
(formerly Stem Cell Therapeutics Corp.)
MANAGEMENT’S DISCUSSION AND ANALYSIS
FOR THE YEARS ENDED
DECEMBER 31, 2015 AND 2014
Dated: March 9, 2016
96 Skyway Avenue
Toronto, Ontario, M9W 4Y9
www.trilliumtherapeutics.com
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
ABOUT THIS MANAGEMENT DISCUSSION AND ANALYSIS
All references in this management’s discussion and analysis, or MD&A to “the Company”, “Trillium”, “we”, “us”, or “our” refer to Trillium Therapeutics Inc. and the subsidiaries through which it conducts its business, unless otherwise indicated.
The following MD&A is prepared as of March 9, 2016 for Trillium Therapeutics Inc. for the years ended December 31, 2015 and 2014, and should be read in conjunction with the audited consolidated financial statements for the years ended December 31, 2015 and 2014, which have been prepared by management in accordance with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or IASB. Our IFRS accounting policies are set out in note 3 of the annual audited consolidated financial statements for the years ended December 31, 2015 and 2014.
On June 1, 2014, we amalgamated with our wholly-owned subsidiary Trillium Therapeutics Inc., or Trillium Privateco and changed its name to Trillium Therapeutics Inc. All amounts are in Canadian dollars, unless otherwise indicated.
CAUTIONARY STATEMENT ABOUT FORWARD-LOOKING STATEMENTS
This MD&A contains forward-looking statements within the meaning of applicable securities laws. All statements contained herein that are not clearly historical in nature are forward-looking, and the words “anticipate”, “believe”, “expect”, “estimate”, “may”, “will”, “could”, “leading”, “intend”, “contemplate”, “shall” and similar expressions are generally intended to identify forward-looking statements. Forward-looking statements in this MD&A include, but are not limited to, statements with respect to:
| | • | our expected future loss and accumulated deficit levels; |
| | • | our projected financial position and estimated cash burn rate; |
| | • | our expectations about the timing of achieving milestones and the cost of our development programs; |
| • | our observations and expectations regarding the relative low binding of SIRPαFc to red blood cells compared to anti-CD47 monoclonal antibodies and proprietary CD47-blocking agents and the potential benefits to patients; |
| | • | our requirements for, and the ability to obtain, future funding on favorable terms or at all; |
| • | our projections for the SIRPαFc development plan and progress of each of our products and technologies, particularly with respect to the timely and successful completion of studies and trials and availability of results from such studies and trials; |
| • | our expectations about the differentiated nature and potential for best-in-class product development programs and discovery research capabilities of Fluorinov Pharma Inc., or Fluorinov; |
| • | our ability to generate future product development programs with improved pharmacological properties using Fluorinov technology; |
| • | our expectations about whether various clinical and regulatory milestones with an existing Fluorinov compound will be achieved; |
| • | our expectations of the final quantum and form of any future contingent milestone payments related to the Fluorinov acquisition; |
| • | our expectations of the ability to secure the requisite approvals (including TSX and NASDAQ approvals) with respect to the issuance of any common shares in satisfaction of future milestone payments; |
| | • | our expectations about our products’ safety and efficacy; |
| | • | our expectations regarding our ability to arrange for the manufacturing of our products and technologies; |
| • | our expectations regarding the progress, and the successful and timely completion, of the various stages of the regulatory approval process; |
| | • | our ability to secure strategic partnerships with larger pharmaceutical and biotechnology companies; |
| • | our strategy to acquire and develop new products and technologies and to enhance the safety and efficacy of existing products and technologies; |
- 2 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
| | • | our plans to market, sell and distribute our products and technologies; |
| | • | our expectations regarding the acceptance of our products and technologies by the market; |
| | • | our ability to retain and access appropriate staff, management, and expert advisers; |
| • | our expectations with respect to existing and future corporate alliances and licensing transactions with third parties, and the receipt and timing of any payments to be made by us or to us in respect of such arrangements; and |
| | • | our strategy with respect to the protection of our intellectual property. |
All forward-looking statements reflect our beliefs and assumptions based on information available at the time the assumption was made. These forward-looking statements are not based on historical facts but rather on management’s expectations regarding future activities, results of operations, performance, future capital and other expenditures (including the amount, nature and sources of funding thereof), competitive advantages, business prospects and opportunities. By its nature, forward-looking information involves numerous assumptions, inherent risks and uncertainties, both general and specific, known and unknown, that contribute to the possibility that the predictions, forecasts, projections or other forward-looking statements will not occur. Factors which could cause future outcomes to differ materially from those set forth in the forward-looking statements include, but are not limited to:
| • | the effect of continuing operating losses on our ability to obtain, on satisfactory terms, or at all, the capital required to maintain us as a going concern; |
| • | the ability to obtain sufficient and suitable financing to support operations, preclinical development, clinical trials, and commercialization of products; |
| • | the risks associated with the development of novel compounds at early stages of development in our intellectual property portfolio; |
| • | the risks of reliance on third-parties for the planning, conduct and monitoring of clinical trials and for the manufacture of drug product; |
| • | the risks associated with the development of our product candidates including the demonstration of efficacy and safety; |
| • | the risks related to clinical trials including potential delays, cost overruns and the failure to demonstrate efficacy and safety; |
| | • | the risks of delays and inability to complete clinical trials due to difficulties enrolling patients; |
| • | risks associated with our inability to successfully develop companion diagnostics for our development candidates; |
| | • | delays or negative outcomes from the regulatory approval process; |
| | • | our ability to successfully compete in our targeted markets; |
| | • | our ability to attract and retain key personnel, collaborators and advisors; |
| • | risks relating to the increase in operating costs from expanding existing programs, acquisition of additional development programs and increased staff; |
| • | risk of negative results of clinical trials or adverse safety events by us or others related to our product candidates; |
| | • | the potential for product liability claims; |
| | • | our ability to achieve our forecasted milestones and timelines on schedule; |
| • | financial risks related to the fluctuation of foreign currency rates and expenses denominated in foreign currencies; |
| | • | our ability to adequately protect proprietary information and technology from competitors; |
| | • | risks related to changes in patent laws and their interpretations; |
| | • | our ability to source and maintain licenses from third-party owners; and |
| | • | the risk of patent-related litigation and the ability to protect trade secrets, |
all as further and more fully described under the heading “Risk Factors” in this MD&A and in our Annual Information Form.
- 3 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
Although the forward-looking statements contained in this MD&A are based upon what our management believes to be reasonable assumptions, we cannot assure readers that actual results will be consistent with these forward looking statements.
Any forward-looking statements represent our estimates only as of the date of this MD&A and should not be relied upon as representing our estimates as of any subsequent date. We undertake no obligation to update any forward-looking statement or statements to reflect events or circumstances after the date on which such statement is made or to reflect the occurrence of unanticipated events, except as may be required by securities legislation.
BUSINESS
Overview
We are an immuno-oncology company developing innovative therapies for the treatment of cancer. Our lead program, SIRPαFc, is a novel, antibody-like protein that harnesses the innate immune system by blocking the activity of CD47, a molecule whose expression is increased on cancer cells to evade the host immune system. Expressed at high levels on the cell surface of a variety of liquid and solid tumors, CD47 functions as a signal that inhibits the destruction of tumor cells by macrophages via phagocytosis. By blocking the activity of CD47, we believe SIRPαFc has the ability to promote the macrophage-mediated killing of tumor cells in a broad variety of cancers both as a monotherapy and in combination with other immune therapies.
The immune system is the body’s mechanism to identify and eliminate pathogens, and can be divided into the innate immune system and the adaptive immune system. The innate immune system is the body’s first line of defense to identify and eliminate pathogens and consists of proteins and cells, such as macrophages, that identify and provide an immediate response to pathogens. The adaptive immune system is activated by, and adapts to, pathogens, creating a targeted and durable response. Cancer cells often have the ability to reduce the immune system’s ability to recognize and destroy them. We believe SIRPαFc could play an important role in enhancing the innate immune system and importantly, because of its ability to target macrophages, also has an important downstream effect on the adaptive immune system.
Our Strategy
Our goal is to become a leading innovator in the field of immuno-oncology by targeting immune-regulatory pathways that tumor cells exploit to evade the host immune system. We believe that SIRPαFc has the potential to improve survival and quality of life for cancer patients.
| • | Rapidly advance the clinical development of SIRPαFc. We have initiated a first-in-human trial of SIRPαFc in patients with relapsed or refractory lymphoma in a dose escalation phase of up to 36 patients, and expect to enroll up to 8 cohorts of 12-15 patients with advanced hematologic malignancies in an expansion phase of the trial after an optimal dose has been determined. |
| • | Expand our SIRPαFc clinical program to include additional cancer indications. Because CD47 is highly expressed by multiple liquid and solid tumors, and high expression is correlated with worse clinical outcomes, we believe SIRPαFc has potential to be effective in a wide variety of cancers. We are carrying out preclinical work necessary to select additional, high potential cancer indications for clinical development. |
| • | Maximize value of SIRPαFc through scientific collaborations. SIRPαFc has broad potential applicability in various cancer indications, and we believe it is well suited for use as both a monotherapy and in combination with other agents. We plan to continue to selectively and opportunistically pursue scientific collaborations with academic researchers as well as with other companies in order to realize the full value proposition of SIRPαFc. |
| • | Initiate IND-enabling studies in 2016 with a Fluorinov product. TTI-281 is an oral bromodomain inhibitor that we believe has strong potency and has shown efficacy in AML xenograft models. We plan to initiate scale up and manufacturing for TTI-281 in 2016. |
- 4 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
SIRPαFc Key Attributes
| • | Potential efficacy in a broad range of cancers. SIRPαFc blocks the tumor’s ability to transmit a “do not eat” signal allowing macrophages to destroy tumor cells; a mechanism that we believe could have broad applicability. |
| • | Potential for use as monotherapy and in combination with other therapies. We intend to develop SIRPαFc as a monotherapy as well as potentially for use in combination with other cancer immuno- therapies. |
| • | Differentiated pharmacokinetic and safety profile. We believe SIRPαFc’s low level of binding to red blood cells lowers the risk of anemia and lowers the loss of drug from circulation. This pharmacokinetic profile potentially allows for lower dosing and an improved safety profile. |
| • | May enhance both innate and adaptive immune response. SIRPαFc may enhance stimulation of tumor attacking T cells since macrophages, in addition to their role in phagocytosis, can also prime T cells through antigen presentation. |
SIRPαFc Clinical development plan
We commenced a phase I study in patients with advanced hematologic malignancies. The two-part clinical trial is designed as a multi-center, open-label Phase 1a/1b trial, evaluating TTI-621 as a single-agent in patients with relapsed or refractory hematologic malignancies. During the dose escalation phase set to enroll up to 36 subjects, the safety, tolerability, pharmacokinetics and pharmacodynamics will be characterized to determine the optimal dose for subsequent enrollment in the expansion phase. To characterize potential changes in hematologic parameters that might occur with blockade of CD47, the dose-escalation portion of the phase I trial will include lymphoma patients with normal hematologic parameters and acceptable marrow function. Once a reasonably well-tolerated dose and schedule of SIRPαFc has been established for further study, safety and antitumor activity will be estimated in expansion cohorts of up to 15 patients with advanced hematologic malignancies including indolent B-cell lymphoma, aggressive B-cell lymphoma, T-cell lymphoma, Hodgkin lymphoma, chronic lymphocytic leukemia, multiple myeloma, acute myeloid leukemia, and myelodysplastic syndrome. We plan to engage five trial sites in the United States for the dose escalation part of the trial.
Blocking the CD47 “do not eat” signal using a SIRPαFc decoy receptor
Macrophages are a type of white blood cell that can ingest and destroy (phagocytose) other cells. They are part of the innate immune system that helps to protect the body from infection. More recently, a role for macrophages in the control of tumors has been described.
Macrophage activity is controlled by both positive “eat” and negative “do not eat” signals. Tumor cells may express “eat” signals (e.g., calreticulin) that make themselves visible to macrophages. To counterbalance this increased visibility the tumor cells often express high levels of CD47, which transmits a “do not eat” signal by binding signal regulatory protein alpha, or SIRPα, on the surface of macrophages. We believe that the higher expression of CD47 on the tumor cell helps it evade destruction by the macrophage by overwhelming any activating “eat” signals.
SIRPαFc is an antibody-like protein that consists of the CD47-binding domain of human SIRPα linked to the Fc region of a human immunoglobulin. It is designed to act as a soluble decoy receptor, preventing CD47 from delivering its inhibitory signal. Neutralization of the inhibitory CD47 signal enables the activation of macrophage anti-tumor effects by the pro-phagocytic “eat” signals. The Fc region of SIRPαFc may, depending on its identity, also assist in the activation of macrophages by engaging Fc receptors.
Figure 1, below, illustrates how SIRPαFc blocks the CD47 “do not eat” signal and engages activating Fc receptors on macrophages, leading to tumor cell phagocytosis.
