SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 6-K
Report of Foreign Private Issuer
Pursuant to Rule 13a-16 or 15d-16 of
the Securities Exchange Act of 1934
For the month of | August | 2015 | |
Commission File Number | 001-37410 | ||
ESSA Pharma Inc. | |||
| (Translation of registrant’s name into English) | |||
Suite 720, 999 West Broadway, Vancouver, British Columbia, Canada, V5Z 1K5 | |||
| (Address of principal executive offices) | |||
Indicate by check mark whether the registrant files or will file annual reports under cover of Form 20-F or Form 40-F:
Form 20-F | X | Form 40-F |
Indicate by check mark if the registrant is submitting the Form 6-K in paper as permitted by Regulation S-T Rule 101(b)(1):
Indicate by check mark if the registrant is submitting the Form 6-K in paper as permitted by Regulation S-T Rule 101(b)(7):
DOCUMENTS INCLUDED AS PART OF THIS REPORT
| Document | |
| 1 | ESSA Pharma Inc. Reports Third Quarter 2015 Financial Results |
| 2 | ESSA Pharma Inc. Third Quarter 2015 Financial Results |
| 3 | ESSA Pharma Inc. Third Quarter 2015 Management Discussion and Analysis |
DOCUMENT 1
ESSA PHARMA INC. REPORTS THIRD QUARTER 2015 FINANCIAL RESULTS
Vancouver, Canada, and Houston, Texas, August 17, 2015 - ESSA Pharma Inc. (”ESSA” or the “Company”) (TSX: EPI, NASDAQ: EPIX) today reported financial results for the third quarter and three months ended June 30, 2015. Amounts, unless specified otherwise, are expressed in Canadian dollars and in accordance with International Financial Reporting Standards (“IFRS”).
Summary Results
ESSA recorded a net loss of $6.2 million ($0.35 per common share) for the quarter ended June 30, 2015 (Q3-2015), compared to a net loss of $1.0 million ($0.07 per common share) for the quarter ended June 30, 2014 (Q3-2014).
Research and Development (“R&D”) expenditures for Q3-2015 were $3.1 million compared to $0.7 million for Q3-2014. The increase was primarily due to increased R&D activity related to preclinical work on the clinical candidate EPI-506 in support of the Investigational New Drug application (the “IND”) to the U.S. Food and Drug Administration.
General and administration expenditures for Q3-2015 were $1.2 million compared to $0.3 million for Q3-2014. The increase was primarily due to increased activity as a corporate entity as the Company successfully completed a listing on the TSX Venture Exchange in January 2015, and a filing of a registration statement on Form 20-F with the United States Securities and Exchange Commission in July 2015 and a listing on the Nasdaq Capital Market (the “Nasdaq”) in July 2015.
Liquidity and Outstanding Share Capital
At June 30, 2015, ESSA had cash and cash equivalents of $7.7 million which included net proceeds of $14.2 million from the issuance of special warrants of the Company in January 2015 (the “2015 Special Warrants”). Working capital as at June 30, 2015 was $8.0 million, which the Company believes is sufficient, together with the anticipated CPRIT advance of US$3.7 million to be received upon clearance of the IND in the fourth fiscal quarter of 2015, to finance operational and capital needs for the following six months.
As of June 30, 2015, the company had 18,332,536 common shares issued and outstanding, 4,283,634 special warrants (“2015 Special Warrants”), 1,768,550 common shares issuable upon the exercise of outstanding stock options at a weighted-average exercise price of $1.33 per share, and 285,590 common shares issuable upon the exercise of outstanding warrants at a weighted-average exercise price of $3.29 per share.
As a result of the Company’s listing on the Nasdaq on July 9, 2015, the 2015 Special Warrants were exercised, without payment of any additional consideration, on July 10, 2015.
Change in Escrow Release Schedule
The Company also announced that as a result of the Company’s listing on the Toronto Stock Exchange in July, 2015, ESSA became an “established issuer” pursuant to the terms of an escrow agreement entered into with Computershare Investor Services Inc. on January 20, 2015 (the “Escrow Agreement”). The Escrow Agreement provides for an early release of ESSA common shares subject to escrow (the “Escrow Shares”) upon the company becoming an “established issuer”, with the following release schedule: ¼ of Escrow Shares released on the original listing date on a Canadian exchange, ¼ released six months after the listing date, ¼ released 12 months after the listing date and the final ¼ released 18 months after the listing date. Pursuant to the new release schedule, 281,256 Escrow Shares are to be released from escrow for the first release date under the new release schedule, being the release which is to occur on the date that is six months from the date the Company listed on the TSX Venture Exchange. The Escrow Shares remain subject to the terms of the Escrow Agreement.
Contact Information:
David Wood
Chief Financial Officer, ESSA Pharma Inc.
T: 778-331-0962
E: dwood@essapharmaceuticals.com
About ESSA Pharma Inc.
ESSA Pharma is a development-stage pharmaceutical company focused on developing novel and proprietary therapies for the treatment of castration resistant prostate cancer (“CRPC”) in patients whose disease is progressing despite treatment with current therapies. ESSA believes that its product candidate, EPI-506, can significantly expand the interval of time in which patients suffering from CRPC can benefit from hormone-based therapies. Specifically, EPI-506 acts by disrupting the androgen receptor (“AR”) signaling pathway, which is the primary pathway that drives prostate cancer growth. We have shown that EPI-002, the primary metabolite of EPI-506, prevents AR activation by binding selectively to the N-terminal domain (“NTD”) of the AR. A functional NTD is essential for activation of the AR. Blocking the NTD prevents activation of the AR by all of the three known mechanisms of activation. In pre-clinical studies, blocking the NTD has demonstrated the capability to overcome the known AR-dependent mechanisms of CRPC. ESSA was founded in 2009 and is headquartered in Vancouver, British Columbia, Canada.
About Prostate Cancer
Prostate cancer is the second-most commonly diagnosed cancer among men and the fifth most common cause of male cancer death worldwide (Globocan, 2012). Adenocarcinoma of the prostate is dependent on androgen for tumour progression and depleting or blocking androgen action has been a mainstay of hormonal treatment for over six decades. Although tumours are often initially sensitive to medical or surgical therapies that decrease levels of testosterone (for example, ADT), disease progression despite castrate levels of testosterone generally represents a transition to the lethal variant of the disease (mCRPC) and most patients ultimately succumb to the illness. The treatment of mCRPC patients has evolved rapidly over the past five years; despite these advances, additional treatment options are needed to improve clinical outcomes in patients, particularly those who fail existing treatments including abiraterone or enzalutamide, or those that have contraindications to receive those drugs. Over time, patients with mCRPC generally experience continued disease progression, worsening pain, leading to substantial morbidity and limited survival rates. In both in vitro and in vivo studies, ESSA's novel approach to blocking the androgen pathway has been shown to be effective in blocking tumour growth when current therapies are no longer effective.
Forward-Looking Statement Disclaimer
This release contains certain information which, as presented, constitutes "forward-looking information" within the meaning of applicable Canadian securities laws. Forward-looking information involves statements that relate to future events and often addresses expected future business and financial performance, containing words such as "anticipate", "believe", "plan", "estimate", "expect", and "intend", statements that an action or event "may", "might", "could", "should", or "will" be taken or occur, or other similar expressions and includes, but is not limited to, statements about the implementation of the Company’s business model and strategic plans.
Forward-looking statements and information are subject to various known and unknown risks and uncertainties, many of which are beyond the ability of ESSA to control or predict, and which may cause ESSA’s actual results, performance or achievements to be materially different from those expressed or implied thereby. Such statements reflect ESSA’s current views with respect to future events, are subject to risks and uncertainties and are necessarily based upon a number of estimates and assumptions that, while considered reasonable by ESSA as of the date of such statements, are inherently subject to
significant medical, scientific, business, economic, competitive, political and social uncertainties and contingencies. In making forward looking statements, ESSA may make various material assumptions, including but not limited to (i) obtaining positive results of clinical trials; (ii) obtaining regulatory approvals; and (iii) general business, market and economic conditions.
Forward-looking information is developed based on assumptions about such risks, uncertainties and other factors set out herein and in ESSA’s prospectus dated December 5, 2014 under the heading “Risk Factors”, a copy of which is available on ESSA’s profile at the SEDAR website at www.sedar.com, and as otherwise disclosed from time to time on ESSA’s SEDAR profile. Forward-looking statements are made based on management's beliefs, estimates and opinions on the date that statements are made and ESSA undertakes no obligation to update forward-looking statements if these beliefs, estimates and opinions or other circumstances should change, except as may be required by applicable Canadian securities laws. Readers are cautioned against attributing undue certainty to forward-looking statements.
DOCUMENT 2

CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS
(Unaudited)
(Expressed in Canadian dollars)
FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014
ESSA PHARMA INC.
CONDENSED CONSOLIDATED INTERIM STATEMENTS OF FINANCIAL POSITION
(Unaudited)
(Expressed in Canadian dollars)
AS AT
June 30, 2015 | September 30, 2014 | |||||||
| ASSETS | ||||||||
| Current | ||||||||
Cash | $ | 7,739,769 | $ | 4,146,938 | ||||
Receivables | 47,231 | 72,295 | ||||||
Prepaids (Note 4) | 1,351,434 | 69,946 | ||||||
| 9,138,434 | 4,289,179 | |||||||
Equipment (Note 5) | 149,119 | 15,806 | ||||||
Intangible assets (Note 6) | 385,437 | 404,430 | ||||||
| Total assets | $ | 9,672,990 | $ | 4,709,415 | ||||
| LIABILITIES AND SHAREHOLDERS' EQUITY | ||||||||
| Current | ||||||||
Accounts payable and accrued liabilities | $ | 1,432,206 | $ | 658,305 | ||||
Derivative liability (Note 8) | 2,796,853 | - | ||||||
Product development and relocation grant (Note 16) | - | 1,838,507 | ||||||
| 4,229,059 | 2,496,812 | |||||||
| Shareholders' equity | ||||||||
Share capital (Note 9) | 9,165,328 | 4,240,917 | ||||||
Preferred shares (Note 9) | - | 3,090,345 | ||||||
Special warrants (Note 9) | 12,763,885 | - | ||||||
Reserves (Note 10) | 1,999,698 | 1,004,467 | ||||||
Accumulated other comprehensive income (loss) | (152,705 | ) | 6,477 | |||||
Deficit | (18,332,275 | ) | (6,129,603 | ) | ||||
| 5,443,931 | 2,212,603 | |||||||
| Total liabilities and shareholders’ equity | $ | 9,672,990 | $ | 4,709,415 | ||||
| On behalf of the Board on August 14, 2015 | ||||||
| “Robert Rieder” | Director | “Franklin Berger” | Director | |||
The accompanying notes are an integral part of these condensed consolidated interim financial statements.
ESSA PHARMA INC.
CONDENSED CONSOLIDATED INTERIM STATEMENTS OF LOSS AND COMPREHENSIVE LOSS
(Unaudited)
(Expressed in Canadian dollars)
Three months ended June 30, 2015 | Three months ended June 30, 2014 | Nine months ended June 30, 2015 | Nine months ended June 30, 2014 | |||||||||||||
| OPERATING EXPENSES | ||||||||||||||||
Research and development, net of recoveries (Note 17) | $ | 3,108,000 | $ | 653,681 | $ | 6,950,487 | $ | 832,685 | ||||||||
Financing costs | 72,748 | 51,938 | 126,021 | 60,896 | ||||||||||||
General and administration, net of recoveries (Note 17) | 1,219,442 | 335,308 | 3,631,102 | 541,061 | ||||||||||||
| Total operating expenses | (4,400,190 | ) | (1,040,927 | ) | (10,707,610 | ) | (1,434,642 | ) | ||||||||
Foreign exchange | (257,941 | ) | (6,142 | ) | 971,362 | (11,465 | ) | |||||||||
Interest income | 185 | - | 185 | 227 | ||||||||||||
Loss on derivative liability (Note 8) | (1,685,743 | ) | - | (2,466,609 | ) | - | ||||||||||
| Net loss for the period | (6,343,689 | ) | (1,047,069 | ) | (12,202,672 | ) | (1,445,880 | ) | ||||||||
| OTHER COMPREHENSIVE INCOME (LOSS) | ||||||||||||||||
Cumulative translation adjustment | 140,773 | 77 | (159,182 | ) | 77 | |||||||||||
| Comprehensive loss for the period | $ | (6,202,916 | ) | $ | (1,046,992 | ) | $ | (12,361,854 | ) | $ | (1,445,803 | ) | ||||
| Basic and diluted loss per common share | $ | (0.35 | ) | $ | (0.07 | ) | $ | (0.71 | ) | $ | (0.09 | ) | ||||
Weighted average number of common sharesoutstanding | 18,163,071 | 15,687,534 | 17,070,193 | 15,687,534 | ||||||||||||
The accompanying notes are an integral part of these condensed consolidated interim financial statements.
| ESSA PHARMA INC. |
| CONDENSED CONSOLIDATED INTERIM STATEMENTS OF CASH FLOWS |
| (Unaudited) |
| (Expressed in Canadian dollars) |
| FOR THE NINE MONTHS ENDED JUNE 30, |
| 2015 | 2014 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES | ||||||||
Loss for the period | $ | (12,202,672 | ) | $ | (1,445,880 | ) | ||
Items not affecting cash: | ||||||||
Accrued interest expense | - | 24,986 | ||||||
Amortization | 28,604 | 18,993 | ||||||
Loss on derivative liability | 2,466,609 | - | ||||||
Product development and relocation grant | (1,894,263 | ) | - | |||||
Unrealized foreign exchange | 704,136 | - | ||||||
Share-based payments (Note 10) | 1,070,889 | 170,062 | ||||||
Changes in non-cash working capital items: | ||||||||
Receivables | 25,064 | (14,104 | ) | |||||
Prepaid expenses | (1,229,392 | ) | (1,260 | ) | ||||
Accounts payable and accrued liabilities | 784,267 | 363,220 | ||||||
Net cash used in operating activities | (10,246,758 | ) | (883,983 | ) | ||||
| CASH FLOWS FROM INVESTING ACTIVITIES | ||||||||
Acquisition of equipment | (136,178 | ) | - | |||||
Net cash used in investing activities | (136,178 | ) | - | |||||
| CASH FLOWS FROM FINANCING ACTIVITIES | ||||||||
Proceeds on private placement | 15,574,435 | - | ||||||
Issuance costs | (1,022,035 | ) | - | |||||
Convertible debenture issued | - | 1,000,000 | ||||||
Financing costs | - | (23,541 | ) | |||||
Options exercised | 96,760 | - | ||||||
Warrants exercised | 203,377 | - | ||||||
Net cash provided by financing activities | 14,852,537 | 976,459 | ||||||
| Effect of foreign exchange on cash | (876,770 | ) | - | |||||
| Change in cash for the period | 3,592,831 | 92,476 | ||||||
| Cash, beginning of period | 4,146,938 | 232,093 | ||||||
| Cash, end of period | $ | 7,739,769 | $ | 324,569 | ||||
Supplemental Cash Flow Information (Note 11)
The accompanying notes are an integral part of these condensed consolidated interim financial statements.
ESSA PHARMA INC.
CONDENSED CONSOLIDATED INTERIM STATEMENT OF CHANGES IN SHAREHOLDERS’ EQUITY
(Unaudited)
(Expressed in Canadian dollars)
| Reserves | ||||||||||||||||||||||||||||||||||||
| Share capital | Preferred shares | Special warrants | Convertible debenture | Share-based payments | Warrants | Cumulative translation adjustment | Deficit | Total | ||||||||||||||||||||||||||||
| Balance, September 30, 2013 | $ | 4,240,917 | $ | - | $ | - | $ | - | $ | 419,991 | $ | - | $ | - | $ | (4,168,097 | ) | $ | 492,811 | |||||||||||||||||
Issuance of convertible debenture | - | - | - | 1,000,000 | - | - | - | - | 1,000,000 | |||||||||||||||||||||||||||
Financing costs – issuance | - | - | - | (39,935 | ) | - | 16,394 | - | - | (23,541 | ) | |||||||||||||||||||||||||
Financing costs | - | - | - | 24,986 | - | - | - | - | 24,986 | |||||||||||||||||||||||||||
Share-based payments | - | - | - | - | 170,062 | - | - | - | 170,062 | |||||||||||||||||||||||||||
Loss for the period | - | - | - | - | - | - | 77 | (1,445,880 | ) | (1,445,803 | ) | |||||||||||||||||||||||||
| Balance, June 30, 2014 | $ | 4,240,917 | $ | - | $ | - | $ | 985,051 | $ | 590,053 | $ | 16,394 | $ | 77 | $ | (5,613,977 | ) | $ | 218,515 | |||||||||||||||||
Financing costs | - | - | - | 10,014 | - | - | - | - | 10,014 | |||||||||||||||||||||||||||
Conversion of debenture | - | 995,065 | - | (995,065 | ) | - | - | - | - | - | ||||||||||||||||||||||||||
Private placement (Note 9(c)) | - | 2,370,800 | - | - | - | - | - | - | 2,370,800 | |||||||||||||||||||||||||||
Share issue costs | - | (275,520 | ) | - | - | - | 50,004 | - | - | (225,516 | ) | |||||||||||||||||||||||||
Share-based payments | - | - | - | - | 348,016 | - | - | - | 348,016 | |||||||||||||||||||||||||||
Loss for the period | - | - | - | - | - | - | 6,400 | (515,626 | ) | (509,226 | ) | |||||||||||||||||||||||||
| Balance, September 30, 2014 | $ | 4,240,917 | $ | 3,090,345 | $ | - | $ | - | $ | 938,069 | $ | 66,398 | $ | 6,477 | $ | (6,129,603 | ) | $ | 2,212,603 | |||||||||||||||||
Private placement (Note 9(b)) | - | - | 1,359,280 | - | - | - | - | - | 1,359,280 | |||||||||||||||||||||||||||
Issuance costs | - | - | (193,921 | ) | - | - | 49,960 | - | - | (143,961 | ) | |||||||||||||||||||||||||
Conversion of special warrants | - | 1,165,359 | (1,165,359 | ) | - | - | - | - | - | - | ||||||||||||||||||||||||||
Private placement (Note 9(a)) | - | - | 14,215,155 | - | - | - | - | - | 14,215,155 | |||||||||||||||||||||||||||
Issuance costs | - | - | (1,212,470 | ) | - | - | - | - | - | (1,212,470 | ) | |||||||||||||||||||||||||
Conversion of preferred shares | 4,255,704 | (4,255,704 | ) | - | - | - | - | - | - | - | ||||||||||||||||||||||||||
Conversion of special warrants | 238,800 | - | (238,800 | ) | - | - | - | - | - | - | ||||||||||||||||||||||||||
Options exercised | 159,094 | - | - | - | (62,334 | ) | - | - | - | 96,760 | ||||||||||||||||||||||||||
Warrants exercised | 270,813 | - | - | - | - | (63,284 | ) | - | - | 207,529 | ||||||||||||||||||||||||||
Share-based payments | - | - | - | - | 1,070,889 | - | - | - | 1,070,889 | |||||||||||||||||||||||||||
Loss for the period | - | - | - | - | - | - | (159,182 | ) | (12,202,672 | ) | (12,361,854 | ) | ||||||||||||||||||||||||
| Balance, June 30, 2015 | $ | 9,165,328 | $ | - | $ | 12,763,885 | $ | - | $ | 1,946,624 | $ | 53,074 | $ | (152,705 | ) | $ | (18,332,275 | ) | $ | 5,443,931 | ||||||||||||||||
The accompanying notes are an integral part of these condensed consolidated interim financial statements.
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
1. NATURE AND CONTINUANCE OF OPERATIONS
ESSA Pharma Inc. (the “Company”) was incorporated under the laws of the Province of British Columbia on January 6, 2009. The Company’s head office address is Suite 720 – 999 West Broadway, Vancouver, BC, V5Z 1K5. The registered and records office address is the 26th Floor at 595 Burrard Street, Three Bentall Centre, Vancouver, BC, V7X 1L3. In the period ended September 30, 2013, the Company modified its fiscal year end from December 31 to September 30.
The Company is focused on the development of small molecule drugs for the treatment of prostate cancer. The Company has acquired a license to certain patents (the “NTD Technology”) which were the joint property of the British Columbia Cancer Agency and the University of British Columbia. As at June 30, 2015, no products are in commercial production or use.
These financial statements have been prepared in accordance with International Financial Reporting Standards (“IFRS”) assuming the Company will continue on a going-concern basis. The Company has incurred losses and negative operating cash flows since inception. The Company incurred a loss of $12,202,672 during the period ended June 30, 2015 and has an accumulated deficit of $18,332,275. The ability of the Company to continue as a going concern in the long-term depends upon its ability to develop profitable operations and to continue to raise adequate financing. As at June 30, 2015, the Company has not advanced its research into a commercially viable product. The Company’s continuation as a going concern is dependent upon the successful development of its NTD Technology to a commercial standard. Management continues to seek sources of additional financing which would assure continuation of the Company’s operations and research programs.
2. BASIS OF PRESENTATION
Statement of Compliance
These condensed consolidated interim financial statements, including comparatives, have been prepared in accordance with International Accounting Standards (“IAS”) 34 ‘Interim Financial Reporting’ (“IAS 34”) using accounting policies consistent with International Financial Reporting Standards (“IFRS”) issued by the International Accounting Standards Board (“IASB”) and Interpretations of the International Financial Reporting Interpretations Committee (“IFRIC”).
The condensed consolidated interim financial statements do not include all the information and disclosures required in the annual consolidated financial statements and should be read in conjunction with the Company’s annual consolidated financial statements for the year ended September 30, 2014.
Estimates
The Company makes estimates and assumptions about the future that affect the reported amounts of assets and liabilities. Estimates and judgments are continually evaluated based on historical experience and other factors, including expectations of future events that are believed to be reasonable under the circumstances. In the future, actual experience may differ from these estimates and assumptions.
The effect of a change in an accounting estimate is recognized prospectively by including it in comprehensive income in the period of the change, if the change affects that period only, or in the period of the change and future periods, if the change affects both. Significant assumptions about the future and other sources of estimation uncertainty that management has made at the statement of financial position date, that could result in a material adjustment to the carrying amounts of assets and liabilities, in the event that actual results differ from assumptions that have been made, relate to the following key estimates:
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
2. BASIS OF PRESENTATION (cont’d…)
Estimates (cont’d…)
Intangible Assets – impairment
The application of the Company’s accounting policy for intangible assets expenditures requires judgment in determining whether it is likely that future economic benefits will flow to the Company, which may be based on assumptions about future events or circumstances. Estimates and assumptions may change if new information becomes available. If, after expenditures are capitalized, information becomes available suggesting that the recovery of expenditures is unlikely, the amount capitalized is written off in profit or loss in the period the new information becomes available.
Intangible Assets – useful lives
Following initial recognition, the Company carries the value of intangible assets at cost less accumulated amortization and any accumulated impairment losses. Amortization is recorded on a straight-line basis based upon management’s estimate of the useful life and residual value. The estimates are reviewed at least annually and are updated if expectations change as a result of technical obsolescence or legal and other limits to use. A change in the useful life or residual value will impact the reported carrying value of the intangible assets resulting in a change in related amortization expense.
Product development and relocation grant
Pursuant to the terms of the Company’s grant from the Cancer Prevention Research Institute of Texas (“CPRIT”), the Company must meet certain terms and conditions to qualify for the grant funding. The Company has assessed its performance relative to these terms as detailed in Note 16 and has judged that there is reasonable assurance the Company will meet the terms of the grant and qualify for the funding. The Company has therefore taken into income a portion of the grant that represents expenses the Company has incurred to date under the grant parameters. The expenses are subject to assessment by CPRIT for compliance with the grant regulations which may result in certain expenses being denied and incurred in a future period.
Share-based payments and compensation
The Company has applied estimates with respect to the valuation of shares issued for non-cash consideration. Shares are valued at the fair value of the equity instruments granted at the date the Company receives the goods or services.
The Company measures the cost of equity-settled transactions with employees by reference to the fair value of the equity instruments at the date at which they are granted. Estimating fair value for share-based payment transactions requires determining the most appropriate valuation model, which is dependent on the terms and conditions of the grant. This estimate also requires determining the most appropriate inputs to the valuation model including the fair value of the underlying common shares, the expected life of the share option, volatility and dividend yield and making assumptions about them. Prior to listing on the TSX-V, the fair value of the underlying common shares was assessed as the most recent issuance price per common share for cash proceeds. Following listing on the TSX-V, the Company makes reference to prices quoted on the TSX-V. The assumptions and models used for estimating fair value for share-based payment transactions are discussed in Note 10.
