As filed with the Securities and Exchange Commission on September 1, 2021.
Registration No. 333-
Delaware | | | 3826 | | | 27-1562193 |
(State or other jurisdiction of incorporation or organization) | | | (Primary Standard Industrial Classification Code Number) | | | (I.R.S. Employer Identification Number) |
E. Thom Rumberger Jr. Glenn R. Pollner Molly Fox Craig Hilts Wilmer Cutler Pickering Hale and Dorr LLP 2600 El Camino Real, Suite 400 Palo Alto, CA 94306 (650) 858 6000 | | | Erica Palsis General Counsel Cue Health Inc. 4980 Carroll Canyon Rd. Suite 100 San Diego, CA 92121 (858) 412-8151 | | | Charles S. Kim Jonie Kondracki Kristin VanderPas Denny Won Cooley LLP 4401 Eastgate Mall San Diego, CA 92121 (858) 550-6000 |
Large accelerated filer | | | ☐ | | | | | Accelerated filer | | | ☐ | |
Non-accelerated filer | | | ☒ | | | | | Smaller reporting company | | | ☐ | |
| | | | | | | Emerging growth company | | | ☒ |
Title of Each Class of Securities to Be Registered | | | Proposed Maximum Aggregate Offering Price(1) | | | Amount of Registration Fee(2) |
Common stock, par value $0.00001 per share | | | $100,000,000 | | | $10,910 |
| (1) | Estimated solely for the purpose of calculating the registration fee pursuant to Rule 457(o) under the Securities Act of 1933, as amended. Includes the aggregate offering price of additional shares that the underwriters have the option to purchase, if any. |
| (2) | Calculated pursuant to Rule 457(o) under the Securities Act of 1933, as amended, based on an estimate of the proposed maximum aggregate offering price. |
| | | Per Share | | | Total | |
Initial public offering price | | | $ | | | $ |
Underwriting discounts and commissions(1) | | | $ | | | $ |
Proceeds, before expenses, to us | | | $ | | | $ |
| (1) | See the section titled “Underwriting” for additional disclosure regarding the estimated underwriting discounts and commissions and estimated offering expenses. |
Goldman Sachs & Co. LLC | | | Morgan Stanley | | | Cowen |
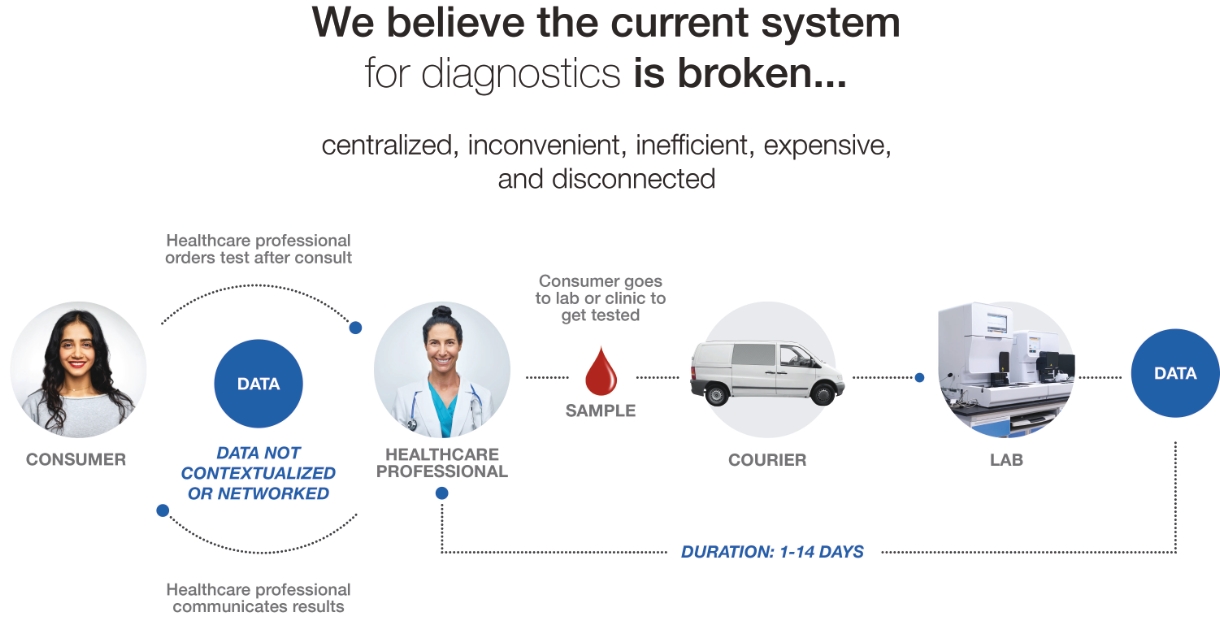
| • | Centralized Care Limits Access: We believe healthcare that is delivered through centralized, physical locations limits access due to the inconvenience and time-consuming nature of visiting hospitals, doctors’ offices, and urgent care clinics. |
| • | The Centralized Diagnostic Testing Framework is Challenged: In the United States today, there are hundreds of thousands of diagnostic access points to serve hundreds of millions of people. The lack of real-time, convenient, and readily accessible diagnostic solutions is a direct result of the legacy central lab testing model. |
| • | Legacy Infrastructure Is Not Built for Virtual Care: The current centralized diagnostic and care infrastructure is even less well suited for the growing virtual care delivery model. For care to truly be virtual, we believe patients need the ability to obtain a diagnostic result from anywhere and at any time, rather than from a central laboratory with high latency. |
| • | Lack of Capabilities to Identify Health Threats: We believe the disconnected and high-latency diagnostic system is not able to deliver the information that public health agencies and other healthcare providers need to identify, mitigate and monitor outbreaks of highly contagious diseases, such as COVID-19 or influenza. |
| • | control over how they manage their acute and chronic conditions as well as their overall health; |
| • | access to actionable clinical insights; |
| • | affordable and transparent pricing; and |
| • | customer-centric user experiences that connect the entire care journey. |
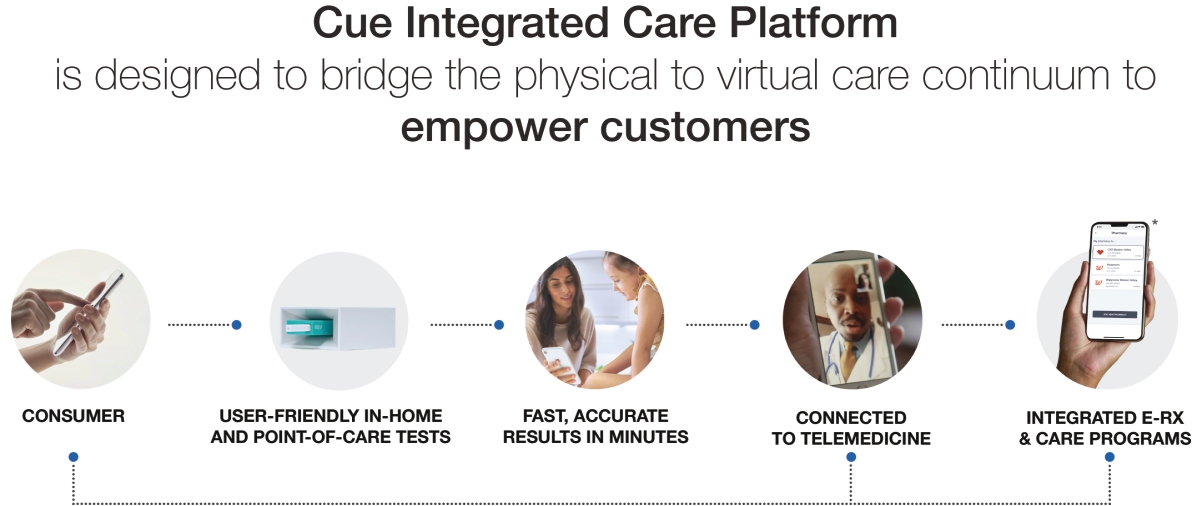
| * | Depicts future product developments. |
| • | Healthcare is Shifting to Consumer-Focused Care and Delivery: Across multiple industries, new disruptors have used technology to transform the consumer experience. We believe a paradigm shift is occurring in healthcare as consumers are both increasingly informed and focused on the user experience. We believe this shift will become one of the most important factors that shapes the next decade of healthcare. |
| • | Diagnostics is at the Center of Healthcare 2.0: We believe diagnostic data is the key to unlocking the full potential of personalized and virtually delivered care. Without an at-home testing solution, we believe telehealth solutions will still be burdened by long turnaround times, and require individuals to visit, or mail samples to, centralized testing laboratories. |
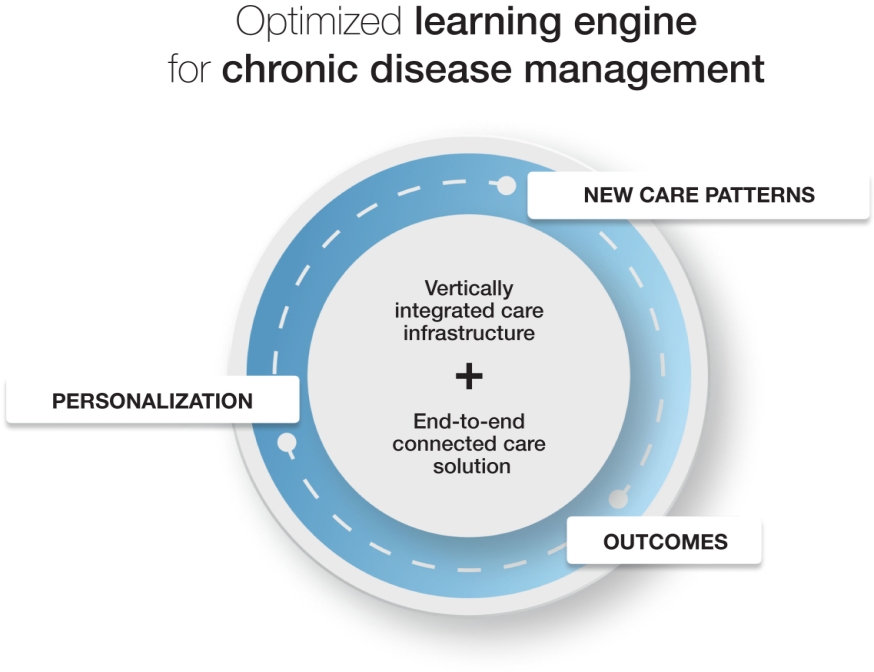
| • | We are well positioned to be at the Center of Healthcare 2.0: We believe we have the potential to become the new standard of care in diagnostics, with the ability to bridge the physical to virtual care continuum and benefit everyone by keeping people healthy and productive. Just as monitoring combined with data-driven insights helps people with chronic conditions live healthier lives, we believe our platform will transform the way people manage their health through real-time, actionable and connected health data. |

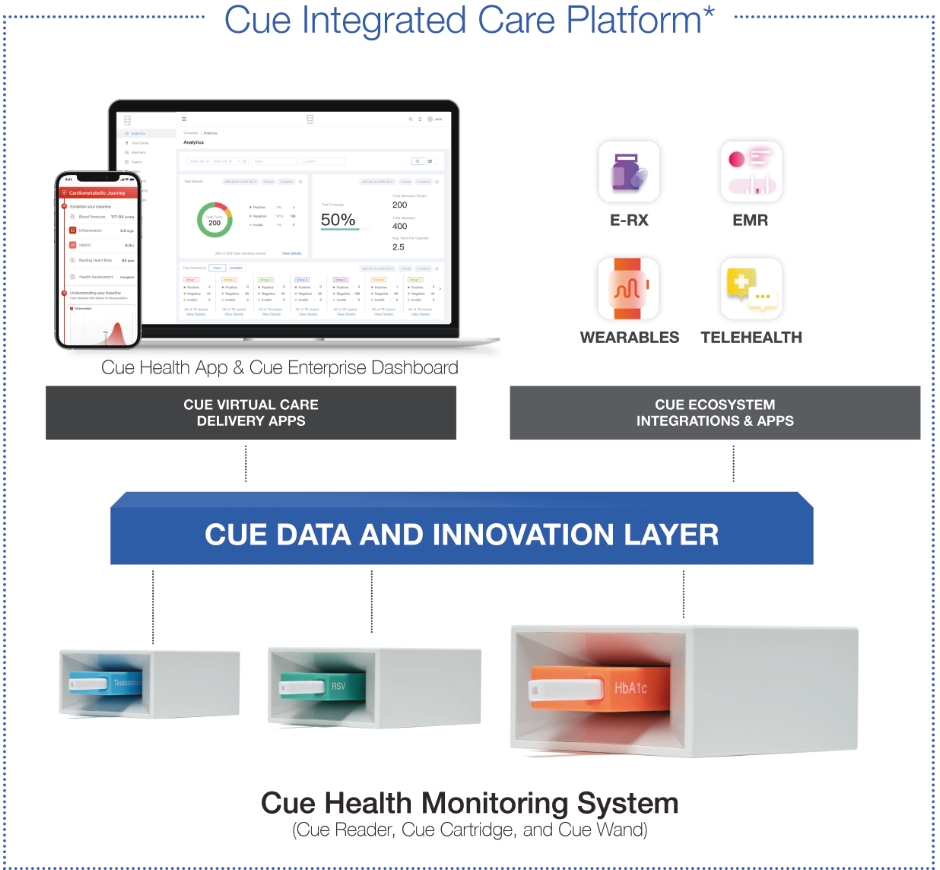
| * | Depicts future product developments. |
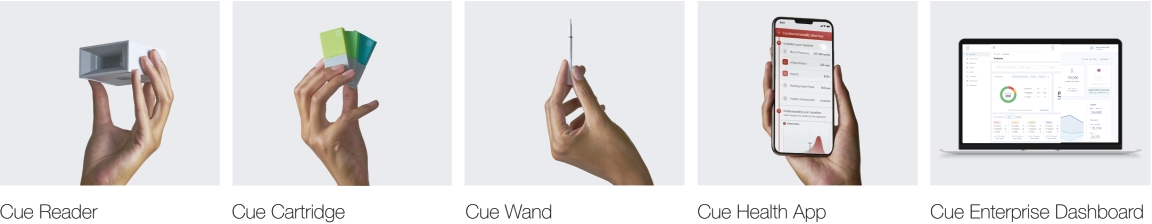
| • | Cue Reader: The Cue Reader is an elegantly designed, automated analyzer of test results and is used with Cue Test Kits and the Cue Health App. The Cue Reader runs the Cue Cartridge and communicates the result of the test digitally via Bluetooth to the Cue Health App. |
| • | Cue Test Kit: Each Cue Test Kit is comprised of a Cue Cartridge and a Cue Wand. |
| ○ | Cue Cartridge: Our sample-specific, single-use cartridges are designed to handle different chemistries, which allows us to create a broad menu of tests. Cue Cartridges are designed to be seamlessly inserted into the Cue Reader. |
| ○ | Cue Wand: Cue Wands are single-use and sterile sample collection devices that are designed to be universally compatible with the Cue Cartridges. The Cue Wand is designed to permit collection of multiple sample types, including saliva, blood, urine and swabs, with only minor modifications. |
| • | Cue Health App: Our mobile app creates a secure interface between the user and their health data. For consumers, it allows a single point of entry for their health data; for healthcare professionals, it is designed to provide a unified platform for managing patient histories and, in the future, is expected to allow for |
| • | Cue Enterprise Dashboard: Our dashboard is designed to allow enterprises, payors, healthcare providers and public health entities to manage population health at the organizational level and has the potential to track the efficacy of various population health programs. Accessible online, the Cue Enterprise Dashboard has the potential to help organizations manage a patient’s journey from onboarding to scheduling, care management and inventory management. The Cue Enterprise Dashboard was built with a focus on user experience, simplifying the sharing of communications, such as results, records, and histories with patients and across providers and streamlining reporting requirements. Powered by our analytics engine and role-based access capabilities, it is designed to provide chief medical officers, environmental health and safety officials, and benefits managers with insight into their organization’s population health, helping to facilitate efficient decision making. As of August 31, 2021, we had 60 active public sector, enterprise and provider accounts on the Cue Enterprise Dashboard. An account on the Cue Enterprise Dashboard is considered to be active if the customer has signed into their account and utilized the programs within the last six months. A customer may have more than one active account on the Cue Enterprise Dashboard. |
| * | Depicts future product developments. |
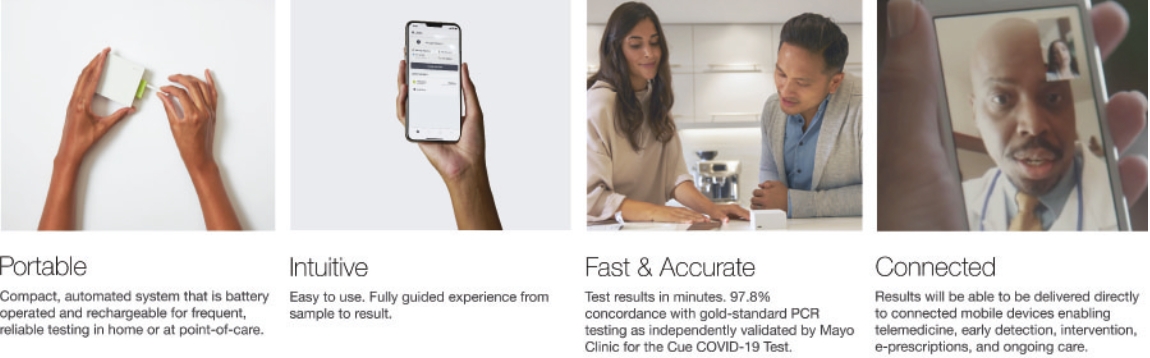
| • | Consumer-centric. The Cue Integrated Care Platform is intended to revolutionize the way individuals and healthcare providers access diagnostic testing at home, at work, or at the point-of-care. Our Cue Integrated Care Platform is designed to deliver a superior user experience in any setting, one that is fully-guided, fast, accurate, and easy to use and that puts the consumer in control of their health data. |
| • | Lab-quality diagnostics anywhere in minutes. Combining the sophistication and accuracy of complex molecular testing systems with the simplicity, convenience and speed of a consumer electronic device, our Cue Health Monitoring System has been developed to deliver highly specific and sensitive results within minutes. |
| • | Extensible platform approach. We designed our technology, platform and infrastructure to be versatile in accommodating a wide range of tests by addressing both main analytical modalities used in diagnostic testing, immunoassays and NAAT. We believe our flexible platform will permit our planned future menu of tests to cover a large portion of diagnostic solutions typically offered by a traditional lab. |
| • | Vertically-integrated, automated and scalable production infrastructure. Our proprietary technology was designed to allow us to optimize our system across the full product life cycle from design to manufacturing. Our integrated cartridge manufacturing and bio-production, including enzymes and chemistry, ensure the quality of our finished product. |
| • | Scaled and growing installed base. We have shipped over 115,000 Cue Readers across the United States as of August 31, 2021, including Cue Readers placed through our agreement with the U.S. DoD and through our other customer agreements, resulting in a broad and active installed base, diversified across industries, locations and end-markets such as schools, essential businesses, nursing homes, hospitals, physicians' offices, dental clinics, sports and other live event venues, and other settings around the country. |
| • | Expand our menu of tests and continue to innovate and enhance our platform. |
| • | Drive ecosystem adoption. |
| • | Continue to expand our installed base and distribution network to enable pull-through of our future extended care offerings. |
| • | Increase adoption through value-based selling and payor reimbursement. |
| • | Continue to build the Cue brand. |
| • | Scale manufacturing capabilities to capitalize on demand. |
| • | Expand our global footprint. |
| • | Public Sector Sales: Our public sector sales team identifies new opportunities within federal, state and local government agencies. While we expect that revenue from other categories of customers will become a larger component of revenue over time, our public sector sales strategy continues to look to identify opportunities with new and existing federal, state and local government agency customers. |
| • | Enterprise Sales: Our enterprise sales team identifies major self-insured enterprises such as Fortune 500 companies with large covered employee populations as well as small-to-medium sized businesses with healthcare plan partners and employee benefits offerings. We believe that enterprise customers will want to utilize our integrated care solutions for their employees and their families, both on-premise and at-home. |
| • | Healthcare Provider Sales: Our healthcare provider sales strategy targets major healthcare systems and healthcare professionals such as hospital systems, private clinics and concierge health systems, and physicians’ offices. Relationships with these customers, such as our current relationship with the Mayo Clinic, help validate our platform, and we believe will help accelerate marketplace adoption of our products. |
| • | Direct-to-Consumer Sales: Our direct-to-consumer sales team identifies opportunities through online and offline retail channels such as e-commerce and in-store sales. |
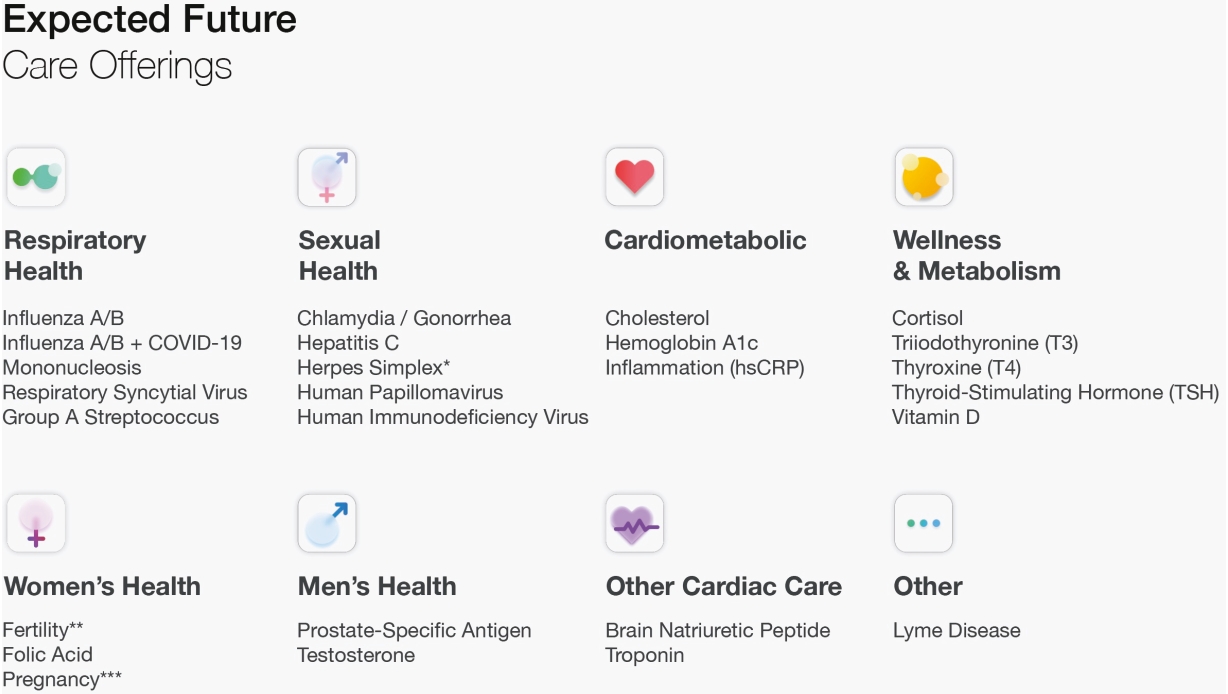
| * | HSV-1 & HSV-2 |
| ** | Luteinizing Hormone (LH) |
| *** | Human chorionic gonadotropin (hCG) |
| • | our technical development capabilities that have led to an authorized COVID-19 test and multiple tests in late-stage technical development; |
| • | our understanding of the regulatory pathways, including FDA authorization or clearance, for the various diagnostic tests; and |
| • | our test-agnostic production capacity that we believe will provide us the flexibility to meet our customers’ needs. |
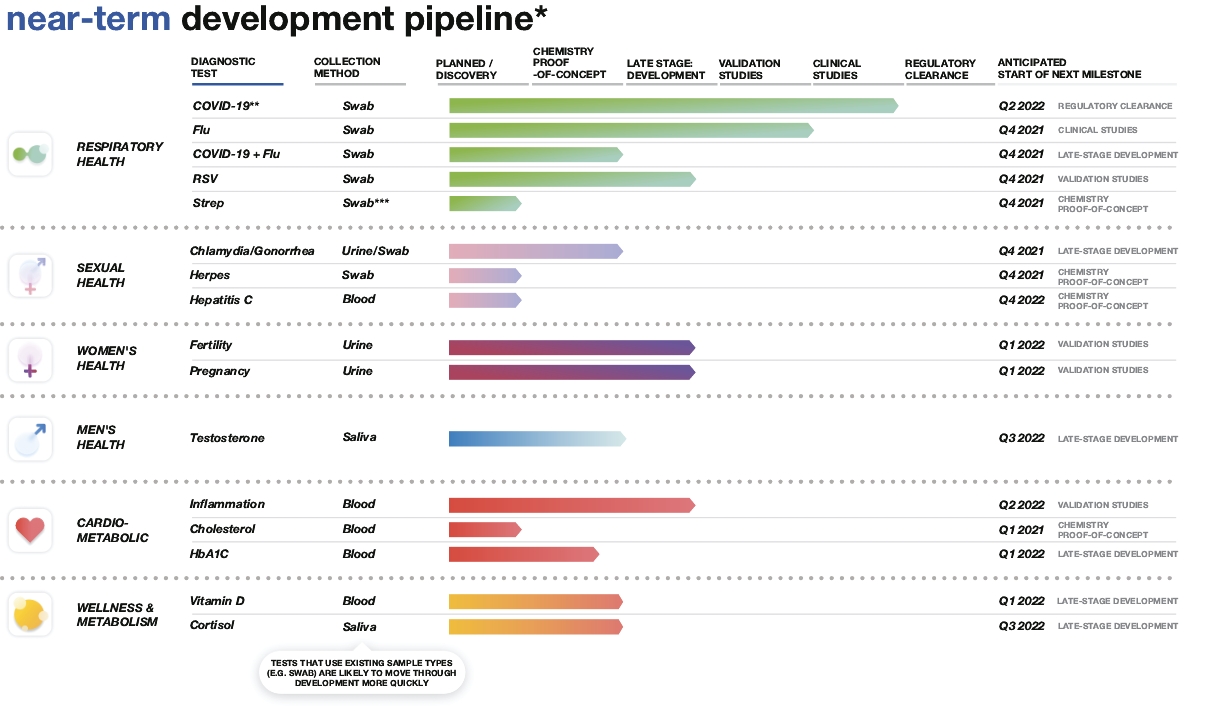
| * | This graphic does not reflect our full development pipeline but rather those of our tests that are furthest along in development. This graphic reflects progress towards 510(k) clearance. |
| ** | Our COVID-19 test has been authorized by the FDA under two EUAs. This graphic reflects progress towards 510(k) clearance. Our COVID-19 test has also received regulatory approval from the CDSCO for professional point-of-care use in India, the CE mark in the European Union and Interim Order authorization from Health Canada. |
| *** | Throat swab sample may be required. |
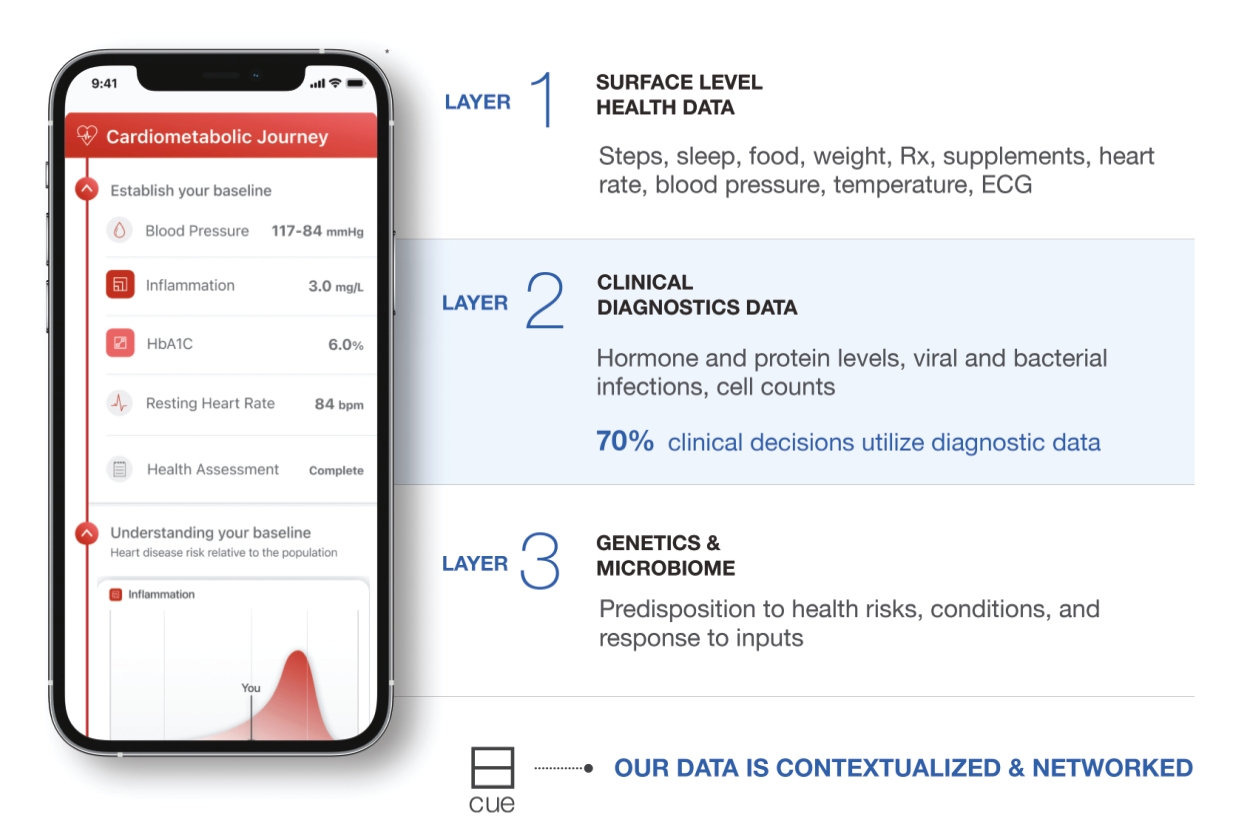
| * | Depicts future product developments. |
| • | We have a limited operating history, which may make it difficult to evaluate our current business and predict our prospects and likelihood of success. |
| • | We have incurred significant losses since our inception, and only recently started generating revenue from commercial sales. We may incur additional significant losses in the future, and we may never become profitable on a sustainable basis. |
| • | If the FDA or other regulatory bodies revoke or terminate our EUAs or other regulatory authorizations for our COVID-19 test, we will be required to stop commercialization of our Cue Readers and COVID-19 Test Kits unless we can obtain 510(k) or other clearance or approval for our COVID-19 test and its currently authorized uses. |
| • | Our near-term success is dependent on the continued commercialization of our COVID-19 test. If our COVID-19 test is unable to attain or maintain market acceptance or be successfully commercialized, our business could be materially adversely affected. |
| • | Our long-term success will depend on the success of our COVID-19 test and a number of other factors, including widespread market adoption of our Cue Health Monitoring System, Cue Virtual Care Delivery Apps and the overall Cue Integrated Care Platform and our ability to introduce new tests for use with our Cue Health Monitoring System. |
| • | Our revenue for at least the near term will almost exclusively depend on sales of our COVID-19 test until we can develop, obtain regulatory clearance or other appropriate authorization for, and commercialize additional tests. |
| • | We currently rely upon the U.S. DoD and a very small number of other customers for almost all of our current product revenue. As a result, unless and until we can further diversify our customer base and sources of revenue, the loss of any of these customers, or a decline in the amount of our COVID-19 tests purchased by or sold to these customers, could materially adversely affect our business, financial condition and results of operations. |
| • | We may encounter difficulties in managing our growth, which could adversely affect our operations. |
| • | The diagnostic testing market is extremely competitive and rapidly evolving, making it difficult to evaluate our business and future prospects. |
| • | If the Cue Health Monitoring System fails to achieve broad adoption by or support from the medical and professional community, key opinion leaders and other key participants in the healthcare system, our business and prospects may be materially adversely affected. |
| • | We have identified material weaknesses in our internal control over financial reporting and may identify material weaknesses in the future or otherwise fail to maintain an effective system of internal controls in the future, as a result of which, we may not be able to accurately report our financial condition or results of operations, which may adversely affect investor confidence in us and, as a result, the value of our common stock. |
| • | The COVID-19 pandemic could materially adversely affect our business, financial condition and results of operations. |
| • | We have limited experience manufacturing our products in commercial quantities; if we are unable to manufacture our products in the required quantities in a timely manner, our business could be materially adversely affected. |
| • | If we, our suppliers or our contract manufacturers experience significant disruptions to our or their manufacturing capabilities or ability to source needed supplies and materials, our business may be materially adversely affected. |
| • | Our patent or other intellectual property protection for the Cue Health Monitoring System, products and Cue Integrated Care Platform may not be sufficient to prevent competitors from developing and commercializing tests and platforms similar to or otherwise comparable to our Cue Test Kits, products and Cue Integrated Care Platform, which could materially adversely affect our business and prospects. |
| • | 9,994,197 shares of common stock issuable upon exercise of stock options outstanding as of June 30, 2021, with a weighted-average exercise price of $4.93 per share; |
| • | 1,049,043 shares of common stock subject to restricted stock units, or RSUs, outstanding as of June 30, 2021; |
| • | 75,744 shares of common stock issuable upon exercise of warrants outstanding as of June 30, 2021 to purchase shares of common stock, with an exercise price of $0.40 per share; |
| • | 79,882 shares of common stock issuable upon exercise of warrants outstanding as of June 30, 2021 to purchase redeemable convertible preferred stock that will automatically become warrants to purchase 79,882 shares of common stock immediately prior to the completion of this offering, with a weighted-average exercise price of $1.12 per share; |
| • | 1,138,635 shares of common stock reserved for future issuance under our 2014 Equity Incentive Plan, as of June 30, 2021, of which our board of directors expects to grant stock awards covering 128,000 shares of common stock to certain of our non-employee directors effective immediately prior to the effectiveness of the registration statement of which this prospectus forms a part; |
| • | additional shares of common stock that will become available for future issuance under our 2021 Stock Incentive Plan, which will become effective immediately prior to the effectiveness of the registration statement of which this prospectus forms a part, as well as any automatic increases in the number of shares of common stock reserved for future issuance under the 2021 Stock Incentive Plan, of which our board of directors expects to grant awards covering shares of common stock to certain of our employees, executive officers and non-employee directors effective prior to the commencement of trading of our common stock on the Nasdaq Stock Market; and |
| • | additional shares of common stock that will become available for future issuance under our 2021 Employee Stock Purchase Plan, which will become effective immediately prior to the effectiveness of the registration statement of which this prospectus forms a part, as well as any automatic increases in the number of shares of common stock reserved for future issuance under the 2021 Employee Stock Purchase Plan. |
| • | the automatic conversion of all outstanding shares of our redeemable convertible preferred stock outstanding as of June 30, 2021 into 83,526,065 shares of our common stock immediately prior to the completion of this offering; |
| • | the automatic conversion of our outstanding $235.5 million in aggregate principal amount of Convertible Notes into shares of common stock upon the closing of this offering, based on accrued interest through , 2021 and a 20% discount to the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus; |
| • | the conversion of outstanding warrants to purchase 79,882 shares of our redeemable convertible preferred stock into warrants to purchase 79,882 shares of common stock, which will occur automatically immediately prior to the completion of this offering; |
| • | no exercise of the outstanding stock options or settlement of outstanding RSUs described above; |
| • | no exercise of the outstanding warrants described above; |
| • | no exercise by the underwriters of their option to purchase additional shares of our common stock; and |
| • | the adoption, filing and effectiveness of our amended and restated certificate of incorporation and our amended and restated bylaws immediately prior to the completion of this offering. |
| | | Year Ended December 31, | | | Six Months Ended June 30, | |||||||
| | | 2019 | | | 2020 | | | 2020 | | | 2021 | |
(in thousands, except share and per share data) | | | | | | | (unaudited) | |||||
Revenue: | | | | | | | | | ||||
Product revenue | | | $— | | | $15,391 | | | $— | | | $201,922 |
Grant and other revenue | | | 6,626 | | | 7,562 | | | 4,960 | | | — |
Total revenue | | | 6,626 | | | 22,953 | | | 4,960 | | | 201,922 |
Operating costs and expenses: | | | | | | | | | ||||
Cost of product revenue(1)(2) | | | — | | | 14,951 | | | — | | | 85,177 |
Sales and marketing(1) | | | 88 | | | 714 | | | 45 | | | 1,959 |
Research and development(1) | | | 21,405 | | | 28,478 | | | 19,680 | | | 12,071 |
General and administrative(1) | | | 5,900 | | | 23,936 | | | 3,764 | | | 23,252 |
Total operating costs and expenses | | | 27,393 | | | 68,079 | | | 23,489 | | | 122,459 |
Income (loss) from operations | | | (20,767) | | | (45,126) | | | (18,529) | | | 79,463 |
Interest expense | | | (152) | | | (974) | | | (788) | | | (9,964) |
Change in fair value of redeemable convertible preferred stock warrants | | | 4 | | | (1,289) | | | (20) | | | (190) |
Change in fair value of convertible notes | | | — | | | — | | | — | | | (23,254) |
Other income (expense), net | | | 309 | | | 47 | | | 59 | | | 61 |
Net income (loss) before income taxes | | | (20,606) | | | (47,352) | | | (19,278) | | | 46,116 |
Income tax expense | | | — | | | — | | | — | | | (13,276) |
Net income (loss) | | | $(20,606) | | | $(47,352) | | | $(19,278) | | | $32,840 |
Basic net income (loss) per share attributable to common stockholders(3) | | | $(1.31) | | | $(2.90) | | | $(1.21) | | | $0.23 |
Weighted-average number of shares of common stock used in basic net income (loss) per share attributable to common stockholders(3) | | | 15,760,246 | | | 16,315,730 | | | 15,909,439 | | | 18,617,247 |
Diluted net income (loss) per share attributable to common stockholders(3) | | | $(1.31) | | | $(2.90) | | | $(1.21) | | | $0.22 |
| | | Year Ended December 31, | | | Six Months Ended June 30, | |||||||
| | | 2019 | | | 2020 | | | 2020 | | | 2021 | |
(in thousands, except share and per share data) | | | | | | | (unaudited) | |||||
Weighted-average number of shares of common stock used in diluted net income (loss) per share attributable to common stockholders(3) | | | 15,760,246 | | | 16,315,730 | | | 15,909,439 | | | 26,036,337 |
Pro forma basic net income (loss) per share attributable to common stockholders (unaudited) | | | | | | | | | ||||
Pro forma weighted-average number of shares of common stock used in basic net income (loss) per share attributable to common stockholders (unaudited) | | | | | | | | | ||||
Pro forma diluted net income (loss) per share attributable to common stockholders (unaudited) | | | | | | | | | ||||
Pro forma weighted-average number of shares of common stock used in diluted net income (loss) per share attributable to common stockholders (unaudited) | | | | | | | | | ||||
| (1) | Includes stock-based compensation expense as follows: |
| | | Year Ended December 31, | | | Six Months Ended June 30, | |||||||
| | | 2019 | | | 2020 | | | 2020 | | | 2021 | |
(in thousands) | | | | | | | (unaudited) | |||||
Cost of product revenue | | | $— | | | $— | | | $— | | | $343 |
Sales and marketing | | | — | | | 1 | | | — | | | 26 |
Research and development | | | 45 | | | 98 | | | 13 | | | 1,444 |
General and administrative | | | 291 | | | 3,064 | | | 84 | | | 3,778 |
Total stock-based compensation expense | | | $336 | | | $3,163 | | | $97 | | | $5,591 |
| (2) | Includes $2.1 million and $10.5 million of depreciation and amortization expense for the year ended December 31, 2020, and for the six months ended June 30, 2021, respectively. |
| (3) | See Note 14 to our audited financial statements and to our unaudited interim condensed financial statements, each included elsewhere in this prospectus, for details on the calculation of basic and diluted net income (loss) per share attributable to common stockholders, and the weighted-average number of shares used in the computation of the per share amounts. |
| | | As of June 30, 2021 | |||||||
| | | Actual | | | Pro Forma(1) | | | Pro Forma As Adjusted(2)(3) | |
(in thousands) | | | (unaudited) | ||||||
Balance Sheet Data: | | | | | | | |||
Cash and cash equivalents | | | $246,326 | | | $ | | | $ |
Working capital(4) | | | 233,965 | | | | | ||
Restricted cash, non-current | | | 6,000 | | | | | ||
Total assets | | | 631,312 | | | | | ||
Redeemable convertible preferred stock warrant liabilities | | | 1,521 | | | | | ||
Convertible notes | | | 258,734 | | | | | ||
Finance lease liabilities, net of current portion | | | 1,694 | | | | | ||
Total liabilities | | | 516,321 | | | | | ||
Redeemable convertible preferred stock | | | 176,323 | | | | | ||
Additional paid-in capital | | | 16,264 | | | | | ||
Accumulated deficit | | | (77,596) | | | | | ||
Total stockholders’ (deficit) equity | | | (61,332) | | | | | ||
| (1) | The pro forma balance sheet data gives effect to (i) the filing and effectiveness of our amended and restated certificate of incorporation, which will be in effect immediately prior to the completion of this offering, (ii) the automatic conversion of all of our outstanding $235.5 million aggregate principal amount convertible promissory notes, or Convertible Notes, into shares of common stock upon the completion of this offering, based on interest accrued through , 2021 and a 20% discount to the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, (iii) the automatic conversion of all outstanding shares of our redeemable convertible preferred stock into an aggregate of 83,526,065 shares of our common stock immediately prior to the completion of this offering, and (iv) the automatic conversion of all of our outstanding warrants to purchase redeemable convertible preferred stock into warrants to purchase common stock, and the related reclassification of our redeemable convertible preferred stock warrant liabilities to additional paid-in capital immediately prior to the completion of this offering. |
| (2) | The pro forma as adjusted balance sheet data reflect: (i) the pro forma adjustments set forth above, and (ii) the issuance and sale of shares of our common stock in this offering at an assumed initial public offering price of $ per share, which is the midpoint of the estimated price range set forth on the cover page of this prospectus, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. The pro forma as adjusted information discussed above is illustrative only and will change based on the actual initial public offering price and other terms of this offering determined at pricing. |
