Item 7.
Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations together with the section of this annual report entitled “Selected Financial Data” and our consolidated financial statements and related notes included elsewhere in this annual report. This discussion and other parts of this annual report contain forward-looking statements that involve risk and uncertainties, such as statements of our plans, objectives, expectations, and intentions. In this annual report, words such as “may,” “will,” “expect,” “anticipate,” “estimate,” “intend,” and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements, as described elsewhere herein. As a result of many factors, including those factors set forth in the “Risk Factors” section of this annual report, our actual results could differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
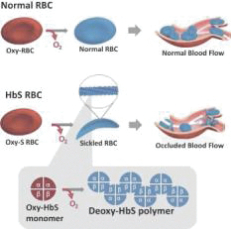
We are a biopharmaceutical company dedicated to the discovery, development and delivery of life-changing treatments that provide hope to underserved patient communities. Founded in 2011, GBT is delivering on its goal to transform the treatment and care of sickle cell disease, or SCD, a lifelong, devastating inherited blood disorder that is marked by red blood cell, or RBC, destruction and occluded blood flow and hypoxia, leading to anemia, stroke, multi-organ failure, severe pain crises, and shortened patient life span. As a result of the historic lack of treatment options, patients with SCD suffer serious morbidity and premature mortality.
It is estimated the prevalence of SCD is approximately 100,000 individuals in the United States, where newborn screening is mandatory, and approximately 60,000 individuals in Europe. The global incidence of SCD is estimated to be 250,000 to 300,000 births annually, and SCD is concentrated in populations of African, Middle Eastern and South Asian descent.
In late November 2019, we received U.S. Food and Drug Administration, or FDA, accelerated approval for our first medicine, Oxbryta (voxelotor) tablets for the treatment of SCD in adults and children 12 years of age and older. Oxbryta, an oral therapy taken once daily, is the first
FDA-approved
treatment that directly inhibits sickle hemoglobin polymerization, the root cause of SCD.
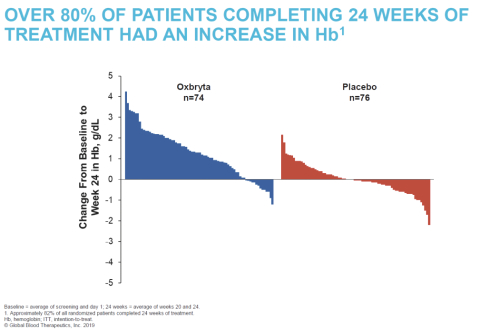
The accelerated approval of Oxbryta is based on clinically meaningful and statistically significant improvements in hemoglobin levels, accompanied by reductions in RBC destruction (hemolysis). Data from the Phase 3 HOPE (Hemoglobin Oxygen Affinity Modulation to Inhibit HbS PolymErization) Study of 274 patients 12 years of age and older with SCD showed that, after 24 weeks of treatment, 51.1% of patients receiving Oxbryta achieved a greater than 1 g/dL increase in hemoglobin compared with 6.5% receiving placebo (p<0.001). The HOPE data also demonstrated corresponding improvements in other markers of hemolysis as well as a favorable safety and tolerability profile for Oxbryta.
In early December 2019, we began to make Oxbryta available to patients through our specialty pharmacy partner network. As part of this product launch, we are focused on securing reimbursement and expanding patient access. As part of our commitment to ensuring patient access, we established GBT Source Solutions, a comprehensive program for patients who are prescribed Oxbryta that provides a wide range of practical, educational and financial support customized to each patient’s needs.
We are conducting and plan to conduct additional studies of Oxbryta, including our ongoing Phase 2a HOPE-KIDS 1 Study, an open-label, single- and multiple-dose Phase 2a study that is evaluating the safety, tolerability, pharmacokinetics and exploratory treatment effect of Oxbryta in pediatric patients aged 4 to 17 years with SCD, and, as a condition of accelerated approval, our Phase 3 HOPE-KIDS 2 Study, a post-approval confirmatory study we initiated in December 2019 that is using transcranial Doppler, or TCD, flow velocity to seek to demonstrate a decrease in stroke risk in children 2 to 15 years of age. We also expect to conduct additional clinical studies of Oxbryta, including to seek to expand the potential approved product label into younger pediatric populations.
Beyond Oxbryta, we are also engaged in other research and development activities, all of which are currently in earlier development stages, including working on new targets to develop the next generation of treatments for SCD. As part of our efforts to build our pipeline, we regularly evaluate opportunities to
in-license,
acquire or invest in new business, technology or assets or engage in related discussions with other business entities.