Derivative financial instruments
Certain broker’s warrants are treated as derivative financial liabilities. The estimated fair value, based on the Black-Scholes model, is adjusted on a quarterly basis with gains or losses recognized in the statement of net loss and comprehensive loss. The Black-Scholes model is based on significant assumptions such as volatility, dividend yield and expected term (Note 8).
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
3. SIGNIFICANT ACCOUNTING POLICIES
The accounting policies adopted in the preparation of the condensed consolidated interim financial statements are consistent with those followed in the preparation of the Company’s annual consolidated financial statements for the year ended September 30, 2014, except for the adoption of new standards and interpretations effective as of October 1, 2014.
Embedded derivatives
Derivatives may be embedded in other financial instruments (the “host instrument”). Embedded derivatives are treated as separate derivatives when their economic characteristics and risks are not clearly and closely related to those of the host instrument, the terms of the embedded derivative are the same as those of a stand-alone derivative, and the combined contract is not held for trading or designated at fair value. These embedded derivatives are measured at fair value with subsequent changes recognized in gains or losses on derivative instruments in the statement of loss and comprehensive loss.
New standards, interpretations and amendments adopted
The following standards, amendments to standards and interpretations have been adopted for the fiscal year beginning October 1, 2014:
| ● | IFRS 2 (Amendment) | Revised definitions for ‘vesting conditions’ and ‘market condition’ related to share based compensation | |||
| ● | IFRS 13 (Amendment) | Revised disclosure requirements for contracts under the scope of IFRS 9/IAS 39 | |||
| ● | IAS 24 (Amendment) | New definitions for ‘related party’ encompassing key management personnel | |||
| ● | IAS 38 (Amendment) | Revised valuation methods for the ‘revaluation model’ for intangible assets | |||
| ● | IAS 39 | New standard for financial instruments including embedded derivatives |
The application of these standards, amendments and interpretations has not had a material impact on the results and financial position of the Company.
New standards not yet adopted
IFRS 9 Financial Instruments
IFRS 9 was issued by the IASB in October 2010. It incorporates revised requirements for the classification and measurement of financial liabilities and carrying over the existing derecognition requirements from IAS 39 Financial instruments: recognition and measurement. The revised financial liability provisions maintain the existing amortized cost measurement basis for most liabilities. New requirements apply where an entity chooses to measure a liability at fair value through profit or loss – in these cases, the portion of the change in fair value related to changes in the entity's own credit risk is presented in other comprehensive income rather than within profit or loss. IFRS 9 is effective for annual periods beginning on or after January 1, 2018. The impact of IFRS 9 on the Company’s consolidated financial instruments has not yet been determined.
IFRS 15 Revenue from Contracts with Customers
IFRS 15 establishes a single five-step model framework for determining the nature, amount, timing and uncertainty of revenue and cash flows arising from a contract with a customer. The standard is effective for annual periods beginning on or after January 1, 2018, with early adoption permitted. The change in accounting standard is unlikely to have a significant impact on the Company’s consolidated financial statements.
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
4. PREPAIDS
June 30, 2015 | September 30, 2014 | |||||||
| Clinical program deposit | $ | 1,271,865 | $ | - | ||||
| Other deposits and prepaid expenses | 79,569 | 69,946 | ||||||
| Balance | $ | 1,351,434 | $ | 69,946 | ||||
During the period ended June 30, 2015, the Company paid $1,271,865 (US$1,018,307) as an initial deposit under a clinical research services agreement with a third party in relation to the Company’s clinical trial activities.
5. EQUIPMENT
Office equipment | ||||
| Cost | ||||
| Balance, September 30, 2013 | $ | - | ||
Additions | 15,806 | |||
| Balance, September 30, 2014 | 15,806 | |||
Additions | 136,178 | |||
Net exchange differences | 7,122 | |||
| Balance, June 30, 2015 | 159,106 | |||
| Accumulated Amortization | ||||
| Balance, September 30, 2013 and 2014 | $ | - | ||
Amortization expense | 9,611 | |||
Net exchange differences | 376 | |||
| Balance, June 30, 2015 | 9,987 | |||
| Net Book Value | ||||
| Balance, September 30, 2014 | $ | 15,806 | ||
| Balance, June 30, 2015 | $ | 149,119 | ||
Amortization expense for the period ended June 30, 2015 has been recorded in “general and administrative expenses” in the statement of loss and comprehensive loss.
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
6. INTANGIBLE ASSETS
NTD Technology | June 30, 2015 | September 30, 2014 | ||||||
| Cost | ||||||||
Balance, beginning and end of period | $ | 500,017 | $ | 500,017 | ||||
| Accumulated amortization | ||||||||
Balance, beginning of period | 95,587 | 70,264 | ||||||
Amortization | 18,993 | 25,323 | ||||||
Balance, end of period | 114,580 | 95,587 | ||||||
Net book value, end of period | $ | 385,437 | $ | 404,430 | ||||
Amortization expense for the period ended June 30, 2015 and year ended September 30, 2014 has been recorded in “general and administrative expenses” in the statement of loss and comprehensive loss.
The NTD Technology is held under a License Agreement signed in fiscal 2010. As consideration for the License Agreement, the Company issued common shares of the Company. The License Agreement contains an annual royalty as a percentage of annual net revenue and a percentage of any annual sublicensing revenue earned with respect to the NTD Technology. The License Agreement stipulates certain minimum advance royalty payments of $40,000 for 2013 and escalating to $85,000 by 2017. In addition, there are certain cumulative milestone payments totaling $2,400,000 for the first compound, to be paid in stages at the start of Phase II and Phase III clinical trials, at application for marketing approval, and with further milestone payments on the second and additional compounds.
In order to maintain the License Agreement in good standing, the Company is required to spend $5,000,000, in cash or in kind, in connection with the commercialization of the NTD Technology by December 20, 2015. This expenditure commitment was completed in the year ended September 30, 2014.
7. DEBENTURE
In April 2014, the Company issued a convertible debenture to the Bloom Burton Healthcare Structured Lending Fund LP (“the Lender”) for $1,000,000. The convertible debenture terms include interest of 12% per annum payable at maturity (24 months from closing), as secured by various security agreements. The Company incurred transaction costs of $23,541 and issued 25,000 warrants to the Lender valued at $16,394, each exercisable into one common share at a price of $2.00 for a period of five years. The warrants were valued using the Black-Scholes model with a risk-free interest rate of 1.63%, term of 5 years, volatility of 80%, and dividend rate of 0%.
The principal and accrued interest are convertible into common shares of the Company at maturity at a conversion price of $1.20 unless the Company opts to settle the balance in cash. The terms of the debenture maintained control of the conversion option in the hands of the Company; therefore, the instrument has been recorded as equity in the statement of financial position.
The Company and Lender agreed to proceed with early conversion of the debenture, including accrued interest, into 517,500 preferred shares of the Company upon the closing of the financing completed in July 2014 (Note 9(c)).
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
7. DEBENTURE (cont’d…)
| Amount | ||||
| Balance, September 30, 2013 | $ | - | ||
| Proceeds on issuance of convertible debenture | 1,000,000 | |||
| Issuance costs | (39,935 | ) | ||
| Interest accrued | 35,000 | |||
| Conversion to preferred shares | (995,065 | ) | ||
| Balance, September 30, 2014 and June 30, 2015 | $ | - | ||
8. DERIVATIVE LIABILITY
In connection with the 2015 Special Warrant financing in January 2015 (Note 9(a)), the Company issued 257,018 broker warrants. Each broker warrant is exercisable to purchase one common share until January 16, 2017 at a price of US$2.75 per broker warrant.
As these broker warrants are denominated in US dollars and are exercisable into the Company’s common shares which are listed in Canadian dollars, the instrument contains an embedded derivative liability. In accordance with IFRS, an obligation to issue shares for a price that is not fixed in the Company’s functional currency, and that does not qualify as a rights offering, must be classified as a derivative liability and measured at fair value with changes recognized in the statement of loss and comprehensive loss as they arise. The embedded derivative is designated as a financial liability carried at fair value through profit and loss.
On issuance of the broker warrants, the Company recorded a derivative liability of $334,396 using the Black-Scholes model (Note 9). During the period ended June 30, 2015, 655 of the broker warrants were exercised, reducing the derivative liability by $4,152. As at June 30, 2015, the derivative liability had a fair value of $2,796,853, using the Black-Scholes model with a risk-free interest rate of 0.49%, term of 1.55 years, volatility of 80.0%, and dividend rate of 0%. The Company has recorded the resulting change in fair value of $2,466,609 in the statement of loss and comprehensive loss for the period ended June 30, 2015.
| Amount | ||||
| Balance, September 30, 2014 | $ | - | ||
| Derivative liability on issuance of broker warrants | 334,396 | |||
| Exercise of broker warrants | (4,152 | ) | ||
| Change in fair value | 2,466,609 | |||
| Balance, June 30, 2015 | $ | 2,796,853 | ||
9. SHAREHOLDERS’ EQUITY
Authorized:
Unlimited common shares, without par value.
Unlimited preferred shares, without par value.
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
9. SHAREHOLDERS’ EQUITY (cont’d…)
| Share capital | Preferred shares | Special warrants | ||||||||||||||||||||||
| Number | Amount | Number | Amount | Number | Amount | |||||||||||||||||||
| Balance, September 30, 2013 and June 30, 2014 | 15,687,534 | $ | 4,240,917 | - | $ | - | - | $ | - | |||||||||||||||
| Balance, September 30, 2014 | 15,687,534 | $ | 4,240,917 | 1,702,900 | $ | 3,090,345 | - | $ | - | |||||||||||||||
Private placement (Note 9(b)) | - | - | - | - | 679,640 | 1,359,280 | ||||||||||||||||||
Financing costs | - | - | - | - | - | (193,921 | ) | |||||||||||||||||
Conversion of special warrants | - | - | 679,640 | 1,165,359 | (679,640 | ) | (1,165,359 | ) | ||||||||||||||||
Private placement - (Note 9(a)) | - | - | - | - | 4,363,634 | 14,215,155 | ||||||||||||||||||
Financing costs | - | - | - | - | - | (1,212,470 | ) | |||||||||||||||||
Conversion of preferred shares | 2,382,540 | 4,255,704 | (2,382,540 | ) | (4,255,704 | ) | - | - | ||||||||||||||||
Conversion of special warrants | 80,000 | 238,800 | - | - | (80,000 | ) | (238,800 | ) | ||||||||||||||||
Options exercised | 81,200 | 159,094 | - | - | - | - | ||||||||||||||||||
Warrants exercised | 101,262 | 270,813 | - | - | - | - | ||||||||||||||||||
| Balance, June 30, 2015 | 18,332,536 | $ | 9,165,328 | - | $ | - | 4,283,634 | $ | 12,763,885 | |||||||||||||||
Listing on the TSX-V
The Company completed its listing on the TSX-V on January 27, 2015 (“Date of Listing”) and began trading under the symbol “EPI”.
Immediately prior to the listing, all of the Company’s 2,382,540 issued and outstanding Preferred Shares were converted into common shares.
Escrow, Lock-Up, and Supplementary Lock-Up Restrictions
The Company’s 15,687,534 common shares outstanding at September 30, 2014 are subject to various trading restrictions.
As at September 30, 2014, 5,092,000 common shares of the Company were subject to a voluntary lock-up agreement. 712,950 common shares were released on the Date of Listing, 763,872 common shares were released on March 5, 2015, and the remaining shares will be released in increments of 875,910 common shares every 3 months beginning October 27, 2015. As at June 30, 2015, 3,615,678 common shares remained subject to lock-up restrictions.
As at September 30, 2014, the remaining 10,595,034 common shares of the Company were subject to an Escrow Agreement with the TSX-V, whereby 10% of the common shares subject to escrow were released upon the Date of Listing with the remaining common shares to be released in 15% increments every 6 months thereafter. Of these common shares, 9,470,000 common shares are subject to supplementary lock-up restrictions, whereby increments of 14%, 11%, and 15% will be released 6, 9, and 12 months after the Date of Listing, respectively, and increments of 15% every 6 months thereafter. As at June 30, 2015, 9,535,530 common shares were held in escrow; however, 947,000 of the common shares released from escrow remained subject to supplementary lock-up restrictions.
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
9. SHAREHOLDERS’ EQUITY (cont’d…)
Private placements:
a) January 2015 Special Warrant Financing
In January 2015, the Company issued 4,363,634 special warrants (the “2015 Special Warrants”) at a price of US$2.75 per 2015 Special Warrant for gross proceeds of approximately US$12,000,000. Each 2015 Special Warrant is exercisable for, without payment of any additional consideration, one common share at any time by the holder thereof and all of the 2015 Special Warrants will be deemed to be exercised on the earlier of: (i) October 16, 2015 and (ii) the date on which the common shares first begin to trade on either (i) the Nasdaq Global Select Market, the Nasdaq Global Market or the Nasdaq Capital Market securities trading platforms of the NASDAQ Stock Market or (ii) the NYSE MKT securities trading platform of the New York Stock Exchange (the “U.S. Listing Date”). Should the U.S. Listing Date not occur on or prior to October 16, 2015, instead of one common share, each 2015 Special Warrant shall entitle the holder thereof to receive 1.5 common shares upon exercise or deemed exercise thereof.
In connection with the 2015 Special Warrant financing, Bloom Burton & Co. and Roth Capital Partners, LLC, as Agents, and selling group members, received cash commission equal to approximately US$706,800 and 257,018 broker warrants. Each broker warrant is exercisable to purchase one common share until January 16, 2017 at a price of US$2.75 per broker warrant. The warrants were valued at $334,396 using the Black-Scholes model with a risk-free interest rate of 0.87%, term of 2 years, volatility of 72.3%, and dividend rate of 0%, and have been recorded as a derivative liability (Note 8).
b) October 2014 Special Warrant Financing
In October 2014, the Company issued 679,640 special warrants (the “2014 Special Warrants”) at a price of $2.00 per 2014 Special Warrant for gross proceeds of $1,359,280. Each 2014 Special Warrant was deemed exercised for, without payment of any additional consideration, one Class A Preferred share in the capital of the Company (each a “Preferred Share”) on December 15, 2014, being the fifth business day after the date on which a receipt for the final prospectus of the Company qualifying the distribution of the Preferred Shares issuable on exercise of the 2014 Special Warrants had been issued. During the period ended June 30, 2015, the Preferred Shares were converted into common shares of the Company.
In connection with the 2014 Special Warrant financing, the Company paid agent and finders’ fees at 7% of proceeds raised by those parties being $40,361, a cash fee to the Agent of $30,000 plus applicable taxes and estimated other expenses of $70,594. In addition, the Agent, and associated selling group, were issued 22,675 special broker warrants (the “Special Broker Warrants”), representing 7% of the number of 2014 Special Warrants sold by the Agent and the finders were issued 2,680 Special Broker Warrants, representing 7% of the number of 2014 Special Warrants sold to purchasers introduced to the Company by such finders. Each Special Broker Warrant was deemed exercised for, without payment of any additional consideration, one broker warrant (the “Broker Warrants”). Each Broker Warrant is exercisable to acquire one common share, subject to adjustment in certain circumstances, at a price of $2.00 until October 22, 2015. The Special Broker Warrants were valued at $49,960 using the Black-Scholes model with a risk-free interest rate of 1.00%, term of 1 year, volatility of 80% and dividend rate of 0%.
c) July 2014 Preferred Shares Financing
On July 29, 2014, the Company completed a brokered private placement of 1,185,400 Preferred Shares at a price of $2.00 per Preferred Share for aggregate gross proceeds of $2,370,800. During the period ended June 30, 2015, the Preferred Shares were converted into common shares of the Company.
In conjunction with the private placement, the Company issued 79,479 warrants to Bloom Burton, the Company’s advisor, and other finders exercisable at a price of $2.00 for a period of one year. The warrants were valued at $50,004 using the Black-Scholes model with a risk-free interest rate of 1.08%, term of 1 year, volatility of 80% and dividend rate of 0%. The Company paid finders’ fees of $158,956 and other share issue costs of $66,560.
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
10. RESERVES
Stock options
The Company has adopted a Stock Option Plan which is a rolling stock option plan whereby a maximum of 10% of the issued shares will be reserved for issuance under the plan. The Stock Option Plan is consistent with the policies and rules of the TSX-V.
Stock option transactions are summarized as follows:
Number of Options | Weighted Average Exercise Price | |||||||
| Balance, September 30, 2013 | 1,119,250 | $ | 0.73 | |||||
Options granted | 1,950,469 | 1.80 | ||||||
| Balance, September 30, 2014 | 3,069,719 | 1.41 | ||||||
Options granted | 425,000 | 4.20 | ||||||
Options exercised | (81,200 | ) | (1.19 | ) | ||||
| Balance outstanding, June 30, 2015 | 3,413,519 | $ | 1.76 | |||||
Balance exercisable, June 30, 2015 | 1,768,550 | $ | 1.33 | |||||
At June 30, 2015, options were outstanding enabling holders to acquire common shares as follows:
| Exercise price | Number of options | Weighted average remaining contractual life (years) | ||||
| $ | 0.50 | 261,000 | 0.86 | |||
0.80 | 1,128,300 | 2.93 | ||||
2.00 | 1,899,219 | 4.17 | ||||
4.65 | 10,000 | 4.60 | ||||
5.15 | 10,000 | 4.68 | ||||
5.35 | 50,000 | 9.68 | ||||
14.90 | 55,000 | 9.53 | ||||
| 3,413,519 | 3.68 | |||||
Escrow, Lock-Up, and Supplementary Lock-Up Restrictions
The Company’s stock options exercisable at $0.50 and $0.80 and 150,000 stock options exercisable at $2.00 are subject to various exercise restrictions.
As at September 30, 2014, 330,500 stock options exercisable at $0.50 and $0.80 were subject to pooling restrictions. 66,100 options were released on the Date of Listing, and the remaining options will be released in increments of 66,100 common shares every 3 months thereafter. As at June 30, 2015, 198,300 options remained subject to pooling restrictions. Subsequent to June 30, 2015, 66,100 options were released.
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
10. RESERVES (cont’d…)
Escrow, Lock-Up, and Supplementary Lock-Up Restrictions (cont’d…)
As at September 30, 2014, 1,110,000 stock options exercisable at $0.50 and $0.80 were subject to an Escrow Agreement with the TSX-V, whereby 10% of the options subject to escrow were released upon the Date of Listing with the remaining options to be released in 15% increments every 6 months thereafter. Of these options, 915,000 options are also subject to lock-up and supplementary lock-up restrictions, whereby increments of 14%, 11%, and 15% will be released 6, 9, and 12 months after the Date of Listing, respectively, and increments of 15% every 6 months thereafter. As at June 30, 2015, 1,090,500 options remained subject to escrow, lock-up, and supplementary lock-up restrictions. Subsequent to June 30, 2015, 206,100 options were released.
As at June 30, 2015, 150,000 stock options exercisable at $2.00 are subject to lock-up and supplementary lock-up restrictions, whereby increments of 14% and 32.2% will be released 6 and 9 months after the Date of Listing, respectively, and increments of 17.2% every 3 months thereafter. Subsequent to June 30, 2015, 21,000 options were released.
Share-based compensation
During the period ended June 30, 2015, the Company granted 425,000 (2014 – 850,469) stock options with a weighted average fair value of $2.67 (2014 – $0.43). The Company recognized share-based payments expense of $1,070,889 (2014 - $170,062) for options granted and vesting during the period.
Share-based payments expense for the nine months ended June 30, 2015 and 2014 is allocated to its functional expense as follows:
| 2015 | 2014 | |||||||
| Research and development expense (Note 17) | $ | 621,363 | $ | 92,723 | ||||
| Financing costs | 69,508 | 35,833 | ||||||
| General and administrative (Note 17) | 380,018 | 41,506 | ||||||
| $ | 1,070,889 | $ | 170,062 | |||||
The following weighted average assumptions were used for the Black-Scholes option-pricing model valuation of stock options granted during the period:
| 2015 | 2014 | |||||||
| Risk-free interest rate | 1.43 | % | 1.35 | % | ||||
| Expected life of options | 5.28 years | 5.00 years | ||||||
| Expected annualized volatility | 79.47 | % | 80.0 | % | ||||
| Dividend | - | - | ||||||
Expected annualized volatility was determined through the comparison of historical share price volatilities used by similar publicly listed companies in the pharmaceuticals / biotechnology industry. The companies chosen for comparison were, Cipher Pharmaceuticals Inc. (June 30, 2015 – 74.57% volatility), Oncolytics Biotech Inc. (June 30, 2015 – 71.05%), Helix BioPharma Corp. (June 30, 2015 – 75.82% volatility), and Cardiome Pharma Corp. (June 30, 2015 – 78.57% volatility). As the Company is less established than these comparable companies, the Company’s stock price is expected to be slightly more volatile than the average of the companies analyzed. As such, the Company used an average annualized volatility of 75%-80%. This is consistent with estimates provided in the period ended June 30, 2015 and provides a reasonable trend for the Company’s expected volatility.
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
10. RESERVES (cont’d…)
Warrants
Warrant transactions are summarized as follows:
Number of Warrants | Weighted Average Exercise Price | |||||||
| Balance, September 30, 2013 | - | $ | - | |||||
Warrants granted | 104,479 | 2.00 | ||||||
| Balance, September 30, 2014 | 104,479 | 2.00 | ||||||
Warrants granted | 282,373 | 3.18 | ||||||
Warrants exercised | (101,262 | ) | 2.01 | |||||
| Balance outstanding and exercisable, June 30, 2015 | 285,590 | $ | 3.29 | |||||
Warrants exercisable in US dollars as at June 30, 2015 are translated at current rates to reflect current weighted average exercise price in Canadian funds for all outstanding warrants.
At June 30, 2015, warrants were outstanding enabling holders to acquire common shares as follows:
Number of Warrants | Exercise Price | Expiry Date | |||
| 25,000 | $ 2.00 | April 15, 2019 | |||
| 1,589 | 2.00 | July 29, 2015* | |||
2,638 | 2.00 | October 22, 2015** | |||
256,363 | US$2.75 | January 16, 2017 | |||
| 285,590 | |||||
* 1,589 warrants were exercised subsequent to June 30, 2015
** 1,512 warrants were exercised subsequent to June 30, 2015
11. SUPPLEMENTAL DISCLOSURE WITH RESPECT TO CASH FLOWS
During the period ended June 30, 2015, the Company:
| (a) | Accrued purchases of equipment of $19,204 through accounts payable and accrued liabilities. |
| (b) | Issued agent warrants valued at $49,960 (Note 9). |
| (c) | Recognized a derivative liability of $334,396 on the issuance of broker warrants denominated in US dollars (Note 8). |
There were no significant non-cash investing and financing transactions for the period ended June 30, 2014.
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
12. RELATED PARTY TRANSACTIONS
Key management personnel of the Company include the Chief Executive Officer, Chief Financial Officer, Chief Medical Officer, Executive VP of Research and Development, and Directors of the Company. Compensation paid to key management personnel for the nine months ended June 30, 2015 and 2014 are as follows:
| 2015 | 2014 | |||||||
| Salaries, consulting fees, and director fees | $ | 1,757,341 | $ | 240,000 | ||||
| Share-based payments | 851,615 | 117,594 | ||||||
| Total compensation | $ | 2,608,956 | $ | 357,594 | ||||
During the nine months ended June 30, 2015, the Company granted 250,000 options (2014 – 270,000) to key management personnel. The vesting of these options and options granted to key management personnel in prior periods were recorded as share-based payments expense in the statement of loss and comprehensive loss at a value of $851,615 (2014 - $117,594).
Included in accounts payable and accrued liabilities at June 30, 2015 is $99,592 (September 30, 2014 – $24,331) due to related parties with respect to key management personnel compensation and expense reimbursements. Amounts due to related parties are non-interest bearing, with no fixed terms of repayment.
Commitments
The CEO is entitled to a payment of one year of base salary upon termination without cause, increasing to two years if the termination without cause occurs after a change of control event or within 60 days prior to a change of control event where such event was under consideration at the time of termination. The CFO is entitled to a payment of one year of base salary upon termination without cause, whether or not the termination was caused by a change of control event. Stock options held by the CEO and CFO vest immediately upon a change of control.
13. SEGMENTED INFORMATION
The Company works in one industry being the development of small molecule drugs for prostate cancer. The Company’s equipment is located in the USA.