- 5 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
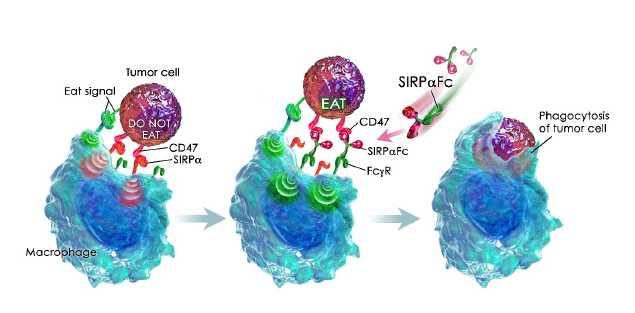
In addition to their direct anti-tumor activity, macrophages can also function as antigen-presenting cells and stimulate antigen-specific T cells. Thus it is possible that increasing tumor cell phagocytosis after SIRPαFc exposure may result in enhanced adaptive immunity. In support of this, CD47 antibody blockade has been recently shown to augment antigen presentation and prime an anti-tumor cytotoxic T cell response in immune-competent mice. CD47 blockade has also been reported to promote tumor-specific T cell responses through a dendritic cell-based mechanism, although the effect of SIRPαFc on dendritic cells is currently unknown.
SIRPαFc has broad clinical potential
We believe that SIRPαFc has broad clinical potential in both hematological and solid tumors. High expression of the CD47 “do not eat” signal on tumor cells has been observed in AML, MDS, chronic myeloid leukemia, or CML, acute lymphoblastic leukemia, or ALL, diffuse large B cell lymphoma, or DLBCL, chronic lymphocytic leukemia, or CLL, follicular lymphoma, mantle cell lymphoma, marginal zone lymphoma, multiple myeloma and in the following solid tumors: bladder, brain, breast, colon, leiomyosarcoma, liver, melanoma, ovarian and prostate. In a number of these cancers high CD47 expression was shown to have negative clinical consequences, correlating with more aggressive disease and poor survival. In normal karyotype AML patients, for example, high CD47 expression was correlated with worse event-free survival (6.8 vs. 17.1 months) and worse overall survival (9.1 vs. 22.1 months) compared to low CD47 expression. These data are consistent with CD47 providing a survival advantage to tumor cells. Furthermore, numerous studies have shown that antibody blockade of CD47 has demonstrated activity in mice engrafted with human tumors. We collaborate with academic investigators to explore the therapeutic potential of SIRPαFc in a variety of solid tumor models.
In vitro studies with primary tumor samples obtained from AML, MDS, multiple myeloma, B cell-ALL and T-cell ALL demonstrated that SIRPαFc frequently triggered significantly macrophage-mediated tumor cell phagocytosis compared to control treatment. Similar results were observed with tumor cell lines established from patients with B lymphoma and CML.
Figure 2, below, shows how SIRPαFc (TTI-621) triggers macrophage-mediated phagocytosis of many different human blood cancer samples.
- 6 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
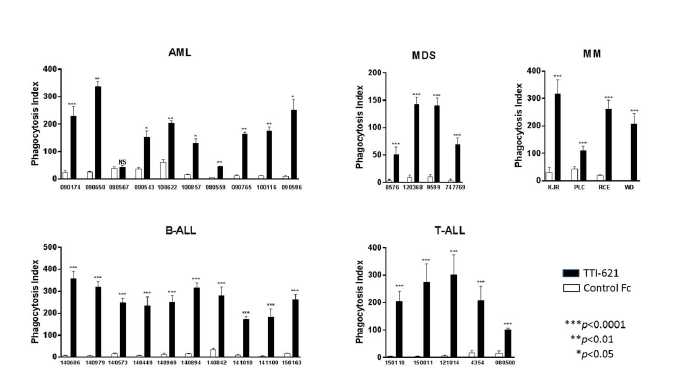
The p value is a probability value, with values <0.05 considered statistically significant versus control treatment. NS indicates p>0.05, which is considered not statistically significant versus control treatment.
SIRPα – Minimal Red Blood Cell Binding - Competition
SIRPαFc competes directly with CD47 blocking antibodies from Forty Seven Inc. (recently licensed from Stanford University) and Celgene Corporation, both of which have entered early clinical development. Novimmune SA has a bispecific (anti-CD47/anti-CD19) antibody program in preclinical development. Vasculox is also developing a CD47 antibody. Alexo Therapeutics is developing mutated, high affinity SIRPα monomers as adjuvants for use with anti-cancer antibodies. Both Vasculox and Alexo Therapeutics are in the preclinical phase of development.
We believe that our approach of using SIRPα, the natural binding partner for CD47, may have important advantages over treatment with CD47-specific antibodies. In April 2015, we presented data at the 106th American Association for Cancer Research annual meeting demonstrating that our SIRPαFc fusion proteins exhibit minimal binding to human red blood cells, or RBCs, compared to anti-CD47 monoclonal antibodies. These data build upon preliminary results announced at the 2014 American Association for Cancer Research annual meeting.
Figure 3, below, shows how SIRPαFc binding to human RBCs (n=43 donors) compares to five CD47-specific antibodies.
- 7 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
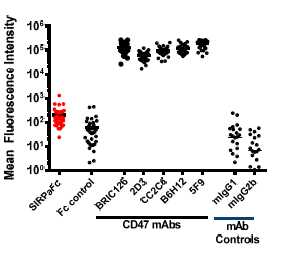
The very low RBC binding profile of our SIRPαFc proteins may provide three important advantages over other CD47 blocking agents. First, there may be a lower risk of experiencing drug-induced anemia with SIRPαFc. This potential advantage may translate into improved patient care, particularly in diseases such as AML where almost all patients experience cytopenias including anemia. Second, SIRPαFc may remain in circulation for longer intervals compared to CD47 antibodies, which have been demonstrated to bind strongly to circulating RBCs and may thus be more likely to be removed from circulation. The ability of RBCs to absorb and remove circulating CD47-specific antibodies, an “antigen-sink effect”, may necessitate high doses of antibody in order to effectively target the tumor cells, which may lead to potentially higher off-target toxicity. Therefore, SIRPαFc may be associated with both improved tolerability and drug kinetics compared with CD47-specific antibodies based on these preclinical analyses. Third, antibodies that bind RBCs have the potential to interfere with laboratory blood typing tests. The minimal binding of TTI-621 to human RBCs may minimize this interference compared to anti-CD47 antibodies.
Combination Therapy
We believe that SIRPαFc enhancement of macrophage activity and possibly T cell responses could be synergistic with other immune-mediated therapies. Published studies conducted by third parties provide evidence that SIRPαFc may be useful in combination with approved anti-cancer antibodies (e.g. Rituxan®, Herceptin®, Campath®, and Erbitux®). Since many cancer antibodies work at least in part by activating cells of the innate immune system, it may be possible to enhance the potency of these agents by blocking the negative “do not eat” CD47 signal that tumor cells deliver to macrophages. We hypothesize that SIRPαFc may act synergistically with other immunological agents, including T cell checkpoint inhibitors (e.g. pembrolizumab and nivolumab), cancer vaccines, oncolytic viruses or chimeric antigen receptor, or CAR T cells.
Small Molecule Program
On January 26, 2016, we acquired all the outstanding shares of Fluorinov, a privately-held oncology company that has developed a proprietary medicinal chemistry platform using unique fluorine chemistry, which permits the creation of new chemical entities from validated drugs and drug candidates with improved pharmacological properties, potentially leading to increased safety and efficacy. Fluorinov’s most advanced preclinical oncology programs include potent, orally-available, bromodomain and proteasome inhibitors, as well as epidermal growth factor receptor antagonists with increased uptake in the brain, all of which have been differentiated from competitors and have potential for best-in-class status. In addition, a number of compounds directed at undisclosed immuno-oncology targets are currently in the discovery phase.
- 8 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
The terms of the acquisition were an upfront payment of $10 million plus up to $35 million of additional future payments that are contingent on us achieving certain clinical and regulatory milestones with an existing Fluorinov compound. We will also have an obligation to pay royalty payments on future sales of such compounds. The upfront payment was subject to adjustment based on the net working capital of Fluorinov and other adjustments at the time of closing. At our discretion, up to 50% of the future contingent payments can be satisfied through the issuance of common shares of Trillium provided that the aggregate number of common shares issuable under such payments will not exceed 1,558,447 common shares unless shareholder approval has first been obtained. In addition, any such future share issuance remains subject to final approval from our board of directors and receipt of any requisite approvals under the applicable rules of the Toronto Stock Exchange and the NASDAQ Stock Market. We have also committed to use commercially reasonable efforts to monetize Fluorinov’s central nervous system, or CNS, assets and share 50% of the net proceeds with Fluorinov shareholders.
Plan of Operations
Over the next 12 months, our primary focus is the advancement of our Phase I clinical trial of SIRPαFc in patients with advanced hematologic malignancies. We also plan to conduct preclinical research in solid tumors and in combination studies to identify future potential clinical indications.
We also plan to advance one, or possibly two, of Fluorinov’s preclinical programs through the Investigational New Drug, or IND-enabling phase using our internal development group, while at the same time focusing our new chemistry group on several immuno-oncology discovery programs.
Capital Markets
We were listed on the TSX Venture Exchange, or TSXV, until April 22, 2014 when we migrated to the Toronto Stock Exchange, or TSX. We traded under the symbol “SSS” until June 6, 2014 when the symbol was changed to “TR”. We were listed on the OTCQX International under the symbol “SCTPF” from May 20, 2013 until we began trading on the NASDAQ Capital Market under the symbol “TRIL” on December 19, 2014.
Legal Proceedings
To our knowledge, there have not been any legal or arbitration proceedings, including those relating to bankruptcy, receivership or similar proceedings, those involving any third party, and governmental proceedings pending or known to be contemplated, which may have, or have had in the recent past, significant effect our financial position or profitability.
Also, to our knowledge, there have been no material proceedings in which any director, any member of senior management, or any of our affiliates is either a party adverse to us or any of our subsidiaries or has a material interest adverse to us or any of our subsidiaries.
- 9 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
RESULTS OF OPERATIONS
For the three months and years ended December 31, 2015 and 2014
Overview
Since inception, we have incurred losses while advancing the research and development of our products. Net loss for the three months ended December 31, 2015 of $2,988,843 was lower than the loss of $4,214,862 for the three months ended December 31, 2014. Net loss for the year ended December 31, 2015 of $14,733,699 exceeded the loss of $12,881,820 for the year ended December 31, 2014. Research and development costs for both 2015 periods were significantly higher than 2014 due mainly to higher costs for our SIRPαFc development program including increased personnel costs. The loss for the three months ended December 31, 2015 was lower than the comparative period due mainly to a net foreign exchange gain of $2,163,429.
Research and Development
Research and development expenses by program for the years ended December 31, 2015 and 2014 were as follows:
| | | Three months | | | Three months | | | | | | | |
| | | ended | | | ended | | | Year ended | | | Year ended | |
| | | December 31, | | | December 31, | | | December 31, | | | December 31, | |
| | | 2015 | | | 2014 | | | 2015 | | | 2014 | |
| | | $ | | | $ | | | $ | | | $ | |
| SIRPαFc | | 4,332,644 | | | 3,828,172 | | | 17,978,930 | | | 9,372,467 | |
| Tigecycline | | - | | | 125 | | | - | | | 1,091,297 | |
| Other | | 32,939 | | | 6,650 | | | 71,161 | | | 132,044 | |
| Total(1) | | 4,365,583 | | | 3,834,947 | | | 18,050,091 | | | 10,595,808 | |
Note:
| (1) | Research and development expenditures in the above table include all direct and indirect costs for the programs, personnel costs, intellectual property related costs net of recoveries, share-based compensation and research and development overhead, and is net of government assistance. Research and development overhead costs have been allocated to the programs based mainly on personnel time spent on the programs. |
During 2015, most of our resources were focused on the development of our SIRPαFc program. For the year ended December 31, 2015, research and development costs increased over the same period in the prior year as we completed IND-enabling toxicology studies, incurred manufacturing costs to supply our clinical trial, completed the IND submission and initiated the Phase I trial in 2015. Personnel costs were also higher in 2015 as we added staff to manage our expanded research and development activities. In 2014, we discontinued our tigecycline program and recorded an impairment charge of $429,763.
Components of research and development expenses for the three months ended December 31, 2015 and 2014 were as follows:
| | | 2015 | | | 2014 | |
| | | $ | | | $ | |
| | | | | | | |
| Research and development programs excluding the below items | | 2,764,541 | | | 2,650,581 | |
| Salaries, fees and short-term benefits | | 1,143,872 | | | 718,714 | |
| Share-based compensation | | 520,853 | | | 258,995 | |
| Amortization of intangible assets | | 84,837 | | | 84,837 | |
| Depreciation of property and equipment | | 32,513 | | | 15,429 | |
| Tax credits | | (181,033 | ) | | 106,391 | |
| | | 4,365,583 | | | 3,834,947 | |
- 10 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
Components of research and development expenses for the years ended December 31, 2015 and 2014 were as follows:
| | | 2015 | | | 2014 | |
| | | $ | | | $ | |
| | | | | | | |
| Research and development programs excluding the below items | | 12,083,797 | | | 5,893,030 | |
| Salaries, fees and short-term benefits | | 4,120,109 | | | 2,311,755 | |
| Share-based compensation | | 1,942,173 | | | 1,626,824 | |
| Amortization of intangible assets | | 339,348 | | | 610,776 | |
| Impairment of intangible assets | | - | | | 429,763 | |
| Depreciation of property and equipment | | 118,394 | | | 47,208 | |
| Tax credits | | (553,730 | ) | | (323,548 | ) |
| | | 18,050,091 | | | 10,595,808 | |
The increase in research and development program expenses for the three months ended December 31, 2015 compared to the three months ended December 31, 2014 was due mainly to higher facility costs and advisory fees related to our IND filing, partially offset by lower program costs. Salaries, fees and short-term benefits increased in the three months ended December 31, 2015 due to higher staffing and salaries compared to the same period in 2014. Share-based compensation increased due mainly to new options granted in 2015 with higher fair values as determined by the Black-Scholes option pricing model.
