BUSINESS Overview We are a commercial-stage biotechnology company focused on developing and commercializing innovative molecularly-targeted and immuno-oncology drugs for the treatment of cancer. Our internally-developed lead drug candidates are currently in late-stage clinical trials, and we are marketing three in-licensed drugs in China from which we have been generating product revenue since September 2017. Our mission is to become a global leader in the discovery, development and commercialization of innovative therapies. We started as a research and development company in Beijing in 2010 focusing on developing best-in-class oncology therapeutics. Over the last eight years, we have developed into a fully-integrated global biotechnology company with a broad portfolio consisting of six internally-developed, clinical-stage drug candidates, including three late-stage clinical drug candidates. We have also in-licensed five drugs and drug candidates, including three marketed drugs in China and two clinical-stage drug candidates for which we have obtained development and commercialization rights in China and other selected countries in the Asia-Pacific region. Our Core Product Candidates include the following: •Zanubrutinib (BGB-3111) — a potentially best-in-class investigational small molecule inhibitor of BTK, that is currently being evaluated in a broad pivotal clinical program in China and in other markets, including the United States and the European Union, which we refer to as globally, for which we expect to file for approval in China in 2018 initially for the treatment of MCL, and submit an NDA in the first half of 2019 an NDA to the FDA to pursue an accelerated approval for the treatment of WM; •Tislelizumab (BGB-A317) — an investigational humanized monoclonal antibody against the immune checkpoint receptor PD-1 that is currently being evaluated in a broad pivotal clinical program globally and in China, for which we expect to file for approval in China in 2018 initially for the treatment of cHL; and •Pamiparib (BGB-290) — an investigational small molecule inhibitor of the PARP1 and PARP2 enzymes that is being evaluated in two pivotal clinical trials in China, with a global Phase 3 trial planned for which we are screening patients in preparation for the first dosing. We are preparing to launch the two lead product candidates from our internal pipeline, zanubrutinib and tislelizumab, which we believe will address major unmet medical needs and have significant commercial potential. In addition to our three late-stage clinical drug candidates, our pipeline also includes three internally-developed drug candidates in Phase 1 clinical development: lifirafenib (BGB-283), an investigational RAF dimer inhibitor, BGB-A333, an investigational humanized monoclonal antibody against the immune checkpoint receptor ligand PD-L1, and BGB-A425, an investigational humanized monoclonal antibody against TIM-3. We entered into a strategic collaboration with Celgene Corporation in August 2017, in which we obtained an exclusive license to market in China Celgene’s approved cancer therapies ABRAXANE®, REVLIMID® and VIDAZA®, as well as rights in China to develop and commercialize avadomide (CC-122), an investigational next-generation Cereblon modulator currently in clinical development by Celgene outside of China for lymphoma and hepatocellular carcinomas, or HCC. As part of the

collaboration, we also granted Celgene an exclusive right to develop and commercialize tislelizumab for solid tumors in the United States, Europe, Japan and the rest of world other than Asia, for which we received US$263 million in upfront license fees and a US$150 million equity investment and are eligible to receive up to US$980 million in milestone payments and royalties on future sales. Our portfolio also includes sitravatinib, an in-licensed, investigational, spectrum-selective kinase inhibitor in clinical development by Mirati Therapeutics, Inc., or Mirati, for the treatment of non-small cell lung cancer, or NSCLC, and other tumors, for which we are planning to initiate clinical development in China. We have strong internal capabilities spanning research, clinical development, manufacturing and commercialization. We have advanced six internally-developed candidates into clinical trials, including three into pivotal trials. With more than 500 clinical development personnel in China, the United States, Australia and Switzerland as of July 20, 2018, we have built internal clinical development capabilities globally which we believe provide a competitive advantage over other biotechnology companies in China. We have an 11,000-square meter facility in Suzhou for the manufacture of small molecule drugs at commercial scale and biologics drugs at pilot scale. We are currently building a 24,000-liter commercial-scale biologics manufacturing factory in Guangzhou. We also have a growing commercial team in China, which provides us with the initial commercial platform for the planned launches of our internally-developed drug candidates as well as current and potentially future in-licensed drug candidates. We have formed collaborations with other biotechnology companies aiming to capture opportunities in China and the broader Asia-Pacific region by leveraging our global clinical development capabilities and China commercial capabilities, as evidenced by our collaborations with Celgene and Mirati. We believe we are well-positioned to capture the significant market opportunities in China, including those created by recent regulatory reforms and new reimbursement policies in China. China is the second largest pharmaceutical market in the world based on revenue, and the oncology sector grew at a 13.7% CAGR from 2013 to 2017, according to the Frost and Sullivan Report. We believe that there is a large and growing opportunity for novel cancer therapeutics in China based on significant unmet medical need, a large target patient population, expanding reimbursement coverage, and increasing treatment affordability and willingness to pay. In addition, the CDA has undertaken significant regulatory reforms that are designed to accelerate the development of new innovative drugs and allow China to be an integral part of global drug development. In addition, innovative oncology drugs have been included in the most recent NDRL, reducing out-of-pocket expenses for patients. We believe that access to the large number of patients in China during clinical development as well as commercialization creates new opportunities for us. Leveraging our strong China presence and commitment to global standards of innovation and quality, we believe we have a unique ability to effectively take advantage of these opportunities.

Our Strengths We believe the following strengths have contributed to our success and differentiate us from our competitors: Fully-Integrated Biotechnology Company with Broad Capabilities in China and Globally Initially started as a research and development company in Beijing in 2010, we have since become a fully-integrated global biotechnology company with broad capabilities spanning research, clinical development, manufacturing and commercialization. Research. As of July 20, 2018, we had a team of approximately 200 researchers based in Beijing. Our scientific advisory board, which may from time to time provide us with assistance upon our request, is comprised of world-renowned experts with extensive expertise in cancer drug research and development, and is led by Dr. Xiaodong Wang, founding director of China’s National Institute of Biological Sciences in Beijing, and member of the U.S. National Academy of Sciences and the Chinese Academy of Sciences. In addition, we have built strong working relationships with key Chinese cancer centers, which give us access to patient biopsy samples that allow us to develop an extensive collection of proprietary cancer models. In eight years, our research team has generated six internally-developed candidates that we have advanced into clinical trials, including three which are currently in pivotal trials. Clinical Development. We believe clinical development capabilities are critical to success in our industry. We have built internal clinical development capabilities globally, which we believe provide a competitive advantage over other biotechnology companies in China. As of July 20, 2018, we had over 200 clinical development staff in the United States and over 300 in China and the broader Asia-Pacific region. We believe that this global capability enables us to take advantage of significant regulatory reforms in China by integrating China and global clinical development, which allows access to a patient base that is as large as the United States and Europe combined. As of July 5, 2018, we had more than 50 clinical trials ongoing or planned for initiation, including 14 pivotal or potentially registration-enabling trials, with more than 3,000 patients and healthy subjects already enrolled in these trials. These trials include clinical sites in the United States, Australia, New Zealand, China and other Asian countries, as well as Europe. We believe that our broad global clinical development program will translate into significant commercial opportunities. In addition, we believe that our investment in research and development of US$269 million during 2017 was the largest in oncology and one of the largest by a China-based biopharmaceutical company. Manufacturing. We have an 11,000-square meter facility in Suzhou for the manufacture of small molecule drugs at commercial scale and biologics drugs at pilot scale. The facility was designed to comply with GMP requirements in China, the European Union and the United States. In January 2018, the facility received a manufacturing license from the Jiangsu Food and Drug Administration, in preparation for the commercial manufacture of zanubrutinib in China. We have another

100,000-square meter facility under construction in Guangzhou for the manufacture of biologics at commercial scale. This facility is planned to have a 24,000-liter capacity, and over US$300 million in funding has been committed for the construction of this facility. We expect the first phase of the facility to be completed in 2019. We also have a commercial supply partner, Boehringer Ingelheim, under an exclusive multi-year arrangement to manufacture our biologic drug candidate, tislelizumab, in its manufacturing facility in Shanghai as part of a MAH trial project. Commercialization. In connection with our collaboration with Celgene, we obtained Celgene’s commercial operations in China and an exclusive license to market three of Celgene’s cancer therapies, ABRAXANE®, REVLIMID® and VIDAZA®. This collaboration provided us with commercial infrastructure and a marketed drug portfolio in China. We believe that our commercial team has established and maintains strong relationships with leading hospitals and medical professionals. We also believe that we have built relationships with key opinion leaders, or KOLs, in oncology through our years of research and clinical development efforts. Recently, we have strengthened our commercial leadership by adding Dr. Xiaobin Wu, who was the Country Manager of Pfizer China and Regional President of Pfizer Essential Health for Greater China, Ivan Yifei Zhu, who was the Vice President of Sales and Marketing at Janssen China, Vivian Xin Bian, who was the Vice President of Innovative Products Division at Janssen China. We believe these commercial leadership additions will further help build our commercial team and drive future product launches. We believe these efforts position us well for the planned launches of our internally-developed drug candidates as well as current and potentially future in-licensed drug candidates. Two Late-Stage Clinical Drug Candidates with Significant Commercial Potential We believe that our two lead drug candidates have significant commercial potential and we are well-positioned to realize these product-based opportunities. Zanubrutinib. We believe that our lead drug candidate, zanubrutinib, is a potentially best-in-class BTK inhibitor based on the clinical data to date. Zanubrutinib has demonstrated higher selectivity against BTK and higher exposure than the first approved BTK inhibitor IMBRUVICA®, or ibrutinib, in pre-clinical models. As of July 5, 2018, we had enrolled more than 1,200 patients in clinical trials of zanubrutinib. The preliminary data reported to date demonstrated favorable response rates, quality and durability in various B-cell malignancies, such as WM and CLL/SLL. We are running a broad pivotal clinical program globally and in China, including a global Phase 3 head-to-head trial against ibrutinib in WM, a global Phase 3 trial in treatment-naïve CLL/SLL, a global pivotal Phase 2 trial in combination with obinutuzumab in FL, and three China pivotal Phase 2 trials in MCL, CLL/SLL and WM, respectively, in which the global Phase 3 WM trial and the three China pivotal Phase 2 trials have completed enrollment.

We have received results from independent review of the Phase 2 study in Chinese patients with R/R MCL. The overall response met the pre-specified criteria for a positive trial. We had a pre-NDA meeting with the CDA earlier this year and based on the feedback we received from the meeting, we currently believe that, subject to the successful completion and satisfactory results of the trials, we are on track to file the NDA for the treatment of R/R MCL in 2018. In July 2018, zanubrutinib was granted Fast Track Designation by the FDA for the treatment of patients with WM. Based on our discussions with the FDA, internal review of available data from our global Phase 1 trial of zanubrutinib in patients with WM, and supported by the Fast Track Designation, we are preparing to submit in the first half of 2019 an NDA to pursue an accelerated approval of zanubrutinib for patients with WM based on results from the global Phase 1 study. The FDA’s Fast Track program is intended to expedite or facilitate the process for reviewing new drugs that are intended to treat a serious or life-threatening disease or condition for which there is no effective treatment and demonstrate the potential to address unmet medical needs for the condition. A drug candidate with a Fast Track designation may be eligible for more frequent communications with the FDA, eligibility for Accelerated Approval and Priority Review (if relevant criteria are met), and rolling review of the NDA. BTK inhibitors reported approximately US$3.2 billion in global sales in 2017 and are projected to reach US$17.8 billion in 2030 according to the Frost & Sullivan Report. According to the same report, the class of BTK inhibitors is projected to represent a US$1.6 billion market in China in 2030. We believe there is significant market opportunity given the potentially best-in-class profile and the broad pivotal clinical program that we are currently conducting for zanubrutinib. Tislelizumab. Tislelizumab is an investigational humanized PD-1 monoclonal antibody that belongs to a class of immuno-oncology agents known as immune checkpoint inhibitors. Tislelizumab is being developed as a monotherapy and in combination with other therapies for the treatment of a broad array of solid tumors and hematologic cancers. As of July 5, 2018, we had enrolled more than 1,500 patients in clinical trials of tislelizumab. The preliminary data reported to date demonstrated that tislelizumab was generally well-tolerated and exhibited anti-tumor activity in a variety of tumor types. We had a pre-NDA meeting with the CDA, and based on the feedback we received from the meeting, we believe we are on track to file the NDA in China in 2018 initially for the treatment for HL. We believe that PD-1/PD-L1 antibody therapies represent a large market opportunity, particularly in the favorably evolving China market. According to the Frost & Sullivan Report, the worldwide annual sales of the PD-1/PD-L1 class reached US$10.1 billion in 2017 and are projected to amount to US$78.9 billion worldwide and US$15.1 billion in China in 2030. We believe the China market is particularly attractive, as currently available clinical data suggest that some of the most prevalent cancers in China, such as lung, gastric, liver and esophageal cancers, are responsive to these PD-1/PD-L1 antibody therapies.

We believe that we are uniquely positioned to capture this market opportunity because of our broad clinical development program and growing commercial capabilities in China. As of July 20, 2018, we had initiated four global pivotal or potentially registration-enabling trials of tislelizumab that are intended to support regulatory submissions globally and in China, and two pivotal trials in China, and we and Celgene expect to commence additional pivotal trials in 2018 and 2019. In China, we are also screening patients in preparation for the first dosing in a new pivotal trial, the Phase 3 trial combined with chemotherapy as a first-line treatment for patients with advanced non-squamous NSCLC. We believe that our broad pivotal clinical program will enable wide reimbursement coverage for patients in China and allow us to maximize the commercial opportunities for tislelizumab. In addition, we believe that our strategic partnership with Celgene will further expand our development programs and help maximize the commercial potential for tislelizumab across product combinations and global markets. Robust Pipeline of Internally-Developed and In-Licensed Product Candidates Beyond zanubrutinib and tislelizumab, we have an extensive pipeline of internally-developed and in-licensed product candidates that we are developing as monotherapies and in combination with other therapies. We have the internal capacity to develop both biologic and small-molecule drugs. Our recent collaboration with Mirati for sitravatinib provides an example of our ability to supplement internal research and development through external collaboration. Pamiparib. We believe that pamiparib, a PARP1 and PARP2 inhibitor, has the potential to be differentiated from other PARP inhibitors because of its potential brain penetration, high selectivity, strong DNA-trapping activity and good oral bioavailability, based on pre-clinical data. In Phase 1/2 trials to date, it was demonstrated that pamiparib was generally well-tolerated and showed promising anti-tumor activity in ovarian cancer, and initial data from the dose-escalation portion of the Phase 1 trial of tislelizumab in combination with pamiparib suggested that the combination was generally well-tolerated and showed anti-tumor activity in multiple solid tumor types. We currently have a pivotal Phase 2 trial in China in patients with germline BRCA-mutated ovarian cancer and a Phase 3 trial in China as a potential maintenance therapy in patients with platinum-sensitive ovarian cancer. We are also screening patients in preparation of the first dosing of a global Phase 3 maintenance trial in patients with platinum-sensitive GC. Other early-stage assets. We are pursuing the clinical development of other early-stage drug candidates, advancing pre-clinical drug candidates toward clinical trials and developing additional novel drug candidates. For example, three internally-developed drug candidates, lifirafenib, BGB-A333, and BGB-A425 are in Phase 1 clinical development. Sitravatinib is in clinical development by Mirati for the treatment of NSCLC and other tumors, and avadomide (CC-122) is currently in clinical development by Celgene for lymphomas and HCC.

Experienced Management Team with Diverse Backgrounds and Skill Sets We have assembled an experienced management team with geographically diverse backgrounds and skillsets to lead our company. Our Co-Founder, Xiaodong Wang, Ph.D., is a highly respected cancer scientist, a member of the U.S. National Academy of Sciences and the Chinese Academy of Sciences, and head of China’s National Institute of Biological Sciences. John Oyler, our Co-Founder, Chief Executive Officer and Chairman of the Board is a serial entrepreneur with a track record of successfully starting and managing companies in several industries including biotechnology. Dr. Xiaobin Wu, our General Manager of China and President, was the Country Manager of Pfizer China and Regional President of Pfizer Essential Health for Greater China and has 17 years of experience leading China operations of multinational companies. Our management team has experience successfully translating scientific visions into tangible drug candidates, solving complex issues in clinical development, progressing drug candidates through regulatory approval, and commercializing innovative therapies. Our Strategy Our mission is to become a global leader in the discovery, development and commercialization of innovative therapies. In the near term, we plan to focus on pursuing the following significant opportunities: Globally Develop and Commercialize Zanubrutinib, a Potentially Best-in-Class BTK Inhibitor Zanubrutinib is an investigational small molecule inhibitor of BTK that is currently being evaluated both as a monotherapy and in combination with other therapies to treat various lymphomas. Our clinical experience to date suggests a potentially best-in-class profile. To pursue this opportunity, we are conducting a broad pivotal clinical program globally and in China, including a global Phase 3 trial head-to-head against ibrutinib in WM, a global Phase 3 trial in treatment-naïve CLL/SLL, a global pivotal Phase 2 trial in combination with obinutuzumab in FL, and three China pivotal Phase 2 trials in MCL, CLL/SLL and WM, respectively. Subject to the successful completion and satisfactory results of the trials, we expect to file for approval in China in 2018 for the treatment of R/R MCL and accelerated approval in the U.S. for the treatment of WM in the first half of 2019. Develop and Commercialize Our Investigational Checkpoint Inhibitor, Tislelizumab, in a Rapidly and Favorably Evolving China Market We believe that there is a large and growing opportunity for novel cancer therapeutics in China and that the market opportunity for PD-1/PD-L1 antibody therapies may be especially attractive, as this class of agents has demonstrated anti-tumor activity in all four of the most common tumors in China: lung, gastric, liver and esophageal cancers. We believe that we are uniquely positioned to capture this opportunity with our strategic collaboration with Celgene, our strong presence in China, and our integrated global and China clinical development capabilities. We have initiated global Phase 3 trials to evaluate tislelizumab as a potential second-or third-line treatment compared to docetaxel

in patients with NSCLC; as a potential first-line treatment compared to sorafenib in patients with HCC; and as a potential second-line treatment compared to investigator-chosen chemotherapy in patients with esophageal squamous cell carcinoma, or ESCC. We also have a global Phase 2 trial in patients with previously treated advanced HCC, as well as a global Phase 2 trial in patients with relapsed or refractory mature T-and NK-cell lymphomas. We and our strategic collaborator Celgene expect to commence additional global pivotal trials in 2018 and 2019. Moreover, we have two additional China pivotal trials ongoing, and subject to the successful completion and satisfactory results of the trials, we expect to file for approval in China for the treatment of cHL in 2018. We are also screening patients in preparation for the first dosing in a new pivotal trial, the Phase 3 trial combined with chemotherapy as a first-line treatment for patients with advanced non-squamous NSCLC. Build A Leadership Position by Further Expanding Our Capabilities Although we believe that we have significant integrated capabilities in research and clinical development, manufacturing and commercialization, we plan to continue to strengthen and expand our platform. In particular, we plan to significantly expand our commercial capabilities in China in preparation for the potential launch of our drug candidates and to support our existing marketed drugs. We have an established commercial team in China, which provides coverage of large hospitals and physician clients. As a result of the improving reimbursement environment in China, which is expected to provide access to innovative medicines for a significantly larger number of patients, we believe that the scale of our commercial organization and the breadth of our market coverage will become even more important. We plan to invest in expanding our teams of sales and marketing, market access, medical and scientific affairs, compliance, and other supporting functions. We aim to become a leading organization in the commercialization of oncology drugs in China. Outside of China, we also plan to build commercial capabilities in the hematology-oncology area ahead of the potential launch of zanubrutinib. In addition, we plan to continue to invest in building our global clinical development capabilities, which we believe will provide a competitive advantage in allowing us to conduct pivotal trials to support approvals globally and in China. Take Advantage of Significant Regulatory Reforms in China to Accelerate Global Drug Development Historically, the regulatory environment in China has been considered highly challenging, with clinical development significantly delayed and regulatory approvals taking much longer than in the United States and Europe. To address these challenges, the CDA has issued a series of reform policies and opinions, which, among many things, are expected to expand access to clinical patients and expedite development and approval by removing delays and creating an environment with international quality standards for drug development, manufacturing and commercialization in China. We expect that these regulatory reforms will allow clinical trials in China to play a major role in global drug development programs. We also believe that the ability to effectively operate in China and integrate trials conducted in China with those in the rest of the world will be of increasing strategic importance. We are already taking advantage of these opportunities by conducting and leading dual-purpose global / China registration trials under our collaboration with Celgene.

