UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 1-K
ANNUAL REPORT PURSUANT TO REGULATION A
For the fiscal year ended:
December 31, 2021
Emerald Health Pharmaceuticals Inc.
(Exact name of issuer as specified in its charter)
Delaware
(State or other jurisdiction of incorporation or organization)
82-0669961
(I.R.S. Employer Identification Number)
5910 Pacific Center Blvd, Suite 320, San Diego, CA 92121
(Full mailing address of principal executive offices)
(858) 352-0622
(Issuer’s telephone number, including area code)
Common stock, par value $0.0001
(Title of each class of securities issued pursuant to Regulation A)
PART II
STATEMENTS REGARDING FORWARD-LOOKING INFORMATION
We make statements in this Annual Report on Form 1-K that are forward-looking statements within the meaning of the federal securities laws. The words “believe,” “estimate,” “expect,” “anticipate,” “intend,” “plan,” “seek,” “may,” and similar expressions or statements regarding future periods are intended to identify forward-looking statements. These forward-looking statements involve known and unknown risks, uncertainties and other important factors that could cause our actual results, performance or achievements, or industry results, to differ materially from any predictions of future results, performance or achievements that we express or imply in this Annual Report or in the information incorporated by reference into this Annual Report.
The forward-looking statements included in this Annual Report on Form 1-K are based upon our current expectations, plans, estimates, assumptions and beliefs that involve numerous risks and uncertainties. Assumptions relating to the foregoing involve judgments with respect to, among other things, future economic, competitive and market conditions and future business decisions, all of which are difficult or impossible to predict accurately and many of which are beyond our control. Although we believe that the expectations reflected in such forward-looking statements are based on reasonable assumptions, our actual results and performance could differ materially from those set forth in the forward-looking statements. Factors which could have a material adverse effect on our operations and future prospects include, but are not limited to:
| ● | The success of our product candidates will require significant capital resources and years of clinical development efforts; |
| ● | The results of clinical testing and trial activities of our products; |
| ● | Our ability to obtain regulatory approval and market acceptance of, and reimbursement for our products; |
| ● | Our ability to protect our intellectual property and to develop, maintain and enhance a strong brand; |
| ● | Our ability to compete and succeed in a highly competitive and evolving industry; |
| ● | Our lack of operating history on which to judge our business prospects and management; |
| ● | Our ability to raise capital and the availability of future financing; |
| ● | Our ability to manage our research, development, expansion, growth and operating expenses; |
| ● | The impact of the coronavirus (COVID-19) pandemic and other unpredictable events, and any business disruptions associated therewith; and |
| ● | Our reliance on third parties to conduct our research, preclinical (non-human) studies, manufacturing and clinical trials. |
Any of the assumptions underlying forward-looking statements could be inaccurate. You are cautioned not to place undue reliance on any forward-looking statements included in this Annual Report. All forward-looking statements are made as of the date of this Annual Report on Form 1-K and the risk that actual results will differ materially from the expectations expressed in this Annual Report will increase with the passage of time. Except as otherwise required by the federal securities laws, we undertake no obligation to publicly update or revise any forward-looking statements after the date of this Annual Report, whether as a result of new information, future events, changed circumstances or any other reason. In light of the significant uncertainties inherent in the forward-looking statements included in this Annual Report, the inclusion of such forward-looking statements should not be regarded as a representation by us or any other person that the objectives and plans set forth in this Annual Report will be achieved.
1
Item 1. Business
Overview
Emerald Health Pharmaceuticals Inc. (the Company, EHP, we, our, and us) was formed on March 2, 2017 under the laws of the State of Delaware, and is headquartered in San Diego, California. We are a clinical stage pharmaceutical company focused on developing drug product candidates currently containing novel, patented small molecules, rationally designed to treat various diseases with unmet medical needs, including neurodegenerative, autoimmune and other diseases. We are currently developing two initial therapeutic product candidates, EHP-101 and EHP-102, that target four initial indications, multiple sclerosis (MS) and systemic sclerosis (SSc, a severe form of scleroderma) with EHP-101, and Parkinson’s disease (PD) and Huntington’s disease (HD) with EHP-102. We believe treatments for these indications represent markets with underserved patient populations.
Our platform technology currently consists of 25 novel, patented molecules which are all new chemical entities (NCE)s with various mechanisms of action (MOA) which make them capable of affecting specific receptors and pathways in the body that are targets for various diseases that currently have unmet medical needs. The novel molecules are currently covered by 28 issued international patents. In addition, we have 21 pending international patent applications. We believe our unique technology platform represents an advancement to existing therapies because our NCEs are designed to act on key biological receptors and pathways in the body to specifically treat the diseases we are targeting.
Our current product development pipeline includes our two initial product candidates, EHP-101 and EHP-102, containing NCEs from our library of molecules. EHP-101, our lead product candidate, is being developed as an oral formulation of one of the molecules in our portfolio, known as VCE-004.8, and is currently in Phase 2 clinical (human) trials. EHP-102 is being developed as an oral formulation of another one of the molecules in our portfolio, known as VCE-003.2, and is currently in preclinical (non-human) development. In addition to the four initial indications being targeted with EHP-101 and EHP-102, other applications and different formulations are also being investigated with these NCEs, and research is ongoing with other molecules within our NCE portfolio, as well as continued discovery of additional unique molecules to add to our portfolio.
In September 2019, we successfully completed a Phase 1 clinical trial on EHP-101 in Australia to establish its safety, tolerability and pharmacokinetics (PK) in healthy volunteers. During 2020, we initiated a Phase 2a safety and efficacy clinical trial with EHP-101 in SSc patients and in 2021 we commenced the initiation of activities for a Phase 2a trial with EHP-101 in MS patients, which is expected to begin enrolling patients in 2022. If such clinical trials are successful, we plan to advance the product candidates into additional clinical trials.
We are currently conducting two clinical trials with EHP-101: a Phase 2a clinical trial in SSc patients in Australia, New Zealand, the United States and Puerto Rico, and a Phase 2a clinical trial in MS patients in Australia and the United States. Prior to initiating Phase 2a, we obtained two separate Investigational New Drug (IND) clearances (essentially “approval” to start a clinical trial) for both trials, as well as Fast Track designation from the FDA for the SSc indication in the United States. See the section “Fast Track Designation, Accelerated Approval, and Breakthrough Therapy Designation” for a description of Fast Track designation. We have also received all regulatory clearances that were required to start the SSc Phase 2a clinical trial in Australia and New Zealand and for the MS Phase 2a trial in Australia. In addition, we have also been granted Orphan Drug Designation (ODD) from the FDA in the United States and from the EMA in Europe for SSc. We may expand our clinical trials to other countries as they progress through Phase 2.
We have completed preclinical proof of concept (POC) studies for EHP-102. We are now in the manufacturing and formulation development stage and are conducting the nonclinical studies required for advancement to clinical trials.
We have been granted ODD from the Food and Drug Administration (FDA) in the United States and from the European Medicines Agency (EMA) in Europe for EHP-101 for the SSc indication and for EHP-102 for the HD indication. We have also received Fast Track designation by the FDA for EHP-101 for the SSc indication.
2
The Technology
Our molecules are synthetically manufactured NCEs, two of which are used as the active pharmaceutical ingredients (API) in our current two product candidates. The 25 patented molecules currently in our portfolio were designed to affect specific biological receptors and pathways in the body related to various diseases, including the four initial indications we are targeting with EHP-101 and EHP-102. Based on the extensive research and discovery work over the past decade, it was determined that our molecules would best be designed based on the molecular structure of cannabidiol (CBD) and cannabigerol (CBG), two non-psychoactive cannabinoids which are believed to have fundamental health and therapeutic benefits and a good safety profile. While CBD and CBG may be classified by the United States Drug Enforcement Administration (DEA) as controlled substances in the United States depending on their origin and purity, since our molecules are NCEs unlike CBD and CBG, in March 2019 we received a decision from the DEA that the API (VCE-004.8) in our lead product candidate, EHP-101, is not a controlled substance. We have also received the same decision from the United Kingdom (UK) Home Office and Canada’s Controlled Substances Directorate. We plan to seek the same determination for the API (VCE-003.2) in our second product candidate, EHP-102, once it gets closer to clinical development.
We believe that one of the competitive advantages of our technology is the effect that the APIs in our lead product candidates have been shown to have in our studies on various biologic receptors and physiologic/biochemical pathways in the body related to the diseases we are targeting. We know of no other molecules which display this combined multi-modal MOA. Our synthetic molecules are designed to improve on the bioactivities of the natural molecules used as the foundational chemical structure of our NCEs and provide opportunities to advance their development into pharmaceutical products.
Our Initial Product Candidates and Clinical Development
Based on the novel MOA of our molecules and the multiple preclinical studies conducted to date, we believe that our initial product candidates have the potential to be disease modifying, while most other compounds approved for many of these diseases are limited to targeting the symptoms of the disease.
EHP-101 (API: VCE-004.8)
Overview
Our lead product candidate, EHP-101, is an oral formulation of our NCE called VCE 004.8, that affects some of the known biologic receptors and physiologic pathways involved in both MS and SSc. Thus, our first two chosen indications for EHP-101 are MS and SSc. These conditions are both autoimmune diseases, which cause inflammation and damage to various parts of the body, including nerve and vascular damage. In SSc, the disease is initiated by endothelial vascular damage followed by inflammation and fibrosis. In MS, brain blood barrier (BBB) disruption and alterations in the neurovascular areas of the body leads to neuroinflammation and demyelination. Thus, EHP-101 acts on specific targets involved in these pathogenic events.
EHP-101 is an orally formulated product containing VCE-004.8, a ligand modulator (partial agonist) of Peroxisome Proliferator-Activated Receptor gamma (PPARγ) and an activator of cannabinoid receptor type-2 (CB2), that can directly bind and activate both receptors as demonstrated by in vitro binding and transcriptional assays. Based on the known effects activators of these receptors have on the body, we believe the combination of activities toward both PPARγ and CB2-dependent signaling pathways could represent an important advancement in the development of therapies for MS and SSc.
In addition to PPARγ and CB2 receptor effects, VCE-004.8 also activates the HIF pathway (via the upstream activation of the B55a regulatory subunit of Protein Phosphatase 2A [PP2A]), which also has potential benefits in both MS and SSc. B55a/PP2A is essential to maintain endothelial vascular homeostasis, thus protecting endothelial damage from harmful stimuli. In addition, it also activates the HIF pathway and different studies have indicated that HIF-1α activation may play a role in inflammatory and remitting phases of MS. For instance, HIF-1α activates many genes whose products exert neuroprotective activities. HIF-1α activation is also implicated in the modulation of the immune system. In addition, there is strong evidence suggesting that activation of the HIF pathway may be linked to neuroprotection and myelination (myelination is important in MS). HIF-1α also activates several genes involved in vascular physiology (especially important in SSc), including VEGF-A and FGF-2, which are mainly produced by vascular endothelial cells. The vascular endothelial cells produce factors that maintain brain and peripheral vascular homeostasis (important in both MS and SSc).
3
Furthermore, using in vitro screening for a number of receptors we have found that EHP-101 has no off-target binding activity, including no CB1 receptor activity, resulting in no psychotropic effects.
EHP-101 is currently formulated for oral administration as a combination of corn oil and long-chain mono-, di, and triglycerides with the API known as VCE-004.8.
To date, the pharmacokinetic (PK) profile of EHP-101 has been studied in a number of mouse, rat and dog studies and has also been evaluated in a Phase 1 clinical (human) trial. PK studies evaluate the absorption, distribution, metabolism, and elimination (ADME) by the body and measure, among other things, the concentration of the substance in plasma.
We have completed extensive animal toxicology studies on EHP-101 that supported a Phase 1 clinical trial, which was completed in September 2019. Additional studies have been completed or are ongoing to support Phase 2 clinical trials worldwide, which have begun for both SSc and MS.
A Good Manufacturing Practices (GMP) process has been developed to manufacture the EHP-101 API (VCE-004.8) and drug product through our contract manufacturing organizations (CMOs). Our CMOs produced several multi-kilogram scale bulk batches of our API for use in our preclinical and clinical studies. We have transferred the manufacturing of the API to a CMO with large scale and commercial capabilities. EHP-101 API is a synthetic molecule and we believe there are readily available supplies of all raw materials needed for the manufacture of EHP-101. We do not own or operate manufacturing facilities for the production of EHP-101. We expect to depend on third-party suppliers and manufacturing organizations for all of our clinical trial quantities of raw materials, drug substance and finished product.
EHP-101 Biologic Receptor and Physiologic Pathway Activity
Cannabinoid Receptor Type-2 (CB2)
The endocannabinoid system (ECS) is an important physiologic system in the body which helps to maintain homeostasis (balance) throughout the body. The ECS is composed of two primary sets of receptors known as cannabinoid receptor type-1 (CB1) and cannabinoid receptor type-2 (CB2). These receptors are affected by several types of molecules known as cannabinoids, which are a complex group of molecules that comprise endocannabinoids (naturally occurring within the body), phytocannabinoids (from plants), and synthetic cannabinoids (made synthetically). Cannabinoids were initially identified by their ability to affect the CB1 and CB2receptors in the body. It has been determined that the psychoactive effects related to certain cannabinoids is through interactions with CB1 cannabinoid receptors. CB2 receptor interaction has not been associated with psychoactive effects. CB2 receptors have been the subject of considerable attention, primarily due to their promising therapeutic potential for treating various pathologies while avoiding the psychoactive effects that can accompany CB1 receptor activation . Neither CBD nor CBG interact with CB1 receptors. We believe the known biologic activity of natural phytocannabinoids (specifically CBD and CBG) on the ECS accounts for their ability to positively modulate several key processes in the body including neuroprotection, inflammation, immunomodulation and vascular responses. In studies conducted to date, our lead novel molecule in EHP-101 (VCE-004.8), which was designed based on the fundamental chemical structure of CBD, has been shown to not interact with CB1 receptors, to enhance the CB2 receptor modulation activity more than CBD, and provide additional interactions with other receptors and pathways in the body beyond what CBD can affect, therefore, having the potential to provide significantly greater therapeutic benefits.
Peroxisome Proliferator-Activated Receptors
One of the additional effects provided by VCE-004.8 compared to CBD is the activation (as a partial agonist) of the PPARg. PPARs play essential roles in the regulation of cellular differentiation, development, and metabolism (carbohydrate, lipid, protein). Three types of PPARs have been identified, alpha (α), gamma (γ), and beta/delta (β/δ). PPARγ is a nuclear receptor originally implicated in the regulation of cell growth, lipid metabolism and blood sugar regulation. However, PPARγ is broadly expressed and has also been recognized to play a key role in inflammatory and connective tissue balance. PPARγ activators have been shown to prevent inflammation, dermal fibrosis and loss of fatty tissue. PPARγ is activated by some endocannabinoids and related signaling lipids, as well as by certain other natural and synthetic cannabinoids. In studies conducted to date, our lead molecule has shown modulation of PPARγ activity, as a partial agonist, providing the potential for immunomodulatory activity, neuroprotection and the promotion of oligodendrocyte progenitor cell differentiation and enhancement of their antioxidant defenses.
4
Hypoxia-Inducible Factor Pathway
Another additional effect provided by VCE-004.8 compared to CBD is the activation of the Hypoxia-Inducible Factor (HIF) pathway via its upstream direct activation of the B55a regulatory subunit of PP2A. HIF plays an important role in the body. HIF-1 is a protein complex that plays an integral role in the body’s response to low oxygen concentrations, or hypoxia. HIF-1 is among the primary genes involved in the homeostatic process and has two subunits, HIF-1α and HIF-1β. HIF operates in all mammalian cell types and responds to changes in oxygen, providing cells with a master regulator that coordinates changes in gene transcription. Hypoxia preconditioning induced by mild hypoxia can be beneficial in a wide number of disorders, including neurologic and inflammatory diseases. Cellular adaptation to severe or mild hypoxia begins immediately with the activation of the HIF pathway and regulates a plethora of genes involved in many biological processes, including red blood cell production, angiogenesis, neuroprotection, myelination, vascular tone and immunity. HIF-1α activation may play a role in the inflammatory and remitting phases of MS. In addition, there is evidence suggesting that activation of the HIF pathway may be also linked to neuroprotection and myelination. The erythropoietin (EPO) gene is HIF-dependent, and EPO is neuroprotective in different animal models of MS. In addition, HIF-1α activates several blood vessel-forming genes, including vascular endothelial growth factor-A (VEGF-A) and fibroblast growth factor-2 (FGF-2), which are mainly produced by vascular endothelial cells. The vascular endothelial cells produce factors that help maintain brain homeostasis within the context of the neurovascular unit. In general, HIF-1α activates many genes whose products exert neuroprotective activities and HIF-1α activation is also implicated in the modulation of the immune system. In studies conducted to date, the API in EHP-101 (VCE-004.8), has shown activation of the HIF pathway, providing the potential for neuroprotection, vascular protection, anti-inflammatory activity and remyelination in MS models. In addition, EHP-101 (oral form of VCE-004.8) prevented vascular disruption and inflammation of peripheral vessels in scleroderma models.
As indicated above, we have identified that VCE-004.8 activates the HIF pathway through a novel mechanism involving activation of the B55a/PP2A holoenzyme phosphatase pathway. It has been demonstrated that this phosphatase specifically inactivates prolyl hydroxylase 2 (PHD2) by dephosphorylation of its serine 125. As its downstream consequence, the HIF pathway is activated, providing neuroprotective and vascular remodeling activities (i.e., prevention of blood brain barrier disruption, which occurs in MS, and peripheral angiogenesis, which is important in SSc).
Initial Indications with EHP-101
Systemic Sclerosis (SSc)
SSc is a rare, heterogeneous, severe and life-threatening form of scleroderma that involves three main hallmarks: fibroblast dysfunction leading to increased deposition of extracellular matrix proteins, small vessel damage resulting in tissue hypoxia and an immune response with autoantibody production. SSc is characterized by progressive thickening and fibrosis of the skin secondary to excessive collagen accumulation, that can be limited to skin areas below, but not above, the elbows and knees, with or without involvement of the face (limited cutaneous SSc) or wider skin areas, including skin on the arms, above and below the elbows, frequently on the legs, above and below the knees, with or without involvement of the face as well as on the torso (diffuse cutaneous SSc, or dcSSc), both with internal organ involvement (e.g., lung, kidney, heart, stomach, bowels).
SSc is initiated by microvascular injury and inflammation followed by fibroblast activation, a key event in fibrosis development. Activated fibroblasts are responsible for the excessive collagen synthesis and transforming growth factor beta (TGFβ) production. TGFβ signaling plays a critical role in the regulation of cell growth, differentiation, and development in a wide range of biological systems. Excessive TGFβ signaling is the hallmark of SSc and different strategies aimed to disrupt this signaling pathway have been proposed for the treatment of SSc and related fibrotic diseases. Different studies prove that PPARγ and CB2 receptors are potential therapeutic targets for the disease because of their involvement in the inhibition of inflammation and fibrosis progression. Vascular damage is also a key pathological event in SSc and therefore the activation of the HIF pathway could provide a benefit for SSc patients.
5
Recent evidence indicates that genetic and pharmacological manipulation of the ECS modulates the fibrotic response. Thus, CB1 and CB2 receptors have shown different patterns in experimental models of dermal fibrosis. While CB1 activation is detrimental for the disease, CB2 activation has shown protection in mice from experimental dermal fibrosis. As stated previously, EHP-101 has been shown to affect CB2 but not CB1 receptors.
SSc is a rare disease with no cure, with a combined prevalence of less than 200,000 patients in North America and Europe. We have been granted ODD by the FDA in the United States and the EMA in the EU. SSc is a condition that is both chronically debilitating and life-threatening. The debilitating nature of the disease manifests itself through the development of painful lesions, disfigurement and the loss of ability to function normally. Currently there are no approved treatments specifically for SSc that could stop the pathological fibrotic transformation in tissues of patients with SSc. Despite recent progress in the understanding of SSc pathophysiology, the current therapeutic recommendations focus on the management of organ specific morbidity and no single therapeutic agent has been proven to be efficacious as a universal disease-modifying agent that provides benefit to SSc patients regardless of which organs are affected by the disease.
