum of (a) 10% of the proceeds received from purchasers sourced by Allied Beacon Partners, Inc. and (b) 5% of the proceeds received from purchasers sourced by the Company. No fee was payable on proceeds received from purchasers who were already stockholders of the Company.The total cash fee that the Company was required to pay in connection with this closing was $882,330. The Company was also obligated to issue to Allied Beacon Partners, Inc. warrants for the purchase of common stock and Series A preferred stock of the Company in connection with the closing. The aggregate number of shares subject to such warrants was the sum of 10% of the number of shares purchased by purchasers sourced by Allied Beacon Partners, Inc. and (b) 5% of the number of shares purchased by purchasers sourced by the Company. No warrants were earned on proceeds received from purchasers who were already stockholders of the Company.
On May 17, 2013, NBI issued an additional1,313,325 shares of Series A preferred stock at $1.00 per share, resulting in gross proceeds of $1,313,325, on which NBI paid the placement agent cash of $131,332 and warrant compensation under the same terms as applied with respect to the February 28, 2013 closing.
The total number of shares subject to warrants that NBI was required to issue in connection with the February and May 2013 closings was988,663, consisting of480,320 shares of common stock and508,343 shares of Series A preferred stock. These warrants have a term of ten years. The strike price for the common stock warrants is $0.01 per share and the strike price for the Series A preferred stock warrants is $1.00 per share.
On August 23, 2013, NBI closed a private placement of11,533,375 additional shares of its Series A preferred stock, for a purchase price of $1.00 per share, for aggregate gross proceeds of $11,533,375 (before deducting placement agent fees and expenses of the offering of $1,103,338). All of the21,920,000 outstanding shares of NBI Series A preferred stock were converted into shares of Neurotrope Inc. Series A convertible preferred stock (the “Series A Preferred Stock”) on a one-for-one basis in the Reverse Merger. This additional financing was in conjunction with the Company’s merger with a publicly reporting shell company (See Note 1 above).
Neurotrope, Inc.
A Development Stage Company
Notes to Condensed Consolidated Financial Statements
December 31, 2013 (audited)
On October 4, 2013, the Company completed its Series A preferred stock offering through a private placement of1,080,000 shares of its Series A preferred stock, for a purchase price of $1.00 per share, for aggregate gross proceeds of $1,080,000 (before deducting placement agent fees and expenses of the offering).As a result of the foregoing, the placement agent was paid an aggregate commission of $108,000 and was issued Agent Warrants to purchase108,000 shares of the Company’s Series A preferred stock.
The Company agreed to pay the placement agent in the offering a cash commission of10% of the gross funds raised from investors in the private placement offering (“PPO”). In addition, the placement agent received(a) for the first $12,000,000 of gross PPO proceeds, (i) warrants exercisable for a period of ten (10) years to purchase a number of shares of Common Stock equal to 7.5% of the number of shares of Series A preferred stock sold to investors introduced by it, with a per share exercise price of $0.01, and (ii) warrants exercisable for a period of ten (10) years to purchase a number of shares of Series A preferred stock equal to 2.5% of the number of shares of Series A preferred stock sold to investors introduced by it, with a per share exercise price of $1.00; and (b) on gross PPO proceeds in excess of $12,000,000, warrants exercisable for a period of ten (10) years to purchase a number of shares of Series A preferred stock equal to 10% of the number of shares of Series A preferred stock sold to investors introduced by it, with an exercise price of $1.00 per share (collectively, the “Agent Warrants”). As a result of the foregoing aggregate placements, the placement agent was issued Agent Warrants to purchase 900,000 shares of the Company’s Common Stock and 1,325,000 shares of the Company’s Series A preferred stock. (See Note 8.)
The Series A preferred stock ranks senior with respect to liquidation preference and dividend rights to the common stock and any other class or series of stock that the Company may issue. The Series A preferred stock accrues a dividend at an annual rate of $0.08 per share, when and if declared by the Board of Directors of the Company. No dividends have been declared on the Series A preferred stock. In the event of any voluntary or involuntary liquidation, dissolution or winding-up of our affairs or similar event, a holder of Series A preferred stock will be entitled to be paid, before any distribution or payment may be made to any holders of common stock or other class or series of stock, the liquidation amount (which shall equal $1.00 per share) and the amount of any accrued and unpaid dividends as of such distribution or payment date. Each share of Series A preferred stock is convertible into common stock at the option of the stockholder at a price of $1.00 per share, subject to adjustment. The Series A shares are subject to mandatory conversion upon the vote of holders of a majority of the outstanding shares of Series A preferred stock at any given time, or upon the closing of a sale of common stock to the public at a price of at least $5.00 per share (subject to adjustment in the case of certain events) in a firm commitment underwritten public offering pursuant to an effective registration statement under the Securities Act, resulting in net proceeds to the Company of at least $30,000,000.The holders of Series A preferred stock are entitled to a class vote on certain Company actions, have the right to elect one of five members of the Company board of directors and have a right of first offer to purchase their pro rata share of equity securities issued by the Company in the future, in addition to certain additional rights and privileges as set forth in the Amended and Restated Certificate of Incorporation and in certain agreements between the Company and the holders thereof.
