UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 1-K
ANNUAL REPORT
ANNUAL REPORT PURSUANT TO REGULATION A OF THE SECURITIES ACT OF 1933
For the fiscal year ended December 31, 2018
| NEURMEDIX, INC. |
| (Exact name of registrant as specified in its charter) |
| Delaware | 47-2860346 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
11601 Wilshire Blvd, Suite 1100 Los Angeles, California | 90025 | |
| (Address of principal executive offices) | (Zip Code) |
(310) 444-4321
Registrant’s telephone number, including area code
Part II.
USE OF MARKET AND INDUSTRY DATA
This Annual Report on Form 1-K includes market and industry data that we have obtained from third-party sources, including industry publications, as well as industry data prepared by our management on the basis of its knowledge of and experience in the industries in which we operate (including our management’s estimates and assumptions relating to such industries based on that knowledge). Management has developed its knowledge of such industries through its experience and participation in these industries. While our management believes the third-party sources referred to in this Annual Report are reliable, neither we nor our management have independently verified any of the data from such sources referred to in this Annual Report or ascertained the underlying economic assumptions relied upon by such sources. Furthermore, internally prepared and third-party market prospective information, in particular, are estimates only and there will usually be differences between the prospective and actual results, because events and circumstances frequently do not occur as expected, and those differences may be material. Also, references in this Annual Report to any publications, reports, surveys or articles prepared by third parties should not be construed as depicting the complete findings of the entire publication, report, survey or article. The information in any such publication, report, survey or article is not incorporated by reference in this Annual Report.
Forward Looking Statements
This Annual Report on Form 1-K (the “Annual Report”) of NeurMedix, Inc., a Delaware corporation (“NeurMedix,” the “Company,” “we,” “us”or “our”), contains certain forward-looking statements that are subject to various risks and uncertainties. Forward-looking statements are generally identifiable by use of forward-looking terminology such as “may,” “will,” “should,” “potential,” “intend,” “expect,” “outlook,” “seek,” “anticipate,” “estimate,” “approximately,” “believe,” “could,” “project,” “predict,” or other similar words or expressions. Forward-looking statements are based on certain assumptions, discuss future expectations, describe future plans and strategies, contain financial and operating projections or state other forward-looking information. Our ability to predict results or the actual effect of future events, actions, plans or strategies is inherently uncertain. Although we believe that the expectations reflected in our forward-looking statements are based on reasonable assumptions, our actual results and performance could differ materially from those set forth or anticipated in our forward-looking statements. Factors that could have a material adverse effect on our forward-looking statements and upon our business, results of operations, financial condition, funds derived from operations, cash flows, liquidity and prospects include, but are not limited to, the factors referenced in the NeurMedix Offering Circular filed pursuant to Regulation A, dated July 19, 2018 (the “Offering Circular”).
Readers are cautioned not to place undue reliance on any of these forward-looking statements, which reflect our views as of the date of this report. The matters summarized below and elsewhere in this report could cause our actual results and performance to differ materially from those set forth or anticipated in forward-looking statements. Accordingly, we cannot guarantee future results or performance. Furthermore, except as required by law, we are under no duty to, and we do not intend to, update any of our forward-looking statements after the date of this report, whether as a result of new information, future events or otherwise.
| 2 |
Use of Market and Industry Data
This Annual Report includes market and industry data that we have obtained from third-party sources, including industry publications, as well as industry data prepared by our management on the basis of its knowledge of and experience in the industry in which we operate (including our management’s estimates and assumptions relating to such industries based on that knowledge). While our management believes the third-party sources referred to in this Annual Report are reliable, neither we nor our management has independently verified any of the data from such sources referred to in this Annual Report or ascertained the underlying economic assumptions relied upon by such sources. Furthermore, internally prepared and third-party market prospective information, in particular, are estimates only, and there will usually be differences between the prospective and actual results because events and circumstances frequently do not occur as expected, and those differences may be material. Also, references in this Annual Report any publications, reports, surveys or articles prepared by third parties should not be construed as depicting the complete findings of the entire publication, report, survey or article. The information in any such publication, report, survey or article is not incorporated by reference in this Annual Report.
Item 1. Business
NeurMedix, Inc., is a “virtual” biotechnology company based in San Diego, California, with experienced leadership, near-term clinical data readouts, a therapeutic and development focus on unmet medical needs in neurological diseases, and a capital efficient business model. We are developing compelling product candidates with novel, first-in-class mechanism of action (“MOA”), human clinical safety database, clinical activity demonstrated in metabolic disease. We are developing multiple therapeutic product opportunities in neurological diseases and have retained all global development and marketing rights. We are currently conducting an offering pursuant to regulation a (Tier 2) to raise up to $49,999,998 in gross proceeds (the “Offering”).
Our product candidate, NE3107, has demonstrated activity in a wide variety of pre-clinical inflammatory disease models, and completed three Phase I, two Phase I/II, and one Phase II clinical studies. NeurMedix has an open IND with the FDA DMEP and intends to initiate studies in cognitively impaired type 2 diabetes subjects, and is positioned to file INDs with the FDA DNP for clinical trials for the treatment of patients with Alzheimer’s disease, Parkinson’s disease, migraine, and post-operative cognitive dysfunction (“POCD”). Further studies are contemplated for amyotrophic lateral sclerosis (also known as “ALS” or “Lou Gehrig’s Disease”), Huntington’s disease, Alzheimer’s disease, and encephalitis. NE3107 is our most developed drug candidate, and its application to various diseases, we believe is a first-in-class therapeutic for neurological disease targeting disorders with significant unmet medical needs. Our focus is on diseases with tremendous unmet medical needs in order to expedite Food and Drug Administration (“FDA”) approvals and commercialization, minimize capital requirements and optimize shareholder value.
A large body of scientific literature has identified neuroinflammation as a critical underlying factor in the initiation and perpetuation of neurodegeneration. The literature also indicates that extracellular signaling-regulated kinase (“ERK”) and nuclear factor kappa-light-chain-enhancer of activated B cells’ (“NF-kB”) hyperactivation (a prolonged state of activation generally associated with aberrant signaling pathological outcome) causes the inflammation. NE3107 binds to ERK and inhibits hyperactivation of inflammatory signaling pathways. The general mechanisms of neuroinflammation and neurodegeneration are common to all neuroinflammatory diseases, although inflammatory signals can arise, often through poorly understood mechanisms, to create unique perturbations to specific areas of the brain, which give rise to the variety of neurological disorders. Moreover, general mechanisms of inflammation and inflammatory signaling pathways are similar in the periphery and the central nervous system (“CNS”). Thus, as inflammation in general is important to neurological disease in general, NE3107’s anti-inflammatory activity in models of systemic inflammatory disease is highly relevant to potential activity against neuroinflammation, and provides mechanistic rationale in support of neurological indications. Glial cell activation and recruitment of peripheral inflammatory cells to sites of neuroinflammation are also general features of neuroinflammation, and are greatly influenced by ERK and NF-kB hyperactivation, and thus potentially susceptible to the modulating activity of NE3107. Both ERK and NF-kB have important homeostatic functions, so that non-specific inhibition of ERK and NF-kB can result in systemic and CNS toxicities. All evidence to date, which includes safety data from animal and human studies and biochemical analysis of animal tissues, indicates NE3107 acts selectively against ERK and NF-kB hyperactivation, and does not impede homeostatic ERK and NF-kB activity. Importantly, in contrast to the majority of approved anti-inflammatory compounds there was no evidence of NE3107 immunosuppressive activity in multiplein vitro and animal studies.
| 3 |
Over the past twenty years, there has been a vast amount of scientific research on the association of type 2 diabetes, insulin resistance, neuroinflammation, and Alzheimer’s disease. A large body of evidence now exists that shows how inflammation causes insulin resistance in the brain. Brain insulin resistance has deleterious effects on many aspects of brain energy homeostasis and neuron function, which include the formation of neurofibrillary tangles, increased oxidative stress, decreased cognition, and increased neurodegeneration. Importantly, type 2 diabetes, i.e. systemic insulin resistance, hyperinsulinemia, and chronic hyperglycemia, also contribute to neuroinflammation and altered glucose homeostasis in the brain through alterations in blood-brain barrier permeability to glucose and inflammatory cells. In obese, inflamed human subjects (the profile of approximately 50% of type 2 diabetics), NE3107 reduced insulin resistance, and improved post-prandial glucose disposal. In this target population of type 2 diabetes, NE3107 also decreased C-reactive protein (“CRP”), a measure of systemic inflammation, and the high variation in glucose disposal parameters that is associated with metabolic dysregulation. In glucose-intolerant pre-diabetics with evidence of systemic inflammation, NE3107 improved glucose disposal parameters, so that the subjects were no longer pre-diabetic while receiving NE3107 treatment. NE3107 activity against type 2 diabetes and insulin resistance has also been observe in animal models, which show that the drug’s effects against insulin resistance and improvements in glucose homeostasis are mediated by anti-inflammatory activity against ERK and NF-kB hyperactivation. NE3107’s activity against type 2 diabetes/ insulin resistance strongly suggests the drug may have use against Alzheimer’s disease and neuroinflammatory diseases in general.
In addition to clinical evaluation, NE3107 has been evaluated in a wide variety of models of systemic inflammation and neuroinflammatory disease. Because ERK and NF-kB signaling mechanisms are generally similar between systemic inflammatory and neuroinflammatory conditions, the anti-inflammatory activity observed in these studies provides supporting rationale for treatment of neuroinflammatory disease. Additionally, the observation of NE3107 anti-inflammatory activityin vitro and in a wide variety of tissue types (hepatic, adipose, muscle, and lymphatic), supports the notion that NE3107’s activity does not have a tissue specific limitation.
NE3107 has been evaluated in 12 models of systemic inflammatory disease in peer-reviewed publications. Major conclusions from the studies are followed by a brief summary.
