Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10‑K
☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended: December 31, 2020
☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission file number 001-38118
DERMTECH, INC.
(Exact name of registrant as specified in its charter)
Delaware (State or other jurisdiction of incorporation or organization) | 84-2870849 (IRS Employer Identification No.) |
| |
11099 N. Torrey Pines Road, Suite 100 La Jolla, CA | 92037 (Zip Code) |
Registrant’s telephone number, including area code: (858) 450‑4222
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
Common Stock, par value $0.0001 per share | DMTK | The Nasdaq Capital Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well‑known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such report(s), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S‑T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non‑accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b‑2 of the Exchange Acts.
Large accelerated filer | | ☐ | | Accelerated filer | | ☐ |
| | | | | | |
Non-accelerated filer | | ☒ | | Smaller reporting company | | ☒ |
| | | | | | |
| | | | Emerging growth company | | ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b‑2 of the Act). Yes ☐ No ☒
The aggregate market value of the registrant’s common stock, $0.0001 par value, held by non‑affiliates of the registrant as of the last business day of the registrant’s most recently completed second fiscal quarter was approximately $150,521,679 (based on the closing price of the registrant’s common stock on June 30, 2020 of $13.23 per share).
The number of shares outstanding of the registrant’s common stock, $0.0001 par value as of March 1, 2021 was 28,755,668.
DOCUMENTS INCORPORATED BY REFERENCE
The registrant intends to file a definitive proxy statement pursuant to Regulation 14A within 120 days after the end of the fiscal year ended December 31, 2020. Portions of such proxy statement are incorporated by reference into Part III of this Form 10‑K.
Table of Contents
DERMTECH, INC
ANNUAL REPORT ON FORM 10‑K
YEAR ENDED DECEMBER 31, 2020
TABLE OF CONTENTS
i
Table of Contents
Special Note Regarding Forward-Looking Statements
This report contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. Forward-looking statements are statements other than historical facts and relate to future events or circumstances or our future performance, and they are based on our current assumptions, expectations and beliefs concerning future developments and their potential effect on our business. Words such as, but not limited to “anticipate,” “aim,” “believe,” “contemplate,” “continue,” “could,” “design,” “estimate,” “expect,” “intend,” “may,” “might,” “plan,” “possible,” “potential,” “predict,” “pro forma,” “project,” “seek,” “should,” “suggest,” “strategy,” “target,” “will,” “would,” and similar expressions or variations thereof are intended to identify forward-looking statements, but are not deemed to represent an all-inclusive means of identifying forward-looking statements as denoted in this report. These statements include, among other things, statements regarding:
| • | our ability to attain profitability; |
| • | our estimates regarding our future performance, including without limitation estimates of potential future revenues; |
| • | our ability to maintain commercial reimbursement for our tests; |
| • | our ability to efficiently bill for and collect revenue resulting from our tests; |
| • | our anticipated need to raise additional capital to fund our operations, commercialize our products, and expand our operations; |
| • | our ability to market and sell our tests to physicians and other clinical practitioners; |
| • | our ability to continue to develop our existing test and develop and commercialize additional novel tests; |
| • | our dependence on third parties for the manufacture of our products; |
| • | our ability to meet market demand for our current and planned future tests; |
| • | our reliance on our sole laboratory facility and the harm that may result if this facility became damaged or inoperable; |
| • | our ability to compete with our competitors and their competing products; |
| • | the importance of our executive management team; |
| • | our ability to retain and recruit key personnel; |
| • | our dependence on third parties for the supply of our laboratory substances, equipment and other materials; |
| • | the potential for us to incur substantial costs resulting from product liability lawsuits against us and the potential for these lawsuits to cause us to suspend sales of our products; |
| • | the possibility that a third party may claim we have infringed or misappropriated our intellectual property rights and that we may incur substantial costs and be required to devote substantial time defending against these claims; |
| • | the potential consequences of our expanding our operations internationally; |
| • | our ability to continue to comply with applicable privacy laws and protect confidential information from breaches; |
| • | how changes in federal health care policy could increase our costs, decrease our revenues and impact sales of and reimbursement for our tests; |
| • | our ability to continue to comply with federal and local laws concerning our business and operations and the consequences resulting from our failure to comply with such laws; |
| • | the possibility that we may be required to conduct additional clinical studies or trials for our tests and the consequences resulting from the delay in obtaining necessary regulatory approvals; |
| • | the harm resulting from the potential loss, suspension, or other restriction on one or more of our licenses, permits, certifications or accreditations, or the imposition of a fine or penalty on us under federal, state, or foreign laws; |
| • | our ability to maintain and our intellectual property protections; |
| • | how recent and potential future changes in tax policy could negatively impact our business and financial condition; |
| • | how recent and potential future changes in healthcare policy could negatively impact our business and financial condition; |
1
Table of Contents
| • | our ability to maintain Nasdaq listing; and |
| • | our ability to manage the increased expenses and administrative burdens as a public company. |
Although forward-looking statements in this report reflect the good faith judgment of our management, such statements can only be based on facts and factors currently known by us. Consequently, forward-looking statements are inherently subject to risks and uncertainties and actual results and outcomes may differ materially from the results and outcomes discussed in or anticipated by the forward-looking statements. Factors that could cause or contribute to such differences in results and outcomes include, without limitation, those specifically addressed under the heading “Risk Factors” below, as well as those discussed elsewhere in this report. Readers are urged not to place undue reliance on these forward-looking statements, which speak only as of the date of this report. We file reports with the Securities and Exchange Commission, or the SEC, and our electronic filings with the SEC (including our quarterly reports on Form 10-Q and current reports on Form 8-K, and any amendments to these reports) are available free of charge on the SEC’s website at http://www.sec.gov.
We undertake no obligation to revise or update any forward-looking statements in order to reflect any event or circumstance that may arise after the date of this report, except as required by law. Readers are urged to carefully review and consider the various disclosures made throughout the entirety of this report, which are designed to advise interested parties of the risks and factors that may affect our business, financial condition and results of operations. We qualify all of our forward-looking statements by this special note.
2
Table of Contents
PART I
Item 1. Business
Unless specifically noted otherwise, as used throughout this Business section, “we,” “our,” or “us” refers to the business, operations and financial results of DermTech Operations prior to, and the Company and its subsidiaries subsequent to, the completion of the Business Combination as the context requires. “Constellation” refers to the Company prior to the completion of the Business Combination.
Business Overview
We are an emerging growth molecular diagnostic company developing and marketing novel non-invasive genomics tests to aid in the diagnosis and management of various skin conditions, including skin cancer, inflammatory diseases, and aging-related conditions. Our technology provides a highly accurate alternative to surgical biopsy, minimizing patient discomfort, scarring, and risk of infection, while maximizing convenience. Our scalable genomics assays have been designed to work with a proprietary “adhesive patch skin sampling kit” utilizing “Smart StickersTM” that provide a tissue sample for analysis non-invasively.
We are initially commercializing tests that will address unmet needs in the diagnostic pathway of pigmented skin lesions, such as moles or dark colored skin spots. Our current test facilitates the clinical assessment of pigmented skin lesions for melanoma. We have initially marketed this test directly to a concentrated group of dermatology clinicians. The simple application of the Smart Stickers to collect a sample may allow us to eventually market the test to primary care physicians and expand our efforts through telemedicine channels. We process our tests in a high complexity molecular laboratory that is certified under the Clinical Laboratory Improvement Amendments of 1988 (“CLIA”), College of American Pathologists (“CAP”) accredited and New York licensed. We also provide laboratory services to several pharmaceutical companies that access our technology on a contract basis within their clinical trials to better advance new drugs. We have a history of net losses since our inception.

Business Combination, Reverse Split and Domestication
On August 29, 2019, the Company, formerly known as Constellation Alpha Capital Corp., or Constellation, and DermTech Operations, Inc., formerly known as DermTech, Inc., or DermTech Operations, consummated the transactions contemplated by the Agreement and Plan of Merger, dated as of May 29, 2019, by and among the Company, DT Merger Sub, Inc., or Merger Sub, and DermTech Operations. We refer to this agreement, as amended by that certain First Amendment to Agreement and Plan of Merger dated as of August 1, 2019, as the Merger Agreement. Pursuant to the Merger Agreement, Merger Sub merged with and into DermTech Operations, with DermTech Operations surviving as our wholly owned subsidiary. We refer to this transaction as the Business Combination. In connection with and two days prior to the completion of the Business Combination, Constellation re-domiciled out of the British Virgin Islands and continued as a company incorporated in the State of Delaware.
On August 29, 2019, immediately following the completion of the Business Combination, we amended and restated our certificate of incorporation, or the Amended and Restated Certificate of Incorporation, to change the name of the Company to DermTech, Inc. Prior to the completion of the Business Combination, the Company was a shell company. Following the Business Combination, the business of DermTech Operations is the business of the Company.
On August 29, 2019, in connection with and immediately following the completion of the Business Combination, we filed a certificate of amendment, or the Certificate of Amendment, to the Amended and Restated Certificate of Incorporation to effect a one-for-two reverse stock split of our common stock on August 29, 2019, or the Reverse Stock Split. As a result of the Reverse Stock
3
Table of Contents
Split, the number of issued and outstanding shares of our common stock immediately prior to the Reverse Stock Split was reduced into a smaller number of shares, such that every two shares of our common stock held by a stockholder immediately prior to the Reverse Stock Split were combined and reclassified into one share of our common stock.
Our Business
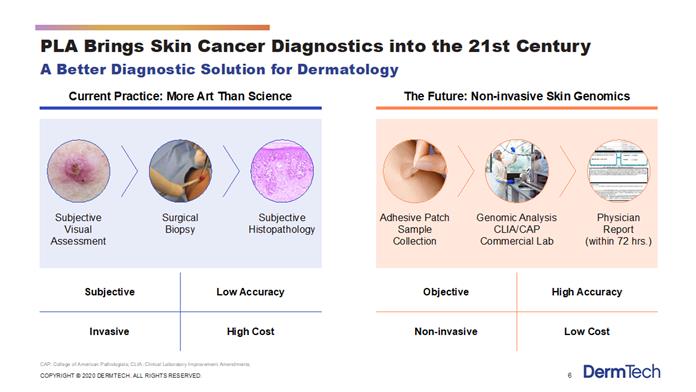
We are an emerging growth molecular diagnostic company developing and marketing novel non-invasive genomics tests that seek to transform the practice of dermatology and related fields. Our platform may change the diagnostic paradigm in dermatology from one that is subjective, invasive, less accurate and higher-cost, to one that is objective, non-invasive, more accurate and lower-cost. Our initial focus is skin cancer. We currently offer a test for the enhanced early detection of melanoma and are developing a product for non-melanoma skin cancer. We are also working on a product to assess skin cancer risk or UV damage. Our scalable genomics platform has been designed to work with a proprietary Smart Sticker adhesive patch sample collection kit that provides a skin sample collected easily and non-invasively, in contrast to the existing standard of care of using a scalpel to biopsy suspicious lesions. We also provide our services and technology platform on a contract basis to large pharmaceutical companies who use the technology in their clinical trials to test for the existence of genomic targets of various diseases and to measure the response of new drugs under development. We process our tests in a CLIA certified, CAP accredited and New York licensed commercial laboratory located in La Jolla, California that is licensed by the State of California and all states requiring out-of-state licensure. As described below, our technology platform is easy to use and integrates seamlessly into the current clinical diagnostic pathway by providing (i) simple and rapid tissue collection and shipping via standard express mail, (ii) sample processing via quantitative polymerase chain reaction, or qPCR, or other technologies and (iii) physician reporting within 48 to 72 hours. In addition, physicians can bill for their services using existing Evaluation and Management, or E&M, codes when our tests are ordered. Physicians can continue to bill for certain other procedures using existing Current Procedural Technology, or CPT, codes.
Dermatology is one of the largest medical markets in the United States. The skin cancer segment alone has over 15 million surgical diagnostic procedures performed each year in the United States, with an average annual spend of $8.1 billion from 2007 to 2011, according to the American Academy of Dermatology, or AAD. Current dermatologic diagnosis is primarily based on subjective visual assessments and subsequent surgical diagnostic procedures. This legacy paradigm is prone to error and results in a substantial number of unnecessary and invasive surgical procedures. Our platform provides a non-invasive alternative that minimizes patient discomfort, scarring, and risk of infection. Further, because our testing results utilize genomic analysis, we provide more accurate, objective diagnostic information than the currently prevailing diagnosis procedures. As described below, our first product, the Pigmented Lesion Assay (“PLA™”) has been demonstrated in a study published in JAMA Dermatology to lower the cost to diagnose melanoma while providing a more accurate and less invasive alternative to current methods based on assessing genomic atypia.
4
Table of Contents
The general genomic testing market is highly saturated with other genomic diagnostic tests that are primarily marketed to pathology and oncology specialists. We are the first company to offer non-invasive genomic tests to the clinical dermatology market. We believe our technology platform will transform the practice of dermatology and will expand the base of clinicians that can practice high quality precision dermatology (e.g., primary care clinicians). As healthcare delivery diverges to more convenient delivery models, such as pharmacy-based/retail clinics and telemedicine, we believe our platform will facilitate the migration of dermatologic care to these alternative models. We believe our platform may allow for expanded consumer-based sample collection shipped directly to our laboratory, positively impacting the ease of use and convenience of providing dermatologic care.
Our PLA assesses pigmented skin lesions, moles or dark skin spots for melanoma and enhances early detection. Of the approximate 4.0 million surgical biopsies performed each year on pigmented skin lesions, over 90% are negative for melanoma and represent avoidable surgical procedures. The PLA improves the assessment of pigmented lesions by reducing the probability of missing melanoma to less than 1.0% (versus approximately 11-17% with the existing standard of care) and by reducing the number of surgical biopsies required to diagnose melanoma by tenfold (from about 25:1 to about 2.5:1). In addition, the PLA improves the positive predictive value (“PPV”) approximately five-fold (from 3-4% with the current surgical techniques to 18.7% with PLA). In March 2019, Medicare’s MolDX program, administered by Palmetto GBA, or MolDX, which performs technology assessments for genomic tests, issued a favorable draft Local Coverage Determination (“LCD”), or Draft LCD, for our PLA. In October 2019, the AMA provided us with a CPT Proprietary Laboratory Analysis code for our PLA of 0089U, or the PLA Code. Pricing of $760 for the PLA Code was published on December 24, 2019 as part of the CMS Clinical Laboratory Fee Schedule, or CLFS, for 2020, which has been confirmed for 2021. The Medicare final LCD, or Final LCD, first made available on December 26, 2019 expanded the coverage proposal in the Draft LCD from one test per date of service to two tests per date of service, and allows clinicians to order our PLA if they have sufficient skill and experience to decide whether a pigmented lesion should be biopsied or assessed by our PLA. Our PLA became eligible for Medicare reimbursement effective on February 10, 2020. Our local Medicare Administrative Contractor, Noridian Healthcare Solutions, LLC, or Noridian, relies upon MolDX for technology assessments of genomic-based tests and has adopted the Final LCD issued by MolDX. Noridian has issued its own LCD announcing coverage of our PLA. Even though the effective date of Noridian’s LCD is June 7, 2020, Noridian began reimbursing us for our PLA as of February 10, 2020.
The performance of the PLA is supported by numerous investigational studies, which enrolled an aggregate of over 7,000 patients and yielded a total of 21 peer-reviewed publications in top-rated medical dermatology journals. A publication in JAMA Dermatology demonstrated that the PLA significantly lowers the cost to diagnose melanoma while providing a more accurate and less invasive alternative to current methods. The current AAD melanoma guidelines indicate that non-invasive gene expression testing can be used as a part of the initial clinical assessment for pigmented lesions. In January 2021, the National Comprehensive Cancer Network® (“NCCN”) updated their NCCN Clinical Practice Guidelines in Oncology (“NCCN Guidelines®”) for cutaneous melanoma to recommend that the use of pre-diagnostic noninvasive genomic patch testing may be helpful to guide biopsy decisions for cutaneous melanoma. The NCCN’s recommendation indicated that there is uniform consensus that the intervention is appropriate. In addition, an independent panel of melanoma experts has produced consensus recommendations for use of our PLA product. We believe the PLA can be used as an alternative for the majority of these surgical biopsy procedures, which could create a total existing market opportunity for melanoma greater than $3.0 billion per year. We have also received Health Canada clearance for use of our platform and have established a non-exclusive licensing partnership with DermTech Canada. We are working with this partner to secure reimbursement coverage with various Canadian provinces.
We initiated the commercialization of our PLA product in the second quarter of 2016. We currently market these tests directly to dermatologists in the United States with a team of approximately 40 sales representatives throughout the United States and plan to expand our team into more regions throughout the United States during 2021. With our recent Medicare coverage and growth of testing volume and physician users, we believe our test is being reviewed for coverage by key United States commercial payors, including Aetna, Cigna Corporation, Humana, United Healthcare, CareCore National, eviCore healthcare, and others. We believe we will achieve successful coverage outcomes from these efforts over the next 24 to 36 months, although no assurances can be given that any reimbursement coverage approvals will be obtained.
In the second quarter of 2018, we introduced our Nevome product, an adjunctive reflex test for the PLA. The Nevome test was used with histopathology to identify additional risk factors for melanoma and to confirm the diagnosis of melanoma in PLA positive tests, which are subjected to surgical biopsy. The Nevome test analyzed early-stage melanoma driver mutations in the v-Raf murine sarcoma viral oncogene homolog B (“BRAF”), neuroblastoma RAS viral oncogene homolog (“NRAS”) and telomerase reverse transcriptase (“TERT”) genes. The Nevome test utilized the same genomic material collected from the initial adhesive patch sample used for the PLA and did not require additional sampling. We discontinued our Nevome product in November 2020, and we expect it to be replaced with the introduction of our second-generation PLA test, PLAplusTM, that we plan to launch in 2021. The PLAplus improves the sensitivity of the melanoma test to up to 97%. The launch of the PLAplus has been postponed due to supply chain related impacts from COVID-19. The timing of the launch of the PLAplus will depend on when we receive the required inventory required to run PCR tests, which is affected by COVID‑19 pandemic. The PLAplus test will add TERT promoter mutation analyses to the current PLA gene expression test further enhancing the test’s performance, and we will no longer test for BRAF or NRAS genes, which we tested for in our Nevome product.
5
Table of Contents
We plan to expand our sales efforts as we obtain reimbursement coverage to provide sales coverage to a majority of over 13,000 healthcare professionals specializing in dermatology in the United States.
We believe the total annual United States market opportunity for our PLA and PLAplus tests exceeds $3.0 billion, and that the select annual worldwide market consisting of Australia, Europe, and Canada exceeds an additional $750 million.
Additional skin cancer product offerings, including for non-melanoma skin cancers (basal cell and squamous cell cancers), are currently under development. In the United States, approximately 12 million surgical biopsies are performed each year to diagnose approximately 5.0 million non-melanoma skin cancers. Many of the initial surgical procedures for these skin cancers are performed on cosmetically sensitive areas of the body, such as the face, neck and chest, creating significant demand for a non-invasive alternative. We believe the total market opportunity for our non-melanoma skin cancer products exceeds $3.0 billion in the United States and $1.0 billion in select world-wide markets.
We are also working on tests to facilitate the assessment of inflammatory skin diseases, such as atopic dermatitis and psoriasis, which will facilitate the appropriate diagnosis and treatment of these inflammatory diseases. The prevalence of atopic dermatitis in the United States is reported to be approximately 12% in children and 7.0% in adults with approximately 6.6 million patients having moderate-to-severe disease. The prevalence of psoriasis in the United States is approximately 2.2% with approximately 1.3 million patients having moderate to severe disease.
We also make our non-invasive molecular skin analysis platform available to pharmaceutical companies to facilitate the development of new targeted therapies in dermatology and cancer, including biologics. These partners use our platform and services to assess treatment response, monitor side effects and identify likely responders to the therapy under development. We have completed and have ongoing research collaborations with large pharmaceutical companies to facilitate their development of new targeted therapeutics in dermatology. We have initiated programs across the spectrum of pharmaceutical development stages from Phase 1 through Phase 3. We believe that some of these collaborations may lead to a complementary or companion diagnostic product for the pharmaceutical partner’s therapeutic candidate, if it reaches the commercial market. We have booked over $2.4 million of orders pursuant to research contracts in 2020, and many of these contracts are multi-year in length.
We offer our genomic tests through our CLIA certified and CAP accredited commercial laboratory located in La Jolla, California, which is licensed by the State of California and all states requiring out-of-state licensure. In the first quarter of 2018, we received our laboratory permit from the New York State Department of Health, the most rigorous licensing process for clinical diagnostic laboratories. We can scale our current facility to approximately 300,000 tests per year, with the ability to scale to over 1,000,000 tests per year with additional facility and capital investments.
Our sample collection technology maximizes collection of relevant tissue with minimal patient discomfort using adhesive patches. We have developed significant intellectual property and know-how around the use of adhesives for non-invasive biopsy and the transportation and handling of this type of sample. We have developed a proprietary process that allows us to extract genomic material from the patches with sufficient quality and quantity to perform gene expression, DNA mutation, transcriptomic analyses and other technologies. We believe our technology can be utilized to assess the microbiome of the skin with superior performance to existing methods that use swabs. The results of these efforts will allow us to introduce our sample collection technology to facilitate the diagnosis of a broad array of dermatologic conditions and other conditions where the skin serves as a surrogate target organ.
6
Table of Contents
Our Competitive Advantages
Enhancing early detection for superior patient care at a lower cost. The PLA is used to assess pigmented lesions that may harbor melanoma at the earliest stages (melanoma in situ or stage 1a), the most difficult lesions to diagnose. In our clinical studies, our PLA test has demonstrated a sensitivity of 91-95% and a specificity of 69-91% in differentiating these early-stage melanomas from non-melanoma using histopathology as the reference standard. This leads to a very high negative predictive value, or NPV, of greater than 99%, which is the probability our PLA test correctly ruled out melanoma. We completed a long-term follow-up study of the PLA that further confirmed the 99% NPV of the PLA by reevaluating and retesting lesions that were PLA negative 12 to 24 months prior to each subject’s enrollment in the study. We also completed a study that demonstrated that the PLA increases the positive predictive value, (“PPV”) for melanoma diagnosis by approximately fivefold, from 3-4% for the current pathway to 18.7% for the PLA. In addition, the PLA has demonstrated an approximate tenfold reduction in unnecessary surgical procedures, relative to the current visual assessment and histopathology standard of care. Such a reduction can result in significant cost savings for the health care system and reduces patient morbidity as compared to other diagnostic approaches. Table 1 below compares our PLA with other techniques and the existing standard of care for assessing early-stage melanoma in pigmented skin lesions.
| | Our PLA | | Visual Assessment & Pathology (Current Standard) |
Mechanism | | Tumor Biology | | Pattern Recognition |
Surgical Procedure Required | | No | | Yes |
Platform Technology | | Yes | | N/A |
Multiple Dermatologic Indications | | Yes | | Yes |
Physician Payment | | Yes | | Yes |
Simple Practice Integration | | Yes | | N/A |
Ease of Use | | Yes | | N/A |
Number Needed to Biopsy(1) | | 2.7 | | >25 |
Number Needed to Excise(2) | | 1.6 | | 5.2 |
Better Performance | | | | |
NPV(3) | | >99% | | >81-89% |
PPV(4) | | 18.7% | | 4% |
Sensitivity(5) | | 91-95% | | 65-84% |
Cost | | $760(6) | | $947 |
Capital Equipment | | No | | No |
Table 1. The data summarized above compares our PLA with the existing standard of care for assessing early-stage melanoma in pigmented skin lesions.
Footnotes to Table 1:
| (1) | Number of surgical biopsies required to diagnose one melanoma. |
| (2) | Number of wide excision surgical procedures per melanoma diagnosed. |
| (3) | NPV measures the probability that a negative result is truly negative. |
| (4) | PPV measures the probability that a positive result is truly positive. |
| (5) | Sensitivity measures the proportion of actual positives that are correctly identified as such. |
| (6) | Figure represents a projected United States reimbursed price, though this price has not yet been negotiated with major United States payors. Pricing of $760 for the PLA Code was published on December 24, 2019 as part of the CMS Laboratory Fee Schedule for 2020 and confirmed for 2021. The Medicare Final Coverage Decision was made available on December 26, 2019 and the PLA became eligible for Medicare reimbursement on February 10, 2020. |
Our technology platform has the potential to transform dermatologic practice. We are the first and only company to offer non-invasive genomic testing to clinicians that practice dermatology. Current dermatologic practice is based on subjective visual assessments of cellular change that are prone to inaccuracy and lead to invasive surgical procedures that drive unnecessary costs. Our technology platform seeks to dramatically transform this paradigm by enhancing early detection at the genome level where cancer
7
Table of Contents
begins providing non-invasive, objective, and more accurate information, thereby broadening the base of clinicians that can practice dermatology while also improving the performance of specialists.
Superior ease of use. Our non-invasive biopsy sample collection procedure can be performed in less than five minutes. All the necessary items, including adhesive patches, instructions, a marking pen for outlining, and a preaddressed and prepaid return shipping label, are contained in our kit. The collection procedure, when a clinician orders the test, can also be performed at the patient’s home with clinician guidance.
Simple integration into clinical practice. Our tests use an adhesive patch that replaces the scalpel traditionally used in the initial clinical assessment. Unlike other technologies, our platform does not require the installation and maintenance of capital equipment. The nursing support, documentation, specimen processing, and requisition post procedure are substantially similar to current practice. These issues are critical in a busy clinical practice where clinicians see patients every five to seven minutes.
Strong intellectual property protection. We have six issued United States patents, one of which is broadly directed to the use of an adhesive to collect samples containing RNA from the skin for analysis. In addition, we have been awarded patents on unique gene expression profiles and classifiers that differentiate melanoma from non-melanoma, one of which will not expire until 2029, and the other will not expire until 2030. Additional efforts to further expand our patent portfolio are ongoing and a number of provisional and non-provisional patent applications have been filed. We have also developed unique know-how and proprietary processes that allow us to extract sufficient quantities of low-quality genomic material from adhesive patch samples suitable for analysis.
Our Strategy
Our goal is to become the global leader in non-invasive genomics testing for dermatologic conditions. We believe our robust intellectual property portfolio, platform technology, first-to-market advantage, and groundbreaking research will facilitate the achievement of this goal. Specifically, we will focus on the following objectives:
Build a specialized sales force to introduce our products into the dermatology market. We intend to expand our existing direct specialty sales force as additional reimbursement coverage is achieved. Consistent with our current sales strategy, we will continue to recruit experienced sales representatives, primarily those from the dermatology sector who have existing physician relationships. We also plan to leverage this sales force by establishing distribution relationships with laboratory companies that do business with the clinical dermatologist or sell molecular tests.
Secure broad reimbursement coverage for our assays. We have targeted regional and national payors to secure favorable coverage decisions for the reimbursement of our tests. The PLA has completed the necessary analytical validity, clinical validity, and clinical utility studies that payors require molecular tests to undertake. We have also published a United States health economic impact study on the PLA in JAMA Dermatology, which shows that the PLA significantly reduces the relative cost to assess a pigmented lesion. The cost to fully adjudicate a pigmented lesion suspicious for melanoma is $947 in the United States. We believe the PLA could lead to cost savings of greater than $650 million per year in aggregate savings, based on approximately 4.0 million surgical biopsies performed per year to rule out melanoma, and assuming the PLA was to become the standard of care in the United States.
In March 2019, MolDX, which performs technology assessments for genomic tests, issued a favorable Draft LCD for the PLA. In late October 2019, the AMA provided us with the PLA Code. Pricing of $760 for the PLA Code was released on December 24, 2019 as part of the CLFS for 2020. The Final LCD, first made available on December 26, 2019, expanded the coverage proposal in the Draft LCD from one test per date of service to two tests per date of service, and allows clinicians to order our PLA if they have sufficient skill and experience to decide whether a pigmented lesion should be biopsied or assessed by our PLA. Our PLA became eligible for Medicare reimbursement on February 10, 2020. Our local Medicare Administrative Contractor, Noridian, relies upon MolDX for technology assessments of genomic-based tests and has adopted the Final LCD issued by MolDX. Noridian has issued its own LCD announcing coverage of our PLA. Even though the effective date of Noridian’s LCD is June 7, 2020, Noridian began reimbursing us for our PLA as of February 10, 2020.
In addition to our demonstrated clinical validity, clinical utility is the most important attribute of a test for establishing coverage policies with payors because it demonstrates how frequently physicians adhere to the recommendation of the test and the resulting improvement in clinical outcomes. In 2020, we completed and published our largest clinical utility study of the PLA based on real-world commercial usage. This most recent clinical utility study on 3,418 cases corroborates earlier utility studies and demonstrates that clinicians adhere to the recommendation of the PLA more than 98% of the time. Our test significantly reduces surgical procedures and improves the diagnostic pathway for pigmented lesion assessment. Lesions clinically suspicious for melanoma have negative PLA results in over 90% of cases, leading to an approximately 90% reduction in surgical biopsies in our 2020 study. In January of 2021, we published additional registry study data highlighting that PLA use enriches biopsied samples for melanoma almost 5-fold. We believe our body of clinical evidence and utility will lead to securing coverage policies from the major commercial payors over the next 24 to 36 months, although no assurances can be given that any reimbursement coverage approvals will be obtained.
8
Table of Contents
We have secured several contracts with major preferred provider networks, including Blue Shield of California, Blue Cross and Blue Shield of Texas, Blue Cross and Blue Shield of Illinois, Carefirst - BCBS of Maryland and Priority Healthcare of Michigan. We have submitted clinical and technology assessment packages to eviCore healthcare, LLC, which provides consultative services for payors. We are in direct discussion with several national commercial payors, including Aetna, Cigna Corporation, UnitedHealthcare, Humana and several independent Blue plans, all of which have the PLA currently under review.
Integrate our products into the standard of care. We conduct rigorous clinical research and basic science research and publish the results of this research in peer-reviewed journals. Overall, our research has yielded 21 publications in top peer-reviewed journals. The PLA’s performance is supported by over ten investigational studies, which enrolled an aggregate of over 7,000 patients. A study published in JAMA Dermatology demonstrated that the PLA significantly lowers the cost to diagnose melanoma while providing a more accurate and less invasive alternative to the current methods. Our research is frequently highlighted at clinical meetings and has several times been accepted for peer-reviewed late-breaking presentations at major medical society meetings.
The AAD melanoma guidelines updated every 5-7 years have indicated that non-invasive gene expression testing can be used as a part of the initial clinical assessment for pigmented lesions. In January 2021, NCCN recommended that there is uniform NCCN consensus to recognize the use of noninvasive genomic patch testing to help guide biopsy decisions for cutaneous melanoma, and it added the intervention to its NCCN Guidelines for cutaneous melanoma. In addition, an independent panel of melanoma experts produced consensus recommendations for use of our PLA product, which were published in 2019.
We have established an extensive board of over a dozen Key Opinion Leaders, or KOLs, in dermatology, including four former presidents of the AAD. These KOLs speak extensively about our technology platform and the PLA at various clinical and research meetings. In addition, these KOLs participate in our clinical studies and publish findings in peer-reviewed journals.
Establish alternate care delivery channels. We plan to expand our efforts in alternate care delivery channels including telemedicine, integrated primary care networks, and on-site and near-site employer health and retail clinics. These channels can help to democratize access to high quality dermatologic care, alleviate certain capacity limitations currently within dermatology and the related long lead times for dermatology specialist appointment availability, and improve the delivery of care to patients with ease of use, improved commute and wait times, and reduced patient fear that can accompany referrals to dermatology specialists.
Establish distribution partnerships for primary care. A substantial portion of dermatology is practiced in primary care. Based on the adoption progress we make within dermatology and integrated primary care networks, we plan to eventually access the primary care market more broadly by potentially establishing distribution relationships with companies that focus on this physician call point. An ideal partner would have several hundred sales professionals in the aggregate who access the primary care market, and ideally have experience selling genomic diagnostic products. Alternatively, we may plan to hire sales representatives to make direct calls to primary care offices.
Expand our product offerings. We have developed a platform that provides genomic analysis of the skin using a non-invasive adhesive patch platform as the sample collection method. This platform can be used to develop multiple products based on the same sample collection method, and it only requires different genomic markers to be assayed in our CLIA-certified laboratory. We are currently working to complete development of additional products, which will assess non-pigmented lesions for basal cell and squamous cell cancers. In addition, we are working to develop tests for inflammatory diseases of the skin.
Expand our marketing of research services to pharmaceutical companies. Our platform is used by several large pharmaceutical companies to facilitate their development of new targeted therapeutics in dermatology. Our PLA product helps identify biomarker treatment responses, track side effects, and identify patients that respond to the therapy. We plan to hire additional business development professionals to sell these services to the pharmaceutical industry. These efforts will include the participation in additional industry conferences and the presentation of our platform and data at additional medical conferences. Additionally, our collaborations with pharmaceutical partners may result in the introduction of complementary or companion diagnostic products for the partners’ therapeutic candidates that reach the commercial market.
Explore reference testing for large integrated dermatology networks and dermatopathology laboratories. Large dermatology practices with multiple clinics and generally more than 50 clinical professionals often have integrated dermatopathology and laboratory testing services for their clinics. For these situations and depending on federal and state regulations, we may plan to explore implementation of reference contracts, whereby the integrated laboratory will accession the PLA samples and bill for these samples, while paying us a contracted price. We estimate that 10-20% of our dermatology market opportunity may be accessed through this model.
9
Table of Contents
Market Opportunity – Skin Cancer
Melanoma is currently one of the fastest growing cancers and the subject of significant attention in the medical community. The incidence of melanoma has doubled since 1973. While there has been a 20% decline in cancer deaths overall since 1991, melanoma is one of three cancers facing increasing death rates. According to a study from the Mayo Clinic, the incidence of melanoma increased eightfold among women under 40 and fourfold among men under 40 from 1970 to 2009.
Melanoma is one of the deadliest forms of skin cancer. On average, melanoma causes more than one death every hour of every day of the year in the United States. The Skin Cancer Foundation projects that more than 7,000 people will die from melanoma in 2021. If diagnosed and removed early in its evolution, when confined to the outermost skin layer and deemed to be “in situ” (Stage 0), patients are expected to have a survival rate of almost 100%. Invasive melanomas that are thin and extend into the uppermost regions of the second skin layer (Stage 1) still have cure rates greater than 90%. However, once the cancer advances into the deeper layers of skin, the risk of it spreading to other parts of the body, or metastasis, and death increases. The table below depicts the survival rate of melanoma based on the stage of the cancer at initial diagnosis.
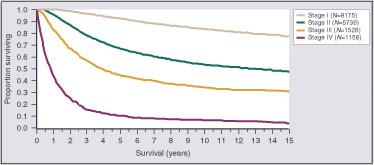
From Balch CM, Buzaid AC, Soong S-j et al: Final Version of the American Joint Committee on Cancer Staging Sysem for Cutaneous Melanoma. Journal of Clinical Oncology, August 2001.
An estimated 207,390 cases of melanoma will be diagnosed in the U.S. in 2021. Of those, it is estimated that 106,110 cases will be in situ (noninvasive), confined to the epidermis (the top layer of skin), and 101,280 cases will be invasive, penetrating the epidermis into the skin’s second layer (the dermis). On average, 25 surgical biopsies are performed per early-stage melanoma diagnosed, creating a total market opportunity of approximately 4.0 million surgical procedures per year. Outside the United States, the incidence of melanoma is highest in Western Europe, Australia, and Canada. We estimate that these select worldwide markets perform over 1.5 million surgical biopsies annually to diagnose approximately 75,000 melanomas, creating additional market opportunity that we believe exceeds $750 million per annum.
Over 5.4 million non-melanoma skin cancers (basal cell and squamous cell carcinomas) are diagnosed in the United States annually. The number of surgical biopsies needed to diagnose one non-melanoma skin cancer is approximately 2.5-3.0 among dermatologists and can be considerably higher when diagnosed by other clinicians such as nurse practitioners and primary care physicians. While these cancers are not as deadly as melanoma, they commonly occur on the face, head, neck, and other cosmetically sensitive areas, creating an important unmet medical need for a non-invasive alternative, and a potential market opportunity of approximately $3.0 billion in the United States per annum based on the approximately 10-12 million surgical biopsies performed to diagnosis of basal and squamous cell skin cancers.
Limitations of Current Melanoma Diagnostic Pathway
The estimated prevalence of pigmented lesions (moles) ranges from 2% to 8% in fair-skinned persons.
Pigmented lesions may be classified as clinically atypical by meeting one or more of the American Cancer Society’s ABCDE criteria, which includes Asymmetric, irregular Border, variegated or dark Color, Diameter greater than 6 mm, or Evolving mole. Atypical pigmented lesions are at risk for harboring melanoma. A meta-analysis of case-control studies found that the relative risk of melanoma is 1.45 in patients with one atypical mole vs. those with none, and this risk increases to 6.36 in those patients with five atypical moles. Management of atypical pigmented lesions involves ruling out melanoma via a visual assessment followed by surgical biopsy and histopathology. Ideally, when melanomas are identified, they are found at the earliest stages (melanoma in situ or stage 1a) when a high cure rate is possible by wide excision. Since a biopsy only partially removes a lesion for histopathologic analysis, early-stage melanomas diagnosed histopathologically from biopsy material are treated with follow-up wide excision procedures (generally with 0.5-1.0 cm margins).
10
Table of Contents
While the purpose of the visual assessment or surgical biopsy is to rule out melanoma, the poor performance metrics of this diagnostic pathway leads to a low NPV for early-stage disease (Table 2 below). This is related to the low specificity of the visual assessment (3-10%), which results in a high number of biopsies on benign atypical nevi. During histopathologic assessment, a small number of melanomas must be identified from this large pool of biopsied atypical nevi. However, there is significant overlap in the histopathologic diagnostic criteria between atypical nevi and early-stage melanoma, invariably leading to false negative diagnoses and a relatively low sensitivity (65-84%). Elmore et al. BMJ (2017) 357:j2183, concluded that the diagnosis of early stage melanoma was not accurate after finding that 35% of slide interpretations for melanoma in situ or stage 1a melanomas by 187 pathologists received a false negative diagnosis as benign. With the prevalence of early-stage melanoma in biopsied lesions at approximately 5%, the negative predictive value ranges from 75-89%.
Welch and colleagues’ most recent N Engl J Med article (2021, 384:72-79) points out that melanoma diagnoses have increased more than 6-fold over the last 40 years. The authors attribute this increase to more frequent enhanced screening, lower pathological thresholds to label the morphologic changes as cancer, and importantly heightened clinical awareness to biopsy pigmented lesions. However, the article fails to address the main limitations of the current care standard for evaluating pigmented lesions relies primarily on visual atypia to guide biopsy decisions. About 4 million pigmented lesions are biopsied in the US alone to diagnose fewer than 200,000 cutaneous melanomas (about 25 biopsies to detect 1 melanoma based on Anderson et al. JAMA Dermatol. (2018) 154(5):569-573. Using non-invasive assessment of genomic atypia offered by the PLA rather than visual atypia alone to guide pigmented lesion biopsy decisions reduces avoidable biopsies while missing fewer melanomas. Precision genomics is currently used in other areas of oncology and has changed the paradigm of treatment. Integrating PLA use and precision genomics to enhance early detection non-invasively into standard practice rather than performing fewer skin examinations appears to be a superior solution to the conundrum highlighted by Welch and colleagues.
According to several published papers, the real NPV of the visual assessment or surgical biopsy pathway is likely 80% to 85%. In a study by Malvehy et al., BJD (2014) 171:1099, 206 in situ and stage 1a (thickness less than 0.75 mm) melanomas were diagnosed with a sensitivity of 81% and a specificity of 10%. The prevalence of early melanoma in the study was about 10%, yielding an NPV of 83%. In addition, the current pathway using visual atypia to guide biopsy decisions suffers from a low PPV of approximately 4% for melanoma diagnosis. The addition of the PLA to the visual assessment by clinicians increased the PPV for a melanoma diagnosis by approximately five-fold to 18.7%.
| | Current Pathway | | | PLA | |
Test Purpose | | Rule-out melanoma | | | Rule-out melanoma | |
Type | | Surgical biopsy/ histopathology | | | Non-invasive gene expression | |
NPV | | 83% | | | 99% | |
Probability of Missed Mel | | 17% | | | 1% | |
Probability of Mel Diagnosis | | 4% | | | 18.7% | |
Number Needed to Biopsy | | 25 | | | 2.7 | |
Number Needed to Excise | | 5.2 | | | 1.6 | |
Cost per Lesion Tested | | $ | 947 | | | $ | 760 | |
Table 2. Data summarized above compares the key performance metrics of the PLA versus the current pathway (visual assessment and surgical biopsy/histopathology) for managing pigmented skin lesions.
This low NPV for the current pathway is accompanied by a high number of unnecessary surgical procedures, again driven by the poor specificity of the visual assessment. The number of surgical biopsies needed to identify one melanoma averages 25 and ranges from eight to greater than 30 depending on the clinical setting. Further, the histopathologic review of biopsied lesions is extremely limited with 2% or less of the lesion sectioned and evaluated, leaving doubt as to what may be occurring in the rest of the lesion. Consequently, lesions that have cellular atypia and positive margins are often clinically managed conservatively and subjected to full excisions with margins. However, only 0.2% to less than 1.0% of lesions with atypia and positive margins that undergo excision are diagnostically upgraded, most commonly to a higher level of atypia and rarely to melanoma in situ, and such excisions can be considered unnecessary. Approximately 5.2 excisions with margins are performed per melanoma identified, emphasizing how the current pathway of surgical biopsy and limited histopathology assessment leads to more complex and invasive excisions.
11
Table of Contents
Our Products
The PLA
The PLA is a gene expression test that enhances early detection of genomic atypia and helps rule out melanoma and the need for a surgical biopsy of atypical pigmented lesions. The performance of the PLA is supported by over ten investigational studies, which enrolled over 7,000 patients and yielded 21 peer-reviewed publications in top rated medical dermatology journals. Key studies and manuscripts are summarized in Table 3 below. The PLA is based on a new platform technology for non-invasive genomic testing of the skin, which allows the molecular analysis of samples collected from adhesive patches. In contrast to the current pathway, the PLA has a very high NPV (greater than 99%) and high sensitivity (91-95%), ensuring a very low probability of missing melanoma. The PLA’s high specificity (69-91%) effectively reduces the number of false positive samples undergoing histopathologic review. This improves the overall sensitivity of the pathway and greatly increases the NPV.
The PLA’s NPV is supported by a 12-month follow-up study of 734 patients, which demonstrated that no melanomas were missed in the 12-month period following initial testing. In the third quarter of 2019 we initiated the TRUST study, which further examined long-term follow up of lesions previously tested negative by the PLA, and incorporated repeat testing of the previously tested lesion. This study more definitively confirmed the high NPV of the PLA test in a real-world setting, and we announced those topline results in December 2020. Of the lesions evaluated by means of repeat testing with the PLA (n=302), none were found to have clinically obvious melanoma upon the subject’s return to the clinic, confirming the results of the initial chart review. Eighty-nine percent of these lesions were negative on repeat testing with the PLA and 11.2% were positive. Positive lesions were biopsied and subjected to a single read histopathologic review. One percent of lesions (n=3) that tested positive on repeat testing were diagnosed as Stage 0, in situ. Photographic review of the three Stage 0 cases identified changes in clinical appearance since the initial test. The pathology reports from the remaining biopsied lesions indicated a variety of non-melanoma diagnoses, including compound nevi with mild to moderate atypia. Given the early stage (in situ) of the melanomas detected on repeat testing, and length of time from the initial test (an average of 15 months), it is difficult to determine whether these melanomas evolved after the initial test or were present at the time of the initial test. In any case, the finding of three melanomas in a cohort of 302 lesions subjected to repeat testing further confirms an NPV of the PLA of at least 99.0% and is consistent with the results from the full long-term follow‑up cohort. These results exemplify how PLA repeat testing of lesions that may have evolved over time after the initial negative PLA test have potential to identify early-stage melanoma and benefit patients. TRUST study findings corroborating the PLA’s high NPV were complemented by most recent registry data on the PLA’s high PPV (Brouha et al., SKIN, January 2021, 5(1):13-18). This data show that 316 lesions clinically suspicious for melanoma that were biopsied based on guidance offered by genomic atypia (positive PLA test results rather than visual atypia alone) were enriched approximately five-fold for histopathologic features of melanoma.
In addition, the non-invasive sampling leads to a dramatic reduction in surgical biopsies and subsequent excisions. Consequently, our studies have shown that the number of surgical biopsies needed to find one melanoma using the PLA is markedly reduced by almost tenfold to approximately 2.7 and the number of excisions needed is reduced to 1.6. Our studies have shown that the PLA can reduce unnecessary surgical biopsies of lesions clinically suspicious for melanoma by 90%, which is consistent with a 2017 review of 18,715 biopsied pigmented lesions that found that approximately 90% of surgical biopsies to rule out melanoma are performed on pigmented lesions that are not melanoma. Non-invasive gene expression testing has been added to the most recent AAD melanoma guidelines as part of the initial clinical assessment for clinically concerning lesions and recommended by the NCCN Guidelines as of January 2021. In addition, an independent expert committee has developed and published consensus use criteria for the PLA.
