Use these links to rapidly review the document
TABLE OF CONTENTS
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 20-F
| | |
o |
|
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR |
ý |
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2018 |
OR |
o |
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to |
OR |
o |
|
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Date of event requiring this shell company report:
Commission File number: 001-38178
ZEALAND PHARMA A/S
(Exact name of Registrant as specified in its charter)
Not Applicable
(Translation of Registrant's name into English)
The Kingdom of Denmark
(Jurisdiction of incorporation or organization)
Smedeland 36
2600 Glostrup (Copenhagen)
Denmark
(Address of principal executive offices)
Mats Blom
Executive Vice President and Chief Financial Officer
Zealand Pharma A/S
Smedeland 36
2600 Glostrup (Copenhagen)
Denmark
Tel: +45 88 77 36 00
Fax: +45 88 77 38 98
(Name, telephone, e-mail and/or facsimile number and address of Company contact person)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
| | |
| Title of each class | | Name of each exchange on which registered |
|---|
| Ordinary Shares | | Nasdaq Copenhagen A/S |
American Depositary Shares, each representing one Ordinary Share |
|
The NASDAQ Global Select Market |
Securities registered or to be registered pursuant to Section 12(g) of the Act.None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act.None
Indicate the number of outstanding shares of each of the issuer's classes of capital or common stock as of the close of the period covered by the annual report.
30,786,827 Ordinary Shares
3,132,086 American Depositary Shares
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ý No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of accelerated filer and large accelerated filer in Rule 12b of the Exchange Act. (Check one):
| | | | | | |
| Large accelerated filer o | | Accelerated filer o | | Non-accelerated filer ý | | Emerging growth company ý |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act. o
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
| | | | |
| o US GAAP | | ý International Financial Reporting Standards as issued by the International Accounting Standards Board | | o Other |
If "Other" has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow o Item 17 o Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No ý
- †
- The term "new or revised financial accounting standard" refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Table of Contents
TABLE OF CONTENTS
| | | | |
INTRODUCTION | | | 3 | |
Part I | | | | |
ITEM 1 IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISORS | | | 6 | |
ITEM 2 OFFER STATISTICS AND EXPECTED TIMETABLE | | | 6 | |
ITEM 3 KEY INFORMATION | | | 6 | |
ITEM 4 INFORMATION ON THE COMPANY | | | 40 | |
ITEM 4A UNRESOLVED STAFF COMMENTS | | | 53 | |
ITEM 5 OPERATING AND FINANCIAL REVIEW AND PROSPECTS | | | 54 | |
ITEM 6 DIRECTORS, EXECUTIVE MANAGEMENT AND EMPLOYEES | | | 60 | |
ITEM 7 MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS | | | 62 | |
ITEM 8 FINANCIAL INFORMATION | | | 63 | |
ITEM 9 THE OFFER AND LISTING | | | 64 | |
ITEM 10 ADDITIONAL INFORMATION | | | 65 | |
ITEM 11 QUALITATIVE AND QUANTITATIVE DISCLOSURES ABOUT MARKET RISKS | | | 73 | |
ITEM 12 DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES | | | 74 | |
ITEM 12A DEBT SECURITIES | | | 74 | |
ITEM 12B WARRANTS AND RIGHTS | | | 74 | |
ITEM 12C OTHER SECURITIES | | | 74 | |
ITEM 12D AMERICAN DEPOSITARY SHARES | | | 74 | |
Part II | | | | |
ITEM 13 DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES | | | 75 | |
ITEM 14 MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS | | | 76 | |
ITEM 15 CONTROLS AND PROCEDURES | | | 77 | |
ITEM 16A AUDIT COMMITTEE FINANCIAL EXPERTS | | | 80 | |
ITEM 16B CODE OF ETHICS | | | 80 | |
ITEM 16C PRINCIPAL ACCOUNTANT FEES AND SERVICES | | | 80 | |
ITEM 16D EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES | | | 81 | |
ITEM 16E PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS | | | 81 | |
ITEM 16F CHANGE IN REGISTRANT'S CERTIFYING ACCOUNTANT | | | 81 | |
ITEM 16G CORPORATE GOVERNANCE | | | 81 | |
ITEM 16H MINE SAFETY DISCLOSURE | | | 82 | |
Part III | | | | |
ITEM 17 FINANCIAL STATEMENTS | | | 83 | |
ITEM 18 FINANCIAL STATEMENTS | | | 84 | |
ITEM 19 EXHIBITS | | | 85 | |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM | | | 89 | |
SIGNATURES | | | 88 | |
2
Table of Contents
INTRODUCTION
In this Annual Report on Form 20-F the terms the "Company", "Zealand Pharma", "Zealand" and the "Group" refer to the parent company Zealand Pharma A/S together with its consolidated subsidiaries. The term "Zealand Pharma A/S" is used when addressing issues specifically related to this legal entity.
Pursuant to Rule 12b-23 of the Securities Exchange Act of 1934, as amended (the"Exchange Act"), we incorporate information for certain items of this Annual Report on Form 20-F by reference to certain pages of the Zealand Pharma A/S statutory Annual Report 2018 (the"Annual Report 2018"), included as Exhibit 99.1 (a) to Form 6-K furnished to the SEC on March 8, 2019. Therefore, the information in this Annual Report on Form 20-F should be read in conjunction with the Annual Report 2018, as furnished to the SEC on March 8, 2019. Items not contained or not specifically referenced to within the Annual Report 2018 should not be deemed to be part of this Annual Report on Form 20-F.
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 20-F contains forward-looking statements concerning our business, operations and financial performance and condition, as well as our plans, objectives and expectations for our business operations and financial performance and condition. Any statements contained herein that are not statements of historical facts may be deemed to be forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as "aim," "anticipate," "assume," "believe," "contemplate," "continue," "could," "due," "estimate," "expect," "goal," "intend," "may," "objective," "plan," "predict," "potential," "positioned," "seek," "should," "target," "will," "would," and other similar expressions that are predictions of or indicate future events and future trends, or the negative of these terms or other comparable terminology.
These forward-looking statements include, but are not limited to, statements about:
- •
- our expectations regarding the potential market size and the size of the patient populations for our product candidates, if approved for commercial use;
- •
- our expectations regarding the potential advantages of our product candidates over existing therapies;
- •
- our development plans with respect to our product candidates;
- •
- our ability to develop, acquire and advance product candidates into, and successfully complete, clinical trials;
- •
- the initiation, timing, progress and results of our preclinical studies and clinical trials, and our research and development programs;
- •
- the timing or likelihood of regulatory filings and approvals for our product candidates;
- •
- the implementation of our business model and strategic plans for our business, product candidates and technology; and
- •
- estimates of our expenses, future revenue, capital requirements, our needs for additional financing and our ability to obtain additional capital.
The forward-looking statements contained herein involve a number of known and unknown risks and uncertainties that could cause our future results, performance or achievements to differ significantly from the results, performance or achievements expressed or implied by such forward-looking statements.
3
Table of Contents
You should understand that many important factors, in addition to those discussed or incorporated by reference in this report, could cause our results to differ materially from those expressed in the forward-looking statements. Potential factors that could affect our results include, in addition to others not described in this report, those described under "Item 3-Key Information—D. Risk Factors." These are factors that we think could cause our actual results to differ materially from expected results.
Forward looking statements speak only as of the date on which they are made, and we undertake no obligation to update any forward-looking statements or other information contained in this report, whether as a result of new information, future events or otherwise. You are advised, however, to consult any additional disclosures we make in our reports on Form 6-K filed with the U.S. Securities and Exchange Commission (the "SEC"). Please also see the cautionary discussion of risks and uncertainties under "Item 3—Key Information—D. Risk Factors". This discussion is provided as permitted by the Private Securities Litigation Reform Act of 1995.
PRESENTATION OF FINANCIAL AND OTHER INFORMATION
The consolidated financial statements contained in this Annual Report on Form 20-F have been prepared in accordance with International Financial Reporting Standards ("IFRS"), as issued by the International Accounting Standards Board ("IASB").
We maintain our books and records in Danish kroner (DKK) and report under IFRS as issued by the IASB. Our consolidated financial statements are not prepared in accordance with accounting principles generally accepted in the United States. Except with respect to U.S. dollar amounts presented as contractual terms or as otherwise indicated, all amounts that are presented in U.S. dollars herein have been translated from DKK solely for convenience at an assumed exchange rate of DKK 6.21 per $1.00, which was the rounded official exchange rate as of December 31, 2018, as reported by Danmarks Nationalbank. We use the symbol "$" to refer to U.S. Dollars and the symbol "€" to refer to Euros herein.
This Annual Report on Form 20-F includes trademarks, tradenames and service marks, certain of which belong to us and others that are the property of other organizations. Solely for convenience, trademarks and tradenames referred to in this prospectus appear without the ® and ™ symbols, but the absence of those references is not intended to indicate, in any way, that we will not assert our rights or that the applicable owner will not assert its rights to these trademarks and tradenames to the fullest extent under applicable law.
Unless otherwise indicated, information contained in this Annual Report on Form 20-F concerning our industry and the markets in which we operate, including our general expectations, market position, market opportunity and market share, is based on information from our own management estimates and research, as well as from industry and general publications, research, surveys and studies conducted by third parties. Management estimates are derived from publicly available information, our knowledge of our industry and assumptions based on such information and knowledge, which we believe to be reasonable. In addition, assumptions and estimates of our and our industry's future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in "Item 3—Key Information—D. Risk Factors". These and other factors could cause our future performance to differ materially from our assumptions and estimates. See "Forward-Looking Statements" above.
Enforceability of civil liabilities
The Company is a Danish corporation and a majority of its directors and officers, as well as certain experts named herein, are non-residents of the United States. A substantial portion of the assets of Zealand Pharma A/S, its subsidiaries and such persons are located outside the United States. As a result, it may be difficult for shareholders of the Company to effect service within the United
4
Table of Contents
States upon directors, officers and experts who are not residents of the United States or to enforce judgments in the United States. In addition, there can be no assurance as to the enforceability in Denmark against the Company or its respective directors, officers and experts who are not residents of the United States, or in actions for enforcement of judgments of United States courts, of liabilities predicated solely upon the federal securities laws of the United States.
5
Table of Contents
PART I
ITEM 1 IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISORS
Not applicable.
ITEM 2 OFFER STATISTICS AND EXPECTED TIMETABLE
Not applicable.
ITEM 3 KEY INFORMATION
A. SELECTED FINANCIAL DATA
The following tables present selected consolidated financial data for our business for the periods indicated. We derived the selected consolidated income statement and balance sheet data as of December 31, 2018 and 2017 and for the years ended December 31, 2018, 2017, and 2016 from our audited consolidated financial statements, incorporated by reference from our Annual Report 2018 on pages 51-87. We derived the selected consolidated income statement and balance sheet data as of December 31, 2016, 2015 and 2014 and for the years ended 2015 and 2014 from our audited consolidated financial statements not included or incorporated by reference herein with the exception for amounts that have been restated in accordance with footnote 1 below. You should read this data in conjunction with our consolidated financial statements and related notes appearing in pages 51 - 87 in our Annual Report 2018. Our historical results are not necessarily indicative of our future results.
The consolidated financial statements represents the consolidated financial statements of Zealand Pharma A/S and its subsidiaries.
Selected financial data
| | | | | | | | | | | | | | | | |
IFRS figure in DKK thousand, except
per share numbers and number of shares | | 2018 | | Restated(1)
2017 | | Restated(1)
2016 | | Restated(1)
2015 | | Restated(1)
2014 | |
|---|
Income statement data | | | | | | | | | | | | | | | | |
Revenue | | | 37,977 | | | 136,322 | | | 230,864 | | | 182,573 | | | 150,633 | |
Operating result | | | 652,390 | | | –249,371 | | | –119,032 | | | –85,742 | | | –76,253 | |
Result before tax | | | 625,056 | | | –280,758 | | | –162,796 | | | –124,247 | | | –75,206 | |
Net result for the year | | | 581,282 | | | –275,258 | | | –157,296 | | | –118,372 | | | –67,706 | |
Earnings per share data | | | | | | | | | | | | | | | | |
Earnings/loss per share—basic (DKK) | | | 18.94 | | | –9.88 | | | –6.47 | | | –5.13 | | | –2.99 | |
Earnings/loss per share—diluted (DKK) | | | 18.94 | | | –9.88 | | | –6.47 | | | –5.13 | | | –2.99 | |
Statement of financial position data | | | | | | | | | | | | | | | | |
Total assets | | | 1,229,797 | | | 721,285 | | | 683,116 | | | 627,621 | | | 593,273 | |
Equity (net assets) | | | 1,116,281 | | | 514,669 | | | 267,381 | | | 244,803 | | | 249,815 | |
Share capital | | | 30,787 | | | 30,751 | | | 26,142 | | | 24,353 | | | 23,193 | |
Treasury shares | | | 64 | | | 64 | | | 564 | | | 564 | | | 564 | |
Dividends per share | | | 0.00 | | | 0.00 | | | 0.00 | | | 0.00 | | | 0.00 | |
Number of shares | | | 30,786,827 | | | 30,751,327 | | | 26,142,365 | | | 24,352,769 | | | 23,193,047 | |
- (1)
- Reference is made to Note 1 on pages 55-63 and 'Consolidated Key Figures', page 11, and the 'Consolidated financial statements', pages 51-87 in our Annual Report 2018 for further data.
6
Table of Contents
Exchange rate information
The following table presents information on the exchange rates between the DKK and the U.S. Dollar for the periods indicated, as published by Danmarks Nationalbank.
| | | | | | | | | | | | | |
DKK per USD | | Monthly
average
rate | | Period
end rate | | High | | Low | |
|---|
2014 | | | 5.65 | | | 6.12 | | | 6.12 | | | 5.35 | |
2015 | | | 6.73 | | | 6.83 | | | 7.08 | | | 6.18 | |
2016 | | | 6.73 | | | 7.05 | | | 7.13 | | | 6.52 | |
2017 | | | 6.55 | | | 6.21 | | | 7.16 | | | 6.17 | |
2018 | | | 6.32 | | | 6.52 | | | 6.63 | | | 5.96 | |
Last 6 months | | |
| | |
| | |
| | |
| |
September 2018 | | | 6.40 | | | 6.44 | | | 6.45 | | | 6.33 | |
October 2018 | | | 6.50 | | | 6.59 | | | 6.59 | | | 6.43 | |
November 2018 | | | 6.56 | | | 6.57 | | | 6.63 | | | 6.49 | |
December 2018 | | | 6.56 | | | 6.52 | | | 6.61 | | | 6.52 | |
January 2019 | | | 6.54 | | | 6.50 | | | 6.58 | | | 6.47 | |
February 2019 | | | 6.57 | | | 6.54 | | | 6.63 | | | 6.51 | |
March 2019 (through March 7) | | | 6.59 | | | 6.62 | | | 6.62 | | | 6.55 | |
On March 7, 2019, the latest available date, Danmarks Nationalbank's daily official exchange rate was 6.62.
B. CAPITALIZATION AND INDEBTEDNESS
Not applicable.
C. REASONS FOR THE OFFER AND USE OF PROCEEDS
Not applicable.
D. RISK FACTORS
Risks Related to Our Business
We have incurred net losses in recent periods and may continue to do so.
Although we made a net profit of DKK 581.3 million in 2018, we recognized net losses of DKK 275.3 million in 2017 and DKK 157.3 million in 2016. Our net profit in 2018 was primarily as a result of the sale of future royalties and milestones from our Sanofi license agreement which may not necessarily be the case for future periods. These losses were primarily the result of our internal and external research expenditures and development costs for conducting preclinical studies and clinical trials in respect of our internal product portfolio. Our ability to generate revenue from our internal product portfolio depends on our ability to successfully develop and commercialize our product candidates and to obtain the regulatory and marketing approvals necessary to commercialize one or more of our product candidates.
Our ability and our collaboration partners' ability to generate future revenue from product sales or pursuant to milestone payments depend heavily on many factors, including, but not limited to:
- •
- completing research activities and preclinical and clinical development of our out-licensed and internal product candidates;
7
Table of Contents
- •
- on our own, or together with our strategic collaboration partners, obtaining regulatory approvals for our product candidates;
- •
- negotiating favorable terms of and entering into further collaboration, licensing or other arrangements;
- •
- the ability of our collaboration partners to successfully commercialize or our ability to commercialize or co-promote our product candidates;
- •
- obtaining market acceptance of our product candidates, if approved;
- •
- addressing any competing technological or market developments;
- •
- identifying, assessing, acquiring, in-licensing or developing new product candidates;
- •
- maintaining, protecting and expanding our portfolio of intellectual property rights, including patents, trade secrets and know-how, and our ability to develop, manufacture and commercialize our product candidates and products without infringing the intellectual property rights of others; and
- •
- attracting, hiring, and retaining qualified personnel.
In cases where we, or our collaboration partners, are successful in obtaining regulatory approvals to market one or more of our product candidates, our revenue will be dependent, in part, upon the size of the markets in the territories for which regulatory approval is granted, the price or prices at which we or our collaboration partners are able to sell such products and our ability to get paid or reimbursed for such products. If the number of individuals suitable for our product candidates is not as significant as we estimate, the indications approved by regulatory authorities are narrower than we expect, or the reasonably accepted population for treatment is narrowed by competition, physician choice or applicable guidelines, we may not generate significant revenue from the sale of such products, even if approved. Our failure to generate revenue from sales of one or more of our product candidates or pursuant to license or milestone payments or if the level of revenue generated therefrom is lower than our or the market's expectations, could have a material adverse effect on our business, financial position, results of operations and future growth prospects.
We expect our expenses to continue to increase and that we will continue to incur losses as we further develop our internal product portfolio. In particular, we anticipate that our expenses and losses will increase substantially if and as we:
- •
- continue the preclinical and clinical development of our internal product candidates;
- •
- expand the scope of or otherwise materially modify our current clinical trials for our internal product candidates;
- •
- begin new clinical trials for our internal product candidates;
- •
- develop our commercial manufacturing capabilities for our internal product candidates;
- •
- seek regulatory and marketing approvals for any internal product candidates that successfully complete clinical trials;
- •
- establish a sales, marketing and distribution infrastructure to commercialize any drugs for which we may obtain marketing approval and for which we have not entered into a commercialization collaboration with a third party;
- •
- seek to identify and validate additional product candidates;
- •
- acquire or in-license product candidates and technologies;
- •
- maintain, protect and expand our intellectual property portfolio;
8
Table of Contents
- •
- attract new and retain existing skilled personnel; and
- •
- create additional infrastructure to support our operations.
Any net losses we incur may fluctuate significantly from year to year, such that a year-to-year comparison of our results of operations may not be a good indication of our future performance. In any particular period or periods, our operating results could be below the expectations of securities analysts or investors, which could cause the price of our American Depository Shares, or ADSs, to decline.
The regulatory approval processes of the FDA, the EMA and other comparable regulatory authorities are lengthy, time consuming and inherently unpredictable, and if we or our collaboration partners are ultimately unable to obtain regulatory approval for our internal or out-licensed product candidates, our business could be substantially harmed.
The time required to obtain approval by the U.S. Food and Drug Administration, or the FDA, the European Medicines Agency, or the EMA and other comparable regulatory authorities is unpredictable, but typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate's clinical development and varies among jurisdictions. We have not obtained regulatory approval in the United States for any product candidate for which we retain full development, commercialization and marketing control, and it is possible that none of our existing product candidates or any product candidates that we may seek to develop in the future will ever obtain regulatory approval.
Our product candidates could fail to receive regulatory approval for many reasons, including, but not limited to, the following:
- •
- the FDA, the EMA or other comparable regulatory authorities may disagree with the design or implementation of our clinical trials;
- •
- we or our collaboration partners may be unable to demonstrate to the satisfaction of the FDA, the EMA or other comparable regulatory authorities that a product candidate is safe and effective for its proposed indications;
- •
- we or our collaboration partners may be unable to demonstrate that a product candidate's clinical and other benefits outweigh its safety risks;
- •
- the FDA, the EMA or other comparable regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials;
- •
- the data collected from clinical trials of our product candidates may not be sufficient to support the submission of a new drug application ("NDA"), supplemental NDA or other submission or to obtain regulatory approval in the United States, Europe or elsewhere;
- •
- the FDA, the EMA or any other comparable regulatory authority may fail to approve the labeled conditions for use that we or our collaboration partners propose for a product candidate;
- •
- the FDA, the EMA or other comparable regulatory authorities may fail to approve the manufacturing processes or facilities of any third party manufacturers with which we may contract for clinical and commercial supplies or such processes or facilities may not pass a preapproval inspection; and
- •
- the approval policies or regulations of the FDA, the EMA or other comparable regulatory authorities may change or differ significantly from one another in a manner rendering our clinical data insufficient for approval.
9
Table of Contents
This lengthy approval process, as well as the unpredictability of ongoing clinical trial results, may result in our or our collaboration partners' failure to obtain regulatory approval to market our product candidates, which would harm our business, financial position, results of operations and future growth prospects significantly. In addition, even if we or our collaboration partners were to obtain approval, regulatory authorities may approve any of our product candidates for fewer or more limited indications than requested, may grant approval contingent on the performance of costly post-marketing clinical trials or may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. In certain jurisdictions, regulatory authorities may not approve the price we or our collaboration partners intend to charge for our products. Any of the foregoing scenarios could materially harm the commercial prospects of our product candidates.
For certain marketed products, product candidates and clinical development programs, we depend on collaboration partners to develop and conduct clinical trials with, obtain regulatory approvals for, and market and sell our product candidates. If such collaboration partners fail to perform as expected, the potential for us to generate future revenue from such product candidates would be significantly reduced and our business would be significantly harmed.
For certain marketed products, product candidates and clinical development programs, we do, and may in the future continue to, rely on our collaboration partners to develop, conduct clinical trials of, and commercialize our product candidates and approved products. We have existing collaborations with Boehringer Ingelheim GmbH, or BI, and, with Beta Bionics Inc., or Beta Bionics, our Licensees. We may also enter into collaboration agreements with other parties in the future relating to product candidates. Ultimately, if such out-licensed product candidates are advanced through clinical trials and receive marketing approval from the EMA, the FDA or similar regulatory authorities, certain of our collaboration partners will be responsible for commercialization of these out-licensed products. The potential for us to obtain future development milestone payments and, ultimately, generate revenue from royalties on sales of such out-licensed products depends on the successful development, regulatory approval, marketing and commercialization by our collaboration partners. If our collaboration partners do not perform in the manner we expect or fail to fulfill their responsibilities in a timely manner or at all, if our agreements with them terminate or if the quality or accuracy of the clinical data they obtain is compromised, the clinical development, regulatory approval and commercialization efforts related to our out-licensed product candidates could be delayed or terminated, and it could become necessary for us to assume the responsibility at our own expense for the clinical development of such product candidates. In that event, we would likely be required to limit the size and scope of efforts for the development and commercialization of such product candidate; we would likely be required to seek additional financing to fund further development or identify alternative strategic collaboration partners; our potential to generate future revenue from royalties and milestone payments from such product candidates would be significantly reduced or delayed; and it could have a material adverse effect on our business, financial position, results of operations and future growth prospects.
Collaborations involving our out-licensed product candidates pose a number of risks, including the following:
- •
- collaboration partners have significant discretion in determining the efforts and resources that they will apply to these partnerships;
- •
- collaboration partners may not perform their obligations as expected;
- •
- collaboration partners may not pursue development and commercialization of our out-licensed product candidates or may elect not to continue or renew development or commercialization programs, based on clinical trial results, changes in the collaboration partners' strategic focus or available funding or external factors, such as an acquisition, that divert resources or create competing priorities;
10
Table of Contents
- •
- collaboration partners may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing;
- •
- collaboration partners may have or could independently develop, or develop with third parties, products that compete directly or indirectly with our out-licensed product candidates;
- •
- disagreements with collaboration partners, including disagreements over proprietary rights, contract interpretation or the conduct of product research, development or commercialization programs, may cause delays or lead to termination of such programs, or require us to assume unplanned expenditures, responsibilities or liabilities with respect to product candidates we have out licensed, or may result in costly and time consuming litigation or arbitration;
- •
- collaboration partners may infringe the intellectual property rights of third parties, which may result in costly and time consuming litigation or arbitration in which we may be involved, as a party or in support of our collaboration partners;
- •
- collaboration partners with marketing and distribution rights to one or more products may not commit sufficient resources to the marketing and distribution of such product or products;
- •
- collaboration partners with marketing and distribution rights may incur costs that have the effect of reducing the base on which royalties are calculated;
- •
- collaboration partners may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; and
- •
- collaboration agreements may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates.