SSc is life-threatening because of its effects on the vital organs: heart, lungs, kidneys and the digestive tract. The life-threatening acute onset of severe hypertension and renal failure occurs in approximately 66% of patients with SSc within the first 12 months of diagnosis. Up to 80% of SSc patients develop interstitial lung disease. Despite advances in early recognition of the disease and availability of efficient treatments for some of its organ complications, the mortality of SSc continues to be high. Although the median survival rate is 21.9 years, it varies significantly among different population groups. Additionally, the side effects of immunosuppressive treatments, which SSc patients typically receive as a standard of care, often result in life-threatening complications requiring intensive care unit admissions. Furthermore, the sites of SSc activity, such as skin, lung and liver, are susceptible to malignant transformation. The incidences of skin, lung and liver cancers are 3 to 4-fold higher in SSc patients compared to the general population.
As described previously, EHP-101 behaves as a dual activator of PPARγ and CB2 receptors and, therefore, inhibits collagen synthesis. Moreover, EHP-101 has been shown to inhibit the TGFβ-associated differentiation of cells (called myofibroblasts) that are responsible for fibrosis.
The anti-fibrotic efficacy in vivo was investigated in two mouse models of scleroderma and fibrosis, namely bleomycin (BLM)-induced fibrosis and angiotensin-II (Ang-II)-induced fibrosis. The BLM model mimics the autoimmune part of the disease and the Ang-II model is useful to investigate the pathogenic role of Ang-II on inflammation and fibrosis in different organs (heart, lung, kidney, skin). EHP-101 reduced dermal thickness, blood vessel collagen accumulation and prevented other negative cell activities in the skin and in several internal organs throughout the body. In addition, it prevented the expression of several key genes associated with fibrosis, qualifying this novel synthetic NCE as a promising compound for the management of systemic sclerosis/scleroderma and, potentially, other fibrotic diseases.
B55a (PP2A/HIF) activators have not been investigated extensively in fibrotic diseases such as SSc. However, based on our preclinical studies, we believe that this class of compounds has the potential to induce vascular protection and address the vasculopathies associated with SSc. Accordingly, we believe our studies have demonstrated that EHP-101 showed efficacy and outperformed ajulemic acid (Lenabasum) in the preclinical models of fibrosis that we have investigated.
6
Summary of Mechanisms of EHP-101 in SSc

EHP-101 has a targeted MOA acting at different molecular targets that are the hallmark of SSc. EHP-101 has potent anti-inflammatory and anti-fibrotic activities by targeting PPARγ and CB2. EHP-101 also inhibits fibroblast to myofibroblast differentiation and collagen synthesis. In addition, EHP-101 activates the B55a/PP2A/HIF pathway and mediates the expression of growth factors that can help vascular remodeling that is impaired in the disease.
In summary, we believe that EHP-101 is a promising product candidate for SSc treatment by ameliorating fibrosis, inflammation, fibroblast migration, collagen synthesis and vasculogenesis through the mechanism described above.
Multiple Sclerosis (MS)
MS is a chronic autoimmune disease of the central nervous system (CNS) that affects over 2.3 million patients worldwide and there is presently no cure for this disease. The hallmarks of MS include neuroinflammation, the loss of myelin (i.e., demyelination) and nerve cell damage. Myelin provides insulation for nerve fibers and is essential to maintain nerve conduction velocity. Disease progression is thought to be composed of two underlying processes: myelin destruction, or demyelination, with failure to remyelinate and progressive nerve cell damage with little capacity for recovery. Exacerbated innate and adaptive immune responses contribute to the pathophysiology of the disease and the majority of current therapies for MS are directed towards inflammation and modulation of the immune response. However, therapies aimed to remyelinate nerve cells are needed.
Cannabinoids such as CBD that do not bind to and activate CB1 receptors, and therefore do not produce psychotropic effects, are considered of special interest as therapeutic agents in CNS diseases. In the CNS, there is evidence that CB2 receptors regulate neurotoxicity in certain cells of the CNS, called microglia. Cannabinoids also activate the nuclear receptor superfamily of PPARs. Three forms of PPARs have been identified (PPARγ, PPARα and PPARβ/δ) and within these receptors, PPARγ can be activated weakly by cannabinoids such as CBD. Due, in part, to their PPARγ-activating properties, we believe these cannabinoids may exert anti-inflammatory activities, thus showing a therapeutic potential for the treatment of inflammatory diseases. PPARγ has been detected in certain nerve cells and participates in mechanisms that control activation of inflammatory response including modulation of cytokines and chemokine expression, neuronal dysfunction, and neurodegeneration. Neuroinflammation is an integral component of disorders such as Alzheimer’s disease, PD, stroke and MS. PPARγ activators have been shown to reduce the incidence and severity of disease in experimental models of MS, such as experimental autoimmune encephalomyelitis (EAE), and a small clinical trial suggested that PPARγ could be a pharmacological target for the management of MS.
7
EHP-101 has been shown to bind to CB2and PPARγ receptors, providing anti-inflammatory activity in the CNS as well as other positive biologic effects. Microglial cells specifically express CB2receptors in EAE tests, and activated microglial cells expressed ten-fold more CB2receptor activity than microglia in the resting state, suggesting that this receptor plays an important role in microglial cell function in the CNS during autoimmune-induced inflammation. In addition, PPARγ is expressed in different CNS cell types and its activation by ligand agonists provides immunomodulatory and neuroprotection and promotes oligodendrocyte progenitor cell differentiation and enhances their antioxidant defenses.
HIF operates in all mammalian cell types and responds to changes in oxygen, providing cells with a master regulator that coordinates changes in gene transcription. Hypoxia preconditioning induced by mild hypoxia can be beneficial in a wide number of disorders including neurologic and inflammatory diseases. Our preliminary studies indicate that VCE-004.8 activates the B55a/PP2A/HIF pathway in different CNS and vascular cells as demonstrated by in vivo and in vitro functional assays. The results of these assays show that EHP-101 up-regulates the expression of neuroprotective genes such as erythropoietin and VEGF-A.
In vivo experiments in two mouse models of MS, EAE and Theiler virus-induced encephalopathy (TMEV) using VCE-004.8, the API in EHP-101, have resulted in the prevention of demyelination, nerve cell damage and immune cell infiltration. VCE-004.8 also downregulated the expression of several genes including chemokines, cytokines and adhesion molecules, which are closely associated with MS pathophysiology. In addition, EHP-101, the oral formulation of VCE-004.8, resulted in the reduction of MS symptoms in both mouse models.
EHP-101 has also been shown to induce statistically significant remyelination in the brain in two different cuprizone (CPZ) mouse model compared to placebo. To evaluate the effect of EHP-101 on myelin damage in a cuprizone-induced demyelination murine model, brain coronal sections from animals after 6 weeks of CPZ 0.2% diet and 2 weeks of EHP-101 treatment were evaluated. In this model, EHP-101 treatment began after CPZ diet removal to directly evaluate the effects of EHP-101 on remyelination. Spontaneous recovery from demyelination was insignificant after 1 and 2 weeks but remyelination was significantly accelerated by EHP-101 treatment. EHP-101 enhanced remyelination in both white and gray matter of the brain, namely the corpus callosum and the cortex.
Furthermore, the potential of oral administration of EHP-101 to promote remyelination was evaluated in a cuprizone/rapamycin (C/R) mouse model (“augmented cuprizone model”), a more aggressive demyelination model since the concomitant administration of rapamycin with cuprizone for 12 weeks reduces spontaneous myelin production by blocking differentiation of oligodendrocyte progenitor cells. In the study, oral administration of EHP-101 once daily for 6 weeks induced statistically significant, dose-dependent remyelination of demyelinated axons in the white matter (corpus callosum) compared to vehicle-treated controls as shown by the enhanced density of myelinated axons.
8
Summary of Mechanisms of EHP-101 in MS
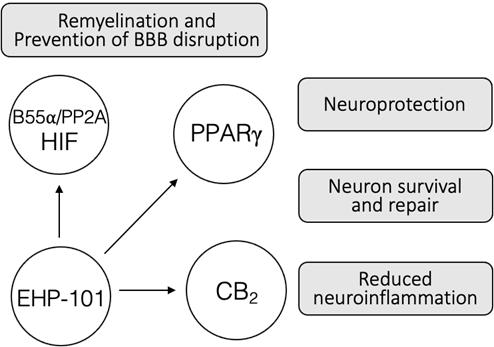
EHP-101 has a targeted MOA acting at different molecular targets involved in the pathophysiology of MS. EHP-101 also has potent anti-inflammatory and neuroprotective activity through effects on PPARγ and CB2. In addition, EHP-101 activates the B55a/PP2A/HIF pathway and mediates the expression of growth factors that play a role in CNS activity, homeostasis, and remyelination.
In summary, we believe that EHP-101 is a promising product candidate for MS treatment, by ameliorating neuroinflammation through PPARγ/CB2receptors and by inducing neuroprotection and remyelination through activation of the B55a/PP2A/HIF pathway through the mechanism described above.
Clinical Development of EHP-101
EHP-101 Phase I Clinical Trial for SSc and MS
Our first-in-human Phase 1 study was a large Phase 1 clinical trial conducted between September 2018 and September 2019, studying 104 healthy volunteers at 12 different dose levels. We designed the completed Phase 1 study so that this single study allowed us to proceed into Phase 2 clinical trials for both SSc and MS. Prior to initiating Phase 1, we sought advice from the FDA on (1) our strategy of pursuing one Phase 1 study in support of subsequent Phase 2 studies for both the SSc and MS indications, (2) the design of the planned Phase 1 study, with the aim of designing the study so that the subsequent Phase 2 studies could be completed wholly or partially in the United States, and (3) the preliminary planned design of our Phase 2 clinical studies. Prior to initiating Phase 2a in SSc and MS, we obtained regulatory approvals, including separate Investigational New Drug (IND) clearances for both the SSc and MS clinical trials, and Fast Track designation and ODD from the US FDA for SSc.
In the Phase 1 study a total of 48 subjects (6 subjects per cohort) received a single dose of either 0.91 mg, 3 mg, 9 mg, 20 mg, 25 mg (fasted and fed), 50 mg, 100 mg or 185 mg of EHP-101. Another 32 subjects (8 subjects per cohort) received daily repeated doses for 7 days of either 20 mg once per day (QD), 25 mg twice per day (BID), 51.9 mg QD, or 50 mg BID of EHP-101. There were 24 subjects that received placebo (2 subjects at each tested dose level in the total of 12 cohorts studied).
9
The half-life of a 25 mg single dose was approximately 2 hours during fasted state and 7 hours during fed state. A mean increase of 1.5-fold in the maximum plasma concentration (Cmax) and area under the curve (AUC, which represents the cumulative plasma concentration over time) was observed post administration with food. No drug accumulation in the blood was observed with QD dosing and minimal accumulation was observed with BID dosing for 7 days.
The Cmax for a predicted anticipated therapeutic dose (ATD) was reached with a 20 mg single dose and the targeted drug exposure based on AUC was approached with a 50 mg single dose and 25 mg BID multiple dosing for 7 days, thus helping the selection of the dosing and treatment regimen for Phase 2 studies.
Preliminary drug-related biomarker analysis in plasma on Day 7 supports the MOA of EHP-101 related to the effects on the B55a/PP2A/HIF pathway, and PPARγ and CB2 receptor activation. Some proteins were upregulated (increased concentrations post-dosing) related to vascular endothelial cell function (HIF pathway activation), lipid metabolism and control of inflammation, whereas other proteins related to CB2 and PPARγ activation (inflammation and immunomodulation) were downregulated (decreased concentrations post-dosing). Quantitative analyses by other techniques have further assessed multiple specific proteins related to EHP-101 MOA.
EHP-101 Phase 2a Clinical Trial in Systemic Sclerosis (SSc)
Our ongoing SSc trial is a randomized, double-blind, intracohort placebo-controlled, multicenter Phase 2a clinical trial designed to evaluate the safety, tolerability, pharmacokinetics and preliminary efficacy of EHP-101 for the treatment of SSc. The trial is designed to include approximately 36 patients (four cohorts of 9 patients each) in clinical sites throughout the United States, Australia, New Zealand and Puerto Rico. The primary trial endpoint (safety) includes the incidence of adverse events vs placebo. Secondary endpoints (preliminary efficacy) include trends in changes from baseline vs placebo based on a variety of analyses, including the CRISS score and its components (modified Rodnan Skin Score [mRSS]), Forced Vital Capacity (FVC), Physician’s and Patient’s Global Assessment, Scleroderma Health Assessment Questionnaire [S-HAQ]), National Institutes of Health Patient-Reported Outcomes Measurement Information-29 Short Form, 5-D Itch Score, UCLA Scleroderma Clinical Trial Consortium Gastrointestinal Tract 2.0, Functional Assessment of Chronic Illness Therapy: Fatigue, European Quality of Life 5 Domain Questionnaire (EQ-5D-3L), drug and disease related biomarkers, and gene expression data from skin biopsy (results 12 weeks after final biopsy).
EHP-101 Phase 2a Clinical Trial in Multiple Sclerosis (MS)
In 2021, we began initiation activities for our Phase 2a clinical trial for the treatment of certain relapsing forms of MS, specifically relapsing-remitting MS (RRMS) and active secondary progressive MS (SPMS). We plan to begin enrolling patients in this international Phase 2a open-label, multicenter dose-finding study during 2022. The trial is designed to include approximately 50 patients with MS in the United States and Australia. The main endpoints of the study include safety, brain lesion activity measured by MRI, disease progression measured by MS Functional Composite (MSFC), disease progression measured by Expanded Disability Status Scale (EDSS), disease progression measured by Symbol Digit Mobilities Test (SDMT), various relapse analyses, microstructural analysis and assessment of potential remyelination measured by Magnetization Transfer Ratio (MTR), assessment of white matter diffusivity and integrity measured by Diffusion Tensor Imaging (DTI), biomarker testing, and other analyses.
EHP-102 (API: VCE-003.2)
Overview
Our second product candidate, EHP-102, is an oral formulation of VCE-003.2, an NCE with a unique MOA that causes it to affect some of the accepted biologic receptors and pathways involved in various neurodegenerative diseases. This MOA includes effects on PPARγ (like EHP-101), as well as other physiologic pathways in the body related to neurodegenerative diseases. Unlike EHP-101, EHP-102 does not affect CB2 receptors or the HIF pathway. In addition to PPARγ, EHP-102 activated pathways involved in neurogenesis, namely through effects on physiologic pathways in the body related to neuroprotection and neurogenesis. Based on preclinical studies to date, we are developing EHP-102 initially for two indications, PD and HD.
10
Preclinical studies have shown that VCE-003.2 is neuroprotective and anti-inflammatory in animal models of PD and HD, as measured by proinflammatory cytokines and behavioral score, respectively. In addition, VCE-003.2 also reduced mutant huntingtin protein aggregates (altered huntingtin protein is associated with HD) detected by confocal microscopy techniques.
We are currently scaling up the manufacturing of VCE-003.2 and selecting an oral dosage form for the further development of EHP-102. Several oral drug product prototypes have been prepared and evaluated for their pharmacokinetics profile and bioavailability in mice, rats and dogs.
An industrial scalable process is being developed to manufacture the EHP-102 API (VCE-003.2) and drug product through our CMOs. We do not own or operate manufacturing facilities for the production of EHP-102. We expect to depend on third-party suppliers and CMOs for all of our preclinical and clinical quantities of raw materials, drug substance and drug product.
We have begun the preclinical studies required to advance to clinical trials. Once the manufacturing and formulation development activities are completed for EHP-102, we plan to initiate Good Laboratory Practice (GLP) animal safety and toxicology studies in support of initiating human clinical development.
EHP-102 Biologic Receptor and Physiologic Pathway Activity
The API (VCE-003.2) in our second product candidate (EHP-102) has been shown to affect PPARγ as a partial agonist, like EHP-101, which can provide the potential for immunomodulatory activity, neuroprotection and the promotion of oligodendrocyte progenitor cell differentiation and enhancement of their antioxidant defenses. Unlike EHP-101, however, it does not affect CB2 receptors or the HIF pathway, but rather has been shown to activate the extracellular signal-regulated kinases (ERK) pathway (a member of the mitogen-activated protein kinases (MAPK) pathway) in hippocampal neuronal progenitor cells as well as a transcription factor involved in nerve cell regeneration (the chicken ovalbumin upstream promoter transcription factor-interacting protein, Ctip2). It has also been shown to reduce the expression of cyclooxygenase-2 (COX-2) in nerve cells (involved in inflammation and pain) and improve activity on various oxidative stress markers which provide potent antioxidant activity in the brain.
Initial Indications with EHP-102
Parkinson’s Disease (PD)
PD is a long-term degenerative disorder of the CNS that mainly affects the motor system. It is a disease where damaged neurons do not produce sufficient dopamine (dopamine helps transmit impulses from the brain to the muscles). Over 10 million people suffer from PD worldwide. The symptoms generally present slowly over time. Early in the disease, the most obvious symptoms are shaking, rigidity, slowness of movement, and difficulty with walking. Thinking and behavioral problems may also occur. Dementia becomes common in the advanced stages of the disease. Depression and anxiety are also common, occurring in more than a third of people with PD. Other symptoms may include sensory, sleep, and emotional problems. The main motor symptoms are collectively called “Parkinsonism,” or “Parkinsonian syndrome.”
Inflammation is a key pathogenic event in PD, so anti-inflammatory strategies are being investigated to limit neuronal deterioration in this disease. Certain cannabinoids have been shown to have anti-inflammatory and neuroprotective properties. In addition, epidemiological data support that the regular use of non-steroidal anti-inflammatory drugs (e.g., ibuprofen) reduces the risk of developing PD. In light of this, different anti-inflammatory agents have been investigated, at preclinical and clinical levels, with variable success in affecting the symptoms of PD.
In addition, another possible rationale for the therapeutic potential in PD may be the possibility of targeting PPARγ receptors. Such conclusion is based on: (i) the relevant role played by these nuclear receptors in the control of inflammation in numerous pathological conditions (ii) the well-described PPARγ-mediated anti-inflammatory activity in different models of central and peripheral inflammation, and (iii) the effects of various PPARγ activators (e.g., thiazolidinediones) in PD. The three different PPAR isotypes (α, β/δ and γ) are expressed in all cell types in the brain and all of them, by functioning in an integrated manner as a complex system – the so-called PPAR triad, have been reported to have neuroprotective properties.
11
Given the activity of EHP-102 on the PPARγ receptor and the ERK pathway, which are important factors involved in the control of inflammation and neurogenesis, we evaluated its anti-inflammatory and neuroprotective properties in a typical in vivo inflammatory model of PD, lipopolysaccharide (LPS)-lesioned mice. Positive results were obtained in all measurements assessed, both qualitative and quantitative. Unlike EHP-101, EHP-102 has no activity on CB2 receptors, however, in this study proinflammatory mediators such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and inducible nitric oxide synthase (iNOS) were strongly reduced by the treatment with EHP-102. Thus, in this model and another validated preclinical PD model (the 6-hydroxydopamine model), EHP-102 reduced inflammatory marker expression and prevented dopaminergic neuronal loss (i.e., the loss of nerve cells that produce dopamine, which is the main pathologic feature of PD). Moreover, we believe our studies have demonstrated that EHP-102 alleviates Parkinsonism symptomatology, neuroinflammation and the loss of dopaminergic neurons induced by overexpression of a mutant form of alfa-synuclein in a third animal model of PD. These data lead us to believe EHP-102 could be a potential treatment for PD as well as other synucleinopathies.