In the second quarter of 2021, we plan to make our second-generation PLA test, PLAplus, commercially available. The commercial launch of the PLAplus has been postponed due to supply chain related impacts from COVID-19. The timing of the commercial launch of the PLAplus will depend on when we receive the required inventory required to run PCR tests, which is affected by the COVID‑19 pandemic. This second-generation test will add a TERT promoter mutation analysis to the current PLA gene expression test. TERT promoter mutations are associated with early-stage melanoma and our validation testing against driver mutations showed in two publications that it can increase the sensitivity of the PLA to 97% with only a minor impact on specificity. Several other independent academic investigators have also shown that TERT promoter mutations have a high sensitivity and specificity for melanoma detection. With the upcoming addition of TERT to the PLA test (PLAplus) we discontinued our Nevome product in November 2020, which was a reflex confirmatory test offered for PLA positive tests.
12
Table of Contents
Study | | Status | | Size (n) | | Publication |
Analytical Validation | | Complete | | 125 | | Yao Z et al. Analytical characteristics of a noninvasive gene expression assay for pigmented skin lesions. Assay Drug Dev Technol. 2016;14(6):355-363. |
Clinical Validation-Pathology | | Complete | | 555 | | Gerami P et al. Development and validation of a noninvasive 2-gene molecular assay for cutaneous melanoma. J Am Acad Dermatol. 2017;76(1):114-120.e2. |
Clinical Validation-Driver Mutations | | Complete | | 626 | | Ferris L et al. Noninvasive analysis of high-risk driver mutations and gene expression profiles in primary cutaneous melanoma. J Invest Dermatol. 2019; 139(5):1127-1134. |
Clinical Utility | | Complete | | 45 Derms | | Ferris L et al. Utility of a noninvasive 2-gene molecular assay for cutaneous melanoma and effect on the decision to biopsy. JAMA Dermatol. 2017;153(7):675-680. |
Real-World Clinical Utility | | Complete | | 381 | | Ferris L et al. Real-world performance and utility of a noninvasive gene expression assay to evaluate melanoma risk in pigmented lesions. Melanoma Res. 2018; 28(5):478-482. |
1-Year Follow Up | | Complete | | 734 | | Ferris L et al. Impact on clinical practice of a non-invasive gene expression melanoma rule-out test: 12-month follow-up of negative test results and utility data from a large US registry study. Dermatology Online Journal. 2019; 25(5). |
Real-World Utility Registry | | Complete | | 1575 | | Ferris L et al. Impact on clinical practice of a non-invasive gene expression melanoma rule-out test: 12-month follow-up of negative test results and utility data from a large US registry study. Dermatology Online Journal. 2019; 25(5). |
Real-World Utility Registry | | Complete | | 3418 | | Brouha B et al. Real-world utility of a non-invasive gene expression test to rule out primary cutaneous melanoma: a large US registry study. J Drugs Dermatol. 2020; 19(3). Brouha B et al. Genomic atypia to enrich melanoma positivity in biopsied lesions: gene expression and pathology findings from a large U.S. registry study. SKIN 2021; 5(1):13-18. |
Adhesive Patch Validation | | Complete | | N/A | | Yao Z et al. An adhesive patch-based skin biopsy device for molecular diagnostics and skin microbiome studies. J Drugs Dermatol. 2017; 16(10):611-618. |
Association With Severe Atypia | | Complete | | 103 | | Jackson S et al. Risk Stratification of Severely Dysplastic Nevi by Non-Invasively Obtained Gene Expression and Mutation Analyses. SKIN. 2020 March; 4(2). |
Recommendations for PLA Use | | Complete | | N/A | | Berman B et al. Appropriate use criteria for the integration of diagnostic and prognostic gene expression profile assays into the management of cutaneous malignant melanoma: an expert panel consensus-based modified Delphi process assessment. SKIN The Journal of Cutaneous Medicine. 2019; 3(5):291-306. National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology. Cutaneous Melanoma. Version 1.2021 at ME-11 Swetter SM et al. Melanoma: Clinical features and diagnosis. UpToDate. Waltham, MA. September 11 2020 |
Health Economics | | Complete | | 319 | | Hornberger J, Siegel D. Clinical and economic implications of a noninvasive molecular pathology assay for early detection of melanoma. JAMA Dermatol. 2018;154(9):1-8. |
Genome Screen | | Complete | | 202 | | Wachsman W et al., Noninvasive genomic detection of melanoma. British Journal of Dermatology. 2011; 164:797-806. |
Table 3. Summarizes key clinical studies and publications supporting the PLA.
13
Table of Contents
Nevome
Our Nevome test was an adjunctive reflex test for the PLA. It was used with histopathology to identify additional risk factors for melanoma and confirm the diagnosis. Approximately 13% of our PLA tests are positive. Lesions that test positively for the PLA are subjected to surgical biopsy and histopathologic review. Due to significant challenges in diagnosing early-stage melanoma by histopathology, additional information can be required to confirm the presence of melanoma and/or identify lesions with significant risk for melanoma that require wide excision. The Nevome test analyzed early-stage melanoma driver mutations in the BRAF, NRAS, and TERT genes, providing additional information and risk factors in the lesion being assessed. The Nevome test utilized the genomic material collected from the initial adhesive patch sample used for the PLA. In November 2020, we discontinued our Nevome test, which will be replaced with our PLAplus test in 2021.
Adhesive Skin Sample Collection Kit
We are the inventor and owner of the intellectual property for the Adhesive Skin Sample Collection Kit (pictured below). We have contracted with a Food and Drug Administration, or FDA, registered supplier to produce our kit under applicable quality systems requirements, and we control the exclusive distribution rights for the kit. Our kit’s adhesive patch allows for the collection of skin samples with minimal patient discomfort. A single kit contains all of the necessary components to complete the sample collection for our analysis, including the adhesive patches, instructions for use, a marking pen for lesion outlining, and a pre-addressed and prepaid return shipping pack. The unique properties of the adhesive maximize the collection of informative cellular material for our PLA. The entire procedure for the kit’s sample collection takes less than five minutes.

Telemedicine Option for the PLA
Telehealth is the provision of health-related services and information via electronic information and telecommunication technologies. Telemedicine is sometimes used interchangeably with telehealth, but some organizations define telemedicine in a more limited sense to describe remote clinical services, such as diagnosis and monitoring. Telemedicine enables patient and clinician interaction when rural settings, lack of transport, a lack of mobility, decreased funding, a lack of staff or other limitations such as social distancing guidelines related to the COVID-19 pandemic restrict or make difficult in-person access to healthcare.
Early detection of melanoma, the most deadly and aggressive form of skin cancer, is critical for best patient outcomes. DermTech’s PLA is the first non-invasive genomic diagnostic test for ruling out melanoma that, in addition to in-office sample collection, allows clinicians to supervise the patient’s sample collection via telemedicine. For patients who cannot easily have an in-office visit, this telemedicine solution enables dermatologists to maintain vigilance with their patients in detecting melanoma at the earliest stages.
Using our telemedicine option, a clinician can choose to assess the patient’s skin and suspicious lesion(s) via a teledermatology appointment and, if indicated, submit a patient-specific order to DermTech for the DermTech PLA. If requested by the clinician, the DermTech PLA Adhesive Skin Collection Kit will then be shipped directly to the patient with support from DermTech customer service. During a follow-up teledermatology appointment, a clinician will instruct and supervise the patient to collect their sample with the easy-to-use DermTech PLA adhesive patch. The patient will then return the collected sample(s) back to DermTech via the pre-labeled shipping envelope for analysis. Test results will be available to the ordering clinician within a few days.
In May 2020, SKIN, the official journal of the National Society for Cutaneous Medicine, published proof-of-concept data demonstrating that patients are able to reliably perform remote self-sampling of concerning moles using the PLA under physician supervision via telemedicine, enabling actionable molecular testing for accurate melanoma detection. As part of the Institutional Review Board (“IRB”) approved pilot study, 258 eligible melanoma survivors were contacted, and of the 211 who expressed interest in the PLA,
14
Table of Contents
there were seven cases of self-identified concerning lesions, which were confirmed by a clinician to be suspicious of melanoma. These patients then conducted sample collections using DermTech’s non-invasive adhesive skin collection kit at home under the supervision of a clinician via telemedicine. Results from the study showed that skin samples collected by patients enabled successful PLA testing to objectively rule out melanoma in all (100%) of the cases evaluated. These findings are in line with sample collection results by licensed providers.
A clinician can assess the patient’s skin and suspicious lesion(s) via teledermatology where permitted by state law. Some state laws impose various restrictions on the practice of telemedicine and reimbursement may not be available under coverage and reimbursement policies governing telemedicine visits issued by third party payors.
Clinical Research Products
Research on the genomic basis of diseases has increased significantly over the last decade. Genomic analysis can facilitate drug development by identifying drug targets and stratifying patients into groups that will maximize drug response. Genomic analysis is part of the effort to personalize medical therapy to patients’ individual needs. Consequently, tools to facilitate this type of research are in high demand.
We offer a suite of products to facilitate clinical research using our technology platform. We have developed a proprietary process that allows us to extract genomic material from the adhesive patch with sufficient quality and quantity to perform gene expression, DNA mutation analysis, DNA methylation, and transcriptomic analyses. In addition, our platform can be utilized to assess the microbiome of the skin with superior performance to existing methods that use swabs. We have developed gene expression assays for the Th1, Th2, IFN-gamma, and Th17 inflammatory pathways. We market these assays to pharmaceutical companies developing drug products in dermatology. In addition, we develop custom gene assays to support development for these pharmaceutical partners. We have completed and have ongoing research collaborations with large pharmaceutical companies to facilitate their development of new targeted therapeutics in dermatology. Our technology platform has been deployed in Phase 1 through Phase 3 clinical programs. These efforts may also lead to the introduction of complementary and companion diagnostic products.
Leveraging Our Platform for Other Indications
We believe our adhesive patch genomic platform is applicable to numerous other indications in dermatology. While we are focused initially on skin cancer products, we believe there are significant business development opportunities in other areas. We have undertaken a number of pilot development activities in inflammatory diseases, and skin aging. This effort will also focus on potential licensing and partnering opportunities for the development of complementary and companion diagnostics for the pharmaceutical partners’ drug product candidates, should they reach the commercial market. In addition, because the processing of samples is the same regardless of the disease indication, our development activities will leverage our existing laboratory operations.
UV Damage DNA Risk Assessment Product (Luminate)
We are developing a UV Damage DNA risk assessment product. This product will assess DNA mutations associated with UV damage and non-melanoma skin cancer risk. Depending on the UV damage level of each individual, there are various treatment options to reduce the current level of DNA damage in their skin and the associated risk of future skin cancer including chemical peels, photodynamic therapy, laser therapy, topical pharmaceuticals, dietary supplements, and increased sunscreen use.
Based upon our market research, there are approximately 84 million Americans between the ages of 30 and 50, of which approximately 70% are concerned with UV damage. Of this population concerned with UV damage, approximately 30% have household incomes of at least $100,000 that would be most likely to consider using this product. We believe this age-group has significant aging anxiety due to the high prevalence of extrinsic signs of aging and are increasingly using anti-aging products to look younger. This UV Damage DNA mutation assessment product will allow individuals to proactively make data driven, fact-based decisions about their skin health.
Non-Melanoma Skin Cancer Diagnostic Products
To complement our melanoma rule-out product PLA, we are also utilizing our platform technology to develop products to rule out non-melanoma skin cancer including squamous cell and basal cell carcinoma. We identified differentially expressed genes that allow the identification of these cancers, and we are currently conducting analytical and clinical validation studies. Nearly 4.5 million basal and squamous cell carcinoma skin cancers are diagnosed each year making skin cancer the most common of all types of cancer. The majority of these cancers occur in cosmetically sensitive areas such as the head, neck and face. The number of skin cancer cases is increasing due to better skin cancer detection, people living longer, and increased sun exposure.
15
Table of Contents
More than 80% of skin cancers are basal cell carcinomas. These cancers usually develop in sun-exposed areas, especially the head and neck, and tend to grow slowly. It is very rare for a basal cell cancer to spread to other parts of the body. If left untreated, basal cell cancers can grow into nearby areas and invade other tissues beneath the skin. If not removed completely, basal cell carcinoma can recur in the same place on the skin. People who have had basal cell skin cancers are also more likely to develop basal cell skin cancers in other places.
About 10% of skin cancers are squamous cell carcinomas. These cancers also commonly appear on sun-exposed areas of the body such as the face, ears, neck, lips, and backs of the hands. These cancers can also develop in scars or chronic skin sores elsewhere. Squamous cell cancers are more likely to grow into deeper layers of skin and spread to other parts of the body than basal cell cancers, although this is still uncommon.
Cutaneous T Cell Lymphoma
We are currently developing a Cutaneous T-cell lymphoma (“CTCL”) rule out test. CTCL is a rare type of skin cancer in which T-cells become immunologically active and attack the skin. CTCL results in rash-like skin redness, slightly raised or scaly round patches on the skin, and, sometimes, skin tumors. These features can resemble much more common inflammatory skin conditions.
Several types of cutaneous T-cell lymphoma exist including mycosis fungoides and Sezary syndrome. Mycosis fungoides is the most common form of CTCL while Sezary syndrome is less common but causes skin redness across larger areas of the body. The definitive diagnosis of CTCL is often challenging because of its nonspecific clinical and pathologic features, which requires integration of clinical, histopathologic, immunophenotyping, and molecular data by the treating physician.
Inflammatory Indications
Atopic dermatitis and psoriasis are chronic inflammatory skin diseases that affect millions of people and are characterized by both local and systemic inflammation. We have investigated gene expression profiles in the skin of atopic dermatitis and psoriasis. Responses to biologic therapy used in moderate to severe forms of these diseases can be variable and may wane over time. For example, only 30-40% of patients have a robust response to either anti-TNF alpha drugs used in psoriasis or the anti-IL-13 drugs used in atopic dermatitis. The low response rate of these drugs creates an unmet need for drug companion and complementary diagnostic products that identify responders to a specific therapy and that monitor responses over time.
Atopic dermatitis and psoriasis are largely characterized by significant epidermal inflammation that can be used to assess benefit of interventional therapies. Due to their existing aberrant and damaged skin barrier, patients are unlikely to consent to repeated surgical biopsy procedures for the purposes of assessing therapy response. Our non-invasive genomics platform is therefore ideal for these types of conditions because it specifically samples tissue from the epidermis. Moreover, we have demonstrated in clinical studies that our platform is superior to surgical biopsy and blood testing for assessing biomarkers related to inflammatory diseases.
In our psoriasis research, for example, we have identified subsets of patients with different gene expression profiles. These different profiles may identify patients that respond more robustly to an expanding group of biologic therapies available for this condition. In addition, we have shown in a pilot clinical investigation that only subsets of patients with atopic dermatitis appear to have high gene expression levels of IL-13. The proportion of patients that are high expressers of IL-13 is approximately 40%, which is consistent with the response rate of approximately 30-40% to the anti-IL4Ralpha (additionally blocks IL-13 signaling) drug dupilumab.
Microbiome Indications
The study of bacterial microbes that inhabit the skin and their relationship to health and disease has been the subject of intense investigation over the last several years. Numerous products are under development that seek to alter the composition and populations of these microbes for therapeutic purposes. We have demonstrated in development studies that our platform can be used to assess the genomics of skin microbes and that the quantity of microbial genomic material and the measurements of microbial variability are superior to the swab-based methods currently in use. In addition, our platform (which simultaneously and non-invasively collects skin host and microbiome samples) has the potential to separate and assess microbial populations at different depth levels in the epidermis. Given the growing interest in this area, we may look to develop products for this market in the future.
Sales and Marketing
The vast majority of molecular diagnostic tests are sold to pathology and oncology practitioners. These markets are quickly becoming saturated with products, services, and sales calls. We believe that we have a unique opportunity as the first company to market a novel non-invasive molecular diagnostic test to dermatologists and other clinical practitioners of dermatology. We believe there are fewer barriers to adoption in this customer base than in other medical markets because our product fits within the current diagnostic and reimbursement pathway for various skin conditions.
16
Table of Contents
We have established a highly experienced team of sales professionals possessing extensive backgrounds in selling dermatology products. Our Chief Commercial Officer spent 24 years at Allergan plc and rose to lead their dermatology and ophthalmology product sales for the entire United States. We expanded our specialty sales force in 2019 and 2020, and plan to continue to expand our specialty sales force in 2021 as we secure reimbursement coverage from additional commercial payors.
There are approximately 13,000 healthcare professionals specializing in dermatology in the United States. We segment these practices into three categories: primarily cosmetic practices (10-15%), mixed medical and cosmetic practices (50-75%), and medical only practices (15-25%). We focus much of our effort on practices that deliver some medical dermatology services. We have initially focused our selling activity on these accounts, which typically have a shorter adoption cycle. We recently completed a review of Medicare and commercial claims for melanoma skin biopsies. From this effort we have identified approximately 4,600 dermatology practitioners that perform the majority of biopsies for melanoma in the U.S. and that treat a majority of the Medicare population. We plan to target these practitioners and have designed our field sales territories around these practices.
Our sales and marketing expansion includes multi-site group practices and integrated dermatology networks. Multi-site group practices and large integrated dermatology networks make up approximately 25% and 15%, respectively, of the remaining dermatology market. We are actively working to integrate our PLA test in large dermatology networks in order to penetrate this market opportunity. As we continue to penetrate these group practices and large integrated networks, we have identified an opportunity to offer reference lab contracts for our PLA test as necessary depending on applicable federal and state regulations. In the reference lab model, the integrated dermatopathology laboratory will accession the PLA samples and bill for these samples, while paying us a contracted price. We believe this reference lab model will be most effective as our reimbursement coverage increases and payments for our tests become more routine.
A portion of dermatology is also practiced in primary care. We may plan to access the primary care market by establishing distribution relationships with companies that focus on this physician call point. These potential partners have should have 400-600 sales professionals in the aggregate who access the primary care market, and ideally have experience offering a diagnostic or genomics product. Alternatively, we may plan to hire sales representative to call on primary care doctors.
Our marketing is focused on a mix of professional targeted campaigns including in person physician education, dermatology symposia, publication distribution, peer to peer education, consumer engagement and education campaigns including a mix of digital platforms. We participate as an exhibitor and sponsor at key dermatology conferences and will expand this effort to primary care conferences. We often submit scientific abstracts for presentation at the conferences we attend. Our KOLs speak on our behalf at various medical conferences, present data from our clinical studies, and chair continuing medical education courses on genomics in dermatology, which include our products.
Our sales and marketing strategy will leverage our extensive network of KOLs in the fields of dermatology, pathology, genomics, biostatistics, healthcare economics, and reimbursement. We use our experts to perform peer-to-peer education, to publish papers utilizing our tests, and to chair continuing medical education courses on genomics in dermatology and our products. These efforts extend to supporting our policy coverage review process with payors. Our KOL group includes four former AAD presidents and numerous melanoma, skin cancer and inflammatory disease experts.
We continuously expand and improve on the validation of our tests by conducting additional clinical trials, and we publish the results of our scientific and clinical work in peer-reviewed medical journals. Through these efforts, we elevated our positioning in the AAD guidelines, obtained a recommendation from NCCN Guidelines, and recent consensus group recommendations. We also utilize advertising in medical journals and social media campaigns to rally the extensive patient advocacy support that exists today for a variety of skin conditions and melanoma sufferers. Because dermatology practitioners often sell cosmetic procedures to their patients, they are very service oriented and responsive to their patient’s requests. We believe direct-to-consumer advertising will engage the patient to request our skin cancer assessment tests and allow us to capitalize on the unique non-invasive benefits our platform provides patients.
We have received Health Canada clearance for our platform and have established a non-exclusive licensing partner, DermTech Canada, for Canada. We are working with this partner to secure reimbursement coverage with various Canadian provinces. We plan to engage in the marketing of our product in other countries outside the United States only after we have established the United States and Canadian markets. We will focus our efforts in regions that have a high incidence of melanoma and skin cancers such as Australia and Western Europe. We will likely seek distribution partners in these select countries for the sales and marketing of our tests. While we have demonstrated that the stability of the skin samples collected with our adhesive patch-based sampling device is suitable for shipping from countries outside the United States, we will likely establish clinical laboratories or laboratory partnerships in some of these countries.
During the COVID‑19 pandemic, we have transitioned our sales teams to make sales calls remotely, with limited in person interaction. We have also participated in various web-based dermatology conferences to highlight the easy-to-use, non-invasive sample collection kit that enables physicians to rule out melanoma without the need to see a patient in person at a clinic.
17
Table of Contents
Reimbursement Strategy
On January 1, 2020, the AMA released a Proprietary Laboratory Analysis code, (0089U), for our PLA test. This code uniquely identifies our PLA test and enables us to bill commercial and government payors when our test is ordered by a clinician.
On February 10, 2020, Medicare Administrative Contractor Palmetto GBA/MolDx issued an LCD for the Pigmented Lesion Assay (L38051). On June 10, 2020 Noridian Medicare, harmonized their coverage determination for the PLA test, in effect making our test nationally covered and available for all Medicare and Medicare Advantage enrollees. The published reimbursement for our PLA code, 0089U, is $760 and was included in the 2020 and 2021 CLFS.
We have developed in-house reimbursement capabilities, including claims submittal, follow-up and appeals functions to bill and collect cash for services provided. We are currently out of network with many commercial payors and our initial claims are commonly denied. In situations where payment is denied, we work through the claims appeals process to secure payment for services performed. The appeals process can require several cycles and can culminate in an independent committee review for blocks of claims. Currently, we are not routinely successful in winning appealed claims.
To improve our allowed claim rate and payment, we are seeking contractual relationships and reimbursement coverage policy decisions from commercial payors. Reimbursement coverage decisions for clinical tests are primarily supported by clinical utility studies, increases in the patient experience and inclusion in guidelines.
The PLA test:
| • | Demonstrates high clinical utility among clinicians. Over 4,500 patients have been included in four (4) clinical utility studies that highlight that clinicians followed the guidance of the test in over 98% of cases. This resulted in 90% fewer biopsies (i.e. avoidable biopsies). Clinical validation and supportive studies were conducted on an additional 3,000 patients. |
| • | Extensively studied. The PLA has been studied in over 7,500 patients and results have been summarized in 21 peer reviewed manuscripts published in leading journals. To date, over 3,500 clinicians and 70,000 patients have benefitted from the test by avoiding over 60,000 surgical biopsies while missing fewer melanomas. |
| • | Saves money and increased the patient experience. The PLA saves money by reducing the costs of unnecessary biopsies and excisions performed on benign lesions. Also, it allows for earlier detection of melanoma which can reduce the early-stage and late-stage costs of treating melanoma. The patient experience is enhanced because in 90% of the cases, they avoid a surgical procedure. |
| • | Included in the NCCN Guidelines Version 1.2021. The NCCN (2a) recommendation listed under “Common Follow-up Recommendation for all Patients” states that our test may be helpful to guide biopsy decisions. A (2a) recommendation from the NCCN indicates there is uniform consensus that the intervention is appropriate. |
| • | Can be used in a telemedicine encounter. Based on a study published in May 2020, patients can reliably perform self-sample collection under remote physician supervision. |
We have currently secured several contracts with major preferred provider networks, including Blue Shield of California, Blue Cross and Blue Shield of Texas, Blue Cross and Blue Shield of Illinois, Carefirst - BCBS of Maryland and Priority Healthcare of Michigan. We have submitted clinical and technology assessment packages to eviCore healthcare, which provides consultative services for payors, and several national commercial payors, including Aetna, Cigna Corporation, UnitedHealthcare, Humana and several independent Blue plans, all of which have the PLA currently under review.
Competition
The molecular diagnostics market is highly competitive. We compete with a number of manufacturers and distributors of molecular diagnostic tests as well as new and traditional medical devices and other technologies that are used to assist physicians with the assessment of pigmented lesions and the diagnosis of skin cancer. We are currently the only company to offer a non-invasive genomics test to clinical dermatology professionals. However, LEO Pharma A/S, a large Danish pharmaceutical company, and Mindera Corporation, a small early-stage start-up, are also working on minimally invasive genomic tests. In the area of pigmented lesions, Myriad Genetics, Inc. and Castle Biosciences, Inc. recently launched gene expression assays as CLIA laboratory tests for surgical biopsy tissue specimens. Castle Biosciences, Inc. also markets a product to determine metastatic potential in later stage melanoma by utilizing surgical tissue samples.
18
Table of Contents
There are several companies that market or are developing medical devices and imaging tools to detect melanoma as skin cancer. In general, medical devices have capital equipment costs and maintenance requirements, do not integrate well into clinical practice, and do not have clear mechanisms to provide physician payment. Strata Sciences, Inc. owns the rights to Melafind, an FDA-approved device that utilizes varying wavelengths of light to capture lesion images at different depths and conducts an algorithmic image analysis to determine the degree of lesion disorganization and the need for biopsy. The clinical trials of this device demonstrated marginal improvement in the assessment of pigmented lesions, and the device has not been adopted in the United States largely due to its specificity of less than 10%, which hampered clinical use. SciBase AB is marketing an epidermal electrical impedance spectrometer to assess pigmented lesions, which received FDA approval in 2018. Verisante Technology, Inc. has received regulatory approval in Europe and Australia to market a device that uses real-time Raman spectroscopy to assess changes in the chemical composition of skin tissue. Welch-Allen, Inc. and various others manufacture dermatoscopes, which provide magnified views of a pigmented lesion during diagnosis. Caliber I.D. and others offer confocal microscopy solutions for enhanced imaging of pigmented skin lesions.
Research and Development
We have expertise in the development of gene expression profiles and other genomic analyses for the diagnosis of dermatologic disease. In addition, we have developed know-how related to the collection of skin samples using adhesives. We have also developed expertise in statistical programs and algorithms that are used to process genomic data.
Our product development process involves several stages. The first stage involves a genome-wide screen for differential gene expression or screens for differences in mutations, methylation patterns, micro-RNAs and other factors. In case of gene expression, differentially expressed genes are then narrowed down to specific gene sets that categorize disease states. These genes sets are then validated by comparison to clinical reference standards to produce a clinical product. We have developed substantial expertise in designing and conducting clinical validation and utility studies.
We have identified additional gene targets that may further improve the performance of our PLA. The qPCR assays for these genes are under development and may be added to our platform in the future if their performance is validated in additional clinical studies. We plan to expand the use of our platform to include products to diagnose or support the diagnosis of non-melanoma skin cancers as well as a variety of inflammatory skin conditions. We have identified gene expression profiles for other conditions, such as psoriasis, atopic dermatitis, and aging of the skin. Should we determine that there are viable market opportunities for products treating these conditions, we plan to consider developing genomic tests for these conditions. Alternatively, we may seek development partners or licensing opportunities for these potential products.
Intellectual Property
We have developed a comprehensive portfolio of intellectual property, comprising six issued or allowed U.S. utility patents, 12 pending U.S. utility patent applications, four pending U.S. design patent applications, six issued foreign patents, 11 pending foreign patent applications, and two PCT applications.
The portfolio includes patents or patent applications directed to aspects of our assays, a sample collection system using adhesive, methods for automated scanning and cutting of cells from skin collection kits, telemedicine methods, methods of detecting nucleic acid expression, methods of quantifying a mutation burden, and methods of diagnosing or treating various skin conditions including melanoma and non-melanoma skin cancers, cutaneous T cell lymphoma, UV damage and autoimmune disorders.
In addition, our intellectual property portfolio includes trademarks, design patents, trade secrets and know-how. We believe our intellectual property adequately protects our products and technology, and may prevent others from commercializing products or laboratory methods substantially similar to ours.
Laboratory Operations
Our CLIA laboratory occupies approximately 13,000 square feet and is divided into an accession area, pre-qPCR-laboratory and post-qPCR-laboratory area as per CLIA standards. Access to all areas is controlled and requires gowning. The laboratory employs commercial state-of-the-art equipment including high-throughput qPCR machines. We use a laboratory information system to track all of our samples. We employ clinical laboratory scientists holding appropriate state licenses to perform the assay.
Our PLA assay utilizes qPCR techniques that requires the extraction and purification of genomic material from the skin adhered to adhesive patches. This extraction process is extremely challenging, and we have developed a proprietary method involving customized reagents and tools to provide suitable material yields reliably. In general the process involves three main steps:
| • | RNA extraction using our proprietary process to maximize the yields and quantity of RNA from the cells on the patch; |
| • | reverse transcription, which converts the RNA into complementary DNA; and |
19
Table of Contents
| • | expression level quantification, using qPCR to determine the expression levels of the target genes in our expression profile. |
After testing is complete, a laboratory report is prepared and reviewed by one of our California-licensed and American Board of Medical Genetics and Genomics-certified Laboratory Directors. This report is made available to the ordering physician by fax or via an internet portal, while adhering to requirements of the Health Insurance Portability and Accountability Act (“HIPAA”). The reports are generated in industry-standard PDF format that allows for high-definition figures to be reproduced clearly.
We continuously work to automate various steps in our end-to-end test processes. Much of this automation will come from purchasing and qualifying off-the-shelf and customized laboratory equipment such as liquid handlers and pipetting robots. We have developed a laser-cutting robot to automate the cutting of the lesion area circumscribed on the adhesive patch by the clinician. We expect these automation efforts to improve assay throughput by reducing processing time compared to manual processing, reducing the need for direct labor, and improving quality by reducing the potential for human error.
Third-Party Suppliers and Manufacturers
We are the owner of intellectual property for the Adhesive Skin Sample Collection Kit with our logo and have contracted with an FDA-registered supplier to produce our kits. We believe this kit is considered a Class I medical device and is exempt from FDA premarket notification requirements. This product is manufactured according to the FDA’s applicable quality system manufacturing requirements. Our FDA-registered supplier conducts the assembly and labeling of this kit. All of our suppliers are high-quality medical component and finished-product suppliers accustomed to working on high volume disposable FDA-regulated products. Our product has a shelf life tested to three years that allows us to build inventory to mitigate against disruptions.
We currently have a sole source provider for our adhesive used in the Skin Sample Collection Kit. We are actively working to identify second source suppliers for this component. We currently have sufficient adhesive supplies and inventory to meet our plans and objectives.
Governmental Regulation
The services that we provide are regulated by federal, state and foreign governmental authorities. Failure to comply with the applicable laws and regulations can subject us to repayment of amounts previously paid to us, significant civil and criminal penalties, loss of licensure, certification, or accreditation, or exclusion from government health care programs.
We believe our Adhesive Skin Sample Collection Kit is a Class I medical device and is manufactured by an FDA-registered supplier according to applicable regulations and is exempt from obtaining premarket approval or clearance from the FDA. The FDA could declare our Sample Collection Kit a Class II device or as non-exempt. This would require us to submit an application for premarket clearance or approval, which may require us to develop additional clinical data to support premarket clearance or approval that could come at substantial expense and could disrupt our current business or affect our results of operations.
Our qPCR gene expression assay is a laboratory developed test, or LDT, that is currently regulated under CLIA. Although the FDA has asserted that it has authority to regulate LDTs, it has generally exercised enforcement discretion and is not otherwise regulating most tests developed and performed within a single high complexity CLIA-certified laboratory. We have commercialized our test as an LDT and will process all tests in our single CLIA-certified central laboratory. We may at some time in the future seek FDA clearance or approval for our qPCR gene expression assay. We believe the data we have collected in the development of our LDT will support any FDA medical device clearance or approval process, but cannot guarantee that the FDA will find these data sufficient to support clearance or approval as a medical device under the applicable FDA regulations. This may require us to collect additional clinical data, which could come at substantial expense and could affect our results of operations.
CLIA and State Regulation of Laboratories
Clinical laboratories must hold certain federal, state, and local licenses, certifications, and permits to conduct business. Laboratories that perform testing on human specimens for the purpose of providing information for the diagnosis, prevention, or treatment of disease are subject to CLIA. CLIA requires such laboratories to be certified by the federal government and mandates compliance with various operational, personnel, facilities administration, quality, and proficiency testing requirements intended to ensure that testing services are accurate, reliable and timely. CLIA certification is also a prerequisite to be eligible to bill state and federal health care programs, as well as many private insurers, for laboratory testing services.
Standards for testing under CLIA vary based on the test and level of test complexity. Laboratories performing high complexity testing must comply with more stringent requirements than laboratories performing waived or moderate complexity testing. In addition, CLIA requires each certified laboratory to enroll in an approved proficiency-testing program if it performs testing in any category for which proficiency testing is required. Such laboratories must periodically test specimens received from an outside proficiency testing organization and then must submit the results back to that organization for evaluation. A laboratory that fails to achieve a passing score
20
Table of Contents
on a proficiency test may lose its right to perform testing in the category at issue. Further, failure to comply with other proficiency testing regulations, such as the prohibition on referral of a proficiency- testing specimen to another laboratory for analysis, can result in revocation of the referring laboratory’s CLIA certification.
As a condition of CLIA certification, our laboratory is subject to survey and inspection every other year, in addition to being subject to additional unannounced inspections. Because we have obtained accreditation by the College of American Pathologists, or CAP, which is a CMS-approved accreditation organization, our biennial survey is conducted by CAP.
Our laboratory must comply with all CLIA requirements as well as with any additional requirements imposed by CAP. We also hold a laboratory permit from New York State Department of Health, which has the most rigorous state licensing process for clinical diagnostic laboratories.
CLIA provides that a state may adopt laboratory regulations that are more stringent than those under federal law, and two states, New York and Washington, have met that standard and therefore substitute for the federal CLIA program. In addition, some, but not all, states require a separate state license or permit, which must be obtained in addition to a CLIA certificate, and some states require a laboratory doing business in its state to be licensed even if the laboratory is located in another state. Our laboratory is licensed by the appropriate state agencies in the states in which we do business, if such licensure is required. If a laboratory is out of compliance with state laws or regulations governing licensed laboratories, penalties for violation vary from state to state but may include suspension, limitation, revocation or annulment of the license, assessment of financial penalties or fines, or imprisonment. We believe that we are in material compliance with all applicable licensing laws and regulations.
We may become aware from time to time of other states that require out-of-state laboratories to obtain licensure to accept specimens from patients within the state, and other states may impose such requirements in the future. If we identify any other state with such requirements, or if we are contacted by any other state advising us of such requirements, we intend to follow all instructions from the state regulators regarding compliance with such requirements.
The FDA
Although the FDA has asserted that it has the authority to regulate LDTs that are validated by the developing laboratory and performed only by that laboratory, it has generally exercised enforcement discretion by not otherwise regulating most tests developed and performed within a single high complexity CLIA-certified laboratory. Nevertheless, the FDA has, for the past decade, been introducing proposals to end enforcement discretion and to bring LDTs clearly under existing FDA regulatory frameworks. In July 2010, the FDA held a two-day public meeting to obtain input from stakeholders on how it should apply its authority to implement a reasonable, risk-based, and effective regulatory framework for LDTs, including genetic tests. Subsequently, FDA issued draft guidance and a 2017 Discussion Paper to allow for further public discussion about an appropriate LDT oversight approach and to give congressional committees the opportunity to develop a legislative solution. Since 2017, Congress has been working on legislation to create an LDT and in vitro diagnostic, or IVD, regulatory framework that would be separate and distinct from the existing medical device regulatory framework. In August 2018, the FDA recommended changes to draft legislation that had been released by Congress in 2017. The agency’s comments addressed the need for a requirement that new tests undergo FDA review to demonstrate analytical and clinical validity and suggested other changes to the draft language. FDA’s recommendations, if included in enacted law, would give the FDA authority to revoke approval, request raw data, and take corrective action against test developers.
In December 2018, legislators released a discussion draft of a bill that incorporated many of FDA’s suggestions and provided opportunities for additional stakeholders to also provide input on the proposed reform legislation. On March 5, 2020, U.S. Representatives Diana DeGette (D-CO) and Dr. Larry Bucshon (R-IN) formally introduced the long-awaited legislation, called the Verifying Accurate, Leading-edge IVCT Development (“VALID”) Act. An identical version of the bill was also introduced in the Senate and is sponsored by U.S. Senators Michael Bennet (D-CO) and Richard Burr (R-NC), demonstrating both bicameral and bipartisan support for the effort to overhaul how the FDA reviews and approves diagnostic tests going forward. The VALID Act would codify into law the term “in vitro clinical test” (“IVCT”), to create new medical product category separate from medical devices that includes products currently regulated as IVDs as well as LDTs. The VALID Act would also create a new system for labs and hospitals to use to submit their tests electronically to the FDA for approval, which is aimed at reducing the amount of time it takes for the agency to approve such tests, and establish a new program to expedite the development of diagnostic tests that can be used to address a current unmet need for patients. It is unclear whether the VALID Act would be passed by Congress in its current form or signed into law by the President. Until the FDA finalizes its regulatory position regarding LDTs, or the VALID Act or other legislation is passed reforming the federal government’s regulation of LDTs, it is unknown how the FDA may regulate our tests in the future and what testing and data may be required to support any required clearance or approval.
Most recently, on August 19, 2020, the United States Department of Health and Human Services, or HHS, published a policy announcement that FDA must go through the formal notice-and-comment rulemaking process before requiring premarket review of LDTs rather than making such changes through guidance documents, compliance manuals, or other informal policy statements. Laboratories may still voluntarily submit LDTs to FDA for premarket review, although the agency does not appear to be prioritizing
21
Table of Contents
such applications for review at the present time, in light of the HHS announcement. Although the ultimate impact of HHS’s policy statement on FDA’s plans for regulating LDTs and its current thinking relating to such diagnostic testing products is unclear, the August 2020 announcement appears to confirm that laboratories may commercialize LDTs for clinical use without submitting such tests for FDA review and marketing authorization. HHS’s policy statement does not affect proposed legislation for the regulation of LDTs, such as the VALID Act described above. It is also unclear whether the Biden Administration, which assumed control of the executive branch on January 20, 2021, would take the same position as the former administration or seek to revoke or revise the HHS policy announcement from August 2020.
If the FDA decides to regulate LDTs, such as our PLA test, as medical devices through notice-and-comment rulemaking or the VALID Act or other new federal legislation is passed reforming the government’s regulation of LDTs, or alternatively, if the FDA disagrees with our assessment that our tests fall within the definition of an LDT, we will be subject to increased regulatory burdens such as registration and listing requirements, medical device reporting requirements and quality control requirements. Any legislation or formal FDA regulatory framework affecting LDTs is also likely to have premarket application requirements prohibiting commercialization without FDA authorization and controls regarding modification to the tests that may require further FDA submissions. The process would likely be costly and time-consuming. We cannot assure that our PLA test, or any new tests that we may develop or new uses for our products that we develop will be cleared or approved by the FDA in a timely or cost-effective manner, if cleared or approved at all. Even if such tests are cleared or approved, the products may not be cleared or approved for all indications. This could significantly limit the market for that product and may adversely affect our results of operations.
The Adhesive Skin Sample Collection Kit we provide for collection and transport of skin samples from a healthcare provider (or in our recently launched telemedicine option, from the patient directly) to our clinical laboratory is a Class I medical device subject to FDA regulations, but it is currently exempt from premarket review by the FDA and manufactured by a third party on our behalf. Class 1 products like our specimen collection kit are required to meet FDA’s general controls for device products, including that they be manufactured in compliance with applicable Quality System Regulations for medical devices, adhere to device labeling requirements, and be listed with FDA upon commercial distribution, among other regulatory controls.
HIPAA and Other Privacy and Data Security Laws
HIPAA established for the first time comprehensive federal protection for the privacy and security of health information. The HIPAA standards apply to three types of organizations, or Covered Entities: health plans, healthcare clearing houses, and healthcare providers that conduct certain healthcare transactions electronically. Title II of HIPAA, the Administrative Simplification Act, contains provisions that address the privacy of health data, the security of health data, the standardization of identifying numbers used in the healthcare system and the standardization of certain healthcare transactions. The privacy regulations protect medical records and other protected health information by limiting their use and release, giving patients a variety of rights, including the right to access their medical records and limiting most disclosures of health information to the minimum amount necessary to accomplish an intended purpose. The HIPAA security standards require the implementation of administrative, physical, and technical safeguards and the adoption of written security policies and procedures. HIPAA requires Covered Entities to enter into business associate agreements with individuals or organizations who provide services to Covered Entities involving the use or disclosure of protected health information, also known as Business Associates.
In 2009, Congress enacted Subtitle D of the Health Information Technology for Economic and Clinical Health Act, or HITECH, provisions of the American Recovery and Reinvestment Act of 2009. HITECH amended HIPAA and, among other things, expanded and strengthened HIPAA, created new targets for enforcement, imposed new penalties for noncompliance and established new breach notification requirements for Covered Entities and Business Associates. Regulations implementing major provisions of HITECH were finalized on January 25, 2013 through publication of the HIPAA Omnibus Rule, or the Omnibus Rule. The Omnibus Rule contained significant changes for Covered Entities and Business Associates with respect to permitted uses and disclosures of Protected Health Information.
Under HITECH’s breach notification requirements, Covered Entities must report breaches of protected health information that has not been encrypted or otherwise secured in accordance with guidance from the Secretary of the United States Department of Health and Human Services, or the Secretary. Required breach notices must be made as soon as is reasonably practicable, but no later than sixty days following discovery of the breach. Reports must be made to affected individuals and to the Secretary and in some cases, they must be reported through local and national media, depending on the size of the breach. We are currently subject to the HIPAA regulations as a Covered Entity and maintain an active compliance program. We are subject to audit under the United States Department of Health and Human Services’ HITECH-mandated audit program. We may also be investigated in connection with a privacy or data security complaint. We are subject to prosecution and/or administrative enforcement and increased civil and criminal penalties for non-compliance, including a new, four-tiered system of monetary penalties adopted under HITECH. These fines are adjusted for inflation each year. We are also subject to enforcement by state attorneys general who were given authority to enforce HIPAA under HITECH. To avoid penalties under the HITECH breach notification provisions, we must ensure that breaches of unsecured protected health information are promptly detected and reported within the company, so that we can make all required notifications to the
22
Table of Contents
government on a timely basis. However, even if we make required reports on a timely basis, we may still be subject to penalties for the underlying breach and at risk of significant reputational harm if we experience a large-scale data breach.
In addition to the federal privacy regulations, there are a number of state laws regarding the privacy and security of health information and personal data that are applicable to clinical laboratories. The compliance requirements of these laws, including additional breach reporting requirements, and the penalties for violation vary widely and new privacy and security laws in this area are evolving. For example, several states, such as California, have implemented comprehensive privacy laws and regulations. The California Confidentiality of Medical Information Act imposes restrictive requirements regulating the use and disclosure of health information and other personally identifiable information. In addition to fines and penalties imposed upon violators, some of these state laws also afford private rights of action to individuals who believe their personal information has been misused. California’s patient privacy laws, for example, provide for penalties of up to $250,000 and permit injured parties to sue for damages. In addition to the California Confidentiality of Medical Information Act, California also recently enacted the California Consumer Privacy Act of 2018, or CCPA, which became effective January 1, 2020. The CCPA has been characterized as the first “GDPR-like” privacy statute to be enacted in the United States because it mirrors a number of the key provisions of the E.U. General Data Protection Regulation (described further below). The CCPA establishes a new privacy framework for covered businesses in the State of California, by creating an expanded definition of personal information, establishing new data privacy rights for consumers imposing special rules on the collection of consumer data from minors, and creating a new and potentially severe statutory damages framework for violations of the CCPA and for businesses that fail to implement reasonable security procedures and practices to prevent data breaches. The regulations issued under the CCPA have been modified several times. There is uncertainty surrounding the application of the CCPA to parts of our business, and amendments to the law and the regulations issued thereunder may have an impact on our operations. In addition to the CCPA, other states are introducing similar legislation which will impact compliance obligations and increase complexity and cost of compliance.
Many states, such as Massachusetts, have also implemented genetic testing and privacy laws imposing specific patient consent requirements and requirements for protecting test results. The interplay of federal and state laws may be subject to varying interpretations by courts and government agencies, creating complex compliance issues for us and potentially exposing us to additional expense, adverse publicity and liability. Further, as regulatory focus on privacy issues continues to increase and laws and regulations concerning the protection of personal information expand and become more complex, these potential risks to our business could intensify.
The applicability and requirements of these laws and penalties for violations vary widely. We believe that we have taken the steps required of us to comply with applicable health information privacy and security statutes and regulations in all jurisdictions, both state and federal. However, we may not be able to maintain compliance in all jurisdictions where we do business. Failure to maintain compliance, or changes in state or federal laws regarding privacy or security, could result in civil and/or criminal penalties and could have a material adverse effect on our business, financial condition, results of operation and cash flows.
We anticipate expanding our business internationally, which would implicate international laws governing the privacy of health information and personal data as well as restrictions on the cross-border transfer of these data. We currently receive samples from Canada and must comply with applicable Canadian federal and provincial laws. Compliance with these laws and with other international regulatory requirements is a complex, time and expense consuming endeavor. Our failure to comply could have a material adverse effect on our business, financial condition, results of operation and cash flows.
Federal and State Self-Referral Prohibitions
We are subject to the federal self-referral prohibitions, commonly known as the Stark Law or the Physician Self-Referral Law, and to similar state restrictions such as California’s Physician Ownership and Referral Act, commonly known as PORA. Together these restrictions generally prohibit us from billing the Medicare or Medicaid program or any patient or commercial payor for a test when the physician ordering the test, or any member of such physician’s immediate family, has an investment interest in or compensation arrangement with us, unless the arrangement meets an exception to the prohibition.
Both the Stark Law and PORA contain an exception for compensation paid to a physician for personal services rendered by the physician, provided that certain conditions are satisfied. We have compensation arrangements with a number of physicians for personal services, such as speaking engagements. We have structured these arrangements with terms intended to comply with the requirements of the personal services exception to the Stark Law and PORA. However, we cannot be certain that regulators would find these arrangements to be in compliance with the Stark Law, PORA or similar state laws.
