Use these links to rapidly review the document
TABLE OF CONTENTS
TABLE OF CONTENTS1
As confidentially submitted to the Securities and Exchange Commission on October 3, 2014.
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form F-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
TiGenix
(Exact name of Registrant as specified in its charter)
| Kingdom of Belgium (State or other jurisdiction of incorporation or organization) | 2834 (Primary Standard Industrial Classification Code Number) | Not Applicable (I.R.S. Employer Identification Number) |
Romeinse straat 12, box 2
3001 Leuven
Belgium
+32 (16) 39 6060
(Address, including zip code, and telephone number, including area code, of Registrant's principal executive offices)
CT Corporation System
111 Eighth Avenue
New York, NY 10011
+1 (212) 894-8800
(Name, address, including zip code, and telephone number, including area code, of agent for service)
| Copies to: | ||
Peter Castellon, Esq. Proskauer Rose LLP Ten Bishops Square London E1 6EG United Kingdom +44 (20) 7539-0600 | Thomas S. Levato, Esq. Goodwin Procter LLP The New York Times Building 620 Eighth Avenue New York, NY 10018 United States +1 (212) 813-8800 | |
Approximate date of commencement of proposed sale to the public:
As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. o
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering: o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
CALCULATION OF REGISTRATION FEE
| Title of Each Class of Securities to be Registered(1) | Proposed Maximum Aggregate Offering Price(2)(3) | Amount of Registration Fee | ||
|---|---|---|---|---|
Ordinary Shares, no nominal value per share | $ | $ | ||
| ||||
- (1)
- American depositary shares, or ADSs, issuable upon deposit of the ordinary shares registered hereby will be registered under a separate registration statement on Form F-6. Each ADS will represent ordinary shares.
- (2)
- Includes ordinary shares that are issuable upon the exercise of the underwriters' option to purchase additional ordinary shares to cover over-allotments, if any.
- (3)
- Estimated solely for the purposes of computing the amount of the registration fee pursuant to Rule 457(o) under the Securities Act of 1933, as amended.
The Registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Securities and Exchange Commission acting pursuant to said Section 8(a) may determine.
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to completion, dated ,
Preliminary Prospectus
American Depositary Shares
Representing Ordinary Shares

$ per American Depositary Share
TiGenix, a Belgian public limited liability company, is offering American Depositary Shares, or ADSs. Each ADS will represent ordinary shares with no nominal value per share.
This is our initial public offering in the United States. We intend to apply to list our ADSs on the NASDAQ Global Market under the symbol "TIG." Prior to this offering our ordinary shares have traded, and subsequent to this offering will continue to trade, on Euronext Brussels under the symbol "TIG." The latest reported closing sale price of our ordinary shares on Euronext Brussels on October 1, 2014 was 0.57 euros per share, or $0.72 per share based on the rate of exchange on that day.
We anticipate that the initial public offering price of our ADSs will be between $ and $ per ADS.
This investment involves a high degree of risk. See "Risk Factors" beginning on page 11.
| | Per ADS | Total | ||
|---|---|---|---|---|
Public offering price | $ | $ | ||
Underwriting discounts and commissions(1) | $ | $ | ||
Proceeds, before expenses, to TiGenix | $ | $ | ||
| ||||
- (1)
- The underwriters will also be reimbursed for certain expenses incurred in this offering. See "Underwriting" for details.
The underwriters have a thirty-day option to purchase up to additional ADSs to cover over-allotments, if any.
We are an "emerging growth company" as that term is defined in the Jumpstart Our Business Startups Act of 2012 and, as such, will be subject to reduced public company reporting requirements for future filings. See "Prospectus Summary—Implications of Being an Emerging Growth Company."
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
Delivery of the ADSs will be made against payment in New York, New York on or about , .
Joint Book-Running Managers
| Canaccord Genuity | Nomura |
Co-Managers
| Maxim Group LLC | Chardan Capital Markets |
The date of this prospectus is , .
| | Page | |||
|---|---|---|---|---|
PROSPECTUS SUMMARY | 1 | |||
RISK FACTORS | 11 | |||
HISTORY AND ORGANIZATIONAL STRUCTURE | 36 | |||
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS | 38 | |||
EXCHANGE RATES | 40 | |||
USE OF PROCEEDS | 41 | |||
DIVIDEND POLICY | 42 | |||
CAPITALIZATION | 43 | |||
DILUTION | 44 | |||
MARKET FOR OUR SHARES | 46 | |||
SELECTED FINANCIAL INFORMATION | 47 | |||
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS | 50 | |||
BUSINESS | 68 | |||
DESCRIPTION OF SHARE CAPITAL | 109 | |||
MANAGEMENT | 120 | |||
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS | 136 | |||
PRINCIPAL SHAREHOLDERS | 137 | |||
DESCRIPTION OF AMERICAN DEPOSITARY SHARES | 139 | |||
SHARES ELIGIBLE FOR FUTURE SALES | 150 | |||
TAXATION | 151 | |||
UNDERWRITING | 162 | |||
EXPENSES RELATED TO THIS OFFERING | 168 | |||
LEGAL MATTERS | 169 | |||
EXPERTS | 170 | |||
SERVICE OF PROCESS AND ENFORCEMENT OF CIVIL LIABILITIES | 171 | |||
WHERE YOU CAN FIND MORE INFORMATION | 173 | |||
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS | F-1 | |||
i
You should rely only on the information contained in this prospectus and any related free-writing prospectus that we authorize to be distributed to you. We have not, and the underwriters have not, authorized any person to provide you with information different from that contained in this prospectus or any related free-writing prospectus that we authorize to be distributed to you. This prospectus is not an offer to sell, nor is it seeking an offer to buy, these securities in any jurisdiction where the offer or sale is not permitted. The information in this prospectus speaks only as of the date of this prospectus unless the information specifically indicates that another date applies, regardless of the time of delivery of this prospectus or of any sale of the securities offered hereby.
No action is being taken in any jurisdiction outside the United States to permit a public offering of the ADSs or possession or distribution of this prospectus in that jurisdiction. Persons who come into possession of this prospectus in jurisdictions outside the United States are required to inform themselves about and to observe any restrictions as to this offering and the distribution of the prospectus applicable to that jurisdiction.
Until , (twenty-five days after the commencement of this offering), all dealers that buy, sell or trade our ADSs, whether or not participating in this offering, may be required to deliver a prospectus. This delivery requirement is in addition to the obligation of dealers to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
Unless otherwise indicated, information contained in this prospectus concerning our industry and the markets in which we operate, including our general expectations and market position, market opportunity and market share, is based on information from our own management estimates and research, as well as from industry and general publications and research, surveys and studies conducted by third parties. Management estimates are derived from publicly available information, our knowledge of our industry and assumptions based on such information and knowledge, which we believe to be reasonable. In addition, assumptions and estimates of our and our industry's future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in "Risk Factors." These and other factors could cause our future performance to differ materially from our assumptions and estimates. See "Special Note Regarding Forward-Looking Statements."
Our registered trademarks, TiGenix and ChondroCelect, the TiGenix logo and other trademarks or service marks of TiGenix appearing in this prospectus are our property. This prospectus contains additional trade names, trademarks and service marks of other companies that are the property of their respective owners.
ii
This summary highlights selected information about us contained elsewhere in this prospectus. This summary does not contain all of the information that you should consider in making your investment decision. Before investing in the ADSs, you should read this entire prospectus carefully, including the sections entitled "Risk Factors" and "Management's Discussion and Analysis of Financial Condition and Results of Operations" and our consolidated financial statements and related notes, for a more complete understanding of our business and this offering.
Except as otherwise required by the context, references to "TiGenix," "Company," "we," "us" and "our" are to TiGenix and its subsidiaries.
We are an advanced biopharmaceutical company focused on developing and commercializing novel therapeutics from our proprietary technology platform of allogeneic, or donor-derived, expanded adipose-derived stem cells, known as eASCs, in inflammatory and autoimmune diseases. Based on our platform, we have developed a pipeline of product candidates, with our most advanced being Cx601, a first-in-class injectable allogeneic stem cell therapy that has been granted orphan designation by the European Medicines Agency, or EMA. We are conducting a single pivotal Phase III trial for Cx601 for the treatment of complex perianal fistulas in patients suffering from Crohn's disease, a painful and debilitating condition affecting approximately 100,000 patients in the United States and Europe. Data from the single pivotal trial should be available in the third quarter of 2015, based on which we plan to submit a marketing authorization application to the EMA in the first half of 2016. We also intend to initiate a Phase III trial for Cx601 in the United States by the second half of 2016. Based on discussions with the U.S. Food and Drug Administration, or FDA, we believe that the U.S. Phase III trial, if successful, could, together with the European Phase III data, serve as supportive evidence for filing for regulatory approval with the FDA.
Our platform has generated other product candidates, including Cx611, for which we have completed a Phase I/IIa trial in rheumatoid arthritis. We are currently developing two clinical trials for Cx611: a Phase Ib trial in severe sepsis in the first quarter of 2015 and a Phase IIb trial in early rheumatoid arthritis in the third quarter of 2015. Our third product candidate, Cx621, completed a Phase I clinical trial for intra-lymphatic administration of allogeneic eASCs. Its mode of administration has the potential to enable applications in other autoimmune diseases. We also developed and commercialized ChondroCelect, the first cell-based medicinal product to receive marketing authorization from the EMA, which is indicated for cartilage repair in the knee.
Our eASC-based product candidates are manufactured at our facility in Madrid, Spain, that has been approved by the Spanish Medicines and Medical Devices Agency as being compliant with current Good Manufacturing Requirements. Through our expansion process, we can generate up to 2,400 doses of Cx601 from cells extracted from a single healthy donor. Drawing on our experience with ChondroCelect, we already have the capacity to scale up the production of our eASC-based products on a late-stage clinical as well as commercial scale.
We have retained the worldwide rights for all of our product candidates. As of September 30, 2014, we owned or co-owned over twenty patent families and had over ninety granted patents in over twenty jurisdictions, including the United States, with expiration dates from 2020 onwards.
Product and Product Candidates
Our approach to cell therapy is to focus on the use of living cells, rather than conventional drugs, for the treatment of inflammatory and autoimmune diseases. Our advanced clinical stage pipeline of stem cell programs is based on a validated platform of eASCs extracted from human adipose, or fat, tissue. Our eASCs are extracted and cultured from tissue sourced from healthy consenting adult donors that can then be administered in patients for the treatment of autoimmune and inflammatory diseases.
1
The following chart summarizes our product candidates and our marketed product:
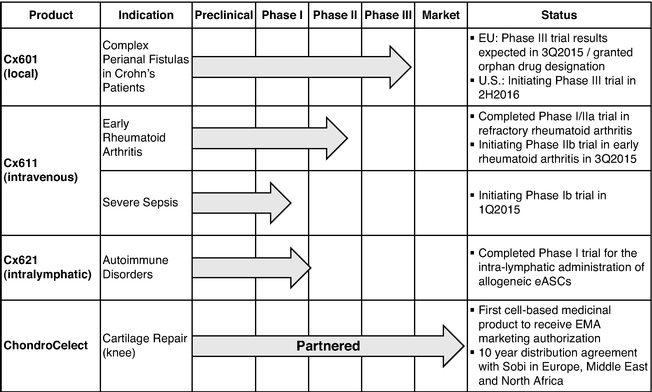
Cx601
Cx601, our lead product candidate, is a first-in-class injectable allogeneic stem cell therapy that is currently in a pivotal Phase III trial in Europe for the treatment of complex perianal fistulas in patients suffering from Crohn's disease. Crohn's disease is a chronic inflammatory disease of the intestine, which frequently leads to the development of abscesses and fistulas. Current therapies have limited efficacy and there is currently no commercially available cell-based therapy for this indication. We received orphan drug designation for the treatment of anal fistulas from the EMA in 2009.
We believe that Cx601, if approved, would fulfill a significant unmet need in the market. We have compelling clinical results that suggest that Cx601 may have clinical utility in treating perianal fistulas in one injectable dose with a more favorable adverse events profile than currently available therapies for this indication. If our pivotal Phase III trial is successful, we expect to file for marketing authorization in Europe by the first half of 2016 and initiate a Phase III trial in the United States by the second half of 2016.
Cx611
Cx611, our second product candidate, is a first-in-class injectable allogeneic stem cell therapy in development for the treatment of early rheumatoid arthritis and severe sepsis. We believe that Cx611, if approved for early rheumatoid arthritis patients, would have advantages over current treatments such as biologics due to its safety profile and higher induction of remission. We have completed a successful Phase I/IIa trial of Cx611 in refractory rheumatoid arthritis patients that illustrated the safety of the therapy and provided indications of therapeutic activity. If Cx611 is approved for severe sepsis, we believe that it would be an add-on therapy that has the potential to reduce mortality. We are planning to advance Cx611 in severe sepsis in a Phase Ib trial and in early rheumatoid arthritis in a Phase IIb trial in the first and the third quarter of 2015, respectively.
2
Cx621
Cx621, our third clinical stage product candidate, has generated safety and feasibility information on the intra-lymphatic administration of allogeneic eASCs. This different route of administration has the potential to enable applications in other autoimmune diseases.
ChondroCelect
ChondroCelect, our commercial product, was the first cell-based product approved in Europe, and received centralized marketing authorization in October 2009 as an advanced therapy medicinal product. Effective June 1, 2014, we have entered into a distribution agreement with Swedish Orphan Biovitrium, or Sobi, for the exclusive marketing and distribution rights with respect to ChondroCelect within the European Union (except for Finland), as well as several other countries, including the Middle East and North Africa.
Technology Platform
Our development programs are based on a proprietary technology platform of eASCs, exploiting their recognized mechanism of action in immune-mediated inflammatory processes. The eASCs target a different pathway than conventional drugs and may be effective in patients who fail to respond to such drugs, including the biologics that are currently used to treat autoimmune conditions. We believe our platform offers significant market opportunities in inflammatory and autoimmune diseases based on the following distinguishing factors:
- •
- Our mechanism of action utilizes two main biological pathways that underlie the efficacy of stem cells in disease treatment: (i) their anti-inflammatory properties and (ii) their secretion of repair and growth promoting molecules. Our eASCs exhibit broad immunomodulatory properties, including the regulation of immune cells such as B lymphocytes, T lymphocytes, natural killer cells, monocytes or macrophages and neutrophils.
- •
- Our experience in enabling both local and systemic routes of administration.
- •
- Our use of eASCs extracted from human adipose tissue sourced from healthy volunteers. We believe that this type of cell offers significant advantages over other cell types, such as stem cells sourced from bone marrow.
- •
- Our use of allogeneic adult stem cells. This enables efficient production of large batches of cells and does not require any patient biopsy or tissue procurement.
Strategy
Key elements of our strategy to provide innovative and safe treatment options for a broad range of inflammatory and autoimmune diseases are as follows:
- •
- Successfully advance the clinical development of Cx601 for the treatment of complex perianal fistulas in patients with Crohn's disease and secure regulatory approval in Europe and the United States.
- •
- Achieve global commercialization of Cx601, leveraging our experience in bringing ChondroCelect to market.
- •
- Advance our product candidates, Cx611 and Cx621, in the United States and the rest of the world.
- •
- Discover, develop and commercialize first-in-class novel therapeutics for areas of high unmet medical need by leveraging our proprietary technology platform of eASCs.
3
- •
- Strengthen our competitive position by leveraging our experienced management team and reinforcing key opinion leader support.
Summary Risk Factors
An investment in the ADSs involves a high degree of risk. You should consider carefully the risks discussed below and under the heading "Risk Factors" beginning on page 11 of this prospectus before purchasing the ADSs. If any of these risks actually occurs, our business, financial condition or results of operations would likely be materially adversely affected. In such case, the trading price of our ADSs would likely decline, and you may lose all or part of your investment.
These risks include the following:
- •
- We may experience delays or failure in the preclinical and clinical development of our product candidates.
- •
- Regulatory approval of our product candidates may be delayed, not obtained or not maintained, and we may be affected by future changes to any pharmaceutical legislation or guidelines.
- •
- We may need substantial additional funding, which may not be available on acceptable terms when required, if at all.
- •
- Our net losses and significant cash used in operating activities have raised substantial doubt regarding our ability to continue as a going concern.
- •
- We may not be able to protect our proprietary technology adequately or enforce any rights related thereto.
- •
- We rely or may rely on third parties for certain of our research, clinical trials, technology, supplies, manufacturing and sales and marketing, and a failure of service by such parties could adversely affect our business and reputation.
Company Information
TiGenix was incorporated in Belgium on February 21, 2000 as a company with limited liability under Belgian corporate law. Our principal executive and registered offices are located at Romeinse straat 12, box 2, 3001 Leuven, Belgium. Our telephone number is +32 (16) 39 60 60. We are registered with the Register of Legal Entities (Leuven) under the enterprise number 0471.340.123. Our internet website is www.tigenix.com. The information contained on, or accessible through, our website is not incorporated by reference into this prospectus and should not be considered a part of this prospectus.
Our agent for service of process in the United States is CT Corporation System, whose registered offices are located at 111 Eighth Avenue, 13th Floor, New York, NY 10011. Their telephone number is +1 (212) 894-8800.
For additional information regarding our Company organizational history, see "History and Organizational Structure."
Recent Developments
On December 20, 2013, we entered into a loan facility agreement of up to 10.0 million euros with Kreos Capital IV (UK), under which we borrowed 7.5 million euros during the first half of 2014. On September 30, 2014, we borrowed the final installment of 2.5 million euros available under the facility.
4
Implications of Being an Emerging Growth Company
As a company with less than $1.0 billion in revenue during its fiscal year, we qualify as an "emerging growth company" as defined in Section 2(a) of the Securities Act of 1933, as amended, as modified by the Jumpstart our Business Startups Act of 2012, or the JOBS Act. As such, we are eligible to take advantage of specified reduced reporting requirements and are relieved of certain other significant requirements that are otherwise generally applicable to public companies. As an emerging growth company:
- •
- We are permitted to present only two years of audited consolidated financial statements and only two years of related Management's Discussion and Analysis of Financial Condition and Results of Operations in the registration statement of which this prospectus forms a part.
- •
- We are exempt from the requirement to obtain an attestation and report from our auditors on the assessment of our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act of 2002.
- •
- We are permitted to provide less extensive disclosure about our executive compensation arrangements.
We expect to remain an "emerging growth company" for up to five years, or until any one of the following occurs:
- •
- The last day of the first fiscal year in which our annual gross revenue exceeds $1 billion.
- •
- The last day of the fiscal year that we become a "large accelerated filer" as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, which would occur if the market value of our common equity held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter and we have been publicly reporting for at least twelve months.
- •
- The date on which we have issued more than $1.0 billion in non-convertible debt during the preceding three-year period.
We may choose to take advantage of some but not all of these reduced burdens. We have taken advantage of reduced reporting requirements in this prospectus. Accordingly, the information contained herein may be different from the information you receive from our competitors that are public companies, or other public companies in which you have made an investment.
5
Issuer | TiGenix | |
ADSs offered | ADSs. | |
Ordinary shares outstanding immediately after this offering | ordinary shares. | |
Over-allotment option | ADSs. | |
The ADSs | Each ADS represents ordinary shares. | |
ADSs may be evidenced by American Depositary Receipts, or ADRs. The depositary will hold the ordinary shares underlying your ADSs. You will have rights of an ADR holder as provided in the deposit agreement. You may cancel your ADSs and withdraw the underlying ordinary shares. The depositary will charge you fees for, among other acts, any cancellation. In certain limited instances described in the deposit agreement, we may amend or terminate the deposit agreement without your consent. If you continue to hold your ADSs, you agree to be bound by the terms of the deposit agreement then in effect. | ||
To better understand the terms of the ADSs, you should carefully read the section in this prospectus entitled "Description of American Depositary Shares." You should also read the deposit agreement, which is an exhibit to the registration statement of which this prospectus forms a part. | ||
Depositary for the ADSs | Deutsche Bank Trust Company Americas. | |
Custodian for the ADSs | Deutsche Bank AG, Amsterdam Branch. | |
Use of proceeds | We expect to receive total net proceeds from this offering of approximately $ , after deducting the underwriting discounts and commissions and estimated offering expenses, assuming an initial public offering price of $ per ADS, the midpoint of the price range set forth on the cover page of this prospectus. We intend to use the net proceeds of this offering to: (i) fund new clinical trials of our product candidates, (ii) discover and develop new product candidates from our proprietary technology platform and (iii) fund research and development activities, working capital and other general corporate purposes, including the costs and expenses of being a U.S.-listed public company. Pending our use of the net proceeds as described above, we may invest the net proceeds in short-term bank deposits or interest-bearing, investment-grade securities. See "Use of Proceeds." | |
Dividend policy | We do not currently pay dividends and we do not anticipate declaring or paying any dividends for the foreseeable future. |
6
Listing | We intend to apply to list our ADSs on the NASDAQ Global Market under the symbol "TIG." Prior to this offering, our ordinary shares have traded, and subsequent to this offering will continue to trade, on Euronext Brussels under the symbol "TIG," and we will timely apply for the listing and admission to trading on Euronext Brussels of the new ordinary shares underlying the ADSs. | |
Risk factors | Investing in our ADSs involves a high degree of risk. You should carefully read the information set forth under "Risk Factors" beginning on page 11 of this prospectus and the other information set forth in this prospectus before deciding to invest in the ADSs. |
The number of our ordinary shares that will be issued and outstanding immediately after this offering is based on 160,476,620 ordinary shares outstanding as of September 30, 2014 and excludes the following:
- •
- ordinary shares represented by the ADSs subject to the underwriters' over-allotment option to purchase additional ADSs.
- •
- 8,696,498 ordinary shares issuable upon exercise of outstanding warrants as of September 30, 2014, at a weighted-average exercise price of 1.36 euros per share.
Unless otherwise indicated, this prospectus assumes no exercise of the underwriters' option to purchase up to an additional ADSs from us.
7
Summary Historical Consolidated Financial Data
The tables below present our summary historical consolidated financial data. Our summary historical consolidated financial data as of December 31, 2013 and 2012 and for the years ended December 31, 2013 and 2012 has been derived from our audited consolidated financial statements, which are included elsewhere in this prospectus. Our summary historical consolidated financial data as of June 30, 2014 and for the periods ended June 30, 2014 and 2013 have been derived from our unaudited interim consolidated financial statements, which are included elsewhere in this prospectus. The consolidated financial statements have been prepared and presented in accordance with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or IASB. The interim consolidated financial statements have been prepared and presented in accordance with International Accounting Standard 34 "Interim Financial Reporting." These interim consolidated financial statements do not include all the information required for full annual financial statements in accordance with IFRS as issued by IASB and should be read in conjunction with our consolidated financial statements as at and for the year ended December 31, 2013.
Our consolidated financial statements are prepared and presented in euros, our presentation currency. Solely for the convenience of the reader our consolidated financial statements as at and for the year ended December 31, 2013 and the period ended June 30, 2014, have been translated into U.S. dollars at 1.00 euro=$1.3779 on December 31, 2013 and 1.00 euro=$1.3690 on June 30, 2014, respectively, based on the certified foreign exchange rates published by the Federal Reserve Bank of New York. Such translation should not be construed as a representation that the euro amounts have been or could be converted into U.S. dollars at these or at any other rate of exchange, or at all.
The following summary historical consolidated financial data should be read in conjunction with our historical consolidated financial statements and the related notes thereto and "Management's Discussion and Analysis of Financial Condition and Results of Operations" and "Selected Financial Information," each of which is included elsewhere in this prospectus. The historical results for any prior period are not necessarily indicative of results to be expected for any future period.
8
Consolidated Income Statement Data:
| | Six-month periods ended June 30, | Years ended December 31, | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2014 | 2013 | 2014 | 2013 | 2012 | 2013 | |||||||||||||
| | In thousands of euros, except per share data (Unaudited) | In thousands of U.S. dollars, except per share data (Unaudited) | In thousands of euros, except per share data | In thousands of U.S. dollars, except per share data (Unaudited) | |||||||||||||||
Continuing Operations | |||||||||||||||||||
Revenues | |||||||||||||||||||
Royalties | — | — | — | — | — | — | |||||||||||||
Grants and other operating income | 821 | 736 | 1,124 | 883 | 1,389 | 1,217 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Total revenues | 821 | 736 | 1,124 | 883 | 1,389 | 1,217 | |||||||||||||
Research and development expenses | (5,097 | ) | (5,314 | ) | (6,978 | ) | (9,705 | ) | (12,136 | ) | (13,373 | ) | |||||||
General and administrative expenses | (2,859 | ) | (2,735 | ) | (3,914 | ) | (5,829 | ) | (6,237 | ) | (8,032 | ) | |||||||
Total operating charges | (7,956 | ) | (8,049 | ) | (10,892 | ) | (15,534 | ) | (18,373 | ) | (21,404 | ) | |||||||
| | | | | | | | | | | | | | | | | | | | |
Operating Loss | (7,135 | ) | (7,313 | ) | (9,768 | ) | (14,651 | ) | (16,984 | ) | (20,188 | ) | |||||||
Financial income | 25 | 5 | 34 | 7 | 35 | 10 | |||||||||||||
Financial expenses | (369 | ) | (28 | ) | (505 | ) | (45 | ) | (58 | ) | (62 | ) | |||||||
Foreign exchange differences | 170 | (38 | ) | 233 | (352 | ) | (142 | ) | (485 | ) | |||||||||
| | | | | | | | | | | | | | | | | | | | |
Loss before taxes | (7,309 | ) | (7,374 | ) | (10,006 | ) | (15,041 | ) | (17,149 | ) | (20,725 | ) | |||||||
Income taxes | — | 42 | — | 59 | (1 | ) | 81 | ||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Loss for the period from continuing operations | (7,309 | ) | (7,332 | ) | (10,006 | ) | (14,982 | ) | (17,150 | ) | (20,644 | ) | |||||||
Discontinued Operations | |||||||||||||||||||
Loss for the period from discontinued operations | (1,842 | ) | (1,505 | ) | (2,522 | ) | (3,408 | ) | (3,243 | ) | (4,696 | ) | |||||||
| | | | | | | | | | | | | | | | | | | | |
Loss for the period | (9,151 | ) | (8,837 | ) | (12,528 | ) | (18,390 | ) | (20,393 | ) | (25,340 | ) | |||||||
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Attributable to equity holders of TiGenix | (9,151 | ) | (8,837 | ) | (12,528 | ) | (18,390 | ) | (20,393 | ) | (25,340 | ) | |||||||
Basic and diluted loss per share | (0.06 | ) | (0.09 | ) | (0.08 | ) | (0.16 | ) | (0.22 | ) | (0.22 | ) | |||||||
Basic and diluted loss per share from continuing operations | (0.05 | ) | (0.07 | ) | (0.07 | ) | (0.13 | ) | (0.19 | ) | (0.18 | ) | |||||||
Basic and diluted loss per share from discontinued operations | (0.01 | ) | (0.02 | ) | (0.01 | ) | (0.03 | ) | (0.04 | ) | (0.04 | ) | |||||||
9
Consolidated Statement of Financial Position Data:
| | As at June 30, | As at June 30, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2014 (Actual) | 2014 (As adjusted)(1) | 2014 (Actual) | 2014 (As adjusted)(1) | |||||||||
| | In thousands of euros (Unaudited) | In thousands of U.S. dollars (Unaudited) | |||||||||||
Assets | |||||||||||||
Non-current assets | 37,732 | 51,655 | |||||||||||
Current assets | 22,399 | 30,664 | |||||||||||
| | | | | | | | | | | | | | |
Total Assets | 60,131 | 82,319 | |||||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Equity and Liabilities | |||||||||||||
Equity attributable to equity holders | 39,340 | 53,856 | |||||||||||
Total equity | 39,340 | 53,856 | |||||||||||
Non-current liabilities | 14,353 | 19,649 | |||||||||||
Current liabilities | 6,438 | 8,814 | |||||||||||
| | | | | | | | | | | | | | |
Total Equity and Liabilities | 60,131 | 82,319 | |||||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
- (1)
- Gives effect to the sale of ADSs by us in this offering at an assumed initial public offering price per ADS of $ , the midpoint of the price range set forth on the cover page of this prospectus, after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
Consolidated Statements of Cash Flow Data—Summary:
| | Six-month periods ended June 30, | Years ended December 31, | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2014 | 2013 | 2014 | 2013 | 2012 | 2013 | |||||||||||||
| | In thousands of euros (Unaudited) | In thousands of U.S. dollars (Unaudited) | In thousands of euros | In thousands of U.S. dollars (Unaudited) | |||||||||||||||
Net cash used in operating activities | (6,093 | ) | (7,817 | ) | (8,341 | ) | (14,427 | ) | (17,627 | ) | (19,879 | ) | |||||||
Net cash used in investing activities | (2,552 | ) | (225 | ) | (3,494 | ) | (1,320 | ) | (721 | ) | (1,819 | ) | |||||||
Net cash provided by financing activities | 6,266 | 707 | 8,580 | 20,237 | 9,647 | 27,885 | |||||||||||||
Cash and cash equivalents at end of period | 13,186 | 3,738 | 18,052 | 15,565 | 11,072 | 21,447 | |||||||||||||
10
Investing in the ADSs involves a high degree of risk. You should carefully consider the risks and uncertainties described below, together with other information contained in this prospectus, before making an investment decision. Any of the following risks and uncertainties could have a material adverse effect on our business, prospects, results of operations and financial condition. The market price of our ADSs could decline due to any of these risks and uncertainties, and you could lose all or part of your investment. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also impair our business operations.
Risks Related to the Clinical Development and Regulatory Approval of Our Product Candidates
We may experience delays or failure in the preclinical and clinical development of our product candidates.
As part of the regulatory approval process, we conduct preclinical studies and clinical trials for each of our unapproved product candidates to demonstrate safety and efficacy. The number of required preclinical studies and clinical trials varies depending on the product, the indication being evaluated, the trial results and the applicable regulations. Clinical testing is expensive and can take many years to be completed, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical studies and initial clinical trials do not necessarily predict the results of later-stage clinical trials, and products may fail to show the desired safety, efficacy and quality despite having progressed through initial clinical trials. The data collected from preclinical studies and clinical trials may not be sufficient to support the U.S. Food and Drug Administration, or FDA, the European Medicines Agency, or EMA, or other regulatory approval or approval by ethics committees in various jurisdictions. In addition, the review of a study by an independent data safety monitoring board or review body does not necessarily indicate that the clinical trial will ultimately be successfully completed.
We cannot accurately predict when our current preclinical studies and clinical trials or future clinical trials will be completed, if at all, nor when planned preclinical studies and clinical trials will begin or be completed. Successful and timely completion of clinical trials will require us to recruit a sufficient number of patient candidates, locate or develop manufacturing facilities with regulatory approval sufficient for production of the product to be tested and enter into agreements with third party contract research organizations to conduct the trials. We may need to engage or further engage in preclinical studies and clinical trials with partners, which may reduce any future revenues from any future products.
Our products may cause unexpected side effects or serious adverse events that could interrupt, delay or halt the clinical trials and could result in the FDA, the EMA or other regulatory authorities denying approval of our products for any or all targeted indications. An independent data safety monitoring board, an institutional review board or ethics board, the FDA, the EMA, any other regulatory authorities or we ourselves may suspend or terminate clinical trials at any time, and none of our product candidates may ultimately prove to be safe and effective for human use.
Regulatory approval of our product candidates may be delayed, not obtained or not maintained.
In the United States, all of our cell-based product candidates are subject to a biologics license application, or BLA, issued by the FDA. In Europe, all of our product candidates require regulatory approval through the centralized marketing authorization procedure coordinated by the EMA for advanced therapy medicinal products.
Besides the marketing authorization, we also need to obtain and maintain specific national licenses to perform our commercial operations, including manufacturing and distribution licenses, as well as authorizations to obtain and handle human cells and tissues.
11
Regulatory approval may be delayed, limited or denied for a number of reasons, most of which are beyond our control, including the following:
- •
- The requirement to perform additional clinical trials.
- •
- The failure of the product to meet the safety or efficacy requirements.
- •
- The failure of the relevant manufacturing processes or facilities to meet the applicable requirements.
Any delay or denial of regulatory approval of our product candidates or any failure to comply with post-approval regulatory policies is likely to have a significant impact on our operations and prospects, in particular on our expected revenues.
Regulatory authorities, including the FDA and the EMA, may disagree with our interpretations of data from preclinical studies and clinical trials, our interpretation of applicable regulations including, without limitations, regulations relating to patent term extensions or restorations. They may also approve a product for narrower spectrum of indications than requested or may grant approval subject to the performance of post-marketing studies for a product. Such post-approval studies, if required, may not corroborate the results of earlier trials. Furthermore, the general use of such products may result in either or both of the safety and efficacy profiles differing from those demonstrated in the trials on which marketing approval was based, which could lead to the withdrawal or suspension of marketing approval for the product. In addition, regulatory authorities may not approve the labelling claims that are necessary or desirable for the successful commercialization of our products.
In addition, a marketed product continues to be subject to strict regulation after approval. Changes in applicable legislation or regulatory policies or discovery of problems with the product, production process, site or manufacturer may result in delays in bringing products to the market, the imposition of restrictions on the product's sale or manufacture, including the possible withdrawal of the product from the market, or may otherwise have an adverse effect on our business.
The failure to comply with applicable regulatory requirements may, among other things, result in criminal and civil proceedings and lead to imprisonment, fines, injunctions, damages, total or partial suspension of regulatory approvals, refusal to approve pending applications, recalls or seizures of products and operating and production restrictions.
We may not receive regulatory clearance for trials at each stage and approval for our products and product candidates still in development without delay or at all. If we fail to obtain or maintain regulatory approval for our products, we will be unable to market and sell such products, and such failure or any delay could prevent us from ever generating meaningful revenues or achieving profitability.
We work in a strict regulatory environment, and future changes in any pharmaceutical legislation or guidelines, or unexpected events or new scientific insights occurring within the field of cell therapy, could affect our business.
Regulatory guidelines may change during the course of a product development and approval process, making the chosen development strategy suboptimal. This may delay development, necessitate additional clinical trials or result in failure of a future product to obtain marketing authorization or the targeted price levels and could ultimately adversely impact commercialization of the authorized product. Market conditions may change, resulting in the emergence of new competitors or new treatment guidelines, which may require alterations in our development strategy. This may result in significant delays, increased trial costs, significant changes in commercial assumptions or the failure of future product candidates to obtain marketing authorization.
12
Although the basic regulatory frameworks appear to be in place in the United States and in Europe for cell-based products, at present regulators have limited experience with such products and the interpretation of these frameworks is sometimes difficult to predict. Moreover, the regulatory frameworks themselves will continue to evolve as the FDA and the EMA issue new guidelines. The interpretation of existing rules or the issuance of new regulations may impose additional constraints on the research, development, regulatory approval, manufacturing or distribution processes of future product candidates.
Unexpected events may occur in the cell therapy field, in particular unforeseen safety issues in ongoing clinical trials of any cell therapy product. Moreover, scientific progress might yield new insights on the biology of stem cells which might in turn impact the requirements of safety and efficacy demonstration for stem cell or other cell therapies. Such events or new insights might change the regulatory requirements and framework, in particular strengthening the required clinical research package and increasing the amount of data required to be provided. This could result in additional constraints on our product development process and lead to significant delays, which could prevent us from ever generating meaningful revenues or achieving profitability.
Risks Related to Our Financial Condition and Capital Requirements
If we fail to obtain additional financing, we may be unable to complete the development and commercialization of our product candidates.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial amounts to continue the clinical development of our product candidates. If our product candidates are approved, we will require significant additional funds in order to launch and commercialize such product candidates in the United States and internationally. We may also need to spend substantial amounts to expand our manufacturing infrastructure.
We believe our currently available cash and cash equivalents, together with our royalties from the sales of ChondroCelect under our distribution agreement with Swedish Orphan Biovitrium, or Sobi, will be sufficient to fund our operations through the third quarter of 2015. However, changing circumstances may cause us to consume capital significantly faster than we currently anticipate, and we may need to spend more money than currently expected because of circumstances beyond our control. As a result, we may require additional capital for the further development and commercialization of our product candidates.
Our future funding requirements, both near and long-term, will depend on many factors, including, but not limited to, the following:
- •
- The initiation, progress, timing, costs and results of clinical trials for our product candidates.
- •
- The clinical development plans we establish for these product candidates.
- •
- The number and characteristics of the product candidates that we develop and for which we seek regulatory approval.
- •
- The outcome, timing and cost of regulatory approvals by the FDA, the EMA and any other comparable foreign regulatory authorities, including the potential for the FDA, the EMA or any other comparable foreign regulatory authorities to require that we perform more studies than those that we currently expect.
- •
- The cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights.
- •
- The effects of competing technological and market developments.
- •
- The cost and timing of completion of commercial-scale manufacturing activities.
13
- •
- The cost of establishing sales, marketing and distribution capabilities for any product candidates for which we may receive regulatory approval in regions where we choose to commercialize our products on our own.
Additional funding may not be available on a timely basis, on favorable terms, or at all, and such funds, if raised, may not be sufficient to enable us to continue to implement our business strategy. If we are unable to raise additional funds through equity or debt financing, we may need to delay, scale back or eliminate expenditures for some of our research, development and commercialization plans, or grant rights to develop and market products that we would otherwise prefer to develop and market ourselves, thereby reducing their ultimate value to us.
We have a history of operating losses and an accumulated deficit and may never become profitable.
We have experienced operating losses since our founding in February 2000. We experienced net losses of 20.4 million euros for the year ended December 31, 2012, 18.4 million euros for the year ended December 31, 2013 and 9.2 million euros for the six-month period ended June 30, 2014. As of June 30, 2014, we had an accumulated deficit of 83.2 million euros. These losses resulted mainly from the preclinical, clinical, manufacturing and regulatory efforts we undertook to advance the product candidates in our pipeline and to obtain marketing authorization from the EMA with respect to ChondroCelect, from our commercial efforts in launching ChondroCelect and from general and administrative costs associated with our operations. Our costs have always exceeded our revenues, which have been historically generated mainly through grants and income from the sale of ChondroCelect.
Our ability to become profitable depends on our ability to develop and commercialize our product candidates, and we do not know when, or if, we will generate significant revenues from their sale in the future. Our revenues to date from sales of ChondroCelect, our approved and commercialized product, have been limited.
Even if we do generate sales from our product candidates in the future, we may never achieve or sustain profitability. We anticipate that our operating losses will substantially increase over the next several years as we execute our plan to expand our research, development and commercialization activities, including the clinical development and planned commercialization of our product candidates, and incur the additional costs of operating as a U.S.-listed public company. In addition, if we obtain regulatory approval of our product candidates, we may incur significant sales and marketing expenses. Because of the numerous risks and uncertainties associated with developing pharmaceutical products, we are unable to predict the extent of any future losses or when we will become profitable, if ever.
Our net losses and significant cash used in operating activities have raised substantial doubt regarding our ability to continue as a going concern.
We have a limited operating history and have experienced net losses and significant cash used in operating activities in each period since inception. We expect to continue to incur net losses and have significant cash outflows for at least the next year. These conditions, among others, raise substantial doubt about our ability to continue as a going concern. As a result, our independent registered public accounting firm included an explanatory paragraph in its report on our financial statements as of and for the year ended December 31, 2013 with respect to this uncertainty. Our ability to continue as a going concern could materially limit our ability to raise additional funds through the issuance of new debt or equity securities or otherwise. Future reports on our financial statements may include an explanatory paragraph with respect to our ability to continue as a going concern. We have not been profitable since inception, and it is possible we will never achieve profitability. None of our product candidates can be marketed until governmental approvals have been obtained. Accordingly, there is no substantial source of revenues, much less profits, to sustain our present activities, and no substantial
14
revenues will likely be available until, and unless, our product candidates are approved by the EMA, FDA or comparable regulatory agencies in other countries and successfully marketed, either by us or a partner, an outcome which may not occur. Based upon our currently expected level of operating expenditures, we expect to be able to fund our operations through the third quarter of 2015. This period could be shortened if there are any significant increases in planned spending on development programs or more rapid progress of development programs than anticipated. Other financing may not be available when needed to allow us to continue as a going concern. The perception that we may not be able to continue as a going concern may cause others to choose not to deal with us due to concerns about our ability to meet our contractual obligations.
Our revenues and operating results may fluctuate and may not be sufficient to cover our fixed costs.
Our revenues and operating results have fluctuated in the past and are likely to do so in the future due to a number of factors, many of which are not under our control. Some of the factors that could cause our operating results to fluctuate include, but are not limited to, those listed below and identified throughout this prospectus:
- •
- The (positive or negative) success rate of our development efforts.
- •
- Our ability to manage future clinical trials, given the regulatory environment.
- •
- The timing of approval, if any, of our products by the appropriate regulatory bodies.
- •
- Our ability to commercialize our products (including our ability to obtain reimbursement from public and private insurers for our products).
There is no direct link between the level of our expenses in connection with developing our pipeline of expanded adipose-derived stem cell-based, or eASC-based, product candidates and our revenues, which will primarily consist of royalties from sales of ChondroCelect under our distribution agreement with Sobi until we are able to bring another product to market. Accordingly, if revenues decline or do not grow as we expect, we may not be able to reduce our operating expenses correspondingly and may suffer losses accordingly.
The allocation of available resources could affect our ability to carry out our business plan.
We have significant flexibility and broad discretion to allocate and use our available resources. If such resources are not wisely allocated, our ability to carry out our business plan could be threatened. Our board of directors and management will determine, in their sole discretion and without the need for approval from the holders of our ordinary shares and ADSs, the amounts and timing of our actual expenditures, which will depend upon numerous factors, including the status of our product development and commercialization efforts, if any, and the amount of cash received resulting from partnerships and out-licensing activities. We constantly evaluate opportunities to acquire businesses and technologies that we believe are complementary to our business activities.
Our international operations pose currency risks, which may adversely affect our operating results and net income.
Our operating results may be affected by volatility in currency exchange rates and our ability to manage effectively our currency transaction risks. We use the euro as our currency for financial reporting purposes. In the future, a significant portion of our operating costs may be in U.S. dollars, because we are entering into research and development collaborations, trial collaborations, and professional services contracts in the United States. We also expect a share of our future revenues to be in U.S. dollars. Our exposure to currency risks could increase over time. We do not manage our foreign currency exposure in a manner that would eliminate the effects of changes in foreign exchange rates. For example, we have not engaged in any active hedging techniques, and we have not employed
15
any derivative instruments to date. Therefore, unfavorable fluctuations in the exchange rate between the euro and U.S. dollars could have a negative impact on our financial results.
Risks Related to Our Business
The manufacturing facilities where our product candidates are made are subject to regulatory requirements, that may affect the development of our product candidates and the successful commercialization of our product candidates.
Our product candidates must be manufactured to high standards in compliance with regulatory requirements. The manufacture of such product candidates is subject to regulatory authorization and to requirements of the current good manufacturing practice, or cGMP requirements, prescribed in the relevant country or territory of manufacture or supply.
The cGMP requirements govern quality control of the manufacturing process and require written documentation of policies and procedures. Compliance with such procedures requires record keeping and quality control to ensure that the product meets applicable specifications and other requirements including audits of vendors, contract laboratories and suppliers. Manufacturing facilities are subject to inspection by regulatory authorities at any time. If an inspection by a regulatory authority indicates that there are deficiencies, we could be required to take remedial actions, stop production or close the relevant facility. If we fail to comply with these requirements, we also may be required to curtail the relevant clinical trials, might not be permitted to sell our product candidates or may be limited as to the countries or territories in which we are permitted to sell them.
Our eASC-based development and clinical stage product candidates are manufactured in our facilities in Madrid, Spain, which have been certified by the Spanish Medicines and Medical Devices Agency under cGMP requirements. However, the certification may be interrupted, suspended or discontinued because of a failure to maintain compliance or for any other reason. In addition, the regulations or policies applied by the relevant authorities may change, and any such change would require us to undertake additional work, which may not be sufficient for us to comply with the revised standards.
Any failure to comply with applicable cGMP requirements and other regulations may result in fines and civil penalties, suspension of production, product seizure or recall, import ban or detention, imposition of a consent decree, or withdrawal of product approval, and may limit the availability of our product candidates. Any manufacturing defect or error discovered after products have been produced and distributed also could result in significant consequences, including adverse health consequences, injury or death to patients, costly recall procedures, damage to our reputation and potential for product liability claims. An inability to continue manufacturing adequate supplies of our product candidates at our facilities in Madrid, Spain, could result in a disruption in the supply of our product candidates.
There may be uncertainty over reimbursement from third parties for newly approved healthcare products or such reimbursement may be refused, which could affect our ability to commercialize our product candidates.
Our ability to commercialize future product candidates will depend, in part, on the availability of reimbursement from government and health administration authorities, private health insurers, managed care programs and other third-party payers. Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. In many countries, medicinal products are subject to a regime of reimbursement by government health authorities, private health insurers or other organizations. Such organizations are under significant pressure to limit healthcare costs by restricting the availability and level of reimbursement. For example, we have not been successful in obtaining certain forms of reimbursement with respect to ChondroCelect, such as the decision of the FrenchHaute Autorité de la Santé that ChondroCelect will not be reimbursed in France. Negative
16
decisions by certain authorities or third-party payers may have an unfavorable spillover effect on pending or future reimbursement applications.
We may not be able to obtain or maintain prices for products sufficient to realize an appropriate return on investment if adequate public health service or health insurance coverage is not available. In addition, rules and regulations regarding reimbursement may change, in some cases at short notice, especially in light of the global cost pressures on healthcare and pharmaceutical markets. Such changes could affect whether reimbursement is available at adequate levels or at all.
Our cell therapy product candidates may not be accepted by patients or medical practitioners.
Our ability to commercialize future product candidates and the ability of our distributors to further commercialize ChondroCelect will depend, in part, on market acceptance, including the willingness of medical practitioners to invest in training programs to use the products. Cell therapy products are a novel treatment, and such products may not be immediately accepted as complementary or alternative treatments to the current standards of care. We may not be able to obtain or maintain recommendations and endorsements from influential physicians, which are an essential factor for market acceptance of our product candidates, or our product candidates may not gain sufficient market recognition in spite of favorable opinions from key leaders.
The public perception of ethical and social issues surrounding the use of tissue-engineered products or stem cells may limit or discourage the use of our product candidates. The use of human cells, such as differentiated cartilage cells, eASCs and other adult stem cells, as starting material for the development of our product candidates could generate negative public perceptions of our product candidates and public expressions of concern could result in stricter governmental regulation, which may, in turn, increase the cost of manufacturing and marketing our product or impede market acceptance of our product candidates.
We face competition and technological change, which could limit or eliminate the market opportunity for our product candidates.
The pharmaceutical industry is characterized by intense competition and rapid innovation. Our competitors may be able to develop other products that are able to achieve similar or better results than our product candidates. Our potential competitors include established and emerging pharmaceutical and biotechnology companies and universities and other research institutions. Many of our competitors have substantially greater financial, technical and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations and well-established sales forces. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated in our competitors. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors may succeed in developing, acquiring or licensing on an exclusive basis products that are more effective or less costly than our product candidates. We believe the key competitive factors that will affect the development and commercial success of our product candidates are efficacy, safety and tolerability profile, reliability, price and reimbursement.
Our success depends on certain key people, and we must continue to attract and retain key employees and consultants to be in a position to continue our activities.
Our future success is substantially dependent on a number of key individuals as well as our ability to attract and retain qualified management, scientific, technical, marketing and sales personnel and consultants and upon the continued contributions of such personnel and consultants. Competition for
17
qualified employees and consultants in scientific research and biotechnology industries is intense and there is a limited number of persons with knowledge appropriate to, and experience within, such industries. We do not maintain "key man" insurance policies on the lives of any of our employees.
Our employees may voluntarily terminate their employment at any time. We may not be successful in attracting and retaining qualified employees and consultants to replace existing employees or consultants or to support our growth strategy further. The process of identifying personnel with the combination of skills required to enable us to carry out our strategy is often lengthy and uncertain as to its outcome.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA regulations, to provide accurate information to the FDA, to comply with manufacturing standards we have established, to comply with federal and state healthcare fraud and abuse laws and regulations, to report financial information or data accurately or to disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent off-label promotion, fraud, kickbacks, self-dealing and other abusive practices in the United States and in jurisdictions outside of the United States where we conduct our business. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. If governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions, up to and including criminal prosecution, fines and imprisonment.
We could face product liability claims, resulting in damages against which we are uninsured or underinsured.
Our business exposes us to potential product liability and professional indemnity risks, which are inherent in the research, development, manufacturing, marketing and use of medical treatments. It is impossible to predict the potential adverse effects that our product candidates may have on humans. The use of our product candidates in human clinical trials may result in adverse effects, and long-term adverse effects may only be identified following clinical trials and approval for commercial sale. In addition, physicians and patients may not comply with any warnings that identify the known potential adverse effects and the types of patients who should not receive our product candidates. We may not be able to obtain necessary insurance at an acceptable cost or at all. In the event of any claim, the level of insurance we carry now or in the future may not be adequate, and a product liability or other claim may materially and adversely affect our business. If we cannot adequately protect ourselves against potential liability claims, we may find it difficult or impossible to commercialize our product candidates. Moreover, such claims may require significant financial and managerial resources, may harm our reputation if the market perceives our drugs or drug candidates to be unsafe or ineffective due to unforeseen side effects, and may limit or prevent the further development or commercialization of our product candidates and future product candidates.
We use various chemical and biological products to conduct our research and to manufacture our medicines. Despite the existence of strict internal controls, these chemical and biological products could be the object of unauthorized use or could be involved in an accident that could cause personal injury to people or damage to the environment, which could result in a claim against us. Our activities are subject to specific environmental regulations that impose obligations which, if not complied with, could
18
give rise to third party or administrative claims and could even result in fines being imposed or, in the worst case scenario, to our operations being suspended or shut down.
Risks Related to Our Intellectual Property
We may not be able to protect adequately our proprietary technology or enforce any rights related thereto.
Our ability to compete effectively with other companies depends, among other things, on the exploitation of our technology. In addition, filing, prosecuting and defending patents on all of our product candidates throughout the world would be prohibitively expensive. Our competitors may, therefore, develop equivalent technologies or otherwise gain access to our technology, particularly in jurisdictions in which we have not obtained patent protection or in which enforcement of such protection is not as strong as it is in the United States.
Patents might not be issued with respect to our pending or future applications. The lack of any such patents may have a material adverse effect on our ability to develop and market our proposed product candidates. We may not be able to develop product candidates that are patentable, or our current or future patents may not be sufficiently broad in their scope to provide commercially meaningful protection against competition from third parties. The validity or scope of any of our patents may be insufficient, claims relating to our patents may be asserted by other parties and, if challenged, our patents may be revoked. Even if competitors do not successfully challenge our patents, they might be able to design around such patents or develop unique technologies or products providing effects similar to our product candidates.
If our intellectual property rights, trade secrets and know-how are infringed, litigation may be necessary to protect our intellectual property rights, trade secrets and know-how, which could result in substantial costs and diversion of efforts with no guarantee of success. Our attempts to obtain patent or other protection for certain of our product candidates or technologies may also be subject to opposition. We may need to incur substantial costs to overcome such opposition with no guarantee of success. We may also decide to engage in costly opposition or interference proceedings to prevent third parties from obtaining relevant patent or other protection, again with no guarantee of success.
Developments in U.S. patent law may prevent us from obtaining or enforcing patents directed to our stem cell technologies, which could have a material adverse effect on our business.
U.S. courts have recently issued decisions limiting the patent eligibility of natural products and natural correlations. On June 13, 2013, inAssociation for Molecular Pathology v. Myriad Genetics, the U.S. Supreme Court held that claims to isolated genomic DNA are not patentable subject matter, but claims to complementary DNA molecules are patentable subject matter. On May 8, 2014, the U.S. Court of Appeals for the Federal Circuit held that claims to cloned animals are not patentable subject matter. Furthermore, on March 20, 2012, inMayo Collaborative Services v. Prometheus Laboratories, the U.S. Supreme Court held that several claims drawn to measuring drug metabolite levels from patient samples and correlating them to drug doses are not patentable subject matter.
The Patent and Trademark Office has signaled that a claim reciting any natural phenomenon or natural product will be treated as ineligible for patenting, unless the claim as a whole recites something significantly different from the natural product. The effect of these decisions on patents for inventions relating to other natural phenomena and natural products, such as stem cells, is uncertain. Because our patent portfolio is largely directed to stem cells and their use, as well as to uses of naturally-occurring biomarkers, these developments in U.S. patent law could affect our ability to obtain new U.S. patents or to enforce our existing patents. In some of our pending U.S. patent applications the Patent and Trademark Office has questioned whether certain of our claims are eligible for patenting. If we are unable to procure additional U.S. patents or to enforce our existing U.S. patents, we would be vulnerable to competition in the United States.
19
Third-party claims of intellectual property infringement may prevent or delay our product discovery and development efforts.
Our commercial success depends in part on avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation involving patents and other intellectual property rights in the biotechnology and pharmaceutical industries, as well as administrative proceedings for challenging patents, including interference and reexamination proceedings before the Patent and Trademark Office or oppositions and other comparable proceedings in foreign jurisdictions. Recently, under U.S. patent reform, new procedures includinginter partes review and post grant review have been implemented. This reform is untried and untested and will bring uncertainty to the possibility of challenge to our patents in the future. Numerous U.S. and non-U.S. issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are developing our product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our product candidates may give rise to claims of infringement of the patent rights of others.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents of which we are currently unaware with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates. Because patent applications can take many years, there may be currently pending patent applications that may later result in issued patents that our product candidates may infringe. In addition, third parties may obtain patents in the future and claim that the use of our technologies infringes upon these patents. If any third-party patents were held by a court of competent jurisdiction to cover the manufacturing process of our product candidates, any molecules formed during the manufacturing process or any final product itself, the holders of any such patents might be able to block our ability to commercialize the product candidate, unless we were to obtain a license under the applicable patents, or until such patents expired or they were finally determined to be invalid or unenforceable. Similarly, if any third-party patent were held by a court of competent jurisdiction to cover aspects of our formulations, processes for manufacture or methods of use, the holders of any such patent might be able to block our ability to develop and commercialize our product candidate unless we were to obtain a license or until such patent expired or was finally determined to be invalid or unenforceable. In either case, such a license might not be available on commercially reasonable terms or at all. If we are unable to obtain a necessary license to a third-party patent on commercially reasonable terms, or at all, our ability to commercialize our product candidates might be impaired or delayed, which could in turn significantly harm our business.
Parties making claims against us may seek and obtain injunctive or other equitable relief, which could effectively block our ability to develop further and commercialize our product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we might have to pay substantial damages, including treble damages and attorneys' fees for willful infringement, obtain one or more licenses from third parties, pay royalties or redesign our infringing products, which might be impossible or require substantial time and monetary expenditure. We cannot predict whether any such license would be available at all or whether it would be available on commercially reasonable terms. Furthermore, even in the absence of litigation, we might need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all.
Our future development may depend on our ability to obtain and maintain licenses to certain technologies.
We might further expand our activities in the future by in-licensing certain technologies. Collaboration and integration may have an important impact on the success of our expansion strategy.
20
In such a case, we might not own the patents or supplementary protection certificates on the basis of which these licenses may be granted. These licenses may generally be terminated by the licensor if we breach certain of our obligations under the license and in other specified circumstances. If any of our license agreements were to be terminated, the further development and commercialization of some of our product candidates could be prevented or delayed, reducing their potential revenues. The scope of our rights under such licenses may be subject to dispute by licensors or third parties. We might not control the filing or the prosecution of all the patents to which we hold licenses and may need to rely upon our licensors to enforce the patents and to prevent or to challenge possible infringement by third parties. We might not be able to obtain licenses for the technologies that we require in the future.
We may be involved in lawsuits to protect or enforce our patents, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe our patents. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that one or more of our patents is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could expose one or more of our patents to the risk of being invalidated, held unenforceable, or interpreted narrowly and could put our patent applications at risk of not issuing. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys' fees for willful infringement, obtain one or more licenses from third parties, pay royalties or redesign our infringing products, which may be impossible or require substantial time and monetary expenditure.
Interference proceedings provoked by third parties or brought by the Patent and Trademark Office may be necessary to determine the priority of inventions with respect to our patents or patent applications. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent misappropriation of our confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our ADSs.
We are currently engaged in proceedings challenging a patent owned by the University of Pittsburgh, the outcome of which may affect our ability to commercialize our expanded adipose-derived stem cell based products in the United States.
On April 1, 2011, Cellerix (the predecessor entity of our subsidiary TiGenix SAU) filed aninter partes re-examination request with the Patent and Trademark Office regarding the patent US6777231, owned by the University of Pittsburgh. The Patent and Trademark Office examiner issued a decision concluding that all ten originally issued and all eighteen newly submitted claims of the patent granted to the University of Pittsburgh were invalid. The University of Pittsburgh then appealed the examiner's
21
decision, but only with respect to two of the newly submitted claims. We cross-appealed the examiner's refusal to reject those two newly submitted claims as anticipated by the prior art. The Patent Trial and Appeal Board issued a decision simultaneously granting both appeals, thus confirming that all claims of the patent were invalid, but with respect to the newly submitted claims, on different grounds than those cited in the decision by the initial examiner. On this basis, the University of Pittsburgh filed a request to reopen prosecution and submitted claim amendments to those newly submitted claims to the Patent and Trademark Office for further consideration in an attempt to overcome the Patent Trial and Appeal Board's institution of a new ground for rejection as anticipated by the prior art. The request to reopen prosecution on the basis of the amended claim has been accepted by the Patent and Trademark Office. We submitted comments to the Patent and Trademark Office arguing that these claim amendments did not overcome the anticipated rejection and as of September 30, 2014, we have not received any decision from the Patent and Trademark Office regarding the amended claims. We do not know when a final decision can be expected, and at this stage, we are not in a position to assess the probable outcome of these proceedings.
This proceeding may take longer than expected and may not ultimately succeed, which may result in unexpected additional costs and may have a material adverse effect on our future business, financial condition, operating results and cash flow. If the re-examination is not successful, we may be required to obtain a license on unfavorable terms, or may not be able to obtain a license at all in order to commercialize our adipose-derived stem cell products in the United States. We would potentially be susceptible to patent infringement or litigation regarding patent infringement while commercializing our eASC products in the United States. We may, therefore, choose to delay the launch of our adipose-derived stem cell products in the U.S. market until the expiration of the patent US6777231 on March 10, 2020. To avoid infringing granted patents equivalent to US6777231 in other countries, we may at any given point in time be forced to develop and utilize alternative technology, to exploit our current technology and products under a royalty-bearing license with respect to the intellectual property rights of other parties or to delay the launch of our adipose-derived stem cell products in the relevant market until patent expiration.
Risks Related to Our Dependence on Third Parties
The manufacturing facilities where our product candidates are made are subject to regulatory requirements, that may affect the development of our product candidates and the successful commercialization of our product candidates.
Our product candidates must be manufactured to high standards in compliance with regulatory requirements. The manufacture of such product candidates is subject to regulatory authorization and to requirements of the current good manufacturing practice, or cGMP requirements, prescribed in the relevant country or territory of manufacture or supply.
The cGMP requirements govern quality control of the manufacturing process and require written documentation of policies and procedures. Compliance with such procedures requires record keeping and quality control to ensure that the product meets applicable specifications and other requirements including audits of vendors, contract laboratories and suppliers. Manufacturing facilities are subject to inspection by regulatory authorities at any time. If an inspection by a regulatory authority indicates that there are deficiencies, we could be required to take remedial actions, stop production or close the relevant facility. If we fail to comply with these requirements, we also may be required to curtail the relevant clinical trials, might not be permitted to sell our product candidates or may be limited as to the countries or territories in which we are permitted to sell them.
Our eASC-based development and clinical stage product candidates are manufactured in our facilities in Madrid, Spain, which have been certified by the Spanish Medicines and Medical Devices Agency under cGMP requirements. However, the certification may be interrupted, suspended or
22
discontinued because of a failure to maintain compliance or for any other reason. In addition, the regulations or policies applied by the relevant authorities may change, and any such change would require us to undertake additional work, which may not be sufficient for us to comply with the revised standards.
Any failure to comply with applicable cGMP requirements and other regulations may result in fines and civil penalties, suspension of production, product seizure or recall, import ban or detention, imposition of a consent decree, or withdrawal of product approval, and may limit the availability of our product candidates. Any manufacturing defect or error discovered after products have been produced and distributed also could result in significant consequences, including adverse health consequences, injury or death to patients, costly recall procedures, damage to our reputation and potential for product liability claims. An inability to continue manufacturing adequate supplies of our product candidates at our facilities in Madrid, Spain, could result in a disruption in the supply of our product candidates.
We rely on third parties to manufacture our product ChondroCelect, and, in the future, we may rely on third parties to manufacture our product candidates; a failure of service by such parties could adversely affect our business and reputation.
PharmaCell, a leading European contract manufacturing organization active in the area of cell therapy, has purchased our former Dutch subsidiary holding our manufacturing facility. Our former subsidiary will continue to manufacture ChondroCelect in that facility under a long-term manufacturing agreement. We are also in the process of entering into an agreement with a U.S.-based contract manufacturing organization and will start the process for technology transfer in connection with a proposed Phase III study with respect to Cx601 in the United States. We are, therefore, exposed to risks relating to the conduct of business of such parties, including the following:
- •
- Their ability to employ and retain suitably qualified staff and maintain good labor relations with their workforce.
- •
- Their ability to meet the required legal, regulatory or quality control standards, including the cGMP requirements prescribed in the relevant country or territory of manufacture or supply.
- •
- Their level of investment in their facilities and equipment and their ability to consistently manufacture our product candidates to the required standard.
In addition, we may face challenges in communicating with such third parties, which could potentially lead to mistakes and difficulties in coordinating activities. We could also face unexpected cost increases that are beyond our control.
Any failure by such parties to meet the required standards could have a materially adverse effect on our reputation or expose us to legal liability, with respect to which we may have limited recourse to the defaulting party. If such a party were to breach its contractual commitments to us, our only option might be to seek a legal remedy, which could be costly or time-consuming and, even if successful, may not fully compensate us for our damages. If we have to terminate our relationship with such a party due to problems with the timeliness or quality of their work, we may not be able to replace them on commercially acceptable terms, or at all, which could delay or threaten our ability to generate meaningful revenue from product sales as a result of which we may have insufficient capital resources to support our operations.
We may need to rely on distributors and other third parties to commercialize our product candidates, and such distributors may not succeed in commercializing our product candidates effectively or at all.
For some market opportunities, we may need to enter into co-development, co-promotion or other licensing arrangements with larger pharmaceutical firms to increase the chances of commercial success of our product candidates. For example, with respect to ChondroCelect we have entered into an
23
exclusive distribution agreement with Sobi for the European Union (excluding Finland) as well as several other countries. In the future, we may enter into additional distribution agreements in other territories. We may not be able to establish sales, marketing and distribution capabilities of our own or to enter into arrangements with contract sales organizations or larger pharmaceutical firms in a timely manner or on acceptable terms. Additionally, building marketing and distribution capabilities may be more expensive than we anticipate and may require us to divert funds from other intended purposes or prevent us from building our own marketing and distribution capabilities to desired levels.
Therefore, the performance of our product candidates will depend in part on our ability to attract and retain suitable partners that will be able to market and support our products effectively. We may lose one or more of our distributors or might not be able to recruit additional or replacement distributors.
Our dependence on third parties may also reduce our profit margins and delay or limit our ability to develop and commercialize our products on a timely and competitive basis.
Our distributors may be faced with hurdles in reimbursement, market acceptance, distribution and competition that delay or even prevent the commercialization of our product candidates. The ability of our distributors to commercialize our product candidates also depends, in part, on the extent to which our competition will react.
We rely on third parties to conduct our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for, or commercialize, our product candidates.
We rely on third-party contract research organizations to conduct clinical trials for our product candidates, and we control only certain aspects of their activities. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with applicable protocol, legal, regulatory and scientific standards, and our reliance on our contract research organizations does not relieve us of our regulatory responsibilities. We and our contract research organizations will be required to comply with current good clinical practices, or cGCP requirements, which are a collection of regulations enforced by the FDA, the EMA and comparable foreign regulatory authorities for product candidates in clinical development. These cGCP requirements are intended to protect the health, safety and welfare of study subjects through requirements such as informed consent. Regulatory authorities enforce these cGCP requirements through periodic inspections of trial sponsors, principal investigators and study sites. If we or any of these contract research organizations fail to comply with applicable cGCP regulations, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, the EMA or a comparable foreign regulatory authority may require us to perform additional clinical trials before approving our marketing applications. Upon inspection, such regulatory authorities might determine that any of our clinical trials do not comply with cGCP regulations. In addition, for biological products, our clinical trials must be conducted with products made under cGMP regulations and will require a large number of test subjects. Our failure or any failure by our contract research organizations to comply with these regulations or to recruit a sufficient number of patients may require us to repeat clinical trials, which would delay the regulatory approval process. Moreover, we may be implicated or subject to civil or criminal liability if any of our contract research organizations violates fraud and abuse or false claims laws and regulations or healthcare privacy and security laws in any jurisdiction in which we conduct our trials.
The contract research organizations will not be employed directly by us and, except for remedies available to us under our agreements with such contract research organizations, we cannot control whether they devote sufficient time and resources to our ongoing preclinical and clinical programs. These contract research organizations may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical studies or other product
24
development activities, which could affect their performance on our behalf. If these contract research organizations do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated, and we may not be able to complete development of, obtain regulatory approval for, or commercialize our product candidates.
Switching or adding contract research organizations involves substantial cost and requires extensive management time and focus. In addition, there is a natural transition period when a new contract research organization commences work. As a result, delays may occur, which could materially affect our ability to meet our desired clinical development timelines. We may encounter challenges in our relationships with our contract research organizations or delays in the future.
We may form or seek strategic alliances in the future, and we might not realize the benefits of such alliances.
We may form or seek strategic alliances, create joint ventures or collaborations or enter into licensing arrangements with third parties that we believe will complement or augment our development and commercialization efforts with respect to our product candidates and any future products that we may develop. Any of these relationships may require us to incur non-recurring and other charges, increase our near and long-term expenditures, issue securities that dilute our existing shareholders or disrupt our management and business. In addition, we face significant competition in seeking appropriate strategic partners, and the negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for our product candidates, because they may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view our product candidates as having the requisite potential to demonstrate safety and efficacy. If we license products or businesses, we may not be able to realize the benefit of such transactions if we are unable to integrate them with our existing operations and company culture. Following a strategic transaction or license, we might not be able to achieve the revenues or specific net income that justifies such transaction. Any delays in entering into new strategic partnership agreements related to our product candidates could delay the development and commercialization of our product candidates in certain geographies for certain indications.
Risks Related to the ADSs and this Offering
There is no established trading market for the ADSs.
This offering constitutes our initial public offering of ADSs, and no public market for the ADSs currently exists. We intend to apply to list the ADSs on the NASDAQ Global Market, subject to completion of customary procedures in the United States. Any delay in the commencement of trading of the ADSs on the NASDAQ Global Market would impair the liquidity of the market for the ADSs and make it more difficult for holders to sell the ADSs.
Even if the ADSs are listed on the NASDAQ Global Market, there is a risk that an active trading market for the ADSs may not develop or be sustained after this offering is completed. The initial offering price will be based, in part, on the price of our ordinary shares on Euronext Brussels, and determined by negotiations among the lead underwriters and us. Among the factors considered in determining the initial offering price are our future prospects and the prospects of our industry in general, our revenue, net income and certain other financial and operating information in recent periods, and the financial ratios, market prices of securities and certain financial and operating information of companies engaged in activities similar to ours. Following this offering, the ADSs may not trade at a price equal to or greater than the offering price.
25
The ADSs may experience price and volume fluctuations.
Stock markets have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of listed companies. Broad market and industry factors may negatively affect the market price of the ADSs, regardless of our actual operating performance. The market price and liquidity of the market for the ADSs that will prevail in the market after this offering may be higher or lower than the price you pay and may be significantly affected by numerous factors, some of which are beyond our control. These factors include:
- •
- Significant volatility in the market price and trading volume of securities of companies in our sector, which is not necessarily related to the operating performance of these companies.
- •
- Delays between our expenditures to develop and market new products and the generation of sales from those products.
- •
- Changes in the amount that we spend to develop, acquire or license new products, technologies or businesses.
- •
- Changes in our expenditures to promote our products and services.
- •
- Success or failure of research and development projects of us or our competitors.
- •
- Announcements of acquisitions by us or one of our competitors.
- •
- The general tendency towards volatility in the market prices of shares of companies that rely on technology and innovation.
- •
- Changes in regulatory policies or tax guidelines.
- •
- Changes or perceived changes in earnings or variations in operating results.
- •
- Any shortfall in revenue or net income from levels expected by investors or securities analysts.
- •
- Disputes or other developments relating to proprietary rights, including patents, and our ability to obtain patent protection for our technologies.
- •
- Departures of key scientific or management personnel.
- •
- Significant lawsuits, including patent litigation.
- •
- General economic trends and other external factors, many of which are beyond our control.
In addition, the stock market in general, and the NASDAQ Global Market and pharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. If the market price of our ADSs after this offering does not exceed the initial public offering price, you may not realize any return on your investment and may lose some or all of your investment. In the past, securities class action litigation has often been instituted against companies following periods of volatility in the market price of a company's securities. This type of litigation, if instituted, could result in substantial costs and a diversion of management's attention and resources.
As a new investor, you will experience substantial dilution as a result of this offering.
The public offering price per ADS will be substantially higher than the net tangible book value per ADS prior to this offering. Consequently, if you purchase ADSs in this offering at an assumed public offering price of $ , the midpoint of the price range set forth on the cover page of this prospectus, you will incur immediate dilution of $ per ADS. For further information regarding the dilution resulting from this offering, please see the section entitled "Dilution" in this prospectus. This dilution is
26
due in large part to the fact that our earlier investors paid substantially less than the assumed initial public offering price when they purchased their ordinary shares.
Raising additional capital may cause additional dilution of the percentage ownership of our shareholders, restrict our operations, require us to relinquish rights to our technologies, products or product candidates and could cause our share price to fall.
We expect that significant additional capital may be needed in the future to continue our planned operations, including conducting clinical trials, commercialization efforts, expanded research and development activities and costs associated with operating as a U.S.-listed public company. To raise capital, we may issue new ordinary shares, ADSs, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we issue new ordinary shares, ADSs, convertible securities or other equity securities, investors may be materially diluted by subsequent sales. Such issuances or sales may also result in material dilution to our existing shareholders, and new investors could gain rights, preferences and privileges senior to the holders of our ordinary shares or ADSs, including ADSs sold in this offering. The incurrence of indebtedness could result in increased fixed payment obligations and could involve certain restrictive covenants, such as limitations on our ability to incur additional debt and other operating restrictions that could adversely impact our ability to conduct our business. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, products or product candidates, or grant licenses on terms unfavorable to us.
Fluctuations in the exchange rate between the U.S. dollar and the euro may increase the risk of holding our ADSs and shares.
