UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-K
(Mark One)
☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
OR
☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number: 001-37725
ViewRay, Inc.
(Exact Name of Registrant as Specified in its Charter)
Delaware | 42-1777485 |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
2 Thermo Fisher Way Oakwood Village, OH | 44146 |
(Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (440) 703-3210
Securities registered pursuant to section 12(b) of the Act:
Title of Each Class | | Name of Each Exchange on Which Registered |
Common Stock, par value $0.01 | | The NASDAQ Global Market |
Securities registered pursuant to Section 12(g) of the Exchange Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer”, “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | | ☐ | | Accelerated filer | | ☐ |
| | | |
Non-accelerated filer | | ☐ (Do not check if a small reporting company) | | Small reporting company | | ☒ |
| | | | | | |
| | | | Emerging growth company | | ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☒
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
At June 30, 2017, the aggregate market value of the registrant’s common stock held by non-affiliates of the registrant was $166,918,728 based on the closing sale price as reported on the Nasdaq Global Market. As of March 2, 2018, the registrant had 67,653,974 shares of common stock, $0.01 par value per share, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement to be delivered to stockholders in connection with the 2018 annual meeting of stockholders are incorporated by reference in Part III of this Form 10-K where indicated.
VIEWRAY, INC.
FORM 10-K
ANNUAL REPORT
TABLE OF CONTENTS
2
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K, or this Report, contains forward-looking statements, including, without limitation, in the sections captioned “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and elsewhere. Any and all statements contained in this Report that are not statements of historical fact may be deemed forward-looking statements. Terms such as “may,” “might,” “would,” “should,” “could,” “project,” “estimate,” “pro forma,” “predict,” “potential,” “strategy,” “anticipate,” “attempt,” “develop,” “plan,” “help,” “believe,” “continue,” “intend,” “expect,” “future” and terms of similar import (including the negative of any of the foregoing) may be intended to identify forward-looking statements. However, not all forward-looking statements may contain one or more of these identifying terms. Forward-looking statements in this Report may include, without limitation, statements regarding: (i) the plans and objectives of management for future operations, including plans or objectives relating to the development of products; (ii) a projection of income (including income/loss), earnings (including earnings/loss) per share, capital expenditures, dividends, capital structure or other financial items; (iii) our future financial performance, including any such statement contained in a discussion and analysis of financial condition by management or in the results of operations included pursuant to the rules and regulations of the Securities and Exchange Commission, or the SEC; and (iv) the assumptions underlying or relating to any statement described in points (i), (ii) or (iii) above.
The forward-looking statements are not meant to predict or guarantee actual results, performance, events or circumstances and may not be realized because they are based upon our current projections, plans, objectives, beliefs, expectations, estimates and assumptions and are subject to a number of risks and uncertainties and other influences, many of which we have no control over. Actual results and the timing of certain events and circumstances may differ materially from those described by the forward-looking statements as a result of these risks and uncertainties. Factors that may influence or contribute to the inaccuracy of the forward-looking statements or cause actual results to differ materially from expected or desired results may include, without limitation:
| • | market acceptance of magnetic resonance imaging (“MRI”)-guided radiation therapy; |
| • | the benefits of MRI-guided radiation therapy; |
| • | our ability to successfully sell and market MRIdian in our existing and expanded geographies; |
| • | the performance of MRIdian in clinical settings; |
| • | competition from existing technologies or products or new technologies and products that may emerge; |
| • | the pricing and reimbursement of MRI-guided radiation therapy; |
| • | the implementation of our business model and strategic plans for our business and MRIdian; |
| • | the scope of protection we are able to establish and maintain for intellectual property rights covering MRIdian; |
| • | our ability to obtain regulatory approval in targeted markets for MRIdian; |
| • | estimates of our future revenue, expenses, capital requirements and our need for additional financing; |
| • | our financial performance; |
| • | our expectations related to the MRIdian linear accelerator technology, or MRIdian Linac; |
| • | developments relating to our competitors and the healthcare industry; and |
| • | other risks and uncertainties, including those listed under the section titled “Risk Factors.” |
Any forward-looking statements in this Report reflect our current views with respect to future events or to our future financial performance and involve known and unknown risks, uncertainties and other factors that may cause actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by these forward-looking statements. Factors that may cause actual results to differ materially from current expectations include, among other things, those listed under Part II, Item 1A, titled “Risk Factors” and discussed elsewhere in this Report. Given these uncertainties, you are cautioned not to place
3
undue reliance on these forward-looking statements. We disclaim any obligation to update the forward-looking statements contained in this Report to reflect any new information or future events or circumstances or otherwise, except as required by law.
This Report also contains estimates, projections and other information concerning our industry, our business, and the markets for certain products, including data regarding the estimated size of those markets. Information that is based on estimates, forecasts, projections, market research or similar methodologies is inherently subject to uncertainties and actual events or circumstances may differ materially from events and circumstances reflected in this information. Unless otherwise expressly stated, we obtained this industry, business, market and other data from reports, research surveys, studies and similar data prepared by market research firms and other third parties, industry, medical and general publications, government data and similar sources.
PART I
Item 1. BUSINESS
Company Overview
We design, manufacture and market the ViewRay MRIdian®, the only clinical MRI-guided radiation therapy system on the market. The MRIdian combines MRI and external-beam radiation therapy to simultaneously image and treat cancer patients. MRI is a broadly used imaging tool that has the ability to clearly differentiate between types of soft tissue. In contrast, X-ray or computed tomography (CT), the most commonly used imaging technologies in radiation therapy today, are often unable to distinguish soft tissues such as the tumor and critical organs. MRIdian integrates MRI technology, radiation delivery and our proprietary software to locate, target and track soft-tissue tumors, while radiation is delivered. These capabilities allow MRIdian to deliver radiation to the tumor more accurately, while reducing the radiation amount delivered to nearby healthy tissue, as compared to other radiation therapy treatments currently available. We believe this will lead to improved patient outcomes and reduced treatment-related side effects.
There are two generations of the MRIdian: the first generation MRIdian with Cobalt-60 based radiation beams and the second generation MRIdian Linac, with more advanced linear accelerator or ‘linac’ based radiation beams. ViewRay’s first generation MRIdian was a breakthrough that integrated high quality radiation therapy with simultaneous magnetic resonance imaging (MRI).
ViewRay first-generation MRIdian System with radiation powered by Cobalt-60 was cleared by the FDA in May 2012. By the end of 2016, the Company had shipped nine of these first generation MRIdian Systems. The MRIdian System demonstrated in clinical practice for the first time the benefits of MRI-guided radiation treatment for cancer, generating a large body of clinical evidence from its use in pancreatic, breast, lung, prostate and other cancers.
ViewRay’s second-generation system, the MRIdian Linac (Linear Accelerator) system received the Conformité Européene, or CE Mark in September 2016 and FDA 510k clearance in February 2017. The MRIdian Linac System has several advantages over the first generation MRIdian System. Linac-based radiation therapy delivery systems allow for higher dose, faster electronic variation of dose and very fast electronic beam activation and deactivation. Linear Accelerator technology obviates the need for the inspection, replacement, and disposal of Cobalt-60, and oversight from the U.S. Nuclear Regulatory Commission (or similar national agency in foreign countries). ViewRay solved two major long-standing problems to integrate a linac beam compactly with an MRI system: 1) linac radiofrequency interference with the operation of the MRI and 2) MRI magnetic interference with the operation of the linac.
• | Linacs utilize high-powered microwave generators similar to equipment used in radar at airports. These “radar stations” inside the linac create noise that can corrupt the delicate signals measured from the patient’s body to generate MR images. ViewRay solved this problem by introducing technology similar to that used in stealth aircraft. Stealth aircraft can hide from radar by using a coating that reflects and absorbs microwaves, thus preventing radar beams that strike the aircraft from bouncing back to the radar station. In a similar manner, ViewRay’s linac based system reflects and absorbs the output of the linac “radar station” to prevent it from interfering with the MRI, producing images as noise-free as MRI images with no integrated linac. |
4
• | MRIs utilize high-powered superconducting magnets that are required to image the patient’s tissues. Many linac components will not operate properly when placed close to or inside these strong magnetic fields. Close placement is necessary to produce a compact system that can fit in existing radiation therapy vaults, as the MRIdian linac does. ViewRay overcame this challenge by creating magnetic shielding shells that create voids in the magnetic field without significantly disturbing the magnetic field used for imaging. This allows the linac to operate on the MRIdian linac gantry as if there were no magnetic field present. |
In addition, ViewRay has applied the same double-focused multi-leaf collimator technology, originally designed for the first generation MRIdian System, to ViewRay’s MRIdian linac technology. This new high-resolution beam-shaping multi-leaf collimator (MLC), called the RayZR® MLC, when combined with the already sharp linac radiation source results in the sharpest linac beam commercially available in the industry. Therefore, we believe that ViewRay’s MRIdian linac technology could be transformative to the standard of care in radiation therapy.
Both generations of the MRIdian have received 510(k) marketing clearance from the US Food and Drug Administration, or FDA, and permission to affix the CE mark.
| • | We received initial 510(k) marketing clearance from the US Food and Drug Administration, or FDA, for our treatment planning and delivery software in January 2011. |
| • | We received 510(k) marketing clearance for MRIdian, with Cobalt-60 as the radiation source, in May 2012. We also received permission to affix the CE mark to MRIdian with Cobalt-60 in November 2014, allowing MRIdian with Cobalt-60 to be sold within the European Economic Area, or EEA. In August 2016, we received regulatory approval from the Japanese Ministry of Health, Labor and Welfare to market MRIdian with Cobalt-60 in Japan. In August 2016, we also received approval from the China Food and Drug Administration to market MRIdian with Cobalt-60 in China. |
| • | In September 2016, we received the CE mark for the second generation MRIdian Linac (with a linear accelerator as the radiation source) in the EEA. In February 2017, we received 510(k) clearance from the FDA to market MRIdian Linac. In June 2017, we received 510(k) clearance to market RayZR™, our high-resolution beam-shaping multi-leaf collimator, or MLC. We also received MRIdian Linac regulatory approval in Taiwan and Canada in August 2017, and in Israel in November 2017. We are also seeking required MRIdian Linac approvals in other countries such as Japan and China. |
Cancer is a leading cause of death globally and the second leading cause of death in the United States. Radiation therapy is a common method used to treat cancer that uses lethal doses of ionizing energy to damage genetic material in cells. Nearly two-thirds of all treated cancer patients in the United States will receive some form of radiation therapy during the course of their illness, according to estimates by the American Society for Radiation Oncology, or ASTRO. In 2016, IMV Medical Information Division, Inc., or IMV, reported that 97% of patients receiving radiation therapy in the United States were treated by a linear accelerator, or linac. The global linac market was estimated at approximately $4.6 billion in 2015 and was expected to grow to approximately $6.3 billion by 2020 according to a 2015 Markets and Markets report. IAEA Human Health Campus reported that there are over 11,500 linacs installed at over 7,800 centers worldwide. We currently estimate the annual market for linacs to be 1,000 units per year globally, the majority of which are replacement units. We believe the addressable market for MRIdian is the annual market for linacs due to MRIdian’s ability to treat a broad spectrum of disease sites. However, we believe that MRIdian may initially be used more frequently for complex cancer cases that may be difficult to treat on a standard linac due to the location of the tumor in relation to the surrounding organs at risk for radiation damage.
Despite the prevalence of MRI for diagnostic purposes and its ability to image soft tissue clearly, the radiation therapy industry has previously been unable to integrate MRI into external-beam radiation therapy systems successfully. Existing radiation therapy systems use X-ray-based imaging technologies, such as CT, which do not clearly differentiate between types of soft tissues or provide a fully accurate view of a tumor’s position in relation to critical organs. X-ray based imaging systems integrated into radiation delivery devices also often suffer from imaging artifacts caused by organ motion related to breathing, and artifacts related to air/fluid levels in the stomach and bowels. In addition, existing systems that offer imaging during the course of a treatment are limited by the rate at which they can image, due to the level of additional radiation to which they expose the patient. These constraints can make it difficult for a clinician to locate a soft tissue tumor accurately, track its motion in real-time or adapt treatment as internal anatomy changes. It is very difficult to both irradiate a tumor and minimize the amount of radiation exposure to critical organs, without the ability to see the exact location and shape of the tumor and
5
surrounding critical organs. If a tumor is insufficiently irradiated, it may not respond to the treatment, resulting in a higher rate of local tumor recurrence and lower probability of overall survival for the patient. Excess radiation exposure to healthy organs and other healthy soft tissues can lead to severe side effects, including organ failure, secondary cancers and in the most serious cases, even death. We believe that the MRIdian’s ability to see the exact location and shape of the tumor and surrounding critical organs will lead to improved patient outcomes and reduced treatment related side effects.
MRIdian is the first radiation therapy solution that enables simultaneous radiation treatment delivery and real-time MRI imaging of a patient’s internal anatomy. It generates high-quality images that differentiate between the targeted tumor, surrounding soft tissue and nearby critical organs. MRIdian also records the level of radiation dose that the treatment area has received, enabling physicians to adapt the prescription between treatments, as needed. We believe this improved visualization and accurate dose recording will enable better treatment, improve patient outcomes and reduce side effects. Key benefits to users and patients include: improved imaging and patient alignment; the ability to adapt the patient’s radiation treatments to changes while the patient is still on the treatment table, or “on-table adaptive treatment planning”; MRI-based motion management; and an accurate recording of the delivered radiation dose. Physicians have already used MRIdian to treat a broad spectrum of radiation therapy patients with more than 45 different types of cancer, as well as patients for whom radiation therapy was previously not an option.
We currently market MRIdian through a direct sales force in the United States and Canada and are developing a sales force to assist distributors in the rest of the world. At December 31, 2017, we had installed or delivered 15 MRIdian systems worldwide and had a backlog with total value of $203.6 million. We generated revenue of $34.0 million, $22.2 million, and $10.4 million for the years ended December 31, 2017, 2016 and 2015, respectively. We had net losses of $72.2 million, $50.6 million and $45.0 million for the years ended December 31, 2017, 2016 and 2015, respectively.
Corporate Information
We were incorporated under the laws of the state of Nevada on September 6, 2013 under the name “Mirax Corp”. Prior to the Merger and Split-Off (each as defined below), Mirax developed and supplied mobile communications accessories.
On July 8, 2015, we completed a 1.185763-for-1 forward stock split of our common stock in the form of a dividend. The result was that the 4,343,339 shares of common stock with a par value of $0.001 per share outstanding immediately prior to the stock split, became 5,150,176 shares of common stock, with a par value of $0.001 per share, outstanding immediately thereafter.
On July 15, 2015, we changed our name to ViewRay, Inc. by filing the Certificate of Amendment to our Articles of Incorporation. Additionally, on July 21, 2015, we changed our domicile from the State of Nevada to the State of Delaware by reincorporation, or the Conversion, and as a result of the Conversion, increased our authorized capital stock from 75,000,000 shares of common stock, par value $0.001 per share, to 300,000,000 shares of common stock, par value $0.01 per share and 10,000,000 shares of “blank check” preferred stock, par value $0.01 per share. Upon effectiveness of the Conversion, our corporate matters and affairs ceased to be governed by the Nevada Revised Statutes and became subject to the General Corporation Law of the State of Delaware. All share and per share numbers in this Report relating to our common stock have been adjusted to give effect to this forward stock split and the Conversion, unless otherwise stated. On July 23, 2015, we amended and restated our certificate of incorporation by filing the Amended and Restated Certificate of Incorporation with the Secretary of State of the State of Delaware and adopted the Amended and Restated Bylaws.
On July 23, 2015, our wholly-owned subsidiary, Vesuvius Acquisition Corp., a corporation formed in the State of Delaware on July 16, 2015, or the Acquisition Sub, merged with and into ViewRay Technologies, Inc. Pursuant to this transaction, or the Merger, ViewRay Technologies, Inc. was the surviving corporation and became our wholly-owned subsidiary. All of the outstanding capital stock of ViewRay Technologies, Inc. was converted into shares of our common stock, as described in more detail below.
Immediately prior to the closing of the Merger, under the terms of a split-off agreement, or the Split-Off Agreement, and a general release agreement, we transferred all of our pre-Merger operating assets and liabilities to our wholly-owned special-purpose subsidiary, Mirax Enterprise Corp., a Nevada corporation, or the Split-Off Subsidiary, formed on July 16, 2015. Thereafter, pursuant to the Split-Off Agreement, we transferred all of the outstanding
6
shares of capital stock of the Split-Off Subsidiary to our pre-Merger majority stockholders, and our former sole officer and director, in consideration of and in exchange for: (i) the surrender and cancellation of an aggregate of 4,150,171 shares of our common stock; and (ii) certain representations, covenants and indemnities, together referred to as the Split-Off.
As a result of the Merger and Split-Off, we discontinued our pre-Merger business, acquired the business of ViewRay Technologies, Inc. and continue the business operations of ViewRay Technologies, Inc. as a publicly-traded company under the name ViewRay, Inc.
On March 31, 2016, our shares of common stock commenced trading on the Nasdaq Global Market under the symbol “VRAY.” Prior to this time, our common stock was quoted on the OTC Markets, OTCQB tier of OTC Markets Group, Inc. under the same symbol.
As a result of the Merger we have ceased to be a “shell company” (as such term is defined in Rule 12b-2 under the Exchange Act).
As used in this Report henceforward, unless otherwise stated or the context clearly indicates otherwise, the terms the “Company,” the “Registrant,” “ViewRay,” “we,” “us” and “our” refer to ViewRay, Inc., incorporated in Delaware, after giving effect to the Merger and the Split-Off.
ViewRay, Inc. is the sole stockholder of ViewRay Technologies, Inc., which commenced operations as a Florida corporation in 2004, subsequently reincorporated in Delaware in in 2007, and changed its name to ViewRay Technologies, Inc. in July 2015.
Our authorized capital stock currently consists of 300,000,000 shares of common stock, and 10,000,000 shares of the preferred stock. Our common stock is listed on The NASDAQ Global Market under the symbol “VRAY.”
Our principal corporate headquarters are located at 2 Thermo Fisher Way, Oakwood Village, Ohio 44146. Our telephone number is (440) 703-3210. Our website address is www.viewray.com. (Any information on ViewRay’s website or which can be accessed through it, are not a part of this Annual Report on Form 10-K.)
Cancer and Radiation Therapy Market
Incidence of Cancer
Cancer is a leading cause of death globally and the second leading cause of death in the United States behind cardiovascular disease. According to the American Cancer Society, nearly 1.7 million people were expected to be diagnosed with cancer in the United States during 2016 and approximately 0.6 million were expected to die from cancer, accounting for nearly one of every four deaths. As a result of a growing and aging population, the World Health Organization’s, or WHO, Global Initiative for Cancer Registry Development estimates that the number of new cancer cases worldwide will grow from 14.1 million in 2008 to 19.3 million in 2025.
Cancer Therapy
The primary goal of cancer therapy is to kill cancerous tissues, while minimizing damage to healthy tissues. There are three main ways to treat cancer: surgery, chemotherapy and radiation therapy. Surgery attempts to remove the tumor from the body, while minimizing trauma to healthy tissue and preventing the spread or translocation of the disease to other parts of the body. Surgery is particularly effective because the surgeon can see the tumor and surrounding healthy tissue directly throughout the course of the procedure and can adapt his or her planned removal approach mid-procedure accordingly. Chemotherapy uses drugs to kill cancer cells. Unlike surgery, most forms of chemotherapy circulate throughout the patient’s body to reach cancer cells almost anywhere in the body systemically. Chemotherapy is most effective at destroying microscopic levels of disease. Radiation therapy is typically used as a local treatment, directed at a tumor and surrounding areas where microscopic cancerous cells are assumed to have spread. Radiation may be used as the primary treatment modality, or in combination with either chemotherapy or surgery or both. Radiation therapy works by damaging genetic material in cells and other cell components through interaction with ionizing energy. Effective radiation therapy balances destroying cancer cells with minimizing damage to normal cells. It can be used at high doses to ablate a tumor, an effect similar to surgery, or at moderate doses to target local microscopic disease, as is done with chemotherapy. Other, more recently developed ways of treating cancer, include hormone therapy and targeted therapy, such as immunotherapy.
7
Radiation Therapy
Radiation therapy has become widespread, with nearly two-thirds of all treated cancer patients in the United States receiving some form of radiation therapy during the course of their cancer treatments, according to estimates by ASTRO. For most cancer types treated with radiation therapy, at least 75% of the patients are treated with the intent to cure the cancer. For lung and brain cancers, that number is somewhat lower, with 59% of lung cancer patients and 50% of brain cancer patients being treated with the goal of curing cancer. The remainder of cases are treated with palliative intent to relieve pain or other tumor related symptoms. The type of radiation therapy delivered by linac or Cobalt 60 based devices is a non-invasive outpatient procedure with little or no recovery time and can be used on patients who are unable to undergo conventional surgery. According to IMV, 97% of patients receiving radiation therapy in the United States are treated using a linac.
Radiation is used to kill cancer cells primarily by damaging their DNA but can also kill healthy cells in the same way or cause them to become cancerous themselves. As a result, the goal of curative radiation therapy is to balance delivery of a sufficiently high dose of radiation to a tumor to kill the cancer cells while, at the same time, minimizing damage to healthy cells, particularly those in critical organs. Normal cells are better able to repair themselves after radiation than tumor cells, so doses of radiation are often fractionated, or delivered in separate sessions with rest periods in between. As a result, standard radiation therapy is often given once a day, five times a week, for one to nine weeks. According to a 2017 IMV report, patients made an estimated 20.2 million radiation therapy treatment visits in the United States from March 2016 to March 2017.
Radiation Therapy Equipment Market
According to a 2015 Markets and Markets report, the global linac market was estimated at approximately $4.6 billion in 2015 and was expected to grow to approximately $6.3 billion by 2020. According to IAEA Human Health Campus, there are more than 11,500 linacs installed at over 7,800 centers worldwide. In the United States, there are approximately 3,600 linacs installed at approximately 2,100 centers. The annual market for linacs is estimated to be 1,000 units per year globally, the majority of which are replacements for older machines.
In the radiation therapy market, new technologies have historically been adopted at a rapid rate. According to IMV, the percentage of centers performing intensity modulated radiation therapy, or IMRT, grew from 30% in 2002 to 96% in 2012. The percentage of sites utilizing image-guided radiation therapy, or IGRT, grew even more quickly: from 15% in 2004 to 83% in 2012. The majority of IGRT procedures use on-board X-ray systems. As leading cancer centers adopt and study MRI-guided radiation therapy, we believe that our next-generation linac based MRI system will also follow a rapid adoption curve in the broader linac replacement market.
Radiation Therapy Treatment Process
Following diagnosis of the disease state, radiation treatment generally consists of the following steps:
| • | Imaging and tumor contouring. To design the treatment plan, physicians obtain initial images of the tumor. This is done most commonly using a CT scan, often supplemented by an MRI, a positron emission tomography, or PET, scan, or both. These images, also known as simulation scans, are then imported into a treatment planning software system and aligned or “registered” to each other. Based on clinical experience, a physician will manually delineate, or “draw”, specific areas on the aligned images to define the location and extent of the tumor highlighting the following: |
| • | Gross tumor volume, or GTV, a volumetric region encompassing the visible tumor. |
| • | Clinical target volume, or CTV, is the GTV plus a larger, surrounding area where cancer cells are already likely to have spread. |
| • | Planning target volume, or PTV, is the CTV plus a further enlarged area to allow for: inexact imaging; patient movement during treatment; tumor movement between planning and treatment; and organ motion caused by breathing. The PTV margin unavoidably includes only normal, healthy tissues and may be many times larger than the CTV. While the PTV margin is necessary to reduce the risk of local tumor recurrence, it does increase the risk of radiation damage to healthy tissue and critical organs. |
8
| • | Treatment planning and dose prescription. Once the clinician has a three-dimensional map of the tumor, surrounding healthy tissues and nearby critical organs, a physician determines a treatment plan using one of the methods below. Creation of these plans typically takes days but can require up to several weeks. A typical curative radiation therapy treatment dose will be delivered over the course of several weeks with 10 to 43 radiation therapy sessions, referred to as fractions, lasting from a few minutes to an hour or more depending on the treatment plan. |
| • | 3D-CRT planning. Using a method called three-dimensional conformal radiation therapy, or 3D-CRT, a clinician will choose both the beam angles and shapes the machine will use to direct the radiation beam towards the tumor, and the time period that each beam will be delivered. A computer will then calculate a prediction of the radiation dose delivered, and the radiation planning team will adjust the treatment plan on an iterative basis to arrive at a clinically acceptable radiation dose plan. |
| • | IMRT planning. Using a method called intensity modulated radiation therapy, or IMRT, a physician will use computer software that calculates hundreds or even thousands of beamlets (small radiation beams) to optimize a treatment plan in order to achieve a more precise dose distribution than 3D-CRT. IMRT plans often allow better radiation coverage of tumors, while simultaneously sparing more healthy tissues from high radiation doses. In select cancers, IMRT has been shown to result in better patient outcomes than 3D-CRT. |
| • | SRS and SBRT planning. Stereotactic radiosurgery, or SRS, and stereotactic body radiation therapy, or SBRT, are methods of delivery using 3D-CRT or IMRT, in a reduced number of sessions. SRS and SBRT deliver precisely targeted radiation in usually one to five fractions delivered in one treatment session on the same day. SRS is frequently used in brain and spine applications, while SBRT is used most often in the rest of the body, and has been shown to be particularly effective in early-stage lung cancer. |
| • | Alignment. Just prior to radiation delivery, clinicians typically take further images to assist with alignment of the patient’s tumor to the radiation beam. Most systems use a form of on-board CT, called “cone-beam CT” or “CBCT” to create this image and then move the patient so that the tumor’s location that day matches the prior planning position. However, cone beam CT may suffer from poor soft-tissue contrast, motion artifacts and may use a higher radiation dose than that available from the types of CT used for diagnosis purposes. Cumulatively, when applied every day prior to radiation delivery, the radiation exposure from CBCT (or other x-ray based image-guidance technologies) may increase the clinically relevant additional radiation dose delivered to the patient and may cause clinicians to adjust the intended radiation treatment plan. |
A less commonly used imaging technology is fluoroscopy, a real-time 2D X-ray system. However, fluoroscopy can expose a patient to even higher doses of radiation than does cone-beam CT.
Because of the limited soft tissue contrast of X-ray-based imaging, clinicians often use registration or fiducial markers to assist with alignment of the patient’s tumor to the treatment beams, such as the patient’s visible bone structures near the tumor or surgically implanted markers which identify the tumor’s location. To minimize motion due to breathing or other normal body activity, patients may also be immobilized by restraining devices, such as abdominal compression or by “respiratory control,” techniques such as cameras that monitory a patient’s breathing during treatments or by asking the patient not to breathe at certain intervals during the treatment delivery. To account for breathing and other body motions during treatment, specific trackers may be used, through a technique known as “4D radiation therapy.
| • | Delivery. Following an assessment of the tumor location relative to the radiation beam geometry, treatment is initiated, and radiation is delivered to the patient. In some cases, additional 2D X-ray images are taken intermittently or registration makers are monitored during treatment to try to account for tumor movement. However, there is no ability for physicians or other clinicians to see the tumor’s location throughout the entire radiation treatment delivery with traditional linacs. |
9
| • | Review. After a treatment session, the physician will review data gathered from the linac system to validate that the treatment it is proceeding according to his or her treatment plan. But, traditional linac systems have no ability to record the actual dose that was delivered to the tumor and nearby critical structures. In certain rare types of cancers, where the tumor is visible simply by looking at the patient without imaging equipment, the physician may decide to adjust the treatment plan during the course of the patient’s overall treatment. However, revising a treatment plan may take several days and will delay completion of the patient’s overall treatment. |
Limitations of Traditional Radiation Therapy
Limitations with traditional radiation therapy result from imaging technologies that make accurate visualization of a tumor and its relation to critical organs difficult or impossible during the treatment delivery. Most current traditional systems take images of the tumor before and after treatments, but, none do so continuously during the treatments in real time. As a result, treatments may not be delivered with the precision assumed by the physician and may not result in the necessary efficacy or reduction in local tumor recurrence. Also, healthy tissues may be exposed to radiation levels different from those predicted by the planning system and can result in patient injury.
| • | Inability to accurately locate a tumor for treatment alignment. To locate a tumor, current radiation therapy systems rely on CT scans taken while the patient is on the delivery unit treatment table, or “on-table.” Because it is difficult for differentiate between the tumor and nearby soft tissues with CT images, clinicians use surrogate registration markers, including existing bone structures, external marks and surgically implanted fiducials, to align a patient’s tumor to the treatment beams prior to commencing treatment. |
10
| • | Comparison of On-Table CT Images to On-Table MRIdian Images |
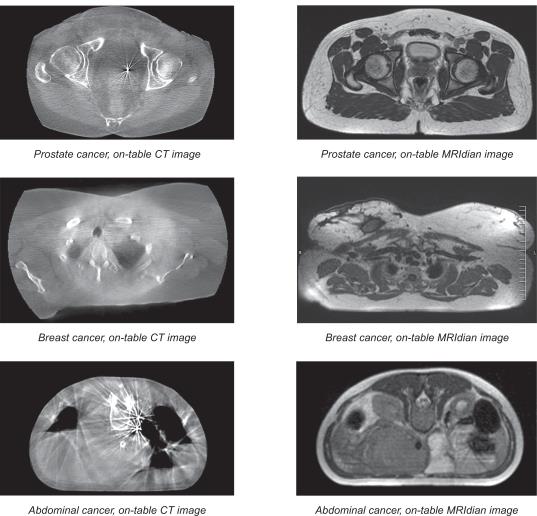
11
However, the spatial relationship between tumors and the registration markers used to locate them often changes between the time of the patient’s initial imaging and the time of his or her first treatment session. This is particularly true for tumors which are located in soft tissue. By relying on a marker as a proxy for the tumor location, rather than on the tumor itself, clinicians risk missing the tumor when they deliver radiation beams into the patient’s body. In addition, placement of surgically implanted fiducial markers comes with inherent risks: the procedures are invasive; there is a risk of pain, infection, bleeding and lung collapse; and fiducials may change location and even migrate inside the body. Despite placement of fiducials, physicians are often unable to track changes in tumor shape. Also, fiducials made of dense metals, such as gold, may cause artifacts which interfere with imaging.
| • | Inability to adapt treatment on-table. A physician designs a treatment plan and dose prescription based on images that are captured days or even weeks prior to initiation of radiation therapy. Creating a treatment plan can take up to several weeks in complex cases, and treatment itself can take up to nine weeks. However, during the course of therapy, tumors often change size, orientation or shape, and patient anatomy can change for reasons such as weight loss or gain. These changes can alter the planned radiation exposure to both the targeted regions and nearby healthy organs; this has the potential to increase the risk of local tumor recurrence and to reduce the safety of the radiation delivery. Adjusting for these changes on conventional delivery units requires re-planning, which includes getting new patient images needed to create a new treatment plan. This process may take several days and is highly resource intensive. As a result of these limitations, re-planning is infrequently performed. |
Due to limitations in imaging technologies, physicians may actually be unaware of changes in the tumor and surrounding anatomy. Consequently, they may continue to administer radiation dose according to the original treatment plan, without realizing its potential to reduce the effectiveness of the tumor treatment and to increase the risk of patient injury.
| • | Inability to track tumor and organ motion accurately. In addition to the difficulty of locating a tumor accurately in a patient’s body at the time treatment begins, a further challenge is accounting for ongoing tumor movement that takes place during treatment. Tumors have been shown to move multiple centimeters relative to surrogate registration markers over the course of only a few seconds. Breathing and other normal bodily functions, such as changes in the bladder or bowel during treatment, can cause significant tumor motion. Although physicians use internal markers, external cameras and blocks placed on the patients’ body to track respiratory and other motion, they are typically unable to track the tumor itself. As a result, physicians usually enlarge the total region to be irradiated. This limitation increases the probability of missing the targeted treatment area and exposing healthy tissues to unnecessary radiation. |
| • | Inability to record cumulative radiation delivered. In order to determine treatment effectiveness, it is important to track how much radiation has been delivered to a tumor and its surrounding healthy tissue. Currently, there are no methods to record the actual dose of radiation that was delivered to the tumor and nearby critical structures. Therefore, physicians must assume that the radiation is delivered according to plan, rather than making decisions based on actual radiation dose delivered. |
Each of these limitations increases the risk of missing a tumor and hitting healthy tissue during treatment. If a tumor is insufficiently irradiated, it may not respond to treatment, resulting in a greater probability of local tumor recurrence and reduced overall survival for the patient. The ability to avoid irradiating healthy tissue has been shown to reduce side effects. If healthy tissues, particularly critical organs, are irradiated, the side effects can be severe, including: scarring of lung tissue; fibrosis and cardiotoxicity in lung and breast cancers; incontinence and sexual dysfunction in pelvic and prostate cancers; infertility in pediatric cancers; memory loss, seizures and necrosis in brain cancer; secondary cancers, and in serious cases, death.
Although MR technology is an imaging tool broadly used to differentiate between types of soft tissue in diagnostic settings, MR technology had not been available in the radiation treatment delivery room before the launch of ViewRay’s MRIdian System. In the past, MR was not used with radiation therapy because the technologies interfered with each other: the magnetic field generated by an MRI interfered with the linac beam, while the radiofrequencies produced by the linac distorted the MR images. Current forms of CT have improved over time, but issues with radiation dose and image quality limit the utility of these technologies. Fluoroscopy and cone-beam CT
12
involve the use of X-rays, a form of ionizing radiation, and pose an increased risk of radiation-induced cancer to the patient.
Our Solution
We developed MRIdian to address the key limitations of existing external-beam radiation therapy technologies. MRIdian employs MRI-based technology to provide real-time imaging that clearly defines the targeted tumor from the surrounding soft tissue and other critical organs, both before and during radiation treatment delivery. MRIdian also allows physicians to record the level of radiation exposure that the tumor has received and adapt the prescription between treatment fractions as needed. We believe this combination of enhanced visualization and accurate dose recording will significantly improve the safety and efficacy of radiation therapy, leading to better outcomes for patients suffering from cancer.
We believe that MRIdian provides the following clinical and commercial benefits to physicians, hospitals and patients:
| • | Improved tumor visibility and patient alignment. The soft-tissue contrast of MRIdian’s on-board MRI enables clinicians to locate, target and track the tumor and healthy tissues and more accurately align a patient to the treatment beams without the use of X-ray, CT or surrogate registration markers. If the clinician prefers, the software has the ability to map the patient’s soft tissue anatomy each treatment session in less than one minute, and clinicians can use that information to align the patient. |
| • | On-table adaptive planning. Due to changes in tumor shape or the patient’s internal anatomy, the clinician may be unable to obtain an optimal setup of the target location during image-guidance using CT-based systems while a patient is on the treatment table. Further, a nearby organ at risk for radiation damage may be exposed to higher radiation doses than anticipated. Using an MR image captured at the beginning of each therapy session, MRIdian software enables clinicians to map each patient’s soft tissue anatomy in 3D. The software also allows for the calculation of the dose that would be delivered to the radiation target and organs at risk using the current treatment plan. If the predicted radiation exposure is not clinically acceptable to the physician, the system provides software tools for tumor and organ at risk re-contouring based on the setup MR-images. The MRIdian system also provides the ability to recalculate the intended radiation dose and adapt the plan to the changed tumor shape or location, and overall anatomy for the respective day of treatment. Utilizing our proprietary algorithm and software, adaptive re-planning can be done with the patient still “on table”. Also, integral to the adaptive planning process are quality assurance protocols, or QA, to enhance the safety of adapted radiation dose delivery. Users in the United States are currently generally reimbursed for the additional time and effort spent by their physicians, medical physicists, dosimetrists and therapists for on-table adaptive planning. When medically necessary, we believe hospitals and physicians will continue to receive additional reimbursement when they perform adaptive radiation therapy. |
| • | Ability to track tumors and manage patient motion. MRIdian can capture dedicated or multiple soft-tissue imaging planes concurrently during the radiation treatment delivery, refreshing the image multiple times per second. This real-time imaging enables physicians to see, watch and track the movement of the tumor and its surrounding healthy tissue directly; they do not need to rely on proxies such as registration markers, existing bones or surgically implanted fiducials. If a tumor or critical organ moves beyond a physician’s defined boundary or allowable range of motion (as individually defined in our dedicated tracking and gating software), MRIdian will automatically detect this and pause the treatment beam. This automatic beam control becomes especially important in the situations where a tumor may be in close proximity to a critical organ, such as the heart during lung and breast cancer treatments, or the rectum during prostate cancer treatments. This ability to actually see the tumor location continuously throughout radiation delivery has enabled physicians to treat patients with greater confidence, including patients who would not have been given radiation therapy for their cancers previously. |
13
| • | Record and evaluate the delivered dose. Using our proprietary algorithm and advanced MR imaging, MRIdian calculates the dose delivered after each treatment, enabling the physician to review and re-optimize the patient’s treatment session, if needed. In addition, MRIdian can utilize diagnostic CT images that are fused with the MR images at each treatment in order to more accurately calculate dose. MRIdian also captures and records a video, known as a MRIdian Movie™, of the delivered treatments, which can be evaluated by the physician or shared with patients. |
| • | Fits into existing treatment paradigms and workflow. MRIdian can treat a broad spectrum of radiation therapy indications and disease sites, because it can perform 3D-CRT, IMRT, IGRT, SBRT and SRS. MRIdian treatments are supported by existing radiation therapy reimbursement codes. In addition, MRIdian fits inside most standard radiation therapy vaults without the need for significant construction costs such as wall or ceiling removal to enable it to be placed inside. In addition, we believe MRIdian’s increased tumor target accuracy will allow physicians to treat patients with higher radiation doses over fewer treatment fractions; this potentially enables the clinic to treat more patients each day and with greater overall efficiency, or patient throughput. |
We believe the ability to image with MRI and treat cancer patients with radiation simultaneously will lead to improved patient outcomes.
Our Strategy
Our objective is to make MRI-guided radiation delivery the standard of care for radiation therapy. To achieve this goal, we intend to do the following:
| • | Invest in Commercialization of the MRIdian Linac. The public response to the clinical release of the second generation MRIdian Linac has been positive as it builds upon and improves the capabilities of the first generation MRIdian. We believe the MRIdian Linac has the potential to broaden our addressable market, accelerate our sales cycle, reduce our backlog conversion time and improve our gross margins. We intend to: |
| • | Broaden awareness of MRIdian’s capabilities and clinical benefits to expand our share of the radiation therapy market. We intend to continue to educate radiation oncologists, medical physicists and radiation oncology administrators about the capabilities and resulting benefits of MRIdian over traditional radiation therapy systems. In order to drive awareness and adoption, we also intend to support the publication of clinical and scientific data and analysis, work with key opinion leaders, present at leading academic conferences and engage in outreach at leading hospitals worldwide. We also plan to leverage our existing customer network as a reference for new potential users to experience our technology in-use in the clinical setting. |
| • | Target top-tier hospitals in initial global sales efforts, followed by their community practice networks. We intend to market MRIdian to a broad range of customers worldwide, including university research and teaching hospitals, private practices, community hospitals, government institutions and freestanding cancer centers. We are focusing initially on the leading hospitals worldwide which are typically early adopters of best-in-class technology, such as MRIdian, and are able to influence and promote adoption by other centers both locally and globally. We plan to continue to work with these institutions to promote broader market awareness of the benefits of MRI-guided radiation therapy and then expand into the community practice networks that many of these leading centers have developed. |
| • | Commercialize MRIdian with a targeted sales force in the United States and through a sales force-assisted distribution network in international markets. We market MRIdian through a combination of direct sales and distributors. We are expanding our sales force for the United States and Canada and are developing a sales force to assist distributors in international markets. We intend to continue to expand our presence in key markets to capitalize on the growing international opportunity for MRIdian. |
14
| • | Perform Clinical Trials to Develop Evidence Supporting the Value of the MRIdian. We have launched the ViewRay Clinical Cooperative Think Tank (C2T2), a group of MRIdian clinical users and customers that are gathering evidence to support MR-guided radiation therapy. ViewRay's C2T2 comprises clinicians from leading institutions around the world who are focused on evidence gathering to support MR-guided radiation therapy. This group includes: |
| • | Dana-Farber/Brigham and Women's Cancer Center in Boston |
| • | Henry Ford Cancer Institute, Detroit |
| • | Institut du Cancer de Montpellier, France |
| • | Institut Paoli Calmettes, Marseille, France |
| • | Loyola Center for Cancer Care and Research at Palos Health South Campus in Illinois |
| • | Moffitt Cancer Center in Tampa, Florida |
| • | Miami Cancer Institute, Baptist Health South Florida |
| • | National Cancer Center (NCC) in Tokyo, Japan |
| • | NewYork-Presbyterian Hospital |
| • | Orlando Health UF Health Cancer Center |
| • | Policlinico Agostino Gemelli, Universita Cattolica del Sacro Cuore, Gemelli ART in Rome, Italy |
| • | Seoul National University Hospital (SNUH) in Seoul, South Korea |
| • | Sylvester Comprehensive Cancer Center, UHealth - University of Miami Health System |
| • | University of California, Los Angeles Health System and Jonsson Comprehensive Cancer Center |
| • | University of Heidelberg, Germany |
| • | University of Wisconsin Carbone Cancer Center in Madison |
| • | VU University Medical Center in Amsterdam, Netherlands |
| • | Washington University and Siteman Cancer Center at Barnes-Jewish Hospital, St. Louis |
At the inaugural meeting of the C2T2 on September 23, 2017, participants formalized the group's first key initiative – a multi-center, prospective, single-arm clinical trial focused on locally advanced unresectable pancreatic cancer. Pancreatic cancer presents considerable radiation targeting challenges given the known limitations of conventional CT image guidance. The novel abilities provided by live MRI guidance combined with daily online treatment adaptation have enabled a new approach in pancreatic cancer therapy. Through this trial the group looks to explore new opportunities to improve survival and quality of life for this deadly disease.
| • | Maintain our competitive lead in MRI-guided radiation therapy through continued innovation. We plan to continue to invest in our technology to maintain our leadership position in the emerging MRI-guided radiation therapy market. We intend to develop and introduce enhancements to the system and software to provide improved capabilities for MRIdian users and patients. In addition, we plan to explore potential benefits of integrating our MRI technology with alternative beam technologies. We believe we have a strong intellectual property portfolio that covers the MRIdian, as well as its critical design elements and key aspects of its subsystem and components. |
15
| • | Continue to work with leading hospitals to optimize efficiency and patient throughput. We strive to maximize the efficiency and effectiveness of the MRIdian system for our customers. We plan to continue to work closely with key opinion leaders, clinicians and hospitals in a proactive manner to determine how best to refine and improve MRIdian’s features, optimize clinical workflow and maximize patient throughput while incorporating our advanced features. |
| • | Drive cost reductions in the design and manufacture of MRIdian and improve our margins. We plan to continue to explore ways to bring down our cost of goods to improve margins for MRIdian. |
The MRIdian System
The MRIdian is comprised of four major components, (i) the MRI system, (ii) the radiation delivery system, (iii) integrated treatment planning and delivery software and (iv) a safety and control system.
| | |
MRIdian Cobalt-60 | | MRIdian Linac |
| |

| | 
|
MRI System
The MRI system is the component of MRIdian that captures soft tissue images of the patient’s body. To address the technical complications that arise from combining an MRI with an external-beam radiation delivery unit, we have designed a proprietary split superconducting magnet that will allow radiation doses to be delivered through a central gap, which eliminates MRI components from the path of the beam. Our MRI system captures and displays live, high-quality images in one plane, four times per second or in three planes, two times per second. These real-time images automatically track selected structures and control radiation treatment beam delivery.
We have engineered our MRI system to be able to produce clear images using a mid-field strength 0.35 Tesla magnet, which enables us to avoid image and radiation dose distortions that result when higher field strength magnets are used. In addition, MRIdian’s 0.35 Tesla field strength prevents over-heating of the patient during uninterrupted imaging, which could occur when a higher field strength magnet is used for fast imaging during radiation delivery. Over-heating can require interruption or termination of the imaging or of the overall treatment.
16
MRI System

MRIdian Radiation Delivery System
In the first-generation MRIdian, radiation is delivered from three Cobalt-60 radiation therapy heads symmetrically mounted on a rotating ring gantry, providing full 360-degree coverage and simultaneous dose delivery. Each head is equipped with a double-focused multi-leaf collimator designed to overcome the wide-beam edge of previous-generation Cobalt-60 systems and to shape the beam for precision radiation therapy treatments. It allows the delivery of treatment plans for 3D-CRT, IMRT and SBRT that are clinically equivalent to those produced on the most advanced linear accelerators available today. Stereotactic procedures are possible with a positioning accuracy of less than one millimeter. Cobalt-60 was used in the first-generation devices because it does not create any radio frequency, which interferes with the MRI.
MRIdian Linear Accelerator Technology Radiation Delivery System
In the second generation MRIdian Linac, we developed solutions to two long-standing problems that had prevented compact integration of a linac beam with an MRI system: 1) linac radiofrequency interference with the operation of the MRI; and 2) MRI magnetic interference with the operation of the linac. First, linacs utilize high-powered microwave generators similar to equipment used in radar at airports. These “radar stations” inside the linac create radiofrequency emissions, or “noise” that can corrupt the delicate signals measured from the patient’s body to generate MR images. ViewRay solved this problem by introducing technology similar to that used in stealth aircraft. Airplanes built with stealth technology can hide from radar by using a coating that absorbs microwaves, thus preventing radar beams that strike the aircraft from bouncing back to the radar station. In a similar manner, we absorb the output of the linac “radar station” to hide it from the MRI, producing images as noise-free as those created without an integrated linac.
Second, MRIs utilize high-powered superconducting magnets required to image the patient’s tissues that must be placed close to the linac components used for radiation therapy. But many linac components will not operate properly when placed close to or inside these strong magnetic fields. ViewRay overcame this challenge by creating magnetic shielding shells that create voids in the magnetic field, without significantly disturbing the magnetic field used for imaging. This allows the linac to operate on the MRIdian gantry as if there were no magnetic field present. MRIdian Linac uses the same split-magnet MRI system used in the first generation MRIdian system. It is specifically designed to fit in standard radiotherapy vaults so that customers do not need to build new vaults in order to replace an X-ray guided linear accelerator with a MRIdian. Existing MRIdian systems currently in use can be upgraded to the MRIdian Linac in the field.
Both MRIdian and MRIdian Linac can provide continuous MR based soft-tissue imaging during radiation beam delivery. Being able to constantly see both the tumor and surrounding organs means physicians can accurately align the tumor to the treatment beams, adapt or reshape the treatment volume to accommodate changes in the shape and
17
location of the tumor and healthy tissues, and track soft tissues in real time to avoid missing a moving tumor or irradiating sensitive internal structures.
Integrated Treatment Planning and Delivery Software
Our proprietary treatment planning and delivery software can create treatment plans and manage the treatment delivery process. It is designed to create optimized 3D-CRT, IMRT, IGRT, SBRT and SRS plans for delivery by MRIdian. Using this software, the on-table adaptive planning process typically takes fifteen additional minutes on average, depending on the treatment plan and includes: auto-contouring, dose prediction and adaptive treatment plan optimization. For contouring, the software will automatically contour the outline of the tumor and nearby organs by matching the MR images with the images used in the original treatment plan. The physician will then make refinements as necessary. Dose prediction can be calculated immediately before treatment, allowing the current state of the patient’s anatomy to be taken into account. If dose parameters for the radiation target or organs at risk no longer meet goals or safety criteria, the software can then generate an optimized adaptive treatment plan, while the patient is on the treatment table. Following physician review and approval, as well as medical physics quality assurance assessment, the adapted plan can be delivered to provide a more accurate treatment.
Independent of the ability to create an adapted treatment plan, the MRIdian system has the ability to use a soft-tissue tracking beam to control or “gate” the radiation beam, by turning it on and off. While the radiation dose is being delivered, our software analyzes images of the patient’s tumor and surrounding anatomy; it can use them to determine tumor or organ location relative to tolerances set by the physician. If the targeted tumor or a critical organ moves beyond a physician-defined boundary, the treatment beams will automatically pause. When the tumor moves back into the target zone, the treatment will automatically resume. Physicians can set both spatial and time thresholds for pausing radiation beam delivery. This enables the system to account for tumor and patient motion during treatment.
The software archives all the information generated during treatment and builds a database of patient-specific planning, delivery and imaging data. It also includes a review tool which provides clinicians with a visual comparison of the delivered treatment versus the treatment as originally planned. At the end of each treatment, the software determines the delivered dose by combining the recorded actions of the radiation delivery system with the daily image and auto-contouring of the patient. With this information, clinicians can fine-tune prescriptions based on the actual dose delivered, rather than estimates. In addition, it provides a MRIdian Movie™ of each delivered treatment, which can be evaluated by the physician or exported and then shared with the patients or their families.
Safety and Control System for MRIdian with Cobalt-60
In addition to complying with the applicable FDA and Nuclear Regulatory Commission, or NRC, requirements, the Cobalt-60 radiation delivery subsystem also meets a double fault tolerant design standard and has redundant safety systems. If any two components in the Cobalt-60 radiation delivery subsystem fail simultaneously, such as power and pneumatics, the system reverts to a safe state. MRIdian also contains redundant computer control for safety and system logging and double encoders on all axes of motion for safety. The control system continuously monitors performance to ensure systems are performing and communicating appropriately.
Installed Base and Clinical Use
At December 31, 2017, we had installed six units at five leading cancer centers in the United States and installed five units outside the United States. One MRIdian with Cobalt-60 has been delivered and is expected to be installed in early 2018 at Edogawa Hospital in Japan. Three MRIdian Linacs have been delivered and are expected to be installed in 2018 at hospitals in Israel, Korea and China.
In January 2014, Washington University in St. Louis, a National Cancer Institute Designated Comprehensive Cancer Center, became the first center to treat patients with MRIdian with Cobalt-60. Washington University in St. Louis has since scaled up its use of MRIdian in its clinical practice. In September 2014, Washington University in St. Louis used MRIdian to perform the first on-table adaptive treatments as part of an ongoing clinical service. Also, in September 2014, the University of Wisconsin–Madison treated its first patients with MRIdian with Cobalt-60 and became the first center to employ the soft-tissue tracking and beam gating control capability unique to MRIdian. In
18
July 2017, Henry Ford Health System in Detroit treated the first cancer patients using the second generation MRIdian Linac. We are working with each of these centers to determine how best to refine and improve MRIdian’s features, optimize workflow and maximize patient throughput.