Sanctions for a violation of the Stark Law include the following:
| • | denial of payment for the services provided in violation of the prohibition; |
| • | refunds of amounts collected by an entity in violation of the Stark Law; |
23
Table of Contents
| • | a civil penalty of up to $25,870 (which reflects the annual inflation adjustment effective as of January 17, 2020) for each service arising out of the prohibited referral; |
| • | exclusion from federal healthcare programs, including the Medicare and Medicaid programs; and |
| • | a civil penalty of up to $172,137 (which reflects the annual inflation adjustment effective as of January 17, 2020) against parties that enter into a scheme to circumvent the Stark Law’s prohibition. |
These prohibitions apply regardless of the reasons for the financial relationship and the referral. No finding of intent to violate the Stark Law is required to commit a violation. In addition, knowing violations of the Stark Law may also serve as the basis for liability under the Federal False Claims Act.
Further, a violation of PORA is a misdemeanor and could result in civil penalties and criminal fines. Finally, other states have self-referral restrictions with which we have to comply that differ from those imposed by federal and California law. While we have attempted to comply with the Stark Law, PORA and similar laws of other states, it is possible that some of our financial arrangements with physicians could be subject to regulatory scrutiny at some point in the future, and we cannot provide an assurance that we will be found to be in compliance with these laws following any such regulatory review.
Federal and State Fraud and Abuse Laws
Because of the significant federal funding involved in Medicare and Medicaid, Congress and the states have enacted, and actively enforce, a number of laws to eliminate fraud and abuse in federal health care programs. Our business is subject to compliance with these laws.
Anti-Kickback Statutes
The federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving, or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing, recommending or arranging for a good or service, for which payment may be made under a federal healthcare program such as Medicare or Medicaid.
The definition of “remuneration” has been broadly interpreted to include anything of value, including, for example, gifts, certain discounts, the furnishing of free supplies, equipment or services, credit arrangements, payment of cash, and waivers of payments. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of services covered by the federal health care programs, the statute has been violated. Penalties for violations include criminal penalties and civil sanctions such as fines, imprisonment, and possible exclusion from Medicare, Medicaid and other federal healthcare programs. In addition, some kickback allegations have been claimed to violate the federal False Claims Act, discussed in more detail below.
The Anti-Kickback Statute is broad and prohibits many arrangements and practices that are otherwise lawful in businesses outside of the healthcare industry. Recognizing that the Anti- Kickback Statute is broad and may technically prohibit many innocuous or beneficial arrangements, Congress authorized the Office of Inspector General, or OIG, of the United States Department of Health and Human Services to issue a series of regulations known as “safe harbors.” These safe harbors set forth provisions that, if all their applicable requirements are met, immunize the parties to the transaction or arrangement from prosecution under the Anti-Kickback Statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal or that prosecution will be pursued. However, transactions and business arrangements that do not fully satisfy an applicable safe harbor may result in increased scrutiny by government enforcement authorities such as OIG.
Many states have adopted laws similar to the Anti-Kickback Statute. Some of these state prohibitions apply to referral of recipients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs. In addition, in October 2018, the Eliminating Kickbacks in Recovery Act of 2018, or EKRA, was enacted as part of the Substance Use-Disorder Prevention that Promotes Opioid Recovery and Treatment for Patients and Communities Act, or SUPPORT Act. EKRA is an all-payor anti-kickback law that makes it a criminal offense to pay any remuneration to induce referrals to, or in exchange for, patients using the services of a recovery home, a substance use clinical treatment facility, or laboratory. Although it appears that EKRA was intended to reach patient brokering and similar arrangements to induce patronage of substance use recovery and treatment, the language in EKRA is broadly written. Further, certain of EKRA’s exceptions, such as the exception applicable to relationships with employees that effectively prohibits incentive compensation, are inconsistent with the Anti-Kickback Statute regulations, which permit payment of employee incentive compensation, a practice that is common in the industry. Significantly, EKRA permits the U.S. Department of Justice to issue regulations clarifying EKRA’s exceptions or adding additional exceptions, but such regulations have not yet been issued. Laboratory industry stakeholders are reportedly seeking clarification regarding EKRA’s scope and/or amendments to its language. Because EKRA is a new law, there is no agency guidance or court precedent to indicate how and to what extent it will be applied and enforced. We cannot assure you that our relationships with physicians, sales representatives, hospitals, customers, or any other party will not be subject to scrutiny or will survive regulatory challenge under such laws. If imposed for any reason, sanctions under the EKRA could have a negative effect on our business.
24
Table of Contents
Government officials have focused their enforcement efforts on the marketing of healthcare services and products, among other activities, and recently have pursued cases against companies, and certain individual sales, marketing, and executive personnel, for allegedly offering unlawful inducements to potential or existing customers in an attempt to procure their business.
Federal False Claims Act
Another development affecting the healthcare industry is the increased use of the federal False Claims Act, and in particular, action brought pursuant to the False Claims Act’s “whistleblower” or “qui tam” provisions. The False Claims Act imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal healthcare program. The qui tam provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government alleging that the defendant has violated the False Claims Act and to share in any monetary recovery. In recent years, the number of suits brought against healthcare providers by private individuals has increased dramatically.
In addition, various states have enacted false claims law analogous to the False Claims Act, many of these state laws apply where a claim is submitted to any commercial payor and not merely a federal healthcare program.
When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties of between $5,500 and $11,000 for each separate instance of false claim, as set by statute. However, the civil penalty amounts are adjusted annually for inflation. For civil penalties assessed after June 19, 2020, where the associated violations occurred after November 2, 2015, the civil penalty amount ranges between $11,665 and $23,331 per claim.
There are many potential bases for liability under the False Claims Act. Liability arises, primarily, when an entity knowingly submits, or causes another to submit, a false claim for reimbursement to the federal government. The federal government has used the False Claims Act to assert liability on a variety of bases, including kickbacks offered or paid to referral sources.
While we are unaware of any current matters, we are unable to predict whether we will be subject to actions under the False Claims Act or a similar state law, or the impact of such actions. However, the costs of defending such claims, as well as any sanctions imposed, could significantly affect our financial performance.
Physician Sunshine Laws
The federal Physician Payments Sunshine Act imposes reporting requirements on manufacturers of certain devices, drugs and biologics for certain payments and transfers of value by them (and in some cases their distributors) to physicians, teaching hospitals and certain advanced non-physician health care practitioners, as well as ownership and investment interests held by physicians and their immediate family members. The reporting program (known as the Open Payments program) is administered by CMS. Because we manufacture our own LDTs solely for use by or within our own laboratory, we believe we are exempt from these reporting requirements. We may become subject to such reporting requirements under the terms of current CMS regulations, however, if the FDA requires us to obtain premarket clearance or approval for our tests.
Corporate Practice of Medicine
Numerous states have enacted laws, prohibiting business corporations, such as us, from practicing medicine and employing or engaging physicians to practice medicine, generally referred to as the prohibition against the corporate practice of medicine. These laws are designed to prevent interference in the medical decision-making process by anyone who is not a licensed physician. For example, California’s Medical Board has indicated that determining the appropriate diagnostic tests for a particular condition and taking responsibility for the ultimate overall care of a patient, including providing treatment options available to the patient, constitutes the unlicensed practice of medicine if performed by an unlicensed person. Violation of these corporate practice of medicine laws may result in civil or criminal fines, as well as sanctions imposed against the business corporation and/or the professional through licensure proceedings. Typically, such laws are only applicable to entities with a physical presence in the applicable state.
Civil Monetary Penalties Law
The federal Civil Monetary Penalties Law, or the CMP Law, prohibits, among other things, (1) the offering or transfer of remuneration to a Medicare or state health care program beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier of services reimbursable by Medicare or a state health care program, unless an exception applies; (2) employing or contracting with an individual or entity that the provider knows or should know is excluded from participation in a federal health care program; (3) billing for services requested by an unlicensed physician or an excluded provider; and (4) billing for medically unnecessary services. The penalties for violating the CMP Law include exclusion, substantial fines, and payment of up to three times the amount billed, depending on the nature of the offense.
25
Table of Contents
Reimbursement and Billing
Reimbursement and billing for diagnostic services is highly complex. Laboratories must bill various payors, such as commercial insurers, including managed care organizations, or MCO, as well as state and federal health care programs, such as Medicare and Medicaid, and each may have different billing requirements. Additionally, the audit requirements laboratories must meet to ensure compliance with applicable laws and regulations, as well as internal compliance policies and procedures, add further complexity to the billing process.
In April 2014, Congress passed the Protecting Access to Medicare Act of 2014, or PAMA, which included substantial changes to the way in which clinical laboratory services are paid under Medicare. Under PAMA, certain clinical laboratories are required to report to CMS commercial payor payment rates and volumes for their tests. Laboratories that fail to report the required payment information may be subject to substantial civil monetary penalties. Further, effective January 1, 2018 under PAMA, Medicare reimbursement for diagnostic tests will be based on the weighted- median of the payments made by commercial payors for these tests, rendering commercial payor payment levels even more significant. As a result, future Medicare payments may fluctuate more often and become subject to the willingness of commercial payors to recognize the value of diagnostic tests generally and any given test individually.
In March 2020, Congress passed the Coronavirus Aid, Relief, and Economic Security Act, which included a provision that delays the next PAMA reporting period for clinical laboratory tests that are not advanced diagnostic tests to January 1, 2022 through March 31, 2022. In addition, the next round of rate cuts will not be implemented until 2022, with tests receiving cuts of up to 15 percent a year from 2022 through 2024.
We cannot predict whether or when these or other recently enacted healthcare initiatives will be implemented at the federal or state level or how any such legislation or regulation may affect us. For instance, the changes to reimbursement amounts paid by Medicare for tests such as ours based on the procedure set forth in PAMA could limit the prices we would be able to charge or the amount of available reimbursement for our tests, which would reduce our revenue. Additionally, these healthcare policy changes could be amended or additional healthcare initiatives could be implemented in the future.
Other Laws Applicable to Our Business
In some cases, we are prohibited from conducting certain tests without a certification of patient consent by the physician ordering the test.
In addition, we are subject to laws and regulations related to the protection of the environment, the health and safety of employees and the handling, transportation and disposal of medical specimens, infectious and hazardous waste, and radioactive materials. For example, the United States Occupational Safety and Health Administration, or OSHA, has established extensive requirements relating specifically to workplace safety for healthcare employers in the United States. This includes requirements to develop and implement multi-faceted programs to protect workers from exposure to blood-borne pathogens, such as HIV and hepatitis B and C, including preventing or minimizing any exposure through needle stick injuries. For purposes of transportation, some biological materials and laboratory supplies are classified as hazardous materials and are subject to regulation by one or more of the following agencies: the United States Department of Transportation, the United States Public Health Service, the United States Postal Service, and the International Air Transport Association. We generally use third-party vendors to dispose of regulated medical waste, hazardous waste, and radioactive materials and contractually requires them to comply with applicable laws and regulations.
Advertising of Laboratory Services or LDTs
Our physician-directed advertising for the PLA, the Adhesive Skin Sample Collection Kit and our laboratory services, as well as our direct-to-consumer advertising and social media presence, are subject to federal truth-in-advertising laws enforced by the Federal Trade Commission, or FTC, as well as comparable state consumer protection laws. Under the Federal Trade Commission Act, the FTC is empowered, among other things, to (a) prevent unfair methods of competition and unfair or deceptive acts or practices in or affecting commerce; (b) seek monetary redress and other relief for conduct injurious to consumers; and (c) gather and compile information and conduct investigations relating to the organization, business, practices, and management of entities engaged in commerce. The FTC has very broad enforcement authority, and failure to abide by the substantive requirements of the FTC Act and other consumer protection laws can result in administrative or judicial penalties, including civil penalties, injunctions affecting the manner in which we would be able to market services or products in the future, or criminal prosecution.
Foreign Corrupt Practices Act
In general, the Foreign Corrupt Practices Act of 1977, as amended, or the FCPA, prohibits offering to pay, paying, promising to pay, or authorizing the payment of money or anything of value to a foreign official in order to influence any act or decision of the foreign official in his or her official capacity or to secure any other improper advantage in order to obtain or retain business for or with, or in
26
Table of Contents
order to direct business to, any person. The prohibitions apply not only to payments made to “any foreign official,” but also those made to “any foreign political party or official thereof,” to “any candidate for foreign political office” or to any person, while knowing that all or a portion of the payment will be offered, given, or promised to anyone in any of the foregoing categories. “Foreign officials” under the FCPA include officers or employees of a department, agency, or instrumentality of a foreign government. The term “instrumentality” is broad and can include state-owned or state-controlled entities. Importantly, United States authorities deem most healthcare professionals and other employees of foreign hospitals, clinics, research facilities and medical schools in countries with public healthcare and/or public education systems to be “foreign officials” under the FCPA. When we interact with foreign healthcare professionals and researchers in testing and marketing our products abroad, we must have policies and procedures in place sufficient to prevent us and agents acting on our behalf from providing any bribe, gift or gratuity, including excessive or lavish meals, travel or entertainment in connection with marketing our products and services or securing required permits and approvals. The FCPA also obligates companies whose securities are listed in the United States to comply with accounting provisions requiring us to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations. We have a policy entitled “Anti-Bribery and Anti-Corruption” that seeks to fully comply with the FCPA.
Foreign Regulations
When we market our tests outside of the United States, we will be subject to foreign regulatory requirements governing laboratory licensure, human clinical testing, use of tissue, privacy and data security, and marketing approval for our tests. These requirements vary by jurisdiction, differ from those in the United States, and may require us to implement additional compliance measures or perform additional pre-clinical or clinical testing. In the European Union, we may be subject to newly enacted legislation that imposes requirements and restrictions on medical devices and in vitro diagnostics; that legislation will become effective in 2020 (for medical devices) and 2022 (for in vitro diagnostics). In light of the ongoing COVID‑19 pandemic, European legislators have voted to delay the effective date of the new Medical Devices Regulation by one year (to May 26, 2021), although to date the May 2022 effective date for the In Vitro Diagnostic Regulation has not been delayed.
In addition, we will also be subject to the E.U. General Data Protection Regulation, or the GDPR, that significantly regulates the possession, use, and disclosure of personal information. In many countries outside of the United States, coverage, pricing, and reimbursement approvals are also required. We are also required to maintain accurate information and control over sales and distributors’ activities that may fall within the purview of the FCPA, our books and records provisions, and our anti-bribery provisions.
Employees
As of December 31, 2020, we had 118 employees, 114 of which were full-time employees, including 10 engaged in research and development, four in clinical operations, 23 in general and administrative, 22 in laboratory operations, and 59 in sales and marketing. We also engage consultants in various areas. None of our employees are represented by a labor union and we believe that our relationships with our employees and contractors are good.
Corporate and Other Information
We incorporated in the British Virgin Islands in 2015 and domesticated in the state of Delaware in 2019. DermTech Operations was incorporated in California in 1995 and reincorporated in the state of Delaware on May 15, 2014. Our principal offices are located at 11099 North Torrey Pines Road, Suite 100, La Jolla, California 92037. Our telephone number is (858) 450-4222 and our website address is www.dermtech.com. The information contained on, or that can be accessed through, our website is not a part of this report, and our reference to the address for our website is intended to be an inactive textual reference only.
27
Table of Contents
Item 1A. Risk Factors
The Company is in a market environment that cannot be predicted and that involves significant risks, many of which are beyond our control. Before making a decision to invest in, hold or sell our common stock, stockholders and potential stockholders should carefully consider the risks and uncertainties described below, in addition to the other information contained in this report, as well as the other information we file with the SEC. If any of the following risks are realized, our business, financial condition, results of operations and prospects could be materially and adversely affected. In that case, the value of our common stock could decline and stockholders may lose all or part of their investment. Furthermore, additional risks and uncertainties of which we are currently unaware, or which we currently consider to be immaterial, could have a material adverse effect on our business, financial condition or results of operations.
Summary Risk Factors
Our business is subject to numerous risks and uncertainties, including those highlighted in the section entitled “Risk Factors,” that represent challenges that we face in connection with the successful implementation of our strategy and growth of our business. The occurrence of one or more of the events or circumstances described in the section entitled “Risk Factors,” alone or in combination with other events or circumstances, may have an adverse effect on our business, financial condition, results of operations, and prospects. Such risks include, but are not limited to:
Risks Relating to Our Financial Condition and Capital Requirements
| • | We are an emerging growth company with a history of net losses; we expect to incur net losses in the future and may never achieve profitability. |
| • | We have a limited operating history and we expect a number of factors to cause our operating results to fluctuate on a quarterly and annual basis, which may make our future performance difficult to predict. |
| • | Our financial condition, commercialization efforts and results of operations could be adversely affected by the ongoing COVID‑19 pandemic. |
| • | Our commercial success could be compromised if customers do not pay our invoices or if commercial payors, including managed care organizations and Medicare, do not provide coverage and reimbursement, breach, rescind, or modify their contracts or reimbursement policies, reimburse at a low rate, or delay payments for our current test and our planned tests. |
| • | We will need to raise additional capital to fund our existing operations, commercialize our products, and expand our operations. |
| • | If clinicians, including dermatologists, decide not to order the PLA or our future tests, we may be unable to generate sufficient revenue to sustain our business. |
| • | We expect to continue to incur significant expenses to develop and market our existing and planned tests, which could make it difficult for us to achieve and sustain profitability. |
| • | We may not be able to generate sufficient revenue from the commercialization of PLA, or successfully develop and commercialize other tests to achieve or sustain profitability. |
| • | If we are unable to execute our marketing strategy for PLA and are unable to gain acceptance in the market, we may be unable to generate sufficient revenue to sustain our business. |
| • | The telemedicine market is immature and unpredictable, and if it does not develop, if it develops more slowly than we expect, if it encounters negative publicity or if limitations on reimbursement or difficulties in obtaining regulatory approvals impede our ability to adopt telemedicine, the growth of our business will be harmed. |
| • | If we cannot develop tests to keep pace with rapid advances in technology, medicine and science, our operating results and competitive position could be harmed. |
| • | Our future success will depend in part upon our ability to enhance PLA, and to develop, introduce, and commercialize other novel innovative and non-invasive diagnostics tests and services; new test development involves a lengthy and complex process and we may be unable to commercialize new or improved tests or any other products we may develop on a timely basis, or at all. |
| • | We rely on a limited number of suppliers and, in some cases, a single supplier, for certain of our laboratory substances, equipment and other materials, and any delays or difficulties securing these materials could disrupt our laboratory operations and materially harm our business. |
| • | Our test employs a novel diagnostic platform and may never be accepted by its intended markets. |
28
Table of Contents
| • | If our current test and our planned tests do not to perform as expected, as a result of human error or otherwise, it could have a material adverse effect on our operating results, reputation, and business. |
| • | If our sole laboratory facility becomes damaged or inoperable, or we are required to vacate the facility, our ability to sell and provide molecular tests and pursue our R&D efforts may be jeopardized. |
| • | If we cannot compete successfully with our competitors, we may be unable to increase or sustain our revenues or achieve and sustain profitability. |
| • | We may encounter manufacturing problems or delays that could result in lost revenue. |
| • | If we cannot support demand for our current test and our planned future tests, including successfully managing the evolution of our technology and manufacturing platforms, our business could suffer. |
| • | If we were to be sued for product or professional liability, we could face substantial liabilities that exceed our resources. |
| • | We may acquire other businesses, form joint ventures, or make investments in other companies or technologies that could harm our operating results, dilute our stockholders’ ownership, increase our debt, or cause us to incur significant expense. |
| • | International expansion of our business would expose us to business, regulatory, political, operational, financial, and economic risks associated with doing business outside of the United States. |
| • | Declining general economic and business conditions as a result of the COVID‑19 pandemic have had a negative impact on our business, and the extent and duration of the effects of the COVID‑19 pandemic and economic downturn are difficult to predict, which makes our future performance more difficult to predict. |
| • | Intrusions into our computer systems could compromise confidential information and our ability to continue operations. |
| • | We rely on FedEx Corporation and United Parcel Service, Inc. to distribute our Adhesive Skin Sample Collection Kits to customers and transport specimens back to our laboratory facility, and any damage to their facilities or inability to deliver our products could have a material adverse effect on our results of operations and business. |
Regulatory Risks Related to Our Business
| • | Changes in health care law and policy may have a material adverse effect on our financial condition, results of operations, and cash flows. |
| • | Our business could be adversely impacted by our failure or clinicians’ failure to comply with the ICD-10-CM Code Set. |
| • | Billing for our test is complex, and we must dedicate substantial time and resources to the billing process to be paid for our test; long payment cycles of Medicare, Medicaid, and/or other commercial payors, or other payment delays, could hurt our cash flows and increase our need for working capital. |
| • | Our business could be harmed by the loss, suspension, or other restriction on a license, certification, or accreditation, or by the imposition of a fine or penalties, under CLIA, its implementing regulations, or other state, federal, and foreign laws and regulations affecting licensure or certification, or by future changes in these laws or regulations. |
| • | If the FDA were to begin requiring approval or clearance of our current test and our planned future tests, or our proprietary specimen collection kit, we could incur substantial costs and time delays associated with meeting the requirements. |
| • | If we were to be required by the FDA to conduct additional clinical studies or trials before continuing to offer tests that we have developed or may develop as LDTs, those studies or trials could lead to delays or failure to obtain necessary regulatory clearance or approval, which could cause significant delays in commercializing any future products and harm our ability to achieve profitability. |
| • | We are subject to numerous federal, local and foreign laws and regulations; complying with laws pertaining to our business is an expensive and time-consuming process, and any failure to comply could result in substantial penalties and a material adverse effect to our business and operations. |
Intellectual Property Risks Related to Our Business
| • | If we are unable to maintain intellectual property protection, our competitive position could be harmed. |
Risks Related to Our Securities
| • | Future issuances of equity securities may dilute the interests of our security holders and reduce the price of our securities. |
29
Table of Contents
Risks Relating to Our Financial Condition and Capital Requirements
We are an emerging growth company with a history of net losses; we expect to incur net losses in the future and may never achieve profitability.
We have historically incurred substantial net losses in each year since our inception, including net losses of $35.2 million for the twelve months ended December 31, 2020. As of December 31, 2020, we had an accumulated deficit of $126.4 million.
We expect our losses to continue as a result of costs relating to ongoing R&D and for increased sales and marketing costs for existing and planned products. These losses have had, and will continue to have, an adverse effect on our working capital, total assets, and stockholders’ equity. Because of the numerous risks and uncertainties associated with our commercialization efforts, we are unable to predict when we will become profitable, and we may never become profitable. Even if we achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our inability to achieve and then maintain profitability would negatively affect our business, financial condition, results of operations, and cash flows.
We have a limited operating history and we expect a number of factors to cause our operating results to fluctuate on a quarterly and annual basis, which may make our future performance difficult to predict.
We are an emerging molecular diagnostics company with a limited operating history. Our operations to date have been primarily focused on developing and market testing our technology. We have not obtained regulatory approvals from the Food and Drug Administration, or FDA, for any of our existing and planned tests as we operate a clinical laboratory under the CLIA guidelines and believe our test are laboratory developed tests, or LDTs, that are not currently being regulated by the FDA. Consequently, if regulatory approval is determined to be necessary or if Congress enacts legislation that alters the regulatory framework for LDTs, any predictions made about our future success or viability may not be as accurate as they could be if we had a longer operating history or more commercialized products. Our financial condition and operating results have varied significantly in the past and will continue to fluctuate from quarter-to-quarter or year-to-year due to a variety of factors, many of which are beyond our control. Factors relating to our business that may contribute to these fluctuations include other factors described elsewhere in this report and also include:
| • | our ability to obtain additional funding to develop and market our existing and planned products and tests; |
| • | the market adoption and demand for our existing and planned tests; |
| • | the existence of favorable or unfavorable clinical guidelines for our existing and planned tests; |
| • | the reimbursement of our existing or planned tests by Medicare and commercial payors; |
| • | our ability to obtain and maintain any necessary regulatory approval for any of our existing and planned tests in the United States and foreign jurisdictions, if required; |
| • | potential side effects of our existing and planned tests that could delay or prevent commercialization, limit the use of our existing and planned tests, or cause any of our commercialized test to be taken off the market; |
| • | our dependence on third-party suppliers and manufacturers, to supply or manufacture our specimen collection products; |
| • | our ability to establish or maintain collaboration, licensing, or other arrangements; |
| • | our ability to maintain and grow an effective sales and marketing infrastructure, either through the expansion of our commercial infrastructure or through strategic collaborations; |
| • | competition from existing and planned tests or new tests that may emerge; |
30
Table of Contents
| • | the ability of patients or healthcare providers to obtain coverage of or sufficient reimbursement for our existing and planned tests; |
| • | our ability to leverage our proprietary technology platform to discover and develop additional test candidates; |
| • | our ability to successfully obtain, maintain, defend, and enforce intellectual property rights important to our business; |
| • | our ability to attract and retain key personnel to manage our business effectively; |
| • | our ability to build our finance infrastructure and improve our accounting systems and controls; |
| • | potential product liability claims; |
| • | potential liabilities associated with hazardous materials; and |
| • | our ability to obtain and maintain adequate insurance policies. |
Accordingly, the results of any quarterly or annual periods should not be relied upon as indications of future operating performance.
Our financial condition, commercialization efforts and results of operations could be adversely affected by the ongoing COVID‑19 pandemic.
Any outbreak of a contagious disease, such as the current COVID‑19 pandemic, or other adverse public health developments, could have a material and adverse effect on our business operations. Such adverse effects could include disruptions or restrictions on the ability of our, our collaborators’, or our suppliers’ personnel to travel, and could result in temporary closures of our facilities or the facilities of our collaborators or suppliers, including our sole laboratory.
As COVID‑19 continues to affect individuals and businesses around the globe, we will likely experience disruptions that could severely impact our business, including:
| • | closure of or reduced access to clinician offices, which would limit our ability to market our test to clinicians and limit clinicians’ ability to offer our test to patients; |
| • | patient concerns about going to clinicians’ offices to have our test administered in person, even if offices are open; |
| • | difficulties in transitioning to marketing our telemedicine option for the PLA or processing test results for our telemedicine option, which we recently initiated on an accelerated basis due to the COVID‑19 environment; |
| • | dependence to a substantial extent on the willingness of clinicians and their patients to use our telemedicine option, as well as on our ability to demonstrate the value of our telemedicine option to payors; |
| • | limitations on reimbursement, which could impede its adoption by clinicians and patients; |
| • | limitations on employee resources that would otherwise be focused on our commercialization and sales efforts, including because of sickness of employees or their families or requirements imposed on employees to avoid contact with large groups of people; |
| • | delays in our third-party suppliers’ ability to manufacture our collection kit, including because of interruptions in shipping that may affect the transport of required materials; |
| • | delays or difficulties marketing our test to new commercial payors, including due to layoffs, furloughs or diversion of attention of payor employees responsible for negotiating coverage contracts for our PLA; |
| • | interruptions in our laboratory operations, including because of the inability of our suppliers to timely obtain laboratory reagents, equipment or other materials due to increased global demand; |
| • | loss of patient insurance coverage due to unemployment caused by COVID‑19, which would likely result in a decline in our sales growth if and as we secure additional insurance contracts; and |
| • | interruption of our clinical studies due to quarantines or other limitations on travel or access to facilities imposed or recommended by federal, state or local governments, employers or others. |
In addition, the continued spread of COVID‑19 globally and implementation of mitigation measures could adversely affect our manufacturing and supply chain. Parts of our direct and indirect supply chain are located overseas and may accordingly be subject to restrictions on export to the U.S. or other disruptions. Additionally, our results of operations have been adversely affected by COVID‑19 and such effects could be expected to worsen to the extent that the COVID‑19 pandemic persists and continues to harm the U.S. economy in general. The extent to which COVID‑19 affects our operations will depend on future developments, which are highly uncertain and cannot be predicted with confidence, including the duration of the pandemic, additional information that may emerge
31
Table of Contents
concerning the severity of COVID‑19 and ongoing actions to contain COVID‑19 or mitigate its impact, among others, which could have a further adverse effect on our business, financial condition, results of operations, and cash flows.
We expect to continue to incur increased costs as a result of operating as a public company and our management will be required to devote substantial time to compliance initiatives and corporate governance practices.
As a public company, we incur and expect to continue to incur additional significant legal, accounting and other expenses in relation to our status as a public reporting company. We expect that these expenses will further increase after we are no longer an “emerging growth company.” We may need to hire additional accounting, finance and other personnel in connection with our continuing efforts to comply with the requirements of being a public company, and our management and other personnel will need to continue to devote a substantial amount of time towards maintaining compliance with these requirements. In addition, the Sarbanes-Oxley Act of 2002 and rules subsequently implemented by the SEC and The Nasdaq Stock Market LLC have imposed various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly.
Pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, we will be required to furnish a report by our management on our internal controls over financial reporting, including an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. However, while we remain an “emerging growth company” and a non-accelerated filer, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. If we identify one or more material weaknesses, this could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our consolidated financial statements.
Our commercial success could be compromised if customers do not pay our invoices or if commercial payors, including managed care organizations and Medicare, do not provide coverage and reimbursement, breach, rescind, or modify their contracts or reimbursement policies, reimburse at a low rate, or delay payments for our current test and our planned future tests.
Clinicians, including dermatologists, may not order our PLA, our PLAplus test when it becomes available, or our planned tests unless commercial payors, such as managed care organizations and government payors (e.g., Medicare and Medicaid), pay a substantial portion of the test price. Coverage and reimbursement by a commercial payor may depend on a number of factors, including a payor’s determination that tests using our technologies are:
| • | not experimental or investigational; |
| • | appropriate for the specific patient; |
| • | supported by peer-reviewed publications; and |
| • | included in clinical practice guidelines. |
Uncertainty surrounds commercial payor reimbursement of any test incorporating new technology, including tests developed using our technologies. Technology assessments of new medical tests conducted by research centers and other entities may be disseminated to interested parties for informational purposes. Commercial payors and health care providers may use such technology assessments as grounds to deny coverage for a test or procedure. Technology assessments can include evaluation of clinical utility studies, which define how a test is used in a particular clinical setting or situation.
32
Table of Contents
Because each payor generally determines for its own enrollees or insured patients whether to cover or otherwise establish a policy to reimburse our test, seeking payor approvals is a time-consuming and costly process. We cannot be certain that coverage for our current test and our planned future tests will be provided in the future by additional commercial payors or that existing policy decisions or reimbursement levels will remain in place or be fulfilled under existing terms and provisions. In addition, the coding procedure used by all commercial payors with respect to establishing payment rates for various procedures, including our test, is complex, does not currently adapt well to the genetic tests we perform and may not enable coverage or adequate reimbursement rates for our test. If we cannot obtain or maintain coverage and reimbursement from commercial payors and governmental payors such as Medicare and Medicaid for our current test, or new tests or test enhancements that we may develop in the future, our ability to generate revenues could be limited, which may have a material adverse effect on our financial condition, results of operations, and cash flows. Measures have been undertaken to reduce payment rates for and decrease utilization of the clinical laboratory testing generally, including the Protecting Access to Medicare Act of 2014, or PAMA, which has resulted in reduced rates on the CLFS. These reductions may also impact our PLA test and tests we develop in the future. Because of the cost-trimming trends, commercial payors that cover and provide reimbursement for our test and our planned tests may suspend, revoke, or discontinue coverage at any time, or may reduce the reimbursement rates payable to us. Any such action could have a negative impact on our revenues, which may have a material adverse effect on our financial condition, results of operations, and cash flows. Additionally, if we are not able to obtain sufficient clinical information in support of our test, commercial payors could designate our test as experimental or investigational and decline to cover and reimburse our test because of this designation. As a result of these factors, obtaining approvals from commercial payors to cover our test and establishing adequate reimbursement levels is an unpredictable, challenging, time-consuming, and costly process, and we may never be successful. Further, we have experienced in the past, and will likely experience in the future, delays and interruptions in the receipt of payments from commercial payors due to missing documentation and/or other issues, which could cause delay in recognizing our revenue.
Additionally, we are currently considered a “non-contracted provider” or “out of network” by most private commercial payors because we have not entered into a specific contract to provide tests to their insured patients at specified rates of reimbursement. We also may be considered now or later to be designated as an “out of network” lab by private commercial payors, who may deny our claims in whole or in part as a result. If we were to become a contracted provider with one or more payors in the future, the amount of overall reimbursement we receive would likely decrease because we could be reimbursed less money per test performed at a contracted rate than at a non-contracted rate, which could have a negative impact on our revenues. Further, we pursue payment of patient co-payments, co-insurance and deductibles, but we typically do not collect substantial payments from patients and therefore experience overall loss to revenue as a result.
We will need to raise additional capital to fund our existing operations, commercialize our products, and expand our operations.
As of December 31, 2020, our cash and cash equivalents totaled approximately $24.2 million and short-term marketable securities totaled $39.5 million. On February 28, 2020, we entered into a securities purchase agreement with certain institutional investors for a private placement, which closed on March 4, 2020, of our equity securities for aggregate gross proceeds of approximately $65.0 million, and net proceeds to the Company of approximately $59.9 million, after deducting estimated offering expenses payable by the Company. On November 10, 2020, we entered into a sales agreement to sell shares of our common stock having aggregate sales proceeds of up to $50.0 million from time to time. In connection with this sales agreement, we raised aggregate gross proceeds of approximately $20.0 million, and net proceeds to the Company of approximately $19.1 million during 2020. On January 11, 2021, the Company completed an underwritten public offering of our common stock for aggregate gross proceeds of approximately $143.7 million, and net proceeds to the Company of approximately $134.6 million, after deducting estimated offering expenses payable by the Company.
Based on our current business operations and the additional financing completed in January 2021, we believe our current cash and cash equivalents, will be sufficient to meet our anticipated cash requirements for at least the next twelve months. We anticipate that we will need to raise additional capital through equity offerings, debt financings, collaborations, or licensing arrangements in the future in order to satisfy our anticipated liquidity requirements. We may also consider raising additional capital in the future to expand our business, to pursue strategic investments, to take advantage of financing opportunities, or for other reasons, including to:
| • | increase our efforts to drive market adoption of our test and address competitive developments; |
| • | fund research and development activities and efforts of commercializing future products; |
| • | acquire, license, or invest in technologies; |
| • | acquire or invest in complementary businesses or assets; and |
| • | finance capital expenditures and general and administrative expenses. |
33
Table of Contents
Our present and future funding requirements will depend on many factors, including:
| • | our revenue growth rate and ability to generate cash flows from operating activities; |
| • | our sales and marketing and R&D activities; |
| • | effects of competing technological and market developments; |
| • | costs of and potential delays in product development; |
| • | changes in regulatory oversight applicable to our test; and |
| • | timing of and costs related to future international expansion. |
The various ways we could raise additional capital carry potential risks. If we raise funds by issuing equity securities, dilution to our stockholders could result. Any equity securities issued also could provide for rights, preferences, or privileges senior to those of holders of our common stock. If we raise funds by issuing debt securities, those debt securities would have rights, preferences, and privileges senior to those of holders of our common stock. The terms of debt securities issued or borrowings pursuant to a credit agreement could impose significant restrictions on our operations. If we raise funds through collaborations and licensing arrangements, we might be required to relinquish significant rights to our platform technologies or products, or grant licenses on terms that are not favorable to us. Additional equity or debt financing might not be available on reasonable terms, if at all. If we cannot secure additional funding when needed, we may have to delay, reduce the scope of, or eliminate one or more R&D programs or sales and marketing initiatives. In addition, we may have to work with a partner on one or more of our development programs, which could lower the economic value of those programs to us. We will also need to raise additional capital to expand our business to meet our long-term business objectives. Additional financing may be from the sale of equity or convertible or other debt securities in a public or private offering, from a credit facility or strategic partnership coupled with an investment in us, or a combination of both. For further discussion of our liquidity requirements as they relate to our long-term plans, see the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations –Liquidity and Capital Resources.”
Our cash, cash equivalents and short-term marketable securities are subject to economic risk.
The Company invests its cash, cash equivalents and short-term marketable securities in domestic bank deposits, money market funds, U.S. Government debt securities, corporate debt, and certificates of deposit. Certain types of these investments are subject to general credit, liquidity, market and interest rate risks. In the event these risks caused a decline in value of any of the Company’s investments, it could adversely affect the Company’s financial condition.
If clinicians, including dermatologists, decide not to order the PLA, or our future tests, we may be unable to generate sufficient revenue to sustain our business.
To generate demand for our current test and our planned tests, we will need to educate dermatologists and other health care professionals on the clinical utility, benefits, and value of the tests we provide through published papers, presentations at scientific conferences, educational programs, and one-on-one education sessions by members of our sales force. In addition, we need to assure dermatologists of their ability to obtain and maintain adequate reimbursement coverage from commercial payors for the adhesive patch sample collection method. Medical professionals are influenced by standard-setting bodies that influence and/or dictate the standard of care. If we are not successful in changing current guidelines from legacy standards to new molecular-based approaches our market adoption will suffer. If we cannot convince medical practitioners to order our current test and our planned tests, we will likely be unable to create demand in sufficient volume for us to achieve profitability or meet our anticipated revenue projections.
We expect to continue to incur significant expenses to develop and market our existing and planned tests, which could make it difficult for us to achieve and sustain profitability.
In recent years, we have incurred significant costs in connection with the development of our existing and planned tests. For the twelve months ended December 31, 2020, our R&D expenses were $5.3 million, our sales and marketing expenses were $16.1 million and our general and administrative expenses were $13.8 million. For the twelve months ended December 31, 2019, our R&D expenses were $2.5 million, our sales and marketing expenses were $6.3 million and our general and administrative expenses were $8.9 million. We expect our expenses to continue to increase for the foreseeable future as we conduct studies of our existing test and planned tests, grow our sales and marketing organization, drive adoption of and reimbursement for our test, and develops new tests. As a result, we need to generate significant revenues in order to achieve profitability.
34
Table of Contents
We may not be able to generate sufficient revenue from the commercialization of PLA and the planned PLAplus test, or successfully develop and commercialize other tests to achieve or sustain profitability.
We launched the PLA assay during the first half of 2016 and anticipate launching the PLAplus test in 2021. We are in varying stages of R&D for other tests that we may offer in the future. We believe that our commercialization success is dependent upon our ability to significantly increase the number of customers who are using our test. In addition, demand for our test may not increase as quickly as planned and we may be unable to increase our revenue levels as expected. We are currently not profitable. Even if we succeed in increasing adoption of PLA test by dermatologists, in maintaining and creating relationships with our existing and new customers, and developing and commercializing additional molecular diagnostic testing products, we may not be able to generate sufficient revenue to achieve or sustain profitability.
If we are unable to execute our marketing strategy for PLA and are unable to gain acceptance in the market, we may be unable to generate sufficient revenue to sustain our business.
Although we believe that our current test and planned future tests represent a promising commercial opportunity, our test may never gain significant acceptance in the marketplace and therefore may never generate substantial revenue or profits for us. We will need to establish a market for our test and build that market through clinician education, awareness programs, and the publication of clinical trial results. Gaining acceptance in medical communities requires publication in leading peer-reviewed journals of results from studies using our current test and/or our planned future tests. The process of publication in leading medical journals is subject to a peer-review process and peer-reviewers may not consider the results of our studies sufficiently novel or worthy of publication. Failure to have our studies published in peer-reviewed journals would limit the adoption of our current test and our planned tests.
Our ability to successfully market the tests that we develop will depend on numerous factors, including:
| • | conducting clinical utility studies of such tests in collaboration with key thought leaders to demonstrate their use and value in important medical decisions such as treatment selection; |
| • | the success of our sales force; |
| • | whether health care providers believe such tests provide clinical utility; |
| • | whether the medical community accepts that such tests are sufficiently sensitive and specific to be meaningful in patient care and treatment decisions; and |
| • | whether health insurers, government health care programs, and other commercial payors will cover and pay for such tests and, if so, whether they will adequately reimburse us. |
Failure to achieve widespread market acceptance of our current test and our planned future tests would materially harm our business, financial condition, and results of operations.
The telemedicine market is immature and unpredictable, and if it does not develop, if it develops more slowly than we expect, if it encounters negative publicity or if limitations on reimbursement or difficulties in obtaining regulatory approvals impede our ability to adopt telemedicine, the growth of our business will be harmed.
With respect to our telemedicine solution for the PLA, the telemedicine market is relatively new and unproven, and it is uncertain whether it will achieve and sustain high levels of demand, consumer acceptance and market adoption. Our success will depend to a substantial extent on the willingness of clinicians and their patients to use our telemedicine solution, as well as on our ability to demonstrate the value of our telemedicine solution to commercial payors and other purchasers of healthcare for beneficiaries. To the extent the COVID-19 pandemic subsides, as a result of the distribution of an effective vaccine or otherwise, and patient access to clinician offices for in-person testing improves, the demand for our telemedicine solution could be adversely affected. Negative publicity concerning our telemedicine solution or the telemedicine market as a whole could limit market acceptance of our solution. If clinicians or their patients do not believe that our telemedicine solution can provide melanoma testing as accurate as our clinical studies have already demonstrated, or if clinicians or their patients are not willing to utilize the clinician-supervised remote collection process then a market for our solution may be slow to develop, or may not develop at all. Changes by state professional licensing boards to the standards of care or other requirements governing the practice of telemedicine, including any such requirements from federal regulatory bodies, could impact the success of our telemedicine solution. Additionally, reimbursement may not be available from government and commercial payors for the teledermatology services or remote collection supervision services that are provided by clinicians as part of our telemedicine solution. Similarly, individual and healthcare industry concerns or negative publicity regarding patient confidentiality and privacy in the context of telemedicine could limit market acceptance of our solution. If any of these events occurs, it could have a material adverse effect on our business, financial condition or results of operations, especially given the ongoing COVID-19 pandemic and patients’ reduced access to clinician offices for testing.
35
Table of Contents
If we cannot develop tests to keep pace with rapid advances in technology, medicine and science, our operating results and competitive position could be harmed.
In recent years, there have been numerous advances in technologies relating to the molecular diagnosis for cancer and other medical conditions. Several new cancer drugs have been approved, including several for melanoma, and a number of new drugs in clinical development may increase patient survival time. There have also been advances in methods used to identify patients likely to benefit from these drugs based on analysis of biomarkers. We must continuously develop new tests and enhance any existing test to keep pace with evolving standards of care. Our current test and our planned tests could become obsolete unless we continually innovate and expand them to demonstrate benefit in the diagnosis, monitoring, or prognosis of patients with cancer and other dermatologic conditions. If we cannot adequately demonstrate the applicability of our current test and our planned future tests to new diagnostic and treatment developments, sales of our test could decline, which would have a material adverse effect on our business, financial condition, results of operations and cash flows.
Our future success will depend in part upon our ability to enhance PLA, and to develop, introduce, and commercialize other novel innovative and non-invasive diagnostics tests and services; new test development involves a lengthy and complex process and we may be unable to commercialize new or improved tests or any other products we may develop on a timely basis, or at all.
Our future success will depend in part upon our ability to enhance PLA, and to develop new innovative products. Our failure to successfully develop new products on a timely basis could have a material adverse effect on our revenue, results of operations, and business.
The development of new or enhanced tests is a complex and uncertain process requiring precise technological execution. In addition, the successful development of new products may depend on the development of new technologies. We may be required to undertake time-consuming and costly development activities. We may experience difficulties that could delay or prevent the successful development, commercialization, and marketing of these new products. Before we can commercialize any new products, we will need to expend significant funds in order to conduct substantial R&D, including validation studies.
Our product development process involves a high degree of risk, and product development efforts may fail for many reasons, including a failure to demonstrate the performance of the product or an inability to obtain any required certification or regulatory approval, if required.
As we develop new tests and other products, we will have to make significant investments in product development, as well as sales and marketing resources. In addition, competitors may develop and commercialize competing products faster than we are able to do so, which could have a material adverse effect on our revenue, results of operations and business.
We rely on a limited number of suppliers and, in some cases, a single supplier, for certain of our laboratory substances, equipment and other materials, and any delays or difficulties securing these materials could disrupt our laboratory operations and materially harm our business.
We rely on a limited number of suppliers for certain of our laboratory substances, including reagents, as well as for the sequencers and various other equipment and materials we use in our laboratory operations. In particular, we rely on Fisher Scientific and VWR for supplies and Adhesive Research for our adhesive tape material. We do not have long-term agreements with any of our suppliers and, as a result, they could cease supplying these materials and equipment to us at any time due to an inability to reach agreement with us on supply terms, disruptions in their operations (including as a result of the ongoing COVID-19 pandemic), a determination to pursue other activities or lines of business, or for other reasons, or they could fail to provide us with sufficient quantities of materials that meet our specifications. Transitioning to a new supplier or locating a temporary substitute, if any are available, would be time-consuming and expensive, could result in interruptions in or otherwise affect the performance specifications of our laboratory operations, or could require that we revalidate our test. In addition, the use of equipment or materials provided by a replacement supplier could require us to alter our laboratory operations and procedures. Moreover, we believe there are currently only a few manufacturers that are capable of supplying and servicing some of the equipment and other materials necessary for our laboratory operations, including sequencers and various associated reagents. As a result, replacement equipment and materials that meet our quality control and performance requirements may not be available on reasonable terms, in a timely manner or at all. If we encounter delays or difficulties securing, reconfiguring or revalidating the equipment, reagents and other materials we require for our test, our operations could be materially disrupted and our business, financial condition, results of operations, and reputation could be adversely affected.
36
Table of Contents
Our test employs a novel diagnostic platform and may never be accepted by its intended markets.
Our future success depends on our ability to successfully commercialize PLA, as well as our ability to develop and market other tests that use our proprietary technology platform. The scientific discoveries that form the basis of our proprietary technology platform and our test is relatively new. We are not aware of any other gene expression tests such as ours and there can be no assurance that clinicians will be willing to use them. If we do not successfully develop and commercialize our test based upon our technological approach, we may not become profitable and the value of our common stock may decline.