Expand Our Product Portfolio and Pipeline Through Collaborations with Other Biopharmaceutical Companies to Complement Our Internal Research We expect to further expand our portfolio of drugs and drug candidates, in oncology as well as potentially in other therapeutic areas, through internal research and external collaborations. We intend to pursue collaborations with other biopharmaceutical companies both in China and globally by leveraging our strong clinical development capabilities. We have pursued and plan to continue to pursue business development opportunities, such as our collaboration with Mirati, in which development in China is expected to contribute to, and potentially accelerate, the global development program. We believe that there will be increasing interest by international biopharmaceutical companies in seeking collaborations in Asia, particularly in oncology, where clinical recruitment is a major bottleneck in new drug development. Our Pipeline and Commercial Products The following table summarizes the status of our pipeline and commercial products: China Global (ex-China) Global (China included) *Some indications will not require a non-pivotal Phase 2 clinical trial prior to beginning pivotal Phase 2 or 3 clinical trials. **Confirmatory clinical trials post approval are required for accelerated approvals. ***Revlimid approved as a combination therapy with dexamethasone. 1 Celgene has the right to develop and commercialize tislelizumab in solid tumors in the United States, European Union, Japan and the rest-of-world outside of Asia. 2 Limited collaboration with Merck KGaA. 3 Partnership with Mirati Therapeutics, Inc. Abbreviations: Dose Esc = dose escalation; Dose Exp = dose expansion; WM = Waldenstrom’s macroglobulinemia; 1L = first line; CLL = chronic lymphocytic leukemia; SLL = small lymphocytic lymphoma; R/R = relapsed / refractory; MCL = mantle cell lymphoma; FL = follicular lymphoma; 2L = second line; NSCLC = non-small cell lung cancer; HCC = hepatocellular carcinoma; ESCC = esophageal squamous cell carcinoma; HL = Hodgkin’s lymphoma; UC = urothelial carcinoma; 3L = third
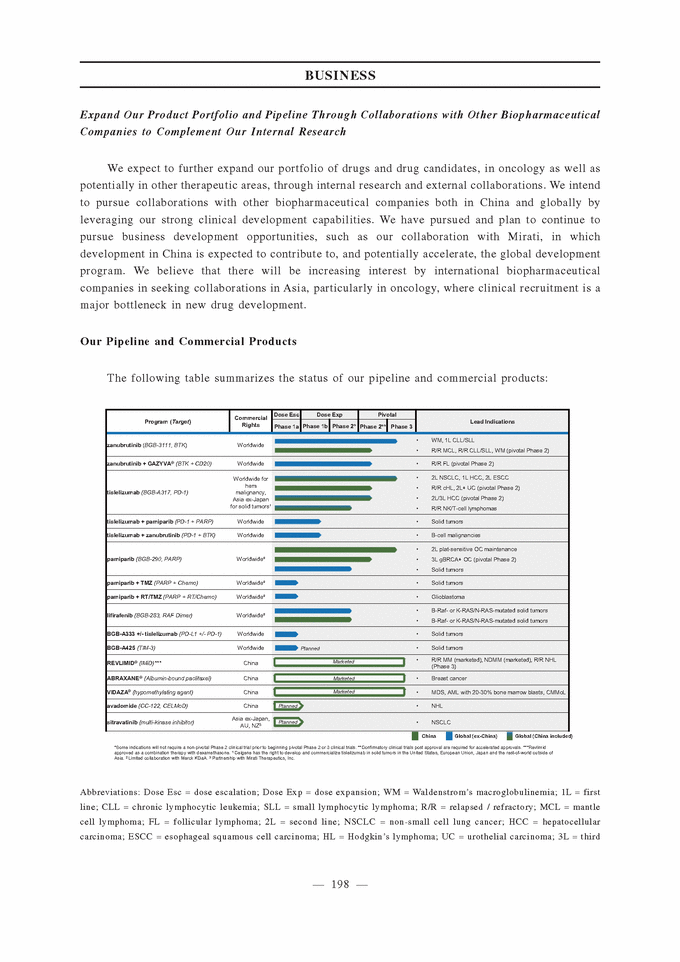
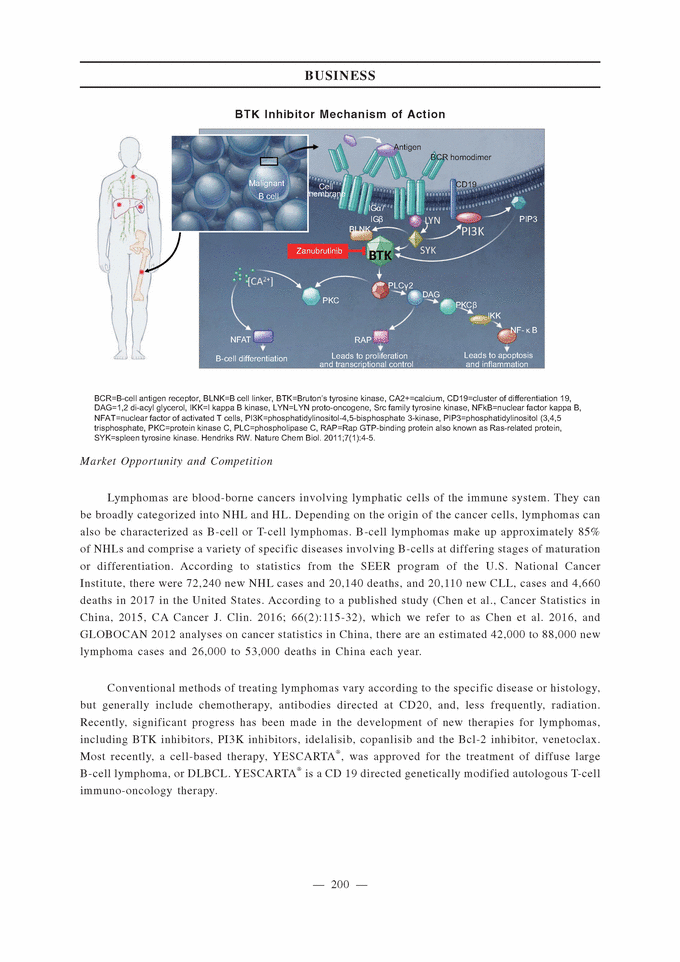
line treatment; gBRCA = germline BRCA; OC = ovarian cancer, TMZ = temozolomide; RT = radiotherapy; IMiD = immunomodulatory drugs; MM = multiple myeloma; ND = newly diagnosed; NHL = non-Hodgkin’s lymphoma; MDS = myelodysplastic syndrome; AML = acute myeloid leukemia; CMMoL = chronic myelomonocytic leukemia; DLBCL = diffuse large B-cell lymphoma; *Some indications will not require a non-pivotal Phase 2 clinical trial prior to beginning pivotal Phase 2 or 3 clinical trials. **Confirmatory clinical trials post-approval are required for accelerated approvals. ***REVLIMID® approved as a combination therapy with dexamethasone. 1 Celgene has the right to develop and commercialize tislelizumab in solid tumors in the United States, Europe, Japan and the rest-of-world outside of Asia. 2 Limited collaboration with Merck KGaA. 3 Partnership with Mirati Therapeutics, Inc. Our drug candidates are subject to NDA approval by the relevant authorities, such as the FDA and the CDA, before commercialization in the relevant jurisdictions. Please refer to the section titled “Regulations — U.S. Regulation — U.S. Government Regulation and Product Approval” and “— PRC Regulation — PRC Drug Regulation” for details. As of the date of this [REDACTED], we believe that we have not received any material comments or concerns raised by the CDA that we are not able to address in a timely manner, and we believe we are on track to file the NDAs related to our Core Product Candidates as described in the section titled “— Our Clinical-Stage Drug Candidates.” Our Clinical-Stage Drug Candidates Zanubrutinib (BGB-3111), a BTK Inhibitor Zanubrutinib is an investigational small molecule inhibitor of BTK that is currently being evaluated in a broad pivotal clinical program globally and in China as a monotherapy and in combination with other therapies to treat various lymphomas. Zanubrutinib has demonstrated higher selectivity against BTK than IMBRUVICA® (ibrutinib), an approved BTK inhibitor, based on our biochemical assays, higher exposure than ibrutinib based on their respective Phase 1 experience in separate studies, and sustained 24-hour BTK occupancy in both the peripheral blood and lymph node compartments. Mechanism of Action BTK is a key component of the BCR signaling pathway and is an important regulator of cell proliferation and cell survival in various lymphomas. BTK inhibitors block BCR-induced BTK activation and its downstream signaling, leading to growth inhibition and cell death in certain malignant white blood cells called B-cells. Zanubrutinib is an orally active inhibitor that covalently binds to BTK, resulting in irreversible inactivation of the enzyme.

BTK Inhibitor Mechanism of Action m BCR homodimer Cell embrane IG / IG CD19 LYN PI3K SYK BTK Zanubrutinib [CA 2+] PKC RAP G IKK NF- B B-cell differentiation Leads to proliferation and transcriptional control Leads to apoptosis and inflammation BCR=B-cell antigen receptor, BLNK=B cell linker, BTK=Bruton’s tyrosine kinase, CA2+=calcium, CD19=cluster of differentiation 19, DAG=1,2 di-acyl glycerol, IKK=I kappa B kinase, LYN=LYN proto-oncogene, Src family tyrosine kinase, NFkB=nuclear factor kappa B, NFAT=nuclear factor of activated T cells, PI3K=phosphatidylinositol-4,5-bisphosphate 3-kinase, PIP3=phosphatidylinositol (3,4,5 trisphosphate, PKC=protein kinase C, PLC=phospholipase C, RAP=Rap GTP-binding protein also known as Ras-related protein, SYK=spleen tyrosine kinase. Hendriks RW. Nature Chem Biol. 2011;7(1):4-5. Market Opportunity and Competition Lymphomas are blood-borne cancers involving lymphatic cells of the immune system. They can be broadly categorized into NHL and HL. Depending on the origin of the cancer cells, lymphomas can also be characterized as B-cell or T-cell lymphomas. B-cell lymphomas make up approximately 85% of NHLs and comprise a variety of specific diseases involving B-cells at differing stages of maturation or differentiation. According to statistics from the SEER program of the U.S. National Cancer Institute, there were 72,240 new NHL cases and 20,140 deaths, and 20,110 new CLL, cases and 4,660 deaths in 2017 in the United States. According to a published study (Chen et al., Cancer Statistics in China, 2015, CA Cancer J. Clin. 2016; 66(2):115-32), which we refer to as Chen et al. 2016, and GLOBOCAN 2012 analyses on cancer statistics in China, there are an estimated 42,000 to 88,000 new lymphoma cases and 26,000 to 53,000 deaths in China each year. Conventional methods of treating lymphomas vary according to the specific disease or histology, but generally include chemotherapy, antibodies directed at CD20, and, less frequently, radiation. Recently, significant progress has been made in the development of new therapies for lymphomas, including BTK inhibitors, PI3K inhibitors, idelalisib, copanlisib and the Bcl-2 inhibitor, venetoclax. Most recently, a cell-based therapy, YESCARTA®, was approved for the treatment of diffuse large B-cell lymphoma, or DLBCL. YESCARTA® is a CD 19 directed genetically modified autologous T-cell immuno-oncology therapy.
The BTK inhibitor ibrutinib was first approved by the FDA in 2013 for the treatment of patients with MCL, who have received at least one prior therapy. Since 2013, ibrutinib has received supplemental FDA approvals for the treatment of patients with CLL, CLL patients with 17p deletion, patients with WM, patients with MZL, who have received at least one prior anti-CD20-based therapy, and patients with chronic graft versus host disease after failure of one or more lines of systemic therapy. Ibrutinib is also approved by the EMA for the treatment of patients with MCL, CLL or WM. Ibrutinib has been approved in over 80 countries and regions, and it was approved and launched in China at the end of 2017. In 2017, global revenues for BTK inhibitors were approximately US$3.2 billion. Another BTK inhibitor, CALQUENCE® (acalabrutinib) was approved by the FDA in 2017 under accelerated approval for the treatment of patients with MCL who have received at least one prior therapy. The table below summarizes the China competitive landscape of zanubrutinib, according to the Frost & Sullivan Report. Abbreviations: CII = Critical Illness Insurance; CTA = Clinical trial application; NA = not applicable Summary of Clinical Results As of July 5, 2018, we have enrolled more than an aggregate of 1,200 patients in clinical trials of zanubrutinib, including trials of zanubrutinib in combination with other therapies, which we refer to as combination trials. A multi-center, open-label Phase 1 trial is being conducted in Australia, New Zealand, the United States, South Korea and European countries to assess the safety, tolerability, pharmacokinetic properties and preliminary activity of zanubrutinib as a monotherapy in patients with different subtypes of B-cell malignancies, such as WM, CLL/SLL, FL, and MCL. The initial results of the dose-escalation phase and dose-expansion phase of this trial, as shown in the chart below, demonstrated that, consistent with zanubrutinib’s pharmacokinetic profile, complete and sustained 24-hour BTK occupancy in the blood was observed in all tested patients, starting at the lowest dose of 40 mg once daily. In addition, sustained full BTK occupancy was observed in the lymph nodes especially for the 160 mg twice daily dosing regimen. There is no guarantee that these results will be reproduced in pivotal trials.
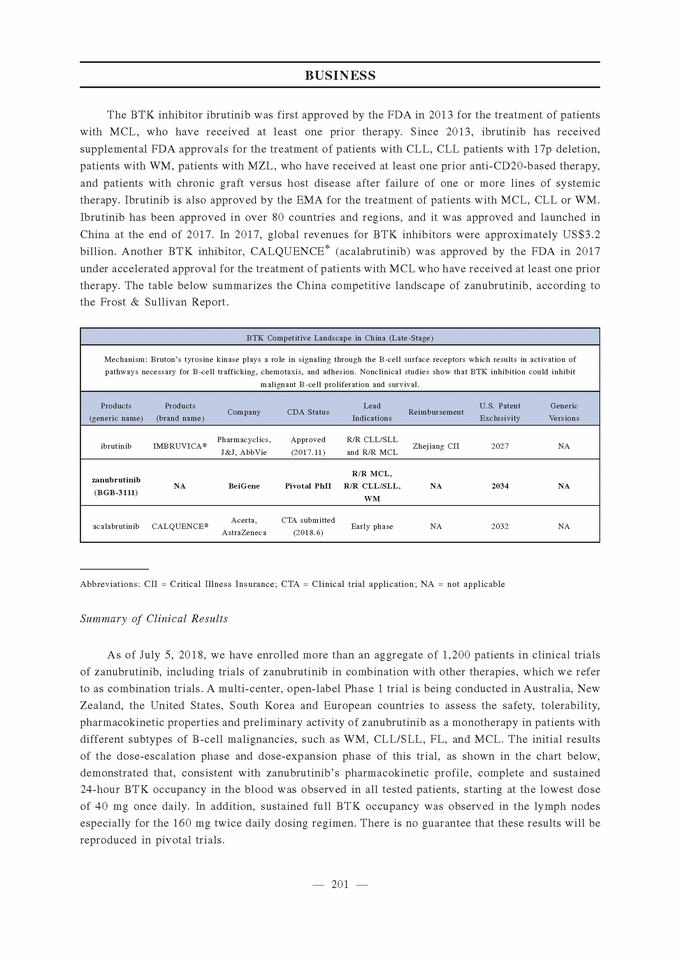
Clinical Trial Data Show Sustained Full BTK Occupancy Peripheral Blood Mononuclear CellLymph Node Note: Data from 20 patients. W1D1 stands for week 1 day 1; W1D2 stands for week 1 day 2; W1D3 stands for week 1 day 3; W2D1 stands for week 2 day 1; Pre stands for pre-dose Note: QD means once daily, BID means twice daily. Paired lymph node biopsies were collected during screening or pre-dose on day 3. Waldenstrom’s Macroglobulinemia On June 15, 2018, we presented data from our Phase 1 trial in patients with WM at the 23rd Congress of the EHA in Stockholm, Sweden. As of the data cutoff of November 3, 2017, 67 WM patients were enrolled in the study. At the time of the data cutoff, 59 patients remained on study treatment. Responses were determined according to the modified Sixth International Workshop on WM Criteria. Zanubrutinib was observed to be generally well-tolerated with no discontinuation for zanubrutinib-related toxicity. AEs were generally mild in severity and self-limited. The most frequent AEs (>15%, all grade 1-2 but one) of any attribution among 67 patients evaluable for safety were petechia/purpura/contusion (37%), upper respiratory tract infection (34%), constipation (18%) and diarrhea (18%). Grade 3-4 AEs of any attribution reported in two or more patients included anemia (7%), neutropenia (6%), basal cell carcinoma (3%), hypertension (3%), squamous cell carcinoma (3%), pyrexia (3%), pneumonia (3%), major hemorrhage (3%), and actinic keratosis (3%). Serious AEs, or SAEs, were seen in 22 patients (33%), with events in five patients (7%) considered possibly related to zanubrutinib treatment: febrile neutropenia, colitis, atrial fibrillation, hemothorax (spontaneous) and headache. Among AEs of special interest, four patients (6%) experienced atrial fibrillation (all grade 1 or 2) and two patients experienced major hemorrhage. Four patients (6%) discontinued study treatment due to AEs, including fatal worsening bronchiectasis, prostate cancer, gastric adenocarcinoma, and acute myeloid leukemia. Two patients (3%) disticontinued study treatment due to disease progression as assessed by investigator and one patient remains on treatment post disease progression.