NE3107 (HE3286) significantly reduced disease, joint destruction and inflammation in the mouse collagen-induced arthritis model
Publication Details: Auci, et al., 2007, A new orally bioavailable synthetic androstene inhibits collagen-induced arthritis in the mouse. Androstene hormones as regulators of regulatory T cells. Ann N Y Acad Sci 1110: 630. This study was initiated in August, 2006. This study was performed under the direction of Prof. Helena Offner, Department of Neurology, Oregon Health and Science University, Portland, Oregon, USA. Arthritis was induced in DBA/1Lac/J mice by immunization with an emulsion of bovine type II collagen and Mycobacterium tuberculosis, and monitored for disease progression. Compared to vehicle control mice, treatment with 25-50 mg HE3286 (beginning at disease onset) resulted in a significant decrease in disease score (evident by day 21 post-onset, p < 0.05).
| 4 |
NE3107 (HE3286) significantly decreased disease, inflammation and joint destruction in the mouse collagen-induced arthritis model
Publication Details: Offner, et al., 2009, An orally Bioavailable synthetic analog of an active dehydroepiandrostene metabolite reduces established disease in rodent models of rheumatoid arthritis. J Pharmacol Exp Ther 329: 1100. This study was initiated in September, 2006. Arthritis was induced in DBA/1Lac/J mice by immunization with an emulsion of bovine type II collagen and Mycobacterium tuberculosis, and monitored for disease progression. Compared to vehicle control mice, treatment with 25-50 mg HE3286 (beginning at disease onset) resulted in a significant decrease in disease score (evident by day 21 post-onset, p < 0.001). HE3286 reduced joint inflammation, synovial proliferation and erosion as evaluated by histology. Treatment with HE3286 compared to vehicle controls was associated with a doubling of T regulatory cells in the spleen. Mice treated with HE3286 showed dramatic decreases in the splenocyte culture production of proinflammatory cytokines tumor necrosis factor alpha, Interleukins-6, -17 and -23 compared to vehicle control mice.
NE3107 (HE3286) significantly decreased inflammatory mediators and reduced disease in the mouse collagen antibody-induced arthritis model
Publication Details: Offner, et al., 2009, An orally Bioavailable synthetic analog of an active dehydroepiandrostene metabolite reduces established disease in rodent models of rheumatoid arthritis. J Pharmacol Exp Ther 329: 1100. This study was initiated in March, 2007, and performed under the direction of Dr. Gary Firestein, Division of Rheumatology, Allergy and Immunology, University of California, San Diego. Male DBA/1Lac/J mice were injected with a bovine collagen type II antibody cocktail and treated with intraperitoneal lipopolysaccharide two days later. The next day, daily treatment with vehicle or HE3286 was initiated. Arthritic severity of each joint was scored, and extra articular tissue was collected for inflammatory mediator RNA analysis. HE3286 treatment significantly reduced the arthritis score in five days (p < 0.05) compared to vehicle, and with 80 mg/kg HE3286, arthritis was virtually absent throughout the experiment. HE3286 showed a dose-dependent decrease in joint interleukin-6 and matrix metalloproteinase-3 inflammatory mediators, compared to vehicle controls.
NE3107 (HE3286) significantly reduced nuclear factor kappa B (NFkB) activation by lipopolysaccharide in mice
Publication Details: Offner, et al., 2009, An orally Bioavailable synthetic analog of an active dehydroepiandrostene metabolite reduces established disease in rodent models of rheumatoid arthritis. J Pharmacol Exp Ther 329: 1100. This study was initiated in September, 2006 and performed under the direction of Dominick Auci, Ph.D. at Hollis- Eden Pharmaceuticals, San Diego, CA. ICR mice were treated with HE3286 or vehicle, followed by intraperitoneal challenge with lipopolysaccharide. Splenocytes were lysed and assayed for phospho- nuclear factor kappa B (NFkB) p65 using an enzyme-linked immunosorbent assay. NFkB activation was reduced by treatment with HE3286 compared to vehicle control (p < 0.001).
NE3107 (HE3286) significantly decreased disease in the Wistar rat model of ulcerative colitis
Publication Details: Ahlem, et al., 2009 A novel synthetic steroid as an oral treatment for autoimmune disease, Ann N Y Ac ad Sci 1173: 781. This study was performed under the direction of Prof. Ferdinando Nicoletti at the Department of Biomedical Sciences, School of Medicine, University of Catania, Italy. The study was initiated in September, 2007. Rats were treated with 2,4-dinitrobenzene sulfonic acid to induce colitis and mucosal necrosis in the colons of all challenged animals. Compared to the vehicle control, animals treated with HE3286 (10 mg/kg, but not 30 mg/kg) had significantly reduced mucosal damage (p = 0.023), and were comparable to the positive control, sulfasalazine (p = 0.037 compared to vehicle control). HE3286 (10 mg/kg)-treated animals showed significant reductions in colon weight compared to the vehicle control (p = 0.005), whereas the positive control did not.
| 5 |
NE3107 (HE3286) decreased disease (statistical trend) in the SJL/J mouse model of experimental autoimmune encephalitis (EAE)
Publication Details: Ahlem, et al., 2009 A novel synthetic steroid as an oral treatment for autoimmune disease, Ann N Y Ac ad Sci 1173: 781. This study was performed under the direction of Prof. Ferdinando Nicoletti at the Department of Biomedical Sciences, School of Medicine, University of Catania, Italy. The study was initiated in November, 2007. Mice were immunized with an emulsion of proteolipid protein 139-151 and Mycobacterium tuberculosis H37RA. Vehicle control mice developed clinical signs of EAE (8 of 8 mice, average disease score 41.5, mortality 2 of 8). Mice treated with HE3286 had a statistical trend for attenuated disease (2 of 8 mice, average score 14.9 p = 0.071, no mortality). After cessation of treatment with HE3286, the treatment effect persisted compared to the vehicle control group.
NE3107 (HE3286) significantly decreased lung myeloperoxidase in a mouse lung injury model
Publication Details: Conrad, et al., 2010, HE3286, an oral synthetic steroid, treats lung inflammation in mice without immune suppression. J Inflammation 7:52. This study was initiated in March, 2006, and performed under the direction of Dr. Douglas Conrad, VA San Diego Healthcare System, San Diego, CA. Mice were treated with HE3286 or vehicle via oral gavage prior to intratrachael lipopolysaccharide challenge. After 48 hours, inflammatory mediators in bronchoaveolar lavage samples were measured by enzyme-linked immunosorbent assay. Treatment with HE3286 significantly decreased myeloperoxidase compared to vehicle control (p = 0.0249).
NE3107 (HE3286) significantly decreased disease in a mouse carrageenan-induced pleurisy model
Publication Details: Conrad, et al., 2010, HE3286, an oral synthetic steroid, treats lung inflammation in mice without immune suppression. J Inflammation 7:52. This study was initiated in October, 2004, and performed under the direction of Prof. Ferdinando Nicoletti at the Department of Biomedical Sciences, School of Medicine, University of Catania, Italy. CD1 mice were pretreated with hE3286 or vehicle, and subsequently challenged with carrageenan injected into the pleural cavity. After 4 hours, the pleural exudate volumes and numbers of neutrophils were measured. HE3286 (40 mg/kg) significantly decreased the pleural neutrophils compared to vehicle control (p < 0.05), and was comparable to the positive control (polyclonal anti-tumor necrosis factor alpha).
NE3107 (HE3286) significantly decreased spontaneous type 1 autoimmune diabetes in a non-obese diabetic (NOD) mouse model
Publication Details: Kosiewicz, et al., 2011, HE3286, an orally bioavailable synthetic analogue of an active DHEA metabolite suppresses spontaneous autoimmune diabetes in the NOD mouse, Eur J Pharmacol 658: 257. This study was initiated in April 2008, and performed under the direction of Prof. Ferdinando Nicoletti at the Department of Biomedical Sciences, School of Medicine, University of Catania, Italy. In this early treatment model, NOD mice were treated with HE3286 or vehicle from 15 to 25 weeks of age. Glucose levels were monitored weekly. HE3286 treated mice had a significantly lower incidence at week 25 (p = 0.007) compared to vehicle controls.
| 6 |
NE3107 (HE3286) significantly decreased type 1 autoimmune diabetes at onset in a NOD mouse model
Publication Details: Kosiewicz, et al., 2011, HE3286, an orally bioavailable synthetic analogue of an active DHEA metabolite suppresses spontaneous autoimmune diabetes in the non-obese diabetic (“NOD”) mouse, Eur J Pharmacol 658: 257. One arm of this study was initiated in January 2007, and performed under the direction of Prof. Ferdinando Nicoletti at the Department of Biomedical Sciences, School of Medicine, University of Catania, Italy. A second arm was initiated in September, 2008, and performed under the direction of Prof. Michelle Kosiewicz, Department of Microbiology and Immunology, Univ. Louisville, Louisville, KY. In this late treatment model, NOD mice were treated with HE3286 or vehicle, daily for 6 weeks, starting two days after the first incidence of autoimmune diabetes in the colony. Glucose levels were monitored weekly. Late treatment with HE3286 significantly decreased the incidence of autoimmune diabetes (p = 0.005) compared to vehicle control. Histological analysis revealed that HE3286 markedly decreased the incidence of insulitis (p = 0.02) compared to vehicle control. Lymphocytes from the pancreatic lymphnodes and spleen were isolated and assayed for T cell phenotypes. HE3286 treatment significantly decreased the destructive Th1 and Th17 response. Interferon-gamma production was decreased in spleen (p = 0.03) and pancreatic lymphocytes (p = 0.08) compared to vehicle control, IL-17 production was decreased in spleen and pancreatic lymphocytes (p = 0.001 each), and serum IL-17 was significantly decreased (p = 0.006) compared to vehicle controls.
NE3107 (HE3286) significantly decreased type 2 diabetes in mouse models of obesity-induced inflammation and diabetes, and reduces inflammatory mediators
Publication Details: Wang, et al., 2010, Amelioration of glucose intolerance by the synthetic androstene HE3286: Link to inflammatory pathways. J Pharmacol Exp Ther 333: 70. This study was initiated in March, 2007, and was performed under the direction of Dr. Jaime Flores-Riveros, Hollis-Eden Pharmaceuticals, Inc., San Diego, CA. Compared to vehicle control, HE3286 significantly suppressed development of hyperglycemia in 8 week old (p < 0.05) and 6 week old (p < 0.01)db/db mice. HE3286 significantly reduced glucose intolerance (glucose and insulin levels, p < 0.05 each compared to vehicle controls) in 6 week olddb/db mice. HE3286 reduced glucose intolerance in obese, insulin-resistant (diet-induced obesity) mice (p < 0.01) and in genetically obeseob/ob mice (p<0.01) compared to vehicle controls. These changes were accompanied by decreases in proinflammatory macrophage activation markers (p < 0.05), compared to vehicle controls. HE3286 decreased serum chemokines and hyperinsulemia inob/ob mice (p<0.05). HE3286 was shown to decrease the activation and nuclear localization of p65 nuclear factor kappa B (NFkB).