The novel nature of our existing and planned tests also means that fewer people are trained in or experienced with products of this type, which may make it difficult to find, hire, and retain capable personnel for research, development, and clinical laboratory positions.
Further, our focus solely on genomic tests, as opposed to multiple, more proven technologies for patient diagnosis, increases the risks associated with the ownership of our common stock. If we do not achieve market acceptance for our test, we may be required to change the scope and direction of our product development activities. In that case, we may not be able to identify and implement successfully an alternative product development strategy.
If our current test and our planned tests do not to perform as expected, as a result of human error or otherwise, it could have a material adverse effect on our operating results, reputation, and business.
Our success depends on the market’s confidence that we can provide reliable, high-quality diagnostic results. There is no guarantee that any accuracy we have demonstrated to date will continue, particularly as the number of tests using our assays increases and as the number of different tests that we develop and commercialize expands. We believe that our customers are likely to be particularly sensitive to test defects and errors. As a result, the failure of our current or planned tests to perform as expected could significantly impair our reputation and the public image of our tests. As a result, the failure or perceived failure of our products to perform as expected could have a material adverse effect on our business, financial condition, results of operations and cash flows.
We may be unable to manage our future growth effectively, which could make it difficult to execute our business strategy.
As part of our strategy, we expect to increase our number of employees as our business grows. This future growth could create strain on our organizational, administrative, and operational infrastructure, including laboratory operations, quality control, customer service, and sales and marketing. Our ability to manage our growth properly will require us to continue to improve our operational, financial, and management controls, as well as our reporting systems and procedures. If our current infrastructure is unable to handle our growth, we may need to further expand our infrastructure and staff and implement new reporting systems. The time and resources required to implement such expansion and systems could adversely affect our operations. Our expected future growth will impose significant added responsibilities on members of management, including the need to identify, recruit, maintain, and integrate additional employees. Our future financial performance and our ability to commercialize our products and to compete effectively will depend, in part, on our ability to manage this potential future growth effectively, without compromising quality.
If our sole laboratory facility becomes damaged or inoperable, or we are required to vacate the facility, our ability to sell and provide molecular tests and pursue our R&D efforts may be jeopardized.
We do not have any clinical reference laboratory facilities outside of our facility in La Jolla, California. Our facilities and equipment could be harmed or rendered inoperable by natural or man-made disasters, including fire, earthquake, flooding, and power outages, which may render it difficult or impossible for us to perform our diagnostic test for some period of time. The inability to perform our current test, our planned tests, or the backlog of tests that could develop if our facility is inoperable for even a short period of time may result in the loss of customers or harm to our reputation or relationships with scientific or clinical collaborators, and we may be unable to regain those customers or repair our reputation in the future. Furthermore, our facilities and the equipment we use to perform our R&D work could be costly and time-consuming to repair or replace.
The San Diego area has recently experienced serious fires and power outages, and is considered to lie in an area with earthquake risk.
Additionally, a key component of our R&D process involves using biological samples as the basis for the development of our diagnostic tests. In some cases, these samples are difficult to obtain. If the parts of our laboratory facility where we store these biological samples were damaged or compromised, our ability to pursue our R&D projects, as well as our reputation, could be jeopardized. We carry insurance for damage to our property and the disruption of our business, but this insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, if at all.
Further, if our CLIA-certified laboratory became inoperable, we may not be able to license or transfer our technology to another facility with the necessary state licensure and CLIA certification under which our current test and our planned future tests could be performed. Even if we find a facility with such qualifications to perform our test, it may not be available to us on commercially reasonable terms. In addition, the use of a third-party laboratory to perform our test could affect their classification as LDTs and require us to seek FDA market authorization for the test prior to the completion of such a transfer.
37
Table of Contents
If we cannot compete successfully with our competitors, we may be unable to increase or sustain our revenues or achieve and sustain profitability.
Our principal competition comes from mainstream clinical diagnostic methods, used by dermatologists for many years, which focus on visual tumor tissue analysis. It may be difficult to change the methods or behavior of dermatologists to incorporate our PLA test and Adhesive Skin Sample Collection Kits into their practices in conjunction with, or instead of, tissue biopsies and analysis. In addition, companies offering capital equipment and kits or reagents to local dermatologists represent another source of potential competition. These tests are used directly by the dermatologists, which can facilitate adoption. We plan to focus our marketing and sales efforts on medical dermatologists rather than pathologists.
We also face competition from companies that offer device products or are conducting research to develop device products for analysis of pigmented lesions. In particular, MELA Sciences, Inc., used to market its MelaFind® device to dermatologists, but we believe they no longer actively market this product. Scibase AB and Verisante Technology, Inc. have devices under development and may market their medical products directly to dermatologists if and when they obtain FDA, approval. In addition to these companies, our competitors also include other device companies selling photographic technologies, whole body photography services, dermatoscopes, or confocal microscopy, such as Fotofinder, Molemate, Canfield Scientific, MedX, and Caliber I.D. Many of these groups, in addition to operating R&D laboratories, are selling equipment and devices.
In addition to these device companies, Myriad Genetics, Inc. and Castle Biosciences, Inc. offer an expression test for melanoma that is used on surgical biopsy specimens. Myriad Genetics, Inc. and Castle Biosciences, Inc. could also try and market their test as a biopsy aid at the point-of-care. Genomic testing is a relatively new area of science, especially in dermatology and we cannot predict what tests others will develop that may compete with or provide results similar or superior to the results we are able to achieve with the tests we develop. There are a number of companies that are focused on the oncology diagnostic market and expression tests including Exact Sciences Corporation, Veracyte, Inc., Genomic Health, Inc. and others.
Additionally, projects related to cancer diagnostics and particularly genomics have received increased government funding, both in the United States and internationally. As more information regarding cancer genomics becomes available to the public, we anticipate that more products aimed at analyzing pigmented lesions and identifying melanoma may be developed and that these products may compete with ours. In addition, competitors may develop their own versions of our current or planned tests in countries where we did not apply for patents or where our patents have not issued or have expired and may compete with us in those countries, including encouraging the use of their test by clinicians or patients in other countries. In addition, one or more competitors may seek to invalidate or render unenforceable any of our patents in a court of competent jurisdiction or at the United States Patent and Trademark Office, or USPTO. If any such proceeding were to be successful and result in the invalidation or unenforceability of one or more patents in our intellectual property portfolio, we may be unable to prevent unlicensed third-party competition in the marketplace with respect to our current and planned future tests.
Some of our present and potential competitors have widespread brand recognition and substantially greater financial and technical resources and development, production, and marketing capabilities than we do. Others may develop lower-priced, less complex tests that payors and dermatologists could view as functionally equivalent to our current or planned tests, which could force us to lower the list price of our tests and impact our operating margins and ability to achieve and maintain profitability. In addition, technological innovations that result in the creation of enhanced diagnostic tools that are more sensitive or specific than ours may enable other clinical laboratories, hospitals, clinicians, or medical providers to provide specialized diagnostic tests similar to ours in a more patient-friendly, efficient, or cost-effective manner than is currently possible. If we cannot compete successfully against current or future competitors, we may be unable to increase or create market acceptance and sales of our current or planned tests, which could prevent us from increasing or sustaining our revenues or achieving or sustaining profitability.
Our competitors may be able to respond more quickly and effectively than we can to new or changing opportunities, technologies, standards, or customer requirements. We anticipate that we will face increased competition in the future as existing companies and competitors develop new or improved products and distribution strategies and as new companies enter the market with new technologies and distribution strategies. We may not be able to compete effectively against these organizations. Our ability to compete successfully and to increase our market share is dependent upon our reputation for providing responsive, professional, and high-quality products and services and achieving strong customer satisfaction. Increased competition in the future could adversely affect our revenue, revenue growth rate, if any, margins and market share.
38
Table of Contents
If we are unable to identify collaborators willing to work with us to conduct clinical utility studies, or the results of those studies do not demonstrate that a test provides clinically meaningful information and value, commercial adoption of our test may be slow, which would negatively impact our business.
We believe clinical utility studies will show how the PLA changes the decision-making of the dermatologist when making a surgical biopsy decision, particularly to avoid performing a surgical biopsy when the test is negative. Clinical utility studies also show the impact of the test results on patient care and management. Clinical utility studies are typically performed with collaborating dermatologists at medical centers and hospitals, analogous to a clinical trial, and generally result in peer-reviewed publications.
We are currently conducting a variety of clinical trials for the PLA and other non-melanoma tests with investigators at multiple sites in the U.S. We will need to conduct additional studies for these tests, as well as other tests we may offer in the future, to drive test adoption in the marketplace and reimbursement. Should we not be able to perform these studies, or should their results not provide clinically meaningful data and value for clinicians, including dermatologists and oncologists, adoption of our existing and planned tests could be impaired and we may not be able to obtain reimbursement for them.
We are undergoing a management transition.
Since the beginning of 2019, we have added a number of new executives. Our management reporting structure may continue to change. Such a management transition subjects us to a number of risks, including risks pertaining to coordination of responsibilities and tasks, creation of new management systems and processes, differences in management style, effects on corporate culture, and the need for transfer of historical knowledge. In addition, our operations will be adversely affected if our management does not work together harmoniously, efficiently allocate responsibilities between themselves, or implement and abide by effective controls.
The loss of key members of our executive management team could adversely affect our business.
Our success in implementing our business strategy depends largely on the skills, experience, and performance of key members of our executive management team and others in key management positions, including John Dobak, M.D., the Company’s Chief Executive Officer. The collective efforts of our executive management team are critical to us as we continue to develop our technologies, tests, and R&D and sales programs. As a result of the difficulty in locating qualified new management, the loss or incapacity of existing members of our executive management team could adversely affect our operations. If we were to lose one or more of these key employees, we could experience difficulties in finding qualified successors, competing effectively, developing our technologies, and implementing our business strategy. Each member of our executive management team has an employment agreements; however, the existence of an employment agreement does not guarantee retention of the members of our executive management team and we may not be able to retain those individuals for the duration of or beyond the end of their respective terms. We do not maintain “key person” life insurance on any of our employees.
In addition, we rely on collaborators, consultants, and advisors, including scientific, clinical and payor advisors, to assist us in formulating our commercialization strategy. Our collaborators, consultants, and advisors are generally employed by employers other than us and may have commitments under agreements with other entities that may limit their availability to us.
The loss of a key employee, the failure of a key employee to perform in his or her current position, or our inability to attract and retain skilled employees could result in our inability to continue to grow our business or to implement our business strategy.
Most of our management has limited experience in operating a public company.
Most of our management team has limited experience in the management of a publicly traded company. Our management team may not successfully or effectively manage our transition to operating as a public company that is subject to significant regulatory oversight and reporting obligations under federal securities laws. Our limited experience in dealing with the increasingly complex laws pertaining to public companies could be a significant disadvantage in that it is likely that an increasing amount of our time may be devoted to these activities which will result in less time being devoted to the management and growth of the Company. It is possible that we will be required to expand our employee base and hire additional employees to support our operations as a public company which will increase our operating costs in future periods.
There is a scarcity of experienced professionals in our industry. If we are not able to retain and recruit personnel with the requisite technical skills, we may be unable to successfully execute our business strategy.
The specialized nature of our industry results in an inherent scarcity of experienced personnel in the field. Our future success depends upon our ability to attract and retain highly skilled personnel, including scientific, technical, laboratory, sales, marketing, business, regulatory, and administrative personnel necessary to support our anticipated growth, develop our business, and perform certain contractual obligations. Given the scarcity of professionals with the scientific knowledge that we require and the competition for qualified personnel among life science businesses, we may not succeed in attracting or retaining the personnel we require to continue and grow our operations.
39
Table of Contents
Our inability to attract, hire, and retain a sufficient number of qualified sales professionals would hamper our ability to launch and increase demand for our PLA, to expand geographically, and to successfully commercialize any other tests or products we may develop.
To succeed in selling our PLA, and any other tests or products that we are able to develop, we must expand our sales force in the United States and/or internationally by recruiting sales representatives with extensive experience in dermatology and close relationships with medical dermatologists, dermatopathologists, and other hospital personnel. To achieve our marketing and sales goals, we will need to substantially build our sales and commercial infrastructure, with which to date we have had little experience. Sales professionals with the necessary technical and business qualifications are in high demand, and there is a risk that we may be unable to attract, hire, and retain the number of sales professionals with the right qualifications, scientific backgrounds, and relationships with decision-makers and potential customers needed to achieve our sales goals. We expect to face competition from other companies in our industry, some of whom are much larger than us and who can pay greater compensation and benefits than we can, in seeking to attract and retain qualified sales and marketing employees. If we are unable to hire and retain qualified sales and marketing personnel, our business will suffer.
We may encounter manufacturing problems or delays that could result in lost revenue.
The Adhesive Skin Sample Collection Kits we distribute are manufactured by a third-party supplier. This manufacturer assembles several components, including the key adhesive patch trifold, into a finished product, then labels, stores, and ships this finished product. The adhesive tape subcomponent of the adhesive patches is provided by a single-source third party. This tape is assembled into the individual adhesive patches by another third-party supplier.
We believe we have arranged for adequate manufacturing capacity for the Adhesive Skin Sample Collection Kits through our third-party manufacturer. If demand for our current test and our planned future tests increases significantly, we will need to either expand manufacturing capabilities through our third-party manufacturer or outsource to other manufacturers. If our third-party or other manufacturers engaged by us fail to manufacture and deliver the Adhesive Skin Sample Collection Kits or certain reagents in a timely manner, or they are unable to fulfil our orders due to regulatory non-compliance or other quality-related issues or due to logistical issues related to the ongoing COVID-19 pandemic, our relationships with our customers could be seriously harmed. We cannot assure you that manufacturing or quality control problems will not arise as we attempt to increase the production of the Adhesive Skin Sample Collection Kit or that we can increase our manufacturing capabilities and maintain quality control in a timely manner or at commercially reasonable costs. If we cannot have the Adhesive Skin Sample Collection Kits manufactured consistently on a timely basis because of these or other factors, it could have a significant negative impact on our ability to perform tests and generate revenues.
If we cannot support demand for our current test and our planned future tests, including successfully managing the evolution of our technology and manufacturing platforms, our business could suffer.
As our test volume grows, we will need to increase our testing capacity, implement automation, increase our scale and related processing, customer service, billing, collection, and systems process improvements, and expand our internal quality assurance program and technology to support testing on a larger scale. We will also need additional technicians, certified laboratory scientists, and other scientific and technical personnel to process these additional tests. Any increases in scale, related improvements and quality assurance may not be successfully implemented and appropriate personnel may not be available. As additional tests are commercialized, we may need to implement new equipment, systems, technology, controls and procedures, and hire personnel with different qualifications. Failure to implement necessary procedures or to hire the necessary personnel could result in a higher cost of processing or an inability to meet market demand. We cannot assure you that we will be able to perform tests on a timely basis at a level consistent with demand, that our efforts to scale our commercial operations will not negatively affect the quality of our test results or that we will respond successfully to the growing complexity of our testing operations. If we encounter difficulty meeting market demand or quality standards for our current test and our planned future tests, our reputation could be harmed and our future prospects and business could suffer, which may have a material adverse effect on our financial condition, results of operations, and cash flows.
If we were to be sued for product liability or professional liability, we could face substantial liabilities that exceed our resources.
The marketing, sale, and use of our current test and our planned future diagnostic tests could lead to the filing of product liability claims against us if someone alleges that our tests failed to perform as designed. We may also be subject to liability for errors in the test results we provide to clinicians or for a misunderstanding of, or inappropriate reliance upon, the information we provide. A product liability or professional liability claim could result in substantial damages and be costly and time-consuming for us to defend.
Although we believe that our existing product and professional liability insurance is adequate, our insurance may not fully protect us from the financial impact of defending against product liability or professional liability claims. Any product liability or professional liability claim brought against us, with or without merit, could increase our insurance rates or prevent us from securing insurance coverage in the future. Additionally, any product liability lawsuit could damage our reputation, result in the recall of tests, or cause current partners to terminate existing agreements and potential partners to seek other partners, any of which could impact our results of operations.
40
Table of Contents
If we use biological and hazardous materials in a manner that causes injury or violates laws or regulations, we could be liable for damages or subject to enforcement actions.
Our activities currently require the controlled use of potentially harmful biological and hazardous materials and chemicals. We cannot eliminate the risk of accidental contamination or injury to employees or third parties from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for any resulting damages, and any liability could exceed our resources or any applicable insurance coverage we may have. Additionally, we are subject to, on an ongoing basis, federal, state, and local laws and regulations governing the use, storage, handling, and disposal of these materials and specified waste products. The cost of compliance with these laws and regulations may become significant and could have a material adverse effect on our financial condition, results of operations and cash flows. In the event of an accident or if we otherwise fail to comply with applicable regulations, we could lose our permits or approvals or be held liable for damages or penalized with fines.
We may acquire other businesses, form joint ventures, or make investments in other companies or technologies that could harm our operating results, dilute our stockholders’ ownership, increase our debt, or cause us to incur significant expense.
As part of our business strategy, we may pursue acquisitions of businesses and assets. We also may pursue strategic alliances and joint ventures that leverage our core technology and industry experience to expand our offerings or distribution. We have no experience with acquiring other companies and limited experience with forming strategic alliances and joint ventures. We may not be able to find suitable partners or acquisition candidates, and we may not be able to complete such transactions on favorable terms, if at all. If we make any acquisitions, we may not be able to integrate these acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. Any future acquisitions also could result in significant write-offs or the incurrence of debt and contingent liabilities, any of which could have a material adverse effect on our financial condition, results of operations and cash flows. Integration of an acquired company also may disrupt ongoing operations and require management resources that would otherwise focus on developing our existing business. We may experience losses related to investments in other companies, which could have a material negative effect on our results of operations. We may not identify or complete these transactions in a timely manner, on a cost-effective basis, or at all, and we may not realize the anticipated benefits of any acquisition, technology license, strategic alliance, or joint venture.
To finance any acquisitions or joint ventures, we may choose to issue shares of our common stock as consideration, which would dilute the ownership of our stockholders. If the price of our common stock is low or volatile, we may not be able to acquire other companies or fund a joint venture project using our stock as consideration. Alternatively, it may be necessary for us to raise additional funds for acquisitions through public or private financings. Additional funds may not be available on terms that are favorable to us, or at all.
International expansion of our business would expose us to business, regulatory, political, operational, financial, and economic risks associated with doing business outside of the United States.
Our business strategy contemplates possible international expansion, including partnering with academic and commercial testing laboratories, and introducing the PLA or other future products outside the United States and exporting the Adhesive Skin Sample Collection Kit. We are currently testing samples through a distributor in Canada. Doing business internationally involves a number of risks, including:
| • | multiple, conflicting, and changing laws and regulations such as tax laws, export and import restrictions, privacy, data security and data transfer laws, employment laws, intellectual property laws, regulatory requirements, and other governmental approvals, permits and licenses; |
| • | failure by us or our distributors to obtain regulatory approvals for the sale or use of our current test and our planned future tests in various countries, if required; |
| • | difficulties in managing foreign operations; |
| • | complexities associated with managing government payor systems, multiple payor-reimbursement regimes, or self-pay systems; |
| • | logistics and regulations associated with shipping blood samples, including infrastructure conditions and transportation delays; |
| • | limits on our ability to penetrate international markets if our current test and our planned future diagnostic tests cannot be processed by an appropriately qualified local laboratory; |
| • | financial risks, such as longer payment cycles, difficulty enforcing contracts and collecting accounts receivable and exposure to foreign currency exchange rate fluctuations; |
| • | reduced protection for intellectual property rights, or lack of them in certain jurisdictions, forcing more reliance on any trade secrets we may have, if such protection is available; |
41
Table of Contents
| • | natural or man-made disasters, political and economic instability, including wars, terrorism and political unrest, outbreak of disease (such as the ongoing COVID‑19 pandemic), boycotts, curtailment of trade, and other business restrictions; and |
| • | failure to comply with the Foreign Corrupt Practices Act, including its books and records provisions and its anti-bribery provisions, by maintaining accurate information and control over sales activities and distributors’ activities, as well as similar foreign anti-bribery and anti-corruption laws that may become applicable to our business. |
Any of these risks, if encountered, could significantly harm our future international expansion and operations and, consequently, have a material adverse effect on our financial condition, results of operations, and cash flows.
Declining general economic and business conditions as a result of the COVID‑19 pandemic have had a negative impact on our business, and the extent and duration of the effects of the COVID‑19 pandemic and economic downturn are difficult to predict, which makes our future performance more difficult to predict.
Economic and business prospects in the United States and other countries have declined rapidly due to the COVID‑19 pandemic and resulting restrictions on individual and business activity to mitigate the pandemic. These factors, coupled with decreased business and consumer confidence and substantial unemployment resulting from the declared global pandemic of COVID‑19 and restrictions on activity, have precipitated a sharp economic slowdown and recession, and the economic climate may deteriorate further. The extent and duration of the effects of the COVID‑19 pandemic and economic downturn are difficult to predict, which makes our future performance more difficult to predict. If the COVID‑19 pandemic and economic downturn persist, or if they worsen, we expect that our business, including our access to patient samples and the addressable market for our tests will continue to be adversely affected, resulting in a further negative impact on our business, financial condition, results of operations and cash flows.
Intrusions into our computer systems could result in compromise of confidential information and our ability to continue operations (in event of a cyber-attack).
Despite the implementation of security measures, our technology or systems that we interface with, including the Internet and related systems, may be vulnerable to physical break-ins, hackers, improper employee or contractor access, computer viruses, programming errors, or similar problems. Any of these might result in confidential medical, business, or payment information, including as may be disclosed as part of a credit card transaction, or other information of other persons or of us, including employees, being revealed to unauthorized persons. Additional use of remote working technology as a result of the COVID‑19 pandemic may increase these vulnerabilities.
We may have to comply with laws governing the use and disclosure of genetic testing information.
Many states have adopted laws governing genetic testing and the use and disclosure of genetic test results. These laws impose specific testing consent requirements and patient authorization requirements for the use and disclosure of test results, and some impose limits on the retention and secondary use of patient samples. Many of these laws are vaguely written and some are overly broad. We must analyze and ensure compliance with the genetic testing laws in the jurisdictions from which we obtain samples and may be required to expend significant capital and other resources to ensure ongoing compliance. Our failure to comply could interfere with our ability to operate and/or lead to sanctions, fines, or other regulatory actions as well as civil claims.
We depend on our information technology and telecommunications systems, and any failure of these systems could harm our business.
We depend on information technology and telecommunications systems for significant aspects of our operations. In addition, our third-party billing and collections provider depends upon telecommunications and data systems provided by outside vendors and information we provide on a regular basis. These information technology and telecommunications systems support a variety of functions, including test processing, sample tracking, quality control, customer service and support, billing and reimbursement, R&D activities, and our general and administrative activities. Information technology and telecommunications systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts, and natural disasters. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses, and similar disruptive problems. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our information technology and telecommunications systems, failures or significant downtime of our information technology or telecommunications systems, or those used by our third-party service providers could prevent us from processing tests, providing test results to oncologists, pathologists, billing payors, processing reimbursement appeals, handling patient or clinician inquiries, conducting R&D activities, and managing the administrative aspects of our business. Any disruption or loss of information technology or telecommunications systems on which critical aspects of our operations depend could have a material effect on our business, financial condition, results of operations and cash flows.
42
Table of Contents
We rely on FedEx Corporation, or FedEx, and United Parcel Service, Inc., or UPS, for the distribution of our Adhesive Skin Sample Collection Kits to customers and to transport specimens back to our laboratory facility and, if FedEx or UPS incurs any damage to their facilities or is unable to deliver our products as needed, it could have a material adverse effect on our results of operations and business.
We rely on FedEx and UPS for the distribution of our Adhesive Skin Sample Collection Kits to customers, as well as to transport patient specimens back to our laboratory facility for processing. The FedEx or UPS facilities involved in such distribution may be harmed or rendered inoperable by natural or man-made disasters, including earthquakes, power outages, communications failure, infectious disease outbreaks, severe weather, or terrorism. Any material destruction to their facilities could adversely affect the ability of FedEx or UPS to meet the needs of our customers. In addition, a disruption or slowdown in the operations of FedEx or UPS, including as a result of the COVID‑19 pandemic and restrictions on business activity, damage to the facilities of FedEx or UPS or a strike by FedEx or UPS employees, could cause delays in our ability to fulfill customer orders and may cause orders to be cancelled, lost, or delivered late, our shipments to be returned, or receipt of shipments to be refused, any of which could adversely affect our business and our results of operations. If our shipping costs were to increase as a result of an increase by FedEx or UPS or as a result of obtaining a new third-party logistics company and if we are unable to pass on these higher costs to our customers, it could have a material adverse effect on our results of operations and business, financial condition, results of operations and cash flows.
Regulatory Risks Related to Our Business
Changes in health care law and policy may have a material adverse effect on our financial condition, results of operations, and cash flows.
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, collectively called the ACA, became law. This law substantially changed the way health care is financed by both governmental and commercial payors, and continues to significantly impact our industry. Since its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA. Members of Congress have expressed their intention to repeal or repeal and replace the ACA, and as a result, certain sections of the ACA have not been fully implemented or were effectively repealed. In December 2019, the Fifth Circuit Court of Appeals upheld a district court’s finding that the individual mandate in the Affordable Care Act is unconstitutional following removal of the penalty provision from the law. However, the Fifth Circuit reversed and remanded the case to the district court to determine if other reforms enacted as part of the Affordable Care Act but not specifically related to the individual mandate or health insurance could be severed from the rest of the Affordable Care Act so as not to have the law declared invalid in its entirety. On March 2, 2020, the United States Supreme Court granted the petitions for writs of certiorari to review this case and allotted one hour for oral arguments, which occurred in on November 10, 2020. A decision from the United States Supreme Court is expected to be issued in spring 2021. It is unclear how such litigation and other efforts to repeal and replace the Affordable Care Act will affect the implementation of that law and our business. The uncertainty around the future of the ACA, and in particular the impact to reimbursement levels and the number of insured individuals, may lead to delay in the purchasing decisions of our customers, which may in turn negatively impact our product sales. Further, if reimbursement levels are inadequate, our business and results of operations could be adversely affected.
In addition to the ACA, there will continue to be proposals by legislators at both the federal and state levels, regulators and commercial payors to reduce costs while expanding individual health care benefits. Certain of these changes could impose additional limitations on the prices we will be able to charge for our test or the amounts of reimbursement available for our test from governmental or commercial payors. Any future changes to legal or regulatory requirements or new cost containment initiatives could have a materially adverse effect on our business, financial condition, results of operation, and cash flows.
Our business could be adversely impacted by our failure or the failure of clinicians to comply with the ICD-10-CM Code Set.
Compliance with ICD-10-CM is required for all claims with dates of service on or after October 1, 2015. We believe we have fully implemented ICD-10-CM. However, our failure to effectively implement and apply the new code set could adversely impact our business. In addition, if clinicians fail to provide appropriate codes for desired tests, we may not be reimbursed for tests we perform.
Billing for our test is complex, and we must dedicate substantial time and resources to the billing process to be paid for our test; long payment cycles of Medicare, Medicaid, and/or other commercial payors, or other payment delays, could hurt our cash flows and increase our need for working capital.
Billing for clinical laboratory testing services is complex, time-consuming, and expensive. Depending on the billing arrangement and applicable law, we will bill various payors, including Medicare, Medicaid, and commercial payors, all of which have different billing requirements. As required by law or contract, we routinely bill patients for co-payments, co-insurance, and deductible amounts owed. We may also face increased risks in our collection efforts, including potential write-offs of doubtful accounts, long collection cycles, and failure by third parties to properly process payment of claims in a timely manner that could adversely affect our business, results of operations, and financial condition. Several factors make the billing practice complex, including:
| • | differences between the list price for our test and the reimbursement rates of payors; |
43
Table of Contents
| • | compliance with complex federal regulations related to Medicare billing; |
| • | disputes among payors as to which party is responsible for payment and resistance by patients to cover any substantial amount of the payment; |
| • | differences in coverage among payors and effect of patient co-payments, co-insurance, or deductibles; |
| • | differences in information and billing requirements among payors; |
| �� | incorrect or missing billing information; and |
| • | the resources required to manage the billing and claims appeals process. |
Additionally, our billing activities require us to implement compliance procedures and oversight, train and monitor our employees, challenge coverage and payment denials, assist patients in appealing claims, and undertake internal audits to evaluate compliance with applicable laws and regulations as well as internal compliance policies and procedures. Payors also conduct external audits to evaluate payments and may seek refunds depending on the audit results, which adds further complexity to the billing process.
Failure to comply with these billing requirements may result in non-payment, refunds, exclusion from government healthcare programs, and civil or criminal liabilities, any of which may have a material adverse effect on our revenues and earnings. These billing complexities and the related uncertainties in obtaining reimbursement could negatively affect our cash flow and our ability to achieve profitability.
Our business could be harmed by the loss, suspension, or other restriction on a license, certification, or accreditation, or by the imposition of a fine or penalties, under CLIA, its implementing regulations, or other state, federal, and foreign laws and regulations affecting licensure or certification, or by future changes in these laws or regulations.
The diagnostic testing industry is subject to extensive laws and regulations, many of which have not been interpreted by the courts. CLIA requires virtually all laboratories to be certified by the federal government and mandates compliance with various operational, personnel, facilities administration, quality, and proficiency testing requirements intended to ensure that testing services are accurate, reliable, and timely. Our clinical laboratory must be certified under CLIA in order for us to perform testing on human specimens. CLIA certification is also a prerequisite to be eligible to bill state and federal health care programs. Further, many commercial payors require CAP accreditation as a condition to contracting with clinical laboratories to cover their tests. In addition, some countries outside the United States require CAP accreditation as a condition to permitting clinical laboratories to test samples taken from their citizens.
We have a current certificate of accreditation from the Centers for Medicare and Medicaid Services, or CMS, to perform high-complexity testing, which is managed by California Laboratory Field Services, or CA LFS. To renew this certificate, we are subject to survey and inspection every two years. We hold a certificate of accreditation because we are accredited by the College of American Pathologists, or CAP, which sets standards than are higher than the CLIA regulations. CAP is an independent, non-governmental organization of board-certified pathologists that accredits laboratories nationwide on a voluntary basis. Because CAP has deemed status with CLIA and hence with CA LFS, our biennial inspections will be performed by teams formed by CAP. Sanctions for failure to comply with CAP or CLIA requirements, including proficiency testing violations, may include suspension, revocation, or limitation of a laboratory’s CLIA certificate, which is necessary to conduct business, as well as the imposition of significant fines or criminal penalties. In addition, we are subject to regulation under state laws and regulations governing laboratory licensure. Two states, one of which is New York, have enacted state licensure laws that are more stringent than CLIA.
Failure to maintain CLIA certification, CAP accreditation, or required state licenses could have a material adverse effect on the sales of our test and the results of our operations. If we were to lose our CLIA certification, CAP accreditation or California laboratory license, whether as a result of a revocation, suspension, or limitation, we would no longer be able to offer our test, which would limit our revenues and harm our business. If we were to lose our license in any other state where we are required to hold a license, we would not be able to test specimens from those states. In addition, state and foreign requirements for laboratory certification may be costly or difficult to meet and could affect our ability to receive specimens from certain states or foreign countries. We receive samples from all 50 U.S. states and certain provinces in Canada. Some states maintain independent licensure, registration, or certification procedures that apply to out-of-state laboratories with which we must maintain compliance in order to receive and test samples from those states. Maintaining compliance with the myriad state and foreign requirements is time consuming and resource intensive and failure to maintain compliance could result in sanctions.
Any sanction imposed under CLIA, its implementing regulations, or state or foreign laws or regulations governing licensure, or our failure to renew a CLIA certificate, a state or foreign license, or accreditation, could have a material adverse effect on our business, financial condition, results of operations and cash flows. If the CLIA certificate of our laboratory is revoked, that could also impact our licensure or certification in the states or in foreign jurisdictions.
44
Table of Contents
If the FDA were to begin requiring approval or clearance of our current test and our planned future tests, or our proprietary specimen collection kit, we could incur substantial costs and time delays associated with meeting requirements for premarket clearance or approval.
The laws and regulations governing the marketing of diagnostic products are evolving, extremely complex and in many instances, there are no significant regulatory or judicial interpretations of these laws and regulations. Pursuant to its authority under the federal Food, Drug, and Cosmetic Act, or FDCA, the FDA has jurisdiction over medical devices, including in vitro diagnostics and, therefore, potentially our clinical laboratory tests. Among other things, pursuant to the FDCA and its implementing regulations, the FDA regulates the research, testing, manufacturing, safety, labeling, storage, recordkeeping, premarket clearance or approval, marketing and promotion, and sales and distribution of medical devices in the United States to ensure that medical products distributed domestically are safe and effective for their intended uses. Although the FDA has asserted that it has authority to regulate the development and use of LDTs, such as our and many other laboratories’ tests, as medical devices, it has generally exercised enforcement discretion and was not otherwise regulating most tests developed and performed within a single high complexity CLIA-certified laboratory. In addition, on August 19, 2020, HHS published a policy announcement that FDA must go through the formal notice-and-comment rulemaking process before requiring premarket review of LDTs rather than making such changes through guidance documents, compliance manuals, or other informal policy statements. Laboratories may still voluntarily submit LDTs to FDA for premarket review, although the agency does not appear to be prioritizing such applications for review at the present time, in light of the HHS announcement. Although the ultimate impact of HHS’s policy statement on FDA’s plans for regulating LDTs and its current thinking relating to such diagnostic testing products is unclear, the August 2020 announcement appears to confirm that laboratories may commercialize LDTs for clinical use without submitting such tests for FDA review and marketing authorization. It is also unclear whether the Biden Administration, which assumed control of the executive branch on January 20, 2021, would take the same position as the former administration or seek to revoke or revise the HHS policy announcement from August 2020.
The FDA could, at any time, engage in notice-and-comment rulemaking with regard to this matter or Congress could take action to amend the law to change the current regulatory framework for in vitro diagnostics and LDTs. For example, the Verifying Accurate, Leading-edge IVCT Development, or VALID, Act introduced in Congress in 2020 has bipartisan support and would codify into law the term “in vitro clinical test” in order to create a new medical product category separate from medical devices that would include products currently regulated as in vitro diagnostics as well as LDTs, as discussed further below.
We believe that our test, as utilized in our clinical laboratory, is and would be LDTs. As a result, we believe that we are not required to obtain regulatory clearances or approvals from the FDA for our LDTs. In addition, we believe the Adhesive Skin Sample Collection Kit we provide for collection and transport of skin samples from a health care provider (or in our recently launched telemedicine option, from the patient directly) to our clinical laboratory is considered a Class I medical device subject to the FDA’s general device controls but exempt from premarket review. However, the FDA could assert the specimen collection kit is non-exempt or is a Class II device, which would subject it to premarket clearance or approval processes, which could be time-consuming and expensive. While we believe that we are currently in material compliance with applicable laws and regulations, we cannot assure you that the FDA, or other regulatory agencies, would agree with our determinations, and any determination by the government that we have violated the FDCA or any FDA regulations, or a public announcement that we are being investigated for possible violations of these laws, could adversely affect our business, prospects, results of operations, or financial condition.
Even though we commercialize our test as LDTs, our test may in the future become subject to more onerous regulation by the FDA. For example, Congress has recently been working on legislation to create an LDT and in vitro diagnostic regulatory framework that would be separate and distinct from the existing medical device regulatory framework. On March 5, 2020, U.S. Representatives Diana DeGette (D-CO) and Dr. Larry Bucshon (R-IN) formally introduced the VALID Act in the House and an identical version of the bill was introduced in the U.S. Senate by Senators Michael Bennet (D-CO) and Richard Burr (R-NC). As anticipated from a discussion draft of the legislation released for stakeholder comment in December 2018, the VALID Act would codify into law the term “in vitro clinical test,” or IVCT, to create a new medical product category separate from medical devices, and bring all such products within the scope of FDA’s oversight. The VALID Act would also create a new system for labs and hospitals to use to submit their tests electronically to the FDA for approval, which is aimed at reducing the amount of time it takes for the agency to approve such tests, and establish a new program to expedite the development of diagnostic tests that can be used to address a current unmet need for patients. It is unclear whether the VALID Act would be passed by Congress in its current form (if reintroduced in the 117th Congress) or signed into law by the President.
Whether as a result of new legislative authority or following formal notice-and-comment rulemaking, if the FDA begins to enforce its medical device requirements for LDTs, or if the FDA disagrees with our assessment that our test is LDTs, our test could for the first time be subject to a variety of regulatory requirements, including registration and listing, medical device reporting, and quality control, and we could be required to obtain premarket clearance or approval for our existing test and any new tests we may develop, which may force us to cease marketing our tests until we obtain the required clearance or approval. The premarket review process for diagnostic products can be lengthy, expensive, time-consuming, and unpredictable. Further, obtaining premarket clearance or approval may involve, among other things, successfully completing clinical trials. Clinical trials require significant time and cash resources and are
45
Table of Contents
subject to a high degree of risk, including risks of experiencing delays, failing to complete the trial or obtaining unexpected or negative results. If we are required to obtain premarket clearance or approval and/or conduct premarket clinical trials, our development costs could significantly increase, our introduction of any new tests we may develop may be delayed, and sales of our existing test could be interrupted or stopped. Any of these outcomes could reduce our revenue or increase our costs and materially adversely affect our business, prospects, results of operations, or financial condition. Moreover, any cleared or approved labeling claims may not be consistent with our current claims or adequate to support continued adoption of and reimbursement for our test. For instance, if we are required by the FDA to label our test as investigational, or if labeling claims the FDA allows us to make are limited, order levels may decline and reimbursement may be adversely affected. As a result, we could experience significantly increased development costs and a delay in generating additional revenue from our existing test or from tests we may develop. Until the FDA finalizes its regulatory position regarding LDTs, or federal legislation is passed concerning regulation of LDTs through notice-and-comment rulemaking, it is unknown how the FDA may regulate our test in the future and what testing and data may be required to support any required clearance or approval as an medical device or an “in vitro clinical test” (as that category is being defined in the VALID Act, as introduced in March 2020).
The requirement of premarket review could negatively affect our business until such review is completed and regulatory clearance or approval is obtained. The FDA could require that we stop selling our test pending premarket clearance or approval. The regulatory authorization process may involve, among other things, successfully completing additional clinical trials and making a premarket submission, such as a 510(k) notification, a premarket approval, or PMA, application or a de novo device classification request to the FDA. If the FDA requires any form of premarket review, our test may not be cleared or approved on a timely basis, if at all. We may also decide voluntarily to pursue FDA premarket review and authorization of our test if we determine that doing so would be appropriate.
Additionally, should future regulatory actions affect any of the reagents we obtain from suppliers and use in conducting our test, our business could be adversely affected in the form of increased costs of testing or delays, limits, or prohibitions on the purchase of reagents necessary to perform our testing. While we qualify all materials used in our products in accordance with the regulations and guidelines of CLIA, the FDA could promulgate regulations or guidance documents impacting our ability to purchase materials necessary for the performance of our test. If any of the reagents we obtain from suppliers and use in our test are affected by future regulatory actions, our business could be adversely affected, including by increasing the cost of testing or delaying, limiting, or prohibiting the purchase of reagents necessary to perform testing with our products. The ongoing COVID-19 pandemic and high demand for laboratory testing services may also have an impact on the supply chain for such reagents and other supplies and cause an adverse effect on our business.
Failure to comply with any applicable FDA requirements could trigger a range of enforcement actions by the FDA, including warning letters, civil monetary penalties, injunctions, criminal prosecution, recall or seizure, operating restrictions, partial suspension or total shutdown of operations and denial of or challenges to applications for clearance or approval, as well as significant adverse publicity.
If we were to be required by the FDA to conduct additional clinical studies or trials before continuing to offer tests that we have developed or may develop as LDTs, those studies or trials could lead to delays or failure to obtain necessary regulatory clearance or approval, which could cause significant delays in commercializing any future products and harm our ability to achieve profitability.
If the FDA decides to require that we obtain 510(k) clearance, premarket approvals pursuant to a PMA, or any other type of premarket authorization in order for us to commercialize our current PLA test or our planned future tests (including PLAplus), whether as a result of new legislative authority or following formal notice-and-comment rulemaking or based on its determination that any of those tests does not meet the definition of an LDT, we may be required to conduct additional clinical testing before submitting a regulatory submission for commercial marketing authorization. In addition, as part of our long-term strategy we may plan to seek FDA clearance or approval for certain genomic tests in order to permit them to be offered by other clinical laboratories in addition to our own; however, we would need to conduct additional clinical validation activities on our test before we could submit an application for FDA approval or clearance. Clinical trials must be conducted in compliance with FDA regulations or the FDA may take certain enforcement actions or reject the data. We believe it would likely take two years or more to conduct the clinical studies and trials necessary to obtain approval from the FDA to commercially launch our current test and our planned future tests outside of our clinical laboratory.
Even if clinical trials are completed as planned, we cannot be certain that their results would be able to support our test claims or that the FDA or foreign authorities will agree with our conclusions regarding the results of our clinical trials. Success in early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior clinical trials and studies. If we are required to conduct clinical trials to support a premarket submission to the FDA, whether using prospectively acquired samples or archival samples, delays in the commencement or completion of clinical testing could significantly increase our test development costs and delay commercialization. Many of the factors that may cause or lead to a delay in the commencement or completion of clinical trials may also ultimately lead to delay or denial of regulatory clearance or approval. The commencement of clinical trials may be delayed due to insufficient patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites and the eligibility criteria for the clinical trial. Moreover, the clinical trial process may fail to demonstrate that our current test and our planned future tests are effective for the proposed indications for use, which could cause us to abandon a test candidate and may delay development of other tests.
46
Table of Contents
We may find it necessary to engage contract research organizations to perform data collection and analysis and other aspects of our clinical trials, which would increase the cost and complexity of our trials. We may also depend on clinical investigators, medical institutions, and contract research organizations to perform the trials properly. If these parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, or if the quality, completeness, or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or for other reasons, our clinical trials may have to be extended, delayed or terminated. Many of these factors would be beyond our control. We may not be able to enter into replacement arrangements without undue delays or considerable expenditures. If there are delays in testing or approvals as a result of the failure to perform by third parties, our R&D costs would increase, and we may not be able to obtain regulatory clearance or approval for our current test and our planned future tests, if needed. In addition, we may not be able to establish or maintain relationships with these parties on favorable terms, if at all. Each of these outcomes would harm our ability to market our test outside of the LDT context or to achieve profitability.
We are subject to numerous federal, local and foreign laws and regulations; complying with laws pertaining to our business is an expensive and time-consuming process, and any failure to comply could result in substantial penalties and a material adverse effect to our business and operations.
Our operations are subject to extensive federal, state, local and foreign laws and regulations, all of which are subject to change. These laws and regulations currently include, among other things:
| • | CLIA, which requires that laboratories obtain certification from the federal government, and state licensure laws; |
| • | FDA laws and regulations; |
| • | Health Insurance Portability and Accountability Act, or HIPAA, which imposes comprehensive federal standards with respect to the privacy and security of protected health information, or PHI, and requirements for the use of certain standardized electronic transactions; amendments to HIPAA under the Health Information Technology for Economic and Clinical Health Act, or HITECH, which strengthened and expanded HIPAA privacy and security compliance requirements, increased penalties for violators, extended enforcement authority to state attorneys general and imposed requirements for breach notification; |
| • | state laws regulating genetic testing and protecting the privacy of genetic test results, as well as state laws protecting the privacy and security of health information and personal data and mandating reporting of breaches to affected individuals and state regulators; |
| • | the federal Anti-Kickback Statute, which prohibits knowingly and willfully offering, paying, soliciting, receiving, or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing, arranging for, or recommending of an item or service that is reimbursable, in whole or in part, by a federal health care program; |
| • | the federal False Claims Act, which imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment to the federal government; |
| • | the federal Civil Monetary Penalties Law, which prohibits, among other things, the offering or transfer of remuneration to a Medicare or state health care program beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier of services reimbursable by Medicare or a state health care program, unless an exception applies; |
| • | other federal and state fraud and abuse laws, such as anti-kickback laws, prohibitions on self-referral, and false claims acts, which may extend to services reimbursable by any commercial payor, including private insurers; |
| • | Section 216 of the PAMA, which requires applicable laboratories to report commercial payor data in a timely and accurate manner every three years (and in some cases annually); |
| • | state laws that impose reporting and other compliance-related requirements; and |
| • | similar foreign laws and regulations that apply to us in the countries in which we operate. |
In addition, in October 2018, the Eliminating Kickbacks in Recovery Act of 2018, or EKRA, was enacted as part of the Substance Use-Disorder Prevention that Promotes Opioid Recovery and Treatment for Patients and Communities Act, or SUPPORT Act. EKRA is an all-payor anti-kickback law that makes it a criminal offense to pay any remuneration to induce referrals to, or in exchange for, patients using the services of a recovery home, a substance use clinical treatment facility, or laboratory. Although it appears that EKRA was intended to reach patient brokering and similar arrangements to induce patronage of substance use recovery and treatment, the language in EKRA is broadly written. Further, certain of EKRA’s exceptions, such as the exception applicable to relationships with employees that effectively prohibits incentive compensation, are inconsistent with the federal anti-kickback statute and regulations, which permit payment of employee incentive compensation, a practice that is common in the industry. Significantly, EKRA permits the U.S.
47
Table of Contents
Department of Justice to issue regulations clarifying EKRA’s exceptions or adding additional exceptions, but such regulations have not yet been issued. Laboratory industry stakeholders are reportedly seeking clarification regarding EKRA’s scope and/or amendments to its language.