As of December 31, 2017, over 2,000 patients with over 3000 on-table adapted fractions have been treated by MRIdian systems. These included cancers of the prostate, breast, lung, colorectal and bladder, which are among the most prevalent types of cancer in the United States, according to the Centers for Disease Control and Prevention, or CDC. MRIdian has also been used to treat liver, stomach, esophagus and pancreatic cancer.
New Orders and Backlog
New orders are defined as the sum of gross product orders, representing MRIdian contract price, recorded during the period. Backlog is the accumulation of all orders for which revenue has not been recognized and which we consider valid. Backlog includes customer deposits or letters of credit, except when the sale is to a customer where a deposit is not deemed necessary or customary. Deposits received are recorded as a liability on the balance sheet. Orders may be revised or cancelled according to their terms or upon mutual agreement between the parties. Therefore, it is difficult to predict with certainty the amount of backlog that will ultimately result in revenue. The determination of backlog includes objective and subjective judgment about the likelihood of an order contract becoming revenue. We perform a quarterly review of backlog to verify that outstanding orders in backlog remain valid, and based upon this review, orders that are no longer expected to result in revenue are removed from backlog. Among other criteria we use to determine whether a transaction to be in backlog, we must possess both an outstanding and effective written agreement for the delivery of a MRIdian signed by a customer with a minimum customer deposit or a letter of credit requirement, except when the sale is to a customer where a deposit is not deemed necessary or customary (i.e. sale to a government entity, a large hospital, group of hospitals or cancer care group that has sufficient credit, sales via tender awards, or indirect channel sales that have signed contracts with end-customers). We decide whether to remove an order from our backlog by evaluating the following criteria: changes in customer or distributor plans or financial conditions; the customer’s or distributor’s continued intent and ability to fulfill the order contract; changes to regulatory requirements; the status of regulatory approval required in the customer’s jurisdiction, if any; and other reasons for potential cancellation of order contracts.
We received new orders for MRIdian systems, totaling $113.6 million, $77.0 million and $40.1 million for fiscal years 2017, 2016 and 2015, respectively. We have two cancellations for fiscal year 2017. At December 31, 2017, we had a backlog with a total value of $203.6 million. There can be no assurance that backlog will result in revenue in any particular time period or at all.
Installation Process
Following execution of a contract, it generally takes nine to 12 months for a customer to prepare an existing facility or construct a new vault, although in some cases customers may request installation for a date later in the future to meet their own clinical or business requirements. After the customer completes its vault customization, it typically takes approximately ninety days to complete the installation and on-site testing of the system, including the completion of acceptance test procedures. MRIdian is designed to fit into a typical radiation therapy vault, similar to other replacement linear accelerators. MRIdian’s components all fit through standard hospital vault entrances for assembly. On-site training takes approximately one week and can be conducted concurrent with installation and acceptance testing.
Our customers are responsible for removing any outgoing linear accelerator and preparing the mounting pad, power and support system connections. Additional room modifications required are consistent with those generally required for MRI systems, such as radio frequency shielding of the room and additional power.
Clinical Development
To date, we have primarily relied on clinical symposia and case studies presented at ASTRO and the European Society for Radiotherapy and Oncology, or ESTRO, to raise awareness of MRI-guided radiation therapy and to market MRIdian to leading cancer centers. In order to promote broader adoption rates at other cancer centers and
19
hospitals, we plan to work with our customers to collect and publish data on clinical efficacy, treatment times and clinical results for patients who have been treated on a MRIdian. Outcomes data presented at the 2017 Annual Meeting of ASTRO highlighted compelling early results using the Company's MRIdian system for the treatment of inoperable, locally advanced pancreatic cancer. These early clinical data suggested nearly 2X prolonged median survival with reduced toxicity for inoperable, locally advanced pancreatic cancer. These results will be tested in a multi-center, prospective, single-arm clinical trial for inoperable, locally advanced or borderline resectable pancreatic cancer. The trial will be conducted by ViewRay's Clinical Cooperative Think Tank (C2T2), a group of MRIdian medical institutions focused on evidence gathering to support MRI-guided radiation therapy. Additionally, Washington University has published a prospective study on Magnetic Resonance Image Guided Radiation Therapy for External Beam Accelerated Partial-Breast Irradiation using a one-week course of treatment. This study demonstrated that on-board MR image-guidance allowed for a greater than 50% reduction of margins while maintaining the same dose to the tumor with patients reporting 100% Excellent/Good Cosmesis.
While we do not currently have statistically significant, prospective evidence that MRIdian improves patient outcomes or decreases healthcare costs relative to CT-based radiotherapy, we believe supporting studies will demonstrate the benefits of MRI-guided radiation therapy and adaptive treatment planning. As data accumulate from the use of MRIdian, we plan to work with professional healthcare organizations to support further global marketing efforts, additional product clearances, approvals and/or registrations and potential improvements in reimbursement.
Selling and Marketing
We currently market MRIdian through a direct sales force in the United States and Canada and are developing a sales force to assist distributors in the rest of the world. We market MRIdian to a broad range of worldwide customers, including university research and teaching hospitals, community hospitals, private practices, government institutions and freestanding cancer centers. As with the traditional linac market, our sales and revenue cycle varies based on the particular customer and can be lengthy, sometimes lasting up to 18 to 24 months (or more) from initial customer contact to order contract execution.
To sell MRIdian globally, we use a combination of sales executives, sales directors and a network of international third-party distributors with internal support from sales operations, product management and application specialists. A targeted group of eight senior sales directors are responsible for selling MRIdian within the United States and Canada. Our product management function helps market MRIdian and works with our engineering group to identify and develop upgrades and enhancements. We also have a team of application specialists who provide post-sales support.
We engage in various physician-targeted advertising efforts, and our selling and marketing practices include participating in trade shows and symposia.
Competition
We compete directly with companies marketing IGRT devices for the treatment of cancer using CT, ultrasound, optical tracking and X-ray imaging. We also compete with companies developing next-generation IGRT devices, specifically those developing MRI-guided devices, amongst others. We expect the following to drive worldwide competitive market dynamics: technological advances, including the ability to provide real-time imaging; clinical outcomes; system size, price, and operational complexity; and operational efficiency.
Our major competitors with devices approved for distribution in the United States or globally include Varian Medical Systems, Inc., or Varian, Elekta AB, or Elekta, and Accuray Incorporated, or Accuray. Many of our direct competitors have greater financial, sales and marketing, service infrastructure and research and development capabilities than we do, as well as more established reputations and current market share. The main limitations of currently approved devices are the lack of real-time, clear images before and during the treatment, as well as the ability to perform on-table adaptive planning.
We are also aware of one commercial and two academic ongoing research efforts to develop radiation therapy systems incorporating MRI. Elekta and Royal Philips have formed a consortium to develop a commercial Elekta-Philips MRI-linac. The University of Sydney, Ingham Institute and the University of Queensland have formed a
20
partnership to develop an MRI-linac and the University of Alberta’s Cross Cancer Institute is working on a MRI-linac as well. Although these academic research centers may not compete directly with us commercially, if they were to form a partnership or other relationship with one of our competitors, it could impact our sales negatively. Of these three, we believe the Elekta-Philips MRI-linac is the most advanced in development, although we believe this combined system may not be commercially available for some time because it has not been cleared or approved by regulatory authorities for patient treatments anywhere in the world. MRIdian is the first and only commercially available MRI-guided radiation therapy device to image and treat cancer patients simultaneously.
The limited capital expenditure budgets of our customers result in all suppliers to these entities competing for a limited pool of funds. Our customers may be required to select between two items of capital equipment. For example, some of our potential customers are considering expensive proton therapy systems, which could consume a significant portion of their capital expenditure budgets.
Manufacturing
We have adopted a model in which we rely on subsystem manufacturing, assembly and testing by our key suppliers. The MRIdian subsystems are then fully integrated at the customer site. Through this approach, we avoid the majority of the fixed cost structure of manufacturing facilities. We purchase major components and subsystems for MRIdian from national and international third-party original equipment manufacturers, or OEM, suppliers and contract manufacturers. These major components include the magnet, MRI electronics, ring gantry, radiation therapy heads, Cobalt-60 sources, linear accelerator, multi-leaf collimators, patient-treatment table and computers. We also purchase minor components and parts directly ourselves. For sales for which we are responsible for installation, we assemble and integrate these components with our proprietary software and perform multiple levels of testing and qualification at the customer site. The system undergoes a final acceptance test, which is performed in conjunction with the customer.
Many of the major subsystems and components of MRIdian are currently procured through single and sole source suppliers. Among these are the magnet, MRI electronics, MRI coils, ring gantry, Cobalt-60 sources, linear accelerator and the patient-treatment table. We have entered into multi-year supply agreements for most of our major components and subsystems. Except for the MRI power, control and image reconstruction subsystem, we own the design of all other major subsystems and components.
We manage our supplier relationships with scheduled business reviews and periodic program updates. We closely monitor supplier quality and delivery performance to ensure compliance with all MRIdian system specifications. We believe our supply chain has adequate capacity to meet our projected sales over the next several years.
Intellectual Property
The proprietary nature of, and protection for, MRIdian components, new technologies, processes and know-how are important to our business. Our policy is to seek patent protection in the United States and in certain foreign jurisdictions for our MRIdian systems and other technology where available and when appropriate. We also in-license technology, inventions and improvements we consider important to the development of our business.
We hold a license to four issued U.S. patents, 19 issued foreign patents (eight of which were issued in Great Britain, Germany, France and the Netherlands as a result of two patent applications filed and allowed through the European Patent Office), one pending U.S. application and five pending foreign applications as of January 15, 2018. We own an additional 20 issued U.S. patents, 36 issued foreign patents (13 of which were issued in Great Britain, Germany, France, Italy and the Netherlands as a result of three patent applications filed and allowed through the European Patent Office), 24 pending U.S. applications and 85 pending foreign applications as of January 15, 2018. Assuming all required fees are paid, individual patents or patent applications owned or licensed by us will expire between 2021 and 2037. We also have a joint ownership interest with Case Western Reserve University in one issued patent and one U.S. application.
Our portfolio includes patents and patent applications directed to system-wide aspects of MRIdian and to key aspects of its subsystems and components. The initial licensed patents for our core technology broadly cover the
21
simultaneous use of MR imaging and isotopic external-beam radiation therapy. These patents have been granted in the United States, Europe, Hong Kong, Australia, China and Japan, and additional related patent applications remain pending in Canada, the United States, Australia and Japan. We have issued U.S. and foreign patents and pending continuation applications of the licensed patents that extend this core technology to alternate beam technologies. Additionally, we have patents and patent applications that cover critical design elements including, among others, our approach to Cobalt IMRT, our methods for integrating MRI with the radiation delivery system, and the design of our disassemblable, or “pop apart,” magnet which enables the MRI sub-system to fit into most standard radiation therapy vaults. The U.S. patent application on our approach to Cobalt IMRT has been issued, the patent application on our split gradient coil has been issued in the United States, Japan, Australia and China and numerous applications on other design elements are pending in the United States and foreign jurisdictions. In addition, we have U.S., Chinese, European and Australian patents and U.S. and foreign patent applications that cover the use of MR- imaging at a frequency sufficient to account for real-time organ motion to provide video-rate tissue tracking in disciplines outside of radiation therapy. Many of the patents and applications in our portfolio covering aspects of the MRIdian with Cobalt-60 system also cover the MRIdian Linac. In addition, we have patents issued in the U.S., Europe, Australia, Japan and China, and additional applications pending in the U.S. and foreign jurisdictions, specifically directed to technology enabling the MRIdian Linac combination of MRI and linear accelerator technology.
We continue to review new technological developments in our system and in the field as a whole, in order to make decisions about what filings would be most appropriate for us. An additional key component of our intellectual property is our proprietary software used in planning and delivering MRIdian’s therapeutic radiation dose.
In December 2004, we entered into a licensing agreement with the University of Florida Research Foundation, Inc., or UFRF, whereby UFRF granted us a worldwide exclusive license to certain of UFRF’s patents in exchange for 33,653 shares of common stock and a royalty from sales of products developed and sold by us utilizing the licensed patents. We were obligated to meet certain product development and commercialization milestones by various dates through December 31, 2014. The significant milestones met prior to December 31, 2013 included: (i) completion of a business plan and Small Business Technology Transfer grant application; (ii) securing a minimum of $20.0 million venture financing; (iii) successful relocation and build out of our headquarters; (iv) receipt of the first magnet from an OEM partner; (v) hiring of a chief executive officer with industry experience in developing and commercializing similar products; and (vi) filing for FDA approval. The final milestone, which required us to recognize the first commercial sale of the MRIdian system to retail customers by December 31, 2014, was met during the year ended December 31, 2013. If these milestones had not been accomplished, UFRF would have had the right to terminate the licensing agreement. Royalty payments are based on 1% of net sales, defined as the amount collected on sales of licensed products and/or licensed processes after deducting trade and/or quantity discounts, credits on returns and allowances, outbound transportation costs paid and sales tax. Minimum quarterly royalty payments of $50,000 commenced with the quarter ended March 31, 2014 and are payable in advance. Minimum royalties paid in any calendar year will be credited against earned royalties for that calendar year. The royalty payments continue until the earlier of (i) the date that no licensed patents remain enforceable; or (ii) the payment of earned royalties, once begun in 2014, cease for more than four consecutive calendar quarters.
In addition to our patents, we also rely upon trade secrets, know-how, trademarks, copyright protection and continuing technological and licensing opportunities to develop and maintain our competitive position. We have periodically monitored and continue to monitor the activities of our competitors and other third parties with respect to their use of intellectual property. We require our employees, consultants and outside scientific collaborators to execute confidentiality and invention assignment agreements upon commencing employment or consulting relationships with us. Despite these safeguards, any of our know-how or trade secrets not protected by a patent could be disclosed to, or independently developed by, a competitor.
Coverage and Reimbursement
We believe that reimbursement rates in the United States have generally supported a favorable return on investment for the purchase of new radiotherapy equipment, including MRIdian. Payments for standard radiation therapy treatments using MRIdian, including 3D-CRT, IMRT and SBRT, are generally covered and reimbursed under existing Current Procedural Terminology, or CPT, codes and coverage policies currently in place. User experience
22
to date indicates that our initial customers have treated a wide spectrum of different patients and treatment modalities using MRIdian. Physicians use the MRIdian system’s on-board MRI for distinct procedures which can be billed by physicians using existing CPT codes, including: complex simulation weekly IMRT or daily for SBRT; special physics consult; and adaptive re-planning. Each of these are distinct procedures which can be billed by physicians using existing CPT codes, so long as these procedures are reimbursed, so long as they meet medical necessity and other documentation and coverage criteria established by government or other third-party payors.
Third-party payors, including governmental healthcare programs such as Medicare and Medicaid, establish coverage policies and reimbursement rates for diagnostic examinations and therapeutic procedures performed by physicians in hospitals and free-standing clinics. Private insurers often model their payment rates and coverage policies based on those established by the government. The U.S. Congress from time to time considers various Medicare and other healthcare reform proposals that could affect both private and public third-party payor coverage and reimbursement for healthcare services provided in hospitals and clinics. In addition, third-party payors regularly update reimbursement amounts, including annual updates to payments to physicians, hospitals and clinics for medical procedures, including radiation treatments using MRIdian.
By way of example, the Medicare Access and CHIP Reauthorization Act of 2015, or MACRA, ended the use of the statutory formula, and provided for a 0.5% annual increase in payment rates under the Medicare Physician Fee Schedule through 2019, but with no annual update from 2020 through 2025. MACRA also introduced a quality payment program under which individual providers with Medicare billings of $30,000 or 100 patient visits per year will be subject to certain incentives or penalties based on new program quality standards. The quality payment program has two tracks, one known as the “merit-based incentive payment system” for providers in the fee-for service Medicare program, and the advanced alternative payment model for providers in specific care models, such as accountable care organizations. Payment adjustments for the Medicare quality payment program will begin in 2019.
The Centers for Medicare & Medicaid Services, or CMS, also publishes annual updates to the hospital outpatient prospective payment system, or HOPPS. These payments are bundled amounts received by our hospital customers for hospital outpatient services, including conventional radiation therapy and IMRT, which may result in lower reimbursement to our customers for procedures performed using MRIdian.
We plan to work with our customers to collect and publish data on clinical results for patients who have undergone procedures on MRIdian. We will support a multi-center, prospective, single-arm clinical trial for inoperable, locally advanced or borderline resectable pancreatic cancer. We plan to continue to support this and further studies to demonstrate the benefits of MRI-guided radiation therapy and adaptive treatment planning. As data accumulate from the use of our system, we plan to work with professional healthcare organizations to further support global marketing efforts, additional product clearances, approvals and/or registrations and potential improvements in reimbursement. Additionally, we currently provide reimbursement support to our customers through a third-party vendor.
Foreign Reimbursement Regulations
Internationally, reimbursement and healthcare payment systems vary from country to country and include single-payor, and government managed systems as well as systems in which private payors and government-managed systems exist side-by-side. In general, the process of obtaining coverage approvals is slower outside of the United States. Our ability to achieve adoption of MRIdian, as well as significant sales volume in international markets we enter will depend in part on the availability of reimbursement for procedures performed using MRIdian.
Research and Development
Continued innovation and development of advanced technologies is critical to our goal of making MRI-guided radiation therapy the standard of care for cancer treatment. Our current development activities include improvements in and expansion of product capabilities, continued clinical workflow refinements, design improvements to reduce system costs and improvements in reliability.
23
The modular design of MRIdian enables the development of new capabilities and performance enhancements by generally allowing each subsystem to evolve within the overall platform design. Access to regular MRIdian upgrades protects customer investment in MRIdian and facilitates the adoption of new features and capabilities among existing installed base customers.
In March 2016, we announced the development of a linear accelerator version of our MRIdian technology. This technology has been used to treat patients at Henry Ford Hospital in Detroit since July 2017 and is being readied for treatment in several other sites. Significant technology incorporated into the MRIdian Linac includes radio frequency “cloaking” technology to preserve the integrity of the MR image quality in the presence of radio frequencies emitted by the linear accelerator, magnetic shielding technology to enable undistorted delivery of radiation, double- focused multi-leaf collimator technology for the delivery of sharp radiation beams and incorporation of the existing MRIdian proprietary “pop-apart” magnet design.
We have designed the linac technology such that our MRIdian with Cobalt-60 systems are able to be modified to incorporate the linac technology. In September 2016, we received CE mark approval in the European Union, and in February 2017, we received 510(k) clearance from the FDA to market the MRIdian Linac system in the United States. MRIdian Linac is now commercially available in the key markets of the EU and the United States.
We believe the MRIdian Linac will broaden our addressable market, accelerate our sales cycle, reduce our backlog conversion time and improve our gross margins.
In addition, we believe our existing and expanding IP portfolio will enable us to continuously develop innovative technologies to further strengthen the differentiation of MRIdian in the marketplace. Magnetic resonance imaging is a powerful and versatile measurement technique and is widely used throughout radiology and medicine because of its ability to generate information about tissues and disease states.
At December 31, 2017, we had a total of 41 employees in our research and development departments. Research and development expenses were $14.7 million, $11.4 million and $10.4 million during the years ended December 31, 2017, 2016 and 2015, respectively. We plan to continue to increase our investment in research and development in future periods.
Government Regulation
U.S. Medical Device Regulation and Nuclear Materials Regulation
As a manufacturer and seller of medical devices and devices that deliver radiation, we and some of our suppliers and distributors are subject to extensive and rigorous regulation by the FDA, the NRC, other federal, state and local authorities in the United States and foreign regulatory authorities. Regulations promulgated by the FDA relating to medical devices and radiation-producing devices govern, among other things, the following activities that we perform or that are performed on our behalf, and that we will continue to perform or have performed on our behalf:
| • | product design, development and testing; |
| • | packaging, labeling and storage; |
| • | marketing, advertising and promotion, sales; |
| • | distribution, including importing and exporting; |
| • | possession and disposal; |
| • | service and surveillance, including post-approval monitoring and reporting; |
24
| • | repair or recall of products and issuance of field safety corrective actions. |
FDA Clearance and Approval of Medical Devices
The FDA regulates the research, testing, manufacturing, safety, labeling, storage, recordkeeping, promotion, distribution, and production of medical devices in the United States to ensure that medical products distributed domestically are safe and effective for their intended uses. Unless an exemption applies, the FDA requires that all new medical devices and all marketed medical devices that have been significantly changed, or that will be marketed with a new indication for use, obtain either clearance via a 510(k) pre-market notification or approval via a Premarket Approval, or PMA, application before the manufacturer may commercially distribute the product in the United States. The type of marketing authorization necessary is generally linked to the classification of the device. The FDA classifies medical devices into one of three classes. Devices deemed to pose the lowest risk are placed in Class I, and most Class I devices are exempt from premarket notification requirements. Class I devices are those for which safety and effectiveness can be reasonably assured by adherence to a set of regulations referred to as General Controls, which require compliance with the applicable portions of the FDA’s Quality System Regulation, or QSR, and regulations regarding facility registration and product listing, reporting of adverse events and malfunctions, and appropriate, truthful and non-misleading labeling and promotional materials. However, some Class I devices, called Class I reserved devices, also require premarket clearance by the FDA through the 510(k) premarket notification process described below.
Moderate risk devices are placed in Class II and are subject to General Controls as well as Special Controls, which can include performance standards, guidelines and post-market surveillance. Most Class II devices are subject to premarket review and clearance by the FDA pursuant to Section 510(k) of the Federal Food, Drug, and Cosmetic Act, or FDCA.
Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) devices are placed in Class III. Class III devices require FDA approval of a PMA prior to marketing.
Both generations of the MRIdian System have been classified as Class II medical devices subject to the 510(k) clearance process.
510(k) clearance process. Most Class II devices are subject to premarket review and clearance by the FDA. Premarket review and clearance by the FDA for Class II devices is accomplished through the 510(k) premarket notification process. Under the 510(k) process, the manufacturer must submit to the FDA a premarket notification, demonstrating that the device is “substantially equivalent” to either
| • | a device that was legally marketed prior to May 28, 1976, the date upon which the Medical Device Amendments of 1976 were enacted; or |
| • | another commercially available, similar device that was cleared through the 510(k) process. |
To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device and either have the same technological characteristics as the predicate device or have different technological characteristics and not raise different questions of safety or effectiveness than the predicate device. Clinical data are sometimes required to support substantial equivalence.
The process of obtaining 510(k) clearance usually takes from three to 12 months from the date the application is filed and generally requires submitting supporting design and test data, which can be extensive and can prolong the process for a considerable period of time. If the FDA agrees that the device is substantially equivalent, it will grant clearance to commercially market the device. We received 510(k) clearances for the treatment planning and delivery software system in January 2011 and for MRIdian in May 2012.
After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a new or major change in the intended use of the device, may require a new 510(k)
25
clearance or, depending on the modification, could require approval of a PMA. The FDA requires each manufacturer to make this determination in the first instance, but the FDA can review any such decision. If the FDA disagrees with the manufacturer’s decision, it may retroactively require the manufacturer to submit a request for 510(k) clearance or PMA approval and can require the manufacturer to cease marketing and/or recall the product until 510(k) clearance or PMA approval is obtained. Since obtaining 510(k) clearances in 2011 and 2012, we have made changes to MRIdian that we believe do not require new 510(k) clearance.
In the fall of 2016, we submitted an application for 510(k) clearance by the FDA for MRIdian Linac, and in February 2017, we received 510(k) clearance from the FDA to market the MRIdian Linac system. An additional 510(k) clearance application for a modification of the MRIdian Linac system was submitted in March 2017, and we received 510(k) clearance from the FDA for that modification in June 2017.
Premarket application approval process. Submission and approval of a PMA is required before marketing of a Class III product may proceed. Under the PMA application process, the applicant must generally conduct at least one clinical investigation and submit extensive data and clinical information demonstrating reasonable assurance of the safety and effectiveness of the device for its intended use to the FDA’s satisfaction. Accordingly, a PMA application typically includes, but is not limited to, extensive technical information regarding device design and development, pre-clinical and clinical trial data, manufacturing information, labeling and financial disclosure information for the clinical investigators in device studies. The PMA process is much more demanding than the 510(k) premarket notification process.
Following receipt of a PMA application, the FDA conducts an administrative review to determine whether the application is sufficiently complete to permit a substantive review. If it is not, the agency will refuse to file the PMA. If it is, the FDA will accept the application for filing and begin the review. The FDA, by statute and by regulation, has 180 days to review a filed PMA application, although the review of an application more often occurs over a significantly longer period of time. During this review period, the FDA may request additional information or clarification of information already provided, and the FDA may issue a major deficiency letter to the applicant, requesting the applicant’s response to deficiencies communicated by the FDA. Before approving or denying a PMA, an FDA advisory committee may review the PMA at a public meeting and provide the FDA with the committee’s recommendation on whether the FDA should approve the submission, approve it with specific conditions, or not approve it. Overall, the PMA application process typically takes between one to three years, but may take significantly longer. The FDA may approve a PMA application with post-approval conditions intended to ensure the safety and effectiveness of the device including, among other things, restrictions on labeling, user training requirements, restrictions on promotion, sale and distribution, and requirements for the collection of long-term follow-up data.
None of our products have been subject to the PMA approval process, and we have no plans for any indication, system improvements or extensions that we believe would require a PMA.
Clinical trials. Clinical trials are generally required to support a PMA application and are sometimes required for 510(k) clearance. Such trials require submission of an investigational device exemption, or IDE, application to the FDA for a specified number of patients and study sites (unless the product is deemed a non-significant risk device eligible for more abbreviated IDE requirements). If an IDE is required, the FDA and the appropriate institutional review boards, or IRBs, at the clinical sites must approve the study before clinical trials may begin. If the device is considered a non-significant risk device, IDE submission to FDA is not required. Instead, only approval from the IRB overseeing the clinical trial is required. Clinical trials are subject to extensive monitoring, record keeping and reporting requirements. Clinical trials must be conducted under the oversight of an IRB for the relevant clinical trial sites and must comply with FDA regulations, including but not limited to those relating to good clinical practices. To conduct a clinical trial, the patient’s informed consent must be obtained in form and substance that complies with both FDA requirements and state and federal privacy and human subject protection regulations.
The clinical trial sponsor, the FDA or the IRB could suspend or terminate a clinical trial at any time for various reasons, including a belief that the subjects are being exposed to an unacceptable health risk. Even if a trial is completed, the results of clinical testing may not adequately demonstrate the safety and effectiveness of the device or may otherwise not be sufficient to obtain FDA clearance or approval to market the product.
26
Continuing FDA regulation. Any devices we manufacture or distribute pursuant to 510(k) clearance or PMA approval by the FDA are subject to pervasive and continuing regulation by the FDA and certain state agencies. These include product listing and establishment registration requirements, which help facilitate FDA inspections and other regulatory actions.
In addition, our manufacturing operations for medical devices and those of our suppliers must comply with the FDA’s Quality System Regulations, or QSR. The QSR requires that each manufacturer, including third party manufacturers, establish and implement a quality system by which the manufacturer monitors the manufacturing process and maintains records that show compliance with FDA regulations and the manufacturer’s written specifications and procedures. Among other things, the QSR requires that manufacturers establish performance requirements before production and follow stringent requirements applicable to the device design, testing, production, control, record keeping, documentation, labeling and installation, as well as supplier/contractor selection, complaint handling and other quality assurance procedures during all aspects of the manufacturing process. Compliance with the QSR is necessary to be able to continue to market medical devices that have received FDA approval or clearance, and to receive FDA clearance or approval to market new or significantly modified medical devices. The FDA makes announced and unannounced inspections of medical device manufacturers, and these inspections may include the manufacturing facilities of subcontractors. Following an inspection, the FDA may issue reports, known as FDA Form 483 reports, listing the investigator’s observations of conditions or practices which indicate the possibility that an FDA-regulated product may be in violation of FDA’s requirements. FDA may also issue warning letters documenting regulatory violations observed during an inspection. The manufacturer’s failure to adequately respond to such reports or warning letters may result in FDA enforcement action against the manufacturer and related consequences, including, among other things, fines, injunctions, civil penalties, recalls or seizures of products, total or partial suspension of production, FDA refusal to grant 510(k) clearance or PMA approval to new devices, withdrawal of existing clearances or approvals, and criminal prosecution.
Manufacturers must also comply with post-market surveillance regulations, including medical device reporting regulations, which require that manufacturers review and report to the FDA any incident in which their device may have caused or contributed to a death or serious injury, or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur. In addition, corrections and removal reporting regulations require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the Federal Food, Drug, and Cosmetic Act that may present a risk to health. The FDA may also order a mandatory recall if there is a reasonable probability that the device would cause serious adverse health consequences or death.
The FDA and the Federal Trade Commission, or FTC, also regulate the promotion and advertising of MRIdian. In general, we may not promote or advertise MRIdian for uses not within the scope of our clearances or approvals or make unsupported safety and effectiveness claims.
Failure to comply with applicable FDA requirements, including delays in or failures to report incidents to the FDA or off-label promotion, can result in enforcement action by the FDA, which can include any of the following sanctions:
| • | warning letters, untitled letters, fines, injunctions, consent decrees and civil penalties; |
| • | customer notifications or repair, replacement, refunds, recall, administrative detention or seizure of our MRIdian systems; |
| • | operating restrictions or partial suspension or total shutdown of production; |
| • | refusing or delaying requests for 510(k) clearance or PMA approval of new or modified products; |
| • | withdrawing 510(k) clearances or PMA approvals that have already been granted; |
| • | refusal to grant export approval for products; or |
Radiological health. We are also regulated by the FDA under the Electronic Product Radiation Control provisions of the FDCA because MRIdian contains radiation producing components, and because we assemble these
27
components during manufacturing and service activities. The Electronic Product Radiation Control provisions require radiation producing products to comply with certain regulations and applicable performance standards. Manufacturers are required to certify in product labeling and reports to the FDA that their products comply with all necessary standards as well as maintain manufacturing, testing and sales records for their products. The Electronic Product Radiation Control provisions also require manufacturers to report product defects and affix appropriate labeling to covered products. Failure to comply with these requirements could result in enforcement action by the FDA, which can include any of the sanctions described above.
Nuclear Regulatory Commission and U.S. State Agencies
In the United States, as a manufacturer of medical devices and devices utilizing radioactive byproduct material (i.e. depleted uranium shielding and Cobalt-60 sources), we are subject to extensive regulation by not only federal governmental authorities, such as the NRC, but also by state and local governmental authorities, such as the Ohio Department of Health, to ensure such devices are safe and effective. In Ohio, the Department of Health, by agreement with the NRC, regulates the possession, use, and disposal of radioactive byproduct material as well as the manufacture of devices containing radioactive sealed sources to ensure compliance with state and federal laws and regulations. We have received sealed source device approval from the Ohio Department of Health for MRIdian and have entered into a standby letter of credit with PNC for $103,000 to provide certification of financial assurance for decommissioning Cobalt-60 radioactive materials in accordance with Ohio Department of Health regulations. We and/or our supplier of radiation sources must also comply with NRC and U.S. Department of Transportation regulations on the labeling and packaging requirements for shipment of radiation sources to hospitals or other users of MRIdian. Compliance with NRC, state and local requirements is required for distribution, installation, use and service within each state that we intend to install MRIdian systems.
Existing radiation therapy facilities practicing nuclear medicine, brachytherapy or Gamma Knife therapy are already required to have necessary NRC and/or state licenses and a radiation safety program requiring compliance to various provisions under NRC regulations at Part 35 of Title 10 of the Code of Federal Regulations (“Medical uses of byproduct material”). Use of MRIdian is regulated under Section 35.1000 of the NRC’s regulations (“Other medical uses of byproduct material or radiation from byproduct material”). In 2013, the NRC released licensing guidance under its regulations to guide our customers in the NRC requirements applicable to the use of MRIdian. We believe that this guidance is favorable in that it is consistent with clinical use of existing image-guided radiation therapy devices.
Moreover, our use, management, and disposal of certain radioactive substances and wastes are subject to regulation by several federal and state agencies depending on the nature of the substance or waste material. We believe that we are in compliance with all federal and state regulations for this purpose.
Outside the United States, various laws apply to the import, distribution, installation and use of MRIdian, in consideration of the nuclear materials within MRIdian. Upon 510(k) clearance and commercialization, we do not expect that the MRIdian Linac would fall under this regulation.
U.S. Privacy and Security Laws
We may also be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and their respective implementing regulations, including the final omnibus rule published on January 25, 2013, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information. Further, “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity are also subject to certain HIPAA privacy and security standards. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many
28
of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
U.S. Fraud and Abuse Laws and Regulations
The healthcare industry is also subject to a number of fraud and abuse laws and regulations, including physician anti-kickback, false claims and physician payment transparency laws. Violations of these laws can lead to civil and criminal penalties, including exclusion from participation in federal healthcare programs and significant monetary penalties, among others. These laws, among other things, constrain the sales, marketing and other promotional activities of manufacturers of medical products, such as us, by limiting the kinds of financial arrangements we may have with hospitals, physicians and other potential purchasers of medical products who may seek reimbursement from a federal or state health care program such as Medicare or Medicaid.
Anti-kickback laws. The federal Anti-Kickback Statute makes it a criminal offense to knowingly and willfully solicit, offer, receive or pay any remuneration in exchange for, or to induce, the referral of business, including the purchase, order, lease of any good, facility, item or service, that are reimbursable by a state or federal health care program, such as Medicare or Medicaid. The term “remuneration” has been broadly interpreted to include anything of value. The Anti-Kickback Statute has been interpreted to apply to the purchase of medical devices from a particular manufacturer or the referral of patients to a particular supplier of diagnostic services utilizing such devices. Although, there are established statutory exceptions and regulatory safe harbors that define certain financial transactions and practices that are not subject to the Anti-Kickback Statute, the exceptions and safe harbors are drawn narrowly. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all its facts and circumstances.
Generally, courts have taken a broad interpretation of the scope of the Anti-Kickback Statute, holding that the statute may be violated if merely one purpose of a payment arrangement is to induce referrals or purchases. Further, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Violations of this law are punishable by up to five years in prison, and can also result in criminal fines, administrative civil money penalties and exclusion from participation in federal healthcare programs. In addition, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act. Many states have also adopted statutes similar to the federal Anti-Kickback Statute, some of which apply to payments in connection with the referral of patients for healthcare items or services reimbursed by any source, not only governmental payor programs.
False Claims Act. The federal civil False Claims Act prohibits anyone from knowingly and willfully presenting, or causing to be presented, claims for payment, that are false or fraudulent, for services not provided as claimed. In addition to actions initiated by the government itself, the statute authorizes actions to be brought on behalf of the federal government by a private party having knowledge of the alleged fraud. Because the complaint is initially filed under seal, the action may be pending for some time before the defendant is even aware of the action. If the government is ultimately successful in obtaining redress in the matter or if the plaintiff succeeds in obtaining redress without the government’s involvement, then the plaintiff will receive a percentage of the recovery. When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties ranging from $10,781 to $21,563 for each separate false claim, and may be excluded from participation in federal health care programs, and, although the federal False Claims Act is a civil statute, violations may also implicate various federal criminal statutes. Several states have also adopted comparable state false claims act, some of which apply to all payors.
Civil monetary penalties laws. The civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent.
29
Other fraud and abuse laws. HIPAA also created new federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Like the federal Anti-Kickback Statute, the intent standard for certain healthcare fraud statutes under HIPAA was amended by the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act, such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Physician payment transparency laws. There has been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers and entities. The Affordable Care Act, among other things, imposed new reporting requirements on certain manufacturers, including certain device manufacturers, for payments provided to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit timely, accurately, and completely the required information may result in civil monetary penalties of up to an aggregate of $165,786 per year and up to an aggregate of $1,105,241 per year for “knowing failures.” Device manufacturers must submit reports by the 90th day of each calendar year.
Certain states also mandate implementation of compliance programs, impose restrictions on device manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to healthcare providers and entities.
The laws and regulations and their enforcement are constantly undergoing change, and we cannot predict what effect, if any, changes may have on our business. In addition, new laws and regulations may be adopted which adversely affect our business. There has been a trend in recent years, both in the United States and internationally, toward more stringent regulation and enforcement of requirements applicable to medical device manufacturers and requirements regarding protection and confidentiality of personal data.
State Certificate of Need Laws
In some states, a certificate of need, or CON, or similar regulatory approval is required by hospitals and other healthcare providers prior to the acquisition of high-cost capital items, including MRIdian, or the provision of new services. These laws generally require appropriate state agency determination of public need and approval prior to the acquisition of such capital items or addition of new services. CON requirements may preclude our customers from acquiring, or significantly delay acquisition of, MRIdian and/or from performing treatments using MRIdian. CON laws are the subject of ongoing legislative activity, and a significant increase in the number of states regulating the offering and use of MRIdian through CON or similar requirements could adversely affect us.
Healthcare Reform
In the United States and foreign jurisdictions, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare system seeking, among other things, to reduce healthcare costs that could affect our results of operations.
30
By way of example, in the United States, the Affordable Care Act was signed into law in March 2010, which is expected to substantially change the way healthcare is delivered and financed by both governmental and private insurers. Among other things, the Affordable Care Act:
| • | imposed an annual excise tax of 2.3% on any entity that manufactures or imports medical devices offered for sale in the United States which, due to subsequent legislative amendments, was suspended from January 1, 2016 to December 31, 2017. This exercise tax was suspended for another two years after the stopgap bill was signed by the President in January 2018; |
| • | established a new Patient-Centered Outcomes Research Institute to oversee and identify priorities in comparative clinical effectiveness research in an effort to coordinate and develop such research; |
| • | implemented payment system reforms including a national pilot program on payment bundling to encourage hospitals, physicians and other providers to improve the coordination, quality and efficiency of certain healthcare services through bundled payment models; and |
| • | created an independent payment advisory board that will submit recommendations to reduce Medicare spending if projected Medicare spending exceeds a specified growth rate. |
We expect that the new presidential administration and U.S. Congress will seek to modify, repeal, or otherwise invalidate all or certain provisions of, the Affordable Care Act. Since taking office, President Trump has continued to support the repeal of all or portions of the Affordable Care Act. In January 2017, the House and Senate passed a budget resolution that authorizes congressional committees to draft legislation to repeal all or portions of the Affordable Care Act and permits such legislation to pass with a majority vote in the Senate. President Trump also issued an executive order in which he stated that it is his administration’s policy to seek the prompt repeal of the Affordable Care Act and directed executive departments and federal agencies to waive, defer, grant exemptions from, or delay the implementation of the provisions of the Affordable Care Act to the maximum extent permitted by law. There is still uncertainty with respect to the impact President Trump’s administration and the U.S. Congress may have, if any, and any changes will likely take time to unfold, and could have an impact on coverage and reimbursement for healthcare items and services covered by plans that were authorized by the Affordable Care Act.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. These changes include the Budget Control Act of 2011, which resulted in reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and will stay in effect through 2025 unless additional Congressional action is taken, as well as, the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several types of providers, including hospitals and imaging centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for MRIdian or additional pricing pressure.
Foreign Regulation of Medical Devices
Our activities outside the United States are subject to regulatory requirements that vary from country to country and frequently differ significantly from those in the United States. Failure to obtain and maintain regulatory approval or clearance in any foreign country in which we market or plan to market MRIdian and MRIdian Linac may have a negative effect on our ability to generate revenue and harm our business.
In general, MRIdian and MRIdian Linac are regulated outside the United States as medical devices by foreign governmental agencies similar to the FDA and the FTC. In addition, in foreign countries where we have operations or sell MRIdian, we are subject to laws and regulations applicable to manufacturers of medical devices, radiation producing devices and to the healthcare industry, and laws and regulation of general applicability relating to environmental protection, safe working conditions, manufacturing practices and other matters. These laws and regulations are often comparable to, or more stringent than U.S. laws and regulations. Our sales of MRIdian in foreign countries are also subject to regulation of matters such as product standards, packaging requirements,
31
labeling requirements, import restrictions, tariff regulations, duties and tax requirements. We rely in some countries on our foreign distributors to assist us in complying with applicable regulatory requirements.
Regulation in the EU
In the European Union, or EU, we are required under the European Medical Device Directive (Council Directive 93/42/EEC) to affix the CE mark to our MRIdian systems in order to sell the MRIdian systems in member countries of the EU. The CE mark is an international symbol that represents adherence to certain essential principles of safety and effectiveness mandated in the European Medical Device Directive (the so-called “essential requirements”). Once affixed, the CE mark enables a product to be sold within the EEA, which is composed of the 28 Member States of the EU plus Norway, Iceland and Liechtenstein.
To demonstrate compliance with the essential requirements, we must undergo a conformity assessment procedure which varies according to the type of medical device and its classification. Except for certain low risk medical devices where the manufacturer can issue an EC Declaration of Conformity based on a self-assessment of the conformity of its products with the essential requirements of the Medical Devices Directive, a conformity assessment procedure requires the intervention of an organization accredited by a Member State of the EEA to conduct conformity assessments, or a Notified Body. Depending on the relevant conformity assessment procedure, the Notified Body would typically audit and examine the technical file and the quality system for the manufacture, design and final inspection of our devices. The Notified Body issues a CE Certificate of Conformity following successful completion of a conformity assessment procedure conducted in relation to the medical device and its manufacturer and their conformity with the essential requirements. This Certificate entitles the manufacturer to affix the CE mark to its medical devices after having prepared and signed a related EC Declaration of Conformity.
We received the CE Certificate of Conformity from our Notified Body in November 2014, allowing us to affix the CE mark to MRIdian in order to sell it throughout the EEA. In September 2016, we received approval for CE mark in the European Union for our MRIdian Linac.
If we modify MRIdian we may need to undergo a new conformity assessment procedure to be able to affix the CE mark to the modified product. Additionally, we will need to undergo new conformity assessments for any new products that we may develop in the future before we are able to affix the CE mark to these new products. We cannot be certain that the outcome of these conformity assessments will be positive and that we will be able to affix the CE mark for modified or new products or that we will continue to meet the quality and safety standards required to maintain the CE marks that we already have or may have in the future. In addition, if we are unable to affix the CE mark to our future products, we would be unable to sell them in EU member countries.
In September 2012, the European Commission published proposals for the revision of the EU regulatory framework for medical devices. The proposals would replace the Medical Devices Directive and the Active Implantable Medical Devices Directive with two new regulations: the Medical Devices Regulation and the In-Vitro Diagnostic Medical Devices Regulation. Unlike directives, which must be implemented into the national laws of the EU Member States, the regulations would be directly applicable, i.e., without the need for adoption of EU Member State laws implementing them, in all EEA Member States and are intended to eliminate current differences in the regulation of medical devices among EEA Member States.
The Medical Devices Regulation will apply to ViewRay starting on May 26, 2020. Once applicable, the new regulation will among other things:
| • | strengthen the rules on placing devices on the market and reinforce surveillance once they are available; |
| • | establish explicit provisions on manufacturers' responsibilities for the follow-up of the quality, performance and safety of devices placed on the market; |
| • | improve the traceability of medical devices throughout the supply chain to the end-user or patient through a unique identification number; and |
32
| • | set up a central database to provide patients, healthcare professionals and the public with comprehensive information on products available in the EU. |
Regulation in Other Countries
We will be subject to additional regulations in foreign countries in which we intend to market, sell and import MRIdian. We or our distributors must receive all necessary approvals or clearance prior to marketing and importing MRIdian in those international markets. We received a license and permission to import MRIdian into the United Arab Emirates in December 2014. We received regulatory approval for MRIdian with Cobalt-60 in Italy in January 2015, Korea in September 2015, as well as Japan and China in August 2016. We also received regulatory approval for MRIdian Linac in Israel in November 2017. We will seek approvals in other countries as may be required in the future.
The International Standards Organization promulgates internationally recognized standards, including those for the requirements of quality systems. We are certified to the ISO 13485:2003 standard, which specify the quality system requirements for medical device manufacturers. To support our ISO certifications, we are subject to surveillance audits by a Notified Body yearly and recertification audits every three years that assess our continued compliance with the relevant ISO standards. Our most recent recertification audit occurred in March 2017.
The ISO 13485:2003 standard is being replaced by a new version, the ISO 13485:2016 with a required conformance date of March 31, 2019. We are modifying our quality system to meet the requirements of the 13485:2016 standard, undergo audits by the Notified Body and to meet the conformance date of March 31, 2019.
Employees
At December 31, 2017, we had 139 full-time employees. Within our workforce at December 31, 2017, 41 employees were engaged in research and development and 98 employees in business development, finance, human resources, facilities and general management and administration. We have no collective bargaining agreements with our employees, and we have not experienced any work stoppages. We consider our relations with our employees to be good.
33
Item 1A. RISK FACTORS
You should consider carefully the risks and uncertainties described below, together with all of the other information in this Annual Report on Form 10-K and other filings we have made and make in the future with the Securities and Exchange Commission, or the SEC. If any of the following risks are realized, our business, financial condition, results of operations and prospects could be materially and adversely affected. The risks described below are not the only risks facing the Company.
Risks Related to Our Business and Strategy
We have incurred significant losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future. These factors may raise substantial doubt about our ability to continue as a going concern.
We have historically incurred substantial net losses, including net losses of $72.2 million, $50.6 million and $45.0 million during the years ended December 31, 2017, 2016 and 2015, respectively. At December 31, 2017, we had an accumulated deficit of $319.9 million. We expect our net losses to continue as a result of ongoing investments in product development and expansion of our commercial operations, including increased manufacturing, and sales and marketing. These net losses have had, and will continue to have, a negative impact on our working capital, total assets and stockholders’ equity. Because of the numerous risks and uncertainties associated with our development and commercialization efforts, we are unable to predict when we will become profitable, and we may never become profitable. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our inability to achieve and then maintain profitability would harm our business, financial condition, results of operations and cash flows.
Further, the net losses we incur may fluctuate significantly from quarter-to-quarter and year-to-year, such that a period-to-period comparison of our results of operations may not be a good indication of our future performance quarter-to-quarter and year-to-year, due to factors including the timing of product approval, commercial ramp, clinical trials, any litigation that we may file or that may be filed against us, the execution of collaboration, licensing or other agreements and the timing of any payments we make or receive under them. These factors may raise substantial doubt about our ability to continue as a going concern.
If clinicians do not widely adopt MRI-guided radiation therapy or MRIdian Linac fails to achieve and sustain sufficient market acceptance, we will not generate sufficient revenue and our growth prospects, financial condition and results of operations could be harmed.
Our MRI-guided radiation therapy system, MRIdian, may never gain significant acceptance in the marketplace and, therefore, may never generate substantial revenue or allow us to achieve or maintain profitability. Widespread adoption of MRI-guided radiation therapy depends on many factors, including: acceptance by clinicians that MRI-guided radiation therapy is clinically-effective and cost-effective in treating a wide range of cancers; demand by patients for MRI-guided treatment; successful education of clinicians on the various aspects of this therapeutic approach; and coverage and adequate reimbursement for procedures performed using MRI-guided radiation therapy. If we are not successful in conveying to clinicians and hospitals that MRI-guided radiation therapy provides equivalent or superior radiation therapy compared to existing technologies, we may experience reluctance or refusal on the part of clinicians and hospitals to order, and third-party payors to pay for, performing a treatment in which MRIdian is utilized. Our ability to achieve commercial market acceptance for MRIdian or any other future products also depends on the strength of our sales, marketing and distribution organizations. In addition, our expectations regarding clinical benefits and cost savings from using MRIdian may not be accurate. These hurdles may make it difficult to demonstrate to physicians, hospitals and other healthcare providers that MRIdian is an appropriate option for radiation therapy, and may be both superior to available radiation therapy systems and more cost-effective than alternative technologies.
Furthermore, we may encounter difficulty in gaining inclusion in cancer treatment guidelines and gaining broad market acceptance by healthcare providers, third-party payors and patients. Healthcare providers may have difficulty in obtaining adequate reimbursement from government and/or third-party payors for cancer treatment, which may negatively impact adoption of MRIdian.
34
We may not be able to generate sufficient revenue from the commercialization of MRIdian Linac and MRIdian with Cobalt-60 to achieve and maintain profitability.
We rely entirely on the commercialization of MRIdian Linac and MRIdian with Cobalt-60 to generate revenue. During the year ended December 31, 2017, we recognized revenue of $34.0 million from product revenue recognized from installation or delivery of six MRIdian Linac systems, service revenue at certain customer sites and distribution rights revenue from Itochu. In order to successfully commercialize MRIdian Linac and MRIdian with Cobalt-60, we will need to: continue to expand our marketing efforts to develop new relationships and expand existing relationships with customers; receive clearance or approval for MRIdian systems in additional countries; achieve and maintain compliance with all applicable regulatory requirements; and develop and commercialize new features for MRIdian systems. We cannot assure you that we will be able to achieve or maintain profitability. If we fail to successfully commercialize MRIdian systems, we may never receive a return on the substantial investments in product development, sales and marketing, regulatory compliance, manufacturing and quality assurance that we have made, as well as further investments we intend to make, which may cause us to fail to generate revenue and gain economies of scale from such investments.
In addition, potential customers may decide not to purchase MRIdian systems, or our customers may decide to cancel orders due to changes in treatment offerings, research and product development plans, difficulties in obtaining coverage or reimbursement for MRI-guided radiation therapy treatment, complications with facility build-outs, utilization of MRI-guided radiation therapy or other cancer treatment methods developed by other parties, lack of financing or the inability to obtain or delay in obtaining a certificate of need from state regulatory agencies or zoning restrictions, all of which are circumstances outside of our control.
In addition, demand for MRIdian systems may not increase as quickly as we predict, and we may be unable to increase our revenue levels as we expect. Even if we succeed in increasing adoption of MRIdian systems by hospitals and other healthcare providers, maintaining and creating relationships with our existing and new customers and developing and commercializing new features for MRIdian systems, we may not be able to generate sufficient revenue to achieve or maintain profitability.
We are an early, commercial-stage company and have a limited history commercializing MRIdian, which may make it difficult to evaluate our current business and predict our future performance.
We are an early, commercial-stage company and have a limited operating history. We commenced operations as a Florida corporation in 2004 and subsequently reincorporated in Delaware in 2007. However, we did not begin commercial operations until 2013. Our limited history commercializing MRIdian may make it difficult to evaluate our current business and predict our future performance. Any assessment as to if or when we may become profitable or predictions about our future success or viability, are subject to significant uncertainty. We have encountered and will continue to encounter risks and difficulties frequently experienced by early, commercial-stage companies in rapidly evolving industries. If we do not address these risks successfully, our business could be harmed.
If MRIdian does not perform as expected, or if we are unable to satisfy customers’ demands for additional product features, our reputation, business and results of operations will suffer.