| (3) | Each $1.00 increase (decrease) in the assumed initial public offering price of $ per share, which is the midpoint of the estimated price range set forth on the cover page of this prospectus, would increase (decrease) the pro forma as adjusted amount of each of cash and cash equivalents, working capital, total assets and total stockholders’ (deficit) equity by $ million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. An increase (decrease) of 1.0 million shares in the number of shares offered by us, as set forth on the cover page of this prospectus, would increase (decrease) the pro forma as adjusted amount of each of cash and cash equivalents, working capital, total assets and total stockholders’ (deficit) equity by $ million, assuming no change in the assumed initial public offering price per share and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
| (4) | We define working capital as current assets less current liabilities, including current finance lease liabilities of $1.3 million. See our financial statements and the related notes included elsewhere in this prospectus for further details regarding our current assets and current liabilities. |
| • | our ability to continue to scale up our manufacturing and commercial capabilities so we can timely manufacture our Cue Readers, Cue Cartridges and Cue Wands in sufficient capacity to meet customer requirements and market demand; |
| • | acceptance by key opinion leaders, healthcare systems and providers, governments and regulatory authorities, enterprise and health plan customers, consumers and others of the convenience, accuracy and other benefits offered by our COVID-19 test and our Cue Integrated Care Platform; |
| • | the ability of our COVID-19 test to accurately detect different strains of SARS-CoV-2, the virus that causes COVID-19, created by genetic mutation or otherwise, such as the five SARS-CoV-2 variants of concern known as the Alpha, Beta, Gamma and Delta variants or other new variants that have emerged or may emerge; |
| • | the ability of consumers and other customers to pay for or otherwise obtain payment coverage or reimbursement from third-party payors for our Cue Readers and/or our COVID-19 Test Kits; |
| • | the length of the COVID-19 pandemic and the extent to which widespread vaccinations in the U.S. reduces demand for our COVID-19 test; |
| • | our ability to maintain our EUAs received from the FDA or otherwise obtain requisite future regulatory approval, as well as our ability to obtain and maintain regulatory authorizations, clearances and approvals in other jurisdictions; and |
| • | our ability to comply with all regulatory requirements applicable to our COVID-19 test, including applicable FDA marketing, manufacturing and post-market surveillance requirements and other requirements of our EUAs. |
| • | the success of our COVID-19 test; |
| • | the successful completion of validation and clinical studies for our anticipated future tests; |
| • | the timely receipt of marketing authorizations, clearances and approvals from the FDA and other similar regulatory authorities for our anticipated future tests and, if required, additional marketing authorizations, clearances and approvals for our COVID-19 test; |
| • | perceptions by the public and members of the medical community, including healthcare stakeholders, as to the convenience, accuracy and the sufficiency of clinical evidence supporting the performance of the Cue Integrated Care Platform; |
| • | demand from the public and members of the medical community for the Cue Health Monitoring System and adoption of our anticipated menu of tests; |
| • | the availability, perceived advantages, relative cost, relative convenience and relative accuracy of the Cue Health Monitoring System compared to products produced by our competitors; |
| • | positive or negative media coverage of the Cue Health Monitoring System or competing products, as to its convenience, accuracy and the sufficiency of clinical evidence supporting its performance; |
| • | the effectiveness of our marketing and sales efforts; |
| • | unanticipated delays in manufacturing our COVID-19 Test Kits; |
| • | our ability to raise additional capital on acceptable terms, or at all, if needed to support the continued growth of our business and the development and commercialization of additional tests; |
| • | unanticipated delays in manufacturing, developing or launching additional tests for our Cue Health Monitoring System; |
| • | our ability to comply with all regulatory requirements applicable to our Cue Health Monitoring Systems and our current and anticipated future tests; |
| • | our ability to price our Test Kits, including our COVID-19 Test Kit, at an acceptable price; |
| • | our ability to obtain, maintain enforce, protect and defend our intellectual property rights; |
| • | our ability to produce a continued supply of Cue Readers and Cue Test Kits; |
| • | our ability to meet the demands and the requirements of our agreements with our largest customers, including the U.S. DoD; |
| • | limitation on use or warnings required by the FDA in our product labeling; and |
| • | availability of, or changes in, coverage or reimbursement rates for any of our current or future tests from government or other enterprise or healthcare payors. |
| • | lack of experience with our company, Cue Integrated Care Platform and products, and concerns about the newness of our technology or that we are relatively new to the industry; |
| • | perceived health, safety or quality risks associated with the use of a new platform and the process of an individual conducting a diagnostic test at home; |
| • | perception that diagnostic testing can only be administered by a healthcare provider; |
| • | traditional or existing relationships between and among healthcare stakeholders that administer, process and sell diagnostic testing; |
| • | concerns about the privacy and security of patient information and data that is available on and that can be shared with or through our Cue Integrated Care Platform; |
| • | competition and negative selling efforts from competitors, including competing tests and platforms and other providers of healthcare technology platforms and services; and |
| • | perception regarding the complexity of using the Cue Health Monitoring System or Cue Virtual Care Delivery Apps. |
| • | our ability to demonstrate the accuracy, ease of use, and affordability of Test Kits using the Cue Health Monitoring System; |
| • | our ability to demonstrate the comparability of test results using the Cue Health Monitoring System to other testing methodologies, including those utilized by centralized labs, such as polymerase chain reaction, or PCR, tests, reverse transcription PCR, or RT-PCR, tests, and loop-mediated isothermal amplification, or LAMP; |
| • | any lack or perceived lack of sufficient clinical evidence supporting the accuracy and performance of our tests; |
| • | a willingness of constituents in the healthcare system to adopt the Cue Integrated Care Platform and our current and future tests over other diagnostic products and tests; |
| • | overcoming any biases these constituencies may have toward the Cue Integrated Care Platform and our current and future tests relative to other diagnostic products and tests; |
| • | the cost and reimbursement from third-party payors or other payment coverage for Cue Readers and Cue Test Kits in relation to other diagnostic products and tests; |
| • | satisfaction with the accuracy and ease of use of the Cue Health Monitoring System and overall customer experience; |
| • | changes in pricing and promotional efforts by competitors; |
| • | demand for point-of-care and over-the-counter diagnostic testing; |
| • | the effectiveness of our sales, marketing and distribution efforts; and |
| • | adverse publicity about the Cue Health Monitoring System, including any current or future developed test kits, competitive products, or the industry as a whole, or favorable publicity about competitive products. |
| • | the level of demand for any of our authorized or approved tests, which may vary significantly; |
| • | authorization, approval and commercialization activities relating to our Cue Test Kits, which may change from time to time; |
| • | the timing and cost of, and level of investment in, research, development, manufacturing, regulatory and commercialization activities related to our tests, which may change from time to time; |
| • | the size, seasonality and customer mix of the COVID-19 diagnostic testing market; |
| • | the effect of the COVID-19 pandemic and the end of the COVID-19 pandemic on our business; |
| • | the effect of current and new therapeutic treatments for COVID-19 and vaccines; |
| • | sales and marketing efforts and expenses; |
| • | the rate at which we grow our sales force and the speed at which newly hired salespeople become effective; |
| • | changes in the productivity of our sales force; |
| • | positive or negative coverage in the media of, or clinical publications about, the Cue Health Monitoring System or any of our current or future tests or competitive products; |
| • | the cost of manufacturing any of the components of the Cue Health Monitoring System; |
| • | the introduction of new tests or enhancements or technologies by us or others in the diagnostic testing industry; |
| • | pricing pressures; |
| • | coverage and reimbursement policies with respect to our tests and products that compete with our tests; |
| • | expenditures that we may incur to acquire, develop or commercialize tests for additional indications, if any; |
| • | the degree of competition in our industry and any change in the competitive landscape of our industry; |
| • | changes in governmental regulations or in the status of our regulatory approvals or applications; |
| • | future accounting pronouncements or changes in our accounting policies; and |
| • | general market conditions and other factors, including factors unrelated to our operating performance or the operating performance of our competitors. |
| • | costs of litigation; |
| • | distraction of management’s attention from our primary business; |
| • | the inability to continue commercializing the Cue Health Monitoring System or other new products; |
| • | decreased demand for our Cue Readers or Cue Test Kits; |
| • | damage to our business reputation; |
| • | product recalls or withdrawals from the market; |
| • | withdrawal of clinical trial participants; |
| • | substantial monetary awards to patients or other claimants; |
| • | loss of sales; or |
| • | termination of existing agreements by our partners and potential partners failing to partner with us. |
| • | failure by us or our distributors to obtain regulatory clearance, authorization or approval for the use of our products in various countries and other jurisdictions; |
| • | multiple, conflicting and changing laws and regulations such as privacy security and data use regulations, tax laws, export and import restrictions, economic sanctions and embargoes, employment laws, anti-corruption laws, regulatory requirements, reimbursement or payor regimes and other governmental approvals, permits and licenses; |
| • | additional potentially relevant third-party patent rights; |
| • | pricing pressures and differing reimbursement regimes; |
| • | complexities and difficulties in obtaining intellectual property protection and maintaining, defending and enforcing our intellectual property; |
| • | difficulties in staffing and managing foreign operations; |
| • | employment risks related to hiring employees outside the United States; |
| • | logistics and regulations associated with shipping samples, including infrastructure conditions and transportation delays; |
| • | limits in our ability to penetrate international markets; |
| • | financial risks, such as longer payment cycles, difficulty collecting accounts receivable, the impact of local and regional financial crises on demand and payment for our products and exposure to foreign currency exchange rate fluctuations; |
| • | regulatory authorities revoking or terminating our authorizations and approvals in Canada, the European Union and India, or other jurisdictions; |
| • | natural disasters, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; |
| • | regulatory and compliance risks related to adherence with foreign privacy and data security laws, including the General Data Protection Regulation 2016/679 and other similar bodies of law; |
| • | regulatory and compliance risks that relate to maintaining accurate information and control over sales and distributors’ activities that may fall within the purview of the U.S. Foreign Corrupt Practices Act, or FCPA, its books and records provisions, or its anti-bribery provisions, or laws similar to the FCPA in other jurisdictions in which we may now or in the future operate, such as the United Kingdom’s Bribery Act of 2010, or U.K. Bribery Act; and |
| • | onerous anti-bribery requirements of several member states in the EU, the United Kingdom, and other countries that are constantly changing and require disclosure of information to which U.S. legal privilege may not extend. |
| • | increase our sales and marketing efforts to facilitate market adoption of our products and address competitive developments; |
| • | fund development and marketing efforts of any future products; |
| • | further expand our operations outside the United States; |
| • | acquire, license or invest in technologies, including information technologies; |
| • | satisfy any outstanding or future debt obligations; |
| • | acquire or invest in complementary businesses or assets; and |
| • | finance capital expenditures and general and administrative expenses. |
| • | our ability to successfully commercialize the Cue Health Monitoring System, including our COVID-19 test; |
| • | the costs of the sales and marketing activities associated with commercializing the Cue Health Monitoring System, including our COVID-19 test; |
| • | the length of the COVID-19 pandemic; |
| • | our ability to secure and maintain domestic and international regulatory authorization, clearance or approval for our products; |
| • | our rate of progress in, and cost of the sales and marketing activities associated with, establishing adoption of our products; |
| • | our rate of progress in, and cost of research and development activities associated with, products in research and early development; |
| • | our ability to control our manufacturing and operating costs; |
| • | our ability to satisfy any outstanding or future debt obligations; |
| • | the effect of competing technological and market developments; |
| • | litigation expenses we incur to defend against claims that we infringe the intellectual property of others or judgments we must pay to satisfy such claims; |
| • | the potential cost of and delays in research and development as a result of any regulatory oversight applicable to our products; and |
| • | the costs of responding to the other risks and uncertainties described in this prospectus. |
| • | production issues that may arise out of the rapid expansion of our manufacturing capacity, including the opening of two new manufacturing facilities within the last 12 months; |
| • | a setback in our anticipated timeline for finalizing the construction of our new production pods, which would result in manufacturing delays; |
| • | key components of our products are provided by a single supplier or limited number of suppliers, and we do not maintain large inventory levels of these components such that, if we experience a shortage or quality issues in any of these components, we would need to identify and qualify new supply sources, which could increase our expenses and result in manufacturing delays; |
| • | a delay in completing assembly of new controlled environment rooms at our manufacturing facility; |
| • | state and federal regulations, including the FDA’s Quality System Regulations, or QSR, for the manufacture of our products, noncompliance with which could cause an interruption in our manufacturing; and |
| • | attraction and retention of qualified employees for our operations in order to significantly increase our manufacturing output. |
| • | reliance on the third party for regulatory compliance and quality assurance; |
| • | the possible breach of the manufacturing agreement by the third party; |
| • | the possible delay or stoppage in production of certain components of the Cue Health Monitoring System that delays shipments of Cue Readers or Cue Test Kits to our customers; |
| • | the possible misappropriation of our proprietary information, including our trade secrets and know-how; and |
| • | the possible termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us. |
| • | others may be able to make systems or tests that are similar to the Cue Health Monitoring System or our current and any future tests or utilize similar technology but that are not covered by the claims of our patents or that incorporate certain technology in the Cue Health Monitoring System or our current and any future tests that is in the public domain; |
| • | we, or our current and future licensors or collaborators, might not have been the first to file patent applications covering certain of our or their inventions; |
| • | we, or our current and future licensors or collaborators, may fail to meet our obligations to the U.S. government regarding any future patents and patent applications funded by U.S. government grants, leading to the loss or unenforceability of patent rights; |
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
| • | it is possible that our current or future pending patent applications will not lead to issued patents; |
| • | it is possible that there are prior public disclosures that could invalidate our patents, or parts of our patents; |
| • | it is possible that there are unpublished applications or patent applications maintained in secrecy that may later issue with claims covering our current and any future test or technology similar to ours; |
| • | it is possible that our patents or patent applications omit people that should be listed as inventors or include people that should not be listed as inventors, which may cause these patents or patents issuing from these patent applications to be held invalid or unenforceable; |
| • | issued patents that we hold rights to may be held invalid or unenforceable, including as a result of legal challenges by our competitors or other third parties; |
| • | the claims of our patents or patent applications, if and when issued, may not cover our current and any future tests or technologies; |
| • | the laws of foreign countries may not protect our proprietary rights or the rights of future licensors or collaborators to the same extent as the laws of the United States; |
| • | the inventors of our patents or patent applications may become involved with competitors, develop test or processes that design around our patents, or become hostile to us or the patents or patent applications on which they are named as inventors; |
| • | our competitors or other third parties might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive platforms or tests for sale in our major commercial markets; |
| • | we have engaged in scientific collaborations in the past and will continue to do so in the future and our collaborators may develop adjacent or competing platforms or tests that are outside the scope of our patents; |
| • | we may not develop additional proprietary technologies that are patentable; |
| • | the patents of others may harm our business; or |
| • | we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third-party may subsequently file a patent covering such intellectual property. |
| • | we may not be able to demonstrate to the FDA’s satisfaction that our tests are safe and effective for their intended uses; |
| • | the data from our pre-clinical studies and clinical trials may be insufficient to support clearance or approval, where required; |
| • | the manufacturing process or facilities we use or contract to use may not meet applicable requirements; and |
| • | disruptions at the FDA caused by funding shortages or global health concerns, including the COVID-19 pandemic. |
| • | untitled letters, warning letters, injunctions, civil penalties and criminal fines; |
| • | customer notifications or repair, replacement, refunds, recall, detention or seizure of our tests; |
| • | operating restrictions or partial suspension or total shutdown of production; |
| • | refusing or delaying our requests for approval of a PMA or 510(k) clearance of new products, modified products or new indications of cleared products; |
| • | withdrawing PMA approvals or reclassifying devices that have 510(k) clearances; |
| • | refusal to grant export certificates for our tests; or |
| • | criminal prosecution. |
| • | civil penalties; |
| • | delays on or denials of pending requests for 510(k) clearance or PMA approval; |
| • | recalls or seizures; |
| • | withdrawals or suspensions of current PMA approvals or reclassification of 510(k) cleared devices, resulting in prohibitions on sales of our tests, if approved; |
| • | warning letters or untitled letters; |
| • | operating restrictions, including a partial or total shutdown of production on our tests for any indication; |
| • | refusal to issue export approvals or certifications; |
| • | obtaining injunctions preventing us from manufacturing or distributing our products; |
| • | commencing criminal prosecutions; and |
| • | total prohibitions on our sales. |
| • | the Federal Acquisition Regulation, or FAR, and agency-specific regulations supplemental to the FAR, which comprehensively regulate the procurement, formation, administration and performance of government contracts; |
| • | the business ethics and public integrity obligations, which govern conflicts of interest and the hiring of former government employees, restrict the granting of gratuities and funding of lobbying activities and incorporate other requirements such as the AKS, the Procurement Integrity Act, the FCA and the FCPA; and |
| • | laws, regulations and executive orders restricting the exportation of certain products and technical data. |
| • | the Anti-Kickback Statute, which prohibits, among other things, knowingly and willingly soliciting, offering, receiving or paying remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward either the referral of a person, or the purchase, order or recommendation of, items or services for which payment may be made, in whole or in part, under a federal healthcare program such as the Medicare and Medicaid programs. The term “remuneration” has been broadly interpreted to include anything of value, and the government can establish a violation of the Anti-Kickback Statute without proving that a person or entity had actual knowledge of the law or a specific intent to violate. In addition, the government may assert that a claim, including items or services resulting from a violation of the Anti-Kickback Statute, constitutes a false or fraudulent claim for purposes of the FCA. There are a number of statutory exceptions and regulatory safe harbors protecting certain business arrangements from prosecution under the Anti-Kickback Statute; however, those exceptions and safe harbors are drawn narrowly, and there may be limited or no exception or safe harbor for many common business activities. Certain common business activities including, certain reimbursement support programs, educational and research grants or charitable donations, and practices that involve remuneration to those who prescribe, purchase or recommend medical devices, including discounts, providing items or services for free or engaging such people as consultants, advisors or speakers, may be subject to scrutiny if they do not fit squarely within any available exception or safe harbor and would be subject to a facts and circumstances analysis to determine compliance with the Anti-Kickback Statute. Our business may not in all cases meet all of the criteria for statutory exception or regulatory safe harbor protection from anti-kickback liability; |
| • | The Eliminating Kickbacks in Recovery Act of 2018, or EKRA, which prohibits payments for referrals to recovery homes, clinical treatment facilities, and laboratories. EKRA’s reach extends beyond federal health care programs to include private insurance (i.e., it is an “all payor” statute); |
| • | the federal false claims and civil monetary penalties laws, including the Civil Monetary Penalties Law and the FCA, which prohibit, among other things, persons or entities from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds and knowingly making, using or causing to be made or used, a false record or statement to get a false claim paid or to avoid, decrease or conceal an obligation to pay money to the federal government. A claim including items or services resulting from a violation of the Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. Actions under the FCA may be brought by the government or as a qui tam action by a private person in the name of the government. These people, sometimes known as “relators” or, more commonly, as “whistleblowers,” may share in any monetary recovery. Many medical device manufacturers have been investigated and have reached substantial financial settlements with the federal government under the FCA for a variety of alleged improper activities, including causing false claims to be submitted as a result of the marketing of their products for unapproved and thus non-reimbursable uses and interactions with prescribers and other customers, including those that may have affected their billing or coding practices and submission of claims to the federal government. FCA liability is potentially significant in the healthcare industry because the statute provides for treble damages |
| • | HIPAA, which imposes criminal and civil liability for, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, or knowingly and willfully falsifying, concealing or covering up a material fact or making a materially false, fictitious or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal healthcare Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH Act, and their implementing regulations, also impose obligations, including mandatory contractual terms, on covered entities subject to the rule, such as health plans, healthcare clearinghouses and certain healthcare providers, as well as their business associates and their subcontractors that perform certain services for them or on their behalf involving the use or disclosure of individually identifiable health information with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| • | various state laws govern the privacy and security of personal information, including the CMIA, which provides for a private right of action for data breaches; |
| • | the federal Physician Payments Sunshine Act, implemented as Open Payments, requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program to report annually, with certain exceptions to CMS, information related to payments or other “transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and requires applicable manufacturers and group purchasing organizations to report annually to CMS ownership and investment interests held by physicians and their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report such information regarding payments and transfers of value provided during the previous year to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, anesthesiologist assistants and certified nurse-midwives; and |