As a clinical laboratory, our business practices may face heightened scrutiny from government enforcement agencies such as the Department of Justice, the U.S. Department of Health and Human Services Office of Inspector General, or OIG, and CMS. The OIG has issued fraud alerts in recent years that identify certain arrangements between clinical laboratories and referring physicians as implicating the Anti-Kickback Statute. The OIG has stated that it is particularly concerned about these types of arrangements because the choice of laboratory, as well as the decision to order laboratory tests, typically are made or strongly influenced by the physician, with little or no input from the patient. Moreover, the provision of payments or other items of value by a clinical laboratory to a referral source could be prohibited under the federal self-referral prohibition, commonly known as the Stark Law or the Physician Self-Referral Law, unless the arrangement meets all criteria of an applicable exception. The government has actively enforced these laws against clinical laboratories in recent years.
These laws and regulations are complex and are subject to interpretation by the courts and by government agencies. Our failure to comply could lead to significant civil or criminal penalties, exclusion from participation in state and federal health care programs, individual imprisonment, disgorgement of profits, contractual damages, reputational harm, diminished profits and future earnings, additional reporting or oversight obligations if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with the law, curtailment or restructuring of our operations, or prohibitions or restrictions on our laboratories’ ability to provide or receive payment for our services, any of which could adversely affect our ability to operate our business and pursue our strategy. We believe that we are in material compliance with all statutory and regulatory requirements, but there is a risk that one or more government agencies could take a contrary position, or that a private party could file suit under the qui tam provisions of the federal False Claims Act or a similar state law. Such occurrences, regardless of their outcome, could damage our reputation and adversely affect important business relationships with third parties, including managed care organizations, and other private commercial payors.
The growth of our business and our expansion outside of the United States may increase the potential of violating similar foreign laws or our internal policies and procedures. The risk of us being found in violation of these or other laws and regulations is further increased by the fact that many have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Any action brought against us for violation of these or other laws or regulations, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. Any of the foregoing consequences could seriously harm our business and our financial results.
We must comply with complex and overlapping laws protecting the privacy and security of health information and personal data.
There are a number of state, federal and international laws protecting the privacy and security of health information and personal data. Under the administrative simplification provisions of HIPAA, the U.S. Department of Health and Human Services, or HHS, has issued regulations which establish uniform standards governing the conduct of certain electronic health care transactions and protecting the privacy and security of PHI used or disclosed by health care providers and other covered entities.
The privacy regulations regulate the use and disclosure of PHI by health care providers engaging in certain electronic transactions or “standard transactions.” They also set forth certain rights that an individual has with respect to his or her PHI maintained by a covered health care provider, including the right to access or amend certain records containing PHI or to request restrictions on the use or disclosure of PHI. The HIPAA security regulations establish administrative, physical, and technical standards for maintaining the integrity and availability of PHI in electronic form. These standards apply to covered health care providers and also to “business associates” or third parties providing services involving the use or disclosure of PHI. The HIPAA privacy and security regulations establish a uniform federal “floor” and do not supersede state laws that are more stringent or provide individuals with greater rights with respect to the privacy or security of, and access to, their records containing PHI. For example, California recently enacted the CCPA, which became effective January 1, 2020. The CCPA establishes a new privacy framework for covered businesses in the State of California, by creating an expanded definition of personal information, establishing new data privacy rights for consumers imposing special rules on the collection of consumer data from minors, and creating a new and potentially severe statutory damages framework for violations of the CCPA and for businesses that fail to implement reasonable security procedures and practices to prevent data breaches. As a result, we may be required to comply with both HIPAA privacy regulations and varying state privacy and security laws.
Moreover, HITECH, among other things, established certain health information security breach notification requirements. In the event of a breach of unsecured PHI, a covered entity must notify each individual whose PHI is breached, federal regulators and in some cases, must publicize the breach in local or national media. Breaches affecting 500 individuals or more are publicized by federal regulators who publicly identify the breaching entity, the circumstances of the breach and the number of individuals affected.
These laws contain significant fines and other penalties for wrongful use or disclosure of PHI. Given the complexity of HIPAA and HITECH and their overlap with state privacy and security laws, and the fact that these laws are rapidly evolving and are subject to changing and potentially conflicting interpretation, our ability to comply with the HIPAA, HITECH and state privacy requirements is
48
Table of Contents
uncertain and the costs of compliance are significant. Adding to the complexity is that our operations are evolving and the requirements of these laws will apply differently depending on such things as whether or not we bill electronically for our services, or provide services involving the use or disclosure of PHI and incur compliance obligations as a business associate. The costs of complying with any changes to the HIPAA, HITECH and state privacy restrictions may have a negative impact on our operations. Noncompliance could subject us to criminal penalties, civil sanctions and significant monetary penalties as well as reputational damage.
We also are required to collect and maintain personal information about our employees, and we collect information about customers as part of some of our marketing programs, as well as receive and transfer certain payment information, to accept payments from our customers, including credit card information. Most states have adopted laws requiring notification of affected individuals and state regulators in the event of a breach of personal information, which is a broader class of information than the health information protected by HIPAA. Many state laws impose significant data security requirements, such as encryption or mandatory contractual terms to ensure ongoing protection of personal information. Activities outside of the United States implicate local and national data protection standards, impose additional compliance requirements, and generate additional risks of enforcement for non-compliance. The collection and use of such information may be subject to contractual obligations as well. If the security and information systems that we or our outsourced third-party providers use to store or process such information are compromised or if we, or such third parties, otherwise fail to comply with these laws, regulations, and contractual obligations, we could face litigation and the imposition of penalties that could adversely affect our financial performance.
We must comply with all applicable privacy and data security laws in order to operate our business and may be required to expend significant capital and other resources to ensure ongoing compliance, to protect against security breaches and hackers or to alleviate problems caused by such breaches. Breaches of health information and/or personal data may be extremely expensive to remediate, may prompt federal or state investigation, fines, civil and/or criminal sanctions and significant reputational damage.
Our services present the potential for embezzlement, identity theft or other similar illegal behavior by our employees, consultants, service providers or commercial partners.
Our operations involve the use and disclosure of personal and business information that could be used to impersonate third parties or otherwise gain access to their data or funds. If any of our employees, consultants, service providers or commercial partners takes, converts or misuses these funds or data, we could be liable for any resulting damages, which could harm our financial condition and damage our business reputation.
Clinical research is heavily regulated and failure to comply with human subject protection regulations may disrupt our research program leading to significant expense, regulatory enforcement, private lawsuits, and reputational damage.
Clinical research is subject to federal, state, and, for studies conducted outside of the United States, international regulation. At the federal level, the Department of Health and Human Services imposes regulations for the protection of human subjects and requirements such as initial and ongoing institutional review board review, informed consent requirements, adverse event reporting and other protections to minimize the risk and maximize the benefit to research participants. Clinical studies done under an investigational device exemption for purposes of an anticipated FDA premarket submission are subject to an additional layer of human subject protection regulations. Many states also impose human subject protection laws that mirror or in some cases exceed federal requirements. HIPAA and other privacy laws also regulate the use and disclosure of PHI in connection with research activities. Research conducted overseas is subject to a variety of national protections such as mandatory ethics committee review, as well as laws regulating the use, disclosure and cross-border transfer of personal data. The costs of compliance with these laws may be significant and compliance with regulatory requirements may result in delay. Noncompliance may disrupt our research and result in data that is unacceptable to regulatory authorities, data lock, or other sanctions that may significantly disrupt our operations.
We could be adversely affected by alleged violations of the Federal Trade Commission Act or other truth-in-advertising and consumer protection laws.
Our advertising for laboratory services and tests is subject to federal truth-in-advertising laws enforced by the Federal Trade Commission, or FTC, as well as comparable state consumer protection laws. Under the Federal Trade Commission Act, the FTC is empowered, among other things, to (a) prevent unfair methods of competition and unfair or deceptive acts or practices in or affecting commerce; (b) seek monetary redress and other relief for conduct injurious to consumers; and (c) gather and compile information and conduct investigations relating to the organization, business, practices, and management of entities engaged in commerce. The FTC has very broad enforcement authority, and failure to abide by the substantive requirements of the FTC Act and other consumer protection laws can result in administrative or judicial penalties, including civil penalties, injunctions affecting the manner in which we would be able to market services or products in the future, or criminal prosecution. Our direct-to-consumer advertising and social media presence, as well as our physician-directed advertising, are subject to these federal and state truth-in-advertising laws. Any actual or perceived non-compliance with those laws could lead to an investigation by the FTC or a comparable state agency, or could lead to allegations of misleading advertising by private plaintiffs. Any such action against us would disrupt our business operations, cause damage to our reputation, and result in a material adverse effects on our business, financial condition, results of operation, and cash flows.
49
Table of Contents
Medical product manufacturers’ use of social media platforms presents new risks.
We believe that our customer base and potential patient populations are active on social media and intend to engage through those platforms to elevate our national marketing presence. Social media practices in the pharmaceutical, biotechnology and medical device industries are evolving, which creates uncertainty and risk of noncompliance with regulations applicable to our business. For example, patients may use social media platforms to comment on the effectiveness of, or adverse experiences with, one of our products, which could result in reporting obligations or the need for us to conduct an investigation. In addition, there is a risk of inappropriate disclosure of sensitive information or negative or inaccurate posts or comments about us or our products on any social networking website. If any of these events were to occur or we otherwise fail to comply with any applicable regulations, we could incur liability, face restrictive regulatory actions or incur other harm to our business.
Intellectual Property Risks Related to Our Business
Our collaborators may assert ownership or commercial rights to inventions we develop from our use of the biological materials which they provide to us, or otherwise arising from the collaboration.
We collaborate with several institutions, clinicians, and researchers in scientific matters. Also, we rely on numerous third parties to provide us with adhesive patch samples and biological materials that we use to develop tests. If we cannot successfully negotiate sufficient ownership, licensing, and/or commercial rights to any inventions that result from our use of a third-party collaborator’s materials, or if disputes arise with respect to the intellectual property developed with the use of a collaborator’s samples, or data developed in a collaborator’s study, our ability to capitalize on the market potential of these inventions or developments may be limited or precluded altogether.
If we are unable to maintain intellectual property protection, our competitive position could be harmed.
Our ability to protect our discoveries and technologies affects our ability to compete and to achieve profitability. Currently, we rely on a combination of U.S. and foreign patents and patent applications, copyrights, trademarks and trademark applications, confidentiality or non-disclosure agreements, material transfer agreements, licenses, consulting agreements, work-for-hire agreements, and invention assignment agreements to protect our intellectual property rights. We also maintain certain company know-how, trade secrets, and technological innovations designed to provide us with a competitive advantage in the marketplace as trade secrets. As of February 19, 2021, we own six issued U.S. patents, 12 pending U.S. patent applications (four provisional and eight non-provisional), several corresponding foreign counterpart patents and applications, and two PCT applications, and four design patent applications, relevant to our testing methodology and expression profiles. While we intend to pursue additional patent applications, it is possible that our pending patent applications and any future applications may not result in issued patents. Even if patents are issued, third parties may independently develop similar or competing technology that avoids our patents. Further, we cannot be certain that the steps we have taken will prevent the misappropriation of our trade secrets and other confidential information as well as the misuse of our patents and other intellectual property, particularly in foreign countries where we have not filed for patent protection.
From time-to-time the U.S. Supreme Court, other federal courts, or the USPTO, may change the standards of patentability, and any such changes could have a negative impact on our business. For instance, in 2008, the Court of Appeals for the Federal Circuit issued a decision that methods or processes cannot be patented unless they are tied to a machine or involve a physical transformation. The U.S. Supreme Court later reversed that decision in Bilski v. Kappos, finding that the “machine-or-transformation” test is not the only test for determining patent eligibility. The Court, however, declined to specify how and when processes are patentable. In 2012, in the case Mayo Collaborative Services v. Prometheus Laboratories, Inc., the U.S. Supreme Court reversed the Federal Circuit’s application of Bilski and invalidated a patent focused on a diagnostic process because the patent claim embodied a law of nature.
In 2013, in Association for Molecular Pathology v. Myriad Genetics, the Supreme Court unanimously ruled that, “[a] naturally occurring DNA segment is a product of nature and not patent eligible merely because it has been isolated,” thereby invalidating Myriad Genetics’ patents on the BRCA1 and BRCA2 breast cancer genes. However, the Supreme Court also held that manipulation of a gene to create something not found in nature, such as a strand of synthetically-produced complementary DNA, or cDNA, could still be eligible for patent protection. The Supreme Court noted that method patents, which concern technical procedures for carrying out a certain process, are not affected by the ruling.
More recently, the Federal Circuit has ruled on several patent cases—such as Univ. of Utah Research Found. v. Ambry Genetics Corp., 774 F.3d 755 (Fed. Cir. 2014), Ariosa Diagnostics, Inc. v. Sequenom, Inc., 788 F.3d 1371 (Fed. Cir. 2015), Genetic Tech. Ltd. v. Merial LLC, 818 F.3d 1369 (Fed. Cir. 2016), and Cleveland Clinic Found. v. True Health Diagnostics, 859 F.3d 1352 (Fed. Cir. 2017)—that some diagnostic method claims are patent ineligible. These decisions have narrowed the scope of patent protection available in certain circumstances or weakened the rights of patent owners in certain situations. Some aspects of our technology involve processes that may be subject to this evolving standard and we cannot guarantee that any of our pending process claims will be patentable as a result of such evolving standards. In addition, this combination of decisions has created uncertainty as to the value of certain issued patents, in particular patents in the molecular biology analysis and diagnostic space. Moreover, there is additional uncertainty around the evolving standard in light of the USPTO Revised Patent Subject Matter Eligibility Guidance issued in Jan. 2019.
50
Table of Contents
It should also be noted that in 2010, the Secretary’s Advisory Committee on Genetics, Health and Society voted to approve a report entitled “Gene Patents and Licensing Practices and Their Impact on Patient Access to Genetic Tests.” That report defines “patent claims on genes” broadly to include claims to isolated nucleic acid molecules as well as methods of detecting particular sequences or mutations. The report also contains six recommendations, including the creation of an exemption from liability for infringement of patent claims on genes for anyone making, using, ordering, offering for sale, or selling a test developed under the patent for patient care purposes, or for anyone using the patent-protected genes in the pursuit of research. The report also recommended that HHS should explore, identify, and implement mechanisms that will encourage more voluntary adherence to current guidelines that promote nonexclusive in-licensing of diagnostic genetic and genomic technologies. It is unclear whether HHS will act upon these recommendations, or if the recommendations would result in a change in law or process that could negatively impact our patent portfolio or future R&D. If acted upon, implementation of such provisions could have a material negative impact on our business.
We may face intellectual property infringement claims that could be time-consuming and costly to defend, and could result in the loss of significant rights, the implementation of an injunction, and the assessment of treble damages.
From time-to-time we may face intellectual property infringement or misappropriation claims from third parties. Some of these claims may lead to litigation. The outcome of any such litigation can never be guaranteed, and an adverse outcome could affect us negatively. For example, were a third party to succeed on an infringement claim against us, we may be required to pay substantial damages, including treble damages if such infringement were found to be willful. In addition, we could face an injunction barring us from conducting the allegedly infringing activity, including an order preventing us from offering our current test and future planned tests in the marketplace. The outcome of the litigation could require us to enter into a license agreement which may not be pursuant to acceptable or commercially reasonable or practical terms or which may not be available at all.
It is also possible that an adverse finding of infringement against us may require us to dedicate substantial resources and time in developing non-infringing alternatives, which may or may not be possible. In the case of diagnostic tests, we would also need to include non-infringing technologies, which would require us to re-validate the test. Any such re-validation, in addition to being costly and time-consuming, may be unsuccessful. Finally, we may initiate claims to assert or defend our own intellectual property against third parties. Any intellectual property litigation, irrespective of whether we are the plaintiff or the defendant, and regardless of the outcome, is expensive and time-consuming, and could divert and distract our management’s attention from our business and negatively affect our operating results or financial condition.
Tax Risks Related to Our Business
Our ability to use our net operating losses to offset future taxable income may be subject to certain limitations.
Our net operating loss, or NOL, carryforwards, may be unavailable to offset future taxable income because of restrictions under U.S. tax law. Our NOLs generated in tax years ending on or prior to December 31, 2017 are only permitted to be carried forward for 20 taxable years under applicable U.S. federal tax law, and therefore could expire unused. Under tax legislation commonly referred to as the Tax Cuts and Jobs Act, or TCJA, as modified by the Coronavirus Aid, Relief, and Economic Security Act, or CARES Act, our federal NOLs generated in tax years ending after December 31, 2017 may be carried forward indefinitely and NOLs arising in taxable years beginning after December 31, 2017 and before January 1, 2021 may be carried back to each of the five taxable years preceding the tax year of such loss, but NOLs arising in taxable years beginning after December 31, 2020 may not be carried back. In addition, under the TCJA, as modified by the CARES Act, for taxable years beginning after December 31, 2020, the deductibility of federal NOLs generated in taxable years beginning after December 31, 2017 is limited to 80% of current year taxable income. It is uncertain if and to what extent various states will conform to the TCJA, as modified by the Cares Act.
In addition, under Section 382 of the Internal Revenue Code of 1986, as amended, or the IRC, a corporation that undergoes an “ownership change” is subject to limitations on its ability to utilize its carryforwards to offset future taxable income. Our existing NOLs may be subject to limitations arising from previous ownership changes, and if we underwent an ownership change in connection with or after the Business Combination, our ability to utilize NOLs could be further limited by Section 382 of the IRC. Future changes in our stock ownership, some of which are outside of our control, could result in an ownership change under Section 382 of the IRC. There is also a risk that due to regulatory changes, such as suspensions on the use of NOLs, or other unforeseen reasons, our existing and any future NOLs could expire or otherwise be unavailable to offset future income tax liabilities. We have not conducted a study to assess whether an ownership change has occurred or whether there have been multiple ownership changes since inception due to the significant complexity and cost associated with such a study.
U.S. federal income tax reform could adversely affect our business and financial condition.
On December 22, 2017, President Trump signed into law the TCJA that significantly reforms the IRC. The TCJA, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, limitation on the deductibility of interest expense to 30% of adjusted earnings (except for certain small businesses), limitation of the deduction for NOLs generated in taxable years beginning after December 31, 2017 to 80% of current year taxable income, elimination of NOL carrybacks, one time taxation of offshore earnings at reduced rates regardless of whether they are repatriated, reduction or elimination of U.S. tax on foreign earnings (subject to certain important exceptions), immediate deductions for
51
Table of Contents
certain new investments instead of deductions for depreciation expense over time, and modifying or repealing many business deductions and credits. The CARES Act modifies certain provisions of the TCJA. Under the CARES Act, NOLs arising in taxable years beginning after December 31, 2017 and before January 1, 2021 may be carried back to each of the five taxable years preceding the tax year of such loss, but NOLs arising in taxable years beginning after December 31, 2020 may not be carried back. In addition, the CARES Act eliminates the limitation on the deduction of NOLs to 80% of current year taxable income for taxable years beginning before January 1, 2021, and increases the amount of interest expense that may be deducted to 50% of adjusted taxable income for taxable years beginning in 2019 or 2020. Notwithstanding the reduction in the corporate income tax rate, the overall impact of the TCJA, as modified by the CARES Act, is uncertain and our business and our financial condition could be adversely affected. In addition, it is uncertain if and to what extent various states will conform to the TCJA, as modified by the CARES Act. The impact of the TCJA, as modified by the CARES Act, on holders of our common stock is also uncertain and could be adverse. You are urged to consult with your legal and tax advisors with respect to such legislation and the potential tax consequences of investing in our common stock.
Risks Related to Our Securities
There is no assurance that we will continue satisfying the listing requirements of the Nasdaq Capital Market.
Our common stock is listed on the Nasdaq Capital Market. To maintain our listing we are required to satisfy continued listing requirements. There can be no assurance we will continue satisfying such continued listing requirements, which include that the closing bid price of our common stock be at least $1 per share, that we have at least 300 round lot holders and at least 500,000 publicly held shares, that the market value of our publicly held securities be at least $1 million, and that we meet one of these standards: stockholders' equity of at least $2.5 million; market value of listed securities of at least $35 million; or net income from continuing operations of $500,000 in the most recently completed fiscal year or in two of the three most recently completed fiscal years. The delisting of our common stock for whatever reason could, among other things, substantially impair our ability to raise additional capital; result in the loss of interest from institutional investors, the loss of confidence in our company by investors and employees, and in fewer financing, strategic and business development opportunities; and result in potential breaches of agreements under which we made representations or covenants relating to our compliance with applicable listing requirements. Claims related to any such breaches, with or without merit, could result in costly litigation, significant liabilities and diversion of our management’s time and attention and could have a material adverse effect on our financial condition, business and results of operations. In addition, the delisting of our common stock for whatever reason may materially impair our stockholders’ ability to buy and sell shares of our common stock and could have an adverse effect on the market price of, and the efficiency of the trading market for, our common stock.
We are an emerging growth company, and a smaller reporting company, and the reduced disclosure requirements applicable to emerging growth companies and smaller reporting companies may make our securities less attractive to investors.
We are an emerging growth company, under the Jumpstart Our Business Startups Act and a smaller reporting company under SEC regulations. For so long as we remain an emerging growth company or smaller reporting company, we will be permitted to and intend to rely on exemptions from certain disclosure requirements applicable to other public companies that are not emerging growth companies or smaller reporting companies. These exemptions include:
| • | for so long as we are an emerging growth company, not being required to comply with the auditor attestation requirements in the assessment of our internal control over financial reporting; |
| • | not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the consolidated financial statements; |
| • | reduced disclosure obligations regarding executive compensation; and |
| • | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved for so long as we are an emerging growth company. |
We may choose to take advantage of some, but not all, of the available exemptions. Emerging growth companies may take advantage of an extended transition period for complying with new or revised accounting standards, allowing emerging growth companies to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies.
We will continue to be an emerging growth company until the earliest to occur of (i) the last day of the fiscal year during which we had total annual gross revenues of at least $1.07 billion, (ii) the day we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700 million, measured as of our most recently completed second fiscal quarter, (iii) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period, and (iv) December 31, 2022. In addition, we are eligible to remain a smaller reporting company for so long as we have a public float (based on our common equity) of less than $250 million measured as of the last business day of our most recently completed second fiscal quarter or, a public float (based on our common equity) of less than $700 million as of this date and annual revenues of less than $100 million during the most recently completed fiscal year.
52
Table of Contents
We cannot predict whether investors will find our securities less attractive if we rely on these exemptions. If some investors find our securities less attractive as a result, there may be a less active trading market for our securities and the price of our securities price may be more volatile.
Future issuances of equity securities may dilute the interests of our security holders and reduce the price of our securities.
Any future issuance of our equity securities could dilute the interests of our then existing security holders and could substantially decrease the trading price of our securities. We may issue equity or equity-linked securities for a number of reasons, including to finance our operations and business strategy, to adjust our ratio of debt to equity, to satisfy our obligations upon the exercise of then-outstanding options or other equity-linked securities, if any, or for other reasons. We currently have the ability to offer and sell up to $36.3 million of common stock, preferred stock, warrants, senior debt, subordinated debt, rights or units under an effective universal shelf registration statement. Sales of substantial amounts of shares of our common stock or other securities under our current universal shelf registration statement could lower the market price of our common stock and impair our ability to raise capital.
We may amend the terms of our publicly traded warrants currently trading on the Pink Market under the ticker symbol “DMTKW,” or the publicly traded warrants, in a manner that may be adverse to holders with the approval by the holders of a majority of the then outstanding publicly traded warrants, and as a result, the exercise price of the publicly traded warrants could be increased, the exercise period could be shortened and the number of shares purchasable upon exercise of a publicly traded warrant could be decreased, all without your approval.
Our publicly traded warrants are subject to the Warrant Agreement. The Warrant Agreement provides that the terms of the publicly traded warrants may be amended without the consent of any holder to cure any ambiguity or correct any defective provision, but requires the approval by the holders of a majority of the then outstanding publicly traded warrants to make any change that adversely affects the interests of the registered holders. Accordingly, we may amend the terms of the publicly traded warrants in a manner adverse to a holder if holders of a majority of the then outstanding publicly traded warrants approve of such amendment. Although our ability to amend the terms of the publicly traded warrants with the consent of a majority of the then outstanding publicly traded warrants is unlimited, examples of such amendments could be amendments to, among other things, increase the exercise price of the publicly traded warrants, shorten the exercise period or decrease the number of shares of common stock purchasable upon exercise of the publicly traded warrants.
We may redeem your unexpired publicly traded warrants prior to their exercise at a time that is disadvantageous to you, thereby making your publicly traded warrants worthless.
We have the ability to redeem our outstanding publicly traded warrants at any time prior to their expiration, at a price of $0.01 per warrant, provided that the last reported sales price of our common stock equals or exceeds $36.00 per share for any 20 trading days within a 30-trading day period ending on the third trading day prior to the date we give notice of redemption. In the first quarter of 2021, the last reported sales price of our common stock has at times equaled or exceeded $36.00 per share for 20 trading days within a 30-trading day period, which would have permitted us to redeem the publicly traded warrants following any such period. To the extent that the publicly traded warrants remain redeemable by us, we may exercise our redemption right even if we are unable to register or qualify the underlying securities for sale under all applicable state securities laws. Redemption of the outstanding publicly traded warrants could force you (i) to exercise your publicly traded warrants and pay the exercise price therefor at a time when it may be disadvantageous for you to do so, (ii) to sell your publicly traded warrants at the then-current market price when you might otherwise wish to hold your publicly traded warrants or (iii) to accept the nominal redemption price which, at the time the outstanding publicly traded warrants are called for redemption, is likely to be substantially less than the market value of your publicly traded warrants.
Because we have no current plans to pay cash dividends on our shares for the foreseeable future, you may not receive any return on investment unless you sell your shares for a price greater than that which you paid for it.
We may retain future earnings, if any, for future operations, expansion and debt repayment and have no current plans to pay any cash dividends for the foreseeable future. Any decision to declare and pay dividends in the future will be made at the discretion of our board of directors and will depend on, among other things, our results of operations, financial condition, cash requirements, contractual restrictions and other factors that our board of directors may deem relevant. In addition, our ability to pay dividends may be limited by covenants of any existing and future outstanding indebtedness we or our subsidiaries incur. As a result, you may not receive any return on an investment in our shares unless you sell your shares of the Company for a price greater than that which you paid for them.
If securities or industry analysts do not publish or cease publishing research or reports about us, our business, or our market, or if they change their recommendations regarding our securities adversely, the price and trading volume of our securities could decline.
The trading market for our securities will be influenced by the research and reports that industry or securities analysts may publish about us, our business, market or competitors. If no securities or industry analysts publish reports about us, our share price and trading volume would likely be negatively impacted. If any of the analysts who may cover us change their recommendation regarding our shares of common stock adversely, or provide more favorable relative recommendations about our competitors, the price of our shares of common stock would likely decline. If any analyst who may cover us were to cease coverage of us or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our share price or trading volume to decline.
53
Table of Contents
Provisions of our charter documents or Delaware law could delay or prevent an acquisition of us, even if the acquisition would be beneficial to our stockholders, and could make it more difficult for you to change our management.
Provisions in our Amended and Restated Certificate of Incorporation and our bylaws may discourage, delay or prevent a merger, acquisition or other change in control that our stockholders may consider favorable, including transactions in which our stockholders might otherwise receive a premium for their shares. In addition, these provisions may frustrate or prevent any attempt by our stockholders to replace or remove our current management by making it more difficult to replace or remove our board of directors. These provisions include:
| • | a classified board of directors so that not all directors are elected at one time; |
| • | a prohibition on stockholder action through written consent; |
| • | no cumulative voting in the election of directors; |
| • | the exclusive right of our board of directors to elect a director to fill a vacancy however created, whether by the expansion of our board of directors, the resignation, death or removal of a director, or otherwise; |
| • | a requirement that special meetings of our stockholders be called only by our board of directors, the chairman of our board of directors, the chief executive officer or, in the absence of a chief executive officer, the president; |
| • | an advance notice requirement for stockholder proposals and nominations; |
| • | the authority of our board of directors to issue preferred stock with such terms as our board of directors may determine; and |
| • | a requirement of approval of at least 75% of all outstanding shares of our capital stock entitled to vote to amend any bylaws by stockholder action, or to amend specific provisions of our certificate of incorporation. |
In addition, Delaware law prohibits a publicly held Delaware corporation from engaging in a business combination with an interested stockholder, generally a person who, together with his, her or its affiliates, owns or within the last three years has owned 15% or more of the company’s voting stock, for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner. Accordingly, Delaware law may discourage delay or prevent a change in control of the Company.
In addition, our Amended and Restated Certificate of Incorporation, to the fullest extent permitted by law, provides that the Court of Chancery of the State of Delaware will be the exclusive forum, or the Delaware Chancery forum provision, for: any derivative action or proceeding brought on our behalf; any action or proceeding asserting a breach of fiduciary duty owed to us, our stockholders, or any of our current or former directors, officers or other employees; any action or proceeding asserting a claim against us or any of our current or former directors, officers or other employees, arising out of or pursuant to the Delaware General Corporation Law, our Amended and Restated Certificate of Incorporation, or our bylaws; any action or proceeding to interpret apply, enforce or determine the validity of our Amended and Restated Certificate of Incorporation or our Bylaws; any action or proceeding as to which the Delaware General Corporation Law confers jurisdiction to the Court of Chancery of the State of Delaware; or any action asserting a claim against us that is governed by the internal affairs doctrine. This exclusive forum provision does not apply to suits brought to enforce a duty or liability created by the Securities Act of 1933, as amended, or the Securities Act, the Securities Exchange Act of 1934, as amended, or the Exchange Act, or any claim for which the federal courts have exclusive jurisdiction.
The Delaware Chancery forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or any of our directors, officers, or other employees, which may discourage lawsuits with respect to such claims. Alternatively, if a court were to find the exclusive forum provisions contained in our Amended and Restated Certificate of Incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could harm our business, results of operations and financial condition.
Further, on March 18, 2020, the Delaware Supreme Court ruled that provisions of a Delaware corporation’s certificate of incorporation that designate a federal forum for securities claims brought pursuant to the Securities Act, or federal forum provisions, are valid and enforceable under Delaware law, or the March 2020 Ruling. Consistent with the March 2020 Ruling, on April 12, 2020, our board of directors approved a Certificate of Amendment to our Amended and Restated Certificate of Incorporation, or the 2020 Certificate of Amendment, which was approved by our stockholders at our 2020 annual meeting of stockholders on May 26, 2020. We filed the 2020 Certificate of Amendment with the Delaware Secretary of State on May 27, 2020. The 2020 Certificate of Amendment added a federal forum provision to our Amended and Restated Certificate of Incorporation, which now provides that, unless we consent in writing to the selection of an alternative forum, the federal district courts of the United States of America shall, to the fullest extent permitted by law, be the sole and exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act. Various U.S. Supreme Court cases offer support for the argument that federal forum provisions do not violate federal policy. However, the March 2020 Ruling applies only to claims brought in Delaware state courts, and it is not binding
54
Table of Contents
on any other state court or the federal courts. Therefore, we are unable to predict whether a state court in any other state or a federal court would enforce a federal forum provision such as the one set forth in the 2020 Certificate of Amendment.
We adopted the 2020 Certificate of Amendment to reduce the costs and inefficiencies to the Company that would result from a Securities Act claim being litigated in both state and federal courts, which was permissible under our Amended and Restated Certificate of Incorporation before the 2020 Certificate of Amendment was adopted. Such simultaneous state and federal litigation could also result in inconsistent judgments and rulings, and the adoption of the 2020 Certificate of Amendment could reduce this risk. However, the federal forum provision set forth in the 2020 Certificate of Amendment may discourage Securities Act claims or limit a stockholder’s ability to submit claims in a judicial forum that the stockholder finds favorable, and may result in additional costs for a stockholder seeking to bring such a claim.
Provisions in our charter and other provisions of Delaware law could limit the price that investors are willing to pay in the future for shares of our common stock.
We expect the price of our common stock may be volatile and may fluctuate substantially.
The stock market in general and the market for life sciences companies in particular, have experienced extreme volatility that has often been unrelated to companies’ operating performance. In addition, the stock market in general has recently experienced relatively large price and volume fluctuations in response to the COVID-19 pandemic. The market price for our common stock may be influenced by many factors, including:
| • | the results of our efforts to develop and commercialize our test; |
| • | actual or anticipated results from, and any delays in, any future clinical trials, as well as results of regulatory reviews relating to the approval of any test candidates we may choose to develop that require such approval; |
| • | commencement or termination of any collaboration or licensing arrangement; |
| • | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technology; |
| • | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures and capital commitments; |
| • | additions or departures of key scientific or management personnel; |
| • | variations in our financial results or those of companies that are perceived to be similar to us; |
| • | new products, product candidates or new uses for existing products introduced or announced by our competitors, and the timing of these introductions or announcements; |
| • | results of clinical trials of product candidates of our competitors; |
| • | general economic and market conditions and other factors that may be unrelated to our operating performance or the operating performance of our competitors, including changes in market valuations of similar companies; |
| • | regulatory or legal developments in the United States and other countries; |
| • | changes in the structure of healthcare payment systems; |
| • | conditions or trends in the life sciences industry; |
| • | actual or anticipated changes in earnings estimates, development timelines or recommendations by securities analysts; |
| • | announcement or expectation of additional financing efforts; |
| • | sales of common stock by us or our stockholders in the future, as well as the overall trading volume of our common stock; and |
| • | other factors described in this “Risk Factors” section. |
In the past, following periods of volatility in companies’ stock prices, securities class-action litigation has often been instituted against such companies. Such litigation, if instituted against us, could result in substantial costs and diversion of management’s attention and resources, which could materially and adversely affect our business and financial condition.
55
Table of Contents
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
We currently occupy approximately 28,655 square feet of leased space at 11099 North Torrey Pines Road, La Jolla, California 92037 under an agreement that expires April 30, 2023, which we have the option to renew for an additional three-year period commencing May 1, 2023.
We believe these facilities are adequate to meet our current and reasonably foreseeable requirements. We believe that we would be able to obtain additional space, if required, on commercially reasonable terms.
Item 3. Legal Proceedings
We are not currently a party to any material legal proceedings.
Item 4. Mine Safety Disclosures
Not applicable.
56
Table of Contents
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock is currently listed on the Nasdaq Capital Market under the symbol “DMTK”.
Holders of Our Common Stock
As of March 1, 2021, there were 28,755,668 shares of our common stock outstanding held by approximately 519 holders of record.
Recent Sales of Unregistered Securities
Between October 23, 2020 and December 8, 2020, we issued 4,110 shares of common stock pursuant to the exercise of warrants that were issued in connection with DermTech Operations’ Series C Convertible Preferred Stock financing and assumed by us in connection with the Business Combination. These warrants had an exercise price of $9.54 per share and were exercised for an aggregate exercise price of $39,209.
On December 31, 2020, we issued 2,449 shares of common stock pursuant to the exercise of placement agent warrants. These warrants had exercise prices of $8.68 or $9.54 per share and were exercised for an aggregate exercise price of $21,588.
Between October 13, 2020 and December 31, 2020, we issued an aggregate of 140,522 shares of common stock upon the cashless exercise of placement agent warrants. The holders who elected to exercise the placement agent warrants on a cashless basis paid the exercise price by surrendering the warrants for that number of shares equal to the quotient obtained by dividing (x) the product of the number of shares underlying the warrants, multiplied by the difference between the exercise price of the warrants ($8.68 or $9.54) and the “fair market value” (defined below) by (y) the fair market value. The “fair market value” for purposes of the placement agent warrants is the closing bid price of our common stock or the closing price quoted on the national securities exchange on which our common stock is listed, as applicable, on the first trading day preceding the date of determination of the fair market value.
The issuances of the above shares were deemed to be exempt from registration under the Securities Act in reliance on Sections 3(a)(9) or 4(a)(2) of the Securities Act. The recipients of the shares represented their intention to acquire the securities for investment only and not with a view to, or for sale in connection with, any distribution thereof, and appropriate legends were affixed to the securities.
Item 6. Selected Financial Data
Not applicable.
57
Table of Contents
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion of our financial condition and results of operations should be read in conjunction with the consolidated financial statements and the related notes thereto included elsewhere in this Annual Report on Form 10-K.
Overview
We are an emerging growth molecular diagnostic company developing and marketing novel non-invasive genomics tests that seek to transform the practice of dermatology and related fields. Our platform may change the diagnostic paradigm in dermatology from one that is subjective, invasive, less accurate and higher-cost to one that is objective, non-invasive, more accurate and lower-cost. Our initial focus is skin cancer. We currently have one clinical commercial test, with a second ready to commercialize, that enhance the early detection of skin cancer and related conditions. Our scalable genomics platform has been designed to work with a proprietary adhesive patch sample collection kit that provides a skin sample collected non-invasively. We process our tests in a Clinical Laboratory Improvement Amendments of 1988, or CLIA, certified and College of American Pathologists accredited commercial laboratory located in La Jolla, California that is licensed by the State of California and all states requiring out-of-state licensure. We also provide our technology platform on a contract basis to large pharmaceutical companies who use the technology in their clinical trials to test for the existence of genetic targets of various diseases and to measure the response of new drugs under development. We have a history of net losses since our inception.
Events, Trends and Uncertainties
The Pigmented Lesion Assay, or PLA, became eligible for Medicare reimbursement on February 10, 2020. In late October 2019, the American Medical Association, or AMA, provided us with a Proprietary Laboratory Analyses Code, or PLA Code. Pricing of $760 for the PLA Code was published on December 24, 2019 as part of the Centers for Medicare and Medicaid Services Laboratory Fee Schedule, or CLFS, for 2020. The Medicare Final Coverage Decision, or Final LCD, expanded the coverage proposal in the Draft LCD from one to two tests per date of service and it allows clinicians to order our PLA if they have sufficient skill and experience to decide whether a pigmented lesion should be biopsied. Our local Medicare Administrative Contractor, Noridian Healthcare Solutions, LLC, or Noridian, has issued its own Local Coverage Decision, or LCD, announcing coverage of our PLA. Even though the effective date of Noridian’s LCD was June 7, 2020, Noridian began reimbursing us for our PLA as of February 10, 2020. With Medicare coverage granted, we have the opportunity to approach commercial payors, and as a result, we believe that the PLA may generate significant revenues in 2021 and 2022.
Despite the grant of Medicare coverage for the PLA, uncertainty surrounds commercial payor reimbursement, including governmental and commercial payors, of any test incorporating new technology, including tests developed using our technologies. Because each payor generally determines for its own enrollees or insured patients whether to cover or otherwise establish a policy to reimburse our tests, seeking payor approvals is a time-consuming and costly process. We cannot be certain that coverage for our current test and our planned future tests will be provided in the future by additional commercial payors or that existing policy decisions or reimbursement levels will remain in place or be fulfilled under existing terms and provisions. If we cannot obtain or maintain coverage and reimbursement from private and governmental payors such as Medicare and Medicaid for our current test, or new tests or test enhancements that we may develop in the future, our ability to generate revenues could be limited. This may have a material adverse effect on our business, financial condition, results of operation, and cash flows.
Revenue Effects Related to COVID-19 Pandemic
Assay Revenue
Beginning in March 2020 and continuing through the end of 2020, the ongoing COVID-19 pandemic has reduced patient access to clinician offices for in-person testing, which has resulted in a reduced volume of billable samples received relative to our pre-pandemic expectations. April 2020 billable sample volume was down by approximately 80%, commensurate with the closure of dermatology offices, compared to the average monthly billable sample volume for the two months preceding the beginning of the COVID-19 stay-at-home orders. Despite the downturn in billable samples in April 2020, we saw a stabilization of billable sample volume throughout the rest of the second quarter and through the fourth quarter as various states and dermatology offices opened throughout the country. Billable sample volume first exceeded pre‑pandemic levels in July 2020 even without all dermatology practices returning to full operations. Billable sample volume for the three months ended December 31, 2020 was 24% higher than billable sample volume for the three months ended September 30, 2020, and billable sample volume for the twelve months ended December 31, 2020 was 75% higher than billable sample volume for the twelve months ended December 31, 2019. Billable sample volume for the three months ended December 31, 2020 was 69% higher than billable sample volume for the three months ended December 31, 2019. Billable sample volumes could continue to be impacted by the ongoing COVID-19 pandemic and further impacted by a resurgence of the virus in the future.
58
Table of Contents
We have made available beginning in late April 2020 a telemedicine option for the PLA, but the telemedicine market is relatively new and unproven, especially within dermatology, and it is uncertain whether it will achieve and sustain high levels of demand, consumer acceptance and market adoption. While the COVID-19 pandemic is ongoing and clinician offices could close again due to the rolling back of reopening plans in various states, or patients could worry about going into the dermatology clinic, we expect that our revenues will depend to an extent on the willingness of clinicians and their patients to use our telemedicine option for the PLA, as well as on our ability to demonstrate the value of our telemedicine option to health plans and other purchasers of healthcare for beneficiaries. We also expect that the duration and extent of the effects of the ongoing COVID-19 pandemic in reducing patient access to clinician offices for in-person testing, and access by our sales force for in-office sales calls will adversely affect our revenues.
Contract Revenue
Contract revenues with major pharmaceutical companies relate to ongoing clinical trial contracts and new contracts. Contract revenue can be highly variable as it is dependent on the pharmaceutical customers’ clinical trial progress which can be difficult to forecast due to variability of patient enrollment, drug safety and efficacy and other factors. Many of our contracts with third parties are structured to contain milestone billing payments, which typically are advance payments on work yet to be performed. These advanced payments are structured to help fund operations and are included in deferred revenue as the work has not yet been performed. These advance payments will remain in deferred revenue until we process the laboratory portion of the contracts allowing us to recognize the revenue.
The ongoing COVID-19 pandemic has negatively affected and will continue to negatively affect our pharmaceutical customers’ clinical trials. The extent of such effect on our future revenue is uncertain and will depend on the duration and extent of the effects of the ongoing COVID-19 pandemic on our pharmaceutical customers’ clinical trials.
Financial Overview
Revenue
We generate revenue through laboratory services that are billed to Medicare, private medical insurance companies and to pharmaceutical companies who order our laboratory services, which can include sample collection kits, assay development, genomic analysis, data analysis and reporting. Our revenue is generated from two revenue streams: contract revenue and assay revenue. Assay revenue can be highly variable as it is based on payments received by private insurance payors that are not under contract and can vary based on patient insurance coverage, deductibles and co-pays. Contract revenue is ordered by customers on projects that may span over several years, which makes this type of revenue highly variable. Segments of these contracts may be increased, delayed or eliminated based on the success of each customers’ clinical trials or other factors.
Operating Expenses
Sales and Marketing Expenses
Sales and marketing expenses are primarily related to our specialty field sales force, market research, reimbursement efforts, trade show attendance, public relations, and general marketing. We expect these expenses to increase significantly as we expand our direct consumer marketing efforts and continue to add to our specialty sales force, marketing and payor access teams throughout 2021.
Research and Development Expenses
Our research and development, or R&D, expenses consist primarily of salaries and fringe benefits, clinical trials, consulting costs, facilities costs, laboratory costs, equipment expense, and depreciation. We also conduct clinical trials to validate the performance characteristics of our tests and to show medical cost benefit in support of our reimbursement efforts. We expect these expenses to increase significantly as we continue to develop new products and expand the use of our existing products.
General and Administrative Expenses
Our general and administrative expenses consist of senior management compensation, consulting, legal, billing and collections, human resources, information technology, accounting, insurance, and general business expenses. We expect our general and administrative expenses, especially employee-related costs, including stock-based compensation, insurance, accounting, and legal fees, to continue to increase due to operating as a publicly traded company.
59
Table of Contents
Financing Activities
Convertible Bridge Notes
On May 23, 2019, DermTech Operations, Inc. (formerly known as DermTech, Inc.), or DermTech Operations, and various holders of its convertible bridge notes agreed to amend DermTech Operations’ then outstanding convertible notes that were issued prior to June 5, 2019. As part of the amendment, the maturity dates of the notes were extended to the earliest of (i) September 24, 2019; (ii) the occurrence of an Event of Default (as defined in such notes); (iii) the consummation of a liquidation or dissolution of DermTech Operations (iv) a Liquidation Transaction (as defined in such notes); or (v) the consummation of a merger with or into the Company or any of its subsidiaries.
Between June 5, 2019 and June 10, 2019, DermTech Operations issued additional convertible bridge notes to its existing investors for aggregate gross proceeds of $2.6 million. These convertible bridge notes carried an interest rate of 10% and matured after the earliest to occur of: (i) September 25, 2019; (ii) the occurrence of an Event of Default (as defined in such notes); (iii) the consummation of a liquidation or dissolution of DermTech Operations; (iv) a Liquidation Transaction (as defined in such notes); or (v) the consummation of a merger of DermTech Operations with DT Merger Sub, Inc., a previous subsidiary of the Company, in accordance with the Merger Agreement (as defined below).
On August 29, 2019, in connection with the completion of the Business Combination (as defined below), all of the outstanding convertible bridge notes of DermTech Operations converted into Company common stock, in accordance with their respective terms.
Business Combination
On August 29, 2019, the Company and DermTech Operations consummated the transactions contemplated by the Agreement and Plan of Merger, dated as of May 29, 2019, by and among the Company, DT Merger Sub, Inc., or Merger Sub, and DermTech Operations. We refer to this agreement, as amended by that certain First Amendment to Agreement and Plan of Merger dated as of August 1, 2019, as the Merger Agreement. Pursuant to the Merger Agreement, Merger Sub merged with and into DermTech Operations, with DermTech Operations surviving as a wholly-owned subsidiary of the Company. We refer to this transaction as the Business Combination.