14. CAPITAL MANAGEMENT
The Company considers its capital to be the components of shareholders’ equity. The Company’s objective when managing capital is to maintain adequate levels of funding to support the development of its business and maintain the necessary corporate and administrative functions to facilitate these activities. This is done primarily through equity financing.
Future financings are dependent on market conditions and the ability to identify sources of investment. There can be no assurance the Company will be able to raise funds in the future.
There were no changes to the Company’s approach to capital management during the period. The Company is not subject to externally imposed capital requirements.
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
15. FINANCIAL INSTRUMENTS AND RISK
The Company’s financial instruments consist of cash, receivables, accounts payable and accrued liabilities and derivative liability. Cash is measured based on level 1 inputs of the fair value hierarchy. The fair value of receivables and accounts payable and accrued liabilities approximates their carrying values due to their short term to maturity. The derivative liability is measured using level 3 inputs.
Fair value estimates of financial instruments are made at a specific point in time, based on relevant information about financial markets and specific financial instruments. As these estimates are subjective in nature, involving uncertainties and matters of judgement, they cannot be determined with precision. Changes in assumptions can significantly affect estimated fair values.
Financial risk factors
The Company’s risk exposures and the impact on the Company’s financial instruments are summarized below:
Credit risk
Financial instruments that potentially subject the Company to a significant concentration of credit risk consist primarily of cash and receivables. The Company’s receivables are primarily due from refundable GST/HST and investment tax credits. The Company limits its exposure to credit loss by placing its cash with major financial institutions. Credit risk with respect to investment tax credits and GST/HST is minimal as the amounts are due from government agencies.
Liquidity risk
The Company’s approach to managing liquidity risk is to ensure that it will have sufficient liquidity to meet liabilities when due. As at June 30, 2015, the Company had a cash balance of $7,739,769 to settle current liabilities of $1,432,206. All of the Company’s current financial liabilities have contractual maturities of 30 days or due on demand and are subject to normal trade terms. The Company does not generate revenue and will be reliant on equity financing and proceeds from the CPRIT grant to fund operations. Equity financing is dependent on market conditions and may not be available on favorable terms. The CPRIT grant is dependent on the Company completing all the milestones (Note 16).
Market risk
Market risk is the risk of loss that may arise from changes in market factors such as interest rates, and foreign exchange rates.
(a) Interest rate risk
The Company has cash balances and no interest-bearing debt and therefore is not exposed to risk in the event of interest rate fluctuations. Interest income is not significant to the Company’s projected operational budget.
(b) Foreign currency risk
The Company is exposed to foreign currency risk on fluctuations related to accounts payable and accrued liabilities that are denominated in United States dollars. As at June 30, 2015, the Company had cash of US$6,004,945 and accounts payable and accrued liabilities of US$708,494. The Company anticipates that, pursuant to the product development and relocation grant disclosed in Note 16, the transactions of the Company will be increasingly subject to fluctuations in the US dollar. Additionally, the Company has broker warrants outstanding which are denominated in United States dollars (Note 8).
A 10% change in the foreign exchange rate between the Canadian and US dollar would result in a fluctuation of $601,975 in the net loss realized for the period.
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
15. FINANCIAL INSTRUMENTS AND RISK (cont’d…)
Financial risk factors (cont’d…)
Market risk (cont’d…)
(b) Foreign currency risk (cont’d…)
The Company does not currently engage in hedging activities.
(c) Price risk
The Company is exposed to price risk with respect to equity prices. The Company closely monitors individual equity movements, and the stock market to determine the appropriate course of action to be taken by the Company.
16. COMMITMENTS
The Company has the following obligations over the next five years:
Contractual obligations | 2016 | 2017 | 2018 | 2019 | 2020 | |||||||||||||||
| Minimum annual royalty per License Agreement (Note 6) | $ | 65,000 | $ | 85,000 | $ | 85,000 | $ | 85,000 | $ | 85,000 | ||||||||||
| Lease on Vancouver office space | 48,510 | 48,510 | 48,510 | 48,510 | 48,510 | |||||||||||||||
| Total | $ | 113,510 | $ | 133,510 | $ | 133,510 | $ | 133,510 | $ | 133,510 | ||||||||||
| Lease on US office space (In USD) | 241,009 | 245,690 | 250,372 | 255,053 | 57,789 | |||||||||||||||
Product Development and Relocation Grant
In February 2014 the Company received notice that it had been awarded a product development and relocation grant by the Cancer Prevention Research Institute of Texas (“CPRIT”) whereby the Company is eligible to receive up to US$12,000,000 on eligible expenditures over a three year period related to the development of the Company’s androgen receptor n-terminus blocker program for prostate cancer. The funding under CPRIT is subject to a number of conditions including negotiation and execution of an award contract which details the milestones that must be met to release the tranched CPRIT funding, proof the Company has raised the 50% matching funds to release CPRIT monies, and relocation of the project to the State of Texas such that the substantial functions of the Company related to the project grant are in Texas and the Company uses Texas-based subcontractor and collaborators wherever possible.
During the year ended September 30, 2014, the Company received US$2,793,533 as an advance on the CPRIT grant, of which US$1,153,181 in qualifying expenses was incurred in that same period. During the nine month period ended June 30, 2015, the Company incurred qualifying expenses of US$6,372,517 and has recognized a reduction in the grant liability in the statement of loss and comprehensive loss. The Company has judged there is reasonable assurance that the funds will be successfully earned under the terms of the grant.
If the Company is found to have used any grant proceeds for purposes other than intended, is in violation of the terms of the grant, or relocates its operations outside of the state of Texas, then the Company is required to repay any grant proceeds received.
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
16. COMMITMENTS (cont’d…)
Product Development and Relocation Grant (cont’d…)
Under the terms of the grant, the Company is also required to pay a royalty to CPRIT, comprised of 4% of revenues until aggregate royalty payments equal US$24,000,000, and 2% of revenues thereafter. The Company has the option to terminate the grant agreement by paying a one-time, non-refundable buyout fee, based on certain factors including the grant proceeds, and the number of months between termination date and the buyout fee payment date.
June 30, 2015 | September 30, 2014 | |||||||
| Opening balance | $ | 1,838,507 | $ | - | ||||
| Advance received | - | 3,048,694 | ||||||
| Recoveries claimed | (1,894,263 | ) | (1,256,621 | ) | ||||
| Effect of foreign exchange | 55,756 | 46,434 | ||||||
| Ending balance | $ | - | $ | 1,838,507 | ||||
Advisory Contract
In February 2014 the Company executed an Engagement Letter with Bloom Burton & Co. (“Bloom Burton”), an investment bank, to retain their services to act as its exclusive agent and financial advisor in connection with a funding strategy for the Company to involve a private financing, that is compatible with the CPRIT grant, followed by an initial public offering on a major North American stock exchange. In exchange for their services, Bloom Burton would receive a percentage of any funds raised and warrants on successful completion of the financing. The Engagement Letter was for a term of nine months and has since expired; however, Bloom Burton retains a right of first refusal on all future financings occurring up to 24 months following the Company’s initial public offering.
The July 2014 Preferred Shares financing, October 2014 Special Warrant financing, and January 2015 Special Warrant financing were completed under the terms of the Advisory Contract (Note 9).
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
17. EXPENSES BY NATURE
General and administrative expenses include the following major expenses by nature for the three and nine month periods ended June 30, 2015 and 2014:
Three months ended June 30, 2015 | Three months ended June 30, 2014 | Nine months ended June 30, 2015 | Nine months ended June 30, 2014 | |||||||||||||
| Amortization | $ | 10,644 | $ | 6,331 | $ | 28,604 | $ | 18,993 | ||||||||
| Consulting and subcontractor fees | 50,683 | 73,496 | 157,568 | 199,091 | ||||||||||||
| Director fees | 47,000 | - | 91,000 | - | ||||||||||||
| Investor relations | 86,866 | - | 174,438 | - | ||||||||||||
| Office, IT and communications | 166,529 | 21,299 | 313,771 | 27,458 | ||||||||||||
| Professional fees | 491,871 | 178,665 | 1,489,020 | 220,922 | ||||||||||||
| Regulatory fees and transfer agent | 19,998 | - | 78,747 | - | ||||||||||||
| Rent | 84,387 | 8,249 | 145,929 | 23,761 | ||||||||||||
| Salaries and benefits | 185,971 | - | 801,991 | - | ||||||||||||
| Share-based payments (Note 10) | 16,235 | 41,506 | 323,505 | 41,506 | ||||||||||||
| Travel and entertainment | 59,258 | 5,762 | 171,839 | 9,330 | ||||||||||||
| CPRIT grant claimed on eligible expenses (Note 16) | - | - | (145,310 | ) | - | |||||||||||
| Total | $ | 1,219,442 | $ | 335,308 | $ | 3,631,102 | $ | 541,061 | ||||||||
Research and development expenses include the following major expenses by nature for the three and nine month period ended June 30, 2015 and 2014:
Three months ended June 30, 2015 | Three months ended June 30, 2014 | Nine months ended June 30, 2015 | Nine months ended June 30, 2014 | |||||||||||||
| Analytical studies, formulation and testing | $ | 696,904 | $ | 153,687 | $ | 2,385,390 | $ | 261,961 | ||||||||
| Consulting | 305,129 | 132,867 | 949,187 | 216,441 | ||||||||||||
| Legal patents and license fees | 231,153 | 99,169 | 542,474 | 239,854 | ||||||||||||
| Manufacturing | 1,190,464 | 203,846 | 2,567,121 | 215,246 | ||||||||||||
| Other | 12,392 | 8,000 | 19,163 | 8,000 | ||||||||||||
| Salaries and benefits | 530,807 | 2,828 | 1,332,877 | 2,828 | ||||||||||||
| Share-based payments (Note 10) | 99,981 | 31,747 | 621,363 | 92,723 | ||||||||||||
| Travel | 105,233 | 21,537 | 345,928 | 31,102 | ||||||||||||
| SRED tax credits | (64,063 | ) | - | (64,063 | ) | (235,470 | ) | |||||||||
| CPRIT grant claimed on eligible expenses (Note 16) | - | - | (1,748,953 | ) | - | |||||||||||
| Total | $ | 3,108,000 | $ | 653,681 | $ | 6,950,487 | $ | 832,685 | ||||||||
| ESSA PHARMA INC. NOTES TO THE CONDENSED CONSOLIDATED INTERIM FINANCIAL STATEMENTS (Unaudited) (Expressed in Canadian dollars) FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014 |
18. SUBSEQUENT EVENTS
Subsequent to June 30, 2015, the Company:
| (a) | Became listed on the NASDAQ Capital Market and began trading on July 9, 2015 under the symbol “EPIX”. As a result of the listing, each of the outstanding special warrants issued on January 16, 2015 was deemed to be exercised into one common share for no additional consideration on July 13, 2015. |
| (b) | Graduated to the TSX Exchange on July 28, 2015, under its existing symbol of “EPI”. |
DOCUMENT 3
FORM 51-102F1
MANAGEMENT DISCUSSION AND ANALYSIS
FOR THE NINE MONTHS ENDED JUNE 30, 2015 AND 2014
ESSA Pharma Inc. Head Office 900 West Broadway – Suite 720 Vancouver, BC V5Z 1K5 Canada | ESSA Pharmaceuticals Corp. 7505 South Main Street – Suite 250 Houston, TX 77030 USA |
1
| Management’s Discussion and Analysis | June 30, 2015 |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS FOR NINE MONTHS ENDED JUNE 30, 2015 AND 2014
This management discussion and analysis (“MD&A”) of ESSA Pharma Inc. (the “Company” or “ESSA”) for the nine months ended June 30, 2015 and 2014 is as of August 17, 2015.
This MD&A has been prepared with reference to National Instrument 51-102 “Continuous Disclosure Obligations” of the Canadian Securities Administrators. This MD&A should be read in conjunction with the unaudited condensed interim consolidated financial statements and notes thereto for the nine months ended June 30, 2015 and 2014 as well as the audited consolidated financial statements for the year ended September 30, 2014, nine months ended September 30, 2013 and year ended December 31, 2012, and the related notes thereto. The consolidated financial statements are prepared in accordance with International Financial Reporting Standards (“IFRS”).
This MD&A may contain certain “forward-looking statements” and certain “forward-looking information” as defined under applicable Canadian securities laws. Please refer to the discussion of forward-looking statements set out under the heading “Forward-Looking Statements”, located at the end of this document. As a result of many factors, our actual results may differ materially from those anticipated in these forward-looking statements.
The Company trades on the Toronto Stock Exchange (“TSX”) under the symbol “EPI” and the NASDAQ under the symbol “EPIX”.
OVERVIEW OF THE COMPANY
We are a development-stage pharmaceutical company focused on developing novel and proprietary therapies for the treatment of prostate cancer in patients whose disease is progressing despite treatment with current therapies, including abiraterone and enzalutamide. We believe our product candidate, EPI-506, can significantly expand the interval of time in which patients suffering from castration resistant prostate cancer (“CRPC”) can benefit from hormone-based therapies. Specifically, EPI-506 acts by disrupting the androgen receptor (“AR”) signaling pathway, which is the primary pathway that drives prostate cancer growth. We have shown that EPI-002, the primary metabolite of EPI-506, prevents AR activation by binding selectively to the N-terminal domain (“NTD”) of the AR. A functional NTD is essential for activation of the AR. Blocking the NTD prevents activation of the AR by all of the known mechanisms of activation. In pre-clinical studies, blocking the NTD has demonstrated the capability to prevent AR activation and overcome the known AR-dependent mechanisms of CRPC.
We have submitted an Investigational New Drug (“IND”) application to the U.S. Food and Drug Administration (“FDA”) for EPI-506 to begin a Phase 1/2 clinical trial. We will explore the safety, tolerability, maximum tolerated dose and pharmacokinetics of EPI-506, in addition to tumor response rates in asymptomatic or minimally symptomatic patients who are no longer responding to either abiraterone or enzalutamide treatments, or both. Efficacy endpoints include prostate specific antigen (“PSA”) reduction, as well as other progression criteria. The IND is currently on clinical hold until we provide the FDA with additional chemistry and stability information relating to EPI-506. We expect to submit this information during the third quarter of 2015.
According to the American Cancer Society, in the United States, prostate cancer is the second most frequently diagnosed cancer among men, behind skin cancer. Approximately one-third of all prostate cancer patients who have been treated for local disease will subsequently have rising serum levels of PSA, which is an indication of recurrent or advanced disease. Patients with advanced disease often undergo androgen ablation therapy using analogues of luteinizing hormone releasing hormone (“LHRH”) or surgical castration. Most advanced prostate cancer patients initially respond to androgen ablation therapy, however many experience a recurrence in tumor growth despite the reduction of testosterone to castrate levels, and at that point are considered to be suffering from CRPC. Following diagnosis of CRPC, patients are often treated with anti-androgens, which block the binding of androgens to the AR.
The growth of prostate tumors is mediated by an activated AR. Generally, there are three means of activating the AR. First, androgens such as dihydrotestosterone can activate AR by binding to its ligand-binding domain (“LBD”). Second, CRPC can be driven by constitutively-active variants of AR (“vAR”) that lack a LBD and do not require androgen for activation. The third mechanism involves certain signaling pathways that activate AR independent of androgen activity. Current drugs for the treatment of prostate cancer work by focusing on the first mechanism and preventing androgen from binding to LBD, but this approach eventually fails and may not block the other two mechanisms of AR activation. By directly and selectively blocking all known means of activating the AR, we believe EPI-506 holds the potential to be effective in cases where current therapies have failed.
2
| Management’s Discussion and Analysis | June 30, 2015 |
According to the Decision Resources Group, in 2014, there were approximately 213,000 prevalent cases of CRPC, and that prevalence is expected to increase to approximately 235,000 in 2023. We expect that EPI-506 could be effective for many of those patients. For the following reasons, we intend to first focus on patients who have failed abiraterone or enzalutamide therapies:
| · | CRPC treatment remains the prostate cancer market segment with the greatest unmet need and is therefore a potentially large market; |
| · | we believe that the unique mechanism of action of our product candidate is well suited to treat patients who have failed LBD focused therapies; and |
| · | we expect the large number of patients with unmet therapeutic needs in this area will facilitate timely enrollment in our clinical trials. |
EPI-506 is a potent pro-drug of EPI-002, a stereoisomer of our discovery compound, EPI-001. A pro-drug is a drug which after administration is converted into an active form through a normal metabolic process. Pro-drugs are typically utilized to administer and more efficiently deliver another drug, which in this case is EPI-002. We believe that EPI-506 can deliver higher concentrations of EPI-002 to the target tissue than EPI-002 itself. In our pre-clinical studies, EPI-001 has been shown to shrink benign prostate tissue in mice. The pro-drug EPI-506 has demonstrated similar biological effects at doses that are lower than those required for EPI-002.
The NTD of AR is flexible with a high degree of intrinsic disorder making it extremely difficult to be used for crystal structure-based drug design. To our knowledge, no crystal structure has been identified in the AR NTD that could facilitate development of drugs which interact with this domain. We are not currently aware of any success by other drug development companies in finding drugs that bind to this drug target.
Once we have been cleared by the FDA to commence clinical trials, we intend to initiate a Phase 1/2 clinical trial with approximately 150 patients, 30 in the Phase 1 dose-escalation group and 120 in the Phase 2 dose expansion group. Key enrollment criteria are progressive, metastatic CRPC for patients who are no longer responding to abiraterone or enzalutamide. Efficacy endpoints include PSA response and radiographic progression criteria. We will also assess biomarkers of resistance including the splice variant status of patients. A biomarker is a measurable biological or chemical change that is believed to be associated with the severity or presence of a disease or condition. If the Phase 1/2 trial is successful, we expect to seek approval from the FDA to commence a Phase 3 trial in a similar patient population.
The British Columbia Cancer Agency (“BCCA”) and the University of British Columbia (“UBC”) are joint owners of the intellectual property that constitutes our primary asset. We have entered into a joint agreement with those two institutions which provides them with exclusive access to the patent and patent applications to our EPI-series compounds, including EPI-506.
Our Strategy
Our therapeutic goal is initially to provide a safe and effective therapy for prostate cancer patients who have failed current therapies, and ultimately to treat all AR-dependent forms of recurrent or advanced prostate cancer. We intend to accomplish those objectives while maximizing shareholder value. Specific components of our strategy include:
Rapidly advancing EPI-506 through clinical development and regulatory approval in CRPC patients
Once we have been cleared by the FDA to commence clinical trials, we intend to initiate a Phase 1/2 trial to determine the safety, tolerability, maximum tolerated dose, pharmacokinetics and potential therapeutic benefits of EPI-506 in CRPC patients. We expect to complete this trial by mid-2017, depending on the FDA’s clearance of our IND. If the Phase 1/2 trial is successful and following authorization by the FDA, we expect to commence a Phase 3 trial in a similar patient population. In order to accelerate the development timeline, we intend to prepare for Phase 3 development concurrently with the execution of the Phase 1/2 trial.
3
| Management’s Discussion and Analysis | June 30, 2015 |
Developing EPI-506 as an essential component of a new standard of care for the treatment of pre-CRPC and expand usage earlier in the disease stage
The activated AR is required for the growth and survival of most prostate cancer; therefore, we believe the AR NTD is an ideal target for next-generation hormone therapy. If EPI-506 is successful in treating CRPC patients, it is reasonable to expect that EPI-506 may be effective in treating earlier stage patients. Therefore, we may conduct additional clinical studies potentially leading to approval of EPI-506 for use in prostate cancer patients at an earlier disease stage.
Identifying new indication areas with high unmet medical need
Several other diseases and conditions are impacted by activated AR, including certain sub-populations of breast cancer, Kennedy’s disease (an orphan neurological condition) and male pattern baldness. While our primary focus will remain the treatment of prostate cancer, we may explore such applications in the future.
Evaluating strategic collaborations to maximize value
We currently retain all commercial rights for our EPI-series drug portfolio. We intend to evaluate potential collaborations that could enhance the value of our prostate cancer program and allow us to leverage the expertise of strategic collaborators. We also intend to explore collaborations in order to develop applications of our product candidate outside prostate cancer.
CORPORATE UPDATE AND OVERALL PERFORMANCE
ESSA is a development stage company and does not currently generate revenue. During the nine months ended June 30, 2015, the Company incurred a comprehensive loss of $12,361,854 (2014 - $1,445,803). As of June 30, 2015, the Company had cash resources of $7,739,769 (September 30, 2014 - $4,146,938) and working capital of $7,706,228 (September 30, 2014 - $3,630,874).
This corporate update highlights significant events and transactions for the three months ended June 30, 2015 and for the subsequent period to the date of this report.
Research and Development Milestones
Filing of Investigational New Drug Application for EPI-506
On March 31, 2015, the Company filed an IND application with the FDA related to the Company’s proposed clinical trial of EPI-506 in prostate cancer patients. Approval of the IND application is required in order for ESSA to commence human testing of EPI-506 in the U.S.
The IND application is a complete description of the chemistry, non-clinical pharmacodynamics and pharmacokinetics, animal toxicology, manufacturing, and other relevant information related to EPI-506 as a potential treatment for patients with advanced prostate cancer.
On April 30, 2015, subsequent to filing the application, the Company received notification from the FDA in the U.S. that the Company’s IND application has been placed on clinical hold pending receipt by the FDA of additional chemistry and pharmaceutical data related to the stability of the drug substance and drug product and a Certificate of Analysis on the drug product.
The Company will submit the required data as soon as available for the FDA’s review. The delay caused by this circumstance is not expected to be significant because the availability of those data had already been built in to the timelines prior to the IND submission.
4
| Management’s Discussion and Analysis | June 30, 2015 |
If the FDA review process results in approval to commence human testing of EPI-506, ESSA expects to recruit the first patient into its proposed Phase 1/2 clinical trial of EPI-506 in the fourth quarter of fiscal 2015. The Company also intends to seek approval from the Health Protection Branch (“HPB”) in Canada to include Canadian sites in the Phase 1/2 study, and plans to file a Clinical Trial Authorization (“CTA”) application with the HPB in order to obtain that approval.
In its upcoming Phase 1/2 clinical trial, ESSA intends to demonstrate the safety, tolerability, maximum tolerated-dose, pharmacokinetics, and efficacy of EPI-506 in metastatic CRPC patients who have failed abiraterone or enzalutamide therapy or both.
Significant Events and Transactions
Appointment of Dr. David R. Parkinson
On June 24, 2015, Dr. Parkinson was appointed to the board of directors of ESSA as an independent director.
David Parkinson is a venture partner at New Enterprise Associates. From 2007 until 2012, Dr. Parkinson served as President and CEO of Nodality, a South San Francisco-based biotechnology company. Until 2007 Dr. Parkinson was Senior Vice President, Oncology Research and Development, at Biogen Idec. Prior to joining industry, Dr. Parkinson worked at the National Cancer Institute from 1990 to 1997, serving as Chief of the Investigational Drug Branch, then as Acting Associate Director of the Cancer Therapy Evaluation Program. Dr. Parkinson is a past Chairman of the FDA’s Biologics Advisory Committee. He currently serves as director on the boards of Threshold Pharmaceuticals and Cerulean Pharma Inc., public biopharma companies focused on the discovery and development of anti-cancer drugs. Dr. Parkinson received his medical degree from the University of Toronto. He has held academic positions both at Tufts and at the University of Texas MD Anderson Cancer Center, and has authored over 100 peer-reviewed publications.
NASDAQ Listing
On July 9, 2015, the Company’s common shares (“Common Shares”) began trading on the NASDAQ Capital Market under the symbol “EPIX”.
As a result of the listing, each outstanding special warrant of the Company issued on January 16, 2015 was deemed to be exercised into one common share for no additional consideration on July 10, 2015.
TSX Graduation
On July 28, 2015, the Company graduated the listing of its Common Shares to the TSX. The Company’s Common Shares continue to trade under the symbol “EPI”.
Events Subsequent to June 30, 2015
Other than as disclosed elsewhere in this report, there were no other significant events subsequent to June 30, 2015 and prior to the date of this report.
5
| Management’s Discussion and Analysis | June 30, 2015 |
DISCUSSION OF OPERATIONS
Programs and Potential Products
Our EPI-Series Drugs
Our product candidate, EPI-506, is a selective, oral small molecule pro-drug that blocks the NTD of the AR. The AR is required for the growth and survival of most prostate cancer; therefore, the NTD of the AR is an ideal target for next-generation hormone therapy. Consistent with the inhibition of AR activity by other EPI compounds, experimentation conducted in a test-tube or in a controlled environment outside a living organism (“in vitro” studies) and experimentation done in or on the living tissue of a whole, living organism (“in vivo” studies) show that EPI-506 selectively blocks AR-dependent proliferation of human prostate cancer cells that express AR and do not inhibit the proliferation of cells that do not express functional AR or do not rely on the AR for growth and survival. By directly inhibiting the NTD of the AR, we believe EPI-506 may be able to overcome resistance mechanisms in CRPC.