The increase in research and development program expenses for the year ended December 31, 2015 compared to the year ended December 31, 2014 was due mainly to completion of IND-enabling toxicology studies, manufacturing costs to supply our clinical trial, costs to prepare and submit our IND and initiation of the Phase I trial in 2015 for SIRPαFc. Salaries, fees, and short-term benefits increased for the year ended December 31, 2015 due mainly to additional research and development personnel hired in 2015. Amortization and impairment of intangible assets was lower in the year ended December 31, 2015 due to the discontinuation of the tigecycline program in 2014. Depreciation expense was higher in 2015 due to higher purchases of new lab equipment.
General and Administrative
Components of general and administrative expenses for the three months ended December 31, 2015 and 2014 were as follows:
| | | 2015 | | | 2014 | |
| | | $ | | | $ | |
| | | | | | | |
| General and administrative expenses excluding the below items | | 460,582 | | | 181,117 | |
| Salaries, fees and short-term benefits | | 119,620 | | | 198,941 | |
| DSU units issued for director compensation | | 240,000 | | | - | |
| Share-based compensation | | 71,339 | | | 59,788 | |
| | | 891,541 | | | 439,846 | |
- 11 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
Components of general and administrative expenses for the years ended December 31, 2015 and 2014 were as follows:
| | | 2015 | | | 2014 | |
| | | $ | | | $ | |
| | | | | | | |
| General and administrative expenses excluding the below items | | 1,521,639 | | | 1,198,181 | |
| Salaries, fees and short-term benefits | | 898,381 | | | 694,849 | |
| DSU units issued for director compensation | | 540,000 | | | 240,000 | |
| Share-based compensation | | 224,327 | | | 444,430 | |
| | | 3,184,347 | | | 2,577,460 | |
General and administrative expenses for the three months ended December 31, 2015 were higher than the comparable prior year period due mainly to expenses related to the Fluorinov acquisition and higher insurance costs related to our NASDAQ stock exchange listing. DSU units issued for director compensation were higher for the three months ended December 31, 2015 as DSUs were not used for director compensation in the comparable 2014 period.
General and administrative expenses for the year ended December 31, 2015 were higher than the comparable prior year period due mainly to higher insurance costs and expenses related to the Fluorinov acquisition, partially offset by lower stock exchange filing fees. Salaries, fees and short-term benefits increased in 2015 over 2014 due mainly to higher administrative staffing. The value of DSUs issued for director compensation increased in 2015, and share-based compensation expense was lower in 2015 due mainly to fewer stock options issued to administrative personnel in the year.
Finance income and costs
Components of finance income for the three months ended December 31, 2015 and 2014 were as follows:
| | | 2015 | | | 2014 | |
| | | $ | | | $ | |
| | | | | | | |
| Interest income | | 135,928 | | | 93,634 | |
| Net foreign currency gain | | 2,163,429 | | | - | |
| | | 2,299,357 | | | 93,634 | |
Components of finance income for the years ended December 31, 2015 and 2014 were as follows:
| | | 2015 | | | 2014 | |
| | | $ | | | $ | |
| | | | | | | |
| Interest income | | 488,486 | | | 378,692 | |
| Net foreign currency gain | | 6,106,703 | | | - | |
| | | 6,595,189 | | | 378,692 | |
- 12 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
Finance income for the three months and year ended December 31, 2015 was higher than the prior year comparable periods due mainly to a net foreign currency gain of $2,163,429 and $6,106,703, respectively, due mainly to holding U.S. dollar denominated cash with a strengthening U.S. dollar. Interest income in 2015 was also higher due to higher average cash balances.
Finance costs for the year ended December 31, 2015 were comparable to the prior year periods.
Liquidity and Capital Resources
Cash, working capital, and debt
Since inception, we have financed our operations primarily from sales of equity, proceeds from the exercise of warrants and stock options, and from interest income on funds available for investment. Our primary capital needs are for funds to support our scientific research and development activities including staffing, facilities, manufacturing, preclinical studies and clinical trials, administrative costs and for working capital.
We have experienced operating losses and cash outflows from operations since incorporation, will require ongoing financing in order to continue our research and development activities, and we have not earned significant revenue or reached successful commercialization of our products. Our future operations are dependent upon our ability to finance our cash requirements which will allow us to continue our research and development activities and the commercialization of our products. There can be no assurance that we will be successful in continuing to finance our operations.
On April 7, 2015, we completed an underwritten public offering of common shares and non-voting convertible preferred shares in the United States. In the offering, we sold 1,750,754 common shares and 1,077,605 Series II Non-Voting Convertible First Preferred Shares at a price of U.S. $19.50 per share, including 228,359 common shares sold pursuant to the full exercise of the underwriters’ option to purchase additional common shares. The gross proceeds from this offering were $68,875,067 (U.S. $55,153,000) before deducting offering expenses of $4,913,443.
The Series II Non-Voting Convertible First Preferred Shares sold in the offering are non-voting and are convertible into common shares, on a one-for-one basis (subject to adjustment), at any time at the option of the holder, subject to certain restrictions on conversion. Holders may not convert Series II Non-Voting Convertible First Preferred Shares into common shares if, after giving effect to the exercise of conversion, the holder and its joint actors would have beneficial ownership or direction or control over common shares in excess of 4.99% of the then outstanding common shares. This limit may be raised at the option of the holder on 61 days’ prior written notice: (i) up to 9.99%, (ii) up to 19.99%, subject to clearance of a personal information form submitted by the holder to the Toronto Stock Exchange, and (iii) above 19.99%, subject to approval by the Toronto Stock Exchange and shareholder approval.
On May 29, 2015, we filed a base shelf prospectus with the British Columbia, Alberta, Manitoba, Ontario and Nova Scotia securities commissions in Canada and a Form F-10 registration statement with the United States Securities and Exchange Commission, or SEC, that provides that we may sell under the prospectus from time to time over the following 25 months up to U.S. $100 million, in one or more offerings, of common shares, First Preferred shares, warrants to purchase common shares, or units comprising a combination of common shares, First Preferred shares and/or warrants.
Our cash totaled $86,770,542 at December 31, 2015 compared to $26,165,056 at December 31, 2014. As at December 31, 2015, our working capital increased to $85,369,945 compared to $23,989,252 at December 31, 2014 due mainly to funds received in the April 7, 2015 offering, warrant exercises, and foreign exchange gains, partially offset by cash used in operations. Accounts payable and accrued liabilities as at December 31, 2015 of $3,233,749 were comparable to the balance of $3,248,984 at December 31, 2014. Amounts receivable as at December 31, 2015 were $974,822 compared to $344,416 at December 31, 2014. The increase in amounts receivable was due mainly to the recording of expected refundable tax credits on research and development activities and refundable withholding taxes in the year ended December 31, 2015.
- 13 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
We are indebted to the Federal Economic Development Agency for Southern Ontario, or FedDev, under a non-interest bearing contribution agreement and are making monthly repayments of $9,586 through November 2019. As at December 31, 2015, the balance repayable is $440,935. The loan payable was discounted using an estimated market interest rate of 15%. Interest expense accretes on the discounted loan amount until it reaches its face value at maturity.
As at December 31, 2015, we have a deferred lease inducement of $348,205 for a new facility lease. The inducement benefit will be recognized over the expected term of the lease.
We have a long-term liability of $60,109 related to certain discontinued technologies. This liability was discounted using an estimated market interest rate of 15% and interest expense is accreting.
Cash flows from operating activities
Cash used in operating activities increased to $18,298,112 for the year ended December 31, 2015, compared to $7,448,068 for the year ended December 31, 2014, due mainly to higher research and development expenses in the current year.
Cash flows from investing activities
Cash used in investing activities totaled $750,382 for the year ended December 31, 2015, compared to cash provided by investing activities of $352,995 for the year ended December 31, 2014. The increase was due to higher purchases of property and equipment compared to the prior year.
Cash flows from financing activities
Cash provided by financing activities totaled $73,642,984 for the year ended December 31, 2015, compared to $803,623 for the year ended December 31, 2014. The increase was due mainly to the completion of an underwritten public offering of common shares and non-voting convertible preferred shares in April 2015.
Contractual Obligations and Contingencies
We enter into research, development and license agreements in the ordinary course of business where we receive research services and rights to proprietary technologies. Milestone and royalty payments that may become due under various agreements are dependent on, among other factors, clinical trials, regulatory approvals and ultimately the successful development of a new drug, the outcome and timing of which is uncertain.
Under the license agreement for SIRPαFc, we have future contingent milestones payable of $35,000 related to successful patent grants, $100,000, $200,000 and $300,000 on the first patient dosed in phase I, II and III trials respectively, and regulatory milestones on their first achievement totalling $5,000,000.We are also required to pay 20% of any sublicensing revenues to the licensors on the first $50 million of sublicensing revenues, and pay 15% of any sublicensing revenues to the licensors after the first $50 million of sublicensing revenue received.
Under two agreements with Catalent pursuant to which we acquired the right to use a proprietary expression system for the manufacture of two SIRPαFc constructs, we have future contingent milestones on pre-marketing approval of up to U.S. $875,000 and aggregate sales milestone payments of up to U.S. $28.8 million for each agreement.
- 14 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
We periodically enter into research and license agreements with third parties that include indemnification provisions customary in the industry. These guarantees generally require us to compensate the other party for certain damages and costs incurred as a result of claims arising from research and development activities undertaken by or on our behalf. In some cases, the maximum potential amount of future payments that could be required under these indemnification provisions could be unlimited. These indemnification provisions generally survive termination of the underlying agreement. The nature of the indemnification obligations prevents us from making a reasonable estimate of the maximum potential amount it could be required to pay. Historically, we have not made any indemnification payments under such agreements and no amount has been accrued in our audited consolidated financial statements with respect to these indemnification obligations.
Other than as disclosed below, we did not have any contractual obligations relating to long-term debt obligations, capital (finance) lease obligations, operating lease obligations, purchase obligations or other long-term liabilities reflected on our balance sheet as at December 31, 2015:
| | | | | | | Payment due by period | | | | |
| | | | | | | Less than | | | 1 to 3 | | | 3 to 5 | | | More than | |
| Contractual Obligations(1)(2) | | | Total | | | 1 year | | | years | | | years | | | 5 years | |
| Long-Term Debt Obligations(3) | | $ | 440,935 | | $ | 105,446 | | $ | 230,064 | | $ | 105,425 | | $ | - | |
| Capital (Finance) Lease Obligations | | | - | | | - | | | - | | | - | | | - | |
| Operating Lease Obligations(4) | | | 2,510,227 | | | 266,435 | | | 457,882 | | | 496,901 | | | 1,289,009 | |
| Purchase Obligations(5) | | | 8,614,808 | | | 4,788,286 | | | 3,826,522 | | | - | | | - | |
| Other Long-Term Liabilities | | | | | | | | | | | | | | | | |
| Reflected on our Balance Sheet(6) | | | 309,634 | | | 249,525 | | | 60,109 | | | - | | | - | |
| | | $ | 11,875,604 | | $ | 5,409,692 | | $ | 4,574,577 | | $ | 602,326 | | $ | 1,289,009 | |
Notes:
| (1) | Contractual obligations in the above table do not include amounts in accounts payable and accrued liabilities on our balance sheet as at December 31, 2015. Annual technology license fees currently approximating $50,000 are not included in the above table. |
| | |
| (2) | Contingent milestones under the UHN license agreement and the Catalent expression system agreements are not included in the above table. |
| | |
| (3) | Amounts due to FedDev repayable in equal monthly installments of $9,586 through November 2019. |
| | |
| (4) | Includes operating lease obligations for laboratory and office facilities. |
| | |
| (5) | Purchase obligations represent significant research and development contracts and other commitments that include the clinical research organization agreement for conducting the Phase I trial, and other preclinical and manufacturing activities. |
| | |
| (6) | Long-term liability related to the cessation of the development of our regenerative medicine products. |
- 15 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
Description of Share Capital
The continuity of the number of our issued and outstanding common and preferred shares for the year ended December 31, 2014, the year ended December 31, 2015, and to the date of this MD&A is presented below:
| | | Number of Series I | | | Number of Series II | | | Number of | |
| | | Preferred Shares(1 | ) | | Preferred Shares(2 | ) | | Common Shares | |
| | | | | | | | | | |
| Balance December 31, 2013 | | 77,895,165 | | | - | | | 121,752,380 | |
| Consolidation 1 for 30 | | - | | | - | | | (117,693,972 | ) |
| Issued on exercise of stock options | | - | | | - | | | 2,614 | |
| Issued on exercise of warrants | | - | | | - | | | 86,540 | |
| Preferred shares converted to common shares | | (8,390,476 | ) | | - | | | 279,682 | |
| Balance at December 31, 2014 | | 69,504,689 | | | - | | | 4,427,244 | |
| Issued on exercise of stock options | | - | | | - | | | 6,666 | |
| Issued on exercise of warrants | | - | | | - | | | 1,087,603 | |
| Issued in public offering | | - | | | 1,077,605 | | | 1,750,754 | |
| Preferred shares converted to common shares | | (15,716,110 | ) | | - | | | 523,870 | |
| Balance at December 31, 2015 | | 53,788,579 | | | 1,077,605 | | | 7,796,137 | |
| Preferred shares converted to common shares | | (562,388 | ) | | - | | | 18,746 | |
| Balance at the date of this MD&A | | 53,226,191 | | | 1,077,605 | | | 7,814,883 | |
Notes:
| (1) | Convertible at a ratio of 30 Series I Preferred Shares for one common share. |
| (2) | Convertible at a ratio of one Series II Preferred Share for one common share. |
Share capital issued –for the year ended December 31, 2015
On April 7, 2015, we completed an underwritten public offering of common shares and non-voting convertible preferred shares in the United States. In the offering, we sold 1,750,754 common shares and 1,077,605 Series II Non-Voting Convertible First Preferred Shares at a price of U.S. $19.50 per share, including 228,359 common shares sold pursuant to the full exercise of the underwriters’ option to purchase additional common shares. The gross proceeds from this offering were $68,875,067 (U.S. $55,153,000) before deducting offering expenses of $4,913,443.