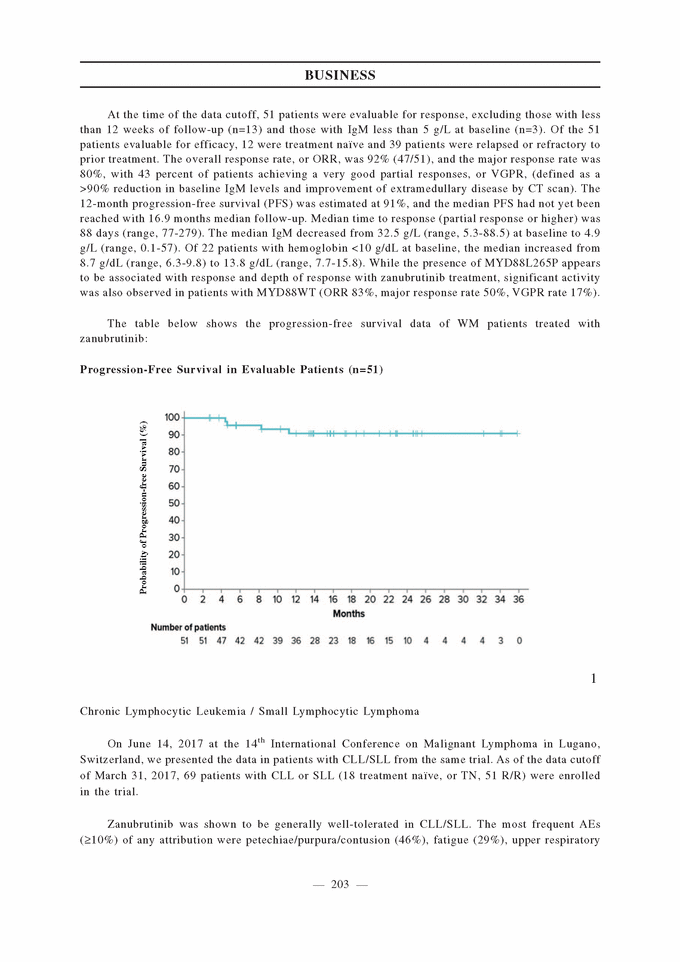
At the time of the data cutoff, 51 patients were evaluable for response, excluding those with less than 12 weeks of follow-up (n=13) and those with IgM less than 5 g/L at baseline (n=3). Of the 51 patients evaluable for efficacy, 12 were treatment naïve and 39 patients were relapsed or refractory to prior treatment. The overall response rate, or ORR, was 92% (47/51), and the major response rate was 80%, with 43 percent of patients achieving a very good partial responses, or VGPR, (defined as a >90% reduction in baseline IgM levels and improvement of extramedullary disease by CT scan). The 12-month progression-free survival (PFS) was estimated at 91%, and the median PFS had not yet been reached with 16.9 months median follow-up. Median time to response (partial response or higher) was 88 days (range, 77-279). The median IgM decreased from 32.5 g/L (range, 5.3-88.5) at baseline to 4.9 g/L (range, 0.1-57). Of 22 patients with hemoglobin <10 g/dL at baseline, the median increased from 8.7 g/dL (range, 6.3-9.8) to 13.8 g/dL (range, 7.7-15.8). While the presence of MYD88L265P appears to be associated with response and depth of response with zanubrutinib treatment, significant activity was also observed in patients with MYD88WT (ORR 83%, major response rate 50%, VGPR rate 17%). The table below shows the progression-free survival data of WM patients treated with zanubrutinib: Progression-Free Survival in Evaluable Patients (n=51) Probability of Progression-free Survival (%) 1 Chronic Lymphocytic Leukemia / Small Lymphocytic Lymphoma On June 14, 2017 at the 14th International Conference on Malignant Lymphoma in Lugano, Switzerland, we presented the data in patients with CLL/SLL from the same trial. As of the data cutoff of March 31, 2017, 69 patients with CLL or SLL (18 treatment naïve, or TN, 51 R/R) were enrolled in the trial. Zanubrutinib was shown to be generally well-tolerated in CLL/SLL. The most frequent AEs (>10%) of any attribution were petechiae/purpura/contusion (46%), fatigue (29%), upper respiratory
tract infection (28%), cough (23%), diarrhea (22%), headache (19%), hematuria (15%), nausea (13%), rash (13%), arthralgia (12%), muscle spasms (12%) and urinary tract infection (12%). All of these events were grade 1 or 2 except for one case of grade 3 purpura (subcutaneous hemorrhage), which was the only major bleeding event. Additional AEs of interest included one case of each grade 2 diarrhea and grade 2 atrial fibrillation. A total of 18 SAEs occurred in 13 patients, with no SAE occurring in more than one patient. Only one patient discontinued treatment due to an AE, a grade 2 pleural effusion. At the time of the data cutoff, 66 patients (16 TN and 50 R/R) had more than 12 weeks of follow-up and were evaluable for efficacy, and three other patients had less than 12 weeks of follow-up. After a median follow-up of 10.5 months (2.2—26.8 months), the ORR was 94% (62/66) with complete responses, or CRs, in 3% (2/66), PRs in 82% (54/66), and PRs with lymphocytosis, or PR-Ls, in 9% (6/66) of patients. Stable disease, or SD, was observed in 5% (3/66) of patients. The patient with pleural effusion discontinued treatment prior to week 12 and was not evaluable for response. There was one instance of Hodgkin’s transformation. In TN CLL/SLL, at a median follow-up time of 7.6 months (3.7—11.6 months), the ORR was 100% (16/16) with CRs in 6% (1/16), PRs in 81% (13/16) and PR-Ls in 13% (2/16) of patients. In R/R CLL/SLL, at a median follow-up time of 14.0 months (2.2—26.8 months), the ORR was 92% (46/50) with CRs in 2% (1/50), PRs in 82% (41/50) and PR-Ls in 8% (4/50) of patients. SD was observed in 6% (3/50) patients. The table below shows the progression-free survival data of CLL/SLL patients treated with zanubrutinib: Zanubrutinib PFS in CLL/SLL Probability of Progression-free Survival (%) 66666253 453727251911964

Other Lymphomas On December 9, 2017, we presented additional data from our Phase 1 trial at the 59th American Society of Hematology, or ASH, Annual Meeting in Atlanta, GA. This dataset included 34 patients in an indolent lymphoma cohort, which consisted of 24 patients with FL and 10 patients with MZL, and 65 patients in an aggressive lymphoma cohort, which consisted of 27 patients with diffuse large B-cell lymphoma, or DLBCL, and 38 patients with MCL. The median follow-up time was 5.6 months (0.3—22.3 months) and 5.1 months (0.1—31.9) for indolent and aggressive lymphoma, respectively. As of the data cutoff of September 15, 2017, the most frequent AEs (occurring in >15% of patients) of any attribution among 34 patients with indolent lymphoma were petechiae/purpura/contusion (24%), upper respiratory tract infection (21%), nausea (18%) and pyrexia (15%). The most frequently reported grade 3 or greater AEs (occurring in >5% of patients) of any attribution were anemia (9%), neutropenia (9%), urinary tract infection (6%) and abdominal pain (6%). SAEs were reported in 11 patients (32%). Of those, four patients had SAEs that were considered possibly related to zanubrutinib, including one case each of nausea, urinary tract infection, diarrhea and creatinine increase. The most frequent AEs (occurring in >15% of patients) of any attribution among 65 patients with aggressive lymphoma were petechiae/purpura/contusion (25%), diarrhea (23%), constipation (22%), fatigue (18%), upper respiratory tract infection (18%), anemia (17%), cough (15%), pyrexia (15%) and thrombocytopenia (15%). The most frequently reported grade 3 or greater AEs (occurring in > 5% of patients) of any attribution were anemia (11%), neutropenia (9%), thrombocytopenia (9%) and pneumonia (6%). SAEs were reported in 26 patients (40%). Of those, three patients had SAEs that were considered possibly related to zanubrutinib, including one case each of peripheral edema and joint effusion (occurring in the same patient), pneumonia and pneumonitis. At the time of data cutoff, 26 patients with indolent lymphoma, including 17 patients with FL and nine patients with MZL, were evaluable for efficacy. In patients with FL, the ORR was 41%, with CRs in 18% and PRs in 24% of patients. SD was observed in 41% of patients. Progressive disease was observed in one patient. In patients with MZL, the ORR was 78%, with no CRs and PRs in 78% of patients. SD was observed in 22% of patients. No progressive disease was observed. 58 patients with aggressive lymphoma, including 26 patients with DLBCL and 32 patients with MCL, were evaluable for efficacy. In patients with DLBCL, the ORR was 31%, with CRs in 15% and PRs in 15% of patients. In patients with MCL, the ORR was 88%, with CRs in 25% and PRs in 63% of patients. Combination with GAZYVA® (obinutuzumab) We are also evaluating zanubrutinib in combination with GAZYVA® (obinutuzumab), an approved anti-CD20 antibody therapy, in patients with B-cell lymphoma in an open label, multi-center Phase 1b trial in Australia, the United States and South Korea. On December 9, 2017, we presented

updated preliminary clinical data from this trial at the 59th ASH Annual Meeting in Atlanta, GA. As of the data cutoff of September 15, 2017, 45 patients with CLL/SLL and 26 patients with FL were enrolled in the trial. The preliminary Phase 1b data demonstrated that the combination was generally well-tolerated and was highly active in patients with FL and TN or R/R CLL/SLL. At the time of data cutoff, the most common AEs were grades 1 and 2. The most common AEs in patients with CLL/SLL (occurring in > 20% of patients) of any attribution were petechiae/purpura/contusion (42%), neutropenia (40%), upper respiratory tract infection (36%), fatigue (24%), thrombocytopenia (24%), diarrhea (20%) and pyrexia (20%). The most common AEs in patients with FL (occurring in > 20% of patients) of any attribution were upper respiratory tract infection (38%), petechia/purpura/contusion (35%), rash (27%) and thrombocytopenia (23%). Grade 3 or 4 AEs of any attribution reported in > 5% of the CLL/SLL patients included neutropenia (24%) and thrombocytopenia (7%). Grade 3 or 4 AEs of any attribution reported in > 5% of the FL patients included neutropenia (12%). There were no cases of serious hemorrhage, which is > grade 3 hemorrhage or central nervous system hemorrhage of any grade, atrial fibrillation, or grade 3 or above diarrhea. Only one patient with CLL/SLL discontinued treatment due to an AE, a case of squamous cell carcinoma, or SCC, who had a prior history of SCC. This was also the only patient in the study who had a fatal AE. 45 patients with CLL/SLL (20 TN and 25 R/R) and 21 patients with R/R FL were evaluable for efficacy. In TN CLL/SLL patients, after a median follow-up of 11.4 months (6.0—17.3 months), the ORR was 95%, with CRs in 35% and PRs in 60% of patients. In R/R CLL/SLL patients, at a median follow-up time of 12.7 months (7.9—19.5 months), the ORR was 92%, with CRs in 20% and PRs in 72% of patients. In R/R FL patients, at a median follow-up time of 12.1 months (0.8—19.7 months), the ORR was 76%, with CRs in 38% and PRs in 38% of patients. Combination with Tislelizumab We are also evaluating zanubrutinib in combination with our investigational anti-PD1 antibody tislelizumab. The open-label, multi-center Phase 1b trial is being conducted in Australia and is currently in a dose-escalation phase to be followed by a dose-expansion phase. On December 11, 2017, we presented initial data from the ongoing Phase 1b trial at the 59th ASH Annual Meeting in Atlanta, GA. The initial dose-escalation data suggested that the combination of zanubrutinib and tislelizumab was generally well-tolerated and exhibited anti-tumor activity in patients with B-cell malignancies. As of September 15, 2017, 25 patients had been enrolled. There were 13 patients with indolent lymphoma, including CLL, FL, MZL and WM, and 12 patients with aggressive lymphoma, including DLBCL, MCL and transformed lymphoma. The median follow-up time was 5.1 months (0.4—14.1 months). Two cases of autoimmune hemolysis occurred in patients with WM in the dose 2 cohort, and one qualified as a dose-limiting toxicity, or DLT. These events were not associated with a positive direct antiglobulin test and were resolved with immunosuppressive therapy, but resulted in the decision to exclude further enrollment of WM patients in the trial. As of the data cutoff date, this autoimmune hemolysis is the only DLT case that was observed.

Among patients with indolent lymphoma, the most common AEs (occurring in > 20% of patients) of any attribution were petechiae/purpura/contusion (31%) and thrombocytopenia (23%). Grade 3 and 4 AEs of any attribution reported in at least two patients included thrombocytopenia, anemia and hemolysis (15% each). In addition to the two cases of autoimmune hemolysis, there was one more immune-related event, a grade 4 autoimmune encephalitis. The patient was treated with aggressive immunosuppressive therapy and gradually improved over time. Among patients with aggressive lymphoma, the most common AEs (occurring in > 20% of patients) of any attribution were diarrhea, fatigue, pyrexia, upper respiratory tract infection (33% each), cough (25%) and nausea (25%). Grade 3 and 4 AEs of any attribution reported in at least two patients included pyrexia (17%). There was one patient with multiple occurrences of grade 2 and 3 pneumonitis. In addition, certain IRAEs may be associated with checkpoint inhibition and the combination of checkpoint inhibitors with other drugs. At the time of data cutoff, the efficacy-evaluable population consisted of 25 patients. Objective responses were observed in 10 patients (40%). By tumor type, two PRs were observed out of five patients with CLL, one CR and one PR were observed out of five patients with FL, one VGPR and one minor response were observed out of two patients with WM, one CR was observed out of five patients with DLBCL, and three PRs were observed out of five patients with transformed lymphoma. Pooled Analysis of Safety Data from Monotherapy Trials We presented at the 2018 EHA meeting the pooled safety data from patients with various B-cell lymphomas in four ongoing zanubrutinib monotherapy studies, totaling 476 patients with a median exposure of seven months. Overall, the data suggest that exposure levels of zanubrutinib resulting in complete and sustained BTK inhibition can be achieved and that zanubrutinib was generally well-tolerated. There are infrequent events of interest with BTK inhibitor therapy, such as atrial fibrillation/flutter (2%), major hemorrhage (2%), and grade 3 and above diarrhea (1%). Treatment discontinuation due to zanubrutinib-related AEs was uncommon (3%). The majority of patients (94%) experienced one or more AE of any attribution, primarily grades 1 or 2. The most common grade 3 or higher AEs of any attribution were neutropenia/neutrophil count decreased/febrile neutropenia (14%), anemia (7%) and thrombocytopenia/platelet count decreased (7%). SAEs were reported in 116 patients (24%), with 38 patients (8%) assessed by the investigator as related to zanubrutinib. The most common SAEs were pneumonia/lung infection (6%), pleural effusion (1%), and febrile neutropenia (1%). The only treatment-related SAE reported in greater than 1% of patients was pneumonia/lung infection (2%). No cases of pneumocystis jiroveci pneumonia (PJP) or cytomegalovirus (CMV) reactivation were reported. The most common bleeding events observed included petechiae/purpura/contusion (26%) and hematuria (11%). Major hemorrhage (2%) included gastrointestinal hemorrhage/melena (n=3), intraparenchymal CNS hemorrhage grade 5, hematuria, purpura, hemorrhagic cystitis, renal hematoma, and hemothorax (one each). The median time to first major hemorrhage was 1.2 months. Amongs patients with emergent atrial fibrillation/flutter (n=8), a majority had known risk factors including hypertension (n=2), pre-existing cardiovascular disease (n=2), and concurrent infection (n=1). The cumulative rates of grade 3 or higher infections were 14 percent at six months, 19% at 12 months and 21% at 18 months. The exposure-adjusted incidence rate was 1.82 per 100 person-months. The most common second primary malignancies included basal cell carcinoma (3%) and squamous cell carcinoma of the skin (1%).

Clinical Development Plan Based on the clinical data to date, we believe that zanubrutinib has a potentially best-in-class profile, and we are running a broad global pivotal program in multiple indications. Globally, we have an ongoing monotherapy head-to-head Phase 3 trial versus ibrutinib in WM, which has met the enrollment target. The trial has closed new patient screening and completed enrollment. We are also conducting an ongoing Phase 3 trial compared to bendamustine and rituximab in patients with TN CLL/SLL; and an ongoing Phase 2 trial in combination with GAZYVA® (obinutuzumab) in patients with FL, which is a pivotal trial for accelerated or conditional approval and will require a confirmatory study. We are also planning a Phase 3 trial for head-to-head comparison versus ibrutinib in patients with R/R CLL/SLL, which is expected to be initiated in 2018. In July 2018, zanubrutinib was granted Fast Track Designation by the FDA for the treatment of patients with WM. Based on our discussions with the FDA, internal review of available data from our global Phase 1 trial of zanubrutinib in patients with WM, and supported by the Fast Track Designation, we are preparing to submit in the first half of 2019 an NDA to pursue an accelerated approval of zanubrutinib for patients with WM based on results from the global Phase 1 study. In China, we are conducting three separate pivotal Phase 2 trials of zanubrutinib as monotherapy in patients with R/R MCL, R/R CLL/SLL, and WM, respectively. We expect to file for regulatory approvals in China for zanubrutinib in these indications based on the results of these Phase 2 trials. If we receive conditional approval instead of full approval, we will be required to conduct one or more confirmatory studies after such conditional approvals. We have completed enrollment in all three pivotal trials. On June 15, 2018, we announced results from the independent review of response data from the 86-patient single-arm pivotal Phase 2 study of zanubrutinib in Chinese patients with relapsed or refractory MCL. The ORR of 84% (59% complete response rate) met the pre-specified criteria for a positive trial, and the median duration of response has not been reached with 8.3 months median follow-up. The safety profile was consistent with previously reported clinical data for zanubrutinib. Subject to the successful completion and satisfactory results of the trials, we plan to submit an NDA in China for patients with R/R MCL later this year. If approved, we plan to commercialize zanubrutinib shortly after approval. In addition, we are also conducting a Phase 2 trial of zanubrutinib in patients with R/R DLBCL. WE MAY NOT BE ABLE TO ULTIMATELY DEVELOP AND MARKET ZANUBRUTINIB SUCCESSFULLY. Tislelizumab (BGB-A317), a PD-1 Antibody Tislelizumab is an investigational humanized monoclonal antibody against the immune checkpoint receptor PD-1 that is currently being evaluated in pivotal clinical trials globally and in China and for which we plan to commence additional pivotal trials as a monotherapy and in combination with standard of care to treat various solid and hematological cancers. Tislelizumab is designed to bind to and block downstream activity of PD-1, a cell surface receptor that plays an important role in downregulating the immune system by preventing the activation of T-cells. Tislelizumab has high affinity and specificity for PD-1. It is differentiated from the currently approved PD-1 antibodies by an engineered Fc region, which we believe may minimize potentially negative interactions with other immune cells based on preclinical data. We have a global strategic collaboration with Celgene for tislelizumab for solid tumors outside of Asia (other than Japan) as further described in “—Collaboration Agreements—Celgene.”

Mechanism of Action Cells called cytotoxic T-lymphocytes, or CTLs, provide an important self-defense mechanism against cancer, patrolling the body, recognizing cancer cells due to immunogenic features that differ from normal cells, and killing cancer cells by injecting deleterious proteins into them. T-lymphocytes have various mechanisms that prevent them from damaging normal cells, among which is a protein called PD-1 receptor, which is expressed on the surface of T-lymphocytes. PD-L1 is an important signaling protein that can engage PD-1. PD-L1 binding to PD-1 sends an inhibitory signal inside the T-lymphocyte and abrogates its cytotoxic effects. Many types of cancer cells have hijacked the PD-L1 expression system that normally exists in healthy cells. By expressing PD-L1, cancer cells protect themselves from being killed by CTLs. Tislelizumab is a monoclonal antibody designed to specifically bind to PD-1, without activating the receptor, thereby blocking engagement of PD-1 by its ligands PD-L1 and PD-L2. Therefore, we believe tislelizumab has the potential to restore the ability of CTLs to kill cancer cells. In addition, tislelizumab was specifically engineered to minimize binding to Fc')'R on macrophages, thereby abrogating antibody-dependent phagocytosis, a potential mechanism of T-cell clearance. Tislelizumab’s Lack of Fc')'R Binding Prevents Macrophage-Mediated T-Cell Clearance FcRI=Fc gamma receptor-1, mAb=monoclonal antibody, MHC=major histocompatibility complex, NK=natural killer, PD-1=programmed cell death receptor-1, PD-L1=programmed cell death-ligand. Arlauckas et al. SciTransl Med. 2017;9(389):eaal3604.

