UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 20-F
| ¨ | REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 FOR THE FISCAL YEAR ENDED DECEMBER 31, 2015 |
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
| ¨ | SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission file number: 001-36565
INNOCOLL HOLDINGS PUBLIC LIMITED COMPANY
(Exact name of Registrant as specified in its charter)
Not Applicable
(Translation of Registrant’s name into English)
Republic of Ireland
(Jurisdiction of incorporation or organization)
Innocoll Holdings plc
Unit 9, Block D
Monksland Business Park
Monksland, Athlone
Ireland
+353 (0) 90 6486834
(Address of principal executive offices)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
| Title of each class | Name of each exchange on which
registered |
| | |
| Ordinary Shares, $0.01 par value per share | NASDAQ Global Market |
Securities registered or to be registered pursuant to Section 12(g) of the Act.
None
(Title of Class)
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act.
None
(Title of Class)
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report.
As of December 31, 2015, the Registrant’s predecessor, Innocoll AG, had outstanding 1,837,493 ordinary shares, notional par value €1.00 per share.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.¨ Yesx No
If the report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.¨ Yesx No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.x Yes¨ No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).x Yes¨ No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer x | Non-accelerated filer ☐ | |
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
| U.S. GAAP ¨ | International Financial Reporting
Standards as issued by the
International Accounting Standards
Board x | Other ¨ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.¨ Item 17¨ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).¨ Yesx No
TABLE OF CONTENTS
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This annual report on Form 20-F contains forward-looking statements that involve substantial risks and uncertainties. All statements, other than statements of historical facts, included in this annual report regarding our strategy, future operations, regulatory process, future financial position, future revenue, projected costs, prospects, plans, objectives of management and expected market growth are forward-looking statements. The words “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “plan,” “predict,” “project,” “will,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words.
The forward-looking statements in this annual report on Form 20-F include, among other things, statements about:
| · | our plans to develop and manufacture XaraColl, Cogenzia and our other product candidates; |
| · | the results of clinical trials for XaraColl, Cogenzia and our other product candidiates; |
| · | the timing of, and our ability to obtain, regulatory approval of XaraColl, Cogenzia and our other product candidates; |
| · | the timing of our anticipated launches of XaraColl, Cogenzia and our other product candidates; |
| · | the rate and degree of market acceptance of XaraColl, Cogenzia and our other product candidates; |
| · | the size and growth of the potential markets for XaraColl, Cogenzia and our other product candidates and our ability to serve those markets; |
| · | our manufacturing and marketing capabilities; |
| · | the timing of, and our ability to obtain, regulatory approvals for the expansion of our manufacturing facility; |
| · | regulatory developments in the United States and foreign countries; |
| · | our ability to obtain and maintain the scope, duration and protection of our intellectual property rights; |
| · | statements concerning our corporate and tax domiciles and potential changes to them, including potential tax charges; |
| · | the accuracy of our estimates regarding expenses and capital requirements; and |
| · | the loss of key scientific or management personnel. |
We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements we make. We have included important factors in the cautionary statements included in this annual report, particularly the factors described in the “Item 3. Key Information—D. Risk Factors” section of this annual report, that could cause actual results or events to differ materially from the forward-looking statements that we make. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments that we may make.
You should read this annual report and the documents that we have filed as exhibits to this annual report, completely and with the understanding that our actual future results may be materially different from what we expect. We do not assume any obligation to update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
SERVICE OF PROCESS AND ENFORCEMENT OF CIVIL LIABILITIES
Innocoll Holdings plc is a public limited company formed under Irish law, and its registered offices and all of its assets are located outside of the United States. In addition, certain members of our board, our senior management and the experts named herein are residents of jurisdictions other than the United States, namely Ireland, the United Kingdom and Germany. As a result, it may not be possible for you to effect service of process within the United States upon Innocoll Holdings plc or these individuals to enforce judgments obtained in U.S. courts based on the civil liability provisions of the U.S. securities laws against Innocoll in the United States.
In addition, it may not be possible to enforce court judgments obtained in the United States against us in Ireland based on the civil liability provisions of the U.S. federal or state securities laws. We have been advised by William Fry, our Irish counsel, that the United States currently does not have a treaty with Ireland providing for the reciprocal recognition and enforcement of judgments in civil and commercial matters.
The following requirements must be met before a judgment of a U.S. court will be deemed to be enforceable in Ireland:
| · | the judgment must be for a definite sum; |
| · | the judgment must be final and conclusive; and |
| · | the judgment must be provided by a court of competent jurisdiction. |
An Irish court may also exercise its right to refuse enforcement if the U.S. judgment was obtained by fraud, if the judgment violates Irish public policy, if the judgment is in breach of natural justice or if it is irreconcilable with an earlier foreign judgment. There is some uncertainty as to whether the courts of Ireland would recognize or enforce judgments of U.S. courts obtained against us or our directors or officers based on the civil liabilities provisions of the U.S. federal or state securities laws or hear actions against us or those persons based on those laws. Therefore, a final judgment for the payment of money rendered by any U.S. federal or state court based on civil liability, whether or not based solely on U.S. federal or state securities laws, would not automatically be enforceable in Ireland.
PART I
Item 1. Identity of Directors, Senior Management and Advisers
Not applicable.
Item 2. Offer Statistics and Expected Timetable
Not applicable.
Item 3. Key Information
Innocoll AG, a German stock corporation, which had American Depository Shares, or ADSs, listed on the Nasdaq Global Market (“Innocoll Germany”), held an Extraordinary General Meeting of Shareholders in Munich, Germany on January 30, 2016, in which its shareholders considered and approved a proposal for a cross-border merger between Innocoll Germany and Innocoll Holdings plc, a public limited company formed under Irish law (“Innocoll Ireland”), with Innocoll Germany being the disappearing entity and Innocoll Ireland being the surviving entity in a merger by acquisition (the “Merger”).
On March 16, 2016, Innocoll Germany merged with Innocoll Ireland by way of a European cross-border merger with Innocoll Ireland being the surviving company. Upon the effectiveness of the Merger, we terminated Innocoll Germany’s ADS facility and each cancelled ADS effectively became an entitlement to receive one ordinary share of Innocoll Ireland. Holders of Innocoll Germany ordinary shares received 13.25 ordinary shares of Innocoll Ireland in respect of each share held of Innocoll Germany. Simultaneous with this transaction, Innocoll Ireland listed its ordinary shares on the Nasdaq Global Market under the symbol “INNL”, which we previously used for Innocoll Germany’s ADSs. The Merger effectively resulted in Innocoll Ireland becoming the publicly-traded parent of the Innocoll group of companies carrying on the same business as that conducted by Innocoll Germany prior to the Merger.
The financial and other information presented in this annual report as of December 31, 2015, including audited financial information, is the information of Innocoll AG, our predecessor. The audited financial statements of Innocoll AG contained in this annual report were prepared in accordance with IFRS as issued by the IASB, audited in accordance with the standards of the Public Company Accounting Oversight Board and approved by the board of Innocoll AG prior to the Merger. Innocoll Holdings plc intends to prepare its financial information in accordance with US GAAP beginning with the first quarter of 2016. As a result of this transition, the Company may report first quarter results at a later date than it has in the past.
Except where indicated to the contrary, or the context suggests otherwise, all share and per share information: (i) not presented in the financial statements or financial statement data refers to ordinary shares of Innocoll Holdings plc, and (ii) included in the financial statements and financial statement data for Innocoll AG refers to ordinary shares of Innocoll Germany prior to the Merger.
In this annual report, unless the context otherwise indicates, Innocoll Holdings plc, a public limited company formed under Irish law, is referred to as Innocoll. Innocoll together with its direct and indirect wholly owned subsidiaries as of the time relevant to the applicable reference are collectively referred to as the Innocoll Group. Notwithstanding that the historical audited financial statements prepared in accordance with IFRS and presented herein is the information of Innocoll AG, our predecessor, Innocoll and its current direct and indirect wholly owned subsidiaries are collectively referred to as “we,” “us,” “our,” “Innocoll” or the “Company.”
| A. | SELECTED FINANCIAL DATA |
Financial information in this annual report is the information of Innocoll AG, our predecessor. The audited financial statements of Innocoll AG contained in this annual report were prepared in accordance with IFRS. Innocoll Holdings plc intends to prepare its financial information in accordance with US GAAP.
We present below our selected historical financial and operating data as of and for each of the years in the three-year period ended December 31, 2015. The financial data as of December 31, 2015 and 2014 and for the years ended December 31, 2015, 2014 and 2013 have been derived from our audited financial statements and the related notes, which are included elsewhere in this annual report and which have been prepared in accordance with IFRS as issued by the IASB and audited in accordance with the standards of the Public Company Accounting Oversight Board (United States). Pursuant to Section 13(a) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), we are permitted to provide fewer than five years of selected financial data.
Our historical results are not necessarily indicative of the financial results to be expected in any future periods. You should read this information in conjunction with “Item 5. Operating and Financial Review and Prospects,” and our financial statements and related notes, each included elsewhere in this annual report.
Amounts presented in U.S. dollars are not audited and have been converted from euros to U.S. dollars solely for the convenience of the reader at an exchange rate of $1.0887 per euro, the exchange rate on December 31, 2015. See “Exchange Rate Information” below.
The financial data have been prepared in accordance with IFRS, unless otherwise noted.
| | | | | | Years Ended December 31, | |
| | | 2015 | | | 2015 | | | 2014 | | | 2013 | |
| | | | | | (in thousands, except for per share data) | |
| Consolidated Statement of Comprehensive Income Data: | | | | | | | | | | | | | | | | |
| Revenue | | | | | | | | | | | | | | | | |
| Revenue – continuing operations | | $ | 2,816 | | | € | 2,587 | | | € | 4,497 | | | € | 3,546 | |
| Cost of sales | | | (5,228 | ) | | | (4,802 | ) | | | (5,573 | ) | | | (4,551 | ) |
| Gross loss | | | (2,412 | ) | | | (2,215 | ) | | | (1,076 | ) | | | (1,005 | ) |
| Operating expense | | | | | | | | | | | | | | | | |
| Research and development expenses | | | (29,262 | ) | | | (26,878 | ) | | | (3,252 | ) | | | (1,663 | ) |
| General and administrative expenses | | | (20,667 | ) | | | (18,983 | ) | | | (11,687 | ) | | | (4,121 | ) |
| Other operating income/(expense) – net | | | 4,220 | | | | 3,876 | | | | (39 | ) | | | (154 | ) |
| Total operating expense – net | | | (45,709 | ) | | | (41,985 | ) | | | (14,978 | ) | | | (5,938 | ) |
| Loss from operating activities – continuing operations | | | (48,121 | ) | | | (44,200 | ) | | | (16,054 | ) | | | (6,943 | ) |
| Finance income/(expense) | | | 1,544 | | | | 1,418 | | | | (4,535 | ) | | | (6,949 | ) |
| Other income | | | - | | | | - | | | | 75 | | | | 16,073 | |
| (Loss)/profit before income tax | | | (46,577 | ) | | | (42,782 | ) | | | (20,514 | ) | | | 2,181 | |
| Income tax expense | | | (400 | ) | | | (367 | ) | | | (152 | ) | | | (72 | ) |
| (Loss)/profit for the period – all attributable to equity holders of the company | | | (46,977 | ) | | | (43,149 | ) | | | (20,666 | ) | | | 2,109 | |
| Currency translation adjustment | | | (566 | ) | | | (520 | ) | | | (623 | ) | | | 155 | |
| Total comprehensive (loss)/income | | $ | (47,543 | ) | | € | (43,669 | ) | | € | (21,289 | ) | | € | 2,264 | |
| (Loss)/earnings per share: | | | | | | | | | | | | | | | | |
| Basic | | | (27.9 | ) | | | (25.6 | ) | | | (28.1 | ) | | | 47.0 | |
| Diluted | | | (27.9 | ) | | | (25.6 | ) | | | (28.1 | ) | | | (9.5 | ) |
| Basic (loss) per ADS(1) | | | (2.1 | ) | | | (1.9 | ) | | | (2.1 | ) | | | | |
| Diluted (loss) per ADS(1) | | | (2.1 | ) | | | (1.9 | ) | | | (2.1 | ) | | | | |
(1) For the years ended December 31, 2015 and 2014 we excluded the dilutive effect of potentially exercisable instruments in issue from the computation of the diluted net loss and diluted weighted-average shares outstanding as the effect would be anti-dilutive.
| | | | | | As of December 31, | |
| | | 2015 | | | 2015 | | | 2014 | | | 2013 | |
| | | | | | (in thousands, except per share data) |
| Consolidated Statement of Financial Position Data: | | | | | | | | | | | | | | | | |
| Current assets | | $ | 48,558 | | | € | 44,602 | | | € | 47,495 | | | € | 4,824 | |
| Total assets | | | 56,977 | | | | 52,335 | | | | 48,733 | | | | 5,556 | |
| Current liabilities | | | (16,913 | ) | | | (15,535 | ) | | | (6,899 | ) | | | (9,048 | ) |
| Long term debt | | | (16,400 | ) | | | (15,064 | ) | | | - | | | | (63,026 | ) |
| Other non-current liabilities | | | (11,547 | ) | | | (10,606 | ) | | | (7,300 | ) | | | (1,055 | ) |
| Total equity attributable to equity holders of the company | | | 12,117 | | | | 11,130 | | | | 34,534 | | | | (67,573 | ) |
| Total equity and liabilities | | | 56,977 | | | | 52,335 | | | | 48,733 | | | | 5,556 | |
Other Data:
The tables below include a reconciliation of our GAAP results to non-GAAP results for the years ended December 31, 2015, 2014 and 2013. We define adjusted non-GAAP earnings per share as basic and diluted earnings per share excluding share based payments, fair value expense on warrants and reversal of impairment of property, plant and equipment. We believe adjusted non-GAAP earnings per share is meaningful to our investors to enhance their understanding of our financial condition and results. We believe that non-GAAP earnings per share excluding these non-cash items may provide securities analysts, investors and other interested parties with a useful measure of our operating performance and cash requirements. Disclosure in this annual report of non-GAAP earnings per share, which is a non-IFRS financial measure, is intended as a supplemental measure of our performance that is not required by, or presented in accordance with, IFRS. Non-GAAP earnings per share should not be considered as an alternative to earnings per share, profit (loss) or any other performance measure derived in accordance with IFRS. Our presentation of adjusted earnings per share should not be construed to imply that our future results will be unaffected by unusual non-cash or non-recurring items.
For the year ended December 31, 2015 the reconciliation primarily relates to non-cash expenses in the amount of €4.8 million with respect to share-based compensation, €3.7 million with respect to fair value expense on warrants and (€3.9) million with respect to the reversal of impairment of property, plant and equipment. On a non-GAAP-basis, the net loss for the year ended December 31, 2015 was €38.5 million, or €22.9 per share of Innocoll AG, compared to a net loss of €9.3 million, or €12.6 per share of Innocoll AG for the year ended December 31, 2014.
| | | Years ended December 31, | |
| | | 2015 | | | 2015 | | | 2014 | | | 2013 | |
| | | (in thousands, except for per share data) | |
| Numerator for non-GAAP (loss)/earnings per share: | | | | | | | | | | | | | | | | |
| Net (loss)/earnings – basic | | $ | (46,977 | ) | | € | (43,149 | ) | | € | (20,666 | ) | | € | 2,109 | |
| Share based payments | | | 5,199 | | | | 4,775 | | | | 5,149 | | | | - | |
| Reversal of impairment of property, plant & equipment | | | (4,220 | ) | | | (3,876 | ) | | | - | | | | - | |
| Fair value expense on warrants | | | 4,066 | | | | 3,735 | | | | 6,265 | | | | 205 | |
| Non-GAAP net (loss)/earnings - basic | | | (41,932 | ) | | | (38,515 | ) | | | (9,252 | ) | | | 2,314 | |
| | | | | | | | | | | | | | | | | |
| Adjustment to net earnings for interest on convertible preferred shares | | | - | | | | - | | | | - | | | | 4,728 | |
| Adjustment to net earnings for interest on convertible promissory notes | | | - | | | | - | | | | - | | | | 1,918 | |
| Adjustment for gain on settlement of promissory notes and preferred stock | | | - | | | | - | | | | - | | | | (15,903 | ) |
| Non-GAAP net (loss) – diluted | | | (41,932 | ) | | | (38,515 | ) | | | (9,252 | ) | | | (6,943 | ) |
| | | | | | | | | | | | | | | | | |
| Denominator – number of shares: | | | | | | | | | | | | | | | | |
| Weighted-average shares outstanding – basic | | | 1,685,088 | | | | 1,685,088 | | | | 735,416 | | | | 44,848 | |
| Dilutive common stock issuable upon conversion of preferred shares(1) | | | | | | | | | | | - | | | | 547,195 | |
| Dilutive common stock issuable upon conversion of promissory notes (1) | | | - | | | | - | | | | - | | | | 160,246 | |
| Weighted-average shares outstanding – diluted | | | 1,685,088 | | | | 1,685,088 | | | | 735,416 | | | | 752,289 | |
| | | | | | | | | | | | | | | | | |
| Non-GAAP (loss)/earnings per share: | | | | | | | | | | | | | | | | |
| Basic | | | (24.9 | ) | | | (22.9 | ) | | | (12.6 | ) | | | 51.6 | |
| Diluted | | | (24.9 | ) | | | (22.9 | ) | | | (12.6 | ) | | | (9.2 | ) |
| | | | | | | | | | | | | | | | | |
| Non-GAAP (loss)/earnings per ADS(2): | | | | | | | | | | | | | | | | |
| Basic | | | (1.9 | ) | | | (1.7 | ) | | | (1.0 | ) | | | | |
| Diluted | | | (1.9 | ) | | | (1.7 | ) | | | (1.0 | ) | | | | |
(1) For the years ended December 31, 2015 and December 31, 2014, we excluded the dilutive effect of potentially exercisable instruments in issue from the computation of the diluted net loss and diluted weighted-average shares outstanding as the effect would be anti-dilutive.
(2) Prior to the termination of the ADS facility, one ordinary share of Innocoll AG represented 13.25 ADSs. Upon consummation of the Merger, each ADS of Innocoll AG was exchanged for one ordinary share of Innocoll Holdings plc. Going forward, Innocoll Holdings plc’s earnings/(loss) per share calculation will be equivalent to the earnings/(loss) per ADS calculation of Innocoll AG.
Exchange Rate Information
Our business to date has been conducted primarily in the European Union, or EU, and we prepare our consolidated financial statements in euros. All references in this annual report to “U.S. dollars” or “$” are to the legal currency of the United States and all references to “€“or “euro” are to the currency introduced at the start of the third stage of the European economic and monetary union pursuant to the treaty establishing the European Community, as amended. Solely for the convenience of the reader, unless otherwise indicated, all amounts in U.S. dollars have been converted from euros to U.S. dollars at an exchange rate of $1.0887 per euro, the official exchange rate quoted as of December 31, 2015 by the European Central Bank. Such U.S. dollar amounts are not necessarily indicative of the amounts of U.S. dollars that could actually have been purchased upon exchange of euros at the dates indicated. Fluctuations in the exchange rate between the U.S. dollar and the euro will affect the U.S. dollar amounts received by owners of our ordinary shares on conversion of dividends, if any, paid in euros on the ordinary shares and will affect the U.S. dollar price of our ordinary shares on the NASDAQ Global Market. The following table presents information on the exchange rates between the U.S. dollar and the euro for the periods indicated. The rates set forth below are provided solely for your convenience and may differ from the actual rates used in the preparation of the financial statements included in this annual report and other financial data appearing in this annual report.
| Year Ended December 31, | | High | | | Low | | | Average | | | Year end | |
| 2011 | | $ | 1.4882 | | | $ | 1.2889 | | | $ | 1.3920 | | | $ | 1.2939 | |
| 2012 | | $ | 1.3454 | | | $ | 1.2089 | | | $ | 1.2848 | | | $ | 1.3194 | |
| 2013 | | $ | 1.3814 | | | $ | 1.2768 | | | $ | 1.3281 | | | $ | 1.3791 | |
| 2014 | | $ | 1.3953 | | | $ | 1.2141 | | | $ | 1.3285 | | | $ | 1.2141 | |
| 2015 | | $ | 1.2043 | | | $ | 1.0552 | | | $ | 1.1095 | | | $ | 1.0887 | |
| Month Ended | | High | | | Low | | | Average | | | Month end | |
| September 2015 | | $ | 1.1419 | | | $ | 1.1138 | | | $ | 1.1221 | | | $ | 1.1203 | |
| October 2015 | | $ | 1.1439 | | | $ | 1.0930 | | | $ | 1.1235 | | | $ | 1.1017 | |
| November 2015 | | $ | 1.1032 | | | $ | 1.0579 | | | $ | 1.0736 | | | $ | 1.0579 | |
| December 2015 | | $ | 1.0990 | | | $ | 1.0600 | | | $ | 1.0877 | | | $ | 1.0887 | |
| January 2016 | | $ | 1.0920 | | | $ | 1.0742 | | | $ | 1.0860 | | | $ | 1.0920 | |
| February 2016 | | $ | 1.1347 | | | $ | 1.0884 | | | $ | 1.1093 | | | $ | 1.0888 | |
On March 16, 2016, the exchange rate was $[ ] per euro.
| B. | CAPITALIZATION AND INDEBTEDNESS |
Not applicable.
| C. | REASONS FOR THE OFFER AND USE OF PROCEEDS |
Not applicable.
Risks Related to Our Financial Position and Capital Requirements
We have a history of operating losses and anticipate that we will continue to incur operating losses in the future and may never sustain profitability.
We have incurred operating losses in each year since inception because our research and development and general and administrative expenses exceeded our revenue. Our operating loss for the years ended December 31, 2013, 2014 and 2015 was, €6.9 million, €16.1 million and €44.2 million, respectively. As of December 31, 2015, we had an accumulated deficit of €149.9 million and our current assets exceeded our current liabilities by €29.1 million.
Our ability to become profitable depends on our ability to develop and commercialize our lead product candidates, XaraColl and Cogenzia. Our lead product candidates are not yet approved for commercial sale in the United States or Europe, and we do not know when, or if, we will generate significant revenues from their sale in the future. Cogenzia has been approved in seven countries outside of the United States and Europe but has not yet been commercialized in any of these countries. Our third late-stage product candidate, CollaGUARD, is approved for commercial sale in 12 countries, but not yet approved for commercial sale in the United States and we do not know when, or if, we if will generate significant revenue from its sale in the United States in the future. We do not anticipate generating revenue from sales of XaraColl until at least the end of 2017 and we will never generate revenue from XaraColl if we do not obtain regulatory approval. While we have products approved or commercialized and available for sale in certain markets, including CollatampG, RegenePro and Septocoll, our revenues to date from these products have been limited.
Even if we do generate product sales, we may never achieve or sustain profitability. We anticipate that our operating losses will substantially increase over the next several years as we execute our plan to expand our research, development and commercialization activities, including the clinical development and planned commercialization of our product candidates, and incur the additional costs of operating as a public company. In addition, if we obtain regulatory approval of our product candidates, we may incur significant sales and marketing expenses. Because of the numerous risks and uncertainties associated with developing pharmaceutical products, we are unable to predict the extent of any future losses or when we will become profitable, if ever.
If we fail to obtain additional financing, we may be unable to complete the development and commercialization of our product candidates.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial amounts to continue the clinical development of our product candidates, including our Phase 3 clinical trials. If our product candidates are approved, we will require significant additional funds in order to launch and commercialize such product candidates in the United States and potentially in the EU. We will also need to spend substantial amounts to significantly expand our manufacturing infrastructure. Finally, we had trade and other payables of €13.2 million and deferred income of €2.0 million (representing products to be delivered for which payment has already been received) as of December 31, 2015.
Our rate of expenses will continue to increase as we advance our planned clinical trials of XaraColl, Cogenzia and CollaGUARD and expand our manufacturing facility in Saal, Germany. As a result, we will be required to seek additional sources of capital during the next 12 months or restrict certain of our expenditures to conserve capital and extend our resources. Our need for additional capital will depend significantly on the level and timing of regulatory approval and product sales, as well as the extent to which we choose to establish collaboration, co-promotion, distribution or other similar agreements for our products and product candidates. Moreover, changing circumstances may cause us to spend cash significantly faster than we currently anticipate, and we may need to spend more cash than currently expected because of circumstances beyond our control. We expect to continue to incur substantial additional operating losses as we seek regulatory approval for and commercialize XaraColl, Cogenzia and CollaGUARD and develop and seek regulatory approval for our other product candidates. If we obtain FDA approval for our products, we will incur significant sales, marketing and manufacturing expenses. In addition, we expect to incur additional expenses to add operational, financial and information systems and personnel, including personnel to support our planned product commercialization and expanded manufacturing efforts for Xaracoll, Cogenzia and CollaGuard in the United States. We also expect to incur significant costs to continue to comply with corporate governance, internal controls and similar requirements applicable to us as a public company.
Our future funding requirements, both near- and long-term, will depend on many factors, including, but not limited to:
| · | the initiation, progress, timing, costs and results of clinical trials for our product candidates, particularly XaraColl and Cogenzia, and the PMA application for CollaGUARD; |
| · | the clinical development plans we establish for these product candidates; |
| · | the number and characteristics of product candidates that we develop and seek regulatory approval for; |
| · | the outcome, timing and cost of regulatory approvals by the U.S. Food and Drug Administration, or FDA, and comparable foreign regulatory authorities, including the potential for the FDA or comparable foreign regulatory authorities to require that we perform more studies than those that we currently expect; |
| · | the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
| · | the effects of competing technological and market developments; |
| · | the timing of, and our ability to obtain, regulatory approvals for the expansion of our manufacturing facility |
| · | the cost and timing of completion of commercial-scale manufacturing activities; and |
| · | the cost of establishing sales, marketing and distribution capabilities for any product candidates for which we may receive regulatory approval in regions where we choose to commercialize our products on our own. |
We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue the development or commercialization of our product candidates or other research and development initiatives. We also could be required to seek collaborators for our product candidates at an earlier stage than would otherwise be desirable or on terms that are less favorable than might otherwise be available or relinquish or license on unfavorable terms our rights to our product candidates in markets in which we would otherwise seek to pursue development or commercialization ourselves.
Any of the above events could significantly harm our business, prospects, financial condition and results of operations and cause the price of our securities to decline.
Our existing and any future indebtedness could adversely affect our ability to operate our business.
Innocoll Germany and our wholly-owned subsidiary, Innocoll Pharmaceuticals Limited, or Innocoll Pharmaceuticals, entered into a Finance Contract with the European Investment Bank, or EIB, in March 2015 whereby the EIB has committed to lend to Innocoll Pharmaceuticals up to €25 million. We drew down €15 million of the loan commitment, and contingent on achieving the primary endpoint on either XaraColl or Cogenzia Phase 3 clinical trials, are entitled to draw down an additional €10 million. We could in the future incur additional debt obligations beyond our borrowings from the EIB. The EIB loan, our existing loan obligations, together with other similar obligations that we may incur in the future, could have significant adverse consequences, including:
| · | requiring us to dedicate a portion of our cash resources to pay for the principal and interest accrued over the 5 years loan payable at the end of the period, which may limit available resource to fund working capital, capital expenditures, product development and other general expenses; |
| · | increasing our vulnerability to adverse changes in general economic, industry and market conditions; |
| · | subjecting us to restrictive covenants that may reduce our ability to take certain corporate actions or obtain further debt or equity financing and could take up management time dealing with any consents required from lenders or other financing sources; |
| · | limiting our flexibility in planning for, or reacting to, changes in our business and the industry in which we compete; and |
| · | placing us at a competitive disadvantage compared to our competitors that have less debt or better debt servicing options. |
We may not have sufficient funds, and may be unable to arrange for additional financing, to pay the amounts due under our existing loan obligations. Failure to make payments or comply with other covenants under our existing debt could result in an event of default and acceleration of amounts due. Under our agreement with the EIB the occurrence of an event which would in the reasonable opinion of EIB have a material adverse effect on our business, operations, property or condition (financial or otherwise) or prospects compared with our condition at the date of the EIB facility agreement is an event of default. If an event of default occurs and the lender accelerates the amounts due, we may not be able to make accelerated payments, and the lender could seek to enforce security interests in the collateral securing such indebtedness, such as the shares in and assets of Innocoll Pharmaceuticals Limited, which include all of our assets. In addition, the covenants under our existing debt, and the pledge of our assets as collateral, could limit our ability to obtain additional debt financing.
Risks Related to the Clinical Development and Regulatory Approval of Our Product Candidates
Our business depends substantially on the success of certain of our lead product candidates, XaraColl and Cogenzia, which are still in development. If we are unable to successfully develop and subsequently commercialize XaraColl and Cogenzia, or experience significant delays in doing so, our business will be materially harmed.
We have invested a significant portion of our efforts and financial resources in the development of XaraColl and Cogenzia, our two lead product candidates, which have not yet been approved for commercial sale in the United States or in Europe. There remains a significant risk that we will fail to successfully develop either XaraColl or Cogenzia, or both. We initiated our Phase 3 efficacy trials for Cogenzia in the second quarter of 2015. We recently received topline data from our pivotal pharmacokinetic study in which we tested both a 200 mg and a 300 mg dose versus standard bupivacaine infiltration and commenced our Phase 3 efficacy trials for XaraColl in the third quarter of 2015. We do not expect to have final pivotal data from our XaraColl Phase 3 trials and from our Cogenzia Phase 3 trials available until the first half of and the third quarter of 2016, respectively. Even if we ultimately obtain statistically significant, positive results from our Phase 3 clinical trials, we do not expect to submit applications for marketing approval for XaraColl and Cogenzia until late 2016. The success of our product candidates will depend on several factors, including:
| · | successful completion of clinical trials; |
| · | receipt of regulatory approvals from applicable regulatory authorities; |
| · | maintaining regulatory compliance for our manufacturing facility; |
| · | manufacturing sufficient quantities in acceptable quality; |
| · | achieving meaningful commercial sales of our product candidates, if and when approved; |
| · | obtaining reimbursement from third-party payors for product candidates, if and when approved; |
| · | sourcing sufficient quantities of raw materials used to manufacture our products; |
| · | successfully competing with other products; |
| · | continued acceptable safety and effectiveness profiles for our product candidates following regulatory approval, if and when received; |
| · | obtaining and maintaining patent and trade secret protection and regulatory exclusivity; and |
| · | protecting our intellectual property rights. |
If we do not achieve one or more of these factors in a timely manner, or at all, we could experience significant delays or an inability to successfully commercialize our product candidates, which would materially harm our business and we may not be able to earn sufficient revenues and cash flows to continue our operations.
Our ability to generate future revenues depends heavily on our success in:
| · | developing and securing U.S. and/or foreign regulatory approvals for our product candidates; |
| · | manufacturing commercial quantities of our product candidates at acceptable costs; |
| · | commercializing our product candidates, assuming we receive regulatory approval; |
| · | achieving broad market acceptance of our product candidates in the medical community and with third-party payors and patients; and |
| · | pursuing clinical development of our product candidates for additional indications. |
Clinical drug development is expensive and involves uncertain outcomes, and results of earlier studies and trials may not be predictive of future trial results. If our Phase 3 clinical trials for XaraColl or Cogenzia are unsuccessful, or significantly delayed, we could be required to abandon development and our business will be materially harmed.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of our Phase 2 clinical trials for XaraColl and Cogenzia may not be predictive of the results of our planned Phase 3 clinical trials. Adverse events may occur or other risks may be discovered in Phase 3 clinical trials that will cause us to suspend or terminate our clinical trials. In some instances, there can be significant variability in safety and/or efficacy results between different trials of the same product candidate due to numerous factors, including changes in or adherence to trial protocols, differences in the size and type of patient populations and the dropout rates among clinical trial participants. Our future clinical trial results, therefore, may not demonstrate efficacy and safety sufficient to obtain regulatory approval for our product candidates.
Flaws in the design of a clinical trial may not become apparent until the clinical trial is well under way. We have limited experience in designing clinical trials and may be unable to design and execute a clinical trial to support regulatory approval. In addition, clinical trials often reveal that it is not practical or feasible to continue development efforts.
We may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to participants. Further, regulatory agencies, institutional review boards or data safety monitoring boards may at any time order the temporary or permanent discontinuation of our clinical trials or request that we cease using certain investigators in the clinical trials if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements or that they present an unacceptable safety risk to participants.
If the results of our clinical trials for our current product candidates or clinical trials for any future product candidates do not achieve their primary efficacy endpoints or raise unexpected safety issues, the prospects for approval of our product candidates will be materially adversely affected. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials have failed to achieve similar results in later clinical trials, or have ultimately failed to obtain regulatory approval of their product candidates. Many products that initially showed promise in clinical trials or earlier stage testing have later been found to cause undesirable or unexpected adverse effects that have prevented their further development. Our upcoming trials for our primary product candidates, XaraColl and Cogenzia, may not produce the results that we expect.
In addition, we may experience numerous unforeseen events that could cause our clinical trials to be delayed, suspended or terminated, or which could delay or prevent our ability to receive regulatory approval or commercialize our product candidates, including:
| · | delay or failure in reaching agreement with the FDA or comparable foreign regulatory authorities on trial designs that we are able to execute; |
| · | the number of patients required for clinical trials of our product candidates may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials at a higher rate than we anticipate; |
| · | clinical trials of our product candidates may produce negative, inconclusive or inconsistent results, and we may decide, or regulators may require us, to conduct additional clinical trials or implement a clinical hold; |
| · | we may elect or be required to suspend or terminate clinical trials of our product candidates, including based on a finding that the participants are being exposed to unacceptable health risks; |
| · | regulators or institutional review boards may not authorize us or our investigators to commence or continue a clinical trial, or conduct or continue a clinical trial at a prospective trial site; |
| · | our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all; |
| · | we may have delays in reaching or fail to reach agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites; |
| · | the cost of clinical trials of our product candidates may be greater than we anticipate; |
| · | changes in government regulation or administrative actions; |
| · | the supply or quality of our product candidates or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate; and |
| · | our product candidates may have undesirable adverse effects or other unexpected characteristics. |
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the size and nature of the patient population, the proximity of subjects to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, ability to obtain and maintain patient consents, risk that enrolled subjects will drop out before completion, competing clinical trials and clinicians’ and patients’ perceptions of the potential advantages of the drug being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating.
If we experience delays in the completion of, or termination of, any clinical trial of our product candidates, the commercial prospects of our product candidates will be materially harmed, and our ability to generate product revenues from any of these product candidates will cease or be delayed. In addition, any termination of, or delays in completing, our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may significantly harm our business, financial condition and prospects. In addition, many of the factors that cause, or lead to a delay in the commencement or completion of or early termination of, clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
We may not receive a general indication of postoperative analgesia for XaraColl, which would have an adverse effect on our ability to market XaraColl for use in surgical procedures other than those studied in our Phase 3 trials and could adversely affect our business and financial results.
The FDA strictly regulates marketing, labeling, advertising and promotion of prescription drugs. These regulations include standards and restrictions for direct-to-consumer advertising, industry-sponsored scientific and educational activities, promotional activities involving the Internet and off-label promotion. The FDA generally does not allow drugs to be promoted for “Off-label” uses — that is, uses that are not described in the product’s labeling and that differ from those that were approved by the FDA. In addition to the FDA approval required for new formulations, any new indication for an approved product also requires FDA approval.
If we are not able to obtain FDA approval for any desired future indications for XaraColl outside of open hernioplasty, our ability to effectively market and sell XaraColl may be limited, and our business may be adversely affected.
While physicians in the United States may choose, and are generally permitted to prescribe drugs for uses that are not described in the product’s labeling, and for uses that differ from those tested in clinical studies and approved by the regulatory authorities, our ability to promote our products is narrowly limited to those indications that are specifically approved by the FDA. “Off-label” uses are common across medical specialties and may constitute an appropriate treatment for some patients in varied circumstances. Regulatory authorities in the United States generally do not regulate the behavior of physicians in their choice of treatments. Regulatory authorities do, however, restrict communications by pharmaceutical companies on the subject of off-label use. Although recent court decisions suggest that certain off-label promotional activities may be protected under the First Amendment, the scope of any such protection is unclear. Moreover, while we intend to promote our products consistent with what we believe to be the approved indication for our drugs, the FDA may disagree. If the FDA determines that our promotional activities fail to comply with the FDA’s regulations or guidelines, we may be subject to warnings from, or enforcement action by, these authorities. In addition, our failure to follow FDA rules and guidelines relating to promotion and advertising may cause the FDA to issue warning letters or untitled letters, bring an enforcement action against us, suspend or withdraw an approved product from the market, require a recall or institute fines or civil fines, or could result in disgorgement of money, operating restrictions, injunctions or criminal prosecution, any of which could harm our reputation and our business.
The results of clinical trials may not support our product candidate claims. Certain of our completed Phase 2 clinical trials failed to meet their primary endpoints and involved small patient populations.
Even if our clinical trials are completed as planned, we cannot be certain that the results will support our product candidate claims or that the FDA or government authorities in other countries will agree with our conclusions regarding such results. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and the results of later clinical trials often do not replicate the results of prior clinical trials and preclinical testing.
In addition, we were unable to achieve certain primary efficacy endpoints in connection with the Phase 2 clinical studies for our two lead product candidates, XaraColl and Cogenzia. For example, in our two Phase 2 trials for XaraColl, which enrolled 53 and 50 patients, respectively, our primary endpoints were total consumption of opioid analgesia and reduction of pain based on patients’ SPI scores, respectively. Over the first 24 hours post operation, XaraColl-treated patients experienced significantly less pain in study 1 with a XaraColl dose of 100 mg (44% reduction; p = 0.001) but showed merely a trend towards significance for pain reduction in study 2 with a XaraColl dose of 200 mg (22% reduction; p = 0.080). Following a change in guidance we received from the FDA in July 2015, we discontinued the use of integrated endpoints in our Phase 3 trials for XaraColl employing a statistical analysis known as the Silverman Method, and instead chose SPI as our primary endpoint for both XaraColl trials. In addition, we plan to conduct both our Phase 3 trials using a 300 mg dose of XaraColl, a dose that was not tested in our Phase 2 trials. Since bupivacaine is believed to work locally by blocking the generation and the conduction of nerve impulses and it is considered dose dependent, we believe a higher dose should increase the local analgesic effect, even though this dose-related response is not initially supported by the results of our Phase 2 trials, which involved small patient populations. We may fail to reach these endpoints and demonstrate efficacy for XaraColl.
In our Phase 2 trial for Cogenzia involving 56 patients, Cogenzia (50 mg or 200 mg) was applied daily for up to four weeks in combination with systemic antibiotic therapy for the treatment of moderately-infected diabetic foot ulcers with the control group receiving systemic therapy alone. The primary efficacy endpoint was the percentage of patients with a clinical outcome of “clinical cure” on a study visit on day 7 of treatment. Efficacy versus the control group was not achieved. However, based on the modified intent-to-treat population, 100% of the patients who received Cogenzia and who completed the trial achieved a clinical cure two weeks after completion of treatment (test-of-cure date), compared to just 70% of patients who received systemic antibiotic therapy alone, which was a statistically significant difference (p = 0.024). Although we have selected a clinical cure measured 10 – 14 days after the last dose of treatment has been administered as our primary endpoint for our COACT Phase 3 trials for Cogenzia, we may fail to reach these endpoints and demonstrate efficacy for Cogenzia.
In addition, our completed clinical trials involved a small patient population. Because of the small sample size, the results of these clinical trials may not be indicative of future results in a larger and more diverse patient population. The clinical trial process may fail to demonstrate that our product candidates are safe for humans and effective for indicated uses. This failure could cause us to abandon a product candidate and may delay development of other product candidates. Any delay in, or termination of, our clinical trials will delay the filing of our NDAs with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues.
If our drug product candidates, such as XaraColl and Cogenzia, receive regulatory approval, we will be subject to ongoing regulatory requirements and we may face future development, manufacturing and regulatory difficulties.
Our drug product candidates, such as XaraColl and Cogenzia, if approved, will be subject to ongoing regulatory requirements for labeling, packaging, storage, advertising, promotion, sampling, record-keeping, submission of safety and other post-market approval information, importation and exportation. In addition, approved products, manufacturers and manufacturers’ facilities are required to comply with extensive FDA and European Medicines Agency, or EMA, requirements and the requirements of other similar agencies, including ensuring that quality control and manufacturing procedures conform to current Good Manufacturing Practices, or cGMP, requirements.
Accordingly, we will be required to expend time, money and effort in all areas of regulatory compliance, including manufacturing, production and quality control. We will also be required to report certain adverse reactions and production problems, if any, to the FDA and EMA and other similar agencies and to comply with certain requirements concerning advertising and promotion for our potential products.
If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or disagrees with the promotion, marketing or labeling of a product, it may impose restrictions on that product or us, including requiring withdrawal of the product from the market. If our potential products fail to comply with applicable regulatory requirements, a regulatory agency may, among other actions:
| · | issue warning letters or untitled letters; |
| · | require product recalls; |
| · | mandate modifications to promotional materials or require us to provide corrective information to healthcare practitioners; |
| · | require us or our potential future collaborators to enter into a consent decree or permanent injunction; |
| · | impose other administrative or judicial civil or criminal actions, including monetary or other penalties, or pursue criminal prosecution; |
| · | withdraw regulatory approval; |
| · | refuse to approve pending applications or supplements to approved applications filed by us or by our potential future collaborators; |
| · | impose restrictions on operations, including costly new manufacturing requirements; or |
| · | seize or detain products. |
Risks Related to Our Business and Strategy
If we fail to manufacture XaraColl, Cogenzia, CollaGUARD or our other marketed products and product candidates in sufficient quantities and at acceptable quality and cost levels, or to fully comply with cGMP or other applicable manufacturing regulations, we may face a bar to, or delays in, the commercialization of our products, breach obligations to our licensing partners or be unable to meet market demand, and lose potential revenues.
The manufacture of our products based on our collagen-based technology platform, including XaraColl and Cogenzia, requires significant expertise and capital investment. Currently, we are manufacturing all commercial and clinical supply for all of our marketed products and product candidates in our sole facility in Saal, Germany without the benefit of any redundant or backup facilities. We need to spend substantial amounts to significantly expand our manufacturing infrastructure in order to satisfy any increases in future demand. Also, substantially all of our inventory of raw material and finished goods is held at this location. We take precautions to safeguard our facility, including acquiring insurance, employing back-up generators, adopting health and safety protocols and utilizing off-site storage of computer data. However, vandalism, terrorism or a natural or other disaster, such as a fire or flood, could damage or destroy our manufacturing equipment or our inventory of raw material or finished goods, cause substantial delays in our operations, result in the loss of key information, and cause us to incur additional expenses. Our insurance may not cover our losses in any particular case. In addition, regardless of the level of insurance coverage, damage to our facilities may have a material adverse effect on our business, financial condition and operating results. In addition, our competitors have substantially greater financial, technical and other resources, such as a larger staff and experienced manufacturing organizations.
We must comply with federal, state and foreign regulations, including FDA regulations governing cGMP enforced by the FDA through its facilities inspection program and by similar regulatory authorities in other jurisdictions where we do business. These requirements include, among other things, quality control, quality assurance and the maintenance of records and documentation. For our medical device products, we are required to comply with the FDA’s Quality System Regulation, or QSR, which covers the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our medical device products.
Our facility has not yet been inspected by the FDA for cGMP compliance. If we do not successfully achieve cGMP compliance for our facility in a timely manner, commercialization of our products could be prohibited or significantly delayed. Even after cGMP compliance has been achieved, the FDA or similar foreign regulatory authorities at any time may implement new standards, or change their interpretation and enforcement of existing standards for manufacture, packaging, testing of or other activities related to our products. For our marketed medical device products, the FDA audits compliance with the QSR through periodic announced and unannounced inspections of manufacturing and other facilities. The FDA may conduct inspections or audits at any time. Similar audit rights exist in Europe and other foreign jurisdictions. Any failure to comply with applicable cGMP, QSR and other regulations may result in fines and civil penalties, suspension of production, product seizure or recall, imposition of a consent decree, or withdrawal of product approval, and would limit the availability of our product. Any manufacturing defect or error discovered after products have been produced and distributed also could result in significant consequences, including adverse health consequences, injury or death to patients, costly recall procedures, re-stocking costs, damage to our reputation and potential for product liability claims. If we are required to find a new manufacturer or supplier, the process would likely require prior FDA and/or equivalent foreign regulatory authority approval, and would be very time consuming. An inability to continue manufacturing adequate supplies of our products at our facility in Saal, Germany, could result in a disruption in the supply of our products. We have licensed the commercial rights in specified foreign territories to market and sell our products. Under those licenses, we have obligations to manufacture commercial product for our commercial partners. If we are unable to fill the orders placed with us by our commercial partners in a timely manner, we may potentially lose revenue and be in breach of our licensing obligations under agreements with them.
We have not obtained regulatory approval for any of our late-stage product candidates in the United States, so we cannot yet generate any revenues from the sales of these products in the United States.
Our late-stage product candidates, XaraColl, Cogenzia and CollaGUARD, have not yet been approved for commercial sale in the United States. We cannot commercialize product candidates in the United States without first obtaining regulatory approval from the FDA to market each product. We initiated our Phase 3 efficacy trials for Cogenzia in the second quarter of 2015. We recently received topline data from our pivotal pharmacokinetic study in which we tested both a 200 mg and a 300 mg dose versus standard bupivacaine infiltration and commenced our Phase 3 efficacy trials for XaraColl in the third quarter of 2015. We do not expect to have final pivotal data from our XaraColl Phase 3 trials and from our Cogenzia Phase 3 trials available until the first half of and the third quarter of 2016, respectively. Even if we ultimately obtain statistically significant, positive results from our Phase 3 clinical trials, we do not expect to submit applications for marketing approval for XaraColl and Cogenzia until late 2016.
Before obtaining regulatory approvals for the commercial sale of any product candidate for a target indication, we must demonstrate in non-clinical, or preclinical, studies and clinical trials, and, with respect to approval in the United States, to the satisfaction of the FDA, that the product candidate is safe and effective for use under the labeled conditions for use and that the manufacturing facilities, processes and controls are adequate. In the United States, we have not submitted an NDA for either XaraColl or Cogenzia. An NDA must include extensive preclinical and clinical data and supporting information to establish the product candidate’s safety and effectiveness for each desired indication. The NDA must also include significant information regarding the chemistry, manufacturing and controls for the product and its components, and draft labeling. Obtaining approval of an NDA is a lengthy, expensive and uncertain process, and approval may not be obtained. If we submit an NDA to the FDA, the FDA must decide whether to accept or reject the submission for filing. We cannot be certain that any of our submissions will be accepted for filing and review by the FDA, or that the FDA will approve the application if it accepts it.
Even though we have a special protocol assessment, or SPA, with the FDA for Cogenzia, the contents of which were fully reaffirmed with the FDA in the fourth quarter of 2014, this SPA is subject to change by the FDA even after we commenced our Phase 3 trials if the FDA determines that a substantial issue essential to determining the safety and effectiveness of the drug was identified after the trial began. Similarly, advice given by the EMA relating to the registrational trial for Cogenzia under the Scientific Advice procedure is only given in the light of the current scientific knowledge, based on the documentation provided by us. The Scientific Advice procedure is designed to avoid major objections regarding the design of the clinical trials being raised during evaluation of the marketing-authorization application but is not legally binding on the EMA.
Regulatory authorities outside of the United States, such as in Europe and in emerging markets, also have requirements for approval of products for commercial sale with which we must comply prior to marketing in those areas. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of our product candidates. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and obtaining regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. Approval processes vary among countries and can involve additional product testing and validation and additional administrative review periods. Seeking foreign regulatory approval could require additional non-clinical studies or clinical trials, which could be costly and time consuming. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval, and potentially may include additional risks.
The process to develop, obtain regulatory approval for, and commercialize product candidates is long, complex and costly both inside and outside of the United States, and approval is not guaranteed. Even if we successfully obtain approval from the regulatory authorities for our product candidates, any approval might significantly limit the approved indications for use, or require that precautions, contraindications or warnings be included on the product labeling that limit its commercialization, or limit its commercialization through a Risk Evaluation and Mitigation Strategy, or REMS, that restricts who may prescribe or dispense the product or imposes other significant limits to assure safe use, or require expensive and time-consuming post-approval clinical studies or surveillance as conditions of approval. Following any approval for commercial sale of our product candidates, certain changes to the product, such as changes in manufacturing processes and additional labeling claims, will be subject to additional regulatory review and approval. In addition, regulatory approval for any of our product candidates may be withdrawn. If we are unable to obtain regulatory approval for our product candidates in one or more jurisdictions, or if any approval we do obtain contains significant limitations, our target market will be reduced and our ability to realize the full market potential of our product candidates will be harmed. Furthermore, we may not be able to obtain sufficient funding or generate sufficient revenue and cash flows to continue the development of any other product candidate in the future.
If we fail to develop and commercialize additional product candidates, we may be unable to grow our business.
If we decide to pursue the development and commercialization of any additional product candidates, we may be required to invest significant resources to acquire or in-license the rights to such product candidates or to conduct product discovery activities. In addition, any other product candidates will require additional, time-consuming development efforts prior to commercial sale, including preclinical studies, extensive clinical trials and approval by the FDA and applicable foreign regulatory authorities. All product candidates are prone to the risk of failure that is inherent in therapeutic product development, including the possibility that the product candidate will not be shown to be sufficiently safe and/or effective for approval by regulatory authorities. In addition, we cannot assure you that we will be able to acquire, discover or develop any additional product candidates, or that any additional product candidates we may develop will be approved, manufactured or produced economically; successfully commercialized; or widely accepted in the marketplace or be more effective than other commercially available alternatives. Research programs to identify new product candidates require substantial technical, financial and human resources whether or not we ultimately identify any candidates. If we are unable to develop or commercialize additional product candidates, our business and prospects will suffer.
We have engaged in only limited sales of our products to date, 87% of which are to one customer for the year ended December 31, 2015.
While we are a global, commercial-stage, specialty pharmaceutical and medical device company, with late-stage development programs targeting areas of significant unmet medical needs, we have engaged in only limited sales of our products to date with approximately 87% of our sales being generated by one customer in the year ended December 31, 2015. Our products may never gain significant acceptance in the marketplace and, therefore, never generate substantial revenue or profits for the company. We must establish a market for our products and build that market through marketing campaigns to increase awareness of, and consumer confidence in, our products. If we are unable to expand our current customer base and obtain market acceptance of our products, our operations could be disrupted and our business may be materially adversely affected. Even if we achieve profitability, we may not be able to sustain or increase profitability.
We face significant competition from other pharmaceutical and medical device companies and our operating results will suffer if we fail to compete effectively.
The pharmaceutical and medical device industry is characterized by intense competition and rapid innovation. Although we believe that we hold a leading position in our understanding of collagen-based therapeutic products, our competitors may be able to develop other products that are able to achieve similar or better results. Our potential competitors include established and emerging pharmaceutical and biotechnology companies and universities and other research institutions. Many of our competitors have substantially greater financial, technical and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations and well-established sales forces. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated in our competitors. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors may succeed in developing, acquiring or licensing on an exclusive basis products that are more effective or less costly than our product candidates. We believe the key competitive factors that will affect the development and commercial success of our product candidates are efficacy, safety and tolerability profile, reliability, price and reimbursement.
We anticipate that XaraColl will compete in the United States with currently marketed bupivacaine and opioid analgesics such as morphine, as well as elastomeric bag/catheter devices intended to provide bupivacaine over several days, which have been marketed by I-FLOW Corporation (owned by Halyard Health) since 2004; and Pacira Pharmaceutical’s Exparel, a liposomal injection of bupivacaine, indicated for single-dose infiltration into the surgical site to produce postsurgical analgesia. While we are not aware of any topically applied antibiotics approved for the treatment of Diabetic Foot Infections, or DFIs, that we anticipate would compete directly with Cogenzia, DFIs are currently treated with systemic antibiotics and physicians may choose not to use Cogenzia in conjunction with these products. Once approved in the United States, CollaGUARD will compete with a number of well-accepted adhesion barriers marketed in the United States and elsewhere by well-established companies, including Sanofi’s Seprafilm®, Baxter’s Adept®, Ethicon’s Interceed® and Mast Biosurgery’s Surgiwrap®.
The financial performance of our medical device products, such as CollaGUARD, may be adversely affected by medical device tax provisions in the healthcare reform laws in the United States.
The the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, the Affordable Care Act, imposes, among other things, an annual excise tax of 2.3% on any entity that manufactures or imports medical devices offered for sale in the United States beginning with tax year 2013. Under these provisions, the Congressional Research Service predicts that the total cost to the medical device industry may be up to $20 billion over the next decade. We do not believe that CollaGUARD is currently subject to this tax based on the retail exemption under applicable Treasury Regulations. However, the availability of this exemption is subject to interpretation by the IRS, and the IRS may disagree with our analysis. In addition, future products that we manufacture, produce or import may be subject to this tax. The financial impact this tax may have on our business is unclear and there can be no assurance that our business will not be materially adversely affected by it.
If we face allegations of noncompliance with the law and encounter sanctions, our reputation, revenues and liquidity may suffer, and our products could be subject to restrictions or withdrawal from the market.
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may significantly and adversely affect our ability to commercialize and generate revenues from our products. If regulatory sanctions are applied or if regulatory approval is withdrawn, the value of our company and our operating results will be adversely affected. Additionally, if we are unable to generate revenues from our product sales, our potential for achieving profitability will be diminished and the capital necessary to fund our operations will be increased.
Even if we obtain regulatory approval for our product candidates, the products may not gain market acceptance among hospitals, physicians, health care payors, patients and others in the medical community.
Even if we obtain regulatory approval for any of our product candidates that we may develop or acquire in the future, the product may not gain market acceptance among hospitals, physicians, health care payors, patients and others in the medical community. Market acceptance of any of our product candidates for which we receive approval depends on a number of factors, including:
| · | the clinical indications for which they are approved; |
| · | the product labeling, including warnings, precautions, side effects, and contraindications that the FDA approves; |
| · | the potential and perceived advantages of our product candidates over alternative products; |
| · | relative convenience and ease of administration; |
| · | the effectiveness of our sales and marketing efforts; |
| · | acceptance by major operators of hospitals, physicians and patients of the product candidate as a safe and effective treatment; |
| · | the prevalence and severity of any side effects; |
| · | product labeling or product insert requirements of the FDA or other regulatory authorities; |
| · | any REMS that the FDA might require; |
| · | the timing of market introduction of our product candidates as well as competitive products; |
| · | the cost of treatment in relation to alternative products; and |
| · | the availability of adequate reimbursement and pricing by third-party payors and government authorities. |
If our product candidates are approved but fail to achieve market acceptance among physicians, patients, payors, or others in the medical community, we will not be able to generate significant revenues, which would have a material adverse effect on our business, prospects, financial condition and results of operations.
If our product candidates are approved, and with respect to our already approved products, we may be subject to healthcare laws, regulation and enforcement. Our failure to comply with those laws could have a material adverse effect on our results of operations and financial conditions.
Although we currently do not have any of our lead products on the market in the United States and several other key jurisdictions, if our lead-product candidates are approved, once we begin commercializing our product candidates, we may be subject to additional healthcare regulation and enforcement by the U.S. federal government and by authorities in the states and foreign jurisdictions in which we conduct our business. In certain jurisdictions outside of the United States where we currently market certain of our products, we are already subject to such regulation and enforcement. Such laws include, without limitation, state and federal anti-kickback, false claims, privacy, security, “sunshine,” and trade regulation and advertising laws and regulations. If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to penalties, including, but not limited to, civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, the exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to operate our business and our financial results.
A recall of our drug or medical device products, or the discovery of serious safety issues with our drug or medical device products, could have a significant negative impact on us.
The FDA and other relevant regulatory agencies have the authority to require or request the recall of commercialized products in the event of material deficiencies or defects in design or manufacture or in the event that a product poses an unacceptable risk to health. Manufacturers may, under their own initiative, recall a product. A government-mandated or voluntary recall by us or one of our distributors could occur as a result of an unacceptable risk to health, component failures, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our products would divert managerial and financial resources and have an adverse effect on our reputation, financial condition and operating results, which could impair our ability to produce our products in a cost-effective and timely manner.
Further, under the FDA’s medical device reporting, or MDR, regulations, we are required to report to the FDA any event which reasonably suggests that our product may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction of the same or similar device marketed by us were to recur, would likely cause or contribute to death or serious injury. The FDA also requires reporting of serious, life-threatening, unexpected and other adverse drug experiences and the submission of periodic safety reports and other information. Product malfunctions or other adverse event reports may result in a voluntary or involuntary product recall and other adverse actions, which could divert managerial and financial resources, impair our ability to manufacture our products in a cost-effective and timely manner and have an adverse effect on our reputation, financial condition and operating results. Similar reporting requirements exist in Europe and other jurisdictions.
Any adverse event involving our products could result in future voluntary corrective actions, such as recalls or customer notifications, or regulatory agency action, which could include inspection, mandatory recall or other enforcement action. Any corrective action, whether voluntary or involuntary, will require the dedication of our time and capital, distract management from operating our business and may harm our reputation and financial results.
Our medical device products, such as CollaGUARD, are subject to extensive governmental regulation, and failure to comply with applicable requirements could cause our business to suffer.
The medical device industry is regulated extensively by governmental authorities, principally the FDA and corresponding state and foreign governmental agencies. The regulations are very complex and are subject to rapid change and varying interpretations. Regulatory restrictions or changes could limit our ability to carry on or expand our operations or result in higher than anticipated costs or lower than anticipated sales. The FDA and other United States or foreign governmental agencies regulate numerous elements of our business, including:
| · | product design and development; |
| · | pre-clinical and clinical testing and trials; |
| · | establishment registration and product listing; |
| · | pre-market clearance or approval; |
| · | advertising and promotion; |
| · | marketing, manufacturing, sales and distribution; |
| · | adverse event reporting; |
| · | servicing and post-market surveillance; and |
| · | recalls and field safety corrective actions. |
Before we can market or sell a new regulated product or a significant modification to an existing product in the United States, we must obtain either marketing clearance under Section 510(k) of the Federal Food, Drug and Cosmetic Act, or FDCA, or approval of a premarket approval application, or PMA, from the FDA, unless an exemption from premarket clearance and approval applies. In the 510(k) clearance process, the FDA must determine that a proposed device is “substantially equivalent” to a device legally on the market, known as a “predicate” device, with respect to intended use, technology and safety and effectiveness, in order to clear the proposed device for marketing. Clinical data are sometimes required to support substantial equivalence. The PMA pathway requires an applicant to demonstrate the safety and effectiveness of the device based on extensive clinical data. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or certain implantable devices. Products that are approved through a PMA application generally need FDA approval before they can be modified. Similarly, some modifications made to products cleared through a 510(k) premarket notification submission may require a new 510(k) submission, including possibly with clinical data. Before we can offer our device products to any of the 31 nations within the EU and the European Free Trade Association, we must first satisfy the requirements for CE Mark clearance, a conformity mark that signifies a product has met all criteria of the relevant EU directives, especially in the areas of safety and performance. The process of obtaining regulatory clearances or approvals to market a medical device can be costly and time-consuming, and we may not be able to obtain these clearances or approvals on a timely basis, or at all for our proposed products. The initial European CE certificate for CollaGUARD was valid from October 7, 2011 to July 26, 2015.
The FDA can delay, limit or deny clearance or approval of a device for many reasons, including:
| · | our inability to demonstrate that our products are safe and effective for their intended uses; |
| · | the data from our clinical trials may not be sufficient to support clearance or approval; and |
| · | the manufacturing process or facilities we use may not meet applicable requirements. |
In addition, the FDA and other regulatory authorities may change their respective clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions which may prevent or delay approval or clearance of our products under development or impact our ability to modify our currently cleared or approved products on a timely basis.
Any delay in, or failure to receive or maintain, clearance or approval for our products under development could prevent us from generating revenue from these products or achieving profitability. Additionally, the FDA and comparable foreign regulatory authorities have broad enforcement powers. Regulatory enforcement or inquiries, or other increased scrutiny of us, could dissuade some customers from using our products and adversely affect our reputation and the perceived safety and efficacy of our products.
Failure to comply with applicable regulations could jeopardize our ability to sell our products and result in enforcement actions such as fines, civil penalties, injunctions, warning letters, recalls of products, delays in the introduction of products into the market, refusal of the FDA or other regulators to grant future clearances or approvals, and the suspension or withdrawal of existing approvals by the FDA or other regulators. Any of these sanctions could result in higher than anticipated costs or lower than anticipated sales and have a material adverse effect on our reputation, business, financial condition and operating results.
Furthermore, we may evaluate international expansion opportunities in the future for our medical device products. As we expand our operations outside of the United States and Europe, we are, and will become, subject to various additional regulatory and legal requirements under the applicable laws and regulations of the international markets we enter. These additional regulatory requirements may involve significant costs and expenditures and, if we are not able comply with any such requirements, our international expansion and business could be significantly harmed.
Modifications to our medical device products, such as CollaGUARD in Europe, may require reclassifications, new CE marking processes or may require us to cease marketing or recall the modified products until new CE marking is obtained.
The initial European CE certificate for CollaGUARD was valid from October 7, 2011 to July 26, 2015. In accordance with normal practice for device recertification, we compiled an updated Design Dossier and submitted this to our European Notified Body (TÜV SÜD, Munich, Germany) in December 2014. As part of our application for recertification, we included an updated literature-based Clinical Evaluation Report and a Post Market Clinical Follow-up (or PMCF) Plan, which rationalized the objectives of our ongoing clinical investigations in accordance with the current 2012 European Guideline for PMCF studies. However, prior to the expiration of the original certificate, the Notified Body requested that we revise our PMCF plan to additionally include a direct clinical comparison of CollaGUARD’s performance and safety compared to another CE-certified adhesion barrier. We do not believe that current European medical device regulations specifically require that our PMCF plan includes such a clinical comparison. In March 2016, we met with the Notified Body to seek clarification. At the meeting, TÜV SÜD agreed that current European medical device regulations do not specifically require that our PMCF plan includes such a clinical comparison, and agreed with our proposal for an updated PMCF based on the pilot clinical study in open myomectomy patients, as was agreed with FDA at the pre-IDE meeting. Provided TÜV SÜD determines that the clinical data from the U.S. pilot study are supportive, then recertification would be possible at that time based on an updated Technical Dossier and Clinical Evaluation. Until a new CE-certificate has been issued, we will not supply CollaGUARD to our marketing partners for sale in Europe or other affected territories.
We are highly dependent on our key personnel, and if we are not successful in attracting and retaining highly qualified personnel, we may be unable to successfully implement our business strategy.
Our ability to compete in the highly competitive pharmaceuticals industry depends upon our ability to attract and retain highly qualified managerial, scientific and medical personnel. We are highly dependent on our management, scientific, medical and operations personnel. The loss of the services of any of our executive officers or other key employees and our inability to find suitable replacements could potentially harm our business, prospects, financial condition or results of operations.
Despite our efforts to retain valuable employees, members of our management, scientific and development teams may terminate their employment with us on short notice or no notice. Although we have employment agreements with our key employees, these employment agreements provide for at-will employment, which means that any of our employees could leave our employment at any time, with or without notice. We do not maintain “key man” insurance policies on the lives of these individuals or the lives of any of our other employees. Our success also depends on our ability to continue to attract, retain and motivate highly skilled junior, mid-level and senior managers as well as junior, mid-level and senior scientific and medical personnel.
Many of the other biotechnology and pharmaceutical companies that we compete against for qualified personnel have greater financial and other resources, different risk profiles and a longer history in the industry than we do. They may also provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high quality candidates than what we can offer. If we are unable to continue to attract and retain high quality personnel, our ability to advance the development of our product candidates, obtain regulatory approval and commercialize our product candidates will be limited.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to: (i) comply with FDA regulations, (ii) provide accurate information to the FDA, (iii) comply with manufacturing standards we have established, (iv) comply with federal and state healthcare fraud and abuse laws and regulations, (v) report financial information or data accurately or (vi) disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices in the United States and in jurisdictions outside of the United States where we conduct our business. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a code of business conduct and ethics and are training our employees to abide by it, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
We will need to grow the size of our organization and we may experience difficulties in managing this growth.
As of December 31, 2015, we, together with our subsidiaries, had 116 total employees, 101 of whom are full-time. As our development and commercialization plans and strategies develop, and as we continue operating as a public company, we expect to need additional managerial, operational, sales, marketing, financial and other personnel. Future growth would impose significant added responsibilities on members of management, including:
| · | identifying, recruiting, integrating, maintaining and motivating additional employees; |
| · | managing our internal development efforts effectively, including the clinical and FDA review process for our product candidates, while complying with our contractual obligations to contractors and other third parties; and |
| · | improving our operational, financial and management controls, reporting systems and procedures. |
Our future financial performance and our ability to commercialize our product candidates will depend, in part, on our ability to effectively manage any future growth, and our management may also have to divert a disproportionate amount of its attention away from day-to-day activities in order to devote a substantial amount of time to managing these growth activities. To date, we have used the services of outside vendors to perform tasks including clinical trial management, statistics and analysis and regulatory affairs. Our growth strategy may also entail expanding our group of contractors or consultants to implement these tasks going forward. Because we rely on numerous consultants, effectively outsourcing many key functions of our business, we will need to be able to effectively manage these consultants to ensure that they successfully carry out their contractual obligations and meet expected deadlines. However, if we are unable to effectively manage our outsourced activities or if the quality or accuracy of the services provided by consultants is compromised for any reason, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for our product candidates or otherwise advance our business. There can be no assurance that we will be able to manage our existing consultants or find other competent outside contractors and consultants on economically reasonable terms, or at all. If we are not able to effectively expand our organization by hiring new employees and expanding our groups of consultants and contractors, we may not be able to successfully implement the tasks necessary to further develop and commercialize our product candidates that we develop and, accordingly, may not achieve our research, development and commercialization goals.
Certain of our employees and patents are subject to foreign laws.
A majority of our employees work in Germany and are subject to German employment law. Ideas, developments, discoveries and inventions made by such employees and consultants are subject to the provisions of the German Act on Employees’ Inventions (Gesetz über Arbeitnehmererfindungen), which regulates the ownership of, and compensation for, inventions made by employees. We face the risk that disputes can occur between us and our employees or ex-employees pertaining to alleged non-adherence to the provisions of this act that may be costly to defend and take up our management’s time and efforts whether we prevail or fail in such dispute. In addition, under the German Act on Employees’ Inventions, certain employees retained rights to patents they invented or co-invented prior to 2009. Although most of these employees have subsequently assigned their interest in these patents to us, to the extent permitted by law, there is a risk that the compensation we provided to them may be deemed to be insufficient and we may be required under German law to increase the compensation due to such employees for the use of the patents. In those cases where employees have not assigned their interests to us, we may need to pay compensation for the use of those patents. If we are required to pay additional compensation or face other disputes under the German Act on Employees’ Inventions, our results of operations could be adversely affected.
We believe that our success depends, in part, upon our ability to protect our intellectual property throughout the world. However, the laws of some foreign countries, including Germany and Ireland, may not be as comprehensive as those of the United States and may not be sufficient to protect our proprietary rights abroad. In addition, we generally do not pursue patent protection outside the United States and certain other key jurisdictions because of cost and confidentiality concerns. Accordingly, our international competitors could obtain foreign patent protection for, and market overseas, products and technologies for which we are seeking patent protection in the United States.
A variety of risks associated with marketing our product candidates internationally could materially adversely affect our business.
We, or our licensing partners, plan to seek regulatory approval for our product candidates outside of the United States and, accordingly, we expect that we will be subject to additional risks related to operating in foreign countries if we obtain the necessary approvals, including:
| · | differing regulatory requirements in foreign countries; |
| · | the potential for so-called parallel importing, which is what happens when a local seller, faced with high or higher local prices, opts to import goods from a foreign market (with low or lower prices) rather than buying them locally; |
| · | unexpected changes in tariffs, trade barriers, price and exchange controls and other regulatory requirements; |
| · | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
| · | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; |
| · | foreign taxes, including withholding of payroll taxes; |
| · | foreign currency fluctuations, which could result in increased operating expenses and reduced revenues, and other obligations incident to doing business in another country; |
| · | difficulties staffing and managing foreign operations; |
| · | workforce uncertainty in countries where labor unrest is more common than in the United States; |
| · | potential liability under the Foreign Corrupt Practices Act of 1977 or comparable foreign regulations; |
| · | challenges enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States; |
| · | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
| · | business interruptions resulting from geo-political actions, including war and terrorism. |
These and other risks associated with our, or our licensing partners’ international operations may materially adversely affect our ability to attain or maintain profitable operations.
Coverage and reimbursement may be limited or unavailable in certain market segments for our product candidates, which could make it difficult for us to sell our product candidates profitably.
Government authorities and third-party payors, such as private health insurers and health maintenance organizations, or, in some jurisdictions, statutory health insurances, decide which products they will cover and the amount of reimbursement. Reimbursement by a third-party payor may depend upon a number of factors, including the third-party payor’s determination that use of a product is:
| · | a covered benefit under its health plan; |
| · | safe, effective and medically necessary; |
| · | appropriate for the specific patient; |
| · | neither experimental nor investigational. |
Obtaining coverage and reimbursement approval for a product from a government or other third-party payor is a time-consuming and costly process that could require us to provide to the payor supporting scientific, clinical and cost-effectiveness data for the use of our products. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. If reimbursement of our future products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, we may be unable to achieve or sustain profitability.
We are approved to market certain of our products in selected foreign jurisdictions and further intend to seek approval to market our product candidates in both the United States and in selected foreign jurisdictions. If we obtain approval in one or more foreign jurisdictions for our product candidates, we will be subject to rules and regulations in those jurisdictions. In some foreign countries, particularly those in the European Union, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after obtaining marketing approval for a product candidate. In addition, market acceptance and sales of our product candidates will depend significantly on the availability of adequate coverage and reimbursement from third-party payors for our product candidates and may be affected by existing and future health care reform measures.
In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory changes to the health care system that could impact our ability to sell our products profitably. In particular, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 revised the payment methodology for many products under Medicare in the United States, which has resulted in lower rates of reimbursement. In 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, the Affordable Care Act, was enacted. This expansion in the government’s role in the U.S. healthcare industry may further lower rates of reimbursement for pharmaceutical products.
Other legislative changes have been proposed and adopted in the United States since the Affordable Care Act was enacted. On August 2, 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2012 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers up to 2% per fiscal year. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or the ATRA, which delayed for another two months the budget cuts mandated by these sequestration provisions of the Budget Control Act of 2011. On March 1, 2013, the President signed an executive order implementing sequestration, and on April 1, 2013, the 2% Medicare payment reductions went into effect. The ATRA also, among other things, reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
In Europe, the European Commission has submitted a Proposal for a Regulation of the European Parliament and the Council on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009, to replace, inter alia, Directive 93/42/EEC and to amend regulations regarding medical devices in the European Union, which could result in changes in the regulatory requirements for medical devices in Europe.
There have been, and likely will continue to be, legislative and regulatory proposals at the federal and state levels directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. We cannot predict the initiatives that may be adopted in the future. The continuing efforts of governments, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare and/or impose price controls may adversely affect:
| · | the demand for our product candidates, if we obtain regulatory approvals; |
| · | our ability to set a price that we believe is fair for our products; |
| · | our ability to generate revenues and achieve or maintain profitability; and |
| · | the level of taxes that we are required to pay. |
Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors, which may adversely affect our future profitability.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems and those of our current and future clinical research organizations, or CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such material system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development and commercialization of our product candidates could be delayed.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize our product candidates or our already commercialized products on a larger scale. For example, we may be sued if our product candidates allegedly cause injury or are found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing; defects in design; a failure to warn of dangers inherent in the product, negligence, strict liability; and a breach of warranties. Claims could also be asserted under state consumer protection acts. In Europe, medical products and medical devices may, under certain circumstances, be subject to no-fault liability. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Even a successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| · | costs to defend litigation and other proceedings; |
| · | a diversion of management’s time and our resources; |
| · | decreased demand for our product candidates; |
| · | injury to our reputation; |
| · | withdrawal of clinical trial participants; |
| · | initiation of investigations by regulators; |
| · | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| · | substantial monetary awards to trial participants or patients; |
| · | exhaustion of any available insurance and our capital resources; |
| · | the inability to commercialize our product candidates; and |
| · | a decline in our share price. |
Our inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products we develop. We currently do not carry product liability insurance covering our clinical trials. If we determine that it is necessary to procure product liability coverage due to the commercial launch of our lead product candidates after approval, we may be unable to obtain such coverage on acceptable terms, or at all. Until we obtain product liability insurance, we will have to pay any amounts awarded by a court or negotiated in a settlement, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
Our business could be adversely affected by animal rights activists.
Our business activities have involved animal testing, including preclinical testing for XaraColl, Cogenzia and CollaGUARD. Animal testing has been the subject of controversy and adverse publicity. Some organizations and individuals have attempted to stop animal testing by pressing for legislation and regulation in these areas. To the extent that the activities of such groups are successful, our business could be adversely affected.
Our international operations pose currency risks, which may adversely affect our operating results and net income.
Our operating results may be affected by volatility in currency exchange rates and our ability to effectively manage our currency transaction risks. In general, we conduct our business, earn revenues and incur costs in the local currency of the countries in which we operate. In 2015, 98% of our revenues were generated and approximately 27% of our costs were incurred in euros. As we execute our strategy to expand internationally, our exposure to currency risks will increase. We do not manage our foreign currency exposure in a manner that would eliminate the effects of changes in foreign exchange rates. Therefore, changes in exchange rates between these foreign currencies and the euro will affect our revenues, cost of goods sold, and operating margins, and could result in exchange losses in any given reporting period.
We incur currency transaction risks whenever we enter into either a purchase or a sale transaction using a different currency from the currency in which we report revenues. In such cases we may suffer an exchange loss because we do not currently engage in currency swaps or other currency hedging strategies to address this risk.
Given the volatility of exchange rates, we can give no assurance that we will be able to effectively manage our currency transaction risks or that any volatility in currency exchange rates will not have an adverse effect on our results of operations.
Failure to comply with the U.S. Foreign Corrupt Practices Act or other applicable anti-corruption legislation could result in fines, criminal penalties and an adverse effect on our business.
We operate in a number of countries throughout the world, including countries known to have a reputation for corruption. We are committed to doing business in accordance with applicable anti-corruption laws. We are subject, however, to the risk that our officers, directors, employees, agents and collaborators may take action determined to be in violation of such anti-corruption laws, including the U.S. Foreign Corrupt Practices Act of 1977, the U.K. Bribery Act 2010 and the European Union Anti-Corruption Act, as well as trade sanctions administered by the Office of Foreign Assets Control and the U.S. Department of Commerce. Any such violation could result in substantial fines, sanctions, civil and/or criminal penalties or curtailment of operations in certain jurisdictions, and might adversely affect our results of operations. In addition, actual or alleged violations could damage our reputation and ability to do business.
Global economic, political and social conditions have adversely impacted our sales and may continue to do so.