Huntington’s Disease (HD)
HD is an autosomal dominant genetic disorder causing the mutation of a protein called the huntingtin protein which causes progressive degeneration of nerve cells in the brain, specifically, cells of the basal ganglia. This devastating and disabling disease affects middle-aged people with typical onset between the ages of 30 and 50. The genetic defect that causes HD is an abnormal repeat of certain DNA sequences on chromosome number 4. With each successive generation, the number of repeats increases. There is a 50% chance that the disease will be passed to offspring.
HD is a rare disease with approximately 30,000 patients annually in the United States. We have been granted ODD by the FDA in the United States and by the EMA in Europe.
One of the hallmark signs and symptoms of this disease is involuntary movements and tics. These involuntary movements and tics begin distally and move proximally as the disease progresses. The involuntary movements and tics are not specific and may involve the hands, feet, and face, with the most prominent movements taking place in the muscles of the back. The involuntary movements lead to the appearance that a patient is inebriated. Swallowing becomes difficult, and patients are at risk of choking. Reduced movement and rigidity are common. Patients may also suffer from flexion contractures and become bed bound. Finally, the patient is completely dependent on others for care. The risk of choking increases and the involuntary movements may become more severe or completely disappear. At this point the patients can no longer walk. Cognitive symptoms may include feelings of low self-esteem, guilt, anxiety, apathy, irritability, aggression, dementia and psychosis with paranoia and auditory hallucinations. The cause of death is usually from secondary causes of the disease such as choking and infection.
There is no known curative treatment for HD. Treatment is mostly directed at symptomatic relief with suppression of the involuntary movements and tics. Dopamine-depleting agents, dopamine activators, benzodiazepines, glutamate antagonists, acetylcholinesterase inhibitors, dopamine antagonists, anti-seizure medications, cannabinoids, lithium, along with deep brain stimulation and fetal cell transplantation are being used to treat the symptoms of HD.
The molecular mechanisms of HD pathophysiology are unclear. The current model of disease progression includes development of mitochondrial dysfunction in the huntingtin protein. PPARγ is believed to play a key role in neurodegenerative diseases as it regulates neural progenitor cell proliferation and differentiation. Studies have demonstrated that there are significant defects in the PPARγ signaling pathway in mutant huntingtin expressing cells as compared to wild-type huntingtin protein cells. PPARγ activators improve mitochondrial function in cells expressing mutant huntingtin. The activation of the PPARγ signaling pathway can help mitochondrial function, a pivotal process in the pathogenesis of HD. Therefore, the PPARγ pathway could be a rational therapeutic target in the treatment of HD.
Preclinical in vitro studies have shown that VCE-003.2 preserves the ability to activate PPARγ and exerts a prosurvival action in progenitor cells during neuronal differentiation. In addition to EHP-102’s effect on PPARγ, our studies indicate effects on other targets involved in neurodegeneration. For example, EHP-102 reduced mutant huntingtin aggregates in striatal cells as noted above. The neuroprotective profile of EHP-102 was also analyzed using three in vivo models of striatal neurodegeneration, which mimic HD in humans. EHP-102 inhibited the upregulation of proinflammatory markers and improved antioxidant defenses in the brain of the test animals.
12
To assess the pathophysiological relevance of the neuroprotective action of EHP-102 in vivo, we employed three mouse models of the disease, namely HD induced by either 3-nitropropionic acid, quinolinic acid or intracranial infection with an Adeno Associated Virus harboring mutated huntingtin.
In summary, these studies suggest that EHP-102 displays neuroprotective and anti-inflammatory activities in different mouse models of HD. For example, in these models EHP-102 has:
| ● | prevented neural damage and neuroinflammation; |
| ● | alleviated motor symptomatology; |
| ● | improved motor symptomatology in mice expressing a mutated form of huntingtin protein in the brain; |
| ● | inhibited the up-regulation of proinflammatory markers such as COX-2, TNF-α, and IL-6; |
| ● | improved oxidative stress markers; and |
| ● | promoted neurogenesis in the striatum. |
These data suggest that EHP-102 could have potential for the treatment of HD and other neurodegenerative diseases with neuroinflammatory traits.
Our Planned Clinical Trials
Since Phase 1 human studies are not expected to begin within the next year, we have not yet begun to develop the full clinical development plan for EHP-102.
Intellectual Property
We believe it is important to our success that we:
| ● | obtain and maintain patent and other legal protections for the proprietary molecules, technology, inventions and improvements we consider important to our business; |
| ● | prosecute our patent applications and defend our issued patents; |
| ● | preserve the confidentiality of our trade secrets; and |
| ● | operate without infringing the patents and proprietary rights of third parties. |
We have sought and intend to continue to seek appropriate patent protection for our product candidates, as well as other proprietary technologies and their uses by filing additional patent applications in the United States and selected other countries.
As of the date of this Annual Report, we owned a total of 28 issued (granted) patents in the following locations: United States and Japan (four patents in each), Europe (three patents), Australia, Canada, China, Hong Kong, India, Israel, Mexico, and the Russian Federation (two patents in each), and Korea (one patent). In addition, we have 21 pending patent applications. These patents and patent applications will expire between 2030 and 2041 and could be eligible for patent term extension for delay caused by regulatory review, thereby further extending their patent terms. Our patent portfolio is not specific to any single indication, which we believe could provide us with patent protection for our developed products for additional patient populations in markets with unmet medical need.
13
Our patent plan is focused on providing patent protection for our NCEs, their formulation and therapeutic applications. The following is a summary of our seven patent families:
| Family Number | Patent Publication/Application Number | Status | Expiry | Title | Description | |||||
001 CBD PPARγ | US8,772,349B EP2551255B1* JP05575324B2 WO2011/117429 *Validated in DE, ES, GB, FR, IT, & NL | Granted Granted Granted Expired | 2030 | Cannabinoid Quinone Derivatives | Cannabinoid quinone derivatives to be used as medicaments, particularly as PPARγ activators for treating diseases which etiology is based on an impaired PPARγ function and can benefit from PPARγ activation. | |||||
001.2 CBD PPARγ | US9,701,618B AU2014390738B2 CA2945867C CN106232570B EP3131874B1* JP06167248B2 KR102256398B1 IN344701 BRPI1623902A2 MX352386B WO2015158381A1 RU2667504C2 IL248030 HK1231041B *Validated in BE, CH DE, DK, ES, FR, GB, IE, IT, LI, & NL | Granted Granted Granted Granted Granted Granted Granted Granted Pending Granted Expired Granted Granted Granted | 2034 | Cannabidiol Quinone Derivatives | CBD quinone derivatives to be used as medicaments in therapy, particularly or treating diseases and conditions responsive to PPARγ modulation due to their high PPARγ activatory effect | |||||
001.3 HIF | WO2018/177516 AU2017406103 CA3058352 EP3600274A1 IL269623 JP6974496B2 US10,919,843B US17/176,743 | Expired Pending Pending Pending Pending Granted Granted Pending | 2037 | Cannabidiol derivatives as inhibitors of the HIF prolyl hydroxylases activity | CBD quinone derivatives to be used as medicaments in therapy, particularly for treating diseases and conditions responsive to HIF-1 activation. | |||||
001.4 CBD Formulations | AU2020219240 CA3128939 CN202080016947.5 EP20753058.5 JP2021-545983 US17428929 | Pending Pending Pending Pending Pending Pending | 2040 | Formulations of Cannabidiol Derivatives | Formulations of CBD derivatives to be used as Modulators of Cannabinoid Receptor Type 2 (CB2) | |||||
001.5 CBD Fibrosis | PCT/US2021/017052 | Pending | 2041 | Compositions of Cannabidiol Derivatives and their use as modulators of Cannabinoid Receptor Type 2 (CB2) Cannabidiol Quinone Derivatives | Composition and Method for the treatment and preventions of cardiac fibrosis Composition and Method for the Treatment and Prevention of Cardiac, Pulmonary, Dermal, and Renal Fibrosis |
14
002 CBG PPARγ | US9,802,880B AU2015222384B CA2937275C CN106061937B EP2913321B1* JP6619349B1 KR2016126006 MX358084B WO2015128200A1 BRPI1619891A2 IN337869 RU2684913C2 IL247149 HK1229783B *Validated in BE, CH, DE, DK, ES, FR, GB,IE, IT, NL | Granted Granted Granted Granted Granted Granted Pending Granted Expired Pending Granted Granted Granted Granted | 2035 | Cannabigerol Derivatives | CBG derivatives to be used as medicaments in therapy particularly for treating PPARγ-related diseases due to their high PPARγ activatory effect. | |||||
003.1 CBGA Salts
| PCT/EP2019/084764 US17413199 AU2019396637 CA3122714 CN2019800078784 EP19831602.8 JP2021-533246 | Expired Pending Pending Pending Pending Pending Pending | 2038 | Cannabigerol Quinone Acid and Salts | CBG quinone acid and its salts, and new methods of synthesis |
Government Regulation and Product Approval
Government regulation and product approval is required in order for a new drug to enter the market. We are currently in the clinical development stage (Phase 2) for EHP-101 and in the preclinical development stage for EHP-102. Since we plan to conduct our Phase 2 clinical studies in Australia, New Zealand and the US, approvals to conduct clinical studies in those countries are required from the Health Research Ethics Committee (HREC) and TGA in Australia, the Director General of Health and the Health and Disabilities Ethics Committee (HDEC) in New Zealand, and the FDA (IND clearance) and a central IRB (WIRB-Copernicus) in the US. All of these approvals required to conduct clinical studies have been granted.
FDA Approval Process
In the US, pharmaceutical products are subject to extensive regulation by the FDA. The Federal Food, Drug, and Cosmetic Act, or the FDC Act, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Failure to comply with applicable US requirements may subject a company to a variety of administrative or judicial sanctions, such as imposition of clinical holds, FDA refusal to approve pending NDAs, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement, civil penalties, and criminal prosecution.
Pharmaceutical product development in the US typically involves preclinical laboratory and animal tests and the submission to the FDA of an IND, which must become effective before clinical testing may commence. For commercial approval, the sponsor must submit adequate tests by all methods reasonably applicable to show that the drug is safe for use under the conditions prescribed, recommended or suggested in the proposed labeling. The sponsor must also submit substantial evidence, generally consisting of adequate, well-controlled clinical trials to establish that the drug will have the effect it purports or is represented to have under the conditions of use prescribed, recommended or suggested in the proposed labeling. In certain cases, the FDA may determine that a drug is effective based on one clinical study plus confirmatory evidence. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease.
15
Preclinical tests include laboratory evaluation of product chemistry, formulation and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements, including the FDA’s GLP regulations and the US Department of Agriculture’s (USDA’s) regulations implementing the Animal Welfare Act. The results of preclinical testing are submitted to the FDA as part of an IND along with other information, including information about product chemistry, manufacturing and controls, and a proposed clinical trial protocol. Long-term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has not imposed a clinical hold on the IND or otherwise commented or questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted: (i) in compliance with federal regulations, (ii) in compliance with Good Clinical Practice (GCP), an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators and monitors, and (iii) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol involving testing on US patients and subsequent protocol amendments must be submitted to the FDA as part of the IND.
The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The trial protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board, or IRB, for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements or may impose other conditions.
Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In general, in Phase 1, the initial introduction of the drug into healthy human subjects or patients, the drug is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses and, if possible, early evidence on effectiveness. Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug for a particular indication, dosage tolerance and optimum dosage, and to identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug. In most cases, the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy of the drug. The FDA may, however, determine that a drug is effective based on one clinical study plus confirmatory evidence. Only a small percentage of investigational drugs complete all three phases and obtain marketing approval. In some cases, the FDA may require post-market studies, known as Phase 4 studies, to be conducted as a condition of approval in order to gather additional information on the drug’s effect in various populations and any side effects associated with long-term use. Depending on the risks posed by the drugs, other post-market requirements may be imposed.
After completion of the required clinical testing, an NDA is prepared and submitted to the FDA. The FDA approval of the NDA is required before marketing of the product may begin in the US. The NDA must include the results of all preclinical, clinical, and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture, and controls. The cost of preparing and submitting an NDA is substantial.
16
The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. Under the statute and implementing regulations, the FDA has 180 days (the initial review cycle) from the date of filing to issue either an approval letter or a complete response letter, unless the review period is adjusted by mutual agreement between the FDA and the applicant or as a result of the applicant submitting a major amendment. In practice, the performance goals established pursuant to the Prescription Drug User Fee Act have effectively extended the initial review cycle beyond 180 days. The FDA’s current performance goals call for the FDA to complete review of 90 percent of standard (non-priority) NDAs within 10 months of receipt and within six months for priority NDAs.
The FDA may also refer applications for novel drug products, or drug products that present difficult questions of safety or efficacy, to an advisory committee, which is typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with cGMP is satisfactory, and the NDA contains data that provide substantial evidence that the drug is safe and effective in the indication studied.
After the FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing 90 percent of resubmissions within two to six months depending on the type of information included.
An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require a risk evaluation and mitigation strategy (REMS) to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for health care professionals, and elements to assure safe use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained, or problems are identified following initial marketing.
The lengthy process of seeking required approvals and the continuing need for compliance with applicable statutes and regulations require the expenditure of substantial resources. There can be no certainty that all required approvals will be granted. If a product receives regulatory approval, the approval may be limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product.
Any drug that receives FDA approval is subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information on an annual basis or as required more frequently for specific events, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements. Failure to comply with FDA requirements can have negative consequences, including the immediate discontinuation of noncomplying materials, adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties. The FDA also may require post-marketing testing, known as Phase 4 testing, risk minimization action plans and surveillance to monitor the effects of an approved product or place conditions on an approval that could otherwise restrict the distribution or use of the product.
17
Disclosure of Clinical Trial Information
Sponsors of clinical trials of certain FDA-regulated products, including prescription drugs, are required to register and disclose certain clinical trial information on a public website maintained by the US National Institutes of Health. Information related to the product, patient population, phase of investigation, study sites and investigator, and other aspects of the clinical trial is made public as part of the registration. Sponsors are also obligated to disclose the results of these trials after completion. Disclosure of the results of these trials can be delayed for up to two years if the sponsor certifies that it is seeking approval of an unapproved product or that it will file an application for approval of a new indication for an approved product within one year. Competitors may use this publicly available information to gain knowledge regarding the design and progress of our development programs.
Fast Track Designation, Accelerated Approval, and Breakthrough Therapy Designation
The FDA is required to facilitate the development, and expedite the review, of drugs that are intended for the treatment of a serious or life-threatening disease or condition for which there is no effective treatment and which demonstrate the potential to address unmet medical needs for the condition. Under the Fast Track Program, the sponsor of a new drug candidate may request that the FDA designate the drug candidate for a specific indication as a Fast Track drug concurrent with, or after, the filing of the IND for the drug candidate. The FDA must determine if the drug candidate qualifies for Fast Track designation within 60 days of receipt of the sponsor’s request.
Under the FDA’s Accelerated Approval regulations, the FDA may approve a drug for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments.
In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A drug candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, will allow the FDA to withdraw the drug from the market on an expedited basis. Unless otherwise informed by the FDA, for an accelerated approval product an applicant must submit to the FDA for consideration during the preapproval review period copies of all promotional materials, including promotional labeling as well as advertisements, intended for dissemination or publication within 120 days following marketing approval. After 120 days following marketing approval, unless otherwise informed by the FDA, the applicant must submit promotional materials at least 30 days prior to the intended time of initial dissemination of the labeling or initial publication of the advertisement.
In addition to other benefits such as the ability to use surrogate endpoints and engage in more frequent interactions with the FDA, the FDA may initiate review of sections of an Accelerated Approval drug’s NDA before the application is complete. This rolling review is available if the applicant provides, and the FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, the FDA’s time period goal for reviewing an application does not begin until the last section of the NDA is submitted. Additionally, the Accelerated Approval designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
Finally, as part of FDA’s expedited programs to facilitate drug development and product approval for serious or life-threatening conditions, Breakthrough Therapy Designation (BTD) requires preliminary clinical evidence that demonstrates the drug may have substantial improvement on at least one clinically significant endpoint over available therapy. A BTD conveys all of the FTD features plus more intensive FDA guidance on an efficient drug development program, an organizational commitment involving senior FDA managers, and eligibility for rolling review and priority review (the NDA review clock will begin earlier).
18
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan drug designation (ODD) to drugs intended to treat a rare disease or condition - generally a disease or condition that affects fewer than 200,000 individuals in the US (or affects more than 200,000 in the US and for which there is no reasonable expectation that the cost of developing and making available in the US a drug for such disease or condition will be recovered from sales in the US of such drug). ODD must be requested before submitting an NDA. After the FDA grants ODD, the generic identity of the drug and its potential orphan use are disclosed publicly by the FDA. ODD does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first NDA applicant to receive FDA approval for a particular active ingredient to treat a particular disease with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the US for that product, for that indication. During the seven-year exclusivity period, the FDA may not approve any other applications to market the same drug for the same disease, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. If the FDA designates an orphan drug based on a finding of clinical superiority, the FDA must provide a written notification to the sponsor that states the basis for ODD, including “any plausible hypothesis” relied upon by the FDA. The FDA must also publish a summary of its clinical superiority findings upon granting orphan drug exclusivity based on clinical superiority. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Among the other benefits of ODD are tax credits for certain research and a waiver of the NDA application user fee.
Controlled Substances Laws
The federal Controlled Substances Act of 1970, or CSA and its implementing regulations establish a “closed system” of distribution for controlled substances. The CSA imposes registration, security, recordkeeping and reporting, storage, manufacturing, distribution, labeling, importation, exportation, disposal and other requirements under the oversight of the DEA. The DEA is the federal agency responsible for regulating controlled substances, and requires those individuals or entities that manufacture, import, export, distribute, research, or dispense controlled substances to comply with the regulatory requirements to prevent the diversion of controlled substances to illicit channels of commerce.
Facilities that research, manufacture, distribute, import or export any controlled substance must register annually with the DEA. The DEA registration is specific to the particular location, activity(ies) and controlled substances utilized. For example, separate registrations are required for importation and manufacturing activities, and each registration authorizes which schedules of controlled substances the registrant may handle. However, certain coincident activities are permitted without obtaining a separate DEA registration, such as distribution of controlled substances by the manufacturer that produces them.
The DEA inspects all manufacturing facilities to review security, record keeping, reporting and compliance with other DEA regulatory requirements prior to issuing a controlled substance registration. The specific security requirements vary by the type of business activity and the schedule and quantity of controlled substances handled.
For drugs manufactured in the United States, the DEA establishes annually an aggregate quota for the number of substances within Schedules I and II that may be manufactured or produced in the United States based on the DEA’s estimate of the quantity needed to meet legitimate medical, scientific, research and industrial needs. The quotas apply equally to the manufacturing of the API and production of dosage forms. The DEA may adjust aggregate production quotas a few times per year, and individual manufacturing or procurement quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments for individual companies.
The states also maintain separate controlled substance laws and regulations, including licensing, recordkeeping, security, distribution, and dispensing requirements. State authorities, including Boards of Pharmacy, regulate use of controlled substances in each state. Failure to maintain compliance with applicable requirements, particularly as manifested in the loss or diversion of controlled substances, can result in enforcement action. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate proceedings to revoke those registrations. In certain circumstances, violations could lead to criminal prosecution.
We currently manufacture the API and formulations, as well as perform most of the preclinical testing for EHP-101 and EHP-102 in Europe, China and Canada. We have completed a Phase 1 trial for EHP-101 in Australia and are enrolling patients in a Phase 2a study in Australia, New Zealand and the US. We may decide to develop, manufacture or commercialize our product candidates in the United States or additional countries in the future. As a result, we may be subject to controlled substance laws and regulations from regulatory agencies in countries where we develop, manufacture or commercialize our product candidates in the future, to the extent the APIs in any of our product candidates are determined to be controlled substances.