| • | analogous state and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require medical device companies to comply with the industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state beneficiary inducement laws, which are state laws that require medical device manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
| • | actual or anticipated fluctuations in our financial condition or results of operations; |
| • | variance in our financial performance from expectations of securities analysts; |
| • | changes in the pricing of our products; |
| • | changes in our projected operating and financial results; |
| • | changes in laws or regulations applicable to our products; |
| • | changes in the number of enterprise customers we are able to partner with; |
| • | the level of market adoption of the Cue Health Monitoring System, including in the over-the-counter and at-home context; |
| • | announcements by us or our competitors of significant business developments, acquisitions, or new offerings; |
| • | changes in the structure of healthcare payment systems; |
| • | significant data breaches of our company, providers, vendors or pharmacies; |
| • | our involvement in litigation; |
| • | future sales of our common stock by us or our stockholders, as well as the anticipation of lock-up releases; |
| • | changes in senior management or key personnel; |
| • | negative publicity, such as whistleblower complaints or unsupported allegations made by short sellers, about us or our products; |
| • | the trading volume of our common stock; |
| • | changes in investor perceptions of us or our industry; |
| • | changes in the anticipated future size and growth rate of our market; |
| • | the effect of the COVID-19 pandemic and the end of the COVID-19 pandemic on our business; |
| • | general economic, political, regulatory, industry, and market conditions; and |
| • | natural disasters or major catastrophic events. |
| • | permit our board of directors to issue shares of preferred stock, with any rights, preferences and privileges as they may designate (including the right to approve an acquisition or other change in our control); |
| • | provide that the authorized number of directors may be changed only by resolution of the board of directors; |
| • | provide that the board of directors or any individual director may only be removed with cause and the affirmative vote of the holders of at least 66 2/3% of the voting power of all of our then outstanding common stock; |
| • | provide that all vacancies, including newly created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum; |
| • | divide our board of directors into three classes; |
| • | require that any action to be taken by our stockholders must be effected at a duly called annual or special meeting of stockholders and not be taken by written consent; |
| • | provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide notice in writing in a timely manner and also specify requirements as to the form and content of a stockholder’s notice; |
| • | do not provide for cumulative voting rights (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose); and |
| • | provide that special meetings of our stockholders may be called only by the chairman of the board, our Chief Executive Officer or by the board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors. |
| • | our expectations regarding our revenue, expenses and other operating results; |
| • | the extent and duration of the COVID-19 pandemic and the impact of the end of the COVID-19 pandemic on our business and our expectations regarding customer and user demand for our COVID-19 test; |
| • | our ability to increase demand for, and the rate of market adoption of, the Cue Health Monitoring System and our platform, tests and other products generally, including with consumers, healthcare professionals, enterprises, insurers and other payors and public health officials; |
| • | our ability to effectively scale our manufacturing capacity and other operations in a timely manner in order to meet contractual obligations, market demand and to be able to successfully operate our business; |
| • | our ability to meet our contractual obligations under our agreement with the U.S. Department of Defense or other customers; |
| • | our ability to successfully develop and commercialize additional tests and other products for use with our Cue Integrated Care Platform; |
| • | our expectations of the reliability, accuracy and performance of our products and services, as well as expectations of the benefits to patients, clinicians and providers of our products and services; |
| • | our ability to obtain and maintain regulatory authorizations, clearances or approvals for our tests, including our existing FDA EUAs for our COVID-19 test; |
| • | our ability to accurately forecast demand for the Cue Health Monitoring System, our tests and other products; |
| • | our ability to successfully build out our sales and marketing infrastructure, the costs and success of our marketing efforts, and our ability to promote our brand; |
| • | our ability to increase demand for our products and services, obtain favorable coverage and reimbursement determinations from third-party payors and expand geographically; |
| • | our intellectual property position and our expectations regarding our ability to obtain and maintain intellectual property protection; |
| • | the performance of our third-party suppliers and our ability to avoid any disruption in sources of supply; |
| • | our ability to effectively manage our growth, including our ability to retain and recruit personnel, and maintain our culture; |
| • | the impact of U.S. and international laws and regulations; |
| • | our competitive position and expectations regarding developments and projections relating to our competitors and any competing products and services; future investments in our business, our anticipated capital expenditures and our estimates regarding our capital requirements, future revenue, expenses, the ability to obtain reimbursement for our products and any needs for additional financing; |
| • | our expectations regarding technology trends and developments in the healthcare industry and our ability to address those trends and developments with our offerings; |
| • | our expectations concerning relationships with third parties, including healthcare professionals, enterprises, insurance companies and other payors, public health officials and other stakeholders in the healthcare system; |
| • | the degree to which we are able to help bring about a new healthcare paradigm, and be a significant participant in any such new paradigm; |
| • | our ability to grow our business internationally, in addition to within the United States; |
| • | our ability to implement, maintain and improve effective internal controls and remediate material weaknesses; |
| • | our expectations related to the use of proceeds from this offering and the sufficiency of such proceeds, together with our existing cash and cash equivalents, to fund our operations; and |
| • | our expectations regarding the time during which we will be an emerging growth company under the JOBS Act. |
| • | approximately $ million for the continued commercial scale up of our activities and build out our corporate infrastructure, other than the scale up of manufacturing facilities and capabilities, including the hiring and training of sales and marketing personnel and to fund marketing initiatives and for the hiring and training of other personnel; |
| • | approximately $ million for the continued scale up of our manufacturing facilities and capabilities; |
| • | approximately $ million for research and development to continue to develop each of our planned tests in our near-term development pipeline, which includes: |
| ○ | approximately $ million for further development and clinical studies for each of our five tests in late-stage technical development (flu, RSV, pregnancy, fertility, and inflammation); and |
| ○ | approximately $ million to further develop our software and other technical capabilities, such as the development of the Cue Data & Innovation Layer and the Cue Ecosystem Integrations and Apps; and |
| • | the remainder, if any, for working capital and other general corporate purposes. |
| • | on an actual basis; |
| • | on a pro forma basis to give effect to (i) the filing and effectiveness of our amended and restated certificate of incorporation, which will be in effect immediately prior to the completion of this offering, (ii) the automatic conversion of all of our outstanding $235.5 million aggregate principal amount Convertible Notes into shares of common stock upon the completion of this offering, based on interest accrued through , 2021 and a 20% discount to the assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, (iii) the automatic conversion of all outstanding shares of our redeemable convertible preferred stock into an aggregate of 83,526,065 shares of our common stock immediately prior to the completion of this offering, and (iv) the automatic conversion of all of our outstanding warrants to purchase redeemable convertible preferred stock into warrants to purchase common stock, and the related reclassification of our redeemable convertible preferred stock warrant liabilities to additional paid-in capital immediately prior to the completion of this offering; and |
| • | on a pro forma as adjusted basis to reflect: (i) the pro forma adjustments set forth above, and (ii) the issuance and sale of shares of our common stock in this offering at an assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
| | | As of June 30, 2021 | |||||||
| | | Actual | | | Pro Forma | | | Pro Forma As Adjusted(1) | |
| | | (unaudited) | |||||||
(in thousands, except share and per share data) | | | | | | | |||
Cash and cash equivalents | | | $246,326 | | | $ | | | $ |
Restricted cash, non-current | | | 6,000 | | | | | ||
Redeemable convertible preferred stock warrant liabilities | | | $1,521 | | | $ | | | $ |
Convertible notes | | | 258,734 | | | | | ||
Finance leases, including current portion | | | 3,043 | | | | | ||
Series A redeemable convertible preferred stock, $0.00001 par value per share; 8,721,437 shares authorized, 8,350,743 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | | | 7,519 | | | | | ||
Series B redeemable convertible preferred stock, $0.00001 par value per share; 46,213,620 shares authorized, 46,176,715 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | | | 66,186 | | | | | ||
Series C-1 redeemable convertible preferred stock, $0.00001 par value per share; 27,308,229 shares authorized, 27,308,227 shares issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | | | 96,436 | | | | | ||
Series C-2 redeemable convertible preferred stock, $0.00001 par value per share; 1,690,380 shares authorized, issued and outstanding, actual; no shares authorized, issued or outstanding, pro forma and pro forma as adjusted | | | 6,182 | | | | | ||
Stockholders’ equity (deficit): | | | | | | | |||
Common stock, $0.00001 par value: 129,030,355 shares authorized 29,128,604 shares issued and outstanding, actual; 500,000,000 shares authorized, shares issued and outstanding, pro forma; 500,000,000 shares authorized, shares issued and outstanding, pro forma as adjusted | | | — | | | | | ||
Preferred stock, $0.00001 par value: no shares authorized, issued or outstanding, actual; 50,000,000 shares authorized, shares issued and outstanding, pro forma; 50,000,000 shares authorized, shares issued and outstanding, pro forma as adjusted | | ||||||||
Additional paid-in capital | | | 16,264 | | | | | ||
Accumulated deficit | | | (77,596) | | | | | ||
Total stockholders’ (deficit) equity | | | (61,332) | | | | | ||
Total capitalization | | | $378,289 | | | $ | | | $ |
| (1) | Each $1.00 increase (decrease) in the assumed initial public offering price of $ per share, the midpoint of the estimated price range set forth on the cover page of this prospectus, would increase (decrease) the pro forma as adjusted amount of each of cash and cash equivalents, additional paid-in capital, total stockholders’ (deficit) equity and total capitalization by approximately $ million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. An increase (decrease) of 1.0 million shares in the number of shares offered by us, as set forth on the cover page of this prospectus, would increase (decrease) the pro forma as adjusted amount of each of cash and cash equivalents, additional paid-in capital, total stockholders’ (deficit) equity and total capitalization by $ million, assuming no change in the assumed initial public offering price per share and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
| • | 9,994,197 shares of common stock issuable upon exercise of stock options outstanding as of June 30, 2021, with a weighted-average exercise price of $4.93 per share; |
| • | 1,049,043 shares of common stock subject to restricted stock units, or RSUs, outstanding as of June 30, 2021; |
| • | 75,744 shares of common stock issuable upon exercise of warrants outstanding as of June 30, 2021 to purchase shares of common stock, with an exercise price of $0.40 per share; |
| • | 79,882 shares of common stock issuable upon exercise of warrants outstanding as of June 30, 2021 to purchase redeemable convertible preferred stock that will automatically become warrants to purchase 79,882 shares of common stock immediately prior to the completion of this offering, with a weighted-average exercise price of $1.12 per share; |
| • | 1,138,635 shares of common stock reserved for future issuance under our 2014 Equity Incentive Plan, as of June 30, 2021, of which our board of directors expects to grant stock awards covering 128,000 shares of common stock to certain of our non-employee directors effective immediately prior to the effectiveness of the registration statement of which this prospectus forms a part; |
| • | additional shares of common stock that will become available for future issuance under our 2021 Stock Incentive Plan, which will become effective immediately prior to the effectiveness of the registration statement of which this prospectus forms a part, as well as any automatic increases in the number of shares of common stock reserved for future issuance under the 2021 Stock Incentive Plan, of which our board of directors expects to grant awards covering shares of common stock to certain of our employees, executive officers and non-employee directors effective prior to the commencement of trading of our common stock on the Nasdaq Stock Market; and |
| • | additional shares of common stock that will become available for future issuance under our 2021 Employee Stock Purchase Plan, which will become effective immediately prior to the effectiveness of the registration statement of which this prospectus forms a part, as well as any automatic increases in the number of shares of common stock reserved for future issuance under the 2021 Employee Stock Purchase Plan. |
Illustrative Initial Public Offering Price Per Share | | | Number of Shares of Common Stock to be Issued upon Conversion of the Convertible Notes |
$ | | | |
$ | | | |
$ | | |
Assumed initial public offering price per share | | | | | $ | |
Historical net tangible book value (deficit) per share as of June 30, 2021 | | | $(2.33) | | | |
Increase in net tangible book value per share attributable to the pro forma adjustments described above | | | | | ||
Pro forma net tangible book value per share as of June 30, 2021, before giving effect to this offering | | | | | ||
Increase in pro forma net tangible book value per share attributable to new investors participating in this offering | | | | | ||
Pro forma as adjusted net tangible book value per share after this offering | | | | | ||
Dilution in pro forma as adjusted net tangible book value per share to new investors participating in this offering | | | | | $ |
| | | Shares Purchased | | | Total Consideration | | | Weighted- Average Price Per Share | |||||||
| | | Number | | | Percentage | | | Amount | | | Percentage | | |||
Existing stockholders(1) | | | | | % | | | $ | | | % | | | $ | |
New investors | | | | | | | | | | | $ | ||||
Total | | | | | 100% | | | $ | | | 100% | | | ||
| (1) | The presentation in this table regarding ownership by existing stockholders does not give effect to any purchases that existing stockholders may make through our directed share program or otherwise purchase in this offering. |
| • | 9,994,197 shares of common stock issuable upon exercise of stock options outstanding as of June 30, 2021, with a weighted average exercise price of $4.93 per share; |
| • | 1,049,043 shares of common stock subject to restricted stock units, or RSUs, outstanding as of June 30, 2021; |
| • | 75,744 shares of common stock issuable upon exercise of warrants outstanding as of June 30, 2021 to purchase shares of common stock, with an exercise price of $0.40 per share; |
| • | 79,882 shares of common stock issuable upon the exercise of warrants outstanding as of June 30, 2021 to purchase redeemable convertible preferred stock that will automatically become warrants to purchase 79,882 shares of common stock immediately prior to the completion of this offering, with a weighted-average exercise price of $1.12 per share; |
| • | 1,138,635 shares of common stock reserved for future issuance under our 2014 Equity Incentive Plan, as of June 30, 2021, of which our board of directors expects to grant stock awards covering 128,000 shares of common stock to certain of our non-employee directors effective immediately prior to the effectiveness of the registration statement of which this prospectus forms a part; |
| • | additional shares of our common stock that will become available for future issuance under our 2021 Stock Incentive Plan, which will become effective immediately prior to the effectiveness of the registration statement of which this prospectus forms a part, as well as any automatic increases in the number of shares of common stock reserved for future issuance under the 2021 Stock Incentive Plan, of which our board of directors expects to grant awards covering shares of common stock to certain of our employees, executive officers and non-employee directors effective prior to the commencement of trading of our common stock on the Nasdaq Stock Market; and |
| • | additional shares of our common stock that will become available for future issuance under our 2021 Employee Stock Purchase Plan, which will become effective immediately prior to the effectiveness of the registration statement of which this prospectus forms a part, as well as any automatic increases in the number of shares of common stock reserved for future issuance under the 2021 Employee Stock Purchase Plan. |
| • | Cue Health Monitoring System |
| ○ | Cue Reader: The Cue Reader is an elegantly designed, automated analyzer of test results and is used with Cue Test Kits and the Cue Health App. The Cue Reader runs the Cue Cartridge and communicates the result of the test digitally via Bluetooth to the Cue Health App. |
| ○ | Cue Test Kit: Each Cue Test Kit is comprised of a Cue Cartridge and a Cue Wand. |
| • | Cue Cartridge: Our sample-specific, single-use cartridges are designed to handle different chemistries, which allows us to create a broad menu of tests. Cue Cartridges are designed to be seamlessly inserted into the Cue Reader. |
| • | Cue Wand: Cue Wands are single-use and sterile sample collection devices that are designed to be universally compatible with the Cue Cartridges. The Cue Wand is designed to permit collection of multiple sample types, including saliva, blood, urine and swabs, with only minor modifications. |
| • | Cue Virtual Care Delivery Apps |
| ○ | Cue Health App: Our mobile app creates a secure interface between the user and their health data. For consumers, it allows a single point of entry for their health data; for healthcare professionals, it is designed to provide a unified platform for managing patient histories and, in the future, is expected to allow for telemedicine and e-prescription services. By connecting the diagnostic test results with interventions and outcomes, we believe the Cue Health App will allow users to be more engaged and satisfied with their healthcare experience, which can ultimately drive better outcomes for users. To run a Test Kit on the Cue Reader, a user will need to download and utilize the Cue Health App. As of August 31, 2021, through our 49 active customers, over 45,000 unique accounts have used the Cue Health App to run our COVID-19 Test Kit. These unique accounts include both organizations and individuals who may take tests episodically, healthcare providers running a large number of tests for multiple patients, and enterprises running a large number of tests for their entire organization, as established by the customer on a customer-by-customer basis. |
| ○ | Cue Enterprise Dashboard: Our dashboard is designed to allow enterprises, payors, healthcare providers and public health entities to manage population health at the organizational level and has the potential to track the efficacy of various population health programs. Accessible online, the Cue Enterprise Dashboard has the potential to help organizations manage a patient’s journey from onboarding to scheduling, care management and inventory management. The Cue Enterprise Dashboard was built with a focus on user experience, simplifying the sharing of communications, such as results, records, and histories with patients and across providers and streamlining reporting requirements. Powered by our analytics engine and role-based access capabilities, it is designed to provide chief medical officers, environmental health and safety officials, and benefits managers with insight into their organization’s population health, helping to facilitate efficient decision making. As of August 31, 2021, we had approximately 60 active public sector, enterprise and provider accounts on the Cue Enterprise Dashboard. An account on the Cue Enterprise Dashboard is considered active if the customer has signed into their account and utilized the programs within the last six months. A customer may have more than one active account on the Cue Enterprise Dashboard. |
| • | Cue Ecosystem Integrations and Apps: We believe that placing our APIs at the core of our integrated care platform will enable us to become foundational within Healthcare 2.0. Our Cue Data and Innovation Layer is designed to be able to securely connect with on-demand services, such as telemedicine, and e-prescription services, which we believe we will enable a truly digital and seamless user experience. In the future, we plan on enhancing our platform to enable third party application development and offerings that complement our solutions. |
| • | Public Sector Sales: Our public sector sales team identifies new opportunities within federal, state and local government agencies. While we expect that revenue from other categories of customers will become a larger component of our revenue over time, our public sector sales strategy continues to look to identify new opportunities within federal, state and local government agency customers. |
| • | Enterprise Sales: Our enterprise sales team identifies major self-insured enterprises, such as Fortune 500 companies with large, covered employee populations, as well as small-to-medium sized businesses with healthcare plan partners and employee benefits offerings. We believe that enterprise customers will want to utilize our integrated care solutions for their employees and their families, both on-premises and at-home. |
| • | Healthcare Provider Sales: Our healthcare provider sales team identifies and targets major healthcare systems and healthcare providers such as hospital systems, clinic networks, concierge health systems and physicians’ offices. Relationships with our healthcare provider customers, such as our current relationship with the Mayo Clinic, help validate our platform, and we believe will help accelerate marketplace adoption of our products. |
| • | Direct-to-Consumer Sales: Our direct-to-consumer sales team identifies opportunities through online and offline retail channels such as e-commerce and in-store sales. |
| • | external costs, including expenses incurred under arrangements with third parties, primarily associated with CROs performing clinical studies and regulatory submissions; and |
| • | internal costs, including: |
| ○ | employee-related expenses, including salaries, benefits, and stock-based compensation; |
| ○ | the costs of laboratory supplies, research materials and Cue Cartridges we produce for research and development purposes; and |
| ○ | facilities, equipment, and information technology, which include depreciation and amortization costs, direct and allocated expenses for rent and maintenance of facilities and equipment. |
| | | Year Ended December 31, | | | Six MonthsEnded June 30, | |||||||
| | | 2019 | | | 2020 | | | 2020 | | | 2021 | |
(dollars in thousands) | | | | | | | (unaudited) | |||||
External costs | | | $1,534 | | | $4,441 | | | $2,859 | | | $1,474 |
Internal costs | | | | |||||||||
Salaries and benefits | | | 8,366 | | | 7,607 | | | 4,428 | | | 3,751 |
Facilities and supplies | | | 11,505 | | | 16,430 | | | 12,393 | | | 6,846 |
Total internal costs | | | 19,871 | | | 24,037 | | | 16,821 | | | 10,597 |
Total research and development expense | | | $21,405 | | | $28,478 | | | $19,680 | | | $12,071 |
| | | Year Ended December 31, | | | Six Months Ended June 30, | |||||||
| | | 2019 | | | 2020 | | | 2020 | | | 2021 | |
(dollars in thousands) | | | | | | | (unaudited) | |||||
Revenue: | | | | | | | | | ||||
Product revenue | | | $— | | | $15,391 | | | $— | | | $201,922 |
Grant and other revenue | | | 6,626 | | | 7,562 | | | 4,960 | | | — |
Total revenue | | | 6,626 | | | 22,953 | | | 4,960 | | | 201,922 |
Operating costs and expenses: | | | | | | | | | ||||
Cost of product revenue(1)(2) | | | — | | | 14,951 | | | — | | | 85,177 |
Sales and marketing(1) | | | 88 | | | 714 | | | 45 | | | 1,959 |
Research and development(1) | | | 21,405 | | | 28,478 | | | 19,680 | | | 12,071 |
General and administrative(1) | | | 5,900 | | | 23,936 | | | 3,764 | | | 23,252 |
Total operating costs and expenses | | | 27,393 | | | 68,079 | | | 23,489 | | | 122,459 |
Income (loss) from operations | | | (20,767) | | | (45,126) | | | (18,529) | | | 79,463 |
Interest expense | | | (152) | | | (984) | | | (788) | | | (9,964) |
Change in fair value of redeemable convertible preferred stock warrants | | | 4 | | | (1,289) | | | (20) | | | (190) |
Change in fair value of convertible notes | | | — | | | — | | | — | | | (23,254) |
Other income (expense), net | | | 309 | | | 47 | | | 59 | | | 61 |
Net income (loss) before income taxes | | | (20,606) | | | (47,352) | | | (19,278) | | | 46,116 |
Income tax expense | | | — | | | — | | | — | | | (13,276) |
Net income (loss) | | | $(20,606) | | | $(47,352) | | | $(19,278) | | | $32,840 |
| (1) | Includes stock-based compensation expense as follows: during the six months ended June 30, 2021, $0.1 million of stock-based compensation expense was capitalized to inventory during the manufacturing process. |
| | | Year Ended December 31, | | | Six Months Ended June 30, | |||||||
| | | 2019 | | | 2020 | | | 2020 | | | 2021 | |
(dollars in thousands) | | | | | | | (unaudited) | |||||