Our shares currently trade on Euronext Brussels in euros, and our ADSs will trade on the NASDAQ Global Market in U.S. dollars. Fluctuations in the exchange rate between the U.S. dollar and the euro may result in temporary differences between the value of our ADSs and the value of our ordinary shares, which may result in heavy trading by investors seeking to exploit such differences.
In addition, as a result of fluctuations in the exchange rate between the U.S. dollar and the euro, the U.S. dollar equivalent of the proceeds that a holder of our ADSs would receive upon the sale in Belgium of any shares withdrawn from the depositary and the U.S. dollar equivalent of any cash dividends paid in euros on our shares represented by the ADSs could also decline.
Holders of ADSs are not treated as shareholders of our Company.
By participating in this offering you will become a holder of ADSs with underlying shares in a Belgian limited liability company. Holders of ADSs are not treated as shareholders of our Company, unless they withdraw our ordinary shares underlying the ADSs. The depositary is the holder of the ordinary shares underlying the ADSs. Holders of ADSs therefore do not have any rights as shareholders of our Company, other than the rights that they have pursuant to the deposit agreement.
You will not have the same voting rights as the holders of our ordinary shares and may not receive voting materials in time to be able to exercise your right to vote.
Except as described in this prospectus and the deposit agreement, holders of ADSs will not be able to exercise voting rights attaching to the ordinary shares evidenced by the ADSs on an individual basis. Under the terms of the deposit agreement, holders of ADSs may instruct the depositary to vote the ordinary shares underlying their ADSs, but only if we ask the depositary to ask for their instructions. Otherwise, holders of ADSs will not be able to exercise their right to vote unless they withdraw ordinary shares underlying their ADSs to vote them in person or by proxy in accordance with Belgian
27
corporate law and our articles of association. Even so, holders of ADSs may not know about a meeting far enough in advance to withdraw those ordinary shares. If we ask for the instructions of holders of ADSs, the depositary, upon timely notice from us, will notify holders of ADSs of the upcoming vote and arrange to deliver our voting materials to them. Upon our request, the depositary will mail to holders of ADSs a shareholder meeting notice that contains, among other things, a statement as to the manner in which voting instructions may be given, including an express indication that such instructions may be given or deemed given to the depositary to give a discretionary proxy to a person designated by us if no instructions are received by the depositary from holders of ADSs on or before the response date established by the depositary. No voting instruction shall be deemed given and no such discretionary proxy shall be given with respect to any matter as to which we inform the depositary that (i) substantial opposition exists, or (ii) such matter materially and adversely affects the rights of shareholders. We cannot guarantee that holders of ADSs will receive the voting materials in time to ensure that they can instruct the depositary to vote their shares. A shareholder is only entitled to participate in, and vote at, the meeting of shareholders, provided that its shares are recorded in its name at midnight (Central European Time) at the end of the fourteenth day preceding the date of the meeting of shareholders. Failure by the depositary to record your shares by the record date, could result in the inability to participate and vote at the relevant meeting of shareholders. In addition, the depositary's liability to holders of ADSs for failing to execute voting instructions or for the manner of executing voting instructions is limited by the deposit agreement. As a result, holders of ADSs may not be able to exercise their right to give voting instructions or to vote in person or by proxy and they may not have any recourse against the depositary or our Company if their shares are not voted as they have requested or if their shares cannot be voted.
We have no present intention to pay dividends on our ordinary shares in the foreseeable future and, consequently, your only opportunity to achieve a return on your investment during that time is if the price of the ADSs appreciates.
We have no present intention to pay dividends in the foreseeable future. Any recommendation by our board of directors to pay dividends will depend on many factors, including our financial condition, results of operations, legal requirements and other factors. Furthermore, pursuant to Belgian law, the calculation of amounts available for distribution to shareholders, as dividends or otherwise, must be determined on the basis of our non-consolidated statutory accounts prepared in accordance with Belgian accounting rules. In addition, in accordance with Belgian law and our articles of association, we must allocate each year an amount of at least 5% of our annual net profit under our non-consolidated statutory accounts to a legal reserve until the reserve equals 10% of our share capital. Therefore, we are unlikely to pay dividends or other distributions in the foreseeable future. If the price of the ADSs or the underlying ordinary shares declines before we pay dividends, you will incur a loss on your investment, without the likelihood that this loss will be offset in part or at all by potential future cash dividends.
As a foreign private issuer, we are exempt from a number of rules under the U.S. securities laws and are permitted to file less information with the SEC than U.S. domestic issuers. This may limit the information available to holders of ADSs.
We are a "foreign private issuer," as defined in the SEC rules and regulations, and, consequently, we are not subject to all of the disclosure requirements applicable to U.S. domestic issuers. For example, we are exempt from certain rules under the Exchange Act that regulate disclosure obligations and procedural requirements related to the solicitation of proxies, consents or authorizations applicable to a security registered under the Exchange Act. In addition, our officers, directors and principal shareholders are exempt from the reporting and "short-swing" profit recovery provisions of Section 16 of the Exchange Act and related rules with respect to their purchases and sales of our securities. Moreover, we are not required to file periodic reports and consolidated financial statements with the
28
SEC as frequently or as promptly as U.S. domestic issuers. Accordingly, there may be less publicly available information concerning our Company than there is for U.S. public companies. As a foreign private issuer, we will file an annual report on Form 20-F within four months of the close of each year ended December 31 and furnish reports on Form 6-K relating to certain material events promptly after we publicly announce these events. However, we are not required to publish quarterly financial information, and, therefore, our shareholders will not be afforded the same information generally available to investors holding shares in public companies organized in the United States.
We are an "emerging growth company," and we intend to take advantage of reduced disclosure and governance requirements applicable to emerging growth companies, which could result in the ADSs being less attractive to investors.
We are an "emerging growth company," as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including the following:
- •
- Exemption from the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002.
- •
- Reduced disclosure obligations regarding executive compensation in this prospectus and our periodic reports and proxy statements
- •
- Exemptions from the requirements to hold nonbinding advisory votes on executive compensation and to seek shareholder approval of any golden parachute payments not previously approved.
We could be an emerging growth company for up to five years following the year in which we complete this offering, although circumstances could cause us to lose that status earlier, including if the market value of our ordinary shares held by non-affiliates exceeds $700.0 million as of any June 30 before that time or if we have total annual gross revenue of $1.0 billion or more during any fiscal year before that time, in which cases we would no longer be an emerging growth company as of the following December 31 or, if we issue more than $1.0 billion in non-convertible debt during any three-year period before that time, we would cease to be an emerging growth company immediately. We cannot predict if investors will find our ADSs less attractive because we may rely on these exemptions. If some investors find our ADSs less attractive as a result, there may be a less active trading market for our ADSs, and our share price may be more volatile.
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to report accurately our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal control over financial reporting and disclosure controls and procedures. In particular, in the future, we will be required, under Section 404 of the Sarbanes-Oxley Act, to perform system and process evaluations and testing of our internal controls over financial reporting to allow management and our independent registered public accounting firm to report on the effectiveness of our internal control over financial reporting. This assessment will need to include disclosure of any material weaknesses in our internal control over financial reporting identified by our management or our independent registered public accounting firm. Section 404 of the Sarbanes-Oxley Act also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting. However, for as long as we remain an emerging growth company as defined in the JOBS Act, we intend to take advantage of the exemption permitting us not to comply with the independent registered public accounting firm attestation requirement. At the time when we are no longer an emerging growth company, our independent registered public accounting firm may issue a report that is adverse in the event it is not satisfied with the level at which our controls are documented, designed or operating. Our remediation efforts may not enable us to avoid a material weakness in the future.
29
Our compliance with Section 404 will require that we incur substantial accounting expense and expend significant management efforts. We currently do not have an internal audit group, and we may need to hire additional accounting and financial staff with the appropriate experience and technical accounting knowledge, and compile the system and process documentation necessary to perform the evaluation needed to comply with Section 404. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of the ADSs could decline, and we could be subject to sanctions or investigations by the NASDAQ Stock Market, the SEC or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
We will incur significant increased costs as a result of operating as a company whose ADSs are publicly traded in the United States, and our management will be required to devote substantial time to new compliance initiatives.
As a company whose ADSs will be publicly traded in the United States, we will incur significant legal, accounting, insurance and other expenses that we have not previously incurred. In addition, the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act and related rules implemented by the SEC and the NASDAQ Stock Market have imposed various requirements on public companies, including requiring establishment and maintenance of effective disclosure and financial controls. These costs will increase at the time when we are no longer an emerging growth company eligible to rely on exemptions under the JOBS Act from certain disclosure and governance requirements. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly. For example, we expect these rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to incur substantial costs to maintain the same or similar coverage. These laws and regulations could also make it more difficult and expensive for us to attract and retain qualified persons to serve on our board of directors or its committees. Furthermore, if we are unable to satisfy our obligations as a U.S.-listed public company, we could be subject to delisting of the ADSs, fines, sanctions and other regulatory action and potentially civil litigation.
You may be subject to limitations on the transfer of your ADSs.
Your ADSs are transferable on the books of the depositary. However, the depositary may close its books at any time or from time to time when it deems doing so expedient in connection with the performance of its duties. The depositary may close its books from time to time for a number of reasons, including in connection with corporate events such as a rights offering, during which time the depositary needs to maintain an exact number of ADS holders on its books for a specified period. The depositary may also close its books in emergencies and on weekends and public holidays. The depositary may refuse to deliver, transfer or register transfers of the ADSs generally when our share register or the books of the depositary are closed, or at any time if we or the depositary thinks that it is advisable to do so because of any requirement of law or of any government or governmental body or of
30
any regulatory authority, or under any provision of the deposit agreement, or for any other reason in accordance with the terms of the deposit agreement.
You may not receive distributions on our ordinary shares represented by the ADSs or any value for them if it is illegal or impractical to make them available to holders of ADSs.
Under the terms of the deposit agreement, the depositary for the ADSs has agreed to pay to you the cash dividends or other distributions it or the custodian receives on our ordinary shares or other deposited securities after deducting its fees and expenses. You will receive these distributions in proportion to the number of our ordinary shares your ADSs represent. However, it may be unlawful or impractical to make a distribution available to holders of ADSs. We have no obligation to take any other action to permit the distribution of the ADSs, ordinary shares, rights or anything else to holders of the ADSs. This means that you may not receive the distributions we make on our ordinary shares or any value from them if it is unlawful or impractical to make them available to you. These restrictions may have a material adverse effect on the value of your ADSs.
If securities or industry analysts do not publish research or reports about our business, or if they adversely change their recommendations regarding the ADSs, the market price for the ADSs and trading volume could decline.
The trading market for the ADSs will be influenced by research or reports that industry or securities analysts publish about our business. If one or more analysts who cover us downgrade the ADSs, the market price for the ADSs would likely decline. If one or more of these analysts cease to cover us or fail to regularly publish reports on us, we could lose visibility in the financial markets, which, in turn, could cause the market price or trading volume for the ADSs to decline.
It may be difficult for investors outside Belgium to serve process on, or enforce foreign judgments against, us or our directors and senior management.
We are a Belgian public limited liability company. Only one member of our board of directors and no member of our executive management is a resident of the United States. All or a substantial portion of the assets of such non-resident persons and most of our assets are located outside the United States. As a result, it may not be possible for investors to effect service of process upon such persons or on us or to enforce against them or us a judgment obtained in U.S. courts. Original actions or actions for the enforcement of judgments of U.S. courts relating to the civil liability provisions of the federal or state securities laws of the United States are not directly enforceable in Belgium. The United States and Belgium do not currently have a multilateral or bilateral treaty providing for reciprocal recognition and enforcement of judgments, other than arbitral awards, in civil and commercial matters. In order for a final judgment for the payment of money rendered by U.S. courts based on civil liability to produce any effect on Belgian soil, it is accordingly required that this judgment be recognized or be declared enforceable by a Belgian court in accordance with Articles 22 to 25 of the 2004 Belgian Code of Private International Law. Recognition or enforcement does not imply a review of the merits of the case and is irrespective of any reciprocity requirement. A U.S. judgment will, however, not be recognized or declared enforceable in Belgium if it infringes upon one or more of the grounds for refusal that are exhaustively listed in Article 25 of the Belgian Code of Private International Law. These grounds mainly require that the recognition or enforcement of the foreign judgment should not be a manifest violation of public policy, that the foreign courts must have respected the rights of the defense, that the foreign judgment should be final, and that the assumption of jurisdiction by the foreign court may not have breached certain principles of Belgian law. In addition to recognition or enforcement, a judgment by a federal or state court in the United States against us may also serve as evidence in a similar action in a Belgian court if it meets the conditions required for the authenticity of judgments according to the law of the state where it was rendered. The findings of a federal or state
31
court in the United States will not, however, be taken into account to the extent they appear incompatible with Belgian public policy.
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us, because pharmaceutical companies have experienced significant share price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management's attention and resources.
We believe that our shares or ADSs should not be treated as stock of a passive foreign investment company, or PFIC, for U.S. federal income tax purposes, but this conclusion is a factual determination that is made annually and thus may be subject to change. If we were to qualify as a PFIC, this could result in adverse U.S. federal income tax consequences to U.S. investors.
Based on the composition of our assets and the nature of our, income, we believe that our shares or ADSs should not be treated as stock of a PFIC for U.S. federal income tax purposes, but this conclusion is a factual determination that is made annually and thus may be subject to change. In general, we will be treated as a PFIC for any taxable year in which either of the following is true:
- •
- At least 75% of our gross income (looking through certain corporate subsidiaries) for the taxable year is "passive income."
- •
- At least 50% of the value, determined on the basis of a quarterly average, of our gross assets (looking through certain corporate subsidiaries) is attributable to assets that produce or are held for the production of "passive income."
Passive income generally includes dividends, interest, royalties, rents (other than certain rents and royalties derived in the active conduct of a trade or business), annuities and gains from assets that produce passive income. If we are treated as a PFIC, and you are a U.S. Holder as defined in "Taxation—U.S. Taxation" that did not make a "mark-to-market election," as described below, you will be subject to potentially adverse U.S. federal income tax consequences in the taxable year in which the share or ADSs are sold or upon receipt of an "excess distribution" with respect to the shares or ADSs. In general, a U.S. Holder would receive an "excess distribution" if the amount of any distribution for U.S. federal income tax purposes in respect of the shares or ADSs is more than 125% of the average distributions made with respect to the shares or ADSs within the three preceding taxable years (or shorter period in which such U.S. Holder held the shares or ADSs). In general, a U.S. Holder would be subject to an additional tax that is equivalent to an interest charge on U.S. taxes that are deemed due during the period the U.S. Holder owned the shares or ADSs computed by assuming that the gain (in the case of a sale) or the "excess distribution" in respect of the shares or ADSs was taxed in equal portions at the highest applicable tax rate throughout the period in which such U.S. Holder owned such shares or ADSs. A "mark-to-market election," if available to and made by a U.S. Holder generally would result in such U.S. Holder taking into account ordinary income or loss in respect of such U.S. Holder's investment in the shares or ADSs by marking the shares or ADSs to market on an annual basis. In addition, as a PFIC, dividends on the shares or ADSs would not be eligible for the special tax rate available to non-corporate U.S. Holders applicable to "qualified dividend income." Prospective U.S. Holders of shares or ADSs should consult their own U.S. tax advisors regarding the potential application of the PFIC rules. See "Taxation—U.S. Taxation—Passive Foreign Investment Company Considerations."
32
We are a Belgian public limited liability company, and shareholders of our Company may have different and in some cases more limited shareholder rights than shareholders of a U.S. listed corporation.
We are a public limited liability company incorporated under the laws of Belgium. Our corporate affairs are governed by Belgian corporate law. The rights provided to our shareholders under Belgian corporate law and our articles of association differ in certain respects from the rights that you would typically enjoy as a shareholder of a U.S. corporation under applicable U.S. federal and state laws.
Under Belgian corporate law, other than certain limited information that we must make public and except in certain limited circumstances, our shareholders may not ask for an inspection of our corporate records, while under Delaware corporate law any shareholder, irrespective of the size of its shareholdings, may do so. Shareholders of a Belgian corporation are also unable to initiate a derivative action, a remedy typically available to shareholders of U.S. companies, in order to enforce a right of our Company, in case we fail to enforce such right ourselves, other than in certain cases of director liability under limited circumstances. In addition, a majority of our shareholders present or represented at our meeting of shareholders may release a director from any claim of liability we may have, including if he or she has acted in bad faith or has breached his or her duty of loyalty, provided, in some cases, that the relevant acts were specifically mentioned in the convening notice to the meeting of shareholders deliberating on the discharge. In contrast, most U.S. federal and state laws prohibit a company or its shareholders from releasing a director from liability altogether if he or she has acted in bad faith or has breached his or her duty of loyalty to the company. Finally, Belgian corporate law does not provide any form of appraisal rights in the case of a business combination.
As a result of these differences between Belgian corporate law and our articles of association, on the one hand, and the U.S. federal and state laws, on the other hand, in certain instances, you could receive less protection as a shareholder of our Company than you would as a shareholder of a listed U.S. company.
Because we are a foreign private issuer and intend to follow certain home country corporate governance practices, our shareholders may not have the same protections afforded to shareholders of companies that are subject to all NASDAQ Global Market corporate governance requirements.
As a foreign private issuer, we have the option to follow Belgian corporate law and the Belgian Corporate Governance Code rather than the corporate governance practices of the NASDAQ Global Market, except to the extent that such laws would be contrary to U.S. securities laws, and provided that we disclose the requirements we are not following and describe the home country practices we follow instead. As a result, our shareholders may not have the same protections afforded to shareholders of companies that are subject to all the NASDAQ Global Market corporate governance requirements. See "Management—Differences between Our Corporate Governance Practices and the Listing Rules of the NASDAQ Stock Market."
Holders of ADSs or ordinary shares have limited rights to call meetings of shareholders or to submit shareholder proposals.
Except under limited circumstances, only the board of directors or the statutory auditor may call a meeting of shareholders. Shareholders that collectively own at least 20% of the share capital of our Company may require the board of directors or the statutory auditor to convene a special or an extraordinary general meeting of shareholders. Provided that certain conditions are satisfied, one or more shareholders holding at least 3% of our share capital may request for items to be added to the agenda of any convened meeting and submit proposed resolutions in relation to existing agenda items or new items to be added to the agenda. As a result, the ability of holders of the ADSs or ordinary shares to participate in and influence the governance of our Company is limited.
33
Holders or beneficial owners of the ADSs have limited recourse if we or the depositary fail to meet our respective obligations under the deposit agreement or if they wish us or the depositary to participate in legal proceedings.
The deposit agreement expressly limits the obligations and liability of us and the depositary. Neither we nor the depositary will be liable to the extent that liability results from the fact that we:
- •
- Are prevented, forbidden from or delayed in doing or performing any obligation under the terms of the deposit agreement by reason of any provision of any present or future law or regulation of the United States, any state thereof, Belgium or any other country, or governmental or regulatory authority or stock exchange, or on account of possible civil or criminal liabilities or by reason of any provision of our constitutional documents or any other provision governing the ordinary shares or by reason of any act of God, war or other circumstances beyond our or their control.
- •
- Exercise or fail to exercise discretion under the deposit agreement or our constitutional documents or any other provision governing the ordinary shares.
- •
- Perform our obligations without gross negligence or willful misconduct or bad faith.
- •
- Take any action or fail to take any action based upon advice of or information from legal counsel, accountants, any person presenting shares for deposit, any holder of the ADSs or any other person believed in good faith to be competent to give such advice or information.
- •
- Rely on any documents we believe to be genuine and properly executed.
In addition, neither we nor the depositary has any obligation to participate in any action, suit or other proceeding in respect of the ADSs which may involve it in expense or liability unless it is indemnified to its satisfaction. Additionally, neither we nor the depositary will incur any liability for any special, consequential, indirect or punitive damages for any breach of the deposit agreement or otherwise. These provisions of the deposit agreement will limit the ability of holders or beneficial owners of the ADSs to obtain recourse if we or the depositary fail to meet our respective obligations under the deposit agreement or if they wish us or the depositary to participate in a legal proceeding.
Investors may not be able to participate in equity offerings, and ADS holders may not receive any value for rights that we may grant.
In accordance with Belgian corporate law, our articles of association provide for preferential subscription rights to be granted to our existing shareholders to subscribe on a pro rata basis for any issue for cash of new shares, convertible bonds or warrants that are exercisable for cash, unless such rights are canceled or limited by resolution of our meeting of shareholders or the board of directors. Our meeting of shareholders or board of directors may cancel or restrict such rights in future equity offerings. In addition, certain shareholders (including those in the United States, Australia, Canada or Japan) may not be entitled to exercise such rights even if they are not canceled unless the rights and related shares are registered or qualified for sale under the relevant legislation or regulatory framework. As a result, there is the risk that investors may suffer dilution of their shareholding should they not be permitted to participate in preference right equity or other offerings that we may conduct in the future.
If rights are granted to our shareholders, as the case may be, but the depositary is unable to sell rights corresponding to shares represented by ADSs that are not exercised by, or distributed to, ADS holders, or if the sale of such rights is not lawful or reasonably practicable, the depositary may allow the rights to lapse, in which case ADS holders will receive no value for such rights.
34
We have broad discretion to determine how to use the net proceeds from this offering and may use them in ways that may not enhance our results of operations or the price of the ADSs.
Our management will have broad discretion over the use of net proceeds from this offering, and we could spend the net proceeds from this offering in ways the holders of the ADSs may not agree with or that do not yield a favorable return. We intend to use the net proceeds of this offering for the following purposes:
- •
- Funding new clinical trials of multiple product candidates.
- •
- Discovering and developing new product candidates from our proprietary technology platform.
- •
- Funding research and development activities, working capital.
- •
- Other general corporate purposes, including the costs and expenses of being a U.S.-listed public company.
Because of the number and variability of factors that will determine our use of the net proceeds from this offering, our use of these proceeds may differ substantially from our current plans. You will not have the opportunity, as part of your investment decision, to assess whether proceeds are being used appropriately. You must rely on the judgment of our management regarding the application of the net proceeds of this offering.
35
HISTORY AND ORGANIZATIONAL STRUCTURE
We were incorporated in Belgium on February 21, 2000, initially to capitalize on technology developed at the universities of Leuven and Ghent for the regeneration of cartilage, bone and other musculoskeletal tissues.
The following chart illustrates our corporate structure as of the date of this prospectus:
TiGenix SAU. On May 3, 2011, we acquired Cellerix, a cell-therapy company based in Madrid, Spain. Cellerix, which was later renamed TiGenix SAU, had an eASC-based technology platform for indications of inflammatory and autoimmune origin that are the basis of our pipeline. The Cellerix team and facilities have been completely integrated into our organization.
Arcarios B.V. On July 8, 2010, we spun off certain drug discovery assets to the Dutch company Arcarios B.V. (formerly named Therosteon B.V.) in which we hold a 3.53% equity stake as of September 30, 2014.
TiGenix Inc. We incorporated TiGenix Inc., a wholly-owned U.S. subsidiary, on February 7, 2006, and on May 8, 2007, TiGenix Inc. and Cognate BioServices entered into a fifty-fifty joint venture with respect to TC CEF LLC, an asset management company. TC CEF LLC subsequently acquired the assets of a fully equipped cell expansion facility from Cell Genesys, Inc., for the manufacture of ChrondroCelect for clinical trials required by the FDA and to serve the U.S. market after obtaining marketing approval for ChondroCelect in the United States. However, after we abandoned our plans to introduce ChondroCelect into the U.S. market independently due to the associated costs and the required time, we withdrew from the joint venture as of November 23, 2010 and terminated our membership interests in TC CEF LLC. As of the date of this prospectus, TiGenix Inc. is a dormant subsidiary.
Other Historical Subsidiaries. On September 24, 2009, we established TiGenix B.V., a wholly-owned Dutch subsidiary. TiGenix B.V. constructed a new European human cell expansion facility in Geleen to increase the manufacturing capacity of ChondroCelect in Europe. On May 30, 2014, we completed the sale of all of the shares of TiGenix B.V. to PharmaCell. ChondroCelect will continue to be manufactured in that facility under a long-term manufacturing agreement with our former subsidiary.
36
On November 30, 2009, we acquired Orthomimetics Limited, a biomaterials company that was later renamed TiGenix Ltd. TiGenix Ltd. designed, developed and manufactured novel, bioresorbable implants for the regenerative repair of articular joint damage resulting from sports injuries and other trauma, including ChondroMimetic, an off-the-shelf biomaterial scaffold for the treatment of small osteochondral defects and small focal chondral lesions with possible underlying subchondral bone plate damage. In view of our exclusive focus on cell therapy since the Cellerix acquisition in 2011, we decided to shut down TiGenix Ltd. The intellectual property related to TiGenix Ltd., which was recognized as part of our intangible assets, was fully impaired in our consolidated financial statements as of December 31, 2011. TiGenix Ltd. was dissolved in May 2014.
37
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements and our estimates with respect to our anticipated future performance and the market in which we operate. Certain of these statements, forecasts and estimates can be recognized by the use of words such as, without limitation, "believes," "anticipates," "expects," "intends," "plans," "seeks," "estimates," "may," "will," "predicts," "projects" and "continue" and similar expressions. Such statements, forecasts and estimates are based on various assumptions and assessments of known and unknown risks, uncertainties and other factors, which may or may not prove to be correct. Actual events are difficult to predict and may depend upon factors that are beyond our control. Therefore, our actual results, financial condition or performance may turn out to be materially different from such statements, forecasts and estimates. Factors that might cause such a difference include, but are not limited to, those discussed in the section "Risk Factors" included elsewhere in this prospectus.
By their nature, forward-looking statements involve risks and uncertainties because they relate to events, competitive dynamics and industry change, and depend on economic circumstances that may or may not occur in the future or may occur on longer or shorter timelines than anticipated. Although we believe that we have a reasonable basis for each forward-looking statement contained in this prospectus, we caution you that forward-looking statements are not guarantees of future performance and involve known and unknown risks, uncertainties and other factors that are in some cases beyond our control. All of our forward-looking statements are subject to risks and uncertainties that may cause our actual results to differ materially from our expectations.
Actual results could differ materially from our forward-looking statements due to a number of factors, including, without limitation, the following:
- •
- We may experience delays or failure in the preclinical and clinical development of our product pipeline.
- •
- Regulatory approval of our products may be delayed, not obtained or not maintained.
- •
- We work in a strict regulatory environment, and future changes to any pharmaceutical legislation or guidelines or unexpected events or new scientific insights occurring within the field of cell therapy, could affect our business.
- •
- If we fail to obtain additional financing, we may be unable to complete the development and commercialization of our product candidates.
- •
- We have a history of operating losses and an accumulated deficit and may never become profitable.
- •
- Our net losses and significant cash used in operating activities have raised substantial doubt about our ability to continue as a going concern.
- •
- The manufacturing facilities at which our product candidates are made are subject to regulatory requirements, which may affect the development of our product pipeline and the successful commercialization of our products.
- •
- Our success depends on our key people and we must continue to attract and retain key employees and consultants to be in a position to continue our activities.
- •
- We may not be able to adequately protect our proprietary technology or enforce any rights related thereto.
- •
- Third party claims of intellectual property infringement may prevent or delay our product discovery and development efforts.
38
- •
- We may need to rely on distributors and other third parties to commercialize our product candidates, and such distributors may not succeed in commercializing our product candidates effectively or at all.
- •
- We rely on third parties to conduct our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates.
- •
- The allocation of available resources could affect our ability to carry out our business plan.
The preceding list is not intended to be an exhaustive list of all of our forward-looking statements. Except as required by law, we undertake no obligation to update publicly any forward-looking statements for any reason after the date of this prospectus or to conform these statements to actual results or to changes in our expectations.
39
The following tables set forth the high, low, average and period end Bloomberg Composite Rate expressed in U.S. dollars per euro. The Bloomberg Composite Rate is a best market calculation, in which, at any point in time, the bid rate is equal to the highest bid rate of all contributing bank indications and the ask rate is set to the lowest ask rate offered by these banks. The Bloomberg Composite Rate is a mid-value rate between the applied highest bid rate and the lowest ask rate.
Year (U.S. dollar per euro) | High | Low | Average Rate(1) | Period End | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
2009 | 1.5094 | 1.2543 | 1.3944 | 1.4331 | |||||||||
2010 | 1.4510 | 1.1952 | 1.3266 | 1.3366 | |||||||||
2011 | 1.4874 | 1.2925 | 1.3922 | 1.2960 | |||||||||
2012 | 1.3463 | 1.2053 | 1.2859 | 1.3197 | |||||||||
2013 | 1.3802 | 1.2780 | 1.3285 | 1.3743 | |||||||||
- (1)
- The average rate for a year means the average of the Bloomberg Composite Rates on the last day of each month during a year.
Month (U.S. dollar per euro) | High | Low | Average Rate(1) | Period End | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
April, 2014 | 1.3886 | 1.3705 | 1.3804 | 1.3824 | |||||||||
May, 2014 | 1.3928 | 1.3591 | 1.3808 | 1.3635 | |||||||||
June, 2014 | 1.3692 | 1.3532 | 1.3600 | 1.3692 | |||||||||
July, 2014 | 1.3679 | 1.3390 | 1.3544 | 1.3390 | |||||||||
August, 2014 | 1.3427 | 1.3132 | 1.3314 | 1.3132 | |||||||||
September, 2014 | 1.3150 | 1.2630 | 1.2895 | 1.2631 | |||||||||
- (1)
- The average rate for a month or for any shorter period, means the average of the daily Bloomberg Composite Rates during that month, or shorter period, as the case may be.
The Bloomberg Composite Rate on September 30, 2014 was $1.2631 per euro.
40
We estimate that the net proceeds to us from this offering, after deducting underwriting discounts and commissions and estimated offering expenses payable by us, will be approximately $ , assuming an initial public offering price of $ per ADS, the midpoint of the price range set forth on the cover page of this prospectus.
A $1.00 increase (decrease) in the assumed initial public offering price of $ per ADS would increase (decrease) the net proceeds from this offering to us by approximately $ , assuming no change to the number of ADSs offered as set forth on the cover page of this prospectus. An increase (decrease) of 500,000 ADSs in the number of ADSs offered by us would increase (decrease) the net proceeds to us by approximately $ , assuming the initial public offering price remains the same.
We intend to use the net proceeds of this offering for the following purposes:
- •
- With respect to Cx601, to accomplish the following objectives:
- •
- Europe. To prepare our marketing and sales infrastructure to commercialize Cx601 in Europe (approximately $ million).
- •
- United States. To complete the process of technology transfer to a U.S.-based contract manufacturing organization to file an investigational new drug application to conduct a pivotal Phase III trial in the United States supporting a biologics license application with the FDA and to commercialize Cx601, whether by ourselves or in collaboration with partners (approximately $ million).
- •
- To advance the clinical development of Cx611 in early rheumatoid arthritis and severe sepsis in the United States and Europe (approximately $ million).
- •
- The remainder for general corporate purposes, including research and development and working capital requirements.
The foregoing represents our current intentions with respect to the use and allocation of the net proceeds of this offering based upon our present plans and business conditions, but our management will have significant flexibility and discretion in applying the net proceeds. The occurrence of unforeseen events or changed business conditions could result in the application of the net proceeds of this offering in a manner other than as described above. Pending our use of the net proceeds as described above, we intend to invest the net proceeds in short-term bank deposits or interest-bearing, investment-grade securities.
41
We do not currently pay dividends, and we do not anticipate declaring or paying any dividends for the foreseeable future.
All of the ordinary shares represented by the ADSs offered by this prospectus will have the same dividend rights as all of our other outstanding ordinary shares. In general, distributions of dividends proposed by our board of directors require the approval of our shareholders at a meeting of shareholders with a simple majority vote, although our board of directors may declare interim dividends without shareholder approval, subject to the terms and conditions of the Belgian Company Code. See "Description of Share Capital."
Pursuant to Belgian law, the calculation of amounts available for distribution to shareholders, as dividends or otherwise, must be determined on the basis of our non-consolidated statutory financial accounts. In addition, under the Belgian Company Code, we may declare or pay dividends only if, following the declaration and issuance of the dividends, the amount of our net assets on the date of the closing of the last financial year according to our statutory annual accounts (i.e., the amount of the assets as shown in the balance sheet, decreased with provisions and liabilities, all as prepared in accordance with Belgian accounting rules), decreased with the non-amortized costs of incorporation and expansion and the non-amortized costs for research and development, does not fall below the amount of the paid-up capital (or, if higher, the called capital), increased with the amount of non-distributable reserves. Finally, prior to distributing dividends, we must allocate at least 5% of our annual net profits (under our non-consolidated statutory accounts prepared in accordance with Belgian accounting rules) to a legal reserve, until the reserve amounts to 10% of our share capital.
For information regarding the Belgian withholding tax applicable to dividends and related U.S. reimbursement procedures, see "Taxation—Belgian Taxation."
42
The following table sets forth our capitalization as of June 30, 2014:
- •
- on an actual basis;
- •
- on a pro forma basis to give effect to the borrowing of the final installment of 2.5 million euros under our loan facility with Kreos Capital IV (UK); and
- •
- on a pro forma basis as adjusted to reflect the sale by us of ADSs in this offering at an assumed initial public offering price of $ per ADS, the midpoint of the price range set forth on the cover page of this prospectus, and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us.
The pro forma as adjusted information below is for illustrative purposes only. Our capitalization following the closing of this offering will be adjusted based on the actual initial public offering price and other terms of this offering determined at pricing.
Solely for the convenience of the reader our pro forma as adjusted capitalization has been translated into U.S. dollars at 1.00 euro=$1.3690 on June 30, 2014 based on the certified foreign exchange rate published by the Federal Reserve Bank of New York. Such translation should not be construed as a representation that the euro amounts have been or could be converted into U.S. dollars at this or at any other rate of exchange, or at all.
This table should be read in conjunction with "Use of Proceeds," "Selected Financial Information," "Management's Discussion and Analysis of Financial Condition and Results of Operations" and our consolidated financial statements and the accompanying notes thereto appearing elsewhere in this prospectus.
| | As at June 30, 2014 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Actual | Pro Forma | Pro Forma As Adjusted | Pro Forma As Adjusted | |||||||||
| | In thousands of euros (Unaudited) | In thousands of euros (Unaudited) | In thousands of euros (Unaudited) | In thousands of U.S. dollars (Unaudited) | |||||||||
Receivables from reverse repurchase agreements(1) | 5,999 | — | 5,999 | 8,213 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Cash and cash equivalents | 13,186 | 2,500 | 15,686 | 21,474 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Financial loans and other payables | 14,238 | 2,500 | 16,738 | 22,914 | |||||||||
Total equity: | |||||||||||||
Share capital | 16,048 | — | 16,048 | 21,970 | |||||||||
Share premium | 100,118 | — | 100,118 | 137,162 | |||||||||
Accumulated deficit | (83,200 | ) | — | (83,200 | ) | (113,901 | ) | ||||||
Other reserves | 6,374 | — | 6,374 | 8,726 | |||||||||
| | | | | | | | | | | | | | |
Total equity | 39,340 | — | 39,340 | 53,856 | |||||||||
| | | | | | | | | | | | | | |
Total capitalization | 53,578 | 2,500 | 56,078 | 76,770 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
- (1)
- Receivables from reverse repurchase agreements are short term non-derivative instruments with a carrying amount approximate to the fair value. Because they can be disposed at any time with no penalty on the principal, they are considered liquid assets.
43
If you invest in the ADSs in this offering, your interest will be diluted immediately to the extent of the difference between the initial public offering price per ADS and the pro forma net tangible book value per ADS after this offering. Dilution results from the fact that the initial public offering price per ADS is substantially in excess of the net tangible book value per ADS attributable to our existing shareholders for our ordinary shares that will be outstanding immediately prior to the closing of this offering. We calculate net tangible book value per ordinary share by dividing the net tangible book value (total assets less intangible assets and total liabilities) by the number of outstanding ordinary shares. Dilution is determined by subtracting net tangible book value per ADS from the initial public offering price per ADS.
Our net tangible book value as of June 30, 2014 was 4.4 million euros ($6.1 million), or 0.03 euros ($0.04) per share. Investors participating in this offering will incur immediate and substantial dilution. Upon the closing of this offering and the sale by us of the ADSs in this offering at an assumed initial public offering price of euros ($ ) per ADS, the midpoint of the price range set forth on the cover page of this prospectus, and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us, our pro forma net tangible book value as of June 30, 2014 would have been approximately euros ($ ), or euros ($ ) per ADS. This amount represents an immediate increase in our pro forma net tangible book value of euros ($ ) per ADS to our existing shareholders and an immediate dilution of $ per ADS to new investors purchasing the ADSs in this offering at the initial public offering price.
The following table illustrates this dilution per ADS:
| | | Per ADS | |||||
|---|---|---|---|---|---|---|---|
| | | (in euros) | |||||
Assumed initial public offering price | |||||||
Historical net tangible book value before the change attributable to investors purchasing ADSs in this offering | |||||||
Pro forma net tangible book value | |||||||
Increase in net tangible book value attributable to investors purchasing ADSs in this offering | |||||||
Pro forma as adjusted net tangible book value after giving effect to this offering | |||||||
Dilution to new investors purchasing in this offering | |||||||
A $1.00 (0.73 euro) increase (decrease) in the assumed initial public offering price of $ ( euros) per ADS, the midpoint of the price range set forth on the cover page of this prospectus, would increase (decrease) our pro forma net tangible book value after this offering by $ ( euros) per ADS/share, and decrease (increase) the dilution in pro forma net tangible book value to new investors by $ ( euros) per ADS/share, assuming that the number of ADSs offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us. Each increase (decrease) of 500,000 ADSs in the number of ADSs offered by us would increase (decrease) our pro forma net tangible book value after this offering by $ ( euros) per ADS/share and decrease (increase) the dilution to investors participating in this offering by approximately $ ( euros) per ADS/share, assuming that the assumed initial public offering price remains the same and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us.
The following table summarizes on a pro forma basis, as of June 30, 2014, the differences between the shareholders as of June 30, 2014 and the new investors with respect to the number of ordinary shares purchased from us, the total consideration paid and the average price per ordinary share paid by existing shareholders and by investors participating in this offering at an assumed initial public offering price of $ ( euros) per ADS, the midpoint of the price range set forth on the cover page of the
44
prospectus, before deducting the underwriting discounts and commissions and estimated offering expenses payable by us:
| | Ordinary Shares Purchased | Total Consideration | | | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Average Price per Ordinary Share | Average Price per ADS | |||||||||||||||||
| | Number | Percent | Amount | Percent | |||||||||||||||
| | | | (in euros) | (in euros) | (in euros) | ||||||||||||||
Existing shareholders | % | ||||||||||||||||||
New investors | % | ||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Total | 100.0 | % | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
A $1.00 (0.73 euro) increase (decrease) in the assumed initial public offering price of $ ( euros) per ADS, the midpoint of the price range set forth on the cover page of this prospectus, would increase (decrease) total consideration paid by new investors by $ million ( million euros), assuming that the number of ADSs offered, as set forth on the cover page of this prospectus, remains the same, and before deducting the underwriting discounts and commissions and estimated offering expenses payable by us.
If the underwriters exercise their over-allotment option in full, our existing shareholders would own ordinary shares, or %, in the aggregate, of our total outstanding share capital and our new investors would own ADSs, or %, in the aggregate, of our total outstanding share capital after this offering. If the underwriters exercise their over-allotment option in full, our pro forma net tangible book value would be $ ( euros) per ADS/share and the dilution to investors participating in this offering would be $ ( euros) per ADS/share.
To the extent that we grant warrants or other equity awards to our directors, executive management or employees in the future, and those warrants or other equity awards are exercised or other issuances of our ordinary shares are made, there will be further dilution to investors participating in this offering. In addition, there will be further dilution to investors participating in this offering in the case of any future capital increase with cancellation of the preferential subscription rights of our existing shareholders and any future offering where U.S. investors are excluded from participation.
45
Our ordinary shares began trading on Euronext Brussels in 2007. The current trading symbol on Euronext Brussels is "TIG."
Our ordinary shares will continue trading on Euronext Brussels under the symbol "TIG" and we expect that our ADSs will trade on the NASDAQ Global Market under the symbol "TIG" after the effective date of the registration statement to which this prospectus relates.
The following table lists the high and low sales prices and the average daily trading volume on Euronext Brussels for our ordinary shares for the last nine months; the last eight fiscal quarters; and the last five fiscal years. Prices indicated below with respect to our ordinary share price include interdealer prices, without retail mark up, mark down or commission and may not necessarily represent actual transactions. All prices are quoted in euros and U.S. dollars using the exchange rate published by the Federal Reserve Bank of New York on the applicable trading date.
| | Euros | U.S. Dollars | | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Average Daily Trading Volume | |||||||||||||||
Period | High | Low | High | Low | ||||||||||||
Monthly | ||||||||||||||||
September 30, 2014 | 0.62 | 0.55 | 0.82 | 0.71 | 202,817 | |||||||||||
August 31, 2014 | 0.66 | 0.49 | 0.87 | 0.66 | 277,729 | |||||||||||
July 31, 2014 | 0.65 | 0.52 | 0.88 | 0.71 | 379,332 | |||||||||||
June 30, 2014 | 0.71 | 0.60 | 0.96 | 0.82 | 350,827 | |||||||||||
May 31, 2014 | 0.75 | 0.63 | 1.03 | 0.86 | 395,855 | |||||||||||
April 30, 2014 | 0.85 | 0.54 | 1.17 | 0.75 | 1,693,967 | |||||||||||
March 31, 2014 | 0.96 | 0.81 | 1.33 | 1.11 | 1,699,356 | |||||||||||
February 28, 2014 | 0.95 | 0.77 | 1.30 | 1.04 | 2,526,978 | |||||||||||
January 31, 2014 | 1.03 | 0.51 | 1.40 | 0.69 | 6,330,813 | |||||||||||
Quarterly | ||||||||||||||||
September 30, 2014 | 0.66 | 0.49 | 0.88 | 0.66 | 288,165 | |||||||||||
June 30, 2014 | 0.85 | 0.54 | 1.17 | 0.75 | 799,349 | |||||||||||
March 31, 2014 | 1.03 | 0.51 | 1.40 | 0.69 | 3,579,427 | |||||||||||
December 31, 2013 | 0.56 | 0.24 | 0.76 | 0.32 | 2,901,565 | |||||||||||
September 30, 2013 | 0.64 | 0.19 | 0.83 | 0.25 | 1,319,410 | |||||||||||
June 30, 2013 | 0.93 | 0.60 | 1.21 | 0.78 | 302,944 | |||||||||||
March 31, 2013 | 1.05 | 0.81 | 1.38 | 1.05 | 453,623 | |||||||||||
December 31, 2012 | 1.02 | 0.77 | 1.35 | 1.00 | 487,177 | |||||||||||
Yearly | ||||||||||||||||
December 31, 2014 (through September 30, 2014) | 1.03 | 0.49 | 1.40 | 0.66 | 1,539,699 | |||||||||||
December 31, 2013 | 1.05 | 0.19 | 1.38 | 0.25 | 1,254,614 | |||||||||||
December 31, 2012 | 1.02 | 0.43 | 1.35 | 0.53 | 314,278 | |||||||||||
December 31, 2011 | 1.45 | 0.57 | 2.13 | 0.81 | 55,974 | |||||||||||
December 31, 2010 | 4.03 | 1.15 | 5.67 | 1.54 | 107,501 | |||||||||||
46
SELECTED FINANCIAL INFORMATION
The tables below present our summary historical consolidated financial data. Our summary historical consolidated financial data as of December 31, 2013 and 2012 and for the years ended December 31, 2013 and 2012 has been derived from our consolidated financial statements, which are included elsewhere in this prospectus. Our summary historical consolidated financial data as of June 30, 2014 and for the periods ended June 30, 2014 and 2013 have been derived from our unaudited interim consolidated financial statements, which are included elsewhere in this prospectus. The consolidated financial statements have been prepared and presented in accordance with IFRS as issued by the IASB. As an emerging growth company, we are not required to present, and have not presented, selected financial data for any period prior to our most recently completed two fiscal years. The interim consolidated financial statements have been prepared and presented in accordance with International Accounting Standard 34 "Interim Financial Reporting." These interim consolidated financial statements do not include all the information required for full annual financial statements in accordance with IFRS as issued by IASB and should be read in conjunction with our consolidated financial statements as at and for the year ended December 31, 2013.
Our consolidated financial statements are prepared and presented in euros, our presentation currency. Solely for the convenience of the reader our consolidated financial statements as at and for the year ended December 31, 2013 and the period ended June 30, 2014 have been translated into U.S. dollars at 1.00 euro=$1.3779 on December 31, 2013, and 1.00 euro=$1.3690 on June 30, 2014, respectively, based on the certified foreign exchange rates published by the Federal Reserve Bank of New York. Such translation should not be construed as a representation that the euro amounts have been or could be converted into U.S. dollars at these or at any other rate of exchange, or at all.
The following summary historical consolidated financial data should be read in conjunction with our historical consolidated financial statements and the related notes thereto and "Management's Discussion and Analysis of Financial Condition and Results of Operations," included elsewhere in this prospectus. The historical results for any prior period are not necessarily indicative of results to be expected for any future period.
47
Consolidated Income Statement Data:
| | Six-month periods ended June 30, | Years ended December 31, | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2014 | 2013 | 2014 | 2013 | 2012 | 2013 | |||||||||||||
| | In thousands of euros, except per share data (Unaudited) | In thousands of U.S. dollars, except per share data (Unaudited) | In thousands of euros, except per share data | In thousands of U.S. dollars, except per share data (Unaudited) | |||||||||||||||
Continuing Operations | |||||||||||||||||||
Revenues | |||||||||||||||||||
Royalties | — | — | — | — | — | — | |||||||||||||
Grants and other operating income | 821 | 736 | 1,124 | 883 | 1,389 | 1,217 | |||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Total revenues | 821 | 736 | 1,124 | 883 | 1,389 | 1,217 | |||||||||||||
Research and development expenses | (5,097 | ) | (5,314 | ) | (6,978 | ) | (9,705 | ) | (12,136 | ) | (13,373 | ) | |||||||
General and administrative expenses | (2,859 | ) | (2,735 | ) | (3,914 | ) | (5,829 | ) | (6,237 | ) | (8,032 | ) | |||||||
Total operating charges | (7,956 | ) | (8,049 | ) | (10,892 | ) | (15,534 | ) | (18,373 | ) | (21,404 | ) | |||||||
| | | | | | | | | | | | | | | | | | | | |
Operating Loss | (7,135 | ) | (7,313 | ) | (9,768 | ) | (14,651 | ) | (16,984 | ) | (20,188 | ) | |||||||
Financial income | 25 | 5 | 34 | 7 | 35 | 10 | |||||||||||||
Financial expenses | (369 | ) | (28 | ) | (505 | ) | (45 | ) | (58 | ) | (62 | ) | |||||||
Foreign exchange differences | 170 | (38 | ) | 233 | (352 | ) | (142 | ) | (485 | ) | |||||||||
| | | | | | | | | | | | | | | | | | | | |
Loss before taxes | (7,309 | ) | (7,374 | ) | (10,006 | ) | (15,041 | ) | (17,149 | ) | (20,725 | ) | |||||||
Income taxes | — | 42 | — | 59 | (1 | ) | 81 | ||||||||||||
| | | | | | | | | | | | | | | | | | | | |
Loss for the period from continuing operations | (7,309 | ) | (7,332 | ) | (10,006 | ) | (14,982 | ) | (17,150 | ) | (20,644 | ) | |||||||
Discontinued Operations | |||||||||||||||||||
Loss for the period from discontinued operations | (1,842 | ) | (1,505 | ) | (2,522 | ) | (3,408 | ) | (3,243 | ) | (4,696 | ) | |||||||
| | | | | | | | | | | | | | | | | | | | |
Loss for the period | (9,151 | ) | (8,837 | ) | (12,528 | ) | (18,390 | ) | (20,393 | ) | (25,340 | ) | |||||||
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Attributable to equity holders of TiGenix | (9,151 | ) | (8,837 | ) | (12,528 | ) | (18,390 | ) | (20,393 | ) | (25,340 | ) | |||||||
Basic and diluted loss per share | (0.06 | ) | (0.09 | ) | (0.08 | ) | (0.16 | ) | (0.22 | ) | (0.22 | ) | |||||||
Basic and diluted loss per share from continuing operations | (0.05 | ) | (0.07 | ) | (0.07 | ) | (0.13 | ) | (0.19 | ) | (0.18 | ) | |||||||
Basic and diluted loss per share from discontinued operations | (0.01 | ) | (0.02 | ) | (0.01 | ) | (0.03 | ) | (0.04 | ) | (0.04 | ) | |||||||
48
Consolidated Statements of Financial Position Data:
| | As at June 30, | As at December 31, | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2014 | 2014 | 2013 | 2012 | 2013 | |||||||||||
| | In thousands of euros (Unaudited) | In thousands of U.S. dollars (Unaudited) | In thousands of euros | In thousands of U.S. dollars (Unaudited) | ||||||||||||
Assets | ||||||||||||||||
Non-current assets | 37,732 | 51,655 | 38,863 | 48,315 | 53,549 | |||||||||||
Current assets | 22,399 | 30,664 | 18,045 | 15,642 | 24,864 | |||||||||||
Assets held for sale | — | — | 6,135 | — | — | |||||||||||
| | | | | | | | | | | | | | | | | |
Total Assets | 60,131 | 82,319 | 63,043 | 63,957 | 86,867 | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Equity and Liabilities | ||||||||||||||||
Equity attributable to equity holders | 39,340 | 53,856 | 48,222 | 48,568 | 66,445 | |||||||||||
Total equity | 39,340 | 53,856 | 48,222 | 48,568 | 66,445 | |||||||||||
Non-current liabilities | 14,353 | 19,649 | 8,378 | 6,306 | 11,544 | |||||||||||
Current liabilities | 6,438 | 8,814 | 5,877 | 9,083 | 8,098 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total Equity and Liabilities | 60,131 | 82,319 | 63,043 | 63,957 | 86,867 | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Consolidated Statements of Cash Flow Data—Summary:
| | Six-month periods ended June 30, | Years ended December 31, | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2014 | 2013 | 2014 | 2013 | 2012 | 2013 | |||||||||||||
| | In thousands of euros (Unaudited) | In thousands of U.S. dollars (Unaudited) | In thousands of euros | In thousands of U.S. dollars (Unaudited) | |||||||||||||||
Net cash used in operating activities | (6,093 | ) | (7,817 | ) | (8,341 | ) | (14,427 | ) | (17,627 | ) | (19,879 | ) | |||||||
Net cash used in investing activities | (2,552 | ) | (225 | ) | (3,494 | ) | (1,320 | ) | (721 | ) | (1,819 | ) | |||||||
Net cash provided by financing activities | 6,266 | 707 | 8,578 | 20,237 | 9,647 | 27,885 | |||||||||||||
Cash and cash equivalents at end of period | 13,186 | 3,738 | 18,052 | 15,565 | 11,072 | 21,447 | |||||||||||||
49
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND
RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with the section entitled "Selected Financial Information" and our consolidated financial statements and accompanying notes included elsewhere in this prospectus. In addition to historical consolidated financial information, this section contains forward-looking statements that reflect our plans, estimates and opinions. Our actual results could differ materially from those discussed in the forward-looking statements. Factors that could cause or contribute to these differences, include, without limitation, those discussed in the sections entitled "Risk Factors," "Special Note Regarding Forward-Looking Statements," "Business" and elsewhere in this prospectus.
Overview
We are an advanced biopharmaceutical company focused on developing and commercializing novel therapeutics from our proprietary platform of allogeneic, or donor-derived, expanded adipose-derived stem cells, known as eASCs, in inflammatory and autoimmune diseases. Based on our platform, we have developed a pipeline of product candidates with our most advanced being Cx601, a first-in-class injectable allogeneic stem cell therapy that has been granted orphan designation by the European Medicines Agency, or EMA. We are conducting a single pivotal Phase III trial for Cx601 for the treatment of complex perianal fistulas in patients suffering from Crohn's disease, a painful and debilitating condition affecting approximately 100,000 patients in the United States and Europe. Data from the single pivotal trial should be available in the third quarter of 2015, based on which we plan to submit a marketing authorization application to the EMA in the first half of 2016. We also intend to initiate a Phase III trial for Cx601 for the treatment of complex perianal fistulas in the United States by the second half of 2016. Based on discussions with the U.S. Food and Drug Administration, or FDA, we believe that this Phase III trial, if successful, could, together with the European Phase III data, serve as supportive evidence for filing for regulatory approval with the FDA.
Our platform has generated other product candidates, including Cx611, for which we have completed a Phase I/IIa trial in rheumatoid arthritis. We are currently developing two clinical trials for Cx611: a Phase Ib trial in severe sepsis in the first quarter of 2015 and a Phase IIb trial in early rheumatoid arthritis in the third quarter of 2015. Our third product candidate, Cx621, completed a Phase I clinical trial for intra-lymphatic administration of allogeneic eASCs; its mode of administration has the potential to enable applications in other autoimmune diseases. We also developed and commercialized ChondroCelect, the first cell-based medicinal product to receive marketing authorization from the EMA, which is indicated for cartilage repair in the knee.
Recent Developments
On December 20, 2013, we entered into a loan facility agreement of up to 10.0 million euros with Kreos Capital IV (UK), under which we borrowed 7.5 million euros during the first half of 2014. On September 30, 2014, we borrowed the final installment of 2.5 million euros available under the facility.
Key Income Statement Items
Revenues
Historically, we have generated revenues from sales of ChondroCelect, our commercialized product, for which we received marketing authorization from the EMA in 2009. During the first half of 2014, we transformed our operations to focus fully on realizing the value of our eASC platform and pipeline by discontinuing our operations in connection with ChondroCelect.
50
Effective June 1, 2014, we entered into an agreement with Swedish Orphan Biovitrium, or Sobi, for the exclusive marketing and distribution rights with respect to ChondroCelect within the European Union (excluding Finland, where we have a pre-existing distribution agreement with Finnish Red Cross Blood Service), Switzerland, Norway, Russia, Turkey and the Middle East and North Africa region (currently excluding certain countries where we have a pre-existing distribution agreement with Genpharm). We also completed the sale of TiGenix B.V., our Dutch subsidiary, which held our manufacturing facility for ChondroCelect, to PharmaCell, a leading European contract manufacturing organization active in the area of cell therapy, for an upfront payment of 3.5 million euros when the sale became effective on May 30, 2014. We will receive a final deferred payment of 0.8 million euros on May 30, 2017.
As a result of the discontinuation of our operations related to ChondroCelect, we have reclassified all ChondroCelect operations as discontinued in our consolidated financial statements for all periods presented, included elsewhere in this prospectus.
Royalties
We expect to receive the majority of our revenues from royalty payments generated from the sale of ChondroCelect by Sobi and any other partners with which we enter into distribution agreements or license agreements. The marketing and distribution agreement with Sobi has a term of ten years. We will receive royalties of 22% on the net sales of ChondroCelect during the first year of the agreement and royalties of 20% on the net sales on an ongoing basis from the second year of the agreement onwards, and such income will be presented in our consolidated financial statements in revenues under the line item "royalties." Any other payments we receive with respect to ChondroCelect,e.g., from Finnish Red Cross Blood Service, will be included under the line item "discontinued operations."
Grants and other operating income
We also receive a portion of our revenues in the form of government grants directly related to our research and development efforts, which are presented in our consolidated income statement under the line item "grants and other operating income."
We do not recognize government grants until we have reasonable assurance that we will be able to comply with their terms and that the grants will be received. Government grants are recognized as income on a systematic basis over the periods in which we recognize expenses for the related costs for which the grants are intended to compensate. Specifically, for grants whose primary condition is the purchase, construction or acquisition of non-current assets, we recognize the grants as deferred revenue in our consolidated balance sheet and transfer them to the income statement on a systematic and rational basis over the useful life of the related assets. Grants that are receivable as compensation for expenses or losses already incurred or for immediate financial support with no related costs are recognized in the period in which they are received. We also treat the benefit of government loans, which we receive at below-market rates of interest, as government grants (measured as the difference between the proceeds received and the fair value of the loan based on prevailing market conditions), only when we have sufficient assurance that we will be able to comply with the terms and conditions of the loan and will not be required to return the funds prematurely. We recognize these loans as grants in our income statement under the line item "grants and other operating income."
In addition, we occasionally receive revenues from certain non-recurring items, which we also present in our income statement under the line item "grants and other operating income."
Research and Development Expenses
Our research and development activities primarily consist of preclinical research; Phase I, Phase II and Phase III of various clinical studies; the production of eASCs used in our preclinical and clinical
51
studies; regulatory activities and intellectual property activities to protect our know-how. Research and development expenses include, among others, employee compensation, including salary, fringe benefits and share-based compensation; expenses related to our manufacturing facilities, including operating costs for such facilities; and regulatory expenses and activities related to the development of our product pipeline.
We recognize expenditure on research activities as an expense in the period in which it is incurred.
An internally-generated intangible asset arising from development is capitalized to the extent that all the following can be demonstrated:
- •
- The technical feasibility of completing the intangible asset so that the asset will be available for use or sale.
- •
- The intention to complete and the ability to use or sell the asset.
- •
- How the asset will generate future economic benefits.
- •
- The availability of resources to complete the asset.
- •
- The ability to measure reliably the expenditure during development.
The amount initially recognized for internally-generated intangible assets is the sum of the various expenses needed to generate the related intangible assets. Amortization starts from the date that the intangible asset first meets the recognition criteria. These intangible assets are amortized on a straight-line basis over their estimated useful life of between five to ten years from the moment they are available for use. Where no internally-generated intangible asset can be recognized, development expenditure is recognized in the income statement in the period in which it is incurred.
Subsequent to initial recognition, internally-generated intangible assets are reported at cost less accumulated amortization and accumulated impairment losses, on the same basis as intangible assets that are acquired.
General and Administrative Expenses
Our general and administrative expenses are costs to support our operations and consist of employee compensation, including salary, fringe benefits and share-based compensation for our core corporate supporting functions including, among others, finance, human resources, legal, information technology, business development and investor relations. Other significant expenses include external corporate costs consisting of outside legal counsel, independent auditors and other outside consultants, insurance, facilities and depreciation.
Discontinued operations
The results of operations disposed during the year are included in our consolidated statement of comprehensive income up to the date of disposal.
A discontinued operation is a component of our business that represents a separate major line of business or geographical area of operations or is a subsidiary acquired exclusively with a view to resale, that has been disposed of, has been abandoned or that meets the criteria to be classified as held for sale.
Discontinued operations are presented in our consolidated statement of comprehensive income as a single line item that is comprised of the post-tax profit or loss of the discontinued operation along with the post-tax gain or loss recognized on the re-measurement to fair value less costs to sell or on disposal of the assets or disposal groups constituting discontinued operations.
52
Results of Operations
Comparison of the Six-Month Periods Ended June 30, 2014 and 2013
The following table summarizes the unaudited results of our operations for the six-month periods ended June 30, 2014 and 2013:
| | Six-month periods ended June 30, | | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2014 | 2013 | % Change | |||||||
| | Thousands of euros (Unaudited) | | ||||||||
Continuing Operations | ||||||||||
Revenues | ||||||||||
Royalties | — | — | — | |||||||
Grants and other operating income | 821 | 736 | 12 | % | ||||||
| | | | | | | | | | | |
Total revenues | 821 | 736 | 12 | % | ||||||
Research and development expenses | (5,097 | ) | (5,314 | ) | (4 | )% | ||||
General and administrative expenses | (2,859 | ) | (2,735 | ) | 5 | % | ||||
Total operating charges | (7,956 | ) | (8,049 | ) | (1 | )% | ||||
| | | | | | | | | | | |
Operating Loss | (7,135 | ) | (7,313 | ) | (2 | )% | ||||
Financial income | 25 | 5 | * | |||||||
Financial expenses | (369 | ) | (28 | ) | * | |||||
Foreign exchange differences | 170 | (38 | ) | * | ||||||
| | | | | | | | | | | |
Loss before taxes | (7,309 | ) | (7,374 | ) | (1 | )% | ||||
Income taxes | — | 42 | ||||||||
| | | | | | | | | | | |
Loss for the period from continuing operations | (7,309 | ) | (7,332 | ) | 0 | % | ||||
Discontinued Operations | ||||||||||
Loss for the period from discontinued operations | (1,842 | ) | (1,505 | ) | 22 | % | ||||
| | | | | | | | | | | |
Loss for the period | (9,151 | ) | (8,837 | ) | 4 | % | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
- *
- Not meaningful.
Royalties. We have not yet received any royalties for the sale of ChondroCelect under our distribution agreement with Sobi, because the transaction only closed in June 2014. We expect to receive the first royalties in the second half of 2014.
Grants and Other Operating Income. Grants increased by 12%, from 0.7 million euros for the six-month period ended June 30, 2013 to 0.8 million euros for the six-month period ended June 30, 2014. In both periods, most of the income was the result of a grant from the Seventh Framework Program for research in connection with the Cx611 program.
Research and Development Expenses. Our research and development expenses decreased by 4%, from 5.3 million euros for the six-month period ended June 30, 2013 to 5.1 million euros for the six-month period ended June 30, 2014. The decrease was mainly related to costs incurred in 2013 in connection with our manufacturing facility in Leuven, which ran in parallel with our Dutch facility for a few months prior to its closing during the first half of 2013.
General and Administrative Expenses. General and administrative costs increased by 5%, from 2.7 million euros for the six-month period ended June 30, 2013 to 2.9 million euros for the six-month
53
period ended June 30, 2014. The increase was mainly due to certain non-recurring legal and advisory expenses of 0.3 million euros.
Financial Income. Financial income increased from 4.9 thousand euros for the six-month period ended June 30, 2013 to 24.8 thousand euros for the six-month period ended June 30, 2014. Financial income mainly includes interest income and varies based on the cash balances in our bank deposits.
Financial Expenses. Financial expenses increased from 28.4 thousand euros for the six-month period ended June 30, 2013 to 368.8 thousand euros for the six-month period ended June 30, 2014. This increase was mainly due to the interest paid on our new credit facility with Kreos Capital IV (UK).
Foreign Exchange Differences. Foreign exchange differences changed from a loss of 37.8 thousand euros for the six-month period ended June 30, 2013 to a gain of 170.0 thousand euros for the six-month period ended June 30, 2014. These amounts arise as a result of our translation of financial information from U.S. dollars and pounds sterling in connection with our subsidiaries TiGenix Inc. and TiGenix Ltd., into our functional currency, which is the euro, using the exchange rate at the balance sheet date, which may differ from the rate in effect at the last measurement date of the item in question and are included in the foreign currency translation reserve.
Income Taxes. For the six-month period ended June 30, 2013, our income taxes were a benefit of 42.0 thousand euros. We did not incur any income taxes for the six-month period ended June 30, 2014.
Loss for the Period from Discontinued Operations. During the first six months of 2014, we completed the discontinuation of our operations in connection with ChondroCelect, our commercialized product, through the combination of the sale of TiGenix B.V., our Dutch subsidiary, that held our production facility for ChondroCelect, to PharmaCell for a total consideration of 4.3 million euros and the entry into an agreement with Sobi for the exclusive marketing and distribution rights for ChondroCelect. Under the terms of the share purchase agreement with PharmaCell, we received an upfront payment of 3.5 million euros when the sale became effective on May 30, 2014 and will receive a final payment of 0.8 million euros on May 30, 2017, which we have recognized at the net present value of 0.5 million euros. At the end of 2013, we conducted an impairment test with respect to the disposal of our Dutch subsidiary and recognized a loss of 0.7 million euros. After the completion of the disposal of the Dutch subsidiary and as a result of entering into the distribution agreement with Sobi, we recognized an additional loss on disposal of 1.1 million euros at June 30, 2014.
On June 1, 2014, we entered into an agreement with Sobi for the exclusive marketing and distribution rights with respect to ChondroCelect. Sobi will continue to market and distribute the product within the European Union (excluding Finland), Switzerland, Norway, Russia, Turkey and the Middle East and North Africa region (currently excluding certain countries where we have a pre-existing distribution agreement with Genpharm). We will receive royalties on the net sales of ChondroCelect, and Sobi will reimburse nearly all of our costs in connection with the product. The agreements with our former subsidiary, now owned by PharmaCell, and Sobi both include commitments for minimum quantities of ChondroCelect that are required to be purchased by us and from us under the respective contracts. If Sobi's actual purchases were to be lower than the required minimum, we would nevertheless be entitled to receive payment from Sobi up to a maximum amount of 5.7 million euros and would be required to pass on such payment to PharmaCell.
The sale of our Dutch subsidiary also included cost relief of up to 1.5 million euros on future purchases of ChondroCelect under the conditions of the long-term manufacturing agreement with our former subsidiary, which is now owned by PharmaCell. We will pass on this cost relief on a like-for-like basis to Sobi, which will purchase ChondroCelect from us at cost.
54
Our loss for the period from discontinued operations increased by 22% from 1.5 million euros for the six-month period ended June 30, 2013 to 1.8 million euros for the six-month period ended June 30, 2014, mainly as a result of the following:
- •
- Revenues increased by 26%, from 2.3 million euros for the six-month period ended June 30, 2013 to 2.9 million euros for the six-month period ended June 30, 2014, driven mainly by sales of ChondroCelect.
- •
- Operating expenses of ChondroCelect decreased by 8% from 3.8 million euros for the six-month period ended June 30, 2013 to 3.5 million euros for the six-month period ended June 30, 2014, mainly due to the fact that as from June 1, 2014 nearly all ChondroCelect expenses are being borne by Sobi.
- •
- Other income and expenses amounted to 0.1 million euros for the six-month period ended June 30, 2014 and were related to translation differences in connection with TiGenix Ltd.
- •
- The loss on the disposal of TiGenix B.V., which amounted to 1.1 million euros for the six-month period ended June 30, 2014 resulting from the closing of both the PharmaCell and the Sobi transactions, is under discontinued operations.
Comparison of the Years Ended December 31, 2013 and 2012
The following table summarizes the audited results of our operations for the years ended December 31, 2013 and 2012:
| | Years ended December 31, | | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2013 | 2012 | % Change | |||||||
| | Thousands of euros | | ||||||||
Continuing Operations | ||||||||||
Revenues | ||||||||||
Royalties | — | — | — | |||||||
Grants and other operating income | 883 | 1,389 | (36 | )% | ||||||
| | | | | | | | | | | |
Total revenues | 883 | 1,389 | (36 | )% | ||||||
Research and development expenses | (9,705 | ) | (12,136 | ) | (20 | )% | ||||
General and administrative expenses | (5,829 | ) | (6,237 | ) | (7 | )% | ||||
Total operating charges | (15,534 | ) | (18,373 | ) | (15 | )% | ||||
| | | | | | | | | | | |
Operating Loss | (14,651 | ) | (16,984 | ) | (13 | )% | ||||
Financial income | 7 | 35 | (80 | )% | ||||||
Financial expenses | (45 | ) | (58 | ) | (23 | )% | ||||
Foreign exchange differences | (352 | ) | (142 | ) | 148 | % | ||||
| | | | | | | | | | | |
Loss before taxes | (15,041 | ) | (17,149 | ) | (14 | )% | ||||
Income taxes | 59 | (1 | ) | * | ||||||
| | | | | | | | | | | |
Loss for the period from continuing operations | (14,982 | ) | (17,150 | ) | (13 | )% | ||||
Discontinued Operations | ||||||||||
Loss for the period from discontinued operations | (3,408 | ) | (3,243 | ) | 5 | % | ||||
| | | | | | | | | | | |
Loss for the period | (18,390 | ) | (20,393 | ) | (10 | )% | ||||
| | | | | | | | | | | |
| | | | | | | | | | | |
- *
- Not meaningful.
55
Royalties. We did not receive any royalties for the sale of ChondroCelect under our distribution agreement with Sobi during 2013 or 2012, because we did not enter into the agreement until 2014.
Grants and Other Operating Income. Revenue from grants and other operating income decreased by 36%, from 1.4 million euros for the year ended December 31, 2012 to 0.9 million euros for the year ended December 31, 2013. The decrease was due to a general decrease in national government grants available to us.
Research and Development Expenses. Our research and development expenses decreased by 20%, from 12.1 million euros for the year ended December 31, 2012 to 9.7 million euros for the year ended December 31, 2013. The decrease was partly related to a decrease of 1.2 million euros in lab fees and other operating expenses due to the completion in 2012 of our Phase I/IIa clinical trial for Cx611 in refractory rheumatoid arthritis and the Phase I clinical trial for Cx621 for intra-lymphatic administration to treat autoimmune disorders and a decrease of a further 1.2 million euros in labor costs mainly due to the internal reorganization of the research and development department in 2012.
General and Administrative Expenses. General and administrative costs decreased by 7%, from 6.2 million euros for the year ended December 31, 2012 to 5.8 million euros for the year ended December 31, 2013. The decrease of 0.4 million euros was mainly driven by the vesting in 2012 of the 2008 equity based incentive plan and the 2010 share based compensation plan, and was partially offset by an increase in service expenses and depreciation and amortization expenses.
Financial Income. Financial income decreased from 35.0 thousand euros for the year ended December 31, 2012 to 7.2 thousand euros for the year ended December 31, 2013. Financial income consists of interest income and varies based on the cash balances in our bank deposits.
Financial Expenses. Financial expenses decreased from 58.2 thousand euros for the year ended December 31, 2012 to 44.8 thousand euros for the year ended December 31, 2013. These expenses represent the interest paid on our credit facilities with ING and BNP Paribas Fortis.
Foreign Exchange Differences. Foreign exchange differences changed, from a loss of 0.1 million euros for the year ended December 31, 2012 to a loss of 0.4 million euros for the year ended December 31, 2013. These losses are related to loans incurred by our subsidiaries, particularly TiGenix Inc., in currencies other than the euro, and the increased loss is due to the strength of the euro against the U.S. dollar in 2013. These amounts arise as a result of our translation of the financial statements from the functional currency, which may be currencies other than the euro, into our presentational currency, which is the euro, using the exchange rate at the balance sheet date, which may differ from the rate in effect at the last measurement date of the item in question and are included in the foreign currency translation reserve.
Income Taxes. For the year ended December 31, 2012, our income taxes were an expense of 0.7 thousand euros. For the year ended December 31, 2013, our income taxes were a credit of 58.7 thousand euros.
The tax losses attributable to our subsidiary TiGenix SAU have an average maturity of fourteen years, and our other tax losses may be held indefinitely. As of December 31, 2012, we had a tax loss carried forward of 113.2 million euros compared to 125.6 million euros as of December 31, 2013, including a potential deferred tax asset of 41.8 million euros. Because it remains uncertain whether we will be able to realize taxable profits in the near future, we did not recognize any deferred tax assets in our balance sheet. In addition to these tax losses, we have unused tax credits amounting to 12.1 million euros as of December 31, 2012 compared to 13.9 million euros as of December 31, 2013 and deductible temporary differences of 8.3 million euros as of December 31, 2012 compared to 7.6 million euros as of December 31, 2013 for which we have not recognized any deferred tax assets in our balance sheet.