19
While the API in our product candidates are based on CBD and CBG architectures, which may be classified by the DEA as controlled substances in the United States depending on their origin and purity, in March 2019 we received a decision from the DEA that the API (VCE-004.8) in our lead product candidate (EHP-101) is not a controlled substance. We have also received the same decision from the UK Home Office and Canada’s Controlled Substances Directorate. VCE-004.8 not being a controlled substance eliminates costs and complexities associated with developing controlled substances, by facilitating the manufacturing and import of the product to the US and simplifying the conduct of nonclinical studies and clinical studies being planned in MS and SSc patients. Once we advance our second product candidate (EHP-102) further in development, we will request a similar decision from the DEA and other countries for this product candidate.
Employees
As of the date of this Annual Report, we have 17 full-time employees and two part-time employees.
Legal Proceedings
We are not currently a party to any legal proceedings that are material to our business or financial condition.
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of our operations together with our financial statements and related notes appearing at the end of this Annual Report. This discussion contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. Actual results and the timing of events may differ materially from those contained in these forward-looking statements due to a number of factors, including those discussed elsewhere in this Annual Report.
Operating Results
Results of Operations for the Years Ended December 31, 2021 and 2020
Revenues
The Company is a pre-revenue development stage pharmaceutical company focused on the development of product candidates to treat diseases with unmet medical needs. We have no products approved for commercial sale and have not generated any revenues from product sales since our inception in March 2017.
Research and Development Expenses
Research and development (R&D) expenses consist primarily of expenses associated with preclinical development and clinical trials, and payments to third-party contract research organizations, or CROs, contract manufacturing organizations, or CMO’s, contractor laboratories and independent contractors, and research and development personnel related expenses, such as salaries, benefits, travel and other related expenses, including stock-based compensation. To date, our research and development expenses have related primarily to the development of, and clinical trials for, our lead product candidate EHP-101, as well as the preclinical development of EHP-102 and related IND-enabling studies.
Our R&D expenses were approximately $10.5 million for the year ended December 31, 2021, compared to approximately $7.8 million for the year ended December 31, 2020. This increase was primarily related to an increase in clinical expenses and related contract manufacturing and development costs of approximately $1.7 million, driven by the advancement of our Phase 2 clinical trials and manufacturing development activities for our product candidate EHP-101. During the year ended December 31, 2021, R&D expenses related to our Phase 2 clinical trials for EHP-101 were approximately $2.9 million, net of approximately $1.4 million recorded as a receivable related to eligible spending incurred under the Australian research and development tax incentive program. In addition, preclinical and related manufacturing and development costs related to the development of EHP-102 increased by approximately $0.7 million, and R&D related stock-based compensation expense and R&D personnel related expenses increased by approximately $0.3 million, compared to the year ended December 31, 2020.
20
We expect R&D expenses to increase in 2022 as compared to 2021 as we advance our Phase 2 clinical trials with EHP-101 and continue our preclinical development work with EHP-102. These expenditures are subject to numerous uncertainties regarding timing and cost to completion. Completion of our clinical trials and preclinical development may take several years or more and the length of time generally varies according to the type, complexity, novelty and intended use of a product candidate.
General and Administrative Expenses
General and administrative (G&A) expenses consist primarily of legal and patent fees, professional service fees, facility and office expenses, and general and administrative personnel related expenses, such as salaries, benefits, travel and other related expenses, including stock-based compensation.
Our G&A expenses were approximately $6.4 million for the year ended December 31, 2021, compared to approximately $4.2 million for the year ended December 31, 2020. This increase was primarily related to an increase in G&A related stock-based compensation expense and G&A personnel related expenses of approximately $1.1 million, an increase in consulting and professional services fees of $0.7 million, and an increase in general corporate expenses of approximately $0.4 million compared to the year ended December 31, 2020.
Other (Income)/Expense
Other (income)/expense consists of interest expense, interest income, sublease income, and foreign currency losses. During the year ended December 31, 2021, we recognized a gain on extinguishment of debt of approximately $0.3 million, related to the forgiveness of our PPP Loan (as defined below) and associated accrued interest payable, which is included in other income.
From inception through March 2019, we received advances from Emerald Health Sciences Inc. (EHS), our significant (former majority) stockholder, to fund our operations under a revolving loan agreement (Related Party Loan). During the year ended December 31, 2021, we recognized approximately $0.1 million in interest expense on the Related Party Loan, compared to approximately $0.4 million in interest expense during the year ended December 31, 2020. In 2021, the Related Party Loan was settled in full and the revolving loan agreement was terminated effective June 1, 2021 pursuant to a Loan Termination Agreement between us and EHS.
During the year ended December 31, 2021, we recognized a foreign currency gain of $65,163, compared to a loss of $41,272 recognized during the year ended December 31, 2020. Foreign currency gains and losses are due primarily to the timing of fluctuations in the exchange rates between the U.S. Dollar and other foreign currencies, related to contracts and other transactions which are denominated in currencies other than the U.S. Dollar.
During the year ended December 31, 2021, we recognized approximately $0.1 million in sublease income related to subleases of our existing non-cancelable building lease.
Net Loss
Our net loss was approximately $16.5 million for the year ended December 31, 2021, compared to approximately $12.4 million for the year ended December 31, 2020.
Liquidity and Capital Resources
Since our inception in 2017, we have devoted most of our cash resources to research and development and general and administrative activities. We have financed our operations to date primarily with the use of the proceeds from the Related Party Loan and with capital raised from a Tier 2 offering (the Regulation A Offering) pursuant to Regulation A (Regulation A+) under the Securities Act of 1933, as amended (the Securities Act). The Regulation A Offering was initially qualified by the SEC in March 2018 and sales commenced in 2019. The Company closed and terminated the Regulation A Offering effective March 28, 2021. In total, from the commencement of the sale of shares pursuant to the Regulation A Offering in March 2019 through the closing and termination on March 28, 2021, we sold an aggregate of 10,422,776 shares of common stock for gross proceeds of approximately $60 million. In June 2021, the Company sold 37,500 shares of common stock at $8.00 per share for gross proceeds of $300,000, in a private placement offering under Regulation D under the Securities Act.
21
To date, we have not generated any revenue from the sale of products, and we do not anticipate generating any revenue from the sale of products for the foreseeable future. We have incurred losses and generated negative cash flows from operations since inception. During the period from March 2, 2017 (inception) through December 31, 2021, we have incurred cumulative net losses of approximately $56.6 million. Our future expenditures and capital requirements will depend on numerous factors, including, among others, the progress of our research and development efforts.
We believe that we have sufficient capital to finance our operations at least into the second quarter of 2023, however, if our operating and development costs are higher than expected, we will need to obtain additional financing prior to that time. Further, we expect that after such period, we will be required to raise additional funds to fund our operations and to further advance clinical development of and to commercially develop our product candidates. There is no assurance that such financing will be available when needed, or that ultimately, we will achieve profitable operations and positive cash flow.
Credit Facilities
In September 2017, we entered into the Related Party Loan with EHS, which was amended in January 2018 and November 2019. Borrowings under the Related Party Loan were drawn down from time to time, and repaid by us in cash, or at the option of EHS, converted into shares of our common stock at a conversion price of $2.00 per share. In November 2019, the Related Party Loan was amended to reduce the interest rate from 12% to 10%, compounded semiannually. The Related Party Loan was payable upon demand and had no expiration date.
During the year ended December 31, 2021, the outstanding balance under the Related Party Loan was settled in full, and the Related Party Loan was terminated effective June 1, 2021 pursuant to a Loan Termination Agreement between us and EHS. In total, EHS advanced approximately $11.3 million to us under the Related Party Loan. Approximately $3.1 million of the total amount advanced by EHS was repaid by us in cash, approximately $5.2 million was offset through cashless discharges (described below) and $3.0 million was converted into 1.5 million shares of our common stock at a conversion price of $2.00 per share. A total of approximately $2.2 million of interest expense was incurred (excluding a non-cash charge of approximately $1.4 million recorded during the year ended December 31, 2019 related to accretion of a beneficial conversion feature) under the Related Party Loan, of which approximately $2.0 million was paid to EHS in cash and approximately $0.2 million was offset through a cashless discharge. We do not have any remaining obligations to EHS under the Related Party Loan as of December 31, 2021.
In May 2019, our Board of Directors authorized a funding arrangement with EHS (Related Party Note Receivable), which was amended in August 2019 and September 2019 to extend the repayment dates, pursuant to which we advanced funds to EHS in the form of interest bearing (12%) short term notes under a Promissory Note between EHS and EHP (the Promissory Note). Advances under the Promissory Note were originally due for repayment with accrued and unpaid interest three months from the date of the advance. A total of $5,000,000 was advanced and $178,933 accrued as interest receivable under the Related Party Note Receivable. During 2019, the Related Party Note Receivable was fully settled through cashless discharges against the unpaid principal and accrued interest payable balances, respectively, under the existing Related Party Loan with EHS.
On January 23, 2020, our Australian subsidiary, EHP Australia, entered into a loan agreement with Rocking Horse Nominees Pty Ltd (Rocking Horse), whereby Rocking Horse advanced $AU1.2 million (approximately $0.8 million) to EHP Australia. The loan was secured by the tax incentive refund anticipated to be received during 2020 for eligible spending incurred under the Australian research and development tax incentive program during 2019. The loan had an upfront establishment fee of 1.2% and bore interest at 1.25% per month compounded daily. The entire principal balance of the loan and approximately $53,000 of accrued interest were repaid to Rocking Horse during the year ended December 31, 2020.
On April 22, 2020, we received loan proceeds of $292,152 (PPP Loan) from Silicon Valley Bank pursuant to the Paycheck Protection Program (PPP) established as part of the Coronavirus Aid, Relief and Economic Security Act (CARES Act). The PPP Loan, which is evidenced by a Note dated April 21, 2020, bore interest at a rate of 1% per annum, and was expected to mature on April 21, 2022. On February 18, 2021, the entire principal balance and accrued interest under the PPP Loan were forgiven by the lender.
22
Going Concern
Our financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. Our ability to continue as a going concern is contingent upon our ability to raise additional capital as required. During the period from March 2, 2017 (inception) through December 31, 2021 we have incurred an accumulated deficit of approximately $56.6 million. Currently, we intend to finance our operations through equity and debt financings and other strategic transactions.
We continually evaluate our plan of operations to determine the manner in which we can most effectively utilize our limited cash resources. The timing of completion of any aspect of our plan of operations is highly dependent upon the availability of cash to implement that aspect of the plan and other factors beyond our control. There is no assurance that we will successfully obtain the required capital or revenues, or, if obtained, that the amounts will be sufficient to fund our ongoing operations.
These circumstances raise substantial doubt on our ability to continue as a going concern. Our financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or amounts and classification of liabilities that might result from this uncertainty.
Capital Expenditures
We do not have any contractual obligations for ongoing capital expenditures at this time.
Contractual Obligations, Commitments and Contingencies
We are required to make future payments to VivaCell Biotechnology España S.L. (VivaCell), a wholly owned subsidiary of Emerald Health Research Inc. (EHR) (which is a wholly owned subsidiary of EHS, our significant stockholder), based on the achievement of milestones set forth in the Intellectual Property Transfer Agreement (IPTA) between the Company and VivaCell. These payments are based on the achievement of development or regulatory milestones, including commencement of various phases of clinical trials, filing of product license applications and approval of product licenses from the United States Food and Drug Administration (FDA) or a foreign regulatory agency. The aggregate amount of future milestone payments that we could be required to pay under our agreement with VivaCell is up to 2.7 million Euros per product and application (approximately $3.1 million, per product and application, based upon the exchange rate at December 31, 2021). These amounts assume that all remaining milestones associated with the milestone payments are met. In the event that product license approval for any of the related products is obtained, we are required to make royalty payments of 2.5% of net revenues from commercial sales of the related products.
During the year ended December 31, 2020, milestone payments of approximately $460,000 were paid to VivaCell in accordance with the IPTA due to the completion of our first Phase 1 clinical study in 2019. Because future milestones are contingent, we are not in a position to reasonably estimate how much, if any, additional milestone payments will ultimately be paid, or when. Many of the remaining milestone events are related to progress in clinical trials which will take several years to achieve.
On May 1, 2018, we entered into a two-year non-cancelable building lease for our corporate headquarters in San Diego, California, which was amended in August 2019, to include additional space at the existing premises and to extend the term of the original lease through August 31, 2022. Under the lease, the Company currently pays a base rent of $21,852 per month. Our remaining obligation under this operating lease from January 1, 2022 through August 31, 2022 is $174,816.
In July 2020, we entered into an agreement to sublease a portion of our existing non-cancelable building lease to a tenant, effective August 1, 2020, and continuing through August 31, 2022. The Company paid commissions of $7,000 related to the execution of the sublease and was expected to incur future losses of approximately $55,000 in conjunction with the sublease. EHS agreed to reimburse the Company for the commission fee and future expected losses, which were offset against the Related Party Loan as of December 31, 2020.
23
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements.
Trend Information
Because we are currently in the research, product development and clinical trial stage, no products are in commercial production or use and we do not currently generate revenue. Accordingly, we are unable to identify any significant recent trends in production, sales and inventory, costs, and selling prices, and, except as set forth below, we are unable to identify any known trends, uncertainties, demands, commitments or events that are reasonably likely to have a material effect on our future net sales or revenues, income from continuing operations, profitability, liquidity or capital resources, or that would cause reported financial information not necessarily to be indicative of future operating results or financial condition.
Our financial success will be dependent upon our ability to continue development of our product candidates through preclinical and clinical stages to commercialization, which ability is, in turn, dependent on our liquidity and capital resources that will be available to fund such development and continuous research. Our liquidity and capital resources may vary substantially from period to period depending on a number of factors, including without limitation, the number of research and development programs being undertaken at any one time, the stage of the development programs, the timing of significant expenditures for clinical trials, manufacturing, toxicology and pharmacology studies and the availability of funding from investors, affiliates, and lenders.
Further, unpredictable events, such as the COVID-19 outbreak, and associated business disruptions including delayed clinical trials and laboratory resources could harm our financial condition, affect our operations, increase our costs and expenses, and impact our ability to raise capital. Our operations could be subject to unpredictable events, such as earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics or pandemics such as the COVID-19 outbreak, and other natural or manmade disasters or business interruptions, for which we may not be insured. We do not carry insurance for all categories of risk that our business may encounter. The occurrence of any of these business disruptions could seriously harm our operations and financial condition, delay our product development and regulatory approvals of clinical trials, and increase our costs and expenses. Additionally, COVID-19 has caused significant disruptions to the global financial markets, which could impact our ability to raise additional capital. The ultimate impact on us and any delays in our research and development is unknown, but our operations and financial condition could suffer in the event of any of these types of unpredictable events. Further, any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our business, results of operations, financial condition and cash flows.
24
Item 3. Directors, Officers and Significant Employees
The table below sets forth our directors, executive officers and significant employees of as of the date of this Annual Report.
| Name | Position | Age | Term of Office | Approximate hours per week for part-time employees | ||||||
| Officers and Significant Employees: | ||||||||||
| James DeMesa | President and Chief Executive Officer | 64 | (1) | |||||||
| Alain Rolland | Chief Operating Officer and Executive Vice President | 62 | (2) | |||||||
| Joachim Schupp | Chief Medical Officer | 69 | (3) | |||||||
| Lisa Sanford | Chief Financial Officer | 55 | (4) | |||||||
| Eduardo Muñoz | Chief Scientific Officer | 63 | Since June 2017 | 25 hours per week (5) | ||||||
| Nancy Coulson | Senior Vice President Regulatory and Quality Affairs | 77 | (6) | 20 hours per week | ||||||
| Directors: | ||||||||||
| James L. Heppell | Director & Chairman | 66 | (7) | |||||||
| James DeMesa | Director | 64 | Since December 2019 | |||||||
| Gaetano A. Morello | Director | 60 | Since March 2017 | |||||||
| Punit S. Dhillon | Director | 41 | Since March 2017 | |||||||
| (1) | James DeMesa has served as Chief Executive Officer since March 2017. He was appointed President in November 2019, subsequent to the resignation of Avtar Dhillon as President in August 2019. |
| (2) | Alain Rolland was appointed Chief Operating Officer and Executive Vice President in September 2019. He served as Chief Development Officer as of May 1, 2018 and from February 1, 2018 through April 20, 2018, he served as Vice President of Product Development. |
| (3) | Joachim Schupp was appointed Chief Medical Officer as of January 1, 2019. From August 1, 2018 through December 31, 2018, he served as Senior Vice President of Medical Affairs. |
| (4) | Lisa Sanford has served as Chief Financial Officer since October 1, 2018. From July 20, 2018 through September 30, 2018, she served as Vice President of Finance. |
| (5) | Dr. Muñoz is a consultant who may be deemed a significant employee and acts as our Chief Scientific Officer pursuant to a consulting agreement with the University of Córdoba where Dr. Muñoz is employed in the Department of Cellular Biology, Physiology and Immunology. |
| (6) | Nancy Coulson was engaged as a senior regulatory advisor, averaging approximately 80 hours per month from March 2017 through January 2020. Effective February 1, 2020, Ms. Coulson was hired as a part-time employee serving as Senior Vice President of Regulatory and Quality Affairs. |
| (7) | James L. Heppell has served as a Director since 2017 and was appointed Chairman as of November 15, 2019. |
There is no arrangement or understanding between the persons described above and any other person pursuant to which the person was selected to his or her office or position.
James M. DeMesa, MD, MBA, President and Chief Executive Officer. Dr. DeMesa has over 30 years of experience in biotech and pharmaceutical leadership, product development, and clinical and regulatory management. He has completed partnerships and collaborations with pharmaceutical, biotech, and medical device companies and has raised more than $250 million to advance product development to clinical stage, regulatory approval, and commercialization. He is a former practicing physician and CEO of two public biotech companies: Migenix, from 2001 to 2008 and GenSci Regeneration Sciences, from 1996 to 2001 (now part of Integra LifeSciences). Dr. DeMesa also currently serves as Director for two biotech companies: OncoSec Medical Incorporated and Induce Biologics. Prior to his CEO roles, Dr. DeMesa was Vice President, Medical and Regulatory Affairs at Biodynamics International (now part of RTI Surgical) and Bentley Pharmaceuticals (now part of Teva Pharmaceuticals). Dr. DeMesa received a bachelor’s degree in Chemistry, and MD, and MBA degrees from the University of South Florida and did his medical residency at the University of North Carolina.
25
Alain Rolland, PharmD, PhD, Chief Operating Officer and Executive Vice President. Dr. Rolland has over 30 years of international leadership experience in pharmaceutical and biotech companies. He has focused on the discovery and development of biologics and small molecules in a variety of therapeutic areas including immuno-oncology, cardiovascular and hematological disorders, dermatology, and infectious disease vaccines. Prior to joining EHP, Dr. Rolland was a co-founder and served as CEO, President and Director of CHIME BioTherapeutics, Inc. He was previously Executive Vice President and Chief Scientific Officer at HUYA Bioscience International, Executive Vice President, Product Development at Vical, and Senior Vice President, Preclinical R&D, Head of the Woodlands Center of Valentis. Dr. Rolland has published over 90 scientific articles and book chapters and is editor of three scientific books. He is a member of several scientific societies, the founding Editor-in-Chief of Current Pharmaceutical Biotechnology, and an editorial board member of several journals. He has been honored by the American Association of Pharmaceutical Scientists as an “AAPS Fellow” for professional excellence and outstanding contributions to the pharmaceutical sciences. Dr. Rolland earned his doctorate degree in Pharmacy (Pharm.D.) and in Pharmaceutical Sciences (Ph.D.) from Rennes University, France.