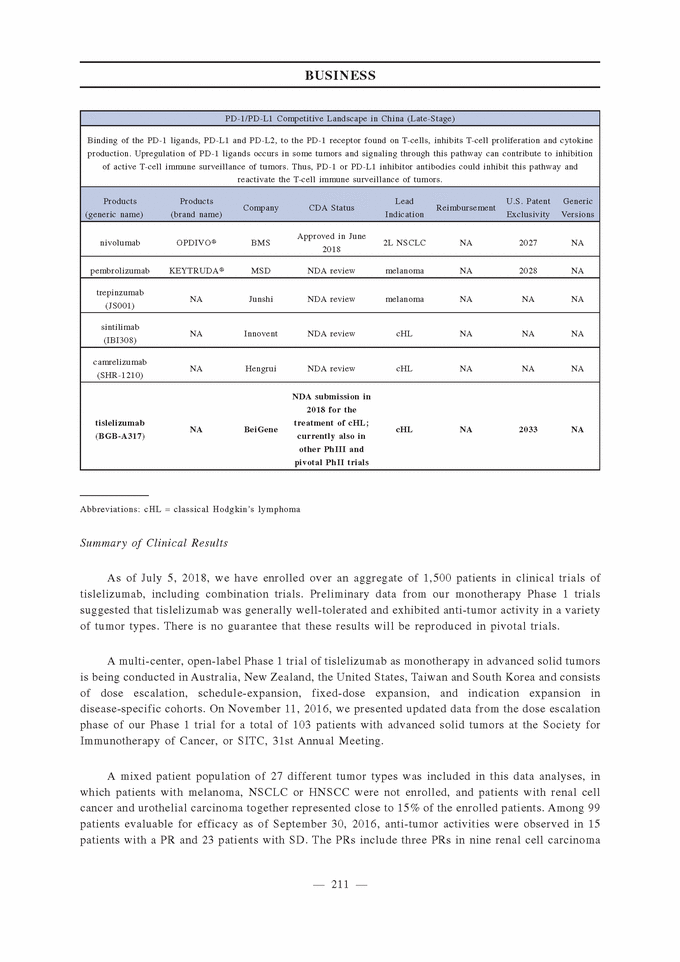
Market Opportunity and Competition A number of PD-1 or PD-L1 antibody drugs have been approved by the FDA. These include Merck’s KEYTRUDA® (pembrolizumab), Bristol-Myers Squibb’s OPDIVO® (nivolumab), Roche’s TECENTRIQ® (atezolizumab), AstraZeneca’s IMFINZI® (durvalumab) and Pfizer and Merck Sereno’s BAVENCIO® (avelumab). In the global setting, several PD-1 or PD-L1 antibody agents are in clinical development besides us, such as Regeneron’s cemiplimab, Novartis’ PDR-001, Tesaro’s TSR042 and Pfizer ’s PF-06801591. According to the Frost & Sullivan Report, in 2017, global sales of the PD-1 class reached US$10.1 billion, which make some of these therapies among the best-selling and fastest launched oncology drugs in history. We believe there is a large commercial opportunity in China for PD-1 or PD-L1 antibody drugs. Currently available clinical data suggest that some of the most prevalent cancers in China, such as lung, gastric, liver and esophageal cancers, are responsive to this class of agents. In 2012, 38%, 45%, 51% and 49% of the worldwide mortalities from lung, gastric, liver and esophageal cancers, respectively, occurred in China, according to the World Health Organization. Collectively, these four tumor types comprised over 2.3 million new cases in 2016 in China alone, according to Chen et al. 2016. In addition, China has a higher proportion of PD-1 responsive tumors in its total annual cancer incidence in comparison to other geographies like the United States and the European Union, or EU. According to Chen et al. 2016, the annual incidence of the top 10 PD-1 responsive tumors in China is estimated to be 3.0 million out of 4.3 million in total annual cancer incidence. In comparison, the estimated annual incidence of the top 10 PD-1 responsive tumors is 0.9 million out of 1.7 million in total annual cancer incidence in the United States, and 0.9 million out of the 1.8 million total in the EU5 countries according to SEER program of the U.S. National Cancer Institute and the World Health Organization. In China, there is only one approved PD-1 antibody agent, Bristol-Myers Squibb’s OPDIVO® (nivolumab) and there are no approved PD-L1 antibody agents yet. The CDE, under the CDA, released guidance in February 2018 on the requirements for NDA submissions of PD-1/L1 agents, specifically for data from single-arm trials on refractory / recurrent advanced cancers without standard-of-care therapies. A pre-NDA meeting is required before NDA submission, and a rolling NDA submission will be accepted for PD-1/L1 therapies. On June 15, 2018, the CDA approved Bristol-Myers Squibb’s OPDIVO® (nivolumab) for the treatment of locally advanced or metastatic NSCLC after platinum-based chemotherapy in adult patients without EGFR or ALK genomic tumor aberration. Merck submitted an NDA for pembrolizumab in February 2018. Among domestic Chinese companies, Junshi submitted an NDA for JS001 (trepinzumab) in March 2018, and Innovent submitted an NDA for IBI308 (sintilimab) and Hengrui submitted an NDA for SHR-1210 (camrelizumab) in April 2018. The table below summarizes the China competitive landscape of tislelizumab, according to the Frost & Sullivan Report.

Abbreviations: cHL = classical Hodgkin’s lymphoma Summary of Clinical Results As of July 5, 2018, we have enrolled over an aggregate of 1,500 patients in clinical trials of tislelizumab, including combination trials. Preliminary data from our monotherapy Phase 1 trials suggested that tislelizumab was generally well-tolerated and exhibited anti-tumor activity in a variety of tumor types. There is no guarantee that these results will be reproduced in pivotal trials. A multi-center, open-label Phase 1 trial of tislelizumab as monotherapy in advanced solid tumors is being conducted in Australia, New Zealand, the United States, Taiwan and South Korea and consists of dose escalation, schedule-expansion, fixed-dose expansion, and indication expansion in disease-specific cohorts. On November 11, 2016, we presented updated data from the dose escalation phase of our Phase 1 trial for a total of 103 patients with advanced solid tumors at the Society for Immunotherapy of Cancer, or SITC, 31st Annual Meeting. A mixed patient population of 27 different tumor types was included in this data analyses, in which patients with melanoma, NSCLC or HNSCC were not enrolled, and patients with renal cell cancer and urothelial carcinoma together represented close to 15% of the enrolled patients. Among 99 patients evaluable for efficacy as of September 30, 2016, anti-tumor activities were observed in 15 patients with a PR and 23 patients with SD. The PRs include three PRs in nine renal cell carcinoma
patients; three in six urothelial cancer patients; two in four gastric cancer patients; two in two Merkel cell carcinoma patients; one in four nasopharyngeal patients; one in one penis squamous cell carcinoma patient; one in one duodenal carcinoma patient; two in two MSI-high patients, one with colorectal cancer among a total of 13 colorectal cancer patients, and one with pancreatic cancer among a total of two pancreatic cancer patients. At the time of the data cutoff for the safety analysis, the most common treatment-related AEs (>5%) were fatigue (19%), diarrhea (13%), rash (11%), pruritus (11%), nausea (8%), hypothyroidism (7%), and infusion related reactions (6%). Treatment-related SAEs included four cases of colitis, two cases of hypotension, and one case each of diarrhea, diabetes mellitus, diabetic ketoacidosis, dyspnea, hypoxia, infusion-related reaction, and pneumonitis. Among these, grade 3 or above treatment-related SAEs included the two cases of hypotension and one case each of colitis, diabetes mellitus, diabetic ketoacidosis, dyspnea, hypoxia, and pneumonitis. Other treatment-related grade 3 or above AEs included two cases each of fatigue and hyperglycemia, and one case each of back pain, elevated alanine aminotransferase, or ALT, and elevated gamma-glutamyl transferase, or GGT. From 2017 to date, we have presented preliminary data from multiple disease-specific subgroups in the ongoing Phase 1 trial of tislelizumab in advanced solid tumors, including patients with HCC, GC, esophageal cancer, or EC, HNSCC, OC and UC. Hepatocellular (Liver) Cancer The data presented on HCC are from 40 patients treated with tislelizumab at a dose of 5 mg/kg every three weeks, or Q3W. The majority of the enrolled patients (34/40 patients) had a hepatitis B virus infection. At the time of the data cutoff on April 28, 2017, the median treatment duration was 64 days (range of 1 to 471 days). AEs assessed by the investigator to be treatment-related occurred in 21 patients (53%). Of those, rash (20%), pruritus (13%), increased aspartate aminotransferase, or AST (8%), fatigue (5%), hypothyroidism (5%) and decreased appetite (5%) were reported in more than one patient. All of the treatment-related AEs were grades 1 or 2, with the exception of one grade 5 event of acute hepatitis assessed by the investigator to be related to tislelizumab. This patient had widely metastatic disease and died five weeks after receiving his first and only dose of tislelizumab and subsequently developing evidence of disease progression. At the time of the data cutoff, the efficacy evaluation was early, and 27 patients were evaluable for response, defined as having measurable disease at baseline and at least one post-baseline tumor assessment, or progression or death. Twelve of the evaluable patients remained on treatment and seven of these had only one tumor assessment at the time of the data cutoff. Confirmed and unconfirmed PRs were observed in three patients, all with hepatitis-B-positive HCC. One PR was confirmed before the cutoff date, one was confirmed one day following the cutoff date, and one was unconfirmed and the patient remained on therapy. Nine patients achieved SD, some of whom also had significant reductions in Alpha-fetoprotein levels.

Gastric and Esophageal Cancers The data presented on GC and EC were from 83 patients, 46 with advanced or metastatic GC and 37 with EC, treated with tislelizumab at 2 mg/kg or 5 mg/kg every two weeks, or Q2W, or Q3W. At the time of the data cutoff on June 8, 2017, median treatment duration was 45 days (range 4—457 days) for patients with GC and 50 days (range 1—246 days) for patients with EC. AEs assessed by the investigator to be treatment-related occurred in 15 patients with GC (33%). Of those, abdominal pain (9%), decreased appetite (9%), fatigue (7%), nausea (7%) and pruritus (4%) were reported in more than one patient, and all of these cases were grades 1 or 2. AEs assessed to be treatment-related occurred in 15 patients with EC (41%). Of those, fatigue (16%), nausea (8%), decreased appetite (5%), infusion-related reaction (5%) and myalgia (5%) occurred in more than one patient, and all of these cases were grades 1 or 2. Only one patient in each cohort reported a treatment-related AE of grade 3 or higher: grade 3 proteinuria in one patient with GC and grade 3 dermatitis in one patient with EC. SAEs considered treatment-related included one case of diarrhea and one case of pyrexia, each occurring in patients with GC. Eight patients (two with GC, six with EC) had a treatment-emergent AE with a fatal outcome; none of which was assessed as treatment-related. The efficacy-evaluable population included 34 GC patients and 31 EC patients. Despite the short median follow-up time, four achieved confirmed PRs and three achieved SD among GC patients. Among EC patients, two achieved a confirmed PR and nine achieved SD. Three of the nine patients with EC who achieved SD also achieved an unconfirmed PR, including one who awaits response confirmation. At the time of the data cutoff, 27 patients remained on treatment. Head and Neck Squamous Cell Cancer The HNSCC data presented were from 18 patients treated with tislelizumab at 5 mg/kg Q3W. At the time of the data cutoff on June 8, 2017, median treatment duration was 104 days (range 30—339 days). AEs assessed by the investigator to be treatment-related occurred in seven patients (39%). Of those, only fatigue (11%, all grade 1 or 2) was reported in more than one patient. One case of grade 3 nausea was the only treatment-related AE of grade 3 or higher in severity. No patient discontinued treatment due to a treatment-related AE, and of the nine deaths reported, none were considered to be treatment-related. The efficacy-evaluable population included 17 HNSCC patients. Despite short median follow-up time, three achieved a confirmed PR and six achieved SD. At the time of the data cutoff, three patients remained on treatment. Ovarian Cancer The OC dataset included 51 patients treated with tislelizumab at different dose levels (0.5 to 10 mg/kg Q2W in dose escalation, 2 or 5 mg/kg Q2W or Q3W or 200 mg Q3W in dose expansion, or 5 mg/kg Q3W in indication expansion). At the time of the data cutoff on June 8, 2017, median treatment duration was 71 days (range 29—540 days).

AEs assessed by the investigator to be treatment-related occurred in 28 patients (55%). Of those, fatigue (18%), pruritus (10%), rash (10%), diarrhea (10%), lethargy (6%), nausea (6%), abdominal pain (4%), dry eye (4%), dry skin (4%), onychoclasis (4%) and maculo-papular rash (4%) were reported in more than one patient, and all, except one case of grade 3 diarrhea, were grades 1 or 2. Two additional treatment-related AEs of grade 3 or higher included one case each of grade 3 pyrexia and stomatitis. SAEs considered to be treatment-related occurred in three patients and included one case each of pyrexia, colitis and mucosal inflammation. The efficacy-evaluable population included 50 OC patients. Two achieved a confirmed PR and 20 achieved SD. At the time of the data cutoff, six patients remained on treatment. Urothelial Cancer The UC dataset included 16 patients. Of these, 12 had one or more prior systemic anticancer treatment for metastatic disease and the remaining four had progressed after receiving platinum-based regimen in the neoadjuvant or adjuvant setting. In addition, five patients had prior radiotherapy. At the time of the data cutoff on August 28, 2017, median treatment duration was 4.3 months (range of 0.7 to 18.3 months). A total of six patients remained on treatment. AEs assessed by the investigator to be treatment-related occurred in 14 patients (88%). Of those, fatigue (31%), rash (19%), infusion-related reactions (13%), nausea (13%), pain in extremity (13%) and proteinuria (13%) occurred in more than one patient. All of the treatment-related AEs were grade 1 or 2 except one case each of fatigue, hyperglycemia and diabetes mellitus. One AE of muscle weakness, which was associated with disease progression and occurred more than one month after the last dose of tislelizumab, had a fatal outcome. This event was considered by the investigator not to be treatment-related. The efficacy-evaluable population included 15 UC patients. One patient had a confirmed CR, four achieved a confirmed PR, and three achieved SD. Nine evaluable patients had PD-L1 status determined. There was one CR, two PR and one SD among six PD-L1 high patients, and one PR among three PD-L1 low or negative patients. We expect to complete enrollment of patients in pivotal Phase 2 trial of urothelial cancer in China in 2018. Hodgkin’s Lymphoma In June 2018, we received preliminary topline results from the independent review of response data from our Phase 2 single-arm pivotal trial of tislelizumab in Chinese patients with R/R cHL. This trial enrolled 70 patients with cHL who either failed autologous stem cell transplantation, or ASCT, or who were ineligible for ASCT. The primary endpoint was overall response rate as defined by the Lugano 2014 criteria. Secondary endpoints included progression-free survival, duration of response, complete response rate, time to response, safety, and tolerability. As of the data cutoff, the median follow-up time was approximately 6.0 months. The overall response rate was 73%, including 50% complete response, and the median duration of response had not been reached. Frequency and severity of adverse events was generally consistent with the previously reported Phase 1 safety and tolerability

data for tislelizumab, or, in the case of certain immune-related events such as hypothyroidism and fever, consistent with previous reports of other PD-1 antibodies for the treatment of cHL. We expect to include these data, along with additional follow-up data from the study, in the NDA that we plan to file with the CDA in China later this year, and we also plan to present full results of the trial at an upcoming medical conference. Combination with Pamiparib On June 5, 2017, we presented initial data from the dose-escalation portion of the Phase 1 trial of tislelizumab in combination with our investigational PARP inhibitor, pamiparib, in patients with advanced solid tumors at the 2017 American Society for Clinical Oncology, or ASCO, Annual Meeting. We presented an updated dataset on January 25, 2018 at the 2018 ASCO-SITC Clinical Immuno-Oncology Symposium. The preliminary data suggested that the combination of tislelizumab and pamiparib was generally well-tolerated and showed anti-tumor activity in multiple solid tumor types. At the data cutoff of July 31, 2017, 49 patients were enrolled in the dose-escalation portion of the trial. Cohorts of six to 13 patients each received treatments at five planned dose levels, or DLs. Tislelizumab was administered at 2 mg/kg Q3W with pamiparib at 20, 40, or 60 mg twice daily, or BID, in DLs 1, 2 and 3, respectively. Tislelizumab was also administered at a fixed dose of 200 mg Q3W with pamiparib at 40 or 60 mg twice daily in DLs 4 and 5, respectively. Duration of treatment was greater than 200 days for 10 patients, and a total of seven patients remained on treatment as of the data cutoff date. Dose-limiting toxicities occurred in four patients; these included one patient with grade 2 nausea, one patient with grade 3 rash at DL 4, one patient with grade 2 nausea or vomiting and one patient with grade 4 autoimmune hepatitis at DL 5. The trial identified the recommended Phase 2 dose to be tislelizumab at 200 mg fixed dose Q3W and pamiparib at 40 mg BID. Grade 3 or 4 non-immune AEs assessed by the investigator to be related to the treatment regimen and reported in more than one patient included anemia (12%), nausea (4%) and fatigue (4%). Immune-related AEs of any grade regardless of causality occurred in 23 patients (47%); those reported in at least two patients included elevated alanine aminotransferase, or ALT, elevated AST, hypothyroidism, auto-immune hepatitis / hepatitis, diarrhea, elevated gamma-glutamyl transferase, or GGT, hyperthyroidism and pruritus. Grade 3 and 4 liver-related AEs regardless of causality were reported in nine patients, including five patients with hepatitis and four patients with ALT, AST and/or GGT elevations. Together, liver-related AEs of any grade regardless of causality were observed in 13 patients; all events were manageable and reversible with corticosteroid treatment. The trial protocol was amended to increase real-time hepatic safety monitoring consistent with new European Society for Medical Oncology, or ESMO, guidance for immune-related treatment-emergent AEs. Certain IRAEs may be associated with checkpoint inhibition and the combination of checkpoint inhibitors with other drugs. At the data cutoff of July 31, 2017, 49 patients were evaluable for efficacy. Best responses included two confirmed CRs, five confirmed PRs, and seven unconfirmed PRs. The clinical benefit rate including CRs, PRs and durable SDs with at least 24 weeks was 31%. With longer follow-up, as

of January 4, 2018, among the 49 evaluable patients, best responses included two confirmed CRs, eight confirmed PRs, and four unconfirmed PRs. The clinical benefit rate was 39%. As of the July 31, 2017 cutoff, 11 patients remained on treatment, the median duration of response was 168.5 days (range: 64-508 days), and duration of treatment was over 200 days in 10 patients. The trial is currently planned to further evaluate the combination’s activity in expansion cohorts of patients with ovarian, triple-negative breast, castration-resistant prostate, lung, gastric or gastro-esophageal junction, urothelial and pancreatic cancers. Clinical Development Plan We are running a broad development program with Celgene including global pivotal trials in Asia-prevalent cancers, such as NSCLC, EC and HCC, which are intended to support regulatory submissions globally and in China. We have initiated Phase 3 trials to evaluate tislelizumab as a potential second-or third-line treatment compared to docetaxel in patients with NSCLC; as a potential first line treatment compared to sorafenib in patients with HCC; and as a potential second-line treatment compared to investigator-chosen chemotherapy in patients with ESCC. We have also recently initiated a global Phase 2 trial in patients with previously treated advanced HCC, as well as a global Phase 2 trial in patients with relapsed or refractory mature T-and NK-cell lymphomas, with the first patient in both trials dosed in April 2018. We and Celgene expect to commence additional pivotal trials in 2018 and 2019, such as a pivotal trial in gastric cancer. We have two additional China pivotal Phase 2 trials ongoing, in patients with R/R cHL and in patients with PD-L1 positive urothelial cancer, and we are screening patients in preparation of the first dosing of one China Phase 3 trial in combination with chemotherapy in patients with non-squamous NSCLC. We expect to file for regulatory approvals in China for tislelizumab in these indications based on the results of these Phase 2 trials. If we receive conditional approval instead of full approval, we will be required to conduct one or more confirmatory studies after such conditional approvals. We received preliminary topline results from the independent review of the Phase 2 pivotal trial in Chinese patients with R/R cHL. The overall response rate was 73%, including 50% complete response, and the frequency and severity of adverse events was generally consistent with the previously reported data for tislelizumab, or, in the case of certain immune-related events such as hypothyroidism and fever, consistent with previous reports of other PD-1 antibodies for the treatment of cHL. We expect to include these data, along with additional follow-up data from the study, in the NDA that we plan to file with the CDA in China later this year, subject to the successful completion and satisfactory results of the trials. If approved, we plan to commercialize tislelizumab shortly after approval. WE MAY NOT BE ABLE TO ULTIMATELY DEVELOP AND MARKET TISLELIZUMAB SUCCESSFULLY.