56
Loss for the Period from Discontinued Operations. Our loss for the period from discontinued operations increased by 5% from 3.2 million euros for the year ended December 31, 2012 to 3.4 million euros for the year ended December 31, 2013.
In 2012, we closed TiGenix Ltd., our biomaterials unit, and stopped all operating activities resulting in operating expenses of 1.9 million euros. During 2013, we decided to sell TiGenix B.V., our subsidiary that held our Dutch production facility. As a result of this decision, we recognized an impairment loss of 0.7 million euros, which reduced the carrying value of the subsidiary to the expected sales price of the asset less the cost of selling at that time. In addition, we incurred 0.8 million euros in operating expenses in TiGenix B.V. in 2012, compared to 1.3 million euros in 2013. In addition, for the six-month period ended June 30, 2014, all ChondroCelect operations, including revenues, production costs, sales and marketing expenses, have been presented as discontinued operations in the consolidated financial statements. For comparability purposes, we have used the same presentation for previous periods.
Critical Accounting Policies
Our financial statements are prepared in accordance with IFRS as issued by the IASB. The preparation of our financial statements in accordance with IFRS as issued by the IASB requires us to make judgments, estimates and assumptions that affect the reported amounts of assets, liabilities, revenue, cost of sales, operating expenses and related disclosures. We consider an accounting policy to be critical if it is important to our financial condition or results of operations, and if it requires significant judgment and estimates on the part of management in its application. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, and we evaluate our estimates on an ongoing basis. Our actual results may differ from these estimates under different assumptions or conditions. If actual results or events differ materially from the judgments and estimates that we have made in reporting our financial position and results of operations, our financial position and results of operations could be materially affected.
Going concern
We have experienced net losses and significant cash used in operating activities since our inception in 2000 and as of June 30, 2014, had an accumulated deficit of 83.2 million euros, a net loss of 9.2 million euros and net cash used in operating activities of 6.1 million euros and as of December 31, 2013 had an accumulated deficit of 74.0 million euros, a net loss of 18.4 million euros and net cash used in operating activities of 14.4 million euros. Our management expects us to continue to incur net losses and have significant cash outflows for at least the next twelve months. These conditions, among others, raise substantial doubt about our ability to continue as a going concern. The consolidated financial statements included elsewhere in this prospectus have been prepared assuming that we will continue as a going concern. This basis of accounting contemplates the recovery of our assets and the satisfaction of liabilities in the normal course of business. A successful transition to attaining profitable operations is dependent upon achieving a level of positive cash flows adequate to support our cost structure.
As at June 30, 2014, we had a liquidity position of 19.2 million euros consisting of cash, cash equivalents and receivables from reverse repurchase agreements. Taking into account this liquidity position and the available loan facility with Kreos Capital IV (UK), our board of directors is of the opinion that our liquidity position is sufficient to continue our current operations at least until the end of the third quarter of 2015.
57
Business combinations and goodwill
We account for business combinations using the acquisition method of accounting, which requires that the assets acquired and liabilities assumed be recorded at the date of acquisition at their respective fair values. Any excess of the fair value of consideration given over the fair values of the identifiable assets and liabilities acquired is recorded as goodwill. The determination of estimated fair values of acquired intangible assets, as well as the useful economic life ascribed to finite lived intangible assets, requires the use of significant judgment. The use of different estimates and assumptions to those we use could result in a materially different valuation of acquired intangible assets, which could have a material effect on our results of operations.
Several methods may be used to determine the estimated fair value of intangible assets acquired in a business combination, all of which require multiple assumptions. We use the relief from royalty method, which is a variant of the income valuation approach. It is based on the principle that ownership of the intangible asset relieves the owner of the need to pay a royalty to another party in exchange for rights to use the asset.
The value of the intangible asset is equal to the present value of the cost savings realized by the owner of the intangible asset as a result of not having to make royalty payments and milestone payments to another party. These cost savings are calculated based on the hypothetical royalty payments and milestone payments that a licensee would be required to pay for use of the asset, reduced by the tax savings realized by the licensee on the royalty payments.
Goodwill is capitalized. Any impairment in carrying amount charged to the consolidated income statement. Where the fair value of identifiable assets and liabilities exceeds the fair value of consideration paid, the excess is credited in full to the consolidated income statement on the acquisition date. Goodwill identified on acquisitions to date has been immaterial.
Acquisition costs incurred are expensed and included in general and administrative expenses.
Impairment of assets
We review the carrying value of intangible assets with indefinite lives for potential impairment on a periodic basis and also whenever events or changes in circumstances indicate the carrying value of an asset may not be recoverable. We review the carrying value of tangible assets and intangible assets with definitive lives for potential impairment whenever events or changes in circumstances indicate the carrying value of an asset may not be recoverable. We determine impairment by comparing the recoverable amount to its carrying value. If impairment is identified, a loss is recorded equal to the excess of the asset's carrying amount over its recoverable amount.
For impaired assets, we recognize a loss equal to the difference between the carrying value of the asset and its estimated recoverable amount. The recoverable amount is based on discounted future cash flows of the asset using a discounted rate commensurate with the risk. Estimates of future costs savings, based on what we believe to be reasonable and supportable assumptions and projections, require management's judgment. Actual results could vary from these estimates. When it is not possible to estimate the recoverable amount of an individual asset, we estimate the recoverable amount of the cash-generating unit to which the asset belongs.
Based on the analysis involved, there was no impairment through December 31, 2013 (except for the impairment recognized upon the presentation of the disposal group held for sale).
Recognition and measurement of internally-generated intangible assets
An internally-generated intangible asset is recognized if it can be documented with sufficient certainty that the future benefits from the development project will exceed the aggregate cost of
58
production, development and the sale and administration of the product. A development project involves a single product candidate undergoing a high number of tests to illustrate its safety profile and the effect on human beings prior to obtaining the necessary approval of the product from the appropriate authorities. The future economic benefits associated with the individual development projects are dependent on obtaining such approval. Considering the significant risk and duration of the development period related to the development of our products, our management has concluded that the future economic benefits associated with a particular project cannot be estimated with sufficient certainty until the project has been finalized and the necessary regulatory final approval of the product has been obtained.
Accordingly, we have capitalized such intangible assets for the development costs related to ChondroCelect with a useful life of between five to ten years. The carrying amount of these assets amounted to 1.5 million euros as at June 30, 2014, as compared to 1.7 million euros as at December 31, 2013 and 1.9 million euros as at December 31, 2012.
Research and Development Costs
Research and development costs are charged to expense as incurred and are typically made up of salaries and benefits, clinical and preclinical activities, drug development and manufacturing costs, and third-party service fees, including for clinical research organizations and investigative sites. Costs for certain development activities, such as clinical trials, are periodically recognized based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations, or information provided by vendors on their actual costs incurred. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the financial statements as prepaid or accrued expenses.
Deferred taxes
Deferred tax assets are recognized for unused tax losses to the extent that it is probable that taxable profit will be available against which the losses can be utilized. Significant management judgment is required to determine the amount of deferred tax assets that can be recognized, based upon the likely timing and the level of future taxable profits together with future tax planning strategies.
At December 31, 2013, we had 147.1 million euros of tax losses carry forward and other tax credits such as investment tax credits and notional interest deduction. This compared to 133.6 million euros in 2012.
These losses relate both to us and our subsidiaries, all of which have a history of losses. Our tax losses do not expire, except for tax losses and tax credits of 37.7 million euros related to TiGenix SAU with an average maturity of fourteen years. These losses may not be used to offset taxable income elsewhere in our group.
With respect to our net operating losses, no deferred tax assets have been recognized, given that there is uncertainly as to the extent to which these tax losses will be used in future years.
Derivative financial instruments
Derivatives are initially recognized at fair value at the date the derivative contracts are entered into and are subsequently re-measured to their fair value at the end of each reporting period. The resulting gain or loss is recognized in profit or loss immediately, unless the derivative is designated and effective as a hedging instrument, in which event the timing of the recognition in profit or loss depends on the nature of the hedge relationship.
59
Pursuant to the terms and conditions of the loan facility agreement that we entered into with Kreos Capital IV (UK), on April 22, 2014, an extraordinary meeting of our shareholders issued and granted 1,994,302 new cash-settled warrants, including a put option to Kreos Capital IV (Expert Fund). These warrants have been designated at fair value through profit and loss. We recognize the warrants, including the put option, as one instrument, because we believe the put option is unconditionally linked to the warrants. Because the issued warrants can be settled in cash, the instrument is considered as a financial derivative liability measured at fair value with changes in fair value recognized immediately in profit or loss.
The measurement of the warrants (and the put option) at fair value is based on the Black-Scholes option pricing model taking into account following variables:
- •
- The share price.
- •
- The strike price.
- •
- The volatility of the share price, which has been determined based on the historical prices of our shares.
- •
- The dividend yield, which has been estimated as zero, because we have never paid a dividend.
- •
- The duration, which has been estimated as the difference between the valuation date of the warrant plans and final exercise date.
- •
- The risk free interest rate, which has been calculated based on the discount curve composed based on liquid euro deposit rates for periods shorter than one year, futures typically for maturities between one and six years and interbank euro swap rates for periods longer than six years.
We will continue to use judgment in evaluating the risk-free interest rate, dividend yield, duration and volatility related to our cash-settled warrant plan on a prospective basis and incorporating these factors into the Black-Scholes option pricing model. If in the future, we determine that other methods are more reasonable and provide better results, or other methods for calculating these assumptions are prescribed by authoritative guidance, we may change or refine our approach, and our share-based payment expense in future periods could change significantly.
Share-based payment arrangements
We used the Black-Scholes model to estimate the fair value of the share-based payment transactions. Using this model requires our management to make assumptions with regard to volatility and expected life of the equity instruments. The assumptions used for estimating fair value for share-based payment transactions are estimated as follows:
- •
- Volatility is estimated based on the average annualized volatility of our share price.
- •
- The estimated life of the warrant is until the first exercise period.
- •
- The dividend return is estimated by reference to our historical dividend payment. Currently, this is estimated to be zero, because no dividend has been paid since our inception.
Non-current assets (disposal groups) held for sale and discontinued operations
Assets held for sale are comprised of non-current assets or disposal groups (together with any liabilities), the carrying amounts of which will be realized principally through a sale transaction expected to conclude within twelve months rather than through continued use.
60
At December 31, 2013, we presented 5.6 million euros (2012: nil) of net assets held for sale in the consolidated statement of financial position, all assets and liabilities within this disposal group relate to the disposal of the Dutch manufacturing facility, which occurred during the first half of 2014.
At the time of their classification as "held for sale" in December 2013, such assets were collectively measured at the lower of their carrying amount and fair value less costs to sell. An impairment charge of 0.7 million euros was recorded reflecting the adjustment of the disposal group's carrying amount to its fair value less cost to sell. Depreciation and amortization ceased at that point.
We exercise significant judgment in assessing at which point all the "held for sale" presentation conditions are met for the disposal group and estimating both the fair value of the disposal group and the incremental costs to transact a sale of the disposal group. If actual events differ from management's estimates, or to the extent that estimates of selling price or costs to sell are adjusted in the future, our financial condition and results of operations could be affected in the period of any such change of estimate.
Liquidity and Capital Resources
We have historically funded our operations principally from equity financing, borrowings, grants and subsidies and cash generated from operations. As we continue to grow our business, we expect to fund our operations through multiple sources, possibly including the expected proceeds from this offering, short and long-term investments, loans or collaborative agreements with corporate partners, future earnings and cash flow from operations and borrowings. This expectation could change depending on how much we spend on our development programs, potential licenses or acquisitions of complementary technologies, products or companies. We may in the future supplement our funding with further debt or equity offerings or commercial borrowings.
On December 20, 2013, we entered into a loan facility agreement of up to 10.0 million euros with Kreos Capital IV (UK), under which we borrowed 7.5 million euros during the first half of 2014. On September 30, 2014, we borrowed the final installment of 2.5 million euros under the facility.
The terms of the loan facility agreement are as follows:
- •
- Term: four years.
- •
- Repayment schedule: repayment of principal amount starts at first anniversary.
- •
- Interest: 12.5% fixed annual interest rate.
- •
- Structure: security over certain of our assets (including a pledge over certain intellectual property and bank accounts); no financial covenants.
In addition, pursuant to the terms of the loan facility agreement, on April 22, 2014, an extraordinary meeting of shareholders issued and granted 1,994,302 new warrants to Kreos Capital IV (Expert Fund).
Our borrowings also include government loans from public and semi-public entities, such as Madrid Network. These loans have an extended repayment period of ten to fifteen years, and are interest-free or have very low interest rates, if all the terms and conditions are met. In addition, there is a grace period of three to five years before any repayment is required under such loans.
On September 30, 2011, our subsidiary TiGenix SAU entered into a loan facility for a total value of 5.0 million euros with Madrid Network, the terms of which were as follows:
- •
- Term: until December 31, 2026.
- •
- Repayment schedule: repayment of interest and principal amount starts at June 30, 2015.
61
- •
- Effective interest: 1.46% fixed annual interest rate.
- •
- Structure: joint and several guarantee from us.
The loan was granted to finance in part our ongoing Phase III trial for Cx601 and also to cover some research and development expenses related to Cx601 from 2012 until the end of 2014. The specific activities covered for this period were agreed with Madrid Network at the time of the grant of the loan, and any change to this plan will need approval from Madrid Network on a semi-annual basis.
On July 30, 2013, our subsidiary TiGenix SAU entered into a second loan facility for a total value of 1.0 million euros with Madrid Network, the terms of which were as follows:
- •
- Term: until December 31, 2025.
- •
- Repayment schedule: repayment of interest and principal amount starts at June 30, 2016.
- •
- Effective interest: 1.46% fixed annual interest rate.
- •
- Structure: first demand bank guarantee for the full amount of the loan provided by Bankinter, where we maintain our primary bank accounts in Spain.
This loan was granted to finance our preparatory work to determine which new indications we should pursue with respect to Cx611, our second product candidate, the development of the clinical protocol for such new indications and the initial expenses of the clinical trials for such indications from July 2013 until the end of 2014. The specific activities covered for this period were agreed with Madrid Network at the time of the grant of the loan and any change to this plan will need approval from Madrid Network on a semi-annual basis.
We have experienced net losses and significant cash used in operating activities in each period since inception. As of June 30, 2014, we had an accumulated deficit of 83.2 million euros, a net loss of 9.2 million euros and net cash used in operating activities of 6.1 million euros. We have only one commercial product that generates limited revenues through royalties and several product candidates in clinical development. We, therefore, expect to incur net losses and have significant cash outflows for at least the next year.
We believe that we will have sufficient liquidity to satisfy the operating requirements of our business until the end of the third quarter of 2015. We have based this estimate on assumptions that may prove to be wrong, and we could use our available capital resources sooner than we currently expect. Because of the numerous risks and uncertainties associated with the development and commercialization of our product candidates, and the extent to which we may enter into collaborations with third parties for development and commercialization of our product candidates, we are unable to estimate the amounts of increased capital outlays and operating expenses associated with completing the development of our current product candidates. Our future capital requirements will depend on many factors, including the following:
- •
- The progress and results of the clinical trials of our lead product candidates, Cx601 and Cx611.
- •
- The scope, progress, results and costs of pre-clinical development, laboratory testing and clinical trials for our other product candidates.
- •
- The extent to which we acquire or in-license other products and technologies.
- •
- The cost, timing and outcome of regulatory review of our product candidates in the United States and Europe.
- •
- The costs of future commercialization activities, including product sales, marketing, manufacturing and distribution, for any of our product candidates for which we receive marketing authorization.
62
- •
- Any additional revenue received from commercial sales of our products, should any of our product candidates receive marketing approval.
- •
- Any premature reimbursement of loans from public or semi public entities if certain terms and conditions were not met.
- •
- The cost of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims.
- •
- Our ability to establish any future collaboration arrangements on favorable terms, if at all.
We will also incur costs as a U.S.-listed public company that we have not previously incurred, including, but not limited to, costs and expenses for increased personnel costs, audit and legal fees, expenses related to compliance with the Sarbanes-Oxley Act of 2002 and the rules implemented by the SEC and NASDAQ and various other costs.
These factors raise uncertainty about our ability to continue as a going concern. The consolidated financial statements included elsewhere in this prospectus have been prepared assuming that we will continue as a going concern. This basis of accounting contemplates the recovery of our assets and the satisfaction of liabilities in the normal course of business. A successful transition to attaining profitable operations is dependent upon achieving a level of positive cash flows adequate to support our cost structure. To improve our financial performance, we discontinued our ChondroCelect operations through several initiatives, including the following:
- •
- The sale of our subsidiary TiGenix B.V. to PharmaCell, for which we received an upfront payment of 3.5 million euros when the sale became effective on May 30, 2014 and will receive a final payment of 0.8 million euros on May 30, 2017.
- •
- Our entry into a distribution agreement with Sobi for the marketing and distribution rights with respect to ChondroCelect, effective June 1, 2014, which will bring us future royalty earnings and allow us to focus on our eASC-based platform of product candidates in inflammatory and autoimmune diseases.
Our liquidity plans are subject to a number of risks and uncertainties, including those described in the section of this prospectus entitled "Risk Factors," some of which are outside of our control.
Cash Flows
The following table summarizes the results of our cash flows for the six-month periods ended June 30, 2014 and 2013 and the years ended December 31, 2013 and 2012:
| | Six-month periods ended June 30, | Years ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2014 | 2013 | 2013 | 2012 | |||||||||
| | Thousands of euros | ||||||||||||
| | (Unaudited) | | | ||||||||||
Net cash generated from (used in): | |||||||||||||
Operating activities | (6,093 | ) | (7,817 | ) | (14,427 | ) | (17,627 | ) | |||||
Investing activities | (2,552 | ) | (225 | ) | (1,320 | ) | (721 | ) | |||||
Financing activities | 6,266 | 707 | 20,237 | 9,647 | |||||||||
| | | | | | | | | | | | | | |
Net increase (decrease) | (2,379 | ) | (7,335 | ) | 4,490 | (8,701 | ) | ||||||
Cash and cash equivalents | 13,186 | 3,738 | 15,565 | 11,072 | |||||||||
63
Comparison of the Six-Month Periods Ended June 30, 2014 and 2013
Net cash outflow from continuing operating activities was 5.0 million euros for the six-month period ended June 30, 2014 compared to 6.4 million euros for the six-month period ended June 30, 2013, a decrease of 21%, mainly due to working capital movements and partly as a result of the decrease in our operating expenses.
Net cash outflow from discontinued operating activities was 1.1 million euros for the six-month period ended June 30, 2014 compared to 1.5 million euros for the six-month period ended June 30, 2013, a decrease of 27%, in line with the increase in ChondroCelect sales.
Net cash outflow from continuing investing activities was 6.1 million euros for the six-month period ended June 30, 2014 compared to 0.2 million euros for the six-month period ended June 30, 2013. This was due to the investment of 6.0 million euros in receivables from reverse repurchase agreements.
Net cash inflow from discontinued investing activities was 3.5 million euros for the six-month period ended June 30, 2014 compared to a cash outflow of 33.3 thousand euros for the six-month period ended June 30, 2013. This is due to the inflow of funds received from the sale of the Dutch manufacturing facility of 3.5 million euro in 2014.
Net cash inflow from continuing financing activities was 6.3 million euros for the six-month period ended June 30, 2014 compared to 0.7 million euros for the six-month period ended June 30, 2013. In the first half of 2014, we borrowed a total of 7.5 million euros in two installments under our loan facility agreement with Kreos Capital IV (UK), which represents the majority of the increase in net cash inflow compared to the first half of 2013. We repaid 0.6 million euros of financial loans and other financial liabilities in June 2014, as well as 0.3 million of interest on such liabilities.
Comparison of Years Ended December 31, 2013 and 2012
Net cash outflow from continuing operating activities was 14.4 million euros for the year ended December 31, 2013 compared to 14.8 million euros for the year ended December 31, 2012, a decrease of 3%. This decrease was mainly related to the completion in 2012 of our Phase I/IIa clinical trial for Cx611 in refractory rheumatoid arthritis and our Phase I clinical trial for Cx621 for intra-lymphatic administration to treat autoimmune disorders as well as a reduction in operating costs due to the synergies resulting from our business combination with Cellerix in 2011.
Net cash inflow from discontinued operating activities was 36.1 thousand euros for the year ended December 31, 2013 compared to net cash outflow of 2.8 million euros for the year ended December 31, 2012. This change was mainly related to the discontinuation of our biomaterials unit, TiGenix Ltd., in 2012.
Net cash outflow from continuing investing activities was 1.3 million euros for the year ended December 31, 2013 compared to 0.2 million euros for the year ended December 31, 2012. In 2013, the primary investment related to a guarantee in connection with a so-called "soft" loan from Madrid Network, an umbrella organization for companies, research centers, universities, technology centers and science and technology parks in the Madrid region of Spain.
Net cash outflow from discontinued investing activities was 0.1 million euros for the year ended December 31, 2013 compared to 0.6 million euros for the year ended December 31, 2012. In 2012, our investing activities primarily included expenses in connection with the construction of our Dutch manufacturing facility, which has now been discontinued.
Net cash inflow from continuing financing activities was 20.2 million euros for the year ended December 31, 2013 compared to 9.6 million euros for the year ended December 31, 2012, an increase of 110%. In 2013, we increased our capital twice, through a private placement in July 2013 in which we raised 6.5 million euros, and a strategic investment by Grifols, a global healthcare company, of
64
12.0 million euros on November 22, 2013. In 2012, we increased our capital through a private placement in December in which we raised 6.7 million euros. We also received financing in the form of proceeds of loans in both 2013 and 2012.
Contractual Obligations
The following table details the remaining contractual maturity for our financial liabilities with agreed repayment periods, including both interest and principal cash flows as at December 31, 2013:
| | Within one year | 1 - 3 years | 3 - 5 years | After 5 years | Total | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Thousands of euros | |||||||||||||||
Financial loans and other payables(1) | 343 | 1,661 | 2,026 | 6,031 | 10,061 | |||||||||||
Other financial liabilities | 874 | — | — | — | 874 | |||||||||||
Operating lease commitments | 843 | 1,107 | 491 | 1,594 | 4,035 | |||||||||||
Guarantee commitments(2) | 131 | 281 | 300 | 300 | 1,012 | |||||||||||
| | | | | | | | | | | | | | | | | |
Total | 2,191 | 3,048 | 2,817 | 7,926 | 15,982 | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
- (1)
- Includes the interest payable on financial loans and other payables.
- (2)
- TiGenix Inc., our wholly owned subsidiary, guarantees the operating lease payments of Cognate BioServices for the building leased in the United States. Cognate BioServices was the party with whom we entered into a joint venture in 2007.
As of June 30, 2014, we had borrowed an additional 7.5 million euros under the loan facility agreement dated December 20, 2013 with Kreos Capital IV (UK). On September 30, 2014, we borrowed the final installment of 2.5 million euros under the facility.
During the first half of 2014, under the long-term manufacturing agreement with our former subsidiary now owned by PharmaCell, we entered into a commitment to purchase minimum binding quantities of ChondroCelect up to a maximum amount of 5.7 million euros for a period starting on June 1, 2014. This commitment has been fully transferred to Sobi under our distribution agreement and will result in zero cash inflow or outflow for the Company.
Under the terms of our roll-over credit agreement with ING Belgium under which 0.1 million euros was outstanding as at December 31, 2013, we were not permitted to grant any pledge on any of our assets without the prior consent of ING Belgium. Since ING Belgium did not consent to the proposed pledge in connection with the loan facility agreement with Kreos Capital IV (UK), we repaid the outstanding amount to ING Belgium on April 4, 2014, and this obligation is represented as a short-term liability in our consolidated financial statements for the year ended December 31, 2013 included elsewhere in this prospectus.
Off-Balance Sheet Arrangements
We do not have any special purpose or limited purpose entity that provides off-balance sheet financing, liquidity or market or credit risk support, and we have not engaged in hedging or other relationships that expose us to liability that is not reflected in our consolidated financial statements.
Quantitative and Qualitative Disclosure about Market Risk
We co-ordinate our access to financial markets and monitor and manage the financial risks relating to our operations through internal risk reports analyzing our exposures by the degree and magnitude of the relevant risk. These risks include: currency risk, interest rate risk, liquidity risk and credit risk.
65
Our risk management policies are more fully described in Note 4 in the notes to our consolidated financial statements included elsewhere in this prospectus.
Currency risk
We are subject to limited currency risk. Our reporting currency is the euro, in addition to which we are exposed to the U.S. dollar. We currently have no commercial revenues denominated in U.S. dollars. We try to match foreign currency cash inflows with foreign currency cash outflows. We have not engaged in hedging of the foreign currency risk via derivative instruments.
Interest rate risk
We are exposed to interest rate risk because we and our subsidiaries borrow funds at both fixed and floating interest rates. We manage this risk by maintaining an appropriate mix between fixed and floating rate borrowings.
As at December 31, 2013 we had two outstanding rollover credit facilities denominated at a floating rate. We entered into these facilities in 2007 to acquire manufacturing equipment in the United States for an original amount of 0.8 million euros. The borrowings have a remaining maturity of four years at a floating interest rate pegged to the three-month Euribor rate. The outstanding amount under these facilities at December 31, 2013 was 0.3 million euros compared to 0.4 million euros at December 31, 2012.
Liquidity risk
We manage our liquidity risk by maintaining adequate reserves, banking facilities and reserve borrowing facilities, by continuously monitoring forecast and actual cash flows, and by matching the maturity profiles of financial assets and liabilities.
Credit risk management
Credit risk refers to the risk that one of our counterparty will default on its contractual obligations resulting in financial loss to us. We have adopted a policy of only dealing with creditworthy counterparties and obtaining sufficient collateral, where appropriate, as a means of mitigating the risk of financial loss from defaults. We continuously monitor our obligations and ensure that we spread the aggregate value of transactions concluded among approved counterparties.
Most of our counterparties are established public health care facilities, and we believe we are exposed to a limited risk of counterparty default.
Jumpstart Our Business Startups Act of 2012
As a company with less than $1.0 billion in revenue during our last fiscal year, we qualify as an "emerging growth company" as defined in Section 2(a) of the Securities Act of 1933, as amended, as modified by the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. As such, we are eligible to take advantage of specified reduced reporting requirements and are relieved of certain other significant requirements that are otherwise generally applicable to public companies. As an emerging growth company:
- •
- We are permitted to present only two years of audited consolidated financial statements and only two years of related Management's Discussion and Analysis of Financial Condition and Results of Operations in the registration statement of which this prospectus forms a part.
66
- •
- We are exempt from the requirement to obtain an attestation and report from our auditors on the assessment of our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act of 2002.
- •
- We are permitted to provide less extensive disclosure about our executive compensation arrangements.
We expect to remain an "emerging growth company" for up to five years, or until any one of the following occurs:
- •
- The last day of the first fiscal year in which our annual gross revenue exceeds $1 billion.
- •
- The last business day of the fiscal year that we become a "large accelerated filer" as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, which would occur if the market value of our common equity held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter and we have been publicly reporting for at least twelve months.
- •
- The date on which we have issued more than $1.0 billion in non-convertible debt during the preceding three-year period.
Once we cease to be an emerging growth company, we will not be entitled to the exemptions provided for under the JOBS Act.
67
Our Company
We are an advanced biopharmaceutical company focused on developing and commercializing novel therapeutics from our proprietary technology platform of allogeneic, or donor-derived, expanded adipose-derived stem cells, known as eASCs, in inflammatory and autoimmune diseases. Based on our platform, we have developed a pipeline of product candidates with our most advanced being Cx601, a first-in-class injectable allogeneic stem cell therapy that has been granted orphan designation by the European Medicines Agency, or EMA, for the treatment of anal fistulas. We are conducting a single pivotal Phase III trial for Cx601 for the treatment of complex perianal fistulas in patients suffering from Crohn's disease, a rare, painful and debilitating condition affecting approximately 100,000 patients in the United States and Europe. Data from the single pivotal trial should be available in the third quarter of 2015, based on which we plan to submit a marketing authorization application to the EMA in the first half of 2016. We also intend to initiate a Phase III trial for Cx601 for the treatment of complex perianal fistulas in the United States by the second half of 2016. Based on discussions with the U.S. Food and Drug Administration, or FDA, we believe that the U.S. Phase III trial, if successful, could, together with the European Phase III data, serve as supportive evidence for filing a biologics license application, or BLA, for regulatory approval with the FDA. Our platform has generated other product candidates, including Cx611, for which we have completed a Phase I/IIa trial in refractory rheumatoid arthritis. We are currently developing two clinical trials for Cx611: a Phase Ib trial in severe sepsis in the first quarter of 2015 and a Phase IIb trial in early rheumatoid arthritis in the third quarter of 2015. Our eASC-based product candidates are manufactured at our facility in Madrid, Spain, which has been approved by the Spanish Medicines and Medical Devices Agency as being compliant with current Good Manufacturing Practices, or cGMP, requirements. Through our expansion process, we can generate up to 2,400 doses of Cx601 from cells extracted from a single healthy donor. We have retained the worldwide rights for all of our product candidates.
Our approach to cell therapy is to focus on the use of living cells, rather than conventional drugs, for the treatment of inflammatory and autoimmune diseases. Cells target a different pathway than conventional drugs and may be effective in patients who fail to respond to such drugs, including the biologics currently used to treat autoimmune conditions. Our pipeline is based on a validated platform of allogeneic eASCs extracted from human adipose, or fat, tissue from healthy adult volunteers. This approach leverages the broad immunomodulatory properties of eASCs. We have conducted a full spectrum of studies analyzing various routes of administration and indications to further the preclinical and clinical development of our platform. We have also had extensive discussions with the EMA regarding our eASC platform through their established procedures for scientific advice regarding chemistry, manufacturing and control packages and preclinical packages as well as a scientific advice meeting with respect to Cx601 that has allowed us to pursue an expedited route to clinical development. In addition, we have had a meeting with the Center for Biologics Evaluation and Research within the FDA on the clinical development of Cx601 in the United States. We already have the capacity to scale up the production of our eASC-based products on a late-stage clinical as well as commercial scale.
As of September 30, 2014, our eASC pipeline portfolio was the most advanced cell therapy platform in Europe, with a product candidate poised to receive pivotal Phase III data in the third quarter of 2015 and two further product candidates in Phases II and I.
- •
- Cx601. Cx601, our lead product candidate, is a first-in-class injectable allogeneic stem cell therapy that is currently in a pivotal Phase III trial for the treatment of complex perianal fistulas in patients suffering from Crohn's disease. We have observed compelling clinical results that suggest that Cx601 may have clinical utility in treating perianal fistulas in one injectable dose with a more favorable adverse events profile than currently available therapies.
68
- •
- Cx611. Cx611, our second product candidate, is a first-in-class injectable allogeneic stem cell therapy intended for the treatment of early rheumatoid arthritis and severe sepsis. We believe that Cx611, if approved for early rheumatoid arthritis, would have advantages over current treatments such as biologics due to its safety profile and higher induction of remission. We have completed a successful Phase I/IIa trial of Cx611 in refractory rheumatoid arthritis patients that illustrated the safety of the therapy and provided indications of therapeutic activity. If it is approved for severe sepsis, we believe that Cx611 would be an add-on therapy that has the potential to reduce mortality. We are planning to advance Cx611 in severe sepsis in a Phase Ib trial and in early rheumatoid arthritis in a Phase IIb trial in the first and the third quarter of 2015, respectively.
- •
- Cx621. Cx621, our third product candidate, has completed a Phase I trial that generated safety and feasibility information on intra-lymphatic administration of allogeneic eASCs. This different route of administration has the potential to enable applications in other autoimmune diseases.
Moreover, Cx601 enjoys significant benefits due to its designation as an orphan drug by the EMA, including the following: (i) research grants and subsidies; (ii) assistance from the EMA in developing our clinical trials, including detailed feedback on proposed study designs; (iii) a streamlined process for obtaining the relevant regulatory approvals in Europe; and (iv) up to ten years of exclusivity in the European market from the date of the product's launch. We have also had a meeting with the FDA to discuss the adequacy of our clinical and non-clinical data to support an investigational new drug, or IND, application for a U.S.-based Phase III trial. We received positive feedback regarding our current pivotal European Phase III trial design for supporting a BLA. Current therapies have limited efficacy, and there is currently no commercially available cell-based therapy for this indication. We believe Cx601, if approved, would fulfill a significant unmet need in the market. If our pivotal Phase III trial is successful, we expect to file for marketing authorization in Europe by the first half of 2016 and initiate a Phase III trial in the United States by the second half of 2016.
ChondroCelect, our commercial product, was the first cell-based product to receive centralized marketing authorization from the EMA in October 2009 as an advanced therapy medicinal product, a new medical product category regulated by the EMA that includes products based on gene therapy, cell therapy or tissue engineering. ChondroCelect, which is indicated for cartilage repair in the knee, is also the first advanced therapy medicinal product to have been approved for reimbursement in Belgium, the Netherlands and Spain. Effective June 1, 2014, we have entered into a distribution agreement with Swedish Orphan Biovitrium, or Sobi, for the exclusive marketing and distribution rights with respect to ChondroCelect within the European Union (excluding Finland, where we have a pre-existing distribution agreement) as well as several other countries.
As of September 30, 2014, we owned or co-owned over twenty patent families and had over ninety granted patents in over twenty jurisdictions, including the United States, with expiration dates from 2020 onwards.
Our Strategy
Our objective is to provide innovative and safe treatment options for a broad range of inflammatory and autoimmune diseases. Key elements of our strategy for achieving this objective are as follows:
- •
- Successfully advance the clinical development of Cx601 and secure regulatory approval in Europe and the United States. Leveraging our experience with ChondroCelect, the first cell-based product to be granted centralized marketing authorization in Europe as an advanced therapy medicinal product, we intend to secure regulatory approval for our eASC-based product candidates, starting with Cx601.
69
- •
- Europe. Based on our discussions with the EMA, we are pursuing a single pivotal Phase III trial for Cx601 with results expected by the third quarter of 2015. If the trial is successful, we expect to file for marketing authorization in Europe by the first half of 2016.
- •
- United States. We received positive feedback in our meeting with the Center for Biologics Evaluation and Research within the FDA, which has agreed to review the results of the European Phase III trial, if successful, as supportive evidence for filing for regulatory approval in the United States. We plan to start the process for a technology transfer to a U.S.-based contract manufacturing organization and the preparation for a special protocol assessment, or SPA, to file an IND application for a Phase III trial in the United States, which, if successful and together with positive Phase III data from the European trial, would enable us to file a BLA with the FDA.
- •
- Achieve global commercialization of Cx601. We intend to commercialize Cx601 independently in certain European markets, where we will leverage our experience in bringing ChondroCelect to market and successfully obtaining national and regional reimbursement in several European countries. Complex perianal fistula in patients with Crohn's disease, for which Cx601 is being developed, is a debilitating condition with a well-defined patient population managed by a limited number of medical specialists, which we believe will allow us to rely on a relatively small and effective commercialization structure to manage the relevant reference centers. We will consider partnerships in the United States, other European markets and elsewhere and will follow a commercial strategy to increase the probability of Cx601's ultimate success. Assuming Cx601 yields positive Phase III data and based on a standard regulatory pathway for advanced therapy medicinal products, we anticipate generating our first revenues from Cx601 within the next three to four years.
- •
- Advance our product candidates, Cx611 and Cx621, in the United States and the rest of the world. As with Cx601, we are focusing on well-defined patient populations with respect to Cx611 and have selected subgroups of patients suffering from early rheumatoid arthritis and severe sepsis within otherwise relatively large indications in the autoimmune and inflammatory disease areas. Based on our successful Phase I/IIa trial in refractory rheumatoid arthritis, we are advancing Cx611 in these two indications with a Phase Ib trial in severe sepsis and a Phase IIb trial in early rheumatoid arthritis in first quarter of 2015 and the third quarter of 2015, respectively. We believe that if Cx611 were approved for these indications, it would supplement existing therapies and would have the potential to reduce mortality in patients with severe sepsis and arrest disease progression in early rheumatoid arthritis patients. With Cx621, our third product candidate, we have established the safety and feasibility of intra-lymphatic administration of eASCs, and may evaluate a potential path forward.
- •
- Discover, develop and commercialize first-in-class novel therapeutics for areas of high unmet medical need by leveraging our proprietary technology platform of eASCs. We intend to advance our position through the continuing discovery and development of new product candidates for multiple indications. We believe that our technology platform as well as our in-house expertise allow us to achieve candidate selection and proof-of-concept in an efficient manner. Our eASC-based product candidates use a novel mechanism of action offering benefits that are expected to be superior to existing treatment options in terms of efficacy and safety in the selected indications, and we believe that they have the potential to be effective in a broad range of indications in autoimmune and inflammatory diseases. We will continue to invest in our technology platform and identify, develop and manufacture additional product candidates. As our subsequent product candidates advance in their development for more prevalent indications, we aim to achieve substantial growth.
- •
- Strengthen our competitive position by leveraging our experienced management team and reinforcing key opinion leader support. Our management team is comprised of highly experienced
70
professionals with track records in the biomedical and pharmaceutical fields. The team has demonstrated its ability to create value by bringing the first cell therapy-based medicinal product in Europe to market and achieving key value enhancing milestones in all other areas of pharmaceutical development, including clinical development, regulatory, manufacturing and commercialization. In doing so, our team has acquired a unique expertise in the field of cell therapy. As a cell therapy pioneer, we have developed and will continue to capitalize on our strong relationships with key opinion leaders who have collaborated and consulted with us in developing our product candidates. As a result, we have established strong scientific advisory boards that share our belief in the therapeutic potential of cell therapies. With respect to Cx601, we have an advisory board in Europe and have appointed an advisory board in the United States, which held its initial meeting on September 4, 2014. For Cx611, we have an advisory board for rheumatoid arthritis and an advisory board for severe sepsis.
Technology Platform
Our product candidates are based on a proprietary technology platform of eASCs, exploiting their recognized mechanism of action in immune-mediated inflammatory processes. Our basic preclinical package for eASCs is based on a full spectrum of studies focusing on three indications—rheumatoid arthritis, inflammatory bowel disease and sepsis—and five possible routes of administration—local (perianal), rectovaginal, intraperitoneal, intravenous and intralymphatic. In these studies, we have found no indications of toxicity, tumorigenicity or ectopic tissue growth. We have extensively characterized our eASC platform to establish the potency, identity and purity of our eASC-based product candidates and had discussions with the EMA via their established procedures for scientific advice regarding our chemistry, manufacturing and control package. Based on these discussions, we have validated our manufacturing process and our platform-associated analytical procedures as per the EMA's guidelines, including the quality guidelines of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. We believe our technology platform offers significant market opportunities in inflammatory and autoimmune diseases based on the following distinguishing factors:
- •
- Our mechanism of action. There are two main biological pathways that underlie the efficacy of stem cells in disease treatment: their anti-inflammatory properties and their secretion of repair and growth promoting molecules. The immunomodulatory properties of these cells offer potential novel treatments for autoimmune and inflammatory diseases, as evidenced by promising preclinical and clinical results. The eASCs exhibit broad immunomodulatory properties, including the regulation of immune cells such as B lymphocytes, T lymphocytes, natural killer cells, monocytes or macrophages and neutrophils. These modulatory effects rely on a direct interaction between eASCs and immune cells as well as paracrine effects through a broad panel of soluble factors, among which interference with tryptophan metabolism through the indoleamine 2,3 dioxygenase enzyme and the enhanced activity of suppressor cells, such as
71
regulatory T cells and anti-inflammatory macrophages, are particularly significant. The following charts demonstrate the mechanisms of action through which eASCs regulate inflammation:
Inhibition of T cell proliferation
Source: De la Rosa et al. Tissue Engineering 2009
Activation of peripheral blood mononuclear cells, or PBMCs, leads to the proliferation of T cells, as shown in the left bar of the chart above. When adipose-derived stem cells, or ASCs, are added or co-cultured with the PBMCs, the T cells are largely inhibited, as indicated in the middle bars. This effect is due to the ASCs' expression of Indoleamine 2,3-dioxygenase, or IDO enzyme, a tryptophan degrading enzyme. The addition of an IDO-inhibitor largely reverses the inhibitory effect, as shown in the right bars. This inhibitory effect is mediated through the medium as demonstrated by the fact that separating the two cell types with a transwell, or semi-permeable membrane, as indicated by the black bars, results in comparable inhibition as when the cells are in contact with each other, as indicated by the white bars.
Reduction of pro-inflammatory cytokines
|
Source: De la Rosa et al. Tissue Engineering 2009
In non-stimulated conditions, as indicated by the above bars titled "PBMC," "ASC" and "PBMC+ASC," there is no secretion of the pro-inflammatory cytokines, interferon-g, or IFN-g, or
72
Tumor Necrosis Factor-a, or TNF-a. Upon stimulation, PBMCs secrete these cytokines, as indicated by the bar "Activated PBMC." In the presence of ASCs, as indicated by the bar "Activated PBMC + ASC," this secretion is strongly reduced. This effect is statistically significant as the p-value is below 0.05.
The following image depicts the mechanism of action of mesenchymal stem cells, or MSCs, a category that includes eASCs:
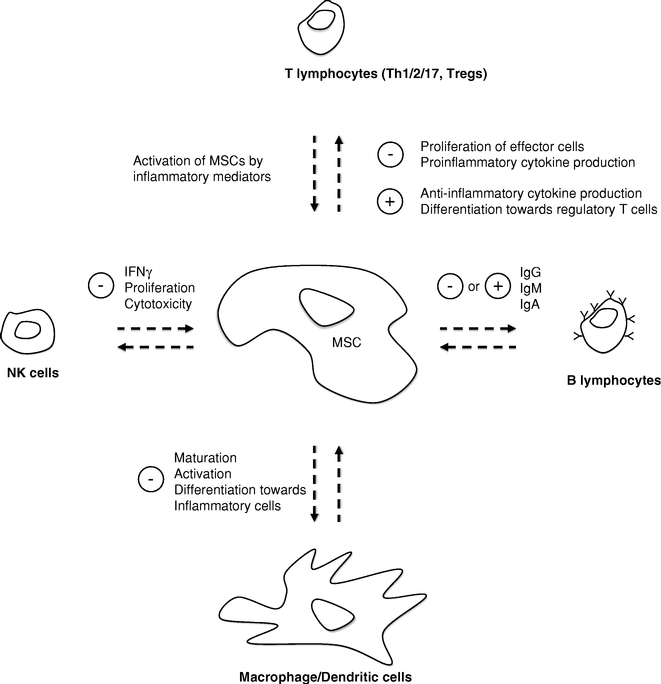
MSCs can interact with the different cells of the immune system, including T cells; B cells, which secrete the immunoglobulins IgG, IgM and IgA; NK cells and macrophages and dendritic cells. The effects of the MSCs on such cells can be decreasing, or inhibitory (-), or increasing, or stimulatory (+). The overall effect of these interactions aims at dampening the inflammatory intensity of the immune reaction.
73
Our product candidates leverage this recognized mechanism of action of MSCs in immune-mediated and inflammatory processes, which should enable us to develop rapidly and bring to market groundbreaking products that have the potential to treat safely a broad range of inflammatory and autoimmune diseases. We have had extensive discussions with the EMA regarding chemistry, manufacturing and control packages and preclinical packages in connection with our eASCs platform, which have allowed us to advance rapidly our clinical development with respect to our pipeline candidates.
- •
- Our experience in enabling multiple routes of administration: Depending on the type of disease and the best means of targeting the immune system, eASCs can be injected into the patients in various ways to optimize the impact on the patient.
- •
- Local administration. For local diseases or tissue damage, we believe that depositing the cells as close as possible to the affected tissue or organ optimizes the effect of the cells, which are not diluted and thus achieve the highest concentration at the site of action. The cells immediately encounter the inflamed environment leading to direct activation of the eASCs and their immunomodulatory actions. Therefore, for diseases like fistulas and inflammatory bowel disease, we locally administer the cells.
- •
- Systemic administration. For systemic diseases like rheumatoid arthritis, where the cells need to act at several places in the body, we believe that systemic administration of the cells, through either the blood or the lymphatic system, is the method of choice. Using this method of administration, the cells are distributed across the body and are able to reach the affected tissues. We believe that the capacity of eASCs to detect inflammation and to accumulate at the site of inflammation will result in an efficient mechanism of action.
- •
- Our use of human-derived adipose tissue. We use eASCs extracted from the human adipose tissue of healthy volunteers. We believe that this type of cell offers significant advantages over other cell types, such as stem cells sourced from bone marrow. The key advantages of this approach are the following:
- •
- Ease and amount of supply. The cells can be collected through standard liposuction.
- •
- Rich supply of stem cells. Stem cells can represent up to 2% of the total cells of the stromal vascular fraction of the fat tissue, a potential yield of 100 to 1,000 times higher than other possible sources of stem cells.
- •
- Robust phenotype. The eASCs do not require overly elaborate growth conditions and can be grown continuously without loss of their primary characteristics. They have also been shown to maintain cell stability during expansion.
- •
- Pharmacological profile. The eASCs have low immunogenicity as defined by the low presence or absence of human leukocyte antigens, co-stimulatory molecules and ligands for neurokinin receptors and are therefore considered to be safe for allogeneic treatment.
- •
- Our use of allogeneic adult stem cells. We have developed our platform using allogeneic stem cells because this approach offers clear advantages over autologous cells,i.e., cells extracted from each individual patient and subsequently processed, which are summarized below:
- •
- Efficient production of large batches of cells. Economies of scale can be applied with respect to manufacturing and quality control tests, reducing the cost of manufacturing and leading to a more consistent end product,i.e., individual lots of a large batch. Up to 360 billion cells can be obtained upon expansion of cells extracted from a single donor. At current scale, this could be used to generate up to 2,400 doses of Cx601.
74
- •
- No patient biopsy/tissue procurement needed. The use of allogeneic cells also benefits physicians and patients, because the treatment can be administered readily in a single procedure, taking less clinical time and resources. The process avoids taking biopsies from patients and allows for the treatment of patients who do not possess sufficient healthy tissue or who for any other medical reason cannot undergo tissue procurement.
- •
- Immediate and consistent availability of cells. The use of allogeneic cells, which are extracted from healthy volunteers and processed in large batches and are therefore available to physicians whenever required, enables the use of eASCs for the immediate treatment of acute conditions, because the additional step of procuring and processing autologous cells, which need to be extracted from each individual patient, is eliminated. This could potentially increase patient throughput significantly, creating a more attractive commercial opportunity than would be possible using autologous cells.
Product and Product Candidates
We have an advanced clinical stage pipeline derived from our validated platform of eASCs. Our eASCs are extracted and cultured from tissue sourced from healthy consenting adult donors and can then be administered in patients for the treatment of autoimmune and inflammatory diseases. Cx601, our lead product candidate, is indicated for the treatment of perianal fistulas in Crohn's Disease patients and has been granted orphan drug designation by the EMA in 2009. Recruitment for our single pivotal European Phase III clinical trial is expected to be completed by the end of 2014, and the primary end-point results are expected to be available in the third quarter of 2015 which would allow filing for marketing authorization by the first half of 2016. The FDA has agreed to review the results of this pivotal Phase III trial, if successful, as supportive evidence for filing for future regulatory approval in the United States.
Cx611, our next most advanced clinical stage product candidate based on our technology platform, has completed a successful Phase I/IIa trial for the treatment of refractory rheumatoid arthritis. We are planning to advance the clinical development of this product candidate in both early rheumatoid arthritis and severe sepsis. Cx621, our third clinical stage product candidate, is a suspension of allogeneic eASCs designed to be administered intra-lymphatically.
We also have one commercial product, ChondroCelect, that is indicated for cartilage repair in the knee and was the first cell-based medicinal product to receive centralized marketing authorization from the EMA.
75
The following chart summarizes our product candidates and our marketed product:
Cx601
Cx601, our lead product candidate, is a suspension of allogeneic eASCs administered locally in the perianal fistula through a single intra-lesional injection. Cx601 is currently in a Phase III trial in Europe, and we are planning to initiate a Phase III trial in the United States for the treatment of complex perianal fistulas in patients suffering from Crohn's disease.
In 2009, the EMA granted Cx601 orphan designation for the treatment of anal fistulas, recognizing the debilitating nature of the disease and the lack of treatment options. We filed for orphan designation for the treatment of anal fistulas in the United States in 2012.
Complex Perianal Fistulas in Crohn's Disease Patients
Crohn's disease is a chronic inflammatory disease of the intestine. It is characterized by focal or segmental transmural inflammation, or inflammation of the intestinal wall, which may occur in any part of the digestive tract with occasional granuloma formation. The transmural inflammation disrupts intestinal mucosal integrity, which frequently leads to the development of abscesses and fistulas. A fistula is an abnormal tract connecting two surfaces; a perianal fistula is defined as a tract between the perianal space and the epithelial surface proximal to the anus.
Although multiple schemes of fistula classification have been proposed, no scheme has been universally adopted. The American Gastroenterology Association recommends classification according to complexity as either simple or complex:
- •
- A simple perianal fistula is a superficial fistula having only a single external opening, without pain or fluctulence to suggest an abscess.
- •
- A complex perianal fistula is a serious condition that typically involves more of the anal sphincters, can have multiple tracts, is associated with a perianal abscess and may be recurrent.
76
Patients with complex fistulas are at an increased risk for incontinence following aggressive surgical intervention and have a smaller chance of healing.
Individuals who suffer from the condition are often unable to carry out ordinary daily activities and have significant decrease in their quality of life due to the recurring nature of the condition. They generally experience severe discomfort, pain and embarrassment and, in many cases, have significant psychological problems, requiring additional treatment and often causing substantial burdens for the health care systems that cover the associated treatment costs. Current treatment options, which include antibiotics, immunosuppressants, biologics and surgical treatment, do not offer a long-term solution and the risk of recurrence is high.
Market Opportunity
Complex perianal fistulas in patients suffering from Crohn's disease tend to occur in individuals between the ages of twenty and forty, though 10-15% of patients are diagnosed before adulthood. We have estimated the worldwide incidence of Crohn's disease in Europe and the United States on the basis of collated scientific publications on the following basis:
- •
- Known Crohn's disease epidemiology.
- •
- Approximately 12% of patients suffering from Crohn's disease will develop a perianal fistula.
- •
- Of these fistulas, 80% will be classified as complex.
The following graphs provide an overview of the estimated population of Crohn's disease patients suffering from complex perianal fistulas in Europe and the United States based on the assumptions stated above:
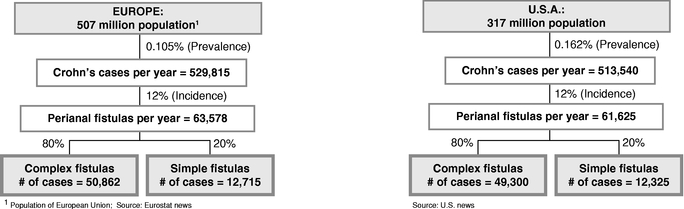
The burden of perianal fistulas in Crohn's disease is high, both to the individual patient and to the health care provider. In financial terms, the most significant portion of the cost burden of diagnosis and treatment can be attributed to the pharmaceutical treatment. In 2010, we commissioned a study by IMS, an independent provider of market research, that concluded that the total median cost of treatment of a patient with complex perianal fistula due to Crohn's disease was approximately $34,500 per patient, of which approximately $25,000 was spent on pharmaceutical treatment alone.
Taking into consideration a target population as described above (approximately 50,000 patients in Europe and approximately 50,000 patients in the United States) and assuming a sales price roughly equivalent to the median cost of pharmaceutical treatment used to treat fistulizing Crohn's disease, we estimate Cx601's target market to be approximately $2.5 billion for Europe and the United States combined.
77
Current Treatment Options
For Crohn's patients with complex perianal fistulas, treatments of choice are antibiotics and azathioprine or 6-mercaptopurine, as first-line therapy, and the biologic Remicade® (Infliximab), as second-line therapy. Both offer limited long-term efficacy and in many instances have notable side effects, such as the reactivation of tuberculosis and increased risk of bacterial infection with Aspergillus, Listeria and Cryptococcus.
The table below gives an overview of the most common drug treatments for perianal fistulas in patients suffering from Crohn's disease:
| | Antibiotics | Immunosuppressants | Antibiotics + immunosuppressants | Biologics | ||||
|---|---|---|---|---|---|---|---|---|
| Use | First-line or adjuvant therapy to treat infections and abscesses from fistulas. | Azathioprine and 6-mercaptopurine used as first-line after antibiotics therapy. | Antibiotics and immunosuppressants often used in combination as first-line therapy. | Remicade® (Infliximab) is the only approved biologic drug for fistulizing Crohn's disease. | ||||
Used as a second-line therapy. |
The standard second-line treatment of complex perianal fistula in patients suffering from Crohn's disease involves the prescription of anti-tumor necrosis factors, or anti-TNFs. As of June 30, 2014, Remicade® (Infliximab), a chimeric monoclonal antibody, is the only biologic approved for the treatment of fistulizing Crohn's by the FDA and the EMA. In a pivotal fifty-four week trial, 306 patients with Crohn's disease with some sort of disease-related fistulas were administered Infliximab at induction at weeks zero, two and six. Patients who had ongoing fistula response to the drug at week fourteen were re-randomized and placed on a maintenance regimen administered every eight weeks thereafter. By the end of the trial, 36% of the patients who went on to receive a maintenance therapy continued to be in complete remission; remission is defined here as complete healing of the fistula. If remission after initial induction is taken into account, efficacy of Infliximab at one year is limited to 23%.
Other biologics used in the treatment of luminal Crohn's but not specifically approved for the treatment of fistulizing Crohn's are the following:
- •
- Humira (adalimumab)—Abbott. Second generation anti-TNF approved for the treatment of Crohn's disease (but not fistulizing Crohn's). Humira has the advantages of requiring only subcutaneous dosing (instead of intravenous infusion) and being a fully human antibody. Fistula healing was studied as a secondary endpoint in the Humira maintenance trial. Efficacy results were a 33% rate of complete closure at fifty-six weeks.
- •
- Cimzia (certolizumab)—UCB. Although not developed for the treatment of fistulizing Crohn's directly, fistula healing was a secondary endpoint in one of Cimzia's maintenance trials and a small number of patients in a second trial also had fistula as a baseline. In none of the two trials did Cimzia outperform the efficacy of the placebo. The EMA refused the marketing authorization for Cimzia to treat active Crohn's disease. Nevertheless, Cimzia received FDA approval for treating adults with moderate to severe Crohn's disease who have not responded to conventional therapies.
The results of these other biologics that have been evaluated for the treatment of perianal fistula in patients suffering from Crohn's disease confirm the limited efficacy of the existing approaches.
78
The following chart summarizes the current treatment algorithm for complex perianal fistulas in patients suffering from Crohn's disease:
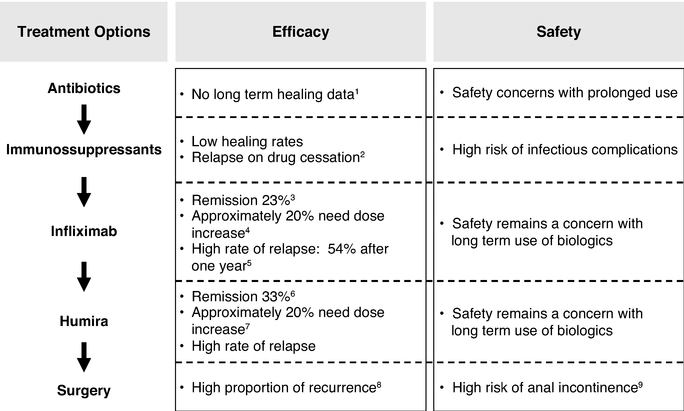
- (1)
- LH Bernstein et al.,Healing of Perianal Crohn's Disease with Metronidazole, 79 GASTROENTEROLOGY 357-65 (1980)
- (2)
- E. S. Goldstein et al.,6 -Mercaptopurine Is Effective in Crohn's Disease Without Concomitant Steroids, 10 INFLAMM BOWEL DIS 79-84 (2004)
- (3)
- ACCENT II clinical trial
- (4)
- L. Katz et al.,Doubling the Infliximab Dose Versus Halving the Infusion Intervals in Crohn's Disease Patients with Loss of Response, 18 INFLAMM BOWEL DIS 2026-33 (2012)
- (5)
- B.E. Sands et al.,Infliximab Maintenance Therapy for Fistulizing Crohn's Disease, 350 N ENGL J MED 876-85 (2004)
- (6)
- CHARM clinical trial
- (7)
- W. J. Sandborn et al.,Dosage adjustment during long-term adalimumab treatment for Crohn's disease: clinical efficacy and pharmacoeconomics, 17 INFLAMM BOWEL DIS 141-51 (2011)
- (8)
- T. Sonoda et al.,Outcome of Primary Repair of Anorectal and Rectovaginal Fistulas Using the Endorectal Advancement Flap, 45 DIS COLON RECTUM 1622-28 (2002); S.J. Van der Hagen,Staged Mucosal Advancement Flap for the Treatment of Complex Anal Fistulas: Pretreatment with Noncutting Setons and in Case of Recurrent Multiple Abscesses a Diverting Stoma, 7 COLORECTAL DIS. 513-18 (2005)
- (9)
- A. Soltani and A. Kaiser,Endorectal Advancement Flap for Crypto Glandular or Crohn's Fistula-in-Ano, 53 DIS COLON RECTUM 486-495 (2010)
79
Clinical Development
We believe that Cx601 can be used to treat complex perianal fistulas in patients suffering from Crohn's disease. Like all the products in our pipeline, Cx601 utilizes eASCs derived from adipose tissue, which we believe have anti-inflammatory and repair and growth-promoting properties and are an effective treatment for fistulas.
Having received positive scientific advice from the Committee for Medicinal Products for Human Use of the EMA, we have initiated a randomized, double-blind, placebo-controlled European Phase III trial with 278 recruited patients in fifty-two centers in eight countries, which is designed to comply with the requirements set by the EMA.
Recruitment for the trial began in mid-2012 and 171 patients had been recruited as of the end of 2013. The whole sample of patients is expected to be recruited by the end of 2014, a period that was prolonged due to a change in the third party contract research organization in charge of conducting the trial. The clinical data and the final clinical report is expected to be available by the third quarter of 2015.
The clinical trial is designed to be a two-group, single-blind placebo controlled trial, in which patients are randomly assigned to either a placebo control group or an active treatment group in a 1:1 ratio. The active treatment group will receive one dose of 100 million eASCs. The study's end-points will be as follows:
- •
- Primary end-point:
- •
- Remission of the fistulous disease, defined as 100% healing of the tracts. Evaluation of healing will include both blinded clinical assessment and MRI confirmation of the lack of abscesses larger than two square centimeters.
- •
- Secondary end-points (among others):
- •
- Response (50% of fistula tracts healing).
- •
- Time to remission.
- •
- Time to response.
- •
- Safety data.
- •
- Tolerability data.
The study includes males and females who will be allowed to maintain their current treatment of their underlying Crohn's disease as long as the dose is not modified during the course of the study and who meet the following criteria:
- •
- Older than eighteen years.
- •
- Have been diagnosed with perianal Crohn's disease with non-active or mildly active luminal disease and have failed previous treatments for the fistulas (antibiotics, immunosuppressants or biologics). Patients refractory to antibiotics will be restricted to less than 25% of patients included in the study.
- •
- Have fistulas with up to two internal orifices and up to three external orifices.
- •
- Were diagnosed with Crohn's disease more than six months prior to their inclusion in the trial.
- •
- Have their fistulas draining for less than six weeks prior to their inclusion in the trial.
The trial will have a first complete analysis of data from at a follow-up visit twenty-four weeks post-treatment, with a second follow-up analysis to be performed at fifty-two weeks post-treatment.
80
The protocol of this Phase III program has been approved by the ethics committees and regulatory agencies in all eight participating countries: Spain, Italy, Austria, Belgium, Germany, France, the Netherlands and Israel. The Committee for Medicinal Products for Human Use of the EMA has indicated that the proposed single pivotal Phase III study, if successful, could suffice to demonstrate the efficacy required to support the marketing authorization application to the EMA. If the results of the study are positive, we intend to submit such a marketing authorization application, and a decision by the EMA could be expected by early 2017. If marketing authorization were to be granted by early 2017, we could start to commercialize Cx601 in Europe in mid-2017.
In addition to allowing us to file for marketing authorization in Europe, this pivotal study, if successful, will serve as a key supportive study in filing for approval in many other jurisdiction, including the United States. We had a Type B meeting with the Center for Biologics Evaluation and Research within the FDA in December 2013, at which we discussed the following issues:
- •
- The adequacy of the existing non-clinical data available from previous trials to support an IND for a pivotal U.S.-based Phase III study.
- •
- Guidance on the design of such pivotal U.S.-based Phase III study.
- •
- Confirmation of the acceptability of using the data from the ongoing European Phase III study to support a BLA filing in the United States.
A Type B meeting is a category of meetings that includes each of the following:
- •
- Pre-IND application meetings.
- •
- Certain end-of-Phase I meetings.
- •
- End-of-Phase II and pre-Phase III meetings.
- •
- Pre-new drug application or BLA meetings.
Based on the positive feedback from the FDA, which has agreed to review the results of the European Phase III trial discussed above as supportive evidence for filing for regulatory approval in the United States, we are considering partnerships in the U.S. market, while preparing in parallel an independent approach. Therefore, we plan to start the process for a technology transfer to a U.S.-based contract manufacturing organization and the preparation for a SPA to file an IND application for a pivotal Phase III study in the United States. In addition to U.S.-based companies, we are starting to contact companies that may be interested in partnering with us to market Cx601 in other regions, most notably Asia (in particular, China and Japan).
Clinical Results
Our Phase II clinical trial for Cx601 was a single arm non-controlled study in which twenty-four patients of both genders suffering from refractory complex perianal fistulas and meeting all of the following criteria were treated:
- •
- They had associated fecal incontinence or risk factors of anal incontinence.
- •
- They had received at least one prior treatment for a fistulous disorder.
- •
- They suffered from Crohn's disease.
Only one fistula tract in each patient was treated, and patients received a maximum of two doses of Cx601.
81
The results of the Phase II clinical trial were as follows:
- •
- Efficacy in treating fistula tracts, defined as the complete closure and re-epithelization of the fistula being treated with absence of drainage, at twenty-four weeks was 56.0%, which is more than twice as high as the anti-TNF, the prevalent standard of care for fistulizing Crohn's disease.
- •
- 69.2% of patients experienced a reduction in the number of initially draining tracts.
- •
- Safety of the use of allogeneic stem cells for the treatment of perianal fistula was demonstrated.
Out of the twenty-four patients who started the trial, we have evaluable results with respect to sixteen patients, who remained at the time of the twenty four-week follow-up. Of the remainder, four patients dropped out due to the exacerbation of their underlying Crohn's disease, for which they needed to resume treatment. This was due to the specific design of the trial, in which patients were required to stop their existing treatment in order to isolate the effect of the therapy. Two patients developed anal abscesses and had to be excluded from the study while two other patients had to be excluded because of significant deviations from the study protocol.
The following chart summarizes some of the results of the trial:
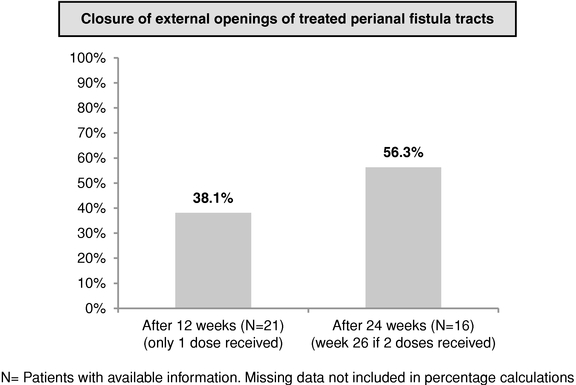
An overview of the adverse event from the trial was as follows (n=24):
Patients with at least one treatment emergent adverse event during the study: | 13 (54.2%) | |
Patients with at least one treatment emergent adverse event possibly related to eASCs during the study: | 5 (20.8%) | |
Serious adverse events reported leading to withdrawal (Events considered to be possibly related to the study treatment; no clinically relevant abnormalities found during physical examination or in vital signs): | 2 (8.3%) (the two adverse events were a local abscess and pyrexia, respectively) |
82
Prior to developing Cx601 as an allogeneic product, we also conducted a Phase II trial with respect to Cx401, an autologous predecessor product candidate,i.e., the cells were extracted from each individual patient and then expanded.
The Cx401 Phase II trial was a randomized controlled study with fifty patients suffering from refractory complex perianal fistula including both Crohn's and non-Crohn's patients. Patients were divided into two groups: one that was treated with fibrin glue, as placebo, and the other that was treated with Cx401 and fibrin glue.
The results of the trial were as follows:
- •
- After eight weeks, the efficacy of Cx401 and fibrin glue in treating complex fistulas was 70.8%, more than four times as high as the efficacy of fibrin glue alone (16.0%).
- •
- The safety of the product was demonstrated.
- •
- Patients who were treated with Cx401 and fibrin glue experienced improvement in physical and emotional functions.
- •
- After one year, the recurrence rate in the patients treated with Cx401 and fibrin glue was 17.6%, and no patient experienced recurrence until at least seven months after the treatment.
After this trial, we conducted a Phase III study with Cx401 in perianal fistulas in non-Crohn's patients. The trial demonstrated the efficacy of Cx401; however, the efficacy of placebo, fibrin glue, was much higher than expected and the trial did not produce statistically significant results. We concluded from this study that while eASCs are a safe and efficacious method of treating complex perianal fistulas, the presence of significant inflammation in the patient is required to activate the eASCs and trigger efficacy, and accordingly, are pursuing the development of Cx601 for complex perianal fistulas in patients suffering from Crohn's disease, who generally display more refractory symptoms, including higher levels of inflammation.
Cx611
Cx611 is an allogeneic cellular suspension of eASCs and injected intravenously. We have completed a Phase I/IIa trial for Cx611 for the treatment of refractory rheumatoid arthritis. We intend to develop Cx611 for patients suffering from early rheumatoid arthritis and from severe sepsis.
Rheumatoid Arthritis
Rheumatoid arthritis is one of the most common autoimmune diseases. It is a chronic systemic disorder characterized by inflammation of the joint tissues, primarily the synovium, the thin layer of tissue that lines the joints and tendon sheaths. The resultant inflammation and build-up of fluid in the joint leads to stiffness, swelling, redness and eventually to debilitating pain. Inflammation of the synovium may progress to degeneration of the joint bone and cartilage as a result of enzymatic actions of the cells involved in the inflammatory process and the resultant joint damage can lead to joint deformity. Rheumatoid arthritis can also affect the lungs, heart, kidneys, eyes, skin, blood cells and peripheral nerves, and can lead to substantial loss of function and mobility if not treated adequately. Within the first three months of presentation of the disease, 25% of patients will have bone erosion. Within three years of diagnosis, bone erosion will be up to 70% or more for most patients.
On a cellular level, rheumatoid arthritis is a multi-pathway disease, in which dendritic cells, T cells, B cells and macrophages and effector cells including neutrophils, mast cells, endothelial cells and synovial fibroblasts are all activated, and there is dysregulated expression of cytokines.
83
Market Opportunity
Rheumatoid arthritis represents the most common form of inflammatory arthritis, and according to a report by Global Data, in 2011, approximately 4 million people in the United States, France, Germany, Italy, Spain, the United Kingdom and Japan had been diagnosed with rheumatoid arthritis. It is three times more common in women than men, and approximately 20 million additional patients had been diagnosed in Australia, India and China. In 2011, the prevalence of rheumatoid arthritis in the adult population in the United States was estimated to be 0.6%.
The economic burden associated with the treatment of rheumatoid arthritis is huge for any healthcare system. In the United States, sales of drugs to treat rheumatoid arthritis were estimated to be approximately $9.5 billion in 2011. The costs associated with rheumatoid arthritis, such as informal care, non-medical costs and loss of productivity, are considerable and increase with disease progression. The estimated direct and indirect cost of rheumatoid arthritis therapy in the United States in 2011 was approximately $19 billion, which equates to about $14,900 per patient.
Current Treatment Options
The treatment of rheumatoid arthritis comprises three general classes of drugs: non-steroidal anti-inflammatory agents, or NSAIDs, corticosteroids and synthetic disease modifying anti-rheumatic drugs, or DMARDs. NSAIDs and corticosteroids have a short onset of action, while DMARDs can take several weeks or months to demonstrate a clinical effect. NSAIDs control acute inflammation, thereby decreasing pain and improving function and are also mild to moderate analgesics, but they are not disease modifiers and do not prevent joint destruction. Corticosteroids have both anti-inflammatory and immunoregulatory effects and are useful in early disease as temporary adjunctive therapy. Only DMARDs have been shown to alter the course of the disease and improve radiographic outcomes, and accordingly, the prevalent pharmacological management of rheumatoid arthritis involves early intervention with DMARDs, either alone or in combination. If inflammation cannot be adequately suppressed by these means, biologic DMARDs targeting the pro-inflammatory cytokine TNF are employed. If the response is inadequate, dose optimization, additional anti-TNFs, or alternatively, biologics using a different mechanism of action can be used. Despite all these treatments, rheumatoid arthritis remains an insufficiently unmet clinical need, and long-term treatments based on biologics have certain shortcomings, as follows:
- •
- Lack of efficacy of biologic treatments in some patients, and non-tolerability or recurrent secondary infections have been identified as factors contributing to the need to develop new therapies.
- •
- Biologic treatments may cause potentially serious adverse effects, especially with long-term use.
- •
- Approximately 20-40% of rheumatoid arthritis patients have been estimated to have an inadequate response to treatment with anti-TNF agents.
- •
- Efficacy declines over time and patients are usually required to switch periodically to another biologic.
Clinical Results
In January 2012, we completed a Phase I/IIa clinical trial using allogeneic eASCs for the intravenous treatment of refractory rheumatoid arthritis.
The Phase I/IIa clinical trial was a twenty-four week, single blind dose-escalating study. Fifty-three patients with moderate to high disease activity (disease activity score in twenty-eight joints, or DAS 28, greater than 3.2), who all were under treatment with one synthetic DMARD participated in the study. Forty-six participants received eASCs, and seven received placebo. All patients received three
84
intravenous infusions on days one, eight and fifteen of the trial. Patients in different cohorts received placebo, low (1 million eASCs per kg), medium (2 million eASCs per kg) and high (4 million eASCs per kg) doses of Cx611.
As follow-up, we conducted a detailed monthly workup of each patient measuring all the pre-defined parameters. We aimed to evaluate the safety, tolerability and optimal dosing over the full six months of the trial, as well as to explore therapeutic activity.
The endpoints of the study were as follows:
- •
- Primary end-points (safety):
- •
- Tolerability.
- •
- Treatment-emergent adverse events, including the following:
- •
- Dose limiting toxicities.
- •
- Serious adverse events.
- •
- Non-serious adverse events.
- •
- Secondary end-points (therapeutic activity):
- •
- American College of Rheumatology scores (known as ACR20/50/70, which measures the percentage of patients who experience 20%, 50% and 70% improvement, respectively, in tender or swollen joint counts as well as three out of five additional parameters identified by the American College of Rheumatology).