Joachim P.H. Schupp, MD, Dr. med, Chief Medical Officer. Dr. Schupp has over 30 years of international pharmaceutical industry experience in all phases of drug development and several therapeutic areas. He directed multiple Phase I – IV clinical studies and led multiple international cross-functional project teams at Ciba-Geigy and Novartis Pharmaceuticals in Switzerland, which resulted in worldwide approval of several New Drug Applications (NDAs), Biologics License Applications (BLAs) and supplemental applications for small molecule drugs, biologics and devices currently on the market. Dr. Schupp also served as Vice President, Clinical & Regulatory Affairs at HUYA Bioscience International, Chief Medical Officer at Imprimis/Transdel Pharmaceuticals, Inc., Vice President, Clinical Development at Apricus Biosciences, Inc., Vice President, Medical Affairs at Adventrx Pharmaceuticals, Inc. and Vice President, Clinical Data Services at ProSanos, Inc. Prior to joining EHP, Dr. Schupp managed his own consulting business (MEQVal), providing services as a medical monitor and drug safety physician. Dr. Schupp received his MD and doctorate (Dr. med.) from Freie Universität Berlin in Germany and practiced medicine in Germany, South Africa, UK and Switzerland.
Lisa Sanford, Chief Financial Officer. Ms. Sanford has over 30 years of diversified experience in finance and accounting in the life sciences, biotechnology, and pharmaceutical industries. From April 2000 through July 2018, Ms. Sanford managed her own consulting business, providing finance and accounting services for both public and private companies. Ms. Sanford also served as an audit senior manager at Ernst & Young LLP, where she worked for 12 years and was involved in multiple IPOs and business combinations. She received her bachelor’s degree in Accounting from Lehigh University and is a Certified Public Accountant.
Eduardo Muñoz, PhD, MD, Chief Scientific Officer. Dr. Muñoz has been a Professor of Immunology in the Department of Cell Biology, Physiology and Immunology of the University of Córdoba (Spain) since 1992 and Director of the Inflammation and Cancer Research Group at the Institute Maimonides for Biomedical Research of Córdoba since 2012. Dr. Muñoz has more than 35 years of experience in biomedical research and is the author of more than 250 articles, patents, and book chapters with more than 9,000 citations. He is experienced in the mechanism of actions of cannabinoids and endocannabinoids as well as the development of cannabinoid-based new chemical entities. Dr. Muñoz belongs to the editorial board of several scientific journals and is a co-founder of three biotech companies, Emerald Health Biotechnology España, S.L.U. (Spain), Glactone Pharma AB (Sweden) and InnnoHealth Group (now part of Evonik Industries AG). He received a PhD in Medicine and Surgery at the University of Córdoba and was an associate researcher at Tufts University in Boston, and at the Institute Pasteur in Paris.
Nancy Coulson, Senior Vice President Regulatory and Quality Affairs. Ms. Coulson has over 30 years of experience in providing strategic counsel for regulatory, clinical, and quality affairs. As a senior advisor for medical device and pharmaceutical companies, she manages United States and international regulatory documents, briefing packages, and global regulatory dossiers across multiple product categories. Ms. Coulson has also completed several successful pre-approval inspections for new drug and device manufacturing facilities. Most recently, she was Worldwide Director, Regulatory Affairs at Cordis, a Johnson & Johnson company, where she provided strategic direction on global regulatory submissions. She also held scientific and/or senior regulatory positions at Bristol-Myers Squibb, Bausch & Lomb, GenSci, and Migenix. She received a Bachelor of Science in Chemistry from LeMoyne College and an MBA from Chapman University.
James L. Heppell, BSc, LLP, Chairman. Mr. Heppell was founder and CEO of the Advantage Life Sciences I Fund, which won the Canadian Venture Capital Deal of the Year Award in 2006 for having the highest realized return of all venture capital funds in Canada. Earlier in his career, he practiced corporate securities law with Fasken Martineau DuMoulin and later served as President and CEO of Catalyst Corporate Finance Lawyers, a boutique corporate finance law firm representing life science and tech companies. He is a past member of the Securities Policy Advisory Committee to the British Columbia Securities Commission and is a Past-Chairman of the Securities Section of the Canadian Bar Association. Over the years, Mr. Heppell has written a number of articles, co-edited the Annotated British Columbia Securities Act and coordinated and taught numerous courses on corporate finance and corporate governance issues. He earned a BSc in microbiology and a law degree from the University of British Columbia.
26
Gaetano A. Morello, ND, Director. Dr. Morello is an accomplished clinician with direct, first-hand experience in the clinical and medical application of cannabinoids. Dr. Morello has practiced at the Complex Chronic Disease Program (CCDP) at Woman’s Hospital in Vancouver, Canada since 2013 and is also a study investigator at the CCDP Clinical Cannabis Trial. He has also served on the Quality Assurance Committee for the College of Naturopathic Physicians of British Columbia since 2010 as well as other health and medical panels. He has authored Cleanse, The Healing Power, of the Endocannabinoid System, Ultimate Inside Out Approach, Whole Body Cleansing, Stress and Anxiety, A Powerful Antioxidant, and was a contributing author to A Textbook of Natural Medicine, and numerous journal publications. He has made more than 500 medical presentations in the United States, Canada, Australia, Germany, and Italy in the last decade. Dr. Morello has a BSc in Cell Biology/ Nutrition from the University of British Columbia and a Doctorate in Naturopathic Medicine from Bastyr University.
Punit S. Dhillon, Director. Mr. Dhillon was appointed CEO and Chair of Skye Bioscience, Inc. (OTCQB: SKYE) in August 2020. He is the co-founder and former President & CEO of OncoSec Medical Incorporated (NASDAQ: ONCS), a leading biopharmaceutical company developing cancer immunotherapies for the treatment of solid tumors, where he served as an Executive until March 2018 and a Director until February 2020. Mr. Dhillon serves as a Director and Audit Committee Chair for Emerald Health Therapeutics, and also serves as Director for Arch Therapeutics Inc. (OTCQB: ARTH). Prior to OncoSec, from 2003-2011, he served as Vice President of Finance and Operations at Inovio Pharmaceuticals, Inc. (NASDAQ: INO). Collectively, he has led and assisted in raising over $500 million through financings, M&A deals and several licensing transactions with large pharma. His management experience spans corporate finance, M&A integration, in-licensing and out-licensing of key intellectual property, strategy implementation, corporate transactions and collaborations with leading universities and global disease specific opinion leaders. Mr. Dhillon also co-founded and is the Director of the YELL Canada, a registered Canadian charity that partners with schools to support entrepreneurial learning. Mr. Dhillon holds a BA (Honours) in Political Science and a minor in Business Administration from Simon Fraser University
Involvement in Certain Legal Proceedings
To our knowledge, except as described below, none of our current directors or executive officers has, during the past ten years:
| ● | been convicted in a criminal proceeding or been subject to a pending criminal proceeding (excluding traffic violations and other minor offenses); | |
| ● | had any bankruptcy petition filed by or against the business or property of the person, or of any partnership, corporation or business association of which he or she was a general partner or executive officer, either at the time of the bankruptcy filing or within two years prior to that time; | |
| ● | been subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction or federal or state authority, permanently or temporarily enjoining, barring, suspending or otherwise limiting, his involvement in any type of business, securities, futures, commodities, investment, banking, savings and loan, or insurance activities, or to be associated with persons engaged in any such activity; |
| ● | been found by a court of competent jurisdiction in a civil action or by the SEC or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated; | |
| ● | been the subject of, or a party to, any federal or state judicial or administrative order, judgment, decree, or finding, not subsequently reversed, suspended or vacated (not including any settlement of a civil proceeding among private litigants), relating to an alleged violation of any federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies including, but not limited to, a temporary or permanent injunction, order of disgorgement or restitution, civil money penalty or temporary or permanent cease-and-desist order, or removal or prohibition order, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or | |
| ● | been the subject of, or a party to, any sanction or order, not subsequently reversed, suspended or vacated, of any self-regulatory organization (as defined in Section 3(a)(26) of the Securities Exchange Act of 1934, as amended [the Exchange Act]), any registered entity (as defined in Section 1(a)(29) of the Commodity Exchange Act), or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
27
Gaetano Morello, one of our non-employee directors, was appointed as Chief Executive Officer of Emerald Health Bioceuticals Inc. (EHB) on February 7, 2020. EHB was a natural health consumer products company that was majority-owned by EHS, our significant stockholder. On October 20, 2020, EHB filed a Chapter 7 bankruptcy petition in the U.S. Bankruptcy Court for the Southern District of California. Mr. Dhillon and Mr. Heppell were directors of EHB when the Chapter 7 bankruptcy petition was filed.
Except as set forth above and in our discussion below in “Interest Of Management And Others In Certain Transactions,” none of our directors or executive officers has been involved in any transactions with us or any of our directors, executive officers, affiliates or associates which are required to be disclosed pursuant to the rules and regulations of the SEC.
Compensation of Executive Officers
The following table represents information regarding the total compensation for the three highest paid executive officers or directors of the Company for the year ended December 31, 2021:
| Capacity in which compensation | Cash Compensation | Other Compensation | Total Compensation | |||||||||||
| Name | was received | ($) | ($)(4) | ($) | ||||||||||
| James M. DeMesa (1) | President & Chief Executive Officer | $ | 470,000 | $ | 226,775 | $ | 696,775 | |||||||
| Alain Rolland (2) | Chief Operating Officer and Executive Vice President | $ | 390,000 | $ | 150,540 | $ | 540,540 | |||||||
| Joachim Schupp (3) | Chief Medical Officer | $ | 350,000 | $ | 135,100 | $ | 485,100 | |||||||
| All (non-executive) directors as a group (3 persons) | Director | $ | 280,000 | $ | - | $ | 280,000 | |||||||
| (1) | Dr. DeMesa’s annual salary is $470,000. He is eligible to earn a bonus of up to 50% of his annual salary, subject to Board approval and achievement of certain milestones and metrics. |
| (2) | Dr. Rolland’s salary is $390,000. He is eligible to earn a bonus of up to 40% of his annual salary, subject to Board approval and achievement of certain milestones and metrics. |
| (3) | Dr. Schupp’s salary is $350,000. He is eligible to earn a bonus of up to 40% of his annual salary, subject to Board approval and achievement of certain milestones and metrics. |
| (4) | Other compensation consists of bonuses paid during 2021. |
Director Compensation
In April 2019, the Board of Directors approved a compensation plan, which was amended effective October 2020 and January 2022, and provides for our non-employee directors to receive compensation for their services. Board compensation consists of annual cash retainers for various responsibilities and annual option grants.
In January 2018, two of our directors were granted a total of 100,000 options to purchase our Common Stock at $5.00 per share. In December 2018, our four directors at the time were granted options to purchase a total of 800,000 shares of our Common Stock at $2.50 per share. In August 2020, our three non-employee directors were granted options to purchase a total of 75,000 shares of our Common Stock at $6.00 per share.
28
Employment Agreements
We have employment agreements with our four executive officers, James DeMesa, Alain Rolland, Joachim Schupp and Lisa Sanford. The initial term of each employment agreement is for a period of three years to be extended automatically for successive one-year periods unless terminated earlier by either party upon written notice at least ninety days prior to the end of that period. The Company may terminate the executive officer’s employment, for cause, as defined in the agreement, at any time, without any advance notice. Further, subject to the terms of the agreement, the executive officer may terminate employment with us, at any time for any reason or no reason at all, upon six weeks’ advance written notice. Subject to the notice provisions described in the agreement, the executive officer may terminate employment with us for good cause as defined in the agreement. Subject to the agreement provisions, in situations where the Company terminates the executive officer’s employment without cause, or the term of the agreement ends without the Company offering to extend the agreement on the same terms, or the executive officer resigns for good cause, then the executive officer will be, under certain conditions, entitled to severance compensation from the Company equal to six months of executive officer’s then current base salary.
Item 4. Security Ownership of Management and Certain Securityholders
Principal Shareholders
The following table shows the beneficial ownership of our Common Stock as of April 29, 2022 held by (i) each person known to us to be the beneficial owner of more than 10% of any class of our voting securities; (ii) each director who is the beneficial owner of more than 10% of any class of our voting securities; (iii) each executive officer who is the beneficial owner of more than 10% of any class of our voting securities; and (iv) all directors and executive officers as a group. As of April 29, 2022, there were 22,073,976 shares of our Common Stock outstanding.
Beneficial ownership is determined in accordance with the rules of the SEC, and generally includes voting power and/or investment power with respect to the securities held. Shares of Common Stock subject to the conversion of a security, or subject to options and warrants currently exercisable or which may become exercisable within 60 days of the date of this Annual Report, are deemed outstanding and beneficially owned by the person holding such convertible securities, options or warrants for purposes of computing the number of shares and percentage beneficially owned by such person, but are not deemed outstanding for purposes of computing the percentage beneficially owned by any other person. The persons or entities named have sole voting and investment power with respect to all shares of Common Stock shown as beneficially owned by them.
The percentages below are based on shares of our Common Stock beneficially owned as of April 29, 2022. Unless otherwise indicated, the business address of each person listed is c/o Emerald Health Pharmaceuticals Inc., 5910 Pacific Center Blvd, Ste 320, San Diego, CA 92121.
| Title of Class | Name and Address of Beneficial Owner: | Amount and Nature of Beneficial Ownership | Amount and Nature of Beneficial Ownership Acquirable by Exercise of Option or Conversion of Security | Percent of Class | ||||||||
| Common Stock | All directors and named executive officers as a group (9 persons) | 382,000 shares owned | 1,768,750 shares acquirable | 9.0 | % | |||||||
| Greater than 10% Beneficial Owners: | ||||||||||||
| Common Stock | Emerald Health Sciences Inc. 8262, 408-55 Water Street, Vancouver, BC, V6B OM9 | 10,500,000 shares owned | no shares acquirable | 47.6 | % | |||||||
29
Item 5. Interest of Management and Others in Certain Transactions
Transactions with Related Persons
Except as described below and except for employment arrangements which are described under “Directors, Officers and Significant Employees” above, since March 2, 2017 (inception), there has not been, nor is there currently proposed, any transaction in which we are or were a participant, the amount involved exceeds the lesser of $120,000 or 1% of the average of the Company’s total assets at year-end for the fiscal years ended December 31, 2020 and December 31, 2021, and any of our directors, executive officers, holders of more than 10% of our common stock or any immediate family member of any of the foregoing had or will have a direct or indirect material interest.
In June 2017, we entered into the IPTA and a Research Agreement with VivaCell. VivaCell is a wholly owned subsidiary of Emerald Health Research Inc. (EHR), which is a wholly owned subsidiary of EHS, our significant stockholder. EHP has no ownership or voting rights related to EHR or VivaCell.
In September 2017, we entered into a revolving loan agreement (the Related Party Loan) with EHS, our significant (former majority) stockholder, which was amended in January 2018 and November 2019. Borrowings under the Related Party Loan were drawn down from time to time, and repaid by us in cash, or at the option of EHS, converted into shares of the Company at $2.00 per share. In November 2019, the Related Party Loan was amended to reduce the interest rate from 12% to 10%, compounded semiannually. The Related Party Loan was payable upon demand and had no expiration date.
In April 2019, we received a written notice of demand (Notice) from EHS for payment of all accrued interest on the Related Party Loan as of March 31, 2019, which resulted in a cash payment of $1,044,901 to EHS. Also in April 2019, we received a second Notice from EHS that called for the following, upon qualification by the SEC of our Form 1-A Post-Qualification Offering Circular Amendment on Form 1-A filed in May 2019 (the Post-Qualification Offering Circular Amendment): (1) repayment of $2,000,000 of the unpaid principal balance under the loan, and (2) the conversion of an additional $2,500,000 of the unpaid principal balance under the loan at a conversion price of $2.00 per share. Our Post-Qualification Offering Circular Amendment was qualified by the SEC on June 7, 2019 and the repayment of the $2,000,000 of unpaid principal was transacted as a cashless discharge and offset between the Related Party Loan and the Related Party Note Receivable (as defined below). Concurrently, 1,250,000 shares of EHP common stock were issued to EHS at a conversion price of $2.00 per share, further reducing the principal balance of the Related Party Loan by $2,500,000.
In May 2019, our Board of Directors authorized a funding arrangement with EHS (the Related Party Note Receivable), which was amended in August 2019 and September 2019 to extend the repayment dates, pursuant to which we advanced funds to EHS in the form of interest bearing (12%) short term notes under a Promissory Note between EHS and EHP (the Promissory Note). Advances under the Promissory Note were originally due for repayment with accrued and unpaid interest three months from the date of the advance. A total of $5,000,000 was advanced and $178,933 accrued as interest receivable under the Related Party Note Receivable, all of which was offset through cashless discharges against the unpaid principal and accrued interest payable balances, respectively, under the Related Party Loan with EHS. In November 2019, we received a written Notice from EHS for payment of $3,000,000 of the unpaid principal balance and $178,933 of accrued interest on the Related Party Loan, which was transacted as a cashless discharge and offset between the Related Party Loan and the remaining unpaid principal and accrued interest balances under the Related Party Note Receivable as of November 15, 2019.
During the year ended December 31, 2020, we received written Notices from EHS for payments of $750,000 of the unpaid principal balance and a total of $822,524 of accrued interest on the Related Party Loan. Additionally, in December 2020, $180,930 of the unpaid principal balance was offset against amounts due from EHS to EHP for reimbursements of expenses related to office expenses, including space allocated to other EHS affiliates.
30
In February 2021, we received a written Notice from EHS, for payment of $500,000 of the unpaid principal balance on the Related Party Loan and in March 2021, we received a written Notice from EHS, for payment of $1,819,771 of the unpaid principal balance and $161,415 of accrued interest on the Related Party Loan. We received a final written Notice from EHS in April 2021, for conversion of the remaining unpaid principal balance under the Related Party Loan in the amount of $500,000 at a conversion price of $2.00 per share into 250,000 shares of EHP Common Stock. Following the issuance of the shares of Common Stock to EHS, all outstanding balances under the Related Party Loan were fully settled. The Related Party Loan was terminated effective June 1, 2021 pursuant to a Loan Termination Agreement between EHP and EHS, and there are no further obligations to EHS under the Related Party Loan as of the date of this Annual Report. There have been no additional funding arrangements with EHS.
In November 2019, we entered into a Board Observer Agreement with EHS, whereby we granted to EHS the right to designate an observer on the Board of Directors for so long as EHS maintains ownership of any securities of the Company.
In December 2019, our Board of Directors approved an Independent Contractor Services Agreement between us and Dr. Avtar Dhillon, co-founder and former Executive Chairperson and President of the Company, to provide corporate advisory services to the Company in exchange for a monthly fee of $10,000. The Independent Contractor Services Agreement had an initial term of one year and renewed automatically thereafter unless terminated earlier by either party. On September 23, 2021, we were provided 30-day written notice from Dr. Dhillon, terminating the Agreement without cause, effective October 23, 2021. During the years ended December 31, 2021 and 2020, the Company paid $97,419 and $130,000, respectively, under the Independent Contractor Services Agreement. There are no remaining obligations, nor amounts due and payable, under the Independent Contractor Services Agreement as of the date of this Annual Report.
In August 2020, our Board of Directors granted Dr. Dhillon the option to purchase 25,000 shares of common stock at $6.00 per share, with vesting occurring 25% on the date of the grant and an additional 25% vesting annually thereafter. Concurrent with the termination of Dr. Dhillon’s Independent Contractor Services Agreement, the remaining 12,500 unvested shares were forfeited.
In September 2021, our Board of Directors approved a modification to the exercise terms of the non-qualified stock option awards previously issued to our founders (including our four current directors and Dr. Dhillon) This modification removed the post-termination exercise period from the award agreements, granting the award holder the ability to exercise the vested portion of the options until the original expiration dates of the awards (ten years following the grant date), if continuous service is terminated for any reason other than for cause, including by death or disability. This modification resulted in the recognition of an additional $1.0 million within stock-based compensation expense during the year ended December 31, 2021, related to the vested portion of each option, with an additional $0.2 million of stock-based compensation expense to be recognized over the remaining vesting periods of the impacted options.