Cost of product revenue | | | $— | | | $— | | | $— | | | $343 |
Sales and marketing | | | — | | | 1 | | | — | | | 26 |
Research and development | | | 45 | | | 98 | | | 13 | | | 1,444(3) |
General and administrative | | | 291 | | | 3,064 | | | 84 | | | 3,778 |
Total stock-based compensation expense | | | $336 | | | $3,163 | | | $97 | | | $5,591 |
| (2) | Includes $2.1 million and $10.5 million of depreciation and amortization expense for the year ended December 31, 2020, and for the six months ended June 30, 2021, respectively. |
| (3) | Includes $1.2 million of stock-based compensation related to a common stock warrant exercised by a vendor. |
| | | Six Months Ended June 30, | ||||||||||
| | | 2020 | | | 2021 | | | $ Change | | | % Change | |
(dollars in thousands) | | | (unaudited) | |||||||||
Revenue: | | | | | | | | | ||||
Product revenue | | | $— | | | $201,922 | | | $201,922 | | | n.m. |
Grant and other revenue | | | 4,960 | | | — | | | (4,960) | | | (100.0)% |
Total revenue | | | 4,960 | | | 201,922 | | | 196,962 | | | n.m. |
Operating costs and expenses: | | | | | | | | | ||||
Cost of product revenue | | | — | | | 85,177 | | | 85,177 | | | n.m. |
Sales and marketing | | | 45 | | | 1,959 | | | 1,914 | | | n.m. |
Research and development | | | 19,680 | | | 12,071 | | | (7,609) | | | (38.7)% |
General and administrative | | | 3,764 | | | 23,252 | | | 19,488 | | | 517.7% |
Total operating costs and expenses | | | 23,489 | | | 122,459 | | | 98,970 | | | 421.3% |
Income (loss) from operations | | | (18,529) | | | 79,463 | | | 97,992 | | | 528.9% |
Interest expense | | | (788) | | | (9,964) | | | (9,176) | | | n.m. |
Change in fair value of redeemable convertible preferred stock warrants | | | (20) | | | (190) | | | (170) | | | n.m. |
Change in fair value of convertible notes | | | — | | | (23,254) | | | (23,254) | | | n.m. |
Other income (expense), net | | | 59 | | | 61 | | | 2 | | | 3.4% |
Net income (loss) before income taxes | | | (19,278) | | | 46,116 | | | 65,394 | | | 339.2% |
Income tax expense | | | — | | | (13,276) | | | (13,276) | | | n.m. |
Net income (loss) | | | $(19,278) | | | $32,840 | | | $52,118 | | | 270.3% |
| | | Year Ended December 31, | ||||||||||
| | | 2019 | | | 2020 | | | $ Change | | | % Change | |
(dollars in thousands) | | | | | | | | | ||||
Revenue: | | | | | | | | | ||||
Product revenue | | | $— | | | $15,391 | | | $15,391 | | | n.m. |
Grant and other revenue | | | 6,626 | | | 7,562 | | | 936 | | | 14.1% |
Total revenue | | | 6,626 | | | 22,953 | | | 16,327 | | | 246.4% |
Operating costs and expenses: | | | | | | | | | ||||
Cost of product revenue | | | — | | | 14,951 | | | 14,951 | | | n.m. |
Sales and marketing | | | 88 | | | 714 | | | 626 | | | 711.4% |
Research and development | | | 21,405 | | | 28,478 | | | 7,073 | | | 33.0% |
General and administrative | | | 5,900 | | | 23,936 | | | 18,036 | | | 305.7% |
Total operating costs and expenses | | | 27,393 | | | 68,079 | | | 40,686 | | | 148.5% |
Loss from operations | | | (20,767) | | | (45,126) | | | (24,359) | | | 117.3% |
Interest expense | | | (152) | | | (984) | | | (832) | | | 547.4% |
Change in fair value of redeemable convertible preferred stock warrants | | | 4 | | | (1,289) | | | (1,293) | | | n.m. |
Other income (expense), net | | | 309 | | | 47 | | | (262) | | | (84.8%) |
Net loss | | | $(20,606) | | | $(47,352) | | | $(26,746) | | | 129.8% |
| | | Six Months Ended June 30, | ||||||||||
| | | 2020 | | | 2021 | |||||||
| | | Dollar Amount | | | Per Diluted Share | | | Dollar Amount | | | Per Diluted Share | |
| | | (unaudited) | ||||||||||
Net income (loss)/diluted EPS | | | $(19,278) | | | $(1.21) | | | $32,840 | | | $0.22 |
Fair value adjustment—convertible notes | | | — | | | — | | | 23,254 | | | 0.19 |
Banking and finance-related items | | | — | | | — | | | 7,998 | | | 0.07 |
Tax effects(1) | | | — | | | — | | | (816) | | | (0.01) |
Adjusted Net Income (Loss)/Adjusted Diluted EPS | | | $(19,278) | | | $(1.21) | | | $63,276 | | | $0.47 |
| (1) | Represents the tax impact with respect to the adjustments noted above. We applied an estimated annual effective tax rate of 24% to amounts deductible for tax purposes to estimate the tax effects. |
| | | Year Ended December 31, | | | Six Months Ended June 30, | |||||||
| | | 2019 | | | 2020 | | | 2020 | | | 2021 | |
(dollars in thousands) | | | | | | | (unaudited) | |||||
Net cash, cash equivalents and restricted cash (used in) provided by operating activities | | | $(12,996) | | | $92,655 | | | $(20,955) | | | $(37,812) |
Net cash, cash equivalents and restricted cash used in investing activities | | | (2,945) | | | (78,148) | | | (1,326) | | | (58,896) |
Net cash, cash equivalents and restricted cash provided by financing activities | | | 3,610 | | | 100,243 | | | 101,723 | | | 219,779 |
Net increase (decrease) in cash, cash equivalents and restricted cash | | | $(12,331) | | | $114,750 | | | $79,442 | | | $123,071 |
| • | Fair Value of Common Stock. See the subsection titled “Common Stock Valuations” below. |
| • | Expected Term. The expected term of options represents the period of time that options are expected to be outstanding. Our historical stock option exercise experience does not provide a reasonable basis upon which to estimate an expected term due to lack of sufficient data. We estimate the expected term by using the simplified method, which calculates the expected term as the average of the time-to-vesting and the contractual life of the options. |
| • | Expected Volatility. As there has been no public market for our common stock to date, and as a result we do not have any trading history of our common stock, expected volatility incorporates the historical volatility over the expected term of the award of comparable companies whose share prices are publicly available. The comparable companies are chosen based on their similar size, stage in the life cycle or area of specialty. |
| • | Risk-Free Interest Rate. The risk-free interest rate is based on the U.S. Treasury zero coupon issues in effect at the time of grant for periods corresponding with the expected term of the stock option grants. |
| • | Expected Dividend Yield. We have never paid dividends on our common stock and have no plans to pay dividends on our common stock. Therefore, we use an expected dividend yield of zero. |
| • | Current Value Method. Under the Current Value Method, our value is determined based on our balance sheet. This value is then first allocated based on the liquidation preference associated with redeemable convertible preferred stock issued as of the valuation date, and then any residual value is assigned to the common stock. |
| • | Option-Pricing Method. Under the option-pricing method, or OPM, shares are valued by creating a series of call options with exercise prices based on the liquidation preferences and conversion terms of each equity class. The estimated fair values of the preferred and common stock are inferred by analyzing these options. |
| • | Probability-Weighted Expected Return Method. The probability-weighted expected return method, or PWERM is a scenario-based analysis that estimates value per share based on the probability-weighted present value of expected future investment returns, considering each of the possible outcomes available to us, as well as the economic and control rights of each share class. |
| • | contemporaneous independent valuations performed by an independent third-party valuation firm; |
| • | the prices at which we sold shares of redeemable convertible preferred stock and the superior rights and preferences of the redeemable convertible preferred stock relative to our common stock at the time of each grant; |
| • | our stage of development and commercialization and our business strategy; |
| • | our actual operating and financial performance; |
| • | our current business conditions and projections; |
| • | external market conditions affecting the diagnostics industry and trends within the diagnostics industry; |
| • | the lack of an active public market for our common stock; and |
| • | the likelihood of achieving a liquidity event, such as an initial public offering or sale of our company in light of prevailing market conditions. |
| • | control over how they manage their acute and chronic conditions as well as their overall health; |
| • | access to actionable clinical insights; |
| • | affordable and transparent pricing; and |
| • | customer-centric user experiences that connect the entire care journey. |

| * | Depicts future product developments. |
| • | A busy parent has few good options for handling a child sick with cold and flu-like symptoms. Currently, they must take time off from work to care for the child and will often make an appointment with a pediatrician or travel to an urgent care facility. An integrated care solution with telemedicine, with diagnostics for the common respiratory threats, such as flu, COVID-19, strep throat, and RSV would allow for testing to be done conveniently and easily at-home with the guidance of a telemedicine visit if desired, and with the results delivered directly to their mobile device in minutes. Prescriptions such as Tamiflu (for influenza) or antibiotics (for strep throat) could be delivered same day to their doorstep, as the telemedicine care provider deems appropriate. Otherwise, appropriate over-the-counter medications for symptom relief could also be delivered. We believe this type of care model for respiratory diseases will become normative for this number one most common reason for a visit to urgent care in the developed world. |
| • | A sexually active adult that wants peace of mind before or after a sexual encounter must currently make an appointment, give a sample, and typically wait days for a result on their sexual transmitted infection, or STI, status. Beyond the inconvenience, the stigma and general friction associated with getting tested is a high barrier for seeking care. We believe an integrated care solution including quick results for some of the most common STIs, consultation with a telemedicine provider through a virtual care delivery app and potential resolution with the appropriate therapy, such as an antibiotic for chlamydia delivered to them – all within hours – is an optimal flow that has the potential to become normative for handling STI related matters. |
| • | For an individual managing a chronic condition such as cardiovascular disease, autoimmune disorder or metabolic disorder such as hypothyroidism, medication adherence and regular diagnostic testing measuring the clinically relevant biomarkers such as cholesterol are critical for effective condition management. Removing significant friction to accessing critical diagnostic information that informs disease management with doctor-informed care could provide an effective way to drive medication adherence and combined with other sensor data to form a more complete picture of health that helps drive engagement and effective disease management. |

| * | Depicts future product developments. |


| * | Depicts future product developments. |
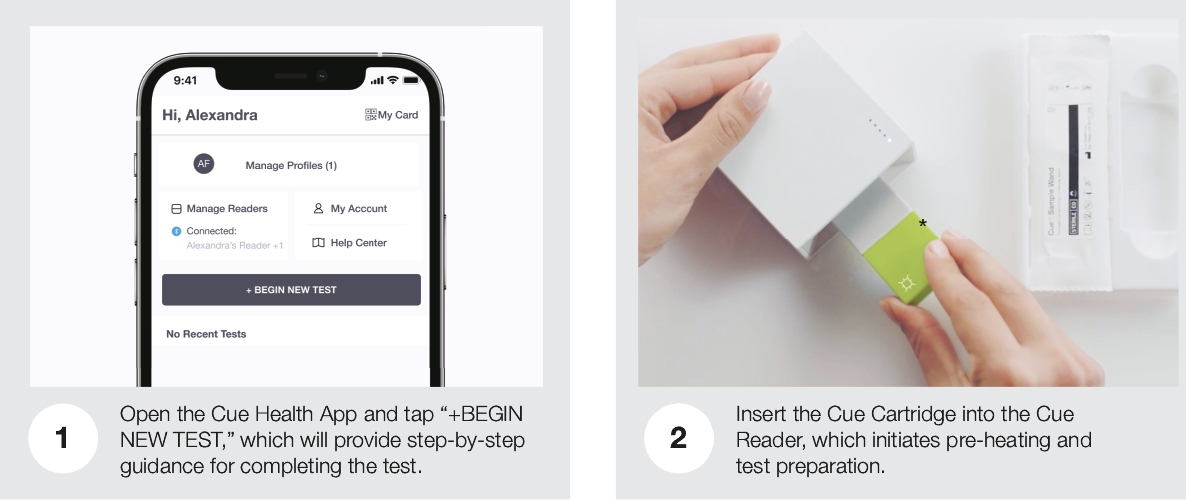
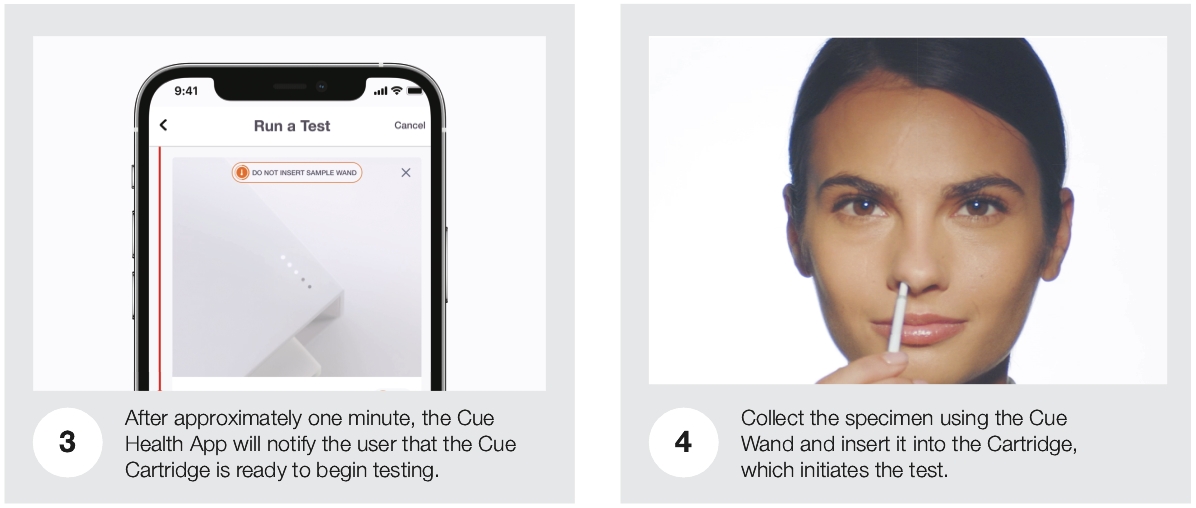
| * | Depicts future product developments. |
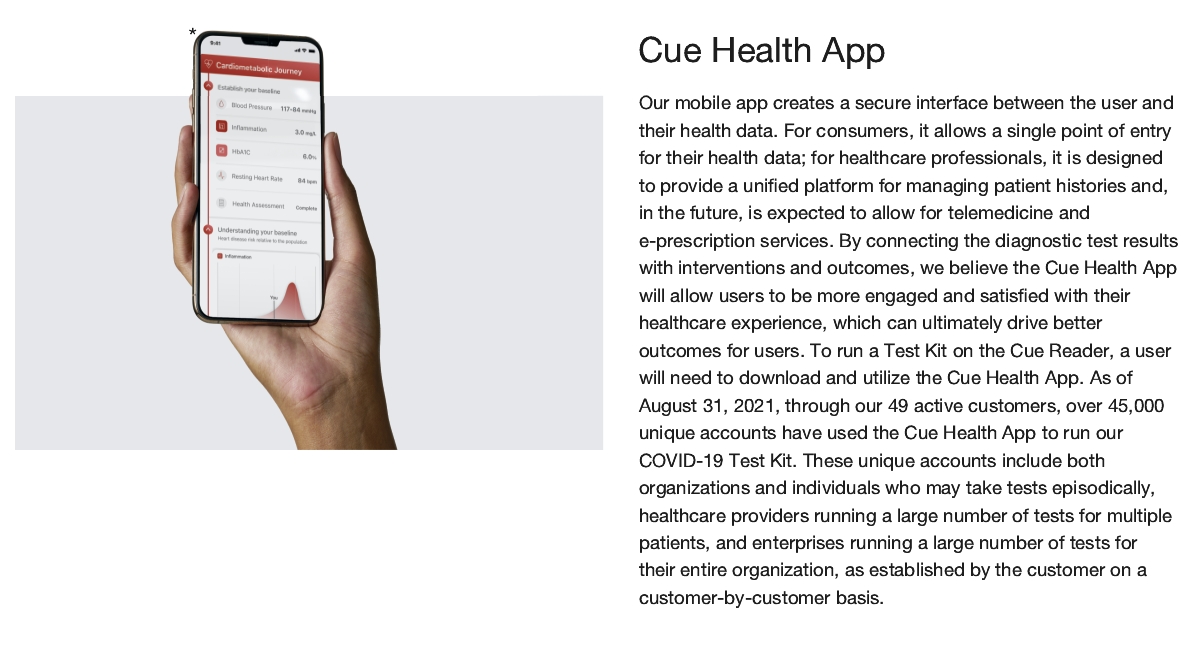

| * | Depicts future product developments. |
| • | Consumer-centric. The Cue Integrated Care Platform is intended to revolutionize the way individuals and healthcare providers access diagnostic testing at home, at work, or at the point-of-care. Our Cue Integrated Care Platform is designed to deliver a superior user experience in any setting, one that is fully-guided, fast, accurate, and easy to use and that puts the consumer in control of their health data. Users only have to take the test and the Platform does the rest, obviating the need for many in-person testing visits and sample shipments, with a focus on at-home testing which we believe is the most consumer-centric and convenient setting. Results are presented in an easy-to-understand format through our Cue Health App and Cue Enterprise Dashboard. The digital nature of our results allows consumers to access their medical data immediately. By connecting this data to the wider healthcare ecosystem, consumers will be able to securely share their data with key stakeholders in their care journey and further streamlining the user experience. This will allow for more testing to be performed at the right point in the care journey, enabling diagnostics to drive care decisions. |
| • | Lab-quality diagnostics anywhere in minutes. By combining the sophistication and accuracy of complex molecular testing platforms with the simplicity, convenience and speed of a consumer electronic device, our Cue Health Monitoring System has been developed to deliver highly specific and sensitive results within minutes. As a result, we believe our tests will provide a better, more convenient user experience compared to traditional lab tests while also delivering “gold standard” molecular testing results - all from a device that fits in the palm of your hand. The accuracy of the Cue Health Monitoring System was confirmed by a recent independent study, conducted by researchers at the Mayo Clinic, that found that the overall concordance between our COVID-19 test and clinical laboratory tests using NAAT was 97.8%. |
| • | Extensible platform approach. We designed our technology, platform and infrastructure to be versatile in accommodating a wide range of tests by addressing both main analytical modalities used in diagnostic testing, immunoassays and NAAT. We believe our flexible platform will permit our planned future menu of tests to cover a large portion of diagnostic solutions typically offered by a traditional lab. The extensibility of our platform is due to the reusability of the Cue Reader, the uniform design of single-use Cue Cartridges and the synergies in chemistry across our pipeline of contemplated future tests, which we believe will allow us to quickly expand and upsell our menu in a cost-efficient manner. We have demonstrated our ability to quickly develop tests, having developed our highly accurate COVID-19 test within weeks. Our digitally native results enable seamless integration into our apps and cloud-based software platform as well as allow for integration with the broader healthcare and partner ecosystem. |
| • | Vertically-integrated, automated and scalable production infrastructure. Our proprietary technology was designed to enable us to optimize our system across the full product life cycle from design to |
| • | Scaled and growing installed base. We have shipped over 115,000 Cue Readers across the United States as of August 31, 2021, including Cue Readers placed through our agreement with the U.S. DoD and through our other customer agreements, resulting in a broad installed base, diversified across industries, locations and end-markets such as schools, essential businesses, nursing homes, hospitals, physicians' offices, dental clinics, sports and other live events, and other settings around the country. With our EUA for at-home and over-the-counter COVID-19 testing, we expect to significantly grow our install base over the coming months and gain a place in more consumer households across the U.S. and internationally. Given our Cue Readers are reusable and universally compatible with our current and planned future Cue Cartridges, we believe this installed base and population of active users will position us well as we expand our testing menu. In addition, our installed base provides us with a wealth of data generation for our own use, and which we intend to use to improve our current and future product offerings. |
| • | Expand our menu of tests and continue to innovate and enhance our platform. We plan to expand our test menu, including in the fields of respiratory health, sexual health, cardiac and metabolic health, women's health, men's health, and chronic disease management, with several of these tests expected to be submitted for FDA authorization or clearance by the end of 2022. Our broad planned future test menu is aimed to appeal to consumers, self-insured employers, and health plans alike and will allow for care that can be personalized to the consumer. We intend to further continue to expand our platform capabilities to provide a comprehensive user experience. |
| • | Drive ecosystem adoption. We have been successful in our ability to integrate our platform into existing enterprise-level health management systems, allowing customers to automate workflows while allowing us to garner long-term commercial partnerships. As we enhance our Cue Integrated Care Platform, we intend to extend our integrations with leading EMR systems and to build-out additional capabilities to integrate with telemedicine and digital health providers, e-prescription, e-commerce, and other connected services, to offer consumers a frictionless, virtual-to-physical care solution that positions them for better outcomes |
| • | Continue to expand our installed base and distribution network to enable pull-through of our future extended care offerings. We believe that the ability of customers to experience our platform for COVID-19 |
| • | Increase adoption through value-based selling and payor reimbursement. Our platform enables enterprise customers and payors to capture consistent, convenient and simple diagnostic information to inform key decisions. We believe this will help create positive outcomes for all stakeholders, especially our customers, as we expand our test menu. For example, helping customers test their HbA1c to manage diabetes or assess their HIV viral load to determine whether their treatment plan is effective would help payors and enterprises incur fewer costs. We believe this strategy will help accelerate our growth and drive further adoption of our platform. |
| • | Continue to build the Cue brand. We believe that there are significant opportunities to drive increased brand awareness, educate consumers and enterprises on the benefits of diagnostics and our connected health platform, and build a lasting consumer brand. As we continue to invest in marketing, we anticipate that many customers who are not aware of our platform or the benefits of continuous, virtual care will begin using our platform. We further intend to increase our brand awareness through our partnership program. We believe the validation of leading institutions, such as the Mayo Clinic, the NBA and others, will help us to become the testing solution of choice in the enterprise and employer, travel, sports and entertainment, education, personal health and wellness, community and population health, and government market. |
| • | Scale manufacturing capabilities to capitalize on demand. In the fall of 2020, we leased two new manufacturing facilities in an effort to scale our capabilities, and we have since commenced construction on a number of new production pods. As of August 31, 2021, we were manufacturing Cue Cartridges at a rate equivalent to over 15 million per year and we anticipate growing our manufacturing capacity to a rate equivalent to tens of millions of Cue Cartridges per year by the end of 2021. |
| • | Expand our global footprint. We believe in the broad suitability of our platform and intend to grow our international customer base. In countries with developed healthcare systems, our value proposition is similar to that of the United States and will offer individuals, enterprises, and healthcare providers with the ability to positively impact health outcomes. In December 2020, our COVID-19 test received the CE mark, clearing it for sale and distribution in the European Union. In April 2021, we received Interim Order authorization from Health Canada to be able to sell and distribute our COVID-19 test and in August 2021 such Interim Order authorization was amended to include both point-of-care and self-testing. In countries with underdeveloped healthcare systems and infrastructure, we believe our platform will be able to provide front-line healthcare providers with access to lab-quality testing to better diagnose and treat underserved patient populations. In June 2021, our COVID-19 test received regulatory approval from the CDSCO for professional point-of-care use in India. |
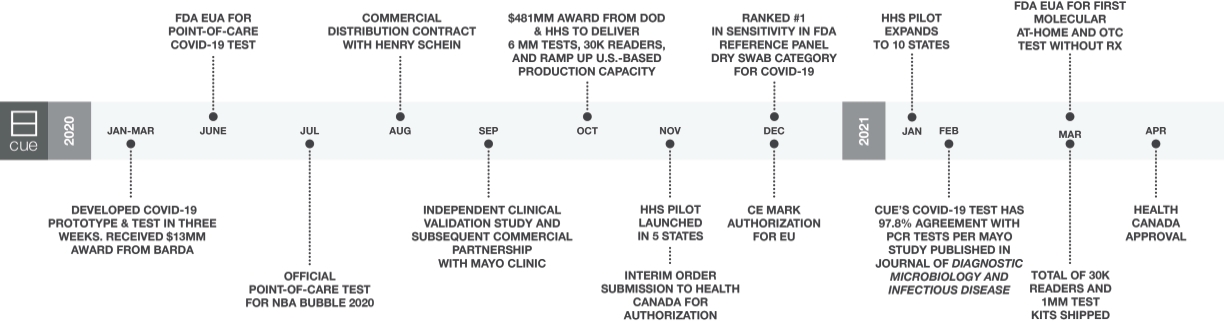
| | | Number of samples with a reference result of | | | |||||
Number of samples with a Cue result of | | | Positive | | | Negative | | | Total |
Positive | | | 22 | | | 4 | | | 26 |
Negative | | | 2 | | | 239 | | | 241 |
Positive percent agreement | | | 91.7%(1) | | | — | | | — |
Negative percent agreement | | | — | | | 98.4% | | | — |
Total | | | 24 | | | 243 | | | 267 |
| (1) | One discrepant positive reference sample did not have a tie-breaker method available, so positive percent agreement would be 22/23 (95.7%) excluding that sample. |
Patient # | | | Cue Result | | | Reference (method) | | | Other performed | | | Reference consensus result |
1 | | | Negative | | | Positive (Hologic) | | | None | | | Positive |
2 | | | Negative | | | Positive (Hologic) | | | Negative (LTD-PCR) | | | Negative |
3 | | | Negative | | | Positive (LTD-PCR) | | | Positive (Hologic) | | | Positive |
4 | | | Positive | | | Negative (Hologic) | | | None | | | Negative |
5 | | | Positive | | | Negative (Hologic) | | | None | | | Negative |
6 | | | Positive | | | Negative (Hologic) | | | None | | | Negative |
7 | | | Positive | | | Negative (Hologic) | | | Negative (Hologic) | | | Negative |
| • | Public Sector Sales: Our public sector sales team identifies new opportunities within federal, state and local government agencies. While we expect that revenue from other categories of customers will become a larger component of revenue over time, our public sector sales strategy continues to look to identify opportunities with new and existing federal, state and local government agency customers. |
| • | Enterprise Sales: Our enterprise sales team identifies major self-insured enterprises such as Fortune 500 companies with large-covered employee populations as well as small to medium sized businesses with healthcare plans partners and employee benefits offerings. We believe that enterprise customers will want to utilize our integrated care solutions for their employees and their families, both on-premise and at-home. |
| • | Healthcare Provider Sales: Our healthcare provider sales strategy targets major healthcare systems and healthcare professionals such as hospital systems, private clinics and concierge health systems, and physicians’ offices. Relationships with our customers, like our current relationship with the Mayo Clinic, help validate our platform, and we believe will help accelerate marketplace adoption of our products. |
| • | Direct-to-Consumer Sales: Our direct-to-consumer sales team identifies opportunities through online and offline retail channels such as e-commerce and in-store sales. |
| • | BARDA - We have partnered with BARDA since June 2018, initially focusing on a molecular influenza test using the Cue Health Monitoring System pursuant to a contract that was originally effective through January 2021 and that provided $14.0 million in base funding. In March 2020, BARDA exercised an option to accelerate development, validation and FDA clearance of our COVID-19 test for a $13.7 million award. This funding enabled us to accelerate the development and validation of our COVID-19 test. In May 2020, our original contract with BARDA was amended to increase the base value from $14.0 million to $21.8 million and to extend the contract term to January 2022. Pursuant to our agreement with BARDA, we agreed to provide regular reports to BARDA regarding our progress and certain customary oversight provisions. BARDA can terminate this agreement for convenience or if we fail to meet our obligations, subject to our opportunity to cure such defaults. |
| • | Department of Defense/Department of Health and Human Services |
| ○ | In October 2020, we entered into an agreement, as amended in March 2021, for an aggregate of $480.9 million, with the U.S. DoD to expand our U.S.-based production capacity, to deploy 6,000,000 Cue COVID-19 Test Kits, 30,000 Cue Readers and 60,000 Cue Control Swab Packs (which is comprised of three positive and three negative control swabs per pack) pursuant to the delivery schedule under the agreement and demonstrate our ability to manufacture an average of approximately 100,000 Cue COVID-19 Cartridges per day over a consecutive seven-day period by October 12, 2021. Included as part of the $480.9 million contract amount was an upfront payment of $184.6 million to scale our manufacturing. This payment was intended to help us onshore our supply chain and rapidly increase our production capacity to enable and support domestic production of critical medical resources. As of August 31, 2021, we have shipped all of the required Cue Readers and over three and a half million COVID-19 Test Kits under the agreement. |
| ○ | In November 2020, as part of our agreement with the U.S. DoD, we started deployment of a pilot program in coordination with the U.S. HHS to assess how to best integrate our diagnostic technology into public health strategies for disease surveillance and infection control in institutions such as nursing homes. Through this program, our COVID-19 test is currently being used in the U.S. in point-of-care settings with high-concern populations and congregate care settings, such as nursing homes, long-term care, assisted living facilities, veterans’ homes, K-12 schools, correctional facilities, homeless populations, essential businesses, remote and tribal communities, and hospitals. In the pilot program, U.S. HHS is using our COVID-19 test to verify antigen test results, which are less sensitive than molecular and PCR tests and occasionally prone to false positives. This pilot program was expanded to ten states in January 2021. As part of this pilot program, we have the ability to directly work with the state or local authorities that decide how to distribute our COVID-19 tests in their jurisdictions, including the ability to offer support and to sell our COVID-19 test directly to such state and local authorities. |
| ○ | During the term of our agreement with the U.S. DoD, we agreed that the U.S. government would be the exclusive purchaser of our entire production until our development obligations under this agreement have been completed, except for previously existing contracts and subject to agreed upon waivers. In April 2021, we received the U.S. DoD Waiver, effective May 1, 2021, which now allows us to distribute commercially up to 50% of our COVID-19 Test production, measured monthly in arrears on a calendar month basis, to non-U.S. federal government customers and other recipients. We expect that the U.S. DoD Waiver will remain in effect for the duration of the U.S. DoD agreement; however, the U.S. government may modify the waiver upon timely written notice to reasonably accommodate changes in U.S. government requirements. We also agreed to provide regular reports as to the status of our production and distribution. We have the right to terminate this agreement without penalty if we cease to undertake development as a result of emerging safety or efficacy data, and the U.S. government can terminate the agreement if we materially fail to comply with our obligations under the agreement. If the agreement is terminated by the U.S. DoD for cause, the U.S. government may be entitled to certain remedies, including grants of licenses and penalty payments. The U.S. government may also terminate the agreement for convenience upon 30 days’ notice, subject to the U.S. government retaining the right to place priority orders for up to a year following termination for other diagnostic tests manufactured using the manufacturing equipment purchased with U.S. government funds under the agreement. |