Pamiparib (BGB-290), a PARP Inhibitor Pamiparib is an investigational small molecule inhibitor of PARP1 and PARP2 that is being evaluated as a potential monotherapy and in combinations for the treatment of various solid tumors. We believe pamiparib has the potential to be differentiated from other PARP inhibitors because of its potential brain penetration, greater selectivity, strong DNA-trapping activity, and good oral bioavailability. Pamiparib has demonstrated pharmacological properties such as brain penetration and PARP—DNA complex trapping in preclinical models. Mechanism of Action PARP family members PARP1 and PARP2 play essential roles in cell survival in response to DNA damage. PARP1 and PARP2 are key base-excision-repair proteins that function as DNA damage sensors by binding rapidly to the site of damaged DNA and modulating a variety of proteins in DNA repair processes. Inhibition of PARPs prevents the repair of common single-strand DNA breaks which leads to formation of double-strand breaks during DNA replication. Double-strand DNA breaks in normal cells are repaired by homologous recombination, and normal cells are relatively tolerant of PARP inhibition. On the other hand, cancer cells with mutations in breast cancer susceptibility gene, or BRCA1/2 genes, which are key players in homologous recombination, are highly sensitive to PARP inhibition. This phenomenon is called “synthetic lethality” and is the foundation of the therapeutic utility of PARP inhibitors as a monotherapy for BRCA mutant cancers. In addition to hereditary BRCA1/2 mutations, the synthetic lethality concept has been broadened to include sporadic tumors that display homologous recombination deficiency, or HRD, a gene expression profile that resembles that of a BRCA deficient tumor. HRD can stem from somatic mutation of BRCA1/2, epigenetic silencing of BRCA genes or genetic or epigenetic loss of function of other genes in homologous recombination DNA damage repair pathways. Third-party clinical studies have published results demonstrating that sensitivity to platinum-based chemotherapies confers sensitivity to PARP inhibitors in OC as well. Thus, the application of PARP inhibitors is likely broader than BRCA or HRD mutations, and there is additional possibility to identify and enrich patient populations for PARP inhibition. Another potential therapeutic utility of PARP inhibitors is combination therapy that has strong scientific rational. PARP proteins are key factors in base-excision-repair, which is critical for the repair of DNA lesions caused by some chemotherapeutic agents and by radiation. PARP inhibitors are hypothesized to potentiate cytotoxicity of DNA-alkylating agents such as platinum compounds, temozolomide and ionizing radiation and may be used in combination with these agents in treating various cancers.

Role of PARP and BRCA in DNA Repair SSB in tumor DNA PARP SSB = single-strand DNA breaks, BRCA=breast cancer gene, PARPi=poly (ADP-ribose) polymerase inhibitor. McLornan et al. N Engl J Med. 2014;371(18):1725–1735, 2. Hoeijm akers et al. Nature. 2001;411:366–374. PARP inhibitors are also considered good potential combination partners with checkpoint inhibitors in part due to increased mutations in tumor cells as a result of the blockade of DNA repair by PARP inhibitors as a higher mutational load of cancers has been shown in clinical studies to correlate with improved response to checkpoint inhibitors. In addition, preclinical data suggest that BRCA mutant tumors which are sensitive to PARP inhibition are likely to be immunogenic and responsive to PD-1 or PD-L1 antibodies. Market Opportunity and Competition We believe that the market opportunity for PARP inhibitors is large and expanding in various tumor histologies, settings and patient segments. Many tumor types have been shown to be responsive to PARP inhibitors, including OC, breast cancer, prostate cancer and GC. PARP inhibitors have demonstrated encouraging activities both in relapsed and refractory patients as well as in the maintenance setting. In the United States, each year there are approximately 22,440 new cases of OC, 252,710 new cases of breast cancer, 161,360 new cases of prostate cancer, and 28,000 new cases of GC, according to the U.S. National Cancer Institute. In China, each year there are approximately 52,000 new cases of OC, 272,000 new cases of breast cancer, 60,000 new cases of prostate cancer, and 680,000 new cases of GC according to Chen et al. 2016. A number of PARP inhibitors have been approved by the FDA. These include AstraZeneca’s LYNPARZA® (olaparib), Clovis Oncology’s RUBRACA® (rucaparib) and Tesaro’s ZEJULA® (niraparib). Several PARP inhibitors are in late-stage clinical development besides pamiparib, including AbbVie’s veliparib and Pfizer ’s talazoparib. In 2017, global sales of the PARP class exceeded US$461 million according to company reports. In China, there were no approved PARP inhibitors as of July 18, 2018. AstraZeneca has submitted an NDA for olaparib. In addition, Zai Lab obtained the development and commercial rights for niraparib in China, and is currently running a Phase 1 pharmacokinetics study and a Phase 3 pivotal trial as a maintenance treatment after two lines

of platinum-containing therapy in patients with OC up to the end of May, 2018. There are some other PARP inhibitors being developed by domestic Chinese companies, including pamiparid and fluzoparib from Hengrui and Hansoh. The table below summarizes the China competitive landscape of pamiparib, according to the Frost & Sullivan Report. Summary of Clinical Data As of July 5, 2018, we have enrolled over 350 patients in clinical trials of pamiparib, including combination trials. A multi-center, open-label Phase 1/2 trial of pamiparib is being conducted in Australia in patients with advanced solid tumors. On September 8, 2017, we presented preliminary clinical data from the ongoing Phase 1/2 trial of pamiparib in patients with advanced solid tumors at the ESMO 2017 Congress. As of June 1, 2017, 68 patients were enrolled in the trial. The median duration of therapy for all patients was 79 days (range 1 to 926 days). At the time of the data cutoff, 20 patients remained on treatment. The safety analysis suggested that pamiparib was generally well-tolerated in patients with advanced solid tumors. AEs assessed to be treatment-related occurred in 78% of patients and were all grade 3 or lower in severity. The most common treatment-related AEs (>10% of patients) were nausea (56%), fatigue (40%), anemia (25%), vomiting (21%), diarrhea (21%), decreased appetite (15%), and neutropenia or neutrophil count decrease (12%). SAEs occurred in 46% of patients, and SAEs considered to be treatment-related and occurring in more than one patient included two cases each of nausea and anemia. Four patients reported dose-limiting toxicity, or DLT. Four patients had a treatment-emergent AE with a fatal outcome, none were assessed as being treatment-related and all of which were associated with disease progression. At the time of the data cutoff, 39 patients with epithelial ovarian cancer, or EOC, or associated tumors such as fallopian tube or primary peritoneal cancers were evaluable for efficacy. Among this group, there were three confirmed CRs, 10 confirmed PRs, and 21 cases of SD. Of the 23 evaluable
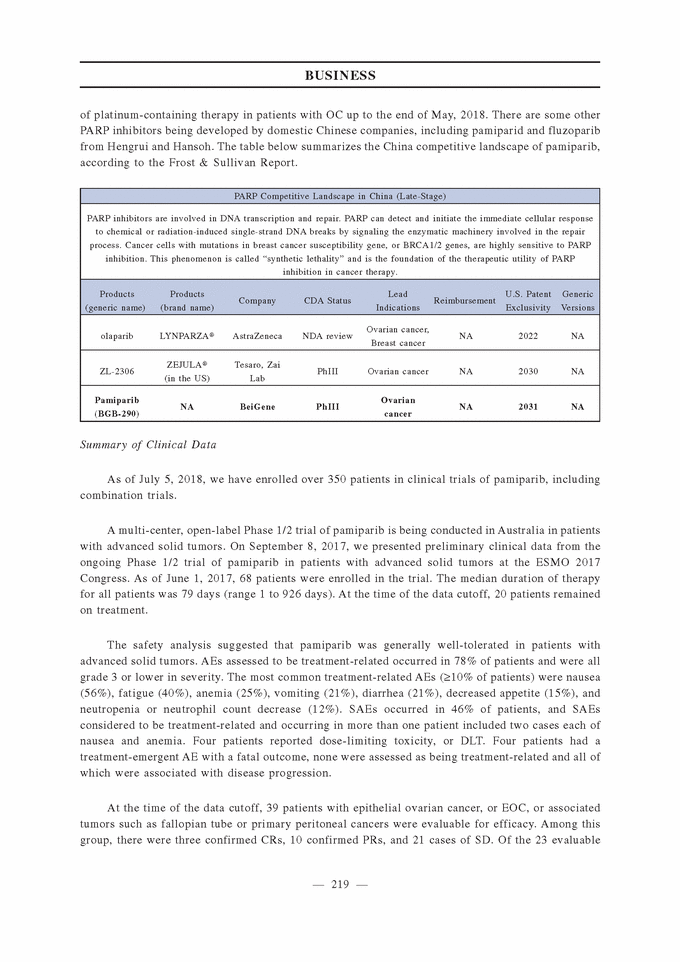
patients with EOC or other associated tumors known to be BRCA-mutated, there were three CRs, seven PRs, and 10 cases of SD. Of the 13 evaluable patients whose BRCA gene types are wild type, there were two PRs. Of the three evaluable patients whose BRCA gene types were unknown, there was one PR. Complete and partial responses were observed in patients known to be platinum-resistant as well as patients with platinum-sensitive disease. There is no guarantee that these results will be reproduced in pivotal trials. On April 16, 2018, we presented the preliminary clinical data from the open-label, multi-center Phase 1 dose-escalation trial of pamiparib in Chinese patients with locally advanced or metastatic high-grade non-mucinous ovarian cancer, or HGOC, including fallopian cancer, or triple-negative breast cancer, or TNBC, who had disease progression following at least one line of chemotherapy at the 2018 American Association for Cancer Research Annual Meeting in Chicago, IL. Patients were dosed at 20mg, 40mg, or 60mg BID. As of September 25, 2017, 15 female patients were enrolled, nine with HGOC and six with TNBC. Nine patients received four or more prior lines of therapies. All nine patients with HGOC were platinum-resistant (n=8) or refractory (n=1). Seven patients had a confirmed BRCA1/2 mutation (BRCAm), including five patients with HGOC and two patients with TNBC and the remaining patients had BRCA 1/2 wildtype (BRCA-WT). The median duration of treatment was 2.5 months (range: 8-260 days). The safety analysis suggested that pamiparib was generally well-tolerated. No dose-limiting toxicities were reported across the dose range, with RP2D confirmed as 60mg BID. Asthenia (n=12) and nausea (n=12) were the most commonly reported treatment-emergent AE. Severity of all AEs was grade 3 or less. Overall, three patients experienced a serious AE (grade 2 abdominal infection, n=1; grade 3 pleural effusion, n=1; grade 3 ileus, n=1), none of which were considered related to treatment. Two of the SAEs led to treatment withdrawal (abdominal infection, n=1; pleural effusion, n=1). As of September 25, 2017, 13 of the 15 patients were evaluable for antitumor activity; five patients remained on treatment. Two of the nine HGOC patients achieved a confirmed PR including one platinum-refractory patient with BRCA wildtype status and one platinum-resistant patient with BRCA1/2 mutation, six HGOC patients had SD (BRCAm, n=4 and BRCA-WT, n=2) and one patient discontinued before the first radiographic assessment. Of the six treated TNBC patients, five (BRCAm, n=1, BRCA-WT, n=4) experienced disease progression and one patient (BRCAm) discontinued before the first radiographic assessment. Four of these evaluable TNBC patients were BRCA-WT and all experienced disease progression during the previous platinum-based chemotherapy. Clinical Development Plan In addition to the ongoing Phase 2 trial of pamiparib in combination with tislelizumab, we are currently conducting two other global combination trials: a Phase 1b/2 trial of pamiparib with radiation therapy and/or temozolomide in patients with glioblastoma and a Phase 1b/2 trial of pamiparib with temozolomide in patients with advanced tumors such as OC, TNBC, small cell lung cancer, prostate cancer and GC. In addition, we are screening patients in preparation of the first dosing of a global Phase 3 maintenance trial in patients with platinum-sensitive GC.

In China, we are conducting a Phase 2 pivotal trial in patients with gBRCA-positive OC who have received at least two prior lines of therapy in advanced or metastatic setting and a Phase 3 trial as a maintenance therapy in patients with platinum-sensitive recurrent OC. WE MAY NOT BE ABLE TO ULTIMATELY DEVELOP AND MARKET PAMIPARIB SUCCESSFULLY. Lifirafenib (BGB-283), a RAF Dimer Inhibitor Lifirafenib is an investigational novel small molecule inhibitor with RAF monomer and dimer inhibition activities. Lifirafenib has shown antitumor activities in preclinical models and in cancer patients with tumors harbouring BRAF V600E mutations, non-V600E BRAF mutations or KRAS/NRAS mutations. We have been developing lifirafenib for the treatment of cancers with aberrations in the mitogen-activated protein kinase, or MAPK, pathway, including BRAF gene mutations and KRAS/NRAS gene mutations where first generation BRAF inhibitors are not effective. The MAPK pathway consists of proteins in the cell that transmit a signal from a receptor on the surface of the cell to the DNA in the nucleus of the cell. This pathway plays an essential role in regulating cell proliferation and survival. We believe that lifirafenib as monotherapy or in combination with other agents may have a potential for treating various malignancies, such as melanoma, NSCLC and endometrial cancer. Roche’s ZELBORAF® (vemurafenib) and Novartis’ TAFINLAR® (dabrafenib) are two of the currently approved BRAF inhibitors for treating late-stage BRAF V600E/K mutant melanoma. In addition, the combination of dabrafenib and GSK’s MEKINIST® (trametinib), an MEK inhibitor, as well as vemurafenib and COTELLIC® (cobimeditinib), another MEK inhibitor, are approved in patients with BRAF V600E/K mutation-positive metastatic melanoma. We are aware of several other BRAF inhibitors in clinical development targeting BRAF V600E/K mutated cancers including melanoma, NSCLC, hairy cell leukemia and thyroid cancer. These BRAF inhibitors include Array Biopharma’s encorafenib, currently in Phase 3 trials, and Takeda’s MLN-2480 (BIIB-024) and TAK-580, Daiichi Sankyo’s PLX-8394, Roche’s RG-6185, Genentech’s HM95573 and Novartis’ LXH254 in Phase 1 trials. Currently approved first-generation BRAF inhibitors, vemurafenib and dabrafenib, are only active against the BRAF monomer. Because lifirafenib is designed to inhibit both the monomer and dimer forms of RAF, we believe lifirafenib has the potential to be a first-in-class RAF dimer inhibitor. Lifirafenib was evaluated in a multicenter, open-label Phase 1 trial conducted in Australia and New Zealand comprised of two parts — dose escalation and dose expansion — in patients with BRAF or KRAS/NRAS mutated solid tumors or patients with pancreatic cancer. Lifirafenib demonstrated antitumor activity in both BRAF and KRAS-mutated tumors in preclinical studies and in the dose-escalation portion of this Phase 1 trial.

We presented data from the dose-expansion portion of the trial at the 2017 American Association for Cancer Research Annual Meeting. The dose-expansion portion of the trial was designed to evaluate the safety and efficacy of lifirafenib at the recommended Phase 2 dose of 30 mg once daily established in the dose-escalation part of the trial. In the dose-expansion portion, lifirafenib was generally well-tolerated at a dose of 30 mg once daily and continued to show antitumor activity in patients with BRAF V600-mutated solid tumors and patients with KRAS-mutated solid tumors. The safety analysis, which included 96 patients as of the September 12, 2016 cutoff, suggested that lifirafenib was generally well-tolerated at 30 mg once daily, with most drug-related AEs being grades 1 or 2 in severity. The most frequent drug-related AEs (>10%) of any grade were fatigue (38.5%), dysphonia (26.0%), decreased appetite (21.9%), palmar-plantar erythrodysaesthesia syndrome (21.9%), thrombocytopenia (19.8%), dermatitis acneiform (17.7%), diarrhea (16.7%), rash (16.7%), nausea (15.6%), hypertension (11.5%) and glossodynia (10.4%). The most frequent drug-related grade 3 and 4 AEs (> 2%, two patients or more) included fatigue (7.3%), hypertension (6.3%), thrombocytopenia (6.3%), pyrexia (3.1%), hyponatremia (2.1%), anemia (2.1%), neutropenia (2.1%), febrile neutropenia (2.1%), decreased platelet count (2.1%), increased alanine aminotransferase (2.1%), increased GGT (2.1%) and sepsis (2.1%). The cutoff for the efficacy analysis was September 17, 2016. In seven patients with BRAF V600-mutated melanoma (including one V600K and one V600R) who were naïve to BRAF or MEK inhibitors, there were three PRs and three cases of SD. In three patients with BRAF V600-mutated PTC, there was one PR and two cases of SD. In six patients with KRAS-mutated NSCLC, there was one PR and two cases of SD. In ten patients with solid tumors with BRAF non-V600 mutations or solid tumors with BRAF V600 mutations that are not included in other cohorts, there were two PRs, in one patient with BRAF V600E-mutated melanoma and one with BRAF V600E-mutated OC, and three cases of SD. In two patients with BRAF V600-mutated NSCLC, there was one unconfirmed PR and one case of SD. Additional cases of SD were observed in four of six melanoma patients with BRAF V600-mutated melanoma who had responses to but developed resistance against BRAF or MEK inhibitors, nine of 13 patients with BRAF V600-mutated CRC, five of five patients with KRAS-mutated endometrial cancer, 12 of 20 patients with KRAS/NRAS-mutated CRC, and 10 of 21 patients with other KRAS/NRAS-mutated solid tumors or pancreatic cancer. In the Phase 1a portion of the trial, confirmed objective responses included a CR in a patient with BRAF V600E-mutated melanoma and two PRs, one in a patient with BRAF V600E-mutated thyroid cancer and one in a patient with KRAS-mutated endometrial cancer. BGB-A333, a PD-L1 Inhibitor BGB-A333 is an investigational humanized IgG1-variant monoclonal antibody against PD-L1, the ligand of PD-1. We intend to develop BGB-A333 either as a monotherapy or in combination with other cancer therapies, such as tislelizumab, to treat various cancers and potentially other indications. BGB-A333 is currently being evaluated in a Phase 1 clinical trial in Australia to test the safety and anti-tumor effect of BGB-A333 alone and in combination with tislelizumab in patients with advanced solid tumors.

BGB-A425, a TIM-3 Inhibitor BGB-A425 is an investigational humanized IgG1-variant monoclonal antibody against T-cell immunoglobulin and mucin-domain containing-3, or TIM-3. We received the investigational new drug clearance in the U.S., and we plan to develop BGB-A425 either as a monotherapy or in combination with other cancer therapies to treat various cancers. Sitravatinib (MGCD-0516), a Multi-Kinase Inhibitor In January 2018, we entered into an exclusive license agreement with Mirati for the development, manufacturing and commercialization of Mirati’s sitravatinib in Asia (excluding Japan and certain other countries), Australia and New Zealand. Sitravatinib is an investigational spectrum-selective kinase inhibitor which potently inhibits receptor tyrosine kinases, including RET, TAM family receptors (TYRO3, Axl, MER), and split family receptors (VEGFR2, KIT). Sitravatinib is being evaluated by Mirati as a single agent in a dose-expansion trial in patients whose tumors harbor specific genetic alterations in NSCLC and other tumors. Sitravatinib has shown encouraging interim results in an ongoing Phase 2 trial in combination with nivolumab in NSCLC patients who have progressed after prior treatment with a checkpoint inhibitor. We plan to investigate sitravatinib in combination with tislelizumab in China and the licensed territory. Under the license agreement, Mirati retains exclusive rights for the development, manufacturing and commercialization of sitravatinib outside of the licensed territory. We made an upfront cash payment of US$10 million to Mirati and agreed to pay up to US$123 million based upon the achievement of certain development, regulatory and sales milestones, as well as royalties at tiered percentage rates ranging from mid-single digits to twenty percent on annual net sales of sitravatinib in the licensed territory, subject to reduction under specified circumstances. Avadomide (CC-122), a Cereblon Modulator Avadomide (CC-122) is an investigational next-generation Cereblon modulator currently in clinical development by Celgene. It is in multiple Phase 1 and Phase 1/2 clinical trials, both as a single agent and in combination, for hematological and solid tumor cancers outside of China. Avadomide (CC-122) has been differentiated from previous compounds (such as thalidomide, lenalidomide and pomalidomide) and has been developed based on the scientific understanding of Cereblon-mediated protein homeostasis. In August 2017, we entered into a license and supply agreement with Celgene, pursuant to which we were granted a license to develop and commercialize avadomide (CC-122) in China. See “—Collaboration Agreements—Celgene Corporation.” Our Commercial Products We commercialize the following cancer drugs in China under an exclusive license from Celgene.