Our success depends on the market’s confidence that MRIdian can provide reliable, high-quality MRI-guided radiation therapy. At December 31, 2017, there were only nine MRIdian with Cobalt-60 and six MRIdian Linacs installed or delivered. Consequently, we have limited statistics regarding the efficacy or reliability of MRIdian. We believe that our customers are likely to be particularly sensitive to product defects and errors, including functional downtime that limits the number of patients that can be treated using the system or a failure that is costly to repair. For example, in January 2014, we initiated a correction of the system at Washington University in St. Louis due to a defect we identified in an advanced software feature in the treatment planning system of MRIdian. We promptly updated our software to resolve this defect and notified the FDA of this correction. We cannot assure that similar product defects or other errors will not occur in the future. This could also include the mistreatment of a patient with MRIdian caused by human error on the part of MRIdian’s operators or prescribing physicians or as a result of a machine malfunction. We may be subject to regulatory enforcement action or legal claims arising from any defects or errors that may occur. Any failure of MRIdian to perform as expected could harm our reputation, business and results of operations.
35
Furthermore, the Cobalt-60 radioactive materials used in MRIdian with Cobalt-60 decay over time, which eventually leads to longer treatment times and may have a negative impact on the number of patients a hospital can treat during a day. U.S. regulations require inspection of Cobalt-60 every five years, at which time customers may consider replacing the Cobalt-60 source. This natural decay or a customer’s failure to replace the Cobalt-60 may have a negative impact on MRIdian performance.
In addition, our customers are technologically well informed and at times have specific demands or requests for additional functionality. If we are unable to meet those demands through the development of new features for MRIdian or future products, or those new features or products do not function at the level that our customers expect, or we are unable to increase patient throughput as expected or we are unable to obtain regulatory clearance or approval of those new features or products, where applicable, our reputation, business and results of operations could be harmed.
The safety and efficacy of MRIdian with Cobalt-60 and MRIdian Linac for certain uses is not currently supported by long-term clinical data, and MRIdian with Cobalt-60 and MRIdian Linac may therefore be less safe and effective than initially anticipated.
MRIdian with Cobalt-60 and MRIdian Linac have received premarket clearance by the FDA under Section 510(k) of the Federal Food, Drug and Cosmetic Act, or FDCA. In the 510(k) clearance process, the FDA must determine that a proposed device is “substantially equivalent” to a device legally on the market, known as a “predicate” device, with respect to intended use, technology and safety and effectiveness, in order to clear the proposed device for marketing. This process is typically shorter and generally requires the submission of less supporting documentation than the FDA’s premarket approval process and does not always require long-term clinical studies. Additionally, to date, we have not been required to complete long-term clinical studies in connection with the sale of MRIdian with Cobalt-60 or MRIdian Linac outside the United States. As a result, we currently lack the breadth of published long-term clinical data supporting the efficacy of MRIdian with Cobalt-60 or MRIdian Linac and the benefits each offers that might have been generated in connection with other marketing authorization processes. In addition, because only a few MRIdian systems have been installed at customer sites, we have limited complication or patient survival rate data with respect to treatments using the systems. If future patient studies or clinical testing do not support our belief that MRIdian with Cobalt-60 or MRIdian Linac offers a more advantageous treatment for a wide variety of cancer types, market acceptance of these systems could fail to increase or could decrease and our business could be harmed.
If we choose to, or are required to, conduct additional studies, the results of these studies or experience could reduce the rate of coverage and reimbursement by both public and private third-party payors for procedures that are performed with MRIdian with Cobalt-60 or MRIdian Linac, slow the market adoption of our product by physicians, significantly reduce our ability to achieve expected revenues and prevent us from becoming profitable. In addition, if future studies and experience indicate that MRIdian with Cobalt-60 or MRIdian Linac causes unexpected or serious complications or other unforeseen negative effects, we could be subject to mandatory product recalls or suspension or withdrawal of FDA clearance, and our reputation with physicians, patients and healthcare providers may suffer.
There have been instances of patients’ severe injury or death due to a variety of factors, including operator error, misuse, radiation therapy product or customer system malfunctions, and others. If our redundant safety systems do not operate as we expect, or any of these or other causes arose in the use of our products, MRIdian with Cobalt-60 or MRIdian Linac could severely injure or kill a patient. This could result in lawsuits, fines or damage to our reputation.
We may be delayed or prevented from implementing our long-term sales strategy if we fail to educate clinicians and patients about the benefits of MRIdian.
In order to increase revenue, we must increase awareness of the range of benefits that we believe MRIdian offers to both existing and potential customers, primarily cancer clinicians. An important part of our sales strategy involves educating and training clinicians to utilize the entire functionality of MRIdian. In addition, we must further educate clinicians about the ability of MRIdian to treat a wide range of cancer types effectively and efficiently. If clinicians are not properly educated about the use of MRIdian for radiation therapy, they may be unwilling or unable to take
36
advantage of the full range of functionality that we believe MRIdian offers, which could have a negative impact on MRIdian sales. Clinicians may decide that certain tumors can be adequately treated using traditional radiation therapy systems, notwithstanding the benefits of MRIdian. We must also succeed in educating customers about the potential for reimbursement for procedures performed using MRIdian. In addition, we need to increase awareness of MRIdian among potential patients, who are increasingly educated about cancer treatment options and therefore impact adoption of new technologies by clinicians. If our efforts to expand sales of MRIdian in the long-term are not successful, our business and results of operations will be harmed.
We may not be able to gain the support of leading hospitals and key opinion leaders, or to publish the results of our clinical trials in peer-reviewed journals, which may make it difficult to establish MRIdian as a standard of care and achieve market acceptance.
Our strategy includes developing relationships with leading hospitals and key opinion leaders in our industry. If these hospitals and key industry thought leaders determine that MRIdian is not clinically effective or that alternative technologies are more effective, or if we encounter difficulty promoting adoption or establishing MRIdian as a standard of care, our ability to achieve market acceptance of MRIdian could be significantly limited.
We believe that publication of scientific and medical results in peer-reviewed journals and presentation of data at leading conferences are critical to the broad adoption of MRIdian. Publication in leading medical journals is subject to a peer-review process, and peer reviewers may not consider the results of studies involving MRIdian sufficiently novel or worthy of publication.
We have a limited history of manufacturing, assembling and installing MRIdian in commercial quantities and may encounter related problems or delays that could result in lost revenue.
The pre-installation manufacturing processes for MRIdian include sourcing components from various third-party suppliers, subassembly, assembly, system integration and testing. We must manufacture and assemble MRIdian in compliance with regulatory requirements and at an acceptable cost in order to achieve and maintain profitability. We have only a limited history of manufacturing, assembling and installing MRIdian and, as a result, we may have difficulty manufacturing, assembling and installing MRIdian in sufficient quantities in a timely manner. To manage our manufacturing and operations with our suppliers, we forecast anticipated product orders and material requirements to predict our inventory needs up to a year in advance and enter into purchase orders on the basis of these requirements. Our limited manufacturing history may not provide us with sufficient data to accurately predict future component demand and to anticipate our costs effectively.
Further, we have experienced and may in the future experience delays in obtaining components from suppliers and installing our systems at customer sites associated with contractor timing delays, which could impede our ability to manufacture, assemble and install MRIdian on our expected timeline. Alternatively, delays or postponements of scheduled customer installations could lead to excess inventory due to our limited flexibility to postpone or delay component shipments from suppliers. Accordingly, we may encounter difficulties in production of MRIdian, including problems with quality control and assurance, component supply shortages or surpluses, increased costs, shortages of qualified personnel and difficulties associated with compliance with local, state, federal and foreign regulatory requirements. In addition, if we are unable to maintain larger-scale manufacturing capabilities, our ability to generate revenue will also be limited and our reputation could be harmed. If we cannot achieve the required level and quality of production, we may need to make changes in our supply chain or enter into licensing and other arrangements with third parties who possess sufficient manufacturing facilities and capabilities in compliance with regulatory requirements. Even if we outsource necessary production or enter into licensing or other third-party arrangements, the associated cost could reduce our gross margin and harm our financial condition and results of operations.
We have limited experience in marketing and selling MRIdian, and if we are unable to adequately address our customers’ needs, it could negatively impact sales and market acceptance of MRIdian and we may never generate sufficient revenue to achieve or sustain profitability.
We have limited experience in marketing and selling MRIdian. We have only been selling MRIdian since 2013 and have only nine MRIdian with Cobalt-60 and six MRIdian Linac installed or delivered at December 31, 2017. We have only treated patients since early 2014. MRIdian is a new technology in the radiation therapy systems sector and
37
our future sales will largely depend on our ability to increase our marketing efforts and adequately address our customers’ needs. We believe it is necessary to maintain a sales force that includes sales representatives with specific technical backgrounds that can address those needs as part of the sales cycle. We will also need to attract and develop sales and marketing personnel with industry expertise. Competition for these types of employees is intense and we may not be able to attract and retain sufficient personnel to maintain an effective sales and marketing force. If we are unable to adequately address our customers’ needs, it could negatively impact sales and market acceptance of MRIdian and we may never generate sufficient revenue to achieve or sustain profitability.
The long sales cycle and low unit volume sales of MRIdian, as well as other factors, may contribute to substantial fluctuations in our operating results and stock price and make it difficult to compare our results of operations to prior periods and predict future financial results.
Because of the relatively small number of systems we expect to install in any period, each installation of a MRIdian will represent a significant percentage of our revenue for a particular period. Additionally, customer site construction, certificate of need and additional zoning and licensing permits are often required in connection with the sale of a MRIdian, any of which may further delay the installation process. When we are responsible for installing a system, we only recognize revenue from the sale of a MRIdian after the system has been installed and accepted by the customer. When a qualified third party is responsible for the installation, we recognize revenue when title is transferred. Therefore, if we do not install a MRIdian or transfer title when anticipated, our operating results will vary significantly from our expectations. We have had experiences with customers postponing installation of MRIdian systems due to delays in facility build-outs, which are often lengthy and costly processes for our existing and potential customers. In addition, if our customers delay or cancel purchases, we may be required to modify or terminate contractual arrangements with our suppliers, which may result in the loss of deposits. Due to future fluctuations in revenue and costs, as well as other potential fluctuations, you should not rely upon our operating results in any particular period as an indication of future performance. In addition to the other risks described, the following factors may also contribute to these fluctuations:
| • | timing of when we are able to recognize revenue associated with sales of MRIdian; |
| • | actions relating to regulatory matters, including regulatory requirements in some states for a certificate of need prior to the installation of a MRIdian; |
| • | delays in shipment due to, for example, unanticipated construction delays at customer locations where MRIdian is to be installed, labor disturbances or natural disasters; |
| • | delays in our manufacturing processes or unexpected manufacturing difficulties; |
| • | timing of the announcements of contract executions or other customer and commercial developments; |
| • | timing of the announcement, introduction and delivery of new products or product features by us and by our competitors; |
| • | timing and level of expenditures associated with expansion of sales and marketing activities and our overall operations; |
| • | fluctuations in our gross margins and the factors that contribute to such fluctuations, as described in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” elsewhere in this Annual Report; |
| • | our ability to effectively execute on our strategic and operating plans; |
| • | the extent to which MRIdian gains market acceptance and the timing of customer demand for MRIdian; |
| • | our ability to protect our proprietary rights and defend against third-party challenges; |
| • | disruptions in the supply or changes in the costs of raw materials, labor, product components or transportation services; and |
| • | changes in third-party coverage and reimbursement, government regulation or in a customer’s ability to obtain financing. |
38
These factors are difficult to forecast and may contribute to fluctuations in our reported revenue and results of operations and variation from our expectations, particularly during the periods in which our sales volume is low. Any fluctuations in our financial results may cause volatility in our stock price.
Each MRIdian is a major capital equipment item and is subject to a lengthy sales cycle. The time from initial customer contact to execution of a contract can take 18 to 24 months or more. Following execution of a contract, it generally takes nine to 12 months for a customer to customize an existing facility or construct a new vault, which is inclusive of the time from when a customer places the order to when the system is delivered. During this time, facilities support and transitioning, as well as permitting, are typically required, which can take several months. The time required to customize an existing facility prior to installation, including modifications of a standard vault to accommodate an MRI, is typically currently two to three months. If a customer does not have an existing vault available, it may take longer to construct a new vault. In some cases, customers may request installation for a date later in the future to meet their own clinical or business requirements. Upon the commencement of installation at a customer’s facility, it typically takes approximately 90 days to complete the installation and on-site testing of the system, including the completion of acceptance test procedures. If a small number of customers defer installation of a MRIdian for even a short period, recognition of a significant amount of revenue may be deferred to a subsequent period. Because our operating costs are relatively fixed, our inability to recognize revenue in a particular period may impact our profitability in that period. As a result, the inability to recognize revenue in a particular period may make it difficult to compare our operating results with prior periods. The price of a MRIdian requires a portion of our target customers to obtain outside financing before committing to purchase a MRIdian. This financing may be difficult for our customers to obtain in any given period, if at all. The requirement of site-specific modifications or construction may also delay adoption or overall demand. In addition, while we believe that our backlog of orders provides a better measure at any particular point in time of the long-term performance prospects of our business than our operating results for a particular period, investors may attribute significant weight to our operating results for a particular period, which may be volatile and as a result, cause fluctuations in our stock price.
A large portion of our revenue in any given reporting period will be derived from a small number of contracts.
Given that a significant portion of the purchase price for MRIdian will generally be recognized as revenue in a single reporting period, we expect a small number of contracts in any given reporting period to account for a substantial portion of our revenue in any period, and we expect this trend to continue. Any decrease in revenue from these contracts could harm our operating results. Accordingly, our revenue and results of operations may vary from period to period. We are also subject to credit risk associated with the concentration of our accounts receivable from our customers. If one or more of our customers at any given time were either to terminate their contracts with us, cease doing business with us or fail to pay us on a timely basis, our business, financial condition and results of operations could be harmed.
The payment structure we use in our customer arrangements may lead to fluctuations in operating cash flows in a given period.
While our customers typically provide a deposit upon entering into a sales contract with us, the substantial majority of the payment owed for a MRIdian is not due until the time of shipment of a MRIdian or following final acceptance by the customer upon installation. If we miss targeted shipments or our customers do not actively work towards completing installation, our receipt of payments and our operating cash flows could be impacted. In addition, if customers do not adhere to our payments terms, our operating cash flows could be impacted in any given period. Due to these fluctuations in operating cash flows and other potential fluctuations, you should not rely upon our operating results in any particular period as an indication of future performance.
Amounts included in backlog may not result in actual revenue and are an uncertain indicator of our future earnings.
We define backlog as the accumulation of all orders for which revenue has not been recognized and we consider valid. The determination of backlog includes, among other factors, our subjective judgment about the likelihood of an order becoming revenue and the regulatory approval required in the customer’s jurisdiction, if any. Our judgments in this area have been, and in the future, may be, incorrect and we cannot assure you that, for any order included in backlog, we will recognize revenue with respect to it. In addition, orders can be delayed for a number of
39
reasons, many of which are beyond our control, including supplier delays, which may cause delays in our manufacturing process, customer delays in commencing or completing construction of its facility, delays in obtaining zoning or other approvals and delays in obtaining financing. We may not be aware of these delays affecting our suppliers and customers and as a result may not consider them when evaluating the contemporaneous effect on backlog. Moreover, orders generally do not have firm dates by when a customer must take delivery or accept our systems, and certain customers may not provide a deposit or letter of credit with the contract, either of which could allow a customer greater flexibility to delay the order without canceling the contract. We believe the introduction of MRIdian Linac will increase the number of orders we receive and accelerate the conversion of orders in backlog; however, customers with orders in backlog currently may delay their installations until MRIdian Linac becomes available in their jurisdiction, or is proven to perform well after installation. Further, our backlog could be reduced due to cancellation of orders by customers. Should a cancellation occur, our backlog and anticipated revenue would be reduced unless we were able to replace it. Reductions in our backlog could negatively impact our future results of operations or the price of our common stock.
We evaluate our backlog at least quarterly to determine if the orders continue to meet our criteria for inclusion in backlog. We may adjust our reported backlog to account for any changes in: customer or distributor plans or financial conditions; the customer’s or distributor’s continued intent and ability to fulfill the order contract; regulatory requirements; the status of regulatory approval required in the customer’s jurisdiction (or other factors); or due to changes in our judgment about the likelihood of completing an order contract. In addition, one or more of our contracts have in the past and may in the future contribute to a material portion of our backlog in any one year. Because revenue will not be recognized until we have fulfilled our obligations to a customer, there may be a significant amount of time from signing a contract with a customer or shipping a system and revenue recognition. We cannot assure you that our backlog will result in revenue on a timely basis or at all, or that any canceled contracts will be replaced.
Our ability to achieve profitability depends substantially on increasing our gross margins by reducing costs of MRIdian and improving our economies of scale, which we may not be able to achieve.
We are not, and never have been, profitable. The MRIdian purchase contracts we have entered into to date have been at a range of selling prices. Generally, earlier contracts have been at lower prices and more recent contracts have been at higher prices. Our earlier contracts resulted in negative gross margins. Our ability to enter into contracts at higher selling prices depends on a number of factors including:
| • | our ability to achieve commercial market acceptance for our system; |
| • | the pricing of competitors’ systems; |
| • | availability of coverage and adequate reimbursement by commercial and government payors; and |
| • | our ability to manufacture and install our systems in a timely and cost-effective manner. |
We bear the risk of warranty claims on all products we supply, including equipment and component parts manufactured by third parties. We cannot assure you that we will be successful in claiming recovery under any warranty or indemnity provided to us by our suppliers or vendors in the event of a successful warranty claim against us by a customer or that any recovery from the vendor or supplier would be adequate. In addition, warranty claims brought by our customers related to third-party components may arise after our ability to bring corresponding warranty claims against the suppliers expires, which could result in additional costs to us. There is a risk that warranty claims made against us will exceed our warranty reserve and our business, financial condition and results of operations could be harmed.
Our customer contracts provide that our customers commit to purchase a MRIdian system for a fixed price, and a MRIdian system will generally not be delivered for 11 to 15 months. In some circumstances, delivery can be postponed several months due to customer delays related to construction, vault preparation or concurrent facility expansion, and the cost of product supplies may increase significantly in the intervening time period. In addition, inflation may generally reduce the real value of the purchase price payable upon the achievement of future progress payment milestones. Either of these occurrences could cause our gross margins to decline or cause us to lose money on the sale of a MRIdian.
40
Moreover, our gross margins may decline in a given period due in part to significant replacement rates for components, resulting in increased warranty expense, negative profit margins on service contracts and customer dissatisfaction. If we are unable to reduce our product costs and improve or maintain quality and reliability, our gross margin may be negatively impacted. Additionally, we may face increased demands for compensation from customers who are not satisfied with the quality and reliability of MRIdian, which could increase our service costs or require us to issue credits against future service payments and negatively impact future product sales. For example, we may have to extend a warranty period due to our failure to meet up-time requirements. We are currently implementing programs to reduce the cost of our MRIdian product; however, we may be unable to reduce our product cost as quickly as we anticipate and in some instances may experience increases in costs from our suppliers.
Even if we are able to implement cost reduction and quality improvement efforts successfully, our service operations may remain unprofitable given the relatively small size and geographic dispersion of our installed base, which prevents us from achieving significant economies of scale for the provision of services. If we are unable to achieve increasingly higher gross margins on our MRIdian systems, we may never become profitable.
We may not be able to develop new products or enhance the capabilities of MRIdian to keep pace with our industry’s rapidly changing technology and customer requirements.
Our industry is characterized by rapid technological changes, new product introductions and enhancements and evolving industry standards. Our business prospects depend on our ability to develop new products and applications for our technology in new markets that develop as a result of technological and scientific advances, while improving the performance and cost-effectiveness of MRIdian. New technologies, techniques or products could emerge that might offer better combinations of price and performance than MRIdian systems. The market for radiation therapy treatment products is characterized by rapid innovation and advancement in technology. It is important that we anticipate changes in technology and market demand, as well as physician, hospital and healthcare provider practices to successfully develop, obtain clearance or approval, if required, and successfully introduce new, enhanced and competitive technologies to meet our prospective customers’ needs on a timely and cost-effective basis. Nevertheless, we must carefully manage our introduction of new products. If potential customers believe that new products will offer enhanced features or be sold for a more attractive price, they may delay purchases until they are available. We may also have excess or obsolete inventory as we transition to new products, and we have no experience in managing product transitions. If we do not successfully innovate and introduce new technology into our anticipated product lines, or effectively manage the transitions of our technology to new product offerings, our business, financial condition and results of operations could be harmed.
We face competition from numerous companies, many of whom have greater resources than we do or offer alternative technologies at lower prices than our MRIdian systems, which may make it more difficult for us to achieve significant market penetration and profitability.
The market for radiation therapy equipment is characterized by intense competition and pricing pressure. In particular, we compete with a number of existing therapy equipment companies, including Elekta AB, Varian Medical Systems, Inc. and Accuray Incorporated. Many of these competitors are large, well-capitalized companies with significantly greater market share and resources than we have. As a result, these companies may be better positioned than we are to spend more aggressively on marketing, sales, intellectual property and other product initiatives and research and development activities. In addition, we may compete with certain MRI-linear accelerator research projects that are currently in development and may be commercialized, including projects by the University of Alberta’s Cross Cancer Institute and a partnership of the University of Sydney, Ingham Institute and the University of Queensland.
Existing technologies may offer certain advantages compared to the MRI technology used by our MRIdian system. For example, computed tomography, or CT, is known to hold certain potential advantages over MRI technology for use in radiation therapy. Diagnostic CT is currently the most widely adopted imaging modality for treatment planning, and can be used to directly measure the electron density of patient tissues, which enables more accurate dose computation. In addition, CT imaging provides superior imaging of bones and boney anatomy than MRI, which is advantageous when imaging those structures for planning and alignment for treatment. Finally, CT is a less expensive technology than MRI and might be preferred by customers seeking a lower cost solution.
41
Our current competitors or other potential competitors may develop new products at any time. In addition, competitors may be able to respond more quickly and effectively than we can to new or changing opportunities, technologies, standards or customer requirements. If we are unable to develop products that compete effectively against the products of existing or future competitors, our future revenue could be negatively impacted. Some of our competitors may compete by changing their pricing model or by lowering the price of their therapy systems. If these competitors’ pricing techniques are effective, it could result in downward pressure on the price of all therapy systems. If we are unable to maintain or increase our selling prices in the face of competition, we may not improve our gross margins.
In addition to the competition that we face from technologies performing similar functions to MRIdian, competition also exists for the limited capital expenditure budgets of our customers. A potential purchaser may be forced to choose between two items of capital equipment. Our ability to compete may also be negatively impacted when purchase decisions are based largely upon price, because MRIdian is a premium-priced system relative to other capital expenditures and alternative radiation therapy technologies. In certain circumstances, a purchaser may decide that an alternative radiation therapy system priced below MRIdian may be sufficient for its patient population given the relative upfront cost savings.
Negative press regarding MRI-guided radiation therapy for the treatment of cancer could harm our business.
The comparative efficacy and overall benefits of MRI-guided radiation therapy are not yet well understood, particularly with respect to certain types of cancer. These types of reports could negatively impact the market’s acceptance of MRI-guided radiation therapy, and therefore our ability to generate revenue could be negatively impacted.
We may acquire other businesses, form joint ventures or make investments in other companies or technologies that could negatively affect our operating results, dilute our stockholders’ ownership, increase our debt or cause us to incur significant expense.
We may pursue acquisitions of businesses and assets. We also may pursue strategic alliances and joint ventures that leverage our proprietary technology and industry experience to expand our offerings or distribution. We have no experience with acquiring other companies and limited experience with forming strategic partnerships. We may not be able to find suitable partners or acquisition candidates, and we may not be able to complete such transactions on favorable terms, if at all. If we make any acquisitions, we may not be able to integrate these acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. Any future acquisitions also could result in the incurrence of debt, contingent liabilities or future write-offs of intangible assets or goodwill, any of which could have a negative impact on our cash flows, financial condition and results of operations. Integration of an acquired company also may disrupt ongoing operations and require management resources that we would otherwise focus on developing our existing business. We may experience losses related to investments in other companies, which could harm our financial condition and results of operations. We may not realize the anticipated benefits of any acquisition, strategic alliance or joint venture.
Foreign acquisitions involve unique risks in addition to those mentioned above, including those related to integration of operations across different cultures and languages, currency risks and the particular economic, political and regulatory risks associated with specific countries.
To finance any acquisitions or joint ventures, we may choose to issue shares of common stock as consideration, which could dilute the ownership of our stockholders. Additional funds may not be available on terms that are favorable to us, or at all. If the price of our common stock is low or volatile, we may not be able to acquire other companies or fund a joint venture project using our stock as consideration.
42
Risks Related to Our Reliance on Third Parties
We rely on a limited number of third-party suppliers and, in some cases, sole suppliers, for the majority of our components, subassemblies and materials and may not be able to find replacements or immediately transition to alternative suppliers.
We rely on several sole suppliers, including Japan Superconductor Technology, Inc., Siemens AG, Best Theratronics Ltd., Tesla Engineering Limited and Quality Electrodynamics, LLC, for certain components of MRIdian. These sole suppliers, and any of our other suppliers, may be unwilling or unable to supply components of MRIdian to us reliably and at the levels we anticipate or are required by the market. For us to be successful, our suppliers must be able to provide us with products and components in substantial quantities, in compliance with regulatory requirements, in accordance with agreed upon specifications, at acceptable costs and on a timely basis. An interruption in our commercial operations could occur if we encounter delays or difficulties in securing these components, and if we cannot then obtain an acceptable substitute. Any such interruption could harm our reputation, business, financial condition and results of operations.
If we are required to transition to new third-party suppliers for certain components of MRIdian, we believe that there are only a few other manufacturers that are currently capable of supplying the necessary components. In addition, the use of components or materials furnished by these alternative suppliers could require us to alter our operations. Furthermore, if we are required to change the manufacturer of a critical component of MRIdian, we will be required to verify that the new manufacturer maintains facilities, procedures and operations that comply with our quality and applicable regulatory requirements, which could further impede our ability to manufacture MRIdian in a timely manner. Transitioning to a new supplier could be time-consuming and expensive, may result in interruptions in our operations and product delivery, could affect the performance specifications of MRIdian or could require that we modify the design of MRIdian. If the change in manufacturer results in a significant change to MRIdian, a new 510(k) clearance from the FDA or similar international regulatory authorization may be necessary, which could cause substantial delays. The occurrence of any of these events could harm our ability to meet the demand for MRIdian in a timely manner or cost-effectively.
We cannot assure you that we will be able to secure alternative equipment and materials and utilize it without experiencing interruptions in our workflow. If we should encounter delays or difficulties in securing, reconfiguring or revalidating the equipment and components we require for MRIdian, our reputation, business, financial condition and results of operations could be negatively impacted.
In addition, we are in early stages of developing suppliers for components that are specific to MRIdian Linac. The inability of these suppliers to produce reliable components and to sufficiently scale up manufacturing could harm our ability to install MRIdian Linac systems in a timely or cost-effective manner.
We depend on third-party distributors to market and distribute MRIdian in international markets.
A significant portion of our backlog is composed of international sales, and we expect a significant amount of our revenue to come from international sales. We depend on a number of distributors for sales in these international markets. We cannot control the efforts and resources our third-party distributors will devote to marketing MRIdian. Our distributors may not be able to successfully market and sell MRIdian and may not devote sufficient time and resources to support the marketing and selling efforts that enable the product to develop, achieve or sustain market acceptance. In some jurisdictions, we rely on our distributors to manage the regulatory process, and we are dependent on their ability to do so effectively. In addition, if a dispute arises with a distributor or if a distributor is terminated by us or goes out of business, it may take time to locate an alternative distributor, to seek appropriate regulatory approvals and to train that distributor’s personnel to market MRIdian; our ability to sell and service MRIdian in the region formerly serviced by the terminated distributor could be harmed. Any of our distributors could become insolvent or otherwise become unable to pay amounts owed to us when due. Any of these factors could reduce our revenue from affected international markets, increase our costs in those markets or damage our reputation. In addition, if we are unable to attract additional international distributors, our international revenue may not grow.
43
Failures by our third-party distributors to deliver or install MRIdian properly and on time could harm our reputation.
We rely on arrangements with third-party distributors for sales and installation of MRIdian in international markets. As a result of our reliance on third-party distributors, we may be subject to disruptions and increased costs due to factors beyond our control, including labor strikes, third-party error and other issues. If the services of any of these distributors become unsatisfactory, including their failure to properly install MRIdian, we may experience delays in meeting our customers’ product demands and we may not be able to find a suitable replacement on a timely basis or on commercially reasonable terms. Any failure to deliver, install or service products in a timely manner may damage our reputation and could cause us to lose current or potential customers.
We rely on third parties to store our inventory and to perform spare parts shipping and other logistics functions on our behalf. A failure or disruption with our logistics providers could harm our business.
Customer service is a critical element of our sales strategy. Third-party logistics providers store most of our spare parts inventory in depots around the world and perform a significant portion of our spare parts logistics and shipping activities. If any of our logistics providers suffers an interruption in its business or experiences delays, disruptions or quality control problems in its operations or we have to change and qualify alternative logistics providers for our spare parts, shipments of spare parts to our customers may be delayed and our reputation, business, financial condition and results of operations could be negatively harmed.
If third-party payors do not provide coverage and adequate reimbursement to our customers, it could negatively impact sales of MRIdian.
In the United States, hospitals and other healthcare providers who purchase MRIdian generally rely on third-party payors to reimburse all or part of the costs and fees associated with the treatments performed with our system. Accordingly, sales of MRIdian depend, in part, on whether coverage and adequate reimbursement for standard planning methodologies are available to our customers from third-party payors, such as government healthcare insurance programs, including the Medicare and Medicaid programs, private insurance plans, health maintenance organizations and preferred provider organizations. In general, third-party payors in the United States have become increasingly cost-conscious, which has limited coverage for, and reimbursement of, certain procedures such as MRI-guided radiation therapy. Third-party payors have also increased utilization controls related to the use of products such as ours by healthcare providers.
Furthermore, there is no uniform policy on coverage and reimbursement for MRI-guided radiation therapy among third-party payors. Payors continue to review their coverage policies carefully for existing and new therapies and can, without notice, deny coverage for treatments that include the use of MRIdian.
44
The Medicare program is increasingly used as a model for how private payors and other governmental payors develop their coverage and reimbursement policies for medical services and procedures. Medicare coverage of advanced and conventional radiation therapies using MRIdian currently varies depending upon the geographic location in which the services are provided. The Centers for Medicare & Medicaid Services, or CMS has not adopted national coverage determination for such therapies that would determine coverage nationally. In the absence of a national coverage determination, Medicare Administrative Contractors, or MACs, with jurisdiction over specific geographic regions have the discretion to determine whether and when the use of MRI-guided radiation therapy will be considered medically necessary and covered in their respective regions. A number of MACs have adopted or proposed local coverage determinations covering MRI-guided radiation therapy. However, these local coverage determinations do not ensure that coverage will be available for MRI-guided radiation therapy for all types of cancer, as the coverage policies may limit coverage to only certain types of cancer.
Even if MRI-guided radiation therapy is covered and reimbursed by third-party payors, adverse changes in payors’ coverage and reimbursement policies that affect MRIdian could harm our ability to market and sell MRIdian. We cannot be sure that third-party payors will reimburse our customers for procedures using MRIdian at a level that will enable us to achieve or maintain adequate sales and price levels for MRIdian. Without coverage and adequate reimbursement from third-party payors, the market for MRIdian may be limited.
Third-party payors regularly update reimbursement amounts and also, from time to time, revise the methodologies used to determine reimbursement amounts. This includes annual updates to payments to physicians, hospitals and ambulatory surgery centers for the radiation treatments performed with MRIdian. Generally, because the cost of MRIdian is recovered by the healthcare provider as part of the payment for performing the treatment and not separately reimbursed, these updates could directly impact the demand for MRIdian. An example of payment updates is the Medicare program’s updates to hospital and physician payments, which are done on an annual basis using a prescribed statutory formula.
Historically, under the Medicare Physician Fee Schedule, or MPFS, when the application of the formula resulted in lower payment, Congress passed interim legislation to prevent the reductions. In April 2015, however, the Medicare Access and CHIP Reauthorization Act of 2015, or MACRA, was signed into law, which repealed and replaced the statutory formula for Medicare payment adjustments to physicians. MACRA provided a permanent end to the annual interim legislative updates that had previously been necessary to delay or prevent significant reductions to payments under the Medicare Physician Fee Schedule. MACRA provided for a 0.5% update from July 1, 2015 through December 31, 2015, and for each calendar year through 2019, after which there will be a 0% annual update each year through 2025. In addition, MACRA required the establishment of the Merit-Based Incentive Payment System, beginning in 2019, under which physicians may receive performance-based payment incentives or payment reductions based on their performance with respect to clinical quality, resource use, clinical improvement activities and meaningful use of electronic health records. MACRA also required CMS, beginning in 2019, to provide incentive payments for physicians and other eligible professionals that participate in alternative payment models, such as accountable care organizations, that emphasize quality and value over the traditional volume-based fee-for-service model. It is unclear what impact, if any, MACRA will have on our business and operating results, but any resulting decrease in payment may result in reduced demand for our services.
CMS also publishes annual updates to HOPPS. These payments are bundled amounts received by our hospital customers for hospital outpatient services, including conventional radiation therapy and IMRT, which may result in lower reimbursement to our customers for procedures performed using MRIdian.
In addition, in 2016, CMS implemented changes to the reimbursement of certain services performed in the freestanding center setting which, to date, have not had any material impact on the services delivered with our products.
Any significant cuts in reimbursement rates or changes in reimbursement methodology or administration for MRI-guided radiation therapy, or concerns or proposals regarding further cuts or changes in methodology or administration, could further increase uncertainty, influence our customers’ decisions, reduce demand for MRIdian, cause customers to cancel orders and impact our revenue and harm our business.
45
Foreign governments also have their own healthcare reimbursement systems, which vary significantly by country and region, and we cannot be sure that adequate reimbursement will be made available with respect to MRIdian under any foreign reimbursement system.
Our employees, consultants and commercial partners may engage in misconduct or other improper activities, including insider trading and non-compliance with regulatory standards and requirements.
We are exposed to the risk that our employees, consultants, distributors, and commercial partners may engage in fraudulent or illegal activity. Misconduct by these parties could include intentional, reckless or negligent conduct or disclosure of unauthorized activities to us that violates the regulations of the FDA and non-U.S. regulators, including those laws requiring the reporting of true, complete and accurate information to such regulators, manufacturing standards, healthcare fraud and abuse laws and regulations in the United States and abroad or laws that require the true, complete and accurate reporting of financial information or data. In particular, sales, marketing and business arrangements in the healthcare industry, including the sale of medical devices, are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. It is not always possible to identify and deter misconduct by our employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could result in the imposition of significant fines or other sanctions, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings and curtailment of operations, any of which could adversely affect our ability to operate our business and our results of operations. Whether or not we are successful in defending against such actions or investigations, we could incur substantial costs, including legal fees, and divert the attention of management in defending ourselves against any of these claims or investigations.
Risks Related to Our Financial Condition and Capital Requirements
We may need to raise additional capital to fund our existing commercial operations, develop and commercialize new features for MRIdian and new products and expand our operations.
Based on our current business plan, we expect that our existing cash and cash equivalents will enable us to conduct our planned operations for at least the next 12 months. If our available cash balances and anticipated cash flow from operations are insufficient to satisfy our liquidity requirements because of lower demand for MRIdian as a result of lower than currently expected rates of reimbursement from commercial third-party payors and government payors or due to other risks described in this Annual Report, we may, from time to time, seek to raise capital through a variety of sources, including the public equity market, private equity financing, and/or public or private debt.
We may consider raising additional capital in the future to expand our business, to pursue strategic investments, to take advantage of financing opportunities or for other reasons, including to:
| • | increase our sales and marketing efforts to increase market adoption of MRIdian and address competitive developments; |
| • | provide for supply and inventory costs associated with plans to accommodate potential increases in demand for MRIdian systems; |
| • | fund development and marketing efforts of any future products and technologies, including MRIdian Linac, or additional features to then-current products; |
| • | acquire, license or invest in new technologies; |
| • | acquire or invest in complementary businesses or assets; and |
| • | finance capital expenditures and general and administrative expenses. |
46
Our present and future funding requirements will depend on many factors, including:
| • | our ability to achieve revenue growth and improve gross margins; |
| • | our rate of progress in establishing coverage and reimbursement arrangements with domestic and international commercial third-party payors and government payors; |
| • | the cost of expanding our operations and offerings, including our sales and marketing efforts; |
| • | our rate of progress in, and cost of the sales and marketing activities associated with, establishing adoption of MRIdian; |
| • | the cost of research and development activities; |
| • | the effect of competing technological and market developments; |
| • | costs related to international expansion; and |
| • | the potential cost of and delays in product development as a result of any regulatory oversight applicable to MRIdian. |
The various ways we could raise additional capital carry potential risks. If we raise funds by issuing equity securities, dilution to our stockholders could result. Any equity securities issued also could provide for rights, preferences or privileges senior to those of holders of our common stock. If we raise funds by issuing debt securities, those debt securities would have rights, preferences and privileges senior to those of holders of our common stock. The terms of debt securities issued or borrowings pursuant to a credit agreement could impose significant restrictions on our operations. If we raise funds through collaborations and licensing arrangements, we might be required to relinquish significant rights to certain components contained within MRIdian, or grant licenses on terms that are not favorable to us.
We have incurred, and will continue to incur significant costs as a result of operating as a public company and our management expects to continue to devote substantial time to public company compliance programs.
As a public company, we have incurred, and will continue to incur significant legal, accounting and other expenses due to our compliance with regulations and disclosure obligations applicable to us, including compliance with the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, as well as rules implemented by the SEC, and the NASDAQ Stock Market. Stockholder activism, the current political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact, in ways we cannot currently anticipate, the manner in which we operate our business. Our management and other personnel have devoted, and will continue to devote a substantial amount of time to these compliance programs and monitoring of public company reporting obligations and as a result of the new corporate governance and executive compensation related rules, regulations and guidelines prompted by the Dodd-Frank Wall Street Reform and Consumer Protection Act, or the Dodd-Frank Act, and further regulations and disclosure obligations expected in the future, we will likely need to devote additional time and costs to comply with such compliance programs and rules. These rules and regulations will continue to cause us to incur significant legal and financial compliance costs and will make some activities more time-consuming and costly.
To comply with the requirements of being a public company, we may need to undertake various actions, including implementing new internal controls and procedures and hiring additional accounting or internal audit staff. The Sarbanes-Oxley Act requires that we maintain effective disclosure controls and procedures and internal control over financial reporting. We are continuing to develop and refine our disclosure controls and other procedures that are designed to ensure that information required to be disclosed by us in the reports that we file with the SEC is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms, and that information required to be disclosed in reports under the Securities Exchange Act of 1934, or the Exchange Act, is accumulated and communicated to our principal executive and financial officers. Our current controls and any new controls that we develop may become inadequate and weaknesses in our internal control over financial reporting may be discovered in the future. Any failure to develop or maintain effective controls could negatively impact the results of periodic management evaluations and annual independent registered public accounting firm attestation reports regarding the effectiveness of our internal control over financial reporting that we may be required to include
47
in our periodic reports we will file with the SEC under Section 404 of the Sarbanes-Oxley Act, harm our operating results, cause us to fail to meet our reporting obligations or result in a restatement of our prior period financial statements. In the event that we are not able to demonstrate compliance with the Sarbanes-Oxley Act, that our internal control over financial reporting is perceived as inadequate or that we are unable to produce timely or accurate financial statements, investors may lose confidence in our operating results and the price of our common stock could decline. In addition, if we are unable to continue to meet these requirements, our common stock may not be able to remain eligible for quotation on The NASDAQ Global Market.
Our independent registered public accounting firm will not be required to formally attest to the effectiveness of our internal control over financial reporting until the first annual report required to be filed with the SEC following the date we are no longer an “emerging growth company” as defined in the JOBS Act. If we are unable to assert that our internal control over financial reporting is effective, or if our independent registered public accounting firm is unable to express an opinion on the effectiveness of our internal control over financial reporting, we could lose investor confidence in the accuracy and completeness of our financial reports, which could harm our business.
Compliance with recently adopted rules of the SEC relating to “conflict minerals” may require us and our suppliers to incur substantial expense and may result in disclosure by us that certain minerals used in products we manufacture or contract to manufacture are not “DRC conflict free.”
Section 1502 of the Dodd-Frank Act required the SEC to promulgate rules requiring disclosure by a public company of any “conflict minerals” (tin, tungsten, tantalum and gold) necessary to the functionality or production of a product manufactured or contracted to be manufactured by the public company. The SEC adopted final rules in 2012 that took effect at the end of January 2013. Because we manufacture or contract to manufacture a product that contains tin, tungsten, tantalum or gold, we will be required under these rules to determine whether those minerals are necessary to the functionality or production of MRIdian and, if so, conduct a country of origin inquiry with respect to all such minerals. If any such minerals may have originated in the Democratic Republic of the Congo, or DRC, or any of its adjoining countries, or covered countries, then we must conduct diligence on the source and chain of custody of those conflict minerals to determine if they originated in one of the covered countries and, if so, whether they financed or benefited armed groups in the covered countries. Disclosures relating to the products that may contain conflict minerals, the country of origin of those minerals and whether they are “DRC conflict free” must be provided in a Form SD (and accompanying conflict minerals report, if required, to disclose the diligence undertaken by us in sourcing the minerals and our conclusions relating to such diligence). Compliance with this disclosure rule may be very time-consuming for management and our supply chain personnel (as well as time-consuming for our suppliers) and could involve the expenditure of significant amounts of money by us and them. Disclosures, mandated by this new rule, which can be perceived by the market to be “negative,” may cause customers to refuse to purchase MRIdian. We cannot assure you that the cost of compliance with the rule will not harm our business, financial condition or results of operations.
Our loan and security agreement with Capital Royalty Partners II L.P., Capital Royalty Partners II - Parallel Fund “A” L.P., Capital Royalty Partners II (Cayman) L.P. and Parallel Investment Opportunities Partners II L.P., or together with their successors by assignment, CRG, contains operating and financial covenants that may restrict our business and financing activities.
At December 31, 2017, we had $45.0 million in outstanding debt to CRG. Borrowings under our loan and security agreement with CRG are secured by substantially all of our personal property, including our intellectual property. Our loan and security agreement restricts our ability to, among other things:
| • | dispose of or sell our assets; |
| • | make material changes in our business; |
| • | merge with or acquire other entities or assets; |
| • | incur additional indebtedness; |
| • | create liens on our assets; |
48
| • | pay off subordinated indebtedness. |
The operating and financial restrictions and covenants in our loan and security agreement, as well as any future financing agreements into which we may enter, may restrict our ability to finance our operations and engage in, expand or otherwise pursue our business activities and strategies. Our ability to comply with these covenants may be affected by events beyond our control, and future breaches of any of these covenants could result in a default under our loan and security agreement. If not waived, future defaults could cause all of the outstanding indebtedness under our loan and security agreement to become immediately due and payable and terminate all commitments to extend further credit.
If we do not have or are unable to generate sufficient cash available to repay our debt obligations when they become due and payable, either upon maturity or in the event of a default, we may not be able to obtain additional debt or equity financing on favorable terms, if at all, which may negatively impact our ability to operate and continue our business as a going concern.
Our ability to use our net operating losses to offset future taxable income may be subject to certain limitations.
At December 31, 2017, we had federal net operating loss carryforwards, or NOLs, of $266.3 million, which begin to expire in the year ending December 31, 2024, and $144.8 million related to state net operating loss carryforwards, which begin to expire in the year ending December 31, 2019. We also had federal and state research and development tax credit carryforwards of $3.8 million and $1.3 million, respectively, which begin to expire in the year ending December 31, 2024. Under Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, a corporation that undergoes an “ownership change” is subject to limitations on its ability to utilize its NOLs to offset future taxable income. We believe we have experienced at least one ownership change in the past. We are currently analyzing the tax impacts of such ownership change on our federal NOLs and credit carryforwards. Future changes in our stock ownership, including this or future offerings, as well as other changes that may be outside of our control, could result in additional ownership changes under Section 382 of the Code. Our NOLs may also be limited under similar provisions of state law. We have recorded a full valuation allowance related to our NOLs and other deferred tax assets due to the uncertainty of the ultimate realization of the future tax benefits of such assets.
We face risks related to the current global economic environment, which could delay or prevent our customers from obtaining financing to purchase MRIdian and implement the required facilities, which could harm our business, financial condition and results of operations.
The state of the global economy continues to be uncertain. The current global economic conditions and uncertain credit markets and concerns regarding the availability of credit pose a risk that could impact customer demand for MRIdian, as well as our ability to manage normal commercial relationships with our customers, suppliers and creditors, including financial institutions. If the current global economic environment deteriorates, our business could be negatively affected.
Risks Related to Administrative, Organizational and Commercial Operations and Growth
We may be unable to manage our future growth effectively, which could make it difficult to execute our business strategy.
We anticipate growth in our business operations. This future growth could create a strain on our organizational, administrative and operational infrastructure, including manufacturing operations, quality control, technical support and customer service, sales force management and general and financial administration. We may not be able to maintain the quality of or installation timelines of MRIdian or satisfy customer demand as it grows. Our ability to manage our growth properly will require us to continue to improve our operational, financial and management controls, as well as our reporting systems and procedures. We may implement new enterprise software systems in a number of areas affecting a broad range of business processes and functional areas. The time and resources required to implement these new systems is uncertain and failure to complete this in a timely and efficient manner could harm our business.
49
If we are unable to support demand for MRIdian and our future products, including ensuring that we have adequate resources to meet increased demand, or we are unable to successfully manage the evolution of our MRI-guided radiation technology, our business could be harmed.
As our commercial operations and sales volume grow, we will need to continue to increase our workflow capacity for manufacturing, customer service, billing and general process improvements and expand our internal quality assurance program, among other things. We will also need to purchase additional equipment, some of which can take several months or more to procure, set up and validate, and increase our manufacturing, maintenance, software and computing capacity to meet increased demand. We cannot assure you that any of these increases in scale, expansion of personnel, purchase of equipment or process enhancements will be successfully implemented.
The loss of our President and Chief Executive Officer or Chief Scientific Officer or our inability to attract and retain highly skilled scientists and salespeople could negatively impact our business.
Our success depends on the skills, experience and performance of our President and Chief Executive Officer, Chris A. Raanes, and our Chief Scientific Officer and founder, James F. Dempsey, Ph.D. The individual and collective efforts of these employees will be important as we continue to develop MRIdian and as we expand our commercial activities. The loss or incapacity of existing members of our executive management team could negatively impact our operations if we experience difficulties in hiring qualified successors. Our executive officers have employment agreements; however, the existence of an employment agreement does not guarantee the retention of the executive officer for any period of time.
Our commercial, manufacturing and research and development programs and operations depend on our ability to attract and retain highly skilled engineers, scientists and technicians. We may not be able to attract or retain qualified managers, engineers, scientists and technicians in the future due to the competition for qualified personnel among medical device businesses, particularly in California and Ohio. We also face competition from universities and public and private research institutions in recruiting and retaining highly qualified scientific personnel. Recruiting and retention difficulties can limit our ability to support our commercial, manufacturing and research and development programs. All of our employees are at-will, which means that either we or the employee may terminate his or her employment at any time.
If we were sued for product liability or professional liability, we could face substantial liabilities that exceed our resources.
The marketing, sale and use of MRIdian could lead to the filing of product liability claims were someone to allege that MRIdian did not effectively treat the conditions its users were intending to target, caused serious medical conditions or injury, or failed to perform as designed. We may also be subject to liability for errors in, a misunderstanding of or inappropriate reliance upon the information we provide in the ordinary course of our business activities, such as customer support or operating instructions. A product liability claim could result in substantial damages and be costly and time-consuming for us to defend.
50
We maintain product liability insurance, but the amounts of insurance coverage may not fully protect us from the financial impact of defending against product liability claims (and we have significant deductibles). Any product liability claim brought against us, with or without merit, could increase our insurance rates or prevent us from securing insurance coverage in the future. Additionally, any product liability lawsuit could lead to regulatory investigations, product recalls or withdrawals, damage our reputation or cause current vendors, suppliers and customers to terminate existing agreements and potential customers and partners to seek other suppliers of radiation therapy systems, any of which could negatively impact our results of operations.
Sanctions against Russia, and Russia’s response to those sanctions, could harm our business, financial condition and results of operations.
Due to Russia’s military intervention in Ukraine and annexation of Crimea, the United States and the European Union, or EU, have imposed sanctions on certain individuals and institutions in Russia and the Ukraine, and have proposed the use of broader economic sanctions. In response, Russia has imposed entry bans on certain U.S. lawmakers and officials. We have engaged a third-party distributor and are currently in discussions with potential customers in Russia. If the United States or the EU were to impose sanctions on Russian businesses, or if Russia were to take retaliatory action against U.S. companies operating in Russia, our sales and marketing efforts in Russia could be harmed.
The results of the United Kingdom’s referendum on withdrawal from the EU may have a negative effect on global economic conditions, financial markets and our business.
In June 2016, a majority of voters in the United Kingdom, or the U.K., elected to withdraw from the EU in a national referendum, also known as Brexit. In March 2017, the U.K. Prime Minister began the process for the U.K. to withdraw from the EU. Negotiations are expected to commence to determine the future terms of the U.K.’s relationship with the EU, including, among other things, the terms of trade between the U.K. and the EU. The effects of Brexit will depend on any agreements the U.K. reaches to retain access to EU markets either during a transitional period or more permanently. Nevertheless, the referendum has created significant uncertainty about the future relationship between the U.K. and the EU, including with respect to the laws and regulations that will apply as the U.K. determines which EU laws to replace or replicate in the event of a withdrawal, including those governing manufacturing, labor, environmental, data protection/privacy, competition, medical sales and advertising and other matters applicable to the medical device industry. The referendum has also given rise to calls for the governments of other EU member states to consider withdrawal. These developments, or the perception that any of them could occur, have had and may continue to have a material adverse effect on global economic conditions and the stability of global financial markets, and may significantly reduce global market liquidity and restrict the ability of key market participants to operate in certain financial markets. Any of these factors could depress economic activity and restrict our access to capital, which could have a material adverse effect on our business, financial condition and results of operations and reduce the price of our securities.
We face risks associated with our international business.