NE3107 (HE3286) significantly attenuated inflammation and insulin resistance in diabetic fatty rats
Publication Details: Lu, et al., 2010, A new antidiabetic compound attenuates inflammation and insulin resistance in Zucker diabetic fatty rats, Am J Physiol Endocrinol Metab 298: E1036. This study was initiated in May, 2007, and performed under the direction of Prof. Jerrold Olefsky, Department of Medicine, Univ. California, San Diego. In primary intraperitoneal mouse macrophages, HE3286 significantly decreased inflammatory pathways stimulated by lipopolysaccharide including activation of nuclear factor kappa B (NFkB) phospho-extracellular receptor kinase (p-ERK), and associated signal transduction pathways compared to vehicle. HE3286 also significantly decreased proinflammatory chemokines and cytokines compared to vehicle. These changes also significantly decreased proinflammatory macrophage chemotaxis. In treated Zucker diabetic fatty rats, HE3286 treatment significantly decreased glucose insulin, glycerol and free fatty acid levels compared to vehicle. Using euglycemic hyperinsulinemic clamp studies, HE3286 was shown to significantly improve glucose sensitivity. In these rats, HE3286 decreased inflammatory macrophage chemotaxis into fat depots and the levels of tissue cytokines tumor necrosis factor alpha and interleukin 1-beta.
| 7 |
NE3107 (HE3286) binds to extracellular kinase-1 and -2
Publication Details: Reading, et al., 2012, Molecular targets for 17a-ethynyl-5-androstene-3b,7b,17b-triol, an anti-inflammatory agent derived from the human metabolome. PLoS ONE 7(2) e32147. This study was initiated in July 2009, and performed under the direction of Steven White, Ph.D., Hollis-Eden Pharmaceuticals, Inc., San Diego, CA. Using extracts from mouse macrophage cells, proteins that bound to immobilized HE3286, but not to a closely related structure, were identified in experiments using stable isotope labeling in culture and mass spec/mass spec analyses. Amongst other proteins, HE3286 was shown to bind to proteins involved in inflammation signaling pathways: extracellular kinase 1 and 2 (ERK1 and ERK2).
The above studies were conducted in rodents, and therefore the translation of the activities to human subjects was not yet demonstrated.
The following two clinical studies were conducted to evaluate the translation of animal data to human subjects:
NE3107 (HE3286) significantly improved insulin sensitivity in obese, inflamed, impaired glucose tolerance human subjects.
Publication Details: Reading, et al., 2013, A synthetic anti-inflammatory sterol improves insulin sensitivity in insulin-resistant obese impaired glucose tolerance subjects. Obesity 21 E343. This Phase I study was initiated in October, 2007, and sponsored by Hollis-Eden Pharmaceuticals, Inc. HE3286 improved insulin sensitivity in insulin-resistant obese impaired glucose tolerance subjects, evidenced by hyperinsulinemic, euglycemic clamp studies (p = 0.009 vs. placebo). This Phase I clinical study demonstrated that animal studies showing HE3286 improved insulin sensitivity in obese, inflamed insulin resistance translated to humans.
NE3107 (HE3286) significantly decreased insulin resistance, increased the frequency of patients with decreased day 84 hemoglobin A1c in metformin-treated type 2 diabetic subjects, and decreased day 112 HbA1c in treatment-naïve diabetic subjects
Publication Details: Reading, et al., 2013, an anti-inflammatory sterol decreased obesity-related inflammation-induced insulin resistance and metabolic dysregulation. Mediators of Inflammation 2013 814989. This Phase II study was initiated in July, 2008, and was sponsored by Hollis-Eden Pharmaceuticals, Inc. In addition, HE3286 improved hematopoietic and metabolic regulation and 1,5-anhydroglucitol retention (a surrogate of postprandial glucose) in type 2 diabetic subjects compared to placebo. This study demonstrated that data obtained in diabetic animals translated to human subjects.
Studies in Animal Models of Neuroinflammatory Disease
In addition to models of systemic inflammatory disease, NE3107 has been evaluated in six models of neuroinflammation, four of which have been published in peer reviewed scientific journals. Two studies were sponsored by the Michael J. Fox Foundation for Parkinson’s Research (“MJFF”). These studies established that NE3107 freely penetrates the blood-brain-barrier (the drug enters the brain), and is active against neuroinflammation.
| 1. | Unpublished Report: Anti-Parkinson and anti-dyskinetic efficacy studies of HE3286 in a MPTP non-human primate model of Parkinson’s disease. This study was sponsored by MJFF, and conducted by Dr. Ingrid Philippens at the Biomedical Primate Research Centre, Rijswijk, The Netherlands. The study was initiated in January 2013. Summary of results: NE3107 decreased clinical signs of Parkinson’s disease, decreased susceptibility to LID, enhanced activity in combination with L-dopa, and decreased neurodegeneration. |
| 8 |
This study evaluated NE3107 in marmoset monkeys treated with the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (“MPTP”), which kills dopamine producing cells and initiates a prolonged neuroinflammatory condition, which produces a disease very similar to Parkinson’s disease in humans. NE3107 (30 mg/kg) was administered orally once daily for 14 weeks (to the end of the study) to one group of 6 monkeys beginning 11 days after completion of MPTP treatments. A second group of 6 MPTP treated monkeys was treated with oral amantadine, and a third group of 6, also MPTP treated, was given placebo. NE3107 activity against Parkinson’s symptoms was evident within the first 24 hours after starting therapy and continued to be observed until the study termination. NE3107 activity against parkinsonism was greater than amantadine in comparison to placebo. Three weeks after starting therapy, all monkeys received oral L-dopa to induce dyskinesia (“LID”). LID was less evident in NE3107 treated monkeys compared to amantadine and placebo treated animals. The animals were sacrificed at the end of the study so that brain tissue could be evaluated for neurodegeneration. NE3107 treated animals had approximately twice as many tyrosine hydroxylase expressing neurons in the dopamine producing region of the brain (substantia nigra) compared to placebo (p=0.0108) and more surviving neurons than amantadine, but the difference was not as great. The results of this study showed that NE3107’s anti-inflammatory activity was effective in reducing clinical signs of Parkinson’s disease, and NE3107 also decreased susceptibility to LID. NE3107 treated animals also had less clinical signs of disease when given L-dopa, compared to the combination of amantadine plus L-dopa, or L-dopa plus placebo. As disease progression is linked to neurodegeneration, the observed decrease in neurodegeneration in NE3107 treated animals suggests NE3107 may slow Parkinson’s disease progression in humans. Overall the results suggest NE3107 may have pro-motoric activity in humans with Parkinson’s, and that NE3107 may decrease LID susceptibility and enhance L-dopa activity and slow disease progression. Some of the early results from this study were presented in a poster by Ingrid Philippens at the Society for Neuroscience meeting in San Diego, November 2013.
| 2. | Publication: Nicoletti, F., et al., 17alpha-Ethynyl-androst-5-ene-3beta,7beta,17beta-triol (HE3286) Is Neuroprotective and Reduces Motor Impairment and Neuroinflammation in a Murine MPTP Model of Parkinson's Disease. Parkinsons Dis, 2012. 2012: p. 969418. This study was sponsored by MJFF and conducted in 2011. Evaluation of NE3107 blood-brain-barrier permeability (“BBB”) was conducted by MPI Research (Mattawan, MI and MicroConstants, San Diego, CA); the MPTP model was conducted by Dr. Ferdinando Nicoletti at Catania University, Catania, IT. Summary of results: NE3107 exhibited pro-motoric activity similar to L-dopa, decreased expression of inflammatory mediators in the brain, and decreased neurodegeneration. |
The evaluation of BBB permeability showed that NE3107 freely penetrated the BBB, with no indication of an efflux mechanism. To evaluate NE3107 activity in this model of Parkinson’s disease, mice were briefly treated with MPTP to kill dopamine producing cells. Groups of mice were treated with oral NE3107 (40 mg/kg), placebo, and L-dopa in various combinations for 4 days, evaluated for drug effects on motor function, and then sacrificed to obtain brain tissue to measure inflammatory mediators and neurodegeneration. MPTP treatment significantly decreased motor activity, and NE3107 restored motor activity to the same extent as treatment with L-dopa. The combination of NE3107 and L-dopa was not more effective than either NE3107 or L-dopa alone. NE3107 significantly decreased expression of inflammatory mediators, interleukin 1b (IL-1b, decreased 33%), tumor necrosis factor alpha (TNF-a, decreased 40%), and inducible nitric oxide (iNOS, decreased 20%) compared to placebo. NE3107 also significantly increased surviving hydroxylase positive neurons by 17%. While the MPTP model of Parkinson’s in mice is not considered as robust in predicting activity in humans as the primate model above, overall, the results of this study show that NE3107 can enter the brain at pharmacologically relevant concentrations and exert activity consistent with results in the monkey MPTP model. The results of the moue model suggest NE3107 may be useful to treat parkinsonism in humans and slow the progression of Parkinson’s disease.
| 9 |
| 3. | Publication: Lambert WS, C.B., Formichella CR, Sappington RM, Ahlem C, Calkins DJ, Oral Delivery of a Synthetic Sterol Reduces Axonopathy and Inflammation in a Rodent Model of Glaucoma. Frontiers in Neuroscience, 2017. 11(45). This study was sponsored by Harbor Therapeutics and conducted in 2013, with immunohistochemical analysis of tissues continuing through 2016. The study was performed in the laboratory of Dr. David Calkins at Vanderbilt University, Nashville, TN. Summary of results: NE3107 decreased neurodegeneration, decreased markers of pathogenic inflammation, but did not inhibit homeostatic NFkB activation. |
This study evaluated NE3107 neuroprotective and anti-inflammatory activity in a model of glaucoma that closely mimics the neurodegenerative and neuroinflammatory response to elevated intraocular pressure (“IOP”), which occurs in the retina and optic nerve of humans with glaucoma. IOP was elevated approximately 30% above normal in one eye of test animals by intraocular injection of polystyrene microbeads, while the other eye was injected with a salt solution, which does not affect IOP. Immediately after microbead injection, the rats were divided into three groups for four weeks of once daily oral treatment with 100 mg/kg NE3107, 20 mg/kg NE3107, or placebo. IOP was measured periodically. Two days before the end of the study, animals were anesthetized and both eyes of each were injected with cholera toxin subunit B (“CTB”). CTB is taken up by retinal ganglia cells and transported through the optic nerve axons to the superior colliculus region of the brain. The amount of CTB transported is a function of neuron health, such that stressed neurons in the early stages of neurodegeneration transport less CTB. This deficit in transport occurs before more overt signs of neurodegeneration can be observed. The animals were sacrificed to obtain tissues from the retina, optic nerve, and superior colliculus. Immunohistochemical methods were used to quantify CTB transport and inflammatory cells and markers of inflammation. NE3107 had no effect on IOP, but countered the inflammatory effects of elevated IOP in the retina and optic nerve. Elevated IOP decreased CTB axonal transport in placebo animals approximately 43% compared to fellow eyes that received saline injections. NE3107 at the highest dose almost completely prevented (94% of microbead saline control) the loss of axonal transport of CTB compare. The lower dose of NE3107 was approximately 85% as effective as the high dose. Compared to vehicle, NE3107 increased brain-derived neurotrophic factor (“BDNF”) in the optic nerve head and retina, while decreasing inflammatory and pathogenic proteins associated with elevated IOP compared to vehicle treatment. Especially important among the results was evidence of decreased microglial activation. Treatment with HE3286also increased nuclear localization of the transcription factor NFkB in collicular and retinal neurons (homeostatic function), but decreased NFkB in glial nuclei in the optic nerve head (anti-inflammatory). Overall, these results indicate that NE3107 decreases neurodegeneration, has anti-inflammatory activity in the CNS, and decreases microglial activation, widely recognized as a major driver of neuroinflammation and neurodegeneration. The results also show that NE3107 decreases the inflammatory activity of NFkB, but does not interfere with NFkB homeostatic function, which in neurons promotes neuron survival.