- •
- The European League against Rheumatism, or EULAR, criteria, which are based on the improvement in the DAS 28.
- •
- A short-form health survey measuring patients' quality of life.
The study was open to rheumatoid arthritis patients under treatment with at least one non-biologic DMARD who had previously failed treatment with at least two biologics (median time since diagnosis ranged from nine to 18.8 years; mean previous treatment with three or more disease-modifying anti-rheumatic drugs and three or more biologics) and was conducted in twenty-three centers.
We reported the final results of the Phase I/IIa study in April 2013, which included positive safety data as well as a first indication of therapeutic activity on standard outcome measures and biologic markers of inflammation, the results of which were as follows:
- •
- Patient and disease characteristics were comparable for all three dose groups.
- •
- There were no major safety signals from the repeated intravenous infusion of eASCs and the dose-limiting safety signal was not identified.
- •
- Three serious adverse events were reported (lacunar infarction, peroneal palsy and fever of unknown origin) of which lacunar infarction was thought to be possibly related to the treatment and led to the discontinuation of the treatment. The patient subsequently recovered.
- •
- The most frequent non-serious adverse effects, occurring in more than 10% of patients treated with eASCs, included the following:
- •
- Pyrexia (19%)
- •
- Headache (13%)
- •
- Urinary tract infection (13%)
- •
- Upper respiratory tract infection (11%)
85
- •
- Nausea (11%)
With respect to the secondary end-points, our findings were as follows:
- •
- A clear dose-response effect was not observed.
- •
- With respect to the American College of Rheumatology scores:
- •
- After three months, 25% of patients achieved a 20% improvement versus no patients in the placebo group; 15% of patients achieved 50% improvement versus no patients in the placebo group and 5% of patients achieved 70% improvement versus no patients in the placebo group.
- •
- With respect to the EULAR criteria based on the improvement in the DAS 28:
- •
- Three months after the treatment, 39% of patients had a good to moderate response compared to no patients in the placebo group.
- •
- With respect to the disease activity score in twenty-eight joints as modified to measure the C-reactive protein value, or DAS 28 (CRP):
- •
- 11% of patients compared to no patients in the placebo group achieved remission through the end of the trial.
The following charts summarize the results of the Phase IIa clinical trial as per the EULAR criteria:
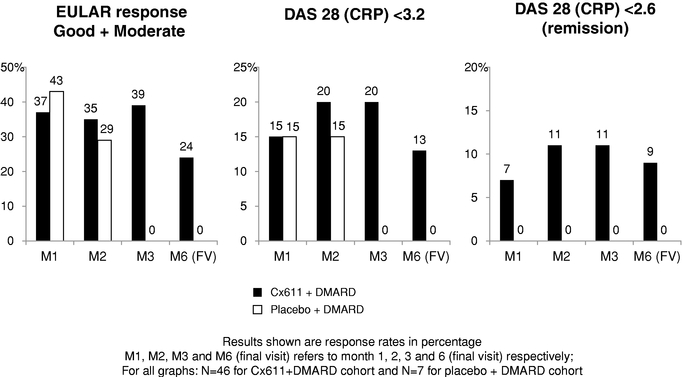
These early clinical results are the first to suggest that intravenous infusion of eASCs is well tolerated along twenty-four weeks and could be associated with clinical benefits in the treatment of refractory rheumatoid arthritis.
These data were submitted to the American College of Rheumatology meeting in San Diego and were among the select few abstracts out of over 3,500 presented at the congress as a whole that were presented at a plenary session on October 29, 2013.
86
Based on the results of this study, on June 30, 2014, we announced our development plan for Cx611 in early rheumatoid arthritis. As early intervention in the disease process is considered key to avoid progression to advanced rheumatoid arthritis, therapeutic approaches that target the early disease process are today a preferred investigational route for treatment. Through their broad immune-regulatory properties, eASCs are expected to positively interfere with the early disease process,i.e., temper the inflammatory rheumatoid arthritis process and could thereby prevent further progression of the disease. This novel therapeutic concept was supported by a steering committee consisting of Professor Mark Genovese, Professor Paul Emery and Professor José María Alvaro-Gracia, who was the principal investigator in our Phase I/IIa study. We are working with a group of leading clinical experts to complete the protocol for a randomized, double-blind comparative Phase IIb study to test the efficacy of Cx611 in rheumatoid arthritis patients who have been diagnosed within the prior year who have failed conventional treatment, but who have not yet been treated with a biologic.
We believe that our Phase I/IIa clinical trial set the stage not only for the further development of Cx611 in rheumatoid arthritis, but also in a wide range of other autoimmune disorders that still represent major clinical unmet needs, including severe sepsis, the second indication we are pursuing with respect to Cx611.
Severe Sepsis
Sepsis is a potentially life-threatening complication of infection that occurs when inflammatory molecules released into the bloodstream to fight the infection trigger systemic inflammation. This inflammation can lead to a cascade of detrimental changes that damage multiple organ systems, causing them to fail. Sepsis simultaneously produces a pro-inflammatory and an anti-inflammatory response. The pro-inflammatory responses lead to organ failure and coagulation leads to tissue hypo-perfusion and tissue injury; the anti-inflammatory responses produce a susceptibility to infection. When sepsis is complicated by organ failure, which may include respiratory compromise, cardiovascular compromise, central nervous system dysfunction or acute kidney injury, it is considered severe. Patients with severe sepsis require close monitoring and treatment in a hospital intensive care unit. If sepsis progresses to septic shock, the patient's blood pressure drops dramatically, potentially leading to death. Mortality increases as the condition progresses, with estimates ranging from 10-20% in sepsis to 20-50% in severe sepsis to 40-80% in patients who progress to septic shock.
Drug therapy is likely to include broad-spectrum antibiotics, corticosteroids, vasopressor drugs to increase blood pressure, plus oxygen and large amounts of intravenous fluids. Supportive therapy may be needed to stabilize breathing and heart function and to replace kidney function.
Current Treatment Options
Severe sepsis represents a high unmet medical need. Current treatments are insufficient and mainly symptomatic, and aim at controlling the infection with antibiotics, improving some of the symptoms, as with vasopressor treatment, or providing supportive treatment such as haemodialysis or mechanical ventilation. Biologics are also used but generally have limited effect. There is a clear need for a product that could impact both the pro-inflammatory and the anti-inflammatory pathways.
Clinical Development
We are working on a Phase Ib development plan for Cx611 in severe sepsis with the assistance of an advisory board. We have finalized the design of a randomized placebo-controlled trial to test the mechanism of action of Cx611 in healthy volunteers challenged with a low dose of bacterial endotoxin (lipopolysaccharide), a potent pro-inflammatory constituent of the outer membrane of gram-negative bacteria, which elicits a strong inflammatory response inducing sepsis-like symptoms. We expect to recruit thirty-two volunteers for the study, who will be divided into four groups of eight patients each.
87
Patients in the first three groups will receive Cx611 in different doses and patients in the final group will receive placebo.
The end-points of the study will include the following:
- •
- Blood pressure
- •
- Temperature
- •
- Heart rate
- •
- Symptom score
- •
- Pro and anti-inflammatory cytokines
- •
- Recruitment and activation of neutrophils
- •
- Macrophages
- •
- T-cells, B-cells and endothelial cells
- •
- C-reactive protein
- •
- (Anti-)coagulation activation and fibrinolysis
- •
- Microarrays
We intend to start recruiting volunteers in early 2015 and to receive the results of the study by the third quarter of 2015. We expect that further clinical trials in severe sepsis will focus on a strictly defined and homogenous target population of patients to demonstrate appropriately the efficacy of the treatment.
Cx621
Cx621 is an allogeneic cellular suspension of eASCs for the potential treatment of a variety of autoimmune diseases via a proprietary technique of intra-lymphatic administration. Autoimmune diseases are a group of more than one hundred conditions caused by disruptions to immune homeostasis. The characteristic immediate result of an autoimmune condition is inflammation, which is the result of the aggregation of cells and molecules associated with the immune pathways in a tissue. While inflammation is a critical component of healing processes, uncontrolled and abnormal inflammatory processes can lead to serious complications such as tissue degeneration. As such autoimmune diseases are often chronic and debilitating conditions that place a huge burden on not only individuals but also their health care providers.
Clinical Development
Based on positive preclinical data on toxicology, biodistribution and efficacy, we conducted a Phase I protocol to assess safety, tolerability and pharmacodynamics of intranodal injected allogeneic eASCs in healthy volunteers. We started the recruitment for this study in the fourth quarter of 2011 and the final results were communicated in 2012.
We conducted a randomized, controlled, single-blind Phase I trial to assess the intra-lymphatic administration of two fixed doses (2.5 and 5 million) of eASCs in two different cohorts. Each dose was administered twice with an interval of seven days and was injected into two inguinal lymph nodes. Two volunteers per cohort received treatment with HypoThermosolTM as a control group. The primary objective was to determine the safety, feasibility and tolerability of intra-lymphatic eASCs administration. The safety assessment was performed over twenty-one days after the second administration. It included signs and symptoms, laboratory tests, chest x-ray and appearance of the
88
injected lymph nodes by ultrasound. Pharmacodynamic parameters were included as an exploratory measure.
Ten healthy volunteers of both genders were included, five in each of the treatment cohorts. All volunteers presented lymph nodes with sufficient size to perform the procedure. No serious or severe adverse events occurred. The volunteers reported local pain using a visual analogue scale after the administration of either eASCs or the placebo without statistically significant differences between them. This inguinal pain was transient, mostly mild and of moderate intensity in two patients. In the highest dose cohort inguinal ultrasound established varying degrees of inflammation of the treated lymph node. No clinically relevant changes were observed in the laboratory tests, vital signs and electrocardiogram. A minor transient, but statistically significant, increase in C-reactive protein was observed in the eASC group, which remained within the normal range. The status of the circulating blood cell subsets in the eASCs' cohort before and after the treatment was comparable to that found in the control cohort.
The confirmation of the safety of intra-lymphatic administration of our eASCs could have significant clinical and commercial implications. This use of a different route of administration has the potential to enable applications in other autoimmune diseases.
ChondroCelect
We have one commercial product: ChondroCelect, a cell-based medicinal product for cartilage repair in the knee. ChondroCelect uses autologous cells, and the treatment involves a two-step surgical procedure in which cells are taken from the patient's own knee, multiplied to reach a sufficient quantity and re-implanted at the site of the defect. It was the first approved cell-based product in Europe that successfully completed the entire development track from research through clinical development to European approval. ChondroCelect received marketing authorization in October 2009 as an advanced therapy medicinal product, a new medical product category regulated by the EMA that includes products based on gene therapy, cell therapy or tissue engineering.
Our marketing authorization from the EMA allowed us to market and sell ChondroCelect across the European Economic Area for an initial period of five years, and we have received renewed authorization for an additional five year period ending in October 2019 based on a review of primarily safety-related data. In parallel to our commercialization efforts, we are conducting an open-label, multicenter, non-interventional study in patients whose symptoms appeared less than three years previously who have been treated with ChondroCelect for single symptomatic cartilage lesions of the knee of between one to five square centimeters. The results from an interim analysis of 153 patients in this study indicate statistically and clinically significant improvement in all knee injury and osteoarthritis outcome subscale scores versus the baseline. These data from treatment in daily clinical practice confirm the positive results from our previous randomized clinical trial. ChondroCelect has also been approved for reimbursement in Belgium in February 2011, in the Netherlands in June 2012 (retroactively applicable through to January 2011) and in Spain in March 2013, and in addition ChondroCelect is available to patients in the U.K. and Finland.
Effective June 1, 2014, we have entered into a distribution agreement with Sobi for the exclusive marketing and distribution rights with respect to ChondroCelect within the European Union (excluding Finland, where we have a pre-existing distribution agreement with Finnish Red Cross Blood Service); Switzerland; Norway; Russia; Turkey and the Middle East and North Africa region (currently excluding certain countries where we have a pre-existing distribution agreement with Genpharm). The distribution agreement with Sobi has a term of ten years during which we will receive royalties of 22% on the net sales of ChondroCelect during the first year of the agreement and 20% on an ongoing basis.
89
Cartilage Repair in the Knee
Various surgical procedures are used for the local treatment of cartilage defects in the knee, including debridement and lavage, microfracture and osteochondral grafting, also called mosaicoplasty.
While microfracture appears to be the accepted standard of care for small-sized cartilage defects, it often leads to scar-like repair tissue, and unlike stable hyaline-like cartilage, which is the most common form of cartilage, as regenerated by ChondroCelect, is not associated with long-term durable outcomes. Various investigators have found that the clinical benefit associated with microfracture reduces after two to three years.
An alternative to such surgical procedures is autologous chondrocyte implantation, a technique to repair articular cartilage by implanting the patient's own expanded cartilage cells, which was developed in order to address the limitations of the surgical procedures which constitute the accepted standard of care.
Product and Technology
ChondroCelect is a cell-based medicinal product indicated for the repair of single symptomatic cartilage defects of the femoral condyle, or the projections of the femur, at the knee in adults. Our cell culture methods have been specifically developed to maintain the phenotypical stability of the cells to promote the formation of stable hyaline-like articular cartilage. Cells are taken from the patient's own knee, multiplied to reach a sufficient quantity, and then finally re-implanted at the site of the defect, a procedure that is known as autologous chondrocyte implantation.
Market Opportunity
The target population with the highest expected benefit consists of adults between eighteen and fifty years with an early onset of symptoms (less than three years) with International Cartilage Repair Society grade III and IV lesions between one to five square centimeters that are located on the femoral condyle. This target population is estimated to be between 17,000 and 28,000 patients per year in Europe, where we are focusing our commercial efforts through our distribution agreement with Sobi. Through our distribution agreement, we will also be able to address other markets, such as the Middle East, North Africa, Russia and Turkey.
Competition
Product Candidates
The biopharmaceutical industry is characterized by intense and dynamic competition to develop new technologies and proprietary therapies. Any product candidates that we successfully develop and commercialize will have to compete with existing therapies and new therapies that may become available in the future. While we believe that our eASC platform and scientific expertise in the field of cell therapy provide us with competitive advantages, we face potential competition from various sources, including larger and better-funded pharmaceutical, specialty pharmaceutical and biotechnology companies, as well as from academic institutions, hospitals, governmental agencies and public and private research institutions.
Cx601 will compete against a variety of therapies in development for perianal fistulas in patients suffering from Crohn's disease, using therapeutic modalities such as biologics and cell therapy, including products under development by Anterogen, Delenex Therapeutics, Novartis and Celgene as well as various hospitals and research centers. In addition, there are products in development for the treatment of Crohn's disease that do not focus on the treatment of fistulas.
90
Likewise, with respect to Cx611, there are a number of competing products under development for the treatment of rheumatoid arthritis, primarily consisting of biologic therapies, which are being developed by Bristol Myers Squibb, Sanofi/Regeneron, Novartis, Johnson & Johnson, GlaxoSmithKline, MorphoSys, Novo Nordisk and Neovacs.
Many of our competitors, either alone or with their strategic partners, have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of product candidates, obtaining FDA, EMA and other regulatory approvals of treatments and commercializing those treatments.
Accordingly, our competitors may be more successful in obtaining approval for treatments and achieving widespread market acceptance. Our competitors' treatments may be more effective, or more effectively marketed and sold, than any treatment we may commercialize and may render our treatments obsolete or non-competitive before we can recover the expenses of developing and commercializing any of our treatments.
Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical study sites and in recruiting patients for clinical studies. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available. We expect any treatments that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, convenience of administration and delivery, price, the level of competition and the availability of reimbursement from government and other third-party payers.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Although we believe that our cell therapy pipeline is the most advanced in Europe as of the date of this prospectus, our competitors also may obtain FDA, EMA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
ChondroCelect
The market for the treatment of cartilage defects is highly fragmented. Treatment options include surgical treatments, cell-based therapies and cell-free products such as scaffolds and gels. The advantage of cell-based therapies is that they offer the possibility of a repair treatment while sparing the osteochondral region.
To date, aside from ChondroCelect, only one autologous chondrocyte implantation product has received approval from the EMA: MACI, which obtained advanced therapy medicinal product status in 2013. Following the acquisition of MACI by Aastrom Biosciences through its acquisition of Sanofi's cell therapy and regenerative medicine business, on June 16, 2014, Aastrom Biosciences announced the immediate discontinuation of European manufacturing and a temporary hiatus in sales of MACI in Europe.
Other companies pursuing an advanced therapy medicinal product approval for autologous chondrocyte implantation products include Tetec, a subsidiary of B. Braun, Co.don and Cellmatrix. In addition to these companies, there are a number of hospitals that produce autologous cartilage for their own patients.
91
Alternative competition may come from cell-free products that also target the cartilage repair market that would generally be brought to market through the medical device regulatory route.
Intellectual Property
Our commercial success depends in part on our ability to obtain and maintain proprietary or intellectual property protection in key markets for certain aspects of our cell therapy products, processes and related technologies to operate without infringing on the proprietary rights of others and to prevent others from infringing our proprietary or intellectual property rights. Our policy is to seek to protect our proprietary and intellectual property position by, among other methods, filing U.S., international and foreign patent applications related to multiple aspects of our proprietary products, processes and technologies.
As of September 30, 2014, we owned or co-owned more than twenty patent families and had more than ninety granted patents in over twenty jurisdictions, including the United States, with expiration dates from 2020 onwards. Of these patents, over ten related to our eASC-based technology platform, with expiration dates from 2024 onwards. Some of our pending patent applications are filed under the Patent Cooperation Treaty, an international patent law treaty that provides a unified procedure for filing a single initial patent application to seek patent protection for an invention simultaneously in each of the 147 member states, followed by the process of entering into national phases in each of the member states, which requires a separate application in each of the member states when continued protection is sought.
A number of our patent families are the result of collaborations with academic parties, and are jointly owned. Co-ownership agreements are in place with respect to all but one of such patent families, and certain types of exploitation of such patents may be subject to the co-owner's approval. We exclusively own the patents and patent applications that form the remainder of our patent portfolio.
Our patent portfolio includes the following:
- •
- Certain key foreign base patents and U.S. and foreign patent applications related to our eASC platform.
- •
- U.S. and foreign patents and patent applications for other cell therapy applications.
- •
- U.S. and foreign patents and patent applications with respect to chrondocyte markers.
- •
- A U.S. patent and U.S. and foreign patent applications for cell therapy delivery mechanisms.
- •
- U.S. and foreign patent applications for technology improvements with respect to our eASC platform.
In addition, we have over fifty registered trademarks and trademark applications.
Finally, several elements of our cell therapy program involve unpatented proprietary technology, processes, know-how or data, including cell isolation, production and release processes, which we consider to be part of our intellectual property. With respect to proprietary technology, know-how and data that are not patentable or potentially patentable, or processes other than production processes for which patents are difficult to enforce, we have chosen to protect our interests by relying on trade secret protection and confidentiality agreements with our employees, consultants and certain contractors and collaborators. All our employees are parties to employment agreements that include such confidentiality provisions.
Partnerships, Licensing and Collaboration
We have entered into partnerships and collaborations in the past and will consider such opportunities in the future.
92
Effective June 1, 2014, we have entered into a distribution agreement with Sobi for the exclusive marketing and distribution rights with respect to ChondroCelect within the European Union, excluding Finland, as well as several other countries. The agreement is for a ten-year term during which we will receive royalties of 22% on the net sales during the first year of the agreement and 20% on the net sales of ChondroCelect on an ongoing basis. In Finland, we have a distribution agreement in place with Finnish Red Cross Blood Service to conduct and facilitate the ChondroCelect business in the Finnish territory.
We have entered into a long-term manufacturing agreement with our former Dutch subsidiary, which we sold to PharmaCell, a leading European contract manufacturing organization active in the area of cell therapy, to continue to manufacture ChondroCelect in its facility. We are also in the process of entering into an agreement with a U.S.-based contract manufacturing organization and will start the process for technology transfer in connection with a proposed Phase III study with respect to Cx601 in the United States. We also rely on third party contract research organizations to conduct our clinical trials.
In addition, a number of our patent families are the result of collaborations with academic parties, including with Universidad Autónoma de Madrid and Consejo Superior de Investigaciones Científicas, and are jointly owned. Co-ownership agreements are in place with respect to all but one of such patent families, and certain types of exploitation of such patents may be subject to the co-owner's approval.
We will consider partnerships in the United States and other markets to rapidly bring Cx601, Cx611 or any of our other future products to market and maximize our value.
Manufacturing and Logistics
Our Product Candidates
Our eASC-based products candidates are considered medicinal products pursuant to the European regulation governing advanced therapy medicinal products and Spanish Order SCO/3461/2003 and therefore must be manufactured in compliance with cGMP requirements in an authorized pharmaceutical establishment. This also applies to the medicinal products manufactured for use in clinical trials.
Our product candidates are allogeneic eASCs that are originally derived from the subcutaneous fat tissue of a healthy donor. The fat biopsy tissue is first enzymatically digested and stem cells are recovered from it through a series of cell culture steps. In this first series of expansion steps, we create a master cell bank and extensively test the quality and safety of these first large cell banks. Once the master cell bank is qualified, it can be used to generate sequentially a large number of so-called final drug substances cell banks. These final drug substances are obtained by expanding the cells of the master cell bank with a new series of cell expansions in cell culture. The final drug substances are then cryopreserved, or frozen at very low temperatures, until final use. When a final product needs to be provided to the physician, the required amount of frozen cells are thawed and recovered in cell culture. These cells are then subsequently collected for final formulation in excipient, or inert, medium. The amounts of cells and excipient volume depend on the particular product and their use in the clinics.
During the entire manufacturing process, there are specific quality controls to guarantee that the product complies with the adequate specifications for use. The controls applied during the process on raw materials and on the finished product before and after it is packaged are particularly important. We also conduct microbiological and environmental controls and process controls to ensure that the manufacturing conditions are compliant for the manufacturing and distribution of the finished product as required by cGMP requirements.
The EMA has established the characterization of eASCs in terms of identity, purity, potency, morphology, viability and cell growth kinetic according to theGuideline on Cell-Based Medicinal
93
Products (EMA/CHMP/410869/2006) and theReflection Paper on Stem Cells (EMA/CAT/571134/2009, adopted on January 14, 2011) in order to set the routine controls that will be applied at final product release as well as those to be performed at different stages of the manufacturing process to guarantee the batch consistency. We obtained scientific advice from the EMA to ensure that our manufacturing process is aligned with their requirements.
Our facilities for the manufacture of eASCs are located in Madrid, Spain, and consist of two separate clean rooms and adjacent support rooms. The facilities have been approved by the Spanish Medicines and Medical Devices Agency as being compliant with cGMP requirements for the manufacture of cellular medicinal products for investigational use (i.e., clinical trials). We believe that the combined capacity of both clean rooms is sufficient to supply the necessary quality of material for our ongoing clinical trial programs.
The logistics for our eASC-based products include the transport of the finished product in a special temperature controlled shipping container. The shipping process has been validated with specialist courier services. Based on our experience with these companies and the proximity of our manufacturing facility to the Madrid international airport of Barajas, we have demonstrated that we can reliably deliver the finished product to treatment sites anywhere in Europe and Israel within twenty-four hours.
ChondroCelect
Cell-based manufacturing products such as ChondroCelect must be manufactured in a facility authorized by the regulatory authorities in compliance with cGMP requirements.
The ChondroCelect expansion process is designed to preserve the integrity and function of the cells and particularly to maintain the ability of cells to produce hyaline cartilage. This method was developed and validated in order to limit the usually observed dedifferentiation of chondrocytes in culture. Critical parameters have therefore been included in process controls routinely to monitor and control the quality of the product. The final product undergoes a series of mandatory quality control tests such as sterility, purity, dosage, potency and visual appearance. Only products that meet these quality control criteria are released and delivered.
On May 30, 2014, we completed the sale of TiGenix B.V., our Dutch subsidiary, which held our manufacturing facility, to PharmaCell, a leading European contract manufacturing organization active in the areas of cell therapy. ChondroCelect will continue to be manufactured in that facility under a long-term manufacturing agreement with our former subsidiary.
In 2012, the site passed an inspection by the Dutch authorities certifying that it was compliant with cGMP requirements, and obtained approval from the EMA for the production of ChondroCelect. To ensure that the manufacturing facility is compliant with cGMP requirements, a stringent quality management system is in place.
Facilities
Our registered office is in Leuven, Belgium. We have facilities in Madrid, Spain, where we lease two adjacent buildings. The first building houses our administrative offices, while the other building hosts our research and development laboratories and a facility compliant with cGMP requirements for the manufacturing of clinical eASC products. The facility contains two separate clean rooms and adjacent support rooms. They have been approved by the Spanish Medicines and Medical Devices Agency as being compliant with cGMP requirements for the manufacture of cellular medicinal products for investigational use,i.e., clinical trials.
94
Employees
We rely on a team of experienced professionals in all areas required to meet our strategic objectives including research and development, medical and regulatory, manufacturing, business development, product development, infrastructure, intellectual property and finance.
On September 30, 2014, we had a total of forty-six employees on a full-time equivalent basis. About 62% of these employees were engaged in research and development activities, including clinical development and manufacturing, and the remainder were engaged in corporate functions, including finance, human resources, legal, information technology, business development, investor relations and intellectual property.
United States Government Regulation
In the United States, the FDA regulates drugs and biologics under the Federal Food, Drug and Cosmetic Act, or the FDCA, and implementing regulations. Failure to comply with the applicable FDA requirements at any time during the product-development process, approval process or after approval may result in administrative or judicial sanctions. These sanctions could include, as applicable, the FDA's imposition of a clinical hold on trials for drugs, devices or biologics, refusal to approve pending applications, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution or any combination of these sanctions. Any agency or judicial enforcement action could have a material adverse effect on us.
The BLA Approval Process
Drugs that are also biological products, including all of our cell-based products, must also satisfy the requirements of the Public Health Service Act, as incorporated in the FDCA, and its implementing regulations. In order for a biological drug product to be legally marketed in the United States, the product must have a BLA approved by the FDA.
The steps for obtaining FDA approval of a BLA to market a biological product in the United States include the following:
- •
- Completion of preclinical laboratory tests, animal studies and formulation studies under the FDA's good laboratory practices regulations.
- •
- Submission to the FDA of an IND application for human clinical studies, which must become effective before human clinical trials may begin and which must include approval by an independent institutional review board, or ethics board, at each clinical site before the trials may be initiated.
- •
- Performance of adequate and well controlled clinical trials in accordance with good clinical practice, as set forth by the International Conference on Harmonization, and FDA, to protect the health and safety of human subjects to establish the safety and efficacy of the product for each indication and assure clinical study data integrity.
- •
- Submission to the FDA of a BLA containing detailed information about the chemistry, manufacturing and controls for the product, reports of the outcomes and full data sets of the preclinical studies and the clinical trials and proposed labeling and packaging for the product.
- •
- Satisfactory review of the contents of the BLA by the FDA, including the satisfactory resolution of any questions raised during the review.
- •
- Satisfactory completion of an FDA Advisory Committee review, if applicable.
95
- •
- Satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with cGMP requirements to assure that the facilities, methods and controls are adequate to ensure the product's identity, strength, quality, purity and potency.
- •
- FDA approval of the BLA, including agreement on post-marketing commitments, if applicable.
Preclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND application. Some preclinical testing may continue after the application is submitted. The investigational new drug application must become effective before human clinical trials may begin. An IND application will automatically become effective thirty days after receipt by the FDA, unless before that time the FDA raises concerns or questions about issues such as the conduct of the trials or supporting preclinical data as outlined in the application. In that case, the IND application sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. In other words, submission of an IND application may not result in the FDA allowing clinical trials to commence.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators. Clinical trials are conducted under strict requirements to ensure the protection of human subjects participating in the trial and protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND application. In addition, an institutional review board (usually, but not necessarily, specific to each study site) must approve the protocol, subject informed consent form and any amendments. All research subjects must be informed, among other things, about the risks and benefits of the investigational product and provide their informed consent in writing. Federal regulations governing the protection of human subjects in clinical trials have remained generally consistent for many years, subject to certain amendments. In July 2011, the U.S. Department of Health and Human Services and the FDA issued an advance notice of proposed rulemaking seeking comments on proposals to substantially change aspects of these regulations, seeking comments, for example, on mandating the use of a single institutional review board for multi-site trials, imposing specified data security and information regulations on trials, imposing new consent requirements with respect to the use of biospecimens that have been stripped of patient-identifiers, and requiring the use of standardized consent forms. The outcome of this regulatory review is not yet certain.
Clinical trials typically are conducted in three sequential phases, but the phases may overlap or be combined.
Phase I trials usually involve the initial introduction of the investigational product into a small group of healthy volunteers. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. These trials may also provide an early indication of efficacy.
Phase II trials usually involve controlled trials in a larger but limited patient population (e.g., a few hundred) to:
- •
- Evaluate dosage tolerance and appropriate dosage.
- •
- Identify possible adverse effects and safety risks.
- •
- Provide a preliminary evaluation of the efficacy of the drug for specific indications.
Phase III trials usually further evaluate clinical efficacy and test further for safety in an expanded patient population (e.g., several hundred to several thousand patients). Phase III trials usually involve
96
comparison with placebo, standard treatments or other active comparators. Usually two well controlled large Phase III or pivotal trials demonstrating safety and efficacy are required. These trials are intended to establish the overall risk-benefit profile of the investigational product and provide an adequate basis for physician labeling. Phase III trials are usually larger, more time consuming, more complex and more costly than Phase I and Phase II trials.
Phase I, Phase II and Phase III clinical trials may not be completed successfully within any specified period, if at all. Furthermore, we or the FDA may suspend or terminate clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk, have experienced a serious and unexpected adverse event, or that continued use in an investigational setting may be unethical. Similarly, an institutional review board or data safety monitoring board can suspend or terminate approval of research if the research is not being conducted in accordance with the institutional review board's requirements or if the research has been associated with unexpected serious harm to patients.
Assuming successful completion of the required clinical testing, the results of the preclinical studies and of the clinical trials, together with other detailed information, including information on the chemistry, manufacture and composition of the product, are submitted to the FDA in the form of a BLA requesting approval to market the product for one or more indications. In most cases, the BLA must be accompanied by a substantial user fee. The FDA will initially review the BLA for completeness before it accepts it for filing. After the BLA submission is accepted for filing, the FDA reviews it to determine, among other things, whether the proposed product is safe and potent, or effective, for its intended use and has an acceptable purity profile, and whether the product is being manufactured in accordance with cGMP requirements to assure and preserve the product's identity, strength, quality, purity and potency. The FDA may refer applications for novel biological products or biological products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and, if so, under what conditions. The FDA is not bound by the recommendations of the advisory committee, but it considers such recommendations carefully when making decisions.
Under the Pediatric Research Equity Act of 2003, BLAs, or BLA supplements, must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The intent of Pediatric Research Equity Act is to compel sponsors whose products have pediatric applicability to study those products in pediatric populations, rather than ignoring pediatric indications for adult indications that could be more economically desirable. By its terms the Pediatric Research Equity Act does not apply to any biological product for an indication for which orphan designation has been granted, unless FDA issues regulations saying otherwise. The FDA may grant deferrals for submission of data or full or partial waivers.
Before approving a BLA, the FDA generally will inspect the facility or the facilities at which the product is manufactured. The FDA will not approve the product if it finds that the facility does not appear to be in compliance with current good manufacturing practice requirements.
The FDA also has authority to require a risk evaluation mitigation strategy from manufacturers to ensure that the benefits of a biological product outweigh its risks. A sponsor may also voluntarily propose a risk evaluation mitigation strategy as part of the BLA submission. The need for a risk evaluation mitigation strategy is determined as part of the review of the BLA. Based on statutory standards, elements of a risk evaluation mitigation strategy may include "dear doctor letters," a medication guide, more elaborate targeted educational programs, and in some cases restrictions on distribution. These elements are negotiated as part of the BLA approval, and in some cases may delay
97
the approval date. Once adopted, risk evaluation mitigation strategies are subject to periodic assessment and modification.
After the FDA completes its initial review of a BLA, it will communicate to the sponsor that the biological product will either be approved, or it will issue a complete response letter to communicate that the BLA will not be approved in its current form. The complete response letter usually describes all of the specific deficiencies in the BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the applicant in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application.
The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. The testing and approval process for a biological product may take several years to complete.
One of the performance goals agreed to by the FDA under the Prescription Drug User Fee Act is to review 90% of standard BLAs in ten months and 90% of priority BLAs in six months, whereupon a review decision is to be made. The FDA does not always meet its goal dates for standard and priority BLAs under the Prescription Drug User Fee Act and its review goals are subject to change from time to time. The review process and the goal data may be extended by three months if the FDA requests or the BLA applicant otherwise provides additional information or clarification regarding information already provided in the submission within the last three months before the goal date under the Prescription Drug User Fee Act.
Even if a product candidate receives regulatory approval, the approval may be limited to specific disease states, patient populations and dosages, or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings, or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval. In addition, the FDA may require Phase IV post-marketing clinical studies, designed to further assess a biological product's safety and effectiveness, and testing and surveillance programs to monitor the safety of approved products that have been commercialized. Even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Delay in obtaining, or failure to obtain, regulatory approval for our product, or obtaining approval but for significantly limited use, would harm our business.
The Biologics Price Competition and Innovation Act of 2009, which was part of the Patient Protection and Affordable Care Act, was signed into law on March 23, 2010 and it introduced an abbreviated approval pathway for biological products shown to be similar to, or interchangeable with, an FDA licensed reference biological product. The biosimilar pathway permits companies to obtain FDA approval of versions of existing biologics that are highly similar to innovator biologics based upon reduced documentation and data requirements deemed sufficient to demonstrate safety and effectiveness than are required for the innovator biologics. Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity and potency, can be shown through analytical studies, animal studies and clinical study or studies The new law provides that a biosimilar application may be submitted as soon as four years after the reference product is first licensed, and that the FDA may not make approval of an application effective until twelve years after the reference product was first licensed. Although drug manufacturers
98
have begun efforts to develop biosimilar drugs for the U.S. market, none have yet been approved by the FDA.
The testing and approval processes require substantial time, effort and financial resources, and each process may take several years to complete. Data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. The FDA may limit the indications for use or place other conditions on any approvals that could restrict the commercial application of the products. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Orphan Drug Designation
The FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the United States, or that affects more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for such a disease or condition will be recovered from sales in the United States for that drug. Orphan drug, or orphan product designation must be requested before submitting an application for marketing approval. Orphan product designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. Orphan product designation can provide opportunities for grant funding towards clinical trial costs, tax advantages and FDA user fee exemptions. In addition, if a product that has an orphan product designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan product exclusivity, which means the FDA may not approve any other application to market the same biological product for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity or a meaningfully different mode of administration. Competitors may receive approval of different drugs or biologics for the indications for which the orphan product has exclusivity. However, if a company with orphan product exclusivity is not able to supply the market, the FDA could allow another company with the same biological product a license to market for said indication.
We filed for orphan designation for Cx601 for the treatment of anal fistulas in the United States in 2012.
Expedited Development and Review Programs
The FDA's expedited review and development programs, one of which is fast track designation, are designed to facilitate the development and review of new drugs and biological products that are intended to treat serious or life-threatening conditions and that demonstrate the potential to address unmet medical needs for the conditions. Fast track designation applies to a combination of the product and the specific indication for which it is being studied. Thus, it is the development program for a specific biological product for a specific indication that receives fast track designation. Unique to a fast track product, FDA may consider for review sections of the marketing application on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the application, the FDA agrees to accept sections of the application and determines that the schedule is acceptable and the sponsor pays any required user fees upon submission of the first section of the application.
99
Any product submitted to the FDA for marketing, including under a fast track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. A product may be eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review in an effort to facilitate the review. Additionally, a product may be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical studies establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical studies. In addition, the FDA requires, as a condition for accelerated approval, pre-submission of all promotional materials prior to the intended date of dissemination or publication, which may, if issues are raised by the FDA, adversely impact the timing of the commercial launch of the product. Fast track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
U.S. patent term restoration and marketing exclusivity Depending upon the timing, duration, and specifics of the FDA approval of the use of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. Patent term restoration can compensate for time lost during product development and the regulatory review process by returning up to five years of patent life for a patent that covers a new product or its use. However, patent term restoration cannot extend the remaining term of a patent beyond a total of fourteen years from the product's approval date. The period of patent term restoration is generally one-half the time between the effective date of an IND (falling after issuance of the patent) and the submission date of a BLA, plus the time between the submission date of the BLA and the approval of that application, provided the sponsor acted with diligence. Only one patent applicable to an approved biological product is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The application for patent term extension is subject to approval by the Patent and Trademark Office in consultation with the FDA.
A patent term extension is only available when the FDA approves a biological product for the first time. We believe that our eASC-based platform and the manner in which it is manufactured and used have not been previously approved by the FDA. However, we cannot be certain that the Patent and Trademark Office and the FDA will agree with our analysis or will grant a patent term extension.
A biological product can obtain pediatric market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued "Written Request" for such a study.
An abbreviated approval pathway for biological products shown to be similar to, or interchangeable with, an FDA-licensed reference biological product was created by the Biologics Price Competition and Innovation Act of 2009, which was part of the Patient Protection and Affordable Care Act signed into law on March 23, 2010. This amendment to the Public Health Service Act attempts to minimize duplicative testing. Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and
100
potency, can be shown through analytical studies, animal studies, and a clinical study or studies. Interchangeability requires that a biological product is biosimilar to the reference biological product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product and, for products administered multiple times, the product and the reference product may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biological product. However, complexities associated with the larger, and often more complex, structure of biological products, as well as the process by which such products are manufactured, pose significant hurdles to implementation that are still being worked out by the FDA.
A reference biological product is granted twelve years of exclusivity from the time of first licensure of the reference product. On April 10, 2013, President Obama released his proposed budget for fiscal year 2014 and proposed to cut this twelve year period of exclusivity down to seven years. He also proposed to prohibit additional periods of exclusivity for brand biological products due to minor changes in product formulation, a practice often referred to as "evergreening." The first biological product submitted under the abbreviated approval pathway that is determined to be interchangeable with the reference product has exclusivity against other biologics submitting under the abbreviated approval pathway for the lesser of (i) one year after the first commercial marketing, (ii) eighteen months after approval if there is no legal challenge, (iii) eighteen months after the resolution in the applicant's favor of a lawsuit challenging the biologic's patents if an application has been submitted, or (iv) forty-two months after the application has been approved if a lawsuit is ongoing within the forty-two-month period.
FDA Post-Approval Requirements
Maintaining substantial compliance with applicable federal, state, local, and foreign statutes and regulations requires the expenditure of substantial time and financial resources. In particular, rigorous and extensive FDA regulation of biological products continues after approval, particularly with respect to cGMP requirements. We will rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of any products that we may commercialize. Manufacturers of our products are required to comply with applicable requirements in the cGMP regulations, including quality control and quality assurance and maintenance of records and documentation. We cannot be certain that we or our present or future suppliers will be able to comply with the cGMP and other FDA regulatory requirements. Other post-approval requirements applicable to biological products include reporting of cGMP deviations that may affect the identity, potency, purity and overall safety of a distributed product, record-keeping requirements, reporting of adverse effects, reporting updated safety and efficacy information, and complying with electronic record and signature requirements. After a BLA is approved, the product also may be subject to official lot release. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer's tests performed on the lot. The FDA also may perform certain confirmatory tests on lots of some products, such as viral vaccines, before releasing the lots for distribution by the manufacturer. In addition, the FDA conducts laboratory research related to the regulatory standards on the safety, purity, potency, and effectiveness of biological products.
Discovery of previously unknown problems or the failure to comply with the applicable regulatory requirements, by us or our suppliers, may result in restrictions on the marketing of a product or withdrawal of the product from the market as well as possible civil or criminal sanctions and adverse publicity. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, clinical hold, warning or untitled letters, product recalls, product seizures, total or partial
101
suspension of production or distribution, injunctions, fines, refusals of government contracts, mandated corrective advertising or communications with doctors, debarment, restitution, disgorgement of profits, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
�� Biological product manufacturers and other entities involved in the manufacture and distribution of approved biological products are required to register their facilities with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP requirements and other laws. In addition, changes to the manufacturing process or facility generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Labeling, Marketing and Promotion
The FDA closely regulates the labeling, marketing and promotion of biological products, including direct-to-consumer advertising, promotional activities involving the internet, and industry-sponsored scientific and educational activities. While doctors are free to prescribe any product approved by the FDA for any use, a company can only make claims relating to safety and efficacy of a biological product that are consistent with FDA approval, and the company is allowed to actively market a biological product only for the particular use and treatment approved by the FDA. In addition, any claims we make for our products in advertising or promotion must be appropriately balanced with important safety information and otherwise be adequately substantiated. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising, injunctions and potential civil and criminal penalties.
Additional U.S. Laws and Regulations
In the United States, the research, manufacturing, distribution, sale and promotion of biological products are potentially subject to regulation by various federal, state and local authorities in addition to the FDA. For example, the Health Insurance Portability and Accountability Act and similar state laws protect the privacy of individually-identifiable information of research subjects and patients. Once biological products are marketed and reimbursed by federal health care programs, such as Medicaid and the 340B Drug Pricing Program, complex price reporting, rebate and discount requirements will apply. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. In addition, sales, marketing, pricing and promotional activities may also be subject to other laws in the United States, such as the federal Physician Payment Sunshine Act, or Open Payments Program, which impose public disclosure requirements with respect to certain financial relationships with certain prescribers and academic medical centers, as well as U.S. anti-bribery laws that regulate the conduct of U.S. businesses occurring outside of the United States, and federal and state consumer protection and unfair competition laws.
Importantly, sales, marketing and scientific or educational grant programs must comply with complex laws and regulations pertaining to healthcare "fraud and abuse," including, but not limited to, the Anti-Kickback Statute, the federal False Claims Act, and other state and federal laws and regulations. The Anti-Kickback Statute makes it illegal for any person, including a biological product manufacturer or a party acting on its behalf to knowingly and willfully solicit, receive, offer, or pay any remuneration that is intended to induce the referral of business, including the purchase or order of an item for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. Violations of this law are punishable by jail sentences, criminal fines, administrative civil money penalties, and exclusion from participation in federal healthcare programs. In addition, many states have adopted laws similar to the Anti-Kickback Statute. Some of these state prohibitions apply to
102
the referral of patients for healthcare services reimbursed by any insurer, not just federal healthcare programs such as Medicare and Medicaid.
The federal False Claims Act prohibits anyone from, among other things, knowingly presenting, or causing to be presented, for payment to federal programs (including Medicare and Medicaid) claims for items or services, including biological products, that are false or fraudulent. Although we would not submit claims directly to payers, manufacturers can be held liable under these laws if they are deemed to "cause" the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers or promoting a product off-label. Penalties for a False Claims Act violation may include three times the actual damages sustained by the government, plus civil penalties of between $5,500 and $11,000 for each separate false claim, the potential for exclusion from participation in federal healthcare programs. In addition, private individuals have the ability to bring actions under the federal False Claims Act under certain circumstances (and can be entitled to receive up to 30% of total recoveries) and certain states have enacted laws modeled after the federal False Claims Act.
Government Regulation in Europe
Medicines can be authorized in the European Union by using either the centralized authorization procedure or national authorization procedures. The EMA is responsible for the centralized authorization procedure.
Centralized Authorization Procedure
The EMA is responsible for the centralized procedure. This procedure results in centralized marketing authorization, the single marketing authorization that is valid across the European Economic Area.
The centralized authorization procedure is required for the following types of products:
- •
- Medicinal products developed by using recombinant DNA technology, the controlled expression of genes coded for biologically active proteins in prokaryotes and eukaryotes, including transformed mammalian cells, or hybridoma or monoclonal antibody methods.
- •
- Advanced therapy medicinal products, such as gene-therapy, somatic cell-therapy or tissue-engineered medicines.
- •
- Medicinal products for human use containing a new active substance that did not receive community marketing authorization when the community authorization procedure was first implemented, for which the therapeutic indication is the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, autoimmune diseases and other immune dysfunctions or viral diseases.
- •
- Officially designated orphan medicines.
The centralized authorization procedure is optional for the following types of products:
- •
- Products containing new active substances for indications other than the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, autoimmune diseases and other immune dysfunctions or viral diseases.
- •
- Products representing significant therapeutic, scientific or technical innovations.
- •
- Products for which the granting of a centralized marketing authorization would be in the interests of EU public health.
103
Applications through the centralized authorization procedure are submitted directly to the EMA. Evaluation by the EMA's relevant scientific committee takes up to 210 days excluding any clock-stops, at the end of which the committee adopts an opinion on whether the medicine should be marketed. This opinion is then transmitted to the European Commission, which has the ultimate authority for granting marketing authorizations in the European Union.
Once centralized marketing authorization has been granted for a medicinal product, the holder of that authorization can make the medicinal product available to patients and healthcare professionals in all European Economic Area countries.
Advanced Therapy Medicinal Products
Advanced therapy medicinal products are new medical products based on genes (gene therapy), cells (cell therapy) or tissues (tissue engineering). These advanced therapies herald revolutionary treatments of a number of diseases and have huge potential for patients and industry.
The lack of an EU-wide regulatory framework in the past led to divergent national approaches which hindered patients' access to products, hampered the growth of this emerging industry and ultimately affected the European Union's competitiveness in a key area of biotechnology.
In 2007, the EU institutions agreed on Regulation (EC) 1394/2007, a regulation on advanced therapies designed to ensure the free movement of advanced therapy products within Europe, to facilitate access of such therapies to the European Economic Area market and to foster the competitiveness of European companies in the field, while guaranteeing the highest level of health protection for patients.
The main elements of the regulation are the following:
- •
- A centralized marketing authorization procedure, to benefit from the pooling of expertise at European level and direct access to the European Economic Area market.
- •
- A new and multidisciplinary expert committee, the Committee for Advanced Therapies, within the EMA, to assess advanced therapy products and follow scientific developments in the field.
- •
- Technical requirements adapted to the particular characteristics of these products.
- •
- Special incentives for small and medium-sized enterprises.
ChondroCelect was the first product to receive centralized authorization as an advanced therapy medicinal product.
Orphan Drug Designation
Applications for designation of orphan medicines are reviewed by the EMA through the Committee for Orphan Medicinal Products. The criteria for orphan designation are as follows:
- 1.
- either (i) the medicinal product is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting no more than five in 10,000 persons in the European Union at the time of submission of the designation applicationor (ii) the medicinal product is intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition, and without incentives it is unlikely that the revenue after marketing of the medicinal product would cover the investment in its developmentand
- 2.
- either (i) no satisfactory method of diagnosis, prevention or treatment of the condition concerned is authorizedor (ii) if such method exists, the medicinal product will be of significant benefit to those affected by the condition.
104
Companies with an orphan designation for a medicinal product benefit from incentives such as the following:
- •
- Protocol assistance (scientific advice for orphan medicines during the product-development phase).
- •
- Direct access to centralized marketing authorization and ten-year marketing exclusivity.
- •
- Financial incentives (fee reductions or exemptions).
- •
- National incentives detailed in an inventory made available by the European Commission.
Since December 2011, orphan medicinal products are eligible for the following level of fee reductions:
- •
- Full (100%) reduction for small- and medium-sized enterprises, or SMEs, for protocol assistance and follow-up, full reduction for non-SME sponsors for pediatric-related assistance and 40% reduction for non-SME sponsors for non-pediatric assistance.
- •
- Full reduction for pre-authorization inspections and 90% reduction for post-authorization inspections for SMEs.
- •
- Full reduction for SMEs for new applications for centralized marketing authorization.
- •
- Full reduction for post-authorization activities including annual fees only to an SME in the first year after granting a marketing authorization.
To qualify for assistance, companies must be established in the European Economic Area, employ fewer than 250 employees and have an annual revenues of not more than 50 million euros or an annual balance sheet total of not more than 43 million euros.
Cx601, our leading therapeutic product candidate, was granted orphan drug designation for the treatment of anal fistulas in 2009.
While the same product can receive centralized marketing authorization for both orphan and "non-orphan" indications, orphan and "non-orphan" indications cannot be covered by the same marketing authorization, and the product would have to go through a second authorization process to receive marketing authorization for the second indication.
Reimbursement
Sales of pharmaceutical products depend, in part, on the extent to which the payments for the products will be covered by third-party payers, such as national health insurance programs, government health programs or private insurance programs or managed health care organizations. Such third-party payers in both the United States and Europe are increasingly challenging the prices charged for medical products and services. Additionally, the containment of health care costs has become a priority, and governments have shown significant interest in implementing cost-containment programs for medicinal products, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures and adoption of more restrictive policies in jurisdictions with existing controls and measures could limit our revenues. If these third-party payers do not consider our products to be cost-effective compared to other available therapies, they may not cover our products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
In some countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing the pricing of medicinal products vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to
105
control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. Countries that have price controls or reimbursement limitations for pharmaceutical products may or may not allow favorable reimbursement and pricing arrangements for any of our products.
Thus, pricing and reimbursement are not harmonized across Europe and are within the exclusive discretion of the national authorities. Reimbursement mechanisms for private and public health insurers vary from country to country and occasionally across different regions of the same country. In public health insurance systems, reimbursement is determined by procedures established by the relevant authority of the EU member state. Inclusion of a product in reimbursement schedules is dependent on many factors, including proof of the product's therapeutic value (efficacy, safety, effectiveness, convenience) and economic value as compared to existing alternatives for a specific disease with a clear medical need. Reimbursement is subject to considerations of cost, use and volume that can vary from country to country.
Historically, products launched in the European Union do not follow price structures of the United States, and generally prices tend to be significantly lower.
Environmental Matters
We use various chemical and biological products to conduct our research and to manufacture our products and are subject to specific environmental and occupational health and safety laws and regulations in the jurisdictions in which we operate. These laws and regulations govern, among other things the generation, storage, handling, use, transportation and disposal of hazardous materials and wastes and the health and safety of our employees. If we violate or fail to comply with these laws and regulations, we could be subject to third-party or administrative claims or fines or other sanctions by regulators. We could also be held responsible for costs and damages arising from any contamination at our past or present facilities or at third party waste disposal sites.
We have established procedures to ensure our compliance with environmental laws and regulations, and such compliance has not had a material impact on our capital expenditures, earnings or competitive position.
Litigation
From time to time, we may be party to litigation that arises in the ordinary course of our business. As of the date of this prospectus, we and our subsidiaries are not involved in any material litigation or legal proceedings, except as disclosed below:
Invalidation of U.S. patent US6777231
On April 1, 2011, Cellerix (the predecessor entity of our subsidiary TiGenix SAU) filed aninter partes re-examination request with the Patent and Trademark Office regarding the patent US6777231, owned by the University of Pittsburgh. The Patent and Trademark Office examiner issued a decision concluding that all ten originally issued and all eighteen newly submitted claims of the patent granted to the University of Pittsburgh were invalid. The University of Pittsburgh then appealed the examiner's decision, but only with respect to two of the newly submitted claims. We cross-appealed the examiner's refusal to reject those two newly submitted claims as anticipated by the prior art. The Patent Trial and Appeal Board issued a decision simultaneously granting both appeals, thus confirming that all claims of the patent were invalid, but with respect to the newly submitted claims, on different grounds than those cited in the decision by the initial examiner. On this basis, the University of Pittsburgh filed a request to reopen prosecution and submitted claim amendments to those newly submitted claims to the Patent and Trademark Office for further consideration in an attempt to overcome the Patent Trial and Appeal
106
Board's institution of a new ground for rejection as anticipated by the prior art. The request to reopen prosecution on the basis of the amended claim has been accepted by the Patent and Trademark Office. We submitted comments to the Patent and Trademark Office arguing that these claim amendments did not overcome the anticipated rejection and as of September 30, 2014, we have not received any decision from the Patent and Trademark Office regarding the amended claims. We do not know when a final decision can be expected, and at this stage, we are not in a position to assess the probable outcome of these proceedings.
Repayment of subsidies
On January 5, 2012, our subsidiary TiGenix SAU lodged an ordinary appeal before the Contentious-Administrative Chamber of the National Appellate Court of Spain (Audiencia Nacional) challenging two decisions taken by the Director General of Technology Transfer and Business Development at the Spanish Ministry of Science and Innovation (the "Administration") on November 16, 2011, which partially revoked and claimed the repayment of two subsidies, granted in 2006 and 2007, respectively.
Both contested subsidies were granted to a consortium of beneficiaries, one of which was TiGenix SAU. TiGenix SAU also acted as representative of the beneficiaries in the consortium.
The Administration claims that (i) the contested subsidies, together with other subsidies granted to TiGenix SAU during the same time period (i.e., 2006 and 2007), exceeded the maximum permitted by law, and has, therefore, requested the reimbursement of the excess amount granted, and that (ii) some of the expenses attributed to the project financed by the contested subsidies had already been financed by other subsidies.
TiGenix SAU contends, among other arguments, that the Administration is not entitled to aggregate all of the subsidies granted to TiGenix SAU (i.e., the contested subsidies and other subsidies granted) for purposes of applying the maximum (i.e., in the particular case of TiGenix SAU, 60% of the eligible cost of the project), because the various subsidies were granted for financing different projects with different purposes and scopes.
The total claim of the Administration, with respect to the full consortium and both contested subsidies, including late payment interest, amounts to 0.9 million euros, and the Administration has claimed the full amount from TiGenix SAU, as the representative of the consortium. However, the claim against TiGenix SAU amounts to 0.3 million euros, and in case the appeal does not succeed, TiGenix SAU would be able to claim the remaining amount from the other members of the consortium.
As an intermediate measure, TiGenix SAU obtained an injunctive decision that the amounts claimed by the Administration do not have to be repaid until a final judgment is received. Instead, TiGenix SAU requested two financial institutions to issue separate guarantees in favor of the Administration guaranteeing the full amount claimed.
On May 20, 2014, TiGenix SAU received the judgment of the Chamber for Contentious Administrative Proceedings of the National High Court of April, 30, 2014. In this judgment, the court partially upheld the claims made by TiGenix SAU throughout the administrative appeal, and declared null the two resolutions on the partial repayment of the two subsidies that were granted in 2006 and 2007, respectively. However, the court also found that there were grounds for a partial repayment of the contested subsidies but ordered the Administration to recalculate the amount of such repayment. It concluded that some of the items included in the Administration's calculations are either wrong or duplicative. Because the court did not calculate the amount to be repaid, the Administration must submit revised calculations of the amounts to be repaid under the contested subsidies. Even though in principle this should have been done within a period of two months, the Administration has not yet
107
submitted such revised calculations as of September 30, 2014. We have no recourse to any further appeals against the judgment of the court.
Insurance
We maintain business liability insurance. In addition, we have obtained liability insurance with respect to our directors and officers, which covers expenses, capped at a certain amount, that our board members and our senior management may incur in connection with their conduct as members of our board of directors or senior management. We also maintain insurance policies with respect to our manufacturing facilities, insurance policies with respect to the clinical trials we conduct as sponsor, group insurance policies for our employees in connection with occupational accidents and a legal expenses insurance policy. We consider our insurance coverage to be adequate in light of the risks we face.
108
The following description is a summary of certain information relating to our share capital, certain provisions of our articles of association and the Belgian Company Code. Because this description is a summary, it may not contain all information important to you. Accordingly, this description is qualified entirely by references to our amended and restated articles of association. Copies of our amended and restated articles of association will be publicly available as an exhibit to the registration statement of which this prospectus forms a part.
The following description includes comparisons of certain provisions of our articles of association and the Belgian Company Code applicable to us and the Delaware General Corporation Law, or the DGCL, the law under which many publicly listed companies in the United States are incorporated. Because such statements are summaries, they do not address all aspects of Belgian law that may be relevant to us and our shareholders or all aspects of Delaware law which may differ from Belgian law, and they are not intended to be a complete discussion of the respective rights.
Share Capital
Share Capital and Shares
Our share capital is represented by ordinary shares without par value. Our share capital is fully paid-up. Our shares are not separated into classes.
As of September 30, 2014 our issued and paid-up share capital amounted to 16,047,662.00 euros represented by 160,476,620 ordinary shares without par value, each representing an identical fraction of our share capital.
As of September 30, 2014, neither we nor any of our subsidiaries held any of our own shares.
Other Outstanding Securities
In addition to the shares already outstanding, we have granted warrants, which upon exercise will lead to an increase in the number of our outstanding shares.
Warrants
A total of 8,696,498 warrants (where each warrant entitles the holder to subscribe to one new share) were outstanding and granted as of September 30, 2014. For further information, see "Management—Stock Options, Warrants and Other Incentive Plans—Our Warrant Plans."
Articles of Association and Other Share Information
Corporate Profile
We are a public limited liability company incorporated in the form of anaamloze vennootschap/société anonyme under Belgian law. We are registered with the Register of Legal Entities (Leuven) under the enterprise number 0471.340.123. Our principal executive and registered offices are located at Romeinse straat 12, box 2, 3001 Leuven, Belgium. Our telephone number is +32 (16) 39 60 60.
We were incorporated in Belgium on February 21, 2000 for an unlimited duration. Our financial year runs from January 1 through December 31.
Corporate Purpose
Our corporate purpose as set forth in Article 3 of our articles of association is as follows: "The company has as its corporate purpose to engage in activities in the field of research and development
109
regarding biological compounds and biomaterials for its own account and for the account of third parties, as well as the industrialisation and commercialisation of the results hereof.
It may engage in all possible commercial, industrial, financial, movable and immovable, transactions, which are, directly or indirectly related to its corporate purpose or which are likely to enhance it. It may, among others, cooperate with, participate in, in any way whatsoever, directly or indirectly, take a stake in each enterprise the corporate purpose of which is similar, analogous or related to its own purpose.
It may mortgage its real estate and may pledge all its other assets, including its entire business, and it may guarantee a bill for all loans, credits and other undertakings, on its own behalf as well as on behalf of third parties, provided that the company itself has an interest thereto."
Board of Directors
Belgian law does not specifically regulate the ability of directors to borrow money from our Company.
Article 523 of the Belgian Company Code provides that if one of our directors directly or indirectly has a personal patrimonial interest that conflicts with a decision or transaction that falls within the powers of our board of directors, the director concerned must inform our other directors before our board of directors makes any decision on such transaction. The statutory auditor must also be notified. The director may neither participate in the deliberation nor vote on the conflicting decision or transaction. A copy of the minutes of the meeting of our board of directors that sets forth the financial impact of the matter on us and justifies the decision of our board of directors must be published in our annual report. The statutory auditors' report on the annual accounts must contain a description of the financial impact on us of each of the decisions of our board of directors where director conflicts arise.
The DGCL generally permits transactions involving a Delaware corporation and an interested director of that corporation if (i) the material facts as to the director's relationship or interest and as to the transaction are disclosed and a majority of disinterested directors consent, (ii) the material facts are disclosed as to the director's relationship or interest and a majority of shares entitled to vote thereon consent or (iii) the transaction is fair to the corporation at the time it is authorized by the board of directors, a committee of the board of directors or the stockholders.
We rely on a provision in the Listing Rules of the NASDAQ Stock Market that allows us to follow Belgian corporate law with respect to certain aspects of corporate governance. This allows us to continue following certain corporate governance practices that differ in significant respects from the corporate governance requirements applicable to U.S. companies listed on the NASDAQ Global Market. In particular, the Listing Rules of the NASDAQ Stock Market require a majority of the directors of a listed U.S. company to be independent, whereas in Belgium, only three directors need to be independent. Nevertheless, our board of directors currently comprises of five independent directors and three non-independent directors. See "Management—Our Board of Directors." The Listing Rules of the NASDAQ Stock Market further require that each of the nominating, compensation and audit committees of a listed U.S. company be comprised entirely of independent directors. However, the Belgian Corporate Governance Code recommends only that a majority of the directors on each of these committees meet the technical requirements for independence under Belgian corporate law. At present, our audit committee is composed of two independent directors and one non-independent director. Our nomination and remuneration committee is composed of three independent directors. Our board of directors will appoint an independent director to replace the current non-independent director within one year of the effective date of the registration statement of which the prospectus forms a part so that all members of our audit committee will be independent as determined under Rule 10A-3 under the Exchange Act and the applicable rules of the NASDAQ Stock Market. Our
110
nomination and remuneration committee is composed of three independent directors. Our board of directors has no plan to change the composition of our nomination and remuneration committee.
Form and Transferability of Our Shares
All of our shares belong to the same class of securities and are in registered form or in dematerialized form.
All of our outstanding shares are fully paid-up and freely transferable, subject to any contractual restrictions.
Currency
Our share capital, which is represented by our outstanding ordinary shares, is denominated in euros.
Changes to Our Share Capital
In principle, changes to our share capital are decided by our shareholders. Our shareholders may at any time at a meeting of shareholders decide to increase or decrease our share capital. Any such resolution of shareholders must satisfy the quorum and majority requirements that apply to an amendment of the articles of association, as described below in"—Description of the Rights and Benefits Attached To Our Shares—Right to Attend and Vote at Our Meeting of Shareholders—Quorum and Majority Requirements." No shareholder is liable to make any further contribution to our share capital other than with respect to shares held by such shareholder that would not be fully paid-up.
Share Capital Increases by Our Board of Directors
Subject to the quorum and majority requirements described below in"—Description of the Rights and Benefits Attached To Our Shares—Right to Attend and Vote at Our Meeting of Shareholders—Quorum and Majority Requirements," our meeting of shareholders may authorize our board of directors, within certain limits, to increase our share capital without any further approval of our shareholders. A capital increase that is authorized in this manner is referred to as authorized capital. This authorization can only be granted for a renewable period of a maximum of five years and may not exceed the amount of the registered share capital at the time of the authorization. On September 8, 2014, our meeting of shareholders renewed the authorization in respect of the authorized capital for an amount equal to the amount of our share capital.
Normally, the authorization of the board of directors to increase our share capital through contributions in cash with cancellation or limitation of the preferential right of the existing shareholders is suspended if we are notified by the Belgian Financial Services and Markets Authority, or the FSMA, of a public takeover bid on the financial instruments of the company. The meeting of shareholders can, however, authorize the board of directors to increase the share capital by issuing shares in an amount of not more than 10% of the existing shares at the time of such a public takeover bid. Such authorization has not been granted to our board of directors.
Preferential Subscription Rights
In the event of a share capital increase for cash through the issuance of new shares, or in the event we issue convertible bonds or warrants, our existing shareholders have a preferential right to subscribe, pro rata, to the new shares, convertible bonds or warrants. These preferential subscription rights are transferable during the subscription period. Our board of directors may decide that preferential subscription rights that were not exercised by any shareholders shall accrue proportionally
111
to the other shareholders that have already exercised their preferential subscription rights and may fix the practical terms for such subscription.
Our shareholders may, at a meeting of shareholders, decide to limit or cancel this preferential subscription right, subject to special reporting requirements. Such decision by the shareholders must satisfy the same quorum and majority requirements as the decision to increase our share capital.
Shareholders may also decide to authorize our board of directors to limit or cancel the preferential subscription right within the framework of the authorized capital, subject to the terms and conditions set forth in the Belgian Company Code. Our board of directors currently has the authority to increase the share capital within the framework of the authorized capital, and the right to limit or cancel the preferential subscription right within the framework of the authorized capital. See also "—Share Capital Increases by Our Board of Directors" above.
Under the DGCL, stockholders of a Delaware corporation have no preemptive rights to subscribe for additional issues of stock or to any security convertible into such stock unless, and to the extent that, such rights are expressly provided for in the corporation's certificate of incorporation.
Purchases and Sales of Our Own Shares
We may only repurchase our own shares pursuant to authorization of our shareholders at a meeting of shareholders taken under the conditions of quorum and majority provided for in the Belgian Company Code. Pursuant to the Belgian Company Code, such a decision requires a quorum of shareholders holding an aggregate of at least 50% of the share capital and approval by a majority of at least 80% of the share capital present or represented. If there is no quorum, a second meeting must be convened. No quorum is required at the second meeting, but the relevant resolution must be approved by a majority of at least 80% of the share capital present or represented.
Within such authorization, we may only repurchase our own shares if the amount that we would use for repurchase is available for distribution. Currently we do not have any funds available for distribution. Currently we do not own any of our own shares.
Under the DGCL, a Delaware corporation may purchase or redeem its own shares, unless the capital of the corporation is impaired or the purchase or redemption would cause an impairment of the capital of the corporation.
Description of the Rights and Benefits Attached To Our Shares
Right to Attend and Vote at Our Meetings of Shareholders
Annual Meeting of Shareholders
Our annual meeting of shareholders will be held on April 20 of each year, at 10 a.m. (Central European Time), at our registered office or at any other place in Belgium mentioned in the notice of the meeting. If this date is a Saturday, Sunday or a public holiday in Belgium, the meeting is held on the next business day in Belgium at the same time.
Special and Extraordinary Meetings of Shareholders
Our board of directors or the statutory auditor (or the liquidators, if appropriate) may, whenever our interests so require, convene a special or extraordinary meeting of shareholders. Such meeting of shareholders must also be convened when one or more shareholders holding at least one-fifth of our share capital so demands.
Under the DGCL, special meetings of the stockholders of a Delaware corporation may be called by such person or persons as may be authorized by the certificate of incorporation or by the bylaws of
112
the corporation, or if not so designated, as determined by the board of directors. Stockholders generally do not have the right to call meetings of stockholders, unless that right is granted in the certificate of incorporation or the bylaws.
Notices Convening Meetings of Shareholders
Notices of our meetings of shareholders contain the agenda of the meeting, indicating the items to be discussed as well as any proposed resolutions that will be submitted at the meeting. One or more shareholders holding at least 3% of our share capital may request for items to be added to the agenda of any convened meeting and submit proposed resolutions in relation to existing agenda items or new items to be added to the agenda, provided that:
- •
- They prove ownership of such shareholding as at the date of their request and record their shares representing such shareholding on the record date.
- •
- The additional items on the agenda and any proposed resolutions have been submitted in writing by these shareholders to the board of directors at the latest on the twenty-second day preceding the day on which the relevant meeting of shareholders is held.
The shareholding must be proven by a certificate evidencing the registration of the relevant shares in the share register of the company or by a certificate issued by the authorized account holder or the clearing organisation certifying the book-entry of the relevant number of dematerialized shares in the name of the relevant shareholder(s).
The notice must be published in the Belgian Official Gazette (Belgisch Staatsblad/Moniteur belge) at least thirty days prior to the meeting of shareholders. In the event a second convening notice is necessary and the date of the second meeting is mentioned in the first convening notice, that period is seventeen days prior to the second meeting of shareholders. The notice must also be published in a national newspaper thirty days prior to the date of the meeting of shareholders, except if the meeting concerned is an annual meeting of shareholders held at the municipality, place, day and hour mentioned in the articles of association and whose agenda is limited to the examination of the annual accounts, the annual report of the board of directors, the annual report of the statutory auditor, the vote on the discharge of the directors and the statutory auditor and the vote on the items referred to in Article 554, paragraphs 3 and 4 of the Belgian Company Code (i.e., in relation to a remuneration report or a severance pay). Notices of all our meetings of shareholders and all related documents, such as specific board and auditor's reports, are also published on our website.
Convening notices must be sent thirty days prior to the meeting of shareholders to the holders of registered shares, holders of registered bonds, holders of registered warrants, holders of registered certificates issued with our cooperation and to our directors and statutory auditor. This communication is made by ordinary letter unless the addressees have individually and expressly accepted in writing to receive the notice by another form of communication, without having to give evidence of the fulfilment of such formality.
Under the DGCL, unless otherwise provided in the certificate of incorporation or by-laws, written notice of any meeting of the stockholders of a Delaware corporation must be given to each stockholder entitled to vote at the meeting not less than ten nor more than sixty days before the date of the meeting and shall specify the place, date, hour and, in the case of a special meeting, the purpose of the meeting.
113
Admission to Meetings
A shareholder is only entitled to participate in and vote at the meeting of shareholders, irrespective of the number of shares he owns on the date of the meeting of shareholders, provided that his shares are recorded in his name at midnight (Central European Time) at the end of the fourteenth day preceding the date of the meeting of shareholders, or the record date:
- •
- In case of registered shares, in our register of registered shares; or
- •
- In case of dematerialized shares, through book-entry in the accounts of an authorized account holder or clearing organisation.
In addition, we (or the person designated by us) must, at the latest on the sixth day preceding the day of the meeting of shareholders, be notified as follows of the intention of the shareholder to participate in the meeting of shareholders:
- •
- In case of registered shares, the shareholder must, at the latest on the above-mentioned date, notify us (or the person designated by us) in writing of his intention to participate in the meeting of shareholders and of the number of shares he intends to participate in the meeting of shareholders with by returning a signed paper form, or, if permitted by the convening notice, by sending an electronic form (signed by means of an electronic signature in accordance with the applicable Belgian law) electronically, to us on the address indicated in the convening notice.
- •
- In case of dematerialized shares, the shareholder must, at the latest on the above-mentioned date, provide us (or the person designated by us), or arrange for us (or the person designated by us) to be provided with, a certificate issued by the authorized account holder or clearing organisation certifying the number of dematerialized shares recorded in the shareholder's accounts on the record date in respect of which the shareholder has indicated his intention to participate in the meeting of shareholders.
Each shareholder has the right to attend a meeting of shareholders and to vote at the meeting of shareholders in person or through a proxy holder. The proxy holder does not need to be a shareholder. A shareholder may only appoint one person as proxy holder for a particular meeting of shareholders, except in cases provided for in the law. Our board of directors may determine the form of the proxies. The appointment of a proxy holder must in any event take place in paper form or electronically, the proxy must be signed by the shareholder (as the case may be, by means of an electronic signature in accordance with the applicable Belgian law) and we must receive the proxy at the latest on the sixth day preceding the day on which the meeting of shareholders is held.
Pursuant to Article 7, section 5 of the Belgian Law of May 2, 2007 on the disclosure of major shareholdings, a transparency declaration has to be made if a proxy holder that is entitled to voting rights above the threshold of 3%, 5%, or any multiple of 5% of the total number of voting rights attached to our outstanding financial instruments on the date of the relevant meeting of shareholders would have the right to exercise the voting rights at his discretion.
Votes
Each shareholder is entitled to one vote per share.
Voting rights can be suspended in relation to shares:
- •
- That were not fully paid up, notwithstanding the request thereto of our board of directors.
- •
- To which more than one person is entitled, except in the event a single representative is appointed for the exercise of the voting right.
114
- •
- That entitle their holder to voting rights above the threshold of 3%, 5%, or any multiple of 5% of the total number of voting rights attached to our outstanding financial instruments on the date of the relevant general meeting of shareholders, except to the extent where the relevant shareholder has notified us and the FSMA at least twenty days prior to the date of the general meeting of shareholders on which he or she wishes to vote of its shareholding reaching or exceeding the thresholds above.
- •
- Of which the voting right was suspended by a competent court or the FSMA.
Quorum and Majority Requirements
Generally, there is no quorum requirement for our meeting of shareholders, except as provided for by law in relation to decisions regarding certain matters. Decisions are made by a simple majority, except where the law provides for a special majority.