In August 2018, we entered into the License Agreement, or Roche Agreement, with F.
Hoffmann-La
Roche Ltd. and
Hoffmann-La
Roche Inc. (together, “Roche”) pursuant to which Roche granted us an exclusive and sublicensable worldwide license under certain patent rights and
know-how
to develop and commercialize inclacumab, a
p-selectin
inhibitor in development to address pain crises associated with the disease, including any modified compounds targeting
p-selectin
and derived from inclacumab, for all indications and uses, except diagnostic use. Roche retained a
non-exclusive,
worldwide, perpetual, royalty-free license to inclacumab solely for any diagnostic use. We are developing inclacumab as a treatment for VOCs in patients with SCD, and we expect to be able to leverage the safety data from Roche’s prior clinical studies, which were not in patients with SCD, as we proceed with our development of inclacumab. We expect to initiate a pivotal clinical study in 2021.
In December 2019, we entered into the License and Collaboration Agreement, or Syros Agreement, with Syros Pharmaceuticals, Inc., or Syros, to discover, develop and commercialize novel therapies for SCD and beta thalassemia. Under the agreement, Syros will use its leading gene control platform to identify therapeutic targets and discover drugs that potentially induce fetal hemoglobin, and we have an option to obtain an exclusive worldwide license to develop, manufacture and commercialize any compounds or products resulting from the collaboration, subject to Syros’ option to
co-promote
the first product in the United States. We will continue to seek the best scientific approaches to transform the treatment of these devastating lifelong diseases.
We own or jointly own and have exclusively licensed rights to Oxbryta and our product candidates in the United States, Europe and other major markets. We are the sole owner of issued U.S. patents covering Oxbryta, including its composition of matter, methods of use, formulations and polymorphs of Oxbryta. These issued U.S. patents covering Oxbryta will expire between 2032 and 2037, absent any applicable patent term extensions. We own or
co-own
additional pending patent applications in the United States and multiple foreign countries relating to Oxbryta.
Since our inception in 2011, we have devoted substantially all of our resources to identifying and developing Oxbryta and product candidates, including conducting clinical trials and nonclinical studies and providing general and administrative support for these operations.
We are not profitable and have incurred losses and negative cash flows from operations each year since our inception. We have financed our operations primarily through sale of equity securities and debt financing. In December 2018, we completed a
follow-on
offering and issued 3,409,090 shares of common stock at a price of $41.54 per share with proceeds of $141.1 million net of underwriting costs and commissions and offering expenses. In addition, in January 2019, we sold an additional 511,363 shares of our common stock directly to the underwriters when they exercised their over-allotment option at the price of $41.54 per share for proceeds of $21.2 million net of underwriting costs and commissions. In June 2019, we completed a
follow-on
offering and issued 3,375,527 shares of common stock at a price of $57.12 per share with proceeds of $192.4 million net of underwriting costs and commissions and offering expenses. In addition, in July 2019, we sold an additional 100,000 shares of our common stock directly to the underwriters when they exercised their over-allotment option at the price of $57.12 per share for proceeds of $5.7 million net of underwriting costs and commissions. In December 2019, we entered into a $150.0 million term loan agreement and drew down proceeds of $72.5 million net of debt issuance costs. We have an option to draw an additional $75.0 million until December 31, 2020.
Our net losses were $266.8 million for the year ended December 31, 2019, $174.2 million for the year ended December 31, 2018 and $117.0 million for the year ended December 31, 2017. As of December 31, 2019, we had an accumulated deficit of $738.9 million. Substantially all of our net losses have resulted from costs incurred in
connection with our research and development programs and from selling, general and administrative costs associated with our operations. We had $302.2 million in cash and cash equivalents and $392.8 million in marketable securities as of December 31, 2019.
Critical Accounting Polices and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with United States generally accepted accounting principles, or U.S. GAAP. The preparation of these consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements, as well as the reported expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. We believe that the accounting policies discussed below are critical to understanding our historical and future performance, as these policies relate to the more significant areas involving management’s judgments and estimates.
Pursuant to Accounting Standards Codification, Topic 606,
Revenue from Contracts with Customers
, or ASC 606
we recognize revenue upon transfer of control of promised products or services to customers in an amount that reflects the consideration we expect to receive in exchange for those products or services.
To determine revenue recognition for arrangements that we determine are within the scope of ASC 606, we perform the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) we satisfy a performance obligation. We only apply the five-step model to contracts when it is probable that we will collect substantially all of the consideration we are entitled to in exchange for the goods or services we transfer to the customer.