During the year ended December 31, 2015, 1,087,603 common shares were issued on the exercise of 32,628,425 warrants for proceeds of $9,515,154 and 6,666 stock options were exercised for proceeds of $49,995.
During the year ended December 31, 2015, 15,716,110 Series I First Preferred Shares were converted into 523,870 common shares.
Share capital issued – for the year ended December 31, 2014
On November 14, 2014, we consolidated our outstanding common shares issuing one post-consolidated share for each 30 pre-consolidated shares. All references in this MD&A to the number of common shares, deferred share units and stock options refer to the post-consolidation amounts.
During the year ended December 31, 2014, 2,596,251 warrants were exercised for 86,540 common shares and for proceeds of $946,813 and 2,614 stock options were exercised for proceeds of $19,600. Also, 909,091 warrants issued in March 2011 expired unexercised.
During the year ended December 31, 2014, 8,390,476 Series I First Preferred Shares were converted into 279,682 common shares.
- 16 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
Warrants
As a result of the November 14, 2014 common share consolidation, the ratio of the number of warrants exercisable for one common share was adjusted from one warrant for one common share to 30 warrants for one common share. The number of warrants outstanding was not adjusted.
The continuity of the number of issued and outstanding warrants for the year ended December 31, 2014, the year ended December 31, 2015, and to the date of this MD&A is presented below:
| | | Number of | |
| | | Warrants | |
| | | | |
| Balance December 31, 2013 | | 142,230,123 | |
| Exercised | | (2,596,251 | ) |
| Expired | | (909,091 | ) |
| Balance at December 31, 2014 | | 138,724,781 | |
| Exercised | | (32,628,425 | ) |
| Balance at December 31, 2015 and the date of this MD&A | | 106,096,356 | |
The following table shows the number of warrants outstanding, the exercise prices, and the number of common shares issuable on exercise of the warrants and the exercise price per common share for 30 warrants at December 31, 2015:
| | | | | | | | | Number of | | | Exercise | |
| | | | | | | | | Common shares | | | Price per | |
| | | Number of | | | Exercise | | | Issuable | | | Common Share | |
| Expiry dates | | Warrants | | | Price | | | on Exercise | | | (30 Warrants | ) |
| | | | | | | | | | | | | |
| March 15, 2018 | | 9,213,780 | | $ | 0.40 | | | 307,126 | | $ | 12.00 | |
| March 27, 2018 | | 300,000 | | $ | 0.40 | | | 10,000 | | $ | 12.00 | |
| December 13, 2018 | | 96,582,576 | | $ | 0.28 | | | 3,219,419 | | $ | 8.40 | |
| Total | | 106,096,356 | | | | | | 3,536,545 | | | | |
Our board of directors authorized or ratified the issuances of the warrants set forth in the table above and the issuance of one common share upon the due exercise of every 30 warrants in accordance with its terms and the receipt by us of the designated exercise price payable in respect of the share prior to the time of expiry on the designated expiry date.
Stock Options
We have a 10% rolling stock option plan, or the 2014 Stock Option Plan, which was approved by our shareholders at our annual general meeting held on May 27, 2014. Pursuant to the 2014 Stock Option Plan, we may grant stock options to purchase up to an aggregate of 10% of our issued and outstanding common shares plus 10% of the total number of common shares into which the outstanding First Preferred Shares may be converted. Options granted under the 2014 Stock Option plan are equity-settled, have a vesting period of four years and have a maximum term of ten years. As at December 31, 2015, the Company was entitled to issue an additional 87,048 stock options under the 2014 Stock Option Plan.
- 17 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
The continuity of the number of issued and outstanding stock options for the year ended December 31, 2014, the years ended December 31, 2015, and to the date of this MD&A is presented below:
| | | Number of | | | Weighted Average | |
| | | Options | | | Exercise Price | |
| | | | | | | |
| Balance December 31, 2013 | | 97,372 | | $ | 9.94 | |
| Granted | | 499,883 | | | 9.78 | |
| Exercised | | (2,614 | ) | | 7.50 | |
| Cancelled/ forfeited | | (4,500 | ) | | 16.58 | |
| Balance at December 31, 2014 | | 590,141 | | | 9.76 | |
| Granted | | 347,359 | | | 21.40 | |
| Exercised | | (6,666 | ) | | 7.50 | |
| Cancelled/forfeited | | (3,000 | ) | | 30.00 | |
| Balance at December 31, 2015 and at the date of this MD&A | | 927,834 | | $ | 14.07 | |
Deferred Share Unit Plan
Our shareholders approved the 2014 DSU Plan on May 27, 2014. The 2014 DSU Plan is intended to promote a greater alignment of long-term interests between our non-executive directors and executive officers and our shareholders through the issuance of DSUs. Since the value of a DSU increases or decreases with the market price of the common shares, DSUs reflect a philosophy of aligning the interests of directors and executive officers with those of the shareholders by tying compensation to share price performance. The Board of Directors intends to use DSUs issued under the 2014 DSU Plan, as well as stock options issued under the 2014 Stock Option Plan, as part of our overall director and executive officer compensation program. A total of 28,777 units were issued during the year ended December 31, 2014 and 23,011 units were issued during the year ended December 31, 2015 for payment of director fees. We had 51,788 units outstanding as at December 31, 2015. We have reserved for issuance up to 66,667 common shares under the 2014 DSU Plan.
Fully Diluted Share Capital
The number of issued and outstanding common shares, Series I First Preferred Shares, Series II First Preferred Shares warrants, stock options and DSUs on a fully converted basis as at December 31, 2015 was as follows:
| | | Number of common | |
| | | share equivalents | |
| | | | |
| Common shares | | 7,796,137 | |
| Series I First Preferred Shares | | 1,792,953 | |
| Series II First Preferred Shares | | 1,077,605 | |
| Warrants | | 3,536,545 | |
| Stock options | | 927,834 | |
| Deferred share units | | 51,788 | |
| Fully diluted common shares as at December 31, 2015 | | 15,182,862 | |
- 18 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
Shareholder Rights Plan
On October 17, 2013 our shareholders adopted a shareholder rights plan, or the 2013 Rights Plan, and approved certain amendments on May 27, 2014, or the Rights Plan Amendment, and which together with the 2013 Rights Plan may be referred to as the Rights Plan. The Rights Plan is designed to provide adequate time for the Board of Directors and the shareholders to assess an unsolicited takeover bid for the Company, to provide the Board of Directors with sufficient time to explore and develop alternatives for maximizing shareholder value if a takeover bid is made, and to provide shareholders with an equal opportunity to participate in a takeover bid and receive full and fair value for their common shares. The Rights Plan will expire at the close of our annual meeting of shareholders in 2016.
The rights issued under the Rights Plan initially attach to and trade with the common shares and no separate certificates will be issued unless an event triggering these rights occurs. The rights will become exercisable only when a person, including any party related to it, acquires or attempts to acquire 20% or more of the outstanding common shares without complying with the “Permitted Bid” provisions of the Rights Plan or without approval of the Board of Directors. Should such an acquisition occur or be announced, each right would, upon exercise, entitle a rights holder, other than the acquiring person and related persons, to purchase common shares at an approximate 50% discount to the market price at the time.
Under the Rights Plan, a Permitted Bid is a bid made to all holders of the common shares and which is open for acceptance for not less than 60 days. If at the end of 60 days at least 50% of the outstanding common shares, other than those owned by the offeror and certain related parties have been tendered, the offeror may take up and pay for the common shares but must extend the bid for a further 10 days to allow other shareholders to tender. The issuance of common shares upon the exercise of the rights is subject to receipt of certain regulatory approvals.
Related Parties
For the years ended December 31, 2015 and 2014, our key management personnel were the Board of Directors, Chief Executive Officer, Chief Financial Officer, Chief Scientific Officer, Chief Medical Officer, and Chief Development Officer.
Compensation for our key management personnel for the years ended December 31, 2015 and 2014 was as follows:
| | | 2015 | | | 2014 | |
| | | $ | | | $ | |
| | | | | | | |
| Salaries, fees and short-term benefits | | 2,595,536 | | | 1,708,717 | |
| Share-based compensation | | 2,433,710 | | | 2,281,561 | |
| | | 5,029,246 | | | 3,990,278 | |
Executive officers and directors participate in the 2014 Stock Option Plan and the 2014 DSU Plan, and officers participate in our benefit plans. Directors receive annual fees for their services. As at December 31, 2015, the key management personnel controlled less than 1% of our voting shares.
Outstanding balances with related parties at the period-end are unsecured, interest free and settlement occurs in cash. There have been no guarantees provided or received for any related party receivables or payables. For the years ended December 31, 2015 and 2014, $0 and $7,916, respectively, was paid to a former director for consulting fees.
- 19 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
Trend Information
Historical patterns of expenditures cannot be taken as an indication of future expenditures. The amount and timing of expenditures and therefore liquidity and capital resources vary substantially from period to period depending on the number of research and development programs being undertaken at any one time, the stage of the development programs, the timing of significant expenditures for manufacturing, toxicology and pharmacology studies and clinical trials, and the availability of funding from investors and prospective commercial partners.
Selected Annual Financial Information
| 2015
$ | 2014
$ | 2013
$ |
| Interest income | 488,486 | 378,692 | 44,113 |
| Net loss | 14,733,699 | 12,881,820 | 4,289,308 |
| Net loss per share | 2.22 | 3.06 | 3.16 |
| Total assets | 90,039,468 | 28,186,032 | 35,087,386 |
| Total long-term liabilities | 678,700 | 353,293 | 446,313 |
In 2013 we acquired the SIRPαFc program and raised capital in December 2013 to take the program forward into IND-enabling studies. In 2014, we significantly increased our research and development activities including manufacturing and preclinical studies resulting in a higher net loss in 2014. We raised additional funds in April 2015 and completed manufacturing, IND-enabling studies and submitted our IND for a first-in-human trial of SIPRaFc in 2015. The net loss for the year ended December 31, 2015 included a net foreign exchange gain of $6,106,703 that resulted mainly from holding U.S. denominated cash with a strengthening U.S. dollar exchange rate. Interest income increased in 2014 and 2015 due mainly to higher average cash balances. Although a higher net loss in 2014 and 2015, the net loss per share decreased due to a higher issued and outstanding number of common shares issued in the two financings. Total assets increased significantly in 2015 as the April 2015 financing raised net proceeds of $64.0 million. Long-term liabilities increased in 2015 due mainly to long-term lease inducements on a new office and laboratory facility that will be recognized over the expected lease term.
Selected Quarterly Financial Information
| 2015 | Q4-2015
$ | Q3-2015
$ | Q2-2015
$ | Q1-2015
$ |
| Revenue | - | - | - | - |
| Research and development expenses | 4,365,583 | 4,954,811 | 4,713,316 | 4,016,381 |
| General and administrative expenses | 891,541 | 721,549 | 966,438 | 604,819 |
| Net loss for the period | 2,988,843 | 1,505,979 | 5,568,564 | 4,670,313 |
| Basic and diluted net loss per share* | 0.40 | 0.21 | 0.80 | 0.98 |
| Cash | 86,770,542 | 89,082,852 | 89,545,097 | 25,702,189 |
- 20 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
| 2014 | Q4-2014
$ | Q3-2014
$ | Q2-2014
$ | Q1-2014
$ |
| Revenue | - | - | - | - |
| Research and development expenses | 3,834,947 | 2,089,977 | 3,105,402 | 1,565,482 |
| General and administrative expenses | 439,846 | 500,072 | 1,075,961 | 561,581 |
| Net loss for the period | 4,214,862 | 2,526,527 | 4,100,371 | 2,040,060 |
| Basic and diluted net loss per share* | 0.97 | 0.60 | 0.99 | 0.50 |
| Cash | 26,165,056 | 27,754,378 | 30,041,001 | 31,864,387 |
*Note: Loss per share has been calculated for all periods on a post-share consolidation basis.