We intend to initiate a Phase 1/2 clinical trial to determine the safety, tolerability, maximum tolerated dose, pharmacokinetics, and efficacy of EPI-506 in CRPC patients. In Phase 1, the trial will evaluate the benefit of once-daily dosing with EPI-506, in single- and multiple-dose escalations. The Phase 2 portion (dose expansion) of the trial will then evaluate activity in three patient cohorts: post-enzalutamide CRPC, post-abiraterone CRPC, and postenzalutamide and -abiraterone CRPC. The trial is expected to enroll approximately 150 patients.
We licensed the EPI- family of drugs from the UBC and BCCA whose initial lead compound was EPI-001. It is a mixture of four stereoisomers, each of which has the same chemical constitution, but different spatial orientation of its constituent atoms. While all the stereoisomers are active against the AR NTD, the most effective stereoisomer of EPI-001 is EPI-002 and substantial experimentation with EPI-002 has been completed and published. EPI-506 is a pro-drug of EPI-002, meaning that EPI-506 metabolizes to EPI-002 in vivo once it is dosed orally.
Pre-clinical Studies
We are focused on developing EPI-506 as our clinical development candidate. EPI-506 has been shown to be more potent than EPI-002 by oral dosing. We believe that the improved potency of EPI-506 relates to its high lipophilicity and other drug product characteristics as compared to EPI-002. The in vivo efficacy of EPI compounds has been demonstrated using a variety of human prostate cancer xenograft models.
Our initial work to support the CRPC indication consisted of pre-clinical pharmacodynamic and pharmacokinetic trials and bioanalytical development, as well as GLP (“Good Laboratory Practices”) and non-GLP toxicology trials in three species. To date, EPI-506 appears to be well-tolerated after daily oral administration. Formulation development work and bioanalytical development for pre-clinical studies have been conducted at BRI Biopharmaceutical Research Inc. in Vancouver, Canada.
To formally assess any potential safety issues related to EPI-506, we have conducted various dose-ranging non-GLP and IND enabling 28-day GLP toxicity trials in rodents and non-rodents, dose-ranging trials that lead to 28-day GLP toxicology trials. Consistent with the development of other oncology therapies at this early stage, no reproductive toxicology trials are required, given the patient population to be treated. The toxicology trials incorporate toxicokinetic data in order to correlate potential toxic effects with EPI-506 exposure. In vitro metabolism data using hepatocytes has been generated. A radiolabeled form of EPI-506 is available and will be used for further metabolism and distribution work in vivo.
We expect to address FDA-mandated Chemistry, Manufacturing and Control requirements by using a combination of in-house expertise and contractual arrangements. We had initially engaged Naeja Pharmaceutical Inc. (“Naeja”) in Edmonton, Alberta to produce non-current good manufacturing practice (“cGMP”) material for its IND-enabling toxicology trials. Chemical processes developed at Naeja, and in the laboratory of ESSA co-founder Dr. Raymond Andersen at UBC, have been transferred to the Southwest Research Institute in San Antonio, Texas for cGMP manufacturing of EPI-506 for early clinical trials. Formulation and cGMP production of the final drug product for clinical trials is performed by Catalent Pharma Solutions, St. Petersburg, Florida.
6
| Management’s Discussion and Analysis | June 30, 2015 |
7
| Management’s Discussion and Analysis | June 30, 2015 |
Planned Clinical Development Program
Phase 1/2 Clinical Trial Design for treating CRPC patients
Once we have been cleared by the FDA to commence clinical trials, we intend to initiate a Phase 1/2 trial to determine the safety, tolerability, maximum tolerated dose, pharmacokinetics and potential therapeutic benefits of EPI-506 in CRPC patients. We have submitted an IND application for EPI-506 with the FDA. The IND is currently on clinical hold until we submit additional chemical and manufacturing information about the stability of EPI-506. We expect to submit this information in the third quarter of 2015, after which the FDA will have up to 30 days to respond. Once the FDA lifts the clinical hold, we intend to initiate the trial. We also expect to submit a CTA for EPI-506 with the Therapeutic Products Directorate of Health Canada (“TPD”) in 2015.
The Phase 1 portion of the trial is expected to enroll approximately 30 patients with CRPC. Following single-dose evaluation, patients are expected to then receive once-daily oral dosing for 28 days to assess safety for dose escalation. Further, patients will continue to receive the trial drug for 12 weeks or longer to assess efficacy. The endpoints of this part of the trial will be to assess safety, tolerability, maximum tolerated dose and pharmacokinetics of EPI-506. Efficacy endpoints include PSA response and radiographic progression criteria. We plan to conduct this Phase 1 portion of the trial at five to six sites and expect it to be completed by approximately mid-2016 depending on the enrollment rate and number of dose escalation steps.
The Phase 2 portion is initially expected to enroll approximately 120 patients with CRPC. Additional patient cohorts may be added to address relevant questions on patients’ tumor response and molecular profile (e.g. splice variant status). This trial is currently expected to focus on CRPC patients with progressive metastatic disease and rising PSA who are no longer responding to abiraterone or enzalutamide, or both. The main outcomes to be measured are expected to be:
| · | PSA response (reduction in blood PSA level of 50% or more); |
| · | PSA progression; |
| · | radiographic progression; and |
| · | objective responses. |
We expect to collect circulating tumor cells so that the status of AR splice variant and other relevant biological markers related to AR signaling can be determined. We expect to conduct this trial in 25 to 30 sites and expect enrollment to be completed by the end of 2016, with the final study report by mid-2017.
Phase 3 Clinical Trial
In order to obtain regulatory approval, we will be required to carry out at least one Phase 3 trial. At this time, we expect that these patients will be a similar population of CRPC patients that were enrolled in the Phase 1/2 trial. However, the results of the Phase 1/2 trial may require modification of the initial patient population based on response and biomarker assessment. In the Phase 3 clinical trials, the key end-point is expected to be overall survival relative to patients receiving the standard-of-care. We expect to conduct the trial at many sites around the world.
8
| Management’s Discussion and Analysis | June 30, 2015 |
Development Timeline
It is currently anticipated that we will accomplish the development of EPI-506 to completion of Phase 1/2 clinical proof-of-concept according to the following timeline:
Expected Development Timeline for EPI-506
Phase 1/2 Clinical Trial Completed in Q2, 2017
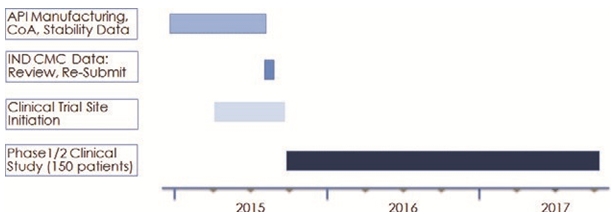
There can be no guarantee that we will complete each stage of development in accordance with the timelines set out above, or at all. Further, there can be no guarantee that the clinical hold on our IND will be removed. See “Risk Factors” elsewhere in this MD&A.
QUARTERLY FINANCIAL INFORMATION
The following table summarizes selected unaudited consolidated financial data for each of the last eight quarters, prepared in accordance with IFRS:
| For the Quarters Ended | ||||||||||||||||
June 30, 2015 | March 31, 2015 | December 31, 2014 | September 30, 2014 | |||||||||||||
| Total assets | $ | 9,672,990 | $ | 13,906,485 | $ | 4,621,182 | $ | 4,709,415 | ||||||||
| Long-term liabilities | 2,796,853 | 1,115,262 | 344,521 | 1,838,507 | ||||||||||||
| Research and development expense | 3,108,000 | 3,121,607 | 720,880 | (126,671 | ) | |||||||||||
| General and administration | 1,219,442 | 1,621,411 | 790,249 | 707,906 | ||||||||||||
| Comprehensive loss | (6,202,916 | ) | (4,621,458 | ) | (1,537,480 | ) | (509,303 | ) | ||||||||
| Basic and diluted loss per share | (0.35 | ) | (0.24 | ) | (0.10 | ) | (0.03 | ) | ||||||||
| For the Quarters Ended | ||||||||||||||||
June 30, 2014 | March 31, 2014 | December 31, 2013 | September 30, 2013 | |||||||||||||
| Total assets | $ | 766,156 | $ | 547,963 | $ | 549,440 | $ | 677,309 | ||||||||
| Long-term liabilities | - | - | - | - | ||||||||||||
| Research and development expense | 653,681 | 31,448 | 147,556 | (5,561 | ) | |||||||||||
| General and administration | 335,308 | 108,282 | 97,471 | 116,367 | ||||||||||||
| Comprehensive loss | (1,046,992 | ) | (152,476 | ) | (246,335 | ) | (112,309 | ) | ||||||||
| Basic and diluted loss per share | (0.07 | ) | (0.01 | ) | (0.02 | ) | (0.04 | ) | ||||||||
9
| Management’s Discussion and Analysis | June 30, 2015 |
Share-based payments expense for prior quarters has been reclassified in the statement of loss and comprehensive loss to be assigned to the functional expense (research and development, general and administrative, or financing) to which the underlying optionee relates. Consequently, the total expense for the functional expense items has been amended from the management’s discussion and analysis filed for quarters prior to March 31, 2015. The allocation of share-based payments expense is detailed in note 10 of the accompanying condensed consolidated interim financial statements.
From the quarters ended June 30, 2013 through June 30, 2014, the Company relied on funds raised in 2012 and tax credit refunds to meet the Company’s operating and research and development plans. There were therefore minimal changes in the capitalization of the Company during that time. In the quarter ended September 30, 2014, the Company received its first tranche of the grant from the Cancer Prevention and Research Institute of Texas (“CPRIT”) of US$2,792,533 which was recorded as a long-term liability for recognition against qualifying expenditures as those expenditures are made. The CPRIT Product Development and Relocation Grant (the “CPRIT Grant”) is detailed in the accompanying unaudited condensed consolidated interim financial statements. The CPRIT Grant agreement was executed by the Chief Executive Officer of CPRIT on July 9, 2014 (the “CPRIT Agreement”).
Also in the three months ended September 30, 2014, the Company completed a financing involving the issuance of 1,185,400 Preferred Shares at a price of $2.00 per Preferred Share for gross proceeds of $2,370,800 which supplemented the Company’s financial resources. In the three months ended December 31, 2014, the Company completed an offering of 679,640 special warrants at $2.00 per special warrant for gross proceeds of $1,359,280 (“2014 Special Warrant Financing”). On January 16, 2015, the Company issued 4,363,634 special warrants at a price of US$2.75 per special warrant for gross proceeds of $14,215,155 (“2015 Special Warrant Financing”). Accordingly, with these additional resources, the Company has accelerated its work relating to the IND filing resulting in a significant increase in comprehensive loss over prior periods. The IND application was filed on March 31, 2015.
Nine months ended June 30, 2015 and 2014
The Company incurred a comprehensive loss of $12,361,854 for the nine months ended June 30, 2015 compared to a comprehensive loss of $1,445,803 for the nine months ended June 30, 2014. Significant changes are as follows:
Research and Development
| · | The overall Research and Development (“R&D”) expense for the nine months ended June 30, 2015 was $6,950,487 compared to $832,685 for the nine months ended June 30, 2014. The gross expense for 2015 was $8,763,503 (2014 - $1,068,155) before recognition of qualifying CPRIT Grant funds of $1,748,953 (2014 - $nil) and Scientific Research & Development (“SRED”) tax credits of $64,063 (2014 - $235,470) from the Canadian government. This signifies a significantly higher investment in research and development activities, inclusive of preclinical work, from the $1,068,155 expended in the comparative period. |
| · | In the fourth quarter of fiscal 2014, the Company established office space and began to hire staff in Houston, Texas in order to undertake the preclinical work needed for the IND submission as well as developing the clinical protocol for the Phase 1/2 study that will be administered from ESSA’s Houston office. Overall, R&D activity is higher than in the comparative period as financing secured in late fiscal 2014 and early fiscal 2015 permitted a more robust research program compared to the prior period when the Company was focusing on achieving the CPRIT Grant and financing objectives. |
| · | Analytical studies, formulation and testing costs of $2,385,390 (2014 - $261,961) and manufacturing costs of $2,567,121 (2014 - $215,246) have increased compared to the nine months ended June 30, 2014. These costs relate to contracted lab facilities to conduct testing and experimentation on the Company’s EPI-series drugs. The investment for the current period was significant as the Company worked with its research facility partners to complete the documentation and information to supplement its IND application as filed at the end of March 2015. Expenditures in the three months ended June 30, 2015 included continuing advancement of chemistry and pharmaceutical data as required by the FDA for approval of the IND. |
10
| Management’s Discussion and Analysis | June 30, 2015 |
| · | Consulting fees have increased to $949,187 (2014 - $216,441) as the Company has engaged qualified professionals to conduct specific R&D services for the Company in addition to regular payments made to the Company’s Chief Scientific Officer and Chief Technical Officer over the two periods. |
| · | Legal patents and license fees have increased to $542,474 (2014 - $239,854) as the Company has submitted a number of patent applications for which the Company owns the rights. The Company has adopted a tiered patent strategy to protect its intellectual property as the pharmaceutical industry places significant importance to patents for the protection of new technologies, products and processes. The Company anticipates that there will be ongoing investment into patent applications. This amount also includes the 2014 $40,000 annual royalty under the license agreement among us, the UBC and the BCCA, dated December 2, 2010, and amended on February 10, 2011 and May 27, 2014, for certain patent rights and technology (the “License Agreement”). |
| · | Salaries and benefits relate to establishment of payroll for the Company’s Chief Medical Officer, Executive VP of Research and additional preclinical and clinical staff in Texas. The Company has invested significantly over the past nine months to develop a team to efficiently advance the IND application and preparation for Phase 1/2 clinical trials. |
Research and development expenses include the following major expenses by nature for the three and nine month period ended June 30, 2015 and 2014:
Three months ended June 30, 2015 | Three months ended June 30, 2014 | Nine months ended June 30, 2015 | Nine months ended June 30, 2014 | |||||||||||||
| Analytical studies, formulation and testing | $ | 696,904 | $ | 153,687 | $ | 2,385,390 | $ | 261,961 | ||||||||
| Consulting | 305,129 | 132,867 | 949,187 | 216,441 | ||||||||||||
| Legal patents and license fees | 231,153 | 99,169 | 542,474 | 239,854 | ||||||||||||
| Manufacturing | 1,190,464 | 203,846 | 2,567,121 | 215,246 | ||||||||||||
| Other | 12,392 | 8,000 | 19,163 | 8,000 | ||||||||||||
| Salaries and benefits | 530,807 | 2,828 | 1,332,877 | 2,828 | ||||||||||||
| Share-based payments | 99,981 | 31,747 | 621,363 | 92,723 | ||||||||||||
| Travel | 105,233 | 21,537 | 345,928 | 31,102 | ||||||||||||
| SRED tax credits | (64,063 | ) | - | (64,063 | ) | (235,470 | ) | |||||||||
| CPRIT Grant claimed on eligible expenses | - | - | (1,748,953 | ) | - | |||||||||||
| Total | $ | 3,108,000 | $ | 653,681 | $ | 6,950,487 | $ | 832,685 | ||||||||
Share-based payments expense of $621,363 (2014 - $92,723) relates to the value assigned to stock options granted to key management and consultants of the Company conducting research and development activities. The expense is recognized in relation to the grant and vesting of these equity instruments as measured by the Black-Scholes pricing model.
General and administrative
General and administration expenses for the nine months ended June 30, 2015 increased to $3,631,102 from $541,061 in 2014. Significant components of the expense in the current period included:
| · | Consulting and subcontractor fees of $157,568 (2014 - $199,091). In the current period, the costs related to ongoing administrative support and intellectual property consulting, and one-time professional recruiting services. In the prior period, costs were related to the CEO and CFO who have since been converted to full time employees included in salaries and benefits. |
11
| Management’s Discussion and Analysis | June 30, 2015 |
| · | Professional fees for legal and accounting services of $1,489,020 (2014 - $220,922) were incurred in conjunction with the corporate activities in fiscal 2015. These services have been engaged to support the Company’s financing activities and work toward listing on the TSX Venture Exchange (“TSXV”) (occurred in January 2015), the NASDAQ (occurred in July 2015) and the TSX (occurred in July 2015). The Company has worked expediently with its professional service providers to develop corporate structures and compliance standards to meet new and developing reporting requirements as a public company. |
| · | Salaries and benefits expense of $801,991 (2014 - $nil) relates primarily to the establishment of the CEO and CFO as full time employees of the organization. |
| · | Other expense categories have increased and been established as overall corporate activity has increased. These expenses predominantly relate to costs toward becoming a reporting issuer and publicly listed company. |
General and administrative expenses include the following major expenses by nature for the three and nine month periods ended June 30, 2015 and 2014:
Three months ended June 30, 2015 | Three months ended June 30, 2014 | Nine months ended June 30, 2015 | Nine months ended June 30, 2014 | |||||||||||||
| Amortization | $ | 10,644 | $ | 6,331 | $ | 28,604 | $ | 18,993 | ||||||||
| Consulting and subcontractor fees | 50,683 | 73,496 | 157,568 | 199,091 | ||||||||||||
| Director fees | 47,000 | - | 91,000 | - | ||||||||||||
| Investor relations | 86,866 | - | 174,438 | - | ||||||||||||
| Office, IT and communications | 166,529 | 21,298 | 313,771 | 27,458 | ||||||||||||
| Professional fees | 491,871 | 178,665 | 1,489,020 | 220,922 | ||||||||||||
| Regulatory fees and transfer agent | 19,998 | - | 78,747 | - | ||||||||||||
| Rent | 84,387 | 8,249 | 145,929 | 23,761 | ||||||||||||
| Salaries and benefits | 185,971 | - | 801,991 | - | ||||||||||||
| Share-based payments | 16,235 | 41,506 | 323,505 | 41,506 | ||||||||||||
| Travel and entertainment | 59,258 | 5,762 | 171,839 | 9,330 | ||||||||||||
| CPRIT Grant claimed on eligible expenses | - | - | (145,310 | ) | - | |||||||||||
| Total | $ | 1,219,442 | $ | 335,308 | $ | 3,631,102 | $ | 541,061 | ||||||||
Share-based payments expense of $323,505 (2014 - $41,506) relates to the value assigned to stock options granted to key management and consultants of the Company. The expense is recognized in relation to the grant and vest of these equity instruments as measured by the Black-Scholes pricing model.
Derivative liability
The 2015 Special Warrant Financing has increased the Company’s net financial assets denominated in U.S. dollars and exposure to fluctuations in the U.S./Canadian exchange rate. In conjunction with the 2015 Special Warrant Financing, the Company issued 257,018 broker warrants exercisable at a price of US$2.75 per Common Share. As these broker warrants are denominated in U.S. dollars and are exercisable into Common Shares which are listed in Canadian dollars, the instrument contains an embedded derivative liability. These warrants are measured at fair value with changes recognized in the statement of net loss and comprehensive loss at each reporting date. During the nine month period ended June 30, 2015, the Company recorded a loss of $2,466,609 with respect to the derivative liability.
12
| Management’s Discussion and Analysis | June 30, 2015 |
Three months ended June 30, 2015 and 2014
The Company incurred a comprehensive loss of $6,202,916 for the three months ended June 30, 2015 compared to a comprehensive loss of $1,046,992 for the three months ended June 30, 2014.
The detailed changes for the research and development and general and administrative expenses for the three months ended June 30, 2015 and 2014 are included in the tables above. Most significantly, the Company made significant increased investment in research and development costs, including analytical studies, formulation and testing costs of $696,904 (2014 - $153,687) and manufacturing costs of $1,190,464 (2014 - $203,846). In the current period, the Company worked toward the commencement of the clinical program. In general, the Company has increased R&D activity as it leverages momentum from pre-clinical development work discussed above and financings completed in the current fiscal year. Expenditures in the three months ended June 30, 2015 included continuing advancement of chemistry and pharmaceutical data as required by the FDA for approval of the IND.
General and administrative expenses have increased over the prior period as the context of the Company has changed significantly. The Company completed its TSXV listing in January 2015, and worked toward a NASDAQ listing, completed in July 2015, which has resulted in a higher overall corporate burden and engagement of professional services. Professional fees of $491,871 in the three months ended June 30, 2015 related to support in achieving a NASDAQ listing and subsequent graduation to the TSX. In the three months ended June 30, 2014, the Company expended $178,665 in professional fees with respect to executing the CPRIT Agreement and preparing for an equity financing completed in July 2014. The Company established formal payroll in August 2014 for its key executives which has led to a recurring salaries and benefits expense.
During the three month period ended June 30, 2015, the Company recorded a loss of $1,685,743 with respect to the derivative liability described above.
USE OF PROCEEDS
During the nine months ended June 30, 2015 and up to the date of this report, the Company received total net proceeds of $14,552,400 from the following financings:
| · | In October 2014, the Company received net proceeds of $1,215,319 in relation to the 2014 Special Warrant Financing. |
| · | In January 2015, the Company received net proceeds of $13,337,081 in relation to the 2015 Special Warrant Financing. |
The following table sets out a comparison of how the Company used the proceeds following the closing dates, an explanation of the variances and the impact of the variance on the ability of the Company to achieve its business objectives and milestones.
| Intended Use of Proceeds | Actual Use of Proceeds |
| To continue the development of EPI-506 Phase 1/2 clinical program through the end of calendar year 2015. | The proceeds have been used as intended to further the development of EPI-506 Phase 1/2 clinical program while meeting administrative requirements. During the nine months ended June 30, 2015, the Company incurred $6,950,487 in research and development costs in relation to the development of the EPI-506 Phase 1/2 clinical program. An additional $3,631,102 has been incurred for general and administrative costs in support of the Company’s research and development activities. The Company intends to use the remaining funds towards the completion of the Phase 1/2 clinical program. |
13
| Management’s Discussion and Analysis | June 30, 2015 |
LIQUIDITY AND CAPITAL RESOURCES
Operational activities during the period ended June 30, 2015 were financed mainly by proceeds from equity financings completed in July 2014, October 2014 and January 2015, and the CPRIT Grant. At June 30, 2015, the Company had available cash reserves of $7,739,769 and $47,231 in accounts receivable related to the refund of GST input tax credits to settle current liabilities of $1,432,206. This compares to cash reserves of $4,146,938 and $72,295 in accounts receivable related to refund of GST input tax credits at September 30, 2014 to settle current liabilities of $658,305.
Cash used in operating activities for the nine months ended June 30, 2015 was $10,246,758 (2014 - $883,983). Working capital items used cash of $420,061 (2014 - $347,856 cash generated), as the Company prepaid $1,229,392 towards a deposit on the upcoming clinical program, offset by an accounts payable increase aligned with the increase of administration and research and development activities.
Cash used in investing activities for the nine months ended June 30, 2015 was $136,178 (2014 - $nil) as the Company invested in equipment in the ongoing establishment of its Houston office.
Cash provided by financing activities for the nine months ended June 30, 2015 was $14,852,537 (2014 - $976,459). The Company received gross proceeds of $1,359,280 and $14,215,155 (US$12,000,000) from the 2014 Special Warrant Financing and 2015 Special Warrant Financing, respectively, each as previously described above, offset by $1,022,035 in share issuance costs. The Company also received $96,760 and $203,377 in proceeds on the exercise of options and warrants during the period, respectively. In the comparative period, the Company issued a convertible debenture for gross proceeds of $1,000,000, offset by $23,541 in issuance costs.
As of June 30, 2015, the Company had working capital of $7,706,228. With the addition of the expected CPRIT advance of US$3.7 million to be received upon clearance of the IND by the FDA in the third quarter of 2015, the Company has assessed that the cash position will be sufficient to finance operational and capital needs for the following six months. Management maintains operating budgets and actively reviews cash flows. While the Company expects that the CPRIT funding will be made available, there is no assurance the IND application will be approved by the FDA (see “Risk Factors”).
Future cash requirements may vary materially from those now expected due to a number of factors, including the costs associated with pre-clinical studies, formulation studies and preparations in order to initiate clinical trials and the ensuing costs associated with Phase 1/2 clinical trials of up to 150 patients in 2015-2016 and to take advantage of strategic opportunities. As a result, in the future it may be necessary to raise additional funds. These funds may come from sources such as entering into strategic collaboration arrangements, the issuance of shares from treasury, or alternative sources of financing. However, there can be no assurance that we will successfully raise funds to continue the development and commercialization of EPI-506 and our operational activities.