| 4. | Publication: Khan, R.S., et al., HE3286 reduces axonal loss and preserves retinal ganglion cell function in experimental optic neuritis. Invest Ophthalmol Vis Sci, 2014. 55(9): p. 5744-51. This study was sponsored by the University of Pennsylvania, and conducted in 2013 in the laboratory of Dr. Kenneth Shindler at the University of Pennsylvania. Summary of results: Compared to placebo, NE3107 decreased vision loss, attenuated inflammation, demyelination, optic nerve axonal loss, and retinal ganglion loss. |
| 10 |
This study evaluated NE3107 in a mouse model of optic neuritis, which in humans is an autoimmune neuroinflammatory condition of the optic nerve that can occur in the early stages of multiple sclerosis. Experimental autoimmune encephalomyelitis (“EAE”) was induced in groups of female mice with myelin oligodendrocyte glycoprotein peptide immunization. Mice were treated daily with intraperitoneal injection of placebo (saline) or 40 mg/kg NE3107 from day 1 (24 h post-immunization) and continued until sacrifice at day 40 post-immunization. Disease severity was monitored by EAE score and visual function (optokinetic responses, OKR). After 40 days the mice were sacrificed and retinal and optic nerve tissue was collected for evaluation of neuroinflammatory markers. NE3107 had a small but statistically significant effect to delay the onset of EAE symptoms compared to vehicle (day 12 vs. day 17, P<0.05). Compared to vehicle, NE3107 significantly improved optokinetic responses (visual function, P<0.001)), decreased inflammatory cell (Iba1 positive) infiltration (P<0.001) into the optic nerve and attendant demyelination (P<0.001). NE3107 also reduced neurodegeneration as assessed by preservation of neurofilament content of the optic nerve (P<0.01) and a small, but significant decrease in the loss of retinal cell ganglia from the temporal retinal quadrant (P<0.05). Overall, these study results are consistent with results in models of Parkinson’s and glaucoma, which demonstrated NE3107 activity against neuroinflammation and neurodegeneration.
| 5. | Unpublished Report: Anti-inflammatory treatment of drug resistant epilepsy with HE3286. This study was sponsored by the Epilepsy Foundation, and conducted in 2013 in the laboratory of Dr. Annamaria Vezzani at the Mario Negri Institute for Pharmacological research in Milan, Italy. Summary of results: in a mouse model of acute seizure, NE3107 significantly reduced the number of seizures and time in ictal activity (time in seizures). NE3107 did not reduce any aspect of seizure frequency or severity in an experimental (unvalidated) model of chronic seizure. |
In the acute seizure model, kainic acid was injected unilaterally into the left hippocampus (7ng/0.5µL) in freely moving C57BL6N mice. NE3107 (40mg/kg) or placebo were injected intraperitoneally 30 minutes before kainic acid. Acute electroencephalogram (“EEG”) seizures were measured in a 3 h period following kainate injection. The mice were not sacrificed after the acute seizure observations, and were preserved for evaluation in model of chronic seizures. NE3107 decreased the number of seizures and time in ictal activity approximately 50% (p<0.01), but did not affect time to seizure onset, average seizure duration, or time between seizures. Mice from the acute seizure phase of the study were used to observe potential NE3107 activity against chronic seizures, which develop approximately 2 months after hippocampal kainic acid injection. Two months after kainic acid injection, mice received twice daily injections of 20 mg/kg NE3107 or placebo for 9 days, during which EEG was continuously monitored. NE3107 had no anti-seizure activity in this model. Overall, the results in the acute seizure model may support additional nonclinical investigations to better understand if any translation to a meaningful clinical indication exists. The results from the unvalidated chronic seizure model suggest that positive data from other seizure models would be needed before contemplating clinical trials in epilepsy.
| 6. | Published Report: Ahlem, C., et al., HE3286: a novel synthetic steroid as an oral treatment for autoimmune disease. Ann N Y Acad Sci, 2009. 1173: p. 781-90. This study was sponsored by Hollis-Eden Pharmaceuticals, and was conducted in 2007-2008 by Dr. Ferdinando Nicoletti in his laboratory at the University of Catania, Catania, IT. Summary of results from experimental autoimmune encephalomyelitis (EAE) investigation: NE3107 in a dose responsive manner significantly delayed onset of disease and reduced clinical signs of EAE. NE3107’s activity was not diminished by co-administration of the estrogen receptor antagonist, ICI 182780. |
| 11 |
This study evaluated NE3107 against EAE in mice, which is an autoimmune disease resembling multiple sclerosis in humans. EAE was induced in mice by injection of myelin sheath peptide (myelin proteolipid, PLP). The autoimmune disease process was allowed to proceed until the appearance of clinical signs (approximately day 8-13 days) before initiating therapy with oral 40 or 4 mg/kg NE3107, or placebo for approximately 4 weeks. Another group of mice treated with 40 mg/kg NE3107 were also treated contemporaneously with injections of 10 mg/kg of an estrogen receptor antagonist, ICI 182780 to see if the estrogen receptor was involved in NE3107’s mechanism of action. Compared to vehicle, the 40 mg/kg dose of NE3107 delayed disease onset by 6 days (18.7 vs. 12.4), reduced incidence (100% vs. 70%) and decreased the cumulative disease score 13.1 vs. 16.9). The ICI compound had no effect on NE3107 activity or intrinsic activity against EAE when administered alone, but was effect as an estrogen receptor antagonist as indicated by decreased uterine weights compared to vehicle. Overall, the results from this study suggest NE3107 is active against inflammatory autoimmune processes in the CNS, and this activity is independent of estrogen receptor activity. This study was not designed to determine if the observed anti-inflammatory activity was directed at glial cells within the CNS or peripheral inflammatory cell infiltrating into the CNS.
Clinical Trials
With the acquisition of assets from Harbor Therapeutics (originally Hollis-Eden Pharmaceuticals), NeurMedix gained all rights, data, and full control of an open IND with the FDA DMEP. Clinical trials of NE3107 activity in pre-diabetic and type 2 diabetes subjects were performed under this IND by Hollis-Eden. NeurMedix has not filed an IND or conducted a clinical trial as of the date this offering was prepared. NE3107, previously designated HE3286, was investigated by Hollis-Eden Pharmaceuticals in a total of 176 subjects in six clinical studies from 2006 to 2010. The cumulative exposure to date totals 20.9 subject/patient years. Six clinical studies have been completed to date. Two Phase I studies and the human clinical safety data from all studies are directly applicable to neuroinflammation clinical trials in the FDA DNP. The two Phase I studies, Study HE3286-100, which measured NE3107 safety, pharmacokinetics, and drug metabolism, is applicable to any clinical development plan, including anticipated neuroinflammation clinical trials, and Study HE3286-102, which measured drug safety, pharmacokinetics, and peripheral anti-inflammatory activity, is also applicable to any clinical development plan, especially involving treatment of neuroinflammatory disease. Clinical trial HE3286-102 measured glucose disposal parameters, insulin resistance, and inflammation in a pre-diabetic population with evidence of systemic inflammation. Clinical trial HE3286-401 measured glucose disposal parameters in type 2 diabetes, with a subset of the patients (approximately 50%) having the obese inflamed phenotype in which NE3107 anti-inflammatory was shown to be active against features of type 2 diabetes. NeurMedix plans to submit data from these two trials to the FDA DNP in support of Alzheimer’s disease investigations, but at the time this document was prepared has not discussed this data or its potential impact on IND submissions or clinical trial design with the FDA DNP. All four of these studies were conducted by the previous owner of the technology, Hollis-Eden Pharmaceuticals. Dr. Christopher Reading and Clarence Ahlem were involved in study design and analysis and publication of results as employees of Hollis-Eden Pharmaceuticals. The drug has been well tolerated and exhibited an excellent safety profile in all HE3286/NE3107 trials conducted to date. Previous clinical experience with NE3107 is summarized below:
| 12 |
Study HE3286-100
Study HE3286-100 was conducted under IND #77,339 (Division of Metabolism and Endocrinology Products), was initiated in May 2007 and completed in July, 2007, and was sponsored by Hollis-Eden Pharmaceuticals, Inc. It was a phase I, single blind, placebo-controlled, dose escalation study of the safety, tolerance and pharmacokinetics of HE3286 when administered orally to healthy adult subjects. It was conducted by dgd (Diabetes and Glandular Disease) Research, Inc. San Antonio, TX, USA. Trial size was 34 subjects. Primary objectives of the study were safety and tolerance of HE3286 and determination of the pharmacokinetic profile of HE3286 during the fasted state and the potential food effect on drug exposure. SAFETY: There were 13 treatment emergent adverse events (AEs) categories observed in two or more subjects receiving HE3286. Eight of these were also observed in placebo subjects. The remaining five were grade 1 blood calcium increased, grade 1 blood creatinine increased, grade 1 blood potassium decreased, grade 1 blood sodium decreased, and grade 1 dizziness, all considered unrelated to study drug by the investigator. No deaths occurred in the study. The results indicated that HE3286 was safe and well tolerated. All safety data was submitted to the IND. Analysis of pharmacokinetics parameters indicated HE3286 was orally bioavailable in humans, with comparable pharmacokinetics in males and females. Drug exposure, approximately 10 ng.h/mL per administered milligram was dose proportional up to 100 mg (highest dose tested), was not decreased by food, or altered by dividing the dose for twice daily administration. The terminal drug half-life was approximately 6-8 h. An extensive analysis of drug metabolites in these subjects indicated that all drug metabolites had been previously observed in nonclinical safety studies in rats and dogs.