In addition, certain collaboration agreements provide our collaboration partners with rights to terminate such agreements and licenses granted under such agreements under various conditions, which, if exercised, would adversely affect our product development efforts, could make it difficult for us to attract new collaboration partners and may adversely affect our reputation. Our collaboration partners may have the right to terminate their respective collaboration agreements with us. Any such termination of any agreement or any future agreement that we may enter into with our collaboration partners could have a material adverse effect on our business, financial position and results of operations.
The timing and amount of any milestone and royalty payments we may receive under our agreements with our collaboration partners will depend on, among other things, the efforts, allocation of resources, and successful development and commercialization of our product candidates. We cannot be certain that any of the development and regulatory milestones will be achieved or that we will receive any future milestone payments under these agreements. In addition, in certain circumstances we may believe that we have achieved a particular milestone and the applicable collaboration partner may disagree with our belief. In that case, receipt of that milestone payment may be delayed or may never be received, which may require us to adjust our operating plans.
Pursuant to our Sale and Purchase Agreement with Royalty Pharma to sell and transfer the royalty streams from the Sanofi License Agreement, we have assigned our right to receive royalty revenue from the sales of Adlyxin/Lyxumia and/or Soliqua 100/33/ Suliqua and, with the exception of certain development milestone payments under our other existing collaboration, we have no ongoing source of revenue.
In 2003 we entered into our global license agreement, or the Sanofi License Agreement, with Sanofi-Aventis Deutschland GmbH, or Sanofi GmbH, a wholly-owned subsidiary of Sanofi S.A., or Sanofi. The Sanofi License Agreement granted Sanofi the exclusive worldwide rights to develop,
11
Table of Contents
manufacture, commercialize and market lixisenatide, both as a stand-alone product and combination therapy. To date, the majority of our revenue has been derived from milestone payments made by Sanofi, as well royalty payments received from Sanofi on sales of these products.
Lixisenatide, is out-licensed to and marketed by Sanofi both as a stand-alone therapy under the brand names Adlyxin in the United States and Lyxumia in the EU and in various other jurisdictions, and as a combination therapy with Lantus, the brand name of insulin glargine developed by Sanofi, under the brand name Soliqua 100/33 in the United States, and in some European countries under the brand name Suliqua.
On September 6, 2018 we, together with two of our wholly-owned subsidiaries, entered into a purchase and sale agreement, or the Royalty Pharma Agreement, with Royalty Pharma Investments ICAV, or Royalty Pharma, to sell and transfer our and our subsidiaries' respective rights to receive royalties and $85 million of potential commercial milestones in respect of global net sales of Soliqua® 100/33/ Suliqua® and Lyxumia®/Adlyxin® from and after July 1, 2018, payable under the Sanofi License Agreement in consideration for an upfront one-time payment of $205 million. We and our subsidiaries also remain eligible to receive payments from Sanofi of up to $15 million, expected in 2020. However, we cannot be certain with regards to the timing and final amount of this payment, as both are dependent on factors that are outside of our control.
With the transfer to Royalty Pharma of all the royalties that we were due to earn under the Sanofi License Agreement, with the exception of the aforementioned payments of up to $15 million together with any certain development milestone payments we may become entitled to under agreements with our other collaboration partners and potential future royalties should any products concerning those collaborations be approved, we currently have no source of revenue and are reliant on our cash on hand and potential capital raising efforts to fund the development of our internal pipeline of product candidates.
Further, while we are no longer directly exposed to the level of any royalty or milestone payments from Sanofi during the term of the Royalty Pharma Agreement, we have certain administrative obligations toward Royalty Pharma which require that we must maintain the intellectual property on lixisenatide and pay the relevant renewals thereon in a timely manner. In addition to this, certain of our obligations to Sanofi under the Sanofi License Agreement will also continue.
A failure by us to comply with the terms of the agreements with Sanofi and/or Royalty Pharma may place us in breach of our contractual obligations with either party and expose us to liability for indemnification of either party and/or may result in arbitration and/or litigation against us. An adverse ruling in such litigation may lead, to an award of significant damages, loss of profits and/or award of attorney fees against us.
The pricing of our out-licensed products and our product candidates, if and when approved for marketing, will depend in part on pricing strategies adopted by our competitors.
The pricing of certain of our products and product candidates, if and when approved for marketing, will depend, in part, on the pricing strategies adopted by our competitors.
We may need to raise additional funding, which may not be available on acceptable terms, or at all, and failure to obtain this capital when needed may force us to delay, limit or terminate our product development efforts or other operations.
We are currently advancing our internal product candidates through clinical development and are conducting preclinical studies with respect to other programs. Developing product candidates is expensive, lengthy and risky, and we expect our Research and Development ("R&D") expenses to
12
Table of Contents
increase in connection with our ongoing activities, particularly as we seek to advance our internal product candidates toward commercialization.
As of December 31, 2018, our cash and cash equivalents and securities were DKK 1,159.2 million. We expect that our existing cash and cash equivalents will be sufficient to fund our current operations for at least the next twenty four months. However, our operating plans may change as a result of a variety of factors, and we may need to seek additional funds sooner than planned through public or private equity or debt financings, government or other third party funding, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements or a combination of these approaches. Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of our shareholders and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of the ADSs to decline. The sale of additional equity or convertible securities could be dilutive to our shareholders. The incurrence of indebtedness would result in increased fixed payment obligations and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. If we are unable to obtain adequate financing, we may be required to delay, reduce or eliminate the number or scope of our projects and internal product candidates (including our preclinical studies and clinical trial programs). We could also be required to seek funds through arrangements with collaboration partners or at an earlier stage than otherwise would be desirable and we may be required to relinquish rights to some of our technologies or internal product candidates or otherwise agree to terms unfavorable to us. If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs or the commercialization of any internal product candidate or be unable to expand our operations or otherwise capitalize on our business opportunities, as desired, which could impair our prospects.
We may not be successful in our efforts to use cash flows from our approved out-licensed products to expand our novel, internal target discovery platform to build a pipeline of product candidates.
A key element of our strategy is to use cash flows from our portfolio of approved, out-licensed drug products to build a pipeline of novel internal product candidates and progress these product candidates through clinical development for the treatment of a variety of diseases. Although our R&D efforts to date have resulted in the development of out-licensed product candidates directed at various diseases, we may not be able to develop additional product candidates in a sufficient timeframe, if at all, to provide for the further development of our pipeline of internal product candidates. Additionally, with our entering into the Royalty Pharma Agreement, we, and two of our wholly-owned subsidiaries, have transferred all the royalties and $85 million of potential commercial milestones that we were due to earn from the Sanofi License Agreement in exchange for a one-time upfront payment of $205 million. As a result we no longer have the rights to receive ongoing royalties in respect of global net sales of Soliqua® 100/33/ Suliqua® and Lyxumia®/Adlyxin® from and after July 1, 2018. Our other ongoing collaborations with BI and Beta Bionics do not have any approved products, and other than milestone payments that may become due under our ongoing collaborations with BI and Beta Bionics upon the achievement of certain clinical milestones, we are not currently entitled to any royalty or other payments thereunder which could be used to help progress our internal pipeline of product candidates through clinical development. Our current internal product candidates are in late stages of clinical development and will require further clinical development and testing, and eventually regulatory approval, prior to commercialization. Even if we are successful in continuing to develop our out-licensed pipeline, the potential product candidates that we identify may not be suitable for clinical
13
Table of Contents
development, including as a result of being shown to have harmful side effects or other characteristics that indicate that they are unlikely to be products that will receive marketing approval and achieve market acceptance. If we do not continue to successfully develop our out-licensed product candidates and if these out-licensed product candidates are not successfully commercialized by our collaboration partners, we will face difficulty in funding our internal pipeline of product candidates and in generally obtaining product revenue in future periods, which could result in significant harm to our financial position and adversely affect the price of the ADSs or our ordinary shares.
Risks Related to Our Products and Product Candidates
We are dependent on the clinical success of our internal product candidates, including glepaglutide and dasiglucagon.
We are dependent on our ability to develop successfully, obtain regulatory approval for, and then successfully commercialize our other internal product candidates, including glepaglutide and dasiglucagon. Our internal product candidates will require additional R&D clinical development, management of clinical and manufacturing activities, regulatory approval in multiple jurisdictions (if regulatory approval can be obtained at all), securing sources of commercial manufacturing supply, building of, or partnering with, a commercial organization, substantial investment and significant marketing efforts before any revenue can be generated from product sales. We are not permitted to market or promote any of our product candidates in any jurisdiction before we receive regulatory approval from the FDA, the EMA or any other comparable regulatory authority in that jurisdiction, and we may never receive such regulatory approval for any of our product candidates in any particular jurisdiction or at all. We cannot assure you that our clinical trials for glepaglutide or dasiglucagon will be completed in a timely manner, or at all, or that we will be able to obtain approval from the FDA, EMA or any other comparable regulatory authority for any of our product candidates. We cannot be certain that we will advance any other product candidates that are part of our early non-clinical pipeline into clinical trials. If any of our advanced product candidates such as glepaglutide and dasiglucagon or any future product candidate is not approved and commercialized in any particular jurisdiction, we may not be able to generate any royalties or product revenue, as the case may be, for that product candidate at all or in such jurisdiction. Moreover, any delay or setback in the development of any product candidate could materially adversely affect our business and cause the price of the ADSs or our ordinary shares to fall.
Our product candidates will need to undergo clinical trials that are time consuming and expensive, the outcomes of which are unpredictable, and for which there is a high risk of failure. If clinical trials of our product candidates fail to satisfactorily demonstrate safety and efficacy to the FDA, the EMA and any other comparable regulatory authority, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development of these product candidates.
The FDA in the United States, the EMA in Europe, and any other comparable regulatory authorities in other jurisdictions must approve new product candidates before they can be marketed, promoted or sold in those territories. We must provide these regulatory authorities with data from preclinical studies and clinical trials that demonstrate that our product candidates are safe and effective for a specific indication before they can be approved for commercial distribution. We cannot be certain that our clinical trials for our product candidates will be successful or that any of our other internal or out-licensed product candidates will receive approval from the FDA, the EMA or any other comparable regulatory authority.
Preclinical studies and clinical trials are long, expensive and unpredictable processes that can be subject to extensive delays. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all. It may take several years and require significant expenditures to complete the preclinical studies and clinical trials necessary to commercialize a product candidate, and
14
Table of Contents
delays or failure are inherently unpredictable and can occur at any stage. Interim results of clinical trials do not necessarily predict final results, and success in preclinical studies and early clinical trials does not ensure that later clinical trials will be successful. A number of companies in the pharmaceutical, biopharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials even after promising results in earlier trials, and we cannot be certain that we will not face similar setbacks. The design of a clinical trial can determine whether its results will support approval of a product, and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced or completed. Changing the design of a clinical trial can be expensive and time consuming. An unfavorable outcome in one or more trials would be a major setback for our product candidates and for us. An unfavorable outcome in one or more trials may require us to delay, reduce the scope of or eliminate one or more product development programs, which could have a material adverse effect on our business, financial position, results of operations and future growth prospects.
In connection with clinical trials of our product candidates, we face a number of risks, including risks that:
- •
- a product candidate is ineffective, inferior to existing approved products for the same indications, unacceptably toxic or has unacceptable side effects;
- •
- patients may die or suffer other adverse effects for reasons that may or may not be related to the product candidate being tested;
- •
- extension studies on long-term tolerance could invalidate the use of our product;
- •
- the results may not confirm the positive results of earlier trials;
- •
- the results may not meet the level of statistical significance required by the FDA, the EMA or other relevant regulatory agencies to establish the safety and efficacy of our product candidates for continued trial or marketing approval; and
- •
- our collaboration partners or contract research organizations, or CROs, are unable or unwilling to perform under their contracts.
The results of preclinical studies do not necessarily predict clinical success, and larger and later-stage clinical trials may not produce the same results as earlier-stage clinical trials. Our and our collaboration partners' clinical trials of our product candidates conducted to date have generated favorable safety and efficacy data. However, we may have different enrollment criteria in our future clinical trials. As a result, we may not observe a similarly favorable safety or efficacy profile as in our prior clinical trials. In addition, we cannot assure you that during the course of potential widespread use of any of our product candidates in future, we will not suffer setbacks in maintaining production quality or stability. In addition, clinical trials of potential products often reveal that it is not possible or practical to continue development efforts for these product candidates. If we do not successfully complete preclinical and clinical development, we will be unable to market and sell our product candidates and generate additional revenue. Even if we successfully complete clinical trials, those results are not necessarily predictive of results of additional trials that may be needed before marketing applications may be submitted to the FDA, the EMA or other regulatory authority, as applicable.
Our two late stage clinical programs are intended to be designated as orphan products. In the case of glepaglutide, its principle indication is intended for the treatment of short bowel disease and for dasiglucagon one of the potential indications is for the treatment of chronic hyperinsulisum (CHI). Products that developed are for the treatment of orphan diseases receive additional assistance, faster processing and reduced application fees from regulatory authorities like the FDA, EMA or comparable authorities in other countries. In addition to these benefits, orphan indications also receive additional exclusivity protections that are not awarded to non-orphan drugs and enable orphan drugs to be
15
Table of Contents
awarded additional market exclusivity. If we are unable to obtain orphan protection for glepaglutide or for daisiglucagon or our other products we may have to pay additional fees to the regulatory authorities, face slower approval time or be unable to prevent other companies from producing very similar products in the same therapeutic area or even competition from generic producers.
Furthermore, we sometimes estimate for planning purposes the timing of the accomplishment of various scientific, clinical, regulatory and other product development objectives. These milestones may include our expectations regarding the commencement or completion of scientific studies, clinical trials, the submission of regulatory filings or commercialization objectives. From time to time, we may publicly announce the expected timing of some of these milestones, such as the completion of an ongoing clinical trial, the initiation of other clinical programs, receipt of marketing approval or a commercial launch of a product. The achievement of many of these milestones may be outside of our control. All of these milestones are based on a variety of assumptions, which may cause the timing of achievement of the milestones to vary considerably from our estimates. If we fail to achieve announced milestones in the timeframes we expect, the commercialization of our product candidates may be delayed, we may not be entitled to receive certain contractual payments, which could have a material adverse effect on our business, financial position, results of operations and future growth prospects.
We selectively rely on third parties to conduct our clinical trials and perform data collection and analysis, which may result in costs and delays that prevent us from successfully commercializing our product candidates.
We currently, and expect to continue to, selectively rely on public and private research institutions, medical institutions, clinical investigators, CROs, contract laboratories and collaboration partners to conduct some of our early-stage product development activities, perform data collection and analysis and carry out our clinical trials. Our development activities or clinical trials conducted in reliance on third parties may be delayed, suspended or terminated if:
- •
- the third parties do not devote a sufficient amount of time or effort to our activities or otherwise fail to successfully carry out their contractual duties or to meet regulatory obligations or expected deadlines;
- •
- we replace a third party; or
- •
- the quality or accuracy of the data obtained by third parties is compromised due to their failure to adhere to clinical protocols, regulatory requirements or for other reasons.
We do not have the ability to control the performance of third parties in their conduct of development activities. Third party performance failures may increase our development costs, delay our ability to obtain regulatory approval and delay or prevent the commercialization of our product candidates. While we believe that there are alternative sources to provide these services, in the event that we seek such alternative sources, we may not be able to enter into replacement arrangements without incurring delays or additional costs.
We rely on third parties to manufacture our preclinical and clinical drug supplies and we intend to rely on third parties to produce commercial supplies of any approved product candidate.
If, for any reason, we were to experience an unexpected loss of supply of our product candidates or placebo or comparator drug used in certain of our clinical trials, whether as a result of manufacturing, supply or storage issues or otherwise, we could experience delays, disruptions, suspensions or terminations of, or be required to restart or repeat, any pending or ongoing clinical trials. We do not currently have, nor do we plan to acquire, the infrastructure or capability internally to manufacture our preclinical and clinical drug supplies and we lack the resources and the capability to manufacture any of our product candidates on a clinical or commercial scale. The facilities used by our
16
Table of Contents
contract manufacturers or other third party manufacturers to manufacture our product candidates are subject to the FDA's, the EMA's and other comparable regulatory authorities' preapproval inspections that will be conducted after we submit our NDA to the FDA or the required approval documents to any other relevant regulatory authority. We do not control the implementation of the manufacturing process of, and are completely dependent on, our contract manufacturers or other third party manufacturers for compliance with the regulatory requirements, known as current good manufacturing practices, or cGMPs, for manufacture of both active drug substances and finished drug products. If our contract manufacturers or other third party manufacturers cannot successfully manufacture material that conforms to applicable specifications and the strict regulatory requirements of the FDA, the EMA or other comparable regulatory authority, we will not be able to secure and/or maintain regulatory approvals for our products manufactured at these facilities. In addition, we have no control over the ability of our contract manufacturers or other third party manufacturers to maintain adequate quality control and quality assurance procedures and qualified personnel. If the FDA, the EMA or another comparable regulatory authority finds deficiencies at these facilities for the manufacture of our product candidates or if it withdraws any approval because of deficiencies at these facilities in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved.
We rely on our manufacturers to purchase from third party suppliers the materials necessary to produce our product candidates for our clinical trials. There are a limited number of suppliers for raw materials that we use to manufacture our drugs and there may be a need to assess alternate suppliers to prevent a possible disruption of the manufacture of the materials necessary to produce our product candidates for our clinical trials, and if approved, for commercial sale. We do not have any control over the process or timing of the acquisition of these raw materials by our manufacturers. Moreover, we currently do not have any agreements in place for the commercial production of these raw materials. Although we generally do not begin a clinical trial unless we believe we have access to a sufficient supply of a product candidate to complete the clinical trial, any significant delay in the supply of a product candidate, or the raw material components thereof, for an ongoing clinical trial due to the need to replace a contract manufacturer or other third party manufacturer could considerably delay completion of our clinical trials, product testing and potential regulatory approval of our product candidates. If our manufacturers or we are unable to purchase these raw materials after regulatory approval has been obtained for our product candidates, the commercial launch of our product candidates would be delayed or there would be a shortage in supply, which would impair our ability to generate revenue from the sale of our product candidates. Additionally, if we receive regulatory approval for our product candidates, we may experience unforeseen difficulties or challenges in the manufacture of our product candidates on a commercial scale compared to the manufacture for clinical purposes.
We face substantial competition from companies with considerably more resources and experience than we have, which may result in others discovering, developing, receiving approval for or commercializing products before or more successfully than us.
The pharmaceutical and biotechnology industries are characterized by intense competition and significant and rapid technological change as researchers learn more about diseases and develop new technologies and treatments. Any product candidates that we successfully develop and commercialize will compete with existing drugs and new drugs that may become available in the future. We have competitors in each of the disease fields in which we compete, many of which have substantially greater name recognition, commercial infrastructure and financial, technical and personnel resources than we have. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with larger and established companies. Significant competitive factors in our industry include product efficacy and safety, quality and breadth of an organization's technology, skill of an organization's employees and its ability to recruit and retain key employees,
17
Table of Contents
timing and scope of regulatory approvals, government reimbursement rates for, and the average selling price of, products, the availability of raw materials and qualified manufacturing capacity, manufacturing costs, intellectual property and patent rights and their protection and sales and marketing capabilities. While we believe that our product and product candidate platform, development expertise and scientific knowledge provide us with competitive advantages, we face potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions and governmental agencies and public and private research institutions.. There can be no assurance that our competitors will not deploy their superior resources to damage our and our drug candidates' prospects. Given the intense competition in our industry, we cannot assure you that any of the products that we successfully develop will be clinically superior or scientifically preferable to products developed or introduced by our competitors.
In addition, significant delays in the development of our product candidates could allow our competitors to succeed in obtaining the FDA, the EMA or other regulatory approvals for their product candidates more rapidly than us, which could place us at a significant competitive disadvantage or deny us marketing exclusivity rights.
Competitors may develop novel products or other technologies that could make our product candidates obsolete or uneconomical. Any of our product candidates that competes with an approved product may need to demonstrate compelling advantages, such as increased efficacy, convenience, pricing, tolerability and/or safety in order to be commercially successful. Any of our product candidates that are approved could also face other competitive factors in the future, including biosimilar competition, which could force us to lower prices or could result in reduced sales. Any failure to compete effectively against our current and future competitors could have a material adverse effect on our business, financial position, results of operations and future growth prospects.
In addition, many of our competitors have significantly greater financial resources and expertise in R&D, manufacturing, conducting preclinical studies and clinical trials, obtaining regulatory approvals and marketing drugs. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of competitors, particularly through partnership arrangements with large established companies. These companies also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Certain of our peptide product candidates are expected to be delivered parenterally by medical devices that may be regulated as combination products that are required to obtain separate FDA clearance or pre-market approval and/or approval by other regulatory authorities.
Certain of our peptide product candidates are intended to be used in combination with a delivery device, such as an injector or other delivery system. Medical products containing a combination of new drugs, biological products or medical devices may be regulated as "combination products" in the United States and Europe. A combination product generally is defined as a product comprised of components from two or more regulatory categories (e.g. , drug/device, device/biologic, drug/biologic). Each component of a combination product is subject to the requirements established by the FDA for that type of component, whether a new drug, biologic or device. In order to facilitate pre-market review of combination products, the FDA designates one of its centers to have primary jurisdiction for the pre-market review and regulation of the overall product based upon a determination by the FDA of the primary mode of action of the combination product. The determination whether a product is a combination product or two separate products is made by the FDA on a case-by-case basis. Our product candidates intended for use with such devices, or expanded indications that we may seek for our products used with such devices, may not be approved or may be substantially delayed in receiving approval if the devices do not gain and/or maintain their own regulatory approvals or clearances.
18
Table of Contents
Where approval of the drug or biologic product and device is sought under a single application, the increased complexity of the review process may delay approval. The FDA review process and criteria is not a well-established area, which could also lead to delays in the approval process. The EMA has a parallel review process in place for combination products, the potential effects of which in terms of approval and timing could independently affect our ability to market our combination products in Europe. In addition, because these delivery devices are provided by unaffiliated third party companies, we are dependent on the sustained cooperation and effort of those third party companies both to obtain regulatory approval and to maintain their own regulatory compliance. Failure of third party companies to assist in the approval process or to maintain their own regulatory compliance could delay or prevent approval of our product candidates, or limit our ability to sell a product once approved.
We currently have no sales function. If we are unable to establish a sales function or enter into sales, marketing and distribution arrangements with third parties, we may not be successful in commercializing our internal product candidates if and when they are approved.
We do not have a sales or marketing infrastructure and have no experience in the sale, marketing or distribution of pharmaceutical products. To achieve commercial success for any internal product candidate for which we obtain marketing approval, we will need to establish a sales and marketing function or make arrangements with third parties to perform sales and marketing functions on our behalf, and we may not be successful in doing so.
If we enter into arrangements with third parties to perform sales, marketing and distribution services on our behalf, our product revenue or the profitability of our drug revenue may be lower, perhaps substantially lower, than if we were to directly market and sell our drugs. Furthermore, we may be unsuccessful in entering into the necessary arrangements with third parties or may be unable to do so on terms that are favorable to us.
Even if we are able to enter into acceptable partnerships, we may have little or no control over such third parties, and our future collaboration partners may fail to devote the necessary resources and attention to sell and market our drugs effectively. Budgeting restrictions or strategy changes of our future collaboration partners could delay or prevent successful clinical development or marketing efforts. Similarly, our future collaboration partners could decide to give priority to the clinical development or marketing of product candidates or develop or seek to develop product candidates in competition with our product candidates.