Immediately following the completion of the Business Combination, the Company changed its name from Constellation Alpha Capital Corp. to DermTech, Inc. and effected a one-for-two reverse stock split of its common stock, or the Reverse Stock Split. Prior to the closing of the Business Combination, the Company’s stock was listed on the Nasdaq Capital Market under the ticker symbol “CNAC.” On August 30, 2019, the Company’s common stock commenced trading on the Nasdaq Capital Market under the ticker symbol “DMTK.”
2019 PIPE Financing
On August 29, 2019, immediately prior to the completion of the Business Combination, the Company issued to certain accredited investors, in a private placement transaction, or the 2019 PIPE Financing, an aggregate of 3,076,925 shares of common stock and 1,231 shares of Series A Convertible Preferred Stock for aggregate gross proceeds of $24.0 million, or $6.50 per share of common stock on an as-converted basis. The 2019 PIPE Financing was conducted pursuant to the terms of separate Subscription Agreements and Amended and Restated Subscription Agreements, dated between May 22, 2019 and August 1, 2019, entered into by the Company and the investors. After giving effect to the Reverse Stock Split, each share of Series A Convertible Preferred Stock was convertible into 500 shares of the Company’s common stock, subject to conditions and adjustment as provided in the Certificate of Designation of Preferences, Rights and Limitations of Series A Convertible Preferred Stock.
On August 10, 2020, entities affiliated with Farallon Capital Management, L.L.C., or the Farallon entities, converted an aggregate of 1,231 shares of Series A Preferred Stock into 615,385 shares of common stock. On September 9, 2020, the Company filed a Certificate of Elimination of Series A Convertible Preferred Stock with the Secretary of State of the State of Delaware to eliminate its Series A Convertible Preferred Stock.
2020 PIPE Financing
On February 28, 2020, the Company entered into a securities purchase agreement with certain institutional investors for a private placement of the Company’s equity securities, or the 2020 PIPE Financing. Cowen and Company, LLC served as lead placement agent for the 2020 PIPE Financing with William Blair & Company, L.L.C. acting as joint placement agent. Lake Street Capital Markets, LLC acted as co-placement agent. The 2020 PIPE Financing closed on March 4, 2020.
Pursuant to the 2020 PIPE Financing, on March 4, 2020 the Company issued an aggregate of 2,467,724 shares of common stock at a purchase price of $10.50 per share, 3,199 shares of Series B-1 Convertible Preferred Stock, or the Series B-1 Shares, at a purchase price
60
Table of Contents
of $10.50 per share of common stock issuable upon conversion thereof, which were convertible into an aggregate of up to 3,198,942 shares of common stock, and 524 shares of Series B-2 Convertible Preferred Stock, or the Series B-2 Shares, at a purchase price of $10.50 per share of common stock issuable upon conversion thereof, which are convertible into an aggregate of up to 523,809 shares of common stock, for aggregate gross proceeds of approximately $65.0 million.
At the Company’s annual meeting held on May 26, 2020, the Company’s stockholders voted to approve the 2020 PIPE Financing, which resulted in the automatic conversion of the Series B-1 Shares into 3,198,949 shares of common stock on May 27, 2020. Each Series B-2 Share was convertible into 1,000 shares of the Company’s common stock, subject to conditions and adjustment as provided in the Certificate of Designation of Preferences, Rights and Limitations of Series B-2 Convertible Preferred Stock. On August 10, 2020, entities affiliated with Farallon Capital Management, L.L.C., or the Farallon entities, converted an aggregate of 524 shares of Series B‑2 Preferred Stock into 523,814 shares of common stock. On September 9, 2020, the Company filed a Certificate of Elimination of Series B-1 Convertible Preferred Stock and Certificate of Elimination of Series B-2 Convertible Preferred Stock with the Secretary of State of the State of Delaware to eliminate its Series B-1 and B-2 Convertible Preferred Stock.
2020 At-The-Market Offering
On November 10, 2020, the Company entered into a sales agreement with Cowen and Company, LLC relating to the sale of shares of the Company’s common stock from time to time with an aggregate offering price of up to $50.0 million. In connection with this sales agreement, the Company issued an aggregate of 951,792 shares of common stock at a weighted average purchase price of $20.97, resulting in aggregate gross proceeds of approximately $20.0 million.
2021 Underwritten Public Offering
On January 6, 2021, the Company, entered into an Underwriting Agreement with Cowen and Company, LLC and William Blair & Company, L.L.C. as representatives of several underwriters, or the Underwriters. The Company agreed to issue and sell up to 4,237,288 shares of its common stock including up to 635,593 shares that could be purchased by the Underwriters pursuant to a 30-day option granted to the Underwriters by the Company.
On January 11, 2021, the Company closed the underwritten public offering of 4,872,881 shares of its common stock, which included the exercise in full by the Underwriters of their option to purchase up to 635,593 additional shares, at a price to the public of $29.50 per share. The Company’s aggregate gross proceeds from the offering, before deducting underwriting discounts and commissions and other offering expenses, were $143.7 million.
Results of Operations
Fiscal Years Ended December 31, 2020 and 2019
Assay Revenue
Assay revenue grew $2.8 million or 202% to $4.2 million for fiscal year 2020 compared to $1.4 million for fiscal year 2019. Billable samples increased to approximately 24,000 for fiscal year 2020 compared to approximately 13,700 for fiscal year 2019. Sample volume is dependent on two major factors: the number of clinicians who order an assay in any given quarter and the number of assays ordered by each clinician during the period. The number of ordering clinicians and the utilization per clinician can vary based on a number of factors including the types of patients presenting skin cancer conditions, clinician reimbursement, office workflow, market awareness, clinician education and other factors. The ongoing COVID-19 pandemic has negatively affected and will continue to negatively affect our assay revenue by, among other things, limiting patient access to clinician offices for in-person testing and limiting access by our sales force for in-office sales calls.
Contract Revenue
Contract revenue with major pharmaceutical companies decreased $0.4 million to $1.6 million for fiscal year 2020, or 16%, compared to $2.0 million for fiscal year 2019. Contract revenue can be highly variable as it is dependent on the pharmaceutical customers’ clinical trial progress, which can be difficult to forecast due to variability of patient enrollment, drug safety and efficacy and other factors. The ongoing COVID-19 pandemic has negatively affected and will continue to negatively affect our pharmaceutical customers’ clinical trials. The extent of such effect on our future revenue is uncertain and will depend on the duration and extent of the effects of the ongoing COVID-19 pandemic on our pharmaceutical customers’ clinical trials. Many of our contracts with third parties are structured to contain milestone billing payments, which typically are advanced payments on work yet to be performed. These advanced payments are structured to help fund operations and are included in deferred revenue as the work has not yet been performed. As of December 31, 2020, the deferred revenue amount for these contracts, which is the advanced payments minus the value of work performed, was $1.5 million. These advanced payments will remain in deferred revenue until we process the laboratory portion of the contracts allowing us to recognize the revenue.
61
Table of Contents
Cost of Revenue
Cost of revenue increased $2.7 million, or 81%, to $6.0 million for fiscal year 2020 compared to $3.3 million for fiscal year 2019. The increase was largely attributable to a higher billable sample volume in 2020, higher fixed facility costs and higher equipment costs. At current capacity, a large portion of the costs of revenue are fixed, and these costs include the CLIA facility, quality assurance, management and supervision and equipment calibration and depreciation. The variable cost of revenue expenses incurred primarily relate to compensation-related costs for our laboratory scientists and technicians, laboratory supplies, shipping costs, equipment maintenance and calibration, and utilities. We remain committed to continuing the automation of our laboratory processes in order to become more cost efficient and productive. We currently operate in a gross loss position due to our current manufacturing capacity, level of sample volume and average selling price (“ASP”). As we increase our ASP and volume, we expect our margin to improve.
Operating Expenses
Sales and Marketing
Sales and marketing expenses increased $9.8 million, or 155%, to $16.1 million for fiscal year 2020 compared to $6.3 million for fiscal year 2019. The increase was primarily attributable to higher compensation-related costs from the expansion of the existing sales force and increased spending on advertising activities to increase market exposure. We expect to add to our specialty sales force, marketing and payor access teams throughout 2021 and 2022, and increase spending on direct-to-consumer marketing campaigns, which collectively would significantly increase our sales and marketing expenses.
Research and Development
R&D expenses increased $2.8 million, or 112%, to $5.3 million for fiscal year 2020 compared to $2.5 million for fiscal year 2020. The increase was due to higher compensation and recruiting costs of expanding the R&D team, including the addition of a new Chief Scientific Officer, increased clinical trial costs, and increased spend on laboratory supplies to support new product development. We expect these expenses to increase as we continue to grow the R&D team and focus on the development of our Luminate test, our basal and squamous cell skin cancer assays and other products in our pipeline.
General and Administrative
General and administrative expenses increased $5.0 million, or 56%, to $13.8 million for fiscal year 2020 compared to $8.9 million for fiscal year 2019. The increase was primarily due to higher payroll-related costs, stock-based compensation and costs required to operate as a publicly traded company. We expect these expenses to continue to increase as we add additional infrastructure such as human resources, information technology and legal resources. Also included in general and administrative expenses fiscal year 2020 was a one-time $1.0 million litigation settlement charge related to the Settlement Agreement with LifeSci.
Gain on Debt Extinguishment
Gain on debt extinguishment decreased $0.9 million, or 100%, to $0 for the fiscal year 2020 compared to $0.9 million for the fiscal year 2019. The decrease in gain on debt extinguishment was due to the extinguishment of the convertible bridge notes that occurred in August 2019 in connection with the Business Combination.
Interest Income/(Expense), net
Interest income for fiscal year 2020 was $40,000 compared to interest expense of $2.7 million for fiscal year 2019. Interest income for fiscal year 2020 consists primarily of interest earned on our short-term investments as a result of the proceeds from the 2020 PIPE Financing in March 2020. The decrease in interest expense was due to the extinguishment of the convertible bridge notes that occurred in August 2019 in connection with the Business Combination. We do not expect to incur significant interest expense for future reporting periods in 2021.
Other Expense
Other expense decreased $0.4 million, or 100%, to $0 for the fiscal year 2020 compared to $0.4 million for fiscal year 2019. The significant decrease in other expense was due to the extinguishment of the convertible bridge notes that occurred in August 2019 in connection with the Business Combination.
62
Table of Contents
Liquidity and Capital Resources
We have never been profitable and have historically incurred substantial net losses, including net losses of $10.0 million for the twelve months ended December 31, 2018, $19.7 million for the twelve months ended December 31, 2019, and $35.2 million for the twelve months ended December 31, 2020. As of December 31, 2020, our accumulated deficit was $126.4 million, and we had negative operating cash flow of $28.7 million. In connection with the Business Combination, we completed the 2019 PIPE Financing that raised a total of $24.0 million in gross proceeds in addition to the $1.8 million in cash the Company had on hand at the closing of the Business Combination. We completed the 2020 PIPE Financing in March 2020, which raised a total of $65.0 million in gross proceeds. In addition, at the end of 2020, we raised approximately $20.0 million in gross proceeds facilitated through our At-The-Market Offering. We have historically financed operations through private placement equity offerings and convertible debt offerings.
We expect our losses to continue as a result of costs relating to ongoing R&D expenses, increased general and administrative expenses and increased sales and marketing costs for existing and planned products. These losses have had, and will continue to have, an adverse effect on our working capital. Because of the numerous risks and uncertainties associated with our commercialization and development efforts, we are unable to predict when we will become profitable, and we may never become profitable. Our inability to achieve and then maintain profitability would negatively affect our business, financial condition, results of operations and cash flows.
As of December 31, 2020, our cash, cash equivalents and marketable securities totaled $63.8 million. On January 11, 2021, we closed a public offering that resulted in aggregate gross proceeds of $143.7 million from the offering, before deducting underwriting discounts and commissions and other offering expenses. Based on our current business operations and the additional 2021 financing, we believe our current cash and cash equivalents will be sufficient to meet our anticipated cash requirements for at least the next twelve months. While we believe we have enough capital to fund anticipated operating costs for at least the next 12 months, we expect to incur significant additional operating losses over at least the next several years. We anticipate that we will raise additional capital through equity offerings, debt financings, collaborations or licensing arrangements in order to support our planned operations and to continue developing and commercializing genomic tests. We may also consider raising additional capital in the future to expand our business, to pursue strategic investments or to take advantage of financing opportunities. Our present and future funding requirements will depend on many factors, including:
| • | our revenue growth rate and ability to generate cash flows from operating activities; |
| • | the willingness of clinicians and their patients to use our telemedicine option for the PLA and the duration and extent of the effects of the ongoing COVID-19 pandemic in reducing patient access to clinician offices for in-person testing; |
| • | the duration and extent of the effects of the ongoing COVID-19 pandemic on our pharmaceutical customers’ clinical trials; |
| • | our sales and marketing and R&D activities; |
| • | effects of competing technological and market developments; |
| • | costs of and potential delays in product development; |
| • | changes in regulatory oversight applicable to our tests; and |
| • | timing of and costs related to future international expansion. |
There can be no assurances as to the availability of additional financing or the terms upon which additional financing may be available to us. If we are unable to obtain sufficient funding at acceptable terms, we may be forced to significantly curtail our operations, and the lack of sufficient funding may have a material adverse impact on our ability to continue as a going concern.
Cash Flow Analysis
Fiscal Year Ended December 31, 2020
Net cash used in operating activities for the twelve months ended December 31, 2020 totaled $28.7 million, primarily driven by the $35.2 million net loss offset by non-cash related items, including $5.0 million in stock-based compensation and $0.5 million in depreciation. In addition, we had a cash inflow of $0.8 million through the increase of accounts payable and accrued compensation as well as a $1.6 million of cash inflow through the increase of accrued liabilities and deferred revenues. This was offset by the cash outflow through the increase of prepaid expenses and other assets of $0.5 million as well as an increase in accounts receivable of $0.8 million.
63
Table of Contents
Net cash used in investing activities totaled $41.3 million for the twelve months ended December 31, 2020, primarily related to the purchase of $41.7 million in short-term marketable securities and $1.8 million in purchases of property and equipment. This was offset by cash inflows from the sale and maturity of marketable securities of $2.2 million. As we scale our sales force, the expected timing of a corresponding increase in assay volume is uncertain due, in part, to challenges presented by the ongoing COVID-19 pandemic, such as related limits on patient access to clinician offices for in-person testing. Additional laboratory equipment investment will be needed to install complex automation systems and other genomic testing equipment needed to expand testing capacity.
Net cash provided by financing activities totaled $78.9 million for the twelve months ended December 31, 2020, which was predominantly driven by the $59.9 million and $19.1 million in net proceeds raised from the 2020 PIPE Financing and At-the-Market Offering, respectively, and $1.3 million from the exercise of stock options and warrants. This was offset by the payment made by the Company of the deferred underwriting fees of $1.4 million.
Fiscal Year Ended December 31, 2019
Net cash used in operating activities for the twelve months ended December 31, 2019 totaled $17.8 million, primarily driven by the $19.7 million net loss offset by non-cash related items, including $2.0 million in amortization of the convertible bridge notes debt discount, $1.3 million in stock-based compensation and $0.4 million in the change in the convertible bridge notes derivative liability, offset by the gain on extinguishment of convertible notes of $0.9 million and a tax payment of $1.6 million related to the release of certain employees’ restricted stock units. In addition, we had $1.3 million of cash inflow through the increase of accounts payables and accrued compensation as well as a $0.5 million of cash inflow through the increase of accrued liabilities and deferred revenues. This was offset by the cash outflow of $1.0 million related to payments for prepaid insurance as well as an increase in accounts receivable of $0.1 million.
Net cash used in investing activities totaled $0.2 million for the twelve months ended December 31, 2019, which related predominantly to the purchase of laboratory equipment. As we scale our sales force and the resulting expected increase to assay volume, additional laboratory equipment investment will be needed to install complex automation systems and other genomic testing equipment.
Net cash provided by financing activities totaled $28.6 million for the twelve months ended December 31, 2019, which was predominantly driven by the $25.6 million in net proceeds raised from the Business Combination and related 2019 PIPE financing and issuing $2.6 million in convertible notes.
Off-Balance Sheet Arrangements
As of December 31, 2020 and 2019, we did not have any off-balance sheet arrangements, as such term is defined under Item 303 of Regulation S-K, that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that is material to investors.
Critical Accounting Policies and Significant Judgments and Estimates
The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles (“U.S. GAAP”) requires that management make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the amounts of revenues and expenses reported during the period. On an ongoing basis, management evaluates these estimates and judgments, including but not limited to those related to revenue, warrants, stock-based compensation, accounts receivable, expense accruals, convertible debt, the realization of deferred tax assets, and common and preferred stock valuations. Actual results may differ from those estimates.
The SEC has defined a company’s critical accounting policies as the ones that are most important to the portrayal of the company’s financial condition and results of operations, and which require the company to make its most difficult and subjective judgments, often as a result of the need to make estimates of matters that are inherently uncertain. While our significant accounting policies are more fully described in Note 1 of our consolidated financial statements included in this report, we believe that the following accounting policies and judgments are most critical to aid in fully understanding and evaluating our reported financial results based upon the SEC’s defined criteria.
64
Table of Contents
Revenue Recognition
Our revenue is generated from two revenue streams, contract revenue and assay revenue. We account for revenue in accordance with Accounting Standards Codification (“ASC”) Topic 606, Revenue from Contracts with Customers (“ASC 606”). The core principle of ASC 606 is that the Company recognizes revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the Company expects to be entitled in exchange for those goods or services. The ASC 606 revenue recognition model consists of the following five steps: (1) identify the contracts with a customer, (2) identify the performance obligations in the contract, (3) determine the transaction price, (4) allocate the transaction price to the performance obligations in the contract and (5) recognize revenue when (or as) the entity satisfies a performance obligation.
We generate revenues from our PLA, and Nevome tests we provide to healthcare clinicians in various states throughout the United States to assist in a clinician’s diagnosis of melanoma. We provide prescribing clinicians with our adhesive sample collection kits to perform non-invasive skin biopsies of clinically ambiguous pigmented skin lesions on patients. The Company also offers clinicians a telemedicine solution where they can request the PLA collection kit be sent to the patient’s home for a clinician-guided remote collection on ambiguous pigmented skin lesions. Once the sample is collected by the healthcare clinician or the patient via the telemedicine solution, it is returned to the Company’s CLIA laboratory for analysis. The patient’s ribonucleic acid (“RNA”) and deoxyribonucleic acid (“DNA”) are extracted from the adhesive patch collection kit and analyzed using gene expression technology to determine if the pigmented skin lesion contains certain genomic features indicative of melanoma. Upon completion of the gene expression analysis, a final report is drafted and provided to the dermatologists detailing the test results for the pigmented skin lesion indicating whether the sample collected is indicative of melanoma or not. A detailed analysis of payments made to us by private health insurance payors for the assays over several quarters is used to estimate the ultimate receipt of funds for payment of billed amounts. These payments can vary widely from payor to payor and can be halted for routine audits or other reasons.
Contract revenue is generated from the sale of laboratory services and adhesive sample collection kits to third party companies through contract research agreements. Revenues are generated from providing gene expression tests to facilitate the development of drugs designed to treat dermatologic conditions. The provision of gene expression services may include sample collection using the Company’s patented adhesive patch collection kits, assay development for research partners, RNA extraction, isolation, expression, amplification and detection, including data analysis and reporting.
See Note 1(k) of our consolidated financial statements for a full discussion of our revenue recognition policy around assay revenue and contract revenue.
Stock-Based Compensation
Compensation costs associated with stock option awards and other forms of equity compensation are measured at the grant-date fair value of the awards and recognized over the requisite service period of the awards on a ratable basis.
We grant stock options to purchase common stock to employees with exercise prices equal to the fair market value of the underlying stock, as determined by the board of directors, management, outside valuation experts and subsequent to the completion of the Business Combination, the closing stock price on the date of grant. The board of directors and outside valuation experts determine the fair value of the underlying stock by considering a number of factors, including historical and projected financial results, the risks we faced at the time, the preferences of our debt holders and preferred stockholders, and the lack of liquidity of our common stock that occurred prior to the Business Combination.
The fair value of each stock option award is estimated using the Black-Scholes-Merton valuation model. Such value is recognized as expense over the requisite service period using the straight-line method. The expected term of options is based on the simplified method which defines the expected term as the average of the contractual term of the options and the weighted average vesting period for all option tranches. The expected volatility of stock options is based upon the historical volatility of a number of related publicly traded companies in similar stages of development as well as the volatility of the Company’s common stock. The risk-free interest rate is based on the average yield of U.S. Treasury securities with remaining terms similar to the expected term of the share-based awards. The assumed dividend yield was based on our expectation of not paying dividends in the foreseeable future.
We account for stock options to non-employees using the fair value approach. The fair value of these options is measured using the Black-Scholes-Merton option pricing model, reflecting the same assumptions applied to employee options, other than expected life, which is assumed to be the remaining contractual life of the award. Options that are granted to employees generally have a requisite service period of three to four years.
65
Table of Contents
Restricted stock units (“RSUs”) are considered restricted stock. The fair value of restricted stock is equal to the fair market value of the underlying stock, as determined by the board of directors, management, input from outside valuation experts and subsequent to the completion of the Business Combination, the closing stock price on the date of grant. We recognize stock-based compensation expense based on the fair value on a ratable basis over the requisite service periods of the awards. RSUs that are granted to employees have a requisite service period typically between two and four years.
Recent accounting pronouncements
See Note 1(s) of our consolidated financial statements for a discussion of the impact of new accounting pronouncements on our consolidated financial statements.
JOBS Act Accounting Election
We are an emerging growth company within the meaning of the JOBS Act. Section 107(b) of the JOBS Act provides that an emerging growth company can use the extended transition period, provided in Section 102(b) of the JOBS Act, for complying with new or revised accounting standards. Thus, an emerging growth company can delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies. We have elected to use this extended transition period and, as a result, our financial statements may not be comparable to companies that comply with public company effective dates. We also intend to rely on other exemptions provided by the JOBS Act, including without limitation, not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act of 2002.
We will remain an emerging growth company until the earliest to occur of (i) the last day of the fiscal year during which we had total annual gross revenues of at least $1.07 billion, (ii) the day we are deemed to be a large accelerated filer as defined in Rule 12b-2 under the Exchange Act, which means the market value of our common stock that is held by non-affiliates exceeds $700 million, measured as of our most recently completed second fiscal quarter, (iii) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period, and (iv) December 31, 2022.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Our cash, cash equivalents, and short-term marketable securities are subject to economic risk which could affect our results of operations, financial condition and cash flows. We manage our exposure to this market risk through our regular operating and financing activities.
Interest Rate Risk
The primary objective of our investment activities is capital preservation to fund operations, while at the same time maximizing investment income without significantly increasing investment risk. To achieve these objectives, our investment policy allows for a portfolio of cash equivalents and investments in a variety of securities, including money market funds, U.S. government debt and corporate debt securities. Due to the short-term and conservative nature of our investments, we do not believe that we have a material exposure to interest rate risk. A 100 basis point change in interest rates would not have a significant impact on the total value of our portfolio.
66
Table of Contents
Item 8. Consolidated Financial Statements and Supplementary Data
DERMTECH, INC.
Index to Consolidated Financial Statements
67
Table of Contents
Report of Independent Registered Public Accounting Firm
To the Stockholders and Board of Directors
DermTech, Inc.:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of DermTech, Inc. and subsidiaries (the Company) as of December 31, 2020 and 2019, the related consolidated statements of operations, comprehensive loss, convertible preferred stock and stockholders’ equity (deficit), and cash flows for each of the years in the two-year period ended December 31, 2020, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2020 and 2019, and the results of its operations and its cash flows for each of the years in the two-year period ended December 31, 2020, in conformity with U.S. generally accepted accounting principles.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ KPMG LLP
We have served as the Company’s auditor since 2016.
San Diego, California
March 5, 2021
68
Table of Contents
DERMTECH, INC.
Consolidated Balance Sheets
(in thousands, except share and per share data)
| | December 31, 2020 | | | December 31, 2019 | |
Assets | | | | | | | | |
Current assets: | | | | | | | | |
Cash and cash equivalents | | $ | 24,248 | | | $ | 15,374 | |
Short-term marketable securities | | | 39,529 | | | | — | |
Accounts receivable, net | | | 1,480 | | | | 680 | |
Inventory | | | 104 | | | | 35 | |
Prepaid expenses and other current assets | | | 1,521 | | | | 1,061 | |
Total current assets | | | 66,882 | | | | 17,150 | |
Property and equipment, net | | | 2,731 | | | | 977 | |
Other assets | | | 167 | | | | 84 | |
Total assets | | $ | 69,780 | | | $ | 18,211 | |
Liabilities, Convertible Preferred Stock and Stockholders’ Equity | | | | | | | | |
Current liabilities: | | | | | | | | |
Accounts payable | | $ | 1,573 | | | $ | 1,609 | |
Accrued compensation | | | 2,075 | | | | 1,142 | |
Accrued liabilities | | | 763 | | | | 218 | |
Short-term deferred revenue | | | 905 | | | | 1,390 | |
Deferred underwriting fees | | | — | | | | 1,363 | |
Current portion of capital lease obligations | | | 109 | | | | — | |
Total current liabilities | | | 5,425 | | | | 5,722 | |
Long-term deferred revenue | | | 639 | | | | — | |
Long-term capital lease obligations, less current portion | | | 226 | | | | — | |
Total liabilities | | | 6,290 | | | | 5,722 | |
Commitments and contingencies | | | | | | | | |
Series A convertible preferred stock, $0.0001 par value per share; zero and 5,000,000 Series A shares authorized as of December 31, 2020 and 2019, respectively; zero and 1,231 shares issued and outstanding at December 31, 2020 and 2019, respectively; zero and $7.6 million liquidation preference at December 31, 2020 and 2019, respectively | | | — | | | | — | |
Stockholders’ equity: | | | | | | | | |
Common stock, $0.0001 par value per share; 50,000,000 shares authorized as of December 31, 2020 and 2019; 20,740,413 and 12,344,818 shares issued and outstanding at December 31, 2020 and 2019, respectively | | | 2 | | | | 1 | |
Additional paid-in capital | | | 189,849 | | | | 103,599 | |
Accumulated other comprehensive loss | | | (1 | ) | | | — | |
Accumulated deficit | | | (126,360 | ) | | | (91,111 | ) |
Total stockholders’ equity | | | 63,490 | | | | 12,489 | |
Total liabilities, convertible preferred stock and stockholders’ equity | | $ | 69,780 | | | $ | 18,211 | |
See accompanying notes to consolidated financial statements.
69
Table of Contents
DERMTECH, INC.
Consolidated Statements of Operations
(in thousands, except share and per share data)
| | Year Ended December 31, | |
| | 2020 | | | 2019 | |
Revenues: | | | | | | | | |
Assay revenue | | $ | 4,241 | | | $ | 1,403 | |
Contract revenue | | | 1,644 | | | | 1,961 | |
Total revenues | | | 5,885 | | | | 3,364 | |
Cost of revenues | | | 5,981 | | | | 3,304 | |
Gross profit/(loss) | | | (96 | ) | | | 60 | |
Operating expenses: | | | | | | | | |
Sales and marketing | | | 16,077 | | | | 6,303 | |
Research and development | | | 5,293 | | | | 2,497 | |
General and administrative | | | 13,823 | | | | 8,865 | |
Total operating expenses | | | 35,193 | | | | 17,665 | |
Loss from operations | | | (35,289 | ) | | | (17,605 | ) |
Other income/(expense): | | | | | | | | |
Gain on debt extinguishment of convertible notes | | | — | | | | 928 | |
Interest income/(expense) | | | 40 | | | | (2,657 | ) |
Other expense | | | — | | | | (355 | ) |
Total other income/(expense) | | | 40 | | | | (2,084 | ) |
Net loss | | $ | (35,249 | ) | | $ | (19,689 | ) |
Weighted average shares outstanding used in computing net loss per share, basic and diluted | | | 16,979,411 | | | | 7,005,037 | |
Net loss per share of common stock outstanding, basic and diluted | | $ | (2.08 | ) | | $ | (2.81 | ) |
See accompanying notes to consolidated financial statements.
70
Table of Contents
DERMTECH, INC.
Consolidated Statements of Comprehensive Loss
(in thousands)
| | Year Ended December 31, | |
| | 2020 | | | 2019 | |
Net loss | | $ | (35,249 | ) | | $ | (19,689 | ) |
Unrealized loss on available-for-sale marketable securities | | | (1 | ) | | | — | |
Comprehensive loss | | $ | (35,250 | ) | | $ | (19,689 | ) |
See accompanying notes to consolidated financial statements.
71
Table of Contents
DERMTECH, INC.
Consolidated Statements of Convertible Preferred Stock and Stockholders’ Equity (Deficit)
(in thousands, except share and per share data)
| | Series A convertible preferred stock | | | | Series B-1 convertible preferred stock | | | | Series B-2 convertible preferred stock | | | | Series C convertible preferred stock | | | | Common stock | | | Additional paid-in | | | Accumulated other comprehensive | | | Accumulated | | | Total stockholders’ | |
| | Shares | | | Amount | | | | Shares | | | Amount | | | | Shares | | | Amount | | | | Shares | | | Amount | | | | Shares | | | Amount | | | capital | | | loss | | | deficit | | | equity (deficit) | |
Balance, December 31, 2018 | | | — | | | $ | — | | | | | — | | | $ | — | | | | | — | | | $ | — | | | | | 1,524,122 | | | $ | — | | | | | 4,411,567 | | | $ | 1 | | | $ | 66,021 | | | $ | — | | | $ | (71,377 | ) | | $ | (5,355 | ) |
Cumulative effect adjustment of accounting method change | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | — | | | | — | | | | (45 | ) | | | (45 | ) |
Issuance of common stock | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | 726,139 | | | | — | | | 934 | | | | — | | | | — | | | 934 | |
Conversion of Series C preferred stock to common stock | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | (1,524,122 | ) | | | — | | | | | 1,524,122 | | | | — | | | | — | | | | — | | | | — | | | | — | |
Conversion of convertible notes to common stock | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | 2,267,042 | | | | — | | | | 12,687 | | | | — | | | | — | | | | 12,687 | |
Additional paid in capital assumed in Business Combination | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | 420 | | | | — | | | | — | | | 420 | |
Issuance of Series A preferred stock at $3,250 per share | | | 1,231 | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | 4,000 | | | | — | | | | — | | | | 4,000 | |
Issuance of common stock at $6.50 per share, net of $0.2 million issuance costs | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | 3,076,923 | | | | — | | | | 19,802 | | | | — | | | | — | | | | 19,802 | |
Restricted stock unit release | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | 339,025 | | | | — | | | | (1,569 | ) | | | | | | | | | | | (1,569 | ) |
Stock-based compensation | | — | | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | | 1,304 | | | — | | | — | | | | 1,304 | |
Net loss | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | — | | | | — | | | | (19,689 | ) | | | (19,689 | ) |
Balance, December 31, 2019 | | | 1,231 | | | $ | — | | | | | — | | | $ | — | | | | | — | | | $ | — | | | | | — | | | $ | — | | | | | 12,344,818 | | | $ | 1 | | | $ | 103,599 | | | $ | — | | | $ | (91,111 | ) | | $ | 12,489 | |
Issuance of common stock at $10.50 per share, net of $2.0 million in issuance costs | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | 2,467,724 | | | | — | | | | 23,889 | | | | — | | | | — | | | | 23,889 | |
Issuance of Series B-1 convertible preferred stock at $10,500 per share, net of $2.6 million in issuance costs | | | — | | | | — | | | | | 3,199 | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | 30,968 | | | | — | | | | — | | | | 30,968 | |
Issuance of Series B-2 convertible preferred stock at $10,500 per share, net of $0.4 million in issuance costs | | | — | | | | — | | | | | — | | | | — | | | | | 524 | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | 5,071 | | | | — | | | | — | | | | 5,071 | |
Issuance of common stock from option exercises and RSU releases | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | 319,522 | | | | — | | | 473 | | | | — | | | | — | | | | 473 | |
Issuance of common stock from warrant exercises | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | 230,619 | | | | — | | | | 842 | | | | — | | | | — | | | 842 | |
Issuance costs in connection with Form S-1 registration statement | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | (77 | ) | | | — | | | | — | | | | (77 | ) |
Conversion of Series B-1 convertible preferred stock to common stock | | | — | | | | — | | | | | (3,199 | ) | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | 3,198,949 | | | | 1 | | | | — | | | | — | | | | — | | | | 1 | |
Conversion of Series A and B-2 convertible preferred stock to common stock | | | (1,231 | ) | | — | | | | — | | | — | | | | | (524 | ) | | — | | | | — | | | — | | | | | 1,139,199 | | | | — | | | | — | | | — | | | — | | | | — | |
Issuance of common stock from Life Sci settlement | | — | | | — | | | | — | | | — | | | | — | | | — | | | | — | | | — | | | | | 87,790 | | | — | | | | 1,011 | | | — | | | — | | | | 1,011 | |
Issuance of common stock at a weighted average price of $20.97 through at-the-market offering, net of $0.9 million in issuance costs | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | 951,792 | | | | — | | | | 19,104 | | | | — | | | | — | | | | 19,104 | |
Unrealized loss on available-for-sale securities | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | — | | | | (1 | ) | | — | | | | (1 | ) |
Stock-based compensation | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | 4,969 | | | | — | | | | — | | | | 4,969 | |
Net loss | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | | — | | | | — | | | | — | | | | — | | | | (35,249 | ) | | | (35,249 | ) |
Balance, December 31, 2020 | | | — | | | $ | — | | | | | — | | | $ | — | | | | | — | | | $ | — | | | | | — | | | $ | — | | | | | 20,740,413 | | | $ | 2 | | | $ | 189,849 | | | $ | (1 | ) | | $ | (126,360 | ) | | $ | 63,490 | |
See accompanying notes to consolidated financial statements.
72
Table of Contents
DERMTECH, INC.
Consolidated Statements of Cash Flows
(in thousands)
| | Year Ended December 31, | |
| | 2020 | | | 2019 | |
Cash flows from operating activities: | | | | | | | | |
Net loss | | $ | (35,249 | ) | | $ | (19,689 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | |
Depreciation | | | 486 | | | | 89 | |
Stock-based compensation | | | 4,969 | | | | 1,304 | |
Amortization of debt discount and issuance costs | | | — | | | | 1,983 | |
Change in fair value of derivative liability | | | — | | | | 355 | |
Gain on extinguishment of convertible notes | | | — | | | | (928 | ) |
Interest income, net | | | (21 | ) | | | — | |
Loss on disposal of equipment | | | 13 | | | | — | |
Payment in connection with restricted stock unit release | | | — | | | | (1,569 | ) |
Changes in operating assets and liabilities: | | | | | | | | |
Accounts receivable, net | | | (800 | ) | | | (100 | ) |
Inventory | | | (69 | ) | | | 5 | |
Prepaid expenses and other assets | | | (487 | ) | | | (1,069 | ) |
Accounts payable and accrued compensation | | | 827 | | | | 1,337 | |
Accrued liabilities and deferred revenue | | | 1,647 | | | | 491 | |
Net cash used in operating activities | | | (28,684 | ) | | | (17,791 | ) |
Cash flows from investing activities: | | | | | | | | |
Purchases of marketable securities | | | (41,706 | ) | | | — | |
Maturities of marketable securities | | | 2,200 | | | | — | |
Purchases of property and equipment | | | (1,834 | ) | | | (210 | ) |
Net cash used in investing activities | | | (41,340 | ) | | | (210 | ) |
Cash flows from financing activities: | | | | | | | | |
Proceeds from issuance of common stock in connection with private placement offering, net | | | 23,889 | | | | — | |
Proceeds from issuance of Series B-1 Convertible Preferred Stock, net | | | 30,968 | | | | — | |
Proceeds from issuance of Series B-2 Convertible Preferred Stock, net | | | 5,071 | | | | — | |
Payments of deferred underwriting fees | | | (1,363 | ) | | | — | |
Payments of issuance costs in connection with Form S-1 registration statement | | | (77 | ) | | | — | |
Proceeds from issuance of common stock in connection with at-the-market offering, net | | | 19,104 | | | | — | |
Proceeds from issuance of common stock | | | — | | | | 19,802 | |
Proceeds from exercise of common stock warrants | | | 842 | | | | 5 | |
Proceeds from exercise of stock options | | | 473 | | | | 929 | |
Proceeds from convertible notes payable | | | — | | | | 2,600 | |
Payments of notes payable | | | — | | | | (516 | ) |
Proceeds from issuance of Series A Convertible Preferred Stock | | | — | | | | 4,000 | |
Principal repayments of capital lease obligations | | | (9 | ) | | | — | |
Proceeds received from close of Business Combination | | | — | | | | 1,802 | |
Net cash provided by financing activities | | | 78,898 | | | | 28,622 | |
Net increase in cash and cash equivalents | | | 8,874 | | | | 10,621 | |
Cash and cash equivalents, beginning of period | | | 15,374 | | | | 4,753 | |
Cash and cash equivalents, end of period | | $ | 24,248 | | | $ | 15,374 | |
Supplemental cash flow information: | | | | | | | | |
Cash paid for interest on capital lease obligations | | $ | 2 | | | $ | — | |
Supplemental disclosure of noncash investing and financing activities: | | | | | | | | |
Issuance of common stock in litigation settlement | | $ | 1,011 | | | $ | — | |
Purchases of property and equipment recorded in accounts payable | | $ | 71 | | | $ | 641 | |
Property and equipment acquired under capital leases | | $ | 342 | | | $ | — | |
Change in unrealized loss on marketable securities, net of tax | | $ | (1 | ) | | $ | — | |
Unpaid deferred issuance costs | | $ | 56 | | | $ | 1,363 | |
Debt discount and derivative liability at issuance of convertible notes payable | | $ | — | | | $ | 270 | |
See accompanying notes to consolidated financial statements.
73
Table of Contents
DERMTECH, INC.
Notes to Consolidated Financial Statements
1. The Company and a Summary of its Significant Accounting Policies
On August 29, 2019, DermTech, Inc., formerly known as Constellation Alpha Capital Corp, (the “Company”), and DermTech Operations, Inc., formerly known as DermTech, Inc., (“DermTech Operations”), consummated the transactions contemplated by the Agreement and Plan of Merger, dated as of May 29, 2019, by and among the Company, DT Merger Sub, Inc., a wholly owned subsidiary of the Company (“Merger Sub”), and DermTech Operations. The Company refers to this agreement, as amended by that certain First Amendment to Agreement and Plan of Merger dated as of August 1, 2019, as the Merger Agreement. Pursuant to the Merger Agreement, Merger Sub merged with and into DermTech Operations, with DermTech Operations surviving as a wholly-owned subsidiary of the Company. The Company refers to this transaction as the Business Combination. In connection with and two days prior to the completion of the Business Combination, the Company domesticated from the British Virgin Islands to Delaware. DermTech Operations changed its name from DermTech, Inc. to DermTech Operations, Inc. shortly before the completion of the Business Combination. On August 29, 2019, immediately following the completion of the Business Combination, the Company changed its name from Constellation Alpha Capital Corp. to DermTech, Inc., and then effected a one-for-two reverse stock split of its common stock (“Reverse Stock Split”).
The Company is an emerging growth molecular diagnostic company developing and marketing its Clinical Laboratory Improvement Amendments of 1988 (“CLIA”) laboratory services including molecular pathology tests to facilitate the diagnosis of dermatologic conditions including melanoma. The Company has developed a proprietary, non-invasive technique for sampling the surface layers of the skin using an adhesive patch in order to collect individual biological information for commercial applications in the medical diagnostic field.
From the end of the first quarter and through the fourth quarter of 2020, there has been a widespread worldwide impact from the COVID-19 pandemic. The Company is considered an essential business due to the importance of early melanoma detection, which has allowed the Company’s CLIA laboratory to remain fully operational. The Company has implemented additional safety measures and social distancing with its CLIA laboratory operations and has transitioned administrative functions to predominantly remote work. Beginning in March 2020 and continuing through the fourth quarter of 2020, the ongoing COVID-19 pandemic has reduced patient access to clinician offices for in-person testing, which has resulted in a reduced volume of billable samples received during the fourth quarter of 2020 relative to the Company’s pre-pandemic expectations. The Company expects the ongoing COVID-19 pandemic to continue to adversely impact billable sample volume until patient access to in-person testing fully resumes or telemedicine options are more widely adopted. Additionally, the ongoing COVID-19 pandemic has negatively affected and will continue to negatively affect the Company’s pharmaceutical customers’ clinical trials. The extent of such effect on the Company’s future revenue is uncertain and will depend on the duration and extent of the effects of the ongoing COVID-19 pandemic on the Company’s pharmaceutical customers’ clinical trials.
The consolidated financial statements include the accounts of DermTech, Inc. and its subsidiaries. All intercompany balances and transactions among the consolidated entity have been eliminated in consolidation. These consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles, (“U.S. GAAP”). In the opinion of management, all adjustments, which include only normal recurring adjustments considered necessary for a fair presentation, have been included.
The preparation of consolidated financial statements in conformity with U.S. GAAP requires that management make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the amounts of revenues and expenses reported during the period. On an ongoing basis, management evaluates these estimates and judgments, including those related, but not limited to, assay revenue, stock-based compensation, short-term marketable securities, accounts receivable, the useful lives and recoverability of property and equipment and the realization of deferred tax assets. Actual results may differ from those estimates.
74
Table of Contents
(d) | Cash and Cash Equivalents |
The Company considers all highly liquid investments with remaining maturities of three months or less when purchased to be cash equivalents. The Company maintains its cash balances at banks and financial institutions. The balances are insured up to the Federal Deposit Insurance Corporation legal limit. The Company maintains cash balances that may, at times, exceed this insured limit.
(e) Marketable Securities
The Company considers securities with original maturities of greater than 90 days to be marketable securities. The Company has the ability, if necessary, to liquidate any of its cash equivalents and marketable securities to meet its liquidity needs in the next 12 months. Accordingly, those investments with contractual maturities greater than one year from the date of purchase are classified as current assets on the accompanying consolidated balance sheets. The Company’s marketable securities consist of U.S. Treasury and agency securities, commercial paper, and corporate debt securities. Marketable securities are recorded at fair value and unrealized gains and losses are recorded within accumulated other comprehensive loss. The estimated fair value of the marketable securities is determined based on quoted market prices or rates for similar instruments. The Company evaluates securities with unrealized losses to determine whether such losses, if any, are other than temporary. Realized gains and losses are calculated using the specific identification method and recorded as interest income or expense. The Company has determined that there were no other-than-temporary declines in fair values of its investments as of December 31, 2020.
(f) Deferred Issuance Costs
The Company capitalizes certain legal and other third-party fees that are direct and incremental costs associated with in process equity financings as deferred offering costs until such financings are consummated. After consummation of the equity financing, these costs are recorded as a reduction of the proceeds generated as a result of the offering. Deferred issuance costs amounted to $0.1 million and zero as of December 31, 2020 and 2019, respectively, and was recorded as a component of prepaid expenses and other current assets on the consolidated balance sheets.
(g) | Property and Equipment |
Property and equipment is recorded at cost less accumulated depreciation. Property includes property we have acquired under build-to-suit arrangements. Equipment includes assets such as office, computer and laboratory equipment, including laboratory equipment acquired under capital lease arrangements. Depreciation is computed using the straight-line method over the estimated useful lives of the assets, which range from two to five years. Leasehold improvements are depreciated over the shorter of the remaining term of the lease or the useful life of the asset. The Company recorded depreciation expense of $0.5 million and $0.1 million, which includes amortization of laboratory equipment acquired under capital leases of $10,000 and zero for the years ended December 31, 2020 and 2019, respectively. Amortization of assets that are recorded under capital leases in depreciation expense is included in cost of revenue on the consolidated statement of operations. Gross assets recorded under capital leases were $0.3 million and zero as of December 31, 2020 and 2019, respectively. Accumulated amortization associated with capital leases was $10,000 and zero as of December 31, 2020 and 2019, respectively. Maintenance and repairs are charged to expense as incurred, and material improvements are capitalized. When assets are retired or otherwise disposed of, the cost and accumulated depreciation are removed from the balance sheet and any resulting gain or loss is reflected in the statements of operations and comprehensive loss in the period realized. $52,000 and zero of equipment was disposed of during the years ended December 31, 2020 and 2019, respectively. The Company assesses its long-lived assets, consisting primarily of property and equipment, for impairment when material events or changes in circumstances indicate that the carrying value may not be recoverable. There were no impairment losses for the years ended December 31, 2020 and 2019.
(h) | Research and Development |
Costs incurred in connection with research and development (“R&D”) activities are expensed as incurred. R&D expenses consist of (i) employee-related expenses, including salaries, benefits, travel and stock-based compensation expense; (ii) and facilities and other expenses, which include direct and allocated expenses for rent and maintenance of facilities and laboratory and other supplies.
The Company expenses all costs as incurred in connection with patent applications (including direct application fees and the legal and consulting expenses related to making such applications), and such costs are included in general and administrative expenses.
75
Table of Contents
(i) | Concentration of Credit Risk |
Financial instruments that potentially subject the Company to significant concentrations of credit risk consist primarily of cash and cash equivalents. As of December 31, 2020, the Company maintained $17.4 million in a sweep account, which maintains cash balances throughout various interest bearing bank accounts under the $250,000 insurance limit provided by the Federal Deposit Insurance Corporation for one federally insured financial institution. Approximately $6.8 million was held in excess of the Federal Deposit Insurance Corporation insured limit as of December 31, 2020. The Company has not experienced any losses in such accounts.
