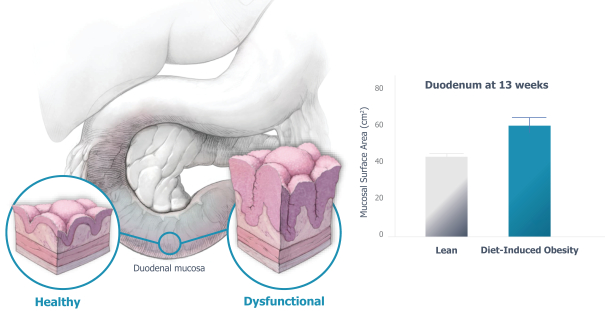
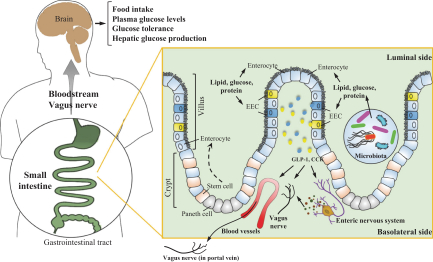
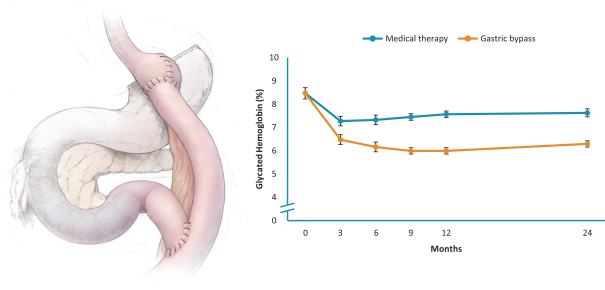
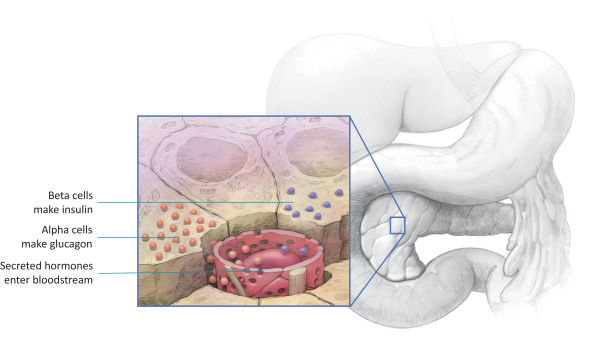
Our ability to utilize our net operating loss carryforwards, research and development tax credit carryforwards, and certain other tax attributes to offset taxable income or taxes may be limited.
As of December 31, 2022, we had U.S. federal and state net operating loss carryforwards of approximately $215.3 million and $207.8 million, respectively, which begin to expire at various dates beginning in 2030. Portions of these net operating loss carryforwards could expire unused and be unavailable to offset future income tax liabilities. Under the legislation enacted in 2017, commonly referred to as the Tax Cuts and Jobs Act, or the Tax Act, as modified by the Coronavirus Aid, Relief, and Economic Security, or the CARES Act, U.S. federal net operating losses incurred in taxable years beginning after December 31, 2017, may be carried forward indefinitely, but the deductibility of such federal net operating losses in taxable years beginning after December 31, 2020, is limited. It is uncertain how various states will respond to the Tax Act and the CARES Act.
In addition, as of December 31, 2022, we had U.S. federal and state research and development tax credit carryforwards of $8.5 million and $3.6 million, respectively. The federal research and development tax credit carryforwards will expire at various dates beginning in 2031. The state research and development tax credit carryforwards will expire at various dates beginning in 2027. We may not be able to utilize these credits for federal and state income tax purposes before they expire.
In addition, under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50% change, by value, in its equity ownership over a three-year period, the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes to offset its post-change income or taxes may be limited. The completion of this offering, together with other transactions that have occurred since our inception, may trigger such an ownership change pursuant to Section 382. To date, we have not completed an analysis under Section 382. We may experience ownership changes as a result of subsequent shifts in our stock ownership, some of which may be outside of our control. If an ownership change occurs and our ability to use our net operating loss carryforwards is materially limited, it would harm our future results of operations by effectively increasing our future tax obligations.
Risks Related to Development, Regulatory Approval and Commercialization
The regulatory approval process of the FDA, comparable foreign regulatory authorities and notified bodies, are lengthy, time-consuming and inherently unpredictable, and even if we complete the necessary clinical studies, we cannot predict when, or if, we will obtain regulatory approval or certification for any of our product candidates, and any such regulatory approval or certification may be for a more narrow indication than we seek.