| ○ | Under the agreement, following completion of our agreement, we have agreed to negotiate in good faith with the U.S. DoD for a new production agreement under which the U.S. DoD would have the right to purchase up to 45% of our quarterly production at a discount to the lowest price offered by us to a commercial customer for the same products, equivalent quantities and comparable terms of sale, subject to a price floor. |
| • | our technical development capabilities that have led to an authorized COVID-19 test and multiple tests in late-stage technical development; |
| • | our understanding of the regulatory pathways, including FDA authorization or clearance, for the various diagnostic tests; and |
| • | our test-agnostic production capacity that we believe will provide us the flexibility to meet our customers’ needs. |
| * | HSV-1 & HSV-2 |
| ** | Luteinizing Hormone (LH) |
| *** | Human chorionic gonadotropin (hCG) |
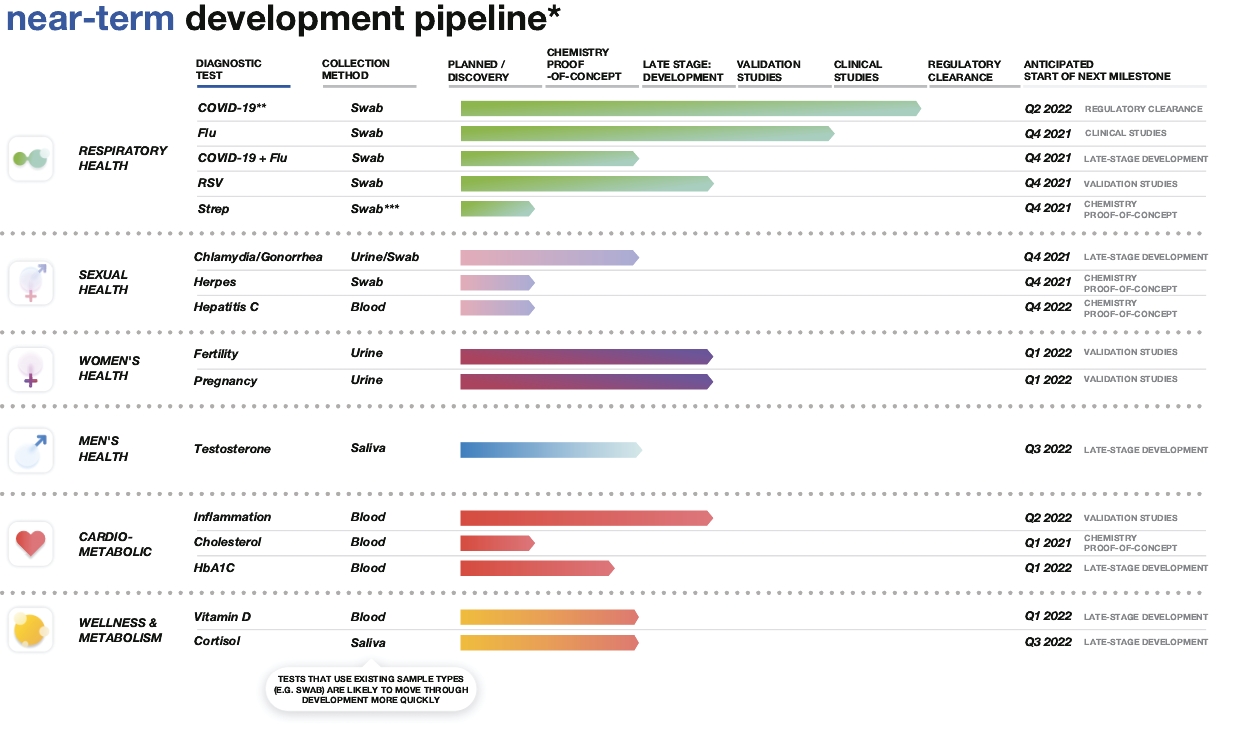
| * | This graphic does not reflect our full development pipeline but rather those of our tests that are furthest along in development. |
| ** | Our COVID-19 test has been authorized by the FDA under two EUAs. This graphic reflects progress towards 510(k) clearance. Our COVID-19 test has also received regulatory approval from the Central Drugs Standard Control Organisation, India’s national regulatory body for pharmaceuticals and medical devices, for professional point-of-care use in India. Internationally, we have also received the CE mark in the European Union, as well as Interim Order authorization from Health Canada. |
| *** | Throat swab sample may be required. |
| * | Depicts future product developments. |
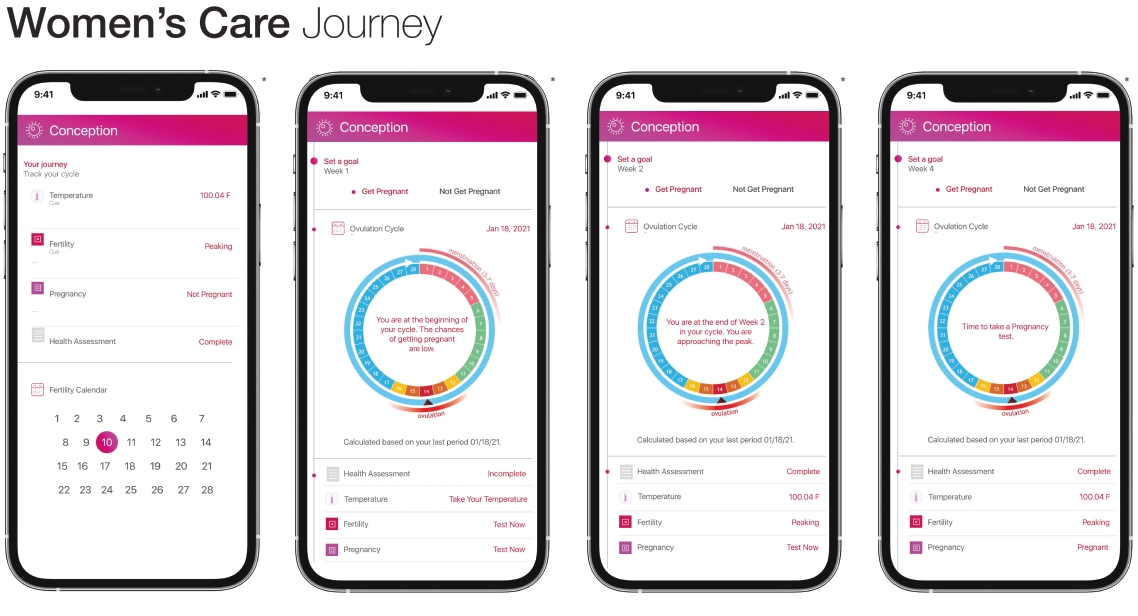
| * | Depicts future product developments. |
| • | establishment registration and device listing with the FDA; |
| • | QSR requirements, which require manufacturers and contract manufacturers, including any third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the design and manufacturing process; |
| • | labeling regulations and FDA prohibitions against the promotion of investigational products, or “off-label” uses of cleared or approved products; |
| • | requirements related to promotional activities; |
| • | clearance or approval of product modifications to 510(k)-cleared devices that could significantly affect safety or effectiveness or that would constitute a major change in intended use of one of our cleared devices; |
| • | medical device reporting regulations, which require that a manufacturer report to the FDA if a device it markets may have caused or contributed to a death or serious injury, or has malfunctioned and the device or a similar device that it markets would be likely to cause or contribute to a death or serious injury, if the malfunction were to recur; |
| • | correction, removal and recall reporting regulations, which require that manufacturers report to the FDA field corrections, product removals or recalls if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health; |
| • | the FDA’s recall authority, whereby the agency can order device manufacturers to recall from the market a product that is in violation of governing laws and regulations; and |
| • | post-market surveillance activities and regulations, which apply when deemed by the FDA to be necessary to protect the public health or to provide additional safety and effectiveness data for the device. |
| • | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| • | unanticipated expenditures to address or defend such actions; |
| • | customer notifications for repair, replacement, refunds; |
| • | recall, withdrawal, administrative detention or seizure of our Cue Health Monitoring System or any of our current or future test cartridges; |
| • | operating restrictions or partial suspension or total shutdown of production; |
| • | refusal of or delay in granting our requests for 510(k) clearance or PMA approval of new tests or modified tests; |
| • | operating restrictions, partial suspension or total shutdown of production; |
| • | withdrawing 510(k) clearance or PMA approvals that are already granted; |
| • | refusal to grant export approval for our Cue Health Monitoring System or any of our current or future tests; or |
| • | criminal prosecution. |
Name | | | Age | | | Position |
Executive Officers and Employee Directors | | | | | ||
Ayub Khattak | | | 36 | | | Chief Executive Officer, Director, Chairman of the Board and Co-Founder |
Chris Achar | | | 36 | | | Chief Strategy Officer and Director |
John Gallagher | | | 48 | | | Chief Financial Officer |
Erica Palsis | | | 36 | | | General Counsel |
Clint Sever | | | 36 | | | Chief Product Officer and Co-Founder |
| | | | | |||
Non-Employee Directors | | | | | ||
Xiangmin “Min” Cui | | | 52 | | | Director |
Scott Stanford(1) | | | 51 | | | Director |
| | ||||||
Director Nominees | | |||||
Joanne Bradford | | | 58 | | | Director Nominee |
Carole Faig(1) | | | 59 | | | Director Nominee |
Maria Martinez(1) | | | 64 | | | Director Nominee |
| (1) | Member of the Audit Committee. |
| (2) | Member of the Compensation Committee. |
| (3) | Member of the Nominating and Corporate Governance Committee. |
| • | the class I directors will be and , and their term will expire at our first annual meeting of stockholders following this offering; |
| • | the class II directors will be and , and their term will expire at our second annual meeting of stockholders following this offering; and |
| • | the class III directors will be and , and their term will expire at our third annual meeting of stockholders following this offering. |
| • | appointing, approving the compensation of, and assessing the independence of our registered public accounting firm; |
| • | overseeing the work of our independent registered public accounting firm, including through the receipt and consideration of reports from that firm; |
| • | reviewing and discussing with management and our independent registered public accounting firm our annual and quarterly financial statements and related disclosures; |
| • | monitoring our internal control over financial reporting, disclosure controls and procedures and code of business conduct and ethics; |
| • | overseeing our internal audit function; |
| • | overseeing our risk assessment and risk management policies; |
| • | establishing policies regarding hiring employees from our independent registered public accounting firm and procedures for the receipt and retention of accounting related complaints and concerns; |
| • | meeting independently with our internal auditing staff, if any, our independent registered public accounting firm and management; |
| • | reviewing and approving or ratifying any related person transactions; and |
| • | preparing the audit committee report required by Securities and Exchange Commission, or SEC, rules. |
| • | reviewing and approving, or making recommendations to our board of directors with respect to, the compensation of our chief executive officer and our other executive officers; |
| • | overseeing an evaluation of our senior executives; |
| • | overseeing and administering our cash and equity incentive plans; |
| • | reviewing and making recommendations to our board of directors with respect to director compensation; |
| • | reviewing and discussing annually with management our “Compensation Discussion and Analysis” disclosure if and to the extent then required by SEC rules; and |
| • | preparing the compensation committee report if and to the extent then required by SEC rules. |
| • | recommending to our board of directors the persons to be nominated for election as directors and to each of our board’s committees; |
| • | reviewing and making recommendations to our board with respect to our board leadership structure; |
| • | reviewing and making recommendations to our board with respect to management succession planning; |
| • | developing and recommending to our board of directors corporate governance principles; and |
| • | overseeing a periodic evaluation of our board of directors. |
Name and Principal Position | | | Year | | | Salary ($) | | | Bonus ($)(1) | | | Stock Awards ($)(2) | | | All Other Compensation | | | Total ($) |
Ayub Khattak Chief Executive Officer | | | 2020 | | | $276,923(3) | | | $250,000 | | | $6,930,773 | | | $8,694(4) | | | $7,466,390 |
Clint Sever Chief Product Officer | | | 2020 | | | $243,235(5) | | | $233,331 | | | $3,465,387 | | | $257,155(6) | | | $4,199,108 |
| (1) | Except where noted otherwise, the amounts reported in the “Bonus” column reflect discretionary annual cash bonuses earned by each of our named executive officers for their performance, as determined by the board of directors in its sole discretion. |
| (2) | The amounts noted above relate to the purchase of shares of common stock in exchange for promissory notes issued by each of Mr. Khattak and Mr. Sever to us, which notes are partially personally recourse and secured by the shares of common stock purchased therewith. Pursuant to ASC 718, these instruments are treated as grants of stock options for accounting purposes and the amount disclosed is the grant date fair value of these instruments. The assumptions used in calculating the grant date fair value of these instruments are set forth in Note 13 to the audited financial statements included elsewhere in this prospectus. |
| (3) | The amount noted above reflects a $150,000 increase in Mr. Khattak’s annual base salary, which took effect as of August 20, 2020. |
| (4) | The amount noted above consists of premiums for medical, vision, dental, and life insurance paid for by us. |
| (5) | The amount noted above reflects a $145,000 increase in Mr. Sever’s annual base salary, which took effect August 20, 2020. |
| (6) | The amount noted above consists of compensation resulting from forgiveness of indebtedness of $246,142 and premiums for medical, dental, and life insurance paid for by us. |
| | | Option Awards | | | Stock Awards | |||||||||||||
Name | | | Number of securities underlying unexercised options (#) exercisable | | | Number of securities underlying unexercised options (#) unexercisable(1) | | | Option exercise price ($) | | | Option expiration date | | | Number of shares of stock that have not vested (#)(2) | | | Market value of shares of stock that have not vested ($)(3) |
Ayub Khattak | | | 295,900 | | | — | | | 0.40 | | | 07/29/2024 | | | 305,517(6) | | | $ |
| | | | | | | | | | | 2,099,304(7) | | | $ | |||||
Clint Sever | | | 729,166 | | | 104,167(4) | | | 0.48 | | | 08/07/2028 | | | 55,000(8) | | | $ |
| | | 989,447 | | | 141,350(5) | | | 0.48 | | | 08/07/2028 | | | 1,075,253(9) | | | $ | |
| | | 295,900 | | | — | | | 0.40 | | | 07/29/2024 | | | | | $ | ||
| | | 880,000 | | | — | | | 0.20 | | | 12/31/2022 | | | | | $ | ||
| | | 400,000(10) | | | — | | | 0.20 | | | 07/11/2021 | | | | | $ | ||
| (1) | Of the unvested options reflected in this table, options are expected to have vested prior to completion of this offering in accordance with their terms. On May 26, 2021, our board of directors approved the accelerated vesting of any remaining unvested options held by Messrs. Khattak and Sever upon the effectiveness of an initial public offering satisfying certain conditions, which conditions may be satisfied in connection with this offering. Accordingly, the remaining unvested options included in this table may vest in connection with the completion of this offering. |
| (2) | Of the unvested shares reflected in this table, shares are expected to have vested prior to completion of this offering in accordance with their terms. On May 26, 2021, our board of directors approved the accelerated vesting of any remaining unvested restricted stock awards held by Messrs. Khattak and Sever upon the effectiveness of an initial public offering satisfying certain conditions, which conditions may be satisfied in connection with this offering. Accordingly, the remaining unvested shares included in this table may vest in connection with the completion of this offering. |
| (3) | The market price of common stock is based on an assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover page of this prospectus. |
| (4) | These options were granted on August 8, 2018 and vest over four years in equal monthly installments, subject to continuous service. On December 29, 2020, our board of directors approved accelerated vesting of 50% of the unvested options as of January 1, 2021, effective as of December 29, 2020. The share numbers in the table reflect this acceleration. |
| (5) | These options were granted on August 8, 2018 and vest over four years, with 25% of the shares having vested on December 31, 2018, and the remainder vesting in equal monthly installments thereafter, subject to continuous service. On December 29, 2020, our board of directors approved accelerated vesting of 50% of the unvested options as of January 1, 2021, effective as of December 29, 2020. The share numbers in the table reflect this acceleration. |
| (6) | These restricted stock awards were granted on August 8, 2018 and vest over four years, with 25% of the shares having vested on December 31, 2018, and the remainder vesting in equal monthly installments thereafter, subject to continuing service. On December 29, 2020, our board of directors approved accelerated vesting of 50% of the unvested shares as of January 1, 2021, effective as of December 29, 2020. The share numbers in the table reflect this acceleration. |
| (7) | These restricted stock awards were granted on July 24, 2020 and vest over four years in equal monthly installments, subject to continuing service. On December 29, 2020, our board of directors approved accelerated vesting of 50% of the unvested shares as of January 1, 2021, effective as of December 29, 2020. The share numbers in the table reflect this acceleration. |
| (8) | These restricted stock awards were granted on August 8, 2018 and vest over four years, with 25% of the shares having vested on |
| (9) | These restricted stock awards were granted on July 24, 2020 and vest over four years in equal monthly installments, subject to continuing service. On December 29, 2020, our board of directors approved accelerated vesting of 50% of the unvested shares as of January 1, 2021, effective as of December 29, 2020. The share numbers in the table reflect this acceleration. |
| (10) | On July 8, 2021, our board of directors approved an amendment to this stock option to permit the option to be “net exercised” with respect to the payment of the exercise price and applicable tax withholding. |
| | | Number of Stock Price Target RSUs Eligible to Vest | ||||
Price Goal | | | Khattak | | | Sever |
$30.07 | | | 17,615 | | | 123,140 |
$37.13 | | | 292,833 | | | 239,591 |
$45.86 | | | 292,833 | | | 239,591 |
$56.63 | | | 292,833 | | | 239,591 |
$69.94 | | | 292,833 | | | 239,591 |
$86.38 | | | 292,833 | | | 239,591 |
$106.68 | | | 292,834 | | | 239,591 |
| • | 292,833 of the Performance-Vesting RSUs granted to Mr. Khattak and 239,591 of the Performance-Vesting RSUs granted to Mr. Sever, or the FY21 Revenue Target RSUs, are eligible to vest based on the achievement of a specified level of our total revenue (as reported under U.S. generally accepted accounting principles on our financial statement), which we refer to as total revenue, for fiscal year 2021 or, if such total revenue for fiscal year 2021 is not achieved, based on the achievement of a specified level of our aggregate total revenue for fiscal years 2021 and 2022. |
| • | 292,833 of the Performance-Vesting RSUs granted to Mr. Khattak and 239,591 of the Performance-Vesting RSUs granted to Mr. Sever, or the FY22 Revenue Target RSUs, are eligible to vest based on the achievement of a specified level of our total revenue for fiscal year 2022 or, if such total revenue for fiscal year 2022 is not achieved, based on the achievement of a specified level of our aggregate total revenue for fiscal years 2022 and 2023. |
| • | 292,834 of the Performance-Vesting RSUs granted to Mr. Khattak and 239,591 of the Performance-Vesting RSUs granted to Mr. Sever, or the Milestone Target RSUs, are eligible to vest based upon the achievement of a specified product milestone by December 31, 2022. If such milestone is not achieved on or before December 31, 2022 but is achieved during the six-month period beginning on January 1, 2023 and ending on June 30, 2023, 50% of the Milestone Target RSUs will be eligible to vest. |
| • | 50% of the Stock Price Target RSUs with respect to any price goal that has not been achieved as of the termination date will be retained by the Founder and may be earned after the termination date as follows: (1) if a price goal is achieved within six months of the termination date, 100% of the retained Stock Price |
| • | 100% of the FY21 Revenue Target RSUs, to the extent then outstanding, will be retained by the Founder and may be earned after the termination date as follows: (1) if the applicable total revenue is achieved within 12 months of the termination date, 100% of the FY21 Revenue Target RSUs will be earned and (2) if the applicable total revenue is achieved more than 12 months following the termination date, 50% of the FY21 Revenue Target RSUs will be earned; |
| • | 100% of the FY22 Revenue Target RSUs, to the extent then outstanding, will be retained by the Founder and may be earned after the termination date as follows: (1) if the applicable total revenue is achieved within 12 months of the termination date, 100% of the FY22 Revenue Target RSUs will be earned and (2) if the applicable total revenue is achieved more than 12 months following the termination date, 50% of the FY22 Revenue Target RSUs will be earned; and |
| • | 100% of the Milestone Target RSUs, to the extent then outstanding, will be retained by the Founder and may be earned after the termination date as follows: (1) if the milestone performance goal is achieved within 12 months of the termination date and on or before December 31, 2022, 100% of the Milestone Target RSUs will be earned, (2) if such goal is achieved within 12 months of the termination date and between January 1, 2023 and June 30, 2023, 50% of the Milestone Target RSUs will be earned, (3) if such goal is achieved more than 12 months following the termination date and on or before December 31, 2022, 50% of the Milestone Target RSUs will be earned, and (4) if such goal is achieved more than 12 months following the termination date and between January 1, 2023 and June 30, 2023, 25% of the Milestone Target RSUs will be earned. |
| • | Our compensation committee will determine whether any price goals that have not previously been achieved are achieved as a result of the change in control, which determination will be based solely on the price to be paid to our stockholders in connection with the transaction, and the Stock Price Target RSUs with respect to any price goal that is achieved as a result of the change in control will vest immediately prior to the closing of the change in control. |
| • | 50% of any Stock Price Target RSUs have not been earned by the Founder prior to the change in control, taking into account any Stock Price Target RSUs that are earned based on the price paid to our stockholders in the change in control as described above, will be retained by the Founder and converted into time-vested RSUs that vest in equal quarterly installments over the two-year period following the closing of the change in control, subject to the Founder’s continued employment on each vesting date. If the Founder’s employment is terminated by us without cause or by the Founder for good reason during the period beginning three months before the change in control (or, in the event we have executed a definitive agreement to effect the change in control as of the termination date, the period beginning six months before such change in control) and ending 24 months following the change in control, the retained Stock Price Target RSUs will vest in full as of the date of termination. If the retained Stock Price Target RSUs are not assumed (or substituted for substantially equivalent awards) by the resulting or acquiring company, the retained Stock Price Target RSUs will vest in full immediately prior to the change in control. |
| • | Any FY21 Revenue Target RSUs, FY22 Revenue Target RSUs, or Milestone Target RSUs outstanding immediately prior to the change in control will be eligible to vest immediately prior to the closing of the |
| • | the participants to receive awards; |
| • | the type or types of awards to be granted to each participant; |
| • | the number of shares of common stock with respect to which an award relates; |
| • | the terms and conditions of any award. |
| • | the number and type of shares of common stock subject to the 2014 Plan, including the number and type of shares of common stock that may be issued pursuant to incentive stock options; |
| • | the number and types of shares of common stock subject to outstanding awards; |
| • | the grant, purchase, or exercise price with respect to any award; and |
| • | the performance goals established under any award. |
| • | in the case of an option or stock appreciation right, the excess of the fair market value as determined by our board of directors of the shares of common stock on the date of the change of control covered by the vested portion of the option or stock appreciation right that has not been exercised over the exercise or grant price of such shares under the award (provided that, if such fair market value does not exceed the exercise or grant price, the option or stock appreciation rate will be cancelled for no consideration); |
| • | in the case of restricted stock and restricted stock units, the fair market value of a share on the date of the change of control multiplied by the number of vested shares or units; and |
| • | in the case of performance shares, the fair market value of a share on the date of the change of control multiplied by the number of earned shares. |
| • | the number of shares of our common stock covered by options and the dates upon which the options become exercisable; |
| • | the type of options to be granted; |
| • | the duration of options, which may not be in excess of ten years; |
| • | the exercise price of options, which must be at least equal to the fair market value of our common stock on the date of grant; and |
| • | the number of shares of our common stock subject to and the terms of any stock appreciation rights, restricted stock awards, restricted stock units or other stock-based awards and the terms and conditions of such awards, including conditions for repurchase, issue price and repurchase price (though the measurement price of stock appreciation rights must be at least equal to the fair market value of our common stock on the date of grant and the duration of such awards may not be in excess of ten years). |
| • | the number and class of securities available under the 2021 Plan, and the number and class of securities available for issuance under the 2021 Plan that may be issued as incentive stock options; |
| • | the share counting rules of the 2021 Plan; |
| • | the number and class of securities and exercise price per share of each outstanding option; |
| • | the share and per-share provisions and the measurement price of each outstanding stock appreciation right; |
| • | the number of shares subject to, and the repurchase price per share subject to, each outstanding award of restricted stock; and |
| • | the share and per-share related provisions and the purchase price, if any, of each outstanding award of restricted stock units and each outstanding other stock-based award. |
| • | provide that outstanding awards will be assumed, or substantially equivalent awards will be substituted, by the acquiring or succeeding corporation (or an affiliate of the acquiring or succeeding corporation); |
| • | upon written notice to a participant, provide that all of the participant's unvested awards will be forfeited immediately prior to the consummation of the reorganization event, and/or that all of the participant’s vested but unexercised awards will terminate immediately prior to the consummation of the reorganization event unless exercised by the participant (to the extent then exercisable) within a specified period following the date of the notice; |
| • | provide that outstanding awards will become exercisable, realizable or deliverable, or restrictions applicable to an award will lapse, in whole or in part, prior to or upon such reorganization event; |
| • | in the event of a reorganization event pursuant to which holders of shares of our common stock will receive a cash payment for each share surrendered in the reorganization event, make or provide for a cash payment to participants with respect to each award held by a participant equal to (1) the number of shares of our common stock subject to the vested portion of the award (after giving effect to any acceleration of vesting that occurs upon or immediately prior to such reorganization event) multiplied by (2) the excess, if any, of the cash payment for each share surrendered in the reorganization event over the exercise, measurement or purchase price of such award and any applicable tax withholdings, in exchange for the termination of such award; and/or |
| • | provide that, in connection with our liquidation or dissolution, awards will convert into the right to receive liquidation proceeds (if applicable, net of the exercise, measurement or purchase price thereof and any applicable tax withholdings). |
| • | amend any outstanding stock option or stock appreciation right granted under the 2021 Plan to provide an exercise or measurement price per share that is lower than the then-current exercise or measurement price per share of such outstanding award; |
| • | cancel any outstanding stock option or stock appreciation right (whether or not granted under the 2021 Plan) and grant a new award under the 2021 Plan in substitution for the cancelled award (other than substitute awards permitted in connection with a merger or consolidation of an entity with us or our acquisition of property or stock of another entity) covering the same or a different number of shares of our common stock and having an exercise or measurement price per share lower than the then-current exercise or measurement price per share of the cancelled award; |
| • | cancel in exchange for a cash payment any outstanding option or stock appreciation right with an exercise or measurement price per share above the then-current fair market value of our common stock (valued in the manner determined by (or in the manner approved by) our board of directors); or |
| • | take any other action that constitutes a “repricing” within the meaning of Nasdaq rules or rules of any other exchange or marketplace on which our common stock is listed or traded. |
| • | such person is customarily employed by us or a designated subsidiary for more than 20 hours a week and for more than five months in a calendar year; |