The uncertain direction and relative strength of the global economy, difficulties in the financial services sector and credit markets, continuing geopolitical uncertainties and other macroeconomic factors all affect spending behavior of potential end-users of our products. The prospects for economic growth in Europe, the United States and other countries remain uncertain and may cause end-users to further delay or reduce technology purchases. In particular, a substantial portion of our sales are made to customers in countries in Europe, which is experiencing a significant economic crisis. If global economic conditions remain volatile for a prolonged period or if European economies experience further disruptions, our results of operations could be adversely affected. The global financial crisis affecting the banking system and financial markets has resulted in a tightening of credit markets, lower levels of liquidity in many financial markets and extreme volatility in fixed income, credit, currency and equity markets. These conditions may make it more difficult for our end-users to obtain financing.
Risks Related to Our Dependence on Third Parties
We rely on third parties to conduct our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be substantially harmed.
We have engaged third-party CROs in connection with our Phase 3 clinical trials for our product candidates and will continue to engage such CROs in the future. We rely heavily on these parties for execution of our clinical trials, and we control only certain aspects of their activities. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with applicable protocol, legal, regulatory and scientific standards, and our reliance on our CROs does not relieve us of our regulatory responsibilities. We and our CROs are required to comply with current Good Clinical Practices, or cGCP requirements, which are a collection of regulations enforced by the FDA or comparable foreign regulatory authorities for product candidates in clinical development in order to protect the health, safety and welfare of patients and assume the integrity of clinical data. cGCP are also intended to protect the health, safety and welfare of study subjects through requirements such as informed consent. Regulatory authorities enforce these cGCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of these CROs fail to comply with applicable cGCP regulations, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, such regulatory authorities will determine that any of our clinical trials comply with the cGCP regulations. In addition, for drugs, our clinical trials must be conducted with products produced under current Good Manufacturing Practice, or cGMP, regulations and will require a large number of test subjects. For our devices, clinical trials must use product manufactured in compliance with design controls under the QSR. Our failure or any failure by our CROs to comply with these regulations or to recruit a sufficient number of patients may require us to repeat clinical trials, which would delay the regulatory approval process. Moreover, we may be implicated if any of our CROs violate federal or state fraud and abuse or false claims laws and regulations, or healthcare privacy and security laws.
The CROs are not employed directly by us and, except for remedies available to us under our agreements with such CROs, we cannot control whether they devote sufficient time and resources to our ongoing preclinical, clinical and nonclinical programs. These CROs may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical studies or other product development activities, which could affect their performance on our behalf. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to complete development of, obtain regulatory approval for or successfully commercialize our product candidates. As a result, our financial results and the commercial prospects for our product candidates would be harmed, our costs could increase and our ability to generate revenues could be delayed.
Switching or adding CROs involves substantial cost and requires extensive management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays may occur, which can materially impact our ability to meet our desired clinical development timelines. Although we plan to carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, prospects, financial condition and results of operations.
We rely on third parties for the supply of specified raw materials and equipment.
We rely on third parties for the timely supply of specified raw materials and equipment for the manufacture of our collagen-based products. Although we actively manage these third-party relationships to provide continuity and quality, some events which are beyond our control could result in the complete or partial failure of these goods and services. Any such failure could have a material adverse effect on our financial condition and operations.
If we are unable to establish effective marketing and sales capabilities or enter into agreements with third parties to market and sell our products, we may be unable to generate revenues from this product candidate.
In order to commercialize our products, we must build our marketing, sales and distribution capabilities. The establishment, development and training of our sales force and related compliance plans to market our products is expensive and time consuming and can potentially delay the commercial launch of our products. In the event we are not successful in developing our marketing and sales infrastructure, we may not be able to successfully commercialize our products, which would limit our ability to generate product revenues.
We currently license the commercialization rights for some of our marketed products outside of the United States, which exposes us to additional risks of conducting business in international markets.
The non-U.S. markets are an important component of existing commercialization strategy for our existing marketed products as well as part of our growth strategy for Xaracoll, Cogenzia and CollaGUARD. We have entered into commercial supply agreements for our three main commercialized products pursuant to which we exclusively supply and our partners exclusively purchase the products from us in their respective territories outside of the United States or worldwide. For Xaracoll and Cogenzia, we have entered into a licensing agreement with EUSA Pharma. For CollaGUARD, we have entered into supply agreements with partners, the most significant of which are with Takeda Pharmaceutical Company Limited, or Takeda, for 15 countries including Canada, Mexico and countries in the Commonwealth of Independent States, or CIS, where distribution of the product is subject to obtaining local marketing approvals, and Pioneer Pharma Co. Ltd., or Pioneer, for nine Asian countries, including China and its territories. For CollatampG, we have entered into an exclusive supply agreement with EUSA Pharma for all territories outside of the United States and for Septocoll, we have entered into an agreement with Biomet Orthopedics for global supply of the product. We are also manufacturing RegenePro, a bioresorbable collagen sponge for dental applications, which we supply to Biomet 3i. Our agreements require us to timely supply products that meet the agreed quality standards and require our customers to purchase products from us, in some cases in specified minimum quantities. If we fail to maintain these agreements and agreements with other partners or to enter into new distribution arrangements with selling parties, or if these parties are not successful, our revenue-generating growth potential will be adversely affected. Moreover, international business relationships subject us to additional risks that may materially adversely affect our ability to attain or sustain profitable operations, including:
| · | efforts to enter into distribution or licensing arrangements with third parties in connection with our international sales, marketing and distribution efforts may increase our expenses or divert our management’s attention from the development of product candidates; |
| · | changes in a specific country’s or region’s political and cultural climate or economic condition; |
| · | differing requirements for regulatory approvals and marketing internationally; |
| · | difficulty of effective enforcement of contractual provisions in local jurisdictions; |
| · | potentially reduced protection for intellectual property rights; |
| · | potential third-party patent rights in countries outside of the United States; |
| · | unexpected changes in tariffs, trade barriers and regulatory requirements; |
| · | economic weakness, including inflation, or political instability, particularly in non-U.S. economies and markets, including several countries in Europe; |
| · | compliance with tax, employment, immigration and labor laws for employees traveling abroad; |
| · | the effects of applicable foreign tax structures and potentially adverse tax consequences; |
| · | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue and other obligations incidental to doing business in another country; |
| · | workforce uncertainty in countries where labor unrest is more common than in the United States; |
| · | the potential for so-called parallel importing, which is what happens when a local seller, faced with high or higher local prices, opts to import goods from a foreign market (with low or lower prices) rather than buying them locally; |
| · | failure of our employees and contracted third parties to comply with Office of Foreign Asset Control rules and regulations and the Foreign Corrupt Practices Act; |
| · | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
| · | business interruptions resulting from geo-political actions, including war and terrorism, or natural disasters, including earthquakes, volcanoes, typhoons, floods, hurricanes and fires. |
These and other risks may materially adversely affect our ability to attain or sustain revenue from international markets.
We may form or seek strategic alliances in the future and we may not realize the benefits of such alliances.
We may form or seek strategic alliances, create joint ventures or collaborations or enter into licensing arrangements with third parties that we believe will complement or augment our development and commercialization efforts with respect to our product candidates and any future products that we may develop. Any of these relationships may require us to incur non-recurring and other charges, increase our near- and long-term expenditures, issue securities that dilute our existing shareholders or disrupt our management and business. In addition, we face significant competition in seeking appropriate strategic partners and the negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for our product candidates because they may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view our product candidates as having the requisite potential to demonstrate safety and efficacy. If we license products or businesses, we may not be able to realize the benefit of such transactions if we are unable to successfully integrate them with our existing operations and company culture and vice versa. We cannot be certain that, following a strategic transaction or license, we will achieve the revenues or specific net income that justifies such transaction. Any delays in entering into new strategic partnership agreements related to our product candidates could delay the development and commercialization of our product candidates in certain geographies for certain indications, which would harm our business prospects, financial condition and results of operations.
Risks Related to Our Intellectual Property
If our efforts to protect the proprietary nature of the intellectual property related to our technologies are not adequate, we may not be able to compete effectively in our market.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our technologies. Any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding our competitive position in our market.
In addition, the patent applications that we own or that we may license may fail to result in issued patents in the United States or in other foreign countries. Even if the patents do successfully issue, third parties may challenge the validity, enforceability or scope thereof, which may result in such patents being narrowed, invalidated or held unenforceable. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property or prevent others from designing around our claims. If the breadth or strength of protection provided by the issued patents and patent applications we hold with respect to our product candidates is threatened, it could dissuade companies from collaborating with us to develop, and threaten our ability to commercialize, our product candidates. Further, if we encounter delays in our clinical trials, the period of time during which we could market our product candidates under patent protection would be reduced. Since patent applications in the United States and most other countries are confidential for a period of time after filing, we cannot be certain that we were the first to file any patent application related to our product candidates. Furthermore, for applications in which all claims are entitled to a priority date before March 16, 2013, an interference proceeding can be provoked by a third-party or instituted by the United States Patent and Trademark Office, or U.S. PTO, to determine who was the first to invent any of the subject matter covered by the patent claims of our applications. For applications containing a claim not entitled to priority before March 16, 2013, there is greater level of uncertainty in the patent law with the passage of the America Invents Act (2012) which brings into effect significant changes to the U.S. patent laws that are yet untried and untested, and which introduces new procedures for challenging pending patent applications and issued patents. A primary change under this reform is creating a “first to file” system in the U.S. This will require us to be cognizant going forward of the time from invention to filing of a patent application.
In addition to the protection afforded by patents, we seek to rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable, processes for which patents are difficult to enforce and any other elements of our product discovery and development processes that involve proprietary know-how, information or technology that is not covered by patents. Although we require all of our employees to assign their inventions to us to the extent permitted by law, and require all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology to enter into confidentiality agreements, we cannot be certain that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Furthermore, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. If we are unable to prevent unauthorized material disclosure of our intellectual property to third parties, we will not be able to establish or maintain a competitive advantage in our market, which could materially adversely affect our business, operating results and financial condition.
Because we have filed a petition for reissuance of our U.S. XaraColl patent, we will be unable to enforce it unless and until the U.S. patent is reissued.
We recently submitted a petition to reissue the U.S. patent directed to XaraColl, mainly for the purposes of submitting prior art references identified in the corresponding European application. We did not submit these prior art references to the U.S. Patent and Trademark Office during the prosecution of the U.S. patent. In addition, this reissue process will allow us to pursue additional claims, e.g., claims within the scope of the originally issued claims but more tailored to our currently proposed XaraColl product. Although we do not believe these prior art references should have any substantial impact on the originally issued claims, especially with respect to the coverage of the proposed XaraColl products, there can be no assurance that any or all of the originally issued claims will be reissued. It is also uncertain whether any or all of the additional claims we have included in the petition will be granted. We anticipate the conclusion of the reissue process before we launch XaraColl in the United States, and if the reissued claims are substantially the same as the originally issued claims, there will be no intervening rights by others during the reissue period. We will be unable to enforce the XaraColl U.S. patent unless and until the U.S. patent is reissued.
There can be no assurance that a patent will reissue from the petition, or that any such patent will be enforceable and will not be challenged, invalidated or circumvented. Nevertheless, we will continue to rely upon our proprietary know-how, techniques, expertise and the decades of collective experience of our team relating to the development and manufacturing of collagen matrix products, as well as the rigorous regulatory barriers applicable to the development, manufacture, distribution and marketing of potential competing products. See “Item 4. History and Development of the Company — B. Business Overview — Intellectual Property and Exclusivity” for further information.
Third-party claims of intellectual property infringement may prevent or delay our product discovery and development efforts.
Our commercial success depends in part on our avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation involving patents and other intellectual property rights in the biotechnology and pharmaceutical industries, as well as administrative proceedings for challenging patents, including interference and reexamination proceedings before the U.S. PTO or oppositions and other comparable proceedings in foreign jurisdictions. Recently, following U.S. patent reform, new procedures includinginter partes review and post grant review have been implemented. As stated above, this reform is untried and untested and will bring uncertainty to the possibility of challenge to our patents in the future. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are developing our product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our product candidates may give rise to claims of infringement of the patent rights of others.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents of which we are currently unaware with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in issued patents that our product candidates may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If any third-party patent is held by a court of competent jurisdiction to cover the manufacturing process of our product candidates, any molecules formed during the manufacturing process or any final product itself, the holders of any such patents may be able to block our ability to commercialize the product candidate unless we obtained a license under the applicable patents, or until such patents expire or they are finally determined to be held invalid or unenforceable. Similarly, if any third-party patent is held by a court of competent jurisdiction to cover aspects of our formulations, processes for manufacture or methods of use product candidates, including combination therapy or patient selection methods, the holders of any such patent may be able to block our ability to develop and commercialize the product candidate unless we obtained a license or until such patent expires or is finally determined to be held invalid or unenforceable. In either case, such a license may not be available on commercially reasonable terms or at all. If we are unable to obtain a necessary license to a third-party patent on commercially reasonable terms, or at all, our ability to commercialize our product candidates may be impaired or delayed, which could in turn significantly harm our business.
Parties making claims against us may seek and obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize our product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, obtain one or more licenses from third parties, pay royalties or redesign our infringing products, which may be impossible or require substantial time and monetary expenditure. We cannot predict whether any such license would be available at all or whether it would be available on commercially reasonable terms. Furthermore, even in the absence of litigation, we may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we would be unable to further develop and commercialize our product candidates, which could harm our business significantly.
We may be involved in lawsuits to protect or enforce our patents, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe our patents. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that one or more of our patents is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated, held unenforceable, or interpreted narrowly and could put our patent applications at risk of not issuing. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, obtain one or more licenses from third parties, pay royalties or redesign our infringing products, which may be impossible or require substantial time and monetary expenditure.
Interference proceedings provoked by third parties or brought by the U.S. PTO may be necessary to determine the priority of inventions with respect to our patents or patent applications. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent misappropriation of our trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our securities.
Obtaining and maintaining our patent protection depends on compliance with various procedures, document submission requests, fee payments and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for noncompliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the U.S. PTO and foreign patent agencies in several stages over the lifetime of the patent. The U.S. PTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Noncompliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. In such an event, our competitors might be able to enter the market, which would have a material adverse effect on our business.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We have received confidential and proprietary information from third parties. In addition, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed confidential information of these third parties or our employees’ former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial cost and be a distraction to our management and employees.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on all of our product candidates throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but where enforcement is not as strong as that in the United States. These products may compete with our products in jurisdictions where we do not have any issued patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
Our trade secrets are difficult to protect.
Confidentiality agreements with employees and others may not adequately prevent disclosure of our trade secrets and other proprietary information and may not adequately protect our intellectual property.
Our success depends upon the skills, knowledge and experience of our scientific and technical personnel, our consultants and advisors as well as our partners, licensors and contractors. Because we operate in a highly competitive technical field of drug development, we rely in part on trade secrets to protect our proprietary technology and processes. However, trade secrets are difficult to protect. We enter into confidentiality agreements with our corporate partners, employees, consultants, sponsored researchers and other advisors. These agreements generally require that the receiving party keep confidential and not disclose to third parties all confidential information developed by the receiving party or made known to the receiving party by us during the course of the receiving party’s relationship with us. Our agreements also provide that any inventions made based solely upon our technology are our exclusive property, and we enter into assignment agreements that are recorded in patent, trademark and copyright offices around the world to perfect our rights.
These confidentiality and assignment agreements may be breached and may not effectively assign intellectual property rights to us. Our trade secrets also could be independently discovered by competitors, in which case we would not be able to prevent use of such trade secrets by our competitors. The enforcement of a claim alleging that a party illegally obtained and was using our trade secrets could be difficult, expensive and time consuming and the outcome would be unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets. The failure to obtain or maintain meaningful trade secret protection could adversely affect our competitive position.
Risks Related to the Merger
The anticipated benefits of the Merger may not be realized.
We may not realize the benefits we anticipate from the Merger. Our failure to realize those benefits could have an adverse effect on our business, results of operations or financial condition.
Your rights as a shareholder have changed as a result of the Merger.
The consummation of the Merger changed the governing law that applies to our shareholders from German law (which applied to the shares of Innocoll Germany) to Irish law (which now applies to Innocoll Ireland’s ordinary shares). Many of the principal attributes of Innocoll Germany’s shares and Innocoll Ireland’s ordinary shares are similar. However, your rights as a shareholder under Irish law differ from your prior rights as a shareholder under German law. In addition, Innocoll Ireland’s constitution differs from Innocoll Germany’s former articles of association. See “Item 14—Material Modifications to the Rights of Security Holders and Use of Proceeds.”
Innocoll Ireland will be exposed to the risk of future adverse changes in Irish and U.S. law, as well as changes in tax rates, which could materially adversely affect us, including by reducing or eliminating the anticipated benefits of the Merger.
Innocoll Ireland is subject to Irish law. As a result, Innocoll Ireland will be subject to the risk of future adverse changes in Irish law (including Irish corporate and tax law). In addition, the tax rates that Innocoll Ireland and its subsidiaries are subject to may be increased in the future.
Innocoll Ireland also is subject to the risk of future adverse changes to U.S. law and German law, as well as changes of law in other countries in which Innocoll Ireland or its subsidiaries operate.
For example, the U.S. Congress may take legislative action that could override tax treaties upon which we rely or could subject Innocoll Ireland or its subsidiaries to U.S. tax. A number of legislative proposals in recent years have sought to deny benefits or impose penalties on companies domiciled outside of the U.S. that conduct substantial business in the U.S. or whose executives with decision-making responsibility are located primarily in the U.S. We cannot predict the outcome of any specific legislative proposal.
We are subject to changes in law and other factors that may not allow us to maintain a worldwide effective corporate tax rate that is competitive in our industry.
While we believe that the Merger will not affect our ability to maintain a worldwide effective corporate tax rate that is competitive in our industry, we cannot give any assurance as to what our effective tax rate will eventually, as the Merger was only recently consummated. Also, the tax laws of Ireland, the United States, Germany, and other jurisdictions could change in the future, and such changes could cause a material change in our worldwide effective corporate tax rate. In particular, legislative action could be taken by Ireland, the United States or Germany which could override tax treaties upon which we expect to rely and adversely affect our effective tax rate. As a result, our actual effective tax rate may be materially different from our expectation.
There is a risk that the IRS will react adversely as a result of our consummation of the Merger.
Although we do not believe our decision to consummate the Merger should increase the likelihood that the IRS will seek to examine any tax years or portions thereof not examined prior to the move to Ireland, we cannot predict how the IRS will react to our decision to consummate the Merger. There can be no assurance that, as a result of the Merger, the IRS will not seek to examine other tax years or portions thereof. In addition, the IRS could seek to challenge our move to Ireland and our ability to receive benefits under the U.S./Ireland Treaty.
As a result of the Merger, we may be treated as a U.S. corporation for U.S. federal income tax purposes, which could result in adverse U.S. federal income tax consequences to us and investors.
For U.S. federal income tax purposes, a corporation generally is considered a U.S. corporation if it is created or organized in the United States or under the law of the United States or of any state thereof or the District of Columbia. Entities treated as U.S. corporations are generally subject to U.S. federal income tax on their worldwide income, and U.S. reporting and withholding tax rules may apply to dividends that they pay. Because we were formed and organized under the law of Germany, we would ordinarily not be treated for U.S. federal income tax purposes as a U.S. corporation. Section 7874 of the Code, however, contains special rules that could result in a non-U.S. corporation being taxed as a U.S. corporation for U.S. federal income tax purposes where the corporation, directly or indirectly, re-domiciles from the U.S. to another country. Under Section 7874 of the Code, as a result of our re-domiciling from the U.S. to Germany in 2013 (the “2013 Transaction”), we would be treated as a U.S. corporation for U.S. federal income tax purposes unless our “expanded affiliated group” (“EAG”) is treated as having “substantial business activities” in Germany. While we believe that we satisfied this “substantial business activities” test at the time of the 2013 Transaction (and, thus, should not be treated as a U.S. corporation for U.S. federal income tax purposes), due to the complexity of certain aspects of the law and the very fact-specific nature of the inquiry, there is no assurance that the IRS will not challenge our determination. Further, the IRS has issued recent guidance in Notice 2015-79 that provides that an EAG cannot meet the substantial business activities test with respect to a foreign country unless the foreign acquiring corporation is a tax resident of that foreign country. This new requirement for the substantial business activities test is effective for acquisitions completed on or after November 19, 2015. However, Notice 2015-79 also states that no inference is intended regarding the treatment under current law of any transaction described in Notice 2015-79 and that the IRS may challenge such transactions under applicable Code sections or judicial doctrines. In addition, there have been proposals to expand the scope of U.S. corporate tax residence and there could be prospective or retroactive legislative changes to Section 7874 of the Code that would result in us being treated as a U.S. corporation. Further, we have re-domiciled in 2016 to Ireland. If the Merger is treated as a related transaction to the 2013 Transaction, then we would be treated as a U.S. corporation for U.S. federal income tax purposes unless our EAG is treated as having substantial business activities in Ireland. In that case, we would not satisfy the substantial business activities test in Ireland, and thus, we would be treated as a U.S. corporation, even if we would have met the substantial business activities test had we remained a German corporation. Due to the complexity of certain aspects of the law and the very fact-specific nature of the inquiry, there is no assurance that the IRS will not successfully challenge our determination. If we are treated as a U.S. corporation for U.S. federal tax purposes, we could be liable for substantial U.S. federal taxes in addition to German and Irish taxes. In addition, our investors could be subject to U.S. withholding tax (including under the Foreign Account Tax Compliance Act) on the receipt of dividends from us. See the US tax discussion for the consequences to an investor if we were treated as a U.S. corporation for U.S. federal income tax purposes.
Dividends you receive may be subject to Irish dividend withholding tax.
In certain circumstances, as an Irish tax resident company, we may be required to deduct Irish dividend withholding tax (currently at the rate of 20%) from dividends paid to our shareholders. Shareholders resident in “relevant territories” (including countries that are European Union Member States (other than Ireland), the United States and other countries with which Ireland has a tax treaty) will generally not be subject to Irish withholding tax provided that, in each case, they complete certain tax forms. However, shareholders residing in other countries will generally be subject to withholding tax.
Please see “Material Tax Considerations Relating to the Transaction — Irish Tax Considerations — Withholding Tax on Dividends.”
We strongly recommend that each shareholder consults his or her own tax advisor as to the tax consequences of holding ordinary shares in and receiving dividends from Innocoll Ireland.
We incurred transaction costs in connection with the Merger.
We incurred transaction costs in connection with the Merger, which have been and will continue to be expensed as incurred. For example, we incurred costs and expenses, including professional fees, to comply with German and Irish corporate and other laws. In addition, incurred attorneys’ fees, accountants’ fees, filing fees, mailing expenses, solicitation fees, transfer agent fees, ADS termination fees and financial printing expenses in connection with the Merger.
If in the future Innocoll Ireland’s ordinary shares are not eligible for deposit and clearing within the facilities of the Depository Trust Company (“DTC”), then transactions in our securities may be disrupted.
The facilities of DTC are a widely-used mechanism that allow for rapid electronic transfers of securities between the participants in the DTC system, which include many large banks and brokerage firms.
Innocoll Ireland will assume the obligation for paying the stamp duty liability with respect to certain transfers of Innocoll Ireland ordinary shares as a result of the arrangement it entered into with DTC. Innocoll Ireland’s post-Merger constitution provides that, if stamp duty resulting from the transfer of Innocoll Ireland ordinary shares, which would otherwise be payable by the transferee, is paid by Innocoll Ireland or any subsidiary of Innocoll Ireland on behalf of or as agent for the transferee, then in those circumstances, Innocoll Ireland shall on its behalf or on behalf of any such subsidiary, be entitled to (i) seek reimbursement of the stamp duty from the transferor or transferee (at Innocoll Ireland’s or relevant subsidiary’s discretion), (ii) set-off the stamp duty against any dividends payable to the transferor or transferee (at Innocoll Ireland’s or relevant subsidiary’s discretion) and (iii) to claim a first and permanent lien on the shares on which stamp duty has been paid by Innocoll Ireland or any subsidiary for the amount of stamp duty paid. Innocoll Ireland’s lien shall extend to all dividends paid on those shares.
If in the future DTC is not obligated to accept the ordinary shares of Innocoll Ireland for deposit and clearing within its facilities at the closing and, even if DTC does initially accept the ordinary shares, it will generally have discretion to cease to act as a depository and clearing agency for the ordinary shares. If in the future DTC determines that Innocoll Ireland ordinary shares are not eligible for continued deposit and clearance within its facilities, then we believe the Innocoll Ireland ordinary shares would not be eligible for continued listing on a U.S. securities exchange and trading in the Innocoll Ireland ordinary shares would be disrupted. While we would pursue alternative arrangements to preserve our listing and maintain trading, any such disruption could have a material adverse effect on the trading price of the Innocoll Ireland ordinary shares.
Innocoll Ireland’s shareholders may be entitled to pre-emptive rights under Irish law, which could limit its ability to raise funds through future issuances of ordinary shares.
Subject to specified exceptions, including the opt-out described in Innocoll Ireland’s constitution, Irish law grants statutory pre-emptive rights to existing shareholders to subscribe for new issuances of shares in exchange for cash. The opt-out described in Innocoll Ireland’s constitution must be renewed every five years by a resolution approved by not less than 75% of the votes cast by our shareholders at a general meeting. Innocoll Ireland expects that it will seek renewal of the opt-out at an annual general meeting within five years from the adoption date of its constitution, which was adopted shortly prior to the completion of the Merger. However, it cannot be guaranteed that the pre-emptive rights opt-out will always be approved. If this opt-out is not renewed, it can make any future equity fundraising more cumbersome, costly and time consuming.
Our board of directors may be limited by the Irish Takeover Rules in its ability to defend an unsolicited takeover attempt.
Upon the listing of our ordinary shares on NASDAQ, we became subject to the Irish Takeover Panel Act, 1997, Irish Takeover Rules 2013 (Irish Takeover Rules), under which we are not be permitted to take certain actions that might “frustrate” an offer for our ordinary shares once our board of directors has received an offer, or has reason to believe an offer is or may be imminent, without the approval of more than 50% of shareholders entitled to vote at a general meeting of our shareholders or the consent of the Irish Takeover Panel. This could limit the ability of our board of directors to take defensive actions even if it believes that such defensive actions would be in our best interests or the best interests of our shareholders.
The operation of the Irish Takeover Rules may affect the ability of certain parties to acquire our ordinary shares.
Under the Irish Takeover Rules if an acquisition of ordinary shares were to increase the aggregate holding of the acquirer and its concert parties to ordinary shares that represent 30% or more of the voting rights of the company, the acquirer and, in certain circumstances, its concert parties would be required (except with the consent of the Irish Takeover Panel) to make an offer for the outstanding ordinary shares at a price not less than the highest price paid for the ordinary shares by the acquirer or its concert parties during the previous 12 months. This requirement would also be triggered by an acquisition of ordinary shares by a person holding (together with its concert parties) ordinary shares that represent between 30% and 50% of the voting rights in the company if the effect of such acquisition were to increase that person’s percentage of the voting rights by 0.05% within a 12 month period. As our ordinary shares are listed on NASDAQ, under the Irish Takeover Rules, certain separate concert parties will be presumed to be acting in concert. The board of directors of Innocoll Ireland and their relevant family members, related trusts and “controlled companies” are presumed to be acting in concert with any corporate shareholder who holds 20% or more of Innocoll Ireland.
The application of these presumptions may result in restrictions upon the ability of any of the concert parties and/or members of our board of directors to acquire more of our securities, including under the terms of any executive incentive arrangements. As our ordinary shares are listed on NASDAQ, we may consult with the Irish Takeover Panel with respect to the application of this presumption and the restrictions on the ability to acquire further securities although, we are unable to provide any assurance as to whether the Irish Takeover Panel will overrule this presumption. Accordingly the application of the Irish Takeover Rules may frustrate the ability of certain of our shareholders and directors to acquire our ordinary shares.
Risks Related to Ownership of our Securities
There has been limited trading volume for our securities.
Even though our ordinary shares have been listed on the NASDAQ Global Market, there has been limited liquidity in the market for our securities, which could make it more difficult for holders to sell our ordinary shares. Prior to the Merger, only Innocoll Germany’s ADSs, and not its ordinary shares, were listed on the NASDAQ Global Market. Upon the consummation of the Merger, we terminated Innocoll Germany’s ADS facility, and now only our ordinary shares are listed on the NASDAQ Global Market.
There can be no assurance that an active trading market for our ordinary shares will be sustained.
In addition, the stock market generally has experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of listed companies. Broad market and industry factors may negatively affect the market price of our ordinary shares, regardless of our actual operating performance. The market price and liquidity of the market for our ordinary shares that will prevail in the market may be higher or lower than the price you pay and may be significantly affected by numerous factors, some of which are beyond our control.
The price of our securities may be volatile, and you could lose all or part of your investment.
Similar to Innocoll Germany’s previously listed ADSs, the trading price of our ordinary shares is likely to be highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control, including limited trading volume. In addition to the factors discussed in this “Risk Factors” section and elsewhere in this annual report, these factors include:
| · | adverse results or delays in clinical trials; |
| · | actual or anticipated variations in our operating results and our financial position; |
| · | our failure to meet the estimates and projections of the investment community or that we may otherwise provide to the public and the publication of research reports about us or our industry; |
| · | adverse regulatory decisions or changes in laws or regulations; |
| · | introduction of new products or services offered by us or our competitors; |
| · | our inability to obtain adequate product supply; |
| · | our inability to establish collaborations, if needed; |
| · | our failure to commercialize our product candidates; |
| · | departures of key scientific or management personnel; |
| · | our ability to successfully manage our growth and enter new markets; |
| · | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| · | significant lawsuits, including patent or shareholder litigation; and |
| · | other events or factors, many of which are beyond our control. |
In addition, the stock market in general, and the NASDAQ Global Market and pharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our ordinary shares, regardless of our actual operating performance. If the market price of our ordinary shares does not exceed your purchase price, you may not realize any return on your investment in us and may lose some or all of your investment. In the past, securities class action litigation has often been instituted against companies following periods of volatility in the market price of a company’s securities. This type of litigation, if instituted, could result in substantial costs and a diversion of management’s attention and resources, which would harm our business, operating results or financial condition.
Our principal shareholders and management own a significant percentage of our ordinary shares and will be able to exert significant control over matters subject to shareholder approval.
Our executive officers, directors, board members, 5% shareholders and their affiliates owned approximately 47.5% of our voting shares as of March 16, 2016. Therefore, these shareholders, as a group, will have the ability to influence us through their ownership position. These shareholders may be able to determine, or significantly influence, all matters requiring shareholder approval. For example, these shareholders may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our shares that you may feel are in your best interest as one of our shareholders. Our board members and their affiliates beneficially owned approximately 9.1% of our outstanding ordinary shares as of March 16, 2016, after giving effect to the exercise of all outstanding options to purchase ordinary shares. See “Item 7 Major Shareholders and Related-Party Transactions—A. Major Shareholders.”
We are an emerging growth company, and we cannot be certain that the reduced reporting requirements applicable to emerging growth companies will make our securities less attractive to investors.
We are an emerging growth company, as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in this annual report and our periodic reports and proxy statements and exemptions from the requirements of holding nonbinding advisory votes on executive compensation and shareholder approval of any golden parachute payments not previously approved. We could be an emerging growth company through December 31, 2019, the last day of our fiscal year following the fifth anniversary of the 2014 date of the first sale of Innocoll Germany’s ADSs in our initial public offering, although circumstances could cause us to lose that status earlier, including if the market value of our ordinary shares held by non-affiliates exceeds $700.0 million as of any June 30 before that time or if we have total annual gross revenue of $1.0 billion or more during any fiscal year before that time, in which cases we would no longer be an emerging growth company as of the following December 31 or, if we issue more than $1.0 billion in non-convertible debt during any three-year period before that time, we would cease to be an emerging growth company immediately. We cannot predict if investors will find our securities less attractive because we may rely on these exemptions. If some investors find our securities less attractive as a result, there may be a less active trading market for our ordinary shares and our share price may be more volatile.
Under the JOBS Act, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. Innocoll AG prepared its financial statements in accordance with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or IASB, which do not have separate provisions for publicly traded and private companies. However, as Innocoll Holdings plc intends to prepare its financial statements in accordance wth U.S. GAAP, as an emerging growth company, we may be able to take advantage of the benefits of this extended transition period.
We have no present intention to pay dividends on our ordinary shares in the foreseeable future and, consequently, your only opportunity to achieve a return on your investment during that time is if the price of our ordinary shares appreciates.
We have no present intention to pay dividends on our ordinary shares in the foreseeable future. In addition, for so long as our loan obligations under the financing contract with EIB are outstanding, our ability to pay dividends is limited to certain circumstances. Any recommendation by our board to pay dividends will depend on many factors, including our financial condition, results of operations, legal requirements and other factors. In addition, if we issue securities at an equivalent value below $7.42 per ordinary share of Innocoll Ireland (based on conversion to U.S. dollars at an exchange rate of $1.1109 per euro, the official exchange rate quoted as of March 15, 2016 by the European Central Bank), we may be required to adjust the exercise price of certain outstanding options pursuant to our obligations under the 2014 Option Agreement (for more details on the 2014 Option Agreement, see “Item 5B. Liquidity and Capital Resources--Financing Activity—2014 Option Agreement”). Accordingly, if the price of our ordinary shares declines in the foreseeable future, you will incur a loss on your investment, without the likelihood that this loss will be offset in part or at all by potential future cash dividends.
Raising additional capital may cause additional dilution of the percentage ownership of our shareholders, restrict our operations, require us to relinquish rights to our technologies, products or product candidates and could cause our share price to fall.
We expect that significant additional capital may be needed in the future to continue our planned operations, including conducting clinical trials, commercialization efforts, expanding our manufacturing facility, expanded research and development activities and costs associated with operating a public company. To raise capital, we may sell ordinary shares, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell ordinary shares, convertible securities or other equity securities, investors may be materially diluted by subsequent sales. Such sales may also result in material dilution to our existing shareholders, and new investors could gain rights, preferences and privileges senior to the holders of our ordinary shares. The incurrence of indebtedness could result in increased fixed payment obligations and could involve certain restrictive covenants, such as limitations on our ability to incur additional debt and other operating restrictions that could adversely impact our ability to conduct our business. In addition, if we issue securities at an equivalent value below $7.42 per ordinary share of Innocoll Ireland (based on conversion to U.S. dollars at an exchange rate of $1.1109 per euro, the official exchange rate quoted as of March 15, 2016 by the European Central Bank), we may be required to adjust the exercise price of certain outstanding options pursuant to our obligations under the 2014 Option Agreement (for more details on the 2014 Option Agreement, see “Item 5B. Liquidity and Capital Resources--Financing Activity—2014 Option Agreement”). If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, products or product candidates, or grant licenses on terms unfavorable to us.
Substantial future sales of our securities in the public market, or the perception that these sales could occur, could cause the price of our securities to decline.
Additional sales of our securities in the public market, or the perception that these sales could occur, could cause the market price of our securities to decline. As of March 16, 2016, we had 24,346,782 ordinary shares outstanding. As of March 16, 2016, after giving effect to the number of our shares following Merger and assuming the post-effective amendment of our registration statement, we also had the resale of an aggregate of up to 12,083,761 ordinary shares registered pursuant to an effective resale registration statement that may be offered by the selling shareholders identified therein. To the extent additional ordinary shares are sold into the market, the market price of our ordinary shares could decline.
Unstable market and economic conditions may have serious adverse consequences on our business, financial condition and share price.
As widely reported, global credit and financial markets have experienced extreme disruptions in the past several years, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates and uncertainty about economic stability. There can be no assurance that further deterioration in credit and financial markets and confidence in economic conditions will not occur. Our general business strategy may be adversely affected by any such economic downturn, volatile business environment or continued unpredictable and unstable market conditions. If the current equity and credit markets deteriorate, or do not improve, it may make any necessary debt or equity financing more difficult, more costly, and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and share price and could require us to delay or abandon clinical development plans. In addition, there is a risk that one or more of our current service providers, manufacturers and other partners may not survive these difficult economic times, which could directly affect our ability to attain our operating goals on schedule and on budget.
At December 31, 2015, we had approximately €38.7 million of cash, cash equivalents and short-term investments. While we are not aware of any downgrades, material losses, or other significant deterioration in the fair value of our cash equivalents since December 31, 2015, no assurance can be given that further deterioration of the global credit and financial markets would not negatively impact our current portfolio of cash equivalents or marketable securities or our ability to meet our financing objectives. Furthermore, our share price may decline due in part to the volatility of the stock market and the general economic downturn.
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because pharmaceutical companies have experienced significant share price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
If securities or industry analysts do not publish research, or publish inaccurate or unfavorable research, about our business, our share price and trading volume could decline.
The trading market for our ordinary shares will depend in part on the research and reports that securities or industry analysts publish about us or our business. Securities and industry analysts do not currently, and may never, publish research on our company. If no securities or industry analysts commence coverage of our company, the trading price for our shares would likely be negatively impacted. In the event securities or industry analysts initiate coverage, if one or more of the analysts who covers us downgrades our shares or publishes inaccurate or unfavorable research about our business, our share price may decline. If one or more of these analysts ceases coverage of our company or fails to publish reports on us regularly, demand for our shares could decrease, which might cause our share price and trading volume to decline.
As a foreign private issuer, we are exempt from a number of rules under the U.S. securities laws and are permitted to file less information with the SEC than U.S. companies. This may limit the information available to holders of ordinary shares.
We are a “foreign private issuer,” as defined in the SEC rules and regulations, and, consequently, we are not subject to all of the disclosure requirements applicable to companies organized within the United States. For example, we are exempt from certain rules under the Exchange Act that regulate disclosure obligations and procedural requirements related to the solicitation of proxies, consents or authorizations applicable to a security registered under the Exchange Act. In addition, members of our board and our principal shareholders are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and related rules with respect to their purchases and sales of our securities. Moreover, we are not required to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. public companies. Accordingly, there may be less publicly available information concerning our company than there is for U.S. public companies.
As a foreign private issuer, we will file an annual report on Form 20-F within four months of the close of each year ended December 31 and furnish reports on Form 6-K relating to certain material events promptly after we publicly announce these events. However, we are not required to issue quarterly financial information because of the above exemptions for foreign private issuers, our shareholders will not be afforded the same protections or information generally available to investors holding shares in public companies organized in the United States.
We expect to lose our foreign private issuer status, which would then require us to comply with the Exchange Act’s domestic reporting regime and cause us to incur significant legal, accounting and other expenses.
We are currently a foreign private issuer and, therefore, we are not required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers. In order to maintain our current status as a foreign private issuer, either (a) a majority of our ordinary shares must be either directly or indirectly owned of record by non-residents of the United States or (b) (i) a majority of our executive officers or directors may not be United States citizens or residents, (ii) more than fifty-percent (50%) of our assets cannot be located in the United States and (iii) our business must be administered principally outside the United States. A foreign private issuer must determine its status on the last business day of its most recently completed second fiscal quarter. We expect to lose our status as a foreign private issuer, effective as of June 30, 2016. If a foreign private issuer no longer satisfies these requirements, it will become subject to U.S. domestic reporting requirements on the first day of its fiscal year immediately succeeding such determination. If we lost this status, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. We may also be required to make changes in our corporate governance practices in accordance with various SEC and NASDAQ Global Market rules. The regulatory and compliance costs to us under U.S. securities laws if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the cost we would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs and would make some activities highly time consuming and costly. We also expect that if we were required to comply with the rules and regulations applicable to U.S. domestic issuers, it would make it more difficult and expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified members to our board.
We may qualify as a passive foreign investment company, or “PFIC,” which could result in adverse U.S. federal income tax consequences to U.S. investors.
The treatment of U.S. holders (as defined in “Item 10. Additional Information—E. Taxation—U.S. Federal Income Tax Considerations”) of Innocoll Ireland ordinary shares could be subject to material adverse tax consequences as described herein if, at any relevant time, Innocoll Germany or Innocoll Ireland were a PFIC (as defined in “Item 10. Additional Information—E. Taxation—U.S. Federal Income Tax Considerations—PFIC Rules”). In general, if you are a U.S. holder, Innocoll Germany and/or Innocoll Ireland will be a PFIC with respect to you if for any taxable year in which you held ADSs or ordinary shares of Innocoll Germany or ordinary shares of Innocoll Ireland: (i) at least 75% of the gross income of Innocoll Germany or Innocoll Ireland, as applicable, for the taxable year is passive income or (ii) at least 50% of the value, determined on the basis of a quarterly average (looking through certain corporate subsidiaries), of the assets of Innocoll Germany or Innocoll Ireland, as applicable, is attributable to assets that produce or are held for the production of passive income. Passive income generally includes dividends, interest, rents and royalties (other than certain rents and royalties derived in the active conduct of a trade or business), annuities and gains from the disposition of assets that produce passive income.
We believe that Innocoll Germany should not be treated as having been a PFIC in any prior taxable year and should not be treated as a PFIC in the taxable year in which the Merger occurred. In addition, we believe that Innocoll Ireland should not be treated as a PFIC immediately after the Merger. However, the tests for determining PFIC status are applied annually, and it is difficult to accurately predict future income and assets relevant to this determination. Accordingly, we cannot assure U.S. holders that Innocoll Germany and Innocoll Ireland will not be or become a PFIC. Moreover, the determination of PFIC status depends, in part, on the application of complex U.S. federal income tax rules, which are subject to differing interpretations. As a result, whether Innocoll Germany or Innocoll Ireland is or will be a PFIC for any relevant taxable year cannot be predicted with certainty, and there can be no assurance that the IRS will not challenge our determination concerning our PFIC status.
If Innocoll Germany prior to the consummation of the Merger, and Innocoll Ireland, after the consummation of the Merger, were to be treated as a PFIC, except as otherwise provided by election regimes described in “Item 10. Additional Information—E. Taxation—U.S. Federal Income Tax Considerations—PFIC Rules” below, a U.S. holder would be subject to special adverse tax rules with respect to (i) “excess distributions” received on our ADS or ordinary shares of Innocoll Germany, and ordinary shares of Innocoll Ireland, as the case may be, and (ii) any gain recognized upon a sale or other disposition (including a pledge) of ADSs or ordinary shares of Innocoll Germany or ordinary shares of Innocoll Ireland, as the case may be. We urge U.S. holders to consult their own tax advisors regarding the possible application of the PFIC rules. See “Item 10. Additional Information—E. Taxation—U.S. Federal Income Tax Considerations—PFIC Rules.”
Exchange rate fluctuations may reduce the amount of U.S. dollars you receive in respect of any dividends or other distributions we may pay in the future in connection with your ordinary shares
Under Irish law, dividends and distributions may only be made from distributable reserves. Distributable reserves, broadly, means the accumulated realized profits of a company less accumulated realized losses of the company on a standalone basis. In addition, no distribution or dividend may be made unless the net assets of Innocoll Ireland are equal to, or in excess of, the aggregate of Innocoll Irelands’s called up share capital plus undistributable reserves and the distribution does not reduce Innocoll Ireland’s net assets below such aggregate. Undistributable reserves include Innocoll Ireland’s undenominated capital and the amount by which Innocoll Ireland’s accumulated unrealized profits, so far as not previously utilized by any capitalization, exceed Innocoll Ireland’s accumulated unrealized losses, so far as not previously written off in a reduction or reorganization of capital. The determination as to whether or not Innocoll Ireland has sufficient distributable reserves to fund a dividend must be made by reference to “relevant entity financial statements” of Innocoll Ireland being either the last set of unconsolidated annual audited financial statements or unaudited financial statements prepared in accordance with the Irish Companies Act, which give a “true and fair view” of Innocoll Ireland’s unconsolidated financial position and accord with accepted accounting practice.
Exchange rate fluctuations may affect the amount in U.S. dollars that our shareholders receive upon the payment of cash dividends or other distributions we declare and pay in euro, if any. Such fluctuations could adversely affect the value of our ordinary shares and, in turn, the U.S. dollar proceeds that holders receive from the sale of our ordinary shares.
U.S. investors may have difficulty enforcing civil liabilities against our company or members of our board and the experts named in this annual report.
The members of our board and certain of the experts named in this annual report are non-residents of the United States, and all or a substantial portion of the assets of such persons are located outside the United States. As a result, it may not be possible, or may be very difficult, to serve process on such persons or us in the United States or to enforce judgments obtained in U.S. courts against them or us based on civil liability provisions of the securities laws of the United States. In addition, it may not be possible to enforce court judgments obtained in the United States against us in Ireland based on the civil liability provisions of the U.S. federal or state securities laws. We have been advised by William Fry, our Irish counsel, that the United States currently does not have a treaty with Ireland providing for the reciprocal recognition and enforcement of judgments in civil and commercial matters.
The following requirements must be met before a judgment of a U.S. court will be deemed to be enforceable in Ireland: the judgment must be for a definite sum; the judgment must be final and conclusive; and the judgment must be provided by a court of competent jurisdiction.
An Irish court will also exercise its right to refuse enforcement if the U.S. judgment was obtained by fraud, if the judgment violates Irish public policy, if the judgment is in breach of natural justice or if it is irreconcilable with an earlier foreign judgment. There is some uncertainty as to whether the courts of Ireland would recognize or enforce judgments of U.S. courts obtained against us or our directors or officers based on the civil liabilities provisions of the U.S. federal or state securities laws or hear actions against us or those persons based on those laws. Therefore, a final judgment for the payment of money rendered by any U.S. federal or state court based on civil liability, whether or not based solely on U.S. federal or state securities laws, would not automatically be enforceable in Ireland.
We will continue to incur significant increased costs as a result of operating as a company whose securities are publicly traded in the United States, and our management will continue to be required to devote substantial time to new compliance initiatives.
As a company whose securities commenced trading in the United States in July 2014, we will continue to incur significant legal, accounting, insurance and other expenses that we did not previously incur. In addition, the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act and related rules implemented by the SEC and the NASDAQ Global Market have imposed various requirements on public companies, including requiring establishment and maintenance of effective disclosure and financial controls. These costs will increase at the time we are no longer an emerging growth company eligible to rely on exemptions under the JOBS Act from certain disclosure and governance requirements. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly. These laws and regulations could also make it more difficult and expensive for us to attract and retain qualified persons to serve on our board or its committees or on our management board. Furthermore, if we are unable to satisfy our obligations as a public company, we could be subject to delisting of our ordinary shares, fines, sanctions and other regulatory action and potentially civil litigation.
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal controls over financial reporting and disclosure controls and procedures. In particular, we are required, beginning with our fiscal year ending December 31, 2015, under Section 404 of the Sarbanes-Oxley Act, to perform system and process evaluations and testing of our internal controls over financial reporting to allow management and our independent registered public accounting firm to report on the effectiveness of our internal control over financial reporting. This assessment will need to include disclosure of any material weaknesses in our internal control over financial reporting identified by our management or our independent registered public accounting firm. A material weakness is a control deficiency, or combination of control deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 of the Sarbanes-Oxley Act also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting. However, for as long as we remain an emerging growth company as defined in the JOBS Act, we intend to take advantage of the exemption permitting us not to comply with the independent registered public accounting firm attestation requirement. At the time when we are no longer an emerging growth company, our independent registered public accounting firm may issue a report that is adverse in the event it is not satisfied with the level at which our controls are documented, designed or operating. Our remediation efforts may not enable us to avoid a material weakness in the future.
Our compliance with Section 404 caused us to incur substantial accounting expenses and expend significant management efforts.We may not always be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of the ordinary shares could decline, and we could be subject to sanctions or investigations by the NASDAQ Global Market, the SEC or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
In order to satisfy our obligations as a public company, we may need to hire qualified accounting and financial personnel with appropriate public company experience.
As a newly public company, we will need to establish and maintain effective disclosure and financial controls and make changes in our corporate governance practices. We may need to hire additional accounting and financial personnel with appropriate public company experience and technical accounting knowledge, and it may be difficult to recruit and retain such personnel. Even if we are able to hire appropriate personnel, our existing operating expenses and operations will be impacted by the direct costs of their employment and the indirect consequences related to the diversion of management resources from research and development efforts.
Item 4. Information on the Company
| A. | HISTORY AND DEVELOPMENT OF THE COMPANY |
The original legal predecessor of our company, Innocoll Holdings, Inc., was incorporated in Delaware under the name Innocoll, Inc. in December 1997 and renamed Innocoll Holdings, Inc. in May 2004. In July 2013, we re-domiciled Innocoll Holdings, Inc. from the United States to Germany pursuant to a contribution in-kind and share-for-share exchange into the newly formed Innocoll GmbH, a German limited liability company.
Pursuant to a notarial deed entered into between the shareholders of Innocoll Holdings, Inc. and Innocoll GmbH in July 2013, the holders of ordinary shares, preferred shares and warrants to purchase ordinary shares of Innocoll Holdings, Inc. contributed their shares and warrants by way of a contribution in kind to Innocoll GmbH in exchange for ordinary shares, preferred shares and options to purchase ordinary shares of Innocoll GmbH and as a result thereof, Innocoll Holdings, Inc. became Innocoll GmbH’s wholly-owned subsidiary. Innocoll Holdings, Inc. was subsequently liquidated and its assets consisting of subsidiary companies Innocoll Pharmaceuticals Ltd., Innocoll Technologies Ltd., both Irish companies, and Innocoll, Inc., a Delaware corporation, were distributed to Innocoll GmbH.
Pursuant to a notarial deed entered into on June 16, 2014, all shareholders of Innocoll GmbH agreed to amend and restate its articles of association and cancel and terminate all preference, redemption and cumulative dividend rights of the preferred shares (other than with respect to the series E shares regarding certain anti-dilution rights) in exchange for ordinary shares of Innocoll GmbH. On July 3, 2014, Innocoll GmbH transformed into a German stock corporation (Aktiengesellschaft or AG) in accordance with the provisions of the German Reorganization Act (Umwandlungsgesetz), and all shares of Innocoll GmbH became ordinary shares of Innocoll AG. Innocoll AG is registered in the commercial register of Regensburg, Germany under the number HRB 14298.
On July 30, 2014, Inocoll AG sold 6,500,000 ADSs of Innocoll Germany representing 490,567 ordinary shares of Innocoll Germany in our initial public offering at a price of $9.00 per ADS of Innocoll Germany, thereby raising $54.4 million after deducting underwriting discounts and commissions. On August 20, 2014, the underwriters in our initial public offering partially exercised their overallotment option to purchase an additional 186,984 ADSs of Innocoll Germany, representing 14,112 ordinary shares of Innocoll Germany, at a public offering price of $9.00 per ADS of Innocoll Germany. The sale of the overallotment option by our underwriters occurred on September 12, 2014, at which we raised additional net proceeds of approximately $1.57 million, after deducting underwriting discounts and commissions. The ADSs of Innocoll Germany sold in the initial public offering represented new shares of Innocoll Germany issued in a capital increase resolved by our shareholders for the purposes of the initial public offering on July 18, 2014.
On March 16, 2016, Innocoll AG, a German stock corporation, with American Depository Shares, or ADSs, listed on the Nasdaq Global Market, merged with Innocoll Holdings plc, a public limited company formed under Irish law, by way of a European cross-border merger with Innocoll Ireland being the surviving company. Prior to the merger Innocoll Holdings plc was a dormant company with minimal legally required share capital and related debtor on the statement of financial position. Upon the effectiveness of the Merger, we terminated Innocoll Germany’s ADS facility and each cancelled ADS effectively became an entitlement to receive one ordinary share of Innocoll Ireland. Holders of Innocoll Germany ordinary shares received 13.25 ordinary shares of Innocoll Ireland in respect of each share held of Innocoll Germany. Simultaneous with this transaction, Innocoll Ireland listed its ordinary shares on the Nasdaq Global Market under the symbol “INNL”, which we previously used for Innocoll Germany’s ADSs. The Merger effectively resulted in Innocoll Ireland becoming the publicly-traded parent of the Innocoll group of companies carrying on the same business as that conducted by Innocoll Germany prior to the Merger.
We are managed and controlled in Ireland and became an Irish tax resident as of January 1, 2014. Our principal executive offices are located at Unit 9, Block D, Monksland Business Park, Monksland, Athlone, Ireland, and our telephone number is +353 (0) 90 648 6834. Our website address is www.innocoll.com. Information contained on our website is not incorporated by reference into this annual report, and you should not consider information contained on our website to be part of this annual report or in deciding whether to purchase our ordinary shares. Our agent for service of process in the United States is Anthony Zook, Cottage 9, 3813 West Chester Pike, Newtown Square, PA 19073. XaraColl®, Cogenzia®, CollaGUARD®, our localized drug delivery technologies trademarked as CollaRx®, CollaFilm®, CollaPress™ and LiquiColl™, the Innocoll logo and other trademarks or service marks of Innocoll appearing in this annual report are our property. This annual report contains additional trade names, trademarks and service marks of other companies.
Principal Capital Expenditure
Our capital expenditures amounted to €2.9 million, €0.9 million, and €0.4 million for the years ended December 31, 2015, 2014 and 2013, respectively. In 2015, our main capital expenditure was for the extension of the production facilities located in Saal, Germany for approximately €2.6 million.
Recent Developments
Overview
We are a global, commercial stage specialty pharmaceutical and medical device company with late stage development programs targeting areas of significant unmet medical need. We utilize our proprietary collagen-based technology platform to develop our biodegradable and fully bioresorbable products and product candidates which can be broken down by the body without the need for surgical removal or applied topically. Using our proprietary processes at our manufacturing facility, we derive and purify bovine and equine collagen and then utilize our technology platform to incorporate the purified collagen into our topical and implantable products. These products combine proven therapeutics, including small molecules and biologics, with highly customized drug release profiles, localized drug delivery and superior handling properties. Innocoll will use code names to refer to its products in development and we expect to seek brand name approval for the names we currently use to refer to our products, which names may change in the future.
Our lead product candidates are XaraColl® for the treatment of post-operative pain and Cogenzia® for the treatment of diabetic foot infections, or DFIs. We initiated our Phase 3 efficacy trials for Cogenzia in both the United States and Europe in the second quarter of 2015. We also initiated our Phase 3 trials for XaraColl in the third quarter of 2015. We expect that data from those Phase 3 efficacy trials for XaraColl will be available in the first half of 2016 and for Cogenzia in the third quarter of 2016. CollaGUARD, which prevents post-surgical adhesions, has been approved in 12 countries in Asia, the Middle East and Latin America and we expect to commence the steps required for approval in the United States in the second quarter of 2016. In 2015, we generated €2.6 million of sales from four marketed products: (i) CollaGUARD, which utilizes our CollaFilm® technology, a transparent, bioresorbable collagen film for the prevention of post-operative adhesions in multiple surgical applications, including digestive, colorectal, gynecological and urological surgeries; (ii) Collatamp® Gentamicin Surgical Implant, or CollatampG, which utilizes our CollaRx® sponge technology, indicated for the treatment or prevention of post-operative infection; (iii) Septocoll®, a bioresorbable, dual-action collagen sponge, indicated for the treatment or prevention of post-operative infection, which we manufacture and supply to Biomet Orthopedics Switzerland GmbH, or Biomet; and (iv) RegenePro, a bioresorbable collagen sponge for dental applications which we manufacture and supply to Biomet 3i. We utilize our proprietary collagen-based technology platform to develop our biodegradable and bioresorbable products and product candidates. We manufacture our products in our own commercial scale facility. We have strategic partnerships in place with large international healthcare companies, such as Takeda, EUSA Pharma and Biomet, which market certain of our approved products in their applicable territory within Asia, Australia, Canada, Europe, Latin America, the Middle East, and the United States.
Our corporate headquarters are located in Athlone, Ireland.
Summaries of Our Product Candidates
XaraColl
Our first lead product candidate, XaraColl, is an implantable, bioresorbable collagen sponge that we designed to provide sustained post-operative pain relief through controlled delivery of bupivacaine at the surgical site. The current standard of care for the treatment of post-operative pain relies heavily on the use of opioids supplemented by other classes of pain medications, the combination of which is known as multi-modal pain therapy. However, 75% of patients receiving standard treatments still report inadequate post-operative pain relief and many patients report adverse events from these medications. Opioid-related adverse events, such as nausea, constipation and respiratory depression, which are potentially severe, may require additional medications or treatments and prolong a patient’s hospital stay, thereby increasing overall treatment costs significantly. Additionally, opioids are highly addictive and induce drug resistance and tolerance. Given the negative side effects and costs associated with opioid use in particular, there is increasing focus from hospitals, payors and regulators on treatments that reduce opioid use in the treatment of post-operative pain.
XaraColl has been studied in one Phase 1 and four completed Phase 2 clinical trials enrolling approximately 184 patients, including 103 patients in two Phase 2 trials in hernia repair at doses of 100 mg and 200 mg of bupivacaine. Results from both trials demonstrated that XaraColl reduces both pain intensity and opioid consumption with the 200 mg dose resulting in an overall greater combined effect at 48 hours. XaraColl-treated patients in the 100 mg dose trial experienced significantly less pain through 24 hours (44% reduction; p = 0.001), 48 hours (37% reduction; p = 0.012) and 72 hours (34% reduction; p = 0.030). In our subsequent 200 mg dose trial, XaraColl demonstrated a statistically significant reduction in opioid consumption through 24 and 48 hours (44% reduction at 24 hours, p = 0.004, and 36% reduction at 48 hours, p = 0.042), and demonstrated a statistical trend in reduction in pain intensity through 24 hours (p = 0.080). After the revised guidance we received from the FDA in July 2015, we determined that we will rely upon a primary endpoint of summed pain intensity, or SPI, in our two Phase 3 trials. Based on the results of our pivotal pharmacokinetic study in which we tested both a 200 mg and a 300 mg dose compared to standard bupivacaine infiltration, are running both Phase 3 trials in parallel, focusing only on the 300 mg dose. The FDA deemed our single-dose approach acceptable in our recent Type C meeting. Because bupivacaine is believed to work locally by blocking the generation and the conduction of nerve impulses and it is considered dose dependent, we believe a higher dose should increase the local analgesic effect. In September 2015, the first patient was dosed in both our MATRIX-1 (Multisite Assessment of PosToperative PainReduction with XaraColl) and MATRIX-2 Phase 3 studies for the treatment of postoperative pain following open hernia repair with mesh using XaraColl, Innocoll’s surgically implantable and bioresorbable bupivacaine-collagen matrix. Our MATRIX Phase 3 studies are two identical randomized, placebo-controlled, double-blinded studies to investigate the safety and efficacy of XaraColl, with pivotal data anticipated in the first half of 2016. We expect to submit an NDA for XaraColl at the beginning of the fourth quarter of 2016. We intend to build commercial capabilities in the U.S. to effectively support our brands, including XaraColl, in the U.S. In Europe, assuming positive data from our MATRIX Phase 3 studies, we plan to work with the European Medicines Agency, or EMA, to finalize our regulatory pathway based on Scientific Advice received from the EMA in late October 2015. We will, however, continue to evaluate the benefits of select partnerships or co-promotion alternatives if we believe it is in the best interest of shareholders, and the success of our brands, now or into the future.
Cogenzia
Our second lead product candidate, Cogenzia, is a topically applied, bioresorbable collagen sponge for the treatment of DFIs. Cogenzia is designed to release a high dose of gentamicin directly at the site of DFIs. There is a significant unmet medical need for more effective treatments of DFIs. Patients suffering from DFIs face a high rate of treatment failure, leading to hospitalization, and potentially limb amputation, which has a five-year mortality rate as high as 80%. Of the approximately 27 million patients globally who suffered a diabetic foot ulcer in 2014, 58%, or approximately 15 million, developed a DFI. DFIs are currently treated with systemic antibiotic therapy. However, peripheral vascular disease, or PVD, a frequent comorbidity of diabetes, leads to reduced blood flow to the extremities thereby rendering systemic antibiotic therapy less effective in this patient population. Published data demonstrates that systemic antibiotics have a treatment failure rate of approximately 30%. Patients with a DFI face hospitalization risk that is more than 55 times higher and risk of amputation more than 150 times higher than diabetic patients with uninfected foot ulcers. The direct cost of an amputation associated with the diabetic foot is estimated to be between $30,000 and $60,000, plus between $43,000 and $60,000 in costs for follow-up care over the next three years. In addition, major amputation is associated with mortality rates as high as 40% within one year and 80% within five years. We believe Cogenzia, when used in combination with standard systemic antibiotic therapy, addresses this significant unmet need and will provide substantially higher infection cure rates than obtained from systemic therapy alone. Cogenzia acts by delivering a high dose of gentamicin directly to the wound site at a concentration that achieves broad eradication of both Gram positive and Gram negative bacteria, including methicillin-resistant Staphylococcus aureus, or MRSA, all of which may be present in DFIs. Delivering gentamicin topically avoids the toxicity side effects associated with systemic dosing and enables the drug to be used in higher concentrations, thus, maximizing its effectiveness across a broader range of bacteria.
Cogenzia has been studied in a multicenter, randomized, placebo-controlled Phase 2 trial involving 56 patients with moderately infected DFIs. Cogenzia, administered in conjunction with systemic antibiotic therapy, achieved a 100% clinical cure rate compared to 70% cure rate for patients who received systemic antibiotic therapy alone, which was a statistically significant difference (p=0.024). In addition, Cogenzia achieved baseline pathogen eradication of 100% of all microbes present at the wound site for all patients treated and a reduced time to pathogen eradication (both statistically significant: p= 0.038, and p≤ 0.001, respectively) when compared to systemic therapy alone. This is a critically important outcome, because, if approved, these results provide practitioners with a comfort level that treatment with Cogenzia results in wounds that not only appear to be free of infection, but actually have achieved complete eradication of the pathogens. Treatment with systemic antibiotic therapy alone frequently results in wounds that appear to have achieved a clinical cure of infection, but still carry residual pathogens, often leading to rapid reinfection. Since a diabetic foot ulcer cannot heal in the presence of pathogens, treatment with Cogenzia has the potential to provide practitioners with a more effective wound healing platform. We have confirmed the regulatory path for Cogenzia with the FDA under an SPA, which we re-affirmed in the fourth quarter of 2014. Our protocols for these trials have also been accepted by the European Medicines Agency, or EMA, under the Scientific Advice procedure. We initiated both our COACT (COgenzia Adjuvant for Complete Therapy) trials in the second quarter of 2015 with pivotal data expected in the third quarter of 2016 if the data is sufficient. We expect to submit an NDA in the fourth quarter of 2016 and we will also seek approval from the EMA for Cogenzia at that time. In June 2015, Cogenzia received Qualified Infectious Disease Product, or QIDP, designation for the adjunctive treatment of moderate and severe diabetic foot infection from the FDA. This designation is a key provision of the Generating Antibiotic Incentives Now Act, or GAIN Act, approved by Congress in 2012 to increase the incentives for drug manufacturers to produce new antibiotics for serious and hard-to-treat bacterial and fungal infections. QIDP designation for a drug adds an additional five years of market exclusivity, which means that the company that brings the drug into commercial use is protected from generic competitors for that period. For Cogenzia, that should result in eight years of data exclusivity in the United States, which would include three years of exclusivity for new clinical investigations that were essential for approval. QIDP designation also provides potential access to priority review of marketing applications and eligibility for fast track and priority review designations by the FDA. We expect to pursue a priority review designation for Cogenzia. A priority review designation means that the FDA’s goal is to take action on an application within 6 months, compared to 10 months under standard review. However, QIDP Designation for Cogenzia does not guarantee priority review designation by the FDA.
We maintain full rights to Cogenzia in the United States and Europe and, upon obtaining marketing approval, intend to commercialize the product in the United States, and potentially in Europe, using our own specialized sales and marketing organization focused on high volume wound treatment centers, primary care physicians and podiatrists. Cogenzia has been approved in Argentina, Australia, Canada, Jordan, Mexico, Russia and Saudi Arabia. We plan to enter into partnerships to market and distribute Cogenzia in countries where we do not intend to establish our own sales force. We have filed five patent applications for Cogenzia in Australia, Canada, Europe, Japan and the United States, all of which are currently in the examination phase. If and when our patents are issued, we expect patent protection for Cogenzia in the United States and Europe to expire at the earliest in 2031.
CollaGUARD
CollaGUARD is our translucent, bioresorbable collagen film for the prevention of post-operative adhesions in multiple surgical applications, including digestive, colorectal, gynecological and urological surgeries. The global market for anti-adhesion products is difficult to accurately measure, but it was estimated at approximately $1.0billion in 2014. We believe that CollaGUARD’s unique combination of features for optimal handling, ease-of-use, hemostatic properties and anti-adhesion performance sets it apart from its competitors. Unlike other competitive products, CollaGUARD can be used in both open and laparoscopic procedures. CollaGUARD is highly robust and can withstand suturing or stapling if required during a procedure and is fully biodegradable and is designed to be safely and completely resorbed over approximately three to five weeks post implantation. CollaGUARD is also translucent which allows for constant visibility of the surgical field. In addition, CollaGUARD is highly stable at room temperature and has up to a five-year shelf life.
CollaGUARD is regulated as a Class III device in the United States and we expect it will require a single pivotal clinical trial to support premarket approval, or PMA, by the FDA. In the first quarter of 2015, we initiated a second pilot efficacy study in patients undergoing gynecological laparoscopic adhesiolysis. However, we decided to terminate the second pilot efficacy study in favor of a planned pilot efficacy study in patients undergoing myomectomy via open laparotomy to be performed in the U.S. under an Investigational Device Exemption, or IDE. We anticipate that data from this pilot study will help us to finalize the design of the U.S. pivotal study protocol. In the fourth quarter of 2015 we completed a pilot clinical study for CollaGUARD, run in the Netherlands, in patients undergoing intrauterine adhesiolysis via operative hysteroscopy. A clinical study report for this study confirmed the ease of use and safety for CollaGUARD in this patient population. We held our pre-IDE submission meeting with the FDA in the first quarter of 2016, in which we agreed on a non-clinical development plan that will be completed and reported prior to our filing a full IDE package, which we expect to do in the third quarter of 2016, with the Pilot (Feasibility) Clinical Study to be initiated immediately after approval. CollaGUARD has been approved in 12 countries in Asia, Latin America and the Middle East and we have launched the product in certain of these territories through our established distribution partners, such as Takeda, which launched and distributed CollaGUARD in Russia in 2014 and received approval in the first quarter of 2015 for the product in Belarus, Ukraine and Kazakhstan.
The initial European CE certificate for CollaGUARD was valid from October 7, 2011 to July 26, 2015. In accordance with normal practice for device recertification, we compiled an updated Design Dossier and submitted this to our European Notified Body (TÜV SÜD, Munich, Germany) in December 2014. As part of our application for recertification, we included an updated literature-based Clinical Evaluation Report and a Post Market Clinical Follow-up (or PMCF) Plan, which rationalized the objectives of our ongoing clinical investigations in accordance with the current 2012 European Guideline for PMCF studies. However, prior to the expiration of the original certificate, the Notified Body requested that we revise our PMCF plan to additionally include a direct clinical comparison of CollaGUARD’s performance and safety compared to another CE-certified adhesion barrier. We do not believe that current European medical device regulations specifically require that our PMCF plan includes such a clinical comparison. In March 2016, we met with the Notified Body to seek clarification. At the meeting, TÜV SÜD agreed that current European medical device regulations do not specifically require that our PMCF plan includes such a clinical comparison, and agreed with our proposal for an updated PMCF based on the pilot clinical study in open myomectomy patients, as was agreed with FDA at the pre-IDE meeting. Provided TÜV SÜD determines that the clinical data from the U.S. pilot study are supportive, then recertification would be possible at that time based on an updated Technical Dossier and Clinical Evaluation. Until a new CE-certificate has been issued, we will not supply CollaGUARD to our marketing partners for sale in Europe or other affected territories.
We submitted a family of patent applications aimed at protecting CollaGUARD on an international basis, including Australia, Canada, Eurasia, Europe, Japan, Mexico, and the United States, which are currently in the examination phase. If issued, these patents are expected to expire at the earliest in 2033 in the United States.
Our Products
Our current late-stage product candidate pipeline is summarized in the table below, including expected milestones:
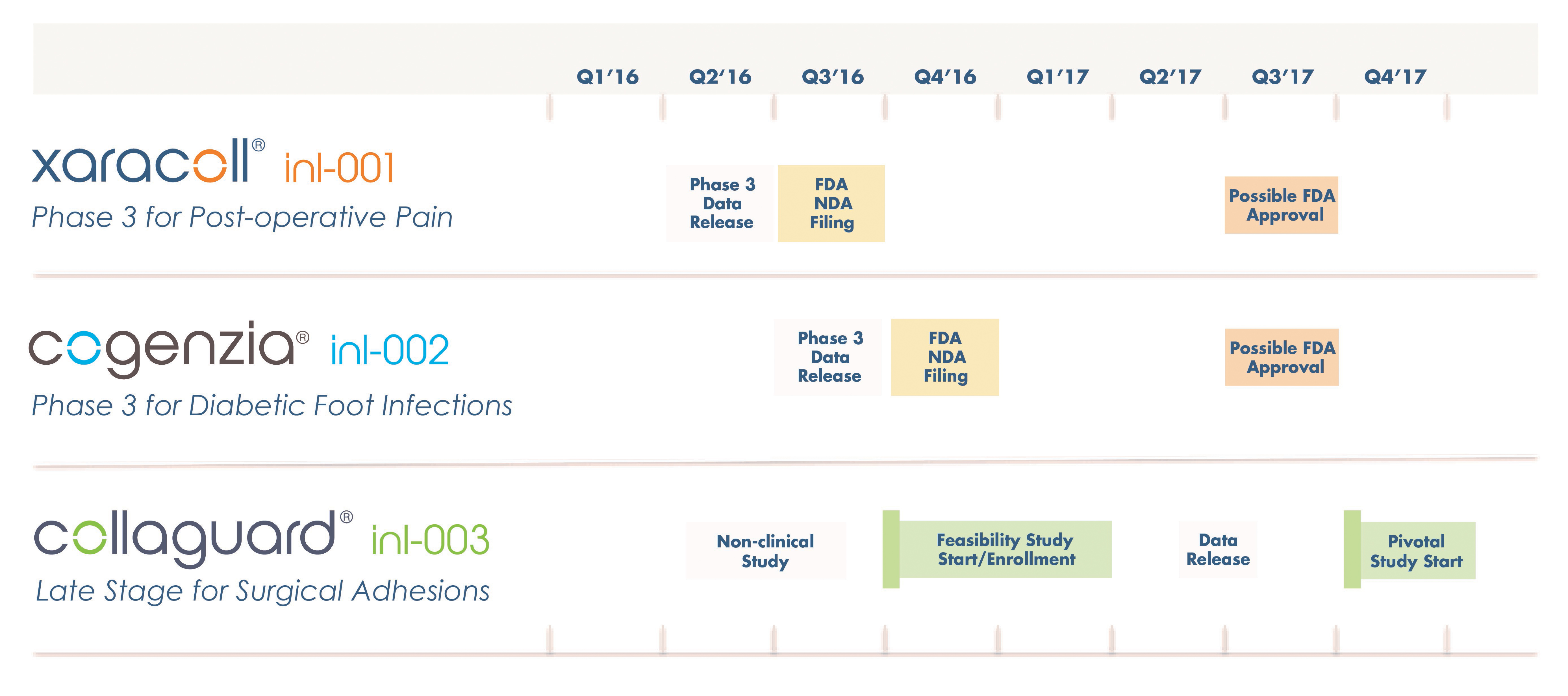
| * | | References to all events in the table denote expected timing of the events. Timing is subject to possible change. Any FDA Approval is subject to positive pivotal trial results as well as other FDA determinations on various matters. |
Innocoll will use code names to refer to its products in development and we expect to seek brand name approval for the names we currently use to refer to our products, which names may change in the future.
XaraColl
XaraColl is an implantable, bioresorbable collagen sponge designed to provide sustained post-operative pain relief through the controlled local delivery of bupivacaine at the surgical site, thereby reducing the need for systemic opioids in the treatment of post-operative pain. Bupivacaine is a local anesthetic that blocks propagation of nerve signals via sodium channel antagonism. We designed XaraColl to:
| · | provide an initial rapid burst of bupivacaine followed by slower, sustained release that delivers effective analgesia over a 48 to 72 hour period, the crucial timeframe that impacts a patient’s quality and duration of recovery; |
| · | provide safe and effective pain relief as part of a multi-modal therapy; |
| · | reduce opioid use and related adverse events; |
| · | target both the incisional and deep visceral pain components associated with moderate and major surgery; |
| · | reduce patient costs, including those associated with length of hospital stay; |
| · | be used in both open and laparoscopic surgery; and |
| · | be easily positioned at different layers within the surgical wound. |
In September 2015, the first patient was dosed in both our MATRIX-1 (Multisite Assessment of PosToperative PainReduction with XaraColl) and MATRIX-2 Phase 3 studies for the treatment of postoperative pain following open hernia repair with mesh using XaraColl, Innocoll’s surgically implantable and bioresorbable bupivacaine-collagen matrix. Our MATRIX Phase 3 studies are two identical randomized, placebo-controlled, double-blinded studies to investigate the safety and efficacy of XaraColl, with pivotal data anticipated in the first half of 2016. If the data is positive, we expect to submit an NDA for XaraColl in the second half of 2016. We intend to build commercial capabilities in the U.S. to effectively support our brands, including XaraColl, in the U.S. In Europe, assuming positive data from our MATRIX Phase 3 studies, we plan to work with the EMA to finalize our regulatory pathway based on Scientific Advice received from the EMA in late October 2015. We will, however, continue to evaluate the benefits of select partnerships or co-promotion alternatives if we believe it is in the best interest of shareholders, and the success of our brands, now or into the future. We are planning to enter into distribution and marketing arrangements with one or more partners to distribute XaraColl in Europe and worldwide.
XaraColl was developed using our proven and commercialized collagen sponge matrix technology, CollaRx. XaraColl is an intraoperative implant that is fully bioresorbable and delivers the local anesthetic, bupivacaine, directly to the surgical site. We designed XaraColl to provide post-operative analgesia for up to 72 hours after surgery. By placing XaraColl directly in contact with the traumatized tissue surfaces, surgeons can sustain an effective drug concentration at both the visceral and incisional sites of surgical wound pain.
Post-operative Pain Market Overview
There are approximately 46 million inpatient and 53 million outpatient surgeries performed in the United States each year. Traditionally, the standard of care for the treatment of post-operative pain relied heavily on the use of opioids supplemented by other classes of pain medications and delivery mechanisms, including non-steroidal anti-inflammatory drugs, or NSAIDs, acetaminophen and wound infiltration with local anesthetics, the combination of which is known as multi-modal pain therapy. However, 75% of patients receiving standard treatments still report inadequate post-operative pain relief and many patients report adverse events from these medications. Unrelieved acute pain can lead to longer post-operative recovery time, delayed ambulation, higher incidence of surgery related complications, increased intensive care unit and hospital length of stays, hospital readmission, progression from acute to chronic pain and reduced levels of daily functioning. The table below summarizes the properties of various options available for the treatment of post-operative pain.