In April 2021, we entered into an agreement with SKYE Bioscience, Inc. (SKYE), to sublease a portion of our existing non-cancelable building lease to SKYE, beginning May 16, 2021 and continuing on a month-to-month basis, for monthly base rent of $4,000 per month, in addition to a portion of common area expenses and utilities. EHS is a significant stockholder of both EHP and SKYE. In August 2021, SKYE provided notice of termination of the month-to-month lease agreement, effective August 31, 2021. During the year ended December 31, 2021, we received a total of $15,453 from SKYE for rent and utilities allocated under the sublease, and there are no remaining obligations, nor amounts receivable, under the sublease as of the date of this Annual Report.
Two of our non-employee directors on our Board of Directors are also on the Board of Directors of our significant (former majority) stockholder, EHS.
We have also entered into indemnification agreements with each of our directors and executive officers. In general, these indemnification agreements require the Company to indemnify a director to the fullest extent permitted by law against liabilities that may arise by reason of his or her service for the Company.
Review, Approval and Ratification of Related Party Transactions
The Audit Committee of the Board of Directors (established in March 2019) reviews and approves all related party transactions.
Item 6. Other Information
None.
31
Item 7. Financial Statements
Emerald Health Pharmaceuticals Inc.
Index to Financial Statements
F-1
To the Board of Directors of
Emerald Health Pharmaceuticals Inc.
5910 Pacific Center Blvd, Suite 320
San Diego, CA 92121
Opinion
We have audited the consolidated financial statements of Emerald Health Pharmaceuticals Inc. and its subsidiaries (the “Company”), which comprise the consolidated balance sheets as of December 31, 2021 and December 31, 2020, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for the years then ended, and the related notes to the consolidated financial statements (collectively referred to as the “financial statements”).
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and December 31, 2020, and the results of its operations and its cash flows for the years then ended in accordance with accounting principles generally accepted in the United States of America.
Basis for Opinion
We conducted our audits in accordance with auditing standards generally accepted in the United States of America (GAAS). Our responsibilities under those standards are further described in the Auditor’s Responsibilities for the Audit of the Financial Statements section of our report. We are required to be independent of the Company and to meet our other ethical responsibilities, in accordance with the relevant ethical requirements relating to our audits. We believe that the audit evidence we have obtained is sufficient and appropriate to provide a basis for our audit opinion.
Substantial Doubt About the Company’s Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has experienced losses and recurring cash outflows from operations since inception and has an accumulated deficit and has stated that substantial doubt exists about its ability to continue as a going concern. Management’s evaluation of the events and conditions and management’s plans regarding these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty. Our opinion is not modified with respect to this matter.
Management’s Responsibility for the Consolidated Financial Statements
Management is responsible for the preparation and fair presentation of the financial statements in accordance with accounting principles generally accepted in the United States of America, and for the design, implementation, and maintenance of internal control relevant to the preparation and fair presentation of financial statements that are free from material misstatement, whether due to fraud or error.
In preparing the financial statements, management is required to evaluate whether there are conditions or events, considered in the aggregate, that raise substantial doubt about the Company’s ability to continue as a going concern for one year after the date that the financial statements are issued.
F-2
Auditors’ Responsibility
Our objectives are to obtain reasonable assurance about whether the financial statements as a whole are free from material misstatement, whether due to fraud or error, and to issue an auditor’s report that includes our opinion. Reasonable assurance is a high level of assurance but is not absolute assurance and therefore is not a guarantee that an audit conducted in accordance with GAAS will always detect a material misstatement when it exists. The risk of not detecting a material misstatement resulting from fraud is higher than for one resulting from error, as fraud may involve collusion, forgery, intentional omissions, misrepresentations, or the override of internal control. Misstatements are considered material if there is a substantial likelihood that, individually or in the aggregate, they would influence the judgment made by a reasonable user based on the financial statements.
In performing an audit in accordance with GAAS, we:
| · | Exercise professional judgment and maintain professional skepticism throughout the audit. |
| · | Identify and assess the risks of material misstatement of the financial statements, whether due to fraud or error, and design and perform audit procedures responsive to those risks. Such procedures include examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. |
| · | Obtain an understanding of internal control relevant to the audit in order to design audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control. Accordingly, no such opinion is expressed. |
| · | Evaluate the appropriateness of accounting policies used and the reasonableness of significant accounting estimates made by management, as well as evaluate the overall presentation of the financial statements. |
| · | Conclude whether, in our judgment, there are conditions or events, considered in the aggregate, that raise substantial doubt about the Company’s ability to continue as a going concern for a reasonable period of time. |
We are required to communicate with those charged with governance regarding, among other matters, the planned scope and timing of the audit, significant audit findings, and certain internal control-related matters that we identified during the audit.
/s/ DELOITTE & TOUCHE LLP
San Diego, California
April 29, 2022
F-3
Emerald Health Pharmaceuticals Inc.
Consolidated Balance Sheets
| December 31, 2021 | December 31, 2020 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 14,912,594 | $ | 17,036,840 | ||||
| Restricted cash | - | 2,752,890 | ||||||
| Incentive and other tax receivables | 1,457,674 | 513,953 | ||||||
| Other current assets | 489,817 | 694,445 | ||||||
| Total current assets | 16,860,085 | 20,998,128 | ||||||
| Property and equipment, net | 31,502 | 35,068 | ||||||
| Other noncurrent assets | 283,464 | 59,136 | ||||||
| Total assets | $ | 17,175,051 | $ | 21,092,332 | ||||
| Liabilities and stockholders’ equity | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 1,373,163 | $ | 428,304 | ||||
| Accrued expenses | 2,479,115 | 2,343,139 | ||||||
| Deposits held in escrow | - | 2,752,890 | ||||||
| Accrued interest payable | - | 97,531 | ||||||
| Related party loan | - | 2,819,771 | ||||||
| Total current liabilities | 3,852,278 | 8,441,635 | ||||||
| Loans payable | - | 292,152 | ||||||
| Total liabilities | 3,852,278 | 8,733,787 | ||||||
| Commitments and contingencies (Note 6) | ||||||||
| Stockholders’ equity | ||||||||
| Common stock, $0.0001 par value, 100,000,000 shares authorized; 22,148,976 shares issued and 22,073,976 shares outstanding at December 31, 2021; 19,585,029 shares issued and 19,510,029 shares outstanding at December 31, 2020 | 2,215 | 1,959 | ||||||
| Additional paid-in-capital | 70,213,689 | 52,648,471 | ||||||
| Accumulated other comprehensive loss | (315,683 | ) | (183,169 | ) | ||||
| Accumulated deficit | (56,577,440 | ) | (40,108,708 | ) | ||||
| Treasury stock, at cost (common stock: 75,000 at December 31, 2021 and December 31, 2020) | (8 | ) | (8 | ) | ||||
| Total stockholders’ equity | 13,322,773 | 12,358,545 | ||||||
| Total liabilities and stockholders’ equity | $ | 17,175,051 | $ | 21,092,332 | ||||
See accompanying Notes to Consolidated Financial Statements.
F-4
Emerald Health Pharmaceuticals Inc.
Consolidated Statements of Operations and Comprehensive Loss
| Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Revenue | $ | - | $ | - | ||||
| Operating expenses: | ||||||||
| Research and development | 10,504,366 | 7,797,908 | ||||||
| General and administrative | 6,392,538 | 4,163,360 | ||||||
| Total operating expenses | 16,896,904 | 11,961,268 | ||||||
| Operating loss | (16,896,904 | ) | (11,961,268 | ) | ||||
| Other (income)/expenses: | ||||||||
| Other income | (429,343 | ) | (48,211 | ) | ||||
| Interest expense | 66,334 | 470,293 | ||||||
| Foreign exchange (gain)/loss | (65,163 | ) | 41,272 | |||||
| Net loss | (16,468,732 | ) | (12,424,622 | ) | ||||
| Other comprehensive loss: | ||||||||
| Foreign currency translation adjustments | (132,514 | ) | (144,445 | ) | ||||
| Comprehensive loss | $ | (16,601,246 | ) | $ | (12,569,067 | ) | ||
| Net loss per share, basic and diluted | $ | (0.76 | ) | $ | (0.77 | ) | ||
| Weighted-average common shares outstanding, basic and diluted | 21,567,180 | 16,154,859 | ||||||
See accompanying Notes to Consolidated Financial Statements.
F-5
Emerald Health Pharmaceuticals Inc.
Consolidated Statements of Stockholders’ Equity
| Common Stock Outstanding | Additional Paid in | Accumulated Other Comprehensive | Accumulated | Treasury Stock | Total Stockholders’ | |||||||||||||||||||||||||||
| Shares | Amount | Capital | Loss | Deficit | Shares | Amount | Equity | |||||||||||||||||||||||||
| Balance at December 31, 2019 | 14,417,085 | $ | 1,449 | $ | 22,546,309 | $ | (38,724 | ) | $ | (27,684,086 | ) | 75,000 | $ | (8 | ) | $ | (5,175,060 | ) | ||||||||||||||
| Issuance of common stock under Regulation A offering, net of issuance costs | 4,972,944 | 498 | 27,524,244 | 27,524,742 | ||||||||||||||||||||||||||||
| Issuance of common stock for services | 100,000 | 10 | 599,990 | 600,000 | ||||||||||||||||||||||||||||
| Issuance of restricted common stock under equity incentive plan | 20,000 | 2 | (2 | ) | - | |||||||||||||||||||||||||||
| Stock-based compensation expense | 1,977,930 | 1,977,930 | ||||||||||||||||||||||||||||||
| Net loss and comprehensive loss | (144,445 | ) | (12,424,622 | ) | (12,569,067 | ) | ||||||||||||||||||||||||||
| Balance at December 31, 2020 | 19,510,029 | $ | 1,959 | $ | 52,648,471 | $ | (183,169 | ) | $ | (40,108,708 | ) | 75,000 | $ | (8 | ) | $ | 12,358,545 | |||||||||||||||
| Issuance of common stock under Regulation A offering, net of issuance costs | 2,273,447 | 227 | 12,914,022 | 12,914,249 | ||||||||||||||||||||||||||||
| Issuance of common stock under Regulation D | 37,500 | 4 | 299,996 | 300,000 | ||||||||||||||||||||||||||||
| Issuance of common stock upon conversion of related party loan | 250,000 | 25 | 499,975 | 500,000 | ||||||||||||||||||||||||||||
| Issuance of restricted common stock under equity incentive plan | 3,000 | - | - | - | ||||||||||||||||||||||||||||
| Stock-based compensation expense | 3,851,225 | 3,851,225 | ||||||||||||||||||||||||||||||
| Net loss and comprehensive loss | (132,514 | ) | (16,468,732 | ) | (16,601,246 | ) | ||||||||||||||||||||||||||
| Balance at December 31, 2021 | 22,073,976 | $ | 2,215 | $ | 70,213,689 | $ | (315,683 | ) | $ | (56,577,440 | ) | 75,000 | $ | (8 | ) | $ | 13,322,773 | |||||||||||||||
See accompanying Notes to Consolidated Financial Statements.
F-6
Emerald Health Pharmaceuticals Inc.
Consolidated Statements of Cash Flows
| Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Operating activities | ||||||||
| Net loss | $ | (16,468,732 | ) | $ | (12,424,622 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 16,036 | 24,596 | ||||||
| Stock-based compensation | 3,851,225 | 2,577,930 | ||||||
| Gain on forgiveness of PPP Loan and accrued interest | (294,603 | ) | - | |||||
| Changes in operating assets and liabilities: | ||||||||
| Incentive and other tax receivables | (943,721 | ) | 906,154 | |||||
| Other current assets | 204,628 | (327,095 | ) | |||||
| Other noncurrent assets | (224,328 | ) | (59,136 | ) | ||||
| Accounts payable | 1,039,892 | (1,604,195 | ) | |||||
| Accrued expenses | 133,170 | 373,398 | ||||||
| Accrued interest payable | (95,080 | ) | (407,758 | ) | ||||
| Net cash used in operating activities | (12,781,513 | ) | (10,940,728 | ) | ||||
| Investing activities | ||||||||
| Purchases of property and equipment | (12,470 | ) | (7,206 | ) | ||||
| Net cash used in investing activities | (12,470 | ) | (7,206 | ) | ||||
| Financing activities | ||||||||
| Issuance of common stock | 13,940,682 | 29,837,664 | ||||||
| Deposits held in escrow | (2,752,890 | ) | 2,752,890 | |||||
| Funds received under loans payable | - | 1,087,373 | ||||||
| Funds repaid under loans payable | - | (795,221 | ) | |||||
| Funds repaid under related party loan | (2,319,771 | ) | (750,000 | ) | ||||
| Stock issuance costs | (818,660 | ) | (2,233,858 | ) | ||||
| Net cash provided by financing activities | 8,049,361 | 29,898,848 | ||||||
| Effect of exchange rate changes on cash | (132,514 | ) | (144,445 | ) | ||||
| Net change in cash and cash equivalents, and restricted cash | (4,877,136 | ) | 18,806,469 | |||||
| Cash, cash equivalents, and restricted cash at beginning of period | 19,789,730 | 983,261 | ||||||
| Cash, cash equivalents, and restricted cash at end of period | $ | 14,912,594 | $ | 19,789,730 | ||||
| Supplemental disclosure of cash flow information: | ||||||||
| Interest paid to related party | $ | 161,415 | $ | 822,524 | ||||
| Interest paid on loans payable | $ | - | $ | 53,183 | ||||
| Non-cash investing and financing activities: | ||||||||
| Conversion of related party loan to common stock | $ | 500,000 | $ | - | ||||
| Gain on forgiveness of PPP Loan and accrued interest | $ | 294,603 | $ | - | ||||
| Discharge between related party loan and related party receivables | $ | - | $ | 180,930 | ||||
| Deferred stock issuance costs in accounts payable and accrued expenses | $ | 3,792 | $ | 96,019 | ||||
See accompanying Notes to Consolidated Financial Statements.
F-7
Emerald Health Pharmaceuticals Inc.
Notes to Consolidated Financial Statements
1. Description of Business
Emerald Health Pharmaceuticals Inc. (EHP, or the Company) was incorporated in the State of Delaware in March 2017. EHP is a clinical stage pharmaceutical company focused on developing drug product candidates currently containing novel, patented new chemical entities (NCEs), rationally designed to treat various diseases with unmet medical needs, including neurodegenerative, autoimmune and other diseases. The Company is currently developing two initial therapeutic product candidates that target four initial indications, multiple sclerosis (MS) and systemic sclerosis (SSc, a severe form of scleroderma), Parkinson’s disease (PD) and Huntington’s disease (HD).
The Company acquired certain intellectual property from VivaCell Biotechnology España S.L. (VivaCell). which became a wholly owned subsidiary of Emerald Health Research Inc. (EHR) in 2018. EHR is a wholly owned subsidiary of Emerald Health Sciences Inc. (EHS), a significant (former majority) stockholder of EHP. EHP has no ownership or voting rights related to VivaCell. See Note 7.
The Company is subject to risks common to other life science companies in the development stage including, but not limited to, uncertainty of product development and commercialization, lack of marketing and sales history, development by its competitors of new technological innovations, dependence on key personnel, market acceptance of products, product liability, protection of proprietary technology, ability to raise additional financing, and compliance with FDA and other government regulations. If the Company does not successfully commercialize any product candidates, it will be unable to generate recurring product revenue or achieve profitability.
The global COVID-19 situation has resulted in and may continue to result in significant disruptions to the global economy, as well as business and capital markets around the world. The ultimate effects of COVID-19 on the Company’s business, operations and financial condition are unknown at this time. To date, the enrollment rate in the Company’s Phase 2a clinical trial has been affected. However, the extent to which COVID-19 impacts the Company’s business will depend on future developments, which are highly uncertain and cannot be predicted, including new information which may emerge concerning the severity of COVID-19 and the actions to contain it or treat its impact, among others.
The Company’s financial statements have been prepared assuming the Company will continue as a going concern, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The Company has experienced losses and recurring cash outflows from operations since inception and has an accumulated deficit of $56,577,440 as of December 31, 2021. As of December 31, 2021, the Company had cash in the amount of $14,912,594. For the years ended December 31, 2021 and 2020, the Company incurred losses from operations of $16,896,904 and $11,961,268, respectively. The Company expects to continue to incur significant losses and negative cash flows from operation through 2022 and into the foreseeable future.
The Company has funded operations with capital raised from a Tier 2 offering (the Regulation A Offering) pursuant to Regulation A (Regulation A+) under the Securities Act of 1933, as amended (the Securities Act), as well as an exempt offshore offering under Regulation S under the Securities Act. The Regulation A Offering commenced in 2019 and terminated in March of 2021. In addition, the Company has received loan proceeds from separate loan arrangements, including a revolving loan (which terminated in June of 2021) with its significant (former majority) stockholder, EHS. See Note 3 and Note 6.
The future viability of the Company is largely dependent upon its ability to raise additional capital to finance its operations. Management expects that future sources of funding may include sales of equity, obtaining loans, or other strategic transactions. Although management continues to pursue these plans, there is no assurance that the Company will be successful in obtaining sufficient financing on terms acceptable to the Company to fund continuing operations, if at all. Based on the Company’s expected cash requirements, without obtaining additional funding by the second quarter of 2023, management believes that the Company will not have enough funds to continue clinical studies. These circumstances raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that the financial statements are issued. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
F-8
2. Significant Accounting Policies
Basis of Presentation
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States (GAAP) and in accordance with the instructions to Form 1-K.
Consolidation
The consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries, Emerald Health Pharmaceuticals Australia Pty Ltd. (EHP Australia) and Emerald Health Pharmaceuticals, España Sociedad Limitada (EHP España). EHP Australia’s functional currency, the Australian dollar, is also its reporting currency, and its financial statements are translated to U.S. dollars prior to consolidation. EHP España’s functional currency, the Euro, is also its reporting currency, and its financial statements are translated to U.S. dollars prior to consolidation. All intercompany accounts and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results could differ from those estimates.
Cash, Cash Equivalents and Restricted Cash
The Company considers all highly liquid investments with an original maturity of three months or less from the date of purchase to be cash equivalents.
Restricted cash consists of cash held in an escrow account, received as deposits from potential investors towards purchases of common stock under the Regulation A Offering which had not yet been fully consummated as of the balance sheet date, as described in Note 4.
The following table provides a reconciliation of cash, cash equivalents and restricted cash, reported within the condensed consolidated statements of cash flows:
| Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Cash and cash equivalents | $ | 14,912,594 | $ | 17,036,840 | ||||
| Restricted cash | - | 2,752,890 | ||||||
| Total cash, cash equivalents and restricted cash presented in the consolidated statements of cash flows | $ | 14,912,594 | $ | 19,789,730 | ||||
Incentive and Tax Receivables
The Company’s subsidiary, EHP Australia, is incorporated in Australia and is eligible to participate in an Australian research and development tax incentive program. As part of this program, EHP Australia is eligible to receive a cash refund from the Australian Taxation Office (ATO) for a percentage (currently 43.5%) of the research and development costs incurred by EHP Australia. The cash refund is available to eligible companies with an annual aggregate revenue of less than $AU20.0 million (Australian Dollars) during the reimbursable period. As of December 31, 2021 and 2020, the Company’s estimate of the amount of cash refunds expected to be received for eligible spending as part of this incentive program was $1.4 million and $0.5 million, respectively, which amounts are included in incentive and other tax receivables. In August 2021, the Company received $0.5 million as a cash refund from the ATO for eligible spending incurred during the year ended December 31, 2020.
In addition, EHP Australia incurs Goods and Services Tax (GST) on services provided by Australian vendors. As an Australian entity, EHP Australia is entitled to a quarterly refund of the GST incurred during the previous quarter. The Company’s estimate of the amount of cash refund expected to be received related to GST incurred as of December 31, 2021 and 2020, was $86,608 and $46,816, respectively, which amounts are included in incentive and other tax receivables.