Under the DGCL, the certificate of incorporation or bylaws of a Delaware corporation may specify the number of shares required to constitute a quorum but in no event shall a quorum consist of less than one-third of shares entitled to vote at a meeting. In the absence of such specifications, a majority of shares entitled to vote shall constitute a quorum.
Matters involving special legal quorum and majority requirements include, among others, amendment to the articles of association, issues of new shares, convertible bonds or warrants and decisions regarding mergers and demergers, which require at least 50% of the share capital to be present or represented and the affirmative vote of the holders of at least 75% of the votes cast. If the quorum is not reached, a second meeting may be convened at which no quorum requirement applies. The special majority requirement for voting, however, remains applicable.
Any modification of our corporate purpose or legal form requires a quorum of shareholders holding an aggregate of at least 50% of the share capital and approval by a majority of at least 80% of the share capital present or represented. If there is no quorum, a second meeting must be convened. At the second meeting, no quorum is required, but the relevant resolution must be approved by a majority of at least 80% of the share capital present or represented.
Right to Ask Questions at our Meetings of Shareholders
Within the limits of Article 540 of the Belgian Company Code, members of the board of directors and the auditor will answer, during the meeting of shareholders, the questions raised by shareholders. Shareholders can ask questions either during the meeting or in writing, provided that we receive the written questions at the latest on the sixth day preceding the meeting of shareholders.
Dividends
All shares participate in the same manner in our profits, if any. Pursuant to the Belgian Company Code, the shareholders can in principle decide on the distribution of profits with a simple majority vote at the occasion of the annual meeting of shareholders, based on the most recent non-consolidated statutory audited annual accounts, prepared in accordance with the generally accepted accounting principles in Belgium and based on a (non-binding) proposal of the board of directors. The articles of association also authorize our board of directors to declare interim dividends subject to the terms and conditions of the Belgian Company Code.
Dividends can only be distributed if following the declaration and issuance of the dividends the amount of the company's net assets on the date of the closing of the last financial year according to the non-consolidated statutory annual accounts (i.e., the amount of the assets as shown in the balance sheet, decreased with provisions and liabilities, all as prepared in accordance with Belgian accounting rules), decreased with the non-amortized costs of incorporation and expansion and the non-amortized
115
costs for research and development, does not fall below the amount of the paid-up capital (or, if higher, the called capital), increased with the amount of non-distributable reserves. In addition, prior to distributing dividends, at least 5% of our annual net profit under our non-consolidated statutory accounts (prepared in accordance with Belgian accounting rules) must be allotted to a legal reserve, until the legal reserve amounts to 10% of the share capital.
The right to payment of dividends expires five years after the board of directors declared the dividend payable.
Under the DGCL, a Delaware corporation may pay dividends out of its surplus (the excess of net assets over capital), or in case there is no surplus, out of its net profits for either or both of the fiscal year in which the dividend is declared and the preceding fiscal year (provided that the amount of the capital of the corporation is not less than the aggregate amount of the capital represented by the issued and outstanding stock of all classes having a preference upon the distribution of assets). Dividends may be paid in the form of shares, property or cash.
Appointment of Directors
Our articles of association provide that our board of directors shall be composed of at least three directors and a maximum of thirteen members, and that:
- •
- two directors shall be appointed among the candidates proposed by a shareholder owning 20% or more of the shares.
- •
- one director shall be appointed among the candidates proposed by a shareholder owning at least 10% but less than 20% of the shares.
Liquidation Rights
Our Company can only be voluntarily dissolved by a shareholders' resolution passed with a majority of at least 75% of the votes cast at an extraordinary meeting of shareholders where at least 50% of the share capital is present or represented. In the event the required quorum is not present or represented at the first meeting, a second meeting needs to be convened through a new notice. The second meeting of shareholders can validly deliberate and decide regardless of the number of shares present or represented.
Under the DGCL, unless the board of directors approves the proposal to dissolve, dissolution of a Delaware corporation must be approved by stockholders holding 100% of the total voting power of the corporation. Only if the dissolution is initiated by the board of directors may it be approved by a simple majority of the corporation's outstanding shares. The DGCL allows a Delaware corporation to include in its certificate of incorporation a supermajority voting requirement in connection with dissolutions initiated by the board.
In the event of the dissolution and liquidation of our Company, (on a non-consolidated basis) the assets remaining after payment of all debts and liquidation expenses will be distributed to the holders of our shares, each receiving a sum on a pro rata basis.
If, as a result of losses incurred, the ratio of our net assets (on a non-consolidated basis, determined in accordance with Belgian legal and accounting rules) to share capital is less than 50%, our board of directors must convene a general meeting of shareholders within two months of the date upon which our board of directors discovered or should have discovered this undercapitalization. At this meeting of shareholders, our board of directors needs to propose either our dissolution or our continuation, in which case our board of directors must propose measures to address our financial situation. Our board of directors must justify its proposals in a special report to the shareholders. Shareholders representing at least 75% of the votes validly cast at this meeting have the right to
116
dissolve us, provided that at least 50% of our share capital is present or represented at the meeting. In the event the required quorum is not present or represented at the first meeting, a second meeting needs to be convened through a new notice. The second meeting of shareholders can validly deliberate and decide regardless of the number of shares present or represented.
If, as a result of losses incurred, the ratio of our net assets to share capital is less than 25%, the same procedure must be followed, it being understood, however, that in the event shareholders representing 25% of the votes validly cast at the meeting can decide to dissolve the company. If the amount of our net assets has dropped below 61,500 euros (the minimum amount of share capital of a Belgian limited liability company), any interested party is entitled to request the competent court to dissolve us. The court can order our dissolution or grant a grace period during which time we must remedy the situation.
Holders of ordinary shares have no sinking fund, redemption or appraisal rights.
Belgian Legislation
Disclosure of Significant Shareholdings
The Belgian Law of May 2, 2007 on the disclosure of significant shareholdings in issuers whose securities are admitted to trading on a regulated market requires each natural or legal person acquiring or transferring our shares (directly or indirectly, by ownership of ADSs or otherwise) to notify us and the FSMA each time their shareholding crosses (upwards or downwards) a threshold of 5% of the total number of outstanding voting rights. Our articles of association provide that such notification is also required each time, as a result of an acquisition or transfer, a threshold of 3% and a multiple of 5% is crossed.
The same disclosure requirement applies if a person transfers or acquires the direct or indirect control of a corporation or other legal entity that itself owns at least 3% of the voting rights attached to our shares. Similarly, if as a result of events changing the breakdown of voting rights, the percentage of the voting rights reaches, exceeds or falls below any of the above thresholds, disclosure is required even when no acquisition or disposal of shares or ADSs has occurred (e.g., as a result of a capital increase or a capital decrease). Finally, disclosure is also required when persons acting in concert enter into, modify or terminate their agreement resulting in their voting rights reaching, exceeding or falling below any of the above thresholds.
The disclosure statements must be addressed to the FSMA and to us at the latest on the fourth trading day following the day on which the circumstance giving rise to the disclosure occurred. Unless otherwise provided by law, a shareholder shall only be allowed to vote at our meeting of shareholders the number of shares such shareholder validly disclosed at the latest twenty days before such meeting.
In accordance with U.S. federal securities laws, holders of our ordinary shares and holders of ADSs will be required to comply with disclosure requirements relating to their ownership of our securities. Any person that, after acquiring beneficial ownership of our ordinary shares or ADSs, is the beneficial owners of more than 5% of our outstanding ordinary shares or ordinary shares underlying ADSs must file with the SEC a Schedule 13D or Schedule 13G, as applicable, disclosing the information required by such schedules, including the number of our ordinary shares or ordinary shares underlying ADSs that such person has acquired (whether alone or jointly with one or more other persons). In addition, if any material change occurs in the facts set forth in the report filed on Schedule 13D (including a more than 1% increase or decrease in the percentage of the total shares beneficially owned), the beneficial owner must promptly file an amendment disclosing such change.
117
Disclosure of Net Short Positions
Pursuant to the Regulation (EU) No. 236/2012 of the European Parliament and the Council on short selling and certain aspects of credit default swaps, any person that acquires or disposes of a net short position relating to our issued share capital, whether by a transaction in shares or ADSs, or by a transaction creating or relating to any financial instrument where the effect or one of the effects of the transaction is to confer a financial advantage on the person entering into that transaction in the event of a decrease in the price of such shares or ADSs is required to notify the FSMA if, as a result of which acquisition or disposal his net short position reaches, exceeds or falls below 0.2% of our issued share capital and each 0.1% above that. If the net short position reaches 0.5%, and also at every 0.1% above that, the FSMA will disclose the net short position to the public.
Public Takeover Bids
The European Takeover Directive 2004/25/EC of 21 April 2004 has been implemented in Belgium through the law of April 1, 2007 on public takeovers, or the Takeover Law, the Royal Decree of April 27, 2007 on public takeovers and the Royal Decree of April 27, 2007 on squeeze-out bids.
Public takeover bids in Belgium for our shares or other securities giving access to voting rights are subject to supervision by the FSMA. The Takeover Law determines when a bid is deemed to be public in Belgium. Public takeover bids must be extended to all of the voting securities, as well as all other securities giving access to voting rights. Prior to making a bid, a bidder must publish a prospectus that has been approved by the FSMA prior to publication.
The Takeover Law provides that a mandatory bid must be launched on all our shares (and our other securities giving access to voting rights), if a person, as a result of its own acquisition or the acquisition by persons acting in concert with it or by persons acting for its account, directly or indirectly holds more than 30% of our voting securities (directly or through ADSs).
Squeeze-out
Pursuant to Article 513 of the Belgian Company Code and the regulations promulgated thereunder, a person or legal entity, or different persons or legal entities acting alone or in concert, that own together with the company 95% of the securities with voting rights in a public company are entitled to acquire the totality of the securities with voting rights in that company following a squeeze-out offer. The securities that are not voluntarily tendered in response to such an offer are deemed to be automatically transferred to the bidder at the end of the procedure. At the end of the procedure, the company is no longer deemed a public company, unless bonds issued by the company are still spread among the public. The consideration for the securities must be in cash and must represent the fair value (verified by an independent expert) in order to safeguard the interests of the transferring shareholders.
The DGCL provides for stockholder appraisal rights, or the right to demand payment in cash of the judicially determined fair value of the stockholder's shares, in connection with certain mergers and consolidations.
Limitations on the Right to Own Securities
Neither Belgian law nor our articles of association impose any general limitation on the right of non-residents or foreign persons to hold our securities or exercise voting rights on our securities other than those limitations that would generally apply to all shareholders.
118
Exchange Controls and Limitations Affecting Shareholders
There are no Belgian exchange control regulations that impose limitations on our ability to make, or the amount of, cash payments to residents of the United States.
We are in principle under an obligation to report to the National Bank of Belgium certain cross-border payments, transfers of funds, investments and other transactions in accordance with applicable balance-of-payments statistical reporting obligations. Where a cross-border transaction is carried out by a Belgian credit institution on our behalf, the credit institution will in certain circumstances be responsible for the reporting obligations.
119
Our Board of Directors
The following table sets forth certain information with respect to the current members of our board of directors as of September 30, 2014:
Name | Position | Age | Term(1) | ||||||
|---|---|---|---|---|---|---|---|---|---|
Innosté SA, represented by Jean Stéphenne(2) | Chairman / Independent director | 65 | 2016 | ||||||
Eduardo Bravo Fernández de Araoz | Managing Director (executive) / CEO | 49 | 2015 | ||||||
Willy Duron(2) | Independent director | 69 | 2015 | ||||||
Greig Biotechnology Global Consulting, Inc., represented by Russell Greig(3) | Independent director | 62 | 2016 | ||||||
Eduard Enrico Holdener(3) | Independent director | 69 | 2015 | ||||||
R&S Consulting BVBA, represented by Dirk Reyn(3) | Independent director | 53 | 2015 | ||||||
Dirk Büscher(2) | Director (non-executive) | 50 | 2016 | ||||||
José Terencio | Director (non-executive) | 47 | 2016 | ||||||
Notes
- (1)
- The term of the mandates of the directors will expire immediately after the annual meeting of shareholders held in the year set forth next to the director's name.
- (2)
- Member of the audit committee.
- (3)
- Member of the nomination and remuneration committee.
The business address of the members of the board of directors is the same as our business address: Romeinse straat 12, box 2, 3001 Leuven, Belgium.
Our board of directors has determined that five out of eight of the members of the board are independent under Belgian law and the NASDAQ Stock Market listing requirements. There are no family relationships between the members of the board.
The following is the biographical information of the members of our board of directors or in case of legal entities being director, their permanent representatives:
Jean Stéphenne, permanent representative of Innosté SA: Chairman and Independent Director
Jean Stéphenne was, until April 2012, a member of the corporate executive team of GlaxoSmithKline and Chairman and President of GSK Biologicals in Wavre, Belgium, which he built into a world leader in vaccines. He currently serves as Chairman of BESIX, Vesalius Biocapital, Nanocyl, Bepharbel, BioWin and Welbio and as board member of BNP Paribas Fortis, VBO/FEB, Groupe Bruxelles Lambert, OncoDNA, Theravectys and Ronveaux. He served as a board member of Auguria Residential Real Estate Fund, which is currently in liquidation.
Eduardo Bravo: Chief Executive Officer and Managing Director (executive)
Mr. Eduardo Bravo has more than twenty years' experience in the biopharmaceutical industry. He has been Chief Executive Officer of TiGenix since May 2011. Previously, he was Chief Executive Officer of Cellerix. He held several senior management positions at Sanofi-Aventis, including Vice President for Latin America. Prior to his tenure at Sanofi-Aventis, Mr. Bravo spent seven years at SmithKline Beecham in commercial positions. Mr. Bravo holds a degree in Business Administration and an MBA (INSEAD). He is a member of the board of EBE.
120
Willy Duron: Independent Director
Mr. Willy Duron has been an independent member of our board of directors since February 2007. He served as Chairman from September 2007 to September 2012. He started his career at ABB Verzekeringen in 1970, becoming a member of the executive committee in 1984. Mr. Duron holds a MSc degree in mathematics from the University of Ghent and a MSc degree in actuarial sciences from the Katholieke Universiteit Leuven. Currently, he is a member of the board of directors of Ravago, Vanbreda Risk & Benefits, Universitaire Ziekenhuizen Leuven, Universitair Centrum St Jozef Kortenberg, Agfa-Gevaert and Van Lanschot Bankiers. In addition, he serves as Chairman of the board of Windvision. Previously, Mr. Duron was Chief Executive Officer of KBC Groep and KBC Bankverzekeringsholding, Chairman of the board of Argosz, Secura, ADD and W&K, as well as member of the board of directors of KBC Asset Management, Synes, CSOB, Warta, FBD and Amonis.
Russell Greig, permanent representative of Greig Biotechnology Global Consulting, Inc.: Independent Director
Dr. Russell Greig worked at GlaxoSmithKline for nearly three decades. More recently he held the office of President of SR One, GSK's corporate venture group. Prior to joining SR One, he served as President of GSK's Pharmaceuticals International from 2003 to 2008 as well as on the GSK corporate executive team. Currently, Dr. Greig holds the office of Chairman of AM Pharma in the Netherlands, and he is a member of the board of directors of Novavax AB in Sweden, Ablynx in Belgium and BioAlliance Pharma in France. He serves as a member of the BioQuarter Board (Scotland, the United Kingdom), the Technology Development Board at the Memorial Sloan Kettering Cancer Centre (New York, NY) and he is venture partner at Kurma Life Sciences (Paris, France).
Eduard Enrico Holdener: Independent Director
Dr. Eduard Enrico Holdener earned his medical degree from the University of Zurich and held the post of Chief Medical Officer & Global Development Head in the Pharma Division of F. Hoffmann-La Roche Pharmaceutical Ltd until February 2008. In that function, Dr. Holdener was also part of the F.Hoffmann-La Roche AG Pharma and Corporate Executive Committee. He began his career in pharmaceuticals in 1986 after fourteen years of working at various hospitals and academic institutions in Switzerland and the United States. During his tenure at Roche, he was credited with winning approval for an important number of new medicines in different therapeutic areas. Dr. Holdener currently serves as Chairman of NovImmune S.A., director of Parexel International Co and HBM Healthcare Investments and member of the Board of Swiss Cancer Research Foundation. Previously, Dr. Holdener was a board member of Syntaxin Ltd. and Cellerix.
Dirk Reyn, permanent representative of R&S Consulting BVBA: Independent Director
Mr. Dirk Reyn obtained his pharmacist degree at the University of Antwerp, and holds an MBA degree from the Handelshogeschool/Northwestern University of Chicago. He founded Movetis NV in 2006 where he served as Chief Executive Officer and Executive Director until the company was acquired by Shire in 2010. He remained with Shire until May 2013. He is currently CEO of Progress Pharma, an asset development company, and leads two projects. Mr. Reyn served as the Head of the GI Strategic Marketing group for many years and then Vice President New Business Development for Janssen-Cilag in Europe. He has more than twenty-five years of experience, having first joined Johnson & Johnson in 1992 and became the driving force behind the global marketing and commercial strategy for such products as PREPULSID and PARIET and other compounds from the Johnson & Johnson GI portfolio. Prior to joining Johnson & Johnson, he served in a number of commercial positions at Eli Lilly in different countries. Mr. Dirk Reyn holds board positions in Flanders Bio, the local industry association, and in non-pharma companies Horizon Pharmaventures, R&R Promotions and BbyB Chocolates and in different charity organizations.
121
Dirk Büscher: Director (non-executive)
Dr. Dirk Büscher is the Chief Executive Officer of Gri-Cel SA. Previously, he served as Vice President R&D of Cellerix. Dr. Büscher obtained his Ph.D. in biology and immunology from the University of Hannover, Germany, and conducted post-doctoral studies in molecular developmental biology and stem cell research at the Salk Institute in La Jolla, California. He also holds an executive MBA from Instituto de Empresa Business School. Dr. Büscher has served as industry expert on mesenchymal stem cells at the EMA. Additionally, he is a member of the board of directors of VCN Biosciences and Araclon Biotech.
José Terencio: Director (non-executive)
Dr. José Terencio is the Chief Operating Officer of Gri-Cel. Previously, he was Director R&D of Laboratorios Grifols. Before that he was at the R&D center of Grupo Ferrer. With more than nineteen years of experience in the pharmaceutical industry, Dr. Terencio has particular expertise in drug discovery and the development of small molecule therapeutics. Dr. Terencio obtained his Ph.D. in CNS Pharmacology from the University of Barcelona and also holds a PDD from IESE Business School and a Master in Financial and Accounting Management from Universidad Pompeu-Fabra. He is a member of the board of directors of VCN Biosciences.
Our Executive Management
The following table sets forth certain information with respect to the current members of our executive management as of September 30, 2014:
Name | Position | Age | ||||
|---|---|---|---|---|---|---|
Eduardo Bravo Fernández de Araoz | Managing Director and Chief Executive Officer | 49 | ||||
Claudia D'Augusta | Chief Financial Officer | 45 | ||||
Wilfried Dalemans | Chief Technical Officer | 56 | ||||
Marie Paule Richard | Chief Medical Officer | 60 | ||||
The business address of the members of the executive management is the same as our business address: Romeinse straat 12, box 2, 3001 Leuven, Belgium.
There is no potential conflict of interest between the private interests or other duties of the members of the executive management listed above and their duties to us. There is no family relationship between any of our directors and members of our executive management.
Below are the biographies of those members of our executive management who do not also serve on our board of directors:
Claudia D'Augusta: Chief Financial Officer
Dr. Claudia D'Augusta has more than fifteen years of experience in the field of corporate finance. After completing her Bachelor's degree and her Ph.D. in Business Administration at the University of Bocconi, Italy, she joined the corporate finance department of Deloitte & Touche in Milan. She later joined Apax Partners in Madrid, where she participated in the origination and execution of M&A transactions. She was subsequently finance director of Aquanima (Santander Group). Dr. D'Augusta was a member of the board of directors of Sensia S.L. from April 2005 until April 2008.
Wilfried Dalemans: Chief Technical Officer
Dr. Wilfried Dalemans holds a Ph.D. in molecular biology from the Universities of Hasselt and Leuven. Before joining TiGenix, Dr. Dalemans held several senior management positions at GlaxoSmithKline Biologicals, Belgium. As director of regulatory strategy and development, he was
122
responsible for the worldwide registration of GlaxoSmithKline's flu franchise. With this firm, he also served as director of molecular biology and research, responsible for the development of nucleic acid and tuberculosis vaccines, as well as immunology research activities. Prior to joining GlaxoSmithKline, Dr. Dalemans worked at Transgène, France, where he was responsible for the cystic fibrosis research program. Dr. Dalemans also served as a supervisory director of Arcarios B.V. and a director of Arcarios NV.
Marie Paule Richard: Chief Medical Officer
Dr. Marie Paule Richard has spent more than twenty-five years in senior executive positions in pharmaceutical and biotechnology companies. She has held international management positions at Bristol-Myers Squibb, Sanofi, GlaxoSmithKline, Sanofi Pasteur and Crucell. Prior to joining TiGenix, Dr. Richard was Chief Medical Officer at AiCuris GmbH, Germany. She has gained global and extensive experience of clinical development strategy and operations across all phases of development, regulatory affairs and pharmacovigilance, involving numerous anti-infective and immunomodulatory drugs and biologicals, as well as the life-cycle management of marketed products. She has obtained several drug approvals and international license renewals in both Europe and the United States. Dr. Richard holds a medical degree from the University of Nancy, France, and, among other qualifications, a certification in Clinical Immunology.
General Information About Our Directors and Our Executive Management
As of the date of this prospectus and except as set out below, none of the directors or members of our executive management or, in case of corporate entities being director, none of their permanent representatives, for at least the previous five years, meet the following criteria:
- •
- Holds any convictions in relation to fraudulent offenses.
- •
- Held an executive function in the form of a senior manager or a member of the administrative, management or supervisory bodies of any company at the time of or preceding any bankruptcy, receivership or liquidation (except for Jean Stéphenne, who was a member of the board of directors of Auguria Residential Real Estate Fund, which is currently in liquidation).
- •
- Has been subject to any official public incrimination and/or sanction by any statutory or regulatory authority (including any designated professional body).
- •
- Has ever been disqualified by a court from acting as member of the administrative, management or supervisory bodies of any company or from acting in the management or conduct of affairs of any company.
Board Practices
The board of directors can set up specialized committees to analyze specific issues and advise the board of directors on those issues.
The committees are advisory bodies only and the decision-making remains within the collegial responsibility of the board of directors. The board of directors determines the terms of reference of each committee with respect to the organisation, procedures, policies and activities of the committee.
Our board of directors has set up and appointed an audit committee and a nomination and remuneration committee.
123
Committees
Audit Committee
The audit committee consists of three members: Willy Duron (Chairman), Innosté SA, represented by Jean Stéphenne, and Dirk Büscher.
Our board of directors has determined that Willy Duron and Jean Stéphenne, the permanent representative of Innosté SA, are independent under Rule 10A-3 of the Exchange Act and the applicable rules of the NASDAQ Stock Market and that each of Willy Duron and Jean Stéphenne qualifies as an "audit committee financial expert" as defined under the Exchange Act.
Dirk Büscher will be a member of our audit committee in reliance on the transition rules of the NASDAQ Stock Market and the Exchange Act for issuers listing in connection with an initial public offering, which permit a non-independent director to serve on the audit committee for up to twelve months following the initial public offering. We expect our board of directors will appoint an independent director to replace Dirk Büscher within one year of the effective date of the registration statement of which the prospectus forms a part so that all members of our audit committee will be independent as determined under Rule 10A-3 under the Exchange Act and the applicable rules of the NASDAQ Stock Market.
The role of the audit committee is to monitor the financial reporting process, the effectiveness of our internal control and risk management systems, the internal audit (if any) and its effectiveness and the statutory audit of the annual and consolidated accounts, and to review and monitor the independence of the external auditor, in particular regarding the provision of additional services to the company. The committee reports regularly to the board of directors on the exercise of its functions. It informs the board of directors about all areas in which action or improvement is necessary in its opinion and produces recommendations concerning the necessary steps that need to be taken. The audit review and the reporting on that review cover us and our subsidiaries as a whole. The members of the committee shall at all times have full and free access to the Chief Financial Officer and to any other employee to whom they may require access in order to carry out their responsibilities.
The audit committee's duties and responsibilities to carry out its purposes include, among others: our financial reporting, internal controls and risk management, and our internal and external audit process. These tasks are further described in the audit committee's terms of reference, as set out in our corporate governance charter and in Article 526bis of the Belgian Company Code.
Nomination and Remuneration Committee
Our nomination and remuneration committee consists of three members: R&S Consulting, represented by Dirk Reyn (Chairman), Greig Biotechnology Global Consulting, represented by Russell G. Greig and Eduard Enrico Holdener.
Our board of directors has determined that R&S Consulting, represented by Dirk Reyn, Greig Biotechnology Global Consulting, represented by Russell G. Greig and Eduard Enrico Holdener are independent under the applicable rules of the NASDAQ Stock Market.
The role of the nomination and remuneration committee is to make recommendations to the board of directors with regard to the election and re-election of directors and the appointment of the Chief Executive Officer and the executive managers, and to make proposals to the board on the remuneration policy for directors, the Chief Executive Officer and the members of the executive management.
The committee's tasks are further described in the nomination and remuneration committee's terms of reference as set out in the company's corporate governance charter and Article 526quater of the Belgian Company Code.
124
Corporate Governance Practices
Along with our articles of association, we adopted a corporate governance charter in accordance with the recommendations set out in the Belgian Corporate Governance Code issued on March 12, 2009 by the Belgian Corporate Governance Committee. The Belgian Corporate Governance Code is based on a "comply or explain" system: Belgian listed companies are expected to follow the Code, but can deviate from specific provisions and guidelines (though not the principles) provided they disclose the justification for such deviations.
Our board of directors complies with the Belgian Corporate Governance Code, but believes that certain deviations from its provisions are justified in view of our particular situation. These deviations include the following:
- •
- As we have only one executive director (the Chief Executive Officer) and there is no executive committee (directiecomité/comité de direction), in accordance with Article 524bis of the Belgian Company Code, we have not drafted specific terms of reference for the executive management, except for the terms of reference for the Chief Executive Officer (and for a Chief Business Officer, although at present we have not appointed a Chief Business Officer);
- •
- Only our independent directors receive a fixed remuneration in consideration of their membership of our board of directors and their attendance at the meetings of committees of which they are members. In principle, under the Belgian Corporate Governance Code, such directors should not receive any performance-related remuneration in their capacity as director. However, upon the advice of the nomination and remuneration committee, our board of directors is permitted to propose at the meeting of our shareholders a deviation from this principle if, in the board's reasonable judgment, performance-related remuneration would be necessary to attract independent directors with the most relevant experience and expertise. On February 26, 2013, our shareholders' meeting approved such a deviation proposed by the board of directors and authorized the grant of warrants to our independent directors.
Our board of directors reviews its corporate governance charter from time to time and makes such changes as it deems necessary and appropriate.
Additionally, the board of directors adopted written terms of reference for each of the audit committee and the nomination and remuneration committee, which are part of the corporate governance charter.
Differences between Our Corporate Governance Practices and the Listing Rules of the NASDAQ Stock Market
The Listing Rules of the NASDAQ Stock Market include certain accommodations in the corporate governance requirements that allow foreign private issuers, to follow "home country" corporate governance practices in lieu of the otherwise applicable corporate governance standards of the NASDAQ Stock Market. The application of such exceptions requires that we disclose each noncompliance with the NASDAQ Stock Market Listing Rules that we do not follow and describe the Belgian corporate governance practices we do follow in lieu of the relevant NASDAQ Stock Market corporate governance standard.
If and when our ADSs are listed on the NASDAQ Global Market, we intend to continue to follow Belgian corporate governance practices in lieu of the corporate governance requirements of the NASDAQ Stock Market in respect of the following:
- •
- Quorum at Shareholder Meetings. NASDAQ Stock Market Listing Rule 5620(c) requires that for any meeting of shareholders, the quorum must be no less than 331/3% of the outstanding ordinary shares. There is no quorum requirement under Belgian law for our meetings of
125
shareholders, except as provided for by law in relation to decisions regarding certain matters. See "Description of Share Capital—Description of the Rights and Benefits Attached to Our Shares—Right to Attend and Vote at Our Meetings of Shareholders—Quorum and Majority Requirements."
Compensation
Compensation of our Board of Directors
The nomination and remuneration committee recommends the level of remuneration for independent directors, including the chairman of the board, subject to approval by our board of directors and, subsequently, by our shareholders at their annual general meeting.
The directors' remuneration was last determined by the meeting of shareholders of February 26, 2013. Currently, each independent director receives a fixed annual fee of 25,000 euros. The chairman receives 40,000 euros. Each independent director that is also a member of a committee receives an additional fixed annual fee of 5,000 euros, or 7,500 euros for each independent director that is also the chairman of a committee. The fixed annual fees are based on the assumption that we will have six board meetings and two committee meetings each year. Directors receive an additional 2,000 euros for each subsequent meeting. Directors are also entitled to a reimbursement of out-of-pocket expenses actually incurred to participate in our board meetings. The level of reimbursement is expected to remain fixed through 2014 and 2015.
On February 26, 2013, the meeting of shareholders approved the principle that our independent directors may receive performance related remuneration and approved the issue and grant of 54,600 warrants (which were effectively issued by the meeting of shareholders on March 20, 2013) to each of the independent directors.
The nomination and remuneration committee benchmarks the compensation of our independent directors against peer companies to ensure that it is competitive. Remuneration is linked to the time committed to the board of directors and its various committees.
We have not made any loans to the members of the board of directors, except that we pre-pay the Belgian salary taxes payable by our Chief Executive Officer, Eduardo Bravo, on the part of his remuneration that is taxable under Belgian law, until such amounts are refunded (on an annual basis) by the Spanish tax authorities, at which time he repays the relevant amounts.
The following table sets forth the fees received by our independent directors for the performance of their mandate as a board member, not as a member of a board committee, during the year ended December 31, 2013:
Name | Fee (in euros) | |||
|---|---|---|---|---|
Willy Duron | 25,000 | |||
Greig Biotechnology Global Consulting, Inc., represented by Russell Greig | 25,000 | |||
Eduard Enrico Holdener | 25,000 | |||
R&S Consulting BVBA, represented by Dirk Reyn | 25,000 | |||
Innosté SA, represented by Jean Stéphenne | 40,000 | |||
| | | | | |
TOTAL | 140,000 | |||
| | | | | |
| | | | | |
126
The following table sets forth the fees received by our independent directors as members of the audit committee for the performance of their mandate during the year ended December 31, 2013:
Name | Position | Fee (in euros) | ||||
|---|---|---|---|---|---|---|
Willy Duron | Chairman of the audit committee; Independent Director | 7,500 | ||||
Innosté SA, represented by Jean Stéphenne | Member of the audit committee; Chairman of the Board of Directors; Independent Director | 5,000 | ||||
| | | | | | | |
TOTAL | 12,500 | |||||
| | | | | | | |
| | | | | | | |
The following table sets forth the fees received by our independent directors as members of the nomination and remuneration committee for the performance of their mandate during the year ended December 31, 2013:
Name | Position | Fee (in euros) | ||||
|---|---|---|---|---|---|---|
R&S Consulting BVBA, represented by Dirk Reyn | Chairman of the nomination and remuneration committee; Independent Director | 7,500 | ||||
Greig Biotechnology Global Consulting, Inc., represented by Russell Greig | Member of the nomination and remuneration committee; Independent Director | 5,000 | ||||
Eduard Enrico Holdener | Member of the nomination and remuneration committee; Independent Director | 5,000 | ||||
| | | | | | | |
TOTAL | 17,500 | |||||
| | | | | | | |
| | | | | | | |
The table below provides an overview as at September 30, 2014 of the shares, options on shares issued under the equity based incentive plans, or EBIPs, of our subsidiary Tigenix SAU, or EBIP Options, and warrants held by the independent and other non-executive directors:
| | Shares | Options on existing shares under EBIPs(4) | Warrants | Total shares, options on existing shares under EBIPs and warrants | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Number | %(1) | Number | %(1) | Number | %(2) | Number | %(3) | |||||||||||||||||
Dirk Büscher | 172,126 | 0.1073 | % | — | — | — | — | 172,126 | 0.1017 | % | |||||||||||||||
Willy Duron | 6,000 | 0.0037 | % | — | — | 54,600 | 0.6278 | % | 60,600 | 0.0358 | % | ||||||||||||||
Greig Biotechnology Global Consulting, Inc., represented by Russell Greig | — | — | — | — | 54,600 | 0.6278 | % | 54,600 | 0.0323 | % | |||||||||||||||
Eduard Enrico Holdener | — | — | 73,989 | 0.0461 | % | 54,600 | 0.6278 | % | 128,589 | 0.0760 | % | ||||||||||||||
R&S Consulting BVBA, represented by Dirk Reyn(5) | 2,500 | 0.0016 | % | — | — | 54,600 | 0.6278 | % | 57,100 | 0.0338 | % | ||||||||||||||
Innosté SA, represented by Jean Stéphenne | — | — | — | — | 54,600 | 0.6278 | % | 54,600 | 0.0323 | % | |||||||||||||||
José Terencio | — | — | — | — | — | — | — | — | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Total | 180,626 | 0.1126 | % | 73,989 | 0.0461 | % | 273,000 | 3.1392 | % | 527,615 | 0.3119 | % | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Notes:
- (1)
- Calculated on the basis of the total number of issued voting financial instruments on September 30, 2014.
127
- (2)
- Calculated on the basis of the total number of outstanding warrants that can be converted into voting financial instruments on September 30, 2014.
- (3)
- Calculated on the basis of the sum of (i) the total number of issued voting financial instruments on September 30, 2014 and (ii) the total number of outstanding warrants that can be converted into voting financial instruments on September 30, 2014.
- (4)
- This column refers to the number of existing shares that the beneficiary of the EBIP options would receive upon exercise of his options with delivery of 2.96 existing TiGenix shares per EBIP option. In this respect for the EBIP 2008 options it has been assumed that they shall all be exchanged for options on existing TiGenix shares.
- (5)
- R&S Consulting BVBA is controlled by Dirk Reyn, who also controls Horizon Pharmaventures BVBA. Horizon Pharmaventures BVBA holds 1,000 shares (0.0006% of the issued and outstanding shares, calculated on the basis of the total number of issued voting financial instruments on September 30, 2014). Therefore Dirk Reyn controls through R&S Consulting BVBA and Horizon Pharmaventures BVBA in aggregate 3,500 shares and 54,600 warrants (0.0343% of the issued and outstanding voting financial instruments, calculated on the basis of the sum of (i) the total number of issued voting financial instruments on September 30, 2014 and (ii) the total number of outstanding warrants that can be converted into voting financial instruments on September 30, 2014).
Compensation of our Executive Management
The remuneration of our executive management is determined by the board of directors based on the recommendation by the nomination and remuneration committee, who receive a recommendation from our Chief Executive Officer.
The remuneration of the members of the executive management currently consists of different components:
- •
- Fixed remuneration: A basic fixed fee designed to fit responsibilities, relevant experience and competences, in line with market rates for equivalent positions. The amount of fixed remuneration is evaluated and determined by the board of directors every year.
- •
- Short-term variable remuneration: A variable fee in cash dependent on the individual, team or company performances in a certain year. The maximum short-term variable remuneration, or maximum bonus, is set at a percentage of the fixed annual remuneration, and is not spread in time. The maximum bonus of the Chief Executive Officer is set at 75% of his yearly fixed remuneration. The maximum bonus of the Chief Financial Officer and the Chief Technical Officer is set at 45% of their fixed annual remuneration. The individual, team or company objectives that determine the amount of the bonus are determined at the beginning of each year and are set on measurable parameters that can include sales, corporate development transactions and clinical trials (e.g., numbers of patients included in a trial, timing of interim or final results). Each member of executive management has a number of objectives and each objective represents a pre-identified percentage of the overall potential bonus (with all objectives together representing 100% of the potential bonus).
- •
- Long-term incentive plan: Warrants have been granted and may be granted in the future, to the members of the executive management team.
- •
- Other benefits: Members of the executive management who are salaried employees may be entitled to a number of perquisites, which may include participating in a defined contribution pension or retirement scheme, disability insurance, a company car, a mobile telephone, a laptop computer or a lump-sum expense allowance according to general company policy, and other collective benefits (such as hospitalization insurance and meal vouchers).
The members of the executive management do not receive any remuneration based on our overall financial results or any long-term variable remuneration in cash.
128
The basis of remuneration of our executive management team is not expected to change in 2014 or 2015.
Eduardo Bravo, our Chief Executive Officer, has been appointed chief executive officer of our subsidiary, TiGenix SAU, on the basis of his corporate responsibility as a member of the board of directors of TiGenix SAU and as managing director (consejero delegado) governed by applicable Spanish Law on capital companies (Ley de Sociedades de Capital). His relationship with TiGenix SAU can be terminated at any time without notice, subject to the payment, in case TiGenix SAU terminates the relationship, of a termination fee equal to his annual remuneration at such time. An additional termination fee of a maximum of two years' annual remuneration is payable in case the relationship is terminated by TiGenix SAU within one year of a corporate transaction involving the company (such as a merger, sale of shares, sale of assets, etc).
Claudia D'Augusta, our Chief Financial Officer, has an employment contract with our subsidiary, TiGenix SAU. Her employment contract is for an indefinite term and may be terminated at any time, subject to a three month notice period and, in case TiGenix SAU terminates the agreement, she is entitled to a severance payment of a minimum of nine month's remuneration. An additional severance payment of a maximum of a year's annual remuneration is payable in certain cases, including unfair or collective dismissal by TiGenix SAU, and termination of the employment agreement by Claudia D'Augusta following certain geographical transfer or substantial modifications to the working conditions made by TiGenix SAU.
Wilfried Dalemans, our Chief Technical Officer, has an employment contract with the company. His employment contract is for an indefinite term and may be terminated at any time by us, subject to a notice period and a severance payment in accordance with applicable law.
Marie Paule Richard, our Chief Medical Officer, has an employment contract with our subsidiary, TiGenix SAU. Her employment contract is for an indefinite term and may be terminated at any time by us, subject to either a three month notice period, or a compensation equal to three months fixed salary, or a combination of both.
The following table sets forth information concerning the compensation earned during the year ended December 31, 2013, by Eduardo Bravo as our Chief Executive Officer:
| | Compensation (in euros) | |||
|---|---|---|---|---|
Fixed remuneration (gross) | 322,000 | |||
Variable remuneration (short term) | 111,573 | |||
Pension/Life | 20,516 | |||
Other benefits | 20,837 | |||
| | | | | |
TOTAL | 474,926 | |||
| | | | | |
| | | | | |
In addition, he was granted (and accepted) 523,740 warrants under the December 16, 2013 warrant plan. The exercise price of the warrants is 0.50 euro. Eduardo Bravo did not exercise any warrants, options on shares or rights to acquire shares in 2013 and none of his warrants, options on shares or rights to acquire shares expired in 2013. For a description of the main characteristics of the December 16, 2013 warrant plan see "—Stock Options, Warrants and Other Incentive Plans."
The following table sets forth information concerning the compensation earned during the year ended December 31, 2013 by the other members of the executive management (including Gil Beyen BVBA, represented by Gil Beyen, who currently is not a member of executive management but who
129
was a member of executive management until May 13, 2013 and excluding Marie Paule Richard who only joined our Company on September 1, 2014):
| | Compensation (in euros) | |||
|---|---|---|---|---|
Fixed remuneration (gross) | 441,217 | |||
Variable remuneration (short term) | 75,563 | |||
Pension/Life | 30,145 | |||
Other benefits | 37,101 | |||
| | | | | |
TOTAL | 584,025 | |||
| | | | | |
| | | | | |
In addition, in 2013, the other members of our executive management were granted (and accepted) the following warrants under the March 20, 2013 and the December 16, 2013 warrant plans. The exercise price of the warrants is 1.00 euro (under the March 20, 2013 warrant plan) and 0.46 euro (under the December 16, 2013 warrant plan). The following table sets forth the number of warrants granted under such plans to the other members of our executive management:
| | Number of warrants under the March 20, 2013 warrant plan | Number of warrants under the December 16, 2013 warrant plan | |||||
|---|---|---|---|---|---|---|---|
Gil Beyen BVBA, represented by Gil Beyen(1) | 160,000 | — | |||||
Claudia D'Augusta | — | 325,080 | |||||
Wilfried Dalemans | — | 270,900 | (2) | ||||
Notes:
- (1)
- Gil Beyen BVBA was a member of the executive management until May 13, 2013.
- (2)
- Warrants not yet accepted as per December 31, 2013; they were accepted on February 28, 2014.
Claudia D'Augusta and Wilfried Dalemans did not exercise any warrants, options on shares or rights to acquire shares in 2013 and none of their warrants, options on shares or rights to acquire shares expired in 2013.
The table below provides an overview, as at September 30, 2014, of the shares, EBIP options and warrants held by the executive management:
| | Shares | Options on existing shares under EBIPs(4) | Warrants | Total shares, options on existing shares under EBIPs and warrants | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Number | %(1) | Number | %(1) | Number | %(2) | Number | %(3) | |||||||||||||||||
Eduardo Bravo, CEO | 150,263 | 0.09 | % | 782,771 | 0.49 | % | 1,883,740 | 21.66 | % | 2,816,774 | 1.67 | % | |||||||||||||
Claudia D'Augusta, CFO | 127,682 | 0.08 | % | 206,492 | 0.13 | % | 805,080 | 9.26 | % | 1,139,254 | 0.67 | % | |||||||||||||
Wilfried Dalemans, CTO | — | — | — | — | 815,900 | 9.38 | % | 815,900 | 0.48 | % | |||||||||||||||
Marie Paule Richard | — | — | — | — | — | — | — | — | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Total | 277,945 | 0.17 | % | 989,263 | 0.62 | % | 3,504,720 | 40.30 | % | 4,771,928 | 2.82 | % | |||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Notes:
- (1)
- Calculated on the basis of the total number of issued voting financial instruments on September 30, 2014.
130
- (2)
- Calculated on the basis of the total number of outstanding warrants that can be converted into voting financial instruments on September 30, 2014.
- (3)
- Calculated on the basis of the sum of (i) the total number of issued voting financial instruments on September 30, 2014 and (ii) the total number of outstanding warrants that can be converted into voting financial instruments on September 30, 2014.
- (4)
- This column refers to the number of existing shares that the beneficiary of the EBIP options would receive upon exercise of his options with delivery of 2.96 existing TiGenix shares per EBIP option. In this respect for the EBIP 2008 options it has been assumed that they shall all be exchanged for options on existing TiGenix shares.
Stock Options, Warrants and Other Incentive Plans
We created several warrants within the context of stock option plans as well as equity incentive plans for our directors, managers, employees as well as other external consultants and collaborators such as scientific advisory board members and clinical advisors.
Our Warrant Plans
We have established a number of warrants plans, under which we have granted warrants free of charge to the recipients. Each warrant entitles its holder to subscribe to one of our ordinary shares at a subscription price determined by the board of directors, within the limits decided upon at the occasion of their issuance.
Generally, unless the board of directors at the time of the grant of the warrant determines a higher exercise price, the exercise price of a warrant will be equal to the lower of the following prices:
- •
- The last closing price of our shares on Euronext Brussels prior to the date on which the warrant is offered.
- •
- The average closing price of our shares on Euronext Brussels over the thirty-day period preceding the date on which the warrant is offered.
For beneficiaries of the warrant plan that are not employees of our group, the exercise price cannot be lower than the average closing price of our shares on Euronext Brussels over the thirty-day period preceding the date of issuance of the warrants.
For our warrants issued in July 2012 and in March 2013, however, our board determined a higher exercise price of 1.00 euro per warrant.
For our December 2013 warrant plan, under which warrants were issued and granted on December 16, 2013, the exercise price was determined as follows:
- •
- For all employees, the exercise price was set at 0.46 euro, the closing price of our ordinary shares on December 13, 2013, the last closing price prior to the grant of the warrants on December 16, 2013, which was lower than the 30-day average price.
- •
- For our CEO, Eduardo Bravo, who is not an employee of TiGenix SAU, the exercise price was set at 0.50 euro, the average closing price of our ordinary shares during 30 calendar days prior to the issuance of the warrants on December 16, 2013.
Since 2007, we have issued 10,777,302 warrants in aggregate (subject to the warrants being granted to and accepted by the beneficiaries) of which 800,000 warrants were issued on February 26, 2007; 400,000 warrants on March 20, 2008; 500,000 warrants on June 19, 2009; 500,000 warrants on March 12, 2010; 4,000,000 warrants on July 6, 2012; 777,000 warrants on March 20, 2013; 1,806,000 warrants on December 16, 2013 and 1,994,302 warrants on April 22, 2014.
131
Of these 10,777,302 warrants:
- •
- 734,800 warrants expired because they were never granted.
- •
- 379,250 warrants expired because they were never accepted by their beneficiaries.
- •
- 964,254 warrants lapsed due to their beneficiaries no longer being employed by the Company.
- •
- 2,500 warrants have been exercised.
As a result, as of September 30, 2014, there are 8,696,498 warrants outstanding, which represent approximately 5.14% of the total number of all our issued and outstanding voting financial instruments.
The warrants issued on February 26, 2007, March 20, 2008, June 19, 2009, March 12, 2010, July 6, 2012 and December 16, 2013 have a term of ten years. The warrants issued on March 20, 2013 and April 22, 2014 have a term of five years. Upon expiration of the ten or five year term, the warrants become null and void.
132
The table below gives an overview (as of September 30, 2014) of the 8,696,498 outstanding warrants:
Issue date | Term | Number of warrants issued | Number of warrants granted | Exercise price (in euros) | Number of warrants no longer exercisable | Number of warrants outstanding | Exercise periods vested warrants | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
February 26, 2007 | From February 26, 2007 to February 25, 2017 | 800,000 | 681,500 | 6.75 (March 24, 2007 grant) 5.23 (September 17, 2007 grant) | 290,187 | 509,813 | From May 1 to 31, and from November 1 to 30. | |||||||||||
March 20, 2008 | From March 20, 2008 to March 19, 2018 | 400,000 | 400,000 | 4.05 for employees and 4.41 for other individuals (March 20, 2008 grant) | 113,500 | 286,500 | From | |||||||||||
June 19, 2009 | From June 19, 2009 to June 18, 2019 | 500,000 | 232,200 | 3.95 | 360,200 | 139,800 | From | |||||||||||
March 12, 2010 | From March 12, 2010 to March 11, 2020 | 500,000 | 495,500 | 3.62 (March 12, 2010 grant) | 247,000 | 253,000 | From | |||||||||||
July 6, 2012 | From July 6, 2012 to July 5, 2022 | 4,000,000 | 4,000,000 | 1.00 | 644,647 | 3,355,353 | From | |||||||||||
March 20, 2013 | From March 20, 2013 to March 19, 2018 | 777,000 | 433,000 | 1.00 | 344,000 | 433,000 | From | |||||||||||
December 16, 2013 | From December 16, 2013 to December 15, 2023 | 1,806,000 | 1,806,000 | 0.46 for employees and 0.50 for other individuals | 81,270 | 1,724,730 | From | |||||||||||
April 22, 2014 | From April 22, 2014 to April 21, 2019 | 1,994,302 | 1,994,302 | 0.75 | — | 1,994,302 | Any time | |||||||||||
| | | | | | | | | | | | | | | | | | | |
TOTAL | 10,777,302 | 8,696,498 | ||||||||||||||||
| | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | |
TiGenix SAU Equity Based Incentive Plans
Summary
On May 3, 2011, we acquired all the shares of Cellerix, a Spanish biotechnology company, which we renamed TiGenix SAU. Cellerix had two Equity Based Incentive Plans, or EBIPs, in place. The contribution in kind, or the Contribution, by the former Cellerix shareholders of all their Cellerix shares into our Company triggered certain consequences which affected both EBIPs.
133
In May 2008, Cellerix had entered into a management agreement with CX EBIP Agreement, a Spanish limited liability company, which we refer to as the EBIP Agreement. The EBIP Agreement was amended and restated in November 2009 and was further amended on May 3, 2011 simultaneously with the completion of the Contribution to establish the procedure for exercise of the EBIP options. In the framework of the Contribution and in accordance with the terms of the EBIP Agreement, CX EBIP Agreement contributed its 642,226 shares of our subsidiary, TiGenix SAU, into TiGenix and received 1,905,144 of our shares in return, which it can only transfer to the beneficiaries of the EBIPs who exercise their options. Pursuant to the agreements reached in relation to the Contribution, the underlying securities of the options are no longer the shares of our subsidiary, TiGenix SAU, but the shares of TiGenix. Therefore, upon the exercise of its options under the EBIPs, a beneficiary will be entitled to receive a number of our shares corresponding to approximately 2.96 shares per option, rounded down to the nearest integer, under any of the EBIPs.
An overview of the EBIPs is provided below.
EBIP 2008
An EBIP for the directors, managers and employees of Cellerix was approved at Cellerix's annual general meeting of shareholders held on November 22, 2007, the conditions of which were definitively approved on May 20, 2008, which we refer to as the EBIP 2008, which was subsequently modified on October 15, 2010.
Options under the EBIP 2008 were granted to employees, executives and independent members of the board of directors of Cellerix prior to the Contribution.
As a result of the Contribution, all EBIP 2008 options have vested except for 32,832 options of employees who terminated their employment relationship with the Company before the Contribution and that were not re-allocated. Beneficiaries must exercise their options prior to August 6, 2015.
The exercise prices of the EBIP 2008 are set at 11 euros, 7 euros or 5.291 euros depending on the date of grant and beneficiary.
The completion of the Contribution gave the beneficiaries the right to opt between:
- •
- Exercising all their options at once to receive either TiGenix SAU shares or our shares in exchange at the relevant exercise price. This right had to be exercised within 60 days following the Contribution date. No beneficiary exercised this right.
- •
- Receiving new options over existing TiGenix shares. As the options retain the same exchange ratio as the Contribution (i.e., 2.96 TiGenix NV shares per TiGenix SAU share contributed to TiGenix), beneficiaries of the EBIP 2008 have the right to receive 2.96 TiGenix shares for each EBIP 2008 option at the time of exercise.
In addition, under the EBIP 2008, the board of directors of our subsidiary, TiGenix SAU, have the option to offer to beneficiaries other exercise options. However, as of the date of this prospectus, the board of directors of TiGenix SAU has not offered beneficiaries of the plan any other exercise alternatives.
EBIP 2010
On October 15, 2010, Cellerix's annual general meeting of shareholders approved an EBIP for the senior management of Cellerix, which we refer to as the EBIP 2010.
The EBIP set the normal exercise price of the options at 5.291 euros per share. However, as a result of the Contribution, the exercise price for all EBIP 2010 options was reduced to 0.013 euros.
134
Cellerix granted 221,508 options under the EBIP 2010. All EBIP 2010 options vested as a result of the Contribution.
Beneficiaries of the plan are required to exercise their options before September 30, 2016. Pursuant to the terms of the EBIP 2010, the board of directors of our subsidiary, TiGenix SAU, opted to exchange all existing options for new options over existing TiGenix shares. As the options retain the same exchange ratio as the Contribution (i.e., 2.96 TiGenix NV shares per TiGenix SAU share contributed to TiGenix), beneficiaries of the EBIP 2010 have the right to receive 2.96 TiGenix shares for each EBIP 2010 option at the time of exercise.
Common characteristics of the TiGenix SAU EBIPs
All options were granted free of charge.
Both EBIPs provide that any options may be ordinarily exercised after each quarterly, half-yearly or annual results announcement.
Under both EBIPs, prior to the Contribution, the options related to existing shares in Cellerix that were held by CX EBIP Agreement. To this effect:
- •
- In June 2008, Cellerix issued 415,700 new shares to CX EBIP Agreement at an issuance price of 0.013 euros per Cellerix share.
- •
- In September 2008, Cellerix issued 37,850 new shares to CX EBIP Agreement at an issuance price of 0.013 euros per Cellerix share.
- •
- In November 2009, Cellerix issued 61,479 new shares to CX EBIP Agreement at an issuance price of 0.013 euros per Cellerix share.
- •
- In May 2010, Cellerix issued 49,446 new shares to CX EBIP Agreement at an issuance price of 0.013 euros per Cellerix share.
- •
- In October 2010, Cellerix issued 77,751 new shares to CX EBIP Agreement at an issuance price of 0.013 euros per Cellerix share.
All Cellerix shares held by CX EBIP Agreement have been exchanged for TiGenix shares as described above.
EBIP options outstanding as of September 30, 2014
In 2013, a total of 31,011 EBIP 2010 options were exercised, as a result of which CX EBIP Agreement transferred 91,992 TiGenix shares to the exercising beneficiaries.
As of September 30, 2014, a total of 611,215 EBIP options (i.e., 420,718 EBIP 2008 options and 190,497 EBIP 2010 options), corresponding to 1,813,152 of our shares, were outstanding.
135
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Article 524 of the Belgian Company Code provides for a special procedure that applies to intra-group or related party transactions with affiliates. The procedure applies to decisions or transactions between us and our affiliates that are not one of our subsidiaries. Prior to any such decision or transaction, our board of directors must appoint a special committee consisting of three independent directors, assisted by one or more independent experts. This committee must assess the business advantages and disadvantages of the decision or transaction, quantify its financial consequences and determine whether the decision or transaction causes a disadvantage to us that is manifestly illegitimate in view of our policy. If the committee determines that the decision or transaction is not illegitimate but will prejudice us, it must analyze the advantages and disadvantages of such decision or transaction and set out such considerations as part of its advice. Our board of directors must then make a decision, taking into account the opinion of the committee. Any deviation from the committee's advice must be justified. Directors who have a conflict of interest are not entitled to participate in the deliberation and vote. The committee's advice and the decision of the board of directors must be notified to our statutory auditor, who must render a separate opinion. The conclusion of the committee, an excerpt from the minutes of the board of directors and the opinion by the statutory auditor must be included in our annual report. This procedure does not apply to decisions or transactions in the ordinary course of business under customary market conditions and security documents, or to transactions or decisions with a value of less than 1% of our net assets as shown in our consolidated annual accounts.
Transactions with Related Companies
From time to time in the ordinary course of our business we may contract for services from companies in which certain of our executive officers or directors may serve as director or advisor. The cost of these services is negotiated on an arm's length basis and none of these arrangements is material to us.
136
The following table sets forth information relating to beneficial ownership of our ordinary shares, as of September 30, 2014, for each person who is known by us to hold beneficially more than 3% of our outstanding ordinary shares, each member of our board of directors, each member of our executive management, and all members of our board of directors and our executive management as a group.
The business address of the members of our board of directors and the members of our executive management is the same as our business address: Romeinse straat 12, box 2, 3001 Leuven, Belgium.
For purposes of the table below, the percentage ownership calculations for beneficial ownership prior to this offering are based on 160,476,620 ordinary shares outstanding as of September 30, 2014.
Beneficial ownership is determined in accordance with the rules and regulations of the SEC. In computing the number of shares beneficially owned by a person and the percentage ownership of that person, shares that the person has the right to acquire within sixty days of September 30, 2014, including through the exercise of any EBIP Option or warrant, have been included. Shares that can be acquired through the exercise of any warrant, however, are not included in the computation of the percentage ownership of any other person.
| | Ordinary shares beneficially owned prior to the offering | Ordinary shares beneficially owned after the offering | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Name of beneficial owner | Number | Percentage | Number | Percentage | |||||||||
3% or greater shareholders | |||||||||||||
Gri-Cel SA | 34,188,034 | (1) | 21.30 | % | |||||||||
Novartis Bioventures Ltd. | 5,534,905 | (1) | 3.45 | % | |||||||||
Roche Finanz AG | 5,261,446 | (1) | 3.28 | % | |||||||||
Members of our board of directors and our executive management | |||||||||||||
Dirk Büscher (director) | 172,126 | 0.11 | % | ||||||||||
Willy Duron (director) | 34,817 | (2) | 0.02 | % | |||||||||
Greig Biotechnology Global Consulting, Inc., represented by Russell G. Greig (director) | 28,817 | (2) | 0.02 | % | |||||||||
Eduard Holdener (director) | 102,806 | (3) | 0.06 | % | |||||||||
R&S Consulting BVBA, represented by Dirk Reyn (director) | 32,317 | (4) | 0.02 | % | |||||||||
Innosté SA, represented by Jean Stéphenne (director) | 28,817 | (2) | 0.02 | % | |||||||||
Jose Terencio (director) | — | — | |||||||||||
Eduardo Bravo (director, CEO) | 2,043,186 | (5) | 1.26 | % | |||||||||
Claudia D'Augusta (CFO) | 740,015 | (6) | 0.46 | % | |||||||||
Wilfried Dalemans (CTO) | 483,201 | (7) | 0.30 | % | |||||||||
Marie Paule Richard (CMO) | — | — | |||||||||||
All members of our board of directors and our executive management as a group (11 persons) | 3,666,102 | 2.27 | % | ||||||||||
- (1)
- At the time of the most recent transparency notification.
- (2)
- This number includes 28,817 warrants that will have vested as of November 30, 2014.
- (3)
- This number includes 73,989 EBIP Options and 28,817 warrants that will have vested as of November 30, 2014.
- (4)
- This number is the sum of (a) the 2,500 shares held by our director R&S Consulting BVBA, represented (and controlled) by Dirk Reyn, (b) the 1,000 shares held by Horizon Pharmaventures
137
BVBA, a company also controlled by Dirk Reyn, and (c) 28,817 warrants that will have vested as of November 30, 2014 held by our director R&S Consulting BVBA.
- (5)
- This number includes 782,771 EBIP Options and 1,110,152 warrants that will have vested as of November 30, 2014.
- (6)
- This number includes 206,492 EBIP Options and 405,841 warrants that will have vested as of November 30, 2014.
- (7)
- This number includes 483,201 warrants that will have vested as of November 30, 2014.
138
DESCRIPTION OF AMERICAN DEPOSITARY SHARES
American Depositary Shares
Deutsche Bank Trust Company Americas, as depositary, will register and deliver the ADSs. Each ADS will represent ownership of ordinary shares deposited with Deutsche Bank AG, Amsterdam Branch, as custodian for the depositary. Each ADS will also represent ownership of any other securities, cash or other property which may be held by the depositary. The depositary's principal office at which the ADSs will be administered is located at 60 Wall Street, New York, NY 10005, USA. The principal executive office of the depositary is located at 60 Wall Street, New York, NY 10005, USA.
The Direct Registration System, or DRS, is a system administered by The Depository Trust Company, or DTC, pursuant to which the depositary may register the ownership of uncertificated ADSs, which ownership shall be evidenced by periodic statements issued by the depositary to the ADS holders entitled thereto.
We will not treat ADS holders as our shareholders and accordingly, you, as an ADS holder, will not have shareholder rights. Belgian law governs shareholder rights. The depositary will be the holder of the ordinary shares underlying your ADSs. As a holder of ADSs, you will have ADS holder rights. A deposit agreement among us, the depositary and you, as an ADS holder, and the beneficial owners of ADSs sets out ADS holder rights as well as the rights and obligations of the depositary. The laws of the State of New York govern the deposit agreement and the ADSs.
The following is a summary of the material provisions of the deposit agreement. For more complete information, you should read the entire deposit agreement and the form of American Depositary Receipt. For directions on how to obtain copies of those documents, see "Where You Can Find More Information."
Holding the ADSs
How will you hold your ADSs?
You may hold ADSs either (i) directly (a) by having an American Depositary Receipt, or ADR, which is a certificate evidencing a specific number of ADSs, registered in your name, or (b) by holding ADSs in DRS or (ii) indirectly through your broker or an other financial institution. If you hold ADSs directly, you are an ADS holder. This description assumes you hold your ADSs directly. ADSs will be issued through DRS, unless you specifically request certificated ADRs. If you hold the ADSs indirectly, you must rely on the procedures of your broker or that other financial institution to assert the rights of ADS holders described in this section. You should consult with your broker or that other financial institution to find out what those procedures are.
Dividends and Other Distributions
How will you receive dividends and other distributions on the shares?
The depositary has agreed to pay to you the cash dividends or other distributions it or the custodian receives on ordinary shares or other deposited securities, after deducting its fees and expenses. You will receive these distributions in proportion to the number of ordinary shares your ADSs represent as of the record date (which will be as close as practicable to the record date for our ordinary shares) set by the depositary with respect to the ADSs.
- •
- Cash. The depositary will convert any cash dividend or other cash distribution we pay on the ordinary shares or any net proceeds from the sale of any ordinary shares, rights, securities or other entitlements into U.S. dollars if it can do so on a practicable basis, and can transfer the U.S. dollars to the United States. If that is not practical or lawful or if any government approval is needed and cannot be obtained, the deposit agreement allows the depositary either to
139
- •
- Shares. The depositary may distribute additional ADSs representing any ordinary shares we distribute as a dividend or free distribution. The depositary will only distribute whole ADSs. It will sell any ordinary shares that would require it to deliver a fractional ADS and distribute the net proceeds in the same way as it does cash. If the depositary does not distribute additional ADSs, the outstanding ADSs will, to the extent permissible by law, also represent the new ordinary shares. The depositary may also sell all or a portion of the ordinary shares that is has not distributed, and distribute the net proceeds in the same way as it does cash. Additionally, the depositary may sell a portion of the distributed ordinary shares sufficient to pay its fees and expenses, and any taxes and governmental charges, in connection with that distribution.
- •
- Elective Distributions in Cash or Shares. If we offer holders of our ordinary shares the option to receive dividends in either cash or shares, the depositary, after consultation with us and having received timely notice as described in the deposit agreement of such elective distribution by us, has discretion to determine to what extent such elective distribution will be made available to you as a holder of the ADSs. We must first instruct the depositary to make such elective distribution available to you and furnish it with satisfactory evidence that it is legal to do so. The depositary could decide it is not legal or reasonably practical to make such elective distribution available to you. In such case, the depositary shall, on the basis of the same determination as is made in respect of the ordinary shares for which no election is made, distribute either cash, in the same way as it does in a cash distribution, or additional ADSs representing ordinary shares, in the same way as it does in a share distribution. The depositary is not obligated to make available to you a method to receive the elective dividend in shares rather than in ADSs. You may not be given the opportunity to receive elective distributions on the same terms and conditions as the holders of our ordinary shares.
- •
- Rights to Subscribe for Additional Shares. If we offer holders of our ordinary shares any rights to subscribe for additional shares or any other rights, the depositary may after consultation with us and having received timely notice as described in the deposit agreement of such distribution by us, make these rights available to you. We must first instruct the depositary to make such rights available to you and furnish the depositary with satisfactory evidence that it is legal to do so. If the depositary decides it is not legal and reasonably practicable to make the rights available, or if rights have been made available but have not been exercised and appear to be about to lapse, the depositary may, if it determines it is lawful and reasonably practicable to do so, endeavour to sell the rights and distribute the net proceeds, in the same way as it does cash. The depositary will allow rights that are not distributed or sold to lapse. In that case, you will receive no value for them.
distribute the foreign currency to the ADS holders or to hold the foreign currency for the account of the ADS holders, in which case it will not invest the foreign currency and it will not be liable for any interest. Before making a distribution, any taxes or other governmental charges, together with fees and expenses of the depositary, that must be paid, will be deducted. See"—Fees and Expenses" and "—Payment of Taxes." It will distribute only whole U.S. dollars and cents and will round fractional cents to the nearest whole cent.If the exchange rates fluctuate during a time when the depositary cannot convert the foreign currency, you may lose some or all of the value of the distribution.
If the depositary makes rights available to you, it will exercise the rights and purchase the shares on your behalf. The depositary will then deposit the shares and deliver ADSs to you. It will only exercise rights if you pay it the exercise price and any other charges the rights require you to pay. The depositary will sell shares that would require it to deliver a fractional ADS and distribute the net proceeds in the same way as it does with cash.
140
- •
- Other Distributions. Subject to receipt of timely notice, as described in the deposit agreement, from us with the request to make any such distribution available to you, and provided the depositary has determined such distribution is lawful and reasonably practicable and in accordance with the terms of the deposit agreement, the depositary will send to you anything else we distribute on deposited securities by any means it thinks is practicable. If the depositary cannot make a distribution in this way, it may endeavor to sell what we distributed and distribute the net proceeds in the same way as it does cash. If the depositary is unable to sell what we distribute, it may dispose of such property in any way it deems reasonably practicable under the circumstances for nominal or no consideration. The depositary may sell a portion of the distributed securities or property sufficient to pay its fees and expenses and any taxes and governmental charges in connection with that distribution.
The depositary may sell a portion of the distributed rights sufficient to pay its fees and expenses, and any taxes and governmental charges, in connection with that distribution.
You may not be given the opportunity to exercise rights on the same terms and conditions as the holders of ordinary shares or be able to exercise such rights. If the depositary is unable to make rights available to you or to arrange for a sale of the rights, the depositary can allow such rights to lapse.
U.S. securities laws may restrict transfers and cancellation of the ADSs represented by shares acquired upon exercise of rights. For example, you may not be able to trade these ADSs freely in the United States. In this case, the depositary may deliver restricted depositary shares that have the same terms as the ADSs described in this section except for changes needed to put the necessary restrictions in place.
The depositary is not responsible if it decides that it is unlawful or impractical to make a distribution available to any ADS holders. We have no obligation to register ADSs, shares, rights or other securities under the Securities Act. We also have no obligation to take any other action to permit the distribution of ADSs, shares, rights or anything else to ADS holders. This means that you may not receive the distributions we make on our shares or any value for them if it is illegal or impractical for us to make them available to you.
Deposit, Withdrawal and Cancellation
How are ADSs issued?
The depositary will deliver ADSs if you or your broker deposit ordinary shares or evidence of rights to receive ordinary shares with the custodian. Upon payment of its fees and expenses and of any taxes or charges, such as stamp taxes or stock transfer taxes or fees, the depositary will register the appropriate number of ADSs in the names you request and will deliver the ADSs to or upon the order of the person or persons entitled thereto. The depositary will only issue ADSs in whole numbers.
How do ADS holders cancel an American Depositary Share?
You may turn in your ADSs at the depositary's principal office or by providing appropriate instructions to your broker. Upon payment of its fees and expenses and of any taxes or charges, such as stamp taxes or stock transfer taxes or fees, the depositary will deliver the ordinary shares and any other deposited securities underlying the ADSs to you or a person you designate in book-entry form in Belgium.
141
Your Right to Receive the Shares Underlying Your ADSs
You have the right to cancel your ADSs and withdraw the underlying ordinary shares at any time except in the following instances:
- •
- When temporary delays arise because: (1) the depositary or relevant settlement system has closed its transfer books or we have closed our transfer books; (2) the transfer of ordinary shares is blocked to permit voting at a shareholders' meeting; or (3) we are paying a dividend on our ordinary shares.
- •
- When you owe money to pay fees, taxes and similar charges.
- •
- When it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of ordinary shares or other deposited securities.
This right of withdrawal may not be limited by any other provision of the deposit agreement.
How do ADS holders interchange between Certificated ADSs and Uncertificated ADSs?
You may surrender your ADR to the depositary for the purpose of exchanging your ADR for uncertificated ADSs. The depositary will cancel that ADR and will send you a statement confirming that you are the owner of uncertificated ADSs. Alternatively, upon receipt by the depositary of a proper instruction from a holder of uncertificated ADSs requesting the exchange of uncertificated ADSs for certificated ADSs, the depositary will execute and deliver to you an ADR evidencing those ADSs.
Voting Rights
How do you vote?
You may instruct the depositary to vote the ordinary shares or other deposited securities underlying your ADSs.Otherwise, you could exercise your right to vote directly if you withdraw the ordinary shares. However, you may not know about the meeting sufficiently enough in advance to withdraw the ordinary shares.
If we ask for your instructions and upon timely notice from us as described in the deposit agreement, the depositary will notify you of the upcoming vote and arrange to deliver our voting materials to you by regular mail delivery or by electronic transmission. The materials will (i) describe the matters to be voted on and (ii) explain how you may instruct the depositary to vote the ordinary shares or other deposited securities underlying your ADSs as you direct. For your voting instructions to be valid, the depositary must receive them in writing on or before the date specified. The depositary will try, as far as practical, subject to applicable law and the provisions of the deposit agreement, the deposited securities and our articles of association, to vote or to have its agents vote the ordinary shares or other deposited securities as you instruct. The depositary will only vote or attempt to vote as you instruct.
We cannot assure you that you will receive the voting materials in time to ensure that you can instruct the depositary to vote the ordinary shares underlying your ADSs. In addition, the depositary and its agents are not responsible for failing to carry out voting instructions or for the manner in which any vote is cast.This means that you may not be able to exercise your right to vote and you may have no recourse if the ordinary shares underlying your ADSs are not voted as you requested.
Under the deposit agreement, a holder of ADSs that gives voting instructions to the depositary must arrange to have such ADSs registered on the record date fixed by the depositary for the meeting, which will be as close as practicable to the record date fixed by us with respect to the ordinary shares. Only shareholders, including persons that have their securities registered on the record date, may attend the meeting and vote in person or by proxy.
142
Persons that hold ADSs through a brokerage account or another financial institution will need to follow the procedures of their broker or that financial institution in order to give voting instructions to the depositary.
Under the deposit agreement, we may request the depositary to provide to us copies of any voting instructions the depositary receives as promptly as practicable after receipt of such instructions. In addition, under the terms of the deposit agreement, holders of ADSs that have delivered voting instructions agree that such voting instructions may, at our request, be disclosed by us, for purposes of compliance with Belgian law, in connection with any meeting of shareholders, whether prior, during or after such meeting of shareholders.
Unless otherwise indicated in the meeting notice distributed by the depositary referred to above, if the depositary receives from an ADS holder (that has otherwise satisfied all conditions to vote) voting instructions that fail to specify the manner in which the depositary is to vote the ordinary shares represented by such holder's ADSs, the depositary will deem such holder (unless otherwise specified in the notice distributed to holders) to have specifically instructed the depositary to vote in favor of all resolutions for which our board of directors recommends approval. In case new items are added to the agenda of a meeting of shareholders after the date of the proxy or new/alternative proposed resolutions are filed with respect to existing agenda items, for which no new specific voting instructions are validly provided to the depositary, the depositary shall abstain from voting on, respectively, the new agenda items and the new/alternative proposed resolutions. In no event will the depositary have the right to exercise voting rights as it will deem fit.
Fees and Expenses
As an ADS holder, you will be required to pay the following service fees to the depositary and certain taxes and governmental charges (in addition to any applicable fees, expenses, taxes and other governmental charges payable on the deposited securities represented by any of your ADSs):
| Service | Fees | |
|---|---|---|
• To any person to which ADSs are issued or to any person to which a distribution is made in respect of ADS distributions pursuant to stock dividends or other free distributions of stock, bonus distributions, stock splits or other distributions (except where converted to cash) | Up to US$0.05 per ADS issued | |
• Cancellation of ADSs, including termination of the deposit agreement | Up to US$0.05 per ADS cancelled | |
• Distribution of cash dividends | Up to US$0.05 per ADS held | |
• Distribution of cash entitlements (other than cash dividends) and/or cash proceeds, including proceeds from the sale of rights, securities and other entitlements | Up to US$0.05 per ADS held | |
• Distribution of ADSs pursuant to exercise of rights. | Up to US$0.05 per ADS held | |
• Depositary services | Up to US$0.05 per ADS held on the applicable record date(s) established by the depositary bank |
As an ADS holder, you will also be responsible to pay certain fees and expenses incurred by the depositary and certain taxes and governmental charges (in addition to any applicable fees, expenses,
143
taxes and other governmental charges payable on the deposited securities represented by any of your ADSs) such as the following:
- •
- Fees for the transfer and registration of ordinary shares charged by the registrar and transfer agent for the ordinary shares in Belgium (i.e., upon deposit and withdrawal of ordinary shares).