Research and development expenses increased through 2014 as the SRIPaFc development program advanced and were higher in 2015 due to higher costs associated with IND-enabling toxicology studies, preparing the IND submission and initiating the Phase I clinical trial. Research and development expenses for the second quarter of 2014 were higher due to higher non-cash share-based compensation and the recognition of an impairment charge on return of the tigecycline rights back to UHN. General and administrative expenses for the second quarter of 2014 were higher due mainly to higher non-cash share-based compensation expenses and costs associated with migrating to the TSX. General and administrative costs for the second quarter of 2015 were higher than the first quarter of 2015 due mainly to the issuance of DSUs for director fees. The net loss for the third and fourth quarters of 2015 were lower due mainly to net foreign exchange gains of $4,019,251 and $2,163,429, respectively, that resulted mainly from holding U.S. denominated cash with a strengthening U.S. dollar exchange rate.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have, or are reasonably likely to have, a current or future effect on our financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that are material to investors.
Quantitative & Qualitative Disclosures About Market Risk
Fair Value
IFRS 13,Fair Value Measurement provides a hierarchy of valuation techniques based on whether the inputs to those valuation techniques are observable or unobservable. Observable inputs are those which reflect market data obtained from independent sources, while unobservable inputs reflects our assumptions with respect to how market participants would price an asset or liability. These two inputs used to measure fair value fall into the following three different levels of the fair value hierarchy:
Level 1 Quoted prices in active markets for identical instruments that are observable.
Level 2 Quoted prices in active markets for similar instruments; inputs other than quoted prices that are observable and derived from or corroborated by observable market data.
Level 3 Valuations derived from valuation techniques in which one or more significant inputs are unobservable.
The hierarchy requires the use of observable market data when available.
- 21 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
We have classified cash and marketable securities as Level 1. The loan payable has been classified as Level 2.
Cash, marketable securities, amounts receivable, accounts payable and accrued liabilities, and other current liabilities, due within one year, are all short-term in nature and, as such, their carrying values approximate fair values. The fair value of the non-current loan payable and long-term liability is estimated by discounting the expected future cash flows at the cost of money to us, which is equal to its carrying value.
Risks
We are exposed to credit risk, liquidity risk, interest rate risk and currency risk. Our Board of Directors has overall responsibility for the establishment and oversight of our risk management framework. The Audit Committee is responsible for reviewing our risk management policies.
Credit risk
Credit risk is the risk of financial loss to the Company if a counterparty to a financial instrument fails to meet its contractual obligations, and arises principally from the Company’s cash and amounts receivable. The carrying amount of these financial assets represents the maximum credit exposure. The Company follows an investment policy to mitigate against the deterioration of principal and to enhance the Company’s ability to meet its liquidity needs. Cash is on deposit with major Canadian chartered banks and the Company invests in high grade short-term instruments. Amounts receivable are primarily comprised of amounts due from the federal government.
Liquidity risk
Liquidity risk is the risk that the Company will not be able to meet its financial obligations as they fall due. The Company is a development stage company and is reliant on external fundraising to support its operations. Once funds have been raised, the Company manages its liquidity risk by investing in cash and short-term instruments to provide regular cash flow for current operations. It also manages liquidity risk by continuously monitoring actual and projected cash flows. The Board of Directors reviews and approves the Company’s operating and capital budgets, as well as any material transactions not in the ordinary course of business. The majority of the Company’s accounts payable and accrued liabilities have maturities of less than three months.
Interest rate risk
Interest rate risk is the risk that the fair value or future cash flows of a financial instrument will fluctuate because of changes in market interest rates. The Company holds its cash in bank accounts or high interest savings accounts which have a variable rate of interest. The Company manages its interest rate risk by holding highly liquid short-term instruments and by holding its investments to maturity, where possible. For the years ended December 31, 2015 and 2014, the Company earned interest income of $488,486 and $378,692, respectively. Therefore, a 1% change in the average interest rate for the years ended December 31, 2015 and 2014, would have a net impact on finance income of $4,885 and $3,787, respectively.
- 22 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
Currency risk
The Company is exposed to currency risk related to the fluctuation of foreign exchange rates and the degree of volatility of those rates. Currency risk is limited to the portion of the Company’s business transactions denominated in currencies other than the Canadian dollar which are primarily expenses in US dollars. As at December 31, 2015 and 2014, the Company held US dollar cash in the amount of US$44,547,591 and US$142,558 and had US dollar denominated accounts payable and accrued liabilities in the amount of US$1,033,319 and US$1,910,430, respectively. Therefore, a 1% change in the foreign exchange rate would have a net impact on finance costs as at December 31, 2015 and 2014 of $435,143 and $17,679, respectively.
US dollar expenses for the years ended December 31, 2015 and 2014 were approximately US$8,700,000 and US$3,260,000, respectively. Varying the US exchange rate for the years ended December 31, 2015 and 2014 to reflect a 5% strengthening of the Canadian dollar would have decreased the net loss by approximately $435,000 and $163,000, respectively, assuming that all other variables remained constant.
Implications of Being an Emerging Growth Company
We are an “emerging growth company” under the U.S. Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and will continue to qualify as an “emerging growth company” until the earliest to occur of: (a) the last day of the fiscal year during which we have total annual gross revenues of $1,000,000,000 (as such amount is indexed for inflation every 5 years by the SEC) or more; (b) the last day of our fiscal year following the fifth anniversary of the date of the first sale of our common shares pursuant to an effective registration statement under the Securities Act; (c) the date on which we have, during the previous 3-year period, issued more than $1,000,000,000 in non-convertible debt; or (d) the date on which we are deemed to be a “large accelerated filer”, as defined in Rule 12b–2 of the Exchange Act.
Generally, a company that registers any class of its securities under Section 12 of the Exchange Act is required to include in the second and all subsequent annual reports filed by it under the Exchange Act, a management report on internal control over financial reporting and, subject to an exemption available to companies that meet the definition of a “smaller reporting company” in Rule 12b-2 under the Exchange Act, an auditor attestation report on management’s assessment of the company’s internal control over financial reporting. However, for so long as we continue to qualify as an emerging growth company, we will be exempt from the requirement to include an auditor attestation report in our annual reports filed under the Exchange Act, even if we do not qualify as a “smaller reporting company”. In addition, Section 103(a)(3) of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, has been amended by the JOBS Act to provide that, among other things, auditors of an emerging growth company are exempt from any rules of the Public Company Accounting Oversight Board requiring mandatory audit firm rotation or a supplement to the auditor’s report in which the auditor would be required to provide additional information about the audit and the financial statements of the company.
Any U.S. domestic issuer that is an emerging growth company is able to avail itself of the reduced disclosure obligations regarding executive compensation in periodic reports and proxy statements, and to not present to its shareholders a non-binding advisory vote on executive compensation, obtain approval of any golden parachute payments not previously approved, or present the relationship between executive compensation actually paid and our financial performance. So long as we are a foreign private issuer, we are not subject to such requirements, and will not become subject to such requirements even if we were to cease to be an emerging growth company.
As a reporting issuer under the securities legislation of the Canadian provinces of Ontario, British Columbia, Manitoba, Nova Scotia and Alberta, we are required to comply with all new or revised accounting standards that apply to Canadian public companies. Pursuant to Section 107(b) of the JOBS Act, an emerging growth company may elect to utilize an extended transition period for complying with new or revised accounting standards for public companies until such standards apply to private companies. We have elected not to utilize this extended transition period.
- 23 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
Critical Accounting Estimates
The preparation of financial statements in conformity with IFRS requires management to make judgments, estimates and assumptions that affect the application of accounting policies and the reported amounts of assets and liabilities, revenue and expenses and the related disclosures of contingent assets and liabilities and the determination of the Company’s ability to continue as a going concern. Actual results could differ materially from these estimates and assumptions. The Company reviews its estimates and underlying assumptions on an ongoing basis. Revisions are recognized in the period in which the estimates are revised and may impact future periods.
The areas involving a higher degree of judgment or complexity, or areas where assumptions and estimates are significant to the financial statements have been set out in Note 2 of our annual audited consolidated financial statements for the year ended December 31, 2015.
Accounting Policies
Our significant accounting policies are outlined in our annual audited consolidated financial statements for the year ended December 31, 2015. This MD&A should be read in conjunction with the annual audited consolidated financial statements for the year ended December 31, 2015.
New standards and interpretations not yet effective
IFRS 9 Financial Instruments
In October 2010, the IASB published amendments to IFRS 9 Financial Instruments, or IFRS 9, which provides added guidance on the classification and measurement of financial liabilities. In July 2014, the IASB issued its final version of IFRS 9, which completes the classification and measurement, impairment and hedge accounting phases of the IASB’s project to replace IAS 39. The final standard is mandatorily effective for annual periods beginning on or after January 1, 2018, with earlier application permitted. We are reviewing the standard to determine the impact that the adoption of this standard may have on the consolidated financial statements.
IFRS 15 Revenue from Contracts with Customers
In May 2014, the IASB issued IFRS 15 Revenue from Contracts with Customers, or IFRS 15, which covers principles for reporting about the nature, amount, timing and uncertainty of revenue and cash flows arising from contracts with customers. IFRS 15 is effective for annual periods beginning on or after January 1, 2018. Entities will transition following either a full or modified retrospective approach. We are reviewing the standard to determine the impact that the adoption of this standard may have on the consolidated financial statements.
IFRS 16 Leases
In January 2016, the IASB has issued IFRS 16 Leases, or IFRS 16, its new leases standard that requires lessees to recognize assets and liabilities for most leases on their balance sheets. Lessees applying IFRS 16 will have a single accounting model for all leases, with certain exemptions. Lessor accounting is substantially unchanged. The new standard will be effective from January 1, 2019 with limited early application permitted. We have not yet begun the process of evaluating the impact of this standard on our consolidated financial statements.
Other accounting standards or amendments to existing accounting standards that have been issued, but have future effective dates, are either not applicable or are not expected to have a significant impact on our consolidated financial statements.
- 24 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
RISK FACTORS
An investment in our common shares involves a high degree of risk and should be considered speculative. An investment in our common shares should only be undertaken by those persons who can afford the total loss of their investment. You should carefully consider the risks and uncertainties described below, as well as other information contained in this MD&A. The risks and uncertainties below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we believe to be immaterial may also adversely affect our business. If any of the following risks occur, our business, financial condition and results of operations could be seriously harmed and you could lose all or part of your investment. Further, if we fail to meet the expectations of the public market in any given period, the market price of our common shares could decline. We operate in a highly competitive environment that involves significant risks and uncertainties, some of which are outside of our control.
Risks Related to our Business and our Industry
We expect to incur future losses and we may never become profitable.
We have incurred losses of $14.7 million, $12.9 million and $4.3 million for the years ended December 31, 2015, 2014, and 2013, respectively, and expect to incur an operating loss for the year ending December 31, 2015. We have an accumulated deficit since inception through December 31, 2015 of $65.3 million. We believe that operating losses will continue as we are planning to incur significant costs associated with the clinical development of SIRPαFc. Our net losses have had and will continue to have an adverse effect on, among other things, our shareholders’ equity, total assets and working capital. We expect that losses will fluctuate from quarter to quarter and year to year, and that such fluctuations may be substantial. We cannot predict when we will become profitable, if at all.
We currently have no product revenue and will not be able to maintain our operations and research and development without additional funding.
To date, we have generated no product revenue and cannot predict when and if we will generate product revenue. Our ability to generate product revenue and ultimately become profitable depends upon our ability, alone or with partners, to successfully develop our product candidates, obtain regulatory approval, and commercialize products, including any of our current product candidates, or other product candidates that we may develop, in-license or acquire in the future. We do not anticipate generating revenue from the sale of products for the foreseeable future. We expect our research and development expenses to increase in connection with our ongoing activities, particularly as we advance our product candidates through clinical trials.
Our prospects depend on the success of our product candidates which are at early stages of development, and we may not generate revenue for several years, if at all, from these products.
Given the early stage of our product development, we can make no assurance that our research and development programs will result in regulatory approval or commercially viable products. To achieve profitable operations, we, alone or with others, must successfully develop, gain regulatory approval, and market our future products. We currently have no products that have been approved by the U.S. Food and Drug Administration, or FDA, Health Canada, or HC, or any similar regulatory authority. To obtain regulatory approvals for our product candidates being developed and to achieve commercial success, clinical trials must demonstrate that the product candidates are safe for human use and that they demonstrate efficacy. While we have commenced a phase I trial for SIRPαFc, we have not yet completed a phase I clinical trial or subsequent required clinical trials.
Many product candidates never reach the stage of clinical testing and even those that do have only a small chance of successfully completing clinical development and gaining regulatory approval. Product candidates may fail for a number of reasons, including, but not limited to, being unsafe for human use or due to the failure to provide therapeutic benefits equal to or better than the standard of treatment at the time of testing. Unsatisfactory results obtained from a particular study relating to a research and development program may cause us or our collaborators to abandon commitments to that program. Positive results of early preclinical research may not be indicative of the results that will be obtained in later stages of preclinical or clinical research. Similarly, positive results from early-stage clinical trials may not be indicative of favorable outcomes in later-stage clinical trials. We can make no assurance that any future studies, if undertaken, will yield favorable results.