In addition to our marketing and sales of MRIdian in the United States, we also market MRIdian in North America, Europe and the Pacific Rim, with contracts signed with customers and distributors in Taiwan, Turkey, Korea, China, the United Arab Emirates, Hong Kong, Japan, Italy, Israel, Germany, Denmark and Russia. Our international business operations are subject to a variety of risks, including:
| • | difficulties in staffing and managing foreign and geographically dispersed operations; |
| • | effective compliance with various U.S. and international laws, including export control laws and the U.S. Foreign Corrupt Practices Act of 1977, or the FCPA, and anti-money laundering laws; |
| • | differing regulatory requirements for obtaining clearances or approvals to market MRIdian and future product enhancements for MRIdian including but not limited to, MRIdian Linac; |
| • | changes in uncertainties relating to foreign rules and regulations that may impact our ability to sell MRIdian, perform services or repatriate profits to the United States; |
51
| • | tariffs, export or import restrictions, restrictions on remittances abroad, imposition of duties or taxes that limit our ability to move MRIdian out of these countries or interfere with the import of essential materials into these countries; |
| • | limitations on our ability to enter into cost-effective arrangements with distributors of MRIdian, or at all; |
| • | fluctuations in foreign currency exchange rates; |
| • | imposition of limitations on production, sale or export of MRI-guided radiation therapy systems in foreign countries; |
| • | imposition of limitations on or increase of withholding and other taxes on remittances and other payments by foreign subsidiaries or joint ventures; |
| • | differing multiple payor reimbursement regimes, government payors or patient self-pay systems; |
| • | imposition of differing labor laws and standards; |
| • | economic, political or social instability in foreign countries and regions; |
| • | an inability, or reduced ability, to protect our intellectual property, including any effect of compulsory licensing imposed by government action; and |
| • | availability of government subsidies or other incentives that benefit competitors in their local markets that are not available to us. |
We expect that we will begin expanding into more target markets; however, we cannot assure you that our expansion plans will be realized, or if realized, be successful. We expect each market to have particular regulatory and funding hurdles to overcome and future developments in these markets, including the uncertainty relating to governmental policies and regulations, could harm our business. If we expend significant time and resources on expansion plans that fail or are delayed, our reputation, business and financial condition may be harmed.
Our results may be impacted by changes in foreign currency exchange rates.
Currently, the majority of our international sales contracts are denominated in U.S. dollars. We pay certain of our suppliers in a foreign currency under the terms of their supply agreements, and we may pay other suppliers in the future in foreign currency. As a result, an increase in the value of the U.S. dollar relative to foreign currencies could require us to reduce our selling price or risk making MRIdian less competitive in international markets or our costs could increase. Also, if our international sales increase, we may enter into a greater number of transactions denominated in non-U.S. dollars, which could expose us to foreign currency risks, including changes in currency exchange rates. We do not currently engage in any hedging transactions. If we are unable to address these risks and challenges effectively, our international operations may not be successful and our business could be harmed.
We could be negatively impacted by violations of applicable anti-corruption laws or violations of our internal policies designed to ensure ethical business practices.
We operate in a number of countries throughout the world, including in countries that do not have as strong a commitment to anti-corruption and ethical behavior that is required by U.S. laws or by corporate policies. We are subject to the risk that we, our U.S. employees or our employees located in other jurisdictions or any third parties such as our sales agents and distributors that we engage to do work on our behalf in foreign countries may take action determined to be in violation of anti-corruption laws in any jurisdiction in which we conduct business, including the FCPA and the Bribery Act of 2010, or the U.K. Anti-Bribery Act. In addition, we operate in certain countries in which the government may take an ownership stake in an enterprise and such government ownership may not be readily apparent, thereby increasing potential anti-corruption law violations. Any violation of the FCPA and U.K. Anti-Bribery Act or any similar anti-corruption law or regulation could result in substantial fines, sanctions, civil and/or criminal penalties and curtailment of operations in certain jurisdictions and might harm our business, financial condition or results of operations. In addition, we have internal ethics policies with which we require our employees to comply in order to ensure that our business is conducted in a manner that our management
52
deems appropriate. If these anti-corruption laws or internal policies were to be violated, our reputation and operations could also be substantially harmed. Further, detecting, investigating and resolving actual or alleged violations is expensive and can consume significant time and attention of our senior management.
We are subject to export restrictions and laws affecting trade and investments, and the future sale of our MRIdian system may be further limited or prohibited in the future by a government agency or authority.
As a global company headquartered in the United States, our MRIdian system is subject to U.S. laws and regulations that may limit, restrict or require a license to export (and re-export from other countries) our MRIdian system and related product and technical information due to MRIdian’s use of hazardous materials, including MRIdian with Cobalt’s use of Cobalt-60, lead and depleted uranium. We are also subject to the export and import laws of those foreign jurisdictions to which we sell or from which we re-export our MRIdian system. Compliance with these laws and regulations could significantly limit our operations and our sales in the future and failure to comply, even indirectly, could result in a range of penalties, including restrictions on exports of our MRIdian system for a specified time period, or forever, and severe monetary penalties. In certain circumstances, these restrictions may affect our ability to interact with any of our future foreign subsidiaries and otherwise limit our trade with third parties, including suppliers and customers, operating inside and outside the United States. In addition, if we introduce new products, we may need to obtain licenses or approvals from the United States and other governments to ship them into foreign countries. Failure to receive the appropriate approvals may mean that our commercial efforts and expenses related to such efforts may not result in any revenue, which could harm our business.
We depend on our information technology systems, and any failure of these systems could harm our business.
We depend on information technology and telecommunications systems for significant elements of our operations. We have developed proprietary software for the management and operation of MRIdian by our customers. We have installed, and expect to expand, a number of enterprise software systems that affect a broad range of business processes and functional areas, including for example, systems handling human resources, financial controls and reporting, contract management, regulatory compliance and other infrastructure operations. In addition to the aforementioned business systems, we intend to extend the capabilities of both our preventative and detective security controls by augmenting the monitoring and alerting functions, the network design and the automatic countermeasure operations of our technical systems. These information technology and telecommunications systems support a variety of functions, including sales and marketing, manufacturing operations, customer service support, billing and reimbursement, research and development activities and general administrative activities.
Information technology and telecommunications systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our information technology and telecommunications systems, failures or significant downtime of our information technology or telecommunications systems or those used by our third-party service providers could prevent us from providing maintenance and support services to our customers, conducting research and development activities and managing the administrative aspects of our business. Any disruption or loss of information technology or telecommunications systems on which critical aspects of our operations depend could harm our business.
Our operations are vulnerable to interruption or loss due to natural or other disasters, power loss, strikes and other events beyond our control.
We conduct a significant portion of our activities, including administration and data processing, at facilities located in California, Ohio and other areas that have experienced major earthquakes, tornadoes and other natural disasters. A major earthquake, tornado or other disaster (such as a major fire, hurricane, flood, tsunami, volcanic eruption or terrorist attack) affecting our facilities, or those of our suppliers, could significantly disrupt our operations, and delay or prevent product shipment or installation during the time required to repair, rebuild or replace our suppliers’ damaged manufacturing facilities; these delays could be lengthy and costly. If any of our customers’ facilities are negatively impacted by a disaster, shipments of MRIdian could be delayed. Additionally, customers may delay purchases of MRIdian until operations return to normal. Even if we are able to quickly respond to a disaster, the
53
ongoing effects of the disaster could create some uncertainty in the operations of our business. In addition, our facilities may be subject to a shortage of available electrical power and other energy supplies. Any shortages may increase our costs for power and energy supplies or could result in blackouts, which could disrupt the operations of our affected facilities and harm our business. Further, MRIdian is typically shipped from a limited number of ports, and any disaster, strike or other event blocking shipment from these ports could delay or prevent shipments and harm our business. In addition, concerns about terrorism, the effects of a terrorist attack, political turmoil or an outbreak of epidemic diseases, such as Ebola or influenza, could have a negative effect on our operations, those of our suppliers and customers and the ability to travel, which could harm our business, financial condition and results of operations.
The recently enacted tax reform bill could adversely affect our business and financial condition.
On December 22, 2017, President Trump signed into law the “Tax Cuts and Jobs Act,” or the TCJA, which significantly amends the Internal Revenue Code of 1986. The TCJA, among other things, reduces the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, limits the tax deduction for interest expense to 30% of adjusted earnings, eliminates net operating loss carrybacks, imposes a one-time tax on offshore earnings at reduced rates regardless of whether they are repatriated, allows immediate deductions for certain new investments instead of deductions for depreciation expense over time, and modifies or repeals many business deductions and credits. We continue to examine the impact these changes may have on our business. Notwithstanding the reduction in the corporate income tax rate, the overall impact of the TCJA is uncertain and our business and financial condition could be adversely affected. The impact of the TCJA on holders of our common stock is also uncertain and could be adverse.
Risks Related to Intellectual Property
Litigation or other proceedings or third-party claims of intellectual property infringement could require us to spend significant time and money and could prevent us from selling MRIdian or impact our stock price.
There is considerable intellectual property litigation and contested patent disputes in the medical device area. Third parties may, in the future, assert claims that we are employing their proprietary technology without authorization, including claims from competitors or from non-practicing entities that have no relevant product revenue and against whom our own patent portfolio may have no deterrent effect. As we continue to commercialize MRIdian in its current or an updated form, launch new products and enter new markets, we expect that competitors may claim that MRIdian infringes their intellectual property rights as part of business strategies designed to impede our successful commercialization and entry into new markets. Although we are presently unaware of any basis by which a third-party would be justified in making such claims, in the future, we may receive letters or other threats or claims from third parties inviting us to take licenses under, or alleging that we infringe, their patents. Third parties may have obtained, and may in the future obtain, patents under which such third parties may claim that the use of our technologies constitutes patent infringement.
Moreover, we may become party to future adversarial proceedings regarding our patent portfolio or the patents of third parties. Such proceedings could include contested post-grant proceedings such as oppositions, inter parties review, reexamination, interference or derivation proceedings before the U.S. Patent and Trademark Office or foreign patent offices. The legal threshold for initiating litigation or contested proceedings is low, so that even lawsuits or proceedings with a low probability of success might be initiated. Litigation and contested proceedings can also be expensive and time-consuming, and our adversaries in these proceedings may have the ability to dedicate substantially greater resources to prosecuting these legal actions than we can.
54
We could incur substantial costs and divert the attention of our management and technical personnel in defending ourselves against any of these claims or in any of such proceedings. Any adverse ruling or perception of an adverse ruling in defending ourselves against these claims could have a negative impact on our cash position and stock price. Furthermore, parties making claims against us may be able to obtain injunctive or other relief, which could block our ability to develop, commercialize and sell products, and could result in the award of substantial damages against us. In the event of a successful claim of infringement or misappropriation against us, we may be required to pay damages, obtain one or more licenses from third parties or be prohibited from selling certain products, all of which could have a negative impact on our cash position, business and financial condition.
In addition, we may be unable to obtain these licenses at a reasonable cost, if at all. We could therefore incur substantial costs related to royalty payments for licenses obtained from third parties, which could negatively affect our gross margins. Moreover, we could encounter delays in product introductions while we attempt to develop alternative methods or products. Defense of any lawsuit or adversarial proceeding or failure to obtain any of these licenses on favorable terms could prevent us from commercializing products, and the prohibition of sale of MRIdian or future products could impact our ability to grow and maintain profitability and could harm our business.
If we are unable to adequately protect our proprietary technology or maintain issued patents that are sufficient to protect MRIdian, others could compete against us more directly, which could harm our business, financial condition and results of operations.
Our commercial success will depend in part on our success in obtaining and maintaining issued patents and other intellectual property rights in the United States and elsewhere and protecting our proprietary technology. If we do not adequately protect our intellectual property and proprietary technology, competitors may be able to use our technologies and erode or negate any competitive advantage we may have, which could harm our business and ability to achieve profitability.
Specifically, we hold a license to four issued U.S. patents, 19 issued foreign patents (eight of which were issued in Great Britain, Germany, France and the Netherlands as a result of two patent applications filed and allowed through the European Patent Office), one pending U.S. patent application and five pending foreign patent applications as of January 15, 2018. We own an additional 20 issued U.S. patents, 36 issued foreign patents (13 of which were issued in Great Britain, Germany, France, Italy and the Netherlands as a result of three patent applications filed and allowed through the European Patent Office), 24 pending U.S. patent applications and 85 pending foreign patent applications as of January 15, 2018. Assuming all required fees are paid, individual patents or patent applications owned or licensed by us will expire between 2021 and 2037. We also have a joint ownership interest with Case Western Reserve University in one issued patent and one U.S. patent application. We cannot provide any assurances that any of our patents have, or that any of our pending patent applications that mature into issued patents will include, claims with a scope sufficient to protect MRIdian, any additional features we develop for MRIdian or any new products. Other parties may have developed technologies that may be related or competitive to our platform, may have filed or may file patent applications and may have received or may receive patents that overlap or conflict with our patent applications, either by claiming the same methods or devices or by claiming subject matter that could dominate our patent position. The patent positions of medical device companies, including our patent position, involve complex legal and factual questions, and, therefore, the issuance, scope, validity and enforceability of any patent claims that we may obtain cannot be predicted with certainty. Patents, if issued, may be challenged, deemed unenforceable, invalidated or circumvented. U.S. patents and patent applications may also be subject to supplemental examination or contested post-grant proceedings such as inter parties review, reexamination, interference or derivation proceedings before the U.S. Patent and Trademark Office and challenges in district court. Patents may be subjected to opposition, post-grant review or comparable proceedings lodged in various foreign, both national and regional, patent offices. These proceedings could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such proceedings may be costly. Thus, any patents that we may own or exclusively license may not provide any protection against competitors. Furthermore, an adverse decision in an interference proceeding can result in a third-party receiving the patent right sought by us, which in turn could affect our ability to commercialize MRIdian.
Furthermore, though an issued patent is presumed valid and enforceable, its issuance is not conclusive as to its validity or its enforceability and it may not provide us with adequate proprietary protection or competitive
55
advantages against competitors with similar products. Competitors may also be able to design around our patents. Other parties may develop and obtain patent protection for more effective technologies, designs or methods. We may not be able to prevent the unauthorized disclosure or use of our technical knowledge or trade secrets by consultants, agents, distributors, suppliers, vendors, former employees and current employees. The laws of some foreign countries do not protect our proprietary rights to the same extent as the laws of the United States, and we may encounter significant problems in protecting our proprietary rights in these countries. If any of these developments were to occur, they each could have a negative impact on our results of operations and business.
Our ability to enforce our patent rights depends on our ability to detect infringement. It is difficult to detect infringers who do not advertise the components that are used in their products. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product. Any litigation to enforce or defend our patent rights, even if we were to prevail, could be costly and time-consuming and would divert the attention of our management and key personnel from our business operations. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded if we were to prevail may not be commercially meaningful.
In addition, proceedings to enforce or defend our patents could put our patents at risk of being invalidated, held unenforceable or interpreted narrowly. Such proceedings could also provoke third parties to assert claims against us, including that some or all of the claims in one or more of our patents are invalid or otherwise unenforceable. If any of our patents covering MRIdian are invalidated or found unenforceable, our financial position and results of operations could be negatively impacted. In addition, if a court found that valid, enforceable patents held by third parties covered MRIdian, our financial position and results of operations could be harmed.
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
| • | any of our patents, or any of our pending patent applications, if issued, will include claims having a scope sufficient to protect MRIdian or any other products; |
| • | any of our pending patent applications will issue as patents; |
| • | we will be able to successfully commercialize MRIdian on a substantial scale before our relevant patents expire; |
| • | we were the first to make the inventions covered by each of our patents and pending patent applications; |
| • | we were the first to file patent applications for these inventions; |
| • | others will not develop similar or alternative technologies that do not infringe our patents; |
| • | any of our patents will be found to ultimately be valid and enforceable; |
| • | any patents issued to us will provide a basis for an exclusive market for our commercially viable products, will provide us with any competitive advantages or will not be challenged by third parties; |
| • | we will develop additional proprietary technologies or products that are separately patentable; or |
| • | our commercial activities or products will not infringe upon the patents of others. |
We rely, in part, upon unpatented trade secrets, unpatented know-how and continuing technological innovation to develop and maintain our competitive position, which we seek to protect, in part, by confidentiality agreements with our employees and our collaborators and consultants. We also have agreements with our employees and selected consultants that obligate them to assign their inventions to us and have non-compete agreements with some, but not all, of our consultants. It is possible that technology relevant to our business will be independently developed by a person that is not a party to such an agreement. Furthermore, if the employees and consultants who are parties to these agreements breach or violate the terms of these agreements, we may not have adequate remedies for any such breach or violation, and we could lose our trade secrets through such breaches or violations. Further, our trade secrets could otherwise become known or be independently discovered by our competitors.
56
If we are not able to meet the requirements of our license agreement with the University of Florida Research Foundation, Inc., we could lose access to the technologies licensed thereunder and be unable to manufacture, market or sell MRIdian.
We license patents and patent applications from the UFRF, covering our combination of MRI and radiation therapy, and other key technologies, incorporated into MRIdian under a license agreement that requires us to pay royalties to UFRF. In addition, the license agreement obligates us to pursue an agreed development plan and to submit periodic reports and restricts our ability to take actions to defend the licensed patents. The license agreement terminates when the underlying patents expire in 2025, although UFRF has the right to unilaterally terminate the agreement if we do not meet our royalty payment obligations, including minimum royalty payments of $50,000 per quarter, or if we fail to satisfy other development and commercialization obligations related to our utilization of the technology. If UFRF were to terminate the agreement or if we were to otherwise lose the ability to exploit the licensed patents, our competitive advantage could be reduced, we may not be able to find a source to replace the licensed technology and we may be prevented from selling MRIdian. The license agreement reserves to UFRF the initial right to defend or prosecute any claim arising with respect to the licensed technology. If UFRF does not vigorously defend the patents, we may be required to engage in expensive patent litigation to enforce our rights and any competitive advantage we have based on the licensed technology may be hampered. Any of these events could harm our business, financial condition and results of operations.
Changes in U.S. patent laws may limit our ability to obtain, defend or enforce our patents.
Past or future patent reform legislation or precedent could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. For example, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted and also affect patent litigation. The first to file provisions of the Leahy-Smith Act limit the rights of an inventor to patent an invention if not the first to file an application for patenting that invention, even if such invention was the first invention.
The Leahy-Smith Act also created an administrative tribunal known as the Patent Trial and Appeal Board, or PTAB, that provides a venue for companies to challenge the validity of a competitor’s patents at a cost that is much lower than district court litigation and on timelines that are much faster. Although it is not clear what, if any, long-term impact the PTAB proceedings will have on the operation of our business, the initial results of patent challenge proceedings before the PTAB since its inception in 2013 have resulted in the invalidation of many U.S. patent claims. The availability of the PTAB as a lower-cost, faster and potentially more potent tribunal for challenging patents could therefore increase the likelihood that our own patents will be challenged, thereby increasing the uncertainties and costs of maintaining and enforcing them. Moreover, if such challenges occur with regard to our UFRF-licensed patents, as indicated above, we have only limited rights to control the defense.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position could be harmed.
In addition to patent protection, we also rely upon copyright and trade secret protection, as well as non-disclosure agreements and invention assignment agreements with our employees, consultants and third parties, to protect our confidential and proprietary information. For example, significant elements of MRIdian are based on unpatented trade secrets and know-how that are not publicly disclosed. In addition to contractual measures, we try to protect the confidential nature of our proprietary information using physical and technological security measures. Such measures may not, for example, in the case of misappropriation of a trade secret by an employee or third party with authorized access, provide adequate protection for our proprietary information. Our security measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse we take against such misconduct may not provide an adequate remedy to protect our interests fully. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, trade secrets may be independently developed by others in a manner that could prevent legal recourse by us. If any of our confidential or proprietary information, such as our trade secrets, were to be disclosed or misappropriated, or if any such information was independently developed by a competitor, our competitive position could be harmed.
57
We may not be able to enforce our intellectual property rights throughout the world.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. This could make it difficult for us to stop the infringement of our patents, if obtained, or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, many countries limit the enforceability of patents against certain third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries.
Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate. In addition, changes in the law and legal decisions by courts in the United States and foreign countries may affect our ability to obtain adequate protection for our technology and the enforcement of intellectual property.
Third parties may assert ownership or commercial rights to inventions we develop.
Third parties may, in the future, make claims challenging the inventorship or ownership of our intellectual property. We have written agreements with collaborators that provide for the ownership of intellectual property arising from our collaborations. These agreements provide that we must negotiate certain commercial rights with collaborators with respect to joint inventions or inventions made by our collaborators that arise from the results of the collaboration. In some instances, there may not be adequate written provisions to address clearly the resolution of intellectual property rights that may arise from a collaboration. If we cannot successfully negotiate sufficient ownership and commercial rights to the inventions that result from our use of a third-party collaborator’s materials where required, or if disputes otherwise arise with respect to the intellectual property developed with the use of a collaborator’s technology, we may be limited in our ability to capitalize on the market potential of these intellectual property rights. In addition, we may face claims by third parties that our agreements with employees, contractors or consultants obligating them to assign intellectual property to us are ineffective or in conflict with prior or competing contractual obligations of assignment, which could result in ownership disputes regarding intellectual property we have developed or will develop and interfere with our ability to capture the commercial value of such intellectual property. Litigation may be necessary to resolve an ownership dispute, and if we are not successful, we may be precluded from using certain intellectual property or may lose our exclusive rights in that intellectual property. Either outcome could harm our business.
Third parties may assert that our employees or consultants have wrongfully used or disclosed confidential information or misappropriated trade secrets.
We employ individuals who were previously employed at universities or other medical device companies, including our competitors or potential competitors. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
A network or data security incident may allow unauthorized access to our products, our network or our data and also that of our customers, resulting in disruption of critical information systems, harm to our reputation and creation of additional liability that could adversely impact our financial results.
58
Increasingly, companies are subject to a wide variety of attacks on their products, networks and systems on an ongoing basis. In addition to traditional computer “hackers,” malicious code (such as viruses and worms), employee theft or misuse, and denial-of-service attacks, sophisticated nation-state and nation-stated supported actors now engage in attacks (including advanced persistent threat intrusions). Despite significant efforts to create security barriers to such threats, it is virtually impossible to entirely mitigate these risks. If we do not allocate and effectively manage the resources necessary to build and sustain the proper infrastructure in our business technology or in our product design, we could be subject to, among other things: transaction errors; processing inefficiencies; the loss of customers; business disruptions; the loss of or damage to intellectual property through a security breach; or the inability to comply with applicable laws.
If a breach of data security were to occur at a customer site through one of our products as a result of third-party action, employee error, malfeasance or otherwise, and the confidentiality, integrity or availability of our customers’ data, including patient health information (PHI) and personally identifiable information (PII) were disrupted, we could incur significant liability to our customers and to individuals or businesses whose information was being stored by our customers. Our systems may be perceived as less desirable, which could negatively affect our business and damage our reputation. In addition, a network or security breach could result in the loss of customers and make it more challenging to acquire new customers. Because techniques used to obtain unauthorized access to, or to sabotage, systems change frequently and generally are not recognized until launched against a target, we may be unable to anticipate these techniques or to implement adequate preventive measures. In addition, security breaches impacting our network could result in a risk of loss or unauthorized disclosure of customers’ data, which, in turn, could lead to litigation, governmental audits and investigations and possible liability, damage our relationships with our existing customers, and have a negative impact on our ability to attract and retain new customers. In addition, the costs associated with the investigation, remediation and potential notification of the breach to customers and counter-parties could be material.
Third parties may attempt to fraudulently induce employees or customers into disclosing sensitive information such as user names, passwords or other information or otherwise compromise the security of our internal networks, electronic systems and/or physical facilities in order to gain access to our data or our customers’ data, which could result in significant legal and financial exposure, interruptions or malfunctions in our operations, and, ultimately, harm to our future business prospects and revenue. We may be required to expend significant capital and financial resources to protect against threats such as these, or to alleviate problems caused by breaches in security.
Risks Related to Regulatory Matters
MRIdian and our operations are subject to extensive government regulation and oversight both in the United States and abroad, and our failure to comply with applicable requirements could harm our business.
MRIdian is a medical device that is subject to extensive regulation in the United States and elsewhere, including by the FDA and its foreign counterparts. The FDA and foreign regulatory agencies regulate, among other things, with respect to medical devices:
| • | design, development and manufacturing; |
| • | testing, labeling, content and language of instructions for use and storage; |
| • | marketing, sales and distribution; |
| • | premarket clearance and approval; |
| • | record keeping procedures; |
| • | advertising and promotion; |
| • | recalls and field safety corrective actions; |
| • | post-market surveillance, including reporting of deaths or serious injuries and malfunctions that, if they were to recur, could lead to death or serious injury; |
59
| • | post-market approval studies; and |
| • | product import and export. |
The regulations to which we are subject are complex and have tended to become more stringent over time. Regulatory changes could result in restrictions on our ability to carry on or expand our operations, higher than anticipated costs or lower than anticipated sales.
In the United States, before we can market a new medical device, or a new use of, new claim for or significant modification to an existing product, we must first receive either clearance under Section 510(k) of the FDCA or approval of a premarket approval, or PMA, application from the FDA, unless an exemption applies. In the 510(k) clearance process, the FDA must determine that a proposed device is “substantially equivalent” to a device legally on the market, known as a “predicate” device, in order to clear the proposed device for marketing. To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device, and either have the same technological characteristics as the predicate device or have different technological characteristics and not raise different questions of safety or effectiveness than the predicate device. Clinical data are sometimes required to support substantial equivalence. In the PMA process, the FDA must determine that a proposed device is safe and effective for its intended use based, in part, on extensive data, including, but not limited to, technical, pre-clinical, clinical trial, manufacturing and labeling data. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices.
Modifications to products that are approved through a PMA application generally require FDA approval. Similarly, certain modifications made to products cleared through a 510(k) may require a new 510(k) clearance, or possible PMA approval. Both the PMA approval and the 510(k) clearance process can be expensive, lengthy and uncertain. The FDA’s 510(k) clearance process usually takes from three to 12 months, but can last longer. The process of obtaining a PMA is much more costly and uncertain than the 510(k) clearance process and generally takes from one to three years, or even longer, from the time the application is filed with the FDA. In addition, PMA generally requires the performance of one or more clinical trials. Despite the time, effort and cost, we cannot assure you that any particular device will be approved or cleared by the FDA. Any delay or failure to obtain necessary regulatory approvals could harm our business.
In the United States, we have obtained 510(k) premarket clearance from the FDA to market MRIdian for the provision of stereotactic radiosurgery and precision radiotherapy for lesions, tumors and conditions anywhere in the body where radiation treatment is indicated. An element of our strategy is to continue to upgrade MRIdian to incorporate new software and hardware enhancements. We expect that such upgrades, as well as other future modifications, may require new 510(k) clearance; however, future upgrades may be subject to the substantially more costly, time-consuming and uncertain PMA process. If the FDA requires us to go through a lengthier, more rigorous examination for future products or modifications to existing products than we had expected, product introductions or modifications could be delayed or canceled, which could cause our sales to decline. In August 2016, we filed for FDA 510(k) clearance for the MRIdian Linac and received FDA clearance in February 2017. In June 2017, we received 510(k) clearance to market RayZR, our high-resolution MLC.
The FDA can delay, limit or deny clearance or approval of a device for many reasons, including:
| • | we may not be able to demonstrate to the FDA’s satisfaction that MRIdian is substantially equivalent to the proposed predicate device or safe and effective for its intended use; |
| • | the data from our pre-clinical studies and clinical trials may be insufficient to support clearance or approval, where required; and |
| • | the manufacturing process or facilities we use may not meet applicable requirements. |
In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions, which may prevent or delay approval or clearance of our future products under development or impact our ability to modify our currently cleared product on a timely basis. For example, in response to industry and healthcare provider concerns regarding the predictability, consistency and rigor of the 510(k) clearance process, the FDA initiated an evaluation, and in January 2011, announced several proposed actions
60
intended to reform the clearance process. The FDA intends these reform actions to improve the efficiency and transparency of the clearance process, as well as bolster patient safety. In addition, as part of the Food and Drug Administration Safety and Innovation Act, or FDASIA, enacted in 2012, Congress reauthorized the Medical Device User Fee Amendments with various FDA performance goal commitments and enacted several “Medical Device Regulatory Improvements” and miscellaneous reforms, which are further intended to clarify and improve medical device regulation both pre- and post-clearance and approval. More recently, the FDA issued guidance (“Deciding When to Submit a 510(k) for a Change to an Existing Device” and “Deciding When to Submit a 510(k) for a Software Change to an Existing Device”) on October 25, 2017 to assist industry in determining when a change to a previously 510(k)-cleared product requires a new premarket notification to be submitted to the FDA. These guidance documents replaced the 1997 guidance on the same topic. These new guidance documents could impose additional regulatory requirements upon us that could: increase the costs of compliance; restrict our ability to maintain our current clearances; and delay our ability to obtain 510(k) clearances.
Even after we have obtained the proper regulatory clearance or approval to market a product, we have ongoing responsibilities under FDA regulations. The failure to comply with applicable regulations could jeopardize our ability to sell MRIdian and result in enforcement actions such as:
| • | termination of distribution; |
| • | recalls or seizures of products; |
| • | delays in the introduction of products into the market; |
| • | total or partial suspension of production; |
| • | refusal to grant future clearances or approvals; |
| • | withdrawals or suspensions of current clearances or approvals, resulting in prohibitions on sales of MRIdian; and |
| • | in the most serious cases, criminal penalties. |
Any of these sanctions could result in higher than anticipated costs or lower than anticipated sales and harm our reputation, business, financial condition and results of operations.
We also cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative or executive action, either in the United States or abroad. For example, certain policies of the Trump administration may impact our business and industry. Namely, the Trump administration has taken several executive actions, including the issuance of a number of Executive Orders, that could impose significant burdens on, or otherwise materially delay, the FDA’s ability to engage in routine regulatory and oversight activities such as implementing statutes through rulemaking, issuance of guidance, and review and approval of marketing applications. Notably, on January 23, 2017, President Trump ordered a hiring freeze for all executive departments and agencies, including the FDA, which prohibits the FDA from filling employee vacancies or creating new positions. This freeze was later lifted in May 2017. Uncertainty at the FDA or if the FDA were under-staffed, could result in delays in FDA’s responsiveness or in its ability to review submissions or applications, issue regulations or guidance, or implement or enforce regulatory requirements in a timely fashion or at all.
Moreover, on January 30, 2017, President Trump issued an Executive Order, applicable to all executive agencies, including the FDA, that requires that for each notice of proposed rulemaking or final regulation to be issued in fiscal year 2017, the agency shall identify at least two existing regulations to be repealed, unless prohibited by law. These requirements are referred to as the “two-for-one” provisions. This Executive Order includes a budget neutrality provision that requires the total incremental cost of all new regulations in the 2017 fiscal year, including repealed
61
regulations, to be no greater than zero, except in limited circumstances. For fiscal years 2018 and beyond, the Executive Order requires agencies to identify regulations to offset any incremental cost of a new regulation and approximate the total costs or savings associated with each new regulation or repealed regulation. In interim guidance issued by the Office of Information and Regulatory Affairs within OMB on February 2, 2017, the administration indicates that the “two-for-one” provisions may apply not only to agency regulations, but also to significant agency guidance documents. In addition, on February 24, 2017, President Trump issued an executive order directing each affected agency to designate an agency official as a “Regulatory Reform Officer” and establish a “Regulatory Reform Task Force” to implement the two-for-one provisions and other previously issued executive orders relating to the review of federal regulations, however it is difficult to predict how these requirements will be implemented, and the extent to which they will impact the FDA’s ability to exercise its regulatory authority. If these executive actions impose constraints on FDA’s ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted.
In order to sell MRIdian in member countries of the European Economic Area, or EEA, MRIdian must comply with the essential requirements of the EU Medical Devices Directive (Council Directive 93/42/EEC). Compliance with these requirements is a prerequisite to be able to affix the CE mark to MRIdian, without which they cannot be sold or marketed in the EEA. To demonstrate compliance with the essential requirements we must undergo a conformity assessment procedure, which varies according to the type of medical device and its classification. Except for low-risk medical devices, where the manufacturer can issue an EC Declaration of Conformity based on a self-assessment of the conformity of its products with the essential requirements of the EU Medical Devices Directive, a conformity assessment procedure requires the intervention of an organization accredited by a Member State of the EEA to conduct conformity assessments, or a Notified Body. Depending on the relevant conformity assessment procedure, the Notified Body would typically audit and examine the technical file and the quality system for the manufacture, design and final inspection of our devices. The Notified Body issues a CE Certificate of Conformity following successful completion of a conformity assessment procedure conducted in relation to the medical device and its manufacturer and their conformity with the essential requirements. This certificate entitles the manufacturer to affix the CE mark to its medical devices after having prepared and signed a related EC Declaration of Conformity.
As a general rule, demonstration of conformity of medical devices and their manufacturers with the essential requirements must be based, among other things, on the evaluation of clinical data supporting the safety and performance of the products during normal conditions of use. Specifically, a manufacturer must demonstrate that the device achieves its intended performance during normal conditions of use, that the known and foreseeable risks, and any adverse events, are minimized and acceptable when weighed against the benefits of its intended performance, and that any claims made about the performance and safety of the device (e.g., product labeling and instructions for use) are supported by suitable evidence. We have the right to affix the CE mark to MRIdian with Cobalt-60 since November 2014 and MRIdian Linac since September 2016. If we fail to remain in compliance with applicable European laws and directives, we would not be able to continue to affix the CE mark to MRIdian with Cobalt-60 and MRIdian Linac, which would prevent us from selling MRIdian with Cobalt-60 or MRIdian Linac within the EEA. We will also need to obtain regulatory approval in other foreign jurisdictions in which we plan to market and sell MRIdian with Cobalt-60 and MRIdian Linac.
Modifications to MRIdian and our future products may require new 510(k) clearances or PMA approvals, or may require us to cease marketing or recall the modified products until clearances are obtained.
In the United States, we have obtained 510(k) premarket clearance from the FDA to market MRIdian for the provision of stereotactic radiosurgery and precision radiotherapy for lesions, tumors and conditions anywhere in the body where radiation treatment is indicated. Any modification to a 510(k)-cleared device that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, design or manufacture, requires a new 510(k) clearance or, possibly, approval of a PMA.
In February 2017, we received a 510(k) premarket clearance from the FDA to market the MRIdian system that contains MRIdian Linac. As we make other changes or enhancements to our MRIdian system, we will need to determine whether additional FDA clearance is required or not. However, the FDA may not agree with our decisions regarding whether new clearances or approvals are necessary. We have made modifications to MRIdian in the past and have determined based on our review of the applicable FDA regulations and guidance that in certain instances new 510(k) clearances or PMA approvals were not required. We may make similar modifications or add additional features in the future that we believe do not require a new 510(k) clearance or approval of a PMA. If the FDA
62
disagrees with our determination and requires us to submit new 510(k) notifications or PMA applications for modifications to our previously cleared products for which we have concluded that new clearances or approvals are unnecessary, we may be required to cease marketing or to recall the modified product until we obtain clearance or approval, and we may be subject to significant regulatory fines or penalties.
Furthermore, the FDA’s ongoing review of the 510(k) clearance process may make it more difficult for us to make modifications to our previously cleared products, either by imposing more strict requirements on when a new 510(k) notification for a modification to a previously cleared product must be submitted, or applying more onerous review criteria to such submissions. For example, the FDA is currently reviewing its guidance describing when it believes a manufacturer is obligated to submit a new 510(k) for modifications or changes to a previously cleared device and issued draft guidance in August 2016 to assist device manufacturers in making this determination. When finalized, this guidance will replace the FDA’s long-standing guidance issued in 1997 on the same topic. We cannot guarantee whether the FDA’s approach in future guidance will result in substantive changes to existing policy and practice regarding the assessment of whether a new 510(k) is required for changes or modifications to existing devices. The FDA continues to review its 510(k) clearance process, which could result in additional changes to regulatory requirements or guidance documents, which could increase the costs of compliance or restrict our ability to maintain current clearances.
If treatment guidelines for cancer radiation therapies change or the standard of care evolves, we may need to redesign and seek new marketing authorization from the FDA for MRIdian.
If treatment guidelines for cancer radiation therapies or the standard of care evolves, we may need to redesign MRIdian and seek new clearances or approvals from the FDA for MRIdian. Our 510(k) clearance from the FDA is based on current treatment guidelines. If treatment guidelines change so that different treatments become desirable, the clinical utility of MRIdian could be diminished and our business could suffer. For example, competition by other forms of cancer treatment, in particular personalized medicine approaches in targeting drugs and biologics, could reduce the use of radiation therapy as a standard of care in certain indications.
The misuse or off-label use of MRIdian with Cobalt-60 or MRIdian Linac may harm our reputation in the marketplace, result in injuries that lead to product liability suits or result in costly investigations, fines or sanctions by regulatory bodies if we are deemed to have engaged in the promotion of these uses, any of which could be costly to our business.
Clinicians or physicians may misuse MRIdian with Cobalt-60 or MRIdian Linac or use improper techniques if they are not adequately trained or otherwise, potentially leading to injury and an increased risk of product liability. If MRIdian with Cobalt-60 or MRIdian Linac is misused or used with improper technique, we may become subject to costly litigation by our customers or their patients. Product liability claims could divert management’s attention from our core business, be expensive to defend and result in sizeable damage awards against us that may not be covered by insurance. In addition, any of the events described above could harm our business and lead to regulatory action.
63
In addition, MRIdian with Cobalt-60 and MRIdian Linac have been cleared by the FDA for specific treatments. We train our marketing and direct sales force to not promote MRIdian with Cobalt-60 and MRIdian Linac for uses outside of the FDA-cleared indications for use, known as “off-label uses.” For example, MRIdian with Cobalt and MRIdian Linac have not been indicated for diagnostic use. We cannot, however, prevent a physician from using MRIdian with Cobalt-60 or MRIdian Linac off-label, when in the physician’s independent professional medical judgment, he or she deems it appropriate. There may be increased risk of injury to patients if physicians attempt to use MRIdian with Cobalt-60 or MRIdian Linac off-label. Furthermore, the use of MRIdian with Cobalt-60 or MRIdian Linac for indications other than those cleared by the FDA or authorized by any foreign regulatory body may not effectively treat such conditions, which could harm our reputation in the marketplace among physicians and patients.
If the FDA or any foreign regulatory body determines that our promotional materials or training constitute promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance or imposition of an untitled letter, which is used for violators that do not necessitate a warning letter, injunction, seizure, civil fine or criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action under other regulatory authority, such as false claims laws, if they consider our business activities to constitute promotion of an off-label use, which could result in significant penalties, including, but not limited to, criminal, civil and administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs and the curtailment of our operations.
Our MRIdian systems may cause or contribute to adverse medical events that we are required to report to regulatory bodies outside of the U.S. and to the FDA, and if we fail to do so, we would be subject to sanctions that could harm our reputation, business, financial condition and results of operations. The discovery of serious safety issues with our MRIdian systems, or a recall of our MRIdian systems either voluntarily or at the direction of the FDA or another governmental authority, could have a negative impact on us.
We are subject to the FDA’s medical device reporting regulations and similar foreign regulations, which require us to report to the FDA when we receive or become aware of information that reasonably suggests that MRIdian may have caused or contributed to a death or serious injury or malfunctioned in a way that, if the malfunction were to recur, it could cause or contribute to a death or serious injury. The timing of our obligation to report is triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events of which we become aware within the prescribed timeframe. We may also fail to recognize that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of MRIdian. If we fail to comply with our reporting obligations, the FDA could take action, including warning letters, untitled letters, administrative actions, criminal prosecution, imposition of civil monetary penalties, revocation of our device clearance, seizure of MRIdian or delay in clearance of future products.
The FDA and foreign regulatory bodies have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture of a product or in the event that a product poses an unacceptable risk to health. The FDA’s authority to require a recall must be based on a finding that there is reasonable probability that the device could cause serious injury or death. We may also choose to voluntarily recall a product if any material deficiency is found. A government-mandated or voluntary recall by us could occur as a result of an unacceptable risk to health, component failures, malfunctions, manufacturing defects, labeling or design deficiencies, packaging defects, repeated misuse or other deficiencies or failures to comply with applicable regulations. For example, in January 2014, we initiated a correction of the system at Washington University in St. Louis due to a defect we identified in an advanced software feature in the treatment planning system of MRIdian. We promptly updated our software to resolve this defect and notified the FDA of this correction, but the FDA has not formally classified this correction as a recall. We cannot assure you that similar or more significant product defects or other errors will not occur in the future. Recalls involving MRIdian could be particularly harmful to our business, financial condition and results of operations because it is currently our only product.
Companies are required to maintain certain records of recalls and corrections, even if they are not reportable to the FDA or other regulatory bodies. We may initiate voluntary withdrawals or corrections for MRIdian in the future that we determine do not require notification of the FDA or other regulators in the US and around the world. If the FDA
64
disagrees with our determinations, it could require us to report those actions as recalls and we may be subject to enforcement action. A future recall announcement could harm our reputation with customers, potentially lead to product liability claims against us and negatively affect our sales.
Any actual or perceived failure by us to comply with legal or regulatory requirements in one or multiple jurisdictions could result in proceedings, actions or penalties against us.
Many jurisdictions have enacted or are considering enacting privacy and/or data security legislation, including laws and regulations applicable to the collection, use, storage, transfer, disclosure and/or processing of personal information. For example, the U.S. Department of Health and Human Services has promulgated rules governing the privacy and security of individually identifiable health information under the Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH. These privacy and security rules protect medical records and other patient health information (PHI) by limiting their use and disclosure, giving individuals the right to access, amend and seek accounting of their own health information, limiting most uses and disclosures of health information to the minimum amount reasonably necessary to accomplish the intended purpose, and requiring administrative, technical and physical safeguards. Although we are not a covered entity under HIPAA, we have entered into agreements with certain covered entity customers, such as health care providers, under which we are considered to be a “business associate” under HIPAA. As a business associate, we are contractually bound and may also be directly responsible under HIPAA, as amended by HITECH, to implement policies, procedures and reasonable and appropriate security measures to protect any individually identifiable health information we may create, receive, maintain or transmit on behalf of covered entities. We may also be subject to state laws protecting the confidentiality of medical records where those state laws have stricter provisions than HIPAA.
The costs of compliance with, and other burdens imposed by, such laws and regulations that are applicable to the businesses of our customers may limit the use and adoption of our products and reduce overall demand for it. These privacy and data security related laws and regulations are evolving and may result in increasing regulatory and public scrutiny and escalating levels of enforcement and sanctions. Although we are working to comply with those federal, state, and foreign laws and regulations, industry standards, contractual obligations and other legal obligations that apply to us, those laws, regulations, standards and obligations are evolving and may be modified, interpreted and applied in an inconsistent manner from one jurisdiction to another, and may conflict with one another, other requirements or legal obligations, our practices or the features of our platform. Any failure or perceived failure by us to comply with federal, state or foreign laws or regulations, industry standards, contractual obligations or other legal obligations, or any actual or suspected security incident, whether or not resulting in unauthorized access to, or acquisition, release or transfer of personal information or other data, may result in governmental enforcement actions and prosecutions, private litigation, fines and penalties or adverse publicity and could cause our customers to lose trust in us, which could have an adverse effect on our reputation and business. Any inability to adequately address privacy and security concerns, even if unfounded, or comply with applicable laws, regulations, policies, industry standards, contractual obligations, or other legal obligations could result in additional cost and liability to us, damage our reputation, inhibit sales, and adversely affect our business.
We also expect that there will continue to be new proposed laws, regulations and industry standards concerning privacy, data protection and information security in the United States, the European Union and other jurisdictions, and we cannot yet determine the impact such future laws, regulations and standards may have on our business. For example, in the E.U., increasingly stringent data protection and privacy rules that may have substantial impact on the use of patient data across the healthcare industry are scheduled to go into effect in May 2018. In addition to government activity, privacy advocacy groups and technology and other industries are considering various new, additional or different self-regulatory standards that my place additional burdens on us. New laws, amendments to or re-interpretations of existing laws and regulations, industry standards, contractual obligations and other obligations may require us to incur additional costs and restrict our business operations. Such laws and regulations may require companies to implement privacy and security policies, inform individuals of security breaches that affect their personal information, and, in some cases, obtain individuals’ consent to use personal information for certain purposes. If we fail to comply with federal, state and international data privacy laws and regulations, our ability to successfully operate our business and pursue our business goals could be harmed.
65
Our failure to comply with applicable laws and regulations, or to protect such data, could result in enforcement action against us, including fines and public censure, claims for damages by customers and other affected individuals, damage to our reputation and loss of goodwill (both in relation to existing customers and prospective customers), any of which could harm our business, results of operations and financial condition.
If we or our distributors do not obtain and maintain international regulatory registrations or approvals for MRIdian, we will not be able to market and sell MRIdian outside of the United States.
Sales of our devices outside the United States are subject to foreign regulatory requirements that vary widely from country to country. In addition, the FDA regulates exports of medical devices from the United States. While the regulations of some countries may not impose barriers to marketing and selling MRIdian or only require notification, others require that we or our distributors obtain the approval of a specified regulatory body. We have applied for and received regulatory approval in Europe, the United Arab Emirates, Taiwan, Korea, Japan, China and Italy, where regulatory approval is required in addition to the CE mark. We currently have orders to deliver MRIdian to customers in the United States, Taiwan, China, Korea, Italy, Germany, Belgium, the Netherlands, the United Kingdom, France and the United Arab Emirates, which we include in our backlog due to the status of each sales order and our regulatory approval processes in these countries. Complying with foreign regulatory requirements, including obtaining registrations or approvals, can be expensive and time-consuming, and we cannot be certain that we or our distributors will receive regulatory approvals in each country in which we plan to market MRIdian or that we will be able to do so on a timely basis. The time required to obtain registrations or approvals, if required by other countries, may be longer than that required for FDA clearance, and requirements for such registrations or approvals may significantly differ from FDA requirements. If we modify MRIdian, we or our distributors may need to apply for additional regulatory approvals before we are permitted to sell the modified product. In addition, we may not continue to meet the quality and safety standards required to maintain the authorizations that we or our distributors have received. If we or our distributors are unable to maintain our authorizations in a particular country, we will no longer be able to sell MRIdian in that country, which could harm our business.
Regulatory clearance or approval by the FDA does not ensure marketing authorization by regulatory authorities in other countries, and authorization for marketing by one or more foreign regulatory authorities does not ensure marketing authorization will be granted by regulatory authorities in other foreign countries or by the FDA. However, a failure or delay in obtaining marketing authorization in one country may have a negative effect on the regulatory process in others.
We must manufacture MRIdian in accordance with federal and state regulations, and we could be forced to recall our installed systems or terminate production if we fail to comply with these regulations.
The methods used in, and the facilities used for, the manufacture of MRIdian must comply with the FDA’s QSR, which is a complex regulatory scheme that covers the procedures and documentation of the design, testing, production, process controls, quality assurance, labeling, packaging, handling, storage, distribution, installation, servicing and shipping of MRIdian. Furthermore, we are required to verify that our suppliers maintain facilities, procedures and operations that comply with our quality and applicable regulatory requirements. The FDA enforces the QSR through periodic announced or unannounced inspections of medical device manufacturing facilities, which may include the facilities of subcontractors. MRIdian is also subject to similar state regulations and various laws and regulations of foreign countries governing manufacturing.
We cannot guarantee that we or any subcontractors will take the necessary steps to comply with applicable regulations, which could cause delays in the delivery of MRIdian. In addition, failure to comply with applicable FDA requirements or later discovery of previously unknown problems with MRIdian or manufacturing processes could result in, among other things:
| • | warning letters or untitled letters; |
| • | fines, injunctions or civil penalties; |
| • | suspension or withdrawal of approvals or clearances; |
66
| • | seizures or recalls of MRIdian; |
| • | total or partial suspension of production or distribution; |
| • | administrative or judicially imposed sanctions; |
| • | FDA’s refusal to grant pending or future clearances or approvals for MRIdian; |
| • | refusal to permit the import or export of MRIdian; and |
| • | criminal prosecution of us or our employees. |
Any of these actions could significantly and negatively impact supply of MRIdian. If any of these events occurs, our reputation could be harmed, we could be exposed to product liability claims and we could lose customers and suffer reduced revenue and increased costs.
Legislative or regulatory reforms in the United States or the EU may make it more difficult and more costly for us to obtain regulatory clearances or approvals for MRIdian or to produce, market or distribute MRIdian after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulation of medical devices or the reimbursement thereof. In addition, the FDA or NRC, regulations and guidance are often revised or reinterpreted by the FDA or NRC in ways that may significantly affect our business and our MRIdian systems. For example, in response to industry and healthcare provider concerns regarding the predictability, consistency and rigor of the 510(k) clearance process, the FDA initiated an evaluation, and in January 2011, announced several proposed actions intended to reform the clearance process. In addition, as part of FDASIA, Congress reauthorized the Medical Device User Fee Amendments with various FDA performance goal commitments and enacted several “Medical Device Regulatory Improvements” and miscellaneous reforms, which are further intended to clarify and improve medical device regulation both pre- and post-clearance or approval. Any new statutes, regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of any future products or make it more difficult to manufacture, market or distribute MRIdian or future products. For example, the FDA issued draft guidance in August 2016 intended to assist the industry in determining when a change to a previously 510(k)-cleared product requires a new premarket notification to the FDA. Once finalized, this guidance will replace the 1997 guidance on the same topic. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require:
| • | additional testing prior to obtaining clearance or approval; |
| • | changes to manufacturing methods; |
| • | recall, replacement or discontinuance of MRIdian or future products; or |
| • | additional record keeping. |
Any of these changes could require substantial time and cost and could harm our business and our financial results.
On April 5, 2017, the European Parliament passed the Medical Devices Regulation, which repealed and replaced the Medical Devices Directive. Unlike directives, which must be implemented into the national laws of the EEA member States, the regulations would be directly applicable, i.e., without the need for adoption of EEA member State laws implementing them, in all EEA member States and are intended to eliminate current differences in the regulation of medical devices among EEA member States. The Medical Devices Regulation, among other things, is intended to establish a uniform, transparent, predictable and sustainable regulatory framework across the EEA for medical devices and ensure a high level of safety and health while supporting innovation.
The Medical Devices Regulation will, however, only become applicable three years after publication. Once applicable, the new regulations will among other things:
67
| • | strengthen the rules on placing devices on the market and reinforce surveillance once they are available; |
| • | establish explicit provisions on manufacturers' responsibilities for the follow-up of the quality, performance and safety of devices placed on the market; |
| • | improve the traceability of medical devices throughout the supply chain to the end-user or patient through a unique identification number; |
| • | set up a central database to provide patients, healthcare professionals and the public with comprehensive information on products available in the EU; |
| • | strengthened rules for the assessment of certain high-risk devices, which may have to undergo an additional check by experts before they are placed on the market. |
These modifications may have an impact on the way we conduct our business in the EEA.