- •
- Expenses incurred for converting foreign currency into U.S. dollars.
- •
- Expenses for cable, telex, fax and electronic transmissions and for delivery of securities.
- •
- Taxes and duties upon the transfer of securities, including any applicable stamp duties, any stock transfer charges or withholding taxes (i.e., when ordinary shares are deposited or withdrawn from deposit).
- •
- Fees and expenses incurred in connection with the delivery of ordinary shares on deposit or the servicing of ordinary shares, deposited securities and/or ADSs.
- •
- Fees and expenses incurred in connection with complying with exchange control regulations and other regulatory requirements applicable to ordinary shares, deposited securities, ADSs and ADRs.
The depositary fees payable upon the issuance and cancellation of ADSs are typically paid to the depositary by the brokers (on behalf of their clients) receiving the newly issued ADSs from the depositary and by the brokers (on behalf of their clients) delivering the ADSs to the depositary for cancellation. The brokers in turn charge these fees to their clients. Depositary fees payable in connection with distributions of cash or securities to ADS holders and the depositary services fee are charged by the depositary bank to the holders of record of ADSs as of the applicable ADS record date.
The depositary fees payable for cash distributions are generally deducted from the cash being distributed or by selling a portion of distributable property to pay the fees. In the case of distributions other than cash (i.e., share dividends, rights), the depositary charges the applicable fee to the ADS record date holders concurrent with the distribution. In the case of ADSs registered in the name of the investor (whether certificated or uncertificated in direct registration), the depositary sends invoices to be paid to the applicable record date ADS holders. In the case of ADSs held in brokerage and custodian accounts (via DTC), the depositary generally collects its fees through the systems provided by DTC (whose nominee is the registered holder of the ADSs held in DTC) from the brokers and custodians holding ADSs in their DTC accounts. The brokers and custodians that hold their clients' ADSs in DTC accounts in turn charge their clients' accounts the amount of the fees paid to the depositary banks.
In the event of refusal to pay the depositary fees, the depositary may, under the terms of the deposit agreement, refuse the requested service until payment is received or may set off the amount of the depositary fees from any distribution to be made to the ADS holder.
The depositary has agreed to reimburse us for certain expenses we incur that are related to establishment and maintenance of the ADR program upon such terms and conditions as we and the depositary may agree from time to time.
Payment of Taxes
You will be responsible for any taxes or other governmental charges payable on your ADSs or on the deposited securities represented by any of your ADSs. The depositary may refuse to register any transfer of your ADSs or allow you to withdraw the deposited securities represented by your ADSs until such taxes or other charges are paid. It may apply payments owed to you or sell deposited securities represented by your ADSs to pay any taxes owed and you will remain liable for any deficiency. If the depositary sells deposited securities, it will, if appropriate, reduce the number of
144
ADSs to reflect the sale and pay to you any net proceeds, or send to you any property, remaining after it has paid the taxes. You agree to indemnify us, the depositary, the custodian and each of our and their respective agents, directors, employees and affiliates for, and hold each of them harmless from, any claims with respect to taxes (including applicable interest and penalties thereon) arising from any refund of taxes, reduced rate or withholding or other tax benefit obtained for you.
Reclassifications, Recapitalizations and Mergers
| If we: | Then: | |
|---|---|---|
• Change the par value of our ordinary shares • Reclassify, split up, subdivide or consolidate any of the deposited securities • Distribute securities on the ordinary shares that are not distributed to you or • Recapitalize, reorganize, merge, (partially) split, liquidate, sell our assets, or take any similar action | • The cash, shares or other securities received by the depositary will become deposited securities, to the extent permitted by law, and each ADS will automatically represent its equal share of the new deposited securities. • The depositary may deliver new ADSs or ask you to surrender your outstanding ADRs in exchange for new ADRs identifying the new deposited securities. • If any securities received by the depositary may not be lawfully distributed to some or all holders of ADSs, the depositary may sell such securities and distribute the net proceeds in the same way as it does cash. |
Amendment and Termination
How may the deposit agreement be amended?
We may agree with the depositary to amend the deposit agreement and the form of ADR without your consent for any reason. If an amendment adds or increases fees or charges, except for taxes and other governmental charges or expenses of the depositary for registration fees, facsimile costs, delivery charges or similar items, including expenses incurred in connection with foreign exchange control regulations, or materially prejudices a substantial existing right of ADS holders, it will not become effective for outstanding ADSs until 30 days after the depositary notifies ADS holders of the amendment.At the time an amendment becomes effective, you are considered, by continuing to hold your ADSs, to agree to the amendment and to be bound by the ADRs and the deposit agreement as amended. If any new laws are adopted that would require the deposit agreement to be amended in order to comply therewith, we and the depositary may amend the deposit agreement in accordance with such laws and such amendment may become effective before notice thereof is given to ADS holders.
How may the deposit agreement be terminated?
The depositary will terminate the deposit agreement if we ask it to do so, in which case the depositary will give notice to you at least ninety days prior to termination. The depositary may also terminate the deposit agreement if the depositary has told us that it would like to resign, or if we have removed the depositary, and in either case we have not appointed a new depositary within ninety days. In either such case, the depositary must notify you at least thirty days before termination.
After termination, the depositary and its agents will do the following under the deposit agreement but nothing else:
- •
- Collect distributions on the deposited securities.
145
- •
- Sell rights and other property.
- •
- Deliver ordinary shares and other deposited securities upon cancellation of ADSs after payment of any fees, charges, taxes or other governmental charges. Six months or more after termination, the depositary may sell any remaining deposited securities by public or private sale.
After that, the depositary will hold the money it received on the sale, as well as any other cash it is holding under the deposit agreement, for the pro rata benefit of the ADS holders that have not surrendered their ADSs. It will not invest the money and has no liability for interest. The depositary's only obligations will be to account for the money and other cash. After termination, our only obligations under the deposit agreement will be to indemnify the depositary and to pay fees and expenses of the depositary that we agreed to pay.
Books of Depositary
The depositary will maintain ADS holder records at its depositary office. You may inspect such records at such office during regular business hours but solely for the purpose of communicating with other holders in the interest of the business of our Company or matters relating to the ADSs or the deposit agreement.
The depositary will maintain facilities in New York to record and process the issuance, cancellation, combination, split-up and transfer of ADRs.
These facilities may be closed from time to time, to the extent not prohibited by law or if any such action is deemed necessary or advisable by the depositary or us, in good faith, at any time or from time to time because of any requirement of law, any government or governmental body or commission or any securities exchange on which the ADRs or ADSs are listed, or under any provision of the deposit agreement or provisions of, or governing, the deposited securities, or any meeting of our shareholders or for any other reason.
Limitations on Obligations and Liability
Limits on our Obligations and the Obligations of the Depositary; Limits on Liability to Holders of ADSs
The deposit agreement expressly limits our obligations and the obligations of the depositary. It also limits our liability and the liability of the depositary as follows:
- •
- We and the depositary are only obligated to take the actions specifically set forth in the deposit agreement without gross negligence or willful misconduct.
- •
- We and the depositary are not liable if either of us is prevented or delayed by law or circumstances beyond our control from performing our obligations under the deposit agreement, including, without limitation, requirements of any present or future law, regulation, governmental or regulatory authority or share exchange of any applicable jurisdiction, any present or future provisions of our memorandum and articles of association, on account of possible civil or criminal penalties or restraint, any provisions of or governing the deposited securities or any act of God, war or other circumstances beyond our control as set forth in the deposit agreement.
- •
- We and the depositary are not liable if either of us exercises, or fails to exercise, discretion permitted under the deposit agreement.
- •
- We and the depositary are not liable for the inability of any holder of ADSs to benefit from any distribution, offering, right or other benefit made available to holders of deposited securities that is not made available to holders of ADSs under the terms of the deposit agreement.
146
- •
- We and the depositary have no obligation to become involved in a lawsuit or other proceeding related to the ADSs or the deposit agreement on your behalf or on behalf of any other party if in our or the depositary's opinion such proceeding may involve us or the depositary in expense or liability, unless satisfactory indemnity against all expenses and liabilities is furnished as often as may be required.
- •
- We and the depositary may rely upon any documents we believe in good faith to be genuine and to have been signed or presented by the proper party.
- •
- We and the depositary disclaim any liability for any action/inaction in reliance on the advice or information of legal counsel, accountants, any person presenting ordinary shares for deposit, holders and beneficial owners (or authorized representatives) of ADSs, or any other person believed in good faith to be competent to give such advice or information.
- •
- We and the depositary disclaim any liability for any indirect, special, punitive or consequential damages for any breach of the terms of the deposit agreement or otherwise.
The depositary and any of its agents also disclaim any liability for any of the following:
- •
- Failure to carry out any instructions to vote.
- •
- Manner in which any vote is cast.
- •
- The effect of any vote.
- •
- Failure to determine that any distribution or action may be lawful or reasonably practicable or for allowing any rights to lapse in accordance with the provisions of the deposit agreement.
- •
- Failure or timeliness of any notice from us, the content of any information submitted to it by us for distribution to you or for any inaccuracy of any translation thereof.
- •
- Any investment risk associated with the acquisition of an interest in the deposited securities.
- •
- The validity or worth of the deposited securities.
- •
- The credit-worthiness of any third party.
- •
- Any tax consequences that may result from ownership of ADSs, ordinary shares or deposited securities. The depositary and its agents shall not be liable for any acts or omissions made by a successor depositary, provided that in connection with any issue out of which a potential liability arises the depositary performed its obligations without gross negligence or willful misconduct while it acted as depositary.
In addition, the deposit agreement provides that each party to the deposit agreement (including each holder, beneficial owner and holder of interests in the ADRs) irrevocably waives, to the fullest extent permitted by applicable law, any right it may have to a trial by jury in any lawsuit or proceeding against the depositary or our Company related to our shares, the ADSs or the deposit agreement.
In the deposit agreement, we and the depositary agree to indemnify each other under certain circumstances.
147
Requirements for Depositary Actions
Before the depositary will issue, deliver or register a transfer of an ADS, split-up, subdivide or combine ADSs, make a distribution on an ADS, or permit withdrawal of ordinary shares, the depositary may require the following:
- •
- Payment of stock transfer or other taxes or other governmental charges and transfer or registration fees charged by third parties for the transfer of any ordinary shares or other deposited securities and payment of the applicable fees, expenses and charges of the depositary.
- •
- Satisfactory proof of the identity and genuineness of any signature or any other matters contemplated in the deposit agreement.
- •
- Compliance with applicable laws and governmental regulations, and such reasonable regulations and procedures as the depositary may establish, from time to time, consistent with the deposit agreement and applicable law, including presentation of transfer documents.
The depositary may refuse to issue and deliver ADSs or register transfers of ADSs generally when the register of the depositary or our transfer books are closed or at any time if the depositary or we think it is necessary or advisable to do so.
Pre-release of ADSs
The deposit agreement permits the depositary to deliver ADSs before deposit of the underlying ordinary shares. This is called a pre-release of the ADSs. The depositary may also deliver ordinary shares upon cancellation of pre-released ADSs (even if the ADSs are cancelled before the pre-release transaction has been closed out). A pre-release is closed out as soon as the underlying ordinary shares are delivered to the depositary. The depositary may receive ADSs instead of ordinary shares to close out a pre-release. The depositary may pre-release ADSs only under the following conditions:
- (i)
- Before or at the time of the pre-release, the person to whom the pre-release is being made represents to the depositary in writing that it or its customer (a) owns the ordinary shares or ADSs to be deposited, (b) agrees to indicate the depositary as owner of such ordinary shares or ADSs in its records and to hold such ordinary shares or ADSs in trust for the depositary until such ordinary shares or ADSs are delivered to the depositary or the custodian, (c) unconditionally guarantees to deliver such ordinary shares or ADSs to the depositary or the custodian, as the case may be and (d) agrees to any additional restrictions or requirements that the depositary deems appropriate.
- (ii)
- The pre-release is fully collateralized with cash, US government securities or other collateral that the depositary considers appropriate.
- (iii)
- The depositary must be able to close out the pre-release on not more than five business days' notice. Each pre-release is subject to further indemnities and credit regulations as the depositary considers appropriate. In addition, the depositary will normally limit the number of ADSs that may be outstanding at any time as a result of pre-release to 30% of the aggregate number of ADSs then outstanding, although the depositary may disregard the limit from time to time if it thinks it is appropriate to do so.
Direct Registration System
The deposit agreement provides that, to the extent available by the depositary, ADSs shall be evidenced by ADRs issued through DRS/Profile, unless certificated ADRs are specifically requested by the ADS holder. DRS is the system administered by DTC pursuant to which the depositary may register the ownership of uncertificated ADSs, which ownership shall be evidenced by periodic statements issued by the depositary to the ADS holders entitled thereto. The Profile Modification
148
System, or Profile, is a required feature of DRS which allows a DTC participant, claiming to act on behalf of an ADS holder, to direct the depositary to register a transfer of those ADSs to DTC or its nominee and to deliver those ADSs to the DTC account of that DTC participant without receipt by the depositary of prior authorization from the ADS holder to register such transfer.
In connection with and in accordance with the arrangements and procedures relating to DRS/Profile, the parties to the deposit agreement understand that the depositary will not verify, determine or otherwise ascertain that the DTC participant claiming to be acting on behalf of an ADS holder in requesting registration of transfer and delivery described in the paragraph above has the actual authority to act on behalf of the ADS holder (notwithstanding any requirements under the Uniform Commercial Code).
149
SHARES ELIGIBLE FOR FUTURE SALES
Upon completion of this offering, we will have outstanding ADSs representing approximately % of our outstanding ordinary shares. All of the ADSs sold in this offering will be freely transferable by persons other than by our "affiliates" without restriction or further registration under the Securities Act. Sales of substantial amounts of the ADSs in the public market could adversely affect prevailing market prices of the ADSs. Prior to this offering our ordinary shares have traded, and subsequent to this offering will continue to trade, on Euronext Brussels under the symbol "TIG."
Lock-Up Agreements
We and the members of our board of directors, our executive management and our principal shareholders, each of whom hold more than 3% of our ordinary shares, have agreed to certain restrictions on our and their ability to sell additional ADSs or ordinary shares for a period of 180 days after the date of this prospectus. We and they have agreed not to directly or indirectly offer for sale, sell, contract to sell, grant any option for the sale of, or otherwise issue or dispose of, any ADSs or ordinary shares, options or warrants to purchase ADSs or ordinary shares, or any related security or instrument, without the prior written consent of Canaccord Genuity and Nomura Securities International. For more information, see "Underwriting."
Rule 144
Beginning ninety days after the effective date of the registration statement of which this prospectus forms a part, a person that is an affiliate of ours and that has beneficially owned "restricted" ordinary shares for at least six months, as measured by SEC rule, including the holding period of any prior owner other than one of our affiliates, is entitled to sell a number of restricted ordinary shares within any three-month period that does not exceed the greater of the following:
- •
- 1% of the number of ordinary shares then outstanding, in the form of ADSs or otherwise, which will equal approximately ordinary shares immediately after this offering.
- •
- The average weekly trading volume of the ordinary shares and ADSs during the four calendar weeks preceding the filing of a notice on Form 144 with respect to the sale.
Sales of restricted ordinary shares under Rule 144 held by our affiliates are also subject to requirements regarding the manner of sale, notice and the availability of current public information about us. Rule 144 also requires that affiliates relying on Rule 144 to sell ordinary shares that are not restricted shares must nonetheless comply with the same restrictions applicable to restricted shares, other than the holding period requirement.
Regulation S
Regulation S under the Securities Act provides that shares owned by any person may be sold without registration in the United States, provided that the sale is effected in an offshore transaction and no directed selling efforts are made in the United States (as these terms are defined in Regulation S), subject to certain other conditions. In general, this means that our shares may be sold outside the United States without registration in the United States being required.
Rule 701
Under Rule 701 under the Securities Act, ordinary shares acquired upon the exercise of options or pursuant to other rights granted under a written compensatory stock or option plan or other written agreement in compliance with Rule 701 may be resold by the following persons:
- •
- Persons other than affiliates, beginning ninety days after the effective date of the registration statement of which this prospectus forms a part, subject only to the manner-of-sale provisions of Rule 144.
- •
- Our affiliates, beginning ninety days after the effective date of the registration statement of which this prospectus forms a part, subject to the manner-of-sale and volume limitations, current public information and filing requirements of Rule 144, in each case, without compliance with the six-month holding period requirement of Rule 144.
150
U.S. Taxation
This section describes the material U.S. federal income tax consequences to U.S. Holders of acquiring owning and disposing of our shares or ADSs. It applies to you only if you acquire your shares or ADSs in this offering and you hold your shares or ADSs as capital assets for U.S. federal income tax purposes. This section does not apply to you if you are a member of a special class of holders subject to special rules, including the following:
- •
- a dealer in securities.
- •
- a real estate investment trust or regulated investment company.
- •
- a trader in securities that elects to use a mark-to-market method of accounting for securities holdings.
- •
- a tax-exempt organization, including an "individual retirement account, or "Roth IRA."
- •
- a bank or other financial institution, or life insurance company.
- •
- a person liable for alternative minimum tax.
- •
- a person that actually or constructively owns 10% or more of our voting stock.
- •
- a person that holds shares or ADSs as part of a straddle or a hedging, integrated or conversion transaction.
- •
- certain former citizens or long-term residents of the United States.
- •
- a U.S. Holder (as defined below) whose functional currency is not the U.S. dollar.
This section is based on the Internal Revenue Code of 1986, as amended or the "Code", its legislative history, existing and proposed regulations, published rulings and court decisions, as well as on the Convention Between the Government of the United States of America and the Government of the Kingdom of Belgium for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion With Respect to Taxes on Income (together with the Protocol thereto, the "Treaty"). These laws are subject to differing interpretations or change, possibly on a retroactive basis. In addition, this section is based in part upon the representations of the Depositary and the assumption that each obligation in the Deposit Agreement and any related agreement will be performed in accordance with its terms.
You are a "U.S. Holder" if you are a beneficial owner of shares or ADSs and you are for U.S. federal income tax purposes, one of the following:
- •
- an individual who is a citizen or resident of the United States.
- •
- a corporation (or other entity that is treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any state thereof, or the District of Columbia,
- •
- an estate whose income is subject to U.S. federal income tax regardless of its source, or
- •
- a trust if (i) a U.S. court can exercise primary supervision over the trust's administration and one or more U.S. persons are authorized to control all substantial decisions of the trust or (ii) such trust has a valid election in effect under applicable U.S. Treasury Regulations to be treated as a U.S. person.
An "eligible U.S. Holder" is a U.S. Holder that:
- •
- is a resident of the United States for purposes of the Treaty;
151
- •
- does not maintain a permanent establishment or fixed base in Belgium to which shares or ADSs are attributable and through which the U.S. Holder carries on or has carried on business (or, in the case of an individual, performs or has performed independent personal services); and
- •
- is otherwise eligible for benefits under the Treaty with respect to income and gain from the shares or ADSs.
A "non-U.S. Holder" is a beneficial owner of shares or ADSs that is not a U.S. person for U.S. federal income tax purposes.
If a partnership (including any entity treated as a partnership for U.S. federal income tax purposes) is a beneficial owner of shares or ADSs, the tax treatment of a partner in the partnership will generally depend upon the status of the partner and the activities of the partnership. A beneficial owner of shares or ADSs that is a partnership, and partners in such partnership, should consult their own tax advisors regarding the tax consequences of acquiring, owning and disposing of the shares or ADSs.
You should consult your own tax advisors regarding the U.S. federal, state and local and other tax consequences of acquiring, owning and disposing of shares or ADSs in your particular circumstances.
This discussion addresses only U.S. federal income taxation. Holders should consult their own tax advisors as to potential application of U.S. state and local tax laws, as well as any other U.S. tax laws (such as the gift, alternative minimum or estate tax) and other U.S. laws, and foreign laws, including the laws of Belgium.
Taxation of U.S. Holders
Treatment of Holders of ADSs. In general, and taking into account the earlier assumptions, for U.S. federal income tax purposes, if you hold ADRs evidencing ADSs, you will be treated as the beneficial owner of the shares represented by those ADSs. Exchanges of ADSs for ordinary shares generally will not be subject to U.S. federal income tax.
Taxation of Dividends. Subject to the passive foreign investment company, or PFIC rules discussed below, the gross amount of any dividend we pay out of our current or accumulated earnings and profits (as determined for U.S. federal income tax purposes) is subject to U.S. federal income taxation. If you are a noncorporate U.S. Holder, dividends paid to you that constitute qualified dividend income may qualify for the preferential rates of taxation under current law applicable to long-term capital gains provided that you hold such shares or ADSs for more than 60 days during the 121-day period beginning 60 days before the ex-dividend date and meet other holding period requirements.
In order for the dividends paid by the Company to be treated as qualified dividend income, the Company must either be eligible for the benefits of a comprehensive income tax treaty with the United States which the Internal Revenue Service has determined is satisfactory and which includes an exchange of information program, or such dividends must be paid with respect to shares or ADSs which are readily tradable on an established securities market in the United States. The Internal Revenue Service has determined that the Treaty is satisfactory for purposes of the qualified dividend rules and that it includes an exchange of information program. The Company expects to qualify as a resident of Belgium for purposes of, and to be eligible for the benefits of, the Treaty by virtue of our shares being traded on Euronext Brussels. Further, we expect the ADSs to be readily tradable on the NASDAQ Global Market. Under a published Internal Revenue Service Notice, common or ordinary shares, or ADSs representing such shares, are considered to be readily tradable on an established securities market in the United States if they are traded on the NASDAQ Global Select Market, as our
152
ADSs are expected to be. As a result, the Company expects that the dividends paid will be treated as qualified dividend income for eligible noncorporate U.S. Holders, provided that the holding period requirement (discussed above) is met. However, if our ADSs cease to be traded on the NASDAQ Global Market, or our ordinary shares cease to be traded on Euronext Brussels, the Company would have to qualify for the benefits of the Treaty under some other provision of the limitation on benefits article of the Treaty in order for dividends we pay to continue to be eligible for treatment as qualified dividend income. U.S. Holders should consult their own tax advisors as to the qualification of dividends paid by the Company as qualified dividend income.
With respect to any dividend we pay, you must include any Belgian tax withheld from the dividend payment in the gross amount of such dividend even though you do not in fact receive it. Dividends are taxable to you when the Depositary receives such dividend, actually or constructively. Such dividends will not be eligible for the dividends-received deduction generally allowed to United States corporations in respect of dividends received from other United States corporations. The amount of a dividend distribution that you must include in your income as a U.S. Holder will be the U.S. dollar value of the euro payments made, determined at the spot euro/U.S. dollar rate on the date the dividend distribution is includible in your income, regardless of whether the payment is in fact converted into U.S. dollars. Generally, any gain or loss resulting from currency exchange fluctuations during the period from the date you include a dividend payment in income to the date you convert the payment into U.S. dollars will be treated as ordinary income or loss and will not be eligible for the special tax rate applicable to qualified dividend income. The gain or loss generally will be income or loss from sources within the United States for foreign tax credit limitation purposes. Distributions in excess of current and accumulated earnings and profits, if any, as determined for U.S. federal income tax purposes, will be treated as a non-taxable return of capital to the extent of your basis in the shares or ADSs and thereafter as capital gain; however, since the Company does not intend to maintain books and records in accordance with U.S. tax principles, a U.S. Holder will effectively be required to treat all amounts the Company distributes as dividends for U.S. federal income tax purposes.
Subject to certain limitations, the Belgian tax withheld in accordance with the Treaty and paid over to Belgium will be creditable against your U.S. federal income tax liability. Special rules apply in determining the foreign tax credit limitation with respect to dividends that are subject to preferential rates of taxation under current law. To the extent a refund of the tax withheld is available to you under Belgian law or under the Treaty, the amount of tax withheld that is refundable will not be eligible for credit against your U.S. federal income tax liability. Each U.S. holder should consult its own tax advisors regarding the foreign tax credit rules.
Dividends will be income from sources outside the United States, and dividends paid will, depending on your circumstances, be "passive" or "general" income which, in either case, is treated separately from other types of income for purposes of computing the foreign tax credit allowable to you.
A U.S. Holder may make an election to treat all foreign taxes paid as deductible expenses in computing taxable income, rather than as a credit against tax, subject to generally applicable limitations. Such an election, once made, applies to all foreign taxes paid for the taxable year subject to the election. The rules governing foreign tax credits are complex and, therefore, U.S. Holders are strongly encouraged to consult their own tax advisors to determine whether they are subject to any special rules that may limit their ability to make effective use of foreign tax credits and whether or not an election would be appropriate based on their particular circumstances.
Taxation of Capital Gains. Subject to the PFIC rules discussed below, if you are a U.S. Holder and you sell or otherwise dispose of your shares or ADSs, you will recognize capital gain or loss for U.S. federal income tax purposes equal to the difference between the U.S. dollar value of the amount that you realize and your tax basis (generally, the amount you pay for your shares or ADSs),
153
determined in U.S. dollars, in your shares or ADSs. Capital gain of a noncorporate U.S. Holder is generally taxed at a preferential rate of taxation under current law where the holder has a holding period greater than one year. Such gain or loss will generally be income or loss from sources within the United States for foreign tax credit limitation purposes.
Passive Foreign Investment Company Considerations. We believe that our shares or ADSs should not be treated as stock of a PFIC for U.S. federal income tax purposes, but this conclusion is a factual determination that is made annually and thus may be subject to change.
In general, if you are a U.S. Holder, we will be a PFIC with respect to you if for any taxable year in which you held shares or ADSs:
- •
- at least 75% of our gross income for the taxable year is "passive income;" or
- •
- at least 50% of the value, determined on the basis of a quarterly average, of our gross assets (which, assuming we are not a controlled foreign company for the year being tested, would be measured by the fair market value of our assets, and for which purpose the total value of our assets may be determined in part by the market value of the shares or ADSs and our shares, which are subject to change) is attributable to assets that produce or are held for the production of "passive income."
Passive income generally includes dividends, interest, royalties, rents (other than certain rents and royalties derived in the active conduct of a trade or business), annuities and gains from assets that produce passive income. If a foreign corporation owns at least 25% by value of the stock of another corporation, the foreign corporation is treated for purposes of the PFIC tests as owning its proportionate share of the assets of the other corporation, and as receiving directly its proportionate share of the other corporation's income.
If we are treated as a PFIC, and you are a U.S. Holder that did not make a mark-to-market election, as described below, you will be subject to special tax rules with respect to:
- •
- any gain you realize on the sale or other disposition of your shares or ADSs; and
- •
- any excess distribution that we make to you (generally, any distributions to you during a single taxable year that are greater than 125% of the average annual distributions received by you in respect of the shares or ADSs during the three preceding taxable years or, if shorter, your holding period for the shares or ADSs).
Under these rules:
- •
- the gain or excess distribution will be allocated ratably over your holding period for the shares or ADSs;
- •
- the amount allocated to the taxable year in which you realized the gain or excess distribution will be taxed as ordinary income;
- •
- the amount allocated to each prior year, with certain exceptions, will be taxed at the highest tax rate in effect for that year; and
- •
- the interest charge generally applicable to underpayments of tax will be imposed in respect of the tax attributable to each such year.
The special PFIC tax rules described above will not apply to you if you make a QEF election, that is, you elect to have us treated as a qualified electing fund and we provide certain requirement information to you. We do not intend to provide U.S. Holders with such information as may be required to make a QEF election effective.
154
Special rules apply for calculating the amount of the foreign tax credit with respect to excess distributions by a PFIC.
If you own shares or ADSs in a PFIC that are treated as marketable stock, you may make a mark-to-market election. If you make this election, you will not be subject to the PFIC rules described above. Instead, in general, you will include as ordinary income each year the excess, if any, of the fair market value of your shares or ADSs at the end of the taxable year over your adjusted basis in your shares or ADSs. These amounts of ordinary income will not be eligible for the favorable tax rates applicable to qualified dividend income or long-term capital gains. You will also be allowed to take an ordinary loss in respect of the excess, if any, of the adjusted basis of your shares or ADSs over their fair market value at the end of the taxable year (but only to the extent of the net amount of previously included income as a result of the mark-to-market election). Your basis in the shares or ADSs will be adjusted to reflect any such income or loss amounts. For purposes of this rule, if you make a mark-to-market election with respect to your shares or ADSs, you will be treated as having a new holding period in your shares or ADSs beginning on the first day of the first taxable year beginning after the last taxable year for which the mark-to-market election applies.
In addition, notwithstanding any election you make with regard to your shares or ADSs, dividends that you receive from us will not constitute qualified dividend income to you if we are a PFIC either in the taxable year of the distribution or the preceding taxable year. Moreover, your shares or ADSs will be treated as stock in a PFIC if we were a PFIC at any time during your holding period in your shares or ADSs, even if we are not currently a PFIC. Dividends that you receive that do not constitute qualified dividend income are not eligible for taxation at the preferential rate of taxation under current law applicable to qualified dividend income. Instead, you must include the gross amount of any such dividend paid by us out of our accumulated earnings and profits (as determined for U.S. federal income tax purposes) in your gross income, and it will be subject to tax at rates applicable to ordinary income.
If we are determined to be a PFIC, the general tax treatment for U.S. Holders described in this section would apply to indirect distributions and gains deemed to be realized by U.S. Holders in respect of any of our subsidiaries that also may be determined to be PFICs.
If you are a U.S. Holder, you must generally file an IRS Form 8621 (Information Return by a Shareholder of a Passive Foreign Investment Company or Qualified Electing Fund) with your U.S. federal income tax return for any taxable year in which we are a PFIC with respect to you. You are urged to consult your own tax advisor concerning the filing of IRS Form 8621.
Net Investment Income Tax. An additional tax is imposed on the "net investment income" of certain U.S. Holders who are citizens and resident aliens, and on the undistributed "net investment income" of certain estates and trusts. Among other items, "net investment income" generally would include dividends paid on shares or ADSs and certain net gain from the sale or other taxable disposition of shares or ADSs, less certain deductions. You should consult your own tax advisor concerning the effect, if any, of this net investment income tax on holding shares or ADSs in your particular circumstances.
Backup Withholding and Information Reporting. If you are a noncorporate U.S. Holder, information reporting requirements, on Internal Revenue Service Form 1099, generally will apply to:
- •
- dividend payments or other taxable distributions made to you within the United States or by a U.S. payor; and
155
- •
- the payment of proceeds to you from the sale of shares or ADSs effected at a United States office of a broker.
Additionally, backup withholding may apply to such payments if you are a noncorporate U.S. Holder that:
- •
- fails to provide an accurate taxpayer identification number;
- •
- is notified by the Internal Revenue Service that you have failed to report all interest and dividends required to be shown on your U.S. federal income tax returns; or
- •
- in certain circumstances, fails to comply with applicable certification requirements.
In addition, a sale of shares or ADSs effected at a foreign office of a broker will be subject to information reporting if the broker is:
- •
- a U.S. person;
- •
- a controlled foreign corporation for U.S. tax purposes;
- •
- a foreign person 50% or more of whose gross income is effectively connected with the conduct of a United States trade or business for a specified three-year period; or
- •
- a foreign partnership, if at any time during its tax year:
- •
- one or more of its partners are "U.S. persons," as defined in U.S. Treasury regulations, who in the aggregate hold more than 50% of the income or capital interest in the partnership, or
- •
- such foreign partnership is engaged in the conduct of a United States trade or business,
unless the broker does not have actual knowledge or reason to know that you are a United States person and the documentation requirements described above are met or you otherwise establish an exemption. Backup withholding will apply if the sale is subject to information reporting and the broker has actual knowledge that you are a U.S. person.
Backup withholding is not an additional tax. You generally may obtain a refund of any amounts withheld under the backup withholding rules that exceed your U.S. federal income tax liability by timely filing a refund claim with the Internal Revenue Service.
Disclosure of Information with respect to Foreign Financial Assets. Certain U.S. Holders who hold any interest in "specified foreign financial assets," including the shares or ADRs during such holder's taxable year must attached to the U.S. federal income tax return for such year certain information with respect to each asset if the aggregate value of all of such assets exceeds $50,000 (or a higher dollar amount prescribed by the Internal Revenue Service). For this purpose, a "specified foreign financial asset" includes any depositary, custodial or other financial account maintained by a foreign financial institution, and certain assets that are not held in an account maintained by a financial institution, including any stock or security issued by a person other than a U.S. person. A U.S. Holder subject to these rules who fails to furnish the required information is subject to a penalty of $10,000, and an additional penalty may apply if the failure continues for more than 90 days after the U.S. Holder is notified of such failure by the Internal Revenue Service; however, these penalties may be avoided if the U.S. Holder demonstrates a reasonable cause for the failure to comply. An accuracy-related penalty of 40 per cent is imposed for an underpayment of tax that is attributable to an "undisclosed foreign financial asset understatement," which for this purpose is the portion of the understatement for any taxable year that is attributable to any transaction involving an "undisclosed foreign financial asset," including any asset that is subject to the information reporting requirements of these rules, which would include the shares or ADSs if the dollar threshold described above were satisfied.
156
The applicable statute of limitations for assessment of U.S. federal income taxes is extended to six years if there is an omission of gross income in excess of $5,000 and the omission of gross income is attributable to a foreign financial asset as to which reporting is required as described above (or would be so required if the requirement for reporting specified foreign financial assets were applied without regard to the dollar threshold specified therein and without regard to certain exceptions that may be specified by the Internal Revenue Service). In addition, the statute of limitations with respect to a U.S. Holder's entire U.S. federal income tax return will be suspended if a U.S. Holder fails to timely provide information with respect to specified foreign financial assets required to be reported or fails to timely provide the annual information reports required for holders of PFIC stock.Holders should consult their own tax adviser concerning any obligation that they may have to furnish information to the Internal Revenue Service as a result of holding the shares or ADSs.
The above discussion is not included to constitute a complete analysis of all the tax consequences relating to the acquisition, ownership and disposition of our shares or ADSs.
Belgian Taxation
The following paragraphs are a summary of material Belgian tax consequences of the ownership of ADSs by an investor. The summary is based on laws, treaties and regulatory interpretations in effect in Belgium on the date of this document, all of which are subject to change, including changes that could have retroactive effect.
The summary only discusses Belgian tax aspects which are relevant to U.S. holders of ADSs, or "Holders." This summary does not address Belgian tax aspects which are relevant to persons who are fiscally resident in Belgium or who avail of a permanent establishment or a fixed base in Belgium to which the ADSs are effectively connected.
This summary does not purport to be a description of all of the tax consequences of the ownership of ADSs, and does not take into account the specific circumstances of any particular investor, some of which may be subject to special rules, or the tax laws of any country other than Belgium. This summary does not describe the tax treatment of investors that are subject to special rules, such as banks, insurance companies, collective investment undertakings, dealers in securities or currencies, persons that hold, or will hold, ADSs in a position in a straddle, share-repurchase transaction, conversion transactions, synthetic security or other integrated financial transactions. Investors should consult their own advisers regarding the tax consequences of an investment in ADSs in the light of their particular circumstances, including the effect of any state, local or other national laws.
In addition to the assumptions mentioned above, it is also assumed in this discussion that for purposes of the domestic Belgian tax legislation, the owners of ADSs will be treated as the owners of the ordinary shares represented by such ADSs. However, the assumption has not been confirmed by or verified with the Belgian Tax Authorities.
Dividend Withholding Tax
As a general rule, a withholding tax of 25% is levied on the gross amount of dividends paid on the ordinary shares represented by the ADSs, subject to such relief as may be available under applicable domestic or tax treaty provisions.
Dividends subject to the dividend withholding tax include all benefits attributed to the ordinary shares represented by the ADSs, irrespective of their form, as well as reimbursements of statutory share capital by us, except reimbursements of fiscal capital made in accordance with the Belgian Company Code. In principle, fiscal capital includes paid-up statutory share capital, and subject to certain conditions, the paid-up issue premiums and the cash amounts subscribed to at the time of the issue of profit sharing certificates.
157
In case of a redemption by us of our own shares represented by ADSs, the redemption distribution (after deduction of the portion of fiscal capital represented by the redeemed shares) will be treated as a dividend which in principle is subject to the withholding tax of 25%, subject to such relief as may be available under applicable domestic or tax treaty provisions. In case of a liquidation of our Company, any amounts distributed in excess of the fiscal capital will also be treated as a dividend, but will in principle be subject to a 25% withholding tax, subject to such relief as may be available under applicable domestic or tax treaty provisions.
For non-residents the dividend withholding tax, if any, will be the only tax on dividends in Belgium, unless the non-resident avails of a fixed base in Belgium or a Belgian permanent establishment to which the ADSs are effectively connected.
Relief of Belgian Dividend Withholding Tax
Under the Belgium-United States Tax Treaty, or the Treaty, under which we are entitled to benefits accorded to residents of Belgium, there is a reduced Belgian withholding tax rate of 15% on dividends paid by us to a U.S. resident which beneficially owns the dividends and is entitled to claim the benefits of the Treaty under the limitation of benefits article included in the Treaty, or Qualifying Holders.
If such Qualifying Holder is a company that owns directly at least 10% of our voting stock, the Belgian withholding tax rate is further reduced to 5%. No withholding tax is however applicable if the Qualifying Holder, is either of the following:
- •
- a company that is a resident of the United States that has owned directly ADSs representing at least 10% of our capital for a twelve-month period ending on the date the dividend is declared, or
- •
- a pension fund that is a resident of the United States, provided that such dividends are not derived from the carrying on of a business by the pension fund or through an associated enterprise.
Under the normal procedure, we or our paying agent must withhold the full Belgian withholding tax, without taking into account the reduced Treaty rate. Qualifying Holders may make a claim for reimbursement for amounts withheld in excess of the rate defined by the Treaty. The reimbursement form (Form 276 Div-Aut.) may be obtained from the Bureau Central de Taxation Bruxelles-Etranger, Boulevard du Jardin Botanique 50 boîte 3429, 1000 Brussels, Belgium or at ctk.db.brussel.buitenland@minfin.fed.be. Qualifying Holders may also, subject to certain conditions, obtain the reduced Treaty rate at source. Qualifying Holders should deliver a duly completed Form 276 Div-Aut. no later than ten days after the date on which the dividend becomes payable.
U.S. holders should consult their own tax advisors as to whether they qualify for reduction in withholding tax upon payment or attribution of dividends, and as to the procedural requirements for obtaining a reduced withholding tax upon the payment of dividends or for making claims for reimbursement.
Withholding tax is also not applicable, pursuant to Belgian domestic tax law, on dividends paid to a U.S. pension fund which satisfies the following conditions:
- (i)
- to be a legal entity with fiscal residence in the United States and without a permanent establishment or fixed base in Belgium,
- (ii)
- whose corporate purpose consists solely in managing and investing funds collected in order to pay legal or complementary pensions,
- (iii)
- whose activity is limited to the investment of funds collected in the exercise of its statutory mission, without any profit making aim and without operating a business in Belgium,
158
- (iv)
- which is exempt from income tax in the United States, and
- (v)
- provided that it (save in certain particular cases as described in Belgian law) is not contractually obligated to redistribute the dividends to any ultimate beneficiary of such dividends for whom it would manage the shares or ADSs, nor obligated to pay a manufactured dividend with respect to the shares or ADSs under a securities borrowing transaction. The exemption will only apply if the U.S. pension fund provides an affidavit confirming that it is the full legal owner or usufruct holder of the shares or ADSs and that the above conditions are satisfied. The organization must then forward that affidavit to us or our paying agent.
Capital Gains and Losses
Pursuant to the Treaty, capital gains and/or losses realized by a Qualifying Holder from the sale, exchange or other disposition of ADSs are exempt from tax in Belgium.
Capital gains realized on ADSs by a corporate Holder who is not a Qualifying Holder are generally not subject to taxation in Belgium unless such Holder is acting through a Belgian permanent establishment or a fixed place in Belgium to which the ADSs are effectively connected (in which case a 33.99%, 25.75%, 0.412% or 0% tax on the capital gain may apply, depending on the particular circumstances). Capital losses are generally not tax deductible.
Private individual Holders which are not Qualifying Holders and which are holding ADSs as a private investment will, as a rule, not be subject to tax in Belgium on any capital gains arising out of a disposal of ADSs. Losses will, as a rule, not be tax deductible.
However, if the gain realized by such individual Holders on ADSs is deemed to be realized outside the scope of the normal management of such individual's private estate and the capital gain is obtained or received in Belgium, the gain will be subject to a final tax of 30.28%.
Moreover, capital gains realized by such individual Holders on the disposal of ADSs for consideration, outside the exercise of a professional activity, to a non-resident corporation (or a body constituted in a similar legal form), to a foreign state (or one of its political subdivisions or local authorities) or to a non-resident legal entity that is established outside the European Economic Area, are in principle taxable at a rate of 16.5% if, at any time during the five years preceding the realization event, such individual Holders own or have owned directly or indirectly, alone or with his/her spouse or with certain other relatives, a substantial shareholding in us (that is, a shareholding of more than 25% of our shares).
Capital gains realized by a Holder upon the redemption of ADSs or upon our liquidation will generally be taxable as a dividend. See "Dividend Withholding Tax."
Potential Application of Article 228, §3 ITC
Under a strict reading of Article 228, §3 of the Belgian Income Tax Code 1992 ("ITC"), capital gains realized on ADSs by non-residents could be subject to Belgian taxation, levied in the form of a professional withholding tax, if the following three conditions are cumulatively met: (i) the capital gain would have been taxable if the non-resident were a Belgian tax resident, (ii) the income is "borne by" a Belgian resident or by a Belgian establishment of a foreign entity (which would, in such a context, mean that the capital gain is realized upon a transfer of ADSs to a Belgian resident or to a Belgian establishment of a foreign entity, together a "Belgian Purchaser"), and (iii) Belgium has the right to tax such capital gain pursuant to the applicable double tax treaty, or, if no such tax treaty applies, the non-resident does not demonstrate that the capital gain is effectively taxed in its state of residence. However, it is unclear whether a capital gain included in the purchase price of an asset can be considered to be "borne by" the purchaser of the asset within the meaning of the second condition
159
mentioned above. Furthermore, applying this withholding tax would require that the Belgian Purchaser is aware of (i) the identity of the non-resident (to assess the third condition mentioned above), and (ii) the amount of the capital gain realized by the non-resident (since such amount determines the amount of professional withholding tax to be levied by the Belgian Purchaser). Consequently, the application of this professional withholding tax on transactions with respect to the ADSs occurring on the stock exchange would give rise to practical difficulties as the seller and purchaser typically do not know each other. In addition to these uncertainties, the parliamentary documents of the law that introduced Article 228, §3 ITC support the view that the legislator did not intend for Article 228, §3 ITC to apply to a capital gain included in the purchase price of an asset, but only to payments for services. On July 23, 2014, formal guidance on the interpretation of Article 228, §3 ITC has been issued by the Belgian tax authorities (published in the Belgian Official Gazette of July 23, 2014). The Belgian tax authorities state therein that Article 228, §3 ITC only covers payments for services, as a result of which no professional withholding tax should apply to capital gains realized by non-residents in the situations described above. It should, however, be noted that a formal guidance issued by the tax authorities does not supersede and cannot amend the law if the latter is found to be sufficiently clear in itself. Accordingly, in case of dispute, it cannot be ruled out that the interpretation of Article 228, §3 ITC made by the tax authorities in their formal guidance is not upheld by the competent courts.
Estate and Gift Tax
There is no Belgium estate tax on the transfer of ADSs on the death of a Belgian non-resident. Donations of ADSs made in Belgium may or may not be subject to gift tax depending on the modalities under which the donation is carried out.
Belgian Tax on Stock Exchange Transactions
A stock market tax is normally levied on the purchase and the sale and on any other acquisition and transfer for consideration in Belgium of ADSs through a professional intermediary established in Belgium on the secondary market, so-called "secondary market transactions." The tax is due from the transferor and the transferee separately. The applicable rate amounts to 0.25% of the consideration paid but with a cap of 740 euros per transaction and per party. Under current Belgian tax law, the applicable rate and the applicable cap will reduce to 0.22% and 650 euros for transactions carried out as from January 1, 2015.
Belgian non-residents who purchase or otherwise acquire or transfer, for consideration, ADSs in Belgium for their own account through a professional intermediary may be exempt from the stock market tax if they deliver a sworn affidavit to the intermediary in Belgium confirming their non-resident status.
In addition to the above, no stock market tax is payable by: (i) professional intermediaries described in Article 2, 9° and 10° of the Law of August 2, 2002 acting for their own account, (ii) insurance companies described in Article 2, §1 of the Law of July 9, 1975 acting for their own account, (iii) professional retirement institutions referred to in Article 2, 1° of the Law of October 27, 2006 relating to the control of professional retirement institutions acting for their own account, or (iv) collective investment institutions acting for their own account.
No stock exchange tax will thus be due by Holders on the subscription, purchase or sale of ADSs, if the Holders are acting for their own account. In order to benefit from this exemption, the Holders must file with the professional intermediary in Belgium a sworn affidavit evidencing that they are non-residents for Belgian tax purposes.
160
Proposed Financial Transactions Tax
The European Commission has published a proposal for a Directive for a common financial transactions tax, or FTT, in Belgium, Germany, Estonia, Greece, Spain, France, Italy, Austria, Portugal, Slovenia and Slovakia, or collectively, the Participating Member States.
The proposed FTT has a very broad scope and could, if introduced in its current form, apply to certain dealings in ADS's in certain circumstances. Under current proposals, the FTT could apply in certain circumstances to persons both within and outside of the Participating Member States. Generally, it would apply to certain dealings in ADSs where at least one party is a financial institution, and at least one party is established in a Participating Member State.
A financial institution may be, or be deemed to be, "established" in a Participating Member State in a broad range of circumstances, including by transacting with a person established in a Participating Member State.
The FTT proposal remains subject to negotiation between the Participating Member States. It may therefore be altered prior to any implementation, the timing of which remains unclear. Additional E.U. Member States may decide to participate. Prospective Holders of ADSs are advised to seek their own professional advice in relation to the FTT.
161
The underwriters named below have agreed to buy, subject to the terms of the underwriting agreement, the number of shares listed opposite their names below. The underwriters are committed to purchase and pay for all of the shares if any are purchased. Canaccord Genuity Inc. and Nomura Securities International, Inc. are acting as the representatives of the underwriters named below.
Underwriters | Number of ADSs | |
|---|---|---|
Canaccord Genuity Inc. | ||
Nomura Securities International, Inc. | ||
Maxim Group LLC | ||
Chardan Capital Markets, LLC | ||
| | | |
| | | |
Total |
The underwriters have advised us that they propose to offer the ADSs to the public at $ per ADS. The underwriters propose to offer the ADSs to certain dealers at the same price less a concession of not more than $ per ADS. The underwriters may allow and the dealers may reallow a concession of not more than $ per ADS on sales to certain other brokers and dealers. After this offering, these figures may be changed by the underwriters.
We have granted to the underwriters an option to purchase up to an additional ADSs, at the same price to the public, and with the same underwriting discount, as set forth in the table below. The underwriters may exercise this option any time during the thirty-day period after the date of this prospectus, but only to cover over-allotments, if any. To the extent the underwriters exercise the option, each underwriter will become obligated, subject to certain conditions, to purchase approximately the same percentage of the additional ADSs as it was obligated to purchase under the underwriting agreement.
We have agreed to pay underwriting discounts and commissions of % of the gross proceeds of this offering. The following table shows the underwriting fees to be paid to the underwriters in connection with this offering. These amounts are shown assuming both no exercise and full exercise of the over-allotment option.
| | No Exercise | Full Exercise | |||||
|---|---|---|---|---|---|---|---|
Per Share | $ | ||||||
Total | $ | ||||||
We have agreed to reimburse the underwriters for their expenses in an amount up to $ .
We have agreed to indemnify the underwriters against certain liabilities, including civil liabilities under the Securities Act, or to contribute to payments that the underwriters may be required to make in respect of those liabilities.
We and the members of our board of directors, our executive management and our principal shareholders, each of whom hold more than 3% of our ordinary shares, have agreed to certain restrictions on our and their ability to sell additional ADSs or ordinary shares for a period of 180 days after the date of this prospectus. We and they have agreed not to offer directly or indirectly for sale, sell, contract to sell, grant any option for the sale of, or otherwise issue or dispose of, any ADSs or ordinary shares, options or warrants to purchase ADSs or ordinary shares, or any related security or instrument, without the prior written consent of Canaccord Genuity Inc. and Nomura Securities International, Inc.
We intend to apply to list our ADSs on the NASDAQ Global Market under the symbol "TIG."
162
The initial public offering price for the ADSs will be based, in part, on the price of our ordinary shares on Euronext Brussels, and determined by negotiations among the representatives and us. Among the factors considered in determining the initial public offering price are our future prospects and the prospects of our industry in general, our revenue, net income and certain other financial and operating information in recent periods, and the financial ratios, market prices of securities and certain financial and operating information of companies engaged in activities similar to ours. However, following this offering the ADSs may not trade at a price equal to or greater than the offering price.
To facilitate this offering, the underwriters may engage in transactions that stabilize, maintain or otherwise affect the price of the ADSs during and after this offering. Specifically, the underwriters may over-allot or otherwise create a short position in the ADSs for their own account by selling more ADSs than have been sold to them by us. The underwriters may elect to cover any such short position by purchasing ADSs in the open market or by exercising the over-allotment option granted to the underwriters. In addition, the underwriters may stabilize or maintain the price of the ADSs by bidding for or purchasing ADSs in the open market and may impose penalty bids. If penalty bids are imposed, selling concessions allowed to syndicate members or other broker-dealers participating in this offering are reclaimed if ADSs previously distributed in this offering are repurchased, whether in connection with stabilization transactions or otherwise. The effect of these transactions may be to stabilize or maintain the market price of the ADSs at a level above that which might otherwise prevail in the open market. The imposition of a penalty bid may also affect the price of the ADSs to the extent that it discourages resales of the ADSs. The magnitude or effect of any stabilization or other transactions is uncertain. These transactions may be effected on the NASDAQ Global Market or otherwise and, if commenced, may be discontinued at any time.
In connection with this offering, some underwriters (and selling group members) may also engage in passive market making transactions in the ADSs on the NASDAQ Global Market. Passive market making consists of displaying bids on the NASDAQ Global Market limited by the prices of independent market makers and effecting purchases limited by those prices in response to order flow. Rule 103 of Regulation M promulgated by the SEC limits the amount of net purchases that each passive market maker may make and the displayed size of each bid. Passive market making may stabilize the market price of the ADSs at a level above that which might otherwise prevail in the open market and, if commenced, may be discontinued at any time.
These activities may have the effect of raising or maintaining the market price of the ADSs or preventing or retarding a decline in the market price of the ADSs, and, as a result, the price of the ADSs may be higher than the price that would otherwise prevail in the open market. If the underwriters commence these activities, they may discontinue them at any time. The underwriters may carry out these transactions on the NASDAQ Global Market or otherwise.
Certain of the underwriters and their affiliates may provide from time to time certain commercial banking, financial advisory, investment banking and other services for us and such affiliates in the ordinary course of their business, for which they may receive customary fees and commissions. In addition, from time to time, certain of the underwriters and their affiliates may effect transactions for their own account or the account of customers, and hold on behalf of themselves or their customers, long or short positions in our debt or equity securities or loans, and may do so in the future.
No action has been taken in any jurisdiction (except in the United States) that would permit a public offering of our ADSs, or the possession, circulation or distribution of this prospectus or any other material relating to us or our ADSs in any jurisdiction where action for that purpose is required.
Accordingly, the shares of ADSs may not be offered or sold, directly or indirectly, and neither this prospectus nor any other offering material or advertisements in connection with our ADSs may be distributed or published, in or from any country or jurisdiction, except in compliance with any applicable rules and regulations of any such country or jurisdiction.
163
Each of the underwriters may arrange to sell the ADSs offered hereby in certain jurisdictions outside the United States, either directly or through affiliates, where they are permitted to do so.
European Economic Area. This document has been prepared on the basis that any offer of ADSs in any member state of the European Economic Area which has implemented the Prospectus Directive, (a Relevant Member State), will be made pursuant to an exemption under Article 3 of the Prospectus Directive from the requirement to publish a prospectus for offers of ADSs. Accordingly any person making or intending to make an offer in that Relevant Member State of shares which are the subject of this offering contemplated in this document may only do so in circumstances in which no obligation arises for us or any of the underwriters to publish a prospectus pursuant to Article 3 of the Prospectus Directive in relation to such offer. Neither we nor the underwriters have authorized, nor do they authorize, the making of any offer of ADSs in circumstances in which an obligation arises for us or the underwriters to publish a prospectus for such offer.
In relation to each Relevant Member State, no offer of ADSs may be made to the public in that Relevant Member State other than:
- •
- to any legal entity which is a "qualified investor" as defined in the Prospectus Directive;
- •
- to fewer than 100 or, if the Relevant Member State has implemented the relevant provision of the 2010 PD Amending Directive, 150 natural or legal persons (other than qualified investors as defined in the Prospectus Directive), as permitted under the Prospectus Directive, subject to obtaining the prior consent of the representatives for any such offer; or
- •
- in any other circumstances falling within Article 3(2) of the Prospectus Directive,
provided that no such offer of ADSs shall require us or any underwriter to publish a prospectus pursuant to Article 3 of the Prospectus Directive.
For purposes of this provision, the expression an "offer of securities to the public" in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and the ADSs to be offered, so as to enable an investor to decide to purchase or subscribe for the ADSs, as the expression may be varied in that Relevant Member State by any measure implementing the Prospectus Directive in that member state, the expression "Prospectus Directive" means Directive 2003/71/EC (and amendments thereto, including the 2010 PD Amending Directive, to the extent implemented in the Relevant Member State) and includes any relevant implementing measure in the Relevant Member State, and the expression "2010 PD Amending Directive" means Directive 2010/73/EU.
United Kingdom. This document is only being distributed to, and is only directed at (i) persons who have professional experience in matters relating to investments falling within Article 19(5) of the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005 as amended, or the Order, (ii) persons falling within Article 49(2)(a) to (d) (high net worth companies, unincorporated associations, etc.) of the Order; or (iii) persons to whom an invitation or inducement to engage in investment activity (within the meaning of section 21 of the Financial Services and Markets Act 2000) in connection with the issue or sale of any securities may otherwise lawfully be communicated or caused to be communicated (all such persons together being referred to as "relevant persons"). Any investment or investment activity to which this document relates is available only to relevant persons and will be engaged in only with relevant persons. Any person who is not a relevant person should not act or rely on this document or any of its contents.
France. Neither this prospectus nor any other offering material relating to the ADSs described in this prospectus has been submitted to the clearance procedures of theAutorité des Marchés Financiers or of the competent authority of another member state of the European Economic Area and notified to theAutorité des Marchés Financiers. The ADSs have not been offered or sold and will not be offered
164
or sold, directly or indirectly, to the public in France. Neither this prospectus nor any other offering material relating to the ADSs has been or will be:
- •
- released, issued, distributed or caused to be released, issued or distributed to the public in France; or
- •
- used in connection with any offer for subscription or sale of the ADSs to the public in France.
Such offers, sales and distributions will be made in France only:
- •
- to qualified investors (investisseurs qualifiés) and/or to a restricted circle of investors (cercle restreint d'investisseurs), in each case investing for their own account, all as defined in, and in accordance with articles L.411-2, D.411-1, D.411-2, D.734-1, D.744-1, D.754-1 and D.764-1 of the FrenchCode monétaire et financier;
- •
- to investment services providers authorized to engage in portfolio management on behalf of third parties; or
- •
- in a transaction that, in accordance with article L.411-2-II-1°-or-2°-or 3° of the FrenchCode monétaire et financier and article 211-2 of the General Regulations (Règlement Général) of theAutorité des Marchés Financiers, does not constitute a public offer (appel public à l'épargne).
The ADSs may be resold directly or indirectly, only in compliance with articles L.411-1, L.411-2, L.412-1 and L.621-8 through L.621-8-3 of the FrenchCode monétaire et financier.
Hong Kong. The ADSs may not be offered or sold in Hong Kong by means of any document other than (i) in circumstances which do not constitute an offer to the public within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong), (ii) to "professional investors" within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder, or (iii) in other circumstances which do not result in the document being a "prospectus" within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong) and no advertisement, invitation or document relating to the ADSs may be issued or may be in the possession of any person for the purpose of issue (in each case whether in Hong Kong or elsewhere), which is directed at, or the contents of which are likely to be accessed or read by, the public in Hong Kong (except if permitted to do so under the laws of Hong Kong) other than with respect to ADSs which are or are intended to be disposed of only to persons outside Hong Kong or only to "professional investors" within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder.
India. This prospectus is for information purposes only and does not constitute an offer or invitation for any investment or subscription for ADSs in India. Any person who is in possession of this prospectus is hereby notified that no action has been or will be taken that would allow an offering of the ADSs in India and neither this prospectus nor any offering material relating to the ADSs has been submitted to the Registrar of Companies or the Securities and Exchange Board of India for prior review or approval. Further, no document filing has been made with the Registrar of Companies, India. Accordingly, the ADSs may not be offered, sold, transferred or delivered and neither this prospectus nor any offering material relating to the ADSs may be distributed or made available (in whole or in part) in India, directly or indirectly in connection with any offer or invitation for any investment or subscription for the ADSs in India. You are advised to read this disclaimer carefully and consult with your advisors before accessing, reading or making any other use of this prospectus.
165
Japan. The ADSs offered in this prospectus have not been registered under the Financial Instruments and Exchange Act of Japan. The ADSs have not been offered or sold and will not be offered or sold, directly or indirectly, in Japan or to or for the account of any resident of Japan, except (i) pursuant to an exemption from the registration requirements of the Financial Instruments and Exchange Act of Japan and (ii) in compliance with any other applicable requirements of Japanese law.
Singapore. This prospectus has not been registered as a prospectus with the Monetary Authority of Singapore. Accordingly, this prospectus and any other document or material in connection with the offer or sale, or invitation for subscription or purchase, of the ADSs may not be circulated or distributed, nor may the ADSs be offered or sold, or be made the subject of an invitation for subscription or purchase, whether directly or indirectly, to persons in Singapore other than (i) to an institutional investor under Section 274 of the Securities and Futures Act, Chapter 289 of Singapore, or the SFA, (ii) to a relevant person pursuant to Section 275(1), or any person pursuant to Section 275(1A), and in accordance with the conditions specified in Section 275 of the SFA or (iii) otherwise pursuant to, and in accordance with the conditions of, any other applicable provision of the SFA, in each case subject to compliance with conditions set forth in the SFA.
Where the ADSs are subscribed or purchased under Section 275 of the SFA by a relevant person which is:
- •
- a corporation (which is not an accredited investor (as defined in Section 4A of the SFA)) the sole business of which is to hold investments and the entire share capital of which is owned by one or more individuals, each of whom is an accredited investor; or
- •
- a trust (where the trustee is not an accredited investor) whose sole purpose is to hold investments and each beneficiary of the trust is an individual who is an accredited investor, shares, debentures and units of shares and debentures of that corporation or the beneficiaries' rights and interest (howsoever described) in that trust shall not be transferred within six months after that corporation or that trust has acquired the ADSs pursuant to an offer made under Section 275 of the SFA except:
- •
- to an institutional investor (for corporations, under Section 274 of the SFA) or to a relevant person defined in Section 275(2) of the SFA, or to any person pursuant to an offer that is made on terms that such shares, debentures and units of shares and debentures of that corporation or such rights and interest in that trust are acquired at a consideration of not less than $200,000 (or its equivalent in a foreign currency) for each transaction, whether such amount is to be paid for in cash or by exchange of securities or other assets, and further for corporations, in accordance with the conditions specified in Section 275 of the SFA;
- •
- where no consideration is or will be given for the transfer; or
- •
- where the transfer is by operation of law.
In addition, investors in Singapore should note that the securities acquired by them are subject to resale and transfer restrictions specified under Section 276 of the SFA, and they, therefore, should seek their own legal advice before effecting any resale or transfer of their securities.
Switzerland. The ADSs may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange, or SIX, or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under Article 652a or Article 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other
166
offering or marketing material relating to the ADSs or this offering may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering or marketing material relating to this offering, the Company or the ADSs have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of ADSs will not be supervised by, the Swiss Financial Market Supervisory Authority, and the offer of ADSs has not been and will not be authorized under the Swiss Federal Act on Collective Investment Schemes, or the CISA. The investor protection afforded to acquirers of interests in collective investment schemes under the CISA does not extend to acquirers of ADSs.
167
EXPENSES RELATED TO THIS OFFERING
The following table sets forth the main expenses we will be required to pay in connection with this offering, other than the underwriting discounts and commissions. All amounts are estimated, except the SEC registration fee, the FINRA filing fee and the NASDAQ listing fee:
Expenses | Amount | |||
|---|---|---|---|---|
SEC registration fee | $ | * | ||
FINRA filing fee | * | |||
NASDAQ listing fee | * | |||
Legal fees and expenses | * | |||
Accounting fees and expenses | * | |||
Printing fees | * | |||
Other fees and expenses | * | |||
Total | $ | * | ||
- *
- To be furnished by amendment.
168
Proskauer Rose LLP has advised the Company on certain U.S. legal matters relating to this offering, and Osborne Clarke BV CVBA has advised the Company on certain Belgian legal matters relating to the offering. Goodwin Procter LLP has advised the underwriters on certain U.S. legal matters relating to the offering, and NautaDutilh has advised the underwriters on certain Belgian legal matters relating to the offering.
169
The consolidated financial statements of TiGenix as of December 31, 2013 and 2012 and for each of the two years in the period ended December 31, 2013 included in this prospectus and in the registration statement of which this prospectus forms a part have been so included in reliance on the report of BDO Bedrijfsrevisoren Burg. CVBA, an independent registered public accounting firm (the report on the consolidated financial statements contains an explanatory paragraph regarding the Company's ability to continue as a going concern), appearing elsewhere herein and in the registration statement, given on the authority of said firm as experts in auditing and accounting.
BDO Bedrijfsrevisoren Burg. CVBA, Zaventem, Belgium, is a member of the Instituut van de Bedrijfsrevisoren / Institut des Réviseurs d'Entreprises.
170
SERVICE OF PROCESS AND ENFORCEMENT OF CIVIL LIABILITIES
We are a Belgian company organized with limited liability. Our registered offices and the majority of our assets are located outside of the United States. In addition, except for one board member, all of our directors and senior management and the experts named herein are residents of jurisdictions other than the United States. As a result, it may not be possible for you to effect service of process within the United States upon these individuals or our Company, to enforce judgments obtained in U.S. courts against these individuals or our Company in courts outside the United States, or to enforce against these individuals and our Company, whether in original actions or in actions for the enforcement of judgments of U.S. courts, civil liabilities based solely upon U.S. federal or state securities laws.
The United States currently does not have a treaty with Belgium providing for the reciprocal recognition and enforcement of judgments, other than arbitral awards, in civil and commercial matters. Consequently, a final judgment rendered by any federal or state court in the United States, whether or not predicated solely upon U.S. federal or state securities laws, would not automatically be enforceable in Belgium. Actions for the enforcement of judgments of U.S. courts are regulated by Articles 22 to 25 of the 2004 Belgian Code of Private International Law. Recognition or enforcement does not imply a review of the merits of the case and is irrespective of any reciprocity requirement. A U.S. judgment will, however, not be recognized or declared enforceable in Belgium, unless (in addition to compliance with certain technical provisions) the Belgian courts are satisfied of the following:
- •
- The effect of the recognition or enforcement of judgment is not manifestly incompatible with (Belgian) public order.
- •
- The judgment did not violate the rights of the defendant.
- •
- The judgment was not rendered in a matter where the parties did not freely dispose of their rights, with the sole purpose of avoiding the application of the law applicable according to Belgian international law.
- •
- The judgment is not subject to further recourse under U.S. law.
- •
- The judgment is not incompatible with a judgment rendered in Belgium or with a prior judgment rendered abroad that might be enforced in Belgium.
- •
- The claim was not filed outside Belgium after a claim was filed in Belgium, if the claim filed in Belgium relates to the same parties and the same purpose and is still pending.
- •
- The Belgian courts did not have exclusive jurisdiction to rule on the matter.
- •
- The U.S. court did not accept its jurisdiction solely on the basis of either the presence of the plaintiff or the location of the disputed goods in the United States.
- •
- The judgment did not concern the deposit or validity of intellectual property rights when the deposit or registration of those intellectual property rights was requested, done or should have been done in Belgium pursuant to international treaties.
- •
- The judgment did not relate to the validity, operation, dissolution, or liquidation of a legal entity that has its main seat in Belgium at the time of the petition of the U.S. court.
- •
- If the judgment relates to the opening, progress or closure of insolvency proceedings, it is rendered on the basis of the European Insolvency Regulation (EC Regulation No. 1346/2000 of May 29, 2000) or, if not, that (a) a decision in the principal proceedings is taken by a judge in the state where the most important establishment of the debtor was located or (b) a decision in territorial proceedings was taken by a judge in the state where the debtor had another establishment than its most important establishment.
- •
- The judgment submitted to the Belgian court is authentic.
171
In addition, with regard to the enforcement by legal proceedings of any claim (including the exequatur of foreign court decisions in Belgium), a registration tax of 3% (to be calculated on the total amount that a debtor is ordered to pay) is due, if the sum of money that the debtor is ordered to pay by a Belgian court judgment, or by a foreign court judgment that is either (i) automatically enforceable and registered in Belgium or (ii) rendered enforceable by a Belgian court, exceeds 12,500 euros. The debtor and the creditor are jointly liable for the payment of the registration tax; however, the liability of the creditor is limited up to a maximum amount of half of the amount he recovers from the debtor. An exemption from such registration tax applies in respect of exequaturs of judgments rendered by courts of states that are bound by European Regulation 44/2001.
172
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form F-1 under the Securities Act, including amendments and relevant exhibits and schedules, covering the underlying ordinary shares represented by the ADSs to be sold in this offering. The depositary will also file with the SEC a related registration statement on Form F-6 to register the ADSs. This prospectus, which constitutes a part of the registration statement on Form F-1, summarizes material provisions of contracts and other documents that we refer to in the prospectus. Since this prospectus does not contain all of the information contained in the registration statement, you should read the registration statement and its exhibits and schedules for further information with respect to us and the ADSs.
Immediately upon the effectiveness of the registration statement, we will become subject to periodic reporting and other informational requirements of the Exchange Act as applicable to foreign private issuers. Our annual reports on Form 20-F for the year ended December 31, 2015 and for all subsequent years will be due within four months after fiscal year end. We are not required to disclose certain other information that is required from U.S. domestic issuers. Also, as a foreign private issuer, we are exempt from the rules of the Exchange Act prescribing the furnishing of proxy statements to shareholders and our directors, senior management and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act.
You may review and copy the registration statement, reports and other information we file at the SEC's public reference room at 100 F Street, N.E., Washington, D.C. 20549. You may also request copies of these documents upon payment of a duplicating fee by writing to the SEC. For further information on the public reference facility, please call the SEC at 1-800-SEC-0330. Our SEC filings, including the registration statement, are also available to you on the SEC's website at http://www.sec.gov.
As a foreign private issuer, we are also exempt from the requirements of Regulation FD (Fair Disclosure) which, generally, are meant to ensure that select groups of investors are not privy to specific information about an issuer before other investors. We are, however, still subject to the anti-fraud and anti-manipulation rules of the SEC, such as Rule 10b-5. Since many of the disclosure obligations required of us as a foreign private issuer are different than those required by other U.S. domestic reporting companies, our shareholders, potential shareholders and the investing public in general should not expect to receive information about us in the same amount and at the same time as information is received from, or provided by, other U.S. domestic reporting companies. We are liable for violations of the rules and regulations of the SEC which do apply to us as a foreign private issuer.
We have filed our amended and restated articles of association and all other deeds that are to be published in the annexes to the Belgian State Gazette with the clerk's office of the Commercial Court of Leuven (Belgium), where they are available to the public. A copy of our amended and restated articles of association will also be publicly available as an exhibit to the registration statement of which this prospectus forms a part. In accordance with Belgian law, we must prepare audited annual statutory and consolidated financial statements. The audited annual statutory and consolidated financial statements and the reports of our board and statutory auditor relating thereto are filed with the Belgian National Bank, where they are available to the public, and are also available on our website.
173
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM | F-2 | |||
CONSOLIDATED INCOME STATEMENTS | F-3 | |||
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME | F-4 | |||
CONSOLIDATED STATEMENTS OF FINANCIAL POSITION | F-5 | |||
CONSOLIDATED STATEMENTS OF CASH FLOWS | F-6 | |||
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY | F-7 | |||
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS | F-8 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Shareholders
Tigenix
Leuven, Belgium
We have audited the accompanying consolidated statements of financial position of Tigenix as at December 31, 2013 and 2012 and the related consolidated income statements and statements of comprehensive income, changes in equity, and cash flows for each of the two years in the period ended December 31, 2013. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Tigenix at December 31, 2013 and 2012, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2013, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As described in Note 2 to the consolidated financial statements, the Company has suffered recurring losses from operations and has a net capital deficiency that raise substantial doubt about its ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty. Our opinion is not modified with respect to this matter.