| • | such person has been employed by us or by a designated subsidiary for at least three months prior to enrolling in the 2021 ESPP; and |
| • | such person was our employee or an employee of a designated subsidiary on the first day of the applicable offering period under the 2021 ESPP. |
| • | provide that options will be assumed, or substantially equivalent options will be substituted, by the acquiring or succeeding corporation (or an affiliate of the acquiring or succeeding corporation); |
| • | upon written notice to employees, provide that all outstanding options will be terminated immediately prior to the consummation of such reorganization event and that all such outstanding options will become exercisable to the extent of accumulated payroll deductions as of a date specified by our board of directors or committee thereof in such notice, which date will not be less than ten days preceding the effective date of the reorganization event; |
| • | upon written notice to employees, provide that all outstanding options will be cancelled as of a date prior to the effective date of the reorganization event and that all accumulated payroll deductions will be returned to participating employees on such date; and/or |
| • | in the event of a reorganization event under the terms of which holders of our common stock will receive upon consummation thereof a cash payment for each share surrendered in the reorganization event, change the last day of the offering period to be the date of the consummation of the reorganization event and make or provide for a cash payment to each employee equal to (1) the cash payment for each share surrendered in the reorganization event times the number of shares of our common stock that the employee's accumulated payroll deductions as of immediately prior to the reorganization event could purchase at the applicable purchase price, where the cash payment for each share surrendered in the reorganization event is treated as the fair market value of our common stock on the last day of the applicable offering period for purposes of determining the purchase price and where the number of shares that could be purchased is subject to the applicable limitations under the 2021 ESPP minus (2) the result of multiplying such number of shares by the purchase price; and/or provide that, in connection with our liquidation or dissolution, options will convert into the right to receive liquidation proceeds (net of the purchase price thereof). |
| • | for any breach of the director’s duty of loyalty to us or our stockholders; |
| • | for acts or omissions not in good faith or that involve intentional misconduct or a knowing violation of law; |
| • | for voting for or assenting to unlawful payments of dividends, stock repurchases or other distributions; or |
| • | for any transaction from which the director derived an improper personal benefit. |
Name | | | Fees earned or paid in cash ($) | | | Stock Awards ($) | | | Option Awards ($) | | | All Other Compensation ($) | | | Total ($) |
Chris Achar(1) | | | — | | | — | | | — | | | — | | | — |
Xiangmin “Min” Cui | | | — | | | — | | | — | | | — | | | — |
Robin Farias-Eisner(2) | | | — | | | — | | | — | | | — | | | — |
Rohan Oza(3) | | | — | | | — | | | — | | | — | | | — |
Scott Stanford | | | — | | | — | | | — | | | — | | | — |
Asish Xavier(4) | | | — | | | — | | | — | | | — | | | — |
| (1) | In July 2021, Mr. Achar entered into an employment agreement with the Company, at which time he became an employee director of the Company. |
| (2) | Dr. Farias-Eisner intends to resign from our board of directors immediately prior to the effectiveness of the registration statement of which this prospectus forms a part. |
| (3) | Mr. Oza intends to resign from our board of directors immediately prior to the effectiveness of the registration statement of which this prospectus forms a part. |
| (4) | Dr. Xavier resigned from our board of directors in April 2021. |
| | | Member Annual Fee | | | Chairman Annual Fee | |
Board of Directors | | | $50,000 | | | $95,000 |
Audit Committee | | | $8,000 | | | $20,000 |
Compensation Committee | | | $5,000 | | | $12,000 |
Nominating and Corporate Governance Committee | | | $4,000 | | | $10,000 |
Purchaser(1) | | | Principal Amount of Convertible Notes |
Decheng Capital China Life Sciences USD Fund III, L.P.(2) | | | $10,000,000 |
Funds managed by ACME, LLC and affiliates(3) | | | 4,696,970 |
JJDC(4) | | | 7,000,000 |
| (1) | See “Principal Stockholders” for additional information about shares held by these entities. |
| (2) | Xiangmin “Min” Cui, a member of our board of directors, is the managing director and founder of Decheng Capital China Life Sciences USD Fund III, L.P |
| (3) | Funds managed by ACME, LLC and affiliates are Sherpa Ventures Fund, LP and Sherpa Ventures Fund II, LP, collectively “ACME Capital.’’ Scott Stanford, a member of our board of directors, is a member of, and has a financial interest in ACME Capital. |
| (4) | Vijay Murthy, a former member of our board of directors, is a former principal at JJDC, and Asish Xavier, a former member of our board of directors, is a principal of JJDC. |
Purchaser(1) | | | Series C-1 Redeemable Convertible Preferred Stock Sold for Cash | | | Cash Purchase Price | | | Series C-2 Redeemable Convertible Preferred Stock Exchange for Convertible Notes | | | Principal Amount of Convertible Notes Cancelled Upon Exchange |
Decheng Capital China Life Sciences USD Fund III, L.P.(2) | | | 8,192,468 | | | $30.0 | | | — | | | $— |
Madrone Opportunity Fund, L.P. | | | 5,461,645 | | | 20.0 | | | — | | | — |
Purchaser(1) | | | Series C-1 Redeemable Convertible Preferred Stock Sold for Cash | | | Cash Purchase Price | | | Series C-2 Redeemable Convertible Preferred Stock Exchange for Convertible Notes | | | Principal Amount of Convertible Notes Cancelled Upon Exchange |
ACME Capital(3) | | | 2,184,658 | | | 8.0 | | | — | | | — |
Entities affiliated with Cove Investors I, LLC(4) | | | 273,082 | | | 1.0 | | | — | | | — |
JJDC(5) | | | 1,042,136 | | | 3.8 | | | 155,571 | | | 0.5 |
| (1) | See “Principal Stockholders” for additional information about shares held by these entities. |
| (2) | Xiangmin “Min” Cui, a member of our board of directors, is the managing director and founder of Decheng Capital China Life Sciences USD Fund III, L.P |
| (3) | Scott Stanford, a member of our board of directors, is a member of, and has a financial interest in ACME Capital. |
| (4) | Robin Farias-Eisner, a member of our board of directors, is an affiliate of Cove Investors I, LP. Dr. Farias-Eisner intends to resign from our board of directors immediately prior to the effectiveness of the registration statement of which this prospectus forms a part. |
| (5) | Vijay Murthy, a former member of our board of directors, is a former principal of JJDC, and Ashish Xavier, a former member of our board of directors, is a principal of JJDC. |
| • | the related person’s interest in the related person transaction; |
| • | the approximate dollar value of the amount involved in the related person transaction; |
| • | the approximate dollar value of the amount of the related person’s interest in the transaction without regard to the amount of any profit or loss; |
| • | whether the transaction was undertaken in the ordinary course of our business; |
| • | whether the terms of the transaction are no less favorable to us than terms that could have been reached with an unrelated third party; |
| • | the purpose, and the potential benefits to us, of the transaction; and |
| • | any other information regarding the related person transaction or the related person in the context of the proposed transaction that would be material to investors in light of the circumstances of the particular transaction. |
| • | interests arising solely from the related person’s position as an executive officer of another entity, whether or not the person is also a director of such entity, that is a participant in the transaction where the related person and all other related persons own in the aggregate less than a 10% equity interest in such entity, the |
| • | a transaction that is specifically contemplated by provisions of our amended and restated certificate of incorporation or bylaws. |
| • | each of our directors; |
| • | each of our named executive officers; |
| • | all of our directors and executive officers as a group; and |
| • | each person, or group of affiliated persons, who is known by us to beneficially own more than 5% of our common stock. |
Name of Beneficial Owner | | | Number of Shares Beneficially Owned | | | Percentage of Shares Beneficially Owned | |||
| | Before Offering (%) | | | After Offering (%) | |||||
5% Stockholders | | | | | | | |||
ACME Capital(1) | | | 14,869,253 | | | 13.20% | | | |
Entities affiliated with Cove Investors I, LLC(2) | | | 12,377,254 | | | 10.99% | | | |
Decheng Capital China Life Sciences USD Fund III, L.P.(3) | | | 8,192,468 | | | 7.27% | | | |
Madrone Opportunity Fund, L.P.(4) | | | 7,078,566 | | | 6.28% | | | |
NVGA I, LLC(5) | | | 6,843,692 | | | 6.08% | | | |
| | | | | | | ||||
Directors, Director Nominees and Named Executive Officers | | | | | | | |||
Ayub Khattak(6) | | | 10,907,055 | | | 9.66% | | | |
Clint Sever(7) | | | 5,728,151 | | | 5.00% | | | |
Chris Achar(8) | | | 1,589,710 | | | 1.41% | | | |
Xiangmin “Min” Cui(3) | | | 8,192,468 | | | 7.27% | | | |
Robin Farias-Eisner(2)(9) | | | 12,421,254 | | | 11.03% | | | |
Rohan Oza(10) | | | 1,104,612 | | | * | | | |
Scott Sanford(1) | | | 14,869,253 | | | 13.20% | | | |
Joanne Bradford | | | — | | | — | | | |
Carole Faig | | | — | | | — | | | |
Maria Martinez | | | — | | | — | | | |
All current executive officers and directors as a group (10 persons) | | | 51,672,073 | | | 45.75% | | | |
| * | Denotes less than 1%. |
| (1) | Consists of (i) 129,354 shares of common stock held by Sherpa Ventures Fund, LP (“ACME I”), (ii) 194,031 shares of common stock held by Sherpa Ventures Fund II, LP (“ACME II”), (iii) 5,450,898 shares of common stock issuable upon conversion of our Series A redeemable convertible preferred stock held by ACME I, (iv) 3,076,224 shares of common stock issuable upon conversion of our shares of Series B redeemable convertible preferred stock held by ACME I, (v) 3,834,088 shares of common stock issuable upon conversion of our Series B redeemable convertible preferred stock held by ACME II, (vi) 1,092,329 shares of common stock issuable upon conversion of our Series C-1 redeemable convertible preferred stock held by ACME I, (vii) 1,092,329 shares of common stock issuable upon conversion of our Series C-1 redeemable convertible preferred stock held by ACME II, and (viii) shares of common stock issuable upon conversion of our Convertible Notes, based on accrued interest through 2021, and a 20% discount to the assumed initial public offering price of $ per share, which is the midpoint of the range set forth on the cover of this prospectus, held by ACME II. Sherpa Ventures Fund GP, LLC (“ACME GP I”) is the manager of ACME I. SherpaVentures Fund II GP, LLC (“ACME GP II”) is the manager of ACME II. Mr. Stanford is a managing member of each of ACME GP I and ACME GP II and may be deemed to have voting and investment power with respect to the shares held by ACME I and ACME II and as a result may be deemed to have beneficial ownership of such shares. Funds managed by ACME, LLC and affiliates of ACME I and ACME II, are collectively defined as “ACME Capital”. Scott Stanford is also a member of our board of directors and a member of, and has a financial interest in, ACME Capital. The address for ACME I and ACME II is 800 Market Street, 8th Floor, San Francisco, California 94102. |
| (2) | Consists of (i) 5,655,540 shares of common stock held by Cove Investors I, LLC (“Cove I”), (ii) 1,090,180 shares of common stock issuable upon conversion of our Series A redeemable convertible preferred stock held by Cove Investors I, LLC, (iii) 5,358,452 shares of common stock issuable upon conversion of our Series B redeemable convertible preferred stock held by Cove Investors II, LLC (“Cove II”) and (iv) 273,082 shares of common stock issuable upon conversion of our Series C-1 redeemable convertible preferred stock held by Cove Investors II, LLC. Dr. Farias-Eisner is a member of Cove I and Cove II. Dr. Farias-Eisner is also currently a member of our board of directors and intends to resign from our board of directors immediately prior to the effectiveness of the registration statement of which this prospectus forms a part. The address for Cove I and Cove II is 865 S. Figueroa Street, Suite 700, Los Angeles, California 90017. |
| (3) | Consists of (i) 8,192,468 shares of common stock issuable upon conversion of our Series C-1 redeemable convertible preferred stock held by Decheng Capital China Life Sciences USD Fund III, L.P. (“Decheng Fund III”) and (ii) shares of common stock issuable upon conversion of our Convertible Notes, based on accrued interest through 2021, and a 20% discount to the assumed initial public offering price of $ per share, which is the midpoint of the range set forth on the cover of this prospectus, held by Decheng Capital Global Healthcare Fund (Master), LP (“Decheng Global”). Decheng Capital Management III (Cayman), LLC (“Decheng Capital Management”) is the general partner of Decheng Fund III, and Decheng Capital Global Healthcare GP, LLC (“Decheng Global GP”) is the general partner of Decheng Global. Dr. Cui is the sole manager of Decheng Capital Management and Decheng Global GP. Dr. Cui may be deemed to have voting and investment power with respect to the shares held by Decheng Fund III and Decheng Global and as a result may be deemed to have beneficial ownership of such shares. Dr. Cui is also a member of our board of directors. The address for Decheng is 3000 Sand Hill Road, Building 2, Suite 110, Menlo Park, California 94025. |
| (4) | Consists of (i) 1,616,921 shares of common stock held by Madrone Opportunity Fund, L.P. (“Madrone”) and (ii) 5,461,645 shares of common stock issuable upon conversion of our Series C-1 redeemable convertible preferred stock held by Madrone. Madrone Capital Partners, LLC (“Madrone Capital”) is the general partner of Madrone. Thomas Patterson, Jameson McJunkin and Gregory Penner are the managers of Madrone Capital and each may be deemed to have voting and investment power with respect to the shares held by Madrone and as a result may be deemed to have beneficial ownership of such shares. The address for Madrone is 1149 Chestnut St, Suite 200, Menlo Park, California 94025. |
| (5) | Consists of 6,843,692 shares of common stock issuable upon conversion of our Series B redeemable convertible preferred stock held by NVGA I, LLC (“NVGA”). TI Manager, LLC (“TI Manager”) is the manager of NVGA. TI Manager is managed by its sole member Tarsadia Enterprises, LLC (“Enterprises”). Tushar Patel is the ultimate indirect beneficial owner of Enterprises and may be deemed to have voting and investment power with respect to the shares held by NVGA and as a result may be deemed to have beneficial ownership of such shares. The address for NVGA is c/o Tarsadia Enterprises, LLC, 520 Newport Center Dr., 21st Floor, Newport Beach, CA 92660. |
| (6) | Consists of 10,611,155 shares of our common stock and options to purchase 295,900 shares of our common stock that are exercisable within 60 days of July 31, 2021. |
| (7) | Consists of (i) 187,017 shares of common stock jointly held by Mr. Sever and his spouse and (ii) 3,140,000 shares of our common stock that are exercisable within 60 days of July 31, 2021. |
| (8) | Consists of (i) 69,710 shares of common stock issuable upon conversion of our Series B redeemable convertible preferred stock held by Mr. Achar and (ii) 1,488,333 shares of our common stock and 31,667 shares of restricted common stock held by Hlth Wrk LLC as of 60 days from July 31, 2021, due to Mr. Achar’s early exercise of his options to purchase shares of our common stock. Mr. Achar is the sole manager of Hlth Wrk LLC and may be deemed to have voting and investment power with respect to the shares held by Hlth Wrk LLC and as a result may be deemed to have beneficial ownership of such shares. |
| (9) | Consists of (i) 16,000 shares of our common stock, and (ii) 28,000 shares of our common stock held in trust by the Robin Farias-Eisner and Therese Farias-Eisner joint trust with rights of survival, or the Farias-Eisner Trust. Dr. Farias-Eisner is the trustee of the Farias-Eisner Trust and may be deemed to indirectly beneficially own such shares. Dr. Farias-Eisner intends to resign from our board of directors immediately prior to the effectiveness of the registration statement of which this prospectus forms a part. |
| (10) | Consists of (i) 280,681 shares of common stock issuable upon conversion of our Series B redeemable convertible preferred stock held by Mr. Oza, (ii) 545,089 shares of common stock issuable upon conversion of our Series A redeemable convertible preferred stock held by RONO, LLC (“RONO”), (iii) 278,842 shares of common stock issuable upon conversion of our Series B redeemable convertible preferred stock held by RONO and (iv) shares of common stock issuable upon conversion of our Convertible Notes, based on accrued interest through 2021, and a 20% discount to the assumed initial public offering price of $ per share, which is the midpoint of the range set forth on the cover of this prospectus, held by Cavu Venture Partners III, L.P. (“Cavu”). Mr. Oza is the managing member of RONO and may be deemed to have voting and investment power with respect to the shares held by RONO and as a result may be deemed to have beneficial ownership of such shares. Mr. Oza is a managing member and co-founder of Cavu and may be deemed to have voting and investment power with respect to the shares held by Cavu and as a result may be deemed to have beneficial ownership of such shares. Mr. Oza intends to resign from our board of directors immediately prior to the effectiveness of the registration statement of which this prospectus forms a part. |
| • | 29,128,604 shares of common stock held by 60 stockholders of record; |
| • | 8,350,743 shares of our Series A redeemable convertible preferred stock held by 22 stockholders of record, convertible into 8,350,743 shares of our common stock; |
| • | 46,176,715 shares of our Series B redeemable convertible preferred stock held by 42 stockholders of record, convertible into 46,176,715 shares of our common stock; |
| • | 27,308,227 shares of our Series C-1 redeemable convertible preferred stock held by 22 stockholders of record, convertible into 27,308,227 shares of our common stock; and |
| • | 1,690,380 shares of our Series C-2 redeemable convertible preferred stock held by 5 stockholders of record, convertible into 1,690,380 shares of our common stock. |
| • | 1% of the number of shares of our common stock then outstanding, which will equal approximately shares immediately after this offering; and |
| • | the average weekly trading volume in our common stock on the Nasdaq Global Stock Market during the four calendar weeks preceding the date of filing of a Notice of Proposed Sale of Securities Pursuant to Rule 144 with respect to the sale. |
| • | offer, pledge, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, lend, or otherwise transfer or dispose of, directly or indirectly, any shares of our common stock beneficially owned (as such term is used in Rule 13d-3 of the Exchange Act) or any other securities so owned convertible into or exercisable or exchangeable for common stock, or make any public announcement of an intention to do any of the foregoing; or |
| • | enter into any hedging, swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of our common stock. |
| • | an individual who is a citizen or resident of the United States; |
| • | a corporation, or other entity treated as a corporation for U.S. federal income tax purposes, created or organized in or under the laws of the United States, any state thereof or the District of Columbia; |
| • | an estate the income of which is subject to U.S. federal income taxation regardless of its source; or |
| • | a trust, if a U.S. court is able to exercise primary supervision over the administration of the trust and one or more U.S. persons has authority to control all substantial decisions of the trust or if the trust has a valid election in effect to be treated as a U.S. person under applicable U.S. Treasury Regulations. |
| • | insurance companies; |
| • | tax-exempt organizations or governmental organizations; |
| • | financial institutions; |
| • | brokers or dealers in securities; |
| • | pension plans; |
| • | controlled foreign corporations; |
| • | passive foreign investment companies; |
| • | corporations that accumulate earnings to avoid U.S. federal income tax; |
| • | “qualified foreign pension funds” as defined in Section 897(1)(2) of the Code and entities of all of the interests of which are held by qualified foreign pension funds; |
| • | persons that own, or are deemed to own, more than 5% if our capital stock; |
| • | owners that hold our common stock as part of a straddle, hedge, conversion transaction, synthetic security, or other integrated investment; and |
| • | certain U.S. expatriates and former citizens or long-term residents of the United States. |
| • | the gain is effectively connected with the non-U.S. holder’s conduct of a trade or business in the United States and, if an applicable income tax treaty so provides, the gain is attributable to a permanent establishment or fixed base maintained by the non-U.S. holder in the United States; in these cases, the non-U.S. holder will be taxed on a net income basis at the same U.S. federal income tax rates applicable to United States persons (as defined in the Code), and if the non-U.S. holder is a foreign corporation, an additional branch profits tax at a 30% rate, or such lower rate as may be specified by an applicable income tax treaty, may also apply; |
| • | the non-U.S. holder is a nonresident alien present in the United States for 183 days or more in the taxable year of the disposition and certain other conditions are met, in which case the non-U.S. holder will be subject to a 30% tax (or such lower rate as may be specified by an applicable income tax treaty) on the net gain derived from the disposition, which may be offset by U.S.-source capital losses of the non-U.S. holder provided the non-U.S. holder has timely filed U.S. federal income tax returns with respect to such losses, if any; or |
| • | we are, or have been at any time during the five-year period preceding such disposition (or the non-U.S. holder’s holding period, if shorter), a “U.S. real property holding corporation,” unless our common stock is regularly traded on an established securities market and the non-U.S. holder held no more than 5% of our outstanding common stock, directly or indirectly, during the shorter of the five year period ending on the date of the disposition or the period that the non-U.S. holder held our common stock. If we are determined to be a U.S. real property holding corporation and the foregoing exception does not apply, then the non-U.S. holder generally will be taxed on its net gain derived from the disposition generally in the same manner as gain that is effectively connected with the conduct of a trade or business in the United States, at the U.S. federal income tax rates applicable to United States persons (as defined in the Code), except that the branch profits tax generally will not apply. Generally, a corporation is a “U.S. real property holding corporation” if the fair market value of its “U.S. real property interests” equals or exceeds 50% of the sum of the fair market value of its worldwide real property interests plus its other assets used or held for use in a trade or business. Although there can be no assurance, we believe that we are not currently, and we do not anticipate becoming, a “U.S. real property holding corporation” for U.S. federal income tax purposes. No assurance can be provided that our common stock will be regularly traded on an established securities market for purposes of the rule described above. |
Underwriters | | | Number of Shares |
Goldman Sachs & Co. LLC | | | |
Morgan Stanley & Co. LLC | | | |
Cowen and Company, LLC | | | |
BTIG, LLC | | | |
Total | | |
| | | No Exercise | | | Full Exercise | |
Per Share | | | $ | | | $ |
Total | | | $ | | | $ |
| (a) | as a bona fide gift or gifts; |
| (b) | to any member of the securityholder’s immediate family or to any trust for the direct or indirect benefit of the securityholder or the immediate family of the securityholder; |
| (c) | by will or other testamentary document or by intestacy; |
| (d) | pursuant to a court order or settlement or other domestic order related to the distribution of assets in connection with the dissolution of a marriage or civil union; |
| (e) | to general or limited partners, members, stockholders, other equity holders or trust beneficiaries of the securityholder or to any investment fund or other entity that controls or manages or serves as investment adviser to, or is under common control or management or shares a common investment adviser with, the securityholder; |
| (f) | in connection with any common stock acquired in this offering (other than any issuer directed shares of common stock purchased in this offering by our officer or director) acquired in open market transactions after the completion of this offering; |
| (g) | to us in connection with the “net” or “cashless” exercise or settlement solely to cover the exercise price and applicable withholding tax obligations in connection with the exercise or settlement of such warrants or stock options, restricted stock units or other equity awards expiring during the restricted period, in each case pursuant to a stock incentive plan, other equity award plan or warrant described in this prospectus (and any transfer to us necessary to generate such amount of cash needed for the payment of withholding tax obligations, and/or payment of estimated taxes, due as a result of such vesting, settlement or exercise whether by means of a “net settlement” or otherwise), provided that if the securityholder is required to file a report reporting a reduction in beneficial ownership of shares of common stock during the restricted period, the securityholder shall clearly indicate in the footnotes thereto that the filing relates to the circumstances described in this clause and that the shares of common stock received upon exercise of the stock option or warrant or vesting event are subject to the lock-up agreement, and no public filing, report or announcement shall be voluntarily made; |
| (h) | pursuant to a bona fide third-party tender offer, merger, consolidation, business combination, stock purchase or other similar transaction or series of related transactions approved by our board of directors and made to all holders of our capital stock involving a change in control, provided that in the event that such tender offer, merger, consolidation, business combination, stock purchase or transaction or series of related transactions is not completed, the securityholder’s securities shall remain subject to the restrictions set forth in the lock-up agreement; |