Our business involves the use of hazardous materials and we and our third-party manufacturers must comply with environmental laws and regulations, which may be expensive and restrict how we do business.
Our third-party manufacturers’ activities and our own activities involve the controlled storage, use and disposal of hazardous materials, including Cobalt-60, lead and depleted uranium. We and our manufacturers are subject to federal, state, local and foreign laws and regulations governing the use, generation, manufacture, storage, handling and disposal of these hazardous materials. We currently carry no insurance specifically covering environmental claims relating to the use of hazardous materials, but we do reserve funds to address these claims at both the federal and state levels. Although we believe that our safety procedures for handling and disposing of these materials and waste products comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental injury or contamination from the use, storage, handling or disposal of hazardous materials. In the event of an accident, state or federal or other applicable authorities may curtail our use of these materials and interrupt our business operations. In addition, if an accident or environmental discharge occurs, or if we discover contamination caused by prior operations, including by prior owners and operators of properties we acquire, we could be liable for cleanup obligations, damages and fines. If such unexpected costs are substantial, this could significantly harm our financial condition and results of operations.
We are subject to federal and state fraud and abuse laws and health information privacy and security laws, which, if violated, could subject us to substantial penalties. Additionally, any challenge to or investigation into our practices under these laws could cause adverse publicity and be costly to respond to, and thus could harm our business.
There are numerous U.S. federal and state laws pertaining to healthcare fraud and abuse, including anti-kickback, false claims and physician transparency laws. Our relationships with providers and hospitals are subject to scrutiny under these laws. We may also be subject to patient privacy regulation by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include:
| • | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce either the referral of an individual or furnishing or arranging for a good or service, for which payment may be made, in whole or in part, under federal healthcare programs, such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. Moreover, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
| • | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent; |
| • | HIPAA, which created federal criminal statutes that prohibit, among other things, executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters. |
68
| | Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation; |
| • | the federal physician sunshine requirements under the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, collectively referred to as the Affordable Care Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies to report annually to the U.S. Department of Health and Human Services information related to payments and other transfers of value to physicians, which is defined broadly to include other healthcare providers and teaching hospitals and ownership and investment interests held by physicians and their immediate family members; |
| • | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers; |
| • | state laws that require device companies to comply with the industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; and |
| • | state laws that require device manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures. |
These laws, among other things, constrain our business, marketing and other promotional activities by limiting the kinds of financial arrangements, including sales programs, we may have with hospitals, physicians or other potential purchasers of medical devices. We have a variety of arrangements with our customers that could implicate these laws. Due to the breadth of these laws, the narrowness of statutory exceptions and safe harbors available, and the range of interpretations to which they are subject, it is possible that some of our current or future practices might be challenged under one or more of these laws. Even an unsuccessful challenge or investigation into our practices could cause adverse publicity, and be costly to respond to, and thus could harm our business, financial condition and results of operations.
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including administrative, civil and criminal penalties, damages, fines, exclusion from participation in government healthcare programs, such as Medicare and Medicaid, imprisonment and the curtailment or restructuring of our operations, any of which could negatively impact our ability to operate our business and our results of operations.
Healthcare policy changes, including recently enacted legislation reforming the U.S. healthcare system, could harm our cash flows, financial condition and results of operations.
In March 2010, the Affordable Care Act was enacted in the United States, which made a number of substantial changes in the way healthcare is financed by both governmental and private insurers. Among other things, the Affordable Care Act:
| • | requires each medical device manufacturer to pay a sales tax equal to 2.3% of the price for which such manufacturer sells its medical devices, which, due to subsequent legislative amendments, has been suspended from January 1, 2016 to December 31, 2017. This exercise tax was suspended for another two years after the stopgap bill was signed by the President in January 2018; |
| • | establishes a new Patient-Centered Outcomes Research Institute to oversee and identify priorities in comparative clinical effectiveness research in an effort to coordinate and develop such research; |
| • | implements payment system reforms including a national pilot program on payment bundling to encourage hospitals, physicians and other providers to improve the coordination, quality and efficiency of certain healthcare services through bundled payment models; and |
| • | establishes an Independent Payment Advisory Board that will submit recommendations to reduce Medicare spending if projected Medicare spending exceeds a specified growth rate. |
We expect that the current presidential administration and U.S. Congress will continue to seek to modify, repeal, or otherwise invalidate all or certain provisions of, the Affordable Care Act. Since taking office, President Trump has
69
continued to support the repeal of all or portions of the Affordable Care Act. In January 2017, the House and Senate passed a budget resolution that authorizes congressional committees to draft legislation to repeal all or portions of the Affordable Care Act and permits such legislation to pass with a majority vote in the Senate. President Trump also recently issued an executive order in which he stated that it is his administration’s policy to seek the prompt repeal of the Affordable Care Act and directed executive departments and federal agencies to waive, defer, grant exemptions from, or delay the implementation of the provisions of the Affordable Care Act to the maximum extent permitted by law. There is still uncertainty with respect to the impact President Trump’s administration and the U.S. Congress may have, if any, and any changes will likely take time to unfold, and could have an impact on coverage and reimbursement for healthcare items and services covered by plans that were authorized by the Affordable Care Act.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. These changes include the Budget Control Act of 2011, which resulted in reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2025 unless additional Congressional action is taken, as well as the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for MRIdian or additional pricing pressure.
Risks Related to Ownership of Our Common Stock
The price of our common stock may be volatile and may be influenced by numerous factors, some of which are beyond our control.
Factors that could cause volatility in the market price of our common stock include, but are not limited to:
| • | actual or anticipated fluctuations in our financial condition and operating results; |
| • | actual or anticipated changes in our growth rate relative to our competitors or market expectations; |
| • | commercial success and market acceptance of MRIdian; |
| • | success of our competitors in discovering, developing or commercializing products; |
| • | ability to commercialize or obtain regulatory approvals for MRIdian, or delays in commercializing or obtaining regulatory approvals; |
| • | strategic transactions undertaken by us; |
| • | additions or departures of key personnel; |
| • | product liability claims; |
| • | prevailing economic conditions; |
| • | disputes concerning our intellectual property or other proprietary rights; |
| • | FDA or other U.S. or foreign regulatory actions affecting us or the healthcare industry; |
| • | healthcare reform measures in the United States; |
| • | sales of our common stock by our officers, directors or significant stockholders; |
| • | future sales or issuances of equity or debt securities by us; |
| • | business disruptions caused by earthquakes, tornadoes or other natural disasters; and |
| • | changes in the manner that investors and securities analysts who provide research on us to the marketplace analyze the value of our common stock. |
70
In addition, the stock markets in general, and the markets for medical device companies in particular, have experienced extreme volatility that have been often unrelated to the operating performance of the issuer. These broad market fluctuations may negatively impact the price or liquidity of our common stock. In the past, when the price of a stock has been volatile, holders of that stock have sometimes instituted securities class action litigation against the issuer. If any of our stockholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the attention of our management would be diverted from the operation of our business.
We are an “emerging growth company” and we cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies,” including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
In addition, Section 102 of the JOBS Act also provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, or the Securities Act, for complying with new or revised accounting standards. An “emerging growth company” can therefore delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. However, we chose to “opt out” of such extended transition period, and as a result, we comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable.
Future sales of our common stock or securities convertible or exchangeable for our common stock may cause our stock price to decline.
If our existing stockholders or option holders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market after any applicable legal restrictions on resale lapse, the price of our common stock could decline. The perception in the market that these sales may occur could also cause the price of our common stock to decline. At December 31, 2017, we have outstanding a total of 67,653,974 shares of common stock.
71
In addition, based on the number of shares subject to outstanding awards under our 2008 Stock Option and Incentive Plan, or 2008 Plan, the number of shares subject to outstanding awards or available for issuance under our 2015 Equity Incentive Award Plan, or 2015 Plan, and our 2015 Employee Stock Purchase Plan, or 2015 ESPP, at December 31, 2017, 3,077,923 shares, 6,634,243 shares and 1,103,481 shares, respectively, of common stock that are either subject to outstanding options, outstanding but subject to vesting or reserved for future issuance under the 2008 Plan, 2015 Plan and 2015 ESPP will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules and Rule 144 and Rule 701 under the Securities Act, which includes, for shares held by directors, executive officers and other affiliates, volume limitations under Rule 144 under the Securities Act. The 2015 Plan contains provisions for the annual increase of the number of shares reserved for issuance under such plan. If the shares we may issue from time to time under the 2008 Plan, 2015 Plan or 2015 ESPP are sold, or if it is perceived that they will be sold, by the award recipients in the public market, the price of our common stock could decline.
You may experience dilution of your ownership interests because of the future issuance of additional shares of our common or preferred stock or other securities that are convertible into or exercisable for our common or preferred stock.
In the future, we may issue our authorized but previously unissued equity securities, resulting in the dilution of the ownership interests of our present stockholders and the purchasers of our common stock. We are authorized to issue an aggregate of 300,000,000 shares of common stock and 10,000,000 shares of “blank check” preferred stock. We may issue additional shares of our common stock or other securities that are convertible into or exercisable for our common stock in connection with hiring or retaining employees, future acquisitions, future sales of our securities for capital raising purposes, or for other business purposes. The future issuance of any such additional shares of our common stock may create downward pressure on the trading price of the common stock. We may need to raise additional capital in the near future to meet our working capital needs, and there can be no assurance that we will not be required to issue additional shares, warrants or other convertible securities in the future in conjunction with these capital raising efforts, including at a price (or exercise prices) below the price you paid for your stock.
Our operating results for a particular period may fluctuate significantly or may fall below the expectations of investors or securities analysts, each of which may cause our stock price to fluctuate or decline.
We expect our operating results to be subject to fluctuations. Our operating results will be affected by numerous factors, including:
| • | variations in the level of expenses related to MRIdian with Cobalt-60, MRIdian Linac or future development programs; |
| • | level of underlying demand for MRIdian and any other products we develop; |
| • | addition or termination of clinical trials or funding support; |
| • | receipt, modification or termination of government contracts or grants, and the timing of payments we receive under these arrangements; |
| • | our execution of any collaborative, licensing or similar arrangements, and the timing of payments we may make or receive under these arrangements; |
| • | any intellectual property infringement lawsuit or opposition, interference or cancellation proceeding in which we may become involved; and |
| • | regulatory developments affecting MRIdian with Cobalt-60, MRIdian Linac or our competitors. |
If our operating results for a particular period fall below the expectations of investors or securities analysts, the price of our common stock could decline substantially. Furthermore, any fluctuations in our operating results may, in turn, cause the price of our common stock to fluctuate substantially. We believe that comparisons of our financial results from various reporting periods are not necessarily meaningful and should not be relied upon as an indication of our future performance.
72
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
Based on the beneficial ownership of our common stock at December 31, 2017, our officers and directors, together with holders of 5% or more of our outstanding common stock and their respective affiliates, beneficially own approximately 63% of our common stock. Accordingly, these stockholders will continue to have significant influence over the outcome of corporate actions requiring stockholder approval, including the election of directors, merger, consolidation or sale of all or substantially all of our assets or any other significant corporate transaction. Further, pursuant to one of the Securities Purchase Agreements related to the October 2017 Direct Registered Offering discussed elsewhere in this Report, we agreed to (a) appoint a representative of Fosun International Limited (“Fosun”) as a non-voting observer to our board of directors and (b) after the date, if ever, that Fosun beneficially owns at least 15% of our then-outstanding shares of common stock, appoint a representative of Fosun as a director upon Fosun’s request. Fosun’s rights expire on the first date that Fosun ceases to own at least 90% of the shares it purchased in the October 2017 Direct Registered Offering discussed elsewhere in this Report. The interests of these stockholders may not be the same as or may even conflict with your interests. For example, these stockholders could delay or prevent a change in control of the Company, even if such a change in control would benefit our other stockholders, which could deprive our stockholders of an opportunity to receive a premium for their common stock as part of a sale of the Company or our assets and might affect the prevailing price of our common stock. The significant concentration of stock ownership may negatively impact the price of our common stock due to investors’ perception that conflicts of interest may exist or arise.
Provisions of our charter documents or Delaware law could delay or prevent an acquisition of the Company, even if such an acquisition would be beneficial to our stockholders, which could make it more difficult for you to change management.
Provisions in our certificate of incorporation and our bylaws may discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. In addition, these provisions may frustrate or prevent any attempt by our stockholders to replace or remove our current management by making it more difficult to replace or remove our board of directors. These provisions include:
| • | a classified board of directors so that not all directors are elected at one time; |
| • | a prohibition on stockholder action through written consent; |
| • | no cumulative voting in the election of directors; |
| • | the exclusive right of our board of directors to elect a director to fill a vacancy created by the expansion of the board of directors or the resignation, death or removal of a director; |
| • | a requirement that special meetings of stockholders be called only by the board of directors, the chairman of the board of directors, the chief executive officer or, in the absence of a chief executive officer, the president; |
| • | an advance notice requirement for stockholder proposals and nominations; |
| • | the authority of our board of directors to issue preferred stock with such terms as our board of directors may determine; and |
| • | a requirement of approval of not less than 66 2/3% of all outstanding shares of our capital stock entitled to vote to amend any bylaws by stockholder action, or to amend specific provisions of our certificate of incorporation. |
In addition, Delaware law prohibits a publicly held Delaware corporation from engaging in a business combination with an interested stockholder, generally a person who, together with its affiliates, owns, or within the last three years has owned, 15% or more of our voting stock, for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner. Accordingly, Delaware law may discourage, delay or prevent a change in control of the Company. Furthermore, our certificate of incorporation specifies that the Court of Chancery of the State of Delaware will be the sole and exclusive forum for most legal actions involving actions brought against us by stockholders. We believe
73
this provision benefits us by providing increased consistency in the application of Delaware law by chancellors particularly experienced in resolving corporate disputes, efficient administration of cases on a more expedited schedule relative to other forums and protection against the burdens of multi-forum litigation. However, the provision may have the effect of discouraging lawsuits against our directors and officers. The enforceability of similar choice of forum provisions in other companies’ certificates of incorporation has been challenged in legal proceedings, and it is possible that, in connection with any applicable action brought against us, a court could find the choice of forum provisions contained in our certificate of incorporation to be inapplicable or unenforceable in such action.
Provisions in our charter documents and other provisions of Delaware law could limit the price that investors are willing to pay in the future for shares of our common stock.
We do not anticipate paying any cash dividends on our common stock in the foreseeable future; therefore, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
We have never declared or paid cash dividends on our common stock. We do not anticipate paying any cash dividends on our common stock in the foreseeable future. We currently intend to retain all available funds and any future earnings to fund the development and growth of our business. In addition, our current loan and security agreement with CRG contains, and our future loan arrangements may contain, terms prohibiting or limiting the amount of dividends that may be declared or paid on our common stock. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
If securities or industry analysts do not publish research, or publish inaccurate or unfavorable research, about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend, in part, on the research and reports that securities or industry analysts publish about us or our business. If one or more of the analysts who cover us downgrade our common stock or publish inaccurate or unfavorable research about our business, our stock price could decline. In addition, if our operating results fail to meet the forecast of analysts, our stock price could decline. If one or more of these analysts cease coverage of the Company or fail to publish reports on us regularly, demand for our common stock could decrease, which might cause our stock price and trading volume to decline.
* * *
The risks above do not necessarily comprise all of those associated with an investment in the Company. This Annual Report contains forward-looking statements that involve unknown risks, uncertainties and other factors that may cause the actual results, financial condition, performance or achievements of the Company to be materially different from any future results, performance or achievements expressed or implied by such forward-looking statements. Factors that might cause such a difference include, but are not limited to, those set out above.
Item 1B. UNRESOLVED STAFF COMMENTS
Not applicable.
Item 2. PROPERTIES
Facilities
Our corporate headquarters are located in Oakwood Village, Ohio, where we lease and occupy approximately 19,800 square feet of office space. The current term of our Oakwood Village lease expires on October 31, 2019, with an option to extend the term through October 31, 2021. We also maintain an office in Mountain View, California, where we lease and occupy approximately 25,500 square feet of office space. The current term of our Mountain View lease expires on November 30, 2019. In connection with our Mountain View, California lease, we entered into a standby letter of credit with PNC Bank, National Association for $0.8 million, which is still outstanding at December 31, 2017.
74
We have analyzed our current facilities in light of our anticipated requirements and have determined to increase our existing space in California to meet the needs of our operations; we are currently seeking additional space on commercially reasonable terms.
Item 3. LEGAL PROCEEDINGS
From time to time, we may become involved in various lawsuits and legal proceedings which arise in the ordinary course of business. However, litigation is subject to inherent uncertainties, and one or more unfavorable outcomes in any claim or litigation against us could have a material adverse effect for the period in which they are resolved and on our business generally. In addition, regardless of their merits or their ultimate outcomes, lawsuits and legal proceedings are costly, divert management attention and may materially adversely affect our reputation, even if resolved in our favor.
The information under the caption “Contingencies” in Note 7 of the consolidated financial statements of this Annual Report on Form 10-K is incorporated herein by reference
Item 4. MINE SAFETY DISCLOSURES
Not applicable.
75
PART II
Item 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock is traded on the NASDAQ Global Market under the symbol “VRAY”, which listing was completed on March 30, 2016. Prior to that, our common stock was quoted on the OTCQB, and there was a limited trading market for our common stock and there were few trades in our common stock. Because our common stock was thinly traded for that period, any reported sale prices may not be a true market-based valuation of our common stock.
The following table sets forth the high and low sale prices per share of our common stock as reported on the NASDAQ Global Market during fiscal 2017 and 2016. The stock prices in the following table prior to March 30, 2016 are based on the high and low bid quotations for our common stock as reported by QTCQB.
| | Common Stock | |
| | High | | | Low | |
| | | |
2017: | | | | | | | | |
First Quarter | | $ | 9.47 | | | $ | 3.04 | |
Second Quarter | | $ | 7.97 | | | $ | 5.80 | |
Third Quarter | | $ | 6.72 | | | $ | 4.60 | |
Fourth Quarter | | $ | 9.90 | | | $ | 5.60 | |
2016: | | | | | | | | |
First Quarter | | $ | 5.50 | | | $ | 3.83 | |
Second Quarter | | $ | 4.86 | | | $ | 3.71 | |
Third Quarter | | $ | 5.31 | | | $ | 2.75 | |
Fourth Quarter | | $ | 4.47 | | | $ | 2.68 | |
Stockholders
At February 12, 2017, we had 6,790 holders of record of our common stock.
Dividend Policy
We have never paid cash dividends on our capital stock and do not anticipate paying cash dividends in the foreseeable future. We intend to retain future earnings to fund ongoing operations and future capital requirements. Any future determination to pay cash dividends will be at the discretion of our board of directors and will be dependent upon financial condition, results of operations, capital requirements and such other factors as the board of directors deems relevant.
Recent Sales of Unregistered Securities
During the year ended December 31, 2017, there were no sales of unregistered equity securities by the Company.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
The Company does not have a stock repurchase program and did not make any share repurchase during the year ended December 31, 2017.
76
Item 6. SELECTED FINANCIAL DATA
The following selected financial data are qualified in their entirety by, and should be read in conjunction with, the more detailed information contained in the consolidated financial statements, the notes thereto and the information set forth in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Annual Report on Form 10-K.
| | Year Ended December 31, | |
| | 2017 | | | 2016 | | | 2015 | | | 2014 | | | 2013 | |
| | (in thousands, except share and per share amounts) | |
Consolidated Statement of Operations Data: | | | | | | | | | | | | | | | | | | | | |
Revenue: | | | | | | | | | | | | | | | | | | | | |
Product | | $ | 30,458 | | | $ | 20,555 | | | $ | 9,620 | | | $ | 5,988 | | | $ | 2,253 | |
Service | | | 3,109 | | | | 1,504 | | | | 530 | | | | 411 | | | | 12 | |
Distribution rights | | | 475 | | | | 178 | | | | — | | | | — | | | | — | |
Grant | | | — | | | | — | | | | 240 | | | | — | | | | 894 | |
Total revenue | | | 34,042 | | | | 22,237 | | | | 10,390 | | | | 6,399 | | | | 3,159 | |
Cost of revenue: | | | | | | | | | | | | | | | | | | | | |
Product | | | 25,488 | | | | 23,897 | | | | 12,673 | | | | 8,176 | | | | 8,173 | |
Service | | | 2,222 | | | | 1,969 | | | | 1,871 | | | | 975 | | | | 14 | |
Total cost of revenue | | | 27,710 | | | | 25,866 | | | | 14,544 | | | | 9,151 | | | | 8,187 | |
Gross margin | | | 6,332 | | | | (3,629 | ) | | | (4,154 | ) | | | (2,752 | ) | | | (5,028 | ) |
Operating expenses: | | | | | | | | | | | | | | | | | | | | |
Research and development(1) | | | 14,709 | | | | 11,442 | | | | 10,449 | | | | 9,404 | | | | 8,780 | |
Selling and marketing(1) | | | 8,412 | | | | 5,601 | | | | 5,139 | | | | 4,681 | | | | 3,781 | |
General and administrative(1) | | | 31,375 | | | | 23,503 | | | | 21,685 | | | | 14,742 | | | | 9,508 | |
Total operating expenses | | | 54,496 | | | | 40,546 | | | | 37,273 | | | | 28,827 | | | | 22,069 | |
Loss from operations | | | (48,164 | ) | | | (44,175 | ) | | | (41,427 | ) | | | (31,579 | ) | | | (27,097 | ) |
Interest income | | 5 | | | 2 | | | | 2 | | | | 1 | | | | 4 | |
Interest expense | | | (7,247 | ) | | | (5,951 | ) | | | (3,452 | ) | | | (2,243 | ) | | | (97 | ) |
Other (expense) income, net | | | (16,770 | ) | | | (512 | ) | | | (117 | ) | | | 21 | | | | (32 | ) |
Loss before provision for income taxes | | $ | (72,176 | ) | | $ | (50,636 | ) | | $ | (44,994 | ) | | $ | (33,800 | ) | | $ | (27,222 | ) |
Provision for income taxes | | | — | | | | — | | | | 1 | | | | — | | | | — | |
Net loss | | $ | (72,176 | ) | | $ | (50,636 | ) | | $ | (44,995 | ) | | $ | (33,800 | ) | | $ | (27,222 | ) |
Cumulative dividends on convertible preferred stock | | | — | | | | — | | | | — | | | | — | | | | (2,898 | ) |
Deemed capital conversion of Series C convertible preferred stock into common stock | | | — | | | | — | | | | — | | | | — | | | | 8,783 | |
Deemed dividend on convertible preferred stock extinguishment | | | — | | | | — | | | | — | | | | — | | | | (6,863 | ) |
Deemed capital contribution on repurchase of Series A preferred stock | | | — | | | | — | | | | — | | | | 9 | | | | — | |
Net loss attributable to common stockholders | | $ | (72,176 | ) | | $ | (50,636 | ) | | $ | (44,995 | ) | | $ | (33,791 | ) | | $ | (28,200 | ) |
Net loss per share attributable to common stockholders, basic and diluted(2) | | $ | (1.23 | ) | | $ | (1.26 | ) | | $ | (2.58 | ) | | $ | (37.87 | ) | | $ | (34.59 | ) |
Weighted-average common shares used in computing net loss per share attributable to common stockholders, basic and diluted(2) | | | 58,457,868 | | | | 40,068,307 | | | | 17,432,434 | | | | 892,315 | | | | 815,340 | |
77
| (1) | Includes stock-based compensation expense as follows: |
| | Year Ended December 31, | |
| | 2017 | | | 2016 | | | 2015 | | | 2014 | | | 2013 | |
| | (in thousands) | |
Research and development | | $ | 952 | | | $ | 593 | | | $ | 262 | | | $ | 85 | | | $ | 29 | |
Selling and marketing | | | 303 | | | | 120 | | | | 50 | | | | 15 | | | | 9 | |
General and administrative | | | 4,064 | | | | 2,194 | | | | 754 | | | | 218 | | | | 181 | |
Total stock-based compensation expense | | $ | 5,319 | | | $ | 2,907 | | | $ | 1,066 | | | $ | 318 | | | $ | 219 | |
| (2) | See Note 17 to our consolidated financial statements for an explanation of the method used to calculate our basic and diluted net loss per share attributable to common stockholders. |
| | At December 31, | |
| | 2017 | | | 2016 | | | 2015 | |
| | (in thousands) | |
Consolidated Balance Sheet Data: | | | | | | | | | | | | |
Cash and cash equivalents | | $ | 57,389 | | | $ | 14,198 | | | $ | 20,667 | |
Total assets | | | 135,711 | | | | 48,764 | | | | 52,157 | |
Deferred revenue, current and noncurrent portion | | | 23,389 | | | | 10,433 | | | | 5,961 | |
Long-term debt | | | 44,504 | | | | 44,290 | | | | 29,016 | |
Total liabilities | | | 133,724 | | | | 92,417 | | | | 59,114 | |
Total stockholders' equity (deficit) | | | 1,987 | | | | (43,653 | ) | | | (6,957 | ) |
78
Item 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following management’s discussion and analysis should be read in conjunction with the historical financial statements and the related notes thereto contained in this Annual Report. The management’s discussion and analysis contains forward-looking statements, such as statements of our plans, objectives, expectations and intentions. Any statements that are not statements of historical fact are forward-looking statements. When used, the words “believe,” “plan,” “intend,” “anticipate,” “target,” “estimate,” “expect” and the like, and/or future tense or conditional constructions (“will,” “may,” “could,” “should,” etc.), or similar expressions, identify certain of these forward-looking statements. These forward-looking statements are subject to risks and uncertainties, including those under “Risk Factors” in this Annual Report that could cause actual results or events to differ materially from those expressed or implied by the forward-looking statements. The Company’s actual results and the timing of events could differ materially from those anticipated in these forward-looking statements as a result of several factors. The Company does not undertake any obligation to update forward-looking statements to reflect events or circumstances occurring after the date of this Annual Report.
References in this section to “ViewRay,” “we,” “us,” “our,” “the Company” and “our Company” refer to ViewRay, Inc. and its consolidated subsidiary, ViewRay Technologies, Inc.
As previously reported, on July 23, 2015, our wholly-owned subsidiary, Vesuvius Acquisition Corp., a corporation formed in the State of Delaware on July 16, 2015, or the Acquisition Sub, merged with and into ViewRay Technologies, Inc., a corporation incorporated in 2004 in the State of Florida originally under the name of ViewRay Incorporated, subsequently reincorporated in the State of Delaware in 2007. Pursuant to this transaction, or the Merger, ViewRay Technologies, Inc. was the surviving corporation and became our wholly-owned subsidiary. All of the outstanding capital stock of ViewRay Technologies, Inc. was converted into shares of our common stock, as described in more detail below.
Also, as previously reported, immediately prior to the closing of the Merger, under the terms of a split-off agreement, or the Split-Off Agreement, and a general release agreement, we transferred all of our pre-Merger operating assets and liabilities to our wholly-owned special-purpose subsidiary, Mirax Enterprise Corp., a Nevada corporation, or the Split-Off Subsidiary, formed on July 16, 2015.
In connection with the Merger and pursuant to the Split-Off Agreement, we transferred all of the outstanding shares of capital stock of the Split-Off Subsidiary to our pre-Merger majority stockholder, in exchange for the surrender and cancellation of 4,150,171 shares of our common stock.
As a result of the Merger and Split-Off, we discontinued our pre-Merger business, acquired the business of ViewRay Technologies, Inc. and continued the business operations of ViewRay Technologies, Inc., as a publicly-traded company under the name ViewRay, Inc.
As a result of the Merger and the change in our business and operations, a discussion of our past financial results is not pertinent, and under applicable accounting principles the historical financial results of ViewRay Technologies, Inc., the accounting acquirer, prior to the Merger are considered our historical financial results.
The following discussion highlights our results of operations and the principal factors that have affected our financial condition as well as our liquidity and capital resources for the periods described, and provides information that management believes is relevant for an assessment and understanding of the statements of financial condition and results of operations presented herein. The following discussion and analysis are based on our consolidated financial statements contained in this Annual Report, which we have prepared in accordance with U.S. generally accepted accounting principles. You should read this discussion and analysis together with such consolidated financial statements and the related notes thereto.
79
Company Overview
We design, manufacture and market MRIdian, an MRI-guided radiation therapy system to simultaneously image and treat cancer patients. MRI is a broadly used imaging tool that has the ability to clearly differentiate between types of soft tissue, unlike X-ray or computed tomography, or CT, which are the most commonly used imaging technologies in radiation therapy today. MRIdian integrates MRI technology, radiation delivery and our proprietary software to locate, target and track the location and shape of soft-tissue tumors while radiation is delivered. These capabilities allow MRIdian to accurately deliver radiation to the tumor while reducing the amount delivered to healthy tissue, as compared to other radiation therapy treatments today. We believe this leads to improved patient outcomes and reduced side effects from off-target radiation delivery.
We received initial 510(k) marketing clearance from the FDA for our treatment planning and delivery software in January 2011 and for MRIdian with Cobalt-60 in May 2012. We also received permission to affix the Conformité Européene, or CE, mark to MRIdian with Cobalt-60 in November 2014, allowing MRIdian with Cobalt-60 to be sold within the European Economic Area, or EEA. In August 2016, we received regulatory approval from the Japanese Ministry of Health, Labor and Welfare to market MRIdian with Cobalt-60 in Japan. In August 2016, we also received approval from the China Food and Drug Administration to market MRIdian with Cobalt-60 in China. In September 2016, we received CE mark approval of MRIdian Linac in the EEA. In February 2017, we received 510(k) clearance from the FDA to market MRIdian Linac. In June 2017, we received 510(k) clearance to market RayZR, our high resolution multi-leaf collimator, or MLC.
MRIdian is a radiation therapy solution that enables treatment and real-time imaging of a patient’s anatomy simultaneously. The high-quality images that it generates differentiate the targeted tumor, surrounding soft tissue and nearby critical organs. MRIdian also records the level of radiation dose that the treatment area has received, enabling physicians to adapt the prescription between treatments as needed. We believe this improved visualization and accurate dose recording will enable better treatment, improve patient outcomes and reduce side effects. Key benefits to users and patients include improved imaging and patient alignment, on-table adaptive treatment planning, motion management and an accurate recording of the delivered radiation dose. Physicians have already used MRIdian to treat a broad spectrum of radiation therapy patients with more than 45 different types of cancer, as well as patients for whom radiation therapy was previously not an option.
At December 31, 2017, we have delivered or installed MRIdian systems at 14 leading cancer centers, including six units in the United States and nine units outside the United States.
We currently market MRIdian through a direct sales force in the United States and distributors in the rest of the world. We market MRIdian to a broad range of worldwide customers, including university research and teaching hospitals, community hospitals, private practices, government institutions and freestanding cancer centers. Our sales and revenue cycle varies based on the customer and can be lengthy, sometimes lasting up to 18 to 24 months or more from initial customer contact to sales contract execution. Following execution of a sales contract, it generally takes nine to 12 months for a customer to customize an existing facility or construct a new vault. After the customer completes their customization, it typically takes approximately ninety days to complete the installation and on-site testing of the system, including the completion of acceptance test procedures.
We generated product, service, distribution rights and grant revenue of $34.0, $22.2 million and $10.4 million, and had net losses of $72.2, $50.6 million and $45.0 million during the years ended December 31, 2017, 2016 and 2015, respectively.
We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. We expect our expenses will increase substantially in connection with our ongoing activities, as we:
| • | add personnel to support our product development and commercialization efforts; |
| • | continue our research and development efforts; |
| • | seek regulatory approval for MRIdian in certain foreign countries; and |
| • | operate as a public company. |
80
Accordingly, we may seek to fund our operations through public or private equity, debt financings or other sources. However, we may be unable to raise additional funds or enter into such other arrangements when needed on favorable terms or at all. Our failure to raise capital or enter into such other arrangements as and when needed would have a negative impact on our financial condition and our ability to develop enhancements to and integrate new technologies into MRI-guided radiation therapy systems.
Merger
On July 23, 2015, ViewRay, Inc. (f/k/a Mirax Corp.), and ViewRay Technologies, Inc. (f/k/a ViewRay Incorporated), consummated an Agreement and Plan of Merger and Reorganization, or Merger Agreement. Pursuant to the Merger Agreement, the stockholders of ViewRay Technologies, Inc. contributed all of their equity interests to ViewRay, Inc. for shares of the ViewRay, Inc.’s common stock and merged with the Company’s subsidiary, which resulted in ViewRay Technologies, Inc. becoming a wholly-owned subsidiary of ViewRay, Inc., or the Merger. Effective as of July 23, 2015, ViewRay, Inc. amended and restated its Certificate of Incorporation to increase its authorized common stock to 300,000,000 shares and 10,000,000 shares of “blank check” preferred stock, par value of $0.01 per share.
Upon closing of the Merger, under the terms of the Split-Off Agreement, dated July 23, 2015 among ViewRay, Inc., ViewRay Technologies, Inc. and Vesuvius Acquisition Sub, Inc., the acquisition subsidiary of Mirax, and a general release agreement dated July 23, 2015, or the General Release Agreement, ViewRay, Inc. transferred all of its pre-Merger operating assets and liabilities to a wholly-owned special-purpose subsidiary incorporated in Nevada, Mirax Enterprise Corp., or the Split-Off Subsidiary. Thereafter, Mirax transferred all of the outstanding shares of capital stock of the Split-Off Subsidiary to certain pre-Merger insiders of Mirax in exchange for the surrender and cancellation of shares of Mirax common stock held by such persons, or the Split-Off.
Together with the Merger, on July 23, 2015, ViewRay Technologies, Inc. effected a 2.975-for-1 stock split of its then outstanding common stock and convertible preferred stock, collectively referred to as Capital Stock, and convertible preferred stock warrants, in which (i) each share of outstanding Capital Stock was increased into 2.975 shares of Capital Stock; (ii) the number of outstanding options to purchase each Capital Stock was proportionately increased on a 2.975-for-1 basis; (iii) number of shares reserved for future option grants under the 2008 Plan were proportionately increased on a 2.975-for-1 basis; (iv) the exercise price of each such outstanding option was proportionately decreased on a 2.975-for-1 basis; and (v) each share of outstanding convertible preferred stock warrant was increased into 2.975 shares of convertible preferred stock warrant. All of the share and per share amounts have been adjusted, on a retroactive basis, to reflect this 2.975-for-1 stock split.
Private Placement
At the closing of the Merger, ViewRay, Inc. conducted a private placement offering, or the Private Placement, of its securities for $26.3 million through the sale of 5,884,504 shares of the common stock of the surviving corporation, at an offering price of $5.00 per share, net of offering cost. Existing ViewRay Technologies, Inc. investors purchased $17.0 million shares of common stock in the Private Placement. Certain shareholders of Mirax retained, after giving effect to the Split-Off, 1,000,005 shares of the common stock of the surviving corporation upon the Private Placement.
The Merger was accounted for as a reverse-merger and recapitalization. ViewRay Technologies, Inc. was the acquirer for financial reporting purposes, and ViewRay, Inc. was the acquired company under the acquisition method of accounting in accordance with the Financial Accounting Standards Board (FASB) Accounting Standards Update (ASU) No. 2014-18, Topic 805, Business Combinations. Consequently, the assets, liabilities and operations that were reflected in the historical financial statements prior to the Merger were those of ViewRay Technologies, Inc. and were recorded at the historical cost basis, and the consolidated financial statements after completion of the Merger included the assets, liabilities and results of operations of ViewRay Technologies, Inc. up to the day prior to the closing of the Merger and the assets, liabilities and results of operations of the combined company from and after the closing date of the Merger.
81
2016 Private Placement
On August 19, 2016, we entered into a Securities Purchase Agreement pursuant to which we sold an aggregate of 5,983,251 shares of common stock which consists of 4,602,506 shares of common stock and warrants to purchase 1,380,745 shares of common stock, or the 2016 Placement Warrants, for aggregate proceeds of $13.2 million, net of offering cost, or the 2016 Private Placement. We completed the initial closing of the 2016 Private Placement on August 22, 2016 with the final closing on September 9, 2016.
January 2017 Private Placement
On January 13, 2017, we entered into a Securities Purchase Agreement pursuant to which we sold an aggregate of 10,323,101 shares of common stock which consists of 8,602,589 shares of common stock and warrants to purchase 1,720,512 shares of common stock, or the January 2017 Placement Warrants, for aggregate gross proceeds of $26.1 million, or the 2017 Private Placement. We completed the closing of the January 2017 Private Placement on January 18, 2017.
October 2017 Direct Registered Offering
On October 23, 2017, we entered into Securities Purchase Agreements with certain investors pursuant to which we sold an aggregate of 8,382,643 shares of common stock for aggregate gross proceeds of $50.0 million, or the October 2017 Direct Registered Offering. We completed the closing of the October 2017 Direct Registered Offering on October 25, 2017.
New Orders and Backlog
New orders are defined as the sum of gross product orders, representing MRIdian contract price, recorded during the period. Backlog is the accumulation of all orders for which revenue has not been recognized and we consider valid. Backlog includes customer deposits or letters of credit, except when the sale is to a customer where a deposit is deemed not necessary or customary. Deposits received are recorded as a liability on the balance sheet. Orders may be revised or cancelled according to their terms or upon mutual agreement between the parties. Therefore, it is difficult to predict with certainty the amount of backlog that will ultimately result in revenue. The determination of backlog includes objective and subjective judgment about the likelihood of an order contract becoming revenue. We perform a quarterly review of backlog to verify that outstanding orders in backlog remain valid, and based upon this review, orders that are no longer expected to result in revenue are removed from backlog. Among other criteria, to consider a transaction to be in backlog we must possess an outstanding and effective written agreement for the delivery of a MRIdian signed by a customer and receipt of a minimum customer deposit or a letter of credit except when the sale is to a customer where a deposit is deemed not necessary or customary (i.e. sale to a government entity, a large hospital, group of hospitals or a cancer care group that has sufficient credit, sales via tender awards, or indirect channel sales that have signed contracts with end-customers). For removal of an order from our backlog, the following criteria are considered: any changes in customer or distributor plans or financial conditions; the customer’s or distributor’s continued intent and ability to fulfill the order contract; changes to regulatory requirements; the status of regulatory approval required in the customer’s jurisdiction, if any; and other reasons for potential cancellation of order contracts.
During the year ended December 31, 2017, 2016 and 2015, our new orders were $113.6 million, $77.0 million and $40.1 million respectively. At December 31, 2017 and 2016, we had backlog with a total value of $203.6 million and $133.2 million, respectively.
Components of Statements of Operations
Revenue
Product Revenue. Product revenue consists of sales of MRIdian systems, as well as optional components, such as additional planning workstations and body coils. Forfeited customer deposits from order cancellations are also included in product revenue.
82
Following execution of a sales contract, it generally takes nine to 12 months for a customer to customize an existing facility or construct a new vault. Upon the commencement of installation at a customer’s facility, it typically takes approximately ninety days to complete the installation and on-site testing of the system, including the completion of acceptance test procedures. On-site training takes approximately one week and can be conducted concurrently with installation and acceptance testing. Sales contracts generally include customer deposits upon execution of the agreement, and in certain cases, additional amounts due at shipment or commencement of installation, and final payment due generally upon customer acceptance.
Revenue recognition for MRIdian systems that we install generally occurs when the customer acknowledges that the system operates in accordance with standard product specifications, the customer accepts the installed unit and title and risk of loss are transferred to the customer. For sales of MRIdian systems that we are not responsible for installation, revenue is recognized when the entire system is delivered and title and risk of loss are transferred to the customer.
Service Revenue. We generally offer maintenance service at no cost to customers to cover parts, labor and maintenance for one to two years. In addition, we offer multi-year, post-installation maintenance and support contracts that provide various levels of service support, which enables our customers to select the level of on-going support services, including parts and labor, which they require. These post-installation contracts are for a period of one to five years and provide services ranging from 24/7 on-site parts and labor, and preventative maintenance to labor only with a longer response time. We also offer technology upgrades to our MRIdian systems, when and if available, for an additional fee. Service revenue is recognized on a straight-line basis over the term during which the contracted services are provided.
Distribution Rights Revenue. We entered into a distribution agreement with Itochu Corporation pursuant to which we appointed Itochu as our exclusive distributor for the promotion, sale and delivery of MRIdian products within Japan. In consideration of the exclusive distribution rights granted, we received $4.0 million which was recorded as deferred revenue and starting in August 2016 was recognized as distribution rights revenue on a straight-line basis over the remaining term of the distribution agreement, which expires in December 2024.
Cost of Revenue
Product Cost of Revenue. Product cost of revenue primarily consists of the cost of materials, installation and services associated with the manufacture and installation of MRIdian systems, as well as medical device excise tax and royalty payments to the University of Florida Research Foundation. Product cost of revenue also includes lower of cost or market inventory, or LCM, adjustments if the carrying value of the inventory is greater than its net realizable value. For strategic reasons, we initially sold our MRIdian systems prior to December 31, 2015 at prices lower than our projected costs to manufacture and install. As we accumulated materials, installation and other costs for these systems, we regularly assessed the carrying value of the related inventory value and recorded charges, or LCM adjustments, to reduce inventory to the lower of cost and net realizable value. The remaining realizable value of inventory was charged to product cost of revenue as those initial sites were completed and accepted. This resulted in LCM charges of $0.9 million, $1.9 million and $2.6 million for the year ended December 31, 2017, 2016 and 2015, respectively.
We expect our materials, installation and service costs to decrease as we continue to scale our operations, improve product designs and work with our third-party suppliers to lower costs. We expect to continue to lower costs and increase sales prices as we transition to the MRIdian Linac.
Service Cost of Revenue. Service cost of revenue is comprised primarily of personnel costs, training and travel expenses to service and maintenance of installed MRIdian systems. Service cost of revenue also includes the costs of replacement parts under maintenance and support contracts.
Operating Expenses
Research and Development. Research and development expenses consist primarily of compensation and related costs for personnel, including stock-based compensation, employee benefits and travel. Other significant research
83
and development costs arise from third-party consulting services, laboratory supplies, research materials, medical equipment, computer equipment and licensed technology, and related depreciation and amortization. We expense research and development expenses as incurred. As we continue to invest in improving MRIdian and developing new technologies, we expect our research and development expenses to increase.
Selling and Marketing. Selling and marketing expenses consist primarily of compensation and related costs for our direct sales force, sales management, and marketing and customer support personnel, and include stock-based compensation, employee benefits and travel expenses. Selling and marketing expenses also include costs related to trade shows and marketing programs. We expense selling and marketing costs as incurred. We expect selling and marketing expenses to increase in future periods as we expand our sales force and our marketing and customer support organizations and increase our participation in trade shows and marketing programs.
General and Administrative. Our general and administrative expenses consist primarily of compensation and related costs for our operations, finance, human resources, regulatory, and other administrative personnel, and include stock-based compensation, employee benefits and travel expenses. In addition, general and administrative expenses include third-party consulting, legal, audit, accounting services, quality and regulatory functions and facilities costs, and gain or loss on the disposal of property and equipment. We expect our general and administrative expenses to increase as our business grows and as we invest in the development of our MRIdian Linac.
Interest Income
Interest income consists primarily of interest income received on our cash and cash equivalents.
Interest Expense
Interest expense consists primarily of interest and amortization of the debt discount related to our long-term debt entered in 2013 from Hercules Technology III, L.P. and Hercules Technology Growth Capital, Inc., or together, Hercules and long-term debt entered in 2015 from Capital Royalty II L.P., Capital Royalty Partners II—Parallel Fund “A” L.P., Capital Royalty Partners II (Cayman) L.P. and Parallel Investment Opportunities Partners II L.P., or together with their successors by assignment, CRG, and such loan the CRG Term Loan.
Other Expense, Net
Other expense, net consists primarily of changes in the fair value of the 2017 and 2016 Placement Warrants and a convertible preferred stock warrant, as well as foreign currency exchange gains and losses.
The outstanding 2017 and 2016 Placement Warrants are re-measured to fair value at each balance sheet date with the corresponding gain or loss from the change in fair value of warrant liabilities recorded as a component of other expense, net.
When the convertible preferred stock warrants were outstanding, the warrants were re-measured to fair value at each balance sheet date with the corresponding gain or loss from the change in fair value of warrant liabilities recorded as a component of other expense, net. In July 2015, upon the closing of the Merger, the convertible preferred stock warrants were converted into warrants to purchase common stock. The aggregate fair value of the convertible preferred stock warrants, upon the closing of the Merger, was reclassified from liabilities to additional paid-in-capital, a component of stockholders’ equity (deficit), and we no longer recorded the change in fair value adjustments.
84
Results of Operations
The following tables set forth our results of operations for the periods presented (in thousands):
| | Year Ended December 31, | |
| | 2017 | | | 2016 | | | 2015 | |
Revenue: | | | | | | | | | | | | |
Product | | $ | 30,458 | | | $ | 20,555 | | | $ | 9,620 | |
Service | | | 3,109 | | | | 1,504 | | | | 530 | |
Distribution rights | | | 475 | | | | 178 | | | | — | |
Grant | | | — | | | | — | | | | 240 | |
Total revenue | | | 34,042 | | | | 22,237 | | | | 10,390 | |
Cost of revenue: | | | | | | | | | | | | |
Product | | | 25,488 | | | | 23,897 | | | | 12,673 | |
Service | | | 2,222 | | | | 1,969 | | | | 1,871 | |
Total cost of revenue | | | 27,710 | | | | 25,866 | | | | 14,544 | |
Gross margin | | | 6,332 | | | | (3,629 | ) | | | (4,154 | ) |
Operating expenses: | | | | | | | | | | | | |
Research and development | | | 14,709 | | | | 11,442 | | | | 10,449 | |
Selling and marketing | | | 8,412 | | | | 5,601 | | | | 5,139 | |
General and administrative | | | 31,375 | | | | 23,503 | | | | 21,685 | |
Total operating expenses: | | | 54,496 | | | | 40,546 | | | | 37,273 | |
Loss from operations | | | (48,164 | ) | | | (44,175 | ) | | | (41,427 | ) |
Interest income | | | 5 | | | | 2 | | | | 2 | |
Interest expense | | | (7,247 | ) | | | (5,951 | ) | | | (3,452 | ) |
Other expense, net | | | (16,770 | ) | | | (512 | ) | | | (117 | ) |
Loss before provision for income taxes | | | (72,176 | ) | | | (50,636 | ) | | | (44,994 | ) |
Provision for income taxes | | | — | | | | — | | | | 1 | |
Net loss | | $ | (72,176 | ) | | $ | (50,636 | ) | | $ | (44,995 | ) |
Comparison of the years ended December 31, 2017 and 2016
Revenue
| | Year Ended December 31, | | | | | |
| | 2017 | | | 2016 | | | Change | |
| | (in thousands) | | | | | |
Product | | $ | 30,458 | | | $ | 20,555 | | | $ | 9,903 | |
Service | | | 3,109 | | | | 1,504 | | | | 1,605 | |
Distribution rights | | | 475 | | | | 178 | | | | 297 | |
Total revenue | | $ | 34,042 | | | $ | 22,237 | | | $ | 11,805 | |
Total revenue during the year ended December 31, 2017 increased $11.8 million compared to the year ended December 31, 2016. The increase was primarily due to revenue from six MRIdian systems during the year ended December 31, 2017, compared to revenue from four MRIdian systems during the year ended December 31, 2016.
Product Revenue. Product revenue increased $9.9 million in fiscal 2017 compared to fiscal 2016. The increase is primarily due to the revenue recognized from six MRIdian systems in fiscal 2017 compared to four units in fiscal 2016.
Service Revenue. Service revenue increased $1.6 million in fiscal 2017 compared to fiscal 2016 due to increased billings to existing customers, as well as the increased install base.
85
Distribution Rights Revenue. Distribution rights revenue increased $0.3 million in fiscal 2017 compared to fiscal 2016. After receipt of Japanese regulatory approval in August 2016, we started recognizing the distribution rights revenue on a straight-line basis over the remaining term of the distribution agreement with Itochu. The increase was due to recognition of revenue for twelve months in fiscal 2017 compared to four and half months for fiscal 2016.
Cost of Revenue
| | Year Ended December 31, | | | | | |
| | 2017 | | | 2016 | | | Change | |
| | (in thousands) | | | | | |
Product | | $ | 25,488 | | | $ | 23,897 | | | $ | 1,591 | |
Service | | | 2,222 | | | | 1,969 | | | | 253 | |
Total cost of revenue | | $ | 27,710 | | | $ | 25,866 | | | $ | 1,844 | |
Product Cost of Revenue. Product cost of revenue increased $1.6 million in fiscal 2017 compared to fiscal 2016. The increase was primarily due to costs of six MRIdian Linac systems in fiscal 2017 compared to costs of four MRIdian with Cobalt-60 in fiscal 2016. The increase was partially offset by the lower cost for MRIdian Linac systems.
Service Cost of Revenue. Service cost of revenue increased $0.3 million in fiscal 2017 compared to fiscal 2016. The increase in service cost of revenue was primarily due to service provided to more installed units in fiscal 2017.
Operating Expenses
| | Year Ended December 31, | | | | | |
| | 2017 | | | 2016 | | | Change | |
| | (in thousands) | | | | | |
Research and development | | $ | 14,709 | | | $ | 11,442 | | | $ | 3,267 | |
Selling and marketing | | | 8,412 | | | | 5,601 | | | | 2,811 | |
General and administrative | | | 31,375 | | | | 23,503 | | | | 7,872 | |
Total operating expenses | | $ | 54,496 | | | $ | 40,546 | | | $ | 13,950 | |
Research and Development. Research and development expenses increased $3.3 million, or 28.5% in fiscal 2017 compared to fiscal 2016. This increase was primarily attributable to a $1.4 million increase in engineering and research expense and projects supplies, a $1.0 million increase in consulting and contract labor expense due to increased usage of consultants and contractors, and a $0.7 million increase in personnel costs due to higher average headcount in fiscal 2017.
Selling and Marketing. Selling and marketing expenses increased $2.8 million, or 50.2% in fiscal 2017 compared to fiscal 2016. This increase was primarily attributable to a $1.4 million increase in trade show costs, a $1.1 million increase in personnel expense due to higher average headcount in fiscal 2017, and a $0.2 million increase in travel expense.
General and Administrative. General and administrative expenses increased $7.9 million, or 33.5% in fiscal 2017 compared to fiscal 2016. This increase was primarily attributable to a $4.0 million increase in personnel and related costs due to higher average headcount, a $2.3 million increase in consulting and contract labor expense, a $0.6 million increase in depreciation expense, and a $0.6 million increase in travel and other general expenses.