Properties of Post-operative Pain Management
| Treatment | | Administration | | Duration of Action | | Reported Adverse Events |
| Opioids | | Constant infusion / Multiple administrations | | 4-6 hours | | Sedation, dizziness, nausea, vomiting, constipation, physical dependence, tolerance and respiratory depression |
| PCA and Elastomeric Bag Systems | | Constant infusion / Multiple administrations | | Dependent on device settings and duration of use | | May introduce catheter-related issues such as infection |
| NSAIDs | | Constant infusion / Multiple administrations | | 4-6 hours | | Increased risk of bleeding and gastrointestinal and renal complications |
| Acetaminophen | | Constant infusion / Multiple administrations | | 6-8 hours | | Liver toxicity |
| Local Anesthetics | | Single | | ≤8 hours | | Mild |
| Injectable Suspensions | | Single | | Up to 24 hours (per label) | | Mild |
| XaraColl | | Single | | Up to 48-72 hours | | Mild |
| · | Opioids. Opioids, such as morphine, are the mainstay of post-operative pain management, but are associated with a variety of unwanted and potentially severe side effects, leading healthcare practitioners to seek opioid-sparing strategies for their patients. Opioid side effects include sedation, nausea, vomiting, urinary retention, headache, itching, constipation, cognitive impairment, respiratory depression and death. Most importantly, opioids are highly addictive and induce drug resistance and tolerance. These side effects may require additional medications or treatments and prolong a patient’s stay in the post-anesthesia care unit and the hospital or ambulatory surgery center, thereby increasing overall treatment costs significantly. |
| · | PCA and Elastomeric Bag Systems.Opioids are often administered intravenously through patient-controlled analgesia, or PCA, systems in the immediate post-operative period. PCA post-operative pain management for three days can cost up to $500, excluding the costs of treating opioid complications. In an attempt to reduce opioid consumption, many hospitals employ elastomeric bag systems designed to deliver bupivacaine to the surgical area through a catheter over a period of time. This effectively extends the duration of bupivacaine in the post-operative site, but has significant shortcomings. PCA systems and elastomeric bag systems are clumsy and difficult to use, which may delay patient ambulation and introduce catheter-related issues, including infection. In addition, PCA systems and elastomeric bags require significant additional hospital resources to implement and monitor. |
| · | NSAIDs. NSAIDs are considered to be useful alternatives to opioids for acute pain relief because they do not produce respiratory depression or constipation. Despite these advantages, the use of injectable NSAIDs, such as ketorolac and ibuprofen, is severely limited in the post-operative period because they increase the risk of bleeding and gastrointestinal and renal complications. |
| · | Acetaminophen. Oral and suppository formulations of acetaminophen have long been used to manage pain, including pain in a post-operative setting. Recently, the FDA approved an IV formulation of acetaminophen, Ofirmev, for the management of post-operative pain to avoid the slow onset of the analgesic effects after oral delivery. Acetaminophen is considered not only a viable alternative to NSAIDs, but also can be combined with NSAIDs for an additive effect. However, in any formulation, acetaminophen increases the risk of liver damage or failure which limits the ability to administer it at high dosages or over an extended period of time. |
| · | Local Anesthetics. Treatment of post-operative pain typically begins at the end of surgery, with local anesthetics, such as bupivacaine, administered by local infiltration. Though this infiltration provides a base platform for the treatment of post-operative pain for the patient, efficacy of conventional bupivacaine and other available local anesthetics is limited, lasting approximately eight hours or less. As local infiltration is not practical after the surgery is complete, and as surgical pain is greatest in the first few days after surgery, additional therapeutics are required to manage post-operative pain. |
| · | Injectable Suspensions. Exparel®, a liposomal injection of bupivacaine, indicated for administration in the surgical site to produce post-operative analgesia, was commercially launched in the United States in 2012. Exparel has demonstrated significant reduction in pain intensity up to 24 hours post surgery. However, between 24 and 72 hours after administration, as per its label, Exparel showed minimal to no difference on mean pain intensity when compared to placebo. Exparel has a reported shelf life of 30 days at room temperature and must be stored under refrigerated conditions. |
| · | XaraColl’s Approach. Given the negative side effects and the costs associated with opioid use in particular, there is an increasing focus from hospitals, payors and regulators on treatments that reduce opioid use for the treatment of post-operative pain. We believe that XaraColl, in combination with NSAIDs, acetaminophen and/or local anesthetics, has the potential to become a cornerstone in the treatment of post-operative pain, focusing on opioid reduction or elimination. |
XaraColl Clinical Data
XaraColl has been studied in one Phase 1 and four completed Phase 2 clinical trials enrolling 184 patients, including 103 patients in two Phase 2 trials in hernia repair at doses of 100 mg and 200 mg of bupivacaine. Results from both trials demonstrated that XaraColl reduces both pain intensity and opioid consumption with the 200 mg dose resulting in an overall greater combined effect at 48 hours.
Phase 2 Clinical Trials in Open Hernia Repair
We conducted two independent, multicenter, double-blind, placebo-controlled trials to evaluate the efficacy and safety of XaraColl in men undergoing unilateral inguinal hernia repair by open laparotomy. Our primary and secondary efficacy endpoints in the trials included (i) SPI and (ii) the total consumption of opioid analgesia, or TOpA (mg of IV morphine equivalent), each analyzed at 24, 48 and 72 hours after surgery, respectively.
In the first trial, we randomized 53 patients to receive either two XaraColl 50 mg sponges for a total dosage of 100 mg bupivacaine (24 patients) or two placebo sponges (29 patients). In all cases, one sponge was placed beneath the hernia repair mesh and the second placed below the laparotomy incision. During patient recovery, immediate post-operative pain was treated with intravenous morphine at incremental doses of 1 to 2 mg, as needed to achieve pain control. Once patients could tolerate oral medication, they were provided with opioid tablets as rescue analgesia and instructed to take only if necessary for breakthrough pain. Patients assessed their post-operative pain intensity after aggravated movement (cough) on a 0 to 100 mm visual analogue scale at regular intervals through 72 hours. Their use of supplementary opioid medication was also recorded. Safety was assessed through 30 days. XaraColl-treated patients in this first trial reported significantly less pain through 24 hours (44% reduction; p = 0.001), 48 hours (37% reduction; p = 0.012) and 72 hours (34% reduction; p = 0.030). They also took less opioid medication through each time point (25%, 16% and 17% reduction, respectively), although these reductions were not statistically significant (p = 0.123, 0.359, and 0.396, respectively).
Based on the safety and efficacy results we observed at the 100 mg dose, we conducted a second multicenter, double-blind, placebo-controlled hernia repair trial, implanting two XaraColl 100 mg sponges for a total dosage of 200 mg. We enrolled 50 patients, 25 randomized to each group, and performed the same efficacy analyses as the first trial. In this second trial, XaraColl-treated patients took significantly less opioid medication through 24 hours (44% reduction; p = 0.004) and 48 hours (36% reduction; p = 0.042), with a trend towards statistical significance at 72 hours (31% reduction; p = 0.094). Of the patients that received XaraColl, 16% did not require any opioid rescue analgesia, compared to 0% in the control group. Although XaraColl-treated patients in the 200 mg also reported less pain (22%, 18%, and 19% reduction through 24, 48, and 72 hours, respectively) these results were not statistically significant. These results are in contrast to the pain intensity results achieved in the 100 mg trial where statistical significance was demonstrated for this parameter.
Results from the efficacy analyses are presented in the table below. p-values of less than or equal to 0.05, representing a less than a 5% probability that the observed difference occurred by chance alone, were considered statistically significant and p-values larger than 0.05, but less than or equal to 0.10, were considered a statistical trend.
Efficacy of XaraColl in Open Hernia Repair
Observed Treatment Effect vs. Placebo Control
Summed Pain Intensity (SPI)
| | | 100 mg trial (n = 53) | | | 200 mg trial (n = 50) |
| Efficacy Endpoint | | Mean
reduction | | | p-value | | | Mean reduction | | | p-value | | | | |
| 24 hours | | | 44 | % | | | 0.001 | * | | | 22 | % | | | 0.080 | ** | | | |
| 48 hours | | | 37 | % | | | 0.012 | * | | | 18 | % | | | 0.178 | | | | |
| 72 hours | | | 34 | % | | | 0.030 | * | | | 19 | % | | | 0.184 | | | | |
Total Use of Opioid Analgesia (TOpA)
| | | 100 mg trial (n = 53) | | | 200 mg trial (n = 50) |
| Efficacy Endpoint | | Mean reduction | | | p-value | | | Mean reduction | | | p-value | | | | |
| 24 hours | | | 25 | % | | | 0.123 | | | | 44 | % | | | 0.004 | * | | | |
| 48 hours | | | 16 | % | | | 0.359 | | | | 36 | % | | | 0.042 | * | | | |
| 72 hours | | | 17 | % | | | 0.396 | | | | 31 | % | | | 0.094 | ** | | | |
*statistically significant
**statistical trend
Patients experiencing an opioid-related adverse event in the United States had a 55% longer hospital stay than those without such events, a 47% higher hospitalization cost, a 36% increased risk of 30-day readmission and a 3.4 times greater risk of in-patient mortality in 2013. We also observed that at the higher 200 mg dose, 16% (4/25) of patients did not require any opioid rescue analgesia throughout the 72 hours, compared to 0% in the corresponding placebo control group. Accompanying the significantly reduced use of rescue analgesia, we also observed a 52% reduction in the number of patients who reported any adverse event commonly associated with consumption of opioids. In addition, there was an increase of 102% in time until patients first used rescue medication following discharge from the post-anesthesia care unit. Furthermore, we found that nausea events reported by XaraColl-treated patients were generally less severe and of shorter duration, while more patients in the control group received antiemetic medication for nausea. We believe that increasing the dosage of XaraColl from 200 mg to 300 mg in Phase 3, may result in a higher number of patients avoiding taking opioid medication than was observed in Phase 2 as well as a further reduction in opioid-related adverse events.
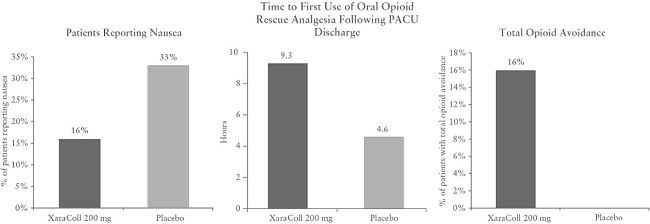
In both trials, XaraColl was generally considered safe and well tolerated. Most adverse events reported by patients were considered mild and none were considered related to XaraColl. The most commonly reported adverse events included headache, rash and gastrointestinal effects such as nausea and constipation.
Phase 2 Clinical Trial in Open Gynecological Surgery
In 2008, for the purpose of comparing the clinical performance of XaraColl with the then-leading, commercially-available product, we designed and conducted a randomized, multicenter trial to compare the efficacy of XaraColl with ON-Q. Prior studies had reported reduced use of opioid analgesia in patients fitted with ON-Q. However, the device is expensive, requires subsequent removal of the indwelling catheter by nursing/hospital staff and is relatively cumbersome.
In our trial, we randomized 27 women undergoing total abdominal hysterectomy or similar open gynecological surgery to receive either three XaraColl 50 mg implants (150 mg total dose divided between the vaginal vault, the peritoneal incision, and along the rectal sheath), or ON-Q (900 mg bupivacaine continuously perfused post-operatively over 72 hours). Following surgery, patients had access to intravenous morphine via a PCA pump as rescue analgesia for the first 24 hours and to oral opioid medication thereafter. Cumulative use of opioid analgesia was compared across treatment groups through 24, 48, 72, and 96 hours after surgery, as described in the table below.
Performance of Three XaraColl 50mg Implants (150 mg) vs. ON-Q Painbuster
900 mg Continuous Perfusion
| | | Mean Total Use of Opioid Analgesia
(TOpA; mg IV morphine equivalent) | | | | | | | |
| Time Post-operative | | On-Q 900 mg
(n=13) | | | XaraColl 150
mg
(n=14) | | | Mean reduction | | | p-value | |
| 24 hours | | | 67.0 | | | | 46.9 | | | | 30 | % | | | 0.067 | * |
| 48 hours | | | 74.9 | | | | 55.4 | | | | 26 | % | | | 0.100 | * |
| 72 hours | | | 85.4 | | | | 62.0 | | | | 27 | % | | | 0.089 | * |
| 96 hours | | | 90.8 | | | | 67.9 | | | | 25 | % | | | 0.129 | |
*statistical trend
Despite delivering only one-sixth (17%) of the total ON-Q bupivacaine dosage, we demonstrated that XaraColl was potentially more effective in providing post-operative analgesia than continuous bupivacaine infusion over 72 hours, with statistical trends towards reduced opioid consumption in favor of XaraColl through 24, 48 and 72 hours. We believe that by implanting XaraColl at different depths within the surgical wound, we may target delivery of anesthetic to the major sites of surgical trauma more efficiently than is possible with continuous infusion via an indwelling catheter.
Phase 1 Trials
We conducted an open-label, single site, Phase 1 pharmacokinetic and safety trial in 12 women undergoing total abdominal hysterectomy for a non-cancerous condition. Pharmacokinetic trials study the interactions of a drug and the body in terms of its absorption, distribution, metabolism and excretion. Three XaraColl 50 mg sponges (150 mg total dose) were implanted; the first over the vaginal vault, the second along the line of peritoneal closure, and the third along the line of rectal sheath closure. Serum samples were obtained through 96 hours for pharmacokinetic analysis and safety was assessed through 30 days. Patients were maintained on a non-opioid oral analgesic regimen according to institutional standards through 96 hours and also given access to intravenous morphine via PCA during the first 24 hours. Pain intensity was assessed at regular intervals using a 100 mm visual analog scale. XaraColl exhibited a biphasic and sustained release pharmacokinetic profile with an early peak typically observed within the first 2 hours followed by a second, generally higher peak up to 24 hours later. The individual maximum serum concentrations ranged from 0.14 to 0.44 μg/ml (mean 0.22 μg/ml), which are well below the accepted neuro- and cardio-toxicity thresholds for bupivacaine. Patient use of PCA morphine for breakthrough pain compared favorably with institutional experience and pain scores were generally low. XaraColl was considered safe and well tolerated at a dose of 150 mg.
Phase 3 Pharmoacokinetic & Safety Trials
In the second half of 2014, we commenced a multicenter, blinded, randomized, single dose study in the United States in subjects scheduled for elective inguinal hernioplasty via open laparotomy. The objectives of this study were to determine the pharmacokinetics and relative bioavailability, and to evaluate the safety of XaraColl 200 mg and XaraColl 300 mg as compared to the bupivacaine 150 mg infiltrate containing epinephrine. All subjects had measurable bupivacaine plasma concentrations 30 minutes after sponge implantation or infiltration. Mean bupivacaine plasma concentrations were comparable between the XaraColl 200 mg group and the bupivacaine 150 mg infiltration group through the first 24 hours after study drug administration. From approximately 25 hours after dosing through the end of the 96-hour observation period, mean bupivacaine plasma concentrations were higher in the XaraColl 200 mg group compared with the bupivacaine 150 mg infiltration group suggesting the availability of more drug over the observation period in the XaraColl group compared with the bupivacaine 150 mg infiltration group.
Mean bupivacaine plasma concentrations were higher in the XaraColl 300 mg group compared with the XaraColl 200 mg and the bupivacaine 150 mg infiltration group throughout the 96-hour observation period. Mean plasma concentrations in the XaraColl 300 mg group peaked at 3 hours, and bupivacaine concentrations in this group were sustained above the peak level attained in both the bupivacaine 150 mg infiltration and XaraColl 200 mg groups from 1 hour through 24 hours after implantation/administration of study drug. At 72 hours and 96 hours, mean plasma concentrations of bupivacaine in the XaraColl 300 mg were nearly double that of the XaraColl 200 mg group. The highest individual bupivacaine plasma concentration in this study was 777 ng/mL for a subject in the XaraColl 300 mg group. This concentration is 2.5 times lower than the threshold concentration that has been associated with signs and symptoms of bupivacaine toxicity (2000 ng/mL). Areas under the plasma concentration time curve (AUC) were dose proportional for XaraColl 200 mg and XaraColl 300 mg relative to the bupivacaine 150 mg infiltration. The relative maximum concentrations (Cmax) for XaraColl 200 mg and XaraColl 300 mg were approximately 80% of the Cmax for the bupivacaine 150 mg infiltrate. These findings suggest slower release of bupivacaine over time from the XaraColl bupivacaine sponges compared with the bupivacaine infiltrate.
XaraColl was well tolerated. No adverse event was considered related to study drug and no adverse event met the criteria for serious. No subject was discontinued due to an adverse event.
The FDA has reviewed the data from this study and agreed with with Innocoll’s decision to proceed with the evaluation of the single 300mg dose of XaraColl in the current Phase 3 program.
Phase 3 Development and Registration Trials
In September 2015, we commenced both our MATRIX-1 and MATRIX-2 Phase 3 studies for the treatment of postoperative pain following open hernia repair with mesh using XaraColl, Innocoll’s surgically implantable and bioresorbable bupivacaine-collagen matrix. The MATRIX Phase 3 studies are two identical randomized, placebo-controlled, double-blind studies to investigate the safety and efficacy of a surgically implantable and resorbable bupivacaine-collagen matrix. Each study is expected to enroll approximately 300 patients aged 18 and older in the United States. Patients with a unilateral inguinal hernia undergoing open hernioplasty with mesh placement will be treated in one of two arms per study with either three 100 mg XaraColl matrices for a total dose of 300 mg of bupivacaine hydrochloride, or three placebo matrices. The matrices are placed at the site of the hernia repair in order to provide local levels of bupivacaine directly at the location of surgical trauma. The primary efficacy endpoint in both Phase 3 studies is SPID (time-weighted sum of pain intensity difference) over 24 hours comparing the XaraColl matrix to placebo. Additional endpoints include SPID at 48 and 72 hours as well as total opioid use at 24, 48 and 72 hours. Safety will be evaluated through the collection of adverse events through 30 days postoperatively. The FDA has approved this study protocol and we commenced testing XaraColl at the 300 mg dose in the third quarter of 2015, with pivotal data anticipated in the first half of 2016. We previously received topline data from our pivotal pharmacokinetic study in which we tested both a 200 mg and a 300 mg dose versus standard bupivacaine infiltration which supports the selection of a 300 mg dose of XaraColl to be tested in our MATRIX Phase 3 efficacy trials. We had initially planned to conduct our Phase 3 trials using a single, integrated endpoint, validated in pain studies, that integrates and weights the patient’s pain score and use of rescue analgesia equally, known as the Silverman method so that the results can take into account a patient’s choice to suffer more pain or take higher dosages of rescue analgesia. However, in accordance with recommendations that we received from the FDA in connection with our recent Type C meeting, we are not using an integrated endpoint in our Phase 3 trials and instead rely upon a primary endpoint of SPIDfor both trials.
We also increased the number of patients from 240 to 300 to ensure that the safety database from our studies will include no less than 500 subjects, as requested by the FDA. In planning our approach for Phase 3, we carefully considered our Phase 2 data to develop a risk-minimized strategy. We decided to conduct our Phase 3 trials exclusively in patients undergoing open hernia repair, where XaraColl has already demonstrated statistically significant efficacy in Phase 2. We believe that, given that bupivacaine is believed to work locally by blocking the generation and the conduction of nerve impulses and is considered dose dependent, testing a higher dose of 300 mg could lead to further reduction in pain intensity and opioid consumption as well as extension of the duration of effective post-operative analgesia.
We intend to build commercial capabilities in the U.S. to effectively support our brands, including XaraColl, in the U.S. We will, however, continue to evaluate the benefits of select partnerships or co-promotion alternatives if we believe it is in the best interest of shareholders, and the success of our brands, now or into the future. We intend to expand XaraColl into markets outside the United States, including Europe, and we will conduct any additional clinical studies required to support such marketing authorization applications.
Cogenzia
Cogenzia, is a topically applied, bioresorbable collagen sponge for the treatment of DFIs. We designed Cogenzia to release a high dose of gentamicin directly at the site of DFIs. After surgical debridement to remove any necrotic tissues, the Cogenzia sponge is applied daily to the DFI and covered with a secondary wound dressing. Cogenzia is highly flexible and will take the shape of the wound bed, minimizing exudate build-up. A high local concentration of gentamicin would penetrate the wound directly, delivering an optimal concentration of drug precisely where it is needed with minimal systemic absorption. The type-1 collagen in Cogenzia, manufactured using our proprietary technologies, is a natural and well-established biocompatible material that can help stimulate the growth of new tissue in the wound bed and accelerate the natural process of wound healing.
Gentamicin is typically delivered systemically which limits maximum possible dosage levels due to its adverse effects and toxicity risks on patients at higher dosage levels. Therefore, it has historically only been used to treat Gram negative bacteria. However, gentamicin’s antibacterial efficacy is concentration dependent, and at higher dose levels has been shown to provide broad spectrum coverage of both Gram positive and Gram negative bacteria including methicillin-resistant Staphylococcus aureus, or MRSA, all of which may be present in DFIs.
The chart below compares published information of the estimated gentamicin concentration levels achievable by delivering gentamicin locally with Cogenzia compared to the peak safe serum levels obtained from systemic delivery. In a published study by Stemberger et al., it was demonstrated that gentamicin begins to achieve broad spectrum microbial coverage at 16 μg/ml and reaches complete broad spectrum coverage at concentrations of >512 μg/ml, both of which exceed the maximum safe peak serum level of gentamicin when delivered systemically (10-12 μg/ml). According to a publication by Dr. Benjamin Lipsky et al., Cogenzia is expected to deliver local concentrations of gentamicin of approximately 1000 μg/ml, which exceeds the concentration required to achieve broad spectrum coverage facilitating eradication of pathogens present at the infected wound site.
Comparison of Achievable
Local vs. Systemic Concentrations of Gentamicin
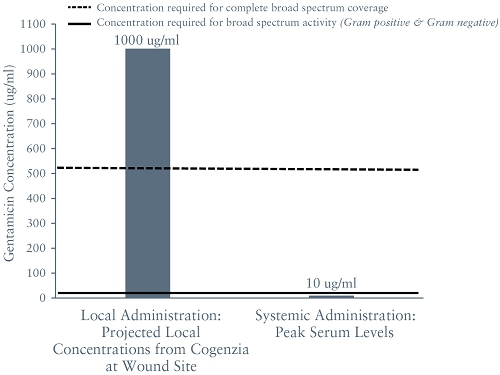
Preliminary data suggests that Cogenzia is able to deliver a higher dose of gentamicin locally at the infection site than can be achieved by systemic delivery, providing broad spectrum coverage of bacteria safely without the side effects and toxicity risks associated with systemic delivery at higher dosage levels, while minimizing the risk of resistance. There are currently no topical agents approved for the treatment of DFIs. We believe this topical route of administration, in combination with systemic antibiotic therapy, has the potential to become the standard of care first line treatment of all types of DFIs, including mild, moderate and severe infections.
We filed an IND for Cogenzia in November 2006. The clinical protocols for our Phase 3 registration trials were agreed upon with the FDA under an SPA, and have also been accepted by the EMA under the Scientific Advice procedure. We initiated the Cogenzia Phase 3 clinical program in both the United States and Europe. In June 2015, Cogenzia received QIDP designation for the adjunctive treatment of moderate and severe diabetic foot infection from the FDA. This designation is a key provision of the GAIN Act approved by Congress in 2012 to increase the incentives for drug manufacturers to produce new antibiotics for serious and hard-to-treat bacterial and fungal infections. QIDP designation for a drug adds an additional five years of market exclusivity, which means that the company that brings the drug into commercial use is protected from generic competitors for that period. For Cogenzia, that should result in eight years of data exclusivity in the United States, which would include three years of exclusivity for new clinical investigations that were essential for approval. QIDP designation also provides potential access to priority review of marketing applications and eligibility for fast track and priority review designations by the FDA. We expect to pursue a priority review designation for Cogenzia. A priority review designation means that the FDA’s goal is to take action on an application within 6 months, compared to 10 months under standard review. However, QIDP Designation for Cogenzia does not guarantee priority review designation by the FDA. We have also developed an expedited path to market in markets that represent a majority of the worldwide diabetes population, and we intend to enter into new commercial partnerships for Cogenzia in these markets. We maintain full rights to Cogenzia in the United States and Europe and, upon obtaining marketing approval, intend to directly commercialize the product in the United States, and potentially in Europe, using our own specialized sales and marketing organization focused on high volume wound treatment centers and podiatrists targeting the highest prescribers of antibiotics for the treatment of DFIs in the United States. We have filed five patent applications for Cogenzia in Australia, Canada, Europe, Japan and the United States, all of which are currently in the examination phase. If and when our patents are issued, we expect patent protection for Cogenzia in the United States and these other territories through 2031.
Infected Diabetic Foot Ulcers Global Market Overview
Global Incidence of Diabetic Foot Ulcers
According to the International Diabetes Federation, there were about 387 million diabetic patients globally in 2014, projected to increase to 592 million by 2035. Of the approximately 27 million patients globally who suffered a diabetic foot ulcer in 2014, 58%, or approximately 15.7 million, developed a DFI. DFIs are currently treated with systemic antibiotic therapy. However, peripheral vascular disease, or PVD, a frequent comorbidity of diabetes, leads to reduced blood flow to the extremities thereby rendering systemic antibiotic therapy less effective in this patient population. Published data demonstrate that systemic antibiotic therapy fails approximately 30% of the time in the treatment of DFIs. Patients with a DFI face hospitalization risk that is more than 55 times higher and risk of amputation more than 150 times higher than diabetic patients with uninfected foot ulcers. The direct cost of an amputation associated with the diabetic foot is estimated to be between $30,000 and $60,000, plus between $43,000 and $60,000 in costs for follow-up care over the next three years. In addition, major amputation is associated with mortality rates as high as 40% within one year and 80% within five years. We believe Cogenzia can offer improvements in patient outcomes and significant costs savings.
Local antibiotic treatment adjunct to a systemic agent can address this major need in DFIs by improving antibacterial efficacy at the wound site leading to a substantially higher success rate than that achieved by systemic antibiotic therapy alone. Since a diabetic foot ulcer cannot heal in the presence of infection, this higher infection cure rate facilitates more effective healing of the ulcer and substantially reduces the potential for amputation. The use of Cogenzia, a gentamicin collagen sponge administered topically, in conjunction with systemic antibiotic therapy, to treat DFIs has been supported by several key opinion leaders and authors affiliated with premier DFI institutions such as VA Puget Sound Healthcare System; University of Washington; Southern Arizona Limb Salvage Alliance; Kings College Hospital, London; Center for Clinical Research, Castro Valley, California; and Pacific Clinical Center, Los Angeles, California.
Cogenzia Clinical Data
Phase 2 Clinical Trial in Diabetic Foot Infections
In our multicenter, randomized, placebo-controlled Phase 2 trial involving 36 patients, Cogenzia (50 mg or 200 mg) was applied daily for up to 28 days in combination with systemic antibiotic therapy for the treatment of moderately-infected diabetic foot ulcers, with the control group receiving systemic therapy alone. Patients were treated for at least 7 days and continued treatment until the investigator determined that all signs and symptoms of infection had resolved, up to a maximum of 28 days. A final test-of-cure and safety assessment for each patient was performed 2 weeks after completion of treatment. The investigator performed clinical assessments at regular study visits while the patient was undergoing antibiotic therapy (i.e., days 3, 7, 10, 14, 21, and 28) and again at the final two-week follow-up visit. The primary efficacy endpoint for this trial was the percentage of patients with a clinical outcome of “clinical cure” (defined as the complete resolution or elimination of infection) at the study visit on day 7 of treatment. This study visit was selected as the primary endpoint because statistical calculations suggested that we would need a substantially larger sample size to test for treatment superiority at the later study visits. Our primary endpoint of clinical cure at 7 days after treatment, however, was not achieved. At the final test-of-cure visit approximately two weeks after completion of treatment, however, all evaluable patients in the treatment group achieved clinical cure. Nevertheless, since 100% of the patients who received Cogenzia and who completed the trial achieved a clinical cure, the trial results demonstrated a statistically significant improvement in cure rate at the final test-of-cure visit, despite the relatively small sample size. Below is a summary of the data from this trial based on the modified intent-to-treat, or mITT, population, modified to include only patients who had been randomized to the Cogenzia and control arms and not earlier terminated from the study for failure to comply with the study inclusion criteria.
Patients with Clinical Outcome of Clinical Cure at Final Test-of-Cure
Modified Intent-to-Treat Population (mITT)
| | | Cogenzia (n=22) | | | Control (n=10) | | | p-value | |
| Completed subjects (n=32) | | | 22 | | | | 100.0 | % | | | 7 | | | | 70.0 | % | | | — | |
| | | Cogenzia (n=26) | | | Control (n=10) | | | p-value | |
| All subjects (n=36) | | | 22 | | | | 84.6 | % | | | 7 | | | | 70.0 | % | | | 0.024 | * |
*statistically significant
Based on the mITT population, a significantly higher proportion of patients reached clinical cure than did the control group (100% versus 70.0%, p = 0.024). Accordingly, clinical cure at test-of-cure, measured approximately 10 to 14 days after the last dose of treatment has been administered, has been set as the primary endpoint of the Phase 3 trials for Cogenzia, as accepted by the FDA under our SPA as well as by the EMA.
Secondary endpoints of the trial included the percentage of patients with pathogen eradication at each time point, and time to eradication of baseline pathogens. Importantly, the Cogenzia group demonstrated a statistically significant higher rate of baseline pathogen eradication at all study visits (p ≤ 0.038) and a significantly reduced time to pathogen eradication (p < 0.001), as shown in the table below.
Baseline Pathogen Persistence
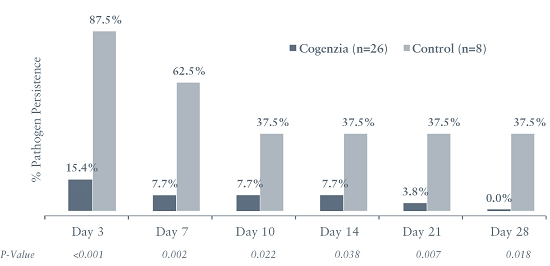
By achieving a complete eradication of all pathogens at the wound site at study visits on day 28, as demonstrated by microbiological testing, the use of Cogenzia in DFIs prepares the underlying wound for healing. Since diabetic foot ulcers generally cannot heal in the presence of infection, a pathogen free wound site provides patients an unmatched opportunity to achieve a complete healing of the ulcer, thereby substantially reducing the risk for both reinfection and potential amputation. Treatment by current systemic antibiotic therapy frequently leads to wounds that ostensibly appear to be free of infection but still prove to have a high level of residual pathogens. It is clinically meaningful that in our Phase 2 trial, Cogenzia achieved both a 100% clinical cure and 100% eradication of all pathogens at the wound site, thereby potentially providing practitioners with a high degree of comfort that a wound which appears to be free of infection is in fact free of infection. This important outcome, we believe, offers the potential for Cogenzia to become the recognized standard of care for the treatment of DFI’s. As the risk of reinfection for patients treated by systemic antibiotic therapy alone is high, this often results in further courses of antibiotic treatment, along with the potential for hospitalization where IV antibiotics are administered. We believe the use of Cogenzia in combination with systemic antibiotic therapy can be much more effective at eradicating the infecting pathogens than systemic antibiotic therapy alone, providing for an overall improved patient outcome at substantially lower costs than those incurred if hospitalization is required.
Our Phase 2 trial also demonstrated that Cogenzia was safe, well tolerated and conducive to ulcer healing. The proportion of patients experiencing any adverse event was similar for the treatment (28.9%) and control (27.8%) groups. The most common adverse events occurring in at least two patients per group were infected skin ulcer, tinea pedis and increased blood creatinine level. There were no clinically or statistically significant changes in laboratory tests values or vital signs. Most adverse events were mild or moderate, but there were six serious adverse events, including five in the Cogenzia treatment group (hypoglycemia, renal failure, cellulitis, tendon rupture and wound hemorrhage), all of which resolved, and one in the control group. Only one patient in the treatment group experienced an adverse event (moderate renal failure) that was considered definitely or probably related to the trial and resolved by the final visit. One clinically improved patient was withdrawn from the trial due to an adverse event (hypoglycemia) that was unrelated to Cogenzia.
United States and European Registration Trials
We have initiated two identical, randomized, placebo-controlled, blinded trials, enrolling approximately 500 patients each, under our SPA with the FDA, in patients with moderate to severe DFIs. Our trial protocols agreed to with the FDA in the SPA, and subsequently reaffirmed in 2014, have also been accepted by the EMA under the Scientific Advice procedure. Each trial consists of three arms, (1) Cogenzia, (2) placebo collagen matrix or (3) no collagen matrix. In each arm, Cogenzia will be used as adjuvant therapy in combination with a systemic antibiotic. Patients will be treated for up to a maximum of 28 days and the investigator will stop the trial treatment if a patient achieves clinical cure on or after day 14. The primary endpoint will be clinical cure at the test-of-cure visit approximately 10 days after the last dose of treatment has been administered with co-primary endpoints, including (i) Cogenzia compared to placebo and a systemic antibiotic, and (ii) Cogenzia compared to a systemic antibiotic alone. Follow-up visits with enrolled patients are scheduled to occur at 10, 30, 60 and 90 days after the last dose of treatment has been administered to assess ulcer closure and re-infection. Planned secondary endpoints include (1) clinical cure time (percentage of patients at each visit), (2) positive clinical response (percentage of patients at each visit), (3) pathogen eradication time (percentage of patients at each visit), (4) microbiological outcomes, (5) surgical intervention, or (6) amputation and re-infection. We commenced the trials in the first quarter of 2015. In addition, we plan to further expand the market opportunity for Cogenzia by conducting additional Phase 2 trials for the prevention of DFIs as well as for the treatment of infected, or at risk of infection, wounds such as venous ulcers, burns and bed sores, among others. Expansion of the market opportunity for Cogenzia into the prevention of DFIs would widen the potential use of the product across the entire diabetic foot ulcer patient population.
Commercialization Strategy for Cogenzia
We maintain full rights to Cogenzia in the United States and Europe and, upon obtaining marketing approval, intend to commercialize the product in the United States and possibly in Europe using our own specialized sales and marketing organization focused on high volume wound treatment centers and podiatrists. This sales force could also market Cogenzia for other indications such as the prevention of DFIs, if it is approved for such indications. Outside of the United States, we have already obtained regulatory approval of Cogenzia in seven countries, Argentina, Australia, Jordan, Mexico, Russia and Saudi Arabia and have filed for approval in India. These 8 countries collectively represent over 20% of the global diabetes population. We intend to enter into a partnership to market and distribute Cogenzia in those markets and in a number of other emerging market countries.
CollaGUARD
CollaGUARD is our translucent, bioresorbable collagen film for the prevention of post-operative adhesions in multiple surgical applications, including digestive, general, colorectal, gynecological and urological surgeries, in both open and laparoscopic approaches. It is approved in 12 countries outside of the United States for the prevention of post-operative adhesions and may be used in patients undergoing laparotomy or laparoscopic surgeries. When tested in vivo, CollaGUARD increased the probability of remaining adhesion-free by more than six fold (p 0.001) and significantly reduced the extent and severity of adhesions (p 0.001) versus no anti-adhesion product. CollaGUARD has been designed and engineered with a unique combination of features for optimal handling, ease-of-use, hemostatic properties and anti-adhesion performance. The film, which is applied directly to tissue surfaces, is translucent allowing constant visibility of the surgical field. It is highly stable at room temperature, does not require any special storage or advanced preparation before use and has up to a five-year shelf life.
The product is non-tacky, non-sticky and can be easily rolled for insertion through a trocar when implanted laproscopically. CollaGUARD is also fully biodegradable and is designed to be safely and completely resorbed over approximately three to five weeks post implantation. CollaGUARD is available in a wide variety of sizes up to 30 x 20 cm and may be cut and sutured if required and, therefore, can be used easily across a broad range of surgeries. CollaGUARD is regulated as a Class III device in the United States and we expect it will require a single pivotal clinical trial for PMA by the FDA. In the first quarter of 2015, we initiated a second pilot efficacy study in patients undergoing gynecological laparoscopic adhesiolysis. However, we decided to terminate the second pilot efficacy study in favor of a planned pilot efficacy study in patients undergoing myomectomy via open laparotomy to be performed in the U.S. under an IDE. We anticipate that data from this pilot study will help us to finalize the design of the U.S. pivotal study protocol. In the fourth quarter of 2015 we completed a pilot clinical study for CollaGUARD, run in the Netherlands, in patients undergoing intrauterine adhesiolysis via operative hysteroscopy. A clinical study report for this study confirmed the ease of use and safety for CollaGUARD in this patient population. We held our pre-IDE submission meeting with the FDA in the first quarter of 2016, in which we agreed on a non-clinical development plan that will be completed and reported prior to our filing a full IDE package, which we expect to do in the third quarter of 2016, with the Pilot (Feasibility) Clinical Study to be initiated immediately after approval. We submitted a family of patent applications aimed at protecting CollaGUARD on an international basis, including Australia, Canada, Eurasia, Europe, Japan, Mexico, and the United States, which is currently in the examination phase. If issued, these patents are expected to expire in 2033 or later in the United States.
Surgical Adhesion Market
Adhesions are fibrous bands of scar tissue that abnormally bind together two anatomic surfaces, and can develop naturally after surgery as part of the healing process. Post-surgical adhesions occur in almost 95% of patients who have had multiple laparotomies. Complications associated with post-operative adhesions can be severe, including chronic abdominal pain, bowl-obstruction, infertility in women, and joint immobilization, among others. Adhesions can also make second surgeries more complicated and even dangerous, depending on their extent and severity, as surgeons may have difficulties reaching and separating tissues, the median abdominal opening time increases threefold for repeat surgery patients, and increased surgical procedure and re-entry time means increased costs and increased risk of infection to the patient. Adhesions cause over 40% of all intestinal obstructions and 60% to 70% of small bowel obstructions. Approximately 35% of patients who underwent open abdominal or pelvic surgery were readmitted due to adhesion-related problems. In the United States alone, there are approximately 350,000 hospitalizations and $2.3 billion spent annually on surgery to remove adhesions formed following gynecologic or abdominal surgeries. According to Global Industry Analysts Inc., the global market for anti-adhesion products was estimated to be $1.3 billion in 2014. The market is mainly driven by increasing surgeon attention towards anti-adhesion products along with the development of new products addressing unmet requirements. The current products available on the market include four basic formulations: gels, films, sprays and liquids. Polymeric film is the most widely used anti-adhesion device to separate as well as isolate wounded tissues following a surgical procedure. However, existing products have a number of disadvantages including poor handling properties, limited efficacy, limitations to applicable surgical settings (i.e., open or laparoscopic procedures) and strict product warnings and contraindications. We believe that CollaGUARD has the ability to address these unmet needs and become a “best-in-class” product.
Although the leading products that compete with CollaGUARD are approved for use in open surgery, only one is approved for use laparoscopically. Accordingly, we believe CollaGUARD offers significant advantages over anti-adhesion products currently on the market, such as Sanofi’s Seprafilm®, Baxter’s Adept®, Ethicon’s Interceed® and Mast Biosurgery’s Surgiwrap®. Each of these products has one or more of the following limitations or contraindications:
| · | tacky, has to be kept in packaging until placed into the surgical site; |
| · | must be brought to temperature, limit to amount used; |
| · | cannot be overlapped on itself or other organs; |
| · | must be sutured in place; |
| · | cannot be used laparoscopically; |
| · | leak potential if wrapped around intestinal anastomosis; |
| · | reports of pulmonary edema / effusion and arrhythmia; or |
| · | risk of damage by excessive activity, requiring removal. |
CollaGUARD Pre-Clinical Data
CollaGUARD is regulated as a Class III device in the United States. In our pre-clinical animal trial with CollaGUARD, we have studied the performance, primary and secondary endpoints and safety of CollaGUARD in rats. This method is a well-established and well-recognized surrogate for human testing and was used to support regulatory approval in Europe and many other countries outside of the United States. The trial was conducted in two stages, comparing the safety and performance of CollaGUARD in stage 1 to an untreated control group and in stage 2 to Prevadh®, a commercially available collagen-based adhesion product approved in Europe. Prevadh, which also consists of collagen among other component materials, was deemed to be the closest comparison to CollaGUARD among marketed products. In the first trial stage, CollaGUARD demonstrated superiority over the control group in both the prevalence and severity of adhesions which was significantly lower compared to the control group (p < 0.001). In stage 2 of this trial, we demonstrated equivalent outcomes in the prevalence (p = 0.625) and severity (p = 0.317) of adhesions between CollaGUARD and Prevadh.
Percentage of Rats Adhesion Free Following
Abdominal Abradement
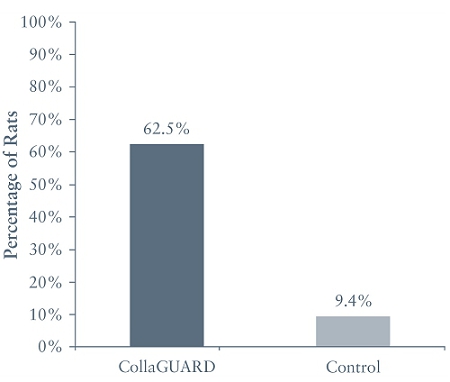
CollaGUARD United States Trial
Because CollaGUARD is regulated as a Class III device in the United States, we expect that it will require a single pivotal clinical trial for PMA by the FDA. We held our pre-IDE submission meeting with the FDA in the first quarter of 2016, in which we agreed on a non-clinical development plan that will be completed and reported prior to our filing a full IDE package, which we expect to do in the third quarter of 2016, with the Pilot (Feasibility) Clinical Study to be initiated immediately after approval.
CollaGUARD Commercialization Strategy
CollaGUARD has been approved in 12 countries within Asia, Latin America and the Middle East and we are preparing for launch in many of these countries through our established distribution arrangements, in place with 18 partners. One of our most significant distribution arrangements is with Takeda, which launched and distributed CollaGUARD in Russia in 2014 and planned to add additional territories in 2015. A second important partnership for CollaGUARD is with Pioneer, to whom we have granted rights to the product in China and several ASEAN countries. Pioneer is in the pre-launch phase for the product in a number of these countries. Further launches with partners in other Asian countries, the Middle East and Europe were planned throughout 2015.
Similar to XaraColl, we plan to commercialize CollaGuard in the United States. Since surgeons represent the primary physician audience for both Xaracoll and CollaGUARD, the same hospital formulary decision makers would be critical purchasers of both products. This complementary customer base will allow us aggregate a single point of sale for both XaraColl and CollaGUARD, and utilize the same sales team to effect these sales, thereby enhancing the efficiency of our potential commercial sales footprint.
Recertification of CollaGUARD in Europe
The initial European CE certificate for CollaGUARD was valid from October 7, 2011 to July 26, 2015. In accordance with normal practice for device recertification, we compiled an updated Design Dossier and submitted this to our European Notified Body (TÜV SÜD, Munich, Germany) in December 2014. As part of our application for recertification, we included an updated literature-based Clinical Evaluation Report and a Post Market Clinical Follow-up (PMCF) Plan, which rationalized the objectives of our ongoing clinical investigations in accordance with the current 2012 European Guideline for PMCF studies. However, prior to the expiration of the original certificate, the Notified Body requested that we revise our PMCF plan to additionally include a direct clinical comparison of CollaGUARD’s performance and safety compared to another CE-certified adhesion barrier. We do not believe that current European medical device regulations specifically require that our PMCF plan includes such a clinical comparison. In March 2016, we met with the Notified Body to seek clarification. At the meeting, TÜV SÜD agreed that current European medical device regulations do not specifically require that our PMCF plan includes such a clinical comparison, and agreed with our proposal for an updated PMCF based on the pilot clinical study in open myomectomy patients, as was agreed with FDA at the pre-IDE meeting. Provided TÜV SÜD determines that the clinical data from the U.S. pilot study are supportive, then recertification would be possible at that time based on an updated Technical Dossier and Clinical Evaluation. Until a new CE-certificate has been issued, we will not supply CollaGUARD to our marketing partners for sale in Europe or other affected territories. Accordingly, no such sales have been assumed in our financial forecasts.
Other Products
In addition to our lead product candidates, we have the following additional products approved or commercialized:
| Marketed and Partnered Products |
| | | |
| Product | Indication | Region (Partner) |
| | | |
| COLLATAMP®G* | Surgical Infections | Ex-US (EUSA) |
| REGENEPRO®** | Dental Wounds | US (Biomet 3i)
Ex-US (Biomet 3i) |
| ZORPREVA®*** | Surgical Hemostat | Ex-US (Pioneer) |
| SEPTOCOLL®E**** | Surgical Infections | Ex-US (Biomet) |
| COLLAGUARD®***** | Surgical Adhesion Prevention | Ex-US (various partners) |
*COLLATAMP®G is a registered trademark of EUSA/Jazz Pharmaceuticals
**REGENEPRO® is a registered trademark of Biomet 3i
***ZORPREVA® is a registered trademark of Innocoll Pharmaceuticals
****SEPTOCOLL®E is a registered trademark of Biomet Deutschland GmbH
*****COLLAGUARD® is a registered trademark of Innocoll Pharmaceuticals
In addition to our late-stage product candidates described above, we develop and manufacture a range of additional biodegradable surgical implants and topically applied pharmaceutical products and medical devices using our proprietary collagen-based technologies. We produce our products and product candidates, such as CollatampG surgical implant, RegenePro and Septocoll, in a range of topical and implantable forms, including sponges, films, membranes and gels, compatible with a variety of therapeutics, including hydrophobic and hydrophilic active ingredients and small molecules and biologics. A number of our products have been marketed for several years. For example, our CollatampG Gentamicin Surgical Implant, a perioperative surgical implant comprised of a lyophilized collagen matrix impregnated with a broad spectrum antibiotic is currently approved for sale in 62 countries across Africa, Asia, Europe, Latin America and the Middle East. EUSA Pharma acquired the rights to distribute CollatampG in all worldwide markets, excluding the United States. In addition, we have an exclusive License and Distribution Agreement with Biomet 3i for our range of CollaCare Dental products, branded RegenePro, covering all global territories outside of China and ASEAN, for which we have partnered with China Pioneer Pharma Holdings Ltd. Biomet 3i launched RegenePro in July 2014 in the United States and, having recently received approvals in the European Union, also has plans for a European launch. We also supply Septocoll, a surgical implant, to Biomet which markets the product in Europe and the Middle East. These agreements with our partner are exclusive manufacturing and supply arrangements (see below “— Commercial Partners and Agreements”) pursuant to which we exclusively supply, and the partner is required to exclusively purchase, the products for the respective territories. The majority of our agreements contain minimum or specified purchase requirements and in addition, pursuant to our agreement with Takeda, we receive payments upon achievement of certain regulatory milestones.
Our Collagen Based Technology Platform
All of our products and product candidates are based on our proprietary collagen technology platform, which includes CollaRx, a lyophilized sponge which is the basis of our XaraColl and Cogenzia products and CollaFilm, a film cast membrane which is the basis of CollaGUARD. We utilize highly purified biocompatible, biodegradable and fully bioresorbable type-1 bovine and equine collagen. Type 1 collagen is the primary fibril-forming collagen in bone, dermis tendons and ligaments and is the most abundant protein in the human body. Our collagen plays an integral role in the repair and replacement of both soft and hard tissue by providing an extracellular scaffold, stimulating certain growth factors and promoting tissue healing. We perform the extraction and purification of collagen from either bovine or equine Achilles tendons using a proprietary process at our manufacturing facility. The purified collagen is then incorporated into our technology platform, to create topical and implantable products that combine proven therapeutics with improved localized drug delivery and superior handling properties. Our proprietary processes and technologies also enable us to finely control the texture, consistency, drug elution dynamics, resorption time and other physical characteristics of the finished product. These characteristics provide meaningful differentiation of our products leading to superior performance and an overall improved user experience, because they:
| · | can be applied to a topical wound, surgically implanted, or injected into a subcutaneous tissue defect or joint; |
| · | are fully biocompatible, bioresorbable and biodegradable; |
| · | are suitable for a wide range of active ingredients (hydrophilic, lipophilic and macromolecules), including combinations thereof; |
| · | allow for versatile drug loading capability from micrograms to grams of single or multiple active ingredients; |
| · | provide for a rate of drug release that can be controlled by formulation and process variations; |
| · | utilize ready-to-use formats for ease of administration - no need for any mixing in the operating theatre; and |
| · | allow certain of our surgical products to be implanted using laparoscopy and are easily manipulated according to the site of application. |
Our technologies have been fully scaled up and in some cases commercialized and our manufacturing processes are well controlled and cost efficient.
Manufacturing
Our wholly-owned subsidiary, Syntacoll GmbH, located in Saal, Germany, is our commercial-scale manufacturing division which exclusively produces both clinical and commercial supply for all our products on a global basis. We believe our ability to manufacture our products allows us to control more effectively the quality and cost of manufacturing, which will enable us to achieve higher operating margins. We have a fully integrated and reliable manufacturing process in Saal, beginning with the extraction and purification of the type-1 collagen (from bovine and equine Achilles tendons), which is further processed using one of our proprietary technologies to produce final finished products in the forms of matrices, films, powders, liquids, and gels. We have qualified multiple sources for bovine and equine tendons, and conduct a rigorous quality control process on the raw materials, locally at our Saal facility. These raw materials are readily available to us from multiple sources at stable pricing. Several of our products, including CollaGUARD, CollatampG, Septocoll and RegenePro, are marketed in countries around the world. Our proprietary technologies have been fully validated. Syntacoll was established in Germany in 1975 and has been manufacturing commercial products based on collagen technology since 1985. Our manufacturing staff is highly qualified and experienced due to Syntacoll’s long history of producing collagen-based products. Our manufacturing facility has been approved in Germany for compliance with cGMPs. Our manufacturing facility is ISO 13485 certified in Europe and Canada. In our 30 years of producing collagen-based products, we have not experienced any significant quality issues or recalls. We believe we currently have adequate production capability to support our current production needs and planned clinical trials for XaraColl and Cogenzia. In addition, we expect to complete a capacity expansion build-out of our production facility to significantly increase capacity in the second half of 2016. Once completed and approved, we anticipate that our production capacity will be sufficient to meet currently forecasted market demand for all our current and late-stage pipeline product candidates.
Intellectual Property and Exclusivity
In the ordinary course of our business, we seek to protect our products, product candidates and technology through a combination of patents, trademarks, processes, proprietary know-how, regulatory exclusivity and contractual restrictions on disclosure.
Our knowledge base and expertise in collagen-based drug delivery and the related trade secrets play an important role in protecting our collagen-based products, product candidates and technology and provide protection apart from patents and regulatory exclusivity. The scale-up and commercial manufacture of XaraColl, Cogenzia and the use of our technology platform involve processes and in-process and release analytical techniques that we believe are unique to us. We have developed the manufacturing processes which we employ in our manufacturing facility for over twenty years, and they include all aspects of the sourcing, extraction and purification of raw source collagen, formulation and cost effective processing of collagen to exhibit the characteristics that are necessary for the effective release of precisely specified amounts of drug product over measured periods of time.
We also employ a strategy of filing patent applications, where possible, to seek patent protection for certain aspects of our compositions, formulations, and processes. We are continually evaluating and refining our patent prosecution strategy and evaluating the defensive strength of our patent position.
Patents and Patent Applications
XaraColl. A U.S. patent directed to XaraColl and entitled “A drug delivery device for providing local analgesia, local anesthesia or nerve blockade” was issued in October 2011 with claims directed to products comprising any amino amide and/or amino ester anesthetic in a collagen matrix intended for the provision of local analgesia or anesthesia over about 24 hours or longer. Its earliest filing date is March 28, 2007. An Intention to Grant notice was received in December 2015 for the corresponding European application, and the corresponding Japanese application was issued as a patent in September 2013, while the corresponding application in Ireland was issued as a patent in September 2011.
In March 2015, we submitted a petition to reissue the U.S. patent directed to XaraColl, mainly for the purposes of submitting prior art references identified in the corresponding European application. We did not submit these prior art references to the U.S. Patent and Trademark Office during the prosecution of the U.S. patent. In addition, this reissue process will allow us to pursue additional claims, e.g., claims within the scope of the originally issued claims but more tailored to our currently proposed XaraColl product. However, there can be no assurance that any or all of the originally issued claims will be reissued. It is also uncertain whether any or all of the additional claims we have included in the petition will be granted. We anticipate the conclusion of the reissue process before we launch XaraColl in the United States, and if the reissued claims are substantially the same as the originally issued claims, there will be no intervening rights by others during the reissue period. We will be unable to enforce the XaraColl U.S. patent unless and until the U.S. patent is reissued.
There can be no assurance that a patent will reissue from the petition, or that any such patent will be enforceable and will not be challenged, invalidated or circumvented. Notwithstanding the application for reissuance, we will continue to rely upon our proprietary know-how, techniques, expertise and the decades of collective experience of our team relating to the development and manufacturing of collagen matrix products, as well as the rigorous regulatory barriers applicable to the development, manufacture, distribution and marketing of potential competing products. See “Item 3. Key Information — D. Risk Factors — Because we have filed a petition for reissuance of our U.S. XaraColl patent, we will be unable to enforce it unless and until the U.S. patent is reissued.”
Cogenzia. We have filed patent applications in each of the United States, Europe, Canada, Australia and Japan specifically related to Cogenzia with an earliest filing date of April 11, 2011. These applications are entitled “Methods for treating bacterial infection” and directed to the local treatment of bacterial infections with an aminogylcoside antibiotic dispersed in a collagen matrix when used in combination with systemic administration of other antibacterial agents. If and when issued, we expect patent protection for Cogenzia in the United States and Europe to expire at the earliest in 2031.
CollaGUARD (US Market). We have also filed patent applications in each of the United States, Europe, Canada, Australia, Japan, Mexico, and Eurasia entitled “A modified collagen” with an earliest filing date January 9, 2012, which is directed to an improved process for manufacturing the next generation of CollaGUARD, e.g., CollaGUARD currently being developed for the U.S. These patent applications are also directed to the current manufacturing process for XaraColl and Cogenzia. If and when issued, these patents are expected to expire in 2033 or later in the United States.
CollaPress Technology. Our proprietary CollaPress technology is described in the issued European patent entitled “Novel collagen-based material with improved properties for use in human and veterinary medicine and the method of manufacturing such,” which expires on March 9, 2020. It has been nationalized and maintained in 6 European countries: France, Germany, Italy, Spain, Sweden and the United Kingdom. The patent is directed to collagen membranes with improved mechanical and fluid-absorption properties which may be produced by thermal compression. The technology patent is currently utilized in our ProColl™ wound management device and may also be used for the development of other proprietary, bioresorbable tissue reinforcement implants and/or as implantable delivery systems for biologically active substances such as hemostatic agents, growth factors, cytokines and drugs.
Trade Secrets and Proprietary Information
Trade secrets play an important role in protecting our collagen-based products, product candidates and technology and provide protection beyond patents and regulatory exclusivity. The scale-up and commercial manufacture of XaraColl, Cogenzia and the use of our technology platform involve processes and in-process and release analytical techniques that we believe are unique to us. We seek to protect our proprietary information, including our trade secrets and proprietary know-how, by requiring our employees, consultants and other advisors to execute proprietary information and confidentiality agreements upon the commencement of their employment or engagement. These agreements generally provide that all confidential information developed or made known during the course of the relationship with us be kept confidential and not be disclosed to third parties except in specific circumstances. In the case of our employees, the agreements also typically provide that all inventions resulting from work performed for us, utilizing our property or relating to our business and conceived or completed during employment shall be our exclusive property to the extent permitted by law. Where appropriate, agreements we obtain with our consultants also typically contain similar assignment of invention obligations. Further, we require confidentiality agreements from entities that receive our confidential data or materials.
Competition
The pharmaceutical and biotechnology industry is characterized by intense competition and rapid and significant innovation and change. Our competitors may be able to develop other drugs or products that are able to achieve similar or better results than our product candidates or marketed products. Our competitors include organizations such as major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical companies and generic drug companies. Many of our competitors have greater financial and other resources than we have, such as more commercial resources, larger research and development staffs and more extensive marketing and manufacturing facilities and organizations. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Our competitors may succeed in developing, acquiring or licensing on an exclusive basis technologies and products that are more effective or less costly than XaraColl, Cogenzia, CollaGUARD, or any other products that we are currently selling through partners or developing or that we may develop, which could render our products obsolete and noncompetitive. We expect any products that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, convenience of administration and delivery, price and the availability of reimbursement from government and other third-party payers. We also expect to face competition in our efforts to identify appropriate collaborators or partners to help commercialize our product candidates in our target commercial markets.
XaraColl Competition
We anticipate that, if approved by the FDA for these indications, XaraColl would be used in conjunction with other pain medications, such as acetaminophen and NSAIDs as part of an advanced multi-modal approach. We believe that XaraColl will primarily be competing with Pacira Pharmaceutical’s Exparel, a liposome injection of bupivacaine, an amide local anesthetic, indicated for single-dose infiltration into the surgical site to produce post-operative analgesia. Both Exparel and XaraColl are focused on opioid reduction or elimination as part of a multi-modal approach to pain relief. Management believes that XaraColl can provide at least comparable pain relief to Exparel with superior convenience. Exparel currently is only indicated for pain relief up to 24 hours post surgery. In addition, we believe our ability to manufacture XaraColl in a very cost-effective manner provides us with a cost of goods advantage over Exparel in the marketplace. In addition, XaraColl will be competing with currently marketed bupivacaine and opioid analgesics such as morphine, as well as elastomeric bag/catheter devices intended to provide bupivacaine over several days, which have been marketed by I-FLOW Corporation (now Halyard Health) since 2004.
Cogenzia Competition
There are currently no topically applied antibiotics approved for the treatment of DFIs, which could compete directly with Cogenzia, if approved for this indication. DFIs are currently treated with systemic antibiotic therapy. However, PVD, a frequent comorbidity of diabetes, leads to reduced blood flow to the extremities thereby rendering systemic antibiotic therapy less effective in this patient population. Compounding the problem, products that treat uninfected diabetic foot ulcers, such as DermaGraft, Apligraf and Regranex, are contraindicated for, or not effective against, DFIs, further limiting the healing process. As an adjuvant therapy, Cogenzia will not compete with any systemic antibiotics currently used to treat DFIs.
In addition to Cogenzia, there are a number of products in Phase 3 clinical trials of which we are aware, such as Pexiganan (Dipexium Pharmaceuticals), an antimicrobial cream with efficacy against Gram-positive and Gram-negative organisms which is currently being tested in mild DFIs. In addition, we believe that two anti-infective products, TaiGen’s Nemonoxacin (investigated in mild to moderate DFIs) and Photopharmica’s PPA904, a photosensitizer gel followed with visible red light exposure, have completed Phase 2 trials.
CollaGUARD Competition
CollaGUARD competes with a number of anti-adhesion products such as Sanofi’s SEPRAFILM, Baxter’s ADEPT, Ethicon’s INTERCEED and Mast Biosurgery’s SURGIWRAP, which have been on the market for many years and have established market share. We believe that CollaGUARD has superior handling properties when compared with competitors and, if and when approved, may include fewer warning and contraindications on the product label. ADEPT is contraindicated for use in procedures with laparotomy incisions and INTERCEED has a black box warning for laparoscopic use.
Commercial Partners and Agreements
EUSA Pharmaceuticals
In August 2007, we entered into a Manufacture and Supply Agreement with EUSA Pharma (Europe) Limited, or EUSA, which was subsequently amended and restated in April 2010. Under this agreement, we agreed to supply to EUSA its total supply of any Gentamicin-Collagen Implant product owned or commercialized by EUSA in finished packaged form for commercial supply. We are supplying EUSA with CollatampG under this agreement. In addition, the parties agreed that EUSA will own and retain all rights to the development data with respect to the product in all countries worldwide, except for the United States and its territories and possessions and Innocoll will have an exclusive, fully-paid perpetual license to use the development data with respect to the product in the United States and its territories and possessions. The agreement has a 10-year term, starting from its original execution date, with automatic renewal for five additional years, unless written notice of non-renewal is received by either party to the other at least three years prior to the expiration of the term of the renewal term. In addition, either party may terminate for breach.
Under another agreement with EUSA, dated April 27, 2010, we are obligated to pay EUSA a royalty on sales of XaraColl in the United States of 5% per year, not to exceed $6.5 million in total for all years, and 10% of sales outside the United States, not to exceed $2.5 million in total for all years. Such payments would accelerate under certain circumstances, including if we enter into a third-party agreement covering the development and commercialization of XaraColl. We also agreed to pay a royalty equal to 10% per year of sales of Cogenzia outside of the United States not to exceed $1.8 million in total for all years, also subject to acceleration if we enter into a third-party agreement.
Takeda
In August 2013, we entered into a 15-year License and Supply Agreement with Takeda GmbH, an affiliate of Takeda Pharmaceutical Company Limited, or Takeda, as expanded on March 24, 2014, pursuant to which we granted Takeda an exclusive license to distribute, promote and sell CollaGUARD Adhesion Barrier in Armenia, Azerbaijan, Belarus, Canada, Georgia, Kazakhstan, Kyrgyzstan, Mexico, Moldova, Mongolia, Russian Federation, Tajikistan, Turkmenistan, Ukraine and Uzbekistan for all current and future approved indications of CollaGUARD. Takeda is obligated to launch the product in the various jurisdictions in its territory, following regulatory approval, where required. Pursuant to the agreement, Takeda is also required to make a milestone payment upon achievement of regulatory approval in Canada. In addition, we are Takeda’s sole supplier for CollaGUARD and Takeda is required to purchase an initial quantity of product from us. We have also agreed on certain annual minimum purchase order requirements and parameters for pricing for future supplies of products. The agreement has a 15-year term, following the first commercial sale of the products in the various countries in the Takeda territory on a country-by-country basis, with automatic renewal for five additional years, unless terminated by either party with 12 months advance notice. In addition, we and Takeda have the right to terminate the agreement for breach and Takeda has the right to terminate the agreement with respect to Canada only in the event the Canadian marketing authorization is different from the indications granted in the European Union with severe restrictions that threaten Takeda’s forecasts in Canada.
Saudi Centre for Pharmaceuticals
In December 2011, we entered into a five-year License, Manufacturing and Supply Agreement with Saudi Centre for Pharmaceuticals, or SCP, pursuant to which we granted SCP an exclusive right to distribute, promote and sell CollaGUARD Adhesion Barrier in Bahrain, Iraq, Jordan, Kuwait, Oman, Qatar, Saudi Arabia and the United Arab Emirates in the field of surgical adhesion barriers. SCP is obligated to launch the product in the various jurisdictions in its territory within three months following regulatory approval, where required. SCP is responsible for compiling, submitting and maintaining the product registrations and associated costs in its territory. We are required to supply product in the required quantity and quality, complying with local law standards. We have also agreed with SCP on certain annual minimum purchase order requirements, following the second year after approval in the territory, and parameters for pricing for future supplies of products. The agreement has a five-year term. In addition, we and SCP have the right to terminate the agreement for breach.
Biomet Orthopedics
In June 2004, our subsidiary, Innocoll Technologies Limited, entered into a Manufacturing and Supply Agreement with Biomet Orthopedics Switzerland GmbH, or Biomet Orthopedics, which was subsequently amended several times, most recently in March 2013. Pursuant to the agreement, we have agreed to exclusively supply to Biomet Orthopedics and Biomet Orthopedics has agreed to exclusively purchase Septocoll® and Septocoll E®, our bioresorbable, dual-action collagen sponge product, from us. The agreement provides that all know-how, manufacturing and technical data, instructions, specifications and experiences as well as all test methods developed in connection with the products, as specified, are owned by Biomet Orthopedics and we receive a limited royalty-free license to the same for the term of the agreement. Biomet Orthopedics also supplies us with gentamicin pursuant to the agreement. The agreement automatically terminates on December 31, 2018 and may be terminated by either party for cause prior to that date. We have also agreed on certain annual minimum purchase order requirements and parameters for pricing for future supplies of products and Biomet Orthopedics has paid to us certain advances for future supplies of products through the current end of the term.
Biomet 3i, LLC
In April 2013, we entered into an Exclusive Distribution Agreement with Biomet 3i, pursuant to which we granted Biomet 3i the exclusive right to distribute and sell our RegenePro range of products in all countries, republics, states, and areas of the world with the exception of Brunei Darussalam, Cambodia, Hong Kong, Indonesia, Laos, Macau, Malaysia, Myanmar, Philippines, Singapore, Taiwan, Thailand, The People’s Republic of China and Vietnam. Pursuant to the agreement, Biomet 3i has agreed to not sell or distribute any competitive products in the Biomet 3i territory and to purchase certain minimum amounts of product. The agreement has a 15-year term, can be terminated by either party during the first two years of the term with six months notice and during the remainder of the term with 18 months notice. The agreement can also be terminated by either party for breach.
Pioneer Pharma Co. Ltd.
In October 2011, we entered into a Licensing, Manufacturing and Supply Agreement with Pioneer Pharma Co Ltd., or Pioneer, pursuant to which we granted Pioneer the exclusive right to distribute and sell CollaGUARD in The People’s Republic of China, including the territories of Hong Kong, Macau and Taiwan for adhesion barrier and any other indication approved by EU regulatory authorities and the FDA. In August 2012, we expanded the territory in which Pioneer has the right to distribute and sell CollaGUARD to include Brunei Darussalam, Cambodia, Indonesia, Laos, Malaysia, Myanmar, Singapore and Vietnam. Pioneer has agreed not to enter into similar arrangements for competitive products in its territory. The agreement provides that Pioneer is responsible for compiling, submitting and maintaining the product’s registration and associated costs in its territory and is required to place agreed-upon minimum purchase orders within a certain time period after the product gains marketing approval in the Pioneer territory. In addition, Pioneer is required to make certain scheduled payments which are creditable against future supply of product. The agreement has a ten-year term and can be terminated by either party for breach.
Government Regulation
Government authorities in the United States (at the federal, state and local level) and in other countries extensively regulate, among other things, the research, development, testing, manufacturing, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug and medical device products such as those we are developing. XaraColl, Cogenzia and our other drug candidates, and CollaGUARD and our other medical device product candidates or products only marketed in certain countries must be approved or cleared by the FDA before they may be legally marketed in the United States and by the appropriate foreign regulatory agency before they may be legally marketed in foreign countries.
United States Drug Development and Review
Drug Development Process
In the United States, the FDA regulates drugs, such as XaraColl and Cogenzia, under the Federal Food, Drug, and Cosmetic Act, or FDCA, and implementing regulations. Drugs are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant to administrative or judicial sanctions. FDA sanctions could include, among other actions, refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning and notice of violation letters, product recalls or withdrawals from the market, product seizures, total or partial suspension of production or distribution injunctions, unfavorable inspections, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us. The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| · | Completion of extensive nonclinical, or preclinical, laboratory trials, preclinical animal trials and formulation trials in accordance with applicable regulations, including the FDA’s Good Laboratory Practice, or GLP, regulations; |
| · | Submission to the FDA of an IND which must become effective before human clinical trials may begin; |
| · | Performance of adequate and well-controlled human clinical trials in accordance with applicable regulations, including cGCP, regulations to establish the safety and efficacy of the proposed drug for its proposed indication; |
| · | Submission to the FDA of an NDA for a new drug product; |
| · | A determination by the FDA within 60 days of its receipt of an NDA to accept the NDA for filing and review; |
| · | Satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the drug is produced to assess compliance with the FDA’s current good manufacturing practice, or cGMP, regulations to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; |
| · | Potential FDA audit of the preclinical and/or clinical trial sites that generated the study data in support of the NDA; and |
| · | FDA review and approval of the NDA. |
Before testing any compounds with potential therapeutic value in humans, the drug candidate enters the preclinical trial stage. Preclinical trials include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal trials to assess the potential safety and activity of the drug candidate. The conduct of the preclinical trials must comply with federal regulations and requirements including GLP. The sponsor must submit the results of the preclinical trials, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. An IND is a request for authorization from the FDA to administer an investigational drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human trials. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions regarding the proposed clinical trials and places the IND on clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a drug candidate at any time before or during clinical trials due to safety concerns, noncompliance with IND requirements, and other deficiencies. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such trial.
Clinical trials involve the administration of the drug candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control, in accordance with GCP, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety and assess efficacy. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers issues such as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. There are also requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| · | Phase 1. The drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism and pharmacologic action of the drug in human distribution and excretion, the side effects associated with increasing dosages, and if possible, to gain early evidence of effectiveness. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human trial is often conducted in patients. |
| · | Phase 2. The drug is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the effectiveness of the drug for a specific indication or indications in patients with the disease or condition under study and to determine dosage tolerance, optimal dosage and dosing schedule. Phase 2 trials are sometimes further divided into Phase 2a and Phase 2b trials. Phase 2a trials are typically smaller and shorter in duration, and generally consist of patient exposure-response trials which focus on proving the hypothesized mechanism of action. Phase 2b trials are typically higher enrolling and longer in duration, and generally consist of patient dose-ranging trials which focus on finding the optimum dosage at which the drug shows clinical benefit with minimal side effects. |
| · | Phase 3. Clinical trials are undertaken after preliminary evidence suggesting effectiveness has been obtained and are intended to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall benefit/risk ratio of the product and provide an adequate basis for product approval. Generally, two adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval of an NDA. Phase 3 clinical trials usually involve several hundred to several thousand participants. |
Post-approval trials, or Phase 4 clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 trials.
The FDCA permits the FDA and an IND sponsor to agree in writing on the design and size of clinical trials intended to form the primary basis of a claim of effectiveness in an NDA. This process is known as a Special Protocol Assessment, or SPA. We have an SPA in place for our Phase 3 registration trial for Cogenzia. An SPA agreement may not be changed by the sponsor or the FDA after the trial begins except with the written agreement of the sponsor and the FDA, or if the FDA determines that a substantial scientific issue essential to determining the safety or effectiveness of the drug was identified after the trial began. For certain types of protocols, including carcinogenicity protocols, stability protocols, and Phase 3 protocols for clinical trials that will form the primary basis of an efficacy claim, the FDA has agreed under its performance goals associated with the Prescription Drug User Fee Act, or PDUFA, to provide a written response on most protocols within 45 days of receipt. However, the FDA does not always meet its PDUFA goals, and additional FDA questions and resolution of issues leading up to an SPA agreement may result in the overall SPA process being much longer, if an agreement is reached at all.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events or any finding from trials in laboratory animals that suggests a significant risk for human subjects. Phase 1, Phase 2 and Phase 3 clinical trials may fail to be completed successfully within any specified period, if at all. The FDA, the IRB, or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. An IRB can suspend or terminate approval of a clinical trial at its institution if, among other things, the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or data monitoring committee. This group provides authorization for whether or not a trial may move forward at designated checkpoints based on access to certain data from the trial. We may also suspend or terminate a clinical trial based on evolving business objectives and/or competitive climate.
Concurrent with clinical trials, companies usually complete additional animal trials and must also develop additional information about the chemistry and physical characteristics of the drug as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final drug. Additionally, appropriate packaging must be selected and tested and stability trials must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life. Labeling of the product must also be developed.
FDA Review and Approval Processes
The results of product development, preclinical trials and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product.
The NDA includes both negative or ambiguous results of preclinical and clinical trials as well as positive findings. Data may come from company-sponsored clinical trials intended to test the safety and effectiveness of a use of a product, or from a number of alternative sources, including trials initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and effectiveness of the investigational drug product to the satisfaction of the FDA. The submission of an NDA is subject to the payment of substantial user fees; a waiver of such fees may be obtained under certain limited circumstances.
For XaraColl, we intend to submit an NDA under Section 505(b)(2) of the FDCA, which allows us to submit an NDA as an application that contains full reports of investigations of safety and effectiveness in which at least some of the information required for approval comes from studies not conducted by or for the 505(b)(2) applicant, but instead from published literature reports and/or the FDA’s findings of safety and/or effectiveness for one or more approved drugs, and for which the 505(b)(2) applicant has not obtained a right of reference or use.
In addition, under the Pediatric Research Equity Act, or PREA, an NDA or supplement to an NDA for any new active ingredient, indication, dosage form, or route of administration must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. Unless otherwise required by regulation, PREA does not apply to any drug for an indication for which orphan designation has been granted. However, if only one indication for a product has orphan designation, a pediatric assessment may still be required for any applications to market that same product for the non-orphan indication(s).
The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA for filing. The FDA must make a decision on accepting an NDA for filing within 60 days of receipt. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the goals and policies agreed to by the FDA under PDUFA, the FDA has ten months from the 60-day filing date in which to complete its initial review of a standard NDA and respond to the applicant, and six months for a priority NDA. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs, and the review process is often significantly extended by FDA requests for additional information or clarification.
After the NDA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. The FDA may refer applications for novel drug or biological products or drug or biological products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical sites to assure compliance with cGCP requirements. After the FDA evaluates the application, manufacturing process and manufacturing facilities and other relevant information, it may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications under specific conditions of use set out in the approved labeling. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter usually describes all of the specific deficiencies in the NDA identified by the FDA. The Complete Response Letter may require additional clinical data and/or an additional pivotal Phase 3 clinical trial(s), and/or other significant and time-consuming requirements related to clinical trials, preclinical trials or manufacturing. If a Complete Response Letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter or withdraw the application. Even if such data and information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data.
If the FDA approves the NDA, the drug’s approved labeling will be limited to specific diseases, dosages and indications and will include certain contraindications, warnings and/or precautions. The FDA may condition the approval of the NDA on changes to the proposed labeling, development of adequate controls and specifications or a commitment to conduct one or more post-market trials or clinical trials. For example, the FDA may require post-approval studies which involve clinical trials designed to further assess a drug’s safety and effectiveness and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized. The FDA may also determine that a risk evaluation and mitigation strategy, or REMS, is necessary to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS; the FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician or patient communication plans, or other elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Following approval of an NDA with a REMS, the sponsor is responsible for marketing the drug in compliance with the REMS and must submit periodic REMS assessments to the FDA. Any of these limitations could restrict the commercial value of the product.
Expedited Development and Review Programs
The FDA has several overlapping programs that are intended to expedite or facilitate the process for reviewing new drug products that meet certain criteria. Specifically, new drugs are eligible for Fast Track designation if they are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. In June 2015, Cogenzia received Qualified Infectious Disease Product, or QIDP, designation for the adjunctive treatment of moderate and severe diabetic foot infection from the FDA. This designation is a key provision of the Generating Antibiotic Incentives Now Act, or GAIN Act, approved by Congress in 2012 to increase the incentives for drug manufacturers to produce new antibiotics for serious and hard-to-treat bacterial and fungal infections. QIDP designation for a drug adds an additional five years of market exclusivity, which means that the company that brings the drug into commercial use is protected from generic competitors for that period. For Cogenzia, that should result in eight years of data exclusivity in the United States, which would include three years of exclusivity for new clinical investigations that were essential for approval. QIDP designation also provides potential access to priority review of marketing applications and eligibility for fast track and priority review designations by the FDA. We expect to pursue a priority review designation for Cogenzia. A priority review designation means that the FDA’s goal is to take action on an application within 6 months, compared to 10 months under standard review. However, QIDP Designation for Cogenzia does not guarantee priority review designation by the FDA.
Any product submitted to the FDA for approval, including a product with a Fast Track designation, may also be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. A product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug designated for priority review in an effort to facilitate the review. Additionally, a product may be eligible for accelerated approval. Drug products studied for their safety and effectiveness in treating serious or life-threatening diseases or conditions may receive accelerated approval upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product.