Zaventem, October 2, 2014
/s/ Gert Claes
BDO Bedrijfsrevisoren Burg. CVBA
Represented by
Gert Claes
F-2
CONSOLIDATED INCOME STATEMENTS
| | | Six-month period ended June 30, | Years ended December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Thousands of euros except per share data | Notes | 2014 | 2013 | 2013 | 2012 | ||||||||||
| | | (Unaudited) | | | |||||||||||
CONTINUING OPERATIONS | |||||||||||||||
Revenues | |||||||||||||||
Royalties | — | — | — | — | |||||||||||
Grants and other operating income | 5 | 821 | 736 | 883 | 1,389 | ||||||||||
| | | | | | | | | | | | | | | | |
Total revenues | 821 | 736 | 883 | 1,389 | |||||||||||
Research and development expenses | 6 | (5,097 | ) | (5,314 | ) | (9,705 | ) | (12,136 | ) | ||||||
General and administrative expenses | 6 | (2,859 | ) | (2,735 | ) | (5,829 | ) | (6,237 | ) | ||||||
Total operating charges | (7,956 | ) | (8,049 | ) | (15,534 | ) | (18,373 | ) | |||||||
| | | | | | | | | | | | | | | | |
Operating Loss | (7,135 | ) | (7,313 | ) | (14,651 | ) | (16,984 | ) | |||||||
Financial income | 7 | 25 | 5 | 7 | 35 | ||||||||||
Financial expenses | 7 | (369 | ) | (28 | ) | (45 | ) | (58 | ) | ||||||
Foreign exchange differences | 7 | 170 | (38 | ) | (352 | ) | (142 | ) | |||||||
| | | | | | | | | | | | | | | | |
Loss before taxes | (7,309 | ) | (7,374 | ) | (15,041 | ) | (17,149 | ) | |||||||
Income taxes | 8 | — | 42 | 59 | (1 | ) | |||||||||
| | | | | | | | | | | | | | | | |
Loss for the period from continuing operations | (7,309 | ) | (7,332 | ) | (14,982 | ) | (17,150 | ) | |||||||
DISCONTINUED OPERATIONS | |||||||||||||||
Loss for the period from discontinued operations | 9 | (1,842 | ) | (1,505 | ) | (3,408 | ) | (3,243 | ) | ||||||
| | | | | | | | | | | | | | | | |
Loss for the period | (9,151 | ) | (8,837 | ) | (18,390 | ) | (20,393 | ) | |||||||
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
Attributable to equity holders of TiGenix | (9,151 | ) | (8,837 | ) | (18,390 | ) | (20,393 | ) | |||||||
Basic and diluted loss per share (euro) | 10 | (0.06 | ) | (0.09 | ) | (0.16 | ) | (0.22 | ) | ||||||
Basic and diluted loss per share from continuing operations (euro) | 10 | (0.05 | ) | (0.07 | ) | (0.13 | ) | (0.19 | ) | ||||||
Basic and diluted loss per share from discontinued operations (euro) | 10 | (0.01 | ) | (0.02 | ) | (0.03 | ) | (0.04 | ) | ||||||
The accompanying notes form an integral part of these consolidated financial statements.
F-3
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME
| | Six-month period ended June 30, | Years ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Thousands of euros | 2014 | 2013 | 2013 | 2012 | |||||||||
| | (Unaudited) | | | ||||||||||
Loss for the period | (9,151 | ) | (8,837 | ) | (18,390 | ) | (20,393 | ) | |||||
Items of other comprehensive income that may be reclassified subsequently to the income statement | |||||||||||||
Currency translation differences | (6 | ) | 61 | 366 | 41 | ||||||||
Other comprehensive income | (6 | ) | 61 | 366 | 41 | ||||||||
| | | | | | | | | | | | | | |
Total comprehensive income | (9,157 | ) | (8,776 | ) | (18,024 | ) | (20,352 | ) | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Attributable to equity holders of TiGenix | (9,157 | ) | (8,776 | ) | (18,024 | ) | (20,352 | ) | |||||
The accompanying notes form an integral part of these consolidated financial statements.
F-4
CONSOLIDATED STATEMENTS OF FINANCIAL POSITION
| | | As at June 30, | As at December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Thousands of euros | Notes | 2014 | 2013 | 2012 | |||||||||
| | | (Unaudited) | | | |||||||||
ASSETS | |||||||||||||
Intangible assets | 12 | 34,912 | 36,407 | 39,205 | |||||||||
Property, plant and equipment | 13 | 735 | 879 | 8,334 | |||||||||
Available-for-sale investments | 14 | 162 | 162 | 278 | |||||||||
Other non current assets | 15 | 1,923 | 1,415 | 498 | |||||||||
| | | | | | | | | | | | | | |
Non-current assets | 37,732 | 38,863 | 48,315 | ||||||||||
Inventories | 16 | 86 | 77 | 105 | |||||||||
Trade and other receivables | 17 | 2,179 | 1,583 | 3,661 | |||||||||
Other current financial assets | 18 | 949 | 820 | 804 | |||||||||
Receivables from reverse repurchase agreements | 19 | 5,999 | — | — | |||||||||
Cash and cash equivalents | 13,186 | 15,565 | 11,072 | ||||||||||
| | | | | | | | | | | | | | |
Current assets | 22,399 | 18,045 | 15,642 | ||||||||||
Assets held for sale | 11 | — | 6,135 | — | |||||||||
| | | | | | | | | | | | | | |
TOTAL ASSETS | 60,131 | 63,043 | 63,957 | ||||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
EQUITY AND LIABILITIES | |||||||||||||
Share capital | 16,048 | 16,048 | 10,030 | ||||||||||
Share premium | 100,118 | 100,125 | 88,852 | ||||||||||
Accumulated deficit | (83,200 | ) | (74,049 | ) | (55,700 | ) | |||||||
Other reserves | 6,374 | 6,098 | 5,386 | ||||||||||
| | | | | | | | | | | | | | |
Equity attributable to equity holders | 39,340 | 48,222 | 48,568 | ||||||||||
Total equity | 20 | 39,340 | 48,222 | 48,568 | |||||||||
Financial loans and other payables | 21 | 14,238 | 8,263 | 6,184 | |||||||||
Deferred tax liability | 22 | 29 | 29 | 27 | |||||||||
Other non-current liabilities | 23 | 86 | 86 | 95 | |||||||||
| | | | | | | | | | | | | | |
Non-current liabilities | 14,353 | 8,378 | 6,306 | ||||||||||
Current portion of financial loans | 21 | 789 | 343 | 388 | |||||||||
Other financial liabilities | 21 | 1,302 | 874 | 1,527 | |||||||||
Trade and other payables | 24 | 2,183 | 3,007 | 4,014 | |||||||||
Other current liabilities | 25 | 2,164 | 1,653 | 3,154 | |||||||||
| | | | | | | | | | | | | | |
Current liabilities | 6,438 | 5,877 | 9,083 | ||||||||||
Liabilities related to non-current assets held for sale | 11 | — | 566 | — | |||||||||
| | | | | | | | | | | | | | |
TOTAL EQUITY AND LIABILITIES | 60,131 | 63,043 | 63,957 | ||||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements.
F-5
CONSOLIDATED STATEMENTS OF CASH FLOWS
| | Six-month period ended June 30, | Years ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Thousands of euros | 2014 | 2013 | 2013 | 2012 | |||||||||
| | (Unaudited) | | | ||||||||||
CASH FLOWS FROM OPERATING ACTIVITIES | |||||||||||||
Operating loss | (7,135 | ) | (7,313 | ) | (14,651 | ) | (16,984 | ) | |||||
Adjustments for: | |||||||||||||
Depreciation and amortisation expense | 1,569 | 2,172 | 3,258 | 3,687 | |||||||||
Gain/(loss) on sale of property, plant and equipment | — | 20 | — | — | |||||||||
Share-based compensation | 275 | 237 | 348 | 612 | |||||||||
Grants income | (817 | ) | (653 | ) | (774 | ) | (887 | ) | |||||
Other | 65 | 61 | 112 | 23 | |||||||||
| | | | | | | | | | | | | | |
| (6,043 | ) | (5,476 | ) | (11,707 | ) | (13,549 | ) | ||||||
Movements in working capital: | |||||||||||||
(Increase)/ decrease in inventories | (9 | ) | (72 | ) | (6 | ) | 230 | ||||||
(Increase)/ decrease in trade and other receivables | (129 | ) | 283 | (52 | ) | (157 | ) | ||||||
(Increase)/ decrease in other financial assets | — | (21 | ) | (16 | ) | (286 | ) | ||||||
(Increase)/decrease in other current assets | 85 | (133 | ) | 19 | 352 | ||||||||
Increase/(decrease) in trade and other payables | (293 | ) | (541 | ) | (975 | ) | (722 | ) | |||||
Increase/(decrease) in other current liabilities | 1,434 | (435 | ) | (1,744 | ) | (709 | ) | ||||||
| | | | | | | | | | | | | | |
Cash used in operations | (4,955 | ) | (6,395 | ) | (14,483 | ) | (14,841 | ) | |||||
Income taxes paid/received | — | 42 | 20 | — | |||||||||
Cash flow from discontinued operations | (1,138 | ) | (1,464 | ) | 36 | (2,786 | ) | ||||||
| | | | | | | | | | | | | | |
Net cash used in operating activities | (6,093 | ) | (7,817 | ) | (14,427 | ) | (17,627 | ) | |||||
| | | | | | | | | | | | | | |
CASH FLOWS FROM INVESTING ACTIVITIES | |||||||||||||
Interests received | 18 | 1 | 4 | 9 | |||||||||
Acquisition of property, plant and equipment | (29 | ) | (37 | ) | (35 | ) | (24 | ) | |||||
Acquisition of intangible assets | (67 | ) | (129 | ) | (323 | ) | (267 | ) | |||||
Proceeds from disposal of property, plant and equipment | — | (23 | ) | 12 | 124 | ||||||||
Increase of other non-current assets | 25 | (4 | ) | (917 | ) | (13 | ) | ||||||
Amounts paid under reverse repurchase agreements | (5,999 | ) | — | — | — | ||||||||
Cash flow from discontinued operations | 3,500 | (33 | ) | (61 | ) | (550 | ) | ||||||
| | | | | | | | | | | | | | |
Net cash used in investing activities | (2,552 | ) | (225 | ) | (1,320 | ) | (721 | ) | |||||
| | | | | | | | | | | | | | |
CASH FLOWS FROM FINANCING ACTIVITIES | |||||||||||||
Net proceeds from issue of equity instruments of the Company | — | (132 | ) | 17,694 | 6,289 | ||||||||
Reimbursements of subordinated loan | — | — | — | (130 | ) | ||||||||
Net proceeds from financial loans | 7,166 | 1,395 | 2,380 | 1,527 | |||||||||
Reimbursements of financial loans | (160 | ) | (605 | ) | (114 | ) | (114 | ) | |||||
Reimbursements of other financial liabilities | (441 | ) | — | — | — | ||||||||
Proceeds from government grants | — | 63 | 324 | 2,123 | |||||||||
Interests paid | (299 | ) | (14 | ) | (47 | ) | (48 | ) | |||||
Cash flow from discontinued operations | — | — | — | — | |||||||||
| | | | | | | | | | | | | | |
Net cash provided by financing activities | 6,266 | 707 | 20,237 | 9,647 | |||||||||
| | | | | | | | | | | | | | |
Net increase/(decrease) in cash and cash equivalents | (2,379 | ) | (7,335 | ) | 4,490 | (8,701 | ) | ||||||
Cash and cash equivalents at beginning of the period | 15,565 | 11,072 | 11,072 | 19,771 | |||||||||
Effect of currency translation on cash and cash equivalents | — | 1 | 3 | 2 | |||||||||
| | | | | | | | | | | | | | |
Cash and cash equivalents at end of period | 13,186 | 3,738 | 15,565 | 11,072 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements.
F-6
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
| | Attributable to equity holders of the Company | | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | | | | | | Other reserves | | ||||||||||||||||||
Thousands of euros except share data | Numbers of shares | Share capital | Share premium | Shares to be issued | Accumulated deficits | Equity-settled employee benefits reserve | Translation reserves | Total Equity | |||||||||||||||||
At January 1, 2012 | 91,122,667 | 89,093 | 81,656 | 2,296 | (115,759 | ) | 5,326 | (593 | ) | 62,019 | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Loss for the period | — | — | — | (20,393 | ) | — | — | (20,393 | ) | ||||||||||||||||
Other comprehensive income | — | — | — | — | — | 41 | 41 | ||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive income | — | — | — | (20,393 | ) | — | 41 | (20,352 | ) | ||||||||||||||||
Capital decrease | — | (80,452 | ) | — | — | 80,452 | — | — | — | ||||||||||||||||
Issuance of shares (contribution in kind) | 536,534 | 526 | 1,770 | (2,296 | ) | — | — | — | — | ||||||||||||||||
Issuance of shares (contribution in cash) | 8,629,385 | 863 | 5,868 | — | — | — | — | 6,731 | |||||||||||||||||
Transaction costs | — | — | (442 | ) | — | — | — | — | (442 | ) | |||||||||||||||
Share-based compensation | — | — | — | — | — | 612 | — | 612 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
At December 31, 2012 | 100,288,586 | 10,030 | 88,852 | — | (55,700 | ) | 5,938 | (552 | ) | 48,568 | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Loss for the period | — | — | — | — | (18,390 | ) | — | — | (18,390 | ) | |||||||||||||||
Other comprehensive income | — | — | — | — | — | — | 366 | 366 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive income | — | — | — | (18,390 | ) | — | 366 | (18,024 | ) | ||||||||||||||||
Issuance of shares | 60,188,034 | 6,018 | 12,481 | — | — | — | — | 18,499 | |||||||||||||||||
Transaction costs | — | — | (1,208 | ) | — | — | — | — | (1,208 | ) | |||||||||||||||
Share-based compensation | — | — | — | — | 41 | 346 | — | 387 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
At December 31, 2013 | 160,476,620 | 16,048 | 100,125 | — | (74,049 | ) | 6,284 | (186 | ) | 48,222 | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Loss for the period | — | — | — | — | (9,151 | ) | — | — | (9,151 | ) | |||||||||||||||
Other comprehensive income | — | — | — | — | — | — | (6 | ) | (6 | ) | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
Total comprehensive income | — | — | — | (9,151 | ) | — | (6 | ) | (9,157 | ) | |||||||||||||||
Transaction costs | — | — | (19 | ) | — | — | — | — | (19 | ) | |||||||||||||||
Share-based compensation | — | — | — | — | — | 282 | — | 282 | |||||||||||||||||
Other | — | — | 12 | — | — | — | — | 12 | |||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
At June 30, 2014 (Unaudited) | 160,476,620 | 16,048 | 100,118 | — | (83,200 | ) | 6,566 | (192 | ) | 39,340 | |||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes form an integral part of these consolidated financial statements.
F-7
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
1. General information
TiGenix (the "Company", and together with its subsidiaries, the "Group", "we" or "us") is an advanced biopharmaceutical company focused on developing and commercializing novel therapeutics from our proprietary platform of allogeneic, or donor-derived, expanded adipose-derived stem cells, known as eASCs, in inflammatory and autoimmune diseases. Based on our proprietary technology platform, we have developed a pipeline of product candidates, including Cx601, which is in Phase III for the treatment of perianal fistulas in Crohn's disease patients, Cx611, for early rheumatoid arthritis in Phase II and for severe sepsis in preparation for Phase I, and Cx621, which completed a Phase I clinical trial for the intra-lymphatic administration of allogeneic eASCs. We also developed and commercialized ChondroCelect, the first cell-based medicinal product to receive marketing authorization from the EMA, which is indicated for cartilage repair in the knee.
TiGenix is a limited liability company incorporated and domiciled in Belgium. The registered office is located at Romeinse straat 12, bus 2, 3001 Leuven, Belgium.
The consolidated financial statements of the Group for the years ended December 31, 2013 and 2012 were approved and authorized for issue on October 2, 2014 in accordance with a resolution of the Company's board of directors.
2. Summary of significant accounting policies
2.1. Basis of preparation
The Group's consolidated financial statements have been prepared in accordance with International Financial Reporting Standards or IFRS, as issued by the International Accounting Standards Board or IASB.
The principal accounting policies applied in the preparation of the consolidated financial statements are set out below. These policies have been consistently applied to all of the periods presented, unless otherwise stated.
The discontinuation of the ChondroCelect operations was successfully completed during the first half of 2014 through the combination of the sale of the Dutch manufacturing facility and a licensing agreement.
On May 30, 2014, the Group completed the sale of TiGenix B.V., our Dutch subsidiary, which held our manufacturing facility, to PharmaCell, a leading European contract manufacturing organization active in the area of cell therapy, for a total consideration of 4.3 million euros. Under the terms of the share purchase agreement with PharmaCell, we received an upfront payment of 3.5 million euros when the sale became effective on May 30, 2014 and will receive a final payment of 0.8 million euros (recognized at its present value of 0.5 million euros) on May 30, 2017. In addition, the sale includes a cost relief of 1.5 million euros under the terms of a long-term manufacturing agreement with our former subsidiary, which is now owned by PharmaCell, to continue manufacturing ChondroCelect at the facility. The 1.5 million euros (total net present value of 1.2 million euros) cost relief has not been included as part of the selling price, because it has been passed on to Sobi. Sobi will purchase all of the ChondroCelect produced by our former subsidiary at cost under the terms of a distribution agreement, as described below. Therefore, the total loss from the TiGenix B.V. disposal recognized as of June 30, 2014 amounts to 1.1 million euros (additional to the impairment of 0.7 million euros recognized at December 31, 2013) included as discontinued operations.
F-8
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
On June 1, 2014, we completed the licensing of the marketing and distribution rights of ChondroCelect to Swedish Orphan Biovitrum, or Sobi, the international specialty healthcare company dedicated to rare diseases. Sobi will continue to market and distribute the product within the European Union (excluding Finland where we have a pre-existing distribution agreement with Finnish Red Cross Blood Service), Switzerland, Norway, Russia, Turkey and the Middle East and North Africa region (currently excluding certain countries where we have a pre-existing distribution agreement with Genpharm). We will receive royalties on the net sales of ChondroCelect, and Sobi will reimburse nearly all of our costs associated with the product.
As a consequence, the ChondroCelect operations, which are deemed as a separate component, have been classified as a discontinued operation. Our financial statements for prior periods have been re-classified in accordance with the requirements of IFRS 5 Non-current Assets Held for Sale and Discontinued Operations. See Note 9.
All amounts are presented in thousands of euros, unless otherwise indicated, rounded to the nearest 1,000 euro.
The financial statements have been prepared on the basis of the historical cost method. Any exceptions to the historical cost method are disclosed in the valuation rules described hereafter.
The preparation of financial statements in compliance with IFRS requires the use of certain critical accounting estimates. It also requires the Group's management to exercise judgment in applying the Group's accounting policies. The areas where significant judgments and estimates have been made in preparing the financial statements and their effect are disclosed in Note 3.
Unaudited interim financial statements
The accompanying interim statements of financial position as at June 30, 2014, the statements of income, comprehensive income, cash flows and changes in equity for the six-month periods ended June 30, 2014 and 2013, are unaudited. These unaudited interim financial statements have been prepared in accordance with International Accounting Standard 34 "Interim Financial Reporting." These interim consolidated financial statements do not include all the information required for full annual financial statements in accordance with IFRS as issued by the IASB and should be read in conjunction with the consolidated financial statements of the Group as at and for the year ended December 31, 2013. In the opinion of the Group's management, the unaudited interim financial statements have been prepared on the same basis as the audited financial statements and include all adjustments, which include only normal recurring adjustments, necessary to present fairly the Group's statement of financial position as at June 30, 2014 and its results of operations and its cash flows for the six-month periods ended June 30, 2014 and 2013. The results for the six-month period ended June 30, 2014 are not necessarily indicative of the results expected for the full year.
Liquidity
The Group has experienced net losses and significant cash outflows from cash used in operating activities since inception, and as at December 31, 2013 had an accumulated deficit of 74.0 million euros, a net loss of 18.4 million euros and net cash used in operating activities of 14.4 million euros and as at June 30, 2014, had an accumulated deficit of 83.2 million euros, a net loss of 9.2 million euros and net cash used in operating activities of 6.1 million euros.
F-9
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
The Group is subject to a number of risks similar to those of other pre-commercial stage companies, including its dependence on key individuals, uncertainty of product development and generation of revenues, dependence on outside sources of capital, risks associated with research, development, testing, and obtaining related regulatory approvals of its pipeline products, dependence on third party manufacturers, suppliers and collaborators, successful protection of intellectual property, competition with larger, better-capitalized companies, successful completion of the Group's development programs. Ultimately, the attainment of profitable operations is dependent on future events, including obtaining adequate financing to fulfill its development activities and generating a level of revenues adequate to support the Group's cost structure.
The Group has sufficient funds to continue operating until the end of the third quarter of 2015, but will require significant additional cash resources to initiate new clinical trials related to its pipeline, to continue seeking regulatory approval of its pipeline and to repay its debt obligations. These conditions, among others, raise substantial doubt about the Group's ability to continue as a going concern. The accompanying consolidated financial statements have been prepared assuming that the Group will continue as a going concern. This basis of accounting contemplates the recovery of the Group's assets and the satisfaction of liabilities in the normal course of business. A successful transition to attaining profitable operations is dependent upon achieving a level of positive cash flows adequate to support the Group's cost structure.
To support the Group's financial performance, management has undertaken several initiatives recently.
Under the loan facility agreement dated December 20, 2013 with Kreos Capital IV (UK), we are permitted to borrow up to an amount of 10.0 million euros, of which 7.5 million euros were drawn as at June 30, 2014 as disclosed in Note 21. Additionally, 2.5 million euros from the credit facility were drawn on September 30, 2014. (See Note 30).
On May 30, 2014, we completed the sale of TiGenix B.V., our Dutch subsidiary, which held our manufacturing facility, to PharmaCell, a leading European contract manufacturing organization, for a total consideration of 4.3 million euros. Under the terms of the share purchase agreement, we received an upfront payment of 3.5 million euros when the sale became effective on May 30, 2014 and will receive a final payment of 0.8 million euros on May 30, 2017. ChondroCelect will continue to be manufactured at the facility under a contract manufacturing agreement with our former subsidiary.
On June 1, 2014, we entered into an agreement with Sobi for the marketing and distribution rights with respect to ChondroCelect. Sobi will continue to market and distribute the product within the European Union (excluding Finland, where we have a pre-existing distribution agreement with Finnish Red Cross Blood Service), Switzerland, Norway, Russia, Turkey and the Middle East and North Africa region, currently excluding certain countries where we have a pre-existing distribution agreement with Genpharm. We will receive royalties on the net sales of ChondroCelect, and Sobi will reimburse nearly all of our costs in connection with the product. Under the agreement, Sobi will purchase ChondroCelect from us at the price at which we purchase it from our former subsidiary, and the agreements with Sobi and our former subsidiary both include corresponding commitments for minimum binding quantities of ChondroCelect that are required to be purchased by us and from us. The agreement with Sobi will result in significant cost savings for us on an ongoing basis, because we have eliminated all expenses in connection with the marketing of ChondroCelect, including all of our marketing personnel and has allowed us to devote our resources and the attention of our management exclusively to the development of our pipeline of eASC-based product candidates.
F-10
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
We will continue to consider additional business opportunities to allow us to develop our pipeline and generate additional revenues, including by accessing the capital markets within the next twelve months. We expect to use any capital obtained from such fund raisings or other arrangements to further develop our eASC-based product candidates.
As at June 30, 2014, the Group had a liquidity position of 19.2 million euros, consisting of cash and cash equivalents and receivables from reverse repurchase agreements. The board of directors of the Company is of the opinion that this liquidity position and the aforementioned actions will enable it to fund its operating expenses and capital expenditure requirements at least until the end of the third quarter of 2015.
The future viability of the Group is dependent on its ability to generate cash from operating activities, to raise additional capital to finance its operations or to successfully obtain regulatory approval to allow marketing of the Group's products. The Group's failure to raise capital as and when needed could have a negative impact on its financial condition and ability to pursue its business strategies.
The consolidated financial statements do not include any adjustments due to this uncertainty relating to the recoverability and classification of recorded asset amounts and classification of liabilities.
- a)
- New and amended standards adopted by the Group
A number of new standards, interpretations and amendments effective for the first time for periods beginning on (or after) January 1, 2013, have been adopted in these financial statements. The nature and effect of each new standard, interpretation and amendment adopted by the Group is detailed below. Not all new standards and interpretations effective for the first time for periods beginning on (or after) January 1, 2013 affect the Group's annual consolidated financial statements.
- —
- IFRS 10Consolidated Financial Statements
IFRS 10 replaces the parts of IAS 27Consolidated and Separate Financial Statements that deal with consolidated financial statements and SIC-12Consolidation—Special Purpose Entities. IFRS 10 changes the definition of control such that an investor has control over an investee when a) it has power over the investee, b) it is exposed, or has rights, to variable returns from its involvement with the investee and c) has the ability to use its power to affect its returns. All three of these criteria must be met for an investor to have control over an investee.
Previously, control was defined as the power to govern the financial and operating policies of an entity so as to obtain benefits from its activities. Additional guidance has been included in IFRS 10 to explain when an investor has control over an investee. Some guidance included in IFRS 10 that deals with whether or not an investor that owns less than 50% of the voting rights in an investee has control over the investee is relevant to the Group.
The adoption of IFRS 10 has not changed the consolidation scope of the Group.
- —
- IFRS 12Disclosures of Interests in Other Entities
IFRS 12 is a new disclosure standard and is applicable to entities that have interests in subsidiaries, joint arrangements, associates and/or unconsolidated structured entities. The standard requires a reporting entity to disclose information that helps users to assess the nature and financial effects of the reporting entity's relationship with other entities.
F-11
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
As the new standard affects only disclosure, there is no effect on the Group's financial position or performance.
- —
- IFRS 13Fair Value Measurement
The Group applied IFRS 13 for the first time in the financial year 2013. IFRS 13 establishes a single source of guidance for fair value measurements and disclosures about fair value measurements. The scope of IFRS 13 is broad; the fair value measurement requirements of IFRS 13 apply to both financial instrument items and non-financial instrument items for which other IFRSs require or permit fair value measurements and disclosures about fair value measurements, except for share-based payment transactions that are within the scope of IFRS 2Share-based Payment, leasing transactions that are within the scope of IAS 17Leases, and measurements that have some similarities to fair value but are not fair value (e.g. net realizable value for the purposes of measuring inventories or value in use for impairment assessment purposes).
IFRS 13 sets out the framework for determining the measurement of fair value and the disclosure of information relating to fair value measurement, when fair value measurements and/or disclosures are required or permitted by other IFRSs.
As a result, the guidance and requirements relating to fair value measurement that were previously located in other IFRSs have now been relocated to IFRS 13.
While there has been some rewording of the previous guidance, there are few changes to the previous fair value measurement requirements. Instead, IFRS 13 is intended to clarify the measurement objective, harmonize the disclosure requirements, and improve consistency in application of fair value measurement.
IFRS 13 defines fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction in the principal (or most advantageous) market at the measurement date under current market conditions. Fair value under IFRS 13 is an exit price regardless of whether that price is directly observable or estimated using another valuation technique. Also, IFRS 13 includes additional disclosure requirements.
Other than the additional disclosure requirements of IFRS 13, as disclosed in Note 4, the application of IFRS 13 has not had any material impact on the amounts recognized in the consolidated financial statements.
- —
- Amendments to IAS 1Presentation of Financial Statements—Presentation of Items of Other Comprehensive Income
The amendments introduce new terminology, the use of which is not mandatory, for the statement of comprehensive income and income statement. The amendments to IAS 1 retain the option to present profit or loss and other comprehensive income in either a single statement or in two separate but consecutive statements. However, the amendments to IAS 1 require items of other comprehensive income to be grouped into two categories in the other comprehensive income section: (a) items that will not be reclassified subsequently to profit or loss and (b) items that may be reclassified subsequently to profit or loss when specific conditions are met. Income tax on items of other comprehensive income is required to be allocated on the same basis—the amendments do not change the option to present items of other comprehensive income either before tax or net of tax. The amendments have been applied retrospectively, and hence the presentation of items of other comprehensive income has been modified to reflect the changes. Other than the above mentioned presentation changes, the application
F-12
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
of the amendments to IAS 1 does not result in any impact on profit or loss, other comprehensive income and total comprehensive income.
The following standards, interpretations and amendments issued by the IASB and effective for annual periods beginning on January 1, 2013 have had no impact on the consolidated financial statements:
- •
- IFRS 11Joint Arrangements
- •
- IAS 19Employee Benefits
- •
- IAS 27Separate Financial Statements
- •
- IAS 28Investments in Associates and Joint Ventures
- •
- Improvements to IFRS (2009-2011)
- •
- Amendments to IFRS 7Financial Instruments: Disclosures—Offsetting Financial Assets and Financial Liabilities
- •
- Amendments to IAS 12Income Taxes—Deferred Tax: Recovery of Underlying Assets
- •
- IFRIC 20Stripping Costs in the Production Phase of a Surface Mine
A number of new standards, interpretations and amendments effective for the first time for periods beginning on (or after) January 1, 2014, have been adopted as of January 1, 2014. The nature and effect of each new standard, interpretation and amendment adopted by the Group is listed below. Not all new standards and interpretations effective for the first time for periods beginning on (or after) January 1, 2014 affect the Group's annual consolidated financial statements.
- —
- Amendments to IAS 32Financial Instruments: Presentation—Offsetting Financial Assets and Financial Liabilities
The amendments clarify the meaning of "currently has a legally enforceable right to set-off". To result in offset of a financial asset and a financial liability, a right to set-off must be available today rather than being contingent on a future event and must be exercisable by any of the counterparties, both in the normal course of business and in the event of default, insolvency or bankruptcy.
In addition, the determination of whether the right meets the legally enforceable criterion will depend on both the contractual terms entered into between the counterparties as well as the law governing the contract and the bankruptcy process in the event of bankruptcy or insolvency.
The offsetting criteria require intending either to settle on a net basis or to realize the asset and settle the liability simultaneously. Only gross settlement mechanisms with features that eliminate or result in insignificant credit and liquidity risk and that process receivables and payables in a single settlement process or cycle would be, in effect, equivalent to net settlement and, therefore, meet the net settlement criterion.
These amendments have no impact the consolidated financial statements of TiGenix.
- —
- Amendments to IAS 36Impairment of Assets—Recoverable Amount Disclosures for Non-Financial Asset
The amendments clarify the disclosure requirements in respect of fair value less costs of disposal. Additional information about the fair value measurement of impaired assets must be disclosed when
F-13
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
the recoverable amount is based on fair value less costs of disposal. In addition, information must be disclosed about the discount rates used when the recoverable amount is based on fair value less costs of disposal using a present value technique.
As the amendments affect only disclosure, there is no effect on the Group's financial position or performance. The amendments have a limited impact on the level of disclosure provided in its financial statements, and have been applied on a retrospective basis.
- b)
- Standards and interpretations issued but not yet effective
The Group elected not to adopt the following new Standards early, Interpretations and Amendments, which have been issued by the IASB but are not yet mandatory:
- —
- IFRS 9Financial Instruments and subsequent amendments
This standard addresses the classification, measurement and recognition of financial assets and liabilities. IFRS 9 was issued in November 2009 and October 2010, and replaces parts of IAS 39 relating to the classification and measurement of financial instruments. IFRS 9 requires the classification of financial assets into two categories: measured at fair value and measured at amortized cost. The classification is determined at the time of initial recognition. The basis for classification depends on the business model of the entity and the contractual characteristics of the cash flow from the financial instruments. With regard to financial liabilities, the standard maintains the majority of the requirements established by IAS 39. The main change is that in cases where the fair value option is adopted for financial liabilities, the part of the change in the fair value due to the credit risk of the entity is registered in other comprehensive income and not in the income statement, except when it results in an accounting mismatch.
The standard is expected to become applicable as of January 1, 2018. The Group has not yet evaluated the impact of the adoption of this new standard.
- —
- IFRS 15Revenue from Contracts with Customers
IFRS 15 specifies how and when a company will recognize revenue as well as requiring such entities to provide users of financial statements with more informative, relevant disclosures. The standard provides a single, principles-based five-step model to be applied to all contracts with customers as follows:
- •
- Identify the contract(s) with a customer
- •
- Identify the performance obligations in the contract
- •
- Determine the transaction price
- •
- Allocate the transaction price to the performance obligations in the contract
- •
- Recognize revenue when (or as) the entity satisfies a performance obligation.
IFRS 15 was issued in May 2014 and replaces IAS 11—Construction Contracts, IAS 18—Revenue, IFRIC 13—Customer Loyalty Programmes, IFRIC 15—Agreements for the Construction of Real Estate, IFRIC 18—Transfers of Assets from Customers and SIC 31—Revenue—Barter Transactions involving Advertising Services. The Standard applies to an annual reporting period beginning on or after January 1, 2017. Earlier adoption is permitted. The Group has not yet evaluated the impact of the adoption of this new standard.
F-14
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
The other standards, interpretations and amendments issued by the IASB, but not yet effective are not expected to have a material impact on the Group's future consolidated financial statements.
2.3. Basis of consolidation
The consolidated financial statements incorporate the financial statements of the Company and entities controlled by the Company. Control is achieved when the Company:
- •
- has power over the investee;
- •
- is exposed, or has rights, to variable returns from its involvement with the investee; and
- •
- has the ability to use its power to affect its returns.
The Company reassesses whether or not it controls an investee if facts and circumstances indicate that there are changes to one or more of the three elements of control listed above.
Consolidation of a subsidiary begins when the Company obtains control over the subsidiary and ceases when the Company loses control of the subsidiary. Specifically, income and expenses of a subsidiary acquired or disposed of during the year are included in the consolidated statement of profit or loss and other comprehensive income from the date the Company gains control until the date when the Company ceases to control the subsidiary.
Profit or loss and each component of other comprehensive income are attributed to the owners of the Company and to the non-controlling interests. Total comprehensive income of subsidiaries is attributed to the owners of the Company and to the non-controlling interests even if this results in the non-controlling interests having a deficit balance.
When necessary, adjustments are made to the financial statements of subsidiaries to bring their accounting policies into line with the Group's accounting policies.
All intragroup assets and liabilities, equity, income, expenses and cash flows relating to transactions between members of the Group are eliminated in full on consolidation.
Changes in the Group's ownership interests in subsidiaries that do not result in the Group losing control over the subsidiaries are accounted for as equity transactions. The carrying amounts of the Group's interests and the non-controlling interests are adjusted to reflect the changes in their relative interests in the subsidiaries. Any difference between the amount by which the non-controlling interests are adjusted and the fair value of the consideration paid or received is recognized directly in equity and attributed to owners of the Company.
When the Company loses control of a subsidiary, a gain or loss is recognized in profit or loss and is calculated as the difference between (i) the aggregate of the fair value of the consideration received and the fair value of any retained interest and (ii) the previous carrying amount of the assets (including goodwill), and liabilities of the subsidiary and any non-controlling interests. All amounts previously recognized in other comprehensive income in relation to that subsidiary are accounted for as if the Company had directly disposed of the related assets or liabilities of the subsidiary (i.e. reclassified to profit or loss or transferred to another category of equity as specified/permitted by applicable IFRSs). The fair value of any investment retained in the former subsidiary at the date when control is lost is regarded as the fair value on initial recognition for subsequent accounting under IAS 39, when applicable, the cost on initial recognition of an investment in an associate or a joint venture.
F-15
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
2.4. Foreign currency translation
In preparing the financial statements of each group entity, transactions in currencies other than the entity's functional currency (foreign currencies) are recognized at the rates of exchange prevailing at the dates of the transactions. At the end of each reporting period, monetary items denominated in foreign currencies are retranslated at the rates prevailing at that date. Non-monetary items carried at fair value that are denominated in foreign currencies are retranslated at the rates prevailing at the date when the fair value was determined. Non-monetary items that are measured in terms of historical cost in a foreign currency are not retranslated.
Exchange differences arising on the settlement of monetary items or on translating monetary items at rates different from those at which they were translated on initial recognition during the period or in previous financial statements are recognized in profit or loss in the period in which they arise.
For the purposes of presenting consolidated financial statements, the assets and liabilities of the Group's foreign operations are translated into euros using exchange rates prevailing at the end of each reporting period. Income and expense items are translated at the average exchange rates for the period. Exchange differences arising, if any, are recognized in other comprehensive income and accumulated in equity (translation reserves).
On the disposal of a foreign operation (i.e., a disposal of the Group's entire interest in a foreign operation), or a disposal involving loss of control over a subsidiary that includes a foreign operation, all of the exchange differences accumulated in equity in respect of that operation attributable to the owners of the Company are reclassified to profit or loss.
2.5. Segment information
The Group's activities are in one segment: biopharmaceuticals. The Group is managed and operated as one business unit, which is reflected in the organizational structure and internal reporting. No separate line of business or separate business entity has been identified with respect to any of the product candidates or geographical markets.
Geographical information is further disclosed in Note 28.
2.6. Business combinations
Acquisitions of businesses are accounted for using the acquisition method. The consideration transferred in a business combination is measured at fair value, which is calculated as the sum of the acquisition-date fair values of the assets transferred by the Group, liabilities incurred by the Group to the former owners of the acquiree and the equity interests issued by the Group in exchange for control of the acquiree. Acquisition-related costs are recognized in profit or loss as incurred, except for costs to issue debt or equity securities, which are recognized in accordance with IAS 32 and IAS 39.
At the acquisition date, the identifiable assets acquired and the liabilities assumed are recognized at their fair value, except for deferred tax assets and liabilities arising from the assets acquired and liabilities assumed (which are recognized and measured in accordance with IAS 12), assets and liabilities relating to employee benefit arrangements (which are recognized and measured in accordance with IAS 19), liabilities or equity-instruments related to the replacement of the acquiree's share-based payment arrangements (which are recognized and measured in accordance with IFRS 2) and assets that are classified as held for sale (which are recognized and measured in accordance with IFRS 5).
F-16
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
Goodwill is measured as the excess of the sum of the consideration transferred, the amount of any non-controlling interests in the acquiree, and the fair value of the acquirer's previously held equity interest in the acquiree (if any) over the net of the acquisition-date amounts of the identifiable assets acquired and the liabilities assumed. If, after reassessment, the net of the acquisition-date amounts of the identifiable assets acquired and liabilities assumed exceeds the sum of the consideration transferred, the amount of any non-controlling interests in the acquiree and the fair value of the acquirer's previously held interest in the acquiree (if any), the excess is recognized immediately in profit or loss as a bargain purchase gain.
2.7. Revenue and other income recognition
Revenue from sale of products is recognized when:
- •
- the ownership of the products is transferred to the buyer;
- •
- the amount of revenue can be measured reliably;
- •
- it is probable that the economic benefits associated with the transaction will flow to the entity; and
- •
- the costs incurred or to be incurred in respect of the transaction can be measured reliably.
Revenue for the sale of the ChondroCelect product is recognized when implantation has occurred. Provisions for rebates, product returns and discounts to customers are provided for as reductions to revenue in the same period as the related sales are recorded. The provisions made at the time of revenue recognition are based on historical experience and updated for changes in facts and circumstances including the impact of new legislation. The provisions are recognized as a reduction to revenue. For all periods presented, revenues from the sales of ChondroCelect products are included in discontinued operations (see Note 9).
Revenue for the royalties related to the sale of the ChondroCelect is recognized when implantation has occurred. Provisions for rebates, product returns and discounts to customers are provided for as reductions to revenue in the same period as the related royalties are recorded.
Government grants are not recognized until there is reasonable assurance that the Group will comply with the conditions attaching to them and that the grants will be received.
Government grants are recognized in profit or loss on a systematic basis over the periods in which the Group recognizes as expenses the related costs for which the grants are intended to compensate. Specifically, government grants whose primary condition is that the Group should purchase, construct or otherwise acquire non-current assets are recognized as deferred revenue in the consolidated statement of financial position and transferred to profit or loss (under "other operating income") on a systematic and rational basis over the useful lives of the related assets.
Government grants that are receivable as compensation for expenses or losses already incurred or for the purpose of giving immediate financial support to the Group with no future related costs are recognized in profit or loss (under "grants and other operating income") in the period in which they become receivable.
The benefit of a government loan at a below-market rate of interest is treated as a government grant, (measured as the difference between proceeds received and the fair value of the loan based on prevailing market interest rates). Only when there is sufficient assurance that the Group will comply
F-17
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
with the conditions attached to it, the grants will be recognized in profit or loss (under "other operating income"). During the period that these benefits cannot be considered as grants due to the insufficient assurance that all the conditions have been meet, these grants will be included in the liabilities as financial loans and other payables.
2.8. Property, plant and equipment
Property, plant and equipment are stated at historical cost less accumulated depreciation and impairment. Repair and maintenance costs are charged to the income statement as incurred. Gains and losses on the disposal of property, plant and equipment are included in other income or expense. Depreciation is charged so as to write off the cost or valuation of assets over their useful lives, using the straight-line method pro rata in the year of purchase, on the following basis:
- •
- (laboratory) equipment: five years
- •
- IT hardware: three years
- •
- furniture: five years
- •
- leasehold improvements: lower of lease term and useful life
- •
- leases: lower of lease term and useful life.
Assets in the course of construction for production, supply or administrative purposes are carried at cost, less any recognized impairment loss. Cost includes professional fees and, for qualifying assets, capitalized borrowing costs. Such assets are classified to the appropriate categories of property, plant and equipment when completed and ready for intended use. Depreciation of these assets, on the same basis as other property assets, commences when the assets are ready for their intended use.
2.9. Intangible assets
Internally-generated intangible assets—research & development expenditure
Expenditure on research activities is recognized as an expense in the period in which it is incurred.
An internally-generated intangible asset arising from development is recognized to the extent that all of the factors for capitalization have been satisfied as specified in IAS 38:
- •
- the technical feasibility of completing the intangible asset so that it will be available for use or sale
- •
- the intention to complete the intangible asset and use or sell it
- •
- the ability to use or sell the intangible asset
- •
- how the intangible asset will generate probable future economic benefits
- •
- the availability of adequate technical, financial and other resources to complete the development and to use or sell the intangible asset
- •
- the ability to measure reliably the expenditure attributable to the intangible asset during its development.
F-18
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
The amount initially recognized for internally-generated intangible assets is the sum of the various expenses needed to generate the related intangible assets. Amortization starts from the date when the intangible asset first meets the recognition criteria listed above. These intangible assets are amortized on a straight-line basis over their estimated useful life (ten years). Where no internally-generated intangible asset can be recognized, development expenditure is recognized in profit or loss in the period in which it is incurred.
Subsequent to initial recognition, internally-generated intangible assets are reported at cost less accumulated amortization and accumulated impairment losses, on the same basis as intangible assets that are acquired separately.
Intangible assets acquired through a business combination
Intangible assets, including in-process research & development projects, acquired in a business combination and recognized separately from goodwill are initially recognized at their fair value at the acquisition date (which is regarded as their cost).
Subsequent to initial recognition, intangible assets (except for in-process research & development projects) acquired in a business combination are reported at cost less accumulated amortization and impairment losses. Such intangible assets are amortized over their useful economic lives, which will depend on their related patent life (up to fifteen years). Goodwill arising from business combinations is not amortized but reviewed annually for impairment. The Company considers that the goodwill arising from past business combinations is not material.
Subsequent to initial recognition, in-process research & development projects acquired in a business combination are reported at cost and are subject to annual impairment tests until the date the projects are available for use, at this moment the in-process research & development projects will be amortized over their remaining useful economic lives, which will depend on their related patent life (generally up to fifteen years).
Patents, licenses and other similar intangible assets acquired separately
Costs related to the register of internally-generated intangible assets (patents) are recognized as intangible assets.
These patents and licenses are amortized over their useful lives on a straight-line basis as from the moment they are available for use. Estimated useful life is based on the lower of the contract life or the economic useful life (five years).
Computer software
Software licenses and software development costs are measured internally at purchase cost and are amortized on a straight-line basis over the economic useful life (three years).
2.10. Leases
Leases are considered finance leases whenever the terms of the lease transfer substantially all the risks and rewards of ownership of the asset to the lessee. All other leases are classified as operating leases.
F-19
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
Assets held under finance leases are recognized at the start of the lease term as assets of the Group at their fair value or, if lower, at the present value of the minimum lease payments, each determined at the inception of the lease. The corresponding liability to the lessor is included in the balance sheet as a finance lease obligation. The financial costs need to be allocated to each term of the lease period so as to achieve a constant rate of interest on the remaining balance of the liability. Finance charges are expensed.
Rentals payable under operating leases are charged to income on a straight-line basis over the term of the relevant lease. Benefits received and receivable as an incentive to enter into an operating lease are also charged to income on a straight-line basis over the lease term.
2.11. Impairment of tangible and intangible assets (other than goodwill)
At each balance sheet date and at each interim reporting date, the Group reviews the carrying amount of its tangible and intangible assets to determine whether there is any indication that those assets have suffered an impairment loss. If any such indication exists, the recoverable amount of the asset is estimated in order to determine the extent of the impairment loss (if any). Where the asset does not generate cash flows that are independent from other assets, the Group estimates the recoverable amount of the cash-generating unit to which the asset belongs. An intangible asset with an indefinite useful life is tested for impairment annually, and whenever there is an indication that the asset might be impaired. The recoverable amount is the higher of fair value less costs to sell and value in use. The estimated future cash flows are discounted to their present value using a pre-tax discount rate that reflects current market assessments of the time value of money and the risks specific to the asset.
If the recoverable amount of an asset or cash generating unit is estimated to be less than the carrying amount, the carrying amount of the asset is reduced to its recoverable amount. An impairment loss is immediately recognized as an expense. Where an impairment loss subsequently reverses, the carrying amount of the asset is increased to the revised estimate of its recoverable amount, but so that the increased carrying amount does not exceed the carrying amount that would have been determined had no impairment loss been recognized for the asset in prior periods. A reversal of an impairment loss is recognized as income.
2.12. Financial assets
Financial assets are classified into the following specified categories: financial assets 'at fair value through profit or loss' (FVTPL), 'held-to-maturity' investments, 'available-for-sale' (AFS) financial assets and 'loans and receivables.' The classification depends on the nature and purpose of the financial assets and is determined at the time of initial recognition.
The Company currently has loans, receivables, financial assets AFS and receivables from reverse repurchase agreements.
Available-for-sale financial assets are non-derivatives that are either designated as AFS or are not classified as (a) loans and receivables, (b) held-to-maturity investments or (c) financial assets at fair value through profit or loss. AFS equity investments that do not have a quoted market price in an active market and whose fair value cannot be reliably measured and derivatives that are linked to and must be settled by delivery of such unquoted equity investments are measured at cost less any identified impairment losses at the end of each reporting period.
F-20
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
Receivables from reverse repurchase agreements are non-derivatives, initially recognized at fair value. Due to the short term character of the instrument, the carrying amount approximates the fair value.
Loans and receivables are non-derivative financial assets with fixed or determinable payments that are not quoted in an active market. Loans and receivables (including trade and other receivables, receivables from reverse repurchase agreements, bank balances and cash) are measured at amortized cost using the effective interest method, less any impairment. For the purposes of the cash flow statements, cash and cash equivalents comprise cash on hand and deposits held on call with banks. In the balance sheet, bank overdrafts, if any, are included in other current financial liabilities.
The effective interest method is a method of calculating the amortized cost of a debt instrument and of allocating interest income over the relevant period. The effective interest rate is the rate that exactly discounts estimated future cash receipts (including all fees and points paid or received that form an integral part of the effective interest rate, transaction costs and other premiums or discounts) through the expected life of the debt instrument, or, where appropriate, a shorter period, to the net carrying amount on initial recognition.
The warrants are considered as a financial derivative liability measured at fair value with changes in fair value recognized immediately in profit or loss.
Financial assets are assessed for indicators of impairment at the end of each reporting period. Financial assets are considered to be impaired when there is objective evidence that, as a result of one or more events that occurred after the initial recognition of the financial asset, the estimated future cash flows of the investment have been affected.
Objective evidence of impairment could include:
- •
- significant financial difficulty of the issuer or counterparty; or
- •
- breach of contract, such as a default or delinquency in interest or principal payments; or
- •
- it becoming probable that the borrower will enter bankruptcy or financial re-organization; or
- •
- the disappearance of an active market for that financial asset because of financial difficulties.
For certain categories of financial assets, such as trade receivables, assets are assessed for impairment on a collective basis even if they were assessed not to be impaired individually. Objective evidence of impairment for a portfolio of receivables could include the Group's past experience of collecting payments, an increase in the number of delayed payments in the portfolio past the average credit period, as well as observable changes in national or local economic conditions that correlate with default on receivables.
For financial assets carried at amortized cost, the amount of the impairment loss recognized is the difference between the asset's carrying amount and the present value of estimated future cash flows, discounted at the financial asset's original effective interest rate.
The carrying amount of the financial asset is reduced by the impairment loss directly for all financial assets with the exception of trade receivables, where the carrying amount is reduced through the use of an allowance account. When a trade receivable is considered uncollectible, it is written off against the allowance account. Subsequent recoveries of amounts previously written off are credited
F-21
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
against the allowance account. Changes in the carrying amount of the allowance account are recognized in profit or loss.
For financial assets measured at amortized cost, if, in a subsequent period, the amount of the impairment loss decreases and the decrease can be related objectively to an event occurring after the impairment was recognized, the previously recognized impairment loss is reversed through profit or loss to the extent that the carrying amount of the investment at the date the impairment is reversed does not exceed what the amortized cost would have been had the impairment not been recognized.
The Group derecognizes a financial asset when the contractual rights to the cash flows from the asset expire, or when it transfers the financial asset and substantially all the risks and rewards of ownership of the asset to another party. If the Group neither transfers nor retains substantially all the risks and rewards of ownership and continues to control the transferred asset, the Group recognizes its retained interest in the asset and an associated liability for amounts it may have to pay. If the Group retains substantially all the risks and rewards of ownership of a transferred financial asset, the Group continues to recognize the financial asset and also recognizes a collateralized borrowing for the proceeds received.
2.13. Inventories
Raw materials, consumables and goods purchased for resale are valued at the lower of their cost determined according to the FIFO-method (first-in-first-out) or their net realizable value.
The costs of finished goods comprises all costs of purchase, costs of conversion and other costs incurred in bringing the inventories to the present location and condition.
2.14. Non-current assets (disposal groups) held for sale and discontinued operations
Non-current assets and disposal groups are classified as held for sale if their carrying amount will be recovered principally through a sale transaction rather than through continuing use. This condition is regarded as met only when the sale is highly probable and the non-current asset (or disposal group) is available for immediate sale in its present condition. Management must be committed to the sale, which should be expected to qualify for recognition as a completed sale within one year from the date of classification.
When the Group is committed to a sale plan involving loss of control of a subsidiary, all of the assets and liabilities of that subsidiary are classified as held for sale when the criteria described above are met, regardless of whether the Group will retain a non-controlling interest in its former subsidiary after the sale.
Non-current assets (and disposal groups) classified as held for sale are measured at the lower of their previous carrying amount and fair value less costs to sell.
The results of operations disposed during the period are included in the consolidated statement of comprehensive income up to the date of disposal.
A discontinued operation is a component of the Group's business that represents a separate major line of business or geographical area of operations or is a subsidiary acquired exclusively with a view to resale, that has been disposed of, has been abandoned or that meets the criteria to be classified as held for sale.
F-22
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
Discontinued operations are presented in the consolidated statement of comprehensive income as a single line which comprises the post-tax profit or loss of the discontinued operation along with the post-tax gain or loss recognized on the re-measurement to fair value less costs to sell or on disposal of the assets or disposal groups constituting discontinued operations.
2.15. Income taxes
Income tax expense represents the sum of the tax currently payable and deferred tax.
The tax currently payable is based on taxable profit for the year. Taxable result differs from "profit/(loss) before tax" as reported in the consolidated income statement because of items of income or expense that are taxable or deductible in other periods and items that are never taxable or deductible. The Group's current tax is calculated using tax rates that have been enacted or substantively enacted by the end of the reporting period.
Deferred taxes are recognized using the "balance sheet liability method" for temporary differences between the carrying amount of assets and liabilities in the consolidated financial statements and the corresponding tax bases used in the computation of taxable profit.
Deferred tax liabilities are recognized for all taxable temporary differences. Deferred tax assets are recognized for all deductible temporary differences to the extent that it is probable that taxable profits will be available against which those deductible temporary differences can be utilized. Such deferred tax assets and liabilities are not recognized if the temporary difference arises from the initial recognition (other than in a business combination) of assets and liabilities in a transaction that affects neither the taxable profit nor the accounting profit.
Deferred tax assets and liabilities are measured based on the expected manner of realization or settlement of assets and liabilities, using tax rates that have been enacted or substantively enacted at the balance sheet date.
2.16. Financial liabilities
The Group classifies its financial liabilities into one of two categories, depending on the purpose for which the liability was acquired. The Group's accounting policy for each category is as follows:
Fair value through profit or loss
This category comprises derivatives with a negative fair value (see "Financial assets" for derivatives with a positive fair value) and financial liabilities designated at fair value through profit or loss.
They are carried in the consolidated statement of financial position at fair value with changes in fair value recognized in the consolidated statement of comprehensive income. The Group does not hold or issue derivative instruments for speculative purposes.
Other than these derivative financial instruments, the Group does not have any liabilities held for trading nor has it designated any financial liabilities as being at fair value through profit or loss. The Group currently has no non-derivative financial liabilities that are accounted for at fair value through profit or loss.
The Group has issued warrants related to one of the Group loans which meet the definition of a derivative financial liability. These warrants were issued in 2014 in connection with the loan facility
F-23
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
agreement with Kreos Capital IV (UK), and contain an option for the holders to put the warrants back to the Company for cash. The warrants are options over the shares of the Company, but are derivatives that must be measured at fair value through profit or loss, and not own equity instruments of the Company, because of the cash settlement alternative. The Group has determined the initial fair value of the warrants using a Black-Scholes valuation model. A portion of the issue amount of the loan corresponding to this initial fair value of the warrants was allocated to the warrants and the remaining balance of the proceeds received were allocated to the loan, which is then measured at amortized cost. The effective interest rate method was applied to determine the effective interest rate on the loan on the basis of the initial carrying amount and the contractual cash flows of the loan (interest payments and repayment of principal). This effective interest rate is 20% compared to the contractual interest rate of 12.5%. The effective interest rate is used to accrue interest in the loan, and to amortize the difference between the initial carrying amounts of the loan to its repayment amount.
Other financial liabilities
Financial liabilities measured at amortized cost, including borrowings, are initially measured at fair value, net of transaction costs. They are subsequently measured at amortized cost using the effective interest method, with interest expense recognized on an effective yield basis.
The effective interest method is a method of calculating the amortized cost of a financial liability and of allocating interest expense over the relevant period. The effective interest rate is the rate that exactly discounts estimated future cash payments through the expected life of the financial liability, or, where appropriate, a shorter period, to the net carrying amount on initial recognition.
The Group's financial liabilities measured at amortized cost comprise financial loans, other current financial liabilities and trade payables.
2.17. Provisions
Provisions are recognized when the Group has a present obligation (legal or constructive) as a result of a past event, it is probable that the Group will be required to settle the obligation, and a reliable estimate can be made of the amount of the obligation.
The amount recognized as a provision is the best estimate of the consideration required to settle the present obligation at the end of the reporting period, taking into account the risks and uncertainties surrounding the obligation. When a provision is measured using the cash flows estimated to settle the present obligation, its carrying amount is the present value of those cash flows (when the effect of the time value of money is material).
When some or all of the economic benefits required to settle a provision are expected to be recovered from a third party, a receivable is recognized as an asset if it is virtually certain that reimbursement will be received and the amount of the receivable can be measured reliably.
2.18. Share capital
Financial instruments issued by the Group are classified as equity only to the extent that they do not meet the definition of a financial liability or financial asset. Ordinary shares are classified as equity.
Incremental costs directly attributable to the issue of new ordinary shares are presented in equity as a deduction, net of tax, from the proceeds.
F-24
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Summary of significant accounting policies (Continued)
2.19. Employee benefits
The Group offers a pension scheme with different premiums depending on job level. The scheme is generally funded through payments to the insurance company. The pension obligations are defined contribution plans. A defined contribution plan is a pension plan under which the Group pays fixed contributions (percentage of annual gross salary). The Group has legal obligations to pay further contributions if the fund does not hold sufficient assets to pay all employees the benefits relating to employees in service. The contributions are recognized as employee benefit expense when they are due.
2.20. Share-based payments
The Group has offered equity-settled share-based payments to employees, directors and business associates. These share-based payments are measured at the fair value of the equity instruments at the grant date.
The fair value determined at the grant date of the equity-settled share-based payments is expensed on a straight-line basis over the vesting period, based on the Group's estimate of equity instruments that will eventually vest, with a corresponding increase in equity.
The estimate of the number of compensation plans which will be vested is revised at each reporting date. The change in estimates will be recorded as expense with a corresponding correction in equity. At the moment of exercise of the compensation plans no adjustments will be made into the share-based compensation reserve.
If a modification of a share-based payment transaction occurs and this modification increases the fair value of the equity instruments granted, measured immediately before and after the modification, the incremental fair value granted shall be included in the measurement of the amount recognized for services received as consideration for the equity instruments granted. The incremental fair value granted is the difference between the fair value of the modified equity instrument and that of the original equity instrument, both estimated as at the date of the modification. If the modification occurs during the vesting period, the incremental fair value granted is included in the measurement of the amount recognized for services received over the period from the modification date until the date when the modified equity instruments vest, in addition to the amount based on the grant date fair value of the original equity instruments, which is recognized over the remainder of the original vesting period. If the modification occurs after vesting date, the incremental fair value granted is recognized immediately, or over the vesting period if the employee is required to complete an additional period of service before becoming unconditionally entitled to those modified equity instruments.
If the terms or conditions of the equity instruments granted are modified in a manner that reduces the total fair value of the share-based payment arrangement, or is not otherwise beneficial to the employee, the services received shall continue to be accounted for as consideration for the equity instruments granted as if that modification had not occurred.
3. Critical accounting judgments and key sources of estimation uncertainty
In the application of the Group's accounting policies, the directors are required to use certain critical accounting estimates, assumptions and judgment about the carrying amounts of certain assets
F-25
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
3. Critical accounting judgments and key sources of estimation uncertainty (Continued)
and liabilities. The areas involving a high degree of judgment or complexity or areas where assumptions and estimates are significant to the consolidated financial statements are the following:
Going concern
The Group has experienced net losses and significant cash used in operating activities since our inception in 2000, and as of June 30, 2014, had an accumulated deficit of 83.2 million euros, a net loss of 9.2 million euros and net cash used in operating activities of 6.1 million euros and as of December 31, 2013 had an accumulated deficit of 74.0 million euros, a net loss of 18.4 million euros and net cash used in operating activities of 14.4 million euros. Management expects the Group to continue to incur net losses and have significant cash outflows for at least the next twelve months. These conditions, among others, raise substantial doubt about our ability to continue as a going concern. These consolidated financial statements have been prepared assuming that the Group will continue as a going concern. This basis of accounting contemplates the recovery of our assets and the satisfaction of liabilities in the normal course of business. A successful transition to attaining profitable operations is dependent upon achieving a level of positive cash flows adequate to support our cost structure.
As at June 30, 2014, the Group had a liquidity position of 19.2 million euros consisting of cash, cash equivalents and receivables from reverse repurchase agreements. Taking into account this liquidity position and the available loan facility with Kreos Capital IV (UK) (Kreos), our board of directors is of the opinion that our liquidity position is sufficient to continue our current operations at least until the end of the third quarter of 2015.
For more information related to the expected cash flows see Section 2.1. Liquidity.
Business combinations and goodwill
The Group accounts for business combinations using the acquisition method of accounting, which requires that the assets acquired and liabilities assumed be recorded at the date of acquisition at their respective fair values. Any excess of the fair value of consideration given over the fair values of the identifiable assets and liabilities acquired is recorded as goodwill. The determination of estimated fair values of acquired intangible assets, as well as the useful economic life ascribed to finite lived intangible assets, requires the use of significant judgment. The use of different estimates and assumptions to those used by the Group could result in a materially different valuation of acquired intangible assets, which could have a material effect on the Group's results of operations.
Several methods may be used to determine the estimated fair value of intangible assets acquired in a business combination, all of which require multiple assumptions. The Group used the relief from royalty method, which is a variant of the income valuation approach. It is based on the principle that ownership of the intangible asset relieves the owner of the need to pay a royalty to another party in exchange for rights to use the asset.
The value of the intangible asset is equal to the present value of the cost savings realized by the owner of the intangible asset as a result of not having to make royalty payments and milestone payments to another party. These cost savings are calculated based on the hypothetical royalty payments and milestone payments that a licensee would be required to pay in exchange for use of the asset, reduced by the tax savings realized by the licensee on the royalty payments.
F-26
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
3. Critical accounting judgments and key sources of estimation uncertainty (Continued)
Goodwill is capitalized. Any impairment in carrying amount is charged to the consolidated income statement. Where the fair value of identifiable assets and liabilities exceeds the fair value of consideration paid, the excess is credited in full to the consolidated income statement on acquisition date. Goodwill identified on acquisitions to date has been deemed immaterial.
Acquisition costs incurred are expensed and included in general and administrative expenses.
Impairment of assets
We review the carrying value of intangible assets with indefinitive lives for potential impairment on a periodic basis and also whenever events or changes in circumstances indicate the carrying value of an asset may not be recoverable. We review the carrying value of tangible assets and intangible assets with definitive lives for potential impairment whenever events or changes in circumstances indicate the carrying value of an asset may not be recoverable. We determine impairment by comparing the recoverable amount to its carrying value. If impairment is identified, a loss is recorded equal to the excess of the asset's carrying amount over its recoverable amount.
For impaired assets, we recognize a loss equal to the difference between the carrying value of the asset and its recoverable amount. The recoverable amount is based on discounted future cash flows of the asset using a discounted rate commensurate with the risk. Estimates of future cost savings, based on what we believe to be reasonable and supportable assumptions and projections, require management's judgment. Actual results could vary from these estimates. When it is not possible to estimate the recoverable amount of an individual asset, the Group estimates the recoverable amount of the cash-generating unit to which the asset belongs.
Based on the analysis performed, there was no impairment through December 31, 2013 (except for the impairment recognized upon the presentation of the disposal group held for sale, see Note 9).
Recognition and measurement of internally-generated intangible assets
An internally-generated intangible asset is recognized if sufficient certainty can be documented that the future benefits from the development project will exceed the aggregate cost of production, development and the sale and administration of the product. A development project involves a single product candidate undergoing a high number of tests to illustrate its safety profile and the effect on human beings prior to obtaining the necessary final approval of the product from the appropriate authorities. The future economic benefits associated with the individual development projects are dependent on obtaining such approval. Considering the significant risk and duration of the development period related to the development of such products, management has concluded that the future economic benefits associated with a particular project cannot be estimated with sufficient certainty until the project has been finalized and the necessary regulatory final approval of the product has been obtained.
Accordingly, the Group has capitalized such intangible assets for the development costs related to ChondroCelect with a useful life of ten years. The carrying amount of these assets amounted to 1.5 million euros as at June 30, 2014 (December 31, 2013: 1.7 million euros, 2012: 1.9 million euros).
Research and Development Costs
Research and development costs are charged to expense as incurred and are typically made up of salaries and benefits, clinical and preclinical activities, drug development and manufacturing costs, and
F-27
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
3. Critical accounting judgments and key sources of estimation uncertainty (Continued)
third-party service fees, including for clinical research organizations and investigative sites. Costs for certain development activities, such as clinical trials, are periodically recognized based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations, or information provided by vendors on their actual costs incurred. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the financial statements as prepaid or accrued expenses.
Deferred taxes
Deferred tax assets are recognized for unused tax losses to the extent that it is probable that taxable profit will be available against which the losses can be utilized. Significant management judgment is required to determine the amount of deferred tax assets that can be recognized, based upon the likely timing and the level of future taxable profits together with future tax planning strategies.
At December 31, 2013, the Group had 147.1 million euros (2012: 133.6 million euros) of tax losses carry forward and other tax credits such as investment tax credits.
These losses relate to the parent and subsidiaries that have a history of losses and do not expire, except for tax losses and other tax credits of 37.7 million euros related to TiGenix SAU. These tax losses may not be used to offset taxable income elsewhere in the Group.
With respect to the net operating losses of the Group, no deferred tax assets have been recognized, given that there is uncertainty as to the extent to which these tax losses will be used in future years.
Derivative financial instruments
Derivatives are initially recognized at fair value at the date the derivative contracts are entered into and are subsequently re-measured to their fair value at the end of each reporting period. The resulting gain or loss is recognized in profit or loss immediately, unless the derivative is designated and effective as a hedging instrument, in which event the timing of the recognition in profit or loss depends on the nature of the hedge relationship.
Pursuant to the terms and conditions of the loan facility agreement that we entered into with Kreos, on April 22, 2014, an extraordinary meeting of our shareholders issued and granted 1,994,302 new cash settled warrants, including a put option to Kreos Capital IV (Export Fund). These warrants have been designated at fair value through profit or loss. The Company recognizes the warrants, including the put option, as one instrument, because the Company believes that the put option is unconditionally linked to the warrant. Because the issued warrants can be settled in cash, the instrument is considered as a financial derivative liability measured at fair value with changes in fair value recognized immediately in profit or loss.
The measurement of the warrant (and the put option) at fair value is based on the Black-Scholes option pricing model taking into account the following variables:
- •
- The share price.
- •
- The strike price.
- •
- The volatility of the share has been determined based on historical stock prices of our shares.
F-28
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
3. Critical accounting judgments and key sources of estimation uncertainty (Continued)
- •
- The dividend yield, which has been estimated as zero, as we have never paid a dividend due to the past experience of losses.
- •
- The duration, which has been estimated as the difference between the valuation date of the warrant plans and final exercise date.
- •
- The risk free interest rate, which has been calculated based on the discount curve composed based on liquid euro deposit rates (for periods shorter than one year), futures (typically for maturities between one and six years) and interbank euro swap rates (for periods longer than six years).
We will continue to use judgment in evaluating the risk-free interest rate, dividend yield, duration and volatility related to our cash-settled warrant plan on a prospective basis and incorporating these factors into the Black-Scholes option pricing model. If in the future we determine that other methods are more reasonable and provide better results, or other methods for calculating these assumptions are prescribed by authoritative guidance, we may change or refine our approach, and our share-based payment expense in future periods could change significantly.
Share-based payment arrangements
The Group used the Black-Scholes model to estimate the fair value of the share-based payment transactions. Using this model requires management to make assumptions with regard to volatility and expected life of the equity instruments. The assumptions used for estimating fair value for share-based payment transactions are further disclosed in Note 26 and are estimated as follows:
- •
- Volatility is estimated based on the average annualized volatility of the TiGenix share price;
- •
- Estimated life of the warrant is estimated to be until the first exercise period;
- •
- The dividend return is estimated by reference to the historical dividend payment of the Group. Currently, this is estimated to be zero, because no dividend has been paid since inception.
Non-current assets (disposal groups) held for sale and discontinued operations
Assets held for sale are comprised of non-current assets or disposal groups (together with any liabilities), the carrying amounts of which will be realized principally through a sale transaction expected to conclude within the next twelve months, rather than through continued use.
At December 31, 2013 the Group presented 5.6 million euros (2012: nil) of net assets as assets held for sale in the consolidated statement of financial position, all assets and liabilities within this disposal group relate to the disposal of the Dutch manufacturing facility as described in Note 11, which occurred during half year 2014.
At the time of their classification as "held for sale" in December 2013, such assets were collectively measured at the lower of their carrying amount and fair value less costs to sell, and depreciation or amortization ceases. An impairment charge of 0.7 million euros was recorded reflecting the adjustment of the disposal group's carrying amount to its fair value less cost to sell.
Significant judgment is employed by the Company in assessing: at which point all of the "held for sale" presentation conditions are met for the disposal group and estimating both the fair value of the disposal group and the incremental costs to transact a sale of the disposal group. If actual events differ from management's estimates, or to the extent that estimates of selling price or costs to sell are
F-29
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
3. Critical accounting judgments and key sources of estimation uncertainty (Continued)
adjusted in the future, the Group's financial condition and results of operations could be affected in the period of any such change of estimate.
4. Financial instruments and financial risk management
The principal financial instruments used by the Group, from which financial risk arises, are as follows:
- •
- Available-for-sale financial assets
- •
- Other non-current assets
- •
- Trade receivables
- •
- Other current financial assets
- •
- Receivables from reverse repurchase agreements
- •
- Derivative financial instruments
- •
- Cash and cash equivalents
- •
- Borrowings and other payables (including financial loans and other financial liabilities)
- •
- Trade payables
4.1. Capital risk management
The Group policy with respect to managing capital is to safeguard the Group's ability to continue as a going concern and to obtain an optimal capital structure over time.
4.2. Categories of financial instruments
| | | | As at December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | | As at June 30, 2014 | ||||||||||
Thousands of euros | Notes | 2013 | 2012 | |||||||||
| | | (Unaudited) | | | ||||||||
Financial assets | ||||||||||||
Loans and receivables | 24,236 | 19,006 | 14,675 | |||||||||
Cash and cash equivalents (including cash balances in disposal group held for sale) | 13,186 | 15,900 | 11,072 | |||||||||
Receivables from reverse repurchase agreements | 5,999 | — | — | |||||||||
Other non-current assets | 15 | 1,923 | 1,415 | 498 | ||||||||
Trade receivables | 2,179 | 1,032 | 2,477 | |||||||||
Other current financial assets | 949 | 659 | 628 | |||||||||
Available-for-sale financial assets | 14 | 162 | 162 | 278 | ||||||||
Financial liabilities |
| |||||||||||
Amortised cost | 12,022 | 5,642 | 5,596 | |||||||||
Financial loans | 21 | 10,591 | 3,467 | 2,983 | ||||||||
Trade payables | 1,431 | 2,175 | 2,613 | |||||||||
Fair value through profit or loss | 799 | — | — | |||||||||
Other financial liabilities | 799 | — | — | |||||||||
F-30
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
4. Financial instruments and financial risk management (Continued)
4.3. Fair value of financial instruments
| | | As at June 30, 2014 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
Thousands of euros | Notes | Carrying amount | Fair value | Fair value hierarchy | |||||||
| | | (Unaudited) | |||||||||
Financial assets | |||||||||||
Loans and receivables | 1,923 | 1,923 | |||||||||
Other non-current assets | 1,923 | 1,923 | Level 2 | ||||||||
Available-for-sale financial assets | 162 | 162 | Level 2 | ||||||||
Financial liabilities | |||||||||||
Amortised cost | 10,591 | 9,982 | |||||||||
Financial loans | 10,591 | 9,982 | Level 2 | ||||||||
Fair value through profit or loss | 799 | 799 | |||||||||
Other financial liabilities | 799 | 799 | Level 2 | ||||||||
| | As at December 31, 2013 | |||||||
|---|---|---|---|---|---|---|---|---|
Thousands of euros | Carrying amount | Fair value | Fair value hierarchy | |||||
Financial assets | ||||||||
Loans and receivables | 1,415 | 1,415 | ||||||
Other non-current assets | 1,415 | 1,415 | Level 2 | |||||
Available-for-sale financial assets | 162 | 162 | Level 2 | |||||
Financial liabilities | ||||||||
Amortised cost | 3,467 | 3,467 | ||||||
Financial loans | 3,467 | 3,467 | Level 2 | |||||
| | As at December 31, 2012 | |||||||
|---|---|---|---|---|---|---|---|---|
Thousands of euros | Carrying amount | Fair value | Fair value hierarchy | |||||
Financial assets | ||||||||
Loans and receivables | 498 | 498 | ||||||
Other non-current assets | 498 | 498 | Level 2 | |||||
Available-for-sale financial assets | 278 | 278 | Level 2 | |||||
Financial liabilities | ||||||||
Amortised cost | 2,983 | 2,983 | ||||||
Financial loans | 2,983 | 2,983 | Level 2 | |||||
The fair values of the financial assets and financial liabilities measured at amortized cost in the statement of financial position have been determined in accordance with generally accepted pricing models based on discounted cash flow analysis, with the most significant inputs being the discount rate that reflects the credit risk. The fair value of the borrowings has been determined based on a discount rate of 21% reflecting the market credit risk for a company such as TiGenix in development stage. The current financial assets and liabilities are not included in the table above as their carrying amounts approximate their fair values.