86
Interest Expense
| | Year Ended December 31, | | | | | |
| | 2017 | | | 2016 | | | Change | |
| | (in thousands) | | | | | |
Interest expense | | $ | (7,247 | ) | | $ | (5,951 | ) | | $ | (1,296 | ) |
Interest expense increased $1.3 million in fiscal 2017, due primarily to higher outstanding CRG loan balances in fiscal 2017.
Other Expense, Net
| | Year Ended December 31, | | | | | |
| | 2017 | | | 2016 | | | Change | |
| | (in thousands) | | | | | |
Other expense, net | | $ | (16,770 | ) | | $ | (512 | ) | | $ | (16,258 | ) |
Other expense, net for fiscal 2017 consisted primarily of a $16.6 million change in fair value of warrant liability related to the 2017 and 2016 Placement Warrants. Other expense, net for fiscal 2016 consisted primarily of a $0.4 million loss on disposal of fixed assets.
Comparison of the Years Ended December 31, 2016 and 2015
Revenue
| | Year Ended December 31, | | | | | |
| | 2016 | | | 2015 | | | Change | |
| | (in thousands) | | | | | |
Product | | $ | 20,555 | | | $ | 9,620 | | | $ | 10,935 | |
Service | | | 1,504 | | | | 530 | | | | 974 | |
Distribution rights | | | 178 | | | | — | | | | 178 | |
Grant | | | — | | | | 240 | | | | (240 | ) |
Total revenue | | $ | 22,237 | | | $ | 10,390 | | | $ | 11,847 | |
Total revenue during the year ended December 31, 2016 increased $11.8 million compared to the year ended December 31, 2015. The increase was primarily due to revenue from four MRIdian systems during the year ended December 31, 2016, compared to revenue from two MRIdian systems during the year ended December 31, 2015.
Product Revenue. Product revenue increased $10.9 million in fiscal 2016 compared to fiscal 2015. The increase is due to the revenue recognized from four units of MRIdian systems in fiscal 2016 compared to two units in fiscal 2015.
Service Revenue. Service revenue increased $1.0 million in fiscal 2016 compared to fiscal 2015 due to increased install base, which was seven MRIdian systems worldwide in fiscal 2016 compared to five MRIdian systems in 2015.
Distribution Rights Revenue. Distribution rights revenue increased $0.2 million in fiscal 2016 compared to fiscal 2015. This increase was due to receipt of Japanese regulatory approval in August 2016, after which we started recognizing the distribution rights revenue on a straight-line basis over the remaining term of the distribution agreement with Itochu.
Grant Revenue. Grant revenue decreased $0.2 million in fiscal 2016 compared to fiscal 2015. This decrease was due to our note payable to the county redevelopment fund in the State of Ohio being forgiven based on meeting certain employment requirements in fiscal 2015, while no such revenue was earned in fiscal 2016.
87
Cost of Revenue
| | Year Ended December 31, | | | | | |
| | 2016 | | | 2015 | | | Change | |
| | (in thousands) | | | | | |
Product | | $ | 23,897 | | | $ | 12,673 | | | $ | 11,224 | |
Service | | | 1,969 | | | | 1,871 | | | | 98 | |
Total cost of revenue | | $ | 25,866 | | | $ | 14,544 | | | $ | 11,322 | |
Product Cost of Revenue. Product cost of revenue increased $11.2 million in fiscal 2016 compared to fiscal 2015. The increase was primarily due to costs of four units of MRIdian systems in fiscal 2016 compared to costs of two units of MRIdian systems in fiscal 2015.
Service Cost of Revenue. Service cost of revenue increased $0.1 million in fiscal 2016 compared to fiscal 2015. The increase in service cost of revenue was due to the provision of services for the MRIdian systems installed at VU University Medical Center, Netherlands, beginning in April 2016.
Operating Expenses
| | Year Ended December 31, | | | | | |
| | 2016 | | | 2015 | | | Change | |
| | (in thousands) | | | | | |
Research and development | | $ | 11,442 | | | $ | 10,449 | | | $ | 993 | |
Selling and marketing | | | 5,601 | | | | 5,139 | | | | 462 | |
General and administrative | | | 23,503 | | | | 21,685 | | | | 1,818 | |
Total operating expenses | | $ | 40,546 | | | $ | 37,273 | | | $ | 3,273 | |
Research and Development. Research and development expenses increased $1.0 million, or 9.5% in fiscal 2016 compared to fiscal 2015. This increase was primarily attributable to a $1.2 million increase in personnel costs due to increased wages and stock-based compensation, and a $0.8 million increase in engineering and research expenses as a result of increased emphasis on research and development projects, partially offset by a $1.0 million decrease in consulting and contract labor expense due to less consultants and contractors used in 2016.
Selling and Marketing. Selling and marketing expenses increased $0.5 million, or 9.0% in fiscal 2016 compared to fiscal 2015. This was a result of an increase of $0.4 million in trade show costs and an increase of $0.3 million in marketing consulting fees and contract labor, partially offset by a $0.2 million decrease in travel expenses, and a $0.1 million decrease in personnel expense due to reduced headcount from ten to nine employees.
General and Administrative. General and administrative expenses increased $1.8 million, or 8.4% in fiscal 2016 compared to fiscal 2015. This increase was primarily attributable to a $3.6 million increase in personnel and related costs due to stock-based compensation and salary increase, a $0.5 million increase in business insurance expense, a $0.3 million increase in accounting and legal fees related to patent and intellectual property as well as public company related SEC expenses, a $0.2 million increase in facility expense as a result of increased utilities expense, partially offset by a $2.9 million write-off of deferred offering costs in June 2015.
Interest Expense
| | Year Ended December 31, | | | | | |
| | 2016 | | | 2015 | | | Change | |
| | (in thousands) | | | | | |
Interest expense | | $ | (5,951 | ) | | $ | (3,452 | ) | | $ | (2,499 | ) |
Interest expense increased $2.5 million in fiscal 2016, due primarily to a higher loan balance from the additional $15.0 million draw down in May 2016 as well as the higher effective interest rate in 2016.
88
Other Expense, Net
| | Year Ended December 31, | | | | | |
| | 2016 | | | 2015 | | | Change | |
| | (in thousands) | | | | | |
Other expense, net | | $ | (512 | ) | | $ | (117 | ) | | $ | (395 | ) |
Other expense, net changed $0.4 million in fiscal 2016, due primarily to loss on disposal of fixed assets.
Liquidity and Capital Resources
Since our inception in 2004, we have incurred significant net losses and negative cash flows from operations. During the years ended December 31, 2017, 2016 and 2015, we had net loss of $72.2 million, $50.6 million and $45.0 million, respectively. At December 31, 2017 and 2016, we had an accumulated deficit of $319.9 and $247.7 million, respectively.
At December 31, 2017 and 2016, we had cash and cash equivalents of $57.4 and $14.2 million, respectively. To date, we have financed our operations principally through placements of our capital stock, issuances of convertible promissory notes, issuances of term loans and receipts of customer deposits for new orders and payments from customers for systems installed. We may, from time to time, seek to raise capital through a variety of sources including the public equity market, private equity financing, and/or public or private debt. In May 2016, we drew down the additional $15.0 million in funds from the CRG term loan. In August and September 2016, we issued common stock and warrants to purchase common stock via the 2016 Private Placement for gross proceeds of $13.8 million. In January 2017, we issued additional common stock and warrants to purchase common stock via the January 2017 Private Placement for gross proceeds of $26.1 million. During fiscal 2017, we also raised aggregate gross proceeds of $40.1 million through our at-the-market offering program in which we sold 6.6 million shares of our common stock at an average sale price of $6.10 per share. In October 2017, we issued more common stock via the October 2017 Direct Registered Offering for gross proceeds of $49.9 million. We expect that our existing cash and cash equivalents, together with cash receipts from sales of MRIdian systems and the plan to raise additional funds from various sources from time to time will enable us to conduct our planned operations for at least the next 12 months.
We could potentially use our available financial resources sooner than we currently expect, and we may incur additional indebtedness to meet future financing needs. Adequate additional funding may not be available to us on acceptable terms or at all. In addition, although we anticipate being able to obtain additional financing, we may be unable to do so. Our failure to raise capital as and when needed could have significant negative consequences for our business, financial condition and results of operations. Our future capital requirements and the adequacy of available funds will depend on many factors, including those set forth in the section titled “Risk Factors.”
The following table summarizes our cash flows for the periods presented (in thousands):
| | Year Ended December 31, | |
| | 2017 | | | 2016 | | | 2015 | |
Cash used in operating activities | | $ | (70,053 | ) | | $ | (28,156 | ) | | $ | (39,849 | ) |
Cash used in investing activities | | | (2,163 | ) | | | (7,243 | ) | | | (4,145 | ) |
Cash provided by financing activities | | | 115,407 | | | | 28,930 | | | | 53,532 | |
Operating Activities
We have historically experienced negative cash outflows as we developed MRIdian with Cobalt-60, MRIdian Linac and expanded our business. Our primary source of cash flow from operating activities is cash receipts from customers including sales of MRIdian systems and, to a lesser extent, up-front payments from customers. Our primary uses of cash from operating activities are amounts due to vendors for purchased components and employee-related expenditures.
During fiscal 2017, cash used in operating activities was $70.1 million as a result of our net loss of $72.2 million and a $26.3 million net change in our operating assets and liabilities, partially offset by aggregated non-cash charges
89
of $28.4 million. The net change in our operating assets and liabilities was primarily a result of an increase in inventory and deposits on purchased inventory, an increase in accounts receivable, an increase in deferred cost of revenue and an increase in prepaid expenses and other assets, partially offset by an increase in customer deposits and deferred revenue, an increase in accounts payable and an increase in accrued expenses and other long-term liabilities. Inventory and deposits on purchased inventory increased $12.3 million and $4.5 million, respectively, in anticipation of upcoming shipments and installation of MRIdian Linac systems. Deferred cost of revenue increased $9.8 million due to the shipment of additional components for MRIdian Linac systems currently being installed. Prepaid expenses and other assets increased $2.0 million, primarily attributable to deferred sales commission on new sales contracts and prepaid insurance premium. The $16.2 million increase in accounts receivable resulted primarily from the timing of collection from shipment and installation of four units of MRIdian Linac in the last quarter of fiscal 2017. The net change in our operating assets and liabilities were partially offset by $11.4 million increase in customer deposits and deferred revenue mainly due to installation in progress. The $6.3 million increase in accounts payable resulted from the timing of payment. The $0.8 million increase in accrued expenses and other long-term liabilities was due primarily to the timing of invoice receipts for services and inventory purchased. Non-cash charges included a $16.6 million change in the fair value of warrant liability related to the 2016 and 2017 Placement Warrants, $3.3 million of amortization of debt discount and interest accrual related to the CRG Term Loan, $5.3 million of stock-based compensation, $2.2 million of depreciation and amortization expense as well as $0.9 million due to LCM adjustments related to the reduction of the carrying value of inventory to its net realizable value.
During fiscal 2016, cash used in operating activities was $28.2 million primarily as a result of our net loss of $50.6 million, partially offset by $12.9 million net increase in our operating assets and liabilities and aggregate non-cash charges of $9.5 million. The net change in our operating assets and liabilities was primarily the result of an increase in customer deposits, deferred revenue and accrued expenses, and a decrease in deferred cost of revenue and deposits on purchased inventory components, offset by an increase in accounts receivable, inventory and prepaid expenses and other current assets. The $11.1 million increase in customer deposits and deferred revenue was the result of 13 new sales contracts and the receipt of payment from Itochu Corporation related to the distribution agreement during the year ended December 31, 2016. The $4.9 million decrease in deferred cost of revenue and the $1.4 million decrease in deposits on purchased inventory components was due to the recognition of four MRIdian systems sales during the year ended December 31, 2016. The $2.2 million increase in accrued expenses is attributable to the timing of invoice receipts for services and inventory purchased, as well as accrued bonuses. The net increase in our operating assets and liabilities was partially offset by a $3.4 million increase in accounts receivable attributable to our increased sales, a $2.1 million increase in inventories due to upcoming shipments and installations of MRIdian systems, and a $1.7 million increase in prepaid expenses and other current assets due to prepayments made for deferred sales commission on new sales contracts. Non-cash charges primarily included $2.9 million of stock-based compensation, $2.6 million due to amortization of debt discount and interest accrual related to the CRG Term Loan, $1.9 million due to LCM adjustments related to the reduction of carrying value of inventory to its net realizable value, and $1.7 million depreciation and amortization expense.
During fiscal 2015, cash used in operating activities was $39.8 million, primarily as a result of our net loss of $45.0 million and $3.7 million net change in our operating assets and liabilities, partially offset by aggregate non-cash charges of $8.8 million. The net change in our operating assets and liabilities was primarily the result of an increase in deferred cost of revenue, purchase of inventory, a decrease in accounts payable and making prepaid payments on inventory components, offset by an increase in customer deposits and accrued expenses. The $4.1 million increase in deferred cost of revenue was the result of new MRIdian systems sales orders awaiting installation. This increase also resulted in an increase of $2.4 million in inventory. The $1.1 million increase in deposits on purchased inventory was due to the growth in our business. The decrease of $2.1 million in accounts payable was primarily due to the timing of payments as a result of the growth in our business. The decrease in our operating assets and liabilities was partially offset by $5.3 million increase in customer deposits and deferred revenue, primarily due to new sales contracts, and a $1.5 million increase in accrued expenses attributable to higher accrued inventory purchase. Non-cash charges primarily included $2.9 million for write-off of deferred offering costs, $2.6 million of inventory lower of cost or market charges related to the expected MRIdian system installation in Miami, $1.3 million of depreciation and amortization charges, $1.1 million for amortization of debt discount and accrued interest related to our debt incurred in December 2013 and June 2015, and $1.1 million of stock-based compensation.
90
Investing Activities
Cash used in investing activities for fiscal 2017 of $2.2 million primarily resulted from capital expenditures to purchase property and equipment.
Cash used in investing activities for fiscal 2016 of $7.2 million primarily resulted from capital expenditures to purchase property and equipment.
Cash used in investing activities for fiscal 2015 of $4.1 million primarily resulted from capital expenditures to purchase property and equipment.
Financing Activities
Cash provided by financing activities for fiscal 2017 of $115.4 million primarily from $49.9 million gross proceeds from the October 2017 Direct Registered Offering, $26.1 million gross proceeds from the January 2017 Private Placement, $40.1 million gross proceeds from our at-the-market offering program, $0.7 million from the exercise of stock options, and $0.1 million from the exercise of warrants, partially offset by offering costs of $1.2 million for our at-the-market offering program and offering costs of $0.4 million for our October 2017 Direct Registered Offering, January 2017 Private Placement and the 2016 Private Placement.
Cash provided by financing activities for fiscal 2016 of $28.9 million primarily resulted from the net proceeds of $15.0 million related to the additional CRG draw down, net proceeds of $13.4 million related to the 2016 Private Placement and $0.5 million from the exercise of stock options.
Cash provided by financing activities for fiscal 2015 of $53.5 million primarily resulted from the net proceeds of $28.9 million related to the draw-down of long-term debt, net of debt issuance cost, the net proceeds of $26.6 million from the Private Placement and the net proceeds of $15.7 million from issuance of Series C convertible preferred stock, which was partially offset by repayments of a term loan of $15.0 million and payments of $2.7 million for costs related to our originally planned initial public offering.
Hercules Term Loan
On June 26, 2015, we paid off in full the $15.0 million outstanding term debt with Hercules using part of the proceeds received from the CRG Term Loan.
CRG Term Loan
In June 2015, we entered the CRG Term Loan for up to $50.0 million, of which $30.0 million was made available to us upon closing with the remaining $20.0 million to be available on or before June 26, 2016 upon meeting certain milestones. We drew down the first $30.0 million on the closing date in June 2015. In March 2016, the CRG Term Loan was amended with regard to the conditions for borrowing the remaining $20.0 million available under the CRG Term Loan. We achieved one milestone at March 31, 2016 and borrowed an additional $15.0 million in May 2016. In April 2017, we executed an amendment to the CRG Term Loan, which included an extension to the availability of the existing $5.0 million tranche at ViewRay’s option through June 30, 2017, added a $15.0 million tranche of borrowing capacity available at ViewRay’s option through September 30, 2017, extended the interest-only and payment in-kind period, decreased the combined 2016 and 2017 revenue covenant and included a 1.75% increase to the facility fee. We did not draw down any amount under the $5.0 million tranche and it has since expired. In October 2017, we executed another amendment to the CRG Term Loan, extending the availability of the existing $15.0 million borrowing capacity through December 31, 2017. In February 2018, the CRG Term Loan was amended to decrease the amount of the minimum combined 2016 and 2017 revenue covenant effective December 31, 2017. We did not draw down any amount under the $15 million tranche and it has since expired. The CRG Term Loan is subject to financial covenants and is collateralized by essentially all our assets and limits our ability with respect to additional indebtedness, investments or dividends, among other things, subject to customary exceptions.
91
At December 31, 2017, we had $45.0 million in outstanding debt to CRG, which is repayable through June 26, 2020. The CRG Term Loan bears cash interest at a rate of 12.5% per annum and has an interest-payment-only period through March 31, 2020. We were in compliance with all financial covenants under the CRG Term Loan at December 31, 2017. Additional details regarding the CRG Term Loan are included in the section entitled “Notes to Consolidated Financial Statements – Note 6 – Debt” in the consolidated financial statements included elsewhere in this Form 10-K.
2016 Private Placement
On August 19, 2016, we entered into a Securities Purchase Agreement pursuant to which we sold an aggregate of 5,983,251 shares of common stock which consists of 4,602,506 shares of common stock and warrants to purchase 1,380,745 shares of common stock, or the 2016 Placement Warrants, for aggregate proceeds of $13.2 million, net of offering cost, or the 2016 Private Placement. We completed the initial closing of the 2016 Private Placement on August 22, 2016 with the final closing on September 9, 2016. The 2016 Placement Warrants have an exercise price of $2.95 per share, are exercisable at any time at the option of the holder and expire seven years from the date of issuance.
January 2017 Private Placement
In January 2017, we entered into a Securities Purchase Agreement pursuant to which we sold an aggregate of 10,323,101 shares of common stock which consists of 8,602,589 shares of common stock and warrants to purchase 1,720,512 shares of common stock, or the 2017 Placement Warrants, for total gross proceeds of $26.1 million, or the January 2017 Private Placement. We completed the closing of the January 2017 Private Placement on January 18, 2017. The 2017 Placement Warrants have a per share exercise price of $3.17 per share, are exercisable after six months and expire seven years from the date of issuance.
At-The-Market Offering of Common Stock
In January 2017, we entered into a sales agreement (the “ATM Sales Agreement”) with FBR Capital Markets & Co., or FBR, under which we may sell up to $25.0 million of our common shares pursuant to an at-the-market offering program in accordance with Rule 415(a)(4) under the Securities Act. FBR acted as sales agent on a best efforts basis and used commercially reasonable efforts to sell on our behalf all of the shares of common stock requested to be sold by us, consistent with its normal trading and sales practices, on mutually agreed terms between FBR and us. There is no arrangement for funds to be received in any escrow, trust or similar arrangement. In April 2017, we agreed to sell up to an additional $25.0 million of our common stock in accordance with the terms of the ATM Sales Agreement with FBR and pursuant to an at-the-market offering program in accordance with Rule 415(a)(4) under the Securities Act.
FBR is entitled to compensation of up to 3.0% of the gross sales price per share sold. We have also agreed to provide indemnification and contribution to FBR with respect to certain liabilities, including liabilities under the Securities Act.
At December 31, 2017, we had sold an aggregate of approximately 6.6 million shares of our common stock at an average market price of $6.10 per share under the at-the-market offering program, resulting in aggregate gross proceeds of approximately $40.1 million.
October 2017 Direct Registered Offering
In October 2017, we entered into Securities Purchase Agreements pursuant to which we sold an aggregate of 8,382,643 shares of common stock for total gross proceeds of $49.9 million, or the October 2017 Direct Registered Offering. We completed the closing of the October 2017 Direct Registered Offering on October 25, 2017.
92
Contractual Obligations
The following summarizes our contractual obligations at December 31, 2017, and the effect such obligations are expected to have on our liquidity and cash flow over the next five years (in thousands):
| | Payment due by period | |
| | Total | | | Less than 1 year | | | 1-3 years | | | 3-5 years | | | More than 5 years | |
CRG Term Loan (1) | | | 45,000 | | | | - | | | | 45,000 | | | | | | | | | |
Interest on CRG Term Loan (1) | | | 26,210 | | | | 4,108 | | | | 22,102 | | | | | | | | | |
Operating leases (2) | | | 2,157 | | | | 1,118 | | | | 1,039 | | | | | | | | | |
Total | | | 73,367 | | | | 5,226 | | | | 68,141 | | | | - | | | | - | |
| (1) | Refer to “Note 6. Debt” |
| (2) | Refer to “Note 7. Commitments and Contingencies” |
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements as of December 31, 2017.
Critical Accounting Policies and Estimates
Our consolidated financial statements are prepared in accordance with generally accepted accounting principles in the United States, or GAAP. The preparation of these consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenue, expenses and related disclosures. We evaluate our estimates and assumptions on an ongoing basis. Our estimates are based on historical experience and various other assumptions that we believe to be reasonable under the circumstances. Our actual results could differ from these estimates.
We believe that the following assumptions and estimates have the greatest potential impact on our consolidated financial statements. Therefore, we consider these to be our critical accounting policies and estimates. For further information on all of our significant accounting policies, see the notes to our consolidated financial statements.
Revenue Recognition
Revenue recognition for systems that we install generally occurs when the customer acknowledges that the system operates in accordance with our standard product specifications, the customer accepts the installed unit and we transfer title and risk of loss to the customer. For sales of MRIdian systems that we are not responsible for installation, revenue is recognized when the entire system is delivered and title and risk of loss are transferred to the customer as qualifying distributors and third-party certified technicians are readily available to perform the installation. Service revenue is recognized on a straight-line basis over the term during which contracted services are provided. We use judgment to estimate revenue allocations from sales arrangements with multiple deliverables between the product and service revenue. In situations where a deliverable in a multi-element arrangement has a value to the customer on a stand-alone basis, we are required to allocate the fair value of the various elements based on the selling price of each element. The principal deliverables consist of (i) sales of MRIdian systems, which generally includes installation, site preparation and software, and (ii) product support, which includes extended service and maintenance. We determine selling prices using vendor specific objective evidence, or VSOE, if it exists, or third-party evidence, or TPE. If neither VSOE nor TPE exists for a deliverable, we use best estimated selling price, or BESP. We allocate revenue to multiple elements generally using the relative fair values as determined by BESP. We regularly review VSOE, TPE and BESP for all of our products and services.
We have, in the past, received payments for cost reimbursement of allowable expenditures and payments for the achievement of certain milestones under government grants in return for qualifying property and equity purchases and research and development activities over a contractually defined period. These payments are nonrefundable. Government grants generally provide us with fixed payments and a contractually defined period of research. Grant
93
revenues were recognized as associated expenses incurred and are billed to grantors in conjunction with the terms of the grants. We do not anticipate grant revenue in the future.
Stock-Based Compensation
Stock-based compensation expense is measured and recognized in the consolidated financial statements based on the fair value of the awards granted. The fair value of each option award is estimated on the grant date using the Black-Scholes option-pricing model. The fair value of Restricted Stock Units, or RSUs, is based on the closing market price of the Company’s common stock on the grant date. Stock-based compensation expense is recognized, net of forfeitures, over the requisite service periods of the awards, which is generally four years. At December 31, 2017, total unrecognized compensation cost related to stock-based awards granted to employees, net of estimated forfeitures, was $9.9 million which is expected to be recognized over a weighted-average period of 2.7 years.
Our use of the Black-Scholes option-pricing model requires the input of highly subjective assumptions, including the options’ expected term and price volatility of the underlying stock. The assumptions used in our option-pricing model represent management’s best estimates. These estimates involve inherent uncertainties and the application of management’s judgment. If factors change and different assumptions are used, our stock-based compensation expense could be materially different in the future.
Common Stock Warrant
In December 2013 in connection with the Hercules Term Loan, we issued a warrant to purchase 128,231 shares of our preferred stock with an exercise price of $5.84 per share, subject to certain adjustments. These warrants were converted to a warrant to purchase our common stock upon the closing of the Merger in July 2015. This warrant is exercisable in whole or in part at any time prior to the expiration date of the warrant, which is the later of (i) December 16, 2023 and (ii) the date that is five years following the effective date of the registration statement of an initial underwritten public offering of our common stock.
Prior to the Merger, the preferred stock warrant was recorded as preferred stock warrant liability and adjusted to fair value at each balance sheet date, with the change in fair value being recorded as a component of other expense, net in the consolidated statements of operations and comprehensive loss.
Upon the closing of the Merger on July 23, 2015, all shares of Series C convertible preferred stock were converted into common stock, and the warrant to purchase Series C convertible preferred stock was converted into the warrant to purchase 128,231 shares of our common stock. Fair value of these warrants at the closing date were reclassified into additional paid-in capital, and we no longer recorded changes in fair value of the converted common stock warrants.
In connection with the Merger and the Private Placement, we issued 198,760 shares of common stock warrants at an exercise price of $5.00 per share to private placement agents as payment for services provided. These placement warrants are exercisable at any time at the option of the holder until the five-year anniversary of its date of issuance. These warrants were accounted for as equity awards.
In connection with the 2016 Private Placement, we issued 1,380,745 shares of common stock warrants at an exercise price of $2.95 per share and these warrants are exercisable at any time at the option of the holder and expire seven years from the date of issuance. In connection with the January 2017 Private Placement, we issued 1,720,512 shares of common stock warrants at an exercise price of $3.17 per share and these warrants are exercisable after six months and expire seven years from the date of issuance. The 2016 and 2017 Placement Warrants were accounted for as a liability with subsequent changes in fair value recorded in other expenses, net at each reporting date until the warrants are exercised or expired.
Inventory Valuation
Inventory consists primarily of purchased components for assembling MRIdian systems and other direct costs associated with MRIdian system installation. Inventory is stated at the lower of cost or market value. When the net
94
realizable value of the inventory is lower than related costs, we reduce the carrying value of the inventory for the difference while recording a corresponding charge to cost of product revenues. The assumptions we used in estimating the net realizable value of the inventory primarily include the total cost to complete the applicable MRIdian system. We recorded an inventory lower of cost and market adjustment of $0.9 million, $1.9 million and $2.6 million during the years ended December 31, 2017, 2016 and 2015, respectively.
Prior to January 1, 2015, our inventory cost was measured on a first-in, first-out basis through specific identification. To support the increasing MRIdian system installations and inventory purchase activities, starting January 1, 2015, we elected to change inventory cost measurement to weighted average basis. The accounting principle change does not have an impact on prior periods’ financial statements, therefore no retrospective adjustment is required. The accounting principle change does not have an impact on product cost of revenue or net loss for the year ended December 31, 2015.
Income Taxes
We are subject to income taxes in the United States, and we use estimates in determining our provision for income taxes. We use the asset and liability method of accounting for income taxes. Under this method, we calculate deferred tax asset or liability account balances at the balance sheet date using current tax laws and rates in effect for the year in which the differences are expected to affect our taxable income.
We estimate actual current tax exposure together with assessing temporary differences resulting from differences in accounting for reporting purposes and tax purposes for certain items, such as accruals and allowances not currently deductible for tax purposes. These temporary differences result in deferred tax assets and liabilities, which are included in our consolidated balance sheets. In general, deferred tax assets represent future tax benefits to be received when certain expenses previously recognized in our consolidated statements of operations and comprehensive loss become deductible expenses under applicable income tax laws or when net operating loss or credit carryforwards are utilized. Accordingly, realization of our deferred tax assets is dependent on future taxable income against which these deductions, losses and credit carryforwards can be utilized.
We assess the likelihood that our deferred tax assets will be recovered from future taxable income, and to the extent we believe that recovery is not likely, establish a valuation allowance. At December 31, 2017, 2016 and 2015, we have a full valuation allowance set up for our net deferred tax assets.
Under federal and similar state tax statutes, changes in our ownership, including ownership changes resulting from the Merger, may limit our ability to use our available net operating loss and tax credit carryforwards. The annual limitation, as a result of a change of ownership, may result in the expiration of net operating losses and credits before utilization. We believe we have experienced at least one ownership change in the past. We are currently analyzing the tax impact of such ownership change on our federal NOLs and credit carryforwards. Our ability to use our remaining net operating loss carryforwards may be further limited if we experience an ownership change or as a result of future changes in our stock ownership.
JOBS Act Accounting Election
We are an “emerging growth company” within the meaning of the JOBS Act. Section 107 of the JOBS Act provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. Thus, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies that are not emerging growth companies.
Recently Issued and Adopted Accounting Pronouncements
We review new accounting standards to determine the expected financial impact, if any, that the adoption of each such standard will have. For the recently issued accounting standards that we believe may have an impact on our
95
consolidated financial statements, see the section entitled “Notes to Consolidated Financial Statements – Note 2 – Summary of Significant Accounting Policies” in the consolidated financial statement.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Not applicable to smaller reporting companies.
96
Item 8. Consolidated Financial Statements and Supplementary Data
97
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of ViewRay, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of ViewRay, Inc. and its subsidiary (the "Company") as of December 31, 2017 and 2016, the related consolidated statements of operations and comprehensive loss, convertible preferred stock and stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the "financial statements"). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Deloitte & Touche LLP
San Francisco, CA
March 12, 2018
We have served as the Company's auditor since 2012.
98
VIEWRAY, INC.
Consolidated Balance Sheets
(In thousands, except share and per share data)
| | December 31, | |
| | 2017 | | | 2016 | |
ASSETS | | | | | | | | |
Current assets: | | | | | | | | |
Cash and cash equivalents | | $ | 57,389 | | | $ | 14,198 | |
Accounts receivable | | | 20,326 | | | | 4,200 | |
Inventory | | | 19,375 | | | | 8,082 | |
Deposits on purchased inventory | | | 7,043 | | | | 2,522 | |
Deferred cost of revenue | | | 13,696 | | | | 3,909 | |
Prepaid expenses and other current assets | | | 4,862 | | | | 3,023 | |
Total current assets | | | 122,691 | | | | 35,934 | |
Property and equipment, net | | | 11,564 | | | | 11,560 | |
Restricted cash | | | 1,143 | | | | 1,143 | |
Intangible assets, net | | | 78 | | | | 97 | |
Other assets | | | 235 | | | | 30 | |
TOTAL ASSETS | | $ | 135,711 | | | $ | 48,764 | |
LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | | | | | | | | |
Current liabilities: | | | | | | | | |
Accounts payable | | $ | 11,014 | | | $ | 4,980 | |
Accrued liabilities | | | 7,207 | | | | 6,334 | |
Customer deposits | | | 17,820 | | | | 19,400 | |
Deferred revenue, current portion | | | 20,151 | | | | 6,515 | |
Total current liabilities | | | 56,192 | | | | 37,229 | |
Deferred revenue, net of current portion | | | 3,238 | | | | 3,918 | |
Long-term debt | | | 44,504 | | | | 44,290 | |
Warrant liability | | | 22,420 | | | | 2,723 | |
Other long-term liabilities | | | 7,370 | | | | 4,257 | |
TOTAL LIABILITIES | | | 133,724 | | | | 92,417 | |
Commitments and contingencies (Note 7) | | | | | | | | |
Stockholders’ equity (deficit): | | | | | | | | |
Convertible preferred stock, par value $0.01 per share; 10,000,000 shares authorized at December 31, 2017 and 2016; no shares issued and outstanding at December 31, 2017 and 2016 | | | — | | | | — | |
Common stock, par value of $0.01 per share; 300,000,000 shares authorized at December 31, 2017 and 2016; 67,653,974 and 43,581,184 shares issued and outstanding at December 31, 2017 and 2016 | | | 666 | | | | 426 | |
Additional paid-in capital | | | 321,174 | | | | 203,598 | |
Accumulated deficit | | | (319,853 | ) | | | (247,677 | ) |
TOTAL STOCKHOLDERS’ EQUITY (DEFICIT) | | | 1,987 | | | | (43,653 | ) |
TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | | $ | 135,711 | | | $ | 48,764 | |
The accompanying notes are an integral part of these consolidated financial statements.
99
VIEWRAY, INC.
Consolidated Statements of Operations and Comprehensive Loss
(In thousands, except share and per share data)
| | Year Ended December 31, | |
| | 2017 | | | 2016 | | | 2015 | |
Revenue: | | | | | | | | | | | | |
Product | | $ | 30,458 | | | $ | 20,555 | | | $ | 9,620 | |
Service | | | 3,109 | | | | 1,504 | | | | 530 | |
Distribution rights | | | 475 | | | | 178 | | | | — | |
Grant | | | — | | | | — | | | | 240 | |
Total revenue | | | 34,042 | | | | 22,237 | | | | 10,390 | |
Cost of revenue: | | | | | | | | | | | | |
Product | | | 25,488 | | | | 23,897 | | | | 12,673 | |
Service | | | 2,222 | | | | 1,969 | | | | 1,871 | |
Total cost of revenue | | | 27,710 | | | | 25,866 | | | | 14,544 | |
Gross margin | | | 6,332 | | | | (3,629 | ) | | | (4,154 | ) |
Operating expenses: | | | | | | | | | | | | |
Research and development | | | 14,709 | | | | 11,442 | | | | 10,449 | |
Selling and marketing | | | 8,412 | | | | 5,601 | | | | 5,139 | |
General and administrative | | | 31,375 | | | | 23,503 | | | | 21,685 | |
Total operating expenses | | | 54,496 | | | | 40,546 | | | | 37,273 | |
Loss from operations | | | (48,164 | ) | | | (44,175 | ) | | | (41,427 | ) |
Interest income | | | 5 | | | | 2 | | | | 2 | |
Interest expense | | | (7,247 | ) | | | (5,951 | ) | | | (3,452 | ) |
Other expense, net | | | (16,770 | ) | | | (512 | ) | | | (117 | ) |
Loss before provision for income taxes | | $ | (72,176 | ) | | $ | (50,636 | ) | | $ | (44,994 | ) |
Provision for income taxes | | | — | | | | — | | | | 1 | |
Net loss and comprehensive loss | | $ | (72,176 | ) | | $ | (50,636 | ) | | $ | (44,995 | ) |
Net loss per share, basic and diluted | | $ | (1.23 | ) | | $ | (1.26 | ) | | $ | (2.58 | ) |
Weighted-average common shares used to compute net loss per share attributable to common stockholders, basic and diluted | | | 58,457,868 | | | | 40,068,307 | | | | 17,432,434 | |
The accompanying notes are an integral part of these consolidated financial statements.
100
VIEWRAY, INC.
Consolidated Statements of Convertible Preferred Stock and Stockholders’ Equity (Deficit)
(In thousands, except share data)
| | Convertible Preferred Stock | | | Common Stock | | | | | | | | | | | | | |
| | Shares | | | Amount | | | Shares | | | Amount | | | 'Additional Paid-in Capital | | | Accumulated Deficit | | | Total Stockholders’ Equity (Deficit) | |
Balance at January 1, 2015 | | | 27,654,928 | | | $ | 145,110 | | | | 907,037 | | | $ | 9 | | | $ | 1,414 | | | $ | (152,046 | ) | | $ | (150,623 | ) |
Issuance of common stock from option exercises | | | — | | | | — | | | | 31,427 | | | | — | | | | 24 | | | | — | | | | 24 | |
Stock-based compensation | | | — | | | | — | | | | — | | | | — | | | | 1,066 | | | | — | | | | 1,066 | |
Issuance of Series C convertible preferred stock (net of issuance costs of $221) | | | 2,727,059 | | | | 15,729 | | | | — | | | | — | | | | — | | | | — | | | | — | |
Conversion of convertible preferred stock into common stock in connection with the Merger | | | (30,381,987 | ) | | | (160,839 | ) | | | 30,381,987 | | | | 304 | | | | 160,535 | | | | — | | | | 160,839 | |
Issuance of common stock upon private placement (net of offering costs of $3,125) | | | — | | | | — | | | | 5,884,504 | | | | 59 | | | | 26,264 | | | | — | | | | 26,323 | |
Issuance of common stock to Mirax | | | — | | | | — | | | | 1,000,005 | | | | — | | | | — | | | | — | | | | — | |
Conversion of convertible preferred stock warrants into common stock warrants in connection with the Merger | | | — | | | | — | | | | — | | | | — | | | | 93 | | | | — | | | | 93 | |
Issuance of common stock warrants to placement agent as payment for services | | | — | | | | — | | | | — | | | | — | | | | 316 | | | | — | | | | 316 | |
Net loss | | | — | | | | | | | | — | | | | — | | | | — | | | | (44,995 | ) | | | (44,995 | ) |
Balance at December 31, 2015 | | | — | | | $ | — | | | | 38,204,960 | | | $ | 372 | | | $ | 189,712 | | | $ | (197,041 | ) | | $ | (6,957 | ) |
Issuance of common stock from option exercises | | | — | | | | — | | | | 773,718 | | | | 8 | | | | 531 | | | | — | | | | 539 | |
Stock-based compensation | | | — | | | | — | | | | — | | | | — | | | | 2,907 | | | | — | | | | 2,907 | |
Issuance of common stock upon private placement (net of offering cost of $529) | | | — | | | | — | | | | 4,602,506 | | | | 46 | | | | 10,448 | | | | — | | | | 10,494 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (50,636 | ) | | | (50,636 | ) |
Balance at December 31, 2016 | | | — | | | $ | — | | | | 43,581,184 | | | $ | 426 | | | $ | 203,598 | | | $ | (247,677 | ) | | $ | (43,653 | ) |
Issuance of common stock from option exercises | | | — | | | | — | | | | 420,377 | | | | 4 | | | | 661 | | | | — | | | | 665 | |
Issuance of common stock from releases of restricted stock units | | | — | | | | — | | | | 57,626 | | | | — | | | | — | | | | — | | | | — | |
Stock-based compensation | | | — | | | | — | | | | — | | | | — | | | | 5,319 | | | | — | | | | 5,319 | |
Issuance of common stock upon private placement (net of offering cost of $111) | | | — | | | | — | | | | 8,602,589 | | | | 86 | | | | 22,530 | | | | — | | | | 22,616 | |
Issuance of common stock upon direct registered offering (net of offering cost of $81) | | | — | | | | — | | | | 8,382,643 | | | | 84 | | | | 49,776 | | | | — | | | | 49,860 | |
Issuance of common stock from at-the-market offering (net of offering cost of 1,147) | | | — | | | | — | | | | 6,575,062 | | | | 66 | | | | 38,913 | | | | — | | | | 38,979 | |
Issuance of common stock from warrant exercises | | | — | | | | — | | | | 34,493 | | | | — | | | | 103 | | | | — | | | | 103 | |
Reclassification of warrant liability to additional paid-in capital upon warrant exercises | | | — | | | | — | | | | — | | | | — | | | | 274 | | | | — | | | | 274 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (72,176 | ) | | | (72,176 | ) |
Balance at December 31, 2017 | | | — | | | $ | — | | | | 67,653,974 | | | $ | 666 | | | $ | 321,174 | | | $ | (319,853 | ) | | $ | 1,987 | |
The accompanying notes are an integral part of these consolidated financial statements.
101
VIEWRAY, INC.
Consolidated Statements of Cash Flows
(In thousands)
| | Year Ended December 31, | |
| | 2017 | | | 2016 | | | 2015 | |
CASH FLOWS FROM OPERATING ACTIVITIES: | | | | | | | | | | | | |
Net loss | | $ | (72,176 | ) | | $ | (50,636 | ) | | $ | (44,995 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
Depreciation and amortization | | | 2,197 | | | | 1,708 | | | | 1,256 | |
Stock-based compensation | | | 5,319 | | | | 2,907 | | | | 1,066 | |
Accretion on asset retirement obligation | | | 40 | | | | 36 | | | | 8 | |
Change in fair value of warrant liability | | | 16,598 | | | | (3 | ) | | | (45 | ) |
Loss on disposal of property and equipment | | | 9 | | | | 358 | | | | 12 | |
Inventory lower of cost and market adjustment | | | 911 | | | | 1,939 | | | | 2,578 | |
Amortization of debt discount and interest accrual | | | 3,321 | | | | 2,629 | | | | 1,129 | |
Write-off of deferred offering cost | | | — | | | | — | | | | 2,920 | |
Changes in operating assets and liabilities: | | | | | | | | | | | | |
Accounts receivable | | | (16,126 | ) | | | (3,370 | ) | | | 74 | |
Inventory | | | (12,329 | ) | | | (2,065 | ) | | | (2,413 | ) |
Deposits on purchased inventory | | | (4,521 | ) | | | 1,414 | | | | (1,138 | ) |
Deferred cost of revenue | | | (9,787 | ) | | | 4,873 | | | | (4,070 | ) |
Prepaid expenses and other assets | | | (2,044 | ) | | | (1,633 | ) | | | (733 | ) |
Accounts payable | | | 6,309 | | | | 381 | | | | (2,053 | ) |
Notes payable | | | — | | | | — | | | | (240 | ) |
Accrued expenses and other long-term liabilities | | | 850 | | | | 2,197 | | | | 1,532 | |
Customer deposits and deferred revenue | | | 11,376 | | | | 11,109 | | | | 5,263 | |
Net cash used in operating activities | | | (70,053 | ) | | | (28,156 | ) | | | (39,849 | ) |
CASH FLOWS FROM INVESTING ACTIVITIES: | | | | | | | | | | | | |
Purchase of property and equipment | | | (2,163 | ) | | | (7,031 | ) | | | (4,151 | ) |
Purchase of intangible and other assets | | | — | | | | (12 | ) | | | (104 | ) |
Change in restricted cash balance | | | — | | | | (200 | ) | | | 110 | |
Net cash used in investing activities | | | (2,163 | ) | | | (7,243 | ) | | | (4,145 | ) |
CASH FLOWS FROM FINANCING ACTIVITIES: | | | | | | | | | | | | |
Proceeds from issuance of convertible preferred stock, net | | | — | | | | — | | | | 15,729 | |
Proceeds from draw down of long-term debt | | | — | | | | 15,000 | | | | 30,000 | |
Payment of debt issuance cost | | | — | | | | (18 | ) | | | (1,132 | ) |
Payments of long-term debt | | | — | | | | — | | | | (15,000 | ) |
Proceeds from common stock private placement, gross | | | 26,100 | | | | 13,750 | | | | 29,447 | |
Payment of offering costs related to common stock private placement | | | (300 | ) | | | (341 | ) | | | (2,808 | ) |
Proceeds from at-the-market offering of common stock, gross | | | 40,126 | | | | — | | | | — | |
Payment of offering costs related to at-the-market offering of common stock | | | (1,147 | ) | | | — | | | | — | |
Proceeds from direct registered offering, gross | | | 49,941 | | | | — | | | | — | |
Payment of offering costs related to direct registered offering | | | (81 | ) | | | — | | | | — | |
Payments of costs related to the initial public offering | | | — | | | | — | | | | (2,728 | ) |
Proceeds from the exercise of stock options | | | 665 | | | | 539 | | | | 24 | |
Proceeds from the exercise of warrants | | | 103 | | | | — | | | | — | |
Net cash provided by financing activities | | | 115,407 | | | | 28,930 | | | | 53,532 | |
NET INCREASE (DECREASE) IN CASH | | | 43,191 | | | | (6,469 | ) | | | 9,538 | |
CASH AND CASH EQUIVALENTS — BEGINNING OF PERIOD | | | 14,198 | | | | 20,667 | | | | 11,129 | |
CASH AND CASH EQUIVALENTS — END OF PERIOD | | $ | 57,389 | | | $ | 14,198 | | | $ | 20,667 | |
SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION: | | | | | | | | | | | | |
Cash paid for interest | | $ | 3,925 | | | $ | 3,310 | | | $ | 2,332 | |
Cash paid for taxes | | $ | 1 | | | $ | — | | | $ | 1 | |
SUPPLEMENTAL NON-CASH INVESTING AND FINANCING ACTIVITIES: | | | | | | | | | | | | |
Fair value of common stock warrants issued to placement agents as payment for service | | $ | — | | | $ | — | | | $ | 316 | |
Fair value of common stock warrants reclassed from liability to additional paid-in capital upon exercise | | $ | 274 | | | $ | — | | | $ | — | |
Transfer of property and equipment from inventory | | $ | 125 | | | $ | 117 | | | $ | — | |
Purchase of property and equipment in accounts payable and accrued expenses | | $ | 96 | | | $ | 193 | | | $ | 1,136 | |
Offering costs included in accounts payable and accrued expenses | | $ | — | | | $ | 189 | | | $ | — | |
Conversion of convertible preferred stock warrants into common stock warrants in connection with the Merger | | $ | — | | | $ | — | | | $ | 160,839 | |
Asset retirement obligation | | $ | — | | | $ | — | | | $ | 258 | |
Conversion of convertible preferred stock warrants into common stock warrants | | $ | — | | | $ | — | | | $ | 93 | |
The accompanying notes are an integral part of these consolidated financial statements.
102
VIEWRAY, INC.
Notes to Consolidated Financial Statements
1. | Background and Organization |
On July 23, 2015, ViewRay, Inc. (f/k/a Mirax Corp.), or the Company, and ViewRay Technologies, Inc. (f/k/a ViewRay Incorporated), consummated an Agreement and Plan of Merger and Reorganization, or Merger Agreement. Pursuant to the Merger Agreement, the stockholders of ViewRay Technologies, Inc. contributed all of their equity interests to the Company for shares of the Company’s common stock and merged with the Company’s subsidiary, which resulted in ViewRay Technologies, Inc. becoming a wholly-owned subsidiary of the Company, or the Merger. Refer to Note 3 for further information on the Merger.
ViewRay, Inc. and its wholly-owned subsidiary ViewRay Technologies, Inc., designs, manufactures and markets the MRIdian system, an MRI-guided radiation therapy system to image and treat cancer patients simultaneously.
Since inception, ViewRay Technologies, Inc. has devoted substantially all of its efforts towards research and development, initial selling and marketing activities, raising capital and the manufacturing and shipment of MRIdian systems. In May 2012, ViewRay Technologies, Inc. was granted clearance from the FDA, to sell MRIdian with Cobalt-60. In November 2013, ViewRay Technologies, Inc. received its first clinical acceptance of a MRIdian with Cobalt-60 at a customer site, and the first patient was treated with that system in January 2014. ViewRay Technologies, Inc. has had the right to affix the CE mark to MRIdian with Cobalt-60 in the European Economic Area since November 2014. In September 2016, the Company received the right to affix the CE mark to MRIdian Linac in the EEA, and in February 2017, the Company received 510(k) clearance from the FDA to market the MRIdian Linac system.
The Company’s consolidated financial statements have been prepared on the basis of the Company continuing as a going concern for a reasonable period of time. The Company’s principal sources of liquidity are cash flows from public and private shares offerings and available borrowings under its Term Loan agreement. These have historically been sufficient to meet working capital needs, capital expenditures, and debt service obligations. During the year ended December 31, 2017, the Company incurred a net loss from operations of $72.2 million and used cash from operations of $70.1 million. The Company believes that its existing cash balance of $57.4 million as of December 31, 2017, and the aggregate $59.1 million of proceeds from the February equity financing (see Note 20), are sufficient to provide liquidity to fund its operations for at least the next 12 months.
2. | Summary of Significant Accounting Policies |
Basis of Presentation
The consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States, or GAAP, and pursuant to the rules and regulation of the Securities and Exchanges Commission, or SEC. The consolidated financial statements include the accounts of ViewRay, Inc. and its wholly-owned subsidiary, ViewRay Technologies, Inc. All inter-company accounts and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of the consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported and disclosed in the consolidated financial statements and accompanying notes. Such estimates include, but are not limited to, allocation of revenue to its multiple deliverable elements, inventory write-downs to reflect net realizable value, assumptions used in the valuation of stock-based awards and warrant liability, and valuation allowances against deferred tax assets. Actual results could differ from those estimates.
103
Cash and Cash Equivalents
The Company considers all highly liquid investments purchased with an original maturity of three months or less to be cash equivalents. The Company deposits its cash primarily in checking and money market accounts.
Restricted Cash
At December 31, 2017 and 2016, the Company had an aggregate of $0.9 million of outstanding letters of credit related to its operating leases and its contractual obligations with distributors and customers. The letters of credit are collateralized by a restricted cash deposit account, which is presented as part of noncurrent assets on the balance sheets because the Company is not certain when the restriction will be lifted on the collateralized letters of credit. At December 31, 2017, and 2016, no amounts were drawn on the letters of credit.
The restricted cash balance as of December 31, 2017 also includes $0.2 million collateral for a credit card account.
Concentration of Credit Risk, Other Risks and Uncertainties
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash and cash equivalents and accounts receivable. Cash and cash equivalents are deposited in checking and money market accounts with various financial institutions. At times, cash balances may be in excess of the amounts insured by the Federal Deposit Insurance Corporation. Management believes the financial risk associated with these balances is minimal and has not experienced any losses to date. The Company performs periodic credit evaluations of its customers’ financial condition and generally requires deposits from its customers. The Company’s accounts receivable was derived from billings to customers. The Company’s customers representing greater than 10% of accounts receivable and revenue for the periods presented were as follows:
| | Revenue | | Accounts Receivables |
| | Year Ended December 31, | | December 31, |
Customers | | 2017 | | 2016 | | 2015 | | 2017 | | 2016 |
Customer A | | 17% | | | | | | | | |
Customer B | | 17% | | | | | | | | |
Customer C | | 16% | | | | 48% | | | | |
Customer D | | 16% | | | | | | 24% | | |
Customer E | | 14% | | | | | | 16% | | |
Customer F | | 10% | | | | | | | | |
Customer G | | | | | | | | 36% | | |
Customer H | | | | 47% | | | | | | 49% |
Customer I | | | | 25% | | | | | | |
Customer J | | | | 23% | | | | | | 41% |
Customer K | | | | | | 43% | | | | |
The Company’s future results of operations involve a number of risks and uncertainties. Factors that could affect the Company’s future operating results and cause actual results to vary materially from expectations include, but are not limited to, rapid technological change, continued acceptance of MRIdian, competition from substitute products and larger companies, protection of proprietary technology, ability to maintain distributor relationships and dependence on key individuals. Furthermore, new products to be developed by the Company require approval from the FDA or other international regulatory agencies prior to commercial sales. There can be no assurance that the Company’s future products will receive the necessary clearances.
The Company relies on a concentrated number of suppliers to manufacture essentially all of the components used in MRIdian. The Company’s suppliers may encounter problems during manufacturing due to a variety of reasons, including failure to comply with applicable regulations, including the FDA’s Quality System Regulation, equipment malfunction and environmental factors, any of which could delay or impede our ability to meet demand.