The FDA may also expedite the approval of a designated breakthrough therapy, which is a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The sponsor of a breakthrough therapy may request the FDA to designate the drug as a breakthrough therapy at the time of, or any time after, the submission of an IND for the drug. If FDA designates a drug as a breakthrough therapy, it must take actions appropriate to expedite the development and review of the application, which may include holding meetings with the sponsor and the review team throughout the development of the drug; providing timely advice to, and interactive communication with, the sponsor regarding the development of the drug to ensure that the development program to gather the nonclinical and clinical data necessary for approval is as efficient as practicable; involving senior managers and experienced review staff, as appropriate, in a collaborative, cross-disciplinary review; assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor; and taking steps to ensure that the design of the clinical trials is as efficient as practicable, when scientifically appropriate, such as by minimizing the number of patients exposed to a potentially less efficacious treatment.
Fast Track designation, priority review and breakthrough therapy designation do not change the standards for approval but may expedite the development or approval process.
Post-Approval Requirements for Approved Drugs
Any drug products for which we receive FDA approvals are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, and complying with FDA promotion and advertising requirements, which include, among other requirements, standards for direct-to-consumer advertising, restrictions on promoting drugs for uses or in patient populations that are not described in the drug’s approved labeling (known as “off-label use”), limitations on industry sponsored scientific and educational activities, and requirements for promotional activities involving the internet. Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such off-label uses.
In addition, quality control and manufacturing procedures must continue to conform to applicable manufacturing requirements after approval. We are relying exclusively on our facility in Saal, Germany, for the production of clinical and commercial quantities of our products in accordance with cGMP regulations, which has not yet been cGMP approved. cGMP regulations require among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP. Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer or holder of an approved NDA, including, among other things, recall or withdrawal of the product from the market. In addition, changes to the manufacturing process are strictly regulated, and depending on the significance of the change, may require prior FDA approval before being implemented and development of and submission of data to support the change. Other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval, as well as, possibly, the development and submission of data to support the change.
The FDA also may require post-approval, sometimes referred to as Phase 4, trials and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product. Discovery of previously unknown problems with a product or the failure to comply with applicable FDA requirements can have negative consequences, including adverse publicity, judicial or administrative enforcement, warning letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties, among others. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures, such as a REMS. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
United States Premarket Clearance and Approval Requirements for Medical Devices
Unless an exemption applies, each medical device, such as CollaGUARD or our other device products or product candidates, we wish to distribute commercially in the United States will require either prior premarket notification (510(k)) clearance or prior approval of a PMA application from the FDA. The FDA classifies medical devices into one of three classes. Devices deemed to pose low to moderate risk are placed in either class I or II, which, absent an exemption, requires the manufacturer to file with the FDA a 510(k) submission requesting permission for commercial distribution. This process is known as 510(k) clearance. Some low-risk devices are exempt from this requirement. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or certain implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device, are placed in class III, requiring approval of a PMA application. CollaGUARD is a Class III device requiring premarket approval. Both premarket clearance and PMA applications are subject to the payment of user fees, paid at the time of submission for FDA review. The FDA can also impose restrictions on the sale, distribution or use of devices at the time of their clearance or approval, or subsequent to marketing.
510(k) Clearance Pathway
To obtain 510(k) clearance, we must file a 510(k) submission demonstrating that the proposed device is substantially equivalent to a previously cleared 510(k) device or a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for the submission of PMA applications. The FDA’s 510(k) clearance pathway usually takes from three to 12 months from the date the application is completed and filed, but it can take significantly longer and clearance is never assured. Although many 510(k) submissions are cleared without clinical data, in some cases, the FDA requires significant clinical data to support substantial equivalence. In reviewing a 510(k) submission, the FDA may request additional information, including clinical data, which may significantly prolong the review process. After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a new or major change in its intended use, will require a new 510(k) clearance or, depending on the modification, could require de novo review or a PMA application. The FDA requires each manufacturer to make this determination initially, but the FDA can review any such decision and can disagree with a manufacturer’s determination. If the FDA disagrees with a manufacturer’s determination that a new 510(k) submission is not required for the modification of an existing device, the FDA can require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance, successful de novo review or approval of a PMA application is obtained. If the FDA requires us to seek 510(k) clearance, de novo review or approval of a PMA application for any modifications to a previously cleared product, we may be required to cease marketing or recall the modified device until we obtain FDA marketing authorization. In addition, in these circumstances, we may be subject to significant regulatory fines or penalties for failure to submit for marketing authorization.
Premarket Approval Pathway
A PMA application must be submitted if the device is not exempt and cannot be cleared through the 510(k) process, which will be the case for CollaGUARD. The PMA application process is generally more costly and time consuming than the 510(k) premarket clearance process and requires proof of the safety and effectiveness of the device to the FDA’s satisfaction. Accordingly, a PMA application must be supported by extensive data including, but not limited to, technical information regarding device design and development, pre-clinical and clinical trials, manufacturing data and labeling to support the FDA’s determination that the device is safe and effective for its intended use. After a PMA application is deemed complete, the FDA will accept the application for filing and begin an in-depth review of the submitted information. By statute, the FDA has 180 days to review the “accepted application,” although, generally, review of the application takes between one and three years, but may take significantly longer. During this review period, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a preapproval inspection of the manufacturing facility to ensure compliance with Quality System Regulation, or QSR, which requires elaborate design development, testing, production control, documentation and other quality assurance procedures and measures upon the design, manufacturing and distribution process. The FDA may approve a PMA application with post-approval conditions intended to ensure the safety and effectiveness of the device including, among other things, restrictions on labeling, promotion, sale and distribution, collection of long-term follow-up data from patients in the clinical trial that supported approval, or new post-approval studies. Failure to comply with the conditions of approval can result in materially adverse enforcement action, including the loss or withdrawal of the approval. PMA supplements are required for modifications that could affect device safety or effectiveness, including, for example, certain types of modifications to the device’s indication for use, manufacturing process, labeling and design. PMA supplements often require submission of the same type of information as an original PMA application, except that the supplement is limited to information needed to support any changes to the device covered by the original PMA application, and may not require as extensive clinical data or the convening of an advisory panel.
Clinical Trials
A clinical trial is almost always required to support a PMA application and may be required for 510(k) premarket clearance. We expect that CollaGUARD, as a Class III device, will require a single pivotal trial for PMA approval. In the United States, absent certain limited exceptions, human clinical trials intended to support product clearance or approval require an IDE application which the FDA reviews. Some types of trials deemed to present “non-significant risk” are deemed to have an approved IDE once certain requirements are addressed and IRB approval is obtained. If the device presents a “significant risk” to human health, as defined by FDA regulations, the sponsor must submit an IDE application to the FDA and obtain IDE approval prior to commencing the human clinical trials. The IDE application must be supported by appropriate data, such as animal and laboratory trial results, showing that it is safe to evaluate the device in humans and that the trial protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of subjects, unless the product is deemed a non-significant risk device and eligible for more abbreviated IDE requirements. Clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and the responsible institutional review boards at the clinical trial sites. There can be no assurance that submission of an IDE will result in the ability to commence clinical trials. Additionally, after a trial begins, the FDA may place it on hold or terminate it if, among other reasons, it concludes that the clinical subjects are exposed to unacceptable health risks that outweigh the benefits of participation in the trial. During a trial, we are required to comply with the FDA’s IDE requirements for investigator selection, trial monitoring, reporting, record keeping and prohibitions on the promotion or commercialization of investigational devices or making safety or efficacy claims for them, among other things. We are also responsible for the appropriate labeling and distribution of investigational devices. Our clinical trials must be conducted in accordance with FDA regulations and federal and state regulations concerning human subject protection, including informed consent and healthcare privacy. The investigators must also obtain patient informed consent, rigorously follow the investigational plan and trial protocol, control the disposition of investigational devices and comply with all reporting and recordkeeping requirements, among other things. The FDA’s grant of permission to proceed with clinical trials does not constitute a binding commitment that the FDA will consider the trial design adequate to support commercial marketing clearance or approval. In addition, there can be no assurance that the data generated during a clinical trial will meet chosen safety and effectiveness endpoints or otherwise produce results that will lead the FDA to grant marketing clearance or approval. Similarly, in Europe, the clinical trial must be approved by the local ethics committee and in some cases, including trials of high-risk devices, by the Ministry of Health in the applicable country.
Pervasive and Continuing FDA Regulation for Medical Devices
After a device is placed on the market, regardless of its classification or premarket pathway, numerous regulatory requirements apply. These include, but are not limited to:
| · | establishing establishment registration and device listings with the FDA; |
| · | Quality System Regulation, or QSR, which requires manufacturers, including third-party manufacturers and certain other parties, to follow stringent design, testing, process control, documentation, CAPA, complaint handling and other quality assurance procedures, as applicable; |
| · | labeling statutes and regulations, which prohibit the promotion of products for uncleared or unapproved, or off-label, uses and impose other restrictions on labeling; |
| · | clearance or approval of product modifications that could affect (or for 510(k) devices, significantly affect) safety or effectiveness or that would constitute a change (or for 510(k) devices, a major change) in intended use; |
| · | medical device reporting regulations, which require that manufacturers report to the FDA if an event reasonably suggests that their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction of the same or a similar device of the manufacturer were to recur; |
| · | corrections and removals reporting regulations, which require that manufacturers report to the FDA field corrections and product removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA, that may present a risk to health. In addition, FDA may order a mandatory recall if there is a reasonable probability that the device would cause serious adverse health consequences or death; and |
| · | post-approval restrictions or conditions, including requirements to conduct post-market surveillance studies to establish additional safety or efficacy data. |
The FDA has broad post-market and regulatory enforcement powers. The agency may conduct announced and unannounced inspections to determine compliance with the QSR and other regulations, and these inspections may include the manufacturing facilities of subcontractors. Failure by us or our suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or other regulatory authorities, which may result in sanctions and related consequences including, but not limited to:
| · | untitled letters or warning letters; |
| · | fines, injunctions, consent decrees and civil penalties; |
| · | recall, detention or seizure of our products; |
| · | operating restrictions, partial suspension or total shutdown of production; |
| · | refusal of or delay in granting our requests for 510(k) clearance or premarket approval of new products or modified products; |
| · | withdrawing 510(k) clearance or premarket approvals that are already granted; |
| · | refusal to grant export approval for our products; |
| · | criminal prosecution; and |
| · | unanticipated expenditures to address or defend such actions. |
We are subject to announced and unannounced device inspections by FDA and other regulatory agencies overseeing the implementation and adherence of applicable local, state and federal statutes and regulations.
Affordable Care Act
| · | In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, the Affordable Care Act, was enacted, which includes measures that have or will significantly change the way health care is financed by both governmental and private insurers. Among the provisions of the Affordable Care Act of greatest importance to the pharmaceutical industry are the following: |
| · | The Medicaid Drug Rebate Program requires pharmaceutical manufacturers to enter into and have in effect a national rebate agreement with the Secretary of the Department of Health and Human Services as a condition for states to receive federal matching funds for the manufacturer’s outpatient drugs furnished to Medicaid patients. Effective in 2010, the Affordable Care Act made several changes to the Medicaid Drug Rebate Program, including increasing pharmaceutical manufacturers’ rebate liability by raising the minimum basic Medicaid rebate on most branded prescription drugs and biologic agents from 15.1% of average manufacturer price (AMP) to 23.1% of AMP and adding a new rebate calculation for “line extensions” (i.e., new formulations, such as extended release formulations) of solid oral dosage forms of branded products, as well as potentially impacting their rebate liability by modifying the statutory definition of AMP. The Affordable Care Act also expanded the universe of Medicaid utilization subject to drug rebates by requiring pharmaceutical manufacturers to pay rebates on Medicaid managed care utilization as of 2010. Per a ruling by the U.S. Supreme Court in 2012, states have the option to expand their Medicaid programs which in turn expands the population eligible for Medicaid drug benefits. The Centers for Medicare and Medicaid Services, or CMS, has proposed to expand Medicaid rebate liability to the territories of the United States as well. In addition, the Affordable Care Act provides for the public availability of retail survey prices and certain weighted average AMPs under the Medicaid program. The implementation of this requirement by the CMS may also provide for the public availability of pharmacy acquisition of cost data, which could negatively impact our sales. |
| · | In order for a pharmaceutical product to receive federal reimbursement under the Medicare Part B and Medicaid programs or to be sold directly to U.S. government agencies, the manufacturer must extend discounts to entities eligible to participate in the 340B drug pricing program. The required 340B discount on a given product is calculated based on the AMP and Medicaid rebate amounts reported by the manufacturer. Effective in 2010, the Affordable Care Act expanded the types of entities eligible to receive discounted 340B pricing, although, under the current state of the law, with the exception of children’s hospitals, these newly eligible entities will not be eligible to receive discounted 340B pricing on orphan drugs when used for the orphan indication. In July 2013, the Health Resources and Services Administration (HRSA) issued a final rule allowing the newly eligible entities to access discounted orphan drugs if used for non-orphan indications. While the final rule was vacated by a federal court ruling, HRSA has stated it will continue to allow discounts for orphan drugs when used for any indication other than for orphan indications. In addition, as 340B drug pricing is determined based on AMP and Medicaid rebate data, the revisions to the Medicaid rebate formula and AMP definition described above could cause the required 340B discount to increase. |
| · | Effective in 2011, the Affordable Care Act imposed a requirement on manufacturers of branded drugs and biologic agents to provide a 50% discount off the negotiated price of branded drugs dispensed to Medicare Part D patients in the coverage gap (i.e., “donut hole”). |
| · | Effective in 2011, the Affordable Care Act imposed an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs, although this fee would not apply to sales of certain products approved exclusively for orphan indications. |
| · | The Affordable Care Act required pharmaceutical manufacturers to track certain financial arrangements with physicians and teaching hospitals, including any “transfer of value” made or distributed to such entities, as well as any ownership or investment interests held by physicians and their immediate family members. Manufacturers were required to begin tracking this information in 2013 and to report this information to CMS by March 2014. |
| · | As of 2010, a new Patient-Centered Outcomes Research Institute was established pursuant to the Affordable Care Actor oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. The research conducted by the Patient-Centered Outcomes Research Institute may affect the market for certain pharmaceutical products. |
| · | The Affordable Care Act created the Independent Payment Advisory Board, IPAB, which, beginning in 2014, will have authority to recommend certain changes to the Medicare program to reduce expenditures by the program that could result in reduced payments for prescription drugs. Under certain circumstances, these recommendations will become law unless Congress enacts legislation that will achieve the same or greater Medicare cost savings. IPAB recommendations are only required when Medicare spending exceeds a target growth rate established by the Affordable Care Act. Members of the IPAB have still not been appointed and Medicare cost growth is below the threshold that would require IPAB recommendations. |
| · | The Affordable Care Act established the Center for Medicare and Medicaid Innovation within CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending. Funding has been allocated to support the mission of the Center for Medicare and Medicaid Innovation from 2011 to 2019. |
Pediatric Exclusivity
Pediatric exclusivity is another type of regulatory market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to any existing exclusivity period or patent term. This six-month exclusivity may be granted by FDA based on the completion of a pediatric trial in accordance with a “Written Request” for such as outlined in §505A(d)(2) of the FDCA. Recently, the Food and Drug Administration Safety and Innovation Act, or FDASIA, amended the FDCA. FDASIA requires that a sponsor who is planning to submit a marketing application for a drug or biological product that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan, or PSP, ideally within sixty days of an end-of-phase 2 meeting or as may be agreed between the sponsor and FDA but no later than 210 days before submission of the NDA or supplement. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. FDA and the sponsor must reach agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from nonclinical studies, early phase clinical trials, and/or other clinical development programs.
United States Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of the FDA approval of the use of our drug or device candidates, some of our U.S. patents or patents issuing based on our pending and future patent applications may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND or IDE and the submission date of an NDA or PMA, respectively, plus the time between the submission date of an NDA or PMA and the approval of that application. Only one patent applicable to an approved drug or device is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we may intend to apply for restoration of patent term for one of our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA.
Market exclusivity provisions under the FDCA can also delay the submission or the approval of certain competing marketing applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to obtain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another drug based on the same active moiety, regardless of whether the drug is intended for the same indication as the original innovative drug or for another indication, where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder. The FDCA also provides three years of marketing exclusivity for an NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, clinical investigations to support new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the modification for which the drug received approval on the basis of the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the active agent for the original indication or condition of use. Five-year and three-year exclusivity will not delay the submission or approval of any full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical and clinical trials necessary to demonstrate safety and effectiveness. An applicant submitting an ANDA would be required to demonstrate bioequivalency in patients comparing their candidate product to our product. Establishment of bioequivalency in patients can be a costly and challenging undertaking.
Other types of non-patent marketing exclusivity include orphan drug exclusivity under the Orphan Drug Act, which may offer a seven-year period of marketing exclusivity as described above, and pediatric exclusivity under the Best Pharmaceuticals for Children Act, which may add six months to existing exclusivity periods and patent terms. This six-month pediatric exclusivity may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial.
The GAIN Act amended the FDCA in 2012 to increase the incentives for drug manufacturers to produce new antibiotics for treating serious and life-threatening bacterial and fungal infections. A qualifying product may be designated by FDA as a QIDP. QIDP designation for an antibiotic drug adds an additional five years of market exclusivity, in addition to the 5-year and 3-year periods discussed above. To obtain the benefit of additional exclusivity, a company must seek and obtain QIDP status before submitting its NDA to the FDA. QIDP designation also provides access to priority review of the NDA and eligibility for Fast Track designation by the FDA. In June 2015, Cogenzia received QIDP designation for the adjunctive treatment of moderate and severe diabetic foot infection from the FDA. This designation is a key provision of the GAIN Act approved by Congress in 2012 to increase the incentives for drug manufacturers to produce new antibiotics for serious and hard-to-treat bacterial and fungal infections. QIDP designation for a drug adds an additional five years of market exclusivity, which means that the company that brings the drug into commercial use is protected from generic competitors for that period. For Cogenzia, that should result in eight years of data exclusivity in the United States, which would include three years of exclusivity for new clinical investigations that were essential for approval. QIDP designation also provides potential access to priority review of marketing applications and eligibility for fast track and priority review designations by the FDA. We expect to pursue a priority review designation for Cogenzia. A priority review designation means that the FDA’s goal is to take action on an application within 6 months, compared to 10 months under standard review. However, QIDP Designation for Cogenzia does not guarantee priority review designation by the FDA.
Non-United States Government Regulation
In addition to regulations in the United States, we will be subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales, promotion and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. We, or our local partners, have filed marketing authorization applications for Cogenzia in Argentina, Australia, India, Mexico and the Russian Federation and have obtained approval for Cogenzia in Canada, Jordan and Saudi Arabia. We plan to submit a Community MA for Cogenzia in the EU in the event our Phase 3 trials for Cogenzia are successful. We, or our local partners, have also filed marketing authorization applications for CollaGUARD in Argentina, Australia, Canada, Hong Kong, Mexico, Belarus, Kazakhstan and Taiwan. We obtained a CE Mark for CollaGUARD in the EU in October 2011 and have received approval for CollaGUARD in India, Israel, the Philippines, the Russian Federation, Saudi Arabia, Singapore, Thailand and Vietnam and require no further registration of approval to market CollaGUARD in New Zealand.
Non-United States Government Regulation Applicable to Drugs
Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like an IND prior to the commencement of human clinical trials. If we fail to comply with applicable foreign regulatory requirements, we may be subject in those countries to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
In the EEA (which is comprised of the 28 Member States of the European Union plus Iceland, Liechtenstein and Norway), for example, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of marketing authorizations:
| · | The Community MA, which is issued by the European Commission through the Centralized Procedure, based on the opinion of the EMA Committee for Medicinal Products for Human Use (CHMP), and which is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, advanced-therapy medicines, orphan medicinal products, and medicinal products containing a new active substance indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the European Union. |
| · | National MAs, which are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA (the Reference Member State, or RMS), this National MA can be recognized in other Member States (the Concerned Member States, or CMS) through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. Under the Decentralized Procedure, an identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the RMS. The competent authority of the RMS prepares a draft assessment report, a draft summary of the product characteristics, or SPC, and a draft of the labeling and package leaflet, which are sent to the CMS for their approval. If the CMS raise no objections, based on a potential serious risk to public health, to the assessment, SPC, labeling, or packaging proposed by the RMS, the product is subsequently granted a national MA in all the Member States (i.e., in the RMS and the CMS). If one or more CMS raise objections based on a potential serious risk to public health, the application is referred to the Coordination group for mutual recognition and decentralized procedure for human medicinal products (the CMDh), which is composed of representatives of the EEA Member States. If a consensus cannot be reached within the CMDh the matters is referred for arbitration to the CHMP, which can reach a final decision binding on all EEA Member States. A similar process applies to disputes between the RMS and the CMS in the Mutual Recognition Procedure. |
As with FDA approval, we may not be able to secure regulatory approvals in Europe in a timely manner, if at all. Additionally, as in the United States, post-approval regulatory requirements, such as those regarding product manufacture, marketing, or distribution, would apply to any product that is approved in Europe, and failure to comply with such obligations could have a material adverse effect on our ability to successfully commercialize any product.
With respect to the conduct of clinical trials in the European Union a clinical trial application, or CTA, must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and IRB requirements in the United States, respectively. Generally, once the CTA is approved in accordance with a country’s requirements, clinical trials may proceed.
In addition to regulations in Europe and the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial distribution of any future products. For other countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with cGCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
Non-United States Government Regulation Applicable to Medical Devices
The advertising and promotion of our products in the EEA is subject to the provisions of the Medical Devices Directive, Directive 2006/114/EC concerning misleading and comparative advertising, and Directive 2005/29/EC on unfair commercial practices, as well as other national legislation in the EEA countries governing the advertising and promotion of medical devices. The European Commission has submitted a Proposal for a Regulation of the European Parliament and the Council on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009, to replace, inter alia, Directive 93/42/EEC and to amend regulations regarding medical devices in the European Union, which could result in changes in the regulatory requirements for medical devices in Europe. In Germany, the advertising and promotion of our products can also be subject to restrictions provided by the German Act Against Unfair Competition (Gesetz gegen den unlauteren Wettbewerb) and the law on the advertising of medicines (Heilmittelwerbegesetz), criminal law, and some codices of conduct with regard to medical products and medical devices among others. These laws may limit or restrict the advertising and promotion of our products to the general public and may impose limitations on our promotional activities with healthcare professionals.
In Ireland, the advertising and promotion of medicinal products to either healthcare professionals or the general public alike are governed by a combination of legislation including consumer protection laws, advertising of human medicinal product laws, competition and criminal laws and various codes of conduct, including in relation to advertising standards. The HPRA is the body responsible for monitoring the advertising of medicinal products and the enforcement of the relevant regulations.
Sales of medical devices are subject to foreign government regulations, which vary substantially from country to country. In order to market our products outside the United States, we must obtain regulatory approvals or CE Certificates of Conformity and comply with extensive safety and quality regulations. The time required to obtain approval by a foreign country or to obtain a CE Certificate of Conformity may be longer or shorter than that required for FDA clearance or approval, and the requirements may differ. In the EEA, we are required to obtain Certificates of Conformity before drawing up an EC Declaration of Conformity and affixing the CE Mark of conformity to our medical devices. Any medical device being advertised in Ireland must have a CE mark and such advertisements must comply with the Advertising Standards Authority for Ireland’s Code of Standards for Advertising, Promotional and Direct Marketing in Ireland. Many other countries, such as Australia, India, New Zealand, Pakistan and Sri Lanka, accept CE Certificates of Conformity or FDA clearance or approval although others, such as Brazil, Canada and Japan require separate regulatory filings.
Reimbursement
Sales of our products will depend, in part, on the extent to which our products will be covered by third-party payors, such as government health care programs, statutory health insurances, commercial insurance and managed healthcare organizations. These third-party payors are increasingly reducing reimbursements for medical products and services. In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, competitive bidding program, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our product candidates or a decision by a third-party payor to not cover our product candidates could reduce physician usage of our products once approved and have a material adverse effect on our sales, results of operations and financial condition.
Fraud and Abuse Laws
We will also be subject to several healthcare regulation and enforcement by the federal government and the states and foreign governments in which we will conduct our business once our products are approved. The laws that may affect our ability to operate include:
| · | the federal healthcare programs’ Anti-Kickback Law, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; |
| · | federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent; |
| · | federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
| · | federal Civil Monetary Penalties Law that prohibits various forms of fraud and abuse involving the Medicare and Medicaid programs; |
| · | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers; |
| · | for Europe, directive 2006/114/EC concerning misleading and comparative advertising, and Directive 2005/29/EC on unfair commercial practices, as well as other national legislation in the European Union governing the advertising and promotion of medical devices; and |
| · | in Germany the advertising and promotion of our products can be subject to restrictions provided by the German Act Against Unfair Competition protecting against commercial practices which unacceptably harass a market participants. |
Healthcare Privacy and Security Laws
We may be subject to, or our marketing activities may be limited by the federal Health Insurance Portability and Accountability Act of 1996, HIPAA, and its implementing regulations, which established uniform standards for certain “covered entities” (healthcare providers, health plans and healthcare clearinghouses) governing the conduct of certain electronic healthcare transactions and protecting the security and privacy of protected health information. The American Recovery and Reinvestment Act of 2009, commonly referred to as the economic stimulus package, included sweeping expansion of HIPAA’s privacy and security standards called the Health Information Technology for Economic and Clinical Health Act, or HITECH, which became effective on February 17, 2010. Among other things, the new law makes HIPAA’s privacy and security standards directly applicable to “business associates,” independent contractors or agents of covered entities that receive or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions.
In Europe and Germany, we may be subject to strict data protection regulations, in particular with regard to health data of individuals, which are categorized as “special categories of personal data” pursuant to Section 3 subsection 9 German Federal Data Protection Act (Bundesdatenschutzgesetz). “Personal data” refers to any information relating to an identified or identifiable natural person (data subject); an identifiable person is one who can be identified, directly or indirectly, in particular by reference to an identification number or to one or more factors specific to his physical, physiological, mental, economic, cultural or social identity. The special categories of data such as health data may only be processed if the data subject consented to such processing or if (i) this is necessary in order to protect vital interests of the data subject or of a third-party, in so far as the data subject is unable to provide consent for physical or legal reasons; (ii) the data concerned have evidently been made public by the data subject; (iii) this is necessary in order to assert, exercise or defend legal claims and there is no reason to assume that the data subject has an overriding legitimate interest in excluding such collection, processing or use; or (iv) this is necessary for the purposes of scientific research, where the scientific interest in carrying out the research project substantially outweighs the data subject’s interest in excluding collection, processing and use and the purpose of the research cannot be achieved in any other way or would otherwise necessitate disproportionate effort. Therefore, we may be subject to and our marketing activities may be limited by the regulations regarding the data protection of individuals (e.g., according to the Directive 95/46/EC of the European Parliament and of the Council of 24 October 1995 on the protection of individuals with regard to the processing of personal data and on the free movement of such data as well as to the German Federal Data Protection Act). These regulations could also restrict the transfer of data from Germany/Europe to the United States. The general transfer of personal data outside of Europe is prohibited according to Section 4b subsection 2 sentence 2 German Federal Data Protection Act (implementing Art. 25 subsection 1 of the Directive 95/46/EC) if the data importer cannot guarantee an appropriate standard of data protection. A transfer of personal data to a non-EU member state (third country) is allowed only if the third country guarantees a reasonable standard of protection. Currently the United States is not regarded to be a country with an appropriate level of data protection meaning that further contractual arrangements have to be adopted to permit the international transfer of personal data to the United States. European data protection law is currently under review. A newly proposed European Data Protection Regulation is currently being negotiated by the European institutions. On March 12, 2014, the European Parliament voted for a new Regulation of the European Parliament and of the Council on the protection of individuals with regard to the processing of personal data and on the free movement of such data (General Data Protection Regulation) which is expected to further strengthen the European data protection law.
| C. | ORGANIZATIONAL STRUCTURE |
The registrant, Innocoll Holdings plc, an Irish corporation, has four direct and indirect subsidiaries which are listed on Exhibit 8.1 hereto. We primarily operate our business out of our Irish subsidiary Innocoll Pharmaceuticals Ltd., an Irish private limited company, and our U.S. subsidiary, Innocoll, Inc. Syntacoll GmbH, a German limited liability company and a wholly-owned subsidiary of Innocoll Pharmaceuticals Ltd. is responsible for the manufacturing of our products and product candidates. See Note 23 to our consolidated financial statements included in this annual report for the proportion of ownership interest in our subsidiaries.
| D. | PROPERTY, PLANT AND EQUIPMENT |
Our manufacturing facility is located on a site in Saal, Germany and consists of two production facilities, which occupy a total of approximately 30,000 square feet, and an office building of approximately 6,500 square feet. We have leased the facilities pursuant to lease agreement dated April 2009 for a 10-year term, which is subject to additional five-year renewal options. Activities in this facility include the manufacture of all our products and product candidates and quality control testing, product storage, development of analytical methods, research and development, the coordination of clinical and regulatory functions, and general administrative functions. We have also leased space in Saal, Germany, including a workshop building, for a 15-year term, which is subject to additional five-year renewal options. This property will be used as an office, a laboratory, a warehouse, and for the production of pharmaceutical products and products under the German Medical Device Act. The monthly rent is €15,494.50 plus the statutory Value Added Tax (“V.A.T.”) applicable from time to time. With the current V.A.T. of 19%, equaling €2,943.96, the gross rent is €18,438.46. We expect to complete a capacity expansion build-out of our production to approximately 50,000 square feet to expand its production capacity to support commercial production of XaraColl and Cogenzia, and the expanded facility is expected to be fully operational by the second half of 2016. Once completed and approved, we anticipate that our production capacity will be sufficient to meet currently forecasted market demand (including those marketed and sold through our partners) for all our current and late-stage pipeline product candidates. We believe that once our factory expansion program has been completed, our facility will be adequate to meet our current needs, and that suitable additional alternative spaces will be available in the future on commercially reasonable terms, if required.
Our corporate headquarters are located in Athlone, Ireland, where we lease an office. We have leased approximately 6,824 square feet of property in Newton Square, PA in the U.S. for a corporate office pursuant to a lease agreement dated October 2015 for a three-year term. The monthly rent charged in the first year under the lease is $15,069.67, which amount shall annually be increased over the three-year term. This lease replaced a previous lease of different property in Newton Square, PA for 2,606 square feet.
Item 4A. Unresolved Staff Comments
None.
Item 5. Operating and Financial Review and Prospects
You should read the following discussion and analysis of our financial condition and results of operations together with the information under “Selected Financial Data” and our consolidated audited financial statements, including the notes thereto, included in this annual report. The following discussion is based on our financial information prepared in accordance with IFRS as issued by the IASB, which might differ in material respects from generally accepted accounting principles in other jurisdictions. Statements in this annual report concerning our business outlook or future economic performance; anticipated revenues, expenses or other financial items; and statements concerning assumptions made or expectations as to any future events, conditions, performance or other matters, are “forward-looking statements” as that term is defined under the United States Federal securities laws. Forward-looking statements are subject to risks, uncertainties and other factors which could cause actual results to differ materially from those stated in such statements. Factors that could cause or contribute to such differences include, but are not limited to, those set forth under “Item 3. Key Information – D. Risk Factors” in this annual report as well as those discussed elsewhere in this annual report and in our other filings with the Securities and Exchange Commission.
Overview
We are a global, commercial stage, specialty pharmaceutical and medical device company, with late-stage development programs targeting areas of significant unmet medical need. We were incorporated in Delaware under the name Innocoll, Inc. in December 1997 and renamed Innocoll Holdings, Inc. in May 2004. Since 2004, our research and business development has been led and coordinated by our Irish subsidiaries. These Irish entities own our intellectual property and will be subject to Irish corporate tax on their future profits after the utilization of their tax losses. The applicable Irish corporate tax rate is 12.5%. In July 2013, we re-domiciled Innocoll Holdings, Inc. from the United States to Germany pursuant to a contribution-in-kind and share for share exchange into Innocoll GmbH, a German limited liability company, as a result of which Innocoll Holdings, Inc. became Innocoll GmbH’s wholly-owned subsidiary. On July 3, 2014, Innocoll GmbH transformed into a German stock corporation, “Innocoll AG.” Innocoll AG was effectively managed and controlled from Ireland and had, therefore, become tax resident in Ireland under the terms of the Ireland-Germany double tax treaty with effect as of January 1, 2014. On March 16, 2016, Innocoll AG merged with Innocoll Holdings plc, a public limited company formed under Irish law, by way of a European cross-border merger with Innocoll Ireland being the surviving company. Prior to the merger Innocoll Holdings plc was a dormant company with minimal legally required share capital and related debtor on the statement of financial position. The Merger effectively resulted in Innocoll Ireland becoming the publicly-traded parent of the Innocoll group of companies carrying on the same business as that conducted by Innocoll Germany prior to the Merger. Accordingly, the consolidated information presented herein refers to Innocoll Holdings, Inc., as the “company,” and with its direct and indirect subsidiaries, collectively, as the “group,” for the period from January 1, 2012 until July 24, 2013 and to Innocoll AG, as the “company,” and with its direct and indirect subsidiaries, collectively, as the “group,” for the period from July 25, 2013 until December 31, 2015. Except where indicated to the contrary, or the context suggests otherwise, all share and per share information: (i) not presented in the financial statements or financial statement data refers to ordinary shares of Innocoll Holdings plc, and (ii) included in the financial statements and financial statement data for Innocoll AG refers to ordinary shares of Innocoll Germany prior to the Merger. Innocoll Ireland, together with its direct and indirect wholly owned subsidiaries as of the time relevant to the applicable reference, are collectively referred to as the Innocoll Group. Innocoll and its current direct and indirect wholly owned subsidiaries are collectively referred to as “we,” “us,” “our,” “Innocoll” or the “Company.”
As of December 31, 2015, we had four marketed products: (i) CollaGUARD, which utilizes our CollaFilm® technology, a transparent, bioresorbable collagen film for the prevention of post-operative adhesions in multiple surgical applications, including digestive, colorectal, gynecological and urological surgeries; (ii) Collatamp Gentamicin Surgical Implant, or CollatampG, which utilizes our CollaRx® sponge technology indicated for the treatment or prevention of post-operative infection; (iii) Septocoll®, a bioresorbable, dual-action collagen sponge, indicated for the treatment or prevention of post-operative infection, which we manufacture and supply to Biomet Orthopedics Switzerland GmbH, or Biomet; and (iv) RegenePro, a bioresorbable collagen sponge for dental applications which we manufacture and supply to Biomet 3i.
All of our marketed products are currently sold by commercial partners. CollatampG has been marketed outside of the United States since 1985, and is currently approved for sale in 62 countries across Africa, Asia, Europe, Latin America and the Middle East. EUSA Pharma has rights to distribute CollatampG in all worldwide markets, excluding the United States. We have been supplying Septocoll to Biomet since 2001, for distribution to markets in Europe and the Middle East. CollaGUARD has been approved in 12 countries within Asia, Europe, Latin America and the Middle East. RegenePro was launched by Biomet 3i in the United States in July 2014.
From 2004 to 2015, we incurred significant operating losses including a total of €75.8 million spent on research and development in relation to our products and product candidates. Our operating losses were €44.2 million, €16.1 million, and €6.9 million for the years ended December 31, 2015, 2014 and 2013, respectively. We do not expect that our currently marketed products alone will generate revenue that is sufficient for us to achieve profitability because we expect to continue to incur significant expenses as we advance the development of XaraColl, Cogenzia, CollaGUARD and our other product candidates, seek FDA and other regulatory approval for our product candidates that successfully complete clinical trials and develop our sales force and marketing capabilities to prepare for the commercial launch of our product candidates. We also expect to incur additional expenses expanding our operational, financial and management information systems and personnel, including personnel to support our product development efforts and our obligations as a public reporting company. For us to become and remain profitable, we believe that we must succeed in commercializing XaraColl, Cogenzia, CollaGUARD or other product candidates with significant market potential.
For further information regarding our business and operations, see Item 4, “Information on the Company.”
Financial Operations Overview
Revenues
In the years ended December 31, 2015, 2014 and 2013, our revenue was derived primarily from the supply of CollaGUARD, CollatampG and Septocoll, our products manufactured by us and sold by our commercial partners. Supply revenue is derived from a contractual supply price paid to us by our commercial partners. In the case of some of our distribution agreements, including for the sale of CollatampG, the supply price is a contractually agreed percentage of the net sales price received by our distribution partner, which is net of discounts, returns, and allowances incurred. In this case, revenue is recognized in two parts: the first amount is recognized for the manufacture and sale of the product at the point of sale, and the final amount is added or deducted when the product is sold by the distributor based upon the net sale price achieved.
Accordingly, the primary factors that determine our revenues derived from our products are:
| · | the level of orders submitted by our commercial partners; |
| · | the level of prescription and institutional demand for our products; |
| · | the amount of gross-to-net sales adjustments realized by our commercial partners. |
The following table sets forth a summary of our revenues by product for the years ended December 31, 2015, 2014 and 2013. The reasons for the decrease in revenues in the years ended December 31, 2015 as compared to December 31, 2014 and the increase in revenues in the year ended December 31, 2014 are more fully described in “—Results of Operations”:
| | | Year Ended
December 31, | | | Increase/ | | | % Increase/ | | | Year Ended December 31, | | | Increase/ | | | % Increase/ | |
| | | 2015 | | | 2014 | | | (Decrease) | | | (Decrease) | | | 2014 | | | 2013 | | | (Decrease) | | | (Decrease) | |
| | | (euros in thousands) | | | | | | | | | (euros in thousands) | | | | | | | |
| CollaGUARD | | € | 3 | | | € | 349 | | | € | (346 | ) | | | (99.0 | )% | | € | 349 | | | € | 151 | | | € | 198 | | | | 131.1 | % |
| CollatampG | | | 2,246 | | | | 3,563 | | | | (1,317 | ) | | | (37.0 | )% | | | 3,563 | | | | 2,840 | | | | 723 | | | | 25.5 | % |
| Septocoll | | | 296 | | | | 515 | | | | (219 | ) | | | (42.5 | ))% | | | 515 | | | | 552 | | | | (37 | ) | | | (6.7 | )% |
| Other(1) | | | 42 | | | | 70 | | | | (28 | ) | | | (40.0 | )% | | | 70 | | | | 3 | | | | 67 | | | | 2,233.3 | % |
| Total | | € | 2,587 | | | € | 4,497 | | | € | (1,910 | ) | | | (42.5 | )% | | € | 4,497 | | | € | 3,546 | | | € | 951 | | | | 26.8 | % |
(1)Includes supply revenue of RegenePro™, Zorpreva™ and Collexa™, insurance proceeds and license fees.
Cost of Sales
Cost of sales consists of the costs associated with producing our products for our commercial partners. In particular, our cost of sales includes:
| · | manufacturing overhead and fixed costs associated with running our manufacturing facilities in Saal, Germany, including salaries and related costs of personnel involved with our manufacturing activities; |
| · | packaging, testing, freight and shipping; and |
| · | the cost of raw materials and active pharmaceutical ingredients. |
All overhead costs associated with operating the manufacturing facility are included in the standard cost of our products as indirect costs and charged to cost of revenue based on sales volume.
Gross margins from supply revenue were (86%), (24%) and (28%) for the years ended December 31, 2015, 2014 and 2013, respectively. Our negative margin is primarily due to absorbing full indirect costs into the standard cost of our products. The amount of indirect costs included in cost of sales was €1.4 million, €2.7 million and €2.2 million for the years ended December 31, 2015, 2014 and 2013, respectively. Excluding these indirect costs, gross margins from supply revenue were (32%), 36% and 34% for the years ended December 31, 2015, 2014 and 2013, respectively.
As we expand our manufacturing capacity in 2016 (as described in “Business—Facilities”) and increase production with the launch of new products, our indirect costs included in cost of sales will remain relatively fixed and therefore will decrease as a proportion of sales. Our direct costs consist primarily of labor associated with our batch production processes, quality control and manual packaging. In our expanded facility we will produce larger batch sizes and further automate our production and packaging processes, resulting in decreased labor costs per unit produced. Accordingly, our gross margins are expected to increase significantly with growth in product sales and expanded production capacity.
Research and Development Expenses
Our research and development expenses consist of expenses incurred in developing, testing, manufacturing and seeking regulatory approval of our product candidates, including:
| · | expenses associated with regulatory submissions, clinical trials and manufacturing, including additional expenses to prepare for the commercial manufacture of XaraColl, Cogenzia and CollaGUARD, such as the hiring and training of additional personnel; |
| · | payments to third-party contract research organizations, contract laboratories and independent contractors; |
| · | payments made to regulatory consultants; |
| · | payments made to third-party investigators who perform clinical research on our behalf and clinical sites where such testing is conducted; |
| · | personnel related expenses, such as salaries, benefits, travel and other related expenses, including share-based compensation; |
| · | expenses incurred to maintain regulatory licenses, patents and trademarks; and |
| · | facility, maintenance, and allocated rent, utilities, and depreciation and amortization, and other related expenses. |
Clinical trial expenses for our product candidates are a significant component of our research and development expenses. Product candidates in late-stage clinical development, such as XaraColl, Cogenzia and CollaGUARD, generally have higher research and development expenses than those in earlier stages of development, primarily due to the increased size and duration of the clinical trials. We coordinate clinical trials through a number of contracted investigational sites and recognize the associated expense based on a number of factors, including actual and estimated subject enrollment and visits, direct pass-through costs and other clinical site fees.
From January 1, 2004 through December 31, 2015, we incurred research and development expenses of €75.8 million net of contributions and collaboration agreements with third parties. We incurred below average research and development expenses of €3.3 million and €1.7 million for the years ended December 31, 2014 and 2013, respectively, because substantially all of our Phase 2 clinical trials were completed prior to 2012 and we did not commence Phase 3 trials for XaraColl until the third quarter of 2014.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries, benefits and other related costs, including share-based compensation, for personnel serving in our executive, finance, business development, clinical and regulatory and administrative functions. Our general and administrative expenses also include facility and related costs not included in research and development expenses and cost of revenues, professional fees for legal, consulting, tax and accounting services, insurance, depreciation and general corporate expenses.
We incurred general and administrative expenses of €19.0 million, €11.7 million and €4.1 million for the years ended December 31, 2015, 2014 and 2013, respectively. General and administrative expenses in the year ended December 31, 2015 included €4.8 million in non-cash charges for stock-based compensation, compared to €5.1 of such charges in the corresponding period in 2014. Excluding stock-based compensation, our general and administrative expenses for the year ended December 31, 2015 were €14.2 million, compared to €6.6 million for the year ended December 31, 2014. The increase in general and administrate expenses, excluding stock-based compensation, was primarily due to our continued infrastructure build-out to support clinical programs, initiation of our pre-commercialization investment and expenses related to our re-domiciliation to Ireland. Additionally, we plan to build a larger commercial infrastructure for the anticipated future launches of XaraColl, Cogenzia and CollaGUARD, and we currently plan to hire our own sales force if Cogenzia is approved by the FDA.
Finance Income/(Expense)
Finance income/(expense) consists of interest income and interest expense, expense from the issue or settlement of options on shares of Innocoll Germany, fair value gain (expense) on investor options outstanding, and foreign exchange gains (losses). Interest income consists of interest earned on our cash and cash equivalents. Finance income in the year ended December 31, 2015 consisted primarily of foreign exchange gains, partially offset by fair value expense of investor options outstanding. Finance expense for the year ended December 31, 2014 consisted primarily of fair value expense of investor options outstanding, partially offset by foreign exchange gains, and for the year ended December 31, 2013 consisted primarily of non-cash interest charges and accruals related to preferred shares and convertible notes issued to certain of our investors. We incurred finance income/(expense) of €1.4 million, (€4.5) million and (€6.9) million in the years ended December 31, 2015, 2014 and 2013, respectively.
Other Income
Other income in the year ended December 31, 2013 consists of non-cash fair value gains on exchange of warrants, and settlement of preferred shares and extinguishment of related liabilities in connection with the debt for equity exchange for preferred shares in June 2013 and the re-domicile of our parent company to Germany in July 2013 (the “Re-domicile of Parent Company,” as described more fully in “—Results of Operations—Financing Activities”). For the year ended December 31, 2013, we had exceptionally high other income of €16.1 million arising from the re-domicile of our parent company. In accordance with IAS 39, the liability associated with the series B convertible stock issued by Innocoll Holdings, Inc. was deemed to be extinguished as the new financial instruments issued had significantly different terms to the instruments they replaced. The difference between the fair value of the new financial instrument, and the carrying value of the extinguished series B convertible stock, was allocated against the carrying value of the liability and equity components with a resulting gain being recognized in profit and loss as noted above. Other income in the year ended December 31, 2014 was €0.1 million representing profit on the sale of a certain property, plant and equipment. Other income in the year ended December 31, 2015 was €0.0.
(Loss)/earnings per Share
The weighted-average number of Innocoll Germany ordinary shares (denominator – basic) was 1,685,088 at December 31, 2015 (2014: 735,416 and 2013: 48,848). The weighted-average number of historic ordinary shares for December 31, 2013 have been adjusted for the effects of the re-domicile of our then parent company, and the Innocoll GmbH conversion of preferred shares into ordinary shares and subsequent transformation into Innocoll AG which took place during the relevant financial period. The weighted-average number of ordinary shares of Innocoll Germany outstanding during the period and for all periods presented is adjusted for events, other than the conversion of potential ordinary shares of Innocoll Germany, that have resulted in a change of the number of ordinary shares of Innocoll Germany outstanding without a corresponding change in resources.
| | | | | | For the Year Ended December 31, | |
| | | 2015 | | | 2015 | | | 2014 | | | 2013 | |
| | | | | | (in thousands, except for per share data) | |
| Numerator: | | | | | | | | | | | | | | | | |
| Net (loss)/earnings – basic | | $ | (46,977 | ) | | € | (43,149 | ) | | € | (20,666 | ) | | € | 2,109 | |
| Adjustment to net earnings for interest on convertible preferred shares(1) | | | - | | | | - | | | | — | | | | 4,728 | |
| Adjustment to net earnings for interest on convertible promissory notes(1) | | | - | | | | - | | | | — | | | | 1,918 | |
| Gain on settlement of promissory notes and preferred stock and extinguishment of related liabilities | | | - | | | | - | | | | — | | | | (15,903 | ) |
| Net loss – diluted | | | (46,977 | ) | | | (43,149 | ) | | | (20,666 | ) | | | (7,148 | ) |
| Denominator – number of shares: | | | | | | | | | | | | | | | | |
| Weighted-average shares outstanding – basic | | | 1,685,088 | | | | 1,685,088 | | | | 734,482 | | | | 44,848 | |
| Dilutive ordinary shares issuable upon conversion of preferred shares(1) | | | - | | | | - | | | | — | | | | 547,195 | |
| Dilutive common stock issuable upon conversion of promissory notes(1) | | | - | | | | - | | | | — | | | | 160,246 | |
| Weighted-average shares outstanding - diluted | | | 1,685,088 | | | | 1,685,088 | | | | 734,482 | | | | 752,289 | |
| (Loss)/profit per share: | | | | | | | | | | | | | | | | |
| Basic | | | (27.9 | ) | | | (25.6 | ) | | | (28.1 | ) | | | 47.0 | |
| Diluted | | | (27.9 | ) | | | (25.6 | ) | | | (28.1 | ) | | | (9.5 | ) |
| Loss per ADS(2): | | | | | | | | | | | | | | | | |
| Basic | | | (2.1 | ) | | | (1.9 | ) | | | (2.1 | ) | | | | |
| Diluted | | | (2.1 | ) | | | (1.9 | ) | | | (2.1 | ) | | | | |
(1) For the years ended December 31, 2015 and 2014, we excluded the dilutive effect of potentially exercisable instruments in issue from the computation of the diluted net loss and diluted weighted-average shares outstanding as the effect would be anti-dilutive.
(2) Prior to the termination of Innocoll Germany’s ADS facility, one ordinary share of Innocoll Germany represented 13.25 ADSs of Innocoll Germany. Upon consummation of the Merger, each ADS of Innocoll AG was exchanged for one ordinary share of Innocoll Holdings plc. Going forward, Innocoll Holdings plc’s earnings/(loss) per share calculation will be equivalent to the earnings/(loss) per ADS calculation of Innocoll AG.
The basic (loss)/earnings per share of Innocoll Germany for the year ended December 31, 2015 was (€25.6) (2014: (€28.1) and 2013: €47.0). For the purpose of calculating diluted loss per share for 2015 and 2014 the potentially exercisable instruments in issue would have the effect of being antidilutive and, as such, the diluted loss per share is the same as the basic loss per share for those periods.
Non-GAAP Earnings per Share
The tables below include a reconciliation of our GAAP results to non-GAAP results for the years ended December 31, 2015, 2014 and 2013. We define adjusted non-GAAP earnings per share as basic and diluted earnings per share excluding share based payments, fair value expense on warrants and the reversal of impairment of property, plant and equipment. We believe adjusted non-GAAP earnings per share is meaningful to our investors to enhance their understanding of our financial condition and results. We believe that non-GAAP earnings per share excluding these non-cash items may provide securities analysts, investors and other interested parties with a useful measure of our operating performance and cash requirements. Disclosure in this annual report of non-GAAP earnings per share, which is a non-IFRS financial measure, is intended as a supplemental measure of our performance that is not required by, or presented in accordance with, IFRS. Non-GAAP earnings per share should not be considered as an alternative to earnings per share, profit (loss) or any other performance measure derived in accordance with IFRS. Our presentation of adjusted earnings per share of Innocoll Germany should not be construed to imply that our future results will be unaffected by unusual non-cash or non-recurring items.
For the year ended December 31, 2015 the reconciliation primarily relates to non-cash expenses in the amount of €4.8 million with respect to share-based compensation, €3.7 million with respect to fair value expense on warrants and (€3.9) million with respect to the reversal of impairment of property, plant and equipment. On a non-GAAP-basis, the net loss for the year ended December 31, 2015 was €38.5 million, or €22.9 per share of Innocoll Germany ($1.88 per ADS of Innocoll Germany), compared to a net loss of €9.3 million, or €12.6 per share of Innocoll Germany ($1.03 per ADS of Innocoll Germany) for the year ended December 31, 2014. The number of ordinary shares of Innocoll Germany outstanding increased from 0.74 million during the year ended December 31, 2014, to 1.69 million during the year ended December 31, 2015, primarily as a result of the conversion of preferred shares into ordinary shares of Innocoll Germany, and the follow-on public offering in the second quarter of 2015.
| | | Years Ended December 31, | |
| | | 2015 | | | 2015 | | | 2014 | | | 2013 | |
| | | (in thousands, except for per share data) | |
| Numerator for non-GAAP (loss)/earnings per share: | | | | | | | | | | | | | | | | |
| Net (loss)/earnings – basic | | $ | (46,977 | ) | | € | (43,149 | ) | | € | (20,666 | ) | | € | 2,109 | |
| Share based payments | | | 5,199 | | | | 4,775 | | | | 5,149 | | | | - | |
| Reversal of impairment of property, plant & equipment | | | (4,220 | ) | | | (3,876 | ) | | | - | | | | - | |
| Fair value expense on warrants | | | 4,066 | | | | 3,735 | | | | 6,265 | | | | 205 | |
| Non-GAAP net (loss)/earnings - basic | | | (41,932 | ) | | | (38,515 | ) | | | (9,252 | ) | | | 2,314 | |
| | | | | | | | | | | | | | | | | |
| Adjustment to net earnings for interest on convertible preferred shares | | | - | | | | - | | | | - | | | | 4,728 | |
| Adjustment to net earnings for interest on convertible promissory notes | | | - | | | | - | | | | - | | | | 1,918 | |
| Adjustment for gain on settlement of promissory notes and preferred stock | | | - | | | | - | | | | - | | | | (15,903 | ) |
| Non-GAAP net loss – diluted | | | (41,932 | ) | | | (38,515 | ) | | | (9,252 | ) | | | (6,943 | ) |
| | | | | | | | | | | | | | | | | |
| Denominator – number of shares: | | | | | | | | | | | | | | | | |
| Weighted-average shares outstanding – basic | | | 1,685,088 | | | | 1,685,088 | | | | 735,416 | | | | 44,848 | |
| Dilutive common stock issuable upon conversion of preferred shares (1) | | | - | | | | - | | | | - | | | | 547,195 | |
| Dilutive common stock issuable upon conversion of promissory notes (1) | | | - | | | | - | | | | - | | | | 160,246 | |
| Weighted-average shares outstanding – diluted | | | 1,685,088 | | | | 1,685,088 | | | | 735,416 | | | | 752,289 | |
| | | | | | | | | | | | | | | | | |
| Non-GAAP (loss)/earnings per share: | | | | | | | | | | | | | | | | |
| Basic | | | (24.9 | ) | | | (22.9 | ) | | | (12.6 | ) | | | 51.6 | |
| Diluted | | | (24.9 | ) | | | (22.9 | ) | | | (12.6 | ) | | | (9.2 | ) |
| | | | | | | | | | | | | | | | | |
| Non-GAAP loss per ADS(2): | | | | | | | | | | | | | | | | |
| Basic | | | (1.9 | ) | | | (1.7 | ) | | | (1.0 | ) | | | | |
| Diluted | | | (1.9 | ) | | | (1.7 | ) | | | (1.0 | ) | | | | |
| | | | | | | | | | | | | | | | | |
(1) For the years ended December 31, 2015 and 2014, we excluded the dilutive effect of potentially exercisable instruments in issue from the computation of the diluted net loss and diluted weighted-average shares outstanding as the effect would be anti-dilutive.
(2) Prior to the termination of Innocoll Germany’s ADS facility, one ordinary share of Innocoll Germany represented 13.25 ADSs of Innocoll Germany. Upon consummation of the Merger, each ADS of Innocoll AG was exchanged for one ordinary share of Innocoll Holdings plc. Going forward, Innocoll Holdings plc’s earnings/(loss) per share calculation will be equivalent to the earnings/(loss) per ADS calculation of Innocoll AG.
Critical Accounting Policies and Use of Estimates
We have based our management’s discussion and analysis of financial condition and results of operations on our financial statements that have been prepared in accordance with International Financial Reporting Standards (IFRS) issued by the International Accounting Standards Board (IASB). The preparation of these financial statements requires us to make estimates that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements as well as the reported revenues and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments. We base our estimates on historical experience and on various other factors we believe to be appropriate under the circumstances. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully discussed in note 1 to our consolidated financial statements included in this annual report, we believe that the following accounting policies are critical to the process of making significant judgments and estimates in the preparation of our financial statements. We have reviewed these critical accounting policies and estimates with the audit committee of our advisory board:
Revenue Recognition
Significant management judgments and estimates must be made and used in connection with the recognition of revenue in each accounting period. Material differences in the amount of revenue in any given period may result if these judgments or estimates prove to be incorrect or if management’s estimates change on the basis of development of the business or market conditions. To date there have been no material differences arising from these judgments and estimates.
Revenue from products is generally recorded as of the date of shipment, consistent with our typical shipment terms. Where the shipment terms do not permit revenue to be recognized as of the date of shipment, revenue is recognized when the group has satisfied all of its obligations to the customer in accordance with the shipping terms.
Revenue is recognized to the extent that it is probable that economic benefit will flow to the group, that the risks and rewards of ownership have passed to the buyer and that the revenue can be measured. No revenue is recognized if there is uncertainty regarding recovery of the consideration due at the outset of the transaction or the possible return of goods.
Revenue is recognized from milestone payments received under collaboration agreements when earned, provided that the milestone event is substantive, its achievability was not reasonably assured at the inception of the agreement, the group has no further performance obligations relating to the event, and collectability is reasonably assured. If these criteria are not met, the group recognizes milestone payments ratably over the remaining period of their performance obligations under the collaboration agreement.
Share-Based Payments
Stock-based compensation expense related to share-based awards granted to employees is measured at the date of grant based on the estimated fair value of the award, net of estimated forfeitures. The charge to comprehensive income in relation to share based payments in the years ending December 31, 2015, 2014 and 2013 were €4.8, €5.1 million and €0.0 million, respectively. With respect to any future stock-based awards, the company plans to re-evaluate each of the fair value assumptions, and revise them as appropriate, on an ongoing basis. We have granted restricted share, phantom share and option awards to members of our historic supervisory and management boards, and employees as follows:
January/March 2014 Grants
Under the 2014 restricted share awards, Innocoll GmbH granted to members of its advisory board, management board and group employees a total of 47,840 restricted shares and 63,256 phantom shares. The total restricted shares and phantom shares represented 12.2% of the diluted share capital of Innocoll GmbH at the time of grant, excluding issued and outstanding options to acquire ordinary shares. Innocoll Germany further created an authorized capital of 22,724 ordinary shares as part of its Authorized Capital II with the intention to grant options to acquire ordinary shares to certain members of Innocoll Germany’s advisory board, management board and employees, and these options were subsequently granted in December 2014 (see “—Management Options”). The restricted shares were issued in exchange for payment of nominal value of €47,840. The phantom shares may be either settled in cash or a new issue of shares, at our option, on the date on which the repurchase right lapses. For purposes of valuing the share-based payment in relation to the restricted share and phantom share grants, management performed the valuation with the assistance of a well-recognized independent third-party valuation consultant, which valued all classes of shares fully diluted for the issue of the restricted shares and phantom shares and options, on a pro-forma basis. The share-based payments will be recognized over a one-year period.
May 2014 Grants
Pursuant to a notarial deed entered into on May 22, 2014, Innocoll GmbH granted to new and existing members of its supervisory board, management board and group employees a total of 43,596 restricted shares, 9,113 phantom shares and 869 unrestricted shares to a former member of our supervisory board (the “May 2014 Grant”). The total restricted shares and phantom shares issued pursuant to the May 2014 grant represented 5.1% of the diluted share capital of Innocoll GmbH at the time of grant, excluding issued and outstanding options to acquire ordinary shares. Innocoll Germany further created an authorized capital of 2,060 ordinary shares as part of its Authorized Capital II with the intention to grant options to acquire ordinary shares to certain members of Innocoll Germany’s advisory board, management board and employees which options were subsequently granted in December 2014 (see “—Management Options”). The restricted shares were issued in exchange for payment of nominal value of €43,596. The phantom shares may be either settled in cash or a new issue of shares, at our option, on the date in which the repurchase right lapses. The May 2014 Grant occurred simultaneously with a new issue of series E preferred shares. The series E preferred shares were purchased in an arms-length transaction by existing investors, new supervisory board members who also received restricted stock grants, and three new investors who are partners of one of the existing shareholders. The pricing of the series E preferred shares at €112.52 per share was based upon our managing directors’ and our advisory board’s estimate, with the assistance of a third-party specialist, who concluded that the price of Innocoll Germany’s ADS in an anticipated initial public offering would be at least equal to such price or higher. To obtain the series E financing, it was necessary to give the investors anti-dilution rights in the event the initial public offering would be priced at a discount to the expected valuation. As a result of the initial public offering price of Innocoll Germany’s ADS of $9.00 per ADS, and the anti-dilution rights granted to the series E preferred investors, such investors have been issued additional shares of Innocoll Germany at €1.00 per share, as a result of which their weighted-average purchase price is equal to €73.76 per share, a 16.7% discount to the effective value per ordinary share of Innocoll Germany in our initial public offering (see “—Financing Events — Innocoll GmbH 2013 and 2014 Equity Financings”). There were no significant valuation events at the company between the May 2014 Grant date and date of our initial public offering in July 2014. Therefore, we have used the Series E preferred share adjusted weighted-average purchase price of €73.76 per share as the value for the share-based payment charge arising out of the May 2014 Grant. The share-based payments will be recognized over a one to three year period as set out above.
July 2014, January 2015 Amendments and Restatements
In July 2014, after the transformation of Innocoll GmbH into Innocoll AG, each of the restricted shares and phantom share awards issued in January 2014 and May 2014 were combined and applied to ordinary shares of Innocoll AG pursuant to amended and restated award agreements entered into with Innocoll AG. In January 2015, the phantom share award agreements were each amended and restated to provide for the definition of Exit Event triggering the lapse of the repurchase right by us to be extended to any trading day within the period beginning on the third trading day after the publication of our quarterly reports for the fourth quarter 2014, and the ending on the trading day immediately preceding the first trading day that is two weeks prior to the end of the first quarter 2015, which may be designated by the company’s management board in its sole discretion. There was no impact to the 2014 or 2015 financial statements as a result of this transaction.
Restricted Stock and Phantom Stock Valuations
The estimated fair market value of the shares issued pursuant to, and underlying the January 2014 grant and May 2014 grant, was determined at the grant date by our historic supervisory board and was deemed appropriate for the valuation of the grants pursuant to IFRS 2: Share-based payments.
The estimated fair value of the shares as of each grant date are based on numerous objective and subjective factors, combined with management’s judgment, including the following:
| · | the assistance of a well-recognized independent third-party valuation consultant in valuing our ordinary shares and series A through D-2 preferred shares, originally issued as of June 30, 2013 in connection with the re-domicile of our parent company, and updated as of December 31, 2013; |
| · | the estimated likelihood of achieving a liquidity event for our shares, such as an initial public offering or an acquisition of our company, given prevailing market conditions and other contingencies affecting the probability or potential timing of such an event; |
| · | the prices at which we sold series D preferred shares in arms-length transactions in October 2013 and November 2013 and the terms of the series D preferred shares relative to the terms of our series A, series B, series C and series D-2 preferred shares and ordinary shares; |
| · | the price at which we sold our shares of series E preferred shares in an arms-length transaction in May 2014 and the terms of the series E preferred shares relative to the terms of our series A, series B, series C, series D and series D-2 preferred shares and ordinary shares, as well as the near term probability that all preferred shares would be converted into ordinary shares pursuant to our planned transformation into Innocoll AG in connection with our initial public offering; |
| · | the subsequent issue of anti-dilution shares to holders of series E preferred and ordinary shares as result of the initial public offering price of our ADS of $9.00 per ADS; and |
| · | the fact that, at the time of the restricted share and phantom share grants, our shares were illiquid securities of a private company. |
Management performed the valuation of our ordinary shares and series A through D-2 preferred shares in the January/March 2014 grant with the assistance of a well-recognized independent third-party valuation consultant.
Valuation Approaches
Management, with the assistance of an independent third-party valuation consultant, considered several valuation approaches as follows: (1) Cost Approach; (2) Market Approach (including both the Guideline Public Company (“GPC”) method and the Guideline Merged and Acquired Company (“GMAC”) method); (3) Income Approach; (4) Current Value Method; (5) Probability Weighted Expected Return Method; (6) Option-Pricing Method; and (7) Back-Solve Method.
Management, with the assistance of an independent third-party valuation consultant, relied upon the income approach to determine the range of fair market value of equity of our company. Because our value is attributable to our business operations, rather than the assets we hold, management, with the assistance of the independent third-party valuation consultant, did not rely upon the cost approach in its conclusion of the range of the fair market value of our equity. Management, with the assistance of the independent third-party valuation consultant, also did not believe that the multiples of established publicly-traded companies provided reliable indicators of value given that we are relatively early-stage; therefore, they considered, but ultimately did not rely on the GPC method. They also considered, but ultimately did not rely on the GMAC method due to the lack of comparable transactions. As a corroborative approach, they utilized the Back-Solve Method to estimate our implied equity value as derived from the Option-Pricing Method, based on the most recent round of financing of the series D preferred shares.
Factors contributing to differences between grant date estimated fair values and the initial public offering price
We believe that the following factors, along with those set forth above with respect to each of the grants, explain the difference between the grant date estimated fair values of our shares on those grant dates and the initial public offering price of $9.00 per ADS of Innocoll Germany.
| · | In January 2014, we knew the initial public offering was a possibility but we were also considering other funding alternatives including the licensing of our products. As a independent third-party valuation was produced in connection with the re-domicile of our parent company in June 2013, updated to December 2013, it was determined that this valuation was a more reasonable estimate of the value than any potential initial public offering price which was far from certain and a potential liquidity event more distant. |
| · | The U.S. markets affecting companies in our industry experienced a significant increase in activities with accompanying increased market multiples and valuations. |
| · | In March 2014, the board determined to proceed with the initial public offering, and the underwriters were subsequently consulted as to the terms of the series E preferred financing, and advised that the valuation and anti-dilution terms reflected a reasonable discount to the expected initial public offering price. |
| · | Simultaneous with the May 2014 grant, we added to our historic supervisory board three prominent and experienced pharmaceutical and business executives: Jonathan Symonds, CBE, David R. Brennan and Shumeet Banerji, Ph.D. This significantly enhanced the value and profile of our company from an investor perspective and was instrumental in bringing new investors into the series E preferred funding round. |
| · | In June 2014, our pipeline product Cogenzia for the treatment of diabetic foot infections was approved in Canada, which was earlier than expected. |
| · | Subsequent to the confidential submission of our registration statement to the U.S. Securities and Exchange Commission on March 26, 2014, we engaged in discussions with potential investors in reliance on Section 5(d) of the Securities Act of 1933, as amended. In connection with such testing the waters meetings, we received positive feedback from potential investors which caused management to increase its expectations regarding the anticipated price range of an initial public offering of Innocoll Germany’s ordinary shares. |
| · | The ADSs of Innocoll Germany issued in the initial public offering were freely tradable in a public market, whereas the estimated fair value of the shares as of all of grant dates described above represents a contemporaneous estimate of the fair value of shares that were then illiquid, might never become liquid and, even if an initial public offering were successfully completed, would remain illiquid at least until the expiration of the 180-day lockup period following the initial public offering. This illiquidity also accounts for a substantial difference between the estimated fair values of the shares from January 2014 and the initial public offering price. |
| · | The holders of preferred shares enjoyed substantial economic rights and preferences over the holders of Innocoll Germany’s ordinary shares, including the right to receive dividends prior to any dividends declared or paid on any shares of Innocoll Germany’s ordinary shares and liquidation payments in preference to holders of ordinary shares. The initial public offering price was determined after the conversion of all of our convertible preferred shares prior to the completion of our initial public offering. The corresponding elimination of the preferences and rights enjoyed by the holders of such preferred shares results in a higher valuation. |
| · | The successful completion of an initial public offering would strengthen our balance sheet, provide access to public equity and debt markets and provide a “currency” of publicly tradable securities to enable us to make strategic acquisitions as our historic supervisory board may deem appropriate, providing enhanced operational flexibility. |
Management Options
In December 2014, the company entered into option agreements with members of its management board and group employees which memorialized options previously promised to them as consideration for past services. Pursuant to the terms of the Management Option Agreements, the members of the management board and group employees have the right to subscribe for 24,784 ordinary shares of Innocoll Germany at an exercise price of $119.25 per ordinary share of Innocoll Germany (equivalent to $9.00 per ADS of Innocoll Germany), which rights are exercisable through June 15, 2019, subject to certain black-out periods and which price is subject to adjustment in accordance with the Merger. Throughout 2015, the company entered into option agreements with members of its management board and group employees. Pursuant to the terms of the Management Option Agreements, the members of the management board and group employees have the right to subscribe for 86,144 ordinary shares of Innocoll Germany at exercise prices ranging from $99.38 to $195.70 per ordinary share of Innocoll Germany (equivalent to from $7.50 to $14.77 per ADS of Innocoll Germany), which rights are exercisable for a ten year period from date of grant, subject to certain black-out periods and which price is subject to adjustment in accordance with the Merger. In January, 2014, the company created an authorized capital to cover, among other issuances, the required share issuances under the Management Option Agreements. On June 15, 2019, any unexercised options expire pursuant to the terms of the Management Option Agreements, unless the shareholders approve the creation of a new authorized capital for an additional five-year period.
The share based payments cost of the options was calculated using a Black Scholes model. The following input assumptions were used to value the options:
| Weighted Average Values: | | 12/31/2015 | | | 12/31/2014 | |
| Expected volatility | | | 66.75 | % | | | 69.68 | % |
| Risk free rate | | | 2.03 | % | | | 1.66 | % |
| Exercise price | | € | 124.67 | | | € | 97.283 | |
| Contractual life | | | 8.78 years | | | | 4.52 years | |
Valuation of financial instruments
The group issued financial instruments during the relevant accounting periods and had financial instruments in issue at both accounting period ends. In conformity with IFRS, the group initially measured these financial instruments at their fair value and thereafter at amortized cost using the effective interest rate method or at fair value through the profit or loss if designated as such upon initial recognition. In order to value these various instruments, the group (and the experts engaged by the group to provide such valuations where applicable) made assumptions and estimates concerning variables such as future cashflows, discount rates, expected volatility, risk free rate and type of valuation models used. The assumptions of future outcomes, and other sources of estimation uncertainty concerning the determination of key inputs to the valuation models, are based on management’s (and relevant experts’) best assessment using the knowledge available, their historical experiences as well as other factors that are considered to be relevant. The estimates and assumptions are reviewed on an ongoing basis.
Taxation
Given the global nature of the business and the multiple taxing jurisdictions in which the group operates, the determination of the group’s provision for income taxes requires significant judgments and estimates, the ultimate tax outcome of which may not be certain. Although estimates are believed to be reasonable, the final outcome of these matters may be different than those reflected in the historical income tax provisions and accruals. Such differences could have a material effect on the income tax provision and results in the period during which such determination is made.
Deferred tax assets and liabilities are determined using enacted tax rates for the effects of net operating losses and temporary differences between the book and tax bases of assets and liabilities. In assessing the reliability of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. While management considers the scheduled reversal of deferred tax liabilities, and projected future taxable income in making this assessment, there can be no assurance that these deferred tax assets may be realizable.
In addition, the group may also be subject to audits in the multiple taxing jurisdictions in which it operates. These audits can involve complex issues which may require an extended period of time for resolution. Management believes that adequate provisions for income taxes have been made in the financial statements.
Allowance for slow-moving and obsolete inventory
The group evaluates the realizability of their inventory on a case-by-case basis and makes adjustments to the inventory provision based on their estimates of expected losses. The group writes off any inventory that is approaching its “use-by” date and for which no further re-processing can be performed. The group also considers recent trends in revenues for various inventory items and instances where the realizable value of inventory is likely to be less than its carrying value. Due to the fact that the allowance is calculated on the basis of the actual inventory on hand at the particular balance sheet date, there were no material changes in estimates made during the years ended December 31, 2015, 2014 or 2013 which would have had an impact on the carrying values of inventory during those periods.
Trade receivables
The group evaluates customer accounts with past-due outstanding balances or specific accounts for which it has information that the customer may be unable to meet its financial obligations. Based upon a review of these accounts and management’s analysis and judgment, the group estimates the future cash flows expected to be recovered from these receivables. The amount of the impairment on doubtful receivables is measured individually and recorded as a specific allowance against that customer’s receivable balance to the amount expected to be recovered. The allowance is re-evaluated and adjusted periodically as additional information is received.
Provisions
Provisions are recognized and measured on the basis of the estimate and probability of future outflows of resources embodying benefits, as well as on the basis of experiential values and the circumstances known at the end of the reporting period. Assumptions also are made as to the probabilities whether and within what ranges the provisions may be used. The assessment of whether a present obligation exists is generally based on assessment of internal experts. Estimates can change on the basis of new information and the actual charges may affect the performance and financial position of the group.
JOBS Act
As a company with less than $1.0 billion in revenues for our fiscal year ended December 31, 2015, we qualify as an “emerging growth company” as defined in Section 2(a) of the U.S. Securities Act of 1933, as amended, or the Securities Act, as modified by the Jumpstart Our Business Startups Act, or the JOBS Act, which was enacted in 2012. An emerging growth company may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including, but not limited to:
| · | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act; |
| · | being permitted to present only two years of audited financial statements and only two years of related Management’s Discussion and Analysis of Financial Condition and Results of Operations; |
| · | reduced disclosure obligations regarding executive compensation; and |
| · | not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements. |
We may choose to take advantage of some or all of the available exemptions and have taken advantage of some of these exemptions in this annual report. Accordingly, the information contained herein may be different from the information you receive from other public companies in which you hold shares. We do not know if some investors will find our ordinary shares less attractive as a result of our utilization of these or other exemptions. The result may be a less active trading market for our ordinary shares and increased volatility in the price of our ordinary shares.
In addition, Section 107 of the JOBS Act provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We currently prepare our financial statements in accordance with IFRS, as issued by the IASB, which do not have separate provisions for publicly traded and private companies. However, in the event we convert to generally accepted accounting principles in the United States, or U.S. GAAP, while we are still an emerging growth company, we may be able to take advantage of the benefits of this extended transition period.
We will remain an emerging growth company until the earliest of (a) the last day of our fiscal year during which we had total annual gross revenues of at least $1.0 billion; (b) December 31, 2019, the last day of our fiscal year following the fifth anniversary of the date of the first sale of Innocoll Germany’s ADSs in our 2014 initial public offering; (c) the date on which we have, during the previous three-year period, issued more than $1.0 billion in non-convertible debt; or (d) the date on which we are deemed to be a “large accelerated filer” under the Securities Exchange Act of 1934, as amended, or the Exchange Act, which would occur if the market value of our shares that are held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter and we have been publicly reporting for at least 12 months. Once we cease to be an emerging growth company, we will not be entitled to the exemptions provided to emerging growth companies in the JOBS Act.
Results of Operations
Comparison of Years Ended December 31, 2015 and 2014
| | | Year Ended December 31, | | | Increase/ | | | % Increase/ | |
| | | 2015 | | | 2014 | | | (Decrease) | | | (Decrease) | |
| | | (in thousands) | |
| Revenue | | € | 2,587 | | | € | 4,497 | | | € | (1,910 | ) | | | (42.5 | )% |
| Cost of sales | | | (4,802 | ) | | | (5,573 | ) | | | (771 | ) | | | (13.8 | )% |
| Research and development expenses | | | (26,878 | ) | | | (3,252 | ) | | | 23,626 | | | | 726.5 | % |
| General and administrative expenses | | | (18,983 | ) | | | (11,687 | ) | | | 7,296 | | | | 62.4 | % |
| Other operating expense | | | 3,876 | | | | (39 | ) | | | 3,915 | | | | 10,038.5 | % |
| Finance expense | | | 1,418 | | | | (4,535 | ) | | | 5,953 | | | | (131.3 | )% |
| Other Income | | | - | | | | 75 | | | | (75 | ) | | | (100.0 | )% |
Revenue. Revenues for the year ended December 31, 2015 were 42.5% lower at €2.6 million, compared to €4.5 million for year ended December 31, 2014. This decrease was primarily due to a decrease in sales to Jazz Pharmaceuticals/EUSA Pharma of CollatampG, our gentamicin implant for the treatment and prevention of post-surgical infection, of €1.3 million, or 37%, and a decrease in CollaGUARD sales of €0.3 million, or 99%, primarily due to partners having purchased sufficient product in 2014 for 2015. CollaGUARD revenues in the year ended December 31, 2015 were net of a €0.0 million charge for free stock due to be shipped per the terms of our distribution partner agreements, leaving a CollaGUARD free stock credit outstanding of €0.1 million as of December 31, 2015. Revenue from the sales of Septocoll decreased 42.5%, which was net of €0.2 million of free stock shipped under an agreement with our distribution partner during the year ended December 31, 2013, leaving a Septocoll free stock credit outstanding of €0.0 million as of December 31, 2015. Biomet end user Septocoll sales based on volume of units shipped decreased by 12.8% in the year ended December 31, 2015 compared to the year ended December 31, 2014.