| (i) | the conversion of outstanding shares of our preferred stock or other securities described in this prospectus and outstanding as of the date of this prospectus into shares of common stock or derivative instruments, as described in this prospectus, provided that the shares of common stock or any derivative instruments received upon conversion shall be subject to the restrictions set forth in the lock-up agreement; |
| (j) | to us pursuant to any contractual arrangement in effect on the date of the lock-up agreement and disclosed in this prospectus that provides for the repurchase of shares of common stock in connection with the termination of the securityholder’s employment with or service to us, provided no public filing, report or announcement reporting a reduction in beneficial ownership of shares of common stock shall be required or shall be voluntarily made during the restricted period within 75 days after the date the securityholder ceases to provide services to us, and after such 75th day, if the securityholder is required to file a report reporting a reduction in beneficial ownership of shares of common stock during the restricted period, the securityholder shall clearly indicate in the footnotes thereto that the filing relates to the circumstances described in this clause and no public filing, report or announcement shall be voluntarily made; |
| (k) | with the prior written consent of Goldman Sachs & Co. LLC and Morgan Stanley & Co. LLC on behalf of the underwriters; |
| (l) | if the securityholder is a corporation, partnership, limited liability company or other business entity, the corporation, partnership, limited liability company or other business entity may effect a transfer to any other corporation, partnership, limited liability company or other business entity that is an affiliate (as defined in Rule 405 promulgated under the Securities Act of 1933, as amended) of the securityholder; provided, however, that in any such case, it shall be a condition to the transfer that the transferee execute an agreement stating that the transferee is receiving and holding such capital stock subject to the provisions of the lock-up agreement and there shall be no further transfer of such capital stock except in accordance with the lock-up agreement, and provided further that any such transfer shall not involve a disposition for value and no public filing under the Exchange Act, or announcement shall be required or shall be made voluntarily; |
| (m) | the securityholder may receive shares of common stock from us in connection with (i) the exercise of options or other rights granted under a stock incentive plan or other equity award plan, limited only to a plan that is described in this prospectus and (ii) the exercise of warrants, which warrants are described in this prospectus; provided that, in each case, any shares of common stock issued upon exercise of such option, warrant or other rights shall continue to be subject to the restrictions set forth herein until the expiration of the restricted period; provided further, that if the securityholder is required to file a report under Section 16 of the Exchange Act reporting such exercise of options or other rights, the securityholder shall include a statement in such report to the effect that any shares of common stock issued upon exercise of such option or other rights remain subject to the restrictions set forth in the lock-up agreement, and provided further that no filing or other public announcement shall be voluntarily made; and |
| (n) | the securityholder may enter into any plan designed to satisfy the requirements of Rule 10b5-1 (a “10b5-1 Plan”) under the Exchange Act (other than the entry into such a plan in such a manner as to allow the sale of shares of common stock, in each case, within the restricted period); provided, however that, no sale of shares of common stock may be made under such 10b5-1 Plan during the restricted period; and provided further that no public filing, report or announcement regarding the establishment of such plan shall be required or shall be voluntarily made during the restricted period. |
| (a) | to any legal entity which is a qualified investor as defined under Article 2 of the Prospectus Regulation; |
| (b) | to fewer than 150 natural or legal persons (other than qualified investors as defined under Article 2 of the Prospectus Regulation), subject to obtaining the prior consent of the representatives for any such offer; or |
| (c) | in any other circumstances falling within Article 1(4) of the Prospectus Regulation, |
| (a) | to any legal entity which is a qualified investor as defined under Article 2 of the UK Prospectus Regulation; |
| (b) | to fewer than 150 natural or legal persons (other than qualified investors as defined under Article 2 of the UK Prospectus Regulation), subject to obtaining the prior consent of the representatives for any such offer; or |
| (c) | in any other circumstances falling within Section 86 of the FSMA. |
| | | ||
Audited Financial Statements as of and for the Years Ended December 31, 2019 and 2020 | | | |
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
Unaudited Interim Condensed Financial Statements for the Six Months Ended June 30, 2020 and 2021 | | | |
| | | ||
| | | ||
| | | ||
| | | ||
| | |
| | | December 31, | ||||
| | | 2019 | | | 2020 | |
Assets | | | | | ||
Current assets: | | | | | ||
Cash and cash equivalents | | | $14,328 | | | $121,578 |
Restricted cash | | | — | | | 6,000 |
Accounts receivable | | | 200 | | | 4,168 |
Inventory | | | — | | | 36,842 |
Prepaid expenses | | | 669 | | | 13,847 |
Other current assets | | | 307 | | | 1,263 |
Total current assets | | | 15,504 | | | 183,698 |
| | | | | |||
Restricted cash, non-current | | | 177 | | | 1,677 |
Property and equipment, net | | | 11,630 | | | 103,683 |
Prepaid rent | | | — | | | 16,771 |
Operating lease right-of-use assets | | | — | | | 8,281 |
Intangible assets, net | | | — | | | 2,038 |
Other non-current assets | | | 50 | | | 180 |
Total assets | | | $27,361 | | | $316,328 |
| | | | | |||
Liabilities, Redeemable Convertible Preferred Stock and Stockholders’ Deficit | | | | | ||
Current liabilities: | | | | | ||
Accounts payable | | | $1,168 | | | $23,847 |
Accrued liabilities | | | 566 | | | 8,822 |
Deferred revenue, current | | | 12 | | | 115,747 |
Deferred rent, current | | | 28 | | | — |
Debt, current | | | 2,555 | | | 5,434 |
Operating lease liabilities, current | | | — | | | 797 |
Finance lease liabilities, current | | | 422 | | | 1,249 |
Total current liabilities | | | 4,751 | | | 155,896 |
| | | | | |||
Redeemable convertible preferred stock warrant liabilities | | | 42 | | | 1,331 |
Deferred revenue, net of current portion | | | — | | | 67,349 |
Deferred rent, net of current portion | | | 2,729 | | | — |
Debt, net of current portion | | | 3,776 | | | — |
Operating leases liabilities, net of current portion | | | — | | | 10,472 |
Finance lease liabilities, net of current portion | | | 497 | | | 1,857 |
Other non-current liabilities | | | — | | | 4,500 |
Total liabilities | | | 11,795 | | | 241,405 |
Commitments and contingencies (Note 16) | | | | | ||
| | | | | |||
Redeemable Convertible Preferred Stock | | | | | ||
Series A redeemable convertible preferred stock, $0.00001 par value; 8,721,437 shares authorized, 8,350,743 issued and outstanding at December 31, 2019 and 2020; liquidation preference of $7,660 at December 31, 2019 and 2020 | | | 7,519 | | | 7,519 |
Series B redeemable convertible preferred stock, $0.00001 par value; 46,213,620 shares authorized, 46,176,715 issued and outstanding at December 31, 2019 and 2020; liquidation preference of $66,240 at December 31, 2019 and 2020 | | | 66,186 | | | 66,186 |
Series C-1 redeemable convertible preferred stock; $0.00001 par value; 27,308,229 shares authorized, 27,308,227 issued and outstanding at December 31, 2020 and none authorized, issued and outstanding at December 31, 2019; liquidation preference of $100,000 at December 31, 2020 | | | — | | | 96,436 |
Series C-2 redeemable convertible preferred stock; $0.00001 par value; 1,690,380 shares authorized, issued and outstanding at December 31, 2020 and none authorized, issued and outstanding at December 31, 2019; liquidation preference of $5,571 at December 31, 2020 | | | — | | | 6,182 |
Total redeemable convertible preferred stock | | | 73,705 | | | 176,323 |
| | | | | |||
Stockholders’ Deficit | | | | | ||
Common stock, $0.00001 par value; 88,778,540 and 129,030,355 shares authorized, 18,704,118 and 27,995,780 issued and outstanding at December 31, 2019 and 2020, respectively | | | — | | | — |
Additional paid-in-capital | | | 4,945 | | | 9,036 |
Accumulated deficit | | | (63,084) | | | (110,436) |
Total stockholders’ deficit | | | (58,139) | | | (101,400) |
Total liabilities, redeemable convertible preferred stock and stockholders’ deficit | | | $27,361 | | | $316,328 |
| | | Year Ended December 31, | ||||
| | | 2019 | | | 2020 | |
Revenue | | | | | ||
Product revenue | | | $— | | | $15,391 |
Grant and other revenue | | | 6,626 | | | 7,562 |
Total revenue | | | 6,626 | | | 22,953 |
| | | | | |||
Operating costs and expenses: | | | | | ||
Cost of product revenue | | | — | | | 14,951 |
Sales and marketing | | | 88 | | | 714 |
Research and development | | | 21,405 | | | 28,478 |
General and administrative | | | 5,900 | | | 23,936 |
Total operating costs and expenses | | | 27,393 | | | 68,079 |
Loss from operations | | | (20,767) | | | (45,126) |
| | | | | |||
Interest expense | | | (152) | | | (984) |
Change in fair value of redeemable convertible preferred stock warrants | | | 4 | | | (1,289) |
Other income | | | 309 | | | 47 |
Net loss | | | $(20,606) | | | $(47,352) |
Net loss per share attributable to common stockholders, basic and diluted | | | $(1.31) | | | $(2.90) |
Weighted-average number of shares used in computation of net loss per share attributable to common stockholders, basic and diluted | | | 15,760,246 | | | 16,315,730 |
| | | Series A Redeemable Convertible Preferred Stock | | | Series B Redeemable Convertible Preferred Stock | | | Series C Redeemable Convertible Preferred Stock | | | Common Stock | | | Additional Paid-In Capital | | | Accumulated Deficit | | | Total Stockholders’ Deficit | |||||||||||||
| | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | |||||||||
| | | | | | | | | | | | | | | | | | | | | | | ||||||||||||
Balance at January 1, 2019 | | | 8,350,743 | | | $7,519 | | | 46,176,715 | | | $66,186 | | | — | | | $— | | | 18,679,868 | | | $ — | | | $4,597 | | | $(42,478) | | | $(37,881) |
Exercise of common stock options | | | — | | | — | | | — | | | — | | | — | | | — | | | 24,250 | | | — | | | 12 | | | — | | | 12 |
Stock-based compensation | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 336 | | | — | | | 336 |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (20,606) | | | (20,606) |
| | | | | | | | | | | | | | | | | | | | | | | ||||||||||||
Balance at December 31, 2019 | | | 8,350,743 | | | 7,519 | | | 46,176,715 | | | 66,186 | | | — | | | — | | | 18,704,118 | | | — | | | 4,945 | | | (63,084) | | | (58,139) |
Issuance of Series C-1 redeemable convertible preferred stock, net of issuance costs of $3,564 | | | — | | | — | | | — | | | — | | | 27,308,227 | | | 96,436 | | | — | | | — | | | — | | | — | | | — |
Conversion of convertible notes to Series C-2 redeemable convertible preferred stock | | | — | | | — | | | — | | | — | | | 1,690,380 | | | 6,182 | | | — | | | — | | | — | | | — | | | — |
Exercise of common stock options | | | — | | | — | | | — | | | — | | | — | | | — | | | 1,918,499 | | | — | | | 669 | | | — | | | 669 |
Vesting of early exercised stock options | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 259 | | | — | | | 259 |
Issuance of common stock per restricted stock purchase agreement | | | — | | | — | | | — | | | — | | | — | | | — | | | 7,373,163 | | | — | | | — | | | — | | | — |
Stock-based compensation | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 3,163 | | | — | | | 3,163 |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (47,352) | | | (47,352) |
Balance at December 31, 2020 | | | 8,350,743 | | | $ 7,519 | | | 46,176,715 | | | $ 66,186 | | | 28,998,607 | | | $ 102,618 | | | 27,995,780 | | | $— | | | $ 9,036 | | | $ (110,436) | | | $ (101,400) |
| | | Year Ended December 31, | ||||
| | | 2019 | | | 2020 | |
Cash flows from operating activities | | | | | ||
Net loss | | | $ (20,606) | | | $ (47,352) |
Adjustments to reconcile net loss to net cash, cash equivalents and restricted cash used in operations | | | | | ||
Depreciation and amortization | | | 3,653 | | | 6,282 |
Change in fair value of warrant liabilities | | | (4) | | | 1,289 |
Stock-based compensation expense | | | 336 | | | 3,163 |
Loss on extinguishment of convertible notes | | | — | | | 610 |
Non-cash lease expense | | | — | | | 568 |
Amortization of debt discount and issuance costs | | | 6 | | | 16 |
Changes in operating assets and liabilities: | | | | | ||
Accounts receivable | | | 4,291 | | | (3,968) |
Inventory | | | — | | | (36,842) |
Prepaid expenses and other current assets | | | (415) | | | (14,207) |
Prepaid rent | | | — | | | (16,771) |
Other non-current assets | | | — | | | (130) |
Accounts payable | | | (253) | | | 4,523 |
Accrued liabilities | | | 263 | | | 8,114 |
Deferred rent | | | (374) | | | — |
Deferred revenue | | | — | | | 183,084 |
Operating leases | | | — | | | (337) |
Other non-current liabilities | | | — | | | 4,500 |
Interest on finance leases | | | 107 | | | 113 |
Net cash, cash equivalents and restricted cash (used in) provided by operating activities | | | (12,996) | | | 92,655 |
Cash flows from investing activities | | | | | ||
Purchase of property and equipment | | | (2,945) | | | (76,034) |
Expenditures for software development | | | — | | | (2,114) |
Net cash, cash equivalents and restricted cash used in investing activities | | | (2,945) | | | (78,148) |
Cash flows from financing activities | | | | | ||
Proceeds from issuance of Series C-1 redeemable convertible preferred stock | | | — | | | 100,000 |
Proceeds from convertible notes | | | — | | | 5,563 |
Payments for issuance costs of Series C redeemable convertible preferred stock | | | — | | | (3,564) |
Exercise of common stock options | | | 12 | | | 1,079 |
Proceeds from debt | | | 4,084 | | | 1,658 |
Repayment of debt | | | — | | | (2,571) |
Payments for finance leases | | | (486) | | | (1,922) |
Net cash, cash equivalents and restricted cash provided by financing activities | | | 3,610 | | | 100,243 |
Net increase (decrease) cash, cash equivalents and restricted cash | | | (12,331) | | | 114,750 |
Cash, cash equivalents and restricted cash, beginning balance | | | 26,836 | | | 14,505 |
Cash, cash equivalents and restricted cash, ending balance | | | $14,505 | | | $129,255 |
Reconciliation of cash, cash equivalents, and restricted cash | | | | | ||
Cash and cash equivalents | | | $14,328 | | | $121,578 |
Restricted cash, current | | | — | | | 6,000 |
Restricted cash, non-current | | | 177 | | | 1,677 |
Total cash, cash equivalents and restricted cash | | | $14,505 | | | $129,255 |
Supplemental disclosure for cash flow information | | | | | ||
Cash paid for interest | | | $152 | | | $340 |
Supplemental disclosure for non-cash investing and financing matters | | | | | ||
Early exercised stock options liability | | | $— | | | $152 |
Conversion of convertible notes to Series C-2 redeemable convertible preferred stock | | | $— | | | $6,182 |
Right-of-use assets obtained in exchange for lease obligations | | | $— | | | $11,269 |
Equipment obtained under capital lease obligations | | | $346 | | | $— |
Purchase of property and equipment included in accounts payable | | | $110 | | | $18,156 |
| Level 1 — | Unadjusted quoted prices in active markets for identical assets or liabilities. |
| Level 2 — | Inputs other than quoted prices included within Level I that are observable, unadjusted quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the related assets or liabilities. |
| Level 3 — | Unobservable inputs that are supported by little or no market activity for the related assets or liabilities. |
| | | Years | |
Leasehold improvements | | | Shorter of the estimated useful life or lease term |
Machinery and equipment | | | 3-7 |
Furniture and fixtures | | | 7 |
| | | December 31, | ||||
| | | 2019 | | | 2020 | |
Raw materials | | | $ — | | | $ 29,948 |
Work-in-process | | | — | | | 4,957 |
Finished goods | | | — | | | 1,645 |
Inventory on consignment | | | — | | | 1,081 |
Reserve | | | — | | | (789) |
Total inventories | | | $— | | | $36,842 |
| | | December 31, | ||||
| | | 2019 | | | 2020 | |
Prepaid expense | | | $669 | | | $5,152 |
Prepaid inventory | | | — | | | 8,695 |
Total prepaid expenses | | | $ 669 | | | $ 13,847 |
| | | December 31, | ||||
| | | 2019 | | | 2020 | |
Construction in progress | | | $614 | | | $83,353 |
Machinery and equipment | | | 13,683 | | | 26,972 |
Leasehold improvements | | | 4,847 | | | 2,897 |
Furniture and fixtures | | | 388 | | | 683 |
Property and equipment | | | 19,532 | | | 113,905 |
Accumulated depreciation | | | (7,902) | | | (10,222) |
Total property and equipment, net | | | $ 11,630 | | | $ 103,683 |
| | | December 31, | ||||
| | | 2019 | | | 2020 | |
Developed software | | | $— | | | $2,114 |
Accumulated amortization | | | — | | | (76) |
Total intangible | | | $ — | | | $ 2,038 |
2021 | | | $705 |
2022 | | | 705 |
2023 | | | 628 |
Total amortization expense | | | $ 2,038 |
| | | | | December 31, 2020 | |||||
| | | Balance Sheet Location | | | Operating Leases | | | Finance Leases | |
Assets | | | | | | | |||
Right-of-use assets operating leases | | | Operating lease right-of-use assets | | | $8,281 | | | |
Right-of-use assets finance leases | | | Property and equipment, net | | | | | $ 4,837 | |
Liabilities | | | | | | | |||
Operating lease liabilities (current) | | | Operating lease liabilities, current | | | 797 | | | |
Finance lease liabilities (current) | | | Finance lease liabilities, current | | | | | 1,249 | |
Operating lease liabilities (non-current) | | | Operating leases liabilities, net of current portion | | | 10,472 | | | |
Finance lease liabilities (non-current) | | | Finance lease liabilities, net of current portion | | | | | 1,857 | |
| | | Year Ended December 31, 2020 | |
Operating lease cost | | | $1,552 |
Finance lease cost: | | | |
Amortization of right-of-use assets | | | 570 |
Interest on lease liabilities | | | 113 |
Total lease cost | | | $ 2,235 |
| | | Operating Leases | | | Finance Leases | |
2021 | | | $1,736 | | | $1,399 |
2022 | | | 1,785 | | | 1,169 |
2023 | | | 1,836 | | | 781 |
2024 | | | 1,889 | | | — |
2025 | | | 1,941 | | | — |
Thereafter | | | 6,944 | | | — |
Total lease payments | | | 16,131 | | | 3,349 |
Less: Imputed interest | | | (4,862) | | | (243) |
Total | | | $ 11,269 | | | $ 3,106 |
| | | Year Ended December 31, 2020 | |
Cash paid for amounts included in the measurement of lease liabilities: | | | |
Operating cash flows from operating leases | | | $ 1,287 |
Operating cash flows from finance leases | | | $113 |
Financing cash flows from finance leases | | | $1,922 |
| | | ||
Right-of-use assets obtained in exchange for lease liabilities: | | | |
Operating leases | | | $8,443 |
Finance leases | | | $2,826 |
| | | December 31, 2020 | ||||
| | | Operating Leases | | | Finance Leases | |
Weighted-average remaining lease term | | | 8.4 years | | | 2.5 years |
Weighted-average discount rate | | | 8.7% | | | 6.5% |
| | | Operating Leases | | | Capital Leases | |
2020 | | | $1,127 | | | $509 |
2021 | | | 1,675 | | | 372 |
2022 | | | 1,725 | | | 142 |
2023 | | | 1,777 | | | 9 |
2024 | | | 1,830 | | | — |
Thereafter | | | 8,551 | | | — |
Total future minimum lease payments | | | $ 16,685 | | | $ 1,032 |
| | | Shares | | | Exercise Price | | | Issuance Date | | | Expiration Date | |
Series A redeemable convertible preferred stock warrants | | | 20,441 | | | $0.91728 | | | May 28, 2015 | | | May 28, 2025 |
Series A redeemable convertible preferred stock warrants | | | 20,441 | | | 0.91728 | | | May 28, 2015 | | | May 28, 2025 |
Series A redeemable convertible preferred stock warrants | | | 7,631 | | | 0.91728 | | | September 6, 2018 | | | September 6, 2028 |
Series B redeemable convertible preferred stock warrants | | | 31,369 | | | 1.4345 | | | November 27, 2018 | | | November 27, 2028 |
| a) | a merger or consolidation in which (i) the Company is a constituent party or (ii) a subsidiary of the Company is a constituent party and the Company issues shares of its capital stock pursuant to such merger or consolidation, except in either case, in respect of any such merger or consolidation involving the Company or a subsidiary in which the shares of capital stock of the Company outstanding immediately |
| b) | the sale, lease, transfer, exclusive license or other disposition, in a single transaction or series of related transactions, by the Company or any subsidiary of the Company of all or substantially all the assets or intellectual property of the Company and its subsidiaries taken as a whole, or the sale or disposition (whether by merger, consolidation or otherwise) of one or more subsidiaries of the Company if substantially all of the assets of the Company and its subsidiaries taken as a whole are held by such subsidiary or subsidiaries, except where such sale, lease, transfer, exclusive license or other disposition is to a wholly owned subsidiary of the Company; or |
| c) | the closing of the transfer (whether by merger, amalgamation, consolidation or otherwise), in one transaction or a series of related transactions, to a person or group of affiliated persons (other than an underwriter of the Company’s securities), of the Company’s securities if, after such closing, such person or group of affiliated persons would hold a majority, by voting power, of the share capital or capital stock of the Company. |
Redeemable convertible preferred stock | | | 83,526,065 |
Warrants to purchase redeemable convertible preferred stock | | | 79,882 |
Common stock option grants issued and outstanding | | | 8,344,752 |
Common stock reserved for future option grants | | | 2,950,871 |
Common stock warrants | | | 75,744 |
Total common shares reserved for future issuance | | | 94,977,314 |
December 31, 2019 | | | Recurring Fair Value Measurements | |||||||||
| | | Level 1 | | | Level 2 | | | Level 3 | | | Total | |
Redeemable convertible preferred stock warrant liabilities | | | $ — | | | $ — | | | $ 42 | | | $ 42 |
December 31, 2020 | | | Recurring Fair Value Measurements | |||||||||
| | | Level 1 | | | Level 2 | | | Level 3 | | | Total | |
Redeemable convertible preferred stock warrant liabilities | | | $ — | | | $ — | | | $ 1,331 | | | $ 1,331 |
| | | Amount | |
Balance, January 1, 2019 | | | $46 |
Issuance | | | — |
Remeasurement | | | (4) |
| | | ||
Balance, December 31, 2019 | | | 42 |
Issuance | | | — |
Remeasurement | | | 1,289 |
| | | ||
Balance, December 31, 2020 | | | $ 1,331 |
| | | December 31, | ||||
| | | 2019 | | | 2020 | |
Expected volatility | | | 41.8% | | | 59.9% |
Expected term (years) | | | 5.92 | | | 4.92 |
Expected dividend yield | | | 0.00% | | | 0.00% |
Risk-free interest rate | | | 1.72% | | | 0.41% |
Fair value per share | | | $ 0.55 | | | $ 16.83 |
| | | December 31, | ||||
| | | 2019 | | | 2020 | |
Expected volatility | | | 37.2% | | | 46.2% |
Expected term (years) | | | 8.91 | | | 7.91 |
Expected dividend yield | | | 0.00% | | | 0.00% |
Risk-free interest rate | | | 1.88% | | | 0.65% |
Fair value per share | | | $ 0.68 | | | $ 16.41 |
| | | Options | | | Weighted Average Exercise Price | | | Weighted Average Remaining Contractual Term (Years) | |
Outstanding at January 1, 2019 | | | 8,227,345 | | | $0.38 | | | |
Granted | | | 375,000 | | | 0.48 | | | |
Exercised | | | (24,250) | | | 0.48 | | | |
Forfeited | | | (215,750) | | | 0.48 | | | |
Expired | | | (117,594) | | | 0.37 | | | |
Outstanding at December 31, 2019 | | | 8,244,751 | | | 0.39 | | | 6.84 |
Granted | | | 2,233,042 | | | 1.41 | | | |
Exercised | | | (1,918,499) | | | 0.56 | | | |
Forfeited | | | (78,043) | | | 0.96 | | | |
Expired | | | (136,499) | | | 0.39 | | | |
Outstanding at December 31, 2020 | | | 8,344,752 | | | $0.61 | | | 6.44 |
Exercisable at December 31, 2020 | | | 6,350,005 | | | $0.43 | | | 5.55 |
Vested and expected to vest at December 31, 2020 | | | 8,176,627 | | | $0.60 | | | 6.36 |
| | | 2019 | | | 2020 | |
Expected volatility | | | 28.4% | | | 39.6% |
Expected term (years) | | | 6.08 | | | 7.04 |
Expected dividend yield | | | 0% | | | 0% |
Risk-free interest rate | | | 1.8% | | | 0.4% |
Grant date fair value | | | $ 0.15 | | | $ 0.57 |
Risk-free interest rate | | | 1.1% |
Expected term (years) | | | 5.86 years |
Exercise price | | | $1.25 |
Expected dividend yield | | | 0.0% |
Expected volatility | | | 37.6% |
Grant date fair value | | | $0.40 |
| | | Number of Shares | |
Outstanding, January 1, 2019 | | | 2,924,130 |
Granted | | | — |
Outstanding, December 31, 2019 | | | 2,924,130 |
Granted | | | 7,373,163 |
Forgiveness of Nonrecourse Notes on vested shares of common stock | | | (425,000) |
Outstanding, December 31, 2020 | | | 9,872,293 |
Vested, December 31, 2020 | | | 6,762,220 |
| | | 2019 | | | 2020 | |
Sales and marketing | | | $— | | | $1 |
Research and development | | | 45 | | | 98 |
General and administrative | | | 291 | | | 3,064 |
Total stock-based compensation expense | | | $ 336 | | | $ 3,163 |
| | | Number of Shares | |
Unvested at beginning of year | | | — |
Early exercised stock options during period | | | 855,000 |
Vested or cancelled | | | (538,334) |
Unvested at end of year | | | 316,666 |
| | | 2019 | | | 2020 | |
Basic: | | | | | ||
Net loss attributable to common stockholders | | | $(20,606) | | | $(47,352) |
Weighted-average common shares outstanding, basic and diluted | | | 15,760,246 | | | 16,315,730 |
Net loss attributable to common stockholders per share, basic and diluted | | | $(1.31) | | | $(2.90) |
| | | As of December 31, | ||||
| | | 2019 | | | 2020 | |
Redeemable convertible preferred stock | | | 54,527,458 | | | 83,526,065 |
Stock options | | | 8,244,751 | | | 8,344,752 |
Early exercised options | | | — | | | 316,666 |
Common stock subject to restricted stock purchase agreements | | | 2,924,130 | | | 9,872,293 |
Common stock warrants | | | 75,744 | | | 75,744 |
Redeemable convertible preferred stock warrants | | | 79,882 | | | 79,882 |
Total | | | 65,851,965 | | | 102,215,402 |
| | | Years Ended December 31, | ||||
| | | 2019 | | | 2020 | |
Expected tax at the federal statutory rate | | | (21.0)% | | | (21.0)% |
State income tax, net of federal benefit | | | (7.0)% | | | (7.6)% |
Permanent items | | | 0.9% | | | 1.3% |
Change in valuation allowance | | | 30.9% | | | 30.8% |
Tax Credits | | | (4.8)% | | | (3.3)% |
Uncertain tax position reserves | | | 1.0% | | | 0.7% |
Stock-based compensation | | | — | | | (0.9)% |
Provision for income taxes | | | — | | | — |
| | | As of December 31, | ||||
| | | 2019 | | | 2020 | |
Deferred tax assets: | | | | | ||
Net operating losses | | | $ 15,755 | | | $ 29,217 |
Research and development credits | | | 2,541 | | | 3,791 |
Operating lease liability | | | — | | | 3,234 |
Share-based compensation | | | 231 | | | 350 |
Accruals | | | 45 | | | 2,963 |
Other | | | 772 | | | 226 |
| | | As of December 31, | ||||
| | | 2019 | | | 2020 | |
Total deferred tax assets | | | 19,344 | | | 39,781 |
Deferred tax liabilities: | | | | | ||