ABRAXANE® ABRAXANE® (paclitaxel albumin-bound particles for injectable suspension) is a solvent-free chemotherapy product which was developed using Celgene’s proprietary nab® technology platform. This protein-bound chemotherapy agent combines paclitaxel with albumin. Globally, ABRAXANE® is approved for uses in breast cancer, NSCLC, pancreatic cancer and GC with geographic differences in labeling. In China, ABRAXANE® is approved for metastatic breast cancer after failure of combination chemotherapy for metastatic disease or relapse within six months of adjuvant chemotherapy. Prior therapy should have included an anthracycline unless clinically contraindicated. According to Chen et al. 2016, there were approximately 4.3 million new cancer cases and 2.8 million cancer deaths in China in 2015, with breast cancer as the most common tumor type in Chinese women. It is estimated that in 2015 breast cancer affected 268,600 women and resulted in 69,500 deaths. Targeted therapy, hormone therapy and chemotherapy are three main strategies to treat different types of breast cancer. Taxane is the backbone chemotherapy to treat triple negative breast cancer, Her2+ or aggressive estrogen-receptor-positive and/or progesterone-receptor-positive breast cancer patients. ABRAXANE® is the only currently approved taxane that does not need pre-medication of dexamethasone to prevent hypersensitivity reactions, and several Phase 3 trials have demonstrated its efficacy and safety compared to solvent-based taxanes in both metastatic breast cancer and neo-adjuvant settings. Unlike other taxanes, ABRAXANE® has demonstrated unique and strong efficacy in pancreatic cancer and has become the backbone of first line standard of care for metastatic pancreatic cancer globally. The taxanes marketed in China include two branded solvent-based paclitaxel (TAXOL® and ANZATAX), one branded docetaxel (TAXOTERE®), one lipsome-paclitaxel (LIPUSU), one albumin-bound paclitaxel (ABRAXANE®) and dozens of generic taxanes. LIPUSU is currently the market leader with approximately one-third of the value share. In 2017, ABRAXANE® held an estimated 5.4% value share in the taxane market in China. In February 2018, a albumin-bound paclitaxel from CSPC Pharmaceutical Group was approved by the CDA. Another form of albumin-bound paclitaxel from Hengrui is under review by the CDA. In 2018, we plan to seek to differentiate and defend ABRAXANE® against generic competition in China, expand our sales force footprint and hospital coverage, and improve patient access through critical illness insurance negotiations and provincial reimbursement listings. As of July 18, 2018, ABRAXANE® is listed on provincial reimbursement drug lists of Fujian, Hubei, Ningxia, and Jiangsu, as well as in critical illness insurance program in Zhejiang and Shandong. ABRAXANE® has recently been added to the PRDL in Hunan, effective September 1, 2018.

REVLIMID® REVLIMID® (lenalidomide) is an oral immunomodulatory drug that was approved by the CDA in China in 2013 for the treatment of multiple myeloma, or MM, in combination with dexamethasone in adult patients who have received at least one prior therapy. On February 2, 2018, REVLIMID® received CDA approval of a new indication for the treatment of MM in combination with dexamethasone in adult patients with previously untreated MM who are not eligible for transplant. MM is a malignant plasma cell disease whose tumor cells originate in plasma cells in the bone marrow, which are cells in which B-lymphocytes develop to the final functional phase. The current World Health Organization classifies it as a B-cell lymphoma, also known as plasma cell myeloma / plasmacytoma. MM is characterized by abnormal proliferation of bone marrow plasma cells accompanied by overproduction of monoclonal immunoglobulin. MM is often accompanied by multiple osteolytic lesions, hypercalcemia, anemia, and kidney damage. Due to the inhibition of normal immunoglobulin production, patients are prone to a variety of bacterial infections. At present, MM is one of the most common malignant tumors in the blood system and occurs frequently in the elderly. The actual incidence increases with age, peaking from 60 to 70 years of age. Men suffer slightly more than women. Globally, the incidence was estimated at 2 to 3 per 100,000, with a male-to-female ratio of 1.6:1, and most patients are over 40 years old, according to Siegel et al., 2011 and IMS analysis. It is estimated that the incidence rate of MM is approximately 1-2 per 100,000 people in China, or approximately 18,000 new patients in 2017, out of which 10,000 are in urban populations, according to Lu et al., 2014, IMS analysis, and local market research. With a growing aging population and improving diagnosis, China has seen a steady increase in MM incidence. Although MM cannot be cured, the progression of the disease can be controlled. The purpose of treatment is to extend patients’ survival and improve quality of life. The main treatments for MM in China include VELCADE®, which is a proteasome inhibitor marketed by Johnson and Johnson in China since 2006, generic thalidomide and REVLIMID®. VELCADE® currently dominates the market in first-line MM treatment in China, while VELCADE® and REVLIMID® share the market in the second line. Chinese guidelines recommend lenalidomide as a standard of care for the treatment of R/R and newly diagnosed MM as well as in the maintenance setting. The first lenalidomide generic and first bortezomib generic in China were approved in November 2017. Another new agent for R/R MM, ixazomib, an oral proteasome inhibitor developed by Takeda, is currently under regulatory review in China. In February 2018, generic lenalidomide from Shuanglu Pharmaceutical Company was approved by the CDA. In 2017, the patient share for REVLIMID® in second-line MM in the top 30 hospitals in China rose from an estimated 36% to 47%. REVLIMID® achieved national reimbursement drug listing through a successful price negotiation with the Ministry of Human Resources and Social Security in June 2017.

VIDAZA® VIDAZA® (azacitidine for injection) is a pyrimidine nucleoside analog that has been shown to reverse the effects of DNA hypermethylation and promote subsequent gene re-expression. VIDAZA was approved in China in April 2017 for the treatment of intermediate-2 and high-risk myelodysplastic syndromes, or MDS, chronic myelomonocyte leukemia, or CMML, and acute myeloid leukemia, or AML, with 20% to 30% blasts and multi-lineage dysplasia. In January 2018, VIDAZA® became commercially available in China. MDS are a group of cancers in which immature blood cells in the bone marrow do not mature and therefore do not become healthy blood cells. Approximately seven per 100,000 people are affected with approximately four per 100,000 people newly acquiring the condition each year globally according to Germing et al., 2013. The typical age of onset is 70 years. The higher-risk MDS (intermediate-2 and high-risk MDS) is fatal because the median overall survival rate is only 0.4-1.1 years and nearly 30% of these patients progress to AML, according to the U.S. National Comprehensive Cancer network, or NCCN, MDS guideline 2013 and MDS Foundation. DNA methylation is an important mechanism of epigenetic gene regulation, but aberrant DNA hypermethylation can result in gene silencing. Silencing of tumor suppressor genes promotes cancer development and progression. MDS patients display aberrant DNA methylation of thousands of genes, which increases with advanced disease and is a poor prognostic factor. In China, the main treatments for intermediate-2 and high-risk MDS are conventional care regimen, or CCR (best supportive care, low-dose cytarabine and intensive chemotherapy), and hypomethylating agents, or HMAs. DACOGEN® (decitabine) marketed by Johnson and Johnson was the first HMA agent approved in China in 2009. In the past several years, at least six decitabine generics have become available. In 2017, decitabine was listed in the NRDL. Nevertheless, there are still over 50% of higher-risk MDS patients treated by CCR and the unmet need remains large. VIDAZA® is the only approved HMA shown to prolong survival for patients with MDS. Besides reversing the effects of DNA hypermethylation, VIDAZA® inhibits protein synthesis via RNA incorporation. VIDAZA® is a Category 1 recommended treatment for patients with intermediate-2 and high-risk MDS, according to the U.S. NCCN guideline. It is also a first-line recommended treatment for patients with intermediate-2 and high-risk MDS, according to the Chinese MDS treatment guidelines. Our Preclinical Programs We have a proprietary cancer biology platform that has also allowed us to develop our clinical-stage drug candidates and several additional preclinical-stage drug candidates in potentially important areas. These currently consist of targeted therapies and immuno-oncology agents, including an additional RAF dimer inhibitor and a BTK inhibitor for non-oncology indications. We anticipate advancing one or more of our preclinical assets into the clinic in the next 12 months. We believe we have the opportunity to combine tislelizumab with our preclinical candidates to target multiple points in the cancer immunity cycle. We also may seek to develop companion diagnostics that will help identify patients who are most likely to benefit from the use of our drug candidates.

Collaboration Agreements Celgene Corporation Exclusive License and Collaboration Agreement On July 5, 2017, we entered into an Exclusive License and Collaboration Agreement, as amended and restated, with Celgene and its wholly-owned subsidiary, Celgene Switzerland LLC, or Celgene Switzerland, which became effective on August 31, 2017, pursuant to which we granted the Celgene parties an exclusive right to develop and commercialize tislelizumab in all fields of treatment, other than hematology, in the United States, Europe, Japan and the rest of world other than Asia, which we refer to as the PD-1 License Agreement. Pursuant to the terms of the PD-1 License Agreement, the Celgene parties made upfront payments of US$263 million to us. In addition, pursuant to a share subscription agreement with Celgene Switzerland dated July 5, 2017, or the Share Subscription Agreement, we issued approximately 32.7 million of our ordinary shares on August 31, 2017 for an aggregate purchase price of US$150 million at $4.58 per ordinary share, or $59.55 per ADS, representing a 35% premium to an 11-day volume-weighted average price of our ADSs. We may also receive up to US$980 million in potential development, regulatory and sales milestone payments and tiered royalties based on percentages of annual net sales, depending on specified terms, in the low double digit to mid-twenties, with customary reductions in specified circumstances. Royalties are payable on a licensed product-by-product and country-by-country basis until the latest of the expiration of the last valid patent claim, the expiration of regulatory exclusivity, or 12 years after the first commercial sale of such licensed product in the country of sale. Each party has the right to develop and commercialize tislelizumab in its respective field and territory, and has also agreed to collaborate through a joint steering committee comprised of an equal number of representatives from each party on, among other things, the conduct of up to eight global pivotal clinical trials, or the Basket Studies. Each Basket Study will be conducted and funded by either us or Celgene in accordance with a mutually agreed development plan and study design. For any Basket Studies conducted and funded by us, Celgene has the right to opt into such program, at which time it will reimburse us for agreed upon development costs based on a multiple of such costs that varies according to the stage of development at which Celgene opts into the program. Celgene has committed to use commercially reasonable efforts to develop at least one licensed product, to seek specified regulatory approvals and to spend at least US$100 million on development for the Basket Studies led by Celgene, subject to specified conditions. In addition, we retain the right to develop tislelizumab in combination therapies with our portfolio compounds, and Celgene has a right of first negotiation for tislelizumab in the hematology field and in our territory, subject to specified conditions. The PD-1 License Agreement contains customary representations, warranties and covenants by us and Celgene. Unless earlier terminated, the agreement will expire on a licensed product-by-product and country-by-country basis upon the expiration of the royalty term in such country for such licensed product. The agreement may be terminated by Celgene upon 30 days’ prior written notice, or by either party upon the other party’s bankruptcy or uncured material breach.

Celgene China Agreement On July 5, 2017, we and a wholly-owned subsidiary of Celgene, Celgene Logistics Sàrl, or Celgene Logistics, entered into a License and Supply Agreement, which we refer to as the China License Agreement and which became effective on August 31, 2017, pursuant to which we were granted the right to exclusively distribute and promote Celgene’s approved cancer therapies, ABRAXANE®, REVLIMID® and VIDAZA®, and its investigational agent avadomide (CC-122) in clinical development in China, excluding Hong Kong, Macau and Taiwan. In addition, if Celgene decides to commercialize a new oncology product through a third-party in the licensed territory during the first five years of the term, we have a right of first negotiation to obtain the right to commercialize the product, subject to certain conditions. We paid an aggregate of US$4.5 million in cash for the license and our acquisition of Celgene Shanghai, as described below. Subsequent to the closing of the arrangements and through the Latest Practicable Date, we had paid Celgene US$17.5 million in total for inventory purchases. The term of the China License Agreement is 10 years and may be terminated by either party upon written notice in the event of uncured material breach or bankruptcy of the other party, or if the underlying regulatory approvals for the covered products are revoked. Celgene Logistics also has the right to terminate the agreement with respect to REVLIMID® at any time upon written notice to the Company. The China License Agreement contains customary representations and warranties and confidentiality and mutual indemnification provisions. On August 31, 2017, our wholly owned subsidiary, BeiGene (Hong Kong) Co., Ltd., acquired 100% of the equity interests of Celgene Pharmaceutical (Shanghai) Co., Ltd., or Celgene Shanghai, a wholly-owned subsidiary of Celgene Holdings East Corporation established under the laws of China. The purchase price of Celgene Shanghai was determined to be approximately US$28.1 million from an accounting perspective, and comprised of a cash consideration of US$4.5 million and non-cash consideration of US$23.6 million. The amount allocated to non-cash consideration, related to the discount on ordinary shares issued to Celgene in connection with the Share Subscription Agreement and was a result of the increase in fair value of our shares between the fixed price of US$59.55 per ADS specified in the Share Subscription Agreement and the fair value per ADS on August 31, 2017, the date the transaction closed. This company, which we subsequently renamed BeiGene Pharmaceutical (Shanghai) Co., Ltd., is in the business of, among other things, providing marketing and promotional services for the pharmaceutical products that we license from Celgene. Prior to closing, Celgene separated out certain business functions, including regulatory and drug safety, that continue to support the business acquired by us. Merck KGaA, Darmstadt Germany Pamiparib On October 28, 2013, we entered into license agreements with Merck KGaA, Darmstadt Germany, which we refer to respectively as the Ex-PRC PARP Agreement and the PRC PARP Agreement, pursuant to which (a) we granted to Merck KGaA, Darmstadt Germany an exclusive

license under certain of our intellectual property rights to develop and manufacture, and, if Merck KGaA, Darmstadt Germany exercised a continuation option, to commercialize and manufacture pamiparib and any other compound covered by the same existing patent rights with primary activity to inhibit PARP 1, 2 or 3 enzymes, or the Licensed PARP Inhibitors, in the Ex-PRC (worldwide except PRC) territory, and (b) Merck KGaA, Darmstadt Germany granted us an exclusive license under certain of its intellectual property rights to develop, manufacture and commercialize the Licensed PARP Inhibitors in the People’s Republic of China, or the PRC, which we refer to as the PRC Territory. On October 1, 2015, pursuant to a purchase of rights agreement, we repurchased all of Merck KGaA, Darmstadt Germany’s rights under the Ex-PRC PARP Agreement, in consideration for, among other things, a one-time payment of US$10 million, which payment has been made, and reduction of future milestone payments we were eligible for under the PRC PARP Agreement. In connection with that repurchase, we also agreed to provide Merck KGaA, Darmstadt Germany with global access to our clinical PARP supplies, including pamiparib, for its combination trials, during the option period. The Ex-PRC PARP Agreement was terminated, except for certain provisions that are needed to effectuate the continuation of the PRC PARP Agreement, including those provisions that were required in the event that Merck KGaA, Darmstadt Germany exercised its PRC Commercialization Option (described below). We repurchased all of Merck KGaA, Darmstadt Germany’s rights under the Ex-PRC PARP Agreement and terminated the Ex-PRC PARP Agreement in order to reacquire the rights to pursue both monotherapy and combination therapy. Pursuant to the PRC PARP Agreement, if we failed to achieve national priority project status in the PRC Territory under its 12th or 13th five-year plan with respect to our pamiparib program in the PRC Territory by July 28, 2017, Merck KGaA, Darmstadt Germany has an option to acquire exclusive commercialization rights under the pamiparib program in the PRC Territory, which we refer to as the PRC Commercialization Option. If, however, we achieved national priority by July 28, 2017, Merck KGaA, Darmstadt Germany only has a right of first negotiation to acquire exclusive commercialization rights under the pamiparib program in the PRC Territory in the event we seek to license our intellectual property rights to a third party. We applied for national priority project status for pamiparib to be effective from the beginning of 2017, and our application is in process and we believe that it will be approved. However, there have been unanticipated governmental delays that have impacted the 2017 applicant pool for national project priority status and we expect that we will now receive formal notification in 2018. As such, we intend to discuss with Merck KGaA, Darmstadt Germany the impact of this delay on the PRC Commercialization Option. If Merck KGaA, Darmstadt Germany exercises the PRC Commercialization Option, it is required to pay us a US$50 million non-refundable payment upon such exercise, and we are eligible for a US$12.5 million milestone payment upon the successful achievement of a certain additional regulatory event in the PRC Territory. Under these agreements, we received US$6 million in non-refundable upfront payments in December 2013 in connection with the signing of the agreements, including US$5 million under the PRC PARP Agreement and US$1 million under the Ex-PRC PARP Agreement. We had also received US$10 million in milestone payments as of March 31, 2018, including US$1 million under the PRC PARP Agreement upon dosing of the first patient in the first Phase 2 trial in the PRC Territory, and US$9 million under the Ex-PRC PARP Agreement upon dosing of the fifth patient in the first Phase 1 trial in the Ex-PRC territory. Pursuant to the terms of the agreements, we are eligible to receive an additional US$7 million and US$2.5 million, respectively, in payments upon the successful

achievement of pre-specified clinical and regulatory milestones in the PRC Territory. Also, in consideration for the licenses granted to us, we are required to pay Merck KGaA, Darmstadt Germany a high single-digit royalty on aggregate net sales of Licensed PARP Inhibitors in the PRC Territory. The PRC PARP Agreement continues unless terminated as permitted by either party. Merck KGaA, Darmstadt Germany has the right to terminate due to our uncured breach or for convenience upon prior written notice. We have the right to terminate these agreements due to Merck KGaA, Darmstadt Germany’s uncured breach or for any challenge brought against our licensed patent rights. Lifirafenib On May 24, 2013, we entered into license agreements with Merck KGaA, Darmstadt Germany for lifirafenib, which were amended in 2013 and 2015 and which we refer to respectively as the Ex-PRC BRAF Agreement and PRC BRAF Agreement. In March 2017, Merck KGaA, Darmstadt Germany informed us that it would not exercise a continuation option in the ex-PRC territory, and thus, the ex-PRC BRAF Agreement terminated in its entirety, except for certain provisions that survive termination. Under the PRC BRAF Agreement, Merck KGaA, Darmstadt Germany granted us an exclusive license under certain of its intellectual property rights to develop, manufacture and commercialize the RAF dimer inhibitor in the PRC, which we refer to as the PRC Territory, subject to certain non-compete restrictions. Further, pursuant to the PRC BRAF Agreement, Merck KGaA, Darmstadt Germany has an exclusive right of first negotiation to acquire exclusive commercialization rights under the lifirafenib BRAF program in the PRC Territory on terms to be mutually agreed in the event we seek to license our intellectual property rights to a third party in the territory. Under these agreements, in December 2013, we received US$13 million in non-refundable payments, including US$10 million under the PRC BRAF Agreement and US$3 million under the Ex-PRC BRAF Agreement. As of March 31, 2018, we had received US$9 million in milestone payments, including US$4 million under the PRC BRAF Agreement upon dosing of the fifth patient in the first Phase 1 trial in the PRC Territory, and US$5 million under the Ex-PRC BRAF Agreement upon dosing of the fifth patient in the first Phase 1 trial in the Ex-PRC territory. We are eligible to receive an additional US$14 million in payments upon the successful achievement of pre-specified clinical milestones in the PRC Territory. We are required to pay Merck KGaA, Darmstadt Germany a high single-digit royalty on aggregate net sales of the licensed compounds in the PRC Territory. The term of the PRC BRAF Agreement continues unless terminated as permitted by either party. Under the PRC BRAF Agreement, Merck KGaA has the right to terminate due to our uncured breach or voluntarily upon prior written notice. We have the right to terminate the PRC BRAF Agreement due to Merck KGaA’s uncured breach or for any challenge brought against our licensed patent rights. Intellectual Property The proprietary nature of, and protection for, our drug candidates and their methods of use are an important part of our strategy to develop and commercialize novel medicines, as described in more detail below. We have obtained patents and filed patent applications in the United States and other countries, such as China, relating to certain of our drug candidates, and are pursuing additional patent protection for them and for other of our drug candidates and technologies. We rely on trade secrets