Cost of Sales. Cost of sales of €4.8 million in the year ended December 31, 2015 decreased by €0.8 million, or 13.8%, compared to €5.6 million in the year ended December 31, 2014. Gross margins from supply revenue were (85)% and (24)% for the year ended December 31, 2015 and 2014, respectively. All overhead costs associated with operating our manufacturing facility are included in the standard cost of our products as indirect costs and charged to cost of sales based on sales volume. Excluding such indirect costs, gross margins from supply revenue were (32%) and 36% for the years ended December 31, 2015 and 2014 respectively.
Research and Development (R&D) Expenses. R&D expenses were €26.9 million for the year ended December 2015 as compared to €3.3 million for the year ended December 31, 2014, a 726.5% increase. R&D expenses in each year consisted of external clinical research costs as well as costs of internal research and development personnel and internal laboratory costs at our Saal, Germany facility. In the year ended December 31, 2015, external clinical research costs were €23.8 million in the year ended December 31, 2015 compared to €1.4 million external clinical research costs in the year ended December 31, 2014, the increase was primarily due to the completion of our pivotal pharmacokinetics and safety study of XaraColl and the initiation and running of our Phase 3 Cogenzia and Xaracoll efficacy trials. From January 1, 2004 through December 31, 2015, we incurred research and development expenses of €75.8 million net of contributions and collaboration agreements with third parties.
General and Administrative (G&A) Expenses. G&A expenses were €19.0 million for the year ended December 31, 2015 as compared to €11.7 million for the year ended December 31, 2014, an increase of 62.4%. G&A expenses in the year ended December 31, 2015 included €4.8 million in non-cash charges for stock-based compensation, compared to €5.1 of such charges in the corresponding period in 2014. Excluding such charges for stock-based compensation, G&A expenses were €14.2 million for the year ended December 31, 2015 as compared to €6.6 million for the year ended December 31, 2014. The increase in G&A, excluding stock-based compensation, was primarily due to our continued infrastructure build-out to support clinical programs, initiation of our pre-commercialization investment and expenses related to our re-domiciliation to Ireland.
Other Operating Income/Expense—Net. Other operating income increased by €3.9 million, or 10,038% to €3.9 million, in the year ended December 31, 2015 compared to €0.04 million in other operating expense in the year ended December 31, 2014. The increase was primarily due to €3.9 million in income arising out of the reversal of impairment of property, plant and equipment.
Finance Income/Expense.Finance income was €1.4 million for the year ended December 31, 2015 as compared to finance expense of €4.5 million in the year ended December 31, 2014, a 131.3% increase. Finance income in the year ended December 31, 2015 consisted primarily of €5.1 million in foreign exchange gains, partially offset by €3.7 million fair value expense of investor options outstanding. Finance expense in the year ended December 2014 included €6.3 million fair value expense of investor options outstanding, and €3.1 million interest on convertible preferred shares, partially offset by €4.7 million in foreign exchange gains.
Other Income:Other income was €0.0 million in the year ended December 31, 2015 compared to €0.1 million in the year ended December 31, 2014, a 100% decrease. Other income in the year ended December 31, 2014 consisted of profit on disposal of property, plant and equipment.
Comparison of Years Ended December 31, 2014 and 2013
| | | Year Ended December 31, | | | Increase/ | | | % Increase/ | |
| | | 2014 | | | 2013 | | | (Decrease) | | | (Decrease) | |
| | | (in thousands) | |
| Revenue | | € | 4,497 | | | € | 3,546 | | | € | 951 | | | | 26.8 | % |
| Cost of sales | | | (5,573 | ) | | | (4,551 | ) | | | 1,022 | | | | 22.5 | % |
| Research and development expenses | | | (3,252 | ) | | | (1,663 | ) | | | 1,589 | | | | 95.6 | % |
| General and administrative expenses | | | (11,687 | ) | | | (4,121 | ) | | | 7,566 | | | | 183.6 | % |
| Other operating expense | | | (39 | ) | | | (154 | ) | | | (115 | ) | | | (74.7 | )% |
| Finance expense | | | (4,535 | ) | | | (6,949 | ) | | | (2,414 | ) | | | (34.7 | )% |
| Other Income | | | 75 | | | | 16,073 | | | | 15,998 | | | | (99.5 | )% |
Revenues. Revenues for the year ended December 31, 2014 were 26.8% higher at €4.5 million, compared to €3.5 million for year ended December 31, 2013. This increase was primarily due to an increase in sales by Jazz Pharmaceuticals of CollatampG, our gentamicin implant for the treatment and prevention of post-surgical infection, of €0.7 million, or 26.2%, and an increase in CollaGUARD sales of €0.2 million, or 131.1%, primarily due to the first shipment of our adhesion barrier CollaGUARD to our partner Takeda in connection with the product’s launch in Russia in the third quarter of 2014. CollaGUARD revenues in the year ended December 31, 2014 were net of a €0.1 million charge for free stock due to be shipped per the terms our distribution partner agreements, leaving a CollaGUARD free stock credit outstanding of €0.1 million as of December 31, 2014. Revenue from the sales of Septocoll decreased 6.6%, which was net of €0.1 million of free stock shipped under an agreement with our distribution partner during the year ended December 31, 2013, leaving a Septocoll free stock credit outstanding of €0.2 million as of December 31, 2014. Biomet end user Septocoll sales based on volume of units shipped decreased by 20% in the year ended December 31, 2014 compared to the year ended December 31, 2013. In addition, during the third quarter of 2014 Biomet 3i launched RegenePro, our product to treat dental wounds in the United States, generating sales of €0.1 million in the year ended December 31, 2014.
Cost of Sales.Cost of sales of €5.6 million in the year ended December 31, 2014 increased by €1.0 million, or 22.5%, compared to €4.6 million in the year ended December 31, 2013. Gross margins from supply revenue were (24)% and (28)% for the year ended December 31, 2014 and 2013, respectively. All overhead costs associated with operating our manufacturing facility are included in the standard cost of our products as indirect costs and charged to cost of sales based on sales volume. Excluding such indirect costs, gross margins for each of the years ended December 31, 2014 and 2013 were positive (as more fully decribed in “—Cost of Sales” above).
Research and Development Expenses. R&D expenses were €3.3 million for the year ended December 2014 as compared to €1.7 million for the year ended December 31, 2013, a 95.6% increase. R&D expenses in each year consisted of external clinical research costs as well as costs of internal research and development personnel and internal laboratory costs at our Saal, Germany facility. In the year ended December 31, 2014, external clinical research costs were €1.4 million in the year ended December 31, 2014 compared to €0.1 million external clinical research costs in the year ended December 31, 2013, the increase was primarily due to the commencement of our pivotal pharmacokinetics and safety study of XaraColl in the third quarter of 2014. From January 1, 2004 through December 31, 2014, we incurred research and development expenses of €48.9 million net of contributions and collaboration agreements with third parties. We incurred below-average research and development expenses in each of the years ended December 31, 2014 and December 31, 2013 due to the fact that substantially all of our Phase 2 clinical trials were completed prior to 2012 and we did not commence Phase 3 trials until the third quarter of 2014.
General and Administrative Expenses. G&A expenses were €11.7 million for the year ended December 31, 2014 as compared to €4.1 million for the year ended December 31, 2013, an increase of 183.6%. G&A expenses in the year ended December 31, 2014 included €5.1 million in non-cash charges for stock-based compensation, compared to €0.0 of such charges in the corresponding period in 2013. Stock-based compensation expense in the year ended ended December, 31, 2014 was exceptionally high due to the timing of share grants and vesting linked to the completion of our initial public offering in the third quarter 2014. Excluding such charges for stock-based compensation, G&A expenses were €6.6 million for the year ended December 31, 2014 as compared to €4.1 million for the year ended December 31, 2013. The increase in G&A excluding stock-based compensation was primarily due to accounting, legal and consulting professional fees, insurance costs and investor relations costs related to becoming a public company.
Other Operating Expense. Other operating expense decreased by €0.1 million, or 74.7% to €0.04 million, in the year ended December 31, 2014 compared to €0.2 million in other operating expense in the year ended December 31, 2013. The decrease was primarily due to a €0.1 million in income arising out of as compensation for the agreement of certain amendments to our CollatampG supply agreement with our distribution partner Jazz Pharmaceuticals in 2012, which was netted off €0.2 impairment of fixed assets and other expense in the year ended December 31, 2014.
Finance Expense. Finance expense was €4.5 million for the year ended December 31, 2014 as compared to €6.9 million in the year ended December 31, 2013, a 34.7% decrease. Finance expensed in the year ended December 31, 2014 included €6.3 million fair value expense of investor options outstanding, and €3.1 million interest on convertible preferred shares, partially offset by €4.7 million in foreign exchange gains. Finance expense in the year ended December 2013 consisted primarily of interest on convertible preferred shares and convertible promissory notes of €6.6 million.
Other Income. Other income was €0.1 million in the year ended December 31, 2014 compared to €16.1 million in the year ended December 31, 2013, a 99.5% decrease. Other income in the year ended December 31, 2013 consisted of exceptionally high non-cash fair value gains on exchange of warrants, and settlement of preferred shares in connection with the re-domicile of the parent company. In accordance with IAS 39, the liability associated with the series B convertible stock issued by Innocoll Holdings, Inc. was deemed to be extinguished as the new financial instruments issued had significantly different terms than the instruments they replaced. The difference between the fair value of the new financial instrument, and the carrying value of the extinguished series B convertible stock, was allocated against the carrying value of the liability and equity components with a resulting gain being recognized in profit and loss as noted above.
B. LIQUIDITY AND CAPITAL RESOURCES
Since 2004, we have devoted a substantial portion of our cash resources to research and development and general and administrative activities primarily related to the development of our products, including XaraColl, Cogenzia and CollaGUARD. We have financed our operations primarily with the proceeds of the sale of preferred shares and convertible notes, supply revenue and collaborative licensing and development revenue, our initial public offering and April 2015 follow-on public offering. Over the course of 2016, we expect to generate revenues from the licensing and sale of CollaGUARD pursuant to our license agreements with Takeda and others, and other product licensing agreements. We expect to generate revenues from the sale of XaraColl and Cogenzia by the end of 2017. We have incurred losses and generated negative cash flows from operations since 2004. As of December 31, 2015, we had an accumulated deficit of €149.9 million and cash and cash equivalents of €38.7 million. As at December 31, 2015 Innocoll Pharmaceuticals Ltd. had Irish tax losses of €101 million which can be carried forward to shelter future Irish trading profits indefinitely. There is no limitation on the use of losses in any particular year (i.e. no minimum tax payment rule). Irish law imposes certain limitations on the application of net operating loss carryforwards and no assurance can be given that these tax losses will be available to the full extent in the future.
The following table summarizes our cash flows from operating, investing and financing activities for the years ended December 31, 2015, 2014 and 2013:
| | | Year Ended December 31, | |
| | | 2015 | | | 2014 | | | 2013 | |
| | | | | | (in thousands) | | | | |
| Consolidated Statement of Cash Flows Data: | | | | | | | | | | | | |
| Net cash provided by (used in): | | | | | | | | | | | | |
| Operating activities | | € | (38,934 | ) | | € | (12,700 | ) | | € | (4,677 | ) |
| Investing activities | | | (2,865 | ) | | | (769 | ) | | | (448 | ) |
| Financing activities | | | 30,077 | | | | 52,158 | | | | 7,965 | |
| Effect of foreign exchange rate changes | | | 4,855 | | | | 4,235 | | | | - | |
| Net (decrease)/increase in cash and cash equivalents | | | (6,867 | ) | | | 42,924 | | | | 2,840 | |
Operating Activities
For the years ended December 31, 2015, 2014 and 2013, our net cash used in operating activities was €38.9, €12.7 million, and €4.7 million, respectively. The increase in net cash used in operating activities in the year ended December 31, 2015 resulted primarily from an increase in both research & development expenses and general & administrative expenses together with an increase in trade and other receivables and an increase in inventory, partially offset by an increase in trade and other payables and an increase in deferred income. The increase in net cash used in operating activities in the year ended December 31, 2014 resulted primarily from an increase in trade and other receivables, a decrease in trade and other payables, and a decrease in deferred income, partially offset by a decrease in inventory.
Investing Activities
For the years ended December 31, 2015, 2014 and 2013, our net cash used in investing activities was €2.9, €0.8 million, and €0.4 million, respectively. The net cash used in investing activities in the years ended December 31, 2015, 2014 and 2013 was primarily for the purchases of plant and machinery at our Saal, Germany facility.
Financing Activities
Our net cash provided by financing activities was €30.1 million for the year ended December 31, 2015 compared to €52.2 million for the year ended December 31, 2014. The cash provided by financing activities for the year ended December 31, 2015 was primarily the result of the issuance of ADS in our follow-on public offering for net proceeds €14.4 million and the drawdown of €15.0 million in loans from the European Investment Bank. Our net cash provided by financing activities was €52.2 million for the year ended December 31, 2014 compared to €8.0 million for the year ended December 31, 2013. The cash provided by financing activities for the year ended December 31, 2014 was primarily the result of the issuance of ADS in our initial public offering for net proceeds of $54.4 million, and the 2014 pre-IPO equity financings for net proceeds for $17.2 million, compared to €8.0 million for the year ended December 31, 2013, primarily the result of the issuance and sale of convertible notes payable for total net proceeds of $1.4 million and from the issue of our series D preferred shares for total net proceeds of $1.0 million plus €6.3 million. All outstanding issues of convertible notes were exchanged for shares of series C and series D preferred stock in Innocoll Holdings, Inc. in June 2013, which were subsequently exchanged for series C and series D preferred shares in Innocoll GmbH in July 2013. On July 3, 2014, all shares of Innocoll GmbH became ordinary shares of Innocoll AG.
Innocoll Holdings, Inc. 2013 Debt Financing
In the year ended December 31, 2013, we received financing of $1.4 million from certain of our investors for which we issued 10% senior convertible promissory notes in aggregate principal of up to $1.5 million, 488 shares of series C-3 preferred stock and 3,491,623 warrants for the purchase of the company’s common stock at an exercise price of $0.30 per share and a contractual life of 10 years. The stated maturity of the convertible promissory notes was September 2013. The January 2013 convertible promissory notes had a liquidation preference of three times principal plus one times interest outstanding.
Innocoll Holdings, Inc. Debt for Equity Exchange, Equity Financing
Pursuant to an exchange agreement entered into in June 2013 between Innocoll Holdings, Inc. and the holders of the convertible notes, an aggregate of $26.7 million of principal and accrued interest on the outstanding convertible notes and shares of series C-3 preferred stock issued from May 2010 to August 2010 were exchanged for 26,687,487 shares of series C preferred stock, and an aggregate of $15.9 million of principal and accrued interest on the outstanding convertible notes and shares of series C-3 preferred stock issued from March 2011 to January 2013 were exchanged for 15,872,592 of shares of series D preferred stock in Innocoll Holdings, Inc. (the “Debt for Equity Exchange”).
In June 2013, we received net proceeds of $1.0 million from certain of our investors from the issue of 1,000,000 shares of series D preferred stock of Innocoll Holdings, Inc., and warrants to purchase an aggregate of 2,500,000 of the company’s common stock with an exercise price of $0.30 per share and a contractual life of 10 years. The Innocoll Holdings, Inc. shares of series D preferred stock issued in this Debt for Equity Exchange and subsequent financing had a liquidation preference of three times stated value plus one times accrued dividends.
Innocoll Holdings, Inc. Share Repurchase
In June 2013, Innocoll Holdings, Inc. repurchased and redeemed restricted shares which had been purchased in December 2007 by certain employees, officers and board members in exchange for promissory notes. Pursuant to the employee share repurchase, we repurchased an aggregate of 5,466,821 shares of common stock (24% of the shares of common stock then outstanding) in exchange for forgiving and writing off loans and accrued interest due in the aggregate amount of €8.6 million. Of this amount, €2.8 million of indebtedness under such promissory notes due and owing from Michael Myers, Ph.D., our former Chief Executive Officer was forgiven in exchange for 1,991,959 shares of common stock owned by Dr. Myers.
Re-Domicile of Parent Company
The shares of series C and series D preferred stock and warrants in Innocoll Holdings, Inc. were subsequently exchanged for series C and series D shares and options in Innocoll GmbH. Pursuant to a notarial deed entered into between the shareholders of Innocoll Holdings, Inc. and Innocoll GmbH in July 2013, the holders of shares of common stock, shares of series A, series B, series C and series D preferred stock and warrants to purchase common stock of Innocoll Holdings Inc. contributed their shares and warrants by way of a contribution-in-kind to Innocoll GmbH in exchange for ordinary shares, series A, series B, series C and series D preferred shares and options to purchase ordinary shares of Innocoll GmbH and as a result thereof, Innocoll Holdings, Inc. became Innocoll GmbH’s wholly-owned subsidiary. Innocoll GmbH issued (i) a total of 738,623 shares, comprised of 38,750 ordinary shares, 316,640 series A preferred shares, 53,234 series B preferred shares, 202,179 series C preferred shares and 127,820 series D preferred shares, each with a nominal value of €1 per share, and (ii) and 158,176 options to purchase ordinary shares at an initial exercise price of €100 per ordinary share (subject to adjustment) and a contractual life of 10 years which options were included in our 2014 Option Agreement. The preferred shares of Innocoll GmbH have similar liquidation preferences and cumulative dividend rights to the preferred shares of the same series of Innocoll Holdings, Inc. after the debt for equity exchange described above under “—Innocoll Holdings, Inc. Debt for Equity Exchange, Equity Financing.” In September 2013, Innocoll Holdings, Inc. filed a certificate of dissolution in Delaware, and Innocoll Pharmaceuticals Limited, Innocoll Technologies Limited, both registered in the Republic of Ireland and Innocoll Inc., a Delaware corporation were distributed to Innocoll GmbH. The above-described transactions including the Debt for Equity Exchange are collectively defined herein and in our financial statements as the “re-domicile” or the “Re-Domicile of Parent Company.”
Innocoll GmbH was effectively managed and controlled from Ireland and, therefore, under the Ireland-Germany double tax treaty, its residence was deemed to have migrated from Germany to Ireland in 2014.
Innocoll GmbH 2013 and 2014 Equity Financings
Subsequent to the re-domicile during the year ended December 31, 2013, Innocoll GmbH received $8.7 million from the sale of our preferred shares to certain of our existing investors, in two separate tranches. In October 2013 we issued $2.8 million of series D preferred shares to certain of our investors and 15,147 options for the purchase of our ordinary shares. In November 2013, we issued $5.9 million of series D preferred shares to certain of our investors and 31,876 options for the purchase of our ordinary shares. The options had an initial exercise price of €100 per share (subject to adjustment) and a contractual life of 10 years and were included in our 2014 Option Agreement.
In May 2014, we issued 77,924 series E preferred shares to certain existing shareholders, three new members of our historic supervisory board and three new investors who are partners of one of the existing shareholders for approximately $12.1 million. We also issued 44,465 restricted and unrestricted shares as described under “Financial Operations Overview—Valuation of financial instruments—May 2014 Grants.” In June 2014, we issued 32,977 new ordinary shares of Innocoll GmbH for $5.1 million to certain existing shareholders under the same terms and conditions and having the same anti-dilution rights as the series E preferred shares. Given that the price of Innocoll Germany’s ADS at our initial public offering in July 2014 was $9.00 per ADS of Innocoll Germany (or €88.52 per ordinary share) in February 2015 we issued 58,953 of our ordinary shares to such holders as a result of these anti-dilution provisions after such issuance was approved at our shareholder meeting on December 4, 2014.
2014 Option Agreement
In January 2014, Innocoll Germany’s predecessor, Innocoll GmbH, entered into an option agreement, as amended on March 20, 2014 with its then-existing shareholders, including certain officers, directors and affiliates in their capacity as shareholders. The option agreement covers all options issued by Innocoll GmbH in connection with its re-domicile and the 2013 equity financing described above under “—Re-Domicile of Parent Company,” and “—Innocoll GmbH 2013 and 2014 Equity Financings,” respectively. Pursuant to the 2014 Option Agreement, these shareholders and Kinabalu Financial Products L.L.P. received the right, at any time and from time to time, to purchase up to an aggregate of 205,199 shares of Innocoll GmbH in the aggregate, with an exercise price of €100 per share (subject to adjustment) and a contractual life of 10 years. In connection with Innocoll Germany’s transformation into a stock corporation, all options under the original 2014 option agreement were cancelled and replaced by a new option agreement on substantially similar terms pursuant to which beneficiaries were entitled to purchase up to 205,199 ordinary shares of Innocoll AG in the aggregate at the same initial exercise price of €100 per share (as so replaced, the “2014 Option Agreement”), which obligations have been assumed by Innocoll Ireland. The 2014 Option Agreement provides that if we issue or sell or are deemed to have issued or sold any of our ordinary shares without consideration or for a consideration per share less than the exercise price in effect immediately prior to the time of such issue or sale, the exercise price is reduced to a price equal to the price per share at which such shares are issued or sold or deemed issued or sold, subject to certain limited exceptions for share option, share purchase or similar plan or arrangement for the benefit of our or our subsidiaries’ employees, officers, consultants or directors, pre-existing options, securities issued as consideration for any acquisition by the us or our subsidiaries, or securities issued to banks, equipment lessors or other financial institutions, or to real property lessors, pursuant to a debt financing, equipment leasing or real property leasing transaction which is approved by the management board. As a result of the $9.00 per ADSs price of Innocoll Germany’s initial public offering, the exercise price was adjusted to €88.52 per ordinary share of Innocoll Germany, which price is subject to further adjustment if we issue securities at an equivalent value below this price, or $7.42 per ordinary share of Innocoll Ireland (based on conversion to U.S. dollars at an exchange rate of $1.1109 per euro, the official exchange rate quoted as of March 15, 2016 by the European Central Bank) through June 15, 2019, on which date any unexercised options expire pursuant to the terms of the 2014 Option Agreement, unless our shareholders approve an additional five-year period.
Innocoll GmbH Conversion of Preferred Shares into Ordinary Shares of Innocoll GmbH and Subsequent Transformation into Innocoll AG
Pursuant to a notarial deed entered into on June 16, 2014, the shareholders of Innocoll GmbH agreed to amend and terminate all preference, redemption and cumulative dividend rights by converting all preferred shares into ordinary shares of Innocoll GmbH (other than, with respect to the holders of series E preferred shares, who retained the anti-dilution right referred to above). On July 3, 2014, subsequent to the recapitalization, Innocoll GmbH transformed into a German stock corporation (Aktiengesellschaft or AG) in accordance with the provisions of the German Reorganization Act (Umwandlungsgesetz). In connection therewith, all 1,004,523 outstanding ordinary shares of Innocoll GmbH became 1,004,523 ordinary shares of Innocoll AG. Accordingly, although we changed our legal form, there was no change in our identity. The following table summarizes the capitalization of Innocoll GmbH before, and giving effect to, its recapitalization and the conversion of all preferred shares into ordinary shares prior to its transformation into Innocoll AG:
| Share Class (in order of preferences)(1) | | Shares
Pre-Conversion(1)(2) | | | Exchange Ratio | | Ordinary
Shares
Upon
Conversion
of Preferred
Shares(2) | | | % | |
| Series E preferred shares | | | 90,286 | | | 1:1 | | | 90,286 | | | | 7.0 | % |
| Series D-2 preferred shares | | | 24,981 | | | 1.88:1 | | | 47,025 | | | | 3.7 | % |
| Series D preferred shares | | | 201,744 | | | 1.88:1 | | | 379,771 | | | | 29.6 | % |
| Series C preferred shares | | | 240,611 | | | 0.67:1 | | | 160,203 | | | | 12.5 | % |
| Series A preferred shares | | | 376,827 | | | 0.67:1 | | | 250,898 | | | | 19.6 | % |
| Series B preferred shares | | | 63,352 | | | 1.83:1 | | | 115,733 | | | | 9.0 | % |
| Ordinary Shares Pre-Conversion(3) | | | 46,115 | | | 0:1 | | | — | | | | 0 | % |
| Ordinary Shares(4) | | | 32,977 | | | 1:1 | | | 32,977 | | | | 2.6 | % |
| Outstanding options(5) | | | 205,199 | | | 1:1 | | | 205,199 | | | | 16.0 | % |
| Total | | | 1,282,092 | | | | | | 1,282,092 | | | | 100.0 | % |
(1)For a complete description of the rights, privileges and preferences of the various securities listed, including liquidation preference and dividend rates, see Note 16 to our audited consolidated financial statements.
(2)The number of each class of preferred shares indicated converted into the corresponding number of ordinary shares at the listed exchange ratio on June 16, 2014. Includes (i) 72,370 shares authorized and issuable upon the settlement of phantom shares and (ii) 85,414 ordinary shares awarded under our 2014 restricted share plan that were subject to repurchase.
(3)Certain ordinary shares outstanding prior to June 16, 2014 were reallocated to holders of preferred shares upon the recapitalization.
(4)In June 2014, immediately prior to the recapitalization, we issued 32,977 ordinary shares to certain investors for $5.1 million.
(5)Represents ordinary shares authorized and issuable upon the exercise of outstanding options. Does not include 24,784 ordinary shares of Innocoll Germany reserved for options that can be awarded in the future.
Initial Public Offering
On July 30, 2014, Innocoll Germany sold 6,500,000 ADSs representing 490,567 ordinary shares of Innocoll Germany in its initial public offering at a price of $9.00 per ADS of Innocoll Germany, thereby raising $54.4 million after deducting underwriting discounts and commissions. On August 20, 2014, the underwriters in our initial public offering partially exercised their overallotment option to purchase an additional 186,984 ADSs of Innocoll Germany, representing 14,112 ordinary shares of Innocoll Germany, at a public offering price of $9.00 per ADS of Innocoll Germany. The sale of the overallotment option by our underwriters occurred on September 12, 2014, at which the company raised additional net proceeds of approximately $1.57 million, after deducting underwriting discounts and commissions. The Innocoll Germany ADSs we sold in the initial public offering represented new ordinary shares of Innocoll Germany issued in a capital increase resolved by our shareholders for the purposes of the initial public offering on July 30, 2014.
EIB Loan
On March 27, 2015, we and our wholly-owned subsidiary, Innocoll Pharmaceuticals, entered into a Finance Contract with the European Investment Bank, or EIB, whereby the EIB committed to lend to Innocoll Pharmaceuticals up to €25 million. In the fourth quarter of 2015, we drew down €15 million of the loan commitment, and contingent on achieving the primary endpoint on either XaraColl or Cogenzia Phase 3 clinical trials, are entitled to draw down an additional €10 million.
Follow-on Public Offering
On April 30, 2015, Innocoll Germany sold 1,999,690 ADSs of Innocoll Germany representing 150,920 ordinary shares of Innocoll Germany in our follow-on public offering at a price of $9.00 per ADS of Innocoll Germany, thereby raising $16.9 million after deducting underwriting discounts and commissions. These ADSs represented new ordinary shares of Innocoll Germany issued in a capital increase resolved by our shareholders for such purposes.
Future Capital Requirements
Our rate of expenses will continue to increase as we advance our planned clinical trials of XaraColl, Cogenzia and CollaGUARD and expand our manufacturing facility in Saal, Germany. As a result, we will be required to seek additional sources of capital during the next 12 months or restrict certain of our expenditures to conserve capital and extend our resources.
Our need for additional capital will depend significantly on the level and timing of regulatory approval and product sales, as well as the extent to which we choose to establish collaboration, co-promotion, distribution or other similar agreements for our products and product candidates. Moreover, changing circumstances may cause us to spend cash significantly faster than we currently anticipate, and we may need to spend more cash than currently expected because of circumstances beyond our control.
We expect to continue to incur substantial additional operating losses as we seek regulatory approval for and commercialize XaraColl, Cogenzia and CollaGUARD and develop and seek regulatory approval for our other product candidates. If we obtain FDA approval for our products, we will incur significant sales, marketing and manufacturing expenses. In addition, we expect to incur additional expenses to add operational, financial and information systems and personnel, including personnel to support our planned product commercialization and expanded manufacturing efforts for Xaracoll, Cogenzia and CollaGuard in the United States. We also expect to incur significant costs to continue to comply with corporate governance, internal controls and similar requirements applicable to us as a public company.
Our future use of operating cash and capital requirements will depend on many forward-looking factors, including the following:
| · | the timing and outcome of the FDA’s review of the NDA for XaraColl and Cogenzia, and the PMA application for CollaGUARD; |
| · | the extent to which the FDA may require us to perform additional clinical trials for XaraColl, Cogenzia and CollaGUARD; |
| · | the costs of our commercialization activities for Xaracoll, Cogenzia and CollaGUARD in the U.S., if approved by the FDA; |
| · | the cost and timing of expanding our manufacturing facilities and purchasing manufacturing and other capital equipment for XaraColl, Cogenzia, CollaGUARD and our other products and product candidates; |
| · | the scope, progress, results and costs of development for additional indications for XaraColl, Cogenzia and CollaGUARD and for our other product candidates; |
| · | the cost, timing and outcome of regulatory review of our other product candidates; |
| · | the cost, timing and outcome of regulatory review of our proposed expansion of our manufacturing facility; |
| · | the extent to which we acquire or invest in products, businesses and technologies; |
| · | the extent to which we choose to establish collaboration, co-promotion, distribution or other similar agreements for our product candidates; and |
| · | the costs of preparing, submitting and prosecuting patent applications and maintaining, enforcing and defending intellectual property claims. |
To the extent that our capital resources are insufficient to meet our future operating and capital requirements, we will need to finance our cash needs through public or private equity offerings, debt financings, corporate collaboration and licensing arrangements or other financing alternatives. Additional equity or debt financing or corporate collaboration and licensing arrangements may not be available on acceptable terms, if at all.
If we raise additional funds by issuing equity securities, our shareholders will experience dilution. Debt financing, if available, would result in increased fixed payment obligations and may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. Any debt financing or additional equity that we raise may contain terms, such as liquidation and other preferences, that are not favorable to us or our shareholders. If we raise additional funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish valuable rights to our technologies, future revenue streams or product candidates or to grant licenses on terms that may not be favorable to us.
| C. | RESEARCH AND DEVELOPMENT, PATENTS AND LICENSES, ETC. |
See “Item 4. Information on the Company—B. Business Overview” and “Item 5. Operating and Financial Review and Prospects—A. Operating Results.”
Other than as disclosed elsewhere in this annual report, we are not aware of any trends, uncertainties, demands, commitments or events for the year ended December 31, 2015 that are reasonably likely to have a material adverse effect on our revenues, profitability, liquidity or capital resources, or that would cause the disclosed financial information to be not necessarily indicative of future operating results or financial conditions.
| E. | OFF-BALANCE SHEET ARRANGEMENTS |
We do not have any off-balance sheet arrangements, or relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities.
| F. | TABULAR DISCLOSURE OF CONTRACTUAL OBLIGATIONS |
The following table summarizes our contractual obligations as of December 31, 2015:
Operating Leases
The group incurred operating lease expenses for the year ended December 31, 2015 of €0.4 million, which are included in general and administrative expenses. At December 31, 2015, the group had outstanding commitments for future minimum rent payments, which will become due as follows:
| | | Year Ended | |
| Thousands of Euros | | December 31,
2015 | |
| Less than one year | | | 437 | |
| Between one and five years | | | 1,029 | |
| More than five years | | | 178 | |
| Total operating lease commitments | | | | |
| | | | 1,644 | |
See “Special Note Regarding Forward Looking Statements” on page 1 of this annual report.
Item 6. Directors, Senior Management and Employees
| A. | DIRECTORS AND SENIOR MANAGEMENT |
Board of Directors
The following table sets forth the names and functions of the current members of our board, their ages, and their principal occupations outside of our company as of March 16, 2016:
| Name | | Age | | | Principal occupation |
| Jonathan Symonds, CBE (Chairperson)(1)(3) | | | 57 | | | Accountant |
| Shumeet Banerji, Ph.D. (Vice Chairperson)(1)(2) | | | 56 | | | Consultant |
| David R. Brennan(1)(3) | | | 62 | | | Retiree |
| A. James Culverwell(2)(3) | | | 59 | | | Consultant |
| Rolf D. Schmidt(3) | | | 83 | | | Investor/Retiree |
| Joseph Wiley, M.D.(1)(2) | | | 45 | | | Investment Manager |
| Anthony P. Zook | | | 55 | | | Chief Executive Officer |
(1)Member of the compensation committee.
(2)Member of the audit committee.
(3)Member of the nominating and corporate governance committee.
The term for each member of our board will expire on the date of the next annual general meeting unless re-elected by shareholders at such meeting.
The business address of the members of our board is the same as our business address: Innocoll Holdings plc, Unit 9, Block D, Monksland Business Park, Monksland, Athlone, Ireland.
The following is a brief summary of the business experience of the members of our board:
Anthony P. Zook is our President and Chief Executive Officer, is a member of our board and has served as the president and chief executive officer of our predecessor since December 7, 2014. Mr. Zook has extensive pharmaceutical executive management, commercialization and marketing experience. He held several executive positions at AstraZeneca including executive vice president of Global Commercial Operations from 2010 to 2013, president and chief executive officer of the North American division from 2007 to 2010 and president of Medimmune from 2008 to 2010. Prior to joining Innocoll, Mr. Zook was chief executive officer and member of the board of directors of Vivus, Inc. in 2013. He has served or continues to serve on several boards including the boards of AltheRx, Inhibikase, Rib-X Pharmaceuticals, the National Pharmaceutical Council, PhRMA, the Pennsylvania Division of the American Cancer Society and his alma mater, Frostburg State University. Mr. Zook earned a B.S. degree from Frostburg State University and an A.A. degree in chemical engineering from Pennsylvania State University. We believe that Mr. Zook’s management and technical experience qualifies him to serve on our board.
Jonathan Symonds,CBE has been the chairperson of our advisory board and our supervisory board, the predecessors to our board, since May 2014. Mr. Symonds has been a director of HSBC Holdings plc since April 2014 and chairman of HSBC Bank plc since April 2014. Since October 2013, Mr. Symonds has served as an independent director and chairman of the audit committee of Genomics England Limited, and since June 2014 he has served as an independent director Proteus Digital Health, Inc. Mr. Symonds was appointed Commander of the British Empire (CBE) for services to business and the pharmaceutical industry in January 2007. Mr. Symonds was previously chief financial officer of Novartis AG from 2009 to 2013. Prior to joining Novartis, he was a partner and managing director of Goldman Sachs from 2007 to 2009, chief financial officer of AstraZeneca plc from 1997 to 2007 and a partner of KPMG from 1992 to 1997. Mr. Symonds has previously served on the board of directors of Diageo plc and Qinetiq plc. Mr. Symonds received a B.A. in Business Finance from the University of Hertfordshire, where he also received an honorary doctorate. We believe that Mr. Symonds’ business experience in the pharmaceutical industry and his service on the boards of directors of other companies qualifies him to serve on our board.
Shumeet Banerji, Ph.D. has been a member of our advisory board and our supervisory board, the predecessors to our board, since May 2014. Dr. Banerji is co-founder and partner of Condorcet, LP, a private investment and advisory firm. He was the founding chief executive officer of Booz & Company, a global management consulting firm, from 2008 to 2012 and served as a senior partner from May 2012 to March 2013. Prior to that, he held multiple roles at Booz Allen Hamilton, a consulting company and predecessor to Booz & Company, including president of the worldwide commercial business, managing director, Europe, and managing director, United Kingdom. He was elected every year for 10 years to the firm’s Board of Directors. Dr. Banerji has been a director of the Hewlett-Packard Company since January 2011. Dr. Banerji received his Ph.D. from Northwestern University, Kellogg School of Management. We believe that Dr. Banerji’s global business experience and his service on several boards of directors qualifies him to serve on our board.
David R. Brennan has been a member of our advisory board and our supervisory board, the predecessors to our board, since May 2014. Mr. Brennan has nearly 40 years’ experience in the pharmaceutical industry. Mr. Brennan served as the chief executive officer of AstraZeneca Plc from January 2006 to June 2012, president and chief executive officer of AstraZeneca Pharmaceuticals LP from 2001 to 2005, president and chief executive officer of AstraZeneca’s North American subsidiary from 2001 to 2006, executive vice president of North America Division of AstraZeneca Plc from 2001 to 2005 and senior vice president of commercialization and portfolio management of AstraZeneca from 1998 to 2000. Prior to the merger between Astra AB and Zeneca Plc, Mr. Brennan served as senior vice president of business planning and development of Astra Pharmaceuticals LP, the American subsidiary of Astra AB. Mr. Brennan has been a director of the Chief Executive Officer Roundtable on Cancer since 2010 and a director of Insmed Incorporated since May 2014. Mr. Brennan served as an executive director of AstraZeneca Plc from 2005 to 2012, chairman of the board for the Southeastern Pennsylvania Chapter of the American Heart Association from 2004 to 2006, an executive board member of the Pharmaceutical Research and Manufacturers of America from 2001 to 2012 and chairman from 2009 to 2010, a member of the executive board and board member of the European Federation for Pharmaceutical Industries and Associations from 2006 to 2012, president of the International Federation of Pharmaceutical Manufacturers and Associations from 2010 to 2012, a member of the European Roundtable of Industrialists from 2006 to 2012 and a member of the National Institute of Health Roundtable on Evidence Based Medicine from 2006 to 2011. He was a participant and member of the International Business Council of the World Economic Forum. Mr. Brennan has been a director of Alexion Pharmaceuticals, Inc. and Insmed Incorporated since 2014. Mr. Brennan holds a B.A. in Business Administration from Gettysburg College, where he currently serves on the board of trustees. We believe that Mr. Brennan’s business experience in the pharmaceutical industry qualifies him to serve on our board.
A. James Culverwell has been a member of our advisory board and our supervisory board, the predecessors to our board, since August 2013. Mr. Culverwell also served on Innocoll Holdings’ board of directors from December 2012 to August 2013. Mr. Culverwell has over 30 years’ experience in the pharmaceutical industry in pharmaceutical company analysis, investment banking and healthcare private equity. Mr. Culverwell joined Hoare Govett in 1982, where he became director of European healthcare research. He joined Merrill Lynch & Co. in 1995, where he became head of European pharmaceutical research and global coordinator for healthcare research and established a top-rated franchise. In 2004, Mr. Culverwell set up Sudbrook Associates, a healthcare corporate advisor specializing in fund raising, corporate advice and due diligence in the private healthcare sector. Mr. Culverwell also sits on the boards of four other private companies in the specialty pharmaceutical, drug development and diagnostic field. Mr. Culverwell received a MSc from the University of Aberdeen. We believe that Mr. Culverwell’s business experience in the pharmaceutical industry and his service on the boards of directors of other companies qualifies him to serve on our board.
Rolf D. Schmidt has been a member of our advisory board and our supervisory board, the predecessors to our board, since August 2013. Mr. Schmidt also served on Innocoll Holdings’ board of directors from 1997 to August 2013. Mr. Schmidt was a co-founder of the medical bio-adhesives product company, Closure Medical Corporation. Mr. Schmidt led the company as chairperson from its early development through its initial public offering in 1996 and until its acquisition by Johnson & Johnson in 2005. He was co-founder of Sharpoint, Inc., a leading developer and manufacturer of ophthalmic surgical needles and sutures prior to its acquisition by Alcon Labs, Inc. in 1986. Mr. Schmidt actively consults with and invests in early-stage healthcare technology companies. We believe that Mr. Schmidt’s business and management experience, primarily in the healthcare industry, and his service on the boards of directors of other companies qualifies him to serve on our board.
Joseph Wiley, M.D. has been a member of our supervisory board, a predecessor to our board, since December 2014. He has been an AMRIT Life Science Investor since April 2015. From April 2012 to April 2015, Dr. Wiley was a principal at Sofinnova Ventures, an affiliate of one of our shareholders. Dr. Wiley has over 20 years of experience in the pharmaceutical, medical and venture capital industries. He was previously a medical director at Astellas Pharma. Prior to joining Astellas, he held investment roles at Spirit Capital, Inventages Venture Capital and Aberdeen Asset Managers (UK). Dr. Wiley trained in general medicine at Trinity College Dublin, specializing in neurology. He is also a Member of the Royal College of Physicians in Ireland. We believe that Dr. Wiley’s business experience in the pharmaceutical industry qualifies him to serve on our board.
Dr. Wiley was nominated to Innocoll Germany’s supervisory board, a predecessor to our board, pursuant to a Supervisory Board Member Nomination Agreement into which Innocoll Germany and certain of our shareholders entered with Sofinnova Venture Partners VIII, L.P., or Sofinnova, and pursuant to which Sofinnova had the right to nominate, in consultation with Innocoll Germany’s Nominating and Corporate Governance Committee, one member for appointment to Innocoll Germany’s supervisory board, subject to the approval of Innocoll Germany’s supervisory board and our shareholders. Innocoll Germany’s shareholders voted in favor of Mr. Wiley’s appointment during our extraordinary shareholder meeting on December 4, 2014.
Senior Management Team
The following table sets forth the names and functions of the Company’s senior management team, their ages and their positions as of March 16, 2016:
The business address of the members of Innocoll Germany’s senior management team is the same as our business address.
Name | | Age | | Position |
| Anthony P. Zook | | 55 | | Chief Executive Officer |
| Jose Carmona | | 43 | | Chief Financial Officer |
| Charles F. Katzer | | 66 | | Vice President, Global Supply / Procurement |
| Rich Fante | | 51 | | Chief Commercial Officer and Head of Business Development |
The following is a brief summary of the business experience of our senior management team:
Mr. Zook’s business experience appears above, See “Item 6. Directors, Senior Management and Employees, A. Directors and Senior Management—Board of Directors.”
Jose (Pepe) Carmona is our Chief Financial Officer and has served as the chief financial officer of our predecessor since September 2015. Mr. Carmona has extensive experience in the pharmaceutical sector with executive positions in the Americas, Europe, Middle East and Africa. Most recently he served as Chief Financial Officer of Alcon Europe, Middle East & Africa, a Division of Novartis, and prior to that he held numerous financial management positions with increasing responsibility in Novartis. Before joining Novartis, Mr. Carmona held senior management positions for several divisions of Procter & Gamble in Latin America. Mr. Carmona received his B.S. in Industrial and Civil Engineering from Universidad Tecnica Federico Santa Maria in Valparaiso, Chile, and his M.B.A. from Columbia Business School in New York City.
Charles F. Katzer has been our Vice President, Global Supply / Procurement since December 2015. Prior to that, Mr. Katzer served on an interim basis as Vice President, Manufacturing Operations at Lannett Company, a developer, manufacturer and distributor of generic prescription pharmaceutical products, between June and December 2015. From 2010 to 2014, Mr. Katzer was Senior Vice President, Technical Operations at Auxilium Pharmaceuticals, Inc., a public biopharmaceutical company focused on developing and commercializing innovative products for specialist audiences. From 2006 to 2010, Mr. Katzer was Senior Vice President, Technical Operations and Chief Technical Officer at Discovery Laboratories Inc. (NASDAQ:DSCO), a specialty biotechnology company focused on developing aerosolized therapies for respiratory diseases. From 2000 to 2006, Mr. Katzer was Vice President Pharmaceutical Operations at Medimmune Vaccines. Prior to joining Medimmune, Mr. Katzer was Vice President, Pharmaceutical Operations at US Bioscience from 1992 to 2000. Mr. Katzer held management and leadership positions of increasing responsibility at three divisions, over 13 years at Rhone-Poulenc Rorer. Earlier in his career, Mr. Katzer held supervisory/management positions in Pharmaceutical R&D at Baxter Travenol and Biological Specialties. Mr. Katzer earned his B.S. degree in Zoology from the University of Wisconsin.
Rich Fante has been our Chief Commercial Officer and Head of Business Development since August 2015. Prior to joining Innocoll, Rich was President and founder of RF Consulting LLC since April 2013. Before founding RF Consulting, Mr. Fante spent over nineteen years at AstraZeneca pharmaceuticals in the United States. He served in a number of roles at AstraZeneca, including most recently as President US, CEO North America and Regional Vice President of the Americas. Prior to his role as President AstraZeneca US, in November 2008, Mr. Fante served as the head of Brand Strategy and Portfolio Operations at AstraZeneca. Before joining AstraZeneca in January 1995, Mr. Fante worked at Lederle Laboratories where he started his career as a sales representative. Mr. Fante served as Board Chairman of the National Pharmaceutical Council (2012) and was a member of the Institute of Medicine of the National Academies of Science Roundtable on Value and Science. Mr. Fante holds a BA in Biology from Princeton University and an MBA from the University of North Carolina, Chapel Hill. Mr. Fante is currently a non-executive Director of the privately held biotech company Inhibikase Therapeutics Inc.
Compensation of Supervisory Board Members
2015 Supervisory Board Member Compensation Table
The following table sets forth information for the year ended December 31, 2015 regarding the compensation awarded to, earned by or paid to our historic supervisory board members who served on Innocoll Germany’s supervisory board during 2015. This table does not include the value of restricted shares and options awarded after December 31, 2015 pursuant to our 2016 Omnibus Incentive Compensation Plan, 2015 Stock Option Plan or 2014 restricted share awards.
| Name(1) | | Fees Earned or
Paid in Cash
($) | | | Share
Awards
($) | | | Option
Awards
($) | | | Non-Equity
Incentive Plan
Compensation
($) | | | All Other
Compensation
($) | | | Total
($) | |
| Jonathan Symonds, CBE | | | 108,870 | | | | 363,010 | | | | — | | | | — | | | | — | | | | 471,880 | |
| Shumeet Banerji, Ph.D. | | | 53,528 | | | | 363,010 | | | | — | | | | — | | | | — | | | | 416,538 | |
| David R. Brennan | | | 48,992 | | | | 219,938 | | | | — | | | | — | | | | — | | | | 268,930 | |
| A. James Culverwell | | | 48,992 | | | | 137,538 | | | | — | | | | — | | | | — | | | | 186,530 | |
| Rolf D. Schmidt | | | 48,992 | | | | — | | | | — | | | | — | | | | — | | | | 48,992 | |
| Joseph Wiley, M.D. | | | 37,198 | | | | — | | | | — | | | | — | | | | — | | | | 37,198 | |
No option awards were made to any of our historic supervisory board members as compensation as of December 31, 2015. As of December 31, 2015, A. James Culverwell and Rolf D. Schmidt held options to acquire 4,320 and 6,461 ordinary shares of Innocoll Germany, respectively, issued pursuant to investments made in the company prior to the transformation of Innocoll GmbH into Innocoll AG (see “Item 7. Major Shareholders and Related-Party Transactions—A. Major Shareholders”).
Each board member is granted a fixed remuneration in the amount of € 35,000 per annum for his activities during the Company’s fiscal year. The chairman of the board is granted a fixed remuneration in the amount of € 100,000 per annum for his activities during the Company’s fiscal year. Except for the chairman of the board each board member who served on a committee receives an additional amount of € 10,000 per annum. Board member compensation is paid pro rata temporis for members beginning or terminating their respective terms during a fiscal year. The remuneration for board activities is due and payable in quarterly installments at the end of each quarter
Our employees who also serve as a member of our board do not receive additional compensation for their performance of services as a board member.
Compensation of Management Board Members
2015 Summary Compensation Table
The following table sets forth information concerning the compensation of our senior managament team and our predecessor’s managament board during the fiscal year ended December 31, 2015:
Name and Principal
Position | | Salary | | | Bonus | | | Share
Awards | | | Option
Awards | | | Non-Equity
Incentive
Plan
Compensation | | | All Other
Compensation | | | Total | |
| Anthony P. Zook | | $ | 500,000 | | | $ | 261,250 | (1) | | $ | 2,313,197 | | | | — | | | | — | | | $ | 28,563 | (2) | | $ | 3,103,010 | |
Chairperson of the Management Board and Chief Executive Officer | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Michael Myers, Ph.D. | | $ | 493,479 | | | $ | 169,282 | (4) | | $ | 144,419 | | | $ | 53,961 | | | | — | | | $ | 43,238 | (5) | | $ | 904,379 | |
Member of the Management Board and Head of Portfolio Operations(3) | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Gordon Dunn | | $ | 301,028 | | | $ | 108,899 | (7) | | $ | 60,952 | | | $ | 53,961 | | | | — | | | $ | 5,700 | (8) | | $ | 530,540 | (9) |
Member of the Management Board and VP/Finance Director Europe(6) | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Jose Carmona Member of the Management Board and Chief Financial Officer(10) | | $ | 133,333 | | | $ | 53,333 | (11) | | | — | | | $ | 281,512 | | | | | | | $ | 9,087 | (12) | | $ | 477,265 | |
Rich Fante
Chief Commercial Officer and Head of Business Development(13) | | $ | 145,641 | | | $ | 60,000 | (14) | | | — | | | $ | 299,438 | | | | — | | | $ | 14,264 | (15) | | $ | 519,343 | |
Charles F. Katzer
Vice President, Global Supply / Procurement(16) | | $ | 17,275 | | | | — | | | | — | | | $ | 14,799 | | | | — | | | $ | 1,410 | (17) | | $ | 33,464 | |
(1) Cash bonus of $261,250 accrued.
(2) Includes term life insurance, short and long-term disability insurance and long-term care insurance.
(3) Dr. Myers previously served as a member of the management board, and as our Head of Portfolio Operations through December 31, 2015.
(4) A cash bonus of $169,282 awarded and accrued. This amount was paid in February 2015.
(5) Includes term life insurance, short and long-term disability insurance, long-term care insurance and matching contributions under the terms of our 401(k) plan.
(6) Mr. Dunn served as a member of the management board through the Merger, our predecessor’s Chief Financial Officer from 2012 through August 2015, and is currently our Vice President, Finance Director Europe.
(7) Cash bonus of $108,899 accrued.
(8) Includes health insurance.
(9) Compensation to Mr. Dunn is denominated in Great Britain Pounds, converted for the table above at a rate of £0.681 to $1.00, the average yearly rate for 2015 published by the United States Internal Revenue Service.
(10) Mr. Carmona has served as a member of the management board and our Chief Financial Officer since September 2015.
(11) Cash bonus of $53,333 accrued.
(12) Includes term life insurance, short and long-term disability insurance and long-term care insurance.
(13) Mr. Fante has served as our Chief Commercial Officer and Head of Business Development since August 20, 2015.
(14) Cash bonus of $60,000 accrued.
(15) Includes term life insurance, short and long-term disability insurances, long-term care insurance and matching contributions under the terms of our 401(k) plan.
(16) Mr. Katzer has served as our Vice President, Global Supply / Procurement since December 2015.
(17) Includes term life insurance, short and long-term disability insurances and long term care insurances.
In the year ended December 31, 2015, Mr. Zook received total compensation of $3,103,010, which included base salary, bonus, share awards and other benefits, Dr. Myers received total compensation of $904,379, which included base salary, bonus, share awards, option awards and other benefits, including our contribution to a direct insurance plan for Dr. Myers, Mr. Dunn received total compensation of $530,540, which included $301,028 in base salary, bonus, share awards, option awards and other benefits, Mr. Carmona received total compensation of $477,265 which included base salary, bonus, option awards and other benefits, Mr. Fante received total compensation of $519,343, which included base salary, bonus, option awards and other benefits and Mr. Katzer received toal compensation of $33,464, which included base salary, option awards and other benefits.
Employment Agreement and Equity Award Agreement with Anthony P. Zook
We entered into an employment agreement with Anthony P. Zook under which he is entitled to receive an initial annual base salary of $500,000 and which provides that Mr. Zook will serve as our chief executive officer (CEO) and as the chairperson of our historic management board. Mr. Zook is further eligible to receive an annual target performance bonus of 55% of his base salary, based on certain annual corporate goals and individual performance goals established annually by our board. The board has discretion to pay the annual target performance bonus to Mr. Zook even if the corporate and individual goals are not fully achieved and it also has the discretion to increase Mr. Zook’s total annual target bonus to up to 150% of the annual target performance bonus if the corporate and individual goals are exceeded.
Our historic supervisory board and shareholders also approved a grant to Mr. Zook, of 37,761 restricted ordinary shares of Innocoll Germany, which represented 2.25% of our outstanding ordinary shares at the time of grant, and entered into a related restricted share award agreement with Mr. Zook. Pursuant to the award agreement, Mr. Zook is required to subscribe to the restricted ordinary shares of Innocoll Germany at their nominal value per share of EUR 1.00 each. The restricted ordinary shares of Innocoll Germany are subject to our repurchase right if Mr. Zook’s employment with us terminates for any reason other than in connection with a change of control, which repurchase right lapses for 33.3% of the restricted ordinary shares upon the first anniversary of the grant date and thereafter, for an additional 8.3325% of the restricted ordinary shares on a quarterly basis. If Mr. Zook’s employment with us terminates within 180 days before or after the occurrence of a change of control, our repurchase right lapses for all the restricted ordinary shares. The award agreement further provides that Mr. Zook may not assign, pledge or otherwise transfer the restricted ordinary shares until the earlier of the fourth anniversary of the date of the award agreement and a change of control.
The employment agreement further provides that, for each calendar year of Mr. Zook’s continuing employment, starting in 2016, our board is required to consider an annual grant of options to purchase ordinary shares to Mr. Zook based upon a variety of factors deemed important to the board including our performance and the competitiveness of Mr. Zook’s compensation within the relevant market, with a target for consideration of 0.5% of the number of ordinary shares issued and outstanding on the date of the grant. The annual equity grant is in the sole discretion of the board’s compensation committee and the board as a whole. The compensation committee and the board further have the discretion to issue additional equity compensation, including but not limited to options, as the board may determine from time to time and will consider changes in our capital when making such decisions. In addition, as soon as practicable after Mr. Zook commences employment, he may, but is not required to, purchase $500,000 in ordinary shares from us based on their then-current market price. After five years of employment, Mr. Zook shall own our ordinary shares, on a continuing basis, having a market value equal to no less than three times his then-current base salary.
The employment agreement also provides that if we terminate Mr. Zook’s employment for reasons other than cause, or as a result of his death or disability or the employment is terminated by Mr. Zook for good reason, Mr. Zook is entitled to continue to receive his base salary and reimbursement for all medical, dental and life insurance benefits for a period of one year after the termination. If Mr. Zook dies during his employment, his estate is entitled to all his compensation and benefits through the date of his death and to the proceeds from any applicable policy of life insurance obtained by us for the benefit of these beneficiaries. In the event Mr. Zook is unable to perform his duties and responsibilities to us to the full extent required by the board by reason of illness, injury or incapacity for 90 consecutive days, or for more than 120 days in the aggregate during any period of 12 consecutive calendar months, he is entitled to all compensation and benefits earned through the date of disability and to the proceeds from any applicable disability insurance policy obtained by us for his benefit. If Mr. Zook’s employment is terminated within 180 days before or after a change in control, (a) by us for any reason other than cause, (b) as a result of his death or disability, or (c) by Mr. Zook for good reason, then any and all of the restricted ordinary shares owned by Mr. Zook that remain subject to forfeiture shall automatically become no longer subject to forfeiture upon the latter to occur of: (i) the occurrence of the change in control, or (ii) the termination of his employment as provided above; provided, however, that Mr. Zook provides the board with written notice of the occurrence of an event constituting good reason within 30 days of the occurrence of such event and we fail to cure or rectify such event within 30 days after receiving such written notice, at Mr. Zook’s option, exercisable within 30 days after the expiration of such cure period, Mr. Zook may resign from the employment relationship established with us, or, if involuntarily terminated without cause, give notice of intention to collect compensation and benefits under the employment agreement by delivering a notice in writing to the board, and in such event, he is entitled to continue to receive his base salary and reimbursement for all medical, dental and life insurance benefits for a period of one year after the termination, as well as, the acceleration of vesting of his equity compensation, subject to limitations imposed under German law.
Pursuant to the employment agreement, Mr. Zook also agreed to a no-solicitation covenant and a covenant not to compete with us worldwide during his employment with us and for a period of 365 days thereafter, which period shall be automatically extended for any period of time during which the he has breached, or threatened to breach the relevant provisions.
Prior Employment Agreement with Michael Myers, Ph.D.
Our subsidiary, Innocoll, Inc. entered into an executive employment agreement with Dr. Myers pursuant to which Dr. Myers served as the Head of Portfolio Operations and as a member of our management board, reporting to our CEO on all matters. The employment agreement provided for a term from January 1, 2015 through December 31, 2015, unless extended. Pursuant to the employment agreement, Dr. Myers received a base salary of $445,479 and a monthly expense allowance and other reimbursements in the aggregate amount of $4,000 as well as other group insurance and fringe benefits provided by Innocoll, Inc. for its other executives. The agreement provided that at the end of 2015, Dr. Myers would be considered, in the normal course of the year-end evaluations for a bonus based on annual corporate goals and individual performance goals established by our supervisory board and, in its discretion, for equity grants. However, the employment agreement provided that for 2015, in any event, Dr. Myers was entitled to a bonus of at least 30% and not more than 60% of his base salary.
The employment agreement also provided that if Dr. Myers’ employment was terminated by Innocoll, Inc. for reasons other than cause, or as a result of his death or disability, Dr. Myers would be entitled to continue to receive his base salary and reimbursement for all medical, dental and life insurance benefits until the later of one year after the termination and December 31, 2016.
Pursuant to the employment agreement, Dr. Myers also agreed to a no-solicitation covenant and a covenant not to compete with us worldwide during his employment with Innocoll, Inc. and for a period ending on December 31, 2017.
Separation Agreement with Michael Myers, Ph.D.
Dr. Myers’ employment with the Company was terminated as of December 31, 2015. The termination was without cause and Dr. Myers is entitled to receive cash payments in accordance with the terms of his employment agreement, including (i) salary continuation, (ii) the acceleration of certain payments in the event of a change of control of company during 2016, (iii) the continuation of certain benefits, (iv) and a 2015 bonus in the amount of $169,282. In addition, pursuant to the separation agreement, Mr. Myers has also agreed to a general release of the Company, confidentiality provisions and certain other customary terms and conditions for agreements of this nature.
Employment Agreement with Jose Carmona
Our subsidiary, Innocoll Inc., entered into an employment agreement with Mr. Carmona, effective as of September 1, 2015, under which he is entitled to receive an initial annual base salary of $400,000, and which provides that Mr. Carmona will serve as chief financial officer and report to the Company’s chief executive officer. Mr. Carmona is further eligible to receive an annual target performance bonus of 40% of his base salary, based on certain annual corporate goals and individual performance goals established annually by the Company’s board. The board has discretion to pay the annual target performance bonus to Mr. Carmona even if the corporate and individual goals are not fully achieved, and it also has the discretion to increase Mr. Carmona’s total annual target bonus to up to 150% of the annual target performance bonus if the corporate and individual goals are exceeded.
Pursuant to the agreement, Mr. Carmona was granted options to purchase 20,980 ordinary shares of Innocoll Germany under the Company’s 2014 Stock Option Plan, and have an exercise price equal to the fair market value of the ordinary shares, which, at the Company’s discretion will be either: (i) the average closing price of the Company’s ordinary shares on the NASDAQ Global Market for the last 10 trading days immediately preceding the grant date, or (ii) the price of the Company’s ordinary shares on the NASDAQ Global Market on the grant date. The options will vest over three (3) years, with one-third of the options vesting after the first year following the effective date of the employment agreement, and thereafter in equal quarterly installments. All such options will be subject to all other terms and conditions of the Company’s 2014 Stock Option Plan, including, but not limited to, a 4-year mandatory waiting period before any option can be exercised, and certain additional performance thresholds.
The employment agreement further provides that, for each calendar year of Mr. Carmona’s continuing employment, starting in 2016, the board is required to consider an annual grant of options to purchase ordinary shares to Mr. Carmona based upon a variety of factors deemed important to the board, including the Company’s performance and the competitiveness of Mr. Carmona’s compensation within the relevant market, with a target for consideration of 0.2% of the number of ordinary shares issued and outstanding on the date of the grant. The annual equity grant is in the sole discretion of the board’s compensation committee and the board as a whole. The compensation committee and the board further have the discretion to issue additional equity compensation, including but not limited to options, as the board may determine from time to time, and will consider changes in the capital of the Company when making such decisions.
The employment agreement also provides that if Mr. Carmona’s employment with the Company is terminated by the Company for reasons other than cause, or as a result of his death or disability or by Mr. Carmona for good reason, and provided that Mr. Carmona executes a separation agreement, Mr. Carmona is entitled to continue to receive his base salary for a period of one year after his termination, and reimbursement for all medical, dental and life insurance benefits for up to the earlier of: (i) one year after his termination, or (ii) the date Mr. Carmona becomes eligible for similar benefits from a third party. If Mr. Carmona dies during his employment with the Company, his estate is entitled to all his compensation and benefits from the Company through the date of his death, and to the proceeds from any applicable policy of life insurance obtained by the Company for the benefit of these beneficiaries. In the event Mr. Carmona is unable to perform his duties and responsibilities to the Company to the full extent required by the board by reason of illness, injury or incapacity for 90 consecutive days, or for more than 120 days in the aggregate, during any period of 12 consecutive calendar months, he will be entitled to all compensation and benefits earned through the date of disability, and to the proceeds from any applicable disability insurance policy obtained by the Company for his benefit. If Mr. Carmona’s employment is terminated within 180 days before or after a change in control, (a) by the Company for any reason other than cause or (b) by Mr. Carmona for good reason, then any and all of the shares and/or options of the Company owned by Mr. Carmona that remain subject to forfeiture shall automatically become no longer subject to forfeiture upon the latter to occur of: (i) the occurrence of the change in control, or (ii) the termination of his employment as provided above; provided, however, that Mr. Carmona provides the board with written notice of the occurrence of an event constituting good reason within 30 days of the occurrence of such event and the Company fails to cure or rectify such event within 30 days after receiving such written notice, and in such event, he will be entitled to continue to receive his base salary for a period of one year after the termination, and reimbursement for all medical, dental and life insurance benefits for up to the earlier of: (i) one year after the termination, or (ii) the date Mr. Carmona becomes eligible for similar benefits from a third party, as well as the acceleration of vesting of his equity compensation.
Pursuant to the employment agreement, Mr. Carmona also agreed to a no-solicitation covenant and a covenant not to compete with the Company worldwide during his employment with the Company and for a period of 365 days thereafter, which period shall be automatically extended for any period of time during which the he has breached, or threatened to breach the relevant provisions.
Prior Employment Agreement with Gordon Dunn
We entered into an employment agreement with Gordon Dunn on June 1, 2013. The agreement entitles Mr. Dunn to receive an initial base salary of £200,000 and provides that Mr. Dunn will serve as chief financial officer for the company and all of its direct and indirect subsidiaries. Mr. Dunn is also entitled to participate in any bonus scheme from time to time determined by us and in any long term equity incentive plan implemented by us. We have agreed, subject to supervisory board and shareholder approval, where required, to set Mr. Dunn’s participation level in any such plan at approximately 50% of the chief executive officer’s level. Both we and Mr. Dunn may terminate his employment agreement with six months’ written notice. In addition, we may terminate Mr. Dunn’s employment agreement for cause. Mr. Dunn has entered into a confidentiality and non-competition agreement with us that prohibits him from disclosing our confidential information and from being an employee or consultant of a company directly competing with us, as well as from soliciting our employees and customers, for one year following a termination of his employment, or from the date he commences any garden leave prior to his actual termination date.
Employment Agreement with Mr. Dunn
Our subsidiary, Innocoll Pharmaceuticals, entered into an employment agreement with Mr. Dunn, dated as of September 3, 2015, under which he is entitled to receive an initial annual base salary of £206,000, and which provides that effective (i) August 31, 2015, Mr. Dunn will cease to be the Company’s Chief Financial Officer, and (ii) September 1, 2015, Mr. Dunn will serve as Vice President, Finance Director, Europe, and report to the company’s Chief Financial Officer. Mr. Dunn is further eligible to receive an annual target performance bonus of 40% of his base salary, based on certain annual corporate goals and individual performance goals established annually by the compensation committee of the Company’s board. Mr. Dunn is entitled to a pro-rata portion of his bonus if terminated during a calendar year.
Pursuant to his employment agreement, Mr. Dunn is entitled to receive grants of options from time to time. In addition, if Mr. Dunn successfully (i) administers the Company’s implementation of an effective system of internal control over financial reporting in compliance with Section 404 of the Sarbanes-Oxley Act and (b) assists the Company in its re-domicile to Ireland, then Mr. Dunn will be entitled to accelerated vesting in the event of a termination with notice such that those unvested options that would vest on or before December 31, 2016 shall become fully vested.
The employment agreement also provides that Mr. Dunn’s employment may be terminated by any party at any time upon six months written notice. At any time after December 1, 2015, Mr. Dunn may terminate his employment with the company on one month’s notice, and at any time after February 1, 2016, the company may terminate Mr. Dunn’s employment on one-month’s written notice. In either event, and subject to his entering into a separation agreement with release, Mr. Dunn will be entitled to receive six months of salary at his base rate, and may be entitled to the continuation of certain health insurance provisions for him and his family for the lesser of six months, or earlier, if Mr. Dunn commences new employment which provides health insurance. Notwithstanding the above, if Mr. Dunn is terminated for cause, prior written notice to Mr. Dunn of such termination shall not be required. Pursuant to the employment agreement, Mr. Dunn also agreed to a confidentiality and non-competition provisions that prohibit him from disclosing our confidential information, a no-solicitation covenant, and a covenant not to compete with the Company worldwide during his employment with the Company and for a period of 12 months (or from the date he commences any garden leave prior to his actual termination date).
Employment Agreement with Rich Fante
Our subsidiary, Innocoll Inc., entered into an employment agreement with Mr. Fante, effective as of August 20, 2015, under which he is entitled to receive an initial annual base salary of $400,000, and which provides that Mr. Fante will serve as Chief Commercial Officer and report to the Company’s chief executive officer. Mr. Fante is further eligible to receive an annual target performance bonus of 40% of his base salary, based on certain annual corporate goals and individual performance goals established annually by the Company’s board. The board has discretion to pay the annual target performance bonus to Mr. Fante even if the corporate and individual goals are not fully achieved, and it also has the discretion to increase Mr. Fante’s total annual target bonus to up to 150% of the annual target performance bonus if the corporate and individual goals are exceeded.
Pursuant to his employment agreement, Mr. Fante received a grant of options to purchase 20,000 ordinary shares of Innocoll Germany under the Company’s 2014 Stock Option Plan. These options have an exercise price equal to the fair market value of the ordinary shares, which, at the Company’s discretion (as indicated in the grant letter), will be either: (i) the average closing price of the Company’s ordinary shares on the NASDAQ Global Market for the last 10 trading days immediately preceding the grant date, or (ii) the Company’s ordinary shares on the NASDAQ Global Market on the grant date. The options vest over three (3) years, with one-third of the options vesting after the first year following the effective date of the employment agreement, and thereafter in equal quarterly installments. All such options are subject to the terms and conditions of the Company’s 2014 Stock Option Plan, including, but not limited to, a 4-year mandatory waiting period before any option can be exercised, and certain additional performance thresholds.
The employment agreement further provides that, for each calendar year of Mr. Fante’s continuing employment, starting in 2016, the board is required to consider an annual grant of options to purchase ordinary shares to Mr. Fante based upon a variety of factors deemed important to the board, including the Company’s performance and the competitiveness of Mr. Fante’s compensation within the relevant market. The annual equity grant is in the sole discretion of the board’s compensation committee and the board as a whole. The compensation committee and the board further have the discretion to issue additional equity compensation, including but not limited to options, as the board may determine from time to time, and will consider changes in the capital of the Company when making such decisions.
The employment agreement also provides that if Mr. Fante’s employment with the Company is terminated by the Company for reasons other than cause, or as a result of his death or disability or by Mr. Fante for good reason, and provided that Mr. Fante executes a separation agreement, Mr. Fante is entitled to continue to receive his base salary for a period of one year after his termination, and reimbursement for all medical, dental and life insurance benefits for up to the earlier of: (i) one year after his termination, or (ii) the date Mr. Fante becomes eligible for similar benefits from a third party. If Mr. Fante dies during his employment with the Company, his estate is entitled to all his compensation and benefits from the Company through the date of his death, and to the proceeds from any applicable policy of life insurance obtained by the Company for the benefit of these beneficiaries. In the event Mr. Fante is unable to perform his duties and responsibilities to the Company to the full extent required by the board by reason of illness, injury or incapacity for 90 consecutive days, or for more than 120 days in the aggregate, during any period of 12 consecutive calendar months, he will be entitled to all compensation and benefits earned through the date of disability, and to the proceeds from any applicable disability insurance policy obtained by the Company for his benefit. If Mr. Fante’s employment is terminated within 180 days before or after a change in control, (a) by the Company for any reason other than cause or (b) by Mr. Fante for good reason, then any and all of the shares and/or options of the Company owned by Mr. Fante that remain subject to forfeiture shall automatically become no longer subject to forfeiture upon the latter to occur of: (i) the occurrence of the change in control, or (ii) the termination of his employment as provided above; provided, however, that Mr. Fante provides the board with written notice of the occurrence of an event constituting good reason within 30 days of the occurrence of such event and the Company fails to cure or rectify such event within 30 days after receiving such written notice, and in such event, he will be entitled to continue to receive his base salary for a period of one year after the termination, and reimbursement for all medical, dental and life insurance benefits for up to the earlier of: (i) one year after the termination, or (ii) the date Mr. Fante becomes eligible for similar benefits from a third party, as well as the acceleration of vesting of his equity compensation.
Pursuant to the employment agreement, Mr. Fante also agreed to a no-solicitation covenant and a covenant not to compete with the Company worldwide during his employment with the Company and for a period of 365 days thereafter, which period shall be automatically extended for any period of time during which the he has breached, or threatened to breach the relevant provisions.
Employment Agreement with Charles F. Katzer
Our subsidiary, Innocoll Pharmaceuticals Ltd., entered into an employment agreement with Mr. Katzer, effective as of December 14, 2015, under which he is entitled to receive an initial annual base salary of $350,000, and which provides that Mr. Katzer will serve as Vice President, Global Supply / Procurement and report to the Company’s chief executive officer. Mr. Katzer is further eligible to receive an annual target performance bonus of 40% of his base salary, based on certain annual corporate goals and individual performance goals established annually by the Company’s board. The board has discretion to pay the annual target performance bonus to Mr. Katzer even if the corporate and individual goals are not fully achieved, and it also has the discretion to increase Mr. Katzer’s total annual target bonus to up to 150% of the annual target performance bonus if the corporate and individual goals are exceeded.
Pursuant to his employment agreement, Mr. Katzer received a grant of options to purchase 11,500 ordinary shares of Innocoll Germany under the Company’s 2014 Stock Option Plan. These options have an exercise price equal to the fair market value of the ordinary shares, which, at the Company’s discretion (as indicated in the grant letter), will be either: (i) the average closing price of the Company’s ordinary shares on the NASDAQ Global Market for the last 10 trading days immediately preceding the grant date, or (ii) the price of the Company’s ordinary shares on the NASDAQ Global Market on the grant date. The options vest over three (3) years, with one-third of the options vesting after the first year following the effective date of the employment agreement, and thereafter in equal quarterly installments. All such options are subject to the terms and conditions of the Company’s 2014 Stock Option Plan, including, but not limited to, a 4-year mandatory waiting period before any option can be exercised, and certain additional performance thresholds.
The employment agreement further provides that, for each calendar year of Mr. Katzer’s continuing employment, starting in 2016, the board is required to consider an annual grant of options to purchase ordinary shares to Mr. Katzer based upon a variety of factors deemed important to the board, including the Company’s performance and the competitiveness of Mr. Katzer’s compensation within the relevant market. The annual equity grant is in the sole discretion of the board’s compensation committee and the board as a whole. The compensation committee and the board further have the discretion to issue additional equity compensation, including but not limited to options, as the board may determine from time to time, and will consider changes in the capital of the Company when making such decisions.
The employment agreement also provides that if Mr. Katzer’s employment with the Company is terminated by the Company for reasons other than cause, or as a result of his death or disability or by Mr. Katzer for good reason, and provided that Mr. Katzer executes a separation agreement, Mr. Katzer is entitled to continue to receive his base salary for a period of one year after his termination, and reimbursement for all medical, dental and life insurance benefits for up to the earlier of: (i) one year after his termination, or (ii) the date Mr. Katzer becomes eligible for similar benefits from a third party. If Mr. Katzer dies during his employment with the Company, his estate is entitled to all his compensation and benefits from the Company through the date of his death, and to the proceeds from any applicable policy of life insurance obtained by the Company for the benefit of these beneficiaries. In the event Mr. Katzer is unable to perform his duties and responsibilities to the Company to the full extent required by the board by reason of illness, injury or incapacity for 90 consecutive days, or for more than 120 days in the aggregate, during any period of 12 consecutive calendar months, he will be entitled to all compensation and benefits earned through the date of disability, and to the proceeds from any applicable disability insurance policy obtained by the Company for his benefit. If Mr. Katzer’s employment is terminated within 90 days before or after a change in control, (a) by the Company for any reason other than cause or (b) by Mr. Katzer for good reason, then any and all of the shares and/or options of the Company owned by Mr. Katzer that remain subject to forfeiture shall automatically become no longer subject to forfeiture upon the latter to occur of: (i) the occurrence of the change in control, or (ii) the termination of his employment as provided above; provided, however, that Mr. Katzer provides the board with written notice of the occurrence of an event constituting good reason within 30 days of the occurrence of such event and the Company fails to cure or rectify such event within 30 days after receiving such written notice, and in such event, he will be entitled to continue to receive his base salary for a period of one year after the termination, and reimbursement for all medical, dental and life insurance benefits for up to the earlier of: (i) one year after the termination, or (ii) the date Mr. Katzer becomes eligible for similar benefits from a third party, as well as the acceleration of vesting of his equity compensation.
Pursuant to the employment agreement, Mr. Katzer also agreed to a no-solicitation covenant and a covenant not to compete with the Company worldwide during his employment with the Company and for a period of 365 days thereafter, which period shall be automatically extended for any period of time during which he has breached, or threatened to breach the relevant provisions.
Equity-based Plans
2016 Omnibus Incentive Compensation Plan
General
The 2016 Omnibus Incentive Compensation Plan, or the 2016 Plan, covers the grant of awards to Innocoll Ireland’s employees (including officers), non-employee consultants and non-employee directors and those of Innocoll Ireland’s affiliates. Under the terms of the 2016 Plan, an aggregate of 2,850,000 ordinary shares will be authorized for delivery in settlement of awards (including incentive stock options).
We expect that the compensation committee of the board of directors will administer the 2016 Plan. The committee may delegate any or all of its administrative authority to our Chief Executive Officer or to a management committee except with respect to awards to executive officers who are subject to Section 16 of the Exchange Act. In addition, the full board of directors must serve as the committee with respect to any awards to our non-employee directors.
The ordinary shares delivered to settle awards made under the 2016 Plan may be authorized and unissued shares or treasury shares. If any shares subject to any award granted under the 2016 Plan (other than a substitute award) is forfeited or otherwise terminated without delivery of such shares, the shares subject to such awards will again be available for issuance under the 2016 Plan. However, any shares that are withheld or applied as payment for shares issued upon exercise of an award or for the withholding or payment of taxes due upon exercise of the award will continue to be treated as having been delivered under the 2016 Plan and will not again be available for grant under the 2016 Plan.