Our failure to establish and maintain successful partnerships could have a material adverse effect on our business, financial position, results of operations and future growth prospects.
Risks Related to Our Operations
There is a risk that our products may have major side effects that may give rise to substantial liability claims.
As a biopharmaceutical company, we operate in a market that is subject to risk of liability. To our knowledge, we are not currently subject to any product liability suits. However, we may be subject to future liability claims alleging adverse effects from the use of our products in clinical trials or medical practice. Any liability claims could have a material adverse effect on our business, financial position, results of operations and future growth prospects.
19
Table of Contents
There is a risk that we may not be able to maintain insurance coverage, and that existing or any future insurance policies or our own resources will not sufficiently cover claims for damages that we may receive in the future.
Our business exposes us to potential product liability and other liability risks that are inherent in clinical development, manufacturing, marketing and use of human therapeutic products. It is generally necessary for us to secure certain levels of insurance as a condition for the conduct of clinical trials and any sale or use of our products. We have taken out product liability insurance with respect to all clinical trials and ongoing trials performed to date for which we were responsible (i.e., in respect of our internal product pipeline).
We may seek to expand our insurance coverage if we obtain marketing approval for any of our internal product candidates or if other risks related to our business increase. We may not be able to obtain or maintain adequate protection against potential liabilities at a cost that is acceptable to us. If we are unable to obtain insurance or other protection against potential product liability claims, we could be exposed to significant liabilities, which may materially and adversely affect our business and financial position. These liabilities could prevent or interfere with our product development and commercialization efforts. If we are sued for any injury caused by our products or processes, our liability could exceed our product liability insurance coverage and our own financial resources and, consequently, could have a material adverse effect on our business, financial position, results of operations and future growth prospects.
Our future success depends on our ability to retain our management team and key employees.
We are highly dependent on the management, development, clinical, financial and business development expertise of our management team and key employees. Recruiting and retaining qualified scientific and clinical personnel will also be critical to our future success. The loss of the services of any of the members of our management team or key employees could impede the achievement of our development and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing any of the members of our management team or key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval for and commercialize drugs. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate the members of our management team or key employees on acceptable terms given the competition among numerous pharmaceutical, biopharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. If we are unable to continue to attract and retain high quality management and employees, our ability to pursue our growth strategy will be limited.
Our R&D activities could be affected or delayed as a result of possible restrictions on animal testing.
Certain laws and regulations require us to test our product candidates on animals before initiating clinical trials involving humans. Animal testing activities have been the subject of controversy and adverse publicity. Animal rights groups and other organizations and individuals have attempted to stop animal testing activities by pressing for legislation and regulation in these areas and by disrupting these activities through protests and other means. To the extent the activities of these groups are successful, our R&D activities may be interrupted, delayed or become more expensive.
If we fail to manage our growth effectively, our ability to develop and commercialize products could suffer.
We expect that if our drug discovery efforts continue to generate product candidates, our clinical product candidates continue to progress in development, and we continue to build our development
20
Table of Contents
and commercial organizations, we will require significant additional investment in personnel, management and resources. Our ability to achieve our research, development and commercialization objectives depends on our ability to respond effectively to these demands and expand our internal organization, systems, controls and facilities to accommodate additional anticipated growth. If we are unable to manage our growth effectively, our business could be harmed and our ability to execute our business strategy could suffer.
Our internal computer systems, or those of our collaboration partners or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs.
Our internal computer systems and those of our current and any future collaboration partners and other contractors or consultants are vulnerable to damage from cyber security breaches, computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we do not believe that we have experienced any such material system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations, whether due to a loss of our trade secrets or other proprietary information or other similar disruptions. For example, the loss of clinical trial data for our product candidates from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications or other data or applications relating to our technology or product candidates, or inappropriate disclosure of confidential or proprietary information, we could incur liabilities, our competitive position could be harmed and the further development and commercialization of our product candidates could be delayed.
Our operations as a global company subject us to various risks, and our failure to manage these risks could adversely affect our results of operations.
We face significant operational risks as a result of doing business internationally, such as:
- •
- fluctuations in foreign currency exchange rates (in particular, U.S. dollars, Euros and Danish kroner);
- •
- potentially adverse and/or unexpected tax consequences, including penalties due to the failure of tax planning or due to the challenge by tax authorities on the basis of transfer pricing and liabilities imposed from inconsistent enforcement;
- •
- potential changes to the accounting standards, which may influence our financial situation and results;
- •
- becoming subject to the different, complex and changing laws, regulations and court systems of multiple jurisdictions and compliance with a wide variety of foreign laws, treaties and regulations;
- •
- reduced protection of, or significant difficulties in enforcing, intellectual property rights in certain countries;
- •
- restrictions imposed by local labor practices and laws on our business and operations, including unilateral cancellation or modification of contracts;
- •
- rapid changes in global government, economic and political policies and conditions, political or civil unrest or instability, terrorism or epidemics and other similar outbreaks or events, and potential failure in confidence of our suppliers or customers due to such changes or events; and
21
Table of Contents
- •
- tariffs, trade protection measures, import or export licensing requirements, trade embargoes and other trade barriers.
We are subject to the Foreign Corrupt Practices Act, or the FCPA, which generally prohibits companies and their intermediaries from making or offering improper payments to non-U.S. officials for the purpose of obtaining or retaining business. The FCPA generally also requires companies listed on a U.S. stock exchange to maintain a system of adequate internal accounting controls and to make and keep books, records and accounts that accurately and fairly reflect transactions and dispositions of assets. Because of the predominance of government-sponsored health care systems around the world, many of our commercial relationships outside of the United States are with governmental entities, and personnel of such entities may be considered non-U.S. officials for purposes of the FCPA. Violations of the FCPA and other applicable anti-bribery laws are punishable by criminal fines and imprisonment, civil penalties, disgorgement of profits, injunctions, debarment from government contracts as well as other remedial measures. We have adopted a written code of business conduct and other policies and procedures to assist us and our personnel in complying with the FCPA and other applicable anti-bribery laws prior to completion of the offering. However, our personnel and others acting on our behalf could take actions that violate these requirements, which could adversely affect our reputation, business, financial condition and results of operations.
We have identified material weaknesses in our internal control over financial reporting. If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results in a timely manner or prevent fraud, which may adversely affect our business, investor confidence in our company and the market price of our shares and ADSs.
Our management has concluded that we had not maintained effective internal control over financial reporting and disclosure controls and procedures as of December 31, 2018 due the material weaknesses we identified in the design and operating effectiveness of our internal controls over financial reporting, including lack of sufficient competencies related to IFRS and SEC reporting knowledge for the purposes of timely and reliable financial reporting in connection with our financial statement preparation process for the years ended December 31, 2018, 2017, 2016 and 2015. Under the standards established by the U.S. Public Company Accounting Oversight Board, or the PCAOB, a material weakness is a deficiency, or a combination of deficiencies, that creates a reasonable possibility that a material misstatement of a company's annual financial statements will not be prevented or detected on a timely basis. The material weaknesses identified by us relate to our processes to assess risk and to design and implement effective control activities over financial reporting. we did not have formalized risk assessment, oversight and compliance processes or formalized control descriptions for all of our key controls. Where control descriptions existed, they did not necessarily include all relevant information to enable the operating effectiveness of such controls and it was not clear whether adequate controls were performed in all areas. Where control activities were dependent on certain information, which is referred to as our IUC, we did not perform or document controls to assess the completeness and accuracy of such information. We did not then monitor control activities and identified control deficiencies; thus, we were unable to evaluate whether other deficiencies, individually or in combination, resulted in a reasonable possibility that a material misstatement of our annual financial statements would not be prevented or detected on a timely basis. Further, restatements resulting from the correction of certain misstatements in our annual financial statements included in this prospectus have been identified. These restatements occurred due to the lack of sufficient overall review of the financial statements and lack of oversight of the application and implementation of accounting policies and accounting standards by the Company.
In addition to the hiring of a Chief Accountant in late 2017 and engaging external experts to improve our competencies with respect to IFRS and SEC reporting, as of December 31, 2018 we have implemented a formalized process for risk assessment, completed the design and documentation of our
22
Table of Contents
key controls to respond to the identified risks, and implemented a formalized process for performing our evaluation of the effectiveness of our internal controls over financial reporting as of the date of our annual report. We have hired additional finance and accounting personnel with appropriate expertise to perform specific functions and intend to hire additional personnel to further assist in the implementation of improved processes and internal controls, build our financial management and reporting infrastructure and further develop and document our accounting policies and financial reporting procedures, including ongoing senior management review and audit committee oversight.
While management believes that the steps that we have taken and plans to continue to take, will improve its overall system of internal control over financial reporting and will remediate identified material weaknesses, the material weaknesses cannot be considered remediated until the applicable relevant controls operate for a sufficient period of time and management has concluded, through testing that these controls are operating effectively. No assurance can be provided at this time that the actions and remediation efforts will effectively remediate the material weaknesses described above or prevent the incidence of other material weaknesses in the Company's internal control over financial reporting in the future. Further, we do not know the specific time frame needed to fully remediate the material weaknesses identified.
Additionally, even if the material weakness in the design and operating effectiveness of our internal controls over financial reporting that had identified was remediated by our actions, we may discover future deficiencies or material weaknesses in our internal controls over financial reporting, including those identified through testing conducted by us pursuant to Section 404(a) of the Sarbanes-Oxley Act or subsequent testing by our independent registered public accounting firm when required pursuant to the Sarbanes-Oxley Act. Such deficiencies may be deemed to be significant deficiencies or material weaknesses and may require changes to our consolidated financial statements or identify other areas for further attention or improvement. Even if we are able to report our financial statements accurately and in a timely manner, if we do not make all necessary improvements to address any outstanding material weaknesses, continued disclosure of such significant deficiencies and material weaknesses may be required in future filings with the SEC, which may adversely affect our business, investor confidence in our company and the market price of our shares and ADSs.
Risks Related to Our Intellectual Property
Our ability to compete may decline if we or our collaboration partners are unable to or do not adequately protect intellectual property rights or if our intellectual property rights are inadequate for our product candidates or future product candidates
Our commercial success and viability depends on our and our collaboration partners' ability to obtain and maintain patent protection in the United States, Europe and other countries with respect to our existing product candidates owned by us and to successfully defend these rights against third party challenges, as well as our ability to maintain adequate intellectual property protection for any future products. If we or our collaboration partners do not adequately protect our intellectual property, competitors may be able to use our technologies and erode or negate any competitive advantage we may have, which could materially harm our business, negatively affect our position in the marketplace, limit our ability to commercialize our product candidates and delay or render impossible our achievement of profitability.
Our strategy and future prospects are based, in particular, on our patent portfolio. We and our collaboration partners or licensees will best be able to protect our product candidates and their uses from unauthorized use by third parties to the extent that valid and enforceable patents, effectively protected trade secrets, or other regulatory exclusivities, cover them. Also, intellectual property rights have limitations and do not necessarily address all potential threats to our competitive advantage. Our ability to obtain patent protection for our product candidates is uncertain and the degree of future
23
Table of Contents
protection afforded by our intellectual property rights is uncertain due to a number of factors, including, but not limited to:
- •
- we or our collaboration partners may not have been the first to make the inventions covered by pending patent applications or issued patents;
- •
- we or our collaboration partners may not have been the first to file patent applications for our product candidates or the compositions we developed or for their uses;
- •
- others may independently develop identical, similar or alternative products or compositions and uses thereof;
- •
- our or our collaboration partners' disclosures in patent applications may not be sufficient to meet the statutory requirements for patentability;
- •
- any or all of our or our collaboration partners' pending patent applications may not result in issued patents;
- •
- we or our collaboration partners may not seek or obtain patent protection in countries that may eventually provide us with a significant business opportunity;
- •
- any patents issued to us or our collaboration partners may not provide a basis for commercially viable products, may not provide any competitive advantages, or may be successfully challenged by third parties;
- •
- our or our collaboration partners' compositions and methods may not be patentable;
- •
- others may design around our or our collaboration partners' patent claims to produce competitive products or uses which fall outside of the scope of our patents;
- •
- others may identify prior art or other bases which could result in the prohibition or limitation of our or our collaboration partners' patents;
- •
- our competitors might conduct R&D activities in the United States and other countries that provide a safe harbor from patent infringement claims for certain R&D activities, as well as in countries where we or our collaboration partners do not have patent rights, and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; or
- •
- we may not develop additional proprietary technologies that are patentable.
Issued patents covering our product candidates could be found invalid or unenforceable if challenged in court.
Even if our patents do successfully issue and even if such patents cover our product candidates and methods of use, third parties may initiate interference, re-examination, post-grant review, inter partes review, or derivation actions in the U.S. Patent and Trademark Office, or theUSPTO, may initiate third party oppositions in the European Patent Office, or the EPO, or similar actions challenging the validity, enforceability or scope of such patents in other patent administrative proceedings worldwide, which may result in our patent claims being narrowed or invalidated. Such proceedings could result in revocation or amendment of our patents in such a way that they no longer cover our product candidates or competitive products. Further, if we initiate legal proceedings against a third party to enforce a patent covering our product candidate or technology, the defendant could counterclaim that the patent covering our product candidate or technology is invalid or unenforceable. In patent litigation in the United States, certain European and other countries worldwide, it is commonplace for defendants to make counterclaims alleging invalidity and unenforceability in the same proceeding, or to commence parallel defensive proceedings such as patent nullity actions to challenge validity and enforceability of asserted patent claims.
24
Table of Contents
In administrative and court actions, grounds for a patent validity challenge may include alleged failures to meet any of several statutory requirements, including lack of novelty, obviousness (lack of inventive step) and in some cases, lack of sufficiently teaching, or non-enablement of, the claimed invention. Grounds for unenforceability assertions include allegations that someone connected with prosecution of the patent withheld relevant information from the Examiner during prosecution in the USPTO, or made a misleading statement during prosecution in the USPTO, the EPO or elsewhere. Third parties may also raise similar claims before administrative bodies in the USPTO or the EPO, even outside the context of litigation. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to validity, for example, we cannot be certain that there is no invalidating prior art, of which we or the patent examiner were unaware during prosecution. Further, we cannot be certain that all of the potentially relevant art relating to our patents and patent applications has been cited in every patent office. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our product candidates.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time consuming and unsuccessful and have a material adverse effect on the success of our business.
Competitors may infringe our patents, trademarks, copyrights or other intellectual property. To counter infringement or unauthorized use, we may be required to file infringement claims on a country-by-country basis, which can be expensive, unpredictable, time consuming and divert the time and attention of our management and scientific personnel. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringe their patents, in addition to counterclaims asserting that our patents are invalid or unenforceable, or both. In any patent infringement proceeding, there is a risk that a court will decide that a patent of ours is invalid or unenforceable, in whole or in part, and that we do not have the right to stop the other party from using the invention at issue. There is also a risk that, even if the validity of such patents is upheld, the court will construe the patent's claims narrowly or decide that we do not have the right to stop the other party from using the invention at issue on the grounds that our patent claims do not cover the invention. An adverse outcome in a litigation or proceeding involving one or more of our patents could limit our ability to assert those patents against those parties or other competitors, and may curtail or preclude our ability to exclude third parties from making and selling similar or competitive products. Similarly, if we assert trademark infringement claims, a court may determine that the marks we have asserted are invalid or unenforceable, or that the party against whom we have asserted trademark infringement has superior rights to the marks in question. In this case, we could ultimately be forced to cease use of such trademarks.
Even if we establish infringement, the court may decide not to grant an injunction against further infringing activity and instead award only monetary damages, which may or may not be an adequate remedy. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could adversely affect the market price of the ADSs. Moreover, there can be no assurance that we will have sufficient financial or other resources to file and pursue such infringement claims, which typically last for years before they are concluded. Even if we ultimately prevail in such claims, the monetary cost of such litigation, any award of damages or loss of profit and the diversion of the attention of our management and scientific personnel could outweigh any benefit we receive as a result of the proceedings.
25
Table of Contents
Claims that our product candidates or their uses infringe the intellectual property rights of third parties could result in costly litigation, and unfavorable outcomes could require us to pay damages or royalties and could limit our R&D activities or our ability to commercialize certain products.
Even if we have or obtain patents covering our product candidates, compositions or uses, we may still be barred from making, using, importing or selling our product candidates or technologies because of the patent rights of others. Others have filed, and in the future may file, patent applications covering compositions or products and uses that are similar or identical to ours. There are many-issued U.S., European and other worldwide patents relating to therapeutic drugs, and some of these relate to compounds we intend to commercialize. Numerous worldwide patents and pending patent applications owned by others exist in the metabolic disease, gastrointestinal disease and cardiovascular disease field and cover, among others, GLP-2 product candidates that we are developing. To the extent that we identify any potential issue with third party patents that may affect our product candidates, we ensure that we have a strategy to deal with such third party patents, either by ensuring that we believe that such patents are invalid, not infringed or that we commercialize our products upon expiry of such patents. Such strategies can include seeking a judicial or administrative revocation of such patents, ensuring that we are in a position to defend a claim for infringement, or seeking a license where that is appropriate. We cannot guarantee that our products, compositions and their uses do not or will not infringe third party patent or other intellectual property rights. Because patent applications can take 18 months to publish and many years to issue, there may be currently pending applications with patent claims unknown to us or which will change over time and may later result in issued patents that purportedly cover our product candidates or compositions and uses. These patent applications may have been filed earlier than or have priority over patent applications filed by us. We may be required to develop or obtain alternative technologies, review product design or, in the case of claims concerning registered trademarks, rename our product candidates.
Claims that our or our collaboration partners' products, compositions or their uses infringe or interfere with the patent rights of third parties, or that we or our collaboration partners have misappropriated third party trade secrets, could result in costly litigation and could require substantial time and money to resolve, even if litigation were avoided. The basis of such litigation could be existing patents or patents that are granted in the future. If we or our collaboration partners were to face infringement claims or challenges by third parties, an adverse outcome could subject us or our collaboration partners to significant liabilities to such third parties. Litigation or threatened litigation could result in significant demands on the time and attention of our management team. A negative outcome could expose us or our collaboration partners to payment of costs, damages and other financial remedies, including in some jurisdictions, increased damages, such as treble damages and attorneys' fees, if found to have willfully infringed a patent. Litigation with third parties concerning alleged infringement of their intellectual property rights could require us and our collaboration partners to bear substantial costs and impose burdens on our and their management and personnel, even if we or our collaboration partners were to ultimately succeed in such proceedings. Costs of patent litigation and awards of damages in patent infringement cases can be significant, and equitable remedies such as temporary restraining orders and injunctions can negatively impact or prevent product development and commercialization. In light of these risks, settlements are often a preferred alternative, to avoid litigation uncertainties and costs, even when there are strong defenses to claims that are made. A negative outcome, potential or actual, could cause us or our collaboration partners to pursue contractual and other remedies against each other; in particular, our license agreements generally allow our collaboration partners to reduce amounts we are owed as royalties and/or milestones by amounts paid to third parties as a result of or in settlement of certain infringement claims, subject to contractual conditions and limitations. We or our collaboration partners could also face equitable remedies, such as being forced, including by court order, to cease developing, manufacturing, importing or commercializing an infringing product candidate or product in one or more jurisdictions. A negative outcome could also lead us or our collaboration partners to delay, curtail or cease the development and
26
Table of Contents
commercialization of some or all of our candidate drugs, or could cause us or our collaboration partners to seek legal or administrative actions against third parties. We or our collaboration partners may need to obtain licenses from third parties and such licenses may not be available on commercially reasonable terms, or at all. Even if we or our collaboration partners are able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same rights licensed to us. In addition, even if we or our collaboration partners were ultimately to succeed in asserting one or more patent defenses in an infringement suit, or to settle at an early stage to avoid litigation uncertainties and costs despite having strong patent defenses, such litigation could burden us and our collaboration partners with substantial unanticipated costs and damages. A negative outcome could cause us or our collaboration partners to pursue contractual remedies against each other, including, for example, over settlement or license related payments or royalty reductions.
Biopharmaceutical patents and patent applications involve highly complex legal and factual questions, which, if determined adversely to us, could negatively impact our patent position.
The patent positions of biopharmaceutical companies can be highly uncertain and involve complex legal and factual questions. The interpretation and breadth of claims allowed in some patents covering biopharmaceutical compositions may be uncertain and difficult to determine, and are often affected materially by the facts and circumstances that pertain to the patented compounds, compositions and related patent claims. The standards of the USPTO, the EPO and other international patent offices are evolving and could change in the future. Consequently, we cannot predict the issuance and scope of patents with certainty. Patents, if issued, may be challenged, invalidated or circumvented. European patents and patents in certain other jurisdictions are subject to third party opposition proceedings. U.S. patents and patent applications may also be subject to interference proceedings, and U.S. patents may be subject to reexamination proceedings, post-grant review and/or inter partes review in the USPTO. The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability and our patents or pending patent applications may be challenged in the courts or patent offices in the United States, Europe and elsewhere worldwide. There is no assurance that all of the potentially relevant prior art relating to our patents and patent applications has been found. For example, publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our owned and licensed patents or pending patent applications, or that we were the first to file for patent protection of such inventions. If such prior art exists, it may be used to invalidate a patent, or may prevent a patent from issuing from a pending patent application. For example, such patent filings may be subject to a third party pre-issuance submission of prior art to the USPTO, EPO or to other patent offices around the world. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights may be uncertain. Our pending and future patent applications may not result in patents being issued that protect our technology or products, in whole or in part, or may not effectively prevent others from commercializing competitive technologies and products. For example, such patent filings may be subject to a third party preissuance submission of prior art to the USPTO, the EPO or to other patent offices around the world. Alternately or additionally, we may become involved in post-grant review procedures, oppositions, derivations, proceedings, reexaminations, inter partes review or interference proceedings, in the United States or elsewhere, challenging patents or patent applications in which we have rights, including patents on which we rely to protect our business. An adverse determination in any such challenges may result in loss of exclusivity or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. European patents or patents in other jurisdictions may be subject also to administrative opposition or comparable proceedings in corresponding worldwide patent offices, which could result in either loss of the patent or denial of the
27
Table of Contents
patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, reexamination, post-grant review, inter partes review and opposition proceedings may be time consuming and costly. Also, given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates may expire before or shortly after such candidates are commercialized. Accordingly, rights under any issued patents may not provide us with sufficient protection against competitive products or processes.
In addition, changes in or different interpretations of patent laws in the United States, Europe and other countries worldwide may diminish the value of our patents or narrow the scope of our patent protection, while patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. For example, changes in or different interpretations of patent laws in the United States, Europe and other countries worldwide may permit others to use our or our collaboration partners' discoveries or to develop and commercialize our technology and products without providing any compensation to us, or may limit the number of patents or claims we can obtain. The laws of some countries may not protect intellectual property rights to the same extent as the laws of the United States or Europe, and those countries may lack adequate rules and procedures for defending our intellectual property rights, or vice versa.
If we fail to obtain and maintain patent protection and trade secret protection for our product candidates, we could lose our competitive advantage and competition we face would increase, reducing any potential revenue and adversely affecting our ability to attain or maintain profitability.
If we are unable to protect the confidentiality of our trade secrets and know-how, our business and competitive position would be harmed.
In addition to seeking patent protection for our product candidates, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, collaboration partners, consultants, advisors, university and/or institutional researchers and other third parties. We also have entered or seek to enter into confidentiality and invention or patent assignment agreements with our employees, advisors and consultants. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Our trade secrets may also be obtained by third parties by other means, such as breaches of our physical or computer security systems. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. Moreover, if any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to, or independently developed by, a competitor, our competitive position would be harmed.
We will not seek to protect our intellectual property rights in all jurisdictions throughout the world, and we may not be able to adequately enforce our intellectual property rights even in the jurisdictions where we seek protection.