to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection including our manufacturing processes. We also rely on know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen and support our development programs. As of July 18, 2018, we owned 17 issued U.S. patents, seven issued China patents, nine pending U.S. patent applications, 11 pending China patent applications, and corresponding patents and patent applications internationally. In addition, we owned 12 pending international patent applications under the Patent Cooperation Treaty, or PCT, which we plan to file nationally in the United States and other jurisdictions, as well as additional priority PCT applications. With respect to any issued patents in the United States and Europe, we may be entitled to obtain a patent term extension to extend the patent expiration date provided we meet the applicable requirements for obtaining such patent term extensions. For example, in the United States, we can apply for a patent term extension of up to five years for one of the patents covering a product once the product is approved by the FDA. The exact duration of the extension depends on the time we spend in clinical studies as well as getting an NDA approval from the FDA. Summaries of patents of our Core Product Candidates and marketed products as of July 18, 2018 are set forth below: Summary of U.S. Patents of Our Core Product Candidates ProductScope of patent protection Patent Expiration Market commercial rights of BeiGene Eligibility for patent extension Zanubrutinib .... Directed to zanubrutinib, a small molecule BTK inhibitor, combinations of zanubrutinib with other therapeutic agents, and its use for the treatment of hematological malignancies 2034All rights in the U.S. Yes Tislelizumab .... Directed to tislelizumab, a humanized monoclonal antibody against PD-1, and its use for the treatment of cancer 2033All rights in the field of hematology in the U.S., and rights to combine with our products in all indications Yes Pamiparib ........ Directed to pamiparib, a small molecule PARP1/2 inhibitor, and its use for the treatment of cancer, including glioblastomas and breast cancer 2031All rights in the U.S. Yes Lifirafenib ....... Directed to lifirafenib, a small molecule BRAF inhibitor, and its use for the treatment of cancer, including BRAF mutated cancers 2031All rights in the U.S. Yes
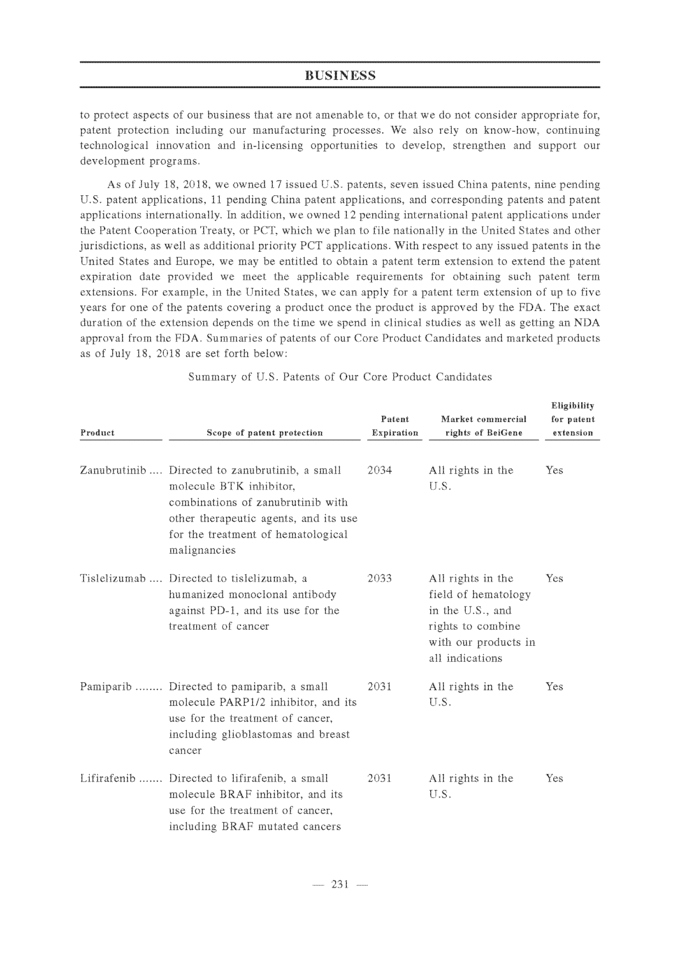
Summary of China Patents for Our Core Product Candidates and Marketed Products ProductScope of patent protection Patent Expiration Market commercial rights of BeiGene Eligibility for patent extension Zanubrutinib ........ Directed to zanubrutinib, a small molecule BTK inhibitor, combinations of zanubrutinib with other therapeutic agents, and its use for the treatment of hematological malignancies 2034All rights in China N/A Tislelizumab ........ Directed to tislelizumab, a humanized monoclonal antibody against PD-1, and its use for the treatment of cancer 2033All rights in China N/A Pamiparib ............ Directed to pamiparib, a small molecule PARP1/2 inhibitor, and its use for the treatment of cancer, including glioblastomas and breast 2031All rights in China, subject to Merck KGaA’s PRC N/A cancer Commercialization Option Lifirafenib ........... Directed to lifirafenib, a small molecule BRAF inhibitor, and its use for the treatment of cancer, including BRAF mutated cancers 2031All rights in China, subject to certain rights of Merck KGaA N/A ABRAXANE® ...... Directed to ABRAXANE®, a nanoparticle albumin-bound paclitaxel, covering its composition, liquid formulation, and use for the treatment of cancer 2018, 2021, 2026, 2031, respectively Marketing and sales in China N/A REVLIMID® ........ Directed to REVLIMID®, covering its use for the treatment of cancer, including MM 2023, 2027, respectively Marketing and sales in China N/A VIDAZA® ............ N/AN/AMarketing and sales in China N/A
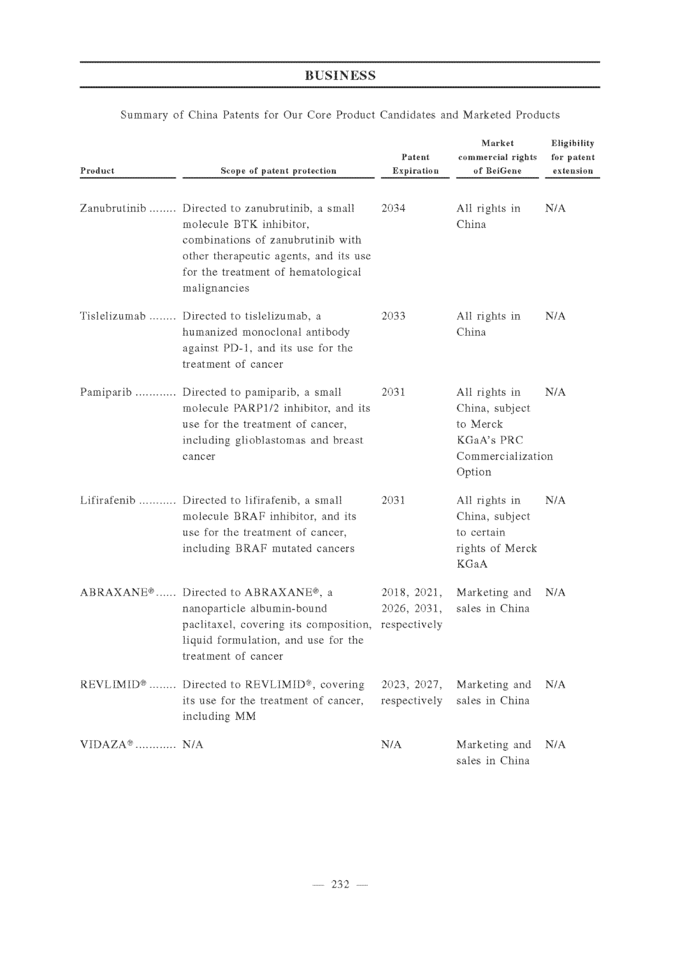
The patent portfolios for our four lead product candidates as of July 18, 2018 are summarized below: Zanubrutinib. We own two issued U.S. patents, one pending U.S. patent application, one issued China patent, two pending PCT applications, and corresponding patent applications in other jurisdictions directed to zanubrutinib, a small molecule BTK inhibitor, combinations of zanubrutinib with other therapeutic agents, and its use for the treatment of hematological malignancies. The expected expiration for the issued U.S. patent is 2034, excluding any additional term for patent term extensions. Any patents that may issue from the currently pending U.S. patent applications would be expected to expire in 2034, not including any patent term adjustments. If a U.S. application is filed based on the pending PCT applications, a patent issuing from these applications, if any, would be expected to expire in 2037. We intend to pursue marketing exclusivity periods that are available under regulatory provisions in certain countries. Tislelizumab. We are the owner of three issued U.S. patents, one pending U.S. application, one pending PCT application, four pending China patent applications, and corresponding pending patent applications in other jurisdictions directed to tislelizumab, a humanized monoclonal antibody against PD-1, and its use for the treatment of cancer. The expected expiration for the issued U.S. patents is 2033, excluding any additional term for patent term extensions. Any patent that may issue from the currently pending U.S. patent application would be expected to expire in 2033, not including any patent term adjustments. We intend to pursue marketing exclusivity periods that are available under regulatory provisions in certain countries. Pamiparib. We own two issued U.S. patents, two pending U.S. patent applications, three pending PCT applications, one issued China patent, two pending China patent applications, and corresponding pending patent applications in other jurisdictions directed to pamiparib, a small molecule PARP1/2 inhibitor, and its use for the treatment of cancer, including glioblastomas and breast cancer. We also own the corresponding pending patent applications in other jurisdictions. The expected expiration for the issued U.S. patents is 2031, excluding any additional term for patent term extensions. Any patent that may issue from the currently pending U.S. patent applications would be expected to expire in 2031 and 2036, not including any patent term adjustments. If a U.S. application is filed based on the pending PCT applications, patents issuing from these applications, if any, would be expected to expire in 2037 and 2038. We intend to pursue marketing exclusivity periods that are available under regulatory provisions in certain countries. Lifirafenib. We own two issued U.S. patents, two pending U.S. patent applications, one pending PCT application, two issued China patents, one pending China patent application, and corresponding pending patent applications in other jurisdictions directed to lifirafenib, a small molecule BRAF inhibitor, and its use for the treatment of cancer, including BRAF mutated cancers. We also own pending patent applications in other jurisdictions corresponding to the U.S. patent applications. In addition, we plan to file nationally in the U.S. and other jurisdictions based on the pending PCT application. The expected expiration for the issued U.S. patents are 2031, excluding any additional term for patent term extensions. Any patents that may issue from the currently pending U.S. patent

applications would be expected to expire in 2031 and 2036, not including any patent term adjustments. If a U.S. application is filed based on the pending PCT application, a patent issuing from this application, if any, would be expected to expire in 2037. We intend to pursue marketing exclusivity periods that are available under regulatory provisions in certain countries. The patent portfolios for our three in-licensed commercial products in China are summarized below: ABRAXANE®. We are the exclusive licensor of five issued Chinese patents and four pending Chinese patent applications directed to ABRAXANE®, a nanoparticle albumin—bound paclitaxel, covering its composition, liquid formulation, and use for the treatment of cancer. The expected expirations for the issued Chinese patents are 2018, 2021, 2026 and 2031, respectively, excluding any additional term for patent term extensions. Any patent that may issue from the currently pending Chinese patent applications would be expected to expire in 2023, 2026 or 2034. In February 2018, a generic version of albumin-bound palclitaxel was approved in China and another is currently under regulatory review. REVLIMID®. We are the exclusive licensor of seven issued Chinese patents directed to REVLIMID®, covering its use for the treatment of cancer, including MM. The expected expirations for the issued Chinese patents are 2023 and 2027 respectively, excluding any additional term for patent term extensions. The first lenalidomide generic in China was approved in November 2017. VIDAZA®. We do not have any rights in any issued China patent or pending China patent applications directed to VIDAZA®, a chemical analog of cytidine, and its use for the treatment of cancer. We are aware of third parties who are seeking to develop and obtain approval for generic forms of this drug. Furthermore, although various extensions may be available, the life of a patent and the protection it affords, is limited. As noted above, ABRAXANE®, REVLIMID®, and VIDAZA® face or are expected to face competition from generic medications, and we may face similar competition for any approved drug candidates even if we successfully obtain patent protection once the patent life has expired for the drug. Manufacturers of generic drugs may challenge the scope, validity or enforceability of our patents in court, and we may not be successful in enforcing or defending those intellectual property rights and, as a result, may not be able to develop or market the relevant product exclusively, which would have a material adverse effect on any potential sales of that product. Under our license agreement with Celgene, Celgene retains the responsibility for, but is not obligated, to prosecute, defend and enforce the patents for these in-licensed products. As such, any issued patents may not protect us from generic competition for these drugs. The term of individual patents may vary based on the countries in which they are obtained. In most countries in which we file, including the United States and China, the term of an issued patent is generally 20 years from the earliest claimed filing date of a non-provisional patent application in the applicable country. In the United States, a patent’s term may be lengthened in some cases by a patent term adjustment, which extends the term of a patent to account for administrative delays by the

USPTO in excess of a patent applicant’s own delays during the prosecution process, or may be shortened if a patent is terminally disclaimed over a commonly owned patent having an earlier expiration date. In addition, in certain instances, a patent term can be extended to recapture a portion of the term effectively lost as a result of the FDA regulatory review period. However, the restoration period cannot be longer than five years and the total patent term including the restoration period must not exceed 14 years following FDA approval. In certain foreign jurisdictions similar extensions as compensation for regulatory delays are also available. The actual protection afforded by a patent varies on a claim by claim and country by country basis and depends upon many factors, including the type of patent, the scope of its coverage, the availability of any patent term extensions or adjustments, the availability of legal remedies in a particular country and the validity and enforceability of the patent. We may rely, in some circumstances, on trade secrets and unpatented know-how to protect aspects of our technology. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with consultants, scientific advisors and contractors and invention assignment agreements with our employees. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. Additionally, we currently own a number of registered trademarks and pending trademark applications. We currently have registered trademarks for BeiGene and our corporate logo in China, the European Union and other jurisdictions and are seeking trademark protection for BeiGene and our corporate logo in the United States and other countries where available and appropriate. Research and Development We are a leader in the research and development of innovative molecularly targeted and immuno-oncology drugs for the treatment of cancer. We have made significant investments identifying, developing and commercializing biologic drug product candidates with significant market potential. Our current research and development activities mainly relate to the clinical advancement of our six internally-developed drug candidates: (1) zanubrutinib, an investigational small molecule inhibitor of BTK; (2) tislelizumab, an investigational humanized monoclonal antibody against PD-1; (3) pamiparib, an investigational small molecule inhibitor of PARP1 and PARP2; (4) lifirafenib, a novel small molecule inhibitor of both the monomer and dimer forms of BRAF; (5) BGB-A333, an investigational humanized monoclonal antibody against PD-L1; and (6) BGB-A425, an investigational humanized monoclonal antibody against TIM-3. In July 2017, we entered into an Exclusive License and Collaboration Agreement, as amended and restated, with Celgene and Celgene Switzerland, pursuant to which we agreed to collaborate to develop and commercialize tiselizumab. For more details see “— Collaboration Agreements — Celgene Corporation — Exclusive License and Collaboration Agreement.”

We had over 500 clinical development staff and approximately 200 research staff as of July 20, 2018. We designate employees in our business units to our research and development projects based on their credentials, areas of expertise and capacity. Recruiting and retaining qualified scientific personnel is critical to our success. We have entered into formal employment agreement with each member of our scientific team. The agreements provide for at-will employment and base salary, cash incentives and equity compensation of various amounts depending on the position of the employee. We also enter into non-disclosure and confidentiality agreements with our scientific personnel. An employment agreement can be terminated by us or the employee in accordance with local employment regulations. While we believe we don’t rely on any single key scientific personnel, replacing key scientific personnel may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize products. The loss of the services of our key research and development personnel could impede the achievement of our research and development objectives. We have our internal succession plan on each critical position in the research team, which helps ensure the continuity of product development. Therefore, in the event of the departure of any of our scientific personnel, we believe his or her duties and responsibilities can easily be shifted to other scientific personnel in similar positions. Customers During the Track Record Period, we derived revenues only from the product distributor in China in connection with our product sales, from Celgene in connection with our strategic collaboration for tislelizumab entered into in 2017 and from Merck KGaA, Darmstadt Germany in connection with our collaboration for pamiparib and lifirafenib. During the year ended December 31, 2017, we had only three customers. We generated 90.0% of our revenues from upfront license fees, reimbursed research and development expenses and milestone payments from our strategic collaboration with Celgene, 9.6% from our product distributor in China in connection with the sales of our drugs licensed from Celgene and 0.4% from Merck KGaA, Darmstadt Germany in connection with our collaboration for pamiparib and lifirafenib. During the year ended December 31, 2016, 100% of our revenues were generated in connection with our collaboration agreements with Merck KGaA, Darmstadt Germany for pamiparib and lifirafenib. See “—Collaboration Agreements” for further details of our collaborations with Celgene and Merck KGaA, Darmstadt Germany. As of the Latest Practicable Date, none of our Directors or any Shareholder, who, to the knowledge of our Directors, owns more than 5% of our issued share capital immediately following completion of the [REDACTED] (but without taking into account the exercise of the [REDACTED]) nor any of their respective associates had any interest in any of our five largest customers.

Raw Materials and Suppliers Raw materials We currently have a facility that may be used as our clinical-scale manufacturing and processing facility. We are also building manufacturing facilities in China. We obtain raw materials for our manufacturing activities from multiple suppliers who we believe have sufficient capacity to meet our demands. We have not yet caused our drug candidates to be manufactured or processed on a commercial scale. Raw materials/starting materials used in manufacturing at our facilities in Suzhou include active pharmaceutical ingredients custom-made by our CROs and excipients, which are commercially available from well-known vendors that meet the requirements of the relevant regulatory agencies. The core raw material to be used in manufacturing at our Guangzhou facility under construction is expected to be a genetically modified cell line that we co-developed and licensed from Boehringer Ingelheim. We typically order raw materials on a purchase order basis and do not enter into long-term dedicated capacity or minimum supply arrangements. We pay for our purchases of raw materials on credit. Credit periods granted to us by our suppliers generally range from 30 to 60 days. Our suppliers are generally not responsible for the defects of our finished products. CROs In line with industry practice, we also engaged certain CROs to conduct preclinical and clinical research as well as clinical trials during the two years ended December 31, 2016 and 2017. We select CROs based on various factors, including their quality, reputation and research experience. Our largest five CROs are all leading global or Chinese CROs. They provide drug discovery and development services to pharmaceutical, biotechnology, medical device, government and academic organizations in China or throughout the world. We generally enter into master contract services agreements with the CROs we engage, which include a statement of work specifying the terms of services provided by CROs. Key terms of an agreement that we typically enter into with a CRO are summarized as follows: •Services. The CRO provides preclinical and clinical research services or clinical trial services as specified in the statement of work. •Term. The CRO is required to provide the services within the prescribed time limit. •Payments. We are required to make payments to the CRO in accordance with the payment schedule set forth in the statement of work. •Intellectual property rights. All intellectual property rights arising from or made in the performance of the services will generally be owned by us.