If a dividend or other distribution (whether in cash, ordinary shares or other property), recapitalization, forward or reverse stock split, subdivision, consolidation or reduction of capital, reorganization, merger, consolidation, scheme of arrangement, split-up, spin-off or combination involving us or repurchase or exchange of our shares or other securities, or other rights to purchase shares of our securities or other similar transaction or event affects our ordinary shares such that the committee determines that an adjustment is appropriate in order to prevent dilution or enlargement of the benefits (or potential benefits) provided to grantees under the 2016 Plan, the committee will make an equitable change or adjustment as it deems appropriate in the number and kind of securities subject to awards (whether or not then outstanding) and the related exercise price relating to an award.
The maximum number of ordinary shares that are subject to awards granted to any individual in a single calendar year may not exceed 400,000 shares. In addition, the maximum value of all awards to be settled in cash or property other than our ordinary shares that may be granted to any individual in a single calendar year may not exceed $2,500,000 million. These limitations apply to the calendar year in which the awards are granted and not the year in which such awards settle.
Substitution of Predecessor Options
As previously disclosed, we are currently exploring the possibility of substituting some or all of the options granted under the January 2015 stock option plan for options governed by 2016 Plan, subject to the consent of the applicable option holder. The terms of any such substitute options will be determined by the Compensation Committee of the Company’s board of directors and will be consistent with similar terms as are expected to be applicable to similar future grants made pursuant to the 2016 Plan.
Types of Awards
The 2016 Plan permits the granting of any or all of the following types of awards to all grantees:
| · | stock options, including incentive stock options, or ISOs; |
| · | stock appreciation rights, or SARs; |
| · | deferred stock and restricted stock units; |
| · | performance units and performance shares; |
| · | other stock-based awards; and |
Generally, awards under the 2016 Plan are granted for no consideration other than prior and future services. Awards granted under the 2016 Plan may, in the discretion of the committee, be granted alone or in addition to, in tandem with or in substitution for, any other award under the 2016 Plan or other plan of ours; provided, however, that if an SAR is granted in tandem with an ISO, the SAR and ISO must have the same grant date and term and the exercise price of the SAR may not be less than the exercise price of the ISO. The material terms of each award will be set forth in a written award agreement between the grantee and us.
Stock Options and SARs
The committee is authorized to grant SARs and stock options (including ISOs except that an ISO may only be granted to an employee of ours or one of our subsidiary corporations). A stock option allows a grantee to purchase a specified number of our ordinary shares at a predetermined price per share (the “exercise price”) during a fixed period measured from the date of grant. An SAR entitles the grantee to receive the excess of the fair market value of a specified number of shares on the date of exercise over a predetermined exercise price per share. The exercise price of an option or an SAR will be determined by the committee and set forth in the award agreement but the exercise price may not be less than the fair market value of an ordinary share on the grant date. The term of each option or SAR is determined by the committee and set forth in the award agreement, except that the term may not exceed 10 years. Options may be exercised by payment of the purchase price through one or more of the following means: payment in cash (including personal check or wire transfer), or, with the approval of the committee, by delivering ordinary shares previously owned by the grantee, by delivery of ordinary shares acquired upon the exercise of such option or by delivering restricted shares. The committee may also permit a grantee to pay the exercise price of an option through the sale of shares acquired upon exercise of the option through a broker-dealer to whom the grantee has delivered irrevocable instructions to deliver sales proceeds sufficient to pay the purchase price to us.
Restricted Shares
The committee may award restricted shares consisting of ordinary shares which remain subject to a risk of forfeiture and may not be disposed of by grantees until certain restrictions established by the committee lapse. The vesting conditions may be service-based (i.e., requiring continuous service for a specified period) or performance-based (i.e., requiring achievement of certain specified performance objectives) or both. A grantee receiving restricted shares will have all of the rights of a shareholder, including the right to vote the shares and the right to receive any dividends, except as otherwise provided in the award agreement. Upon termination of the grantee’s affiliation with us during the restriction period (or, if applicable, upon the failure to satisfy the specified performance objectives during the restriction period), the restricted shares will be forfeited as provided in the award agreement.
Restricted Stock Units and Deferred Stock
The committee may also grant restricted stock unit awards and/or deferred stock awards. A deferred stock award is the grant of a right to receive a specified number of our ordinary shares at the end of specified deferral periods or upon the occurrence of a specified event, which satisfies the requirements of Section 409A of the Internal Revenue Code. A restricted stock unit award is the grant of a right to receive a specified number of our ordinary shares upon lapse of a specified forfeiture condition (such as completion of a specified period of service or achievement of certain specified performance objectives). If the service condition and/or specified performance objectives are not satisfied during the restriction period, the award will lapse without the issuance of the shares underlying such award.
Restricted stock units and deferred stock awards carry no voting or other rights associated with stock ownership. The award agreement will provide whether grantees may receive dividend equivalents with respect to restricted stock units or deferred stock, and if so, whether such dividend equivalents are distributed when credited or deemed to be reinvested in additional shares of restricted stock units or deferred stock.
Performance Units
The committee may grant performance units, which entitle a grantee to cash or shares conditioned upon the fulfillment of certain performance conditions and other restrictions as specified by the committee and reflected in the award agreement. The initial value of a performance unit will be determined by the committee at the time of grant. The committee will determine the terms and conditions of such awards, including performance and other restrictions placed on these awards, which will be reflected in the award agreement.
Performance Shares
The committee may grant performance shares, which entitle a grantee to a certain number of ordinary shares, conditioned upon the fulfillment of certain performance conditions and other restrictions as specified by the committee and reflected in the award agreement. The committee will determine the terms and conditions of such awards, including performance and other restrictions placed on these awards, which will be reflected in the award agreement.
Bonus Shares
The committee may grant fully vested ordinary shares as bonus shares on such terms and conditions as specified in the award agreement.
Dividend Equivalents
The committee is authorized to grant dividend equivalents which provide a grantee the right to receive payment equal to the dividends paid on a specified number of our ordinary shares. Dividend equivalents may be paid directly to grantees or may be deferred for later delivery under the 2016 Plan. If deferred such dividend equivalents may be credited with interest or may be deemed to be invested in our ordinary shares or in other property. No dividend equivalents may be granted in conjunction with any grant of stock options or SARs.
Cash Incentive Awards
The committee may grant cash incentive awards to any eligible person in such amounts and upon such terms, including the achievement of specific performance goals during the applicable performance period, as the committee may determine. An eligible person may have more than one cash incentive award outstanding at any time. For instance, the committee may grant an eligible employee one cash incentive award with a calendar year performance period as an annual incentive bonus and a separate cash incentive award with a multi-year performance period as a long-term cash incentive bonus.
The committee shall establish performance goals applicable to each cash incentive award in its discretion and the amount that will be paid to the grantee pursuant to such cash incentive award if the applicable performance goals for the performance period are met. If an eligible person earns the right to receive a payment with respect to a cash incentive award, such payment will be made in cash in accordance with the terms of the award agreement. If the award agreement does not specify a payment date with respect to a cash incentive award, payment of the cash incentive award will be made no later than the 15th day of the third month following the end of the taxable year of the grantee or our fiscal year during which the performance period ends.
Other Stock-Based Awards
In order to enable us to respond to material developments in the area of taxes and other legislation and regulations and interpretations thereof, and to trends in executive compensation practices, the 2016 Plan authorizes the committee to grant awards that are valued in whole or in part by reference to or otherwise based on our securities. The committee determines the terms and conditions of such awards, including consideration paid for awards granted as share purchase rights and whether awards are paid in shares or cash.
Performance-Based Awards
The committee may require satisfaction of pre-established performance goals, consisting of one or more business criteria and a targeted performance level with respect to such criteria, as a condition of awards being granted or becoming exercisable or payable under the 2016 Plan, or as a condition to accelerating the timing of such events. The committee has the discretion to adjust the determinations of the degree of attainment of the pre-established performance goals.
Awards may be settled in cash, ordinary shares, other awards or other property, in the discretion of the committee.
Change of Control
If there is a merger or consolidation of us with or into another corporation or a sale of substantially all of our ordinary shares (a “Corporate Transaction”), and the outstanding awards are not assumed by surviving company (or its parent company) or replaced with economically equivalent awards granted by the surviving company (or its parent company), the committee will cancel any outstanding awards that are not vested and nonforfeitable as of the consummation of such Corporate Transaction (unless the committee accelerates the vesting of any such awards) and with respect to any vested and nonforfeitable awards, the committee may either (i) allow all grantees to exercise options and SARs within a reasonable period prior to the consummation of the Corporate Transaction and cancel any outstanding options or SARs that remain unexercised upon consummation of the Corporate Transaction, or (ii) cancel any or all of such outstanding awards (including options and SARs) in exchange for a payment (in cash, or in securities or other property) in an amount equal to the amount that the grantee would have received (net of the exercise price with respect to any options or SARs) if the vested awards were settled or distributed or such vested options and SARs were exercised immediately prior to the consummation of the Corporate Transaction. If an exercise price of the option or SAR exceeds the fair market value of our ordinary shares and the option or SAR is not assumed or replaced by the surviving company (or its parent company), such options and SARs will be cancelled without any payment to the grantee.
Amendment to and Termination of the 2016 Plan
The 2016 Plan may be amended, altered, suspended, discontinued or terminated by our board of directors without further shareholder approval, unless such approval of an amendment or alteration is required by law or regulation or under the rules of any stock exchange or automated quotation system on which the ordinary shares are then listed or quoted. Thus, shareholder approval will not necessarily be required for amendments which might increase the cost of the 2016 Plan or broaden eligibility. Shareholder approval will not be deemed to be required under laws or regulations that condition favorable treatment of grantees on such approval, although our board of directors may, in its discretion, seek shareholder approval in any circumstance in which it deems such approval advisable.
In addition, subject to the terms of the 2016 Plan, no amendment or termination of the 2016 Plan may materially and adversely affect the right of a grantee under any award granted under the 2016 Plan.
Unless earlier terminated by our board of directors, the 2016 Plan will terminate when no ordinary shares remain reserved and available for issuance or, if earlier, on the tenth anniversary from the date of adoption.
U.S. Federal Income Tax Consequences
The grant of an option or SAR will create no tax consequences for the participant or us at the time of the grant. A participant will have no taxable income upon exercise of an incentive stock option except that a participant must recognize income equal to the fair market value of the ordinary shares acquired minus the exercise price for alternative minimum tax purposes. Upon exercise of an option (other than an incentive stock option) or a SAR, a participant generally must recognize ordinary income equal to the fair market value of the ordinary shares acquired minus the exercise or grant price. Upon a disposition of shares acquired by exercise of an incentive stock option on or before the earlier of the second anniversary of the grant of such incentive stock option or the first anniversary of the exercise of such option, the participant generally must recognize ordinary income equal to the lesser of (1) the fair market value of the shares at the date of exercise minus the exercise price or (2) the amount realized upon the disposition of the incentive stock option shares minus the exercise price. Otherwise, a participant’s disposition of shares acquired upon the exercise of an option (including an incentive stock option for which the incentive stock option holding periods are met) generally will result in only capital gain or loss. Other awards under the 2016 Plan, including restricted stock and restricted stock units will generally result in ordinary income to the participant equal to the cash or the fair market value of the ordinary shares or other property (minus the amount, if any, paid by the participant for shares or other property) at the time such cash, ordinary shares or other property is received by the participant or the time that the risk of forfeiture of such ordinary shares or other property lapses.
We are generally entitled to claim a tax deduction with respect to an award granted under the Plan when the participant recognizes ordinary income with respect to the award in an amount equal to the ordinary income that is recognized by the participant. We are not entitled to claim any tax deduction for any amount recognized by a participant as capital gains.
Section 83(b) of the Code
A participant may elect under Section 83(b) of the Code to be taxed at the time of grant of restricted stock or other restricted property on the fair market value of the ordinary shares or other property at that time rather than to be taxed when the risk of forfeiture lapses on the value of the property at that time, and we would have a deduction available at the same time and in the same amount as the participant recognizes income. If a participant files an election under Section 83(b) of the Code and the participant subsequently forfeits the restricted shares or other restricted property, he or she would not be entitled to any tax deduction, including as a capital loss, for the value of the ordinary shares or property on which he or she previously paid tax. Except as discussed below, we generally will be entitled to a tax deduction at the time and equal to the amount recognized as ordinary income by the participant in connection with an option, stock appreciation right, or other award, but will be entitled to no tax deduction relating to amounts that represent a capital gain to a participant. Thus, we will not be entitled to any tax deduction with respect to an incentive stock option if the participant holds the shares for the incentive stock option holding periods.
Section 162(m) of the Code
Section 162(m) of the Code limits the amount of compensation we may deduct with respect to our Chief Executive Officer and each of the other three highest paid named executive officers (other than a chief financial officer) to $1 million per year. This deduction limit generally applies to companies that have any class of equity securities that is publicly held. This limitation does not apply, however, to performance-based compensation that satisfies certain requirements, including approval of the material terms of the plan by the company’s shareholders. In addition, the deduction limitations imposed under Section 162(m) of the Code do not apply to options, SARs and restricted shares granted prior to the expiration of the transition period (as described below) if such equity awards are granted under a plan that was adopted before any class of the company’s equity securities became publically traded. The transition period ends on the earliest of (i) the date the plan expires, (ii) the date on which the terms of the plan are materially modified, (iii) the date all of the ordinary shares issuable under the plan have been issued, or (iv) the first meeting of the shareholders of the Corporation at which directors are elected that occurs after the close of the third calendar year following the calendar year in which the initial public offering occurs (or, if the company’s stock becomes publicly held without an initial public offering, the first calendar year following the calendar year in which the company’s stock becomes publicly held). Pursuant to this transition relief, compensation attributable to stock options, SARs and restricted shares granted under the 2016 Plan prior to the expiration of the transition period should be exempt from the deduction limitations under Section 162(m) of the Code.
Section 409A of the Code
Some restricted stock units and other awards subject to deferral features may be subject to Section 409A of the Code, which regulates deferral arrangements. In such cases, the timing of the settlement of the award would have to meet certain restrictions in order for the participant not to be subject to accelerated tax and a tax penalty at the time of vesting rather than at the time of settlement. One significant restriction would be a requirement that the timing of the settlement not be controlled by the participant’s exercise of discretion. If the participant is subject to accelerated tax at the time of vesting (instead of the time of settlement), our deduction would also be accelerated. If we grant awards under the 2016 Plan that constitute deferred compensation within the meaning of Section 409A of the Code, such awards will generally be structured to comply with the applicable requirements imposed under Section 409A.
2014 Restricted Share Awards
In January 2014 (as amended on March 20, 2014), Innocoll Germany adopted a restricted share plan pursuant to which certain restricted shares of Innocoll Germany have been issued to members of our management board and certain of our employees and employees of our subsidiaries, which restricted shares were converted into Innocoll Germany’s ordinary shares or rights to receive Innocoll Germany’s ordinary shares or cash in the course of our transformation into a stock corporation. Pursuant to the restricted share plan and the award agreements entered into in connection therewith, the restricted shares are forfeited and subject to a repurchase right by us in the case of a bad leaver event, which is (i) a voluntary termination by the employee, other than in the context of a constructive termination, and (ii) a termination of the grantee by us for cause. If a bad leaver event occurs, the restricted shares and phantom shares can be repurchased by us at any time without further notice or action of the grantee at a price equal to the lesser of the amount paid by the grantee for such shares, and the fair market value per share on the date of such forfeiture. However, if no bad leaver event occurs before the occurrence of the earlier of (i), in the case of members of our management board and certain members of our advisory board, (a) the 183rd day after our initial public offering, and (b) a so-called liquidity event, or (ii) in the case of certain members of our advisory board, (a) upon a so called liquidity event or (b) in the case of an initial public offering, in relation to 33.3% and 66.7% and 100% of the shares subject to the grant, on the date which is one, two and three years after the grant date respectively, or in each case the 183rd day after the completion of an initial public offering, whichever is later, our repurchase right terminates and the ordinary shares or phantom shares held by the grantee are no longer subject to any restrictions. A liquidity event occurs in the event we merge or consolidate into or with another entity or vice versa (subject to certain limited exceptions), of the sale, conveyance, mortgage, pledge or lease of all or substantially all our assets, or of the disposition of securities representing a majority of our voting power through a transaction or series of related transactions. For certain of the grantees holding restricted shares, their award agreement specifies that our repurchase right also terminates on the day prior to date that is the fifth anniversary of the date the restricted shares were granted to such person, if earlier than the other termination events described above. While the restricted shares are still subject to our repurchase right, they may not be transferred by the grantee other than by way of inheritance or for estate planning purposes. The January 2014 restricted share plan and the award agreements granted thereunder as well as the phantom share award agreements were terminated with effect as of the effective date of our transformation into Innocoll AG and were replaced by substantially similar award agreements.
Innocoll Germany also entered into certain phantom share award agreements with certain of our executives who did not receive restricted shares under the restricted share plan, which phantom share award agreements were subsequently amended and restated following our initial public offering. Pursuant to the amended and restated phantom share award agreement entered into in January 2015, the grantee has a contractual claim against us to receive a bonus payment in case of a so-called exit event, which is the earlier of (i) the occurrence of a liquidity event, as described above for the restricted share plan and (ii) any trading day of our securities on Nasdaq within the period beginning on the third trading day after the publication of our quarterly reports for the fourth quarter of 2014 and ending on the trading day immediately preceding the first trading day that is two weeks prior to the end of the first quarter of 2015, which has been designated by our management board in its free discretion. The bonus payment will be an amount equal to the fictitious value of the phantom shares held by the grantee to be calculated on the basis of the value of our company on a cash free/debt free basis at the time and as a result of an exit event, divided by the real number of shares of our company issued at the time of the exit event, in each case treating phantom shares as if they were actual restricted shares. We have the option, but not the obligation, to issue restricted shares to the grantee in lieu of a cash bonus payment. The phantom shares are subject to our repurchase right if a bad leaver event occurs under the same terms and conditions as those applicable to the restricted shares, described above. While the restricted shares or the phantom shares are still subject to our repurchase right, they may not be transferred by the grantee other than by way of inheritance or for estate planning purposes.
Management Option Agreements
In December 2014, we entered into option agreements with each of Dr. Myers and Mr. Dunn (the “Management Option Agreements”) which memorialized options previously promised to Dr. Myers and Mr. Dunn as consideration for past services. Pursuant to the terms of the Management Option Agreements, Dr. Myers and Mr. Dunn have the right to subscribe for 10,744 and 4,672 ordinary shares of Innocoll Germany, respectively, at an exercise price of $119.25 per ordinary share of Innocoll Germany, which rights are exercisable through June 15, 2019, subject to certain black-out periods. We entered into option agreements on substantially similar terms with certain of our members of management and employees pursuant to which the optionees have the right to subscribe for a total of 9,368 ordinary shares of Innocoll Germany at an at the same exercise price of $119.25 per ordinary share of Innocoll Germany. Throughout 2015, the company entered into option agreements with members of its management board and group employees. Pursuant to the terms of the Management Option Agreements, the members of the management board and group employees have the right to subscribe for 81,144 ordinary shares of Innocoll Germany at exercise prices ranging from $99.38 to $195.70 per ordinary share of Innocoll Germany (equivalent to from $7.50 to $14.77 per ADS of Innocoll Germany), which rights are exercisable for a ten year period from date of grant, subject to certain black-out periods.
Stock Option Plan
In January 2015 Innocoll Germany established a stock option plan pursuant to which members of our and our subsidiaries’ management board(s) and employees are eligible to acquire our ordinary shares pursuant to stock options. During our extraordinary shareholder meeting on December 4, 2014, our shareholders approved a contingent capital which provides for an increase of our share capital by up to €150,920 by issuance of up to 150,920 new ordinary shares of Innocoll Germany issuable solely upon exercise of options granted under the plan. Stock options under the plan may be granted at an exercise price determined, at our sole discretion, either (i) on the basis of the average closing price for our ordinary shares on the NASDAQ Global Market on the last 10 trading days immediately preceding the date of grant or (ii) the price of our ordinary shares on the NASDAQ Global Market on the date of grant. The exercise price is subject to certain adjustments upon changes in our capitalization. Any granting of stock options is subject to the recommendation of our compensation committee and subject to this recommendation, the management board, and, to the extent that our management board is concerned, our supervisory board, selects the eligible persons to whom options will be granted and determines the grant date, amounts, exercise price and other relevant terms of the stock option grants in accordance with the provisions of the plan. Stock options may be granted at any time during the year to new management board members and employees. Stock options for existing employees and existing management board members may be granted each year, during the month of December or during the first quarter of the following year, and will be deemed to be granted as of the last trading day on NASDAQ prior to the date of grant.
A stock option granted under the plan may not be exercised until the occurrence of each of the following events: (i) the expiration of a four-year waiting period (measured from the date of grant), and (ii) during the period between the end of the date of grant and the first day of the relevant exercise period, the price of our securities on the NASDAQ Global Market has risen by a higher percentage than the performance of the MSCI World Index has increased during the same period. The relevant factors for the comparison of the development of the price of our securities on the NASDAQ Global Market and the MSCI World Index is the difference, in percentages, between both during the period which starts at the end of the date of grant and ends with the determination of the arithmetic average of the price of our securities on the NASDAQ Global Market during the 20 trading days preceding the first day of the relevant exercise period and the determination of the final value of the arithmetic average of the MSCI World Index during the 20 trading days preceding the first day of the relevant exercise period.
In addition a stock option may not be exercised during any of the following periods: (i) from the end of the seventh trading day before up to the third trading day after, our general meeting;(ii) between the first trading day on which we have published an offer to acquire new shares, bonds or option rights, up to the end of the last day of the subscription period for such offer; and (iii) beginning at the opening of trading on the first trading day that is two weeks prior to the end of each quarter and ending at the close of trading on the second trading day after the publication of our quarterly reports.
In the event of any termination of service of an optionee, other than for cause and not occurring either 180 days before or after a change of control, the optionee has the right to retain such percentage of his or her options equal to the number of months of service of the optionee from the date of grant to the date of termination divided by a factor of 36 so that those stock options vest pro rata over a period of 3 years, beginning with the date of grant. Any vested stock options for which the four-year waiting period has not expired at the time of termination can be exercised with in 12 months after the waiting period has expired. Any unvested stock options expire without compensation upon such termination of service. In the event of any termination of service of an optionee by us for cause, all stock options held by the optionee expire without any compensation at the time that his or her termination becomes effective.
In the event of any termination of service of an optionee, other than for cause, occurring within 180 days after a change of control, the optionee has the right to exercise all stock options for which the four-year waiting period has expired at the time that the termination becomes effective within the first 12 months following the termination, or, the optionee is terminated other than for cause within 180 days before a change of control, within the first 12 months following the change of control. Any stock options for which the waiting period has not yet expired at the time of termination or the change of control, as applicable, can be exercised within the first 12 months after expiration of the waiting period. Any stock option not exercised within this 12 month period expires without any compensation.
In extraordinary circumstances, our board may abstain from terminating the stock options.
We have the right to make a cash payment upon exercise of the stock options instead of delivering ordinary shares. Such cash payment will be equivalent to the closing price of our securities on the NASDAQ Global Market on the day of exercise, multiplied by the number of ordinary shares for which stock options are exercised. Any payment will be set off against the exercise price to be paid by the optionee.
Notwithstanding the foregoing, a stock option may only be exercised within ten years from the date of grant and expires thereafter if not exercised. A stock option is generally not transferable during the life of the optionee, but is inheritable upon the death of the optionee.
As a result of the Merger, the stock options under this plan have been assumed by Innocoll Ireland. Certain terms may be subject to amendment in accordance with Irish law.
Board of Directors
For additional information about our Board of Directors, please see “Item 6A — Directors and Senior Management.”
Board Committees and Independence
Our board has determined that all of our board members are independent directors in accordance with the listing requirements of the NASDAQ Global Market except for Anthony P. Zook, our Chief Executive Officer. The NASDAQ independence definition includes a series of objective tests, including that the board member is not, and has not been for at least three years, one of our employees and that neither the board member nor any of his family members has engaged in various types of business dealings with us. In addition, as required by NASDAQ rules, our board has made a subjective determination as to each independent director that no relationships exist, which, in the opinion of our board, would interfere with the exercise of independent judgment in carrying out the responsibilities of a board member. In making these determinations, our board reviewed and discussed information provided by the members of the board and us with regard to each board member’s business and personal activities and relationships as they may relate to us and our management. There are no family relationships among any of the members of our board or our executive officers. In addition, all members of the audit committee meet the independence requirements contemplated by Rule 10A-3 under the Exchange Act.
Audit Committee
The audit committee’s main function is to oversee our accounting and financial reporting processes and the audits of our financial statements. This committee’s responsibilities include, among other things:
| · | evaluating the qualifications, independence and performance of our independent registered public accounting firm; |
| · | approving the audit and non-audit services to be performed by our independent registered public accounting firm; |
| · | reviewing the design, implementation, adequacy and effectiveness of our internal accounting controls and our critical accounting policies; |
| · | discussing with management and the independent registered public accounting firm the results of our annual audit and the review of our quarterly unaudited financial statements; |
| · | reviewing, overseeing and monitoring the integrity of our financial statements and our compliance with legal and regulatory requirements as they relate to financial statements or accounting matters; |
| · | reviewing on a periodic basis, or as appropriate, any investment policy and recommending to our board any changes to such investment policy; |
| · | reviewing with management and our auditors any earnings announcements and other public announcements regarding our results of operations; |
| · | reviewing and approving any related-party transactions and reviewing and monitoring compliance with our code of conduct and ethics; and |
| · | reviewing and evaluating, at least annually, the performance of the audit committee and its members including compliance of the audit committee with its charter. |
The members of our audit committee are Dr. Banerji, Mr. Culverwell and Dr. Wiley. Mr. Culverwell serves as the chairperson of the committee. All members of our audit committee meet the requirements for financial literacy under the applicable rules and regulations of the SEC and the NASDAQ Global Market. Our board has determined that Mr. Culverwell is an “audit committee financial expert,” as defined by applicable SEC rules, and has the requisite financial sophistication as defined under the applicable NASDAQ rules and regulations. Our board has determined that Dr. Banerji, Mr. Culverwell and Dr. Wiley are independent under the applicable rules of the SEC and the NASDAQ Global Market. The audit committee operates under a written charter that satisfies the applicable standards of the SEC and the NASDAQ Global Market.
Compensation Committee
Our compensation committee reviews and approves policies relating to compensation and benefits of our senior management team and our employees. The compensation committee reviews and approves corporate goals and objectives relevant to the compensation of our chief executive officer and other executive officers, evaluates the performance of these officers in light of those goals and objectives and approves the compensation of these officers based on such evaluations. The compensation committee also reviews and approves the issuance of share options and other awards under our equity plans. The compensation committee reviews and evaluates, at least annually, the performance of the compensation committee and its members, including compliance by the compensation committee with its charter.
The members of our compensation committee are Dr. Banerji, Mr. Brennan, Mr. Symonds and Dr. Wiley. Mr. Brennan serves as the chairperson of the committee. Our board has determined that Dr. Banerji, Mr. Brennan, Mr. Symonds and Dr. Wiley are independent under the applicable rules and regulations of the NASDAQ Global Market, is a “non-employee director” as defined in Rule 16b-3 promulgated under the Exchange Act and is an “outside director” as that term is defined in Section 162(m) of the U.S. Internal Revenue Code of 1986, as amended. The compensation committee operates under a written charter, which the compensation committee reviews and evaluates at least annually.
Nominating and Corporate Governance Committee
The nominating and corporate governance committee is responsible for assisting our board in discharging the board’s responsibilities regarding the identification of qualified candidates to become board members, the selection of nominees for election as members of the board at our annual meetings of shareholders (or special meetings of shareholders at which board members are to be elected), and the selection of candidates to fill any vacancies on our board and any committees thereof. In addition, the nominating and corporate governance committee is responsible for overseeing our corporate governance policies, reporting and making recommendations to our board concerning governance matters and oversight of the evaluation of our board.
The members of our nominating and corporate governance committee are Mr. Brennan, Mr. Culverwell, Mr. Schmidt and Mr. Symonds. Mr. Schmidt serves as the chairperson of the committee. Our board has determined that Mr. Brennan, Mr. Culverwell, Mr. Schmidt and Mr. Symonds are independent under the applicable rules and regulations of the NASDAQ Global Market relating to nominating and corporate governance committee independence. The nominating and corporate governance committee operates under a written charter, which the nominating and corporate governance committee reviews and evaluates at least annually.
Compensation Committee Interlocks and Insider Participation
None of the members of our compensation committee has ever been one of our officers or employees. None of our executive officers currently serves, or has served, as a member of the compensation committee of any entity that has one or more executive officers serving as a member of our board or compensation committee.
Board Diversity
Our nominating and corporate governance committee is responsible for reviewing with the board, on an annual basis, the appropriate characteristics, skills and experience required for our board and their individual members. In evaluating the suitability of individual candidates (both new candidates and current members), the nominating and corporate governance committee, in recommending candidates for election, and the board, in approving (and, in the case of vacancies, appointing) such candidates, will take into account many factors, including the following:
| · | personal and professional integrity, ethics and values; |
| · | experience in corporate management, such as serving as an officer or former officer of a publicly-held company; |
| · | experience as a board member or executive officer of another publicly-held company; |
| · | strong finance experience; |
| · | diversity of expertise and experience in substantive matters pertaining to our business relative to other board members; |
| · | diversity of background and perspective, including, but not limited to, with respect to age, gender, race, place of residence and specialized experience; |
| · | experience relevant to our business industry and with relevant social policy concerns; and |
| · | relevant academic expertise or other proficiency in an area of our business operations. |
Currently, our board evaluates each individual in the context of the board, with the objective of assembling a group that can best maximize the success of the business and represent shareholder interests through the exercise of sound judgment using its diversity of experience in these various areas.
At the end of each of the past three years, the breakdown of employees, including our subsidiaries by main categories of activity was as follows:
| At December 31, | | 2013 | | | 2014 | | | 2015 | |
| Senior Management Team | | | 2 | | | | 3 | | | | 4 | |
| Research and Development & Clinical | | | 11 | | | | 13 | | | | 22 | |
| Management, Financial & Business Development | | | 14 | | | | 15 | | | | 20 | |
| Manufacturing | | | 64 | | | | 65 | | | | 70 | |
| Total | | | 91 | | | | 96 | | | | 116 | |
At the end of each of the past three years, the breakdown of employees, including our subsidiaries by geographic location was as follows:
| At December 31, | | 2013 | | | 2014 | | | 2015 | |
| Germany(1) | | | 76 | | | | 78 | | | | 91 | |
| Ireland(2) | | | 10 | | | | 11 | | | | 11 | |
| United States(3) | | | 3 | | | | 5 | | | | 11 | |
| United Kingdom | | | 1 | | | | 1 | | | | 2 | |
| Serbia | | | 1 | | | | 1 | | | | 1 | |
| Total | | | 91 | | | | 96 | | | | 116 | |
(1)Industriegebiet Saal, Donaustraße 24, 93342 Saal/Donau, Germany.
(2)Unit 9, Block D, Monksland Business Park, Monksland, Athlone, Co. Roscommon, Ireland.
(3)3813 West Chester Pike, Newtown Square, Pennsylvania 19073, United States.
See “Item 7 –Major Shareholders and Related-Party Transactions” A. Major Shareholders, and B. Related-Party Transactions.
Item 7. Major Shareholders and Related-Party Transactions
The following table sets forth information with respect to the beneficial ownership of our ordinary shares as of the date of this annual report by:
| · | members of our senior management team; |
| · | members of our board and senior management team as a group; |
| · | members of our predecessor’s supervisory and management boards; |
| · | each person who is known by us to own beneficially more than 5% of our outstanding ordinary shares as of the date of this annual report. |
The number of shares beneficially owned by each shareholder is determined under rules issued by the SEC. Under these rules, beneficial ownership includes any shares as to which a person has sole or shared voting power or investment power. Applicable percentage ownership is based on 24,346,782 ordinary shares outstanding on March 16, 2016 but does not include the up to 13,758,203 additional ordinary shares the board is entitled to issue.
In computing the number of shares beneficially owned by a person and the percentage ownership of that person, ordinary shares subject to options, and other rights held by such person that are currently exercisable or will become exercisable within 60 days of the date of this annual report are considered outstanding, although these shares are not considered outstanding for purposes of computing the percentage ownership of any other person. None of our shareholders has different voting rights from other shareholders. As of March 16, 2016, we had 20 holders of record.
Unless otherwise indicated, the address of each beneficial owner who is a member of our board or senior management team listed below is c/o Innocoll Holdings plc, Unit 9, Block D, Monksland Business Park, Monksland, Athlone, Ireland. We believe, based on information provided to us, that each of the shareholders listed below has sole voting and investment power with respect to the shares beneficially owned by the shareholder, unless noted otherwise, subject to community property laws where applicable.
| | | Shares Beneficially Owned | |
| Name of Beneficial Owner | | Number | | | Percentage | |
| Major Shareholder (5% or Greater Shareholders) | | | | | | | | |
| Cam Investment Cayman Holdings L.P.(1) | | | 5,014,626 | | | | 19.8 | % |
| Morgan Stanley & Co. LLC(2) | | | 4,165,358 | | | | 16.6 | % |
| Sofinnova Venture Partners VIII, L.P.(3) | | | 1,888,889 | | | | 7.8 | % |
| Members of Our Board and Senior Management Team and Our Predecessor’s Supervisory and Management Boards | | | | | | | | |
| Jonathan Symonds, CBE(4) | | | 463,822 | | | | 1.9 | % |
| Shumeet Banerji, Ph.D.(5) | | | 308,660 | | | | 1.3 | % |
| David R. Brennan(6) | | | 237,440 | | | | 1.0 | % |
| A. James Culverwell(7) | | | 338,806 | | | | 1.4 | % |
| Rolf D. Schmidt(8) | | | 472,195 | | | | 1.9 | % |
| Joseph Wiley M.D. (9) | | | — | | | | — | |
| Anthony P. Zook(10) | | | 528,111 | | | | 2.2 | % |
| Jose Carmona(11) | | | — | | | | — | |
| Gordon Dunn(12) | | | 549,915 | | | | 2.2 | % |
| Michael Myers, Ph.D.(13) | | | 551,385 | | | | 2.3 | % |
| Rich Fante(14) | | | 8,650 | | | | — | % |
| Charles F. Katzer(15) | | | — | | | | — | % |
| All members of our board and senior management team as a group (10 persons) | | | 2,357,684 | | | | 9.6 | % |
(1) Based on information provided on Form 13D filed with the SEC on August 12, 2014 and gives effect to the issuance of 13.25 ordinary shares of Innocoll Ireland in respect of each ordinary share of Innocoll AG. Consists of 4,085,465 ordinary shares and 929,156 ordinary shares issuable upon the exercise of options that have vested or will vest within 60 days of the date of this annual report.
(2) Based on information provided on Form 13D filed with the SEC on May 8, 2015 and gives effect to the issuance of 13.25 ordinary shares of Innocoll Ireland in respect of each ordinary share of Innocoll AG. Consists of 3,369,382 ordinary shares and 795,980 ordinary shares issuable upon the exercise of options that have vested or will vest within 60 days of the date of this annual report.
(3)Based on information provided on Form 13D filed with the SEC on August 4, 2014 and gives effect to the issuance of 13.25 ordinary shares of Innocoll Ireland in respect of each ordinary share of Innocoll AG.
(4)Mr. Symonds was the chairperson of the supervisory board of Innocoll AG and is chairperson of the board of Innocoll Holdings plc.
(5)Dr. Banerji was a member of the supervisory board of Innocoll AG and is a member of the board of Innocoll Holdings plc.
(6)Mr. Brennan was a member of the supervisory board of Innocoll AG and is a member of the board of Innocoll Holdings plc.
(7)Mr. Culverwell was a member of the supervisory board of Innocoll AG and is a member of the board of Innocoll Holdings plc. Consists of 281,566 ordinary shares and 57,240 ordinary shares issuable upon the exercise of options that have vested or will vest within 60 days of the date of this annual report.
(8) Mr. Schmidt was a member of the supervisory board of Innocoll AG and is a member of the board of Innocoll Holdings plc. Consists of 386,587 ordinary shares and 85,608 ordinary shares issuable upon the exercise of options that have vested or will vest within 60 days of the date of this annual report.
(9)Dr. Wiley was a member of the supervisory board of Innocoll AG and is a member of the board of Innocoll Holdings plc.
(10)Mr. Zook is our Chief Executive Officer and was the chairperson of the management board of Innocoll AG and is a member of the board of Innocoll Holdings plc.
(11)Mr. Carmona is our Chief Financial Officer and was a member of the management board of Innocoll AG. Does not include 277,985 shares issuable upon the exercise of options that have not vested.
(12)Mr. Dunn was a member of the management board of Innocoll AG and is our Vice President, Finance Director of Europe. Consists of 115,526 ordinary shares held beneficially by the Rebecca F. Dunn 2011 Irrevocable Trust, 38,080 ordinary shares issuable upon the exercise of options that have vested or will vest within 60 days of the date of this annual report held by the Rebecca F. Dunn 2011 Irrevocable Trust, of 55,557 ordinary shares held beneficially by the George J. Dunn Trust, 278,848 ordinary shares and 61,904 ordinary shares issuable upon exercise of options that have vested or will vest within 60 days of the date of this annual report. Does not include 33,125 shares issuable upon the exercise of options that have not vested. Mr. Dunn is joint trustee and beneficiary of each of the Rebecca F. Dunn 2011 Irrevocable Trust and the George J. Dunn Trust and has a shared power to vote, acquire, hold and dispose of the shares and options held by each trust.
(13)Dr. Myers was previously a member of Innocoll Germany’s management board and our Chief Executive Officer through December 2014. Consists of 395,857 ordinary shares and 155,528 ordinary shares issuable upon the exercise of options that have vested or will vest within 60 days of the date of this annual report. Does not include 33,125 shares issuable upon the exercise of options that have not vested.
(14)Mr. Fante is our Chief Commercial Officer and Head of Business Development. Does not include 265,000 shares issuable upon the exercise of options that have not vested.
(15)Mr Katzer is our Vice President, Global Supply/Procurement. Does not include 152,375 shares issuable upon the exercise of options that have not vested.
In so far as is known to us, there was no person who, directly or indirectly, joint or severally, exercised or could exercise control over us and we are not aware of any arrangements which might result in a change of control of Innocoll.
| B. | RELATED-PARTY TRANSACTIONS |
Other than as referred to below, none of our directors, officers or major shareholders or, to our knowledge, their families, had any interest, direct or indirect, in any transaction during the last fiscal year or in any proposed transaction which has affected or will materially affect us or our investment interests or subsidiaries.
Innocoll AG 2015 Follow-On Public Offering
On April 30, 2015, Innocoll Germany sold 1,999,690 ADSs of Innocoll Germany to the public in an underwritten offering, representing 150,920 ordinary shares of Innocoll Germany, at a price of $9.00 per ADS, thereby raising $16.9 million after deducting underwriting discounts and commissions. In the same offering certain selling shareholders offered 1,321,979 ADSs of Innocoll Germany, representing 99,772 ordinary shares of Innocoll Germany. We received no proceeds from the shares sold by the selling shareholders. In the same offering, one of our existing shareholders and certain members of Innocoll Germany’s management and supervisory boards purchased an aggregate of $3.8 million of Innocoll Germany ADSs at the public offering price of $9.00 per ADS, consisting of an aggregate of 420,000 of the 3,321,669 ADSs then sold. Upon the effectiveness of the Merger, we terminated Innocoll Germany’s ADS facility and each cancelled ADS effectively became an entitlement to receive one ordinary share of Innocoll Ireland.
The following sets forth the ordinary shares of Innocoll AG acquired by related persons as a result of the April 2015 follow-on public offering:
| Participants | | Innocoll AG
Ordinary
Shares(2) |
| 5% or Greater Shareholders(1) | | | | |
| Sofinnova Venture Partners VIII, L.P. | | | 16,771 | |
| | | | | |
| Executive Officers and Directors(1) | | | | |
| Shumeet Banerji | | | 3,773 | |
| A. James Culverwell | | | 1,509 | |
| Jonathan Symonds | | | 7,547 | |
| Anthony Zook | | | 2,096 | |
| Joe Wiley | | | — | |
(1)Additional details regarding these shareholders and their equity holdings are provided in “Principal Shareholders.”
(2)Consists of Innocoll AG ordinary shares underlying ADSs acquired in our April 2015 follow-on public offering, and disregards fractional shares.
Some of our historic supervisory board and management board members are associated with our principal shareholders as indicated in the table below:
Board Members | | Principal Shareholder |
Joe Wiley | | Sofinnova Venture Partners VIII, L.P. |
Employment Agreements
We have entered into employment agreements with each of the members of the management board. For more information regarding these agreements, see “Item 6. Directors, Senior Management and Employees—B. Compensation.”
Management Option Agreements
In December 2014 and throughout 2015, we entered into the Management Option Agreements with certain of our management which memorialized option previously promised to Dr. Myers and Mr. Dunn as consideration for past services. See “Item 6. Directors, Senior Management and Employees—B. Compensation—Equity-based Plans—Management Option Agreements.”
Policies and Procedures for Related Person Transactions
Our board has adopted a written related person transaction policy, setting forth the policies and procedures for the review and approval or ratification of related-person transactions. This policy will cover, with certain exceptions set forth in Item 404 of Regulation S-K under the Securities Act, any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships in which we were or are to be a participant, where the amount involved exceeds $120,000 and a related person had or will have a direct or indirect material interest, including, without limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness and employment by us of a related person. In reviewing and approving any such transactions, our audit committee is tasked to consider all relevant facts and circumstances, including, but not limited to, whether the transaction is on terms comparable to those that could be obtained in an arm’s length transaction and the extent of the related person’s interest in the transaction. All of the transactions described in this section occurred prior to the effectiveness of this policy.
| C. | INTERESTS OF EXPERTS AND COUNSEL |
Not applicable.
Item 8. Financial Information
| A. | CONSOLIDATED STATEMENTS AND OTHER FINANCIAL INFORMATION |
See “Item 18. Financial Statements.”
Legal Proceedings
From time to time, we may be subject to various claims or legal, arbitral or administrative proceedings that arise in the ordinary course of our business. We are currently not a party to, and we are not aware of any threat of, any legal, arbitral or administrative proceedings which, in the opinion of our management, is likely to have a material adverse effect on our business, financial condition or results of operations.
Dividend Policy
Neither we nor our legal predecessors, Innocoll AG or Innocoll GmbH, have ever declared or paid any cash dividends on our ordinary shares, and we have no present intention of declaring or paying any dividends in the foreseeable future. In addition, for so long as our loan obligations under the financing contract with EIB are outstanding, our ability to pay dividends is limited to certain circumstances. Any recommendation by our board to pay dividends, subject to compliance with applicable law and any contractual provisions that restrict or limit our ability to pay dividends, including under agreements for indebtedness that we may incur, will depend on many factors, including our financial condition, results of operations, legal requirements, capital requirements, business prospects and other factors that our board deems relevant.
All of our shares have the same dividend rights as all of our other outstanding shares. Any distribution of dividends proposed by our board requires the approval of our shareholders at a shareholders’ meeting. See “Item 10. Additional Information—B. Memorandum and Articles of Association” that explain in more detail the procedures we must follow and the German law provisions that determine whether we are entitled to declare a dividend.
For information regarding the German withholding tax applicable to dividends and related United States refund procedures, see “Item 10. Additional Information—E. Taxation.”
Except as set forth elsewhere in this annual report, no significant changes have occurred since December 31, 2015.
Item 9. The Offer and Listing
| A. | OFFER AND LISTING DETAILS |
Historically, Innocoll Germany’s ADSs represented 1/13.25 of one ordinary share of Innocoll AG. Innocoll Germany’s ADSs were listed on the NASDAQ Global Market from July 25, 2014 to March 15, 2016. Upon the effectiveness of the Merger, we terminated Innocoll Germany’s ADS facility, each cancelled ADS was effectively exchanged for one ordinary share of Innocoll Ireland and holders of Innocoll Germany ordinary shares received 13.25 ordinary shares of Innocoll Ireland in exchange for each share held of Innocoll Germany. Simultaneous with this transaction, Innocoll Ireland listed its ordinary shares on the NASDAQ Global Market under the symbol “INNL”, which we previously used for Innocoll Germany’s ADS. Our ordinary shares, par value $0.01 per share, have been listed on the NASDAQ Global Market since March 16, 2016.
The following table sets forth for the periods indicated the reported high and low sale prices of Innocoll Germany’s ADSs through March 15, 2016, and Innocoll Ireland’s ordinary shares on March 16, 2016, each on the NASDAQ Global Market.
| Year Ended December 31, | | High | | | Low | |
| 2014 (from July 25, 2014) | | $ | 9.51 | | | $ | 4.45 | |
| 2015 | | $ | 16.46 | | | $ | 6.00 | |
| | | | | | | | | |
| Quarter Ended | | | | | | | | |
| September 30, 2014 (from July 25, 2014) | | $ | 9.51 | | | $ | 6.15 | |
| December 31, 2014 | | $ | 8.91 | | | $ | 4.45 | |
| March 31, 2015 | | $ | 8.98 | | | $ | 6.00 | |
| June 30, 2015 | | $ | 16.46 | | | $ | 6.98 | |
| September 30, 2015 | | $ | 15.19 | | | $ | 10.98 | |
| December 31, 2015 | | $ | 13.72 | | | $ | 6.30 | |
| March 31, 2016 (through March 16, 2016) | | $ | 9.35 | | | $ | 7.07 | |
| | | | | | | | | |
| Month Ended | | | | | | | | |
| September 2015 | | $ | 14.00 | | | $ | 11.51 | |
| October 2015 | | $ | 13.72 | | | $ | 11.50 | |
| November 2015 | | $ | 12.68 | | | $ | 6.70 | |
| December 2015 | | $ | 9.25 | | | $ | 6.30 | |
| January 2016 | | $ | 9.35 | | | $ | 7.07 | |
| February 2016 | | $ | 9.00 | | | $ | 7.99 | |
During the first quarter of 2016 (through March 15, 2016) the reported high and low sale prices of Innocoll Germany’s ADSs on the NASDAQ Global Market was $9.35 and $7.07, respectively. During the first quarter of 2016 (on March 16, 2016) the reported high and low sale prices of Innocoll Ireland’s ordinary shares on the NASDAQ Global Market was $8.05 and $8.05, respectively.
Not applicable.
Through March 15, 2016, Innocoll Germany’s ADSs were listed for trading on the NASDAQ Global Market under the symbol “INNL.” Since March 16, 2016, our ordinary shares have been listed for trading on the NASDAQ Global Market under the symbol “INNL.”
Not applicable.
Not applicable.
Not applicable.
Item 10. Additional Information
Merger
On March 16, 2016, Innocoll AG merged with Innocoll Holdings plc by way of a European cross-border merger with Innocoll Holdings plc being the surviving company. Upon the effectiveness of the Merger, we terminated Innocoll AG ADS facility and each cancelled ADS effectively became an entitlement to receive one ordinary share of Innocoll Holdings plc. Holders of Innocoll AG ordinary shares received 13.25 ordinary shares of Innocoll Holdings plc in respect of each share held of Innocoll AG. Simultaneous with this transaction, Innocoll Holdings plc listed its ordinary shares on the Nasdaq Global Market under the symbol “INNL”, which we previously used for Innocoll Germany’s ADSs. The Merger effectively resulted in Innocoll Holdings plc becoming the publicly-traded parent of the Innocoll group of companies carrying on the same business as that conducted by Innocoll AG prior to the Merger.
Potential Claims Related to the Merger
Nine shareholders of record as of January 23, 2016, collectively holding 176 ordinary shares of Innocoll Germany as of March 15, 2016, have recorded their objection to the Merger in the minutes of Innocoll Germany’s extraordinary general meeting, held on January 30, 2016. As a result, the objecting shareholders are entitled to accept the cash offer to acquire the ordinary shares held by them, as set forth in the Merger Proposal, which offer has not yet been accepted. Following the Merger, and the receipt of 13.25 ordinary shares of Innocoll Ireland for each previously outstanding ordinary share of Innocoll Germany, Innocoll Ireland would be required to acquire the 2,330 ordinary shares of Innocoll Ireland now held by the objecting shareholders for cash, provided such offer is accepted by the objecting shareholders within the applicable time.
Under the terms of the Merger Proposal, these objecting shareholders would be entitled to cash compensation in an aggregate amount of €21,688. However, under a special procedure available under German law, these shareholders may contest the adequacy of such cash compensation. If a German court agrees that the offered compensation was undervalued, or if we enter into a binding settlement, the per share cash compensation ultimately payable to the objecting shareholders may exceed the amount contemplated by the Merger Proposal.
Not Applicable.
| B. | MEMORANDUM AND ARTICLES OF ASSOCIATION |
For a desciption of the information required by this Item, see the description included in our registration statement on Form F-4 (Registration No. 333-208438) under the heading “DESCRIPTION OF THE SHARE CAPITAL OF INNOCOLL IRELAND”.
We have not entered into any material contracts other than in the ordinary course of business and other than those described elsewhere in “Item 4. Information on the Company—B. Business Overview”, “Item 6. Directors, Senior Management and Employees” and “Item 7. Major Shareholders and Related-Party Transactions”, or elsewhere in this annual report.
Exchange Controls and Other Limitations Affecting Security Holders
Irish exchange control regulations ceased to apply from and after December 31, 1992. Except as indicated below, there are no restrictions on non-residents of Ireland dealing in domestic securities, which includes shares or depositary receipts of Irish companies such as Innocoll Ireland. Except as indicated below, dividends and redemption proceeds also continue to be freely transferable to non-resident holders of such securities. The Financial Transfers Act, 1992 gives power to the Minister for Finance of Ireland to make provision for the restriction of financial transfers between Ireland and other countries and persons. Financial transfers are broadly defined and include all transfers that would be movements of capital or payments within the meaning of the treaties governing the member states of the European Union. The acquisition or disposal of ordinary shares issued by an Irish incorporated company and associated payments falls within this definition. In addition, dividends or payments on redemption or purchase of shares and payments on a liquidation of an Irish incorporated company would fall within this definition.
Irish exchange control regulations ceased to apply from and after December 31, 1992. Except as indicated below, there are no restrictions on non-residents of Ireland dealing in domestic securities, which includes shares or depositary receipts of Irish companies such as Innocoll Holdings plc. Except as indicated below, dividends and redemption proceeds also continue to be freely transferable to non-resident holders of such securities.
The Financial Transfers Act, 1992 gives power to the Minister for Finance of Ireland to make provision for the restriction of financial transfers between Ireland and other countries and persons. Financial transfers are broadly defined and include all transfers that would be movements of capital or payments within the meaning of the treaties governing the member states of the European Union. The acquisition or disposal of shares issued by an Irish incorporated company and associated payments falls within this definition. In addition, dividends or payments on redemption or purchase of shares and payments on a liquidation of an Irish incorporated company would fall within this definition.
At present the Financial Transfers Act, 1992 prohibits financial transfers involving: certain persons and activities in Ukraine, Democratic Republic of Congo, Belarus, Liberia, Tunisia, Zimbabwe, Somalia, Democratic People’s Republic of Korea, Iraq, Libya, Afghanistan, Republic of Guinea-Bissau, Burma, Cote d’Ivoire, Iran, Republic of Guinea, Syria, Egypt, Eritrea, Lebanon, Uzbekistan, Sudan; the late Slobodan Milosevic and associated persons; certain persons indicted by the International Criminal Tribunal for the former Yugoslavia; the late Osama bin Laden and Al-Qaida.
Any transfer of, or payment in respect of, shares involving the government of any country that is currently the subject of United Nations or European Union sanctions, any person or body controlled by any of the foregoing, or by any person acting on behalf of the foregoing, may be subject to restrictions pursuant to such sanctions as implemented into Irish law. We do not anticipate that orders under the Financial Transfers Act, 1992 or United Nations or European Union sanctions implemented into Irish law will have a material effect on our business.
Taxation in Ireland
Scope of Discussion
The following is a general summary of the main Irish tax considerations applicable to certain investors who are the owners of our ordinary shares and is the opinion of William Fry insofar as it relates to legal conclusions with respect to matters of Irish tax law. It is based on existing Irish law and our understanding of the practices of the Irish Revenue Commissioners on the date of this annual report. Legislative, administrative or judicial changes may modify the tax consequences described below, possibly with retrospective effect. Furthermore, we can provide no assurances that the consequences contained in this summary will not be challenged by the Irish Revenue Commissioners or will be sustained by a court if challenged.
The statements do not constitute tax advice and are intended only as a general guide. Furthermore, this information applies only to our ordinary shares that are held as capital assets and does not apply to all categories of shareholders, such as dealers in securities, trustees, insurance companies, collective investment schemes or shareholders who have, or who are deemed to have, acquired their shares by virtue of an office or employment. This summary is not exhaustive and shareholders should consult their own tax advisors as to the tax consequences in Ireland, or other relevant jurisdictions, including the acquisition, ownership and disposition of our ordinary shares.
Tax on Chargeable Gains
The rate of capital gains tax in Ireland is currently 33%.
A disposal of our ordinary shares by a shareholder who is not resident or ordinarily resident for tax purposes in Irelandand who does not hold their shares in connection with a trade carried on by such holder in Ireland through a branch or agency should not give rise to Irish tax on any chargeable gain realized on such disposal provided at the time of disposal: (i) the ordinary shares are quoted on a recognized stock exchange; or (ii) the ordinary shares do not derive the greater part of their value from land, buildings, minerals, or mineral or exploration rights in Ireland.
A disposal of our ordinary shares by a shareholder who is not resident or ordinarily resident for tax purposes in Ireland or a shareholder that holds their shares in connection with a trade carried on by such holder in Ireland through a branch or agency will, subject to the availability of exemptions and reliefs, be within the charge to Irish tax on chargeable gains and may, depending on the circumstances, give rise to a chargeable gain or allowable loss for that shareholder.
A holder of our ordinary shares who is an individual and who is temporarily not resident in Ireland may, under Irish anti-avoidance legislation, be liable to Irish tax on any chargeable gain realized on a disposal during the period in which such individual is not resident.
Dividend Withholding Tax
Distributions made by us will generally be subject to dividend withholding tax (“DWT”) at the standard rate of income tax (currently 20%) unless one of the exemptions described below applies. For DWT purposes, a dividend includes any distribution made by us to the shareholders, including cash dividends, non-cash dividends and additional stock or units taken in lieu of a cash dividend. The company responsible for withholding DWT at source and forwarding the relevant payment to the Irish Revenue Commissioners.
Certain shareholders (both individual and corporate) are entitled to an exemption from DWT. In particular, dividends to a non-Irish resident shareholder should not be subject to DWT if the shareholder is beneficially entitled to the dividend and is:
| · | an individual shareholder resident for tax purposes in a “relevant territory” and the individual is neither resident nor ordinarily resident in Ireland; |
| · | a corporate shareholder resident for tax purposes in a “relevant territory” provided that the corporate shareholder is not under the control, whether directly or indirectly, of a person or persons who is or are resident in Ireland; |
| · | a corporate shareholder that is not resident for tax purposes in Ireland that is ultimately controlled, directly or indirectly, by persons resident in a “relevant territory” and that is not controlled directly or indirectly, by persons who are not resident in a “relevant territory”; |
| · | a corporate shareholder that is not resident for tax purposes in Ireland and whose principal class of shares (or those of its 75% parent) is substantially and regularly traded on a recognized stock exchange either in a “relevant territory” or on such other stock exchange approved by the Irish Minister for Finance; or |
| · | a corporate shareholder that is not resident for tax purposes in Ireland and is wholly owned, directly or indirectly, by two or more companies where the principal class of shares of each of such companies is substantially and regularly traded on a recognized stock exchange in a “relevant territory” or on such other stock exchange approved by the Irish Minister for Finance; |
and, provided that, in all cases noted above (but subject to the exception in the paragraph below regarding ’U.S. Resident Shareholders’), the shareholder has provided a relevant Irish DWT declaration form to his or her broker before the record date for the dividend (in the case of ordinary shares held through the Depositary Trust Company, or DTC), and the relevant information is further transmitted to us (in the case of ordinary shares held through DTC) or to our transfer agent (in the case of shares held outside of DTC).
A list of “relevant territories” for the purposes of DWT is set forth below.
| Albania | China | Hong Kong | Macedonia | Portugal | Switzerland |
| Armenia | Croatia | Hungary | Malaysia | Qatar | Thailand |
| Australia | Cyprus | Iceland | Malta | Romania | Turkey |
| Austria | Czech | India | Mexico | Russia | United Arab Emirates |
| Bahrain | Republic | Israel | Moldova | Saudi Arabia |
| Belarus | Denmark | Italy | Montenegro | Serbia | Ukraine |
| Belgium | Egypt | Japan | Morocco | Singapore | United Kingdom |
| Bosnia & | Estonia | Republic of | Netherlands | Slovak | United States of America |
| Herzegovina | Ethiopia | Korea | New Zealand | Republic |
| Botswana | Finland
France | Kuwait | Norway | Slovenia | Uzbekistan |
| Bulgaria | Georgia | Latvia | Pakistan | South Africa | Vietnam |
| Canada | Germany | Lithuania | Panama | Spain | Zambia |
| Chile | Greece | Luxembourg | Poland | Sweden | |
Prior to paying any dividend, we will put in place an agreement with an entity which is recognized by the Irish Revenue Commissioners as a “qualifying intermediary” which satisfies one of the Irish requirements for dividends to be paid free of DWT to certain shareholders who hold their ordinary shares through DTC.
We will rely on information received directly or indirectly from brokers and our transfer agent in determining where shareholders reside, whether they have provided the required U.S. tax information and whether they have provided the required Irish dividend withholding tax forms, as described below. Shareholders who are required to file Irish forms in order to receive their dividends free of DWT should note that such forms are generally valid until December 31st of the fifth year after the year of issue of the forms and new forms must be filed before the expiration of that period in order to continue to enable them to receive dividends without DWT. Links to the Irish Revenue forms are available athttp://www.revenue.ie/en/tax/dwt/forms/index.html.
For shareholders that cannot avail themselves of one of Ireland’s domestic law exemptions from DWT, it may be possible for such shareholders to rely on the provisions of a double tax treaty to which Ireland is a party to reduce the rate of DWT.
U.S. Resident Shareholders
Dividends paid in respect of shares in an Irish resident company that are owned by residents of the United States and held through DTC will not be subject to DWT provided that the address of the beneficial owner of the shares in the records of the broker is in the U.S. We strongly recommend that such shareholders ensure that their information has been properly recorded by their brokers (so that such brokers can provide the relevant information to a qualifying intermediary appointed by us).
Dividends paid in respect of shares in an Irish resident company that are owned by residents of the U.S. and held outside of DTC will not be subject to DWT provided that the shareholder has provided a valid Irish DWT form showing a U.S. address to our transfer agent at least seven business days before the record date for the first dividend payment to which they are entitled.
If any shareholder who is resident in the U.S. receives a dividend subject to DWT, he or she may be able to make an application for a refund from the Irish Revenue Commissioners on the prescribed form, subject to certain time limits.
Residents of “Relevant Territories” other than the United States
Shareholders who are residents of “relevant territories” other than the United States (regardless of when such shareholders acquired their ordinary shares) must satisfy the conditions of one of the exemptions referred to above including the requirement to complete the appropriate Irish DWT declaration form in order to receive dividends without DWT.
Shareholders must provide the appropriate Irish DWT form to their brokers (so that such brokers can provide the relevant information to a qualifying intermediary appointed by us) before the record date for the first dividend to which they are entitled (in the case of ordinary shares held through DTC), or to our transfer agent at least seven business days before such record date (in the case of ordinary shares held outside of DTC). We strongly recommend that such shareholders complete the appropriate Irish DWT declaration form and provide them to their brokers or our transfer agent as soon as possible.
If any shareholder who is resident in a “relevant territory” receives a dividend subject to DWT, he or she may be able to make an application for a refund from the Irish Revenue Commissioners on the prescribed form, subject to certain time limits.
Irish Resident Shareholders
Irish tax resident or ordinarily resident shareholders will generally be subject to DWT in respect of dividends or distributions received from an Irish resident company unless an exemption applies.
Irish tax resident or ordinarily resident shareholders that are entitled to receive dividends without DWT must complete the relevant Irish DWT form and provide them to their brokers (so that such brokers can provide the relevant information to a qualifying intermediary appointed by us) before the record date for the first dividend to which they are entitled (in the case of ordinary shares held through DTC), or to our transfer agent at least seven business days before such record date (in the case of ordinary shares held outside of DTC).
Shareholders who are resident or ordinarily resident in Ireland or are otherwise subject to Irish tax should consult their own tax advisor.
Other Persons
Shareholders who do not reside in “relevant territories” or in Ireland will be subject to DWT, but there are a number of other exemptions that could apply on a case-by-case basis. Dividends paid to such shareholders will be paid subject to DWT unless the relevant shareholder has provided the appropriate Irish DWT form to his or her broker (so that such broker can further transmit the relevant information to our qualifying intermediary) prior to the record date for the first dividend to which they are entitled (in the case of ordinary shares held through DTC), or to our transfer agent at least seven business days before such record date (in the case of shares held outside DTC).
We strongly recommend that such shareholders to whom an exemption applies complete the appropriate Irish forms and provide them to their brokers or our transfer agent, as the case may be, as soon as possible.
If any shareholder who is not a resident of a “relevant territory” or Ireland but is exempt from withholding receives a dividend subject to DWT, he or she may be able to make an application for a refund from the Irish Revenue Commissioners on the prescribed form, subject to certain time limits.
Income Tax on Dividends
Non-Irish Resident Shareholders
A shareholder who is not resident or ordinarily resident for tax purposes in Ireland and who is entitled to an exemption from DWT, generally has no liability to Irish income tax or the universal social charge on a dividend from an Irish resident company unless that shareholder holds their ordinary shares through a branch or agency which carries on a trade in Ireland.
A shareholder who is not resident or ordinarily resident for tax purposes in Ireland and who is not entitled to an exemption from DWT, generally has no additional liability to Irish income tax or the universal social charge unless that shareholder holds their ordinary shares through a branch or agency which carries on a trade in Ireland. The shareholder’s liability to Irish income tax and the universal social charge is effectively limited to the amount of DWT already deducted by the company.
Irish Resident Shareholders
Irish resident or ordinarily resident individual shareholders may be subject to Irish income tax and the universal social charge charges and in certain circumstances pay related social insurance (PRSI) on dividends received from us. Such shareholders should consult their own tax advisor. Irish resident corporate shareholders should not be subject to tax on dividends from the company on the basis that the dividend is not in respect of preferred shares.
Capital Acquisitions Tax
Irish capital acquisitions tax (“CAT”) comprises principally of gift tax and inheritance tax. CAT could apply to a gift or inheritance of our ordinary shares irrespective of the place of residence, ordinary residence or domicile of the deceased or donor of the shares (collectively referred to as the “donor”) or the successor or donee of the shares (collectively referred to as the “donee”). This is because our ordinary shares are regarded as property situated in Ireland as the share register of the company must be held in Ireland. The person who receives the gift or inheritance has primary liability for CAT. However there are certain circumstances where another person such as an agent or personal representative may become accountable for the CAT.
CAT is currently levied at a rate of 33% above certain tax-free thresholds. The appropriate tax-free threshold is dependent upon (1) the relationship between the donor and the donee and (2) the aggregation of the values of previous gifts and inheritances received by the donee from persons within the same group threshold. Gifts and inheritances passing between spouses are exempt from CAT. Shareholders should consult their own tax advisor as to whether CAT is creditable or deductible in computing any domestic tax liabilities.
Stamp Duty
The rate of stamp duty (where applicable) on transfers of shares of Irish incorporated companies is 1% of the price paid or the market value of the shares acquired, whichever is greater. Where Irish stamp duty arises it is generally a liability of the transferee.
Irish stamp duty may, depending on the manner in which the our ordinary shares are held, be payable in respect of transfers of our ordinary shares.
We have entered into arrangements with DTC to allow our ordinary shares to be settled through the facilities of DTC. As such, the summary below deals with shareholders who hold their shares through DTC and shareholders who hold shares outside of DTC.
Shares Held Through DTC
A transfer of our ordinary shares effected by means of the transfer of book-entry interests in DTC will not be subject to Irish stamp duty.
Shares Held Outside of DTC or Transferred Into or Out of DTC
A transfer of our ordinary shares where any party to the transfer holds such shares outside of DTC may be subject to Irish stamp duty.
Shareholders wishing to transfer their shares into (or out of) DTC may do so without giving rise to Irish stamp duty, provided:
| · | there is no change in the ultimate beneficial ownership of such shares as a result of the transfer; and |
| · | the transfer into (or out of) DTC is not on a sale or in contemplation of a sale. |
Because of the potential Irish stamp duty on transfers of our ordinary shares, we strongly recommend that any person who wishes to acquire our ordinary shares acquires such shares through DTC.
We will be required to assume the obligation for paying the stamp duty liability with respect to certain transfers of our ordinary shares as a result of the arrangements we enter into with DTC. Our constitution provides that, if stamp duty resulting from the transfer of our ordinary shares, which would otherwise be payable by the transferee, is paid by us or any of our subsidiaries on behalf of or as agent for the transferee, then in those circumstances, we shall on our behalf or on behalf of any such subsidiary, be entitled to (i) seek reimbursement of the stamp duty from the transferor or transferee (at our or our relevant subsidiary’s discretion), (ii) set-off the stamp duty against any dividends payable to the transferor or transferee (at our or our relevant subsidiary’s discretion) and (iii) to claim a first and permanent lien on the shares on which stamp duty has been paid by us or any of our subsidiaries for the amount of stamp duty paid. Our lien shall extend to all dividends paid on those shares.
U.S. Federal Income Tax Considerations
Scope of Discussion
The following discussion summarizes (i) the material U.S. federal income tax consequences to Innocoll Germany, Innocoll Ireland, and our shareholders of the Merger and (ii) the material U.S. federal income tax consequences to our shareholders of owning and disposing of Innocoll Ireland ordinary shares. This discussion does not generally address any aspects of U.S. taxation other than U.S. federal income taxation, is not a complete analysis or listing of all of the possible tax consequences of the consummation of the Merger or of holding and disposing of Innocoll Ireland ordinary shares and does not address all tax considerations that may be relevant to you. Special rules that are not discussed in the general descriptions below may also apply to you. In particular, this discussion deals only with holders that hold their Innocoll Germany ADSs or ordinary shares and will hold their Innocoll Ireland ordinary shares as capital assets and, except as otherwise indicated below, does not address the tax treatment of special classes of holders, such as:
| · | a holder of Innocoll Germany ADSs or ordinary shares who, at any time within the five year period ending on the date of the Merger, has actually and constructively owned 10% or more of the total combined voting power of all classes of stock entitled to vote of Innocoll Germany, after taking into account any voting restrictions on treasury shares or otherwise imposed under German law; |
| · | a holder of Innocoll Germany ADSs or ordinary shares who, immediately before the Merger, actually and constructively owns at least 5% of either the total combined voting power or the total value of all classes of stock of Innocoll Germany; |
| · | a bank or other financial institution; |
| · | a person holding shares as part of a “straddle,” “hedge,” “wash sale,” “integrated transaction,” or “conversion transaction”; |
| · | a person holding shares through a partnership or other pass-through entity (including arrangements treated as partnerships under U.S. federal income tax law); |
| · | a former citizen or resident of the United States under Section 877 or Section 877A of the Code; |
| · | a non-resident alien who has elected to be treated as a resident of the United States or is a bona fide resident of Puerto Rico, Guam, American Samoa or the Northern Mariana Islands; |
| · | a person who is liable for alternative minimum tax; |
| · | a broker-dealer or trader in securities or currencies; |
| · | a U.S. holder whose “functional currency” is not the U.S. dollar; |
| · | a regulated investment company or real estate investment trust; |
| · | a trader in securities who has elected the mark-to-market method of accounting for its securities; |
| · | a holder that has filed a gain recognition agreement with respect to Innocoll Germany ADSs or ordinary shares; |
| · | a holder who received Innocoll Germany ordinary shares through the exercise of employee stock options or otherwise as compensation or through a tax qualified retirement plan; or |
| · | a non-corporate holder of Innocoll Ireland ordinary shares who, because of limitations under the U.S. securities laws or other legal limitations, is not free to dispose of those shares without restriction. |
This discussion is based on the laws of the United States, including the U.S. Internal Revenue Code of 1986, as amended (“Code”), its legislative history, existing and proposed Treasury regulations promulgated thereunder (“Regulations”), judicial decisions, published rulings, administrative pronouncements and income tax treaties to which the United States is a party, each as in effect on the date hereof and all of which are subject to change or differing interpretations, possibly with retroactive effect. There can be no assurance that the U.S. Internal Revenue Service (“IRS”) will not disagree with or will not successfully challenge any of the conclusions reached and described in this discussion.
For purposes of this discussion, a “U.S. holder” means (a) a beneficial owner of Innocoll Germany ADSs or ordinary shares or (b), after the completion of the Merger, a beneficial owner of Innocoll Ireland ordinary shares that for U.S. federal income tax purposes is:
| · | an individual citizen or resident alien of the United States; |
| · | a corporation or other entity (or arrangement) taxable as a corporation organized under the laws of the United States, any state thereof or the District of Columbia; |
| · | an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or |
| · | a trust, if it (i) is subject to the primary supervision of a U.S. court and the control of one or more U.S. persons or (ii) has a valid election in effect under applicable Regulations to be treated as a U.S. person. |
A “non-U.S. holder” means a holder (other than an entity or arrangement treated as a partnership for U.S. federal income tax purposes) of Innocoll Germany ADSs or ordinary shares or, after the completion of the Merger, Innocoll Ireland ordinary shares that is not a U.S. holder. For purposes of this summary, “holder” or “shareholder” means either a U.S. holder or a non-U.S. holder or both, as the context may require.
If a partnership (including any entity or arrangement treated as a partnership for U.S. federal income tax purposes) is a beneficial owner of Innocoll Germany ADSs or ordinary shares or Innocoll Ireland ordinary shares, the tax treatment of a partner in that partnership will generally depend on the status of the partner and the activities of the partnership. Holders of Innocoll Germany ADSs or ordinary shares or Innocoll Ireland ordinary shares that are partnerships and partners in these partnerships are urged to consult their tax advisors regarding the U.S. federal income tax consequences to them of the Merger and the ownership and disposition of the Innocoll Ireland ordinary shares.
Innocoll intends to take the position that, after the Merger, Innocoll Ireland will be a foreign corporation for U.S. tax purposes (a “non-U.S. corporation”). Except as provided below in “Certain Tax Consequences if Innocoll Ireland and/or Innocoll Germany are Treated as a U.S. Corporation for U.S. Federal Income Tax Purposes,” this summary assumes that Innocoll Germany, and after the completion of the Merger, Innocoll Ireland, is a non-U.S. corporation.
Consequences of the Merger
For U.S. federal income tax purposes, (i) the Merger will qualify as a “reorganization” under Section 368(a) of the Code and (ii) no gain or loss will be recognized by us, Innocoll Ireland, or our shareholders as a result of the Merger, except to the extent relating to the exercise of withdrawal rights or with respect to cash received in lieu of fractional ordinary shares of Innocoll Ireland or as provided in the following sentence. In the event that (i) the Merger does not qualify as an “F” reorganization, (ii) Innocoll Germany is or was a PFIC (as defined below) during a U.S. holder’s holding period of the ordinary shares or ADSs of Innocoll Germany, and (iii) Innocoll Ireland is not a PFIC (as defined below) on the effective date of the Merger, all or a portion of a U.S. holder’s gain may be subject to tax as ordinary income notwithstanding the Merger qualifying as a nontaxable reorganization. See discussion under “— PFIC Rules.” For those U.S. holders that do not recognize gain as a result of the Merger, the basis of a shareholder’s Innocoll Ireland ordinary shares received in exchange for such shareholder’s Innocoll Germany ADSs or ordinary shares will be equal to the basis of the Innocoll Germany ADSs or ordinary shares exchanged, reduced by an amount allocable to a fractional ordinary share of Innocoll Ireland. The holding period of the Innocoll Ireland ordinary shares will include the period such shareholders held the Innocoll Germany ADSs or ordinary shares exchanged. Shareholders who hold their Innocoll Germany ADSs or ordinary shares with differing bases or holding periods should consult their tax advisors as to the determination of the bases and holding periods of the Innocoll Ireland ordinary shares received in the Merger.
Taxation of Holders Who Exercised Withdrawal Rights or Who Received Cash in Lieu of Fractional Innocoll Ireland ordinary shares.
The discussion above in “— Consequences of the Merger” does not apply to shareholders of Innocoll Germany ADSs or ordinary shares who properly exercised withdrawal rights with respect to the such ADSs or shares, or who received cash in lieu of fractional ordinary shares of Innocoll Ireland. Generally, a shareholder who exercises withdrawal rights and receives cash compensation and has its Innocoll Ireland ordinary shares converted into Innocoll Ireland deferred shares will be treated as redeeming such Innocoll Ireland ordinary shares. Principles similar to the principles described in “Subsequent Dispositions of Innocoll Ireland Ordinary Shares” in the “U.S. holder” section and “Subsequent Dispositions of Innocoll Ireland Ordinary Shares” in the “non-U.S. holder” section shall apply to U.S. holders and non-U.S. holders, respectively. Shareholders who received cash in lieu of fractional ordinary shares of Innocoll Ireland are deemed to receive such shares and then Innocoll Ireland is treated as redeeming such shares for the cash received in exchange therefor. Principles similar to the principles described in “Subsequent Dispositions of Innocoll Ireland Ordinary Shares” in the “U.S. holder” section and “Subsequent Dispositions of Innocoll Ireland Ordinary Shares” in the “non-U.S. holder” section shall apply to U.S. holders and non-U.S. holders, respectively.
Certain Consequences of Owning and Disposing of Innocoll Ireland Ordinary Shares.
U.S. HOLDERS
Taxation of Distributions on Innocoll Ireland Ordinary Shares.
Under the U.S. federal income tax laws and subject to the passive foreign investment company (“PFIC”) rules discussed below in “— PFIC Rules”, the gross amount of any distribution that is actually or constructively received (as described below, including any Irish withholding tax or other withholding tax on such amount) by a U.S. holder with respect to Innocoll Ireland ordinary shares, will be a dividend for U.S. federal income tax purposes to the extent of Innocoll Ireland’s current or accumulated earnings and profits (as determined for U.S. federal income tax purposes). To the extent that the amount of such distribution exceeds Innocoll Ireland’s current and accumulated earnings and profits as so computed, it will be treated first as a non-taxable return of capital to the extent of such U.S. holder’s adjusted tax basis in its Innocoll Ireland ordinary shares, and to the extent the amount of such distribution exceeds such adjusted tax basis, will be treated as gain from the sale of Innocoll Ireland ordinary shares. Because we do not compute our earnings and profits under U.S. tax principles, however, we expect to report the full amount of any distribution as a dividend for U.S. tax purposes if and when we pay a dividend to our shareholders. With respect to non-corporate U.S. holders, certain dividends received from a qualified foreign corporation will be subject to U.S. federal income tax at a maximum rate of 20%. As long as Innocoll Ireland ordinary shares are listed on NASDAQ or certain other exchanges and/or Innocoll Ireland qualifies for benefits under the income tax treaty between the United States and Ireland or another applicable income tax treaty, Innocoll Ireland will be treated as a qualified foreign corporation for this purpose. This reduced rate will not be available in all situations, and U.S. holders should consult their own tax advisors regarding the application of the relevant rules to their particular circumstances. If Innocoll Ireland is a PFIC as discussed below under “— PFIC Rules,” distributions paid by Innocoll Ireland with respect to its ordinary shares will not be eligible for the preferential income tax rate.