Operating right-of-use asset | | | — | | | (2,376) |
Depreciation and amortization | | | (41) | | | (3,511) |
Total deferred tax liabilities | | | (41) | | | (5,887) |
Gross deferred tax assets | | | 19,303 | | | 33,894 |
Less: Valuation allowance | | | (19,303) | | | (33,894) |
Net deferred income taxes | | | $— | | | $— |
| | | Years Ended December 31, | ||||
| | | 2019 | | | 2020 | |
Beginning balance | | | $489 | | | $705 |
Increases related to current year tax positions | | | 216 | | | 340 |
Ending Balance | | | $ 705 | | | $ 1,045 |
| | | December 31, 2020 | | | June 30, 2021 | |
Assets | | | | | ||
Current assets: | | | | | ||
Cash and cash equivalents | | | $121,578 | | | $246,326 |
Restricted cash | | | 6,000 | | | 6,000 |
Accounts receivable | | | 4,168 | | | 40,277 |
Inventory | | | 36,842 | | | 63,263 |
Prepaid expenses | | | 13,847 | | | 35,784 |
Other current assets | | | 1,263 | | | 827 |
Total current assets | | | 183,698 | | | 392,477 |
Restricted cash, non-current | | | 1,677 | | | — |
Property and equipment, net | | | 103,683 | | | 160,182 |
Prepaid rent | | | 16,771 | | | 1,648 |
Operating lease right-of-use assets | | | 8,281 | | | 69,511 |
Intangible assets, net | | | 2,038 | | | 2,728 |
Other non-current assets | | | 180 | | | 4,766 |
Total assets | | | $316,328 | | | $631,312 |
| | | | | |||
Liabilities, Redeemable Convertible Preferred Stock and Stockholders’ Deficit | | | | | ||
Current liabilities: | | | | | ||
Accounts payable | | | $23,847 | | | $25,824 |
Accrued liabilities and other current liabilities | | | 8,822 | | | 34,424 |
Deferred revenue, current | | | 115,747 | | | 93,745 |
Debt, current | | | 5,434 | | | — |
Operating lease liabilities, current | | | 797 | | | 3,170 |
Finance lease liabilities, current | | | 1,249 | | | 1,349 |
Total current liabilities | | | 155,896 | | | 158,512 |
Redeemable convertible preferred stock warrant liabilities | | | 1,331 | | | 1,521 |
Deferred revenue, net of current portion | | | 67,349 | | | 46,748 |
Convertible notes | | | — | | | 258,734 |
Operating leases liabilities, net of current portion | | | 10,472 | | | 46,274 |
Finance lease liabilities, net of current portion | | | 1,857 | | | 1,694 |
Other non-current liabilities | | | 4,500 | | | 2,838 |
Total liabilities | | | 241,405 | | | 516,321 |
Commitments and contingencies (Note 16) | | | | | ||
| | | | | |||
Redeemable Convertible Preferred Stock | | | | | ||
Series A redeemable convertible preferred stock, $0.00001 par value; 8,721,437 shares authorized, 8,350,743 issued and outstanding at December 31, 2020 and June 30, 2021; liquidation preference of $7,660 at December 31, 2020 and June 30, 2021 | | | 7,519 | | | 7,519 |
Series B redeemable convertible preferred stock, $0.00001 par value; 46,213,620 shares authorized, 46,176,715 issued and outstanding at December 31, 2020 and June 30, 2021; liquidation preference of $66,240 at December 31, 2020 and June 30, 2021 | | | 66,186 | | | 66,186 |
| | | December 31, 2020 | | | June 30, 2021 | |
Series C-1 redeemable convertible preferred stock, $0.00001 par value; 27,308,229 shares authorized, 27,308,227 issued and outstanding at December 31, 2020 and June 30, 2021, respectively; liquidation preference of $100,000 at December 31, 2020 and June 30, 2021 | | | 96,436 | | | 96,436 |
Series C-2 redeemable convertible preferred stock, $0.00001 par value; 1,690,380 shares authorized, issued and outstanding at December 31, 2020 and June 30, 2021; liquidation preference of $5,571 at December 31, 2020 and June 30, 2021 | | | 6,182 | | | 6,182 |
Total redeemable convertible preferred stock | | | 176,323 | | | 176,323 |
| | | | | |||
Stockholders’ Deficit | | | | | ||
Common stock, $0.00001 par value; 129,030,355 shares authorized, 27,995,780 and 29,128,604 issued and outstanding at December 31, 2020 and June 30, 2021, respectively | | | — | | | — |
Additional paid-in-capital | | | 9,036 | | | 16,264 |
Accumulated deficit | | | (110,436) | | | (77,596) |
Total stockholders’ deficit | | | (101,400) | | | (61,332) |
Total liabilities, redeemable convertible preferred stock and stockholders’ deficit | | | $316,328 | | | $631,312 |
| | | Six Months Ended June 30, | ||||
| | | 2020 | | | 2021 | |
Revenue | | | | | ||
Product revenue | | | $— | | | $201,922 |
Grant and other revenue | | | 4,960 | | | — |
Total revenue | | | 4,960 | | | 201,922 |
| | | | | |||
Operating costs and expenses: | | | | | ||
Cost of product revenue | | | — | | | 85,177 |
Sales and marketing | | | 45 | | | 1,959 |
Research and development | | | 19,680 | | | 12,071 |
General and administrative | | | 3,764 | | | 23,252 |
Total operating costs and expenses | | | 23,489 | | | 122,459 |
Income (loss) from operations | | | (18,529) | | | 79,463 |
Interest expense | | | (788) | | | (9,964) |
Change in fair value of redeemable convertible preferred stock warrants | | | (20) | | | (190) |
Change in fair value of convertible notes | | | — | | | (23,254) |
Other income (expense), net | | | 59 | | | 61 |
Net income (loss) before income taxes | | | (19,278) | | | 46,116 |
Income tax expense | | | — | | | (13,276) |
Net income (loss) | | | $(19,278) | | | $32,840 |
Basic net income (loss) per share attributable to common stockholders | | | $(1.21) | | | $0.23 |
Weighted-average number of shares used in computation of basic net income (loss) per share attributable to common stockholders | | | 15,909,439 | | | 18,617,247 |
Diluted net income (loss) per share attributable to common stockholders | | | $(1.21) | | | $0.22 |
Weighted-average number of shares used in computation of diluted net income (loss) per share attributable to common stockholders | | | 15,909,439 | | | 26,036,337 |
| | | Series A Redeemable Convertible Preferred Stock | | | Series B Redeemable Convertible Preferred Stock | | | Series C Redeemable Convertible Preferred Stock | | | Common Stock | | | Additional Paid-In Capital | | | Accumulated Deficit | | | Total Stockholders’ Deficit | |||||||||||||
| | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | |||||||||
Balance at December 31, 2019 | | | 8,350,743 | | | $7,519 | | | 46,176,715 | | | $66,186 | | | — | | | $— | | | 18,704,118 | | | $— | | | $4,945 | | | $(63,084) | | | $(58,139) |
Issuance of Series C-1 preferred stock | | | — | | | — | | | — | | | — | | | 27,308,227 | | | 96,963 | | | — | | | — | | | — | | | — | | | — |
Conversion of convertible notes to Series C-2 preferred stock | | | — | | | — | | | — | | | — | | | 1,690,380 | | | 6,182 | | | — | | | — | | | — | | | — | | | — |
Exercise of common stock options | | | — | | | — | | | — | | | — | | | — | | | — | | | 1,520,000 | | | — | | | — | | | — | | | — |
Vesting of early exercised stock options | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 729 | | | — | | | 729 |
Stock-based compensation | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 97 | | | — | | | 97 |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (19,278) | | | (19,278) |
Balance at June 30, 2020 | | | 8,350,743 | | | $7,519 | | | 46,176,715 | | | $66,186 | | | 28,998,607 | | | $103,145 | | | 20,224,118 | | | $— | | | $5,771 | | | $(82,362) | | | $(76,591) |
| | | Series A Redeemable Convertible Preferred Stock | | | Series B Redeemable Convertible Preferred Stock | | | Series C Redeemable Convertible Preferred Stock | | | Common Stock | | | Additional Paid-In Capital | | | Accumulated Deficit | | | Total Stockholders’ Deficit | |||||||||||||
| | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | |||||||||
Balance at December 31, 2020 | | | 8,350,743 | | | $7,519 | | | 46,176,715 | | | $66,186 | | | 28,998,607 | | | $102,618 | | | 27,995,780 | | | $— | | | $9,036 | | | $(110,436) | | | $(101,400) |
Exercise of common stock options | | | — | | | — | | | — | | | — | | | — | | | — | | | 1,048,706 | | | — | | | 258 | | | — | | | 258 |
Stock-based compensation expense from issuance of a fully vested warrant to vendor | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 1,239 | | | — | | | 1,239 |
Exercise of common stock warrant | | | — | | | — | | | — | | | — | | | — | | | — | | | 84,118 | | | — | | | 77 | | | — | | | 77 |
Vesting of early exercised stock options | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 63 | | | — | | | 63 |
Stock-based compensation | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 5,591 | | | — | | | 5,591 |
Net income | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 32,840 | | | 32,840 |
Balance at June 30, 2021 | | | 8,350,743 | | | $7,519 | | | 46,176,715 | | | $66,186 | | | 28,998,607 | | | $102,618 | | | 29,128,604 | | | $— | | | $16,264 | | | $(77,596) | | | $(61,332) |
| | | Six Months Ended June 30, | ||||
| | | 2020 | | | 2021 | |
Cash flows from operating activities | | | | | ||
Net income (loss) | | | $(19,278) | | | $32,840 |
Adjustments to reconcile net income (loss) to net cash, cash equivalents and restricted cash used in operations | | | | | ||
Depreciation and amortization | | | 3,399 | | | 14,500 |
Inventory reserve | | | — | | | 353 |
Change in fair value of redeemable convertible preferred stock warrant liabilities | | | 20 | | | 190 |
Change in fair value of convertible notes | | | — | | | 23,254 |
Stock-based compensation expense | | | 97 | | | 5,591 |
Loss on extinguishment of debt | | | 610 | | | 1,998 |
Non-cash lease expense | | | 277 | | | 1,822 |
Amortization of debt issuance costs | | | 16 | | | 130 |
Convertible notes issuance costs | | | — | | | 6,000 |
Deferred income taxes | | | — | | | 588 |
Interest on finance leases | | | 39 | | | 100 |
Stock-based compensation expense from issuance of fully vested warrant to vendor | | | — | | | 1,239 |
Changes in operating assets and liabilities: | | | | | ||
Accounts receivable | | | (2,579) | | | (36,109) |
Inventory | | | (664) | | | (26,774) |
Prepaid expenses and other current assets | | | (4,684) | | | (22,818) |
Other non-current assets | | | — | | | (676) |
Accounts payable, accrued liabilities and other current liabilities | | | 1,741 | | | 11,473 |
Deferred revenue | | | — | | | (42,602) |
Operating lease liabilities | | | 51 | | | (8,911) |
Net cash, cash equivalents and restricted cash used in operating activities | | | (20,955) | | | (37,812) |
Cash flows from investing activities | | | | | ||
Purchase of property and equipment | | | (1,326) | | | (56,545) |
Expenditures for software development | | | — | | | (2,351) |
Net cash, cash equivalents and restricted cash used in investing activities | | | (1,326) | | | (58,896) |
Cash flows from financing activities | | | | | ||
Proceeds for Series C-1 redeemable convertible preferred stock | | | 100,000 | | | — |
Proceeds from convertible notes | | | 5,563 | | | 235,480 |
Payments for issuance costs of Series C redeemable convertible preferred stock | | | (3,037) | | | — |
Payments of issuance costs of convertible notes | | | — | | | (6,000) |
Proceeds from exercise of common stock options | | | 729 | | | 258 |
Proceeds from exercise of common stock warrant | | | — | | | 77 |
Proceeds from debt | | | — | | | 82,250 |
Debt issuance and prepayment costs | | | — | | | (2,128) |
Repayment of debt | | | (1,286) | | | (87,684) |
Payments for deferred initial public offering costs | | | — | | | (1,610) |
Payments for finance leases | | | (246) | | | (864) |
Net cash, cash equivalents and restricted cash provided by financing activities | | | 101,723 | | | 219,779 |
| | | Six Months Ended June 30, | ||||
| | | 2020 | | | 2021 | |
| | | | | |||
Net increase in cash, cash equivalents and restricted cash | | | 79,442 | | | 123,071 |
Cash, cash equivalents and restricted cash, beginning balance | | | 14,505 | | | 129,255 |
Cash, cash equivalents and restricted cash, ending balance | | | $93,947 | | | $252,326 |
| | | | | |||
Reconciliation of cash, cash equivalents, and restricted cash | | | | | ||
Cash and cash equivalents | | | $92,270 | | | $246,326 |
Restricted cash, current | | | — | | | 6,000 |
Restricted cash, non-current | | | 1,677 | | | — |
Total cash, cash equivalents and restricted cash | | | $93,947 | | | $252,326 |
| | | | | |||
Supplemental disclosure for cash flow information | | | | | ||
Cash paid for interest | | | $72 | | | $760 |
| | | | | |||
Supplemental disclosure for non-cash investing and financing matters | | | | | ||
Early exercised stock options liability | | | $— | | | $63 |
Right-of-use assets obtained in exchange for lease obligations | | | $8,849 | | | $38,717 |
Prepaid rent reclassified to right-of-use assets | | | $— | | | $15,966 |
Purchase of property and equipment included in accounts payable | | | $393 | | | $11,618 |
Deferred initial public offering costs included in accounts payable and accrued liabilities and other current liabilities | | | $— | | | $2,301 |
| Level 1 — | Unadjusted quoted prices in active markets for identical assets or liabilities. |
| Level 2 — | Inputs other than quoted prices included within Level I that are observable, unadjusted quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the related assets or liabilities. |
| Level 3 — | Unobservable inputs that are supported by little or no market activity for the related assets or liabilities. |
| | | Amount | |
Balance, December 31, 2020 | | | $— |
Provision for warranties | | | 4,611 |
Settlements | | | (101) |
Change in warranty estimates | | | — |
Balance, June 30, 2021 | | | $4,510 |
| | | Six Months Ended June 30, | ||||
| | | 2020 | | | 2021 | |
Public sector entities | | | $— | | | $167,120 |
Other customers | | | — | | | 34,802 |
Total product revenue | | | $— | | | $201,922 |
| | | Six Months Ended June 30, | ||||
| | | 2020 | | | 2021 | |
Product revenue | | | $— | | | $201,922 |
Cost of product revenue | | | — | | | 85,177 |
Product gross profit | | | $— | | | $116,745 |
Product gross profit margin | | | 0% | | | 58% |
| | | Amount | |
Balance, December 31, 2020 | | | $183,096 |
Recognition of U.S. DoD Advance | | | (42,208) |
Recognition of non-refundable customer deposits | | | (395) |
Balance, June 30, 2021 | | | $140,493 |
| | | December 31, 2020 | | | June 30, 2021 | |
Raw materials | | | $29,948 | | | $36,715 |
Work-in-process | | | 4,957 | | | 12,312 |
Finished goods | | | 1,645 | | | 14,600 |
Inventory on consignment | | | 1,081 | | | 1,106 |
Reserve | | | (789) | | | (1,470) |
Total inventories | | | $36,842 | | | $63,263 |
| | | December 31, 2020 | | | June 30, 2021 | |
Prepaid expenses | | | $5,152 | | | $13,834 |
Prepaid inventory | | | 8,695 | | | 21,950 |
Total prepaid expenses | | | $13,847 | | | $35,784 |
| | | December 31, 2020 | | | June 30, 2021 | |
Construction in progress | | | $83,353 | | | $38,467 |
Machinery and equipment | | | 26,972 | | | 129,083 |
Leasehold improvements | | | 2,897 | | | 15,872 |
Furniture and fixtures | | | 683 | | | 750 |
Property and equipment | | | 113,905 | | | 184,172 |
Accumulated depreciation and amortization | | | (10,222) | | | (23,990) |
Total property and equipment, net | | | $103,683 | | | $160,182 |
| | | December 31, 2020 | | | June 30, 2021 | |
Capitalized software | | | $2,114 | | | $4,465 |
Accumulated amortization | | | (76) | | | (1,737) |
Total intangible assets | | | $2,038 | | | $2,728 |
2021 (excluding the six months ended June 30, 2021) | | | $509 |
2022 | | | 1,018 |
2023 | | | 970 |
2024 | | | 231 |
Total amortization expense | | | $2,728 |
| | | | | June 30, 2021 | |||||
| | | Balance Sheet Location | | | Operating Leases | | | Finance Leases | |
Assets | | | | | | | |||
Right-of-use assets operating leases | | | Operating lease right-of-use assets | | | $69,511 | | | |
Right-of-use assets finance leases | | | Property and equipment, net | | | | | $5,456 | |
| | | | | | | ||||
Liabilities | | | | | | | |||
Operating lease liabilities (current) | | | Operating lease liabilities, current | | | 3,170 | | | |
Finance lease liabilities (current) | | | Finance lease liabilities, current | | | | | 1,349 | |
Operating lease liabilities (non-current) | | | Operating lease liabilities, net of current portion | | | 46,274 | | | |
Finance lease liabilities (non-current) | | | Finance lease liabilities, net of current portion | | | | | 1,694 | |
| | | Six Months Ended June 30, | ||||
| | | 2020 | | | 2021 | |
Operating lease cost | | | $766 | | | $2,967 |
Finance lease cost: | | | | | ||
Amortization of right-of-use assets | | | 144 | | | 706 |
Interest on lease liabilities | | | 39 | | | 100 |
Total lease cost | | | $949 | | | $3,773 |
| | | Shares | | | Exercise Price | | | Issuance Date | | | Expiration Date | |
Series A redeemable convertible preferred stock warrants | | | 20,441 | | | $0.91728 | | | May 28, 2015 | | | May 28, 2025 |
Series A redeemable convertible preferred stock warrants | | | 20,441 | | | 0.91728 | | | May 28, 2015 | | | May 28, 2025 |
Series A redeemable convertible preferred stock warrants | | | 7,631 | | | 0.91728 | | | September 6, 2018 | | | September 6, 2028 |
Series B redeemable convertible preferred stock warrants | | | 31,369 | | | 1.4345 | | | November 27, 2018 | | | November 27, 2028 |
December 31, 2020 | | | Recurring Fair Value Measurements | |||||||||
| | | Level 1 | | | Level 2 | | | Level 3 | | | Total | |
Redeemable convertible preferred stock warrant liabilities | | | $— | | | $— | | | $1,331 | | | $1,331 |
June 30, 2021 | | | Recurring Fair Value Measurements | |||||||||
| | | Level 1 | | | Level 2 | | | Level 3 | | | Total | |
Redeemable convertible preferred stock warrant liabilities | | | $— | | | $— | | | $1,521 | | | $1,521 |
Convertible Notes | | | $— | | | $— | | | $258,734 | | | $258,734 |
| | | Conversion Event | | | Delayed Conversion Event | | | Repayment at Maturity | |
Expected event date | | | September 30, 2021 | | | December 30, 2021 | | | May 6, 2023 |
Term (years) | | | 0.25 | | | 0.50 | | | 1.85 |
Discount rate | | | 44.1% | | | 44.1% | | | 44.1% |
Probability | | | 90% | | | 5% | | | 5% |
| | | Redeemable Convertible Preferred Stock Warrants | | | Convertible Notes | |
Balance, December 31, 2020 | | | $1,331 | | | $— |
Issuance | | | — | | | 235,480 |
Remeasurement | | | 190 | | | 23,254 |
Balance, June 30, 2021 | | | $1,521 | | | $258,734 |
| | | December 31, 2020 | | | June 30, 2021 | |
Expected volatility | | | 59.9% | | | 70.5% |
Expected term (years) | | | 4.92 | | | 4.43 |
Expected dividend yield | | | 0.00% | | | 0.0% |
Risk-free interest rate | | | 0.41% | | | 0.75% |
Fair value per share | | | $16.83 | | | $19.07 |
| | | December 31, 2020 | | | June 30, 2021 | |
Expected volatility | | | 46.2% | | | 41.2% |
Expected term (years) | | | 7.91 | | | 7.41 |
Expected dividend yield | | | 0.00% | | | 0.00% |
Risk-free interest rate | | | 0.65% | | | 1.21% |
Fair value per share | | | $16.41 | | | $18.99 |
| | | Six Months Ended June 30, | ||||
| | | 2020 | | | 2021 | |
Cost of revenue | | | $— | | | $343 |
Sales and marketing | | | — | | | 26 |
Research and development | | | 13 | | | 1,444 |
General and administrative | | | 84 | | | 3,778 |
Total stock-based compensation expense | | | $97 | | | $5,591 |
| | | Options | | | Weighted Average Exercise Price | | | Weighted Average Remaining Contractual Term (Years) | |
Outstanding at January 1, 2021 | | | 8,344,752 | | | $0.61 | | | 6.48 |
Granted | | | 2,975,821 | | | 15.56 | | | |
Exercised | | | (1,048,706) | | | 0.26 | | | |
Forfeited | | | (219,878) | | | 9.47 | | | |
Expired | | | (107,792) | | | 0.37 | | | |
Outstanding at June 30, 2021 | | | 9,944,197 | | | $4.93 | | | 7.26 |
Exercisable at June 30, 2021 | | | 5,568,512 | | | $0.69 | | | 5.62 |
Vested and expected to vest at June 30, 2021 | | | 9,507,169 | | | $4.68 | | | 7.16 |
| | | 2020 | | | 2021 | |
Expected volatility | | | 39.6% | | | 40.9% |
Expected term (years) | | | 7.04 | | | 7.71 |
Expected dividend yield | | | 0.0% | | | 0.0% |
Risk-free interest rate | | | 0.41% | | | 0.83% |
Grant date fair value | | | $0.57 | | | $6.93 |
| | | Number of shares | |
Outstanding, December 31, 2020 | | | 9,872,293 |
Granted | | | — |
Outstanding, June 30, 2021 | | | 9,872,293 |
Vested at June 30, 2021 | | | 8,044,384 |
| | | Number of Shares | |
Unvested at January 1, 2021 | | | 316,666 |
Early exercised stock options during period | | | — |
Vested or cancelled | | | (189,998) |
Unvested at June 30, 2021 | | | 126,668 |
| | | Six Months Ended June 30, | ||||
| | | 2020 | | | 2021 | |
Numerator: | | | | | ||
Net income (loss) | | | $(19,278) | | | $32,840 |
Minus: Income allocated to participating securities | | | — | | | 28,565 |
Net income (loss) attributable to common stockholders – basic | | | $(19,278) | | | $4,275 |
| | | | | |||
Plus: Income allocated to non-participating securities | | | — | | | 1,332 |
Net income (loss) attributable to common stockholders - diluted | | | $(19,278) | | | $5,607 |
Denominator: | | | | | ||
Basic weighted-average common shares outstanding | | | 15,909,439 | | | 18,617,247 |
Dilutive potential common stock issuable: | | | | | ||
Common stock warrants | | | — | | | 89,551 |
Preferred stock warrants | | | — | | | 74,149 |
Stock options | | | — | | | 7,255,390 |
Diluted weighted-average shares outstanding | | | 15,909,439 | | | 26,036,337 |
| | | | | |||
Net income (loss) attributable to common stockholders per share | | | | | ||
Basic | | | $(1.21) | | | $0.23 |
Diluted | | | $(1.21) | | | $0.22 |
| | | Six Months Ended June 30, | ||||
| | | 2020 | | | 2021 | |
Redeemable convertible preferred stock | | | 54,527,458 | | | — |
Stock options | | | 8,244,751 | | | 2,838,421 |
Common stock subject to restricted stock purchase agreements | | | 2,924,130 | | | — |
Common stock warrants | | | 75,744 | | | — |
Redeemable convertible preferred stock warrants | | | 79,882 | | | — |
Total | | | 65,851,965 | | | 2,838,421 |
Goldman Sachs & Co. LLC | | | Morgan Stanley | | | Cowen |
| Item 13. | Other Expenses of Issuance and Distribution. |
| | | Amount | |
SEC registration fee | | | $10,910 |
FINRA filing fee | | | 15,500 |
Nasdaq initial listing fee | | | * |
Accountants’ fees and expenses | | | * |
Legal fees and expenses | | | * |
Transfer agent’s fees and expenses | | | * |
Printing and engraving expenses | | | * |
Miscellaneous | | | * |
Total expenses | | | $ * |
| * | To be filed by amendment. |
| Item 14. | Indemnification of Directors and Officers. |
| Item 15. | Recent Sales of Unregistered Securities. |
| (a) | Issuances of Preferred Stock. |
| (b) | Issuances of Preferred Stock Warrants. |
| (c) | Issuances of Convertible Notes. |
| (d) | Issuances of Common Stock. |
| (e) | Stock Option Grants and Option Exercises |
| (f) | Restricted Stock Units |
| (g) | Warrant Exercises |
| Item 16. | Exhibits and Financial Statement Schedules. |
Exhibit Number | | | Description of Exhibit |
1.1* | | | Form of Underwriting Agreement |
| | | Amended and Restated Certificate of Incorporation of the Registrant | |
| | | Bylaws of the Registrant | |
3.4* | | | Form of Restated Certificate of Incorporation of the Registrant (to be effective immediately prior to the completion of this offering) |
3.5* | | | Form of Amended and Restated Bylaws of the Registrant (to be effective immediately prior to the completion of this offering) |
| | | Specimen Stock Certificate evidencing the shares of common stock |
Exhibit Number | | | Description of Exhibit |
| | | Amended and Restated Investors’ Rights Agreement, dated June 1, 2020 by and among the Registrant and the investors party thereto | |
| | | Common Stock Warrant | |
5.1* | | | Opinion of Wilmer Cutler Pickering Hale and Dorr LLP |
| | | Form of Stock Option Award Agreement | |
| | | 2014 Equity Incentive Plan | |
| | | Form of Stock Option Agreement under 2014 Equity Incentive Plan | |
| | | Form of Restricted Stock Agreement under 2014 Equity Incentive Plan | |
10.5* | | | 2021 Stock Incentive Plan |
10.6* | | | Form of Stock Option Agreement under the 2021 Stock Incentive Plan |
10.7* | | | Form of Restricted Stock Agreement under the 2021 Stock Incentive Plan |
10.8* | | | Form of Restricted Stock Unit Agreement under the 2021 Stock Incentive Plan |
10.9* | | | 2021 Employee Stock Purchase Plan |
| | | Summary of Non-Employee Director Compensation Program | |
| | | Award/Contract No. W911NF2190001, dated October 13, 2020, by and between the Registrant and the US Army, as amended through April 19, 2021 | |
| | | Loan and Security Agreement, dated February 5, 2021, by and between the Registrant and East West Bank, Comerica Bank and Silicon Valley Bank | |
| | | Standard Industrial/Commercial Multi-tenant Lease, dated January 20, 2021, by and between the Registrant and Nancy Ridge Technology Center, L.P. | |
| | | Lease, dated December 4, 2018, by and between the Registrant and BMR-MODA Sorrento LP | |
| | | Lease Agreement, dated January 16, 2017, by and between the Registrant and ARE-SD Region No. 25, LLC | |
| | | Standard Form Industrial Net Lease, dated October 9, 2020, by and between the Registrant and Westcore CG Commerce, LLC | |
| | | Lease Agreement, dated June 4, 2020, by and between the Registrant and ARE-SD Region No. 67, LLC | |
| | | Employment Agreement, dated January 20, 2021, by and between the Registrant and Erica Palsis | |
| | | Employment Agreement, dated February 23, 2021, by and between the Registrant and John Gallagher | |
| | | Form of Indemnification Agreement between the Registrant and each of its Executive Officers and Directors | |
| | | Employment Agreement, dated July 8, 2021, by and between the Registrant and Ayub Khattak | |
| | | Employment Agreement, dated July 8, 2021, by and between the Registrant and Clint Sever | |
| | | Employment Agreement, dated July 8, 2021, by and between the Registrant and Chris Achar | |
10.24* | | | Form of Restricted Stock Unit Agreement, by and between the Registrant and Ayub Khattak |
10.25* | | | Form of Restricted Stock Unit Agreement, by and between the Registrant and Clint Sever |
| | | Consent of BDO USA, LLP, independent registered public accounting firm | |
23.2* | | | Consent of Wilmer Cutler Pickering Hale and Dorr LLP (included in Exhibit 5.1) |
| | | Power of Attorney (included on signature page) | |
| | | Consent of Joanne Bradford | |
| | | Consent of Carole Faig | |
| | | Consent of Maria Martinez |
| * | To be filed by amendment. |
| † | Portions of this exhibit have been omitted pursuant to Item 601(b)(10)(iv) of Regulation S-K. |
| Item 17. | Undertakings. |
| (a) | Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or |
| (b) | The undersigned registrant hereby undertakes that: |
| (1) | For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. |
| (2) | For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| | | CUE HEALTH INC. | ||||
| | | | | |||
| | | By: | | | /s/ Ayub Khattak | |
| | | | | Ayub Khattak Chief Executive Officer | ||
Signature | | | Title | | | Date |
| | | | | |||
/s/ Ayub Khattak | | | Chief Executive Officer, Director, Chairman of the Board (Principal Executive Officer) | | | September 1, 2021 |
Ayub Khattak | | |||||
| | | | | |||
/s/ John Gallagher | | | Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | | | September 1, 2021 |
John Gallagher | | |||||
| | | | | |||
/s/ Chris Achar | | | Director | | | September 1, 2021 |
Chris Achar | | |||||
| | | | | |||
/s/ Xiangmin Cui | | | Director | | | September 1, 2021 |
Xiangmin Cui | | |||||
| | | | | |||
/s/ Robin Farias-Eisner | | | Director | | | September 1, 2021 |
Robin Farias-Eisner | | |||||
| | | | | |||
/s/ Rohan Oza | | | Director | | | September 1, 2021 |
Rohan Oza | | |||||
| | | | | |||
/s/ Scott Stanford | | | Director | | | September 1, 2021 |
Scott Stanford | |