Obtaining and maintaining a patent portfolio entails significant expense and resources. Part of the expense includes periodic maintenance fees, renewal fees, annuity fees, various other governmental fees on patents or applications due in several stages over the lifetime of patents or applications, as well as the cost associated with complying with numerous procedural provisions during the patent application
28
Table of Contents
process. Filing, prosecuting and defending patents on our product candidates in all countries and jurisdictions throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States and Europe could be less extensive than those in the United States and in Europe, assuming that rights are obtained in the United States or in Europe. We may choose not to pursue or maintain protection for particular inventions. In addition, there are situations in which failure to make certain payments or noncompliance with certain requirements in the patent process can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If we choose to forego patent protection or allow a patent application or patent to lapse purposefully or inadvertently, our competitive position could suffer. Competitors may use our technologies in jurisdictions where we do not pursue and obtain patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States or in Europe. These products may compete with our product candidates and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. Even if we pursue and obtain issued patents in particular jurisdictions, our patent claims or other intellectual property rights may not be effective or sufficient to prevent third parties from so competing.
In addition, the laws of some countries do not protect intellectual property rights to the same extent as the federal and state laws in the United States and Europe. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to biopharmaceuticals or biotechnologies. This could make it difficult for us to stop the infringement of our patents, if obtained, or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries. Proceedings and legal actions to enforce our patent rights in the United States or in Europe and in foreign jurisdictions can be expensive, could result in substantial costs, and could divert management time and our efforts and attention from other aspects of our business. In addition, such proceedings or legal actions could put our patents at risk of being invalidated, found unenforceable or interpreted narrowly, could put our patent applications at risk of not being issued and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. We may or may not choose to pursue litigation or other actions against those that have infringed our patents, or used them without authorization, due to the associated expense and time commitment of monitoring these activities. If we fail to protect or to enforce our intellectual property rights successfully, our competitive position could suffer, which could harm our results of operations.
In addition, changes in the law and legal decisions by courts in the United States, Europe and foreign countries may affect our ability to obtain adequate protection for our technology and the enforcement of intellectual property. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
29
Table of Contents
Patent terms and regulatory exclusivities may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. We expect to seek extensions of patent terms in the United States and, if available, in other countries where we have patents.
Depending upon the timing and duration of the U.S. regulatory review process and patent life considerations, certain of our U.S. patents may be eligible for patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Amendments. The Hatch-Waxman Amendments provide up to five years of patent term extension, or PTE, on a patent that covers an approved product or method of use as compensation for patent term lost during the FDA regulatory review process. Patent term restoration cannot extend the term of a patent beyond a total of 14 years from the product's approval date. Only one patent with a claim covering an approved drug or method is eligible for the extension, and the extension must be applied for prior to the patent expiration date (which due date may be extended by submission of one or more applications for interim extensions for periods of up to one year each and cannot be extended longer than the maximum period of patent term extension). The USPTO, in consultation with the FDA, reviews and approves a request for patent term extension or restoration and calculates the PTE period that will be awarded. PTE only extends patent coverage on the approved product or method of use.
In certain Member States of the EU, patent term extensions may be obtained through a Supplementary Protection Certificate ("SPC"), to recover some of the time lost between the patent application filing date and the date of first regulatory approval, up to a maximum term of five years. Up to five years of patent term extension are also available in Japan for patent term recovery related to the pharmaceutical regulatory review and approval process.
Applicable authorities, including the FDA/USPTO in the United States, and comparable regulatory authorities and intellectual property offices in other EU countries and worldwide, may not agree with our assessment of whether such extensions are available, and may refuse to grant extensions to our patents, or may grant more limited extensions than we request. If this occurs, our competitors may be able to take advantage of our investment in development and clinical trials by referencing our clinical and preclinical data and launch their product earlier than might otherwise be the case.
Third parties may challenge the inventorship of our patent filings and other intellectual property or may assert ownership or commercial rights to inventions we develop.
Third parties may in the future make claims challenging the inventorship or ownership of our intellectual property. We have written agreements with collaboration partners that provide for the ownership of intellectual property arising from our collaborations. These agreements provide that we or our licensees must negotiate certain commercial rights with collaboration partners with respect to joint inventions or inventions made by our collaboration partners that arise from the results of the collaboration. In addition, our standard employment contracts ensure that any inventions are ours by right and this contractual position is in addition to our rights to any invention by the operation of law where we conduct research and development ourselves.
In some instances, there may not be, or parties may dispute that there are, adequate written provisions to address clearly the resolution of intellectual property rights that may arise from collaboration. If we or our licensees cannot successfully negotiate sufficient ownership and commercial rights to the inventions that result from our use of a third party collaboration partner's materials where required, or if disputes otherwise arise with respect to the intellectual property developed with the use of a collaboration partner's samples, we may be limited in our ability to capitalize on the market potential of these inventions. In addition, we may face claims by third parties that our agreements with
30
Table of Contents
employees, contractors or consultants obligating them to assign intellectual property to us are ineffective, or in conflict with prior or competing contractual obligations of assignment, which could result in ownership disputes regarding intellectual property we have developed or will develop and interfere with our ability to capture the commercial value of such inventions. Litigation may be necessary to resolve an ownership dispute, and if we are not successful, we may be precluded from using certain intellectual property, or may lose our exclusive rights in that intellectual property. Either outcome could have an adverse impact on our business, financial position, results of operations and future growth prospects.
Third parties may assert that our employees or consultants or we have wrongfully used or disclosed confidential information or misappropriated trade secrets, or claim ownership of what we regard as our own intellectual property.
We employ individuals who were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, and no such claims against us are currently pending, we may be subject to claims that we or our employees, consultants or independent contractors have used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and applications are required to be paid to the USPTO and various governmental patent agencies outside the United States in several stages over the lifetime of the patents and applications. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process and after a patent has issued. There are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction.
Risks Related to Government Regulation
Government restrictions on pricing and reimbursement, as well as other healthcare payor cost-containment initiatives, may negatively impact our ability to generate revenue.
Sales of certain of our out-licensed products and our product candidates, if and when approved for marketing, has and will depend, in part, on the extent to which our products will be covered by third party payors, such as government health care programs like Medicare and Medicaid, commercial insurance and managed healthcare organizations. These third party payors play an important role in determining the extent to which new drugs, biologics and medical devices will be covered. The Medicare and Medicaid programs increasingly are used as models for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs, biologics and medical devices. It is difficult to predict at this time what third party payors will decide with respect to the coverage and reimbursement for our product candidates. The primary trend in the U.S. healthcare industry and elsewhere has been cost containment, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products and/or biosimilars. Adoption
31
Table of Contents
of price controls, cost containment measures and adoption of more restrictive policies in jurisdictions with existing controls and measures, could limit our net revenue and results.
Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for medical products, drugs and services. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for drugs. We cannot be sure that coverage will be available for any product candidate that we commercialize and, if coverage is available, the level of reimbursement. Because coverage and reimbursement determinations are made on a payor-by-payor basis, obtaining coverage and adequate reimbursement from a third party payor does not guarantee that we will obtain similar coverage or reimbursement from another third party payor. Reimbursement may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. If reimbursement is not available or is available only to limited levels, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval. Decreases in third party reimbursement for our product candidates or a decision by a third party payor not to cover our product candidates or provide only limited reimbursement for our product candidates could reduce physician usage of our products once approved and have a material adverse effect on our sales, results of operations and financial condition. Further, the adoption and implementation of any future governmental cost containment or other health reform initiative may result in additional downward pressure on the price that we may receive for any approved product.
Outside the United States, international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe, Canada, and other countries has and will continue to put pressure on the pricing and usage of our product candidates. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. Other countries allow companies to fix their own prices for medical products, but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside the United States, the reimbursement for our products may be reduced compared with the United States and may be insufficient to generate commercially reasonable revenue and profits.
Moreover, increasing efforts by governmental and third party payors in the United States and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for new products approved and, as a result, they may not cover or provide adequate payment for our product candidates. We expect to experience pricing pressures in connection with the sale of any of our product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations, and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs, medical devices and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the successful commercialization of new products.
We may face difficulties from changes to current regulations and future legislation.
Existing regulatory policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
32
Table of Contents
The current presidential administration and U.S. Congress have also recently attempted to repeal or "repeal and replace" the Affordable Care Act, or the ACA. Although those efforts did not succeed, the presidential administration may continue to seek to modify, repeal, or otherwise invalidate all, or certain provisions of, the ACA. There is still uncertainty with respect to the impact President Trump's administration and the U.S. Congress may have on the ACA, if any, and any changes will likely take time to unfold. Additionally, since its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA. For example, on December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, or Texas District Court Judge, ruled that the entire ACA is invalid based primarily on the fact that the Tax Cuts and Jobs Act of 2017 repealed the tax-based shared responsibility payment imposed by the ACA, on certain individuals who fail to maintain qualifying health coverage for all or part of a year, which is commonly referred to as the "individual mandate". While the Texas District Court Judge, as well as the current presidential administration and the Centers for Medicare and Medicaid Services, have stated that this ruling will have no immediate effect, it is unclear how this decision and subsequent appeals will impact the law and the effect such impact could have on coverage and reimbursement for healthcare items and services covered by plans that were authorized by the ACA.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. These changes included aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, effective April 1, 2013, which, due to subsequent legislative amendments, will stay in effect through 2025 unless additional Congressional action is taken. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers and increased the statute of limitations period for the U.S. government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on customers for our out-licensed products and product candidates (if and when approved) and accordingly, our financial results.
Likewise, the annual Medicare Physician Fee Schedule update, which, until recently, was based on a target-setting formula system called the Sustainable Growth Rate, or SGR, , was adjusted to reflect the comparison of actual expenditures to target expenditures. Because one of the factors for calculating the SGR was linked to the growth in the U.S. gross domestic product, or GDP, the SGR formula often resulted in a negative payment update when growth in Medicare beneficiaries' use of services exceeded GDP growth. Congress repeatedly intervened to delay the implementation of negative SGR payment updates. For example, on April 1, 2014, with the enactment of the Protecting Access to Medicare Act of 2014, Congress prevented the 24% cut that was to occur by continuing the previously implemented 0.5% payment increase through December 31, 2014 and maintaining a 0% payment update from January 1, 2015 through March 31, 2015. However, on April 14, 2015, Congress passed the Medicare Access and CHIP Reauthorization Act of 2015, which was signed into law by President Obama on April 16, 2015. This law repeals the SGR methodology from the physician payment formula, institutes a 0% update to the Medicare Physician Fee Schedule for the January 1 to July 1, 2015 period, a 0.5% payment update for July 2015 through the end of 2019, and a 0% payment update for 2020 through 2025, along with a merit-based incentive payment system beginning January 1, 2019, that will replace current incentive programs. For 2026 and subsequent years, the payment update will be either 0.75% or 0.25%, depending on which Alternate Payment Model the physician participates.
We expect more rigorous coverage criteria in the future in the U.S. healthcare market and an additional downward pressure on the prices that we receive for approved products. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our out-licensed products and product candidates.
33
Table of Contents
In addition, it is not currently possible to predict how, if at all, the FDA's approved process or regulation will change as a result of the Trump Administration or what impact any such changes will have on us.
Our operations involve hazardous materials and we and third parties with whom we contract must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
As a pharmaceutical company, we are subject to environmental and safety laws and regulations, including those governing the use of hazardous materials. The cost of compliance with health and safety regulations is substantial. Our business activities involve the controlled use of hazardous materials. Our R&D activities involve the controlled storage, use and disposal of hazardous materials, including the components of our product candidates and other hazardous compounds. We and manufacturers and suppliers with whom we may contract are subject to laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. In some cases, these hazardous materials and various wastes resulting from their use are stored at our and our manufacturers' facilities pending their use and disposal. We cannot eliminate the risk of accidental contamination or injury from these materials, which could cause an interruption of our commercialization efforts, R&D efforts and business operations, environmental damage resulting in costly clean-up and liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. We cannot guarantee that that the safety procedures utilized by third party manufacturers and suppliers with whom we may contract will comply with the standards prescribed by laws and regulations or will eliminate the risk of accidental contamination or injury from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources and European, U.S. federal and state or other applicable authorities may curtail our use of certain materials and/or interrupt our business operations. Furthermore, environmental laws and regulations are complex, change frequently and have tended to become more stringent. We cannot predict the impact of such changes and cannot be certain of our future compliance. We do not currently carry biological or hazardous waste insurance coverage. In the event of an accident or environmental discharge, we may be held liable for any consequential damage and any resulting claims for damages, which may exceed our financial resources and may materially adversely affect our business, results of operations and prospects, and the value of our shares.
We are subject to healthcare laws and regulations, which may require substantial compliance efforts and could expose us to criminal sanctions, civil penalties, exclusion from government healthcare programs, contractual damages, reputational harm and diminished profits and future earnings, among other penalties.
If we market products ourselves, healthcare providers, such as physicians, and other health care entities and organizations, will play a primary role in the recommendation and prescription of our products, if approved. Our arrangements with such persons, entities and third party payors and our general business operations will expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we research, market, sell and distribute our products. Restrictions under applicable U.S. federal, state, local and non-U.S. healthcare laws and regulations include, but are not limited to, the following:
- •
- the federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, including any kickback, bribe or rebate, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase or lease, order or recommendation of, any item, good, facility or service, for which payment may be made under federal healthcare programs such as Medicare and Medicaid;
34
Table of Contents
- •
- the federal Beneficiary Inducement Statute, which prohibits giving anything of value to a government insurance beneficiary that could influence the choice of provider or reimbursable covered product;
- •
- federal civil and criminal false claims laws and civil monetary penalties laws, including the civil False Claims Act, which impose criminal and civil penalties, including those from civil whistleblower or qui tam actions, against individuals or entities for, among other things, knowingly presenting, or causing to be presented, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government;
- •
- the anti-inducement law prohibits, among other things, the offering or giving of remuneration, which includes, without limitation, any transfer of items or services for free or for less than fair market value (with limited exceptions), to a Medicare or Medicaid beneficiary that the person knows or should know is likely to influence the beneficiary's selection of a particular supplier of items or services reimbursable by a federal or state governmental program;
- •
- the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal statutes that impose criminal and civil liability for, among other things, executing or attempting to execute a scheme to defraud any healthcare benefit program or knowingly and willingly falsifying, concealing or covering up a material fact or making false statements relating to healthcare matters;
- •
- HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, which impose certain requirements on covered entities and their business associates, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information;
- •
- the federal transparency requirements under the Physician Payments Sunshine Act, enacted as part of the Patient Protection and ACA that require applicable manufacturers of covered drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid, or the Children's Health Insurance Program, with specific exceptions, to track and annually report to the Centers for Medicare & Medicaid Services, or CMMS, payments and other transfers of value provided to physicians and teaching hospitals, and certain ownership and investment interests held by physicians or their immediate family members;
- •
- analogous state, local and non-U.S. laws and regulations, such as state anti-kickback and false claims laws, which may apply to items or services reimbursed by any third party payor, including commercial insurers; state, local and non-U.S. marketing and/or transparency laws applicable to manufacturers that may be broader in scope than the federal requirements; state laws that require biopharmaceutical companies to comply with the biopharmaceutical industry's voluntary compliance guidelines, relevant compliance guidance promulgated by the federal government, implementation of compliance programs, and compliance with the state's code of conduct; state and local laws that require a pharmaceutical company's sales representatives to be registered or licensed by the state or local governmental entity; and state and non-U.S. laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may be more stringent than HIPAA, thus complicating compliance efforts; and
- •
- rules/legislation covering more or less the same subject matter are found in numerous other countries, including in Denmark, which sometimes result in lower or higher exposures in those countries than in the United States.
Ensuring that our business arrangements with third parties comply with applicable healthcare laws and regulations will likely be costly. It is possible that governmental authorities will conclude that our
35
Table of Contents
business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations were found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement, individual imprisonment, possible exclusion from government funded healthcare programs, such as Medicare and Medicaid, integrity obligations, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could substantially disrupt our operations. If the physicians or other providers or entities with whom we expect to do business are found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusion from government funded healthcare programs.
The risk of us being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. For example, the definition of the "remuneration" under the federal Anti-Kickback Statute has been interpreted to include anything of value. Further, courts have found that if "one purpose" of remuneration is to induce referrals, the federal Anti-Kickback Statute is violated.
Additionally, recent healthcare reform legislation has strengthened federal and state healthcare fraud and abuse laws. For example, the ACA amends the intent requirement of the federal Anti-Kickback Statute and criminal healthcare fraud statutes to clarify that liability under these statutes does not require a person or entity to have actual knowledge of the statutes or a specific intent to violate them. Moreover, the ACA provides that the government may assert that a claim that includes items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act. Furthermore, on January 31, 2019, the Department of Health and Human Services (HHS) and HHS Office of Inspector General (OIG) proposed an amendment to one of the existing Anti-Kickback safe harbors (42 C.F.R. 1001.952(h)) which would prohibit certain pharmaceutical manufacturers from offering rebates to pharmacy benefit managers ("PBMs") in the Medicare Part D and Medicaid managed care programs. The proposed amendment would remove protection for "discounts" from Anti-Kickback enforcement action, and would include criminal and civil penalties for knowingly and willfully offering, paying, soliciting, or receiving remuneration to induce or reward the referral of business reimbursable under federal health care programs. At the same time, HHS also proposed to create a new safe harbor to protect point-of-sale discounts that drug manufacturers provide directly to patients, and adds another safe harbor to protect certain administrative fees paid by manufacturers to PBMs. If this proposal is adopted, in whole or in part, it could affect the pricing and reimbursement for any products for which we receive approval in the future. Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws.
Our employees and collaboration partners may engage in misconduct or other improper activities, including violating applicable regulatory standards and requirements or engaging in insider trading, which could significantly harm our business.
We are exposed to the risk of employee fraud or other misconduct and the fraud and misconduct of our collaboration partners. Misconduct by our employees or our collaboration partners could include intentional failures to:
- •
- comply with legal requirements or the requirements of the FDA, the EMA, the CMMS and other comparable regulatory authorities;
- •
- provide accurate information to applicable government authorities;
36
Table of Contents
- •
- comply with fraud and abuse and other healthcare laws and regulations in the United States, or similar laws in Denmark and elsewhere;
- •
- comply with the FCPA and other applicable anti-bribery laws;
- •
- report financial information or data accurately; or
- •
- disclose unauthorized activities to us.
In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing, bribery and other abusive practices. These laws and regulations restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee or collaboration partner misconduct could also involve the improper use of, including trading on, information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a code of conduct, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may be ineffective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we or such collaboration are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions. Further, if any actions are instituted against any of our collaboration partners and such partner fails to defend itself or assert its rights and as a result, is subjected to criminal, civil or administrative sanctions, including exclusion from government funded healthcare programs, such actions and outcomes could have a significant impact on our business.
Changes in Danish, U.S. or other foreign tax laws or compliance requirements, or the practical interpretation and administration thereof, could have a material adverse effect on our business, financial condition and results of operations.
We are affected by various Danish, U.S. and foreign taxes, including direct and indirect taxes imposed on our global activities, such as corporate income, withholding, customs, excise/energy, value added, sales, environmental and other taxes. Significant judgment is required in determining our provisions for taxes and there are many transactions and calculations where the ultimate tax determination is uncertain.
In recent years, tax authorities around the world have increased their scrutiny of company tax filings, and have become more rigid in exercising any discretion they may have. As part of this, the Organization for Economic Co-operation and Development, or OECD, has proposed a number of tax law changes under its Base Erosion and Profit Shifting, or BEPS, Action Plans to address issues of transparency, coherence and substance.
At the same time, the European Commission is finalizing its Anti Tax Avoidance Directive, which seeks to prevent tax avoidance by companies and to ensure that companies pay appropriate taxes in the markets where profits are effectively made and business is effectively performed. The European Commission also continues to extend the application of its policies seeking to limit fiscal aid by Member States to particular companies, and the related investigation of the Member States' practices regarding the issuance of rulings on tax matters relating to individual companies.
These OECD and EU tax reform initiatives also need local country implementation, including in our home country of Denmark, which may result in significant changes to established tax principles. Although we have taken steps to be in compliance with the evolving OECD and EU tax initiatives, and will continue to do so, significant uncertainties remain as to the outcome of these efforts.
37
Table of Contents
In addition, in the United States, on December 22, 2017, President Trump signed into law the tax reform legislation commonly referred to as the Tax Cuts and Jobs Act of 2017, which includes substantial changes to the US taxation of individuals and businesses. Although the new law substantially decreased tax rates applicable to corporations in the United States, we do not yet know what all of the consequences of this new statute will be, including whether the law will have any unintended consequences. In particular, significant uncertainties remain as to how the U.S. government will implement the new law, including with respect to the tax qualification of interest deductions, the concept of a territorial tax regime, royalty payments and cost of goods sold.
In general, such tax reform efforts, including with respect to tax base or rate, transfer pricing, intercompany dividends, cross border transactions, controlled corporations, and limitations on tax relief allowed on the interest on intercompany debt, will require us to continually assess our organizational structure against tax policy trends, and could lead to an increased risk of international tax disputes and an increase in our effective tax rate, and could adversely affect our financial results.
Changes in Danish or foreign direct or indirect tax laws or compliance requirements, including the practical interpretation and administration thereof, including in respect to market practices, or otherwise, could have a material adverse effect on our business, financial position, results of operations and future growth prospects.
The UK's pending withdrawal from the EU could result in increased regulatory and legal complexity, which may make it more difficult for us to do business in the EU and the rest of Europe and impose additional challenges in securing regulatory approval of our product candidates in the EU and the rest of Europe.
The UK is a major market for pharmaceutical products, in general, and for products that treat diabetes, in particular. On June 23, 2016, the UK voted to leave the EU in an advisory referendum, which is generally referred to as Brexit. On March 29, 2017, the UK delivered notice under Article 50 of the Lisbon Treaty of its intent to leave the EU. The UK is currently scheduled to leave the EU on May 29, 2019, unless this date is extended. To date there has been no agreement between the EU and the U.K. on the terms of the exit.
Brexit, the subsequent high-profile failures of the UK to agree on an exit strategy, and the pending withdrawal of the UK, may lead to legal uncertainty and potentially divergent laws and regulations between the UK and the EU, as the UK determines which EU laws to replicate or replace and this uncertainty may persist for years. We cannot predict whether or not the UK will significantly alter its current laws and regulations in respect of the pharmaceutical industry and, if so, what impact any such alteration would have on us or our business. Moreover, we cannot predict the impact that Brexit will have on (i) the marketing of pharmaceutical products, (ii) the process to obtain regulatory approval in the United Kingdom for product candidates or (iii) the award of exclusivities that are normally part of the EU legal framework (for instance Supplementary Protection Certificates, Pediatric Extensions or Orphan exclusivity).
Brexit may also result in a reduction of funding to the EMA if the UK no longer makes financial contributions to European institutions, such as the EMA. If UK funding is so reduced, it could create delays in the EMA issuing regulatory approvals for our product candidates and, accordingly, have a material adverse effect on our business, financial position, results of operations and future growth prospects.
As a result of Brexit, other European countries may seek to conduct referenda with respect to their continuing membership with the EU. Given these possibilities and others we may not anticipate, as well as the absence of comparable precedent, it is unclear what financial, regulatory and legal implications the withdrawal of the United Kingdom from the EU would have and how such withdrawal would affect us, and the full extent to which our business could be adversely affected.
38
Table of Contents
In addition, following the Brexit vote, the EU has decided to move the headquarters of the EMA from the UK to the Netherlands by March 2019. It is expected that a significant percentage of the current employees of the EMA will decide not to make the move to the Netherlands. This raises the possibility that new drug approvals in the EU could be delayed as a result.
PCAOB inspection of our independent auditors
With Zealand Pharma being a public company listed in the United States, our independent public accounting firm, Deloitte Statsautoriseret Revisionspartnerselskab (CVR no 33 96 35 56), is registered with the PCAOB and therefore required to undergo regular PCAOB inspections to assess the registered accounting firm's compliance with United States law and professional standards in connection with its audits of financial statements filed with the SEC.
39
Table of Contents
ITEM 4 INFORMATION ON THE COMPANY
A. HISTORY AND DEVELOPMENT OF THE COMPANY
We were founded in 1998 and are a biotechnology company focused on the discovery, design and development of innovative peptide-based medicines. We intend to be a leader in specialty medicines focusing on metabolic and gastrointestinal diseases and other specialty disease areas with significant unmet medical needs.