The Company provides for federal and state income taxes on the asset and liability approach which requires deferred tax assets and liabilities to be recognized based on temporary differences between the consolidated financial statement and tax bases of assets and liabilities using enacted tax rates in effect for the year in which the temporary differences are expected to reverse.
Deferred tax assets are reduced by a valuation allowance when, in management’s opinion, it is more likely than not that some portion or all of the deferred tax assets will not be realized. The Company considers the scheduled reversal of deferred tax liabilities, projected future taxable income, and tax planning strategies in making this assessment. The Company’s valuation allowance is based on available evidence, including its current year and prior year operating losses, evaluation of positive and negative evidence with respect to certain specific deferred tax assets including evaluation sources of future taxable income to support the realization of the deferred tax assets. The Company has established a full valuation allowance on the deferred tax assets as of December 31, 2020.
Current and deferred tax assets and liabilities are recognized based on the tax positions taken or expected to be taken in the Company’s income tax returns. U.S. GAAP requires that the tax benefits of an uncertain tax position can only be recognized when it is more likely than not that the tax position will be sustained upon examination by the relevant taxing authority. Tax benefits related to tax positions that do not meet this criterion are not recognized in the consolidated financial statements, of which there are none.
The Company recognizes interest and penalties related to income tax matters in income tax expense.
The Company’s revenue is generated from two revenue streams: contract revenue and assay revenue. The Company accounts for revenue in accordance with Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers (Topic 606), as amended, and accounts for revenue in accordance with Accounting Standards Codification Topic 606 (“ASC 606”). The core principle of ASC 606 is that the Company recognizes revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the Company expects to be entitled in exchange for those goods or services. The ASC 606 revenue recognition model consists of the following five steps: (1) identify the contracts with a customer, (2) identify the performance obligations in the contract, (3) determine the transaction price, (4) allocate the transaction price to the performance obligations in the contract and (5) recognize revenue when (or as) the entity satisfies a performance obligation.
The Company recognizes revenue from its assay and contract services in accordance with the core principles and key aspects considered by the Company. These considerations are described in detail below, first for Assay Revenue and then for Contract Revenue.
Assay Revenue
The Company generates revenues from its PLA and Nevome tests it provides to healthcare clinicians in various states throughout the United States to assist in a clinician’s diagnosis of melanoma. The Company provides prescribing clinicians with its Smart Sticker adhesive sample collection kits to perform non-invasive skin biopsies of clinically ambiguous pigmented skin lesions on patients. The Company also offers clinicians a telemedicine solution where they can request the PLA collection kit be sent to the patient’s home for a clinician-guided remote collection on ambiguous pigmented skin lesions. Once the sample is collected by the healthcare clinician or the patient via the telemedicine solution, it is returned to the Company’s CLIA laboratory for analysis. The patient’s RNA and DNA are extracted from the Smart Sticker adhesive patch collection kit and analyzed using gene expression technology to determine if the pigmented skin lesion contains certain genomic features indicative of melanoma. Upon completion of the gene expression analysis, a final report is drafted and provided to the dermatologists detailing the test results for the pigmented skin lesion indicating whether the sample collected is indicative of melanoma or not.
76
Table of Contents
Contracts
The Company’s customer is the patient. However, the Company does not enter into a formal reimbursement agreement with a patient, as formal reimbursement agreements are more commonly established with insurance payors. Accordingly, the Company establishes an agreement with a patient in accordance with other customary business practices.
| • | Approval of an agreement is established by the use of the Company’s adhesive sample collection kit on a patient by an ordering physician, which is then sent to the Company’s central lab for testing. |
| • | The Company is obligated to perform the Company’s laboratory services upon receipt of a sample from a physician, and the patient and/or applicable payor are obligated to reimburse us for services rendered based on the patient’s insurance benefits. |
| • | Payment terms are a function of a patient’s existing insurance benefits. |
| • | Once the Company delivers a patient’s test result to the ordering physician, the Company is legally able to collect payment and bill an insurer and/or patient, depending on payor agreement status or patient insurance benefit status. |
| • | The Company’s consideration is deemed to be variable, and the Company considers collection of such consideration to be probable to the extent that it is unconstrained. |
Performance Obligations
A performance obligation is a promise in an agreement to transfer a distinct good or service (or a bundle of goods or services) to the customer. The customer is able to order a PLA test. However, a Nevome test cannot be ordered separately from the PLA test and it is contingent on being run only when a PLA test comes back positive on a sample. The Nevome test would not qualify as a distinct service. Therefore, the PLA test is recognized as a single performance obligation and the Nevome test, if rendered, is bundled with the single PLA performance obligation.
Transaction Price
The transaction price is the amount of consideration that the Company expects to collect in exchange for transferring promised goods or services to a customer, excluding amounts collected on behalf of third parties (for example, some sales taxes). The consideration expected from an agreement with a customer may include fixed amounts, variable amounts, or both.
The consideration derived from the Company’s agreements is deemed to be variable, though the variability is not explicitly stated in any agreement. Rather, the implied variability is due to several factors, such as the amount of contractual adjustments, any patient co-payments, deductibles or patient compliance incentives, the existence of secondary payors and claim denials.
The Company estimates the amount of variable consideration using the expected value method, which represents the sum of probability-weighted amounts in a range of possible consideration amounts. When estimating the amount of variable consideration, the Company considers several factors, such as historical collections experience, patient insurance eligibility and payor reimbursement agreements.
The Company limits the amount of variable consideration included in the transaction price to the unconstrained portion of such consideration. In other words, the Company recognizes revenue up to the amount of variable consideration that is not subject to a significant reversal until additional information is obtained or the uncertainty associated with the additional payments or refunds is subsequently resolved. Differences between original estimates and subsequent revisions, including final settlements, represent changes in the estimate of variable consideration and are included in the period in which such revisions are made. Revenue recognized from changes in transaction prices was not material for the years ended December 31, 2020 and 2019, respectively.
The Company monitors its estimates of transaction price to depict conditions that exist at each reporting date. If the Company subsequently determines that it will collect more consideration than it originally estimated for an agreement with a patient, it will account for the change as an increase in the estimate of the transaction price (i.e., an upward revenue adjustment) in the period identified. Similarly, if the Company subsequently determines that the amount it expects to collect from a patient is less than it originally estimated, it will generally account for the change as a decrease in the estimate of the transaction price (i.e., a downward revenue adjustment), provided that such downward adjustment does not result in a significant reversal of cumulative revenue recognized.
77
Table of Contents
When the Company does not have significant historical experience or that experience has limited predictive value, the constraint over estimates of variable consideration may result in no revenue being recognized upon delivery of a patient’s test result to the ordering physician, with recognition, generally occurring at the date of cash receipt.
Allocate the Transaction Price
The entire transaction price is allocated entirely to the single performance obligation contained within the agreement with a patient.
Recognize Revenue
The Company’s single performance obligation is satisfied at a point in time, and that point in time is defined as the date a patient’s successful test result is delivered to the patient’s ordering physician. The Company considers this date to be the time at which the patient obtains control of the final results of the promised test service.
If a Nevome test service is ordered and completed in conjunction with the Company’s PLA service, then the Company will recognize revenue upon the delivery of both final reports to the physician. The delivery of the Company’s Nevome test results are typically after the Company’s PLA results are delivered due to the circumstances of how the Company processes the Nevome test. However, this length in time is determined to not materially impact the final overall revenue recognition timing.
Contract Revenue
Contract revenue is generated from the sale of laboratory services and adhesive sample collection kits to third party companies through contract research agreements. Revenues are generated from providing gene expression tests to facilitate the development of drugs designed to treat dermatologic conditions. The provision of gene expression services may include sample collection using the Company’s patented adhesive patch collection kits, assay development for research partners, RNA extraction, isolation, expression, amplification and detection, including data analysis and reporting.
Contracts
As part of the Company’s contract revenue, the Company has established agreements and work orders with the Company’s third-party partners that fall under the scope of ASC 606.
Performance Obligations
ASC 606 requires an entity to assess the goods or services promised in a contract and identify as a performance obligation each promise to transfer to the customer either a good or service (or a bundle of goods or services) that is distinct, or a series of distinct goods or services that are substantially the same and that have the same pattern of transfer to the customer. Based upon review of existing contracts, a majority of the Company’s contract revenue agreements contain three performance obligations:
| (1) | Adhesive sample collection kits |
| (2) | RNA extractions and analysis |
| (3) | Certain project management fees |
Many of the Company’s contract revenue agreements contain promises such as start-up activities and quality system setup fees, which are activities that the Company performs to fulfill the agreement and they do not transfer any good or service to the customer. These promises encompass the administrative tasks associated with beginning and initiating a new project or study with a third-party company. In accordance with ASC 606, an entity does not account for these activities as a promised good or service within the agreement nor evaluate whether they are a performance obligation.
Transaction Price
The transaction price is the amount of consideration to which an entity expects to be entitled in exchange for transferring promised goods or services to a customer. The consideration promised in an agreement with a customer may include fixed amounts, variable amounts, or both.
78
Table of Contents
The transaction prices of the Company’s performance obligations are listed in its agreements on a per unit basis and are fixed for adhesive sample collection kits and RNA extractions and analysis. The project management fees are assessed based on a monthly service fee which range within the agreements depending on certain factors which include length of the project and the amount of kits or RNA extractions and analysis promised within the agreement. The fixed and variable rates are materially consistent within the Company’s agreements. Therefore, the Company utilizes the prices listed in our agreements as the transaction price for each performance obligation.
In determining the transaction price, ASC 606 requires an entity to adjust the promised amount of consideration for the effects of the time value of money if the agreement contains a significant financing component. The Company’s agreements state fixed transaction prices for each deliverable associated with the agreement and do not qualify for the significant financing component of ASC 606.
Allocate the Transaction Price
The Company’s contracts have a directly observable transaction price pertaining to each promised good or service. Those prices are consistent across agreements for adhesive sample collection kits and RNA extractions and analysis, with the exception of the Company’s project management fees, which the Company’s believes encompass a sufficiently narrow range of prices that are dictated upon factors of each agreement previously discussed above. Therefore, the Company’s relies on those transaction prices as the basis to allocate the stand-alone selling prices to the performance obligations of the agreement.
Most of the Company’s agreements contain a discount that is allocated to items within the agreement, whether they are performance obligations or not. Those items that are not performance obligations (e.g. quality system setup and start up fees) have the associated discount allocated to the transaction prices of the performance obligations evenly.
Recognize Revenue
An entity should recognize revenue when (or as) it satisfies a performance obligation by transferring a promised good or service to a customer. A good or service is transferred when (or as) the customer obtains control of that good or service. The adhesive sample collection kits are recognized at a point in time when shipped to the customer. The RNA extraction and analysis are recognized at a point in time when the extraction and analysis process is complete and the results are sent to the customer. The Company provides its project management service over the life of the agreement, providing equal benefit to the customer throughout the life of the project or study. Therefore, the revenue related to the Company’s project management fees is recognized straight-line over the life of the agreement.
(a) | Disaggregation of Revenue |
The following tables present the Company’s revenues disaggregated by revenue source during the years ended December 31, 2020 and 2019, respectively (in thousands):
| | Year Ended December 31, | |
| | 2020 | | | 2019 | |
Assay Revenue | | | | | | | | |
PLA Test | | $ | 4,241 | | | $ | 1,403 | |
Contract Revenue | | | | | | | | |
Adhesive patch kits | | | 213 | | | | 476 | |
RNA extractions | | | 1,172 | | | | 626 | |
Project management fees | | | 258 | | | | 336 | |
Other | | | 1 | | | | 523 | |
Total revenues | | $ | 5,885 | | | $ | 3,364 | |
79
Table of Contents
In 2020, there were two payors and one customer that each accounted for more than 10% of our total revenue. These two payors and one customer combined accounted for 66% of our total revenue for the twelve months ended December 31, 2020. There were no other payors or customers that individually accounted for more than 10% of our total revenue for the twelve months ended December 31, 2020. In addition, the Company had two payors and one customer that each accounted for more than 10% of accounts receivable at December 31, 2020. These two payors and one customer combined accounted for 38% of accounts receivable at December 31, 2020. There were no other payors or customers that individually accounted for more than 10% of accounts receivable at December 31, 2020.
(b) | Deferred Revenue and Remaining Performance Obligations |
The timing of revenue recognition, billings and cash collections results in billed accounts receivable and deferred revenue on the consolidated balance sheets.
In a majority of agreements that produce contract revenue, the Company receives a substantial up-front payment and additional payments upon the achievement of various milestones over the life of the agreement. This results in deferred revenue and is relieved upon delivery of the applicable adhesive patch kits or RNA extraction results. Changes in accounts receivable and deferred revenue were not materially impacted by any other factors.
The Company records a deferred revenue liability if a customer pays consideration before the Company transfers a good or service to the customer. Deferred revenue primarily represents upfront milestone payments, for which consideration is received prior to when goods/services are completed or delivered. Upfront fees that are estimated to be recognized as revenue more than one year from the date of collection are classified as long-term deferred revenue. Short-term deferred revenue as of December 31, 2020 and December 31, 2019 was $0.9 million and $1.4 million, respectively. Long-term deferred revenue as of December 31, 2020 and December 31, 2019 was $0.6 million and zero, respectively.
Remaining performance obligations include deferred revenue and amounts the Company expects to receive for goods and services that have not yet been delivered or provided under existing agreements. For agreements that have an original duration of one year or less, the Company has elected the practical expedient applicable to such agreements and does not disclose the remaining performance obligations at the end of each reporting period and when the Company expects to recognize this revenue. As of December 31, 2020, the estimated revenue expected to be recognized in future periods related to performance obligations that are unsatisfied for executed agreements with an original duration of one year or more was approximately $1.8 million. The Company expects to recognize revenue on the majority of these remaining performance obligations over the next two to three years.
Assay Accounts Receivable
Due to the nature of the Company’s assay revenue, it can take a significant amount of time to collect upon billed PLA tests. The Company prepares an analysis on reimbursement collections and data obtained for each financial reporting period to determine the amount of receivables to be recorded relating to PLA tests performed in the applicable period. The Company generally does not perform evaluations of customers’ financial condition and generally does not require collateral. Accounts receivable are written off when all efforts to collect the balance have been exhausted. Adjustments for implicit price concessions attributable to variable consideration are incorporated into the measurement of the accounts receivable balances. The Company recorded $1.0 million and $0.5 million of gross assay accounts receivable as of December 31, 2020 and 2019, respectively.
Contract Accounts Receivable
Contract accounts receivable are recorded at the net invoice value and are not interest bearing. The Company reserves specific receivables if collectability is no longer reasonably assured, and as of December 31, 2020, the Company did not maintain any reserve over contract receivables as they relate to large established credit worthy customers. The Company re-evaluates such reserves on a regular basis and adjusts its reserves as needed. Once a receivable is deemed to be uncollectible, such balance is charged against the reserve. The Company recorded $0.5 million and $0.3 million of contract accounts receivable as of December 31, 2020 and 2019, respectively.
(m) | Freight and Shipping Costs |
The Company records outbound freight and shipping costs for its contract and assay revenues in cost of revenues.
80
Table of Contents
Comprehensive loss is defined as a change in equity during a period from transactions and other events and circumstances from non-owner sources. We report net loss and the components of other comprehensive loss, including unrealized gains and losses on marketable securities, net of their related tax effect to arrive at total comprehensive loss.
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker, or decision-making group, in making decisions on how to allocate resources and assess performance. The Company views its operations and manages its business as one operating segment.
Basic and diluted net loss per common share is determined by dividing net loss applicable to common shareholders by the weighted average common shares outstanding during the period. Because there is a net loss attributable to common shareholders during the years ended December 31, 2020 and 2019, the outstanding common stock warrants, stock options, restricted stock units and preferred stock have been excluded from the calculation of diluted loss per common share because their effect would be anti-dilutive. Therefore, the weighted average shares used to calculate both basic and diluted loss per share are the same. Diluted net loss per common share for the year ended December 31, 2020 excludes the effect of anti-dilutive equity instruments including zero shares of common stock issuable upon conversion of the Company’s preferred stock, 3,885,311 shares of common stock issuable upon the exercise of outstanding common stock warrants and 2,112,980 shares of common stock issuable upon the exercise stock options. Diluted net loss per common share for the year ended December 31, 2019 excludes the effect of anti-dilutive equity instruments including 615,385 shares of common stock issuable upon conversion of the Company’s preferred stock, 4,200,497 shares of common stock issuable upon the exercise of outstanding warrants and 443,547 shares of common stock issuable upon the exercise stock options and release of restricted stock units. The Company did not consider a two-class method of earnings (loss) per share given that the Company’s convertible participating securities do not participate in losses.
(q) | Stock-Based Compensation |
Effective January 1, 2020, the Company elected an accounting policy change to no longer estimate forfeitures in connection with expense recognition of stock options and RSUs. All stock options and RSUs granted on or subsequent to January 1, 2020 will recognize forfeitures when they occur in accordance with ASU 2016-09, Compensation - Stock Compensation (Topic 718). In addition, effective January 1, 2020, the Company has elected to recognize stock-based compensation expense over the requisite service period of options and awards on a ratable basis. The Company believes that the recognition of stock-based compensation on a ratable basis is more aligned with their business practices of granting options and awards. The accounting policy change does not have a material effect on the Company’s consolidated financial statements.
Compensation costs associated with stock option awards and other forms of equity compensation are measured at the grant-date fair value of the awards and recognized over the requisite service period of the awards on a ratable basis.
The Company grants stock options to purchase common stock to employees with exercise prices equal to the fair market value of the underlying stock, as determined by the board of directors, management and outside valuation experts prior to the Business Combination. The board of directors and outside valuation experts determined the fair value of the underlying stock by considering a number of factors, including historical and projected financial results, the risks the Company faced at the time, the preferences of the Company’s debt holders and preferred stockholders, and the lack of liquidity of the Company’s common stock. Subsequent to the close of the Business Combination, the fair market value of stock options is based on the closing stock price on the grant date.
The fair value of each stock option award is estimated using the Black-Scholes-Merton valuation model. Such value is recognized as expense over the requisite service period using the ratable method. The expected term of options is based on the simplified method which defines the expected term as the average of the contractual term of the options and the weighted average vesting period for all option tranches. The expected volatility of stock options is based upon the historical volatility of a number of related publicly traded companies in similar stages of development as well as the volatility of the Company’s common stock. The risk-free interest rate is based on the average yield of U.S. Treasury securities with remaining terms similar to the expected term of the share-based awards. The assumed dividend yield was based on the Company’s expectation of not paying dividends in the foreseeable future.
The Company accounts for stock options to non-employees using the fair value approach. The fair value of these options is measured using the Black-Scholes-Merton option pricing model, reflecting the same assumptions applied to employee options, other than expected life, which is assumed to be the remaining contractual life of the award. Options that are granted to employees generally have a requisite service period of three to four years.
81
Table of Contents
RSUs are considered restricted stock. The fair value of restricted stock is equal to the fair market value of the underlying stock, as determined by the board of directors, management and input from outside valuation experts prior to the Business Combination. Subsequent to the close of the Business Combination, the fair market value of RSUs is based on the closing stock price on the grant date. The Company recognizes stock-based compensation expense based on the fair value on a ratable basis over the requisite service periods of the awards. RSUs that are granted to employees have a requisite service period typically between two and four years.
All stock options and RSUs granted prior to January 1, 2020 will maintain the estimated forfeiture approach and will be recognized over the requisite service period using the straight-line method.
The fair value of each option for employees was estimated on the date of grant using the following assumptions:
| | Year Ended December 31, | |
| | 2020 | | | 2019 | |
Assumed risk-free interest rate | | 0.36% - 1.69% | | | 1.68% - 2.50% | |
Assumed volatility | | 64.03% - 73.44% | | | 72.30% - 73.50% | |
Expected option term | | 5.04 - 6.25 | | | 6.02 - 6.08 | |
Expected dividend yield | | | — | | | | — | |
The following table sets forth assumptions used to determine the fair value of the purchase rights issued under the ESPP:
| | Year Ended December 31, | |
| | 2020 | |
Assumed risk-free interest rate | | 0.18% | |
Assumed volatility | | 68.44% | |
Expected option term | | 0.49 years | |
Expected dividend yield | | | — | |
82
Table of Contents
The Company recorded stock-based compensation expense for employee options, RSUs, and consultant options of $5.0 million and $1.3 million for the years ended December 31, 2020 and 2019. The total compensation cost related to non-vested awards not yet recognized at December 31, 2020 was $14.1 million, which is expected to be recognized over a weighted average term of 2.95 years.
(r) | Fair Value Measurements |
The Company measures certain financial assets at fair value on a recurring basis. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability (an exit price) in an orderly transaction between market participants at the measurement date. The Company uses a three-tier fair value hierarchy to prioritize the inputs used in the Company’s fair value measurements. These tiers include: Level 1, defined as observable inputs such as quoted prices in active markets for identical assets; Level 2, defined as inputs other than quoted prices in active markets that are either directly or indirectly observable; and Level 3, defined as unobservable inputs in which little or no market data exists, therefore requiring an entity to develop its own assumptions. The following table provides a summary of the assets that are measured at fair value on a recurring basis as of December 31, 2020 (in thousands):
Fair Value Measurements at Reporting Date
| | December 31, 2020 | |
| | Level 1 | | | Level 2 | | | Level 3 | | | Total | |
Assets: | | | | | | | | | | | | | | | | |
Cash equivalents | | $ | 448 | | | $ | — | | | $ | — | | | $ | 448 | |
| | | | | | | | | | | | | | | | |
Marketable securities, available for sale: | | | | | | | | | | | | | | | | |
Corporate debt | | | — | | | | 8,940 | | | | — | | | | 8,940 | |
Municipal securities | | | — | | | | 7,324 | | | | — | | | | 7,324 | |
U.S. government debt securities | | | — | | | | 23,265 | | | | — | | | | 23,265 | |
Total marketable securities, available for sale | | | — | | | | 39,529 | | | | — | | | | 39,529 | |
Total assets measured at fair value on a recurring basis | | $ | 448 | | | $ | 39,529 | | | $ | — | | | $ | 39,977 | |
The Company’s marketable debt securities are classified as available-for-sale securities based on management's intentions and are at level 2 of the fair value hierarchy, as these investment securities are valued based upon quoted prices for identical or similar instruments in markets that are not active. The Company has classified marketable securities with original maturities of greater than one year as short-term investments based upon the Company’s ability to use all of those marketable securities to satisfy the liquidity needs of the Company’s current operations.
There were no assets or liabilities that were measured at fair value on a recurring basis as of December 31, 2019. The Company believes the carrying amount of cash and cash equivalents, accounts payable and accrued expenses approximate their estimated fair values due to the short-term nature of these accounts.
(s) | Accounting Pronouncement Recently Adopted |
In June 2019, the Financial Accounting Standards Board (“FASB”) issued ASU 2018-07, Compensation-Stock Compensation (Topic 718) – Improvements to Nonemployee Share-Based Payment Accounting, which simplifies accounting for nonemployee share-based payment transactions to now include share-based payment transactions for acquiring goods and services from nonemployees. This new standard is effective for interim and annual periods beginning December 15, 2019 and early adoption is permitted. The Company adopted this guidance on January 1, 2020, and it did not have a material impact on the consolidated financial statements.
In August 2018, the FASB issued ASU 2018-13, Fair Value Measurement (Topic 820): Disclosure Framework—Changes to the Disclosure Requirements for Fair Value Measurement, which modified the disclosure requirements on fair value measurements in Topic 820, Fair Value Measurement, based on the concepts in the Concepts Statement, including the consideration of costs and benefits. This new standard is effective for interim and annual periods beginning after December 15, 2019 and early adoption is permitted. The Company adopted this guidance on January 1, 2020, and it did not have a material impact on the consolidated financial statements.
83
Table of Contents
(t) | Accounting Pronouncements Issued But Not Yet Effective |
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842) (“ASU 2016-02”), which supersedes FASB ASC Topic 840, Leases (“Topic 840”), and provides principles for the recognition, measurement, presentation and disclosure of leases for both lessees and lessors. The new standard requires lessees to apply a dual approach, classifying leases as either finance or operating leases based on the principle of whether or not the lease is effectively a financed purchase by the lessee. This classification will determine whether lease expense is recognized based on an effective interest method for finance leases or on a straight-line basis over the term of the lease for operating leases. A lessee is also required to record a right-of-use asset and a lease liability for all leases with a term of greater than 12 months regardless of classification. Leases with a term of 12 months or less will be accounted for similar to existing guidance for operating leases. For companies that are not emerging growth companies (‘‘EGCs’’), ASU 2016-02 is effective for fiscal years beginning after December 15, 2018. For EGCs, the ASU was to be effective for fiscal years beginning after December 15, 2019. However, in November 2019, the FASB issued ASU No. 2019-10, Financial Instruments—Credit Losses (Topic 326), Derivatives and Hedging (Topic 815) and Leases (Topic 842): Effective Dates (“ASU 2019-10”), which included a one-year deferral of the effective date of ASU 2016-02 for certain entities. In June 2020, the FASB issued ASU No. 2020-05, Revenue from Contracts with Customers (Topic 606) and Leases (Topic 842): Effective Dates for Certain Entities (“ASU 2020-05”), which further defers the effective date for certain entities. As a result, the ASU is now effective for EGCs for fiscal years beginning after December 15, 2021, and interim periods within fiscal years beginning after December 15, 2022. Assuming we remain an EGC, we intend to adopt the new standard in the fourth quarter of 2022 using the modified retrospective method, under which the Company will apply Topic 842 to existing and new leases as of January 1, 2022, but prior periods will not be restated and will continue to be reported under Topic 840 guidance in effect during those periods. The Company expects to elect certain optional practical expedients. The Company anticipates that the adoption will not have a material impact on its statements of operations, statements of comprehensive loss or its statements of cash flows but expects to recognize right-of-use assets and liabilities for lease obligations associated with its operating leases.
The Company is currently evaluating the impact of this standard on its consolidated financial statements. In June 2016, the FASB issued ASU 2016-13, Financial Instruments—Credit Losses: Measurement of Credit Losses on Financial Instruments (Topic 326), which amends the impairment model by requiring entities to use a forward-looking approach based on expected losses to estimate credit losses on certain types of financial instruments, including trade receivables and available-for-sale debt securities. This standard covers the Company’s financial instruments, such as debt securities that are available for sale. Previously, when credit losses were measured under U.S. GAAP, an entity generally only considered past events and current conditions in measuring the incurred loss. The new guidance requires companies to identify, analyze, document and support new methodologies for quantifying expected credit loss estimates for financial instruments, using information such as historical experience and current economic conditions, plus the use of reasonable supportable forecast information. In November 2018, the FASB issued ASU No. 2018-19, Codification Improvements to Topic 326, Financing Instruments—Credit Losses, which included an amendment of the effective date for nonpublic entities. For non-EGCs, ASU 2016-13 is effective for fiscal years beginning after December 15, 2019. For EGCs, ASU 2016-13 was to be effective for fiscal years beginning after December 15, 2021. However, in November 2019, the FASB issued ASU 2019-10, which included a one-year deferral of the effective date of ASU 2016-13 for certain entities. As a result, ASU is now effective for EGCs for fiscal years beginning after December 15, 2022, including interim periods within those fiscal years. Early adoption is permitted, and the standard is adopted using a modified retrospective transition method through a cumulative-effect adjustment to retained earnings as of the beginning of the first reporting period in which the guidance is effective. Assuming the Company remains an EGC, it intends to adopt ASU 2016-13 at the beginning of its fiscal year ending December 31, 2022. Adoption will require a modified retrospective transition. We are currently evaluating the impact the adoption of this standard will have on our consolidated financial statements.
In December 2019, the FASB issued ASU No. 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes (“ASU 2019-12”), which eliminates certain exceptions to the general principles in Topic 740 and simplifies other areas of the existing guidance. For non-EGCs, ASU 2019-12 is effective for fiscal years beginning after December 15, 2020, and interim periods within those fiscal years. For EGCs, the standard is effective for fiscal years beginning after December 15, 2021, and interim periods within fiscal years beginning after December 15, 2022. Early adoption is permitted. The Company is currently evaluating the impact of ASU 2019-12 on its financial statements.
2. Balance Sheet Details
Short-Term Marketable Securities
84
Table of Contents
The amortized cost, gross unrealized holding gains, gross unrealized holding losses, and fair value debt securities classified as available-for-sale securities by major security type and class of security at December 31, 2020 were as follows (in thousands):
| | December 31, 2020 | |
| | Amortized Cost | | | Gross Unrealized Gain | | | Gross Unrealized Loss | | | Estimated Market Value | |
Short-term marketable securities, available-for-sale: | | | | | | | | | | | | | | | | |
Corporate debt | | $ | 8,946 | | | $ | — | | | $ | (6 | ) | | $ | 8,940 | |
Municipal securities | | | 7,325 | | | | 1 | | | | (2 | ) | | | 7,324 | |
U.S. government debt securities | | | 23,259 | | | | 6 | | | | — | | | | 23,265 | |
Total short-term marketable securities, available-for-sale | | $ | 39,530 | | | $ | 7 | | | $ | (8 | ) | | $ | 39,529 | |
As of December 31, 2019, there were no marketable securities maintained by the Company.
As of December 31, 2020, the estimated market value of debt securities with contractual maturities of less than 12 months was $37.3 million; the remaining debt securities that we held at that date had an estimated market value of $2.3 million and contractual maturities of up to 14 months.
Gross realized gains and losses on our debt securities for the twelve months ended December 31, 2020 were not significant.
Prepaid Expenses and PP&E
Consolidated balance sheet details are as follows (in thousands):
| | December 31, 2020 | | | December 31, 2019 | |
Prepaid expenses and other current assets: | | | | | | | | |
Prepaid insurance | | $ | 1,172 | | | $ | 951 | |
Prepaid trade shows | | | — | | | | 85 | |
Prepaid software development fees | | | 214 | | | | — | |
Deferred issuance costs | | | 56 | | | | — | |
Other current assets | | | 79 | | | | 25 | |
Total prepaid expenses and other current assets | | $ | 1,521 | | | $ | 1,061 | |
Property and equipment, gross: | | | | | | | | |
Laboratory equipment | | $ | 2,544 | | | $ | 1,135 | |
Computer equipment | | | 38 | | | | 15 | |
Furniture and fixtures | | | 109 | | | | 34 | |
Leasehold improvements | | | 727 | | | | 32 | |
Total property and equipment, gross | | | 3,418 | | | | 1,216 | |
Less accumulated depreciation | | | (687 | ) | | | (239 | ) |
Total property and equipment, net | | $ | 2,731 | | | $ | 977 | |
85
Table of Contents
Accrued Liabilities and Accrued Compensation
Consolidated balance sheet details are as follows (in thousands):
| | December 31, 2020 | | | December 31, 2019 | |
Accrued liabilities: | | | | | | | | |
Accrued consulting services | | $ | 285 | | | $ | 37 | |
Accrued printing fees | | | — | | | | 55 | |
Deferred rent | | | 300 | | | | 88 | |
Other accrued expenses | | | 178 | | | | 38 | |
Total accrued liabilities | | $ | 763 | | | $ | 218 | |
Accrued compensation: | | | | | | | | |
Accrued paid time off | | $ | 606 | | | $ | 309 | |
Accrued bonus and deferred compensation | | | 1,469 | | | | 465 | |
Accrued severance | | | — | | | | 368 | |
Total accrued compensation | | $ | 2,075 | | | $ | 1,142 | |
3. Debt
2018 Convertible Bridge Notes
From August to November 2018, DermTech Operations issued $6.8 million aggregate principal amount of convertible bridge notes (“2018 Bridge Notes”), resulting in $6.6 million in net proceeds. The 2018 Bridge Notes carried a 10% interest rate and matured on March 31, 2019. Since the 2018 Bridge Notes were not paid or converted by March 31, 2019, the interest rate increased to 15%.
The 2018 Bridge Notes were subject to automatic conversion into equity securities of DermTech Operations at the closing of a single capital raising transaction or series of related capital raising transactions in which DermTech Operations issued equity securities with aggregate gross proceeds to DermTech Operations of at least $20 million (“Qualified Financing”) that occurred on or prior to the maturity date. Upon automatic conversion of these 2018 Bridge Notes, the note holders were entitled to receive shares of DermTech Operations’ equity securities equal to the quotient obtained by dividing the unpaid principal amount of these 2018 Bridge Notes plus interest accrued but unpaid by the lesser of:
| 1) | the lowest price per share of the new stock paid in the Qualified Financing by investors multiplied by 70%. |
| 2) | the price per share obtained by dividing $45 million by DermTech Operations’ fully-diluted capitalization immediately prior to such Qualified Financing assuming exercise or conversion of all outstanding options and issuance of all outstanding restricted stock unit awards, including all shares of common stock reserved and available for future grant under any equity incentive plan of the Company, and/or any equity incentive or similar plan to be created or increased in connection with the Qualified Financing, but excluding any shares issuable upon exercise of the DermTech Operations’ outstanding common stock warrants or conversion of the 2018 Bridge Notes. |
Several of the embedded features of the 2018 Bridge Notes were identified as meeting the criteria of a derivative and ultimately bifurcated from the host contract. DermTech Operations accounted for this by separating the derivative component of the 2018 Bridge Notes as a derivative liability on the consolidated balance sheet. DermTech Operations assigned a value to the debt component of the 2018 Bridge Notes equal to the difference between the estimated fair value of the 2018 Bridge Notes with and without the conversion features, which resulted in DermTech Operations recording the 2018 Bridge Notes at a discount. The total debt discount amount as of the respective date of issuance of the 2018 Bridge Notes was determined to be $2.5 million. DermTech Operations amortized the debt discount over the contractual life (i.e., March 31, 2019) of the 2018 Bridge Notes as additional non-cash interest expense utilizing the effective interest method. At each financial reporting period, DermTech Operations remeasured the fair value of the embedded features bifurcated from the 2018 Bridge Notes (i.e., the derivative liability) and changes in the fair value are recognized in earnings. Losses relating to the change in fair value of the derivative liability recognized as other expense on the Statement of Operations were zero and $0.4 million for the years ended December 31, 2020 and 2019, respectively.
On May 23, 2019, DermTech Operations and the various convertible 2018 Bridge Note holders agreed to amend the outstanding convertible notes that were issued in the last half of 2018. As part of the amendment, the maturity dates of the notes were extended to the earliest of (i) September 24, 2019; (ii) the occurrence of an Event of Default (as defined in the 2018 Bridge Notes); (iii) the
86
Table of Contents
consummation of a liquidation or dissolution of DermTech Operations (iv) a Liquidation Transaction (as defined in the 2018 Bridge Notes); or (v) the consummation of a merger with or into the Company or any of its subsidiaries.
In addition, immediately prior to the consummation of a DermTech Operations merger with or into the Company or any of its subsidiaries substantially on the terms contemplated as of the date of the amendment to the outstanding convertible notes on or before September 24, 2019 (a “Qualifying Merger”), the outstanding principal amount of and all accrued but unpaid interest on each of the convertible notes would automatically be converted into shares of the DermTech Operations’ common stock at a price per share equal to 70% of the Merger Consideration. For purposes of the preceding sentence, the “Merger Consideration” means (i) the lesser of $6.46 and (ii) the offering price per share of the private investment in public equity (“PIPE”) transaction to be consummated concurrently with the consummation of the Qualifying Merger multiplied by the Conversion Ratio. For the purposes of the preceding sentence, the “Conversion Ratio” means the quotient resulting from dividing 8,000,000 by the number of fully diluted shares of the Company as of immediately after the conversion of the notes.
This new embedded Qualifying Merger feature of the 2018 Bridge Notes was identified as meeting the criteria of a derivative and ultimately bifurcated from the host contract with the previously identified embedded features that met the criteria of being a derivative. In addition, this amendment was accounted for as a debt modification of the existing 2018 Bridge Notes.
2019 Convertible Bridge Notes
Between June 5th and June 10th, 2019, DermTech Operations issued additional convertible bridge notes (the “2019 Bridge Notes”) to existing investors for aggregate gross proceeds of $2.6 million. These convertible bridge notes carried an interest rate of 10% and matured after the earliest to occur of: (i) September 25, 2019; (ii) the occurrence of an Event of Default; (iii) the consummation of a liquidation or dissolution of DermTech Operations; (iv) a Liquidation Transaction; or (v) the consummation of a merger of DermTech Operations with Merger Sub, a subsidiary of the Company, in accordance with the Merger Agreement.
The unpaid principal amount of these convertible bridge notes together with any interest accrued but unpaid thereon, would automatically be converted into shares of DermTech Operations’ common stock immediately prior to the consummation of a Qualifying Merger. Upon the conversion of these notes, the note holders were entitled to receive a number of shares of DermTech Operations’ common stock equal to the quotient obtained by dividing (i) the unpaid principal amount of these notes plus interest accrued but unpaid thereon, by (1) if the Qualifying Merger consummates prior to the maturity date, the lesser of (x) $5.80 and (y) 90% of the Merger Consideration (as defined below), or (2) if the Qualifying Merger consummates on or after the maturity date, the lesser of (x) $4.51 and (y) 70% of the Merger Consideration. For purposes of the preceding sentence, the “Merger Consideration” means the offering price per share of the PIPE transaction between Constellation and the investors thereto, consummated substantially concurrently with the consummation of the Qualifying Merger, multiplied by the Conversion Ratio (as defined below). For purposes of the preceding sentence, the “Conversion Ratio” means the quotient resulting from dividing 8,000,000 by the number of the Company’s fully diluted shares immediately prior to the consummation of the Qualifying Merger, assuming exercise of all outstanding options, issuance of all common stock underlying outstanding restricted stock unit awards, exercise of all outstanding warrants, and conversion of all outstanding convertible promissory notes, including these notes and any other note of substantially the same form, but excluding all shares of DermTech Operations’ common stock reserved and available for future grant under any equity incentive or similar plan of DermTech Operations, and in each case as adjusted for stock splits, combinations and similar transactions, all calculated in accordance with the final allocation schedule delivered in connection with the Qualifying Merger.
Several of the embedded features of the 2019 Bridge Notes were identified as meeting the criteria of a derivative and ultimately bifurcated from the host contract. DermTech Operations accounted for this by separating the derivative component of the 2019 Bridge Notes as a derivative liability on the consolidated balance sheet. The Company assigned a value to the debt component of the 2019 Bridge Notes equal to the difference between the estimated fair value of the 2019 Bridge Notes with and without the conversion features, which resulted in DermTech Operations recording the 2019 Bridge Notes at a discount. The total debt discount amount as of the respective date of issuance of the 2019 Bridge Notes was determined to be $0.3 million. DermTech Operations amortized the debt discount over the contractual life (i.e., September 25, 2019) of the 2019 Bridge Notes as additional non-cash interest expense utilizing the effective interest method. At each financial reporting period, DermTech Operations remeasured the fair value of the embedded features bifurcated from the 2019 Bridge Notes (i.e., the derivative liability) and changes in the fair value were recognized in earnings. Losses relating to the change in fair value of the derivative liability recognized as other expense on the Statement of Operations were of zero and $14,000 for the years ended December 31, 2020 and 2019, respectively.
87
Table of Contents
Exchange of Convertible Debt for Common Shares
On August 29, 2019, immediately prior to the completion of the Business Combination, all unpaid principal and interest on the 2019 Bridge Notes and the 2018 Bridge Notes (collectively, the “Bridge Notes”) was converted into 2,267,042 common shares of DermTech Operations.
The conversion of the Bridge Notes debt for common shares of DermTech Operations was accounted for as an extinguishment of the Bridge Notes. The conversion resulted in DermTech Operations having legally settled the debt obligations. DermTech Operations’ equity was increased by the settlement-date fair value of the common shares issued. Certain bifurcated embedded derivative instruments also were settled as part of the transaction.
The net carrying amounts of the Bridge Notes, including remaining unamortized debt discount and issuance costs, and the bifurcated embedded derivative liability were extinguished on the date of the Business Combination. A gain on debt extinguishment of $0.9 million was recognized, which represented the unamortized debt discounts and issuance costs remaining at the time of the debt extinguishment.
There was no liability balance for the Company’s 2019 Bridge Notes or 2018 Bridge Notes as of December 31, 2020 and 2019.
4. Convertible Preferred Stock and Stockholders’ Equity
The Company’s amended and restated certificate of incorporation authorizes it to issue 50,000,000 shares of common stock and 5,000,000 shares of preferred stock. Both classes of stock have a par value of $0.0001 per share.
Pursuant to the Business Combination, the Company issued shares of its common stock to DermTech Operations common stockholders, at an exchange ratio of approximately 1.16 shares of the Company’s common stock for each share of DermTech Operations common stock. In connection with and immediately following the Business Combination, the Company filed a certificate of amendment to its amended and restated certificate of incorporation to affect a one-for-two reverse stock split of its common stock. All stock information presented throughout this document have been adjusted to reflect these capital structure changes.
(b) | DermTech Operations, Inc. Series C Convertible Preferred Stock Financing |
DermTech Operations conducted a Series C Convertible Preferred Stock private offering in August of 2016 for a total offering amount of $15 million at a price per share of $9.54. During 2017, 559,849 shares of Series C Convertible Preferred Stock were issued for gross cash proceeds of $5.3 million, reduced by issuance costs of $0.4 million. In addition, 102,740 common stock warrants were issued with this offering, exclusive of compensatory warrants issued to the placement agent. During 2018, 506,539 shares of Series C Convertible Preferred Stock were issued for gross cash proceeds of $4.8 million, reduced by issuance costs of $0.3 million.
On May 23, 2019, DermTech Operations agreed to an amendment with the Series C Convertible Preferred Stockholders that immediately prior the consummation of a merger with or into the Company or any of its subsidiaries on or before September 24, 2019, the outstanding Series C Convertible Preferred Stock would convert into common stock at a one-to-one ratio in accordance with DermTech Operations’ amended and restated certificate of incorporation. Immediately prior to the completion of the Business Combination, each share of Series C Convertible Preferred Stock of DermTech Operations outstanding as of such time was automatically converted into one share of common stock of DermTech Operations.
(c) | Series A Convertible Preferred Stock Financing |
In connection with the PIPE transaction and on August 29, 2019, immediately following the completion of the Business Combination, the Company filed a Certificate of Designation of Preferences, Rights and Limitations for the Company’s Series A Convertible Preferred Stock (the “Series A Certificate of Designation”). An aggregate of 1,231 shares of Series A Convertible Preferred Stock for an aggregate purchase price of $4.0 million were issued to certain accredited investors. On August 10, 2020, entities affiliated with Farallon Capital Management, L.L.C. converted an aggregate of 1,231 shares of Series A Preferred Stock into 615,385 shares of common stock. On September 9, 2020, the Company filed a Certificate of Elimination of Series A Convertible Preferred Stock with the Secretary of State of the State of Delaware to eliminate its Series A Convertible Preferred Stock.
88
Table of Contents
Preferred Dividends
Holders of the Company’s Series A Convertible Preferred Stock (the “Series A Convertible Preferred Stock”) were entitled to receive dividends on an as-converted basis equal to and in the same form as dividends paid on shares of the Company’s common stock when, as and if these dividends were paid on the Company’s common stock.
Preferred Liquidation Preference
Holders of the Series A Convertible Preferred Stock were to participate pari passu with the holders of the Company’s common stock on an as-converted basis in the event of dissolution, liquidation or winding up of the Company.
Redemption
The Series A Convertible Preferred Stock did not contain any mandatory redemption features. The Series A Convertible Preferred Stock were classified as temporary equity in the accompanying consolidated balance sheets in accordance with authoritative guidance for the classification and measurement of potentially redeemable securities whose redemption is based upon certain change in beneficial ownership events outside of the Company’s control. The Company determined not to adjust the carrying values of the convertible preferred stock to the liquidation preferences of such shares because of the uncertainty of whether or when such events would occur.
Conversion
Each share of the Company’s Series A Convertible Preferred Stock was convertible into 500 shares of the Company’s common stock at a conversion price, as adjusted for the Reverse Stock Split, of $6.50 per share, subject to adjustment as set forth in the Series A Certificate of Designation, and provided that in no event may any shares of the Series A Convertible Preferred Stock be convertible if the conversion would result in the holder beneficially owning more than 9.99% of the Company’s then-outstanding shares of common stock.
Voting Rights
The shares of the Series A Convertible Preferred Stock had no voting rights, except with respect to certain protective provisions set forth in the Series A Certificate of Designation relating to the powers, preferences and rights of such shares.
On February 28, 2020, the Company entered into a securities purchase agreement with certain institutional investors for a private placement of the Company’s equity securities (the “2020 PIPE Financing”). Cowen and Company, LLC served as lead placement agent for the 2020 PIPE Financing, with William Blair & Company, L.L.C. acting as joint placement agent. Lake Street Capital Markets, LLC acted as co-placement agent. The 2020 PIPE Financing closed on March 4, 2020.
The 2020 PIPE Financing consisted of 2,467,724 shares of common stock at a price of $10.50 per share, 3,199 shares of Series B-1 Convertible Preferred Stock (the “Series B-1 Shares”) at a price of $10,500 per share, and 524 shares of Series B-2 Convertible Preferred Stock (the “Series B-2 Shares”) at a price of $10,500 per share, for aggregate gross proceeds of approximately $65.0 million, reduced by $5.1 million in issuance costs.
Prior to the closing of the 2020 PIPE Financing, the Company designated (i) 3,200 shares of its authorized and unissued preferred stock as Series B-1 Convertible Preferred Stock by filing the Series B-1 Certificate of Designation with the Delaware Secretary of State and (ii) 525 shares of its authorized and unissued preferred stock as Series B-2 Convertible Preferred Stock by filing the Series B-2 Certificate of Designation with the Delaware Secretary of State.