F-9
Property and Equipment
Property and equipment generally consist of computer equipment and software, manufacturing equipment, and office furniture and are recorded at cost and depreciated over the estimated useful lives of the assets (generally three to five years) using the straight-line method. Leasehold improvements are stated at cost and are amortized on a straight-line basis over the lesser of the remaining term of the related lease or the estimated useful lives of the assets. Repairs and maintenance costs are charged to expense as incurred and improvements and betterments are capitalized. When assets are retired or otherwise disposed of, the cost and accumulated depreciation are removed from the consolidated balance sheets and any resulting gain or loss is reflected in the consolidated statements of operations in the period realized.
Impairment of Long-lived Assets
The Company reviews property and equipment for impairment on an annual basis and whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. An impairment loss would be recognized when estimated future undiscounted cash flows relating to the asset or asset group are less than its carrying amount. An impairment loss is measured as the amount by which the carrying amount of an asset or asset group exceeds its fair value. While the Company’s current and historical operating losses and negative cash flows are possible indicators of impairment, management believes that future cash flows to be generated by these assets support the carrying value of its long-lived assets and, accordingly, did not recognize any impairment losses during the years ended December 31, 2021 and 2020.
Research and Development
Research and development costs are charged to expense as incurred and consist primarily of contract research fees, contract manufacturing costs, consultant fees, preclinical studies, clinical trials and related costs, compensation and related benefits, and non-cash stock-based compensation. At the end of each reporting period, the Company compares the payments made to its vendors, clinical research organizations and consultants to the estimated progress towards completion of the related project. Factors that the Company considers in preparing these estimates include the number of patients enrolled in clinical trials, milestones achieved, and other criteria related to the efforts of its vendors. These estimates will be subject to change as additional information becomes available. Depending on the timing of payments to vendors and estimated services provided, the Company will record net prepaid or accrued expenses related to these costs. Research and development expenses are recorded net of expected refunds of eligible research and development costs paid pursuant to the Australian research and development tax incentive program and GST incurred on services provided by Australian vendors.
Income Taxes
The Company has incurred net operating losses from inception through December 31, 2021. Therefore, no United States federal, state, or foreign income taxes are expected to be paid for 2021 or 2020 and no amounts payable have been recorded as of December 31, 2021 and 2020.
Due to the Company’s history of losses since inception, there is not enough evidence at this time to support the conclusion that it will generate future income of a sufficient amount and nature to utilize the benefits of the Company’s net deferred tax assets. Accordingly, the Company fully reduced its net deferred tax assets by a valuation allowance, since it has been determined that it is more likely than not that all of the deferred tax assets will not be realized.
The Tax Reform Act of 1986 contains provisions which limit the ability to utilize the net operating loss carryforwards in the case of certain events including significant changes in ownership interests. If the Company’s net operating loss carryforwards are limited, and the Company has taxable income which exceeds the permissible yearly net operating loss carryforwards, the Company would incur a federal income tax liability even though net operating loss carryforwards would be available in future years.
F-10
Fair Value Measurements
The Company does not have any financial assets and liabilities reported at fair value on a recurring basis. The carrying amounts of the Company’s financial instruments including cash and cash equivalents, restricted cash, incentive and other tax receivables, other current assets, property and equipment, net, accounts payable, accrued expenses, and deposits held in escrow, approximate fair value due to the short-term nature of those instruments. The Company’s related party loan and the associated accrued interest payable was carried at amortized cost. Due to the related party nature of these advances with the significant (former majority) shareholder, management concluded that its fair value was not reasonably determinable. See Note 3.
The Company determines fair value based upon the exit price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants, as determined by either the principal market or the most advantageous market. Inputs used in the valuation techniques to derive fair values are classified based on a three-level hierarchy. These levels are:
Level 1—Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs.
Level 2—Observable prices that are based on inputs not quoted on active markets but corroborated by market data.
Level 3—Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs.
As of December 31, 2021 and 2020 the carrying amount of the Company’s cash and cash equivalents and restricted cash were determined using Level 1 inputs.
Business Segments
The Company operates within the United States, Europe, and Australia, in one business segment, which is dedicated to research and development of drug candidates based on patented NCEs with a unique mechanism of action, to treat diseases with unmet medical needs.
Stock-Based Compensation
The Company accounts for stock option awards issued to employees and non-employees in accordance with Financial Accounting Standards Board Accounting Standards Codification (ASC) Topic No. 718, Compensation-Stock Compensation. Under FASB ASC Topic No. 718, compensation expense related to stock-based payments is recorded over the requisite service period based on the grant date fair value of the awards. Forfeitures are accounted for as they occur. The Company uses the Black-Scholes option pricing model for determining the estimated fair value for stock-based awards. The Black-Scholes model requires the use of assumptions which determine the fair value of stock-based awards, including the option’s expected term and the price volatility of the underlying stock. See Note 5.
Net Loss per Share
The Company computes basic net loss per common share by dividing the applicable net loss by the weighted average number of common shares outstanding during the period. Diluted earnings per share is computed using the weighted average number of common shares outstanding during the period, plus additional shares to account for the dilutive effect of potential future issuances of common stock relating to stock options and other potentially dilutive securities using the treasury stock method. For the years ended December 31, 2021 and 2020, there were 3,553,500 and 2,892,500 options, respectively, excluded from the computation of diluted earnings per share, as the effect would be anti-dilutive.
F-11
Comprehensive Loss
Comprehensive loss includes foreign currency translation adjustments related to the Company’s subsidiaries in Australia and Spain.
Recently Issued Accounting Pronouncements
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842) (ASU No. 2016-02), which changes the presentation of assets and liabilities relating to leases. The core principle of ASU No. 2016-02 is that a lessee should recognize the assets and liabilities that arise from leases. In June 2020, the FASB issued ASU No. 2020-05, Revenue from Contracts with Customers (Topic 606) and Leases (Topic 842) (ASU No. 2020-05), which further deferred the effective date of ASU No. 2016-02 for the Company from January 1, 2021 to January 1, 2022. The Company adopted the new standard effective January 1, 2022, using the modified retrospective transition approach, and will not recast prior periods. In addition, the Company did not record a cumulative effect adjustment to the opening balance of retained earnings upon adoption. As permitted by the standard, the Company has elected all practical expedients and the transition practical expedient package, which among other things, allows the carryforward of historical lease classifications. Accordingly, the Company will continue to apply FASB ASC Topic 840, Leases (Topic 840), prior to January 1, 2022, including Topic 840 disclosure requirements, in the comparative periods presented. Due to the short-term nature of the Company’s existing leases, implementation of the standard did not have a material impact on its consolidated financial statements.
In November 2019, the FASB issued ASU No. 2019-10, Financial Instruments - Credit Losses (Topic 326), Derivatives and Hedging (Topic 815), and Leases (Topic 842) (ASU No. 2019-10), which deferred the effective date of ASU No. 2016-02 for the Company from January 1, 2020 to January 1, 2021, and deferred the effective date of Topic 326 for the Company from January 1, 2022 to January 1, 2023. The Company is evaluating the adoption of Topic 326 as of the date of the financial statements.
3. Related Party Transactions
Since inception, the Company has received advances from EHS to fund its operations. In September 2017, the Company and EHS entered a revolving loan agreement (Related Party Loan), which was amended in January 2018 and November 2019. Borrowings under the Related Party Loan were drawn down from time to time, and repaid by the Company in cash, or at the option of EHS, converted into shares of the Company at $2.00 per share, or at a price to be equally agreed to between EHS and the Company (Conversion Feature). In November 2019, the Related Party Loan was amended to reduce the interest rate from 12% to 10%, compounded semiannually. The Related Party Loan was payable upon demand and had no expiration date.
During the year ended December 31, 2021, the outstanding balance under the Related Party Loan was settled in full, and the Related Party Loan was terminated effective June 1, 2021, pursuant to a Loan Termination Agreement between the Company and EHS. In total, EHS advanced approximately $11.3 million to the Company under the Related Party Loan. The Company repaid approximately $3.1 million of the total advanced by EHS in cash, of which approximately $2.3 million was repaid during the year ended December 31, 2021, and approximately $5.2 million was offset through cashless discharges (described below). The remaining $3.0 million was converted into 1.5 million shares of the Company’s common stock at a conversion price of $2.00 per share, of which $0.5 million was converted into 250,000 shares of the Company’s common stock during the year ended December 31, 2021. A total of approximately $2.2 million of interest expense was incurred (excluding a non-cash charge of approximately $1.4 million recorded during the year ended December 31, 2019 related to accretion of the beneficial Conversion Feature) under the Related Party Loan, of which approximately $0.1 million was incurred during the year ended December 31, 2021. Approximately $2.0 million of the total interest expense was paid to EHS in cash, of which approximately $0.2 million was paid during the year ended December 31, 2021, and approximately $0.2 million was offset through a cashless discharge during the year ended December 31, 2019. The Company does not have any remaining obligations to EHS under the Related Party Loan as of December 31, 2021.
F-12
The Conversion Feature of the loan agreement was not considered an embedded derivative under FASB Accounting Standards Codification (ASC) Topic 815, Derivatives and Hedging, since there were no provisions for net settlement nor was there a means for EHS to receive an asset that put EHS in a position not substantially different from net settlement.
In May 2019, the Company’s Board of Directors authorized a funding arrangement with EHS (Related Party Note Receivable), which was amended in August 2019 and September 2019 to extend the repayment dates, pursuant to which the Company advanced funds to EHS in the form of interest bearing (12%) short term notes under a Promissory Note between EHS and EHP (the Promissory Note). Advances under the Promissory Note were originally due for repayment with accrued and unpaid interest three months from the date of the advance. A total of $5,000,000 was advanced and $178,933 accrued as interest receivable under the Related Party Note Receivable. During 2019, the Related Party Note Receivable was settled through cashless discharges against the unpaid principal and accrued interest payable balances, respectively, under the existing Related Party Loan with EHS.
During the years ended December 31, 2021 and 2020, the Company billed EHS for operating expenses amounting to $5,938 and $82,003, respectively.
On November 15, 2019, Dr. Avtar Dhillon, a co-founder of the Company, resigned as Chairman of the Board of Directors. The Company and EHS concurrently entered into a Board Observer Agreement, whereby the Company granted to EHS the right to designate any director or officer to be an observer on the Board of Directors for so long as EHS maintains ownership of any securities of the Company. Dr. Dhillon, who was at that time CEO and a director of EHS initially served as the EHS Board observer.
In December 2019, the Board of Directors approved an Independent Contractor Services Agreement between the Company and Dr. Dhillon, to provide corporate advisory services to the Company in exchange for a monthly fee of $10,000 (the “Agreement”). The Agreement had an initial term of one year and renewed automatically thereafter unless terminated earlier by either party. On September 23, 2021, the Company was provided 30-day written notice from Dr. Dhillon, terminating the Agreement without cause, effective October 23, 2021. During the years ended December 31, 2021 and 2020, the Company paid $97,419 and $130,000, respectively, under the Agreement. There are no remaining obligations, nor amounts due and payable, under the Agreement as of December 31, 2021.
In August 2020, the Company’s Board of Directors granted Dr. Dhillon the option to purchase 25,000 shares of common stock at $6.00 per share under the Plan (as defined in Note 7), with vesting occurring 25% on the date of the grant and an additional 25% vesting annually thereafter. Concurrent with the termination of the Agreement, the remaining 12,500 unvested shares were forfeited.
In September 2021, the Company’s Board of Directors approved a modification to the exercise terms of the non-qualified stock option awards previously issued to Dr. Dhillon, resulting in the recognition of an additional $0.3 million within stock-based compensation expense during the year ended December 31, 2021. See Note 7.
In April 2021, the Company entered into an agreement with SKYE Bioscience, Inc. (SKYE), to sublease a portion of its existing non-cancelable building lease to SKYE, beginning May 16, 2021 and continuing on a month-to-month basis. EHS is a significant stockholder of both EHP and SKYE. In August 2021, SKYE provided notice of termination of the month-to-month lease agreement, effective August 31, 2021. Under the sublease agreement, SKYE paid monthly base rent of $4,000 per month, in addition to a portion of common area expenses and utilities. During the year ended December 31, 2021, the Company received a total of $15,453 from SKYE for rent and utilities allocated under the sublease.
F-13
4. Common Stock
The Company has 100,000,000 shares of common stock authorized with a par value of $0.0001 per share. The Company has issued 9,000,000 shares of common stock at $0.0001 per share to EHS for proceeds of $900, and 1,000,000 shares to the founders of the Company for total proceeds of $100. The shares issued to founders vested 25% on the date of issuance and vested 25% annually thereafter until fully vested. In total, 925,000 of these shares have fully vested as of December 31, 2021 and 2020. The Company repurchased 75,000 unvested shares at par value from a founding member, which are currently held by the Company as treasury stock.
The Company has issued a total of 1,250,000 additional shares of common stock to EHS upon the conversion of $2,500,000 of unpaid principal balance under the Related Party Loan at a conversion price of $2.00 per share. See Note 3.
The Company sold 65,700 shares of common stock at $5.00 per share for gross proceeds of $328,500, less issuance costs of $51,776, in an exempt offshore offering under Regulation S under the Securities Act.
From March 2019 through March 2021, the Company sold shares of common stock under a Tier 2 offering (the Regulation A Offering) pursuant to Regulation A (Regulation A+) under the Securities Act. The Regulation A Offering was qualified by the SEC in March 2018 with subsequent post-qualification offering circular amendments qualified by the SEC in June 2019, and November 2020. The Company closed and terminated the Regulation A Offering effective March 28, 2021. During the year ended December 31, 2021, the Company sold 2,273,447 shares of common stock under the Offering, for gross proceeds of $13.6 million, less issuance costs incurred of $0.7 million; and during the year ended December 31, 2020, the Company sold 4,972,944 shares of common stock under the Offering, for gross proceeds of $29.8 million, less issuance costs of $2.3 million.
In total, from the commencement of the sale of shares pursuant to the Regulation A Offering in March 2019 through the closing and termination of the Regulation A Offering on March 28, 2021, the Company sold an aggregate of 10,422,776 shares of common stock pursuant to the Regulation A Offering for gross proceeds of approximately $60 million.
The Company had a Broker-Dealer Agreement with Dalmore Group, LLC (Dalmore), a broker-dealer registered with the SEC and a member of the Financial Industry Regulatory Authority (FINRA), to perform administrative, compliance and placement agent related functions in connection with the Offering. Pursuant to this agreement, the Company paid Dalmore $28,000 in one-time set up fees, consisting of a $20,000 agreement fee and $8,000 for fees paid to FINRA, plus 1.0% commission on the sale of common stock under the Offering, commencing with sales following regulatory approval by FINRA, which occurred on July 25, 2019. During the years ended December 31, 2021 and 2020, the Company incurred approximately $0.1 million and $0.3 million, respectively, under this agreement, related to commissions on the sale of common stock under the Regulation A Offering.
The Company had an Escrow Services Agreement with Prime Trust, LLC in connection with the Regulation A Offering. Under this agreement, the proceeds received from the Offering were deposited into an escrow account prior to distribution to the Company, which was recorded as restricted cash, and offset by deposits held in escrow liability. The Company terminated the Escrow Services Agreement in September 2021, and there are no remaining deposits held in the escrow account as of December 31, 2021.
In June 2020, the Company issued 100,000 shares of common stock to a consultant as payment for services. At the time of issuance, the Company recognized $600,000 of stock-based compensation expense, of which $300,000 was for research and development and $300,000 was for general and administrative services.
During the years ended December 31, 2021 and 2020, the Company issued 3,000 shares and 20,000 shares, respectively, of restricted common stock under the Plan (as defined below), to consultants as payment for services.
In July 2020, the Company entered into a consulting agreement with a third party to provide business advisory services in connection with strategic development and private financing matters. Pursuant to this agreement, during the years ended December 31, 2021 and 2020, the Company incurred and paid consulting fees of approximately $0.4 million and $1.2 million, respectively, which were recorded as stock issuance costs within equity as the services performed were directly related to the Regulation A Offering. The agreement expired on March 31, 2021.
In April 2021, the Company issued 250,000 shares of common stock to EHS in accordance with a written notice received from EHS, for the conversion of $500,000 of the unpaid principal balance under the Related Party Loan at a conversion price of $2.00 per share. See Note 3.
In June 2021, the Company sold 37,500 shares of common stock at $8.00 per share for gross proceeds of $300,000, in a private placement offering under Regulation D under the Securities Act.
F-14
5. Equity Incentive Plan
In January 2018, the Company adopted the 2018 Equity Incentive Plan, which was amended on December 13, 2018 and August 12, 2020 (the Plan). On August 12, 2020, the Company adopted an amendment to the Plan which increased the number of shares of Common Stock authorized to be issued under the Plan to equal 18% of the number of issued and outstanding shares of common stock of the Company as of the applicable date of issuance. As of December 31, 2021, there were 3,973,315 shares of Common Stock reserved for issuance pursuant to awards under the Plan, and 396,815 shares of Common Stock available for grant under the Plan.
The Plan provides incentives to eligible employees, consultants, officers, and directors in the form of incentive stock options and non-qualified stock options, stock appreciation rights, restricted stock, restricted stock units and other rights or benefits. Recipients of stock options are eligible to purchase shares of the Company’s common stock at an exercise price equal to no less than the estimated fair market value of such stock on the date of grant. The maximum term of options granted under the Plan is ten years. Vesting schedules are determined by the Board of Directors.
The following table summarizes stock-based compensation expense related to stock options and restricted stock granted to employees and non-employees included in the consolidated statements of operations as follows:
| Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Research and development | $ | 1,313,152 | $ | 758,670 | ||||
| General and administrative | 2,538,073 | 1,219,260 | ||||||
| Total | $ | 3,851,225 | $ | 1,977,930 | ||||
Stock-based compensation for the years ended December 31, 2021 and 2020, includes expense of $1,251,371 and $654,926, respectively, related to option grants and restricted stock issued to non-employees.
Stock Options
The following table summarizes stock option activity:
| Options | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term (in Years) | Aggregate Intrinsic Value (1) | |||||||||||||
| Outstanding as of December 31, 2020 | 2,892,500 | $ | 3.77 | |||||||||||||
| Granted | 733,500 | $ | 6.18 | |||||||||||||
| Exercised | - | |||||||||||||||
| Forfeited/Expired | (72,500 | ) | $ | 4.55 | ||||||||||||
| Outstanding as of December 31, 2021 | 3,553,500 | $ | 4.25 | 7.71 | $ | 13,324,500 | ||||||||||
| Options Exercisable as of December 31, 2021 | 2,654,625 | $ | 3.62 | 7.29 | $ | 11,624,250 | ||||||||||
| Options Vested and Expected to Vest as of December 31, 2021 | 3,553,500 | $ | 4.25 | 7.71 | $ | 13,324,500 | ||||||||||
| (1) | As of December 31, 2021, the fair value of the Company’s common shares as determined by its Board of Directors, based upon the Company’s most recent sale under Regulation D under the Securities Act, was $8.00 per share. |
F-15
The following table summarizes certain information regarding stock options for the years ended December 31, 2021 and 2020:
| 2021 | 2020 | |||||||
| Weighted average grant date fair value per share of options granted during the period | $ | 4.48 | $ | 4.33 | ||||
| Fair value per share of options vested during the period | $ | 3.26 | $ | 2.80 | ||||
| Cash received from options exercised during the period | $ | - | $ | - | ||||
| Intrinsic value of options exercised during the period | $ | - | $ | - | ||||
As of December 31, 2021, unrecognized stock-based compensation expense for employee and non-employee stock options was approximately $3.3 million, which the Company expects to recognize over a weighted-average remaining period of 1.9 years, assuming all unvested options become fully vested.