Our current pipeline of internal product candidates focuses on specialty gastrointestinal and metabolic diseases. Our portfolio also includes two clinical license collaborations with Boehringer Ingelheim.
Since 2003 we have had a license collaboration with Sanofi, or the Sanofi License Agreement, in the diabetes field pertaining to the development and commercialization of lixisenatide, both as a standalone therapy and as a combination therapy. Pursuant to the Sanofi License Agreement, we were entitled to receive certain royalties and commercial milestones in respect of global net-sales of Soliqua® 100/33/ Suliqua® and Lyxumia®/Adlyxin® . On September 6, 2018 we, together with two of our wholly-owned subsidiaries, entered into a purchase and sale agreement with Royalty Pharma, or the Royalty Pharma Agreement, pursuant to which we sold and transferred our and our subsidiaries' respective rights to receive royalties and USD 85 million of potential commercial milestones in respect of global net sales of Soliqua® 100/33/ Suliqua® and Lyxumia®/Adlyxin® from and after July 1, 2018. On September 17, 2018 we received DKK 1,310.2 million, or $205.0 million, upon closing of the transactions contemplated by the Royalty Pharma Agreement and redeemed our ZP SPV Notes issued in 2014 and related debt, making us debt free. The net gain from the transaction amounted to DKK 1,098.9 million, or $170.6 million. In addition, we also remained eligible for a payment from Sanofi of potentially up to USD 15.0 million related to an offset from a settlement of an intellectual property dispute. However, we cannot be certain with regards to the timing and final amount of this payment, as both are dependent on factors that are outside of our control.
Under the terms of the Royalty Pharma Agreement, we remain required to perform certain of our obligations under the Sanofi License Agreement and to perform cetain administrative obligations under the Royalty Pharma Agreement, which include that retain the underlying intellectual property and pay the relevant renewals thereon in a timely manner. A failure by us to comply with the terms of the Royalty Pharma Agreement or the Sanofi License Agreement may place us in breach of our contractual obligations with either party, expose us to liability for indemnification of either party and/or may result in arbitration and/or litigation against us that may lead, in the event of an adverse finding against us, to an award of significant damages, loss of profits and/or award of attorney fees against us. For a further discussion of the Royalty Pharma Agreement and the Sanofi License Agreement, see note 7 in the consolidated financial statements of our Annual Report 2018.
Our pipeline focuses on gastrointestinal, metabolic and other specialty diseases where we believe that the present standard of care is inadequate and where we have the resources to advance our peptide-based product candidates into the later stages of clinical development, including registration and commercialization, while opportunistically considering partnership relationships that may arise.
We have a track record of successfully inventing and developing novel peptide-based product candidates. This success is based on our deep understanding of peptide chemistry and extensive experience in improving the therapeutic characteristics of naturally-occurring peptides by modifying and optimizing their structures. The modifications we make are designed to improve upon naturally occurring peptides so that their therapeutic benefit, duration of action, stability and/or convenience of use favorably compare to other treatment options.
Our research and development, organization is structured to enable dynamic collaboration across various functions and project teams at each stage of discovery and development, allowing us to advance
40
Table of Contents
promising opportunities quickly and take advantage of our extensive knowledge of peptide design and product development. We are looking to focus our efforts on drug candidates that may qualify for orphan/rare disease status.
Our shares are listed on Nasdaq Copenhagen (ZEAL). Our American Depositary Shares (ADS) are listed on the Nasdaq Global Select Market ("NASDAQ") in the United States (ZEAL).
Legal name: Zealand Pharma A/S
Commercial name: Zealand Pharma
Domicile: Smedeland 36, 2600 Glostrup, Denmark
Tel: +45 8877 3600
Fax: +45 8877 3898
Website: zealandpharma.com
The SEC maintains an Internet site at www.sec.gov, that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. Our website address iswww.zealandpharma.com.
(The contents of this website are not incorporated by reference into this Annual Report on Form 20-F.)
Date of incorporation: December 22, 1998
Legal form of the Company: A Danish limited liability company
Legislation under which the Company operates: Danish law
Country of incorporation: Denmark
Emerging Growth Company
As a company with less than $1.07 billion in revenue during our last fiscal year, we qualify as an "emerging growth company," as defined in the Jumpstart Our Business Startups Act ("JOBS Act"). An emerging growth company may take advantage of specified reduced reporting and other burdens that are otherwise applicable generally to public companies. These provisions include:
- •
- a requirement to have only two years of audited financial statements and only two years of related Management's Discussion and Analysis of Financial Condition and Results of Operations disclosure; and
- •
- an exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting pursuant to the Sarbanes-Oxley Act.
We may take advantage of these provisions for up to five years or such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company as of the end of any fiscal year following the fifth anniversary of our initial public offering or if, prior to that date, we have more than $1.07 billion in annual revenue or more than $700 million in market value of the equity securities held by non-affiliates as of the end of our second fiscal quarter, or on the date that we have issued more than $1 billion of non-convertible debt over a three-year period. We may choose to take advantage of some but not all of these reduced burdens, and therefore the information that we provide holders of shares and ADSs may be different than the information available from other public companies. In addition, Section 107 of the JOBS Act also provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards applicable to public companies. We currently prepare our consolidated financial statements in
41
Table of Contents
accordance with IFRS as issued by the IASB, which do not have separate provisions for publicly traded and private companies. However, in the event that we convert to accounting principles generally accepted in the United States (which we do not currently intend to do) while we remain an emerging growth company, we have irrevocably elected to opt out of such extended transition period.
Important events in 2018
Reference is made to '2018 Achievements', page 9 in our Annual Report 2018 for a description of important events in 2018. Further, reference is made to Note 7 'Other operating income' and Note 20 'Royalty bond' to the consolidated financial statements on page 75 and 82-83 in our Annual Report 2018 for discussion regarding the sale of future royalties and the repayment of the Royalty bond.
Capital expenditure
Capital expenditures primarily relate to purchase of lab equipment at our location in Glostrup, Denmark. All capital expenditures are expected to be financed internally through cash flows from operations.
We have no capital expenditure for property, plant and equipment for the interim period of 2019 prior to the publication of this Annual Report on Form 20-F.
Public takeover offers in respect of the Company's shares
No such offers occurred during 2018 or 2019 to date.
B. BUSINESS OVERVIEW
We were founded in 1998 and is a biotechnology company focused on the discovery and development of innovative peptide-based medicines. More than 10 drug candidates invented by us have advanced into clinical development, of which two have reached the market. Our current pipeline of internal product candidates focus on specialty gastrointestinal and metabolic diseases. Our portfolio also includes two clinical license collaborations with Boehringer Ingelheim reference is made to 'Partnered Programs:Obesity/type 2 diabetes, page 29 in our Annual Report 2018.
We have four fully owned programs in late clinical development:
1 Glepaglutide, a long-acting GLP-2 analog in development for the treatment of short bowel syndrome (SBS).
The pivotal Phase 3 trial in 129 patients was initiated in Q4 2018 with expected results by mid- 2020.
Dasiglucagon, a Zealand-invented proprietary glucagon analog currently in development for three different indications:
2 Dasiglucagon in Dual-hormone pump therapy for diabetes treatment
We have already reported positive results from two Phase 2a trials during the second quarter of 2017, and the initiation of a small Phase 2b trial in iLet™ dual-hormone artificial pancreas system is planned for 2019.
3 Dasiglucagon Hypoplal ® Rescue Pen for severe hypoglycemia
Ready-to-use dasiglucagon may offer diabetes patients and their families a fast treatment solution for severe hypoglycemia that is easier to use than currently marketed glucagon kits. The pivotal Phase 3
42
Table of Contents
trial with dasiglucagon for the treatment of severe hypoglycemia was completed with good results in 2018. A pediatric Phase 3 trial was initiated in the end of 2018, with results expected in H2 2019.
4 Dasiglucagon for Congenital hyperinsulinism
Congenital hyperinsulinism, or CHI, is an ultra-rare but devastating disease caused by inappropriately elevated insulin secretion irrespective of glucose levels. This leads to frequent and often severe hypoglycemia and long-term irreversible damage to health. In 2017, the FDA in the U.S. and the Committee for Orphan Medicinal Products in the EU issued a positive opinion on an orphan medicinal product application for our glucagon analog. In January 2018, the FDA issued a safe-to-proceed letter, and a first Phase 3 trial started in Q1 2019.
In addition to the late stage clinical programs we also have a pipeline of pre-clinical programs with the potential to enter into the clinic in 2019 and the years to come.
Our Product Pipeline
We operate within the global market for peptide based medicines. All revenues are generated from milestone and royalty payments related to our license agreements. See Note 2 'Revenue' to the consolidated financial statements on pages 64-66 in our Annual Report 2018 for disclosure of revenue by category. We do not sell products to specific geographic markets.
The chart below summarizes the development stage and status of our portfolio of product candidates:
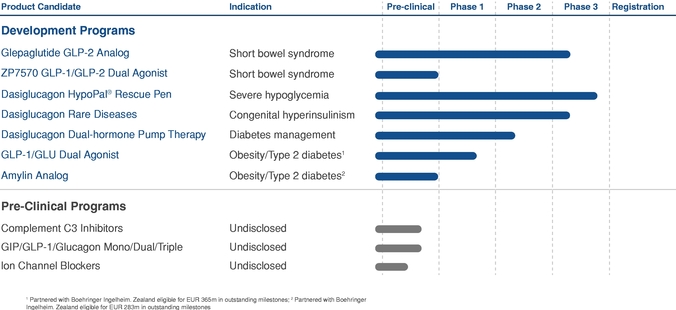
- (1)
- Partnered with Boehringer Ingelheim. Zealand eligible for EUR 365m in outstanding milestones;
- (2)
- Partnered with Boehringer Ingelheim. Zealand eligible for EUR 283m in outstanding milestones
43
Table of Contents
In-house inventions are the basis of our portfolio, demonstrating our ability to discover and develop innovative peptide-based product candidates with favorable therapeutic profiles.
Our Focus on Peptide-Based Medicines
We currently focus on specialty gastrointestinal and metabolic diseases where we believe that the present standard of care is inadequate and where we believe that we have the resources to advance our peptide-based product candidates into the later stages of clinical development, including registration and, potentially, commercialization, while opportunistically considering partnership relationships that may arise. In addition, we are looking to focus our efforts on drug candidates that may qualify for orphan/rare disease status. Our R&D organization is structured to enable dynamic collaboration across various functions and project teams at each stage of discovery and development, allowing us to advance promising opportunities quickly and take advantage of our extensive knowledge of peptide design and product development.
We have a track record of successfully inventing and developing novel peptide-based product candidates. This success is based on our deep understanding of peptide chemistry and extensive experience in improving the therapeutic characteristics of naturally-occurring peptides by modifying and optimizing their structures. The modifications we make are designed to improve upon naturally occurring peptides so that their therapeutic benefit, duration of action, stability and convenience of use favorably compare to other treatment options.
Peptides generally have a number of advantages as drug candidates that can provide specific therapeutic benefits including: high selectivity with effects only on the intended target thereby providing specific therapeutic benefits; lower risks of toxicity with limited, or no, off-target effects; high potency with strong effects even at low concentrations; favorable safety profiles (including fewer side effects) with minimal drug-to-drug interactions, tailored half-lives and binding affinity; and high regulatory approval rates, with approval rates of 20%, as compared to 10% for small molecule medications. Peptides are also smaller than proteins on a molecular level, which can offer potential advantages in terms of therapeutic administration.
Our peptide chemistry and pharmaceutical development expertise is complemented by strong downstream development competencies, including a clinical development team with experience in quality assurance and in regulatory matters. We believe that we have the requisite in-house capabilities to advance product candidates in our selected disease areas from preclinical Investigational New Drug, orIND enabling studies to late-stage clinical development, including registration.
Reference is made to the sections 'Our programs on pages 18-31 in our Annual Report 2018.
Segment information
The Group is managed by a corporate management team reporting to the Chief Executive Officer. The corporate management team, including the Chief Executive Officer, represents the chief operating decision maker (CODM). No separate business areas or separate business units have been identified in connection with product candidates or geographical markets. As a consequence of this, no segment reporting is made concerning business areas or geographical areas.
Seasonality
The Company's financial performance, financial position and cash flows are not subject to significant seasonality.
44
Table of Contents
Raw materials
A number of raw materials are used to produce our proprietary product candidates. The bulk of the raw materials are items that are also used by other pharmaceutical producers, so are generally not difficult for us to obtain. We are dependent only on suppliers of raw materials solely for use in the preclinical and clinical development stages of our product candidates. The raw materials have relatively low price volatility.
Market and competition
The pharmaceutical and biotechnology industries are characterized by intense competition and significant and rapid technological change as researchers learn more about diseases and develop new technologies and treatments. Significant competitive factors in our industry include: (i) product safety and efficacy; (ii) quality and breach of an organization's technology; (iii) skill of an organization's employees and its ability to recruit and retain key employees; (iv) timing and scope of regulatory approvals; (v) government reimbursement rates for, and the average settling price of, products; (vi) the availability of raw materials and qualified manufacturing capacity; (vii) manufacturing costs; (viii) intellectual property and patent rights and their protection; and (ix) sales and marketing capabilities. While we believe that our product and product candidate platform, development expertise and scientific knowledge provide us with competitive advantages, we face potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions and governmental agencies and public and private research institutions.
Any product candidates that we successfully develop and commercialize will compete with existing drugs and new drugs that may become available in the future.
We compete with companies that are producing drugs for, among other disease indications, SBS, such as Takeda plc which currently markets and distributes Gattex, and hypoglycemia, such as Novo Nordisk and Eli Lilly which each market and distribute glucagon rescue kits.
Our competitors may also succeed in obtaining FDA, EMA or other regulatory approvals more rapidly than us, which could place us at a significant competitive disadvantage or deny us marketing exclusivity rights. Market acceptance of our product candidates will depend on a number of factors, including:
- •
- potential advantages over existing or alternative therapies or tests;
- •
- the actual or perceived safety of similar classes of products;
- •
- the effectiveness of our sales, marketing and distribution capabilities; and
- •
- the scope of any approval provided by the FDA, the EMA or other comparable regulatory authorities.
Although we believe our drugs and product candidates possess attractive attributes, we cannot ensure that our product candidates will achieve regulatory or market acceptance, or that we will be able to compete effectively in the market.
If our product candidates fail to gain regulatory approvals and acceptance in their intended markets, we may not generate meaningful revenue or achieve profitability.
In addition, many of our competitors have significantly greater financial resources and expertise in R&D, manufacturing, preclinical studies, conducting clinical trials, obtaining regulatory approvals and marketing drugs. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of competitors, particularly through partnership arrangements with large established companies. These companies also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
45
Table of Contents
Patents
Patent Strategy
Our strategy for filing patent applications is to file early in the drug discovery process, typically before a lead compound has been selected. Before filing an initial patent application, we conduct searches of patents and publications based on keywords, patent classification codes and/or sequences to verify patentability of the compounds identified to date. A more focused, structure-based search is conducted once a lead compound is selected.
Patent applications are generally prepared by our in-house patent professionals in collaboration with outside patent counsel. Patent applications drafted before a lead compound is chosen typically disclose a large number of structurally related compounds. Our patent applications cover compositions of matter and methods of use, and may additionally cover dosing regimens and methods of making the compounds. Later-filed patent applications typically cover next-generation compounds having for example, structural differences that might confer improved properties. Initially, we file one or more patent applications that establish priority to be claimed in later-filed applications. For most patent families, we file a patent application under the International Patent System ("PCT") which can be entered for examination into the patent office in any of the countries that are signatories to the PCT. In some cases, we file national applications directly in the major jurisdictions, which include Europe, the United States and Japan. For certain patent families, we file in parallel an application under the PCT and national applications in certain jurisdictions, such as the United States. Our patent strategy includes an evaluation of the of third party patents that may be infringed by our drug candidate products and development programs, and we prepare our development and commercialization plans to avoid claims of infringement. To the extent that we identify any potential issue with third party patents that may affect any of our product candidates, we develop a strategy to deal with such third party patents by ensuring that we are satisfied that such patents are invalid, not infringed, or that we commercialize our products after the expiry of such patents. Such strategies can include seeking a judicial or administrative revocation of such patents, ensuring that we are in a position to defend a claim for infringement, or seeking a license where that is appropriate.
We or our outside patent counsel handle the prosecution of our patent applications. If we enter into a licensing arrangement with a collaboration partner, we typically retain ultimate control of patent prosecution of patent applications for our inventions. For new inventions arising from collaboration under the license agreement, the collaboration partner may, depending on the identity of the inventors, file patent applications that are owned either by the collaboration partner alone or jointly with us.
Patent and Patent Application Portfolio
We own one patent family covering lixisenatide, including 45 non-US patents in 10 non-US jurisdictions, all licensed exclusively to Sanofi. We own two patent families covering two related proprietary GLP-2 analogs, glepaglutide and elsiglutide or backup candidates. These two patent families include 59 non-US patents in 16 non-US jurisdictions and 7 pending patent applications in 4 jurisdictions. Although the disclosures of one of these two patent families encompass both elsiglutide and glepaglutide, it has been possible to claim the subject matter relating to elsiglutide and glepaglutide in separate patents in the United States. For our internal compound dasiglucagon, a glucagon analog that has a favorable stability profile, we own one patent family including 19 pending non-US patent applications and in 18 non-US jurisdictions.
We also possess Intellectual Property ("IP") with respect to certain technologies we employ when designing novel peptide drug candidates. An example of one of our internal peptide enhancing technologies is the SIP technology. The SIP technology adds a number of specific amino acids to a peptide thereby strengthening or tightening the molecular structure to make the peptide less susceptible to biological degradation. This may help maintain the peptide in the blood for a longer period of time
46
Table of Contents
before the peptide is degraded and may permit less frequent dosing of the peptide. The SIP technology has been employed for the development of lixisenatide, glepaglutide and elsiglutide.
Other proprietary technologies we use involve the addition of a fatty acid to the amino acid chain of a given peptide as another technique to increase the half-life of the peptide in the blood stream.
Although specific reference is made to the status of patents granted or pending in the USPTO, the EPO, and Japan, in many cases the patent families also include patents or applications in a number of additional jurisdictions, including Australia, Canada, China, and India. Nominal expiration dates mentioned below are based on the statutory patent terms. Upon marketing approval, patent term extensions or supplementary protection certificates may be obtainable in various jurisdictions, including the United States, certain European jurisdictions, and Japan, with respect to certain patents claiming compositions of matter, methods of use or methods of manufacturing the products, with a maximum of five years of extension potentially available. U.S. patents may also be entitled to adjustments to their statutory patent term depending on the length of the delay to the issuance of the patents caused by the USPTO.
We (or our wholly owned subsidiary, ZP Holding, in the case of lixisenatide) own all patents and applications described below, as relating to lixisenatide, elsiglutide, glepaglutide and dasiglucagon.
Lixisenatide
Our wholly owned subsidiary, ZP Holding, owns two patent families covering lixisenatide compound and compositions, methods of using lixisenatide and dosing regimens for administering lixisenatide. The first patent family includes granted patents in Europe (one European patent validated in 25 countries and a second European patent validated in 18 countries), the United States, Australia (two), Canada, China, Hong Kong (two), India, Israel (three), Japan (three), New Zealand and South Africa. The patents include composition of matter claims covering both the peptide and composition of lixisenatide, originally known as ZP10. The granted U.S. patent originally claimed lixisenatide and closely related analogs of lixisenatide, but was reissued with claims covering lixisenatide only. The closely related analogs may be pursued in one or more continuing applications. The granted European patents include claims drawn to the composition of matter of a genus of compounds that encompasses lixisenatide and of lixisenatide, compositions comprising lixisenatide, and uses of lixisenatide to treat various diseases, including type 2 diabetes, type 1 diabetes and obesity. The patents in this patent family have a nominal expiration date in July 2020 and may be eligible for up to 5 years of regulatory patent term extension in several jurisdictions, including certain European jurisdictions, Japan, Israel and the United States.
Elsiglutide and glepaglutide
We own two patent families, which include 59 non-US patents and 16 pending non-US applications in a total of 54 non-US jurisdictions. The first patent family discloses both the elsiglutide compound and the glepaglutide compound, which are structurally-related GLP-2 receptor agonists developed using our SIP technology.
The first patent family includes six granted U.S. patents and one granted European patent (validated in 30 countries). The first patent family also includes granted patents in Australia, Canada, China (two), Eurasia (designated in 9 countries), Hong Kong, Israel (three), India(two), Japan (thre), South Korea (three), Mexico, New Zealand (three), Ukraine and South Africa(two). The granted U.S. patents include composition of matter claims covering both the peptide and composition of elsiglutide and glepaglutide, and claims drawn to a method of treating CID by administering elsiglutide and a method of treating inflammatory bowel disease by administering glepaglutide. The granted European patent includes claims drawn to the composition of matter of a genus of compounds that encompasses elsiglutide and glepaglutide and of elsiglutide and glepaglutide, as well as analogs thereof and methods
47
Table of Contents
of using elsiglutide and glepaglutide for the preparation of a medicament to treat or prevent side effects of chemotherapy, including diarrhea. Twelve applications are pending in this family in the US and non-US jurisdictions, including Brazil, Europe, Hong Kong, India, Japan, Norway and South Africa. The patents in this patent family have a nominal expiration date in May 2026, with the exception of one of the granted U.S. patents, which has an adjusted expiration date in June 2026.
The granted European patent in this first patent family was opposed in the EPO by a competitor. The Opposition Division of the EPO maintained all original claims at a hearing in June 2014. The decision to maintain the patent with narrow claims covering glepaglutide and elsiglutide was made final at an oral hearing November 2018 before the Technical Board of Appeals of the EPO.
A second patent family covers backup compounds for elsiglutide. This family includes granted patents in Australia, China, Hong Kong, Israel, Japan, South Korea, Mexico, New Zealand (two), Ukraine and the United States. The second family also includes seven pending patent applications in the United States, Brazil, Canada, Eurasia, Europe, Norway and South Africa. The patents in this second family have a nominal expiration date in November 2027, with the exception of the granted U.S. patent, which has an adjusted expiration date in February 2030.
The patents in each of these patent families were licensed to Helsinn for elsiglutide until June 2017. With the termination of the license agreement with Helsinn, we are now responsible for the maintenance of both patent families.
Dasiglucagon
We own one patent family covering dasiglucagon, including granted patents in 13 European countries and 19 patent applications outstanding in 18 jurisdictions. The pending claims in this patent family cover the dasiglucagon compound and a group of structurally related compounds having glucagon agonist activity and increased solubility and/or stability relative to the native glucagon. The pending claims also cover pharmaceutical compositions comprising such compounds and related uses for treating a variety of diseases including hypoglycemia, type 1 and 2 diabetes and other metabolic conditions, and nucleic acid molecules for expression of the compounds in host cells. The patents in this family, when issued, will have a nominal expiration date in July 2033.
Government Regulation
The FDA and comparable regulatory authorities in state and local jurisdictions and in other countries impose substantial and burdensome requirements upon companies involved in the clinical development, manufacture, marketing and distribution of drugs, such as those we are developing. These agencies and other federal, state and local entities regulate, among other things, the R&D, testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion, distribution, post-approval monitoring and reporting, sampling and export and import of our product candidates.
U.S. Government Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act ("FDCA") and its implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA's refusal to approve pending NDAs, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution,
48
Table of Contents
injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
- •
- completion of preclinical laboratory studies, animal studies and formulation studies in compliance with the FDA's good laboratory practice regulations;
- •
- submission to the FDA of an IND, which must become effective before human clinical trials may begin;
- •
- approval by the institutional review board, or IRB, at each clinical site before each trial may be initiated;
- •
- performance of adequate and well-controlled human clinical trials in accordance with applicable IND and other clinical trial-related regulations, sometimes referred to as good clinical practices to establish the safety and efficacy of the proposed product candidate for its proposed indication;
- •
- submission to the FDA of an NDA;
- •
- satisfactory completion of an FDA pre-approval inspection of the production facility or facilities where the product is produced to assess compliance with the FDA's cGMP requirements to assure that the facilities, methods and controls are adequate to preserve the product's identity, strength, quality, purity and potency;
- •
- potential FDA audit of the preclinical and/or clinical trial sites that generated the data in support of the NDA; and
- •
- FDA review and approval of the NDA prior to any commercial marketing or sale of the product in the United States.