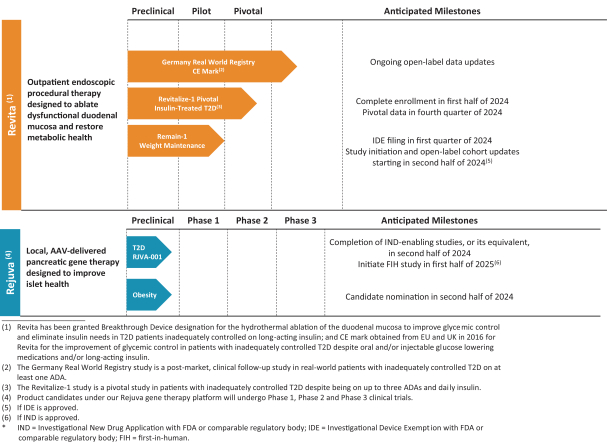
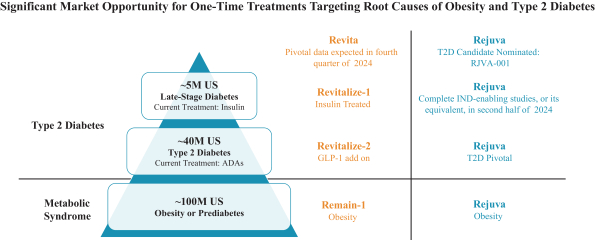
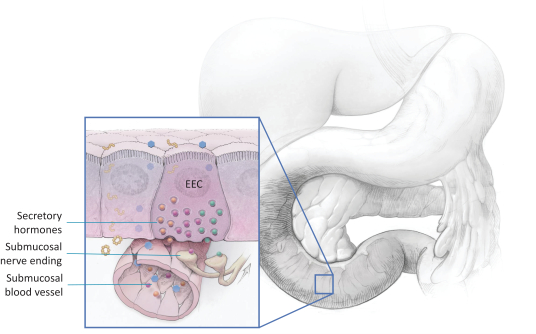
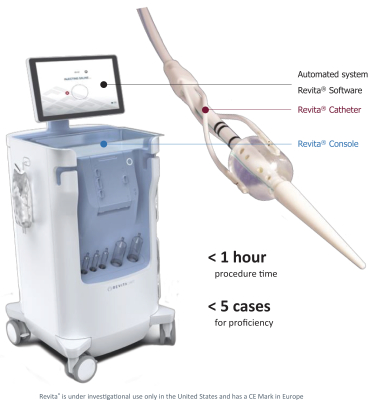
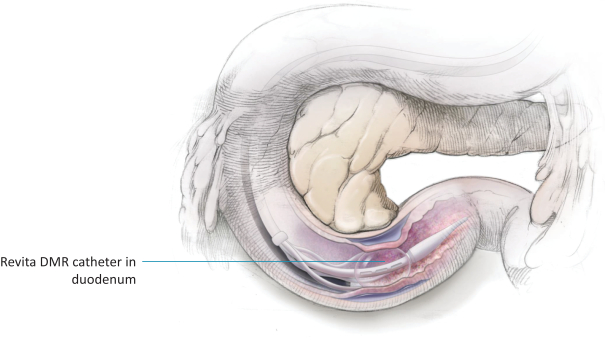
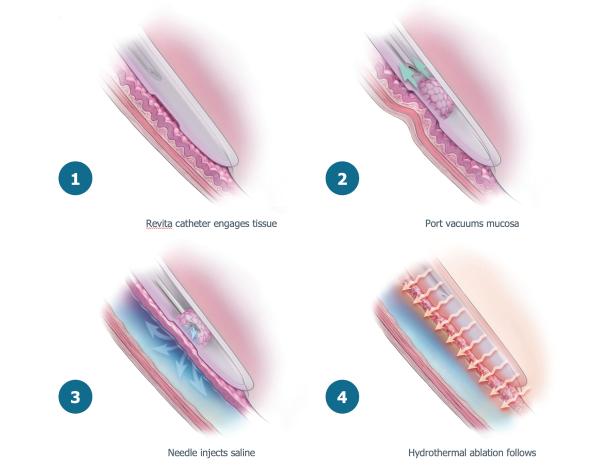
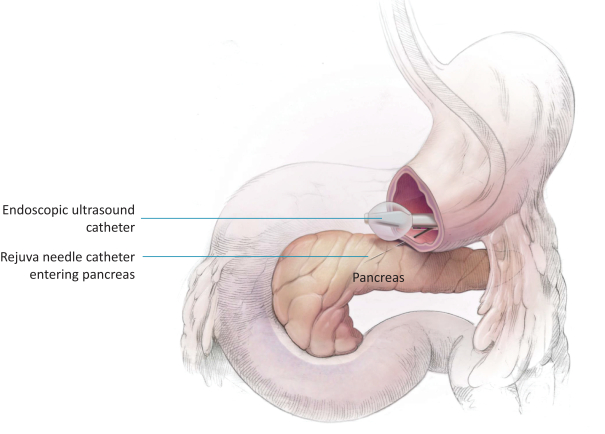
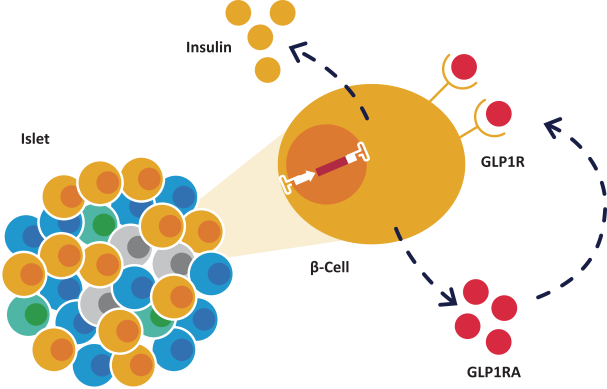
The research, testing, manufacturing, labeling, approval, certification, selling, import, export, marketing, and distribution of medical devices and biopharmaceutical products are subject to extensive regulation by the FDA and other regulatory authorities in and outside the United States. We are currently in clinical-stage development of Revita, which is an investigational medical device, and are conducting preclinical development of our Rejuva PGTx candidates along with a device delivery system, which together with the gene therapy candidate, we anticipate will be regulated as a combination biologic-device.
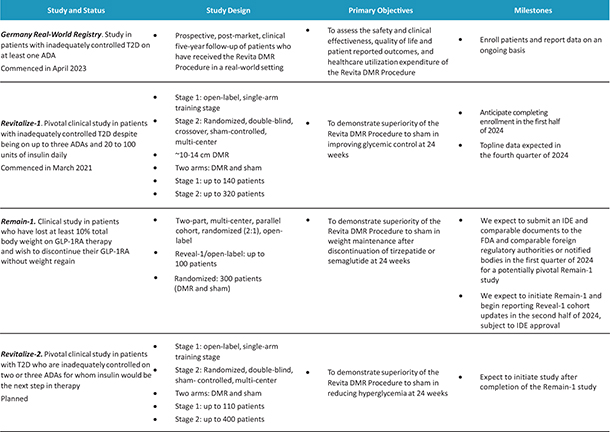