Study HE3286-102
Study HE3286-102 was conducted under IND #77,339 (Division of Metabolism and Endocrinology Products), was initiated in October 2007 and completed in October, 2009, and was sponsored by Hollis-Eden Pharmaceuticals, Inc. It was a Phase I, double-blind, placebo-controlled, dose ranging study of the safety, tolerance, pharmacokinetics and potential activity of HE3286 when administered orally to obese adult subjects for 28 days. An additional cohort of type 2 diabetic subjects was added by amendment for safety evaluation. The study was conducted at two sites: dgd Research, San Antonio, TX, USA, and Pennington Biomedical Research Center, Baton Rouge, LA, USA. Trial size was 35 obese healthy subjects and 13 type 2 diabetic subjects. The primary objectives were safety and tolerance of placebo and five dose levels of HE3286 (4 mg, 5 mg, 10 mg, 20 mg and 40 mg) when administered orally over 28 days, pharmacokinetic (“PK”) profiles of HE3286 at different dose levels after 28 days of dosing, potential activity of HE3286 to decrease insulin resistance, and safety and tolerance of HE3286 (10 mg) when administered orally over 28 days to subjects with type 2 diabetes mellitus. SAFETY: There were two SAEs in the obese healthy subjects receiving HE3286. Blood amylase was increased (grade 3, considered possibly related to study drug by the investigator) two weeks after the last 20 mg HE3286 dose, in a subject that had elevated amylase prior to treatment, and this SAE resolved by the next assessment. Lymphocyte count was decreased in a subject two weeks after the last 20 mg HE3286 dose (grade 3, considered possible related to study drug by the investigator) and this SAE resolved by the next assessment. Three type 2 diabetes subjects experienced SAEs. Lipase was elevated (grade 3 considered, unrelated to study drug by the investigator) in a subject four weeks after the last 10 mg HE3286 dose. One subject had elevated blood glucose increased (grade 3, considered unrelated to study drug by the investigator) after 10 mg HE3286 treatment; this subject had an episode of elevated blood glucose (grade 3) prior to the first HE3286 treatment. This subject also had glucose in urine (grade 3, considered unrelated to study drug) two weeks after the last 10 mg HE3286 dose; this subject had an episode of glucose in urine (grade 3) prior to the first HE3286 treatment. In addition to the above SAEs, there were 26 categories of treatment emergent AEs observed in two or more obese healthy subjects receiving HE3286. Twenty of these were also observed in the obese placebo subjects. The remaining six were Blood cholesterol increased in four subjects (grade 1 in two subjects in the 5 mg HE3286 cohort and two subjects in the 10 mg HE3286 cohort), Red blood cells urine in three subjects (grade 1 in two subjects and grade 2 in one subject in the 10 mg HE3286 cohort), Toothache in two subjects (grade 1 in two subjects in the 20 mg cohort), Dizziness in two subjects (one grade 1 in the 5 mg cohort and one grade 2 in the 10 mg cohort subjects), and Sinus congestion in two subjects (grade 1 in two subjects in the 5 mg cohort). In addition to the above SAEs, there were 14 categories of treatment emergent AEs observed in 2 or more T2DM subjects in this open label 10 mg HE3286 cohort. Twelve of these were also observed in the obese healthy placebo subjects in this study. The remaining two were Blood cholesterol increased in 2 subjects (grade 1), Glucose urine in four subjects (grade 1 in two subjects and grade two in one subject). HE3286 was considered to be safe and well tolerated when administered to obese healthy subjects or subjects with type 2 diabetes. There was no trend in AEs to differentiate between placebo- and HE3286-treated subjects, nor was there an increase in AEs with dose escalation. No patient died while on study. All safety data was submitted to the IND. Pharmacokinetics in this population did not differ from subjects in trial HE3286-100. HE3286 was shown to improve insulin sensitivity in obese subjects in this study (see publication details above).
| 13 |
Study HE3286-0103
Study HE3286-0103 was conducted under IND #77,339 (Division of Metabolism and Endocrinology Products), was initiated in July 2009 and completed in July, 2010, and was sponsored by Hollis-Eden Pharmaceuticals, Inc. It was a Phase I, open label study of the safety tolerance and assessment of HE3286 in Insulin sensitivity and hepatic glucose production when administered orally to obese insulin-resistant adult subjects for 28 days. The study was conducted at Pennington Biomedical Research Center, Baton Rouge, LA, USA. The trial size was 6 subjects. The primary objectives were safety and tolerance of 29 mg (10 mg BID) of HE3286 and the activity of HE3286 on insulin sensitivity and hepatic glucose production in obese insulin-resistant subjects. SAFETY: Blood triglycerides were increased in one subject (grade 3, assessed as unrelated to study drug by the investigator) in one subject four weeks after the last 20 mg HE3286 dose. There were 6 categories of treatment emergent adverse events occurring in two or more subjects receiving HE3286 (increases in blood bicarbonate, calcium, glucose, triglycerides and uric acid, and decreases in blood sodium and in haemoglobin). No deaths occurred in the study. Administration of HE3286 was found to be safe and well tolerated in the study. All safety data was submitted to the IND. Technical difficulties in two-stage hyperinsulinemic euglycemic isotopic glucose clamp studies did not allow firm conclusions regarding hepatic glucose production and insulin resistance.
Study HE3286-0201
Study HE3286-0201 was conducted under IND #79,315 (Division of Anesthesia, Analgesia and Rheumatology Products), was initiated in August 2008 and completed in June 2009, and was sponsored by Hollis-Eden Pharmaceuticals, Inc. It was a Phase I/II, open label, dose ranging study of the safety, tolerance, pharmacokinetics and potential activity of HE3286 when administered orally for 29 days to patients with rheumatoid arthritis on a stable dose of methotrexate. The study was conducted by Impact Clinical Trials, Beverly Hills, CA, USA. The trial size was 14 subjects. The primary objectives of the study were safety and tolerance of HE3286 (10 mg, 20 mg, 40 mg) when administered orally daily over 29 days to patients with Rheumatoid Arthritis receiving a stable dose of methotrexate, pharmacokinetic and metabolism profiles of methotrexate and HE3286 at different dose levels during 29 days of dosing, and potential anti-inflammatory activity of HE3286 in patients with rheumatoid arthritis. SAFETY: Abdominal upper pain (grade 3, considered unrelated to study drug by the investigator) occurred in a subject with history of gastrointestinal reflux disease. Lipase was increased (grade 3, considered possibly related to study drug by the investigator) in a subject 9 days after the last 40 mg dose of HE3286. Blood creatinine phosphokinase was increased (grade 3, considered unrelated to study drug by the investigator) in a subject four weeks after the last 10 mg HE3286 dose with an episode of increased creatinine phosphokinase (grade 3) prior to the first HE3286 dose. The most frequent treatment emergent AEs occurring in two or more subjects in this study were grade 1 blood increases in cholesterol (one at 10 mg, three at 20 mg, and one at 40 mg), and in lactate dehydrogenase (two at 10 mg, one at 20 mg and one at 40 mg). No deaths occurred in the study. Administration of HE3286 was found to be safe and well tolerated in the study. All safety data was submitted to the IND. There was no evidence of drug interaction between HE3286 and methotrexate. HE3286 pharmacokinetics in subjects receiving methotrexate were similar to normal subjects; HE3286 did not alter methotrexate pharmacokinetics. There was insufficient information in this small drug-drug interaction study to reach a conclusion regarding anti-inflammatory activity in rheumatoid arthritis subjects.
| 14 |
Study HE3286-0301
Study HE3286-0301 was conducted under IND #79,334 (Division of Gastroenterology Products), was initiated in January 2008 and completed in July, 2009, and was sponsored by Hollis-Eden Pharmaceuticals, Inc. It was a Phase I/II double blind, randomized, placebo-controlled, dose ranging study of the safety, tolerance, pharmacokinetics and activity of HE3286 when administered orally to patients with active, mild-to-moderate ulcerative colitis. The study was conducted in 11 study centers: Univ. Louisville, Louisville, KY; Scripps Clinic Torrey Pines, La Jolla, CA; Shafran Gastroenterology Center, Winter Park, FL; Atlanta Gastroenterology Assoc., Atlanta, GA; Rocky Mountain Gastroenterology, (Dr. Golf), Golden CO; Rocky Mountain Gastroenterology Center, (Dr. Trouillet), Golden, CO; Adv. Clin. Res., Anaheim, CA; Texas Tech U HSC, Lubbock, TX; Adobe Gastroenterology Res. Tucson AZ; Capital Gastro Medical Group, Roseville, CA; and Rocky Mountain Clin Res. Golden, CO. The trial size was 27 subjects. The primary objectives were safety, tolerance and pharmacokinetics of placebo and four dose levels of HE3286 when administered orally for 28 days, and activity of HE3286 on the signs and symptoms of active mild-to-moderate ulcerative colitis. SAFETY: One subject receiving 5 mg HE3286 developed chest pain and pneumonia (grade 2, considered unrelated to study drug by the investigator). One subject receiving 5 mg HE3286 exhibited decreased lymphocyte count (grade 3, considered unrelated to study drug by the investigator) that resolved before the next assessment. One subject had a Lipase increase (grade 3, considered possibly related to study drug by the investigator) that resolved before the next assessment. One subject receiving 5 mg HE3286 developed a grade 2 ulcerative colitis flare (grade 2, considered possibly related to the study drug by the investigator) that resolved completely. Of the 16 categories of treatment emergent AEs in two or more HE3286 subjects, nine occurred in placebo. In addition to the SAEs above, the remaining seven were increased blood amylase in two subjects (grade 1 [10 and 20 mg HE3286]), lipase in four subjects (grade 1 [10 mg and two 20 mg]), decreased sodium in two subjects (grade 1 [10 and 20 mg HE3286]) and lymphocytes in three subjects (grade 1 [5 mg], grade 2 [10 mg]), red blood cells urine in three subjects (grade 1 [10 mg] and grade 2 [10 mg HE3286]), colitis ulcerative in three subjects (grade 1 [20 mg], grade 2 [5 and 10 mg HE3286]), and upper respiratory tract infection in two subjects (grade 1 [5 mg] and grade 2 [10 mg]). Of these, the increased blood amylase in the 10 mg HE3286, and increased lipase in the 10 mg, HE3286 subject were considered possibly related to HE3286. There were no deaths in this study. Administration of HE3286 was found to be safe and well tolerated in this study. All safety data was submitted to the IND. Estimates of drug exposure with a sparse sampling schedule indicated that pharmacokinetics in this population was similar normal subjects. Due to the failure to detect early ulcerative colitis flares at screening, the population was not optimal for analysis of activity of HE3286 in mild-to moderate UC.