Preclinical Studies
Preclinical studies include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies to assess potential safety and efficacy. An IND sponsor must submit the results of the preclinical studies, together with manufacturing information, analytical data and any available clinical data or literature, among other things, to the FDA as part of an IND. Some preclinical studies may continue even after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical Trials
Clinical trials involve the administration of the IND to human patients under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research patients provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution. Information about
49
Table of Contents
certain clinical trials must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their www.clinicaltrials.gov website.
Human clinical trials are typically conducted in three sequential phases, which may overlap or be combined:
- •
- Phase 1 clinical trial: the product candidate is initially introduced into healthy human patients or patients with the target disease or condition and tested for safety, dosage tolerability, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness;
- •
- Phase 2 clinical trial: the product candidate is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerability and optimal dosage; and
- •
- Phase 3 clinical trial: the product candidate is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Each of Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB's requirements or if the drug has been associated with unexpected serious harm to patients.
Marketing Approval
Assuming successful completion of the required clinical testing, the results of the preclinical studies and clinical trials, together with detailed information relating to the product's chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. In most cases, the submission of an NDA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act, or PDUFA, guidelines that are currently in effect, the FDA has a goal of ten months from the date of "filing" of a standard NDA for a new molecular entity to review and act on the submission. This review typically takes twelve months from the date the NDA is submitted to the FDA because the FDA has approximately two months to make a "filing" decision.
In addition, under the Pediatric Research Equity Act of 2003, or PREA, as amended and reauthorized, certain NDAs or supplements to an NDA must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements.
The FDA also may require submission of a Risk Evaluation and Mitigation Strategies, or REMS, plan to ensure that the benefits of the drug outweigh its risks. The REMS plan could include medication guides, physician communication plans, assessment plans, or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
50
Table of Contents
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing.
Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product's continued safety, quality and purity.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical trial sites to assure compliance with GCP requirements.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA and may require additional clinical or preclinical testing in order for the FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA's satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a drug's safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Post-Approval Requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the
51
Table of Contents
establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product's safety and effectiveness after commercialization. In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP requirements and impose reporting and documentation requirements upon the sponsor and any third party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
- •
- restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
- •
- fines, warning letters or holds on post-approval clinical trials;
- •
- refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product approvals;
- •
- product seizure or detention, or refusal to permit the import or export of products; or
- •
- injunctions or the imposition of civil or criminal penalties.
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
C. ORGANIZATIONAL STRUCTURE
For information regarding our organizational structure, please refer to the section 'Business overview' on page 54 in our Annual Report 2018.
D. PROPERTY, PLANT AND EQUIPMENT
We lease approximately 5,344 square meters of office space at Smedeland 36 and Smedeland 26B, 2600 Glostrup (Copenhagen), Denmark for our corporate headquarters and other administrative functions. The lease for this facility may, subject to certain Danish law restrictions, be terminated by us or the lessor upon six months' prior written notice. We have recently signed a new lease of approximately 7,181 square meters of office and laboratory space at Sydmarken 5, 2860 Søborg, Denmark. The lease is interminable for 13 years for Zealand and 15 years for Ejendomsselskabet
52
Table of Contents
Sydmarken 5 A/S ("the landlord") from the date when Zealand moves in to the facilities, which is expected to be around September 1 2019. After said periods Zealand can, without restrictions, and the landlord, subject to certain restrictions under Danish law, terminate the lease upon 12 months written notice.
ITEM 4A UNRESOLVED STAFF COMMENTS
Not applicable.
53
Table of Contents
ITEM 5 OPERATING AND FINANCIAL REVIEW AND PROSPECTS
Critical accounting estimates
Reference is made to Note 1 'Significant accounting policies and significant accounting estimates and assessments' to the consolidated financial statements on pages 55-63 in our Annual Report 2018.
New accounting pronouncements
Reference is made to Note 1 'Significant accounting policies and significant accounting estimates and assessments' to the consolidated financial statements on pages 55-63 in our Annual Report 2018.
A. OPERATING RESULTS
Reference is made to the section 'Financial review' contained on pages 41-43 in our Annual Report 2018 and "Item 3—Key Information—D. Risk Factors". Reference is further made to 'Risk management and internal control' on pages 38-39 in our Annual Report 2018.
The financial condition of the Group and its development is described in our Annual Report 2018. The information in this section is based on these reports and should be read in conjunction with our Annual Report 2018. The analysis and discussion included in our Annual Report 2018 is primarily based on the consolidated financial statements, which are prepared in accordance with IFRS as issued by the IASB.
2018 compared with 2017
The section 'Financial review' contained on pages 41-43 in our Annual Report 2018 constitutes the Board of Directors' and Executive Management's discussion and analysis of results of operations.
2017 compared with 2016
Revenue for the year ended December 31, 2017 was DKK 136.3 million, as compared to DKK 230.9 million for the year ended December 31, 2016 reflecting a decrease of 41% (or DKK 94.5 million) in 2017, as compared to 2016.
Revenue from milestone payments was DKK 101.0 million for the year ended December 31, 2017, as compared to DKK 210.4 million for the year ended December 31, 2016, reflecting a decrease of 52% (or DKK 109.4 million) in 2017, as compared to 2016. Milestones comprise a payment of DKK 69.6 million from Sanofi in connection with the EU approval of Suliqua® and a payment of DKK 29.8 million from Boehringer Ingelheim related to the initiation of Phase 1 trials for the long-acting amylin analog. A milestone payment of DKK 1.7 million was also received from a license agreement with Protagonist Therapeutics Inc. Milestones in 2016 relate to the FDA approval of Adlyxin and Soliqua® 100/33 in the United States in July 2016 and November 2016, triggering milestone payments from Sanofi of DKK 33.5 million and DKK 175.2 million, respectively. Royalty income was DKK 35.3 million for the year ended December 31, 2017, as compared to DKK 20.4 million for the year ended December 31, 2016, reflecting an increase of 73% (or DKK 14.9 million) in 2017, as compared to 2016. The increase in royalty income was due to decreased sales of Lyxumia offset by royalty income from first year sales of Soliqua® 100/33.
Royalty expenses for the year ended December 31, 2017 were DKK 14.2 million, as compared to DKK 30.9 million for the year ended December 31, 2016. Royalty expenses for the years ended December 31, 2017 and 2016 were comprised of payments that we made with respect to royalty payments received by us from the sales of Lyxumia and Soliqua® 100/33 plus milestone payments received from Sanofi. We pay royalties of 13% on all revenue related to lixisenatide under the Sanofi
54
Table of Contents
License Agreement to Alkermes and 0.5% of the total amounts we receive in connection with our SIP modified peptides to one of the inventors of our SIP technology.
R&D expenses for the year ended December 31, 2017 were DKK 324.7 million, as compared to DKK 268.2 million for the year ended December 31, 2016 reflecting an increase of 21% (or DKK 56.5 million) in 2017, as compared to 2016. The increase in R&D expenses for the year ended December 31, 2017 was primarily related to external costs of DKK 30.6 million from accelerated development activities. This figure comprises costs for the three dasiglucagon programs, including the two Phase 3 trials relating to the rescue pen for severe hypoglycemia and the two Phase 2b trials for dasiglucagon to be used in a dual-hormone artificial pancreas. It also includes costs for finalizing the Phase 2 trial with glepaglutide as well as costs relating to preclinical activities. The R&D share of the personnel expenses for the year ended December 31, 2017 amounted to DKK 119.5 million, as compared to DKK 109.5 million for the year ended December 31, 2016, reflecting an increase of 9.1% (or DKK 10.0 million) in 2017, as compared to 2016. The increase in R&D share of the personnel expenses in the year ended December 31, 2017, as compared to the year ended December 31, 2016, was primarily due to an increase in the number of employees in the clinical development function.
As of December 31, 2017 and 2016, we have not capitalized any development expenses, and, accordingly, all such costs have been recognized within the income statement.
Administrative expenses for the year ended December 31, 2017 were DKK 47.5 million, as compared to DKK 52.5 million for the year ended December 31, 2016, reflecting a decrease of 9.5% (or DKK 5.0 million) in 2017, as compared to 2016.
The decrease in administrative expenses in the year ended December 31, 2017 was primarily related to a change in the composition of employees working in R&D and Administration in comparison to previous year.
Expenses related to staff and remuneration for the year ended December 31, 2017 were DKK 152.0 million, as compared to DKK 145.7 million for the year ended December 31, 2016. The increase in our staff and remuneration costs was primarily due to an increase in salaries as a consequence of an increases in the number of employees. A total of DKK 119.5 million of expenses related to staff and remuneration for the year ended December 31, 2017 was charged to R&D expenses and a total of DKK 32.6 million of expenses related to staff and remuneration for the year ended December 31, 2017 was charged to administrative expenses.
Other operating income for the year ended December 31, 2017 was DKK 0.6 million, as compared to DKK 1.7 million for the year ended December 31, 2016. Other operating income for was primarily comprised of Government grants.
As a result of the foregoing, operating losses for the year ended December 31, 2017 were DKK 249.4 million, as compared to DKK 119.0 million for the year ended December 31, 2016.
Financial income for the year ended December 31, 2017 was DKK 2.1 million, as compared to DKK 0.6 million for the year ended December 31, 2016. The increase in financial income for the year ended December 31, 2017 as compared to the year ended December 31, 2016 was primarily due to interest income which was DKK 2.0 million for the year ended December 31, 2017, as compared to DKK 0.6 million for the year ended December 31, 2016. In each of the years ended December 31, 2016 and 2015, interest income related to our cash and cash equivalents as well as our restricted cash.
Financial expenses for the year ended December 31, 2017 were DKK 33.5 million, as compared to DKK 44.4 million for the year ended December 31, 2016. The decrease in financial expenses in the year ended December 31, 2017 was primarily attributable to interest expenses and amortization on the Royalty bond since half of the outstanding amount was repaid in March 2017.
55
Table of Contents
Pursuant to Danish tax legislation, we have received DKK 5.5million in respect of the tax losses for 2016 and are eligible to receive DKK 5.5 million in respect of tax losses for 2017, which losses originated from qualifying research and development expenditures. These tax benefits comprised the entire current tax benefit in 2017 and 2016.
No deferred tax asset has been recognized in the statement of financial position due to an uncertainty as to when and if tax losses can be utilized against future taxable income.
As a result of the foregoing, our net loss for the year ended December 31, 2017 was DKK 275.3 million, as compared to DKK 157.3 million for the year ended December 31, 2016.
Segment information
The Group is managed by a Corporate Management Team reporting to the Chief Executive Officer. The Corporate Management Team, including the Chief Executive Officer, represents the chief operating decision maker (CODM). No separate business areas or separate business units have been identified in connection with product candidates or geographical markets. As a consequence of this, no segment reporting is made concerning business areas or geographical areas.
Inflation
Inflation for the three most recent fiscal years has not had a material impact on the Group's revenue or net loss.
Foreign currencies
Reference is made to Note 23 'Financial risks' to the consolidated financial statements on pages 84-86 in our Annual Report 2018.
Governmental policies
Please refer to "Item 4—Information on the Company—Government Regulation".
B. LIQUIDITY AND CAPITAL RESOURCES
It is our aim to have an adequate capital structure in relation to the underlying operating results and research and development projects, so that it is always possible to provide sufficient capital to support operations and its long-term growth targets.
The Board of Directors finds that the current capital and share structure is appropriate for the shareholders and the Group and for the Company's present requirements.
As of December 31, 2018, we had cash, cash equivalents and securities of DKK 1,159.2 million. We require cash to meet our operating expenses and capital expenditures. We have funded our cash requirements since our incorporation, primarily with equity financing and milestone and royalty payments from our collaboration partners along with the proceeds of ZP SPV Notes issued in 2014. In September 2018, we entered into the Royalty Pharma Agreement, pursuant to which we and two of our wholly-owned subsidiaries agreed to sell and transfer our and our subsidiaries' respective rights to receive royalties and USD 85 million of potential commercial milestones in respect of global net sales of Soliqua® 100/33/ Suliqua® and Lyxumia®/Adlyxin® from and after July 1, 2018. We received DKK 1,310.2 million, or $205.0 million upon closing of the transaction. The net gain from the transaction amounted to DKK 1,098.9 million / USD 170.6 million.
The overall objectives and policies for our financial risk management are outlined in the Finance Policy, which is approved by the Board of Directors. For further information, reference is made to "Item 11—Qualitative and Quantitative Disclosures about Market Risks".
56
Table of Contents
Financial resources
Reference is made to page 52 'Consolidated statements of financial position' and page 53 'Consolidated statements of cash flows' in our Annual Report 2018.
We believe our financial resources are sufficient to meet its requirements for at least the next 12 months.
Cash flow in 2018, 2017 and 2016
Reference is made to 'Consolidated statements of cash flows' on page 53 and 'Financial review' contained on page 41-43 in our Annual Report 2018 for a discussion of cash flows for the year ended December 31, 2018 to the comparable period in 2017.
Cash outflow from operating activities for the year ended December 31, 2017 was DKK 278.7 million, as compared to an inflow of DKK 40.9 million for the year ended December 31, 2016. The decrease in cash outflow from operating activities for the year ended December 31, 2017 was mainly as a result of the net loss for the year adjusted for non-cash items.
Cash inflow from investing activities for the year ended December 31, 2017 was DKK 221.4 million, as compared to an outflow of DKK 300.0 million for the year ended December 31, 2016. The change in cash inflow in 2017, as compared to 2016, was primarily due to the transfer of milestone payments received from Sanofi during 2017 from restricted cash to cash and cash equivalents in conjunction with the repayment of 50.4% of the royalty bond.
Cash inflow from financing activities for the year ended December 31, 2017 was DKK 337.9 million, as compared to an outflow of DKK 157.1 million for the year ended December 31, 2016. The increase in cash inflow from financing activities for the year ended December 31, 2017 was related to the net proceeds of DKK 507.5 from the initial public offering and a capital increase of DKK 6.8 million due to the exercise of warrants.
The total cash inflow for the year ended December 31, 2017 was DKK 280.5 million as compared to an outflow of DKK 101.9 million for the year ended December 31, 2016.
There are no material restrictions on the ability of subsidiaries with material cash amounts to transfer funds to the Parent Company.
Debt financing
Reference is made to 'Consolidated statements of financial position' on page 52 and Note 20 'Royalty bond' to the consolidated financial statements on pages 82-83 in our Annual Report 2018 for information on current debt financing. In connection with the closing of the Royalty Pharma Agreement, we redeemed our ZP SPV Notes issued in 2014 and related debt, making us debt free.
Financial instruments
Reference is made to Note 23 'Financial risks' to the consolidated financial statements on pages 84-86 in our Annual Report 2018.
Commitments for capital expenditure etc.
Contractual obligations for capital expenditure and other contingent liabilities as of December 31, 2018, respectively, are shown in Note 22 'Contingent assets, liabilities and other contractual obligations' on pages 83-84 to the consolidated financial statements in our Annual Report 2018.
The Executive Management of the Group believes that the obligations are covered by the Group's financial resources as well as expected future cash flows from operating activities.
57
Table of Contents
C. RESEARCH AND DEVELOPMENT, PATENTS AND LICENSES, ETC.
We currently focus on gastrointestinal, metabolic and other specialty diseases where we believe that the present standard of care is inadequate and where we believe that we have the resources to advance our peptide-based product candidates into the later stages of clinical development, including registration and, potentially, commercialization, while opportunistically considering partnership relationships that may arise. In addition, we are looking to focus our efforts on drug candidates that may qualify for orphan/rare disease status. Our R&D organization is structured to enable dynamic collaboration across various functions and project teams at each stage of discovery and development, allowing us to advance promising opportunities quickly and take advantage of our extensive knowledge of peptide design and product development.
Reference is made to 'Research and development expenses' in 'Financial review' on pages 41-43 in our Annual Report 2018 for Research and development expenses in 2018, 2017 and 2016, respectively.
Information related to selected research and development projects can be found under 'Our programs' on pages 18-31 in our Annual Report 2018.
Reference is made to "Item 3—Key Information—D. Risk Factors".
D. TREND INFORMATION
We do not currently produce, hold inventory or sell any products by ourselves or through partnerships.
Information about expectations for the financial year 2019 can be found on page 10 in the subsection 'Financial highlights and 2019 guidance" in our Annual Report 2018.
E. OFF-BALANCE SHEET ARRANGEMENTS
Reference is made to Note 22 'Contingent assets, liabilities and other contractual obligations' to the consolidated financial statements pages 83-84 in our Annual Report 2018.
F. TABULAR DISCLOSURE OF CONTRACTUAL OBLIGATIONS
2018
| | | | | | | | | | | | | | | | |
DKK thousand | | Within 1 year | | 1 - 3 years | | 3 - 5 years | | More
than
5 years | | Total | |
|---|
Operating leases | | | 6,945 | | | 31,098 | | | 29,464 | | | 0 | | | 67,507 | |
Contractual obligations related to agreements with contract research organizations | | | 156,369 | | | 88,621 | | | 618 | | | 0 | | | 245,608 | |
| | | | | | | | | | | | | | | | | |
Total obligations not recognized in the statement of financial position | | | 163,314 | | | 119,719 | | | 30,082 | | | 0 | | | 313,115 | |
| | | | | | | | | | | | | | | | | |
Total contractual obligations | | | 163,314 | | | 119,719 | | | 30,082 | | | 0 | | | 313,115 | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
58
Table of Contents
2017
| | | | | | | | | | | | | | | | |
DKK thousand | | Within 1 year | | 1 - 3 years | | 3 - 5 years | | More
than
5 years | | Total | |
|---|
Royalty bond | | | 2,748 | | | 6,284 | | | 144,769 | | | 0 | | | 153,801 | |
| | | | | | | | | | | | | | | | | |
Total obligations recognized in the statement of financial position | | | 2,748 | | | 6,284 | | | 144,769 | | | 0 | | | 153,801 | |
| | | | | | | | | | | | | | | | | |
Operating leases | | | 4,292 | | | 2,593 | | | 117 | | | 0 | | | 7,002 | |
Contractual obligations related to agreements with contract research organizations | | | 52,589 | | | 22,742 | | | 1,305 | | | 0 | | | 76,636 | |
| | | | | | | | | | | | | | | | | |
Total obligations not recognized in the statement of financial position | | | 56,881 | | | 25,335 | | | 1,422 | | | 0 | | | 83,638 | |
| | | | | | | | | | | | | | | | | |
Total contractual obligations | | | 59,629 | | | 31,619 | | | 146,191 | | | 0 | | | 237,439 | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Reference is made to Note 22 'Contingent assets, liabilities and other contractual obligations' to the consolidated financial statements on pages 83-84 in our Annual Report 2018.
G. SAFE HARBOR
Forward-looking information discussed in this Item 5 includes assumptions, expectations, projections, intentions and beliefs about future events. These statements are intended as "forward-looking statements". We caution that assumptions, expectations, projections, intentions and beliefs about future events may and often do vary from actual results and the differences can be material. Please see the section entitled "Forward-Looking Statements" at the beginning of this Annual Report.
59
Table of Contents
ITEM 6 DIRECTORS, EXECUTIVE MANAGEMENT AND EMPLOYEES
A. DIRECTORS AND EXECUTIVE MANAGEMENT
Reference is made to 'Board of Directors and Corporate Management' on pages 46-47 in our Annual Report 2018 for name, position and period of service as director for the members of our Board of Directors.
Reference is made to 'Corporate Management' on page 48 in our Annual Report 2018 for name, position, age, year of appointment and year of joining Zealand for the members of Corporate Management.
The Board of Directors has the overall responsibility for the affairs of the Company.
The activities of the members of Board of Directors and members of Corporate Management outside the Company are included in Board of Directors and Corporate Management' on pages 46-48 in our Annual Report 2018.
There are no family relationships between the Board of Directors and Corporate Management. No Director or member of Corporate Management have been elected according to an arrangement or understanding with shareholders, customers, suppliers or others.
As required by the Danish Companies Act, directors are elected at General Meetings by simple majority vote. In addition, three employee representatives are elected for four-year terms by the employees of Zealand Pharma A/S.
B. COMPENSATION
Reference is made to Note 6 'Information on staff and remuneration' to the consolidated financial statements on pages 67-74 in our Annual Report 2018.
C. BOARD PRACTICES
Reference is made to 'Corporate governance' on pages 33-35 in our Annual Report 2018 regarding board practices. The year of election for each member of the Board of Directors and the year of appointment for each member of Corporate Management is included in 'Board of Directors and Corporate Management' on pages 46-48 in our Annual Report 2018.
The terms of office of all the members of our board of directors elected by the general meeting expire at the next annual general meeting to be held in April 2019. All members of the board of directors elected by the general meeting are eligible for re-election. Employee elected board members are elected for a period of four years.
None of our directors has a service contract with us or any of our subsidiaries providing for benefits upon termination of employment.
D. EMPLOYEES
Reference is made to the section entitled 'Our people' on page 37 and 'Consolidated key figures' on page 11 in our Annual Report 2018 regarding the total number of our full-time employees at year-end for the years 2014-2018. Labor unions currently representing our employees include HK it, medie & Industri Hovedstaden. We negotiate a collective agreement in good faith every three years, with the next negotiation scheduled to take place in 2020.
60
Table of Contents
E. SHARE OWNERSHIP
For information on the Board of Directors' and Corporate Management's individual holdings of shares and granting of shares, reference is made to the sections 'Board of Directors' on pages 46-47 and 'Corporate Management' on page 48, and Note 6 'Information on staff and remuneration' to the consolidated financial statements on pages 67-74 in our Annual Report 2018 . The members of our Board of Directors and Corporate Management in the aggregate hold less than 1% of the beneficial ownership of the Company.
For information on the Board of Directors' and Corporate Management's individual holdings of and trading in our shares during 2018, reference is made to the sections 'Board of Directors' on pages46-47 and 'Corporate Management' on page 48, and Note 6 'Information on staff and remuneration' to the consolidated financial statements on pages 67-74 in our Annual Report 2018. As of March 7, 2019 the Board of Directors and Executive Management owned 177,734 shares.
In the period from January 1, 2018 until March 7, 2019, no shares were sold or purchased by the members of the Board of Directors or Executive Management. The internal rules on trading in our shares by members of the Board of Directors and Executive Management only permit trading in the 30 calendar-day period following each quarterly earnings announcement.
We granted warrants to our executive management and selected employees of the company in 2005, 2007 and in each of the years between 2009 and 2018. Since our Annual General Meeting in 2012, it has been part of our remuneration policy that members of the Board of Directors are not permitted to participate in the warrant incentive program in their capacity as board members.
Warrants are, and have been, granted pursuant to shareholder authorizations provided to our board of directors under our articles of association. The terms of the warrants, including the exercise price and the size of the grants, and our guidelines for incentive pay in force at the time of grant are fixed in accordance with this authorization. Warrants are granted for employee services and will typically become exercisable between approximately one to five years after the date of grant and may be exercised to subscribe for shares in a number of pre-defined exercise windows against payment of the exercise price. Unexercised warrants will lapse. Granted warrants to employees are generally subject to provisions reflecting the principles of the Danish Stock Option Act (Aktieoptionsloven), which allows for the forfeiture of unexercised warrants if the grantee separates from the company or one of our subsidiaries under circumstances in which the warrant holder is considered a "bad-leaver", understood as, for example, being dismissed for cause or resigning without us having materially breached the employment contract. Warrant holders may maintain all granted warrants if they separate from the company or one of our subsidiaries under circumstances where they are considered as "good-leavers", such as dismissal without cause, leaving us pursuant to an agreed severance agreement or retirement, warrant holder's resignation due to our material breach of contract or the warrant holder's death. Warrants granted to Britt Meelby Jensen are, however, subject to leaver provisions, which deviate from the principles of the Danish Stock Option Act (Aktieoptionsloven).