•Audit. The service provider shall allow us to inspect their facilities. •Insurance. During the term of the agreement and for a period of at least two (2) years after termination or expiration of the agreement, the service provider will maintain certain minimum insurance and name us as an additional insured. For the years ended December 31, 2016 and 2017, purchases from our five largest suppliers were approximately US$50.3 million and US$113.6 million in terms of expenses, accounting for approximately 73% and 62% of our total purchases, respectively. During the same period, purchases from our largest supplier were approximately US$14.9 million and US$35.1 million, respectively, in terms of expenses, accounting for approximately 22% and 19% of our total purchases, respectively. As of the Latest Practicable Date, none of our Directors or any Shareholder, who to the knowledge of our Directors, owns more than 5% of our issued share capital immediately following completion of the [REDACTED] (but without taking into account the exercise of the [REDACTED]) nor any of their respective associates had any interest in any of our five largest suppliers. Production We have an approximately 11,000 square meter manufacturing facility in Suzhou, China, where we produce small molecule and biologics drug candidates for clinical supply and which we plan to use for commercial supply of our small molecule drug candidates, if approved. This facility consists of one oral-solid-dosage production line for small molecule drug products and one pilot plant for monoclonal antibody drug substances. In January 2018, the facility received a manufacturing license from Jiangsu Food and Drug Administration, which is required for the commercial manufacture of zanubrutinib in China following NDA approval. In addition, we have formed a joint venture with Guangzhou GET Technology Development Co., Ltd., an affiliate of Guangzhou Development District, to build a 24,000-liter commercial-scale biologics manufacturing facility in Guangzhou, China. Over US$300 million in funding has been committed for the construction of the 100,000 square meter manufacturing site. We have contracted with General Electric for the purchase of its state-of-the-art KuBioM prefabricated biomanufacturing equipment and commenced construction in 2017. We expect the first phase of the facility to be completed in 2019. We also have an approximately 140 square meter manufacturing facility at our research and development facilities in Beijing, China, which produces preclinical and clinical trial materials for some of our small molecule drug candidates. Manufacturing is subject to extensive regulations that impose various procedural and documentation requirements governing recordkeeping, manufacturing processes and controls, personnel, quality control and quality assurance, among others. Our manufacturing facilities and the contract manufacturing organizations we use to manufacture our drugs and drug candidates operate under cGMP conditions. cGMP are regulatory requirements for the production of pharmaceuticals that will be used in humans.

CMOs We outsource to a limited number of external contract manufacturers the production of some drug substances and drug products, and we expect to continue to do so to meet the preclinical, clinical and potential commercial requirements of our drugs and drug candidates. By outsourcing our manufacturing activities, we can increase our focus on core areas of competence such as drug candidate development, commercialization and research. We have adopted procedures to ensure that the production qualifications, facilities and processes of our third-party outsourced suppliers comply with the relevant regulatory requirements and our internal guidelines. We select our third-party suppliers carefully by taking into account a number of factors, including their qualifications, relevant expertise, production capacity, geographic proximity, reputation, track record, product quality, reliability in meeting delivery schedules, and terms offered by such third-party outsourced suppliers. As of the Latest Practicable Date, we had engaged approximately 30 outsourced suppliers, who are Independent Third Parties and most of them had established business relationships with us for more than three years. We have framework agreements with most of our external service providers, under which they generally provide services to us on a short-term and project-by-project basis. For example, we have an agreement with a contract manufacturer for clinical supply of zanubrutinib and expect to enter into a commercial supply agreement for zanubrutinib in the future. In addition, in January 2018, we entered into a commercial supply agreement with Boehringer Ingelheim Biopharmaceuticals (China) Ltd., or Boehringer Ingelheim, for our investigational anti-PD-1 antibody therapy, tislelizumab, which will be manufactured at Boehringer Ingelheim’s facility in Shanghai, China as part of a MAH trial project pioneered by us and Boehringer Ingelheim. We believe the MAH status will be maintained after the expiration of the MAH pilot program in November 2018, based on confirmation from the relevant governmental authority, and therefore we believe that the expiration of the MAH pilot program will not impact our drug candidates. Under the terms of the commercial supply agreement, Boehringer Ingelheim will manufacture tislelizumab in China under an exclusive multi-year arrangement, with contract extension possible. In addition, we also obtained certain preferred rights for future capacity expansion by Boehringer Ingelheim in China. For our commercial products licensed from Celgene, we rely on Celgene and its contract manufacturers outside of China for the supply of these drugs. Agreements with outsourced suppliers generally set out terms, including product quality or service details, technical standards or methods, delivery terms, agreed price and payment, and product inspection and acceptance criteria. We are general allowed to return any products that fail to meet our quality standards. Our outsourced suppliers procure raw materials themselves. Typically, outsourced suppliers request settlement of payment within 30 days from the date of invoice. Either party may terminate the agreements by serving notice to the other party under certain circumstances. Sales and Marketing As of the Latest Practicable Date, we had no internally-developed products approved for commercial sale. In connection with our strategic collaboration with Celgene, we were granted an exclusive license in China, excluding Hong Kong, Macau and Taiwan, to commercialize Celgene’s approved cancer therapies, ABRAXANE®, REVLIMID® and VIDAZA®, and Celgene’s investigational

agent avadomide (CC-122) in clinical development. We rely on an independent third-party distributor to sell these drugs. As of the Latest Practicable Date, we had sold our products to one distributor in China. See “Risk Factors—If we fail to maintain an effective distribution channel for our products, our business and sales of the relevant products could be adversely affected.” We selected our distributor based on its business qualifications and marketing capabilities, such as distribution network coverage, quality, number of personnel, cash flow conditions, creditworthiness, logistics, compliance standard and past performance, and its capacities in customer management. As of the Latest Practicable Date, we were not aware of any potential abuses or improper use of our name by our distributor which could adversely affect our reputation, business operation or financial condition. We have entered into a written distribution agreement with our distributor. The principal terms are as follows: DurationThe distribution agreement will remain effective unless terminated by either party upon six months’ prior written notice. Geographic or other exclusivity Our distributor shall not sell or otherwise distribute the products outside the PRC, unless otherwise agreed by us in writing. We grant our distributor a non-sublicensable, non-transferable and non-assignable: •non-exclusive limited right to use the know-how and other confidential information in the PRC. •exclusive right to sell the commercial pack in the PRC. The rights and obligations of parties involved We offer rebates to our distributor, consistent with pharmaceutical industry practice. We retain no ownership control over the products sold to our distributor, and all significant risks and rewards associated with the products are generally transferred to the distributor upon delivery to and acceptance by the distributor. Sales and pricing policies Our distributor retains the discretion to determine the retail prices with reference to local market conditions, competition and customer demand in the regions where it operates, whether greater or lesser than any prices listed, referred or charged by us. Obsolete stock arrangementsThere is no obsolete stock arrangements condition. Goods return arrangementsThere is no goods return arrangements condition.

Sales and expansion targetsBased on sale forecasts we provide, our distributor shall provide its required amounts of the products to us. Sales and inventory reports and estimates Our distributor shall provide to us daily and monthly reports containing full details about the inventory, forecasts, shipping and, returns of the products. Our distributor shall also provide to us daily and monthly reports containing full details about the local sales of the products. Any minimum purchase amounts There is no binding minimum purchase condition. Payment and credit termsCredit term is generally 90 days following the invoice date. Conditions for terminating and renewing the agreements Either party has the right to terminate the agreement on a product-by-product basis with immediate effect upon written notice, if the other party breaches any of the material provisions in the agreement applicable to it and fails to rectify such breach within 30 days of written notice from the other party. Use of the trademark Our distributor shall have a non-sublicensable, non-transferable, non-assignable and non-exclusive right to use our trademark for selling our products in the PRC. The use of trademark shall be subject to our prior written approval. Our distributor shall not use the trademark within the PRC for any other product and shall use the trademark only for the purpose of selling our products in the PRC under the agreement. We rely on our internal sales team for the sales and marketing of our products. As of July 20, 2018, our commercial team consisted of 285 sales and marketing personnel. In anticipation of our business expansion and as our internally-developed drugs become available for sale, if approved, we plan to further expand our sales and marketing force in the next few years. We also actively attend trade shows, symposia, conventions, seminars and other notable events to promote our brand at the forefront of the industry. We frequently conduct technical seminars at well-recognized academic institutes and pharmaceutical companies to promote our products. In China, prices of pharmaceutical products are regulated by the government to ensure that drugs are offered at affordable prices. In June 2015, the Chinese government abolished the 15-year-old government-led pricing system for drugs, and lifted the maximum retail price requirement for most drugs, including drugs reimbursed by government medical insurance funds, patented drugs, and some other drugs. The government regulates prices mainly by establishing a consolidated procurement mechanism, restructuring medical insurance reimbursement standards and strengthening regulation of medical and pricing practices. See the section titled “Regulations—PRC Regulation—PRC Drug Regulation—Post-Marketing Surveillance—Government price controls” for further details on pricing regulations.

Our commercial products need to go through the centralized procurement process in the form of public tenders operated by provincial-level government agencies, in order to be commercially available at public medical institutions owned by the government or owned by state-owned or controlled enterprises. Assessment of the bids takes a number of factors into consideration, including but not limited to bid price, product quality, clinical effectiveness, product safety, level of technology, qualifications and reputation of the manufacturer, after-sale services and innovation. As a result, the prices of our commercial products are affected by the bidding process. In addition, in order for our commercial products to be included in the NDRL and critical illness insurance reimbursement listings, we are subject to price negotiation with the Ministry of Human Resources and Social Security and the relevant authorities at provincial level. As of July 18, 2018, REVLIMID® is included in the NDRL and ABRAXANE® is included in various provincial drug or critical illness insurance reimbursement listings. Employees As of July 20, 2018, we had a total of 1,335 full-time employees, which increased from 321 full-time employees as of December 31, 2016. Approximately 1,000 of our employees are based in China, and approximately 300 employees are based in the United States. The remaining employees are based in Australia and Switzerland. The following table sets out the breakdown of our full-time employees by function as of July 20, 2018: Function Number of employees We primarily recruit our employees through recruitment agencies, on-campus job fairs and online channels including our corporate website and social networking platforms, as well as industry referrals. We have adopted a training policy, pursuant to which management, technology and other training are regularly provided to our employees by internally sourced speakers or externally hired consultants. Our employees may also attend external trainings upon their supervisors’ approvals. As required under PRC regulations, we participate in housing fund and various employee social security plans that are organized by applicable local municipal and provincial governments, including housing, pension, medical, work-related injury and unemployment benefit plans, under which we make contributions at specified percentages of the salaries of our employees. We also purchase

commercial health and accidental insurance for our employees. We have granted performance-based cash bonuses to our executive officers. In addition, we have granted and plan to continue to grant share-based incentive awards to our employees in the future to incentivize their contributions to our growth and development. None of our employees are represented by a labor union or covered by a collective bargaining agreement. We have never experienced any employment-related work stoppages, and we consider our relations with our employees to be good. Insurance We maintain property damage and business interruption insurance coverage on our corporate, development, research and manufacturing facilities in amounts we believe are reasonable. We hold product liability coverage for our internally-develop drugs as well as public liability, and products and completed operations liability coverage for our commercial products. In addition, we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of or exposure to hazardous materials. We do not maintain key-man life insurance, or insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage, use or disposal of biological or hazardous. We believe the coverage of the insurance obtained by us is adequate and consistent with market practice in China and in the United States for our business and operations. During the Track Record Period, we did not make any material insurance claims in relation to our business. See “Risk Factors — Risks Related to Our Industry, Business and Operations — Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.” Properties As of the Latest Practicable Date, through BeiGene Guangzhou Manufacturing, which is a wholly owned subsidiary of our joint venture with Guangzhou Development District and its affiliate, GET, we jointly owned one property in Guangzhou. The property is being used for the construction of a 24,000 liter commercial-scale biologics manufacturing facility with a total of gross floor area of approximately 100,000 square meters. As of the Latest Practicable Date, we operated our businesses through 12 leased properties in Beijing, Shanghai, Guangzhou and Suzhou, China, and in Emeryville and San Mateo, California, Cambridge, Massachusetts, Fort Lee and Ridgefield, New Jersey and Basel, Switzerland. These properties are used for non-property activities as defined under Rule 5.01(2) of the Listing Rules and are principally used as office and manufacturing premises and for our business operations. We believe that there is sufficient supply of properties in China. Furthermore, even if we experience temporary interruption to our usage of any of our leased office, laboratory or manufacturing space, we believe that our employees can continue to perform the material aspects of their duties remotely given that our offices in other locations can adequately support the functioning of our business operations in areas where we experience temporary office space interruptions. Therefore, we do not rely on the existing leases for our business operations, and we do not believe a contingency relocation plan is required.

As of the Latest Practicable Date, our leased properties have a total gross floor area of approximately 44,639 square meters, and each leased property ranges from a gross floor area of approximately 73 square meters to 11,290 square meters. The relevant lease agreements have lease expiration dates ranging from August 21, 2018 to January 31, 2024. As of the Latest Practicable Date, the lessor of two of our leased properties in China had not provided us with valid title certificate, valid title certificate for commercial purpose or relevant authorization documents evidencing its rights to lease the property to us. As a result, this lease may not be valid, and there are risks that we may not be able to continue to use such property. Pursuant to the applicable PRC laws and regulations, property lease contracts must be registered with the local branch of the Ministry of Housing and Urban-Rural Development of the PRC. As of the Latest Practicable Date, except for one lease that is (located in Room 1506, Tower A, NO. 33, Zhongshan No.3 Road, Yuexiu District, Guangzhou) renewing the lease registration, we had not obtained any lease registration for the rest of the properties we leased in China, primarily due to the difficulty of procuring our lessors’ cooperation to register such leases. The registration of such leases will require the cooperation of our lessors. We will take all practicable and reasonable steps to ensure that the unregistered leases are registered. Our PRC Legal Advisor has advised us that the lack of registration of the lease contracts will not affect the validity of the lease agreements under PRC laws. Health, Safety and Environmental Matters We and third parties, such as our CROs and CMOs, are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. During the Track Record Period and up to the Latest Practicable Date, we had not been subject to any fines or other penalties due to material non-compliance with health, safety or environmental regulations. Legal Proceedings and Compliance From time to time we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. As of the Latest Practicable Date, we were not aware of any current, pending or threatened material litigation arbitration proceedings or administrative proceedings against us that could have a material adverse effect on our business, results of operations, financial condition or cash flows. On June 8, 2017, Nasdaq notified us that the resignation from our Board of Ke Tang, one of our audit committee members caused us to no longer comply with Nasdaq’s audit committee requirements in Nasdaq listing rule 5605, which require that audit committees have at least three members. Ke Tang had served as a member of the Board since October 2014, and his resignation was due to his decision not to stand for re-election to the Board when his term expired at our annual general meeting of

shareholders held in June 2017. This decision not to stand for re-election did not involve any disagreement with us on any matter relating to our operations, policies or practices. Under the Nasdaq rules, we had a cure period in order to regain compliance with this rule until the earlier of our next annual general shareholders’ meeting or June 1, 2018. Effective April 1, 2018, our Board appointed Mr. Timothy Chen, one of our existing independent directors, to serve as a member of the audit committee, resulting in us regaining compliance with the Nasdaq’s audit committee requirements in the Nasdaq listing rule 5605. Risk Management and Internal Control We have devoted ourselves to establishing and maintaining risk management and internal control systems consisting of policies and procedures that we consider to be appropriate for our business operations, and we are dedicated to continuously improving these systems. We have adopted and implemented comprehensive risk management policies in various aspects. Financial Reporting Risk Management As a public company in the United States, we are subject to the Sarbanes-Oxley Act, together with rules implemented by the U.S. Securities and Exchange Commission, or the SEC, and applicable market regulators. The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal control for financial reporting and disclosure controls and procedures. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. generally accepted accounting principles. Management is responsible for establishing and maintaining adequate internal control over our financial reporting process, and the audit committee oversees our financial reporting process on behalf of the Board. We perform system and process evaluations and testing of our internal control over financial reporting based on the framework in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission, in order to allow management to report on the effectiveness of our internal control over financial reporting and describe any material weakness in internal control over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act. The effectiveness of our internal control over financial reporting is also tested by our independent registered public accounting firm on an annual basis. Information System Risk Management In the ordinary course of our business, we collect and store sensitive data, including, among other things, legally protected patient health information, personally identifiable information about our employees, intellectual property, and proprietary business information. We manage and maintain our applications and data utilizing on-site systems and outsourced vendors. These applications and data encompass a wide variety of business critical information including research and development information, commercial information and business and financial information. We have implemented

relevant internal procedures and controls to ensure that such sensitive data is protected and that leakage and loss of such data is avoided. During the Track Record Period and up to the Latest Practicable Date, we do not believe that we have experienced any material information leakage or loss of sensitive data. Human Resources Risk Management We provide regular and specialized training tailored to the needs of our employees in different departments. We regularly organize internal training sessions conducted by senior employees or outside consultants on topics of interest. The human resources team, run by senior leaders and experienced human resource professionals, create, schedule and deliver the training. The long term goal is to further increase the number of trainings available to all employees as well as measure the success of the trainings. In China, we have in place an employee handbook approved by our management and distributed to all our employees, which contains internal rules and guidelines regarding best commercial practice, work ethics, fraud prevention mechanism, negligence and corruption. A similar handbook for the U.S. employee base is in production. We also have in place an FCPA Policy to safeguard against any corruption within our company. The policy explains potential corruption conducts and our anti-corruption measures. We make our internal reporting channel open and available for our staff to report any corruption acts, and our staff can also make anonymous reports to our internal audit department. Our internal audit department is responsible for investigating the reported incidents and taking appropriate measures. Investment Risk Management and Treasury Policy With our surplus cash on hand, we make short-term investments comprised primarily of U.S. treasury securities, U.S. agency securities and time deposits with original maturities between three and twelve months. The primary objective of short-term investments is to preserve principle, provide liquidity and maximize income without significant increasing risk. Our investment decisions are made on a case-by-case basis and after due and carefully consideration of a number of factors, including but not limited to the market conditions, the anticipated investment conditions, the investment costs, the duration of the investment and expected benefit and potential loss of the investment. Our finance department, under the supervision of our Chief Financial Officer, is responsible for managing our short-term investment activities. Before making a proposal to invest in wealth management products, our financial department must assess our cash flow and operational needs and capital expenditures. We operate under a Board approved investment policy which governs the investment of our funds. The investment policy is reviewed annually by the Board and is circulated to the investment advisors to ensure compliance of investments. Our investments to date have been limited to U.S. Treasury securities, U.S. agency securities, and time deposits at reputable banks. Any deviations from the investment policy, would require consent by the Board. There have been no cases of deviation from our investment policy to date.

In assessing a proposal to invest in wealth management products, a number of criteria must be met, including but not limited to: •investments in high risk products being prohibited; •the primary objectives of investment activities are safety, liquidity and reasonable yield; •the proposed investment must not interfere with our business operation or capital expenditures; and •the wealth management products should be issued by a reputable bank. We believe that our internal policies regarding investment in wealth management products and the related risk management mechanism are adequate. We may make investments in wealth management products that meet the above criteria, after consultation and approval by our Board, as part of our treasury management where we believe it is prudent to do so after the [REDACTED]. Audit Committee Experience and Qualification and Board Oversight We have established an audit committee to review the adequacy of our internal control over financial reporting to ensure that our internal control system is effective in identifying, managing and mitigating risks involved in our business operations. The audit committee consists of three members, namely Thomas Malley, Qingqing Yi and Timothy Chen. Each of our audit committee members is an independent non-executive director. Thomas Malley is the chairman of the audit committee. For the professional qualifications and experiences of the members of our audit committee, see “Directors and Senior Management” in this [REDACTED]. We also maintain an internal audit department which is responsible for reviewing the effectiveness of internal control and reporting to the audit committee on any issues identified. Ongoing Measures to Monitor the Implementation of Risk Management Policies Our audit committee, internal audit department and management together monitor the implementation of our risk management policies on an ongoing basis to ensure our policies and implementation are effective and sufficient. Licenses and Permits As of the Latest Practicable Date, we believe that we have obtained all requisite licenses, approvals and permits from relevant authorities that are material to our operations. The following table sets out a list of material licenses and permits currently held by us: No.EntityName of the LicenseExpiry Date 1.BeiGene (Suzhou) Co., Ltd.Drug Production License2020/12/31

Awards and Recognition We have received recognition for our research and development achievements and our global collaborations. Some of the significant awards and recognition we have received are set forth below. Award/RecognitionAward YearAwarding Institution/Authority Deal of the Year: BeiGene/Celgene collaboration ............................................. 2017The BayHelix Group Leading Innovative Enterprise - Beijing Bio-pharmaceutical Industry Leaping Development Project (G20 Project) ......... 2016Beijing Municipal Science and Technology Commission Alliance of the Year: BeiGene Ltd./Merck KGaA, for a global licensing, co-development and commercialization agreement for BeiGene-283, a second-generation BRAF inhibitor for the treatment of cancer. 2013The BayHelix Group and Elsevier Business Intelligence