| 15 |
Study HE3286-0401
Study HE3286-0401 was conducted under IND #77,339 (Division of Metabolism and Endocrinology Products), was initiated in July 2008 and completed in January, 2010, and was sponsored by Hollis-Eden Pharmaceuticals, Inc. It was a Phase II, double blind, randomized, placebo-controlled study of the safety, tolerance and activity of HE3286 when administered orally for 12 weeks to adult patients with type 2 diabetes mellitus. The study was conducted in 21 study centers: Dgd Research, Inc. San Antonio, TX, USA; Impact Clinical Trials (Dr. Hazan), Beverly Hills, CA, USA; Impact Clinical Trials (Dr. Madoff), Los Angeles, CA, USA; Impact Clinical Trials (Dr. Lowe), Los Angeles, CA, USA; National Research Center, Los Angeles, CA, USA; Novellus Research Sites (Dr. Heller), La Jolla, CA, USA; NuLife Clinical Research, Inc., Anaheim, CA, USA; Associated Pharmaceutical Research Center, Inc., Buena Park, CA, USA; MedCenter Investigation, Inc., Fair Oaks, CA, USA; Medical Research Incorporated of Las Vegas, Las Vegas, NV, USA; Research Center of Fresno, Inc., Orange, CA, USA; Texas Tech University Health Sciences Center, El Paso, TX, USA; Clinical Trials Research, Sacramento, CA, USA; Synergy Clinical Research, National City, CA, USA; Time Clinical Research, La Palma, CA, USA; Translational Research Grp. Inc., Philadelphia, PA, USA; Novellus Research Sites (Dr. Ratniewski), La Jolla, CA, USA; Radiant Research (Dr. Berwald), Seattle, WA, USA; Radiant Research (Dr. Adams), Seattle, WA, USA; Clinical Investigation Specialists, Gumee, IL, USA; Apex Medical Research; Chicago, IL, USA. The trial size was 94 subjects. The primary objectives were change in hemoglobin A1c (HbA1c) from baseline to week 12 in the HE3286 treated group when compared to the placebo group; safety and tolerance of HE3286 10 mg per day (5 mg twice daily (BID)) compared to placebo from baseline to week 12. The secondary objectives were the effect of HE3286 on fasting blood glucose over time; the effect of HE3286 on lipids (cholesterol, HDL, LDL, TG, etc.) over time; the effect of HE3286 on insulin sensitivity (insulin, C-peptide, HOMA2, fructosamine, etc.) over time; and the effect of HE3286 on serum cytokines, adipokines and chemokines and LPS stimulated PBMC inflammatory cytokine expression. SAFETY: There were four SAEs in HE3286 subjects, one of which was a lab error, and another due to a criminal attack. One (Grade 3 Blood amylase increased) was considered possibly related to HE3286 by the investigator. One subject experienced a blood amylase increase (grade 3, considered unrelated to study drug by the investigator) in a 10 mg HE3286 subject with elevated amylase (grade 1) prior to HE3286 administration, and returned to grade 1 at the next assessment. There were 12 treatment emergent AE categories observed in two or more subjects receiving HE3286. Ten of these were also observed in placebo subjects. The remaining two categories were: two grade 1 and one grade 2 hemoglobin decreased in three metformin + 10 mg HE3286 subjects, and grade 2 back pain in two metformin + 10 mg HE3286 subjects. Each of these AEs were considered as unrelated to the study drug by the investigators. No deaths occurred in the study. Administration of HE3286 was found to be safe and well tolerated in the study. All safety data was submitted to the IND. Although the existing data at the time of the initiation of this study argued that HE3286 would improve insulin sensitivity only in obese, inflamed, insulin-resistant subjects, the sponsor decided to include all subjects (obese and non-obese, inflamed and non-inflamed, and insulin resistant and insulin-sensitive) in this study. Because of this, the primary objective of decreasing hemoglobin A1c in all HE3286-treated subjects was not obtained. The primary objective of safety and tolerability was met. Analysis of the clinical data revealed that HE3286 did have a significant effect on 1,5-anhydroglucitol (a surrogate of postprandial glucose), meeting the secondary objective of activity on glucose levels, and significant effects on insulin sensitivity in the target populations (obese and inflamed). Due to financial constraints of the sponsor, the collection and analysis of cytokines, adipokines, chemokines and LPS stimulated peripheral blood inflammatory cytokine expression was curtailed. See the publication details above for more information.
We believe that we have obtained all worldwide patent and related intellectual property rights to the NE3107 therapies, which we believe will significantly enhance shareholder value . Our patent protection extends to 2034, and will be lengthened by extensions and successor molecules in development.
NE3107, administered orally in a pill, penetrates the blood-brain barrier, stays in the brain long enough to register its desired effect, and to date has not demonstrated significant toxicity in animal studies at up to 40 times the human dose. No drug related, clinically significant side effects have been observed in human studies. NE3107 has no intrinsic interaction with neurotransmitter receptors, but instead acts on inflammatory signaling pathways that influence or modify the course of neuroinflammatory diseases. In this manner NE3107 acts indirectly against the manifestation of disease symptoms, in contrast to drugs that directly interfere with nerve signal transmission (such as a narcotic pain reliever) or directly stimulate or block the activity of neurotransmitter receptors (such as L-dopa acting to directly aid movement in Parkinson’s disease). The general term for this indirect mechanism of activity is disease modifying. Disease modifying agents for neurological conditions are desirable treatment options because they are generally believed to alter disease progression, and associated with less toxicity and fewer side effects than conventional neuro-active drugs. Neuroinflammation promotes neurological disease and is a major factor in neurodegeneration. NE3107 decreases neuroinflammation, and the scientific literature strongly supports the belief that this may slow disease progression for diseases such as Alzheimer’s and Parkinson’s disease, although this has not been demonstrated for NE3107 in humans.
| 16 |
Based on ’NE3107's anti-inflammatory activity and MOA, we are currently targeting four distinct diseases. One of these, namely, Post-Operative Cognitive Dysfunction (“POCD”), may receive the FDA’s “Breakthrough” designation. This designation is applicable when there aren’t any drugs approved to treat the disease, and the disease represents a significant unmet medical need. It is possible that demonstration of efficacy in a single study could lead to commercial approval with a requirement for follow-up studies post approval. The third, L-dopa Induced Dyskinesia (“LID”), a highly debilitating aspect of Parkinson’s disease, has received “Orphan drug” status. ERK’s hyper activation is necessary for LID; hence NE3107’s efficacy in LID in the primate study. The fourth disease we are targeting is Migraine Prophylaxis, and we are currently planning to conduct a Phase II prophylactic migraine study. ERK’s hyper activation is also implicated in the scientific literature to cause migraines. If efficacious, NE3107 will fulfill a great need for a safe, oral, prophylactic migraine medication, which remains largely unmet by current medications.
We currently have three early pre-clinical stage product candidates in addition to NE3107, all of which will require extensive preclinical and clinical evaluation, regulatory review and approval, significant marketing efforts and substantial investment before such product candidates and any successors could provide us with any revenue. As a result, if we do not successfully develop, achieve regulatory approval, and commercialize NE3107, we will be unable to generate any revenue for many years, if at all. We do not anticipate that we will generate revenue for at least several years, and we do not anticipate achieving profitability for at least several years after generating material revenue, if at all. If we are unable to generate revenue, we will not become profitable, and we may be unable to continue our operations.
NE3107 for Alzheimer’s Disease
Alzheimer's disease is a progressive neurodegenerative disease characterized by the progressive decline of memory, cognitive functions, and changes in behavior and personality. Alzheimer’s disease is the one of the leading causes of death in the United States, especially for those aged 65 and older. The cost of patient care for Alzheimer’s disease is estimated near $260 billion for 2017, and that amount is expected to rise dramatically in the next decade.
Alzheimer’s disease is associated with type 2 diabetes and insulin resistance epidemiologically, and scientific research has revealed a complex intertwining of altered insulin signaling and glucose utilization in type 2 diabetes that has many deleterious effects on brain energy production, cognition, and neuron survival. Although glucose transport into neurons is not entirely insulin dependent (within the brain, basal glucose transport is constitutively active), insulin resistance reduces signaling in several downstream pathways that are important to the regulation of stress kinase activation, cell survival, mitochondria glucose utilization, oxidative stress and cognition. Systemic type 2 diabetes effects also alter blood-brain barrier permeability to decrease glucose availability to neurons and increase infiltration of inflammatory cells. There are many scientific reports describing decreased neurodegeneration and enhanced cognition though improved insulin signaling in Alzheimer’s disease animal models. Importantly, there are recent reports suggesting that the mechanism of action of one of the most widely used drugs for type 2 diabetes, metformin, may actually increase susceptibility to neurodegenerative disease. The clinical observation of this was reported from a large study in Taiwan, which if confirmed elsewhere would suggest an even greater need for new, safe, insulin-sensitizing drugs to combat Alzheimer’s disease and neuroinflammation.
| 17 |
Because NE3107 has already demonstrated clinical activity against type 2 diabetes and insulin resistance, it is well positioned for clinical investigation of its potential to slow disease progression and improve cognition in insulin resistant subjects with Alzheimer’s disease, or at risk of developing Alzheimer’s disease or dementia of a less specific nature. NE3107’s prior experience with the FDA DMEP provides an opportunity to rapidly initiate trials in Alzheimer’s disease susceptible subjects. We note that NE3107’s safety, side effect, and drug-drug interaction profile is favorable, but NE3107’s experience in subjects greater than 65 years of age is limited, and thus a conservative clinical trial model for accessing older subjects, while simultaneously collecting safety data, should be expected, and access to subjects older than 65 YOA may be limited by the FDA until sufficient safety data is collected. We have no evidence in our prior clinical experience or animal studies to suggest accumulating the required safety information will be problematic.
We have not yet discussed Alzheimer’s disease clinical trial design with the FDA DNEP or DNP. We anticipate pursuing a multi-center, placebo controlled, Phase II/a trial design to evaluate several hundred subjects with type 2 diabetes with mild cognitive impairment (MCI) or at immediate risk for developing Alzheimer’s disease, treated for 6 to 12 months, measuring safety and comparing one or two dose levels of NE3107 to placebo for effects on glucose disposal, insulin resistance, and cognitive parameters as a combination of primary and secondary endpoints. The details of the trial will be determined through consultations with the FDA and our medical, biostatistical, and scientific advisors. The large size of the Alzheimer’s disease market and the enormous socioeconomic need to prevent and slow Alzheimer’s disease can be expected to drive a major increase in valuation if NeurMedix can demonstrate significant clinical activity.