Reference is made to Note 6 'Information on staff and remuneration' to the consolidated financial statements on pages 67 –74 in our Annual Report 2018.
61
Table of Contents
ITEM 7 MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
A. MAJOR SHAREHOLDERS
As of November 2, 2010, we had one class of shares (prior to this date we had multiple classes of shares). As of December 31, 2018 our registered, issued and outstanding share capital was DKK 30,786,827 distributed into 30,786,827 shares of nominal value DKK 1 each.
Below is information as of March 7, 2019 with respect to (a) any shareholder who is known to the Company to be the owner of more than 5% of the Company's shares and (b) the total amount of any class owned by Zealand Pharma A/S and its affiliates (treasury shares) and by the Board of Directors and Executive Management as a group:
| | | | | | | | | | |
Identify of person or group | | Shares
owned | | Percent
of class | | Percent
of total
votes | |
|---|
Wellington Management Company LLP, Boston, U.S. | | | 2,437,467 | | | 7.9 | % | | 7.9 | % |
Sunstone Capital A/S, Copenhagen, Denmark | | | 2,104,257 | | | 6.8 | % | | 6.8 | % |
Van Herk Investments, Rotterdam, Netherlands | | | 1,962,535 | | | 6.4 | % | | 6.4 | % |
Bank Julius Bär & Co. AG, Zurich, Switzerland | | | 1,910,921 | | | 6.2 | % | | 6.2 | % |
Zealand Pharma A/S and subsidiaries (treasury shares) | | | 64,223 | | | 0.2 | % | | 0.2 | % |
Board of Directors and Executive Management | | | 195,339 | | | 0.6 | % | | 0.6 | % |
As of December 31, 2018, approximately 45% of our share capital was held in Denmark and approximately 22% of its share capital was held in North America. The total number of shareholders is more than 16,000 of whom more than 85% are estimated to be Danish residents and 0.5% are estimated to be resident in the United States.
No shareholders of the Company have different voting rights from any other shareholder.
For further information reference is made to 'Shareholder information' on pages 44-45 in our Annual Report 2018.
B. RELATED PARTY TRANSACTIONS
We have no related parties with controlling interest.
Our other related parties comprise of the Company's Board of Directors and Corporate Management.
For further information reference is made to Note 24 'Related parties' to the consolidated financial statements on page 86 in our Annual Report 2018.
There have not been and there are no loans to members of the Board of Directors or Corporate Management in 2018, 2017 or 2016.
C. INTERESTS OF EXPERTS AND COUNSEL
Not applicable.
62
Table of Contents
ITEM 8 FINANCIAL INFORMATION
A. CONSOLIDATED STATEMENTS AND OTHER FINANCIAL INFORMATION
The consolidated financial statements required by this item accompany this annual report in the form of our Annual Report 2018 (see Exhibit no. 15.1).
Legal proceedings
There are no legal proceedings at December 31, 2018 or December 31, 2017.
Dividends
We have never declared or paid any cash dividends on our shares and we do not anticipate paying any cash dividends on our shares in the foreseeable future. We intend to retain all available funds and any future earnings to fund the development and expansion of our business. Any future determination related to our dividend policy and the declaration of any dividends will be made at the discretion of our board of directors and will depend on a number of factors, including our results of operations, financial condition, future prospects, contractual restrictions, restrictions imposed by applicable law and other factors our board of directors deems relevant.
In accordance with the Danish Companies Act ("DCA") dividends, if any, are declared with respect to a financial year at the annual general meeting of shareholders in the following year, where the statutory annual report (which includes the audited financial statements) for that financial year is approved. Further, our shareholders may resolve at a general meeting to distribute interim dividends and our Board of Directors may, pursuant to an authorization that will be granted to it by our shareholders prior to completion of this offering, resolve to distribute interim dividends. Any resolution to distribute interim dividends within six months of the date of the statement of financial position as set out in our latest adopted annual report must be accompanied by the statement of financial position from our latest annual report or an interim statement of financial position which must be reviewed by our auditor. If the decision to distribute interim dividends is passed more than six months after the date of the statement of financial position as set out in our latest adopted annual report, an interim statement of financial position must be prepared and reviewed by our auditor. The statement of financial position or the interim statement of financial position, as applicable, must show that sufficient funds are available for distribution. Dividends may not exceed the amount recommended by the board of directors for approval by the general meeting of shareholders. Moreover, dividends and interim dividends may only be made out of distributable reserves and may not exceed what is considered sound and adequate with regard to our financial condition or be to the detriment of our creditors and such other factors as the board of directors may deem relevant.
In accordance with the DCA, share buybacks, if any, may only be carried out by the board of directors using funds that could have been distributed as dividends at the latest annual general meeting of shareholders. Any share buyback must be conducted in accordance with an authorization obtained at a general meeting of our shareholders. The authorization must be granted for a defined period of time not exceeding five years. In addition, the authorization must specify the maximum permitted value of treasury shares as well as the minimum and maximum amount that we may pay as consideration for such shares. A decision by our board of directors to engage in share buybacks, if any, will be made in accordance with the factors applicable to dividend payments set forth above.
For further information reference is made to 'Shareholder information', on pages 44-45 in our Annual Report 2018.
B. SIGNIFICANT CHANGES
No significant events have occurred since December 31, 2018. Reference is made to Note 27 'Significant events after the balance sheet date' to the consolidated financial statements on page 87 in our Annual Report 2018.
63
Table of Contents
ITEM 9 THE OFFER AND LISTING
A. OFFER AND LISTING DETAILS
The table below sets forth for the calendar periods indicated, in the first two columns, high and low prices for the shares as reported by the Nasdaq Copenhagen and, in the third and fourth columns, high and low ADS prices as reported by the NASDAQ Global Select Market since the Company's listing in August 2017.
| | | | | | | | | | | | | |
| | DKK per share | | USD per ADS | |
|---|
SHARE PRICE | | High | | Low | | High | | Low | |
|---|
2014 | | | 83.50 | | | 59.00 | | | N/A | | | N/A | |
2015 | | | 168.50 | | | 80.00 | | | N/A | | | N/A | |
2016 | | | 154.50 | | | 87.00 | | | N/A | | | N/A | |
2017 | | | 140.00 | | | 79.00 | | | 20.15 | | | 12.46 | |
2018 | | | 127.00 | | | 78.60 | | | 18.91 | | | 11.51 | |
2017 | | |
| | |
| | |
| | |
| |
1st quarter | | | 124.00 | | | 105.00 | | | N/A | | | N/A | |
2nd quarter | | | 130.50 | | | 115.00 | | | N/A | | | N/A | |
3rd quarter | | | 140.00 | | | 112.00 | | | 20.15 | | | 18.07 | |
4th quarter | | | 122.00 | | | 79.00 | | | 19.36 | | | 12.46 | |
2018 | | |
| | |
| | |
| | |
| |
1st quarter | | | 105.00 | | | 85.50 | | | 17.78 | | | 13.39 | |
2nd quarter | | | 103.00 | | | 83.00 | | | 16.7 | | | 12.17 | |
3rd quarter | | | 127.00 | | | 80.00 | | | 18.91 | | | 12.01 | |
4th quarter | | | 107.80 | | | 78.60 | | | 16.68 | | | 11.51 | |
Reference is made to 'Shareholder information' on pages 44-45 in our Annual Report 2018.
B. PLAN OF DISTRIBUTION
Not applicable.
C. MARKETS
The shares have been publicly traded since 2010 and have been listed on Nasdaq Copenhagen since that time.
ADSs representing the shares, as evidenced by American Depository Shares issued by The Bank of New York Mellon, as the Depository, have been listed on the NASDAQ Global Select Market since August 2017.
D. SELLING SHAREHOLDERS
Not applicable.
E. DILUTION
Not applicable.
F. EXPENSES OF THE ISSUE
Not applicable.
64
Table of Contents
ITEM 10 ADDITIONAL INFORMATION
A. SHARE CAPITAL
Not applicable.
B. MEMORANDUM AND ARTICLES OF ASSOCIATION
This section summarizes certain material provisions of our Articles of Association, certain other constitutive documents and relevant Danish corporate law.
General
We are a limited liability company organized under the laws of Denmark and registered with the Danish Business Authority under CVR number 20045078. We have been established with the objective of engaging in research, manufacture trade and related activities primarily within the pharmaceutical industry. Our objectives are set out in Article 2 of our Articles of Association.
Powers of the Board of Directors
Unless otherwise directed by the Board of Directors all members of the Board of Directors have equal voting rights, and all resolutions are passed by a simple majority of votes. In the event of a tie, the Chairman shall have the casting vote. The Board of Directors forms a quorum when at least a majority of its members is present.
According to the Danish Companies Act, no member of the Board of Directors or the Executive Management may take part in the consideration of any business involving agreements between any member of the group and himself, legal actions brought against himself, or any business involving agreements between any member of the Group and any third party or legal actions brought against any third party, if he has a major interest therein that might conflict with our interests.
The Danish Companies Act prohibits us from granting loans or providing securities to any member of the Board of Directors and anyone particularly close to such a member of the Board of Directors.
Under our Articles of Association, no person can be elected to the board having reached the age of 70. Further a Board Member shall retire at the end of the first annual general meeting held after the relevant Board Member reaches the age of 70.
The remuneration of the Board of Directors must be approved by our shareholders at the Annual General Meeting.
Rights, restrictions and preferences attaching to the shares
No share carries any special rights. Each share confers the right to cast one vote at the general meeting of shareholders, unless the articles of association provide otherwise. Each holder of shares may cast as many votes as it holds shares. Voting instructions may be given only in respect of a number of ADSs representing an integral number of shares or other deposited securities. Shares that are held by the company or direct or indirect subsidiaries do not confer the right to vote.
ADS holders may only exercise voting rights with respect to the shares underlying their respective ADSs. In accordance with the provisions of the deposit agreement, which provides that a holder may vote the shares underlying any ADSs for any particular matter to be voted on by our shareholders either by withdrawing the shares underlying the ADSs or, to the extent permitted by applicable law and as permitted by the depositary, by requesting temporary registration as shareholder and authorizing the depositary to act as proxy. The depositary will try, as far as practical, to vote the shares underlying the ADSs as instructed by the ADS holders.
65
Table of Contents
Changes to Shareholders' rights
Any change to the rights of the shareholders would require an amendment to the Articles of Association.
Where we elect to pay a Dividend these are accrued to the company where they have not been claimed after 3 years.
General Meetings
General Meetings of the company are held in the greater Copenhagen area and must be held not later than four months from the closing of the financial year. Notice of the Annual General Meeting shall be sent to shareholders not later than eight weeks before the date of that meeting together with a date by which any shareholders wishing to have any specific item included on the agenda of the meeting should submit that item.
Ownership restrictions
There are no limitations on the rights of non-resident or foreign owners to hold or vote the shares imposed by the laws of Denmark, our Association, or any other of its constituent documents.
Change of control
There is no provision in the Articles of Association, nor any other constituent document, that would have an effect of delaying, deferring or preventing a change in control of Zealand Pharma A/S and that would operate only with respect to a merger, acquisition or corporate restructuring involving the company (or any of its subsidiaries).
Ownership disclosure
Pursuant to section 29 of the Danish Securities Trading Act (Værdipapirhandelsloven), shareholders in a company incorporated in Denmark with its shares admitted to trading and official listing are required to immediately (meaning within the same trading day as the transaction) and simultaneously notify the company and the Danish Financial Supervisory Authority ("FSA"), when the shareholder's stake (i) represents 5% or more of the voting rights in the company or the nominal value of its share capital, and (ii) when a change in a holding already notified implies that the limits of 5%, 10%, 15%, 20%, 25%, 50% or 90% and the limits of one-third and two-thirds of the voting rights or the nominal value are reached.
This duty to notify also applies to anyone, who directly or indirectly holds (a) financial instruments that afford the holder a right to purchase existing shares, e.g., share options; and/or (b) financial instruments based on existing shares and with an economic effect equal to that of the financial instruments mentioned under (a), regardless of them not affording the right to purchase existing shares, e.g., the ADSs or, under the circumstances, cash-settled derivatives linked to the value of our shares or ADSs. Holding these kinds of financial instruments counts towards the thresholds mentioned above and may thus trigger a duty to notify by themselves or when accumulated with a holding of shares or ADSs.
The notifications must comply with the requirements for the contents thereof set out in sections 16 and 17 of the Danish executive order on major shareholders (Storaktionærbekendtgørelsen), including the identity of the shareholder and the date when a limit is reached or no longer reached. Failure to comply with the duties of disclosure is punishable by fine or suspension of voting rights in instances of gross or repeated non-compliance. The FSA will in certain cases publish information concerning sanctions imposed, including, as a general rule, the name of the shareholder in question, as a consequence of non-compliance with the above rules. When we receive a notification pursuant to section 29 of the Danish Securities Trading Act, we must publish its contents as soon as possible.
66
Table of Contents
Furthermore, the general duty of notification pursuant to the DCA applies, which implies that shareholders must notify the company when the limit of 100% of the voting rights or nominal value of the shares is reached or no longer reached. This also applies to holders of the ADSs.
Changes in capital
Our Articles of Association do not contain conditions governing changes in the capital more stringent than those contained in the Danish Companies Act.
C. MATERIAL CONTRACTS
We have not entered into any material contracts other than in the ordinary course of business and other than those described in "Item 4. Information on the Company," "Item 6. Directors, Senior Management and Employees" or elsewhere in this Annual Report.
D. EXCHANGE CONTROLS
There are no governmental laws, decrees, or regulations in Denmark (including, but not limited to, foreign exchange controls) that restrict the export or import of capital, or that affect the remittance of dividends, interest or other payments to non-resident holders of the shares or the ADSs. There are no limitations on the right of non-resident or foreign owners to hold or vote the shares or the ADSs imposed by the laws of Denmark or the Articles of Association of the Company.
E. TAXATION
The following is a description of the material U.S. federal income tax consequences to the U.S. Holders described below of owning and disposing of the ADSs. It is not a comprehensive description of all tax considerations that may be relevant to a particular person's decision to acquire securities. This discussion applies only to a U.S. Holder that holds the ADSs as capital assets for tax purposes. In addition, it does not describe all of the tax consequences that may be relevant in light of a U.S. Holder's particular circumstances, including foreign, state and local tax consequences, U.S. federal gift, estate and alternative minimum tax consequences, and tax consequences applicable to U.S. Holders subject to special rules, such as:
- •
- certain financial institutions;
- •
- dealers or traders in securities who use a mark-to-market method of tax accounting;
- •
- persons holding the ADSs as part of a hedging transaction, "straddle," wash sale, conversion transaction or integrated transaction or persons entering into a constructive sale with respect to the ADSs;
- •
- regulated investment companies;
- •
- insurance companies;
- •
- real estate investment trusts, grantor trusts or other trusts;
- •
- persons whose "functional currency" for U.S. federal income tax purposes is not the U.S. dollar;
- •
- expatriates of the United States;
- •
- tax exempt entities, including "individual retirement accounts" and "Roth IRAs";
- •
- entities classified as partnerships or other pass-through entities for U.S. federal income tax purposes;
67
Table of Contents
- •
- persons that own or are deemed to own shares representing ten percent or more of our vote or value;
- •
- persons who acquired the ADSs in exchange for services or otherwise as compensation; and
- •
- persons holding the ADSs in connection with a trade or business conducted outside the United States.
If an entity that is classified as a partnership for U.S. federal income tax purposes holds the ADSs, the U.S. federal income tax treatment of a partner generally will depend on the status of the partner and the activities of the partnership. Partnerships holding the ADSs and partners in such partnerships are encouraged to consult their own tax advisers as to the particular U.S. federal income tax consequences of holding and disposing of the ADSs.
For U.S. federal income tax purposes, U.S. Holders of ADSs will be treated as the beneficial owners of the underlying shares represented by the ADSs and an exchange of ADSs for our shares generally will not be subject to U.S. federal income tax.
The discussion is based on the Code, administrative pronouncements, judicial decisions, and final, temporary and proposed U.S. Treasury Regulations, all as of the date hereof, changes to any of which may affect the tax consequences described herein—possibly with retroactive effect.
A "U.S. Holder" is a holder who, for U.S. federal income tax purposes, is a beneficial owner of the ADSs who is eligible for the benefits of the Treaty and is:
- (1)
- an individual who is a citizen or resident of the United States;
- (2)
- a corporation, or other entity taxable as a corporation, created or organized in or under the laws of the United States, any state therein or the District of Columbia;
- (3)
- an estate the income of which is subject to U.S. federal income tax regardless of its source; or
- (4)
- a trust, if (A) a U.S. court is able to exercise primary supervision over the trust's administration and one or more United States persons (as such term is defined under the Code) have authority to control all substantial decisions of the trust, or (B) the trust has a valid election in place under all applicable U.S. Treasury regulations to treat the trust as a United States person (as such term is defined under the Code).
U.S. Holders are encouraged to consult their own tax advisers concerning the U.S. federal, state, local and foreign tax consequences of owning and disposing of the ADSs in their particular circumstances.
Taxation of distributions
Subject to the Passive Foreign Investment Company ("PFIC") rules described below, distributions paid on the ADSs, other than certain pro rata distributions of the ADSs, generally will be treated as dividends to the extent paid out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). Because we do not maintain calculations of our earnings and profits under U.S. federal income tax principles, we expect that distributions generally will be reported to U.S.
Holders as dividends. Subject to applicable limitations, dividends paid to certain non-corporate U.S. Holders may be taxable at preferential rates applicable to long-term capital gain. However, if we are a PFIC for the taxable year in which the dividend is paid or the preceding taxable year (see discussion below under "—Passive Foreign Investment Company rules"), the preferential rate will not apply. Each U.S. Holder is advised to consult its tax advisors regarding the availability of the reduced tax rate on dividends with regard to its particular circumstances.
68
Table of Contents
The amount of a dividend will include any amounts withheld by us in respect of Danish income taxes. The amount of the dividend will be treated as foreign-source dividend income to U.S. Holders and will not be eligible for the dividends-received deduction generally available to U.S. corporations under the Code. Dividends will be included in a U.S. Holder's income on the date of the U.S. Holder's receipt of the dividend. The amount of any dividend income paid in DKK will be the U.S. dollar amount calculated by reference to the exchange rate in effect on the date of actual or constructive receipt, regardless of whether the payment is in fact converted into U.S. dollars. If the dividend is converted into U.S. dollars on the date of receipt, a U.S. Holder should not be required to recognize foreign currency gain or loss in respect of the dividend income. A U.S. Holder may have foreign currency gain or loss if the dividend is converted into U.S. dollars after the date of receipt.
Subject to applicable limitations, some of which vary depending upon the U.S. Holder's particular circumstances, Danish income taxes withheld from dividends on the ADSs (or shares underlying the ADSs) will be creditable against the U.S. Holder's U.S. federal income tax liability. The rules governing foreign tax credits are complex and U.S. Holders should consult their tax advisers regarding the creditability of foreign taxes in their particular circumstances. In lieu of claiming a foreign tax credit, U.S. Holders may, at their election, deduct foreign taxes, including any Danish income tax, in computing their taxable income, subject to generally applicable limitations under U.S. law. An election to deduct foreign taxes instead of claiming foreign tax credits applies to all foreign taxes paid or accrued in the taxable year.
Sale or other taxable disposition of the ADSs
Subject to the PFIC rules described below, gain or loss realized on the sale or other taxable disposition of the ADSs will be capital gain or loss, and will be long-term capital gain or loss if the U.S. Holder held the ADSs for more than one year. The amount of the gain or loss will equal the difference between the U.S. Holder's tax basis in the ADSs disposed of and the amount realized on the disposition, in each case as determined in U.S. dollars. This gain or loss generally will be U.S.-source gain or loss for foreign tax credit purposes. Capital gain from the sale, exchange or other disposition of ADSs by a non-corporate U.S. Holder generally is eligible for preferential rates of taxation if the non-corporate U.S. Holder's holding period for such ADSs determined at the time of such sale, exchange, or other taxable disposition exceeds one year (i.e., such gain is long-term capital gain). The deductibility of capital losses is subject to limitations.
Passive Foreign Investment Company rules
Under the Code, we will be a PFIC for any taxable year in which, after the application of certain "look-through" rules with respect to subsidiaries, either (i) 75% or more of our gross income consists of "passive income," or (ii) 50% or more of the average quarterly value of our assets, including cash, consist of assets that produce, or are held for the production of, "passive income." Passive income generally includes interest, dividends, rents, certain non-active royalties and capital gains. Whether we will be a PFIC in any year depends on the composition of our income and assets, and the relative fair market value of our assets from time to time, which we expect may vary substantially over time. If we are a PFIC for any year during which a U.S. Holder holds the ADSs, we generally would continue to be treated as a PFIC with respect to that U.S. Holder for all succeeding years during which the U.S. Holder holds the ADSs, even if we ceased to meet the threshold requirements for PFIC status in any particular year.
Based on certain estimates of our gross income and gross assets, the latter determined by reference to the value of the ADSs and shares, we believe that we will not be classified as a PFIC for the taxable year ending December 31, 2018. However, because PFIC status is based on our income, assets and activities for the entire taxable year, it is not possible to determine whether we will be classified as a PFIC for the 2018 taxable year (or any later year) until after the close of the relevant
69
Table of Contents
year. The determination of whether we are a PFIC is made annually for each of our taxable years. As a result, our PFIC status may change. In particular, for purposes of the asset test described above, the total value of our assets will be treated as equal to the sum of the aggregate value of the ADSs and shares plus the Company's liabilities. Therefore, for purposes of the asset test, the total value of our assets will depend on the market price of the ADSs. However, the value of our passive assets generally will be equal to the actual fair market value of such assets. A decrease in the market price of the ADSs would cause a decrease in the deemed total value of our assets for purposes of the asset test but generally would not cause a corresponding decrease in the actual value of our passive assets. Accordingly, fluctuations in the market price of the ADSs may result in us being a PFIC. In addition, the composition of our income and assets will be affected by how, and how quickly, we spend the cash raised in this offering. Because of the fact specific nature of the inquiry, it cannot be predicted at this time what portion of the net proceeds from this offering we would need to spend in order to avoid PFIC status.
For purposes of the income test, we believe that we are engaged in an active trade or business of discovering and developing peptide drugs and that the royalties and milestone payments we receive from unrelated parties should be treated as derived in the active conduct of a trade or business and not characterized as passive income. However, we have no assurance that these anticipated milestone payments and royalties will be paid when expected. If any such payments are delayed or not received then, depending on the amount of passive income we receive from other sources, the relative percentage of our income that is passive could increase and potentially cause us to be classified as a PFIC. There can be no assurances that we will not be classified as a PFIC for the current taxable year or for any future taxable year.
If we are a PFIC for any taxable year during which a U.S. Holder holds the ADSs, the U.S. Holder may be subject to adverse tax consequences. Generally, gain recognized upon a disposition (including, under certain circumstances, a pledge) of the ADSs by the U.S. Holder would be allocated ratably over the U.S. Holder's holding period for such shares. The amounts allocated to the taxable year of disposition and to years before we became a PFIC would be taxed as ordinary income. The amount allocated to each other taxable year would be subject to tax at the highest rate in effect for that taxable year for individuals or corporations, as appropriate, and would be increased by an additional tax equal to interest on the resulting tax deemed deferred with respect to each such other taxable year. Further, to the extent that any distribution received by a U.S. Holder on its ADSs exceeds 125% of the average of the annual distributions on such ADSs received during the preceding three years or the U.S. Holder's holding period, whichever is shorter, that distribution would be subject to taxation in the same manner described immediately above with respect to gain on disposition.