We recently acquired several preclinical and discovery research programs in our acquisition of Fluorinov, including certain assets relating to the treatment of central nervous system disorders. While we conducted extensive due diligence before making this acquisition, our assessment of the Fluorinov technologies may not be accurate. Therefore, our expectations about whether various clinical and regulatory milestones with an existing Fluorinov compound or development of a future program on the Fluorinov development platform will be achieved may not be borne out fully or at all. We have made a commitment to attempt to monetize the Fluorinov central nervous system assets and, if successful, to share the net proceeds with the Fluorinov vendors. As this is not a core competency of the Company, our efforts to monetize these assets or any other Fluorinov assets may not be successful. We can make no assurances that toxicology, or other preclinical, studies will yield results that will allow us to proceed with clinical trials in humans.
- 25 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
The early stage of our product development makes it particularly uncertain whether any of our product development efforts will prove to be successful and meet applicable regulatory requirements, and whether any of our product candidates will receive the requisite regulatory approvals, be capable of being manufactured at a reasonable cost or be successfully marketed. If we are successful in developing our current and future product candidates into approved products, we will still experience many potential obstacles such as the need to develop or obtain manufacturing, marketing and distribution capabilities. If we are unable to successfully commercialize any of our products, our financial condition and results of operations may be materially and adversely affected.
We rely and will continue to rely on third parties to plan, conduct and monitor our preclinical studies and clinical trials, and their failure to perform as required could cause substantial harm to our business.
We rely and will continue to rely on third parties to conduct a significant portion of our preclinical and clinical development activities. Preclinical activities include in vivo studies providing access to specific disease models, pharmacology and toxicology studies, and assay development. Clinical development activities include trial design, regulatory submissions, clinical patient recruitment, clinical trial monitoring, clinical data management and analysis, safety monitoring and project management. If there is any dispute or disruption in our relationship with third parties, or if they are unable to provide quality services in a timely manner and at a feasible cost, our active development programs will face delays. Further, if any of these third parties fails to perform as we expect or if their work fails to meet regulatory requirements, our testing could be delayed, cancelled or rendered ineffective.
We rely on contract manufacturers over whom we have limited control. If we are subject to quality, cost or delivery issues with the preclinical and clinical grade materials supplied by contract manufacturers, our business operations could suffer significant harm.
We have limited manufacturing experience and rely on contract manufacturing organizations, or CMOs to manufacture our product candidates for larger preclinical studies and clinical trials. We produce small quantities of our product candidates at bench scale in our laboratory facilities for use in smaller preclinical studies. We rely on CMOs for manufacturing, filling, packaging, storing and shipping of drug product in compliance with cGMP regulations applicable to our products. The FDA ensures the quality of drug products by carefully monitoring drug manufacturers’ compliance with cGMP regulations. The cGMP regulations for drugs contain minimum requirements for the methods, facilities and controls used in manufacturing, processing and packing of a drug product.
We contracted with Catalent for the manufacture of the SIRPαFc protein to supply drug product for our phase I clinical trial. The manufacture of recombinant proteins uses well established processes including a protein expression system. Catalent is producing SIRPαFc using their proprietary GPEx® expression system. We believe that Catalent has the capacity, the systems, and the experience to supply SIRPαFc for our phase I clinical trial and we may consider using Catalent for manufacturing for later clinical trials. However, since the Catalent manufacturing facility where SIRPαFc is being produced was only recently established, it has not been inspected by the FDA. Any manufacturing failures or delays or compliance issues could cause delays in the conduct of SIRPαFc preclinical studies and clinical trials.
There can be no assurances that CMOs will be able to meet our timetable and requirements. We have not contracted with alternate suppliers in the event Catalent is unable to scale up production, or if Catalent otherwise experiences any other significant problems. If we are unable to arrange for alternative third-party manufacturing sources on commercially reasonable terms or in a timely manner, we may be delayed in the development of our product candidates. Further, contract manufacturers must operate in compliance with cGMP and failure to do so could result in, among other things, the disruption of product supplies. Our dependence upon third parties for the manufacture of our products may adversely affect our profit margins and our ability to develop and deliver products on a timely and competitive basis.
- 26 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
If clinical trials of our product candidates fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or do not otherwise produce positive results, we would incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we must conduct preclinical studies in animals and extensive clinical trials in humans to demonstrate the safety and efficacy of the product candidates. Clinical testing is expensive and difficult to design and implement, can take many years to complete and has uncertain outcomes. The outcome of preclinical studies and early clinical trials may not predict the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety profiles, notwithstanding promising results in earlier trials. We do not know whether the clinical trials we may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market any of our product candidates in any jurisdiction. A product candidate may fail for safety or efficacy reasons at any stage of the testing process. A major risk we face is the possibility that none of our product candidates under development will successfully gain market approval from the FDA or other regulatory authorities, resulting in us being unable to derive any commercial revenue from them after investing significant amounts of capital in multiple stages of preclinical and clinical testing.
If we experience delays in clinical testing, we will be delayed in commercializing our product candidates, and our business may be substantially harmed.
We cannot predict whether any clinical trials will begin as planned, will need to be restructured, or will be completed on schedule, or at all. Our product development costs will increase if we experience delays in clinical testing. Significant clinical trial delays could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before us, which would impair our ability to successfully commercialize our product candidates and may harm our financial condition, results of operations and prospects. The commencement and completion of clinical trials for our products may be delayed for a number of reasons, including delays related, but not limited, to:
| | • | failure by regulatory authorities to grant permission to proceed or placing the clinical trial on hold; |
| | • | patients failing to enroll or remain in our trials at the rate we expect; |
| • | suspension or termination of clinical trials by regulators for many reasons, including concerns about patient safety or failure of our contract manufacturers to comply with cGMP requirements; |
| | • | any changes to our manufacturing process that may be necessary or desired; |
| • | delays or failure to obtain clinical supply from contract manufacturers of our products necessary to conduct clinical trials; |
| | • | product candidates demonstrating a lack of safety or efficacy during clinical trials; |
| • | patients choosing an alternative treatment for the indications for which we are developing any of our product candidates or participating in competing clinical trials; |
| • | patients failing to complete clinical trials due to dissatisfaction with the treatment, side effects or other reasons; |
| | • | reports of clinical testing on similar technologies and products raising safety and/or efficacy concerns; |
| | • | competing clinical trials and scheduling conflicts with participating clinicians; |
| • | clinical investigators not performing our clinical trials on their anticipated schedule, dropping out of a trial, or employing methods not consistent with the clinical trial protocol, regulatory requirements or other third parties not performing data collection and analysis in a timely or accurate manner; |
- 27 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
| • | failure of our contract research organizations, or CROs, to satisfy their contractual duties or meet expected deadlines; |
| • | inspections of clinical trial sites by regulatory authorities or Institutional Review Boards, or IRBs, or ethics committees finding regulatory violations that require us to undertake corrective action, resulting in suspension or termination of one or more sites or the imposition of a clinical hold on the entire study; |
| • | one or more IRBs or ethics committees rejecting, suspending or terminating the study at an investigational site, precluding enrollment of additional subjects, or withdrawing its approval of the trial; or |
| | • | failure to reach agreement on acceptable terms with prospective clinical trial sites. |
Our product development costs will increase if we experience delays in testing or approval or if we need to perform more or larger clinical trials than planned. Additionally, changes in regulatory requirements and policies may occur, and we may need to amend study protocols to reflect these changes. Amendments may require us to resubmit our study protocols to regulatory authorities or IRBs or ethics committees for re-examination, which may impact the cost, timing or successful completion of that trial. Delays or increased product development costs may have a material adverse effect on our business, financial condition and prospects.
If we have difficulty enrolling patients in clinical trials, the completion of the trials may be delayed or cancelled.
As our product candidates advance from preclinical testing to clinical testing, and then through progressively larger and more complex clinical trials, we will need to enroll an increasing number of patients that meet our eligibility criteria. There is significant competition for recruiting cancer patients in clinical trials, and we may be unable to enroll the patients we need to complete clinical trials on a timely basis or at all. The factors that affect our ability to enroll patients are largely uncontrollable and include, but are not limited to, the following:
| | • | size and nature of the patient population; |
| | • | eligibility and exclusion criteria for the trial; |
| | • | design of the study protocol; |
| | • | competition with other companies for clinical sites or patients; |
| | • | the perceived risks and benefits of the product candidate under study; |
| | • | the patient referral practices of physicians; and |
| | • | the number, availability, location and accessibility of clinical trial sites. |
If we are unable to successfully develop companion diagnostics for our therapeutic product candidates, or experience significant delays in doing so, we may not achieve marketing approval or realize the full commercial potential of our therapeutic product candidates.
We may develop companion diagnostics for our therapeutic product candidates. We expect that, at least in some cases, regulatory authorities may require the development and regulatory approval of a companion diagnostic as a condition to approving our therapeutic product candidates. We have limited experience and capabilities in developing or commercializing diagnostics and plan to rely in large part on third parties to perform these functions. We have not begun to develop companion diagnostics for any of our therapeutic product candidates.
Companion diagnostics are subject to regulation by the FDA, HC, and comparable foreign regulatory authorities as medical devices and may require separate regulatory approval or clearance prior to commercialization. If we, or any third parties that we engage to assist us, are unable to successfully develop companion diagnostics for our therapeutic product candidates, or experience delays in doing so, our business may be substantially harmed.
Regulatory approval processes are lengthy, expensive and inherently unpredictable. Our inability to obtain regulatory approval for our product candidates would substantially harm our business.
Our development and commercialization activities and product candidates are significantly regulated by a number of governmental entities, including the FDA, HC, and comparable authorities in other countries. Regulatory approvals are required prior to each clinical trial and we may fail to obtain the necessary approvals to commence or continue clinical testing. We must comply with regulations concerning the manufacture, testing, safety, effectiveness, labeling, documentation, advertising, and sale of products and product candidates and ultimately must obtain regulatory approval before we can commercialize a product candidate. The time required to obtain approval by such regulatory authorities is unpredictable but typically takes many years following the commencement of preclinical studies and clinical trials. Any analysis of data from clinical activities we perform is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. Even if we believe results from our clinical trials are favorable to support the marketing of our product candidates, the FDA or other regulatory authorities may disagree. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. We have not obtained regulatory approval for any product candidate and it is possible that none of our existing product candidates or any future product candidates will ever obtain regulatory approval.
- 28 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
We could fail to receive regulatory approval for our product candidates for many reasons, including, but not limited to:
| | • | disagreement with the design or implementation of our clinical trials; |
| | • | failure to demonstrate that a product candidate is safe and effective for its proposed indication; |
| | • | failure of clinical trials to meet the level of statistical significance required for approval; |
| | • | failure to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
| | • | disagreement with our interpretation of data from preclinical studies or clinical trials; |
| • | the insufficiency of data collected from clinical trials of our product candidates to support the submission and filing of a BLA or other submission to obtain regulatory approval; |
| • | deficiencies in the manufacturing processes or the failure of facilities of CMOs with whom we contract for clinical and commercial supplies to pass a pre-approval inspection; or |
| • | changes in the approval policies or regulations that render our preclinical and clinical data insufficient for approval. |
A regulatory authority may require more information, including additional preclinical or clinical data to support approval, which may delay or prevent approval and our commercialization plans, or we may decide to abandon the development program. If we were to obtain approval, regulatory authorities may approve any of our product candidates for fewer or more limited indications than we request, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. Moreover, depending on any safety issues associated with our product candidates that garner approval, the FDA may impose a risk evaluation and mitigation strategy, thereby imposing certain restrictions on the sale and marketability of such products.
We will require additional capital to finance our operations, which may not be available to us on acceptable terms, or at all. As a result, we may not complete the development and commercialization of our product candidates or develop new product candidates.
As a research and development company, our operations have consumed substantial amounts of cash since inception. We expect to spend substantial funds to continue the research, development and testing of our product candidates and to prepare to commercialize products subject to FDA approval in the U.S. and similar approvals in other jurisdictions. We will also require significant additional funds if we expand the scope of our current clinical plans for the SIRPαFc program or if we were to acquire any new assets and advance their development. Therefore, for the foreseeable future, we will have to fund all of our operations and development expenditures from cash on hand, equity or debt financings, through collaborations with other biotechnology or pharmaceutical companies or through financings from other sources. We expect that our existing cash at December 31, 2015 of $86,770,542 will enable us to fund our current operating plan requirements for at least the next twelve months. Additional financing will be required to meet our long term liquidity needs. If we do not succeed in raising additional funds on acceptable terms, we might not be able to complete planned preclinical studies and clinical trials or pursue and obtain approval of any product candidates from the FDA and other regulatory authorities. It is possible that future financing will not be available or, if available, may not be on favorable terms. The availability of financing will be affected by the achievement of our corporate goals, the results of scientific and clinical research, the ability to obtain regulatory approvals, the state of the capital markets generally and with particular reference to drug development companies, the status of strategic alliance agreements and other relevant commercial considerations. If adequate funding is not available, we may be required to delay, reduce or eliminate one or more of our product development programs, or obtain funds through corporate partners or others who may require us to relinquish significant rights to product candidates or obtain funds on less favorable terms than we would otherwise accept. To the extent that external sources of capital become limited or unavailable or available on onerous terms, our intangible assets and our ability to continue our clinical development plans may become impaired, and our assets, liabilities, business, financial condition and results of operations may be materially or adversely affected.