(e) | Series B-1 Convertible Preferred Stock Issued in Connection with 2020 PIPE Financing |
In connection with the 2020 PIPE Financing transaction and on March 2, 2020, the Company filed a Certificate of Designation of Preferences, Rights and Limitations for the Company’s Series B-1 Convertible Preferred Stock (the “Series B-1 Certificate of Designation”). An aggregate of 3,199 shares of Series B-1 Convertible Preferred Stock for an aggregate purchase price of $33.6 million were issued to certain accredited investors.
89
Table of Contents
At the Company’s annual meeting held on May 26, 2020, the Company’s stockholders voted to approve the 2020 PIPE Financing. As a result, on May 27, 2020 the 3,199 outstanding shares of Series B‑1 Convertible Preferred Stock were automatically converted into an aggregate of 3,198,949 shares of common stock. On September 9, 2020, the Company filed a Certificate of Elimination of Series B-1 Convertible Preferred Stock with the Secretary of State of the State of Delaware to eliminate its Series B-1 Convertible Preferred Stock.
Preferred Dividends
Holders of the Company’s Series B-1 Convertible Preferred Stock (the “Series B-1 Convertible Preferred Stock”) were entitled to receive dividends on an as-converted basis equal to and in the same form as dividends paid on shares of the Company’s common stock when, as and if these dividends were paid on the Company’s common stock.
Preferred Liquidation Preference
Holders of the Series B-1 Convertible Preferred Stock were to participate pari passu with the holders of the Company’s common stock on an as-converted basis in the event of dissolution, liquidation or winding up of the Company.
Redemption
The Series B-1 Convertible Preferred Stock did not contain any mandatory redemption features. The Series B-1 Convertible Preferred Stock was classified as temporary equity in the accompanying consolidated balance sheets in accordance with authoritative guidance for the classification and measurement of potentially redeemable securities whose redemption is based upon certain change in beneficial ownership events outside of the Company’s control. The Company previously determined not to adjust the carrying values of the convertible preferred stock to the liquidation preferences of such shares because of the uncertainty of whether or when such events would occur.
Conversion
Each Series B-1 Share was converted into 1,000 shares of the Company’s common stock at a conversion price of $10.50 on May 27, 2020, which was the first trading day after the approval of the 2020 PIPE Financing by the stockholders of the Company (the “Stockholder Approval”).
Voting Rights
The Series B-1 Shares had no voting rights, except with respect to certain protective provisions set forth in the Series B-1 Certificate of Designation relating to the powers, preferences and rights of such shares.
(f) | Series B-2 Convertible Preferred Stock Issued in Connection with 2020 PIPE Financing |
In connection with the 2020 PIPE Financing transaction and on March 2, 2020, the Company filed a Certificate of Designation of Preferences, Rights and Limitations for the Company’s Series B-2 Convertible Preferred Stock (the “Series B-2 Certificate of Designation”). An aggregate of 524 shares of Series B-2 Convertible Preferred Stock for an aggregate purchase price of $5.5 million were issued to certain accredited investors. On August 10, 2020, entities affiliated with Farallon Capital Management, L.L.C. converted an aggregate of 524 shares of Series B‑2 Preferred Stock into 523,814 shares of common stock. On September 9, 2020, the Company filed a Certificate of Elimination of Series B-2 Convertible Preferred Stock with the Secretary of State of the State of Delaware to eliminate its Series B-2 Convertible Preferred Stock.
Preferred Dividends
Holders of the Company’s Series B-2 Convertible Preferred Stock (the “Series B-2 Convertible Preferred Stock”) were entitled to receive dividends on an as-converted basis equal to and in the same form as dividends paid on shares of the Company’s common stock when, as and if these dividends are paid on the Company’s common stock.
Preferred Liquidation Preference
Holders of the Series B-2 Convertible Preferred Stock were to participate pari passu with the holders of the Company’s common stock on an as-converted basis in the event of dissolution, liquidation or winding up of the Company.
90
Table of Contents
Redemption
The Series B-2 Convertible Preferred Stock did not contain any mandatory redemption features. The Company’s Series B-2 Convertible Preferred Stock was classified as temporary equity in the accompanying consolidated balance sheets in accordance with authoritative guidance for the classification and measurement of potentially redeemable securities whose redemption is based upon certain change in beneficial ownership events outside of the Company’s control. The Company determined not to adjust the carrying values of the convertible preferred stock to the liquidation preferences of such shares because of the uncertainty of whether or when such events would occur.
Conversion
Each Series B-2 Share was convertible into 1,000 shares of the Company’s common stock at a conversion price equal to $10.50, subject to adjustment as provided in the Series B-2 Certificate of Designation. Each Series B-2 Share was convertible into Company common stock at the option of the holder, provided that conversion will be prohibited (i) until the first trading day after the Stockholder Approval, which occurred on May 27, 2020, and (ii) following the Stockholder Approval, if, as a result of any such conversion, the holder would beneficially own in excess of 9.99% of the total number of shares of Company common stock outstanding immediately after giving effect to such conversion (the “Beneficial Ownership Limitation”). A holder of Series B-2 Shares may reset the Beneficial Ownership Limitation to a higher or lower number upon providing written notice to the Company. Any such notice providing for an increase to such Holder’s Beneficial Ownership Limitation will be effective on the 61st day after its delivery to the Company.
Voting Rights
The Series B-2 Shares had no voting rights, except with respect to certain protective provisions set forth in the Series B-2 Certificate of Designation relating to the powers, preferences and rights of such shares.
(g) | At-The Market Offering |
On November 10, 2020, the Company entered into a sales agreement with Cowen and Company, LLC relating to the sale of shares of the Company’s common stock from time to time with an aggregate offering price of up to $50.0 million. In connection with this sales agreement, the Company issued an aggregate of 951,792 shares of common stock at a weighted average purchase price of $20.97 resulting in aggregate gross proceeds of approximately $20.0 million, reduced by $0.9 million in issuance costs, resulting in net proceeds to the Company of approximately $19.1 million.
(h) | Accelerated Vesting in Association with Business Combination |
On January 4, 2019, in contemplation of the Business Combination (refer to Note 8), DermTech Operations modified certain provisions of its stock-based compensation awards to all employees and certain non-employees to accelerate the vesting period for various outstanding stock awards.
In connection with the modifications, the incremental fair value of certain unvested stock option grants were measured at the date of the modification. For any options in which the fair value immediately after the modification was lower than the fair value immediately prior to the modification, no additional compensation expense was recognized. For options in which the fair value increased as a result of the modification and the award was not fully vested, the incremental fair value is being recognized as an expense over the remaining service period. For options that were modified and became fully vested as a result of the accelerated vesting, the Company recognized an expense for the remaining unrecognized grant date fair value. As a result of the accelerated vesting, the Company recognized stock-based compensation expense of $0.4 million related to this modification.
Public Warrants
The Company previously issued 14,936,250 warrants to purchase common stock in a public offering and a private placement which were each consummated on June 23, 2017 (the “Public Warrants”). The Public Warrants have a five year life from the date the Business Combination was consummated and every four Public Warrants entitle the holder to purchase one share at an exercise price of $23.00 per whole share (as adjusted for the Reverse Stock Split). Outstanding Public Warrants totaled 14,936,250 at both December 31, 2020 and 2019.
91
Table of Contents
Series C Warrants
In connection with DermTech Operations’ Series C Preferred Stock financing that took place between 2016 and 2018, each investor that purchased at least $1 million of Series C Convertible Preferred Stock in a single closing received a three-year warrant to purchase shares of common stock at an exercise price of $9.54 per share in the amount equal to 20% of shares of Series C Preferred Stock purchased. Outstanding Series C warrants totaled 97,563 and 202,897 at December 31, 2020 and 2019, respectively.
Placement Agent Warrants
In connection with several of DermTech Operations’ financings that took place between 2015 and 2018, DermTech Operations engaged a registered placement agent to assist in marketing and selling of common and preferred units. From 2015 to 2016, DermTech Operations issued 168,522 seven-year warrants to purchase one share of common stock at an exercise price of $8.68 per share. From 2016 to 2018, DermTech Operations issued 72,695 seven-year warrants to purchase one on share of common stock at an exercise price of $9.54 per share. In 2020, the Company issued 15,724 seven-year warrants to purchase one share of common stock at an exercise price of $9.54 per share in connection with the Company’s 2018 Bridge Note financing. Outstanding placement agent warrants totaled and 31,365 and 241,217 at both December 31, 2020 and 2019, respectively.
(j) | Stock-Based Compensation |
2010 Stock Option Plan
In connection with the Business Combination, the Company assumed the DermTech Operations’ Amended and Restated 2010 Stock Option Plan (the “2010 Plan”), which provided for the granting of incentive and non-statutory stock options and restricted stock purchase rights and bonus awards. Under the 2010 Plan, incentive and non-statutory stock options were granted at not less than 100% of the fair market value of the Company’s common stock on the date of grant. For incentive stock options granted to a ten percent shareholder under the 2010 Plan, the exercise price was not less than 110% of the fair market value of a share of stock on the effective date of grant. DermTech Operations initially reserved 1.0 million shares of common stock for issuance to its employees, non-employee directors and consultants. The 2010 Plan included a provision which annually increased the amount of common stock reserved for issuance under the 2010 Plan. The contractual term of options granted under the 2010 Plan was ten years. Vesting provisions varied based on the specific terms of the individual option awards. At the Company’s annual meeting held on May 26, 2020, the Company’s shareholders voted to approve the DermTech, Inc. 2020 Equity Incentive Plan (the “2020 Plan”), which terminated the 2010 Plan. All outstanding awards under the 2010 Plan remain in effect under the 2020 plan. Zero and 0.1 million options remained available for future grant under the 2010 Plan as of December 31, 2020 and 2019, respectively.
2020 Equity Incentive Plan
On May 26, 2020, the Company’s stockholders approved the adoption of the 2020 Plan, which provides for the granting of incentive and non-qualified stock options, restricted stock and stock-based awards. Under the 2020 Plan, incentive and non-qualified stock options may be granted at not less than 100% of the fair market value of the Company’s common stock on the date of grant. If an incentive stock option is granted to an individual who owns more than 10% of the combined voting power of all classes of the Company’s capital stock, the exercise price may not be less than 110% of the fair market value of the Company’s common stock on the date of grant and the term of the option may not be longer than five years.
The 2020 Plan authorizes the Company to issue up to 1,900,000 shares of the Company’s common stock pursuant to awards granted under the 2020 Plan, plus the number of shares underlying any stock option and other stock-based awards previously granted under the 2010 Plan that are forfeited, canceled, or terminated (other than by exercise) on or after May 26, 2020; provided that no more than 1,400,000 shares may be added to the 2020 Plan pursuant to such forfeitures, cancellations and terminations. In addition, the number of shares available for issuance under the 2020 Plan will automatically increase on the first day of each fiscal year beginning in fiscal year 2021 and ending on the second day of fiscal year 2025, by an amount equal to the smaller of (i) 3.5% of the number of shares of common stock outstanding on such date and (ii) an amount determined by the administrator of the 2020 Plan. The 2020 Plan will expire on April 12, 2030 or an earlier date approved by a vote of the Company’s stockholders or board of directors. The contractual term of options granted under the 2020 Plan is not more than ten years. Vesting provisions vary based on the specific terms of the individual option awards. 934,538 shares remained available for future grant under the 2020 Plan as of December 31, 2020.
92
Table of Contents
The following table summarizes stock option transactions for the years ended December 31, 2020 and 2019:
| | Total options | | | Weighted average exercise price | | | Weighted average remaining contractual term (in years) | | | Aggregate intrinsic value (in thousands) | |
Outstanding at December 31, 2018 | | | 535,051 | | | $ | 3.25 | | | | 6.86 | | | $ | 8 | |
Granted | | | 662,470 | | | | 1.45 | | | | | | | | | |
Exercised | | | (725,719 | ) | | | 1.28 | | | | | | | | | |
Forfeited | | | (28,255 | ) | | | 2.63 | | | | | | | | | |
Outstanding at December 31, 2019 | | | 443,547 | | | $ | 3.84 | | | | 7.80 | | | $ | 3,796 | |
Granted | | | 1,285,183 | �� | | | 12.25 | | | | | | | | | |
Exercised | | | (143,995 | ) | | | 3.29 | | | | | | | | | |
Forfeited | | | (32,252 | ) | | | 8.28 | | | | | | | | | |
Outstanding at December 31, 2020 | | | 1,552,483 | | | $ | 10.76 | | | | 8.91 | | | $ | 33,656 | |
Options vested and expected to vest as of December 31, 2020 | | | 1,552,483 | | | $ | 10.76 | | | | 8.91 | | | $ | 33,656 | |
Options exercisable as of December 31, 2020 | | | 382,152 | | | $ | 5.99 | | | | 7.34 | | | $ | 10,108 | |
The following table summarizes RSU transactions for the years ended December 31, 2020 and 2019:
| | Total RSUs | | | Weighted average grant date fair value per share | |
Outstanding at December 31, 2018 | | | 465,567 | | | $ | 4.15 | |
Released | | | (339,025 | ) | | | 4.16 | |
Forfeited | | | (126,542 | ) | | | 4.11 | |
Outstanding at December 31, 2019 | | | — | | | $ | — | |
Granted | | | 739,962 | | | | 12.47 | |
Released | | | (175,527 | ) | | | 11.60 | |
Forfeited | | | (3,938 | ) | | | 11.41 | |
Outstanding at December 31, 2020 | | | 560,497 | | | $ | 12.75 | |
RSUs vested and expected to vest as of December 31, 2020 | | | 560,497 | | | $ | 12.75 | |
RSUs vested, but not yet issued as of December 31, 2020 | | | 5,000 | | | $ | 13.98 | |
2020 Employee Stock Purchase Plan
On May 26, 2020, the Company’s stockholders approved the adoption of the Company’s 2020 Employee Stock Purchase Plan (the “ESPP”), which allows for full-time and certain part-time employees of the Company to purchase shares of common stock at a discount to fair market value. Eligible employees enroll in a six-month offering period during the open enrollment period prior to the start of that offering period. A new offering period begins approximately every March 1 and September 1. At the end of each offering period, the accumulated contributions are used to purchase shares of the Company’s common stock. Shares are purchased at a price equal to 85% of the lower of: (i) the fair market value of our common stock on the first business day of an offering period or (ii) the fair market value of our common stock on the last business day of an offering period.
The ESPP authorizes the Company to issue up to 400,000 shares of the Company’s common stock. In addition, the number of shares available for issuance under the ESPP will automatically increase on the first day of each of the Company’s fiscal years beginning in 2021 and ending on the first day of 2030, in an amount equal to the lesser of (i) 300,000 shares, (ii) 1% of the shares of Company common stock outstanding on the last day of the immediately preceding fiscal year, or (iii) such lesser number of shares as is determined by the Board of Directors, subject to adjustment upon changes in capitalization of the Company. 400,000 shares remained available for future grant under the ESPP as of December 31, 2020.
93
Table of Contents
Management Warrants
Warrants to purchase DermTech Operations common stock were issued to executive officers of DermTech Operations in lieu of issuing certain stock options (the “Management Warrants”). The Management Warrants were assumed by the Company in connection with the Business Combination. The Management Warrants have a ten year life and are exercisable for Company common stock at $1.08 per common share. The Management Warrants vested monthly over a four-year period. Outstanding Management Warrants totaled 22,320 at December 31, 2020 and 2019.
Common Stock Reserved for Future Issuance
Common stock reserved for future issuance consists of the following at December 31, 2020 and December 31, 2019 (in thousands):
| | December 31, 2020 | | | December 31, 2019 | |
Warrants to purchase common stock | | | 151 | | | | 466 | |
Public Warrants to purchase common stock* | | | 3,734 | | | | 3,734 | |
Stock options issued and outstanding | | | 1,552 | | | | 444 | |
Restricted stock units issued and outstanding | | | 560 | | | | — | |
Authorized for future equity grants | | | 935 | | | | 143 | |
Authorized for future ESPP purchases | | | 400 | | | | — | |
Total common stock reserved for future issuance | | | 7,332 | | | | 4,787 | |
* | Four Public Warrants are needed to purchase one share of common stock. The figures presented above reflect the number of shares of common stock underlying Public Warrants. |
5. Income Taxes
The Company has reported net losses since inception and maintains a full valuation allowance. Therefore, the Company’s effective tax rate is 0% for the periods ended December 31, 2020 and 2019. The following table provides a reconciliation between income taxes computed at the federal statutory rate of 21% at both December 31, 2020 and 2019, respectively, and the Company’s provision for income taxes.
| | Year ended December 31 | |
| | 2020 | | | 2019 | |
Income tax at statutory rate | | | 21.0 | % | | | 21.0 | % |
State tax, net of federal tax benefit | | | 4.9 | | | | — | |
Permanent items | | | (0.1 | ) | | | (0.8 | ) |
Tax credits | | | 0.4 | | | | 0.2 | |
Other | | | (0.5 | ) | | | — | |
Valuation allowance (decrease) increase | | | (25.7 | ) | | | (20.4 | ) |
Income tax expense | | | — | % | | | — | % |
94
Table of Contents
Significant components of the Company’s deferred tax assets and liabilities from federal and state income taxes as of December 31, 2020 and 2019 are shown below (in thousands):
| | December 31, 2020 | | | December 31, 2019 | |
Deferred tax assets: | | | | | | | | |
Net operating loss | | $ | 28,422 | | | $ | 20,336 | |
Research and development credits | | | 1,631 | | | | 1,400 | |
Depreciation and amortization | | | 14 | | | | 33 | |
Stock based compensation | | | 653 | | | | 119 | |
Accruals and other | | | 422 | | | | 194 | |
| | | 31,142 | | | | 22,082 | |
Less valuation allowance | | | (31,142 | ) | | | (22,082 | ) |
Net deferred tax assets | | $ | — | | | $ | — | |
The Company maintains a full valuation allowance against its net deferred tax assets as realization of such assets is not more likely than not.
At December 31, 2020 and 2019, the Company had federal tax net operating loss (“NOL”) carryforwards of approximately $110.8 million and $79.4 million, respectively, as well as state tax NOL carryforwards at December 31, 2020 and 2019 of approximately $84.3 million and $53.4 million, respectively. Federal NOL carryforwards began to expire during 2020 while the Company’s state NOL carryforwards begin to expire during various years, dependent on the jurisdiction.
The Company also had federal research and development (“R&D”) tax credit carryforwards at December 31, 2020 and 2019 of approximately $0.9 million and $0.8 million, respectively, and state R&D tax credits of approximately $0.9 million and $0.8 million at December 31, 2020 and 2019, respectively. The federal and state R&D tax credit carryforwards will begin to expire during 2021 and do not expire, respectively. The Company has not performed a formal study validating its federal and state R&D tax credits and upon preparation, such tax credit carryforwards could vary from what was originally claimed on applicable income tax returns.
Per Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, (“IRC”), a corporation that undergoes an ownership change may be subject to limitations on its ability to utilize pre-change NOLs and other tax attributes otherwise available to offset future taxable income and/or tax liability. An ownership change, per IRC Section 382, is defined as a cumulative change of 50% or more in the ownership positions of certain stockholders during a rolling three-year testing period. The Company has not completed a formal study to determine if any ownership changes within the meaning of IRC Section 382 and 383 have occurred. If an ownership change has occurred, the Company’s ability to use NOL or tax credit carryforwards may be restricted, which could require the Company to pay federal or state income taxes earlier than would be required if such limitations were not in effect.
Because the Company has incurred NOLs, since inception, the federal and state income tax returns are open to examination for all taxable years.
The Company records uncertain tax positions on the basis of a two-step process in which it determines whether it is more likely than not tax positions will be sustained on the basis of the technical merits of the position and for those tax positions that meet the more likely than not recognition threshold the Company would recognize the largest amount of tax benefit that is more than 50% likely to be realized upon ultimate settlement with the related tax authority. The Company has determined it has no uncertain tax positions as of December 31, 2020 and 2019. The Company classifies interest and penalties recognized on uncertain tax positions as a component of income tax expense.
On March 27, 2020, President Trump signed into law the Coronavirus Aid, Relief, and Economic Security (“CARES”) Act (H.R. 748) which includes a number of provisions relating to refundable payroll tax credits, deferment of employer portion of social security payments, NOL carryback periods, alternative minimum tax credit refunds, modifications to IRC Section 163(j) and technical corrections to tax depreciation methods for qualified improvement property. Under ASC 740, the effects of new legislation are recognized upon enactment. Accordingly, the effects of the CARES Act have been incorporated into the income tax provision for the year ended December 31, 2020. These provisions did not have a material impact on the income tax provision.
On December 27, 2020, President Trump signed into law the Consolidated Appropriations Act, 2021 (“CAA 2021”), which included a number of provisions including, but not limited to the extension of numerous employment tax credits, the extension of the Section 179D deduction, enhanced business meals deductions, and the deductibility of expenses paid with Paycheck Protection Program
95
Table of Contents
(“PPP”) loan funds that are forgiven. Accordingly, the effects of the CAA 2021 have been incorporated into the income tax provision for the year ended December 31, 2020. These provisions did not have a material impact on the income tax provision.
Business Combination Tax Implications
In connection with the Business Combination, the Company changed its jurisdiction of incorporation from the British Virgin Islands to the State of Delaware. This reincorporation constituted a tax-free reorganization within the meaning of Section 368(a)(1)(F) of the IRC. The IRC provides that corporations and shareholders do not recognize gain with respect to certain qualifying reorganizations. To satisfy the requirements for this nonrecognition benefit, a transaction must meet one of the statutory definitions of a “reorganization” set forth in IRC Section 368(a)(1). IRC Section 368(a)(1)(F) provides that a reorganization includes a mere change in identity, form, or place of organization. As a result of the reincorporation, the Company will be treated as a U.S. corporation for federal income tax purposes.
For federal income tax purposes, the Business Combination qualified as a reverse triangular merger within the meaning IRC Sections 368(a) and 368(a)(2)(E). Additionally, the Company, Merger Sub, and DermTech Operations are all parties to the reorganization under IRC Section 368(b). As the transaction qualifies as reorganization under IRC Section 368(a), there are no tax consequences to either DermTech Operations or the Company and all tax attributes retain carryover basis.
6. Commitments and Contingencies
Capital Leases
Certain laboratory equipment has been acquired under a capital lease. The Company determined the interest rate implicit in the lease arrangement for the purpose of calculating the interest and principal components of each lease payment was 5.54%. Total capital lease interest expense was approximately $3,000 and zero for the years ended December 31, 2020 and 2019, respectively, and is included within Interest income/(expense) on the consolidated statements of operations. Long-term capital lease obligations are as follows (in thousands):
| | December 31, 2020 | |
Gross capital lease obligations | | $ | 362 | |
Less: imputed interest | | | (27 | ) |
Present value of net minimum lease payments | | | 335 | |
Less: current portion of capital lease obligations | | | (109 | ) |
Total long-term capital lease obligations | | $ | 226 | |
Operating Leases
In January 2013, DermTech Operations entered into a non-cancelable lease agreement for its operating facilities. In January 2014, DermTech Operations signed an amendment to the lease to extend the term through January 2017. In November 2016, DermTech Operations signed a second amendment to the lease to extend the term through March 2022. In August 2019, DermTech Operations signed a third amendment to the lease to add additional space, and in September 2019, the Company signed a fourth amendment to the lease to add additional space. In February 2020, the Company signed a fifth amendment to the lease to add additional space. In connection with the Business Combination, the Company assumed all obligations under the lease, as amended, from DermTech Operations. As part of the fifth amendment, the Company was entitled to a tenant improvement allowance for certain costs incurred while performing these improvements in the amount of $0.3 million, which amount may be increased by up to $0.1 million at the Company’s election and subject to corresponding increase in rent. The Company records rent expense on a straight-line basis over the life of the lease and the difference between the average rent expense and cash payments for rent is recorded as deferred rent and is included in accrued liabilities on the consolidated balance sheet. Rent and associated common area maintenance expense totaled $1.8 million and $0.7 million for the years ended December 31, 2020 and 2019, respectively.
Future minimum operating lease and capital lease payments for the operating facilities and laboratory equipment as of December 31, 2020 were (in thousands):
| | 2021 | | | 2022 | | | 2023 | | | Total | |
Operating lease obligations | | $ | 1,370 | | | $ | 1,411 | | | $ | 478 | | | $ | 3,259 | |
Capital lease obligations, including interest | | | 124 | | | | 124 | | | | 114 | | | | 362 | |
Total future minimum lease payments | | $ | 1,494 | | | $ | 1,535 | | | $ | 592 | | | $ | 3,621 | |
| | | | | | | | | | | | | | | | |
96
Table of Contents
Deferred Underwriting Fees
In connection with the execution of the Merger Agreement, the Company, DermTech Operations and Cowen and Company, LLC (“Cowen”) entered into a letter agreement, dated May 29, 2019, (the “Deferred Underwriting Fee Assignment Agreement”), pursuant to which the Company agreed to assign to DermTech Operations, and DermTech Operations agreed to assume, the Company’s obligations under the Underwriting Agreement, dated as of June 19, 2017 (the “Underwriting Agreement”), by and among the Company and Cowen. On September 4, 2019, the Company, DermTech Operations and Cowen amended the Deferred Underwriting Fee Assignment Agreement, pursuant to which the Company paid Cowen $0.8 million for the reduction of the balance owed by the Company to Cowen under the Underwriting Agreement to $1.4 million.
Pursuant to the terms of the Deferred Underwriting Fee Assignment Agreement, as amended, if the Company raises at least $15.0 million in proceeds received from equity financings consummated prior to the one-year anniversary of the Business Combination, excluding the proceeds received from any financing consummated prior to or simultaneous with the Business Combination, then the Company will pay to the underwriters $1.4 million within one week of the one-year anniversary of the Business Combination. In connection with the Company’s 2020 PIPE Financing, the Company raised $65.0 million in gross proceeds, which satisfied this condition of the Deferred Underwriting Fee Assignment Agreement. On September 2, 2020, the Company paid the underwriters $1.4 million in satisfaction of the Company’s obligation of the deferred underwriting fees in full. No further payment will be required of the Company in connection with the deferred underwriting fees.
Legal Proceedings
The Company is not currently party to any material legal proceedings.
7. Retirement Plan
The Company has an IRC Section 401(k) retirement plan, covering all employees. The Company does not offer a contribution percentage match.
8. Business Combination with DermTech Operations
On August 29, 2019, the Company completed the Business Combination with DermTech Operations. Upon the closing of the Business Combination, DermTech Operations became a wholly-owned subsidiary of the Company.
The Business Combination was accounted for as a reverse acquisition in accordance with ASC 805-40, Business Combinations, Reverse Acquisitions, as the stockholders of DermTech Operations obtained effective control of the Company through (1) a majority of the voting common stock of the post-merger company, (2) appointment of a majority of the board of directors, (3) continued business operations of DermTech Operations, including certain directors and management, and (4) the ability to appoint the executive officers of the combined company. Accordingly, the assets, liabilities and results of operations prior periods presented before the Business Combination reflect those of DermTech Operations. Since the Business Combination, the assets, liabilities, and results of operations have been presented on a consolidated basis. Historical stockholders’ (deficit) equity of the Company prior to the Business Combination has been retroactively adjusted for the equivalent number of shares received by the stockholders of DermTech Operations after giving effect to any difference in par value of the Company and the DermTech Operations’ stock, with any such difference recognized as additional paid-in capital. Retained earnings and other equity balances of the Company/DermTech Operations have been carried forward after the Business Combination. Certain direct costs incurred in connection with the Business Combination were expensed in the period that such costs were incurred and services were received. Approximately $0.2 million in printer fees related to the Business Combination were treated as a reduction of the total amount of equity raised as an offset to additional paid in capital.
9. Related Party Transactions
During 2019 and 2020, the Company engaged EVERSANA Life Science Services, LLC, or EVERSANA, to provide certain marketing services to the Company. Leana Wood, the spouse of Todd Wood, the Company’s Chief Commercial Offer, is an employee of EVERSANA. The Company incurred $1.3 million and $0.4 million in costs for the year ended December 31, 2020 and 2019, respectively.
97
Table of Contents
On October 1, 2019, we entered into a consulting agreement with Michael Dobak pursuant to which we will compensate Michael Dobak, in an amount not to exceed $100,000, for certain public relations and marketing services. On July 28, 2020, the Company and Michael Dobak entered into an amendment to such consulting agreement to modify the terms of Michael Dobak’s compensation. The amended consulting agreement compensates Michael Dobak $15,000 per month for the period May 11, 2020 through September 30, 2020 and also grants him a restricted stock unit award that fully vests in a single installment on August 31, 2020 and represents the contingent right to receive 5,000 shares of common stock on January 2, 2021. On November 11, 2020, the Company and Michael Dobak entered into an amendment to such consulting agreement to extend the term through December 31, 2020 with a continued monthly payment of $15,000. Michael Dobak is the brother of Dr. John Dobak, the Company’s Chief Executive Officer. The Company incurred $0.2 million and $20,000 in costs for the year ended December 31, 2020 and 2019, respectively.
There were no other related party transactions identified for the years ended December 31, 2020 or 2019.
10. Subsequent Events
2021 Underwritten Public Offering
On January 6, 2021, the Company, entered into an Underwriting Agreement with Cowen and Company, LLC and William Blair & Company, L.L.C. as representatives of several underwriters, or the Underwriters. The Company agreed to issue and sell up to 4,237,288 shares of its common stock including up to 635,593 shares that could be purchased by the Underwriters pursuant to a 30-day option granted to the Underwriters by the Company. On January 11, 2021, the Company closed the underwritten public offering of 4,872,881 shares of its common stock, which included the exercise in full by the Underwriters of their option to purchase up to 635,593 additional shares, at a price to the public of $29.50 per share. The Company’s aggregate gross proceeds from the offering, before deducting underwriting discounts and commissions and other offering expenses, were $143.7 million.
Public Warrant Exercises
Through March 1, 2021, a total of 12,059,171 public warrants were exercised resulting in the issuance of 3,014,786 common shares. The Company has received a total of $69.3 million in total proceeds from these public warrant exercises.
The Company considered subsequent events through March 5, 2021, the date the consolidated financial statements were available to be issued.
98
Table of Contents
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures.
As required by Rule 13a‑15(b) under the Securities Exchange Act of 1934 (the Exchange Act), our management, including our principal executive officer and principal financial officer, conducted an evaluation as of the end of the period covered by this report, of the effectiveness of our disclosure controls and procedures as defined in Rule 13a‑15(e) under the Exchange Act.
Based on that evaluation, our principal executive officer and principal financial officer have concluded that these disclosure controls and procedures were effective as of December 31, 2020 to provide reasonable assurance that information required to be disclosed by us in reports that we file under the Exchange Act is recorded, processed, summarized, and reported, within the time periods specified in Securities and Exchange Commission rules and forms and that material information relating to the Company is accumulated and communicated to management, including our principal executive officer and our principal financial officer, as appropriate to allow timely decisions regarding required disclosures
Changes in Internal Control over Financial Reporting.
There were no changes in our internal control over financial reporting identified in connection with the evaluation required by Rule 13a-15(d) under the Exchange Act during the quarter ended December 31, 2020, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control over Financial Reporting.
Management of the Company is responsible for establishing and maintaining effective internal control over financial reporting as defined in Rule 13a‑15(f) under the Exchange Act. The Company’s internal control over financial reporting is designed to provide reasonable assurance to the Company’s management and board of directors regarding the preparation and fair presentation of published financial statements in accordance with generally accepted accounting principles.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2020. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in Internal Control—Integrated Framework (2013). Based on our assessment, we concluded that, as of December 31, 2020, our internal control over financial reporting was effective based on those criteria.
Item 9B. Other Information
None.
99
Table of Contents
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The information required under this item is incorporated by reference to the following sections of our proxy statement for our 2021 Annual Meeting of Stockholders: “Management and Corporate Governance,” “Delinquent Section 16(a) Reports,” and “Code of Conduct and Ethics.”
Item 11. Executive Compensation
The information required under this item is incorporated by reference to the following section of our proxy statement for our 2021 Annual Meeting of Stockholders: “Executive Officer and Director Compensation.”
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required under this item is incorporated by reference to the following sections of our proxy statement for our 2021 Annual Meeting of Stockholders: “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information.”
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required under this item is incorporated by reference to the following sections of our proxy statement for our 2021 Annual Meeting of Stockholders: “Certain Relationships and Related Person Transactions” and “Management and Corporate Governance.”
Item 14. Principal Accountant Fees and Services
The information required under this item is incorporated by reference to the following section of our proxy statement for our 2021 Annual Meeting of Stockholders: “Ratify the Selection of our Independent Registered Public Accounting Firm.”
100
Table of Contents
PART IV
Item 15. Exhibits and Financial Statement Schedules
(a) | The following documents are filed as part of this Form 10‑K: |
| (1) | Financial Statements (see “Consolidated Financial Statements and Supplementary Data” at Item 8 and incorporated herein by reference). |
| (2) | Financial Statement Schedules (Schedules to the Financial Statements have been omitted because the information required to be set forth therein is not applicable or is shown in the accompanying Financial Statements or notes thereto). |
Exhibit Number | | Exhibit Description | | Filed with this Report | | Incorporated by Reference herein from Form or Schedule | | Filing Date | | SEC File/ Registration Number |
2.1 | | Agreement and Plan of Merger, dated as of May 29, 2019, by and among the Company, DermTech Operations, Inc. and DT Merger Sub, Inc., as amended, included as Annex A to the proxy statement/prospectus/information statement forming a part of the referenced filing | | | | S-4/A | | 8/7/2019 | | 333-232181 |
2.2 | | First Amendment to Agreement and Plan of Merger, dated as of August 1, 2019, by and among the Company, DermTech Operations, Inc. and DT Merger Sub, Inc. | | | | S-4/A | | 8/2/2019 | | 333-232181 |
3.1 | | Amended and Restated Certificate of Incorporation of the Company, as amended | | | | 10-Q | | 11/10/2020 | | 001-38118 |
3.2 | | Bylaws of the Company | | | | 10-K | | 3/11/2020 | | 001-38118 |
4.1 | | Specimen Stock Certificate | | | | 10-Q | | 8/5/2020 | | 001-38118 |
4.2 | | Specimen Warrant Certificate of the Company | | | | S-1/A | | 6/9/2017 | | 333-218093 |
4.3 | | Warrant Agreement, dated June 19, 2017, between the Company and Continental Stock Transfer & Trust Company | | | | 8-K | | 6/23/2017 | | 001-38118 |
4.4* | | Form of Management Warrant | | | | 8-K | | 9/5/2019 | | 001-38118 |
4.5 | | Form of Series C Warrant | | | | 8-K | | 9/5/2019 | | 001-38118 |
4.6 | | Form of Placement Agent Warrant 2015 and July 2016 | | | | 8-K | | 9/5/2019 | | 001-38118 |
4.7 | | Form of Placement Agent Warrant December 2016 | | | | S-1 | | 5/4/2020 | | 333-237991 |
4.8 | | Form of Placement Agent Warrant 2017 and 2018 | | | | S-1 | | 5/4/2020 | | 333-237991 |
4.9 | | Form of 2020 Placement Agent Warrant | | | | S-1/A | | 2/6/2020 | | 333-235780 |
4.10 | | Form of Omnibus Warrant Amendment for 2015 and July 2016 Placement Agent Warrants | | | | S-1 | | 5/4/2020 | | 333-237991 |
4.11 | | Omnibus Warrant Amendment for December 2016, 2017 and 2018 Placement Agent Warrants, dated as of March 30, 2020 by and between the Company and Paulson Investment Company, LLC | | | | S-1 | | 5/4/2020 | | 333-237991 |
4.12 | | Description of Securities | | X | | | | | | |
10.1 | | Sales Agreement, dated November 10, 2020, by and between the Company and Cowen and Company, LLC | | | | 8-K | | 11/10/2020 | | 001-38118 |
10.2 | | Securities Purchase Agreement, dated February 28, 2020, by and among the Company and the Purchasers identified on the signature pages thereto | | | | 8-K | | 3/2/2020 | | 001-38118 |
10.3 | | Form of Registration Rights Agreement, dated March 4, 2020, by and among the Company and the Purchasers | | | | 8-K | | 3/2/2020 | | 001-38118 |
10.4 | | Registration Rights Agreement, dated August 29, 2019, by and among the Company, certain stockholders of the Company and certain stockholders of DermTech Operations, Inc. | | | | 8-K | | 9/5/2019 | | 001-38118 |
101
Table of Contents
10.5 | | Amended and Restated Subscription Agreement, dated August 1, 2019, between the Company and Farallon Capital (AM) Investors, L.P. | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.6 | | Amended and Restated Subscription Agreement, dated August 1, 2019, between the Company and Farallon Capital F5 Master I, L.P. | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.7 | | Amended and Restated Subscription Agreement, dated August 1, 2019, between the Company and Farallon Capital Institutional Partners, L.P. | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.8 | | Amended and Restated Subscription Agreement, dated August 1, 2019, between the Company and Farallon Capital Institutional Partners II, L.P. | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.9 | | Amended and Restated Subscription Agreement, dated August 1, 2019, between the Company and Farallon Capital Institutional Partners III, L.P. | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.10 | | Amended and Restated Subscription Agreement, dated August 1, 2019, between the Company and Farallon Capital Offshore Investors II, L.P. | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.11 | | Amended and Restated Subscription Agreement, dated August 1, 2019, between the Company and Farallon Capital Partners, L.P. | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.12 | | Amended and Restated Subscription Agreement, dated August 1, 2019, between the Company and Four Crossings Institutional Partners V, L.P. | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.13 | | Subscription Agreement, dated May 22, 2019, between the Company and Victory RS Science and Technology Fund | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.14 | | Subscription Agreement, dated May 22, 2019, between the Company and The Irwin Mark and Joan Klein Jacobs Family Trust UA DTD 6/20/80 | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.15 | | Subscription Agreement, dated May 23, 2019, between the Company and Jacobs Investment Company LLC | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.16 | | Subscription Agreement, dated May 23, 2019, between the Company and RTW Master Fund, Ltd. and RTW Innovation Master Fund, Ltd. | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.17 | | Omnibus Common Share Subscription Agreement Amendment, dated as of August 1, 2019, by and among the Company and the Common Share Purchasers | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.18 | | Subscription Agreement, dated August 1, 2019, between the Company and HLM Venture Partners IV, L.P. | | | | S-4/A | | 8/2/2019 | | 333-232181 |
10.19 | | Letter Agreement, dated June 19, 2017, by and among the Company, Centripetal, LLC, and certain former directors and officers of the Company | | | | 8-K | | 6/23/2017 | | 001-38118 |
10.20 | | Amendment No. 1 to Letter Agreement, dated August 28, 2019 by and among the Company, Centripetal, LLC, and certain former directors and officers of the Company | | | | 10-Q | | 11/7/2019 | | 001-38118 |
10.21 | | Amended and Restated Unit Purchase Agreement, dated June 2017, between the Company and Cowen Investments LLC | | | | S-1/A | | 6/14/2017 | | 333-218093 |
10.22* | | Employment Agreement, dated June 26, 2012, between DermTech Operations and John Dobak | | | | S-4 | | 6/18/2019 | | 333-232181 |
10.23* | | Amendment to Employment Agreement, dated February 28, 2014, between DermTech Operations and John Dobak | | | | S-4 | | 6/18/2019 | | 333-232181 |
10.24* | | Offer of Employment Letter, dated October 1, 2015, from DermTech Operations to Burkhard Jansen | | | | S-4 | | 6/18/2019 | | 333-232181 |
102
Table of Contents
10.25* | | Offer of Employment Letter, dated December 7, 2018, from DermTech Operations to Todd Wood | | | | S-4 | | 6/18/2019 | | 333-232181 |
10.26* | | Offer of Employment Letter, dated August 14, 2019, from the Company to Kevin Sun | | | | 8-K | | 9/17/2019 | | 001-38118 |
10.27* | | Offer of Employment Letter, dated September 23, 2019, from the Company to Claudia Ibarra | | | | 8-K | | 3/24/2020 | | 001-38118 |
10.28* | | Offer of Employment Letter, dated October 14, 2020, from the Company to Ray Akhavan | | X | | | | | | |
10.29 | | Amendment Number 1 to Deferred Underwriting Fee Assignment Agreement, dated September 4, 2019, by and among the Company, DermTech Operations and Cowen and Company, LLC | | | | 8-K | | 9/5/2019 | | 001-38118 |
10.30 | | Amendment Number 2 to the Deferred Underwriting Fee Assignment Agreement, dated July 14, 2020, by and among the Company, DermTech Operations and Cowen and Company, LLC | | | | 10-Q | | 8/5/2020 | | 001-38118 |
10.31 | | Standard Multi-Tenant Officer Lease–Net and Addendum to Lease, dated January 25, 2013, by and between DermTech Operations and AG/Touchstone TP, LLC | | | | 8-K | | 9/5/2019 | | 001-38118 |
10.32 | | First Amendment to Standard Rental Lease, Storage Lease and Signage to Expand and Extend Term, dated January 30, 2014, by and between DermTech Operations and AG/Touchstone TP, LLC | | | | 8-K | | 9/5/2019 | | 001-38118 |
10.33 | | Assignment, Consent to Assignment, and Second Amendment to Standard Multi-Lease–Net, dated November 21, 2016, by and between DermTech Operations and AG/Touchstone TP, LLC | | | | 8-K | | 9/5/2019 | | 001-38118 |
10.34 | | Third Amendment to Lease, dated August 6, 2019, by and between DermTech Operations and HCP Torrey Pines, LLC | | | | 8-K | | 9/5/2019 | | 001-38118 |
10.35 | | Fourth Amendment to Lease, dated as of September 10, 2019, by and between the Company and HCP Torrey Pines, LLC | | | | 8-K | | 9/23/2019 | | 001-38118 |
10.36 | | Fifth Amendment to Lease and Signage Lease, dated February 5, 2020, by and between the Company and HCP Torrey Pines, LLC | | | | S-1/A | | 2/6/2020 | | 333-235780 |
10.37* | | DermTech, Inc. 2020 Equity Incentive Plan | | | | 8-K | | 5/27/2020 | | 001-38118 |
10.38* | | DermTech, Inc. 2020 Employee Stock Purchase Plan | | | | 8-K | | 5/27/2020 | | 001-38118 |
10.39* | | Form of Stock Option Agreement and Forms of Stock Option Grant Notice under the DermTech, Inc. 2020 Equity Incentive Plan | | | | 8-K | | 5/27/2020 | | 001-38118 |
10.40* | | Form of Restricted Stock Unit Agreement and Forms of Restricted Stock Unit Award Grant Notice under the DermTech, Inc. 2020 Equity Incentive Plan | | | | 8-K | | 5/27/2020 | | 001-38118 |
10.41* | | Amended and Restated 2010 Stock Plan of the Company, included as Annex E to the proxy statement/prospectus/information statement forming a part of the referenced filing | | | | S-4/A | | 8/7/2019 | | 333-232181 |
10.42* | | Form of Stock Option Grant Notice and Stock Option Agreement under the Amended and Restated 2010 Stock Plan of the Company | | | | S-1 | | 1/3/2020 | | 333-235780 |
10.43* | | Form of Restricted Stock Unit Award Grant Notice and Restricted Stock Unit Agreement under the Amended and Restated 2010 Stock Plan of the Company | | | | S-1 | | 1/3/2020 | | 333-235780 |
10.44* | | 2020 Form of Stock Option Agreement and Forms of Stock Option Grant Notice under Amended and Restated 2010 Stock Plan | | | | 8-K | | 1/21/2020 | | 001-38118 |
103
Table of Contents
* | Management contract or compensatory plan or arrangement. |
Item 16. Form 10-K Summary
None.
104
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| DERMTECH, INC. |
| | | |
Date: March 5, 2021 | By: | | /s/ John Dobak, M.D. John Dobak, M.D.
Chief Executive Officer |
POWER OF ATTORNEY AND SIGNATURES
We, the undersigned officers and directors of DermTech, Inc., hereby severally constitute and appoint John Dobak, M.D. and Kevin Sun, and each of them singly (with full power to each of them to act alone), our true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution in each of them for her or him and in her or his name, place and stead, and in any and all capacities, to sign any and all amendments to this Annual Report on Form 10‑K, and generally to do all things in our names and on our behalf in such capacities to enable DermTech, Inc. to comply with the provisions of the Securities Exchange Act of 1934, as amended, and all the requirements of the Securities Exchange Commission.
Pursuant to the requirements of the Securities and Exchange Act of 1934, as amended, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Name | | Title | | Date |
| | | | |
/s/ John Dobak, M.D. John Dobak, M.D. | | Chief Executive Officer and Director (Principal Executive Officer) | | March 5, 2021 |
| | | | |
/s/ Kevin Sun Kevin Sun | | Chief Financial Officer and Treasurer (Principal Financial and Accounting Officer) | | March 5, 2021 |
| | | | |
/s/ Cynthia Collins Cynthia Collins | | Director | | March 5, 2021 |
| | | | |
/s/ Gary Jacobs Gary Jacobs | | Director | | March 5, 2021 |
| | | | |
/s/ Scott Pancoast Scott Pancoast | | Director | | March 5, 2021 |
| | | | |
/s/ Enrico Picozza Enrico Picozza | | Director | | March 5, 2021 |
| | | | |
/s/ Matthew Posard Matthew Posard | | Director | | March 5, 2021 |
| | | | |
/s/ Herm Rosenman Herm Rosenman | | Director | | March 5, 2021 |
| | | | |
| | | | |
105