The Company uses a Black-Scholes option-pricing model to value the Company’s option awards. Using this option-pricing model, the fair value of each employee and non-employee award is estimated on the grant date. The fair value is expensed on a straight-line basis over the vesting period. In general, the option awards vest partially upfront and then pro-rata annually thereafter. The expected volatility assumption is based on the volatility of the share price of comparable public companies. The expected life is determined using the “simplified method”. The risk-free interest rate is based on the implied yield on a U.S. Treasury security at a constant maturity with a remaining term equal to the expected term of the option granted. The dividend yield is zero, as the Company has never declared a cash dividend.
The fair value of the stock options granted was estimated on the date of grant using the Black-Scholes option pricing model with the following weighted-average assumptions for the periods indicated:
Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Expected term (in years) | 5.75 | 5.71 | ||||||
| Stock price volatility | 90 | % | 90 | % | ||||
| Risk-free interest rate | 0.69 | % | 0.40 | % | ||||
| Dividend yield | 0 | % | 0 | % | ||||
In September 2021, the Company’s Board of Directors approved a modification to the exercise terms of certain stock options previously issued under the Plan. This modification removed the post-termination exercise period from the award agreements, granting the award holders the ability to exercise the vested portion of the options until the original expiration dates of the awards, if continuous service is terminated for any reason other than for cause, including by death or disability. The modification applied to all non-qualified stock option awards previously issued to the Company’s three non-employee Directors, the Company’s President, Chief Executive Officer and Director, and the Company’s co-founder and former Executive Chairperson and President, Dr. Dhillon. During the year ended December 31, 2021, stock-based compensation expense includes an additional $1.0 million related to the vested portion of each option, with an additional $0.2 million of stock-based compensation expense to be recognized over the remaining vesting periods of the impacted options.
Restricted Stock
During the years ended December 31, 2021 and 2020, there were 3,000 shares and 20,000 shares, respectively, of restricted common stock granted and vested under the Plan. The shares were issued to non-employees, with vesting periods ranging from 3 to 6 months for each grant, and a weighted average grant date price of $6 per share for each period. The Company recognized $18,000 and $120,000 of stock-based compensation expense related to restricted stock during the years ended December 31, 2021 and 2020, respectively, and there were no unvested shares outstanding as of the end of each period.
F-16
6. Commitments and Contingencies
On May 1, 2018, the Company entered into a two-year non-cancelable building lease for its corporate headquarters in San Diego, California. Effective August 15, 2019, the lease was amended to include additional space at the existing premises and to extend the term of the original lease through August 31, 2022. Under the lease, the Company pays a base rent of $21,852 per month. The Company has paid a $41,503 security deposit related to the lease, which is recorded within other current assets. Rent expense for the years ended December 31, 2021 and 2020, was $256,558 and $197,128, respectively.
Future minimum payments under the non-cancelable operating lease as of December 31, 2021 were as follows:
| 2022 | $ | 174,816 | ||
| Total | $ | 174,816 |
In July 2020, the company entered into an agreement to sublease a portion of its existing non-cancelable building lease to a tenant, effective August 1, 2020, and continuing through August 31, 2022. The Company paid commissions of $7,000 related to the execution of the sublease and was expected to incur future losses of approximately $55,000 in conjunction with the sublease. EHS agreed to reimburse the Company for the commission fee and future expected losses, which were offset against the Related Party Loan as of December 31, 2020.
Loans payable
In January 2020, EHP Australia entered into a loan agreement with Rocking Horse Nominees Pty Ltd (Rocking Horse), whereby Rocking Horse advanced $AU1.2 million (approximately $0.8 million) to EHP Australia. The loan was secured by the tax incentive refund anticipated to be received during 2020 for eligible spending incurred under the Australian research and development tax incentive program during 2019. The loan had an upfront establishment fee of 1.2%, bearing interest at 1.25% per month compounded daily. Approximately $53,000 in interest expense was incurred under this loan during the year ended December 31, 2020, and the loan was repaid in June 2020.
On April 22, 2020, The Company received loan proceeds of $292,152 from Silicon Valley Bank pursuant to the Paycheck Protection Program established as part of the Coronavirus Aid, Relief and Economic Security Act (PPP Loan). The PPP Loan, which is evidenced by a note dated April 21, 2020, bore interest at a rate of 1% per annum, and was expected to mature on April 21, 2022. On February 18, 2021, the PPP Loan, including accrued interest of $2,451, was forgiven in full by the lender. During the year ended December 31, 2021, the income from the forgiveness of the amount borrowed and the accrued interest was recognized in the statement of operations in other income as a gain on the extinguishment of debt, in accordance with ASC 405-20, Extinguishment of Liabilities.
7. Intellectual Property Transfer and Research Agreements
In June 2017, upon the execution of the Intellectual Property Transfer Agreement (IPTA), EHP paid VivaCell approximately $112,000 for the purchase of three United States patents, two Japanese patents, one European patent and fourteen pending patent applications covering two series of molecules which are based on the molecular structure of CBD and CBG. Future payments of up to 2.7 million Euro (approximately $3.1 million, based upon the exchange rate at December 31, 2021) per product and application are due upon completion of certain development milestones. As further consideration, the Company will pay VivaCell a 2.5% royalty on all net revenues of any drug developed from the transferred compounds. During the year ended December 31, 2020, the Company paid approximately $0.5 million related to the first milestone payments due to VivaCell for the Company’s completion of a Phase 1 clinical study for MS and SSc.
F-17
Concurrent with the execution of the IPTA, the Company signed a Research Agreement with VivaCell for an initial term of 5 years. Under the terms of the Research Agreement, VivaCell is providing research services under the Company’s direction for consideration of cost plus a standard mark-up. Thereafter, the agreement will renew for successive one-year terms and may be terminated by either party on the expiration of the original term or any renewal term by delivering written notice at least 90 days prior to expiration. During the years ended December 31, 2021 and 2020, the Company recorded $145,543 and $192,469, respectively, in research and development expense for services performed by VivaCell under the Research Agreement. As of December 31, 2021 and 2020, $9,375 and $14,876, respectively, are included in accrued expenses, for amounts due to VivaCell under the Research Agreement.
The Company performed a qualitative analysis to determine whether a variable interest in another entity represents a controlling financial interest in a variable interest entity. A controlling financial interest in a variable interest entity is characterized by having both the power to direct the most significant activities of the entity and the obligation to absorb losses or the right to receive benefits of the entity. Since EHP does not have voting control or other forms of control over the operations and decision making at VivaCell, the Company determined that it does not have a variable interest in VivaCell. This guidance requires on-going reassessments of variable interests based on changes in facts and circumstances. The Company continues to assess its variable interests and has determined that no significant changes have occurred as of April 29, 2022.
8. Balance Sheet Details
Other current assets consisted of the following:
December 31, | ||||||||
| 2021 | 2020 | |||||||
| Prepaid contracts, expenses and deferred costs | $ | 423,200 | $ | 644,876 | ||||
| Other | 66,617 | 49,569 | ||||||
| Total | $ | 489,817 | $ | 694,445 | ||||
Property and equipment consisted of the following:
December 31, | ||||||||
| 2021 | 2020 | |||||||
| Furniture and fixtures | $ | 61,177 | $ | 62,228 | ||||
| Manufacturing equipment | 12,470 | - | ||||||
| Office equipment | 19,480 | 19,480 | ||||||
| Leasehold improvements | 20,638 | 20,638 | ||||||
| Property and equipment, gross | 113,765 | 102,346 | ||||||
| Accumulated depreciation | (82,263 | ) | (67,277 | ) | ||||
| Property and equipment, net | $ | 31,502 | $ | 35,069 | ||||
Depreciation expense for the years ended December 31, 2021 and 2020 was $16,036 and $24,596, respectively.
Other noncurrent assets include upfront deposits paid to clinical research organizations or other contract research providers, which are expected to be retained by the service provider until at least 12 months beyond the balance sheet date or until the expected completion of services.
Accrued expenses are comprised of the following:
December 31, | ||||||||
| 2021 | 2020 | |||||||
| Research and development liabilities | $ | 225,019 | $ | 49,290 | ||||
| Clinical trial related liabilities | 1,331,233 | 941,993 | ||||||
| Accrued payroll liabilities | 764,941 | 1,180,751 | ||||||
| Related party liabilities | 12,593 | 14,876 | ||||||
| Other liabilities | 145,329 | 156,229 | ||||||
| Total | $ | 2,479,115 | $ | 2,343,139 | ||||
F-18
9. Defined Contribution Plan
Effective January 1, 2018, the Company adopted a defined contribution savings plan pursuant to Section 401(k) of the Internal Revenue Code. The plan is for the benefit of all qualifying employees and permits voluntary contributions by employees up to 100% of eligible compensation, subject to the Internal Revenue Service imposed maximum limits. The terms of the plan allow for discretionary employer contributions.
On January 1, 2020, the Company commenced a safe harbor contribution of 3% of each eligible employee’s gross earnings, subject to Internal Revenue Service limitations. Employer safe harbor contributions vest immediately.
10. Income Taxes
The Company has incurred net operating losses from inception through December 31, 2021. Therefore, no United States federal, state, or foreign income taxes are expected to be paid for 2021 or 2020 and no amounts payable have been recorded as of December 31, 2021 and 2020.
The Company’s loss before income taxes for the years ended December 31, 2021 and 2020, respectively, was generated in the following jurisdictions:
Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Domestic | $ | (13,142,022 | ) | $ | (9,968,009 | ) | ||
| Foreign | (3,326,710 | ) | (2,456,613 | ) | ||||
| Worldwide | $ | (16,468,732 | ) | $ | (12,424,622 | ) | ||
A reconciliation of income tax expense (benefit) to the amount computed by applying the statutory federal income tax rate to the loss from operations is summarized for the years ended December 31, 2021 and 2020, respectively, as follows:
| Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Expected income tax benefit at U.S. federal statutory rate | $ | (3,458,434 | ) | $ | (2,609,171 | ) | ||
| State income tax benefit, net of federal benefit | (688,817 | ) | (597,772 | ) | ||||
| Tax effect of: | ||||||||
| Change in valuation allowance | 3,372,961 | 2,233,775 | ||||||
| Uncertain tax positions | 787,030 | 537,386 | ||||||
| Australian tax incentive | 459,470 | 149,752 | ||||||
| Stock-based compensation | 160,687 | 209,196 | ||||||
| Foreign rate differential | (111,258 | ) | (132,784 | ) | ||||
| Return to provision true-up | (269,229 | ) | 89,209 | |||||
| Other | (252,410 | ) | 120,409 | |||||
| Provision for income taxes | $ | - | $ | - | ||||
F-19
The Company’s net deferred tax assets are comprised of the following as of December 31, 2021 and 2020, respectively:
| As of December 31, | ||||||||
| 2021 | 2020 | |||||||
| Deferred tax assets: | ||||||||
| Tax loss carryforwards | $ | 8,826,831 | $ | 6,217,722 | ||||
| Stock-based compensation | 1,387,093 | 759,549 | ||||||
| Accrued expenses | 154,107 | 37,411 | ||||||
| Intangible assets | 104,804 | 118,010 | ||||||
| Other | 7,373 | 6,192 | ||||||
| Gross deferred tax assets | 10,480,208 | 7,138,884 | ||||||
| Less: valuation allowance | (10,480,208 | ) | (7,138,884 | ) | ||||
| Total deferred tax assets | - | - | ||||||
| Deferred tax liabilities: | ||||||||
| - | - | |||||||
| Net deferred tax assets | $ | - | $ | - | ||||
At December 31, 2021, the Company had federal, state, and foreign net operating loss (NOL) carryforwards of approximately $38.5 million, $40 million and $2.6 million, respectively. The federal and certain state loss carryforwards generated in 2018 onwards of $36.5 million and $1.7 million, respectively, will carry forward indefinitely and can be used to offset up to 80% of future annual taxable income. Federal loss carryforwards generated prior to 2018 begin expiring in 2037, unless previously utilized. State loss carryforwards begin expiring in 2037, unless previously utilized, while the Company’s foreign loss carryforwards do not expire.
Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Management assesses the available positive and negative evidence to estimate if sufficient future taxable income will be generated to use existing deferred tax assets. Based on the weight of evidence, including a history of operating losses, management has determined that it is more likely than not that the Company’s net deferred tax assets will not be realized. Accordingly, a valuation allowance of $10.5 million and $7.1 million has been established by the Company to fully offset these net deferred tax assets as of December 31, 2021 and 2020, respectively. The valuation allowance increased by $3.3 million during the year ended December 31, 2021.
Future utilization of the Company’s NOL carryforwards to offset taxable income may be subject to a substantial annual limitation as a result of ownership changes that may have occurred or that could occur in the future pursuant to Internal Revenue Code Sections 382 and 383. These ownership changes may limit the amount of NOL carryforwards that can be utilized to offset future taxable income and tax, respectively. In general, an ownership change, as defined by the tax code, results from a transaction or series of transactions over a three-year period resulting in an ownership change of more than 50 percent of the outstanding stock of a company by certain stockholders or public groups. The Company has not completed an analysis regarding the limitation of NOL carryforwards.
The Company applies the two-step approach to recognizing and measuring uncertain tax positions. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates it is more likely than not that the position will be sustained on audit, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount, which is more than 50% likely of being realized upon ultimate settlement. Income tax positions must meet a more likely than not recognition threshold at the effective date to be recognized upon the adoption of ASC 740 and in subsequent periods. This interpretation also provides guidance on measurement, derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition. The following table summarizes the activity related to the Company’s gross unrecognized tax benefits for the years ended December 31, 2021 and 2020:
| Years Ended December 31, | ||||||||
| 2021 | 2020 | |||||||
| Balance at beginning of year | $ | 2,378,723 | $ | 1,698,488 | ||||
| Increases/(decreases) related to prior year tax positions | 148,360 | (55,939 | ) | |||||
| Increases related to current year tax positions | 847,881 | 736,174 | ||||||
| Balance at end of year | $ | 3,374,964 | $ | 2,378,723 | ||||
F-20
At December 31, 2021 and 2020, the amount of unrecognized tax benefits that would affect the effective tax rate was $0. The Company’s policy is to recognize interest and penalties related to uncertain tax positions in income tax expense. The Company has not accrued any interest or penalties related to uncertain tax positions since inception. The Company does not anticipate that there will be a significant change in the amount of unrecognized tax benefits over the next twelve months.
The Company is subject to tax in the U.S. federal jurisdiction as well as various state and foreign jurisdictions. The Company’s federal tax returns from 2018 onwards, and state and foreign income tax returns from 2017 onwards are subject to examination by tax authorities; however, no such examinations have taken place.
CARES Act & Consolidated Appropriations Act
On March 27, 2020, the CARES Act was enacted in response to the COVID-19 pandemic. The CARES Act, among other things, permits NOL carryovers and carrybacks to offset 100% of taxable income for taxable years beginning before 2021. In addition, the CARES Act allows NOLs incurred in 2018, 2019, and 2020 to be carried back to each of the five preceding taxable years to generate a refund of previously paid income taxes. Due to the Company’s history of net operating losses, the CARES Act is not expected to have a material impact on the Company’s financial statements.
In accordance with the Consolidated Appropriations Act, 2021 enacted on December 27, 2020, certain qualified expenses used with the funds of the PPP Loan are fully deductible for Federal income tax purposes. Forgiveness of the PPP loan is not considered taxable for Federal and certain state income tax purposes.
California NOL Suspension
California Assembly Bill No. 85 (“AB 85”) (as updated) suspended California net operating loss utilization and imposed a cap on the amount of business incentive tax credits companies can utilize, effective for tax years 2020 and 2021. There was no material impact from the provisions of AB 85 in 2021.
11. Subsequent Events
No subsequent events have occurred subsequent to the balance sheet date through April 29, 2022.
F-21
Item 8. Exhibits
| † | Filed herewith. |
32
| # | Filed as an exhibit to the Emerald Health Pharmaceuticals Inc. Regulation A Offering Statement on Form 1-A filed with the United States Securities and Exchange Commission (Commission) (Commission File No. 024-10810) on January 29, 2018 and incorporated herein by reference. |
| + | Filed as an exhibit to the Emerald Health Pharmaceuticals Inc. Regulation A Offering Statement on Form 1-A filed with the United States Securities and Exchange Commission (Commission) (Commission File No. 024-10810) on March 5, 2018, and incorporated herein by reference. |
| * | Filed as an exhibit to the Emerald Health Pharmaceuticals Inc. Post-Qualification Offering Circular Amendment No. 1 filed with the United States Securities and Exchange Commission (Commission) (Commission File No. 024-10810) on March 29, 2019 and incorporated herein by reference. |
| α | Filed as an exhibit to the Emerald Health Pharmaceuticals Inc. Semiannual Report on Form 1-SA filed with the United States Securities and Exchange Commission (Commission) (Commission File No. 024-10810) on September 30, 2019 and incorporated herein by reference. |
| ^ | Filed as an exhibit to the Emerald Health Pharmaceuticals Inc. Current Report on Form 1-U filed with the United States Securities and Exchange Commission on November 21, 2019 and incorporated herein by reference. |
| ± | Filed as an exhibit to the Emerald Health Pharmaceuticals Inc. Annual Report on Form 1-K filed with the SEC on April 28, 2020 and incorporated herein by reference. |
| π | Filed as an exhibit to the Emerald Health Pharmaceuticals Inc. Regulation A Post-Qualification Offering Circular Amendment on Form 1-A filed with the SEC (Commission File No. 024-10810) on July 10, 2020 and incorporated herein by reference. |
| β | Filed as an exhibit to the Emerald Health Pharmaceuticals Inc. Semiannual Report on Form 1-SA filed with the SEC on September 28, 2020 and incorporated herein by reference. |
| € | Filed as an exhibit to the Emerald Health Pharmaceuticals Inc. Regulation A Post-Qualification Offering Circular Amendment on Form 1-A filed with the SEC (Commission File No. 024-10810) on November 6, 2020 and incorporated herein by reference.
|
| § | Filed as an exhibit to the Emerald Health Pharmaceuticals Inc. Annual Report on Form 1-K filed with the SEC on April 30, 2021 and incorporated herein by reference. |
| £ | Filed as an exhibit to the Emerald Health Pharmaceuticals Inc. Regulation A Offering Statement on Form 1-A filed with the SEC on July 26, 2021 and incorporated herein by reference. |
| ‡ | Portions of this exhibit containing confidential information have been omitted pursuant to a request for confidential treatment filed with the SEC pursuant to Rule 406 under the Securities Act. Confidential information has been omitted from the exhibit in places marked “[*****]” and has been filed separately with the SEC. |
33
SIGNATURES
Pursuant to the requirements of Regulation A, the issuer had duly caused this Annual Report on Form 1-K to be signed on its behalf by the undersigned, thereunto duly authorized on April 29, 2022.
| Emerald Health Pharmaceuticals Inc. | |||
| By: | /s/ James M. DeMesa | ||
| Name: | James M. DeMesa, M.D. | ||
| Title: | President & Chief Executive Officer | ||
Pursuant to the requirements of Regulation A, this report has been signed below by the following persons on behalf of the issuer and in the capacities and on the dates indicated.
| /s/ James M. DeMesa | Date: April 29, 2022 | |||
Name: James M. DeMesa, M.D. Title: President & Chief Executive Officer | ||||
| /s/ Lisa Sanford | Date: April 29, 2022 | |||
Name: Lisa Sanford Title: Chief Financial Officer | ||||
| /s/ James L. Heppell | Date: April 29, 2022 | |||
| Name: James L. Heppell, LLB | ||||
| Title: Director and Chairman of the Board | ||||
| /s/ Gaetano A. Morello | Date: April 29, 2022 | |||
| Name: Gaetano A. Morello, ND | ||||
| Title: Director | ||||
| /s/ Punit S. Dhillon | Date: April 29, 2022 | |||
| Name: Punit S. Dhillon | ||||
| Title: Director |
34