14
| Management’s Discussion and Analysis | June 30, 2015 |
CONTRACTUAL OBLIGATIONS
As of June 30, 2015, and in the normal course of business, we have the following obligations to make future payments, representing contracts and other commitments that are known and committed.
Contractual obligations | 2016 | 2017 | 2018 | 2019 | 2020 | |||||||||||||||
Minimum annual royalty per License Agreement (1) | $ | 65,000 | $ | 85,000 | $ | 85,000 | $ | 85,000 | $ | 85,000 | ||||||||||
| Lease on Vancouver office space | 48,510 | 48,510 | 48,510 | 48,510 | 48,510 | |||||||||||||||
| Total | $ | 113,510 | $ | 133,510 | $ | 133,510 | $ | 133,510 | $ | 133,510 | ||||||||||
| Lease on US office space (In USD) | 241,009 | 245,690 | 250,372 | 255,053 | 57,789 | |||||||||||||||
Notes:
| (1) | ESSA has the worldwide, exclusive right to develop products based on Licensed IP, as defined in, and pursuant to, the License Agreement. The Company must pay a minimum annual royalty of $65,000 in the 2015 and 2016 calendar years, increasing to $85,000 in 2017 and for each year thereafter. |
OFF-BALANCE SHEET ARRANGEMENTS & PROPOSED TRANSACTIONS
We have no material undisclosed off-balance sheet arrangements that have, or are reasonably likely to have, a current or future effect on our results of operations, financial condition, revenues or expenses, liquidity, capital expenditures or capital resources that is material to investors.
We have no material proposed transactions that have, or are reasonably likely to have, a current or future effect on our results of operations, financial condition, revenues or expenses, liquidity, capital expenditures or capital resources that is material to investors.
RELATED PARTY TRANSACTIONS
Key management personnel of the Company include Robert Rieder, the Chief Executive Officer, David Wood, Chief Financial Officer, Dr. Frank Perabo, Chief Medical Officer, Paul Cossum, Executive VP of Research and Development, Dr. Marianne Sadar, Chief Scientific Officer and Director, Dr. Raymond Andersen, Chief Technology Officer and Director, Richard Glickman, Director and Chairman of the Board, Gary Sollis, Director, Franklin Berger, Director, and David Parkinson, Director. Compensation paid to key management personnel for the nine months ended June 30, 2015 and 2014 are as follows:
| 2015 | 2014 | |||||||
| Salaries, consulting fees, and director fees | $ | 1,757,341 | $ | 240,000 | ||||
| Share-based payments | 851,615 | 117,594 | ||||||
| Total compensation | $ | 2,608,956 | $ | 357,594 | ||||
During the nine months ended June 30, 2015, the Company granted 250,000 options (2014 – 270,000) to key management personnel. The vesting of these options and options granted to key management personnel in prior periods were recorded as share-based payments expense in the statement of loss and comprehensive loss at a value of $851,615 (2014 - $117,5954). The balance of the share-based payments expense included in related party compensation in the period relates to the vesting of stock options granted in prior periods.
15
| Management’s Discussion and Analysis | June 30, 2015 |
Included in accounts payable and accrued liabilities at June 30, 2015 is $99,592 (September 30, 2014 – $24,331) due to related parties with respect to the transactions detailed above and expense reimbursements. Amounts due to related parties are non-interest bearing, with no fixed terms of repayment.
Mr. Rieder is entitled to a payment of one year of base salary upon termination without cause, increasing to two years if the termination without cause occurs after a change of control event or within 60 days prior to a change of control event where such event was under consideration at the time of termination. Mr. Wood is entitled to a payment of one year of base salary upon termination without cause, whether or not the termination was caused by a change of control event. Stock options held by the CEO and CFO vest immediately upon a change of control.
CHANGES IN OR ADOPTION OF ACCOUNTING POLICIES
The accounting policies adopted in the preparation of the condensed consolidated interim financial statements are consistent with those followed in the preparation of the Company’s annual consolidated financial statements for the year ended September 30, 2014, except for the adoption of new standards and interpretations effective as of October 1, 2014.
Embedded derivatives
Derivatives may be embedded in other financial instruments (the “host instrument”). Embedded derivatives are treated as separate derivatives when their economic characteristics and risks are not clearly and closely related to those of the host instrument, the terms of the embedded derivative are the same as those of a stand-alone derivative, and the combined contract is not held for trading or designated at fair value. These embedded derivatives are measured at fair value with subsequent changes recognized in gains or losses on derivative instruments in the statement of loss and comprehensive loss.
New standards, interpretations and amendments adopted
The following standards, amendments to standards and interpretations have been adopted for the fiscal year beginning October 1, 2014:
| IFRS 2 (Amendment) | Revised definitions for ‘vesting conditions’ and ‘market condition’ related to share based compensation | |
| IFRS 13 (Amendment) | Revised disclosure requirements for contracts under the scope of IFRS 9/IAS 39 | |
| IAS 24 (Amendment) | New definitions for ‘related party’ encompassing key management personnel | |
| IAS 38 (Amendment) | Revised valuation methods for the ‘revaluation model’ for intangible assets | |
| IAS 39 | New standard for financial instruments including embedded derivatives |
The application of these standards, amendments and interpretations has not had a material impact on the result and financial position of the Company.
New standards not yet adopted
IFRS 9 Financial Instruments (Revised)
IFRS 9 was issued by the IASB in October 2010. It incorporates revised requirements for the classification and measurement of financial liabilities and carrying over the existing derecognition requirements from IAS 39 Financial instruments: recognition and measurement. The revised financial liability provisions maintain the existing amortized cost measurement basis for most liabilities. New requirements apply where an entity chooses to measure a liability at fair value through profit or loss – in these cases, the portion of the change in fair value related to changes in the entity's own credit risk is presented in other comprehensive income rather than within profit or loss. IFRS 9 is effective for annual periods beginning on or after January 1, 2018. The impact of IFRS 9 on the Company’s consolidated financial instruments has not yet been determined.
16
| Management’s Discussion and Analysis | June 30, 2015 |
IFRS 15 Revenue from Contracts with Customers
IFRS 15 establishes a single five-step model framework for determining the nature, amount, timing and uncertainty of revenue and cash flows arising from a contract with a customer. The standard is effective for annual periods beginning on or after January 1, 2018, with early adoption permitted. The change in accounting standard is unlikely to have a significant impact on the Company’s consolidated financial statements.
CRITICAL ACCOUNTING ESTIMATES
The Company makes estimates and assumptions about the future that affect the reported amounts of assets and liabilities. Estimates and judgments are continually evaluated based on historical experience and other factors, including expectations of future events that are believed to be reasonable under the circumstances. In the future, actual experience may differ from these estimates and assumptions.
The effect of a change in an accounting estimate is recognized prospectively by including it in comprehensive income in the period of the change, if the change affects that period only, or in the period of the change and future periods, if the change affects both. Significant assumptions about the future and other sources of estimation uncertainty that management has made at the statement of financial position date, that could result in a material adjustment to the carrying amounts of assets and liabilities, in the event that actual results differ from assumptions that have been made, relate to the following key estimates:
Intangible Assets – impairment
The application of the Company’s accounting policy for intangible assets expenditures requires judgment in determining whether it is likely that future economic benefits will flow to the Company, which may be based on assumptions about future events or circumstances. Estimates and assumptions may change if new information becomes available. If, after expenditures are capitalized, information becomes available suggesting that the recovery of expenditures is unlikely, the amount capitalized is written off in profit or loss in the period the new information becomes available.
Intangible Assets – useful lives
Following initial recognition, the Company carries the value of intangible assets at cost less accumulated amortization and any accumulated impairment losses. Amortization is recorded on a straight-line basis based upon management’s estimate of the useful life and residual value. The estimates are reviewed at least annually and are updated if expectations change as a result of technical obsolescence or legal and other limits to use. A change in the useful life or residual value will impact the reported carrying value of the intangible assets resulting in a change in related amortization expense.
Product development and relocation grant
Pursuant to the terms of the Company’s CPRIT Grant, the Company must meet certain terms and conditions to qualify for the grant funding. The Company has assessed its performance relative to these terms as detailed in the accompanying unaudited condensed consolidated financial statements (Note 16) and has judged that there is reasonable assurance the Company will meet the terms of the grant and qualify for the funding. The Company has therefore taken into income a portion of the grant that represents expenses the Company has incurred to date under the grant parameters. The expenses are subject to assessment by CPRIT for compliance with the grant regulations which may result in certain expenses being denied and incurred in a future period.
17
| Management’s Discussion and Analysis | June 30, 2015 |
Share-based payments and compensation
The Company has applied estimates with respect to the valuation of shares issued for non-cash consideration. Shares are valued at the fair value of the equity instruments granted at the date the Company receives the goods or services.
The Company measures the cost of equity-settled transactions with employees by reference to the fair value of the equity instruments at the date at which they are granted. Estimating fair value for share-based payment transactions requires determining the most appropriate valuation model, which is dependent on the terms and conditions of the grant. This estimate also requires determining the most appropriate inputs to the valuation model including the fair value of the underlying common shares, the expected life of the share option, volatility and dividend yield and making assumptions about them. Prior to listing on the TSX-V, the fair value of the underlying common shares was assessed as the most recent issuance price per common share for cash proceeds. Following listing on the TSX-V, the Company makes reference to prices quoted on the TSX-V. The assumptions and models used for estimating fair value for share-based payment transactions are discussed in Note 10 of the accompanying unaudited condensed consolidated interim financial statements.
Derivative financial instruments
Certain broker’s warrants are treated as derivative financial liabilities. The estimated fair value, based on the Black-Scholes model, is adjusted on a quarterly basis with gains or losses recognized in the statement of net loss and comprehensive loss. The Black-Scholes model is based on significant assumptions such as volatility, dividend yield and expected term as detailed in Note 8 of the accompanying unaudited condensed consolidated interim financial statements.
FINANCIAL INSTRUMENTS AND RISKS
The Company’s financial instruments consist of cash, receivables, accounts payable and accrued liabilities and derivative liability. Cash is measured based on level 1 inputs of the fair value hierarchy. The fair value of receivables and accounts payable and accrued liabilities approximates their carrying values due to their short term to maturity. The derivative liability is measured using level 3 inputs.
Fair value estimates of financial instruments are made at a specific point in time, based on relevant information about financial markets and specific financial instruments. As these estimates are subjective in nature, involving uncertainties and matters of judgement, they cannot be determined with precision. Changes in assumptions can significantly affect estimated fair values.
Financial risk factors
The Company’s risk exposures and the impact on the Company’s financial instruments are summarized below:
Credit risk
Financial instruments that potentially subject the Company to a significant concentration of credit risk consist primarily of cash and receivables. The Company’s receivables are primarily due from refundable GST/HST and investment tax credits. The Company limits its exposure to credit loss by placing its cash with major financial institutions. Credit risk with respect to investment tax credits and GST/HST is minimal as the amounts are due from government agencies.
18
| Management’s Discussion and Analysis | June 30, 2015 |
Liquidity risk
The Company’s approach to managing liquidity risk is to ensure that it will have sufficient liquidity to meet liabilities when due. As at June 30, 2015, the Company had a cash balance of $7,739,769 to settle current liabilities of $1,432,206. All of the Company’s current financial liabilities have contractual maturities of 30 days or due on demand and are subject to normal trade terms. The Company does not generate revenue and will be reliant on equity financing and proceeds from the CPRIT Grant to fund operations. Equity financing is dependent on market conditions and may not be available on favorable terms. The CPRIT Grant is dependent on the Company completing all the milestones (see accompanying unaudited condensed consolidated interim financial statements for details with respect to the CPRIT Grant terms).
Market risk
Market risk is the risk of loss that may arise from changes in market factors such as interest rates, and foreign exchange rates.
| (a) | Interest rate risk | |
| The Company has cash balances and no interest-bearing debt and therefore is not exposed to risk in the event of interest rate fluctuations. | ||
| (b) | Foreign currency risk | |
| The Company is exposed to foreign currency risk on fluctuations related to accounts payable and accrued liabilities that are denominated in United States dollars. As at June 30, 2015, the Company had cash of US$6,004,945 and accounts payable and accrued liabilities of US$708,494. The Company anticipates that, pursuant to the product development and relocation grant disclosed in Note 14 of the accompanying unaudited condensed consolidated interim financial statements, the transactions of the Company will be increasingly subject to fluctuations in the U.S. dollar. Additionally, the Company has broker warrants outstanding which are denominated in United States dollars (Note 8 of the accompanying unaudited condensed consolidated interim financial statements). | ||
| A 10% change in the foreign exchange rate between the Canadian and U.S. dollar would result in a fluctuation of $601,975 in the net loss realized for the period. | ||
| The Company does not currently engage in hedging activities. | ||
| (c) | Price risk | |
| The Company is exposed to price risk with respect to equity prices. The Company closely monitors individual equity movements, and the stock market to determine the appropriate course of action to be taken by the Company. |
ADDITIONAL INFORMATION
Additional information can be found on Sedar at www.sedar.com, the website of the United States Securities and Exchange Commission at www.sec.gov and the Company’s website at www.essapharma.com.
OUTSTANDING SHARE CAPITAL
Equity instruments outstanding as of the date of this MD&A: | ||||
| Common shares | 22,629,271 | |||
| Stock options | 3,403,519 | |||
| Warrants | 282,489 | |||
19
| Management’s Discussion and Analysis | June 30, 2015 |
RISK FACTORS
An investment in our Common Shares involves a high degree of risk. You should carefully consider the risks and uncertainties described below, together with all of the other information, including our financial statements and related notes thereto, before deciding to invest in our Common Shares. The occurrence of any of the following risks could have a material adverse effect on our business, financial condition, results of operations and future prospects. In these circumstances, the market price of our Common Shares could decline, and you may lose all or part of your investment. This document also contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in the forward-looking statements as a result of a number of factors, including the risks described below. See “Cautionary Note Regarding Forward-Looking Statements.”
Risks Related to Our Financial Position and Capital Needs
We have incurred significant losses in every quarter since our inception and anticipate that we will continue to incur significant losses in the future and may never generate profits from operations or maintain profitability.
We are a development stage pharmaceutical company with a limited operating history. Investment in pharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk that any potential product candidate will fail to demonstrate adequate effect or an acceptable safety profile, gain regulatory approval or become commercially viable. We do not have any products approved by regulatory authorities for marketing or commercial sale and have not generated any revenue from product sales, or otherwise, to date, and we continue to incur significant research, development and other expenses related to our ongoing operations. As a result, we are not profitable and have incurred losses in every reporting period since inception in 2009.
We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate these losses to increase as it continues the research and development of, and seeks regulatory approvals for, any of our future product candidates and potentially begin to commercialize any products that may achieve regulatory approval. We may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our financial condition. The size of our future net losses will depend, in part, on the rate of future growth of our expenses and ability to generate revenues. Our prior losses and expected future losses have had and will continue to have an adverse effect on our financial condition.
Even if we are able to commercialize any product candidate, there can be no assurance that we will generate significant revenues or ever achieve profitability.
We expect to continue to incur substantial losses for the foreseeable future and these losses may increase over time. We are uncertain about when or if we will be able to achieve or sustain profitability. If we achieve profitability in the future, it may not be able to sustain profitability in subsequent periods. Failure to become and remain profitable would impair our ability to sustain operations and adversely affect the price of the Common Shares and our ability to raise capital.
We currently have no commercial revenue and may never become profitable.
To date, the only revenue we have generated has been from the receipt of research grants. Our ability to generate revenue and become profitable depends upon our ability to obtain regulatory approval for, and successfully commercialize, EPI-506 or other product candidates that we may develop, in-license or acquire in the future.
We have submitted an IND application to the FDA for EPI-506 to begin a Phase 1/2 clinical trial, which is currently on clinical hold pending additional information. Even if we are able to successfully achieve regulatory approval for EPI-506, we do not know when this product will generate revenue for us, if at all. We have not generated, and do not expect to generate, any product revenue for the foreseeable future, and we expect to continue to incur significant operating losses for the foreseeable future due to the cost of research and development, preclinical studies and clinical trials and the regulatory approval process for our product candidates. The amount of future losses is uncertain and will depend, in part, on the rate of growth of our expenses.
20
| Management’s Discussion and Analysis | June 30, 2015 |
Our ability to generate revenue from our product candidates also depends on a number of additional factors, including our ability to:
| · | successfully complete development activities, including the remaining preclinical studies and planned clinical trials for our product candidates; |
| · | complete and submit New Drug Applications (“NDAs”) to the FDA; |
| · | complete and submit applications to, and obtain regulatory approval from, foreign regulatory authorities; |
| · | manufacture any approved products in commercial quantities and on commercially reasonable terms; |
| · | develop a commercial organization, or find suitable partners, to market, sell and distribute approved products in the markets in which we have retained commercialization rights; |
| · | achieve acceptance among patients, clinicians and advocacy groups for any products we develop; |
| · | obtain coverage and adequate reimbursement from third parties, including government payors; and |
| · | set a commercially viable price for any products for which we may receive approval. |
We will have significant additional future capital needs and there are uncertainties as to our ability to raise additional funding.
We will require significant additional capital resources to expand our business, in particular the further development of our product candidates. Advancing our product candidates or the acquisition and development of any new products or product candidates may require considerable resources and additional access to capital. In addition, our future cash requirements may vary materially from those now expected. For example, our future capital requirements may increase if:
| · | we experience generic competition from other life sciences companies or in more markets than anticipated; |
| · | we experience delays or unexpected increases in costs in connection with obtaining regulatory approvals in the various markets where we hope to sell our products; |
| · | we experience unexpected or increased costs relating to preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, or other lawsuits, brought by either us or our competitors; |
| · | we experience scientific progress sooner than expected in our discovery, research and development projects, if we expand the magnitude and scope of these activities, or if we modify our focus as a result of discoveries; |
| · | we experience setbacks or delays in our pre-clinical studies or clinical trials; |
| · | we are required to perform additional pre-clinical studies or clinical trials; or |
| · | we elect to develop, acquire or license new technologies, products or businesses. |
We could potentially seek additional funding through strategic collaborations, alliances and licensing arrangements, through public or private equity or debt financing, or through other transactions. However, if capital market conditions in general, or with respect to life sciences companies such as ours, are unfavorable, our ability to obtain significant additional funding on acceptable terms, or at all, will be negatively affected.
If sufficient capital is not available, we may be required to delay our business expansion or our research and development projects, either of which could have a material adverse effect on our business, financial condition, prospects and results of operations.
Raising additional capital may cause dilution to our existing shareholders, restrict our operations or require us to relinquish rights to technologies or any future product candidates.
Until we can generate substantial revenue from product sales, if ever, we expect to finance future cash needs through a combination of private and public equity offerings, debt financings, strategic collaborations and alliances and licensing arrangements. Additional financing that we may pursue may involve the sale of our Common Shares or financial instruments that are exchangeable for, or convertible into, our Common Shares, which could result in significant dilution to our shareholders and the terms may include liquidation or other preferences that adversely affect the rights of existing shareholders. Additional capital may not be available on reasonable terms, if at all. Furthermore, these securities may have rights senior to those of our Common Shares and could include restrictive covenants limiting our ability to take important actions and potentially impair our competitiveness, such as limitations on our ability to incur additional debt, make capital expenditures, acquire, sell or license intellectual property rights or declare dividends. If we raise additional funds through strategic collaborations and alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to technologies or future
21
| Management’s Discussion and Analysis | June 30, 2015 |
product candidates, or grant licenses on terms that are not favorable to us. If we are unable to raise additional funds when needed, we may be required to delay, limit, reduce or terminate our product development or commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
We remain subject to the restrictions and conditions of the CPRIT Agreement. Failure to comply with the CPRIT Agreement may adversely affect our financial condition and results of operations.
We have relied on the CPRIT Grant to fund a portion of our operations to date and expect to receive additional proceeds from the CPRIT Grant, including $3.7 million expected in the third quarter of 2015 upon clearance of the IND by the FDA, and a further $5.5 million expected in mid-2016 upon determining the maximum tolerated dose in the Phase 1 portion of the clinical trial. The CPRIT Grant is subject to our compliance with the scope of work outlined in the CPRIT Agreement and demonstration of our progress towards achievement of the milestones set forth in the CPRIT Agreement. If we fail to comply with the terms of the CPRIT Agreement, we may not receive the remaining tranches of the CPRIT Grant or we may be required to reimburse some or the entire CPRIT Grant. Further, the CPRIT Grant may only be applied to a limited number of allowable expenses. Failure to obtain the remaining tranches of the CPRIT Grant or being required to reimburse all or a portion of the CPRIT Grant may cause a halt or delay in our ongoing operations, which may adversely affect our financial condition and results of operations.
If we fail to comply with the terms of the CPRIT Agreement, CPRIT will have the option to pursue the transfer and assignment of our rights, title and interest in the intellectual property rights and technologies developed as a result of the CPRIT Grant. Failure to maintain ownership over our intellectual property and technologies may adversely affect our financial condition and results of operations.
We have a limited operating history, which may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
Our operations to date have been primarily limited to organizing and staffing, in-licensing intellectual property, discovering and developing small molecule product candidates and conducting pre-clinical research. We have not yet obtained regulatory approval for any of our product candidates. Consequently, evaluating our performance, viability or future success will be more difficult than it would be if we had a longer operating history or approved products on the market.
Risks Related to Our Business
Our future success is dependent primarily on the regulatory approval and commercialization of a single product candidate, EPI-506, which is still in pre-clinical development and will require significant capital resources and years of clinical development efforts.
We do not have any products that have obtained regulatory approval. Currently, our only product candidate is EPI-506, and once we have been cleared by the FDA to commence clinical trials, we expect to execute a Phase 1/2 clinical trial starting in 2015 to determine the safety and potential therapeutic benefits of EPI-506 in CRPC patients. We have submitted an IND application to the FDA for EPI-506 to begin the Phase 1/2 clinical trial. The IND application is currently on clinical hold until we provide the FDA with additional chemical and manufacturing information about the stability of EPI-506. We expect to submit this information in the third quarter of 2015, after which the FDA will have up to 30 days to respond. We cannot guarantee that the FDA will lift the clinical hold and allow the trial to proceed on the timeline expected, or at all. As a result, our near-term prospects, including our ability to finance our operations and generate revenue, are substantially dependent on our ability to obtain regulatory approval for, and, if approved, to successfully commercialize EPI-506 in a timely manner. We cannot commercialize EPI-506 or other future product candidates in the United States without first obtaining regulatory approval for the product from the FDA; similarly, we cannot commercialize EPI-506 or other future product candidates outside of the United States without obtaining regulatory approval from comparable foreign regulatory authorities. The FDA review process typically takes years to complete and approval is never guaranteed.
22
| Management’s Discussion and Analysis | June 30, 2015 |
As an organization, we have never previously conducted a clinical trial or submitted a New Drug Application/New Drug Submission or a Clinical Trial Application and may be unable to do so for our current or future product candidates.
Our product candidates are currently at the pre-clinical stage and still need to complete all levels of clinical trials. The conduct of Phase 3 clinical trials and the submission of a successful IND CTA and NDA/New Drug Submission (“NDS”) to the applicable regulatory authority is a complicated process. As an organization, we have not conducted a clinical trial before, have limited experience in preparing, submitting and prosecuting regulatory filings and have not submitted an NDA/NDS or CTA before. We also have had limited interactions with the FDA and regulatory authorities in other jurisdictions. Consequently, even if our initial clinical trials are successful, we may be unable to successfully and efficiently execute and complete necessary clinical trials in a way that leads to NDA/NDS submission and approval of our proposed products or any other future product candidate we may develop. We may require more time and incur greater costs than competitors and may not succeed in obtaining regulatory approvals of products that we develop. Failure to commence or complete, or delays in, our planned clinical trials, would prevent us from or delay us in commercializing proposed products or any other future product candidate we develop.
Clinical drug development involves a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results and our current and future product candidates may not have favorable results in later trials or in the commercial setting.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of pre-clinical studies and early clinical trials may not be predictive of the results of later-stage clinical trials. Pre-clinical tests and Phase 1 and Phase 2 clinical trials are primarily designed to test safety, to study pharmacokinetics and pharmacodynamics and to understand the side effects of product candidates at various doses and schedules. Success in pre-clinical or animal studies and early clinical trials does not ensure that later large scale efficacy trials will be successful nor does it predict final results. Favorable results in early trials may not be repeated in later trials. We cannot assure you that the TPD/FDA or other similar government bodies will view the results as we do or that any future trials of our proposed products for other indications will achieve positive results. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through pre-clinical studies and initial clinical trials.