Our product sales consist of U.S. sales of Oxbryta, which we began shipping to customers in December 2019. Prior to December 2019, we had no product sales. We sell Oxbryta to a limited number of specialty pharmacies and a specialty distributor, or collectively, Customers. These agreements with our Customers provide for transfer of title to the product at the time the product has been delivered to the Customers. The Customers subsequently dispense our product directly to a patient or resell our product to hospitals and certain pharmacies.
We recognize revenue on product sales when the Customers obtain control of our product, which occurs at a point in time, typically upon delivery to our Customers. It is at that point that we have a right to payment and that our Customers obtain title and the risks and rewards of ownership. Shipping and handling activities are considered to be fulfillment activities rather than a separate performance obligation. Payment terms are typically
30-60
days following delivery to our Customers. As allowed under ASC 606 via practical expedient, because payment is expected shortly after delivery, we do not adjust the amount of consideration expected to be received for the effects of a significant financing component.
We consider the effects of items that can decrease the transaction price such as variable consideration and consideration payable to Customers or payer. Amounts related to such items are estimated at contract inception and updated at the end of each reporting period as additional information becomes available. The amount of variable consideration may be constrained and is included in the transaction price only to the extent it is probable
that a significant reversal of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is resolved. Revenue from product sales is recorded after considering the impact of the following variable consideration amounts along with the constraint at the time of revenue recognition:
We are subject to government mandated rebates for Medicaid Drug Rebate Program, Medicare Part D Prescription Drug Benefit Program, and other government health care programs in the United States. Rebate amounts are based upon contractual agreements or legal requirements with public sector benefit providers. We use the expected-value method for estimating these rebates based on statutory discount rates and expected utilization. The expected utilization of rebates is estimated based on third party market research data and data received from the specialty pharmacies and specialty distributor. Estimates for these rebates are adjusted quarterly to reflect the most recent information. We record an accrued liability for unpaid rebates related to products for which control has been transferred to Customers.
: We provide discounts to our Customers if they pay for our products within a defined period of time after title transfers, which terms are explicitly stated in the contract. We use the most-likely-amount method for estimating prompt payment discounts. We expect that our Customers will earn prompt payment discounts. As a result, we deduct the full amount of those discounts from total product sales when revenues are recognized and record these discounts as a reduction of accounts receivable.
We provide
co-payment
assistance to patients who have commercial insurance and meet certain eligibility requirements. We use the expected-value method for estimating
co-payment
assistance based on estimates of program redemption using data provided by third-party administrators. Estimates for the
co-payment
assistance are adjusted quarterly to reflect actual experience. We record an accrued liability for unredeemed
co-payment
assistance related to products for which control has been transferred to Customers.
Medicare Part D Coverage Gap:
The Medicare Part D coverage gap is a federal program to subsidize the costs of prescription drugs for Medicare beneficiaries in the United States, which mandates manufacturers to fund a portion of the Medicare Part D insurance coverage gap for prescription drugs sold to eligible patients. Funding of the coverage gap is generally invoiced and paid in arrears. We estimate the impact of the Medicare Part D coverage gap using the expected-value method based on an amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarters. Estimates for the impact of the Medicare Part D coverage gap are adjusted quarterly to reflect actual experience. We record an accrued liability for unpaid reserves related to the Medicare Part D coverage gap.
Consistent with industry practice, we offer limited product return rights and generally allow for the return of product that is damaged or defective, or within a few months prior to and up to a few months after the product expiration date. We consider several factors in the estimation of potential product returns, including expiration dates of the product shipped, the limited product return rights, third-party data in monitoring channel inventory levels, shelf life of the product, prescription trends, and other relevant factors. We expect product returns to be immaterial. Other than these limited returns, we do not provide any product warranties.
: Chargebacks are discounts that occur when contracted parties purchase directly from a specialty distributor. Contracted parties, which currently consist primarily of Public Health Service Institutions and federal government entities purchasing via the Federal Supply Schedule, generally purchase the product at a discounted price. The specialty distributor, in turn, charge back the difference between the price initially paid by the specialty distributor and the discounted price paid to the specialty distributor by the contracted parties to us. The reserves for chargeback are based on known sales to contracted parties. We establish the reserves for chargebacks in the same period that the related revenue is recognized, resulting in a reduction of product revenue and receivables.
Our specialty distributor provides distribution services to us for a fee, based on a contractually determined fixed percentage of sales. We estimate these distributor fees and record such estimates