On August 21, 2013, NBI increased its authorized shares of preferred stock to50,000,000 shares. As a result, the Company had a sufficient number of preferred shares to close the August 23, 2013 private placement.
Neurotrope, Inc.
A Development Stage Company
Notes to Condensed Consolidated Financial Statements
December 31, 2013 (audited)
Note 8 – Warrants:
During the twelve months ended December 31, 2013, in conjunction with the Series A preferred stock financing (see Note 7), the Company issued warrants for2,225,000 underlying shares, consisting of warrants for900,000 underlying shares of common stock and warrants for1,325,000 underlying shares of Series A preferred stock to the placement agents. Both warrants have a term of ten years. The strike price for the common stock warrants is $0.01 per share and the strike price for the Series A preferred stock warrants is $1.00 per share.
The2,225,000 warrants issued during the twelve months ended December 31, 2013 were valued using the Black-Scholes option pricing model. The following weighted average assumptions were used for these warrants; risk free interest rates of2.70%; expected dividend yield of0%; expected term of10 years and expected volatility of105.5%. The weighted average remaining life of these warrants at December 31, 2013 was assumed to be10 years. The total calculated fair value of these warrants as of December 31, 2013 is $2,020,813.
Of the $2,020,813 total fair value of warrants issued during the twelve months ended December 31, 2013, $806,428 has decreased Preferred Stock and increased paid-in-capital in the balance sheet and $1,214,385has beenrecorded as an increase and decrease to preferred stock.
Note 9 – Stock Options:
Before the Reverse Merger, our Board of Directors adopted, and our stockholders approved, our 2013 Equity Incentive Plan (the “2013 Plan”), which provides for the issuance of incentive awards of up to7,000,000 shares of the Company’s common stock to officers, key employees, consultants and directors. Upon the closing of the Reverse Merger, options to purchase an aggregate of5,154,404 shares of the Company’s common stock were issued. Of these options: 1.3,500,000 options were issued to founding stockholders and54,404 were granted to a consultant, each with a term of ten years, exercisable at $1.75 per common share,100% of these options vested on date of grant; 2.1,050,000 options were issued to four directors (or their affiliates) and250,000 to a consultant with a term of ten years, exercisable at $1.00 per common share, which vest20% per year for each of five years after the date of grant, and; 3.300,000 options were granted to a consultant with a term of ten years, exercisable at $1.00 per common share which vested20% on date of grant and vest 20% per year over the next four years. These options were issued in conversion of options issued by NBI on February 28, 2013.
A summary of the Company’s stock option activity from inception to December 31, 2013 is as follows:
| | | | Weighted-Average | |
| | | Options Outstanding | | Exercise Price | |
| Outstanding at December 31, 2012 | | | 0 | | | | |
| Options granted | | | 6,489,404 | | $ | 1.47 | |
| Options forfeited | | | (239,452) | | $ | (1.00) | |
| Options exercised | | | 0 | | | 0 | |
| Outstanding at December 31, 2013 | | | 6,249,952 | | $ | 1.48 | |
| Options exercisable | | | 3,764,733 | | $ | 1.71 | |
Neurotrope, Inc.
A Development Stage Company
Notes to Condensed Consolidated Financial Statements
December 31, 2013 (audited)
The weighted-average remaining contractual term of options exercisable and outstanding at December 31, 2013 was approximately9.64 and9.68 years, respectively.
The Company used the Black-Scholes valuation model to calculate the fair value of stock options. Stock-based compensation expense is recognized over the vesting period using the straight-line method.
The fair value of stock options issued for the twelve months ended December 31, 2013 was estimated at the grant date using the following weighted average assumptions: Dividend yield0%; Volatility104.7%; Risk-free interest rate2.71%; weighted average grant date fair value of $0.84 per common share.
The total stock option-based compensation recorded as operating expense was approximately $3.1 million for the twelve months ended December 31, 2013 and from inception to December 31, 2013. Approximately $0.2 million of the expense is classified as research and development and the remaining approximately $2.9 million of stock option-based compensation expense was classified as general and administrative expense.