A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials. Any future clinical trial results for our proposed products may not be successful. A number of factors could contribute to a lack of favorable safety and efficacy results for our proposed products for other indications. For example, such trials could result in increased variability due to varying site characteristics, such as local standards of care, differences in evaluation period and due to varying patient characteristics including demographic factors and health status. There can be no assurance that our clinical trials will demonstrate sufficient safety and efficacy for TPD or the FDA to approve our potential products for the treatment of castrate-resistant prostate cancer CRPC, or any other indication that we may consider in any additional NDA/NDS submissions for our potential products.
We will be required to demonstrate through larger-scale clinical trials that any potential future product is safe and effective for use in a diverse population before we can seek regulatory approvals for its commercial sale. There is typically an extremely high rate of attrition from the failure of product candidates proceeding through clinical and post-approval trials. If our potential products fail to demonstrate sufficient safety and efficacy in ongoing or future clinical trials, we could experience potentially significant delays in, or be required to abandon development of, our product candidates currently under development.
In addition, clinical trials and nonclinical studies performed by research organizations and other independent third parties may yield negative results regarding the effect of our potential products on CRPC, either in absolute terms or relative to other products.
23
| Management’s Discussion and Analysis | June 30, 2015 |
We may not be able to obtain required regulatory approval of our product candidate, EPI-506, or other future product candidates.
The research, testing, manufacturing, labeling, packaging, storage, approval, sale, marketing, advertising and promotion, pricing, export, import and distribution of drug products developed by us or our future collaborative partners, if any, is subject to extensive regulation by federal, provincial, state and local governmental authorities and those regulations differ from country to country. Our potential product candidates will be principally regulated in the United States by the FDA, in Canada by the TPD, in the European Union by the European Medicines Agency (“EMA”) and the regulators in the individual European Union member countries and by other similar regulatory authorities in Japan and other jurisdictions. Government regulation substantially increases the cost and risk of researching, developing, manufacturing and selling products. Following several widely publicized issues in recent years, the FDA and similar regulatory authorities in other jurisdictions have become increasingly focused on product safety. This development has led to requests for more clinical trial data, for the inclusion of a significantly higher number of patients in clinical trials and for more detailed analysis of trial results. Consequently, the process of obtaining regulatory approvals, particularly from the FDA, has become more costly, time consuming and challenging than in the past. Any product developed by us or our future collaborative partners, if any, must receive all relevant regulatory approvals or clearances from the applicable regulatory authorities before it may be marketed and sold in a particular country.
We will not be permitted to market any potential products in the United States, Canada or in other countries where we intend to market our potential products until the product candidate receives approval of a NDA from the FDA or similar approval in other countries as restrictions apply. In the United States, the FDA generally requires the completion of pre-clinical testing and clinical trials of each drug to establish its safety and efficacy and extensive pharmaceutical development to ensure its quality before an NDA is approved. This process can take many years and require the expenditure of substantial resources and may include post-marketing studies and surveillance. Regulatory authorities in other jurisdictions impose similar requirements. Of the large number of drugs in development, only a small percentage result in the submission of an NDA to the FDA and even fewer are approved for commercialization. To date, we have not submitted an NDA for any of our potential products to the FDA or comparable applications to other regulatory authorities. If our development efforts for potential products are not successful for the treatment of CRPC and regulatory approval is not obtained in a timely fashion or at all, our business will be adversely affected.
The receipt of required regulatory approvals for our potential products is uncertain and subject to a number of risks, including the following:
| · | the FDA, Institutional Review Boards (“IRBs”) or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
| · | we may not be able to provide acceptable evidence of the safety, efficacy, and quality of our potential products; |
| · | the results of our clinical trials may not meet the level of statistical or clinical significance required by the FDA or other regulatory agencies for marketing approval; |
| · | the dosing of our potential products in a particular clinical trial may not be at an optimal level; |
| · | patients in our clinical trials may suffer adverse effects for reasons that may or may not be related to our potential products; |
| · | the data collected from our clinical trials may not be sufficient to support the submission of an NDA for our potential products or to obtain regulatory approval in Canada, the United States or elsewhere; |
| · | the FDA or comparable foreign regulatory authorities may find deficiencies in the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and |
| · | the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
The FDA and other regulators have substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional clinical trials or other studies. In addition, varying interpretations of the data obtained from pre-clinical studies and clinical trials could delay, limit or prevent regulatory approval of our potential products. We, or our future collaborative partner, if any, must obtain and maintain regulatory authorization to conduct clinical trials. Our pre-clinical research is subject to GLP and other requirements and our clinical research is subject to good clinical practice and other requirements. Failure to adhere to these requirements could invalidate our data. In addition, the relevant regulatory authority or
24
| Management’s Discussion and Analysis | June 30, 2015 |
independent review board may modify, suspend or terminate a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the benefits. Further, the process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially based upon, among other things, the type, complexity and novelty of the product candidates involved, the jurisdiction in which regulatory approval is sought and the substantial discretion of the regulatory authorities. If regulatory approval is obtained in one jurisdiction, that does not necessarily mean that our product candidates will receive regulatory approval in all jurisdictions in which we may seek approval, or any regulatory approval obtained may not be as broad as what was obtained in other jurisdictions. However, the failure to obtain approval for our product candidates in one or more jurisdictions may negatively impact our ability to obtain approval in a different jurisdiction. Accordingly, despite our expenditures and investment of time and effort, we may be unable to receive required regulatory approvals for product candidates developed by us. If a significant portion of these development efforts are not successfully completed, required regulatory approvals are not obtained, or any approved products are not commercially successful, our business, financial condition and results of operations may be materially harmed.
Administering any of our potential product candidates to humans may produce undesirable side effects. These side effects could interrupt, delay or halt clinical trials and could result in the applicable regulatory authorities denying approval of our product candidates for any or all of the targeted indications. If regulatory approval for a product candidate is granted, the approval will be limited to those disease states, conditions, and populations for which the product is demonstrated through clinical trials to be safe and effective, and any approval granted may be too narrow to be commercially viable.
We have no experience manufacturing our product candidates on a large clinical or commercial scale and have no manufacturing facility. As a result, we are dependent on third-party manufacturers for the manufacture of our product candidates as well as on third parties for our supply chain, and if we experience problems with any such third parties, the manufacturing of our product candidates or products could be delayed.
We do not own or operate facilities for the manufacture of future potential product candidates. We currently have no plans to build internal clinical or commercial scale manufacturing capabilities. As a result, we will rely on third party contract manufacturing organizations (“CMOs”), in the future, for the chemical manufacture of active pharmaceutical ingredients for our potential products. Also, we may rely on another CMO for the production of the final product formulation. To meet our projected potential needs for clinical supplies to support our activities through regulatory approval and commercial manufacturing, the CMOs with which we may work will need to increase the scale of production. We may need to identify additional CMOs for continued production of supply for product candidates in the event the CMOs we choose to utilize are unable to scale production, or if we otherwise experiences any problems with them. Although alternative third-party suppliers with the necessary manufacturing and regulatory expertise and facilities exist, it could be expensive and take a significant amount of time to arrange for alternative suppliers. We may encounter technical difficulties or delays in the transfer of any future potential product manufacturing on a commercial scale to additional third-party manufacturers. We may be unable to enter into agreements for commercial supply with third party manufacturers, or may be unable to do so on acceptable terms. If we are unable to arrange for alternative thirdparty manufacturing sources or to do so on commercially reasonable terms or in a timely manner, we may not be able to complete development of our potential product candidates, market or distribute them.
Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured product candidates or products ourselves, including reliance on the third party for regulatory compliance and quality assurance, the possibility of breach of the manufacturing agreement by the third party because of factors beyond our control, including a failure to synthesize and manufacture product candidates or any products we may eventually commercialize in accordance with our specifications and the possibility of termination or nonrenewal of the agreement by the third party, based on our own business priorities, at a time that is costly or damaging to us. In addition, the FDA and other regulatory authorities require that our product candidates and any products that we may eventually commercialize be manufactured according to cGMP and similar foreign standards. Any failure by our third-party manufacturers to comply with cGMP or failure to scale up manufacturing processes, including any failure to deliver sufficient quantities of product candidates in a timely manner, could lead to a delay in, or failure to obtain, regulatory approval of any of our potential product candidates and could cause us to incur higher costs and prevent us from commercializing product candidates successfully. In addition, such failure could be the basis for the FDA to issue a warning letter, withdraw approvals for product candidates previously granted to us, or take other regulatory or legal action, including recall or seizure of outside supplies of the product candidate, total or partial
25
| Management’s Discussion and Analysis | June 30, 2015 |
suspension of production, suspension of ongoing clinical trials, refusal to approve pending applications or supplemental applications, detention or product, refusal to permit the import or export of products, injunction, or imposing civil and criminal penalties.
Any significant disruption in our supplier relationships could harm our business. Any significant delay in the supply of a product candidate or its key materials for a potential ongoing clinical trial could considerably delay completion of our potential clinical trials, product testing and regulatory approval of our potential product candidates. If our manufacturers or we are unable to purchase these key materials after regulatory approval has been obtained for our product candidates, the commercial launch of our product candidates would be delayed or there would be a shortage in supply, which would impair our ability to generate revenues from the sale of our product candidates. It may take several years to establish an alternative source of supply for our product candidates and to have any such new source approved by the FDA.
We may not be able to successfully commercialize EPI-506 or other future product candidates.
Even if EPI-506 or other future product candidates were to successfully obtain approval from the FDA and comparable foreign regulatory authorities, any approval might contain significant limitations related to use restrictions for specified age groups, warnings, precautions or contraindications, may be subject to burdensome post-approval trial or risk management requirements, or may be limited to a subset of CRPC patients with limited commercial value. If we are unable to obtain regulatory approval for EPI-506 in one or more jurisdictions, or any approval contains significant limitations, we may not be able to obtain sufficient funding or generate sufficient revenue to continue the development of any other future product candidates that we may discover, in-license, develop or acquire in the future. Also, any regulatory approval of EPI-506 or any future product candidates, once obtained, may be withdrawn. Furthermore, even if we obtain regulatory approval for EPI-506, the commercial success of EPI-506 will depend on a number of factors, including the following:
| · | development of a commercial organization or establishment of a commercial collaboration with a third party that has a commercial infrastructure; |
| · | establishment of commercially viable pricing and approval for adequate reimbursement from third-party and government payors; |
| · | the ability of our third-party manufacturers to manufacture quantities of EPI-506 using commercially sufficient processes and at a scale sufficient to meet anticipated demand and enable us to reduce our cost of manufacturing; |
| · | our success in educating physicians and patients about the benefits, administration and use of EPI-506; |
| · | the availability, perceived advantages, relative cost, relative safety and relative efficacy of alternative and competing treatments; |
| · | the effectiveness of our own or our potential strategic collaborators’ marketing, sales and distribution strategy and operations; |
| · | acceptance of EPI-506 as safe and effective by patients and the medical community; and |
| · | a continued acceptable safety profile of EPI-506 following approval. |
Many of these factors are beyond our control. If we, or our potential commercialization collaborators, are unable to successfully commercialize EPI-506, we may not be able to earn sufficient revenues to continue our business.
If we are unable to enroll patients in clinical trials, we will be unable to complete these trials on a timely basis.
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the size and nature of the patient population, the proximity of subjects to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, ability to obtain and maintain patient consents, risk that enrolled subjects will drop out before completion, competing clinical trials and clinicians’ and patients’ perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating. Furthermore, we rely on Contract Research Organizations (“CROs”) and clinical trial sites to ensure the proper and timely conduct of our clinical trials, and while we have agreements governing their committed activities, we have limited influence over their actual performance.
If we experience delays in the completion or termination of any clinical trial of our proposed products or any future product candidates, the commercial prospects of product candidates will be harmed, and our ability to generate product revenues from any of these product candidates will be delayed. In addition, any delays in completing our
26
| Management’s Discussion and Analysis | June 30, 2015 |
clinical trials will increase costs, slow down product candidate development and approval process and could shorten any periods during which we may have the exclusive right to commercialize product candidates or allow competitors to bring products to market before we do, and jeopardize our ability to commence product sales, which would impair our ability to generate revenues and may harm our business, results of operations, financial condition and cash flows and future prospects. In addition, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our proposed products or future product candidates.
We intend to conduct clinical trials for existing or future product candidates at sites outside the United States and the FDA may not accept data from trials conducted in such locations.
We intend to file a Clinical Trial Application (“CTA”) application in Canada and we may in the future choose to conduct one or more clinical trials outside the United States. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of this data is subject to certain conditions imposed by the FDA. For example, the clinical trial must be well designed and conducted and performed by qualified investigators in accordance with ethical principles. The trial population must also adequately represent the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. In addition, while these clinical trials are subject to the applicable local laws, FDA acceptance of the data will be dependent upon its determination that the studies also complied with all applicable U.S. laws and regulations. There can be no assurance the FDA will accept data from trials conducted outside of the United States If the FDA chooses to not accept data collected outside the United States, it would likely result in the need for additional trials, which would be costly and time-consuming and delay or permanently halt the development of our proposed products or any future product candidates.
Even if we obtain marketing approval for any product candidate, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense.
Even if we obtain U.S. or Canadian regulatory approval for any of our product candidates for the treatment of CRPC, which would not occur until we successfully complete Phase 3 clinical trials, the FDA or TPD may still impose significant restrictions on its indicated uses or marketing or the conditions of approval, or impose ongoing requirements for potentially costly and time-consuming post-approval studies, including Phase 4 clinical trials or clinical outcome studies and post-market surveillance to monitor the safety and efficacy of our potential products. Even if we secure U.S. or Canadian regulatory approval, we would continue to be subject to ongoing regulatory requirements governing manufacturing, labeling, packaging, storage, distribution, safety surveillance, advertising, promotion, recordkeeping and reporting of adverse events and other post-market information. These requirements include registration with the FDA, as well as continued compliance with good clinical practice obligations, for any clinical trials that we conduct post-approval. In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP requirements relating to quality control, quality assurance and corresponding maintenance of records and documents.
With respect to any drug candidates for which we obtain regulatory approval, we will be subject to post-marketing regulatory obligations, including the requirements by the FDA, EMA and similar agencies in other jurisdictions to maintain records regarding product safety and to report to regulatory authorities serious or unexpected adverse events. Compliance with extensive post-marketing record-keeping and reporting requirements requires a significant commitment of time and funds, which may limit our ability to successfully commercialize approved products.
In addition, manufacturing of approved drug products must comply with extensive regulations governing cGMP. Manufacturers and their facilities are subject to continual review and periodic inspections. As we will be dependent on third parties for manufacturing, we will have limited ability to ensure that any entity manufacturing products on our behalf is doing so in compliance with applicable cGMP requirements. Failure or delay by any manufacturer of our potential products to comply with cGMP regulations or to satisfy regulatory inspections could have a material adverse effect on us, including potentially preventing us from being able to supply products for clinical trials or commercial sales. In addition, manufacturers may need to obtain approval from regulatory authorities for product, manufacturing, or labeling changes, which requires time and money to obtain and can cause delays in product availability. We are also required to comply with good distribution practices such as maintenance of storage and shipping conditions, as well as security of products, in order to ensure product quality determined by cGMP is
27
| Management’s Discussion and Analysis | June 30, 2015 |
maintained throughout the distribution network. In addition, we are subject to regulations governing the import and export of our products.
Sales and marketing of pharmaceutical products are subject to extensive federal and provincial or state laws governing on-label and off-label advertising, scientific/educational grants, gifts, consulting and pricing and are also subject to consumer protection and unfair competition laws. Compliance with extensive regulatory requirements requires training and monitoring of the sales force, which imposes a substantial cost on us and our collaborators. To the extent our products are marketed by collaborators, our ability to ensure the collaborator’s compliance with applicable regulations will be limited. In addition, we are subject to regulations governing the design, testing, control, manufacturing, distribution, labeling, quality assurance, packaging, storage, shipping, import and export of our potential products and product candidates.
Failure to comply with applicable legal and regulatory requirements may result in administrative or judicial sanctions.
If we or a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, lack of efficacy, problems with the facility where the product is manufactured, or we or our manufacturers fail to comply with applicable regulatory requirements, we may be subject to the following administrative or judicial sanctions:
| · | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
| · | issuance of warning letters or untitled letters; |
| · | clinical holds; |
| · | injunctions or the imposition of civil or criminal penalties or monetary fines; |
| · | suspension or withdrawal of any regulatory approval; |
| · | suspension of any ongoing clinical trials; |
| · | refusal to approve pending applications or supplements to approved applications filed by us, or suspension or revocation of product license approvals; |
| · | suspension or imposition of restrictions on operations, including costly new manufacturing requirements; or |
| · | product seizure or detention or refusal to permit the import or export of product. |
The occurrence of any event or penalty described above may inhibit our ability to commercialize potential products and generate revenue. Adverse regulatory action, whether pre- or post-approval, can also potentially lead to product liability claims and increase our product liability exposure.
In the future, the regulatory climate might change due to changes in the FDA and other regulatory authorities’ staffing, policies or regulations and such changes could impose additional post-marketing obligations or restrictions and related costs. While it is impossible to predict future legislative or administrative action, if we are not able to maintain regulatory compliance, we will not be able to market our products and our business could suffer.
Termination or suspension of, or delays in the commencement or completion of, any necessary future clinical trials of our product candidates for any indications could occur.
The commencement and completion of any potential clinical trials for future products can be delayed for a number of reasons, including delays related to:
| · | the FDA, TPD or similar regulatory authorities not granting permission to proceed and placing the clinical trial on hold or not reviewing and responding to IND/CTA application requests; |
| · | patients failing to enroll or remain in our trials at the rate we expect; |
| · | a facility manufacturing a product being ordered by the FDA or other government or regulatory authorities to temporarily or permanently shut down due to violations of cGMP requirements or other applicable requirements, or cross-contaminations of product candidates in the manufacturing process; |
| · | any changes to our potential manufacturing processes that may be necessary or desired; |
| · | patients experiencing severe or unexpected drug-related adverse effects; |
| · | reports from clinical testing as well as post-market reports on similar technologies and products raising safety or efficacy concerns; |
28
| Management’s Discussion and Analysis | June 30, 2015 |
| · | third-party clinical investigators losing their license or permits necessary to perform our clinical trials, not performing our clinical trials on our anticipated schedule or employing methods not consistent with the clinical trial protocol, cGMP requirements, or other third parties not performing data collection and analysis in a timely or accurate manner; |
| · | inspections of clinical trial sites by the FDA, TPD or similar regulatory authorities, or IRBs or similar research ethics boards, finding regulatory violations that require us to undertake corrective action, result in suspension or termination of one or more sites or the imposition of a clinical hold on the entire clinical trial, or that prohibit us from using some or all of the data in support of our marketing applications; |
| · | third-party contractors becoming debarred or suspended or otherwise penalized by the FDA, TPD or other government or regulatory authorities for violations of regulatory requirements, in which case we may need to find a substitute contractor and we may not be able to use some or any of the data produced by such contractors in support of our marketing applications; |
| · | one or more IRBs refusing to approve, suspending or terminating the trial at an investigational site, precluding enrolment of additional subjects, or withdrawing its approval of the trial; reaching agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| · | deviations of the clinical sites from trial protocols or dropping out of a trial; |
| · | the addition of new clinical trial sites; and |
| · | the inability of the CRO to execute any clinical trials for any reason. |
Product development costs for any of our product candidates will increase if we have delays in testing or approval or if we need to perform more or larger clinical studies than planned. Additionally, changes in regulatory requirements and policies may occur and we may need to amend trial protocols to reflect these changes. Amendments may require us to resubmit our trial protocols to the FDA, TPD or similar regulatory authorities or IRBs for re-examination, which may impact the costs, timing or successful completion of that trial. Any delays in completing our future clinical trials will increase our costs, slow down our development and approval process and jeopardize our ability to generate revenues. Any of these occurrences may have a material adverse effect on our business, financial condition and prospects.
In order to establish our sales and marketing infrastructure, we will need to expand the size of our organization and we may experience difficulties in managing this growth.
As our development and commercialization plans and strategies develop, we expect that we will need to expand the size of our employee base for managerial, operational, sales, marketing, financial and other resources. Future growth would impose significant added responsibilities on members of management, including the need to identify, recruit, maintain, motivate and integrate additional employees. In addition, our management may have to divert a disproportionate amount of its attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. Our future financial performance and our ability to commercialize our potential products and any other future product candidates and our ability to compete effectively will depend, in part, on our ability to effectively manage any future growth.
We face intense competition from other biotechnology and pharmaceutical companies and our operating results will suffer if we fail to compete effectively.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. Our potential competitors in Canada, the United States and globally include large, wellestablished pharmaceutical companies, specialty pharmaceutical sales and marketing companies and specialized cancer treatment companies. Many companies, as well as research organizations, currently engage in, or have in the past engaged in, efforts related to the development of products in the same therapeutic areas as we do. Due to the size of the prostate cancer treatment market and the large unmet medical need for products that treat CRPC, a number of the world’s largest pharmaceutical companies are developing, or could potentially develop, products that could compete with our product candidates.
Many of the companies developing competing technologies and products in our field have significantly greater financial resources and expertise in discovery, research and development, manufacturing, pre-clinical studies and clinical testing, obtaining regulatory approvals and marketing than we do. Other smaller companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
29
| Management’s Discussion and Analysis | June 30, 2015 |
Academic institutions, government agencies and other public and private research organizations may also conduct research, seek patent protection and establish collaborative arrangements for discovery, research, clinical development and marketing of products similar to ours. There is a risk that one or more of our competitors may develop more effective or more affordable products than ours and that such competitors will commercialize products that will render our product candidates obsolete. We face competition with respect to product efficacy and safety, ease of use and adaptability to various modes of administration, acceptance by physicians, the timing and scope of regulatory approvals, availability of resources, reimbursement coverage, price and patent positions of others. In addition, these companies and institutions also compete with us in recruiting and retaining qualified personnel. If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition and operations will suffer materially adverse effects.
If we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
Our ability to compete in the highly competitive pharmaceuticals industry depends in large part upon our ability to attract and retain highly qualified managerial, scientific and medical personnel. Competition affects our ability to hire and retain highly qualified personnel on acceptable terms. We are highly dependent on our management, scientific and medical personnel, such as Robert Rieder, Marianne Sadar, Raymond Andersen and Frank Perabo, who have substantial knowledge in many different aspects of drug development and commercialization. Despite our efforts to retain valuable employees, members of our management, scientific and medical teams may terminate their employment with us on short notice or, potentially, without any notice at all. The loss of the services of any of our executive officers or other key employees could potentially harm our business, operating results or financial condition. Our success may also depend on our ability to attract, retain and motivate highly skilled junior, mid-level, and senior managers and scientific personnel. Other pharmaceutical and biotechnology companies with which we compete for qualified personnel have greater financial and other resources, different risk profiles, and a longer history in the industry than the we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high-quality candidates than what we have to offer. If we are unable to continue to attract and retain high-quality personnel, the rate and success at which we can develop and commercialize product candidates would be limited.
Our directors and officers may be subject to conflicts of interest.
Some of our directors and officers are engaged and will continue to be engaged in the search for additional business opportunities on behalf of other corporations, and situations may arise in which these directors and officers will be in direct competition with us. Not all of our directors or officers are subject to any non-competition agreements. Some of our directors and officers are or may become directors or officers of the other companies engaged in other business ventures whose operations may, from time to time, be in direct competition with our operations. Conflicts, if any, will be dealt with in accordance with the relevant provisions of the Business Corporations Act (British Columbia) and under our articles of incorporation.
Failure to obtain regulatory approval in international jurisdictions would prevent any future product candidates from being marketed outside Canada or the United States.
In order to market and sell our potential future products in the European Union and many other jurisdictions, we must obtain separate marketing approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ substantially from that required to obtain TPD or FDA approval. The regulatory approval process outside of Canada and the United States generally includes all of the risks associated with obtaining TPD or FDA approval. In addition, in many countries outside Canada and the United States, it is required that the product candidate be approved for reimbursement before the product can be approved for sale in that country. Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our potential products in certain countries. We may not obtain approvals from regulatory authorities outside Canada or the United States on a timely basis, if at all. A failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory approval process in others. We may not be able to file for marketing approvals and may not receive necessary approvals to commercialize our potential products in any market. If we are unable to obtain approval of any of our
30
| Management’s Discussion and Analysis | June 30, 2015 |
future product candidates by regulatory authorities in the European Union or another jurisdiction, the commercial prospects of that product candidate may be significantly diminished and our business prospects could decline.