NE3107 for Post-Operative Cognitive Dysfunction
Post-Operative Cognitive Dysfunction (“POCD”) is a syndrome of durable cognitive impairment commonly observed in the elderly following major surgery, which is typically several months to several years in duration. POCD is defined by a drop in cognitive performance on a set of neuropsychological tests from before to after surgery, and is usually associated with post-operative mortality and rapid progression of Alzheimer’s disease. While patient age is best predictor of susceptibility (with the most concern for patients over sixty years of age), the type of anesthesia, intraoperative stress, and underlying neurodegenerative disease may be additional risk factors. Accumulating research indicates POCD is driven by neuroinflammation and major surgery greatly increases systemic and neuroinflammation. Few anti-inflammatory agents have an appropriate side effect profile for application to POCD, where immunosuppression, drug-drug interactions and potential for anti-coagulant activity are critical considerations, and glucocorticoids are not effective and are associated with cognitive decline.
We believe effective treatment for POCD is a major unmet medical need, particularly in the United States where more than 16 Million people over the age of sixty undergo major surgery each year. Since up to 40% of the population may be susceptible to POCD and there are no approved medications, and existing drugs do not provide significant protection or lack broad applicability, treatments for POCD, such as NE3107, may be eligible for accelerated approval from the FDA. It is our belief that a POCD treatment would be initiated prior to surgery and is envisioned to be continued for at least several months, possibly years. We believe NE3107’s activity and safety profile is well suited to perioperative and chronic use in a geriatric population, because NE3107 has anti-inflammatory activity against peripheral and central inflammation, the drug also has very low potential for toxicity, is not immunosuppressive, has no intrinsic neuropharmacological activity, has very low potential for drug-drug interactions, and NE3107 has no effect on coagulation.
| 18 |
Clinically, we believe there is a market opportunity for NE3107 as a treatment or POCD for a number of reasons. The characterization and prevention of POCD is a major clinical research effort at Duke University Medical Center (“DUMC”), which has ongoing non-interventional studies of the relationship between peripheral and central nervous system (“CNS”) inflammatory markers and POCD. The contemplated trial designs would add NE3107 intervention to a POCD characterization study in non-cardiac major surgery, using the oldest subjects possible to increase POCD susceptibility and decrease the number of subjects needed to decrease cost and time. In addition to supporting the use of NE3107 in POCD, the clinical evaluation would yield objective anti-inflammatory data that would be valuable for development of other neuroinflammatory indications and partnering and should cause a major valuation inflection.
NE3107 for Parkinson’s Disease
Our rationale for NE3107 to treat Parkinson’s disease is based on the premise that immunomodulatory mechanisms that are attenuated by NE3107 drive pathophysiology in Parkinson’s disease. We initiated investigations in Parkinson’s disease because of the well-established link between Parkinson’s disease and activated microglia and neuroinflammation. Our research has demonstrated that NE3107 is efficacious in rodent and primate Parkinson’s disease models. We obtained a Michael J. Fox Foundation (“MJFF”) grant to study BBB permeability, motor activity, and neurodegeneration in mice (4-day MPTP model), and another MJFF grant to study activity against parkinsonism, LID, and neurodegeneration in monkeys (14-week MPTP model). Our study in mice showed excellent BBB penetration, mobility improvement equal to L-dopa, decreased brain inflammatory markers, and decreased neuron stress/death. Our study in monkeys showed decreased parkinsonism, decreased LID development, and decreased neurodegeneration. The fact that NE3107 showed a decreased development of LID, is extremely promising because LID is a highly debilitating aspect of Parkinson’s disease. Furthermore, in a marmoset model, NE3107 decreased the development of dyskinesia (abnormal involuntary movement scale or “AIMS”), without decreasing the beneficial activity of L-dopa. Our research has shown that:
| ● | NE3107 monotherapy improves clinical scores, comparable to L-dopa in mice; |
| ● | NE3107 + L-dopa improves clinical score, mobility, and apathy greater than L-dopa alone or amantadine + L-dopa in marmosets; |
| ● | NE3107 decreases the development of L-dopa induced dyskinesias in marmosets; and |
| ● | NE3107 decreases neuron death (neuroprotection) in marmosets, which correlates to slowing clinical progression. |
We believe the marketplace opportunity for NE3107 in the treatment of Parkinson’s disease to be significant. There is currently an unmet medical need for safe, efficacious interventions to: (i) improve motor symptomatic therapies with lower dyskinesia induction liability; (ii) prevent development of dyskinesias; (iii) slow disease progression; and (iv) treat cognitive impairment. Statistics indicate there are approximately 2.5 million people with Parkinson’s disease in the United States, Europe and Japan, and the expectation is that the number of people with Parkinson’s disease is expected to double in the next fifteen (15) years or so. As a result, we believe the treatment of Parkinson’s disease to be a multi-billion-dollar market opportunity. Set forth below is a summary of our current Parkinson’s disease development plan:
| Parkinson’s Disease Development Plan | ||
| Therapeutic | Clinical Indication | Trial Phase & Duration |
| NE3107 | L-dopa Interaction and motoric activity study | Phase IB, Trial duration: 6 – 9 months |
| Dyskinesia prevention | Phase II, Trial duration: 12 – 18 months | |
| Disease modification | Phase III/IV, Trial duration: 2 – 3 years | |
| 19 |
NE3107 for Migraine Prophylaxis
Migraine is a neuroinflammatory condition. Migraine pathophysiology is mediated, in part, by the neuro-active peptide, calcitonin-gene related peptide (“CGRP”), which exerts its activity through inflammatory signaling mechanisms that can be diminished by NE3107’s activity against ERK hyperactivation (“p-ERK”). Migraine headache is experienced by 17% of women and 6% of men and migraine prevention is the ultimate migraine therapy. Both large and small pharmaceutical companies are pursuing prevention strategies based on anti-CGRP antibodies, which are high cost, must be injected and achieve about 50% reduction in 50% of the population, leaving a large opportunity for an oral, moderately priced therapy with a safe and unique mechanism of action. The total migraine market was estimated to be in excess of $4.5 Billion in 2016, and there’s been significant merger and acquisition activity in the migraine therapy space in recent years.
NE3107 for Hematological Cancers
Scientific literature suggests NE3107’s activity to decrease inflammation, production of inflammatory cytokines, and ERK hyperactivation may be useful to decrease growth or metastasis and to increase killing of certain cancers. Inflammatory factors have long been known to promote cancer, but anti-inflammatory agents are generally immunosuppressive and decrease important immune responses against cancer cells. NE3107 is not immunosuppressive, and NE3107 has a safety, drug-drug interaction, and tolerability profile that would promote acceptance if active. The potential for NE3107 anti-cancer activity has the strongest foundation in hematological cancers, where NE3107 may act directly against the cancer cell to decrease uncontrolled growth and promote cancel cell death (apoptosis) as well as decreasing inflammatory cytokine production (growth factors for the cancer) in surrounding cells. According to the Research and Markets report on global hematological cancer market (June 1, 2016), the 2022 global market for hematological cancers is estimated near $70 Billion. Another potential opportunity in the cancer arena is castration resistant prostate cancer (CRPC.) The potential use of NE3107 for this indication was brought to light by a 2018 publication inNature (Calcinotto et al,Nature 19 July 2018 vol 559 pg 363) describing the inflammatory cytokine, IL-23, as a CRPC-growth stimulating factor. NE3107 has been shown to decrease IL-23 production in a model of systemic inflammation, and thus has the potential to decrease IL-23-dependent growth of CPRC. Clinical studies of NE3107 in any cancer indication would necessarily be preceded by studies in the appropriate cancer-specific animal model.
Intellectual Property
Our most developed intellectual property asset, NE3107, is a first-in-class therapeutic for neurological disease targeting disorders with significant unmet medical needs. We also own additional intellectual property assets which we have not yet elected to develop further, including NE3291 (“NE3291”), NE3789 (“NE3789”) and NE3413 (“NE3413”). NE3291, NE3789 and NE3413 are also potential therapeutics for neurological diseases targeting Alzheimer’s disease and Encephalitis, respectively. We may elect to further develop NE 3789 and/or NE 3413 in the future. We have dedicated significant resources to obtain patent protection on our intellectual property for NE3107 until 2034, which includes:
| ● | Compound and formulations patents issued in United States, Europe and Canada (expiration September 2020); |
| 20 |
| ● | Medical treatment patents (Parkinson’s disease, inflammation) issued in the United States, Europe, Japan, Canada, and Australia (expiration April 2027); |
| ● | Solid state form patents issued or allowed in the United States, Europe, Canada, Australia, South Korea, and patent pending in Japan (expiration April 2029; United States expiration April 2030; maximum term extension until April 2034); and |
| ● | Large-scale synthesis patents issued or allowed in the United States, Europe, Japan, Canada, and patents pending in Israel and India (expiration June 2029). |
In addition to the foregoing, we generally rely on patent, trademark, copyright and trade secret laws and employee and third-party non-disclosure agreements to protect our intellectual property and proprietary rights. We are currently in the process of pursuing trademark protection for our name and logos in the United States. Although we believe that our pending trademark applications will be granted by the United States Patent and Trademark Office, there can be no assurance that any trademarks will be granted or that any trademark relied upon by us in the future, if any, will not be challenged, invalidated or circumvented or that the rights granted thereunder or under licensing agreements will provide competitive advantages to the Company.
We also rely heavily on patents to protect our intellectual property and proprietary technology (i.e. pharmaceuticals and attendant methods and uses). We currently have eight (8) issued patents in the United States and eighty-three (83) issued patents in Europe, the United Kingdom, Canada and other countries, covering our product candidates based on our lead drug candidate, NE3107. Our current NE3107 patent portfolio includes the following: (a) compound formulation patents issued in the U.S., Europe and Canada with an expiration date in September 2020; (b) medical treatment patents (including clinical indication of Parkinson’s disease and inflammation myopathy) issued in the U.S., Europe, Japan, Canada and Australia, expiring in April 2027; (c) solid state form patents issued in the U.S. (expiring in April 2030 with a maximum term extension until April 2034), Europe, Canada, Australia, South Korea, expiring in April 2029, and patent pending in Japan; and (d) large-scale synthesis patents issued or allowed in the U.S., Europe, Japan, Canada, expiring in June 2029, and patent pending in Israel and India. Although we believe that the majority of our intellectual property may be patentable, there can be no assurance that any pending patents will be granted or that any patent relied upon by us in the future, will not be challenged, invalidated or circumvented or that the rights granted thereunder or under licensing agreements will provide competitive advantages to the Company.