Alternatively, if we are a PFIC and if the ADSs are "regularly traded" on a "qualified exchange," a U.S. Holder could make a mark-to-market election that would result in tax treatment different from the general tax treatment described in the preceding paragraph. The ADSs would be treated as "regularly traded" in any calendar year in which more than a deminimis quantity of the ADSs are traded on a qualified exchange on at least 15 days during each calendar quarter. NASDAQ is a qualified exchange for this purpose. If a U.S. Holder makes the mark-to-market election, the U.S. Holder generally will recognize as ordinary income any excess of the fair market value of the ADSs at the end of each taxable year over their adjusted tax basis, and will recognize an ordinary loss in respect of any excess of the adjusted tax basis of the ADSs over their fair market value at the end of the taxable year (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). If a U.S. Holder makes the election, the U.S. Holder's tax basis in the ADSs will be adjusted to reflect these income or loss amounts. Any gain recognized on the sale or other disposition of ADSs in a year when we are a PFIC will be treated as ordinary income and any loss will be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark-to-market election).
70
Table of Contents
A timely election to treat a PFIC as a qualified electing fund under Section 1295 of the Code would result in alternative treatment. U.S. Holders should be aware, however, that we do not intend to satisfy the record-keeping and other requirements that would permit U.S. Holders to make qualified electing fund elections if we were a PFIC.
In addition, if we are a PFIC or, with respect to particular U.S. Holders, are treated as a PFIC for the taxable year in which we paid a dividend or for the prior taxable year, the preferential rates discussed above with respect to dividends paid to certain non-corporate U.S. Holders would not apply.
U.S. Holders should consult their tax advisers regarding whether we are or may become a PFIC and the potential application of the PFIC rules.
Medicare Tax
In general, a U.S. Holder that is an individual or estate, or a trust is subject to a 3.8% Medicare tax on the lesser of (1) the U.S. Holder's "net investment income" for the relevant taxable year and (2) the excess of the U.S. Holder's modified adjusted gross income for the taxable year over a certain threshold (which in the case of individuals will be between $125,000 and $250,000, depending on the individual's circumstances). A U.S. Holder's net investment income will include its gross dividend income and its net gains from the disposition of ADSs, unless such dividends or net gains are derived in the ordinary course of the conduct of a trade or business (other than a trade or business that consists of certain passive or trading activities). If you are a U.S. Holder that is an individual, estate or trust, you are encouraged to consult your tax advisors regarding the applicability of this tax to income and gains in respect of your investment in the ADSs.
Information reporting and backup withholding
Payments of dividends and sales proceeds that are made within the United States or through certain U.S.-related financial intermediaries generally are subject to information reporting, and may be subject to backup withholding, unless (i) the U.S. Holder is a corporation or other exempt recipient or (ii) in the case of backup withholding, the U.S. Holder provides a correct taxpayer identification number and certifies that it is not subject to backup withholding.
Backup withholding is not an additional tax. The amount of any backup withholding from a payment to a U.S. Holder will be allowed as a credit against the holder's U.S. federal income tax liability and may entitle it to a refund, provided that the required information is timely furnished to the IRS.
If a U.S. Holder owns ADS during any year in which we are a PFIC, such U.S. Holder (including, potentially, indirect holders) will generally be required to file an IRS Form 8621 with such holder's federal income tax return for that year.
Certain U.S. Holders who are individuals may be required to report information relating to their ownership of an interest in certain foreign financial assets, including shares of a non-U.S. person, generally on Form 8938, subject to exceptions (including an exception for shares held through a U.S. financial institution). In addition, certain U.S. Holders may be required to file a FinCEN Form 114 (Report of Foreign Bank and Financial Accounts) with the U.S. Treasury Department each year to report their interests in the ADSs. U.S. Holders should consult their tax advisers regarding their reporting obligations with respect to the ADSs.
F. DIVIDENDS AND PAYING AGENTS
Not applicable.
71
Table of Contents
G. STATEMENT BY EXPERTS
Not applicable.
H. DOCUMENTS ON DISPLAY
Documents referred to and filed with the SEC together with this Annual Report on Form 20-F can be read and copied at the SEC's public reference room located at 100 F Street, NE, Washington, DC 20549. Please call the United States Securities and Exchange Commission at 1-800-SEC-0330 for further information on the public reference rooms.
Copies of this Annual Report on Form 20-F as well as our Annual Report 2018 can be downloaded from the Investors page at zealandpharma.com. The contents of our website are not incorporated by reference into this Annual Report on Form 20-F. This Annual Report on Form 20-F is also filed and can be viewed via EDGAR on www.sec.gov.
I. SUBSIDIARY INFORMATION
Not applicable.
72
Table of Contents
ITEM 11 QUALITATIVE AND QUANTITATIVE DISCLOSURES ABOUT MARKET RISKS
Financial exposure and financial risk management
For a description and discussion of the Company's foreign exchange rate risk management, interest rate risk management, credit risk management and liquidity risk management, reference is made to Note 23 'Financial risks' to the consolidated financial statements on 84-86 in our Annual Report 2018 and 'Risk management and internal control' on pages 38-39 in our Annual Report 2018.
Sensitivity analysis
When conducting a sensitivity analysis, the Group assesses the change in fair value on the market-sensitive instruments following hypothetical changes in interest and exchange rates. The rates used to mark-to-market the instruments are market data as of December 28, 2018.
Interest rate sensitivity analysis
For information on Interest rate sensitivity analysis in the financial year 2018, reference is made to Note 23 'Financial risks' to the consolidated financial statements on pages 84-86 in our Annual Report 2018.
Foreign exchange sensitivity analysis
For information on Foreign exchange sensitivity analysis in the financial year 2018, reference is made to Note 23 'Financial risks' to the consolidated financial statements on pages 84-86 in our Annual Report 2018.
73
Table of Contents
ITEM 12 DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES
ITEM 12A DEBT SECURITIES
Not applicable.
ITEM 12B WARRANTS AND RIGHTS
Not applicable.
ITEM 12C OTHER SECURITIES
Not applicable.
ITEM 12D AMERICAN DEPOSITARY SHARES
Our ADS program is administrated by The Bank of New York Mellon. Each ADS represents one ordinary share (or a right to receive one ordinary share) deposited with the Copenhagen office of Danske Bank A/S, as custodian for the depositary in the United States. Each ADS also represents any other securities, cash or other property which may be held by the depositary. The depositary's office at which the ADSs is administered is located at 101 Barclay Street, New York, New York 10286. The Bank of New York Mellon's principal executive office is located at 225 Liberty Street, New York, New York 10286.
You may hold ADSs either (a) directly (i) by having an American Depositary Receipt, also referred to as an ADR, which is a certificate evidencing a specific number of ADSs, registered in your name, or (ii) by having uncertificated ADSs registered in your name, or (b) indirectly by holding a security entitlement in ADSs through your broker or other financial institution that is a direct or indirect participant in The Depository Trust Company ("DTC"). If you hold ADSs directly, you are a registered ADS holder, also referred to as an ADS holder. This description assumes you are an ADS holder. If you hold the ADSs indirectly, you must rely on the procedures of your broker or other financial institution to assert the rights of ADS holders described in this section. You should consult with your broker or financial institution to find out what those procedures are.
Registered holders of uncertificated ADSs will receive statements from the depositary confirming their holdings.
As an ADS holder, we will not treat you as one of our shareholders and you will not have shareholder rights. Danish law governs shareholder rights. The depositary will be the holder of the shares underlying your ADSs. As a registered holder of ADSs, you will have ADS holder rights. A deposit agreement among us, the depositary, ADS holders and all other persons indirectly or beneficially holding ADSs sets out ADS holder rights as well as the rights and obligations of the depositary. New York law governs the deposit agreement and the ADSs.
74
Table of Contents
PART II
ITEM 13 DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES
None.
75
Table of Contents
ITEM 14 MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS
None.
76
Table of Contents
ITEM 15 CONTROLS AND PROCEDURES
A. Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a- 15(e) and 15d- 15(e) under the Securities Exchange Act of 1934, as amended), as of the end of the period covered by this Annual Report on Form 20-F. Based on such evaluation, our principal executive officer and principal financial officer have concluded that as of such date, our disclosure controls and procedures were not effective as of December 31, 2018 due to the material weaknesses described below.
B. Management's Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting.
Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and preparation of financial statements for external purposes in accordance with IFRS.
Internal control over financial reporting is defined in rules 13a-15(f) and 15d-15(f) under the Exchange Act as a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the Company's assets that could have a material effect on the audited consolidated financial statements.
Management, under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, conducted an evaluation of the effectiveness of the internal control over financial reporting as of December 31, 2018, using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission ("COSO") in Internal Control—Integrated Framework (2013). Based on the evaluation performed, management concluded that material weaknesses existed as of December 31, 2018 as described below.
Material weaknesses as of December 31, 2018
As disclosed in our 2017 Annual Report, in connection with our financial statement preparation process for the year ended December 31, 2017, we identified material weaknesses, including lack of sufficient competencies related to IFRS and SEC reporting knowledge for the purposes of timely and reliable financial reporting. Under the standards established by the PCAOB, a material weakness is a deficiency, or a combination of deficiencies, that creates a reasonable possibility that a material misstatement of a Company's annual financial statements will not be prevented or detected on a timely basis.
The material weaknesses we identified related to our existing processes to assess risks and to design and implement effective control activities over financial reporting. In particular, we did not have formalized risk assessment, oversight and compliance processes or formalized control descriptions for
77
Table of Contents
all of our key controls. Where control descriptions existed, they did not necessarily include all relevant information to enable the operating effectiveness of such controls and it was not clear whether adequate controls were performed in all areas. Where control activities were dependent on certain information, which is referred to as our IUC, we did not perform or document controls to assess the completeness and accuracy of such information. We did not then monitor control activities and identified control deficiencies; thus, we were unable to evaluate whether other deficiencies, individually or in combination, resulted in a reasonable possibility that a material misstatement of our annual financial statements would not be prevented or detected on a timely basis.
We initiated several steps to begin to remediate the material weaknesses identified by us. In late 2017, we hired a Chief Accountant and along with external consultants, who have worked to further develop and implement formal policies, processes, internal controls and documentation relating to our financial reporting. However, despite efforts taken to remediate the material weaknesses, the newly designed controls were only able to be introduced shortly before the 2018 year end and had not been operating for a sufficient period to allow sufficient testing to demonstrate that such controls were operating effectively. For certain testing that were performed, management identified control deficiencies which in the aggregate constituted material weaknesses.
As such, management has not been able to obtain sufficient evidence that the newly implemented controls would be effective in remediating the material weaknesses previously identified in 2017. Accordingly, a reasonable possibility exists that a material misstatement of the Company's annual or interim financial statements will not be prevented or detected on a timely basis by the Company's internal control. Management, including Chief Executive Officer and Chief Financial Officer, thus concluded that internal controls over financial reporting were not effective as of December 31, 2018.
As we continue to evaluate and work to improve internal control over financial reporting during 2019, management may determine to take additional measures to address control deficiencies. We have hired additional finance and accounting personnel with appropriate expertise to perform specific functions and intend to hire additional personnel to further assist in the implementation of improved processes and internal controls, build our financial management and reporting infrastructure and further develop and document our accounting policies and financial reporting procedures, including ongoing senior management review and audit committee oversight
While management believes that the steps that we have taken and plans to continue to take, will improve the overall system of internal control over financial reporting and will remediate identified material weaknesses, the material weaknesses cannot be considered remediated until the applicable relevant controls operate for a sufficient period of time and management has concluded, through testing that these controls are operating effectively. No assurance can be provided at this time that the actions and remediation efforts will effectively remediate the material weaknesses described above or prevent the incidence of other material weaknesses in the Company's internal control over financial reporting in the future. Further, we do not know the specific time frame needed to fully remediate the material weaknesses identified above. See "Risk Factors." Management, including the Chief Executive Officer and Chief Financial Officer, does not expect that disclosure controls and procedures or internal control over financial reporting will prevent all misstatements, even as the remediation measures are implemented and further improved to address the material weaknesses. The design of any system of internal controls is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving the stated goals under all potential future conditions.
78
Table of Contents
C. Attestation Report of the Registered Public Accounting Firm
This annual report does not include an attestation report of our independent registered public accounting firm regarding our internal control over financial reporting due to an exemption provided by the JOBS Act for emerging growth companies.
D. Changes in Internal Control over Financial Reporting
As noted above, management have begun to undertake steps to remediate the material weaknesses identified and reported in the annual report for 2017 and this Annual Report, albeit such controls are not yet deemed to be operating effectively. There have been no other changes in our internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) that occurred during the period covered by this Annual Report that has materially affected, or is reasonably likely to materially affect, internal control over financial reporting.
79
Table of Contents
ITEM 16A AUDIT COMMITTEE FINANCIAL EXPERTS
Our Audit Committee consists of Catherine Moukheibir, Martin Nicklasson and Rosemary Crane, and is chaired by Catherine Moukheibir. The Audit Committee reviews and considers matters relating to accounting, audit and regulatory control with our auditors and executive management in accordance with the working terms of reference of the Audit Committee. Our Audit Committee oversees our accounting and financial reporting processes and the audits of our consolidated financial statements.
Each member satisfies the independence requirements of the corporate governance standards of NASDAQ, and Catherine Moukheibir qualifies as an "Audit Committee financial expert," as defined in NASDAQ Rule 5605(c)(2)(A) and as determined by our board of directors.
ITEM 16B CODE OF ETHICS
We have adopted a written code of business conduct, or code of conduct, which outlines the principles of legal and ethical business conduct under which we do business. The code of conduct applies to all of our board members and employees. This document is available under the "Corporate governance" tab in the "About Zealand" section of our website (www.zealandpharma.com). We undertake to provide to any person, without charge, upon request, a copy of the code of business conduct. Requests shall be made on mail info@zealandpharma.com. For the full text of the code of conduct please view exhibit 11.1.
ITEM 16C PRINCIPAL ACCOUNTANT FEES AND SERVICES
Reference is made to Note 5 'Fees to auditors appointed at the Annual General Meeting' to the consolidated financial statements on page 67 in our Annual Report 2018 regarding fees paid to our statutory auditors.
Audit fees
Audit fees consist of fees billed for professional services rendered by the principal accountant for the audit of the registrant's annual financial statements or services that are normally provided by the accountant in connection with statutory and regulatory filings or engagements for those fiscal years.
Audit-Related fees
Audit-Related fees consist of assurance and related services by the principal accountant that are reasonably related to the performance of the audit or review of the registrant's financial statements and are not reported under "Audit fees". Fees for audit-related services include consultations concerning financial accounting reporting standards.
Tax fees
Tax fees consist of fees billed for professional services rendered by the principal accountant for tax compliance, tax advice, and tax planning, including tax fees billed for tax consultations.
All other fees
All other fees consist of products and services provided by the principal accountant, other than the services reported in "Audit fees", "Audit-Related fees" and "Tax fees".
Fees for other services comprise fees billed for other permitted services such as review of IT security plans.
80
Table of Contents
Pre-approval policies
The Audit Committee assesses and pre-approves all audit and non-audit services provided by the statutory auditors. The pre-approval includes the type of service and a fee budget.
ITEM 16D EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES
Not applicable.
ITEM 16E PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED
Not applicable.
ITEM 16F CHANGE IN REGISTRANT'S CERTIFYING ACCOUNTANT
Not applicable.
ITEM 16G CORPORATE GOVERNANCE
As a foreign private issuer and as permitted by the listing requirements of NASDAQ, we rely on certain home country corporate governance practices rather than the corporate governance requirements of NASDAQ.
We qualify as a foreign private issuer and our ADSs are listed on NASDAQ. As a result, in accordance with the listing requirements of NASDAQ, we rely on home country governance requirements and certain exemptions thereunder rather than relying on the corporate governance requirements of NASDAQ. For example, we are exempt from certain rules under the Exchange Act that regulate disclosure obligations and procedural requirements related to the solicitation of proxies, consents or authorizations applicable to a security registered under the Exchange Act, including the U.S. proxy rules under Section 14 of the Exchange Act. In addition, our officers and directors are exempt from the reporting and "short-swing" profit recovery provisions of Section 16 of the Exchange Act and related rules with respect to their purchases and sales of our securities. Moreover, while we currently publish annual and quarterly reports on our website pursuant to the rules of Nasdaq Copenhagen and expect to file such financial reports on an annual and quarterly basis with the SEC, we are not required to file such reports with the SEC as frequently or as promptly as U.S. public companies and are not required to file quarterly reports on Form 10-Q or current reports on Form 8-K that a domestic company would be required to file under the Exchange Act. Accordingly, there may be less publicly available information concerning our company than there would be if we were not a foreign private issuer.
In addition, the Listing Rules for the Nasdaq Stock Market (the "Nasdaq Listing Rules"), for domestic U.S. issuers require listed companies to have, among other things, a majority of their board members be independent, and to have independent director oversight of executive compensation, nomination of board members and corporate governance matters. While we intend to comply with these requirements, we are permitted to follow home country practice in lieu of the above requirements. Danish law does not require that a majority of our board consist of independent directors or the implementation of a nominating and corporate governance committee, and our board may thus in the future not include, or include fewer, independent directors than would be required if we were subject to the Nasdaq Listing Rules, or they may decide that it is in our interest not to have a compensation committee or nominating and corporate governance committee, or have such committees governed by practices that would not comply with the Nasdaq Listing Rules. We follow home country practice with regard to, among other things, quorum requirements generally applicable to general meetings of shareholders. Danish law does not have a regulatory regime for the solicitation of proxies and the solicitation of proxies is not a generally accepted business practice in Denmark, thus our
81
Table of Contents
practice will vary from the requirement of Nasdaq Listing Rule 5620(b). In addition, our shareholders have authorized our board of directors to issue securities, including in connection with certain events such as the acquisition of shares or assets of another company, the establishment of or amendments to equity-based compensation plans for employees, rights issues at or below market price, certain private placements, directed issues at or above market price, and issuance of convertible notes. To this extent, our practice varies from the requirements of Nasdaq Listing Rule 5635, which generally requires an issuer to obtain shareholder approval for the issuance of securities in connection with such events.
ITEM 16H MINE SAFETY DISCLOSURE
Not applicable.
82
Table of Contents
PART III
ITEM 17 FINANCIAL STATEMENTS
See response to Item 18.
83
Table of Contents
ITEM 18 FINANCIAL STATEMENTS
The financial statements required by this item accompany this Annual Report on Form 20-F in the form of our Annual Report 2018 on pages 51-87 (see Item 19).
Reconciliation of non-IFRS financial measures
In the consolidated financial statements, Zealand discloses non-IFRS financial measures of the Group that reflect adjustments to the most directly comparable measure calculated and presented in accordance with IFRS. The inclusion of non-IFRS measures has been expressly permitted by the Danish Business Authorities and thereby exempted from the prohibition in Item 10(e)(1)(ii)(C) of Regulation S-K. However, the non-IFRS financial measures may not be defined and calculated by other companies in the same manner and may not be comparable with such measure.
Reference is made to the section 'Alternative performance measures for the Group (non-audited)' on page 96 in our Annual Report 2018.
84
Table of Contents
ITEM 19 EXHIBITS
The following pages from our Annual Report 2018, furnished to the SEC on Form 6-K, dated March 8, 2019, are incorporated by reference into this Annual Report on Form 20-F. The content of websites and other sources referenced on these pages are not incorporated by reference into this Annual Report on Form 20-F.
Page(s) incorporated by reference from our Annual Report 2018
| | |
Management review | | |
2018 Achievements | | 9 |
Financial highlights and 2019 guidance | | 10 |
Consolidated key figures | | 11 |
2019 Objectives | | 12 |
Our Ambition and Business Model | | 14-16 |
Our Programs | | 18-31 |
Corporate governance | | 33-35 |
Corporate social responsibility (CSR) | | 36 |
Our People | | 37 |
Risk management and internal control | | 38-39 |
Financial review | | 41-43 |
Shareholder information | | 44-45 |
Board of Directors and Corporate Management | | 46-48 |
Consolidated Financial Statements | | |
Consolidated Income statements | | 51 |
Consolidated statements of comprehensive income | | 51 |
Consolidated statements of financial position | | 52 |
Consolidated statements of cash flows | | 53 |
Consolidated statements of changes in equity | | 53 |
Notes to the Consolidated financial statements | | 55 |
Statement of the Board of Directors and Executive Management | | 89 |
85
Table of Contents
List of exhibits:
| | | | | |
| Exhibit No. | | Description | | Method of filing |
|---|
| | 1.1 | | Articles of Association | | Incorporated by reference to the Registrant's Report furnished to the SEC on Form 6-K on February 28, 2019. |
|
2.1 |
|
Deposit Agreement |
|
Incorporated by reference to Exhibit 4.1 to the Registrant's Registration Statement on Form F-1/A filed with the SEC August 3, 2017. |
|
4.1 |
|
Purchase and Sale Agreement, dated as of September 6, 2018 among ZP Holding SPV K/S, ZP SPV 1 K/S, Zealand Pharma A/S and Royalty Pharma Investments ICAV. |
|
Filed together with this Form 20-F 2018 |
|
8.1 |
|
List of subsidiaries |
|
Incorporated by reference to our Annual Report 2018, furnished to the SEC on Form 6-K dated March 8,2019, as exhibit 99.1(a). |
|
12.1 |
|
Certification of the principal Executive Officer |
|
Filed together with this Form 20-F 2018 |
|
12.2 |
|
Certification of the Financial Officer |
|
Filed together with this Form 20-F 2018 |
|
13.1 |
|
Principal Executive Officer certification pursuant to 18 U.S.C. section 1350 |
|
Furnished together with this Form 20-F 2018 |
|
13.2 |
|
Principal Financial Officer certification pursuant to 18 U.S.C. section 1350 |
|
Furnished together with this Form 20-F 2018 |
|
15.1 |
|
Extracts from the Registrant's Annual Report 2018 |
|
Incorporated by reference to our Annual Report 2018, furnished to the SEC on Form 6-K dated March 8,2019, as exhibit 99.1(a) identified in Item 19.a of this Form 20-F. |
|
EX-101.INS |
|
XBRL Instance Document |
|
Incorporated by reference to the Registrant's report furnished to the SEC on Form 6-K on March 8, 2019 as exhibit 99.1 (a) |
|
EX-101.SCH |
|
XBRL Taxonomy Extension Schema Document |
|
Incorporated by reference to the Registrant's report furnished to the SEC on Form 6-K on March 8, 2019 as exhibit 99.1 (a) |
|
EX-101.CAL |
|
XBRL Taxonomy Extension Calculation Linkbase Document |
|
Incorporated by reference to the Registrant's report furnished to the SEC on Form 6-K on March 8, 2019 as exhibit 99.1 (a) |
86
Table of Contents
- †
- Portions of this exhibit have been omitted pursuant to a request for confidential treatment by the Securities and Exchange Commission and the non-public information has been filed separately with the Securities and Exchange Commission.
87
Table of Contents
SIGNATURES
The Registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this Annual Report on its behalf.
| | | | | | |
| Zealand Pharma A/S | | | | |
/s/ ADAM STEENSBERG
|
|
/s/ MATS BLOM
|
| Name: | | Adam Steensberg | | Name: | | Mats Blom |
| Title: | | Interim Chief Executive Officer, Executive Vice President and Chief Medical and Development Officer | | Title: | | Executive Vice President and Chief Financial Officer |
Denmark
Dated: March 15, 2019
88
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the shareholders and the Board of Directors of Zealand Pharma A/S
Opinion on the Financial Statements
We have audited the consolidated statements of financial position of Zealand Pharma A/S and subsidiaries (the "Company") as of December 31, 2018 and 2017, the related consolidated income statements and consolidated statements of comprehensive income (loss), changes in equity and cash flow for each of the three years in the period ended December 31, 2018, and the related notes (collectively referred to as the "financial statements") included in Exhibit 15.1 on pages 51-87. In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2018 and 2017, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2018, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
As discussed in Note 1 to the financial statements, the 2017 and 2016 financial statements have been restated to correct a misstatement in relation to royalty revenue and royalty expenses.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles
89
Table of Contents
used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Deloitte
Statsautoriseret Revisionspartnerselskab
CVR no: 33963556
| | |
/s/ Martin Norin Faarborg

State Authorized
Public Accountant | | /s/ Sumit Sudan
State Authorized
Public Accountant
|
Copenhagen, Denmark
March 14, 2019
We have served as the Company's auditor since 2014.
90