31
| Management’s Discussion and Analysis | June 30, 2015 |
Recently enacted and future legislation in the United States may increase the difficulty and cost for us to obtain marketing approval of, and commercialize, our potential future products and affect the prices we may obtain.
In the United States, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval for our potential future products, restrict or regulate post-approval activities and affect our ability to profitably sell potential future products. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We do not know whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our potential future products, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
In the United States, the Medicare Modernization Act (the “MMA”) changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for drugs. In addition, this legislation authorized Medicare Part D prescription drug plans to use formularies where they can limit the number of drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for our potential products and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private health insurance companies often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private health insurance companies.
In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 or, collectively, the “Health Care Reform Law”, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. The Health Care Reform Law revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states. Further, the new law imposed a significant annual fee on companies that manufacture or import branded prescription drug products. Substantial new provisions affecting compliance have also been enacted, which may possibly require us to modify our business practices with healthcare practitioners. We will not know the full effects of the Health Care Reform Law until applicable federal and state agencies issue regulations or guidance under the new law. Although it is too early to determine the full effect of the Health Care Reform Law, the law appears likely to continue the pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs.
Our business may be materially adversely affected by new legislation, new regulatory requirements and the continuing efforts of governmental and third party payors to contain or reduce the costs of healthcare through various means.
Governments and regulatory authorities in Europe and other markets in which we intend to sell our products may propose and adopt new legislation and regulatory requirements relating to pharmaceutical approval criteria and manufacturing requirements. Such legislation or regulatory requirements, or the failure to comply with such, could adversely impact our operations and could have a material adverse effect on our business, financial condition and results of operations.
In recent years, national, federal, provincial, state, and local officials and legislators have proposed, or are reportedly considering proposing, a variety of price-based reforms to the healthcare systems in the European Union, the United States and other countries. Some proposals include measures that would limit or eliminate payments for certain medical procedures and treatments or subject the pricing of pharmaceuticals to government control. Furthermore, in certain foreign markets, the pricing or profitability of healthcare products is subject to government controls and other measures that have been prepared by legislators and government officials. While we cannot predict whether any such legislative or regulatory proposals or reforms will be adopted, the adoption of any such proposals or reforms could adversely affect the commercial viability of our existing and potential products. Significant changes in
32
| Management’s Discussion and Analysis | June 30, 2015 |
the healthcare system in the European Union and other countries may have a substantial impact on the manner in which we conducts business. Such changes could also have a material adverse effect on our business, financial condition and results of operations.
We are subject to risks inherent in foreign operations.
We intend to pursue international market growth opportunities, such that international sales may account for a significant portion of our revenue. We are subject to a number of risks associated with our potential international business operations, sales and marketing activities that may increase liability, costs, lengthen sales cycles and require significant management attention. These risks include:
| · | compliance with the laws of the United States, Canada, the European Union and other jurisdictions where we may conduct business, including import and export legislation; |
| · | increased reliance on third parties to establish and maintain foreign operations; |
| · | the complexities and expenses of administering a business abroad; |
| · | complications in compliance with, and unexpected changes in, foreign regulatory requirements; |
| · | instability in economic or political conditions, including inflation, recession and actual or anticipated military conflicts, social upheaval or political uncertainty; |
| · | foreign currency fluctuations; |
| · | foreign exchange controls and cash repatriation restrictions; |
| · | tariffs and other trade barriers; |
| · | difficulties in collecting accounts receivable; |
| · | differing tax structures and related potential adverse tax consequences; |
| · | uncertainties of laws and enforcement relating to the protection of intellectual property or secured technology; |
| · | litigation in foreign court systems; |
| · | unauthorized copying or use of our intellectual property; |
| · | cultural and language differences; |
| · | difficulty in managing a geographically dispersed workforce in compliance with local laws and customs that vary from country to country; and |
| · | other factors, depending upon the country involved. |
There can be no assurance that the policies and procedures we implement to address or mitigate these risks will be successful, that our personnel will comply with them or that we will not experience these factors in the future or that they will not have a material adverse effect on our business, results of operations and financial condition.
Compulsory licensing or generic competition may affect our business in certain countries.
In a number of countries, governmental authorities and other groups have suggested that companies which manufacture medical products (e.g. pharmaceuticals) should make products available at a low cost. In some cases, governmental authorities have held that where a pharmaceutical company does not do so, its patents might not be enforceable to prevent generic competition. Alternatively, some governmental authorities could require that we grant compulsory licenses to allow competitors to manufacture and sell their own versions of our products, thereby reducing our sales or the sales of our licensees. In all of these situations, the results of future operations in these countries if any, could be adversely affected.
We may face exposure to adverse movements in foreign currency exchange rates.
Our business may expand internationally and as a result, a significant portion of our revenues, expenses, current assets and current liabilities may be preliminary denominated in U.S. dollars, Euros, and other foreign currencies, while our financial statements are expressed in Canadian dollars. In addition, the CPRIT Grant of US$12,000,000 to help fund the clinical development of our program is payable in U.S. dollars and the formula for determining the funds we must dedicate to our development of EPI-506 towards completion of Phase 1/2 clinical proof-of-concept in order to receive CPRIT Grant funds each year, as set out in the CPRIT Grant Agreement, is calculated in U.S. dollars. A decrease in the value of such foreign currencies, in particular the U.S. dollar, relative to the Canadian dollar could result in losses in revenues from currency exchange rate fluctuations. To date, we have not hedged against risks associated with foreign exchange rate exposure. We cannot be sure that any hedging techniques we
33
| Management’s Discussion and Analysis | June 30, 2015 |
may implement in the future will be successful or that our business, financial condition, and results of operations will not be materially adversely affected by exchange rate fluctuations.
Laws and regulations governing international operations may preclude us from developing, manufacturing and selling certain product candidates outside of the United States and Canada and require us to develop and implement costly compliance programs.
We must comply with numerous laws and regulations in each jurisdiction in which we plan to operate. We must also comply with U.S. laws applicable to the foreign operations of U.S. registrants, such as the U.S. Foreign Corrupt Practices Act (“FCPA”), and Canadian laws applicable to the foreign operations of Canadian businesses and individuals, such as the Canadian Corruption of Foreign Public Officials Act (“CFPOA”). The creation and implementation of international business practices compliance programs is costly and such programs are difficult to enforce, particularly where reliance on third parties is required.
The CFPOA prohibits us from giving or offering or agreeing to give a benefit of any kind to a foreign public official, or any other person for the benefit of the foreign public official, where the ultimate purpose is to obtain or retain a business advantage. Furthermore, a company may be found liable for violations by not only its employees, but also by its third-party agents. Any failure to comply with the CFPOA, as well as applicable laws and regulations in foreign jurisdictions, could result in substantial penalties or restrictions on our ability to conduct business in certain foreign jurisdictions, which may have a material adverse impact on us and our share price.
The FCPA prohibits us from paying, offering, authorizing payment or offering of anything of value, directly or indirectly, to any foreign official, political party or candidate for the purpose of influencing any act or decision of the foreign entity in order to assist the individual or business in obtaining or retaining business. The FCPA also obligates companies whose securities are listed in the United States to comply with certain accounting provisions requiring us to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations. The anti-bribery provisions of the FCPA are enforced primarily by the Department of Justice. The Securities and Exchange Commission (“SEC”) is involved with enforcement of the books and records provisions of the FCPA.
Compliance with the FCPA is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, the FCPA presents particular challenges in the pharmaceutical industry, because, in many countries, hospitals are operated by the government, and doctors and other hospital employees are considered foreign officials. Certain payments to hospitals in connection with clinical studies and other work have been deemed to be improper payments to government officials and have led to FCPA enforcement actions.
Violation of the FCPA can result in significant civil and criminal penalties. Indictment alone under the FCPA can lead to suspension of the right to do business with the U.S. government until the pending claims are resolved. Conviction of a violation of the FCPA can result in long-term disqualification as a government contractor. The termination of a government contract or relationship as a result of our failure to satisfy any of our obligations under laws governing international business practices would have a negative impact on our operations and harm our reputation and ability to procure government contracts. The SEC also may suspend or bar issuers from trading securities on U.S. exchanges for violations of the FCPA’s accounting provisions.
Our employees or other agents may, without our knowledge and despite our efforts, engage in prohibited conduct under our policies and procedures and the CFPOA, FCPA or other anti-bribery laws that we may be subject to for which we may be held responsible. If our employees or other agents are found to have engaged in such practices, we could suffer severe penalties and other consequences that may have a material adverse effect on our business, financial condition and results of operations.
Various laws, regulations and executive orders also restrict the use and dissemination outside of the United States, or the sharing with certain non-U.S. nationals, of information classified for national security purposes, as well as certain products and technical data relating to those products. If we expand our presence outside of the United States in the future, we will be required to dedicate additional resources to comply with these laws, and these laws may preclude us from developing, manufacturing, or selling certain products and product candidates outside of the United States, which could limit our growth potential and increase development costs.
34
| Management’s Discussion and Analysis | June 30, 2015 |
Third-party coverage and reimbursement and health care cost containment initiatives and treatment guidelines may constrain our future revenues.
In many of the markets in which we hope to sell future products, sales of healthcare products are dependent in part on the availability of reimbursement to the consumer from third party payors, such as government and private insurance plans. Third party payors are increasingly challenging the effectiveness of, and prices charged for, medical products and services and therefore uncertainty exists as to the reimbursement of existing and newly approved healthcare products. The prices of our future products may be subject to direct price controls by law and to drug reimbursement programs with varying price control mechanisms. In addition, as drug costs have increased, there have been more cost containment measures taken by government and third-party private payors, including limitations on both the number of products they list for reimbursements, the conditions under which they will reimburse, and the reimbursement drug prices. Some payors require manufacturers to enter into agreements with them in order for drugs to be reimbursed by the payor, and the agreements may contain cost sharing or other onerous provisions. Also, the current conditions and rules relating to the listing submissions to public and private formulary listings may change or become more onerous in the future. If we fail to achieve the listing of our products, we will affect the physicians’ decisions regarding the use of our products.
Our ability to successfully market any of our future products will depend in part on the level of reimbursement that government health administration authorities, private health coverage insurers and other organizations provide for the cost of our potential products and related treatments. Countries in which our potential products may in the future be sold through reimbursement schemes under national health insurance programs frequently require that manufacturers and sellers of pharmaceutical products obtain governmental approval of initial prices and any subsequent price increases. In certain countries, including the United States, government-funded and private medical care plans can exert significant indirect pressure on prices. We may not be able to sell any potential products profitably if our prices are not approved or coverage and reimbursement is unavailable or limited in scope.
If we are not able to convince public payors and hospitals to include our potential future products on their approved formulary lists, revenues may not meet expectations and our business, results of operations and financial condition may be adversely affected.
Hospitals establish formularies, which are lists of drugs approved for use in the hospital. If a drug is not included on the hospital’s formulary, the ability to promote and sell our potential future products may be limited or denied. If we fail to secure and maintain formulary inclusion for potential future products on favorable terms or are significantly delayed in doing so, we may have difficulty achieving market acceptance of potential future products and our business, results of operations and financial condition could be materially adversely affected.
We have never marketed a drug before, and if we are unable to establish an effective sales force and marketing infrastructure, or enter into acceptable third-party sales and marketing or licensing arrangements, we may be unable to generate any revenue.
We do not currently have an infrastructure for the sales, marketing and distribution of pharmaceutical drug products and the cost of establishing and maintaining such an infrastructure may exceed the cost-effectiveness of doing so. In order to market any products that may be approved by the FDA and comparable foreign regulatory authorities, we must build our sales, marketing, managerial and other nontechnical capabilities or make arrangements with third parties to perform these services. If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, we may not be able to generate product revenue and may not become profitable. We will be competing with many companies that currently have extensive and well-funded sales and marketing operations. Without an internal commercial organization or the support of a third party to perform sales and marketing functions, we may be unable to compete successfully against these more established companies.
Our products may, if approved for sale, not achieve or maintain expected levels of market acceptance, which could have a material adverse effect on our business, financial condition and results of operations and could cause the market value of our securities to decline.
35
| Management’s Discussion and Analysis | June 30, 2015 |
Even if we are able to obtain regulatory approvals for our product candidates, the success of those products is dependent upon achieving and maintaining market acceptance. New product candidates that appear promising in development may fail to reach the market or may have only limited or no commercial success. Levels of market acceptance for our potential products could be impacted by several factors, many of which are not within our control, including but not limited to:
| · | demonstration of clinical safety and efficacy of our product candidates and other possible AR NTD inhibitors generally; |
| · | safety, efficacy, convenience and cost-effectiveness of our potential products compared to products of our competitors; |
| · | the prevalence and severity of any adverse side effects; |
| · | scope of approved uses and marketing approval; |
| · | limitations or warnings contained in FDA-approved labeling; |
| · | timing of market approvals and market entry; |
| · | the willingness of physicians to prescribe our potential products and of the target patient population to try new therapies; |
| · | the inclusion of AR NTD inhibitor products in applicable treatment guidelines; |
| · | new procedures or methods of treatment that may reduce the incidences of any of the indications for which our potential products shows utility; |
| · | difficulty in, or excessive costs to, manufacture; |
| · | infringement or alleged infringement of the patents or intellectual property rights of others; |
| · | the introduction of any new products, including generic AR NTD inhibitor products, that may in the future become available to treat indications for which our potential product may be approved; |
| · | availability of alternative products from our competitors; |
| · | acceptance of the price of our potential products; and |
| · | ability to market our potential products effectively at the retail level. |
In addition, the success of any new product will depend on our ability to either successfully build our in-house sales capabilities or to secure new, or to realize the benefits of existing arrangements with third-party marketing or distribution partners. Seeking out, evaluating and negotiating marketing or distribution agreements may involve the commitment of substantial time and effort and may not ultimately result in an agreement. In addition, any third party marketing or distribution partners may not be successful in promoting our potential products. If we are unable to commercialize new products successfully, whether through a failure to achieve market acceptance, a failure to build our own in-house sales capabilities, a failure to secure new marketing partners or to realize the benefits of arrangements with any marketing partners, it may result in a material adverse effect on our business, financial condition and results of operations and it could cause the market value of our securities to decline.
In addition, by the time any products are ready to be commercialized, the market anticipated for these products may have changed. Our estimates of the number of patients who have received or might have been candidates to use a specific product may not accurately reflect the true market or market prices for such products or the extent to which such products, if successfully developed, will actually be used by patients. Our failure to successfully introduce and market our products that are under development would have a material adverse effect on our business, financial condition and results of operations.
We are subject to U.S. laws relating to fraud and abuse and patients’ rights.
As a pharmaceutical company, we do not and will not bill directly to Medicare, Medicaid or other third-party payors, federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our future arrangements with third-party payors and customers who are in a position to purchase, recommend or prescribe our product candidates for which we obtain marketing approval. These broadly applicable fraud and abuse and other healthcare laws and regulations may constrain our future business or financial arrangements and relationships with healthcare professionals, principal investigators, consultants, customers, and third-party payors and other entities, including our marketing practices, educational programs and pricing policies. Restrictions under applicable federal and state healthcare laws and regulations that may affect our ability to operate include, but are not limited to, the following:
| · | the U.S. Anti-Kickback Statute, among other things, prohibits persons from knowingly and willfully soliciting, offering, receiving or providing paying any remuneration (including any kickback, bribe, or |
36
| Management’s Discussion and Analysis | June 30, 2015 |
| rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid; |
| · | civil and criminal false claims laws and civil monetary penalty laws impose criminal and civil penalties, including through civil whistleblower or qui tam actions, and, among other things, prohibit individuals or entities from knowingly presenting, or causing to be presented, to the U.S. federal government, including the Medicare and Medicaid programs, claims for payment or approval that are false or fraudulent or from knowingly making a false statement to improperly avoid, decrease or conceal an obligation to pay money to the federal government; |
| · | the U.S. Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), among other things, imposes criminal liability for knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or to obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g. public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of or payment for healthcare benefits, items or services relating to healthcare matters; |
| · | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, and its implementing regulations, also imposes certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information without the appropriate authorization by entities subject to the law, such as health plans, healthcare clearinghouses and healthcare providers; |
| · | the federal Physician Payment Sunshine Act, created under Section 6002 of the Affordable Care Act and its implementing regulations, requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the Centers for Medicare & Medicaid Services (“CMS”), information related to “payments or other transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and applicable manufacturers and applicable group purchasing organizations to report annually to CMS ownership and investment interests held by physicians (as defined above) and their immediate family members and payments or other “transfers of value” to such physician owners and their immediate family members; and |
| · | analogous state and foreign laws and regulations, including: state anti-kickback and false claims laws which may apply to our business practices, including, but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by state governmental and non-governmental third-party payors, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government; state laws that require drug manufacturers to track gifts and other remuneration and items of value provided to healthcare professionals and entities and file reports relating to pricing and marketing information; and state and foreign laws that govern the privacy and security of health information in specified circumstances, many of which differ from each other in significant ways and often are not pre-empted by HIPAA, thus complicating compliance efforts. |
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations, agency guidance, or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to penalties, including without limitation, significant civil, criminal and administrative penalties, damages, fines, disgorgement, exclusion from government funded healthcare programs, such as Medicare and Medicaid, contractual damages, reputational harm, administrative burdens and diminished profits and future earnings, and the curtailment or restructuring of our operations. If any physicians or other healthcare providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs. Moreover, we expect there will continue to be federal and state laws and regulations, proposed and implemented, that could impact our operations and business. The extent to which future legislation or regulations, if any, relating to healthcare fraud
37
| Management’s Discussion and Analysis | June 30, 2015 |
abuse laws or enforcement, may be enacted or what effect such legislation or regulation would have on our business remains uncertain.
We may acquire businesses or products or form strategic alliances in the future and we may not realize the benefits of such acquisitions.
We may acquire additional businesses or products, form strategic alliances or create joint ventures with third parties that we believe will complement or augment our existing business.
If we acquire businesses in the future, we may not be able to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and company culture. We may encounter numerous difficulties in developing, manufacturing and marketing any new products resulting from a strategic alliance or acquisition that delay or prevent us from realizing their expected benefits. The potential failure of the due diligence processes to identify significant problems, liabilities or other shortcomings or challenges with respect to intellectual property, product quality, revenue recognition or other accounting practices, taxes, corporate governance and internal controls, regulatory compliance, employee, customer or partner disputes or issues and other legal and financial contingencies could decrease or eliminate the anticipated benefits and synergies of any acquisition and could negatively affect our future business and financial results.
As part of our business strategy, we may also continue to acquire additional companies, products or technologies principally related to, or complementary to, our current operations. Any such acquisitions will be accompanied by certain risks including but not limited to:
| · | exposure to unknown liabilities of acquired companies and the unknown issues with any associated technologies or research; |
| · | higher than anticipated acquisition costs and expenses; |
| · | the difficulty and expense of integrating operations, systems and personnel of acquired companies; |
| · | disruption of our ongoing business; |
| · | inability to retain key customers, distributors, vendors and other business partners of the acquired company; |
| · | diversion of management’s time and attention; and |
| · | possible dilution to shareholders. |
We may not be able to successfully overcome these risks and other problems associated with acquisitions and this may adversely affect our business, financial condition or results of operations.
We may seek to enter into collaborations with third parties for the development and commercialization of our product candidates. If we fail to enter into such collaborations, or such collaborations are not successful, we may not be able to capitalize on the market potential of our product candidates.
We may seek third-party collaborators for development and commercialization of our product candidates. We are not currently party to any such arrangement. However, if we do enter into any such arrangements with any third parties in the future, we will likely have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our product candidates. Our ability to generate revenues from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements.
Collaborations involving our product candidates would pose the following risks to us:
| · | collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations; |
| · | collaborators may not pursue development and commercialization of our product candidates or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the collaborators’ strategic focus or available funding, or external factors such as an acquisition that diverts resources or creates competing priorities; |
| · | collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing; |
38
| Management’s Discussion and Analysis | June 30, 2015 |
| · | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; |
| · | collaborators with marketing and distribution rights to one or more of our products may not commit sufficient resources to the marketing and distribution of such product or products; |
| · | collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation; |
| · | collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; |
| · | disputes may arise between the collaborators and us that result in the delay or termination of the research, development or commercialization of our products or product candidates or that result in costly litigation or arbitration that diverts management attention and resources; and |
| · | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates. |
Collaboration agreements may not lead to development or commercialization of product candidates in the most efficient manner or at all. If a collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program could be delayed, diminished or terminated.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could cause significant liability for us and harm our reputation.
We are exposed to the risk of employee fraud or other misconduct, including intentional failures to comply with FDA regulations or similar regulations of comparable foreign regulatory authorities, provide accurate information to the FDA or comparable foreign regulatory authorities, comply with manufacturing standards we have established, comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities, report financial information or data accurately or disclose unauthorized activities to us. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, results of operations, financial condition and cash flows from future prospects, including the imposition of significant fines or other sanctions.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to cease the sale, marketing and distribution of our future products.
We could face a potential risk of product liability as a result of our potential sales, marketing and distribution activities relating to any future commercialization of any future product. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties.
Claims could also be asserted under U.S. state or Canadian provincial or other foreign consumer protection legislation. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to cease the sale, marketing and distribution of our products. Even successful defense against product liability claims would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| · | decreased demand for any future products that we may develop; |
| · | injury to our reputation; |
| · | withdrawal of clinical trial participants; |
| · | costs to defend the related litigation; |
| · | a diversion of management’s time and our resources; |
39
| Management’s Discussion and Analysis | June 30, 2015 |
| · | substantial monetary awards to consumers, trial participants or patients; |
| · | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| · | loss of revenue; |
| · | the inability to commercialize; |
| · | the inability to continue the sale, marketing and distribution of our potential future products; and |
| · | a decline in the price of the Common Shares. |
The insurance we currently maintain may not be sufficient to protect against the liability risks discussed above. In addition, if we are unable to obtain and retain sufficient product liability insurance in the future at an acceptable cost to protect against potential product liability claims, the commercialization of products we develop could be hindered or prevented.
Our business and operations would suffer in the event of computer system failures.
Despite the implementation of security measures, our internal computer systems and those of other third parties on which we rely are vulnerable to damage from computer viruses, unauthorized access, natural disasters, fire, terrorism, war and telecommunication and electrical failures. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. To the extent that any disruption or security breach results in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of our future product candidates could be delayed.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations in Canada and in the United States, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and waste. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
The workers’ compensation insurance we maintain to cover costs and expenses we may incur due to injuries to employees resulting from the use of hazardous materials may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological or hazardous materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Business disruptions could seriously harm our future revenues and financial condition and increase costs and expenses.
Our operations could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or manmade disasters or business interruptions, for which we are predominantly self-insured. We do not carry insurance for all categories of risk that our business may encounter. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase costs and expenses. Further, any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our business, results of operations, financial condition and cash flows from future prospects.
Our business depends heavily on the use of information technologies.
40
| Management’s Discussion and Analysis | June 30, 2015 |
Several key areas of our business depend on the use of information technologies. Despite our best efforts to prevent such behavior, third parties may nonetheless attempt to hack into our systems and obtain data relating to our preclinical studies or proprietary information on potential products. If we fail to maintain or protect our information systems and data integrity effectively, we could lose customers, have difficulty attracting customers, have difficulty preventing, detecting, and controlling fraud, have regulatory sanctions or penalties imposed, have increases in operating expenses, incur expenses or lose revenues as a result of a data privacy breach, or suffer other adverse consequences. While we have invested in the protection of data and information technology, there can be no assurance that our efforts, or those of our third-party collaborators, if any, to implement adequate security and quality measures for data processing would be sufficient to protect against data deterioration or loss in the event of a system malfunction, or to prevent data from being stolen or corrupted in the event of a security breach. Any such loss or breach could have a material adverse effect on our business, operating results and financial condition.
If we are unable to implement and maintain effective internal control over financial reporting in the future, we may not be able to report financial results accurately or prevent fraud. In that case, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our Common Shares may be negatively affected.
Maintaining effective internal control over financial reporting is necessary for us to produce reliable financial reports and is important in helping to prevent financial fraud. If we are unable to maintain adequate internal controls, our business and operating results could be harmed. We are not currently required to comply with Section 404 of the Sarbanes-Oxley Act of 2002 (“Sarbanes-Oxley”), or National Instrument 52-109—Certification of Disclosure in Issuers’ Annual and Interim Filings of the Canadian Securities Administrators (“NI 52-109”). As a result, we are not currently required to make an assessment of the effectiveness of our internal controls, or to deliver a report that assesses the effectiveness of our internal control over financial reporting. We has begun to evaluate how to document and test internal control procedures to satisfy the requirements of Section 404 of Sarbanes-Oxley and the related rules of the SEC and NI 52-109, which require, among other things, our management to assess annually the effectiveness of our internal control over financial reporting. During the course of this documentation and testing, we may identify weaknesses or deficiencies that we may be unable to remedy before the requisite deadline for those reports.