You must include any Irish tax withheld from the dividend payment in this gross amount even though you do not in fact receive it. The gross amount of the dividend is taxable to you when you receive the dividend, actually or constructively. Dividends paid on Innocoll Ireland ordinary shares generally will constitute income from sources outside the United States and will generally not be eligible for the dividends-received deduction generally available to corporate U.S. holders. Because we expect to pay any future dividends in a non-U.S. currency, the gross amount of any such dividend will be included in the gross income of a U.S. holder in an amount equal to the U.S. dollar value of the non-U.S. currency calculated by reference to the exchange rate in effect on the date the dividend distribution is includable in the U.S. holder’s income, regardless of whether the payment is in fact converted into U.S. dollars. If the non-U.S. currency is converted into U.S. dollars on the date of receipt by the U.S. holder, a U.S. holder generally should not be required to recognize non-U.S. currency gain or loss in respect of the dividend. If the non-U.S. currency received is not converted into U.S. dollars on the date of receipt, a U.S. holder will have a basis in the non-U.S. currency equal to its U.S. dollar value on the date of receipt. Any gain or loss on a subsequent conversion or other disposition of the non-U.S. currency will be treated as ordinary income or loss, and will generally be income or loss from sources within the United States for foreign tax credit limitation purposes. The amount of any distribution of property other than cash will be the fair market value of the property on the date of the distribution, less the sum of any encumbrance assumed by the U.S. holder.
Subject to applicable limitations that may vary depending upon a U.S. holder’s circumstances, a U.S. holder will be entitled to a credit against its U.S. federal income tax liability for any Irish withholding taxes withheld in respect of our dividend distributions not in excess of the applicable rate under the income tax treaty between the United States and Ireland. For purposes of calculating the foreign tax credit, dividends paid on Innocoll Ireland ordinary shares generally will be treated as income from sources outside the United States, except that a portion of such dividends may be treated as income from U.S. sources. In addition, the limitation on foreign taxes eligible for credit is calculated separately with respect to specific classes of income, such as “passive” or “general” income. In addition, the amount of the qualified dividend income, if any, paid to a U.S. holder that is subject to the reduced dividend income tax rate and that is taken into account for purposes of calculating the U.S. holder’s U.S. foreign tax credit limitation must be reduced by the rate differential portion of the dividend. The rules governing foreign tax credits are complex. Investors should consult their own tax advisors regarding the availability of foreign tax credits in their particular situation. In lieu of claiming a foreign tax credit, U.S. holders may elect to deduct all non-U.S. taxes paid or accrued in a taxable year in computing their taxable income, subject to generally applicable limitations under U.S. federal income tax law.
Subsequent Dispositions of Innocoll Ireland Ordinary Shares.
Subject to the discussion below under “— PFIC Rules,” U.S. holders of Innocoll Ireland ordinary shares generally should recognize capital gain or loss for U.S. federal income tax purposes on the sale, exchange or other disposition of Innocoll Ireland ordinary shares in the same manner as on the sale, exchange or other disposition of any other shares held as capital assets. Such capital gain or loss will be long-term capital gain or loss if the U.S. holder’s holding period for the Innocoll Ireland ordinary shares exceeds one year. Under current law, long-term capital gain of non-corporate shareholders is subject to tax at a maximum rate of 20%. The gain or loss will generally be income or loss from sources within the United States for foreign tax credit limitation purposes. There are limitations on the deductibility of capital losses.
A U.S. holder that receives non-U.S. currency on the sale or other disposition of Innocoll Ireland ordinary shares will realize an amount equal to the U.S. dollar value of the non-U.S. currency on the date of sale (or, in the case of cash basis and electing accrual basis taxpayers, the U.S. dollar value of the non-U.S. currency on the settlement date) provided that the Innocoll Ireland ordinary shares are treated as being “traded on an established securities market.” If a U.S. holder receives non-U.S. currency upon a sale or exchange of Innocoll Ireland ordinary shares, gain or loss, if any, recognized on the subsequent sale, conversion or disposition of such non-U.S. currency will be ordinary income or loss, and will generally be income or loss from sources within the United States for foreign tax credit limitation purposes. However, if such non-U.S. currency is converted into U.S. dollars on the date received by the U.S. holder, a cash basis or electing accrual U.S. holder should not recognize any gain or loss on such conversion. In addition, depending on the circumstances, a redemption of Innocoll Ireland ordinary shares by Innocoll Ireland will be treated as a sale of the redeemed shares by the U.S. holder or as a distribution to the U.S. holder (which is taxable as described above under “— Taxation of Distributions on Innocoll Ireland Ordinary Shares”).
Controlled Foreign Corporation Rules.
Generally, a non-U.S. corporation, such as Innocoll Ireland, will be classified as a controlled foreign corporation (“CFC”) if more than 50% (by vote or value) of the shares of the corporation are held directly, indirectly, or constructively, by “U.S. Shareholders.” For this purpose, a U.S. Shareholder is generally any U.S. holder that possesses, directly, indirectly or constructively, 10% or more of the combined voting power of all classes of shares of the corporation. Based on our current and anticipated ownership structure, we do not expect that Innocoll Ireland will be classified as a CFC. However, we can offer no assurances in this regard.
If Innocoll Ireland were classified as a CFC, however, each of Innocoll Ireland’s U.S. Shareholders generally would be required to include in gross income (as ordinary income) at the end of each of Innocoll Ireland’s taxable years an amount equal to the U.S. Shareholder’s pro rata share of our “subpart F income.” Subpart F income generally includes dividends, interest, rents and royalties, gains from the sale of securities, and income from certain transactions with related parties. If Innocoll Ireland is classified as both a CFC and a PFIC, it generally will not be treated as a PFIC with respect to those U.S. holders that meet the definition of a U.S. Shareholder.
PFIC Rules.
The treatment of U.S. holders of Innocoll Ireland ordinary shares could be materially different from that described above if, at any relevant time, Innocoll Germany or Innocoll Ireland were a PFIC. In general, if you are a U.S. holder, Innocoll Germany and/or Innocoll Ireland will be a PFIC with respect to you if for any taxable year in which you held ADSs or ordinary shares of Innocoll Germany or ordinary shares of Innocoll Ireland: (i) at least 75% of the gross income of Innocoll Germany or Innocoll Ireland, as applicable, for the taxable year is passive income or (ii) at least 50% of the value, determined on the basis of a quarterly average, of the assets of Innocoll Germany or Innocoll Ireland, as applicable, is attributable to assets that produce or are held for the production of passive income. The determination of whether a non-U.S. corporation is a PFIC is made annually. Accordingly, it is possible that we may become a PFIC in the current or, in the case of Innocoll Ireland, any future taxable year due to changes in Innocoll Ireland’s asset or income composition.
Passive income generally includes dividends, interest, rents and royalties (other than certain rents and royalties derived in the active conduct of a trade or business), annuities and gains from the disposition of assets that produce passive income. Any cash held generally will be treated as held for the production of passive income for the purpose of the PFIC test, and any income generated from cash or other liquid assets generally will be treated as passive income for such purpose. If a non-U.S. corporation owns at least 25% by value of the shares of another corporation, the non-U.S. corporation is treated for purposes of the PFIC tests as owning its proportionate share of the assets of the other corporation, and as receiving directly its proportionate share of the other corporation’s income. We believe that Innocoll Germany should not be treated as having been a PFIC in any prior taxable year and should not be treated as a PFIC in the taxable year in which the Merger occurred. In addition, we believe that Innocoll Ireland should not be treated as a PFIC immediately after the Merger. However, the tests for determining PFIC status are applied annually, and it is difficult to accurately predict future income and assets relevant to this determination. Accordingly, we cannot assure U.S. holders that Innocoll Germany and Innocoll Ireland will not be or become a PFIC. Moreover, the determination of PFIC status depends, in part, on the application of complex U.S. federal income tax rules, which are subject to differing interpretations. As a result, whether Innocoll Germany or Innocoll Ireland is or will be a PFIC for any relevant taxable year cannot be predicted with certainty, and there can be no assurance that the IRS will not challenge our determination concerning our PFIC status.
If Innocoll Germany prior to the consummation of the Merger, and Innocoll Ireland, after the consummation of the Merger, were to be treated as a PFIC, except as otherwise provided by election regimes described below, a U.S. holder would be subject to special adverse tax rules with respect to (i) “excess distributions” received on our ADSs or ordinary shares of Innocoll Germany, and ordinary shares of Innocoll Ireland, as the case may be, and (ii) any gain recognized upon a sale or other disposition (including a pledge) of ADSs or ordinary shares of Innocoll Germany or ordinary shares of Innocoll Ireland, as the case may be. A U.S. holder would be treated as if it had realized such gain and certain “excess distributions” ratably over its holding period for such shares. The amounts allocated to the current taxable year and to any taxable year in the holding period prior to the first taxable year in which we were a PFIC would be taxed as ordinary income. The amounts allocated to any other taxable year would be taxed at the highest tax rate in effect for each such year to which the gain was allocated, together with an interest charge in respect of the tax attributable to each such year. Special rules apply for calculating the amount of the foreign tax credit with respect to “excess distributions” by a PFIC.
Dividends that a U.S. holder receives from Innocoll Ireland will not be eligible for the special tax rates applicable to qualified dividend income if Innocoll Germany and/or Innocoll Ireland are treated as a PFIC either in the taxable year of the distribution or the preceding taxable year, but instead will be taxable at rates applicable to ordinary income, or if an excess distribution treated as discussed above.
If a U.S. holder owns ordinary shares in a PFIC that are treated as “marketable stock,” the U.S. holder may make a mark-to-market election. If a U.S. holder makes this election, the U.S. holder will not be subject to all of the PFIC rules described above. Instead, in general, the U.S. holder will include as ordinary income the excess, if any, of the fair market value of its Innocoll Ireland ordinary shares at the end of the taxable year over the U.S. holder’s adjusted basis in its Innocoll Ireland ordinary shares. Similarly, any gain realized on the sale, exchange or other disposition of the Innocoll Ireland ordinary shares will be treated as ordinary income, and will not be eligible for the favorable tax rates applicable to qualified dividend income or long-term capital gains. The U.S. holder will also be allowed to take an ordinary loss in respect of the excess, if any, of the adjusted basis of its Innocoll Ireland ordinary shares over the fair market value at the end of the taxable year (but only to the extent of the net amount of previously included income as a result of the mark-to-market election). A U.S. holder’s basis in the Innocoll Ireland ordinary shares will be adjusted to reflect any such income or loss amount.
A U.S. holder may in certain circumstances also mitigate adverse tax consequences of the PFIC rules by filing an election to treat the PFIC as a qualified electing fund (“QEF”), if the PFIC complies with certain reporting requirements. However, in the event that we are or become a PFIC, Innocoll Ireland does not intend to comply with such reporting requirements necessary to permit U.S. holders to elect to treat us as a QEF.
U.S. holders should consult their own tax advisors regarding the application of the CFC rules and PFIC rules to their investment and the elections discussed above.
Tax on Net Investment Income.
An additional 3.8% tax generally will be imposed on the “net investment income” of individuals (other than nonresident aliens) with a modified adjusted gross income over $200,000 ($250,000 in the case of joint filers) and on the undistributed net investment income of certain estates and trusts. For this purpose, “net investment income” generally includes interest, dividends (including dividends paid with respect to Innocoll Ireland ordinary shares), annuities, royalties, rents, net gain attributable to the disposition of property not held in a trade or business (including net gain from the taxable disposition of the Innocoll Ireland ordinary shares) and certain other income, as reduced by any deductions properly allocable to such income or gain. If you are a U.S. holder that is an individual, estate or trust, you are urged to consult your tax advisors regarding the applicability of the tax on net investment income to your income and gains in respect of your investment in Innocoll Ireland ordinary shares.
Information Reporting and Backup Withholding on Distributions and Disposition Proceeds with Respect to Innocoll Ireland Ordinary Shares.
Dividends on Innocoll Ireland ordinary shares paid within the United States or through certain U.S.-related financial intermediaries are subject to information reporting and may be subject to backup withholding (currently at a 28% rate) unless the holder (1) is a corporation or other exempt recipient or (2) provides a taxpayer identification number and satisfies certain certification requirements. Information reporting requirements and backup withholding may also apply to the cash proceeds of a sale of Innocoll Ireland ordinary shares. If the sale is made through a non-U.S. office of a non-U.S. broker, however, the sale will generally not be subject to either backup withholding or information reporting. This exception may not apply if the non-U.S. broker is owned or controlled by U.S. persons, or is engaged in a U.S. trade or business.
In addition to being subject to backup withholding, if a U.S. holder of Innocoll Ireland ordinary shares does not provide us (or our paying agent) with the holder’s correct taxpayer identification number or other required information, the holder may be subject to penalties imposed by the IRS. Any amounts withheld under the backup withholding rules may be allowed as a refund or a credit against the holder’s U.S. federal income tax liability, provided that the holder furnishes certain required information to the IRS.
Certain specified individuals and, to the extent provided by future guidance, certain U.S. entities, who, at any time during the taxable year, hold interests in specified foreign financial assets that are not held in an account maintained by a financial institution and that have an aggregate value in excess of applicable reporting thresholds (which depend on the individual’s filing status and tax home, and begin at a low of more than $50,000 on the last day of the taxable year or more than $75,000 at any time during the taxable year) are required to attach a disclosure statement on Form 8938 (Statement of Specified Foreign Financial Assets) to their U.S. federal income tax return. A specified person who reports its ordinary shares on a Form 8621 does not have to report the ordinary shares on the Form 8938 if the person identifies the Form 8621 which includes the ordinary shares on the Form 8938. No Form 8938 is required to be filed by a specified person who is not required to file a U.S. federal income tax return for the taxable year. Investors are urged to consult their own tax adviser regarding these reporting requirements.
NON-U.S. HOLDERS
Taxation of Distributions on Innocoll Ireland Ordinary Shares.
A non-U.S. holder generally will not be subject to U.S. federal income or withholding tax on dividends received on its Innocoll Ireland ordinary shares, unless the dividends are effectively connected with the holder’s conduct of a trade or business in the United States and, if an income tax treaty applies, the dividends are attributable to a permanent establishment or fixed place of business maintained by the holder in the United States or such holder is subject to backup withholding as discussed below.
Except to the extent otherwise provided under an applicable income tax treaty, a non-U.S. holder generally will be taxed in the same manner as a U.S. holder on dividends paid and gains recognized that are effectively connected with the holder’s conduct of a trade or business in the United States. Effectively connected dividends received and gains recognized by a corporate non-U.S. holder may also, under certain circumstances, be subject to an additional “branch profits tax” at a 30% rate (or, if applicable, a lower treaty rate), subject to certain adjustments.
Subsequent Disposition of Innocoll Ireland Ordinary Shares.
In general, a non-U.S. holder of Innocoll Ireland ordinary shares will not be subject to U.S. federal income or withholding tax on any gain recognized on a subsequent disposition of the Innocoll Ireland ordinary shares, unless: (1) such gain is effectively connected with the conduct by the holder of a trade or business within the United States and, if a tax treaty applies, is attributable to a permanent establishment or fixed place of business maintained by such holder in the United States, (2) in the case of capital gain of a holder who is an individual, such holder is present in the United States for 183 days or more during the taxable year in which the capital gain is recognized and certain other conditions are met, or (3) such holder is subject to backup withholding as discussed below. In addition, depending on the particular circumstances, a redemption of Innocoll Ireland ordinary shares by Innocoll Ireland will be treated as a sale of the redeemed shares by the non-U.S. holder or as a distribution to the non-U.S. holder (which is taxable as described in the preceding paragraph under “— Taxation of Distributions on Innocoll Ireland ordinary shares.”
Information Reporting and Backup Withholding on Distributions and Disposition Proceeds with Respect to Innocoll Ireland Ordinary Shares.
In order not to be subject to backup withholding tax on distributions and disposition proceeds with respect to Innocoll Ireland ordinary shares, a non-U.S. holder may be required to provide a taxpayer identification number, certify the holder’s foreign status, or otherwise establish an exemption. Non-U.S. holders of Innocoll Ireland ordinary shares should consult their tax advisors regarding the application of information reporting and backup withholding in their particular situations, the availability of exemptions, and the procedure for obtaining such an exemption, if available. Any amount withheld from a payment to a non-U.S. holder under the backup withholding rules may be allowed as a refund or credit against the holder’s U.S. federal income tax, provided that the required information is furnished to the IRS.
Certain Tax Consequences if Innocoll Ireland and/or Innocoll Germany are Treated as a U.S. Corporation for U.S. Federal Income Tax Purposes
For U.S. federal income tax purposes, a corporation generally is considered a domestic corporation for U.S. federal tax purposes (a “U.S. corporation”) if it is created or organized in the United States or under the law of the United States or of any state thereof or the District of Columbia. Entities treated as U.S. corporations are generally subject to U.S. federal income tax on their worldwide income, and U.S. reporting and withholding tax rules may apply to dividends that they pay. Because Innocoll Germany was formed and organized under the law of Germany, Innocoll Germany would ordinarily not be treated for U.S. federal income tax purposes as a U.S. corporation. Section 7874 of the Code, however, contains special rules that could result in a non-U.S. corporation being taxed as a U.S. corporation for U.S. federal income tax purposes where the corporation, directly or indirectly, re-domiciles from the U.S. to another country. Under Section 7874 of the Code, as a result of the re-domiciling from the U.S. to Germany in 2013 (the “2013 Transaction”), we would be treated as a U.S. corporation for U.S. federal income tax purposes unless our “expanded affiliated group” (“EAG”) is treated as having “substantial business activities” in Germany. While we believe that we satisfied this “substantial business activities” test at the time of the 2013 Transaction (and, thus, should not be treated as a U.S. corporation for U.S. federal income tax purposes), due to the complexity of certain aspects of the law and the very fact-specific nature of the inquiry, there is no assurance that the IRS will not challenge our determination. Further, the IRS has issued recent guidance in Notice 2015-79 that provides that an EAG cannot meet the substantial business activities test with respect to a foreign country unless the foreign acquiring corporation is a tax resident of that foreign country. This new requirement for the substantial business activities test is effective for acquisitions completed on or after November 19, 2015. However, Notice 2015-79 also states that no inference is intended regarding the treatment under current law of any transaction described in Notice 2015-79 and that the IRS may challenge such transactions under applicable Code sections or judicial doctrines. In addition, there have been proposals to expand the scope of U.S. corporate tax residence and there could be prospective or retroactive legislative changes to Section 7874 of the Code that would result in Innocoll Germany and/or Innocoll Ireland being treated as a U.S. corporation.
In addition to the issues discussed above regarding the 2013 Transaction, we have re-domiciled in 2016 to Ireland. If the Merger is treated as a related transaction to the 2013 Transaction, then Innocoll Germany and/or Innocoll Ireland would be treated as a U.S. corporation as of the 2013 Transaction unless our EAG is treated as having substantial business activities in Ireland. In that case, we would not satisfy the substantial business activities test in Ireland, and thus, we would be treated as a U.S. corporation, even if we would have met the substantial business activities test had we remained a German corporation. Due to the complexity of certain aspects of the law and the very fact-specific nature of the inquiry, there is no assurance that the IRS will not successfully challenge our position that we are a non-U.S. corporation. If Innocoll Germany and/or Innocoll Ireland are treated as U.S. corporations, Innocoll Germany and Innocoll Ireland would be liable for U.S. federal income taxes in addition to German and Irish taxes. In addition, our non-U.S. holders would be subject to U.S. withholding tax (including under the Foreign Account Tax Compliance Act, as defined below) on the receipt of dividends from Innocoll Ireland.
Certain U.S. Federal Income Tax Consequences to Innocoll Germany and Innocoll Ireland
If we are treated as a U.S. corporation, Innocoll Germany and Innocoll Ireland would be treated as a U.S. Shareholder of the non-U.S. subsidiaries owned by Innocoll Germany prior to the Merger, and Innocoll Ireland, after the Merger, and such subsidiaries would be “controlled foreign corporations”. See the discussion above in “— Controlled Foreign Corporation Rules” that could apply to Innocoll Germany or Innocoll Ireland. Regardless of whether Innocoll Germany or Innocoll Ireland is treated as a U.S. corporation, it is expected that it will continue to be treated as an Irish tax resident for Irish and German tax purposes, and therefore it could be liable for both Ireland and U.S. taxes, which could have a material adverse effect on its financial condition and results of operations. In addition, any loans made to Innocoll Germany or Innocoll Ireland by a non-U.S. subsidiary corporation of Innocoll Germany or Innocoll Ireland, could result in adverse U.S. federal income tax consequences. Further, any determination by the IRS or a U.S. court that we are a U.S. corporation would apply retroactively, and so we could face substantial U.S. federal income tax, interest, and penalties for taxable years prior to such determination.
Certain U.S. Federal Income Tax Consequences to U.S. Holders
If Innocoll Ireland is treated as a U.S. corporation, then U.S. holders, among other tax consequences, would not be eligible to obtain a U.S. foreign tax credit with respect to any Irish tax withheld from any dividend distributions. This is because dividend distributions from U.S. corporations are treated as U.S. source income for U.S. federal income tax purposes, and therefore such Irish withholding taxes will likely not be creditable for U.S. foreign tax credit purposes. Thus, to the extent a U.S. holder receives dividend distributions, such holder could be liable for both Irish and U.S. taxes on the receipt of such dividends without the ability to take a foreign tax credit for the Irish taxes. The interaction of Section 7874 of the Code to certain other U.S. federal income tax provisions is not yet well defined under U.S. federal income tax law. This summary does not discuss all of the U.S. federal income tax considerations that could affect a U.S. holder if Innocoll Ireland is treated as a U.S. corporation for U.S. federal income tax purposes. Each U.S. holder is urged to contact its own tax advisor regarding U.S. federal income tax consequences if Innocoll Ireland is treated as a U.S. corporation.
Certain U.S. Federal Income Tax Consequences to Non-U.S. Holders
If Innocoll Ireland is treated as a U.S. corporation for U.S. federal income tax purposes, the following additional U.S. federal income tax consequences could arise.
Taxation of Distributions on Innocoll Ireland Ordinary Shares.
If distributions are made with respect to Innocoll Ireland ordinary shares, such distributions will be treated as dividends to the extent of Innocoll Ireland’s current and accumulated earnings and profits as determined for U.S. federal income tax purposes and will be subject to withholding as described below. Any portion of a distribution that exceeds Innocoll Ireland’s current and accumulated earnings and profits will be applied first to reduce the non-U.S. holder’s tax basis in Innocoll Ireland’s ordinary shares, and, to the extent such portion exceeds the non-U.S. holder’s basis, the excess will be treated as gain from the disposition of the Innocoll Ireland’s ordinary shares, the tax treatment of which is discussed below under “Subsequent Disposition of Innocoll Ireland ordinary shares.” In addition, special adverse rules may apply if Innocoll Ireland is a U.S. real property holding corporation within the meaning of Section 897(c) of the Code (“USRPHC”). We would not, however, expect to be a USRPHC.
Dividends paid to a non-U.S. holder of Innocoll Ireland’s ordinary shares will generally be subject to withholding of U.S. federal income tax at a 30% rate or such lower rate as may be specified by an applicable income tax treaty. Special rules could apply if the dividends would be effectively connected with the conduct of a trade or business by the non-U.S. holder within the United States. The interaction of Section 7874 of the Code to certain other U.S. federal income tax provisions is not yet well defined under U.S. federal income tax law. This summary does not discuss all of the U.S. federal income tax considerations that could affect a non-U.S. holder if Innocoll Ireland is treated as a U.S. corporation for U.S. federal tax purposes. Each non-U.S. holder is urged to contact its own tax advisor regarding U.S. federal income tax consequences if Innocoll Ireland is treated as a U.S. corporation.
Subsequent Disposition of Innocoll Ireland Ordinary Shares.
If a non-U.S. holder were to dispose of ordinary shares of Innocoll Ireland, the same U.S. federal income tax considerations that are applicable to non-U.S. holders as described above in “— Subsequent Disposition of Innocoll Ireland Ordinary Shares” shall apply. However, if Innocoll Ireland were treated as a USRPHC within the meaning of Section 897(c) of the Code, in certain cases, U.S. federal income tax could apply. Nonetheless, even if Innocoll Ireland were treated as a U.S. corporation, we do not anticipate that it would be treated as a USRPHC.
Foreign Account Tax Compliance Act.
Under the Foreign Account Tax Compliance Act (“FATCA”), a person who makes a withholdable payment (as defined in Section 1473 of the Code) to a foreign financial institution (“FFI”) or a non-financial foreign entity (“NFFE”) must withhold at a 30% rate unless the FFI or NFFE meets certain requirements or provides certain information to the person making the payment. Withholdable payments generally include fixed or determinable annual or periodical (“FDAP”) payments (such as dividends) from U.S. sources and gross proceeds from the sale or other disposition of any property of a type which can produce U.S.-source interest or dividends (e.g., Innocoll Ireland ordinary shares, if Innocoll Ireland was treated as a U.S. corporation). Thus, if Innocoll Ireland is treated as a U.S. corporation, Innocoll Ireland ordinary shares will be subject to the requirements imposed under FATCA. FATCA withholding on U.S.-source FDAP payments generally commenced on July 1, 2014, and FATCA withholding on payments of gross proceeds is generally scheduled to commence on January 1, 2019.
Because we are taking the position that neither Innocoll Germany nor Innocoll Ireland should be treated as a U.S. corporation, we do not believe that we have obligations to report or withhold under FATCA. If FATCA were to apply to Innocoll Ireland as a result of its being treated as a U.S. corporation, however, Innocoll Ireland would have withholding tax obligations, possibly retroactively, for FATCA and regular U.S. withholding tax purposes. In such an event, there can be no assurance that Innocoll Ireland or a paying agent will be able to comply with the relevant requirements, or that it or an intermediary financial institution would not be required to deduct FATCA withholding from payments on Innocoll Ireland ordinary shares.
FATCA is particularly complex and its application is uncertain at this time. The above description is based in part on regulations and official guidance, all of which is subject to change or may be implemented in a materially different form. Investors should consult their tax advisors on how these rules may apply to us or other party and to payment they may receive in connection with Innocoll Ireland ordinary shares.
| F. | DIVIDENDS AND PAYING AGENTS |
Not applicable.
Not applicable.
We previously filed with the SEC our registration statement on Form F-4 (Registration No. 333-208438), as amended, to register the issuance of our ordinary shares in connection with the merger.
We are subject to the periodic reporting and other informational requirements of the Exchange Act. Under the Exchange Act, we are required to file reports and other information with the SEC. Specifically, we are required to file annually a Form 20-F within four months after the end of each fiscal year, which is December 31. Copies of reports and other information, when so filed, may be inspected without charge and may be obtained at prescribed rates at the public reference facilities maintained by the Securities and Exchange Commission at 100 F Street, N.E., Room 1580, Washington, D.C. 20549. The public may obtain information regarding the Washington, D.C. Public Reference Room by calling the Commission at 1-800-SEC-0330. The SEC also maintains a website at www.sec.gov that contains reports, proxy and information statements, and other information regarding registrants that make electronic filings with the SEC using its EDGAR system. As a foreign private issuer, we are exempt from the rules under the Exchange Act prescribing the furnishing and content of quarterly reports and proxy statements, and officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act.
A copy of each document (or a translation thereof to the extent not in English) concerning us that is referred to in this annual report, is available for public view at our principal executive offices at Innocoll Holdings plc, Unit 9, Block D, Monksland Business Park, Monksland, Athlone, Ireland, telephone +353 (0) 90 6486834.
Not applicable.
Item 11. Quantitative and Qualitative Disclosures About Market Risk
We are exposed to various risks in relation to financial instruments including credit risk, liquidity risk and currency risk. Our risk management is coordinated by our managing directors. We do not engage in the trading of financial assets for speculative purposes. The most significant financial risks to which we are exposed include the following:
Credit risk
Our sales are currently concentrated with two customers and accordingly we are exposed to the possibility of loss arising from customer default. We are addressing this risk by monitoring our commercial relationships with these customers and by seeking to develop additional products for sale and entering into new partnerships.
Liquidity risk
We have been dependent on our shareholders to fund our operations. As described in note 1 of our financial statements, our ability to continue as a going concern is dependent on our ability to raise additional finance by way of debt and/or equity offerings to enable us to fund our clinical trial programs.
Currency risk
We are subject to currency risk, as our income and expenditures are denominated in euro and the U.S. dollar. As such we are exposed to exchange rate fluctuations between the U.S. dollar and the euro. We aim to match foreign currency cash inflows with foreign cash outflows where possible. We do not hedge this exposure. A 10% movement in the U.S. dollar versus euro exchange rate at December 31, 2015 would have the effect of increasing/decreasing net liabilities by approximately €1.5 million. As we incur clinical trial expenses in the United States in U.S. dollars, raise funds through licensing and collaboration revenue in U.S. dollars and commence sales of our products in the United States, we expect to have significant increases in cash balances, revenues and research and development costs denominated in U.S. dollars, while the majority of our cost of sales and operating costs are expected to remain denominated in euro. Accordingly, our exposure to exchange rates between the U.S. dollar and the euro has increased significantly commencing in 2014 compared to 2013. Between January 2013 and December 2015, the exchange rate between the U.S. dollar and the euro ranged between $1.3953 per euro and $1.0552 per euro.
Item 12. Description of Securities Other Than Equity Securities
Not applicable.
Not applicable.
Not applicable.
| D. | AMERICAN DEPOSITARY SHARES |
Termination of ADS Facility
Upon the effectiveness of the Merger, we terminated the Innocoll Germany American Depositary Share (“ADS”) facility. Prior to its cancellation, each Innocoll Germany ADS represented 1/13.25 of an ordinary share of Innocoll Germany. Following the Merger, each cancelled Innocoll Germany ADS effectively became an entitlement to receive one ordinary share of Innocoll Ireland. Additionally, each holder of one Innocoll Germany ordinary share received 13.25 ordinary shares of Innocoll Ireland. Simultaneous with the Merger, Innocoll Ireland listed its ordinary shares on the Nasdaq Global Market under the symbol “INNL”, which Innocoll Germany previously used for its ADSs.
Innocoll Germany entered into a termination agreement with Citibank N.A., the former depositary (“Depositary”) for Innocoll Germany’s ADS facility, in which it agreed to undertake any and all tax reporting, and to pay any and all stock transfer taxes that may be applicable to the termination and the transfer of shares in respect of the termination. In accordance with the termination agreement, on March 15, 2016, Innocoll Germany paid the Depositary an aggregate amount of approximately $0.8 million, representing (i) a fee to the Depositary of US$0.05 per ADS (including Restricted ADSs) cancelled in connection with the exchange of the Innocoll Ireland ordinary shares for the Innocoll Germany ADSs in accordance with the termination, and (ii) reimbursement to the Depositary for certain fees and expenses.
PART II
Item 13. Defaults, Dividend Arrearages and Delinquencies
There have been no material defaults in the payment of principal, interest, a sinking fund or purchase fund installment or any other material default with respect to any of our indebtedness.
Item 14. Material Modification to the Rights of Security Holders and Use of Proceeds
| A. | Material Modifications to the Rights of Security Holders |
The rights of holders of Innocoll Ireland ordinary shares are governed by Innocoll Ireland’s constitution. A description of the Innocoll Ireland ordinary shares and the rights of holders of Innocoll Ireland ordinary shares is included in our Report of Foreign Private Issuer on Form 6-K filed with the SEC on March 16, 2016, and is incorporated herein by reference.
A description of the material terms of our constitution is discussed in “Item 10. Additional Information—B. Memorandum and Articles of Association.”
On July 30, 2014, we completed an initial offering of 6,500,000 ADSs representing 490,567 ordinary shares, pursuant to a Registration Statement on Form F-1, as amended (File No. 333-196910), which became effective on July 24, 2014. Piper Jaffray, Stifel and JMP Securities acted as representatives of the several underwriters. The ordinary shares were sold at a price of $9.00 per ADS, thereby raising $54.4 million after deducting underwriting discounts and commissions. On August 20, 2014, the underwriters in our initial public offering partially exercised their overallotment option to purchase an additional 186,984 ADSs, representing 14,112 Ordinary Shares, at a public offering price of $9.00 per ADS. The sale of the overallotment option by our ADSs occurred on September 12, 2014, at which we raised additional net proceeds of approximately $1.57 million, after deducting underwriting discounts and commissions. We paid, out of company proceeds, all of our fees, costs and expenses in connection with our initial public offering (excluding, in the case of the selling shareholder, underwriting discounts and commissions and similar brokers’ fees and transfer taxes), which expenses totaled approximately €2.0 million.
None of the payments described in this Item 14 were direct or indirect payments to our directors, officers, general partners or their associates, or any persons owning 10% or more of our ordinary shares, or our affiliates.
There has been no material change in the planned use of proceeds from our initial public offering, as described in our final prospectus filed with the SEC pursuant to rule 424(b) under the Securities Act on July 25, 2014.
In March 2015, we and our wholly-owned subsidiary, Innocoll Pharmaceuticals Limited, or Innocoll Pharmaceuticals, entered into a Finance Contract with the European Investment Bank, or EIB, whereby the EIB committed to lend to Innocoll Pharmaceuticals up to €25 million. In the fourth quarter of 2015, we drew down €15 million of the loan commitment. The proceeds of the loan have been and will continue to be used in accordance with the terms of the Finance Contract.
On April 30, 2015, Innocoll Germany sold 1,999,690 ADSs to the public in an underwritten offering, representing 150,920 ordinary shares of Innocoll Germany, at a price of $9.00 per ADS, thereby raising $16.9 million after deducting underwriting discounts and commissions. In the same offering certain selling shareholders offered 1,321,979 ADSs, representing 99,772 ordinary shares of Innocoll Germany. We received no proceeds from the shares sold by the selling shareholders.
There has been no material change in the planned use of proceeds from our follow-on public offering, as described in our final prospectus filed with the SEC pursuant to rule 424(b) under the Securities Act on April 27, 2015.
Item 15. Controls and Procedures
(a) Disclosure Controls and Procedures: Our principal executive officer and principal financial officer, after evaluating the effectiveness of our disclosure controls and procedures (as defined in Exchange Act Rule 13a-15(e)) as of the end of the period covered by this Form 20-F, have concluded that, as of such date, our disclosure controls and procedures were effective to ensure that material information relating to Innocoll AG was timely made known to them by its subsidiaries.
(b) Management’s Annual Report on Internal Control over Financial Reporting: Management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Management assessed the effectiveness of internal control over financial reporting as of December 31, 2015 based on the framework in “Internal Control – Integrated Framework” (2013 framework) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
Based on that assessment, management has concluded that the Company’s internal control over financial reporting was effective as of December 31, 2015 to provide reasonable assurance regarding the reliability of its financial reporting and the preparation of its financial statements for external purposes, in accordance with IFRS.
Due to its inherent limitations, internal control over financial reporting may not prevent or detect misstatements, and can only provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
(c) Attestation Report of the Registered Public Accounting Firm: An attestation report of our registered public accounting firm is not required as we qualify as an emerging growth company as defined in the Exchange Act, and are therefore exempt from the attestation requirement.
(d) Changes in Internal Control Over Financial Reporting: There have been no changes in the company’s internal control over financial reporting identified in connection with the evaluation that occurred during the year ended December 31, 2015 that have materially affected, or are reasonably likely to materially affect the company’s internal control over financial reporting.
Item 16. Reserved
Item 16A. Audit Committee Financial Expert
Our board has determined that that Mr. Culverwell, the current chairman of the audit committee, was an “audit committee financial expert” as defined in Item 16A of Form 20-F. Mr. Culverwell and each of the other members of the audit committee (being Dr. Banerji and Dr. Wiley) are independent non-executive directors. All three members of the committee have considerable financial knowledge and experience to assist in overseeing and guiding the board and the company in respect of audit and corporate governance disciplines.
Item 16B. Code of Ethics
We have adopted a written code of business conduct and ethics that applies to members of our management board, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. Our code of business conduct and ethics is available under the Investor Relations—Corporate Governance section of our website at www.innocoll.com. In addition, we intend to post on our website all disclosures that are required by law or the listing standards of the NASDAQ Global Market concerning any amendments to, or waivers from, any provision of the code. The reference to our website address does not constitute incorporation by reference of the information contained at or available through our website, and you should not consider it to be a part of this annual report.
No waivers have been granted to the code of conduct since its adoption.
Item 16C. Principal Accountant Fees and Services
Grant Thornton has served as our independent registered public accounting firm for the financial years ended December 31, 2015, 2014 and 2013.
The following table sets forth the fees billed to us by our independent auditors during the fiscal years ended December 31, 2015 and 2014:
| | | Year ended
December 31, | |
| | | 2015 | | | 2014 | |
| | | (€ in thousands) | |
| Audit fees | | | 256 | | | | 235 | |
| Audit-related fees | | | — | | | | — | |
| Tax fees | | | — | | | | — | |
| All other fees | | | — | | | | — | |
| Total | | | 256 | | | | 235 | |
Audit services include the audit of our financial statements, the review of interim financial information and SEC registration statements, and statutory audits.
Audit Committee Pre-Approval Policies and Procedures
The advance approval of our audit committee is required for all audit services to be provided to the company, whether provided by the principal auditor or other firms, and all other services (review, attest and non-audit) to be provided to the company by the independent auditor; provided, however, that the minimums non-audit services may instead be approved in accordance with paragraph (c)(7)(i)(C) of Rule 2-01 of Regulation S-X.
Item 16D. Exemptions from the Listing Standards for Audit Committees
Not Applicable.
Item 16E. Purchases of Equity Securities by the Issuer and Affiliated Purchasers
Neither the issuer nor any affiliate of the issuer purchased any of our shares during 2015.
Item 16F. Change in Registrant’s Certifying Accountant
Not Applicable.
Item 16G. Corporate Governance
Please refer to “Item 6. Directors, Senior Management and Employees”.
Item 16H. Mine Safety Disclosure
Not Applicable.
Item 17. Financial Statements
Not Applicable.
Item 18. Financial Statements
The audited consolidated financial statements as required under Item 18, are attached hereto starting on page F-1 of this annual report. The audit report of Grant Thornton, an independent registered public accounting firm, is included herein preceding the audited consolidated financial statements.
Item 19. Exhibits
See the Exhibit Index immediately following the signature page of this Annual Report on Form 20-F.
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
| | INNOCOLL AG |
| | | |
| | By: | /s/ Anthony P. Zook |
| | | Name: Anthony P. Zook |
| | | Title: Chief Executive Officer |
| | | Date: March 17, 2016 |
Exhibit Index
| ExhibitNo. | | Exhibit |
| 1.1 | | Innocoll Holdings plc Constitution(1) |
| 4.1# | | License and Supply Agreement, dated August 14, 2013, between Innocoll Pharmaceuticals Ltd. and Takeda GmbH, an affiliate of Takeda Pharmaceutical Company Limited, as amended on March 19, 2014(2) |
| 4.2# | | Licensing, Manufacturing and Supply Agreement, dated October 12, 2011, between Innocoll Pharmaceuticals Ltd. and Pioneer Pharma Co. Ltd., as amended on August 6, 2012(2) |
| 4.3# | | Exclusive Distribution Agreement, dated April 3, 2013, between Innocoll Pharmaceuticals Ltd. and Biomet 3i, LLC(2) |
| 4.4# | | Manufacture and Supply Agreement, dated August 17, 2007, among Innocoll Pharmaceuticals Ltd., Syntacoll AG and EUSA Pharma (Europe) Limited (later acquired by Jazz Pharmaceuticals), as amended and restated on April 27, 2010(2) |
| 4.5# | | Manufacturing and Supply Agreement, dated June 1, 2004, between Innocoll Technologies Ltd., and Biomet Orthopedics Switzerland GmbH, as amended on January 1, 2006; December 15, 2009; September 1, 2010; April 1, 2011; March 15, 2012; March 1, 2013 and July 26, 2013(2) |
| 4.6# | | Licensing, Manufacturing and Supply Agreement, dated December 5, 2011, between Innocoll Pharmaceuticals Ltd. and Saudi Centre for Pharmaceuticals(2) |
| 4.7+ | | Innocoll AG - Stock Option Plan(3) |
| 4.8+ | | Form of Management Option Agreement(3) |
| 4.9+ | | Form of Innocoll GmbH Option Agreement, as amended and restated(2) |
| 4.10+ | | Form of Innocoll AG Restricted Share Award Agreement(2) |
| 4.11+ | | 2016 Omnibus Incentive Compensation Plan(4) |
| 4.12+ | | Employment Agreement by and between Anthony Zook and Innocoll AG, dated as of December 2014 |
| 4.13+ | | Restricted Share Award Agreement by and between Anthony Zook and Innocoll AG, dated December 7, 2014 |
| 4.14+* | | Employment Agreement by and between Rich Fante and Innocoll, Inc., dated as of August 20, 2015 |
| 4.15+* | | Employment Agreement by and between Jose Carmona and Innocoll, Inc., dated as of September 1, 2015. |
| 4.16+* | | Employment Agreement by and between Charles F. Katzer and Innocoll Pharmaceuticals Ltd., dated as of December 14, 2015. |
| 4.17# | | Lease Agreement between Karl Sipmeier and Syntacoll GmbH, dated December 17, 2009 (English translation)(2) |
| 4.18 | | Lease Agreement between Athlone Institute of Technology and Innocoll Technologies Ltd., dated November 24, 2008(2) |
| 4.19* | | Lease Agreement between Thomas Angerer and Syntacoll GmbH, effective as of February 1, 2016. |
| 4.20* | | Lease Agreement between Campus Investors H Building, L.P. and Innocoll AG, dated October 31, 2015. |
| 4.21 | | Finance Contract between the European Investment Bank and Innocoll Pharmaceuticals Ltd. and Innocoll AG, dated March 27, 2015(5) |
| 8.1 | | List of Subsidiaries(2) |
| 12.1* | | Certification of Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| 12.2* | | Certification of Chief Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| 13.1* | | Certification of Chief Executive Officer and Chief Financial Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| 15.1* | | Consent of Independent Registered Public Accounting Firm |
| * | Filed herewith. |
| + | Indicates a management contract or compensatory plan. |
| # | Portions of this exhibit (indicated by asterisks) have been omitted pursuant to a request for confidential treatment pursuant to Rule 406 under the Securities Act of 1933. |
| (1) | Incorporated by reference to our report of foreign private issuer on Form 6-K filed with the Commission on March 16, 2016. |
| (2) | Incorporated by reference to our registration statement on Form F-1 (file no. 333-196910), as amended, initially filed with the Commission on June 19, 2014. |
| (3) | Incorporated by reference to our annual report on Form 20-F filed with the Commission on March 19, 2015. |
| (4) | Incorporated by reference to our registration statement on Form F-4 (file no. 333-208438), as amended, initially filed with the Commission on December 10, 2015. |
| (5) | Incorporated by reference to our registration statement on Form F-1 (file no. 333-23362), as amended, initially filed with Commission on April 10, 2015. |
INNOCOLL AG
INDEX TO FINANCIAL STATEMENTS
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders
Innocoll AG
We have audited the accompanying consolidated statements of the financial position of Innocoll AG and subsidiaries (“the Company”) as of December 31, 2015 and 2014, and the related consolidated statements of comprehensive (loss)/income, changes in equity, and cash flows for each of the three years in the period ended December 31, 2015. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Innocoll AG and subsidiaries as of December 31, 2015 and 2014, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2015, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Grant Thornton
/s/ Grant Thornton
Dublin, Ireland
March 15, 2016
INNOCOLL AG
CONSOLIDATED STATEMENT OF COMPREHENSIVE (LOSS)/INCOME
for the years ended December 31, 2015, 2014 and 2013
| Thousands of Euros (except share and share data) | | Notes | | | 2015 | | | 2014 | | | 2013 | |
| Revenue | | | 2 | | | € | 2,587 | | | € | 4,497 | | | € | 3,546 | |
| Cost of sales | | | | | | | (4,802 | ) | | | (5,573 | ) | | | (4,551 | ) |
| Gross loss | | | | | | | (2,215 | ) | | | (1,076 | ) | | | (1,005 | ) |
| Research and development expenses | | | 3 | | | | (26,878 | ) | | | (3,252 | ) | | | (1,663 | ) |
| General and administrative expenses | | | 4 | | | | (18,983 | ) | | | (11,687 | ) | | | (4,121 | ) |
| Other operating income/(expense) – net | | | 5 | | | | 3,876 | | | | (39 | ) | | | (154 | ) |
| Loss from operating activities – continuing operations | | | | | | | (44,200 | ) | | | (16,054 | ) | | | (6,943 | ) |
| Finance income/(expense) | | | 6 | | | | 1,418 | | | | (4,535 | ) | | | (6,949 | ) |
| Other income | | | 7 | | | | - | | | | 75 | | | | 16,073 | |
| (Loss)/profit before income tax | | | | | | | (42,782 | ) | | | (20,514 | ) | | | 2,181 | |
| Income tax | | | 8 | | | | (367 | ) | | | (152 | ) | | | (72 | ) |
| (Loss)/profit for the year – all attributable to equity holders of the company | | | | | | € | (43,149 | ) | | € | (20,666 | ) | | € | 2,109 | |
| Other comprehensive income: | | | | | | | | | | | | | | | | |
| Currency translation adjustment | | | | | | | (520 | ) | | | (623 | ) | | | 155 | |
| Total comprehensive (loss)/income for the year | | | | | | € | (43,669 | ) | | € | (21,289 | ) | | € | 2,264 | |
| (Loss)/earnings per share: | | | 9 | | | | | | | | | | | | | |
| Basic | | | | | | | (25.6 | ) | | | (28.1 | ) | | | 47.0 | |
| Diluted | | | | | | | (25.6 | ) | | | (28.1 | ) | | | (9.5 | ) |
See accompanying notes to consolidated financial information.
INNOCOLL AG
CONSOLIDATED STATEMENT OF FINANCIAL POSITION
at December 31, 2015 and 2014
| Thousands of Euros | | Notes | | 12/31/2015 | | | 12/31/2014 | |
| Assets | | | | | | | | | | |
| Property, plant and equipment | | 10 | | € | 7,733 | | | € | 1,238 | |
| Total non-current assets | | | | | 7,733 | | | | 1,238 | |
| Inventories | | 11 | | | 1,661 | | | | 1,118 | |
| Trade and other receivables | | 12 | | | 4,192 | | | | 761 | |
| Cash and cash equivalents | | | | | 38,749 | | | | 45,616 | |
| Total current assets | | | | | 44,602 | | | | 47,495 | |
| Total assets | | | | € | 52,335 | | | € | 48,733 | |
| Equity | | | | | | | | | | |
| Share capital | | | | | 1,785 | | | | 1,503 | |
| Share premium | | | | | 137,292 | | | | 122,084 | |
| Capital contribution | | | | | 723 | | | | 723 | |
| Other reserves | | | | | 12,415 | | | | 12,415 | |
| Currency translation reserve | | | | | (1,142 | ) | | | (622 | ) |
| Accumulated share compensation reserve | | | | | 9,924 | | | | 5,149 | |
| Accumulated deficit | | | | | (149,867 | ) | | | (106,718 | ) |
| Total equity attributable to equity holders of the company | | 16 | | | 11,130 | | | | 34,534 | |
| Liabilities | | | | | | | | | | |
| Interest bearing loans and borrowings | | 14 | | | 15,064 | | | | — | |
| Warrant liability | | 15 | | | 10,561 | | | | 7,239 | |
| Defined benefit pension liability | | 22 | | | 45 | | | | 61 | |
| Total non-current liabilities | | | | | 25,670 | | | | 7,300 | |
| Trade and other payables | | 13 | | | 13,237 | | | | 5,055 | |
| Deferred income | | 2 | | | 2,038 | | | | 1,835 | |
| Deferred taxation | | 8 | | | 242 | | | | — | |
| Current taxes payable | | | | | 18 | | | | 9 | |
| Total current liabilities | | | | | 15,535 | | | | 6,899 | |
| Total liabilities | | | | | 41,205 | | | | 14,199 | |
| Total equity and liabilities | | | | € | 52,335 | | | € | 48,733 | |
See accompanying notes to consolidated financial information.
INNOCOLL AG
CONSOLIDATED STATEMENT OF CHANGES IN EQUITY
at December 31, 2015, 2014 and 2013
| Thousands of Euros | | Share
capital | | | Share
premium | | | Capital
contribution | | | Other
reserves | | | Currency
translation
reserve | | | Share
compensation
reserve | | | Accumulated
deficit | | | Total | |
| Balance at January 1, 2013 | | € | 39 | | | € | 7,074 | | | € | 723 | | | € | 8,800 | | | € | (154 | ) | | € | 906 | | | € | (91,317 | ) | | € | (73,929 | ) |
| Total comprehensive income | | | — | | | | — | | | | — | | | | — | | | | 155 | | | | — | | | | 2,109 | | | | 2,264 | |
| Share based payment | | | — | | | | — | | | | — | | | | — | | | | — | | | | (906 | ) | | | 906 | | | | — | |
| Equity arising on convertible debt | | | — | | | | — | | | | — | | | | 38 | | | | — | | | | — | | | | — | | | | 38 | |
| Derecognition of convertible instruments and warrants | | | — | | | | — | | | | — | | | | (8,861 | ) | | | — | | | | — | | | | — | | | | (8,861 | ) |
| Equity recognized on issue of preferred stock | | | — | | | | — | | | | — | | | | 11,426 | | | | — | | | | — | | | | — | | | | 11,426 | |
| Gain on settlement of B Preferred stock | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 2,250 | | | | 2,250 | |
| Foreign exchange on reorganization | | | — | | | | — | | | | — | | | | (761 | ) | | | — | | | | — | | | | — | | | | (761 | ) |
| Balance at December 31, 2013 | | € | 39 | | | € | 7,074 | | | € | 723 | | | € | 10,642 | | | € | 1 | | | € | — | | | € | (86,052 | ) | | € | (67,573 | ) |
| Balance at January 1, 2014 | | | 39 | | | | 7,074 | | | | 723 | | | | 10,642 | | | | 1 | | | | — | | | | (86,052 | ) | | | (67,573 | ) |
| Total comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | (623 | ) | | | — | | | | (20,666 | ) | | | (21,289 | ) |
| Equity arising on convertible debt | | | — | | | | — | | | | — | | | | 1,773 | | | | — | | | | — | | | | — | | | | 1,773 | |
| Share based payment | | | — | | | | — | | | | — | | | | — | | | | — | | | | 5,149 | | | | — | | | | 5,149 | |
| Conversion of preference into ordinary shares | | | 841 | | | | 72,194 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 73,035 | |
| Issue of ordinary shares net of issue costs | | | 623 | | | | 42,816 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 43,439 | |
| Balance at December 31, 2014 | | € | 1,503 | | | € | 122,084 | | | € | 723 | | | € | 12,415 | | | € | (622 | ) | | € | 5,149 | | | € | (106,718 | ) | | € | 34,534 | |
| Balance at January 1, 2015 | | | 1,503 | | | | 122,084 | | | | 723 | | | | 12,415 | | | | (622 | ) | | | 5,149 | | | | (106,718 | ) | | | 34,534 | |
| Total comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | (520 | ) | | | — | | | | (43,149 | ) | | | (43,669 | ) |
| Share based payment | | | — | | | | — | | | | — | | | | — | | | | — | | | | 4,775 | | | | — | | | | 4,775 | |
| Fair value movement on warrants exercised in the period | | | — | | | | 413 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 413 | |
| Issue of ordinary shares net of issue costs | | | 282 | | | | 14,795 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 15,077 | |
| Balance at December 31, 2015 | | € | 1,785 | | | € | 137,292 | | | € | 723 | | | € | 12,415 | | | € | (1,142 | ) | | € | 9,924 | | | € | (149,867 | ) | | € | 11,130 | |
See accompanying notes to consolidated financial information.
INNOCOLL AG
CONSOLIDATED STATEMENT OF CASH FLOWS
for the years ended December 31, 2015, 2014 and 2013
| Thousands of Euros | | 2015 | | | 2014 | | | 2013 | |
| Operating activities | | | | | | | | | | | | |
| (Loss)/profit for the year | | € | (43,149 | ) | | € | (20,666 | ) | | € | 2,109 | |
| Adjustments for: | | | | | | | | | | | | |
| Finance (income)/expense | | | (1,418 | ) | | | 4,535 | | | | 6,949 | |
| Depreciation/impairment, (reversal of impairment) of property, plant & equipment | | | (3,557 | ) | | | 403 | | | | 386 | |
| Income tax expense/(credit) | | | 367 | | | | 152 | | | | (17 | ) |
| Gains on financial instruments/liquidation of subsidiaries | | | — | | | | — | | | | (16,073 | ) |
| Profit on disposals of property, plant & equipment | | | — | | | | (75 | ) | | | — | |
| Share based payment | | | 4,775 | | | | 5,149 | | | | — | |
| Foreign exchange (gains)/losses | | | (231 | ) | | | (129 | ) | | | 50 | |
| Operating cash outflows before movements in working capital | | | (43,213 | ) | | | (10,631 | ) | | | (6,596 | ) |
| (Increase)/decrease in inventory | | | (543 | ) | | | 605 | | | | (202 | ) |
| Increase in trade and other receivables | | | (3,431 | ) | | | (352 | ) | | | (100 | ) |
| Increase/(decrease) in trade and other payables | | | 8,182 | | | | (1,334 | ) | | | 725 | |
| Increase/(decrease) in deferred income and defined benefit pension liability | | | 187 | | | | (792 | ) | | | 1,508 | |
| Income taxes paid | | | (116 | ) | | | (196 | ) | | | (12 | ) |
| Net cash used in operating activities | | | (38,934 | ) | | | (12,700 | ) | | | (4,677 | ) |
| Cash flows from investing activities: | | | | | | | | | | | | |
| Interest received | | | 209 | | | | 65 | | | | — | |
| Interest paid | | | (136 | ) | | | — | | | | — | |
| Proceeds from disposals of property, plant and equipment | | | 2 | | | | 75 | | | | — | |
| Purchases of property, plant and equipment | | | (2,940 | ) | | | (909 | ) | | | (448 | ) |
| Net cash used in investing activities | | | (2,865 | ) | | | (769 | ) | | | (448 | ) |
| Cash inflows from financing activities: | | | | | | | | | | | | |
| Proceeds from interest bearing loans & borrowings | | | 15,000 | | | | — | | | | — | |
| Proceeds from issue of ordinary shares | | | 16,904 | | | | 45,463 | | | | — | |
| Issuance costs allocated against share premium | | | (1,827 | ) | | | (2,023 | ) | | | — | |
| Proceeds from issue of preferred stock and convertible promissory notes | | | — | | | | 8,718 | | | | 7,965 | |
| Net cash inflows from financing activities | | | 30,077 | | | | 52,158 | | | | 7,965 | |
| Net (decrease)/increase in cash and cash equivalents | | | (11,722 | ) | | | 38,689 | | | | 2,840 | |
| Cash and cash equivalents at the beginning of the year | | | 45,616 | | | | 2,692 | | | | (148 | ) |
| Effect of foreign exchange rate changes on cash and cash equivalents | | | 4,855 | | | | 4,235 | | | | — | |
| Cash and cash equivalents at the end of the year | | € | 38,749 | | | € | 45,616 | | | € | 2,692 | |
See accompanying notes to consolidated financial information.
INNOCOLL AG
Notes to the consolidated financial information
| 1 | Summary of significant accounting policies |
Reporting entity
Innocoll AG, a German stock corporation, is a global, commercial stage, specialty pharmaceutical company, with late stage development programs targeting areas of significant unmet medical need.
The consolidated financial information for the year ended December 31, 2015 comprises the financial information of Innocoll AG, the “company”, and its direct and indirect subsidiaries, the “group”, (whose subsidiaries are described in more detail in note 23). As further described in note 14, during 2013, Innocoll Holdings, Inc. re-domiciled from the United States to Germany pursuant to a contribution in kind and share for share exchange into the recently formed Innocoll GmbH as a result of which Innocoll Holdings, Inc. became Innocoll GmbH’s wholly-owned subsidiary and all of Innocoll Holdings, Inc. assets and operations (including all of its subsidiaries) were transferred to Innocoll GmbH with effect as of July 25, 2013. Accordingly, the consolidated financial information presented herein refers to Innocoll Holdings, Inc., as the “company,” and with its direct and indirect subsidiaries, collectively, as the “group,” for the period from January 1, 2012 until July 24, 2013, and to Innocoll AG (formerly known as Innocoll GmbH), as the “company,” and with its direct and indirect subsidiaries, collectively, as the “group,” for the period from July 25, 2013 until December 31, 2015.
Basis of preparation
Statement of compliance
The consolidated financial information has been prepared in accordance with International Financial Reporting Standards (IFRS) as issued by the International Accounting Standards Board (IASB) and interpretations of the IFRS Interpretations Committee (IFRIC). The designation IFRS also includes all valid International Accounting Standards (IAS); the designation IFRIC also includes all valid interpretations of the Standing Interpretations Committee (SIC). The consolidated financial information was approved by the managing directors on 15 March, 2016.
The consolidated financial information has been prepared on the historical cost basis except for defined benefit pension liability and warrants determined as meeting the criteria for recognition as financial liabilities, which have been recorded at fair value. The consolidated financial information is presented in euro (’€’), rounded to the nearest thousand, except where otherwise stated. The accounting policies set out below have been applied consistently to all periods presented in this consolidated financial information. Certain comparative amounts have been reclassified to conform with the current year presentation.
Going concern
The consolidated financial information has been prepared on the basis that the group is a going concern. The group’s rate of expenses will continue to increase as it advances its planned clinical trials of XaraColl, Cogenzia and CollaGUARD and expands its manufacturing facility in Saal, Germany. As a result, the group will be required to seek additional sources of capital during the next 12 months or restrict certain of its expenditures to conserve capital and extend its resources.
Basis of consolidation
The consolidated financial information includes all of the subsidiaries that are controlled by the group. Control exists when the group has the power to govern the financial and operating policies and obtains the benefits from an entity’s activities. Control is generally presumed to exist when the group owns, directly or indirectly, more than 50% of an entity’s voting rights. The existence and effect of potential voting rights that are currently exercisable or convertible are considered when assessing whether the group controls another entity. Inter-company transactions, balances and unrealized gains and losses on transactions between group companies are eliminated in preparing the consolidated financial information.
Subsidiaries
Subsidiaries are entities controlled by the group. The financial information of subsidiaries is included in the consolidated financial information from the date that control commences until the date that control ceases. Details of the group’s subsidiaries are included in note 23.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Foreign currency
Functional and presentation currency
Items included in the consolidated financial information of each of the group’s entities are measured using the currency of the primary economic environment in which each entity operates (‘functional currency’). The consolidated financial information is presented in euro (’€’). All companies within the group currently have the euro as their functional currency, except for a US subsidiary entity which has a US dollar functional currency. On consolidation, the assets and liabilities of the group’s foreign operations are translated at exchange rates prevailing at the balance sheet date. Income and expense items are translated to euro at rates at the dates of the transactions. Exchange differences arising, if any, are classified as equity and transferred to the group’s translation reserve. On disposal, in part or in full, the relevant amount of the currency translation reserve is transferred to the income statement.
Transactions and balances
Transactions in currencies other than the functional currency of the group entities are recorded at the rates of exchange prevailing on the dates of the transaction. At each balance sheet date, monetary assets and liabilities that are denominated in foreign currencies are translated to the respective functional currencies of group entities at the rates prevailing on the relevant balance sheet date.
Property, plant and equipment
Property, plant and equipment are carried at historical cost less accumulated depreciation and impairment losses. Subsequent costs are included in the asset’s carrying amount or recognized as a separate asset as appropriate only when it is probable that future economic benefits associated with the item will flow to the group and the cost of the item can be measured reliably. All other repair and maintenance costs are charged to the income statement as incurred. The cost of assets retired or otherwise disposed of and the related accumulated depreciation are recognized in the income statement as part of the gain or loss on disposal in the year of disposal. Gains and losses on disposals of property, plant and equipment are included in other income or expense.
Depreciation
Depreciation is calculated using the straight-line method to allocate the cost of property, plant and equipment over their estimated useful lives, less their estimated residual values, as follows:
| Leasehold improvements | | in line with the term of the rental agreement | |
| Plant and machinery | | 3 to 10 years | |
| Furniture and fittings | | 5 years | |
Depreciation methods, useful lives and residual values are reassessed at each reporting date. Assets that are subject to amortization or depreciation are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount may not be recoverable. An impairment loss is recognized for the amount by which the asset’s carrying amount exceeds its recoverable amount. For assets where an impairment loss subsequently reverses, the carrying amount of the asset (cash-generating unit) is increased to the revised estimate of its recoverable amount, but so that the increased carrying amount does not exceed the carrying amount that would have been determined had no impairment loss been recognized for the asset (cash-generating unit) in prior years. A reversal of an impairment loss is recognized immediately in the income statement.
Leased assets
Rentals payable under operating leases are charged to the income statement on a straight-line basis over the relevant lease term.
Inventories
Inventories are stated at the lower of cost and net realizable value. The cost of inventories is based on the weighted average cost method and includes expenditure in acquiring the inventories and bringing them to their existing location and condition. In the case of work in progress and finished goods, cost includes an appropriate share of overhead based on normal operating capacity. Net realizable value is the estimated selling price less the estimated costs of completion and the estimated costs necessary to make the sale.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Financial instruments
Non-derivative financial assets
Financial assets are initially recognized on the date they are originated and when the group obtains contractual rights to receive cash flows. The group derecognizes financial assets when the contractual rights to cash flows expire or it transfers the right to receive cash flows in a transaction which transfers substantially all the risks and rewards of ownership of the asset.
Trade receivables
Such assets are initially recognized at fair value and subsequently measured at amortized cost less accumulated impairment losses.
Cash and cash equivalents
Cash and cash equivalents comprise cash on hand and call deposits (with less than 3 months maturity) and are subject to an insignificant risk of changes in value.
Non-derivative financial liabilities
The group’s non-derivative financial liabilities comprised the following categories:
Convertible notes
Convertible notes that can be converted into share capital at the option of the holder are accounted for as compound financial instruments and include a liability and an equity component. The liability components are recognized initially at fair value and thereafter, measured at amortized cost using the effective interest rate method. Transaction costs that relate to the issue of the compound financial instrument are allocated to the liability and equity components in proportion to the allocation of proceeds. The equity component of the convertible notes is calculated as the excess of the issue proceeds over the present value of the future interest and principal payments, discounted at the market rate of interest applicable to similar liabilities that do not have a conversion option. The interest expense recognized in the income statement is calculated using the effective interest rate method.
Where the group extinguishes a convertible instrument before maturity, the group allocates the consideration and any transaction costs for the extinguishment to the liability and equity components of the instrument at the date of the transaction. In accordance with IAS 32 “Financial instruments: Presentation and disclosure”, the method used in allocating the consideration and transaction costs to the separate components is consistent with that used in the original allocation to the separate components of the proceeds received by the group when the convertible instrument was first issued.
Preferred shares
Preferred shares may include conversion rights which may be settled by ordinary shares. Preferred shares that can be converted into share capital at the option of the holder are accounted for as compound financial instruments and include a liability and an equity component. The liability components are recognized initially at fair value and thereafter, measured at amortized cost using the effective interest rate method. Transaction costs that relate to the issue of the compound financial instrument are allocated to the liability and equity components in proportion to the allocation of proceeds. The equity component of the convertible preferred shares is calculated as the excess of the issue proceeds over the present value of the future interest and principal payments, discounted at the market rate of interest applicable to similar liabilities that do not have a conversion option. The interest expense recognized in the income statement is calculated using the effective interest rate method.
The managing directors have reviewed the impact of IFRS 7 “Financial Instruments: Disclosure” on the financial information and have determined that the group can avail itself of an exemption under the standard requiring the fair values of its financial instruments to be disclosed on the basis that the fair values do not materially differ from their carrying amounts.
On this basis, no additional disclosures have been included in the financial statements in respect of the fair values of such financial instruments other than as set out above and within notes 14, 15 and 17.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Trade payables
Trade payables are initially measured at amortized cost which equates to fair value.
Equity instruments
Equity instruments issued by the group are recorded at the proceeds received, net of direct issue costs.
Warrants
The group calculates the fair value of warrants issued as part of fundraising activities at the date of issue taking the amount directly to equity where no cash settlement option exists and where a fixed number of warrants are issued at a fixed rate. The fair value is calculated using a recognized valuation methodology for the valuation of financial instruments (either the Black Scholes model or Monte Carlo simulation model dependant on the terms of the warrants issued). Fair value, which is assessed at the grant date, or, in the case of warrants classified as financial liabilities, at the end of each period, and is calculated on the basis of the contractual terms of the warrants.
Warrants containing either a cash settlement option or which have a variable exercise price are accounted for as financial liabilities in line with the requirements of IAS 32. These derivative financial instruments are designated as at fair value through profit or loss, as this category includes derivative financial instruments entered into that are not designated as hedging instruments in hedge relationships as defined by IAS 39 “Financial Instruments: Recognition and measurement”.
Financial liabilities designated upon initial recognition at fair value through profit or loss are designated at the initial date of recognition, and only if the criteria in IAS 39 are satisfied.
Transactions with individuals in their capacity as equity instruments holders, as described above, do not fall under the scope of IFRS 2 “Share-based payment” and as a result are not accounted for in accordance with the standard.
Provisions
A provision is recognized if, as a result of a past event, the group has a present obligation that it is probable, will result in an outflow of resources and can be estimated reliably.
Employee benefit plans
Pension plans
The group operates a number of defined contribution retirement benefit plans, the assets of which are held in separate trustee-administered funds. Payments to defined contribution benefit plans are charged as an expense to the income statement as they fall due.
The group operates a defined benefit pension plan within its German subsidiary. A defined benefit plan is a pension plan that is not a defined contribution plan. Typically defined benefit plans define the amount of pension benefit that an employee will receive on retirement, usually dependent on one or more factors such as age, years of service and compensation. Obligations for contributions to defined benefit pension plans are recognized as an expense in the income statement as service is received from the relevant employees.
Share-based compensation
The group from time to time has granted restricted stock units to individual employees and non employee directors. The employees purchase the stock at an agreed price which may be below the then fair value of the stock unit. The difference between the purchase price and the fair value is expensed over the vesting period of the restricted stock unit.
The group also operates equity-settled, share-based compensation plans through which it grants options to subscribe to a specific number of shares in accordance with the share option plan. The fair value of the employee services received in exchange for the grant of the option is recognized as an expense with a corresponding increase in equity.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
The total amount expensed over the vesting period is determined by reference to the fair value of the option granted, measured using the Black Scholes model, taking into account the terms and conditions upon which the option was granted excluding the impact of any non-market vesting conditions. At each balance sheet date, the entity revises its estimates of the number of options that are expected to become exercisable. It recognizes the impact of the revision of original estimates, if any, in the consolidated statement of comprehensive (loss)/income, and a corresponding adjustment to equity over the remaining vesting period. The proceeds received net of any directly attributable transaction costs are credited to share capital (par value) and share premium when the options are exercised.
The assumptions used in measuring the fair value of the stock granted, using the Black Scholes model were determined as follows:
| • | Prior to the shares being publicly traded, the current market value of shares was based on the valuation of the company by the managing directors at the share option grant date; |
| • | Subsequent to the shares being publically traded, the current market value of shares was based on the share price at the share option grant date; |
| • | The estimated volatility is based on the historical volatility of biotech companies that operate in the same therapeutic areas as the group, or that are of a similar size; |
| • | The expected duration is calculated as the estimated duration until exercise, taking into account the specific features of the plans; and |
| • | The weighted average risk-free interest rates used are based on government treasury bills at the date of grant with a term equal to the expected life of the options. |
Prior to the shares being publically traded, the valuation of our ordinary shares required us to make highly complex and subjective estimates.
Revenue recognition
Revenue from sales of products is measured at the fair value of the consideration received or receivable net of returns and allowances, trade discounts and volume rebates. Revenue is recognized when all of the following conditions are met:
| i. | The significant risks and rewards of the ownership of goods are transferred to the buyer. This usually occurs when the goods have been delivered; |
| ii. | The group retains neither continuing managerial involvement to the degree usually associated with ownership nor effective control over the goods sold; |
| iii. | The amount of revenue can be measured reliably; |
| iv. | It is probable that the economic benefits associated with the transaction will flow to the entity; and |
| v. | The costs incurred or to be incurred in respect of the transaction can be measured reliably. |
Deferred revenue is calculated when cash is received from a customer for a product which at the time of receipt has not yet been delivered.
Expenses
Research and development expenses
Research and development expenses are charged to the income statement as incurred. The group has determined that the regulatory, clinical or field trial risks inherent in the development of its products currently preclude it from capitalizing its development costs.
Financing costs and income
Financing costs consist of interest payable on borrowings and finance income of interest receivable on funds invested. Both are calculated using the effective interest rate method. Foreign exchange gains and losses arising on the retranslation of foreign currency balances are also included here.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Income taxes
Tax expense comprises current and deferred tax.
Current tax is the expected tax payable or receivable on the taxable result for the year and any adjustments in relation to tax payable or receivable in respect of the previous years.
Deferred tax is recognized in respect of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for taxation purposes. Deferred tax is not recognized for:
| • | temporary differences on the initial recognition of assets and liabilities in a transaction that is not a business combination and that affects neither accounting nor taxable profit; and |
| • | temporary differences related to subsidiaries to the extent that it is probable that they will not reverse in the foreseeable future. |
Deferred tax is measured at the tax rates at which the temporary differences are expected to reverse, using tax rates enacted or substantively enacted at the reporting date. Deferred tax assets and liabilities are offset where the entity has a legally enforceable right to set off current tax assets against current tax liabilities and the deferred tax assets and liabilities relate to the same taxation authority. Deferred tax assets are recognized to the extent that it is probable that there will be taxable profits in the foreseeable future against which they can be utilized.
Government Grants
Grants for the purchase of plant and equipment are recognised as receivable when there is reasonable assurance that they will be received and the conditions to obtain them have been complied with. Grants are initially credited to deferred income and released to profit and loss over the same useful life as the plant and equipment they relate to.
Earnings per ordinary share
Basic earnings per share is computed by dividing the (loss)/profit for the financial year attributable to ordinary shareholders of the company by the weighted average number of ordinary shares outstanding during the financial period.
Diluted earnings per share is computed by dividing the (loss)/profit for the financial year attributable to ordinary shareholders of the company by the weighted average number of ordinary shares in issue after adjusting for the effects of all potential dilutive ordinary shares that were outstanding during the financial period.
Earnings are adjusted for the after-tax amounts of preference dividends, differences arising on the settlement of preferred shares, and other similar effects of preferred shares classified as equity.
Critical accounting estimates and judgments
The preparation of the financial information in conformity with IFRS requires management to make judgements, estimates and assumptions. Estimates are reviewed on an ongoing basis.
Estimates and judgments are based on historical experience and on other factors that are reasonable under current circumstances. Actual results may differ from these estimates if these assumptions prove to be incorrect or if conditions develop other than as assumed for the purposes of such estimates. The following are the critical areas requiring estimates and judgments by management:
Revenue recognition
Significant management judgments and estimates must be made and used in connection with the recognition of revenue in each accounting period. Material differences in the amount of revenue in any given period may arise if these judgments or estimates prove to be incorrect or if management’s estimates change on the basis of development of the business or market conditions. To date there have been no material differences arising from these judgments and estimates.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Revenue from products is generally recorded as of the date of shipment, consistent with our typical shipment terms. Where the shipment terms do not permit revenue to be recognized as of the date of shipment, revenue is recognized when the group has satisfied all of its obligations to the customer in accordance with the shipping terms. Revenue is recognized to the extent that it is probable that economic benefit will flow to the group, that the risks and rewards of ownership have passed to the buyer and the revenue can be measured. No revenue is recognized if there is uncertainty regarding recovery of the consideration due at the outset of the transaction or the possible return of goods.
Revenue is recognized from milestone payments received under collaboration agreements when earned, provided that the milestone event is substantive, its achievability was not reasonably assured at the inception of the agreement, the group has no further performance obligations relating to the event, and collectability is reasonably assured. If these criteria are not met, the group recognizes milestone payments ratably over the remaining period of their performance obligations under the collaboration agreement.
Valuation of financial instruments
The group issued financial instruments during the relevant accounting periods and had financial instruments in issue at both accounting period ends. In conformity with IFRS, the group initially measured these financial instruments at their fair value and thereafter at amortized cost using the effective interest rate method or at fair value through profit or loss if designated as such upon initial recognition. In order to value these various instruments, the group (and the experts engaged by the group to assist with such valuations where applicable) made assumptions and estimates concerning variables such as future cashflows, discounts rates, expected volatility, risk free rate and type of valuation models used. The assumptions of future outcomes, and other sources of estimation uncertainty concerning the determination of key inputs to the valuation models, are based on management’s (and the relevant experts’) best assessment using the knowledge available, their historical experiences as well as other factors that are considered to be relevant. The estimates and assumptions are reviewed on an ongoing basis.
Taxation
Given the global nature of the business and the multiple taxing jurisdictions in which the group operates, the determination of the group’s provision for income taxes requires significant judgments and estimates, the ultimate tax outcome of which may not be certain. Although estimates are believed to be reasonable, the final outcome of these matters may be different than those reflected in the historical income tax provisions and accruals. Such differences could have a material effect on the income tax provision and results in the period during which such determination is made.
Deferred tax assets and liabilities are determined using enacted tax rates for the effects of net operating losses and temporary differences between the book and tax bases of assets and liabilities. In assessing the realizability of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. While management considers the scheduled reversal of deferred tax liabilities, and projected future taxable income in making this assessment, there can be no assurance that these deferred tax assets may be realizable.
In addition, the group may also be subject to audits in the multiple taxing jurisdictions in which it operates. These audits can involve complex issues which may require an extended period of time for resolution. Management believes that adequate provisions for income taxes have been made in the financial statements.
Allowance for slow-moving and obsolete inventory
The group evaluates the realizability of its inventory on a case-by-case basis and makes adjustments to the inventory provision based on management’s estimates of expected losses. The group writes off any inventory that is approaching its “use-by” date and for which no further re-processing can be performed. The group also considers recent trends in revenues for various inventory items and instances where the realizable value of inventory is likely to be less than its carrying value. Given the allowance is calculated on the basis of the actual inventory on hand at the particular balance sheet date, there were no material changes in estimates made during 2014 or 2015 which would have an impact on the carrying values of inventory during those periods.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Trade receivables
The group evaluates customer accounts with past-due outstanding balances or specific accounts for which it has information that the customer may be unable to meet its financial obligations. Based upon a review of these accounts and management’s analysis and judgment, the group estimates the future cash flows expected to be recovered from these receivables. The amount of the impairment on doubtful receivables is measured individually and recorded as a specific allowance against that customer’s receivable balance to the amount expected to be recovered. The allowance is re-evaluated and adjusted periodically as additional information is received.