As of December 31, 2013 and 2012, there was approximately $2.6 million and $0, respectively, of unrecognized compensation cost related to non-vested options under the plans.
Note 10 – Common and Preferred Stock Reserved for Future Issuances:
Common stock and preferred stock reserved for future issuances consisted of the following at December 31, 2013:
| | | Common Stock | | Preferred Stock | |
| | | Reserved | | Reserved | |
| Common stock warrants outstanding | | 900,000 | | | |
| Preferred stock warrants outstanding | | 1,217,000 | | 1,217,000 | |
| Common stock options outstanding | | 6,249,952 | | 0 | |
| Conversion of Series A preferred stock | | 23,000,000 | | 0 | |
| Total | | 31,366,952 | | 1,217,000 | |
Note 11 – Subsequent Events:
The Company’s management has evaluated the effects of events occurring through April15,2014, the date these financial statements were available to be issued.
On January 2, 2014, the Company executed a one-year employment letter which is renewable annually, with NBI’s Vice President – Regulatory Affairs. This agreement provides for an annual salary of $195,000 which shall increase at the discretion of the Company’s Compensation Committee. In addition, the offer letter provides for a discretionary bonus of up to20% of base salary for all years. In addition, on January 23, 2014, the Company’s Board of Directors granted a qualified stock option to its VP – Regulatory Affairs under the Company’s 2013 Equity Incentive Plan to purchase75,000 shares of the Company’s common stock, with a term of ten years, exercisable at $1.76 per share. These options vest20% per year over five years.
Neurotrope, Inc.
A Development Stage Company
Notes to Condensed Consolidated Financial Statements
December 31, 2013 (audited)
On January 16, 2014, the Company executed a four-year employment letter, with NBI’s Vice President – Commercial Operations. This agreement provides for an annual salary of $210,000 which shall increase at the discretion of the Company’s Compensation Committee. In addition, the offer letter provides for a discretionary bonus of up to35% of base salary for all years. In addition, on January 23, 2014, the Company’s Board of Directors granted a qualified stock option to its VP – Commercial Operations under the Company’s 2013 Equity Incentive Plan to purchase100,000 shares of the Company’s common stock, with a term of ten years, exercisable at $1.76 per share. These options vest20% per year over five years.
On January 22, 2014, the Company executed a four-year employment agreement, with NBI’s Vice President and Chief Medical Officer. This agreement provides for an annual salary of $210,000 which shall increase at the discretion of the Company’s Compensation Committee. In addition, the offer letter provides for a discretionary bonus of up to35% of base salary for all years. In addition, on January 23, 2014, the Company’s Board of Directors granted a qualified stock option to its VP – Commercial Operations under the Company’s 2013 Equity Incentive Plan to purchase125,000 shares of the Company’s common stock, with a term of ten years, exercisable at $1.76 per share. These options vest25% per year over four years, with accelerated vesting of 25% upon a termination without cause or for good reason.
On March 12, 2014, the Company signed an SOW with BRNI to continue pre-clinical activities relating to the commercialization of the Company’s therapeutic product. The Company is obligated to pay BRNI a total of $465,000. Of this amount, the Company has paid $358,470(see “Related Party Transactions” above), the remaining $106,530to be paid upon the completion, by BRNI, of certain activities relating to: transferring test materials; bio-analytical testing; contracting with a suitable contract research organization; completion of testing assays, and; finalizing a clinical study protocol.
After December 31, 2013,149,000 shares of Series A Preferred Stock were converted at the option of their holders into149,000 shares of common stock.
EXHIBIT INDEX
14.1 Registrant’s Code of Business Conduct and Ethics
21.1 Subsidiaries of the Registrant
31.1 Rule 13(a)-14(a)/15(d)-14(a) Certification of Principal Executive Officer
31.2 Rule 13(a)-14(a)/15(d)-14(a) Certification of Principal Financial And Accounting Officer
32.1 Section 1350 Certification of Principal Executive Officer (This certification is being furnished and shall not be deemed “filed” with the SEC for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of that section, and shall not be deemed to be incorporated by reference into any filing under the Securities Act or the Exchange Act, except to the extent that the registrant specifically incorporates it by reference.)
32.2 Section 1350 Certification of Principal Financial And Accounting Officer (This certification is being furnished and shall not be deemed “filed” with the SEC for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of that section, and shall not be deemed to be incorporated by reference into any filing under the Securities Act or the Exchange Act, except to the extent that the registrant specifically incorporates it by reference.)