Preparing our consolidated financial statements involves a number of complex manual and automated processes which are dependent on individual data input or review and require significant management judgment. One or more of these elements may result in errors that may not be detected and could result in a material misstatement of our consolidated financial statements. Additionally, Section 404 will eventually require our auditors to deliver an attestation report on the effectiveness of our internal control over financial reporting.
The process of designing and implementing effective internal controls and procedures, and expanding our internal accounting capabilities, is a continuous effort that requires us to anticipate and react to changes in our business and the economic and regulatory environments and expend significant resources to establish and maintain a system of internal controls that is adequate to satisfy our reporting obligations as a public company. The standards that must be met for management to assess the internal control over financial reporting as effective are complex, and require significant documentation, testing and possible remediation to meet the detailed standards. We cannot be certain at this time whether we will be able to successfully complete the continuing implementation of controls and procedures or the certification and attestation requirements of Section 404 and NI 52-109.
If a material misstatement occurs in the future, we may fail to meet our future reporting obligations, we may need to restate financial results and the price of our Common Shares may decline. Any failure of our internal controls could also adversely affect the results of the periodic management evaluations and annual independent registered public accounting firm attestation reports regarding the effectiveness of our internal control over financial reporting that will be required when Section 404 of Sarbanes-Oxley or NI 52-109 become fully applicable to us. Effective internal controls are necessary for us to produce reliable financial reports and are important to helping prevent financial fraud. If we cannot provide reliable financial reports or prevent fraud, our business and results of operations could be harmed, investors could lose confidence in our reported financial information, and the trading price of our Common Shares could drop significantly.
We will incur significantly increased costs and devote substantial management time as a result of operating as a U.S. public company.
41
| Management’s Discussion and Analysis | June 30, 2015 |
We recently became subject to U.S. continuous reporting requirements as a result of the registration of our Common Shares under Section 12(b) of the Exchange Act. As a U.S. public company, we will incur significant legal, accounting and other expenses that we did not incur as a private company or as a Canadian public company. For example, we will be subject to the reporting requirements of the Exchange Act and will be required to comply with the applicable requirements of Sarbanes-Oxley and the Dodd-Frank Wall Street Reform and Consumer Protection Act, as well as rules and regulations implemented by the SEC and the including the establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. We expect that compliance with these requirements will increase our legal and financial compliance costs and will make some activities more time consuming and costly. In addition, we expect that management and other personnel will need to divert attention from operational and other business matters to devote substantial time to these public company requirements. In particular, we expect to incur significant expenses and devote substantial management effort toward ensuring compliance with the requirements of Section 404 of Sarbanes-Oxley, which involve annual assessments of a company’s internal controls over financial reporting. We will need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge and may need to establish an internal audit function. We cannot predict or estimate the amount of additional costs we may incur as a result of becoming a U.S. public company or the timing of such costs.
We may lose our “foreign private issuer” status in the future, which could result in significant additional costs and expenses to us.
We are a “foreign private issuer,” as such term is defined in Rule 405 under the Securities Act of 1933, as amended (the “U.S. Securities Act”) and are not subject to the same requirements that are imposed upon U.S. domestic issuers by the SEC. We may in the future lose foreign private issuer status if a majority of our Common Shares are held in the United States and we fail to meet the additional requirements necessary to avoid loss of foreign private issuer status, such as if: (i) a majority of our directors or executive officers are U.S. citizens or residents; (ii) a majority of our assets are located in the United States; or (iii) our business is administered principally in the United States. The regulatory and compliance costs to us under U.S. securities laws as a U.S. domestic issuer will be significantly more than the costs incurred as a Canadian foreign private issuer. If we are not a foreign private issuer, we would be required to file periodic and current reports and registration statements on U.S. domestic issuer forms with the SEC, which are generally more detailed and extensive than the forms available to a foreign private issuer.
In addition, we may lose the ability to rely upon exemptions from corporate governance requirements that are available to foreign private issuers. Further, if we engage in capital raising activities after losing foreign private issuer status, there is a higher likelihood that investors may require us to file resale registration statements with the SEC as a condition to any such financing.
Risks Related to our Intellectual Property
If we breach any of the agreements under which we license rights to our product candidates or technology from third parties, we could lose license rights that are important to our business. Our current license agreement may not provide an adequate remedy for its breach by the licensor.
We are developing our EPI-series drug candidates pursuant to the License Agreement. We are subject to a number of risks associated with our collaboration with UBC and the BCCA, including the risk that UBC or the BCCA may terminate the License Agreement upon the occurrence of certain specified events. Our License Agreement requires, among other things, that we make certain payments and use reasonable commercial efforts to meet certain clinical and regulatory milestones. If we fail to comply with any of these obligations or otherwise breach this or similar agreements, UBC, the BCCA or any future licensors may have the right to terminate the license. We could also suffer the consequences of non-compliance or breaches by licensors in connection with our license agreements. Such non-compliance or breaches by such third parties could in turn result in our breaches or defaults under our agreements with our other collaboration partners, and we could be found liable for damages or lose certain rights, including rights to develop or commercialize a product or product candidate. Loss of our rights to the patents and know-how related to compounds that modulate AR activity created through research done by the BCCA and UBC under direction of Drs. Marianne Sadar and Raymond Andersen, respectively (the “Licensed IP”), or any similar license granted to us in the future, or the exclusivity rights provided therein, could harm our financial condition and operating results.
42
| Management’s Discussion and Analysis | June 30, 2015 |
We rely on proprietary technology, the protection of which can be unpredictable and costly.
Our activities depend, in part, on our ability to (i) obtain and maintain patents, trade secret protection and operate without infringing the intellectual proprietary rights of third parties, (ii) successfully defend these patents (including patents owned by or licensed to us) against third-party challenges, and (iii) successfully enforce these patents against third party competitors. There is no assurance that we will be granted such patents or proprietary technology or that such granted patents or proprietary technology will not be circumvented through the adoption of a competitive, though non-infringing, process or product. The patent positions of pharmaceutical companies can be highly uncertain and involve complex legal, scientific and factual questions for which important legal principles remain unresolved. Changes in either the patent laws or in interpretations of patent laws may diminish the value of the our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowable or enforceable in our patents (including patents owned by or licensed to us). Failure to protect our existing and future intellectual property rights could seriously harm our business and prospects and may result in the loss of our ability to exclude others from using our technology or our own right to use the technologies. If we do not adequately ensure the right to use certain technologies, we may have to pay others for the right to use their intellectual property, pay damages for infringement or misappropriation or be enjoined from using such intellectual property. Our patents do not guarantee the right to use the technologies if other parties own intellectual property rights that are necessary in order to use such technologies. Our patent position is subject to complex factual and legal issues that may give rise to uncertainty as to the validity, scope and enforceability of a particular patent.
In addition, there is a risk that improved versions of our own product developed by third parties will be granted patent protection and compete with our products. For example, any patents we obtain may not be sufficiently broad to prevent others from utilizing our technologies or from developing competing products and technologies. Third parties may attempt to circumvent our patents by means of alternative designs and processes or may independently develop similar products, duplicate any of our potential products not under patent protection, or design around the inventions we claim in any of our existing patents, existing patent applications or future patents or patent applications. The actual protection afforded by a patent varies on a product-by-product basis, from country to country and depends upon many factors, including the type of patent, the scope of our coverage, the availability of regulatory related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patents. It is impossible to anticipate the breadth or degree of protection that patents will afford products developed by us or their underlying technology.
In any case, there can be no assurance that:
| · | any rights under Canadian, U.S. or foreign patents owned by us or other patents that third parties license to us will not be curtailed; |
| · | we were the first inventor of inventions covered by our issued patents or pending applications or that we were the first to file patent applications for such inventions; |
| · | our pending or future patent applications will be issued with the breadth of claim coverage sought by us, or be issued at all; |
| · | our competitors will not independently develop or patent technologies that are substantially equivalent or superior to our technologies; |
| · | third parties will not attempt to circumvent our patents by means of alternative designs and processes or that third parties will not also independently develop similar products, duplicate any of our products not under patent protection, or design around the inventions we claim in any of our existing patents, existing patent applications or future patents or patent applications; |
| · | any of our trade secrets will not be learned independently by our competitors; or |
| · | the steps we take to protect our intellectual property will be adequate. |
In addition, effective patent, trademark, copyright and trade secret protection may be unavailable, limited or not sought in certain foreign countries. Further, the laws of other countries may not protect our intellectual property to the same extent as the laws of the United States, Canada or Europe, and may lack rules and procedures required for defending our patents.
There is a risk that any patents issued relating to our potential products or any patents licensed to us may be successfully challenged or that the practice of our products might infringe the patents of third parties. If the practice
43
| Management’s Discussion and Analysis | June 30, 2015 |
of our potential products infringes the patents of third parties, we may be required to design around such patents, potentially causing increased costs and delays in product development and introduction or precluding us from developing, manufacturing or selling our planned products. In addition, disputes may arise as to the rights to knowhow and inventions among our employees and consultants who use intellectual property owned by others for the work performed for us. The scope and validity of patents which may be obtained by third parties, the extent to which we may wish or need to obtain patent licenses and the cost and availability of such licenses are currently unknown. If such licenses are obtained, it is likely they would be royalty bearing, which could reduce our income. If licenses cannot be obtained on an economical basis or at all, delays in market introduction of our planned products could occur or introduction could be prevented, in some cases causing the expenditure of substantial funds.
In certain instances, we may elect not to seek patent protection but instead rely on the protection of our technology through confidentiality agreements or trade secrets. There can be no assurance that these agreements will not be breached, that we will have adequate remedies for any breach or that such persons or institutions will not assert rights to intellectual property arising out of these relationships. The value of our assets could also be reduced to the extent that third parties are able to obtain patent protection with respect to aspects of our technology or products or that confidential measures we have in place to protect our proprietary technology are breached or become unenforceable. However, third parties may independently develop or obtain similar technology and such third parties may be able to market competing products and obtain regulatory approval through a showing of equivalency to one of our products which has obtained regulatory approval, without being required to undertake the same lengthy and expensive clinical trials that we would have already completed. The cost of enforcing our patent rights or defending rights against infringement charges by other patent holders may be significant and could limit operations.
Litigation may also be necessary to enforce patents issued or licensed to us or to determine the scope and validity of a third party’s proprietary rights. We could incur substantial costs if we are required to defend ourselves in patent suits brought by third parties, if we participate in patent suits brought against or initiated by our corporate collaborators or if we initiate such suits. We may not have the necessary resources to participate in or defend any such activities or litigation. Even if we did have the resources to vigorously pursue our interests in litigation, because of the complexity of the subject matter, it is impossible to predict whether we would prevail in any such action. Any claims of patent infringement asserted by third parties may:
| · | divert the time and attention of our technical personnel and management; |
| · | cause product development or commercialization delays; |
| · | require us to cease or modify our use of the technology or develop non-infringing technology; or |
| · | require us to enter into royalty or licensing agreements. |
An adverse outcome in litigation or an interference to determine priority or other proceeding in a court or patent or selling office could subject us to significant liabilities, require disputed rights to be licensed from third parties or require us to cease using certain technology or products, any of which may have a material adverse effect on the our business, financial condition and results of operations.
We may be subject to claims by third parties asserting that we, or our employees have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
Certain of our employees, including senior management, were previously employed, or continue to be employed, at universities or other public institutions, or at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Some of these employees, executed proprietary rights, nondisclosure and noncompetition agreements, in connection with such previous employment. We may be subject to claims that we, or these employees, have used or disclosed confidential information or intellectual property, including trade secrets or other proprietary information, of any such employee’s former employer. If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel or sustain damages. Such intellectual property rights could be awarded to a third party, and we could be required to obtain a license from such third party to commercialize our technology or products. Such a license may not be available on commercially reasonable terms or at all. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
Risks Related to Ownership of our Common Shares
44
| Management’s Discussion and Analysis | June 30, 2015 |
We have never declared dividends and may not do so in the future.
We have not declared or paid any cash dividends on Common Shares to date. The payment of dividends in the future will be dependent on our earnings and financial condition and on such other factors as our board of directors considers appropriate. Unless and until we pay dividends, shareholders may not receive a return on their shares. There is no present intention by our board of directors to pay dividends on the Common Shares.
We are a “foreign private issuer” and have disclosure obligations that are different from those of U.S. domestic reporting companies. As a foreign private issuer, we are subject to different U.S. securities laws and rules than a domestic U.S. issuer, which may limit the information publicly available to our shareholders.
As a “foreign private issuer”, we are subject to reporting obligations that, in certain respects, are less detailed and less frequent than those of U.S. domestic reporting companies. We are required to file or furnish to the SEC the continuous disclosure documents that we are required to file in Canada under Canadian securities laws. For example, we are not required to issue quarterly reports, proxy statements that comply with the requirements applicable to U.S. domestic reporting companies, or individual executive compensation information that is as detailed as that required of U.S. domestic reporting companies. We will also have four months after the end of each fiscal year to file our annual reports with the SEC and will not be required to file current reports as frequently or promptly as U.S. domestic reporting companies. Furthermore, our officers, directors and principal shareholders are exempt from the insider reporting and short-swing profit recovery requirements in Section 16 of the Exchange Act. Accordingly, our shareholders may not know on as timely a basis when our officers, directors and principal shareholders purchase or sell their Common Shares, as the reporting deadlines under the corresponding Canadian insider reporting requirements are longer. As a foreign private issuer, we are also exempt from the requirements of Regulation FD (Fair Disclosure) which, generally, are meant to ensure that select groups of investors are not privy to specific information about an issuer before other investors. As a result of such varied reporting obligations, shareholders should not expect to receive the same information at the same time as information provided by U.S. domestic companies.
In addition, as a foreign private issuer, we have the option to follow certain Canadian corporate governance practices rather than those of the United States, except to the extent that such laws would be contrary to U.S. securities laws, provided that we disclose the requirements we are not following and describe the Canadian practices we follow instead. As a result, our shareholders may not have the same protections afforded to shareholders of companies that are subject to all domestic U.S. corporate governance requirements.
There is a risk that we are or could become a “passive foreign investment company” which would likely result in materially adverse U.S. federal income tax consequences for U.S. investors.
We believe that we may have been classified as a passive foreign investment company (“PFIC”) for the taxable year ending September 30, 2014, and we believe we may be classified as a PFIC for the current taxable year and in future taxable years. However, the determination as to whether we are a PFIC for any taxable year is based on the application of complex U.S. federal income tax rules that are subject to differing interpretations. If we are a PFIC for any taxable year during which a U.S. Holder (as defined below) holds the Common Shares, it would likely result in adverse U.S. federal income tax consequences for such U.S. Holder. U.S. Holders should consult their own tax advisors regarding the likelihood and consequences of us being treated as a PFIC for U.S. federal income tax purposes, including the advisability of making a qualified electing fund election (including a protective election), which may mitigate certain possible adverse U.S. federal income tax consequences but may result in an inclusion in gross income without receipt of such income.
For the purposes of this Risk Factor, a “U.S. Holder” is a beneficial owner of Common Shares who, for U.S. federal income tax purposes, is (i) a citizen or individual resident of the United States, (ii) a corporation (or other entity that is classified as a corporation for U.S. federal income tax purposes) that is created or organized in or under the laws of the United States, any State thereof or the District of Columbia, (iii) an estate whose income is subject to U.S. federal income tax regardless of its source, or (iv) a trust if (A) a U.S. court can exercise primary supervision over the trust’s administration and one or more U.S. persons are authorized to control all substantial decisions of the trust, or (B) the trust has a valid election in effect to be treated as a U.S. person for U.S. federal income tax purposes.
45
| Management’s Discussion and Analysis | June 30, 2015 |
Our status as an “Emerging Growth Company,” and the reduced disclosure requirements applicable to Emerging Growth Companies, may make the Common Shares less attractive to investors.
We are an “emerging growth company,” as defined in Section 2(a)(19) of the U.S. Securities Act, as modified by the JOBS Act. As such, we are eligible to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act.
In addition, Section 107 of the JOBS Act also provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the U.S. Securities Act for complying with new or revised accounting standards, and delay compliance with new or revised accounting standards until those standards are applicable to private companies. We will not take advantage of the extended transition period for complying with new or revised accounting standards. This election is irrevocable.
We may take advantage of some or all of the reduced regulatory and reporting requirements that will be available to it so long as we qualify as an “emerging growth company” and thus the level of information provided may be different than that of other U.S. public companies. If we do take advantage of any of these exemptions, some investors may find our securities less attractive, which could result in a less active trading market for our Common Shares, and our share price may be more volatile as a result.
We could be an emerging growth company until the last day of the first fiscal year following the fifth anniversary of our first common equity offering, although circumstances could cause us to lose that status earlier if annual revenues exceed US$1.0 billion, if we issue more than US$1.0 billion in non-convertible debt in any three-year period or if we become a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act.
It may be difficult for United States investors to effect services of process or enforcement of actions against us or certain of our directors and officers under U.S. federal securities laws.
We are incorporated under the laws of the Province of British Columbia, Canada. A majority of our directors and officers reside in Canada and all or a substantial portion of our assets and these persons are located outside the United States. Although we have agreed to accept service of process through our agent designated for that purpose, it may be difficult for United States investors to effect service of process in the United States upon us or our directors or officers to enforce against us or these persons judgments obtained in United States courts predicated upon civil liabilities under the Exchange Act or other United States laws. There is substantial doubt as to whether an original action could be brought successfully in Canada against any of such persons or us predicated solely upon such civil liabilities and whether a judgment of a United States court predicated solely upon such civil liabilities would be enforceable in Canada by a Canadian court.
An active trading market for the Common Shares may not be sustained.
Although we have listed the Common Shares on the Nasdaq and the TSX, an active trading market for the Common Shares may not be sustained. In addition, the Common Shares only began trading on Nasdaq on July 9, 2015 and it is too soon to determine whether an active trading market in the United States will develop. Accordingly, if an active trading market for the Common Shares is not maintained, the liquidity of the Common Shares, your ability to sell the Common Shares when desired and the prices that you may obtain for the Common Shares will be adversely affected.
The market price and trading volume of our Common Shares may be volatile, which could result in rapid and substantial losses for our stockholders.
The market price of our Common Shares may be highly volatile and could be subject to wide fluctuations. In addition, the trading volume in the Common Shares may fluctuate and cause significant price variations to occur. Adding further uncertainty to the market price of our Common Shares is our limited trading history on the Nasdaq, where we began trading on July 9, 2015. If the market price of the Common Shares declines significantly, you may be unable to resell your shares at or above your purchase price, if at all. The market price of the Common Shares may fluctuate or decline significantly in the future. Some of the factors that could negatively affect our share price or result in fluctuations in the price or trading volume of the Common Shares include:
46
| Management’s Discussion and Analysis | June 30, 2015 |
| · | quarterly variations in operating results; |
| · | operating results that vary from the expectations of securities analysts and investors; |
| · | change in valuations; |
| · | changes in our operations; |
| · | expenses we incur related to future research; |
| · | regulatory approvals; |
| · | fluctuations in the demand for our product candidates; |
| · | changes in the industry in which we operate; |
| · | announcements by us or other companies of significant contracts, acquisitions, dispositions, strategic partnerships, joint ventures, capital commitments, plans, prospects, service offerings or operating results; |
| · | additions or departures of key personnel; |
| · | future sales of our securities; |
| · | other risk factors discussed herein; and |
| · | other unforeseen events. |
Stock markets in the United States and Canada have experienced extreme price and volume fluctuations. Market fluctuations, as well as general political and economic conditions such as acts of terrorism, prolonged economic uncertainty, a recession or interest rate or currency rate fluctuations, could adversely affect the market price of our Common Shares.
Substantial future sales of our Common Shares, or the perception that these sales could occur, may cause the price of our Common Shares to drop significantly, even if our business is performing well.
A large volume of sales of our Common Shares could decrease the prevailing market price of our Common Shares and could impair our ability to raise additional capital through the sale of equity securities in the future. Even if a substantial number of sales of shares of our Common Shares do not occur, the mere perception of the possibility of these sales could depress the market price of our Common Shares and have a negative effect on our ability to raise capital in the future.
FORWARD-LOOKING AND OTHER STATEMENTS
This MD&A, including the documents incorporated by reference herein, contains forward-looking statements or forward-looking information within the meaning of the U.S. Private Securities Litigation Reform Act and applicable Canadian securities laws. All statements in this MD&A, other than statements of historical facts, are forward-looking statements. These statements appear in a number of different places in this MD&A and can be identified by words such as “anticipates”, “estimates”, “projects”, “expects”, “intends”, “believes”, “plans”, “will”, “could”, “may”, or their negatives or other comparable words. Such forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements that may be expressed or implied by such forward-looking statements. Examples of such forward looking statements include, but are not limited to:
| · | our expectations with respect to the occurrence, scope and timing of the FDA’s clearance of our IND application; |
| · | the intention to file a CTA (as defined above) application in Canada, and expectations regarding the timing of such applications; |
| · | the initiation, timing, cost, progress and success of our research and development programs, pre-clinical studies and clinical trials; |
| · | our ability to advance product candidates into, and successfully complete, clinical trials; |
| · | our ability to achieve profitability; |
| · | our ability to obtain funding for our operations, including research funding; |
| · | our ability to recruit sufficient numbers of patients for our future clinical trials; |
| · | our ability to establish and maintain relationships with collaborators with acceptable development, regulatory and commercialization expertise and the benefits to be derived from such collaborative efforts; |
| · | the implementation of our business model and strategic plans; |
| · | our ability to develop and commercialize product candidates; |
| · | our commercialization, marketing and manufacturing capabilities and strategy; |
47
| Management’s Discussion and Analysis | June 30, 2015 |
| · | our ability to protect our intellectual property and operate our business without infringing upon the intellectual property rights of others; |
| · | our expectations regarding federal, provincial and foreign regulatory requirements; |
| · | whether the Company will receive, and the timing and costs of obtaining, regulatory approvals in the U.S., Canada, the European Union and other jurisdictions; |
| · | the therapeutic benefits, effectiveness and safety of our product candidates; |
| · | the accuracy of our estimates of the size and characteristics of the markets that may be addressed by our products and product candidates; |
| · | the rate and degree of market acceptance and clinical utility of our future products, if any; |
| · | the timing of, and our ability and our collaborators’ ability, if any, to obtain and maintain regulatory approvals for our product candidate; |
| · | our expectations regarding market risk, including interest rate changes and foreign currency fluctuations; |
| · | our ability to engage and retain the employees required to grow our business; |
| · | the compensation that is expected to be paid to employees of the Company; |
| · | our future financial performance and projected expenditures; |
| · | developments relating to our competitors and our industry, including the success of competing therapies that are or become available; and |
| · | estimates of our expenses, future revenue, capital requirements and our needs for additional financing. |
Such statements reflect our current views with respect to future events, are subject to risks and uncertainties and are necessarily based upon a number of estimates and assumptions that are inherently subject to significant medical, scientific, business, economic, competitive, political and social uncertainties and contingencies. Many factors could cause our actual results, performance or achievements to be materially different from any future results, performance, or achievements that may be expressed or implied by such forward-looking statements, including those described under “Risk Factors”. In making the forward looking statements included in this MD&A, the Company has made various material assumptions, including but not limited to:
| · | our ability to obtain positive results of clinical trials; |
| · | our ability to obtain required regulatory approvals; |
| · | our ability to successfully out-license or sell future products, if any, and in-license and develop new products; |
| · | favourable general business and economic conditions; |
| · | the availability of financing on reasonable terms; |
| · | our ability to attract and retain skilled staff; |
| · | market competition; |
| · | the products and technology offered by the Company’s competitors; and |
| · | our ability to protect patents and proprietary rights. |
If one or more of these risks or uncertainties occur, of if our underlying assumptions prove to be incorrect, actual results may vary significantly from those expressed or implied by forward-looking statements. The forward-looking statements represent our views as of the date of this document. While we may elect to update these forward-looking statements In the future, we have no current intention to do so except as to the extent required by applicable law.
48
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Essa Pharma Inc. | ||||||
| (Registrant) | ||||||
| Date: | August 18, 2015 | By: | /s/ David Wood | |||
| Name: | David Wood | |||||
| Title: | Chief Financial Officer | |||||