104
Accounts Receivables and Allowance for Doubtful Accounts
Accounts receivable are recorded at the invoiced amount, net of any allowance for doubtful accounts, and do not bear interest. The allowance for doubtful accounts, if any, is based on the assessment of the collectability of customer accounts.
Based on the specific customers and the current economic conditions, there was no allowance for doubtful accounts recorded at December 31, 2017 and 2016.
Fair Value of Financial Instruments
Financial instruments consist of cash and cash equivalents, accounts receivable, restricted cash, prepaid expenses and other current assets, accounts payable, accrued liabilities, warrant liability and long-term debt. Cash equivalents are stated at amortized cost, which approximates fair value at the balance sheet dates, due to the short period of time to maturity. Accounts receivable, prepaid expenses and other current assets, accounts payable and accrued liabilities are stated at their carrying value, which approximates fair value due to the short time to the expected receipt or payment date. The warrant liability is carried at fair value. The carrying amount of the Company’s long-term debt approximates fair value as the stated interest rate approximates market rates currently available to the Company.
Inventory and Deposits on Purchased Inventory
Inventory consists of purchased components for assembling MRIdian systems and other direct and indirect costs associated with MRIdian system installation. Inventory is stated at the lower of cost (on a weighted average cost basis) or market value. All inventories expected to be placed in service during the normal operating cycle of the Company for the delivery and assembly of MRIdian systems, including items expected to be on hand for more than one year, are classified as current assets.
The Company reduces the carrying value of its inventory for the difference between cost and net realizable value and records a charge to cost of product revenues for the amount required to reduce the carrying value of inventory to net realizable value. The Company recorded an inventory lower of cost and market adjustment of $0.9 million, $1.9 million and $2.6 million during the years ended December 31, 2017, 2016 and 2015, respectively.
The Company records inventory items which have been paid for but not yet received and title has not yet transferred to the Company as deposits on purchased inventory. Deposits on purchased inventory are included within current assets as the related inventory items are expected to be received and used in MRIdian systems within the Company’s normal operating cycle. The Company assesses the recoverability of deposits on purchased inventory based on credit assessments of the vendors and their history supplying these assets. At December 31, 2017, the Company did not have any instances whereby deposits for purchased inventory were written off or the purchased inventory was not delivered.
Shipping and Handling Costs
Shipping and handling costs for product shipments to customers are included in cost of product revenue. Shipping and handling costs incurred for inventory purchases are capitalized in inventory and expensed in cost of product revenue. These costs are not passed on to customers.
Property and Equipment
Property and equipment are recorded at cost. Depreciation is computed over estimated useful lives, ranging from two to 15 years, of the related assets using the straight-line method. Acquired software is recorded at cost. Amortization of acquired software generally occurs over three years using the straight-line method. Leasehold improvements are amortized on a straight-line basis over the shorter of the useful life or term of the lease. Demonstration units, which are the Company products used for demonstration purpose for customers and/or potential customers, and generally not intended to be sold, are amortized by the straight-line method. Upon retirement or sale, the cost and related accumulated depreciation are removed from the balance sheet and the
105
resulting gain or loss is recorded to general and administrative expense in the accompanying statements of operations and comprehensive loss. Routine expenditures for maintenance and repairs are expensed as incurred.
Depreciation and amortization periods for property and equipment are as follows:
Property and Equipment | | Estimated Useful Life |
Prototype | | 2 – 10 years |
Machinery and equipment | | 5 – 15 years |
Furniture and fixture | | 5 – 10 years |
Software | | 3 years |
Leasehold improvements | | Lesser of estimated useful life or remaining lease term |
Asset Retirement Obligation
In connection with certain lease agreements entered into in October 2015, the Company has a legal requirement to remove long-lived assets constructed on leased property and to restore the leased property to its original condition. The Company records the fair value of the liability for a legal obligation to retire an asset in the period in which the obligation is incurred if a reasonable estimate of fair value can be made. The Company measures the fair value of the asset retirement obligation based upon the present value of the expected future payments, and recognized asset retirement obligation of $250,000 at inception. The liability is accreted to its present value each period and the capitalized cost is depreciated over the remaining lease term. Accretion expense is calculated by applying the effective interest rate to the carrying amount of the liability at the beginning of each period. The effective interest rate is the credit-adjusted risk-free rate applied when the liability was initially measured and recognized.
At December 31, 2017, the Company had outstanding asset retirement obligations of $334,000, which was included in other long-term liabilities in the accompanying consolidated balance sheets. For the years ended December 31, 2017, 2016 and 2015, the Company recognized accretion expenses of $40,000, $36,000 and $8,000 in the accompanying statements of operations and comprehensive loss.
Intangible Assets
Intangible assets consist primarily of patents and license acquisition costs associated with certain technology components incorporated into the Company’s MRIdian systems. The Company capitalizes the cost and amortizes it on a straight-line basis over the estimated useful lives, which is generally three years for license cost and five to seven years for patents.
Impairment of Long-Lived Assets
The Company reviews the recoverability of long-lived assets, including equipment, leasehold improvements, software and intangible assets when events or changes in circumstances occur that indicate that the carrying value of the asset may not be recoverable. The assessment of possible impairment is based on the ability to recover the carrying value of the assets from the expected future cash flows (undiscounted and without interest charge) of the related operations. If these cash flows are less than the carrying value of such assets, an impairment loss for the difference between the estimated fair value and carrying value is recorded. There was no impairment loss recognized during the years ended December 31, 2017, 2016 and 2015.
Deferred Offering Costs
ViewRay Technologies, Inc. capitalized qualified legal, accounting and other direct costs related to its efforts to raise capital through a public sale of its common stock in its planned IPO. These costs were recorded in deferred offering costs in the accompanying balance sheets. During the year ended December 31, 2015, ViewRay Technologies, Inc. terminated its plan for IPO, and wrote off deferred offering costs of $2.9 million in the accompanying consolidated statements of operations and comprehensive loss. The balance of deferred offering costs was zero at December 31, 2017 and 2016.
106
Comprehensive Loss
Comprehensive loss is the change in equity of a company during a period from transactions and other events and circumstances, excluding transactions resulting from investment owners and distribution to owners. For the periods presented, comprehensive loss did not differ from net loss.
Revenue Recognition
The Company derives revenue primarily from the sale of the systems and related services, which are sales of MRIdian, as well as support and maintenance services on sold systems. In all sales arrangements, the Company recognizes revenues when there is persuasive evidence of an arrangement, the fee is fixed or determinable, collection of the fee is reasonably assured and delivery has occurred. For sales of MRIdian systems that the Company is required to install at the customer site, product revenue is recognized upon receipt of customer acceptance. For sales of MRIdian systems that the Company is not responsible for installation, product revenue is recognized when the entire system is delivered and title and risk of loss are transferred to the customer. For sales of the related support and maintenance services, the Company recognizes service revenue on a straight-line basis over the service contract term, which is typically 12 months.
Multiple Elements
Based on the nature of the Company’s business, it frequently enters into sales arrangements with customers that contain multiple elements or deliverables. The principal deliverables consist of (i) sale of MRIdian systems, which generally includes installation, site preparation and software, and (ii) product support, which includes extended service and maintenance.
The Company determines selling prices of each deliverable using vendor specific objective evidence, or VSOE, if it exists, or third-party evidence, or TPE. If neither VSOE nor TPE exists for a deliverable, the Company uses best estimated selling price, or BESP. The Company allocates revenue to each standalone deliverable using the relative fair values for each deliverable as determined by BESP. The Company regularly reviews VSOE, TPE and BESP for all of its MRIdian systems and services.
Product Revenue
Product revenue is derived primarily from the sales of MRIdian. The system contains both software and non-software components that together deliver essential functionality. However, because MRIdian includes hardware products as well as software components that function together with the hardware components to deliver MRIdian’s essential functionality, the revenue from the sale of MRIdian systems does not fall within the scope of the software revenue recognition rules.
The Company’s customer contracts generally call for on-site assembly of the system components and system integration. Once the system installation is completed, the Company performs a detailed demonstration with the customer showing that MRIdian meets the standard product specifications. After successful demonstration, the customer signs a document indicating customer’s acceptance. For sales of MRIdian systems that the Company is required to install at the customer site, revenue recognition occurs when the customer acknowledges that the system operates in accordance with standard product specifications, the customer accepts the installed unit and title and risk of loss are transferred to the customer.
Certain customer contracts with distributors do not require installation at the customer site, and the distributors typically have its own or engage a qualifying third-party certified technician to perform the installation. For sales of MRIdian systems when the Company is not responsible for installation, revenue recognition occurs when the entire system is delivered and title and risk of loss are transferred to the customer.
All contracts include customer deposits upon signing of the agreement with final payment generally due upon customer acceptance.
107
Service Revenue
Service revenue is derived primarily from maintenance services. Service revenue is recognized ratably over the service period.
Distribution Rights Revenue
The Company entered into a distribution agreement with Itochu Corporation pursuant to which it appointed Itochu as its exclusive distributor for the promotion, sale and delivery of MRIdian products within Japan. In consideration of the exclusive distribution rights granted, the Company received $4.0 million which was recorded as deferred revenue and starting in August 2016 was recognized as distribution rights revenue on a straight-line basis over the remaining term of the distribution agreement of approximately 8.5 years.
Customer Deposits
Customer deposits represent payments received in advance of system installation. For domestic sales, advance payments received prior to inventory shipments and customer acceptance are recorded as customer deposits. For international sales, advance payments are initially recorded as customer deposits and are subsequently reclassified to deferred revenue upon inventory shipment when the title and risk of loss of inventory items transfer to customers. All customer deposits, including those that are expected to be a deposit for more than one year, are classified as current liabilities based on consideration of the Company’s normal operating cycle (the time between acquisition of the inventory components and the final cash collection from customers on these inventory components) which is in excess of one year.
Deferred Revenue and Deferred Cost of Revenue
Deferred revenue consists of deferred product revenue and deferred service revenue. Deferred product revenue arises from timing differences between the fulfillment of other contract deliverables and satisfaction of all revenue recognition criteria consistent with the Company’s revenue recognition policy. Deferred service revenue results from the advance billing for services to be delivered over a period of time. Deferred revenues expected to be realized within one year are classified as current liabilities.
Deferred cost of revenue consists of cost for inventory items that have been shipped with title and risk of loss transferred to the customer but the customer acceptance has not been received. Deferred cost of revenue is included as part of current assets as the corresponding deferred product revenue is expected to be realized within one year. The inventories recorded in deferred cost of revenue are also included in the inventory lower of cost or market analysis. At December 31, 2017 and 2016, no reserve was required for deferred cost of revenue.
Research and Development Costs
Expenditures, including payroll, contractor expenses and supplies, for research and development of products and manufacturing processes are expensed as incurred.
Software development costs incurred subsequent to establishing technological feasibility are capitalized through the general release of MRIdian systems that contain the embedded software elements. Technological feasibility is demonstrated by the completion of a working model. The Company has not capitalized any software development costs at December 31, 2017 or 2016, since the costs incurred subsequent to achieving technological feasibility and completing the research and development for the software components were immaterial.
Stock-Based Compensation
The Company uses the Black-Scholes option-pricing model as the method for estimating the fair value of stock options. The Black-Scholes option-pricing model requires the use of highly subjective and complex assumptions that determine the fair value of share-based awards, including the options’ expected term and the price volatility of the underlying stock. The fair value of Restricted Stock Units, or RSUs, is based on the closing market price of the Company’s common stock on the grant date. The fair value of the portion of the award that is ultimately expected to
108
vest is recognized as compensation expense over the awards’ requisite service periods in the consolidated statements of operations and comprehensive loss. The Company attributes the value of share-based compensation to expense using the straight-line method.
Medical Device Excise Tax
Medical Device Excise Tax, or MDET, Section 4191 of the Internal Revenue Code enacted by the Health Care and Education Reconciliation Act of 2010, in conjunction with the Patient Protection and Affordable Care Act, established a 2.3% excise tax on medical devices sold domestically which, due to subsequent legislative amendments, was suspended from January 1, 2016 to December 31, 2017. MDET was suspended for another two years after the stopgap bill was signed by the President in January 2018. The Company included MDET in cost of product revenue during the year ended December 31, 2015, net of amounts directly billed to the customer for this tax, if any.
Deferred Commissions
Deferred commissions are the direct and incremental costs directly associated with the MRIdian system contracts with customers, which primarily consist of sales commissions to our direct sales force. The commissions are deferred and expensed in proportion to the revenue recognized upon the acceptance of the MRIdian system. At December 31, 2017 and 2016, the Company had $3.5 million and $2.6 million deferred commissions recorded as part of prepaid expenses and other current assets on the accompanying consolidated balance sheets.
Income Taxes
The Company uses the asset and liability method of accounting for income taxes. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. Deferred tax expense or benefit is the result of changes in the deferred tax assets and liabilities. Valuation allowances are established when necessary to reduce deferred tax assets where, based upon the available evidence, management concludes that it is more-likely-than not that the deferred tax assets will not be realized. Because of the uncertainty of the realization of the deferred tax assets, the Company has recorded a full valuation allowance against its net deferred tax assets.
In evaluating the ability to recover its deferred income tax assets, the Company considers all available positive and negative evidence, including its operating results, ongoing tax planning and forecasts of future taxable income on a jurisdiction-by-jurisdiction basis. In the event the Company was to determine that it would be able to realize its deferred income tax assets in the future in excess of their net recorded amount, it would make an adjustment to the valuation allowance which would reduce the provision for income taxes.
Reserves are provided for tax benefits for which realization is uncertain. Such benefits are only recognized when the underlying tax position is considered more likely than not to be sustained on examination by a taxing authority, assuming they possess full knowledge of the position and facts. It is the Company’s policy to include any penalties and interest related to income taxes in its income tax provision; however, the Company currently has no penalties or interest related to income taxes. The earliest year that the Company is subject to examination is the year ended December 31, 2004.
Warrant Liability
Convertible Preferred Stock Warrant Liability
The Company’s warrant to purchase convertible preferred stock was classified as a liability on the consolidated balance sheets at fair value upon issuance because the warrant is exercisable for contingently redeemable preferred stock which is classified outside of stockholders’ equity (deficit). The warrant was subject to re-measurement to fair value at each balance sheet date, and any change in fair value was recognized in the consolidated statements of operations and comprehensive loss as other expense, net. In July 2015, upon the Merger of the Company and
109
ViewRay Technologies, Inc., and the Private Placement, the convertible preferred stock warrants were converted into warrants to purchase the Company’s common stock, and the fair value of the preferred stock warrant liability was reclassified to additional paid-in capital.
Common Stock Warrant Liability
Certain warrants to purchase common stock provide for cash settlement in the event of change in control, and are classified as liabilities on the balance sheets at fair value upon issuance (see Note 14). These warrants are subject to re-measurement to fair value at each balance sheet date, and any change in fair value are recognized in the consolidated statements of operations and comprehensive loss as other expense, net. Upon exercise or expiration of the warrants, the related warrant liability will be reclassified to additional paid-in capital.
Net Loss per Share
The Company’s basic net loss per share is calculated by dividing the net loss by the weighted-average number of shares of common stock outstanding for the period. Contingently issuable shares are included in the computation of basic net loss per share as of the date that all necessary conditions have been satisfied and issuance of the shares is no longer contingent. The diluted net loss per share is computed by giving effect to all potential common stock equivalents outstanding for the period determined using the treasury stock method. For purposes of this calculation, stock options, restricted stock units and warrants to purchase common stock are considered to be common stock equivalents but have been excluded from the calculation of diluted net loss per share as their effect is anti-dilutive.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”) issued ASU No. 2014-09, Revenue from Contracts with Customers (Topic 606), which supersedes the revenue recognition requirements in Accounting Standards Codification 605, Revenue Recognition. This ASU, herein referred to as Topic 606, is based on the principle that revenue is recognized to depict the transfer of goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The ASU also requires additional disclosure about the nature, amount, timing and uncertainty of revenues and cash flows arising from customer contracts, including significant judgments and changes in judgments and assets recognized from costs incurred to obtain or fulfill a contract. In August 2015, the FASB issued ASU No. 2015-14 to defer the effective date by one year with early adoption permitted as of the original effective date. ASU No. 2014-09 will be effective for the Company’s fiscal year beginning after December 15, 2017, and the interim periods thereafter. In addition, the FASB issued ASU No. 2016-08, 2016-10, 2016-12, 2016-20, 2017-13 and 2017-14 in March 2016, April 2016, May 2016, December 2016, September 2017 and November 2017, respectively, to help provide interpretive clarification on the new guidance in Topic 606. ASU No. 2016-08, 2016-10, 2016-12, 2017-13 and 2017-14 are all effective during the same period as ASU No. 2014-09.
The Company will adopt Topic 606 on January 1, 2018 using the full retrospective method which requires the Company to restate each prior reporting period presented. Based on the nature of its sales arrangements, the Company does not believe the adoption of the new standards will have a material impact on the amount or timing of its revenue recognition, and the Company’s product revenue, service revenue and distribution rights revenue will remain substantially unchanged. As a result, the adoption of the new standards will have no material impact on the Company’s prior period financial statements. The Company expects to update the related disclosures upon adoption of the new standards in the first quarter of fiscal year 2018.
110
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842) and issued subsequent amendments to the initial guidance in September 2017 within ASU 2017-13 (collectively, Topic 842). Topic 842 supersedes the Accounting Standards Codification 840, Leases, and requires lessees to recognize all leases, with exception of short-term leases, as a lease liability on the balance sheet. Under this ASU, a lease is defined as a lessee’s obligation to make lease payments arising from a lease, measured on a discounted basis, and a right-of-use asset which is an asset that represents the lessee’s right to use, or control the use of, a specified asset during the lease term. The ASU also requires additional disclosure about the amount, timing and uncertainty of cash flow from leases. The new standard is effective for fiscal years beginning after December 15, 2018, and interim periods therein. Early adoption is permitted. As disclosed in Note 7, future minimum payments under noncancelable operating leases are approximately $2.2 million. This new standard will require the present value of these leases to be recorded in the consolidated balance sheets as a right of use asset and lease liability. The Company is continuing to evaluate the impact of this guidance on its consolidated financial statements and related disclosures.
In August 2016, the FASB issued ASU No. 2016-15, Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments, which eliminates the diversity in practice related to the classification of certain cash receipts and payments for debt prepayment or extinguishment costs, the maturing of a zero coupon bond, the settlement of contingent liabilities arising from a business combination, proceeds from insurance settlements, distributions from certain equity method investees and beneficial interests obtained in a financial asset securitization. ASU No. 2016-15 designates the appropriate cash flow classification, including requirements to allocate certain components of these cash receipts and payments among operating, investing and financing activities. In November 2016, the FASB issued ASU No. 2016-18, Statement of Cash Flows (Topic 230): Restricted Cash, which requires that the statement of cash flows explain the change during the period in the total of cash, cash equivalents, and amounts generally described as restricted cash or restricted cash equivalents. ASU No. 2016-15 and ASU No. 2016-18 should be applied using the retrospective transition method, requiring adjustment to all comparative periods presented, unless it is impracticable for some of the amendments, in which case those amendments would be made prospectively as of the earliest date practicable. The amendments in ASU No. 2016-15 and ASU No. 2016-18 are effective for fiscal years beginning after December 15, 2017, and interim periods therein. Early adoption is permitted, including adoption in an interim period. The Company had restricted cash of $1.1 million at both December 31, 2017 and December 31, 2016. The adoption of ASU No. 2016-15 and ASU No. 2016-18 will not have a material impact on the Company’s consolidated financial statements and related disclosures.
In May 2017, the FASB issued ASU No. 2017-09, Compensation-Stock Compensation (Topic 718): Scope of Modification Accounting, which provides clarified guidance on applying modification accounting to changes in the terms or conditions of a share-based payment award. Changes that do not impact the award’s fair value, vesting conditions, or classification as an equity or liability instrument will not be subject to modification accounting. ASU No. 2017-09 is effective prospectively for annual periods beginning after December 15, 2017 and interim periods therein. Early adoption is permitted, including adoption in an interim period. The Company does not believe that the adoption of ASU No. 2017-09 will have a material impact on its consolidated financial statements and related disclosures.
111
Recently Adopted Accounting Pronouncements
In July 2015, the FASB issued ASU No. 2015-11, Simplifying the Measurement of Inventory, which requires entities to measure most inventory at the lower of cost and net realizable value, thereby simplifying the current guidance under which an entity must measure inventory at the lower of cost or market. ASU No. 2015-11 is effective prospectively for annual periods beginning after December 15, 2016 and interim periods therein. Early application is permitted. The Company adopted ASU No. 2015-11 as required in the first quarter of fiscal year 2017. The adoption of the new guidance did not have a material impact on its consolidated financial reporting statements and related disclosures.
In March 2016, the FASB issued ASU No. 2016-09, Compensation – Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting, which simplifies several aspects of the accounting for share-based payment transactions, including the income tax consequences, application of award forfeitures to expense, classification of awards as either equity or liabilities, and classification on the statement of cash flows. ASU No. 2016-09 is effective for fiscal years beginning after December 15, 2016, and interim periods therein. Early adoption is permitted. The Company adopted ASU No. 2016-09 as required in the first quarter of fiscal year 2017, and there was no material impact on the financial statements given the full valuation allowance position of its deferred tax assets.
On July 23, 2015, ViewRay, Inc. (f/k/a Mirax Corp.), or the Company, and ViewRay Technologies, Inc. (f/k/a ViewRay Incorporated), consummated an Agreement and Plan of Merger and Reorganization, or Merger Agreement. Pursuant to the Merger Agreement, the stockholders of ViewRay Technologies, Inc. contributed all of their equity interests to the Company for shares of the Company’s common stock and merged with the Company’s subsidiary, which resulted in ViewRay Technologies, Inc. becoming a wholly-owned subsidiary of the Company, or the Merger. Effective as of July 23, 2015, the Company amended and restated its Certificate of Incorporation to increase its authorized common stock to 300,000,000 shares and 10,000,000 shares of “blank check” preferred stock, par value of $0.01 per share.
Upon the closing of the Merger, under the terms of the Split-Off Agreement, dated July 23, 2015 among the Company, ViewRay Technologies, Inc. and Vesuvius Acquisition Sub, Inc., the acquisition subsidiary of the Company, or the Split-Off Agreement, and a general release agreement dated July 23, 2015, or the General Release Agreement, the Company transferred all of its pre-Merger operating assets and liabilities to wholly- owned special-purpose subsidiary incorporated in Nevada, Vesuvius Acquisition Sub, Inc. or the Split-Off Subsidiary. Thereafter, the Company transferred all of the outstanding shares of capital stock of the Split-Off Subsidiary to certain pre-Merger insiders of the Company in exchange for the surrender and cancellation of shares of the Company’s common stock held by such persons.
Together with the Merger, on July 23, 2015, ViewRay Technologies, Inc. effected a 2.975-for-1 stock split of its then outstanding common stock and convertible preferred stock, collectively referred to as Capital Stock, and convertible preferred stock warrants, in which (i) each share of outstanding Capital Stock was increased into 2.975 shares of Capital Stock; (ii) the number of outstanding options to purchase each Capital Stock was proportionately increased on a 2.975-for-1 basis; (iii) number of shares reserved for future option grants under the 2008 Plan were proportionately increased on a 2.975-for-1 basis; (iv) the exercise price of each such outstanding option was proportionately decreased on a 2.975-for-1 basis; and (v) each share of outstanding convertible preferred stock warrant was increased into 2.975 shares of convertible preferred stock warrant. All of the share and per share amounts have been adjusted, on a retroactive basis, to reflect this 2.975-for-1 stock split.
At the closing of the Merger, the Company conducted a private placement offering, or the Private Placement, of its securities for $26.3 million, net of offering cost, through the sale of 5,884,504 shares of the common stock of the surviving corporation, at an offering price of $5.00 per share. Investors in ViewRay Technologies, Inc. purchased $17.0 million of shares in the Private Placement. Certain shareholders of the Company retained, after giving effect to the Split-Off, 1,000,005 shares of the common stock of the surviving corporation upon the Private Placement. The former stockholders of ViewRay Technologies Inc. collectively own approximately 90.9% of the outstanding shares of the Company’s common stock.
112
Immediately following the closing of the Merger, the Company’s outstanding shares of common stock (on a fully diluted basis) were owned as follows:
| • | Former holders of the ViewRay Technologies, Inc.’s capital stock hold an aggregate of 34,715,582 shares of the Company’s common stock, or approximately 72.7% on a fully diluted basis; |
| • | The Private Placement, resulted in an aggregate of 5,884,504 shares of the Company’s common stock, consisting of 3,400,003 shares held by ViewRay Technologies, Inc. shareholders and 2,484,501 shares issued to new shareholders, or together approximately 12.3% on a fully diluted basis; |
| • | 128,231 shares of ViewRay Technologies, Inc.’s preferred stock warrants were converted to the Company’s common stock warrant, or approximately 0.3% on a fully diluted basis; |
| • | 198,760 shares of common stock issued as warrants to placement agents as payment for services provided, or approximately 0.4% on a fully diluted basis; |
| • | Holders of the Company’s common stock prior to the closing of the Merger hold an aggregate of 1,000,005 shares of the Company’s common stock, or approximately 2.1% on a fully diluted basis; and |
| • | 9,225,397 shares of common stock are reserved for issuance under the 2008 Stock Incentive Plan, or the 2008 Plan, and the 2015 Equity Incentive Plan of ViewRay, or the 2015 Plan, collectively representing approximately 19.3% on a fully diluted basis. Upon closing, 1,507,147 options to purchase shares of the Company’s common stock are granted to employees under the 2015 Plan. In addition, the Board of Directors of the Company has adopted a 285,621-share reserve under the 2015 ESPP. |
The Merger was accounted for as a reverse-merger and recapitalization. ViewRay Technologies, Inc. was the acquirer for financial reporting purposes, and ViewRay, Inc. was the acquired company under the acquisition method of accounting in accordance with FASB ASC Topic 805, Business Combination. Consequently, the assets, liabilities and operations that will be reflected in the historical consolidated financial statements prior to the Merger will be those of ViewRay Technologies, Inc. and will be recorded at the historical cost basis, and the consolidated financial statements after completion of the Merger will include the assets, liabilities and results of operations of ViewRay Technologies, Inc. up to the day prior to the closing of the Merger and the assets, liabilities and results of operations of the combined company from and after the closing date of the Merger.
4. | Balance Sheet Components |
Property and Equipment
Property and equipment consisted of the following (in thousands):
| | December 31, | |
| | 2017 | | | 2016 | |
Prototype | | $ | 11,929 | | | $ | 6,405 | |
Machine and equipment | | | 7,831 | | | | 6,057 | |
Leasehold improvements | | | 4,438 | | | | 4,371 | |
Furniture and fixtures | | | 558 | | | | 368 | |
Software | | | 1,142 | | | | 1,028 | |
Construction in progress | | | - | | | | 5,498 | |
Property and equipment, gross | | | 25,898 | | | | 23,727 | |
Less: accumulated depreciation and amortization | | | (14,334 | ) | | | (12,167 | ) |
Property and equipment, net | | $ | 11,564 | | | $ | 11,560 | |
Depreciation and amortization expense related to property and equipment was $2.2 million, $1.6 million and $1.1 million during the years ended December 31, 2017, 2016 and 2015, respectively.
113
Intangible Assets
Intangible assets consisted of the following (in thousands):
| | December 31, | |
| | 2017 | | | 2016 | |
License cost | | $ | 512 | | | $ | 512 | |
Patents | | | 104 | | | | 104 | |
Intangible assets, gross | | | 616 | | | | 616 | |
Accumulated amortization | | | (538 | ) | | | (519 | ) |
Intangible assets, net | | $ | 78 | | | $ | 97 | |
Intangible amortization expense was $19 thousand, $115 thousand and $168 thousand during the years ended December 31, 2017, 2016 and 2015, respectively, which were recorded in general and administrative expenses in the consolidated statements of operations and comprehensive loss.
At December 31, 2017, the estimated future amortization expense of purchased intangible assets was as follows (in thousands):
Year Ended December 31, | | Estimated Future Amortization Expense | |
2018 | | $ | 19 | |
2019 | | | 19 | |
2020 | | | 19 | |
2021 | | | 10 | |
2022 | | | 3 | |
Thereafter | | | 8 | |
Total amortization expense | | $ | 78 | |
Accrued Liabilities
Accrued liabilities consisted of the following (in thousands):
| | December 31, | |
| | 2017 | | | 2016 | |
Accrued payroll and related benefits | | $ | 3,944 | | | $ | 4,274 | |
Accrued accounts payable | | | 2,671 | | | | 1,202 | |
Tax payable | | | 149 | | | | 13 | |
Accrued legal and accounting | | | 322 | | | | 509 | |
Other | | | 121 | | | | 336 | |
Total accrued liabilities | | $ | 7,207 | | | $ | 6,334 | |
114
Deferred Revenue
Deferred revenue consisted of the following (in thousands):
| | December 31, | |
| | 2017 | | | 2016 | |
Deferred revenue: | | | | | | | | |
Product | | $ | 18,861 | | | $ | 5,050 | |
Services | | | 1,182 | | | | 1,561 | |
Distribution rights | | | 3,346 | | | | 3,822 | |
Total deferred revenue | | | 23,389 | | | | 10,433 | |
Less: current portion of deferred revenue | | | (20,151 | ) | | | (6,515 | ) |
Noncurrent portion of deferred revenue | | $ | 3,238 | | | $ | 3,918 | |
5. | Fair Value of Financial Instruments |
Assets and liabilities recorded at fair value on a recurring basis in the balance sheets are categorized based upon the level of judgment associated with the inputs used to measure their fair values. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The standard describes a fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value which are the following:
Level 1—Quoted prices in active markets for identical assets or liabilities.
Level 2—Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
The assets’ or liabilities’ fair value measurement level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement.
The Company’s financial instruments that are carried at fair value mainly consist of Level 1 assets and Level 3 liabilities. Level 1 assets include highly liquid bank deposits and money market funds, which were not material at December 31, 2017 or 2016. Level 3 liabilities that are measured on a recurring basis consists of convertible preferred stock warrants and common stock warrants. Preferred stock warrant and common stock warrant liabilities are valued using the Black-Scholes option-pricing model. Generally, increases (decreases) in the fair value of the underlying stock, estimated term and volatility would result in a directionally similar impact to the fair value of the warrant (see Note 13).
The convertible preferred stock warrants were issued in December 2013 and were converted into warrants to purchase common stock upon the Merger of the Company and ViewRay Technologies, Inc. The aggregate fair value of these warrants upon the closing of the Merger is $93 thousand which was reclassified from liabilities to additional paid-in-capital, and the Company no longer recorded change in fair value adjustments in relation to convertible preferred stock warrants.
The Company’s common stock warrants liabilities consist of the 2017 and 2016 Placement Warrants, as described in Note 13. The 2016 Placement Warrants were issued in August and September 2016, and the 2017 Placement Warrants were issued in January 2017. In December 2017, 25,014 shares of 2016 Placement Warrants and 9,389 shares of 2017 Placement Warrants were exercised and the aggregate fair value of these warrants upon exercise of
115
$274 thousand was reclassified from liabilities to additional paid-in-capital. At December 31, 2017, 1,355,641 shares of 2016 Placement Warrants and 1,711,123 shares of 2017 Placement Warrants were outstanding.
The gains and losses from re-measurement of Level 3 financial liabilities are recorded as part of other income (expense), net in the consolidated statements of operations and comprehensive loss. During the year ended December 31, 2017 and 2016, the Company recorded a loss of $16.6 million and a gain of $3 thousand, respectively, related to the change in fair value of the 2016 and 2017 Placement Warrants. There have been no transfers between Level 1, Level 2 and Level 3 in any periods presented.
The following table sets forth the fair value of the Company’s financial liabilities by level within the fair value hierarchy (in thousands):
| | At December 31, 2017 | |
| | Level 1 | | | | | Level 2 | | | Level 3 | | | Total | |
2017 Placement Warrants Liability | | $ | — | | | | | $ | — | | | $ | 12,487 | | | $ | 12,487 | |
2016 Placement Warrants Liability | | | — | | | | | | — | | | | 9,933 | | | | 9,933 | |
Total Warrant Liability | | $ | — | | | | | $ | — | | | $ | 22,420 | | | $ | 22,420 | |
| | At December 31, 2016 | |
| | Level 1 | | | Level 2 | | | Level 3 | | | Total | |
2016 Placement Warrants Liability | | $ | — | | | $ | — | | | $ | 2,723 | | | $ | 2,723 | |
The following table sets forth a summary of the changes in the fair value of the Company’s Level 3 financial liabilities (in thousands):
| | Year Ended December 31, | |
| | 2017 | | | 2016 | | | 2015 | |
Fair value, beginning of period | | $ | 2,723 | | | $ | — | | | $ | 138 | |
Issuance of 2017 Placement Warrants | | | 3,373 | | | | — | | | | — | |
Issuance of 2016 Placement Warrants | | | — | | | | 2,726 | | | | — | |
Change in fair value of Level 3 financial liabilities | | | 16,598 | | | | (3 | ) | | | (45 | ) |
Conversion of convertible preferred stock warrants to common stock warrants | | | — | | | | — | | | | (93 | ) |
Fair value of 2017 Placement Warrants at exercise | | | (74 | ) | | | — | | | | — | |
Fair value of 2016 Placement Warrants at exercise | | | (200 | ) | | | — | | | | — | |
Fair value, end of period | | $ | 22,420 | | | $ | 2,723 | | | $ | — | |
Hercules Term Loan
In December 2013, ViewRay Technologies, Inc. entered into a Loan and Security Agreement, or the Hercules Term Loan, with Hercules Technology Growth Capital, Inc. and Hercules Technology III, L.P., or together, Hercules, for $15.0 million that was outstanding at December 31, 2014. Borrowings under the Hercules Term Loan bear cash interest at the greater of the annual prime rate plus 7.0% or 10.25%. In addition, borrowings under the Hercules Term Loan bear deferred payment in-kind interest at 1.5% per annum. Interest only payments began in January 2014, with monthly principal and interest payments beginning on January 1, 2015 and the entire balance of the Hercules Term Loan are to be paid in full by the June 1, 2017 maturity date. The Hercules Term Loan is subject to a prepayment penalty of 5% on the outstanding balance during the first 12 months following the funding of the loan and 1% on the outstanding balance thereafter until maturity. The Hercules Term Loan was issued at a discount of $466 thousand, which was amortized to interest expense during the life of the loan using the effective interest method. The discount included the fair value of a convertible preferred stock warrant that was issued with the
116
Hercules Term Loan, as discussed in the following paragraph, and the related transaction costs. The Hercules Term Loan is collateralized by essentially all the assets of ViewRay Technologies, Inc. and limits its ability with respect to additional indebtedness, investments or dividends, among other things, subject to customary exceptions.
In connection with the issuance of the Hercules Term Loan, ViewRay Technologies, Inc. entered into a Warrant Agreement with Hercules to issue a fully vested and exercisable warrant to purchase 128,231 shares of Series C convertible preferred stock with an exercise price of $5.84 per share. The warrant is exercisable any time before the later of 10 years from issuance or five years after an IPO. The warrant provides for anti-dilution rights on the Series C convertible preferred stock, which includes one-time down-round protection. The fair value of the warrant upon issuance of $158 thousand was recorded as convertible preferred stock warrant liability and a discount to the carrying value of the Hercules Term Loan. The fair value of the warrant at the time of issuance was estimated using the Black-Scholes option-pricing model with the following assumptions: expected term of two years, expected volatility of 30%, risk-free interest rate of 0.4% and expected dividend yield of 0%. The convertible preferred stock warrants were converted into warrants to purchase the Company’s common stock upon the consummation of the Merger in July 2015 as disclosed in Notes 1 and 5. See Note 14 for assumptions used to estimate the fair value of convertible preferred stock warrant liability upon conversion into warrants to purchase common stock on July 23, 2015.
In June 2015, ViewRay Technologies, Inc. paid off in full the outstanding balances on Hercules Term Loan, including the related interest and other penalty fee, using part of the proceeds received from the CRG Term Loan discussed below.
CRG Term Loan
In June 2015, ViewRay Technologies, Inc. entered into a Term Loan Agreement, or the CRG Term Loan, with Capital Royalty Partners II L.P., Capital Royalty Partners II – Parallel Fund “A” L.P., Capital Royalty Partners II (Cayman) L.P. and Parallel Investment Opportunities Partners II L.P. or together with their successors by assignment, CRG, for up to $50.0 million of which $30.0 million was made available to the Company upon closing with the remaining $20.0 million available on or before June 26, 2016 at its option upon the occurrence of either (i) an initial public offering of its common stock on a nationally recognized securities exchange that raises a minimum of $40.0 million in net cash proceeds with a minimum of $120.0 million post money valuation, or Qualifying IPO, or (ii) achievement of a minimum of $25.0 million gross revenue from the sales of the MRIdian system during any consecutive 12 months before March 31, 2016. The Company drew down the first $30.0 million on the closing date. The CRG Term Loan has a maturity date of June 26, 2020 and bears cash interest at a rate of 12.5% per annum to be paid quarterly during the interest-payment-only period of three years. In April 2017, the CRG Term Loan was amended to allow for interest-payment-only until March 31, 2020. During the interest-payment-only period, the Company has the option to elect to pay only 8% of the 12.5% per annum interest in cash, and the remaining 4.5% of the 12.5% per annum interest as compounded interest, or deferred payment in-kind interest, added to the aggregate principal amount of the CRG Term Loan. Principal payment and any deferred payment in-kind interest will be paid quarterly in equal installments following the end of the interest-payment-only period through maturity date.
The CRG Term Loan is subject to a prepayment penalty of 3% on the outstanding balance during the first 12 months following the funding of the loan, 2% on the outstanding balance after year 1 but on or before year 2, 1% on the outstanding balance after year 2 but on or before year 3, and 0% on the outstanding loan if prepaid after year 3 thereafter until maturity. The Term Loan is also subject to a facility fee of 7% based on the sum of the amount drawn and any outstanding payment in-kind interest payable on the maturity date or the date such loan becomes due. All direct financing costs were accounted for as a discount on the CRG Term Loan and will be amortized to interest expense during the life of the loan using the effective interest method. The CRG Term Loan is subject to financial covenants and is collateralized by essentially all assets of the Company and limits its ability with respect to additional indebtedness, investments or dividends, among other things, subject to customary exceptions.
In March 2016, the Company and CRG executed an amendment to the original terms of the CRG Term Loan such that, with regard to the conditions for borrowing the remaining $20.0 million available under the CRG Term Loan, the Company may, at its election, draw down (i) an amount of either $10.0 million or $15.0 million in up to two advances upon achievement of a minimum of $15.0 million of aggregate product and service revenue during any consecutive 12 month period ending on or before March 31, 2016 and (ii) an additional $5.0 million (or $10.0
117
million, if the previous draw made was only in an amount of $10.0 million) upon achievement of a minimum of $25.0 million of aggregate product and service revenue during any consecutive 12 month period ending on or before December 31, 2016 and upon execution of the first sales contract of the Company’s second generation product. The Company achieved the minimum of $15.0 million gross revenue requirement in March 2016 which made the first $15.0 million of the remaining $20.0 million credit facility immediately available for draw down. In May 2016, the Company drew down the additional $15.0 million available amount.
In April 2017, the Company and CRG executed an amendment to the terms of its CRG Term Loan, as amended in March 2016. Amendments to the CRG Term Loan include availability of the existing $5.0 million tranche at ViewRay’s option through June 30, 2017, the addition of a $15.0 million tranche of borrowing capacity available at ViewRay’s option through September 30, 2017, extension of the interest-only and payment in-kind period, a decrease to the combined 2016 and 2017 revenue covenant and a 1.75% increase to the facility fee. The Company did not draw down any amounts under the $5.0 million tranche and it has since expired.
In October 2017, the Company and CRG executed another amendment to the terms of its CRG Term Loan, as amended in March 2016 and April 2017. This amendment extends the availability of the $15.0 million borrowing capacity through December 31, 2017. The Company did not draw down any amount under the $15.0 million tranche and it has since expired.
In February 2018, the Company and CRG executed an amendment to the terms of its CRG Term Loan, as amended in March 2016, April 2017 and October 2017, to decrease the amount of the minimum combined 2016 and 2017 revenue covenant.
At December 31, 2017, the Company had $45.0 million in outstanding debt and $4.8 million in deferred payment in- kind interest to CRG, and was in compliance with all financial covenants under the CRG Term Loan.
The Company’s scheduled future payment on the CRG Term Loan at December 31, 2017 are as follows (in thousands):
Year Ended December 31, | | | | |
2018 | | $ | 4,108 | |
2019 | | | 4,299 | |
2020 | | | 62,803 | |
Total future payments | | | 71,210 | |
Less: amount representing interest and end-of-term facility fee | | | (26,210 | ) |
Total principal amount | | | 45,000 | |
Less: unamortized debt discount | | | (496 | ) |
Carrying value of long-term debt | | | 44,504 | |
Less: current portion | | | — | |
Long-term portion | | $ | 44,504 | |
118
7. | Commitments and Contingencies |
Operating Leases
The Company leases office space in Oakwood Village, Ohio and Mountain View, California under non-cancellable operating leases. At December 31, 2017, the future minimum payments for the operating leases are as follows (in thousands):
Year Ended December 31, | | | | |
2018 | | $ | 1,118 | |
2019 | | | 1,039 | |
Total future minimum payments | | $ | 2,157 | |
Rent expense incurred under operating leases was $1.3 million in each of the years ended December 31, 2017, 2016 and 2015, respectively.
Contingencies
The Company is subject to claims and assessments from time to time in the ordinary course of business. The Company records a provision for a liability when it believes that it is both probable that a liability has been incurred and the amount can be reasonably estimated. Significant judgment is required to determine both probability and the estimated amount.
In the normal course of business, the Company may become involved in legal proceedings. The Company will accrue a liability for such matters when it is probable that a liability has been incurred and the amount can be reasonably estimated. When only a range of possible loss can be established, the most probable amount in the range is accrued. If no amount within this range is a better estimate than any other amount within the range, the minimum amount in the range is accrued. The accrual for a litigation loss contingency might include, for example, estimates of potential damages, outside legal fees and other directly related costs expected to be incurred. The Company was subject to an arbitration claim that arose in the ordinary course of business at September 30, 2017. This claim was settled in the fourth quarter of fiscal year 2017. At December 31, 2017, the Company was not involved in any material legal proceedings.
Purchase Commitments
At December 31, 2017 and 2016, the Company had no outstanding firm purchase commitments.
In December 2004, ViewRay Technologies, Inc. entered into a licensing agreement with the University of Florida Research Foundation, Inc., or UFRF, whereby UFRF granted the Company a worldwide exclusive license to certain of UFRF’s patents in exchange for 33,652 shares of common stock and a royalty from sales of products developed and sold by the Company utilizing the licensed patents. ViewRay Technologies, Inc. met all of the product development and commercialization milestones at December 31, 2013 and started to make quarterly royalty payments in 2014. Royalty payments are based on 1% of net sales, defined as the amount collected on sales of licensed products and/or licensed processes after deducting trade and/or quantity discounts, credits on returns and allowances, outbound transportation costs paid and sales tax. Minimum quarterly royalty payments of $50 thousand commenced with the quarter ended March 31, 2014 and are payable in advance. Minimum royalties paid in any calendar year will be credited against earned royalties for such calendar year. The royalty payments continue until the earlier of (i) the date that no licensed patents remain enforceable or (ii) the payment of earned royalties, once begun in 2014, cease for more than four consecutive calendars quarters. Royalty expenses based on 1% of net sales were $274.4 thousand, $206 thousand and $49 thousand during the years ended December 31, 2017, 2016 and 2015, respectively, and were recorded as product cost of revenue in the accompanying consolidated statements of operations and comprehensive loss. The minimum royalty payments in excess of 1% of net sales were $25 thousand, $57 thousand and $102 thousand during the years ended December 31, 2017, 2016 and 2015, respectively, and were
119
recorded as general and administrative expenses in the accompanying consolidated statements of operations and comprehensive loss.
In December 2014, the Company entered into a distribution agreement with Itochu Corporation, or Itochu, a Japanese entity, pursuant to which the Company appointed Itochu as its exclusive distributor for the sale and delivery of its MRIdian products within Japan. The exclusive distribution agreement has an initial term of 10 years from December 2014, and contains features customary in such distribution agreements. Under this distribution agreement, the Company will supply its products and services to Itochu based upon the Company’s then-current pricing. In consideration of the exclusive distribution rights granted, ltochu agreed to pay a distribution fee of $4.0 million in three installments: (i) the first installment of $1.0 million was due upon execution of the distribution agreement; (ii) the second installment of $1.0 million was due within 10 business days following submission of the application for regulatory approval of the Company’s product to the Japan regulatory authority; and (iii) the final installment of $2.0 million was due within 10 business days following receipt of approval for the Company’s product from the Japanese Ministry of Health, Labor and Welfare. The distribution fee paid by Itochu was refundable if the Company failed to obtain the approval from the Japan regulatory authority before December 31, 2017. The first and second installments of $2.0 million in aggregate were received in December 2014 and December 2015, respectively. In August 2016, the Company received the third and final $2.0 million installment upon the receipt of regulatory approval to market MRIdian in Japan. The entire $4.0 million distribution fee received was reclassified to deferred revenue as it was no longer refundable. In August 2016, the Company started recognizing distribution rights revenue on a straight-line basis over the remaining term of the exclusive distribution agreement of approximately 8.5 years. The distribution rights revenue was $475 thousand and $178 thousand during the years ended December 31, 2017 and 2016, respectively.
Private Placements
In September 2016, the Company completed the final closing of a private placement offering, or the 2016 Private Placement, through which it sold (i) 4,602,506 shares of its common stock and (ii) warrants that provide the warrant holders the right to purchase 1,380,745 shares of common stock, or the 2016 Placement Warrants, and raised total gross proceeds of $13.8 million. The 2016 Placement Warrants have an exercise price of $2.95 per share, are exercisable at any time at the option of the holder and expire seven years from the date of issuance.
In January 2017, the Company completed the final closing of a private placement offering, or the January 2017 Private Placement, through which it sold (i) 8,602,589 shares of its common stock and (ii) warrants that provide the warrant holders the right to purchase 1,720,512 shares of common stock, or the 2017 Placement Warrants, and raised total gross proceeds of $26.1 million. The 2017 Placement Warrants have an exercise price of $3.17 per share, became exercisable in July 2017 and expire in January 2024.
Direct Registered Offering
In October 2017, the Company completed the final closing of a direct registered offering, or the October 2017 Direct Registered Offering, through which it sold 8,382,643 shares of its common stock and raised total gross proceeds of $50.0 million.
At-The-Market Offering of Common Stock
In January 2017, the Company filed a shelf registration statement on Form S-3 with the SEC, which included a base prospectus covering the offering, issuance and sale of up to a maximum aggregate offering of $75.0 million of the Company’s common stock, preferred stock, debt securities, warrants, purchase contracts and/or units; and the Company entered into a sales agreement with FBR Capital Markets & Co., or FBR, under which it may sell up to $25.0 million of its common shares pursuant to an at-the-market offering program in accordance with Rule 415(a)(4) under the Securities Act. FBR acted as sales agent on a best efforts basis and used commercially reasonable efforts to sell on behalf of the Company all of the shares of common stock requested to be sold by the Company, consistent with its normal trading and sales practices, on mutually agreed terms between FBR and the Company. There is no
120
arrangement for funds to be received in any escrow, trust or similar arrangement. In April 2017, the Company agreed to sell up to an additional $25.0 million of the Company’s common stock in accordance with the terms of a sales agreement with FBR and pursuant to an at-the-market offering program in accordance with Rule 415(a)(4) under the Securities Act.
FBR is entitled to compensation of up to 3.0% of the gross sales price per share sold. In connection with the sale of the Company’s common stock on the Company’s behalf, FBR is deemed to be an “underwriter” within the meaning of the Securities Act and the compensation of FBR is deemed to be underwriting commissions or discounts. The Company has also agreed to provide indemnification and contribution to FBR with respect to certain liabilities, including liabilities under the Securities Act.
At December 31, 2017, the Company sold an aggregate of 6,575,062 shares of its common stock under the at-the-market offering program at an average market price of $6.10 per share, resulting in aggregate gross proceeds of approximately $40.1 million.
In April 2017, the Company filed another shelf registration statement on Form S-3, which included a base prospectus covering the offering, issuance and sale of up to a maximum aggregate offering of $100.0 million of the Company’s common stock, preferred stock, debt securities, warrants, purchase contracts and/or units. At December 31, 2017, no securities had been sold pursuant to this registration statement.
11. | Common Stock Reserved for Issuance |
The common stock reserved for future issuance at December 31, 2017 and 2016 was as follows:
| | December 31, | |
| | 2017 | | | 2016 | |
Shares underlying outstanding stock options | | | 8,592,747 | | | | 6,127,291 | |
Shares available for future stock option grants | | | 969,783 | | | | 2,168,391 | |
Shares issuable upon settlement of restricted stock units outstanding | | | 149,636 | | | | 151,240 | |
ESPP shares available for issuance | | | 1,103,481 | | | | 667,670 | |
Warrant to purchase common stock | | | 3,393,755 | | | | 1,707,736 | |
Total shares of common stock reserved | | | 14,209,402 | | | | 10,822,328 | |
12. | Convertible Preferred Stock |
In January 2015, the Company issued an aggregate of 162,407 shares of Series C convertible preferred stock to a new investor at a price of $5.84 per share for a total gross consideration of $950 thousand.
In February 2015, the Company issued 2,564,652 shares of Series C convertible preferred stock to another investor at a price of $5.84 per share for total gross consideration of $15.0 million.
In July 2015, upon the closing of the Merger, all of ViewRay Technologies, Inc.’s 30,381,987 shares of outstanding convertible preferred stock were converted into the Company’s common stock at a 1:1 conversion rate. As a result, the Company had no convertible preferred stock issued and outstanding at December 31, 2017, 2016 and 2015.
Preferred Stock Warrants
In connection with a 2013 debt financing (see Note 6), the Company issued a warrant to purchase 128,231 shares of Series C convertible preferred stock. These warrants have an exercise price of $5.84 per share, and are exercisable any time at the option of the holder until December 16, 2023. The convertible preferred stock warrant was recorded as a liability and is adjusted to fair value at each balance sheet date, with the change in fair value being recorded as a component of other expense, net in the consolidated statements of operations and comprehensive loss. For the year
121
ended December 31, 2015, the Company recognized a gain of $45 thousand related to the change in fair value of the warrant in the accompanying consolidated statements of operations and comprehensive loss.
Upon the consummation of the Merger in July 2015, the warrant to purchase Series C convertible preferred stock was converted into the warrant to purchase 128,231 shares of the Company’s common stock. As a result, the fair value of the preferred stock warrant liability of $93 thousand was reclassified into additional paid-in capital.