- 29 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
We face competition from other biotechnology and pharmaceutical companies and our financial condition and operations will suffer if we fail to effectively compete.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. Our competitors include large, well-established pharmaceutical companies, biotechnology companies, and academic and research institutions developing cancer therapeutics for the same indications we are targeting and competitors with existing marketed therapies. Many other companies are developing or commercializing therapies to treat the same diseases or indications for which our product candidates may be useful. Although there are no approved therapies that specifically target the CD47 pathway, some competitors use therapeutic approaches that may compete directly with our product candidates. For example, SIRPαFc is in direct competition with CD47 blocking antibodies from Stanford University, Celgene Corporation, and Novimmune SA.
Many of our competitors have substantially greater financial, technical and human resources than we do and have significantly greater experience than us in conducting preclinical testing and human clinical trials of product candidates, scaling up manufacturing operations and obtaining regulatory approvals of products. Accordingly, our competitors may succeed in obtaining regulatory approval for products more rapidly than we do. Our ability to compete successfully will largely depend on:
| • | the efficacy and safety profile of our product candidates relative to marketed products and other product candidates in development; |
| • | our ability to develop and maintain a competitive position in the product categories and technologies on which we focus; |
| • | the time it takes for our product candidates to complete clinical development and receive marketing approval; |
| | • | our ability to obtain required regulatory approvals; |
| | • | our ability to commercialize any of our product candidates that receive regulatory approval; |
| • | our ability to establish, maintain and protect intellectual property rights related to our product candidates; and |
| • | acceptance of any of our product candidates that receive regulatory approval by physicians and other healthcare providers and payers. |
Competitors have developed and may develop technologies that could be the basis for products that challenge the differentiated nature and potential for best-in-class product development programs and discovery research capabilities of Fluorinov. Some of those products may have an entirely different approach or means of accomplishing the desired therapeutic effect than our products and may be more effective or less costly than our products. The success of our competitors and their products and technologies relative to our technological capabilities and competitiveness could have a material adverse effect on the future pre-clinical and clinical trials of our products, including our ability to obtain the necessary regulatory approvals for the conduct of such trials. This may further negatively impact our ability to generate future product development programs with improved pharmacological properties using Fluorinov technology.
- 30 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition and operations will substantially suffer.
We heavily rely on the capabilities and experience of our key executives and scientists and the loss of any of them could affect our ability to develop our products.
The loss of Dr. Niclas Stiernholm, our President and Chief Executive Officer, or other key members of our staff, including Dr. Robert Uger, our Chief Scientific Officer, Dr. Eric Sievers, our Chief Medical Officer, James Parsons, our Chief Financial Officer, Dr. Penka Petrova, our Chief Development Officer, or Dr. Malik Slassi, our Senior Vice President, Discovery Research could harm us. We have employment agreements with Drs. Stiernholm, Uger, Sievers, Petrova and Slassi and Mr. Parsons, although such employment agreements do not guarantee their retention. We also depend on our scientific and clinical collaborators and advisors, all of whom have outside commitments that may limit their availability to us. In addition, we believe that our future success will depend in large part upon our ability to attract and retain highly skilled scientific, managerial, medical, clinical and regulatory personnel, particularly as we expand our activities and seek regulatory approvals for clinical trials. We enter into agreements with our scientific and clinical collaborators and advisors, key opinion leaders and academic partners in the ordinary course of our business. We also enter into agreements with physicians and institutions who will recruit patients into our clinical trials on our behalf in the ordinary course of our business. Notwithstanding these arrangements, we face significant competition for these types of personnel from other companies, research and academic institutions, government entities and other organizations. We cannot predict our success in hiring or retaining the personnel we require for continued growth. The loss of the services of any of our executive officers or other key personnel could potentially harm our business, operating results or financial condition.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have a material adverse effect on our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include failures to comply with FDA regulations, provide accurate information to the FDA, comply with manufacturing standards we have established, comply with federal and state health-care fraud and abuse laws and regulations, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing, and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a substantial impact on our business and results of operations, including the imposition of substantial fines or other sanctions.
We may expand our business through the acquisition of companies or businesses or by entering into collaborations or by in-licensing product candidates, each of which could disrupt our business and harm our financial condition.
We have in the past and may in the future seek to expand our pipeline and capabilities by acquiring one or more companies or businesses, entering into collaborations, or in-licensing one or more product candidates. For example, in January 2016, we acquired Fluorinov, a small molecule medicinal chemistry company with preclinical oncology assets and a potential discovery platform. In April 2013, we in-licensed tigecycline which was subsequently returned to the UHN in 2014. Acquisitions, collaborations and in-licenses involve numerous risks, including, but not limited to:
| | • | substantial cash expenditures; |
| | • | technology development risks; |
| | • | potentially dilutive issuances of equity securities; |
- 31 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
| • | incurrence of debt and contingent liabilities, some of which may be difficult or impossible to identify at the time of acquisition; |
| | • | difficulties in assimilating the operations of the acquired companies; |
| | • | potential disputes regarding contingent consideration; |
| | • | diverting our management’s attention away from other business concerns; |
| | • | entering markets in which we have limited or no direct experience; and |
| | • | potential loss of our key employees or key employees of the acquired companies or businesses. |
We have experience in making acquisitions, entering collaborations, and in-licensing product candidates, however, we cannot provide assurance that any acquisition, collaboration or in-license will result in short-term or long-term benefits to us. We may incorrectly judge the value or worth of an acquired company or business or in-licensed product candidate. In addition, our future success would depend in part on our ability to manage the rapid growth associated with some of these acquisitions, collaborations and in-licenses. We cannot provide assurance that we would be able to successfully combine our business with that of acquired businesses, manage a collaboration or integrate in-licensed product candidates. Furthermore, the development or expansion of our business may require a substantial capital investment by us.
Negative results from clinical trials or studies of others and adverse safety events involving the targets of our products may have an adverse impact on our future commercialization efforts.
From time to time, studies or clinical trials on various aspects of biopharmaceutical products are conducted by academic researchers, competitors or others. The results of these studies or trials, when published, may have a significant effect on the market for the biopharmaceutical product that is the subject of the study. The publication of negative results of studies or clinical trials or adverse safety events related to our product candidates, or the therapeutic areas in which our product candidates compete, could adversely affect our share price and our ability to finance future development of our product candidates, and our business and financial results could be materially and adversely affected.
We face the risk of product liability claims, which could exceed our insurance coverage and produce recalls, each of which could deplete our cash resources.
We are exposed to the risk of product liability claims alleging that use of our product candidates caused an injury or harm. These claims can arise at any point in the development, testing, manufacture, marketing or sale of our product candidates and may be made directly by patients involved in clinical trials of our product candidates, by consumers or healthcare providers or by individuals, organizations or companies selling our products. Product liability claims can be expensive to defend, even if the product or product candidate did not actually cause the alleged injury or harm.
Insurance covering product liability claims becomes increasingly expensive as a product candidate moves through the development pipeline to commercialization. We currently maintain clinical trial liability insurance coverage of $10 million. However, there can be no assurance that such insurance coverage is or will continue to be adequate or available to us at a cost acceptable to us or at all. We may choose or find it necessary under our collaborative agreements to increase our insurance coverage in the future. We may not be able to secure greater or broader product liability insurance coverage on acceptable terms or at reasonable costs when needed. Any liability for damages resulting from a product liability claim could exceed the amount of our coverage, require us to pay a substantial monetary award from our own cash resources and have a material adverse effect on our business, financial condition and results of operations. Moreover, a product recall, if required, could generate substantial negative publicity about our products and business, inhibit or prevent commercialization of other products and product candidates or negatively impact existing or future collaborations.
In addition, some of our licensing and other agreements with third parties require or might require us to maintain product liability insurance. If we cannot maintain acceptable amounts of coverage on commercially reasonable terms in accordance with the terms set forth in these agreements, the corresponding agreements would be subject to termination, which could have a material adverse impact on our operations.
- 32 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
We may not achieve our publicly announced milestones according to schedule, or at all.
From time to time, we may announce the timing of certain events we expect to occur, such as the anticipated timing of results from our clinical trials. These statements are forward-looking and are based on the best estimates of management at the time relating to the occurrence of such events. However, the actual timing of such events may differ from what has been publicly disclosed. The timing of events such as initiation or completion of a clinical trial, filing of an application to obtain regulatory approval, or announcement of additional clinical trials for a product candidate may ultimately vary from what is publicly disclosed. These variations in timing may occur as a result of different events, including the nature of the results obtained during a clinical trial or during a research phase, problems with a CMO or a CRO or any other event having the effect of delaying the publicly announced timeline. We undertake no obligation to update or revise any forward-looking information, whether as a result of new information, future events or otherwise, except as otherwise required by law. Any variation in the timing of previously announced milestones could have a material adverse effect on our business plan, financial condition or operating results and the trading price of common shares.
We are exposed to the financial risk related to the fluctuation of foreign exchange rates and the degrees of volatility of those rates.
We may be adversely affected by foreign currency fluctuations. To date, we have been primarily funded through issuances of equity, proceeds from the exercise of warrants and stock options and from interest income on funds available for investment, which are all denominated both in Canadian and U.S. dollars. Also, a growing portion of our expenditures are in U.S. dollars, and we are therefore subject to foreign currency fluctuations which may, from time to time, impact our financial position and results of operations.
Risks Related to Intellectual Property
If we are unable to adequately protect and enforce our intellectual property, our competitors may take advantage of our development efforts or acquired technology and compromise our prospects of marketing and selling our key products.
We control two patent families relating to SIRPα. One family relates to the use of SIRPα to treat cancer. The other family relates our drug as a composition of matter, SIRPαFc.
More recently, we acquired the patent portfolio of Fluorinov, which embraces patent filings that cover eleven different inventions. With the exception of one process scheme, these patent filings each claim a family of small molecule drugs as compositions of matter, together with claims for their production and their medical uses. These drugs target cancer for the most part, and some related medical end-uses.
Our success will depend in part upon our ability to protect our intellectual property and proprietary technologies and upon the nature and scope of the intellectual property protection we receive. For example, some of our patent portfolio covers primarily methods of medical use but not compositions of matter. The ability to compete effectively and to achieve partnerships will depend on our ability to develop and maintain proprietary aspects of our technology and to operate without infringing on the proprietary rights of others. The presence of such proprietary rights of others could severely limit our ability to develop and commercialize our products, to conduct our existing research and could require financial resources to defend litigation, which may be in excess of our ability to raise such funds. There is no assurance that our pending patent applications or those that we intend to acquire will be approved in a form that will be sufficient to protect our proprietary technology and gain or keep any competitive advantage that we may have.
- 33 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
The patent positions of pharmaceutical companies can be highly uncertain and involve complex legal, scientific and factual questions for which important legal principles remain unresolved. Patents issued to us or our respective licensors may be challenged, invalidated or circumvented. To the extent our intellectual property, including licensed intellectual property, offers inadequate protection, or is found to be invalid or unenforceable, we are exposed to a greater risk of direct competition. If our intellectual property does not provide adequate protection against our competitors’ products, our competitive position could be adversely affected, as could our business, financial condition and results of operations. Both the patent application process and the process of managing patent disputes can be time consuming and expensive, and the laws of some foreign countries may not protect our intellectual property rights to the same extent as do the laws of Canada and the United States.
We will be able to protect our intellectual property from unauthorized use by third parties only to the extent that our proprietary technologies, key products, and any future products are covered by valid and enforceable intellectual property rights including patents or are effectively maintained as trade secrets, and provided we have the funds to enforce our rights, if necessary.
If we lose our licenses from third-party owners we may be unable to continue a substantial part of our business.
We are party to licenses that give us rights to intellectual property that is necessary or useful for a substantial part of our business. Pursuant to our exclusive license agreement with UHN and the Hospital for Sick Children, or HSC, under which we license certain patent rights for our key products and their uses, we are required to use commercially reasonable efforts to commercialize products based on the licensed rights and pay certain royalties and sublicensing revenue to UHN and HSC. These licenses require that we pay development milestone payments, regulatory milestone payments, royalties on net sales, and sublicensing revenues, as well as annual maintenance fees.
We have also entered into agreements allowing us to manufacture SIRPαFc using Catalent’s proprietary GPEx® expression system. The consideration includes payments at the time we successfully reach a series of development and sales milestones. We may also enter into licenses in the future to access additional third-party intellectual property.
If we fail to pay annual maintenance fees, development and sales milestones, or it is determined that we did not use commercially reasonable efforts to commercialize licensed products, we could lose our licenses which could have a material adverse effect on our business and financial condition.
We may require additional third-party licenses to effectively develop and manufacture our key products and are currently unable to predict the availability or cost of such licenses.
A substantial number of patents have already been issued to other biotechnology and pharmaceutical companies. To the extent that valid third-party patent rights cover our products or services, we or our strategic collaborators would be required to seek licenses from the holders of these patents in order to manufacture, use or sell these products and services, and payments under them would reduce our profits from these products and services. We are currently unable to predict the extent to which we may wish or be required to acquire rights under such patents, the availability and cost of acquiring such rights, and whether a license to such patents will be available on acceptable terms or at all. There may be patents in the U.S. or in foreign countries or patents issued in the future that are unavailable to license on acceptable terms. Our inability to obtain such licenses may hinder or eliminate our ability to manufacture and market our products.
- 34 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
Changes in patent law and its interpretation could diminish the value of patents in general, thereby impairing our ability to protect our product candidates.