F-31
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
4. Financial instruments and financial risk management (Continued)
4.4. Financial risk management objectives
The Group coordinates access to financial markets, monitors and manages the financial risks relating to the operations through internal risk reports that analyze exposures by degree and magnitude of risks. These risks include market risk (including currency risk, interest rate risk and other price risk), credit risk and liquidity risk.
The Group does not use any derivative financial instruments to hedge risk exposures.
Currency risk
The Group may be subject to limited currency risk. The Group's reporting currency is the euro, in addition to which we are exposed to the U.S. dollar. The Company tries to match foreign currency cash inflows with foreign currency cash outflows. The Company has no commercial revenues denominated in U.S. dollars. The Company has not engaged in hedging of the foreign currency risk via derivative instruments.
The Group's financial assets and financial liabilities were denominated in the following currencies:
| | EUR | USD | GBP | Other | Total | |||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | | As at December 31 | | As at December 31 | | As at December 31 | | As at December 31 | | As at December 31 | ||||||||||||||||||||||||||||||||||||
| | As at June 30 2014 | As at June 30 2014 | As at June 30 2014 | As at June 30 2014 | As at June 30 2014 | |||||||||||||||||||||||||||||||||||||||||
Thousands of euros | 2013 | 2012 | 2013 | 2012 | 2013 | 2012 | 2013 | 2012 | 2013 | 2012 | ||||||||||||||||||||||||||||||||||||
| | (Unaudited) | | | (Unaudited) | | | (Unaudited) | | | (Unaudited) | | | (Unaudited) | | | |||||||||||||||||||||||||||||||
Financial assets | ||||||||||||||||||||||||||||||||||||||||||||||
Cash and cash equivalents (including held for sale) | 13,178 | 15,790 | 11,008 | 7 | 7 | 9 | 1 | 103 | 55 | — | — | — | 13,186 | 15,900 | 11,072 | |||||||||||||||||||||||||||||||
Trade receivables | 1,248 | 1,032 | 2,141 | 30 | — | — | 297 | — | 336 | — | — | — | 1,575 | 1,032 | 2,477 | |||||||||||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Total Financial assets | 14,426 | 16,822 | 13,149 | 37 | 7 | 9 | 298 | 103 | 391 | — | — | — | 14,761 | 16,932 | 13,549 | |||||||||||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Financial liabilities | ||||||||||||||||||||||||||||||||||||||||||||||
Trade payables | 1,120 | 2,156 | 2,397 | 108 | 5 | 5 | 104 | 14 | 210 | 99 | — | 1 | 1,431 | 2,175 | 2,613 | |||||||||||||||||||||||||||||||
Borrowings | 16,329 | 9,480 | 8,099 | — | — | — | — | — | — | — | — | — | 16,239 | 9,480 | 8,099 | |||||||||||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Total financial liabilities | 17,449 | 11,636 | 10,496 | 108 | 5 | 5 | 104 | 14 | 210 | 99 | — | 1 | 17,760 | 11,655 | 10,712 | |||||||||||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
The Group's exposure is only limited to pounds sterling and U.S. dollars.
Due to the limited currency exposure, no sensitivity analysis has been performed.
Interest rate risk
The Group is exposed to interest rate risk, because entities in the Group borrow funds at both fixed and floating interest rates. The risk is managed by the Group by maintaining an appropriate mix between fixed and floating rate borrowings. The Group's exposures to interest rates on financial assets and financial liabilities are detailed in the liquidity risk management section of this note.
The sensitivity analysis has been determined based on the exposure to interest rates for borrowings at the end of the reporting period. For floating rate liabilities, the analysis is prepared assuming the amount of the liability outstanding at the end of the reporting period was outstanding for the whole year. A fifty basis point increase or decrease is used when reporting interest rate risk internally to key management personnel and represents management's assessment of the reasonably possible change in interest rates.
F-32
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
4. Financial instruments and financial risk management (Continued)
The Group has two debts with a floating rate. It concerns two roll-over credit facilities (from 2007) for an original amount of 0.8 million euros used for the acquisition of manufacturing equipment in the United States. The borrowings have a remaining maturity of four years and carry a floating interest rate of three-month Euribor + 1.40%. The outstanding amount for these borrowings per December 31, 2013 was 0.3 million euros (2012: 0.4 million euros). (See Note 21).
If interest rates had been fifty basis points higher/lower and all other variables were held constant, the impact on the Group's profit/(loss) for the year ended December 31, 2013 would be very limited, because the total interest expense relating to these borrowings at floating rate amount to 5 thousand euros (2012: 9 thousand euros).
Liquidity risk
The Group manages liquidity risk by maintaining adequate reserves, banking facilities and reserve borrowing facilities, by continuously monitoring forecast and actual cash flows, and by matching the maturity profiles of financial assets and liabilities.
The following table details the Group's remaining contractual maturity for its financial liabilities with agreed repayment periods. The table has been drawn up based on the undiscounted cash flows of financial liabilities based on the earliest date on which the Group can be required to pay. The table includes both interest and principal cash flows.
Thousands of euros | Interest rate | Within one year | 1 - 5 years | After 5 years | Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
As at December 31, 2013 | |||||||||||||||
Non-interest bearing | N/A | 112 | 1,551 | 1,315 | 2,978 | ||||||||||
Floating interest rate borrowings | Euribor 3M + 1.40% | 180 | 100 | — | 280 | ||||||||||
Fixed interest rate borrowings | 1.46% | — | 3,039 | 3,713 | 6,752 | ||||||||||
Other financial liabilities | N/A | 874 | — | — | 874 | ||||||||||
| | | | | | | | | | | | | | | | |
Total | 1,166 | 4,690 | 5,028 | 10,884 | |||||||||||
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
As at December 31, 2012 |
| ||||||||||||||
Non-interest bearing | N/A | 46 | 1,184 | 1,390 | 2,620 | ||||||||||
Floating interest rate borrowings | Euribor 3M + 1.40% | 80 | 280 | — | 360 | ||||||||||
Fixed interest rate borrowings | 1.46% | — | 1,817 | 2,725 | 4,542 | ||||||||||
Other financial liabilities | N/A | 1,527 | — | — | 1,527 | ||||||||||
| | | | | | | | | | | | | | | | |
Total | 1,653 | 3,281 | 4,115 | 9,049 | |||||||||||
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
On December 20, 2013, the Group entered into a loan facility agreement of up to 10.0 million euros with Kreos. The loan can be drawn in three tranches (5.0 million euros by February 3, 2014; 2.5 million euros by May 31, 2014; and 2.5 million euros by September 30, 2014). As per December 31, 2013, no money had been drawn under the facility loan agreement.
As part of the consideration for this debt financing agreement, in April 2014 the Group issued a warrant plan to Kreos Capital IV (Expert Fund). The warrant plan consisted of 1,994,302 warrants that were issued with an exercise price of 0.75 euros exercisable immediately and which expire in April
F-33
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
4. Financial instruments and financial risk management (Continued)
2019. The warrants also include a put option that authorizes Kreos Capital IV (Expert Fund) to return the warrants to the Company and to settle the warrants in cash under certain circumstances.
The loan is measured at amortized cost in accordance with IAS 39. At initial recognition of the loan, the nominal amount of the loan is decreased with the transactions costs related to the loan which also includes the amount of the warrants allocated to the tranches. The interest rate is the effective interest rate (20.16%).
The warrants, including the put option, are accounted for as one instrument (not separating the put option from the warrants) and at issuance had a fair value of 0.7 million euros. Since Kreos Capital IV (Expert Fund) has the option to settle the warrants in cash, the instrument is considered as a financial derivative liability measured at fair value with changes in fair value recognized immediately in profit or loss. The measurement of the warrant (including the put option) at June 30, 2014 at fair value is based on a Black-Scholes valuation model taking into account following inputs: share price (0.6 euros), strike price (0.74 euros), volatility of the share (64.8%), duration (4.8 years) and risk free interest rate (0.62%).
More information regarding the Kreos loan facility is presented in Note 30 of these consolidated financial statements.
Under the terms of a roll-over credit agreement between the Company and ING Belgium (the outstanding amount of which amounted to 0.1 million euros as at December 31, 2013), the Company could not grant any pledge on any of its assets without the prior consent of ING Belgium. Because ING Belgium did not consent to the pledge as proposed to be entered into between the Company and Kreos, the Company and ING Belgium agreed that the Company would repay to ING Belgium the outstanding amount of 0.1 million euros in early 2014. Therefore, the ING roll-over credit has been presented as a short-term liability in these consolidated financial statements.
Credit risk management
Credit risk refers to the risk that a counterparty will default on its contractual obligations resulting in financial loss to the Group. The Group has adopted a policy of only dealing with creditworthy counterparties and obtaining sufficient collateral, where appropriate, as a means of mitigating the risk of financial loss from defaults. The Group's exposure is continuously monitored, and the aggregate value of transactions concluded is spread among approved counterparties.
Most of the counterparties of the Group are established public health care facilities that do not present specific risks. Therefore, the Company is only exposed to a limited risk of counterparty default.
The maximum exposure to credit risk at the reporting date is the carrying amount of each class of financial asset. The Group does not hold any collateral as security.
More information on the trade receivables can be found in Note 17 to the consolidated financial statements.
F-34
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
5. Grants and other operating income
| | Years ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Grant revenues | 774 | 1,227 | |||||
Subcontracting | 67 | 34 | |||||
Other income | 42 | 128 | |||||
| | | | | | | | |
Total Grants and other operating income | 883 | 1,389 | |||||
| | | | | | | | |
| | | | | | | | |
6. Operating expenses
The operating expenses consist of the following elements:
Research and development expenses
| | Years ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Employee benefits expenses | 1,782 | 2,959 | |||||
Depreciations, amortisations and impairment losses | 3,327 | 3,492 | |||||
Lab fees and other operating expenses | 3,095 | 4,267 | |||||
Other expenses | 1,501 | 1,418 | |||||
| | | | | | | | |
Total | 9,705 | 12,136 | |||||
| | | | | | | | |
| | | | | | | | |
General and administrative expenses
| | Years ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Employee benefits expenses | 3,028 | 3,440 | |||||
Depreciation and amortisation expense | 318 | 235 | |||||
Services and other sundry expenses | 1,667 | 1,566 | |||||
Other expenses | 816 | 996 | |||||
| | | | | | | | |
Total | 5,829 | 6,237 | |||||
| | | | | | | | |
| | | | | | | | |
Employee benefits expenses and mandate contractors
| | Years ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Wages, salaries, fees and bonuses | 5,511 | 6,795 | |||||
Social security cost | 1,097 | 1,213 | |||||
Group & Hospitalisation insurance | 210 | 161 | |||||
Share-based compensation | 398 | 612 | |||||
Other expenses | 195 | 84 | |||||
| | | | | | | | |
Total | 7,411 | 8,865 | |||||
| | | | | | | | |
| | | | | | | | |
of which included in discontinued operations | 2,376 | 2,037 | |||||
F-35
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
6. Operating expenses (Continued)
The Group operates a pension plan with different premiums depending on the job level. The assets of the plans are held separately from those of the Group in designated funds. In 2013, a total cost of 0.1 million euros (2012: 0.1 million euros) represents contributions payable to these plans by the Group at rates specified in the rules of the plans (the insurance plan guarantees an interest rate of 3.25% on the premiums and reserves until January 31, 2013 and as of February 1, 2013 there is a guaranteed interest rate of 1.75% on the 'increase' of premiums and reserves of the existing contracts and a rate of 1.75% for the new contracts as from that date).
At year-end, the number of employees (full-time equivalents) from continuing operations was as follows:
| | As at December 31, | ||||||
|---|---|---|---|---|---|---|---|
Number of employees and mandate contractors | 2013 | 2012 | |||||
Research and development staff | 34 | 28 | |||||
Sales and marketing staff | — | — | |||||
General and administrative staff | 15 | 19 | |||||
| | | | | | | | |
Total | 49 | 47 | |||||
| | | | | | | | |
| | | | | | | | |
For further details about the share-based compensation plans, see Note 26.
7. Financial result
| | Years ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Interest income on bank deposits | 1 | 9 | |||||
Other interest income | 6 | 26 | |||||
| | | | | | | | |
Total financial income | 7 | 35 | |||||
Interest on borrowings | (28 | ) | (14 | ) | |||
Interest on subordinated loan | — | (24 | ) | ||||
Interest on obligations under finance leases | — | (3 | ) | ||||
Other finance costs | (17 | ) | (17 | ) | |||
| | | | | | | | |
Total financial expenses | (45 | ) | (58 | ) | |||
Net foreign exchange differences | (352 | ) | (142 | ) | |||
| | | | | | | | |
Financial result | (390 | ) | (165 | ) | |||
| | | | | | | | |
| | | | | | | | |
See also Note 21 to these consolidated financial statements.
8. Income tax expense
The income tax of 58.7 thousand euros consists mainly of a settlement of current income taxes for prior periods of 60.7 thousand euros and deferred tax expenses of 2.0 thousand euros.
F-36
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
8. Income tax expense (Continued)
The income tax expense for the year can be reconciled to the accounting profit as follows:
| | Years ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Profit/(Loss) before taxes | (15,041 | ) | (17,149 | ) | |||
Income tax expense calculated at 33.99% | (5,112 | ) | (5,829 | ) | |||
Effect of income that is exempt from taxation | (885 | ) | 602 | ||||
Effect of expenses that are not deductible | 1,529 | 316 | |||||
Effect of unused tax losses and tax offsets not recognised as deferred tax assets | 4,068 | 4,616 | |||||
Effect of different tax rates in foreign jurisdictions | 399 | 296 | |||||
Other | (58 | ) | — | ||||
| | | | | | | | |
Total | (59 | ) | 1 | ||||
| | | | | | | | |
| | | | | | | | |
The deferred taxes are further detailed in Note 22.
9. Discontinued operations
At the end of 2013, the board of directors decided to discontinue the ChondroCelect operations, as such and as negotiations to sell the Dutch manufacturing facility were significantly advanced, the Group recognized an impairment of 0.7 million euros at December 31, 2013, which is included in Loss for the period from discontinued operations.
During the first half of 2014, the discontinuation of the ChondroCelect operations was successfully completed through the combination of the sale of the Dutch manufacturing facility and a licensing agreement on the marketing and distribution rights of the ChondroCelect operations.
On May 30, 2014, the Group completed the sale of TiGenix B.V., our Dutch subsidiary, which held our manufacturing facility, to PharmaCell, a leading European contract manufacturing organization active in the area of cell therapy, for a total consideration of 4.3 million euros. Under the terms of the share purchase agreement with PharmaCell, we received an upfront payment of 3.5 million euros when the sale became effective on May 30, 2014 and will receive a final payment of 0.8 million euros (recognized at its present value of 0.5 million euros) after three years. At the end of 2013 an impairment test in respect of the Dutch manufacturing facility was conducted and 0.7 million euros were recognized as a loss. During the first half of 2014 and after the sale of the plant was completed, the Company registered an additional loss on disposal of 1.1 million euros which was included in Loss for the period from discontinued operations.
On June 1, 2014, TiGenix completed the licensing of the marketing and distribution rights of ChondroCelect to Sobi, the international specialty healthcare company dedicated to rare diseases. Sobi will continue to market and distribute the product within the European Union (excluding Finland, where we have a pre-existing distribution agreement with Finnish Red Cross Blood Service), Switzerland, Norway, Russia, Turkey and the Middle East and North Africa region (currently excluding certain countries where we have a pre-existing distribution agreement with Genpharm). TiGenix will receive in return royalties on the net sales of ChondroCelect, and Sobi will reimburse of nearly all of TiGenix's costs associated with the product.
F-37
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
9. Discontinued operations (Continued)
Based on a contract manufacturing agreement with our former subsidiary, now owned by PharmaCell, the Company is entitled to a cost relief amounting up to a maximum of 1.5 million euros on future purchases during the first three years since the effective date. Based on the distribution contract with Sobi, this cost relief will be transferred to Sobi on future ChondroCelect sales with the same maximum of 1.5 million euros during the same period. Both the contract manufacturing agreement with our former subsidiary now owned by PharmaCell and the distribution agreement with Sobi include commitments for minimum binding quantities of ChondroCelect that are required to be purchased by us and from us under the respective agreements. If Sobi's actual purchases were to be lower than the required minimum, we would nevertheless be entitled to receive payment from Sobi up to a maximum amount of 5.7 million euros and would be required to pass on such payment to PharmaCell.
At the end of 2012, the Group stopped all operating activities of TiGenix Ltd., its biomaterials unit, to allow the Group to focus on further progressing in the commercial roll-out of ChondroCelect and its cell therapy product development pipeline. During May 2014 TiGenix Ltd. was formally dissolved. As such, TiGenix Ltd. has been deconsolidated and presented as part of our discontinued operations.
Analysis of loss for the period from discontinued operations
| | Six-month period ended June 30, | Years ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Thousands of euros | 2014 | 2013 | 2013 | 2012 | |||||||||
| | (Unaudited) | | | ||||||||||
Revenue | 2,916 | 2,315 | 4,324 | 4,190 | |||||||||
Expenses | (4,618 | ) | (3,819 | ) | (7,729 | ) | (7,429 | ) | |||||
Operating expenses | (3,513 | ) | (3,819 | ) | (7,042 | ) | (7,429 | ) | |||||
Impairment losses | — | — | (687 | ) | — | ||||||||
Loss on disposal | (1,105 | ) | — | — | — | ||||||||
Other income and expenses | (140 | ) | (1 | ) | (3 | ) | (4 | ) | |||||
| | | | | | | | | | | | | | |
Loss before taxes | (1,842 | ) | (1,505 | ) | (3,408 | ) | (3,243 | ) | |||||
| | | | | | | | | | | | | | |
Attributable income tax expense | — | — | — | — | |||||||||
| | | | | | | | | | | | | | |
Total | (1,842 | ) | (1,505 | ) | (3,408 | ) | (3,243 | ) | |||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Basic and diluted loss per share from discontinued operations (in euro) | (0.01 | ) | (0.02 | ) | (0.03 | ) | (0.04 | ) | |||||
The loss on disposal included in the discontinued operations at June 30, 2014 of 1.1 million euros is composed of the following (thousands of euros):
Consideration received in cash | 3,500 | ||||
Deferred consideration | 534 | ||||
Net assets disposed of | (5,139 | ) | |||
| | | | | | |
Loss on disposal | (1,105 | ) | |||
| | | | | | |
| | | | | | |
F-38
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
9. Discontinued operations (Continued)
Cash flows from discontinued operations
| | Six-month period ended June 30, | Years ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Thousands of euros | 2014 | 2013 | 2013 | 2012 | |||||||||
| | (Unaudited) | | | ||||||||||
Cash flows from operating activities | (1,138 | ) | (1,464 | ) | 36 | (2,786 | ) | ||||||
Cash flows from investing activities | 3,500 | (33 | ) | (61 | ) | (550 | ) | ||||||
Cash flows from financing activities | — | — | — | — | |||||||||
| | | | | | | | | | | | | | |
Net cash flows from discontinued operations | 2,362 | (1,497 | ) | (25 | ) | (3,336 | ) | ||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
10. Loss per share
The calculation of the basic net loss per share is based on the loss attributable to the holders of ordinary shares and the weighted average number of ordinary shares outstanding during the period.
The Group offers its employees share-based compensation benefits (see Note 26), which may have a dilutive effect on the basic loss per share. For the purpose of calculating diluted loss per share, the number of ordinary shares shall be the weighted average number of ordinary shares plus the weighted average number of ordinary shares that would be issued in case of conversion into ordinary shares of all instruments that can be converted into ordinary shares.
However, due to the losses incurred by the Group, these instruments have an anti-dilutive effect on the loss per share. Instruments that can be converted into ordinary shares shall only be treated as dilutive when their conversion into ordinary shares would decrease earnings per share or increase loss
F-39
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
10. Loss per share (Continued)
per share from continuing operations. As there was a loss in all periods presented, the dilutive loss is the same as the basic loss per share.
| | Six-month period ended June 30, | Years ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Thousands of euros except share and per share data | 2014 | 2013 | 2013 | 2012 | |||||||||
| | (Unaudited) | | | ||||||||||
CONTINUING AND DISCONTINUED OPERATIONS | |||||||||||||
Loss for the period for the purpose of basic earnings per share | (9,151 | ) | (8,837 | ) | (18,390 | ) | (20,393 | ) | |||||
Weighted average number of shares for the purpose of basic earnings per share | 160,476,620 | 100,288,586 | 115,237,304 | 91,596,484 | |||||||||
Basic loss per share from continuing and discontinued operations (in euros) | (0.06 | ) | (0.09 | ) | (0.16 | ) | (0.22 | ) | |||||
CONTINUING OPERATIONS | |||||||||||||
Loss for the period for the purpose of basic earnings per share | (7,309 | ) | (7,332 | ) | (14,982 | ) | (17,150 | ) | |||||
Weighted average number of shares for the purpose of basic earnings per share | 160,476,620 | 100,288,586 | 115,237,304 | 91,596,484 | |||||||||
Basic loss per share from continuing operations (in euros) | (0.05 | ) | (0.07 | ) | (0.13 | ) | (0.19 | ) | |||||
DISCONTINUED OPERATIONS | |||||||||||||
Loss for the period for the purpose of basic earnings per share | (1,842 | ) | (1,505 | ) | (3,408 | ) | (3,243 | ) | |||||
Weighted average number of shares for the purpose of basic earnings per share | 160,476,620 | 100,288,586 | 115,237,304 | 91,596,484 | |||||||||
Basic loss per share from discontinued operations (in euros) | (0.01 | ) | (0.02 | ) | (0.03 | ) | (0.04 | ) | |||||
POTENTIAL DILUTIVE INSTRUMENTS | |||||||||||||
Number of share-based options (out-of-the-money) | 4,822,410 | 5,890,683 | 6,570,285 | 5,617,683 | |||||||||
F-40
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
11. Disposal group held for sale
As at December 2013, the disposal group held for sale relates to the classification of TiGenix B.V., a 100% subsidiary of TiGenix, as held for sale. Details of the figures presented on the statement of financial position are presented below. For further information see Note 9.
Thousands of euros | As at December 31, 2013 | |||
|---|---|---|---|---|
DISPOSAL GROUP ASSETS HELD FOR SALE | ||||
Property, plant and equipment | 5,651 | |||
Inventories | 104 | |||
Trade and other receivables | 45 | |||
Cash and cash equivalents | 335 | |||
| | | | | |
Total assets | 6,135 | |||
LIABILITIES RELATED TO DISPOSAL GROUP HELD FOR SALE | ||||
Trade and other payables | (162 | ) | ||
Other current liabilities | (404 | ) | ||
| | | | | |
Total liabilities | (566 | ) | ||
| | | | | |
Net assets of disposal group held for sale | 5,569 | |||
| | | | | |
| | | | | |
F-41
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
12. Intangible assets
Thousands of euros | Development | Intellectual property | Patents and licences | Software | Total | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
COST | ||||||||||||||||
Balance at January 1, 2012 | 2,458 | 41,117 | 780 | 1,118 | 45,474 | |||||||||||
| | | | | | | | | | | | | | | | | |
Additions—separately acquired | — | — | 276 | 3 | 279 | |||||||||||
Reclassification to/from held for sale | 50 | — | — | — | 50 | |||||||||||
| | | | | | | | | | | | | | | ��� | | |
Balance at December 31, 2012 | 2,508 | 41,117 | 1,056 | 1,122 | 45,803 | |||||||||||
Additions—separately acquired | — | — | 324 | — | 324 | |||||||||||
Effect of foreign exchange differences | (1 | ) | — | — | — | (1 | ) | |||||||||
| | | | | | | | | | | | | | | | | |
Balance at December 31, 2013 | 2,507 | 41,117 | 1,380 | 1,122 | 46,126 | |||||||||||
ACCUMULATED AMORTISATION AND IMPAIRMENT | ||||||||||||||||
Balance at January 1, 2012 | (293 | ) | (1,827 | ) | (274 | ) | (1,053 | ) | (3,447 | ) | ||||||
| | | | | | | | | | | | | | | | | |
Amortisation expense | (247 | ) | (2,741 | ) | (77 | ) | (35 | ) | (3,101 | ) | ||||||
Disposals or reclassified to/from held for sale | (50 | ) | — | — | — | (50 | ) | |||||||||
| | | | | | | | | | | | | | | | | |
Balance at December 31, 2012 | (590 | ) | (4,569 | ) | (352 | ) | (1,088 | ) | (6,598 | ) | ||||||
Amortisation expense | (249 | ) | (2,741 | ) | (102 | ) | (30 | ) | (3,122 | ) | ||||||
Effect of foreign exchange differences | 1 | — | — | — | 1 | |||||||||||
| | | | | | | | | | | | | | | | | |
Balance at December 31, 2013 | (837 | ) | (7,310 | ) | (454 | ) | (1,118 | ) | (9,719 | ) | ||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Carrying amount at December 31, 2012 | 1,918 | 36,549 | 704 | 34 | 39,205 | |||||||||||
Carrying amount at December 31, 2013 | 1,670 | 33,808 | 927 | 4 | 36,407 | |||||||||||
Intellectual property and other intangibles recognized relate to the acquisition of TiGenix SAU in May 2011 and consist of the technology platform, in-process research & development and goodwill. These intangible assets were recognized at fair value in accordance with IFRS 3—Business Combinations. The technology platform's carrying value of 31.2 million euros at December 31, 2013 (34.0 million euros at December 31, 2012) is amortized over its useful life of fifteen years. The remaining useful life is thirteen years at the end of 2013. In-process research & development of 2.6 million euros is currently not amortized, because it is not yet available for use and is, therefore, subject to an annual test for impairment. Goodwill from the acquisition of TiGenix SAU is deemed to be immaterial.
The Company has also recognized during 2011 and 2010 development costs for ChondroCelect. They are amortized over their useful life of ten years. No additional development costs for ChondroCelect were capitalized during 2013 or 2012. The carrying amount of these development costs amounted to 1.7 million euros at December 31, 2013 (2012: 1.9 million euros). The remaining useful life is seven years at December 31, 2013.
A portion of our intangible assets has been pledged to secure the new Kreos credit facilities. The Group is not allowed to pledge these assets as security for other borrowings or to sell them.
At December 31, 2013, no commitments (2012: nil) were signed to acquire intangible assets.
F-42
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
13. Property, plant and equipment
Thousands of euros | IT & machinery | Furniture | Laboratory equipment | Leasehold improvements | TOTAL | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
COST | ||||||||||||||||
Balance at January 1, 2012 | 2,022 | 395 | 1,256 | 7,259 | 10,933 | |||||||||||
| | | | | | | | | | | | | | | | | |
Additions | 343 | — | 7 | 261 | 611 | |||||||||||
Disposals | (605 | ) | (17 | ) | (27 | ) | — | (649 | ) | |||||||
Reclassification to/from held for sale | 508 | 100 | — | — | 608 | |||||||||||
Effect of foreign exchange differences | 9 | 2 | 1 | — | 12 | |||||||||||
| | | | | | | | | | | | | | | | | |
Balance at December 31, 2012 | 2,277 | 481 | 1,237 | 7,520 | 11,515 | |||||||||||
Additions | 61 | — | 40 | 16 | 116 | |||||||||||
Disposals | (14 | ) | — | — | — | (14 | ) | |||||||||
Reclassification to/from held for sale | (166 | ) | (31 | ) | (578 | ) | (6,321 | ) | (7,096 | ) | ||||||
Effect of foreign exchange differences | 7 | 2 | 5 | — | 14 | |||||||||||
| | | | | | | | | | | | | | | | | |
Balance at December 31, 2013 | 2,164 | 451 | 704 | 1,215 | 4,535 | |||||||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
ACCUMULATED DEPRECIATION AND IMPAIRMENT | ||||||||||||||||
Balance at January 1, 2012 | (1,243 | ) | (167 | ) | (399 | ) | (462 | ) | (2,275 | ) | ||||||
| | | | | | | | | | | | | | | | | |
Depreciation expense | (409 | ) | (76 | ) | (164 | ) | (296 | ) | (945 | ) | ||||||
Impairment losses | (67 | ) | (1 | ) | — | — | (68 | ) | ||||||||
Eliminated on disposals | 555 | 16 | 27 | — | 598 | |||||||||||
Eliminated on reclassification as held for sale | (386 | ) | (96 | ) | — | — | (481 | ) | ||||||||
Effect of foreign exchange differences | (8 | ) | (2 | ) | 1 | — | (9 | ) | ||||||||
| | | | | | | | | | | | | | | | | |
Balance at December 31, 2012 | (1,557 | ) | (326 | ) | (535 | ) | (759 | ) | (3,180 | ) | ||||||
Depreciation expense | (282 | ) | (48 | ) | (160 | ) | (469 | ) | (959 | ) | ||||||
Impairment losses | (60 | ) | (6 | ) | (47 | ) | (847 | ) | (960 | ) | ||||||
Eliminated on disposals | 13 | — | — | — | 13 | |||||||||||
Eliminated on reclassification as held for sale | 69 | 18 | 201 | 1,157 | 1,445 | |||||||||||
Effect of foreign exchange differences | (7 | ) | (2 | ) | (5 | ) | — | (14 | ) | |||||||
| | | | | | | | | | | | | | | | | |
Balance at December 31, 2013 | (1,825 | ) | (365 | ) | (547 | ) | (918 | ) | (3,655 | ) | ||||||
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Carrying amount at December 31, 2012 | 720 | 155 | 702 | 6,761 | 8,334 | |||||||||||
Carrying amount at December 31, 2013 | 339 | 86 | 157 | 297 | 879 | |||||||||||
In 2013, TiGenix BV is classified as held for sale. Therefore, all related property, plant and equipment were transferred to held for sale.
A portion of our property, plant and equipment have been pledged to secure the new Kreos credit facility. The Group is not allowed to pledge these assets as security for other borrowings or to sell them.
At December 31, 2013, no commitments (2012: nil) were signed to acquire property, plant and equipment.
F-43
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
14. Available-for-sale investments
The available-for-sale investments consist of the participation of TiGenix in Arcarios B.V., a spin-off established jointly with Therosteon in which the Company at December 31, 2013 holds 3.97% of the shares. The participation is classified as a financial asset available for sale in accordance with IAS 39—Financial Instruments: Recognition and Measurement. However, due to the fact that Arcarios B.V. is not traded on an active market and the Group is not able to measure fair value in an alternative way, the investment is carried at cost.
As a result of a capital increase in Arcarios B.V. in two tranches in 2013, the participation of the Company in Arcarios B.V. diluted from 14% to 3.97%. The Company recognized an impairment loss of 0.1 million euros.
15. Other non-current assets
The other non-current assets include guaranteed deposits in relation to operating lease commitments of TiGenix and guaranteed deposits in relation to a soft loan obtained in 2013 through the Madrid Network.
16. Inventories
The carrying amounts of the different components of the inventory are as follows:
| | As at December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Raw materials and consumables | 77 | 91 | |||||
Finished goods and goods for resale | — | 14 | |||||
| | | | | | | | |
Total | 77 | 105 | |||||
| | | | | | | | |
| | | | | | | | |
17. Trade and other receivables
| | As at December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Trade receivables | 1,032 | 2,477 | |||||
Other receivables | 551 | 1,184 | |||||
Recoverable taxes | 474 | 849 | |||||
Other | 77 | 335 | |||||
| | | | | | | | |
Total | 1,583 | 3,661 | |||||
| | | | | | | | |
| | | | | | | | |
The trade receivables can be detailed as follows:
| | As at December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Trade receivables | 1,146 | 2,489 | |||||
Allowance for doubtful debts | (114 | ) | (12 | ) | |||
| | | | | | | | |
Total | 1,032 | 2,477 | |||||
| | | | | | | | |
| | | | | | | | |
F-44
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
17. Trade and other receivables (Continued)
The aging analysis of the Group's trade receivables at year-end is as follows:
| | As at December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Not past due | 999 | 963 | |||||
Up to three months | — | 831 | |||||
Three to 6 months | — | 106 | |||||
Six to twelve months | 33 | 560 | |||||
More than one year | — | 17 | |||||
| | | | | | | | |
Total | 1,032 | 2,477 | |||||
| | | | | | | | |
| | | | | | | | |
The movement in the allowance for doubtful debts is detailed below:
| | As at December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Balance at January 1 | 12 | — | |||||
| | | | | | | | |
Impairment losses recognised | 102 | 12 | |||||
Amounts written off during the year as uncollectible | — | — | |||||
Amounts recovered during the year | — | — | |||||
Impairment losses reversed | — | — | |||||
Other | — | — | |||||
| | | | | | | | |
Balance at December 31 | 114 | 12 | |||||
| | | | | | | | |
| | | | | | | | |
The credit risk management is described in section 4 of the consolidated financial statements.
18. Other current financial assets
The other current financial assets primarily include bank deposits that were pledged to guarantee the potential repayment of part of certain subsidies granted to TiGenix SAU in 2006 and 2007 for a total amount of 0.3 million euros, not including interest. These bank balances cannot be used for other purposes than the ones defined in the grants. (See Note 29).
19. Receivables from reverse repurchase agreement
In March, 2014, the Group entered into reverse repurchase agreements that were due for repayment to the Group over terms of less than nine months.
20. Equity
- 20.1.
- Share Capital
The share capital of TiGenix amounts to 16.0 million euros at December 31, 2013 (2012: 10.0 million euros), represented by 160,476,620 shares (2012: 100,288,586 shares). The Company's shares have no par value. The holders of TiGenix shares are entitled to receive dividends as declared and to one vote per share at the shareholders' meeting of the Company. All shares issued are fully paid.
F-45
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
20. Equity (Continued)
The Company has never declared or paid any dividend on its shares. In the future, the Company's dividend policy will be determined by its board of directors and may change from time to time. Any declaration of dividends will be based upon the Company's earnings, financial condition, capital requirements and other factors considered important by the board of directors. Belgian law and the Company's articles of association do not require the Company to declare dividends. Currently, the board of directors expects to retain all earnings, if any, generated by the Company's operations for the development and growth of its business and does not anticipate paying any dividend in the near future.
The change in the number of shares during the period is as follows:
Number of shares | 2013 | 2012 | |||||
|---|---|---|---|---|---|---|---|
Balance at January 1, | 100,288,586 | 91,122,667 | |||||
| | | | | | | | |
Exercise of warrants | — | — | |||||
Capital increase—contribution in kind | — | 536,534 | |||||
Capital increase—contribution in cash | 60,188,034 | 8,629,385 | |||||
| | | | | | | | |
Balance at December 31, | 160,476,620 | 100,288,586 | |||||
| | | | | | | | |
| | | | | | | | |
During 2013, the share capital of the Company was increased twice:
- •
- 26,000,000 shares were issued pursuant to a contribution in cash on July 24 and 26, 2013 (6.5 million euros); and
- •
- 34,188,034 shares were issued pursuant to a contribution in cash on November 22, 2013 (12.0 million euros).
Transaction costs related to these capital increases amounted to 1.2 million euros.
The 9,165,919 shares that were issued in 2012 were issued as follows:
- •
- 536,534 shares were issued pursuant to a contribution in kind on April 17, 2012; and
- •
- 8,629,385 shares were issued pursuant to a contribution in cash on December 27, 2012.
Transaction costs related to this capital increase amounted to 0.4 million euros.
- 20.2.
- Equity-settled employee benefits reserve
The equity-settled employee benefits reserve relates to share options granted by the Group to its employees under its employee share option plan. Further information about share-based payments to employees is set out in Note 26.
- 20.3.
- Translation reserves
Exchange differences relating to the translation of the results and net assets of the Group's foreign operations from their functional currencies to the Group's presentation currency (the euro) are recognized directly in other comprehensive income and accumulated in the foreign currency translation reserve. Exchange differences previously accumulated in the foreign currency translation reserve (in respect of translating the net assets of foreign operations) are reclassified to profit or loss on the disposal of the foreign operation.
F-46
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
21. Financial loans and other payables
| | | As at December 31, | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | As at June 30, 2014 | |||||||||
Thousands of euros | 2013 | 2012 | ||||||||
| | (Unaudited) | | | |||||||
Non-current | ||||||||||
Financial loans | 9,802 | 3,124 | 2,595 | |||||||
Other payables | 4,436 | 5,139 | 3,589 | |||||||
Financial loans and other payables | 14,238 | 8,263 | 6,184 | |||||||
| | | | | | | | | | | |
Non-current borrowings | 14,238 | 8,263 | 6,184 | |||||||
Current | ||||||||||
Current portion of financial loans | 789 | 343 | 388 | |||||||
Other financial liabilities | 1,302 | 874 | 1,527 | |||||||
| | | | | | | | | | | |
Current borrowings | 2,091 | 1,217 | 1,915 | |||||||
| | | | | | | | | | | |
Total | 16,329 | 9,480 | 8,099 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
The Company borrowings include financial loans as follows:
- •
- Roll-over credit facilities (from 2007) for an original amount 0.8 million euros used for the acquisition of manufacturing equipment in the United States. The borrowings have a remaining maturity of four years and carry a variable interest of three-month Euribor + 1.40%.
- •
- Two loans received in different tranches over 2011 and 2013 from Madrid Network for an original amount of 5.9 million euros to finance the TiGenix SAU Phase III study for complex perianal fistulas in Crohn's disease patients and to develop the potential of the stem cells in autoimmune inflammatory diseases. The loans will be reimbursed over a period of ten years starting in 2015 with an annual fixed interest rate of 1.46%.
- •
- Interest-free loans maturing in 2025 received from the Spanish Government. These loans have an original amount of 3.2 million euros.
- •
- Kreos loan received in 2 tranches over the first half of 2014 of 5.0 million euros and 2.5 million euros respectively. The loan will be repaid as from the first anniversary over a period of four years and has a fixed interest rate of 12.5%.
The borrowings were granted subject to the condition of maintaining specific covenants. As at December 31, 2013, the Group was in breach of one of these covenants: under the terms of a roll-over credit agreement between the Company and ING Belgium (the outstanding amount of which amounted to 0.1 million euros as per December 31, 2013), the Company could not grant any pledge on any of its assets without the prior consent of ING Belgium. As ING Belgium did not consent to the pledge as proposed to be entered into between the Company and Kreos, the Company and ING Belgium agreed that the Company would repay to ING Belgium the outstanding amount of 0.1 million euros in early 2014. Therefore the ING roll-over credit has been presented as a short-term liability in the financial statements.
Other payables consist of deferred income related to government grants received in the form of loans obtained at below market rate interest. These government grants will be recognized in profit and loss once all of the related conditions are met.
F-47
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
21. Financial loans and other payables (Continued)
Other financial liabilities are explained by the factoring of trade receivables. As the trade receivables are not paid until their maturity, the bank reserves the right to request the Group to pay for the unsettled balance. As a consequence, the Group recognizes the full carrying amount of the trade receivables, as well as the cash received on the transfer, as a secured borrowing due to the fact that it has not transferred the significant risks and rewards relating to these trade receivables to the bank.
At December 31, 2013, the carrying amount of the trade receivables that have been transferred but have not been derecognized amounted to 1.0 million euros and the carrying amount of the associated liability is 0.9 million euros.
22. Deferred taxes
| | As at December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Deferred tax assets | — | — | |||||
Deferred tax liabilities | (29 | ) | (27 | ) | |||
| | | | | | | | |
Total | (29 | ) | (27 | ) | |||
| | | | | | | | |
| | | | | | | | |
The variation in the deferred tax balances presented in the consolidated statement of financial position is as follows:
Thousands of euros | Intangible assets | Tax losses | Other | Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Balance at January 1, 2012 | (11,787 | ) | 11,787 | (27 | ) | (27 | ) | ||||||
| | | | | | | | | | | | | | |
Recognised in income statement—continuing operations | 822 | (822 | ) | — | — | ||||||||
Recognised in income statement—discontinued operations | — | — | — | — | |||||||||
Recognised in other comprehensive income | — | — | — | — | |||||||||
Business combinations | — | — | — | — | |||||||||
| | | | | | | | | | | | | | |
Balance at December 31, 2012 | (10,965 | ) | 10,965 | (27 | ) | (27 | ) | ||||||
Recognised in income statement—continuing operations | 822 | (822 | ) | (2 | ) | (2 | ) | ||||||
Recognised in income statement—discontinued operations | — | — | — | — | |||||||||
Recognised in other comprehensive income | — | — | — | — | |||||||||
Business combinations | — | — | — | — | |||||||||
| | | | | | | | | | | | | | |
Balance at December 31, 2013 | (10,143 | ) | 10,143 | (29 | ) | (29 | ) | ||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
In the context of the business combination with TiGenix SAU, the Group recognized a deferred tax liability of 12.3 thousand euros relating to the recognition of the intangible assets of TiGenix SAU at the acquisition date. At the same time (i.e., the acquisition date), a deferred tax asset was recognized for the tax losses carried forward of TiGenix SAU to the extent of the deferred tax liabilities recognized.
F-48
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
22. Deferred taxes (Continued)
Deductible temporary differences, unused tax losses and unused tax credits for which no deferred tax assets have been recognized, are attributable to the following:
| | As at December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Unused Tax losses | 125,585 | 113,281 | |||||
Unused tax credits | 13,994 | 12,062 | |||||
Deductible temporary differences | 7,570 | 8,302 | |||||
| | | | | | | | |
Total | 147,149 | 133,645 | |||||
| | | | | | | | |
| | | | | | | | |
The tax losses attributable to TiGenix SAU (23.7 million euros) have an average maturity of fourteen years. The other tax losses do not have an expiration date. The tax credits have an average remaining maturity of eleven years.
Due to the losses of the Group, no income taxes were payable. On December 31, 2013 the Group had a loss carried forward amounting to 125.6 million euros (2012: 113.2 million euros), including a potential deferred tax asset of 41.8 million euros. Due to the uncertainty surrounding TiGenix's ability to realize taxable profits in the near future, the Company did not recognize any deferred tax assets on its balance sheet.
In addition to tax losses, the Group has unused tax credits (2013: 13.9 million euros; 2012: 12.1 million euros) and deductible temporary differences (2013: 7.6 million euros; 2012: 8.3 million euros) for which no deferred tax assets have been recognized.
23. Other non-current liabilities
The other non-current liabilities include the capital grants of 0.1 million euros at December 31, 2013 (2012: 0.1 million euros) received by TiGenix SAU, which are deferred.
24. Trade and other payables
| | As at December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Trade payables | 2,175 | 2,613 | |||||
Other payables | 832 | 1,401 | |||||
Payables relating to personnel | 683 | 1,189 | |||||
Other | 149 | 212 | |||||
| | | | | | | | |
Total | 3,007 | 4,014 | |||||
| | | | | | | | |
| | | | | | | | |
25. Other current liabilities
The other current liabilities consist principally of accrued expenses and deferred grant income.
F-49
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
26. Share-based payments
TiGenix—Stock options
On May 14, 2004 (135,802), April 20, 2005 (45,268), November 3, 2005 (454,570), February 26, 2007 (800,000), March 20, 2008 (400,000), June 19, 2009 (500,000), March 12, 2010 (500,000) July 6, 2012 (4,000,000), March 20, 2013 (777,000) and December 16, 2013 (1,806,000) in the aggregate 9,418,640 warrants were issued, subject to the warrants being granted to and accepted by the beneficiaries. Of these 9,418,640 warrants, (i) 889,683 warrants expired as they have not been granted, (ii) 379,250 warrants have expired as they have not been accepted by their beneficiaries, (iii) 721,312 warrants have lapsed due to their beneficiaries leaving the Company, (iv) 9,290 warrants have been exercised, and (v) 848,820 warrants were granted but not yet accepted on December 31, 2013. As a result, as at December 31, 2013, there are 6,570,285 warrants outstanding (2012: 5,617,683).
The warrants are granted to employees, consultants and directors of the Company and its subsidiaries, as well as to other persons who in the scope of their professional activity have made themselves useful to the Group, including but not limited to the members of the scientific advisory board and the clinical advisors. The warrants have been granted free of charge. Each warrant entitles its holder to subscribe to one common share of the Company at a subscription price determined by the board of directors, within the limits decided upon at the occasion of their issuance.
The warrants issued on May 14, 2004, April 20, 2005 and November 3, 2005 had a term of five years, but their term was extended until May 13, 2014 by decision of the extraordinary shareholders' meeting held May 13, 2009. The warrants issued on February 26, 2007, March 20, 2008, June 19, 2009, March 12, 2010, July 6, 2012 and December 16, 2013 have a term of ten years. The warrants issued on March 20, 2013 have a term of five years. Upon expiration of the ten or five year term, the warrants become null and void.
The warrants issued on May 14, 2004, April 20, 2005, November 3, 2005, February 26, 2007, March 20, 2008, June 19, 2009, March 12, 2010 vest, in principle, in cumulative tranches of 25% per year,i.e., 25% as of the first anniversary date of their granting, 50% as of the second anniversary date of their granting, 75% as of the third anniversary date of their granting, 100% as of the fourth anniversary date of their granting provided that the cooperation between the Company and the warrant holder has not yet ended, unless the board of directors approved a deviation from this vesting schedule. As to the warrants issued on July 6, 2012 and March 20, 2013, in principle, (i) one-third of the warrants granted will vest on the first anniversary of the granting of the warrants and (ii) one-twenty fourth of the remaining two-thirds of the warrants granted will vest on the last day of each of the twenty-four months following the month of the first anniversary of the granting of the warrants. As to the warrants issued on December 16, 2013, in principle, (i) 10% of the warrants granted will vest on the date of acceptance of the warrants, (ii) 25% of the warrants granted will vest on the first anniversary of the granting of the warrants and (iii) 65% of the warrants granted will only vest (one-twenty fourth on the last day of each of the months included in the period January 2015 to December 2016) if the Company effectively enters into certain business transactions. The warrants can only be exercised by the warrant holder if they have effectively vested.
F-50
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
26. Share-based payments (Continued)
The table below provides an overview as at December 31, 2013 of all outstanding warrant pools remaining, together with the activities under the different pools of warrants during 2013.
| | | | Options issued in | |||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Weighted average exercise price | | ||||||||||||||||||||||||||||||||||||||
Number of options Grant date | Total | December 16, 2013 | March 20, 2013 | March 20, 2013 | July 6, 2012 | March 12, 2010 | June 19, 2009 | March 20, 2008 | Febr 26, 2007 | Nov 03, 2005 | April 20, 2005 | May 14, 2004 | ||||||||||||||||||||||||||||
Number of options created | 1,806,000 | 160,000 | 273,000 | 4,000,000 | 500,000 | 500,000 | 400,000 | 800,000 | 454,570 | 45,268 | 135,802 | |||||||||||||||||||||||||||||
Weighted average exercise price (euros) | 0.47 | 1.00 | 0.91 | 1.00 | 2.74 | 3.98 | 4.10 | 5.49 | 3.50 | 3.18 | 3.10 | |||||||||||||||||||||||||||||
Fair value at grant date (euros) | 0.35 | 0.20 | 0.43 | 0.17 | 2.00 | 3.53 | 2.56 | 2.64 | 1.29 | 1.15 | 1.08 | |||||||||||||||||||||||||||||
Expiration date | 11/30/2024 | 11/30/2019 | 11/30/2019 | 05/31/2022 | 11/30/2019 | 05/31/2019 | 11/30/2017 | 03/31/2017 | 03/31/2014 | 03/31/2014 | 03/31/2014 | |||||||||||||||||||||||||||||
Balance at January 1, 2012 | 4.31 | 1,727,683 | — | — | — | — | 342,750 | 144,050 | 287,375 | 509,813 | 293,663 | 45,268 | 104,764 | |||||||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Granted | 1.00 | 3,948,000 | 3,948,000 | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||||
Forfeited | 2.03 | (58,000 | ) | (26,000 | ) | (30,000 | ) | (1,125 | ) | (875 | ) | — | — | — | ||||||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2012 | 2.01 | 5,617,683 | — | — | — | 3,922,000 | 312,750 | 142,925 | 286,500 | 509,813 | 293,663 | 45,268 | 104,764 | |||||||||||||||||||||||||||
Granted | 0.62 | 1,390,180 | 957,180 | 160,000 | 273,000 | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
Forfeited | 1.14 | (437,578 | ) | (374,703 | ) | (59,750 | ) | (3,125 | ) | — | — | — | — | — | ||||||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2013 | 1.77 | 6,570,285 | 957,180 | 160,000 | 273,000 | 3,547,297 | 253,000 | 139,800 | 286,500 | 509,813 | 293,663 | 45,268 | 104,764 | |||||||||||||||||||||||||||
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
The fair value of each warrant is estimated on the date of grant using the Black-Scholes model with the following assumptions:
- •
- The historic volatility of the Company (determined at 67% for the 2013 warrant plan, 52.8% for the 2012 warrant plan and 60% for the previous plans), which was determined based on past (three years) volatility of the TiGenix share;
- •
- The expected dividends are assumed to be zero in the model;
- •
- Weighted average risk-free interests rates based on Belgian Sovereign Strips at the date of grant with a term equal to the expected life of the warrants, ranging between 1.7% and 4.6%;
- •
- Weighted average share price (determined at 0.47 euros for the latest warrant plan); and
- •
- The expected lifetime of the warrants, which on average is about seven years for the warrants with a maximum duration of ten years.
The remaining weighted average life of these options was 7.31 years at December 31, 2013 (2012: 7.86 years)
TiGenix SAU—Stock options
Prior to the business combination, TiGenix SAU (formerly Cellerix) had created two equity based incentive plans, or EBIPs. The completion of the business combination triggered certain consequences outlined below which affect both EBIPs. A summary overview of some of the conditions of both EBIPs is given below.
Options under the EBIP 2008 were granted to employees, executives and independent members of the board of directors of TiGenix SAU prior to the business combination. Options under the EBIP 2008 were granted to each beneficiary through individual letters. As a result of the business combination, all EBIP 2008 options have vested except for 32,832 options of employees who terminated their employment with TiGenix SAU before the business combination and that were not re-allocated.
F-51
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
26. Share-based payments (Continued)
The exercise prices of the EBIP 2008 are set at 11.0 euros, 7.0 euros and 5.291 euros depending on the date of grant and beneficiary. TiGenix SAU granted 453,550 options under the EBIP 2008 of which 420,718 are vested. As a result of the business combination, all TiGenix SAU options were exchanged into TiGenix stock options.
Options under the EBIP 2010 were only granted to senior management of TiGenix SAU. The EBIP provides that the normal exercise price of the options is set at 5.291 euros. However, as a result of the business combination the exercise price for all EBIP 2010 options has been reduced to 0.013 euros. TiGenix SAU has granted 221,508 options under the EBIP 2010. As a result of the business combination, all EBIP 2010 options have vested. Beneficiaries must exercise their options before September 30, 2016. Pursuant to the terms of the EBIP 2010 the board of directors of TiGenix SAU has opted to exchange all existing options for new options over existing TiGenix shares. As the options keep the same exchange rate of the Contribution (i.e., 2.96 shares per TiGenix SAU share contributed to TiGenix), each EBIP 2010 option shall give the EBIP 2010 beneficiaries the right to receive 2.96 shares at the time of exercise.
As of December 31, 2013, all EBIP 2008 and EBIP 2010 options were vested. The table below provides an overview as per December 31, 2013 of all outstanding options remaining:
| | | Options issued in | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Number of options Grant date | Total | 2010 | 2008 | |||||||
Number of options created | 642,226 | 221,508 | 420,718 | |||||||
Weighted average exercise price (euros) | 0.01 | 5.29 | ||||||||
Fair value at grant date (euros) | 2.30 | 6.36 | ||||||||
Expiration date | 9/30/2016 | 9/30/2016 | ||||||||
| | | | | | | | | | | |
Balance at January 1, 2012 | 642,226 | 221,508 | 420,718 | |||||||
Granted | — | — | — | |||||||
Forfeited | — | — | — | |||||||
Exercised | — | — | — | |||||||
Expired | — | — | — | |||||||
| | | | | | | | | | | |
Balance at December 31, 2012 | 642,226 | 221,508 | 420,718 | |||||||
Granted | — | — | — | |||||||
Forfeited | — | — | — | |||||||
Exercised | (31,011 | ) | (31,011 | ) | — | |||||
Expired | — | — | — | |||||||
| | | | | | | | | | | |
Balance at December 31, 2013 | 611,215 | 190,497 | 420,718 | |||||||
| | | | | | | | | | | |
| | | | | | | | | | | |
| �� | ||||||||||
The fair value of each stock option is estimated on the date of grant using the Black-Scholes model with the following assumptions:
- •
- The volatility of TiGenix SAU (determined at 55%).
- •
- Weighted average risk-free interests rates based on German Sovereign bond at the date of grant with a term equal to the expected life of the stock option (i.e., 7.3 years), ranging between 0.85% and 1.95%.
F-52
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
27. Related party transactions
Transactions between the Group and its employees, consultants or directors are disclosed below.
There were no other related party transactions.
Compensation of key management personnel
Key management personnel are identified as being the CEO, CFO, CBO and CTO.
The combined remuneration package of key management was as follows:
| | Years ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Short-term benefits | 1,075 | 1,250 | |||||
Post-employment benefits | 57 | 16 | |||||
Share-based payments | 240 | 318 | |||||
Other employee benefits | — | — | |||||
| | | | | | | | |
Total | 1,372 | 1,584 | |||||
| | | | | | | | |
| | | | | | | | |
No loan, quasi-loan or other guarantee is outstanding with members of the management team.
Transactions with non-executive directors
Non-executive directors that represent shareholders of the Company receive no compensation for their position as directors.
The independent directors receive a fee for attending and preparing the meetings of the board of directors and they receive reimbursement for expenses directly related to the board meetings. In 2013, an amount of 0.1 million euros (2012: 0.1 million euros) in total was paid as fees and expense reimbursement to independent members of the board of directors.
No advances or credits have been granted to any member of the board of directors. None of the members of the board of directors has received any non-monetary remuneration other than warrants.
28. Segment information
The Group's activities are managed and operated in one segment, biopharmaceuticals. There is no other significant class of business, either individual or in aggregate. As such, the chief operating decision maker (i.e., the CEO) reviews the operating results and operating plans and makes resource allocation decisions on a company-wide basis.
Geographical information
All sales are related to the product ChondroCelect, which has been disclosed as a discontinued operation.
F-53
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
28. Segment information (Continued)
The Group's sales from discontinued operations from external customers by market location are detailed below:
| | Years ended December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Belgium | 2,023 | 1,653 | |||||
The Netherlands | 1,786 | 1,949 | |||||
United Kingdom | 427 | 368 | |||||
Other | 65 | 114 | |||||
| | | | | | | | |
Total | 4,301 | 4,084 | |||||
| | | | | | | | |
| | | | | | | | |
The Group's non-current assets (excluding non-current assets held for sale) by location are presented below:
| | As at December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Belgium | 2,467 | 3,210 | |||||
The Netherlands | — | 6,805 | |||||
United Kingdom | — | — | |||||
Spain | 36,396 | 38,298 | |||||
Other | — | 2 | |||||
| | | | | | | | |
Total | 38,863 | 48,315 | |||||
| | | | | | | | |
| | | | | | | | |
29. Commitments and contingencies
Operating lease commitments
The operating leases of the Group relate to leases of buildings between one and seventeen years and leases of cars for four years. The Group does not have an option to purchase the leased assets.
In 2013, the Group made operating minimum lease payments for a total amount of 0.9 million euros (2012: 1.2 million euros).
The operating lease commitments for future periods are presented in the table below:
| | As at December 31, | ||||||
|---|---|---|---|---|---|---|---|
Thousands of euros | 2013 | 2012 | |||||
Within one year | 843 | 1,018 | |||||
In the second to fifth year | 1,598 | 2,308 | |||||
After five years | 1,594 | 2,281 | |||||
| | | | | | | | |
Total | 4,035 | 5,607 | |||||
| | | | | | | | |
| | | | | | | | |
Of the above presented commitments, 2.7 million euros related to TiGenix B.V. (sold in 2014) in 2013 and 3.5 million euros in 2012.
F-54
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
29. Commitments and contingencies (Continued)
Other commitments
TiGenix Inc. guarantees the operating lease payments of Cognate for the building leased in the United States. Total remaining operating lease commitments at December 31, 2013 for which TiGenix Inc. was a guarantor were 1.0 million euros. Cognate was the party with whom TiGenix had a joint venture, TC CEF LLC, in the past.
Both the contract manufacturing agreement with our former subsidiary now owned by PharmaCell and the distribution agreement with Sobi include commitments for minimum binding quantities of ChondroCelect that are required to be purchased by us and from us under the respective agreements. If Sobi's actual purchases were to be lower than the required minimum, we would nevertheless be entitled to receive payment from Sobi up to a maximum amount of 5.7 million euros and would be required to pass on such payment to PharmaCell.
Legal proceedings
TiGenix SAU is involved in the following legal proceedings.
Invalidation of U.S. patent US6777231
On April 1, 2011, Cellerix (the predecessor entity of our subsidiary TiGenix SAU) filed an inter partes re-examination request with the Patent and Trademark Office regarding the patent US6777231, owned by the University of Pittsburgh. The Patent and Trademark Office examiner issued a decision concluding that all ten originally issued and all eighteen newly submitted claims of the patent granted to the University of Pittsburgh were invalid. The University of Pittsburgh then appealed the examiner's decision, but only with respect to two of the newly submitted claims. We cross-appealed the examiner's refusal to reject those two newly submitted claims as anticipated by the prior art. The Patent Trial and Appeal Board issued a decision simultaneously granting both appeals, thus confirming that all claims of the patent were invalid, but with respect to the newly submitted claims, on different grounds than those cited in the decision by the initial examiner. On this basis, the University of Pittsburgh filed a request to reopen prosecution and submitted claim amendments to those newly submitted claims to the Patent and Trademark Office for further consideration in an attempt to overcome the Patent Trial and Appeal Board's institution of a new ground for rejection as anticipated by the prior art. We submitted comments to the Patent and Trademark Office arguing that these claim amendments did not overcome the anticipated rejection and as of June 30, 2014, we have not received any decision from the Patent and Trademark Office regarding the amended claims. We do not know when a final decision can be expected, and at this stage, we are not in a position to assess the probable outcome of these proceedings.
Repayment of subsidies
On January 5, 2012, TiGenix SAU lodged an ordinary appeal before the Contentious-Administrative Chamber of the National Appellate Court of Spain (Audiencia Nacional) challenging two decisions taken by the Director General of Technology Transfer and Business Development at the Spanish Ministry of Science and Innovation (the "Administration") on November 16, 2011, which partially revoked and claimed the repayment of two subsidies granted in 2006 and 2007, respectively.
Both contested subsidies were granted to a consortium of beneficiaries, one of which was TiGenix SAU. TiGenix SAU also acted as representative of the beneficiaries in the consortium.
F-55
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
29. Commitments and contingencies (Continued)
The Administration claims that (i) the contested subsidies, together with other subsidies granted to TiGenix SAU during the same time period (i.e., 2006 and 2007), exceeded the maximum permitted by law, and has, therefore, requested the reimbursement of the excess amount granted, and that (ii) some of the expenses attributed to the project financed by the contested subsidies had already been financed by other subsidies.
TiGenix SAU contends, among other arguments, that the Administration is not entitled to aggregate all of the subsidies granted to TiGenix SAU (i.e., the contested subsidies and other subsidies granted) for purposes of applying the maximum (i.e., in the particular case of TiGenix SAU, 60.0% of the eligible cost of the project), because the various subsidies were granted for financing different projects with different purposes and scopes
The total claim of the Administration, with respect to the full consortium and both contested subsidies, including late payment interest, amounts to 0.9 million euros, and the Administration has claimed the full amount from TiGenix SAU, as the representative of the consortium. However, the claim against TiGenix SAU amounts to 0.3 million euros, and in case the appeal does not succeed, TiGenix SAU would be able to claim the remaining amount from the other members of the consortium.
As an intermediate measure, TiGenix SAU obtained an injunctive decision that the amounts claimed by the Administration do not have to be repaid until a final judgment is received. Instead, TiGenix SAU requested two financial institutions to issue separate guarantees in favor of the Administration guaranteeing the full amount claimed.
On May 20, 2014, TiGenix SAU received the judgment of the Chamber for Contentious Administrative Proceedings of the National High Court of April, 30, 2014. In this judgment, the court partially upheld the claims made by TiGenix SAU throughout the administrative appeal, and declared null the two resolutions on the partial repayment of the two subsidies that were granted in 2006 and 2007, respectively. However, the court also found that there were grounds for a partial repayment of the contested subsidies but ordered the Administration to recalculate the amount of such repayment. It concluded that some of the items included in the Administration's calculations are either wrong or duplicative. Because the court did not calculate the amount to be repaid, the Administration must submit revised calculations of the amounts to be repaid under the contested subsidies. Even though in principle this should have been done within a period of two months, the Administration has not yet submitted such revised calculations as of June 30, 2014. We have no recourse to any further appeals against the judgment of the court.
30. Subsequent events
On September 30, 2014, we drew down the final installment of 2.5 million euros available under the loan facility with Kreos Capital IV (UK).
F-56
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS (Continued)
31. Consolidation scope
| | | | Ownership interest | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | | | As at June 30, | As at December 31, | ||||||||||
| | | Place of incorporation | ||||||||||||
Legal Entity | Principal activity | 2014 | 2013 | 2012 | ||||||||||
TiGenix | Biopharmaceutical company | Belgium | 100 | % | 100 | % | 100 | % | ||||||
TiGenix SAU | Biopharmaceutical company | Spain | 100 | % | 100 | % | 100 | % | ||||||
TiGenix Inc. | Biopharmaceutical company | U.S.A. | 100 | % | 100 | % | 100 | % | ||||||
TiGenix B.V. | Biopharmaceutical company | Netherlands | — | % | 100 | % | 100 | % | ||||||
TiGenix Ltd. | Biopharmaceutical company | England and Wales | — | % | 100 | % | 100 | % | ||||||
F-57
American Depositary Shares
Representing Ordinary Shares
PROSPECTUS
Canaccord Genuity
Nomura
Maxim Group LLC
Chardan Capital Markets
,
PART II INFORMATION NOT REQUIRED IN PROSPECTUS.
Item 6. Indemnification of Directors and Officers.
Under Belgian law, the directors of a company may be liable for damages to the company in case of improper performance of their duties. Our directors may be liable to our Company and to third parties for infringement of our articles of association or Belgian company law. Under certain circumstances, directors may be criminally liable. We maintain liability insurance for the benefit of our directors and executive management.
In the underwriting agreement, the form of which is filed as Exhibit 1.1 to this registration statement, the underwriters will agree to indemnify, under certain conditions, us, the members of our board of directors and persons who control our Company within the meaning of the Securities Act against certain liabilities, but only to the extent that such liabilities are caused by information relating to the underwriters furnished to us in writing expressly for use in this registration statement and certain other disclosure documents.
Item 7. Recent Sales of Unregistered Securities.
During the past three years, we issued securities in transactions that have not been registered under the Securities Act as set forth below. We believe that each such issuance was exempt from registration under the Securities Act in reliance on Regulation S or Regulation D under the Securities Act or Section 4(a)(2) of the Securities Act regarding transactions by an issuer not involving a public offering or involving offers and sales of securities outside the United States.
On May 3, 2011, we issued 44,814,402 ordinary shares at a price per share of 1.2977 euros pursuant to a capital increase in kind of the share capital.
On June 6, 2011, we issued 15,187,111 ordinary shares at a price per share of 1.00 euros pursuant to a capital increase in cash of the share capital.
On April 17, 2012, we issued 536,534 ordinary shares at a price per share of 4.28 euros pursuant to a capital increase in kind of the share capital.
On December 27, 2012, we issued 8,629,385 ordinary shares at a price per share of 0.78 euros pursuant to a capital increase in cash of the share capital.
On July 24, 2013, we issued 21,259,092 ordinary shares at a price per share of 0.25 euros pursuant to a capital increase in cash of the share capital.
On July 26, 2013, we issued 4,740,908 ordinary shares at a price per share of 0.25 euros pursuant to a capital increase in cash of the share capital.
On November 22, 2013, we issued 34,188,034 ordinary shares at a price per share of 0.351 euros pursuant to a capital increase in cash of the share capital.
On July 6, 2012, we issued and granted 4,000,000 warrants to members of executive management and employees at an exercise price of 1.00 euro per share.
On March 20, 2013, we issued 777,000 warrants, 433,000 of which were effectively granted to directors on April 3, 2013 and one member of executive management on May 7, 2013 at an exercise price of 1.00 euro per share.
On December 16, 2013, we issued and granted 1,806,000 warrants to members of executive management and employees at an exercise price of 0.46 euros per share for employees and 0.50 euros per share for non-employees.
II-1
On April 22, 2014, we issued and granted 1,994,302 warrants to Kreos Capital IV (Expert Fund) at an exercise price of 0.75 euros per share.
Item 8. Exhibits and Financial Statement Schedules.
| Exhibit Number | Description of Exhibit | ||
|---|---|---|---|
| *1.1 | Form of Underwriting Agreement | ||
3.1 | Articles of Association of TiGenix, as amended and currently in effect (English translation) | ||
*4.1 | Form of Deposit Agreement | ||
*4.2 | Form of American Depositary Receipt (included in Exhibit 4.1) | ||
*5.1 | Opinion of Proskauer Rose LLP | ||
*8.1 | Opinion of Proskauer Rose LLP as to U.S. tax matters | ||
*8.2 | Opinion of Osborne Clarke BV CVBA as to Belgian tax matters | ||
†10.1 | Distribution Agreement dated April 2, 2014 between TiGenix and Swedish Orphan Biovitrium AB as restated on April 23 and on May 28, 2014 | ||
†10.2 | Loan Facility Agreement dated December 20, 2013 between TiGenix and Kreos Capital IV (UK) Limited | ||
†10.3 | Share Purchase Agreement dated January 23, 2014 between TiGenix and PharmaCell B.V. | ||
10.4 | Warrants Plan 2012 (English translation) | ||
10.5 | Warrants Plan 2013 (English translation) | ||
10.6 | Second Warrants Plan 2013 (English translation) | ||
10.7 | Kreos Warrants Plan (English translation) | ||
21.1 | Subsidiaries of TiGenix | ||
*23.1 | Consent of BDO Bedrijfsrevisoren Burg. CVBA, independent registered public accounting firm | ||
*23.2 | Consents of Proskauer Rose LLP (included in Exhibits 5.1 and 8.1) | ||
*23.3 | Consent of Osborne Clarke BV CVBA (included in Exhibit 8.2) | ||
*24.1 | Powers of Attorney (included on the signature page) | ||
- *
- To be filed by amendment.
- †
- Confidential treatment has been sought with respect to portions of the exhibit, which has been separately submitted to the SEC.
(a) The undersigned registrant hereby undertakes to provide to the underwriter at the closing specified in the underwriting agreements, certificates in such denominations and registered in such names as required by the underwriter to permit prompt delivery to each purchaser.
(b) Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange
II-2
Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
(c) The undersigned registrant hereby undertakes that:
(i) For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(ii) For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-3
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form F-1 and has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in Leuven, Belgium on the day of October, 2014.
| TIGENIX | ||||||
By: | ||||||
| Name: | Eduardo Bravo Fernández de Araoz | |||||
| Title: | Chief Executive Officer | |||||
Each person whose signature appears below hereby constitutes and appoints Eduardo Bravo as his attorney-in-fact and agent, with full power of substitution and resubstitution for him in any and all capacities, to sign any or all amendments or post-effective amendments to this registration statement, or any registration statement for the same offering that is to be effective upon filing pursuant to Rule 462(b) under the Securities Act of 1933, as amended, and to file the same, with exhibits thereto and other documents in connection therewith or in connection with the registration of the shares of common stock under the Securities Exchange Act of 1934, as amended, with the Securities and Exchange Commission, granting unto such attorney-in-fact and agent full power and authority to do and perform each and every act and thing requisite and necessary in connection with such matters and hereby ratifying and confirming all that such attorney-in-fact and agent or his substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, as amended, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
Signatures | Title | Date | ||||
|---|---|---|---|---|---|---|
| By: | Eduardo Bravo Fernández de Araoz | Chief Executive Officer and Managing Director (Principal Executive Officer) | , 2014 | |||
By: | Claudia D'Augusta | Chief Financial Officer (Principal Financial and Accounting Officer) | , 2014 | |||
By: | Innosté SA, represented by Jean Stéphenne | Director | , 2014 | |||
By: | Dirk Büscher | Director | , 2014 | |||
II-4
Signatures | Title | Date | ||||
|---|---|---|---|---|---|---|
| By: | Willy Duron | Director | , 2014 | |||
By: | Greig Biotechnology Global Consulting, Inc., represented by Russell Greig | Director | , 2014 | |||
By: | Eduard Enrico Holdener | Director | , 2014 | |||
By: | R&S Consulting BVBA, represented by Dirk Reyn | Director | , 2014 | |||
By: | José Terencio | Director | , 2014 | |||
II-5
SIGNATURE OF AUTHORIZED REPRESENTATIVE IN THE UNITED STATES
Pursuant to the requirements of the Securities Act of 1933, as amended, the undersigned, the registrant's duly authorized representative in the United States, has signed this registration statement in on the day of , 2014.
| By: | | |||||
| Name: | Donald J. Puglisi | |||||
II-6
| Exhibit Number | Description of Exhibit | ||
|---|---|---|---|
| *1.1 | Form of Underwriting Agreement | ||
3.1 | Articles of Association of TiGenix, as amended and currently in effect (English translation) | ||
*4.1 | Form of Deposit Agreement | ||
*4.2 | Form of American Depositary Receipt (included in Exhibit 4.1) | ||
*5.1 | Opinion of Proskauer Rose LLP | ||
*8.1 | Opinion of Proskauer Rose LLP as to U.S. tax matters | ||
*8.2 | Opinion of Osborne Clarke BV CVBA as to Belgian tax matters | ||
†10.1 | Distribution Agreement dated April 2, 2014 between TiGenix and Swedish Orphan Biovitrium AB as restated on April 23 and on May 28, 2014 | ||
†10.2 | Loan Facility Agreement dated December 20, 2013 between TiGenix and Kreos Capital IV (UK) Limited | ||
†10.3 | Share Purchase Agreement dated January 23, 2014 between TiGenix and PharmaCell B.V. | ||
10.4 | Warrants Plan 2012 (English translation) | ||
10.5 | Warrants Plan 2013 (English translation) | ||
10.6 | Second Warrants Plan 2013 (English translation) | ||
10.7 | Kreos Warrants Plan (English translation) | ||
21.1 | Subsidiaries of TiGenix | ||
*23.1 | Consent of BDO Bedrijfsrevisoren Burg. CVBA, independent registered public accounting firm | ||
*23.2 | Consents of Proskauer Rose LLP (included in Exhibits 5.1 and 8.1) | ||
*23.3 | Consent of Osborne Clarke BV CVBA (included in Exhibit 8.2) | ||
*24.1 | Powers of Attorney (included on the signature page) | ||
- *
- To be filed by amendment.
- †
- Confidential treatment has been sought with respect to portions of the exhibit, which has been submitted separately with the SEC.
II-7