The Company used the Black-Scholes option-pricing model to estimate the fair value of the convertible preferred stock warrant upon conversion with the following assumptions:
| | Upon the Closing of the Merger on July 23, 2015 | |
Common Stock Warrants: | | | | |
Expected term (in years) | | | 5.0 | |
Expected volatility (%) | | 31.8% | |
Risk-free interest rate (%) | | 1.7% | |
Expected dividend yield (%) | | 0% | |
Equity Classified Common Stock Warrants
In connection with the Merger and the Private Placement, in July and August 2015, the Company issued warrants, or 2015 Placement Warrants, that provide the warrant holder the right to purchase 198,760 shares of common stock at an exercise price of $5.00 per share. These warrants were issued to private placement agents as payment for services provided. The 2015 Placement Warrants are exercisable at any time at the option of the holder until the five-year anniversary of their date of issuance.
The Company estimated the aggregate fair value of 2015 Placement Warrants on issuance date to be $316 thousand which was recorded in additional paid-in capital as an offering cost against the total proceeds from the Private Placement.
The fair value of the placement warrants was measured at their grant dates using the Black-Scholes pricing model and the following weighted average assumptions:
| | Upon Issuance | |
Common Stock Warrants: | | | | |
Expected term (in years) | | | 5.0 | |
Expected volatility (%) | | 31.8% | |
Risk-free interest rate (%) | | 1.6% | |
Expected dividend yield (%) | | 0% | |
At December 31, 2017 and 2016, all of these equity classified warrants had not been exercised and remain outstanding.
Liability Classified Common Stock Warrants
In connection with the 2016 Private Placement, in August and September 2016, the Company issued warrants, the 2016 Placement Warrants, that provide the warrant holder the right to purchase 1,380,745 shares of common stock at an exercise price of $2.95 per share.
These 2016 Placement Warrants are exercisable at any time at the option of the holder until the seven-year anniversary of their date of issuance. The 2016 Placement Warrants also contain protection whereby the warrants will expire immediately prior to the consummation of a change of control, as defined in the agreement, and holders have the right to receive cash in the amount equal to the Black-Scholes value of warrants. The 2016 Placement Warrants were accounted for as a liability at the date of issuance and are adjusted to fair value at each
122
balance sheet date, with the change in fair value recorded as a component of other expense, net in the consolidated statements of operations and comprehensive loss.
As separate classes of securities were issued in a bundled transaction, the gross proceeds from the 2016 Private Placement of $13.8 million was allocated first to the 2016 Placement Warrants based on their fair value upon issuance, and the residual was allocated to the common stock. The fair value upon issuance of $2.7 million for the 2016 Placement Warrants was estimated using the Black-Scholes option-pricing model with the following weighted-average assumptions: expected term of seven years, expected volatility of 61.6%, risk-free interest rate of 1.4% and expected dividend yield of 0%.
During the year ended December 31, 2017 and 2016, the Company recorded loss of $7.4 million and a gain of $3 thousand, respectively, related to the change in fair value of the 2016 Placement Warrants. The fair value of the 2016 Placement Warrants of $9.9 million and $2.7 million at December 31, 2017 and 2016, respectively, was estimated using the Black-Scholes option pricing model and the following weighted-average assumptions:
| | December 31, | |
| | 2017 | | | 2016 | |
2016 Placement Warrants: | | | | | | | | |
Expected term (in years) | | | 5.7 | | | | 6.7 | |
Expected volatility (%) | | 62.1% | | | 63.6% | |
Risk-free interest rate (%) | | 2.2% | | | 2.3% | |
Expected dividend yield (%) | | 0% | | | 0% | |
In connection with the January 2017 Private Placement, the Company issued warrants, the 2017 Placement Warrants, that provide the warrant holder the right to purchase 1,720,512 shares of common stock at an exercise price of $3.17 per share. These 2017 Placement Warrants became exercisable in July 2017 and expire in January 2024. The 2017 Placement Warrants also contain protection whereby warrants will expire immediately prior to the consummation of a change of control, as defined in the agreement, and holders have the right to receive cash in the amount equal to the Black-Scholes value of the warrants. The 2017 Placement Warrants were accounted for as a liability at the date of issuance and are adjusted to fair value at each balance sheet date, with the change in fair value recorded as a component of other expense, net in the consolidated statements of operations and comprehensive loss.
As separate classes of securities were issued in a bundled transaction, the gross proceeds from the January 2017 Private Placement of $26.1 million was allocated first to the 2017 Placement Warrants based on its fair value upon issuance, and the residual was allocated to the common stock. The fair value upon issuance of $3.4 million for the 2017 Placement Warrants was estimated using the Black-Scholes option-pricing model with the following weighted-average assumptions: expected term of seven years, expected volatility of 62.9%, risk-free interest rate of 2.2% and expected dividend yield of 0%.
During the year ended December 31, 2017, the Company recorded a loss of $9.2 million related to the change in fair value of the 2017 Placement Warrants. The fair value of the 2017 Placement Warrants of $12.5 million at December 31, 2017 was estimated using the Black-Scholes option pricing model and the following weighted-average assumptions:
| | December 31, 2017 | |
2017 Placement Warrants: | | | | |
Expected term (in years) | | | 6.1 | |
Expected volatility (%) | | 62.3% | |
Risk-free interest rate (%) | | 2.3% | |
Expected dividend yield (%) | | 0% | |
In December 2017, 25,104 shares of 2016 Placement Warrant and 9,389 shares of 2017 Placement Warrant were exercised and the related warrant liability of $274 thousand was reclassified into additional paid-in capital upon
123
exercise. At December 31, 2017, 1,355,641 shares of the 2016 Placement Warrant and 1,711,123 shares of the 2017 Placement Warrant were outstanding.
14. | Stock-Based Compensation |
The Company adopted 2008 Stock Option and Incentive Plan, or 2008 Plan, and 2015 Equity Incentive Award Plan, or 2015 Plan, to its employees, officers, directors, advisors and consultants. With the establishment of the 2015 Plan, the Company no longer grants stock options under the 2008 Plan, and the shares available for future grants under the 2008 Plan were transferred to the 2015 Plan.
Only stock options were granted under the 2008 Plan. The 2015 Plan provides for the grant of stock and stock-based awards including stock options, restricted stock awards, restricted stock units and stock appreciation rights.
Options granted may be either incentive stock options or non-statutory stock options. Under the 2008 Plan, incentive stock options could only have been granted to employees with exercise prices of no less than the fair value of the common stock on the grant date and non-statutory options may be granted to employees or consultants at exercise prices of no less than 85% of the fair value of the common stock on the grant date, as determined by the board of directors. Under the 2015 Plan, for both inventive stock options and nonstatutory options, the exercise price should not be less than the fair value of the common stock on the date of grant. Under both the 2008 Plan and the 2015 Plan, if, at the time of grant, the optionee owns stock representing more than 10% of the voting power of all classes of stock of the Company, a 10% shareholder, the exercise price must be at least 110% of the fair value of the common stock on the grant date as determined by the board of directors. Options become exercisable generally ratably over four years, and expire in 10 years from the date of grant, or five years from the date of grant for 10% shareholders.
In July 2015, the Company adopted the 2015 Employee Stock Purchase Plan, or 2015 ESPP, and 667,670 shares were reserved for issuance under the 2015 ESPP. At December 31, 2017 and 2016, no shares have been issued under the 2015 ESPP.
A summary of the Company’s stock option activity and related information is as follows:
| | | | | | Options Outstanding | |
| | Shares Available for Grant | | | Number of Stock Options Outstanding | | | Weighted- Average Exercise Price | | | Weighted- Average Remaining Contractual Life (Years) | | | Aggregate Intrinsic Value | |
| | | | | | | | | | | | | | | | | | (In thousands) | |
Balance at December 31, 2016 | | | 2,168,391 | | | | 6,127,291 | | | | 2.60 | | | | 7.3 | | | | 7,800 | |
Additional authorized | | | 1,743,247 | | | | | | | | | | | | | | | | | |
Granted | | | (2,986,244 | ) | | | 2,986,244 | | | | 5.65 | | | | | | | | | |
Exercised | | | — | | | | (420,377 | ) | | | 1.58 | | | | | | | | | |
Cancelled | | | 100,411 | | | | (100,411 | ) | | | 3.95 | | | | | | | | | |
RSUs granted | | | (56,022 | ) | | | — | | | | | | | | | | | | | |
Balance at December 31, 2017 | | | 969,783 | | | | 8,592,747 | | | $ | 3.69 | | | | 7.4 | | | $ | 47,864 | |
Vested and exercisable at December 31, 2017 | | | | | | | 5,011,207 | | | $ | 2.65 | | | | 6.5 | | | $ | 33,130 | |
Vested and expected to vest at December 31, 2017 | | | | | | | 8,303,413 | | | $ | 3.64 | | | | 7.4 | | | $ | 46,655 | |
The weighted-average grant date fair value of options granted to employees was $3.38, $2.72 and $3.13 per share for the year ended December 31, 2017, 2016 and 2015. The grant date fair value of options vested was $4.8 million, $2.4 million and $782 thousand, respectively, during the year ended December 31, 2017, 2016 and 2015.
124
Aggregate intrinsic value represents the difference between the estimated fair value of the underlying common stock and the exercise price of outstanding, in-the-money options. The aggregate intrinsic value of options exercised was $2.6 million and $2.3 million for the year ended December 31, 2017 and 2016. The aggregate intrinsic value of options exercised was insignificant for the year ended December 31, 2015.
At December 31, 2017, total unrecognized compensation cost related to stock-based awards granted to employees, net of estimated forfeitures, was $9.9 million which is expected to be recognized over a weighted-average period of 2.7 years.
Determination of Fair Value
The determination of the fair value of stock options on the date of grant using an option-pricing model is affected by the estimated fair value of the Company’s common stock, as well as assumptions regarding a number of complex and subjective variables. The variables used to calculate the fair value of stock options using the Black-Scholes option-pricing model include actual and projected employee stock option exercise behaviors, expected price volatility of the Company’s common stock, the risk-free interest rate and expected dividends. Each of these inputs is subjective and generally requires significant judgment to determine.
Fair Value of Common Stock
Prior to the Merger, the fair value of the common stock underlying the stock-based awards was determined by ViewRay Technologies, Inc.’s board of directors, with input from management and third-party valuations. Post-Merger and up through March 30, 2016, the Company’s common stock shares were listed on the OTC Bulletin Board. Beginning March 31, 2016, the Company’s common stock shares were listed on The NASDAQ Global Market, or NASDAQ. Fair value of the common stock is the adjusted closing price of the Company’s common stock on the trading date on these stock exchanges.
Expected Term
The expected term represents the period that the Company’s option awards are expected to be outstanding. The Company considers several factors in estimating the expected term of options granted, including the expected lives used by a peer group of companies within the Company’s industry that the Company considers to be comparable to its business and the historical option exercise behavior of its employees, which the Company believes is representative of future behavior.
Expected Volatility
As the Company does not have a sufficient trading history for its common stock, the expected stock price volatility for the Company’s common stock was estimated by taking the average historic price volatility for industry peers based on daily price observations over a period equivalent to the expected term of the stock option grants. Industry peers consist of several public companies in the Company’s industry which were the same as the comparable companies used in the common stock valuation analysis. The Company intends to continue to consistently apply this process using the same or similar public companies until a sufficient amount of historical information regarding the volatility of its own share price becomes available, or unless circumstances change such that the identified companies are no longer similar to the Company, in which case, more suitable companies whose share prices are publicly available would be used in the calculation.
Risk-Free Interest Rate
The risk-free interest rate is based on the zero-coupon U.S. Treasury notes, with maturities similar to the expected term of the options.
125
Expected Dividend Yield
The Company does not anticipate paying any dividends in the foreseeable future and, therefore, uses an expected dividend yield of zero in the Black-Scholes option-valuation model.
In addition to the Black-Scholes assumptions discussed immediately above, the estimated forfeiture rate also has a significant impact on the related stock-based compensation. The forfeiture rate of stock options is estimated at the time of grant and revised in subsequent periods if actual forfeitures differ from those estimates. The Company uses historical data to estimate pre-vesting option forfeitures and records stock-based compensation expense only for those awards that are expected to vest.
The fair value of employee stock options was estimated at the date of grant using a Black-Scholes option-pricing model with the following weighted-average assumptions:
| | Year Ended December 31, | |
| | 2017 | | | 2016 | | | 2015 | |
Expected term (in years) | | | 5.9 | | | | 6.0 | | | | 5.9 | |
Expected volatility (%) | | 66.0% | | | 67.1% | | | 68.7% | |
Risk-free interest rate (%) | | 2.1% | | | 1.3% | | | 1.8% | |
Expected dividend yield (%) | | 0.0% | | | 0.0% | | | 0.0% | |
Restricted Stock Units
From time to time, the Company grants Restricted Stock Units, or RSUs, to its board of directors for their services. These RSUs were fully vested upon issuance and will be released and settled upon termination of the board services or the occurrence of a change in control event. In September 2016 and November 2017, the Company granted 112,578 shares and 43,554 shares of RSUs to its board members, respectively, and 18,964 shares of these RSUs were released in December 2017 upon termination of one board member.
In December 2016, the Company granted 18,017 shares of RSUs to certain executive officers for bonus and 20,645 shares of RSUs to a consultant for service. These RSUs were fully vested upon issuance and released in fiscal year 2017.
In November 2017, the Company granted 12,468 shares of RSUs to one executive officer upon his termination. These RSUs were fully vested upon issuance but not released in fiscal year 2017, although the conditions to release these RSUs were satisfied at December 31, 2017.
The fair value of RSUs is based on the closing market price of the Company’s common stock on the grant date. The weighted-average grant date fair value of RSUs granted in fiscal year 2016 and 2017 was $3.52 per share and $8.02 per share, respectively, and the Company recorded stock based compensation expense related to RSUs of $532 thousand and $449 thousand during the year ended December 31, 2016 and 2017, which was included in general and administrative expenses in the accompanying statements of operations and comprehensive loss. There was no stock based compensation expense related to RSUs during the year ended December 31, 2015.
Stock-Based Compensation Expense
Total stock-based compensation expense recognized in the Company’s consolidated statements of operations and comprehensive loss is classified as follows (in thousands):
| | Year Ended December 31, | |
| | 2017 | | | 2016 | | | 2015 | |
Research and development | | $ | 952 | | | $ | 593 | | | $ | 262 | |
Selling and marketing | | | 303 | | | | 120 | | | | 50 | |
General and administrative | | | 4,064 | | | | 2,194 | | | | 754 | |
Total stock-based compensation expense | | $ | 5,319 | | | $ | 2,907 | | | $ | 1,066 | |
126
During the years ended December 31, 2017, 2016 and 2015 there were no stock-based compensation expenses capitalized as a component of inventory or recognized in cost of revenue. Stock-based compensation relating to stock-based awards granted to consultants was insignificant for the years ended December 31, 2017, 2016 and 2015.
On December 22, 2017, the Tax Cuts and Jobs Act (the “2017 Tax Act”) was enacted. The 2017 Tax Act includes a number of changes to existing U.S. tax laws that impact the Company, most notably a reduction of the top U.S. corporate income tax rate from 35 percent to 21 percent for tax years beginning after December 31, 2017.
The Company recognized the income tax effects of the 2017 Tax Act in its 2017 financial statements in accordance with SEC Staff Accounting Bulletin No. 118 (“SAB 118”), which provides guidance for the application of ASC Topic 740, Income Taxes, in the reporting period in which the 2017 Tax Act was signed into law. As such, the Company’s financial results reflect the income tax effects of the 2017 Tax Act for which the accounting under ASC Topic 740 is complete and provisional amounts for those specific income tax effects of the 2017 Tax Act for which the accounting under ASC Topic 740 is incomplete but a reasonable estimate could be determined. Upon completion of our 2017 U.S. income tax return in 2018 we may identify additional remeasurement adjustments to our recorded deferred tax assets. We will continue to assess our provision for income taxes as future guidance is issued, but do not currently anticipate significant revisions will be necessary. Any such revisions will be treated in accordance with the measurement period guidance outlined in SAB 118.
The changes to existing U.S. tax laws as a result of the 2017 Tax Act, which we believe have the most significant impact on the Company’s federal income taxes are as follows:
Reduction of the U.S. Corporate Income Tax Rate
The Company measures deferred tax assets and liabilities using enacted tax rates that will apply in the years in which the temporary differences are expected to be recovered or paid. Accordingly, the Company’s deferred tax assets and liabilities were remeasured to reflect the reduction in the U.S. corporate income tax rate from 35 percent to 21 percent. This resulted in a $38.7 million decrease in net deferred tax assets and a corresponding $38.7 million decrease to the valuation allowance as of December 31, 2017.
Income Tax Expense
The following reconciles the differences between income taxes computed at the federal income tax rate and the provision for income taxes:
| | Year Ended December 31, |
| | 2017 | | | 2016 | | | 2015 | | |
Expected income tax benefit at the federal statutory rate | | | 34.0 | | % | | 34.0 | | % | | 34.0 | | % |
State taxes, net of federal benefit | | | 0.0 | | | | 0.0 | | | | (0.8 | ) | |
Change in effective tax rate | | | (54.1 | ) | | | 0.0 | | | | (0.9 | ) | |
Non-deductible items and other | | | 0.5 | | | | (0.7 | ) | | | (0.5 | ) | |
Federal and state credits | | | 0.5 | | | | 0.6 | | | | (0.7 | ) | |
Change in valuation allowance | | | 19.1 | | | | (33.9 | ) | | | (31.1 | ) | |
Total | | | 0.0 | | % | | 0.0 | | % | | 0.0 | | % |
127
Deferred income taxes reflect the net effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The principal components of the Company’s net deferred tax assets consisted of the following at December 31, 2017 and 2016 (in thousands):
| | Year Ended December 31, | |
| | 2017 | | | 2016 | |
Net operating loss carryforwards | | $ | 61,049 | | | $ | 75,036 | |
Research and development tax credits | | | 3,731 | | | | 2,131 | |
Reserves and accruals | | | 1,168 | | | | 1,791 | |
Other | | | 6,611 | | | | 4,594 | |
Total deferred tax assets | | | 72,559 | | | | 83,552 | |
Valuation allowance | | | (72,559 | ) | | | (83,552 | ) |
Net deferred tax assets | | $ | — | | | $ | — | |
The Company maintains a valuation allowance related to its deferred tax asset position when management believes it is more likely than not that the net deferred tax assets will not be realized in the future. The Company’s valuation allowance decreased by $11.0 million and increased by $17.4 million during the year ended December 31, 2017 and 2016.
At December 31, 2017, the Company had federal net operating loss carryforwards of $266.3 million, which begin to expire in the year ending December 31, 2024, and $144.8 million related to state net operating loss carryforwards, which begin to expire in the year ending December 31, 2019. The Company had federal research and development tax credit carryforwards of $3.8 million, and state carryforwards of $1.3 million at the year ended December 31, 2017. These credits begin to expire in the year ending December 31, 2024.
Under the provisions of the Internal Revenue Code, or IRC, net operating loss and credit carryforwards and other tax attributes may be subject to limitation if there has been a significant change in ownership of the Company, as defined by the IRC. The Company believes it has experienced at least one ownership change in the past. The Company is currently analyzing the tax impact of such ownership change on its federal net operating loss and credit carryforwards. Future change in the Company’s ownership could result in limitations on net operating loss and credit carryforwards.
Because of the net operating loss and credit carryforwards, all of the Company’s federal tax returns and state returns since the year ended December 31, 2004 remain subject to federal and California examination.
The Company accounts for uncertain tax positions using a “more-likely-than-not” threshold. The evaluation of uncertain tax positions is based on factors including, but not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. The Company evaluates these tax positions on an annual basis. In addition, the Company also accrues for potential interest and penalties related to unrecognized tax benefits in income tax expense.
At December 31, 2017 and 2016, the Company’s unrecognized tax benefits consist of the following:
| | Year Ended December 31, | |
| | 2017 | | | 2016 | |
Unrecognized tax benefit, beginning of period | | $ | 940 | | | $ | 742 | |
Gross increases — current year tax positions | | | 327 | | | | 198 | |
Gross increases — prior year tax positions | | | 73 | | | | — | |
Gross decreases — prior year tax positions | | | (205 | ) | | | — | |
Unrecognized tax benefit, end of period | | $ | 1,135 | | | $ | 940 | |
128
The Company has a 401(k) Plan which covers its eligible employees. The 401(k) Plan permits the participants to defer a portion of their compensation in accordance with the provisions of Section 401(k) of the IRC. At its discretion, the Company can match a portion of the participants’ contributions or make profit-sharing contributions. There was no matching or profit-sharing contributions during the years ended December 31, 2017, 2016 or 2015.
The following table sets forth the computation of the Company’s basic and diluted net loss per share for the periods presented (in thousands, except share and per share data):
| | Year Ended December 31, | |
| | 2017 | | | 2016 | | | 2015 | |
Net loss | | $ | (72,176 | ) | | $ | (50,636 | ) | | $ | (44,995 | ) |
Weighted-average common shares used in computing net loss per share, basic and diluted | | | 58,457,868 | | | | 40,068,307 | | | | 17,432,434 | |
Net loss per share, basic and diluted | | $ | (1.23 | ) | | $ | (1.26 | ) | | $ | (2.58 | ) |
The following weighted-average common stock equivalents were excluded from the calculation of diluted net loss per share for the periods presented because including them would have had an anti-dilutive effect:
| | Year Ended December 31, | |
| | 2017 | | | 2016 | | | 2015 | |
Convertible preferred stock (if converted) | | | — | | | | — | | | | 16,558,330 | |
Options to purchase common stock | | | 7,914,067 | | | | 6,181,015 | | | | 5,032,768 | |
Convertible preferred stock warrant | | | — | | | | — | | | | 71,318 | |
Common stock warrant | | | 3,345,674 | | | | 804,248 | | | | 142,513 | |
Restricted stock units | | | 108,107 | | | | 33,835 | | | | — | |
18. | Segment and Geographic Information |
The Company has one business activity, which is radiation therapy technology combined with magnetic resonance imaging, and operates in one reportable segment. The Company’s chief operating decision-maker, its chief executive officer, reviews its operating results on an aggregate basis for purposes of allocating resources and evaluating financial performance. Also, the Company does not have segment managers as the Company manages its operations as a single operating segment.
The following table sets forth revenue by geographic area on the customers’ location (in thousands):
| | Year Ended December 31, | |
| | 2017 | | | 2016 | | | 2015 | |
United States | | $ | 11,506 | | | $ | 1,106 | | | $ | 5,332 | |
UAE | | | 5,675 | | | | — | | | | — | |
Korea | | | 5,504 | | | | 182 | | | | 4,988 | |
Israel | | | 5,309 | | | | — | | | | — | |
China | | | 4,680 | | | | — | | | | — | |
Japan | | | 753 | | | | 10,375 | | | | — | |
Netherlands | | | 317 | | | | 5,486 | | | | — | |
Italy | | | 298 | | | | 5,088 | | | | — | |
Rest of world | | | — | | | | — | | | | 70 | |
Total revenue | | $ | 34,042 | | | $ | 22,237 | | | $ | 10,390 | |
At December 31, 2017 and 2016, all long-lived assets are located in the United States.
129
19. | Related Party Transactions |
As discussed in Note 8, the Company pays a royalty to UFRF, a common stockholder, related to a licensing agreement.
In January 2017, the Company entered into a sales consulting agreement with Puissance Capital Management, or PCM, to assist with business development activities in a key market in Asia. PCM is the investment manager of Puissance Cross Board Opportunities LLP, a stockholder in the Company. Theodore T. Wang, Ph.D., a member of the Company’s board of directors, is the managing member of the general partners of PCM. The sales consulting agreement has a term of one year with a total consideration of $1.3 million.
In February 2018, the Company entered into a Securities Purchase Agreement pursuant to which it sold 4.1 million shares of common stock, 3.0 million shares of Series A convertible preferred stock and warrants to purchase 1.4 million shares of common stock for total gross proceeds of $59.1 million. These warrants have an exercise price of $8.31 per share, became exercisable upon issuance at the closing and expire seven years from the date of issuance. The Company completed the closing of this equity financing on March 5, 2018.
130
Item 9. Changes in and Disagreements with Accountants and Financial Disclosure and Supplementary Data
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the timelines specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
As required by Rule 13a-15(b) under the Exchange Act, we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures, as of the end of the period covered by this report. Based on the foregoing, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective at December 31, 2017 at the reasonable assurance level.
Changes in Internal Control
There were no changes in our internal control over financial reporting identified in management's evaluation pursuant to Rules 13a-15(d) or 15d-15(d) of the Exchange Act during the fourth quarter of 2017 that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Limitations on Effectiveness of Controls and Procedures
In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives. In addition, the design of controls and procedures must reflect the fact that there are resource constraints and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) and 15d-15(f) of the Exchange Act. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2017 based on the framework established in “Internal Control – Integrated Framework (2013)” issued by the Committee of Sponsoring Organizations of the Treadway Commission. Our management concluded that our internal control over financial reporting was effective as of that date.
131
As a smaller reporting company, pursuant to the rules of the SEC, this Annual Report on Form 10-K does not include an attestation report of the Company’s independent registered public accounting firm.
Item 9B. Other Information.
None.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Directors, Executive Officers and Corporate Governance
The information in our Proxy Statement for the 2018 Annual Meeting of stockholders regarding directors and executive officers appearing under the headings "Proposal One—Election of Directors," "Executive Officers" and "Section 16(a) Beneficial Ownership Reporting Compliance" is incorporated herein by reference.
In addition, the information in our Proxy Statement for the 2018 Annual Meeting of stockholders regarding the director nomination process, the Audit Committee financial expert and the identification of the Audit Committee members appearing under the heading "Corporate Governance and Board of Directors Matters" is incorporated herein by reference.
Code of Conduct and Ethics
We have adopted a Code of Conduct and Ethics that applies to all employees including our principal executive officer and principal financial officer. The full text of our Code of Business Conduct and Ethics is posted on our website at http://investors.viewray.com/corporate-governance/highlights.We intend to disclose future amendments to certain provisions of our code, or waivers of such provisions granted to executive officers and directors, on our website within four business days following the date of such amendment or waiver. Any information on ViewRay’s website or which can be accessed through it is not a part of this Annual Report on Form 10-K).
Item 11. Executive Compensation
We maintain employee compensation programs and benefit plans in which our executive officers are participants. Copies of these plans and programs are set forth or incorporated by reference as Exhibits to this Annual Report. The information in our Proxy Statement for the 2018 Annual Meeting of stockholders appearing under the heading “Executive Compensation” is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management Related Stockholder Matters
The information in our Proxy Statement for the 2018 Annual Meeting of stockholders appearing under the heading "Security Ownership of Certain Beneficial Owners and Management" and "Equity Compensation Plan Information" is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information in our Proxy Statement for the 2018 Annual Meeting of stockholders appearing under the headings "Certain Relationships and Related Party Transactions" and "Corporate Governance—Director Independence" is incorporated herein by reference.
Item 14. Principal Accountant Fees and Services
The information in our Proxy Statement for the 2018 Annual Meeting of stockholders appearing under the headings "Proposal Three—Ratification of Appointment of Independent Registered Public Accounting Firm—Audit and Non-Audit Services" and "Proposal Three—Ratification of Appointment of Independent Registered Public Accounting Firm—Audit Committee Pre-Approval Policies and Procedures" is incorporated herein by reference.
132
PART IV
Item 15. Exhibits and Financial Statement Schedules
(a) (1) The financial statements required by Item 15(a) are filed in Item 8 of this Report.
(2) The financial statement schedules required by Item 15(a) are omitted because they are not applicable, not required or the required information is included in the financial statements or notes thereto as filed in Item 8 of this Report.
(3) We have filed, or incorporated into this report by reference, the exhibits listed below.
| | | | Incorporated by Reference | | |
Exhibit Number | | Description | | Form | | Exhibit | | Date Filed | | Filed Herewith |
| | | | | | | | | | |
2.1 | | Agreement and Plan of Merger and Reorganization, dated as of July 23, 2015, by and among ViewRay Inc., Acquisition Sub and ViewRay Technologies, Inc. | | S-1/A | | 2.1 | | 12/16/15 | | |
| | | | | | | | | | |
3.1 | | Amended and Restated Certificate of Incorporation. | | S-1/A | | 3.1 | | 12/16/15 | | |
| | | | | | | | | | |
3.2 | | Amended and Restated Bylaws. | | S-1/A | | 3.2 | | 12/16/15 | | |
| | | | | | | | | | |
3.3 | | Certificate of Merger of Acquisition Sub with and into ViewRay Technologies, Inc. | | S-1/A | | 3.3 | | 12/16/15 | | |
| | | | | | | | | | |
3.4 | | Certificate of Designations, Preferences and Rights of Series A Convertible Preferred Stock of ViewRay, Inc. | | | | | | | | X |
| | | | | | | | | | |
4.1 | | Form of Common Stock Certificate. | | S-1/A | | 4.1 | | 12/16/15 | | |
| | | | | | | | | | |
4.2 | | Form of Placement Agent Warrant for Common Stock of ViewRay, Inc. | | S-1/A | | 10.6 | | 12/16/15 | | |
| | | | | | | | | | |
4.3 | | Form of Warrants issued pursuant to that certain Securities Purchase Agreement, dated as of August 19, 2016, by and among ViewRay, Inc. and the Purchasers named therein. | | S-1 | | 10.3 | | 9/26/16 | | |
| | | | | | | | | | |
4.4 | | Form of Warrants issued pursuant to that certain Securities Purchase Agreement, dated as of January 13, 2017, by and among ViewRay, Inc. and the Purchasers named therein. | | 10-K | | 4.4 | | 3/17/17 | | |
| | | | | | | | | | |
10.1 | | Split-Off Agreement, dated as of July 23, 2015, by and among ViewRay, Inc., Mirax Enterprise Corp. and Dinara Akzhigitova. | | S-1/A | | 10.1 | | 12/16/15 | | |
| | | | | | | | | | |
10.2 | | General Release Agreement, dated as of July 23, 2015, by and among ViewRay, Inc., Mirax Enterprise Corp. and Dinara Akzhigitova. | | S-1/A | | 10.2 | | 12/16/15 | | |
| | | | | | | | | | |
10.3 | | Form of Lock-Up and No Short Selling Agreement between ViewRay, Inc., and the officers, directors and shareholders party thereto. | | S-1/A | | 10.3 | | 12/16/15 | | |
| | | | | | | | | | |
10.4 | | Form of Securities Purchase Agreement between ViewRay, Inc., and the investors party thereto. | | S-1/A | | 10.4 | | 12/16/15 | | |
| | | | | | | | | | |
10.5 | | Engagement Letter, dated June 9, 2015, among ViewRay, Inc. and the Placement Agents as defined therein. | | S-1/A | | 10.5 | | 12/16/15 | | |
| | | | | | | | | | |
133
| | | | Incorporated by Reference | | |
Exhibit Number | | Description | | Form | | Exhibit | | Date Filed | | Filed Herewith |
10.6 | | Form of Registration Rights Agreement, by and among ViewRay, Inc. and certain investors named therein. | | S-1/A | | 4.2 | | 12/16/15 | | |
| | | | | | | | | | |
10.7(a) | | Office Lease, effective April 17, 2008, by and between Cleveland Industrial Portfolio, LLC and ViewRay Incorporated. | | S-1/A | | 10.7(a) | | 12/16/15 | | |
| | | | | | | | | | |
10.7(b) | | First Amendment to the Office Lease, effective April 16, 2013 by and between Cleveland Industrial Portfolio, LLC and ViewRay Incorporated. | | S-1/A | | 10.7(b) | | 12/16/15 | | |
| | | | | | | | | | |
10.7(c) | | Second Amendment to the Office Lease, effective August 15, 2014 by and between Cleveland Industrial Portfolio, LLC and ViewRay Incorporated. | | S-1/A | | 10.7(c) | | 12/16/15 | | |
| | | | | | | | | | |
10.8 | | Office Lease, effective June 19, 2014, by and between BXP Research Park LP and ViewRay Incorporated. | | S-1/A | | 10.8 | | 12/16/15 | | |
| | | | | | | | | | |
10.9† | | Employment Agreement, effective January 18, 2013, by and between ViewRay Incorporated and Chris A. Raanes. | | S-1/A | | 10.9 | | 12/16/15 | | |
| | | | | | | | | | |
10.10† | | Offer Letter, effective November 11, 2010, by and between ViewRay Incorporated and D. David Chandler. | | S-1/A | | 10.10 | | 12/16/15 | | |
| | | | | | | | | | |
10.11† | | First Amended and Restated Offer Letter, dated October 6, 2010, by and between ViewRay Incorporated and James F. Dempsey, Ph.D. | | S-1/A | | 10.11 | | 12/16/15 | | |
| | | | | | | | | | |
10.12† | | Offer Letter, dated December 9, 2011, by and between ViewRay Incorporated and Michael Brandt. | | S-1/A | | 10.12 | | 12/16/15 | | |
| | | | | | | | | | |
10.13# | | Manufacturing and Supply Agreement, effective September 18, 2013, by and between ViewRay Incorporated and Japan Superconductor Technology, Inc. | | S-1/A | | 10.13 | | 12/16/15 | | |
| | | | | | | | | | |
10.14(a)# | | Development and Supply Agreement, effective May 29, 2008, by and between ViewRay Incorporated and Siemens Aktiengesellschaft, Healthcare Sector. | | S-1/A | | 10.14(a) | | 12/16/15 | | |
| | | | | | | | | | |
10.14(b)# | | Amendment No. 1 to the Development and Supply Agreement, effective December 1, 2009, by and between ViewRay Incorporated and Siemens Aktiengesellschaft, Healthcare Sector. | | S-1/A | | 10.14(b) | | 12/16/15 | | |
| | | | | | | | | | |
10.14(c)# | | Amendment No. 2 to the Development and Supply Agreement, effective May 4, 2010, by and between ViewRay Incorporated and Siemens Aktiengesellschaft, Healthcare Sector. | | S-1/A | | 10.14(c) | | 12/16/15 | | |
| | | | | | | | | | |
10.14(d)# | | Amendment No. 3 to the Development and Supply Agreement, effective February 9, 2011, by and between ViewRay Incorporated and Siemens Aktiengesellschaft, Healthcare Sector. | | S-1/A | | 10.14(d) | | 12/16/15 | | |
| | | | | | | | | | |
10.14(e)# | | Amendment No. 4 to the Development and Supply Agreement, effective May 11, 2012, by and between ViewRay Incorporated and Siemens Aktiengesellschaft, Healthcare Sector. | | S-1/A | | 10.14(e) | | 12/16/15 | | |
134
| | | | Incorporated by Reference | | |
Exhibit Number | | Description | | Form | | Exhibit | | Date Filed | | Filed Herewith |
| | | | | | | | | | |
10.14(f)# | | Amendment No. 5 to the Development and Supply Agreement, effective May 30, 2012, by and between ViewRay Incorporated and Siemens Aktiengesellschaft, Healthcare Sector. | | S-1/A | | 10.14(f) | | 12/16/15 | | |
| | | | | | | | | | |
10.14(g)# | | Amendment No. 6 to the Development and Supply Agreement, effective February 21, 2014, by and between ViewRay Incorporated and Siemens Aktiengesellschaft, Healthcare Sector. | | S-1/A | | 10.14(g) | | 12/16/15 | | |
| | | | | | | | | | |
10.15# | | Cobalt-60 Source Supply and Removal Agreement, effective December 19, 2013, by and between ViewRay Incorporated and Best Theratronics, Ltd. | | S-1/A | | 10.15# | | 12/16/15 | | |
| | | | | | | | | | |
10.16# | | Development and Supply Agreement, effective June 24, 2009, by and between ViewRay Incorporated and Manufacturing Sciences Corporation. | | S-1/A | | 10.16# | | 12/16/15 | | |
| | | | | | | | | | |
10.17(a)# | | Development and Supply Agreement, effective July 9, 2009, by and between ViewRay Incorporated and Tesla Engineering Limited. | | S-1/A | | 10.17(a) | | 12/16/15 | | |
| | | | | | | | | | |
10.17(b)# | | Amendment No. 1 to the Development and Supply Agreement, effective January 20, 2015, by and between ViewRay Incorporated and Tesla Engineering Limited. | | S-1/A | | 10.17(b) | | 12/16/15 | | |
| | | | | | | | | | |
10.18# | | Development and Supply Agreement, effective July 2, 2010, by and between ViewRay Incorporated and PEKO Precision Products, Inc. | | S-1/A | | 10.18 | | 12/16/15 | | |
| | | | | | | | | | |
10.19(a)# | | Amended and Restated Joint Development and Supply Agreement, effective May 15, 2008, by and between ViewRay Incorporated and 3D Line GmbH. | | S-1/A | | 10.19(a) | | 12/16/15 | | |
| | | | | | | | | | |
10.19(b)# | | Amendment No. 1 to the Amended and Restated Joint Development and Supply Agreement, effective August 13, 2008, by and between ViewRay Incorporated and Euromechanics Medical GmbH. | | S-1/A | | 10.19(b) | | 12/16/15 | | |
| | | | | | | | | | |
10.19(c)# | | Amendment No. 2 to the Amended and Restated Joint Development and Supply Agreement, effective November 27, 2009, by and between ViewRay Incorporated and Euromechanics Medical GmbH. | | S-1/A | | 10.19(c) | | 12/16/15 | | |
| | | | | | | | | | |
10.20# | | Development and Supply Agreement, effective June 1, 2010, by and between ViewRay Incorporated and Quality Electrodynamics, LLC. | | S-1/A | | 10.20 | | 12/16/15 | | |
| | | | | | | | | | |
10.21(a)# | | Standard Exclusive License Agreement with Sublicensing Terms, effective December 15, 2004, by and between ViewRay Incorporated and the University of Florida Research Foundation, Inc. | | S-1/A | | 10.21(a) | | 12/16/15 | | |
| | | | | | | | | | |
10.21(b)# | | Amendment No. 1 to the Standard Exclusive License Agreement with Sublicensing Terms, effective December 6, 2007, by and between ViewRay Incorporated and the University of Florida Research Foundation, Inc. | | S-1/A | | 10.21(b) | | 12/16/15 | | |
135
| | | | Incorporated by Reference | | |
Exhibit Number | | Description | | Form | | Exhibit | | Date Filed | | Filed Herewith |
| | | | | | | | | | |
10.22 | | Warrant Agreement, effective December 16, 2013, by and between ViewRay Incorporated and Hercules Technology III, L.P. | | S-1/A | | 10.23 | | 12/16/15 | | |
| | | | | | | | | | |
10.23(a)# | | Term Loan Agreement, effective June 26, 2015, by and among ViewRay Incorporated, the Subsidiary Guarantors (as defined therein), Capital Royalty Partners II L.P., Capital Royalty Partners II—Parallel Fund “A” L.P., Capital Royalty Partners II (Cayman) L.P. and Parallel Investment Opportunities Partners II L.P. | | S-1/A | | 10.22 | | 12/16/15 | | |
| | | | | | | | | | |
10.23(b) | | Amendment No. 1 to Term Loan Agreement effective March 24, 2016, by and among ViewRay Technologies, Inc. (formerly known as ViewRay Incorporated), the Subsidiary Guarantors (as defined therein), Capital Royalty Partners II L.P., Capital Royalty Partners II—Parallel Fund “A” L.P., Capital Royalty Partners II (Cayman) L.P. and Parallel Investment Opportunities Partners II L.P. | | 10-K | | 10.23(b) | | 3/28/16 | | |
| | | | | | | | | | |
10.23(c) | | Amendment No. 2 to Term Loan Agreement dated April 12, 2017, by and among ViewRay Technologies, Inc. (formerly known as ViewRay Incorporated), the Subsidiary Guarantors (as defined therein), Capital Royalty Partners II L.P., Capital Royalty Partners II—Parallel Fund “A” L.P., Capital Royalty Partners II (Cayman) L.P. and Parallel Investment Opportunities Partners II L.P. | | 10-Q | | 10.1 | | 8/7/17 | | |
| | | | | | | | | | |
10.23(d) | | Amendment No. 3 to Term Loan Agreement effective September 30, 2017, by and among ViewRay Technologies, Inc. (formerly known as ViewRay Incorporated), the Subsidiary Guarantors (as defined therein), Capital Royalty Partners II L.P., Capital Royalty Partners II—Parallel Fund “A” L.P., Capital Royalty Partners II (Cayman) L.P. and Parallel Investment Opportunities Partners II L.P. | | 10-Q | | 10.1 | | 11/13/17 | | |
| | | | | | | | | | |
10.23(e) | | Amendment No.4 to Term Loan Agreement effective December 31, 2017, by and among ViewRay Technologies, Inc. (formerly known as ViewRay Incorporated), the Subsidiary Guarantors (as defined therein), Capital Royalty Partners II L.P., Capital Royalty Partners II—Parallel Fund “A” L.P., Capital Royalty Partners II (Cayman) L.P. and Parallel Investment Opportunities Partners II L.P | | | | | | | | X |
| | | | | | | | | | |
10.24(a)† | | ViewRay Incorporated 2008 Stock Incentive Plan. | | S-1/A | | 10.24(a) | | 12/16/15 | | |
| | | | | | | | | | |
10.24(b)† | | Form of Incentive Stock Option and Reverse Vesting Agreement (Change of Control) under the 2008 Plan. | | S-1/A | | 10.24(b) | | 12/16/15 | | |
| | | | | | | | | | |
10.24(c)† | | Form of Incentive Stock Option and Reverse Vesting Agreement under the 2008 Plan. | | S-1/A | | 10.24(c) | | 12/16/15 | | |
| | | | | | | | | | |
136
| | | | Incorporated by Reference | | |
Exhibit Number | | Description | | Form | | Exhibit | | Date Filed | | Filed Herewith |
10.24(d)† | | Form of Nonstatutory Stock Option and Reverse Vesting Agreement under the 2008 Plan. | | S-1/A | | 10.24(d) | | 12/16/15 | | |
| | | | | | | | | | |
10.25† | | Contingent Equity Agreement, effective January 8, 2008, by and among ViewRay Incorporated, James F. Dempsey, Ph.D., Russell S. Donda, Jim Carnall and William Wells. | | S-1/A | | 10.25 | | 12/16/15 | | |
| | | | | | | | | | |
10.26(a)† | | ViewRay, Inc. 2015 Equity Incentive Award Plan. | | S-1/A | | 10.26(a) | | 12/16/15 | | |
| | | | | | | | | | |
10.26(b)† | | Form of Option Agreement under the 2015 Plan. | | S-1/A | | 10.26(b) | | 12/16/15 | | |
| | | | | | | | | | |
10.26(c)† | | Form of Restricted Stock Agreement under the 2015 Plan. | | S-1/A | | 10.26(c) | | 12/16/15 | | |
| | | | | | | | | | |
10.26(d)† | | Form of Restricted Stock Unit Agreement under the 2015 Plan. | | S-1/A | | 10.26(d) | | 12/16/15 | | |
| | | | | | | | | | |
10.27† | | Form of Indemnification Agreement for directors and executive officers. | | S-1/A | | 10.27 | | 12/16/15 | | |
| | | | | | | | | | |
10.28† | | Agreement, effective June 11, 2008, by and among ViewRay Incorporated, James F. Dempsey, Ph.D., William W. Wells, James D. Carnall and Russell S. Donda. | | S-1/A | | 10.28 | | 12/16/15 | | |
| | | | | | | | | | |
10.29† | | ViewRay, Inc. 2015 Employee Stock Purchase Plan. | | S-1/A | | 10.29 | | 12/16/15 | | |
| | | | | | | | | | |
10.30† | | Offer Letter, dated April 30, 2015, between ViewRay, Inc. and Doug Keare. | | S-1/A | | 10.30 | | 12/16/15 | | |
| | | | | | | | | | |
10.31 | | Securities Purchase Agreement, dated as of August 19, 2016, by and among ViewRay, Inc. and the Purchasers named therein. | | S-1 | | 10.1 | | 9/26/16 | | |
| | | | | | | | | | |
10.32 | | Registration Rights Agreement, dated as of August 22, 2016, by and among ViewRay, Inc. and the Purchasers named therein. | | S-1 | | 4.3 | | 9/29/16 | | |
| | | | | | | | | | |
10.33 | | Securities Purchase Agreement, dated as of January 13, 2017, by and among ViewRay, Inc. and the Purchasers named therein. | | 10-K | | 10.33 | | 3/17/17 | | |
| | | | | | | | | | |
10.34 | | Stockholders’ Agreement, dated as of January 13, 2017, by and among ViewRay, Inc. and the Purchasers named therein. | | 10-K | | 10.34 | | 3/17/17 | | |
| | | | | | | | | | |
10.35 | | Agreement for Consulting Services by and among ViewRay, Inc. and Puissance Capital Management dated January 13, 2017. | | 10-Q | | 10.3 | | 5/15/17 | | |
| | | | | | | | | | |
10.36 | | Securities Purchase Agreement, dated as of October 23, 2017, by and among ViewRay, Inc. and Fosun International Limited named therein. | | 8-K | | 10.1 | | 10/25/17 | | |
| | | | | | | | | | |
10.37 | | Securities Purchase Agreement, dated as of October 23, 2017, by and among ViewRay, Inc. and the Purchasers named therein. | | 8-K | | 10.2 | | 10/25/17 | | |
| | | | | | | | | | |
10.38 | | Registration Rights Agreement, dated as of October 23, 2017, by and among ViewRay, Inc. and Strong Influence Limited. | | 8-K | | 10.3 | | 10/25/17 | | |
| | | | | | | | | | |
137
| | | | Incorporated by Reference | | |
Exhibit Number | | Description | | Form | | Exhibit | | Date Filed | | Filed Herewith |
10.39 | | Registration Rights Agreement, dated as of October 23, 2017, by and among ViewRay, Inc. and KVP Capital, LP. | | 8-K | | 10.4 | | 10/25/17 | | |
| | | | | | | | | | |
10.40 | | Amended and Restated Securities Purchase Agreement, dated as of March 5, 2018, by and among ViewRay, Inc. and Fosun International Limited named therein | | | | | | | | X |
| | | | | | | | | | |
10.41 | | Amended and Restated Registration Rights Agreement, dated as of March 5, 2018, by and among ViewRay, Inc. and Strong Influence Limited. | | | | | | | | X |
| | | | | | | | | | |
10.42 | | Warrant Agreement, effective February 25, 2018, by and between ViewRay Inc. and Strong Influence Limited. | | | | | | | | X |
| | | | | | | | | | |
| | | | | | | | | | |
21 | | List of Subsidiaries. | | | | | | | | X |
| | | | | | | | | | |
23.1 | | Consent of Deloitte & Touche LLP | | | | | | | | X |
| | | | | | | | | | |
24 | | Power of Attorney (contained on the signature page hereto). | | | | | | | | X |
| | | | | | | | | | |
31.1 | | Certification of Principal Executive Officer Required under Securities Exchange Act Rule 13a-14(a) and 15d-14(a). | | | | | | | | X |
| | | | | | | | | | |
31.2 | | Certification of Principal Financial Officer under Securities Exchange Act Rule 13a-14(a) and 15d-14(a). | | | | | | | | X |
| | | | | | | | | | |
32.1 | | Certification of Principal Executive Officer and Principal Financial Officer pursuant to 18 U.S.C. 1350 and Securities Exchange Act Rule 13a-14(b). | | | | | | | | X |
| | | | | | | | | | |
101 | | Interactive Data Files of Financial Statements and Notes. | | | | | | | | X |
| | | | | | | | | | |
101.INS | | Instant Document. | | | | | | | | X |
| | | | | | | | | | |
101.SCH | | XBRL Taxonomy Schema Document. | | | | | | | | X |
| | | | | | | | | | |
101.CAL | | XBRL Taxonomy Calculation Linkbase Document. | | | | | | | | X |
| | | | | | | | | | |
101.DEF | | XBRL Taxonomy Definition Linkbase Document. | | | | | | | | X |
| | | | | | | | | | |
101.LAB | | XBRL Taxonomy Label Linkbase Document. | | | | | | | | X |
| | | | | | | | | | |
101.PRE | | XBRL Taxonomy Presentation Linkbase Document. | | | | | | | | X |
# | Portions of this exhibit (indicated by asterisks) have been omitted pursuant to a request for confidential treatment and this exhibit has been filed separately with the SEC. |
† | Indicates management contract or compensatory plan. |
Item 16. Form 10-K Summary
None.
138
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on March 12, 2018.
VIEWRAY, INC. |
| | |
By: | | /s/ Chris A. Raanes |
| | Chris A. Raanes |
| | Chief Executive Officer |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints Chris A. Raanes and Ajay Bansal, and each of them, with full power of substitution and full power to act without the other, his or her true and lawful attorney-in-fact and agent to act for him or her in his or her name, place and stead, in any and all capacities, to sign any amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the U.S. Securities and Exchange Commission, hereby ratifying and confirming all that said attorney-in-fact, or his substitute, may do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed by the following persons in the capacities and on the dates indicated.
Signature | Title | Date |
| | |
/s/ Chris A. Raanes | Director, President and Chief Executive Officer | March 12, 2018 |
Chris A. Raanes | (Principal Executive Officer) | |
| | |
/s/ Ajay Bansal | Chief Financial Officer | March 12, 2018 |
Ajay Bansal | (Principal Financial and Accounting Officer) | |
| | |
| | |
/s/ David Bonita, M.D. | Director | March 12, 2018 |
David Bonita, M.D. | | |
| | |
/s/ Caley Castelein, M.D. | Director | March 12, 2018 |
Caley Castelein, M.D. | | |
| | |
/s/ James F. Dempsey, Ph.D. | Director and Chief Scientific Officer | March 12, 2018 |
James F. Dempsey, Ph.D. | | |
| | |
/s/ Mark S. Gold, M.D. | Director | March 12, 2018 |
Mark S. Gold, M.D. | | |
| | |
/s/ Aditya Puri | Director | March 12, 2018 |
Aditya Puri | | |
| | |
/s/ Henry A. McKinnell, Jr., Ph.D. | Director | March 12, 2018 |
Henry A. McKinnell, Jr., Ph.D | | |
| | |
/s/ Brian K. Roberts | Director | March 12, 2018 |
Brian K. Roberts | | |
| | |
/s/ Theodore T. Wang, Ph.D. | Director | March 12, 2018 |
Theodore T. Wang, Ph.D. | | |
| | |
/s/ Scott Huennekens, MBA | Director | March 12, 2018 |
Scott Huennekens, MBA | | |
| | |
139
/s/ Daniel Moore, MBA | Director | March 12, 2018 |
Daniel Moore, MBA | | |
| | |
140