As is the case with other biotechnology and pharmaceutical companies, our success is heavily dependent on intellectual property rights, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves technological and legal complexity, and obtaining and enforcing biopharmaceutical patents is costly, time-consuming and inherently uncertain. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our and our licensors’ or collaborators’ ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the U.S. Patent and Trademark Office, or USPTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our and our licensors’ or collaborators’ ability to obtain new patents or to enforce existing patents and patents we and our licensors or collaborators may obtain in the future.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our and our licensors’ or collaborators’ patent applications and the enforcement or defense of our or our licensors’ or collaborators’ issued patents. On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted and may also affect patent litigation. The USPTO recently developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective on March 16, 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our or our licensors’ or collaborators’ patent applications and the enforcement or defense of our or our licensors’ or collaborators’ issued patents, all of which could have a material adverse effect on our business and financial condition.
Litigation regarding patents, patent applications, and other proprietary rights may be expensive, time consuming and cause delays in the development and manufacturing of our key products.
Our success will depend in part on our ability to operate without infringing the proprietary rights of third parties. The pharmaceutical industry is characterized by extensive patent litigation. Other parties may have, or obtain in the future, patents and allege that the use of our technologies infringes these patent claims or that we are employing their proprietary technology without authorization.
In addition, third parties may challenge or infringe upon our existing or future patents. Proceedings involving our patents or patent applications or those of others could result in adverse decisions regarding:
| | • | the patentability of our inventions relating to our key products; and/or |
| | • | the enforceability, validity, or scope of protection offered by our patents relating to our key products. |
If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, defend an infringement action, or challenge the validity of the patents in court. Regardless of the outcome, patent litigation is costly and time consuming. In some cases, we may not have sufficient resources to bring these actions to a successful conclusion. In addition, if we do not obtain a license, develop or obtain non-infringing technology, fail to defend an infringement action successfully or have infringed patents declared invalid, we may:
| | • | incur substantial monetary damages; |
| | • | encounter significant delays in bringing our key products to market; and/or |
- 35 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
| • | be precluded from participating in the manufacture, use or sale of our key products or methods of treatment requiring licenses. |
Even if we are successful in these proceedings, we may incur substantial costs and divert management time and attention in pursuing these proceedings, which could have a material adverse effect on us.
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them.
Because we rely on third parties to develop our products, we must share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically restrict the ability of our collaborators, advisors, employees and consultants to publish data potentially relating to our trade secrets. Our academic collaborators typically have rights to publish data, provided that we are notified in advance and may delay publication for a specified time in order to secure our intellectual property rights arising from the collaboration. In other cases, publication rights are controlled exclusively by us, although in some cases we may share these rights with other parties. We also conduct joint research and development programs which may require us to share trade secrets under the terms of research and development collaboration or similar agreements. Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of these agreements, independent development or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication. A competitor’s discovery of our trade secrets may impair our competitive position and could have a material adverse effect on our business and financial condition.
Risks Related to Our Common Shares
Our common share price has been volatile in recent years, and may continue to be volatile.
The market prices for securities of biopharmaceutical companies, including ours, have historically been volatile. In the year ended December 31, 2015, our common shares traded on the TSX, at a high of $37.27 and a low of $10.50 per share. In the year ended December 31, 2014, on the TSXV/TSX our common shares traded at a high of $22.20 and a low of $6.30 per share. A number of factors could influence the volatility in the trading price of our common shares, including changes in the economy or in the financial markets, industry related developments, the results of product development and commercialization, changes in government regulations, and developments concerning proprietary rights, litigation and cash flow. Our quarterly losses may vary because of the timing of costs for manufacturing, preclinical studies and clinical trials. Also, the reporting of adverse safety events involving our products and public rumors about such events could cause our share price to decline or experience periods of volatility. Each of these factors could lead to increased volatility in the market price of our common shares. In addition, changes in the market prices of the securities of our competitors may also lead to fluctuations in the trading price of our common shares.
We have never paid dividends and do not expect to do so in the foreseeable future.
We have not declared or paid any cash dividends on our common or preferred shares to date. The payment of dividends in the future will be dependent on our earnings and financial condition in addition to such other factors as our board of directors considers appropriate. Unless and until we pay dividends, shareholders may not receive a return on their shares. There is no present intention by our board of directors to pay dividends on our shares.
- 36 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
Future sales or issuances of equity securities and the conversion of outstanding securities to common shares could decrease the value of the common shares, dilute investors’ voting power, and reduce our earnings per share.
We may sell additional equity securities in future offerings, including through the sale of securities convertible into equity securities, to finance operations, acquisitions or projects, and issue additional common shares if outstanding warrants or stock options are exercised, or preferred shares are converted to common shares, which may result in dilution. See the information in the section of this MD&A entitled “Description of Share Capital” for details of our outstanding securities convertible into common shares. We filed a base shelf prospectus with securities commissions in Canada and a Form F-10 registration statement with the SEC on May 29, 2015 that provides that we may sell under the prospectus from time to time over the following 25 months up to U.S. $100 million, in one or more offerings, of common shares, First Preferred shares, warrants to purchase common shares, or units comprising a combination of common shares, First Preferred shares and/or warrants. Subject to receipt of any required regulatory approvals, subscribers of the December 2013 private placement who purchased a minimum of 10% of the securities sold under the offering received rights to purchase our securities in future financings to enable each such shareholder to maintain their percentage holding in our common shares for so long as the subscriber holds at least 10% of the outstanding common shares on a fully-diluted basis. Shareholders who do not have this future financing participation right may be disadvantaged in participating in such financings.
Our board of directors has the authority to authorize certain offers and sales of additional securities without the vote of, or prior notice to, shareholders. Based on the need for additional capital to fund expected expenditures and growth, it is likely that we will issue additional securities to provide such capital. Such additional issuances may involve the issuance of a significant number of common shares at prices less than the current market price for our common shares.
Sales of substantial amounts of our securities, or the availability of such securities for sale, as well as the issuance of substantial amounts of our common shares upon conversion of outstanding convertible equity securities, could adversely affect the prevailing market prices for our securities and dilute investors’ earnings per share. A decline in the market prices of our securities could impair our ability to raise additional capital through the sale of securities should we desire to do so.
We are likely a “passive foreign investment company,” which may have adverse U.S. federal income tax consequences for U.S. shareholders.
U.S. investors should be aware that we believe we were classified as a passive foreign investment company, or PFIC, during the tax years ended December 31, 2015 and 2014, and based on current business plans and financial expectations, we expect that we will be a PFIC for the current tax year and may be a PFIC in future tax years. If we are a PFIC for any year during a U.S. shareholder’s holding period of our common shares, then such U.S. shareholder generally will be required to treat any gain realized upon a disposition of our common shares, or any so-called “excess distribution” received on our common shares, as ordinary income, and to pay an interest charge on a portion of such gain or distributions, unless the shareholder makes a timely and effective “qualified electing fund” election, or QEFElection, or a “mark-to-market” election with respect to our shares. A U.S. shareholder who makes a QEF Election generally must report on a current basis its share of our net capital gain and ordinary earnings for any year in which we are a PFIC, whether or not we distribute any amounts to our shareholders. A U.S. shareholder who makes the mark-to-market election generally must include as ordinary income each year the excess of the fair market value of the common shares over the shareholder’s adjusted tax basis therein. Each U.S. shareholder should consult its own tax advisors regarding the PFIC rules and the U.S. federal income tax consequences of the acquisition, ownership and disposition of our common shares.
- 37 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
It may be difficult for non-Canadian investors to obtain and enforce judgments against us because of our Canadian incorporation and presence.
We are a corporation existing under the laws of the Province of Ontario, Canada. Several of our directors and officers, and several of the experts are residents of Canada, and all or a substantial portion of their assets, and a substantial portion of our assets, are located outside the United States. Consequently, although we have appointed an agent for service of process in the United States, it may be difficult for holders of our securities who reside in the United States to effect service within the United States upon those directors and officers, and the experts who are not residents of the United States. It may also be difficult for holders of our securities who reside in the United States to realize in the United States upon judgments of courts of the United States predicated upon our civil liability and the civil liability of our directors, officers and experts under the United States federal securities laws. Investors should not assume that Canadian courts (i) would enforce judgments of United States courts obtained in actions against us or such directors, officers or experts predicated upon the civil liability provisions of the United States federal securities laws or the securities or “blue sky” laws of any state or jurisdiction of the United States or (ii) would enforce, in original actions, liabilities against us or such directors, officers or experts predicated upon the United States federal securities laws or any securities or “blue sky” laws of any state or jurisdiction of the United States. In addition, the protections afforded by Canadian securities laws may not be available to investors in the United States.
If there are substantial sales of our common shares, the market price of our common shares could decline.
Sales of substantial numbers of our common shares could cause a decline in the market price of our common shares. Any sales by existing shareholders or holders who exercise their warrants or stock options may have an adverse effect on our ability to raise capital and may adversely affect the market price of our common shares.
We are an “emerging growth company,” and we cannot be certain if the reduced reporting requirements applicable to emerging growth companies will make our common shares less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We could be an emerging growth company for up to five years, although circumstances could cause us to lose that status earlier. We cannot predict if investors will find our common shares less attractive because we may rely on these exemptions. If some investors find our common shares less attractive as a result, there may be a less active trading market for our common shares and our share price may be more volatile.
Any failure to maintain an effective system of internal controls may result in material misstatements of our consolidated financial statements or cause us to fail to meet our reporting obligations or fail to prevent fraud; and in that case, our shareholders could lose confidence in our financial reporting, which would harm our business and could negatively impact the price of our common shares.
Effective internal controls are necessary for us to provide reliable financial reports and prevent fraud. If we fail to maintain an effective system of internal controls, we might not be able to report our financial results accurately or prevent fraud; and in that case, our shareholders could lose confidence in our financial reporting, which would harm our business and could negatively impact the price of our common shares. While we believe that we have sufficient personnel and review procedures to allow us maintain an effective system of internal controls, we cannot provide assurance that we will not experience potential material weaknesses in our internal control. Even if we conclude that our internal control over financial reporting provides reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with International Financial Reporting Standards, as issued by the International Accounting Standards Board, because of its inherent limitations, internal control over financial reporting may not prevent or detect fraud or misstatements. Failure to implement required new or improved controls, or difficulties encountered in their implementation, could harm our results of operations or cause us to fail to meet our future reporting obligations.
- 38 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
If we fail to timely achieve and maintain the adequacy of our internal control over financial reporting, we may not be able to produce reliable financial reports or help prevent fraud. Our failure to achieve and maintain effective internal control over financial reporting could prevent us from complying with our reporting obligations on a timely basis, which could result in the loss of investor confidence in the reliability of our consolidated financial statements, harm our business and negatively impact the trading price of our common shares.
As a foreign private issuer, we are not subject to certain United States securities law disclosure requirements that apply to a domestic United States issuer, which may limit the information which would be publicly available to our shareholders.
As a foreign private issuer, we are not required to comply with all the periodic disclosure requirements of the Exchange Act, and therefore, there may be less publicly available information about us than if we were a United States domestic issuer. For example, we are not subject to the proxy rules in the United States and disclosure with respect to our annual meetings will be governed by Canadian requirements.
Our charter documents and certain Canadian legislation could delay or deter a change of control, limit attempts by our shareholders to replace or remove our current management and limit the market price of our common shares.
Our authorized preferred shares are available for issuance from time to time at the discretion of our board of directors, without shareholder approval. Our articles grant our board of directors the authority to determine the special rights and restrictions granted to or imposed on any unissued series of preferred shares, and those rights may be superior to those of our common shares. Further, the Investment Canada Act subjects any acquisition of control of a company by a non-Canadian to government review if the value of the assets as calculated pursuant to the legislation exceeds a threshold amount or in other circumstances determined at the discretion of the Canadian government. A reviewable acquisition may not proceed unless the relevant minister is satisfied that the investment is likely to be of net benefit to Canada and the Canadian government is satisfied that no other important concerns arise from the acquisition of control. Any of the foregoing could prevent or delay a change of control and may deprive or limit strategic opportunities to our shareholders to sell their shares.
DISCLOSURE CONTROLS AND INTERNAL CONTROLS OVER FINANCIAL REPORTING
We have implemented a system of internal controls that we believe adequately protects our assets and is appropriate for the nature of our business and the size of our operations. Our internal control system was designed to provide reasonable assurance that all transactions are accurately recorded, that transactions are recorded as necessary to permit preparation of financial statements in accordance with IFRS, and that our assets are safeguarded.
These internal controls include disclosure controls and procedures designed to ensure that information required to be disclosed by us is accumulated and communicated as appropriate to allow timely decisions regarding required disclosure.
Internal control over financial reporting means a process designed by or under the supervision of the Chief Executive Officer and the Chief Financial Officer to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with IFRS as issued by the IASB. The internal controls are not expected to prevent and detect all misstatements due to error or fraud.
There were no changes in our internal control over financial reporting that occurred during the three months ended December 31, 2015 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting. As at December 31, 2015, we have assessed the effectiveness of our internal control over financial reporting and disclosure controls and procedures using the Committee of Sponsoring Organizations of the Treadway Commission’s 2013 framework. Based on their evaluation, the Chief Executive Officer and the Chief Financial Officer have concluded that these controls and procedures are effective.
- 39 -
| TRILLIUM THERAPEUTICS INC. |
| |
| Management’s Discussion and Analysis |
ADDITIONAL INFORMATION
Additional information for Trillium can be found on SEDAR at www.sedar.com, on EDGAR at www.sec.gov/edgar.shtml.
- 40 -