Provisions
Provisions are recognized and measured on the basis of the estimate and probability of future outflows of resources, as well as on the basis of experiential values and the circumstances known at the end of the reporting period. The assessment of whether a present obligation exists is generally based on assessment of internal experts. Estimates can change on the basis of new information and the actual charges may affect the performance and financial position of the group.
Share based payments
The company recognizes stock-based compensation on the fair value of the related awards. Under the fair value recognition guidance of stock-based compensation accounting rules, stock-based compensation expense is estimated at the grant date based on the fair value of the award and is recognized as an expense over the requiste vesting period of the award. The fair value of the service-based awards are determined using the Black Scholes valuation model. The use of this valuation model involves assumptions that are judgmental and highly sensitive in the determination of compensation expense and include the expected life of the option, stock price, volatility, risk-free interest rate and exercise price.
Impairment reversal
With the exception of goodwill, a previously recognized impairment loss will be reversed insofar as estimates change as a result of an event occurring after the impairment loss was recognized. An impairment loss is reversed only to the extent that the asset’s carrying amount does not exceed the carrying amount that would have been determined had no impairment loss been recognized. A reversal of an impairment loss is recognized in the income statement.
Standards and amendments to existing standards effective January 1 2015
The following standards, amendments and interpretations which become effective in 2015 are of relevance to the Group. The application of the standards has had no material impact on the disclosures in the Group’s financial statements.
| Standard/Interpretation | | Content | | Applicable for periods beginning on/after |
| | | | | |
| IFRS 2 | | Share Based Payment | | July 1 2014 |
| IFRS 3 | | Business Combinations | | July 1 2014 |
| IFRS 8 | | Operating Segments | | July 1 2014 |
| IAS 24 | | Related Party Disclosure | | July 1 2014 |
Standards, amendments and interpretations to existing standards that are not effective and have not been adopted early by the Group.
| Standard/Interpretation | | Content | | Applicable for periods beginning on/after |
| | | | | |
| IFRS 5 | | Non-current Assets Held for Sale | | January 1 2016 |
| IFRS 7 | | Financial Instruments: Disclosures Amended | | January 1 2016 |
| IFRS 10 | | Consolidated Financial Statements | | January 1 2016 |
| IFRS 11 | | Joint Arrangements | | January 1 2016 |
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
| IAS 1 | | Presentation of Financial Statements | | January 1 2016 |
| IAS 16 | | Property, Plant and Equipment | | January 1 2016 |
| IAS 28 | | Investments in Associates and Joint Ventures | | January 1 2016 |
| IAS 7 | | Statement of Cash Flows | | January 1 2017 |
| IAS 12 | | Income Taxes | | January 1 2017 |
| IFRS 9 | | Financial Instruments | | January 1 2018 |
| IFRS 15 | | Revenue from Contracts with Customers | | January 1 2018 |
| IFRS 16 | | Leases | | January 1 2019 |
In 2015, the Group did not early adopt any new or amended standards and does not plan to early adopt any of the standards issued but not yet effective. The group does not anticipate that the adoption of these standards and interpretations will have a material effect on its financial statements on initial adoption.
Due to the nature of the group’s current activities, the managing directors consider there to be one operating segment, the manufacture and sale of collagen-based pharmaceutical products. The entire group’s revenue is derived from this operating segment which can be spread among five geographical regions. The group principally sells four products; CollatampG globally outside of the United States, Septocoll within Europe and the Middle East, CollaGUARD within Europe, the Middle East and Asia and RegenePro in the United States. The results of the group are reported on a consolidated basis to the chief operating decision maker of the group, the chief executive officer. There are no reconciling items between the group’s reported income statement and statement of financial position and the results and financial position, respectively, of the above segment.
The majority of the product revenue, €2.6 million, relates to sales of CollatampG and Septocoll and is split between two customers; in 2015 the split was 88% and 12%. The group receives a contractually agreed percentage of the net in-market sales of CollatampG from one of its customers which distributes the product. This is recognized in two parts; the first amount is recognized for the manufacture and sale of product at the point of sale; and the final amount when the product is sold.
As of December 31, 2015, the group had deferred income in the amount of €2.0 million (2014: €1.8 million) relating to upfront payments in the amount of €0.5 million in respect of Septocoll customers, €1.1 million in respect of CollaGUARD and the remaining €0.4 million mainly relating to its Regenepro product. Innocoll expects to deliver products to these customers in the period from 2016 to 2017 in settlement of these advance payments respectively.
As mentioned above, the group has determined that all revenue is derived from one business segment. The managing directors have reviewed the impact of IFRS 8 “Operating Segments” on the financial statements with the above in mind. Given that the group only has one business segment, the managing directors have concluded that it is not necessary to show the full requirements of the standard within this note as the key information is displayed in other areas of the financial information.
The distribution of revenue by customers’ geographical area was as follows:
| Thousands of Euros | | 2015 | | | 2014 | | | 2013 | |
| Europe | | | 2,425 | | | | 4,340 | | | | 3,459 | |
| Middle East | | | — | | | | — | | | | 10 | |
| Asia | | | 43 | | | | 87 | | | | 73 | |
| United States | | | 119 | | | | 70 | | | | — | |
| ROW | | | — | | | | — | | | | 4 | |
| Gross revenue | | | 2,587 | | | | 4,497 | | | | 3,546 | |
All non-current assets are located in Germany and Ireland.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
| 3 | Research and development expenses |
| Thousands of Euros | | 2015 | | | 2014 | | | 2013 | |
| Employee compensation | | | 2,199 | | | | 1,495 | | | | 1,441 | |
| External clinical research costs | | | 23,789 | | | | 1,423 | | | | 110 | |
| General operating costs | | | 890 | | | | 334 | | | | 200 | |
| Research and development tax credit received | | | — | | | | — | | | | (88 | ) |
| Total research and development expenses | | | 26,878 | | | | 3,252 | | | | 1,663 | |
Research and development expenses include labor, materials and direct overheads associated with the various research programmes.
| 4 | General and administrative expenses |
| Thousands of Euros | | 2015 | | | 2014 | | | 2013 | |
| Employee compensation | | | 4,880 | | | | 2,728 | | | | 2,407 | |
| Depreciation | | | 319 | | | | 248 | | | | 238 | |
| Share based payments | | | 4,775 | | | | 5,149 | | | | — | |
| Other | | | 9,009 | | | | 3,562 | | | | 1,476 | |
| Total general and administrative expenses | | | 18,983 | | | | 11,687 | | | | 4,121 | |
| 5 | Other operating (income)/expense — net |
| Thousands of Euros | | 2015 | | | 2014 | | | 2013 | |
| Impairment of property, plant and equipment | | | (3,876 | ) | | | 155 | | | | 148 | |
| Compensation for amendments to supply agreements | | | — | | | | (118 | ) | | | — | |
| Other expense | | | — | | | | 2 | | | | 6 | |
| Total other operating expense | | | (3,876 | ) | | | 39 | | | | 154 | |
| 6 | Finance (income)/expense |
| Thousands of Euros | | 2015 | | | 2014 | | | 2013 | |
| Interest on convertible preferred shares | | | — | | | | 3,063 | | | | 4,728 | |
| Interest on convertible promissory notes | | | — | | | | — | | | | 1,918 | |
| Interest received | | | (209 | ) | | | (65 | ) | | | — | |
| Warrant expense | | | 3,735 | | | | 6,265 | | | | 205 | |
| Foreign exchange (gain)/loss | | | (5,145 | ) | | | (4,735 | ) | | | 84 | |
| Other expense | | | 201 | | | | 7 | | | | 14 | |
| Total finance (income)/expense | | | (1,418 | ) | | | 4,535 | | | | 6,949 | |
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
| Thousands of Euros | | 2015 | | | 2014 | | | 2013 | |
| Fair value gain on warrant exchanges | | | — | | | | — | | | | 170 | |
| Gain on settlement of promissory notes and preferred stock | | | — | | | | — | | | | 973 | |
| Gain on extinguishment of liabilities component of series B convertible preferred stock | | | — | | | | — | | | | 14,930 | |
| Profit on disposal of property, plant and equipment | | | — | | | | 75 | | | | — | |
| Total other income | | | — | | | | 75 | | | | 16,073 | |
In 2013 and in accordance with IAS 39, the liability associated with the series B convertible stock issued by Innocoll Holdings, Inc. was deemed to be extinguished as the new financial instruments issued had significantly different terms to the instruments they replaced. The difference between the fair value of the new financial instrument, and the carrying value of the extinguished series B convertible stock, was allocated against the carrying value of the liability and equity components with a resulting gain being recognized in profit and loss as noted above.
| Thousands of Euros | | 2015 | | | 2014 | | | 2013 | |
| Current tax expense | | | | | | | | | | | | |
| Tax charge | | | 125 | | | | 101 | | | | 72 | |
| Under provision in prior year | | | — | | | | 51 | | | | — | |
| Total current income tax | | | 125 | | | | 152 | | | | 72 | |
| Deferred tax expense | | | | | | | | | | | | |
| Deferred tax charge in year | | | 242 | | | | — | | | | — | |
| Total income tax charge | | | 367 | | | | 152 | | | | 72 | |
A reconciliation of the expected income tax of the group and the actual income tax charge is as follows:
| Thousands of Euros | | 2015 | | | 2014 | | | 2013 | |
| (Loss)/profit before taxation for the period | | | (42,782 | ) | | | (20,514 | ) | | | 2,181 | |
| Expected income tax (credit)/charge, computed by applying the Irish tax rate 12.5% (2013: German tax rate 28%) | | | (5,348 | ) | | | (2,564 | ) | | | 611 | |
| Effect of different tax rates of subsidiaries operating in foreign jurisdictions | | | 145 | | | | 279 | | | | 1,421 | |
| Unrecognized tax losses carried forward | | | 4,458 | | | | 1,082 | | | | 2,213 | |
| Non-deductible expenses/non-taxable income | | | 870 | | | | 1,304 | | | | (4,173 | ) |
| Under provision in prior year | | | — | | | | 51 | | | | — | |
| Current income taxes | | | 125 | | | | 152 | | | | 72 | |
Innocoll AG became tax resident in Ireland with effect as of January 1, 2014.
One of the main differences between expected tax and effective tax is explained by unrecognized deferred tax assets on tax losses carried forward amounting to €12.6 million (2014: €8.9 million, 2013: €7.3 million).
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Deferred tax assets and liabilities
The following temporary differences which might give rise to deferred taxes relate to:
| Thousands of Euros | | 2015 | | | 2014 | | | 2013 | |
| Net tax losses carry forward | | | 100,640 | | | | 69,816 | | | | 57,897 | |
| Excess capital allowances and other timing differences | | | (132 | ) | | | 537 | | | | 434 | |
| Total deductible differences | | | 100,508 | | | | 70,353 | | | | 58,331 | |
| Unrecognized deferred tax asset | | | 12,564 | | | | 8,905 | | | | 7,291 | |
All of the unrecognized deferred tax assets arise on operations in Ireland. Losses arising on operations in Ireland can be carried forward indefinitely, but are limited to the same trade/trades. The group has not recognized any deferred tax assets in the period as management does not consider the realization of these deferred tax balances to be probable in the near future due to the significant costs the group is likely to incur in the short term in advancing its product pipeline.
The deferred tax liability of €0.2 million (2014: €0) relates to timing differences of unrealized foreign exchange gains and losses totalling €0.5 million less tax losses arising in the United States of €0.3 million.
| 9 | (Loss)/Earnings per share |
The weighted average number of ordinary shares (denominator - basic) amounted to 1,685,088 in 2015 (2014: 735,416 and 2013: 44,848). The weighted average number of ordinary shares for 2013 has been adjusted for the effects of the re-domicile which took place during the financial period ended December 31, 2013 (note 14).
| | | 2015 | | | 2014 | | | 2013 | |
| Numerator – Thousands of Euros: | | | | | | | | | | | | |
| Net (loss)/earnings – basic | | € | (43,149 | ) | | € | (20,666 | ) | | € | 2,109 | |
| Adjustment to net earnings for interest on convertible preferred shares | | | — | | | | — | | | | 4,728 | |
| Adjustment to net earnings for interest on convertible promissory notes | | | — | | | | — | | | | 1,918 | |
| Adjustment for gain on settlement of promissory notes and preferred stock | | | — | | | | — | | | | (15,903 | ) |
| Net loss – diluted | | | (43,149 | ) | | | (20,666 | ) | | | (7,148 | ) |
| | | | | | | | | | | | | |
| Denominator – number of shares: | | | | | | | | | | | | |
| Weighted-average shares outstanding – basic | | | 1,685,088 | | | | 735,416 | | | | 44,848 | |
| Dilutive common stock issuable upon conversion of preferred shares | | | — | | | | — | | | | 547,195 | |
| Dilutive common stock issuable upon conversion of promissory notes | | | — | | | | — | | | | 160,246 | |
| Weighted-average shares outstanding – diluted | | | 1,685,088 | | | | 735,416 | | | | 752,289 | |
| | | | | | | | | | | | | |
| (Loss)/Earnings per share: | | | | | | | | | | | | |
| Basic | | | (25.6 | ) | | | (28.1 | ) | | | 47.0 | |
| Diluted | | | (25.6 | ) | | | (28.1 | ) | | | (9.5 | ) |
The basic loss per share for the year ended December 31, 2015 was €(25.6) (2014: €(28.1) loss per share and 2013: €47.0 earnings per share).
For the purpose of calculating diluted loss per share for 2015 and 2014 the potentially exercisable instruments in issue would have the effect of being antidilutive and, as such, the diluted loss per share is the same as the basic loss per share for those periods.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
| 10 | Property, plant and equipment |
| Thousands of Euros | | Leasehold
improvements | | | Plant &
machinery | | | Furniture
& fittings | | | Total | |
| Cost | | | | | | | | | | | | | | | | |
| Balance January 1, 2014 | | | 673 | | | | 11,102 | | | | 1,657 | | | | 13,432 | |
| Additions | | | 264 | | | | 623 | | | | 22 | | | | 909 | |
| Disposals | | | — | | | | (236 | ) | | | — | | | | (236 | ) |
| Balance December 31, 2014 | | | 937 | | | | 11,489 | | | | 1,679 | | | | 14,105 | |
| Additions | | | 992 | | | | 1,812 | | | | 136 | | | | 2,940 | |
| Disposals | | | (12 | ) | | | — | | | | (110 | ) | | | (122 | ) |
| Disposal of capitalized borrowings | | | — | | | | (634 | ) | | | — | | | | (634 | ) |
| Balance December 31, 2015 | | | 1,917 | | | | 12,667 | | | | 1,705 | | | | 16,289 | |
| Thousands of Euros | | Leasehold
improvements | | | Plant &
machinery | | | Furniture
& fittings | | | Total | |
| Depreciation | | | | | | | | | | | | | | | | |
| Balance January 1, 2014 | | | 479 | | | | 10,615 | | | | 1,606 | | | | 12,700 | |
| Depreciation charge for year | | | 40 | | | | 202 | | | | 6 | | | | 248 | |
| Impairment charge for year | | | — | | | | 155 | | | | — | | | | 155 | |
| Disposals | | | — | | | | (236 | ) | | | — | | | | (236 | ) |
| Balance December 31, 2014 | | | 519 | | | | 10,736 | | | | 1,612 | | | | 12,867 | |
| Depreciation charge for year | | | 63 | | | | 235 | | | | 21 | | | | 319 | |
| Reversal of impairment for period | | | — | | | | (3,876 | ) | | | — | | | | (3,876 | ) |
| Disposals | | | (11 | ) | | | — | | | | (109 | ) | | | (120 | ) |
| Disposal of capitalized borrowings | | | — | | | | (634 | ) | | | — | | | | (634 | ) |
| Balance December 31, 2015 | | | 571 | | | | 6,461 | | | | 1,524 | | | | 8,556 | |
| Net book value | | | | | | | | | | | | | | | | |
| At December 31, 2014 | | | 418 | | | | 753 | | | | 67 | | | | 1,238 | |
| At December 31, 2015 | | | 1,346 | | | | 6,206 | | | | 181 | | | | 7,733 | |
The impairment charge included in plant and machinery for 2014 represents a write-down of equipment which was capitalized but not brought into use. The managing directors have reversed the impairment of plant and machinery by €3.9 million in 2015.
In 2011 and 2012, the group impaired property, plant and equipment relating to their manufacturing facility in Saal, Germany by €4.8 million. The group recommenced their facility expansion during 2015. As the initial conditions reversed from when the initial decision was taken to impair the property, plant and equipment, the impairment was reversed. The value of the property, plant and equipment was assessed against current market values and the reversal of the impairment was recorded to the extent that each asset’s carrying amount did not exceed the carrying amount that would have been determined had no impairment loss been recognised.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
| Thousands of Euros | | 12/31/2015 | | | 12/31/2014 | |
| Raw materials | | | 638 | | | | 511 | |
| Work in progress | | | 882 | | | | 505 | |
| Finished goods | | | 141 | | | | 102 | |
| Total inventories | | | 1,661 | | | | 1,118 | |
The replacement cost of inventory does not differ materially from its carrying value. The impairment provision against inventory amounted to €0.5 million (2014: €0.6 million).
In 2015, raw materials, changes in work and progress and finished goods included in cost of sales amounted to €4.8 million (2014: €5.6 million).
| 12 | Trade and other receivables |
| Thousands of Euros | | 12/31/2015 | | | 12/31/2014 | |
| Trade receivables, net | | | 515 | | | | 326 | |
| Sales taxes receivable | | | 354 | | | | 159 | |
| Prepaid expenses and other current assets | | | 3,323 | | | | 276 | |
| Total trade and other receivables | | | 4,192 | | | | 761 | |
The impairment provision against trade receivables amounted to €0.04 million at December 31, 2015 (2014: €0.04 million).
| 13 | Trade and other payables |
| Thousands of Euros | | 12/31/2015 | | | 12/31/2014 | |
| Trade payables | | | 5,162 | | | | 2,525 | |
| Accrued expenses | | | 8,075 | | | | 2,530 | |
| Total trade and other payables | | | 13,237 | | | | 5,055 | |
| 14 | Interest-bearing loans and borrowings |
Gross liabilities at December 31, 2014:
| Thousands of Euros | | Series A
Convertible
Stock | | | Series B
Convertible
Stock | | | Series C
Convertible
Stock | | | Series D
Convertible
Stock | | | Series E
Convertible
Stock | | | Total | |
| Balance as at January 1, 2014 | | | 31,664 | | | | 5,323 | | | | 20,218 | | | | 19,052 | | | | — | | | | 76,257 | |
| Cash received for issue of E preferred shares | | | — | | | | — | | | | — | | | | — | | | | 8,768 | | | | 8,768 | |
| Settlement on conversion to ordinary shares | | | (31,664 | ) | | | (5,323 | ) | | | (20,218 | ) | | | (19,052 | ) | | | (8,768 | ) | | | (85,025 | ) |
| | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | |
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Reconciliation of gross proceeds to carrying value:
Year ended December 31, 2014
Thousands of Euros | | Series A
Convertible
Stock | | | Series B
Convertible
Stock | | | Series C
Convertible
Stock | | | Series D
Convertible
Stock | | | Series E
Convertible
Stock | | | Total | |
| Gross Proceeds | | | 31,664 | | | | 5,323 | | | | 20,218 | | | | 19,052 | | | | 8,768 | | | | 85,025 | |
| Recognised in equity | | | (4,295 | ) | | | (105 | ) | | | (5,209 | ) | | | (1,817 | ) | | | (1,773 | ) | | | (13,199 | ) |
| Fair value adjustment | | | — | | | | (4,789 | ) | | | — | | | | — | | | | — | | | | (4,789 | ) |
| Transaction costs | | | — | | | | — | | | | — | | | | (87 | ) | | | (50 | ) | | | (137 | ) |
| Accrued interest | | | 1,692 | | | | 325 | | | | 2,520 | | | | 1,540 | | | | 58 | | | | 6,135 | |
| Conversion to ordinary shares | | | (317 | ) | | | (53 | ) | | | (202 | ) | | | (191 | ) | | | (78 | ) | | | (841 | ) |
| Transfer to share premium on conversion to ordinary shares | | | (28,744 | ) | | | (701 | ) | | | (17,327 | ) | | | (18,497 | ) | | | (6,925 | ) | | | (72,194 | ) |
| | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | |
Financial instruments in issue as at January 1, 2013:
2007 series A convertible preferred stock
On December 5, 2007, the company issued $30.0 million in shares of series A convertible preferred stock to new private equity investors. The holders were entitled to receive a dividend per share of 6% per annum. Dividends were cumulative, compounded quarterly and were payable quarterly in arrears on March 31, June 30, September 30 and December 31. Dividends were payable in additional shares of series A convertible preferred stock through to December 31, 2013 and thereafter were to be paid in cash. Each share of series A convertible preferred stock could be converted at any time, at the option of the holder, into common stock at a rate of one share of common stock per $2.124 of series A convertible preferred stock. In addition the holders had rights in respect of appointment of directors, liquidation, anti-dilution, drag and tag rights and other rights normally associated with a similar investment in the United States.
2007 series B convertible preferred stock
On December 5, 2007, the company issued $16.8 million in shares of series B convertible preferred stock to existing shareholders. The holders were entitled to receive dividends per share of 6% per annum, after and subject to prior payment of dividends to the holders of series A convertible preferred stock. Dividends were cumulative, compounded quarterly and were payable quarterly in arrears on March 31, June 30, September 30 and December 31. Dividends were payable in additional shares of series B convertible preferred stock through to December 31, 2013 and thereafter were to be paid in cash. Each share of series B convertible preferred stock could be converted at any time, at the option of the holder, into common stock at a rate of one share of common stock per $2.124 of series B convertible preferred stock. In addition the holders had rights, subordinate in some respects to the holders of series A convertible preferred stock, in respect of liquidation, anti-dilution, drag and tag rights and other rights normally associated with a similar investment.
Also during 2007, the company issued 502,369 share warrants for the purchase of the company’s common stock as part of the above fundraising activities with an exercise price of $2.124 and a contractual life of 5 years.
2009 convertible promissory notes
On April 21, 2009, the company received bridge financing of $4.0 million from certain of its investors. The financing was provided in the form of convertible promissory notes in aggregate principal of up to $4.0 million; and 1,946,416 warrants for the purchase of the company’s common stock at a price of $2.124 per share with a contractual life of 10 years. The maturity of the convertible promissory notes was April 2015 and the notes could be converted at any time, at the option of the holder, into common stock at a rate of one share of common stock per $2.124 of unpaid principal plus any accrued and unpaid interest thereon, subject to adjustments. The holders of convertible promissory notes were entitled to receive interest at a rate of 20% per annum increasing to 25% per annum depending on circumstances. Interest was cumulative, compounded quarterly and was payable in arrears on March 31, June 30, September 30 and December 31.
On December 21, 2009, the company received further bridge financing of $4.0 million under similar terms to the April 2009 financing. The number of warrants for the purchase of the company’s common stock granted in the December financing was 2,068,012, again with an exercise price of $2.124 per share and a contractual life of 10 years.
On May 1, 2010, the 2009 convertible promissory notes were amalgamated and refinanced. The primary change was an amendment to the interest rate which reduced the rate to a fixed 10%.
2010 convertible promissory notes
On May 13, 2010, the company received bridge financing of $5.0 million from certain of its investors. The financing was provided in the form of convertible promissory notes in aggregate principal of up to $5.3 million; and 3,750,000 warrants for the purchase of the company’s common stock at a price of $1.00 per share and a contractual life of 10 years. The maturity of the convertible promissory notes was April 2015 and the notes could be converted at any time, at the option of the holder, into common stock at a rate of one share of common stock per $1.00 of unpaid principal plus any accrued and unpaid interest thereon, subject to adjustments.
The holders of convertible promissory notes were entitled to receive interest at a rate of 10% per annum. Interest was cumulative, compounded quarterly and was payable in arrears on March 31, June 30, September 30 and December 31.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
On August 11, 2010, the company received further bridge financing of $5.0 million under similar terms to the May financing. The number of warrants for the purchase of the company’s common stock granted in the August 2010 financing was 3,750,000, again with an exercise price of $1.00 per share and a contractual life of 10 years.
2011 convertible promissory notes
On March 22, 2011, the company received bridge financing of $3.3 million from certain of its investors. The financing was provided in the form of senior convertible promissory notes in aggregate principal of up to $3.4 million, 1,140 shares of series C-3 preferred stock and 8,125,000 warrants for the purchase of the company’s common stock at a price of $0.30 per share and a contractual life of 10 years.
The maturity of the convertible promissory notes was September, 2013 and the notes could be converted at any time, at the option of the holder, into common stock at a conversion ratio of 3.33 shares for each $1.00 of the unpaid principal and accrued interest thereon, subject to adjustments. The holder of convertible promissory notes were entitled to receive interest at a rate of 10% per annum. Interest was cumulative, compounded quarterly and was payable in arrears on March 31, June 30, September 30 and December 31.
On June 28, 2011, the company received further bridge financing of $3.3 million under similar terms to the March 2011 financing. The number of warrants for the purchase of the company’s common stock granted in the June 2011 financing was 8,125,000, again with an exercise price of $0.30 per share and a contractual life of 10 years along with 1,140 shares of series C-3 preferred stock.
These convertible promissory notes had a liquidation preference of three times principal plus one times interest outstanding.
2012 convertible promissory notes
On January 23, 2012 the company received bridge financing of $1.1 million from certain of its investors. The financing was provided in the form of senior convertible promissory notes in aggregate principal of up to $1.6 million, 386 shares of series C-3 preferred stock and 2,750,077 warrants for the purchase of the company’s common stock at an exercise price of $0.30 per share and a contractual life of 10 years. The maturity of the convertible promissory notes was September 2013, and the notes could be converted, at the option of the holder, into common stock at a conversion ratio of 3.33 shares for each $1.00 of the unpaid principal and accrued interest.
On June 26, 2012, the company received further bridge financing of $2.0 million under similar terms to the January 2012 financing. The number of warrants for the purchase of the company’s common stock granted in the June 2012 financing was 5,000,000, again with an exercise price of $0.30 per share of common stock and a contractual life of 10 years, along with 702 shares of series C-3 preferred stock.
On November 1, 2012, the company received further bridge financing of $2.0 million under similar terms to the January 2012 financing. The number of warrants for the purchase of the company’s common stock granted in the June 2012 financing was 5,000,000, again with an exercise price of $0.30 per share of common stock and a contractual life of 10 years, along with 695 shares of series C-3 preferred stock.
These convertible promissory notes had a liquidation preference of three times principal plus one times interest outstanding.
Financial instruments issued in 2013 prior to re-domicile of parent and share for share exchange:
2013 convertible promissory notes
On January 22, 2013 the company received bridge financing of $1.4 million from certain of its investors. The financing was provided in the form of senior convertible promissory notes in aggregate principal of up to $1.5 million, 488 shares of series C-3 preferred stock and 3,491,623 warrants for the purchase of the company’s common stock at an exercise price of $0.30 per share and a contractual life of 10 years. The maturity of the convertible promissory notes was September 2013, and the notes could be converted, at the option of the holder, into common stock at a conversion ratio of 3.33 shares for each $1.00 of the unpaid principal and accrued interest.
These convertible promissory notes had a liquidation preference of three times principal plus one times interest outstanding.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
2013 series D convertible preferred stock
On June 21, 2013 the company issued $1.0 million in shares of series D convertible preferred stock to certain of its investors. The financing was provided in the form of 10% series D convertible preferred stock and 2,500,000 warrants for the purchase of the company’s common stock at a price of $0.30 per share and a contractual life of 10 years. The series D convertible preferred stock, with a maturity date of September 2013, could be converted, at the option of the holder, into common stock at a conversion ratio of 3.33 shares for each $1.00 of the unpaid principal and accrued interest. The series D convertible preferred stock had a liquidation preference of three times stated value plus one times accrued and unpaid dividends.
Re-domicile of parent company
In June 2013, Innocoll Holdings, Inc., and the holders of its convertible promissory notes executed a debt-for-equity agreement (“the Agreement”), pursuant to which all outstanding convertible promissory notes were converted into shares of series C and D convertible preferred stock of the company in the amounts and on the terms as set forth in the Agreement.
Holders of the 2009 – 2010 promissory notes agreed that in exchange for the delivery and cancellation of their notes, they would receive shares of series C convertible preferred stock.
Holders of the 2011 – 2013 promissory notes agreed that in exchange for the delivery and cancellation of their notes, they would receive shares of series D convertible preferred stock.
All exchanges were made on a 1:1 basis.
Share for share exchange
Pursuant to a notarial deed entered into between the stockholders of Innocoll Holdings, Inc. and Innocoll AG in July 2013, the holders of shares of common stock, shares of series A, series B, series C and series D preferred stock and warrants to purchase shares of common stock of Innocoll Holdings, Inc. contributed their shares and warrants by way of a contribution in kind to Innocoll AG in exchange for ordinary shares, series A, series B, series C and series D preferred shares and options to purchase ordinary shares of Innocoll AG and as a result thereof, Innocoll Holdings, Inc. became Innocoll AG’s wholly-owned subsidiary. In September 2013, Innocoll Holdings, Inc. transferred substantially all of its operations, assets and liabilities, including its interests in Innocoll Pharmaceuticals Limited, Innocoll Technologies Limited, both registered in the Republic of Ireland and Innocoll Inc., a Delaware corporation to Innocoll AG. Innocoll Holdings, Inc. filed a certificate of dissolution in Delaware, USA on the next day.
As part of the re-domicile, Innocoll AG issued 38,750 ordinary shares with a nominal value of €1 per share, 316,640 series A preferred shares with a nominal value of €1 per share with a premium of €99; 53,234 series B preferred shares with a notional value of €1 per share with a €99 premium; 202,179 series C preferred shares with a nominal value of €1 per share with a premium of €99; 127,820 shares of series D preferred stock with a nominal value of €1 per share with a premium of €99; and options to purchase ordinary shares at an exercise price of €100 per share. In accordance with IFRS, these shares and the associated share premium were recorded in the Statement of Financial Position at the fair value of the consideration received on the date of their issue. Where the carrying value of the financial instruments settled exceeded the fair value of the instruments issued in exchange, the resulting gain was recognized in the income statement in accordance with IAS 39. Details of inputs into the fair value calculations related to share transactions above are provided below:
| | 12/31/2014 | | | 12/31/2013 | |
| Expected volatility | | | — | | | | 60.00 | % |
| Discount rate | | | — | | | | 15.00 | % |
| Risk free rate | | | — | | | | 0.96 | % |
Furthermore, in October and November 2013, Innocoll AG (formerly known as Innocoll GmbH) issued series D preferred shares for consideration of €6.3 million to existing shareholders.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Owing to the nature of the re-domicile, there was no change in the substance of the reporting entity and therefore, a business combination, in accordance with IFRS 3, was not deemed to have occurred. IFRS 3 does not apply to a business combination involving entities or businesses under common control. The introduction therefore of Innocoll AG (formerly known as Innocoll GmbH) as a new holding company for the group was accounted for as a continuation of the business previously carried out by Innocoll Holdings, Inc. Consequently, even though Innocoll AG (formerly known as Innocoll GmbH) was only incorporated on July 25, 2013 and was not a group company as at January 1, 2013, or for the period from January 1, 2013 to the date of the re-domicile, the disclosures in the consolidated financial information for those periods are those of Innocoll Holdings, Inc. The 2013 financial information has consequently been presented as if the previous business and operations of Innocoll Holdings, Inc. continued as before through each of the accounting periods presented, despite the fact that Innocoll AG (formerly known as Innocoll GmbH) became the principal operating and parent entity with effect from July 25, 2013 and was registered in the commercial register on August 23, 2013.
The following table sets out the classes of shares before and after the above-mentioned reorganization.
| Pre-reorganization | | No. of shares | |
| Common stock | | | 17,050,000 | |
| Series A preferred stock | | | 19,678,194 | |
| Series B preferred stock | | | 11,008,366 | |
| Series C preferred stock | | | 26,687,487 | |
| Series D preferred stock | | | 16,872,592 | |
| Total number of shares | | | 91,296,639 | |
| Post-reorganization | | No. of shares | |
| Ordinary shares | | | 38,750 | |
| Series A preferred shares | | | 316,640 | |
| Series B preferred shares | | | 53,234 | |
| Series C preferred shares | | | 202,179 | |
| Series D preferred shares | | | 127,820 | |
| Total number of shares | | | 738,623 | |
2013 series D preferred shares (post re-domicile)
In October 2013 the company issued €2.0 million of series D preferred shares to certain of its investors. The financing was provided in the form of 20,194 10% series D preferred shares and 15,147 warrants for the purchase of the company’s ordinary shares at a price of €100 per share (subject to adjustment) and a contractual life of 10 years.
On November 29, 2013 the company issued €4.3 million of series D preferred shares to certain of its investors. The financing was provided in the form of 42,500 series D preferred shares and 31,876 warrants for the purchase of the company’s ordinary shares at a price of €100 per share (subject to adjustment) and a contractual life of 10 years.
Financial instruments issued in 2014:
Issue of Series E Preferred Shares
On May 22, 2014, the Company’s shareholders approved the issuance of 77,924 series E preferred shares, including 6,103 restricted shares, to certain existing shareholders, three new members of our supervisory board and three new investors who are partners of one of the existing shareholders. The Company also issued 44,465 restricted and unrestricted shares The terms of the series E preferred share issue provided an anti-dilution right such that, in the case of an initial public offering in which the price per ordinary share equivalent of American Depositary Shares (ADSs) is less than 1.2 times the series E preferred price per share (the “IPO Premium Requirement’’), the shareholders had agreed to approve a further capital increase in the course of which holders of series E preferred shares, or ordinary shares issued after our transformation into Innocoll AG, would be issued newly issued ordinary shares in Innocoll AG at notional value of €1.00 per share in an amount such that the weighted average price per share of the series E preferred shares and the newly issued ordinary shares satisfied the IPO Premium Requirement. On the same day, the Company’s articles were also restated to reflect the increased share capital and the introduction of series E preferred shares. Both the capital increase and the articles restatement became effective upon registration with the commercial register on June 17, 2014.
The series E preferred shares were entitled to vote. The series E preferred shares had a dividend preference pursuant to Section 32 of the Company’s articles of association, under which they were entitled to receive an annual dividend in the amount of 10% of the stated value of €112.52 per share, out of profit in that year. If the profit for the year was not sufficient, the dividends for each year accrue and were payable, in whole or in part, in the first year that the profit was sufficient.
The series E preferred shares had a right of withdrawal and redemption compensation (the “Series E Redemption”) pursuant to Sections 30 and 31 of the Company’s articles of association, under which the series E preferred shares were entitled to terminate their shares and withdraw from the Company in exchange for redemption compensation of the stated value per share. The Series E Redemption was senior in preference to the Series D Liquidation Preference and the withdrawal and redemption compensation rights of the series C, A and B preferred shares. The Series E Redemption right could be exercised after June 30, 2018 with six months’ notice, or without a notice period upon (i) a sale or change of control of the Company, or (ii) an initial public offering of the Company.
Innocoll GmbH Conversion into Ordinary Shares, Capital Increase, Transformation to Innocoll AG
Pursuant to a notarial deed entered into on June 16, 2014 the shareholders of Innocoll GmbH agreed to amend and restate its articles of association and to cancel and terminate all preference, redemption and cumulative dividend rights in exchange for ordinary shares in Innocoll GmbH (except for certain anti-dilution rights relating to the series E preferred shares). Innocoll GmbH further issued €3.7 million of new ordinary shares (including share premium) to certain existing shareholders, with the same anti-dilution rights as the series E preferred shares. The shareholders further agreed that upon completion of conversion into ordinary shares and capital increase by Innocoll GmbH, Innocoll GmbH will transform into a German stock corporation (Aktiengesellschaft or AG) in accordance with the provisions of the German Reorganization Act (Umwandlungsgesetz), upon registration of the reorganization in the commercial register.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Gross liabilities at December 31, 2015:
| Thousands of Euros | | Interest
bearing loans | | | Tota1 | |
| Balance as at January 1, 2015 | | | — | | | | — | |
| Loan received | | | 15,000 | | | | 15,000 | |
| Balance as at December 31, 2015 | | | 15,000 | | | | 15,000 | |
Reconciliation of gross proceeds to carrying value:
Year ended December 31, 2015 Thousands of Euros | | Interest
bearing loans | | | Tota1 | |
| Gross Proceeds | | | 15,000 | | | | 15,000 | |
| Accrued interest | | | 64 | | | | 64 | |
| | | | 15,064 | | | | 15,064 | |
On March 27, 2015, the group approved the conclusion of the Finance Contract between Innocoll Pharmaceuticals Limited and the European Investment Bank for a loan of up to €25 million.
The loan terms specify that the amounts drawn will be used to finance up to 50% of certain research and development and capital expenditures forecast to be made by the company over a three year period from 2015 through 2017. The first €15 million of the loan commitment may be drawn at any time up to fifteen months from the date of the Finance Contract. The second €10 million may be drawn at any time up to eighteen months from the date of the Finance Contract subject to the delivery of clinical data, certified by the management board of Innocoll AG, confirming to the satisfaction of EIB that the primary clinical endpoints for either Cogenzia or XaraColl Phase 3 trials have been achieved and therefore it can be concluded that the Phase 3 clinical trial has successfully been completed. The loan bears interest at a fixed rate of 12% per annum, compounded annually. There are no cash payments of interest required during the term of the loan, interest on the outstanding loan balance accrues daily, and is payable only on the maturity date or prepayment date of the loan, whichever is earlier. The maturity date of each tranche is three to five years from the draw down date, as may be determined by the company in each draw down request. Principal of the loan is payable on the maturity date or the prepayment date, whichever is earlier.
The loan contains an expenditure covenant requiring the company to spend at least 75% of our forecast research and development, and capital expenditures during the period from 2015 to 2020 as set out in a business plan agreed with the EIB. If, upon completion of the Project, it is determined that the total principal amount of the loan drawn by the company exceeds 50% of the total cost of the Project, EIB may cancel the undisbursed portion of the loan and demand prepayment of the loan up to the amount by which the loan, excluding accrued interest, exceeds 50% of the total cost of the Project.
On December 18, 2015 Innocoll Pharmaceuticals Limited drew down the first tranche of €15 million.
The security on the Finance Contract includes a guarantee from the company on 100% of the outstanding principal, interest, expense, commission, indemnity and other sum owed by Innocoll Pharmaceuticals Limited, together with a fixed charge over all the shares in Innocoll Pharmaceuticals Limited and a floating charge over all the assets of Innocoll Pharmaceuticals Limited. As of December 31, 2015 no violations of the covenants occurred.
| Number | | Exercise
price | | | Contractual
life (years) | | | 12/31/2015 | | | 12/31/2014 | |
| Outstanding at January 1 | | € | 88.52 | | | | 10 | | | | 205,199 | | | | 205,199 | |
| Exercised during the year | | € | 88.52 | | | | 10 | | | | (8,287 | ) | | | — | |
| Outstanding at December 31 | | | | | | | | | | | 196,912 | | | | 205,199 | |
| Exercisable at December 31 | | | | | | | | | | | 196,912 | | | | 205,199 | |
All warrants have been classified as a liability due to certain provisions pursuant to which the exercise price of the warrants may be reduced in the event that the company issues or sells any of its ordinary shares at a price per share less than the exercise price in effect immediately prior to such issue or sale. Pursuant to a notarial deed entered into on June 16, 2014 the shareholders of Innocoll GmbH agreed to amend and terminate all preference, redemption and cumulative dividend rights by converting all preferred shares into ordinary shares of Innocoll GmbH, which had the effect of increasing the value of the warrants, which were now exercisable over ordinary shares. On July 3, 2014 Innocoll GmbH transformed into a German stock corporation Innocoll AG, and Innocoll AG subsequently entered into an amended and restated Option Agreement with the warrant holders on July 10, 2014. The warrants issued by Innocoll AG have an initial expiration date of June 15, 2019, subject to possible extension for an additional 54 month period subject to a shareholder resolution. Innocoll AG issued ADS in its initial public offering priced on July 24, 2014 at a price of $9.00 per ADS, or the equivalent of €88.52 per ordinary share. This resulted in the reduction of the exercise price from €100.00 per ordinary share to €88.52 per ordinary share pursuant to the above-described exercise price adjustment provision. A follow-on public offering in April 2015 was also priced at $9.00 per ADS and therefore the exercise price of the options remains unaffected. In addition, as Innocoll AG is a public company post the completion of its initial public offering, the shareholder resolution required for the term extension is uncertain, therefore the expiration date for purposes of valuation is assumed to be June 15, 2019. As a result of the above described provisions and events, the warrant liability has been valued at €10.6m as of December 31, 2015 as compared to €7.2m as of December 31, 2014.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
The carrying value of warrants was as follows:
| Thousands of Euros | | 12/31/2015 | | | 12/31/2014 | |
| Liability | | | | | | | | |
| Fair value at January 1 | | | 7,239 | | | | 974 | |
| Warrants exercised in the period | | | (413 | ) | | | — | |
| Fair value movement on warrants in issue | | | 3,735 | | | | 6,265 | |
| Fair value at December 31 | | | 10,561 | | | | 7,239 | |
The following input assumptions were used to fair value the warrants:
| | | 12/31/2015 | | | 12/31/2014 | |
| Expected volatility | | | 66.09 | % | | | 69.68 | % |
| Risk free rate | | | (0.05 | )% | | | 0.02 | % |
| Exercise price | | € | 88.52 | | | € | 88.52 | |
| Contractual life | | | 3.46 years | | | | 4.46 years | |
| Stock price on valuation date | | € | 101.01 | | | € | 64.83 | |
| 16 | Share capital and reserves |
| Number | | 12/31/2015 | | | 12/31/2014 | |
| Authorized number of shares: | | | | | | | | |
| Ordinary shares of no par value | | | 1,837,493 | | | | 1,568,155 | |
| Authorized Capital I | | | 196,912 | | | | 205,199 | |
| Authorized Capital II | | | 24,784 | | | | 97,154 | |
| Authorized Capital III | | | 665,739 | | | | 452,248 | |
| Contingent Capital | | | 150,920 | | | | 150,920 | |
| | | | 2,875,848 | | | | 2,473,676 | |
| Issued, called up and fully paid number of shares: | | | | | | | | |
| Ordinary shares at €1 nominal value | | | 1,785,009 | | | | 1,444,318 | |
| Issued, called up but not fully paid number of shares: | | | | | | | | |
| Ordinary shares at €1 nominal value | | | — | | | | 58,953 | |
Movement on issued ordinary shares during the year
On April 16, 2015 the management board approved the issuance of 72,370 ordinary shares for a cash contribution of €1.00 per share pursuant to a notarial deed entered into on June 16, 2014.
Following a follow on offering the company issued 150,920 ordinary shares on April 24, 2015.
On November 20, 2015 the management board approved the issuance of 8,287 ordinary shares for a cash contribution of €87.52 per share pursuant to a notarial deed entered into on June 16, 2014.
The company also included in share capital 50,161 ordinary shares held by members of the supervisory board and management board who had previously been granted restricted shares and a portion of same vested during the year.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Authorized and Contingent Capital
Authorized Capital I
On November 29, 2013 the company entered into an agreement whereby the management board shall be entitled to increase the company’s share capital, with the approval of the supervisory board, until June 15, 2019 against contribution in cash or in kind once or several times by issuing new non-par value shares registered by up to 205,199 (“Authorized Capital I”). In November 2015 the management board, with the approval of the supervisory board, increased the company’s share capital by 8,287 ordinary shares reducing the entitlement to increase the company’s share capital, with the approval of the supervisory board, until June 15, 2019 against contribution in cash or in kind once or several times by issuing new non-par value shares registered by up to 196,912 (“Authorized Capital I”). The management board shall be entitled, with the approval of the supervisory board, to exclude the subscription right of shareholders. The management board determines the subscription amount of new shares and may fix the commencement of their entitlement to profit.
Authorized Capital II
On May 22, 2014 the company entered into an agreement whereby the management board shall be entitled to increase the company’s share capital, with the approval of the supervisory board, until June 15, 2019 against contribution in cash or in kind once or several times by issuing new non-par value shares registered by up to 97,154 (“Authorized Capital II”). In the course of an offering in April, 2015 the management board increased the company’s share capital by 72,370 ordinary shares reducing the entitlement to increase the company’s share capital, until June 15, 2019 against contribution in cash or in kind once or several times by issuing new non-par value shares registered by up to 24,784 (“Authorized Capital II”).The management board shall be entitled, with the approval of the supervisory board, to exclude the subscription right of shareholders. The management board determines the subscription amount of new shares and may fix the commencement of their entitlement to profit.
Authorized Capital III
On December 4, 2014 the company entered into an agreement whereby the management board shall be entitled to increase the company’s share capital, with the approval of the supervisory board, until June 15, 2019 against contribution in cash or in kind once or several times by issuing new non-par value shares registered by up to 452,248. In the course of an offering in April, 2015 the management board increased the company’s share capital by 150,920 ordinary shares reducing the entitlement to increase the company’s share capital, until December 3, 2019 against contribution in cash or in kind once or several times by issuing new non-par value shares registered by up to 301,328 (“Authorized Capital III”). At the company’s AGM on August 24, 2015 the agreement whereby the management board shall be entitled to increase the company’s share capital, with the approval of the supervisory board, until December 3, 2019 against contribution in cash or in kind once or several times by issuing new non-par value shares registered by up to 301,328 was cancelled. Subsequently a new agreement was entered into whereby the management board shall be entitled to increase the company’s share capital, with the approval of the supervisory board, until August 23, 2020 against contribution in cash or in kind once or several times by issuing new non-par value shares registered by up to 665,739. New shares may be issued, inter alia, in order to fulfill an option to receive more shares granted to the underwriters in connection with the initial public offering of the company (“Authorized Capital III”). The management board is further authorized to exclude the subscription right of the shareholders with the approval of the supervisory board.
Contingent Capital
The company has Contingent Capital of up to 150,920 shares exclusively for the purpose of granting option rights to members of the management board and employees, respectively, of the company and its subsidiaries within the framework of the company’s stock option plan. The Contingent Capital shall be implemented to the extent that the option rights are exercised by the grantees, and the company does not use own shares to fulfill the option rights.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Rights attached to various classes of preferred shares converted to ordinary shares in the prior year
Series E Preferred Shares
The series E preferred shares were entitled to vote.
The series E preferred shares had a dividend preference pursuant to Section 32 of the company’s articles of association, under which they were entitled to receive an annual dividend in the amount of 10% of the stated value of €112.52 per share, out of profit in that year. If the profit for the year was not sufficient, the dividends for each year accrue and were payable, in whole or in part, in the first year that the profit is sufficient.
The series E preferred shares had a right of withdrawal and redemption compensation (the “Series E Redemption”) pursuant to Sections 30 and 31 of the company’s articles of association, under which the series E preferred shares were entitled to terminate their shares and withdraw from the company in exchange for redemption compensation of the stated value per share. The Series E Redemption was senior in preference to the Series D Liquidation Preference and the withdrawal and redemption compensation rights of the series C, A and B preferred shares. The Series E Redemption right could be exercised after June 30, 2018 with six months’ notice, or without a notice period upon (i) a sale or change of control of the company, or (ii) an initial public offering of the company.
The notarial deed executed in connection with the issue of the series E preferred share issue provided an anti-dilution right such that, in the case of an IPO Premium Requirement, the shareholders had agreed to approve a further capital increase in the course of which the holders of series E preferred shares, or ordinary shares issued on the conversion thereof, would be issued new ordinary shares in Innocoll AG at notional value at €1.00 per share in an amount such that the weighted average price per share of the series E preferred shares and the newly issued ordinary shares satisfied the IPO Premium Requirement.
Series D Preferred Shares
The series D preferred shares were entitled to vote.
The series D preferred shares had a dividend preference pursuant to Section 30 of the Articles of Association of the company (the “Articles”), under which they were entitled to receive an annual dividend in the amount of 10% of the stated value of €100 per share, out of profit in that year. If the profit for the year was not sufficient, the dividends for each year accrue and were payable, in whole or in part, in the first year that the profit is sufficient.
The series D preferred shares had a liquidation preference (the “Series D Liquidation Preference”) pursuant to Sections 28 and 31 of the Articles, under which the D preferred shares were entitled to payment of three times stated value plus any accrued but unpaid dividends, in preference to the withdrawal and redemption compensation rights of the series C, A and B preferred shares. The Series D Liquidation Preference was payable on the liquidation of the company or the earlier of (i) change of control of the company, (ii) an initial public offering of the company, or (iii) June 30, 2018. The series D preferred shares were not entitled to a right of withdrawal and redemption compensation.
Series C Preferred Shares
The series C preferred shares were entitled to vote.
The series C preferred shares had a dividend preference pursuant to Section 30 of the Articles, under which they were entitled to receive an annual dividend in the amount of 10% of the stated value of €100 per share, after payment of all outstanding dividends on the series D preferred shares. The dividends were payable out of the profit in that year. If the profit for the year was not sufficient, the dividends for each year accrue and were payable, in whole or in part, in the first year that the profit is sufficient.
The series C preferred shares had a right of withdrawal and redemption compensation (the “Series C Redemption”) pursuant to Sections 28 and 29 of the Articles, under which the series C preferred shares were entitled to terminate their shares and withdraw from the company in exchange for redemption compensation of the stated value per share. The Series C Redemption right was exercisable at any time after the holders of the series D preferred shares had received payment of the Series D Liquidation Preference in full.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Series A Preferred Shares
The series A preferred shares were entitled to vote.
The series A preferred shares had a dividend preference pursuant to Section 30 of the Articles, under which they were entitled to receive an annual dividend in the amount of 6% of the stated value of €100 per share, after payment of all outstanding dividends on the series D and series C preferred shares. The dividends were payable out of the profit in that year. If the profit for the year was not sufficient, the dividends for each year accrue and were payable, in whole or in part, in the first year that the profit is sufficient.
The series A preferred shares had a right of withdrawal and redemption compensation (the “Series A Redemption”) pursuant to Sections 28 and 29 of the Articles, under which the series A preferred shares were entitled to terminate their shares and withdraw from the company in exchange for redemption compensation of the stated value per share. The Series A Redemption right was exercisable at any time after the holders of the series D preferred shares had received payment of the Series D Liquidation Preference and the holders of the series C preferred shares had received payment of the Series C Redemption in full.
Series B Preferred Shares
The series B preferred shares were entitled to vote.
In addition, the holders of the series B preferred shares by resolution of a simple majority of the series B preferred shares were entitled to appoint a member to the advisory board of the company.
The series B preferred shares had a dividend preference pursuant to Section 30 of the Articles, under which they were entitled to receive an annual dividend in the amount of 6% of the stated value of €100 per share, after payment of all outstanding dividends on the series D, series C and series A preferred shares. The dividends were payable out of the profit in that year. If the profit for the year was not sufficient, the dividends for each year accrue and were payable, in whole or in part, in the first year that the profit is sufficient.
The series B preferred shares had a right of withdrawal and redemption compensation (the “Series B Redemption”) pursuant to Sections 28 and 29 of the Articles, under which the series B preferred shares were entitled to terminate their shares and withdraw from the company in exchange for redemption compensation of 3.327 times the stated value. The Series B Redemption right was exercisable at any time after the holders of the series D preferred shares had received payment of the Series D Liquidation Preference and the holders of the series C and series A preferred shares had received payment of the Series C Redemption and Series A Redemption in full.
Summary of line items contained in reserves
| Thousands of Euros | | 12/31/2015 | | | 12/31/2014 | |
| Share capital and reserves | | | | | | | | |
| Share capital | | | 1,785 | | | | 1,503 | |
| Share premium | | | 137,292 | | | | 122,084 | |
| Capital contribution | | | 723 | | | | 723 | |
| Other reserves | | | 12,415 | | | | 12,415 | |
| Currency translation reserve | | | (1,142 | ) | | | (622 | ) |
| Share compensation reserve | | | 9,924 | | | | 5,149 | |
| Accumulated deficit | | | (149,867 | ) | | | (106,718 | ) |
| | | | 11,130 | | | | 34,534 | |
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Share premium
Share premium reflects the excess of consideration received, net of issue costs, over par value of shares issued.
Capital contribution reserve
The capital contribution reserve relates to amounts contributed by shareholders of the company in previous years.
Share compensation reserve
Share compensation reserve reflects the fair value of stock based compensation issued in accordance with IFRS 2.
Other reserves
Other reserves relate to amounts recognized in equity on issuance of preferred stock and amounts in relation to the 2013 re-domicile.
Currency translation reserve
Translation reserve comprises all foreign exchange differences arising from the translation of the financial statements of foreign operations.
| 17 | Financial instruments and risk management |
The group is exposed to various risks in relation to financial instruments including credit risk, liquidity risk and currency risk. The group’s risk management is coordinated by its managing directors. The group does not actively engage in the trading of financial assets for speculative purposes.
The most significant financial risks to which the group is exposed are described below:
Credit risk
The group’s sales are currently concentrated with two customers and accordingly the group is exposed to the possibility of loss arising from customer default. The group is addressing this risk by monitoring our commercial relationships with these customers by seeking to develop additional products for sale and entering into new partnerships.
Liquidity risk
The group’s operations are not cash generating, however the group has excess short term cash resources from the initial public offering in July 2014, a follow-on offering in April 2015, the drawdown of €15 million from the European Investment Bank and a further €10 million available from the European Investment Bank. Short term flexibility is achieved through the management of the group’s short term deposits.
Market risk
The group is exposed to market risk through its use of financial instruments and specifically to currency risk, interest rate risk and certain other price risks, which result from both its operations and financial activities.
— Currency risk: The group is subject to currency risk, as its income and expenditures are denominated in euro and US dollar. As such the group is exposed to exchange rate fluctuations between the US dollar and the euro. The group aims to match foreign currency cash inflows with foreign cash outflows where possible. The group does not hedge this exposure. A 10% movement in the US dollar versus euro exchange rate at the year end would have the effect of increasing/decreasing net liabilities by approximately €1.5 million (2014: €4.0 million).
— Interest rate risk: The group has third-party indebtedness of €15 million in 2015 (2014: €Nil) with a fixed interest rate of 12% per annum. See note 14 and below for further details.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Terms of debt and repayment schedule
The terms and conditions of outstanding loans were as follows:
| | | | | | | | | | | 12/31/2015 | | | 12/31/2014 | |
| | | Currency | | Nominal
interest
rate | | | Year of
maturity | | | Face
value | | | Liability
carrying
amount | | | Face
value | | | Liability
carrying
amount | |
| Interest bearing loans | | Euro (€) | | | 12 | % | | | 2020 | | | | 15,000 | | | | 15,064 | | | | — | | | | — | |
| | | | | | | | | | | | | | 15,000 | | | | 15,064 | | | | — | | | | — | |
Credit risk
Exposure to credit risk:
The carrying amount of financial assets represents the maximum credit exposure. The maximum exposure to credit risk at the reporting date was:
| | | Carrying amount | |
| Thousands of Euros | | 12/31/2015 | | | 12/31/2014 | |
| Trade and other receivables | | | 4,192 | | | | 761 | |
| Cash and cash equivalents | | | 38,749 | | | | 45,616 | |
| | | | 42,941 | | | | 46,377 | |
The maximum exposure to credit risk for trade receivables at the reporting date by geographic region was:
| | | Carrying amount | |
| Thousands of Euros | | 12/31/2015 | | | 12/31/2014 | |
| Euro-zone countries | | | 1,122 | | | | 702 | |
| Middle East | | | — | | | | 59 | |
| United States | | | 2,888 | | | | — | |
| Australia | | | 107 | | | | — | |
| Asia | | | 75 | | | | — | |
| | | | 4,192 | | | | 761 | |
Impairment losses
The aging of trade receivables and other assets at the reporting date was:
| Thousands of Euros | | Gross
12/31/2015 | | | Impairment
12/31/2015 | | | Gross
12/31/2014 | | | Impairment
12/31/2014 | |
| Not past due | | | 4,192 | | | | — | | | | 761 | | | | — | |
| Past due 0 – 30 days | | | — | | | | — | | | | — | | | | — | |
| Past due 31 –120 days | | | — | | | | — | | | | — | | | | — | |
| Past due 121 – 365 days | | | — | | | | — | | | | — | | | | — | |
| More than one year | | | 39 | | | | 39 | | | | 39 | | | | 39 | |
| Total | | | 4,231 | | | | 39 | | | | 800 | | | | 39 | |
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
The movement in the allowance for impairment in respect of trade receivables during the year was as follows:
| Thousands of Euros | | 12/31/2015 | | | 12/31/2014 | |
| Balance at January 1, | | | 39 | | | | 74 | |
| Decrease in provision | | | — | | | | (35 | ) |
| Balance at December 31, | | | 39 | | | | 39 | |
Based on past experience, management believes that no impairment allowance is necessary in respect of trade receivables not past due and past due 0 – 30 days.
Liquidity risk
The following are the contractual maturities of financial liabilities, including interest payments and excluding the impact of netting agreements:
| Thousands of Euros | | Carrying
Amount | | | Contractual
cash flow | | | 1 year
or less | | | 1 – 2
years | | | 2 – 5
years | | | More
than
5 years | |
| December 31, 2014 | | | | | | | | | | | | | | | | | | | | | | | | |
| Trade and other payables | | | 5,055 | | | | (5,055 | ) | | | (5,055 | ) | | | — | | | | — | | | | — | |
| Defined pension liability | | | 61 | | | | (61 | ) | | | — | | | | — | | | | (61 | ) | | | — | |
| | | | 5,116 | | | | (5,116 | ) | | | (5,055 | ) | | | — | | | | (61 | ) | | | — | |
| December 31, 2015 | | | | | | | | | | | | | | | | | | | | | | | | |
| Interest bearing loans & borrowings | | | 15,064 | | | | (26,435 | ) | | | — | | | | — | | | | (26,435 | ) | | | — | |
| Trade and other payables | | | 13,237 | | | | (13,237 | ) | | | (13,237 | ) | | | — | | | | — | | | | — | |
| Defined pension liability | | | 45 | | | | (45 | ) | | | — | | | | (45 | ) | | | — | | | | — | |
| | | | 28,346 | | | | (39,717 | ) | | | (13,237 | ) | | | (45 | ) | | | (26,435 | ) | | | — | |
| 18 | Financial instruments measured at fair value through profit and loss |
Financial liabilities measured at fair value in the statement of financial position are grouped into three levels of fair value hierarchy. This grouping is determined based on the lowest level of significant inputs used in fair value measurement, as follows:
—Level 1 — quoted prices in active markets for identical assets or liabilities. No such Level 1 financial instruments were held.
— Level 2 — inputs other than quoted prices included within Level 1 that are observable for the instrument, either directly (i.e. as prices) or indirectly (i.e., derived from prices). No such Level 2 financial instruments were held.
— Level 3 — inputs for instrument that are not based on observable market data (unobservable inputs). The value of derivative financial instruments at fair value measurement using significant unobservable inputs (i.e. level 3) was €10.6 million (2014: €7.2 million).
There have been no transfers between Level 1, Level 2 and Level 3 during the period.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
At the balance sheet date the group had unprovided contractual capital commitments of €5.1 million (2014: €0). The €5.1 million is committed for the expansion of the group’s German manufacturing facility.
Operating leases
The group incurred operating lease expenses for the year ended December 31, 2015 of €0.4 million (2014: €0.3 million) which are included in general and administrative expenses. At the balance sheet date, the group had outstanding commitments for future minimum rent payments, which fall due as follows:
| Thousands of Euros | | 12/31/2015 | | | 12/31/2014 | |
| Less than one year | | | 437 | | | | 239 | |
| Between one and five years | | | 1,029 | | | | 714 | |
| More than five years | | | 178 | | | | — | |
| Total operating lease commitments | | | 1,644 | | | | 953 | |
The number and weighted average exercise price of share options outstanding at December 31, 2014 and 2015 is as follows:
| | | 2015 | | | 2014 | |
| | | Number of
Options | | | Weighted
average
exercise
price | | | Number of
Options | | | Weighted
average
exercise
price | |
| Outstanding at January 1 | | | 162,037 | | | € | 15.73 | | | | — | | | | — | |
| Granted | | | 145,081 | | | € | 93.85 | | | | 189,458 | | | € | 13.60 | |
| Exercised during the year | | | (122,530 | ) | | € | (1.00 | ) | | | (27,421 | ) | | € | (1.00 | ) |
| Forfeited during the year | | | (21,176 | ) | | | 102.04 | | | | — | | | | — | |
| Outstanding at December 31 | | | 163,412 | | | € | 84.94 | | | | 162,037 | | | € | 15.73 | |
| Exercisable at December 31 | | | 24,784 | | | € | 97.28 | | | | 24,784 | | | € | 97.28 | |
Innocoll GmbH 2014 Restricted Stock Plan
Pursuant to a notarial deed entered into on January 28, 2014, as amended on March 20, 2014, the company adopted the Innocoll GmbH 2014 Restricted Stock Plan. Under the Plan, the company granted to members of its supervisory board, management board and group employees a total of 47,840 restricted shares (“Restricted Shares”) and 63,256 phantom shares (“Phantom Shares”). The Restricted Share and Phantom Share grants were split among the company’s ordinary shares,series A, series B, series C, and series D preferred shares, and a newly created class Series D-2 preferred shares.
Each of the Restricted Shares and the Phantom Shares are subject to a right of repurchase by the company at nominal value in the case of a Bad Leaver Event prior to an Exit Event. A Bad Leaver Event is defined as the grantee’s termination of affiliation with the company under certain circumstances. An Exit Event is defined as the earlier of the 183rd day after the company successfully completes an IPO, or a liquidity event. A liquidity event occurs in the event the company merge or consolidate into or with another entity or vice versa (subject to certain limited exceptions), of the sale, conveyance, mortgage, pledge or lease of all or substantially all the company’s assets, or of the disposition of securities representing a majority of the company’s voting power through a transaction or series of related transactions. The Restricted Shares were issued as of January 28, 2014 in exchange for payment of nominal value, of €47,840. The Phantom Shares may be either settled in cash or a new issue of shares, at the company’s option, on the date in which the repurchase right lapses.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
For purposes of valuing the share-based payment in relation to the Restricted Share and Phantom Share grants, management performed the valuation with the assistance of a well-recognized independent third-party valuation consultant, which valued all classes of shares fully diluted for the issue of the Restricted Shares and Phantom Shares and options, on a pro-forma basis. The share-based payments are recognized over a one-year period, on the basis that as of the grant date the company estimated that the period in which the vesting conditions are to be satisfied is likely to be the Exit Event defined as the 183rd day after it completes its IPO.
Pursuant to a notarial deed entered into on May 22, 2014, Innocoll GmbH granted to new and existing members of its supervisory board, management board and group employees a total of 44,465 Restricted Shares and 9,113 Phantom Shares.
Each of the Restricted Shares and the Phantom Shares are subject to a right of repurchase by the company at nominal value in the case of a Bad Leaver Event prior to an Exit Event. A Bad Leaver Event is defined as the grantee’s termination of affiliation with the company under certain unfavourable circumstances. However, if no Bad Leaver event occurs before the occurrence of the earlier of:
(i) in the case of members of our management board and certain members of our advisory board
(a) the 183rd day after our initial public offering, and
(b) a so-called liquidity event, or
(ii) in the case of certain members of our supervisory board:
(a) upon a so called liquidity event or
(b) in the case of an initial public offering, in relation to 33.3% and 66.7% and 100% of the shares subject to the grant, on the date which is one, two and three years after the grant date respectively, or in each case the 183rd day after the completion of an initial public offering, whichever is later, our repurchase right terminates and the ordinary shares or phantom shares held by the grantee are no longer subject to any restrictions.
A liquidity event occurs in the event we merge or consolidate into or with another entity or vice versa (subject to certain limited exceptions), of the sale, conveyance, mortgage, pledge or lease of all or substantially all our assets, or of the disposition of securities representing a majority of our voting power through a transaction or series of related transactions. The Restricted Shares were issued in exchange for payment of nominal value of €43,596. The Phantom Shares may be either settled in cash or a new issue of shares, at the company’s option, on the date in which the repurchase right lapses.
The May 2014 Grant occurred simultaneously with a new issue of our series E preferred shares. The series E preferred shares were purchased in an arms length transaction by existing investors, new supervisory board members who also received restricted stock grants, and three new investors who are partners of one of the existing shareholders. The pricing of the series E preferred shares at €112.52 per share was based on third party advice obtained by the company’s advisory board and managing directors that the price of the company’s ADS in an anticipated initial public offering would be at least equal to such price or higher. To obtain the series E financing, it was necessary to give the investors anti-dilution rights in the event the initial public offering would be priced at a discount to the expected valuation. As a result of the initial public offering price of the company’s ADS of $9.00 per ADS (which equates to €82.52 per share), and the anti-dilution rights granted to the series E preferred investors, such investors have been issued additional shares at €1.00 per share, as a result of which their weighted average purchase price is equal to €73.76 per share, a 16.7% discount to the effective value per ordinary share in our initial public offering. There were no significant valuation events at the company between the May 2014 Grant date and date of our initial public offering in July 2014. Therefore we have used the E preferred share adjusted weighted average purchase price of €73.76 per share as the value for the share-based payment charge arising out of the May 2014 Grant. The share-based payments will be recognized over a one to three year period as set out above.
In July 2014, after the transformation of Innocoll GmbH into Innocoll AG, each of the Restricted Shares and Phantom Shares awards issued in January 2014 and May 2014 were combined and applied to ordinary shares of Innocoll AG pursuant to amended and restated award agreements entered into with Innocoll AG. In January 2015, the Phantom Share award agreements were each amended and restated to provide for the definition of Exit Event triggering the lapse of the repurchase right by the company to be extended to any trading day within the period beginning on the third trading day after the publication of the quarterly reports of the company for the fourth quarter 2014, and the ending on the trading day immediately preceding the first trading day that is two weeks prior to the end of the first quarter 2015, which may be designated by the company’s management board in its free discredtion.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Management Option Agreements
In December 2014, the company entered into option agreements with members of its management board and group employees. Pursuant to the terms of the 2014 Management Option Agreements, the members of the management board and group employees have the right to subscribe for 24,784 ordinary shares at an exercise price of $119.25 per ordinary share (equivalent to $9.00 per ADS), which rights are exercisable through June 15, 2019, subject to certain black-out periods.
Throughout 2015, the company entered into option agreements with members of its management board and group employees. Pursuant to the terms of the 2014 Management Option Agreements, the members of the management board and group employees have the right to subscribe for 86,144 ordinary shares at exercise prices ranging from $99.38 to $195.70 per ordinary share (equivalent to from $7.50 to $14.77 per ADS), which rights are exercisable for a ten year period from date of grant, subject to certain black-out periods.
The share based payments cost of the options was calculated using a Black Scholes model. The following input assumptions were used to value the options:
| Weighted average values: | | 12/31/2015 | | | 12/31/2014 | |
| Expected volatility | | | 66.75 | % | | | 69.68 | % |
| Risk free rate | | | 2.03 | % | | | 1.66 | % |
| Exercise price | | € | 124.67 | | | € | 97.283 | |
| Contractual life | | | 8.78 years | | | | 4.52 years | |
Share based payment charge
The charge to the statement of comprehensive income in 2015 in relation to share based payments was €4.8million (2014: €5.1 million).
Identity of related parties
The group has related party relationships with its directors, executive officers and shareholders.
In June 2013, Innocoll Holdings, Inc. repurchased and redeemed restricted shares which had been purchased in December 2007, by certain employees and board members in exchange for promissory notes. Pursuant to the employee share repurchase, the company purchased an aggregate of 5,466,821 shares of common stock (24% of the shares of common stock then outstanding) in exchange for forgiving and writing off loans and accrued interest due in aggregated amount of €8.6 million. This amount primarily related to key management personnel as defined by IAS 24 “Related Party Disclosures”
Remuneration of key management personnel
The remuneration of executive officers and directors of the group is set out below. These amounts reflect the costs for the group. There were fifteen executive officers and directors at December 31, 2015 (2014: eleven, 2013: ten).
| Thousands of Euros | | 12/31/2015 | | | 12/31/2014 | | | 12/31/2013 | |
| Short-term employee benefits | | | 3,324 | | | | 1,695 | | | | 1,557 | |
| Post-employment benefits – defined contribution pension plans | | | 60 | | | | 34 | | | | 32 | |
| Share based payments | | | 4,341 | | | | 3,745 | | | | — | |
| Total key management costs | | | 7,725 | | | | 5,474 | | | | 1,589 | |
| | | 12/31/2015
Number | | | 12/31/2014
Number | | | 12/31/2013
Number | |
| Number of restricted stock units/share options outstanding at end of year | | | 163,412 | | | | 162,037 | | | | — | |
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Directors’ interests
| Shares held | | 2015
Common
Stock | | | 2015
Options | | | 2014
Common
Stock | | | 2014
Options | |
| Jonathon Symonds | | | 25,840 | | | | 9,165 | | | | 13,710 | | | | 13,748 | |
| Shumeet Banerji | | | 14,131 | | | | 9,165 | | | | 5,774 | | | | 13,748 | |
| David Brennan | | | 12,318 | | | | 5,602 | | | | 9,517 | | | | 8,403 | |
| Rolf Schmidt | | | 29,176 | | | | — | | | | 34,476 | | | | — | |
| James Culverwell | | | 17,875 | | | | 3,375 | | | | 13,133 | | | | 6,888 | |
| Anthony Zook | | | 14,683 | | | | 25,174 | | | | — | | | | — | |
| Pepe Carmona | | | — | | | | 20,980 | | | | — | | | | — | |
| Michael Myers | | | 29,876 | | | | 13,244 | | | | 2,654 | | | | 58,764 | |
| Gordon Dunn | | | 33,956 | | | | 7,173 | | | | 12,912 | | | | 25,551 | |
| Total stock held | | | 177,855 | | | | 93,878 | | | | 92,176 | | | | 127,102 | |
Defined Contribution Schemes
Certain employees of the group are eligible to participate in a defined contribution plan. Participants in the defined contribution plan may elect to defer a portion of their pre-tax earnings into a pension plan, which is administered by an independent party. The group matches each participant’s contributions typically at 5% of the participant’s annual compensation. Contributions to this plan are recorded, as a remuneration expense in the consolidated statement of comprehensive (loss)/income. Contributions for the years ended December 31, 2015 and 2014 were €0.08 million and €0.07 million respectively.
Defined benefit plan
One of the group’s subsidiaries, Syntacoll GmbH, operates a defined benefit pension plan in Germany for one of its former employee’s who is now deceased. Following the death of the plan’s only member in 2009, the amounts payable until September 30, 2017 were transferred to the member’s widow. The total amounts payable relate to the full amount of the deceased employee’s life annuity from Syntacoll GmbH. This plan is now closed to new members. The plan is managed externally and the related pension costs and liabilities are assessed in accordance with the advice of a professionally qualified actuary. Plan assets at December 31, 2015 and December 31, 2014 consisted of units held in independently administered funds. The most recent valuation of plan obligations was carried out as at December 31, 2015, using the projected unit credit method.
Interest rate risk
The present value of the defined benefit liability is calculated using a discount rate determined by reference to market yields of high quality corporate bonds. The estimated term of the bonds is consistent with the estimated term of the defined benefit obligation. A decrease in market yield on high quality corporate bonds will increase the group’s defined benefit liability, although it is expected that this would be offset partially by an increase in the fair value of certain of the plan assets.
Longevity risk
There is no longevity risk given that the member is deceased and there is a set end date.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
Financial assumptions
The following assumptions were used in determining the fair value of the plan assets and the present value of the projected benefit obligation at December 31, 2015:
| | | 12/31/2015 | | | 12/31/2014 | |
| Discount rate | | | 2.1 | % | | | 2.0 | % |
| Retirement age | | | 60 yrs | | | | 60 yrs | |
| Inflation rate | | | 1.5 | % | | | 1.5 | % |
| Future salary increases | | | 0 | % | | | 0 | % |
Consolidated Financial Statements
Movement in the net benefit obligation recognized in non-current other liabilities was as follows:
| Thousands of Euros | | 12/31/2015 | | | 12/31/2014 | |
| Projected benefit obligation at start of year | | | 147 | | | | 196 | |
| Current service cost | | | (54 | ) | | | (54 | ) |
| Finance cost | | | 3 | | | | 5 | |
| Projected benefit obligation at end of year | | | 96 | | | | 147 | |
| Fair value of plan assets at start of year | | | 86 | | | | 115 | |
| Distributions of assets | | | (35 | ) | | | (29 | ) |
| Fair value of plan assets at end of year | | | 51 | | | | 86 | |
| Net benefit obligation | | | (45 | ) | | | (61 | ) |
| Name of subsidiary | | Place of
incorporation
and operation | | Proportion
of
ownership
interest | | | Principal activity |
| Innocoll Inc. | | US | | | 100 | % | | Administration |
| Syntacoll GmbH | | Germany | | | *99 | % | | Manufacturing |
| Innocoll Pharmaceuticals Limited | | Ireland | | | 100 | % | | Selling & distribution |
| Innocoll Technologies Limited | | Ireland | | | 100 | % | | Dormant |
*The 1% non-controlling interest has no beneficial interest in the company. These shares are held in trust.
The registered office of Innocoll AG is Donaustr. 24, 93342 Saal, Germany and its main place of business is Unit 9, Block D, Monksland Business Park, Monksland, Athlone, Co. Roscommon, Ireland.
INNOCOLL AG
Notes to the consolidated financial information — (Continued)
| 24 | Post balance sheet events |
On March 16, 2016, following approval in the Innocoll AG extraordinary meeting on January 30, 2016, registration with the German Commercial Register on March 8, 2016 and approval by the Irish High Court on March 14, 2016, Innocoll Holdings plc (“Innocoll Ireland”) will become the parent company of the Innocoll group of companies as a result of Innocoll AG (“Innocoll Germany”) merging (the “merger”) with and into Innocoll Ireland. The change in place of incorporation of the parent company of the Innocoll group will be effected pursuant to the common draft terms of a cross-border merger proposal dated December 20, 2015 between Innocoll Germany and Innocoll Ireland (the “Merger Proposal”). At the effective time of the Merger and pursuant to the Merger Proposal (i) Innocoll Germany will be merged into Innocoll Ireland, with Innocoll Germany being the disappearing entity and Innocoll Ireland being the surviving entity in a merger by acquisition and (ii) each shareholder of Innocoll Germany will be issued, as consideration in the Merger, 13.25 Innocoll Ireland ordinary shares, par value $0.01 per share (the “Innocoll Ireland Ordinary Shares”), for each ordinary share of Innocoll Germany held by it immediately prior to the Merger, (iii) the Innocoll Germany American Depositary Share (“ADS”) facility will be terminated and (iv) each outstanding ADS, which formerly represented 1/13.25 of an ordinary share of Innocoll Germany, will be cancelled and will represent the right to receive one Innocoll Ireland Ordinary Share.
Effective January 1, 2016 the Group began preparing its financial information in accordance with US GAAP.