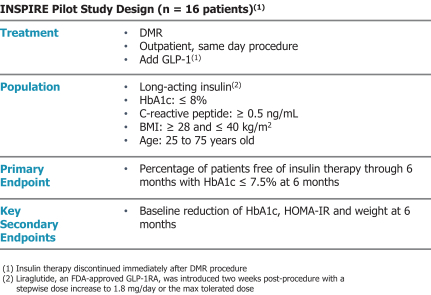
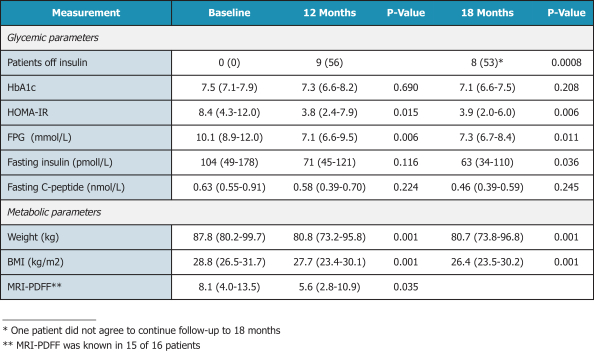
In the United States, before we can market a new medical device, we must first receive either clearance under Section 510(k) of the Federal Food, Drug, and Cosmetic Act, or the FDCA, or approval of a PMA, from the FDA, unless an exemption applies. We expect Revita to be subject to the requirement for approval of a PMA. In the process of obtaining PMA approval, the FDA must determine that a proposed device is safe and effective for its intended use based, in part, on extensive data, including, but not limited to, technical, preclinical, clinical, manufacturing and labeling data. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as life sustaining, life supporting or implantable devices. We plan to seek approval of a PMA from the FDA for the Revita DMR Procedure to improve glycemic control and eliminate insulin needs in T2D patients who are inadequately controlled on insulin.
24