Product Development Pipeline
Our product pipeline includes the application of NE3107 to various diseases. As of the date of this Annual Report, we have retained all global development and marketing rights with respect to our product pipeline. The order of clinical indications for NE3107 presented in the table below is not intended to reflect corporate priority or an order in which activities will occur.
| 21 |
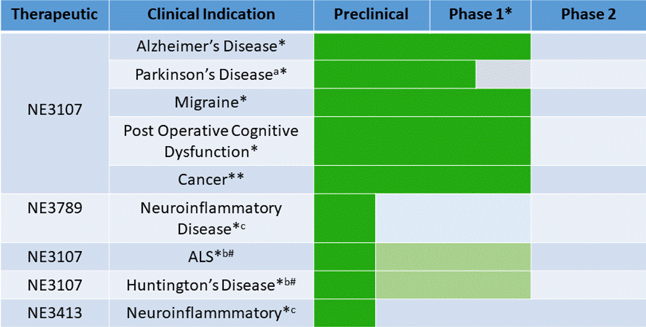
aIn addition to Phase I studies HE3286-100 and HE3286-102 previously conducted by Hollis-Eden Pharmaceuticals, a Phase 1B drug-drug interaction study of NE3107 with L-dopa is required before Phase 2 studies in Parkinson’s disease. NE3107 and HE3286 are codes names or product codes for the same compound, 17a-ethynyl-androst-5-ene-3b,7b,17b-triol. HE3286 was the product code used by the previous owner of the technology, Hollis-Eden Pharmaceuticals. NE3107 is the product code used by NeurMedix.
* Phase 1 studies HE3286-100 and HE3286-102 previously conducted by Hollis-Eden support Phase 1 requirements for these indications.
bPhase 2 ready after preclinical activity confirmation.
#Orphan Indication with a favorable regulatory and commercialization path.
cSpecific clinical indications for these drug candidates have not been made. In addition, the Company has not yet determined whether it will further develop NE3789 and NE3413.
**Phase I requirements complete; nonclinical cancer specific model needed prior clinical efficacy study.
Our clinical and regulatory advisors have indicated that NE3107’s previous Phase I data is sufficient to support Phase II IND applications for Alzheimer’s disease, migraine, and POCD, and our correspondence from the FDA DNP regarding NE3107 development for Parkinson’s is consistent with our advisors’ counsel. However, we have not yet communicated with the FDA about specific plans for Alzheimer’s disease, migraine, and POCD development, and therefore have not received any formal FDA feedback regarding the potential need for additional Phase I studies. Parkinson’s disease requires an additional Phase 1 drug-drug interaction study of NE3107 with L-dopa. NeurMedix has not initiated a Phase 2 study for any indication.
| 22 |
Manufacturing and Distribution
We currently rely on, and expect to continue to rely on, contract manufacturers to produce sufficient quantities of our product candidates for use in our preclinical and clinical trials. In addition, we intend to rely on third parties to manufacture any products that we may commercialize in the future. We have established an internal pharmaceutical development group to develop manufacturing methods for our product candidates, to optimize manufacturing processes, and to select and transfer these manufacturing technologies to our suppliers. We contract with multiple manufacturers to ensure adequate product supply and to mitigate risk. There currently are a limited number of these manufacturers. Furthermore, some of the contract manufacturers that we have identified to date only have limited experience at manufacturing, formulating, analyzing and packaging our product candidates in quantities sufficient for conducting clinical trials or for commercialization.
We currently have no distribution capabilities. In order to independently commercialize any of our product candidates, we must either internally develop distribution capabilities or make arrangements with third parties to perform these services.
Marketing and Sales
We currently have limited experience in marketing or selling pharmaceutical products. To market any of our products independently would require us to develop a sales force with technical expertise along with establishing commercial infrastructure and capabilities.
Employees & Consultants
We currently rely heavily on consultants and service providers to assist us in the execution of our business plans. Upon the closing of the Offering, we expect to employ four senior executives to commence full-time employment.
Property & Facilities
We lease and maintain our primary offices and headquarters at 6165 Greenwich Drive, Suite 150, San Diego, California 92122. The thirty-eight (38) month non-cancellable lease agreement for our headquarter office space entered into on October 20, 2015 was extended to August 2019. The lease agreement provides for escalation of rent payments each year. We do not currently own any real estate.
Insurance
We do not currently maintain product liability insurance for our clinical trials. However, we intend to obtain product liability insurance for our clinical trials as soon as reasonably practicable following the successful completion of our fund raising efforts. We also intend to expand such insurance coverage to include the sale of commercial products if marketing approval is obtained for products in development. However, insurance coverage is becoming increasingly expensive, and we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. In addition, we may not be able to obtain commercially reasonable product liability insurance for any products approved for marketing.
Key Development Partners
We have developed relationships with certain strategic development partners who we believe will be essential to the development and commercialization of our products, including NE3107. Our key development partners include:
| 23 |
| ● | Clintrex, Inc. |
| ○ | Premier Parkinson’s Disease Advisors/CRO; |
| ○ | Dr. C. Warren Olanow, M.D., Fellow of The Royal College of Physicians of Canada, Professor and Past Chair of Neurology at Mt. Sinai, and President of Movement Disorder Society; |
| ○ | Dr. Karl Keiburtz, M.D., Master of Public Health - Professor of Neurology at University of Rochestor, and FDA Advisory Committee Chair for Neurology |
| ○ | Dr. Russel Katz, M.D., Former Director of Neurology Division at FDA |
| ○ | Parkinson’s Study Group |
| ● | Movement Disorders Clinical Trials Network |
| ○ | Dr. Hubert Fernandez, M.D. – Professor of Neurology at Cleveland Clinic |
| ○ | Dr. Michael Schwarzschild, M.D., Ph.D. – Professor of Neurology at Harvard/Mass General |
| ● | NIH/Motac |
| ○ | Pre-clinical partners on dyskinesia and target engagement studies |
| ○ | Dr. Charles Gerfen, Ph.D. - Tenured Investigator at the National Institutes of Health |
| ○ | Dr. Erwan Bezard, Ph.D. – University of Strasbourg |
| ● | Knobbe Martens |
Government Regulation
Clinical trials, the pharmaceutical approval process, and the marketing of pharmaceutical products, are intensively regulated in the U.S. and in all major foreign countries. Regulation by government authorities in the United States and foreign countries is a significant factor in the development, manufacture, distribution, marketing and sale of our proposed products and in our ongoing research and product development activities. All of our products will require regulatory approval by government agencies prior to commercialization. In particular, human therapeutic products are subject to rigorous preclinical studies and clinical trials and other approval procedures of the FDA and similar regulatory authorities in foreign countries. The process of obtaining these approvals and the subsequent compliance with appropriate federal and state statutes and regulations require the expenditure of substantial time and financial resources.
In the United States, various federal and state statutes and regulation also govern or influence testing, manufacturing, safety, labeling, storage, and record-keeping of human therapeutic products and their marketing. Recent federal legislation imposes additional obligations on pharmaceutical manufacturers regarding product tracking and tracing. In addition, federal and state healthcare laws restrict business practices in the pharmaceutical industry. These laws include, without limitation, federal and state fraud and abuse laws, false claims laws, data privacy and security laws, as well as transparency laws regarding payments or other items of value provided to healthcare providers.
| ● | The federal Anti-Kickback Statute makes it illegal for any person or entity, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully, directly or indirectly, solicit, receive, offer, or pay any remuneration that is intended to induce the referral of business, including the purchase, order, lease of any good, facility, item or service for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. For these purposes, the term “remuneration” has been broadly interpreted to include anything of value. |
| 24 |
| ● | Federal false claims and false statement laws, including the federal civil False Claims Act, prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, for payment to, or approval by, federal programs, including Medicare and Medicaid, claims for items or services, including drugs, that are false or fraudulent or not provided as claimed. |
| ● | The federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) created additional federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Also, many states have similar fraud and abuse statutes or regulations that may be broader in scope and may apply regardless of payor, in addition to items and services reimbursed under Medicaid and other state programs. Additionally, to the extent that our products are sold in a foreign country, we may be subject to similar foreign laws. |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and their implementing regulations, requires certain types of individuals and entities to abide by standards relating to the privacy and security of individually identifiable health information, including the adoption of administrative, physical and technical safeguards to protect such information. In addition, certain state laws govern the privacy and security of health information in certain circumstances, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
| ● | The federal Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services (“CMS”) information related to payments or other transfers of value made to physicians and teaching hospitals, and applicable manufacturers and applicable group purchasing organizations to report annually to CMS ownership and investment interests held by the physicians and their immediate family members. |
| ● | There are also an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the laws. In addition, beginning in 2013, a similar federal law requires manufacturers to track and report to the federal government certain payments made to physicians and teaching hospitals made in the previous calendar year. These laws may affect our sales, marketing, and other promotional activities by imposing administrative and compliance burdens on us. In addition, given the lack of clarity with respect to these laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state, and soon federal, authorities. |
Failure to comply with these laws, where applicable, can result in significant penalties, including the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgement, individual imprisonment, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
| 25 |
Preclinical Studies & Clinical Trials
Preclinical studies generally are conducted in laboratory animals to evaluate the potential safety and efficacy of a product. Drug developers submit the results of preclinical studies to the FDA as a part of an Investigational New Drug (“IND”) application before clinical trials can begin in humans. Typically, clinical evaluation involves a time consuming and costly three-phase process.
| Phase I | Clinical trials are conducted with a small number of subjects to determine the early safety profile, maximum tolerated dose and pharmacological properties of the product in human volunteers. |
| Phase II | Clinical trials are conducted with groups of patients afflicted with a specific disease in order to determine preliminary efficacy, optimal dosages and expanded evidence of safety. |
| Phase III | Large-scale, multi-center, comparative clinical trials are conducted with patients afflicted with a specific disease in order to determine safety and efficacy as primary support for regulatory approval by the FDA to market a product candidate for a specific disease. |
The FDA closely monitors the progress of each of the three phases of clinical trials that are conducted in the United States and may, at its discretion, re-evaluate, alter, suspend or terminate the testing based upon the data accumulated to that point and the FDA’s assessment of the risk/benefit ratio to the patient. Clinical trials conducted in foreign countries are also subject to oversight by regulatory authorities in those countries.
The FDA interacts with sponsors throughout the drug development and approval process in order to guide and regulate testing procedure. Official review of a new drug’s safety and effectiveness, however, only begins after clinical trials have been concluded, with the submission of a New Drug Application (“NDA”). An NDA contains all of the data that researchers have gathered about a drug through clinical trials, plus manufacturing, labeling, biological, and chemical information to determine the drug’s appropriate use. The FDA uses this information to determine whether or not a drug is sufficiently safe and effective to be approved.
New Drug Applications
In order to obtain approval to market a pharmaceutical in the U.S., a marketing application must be submitted to the FDA that provides data establishing to the FDA’s satisfaction the safety and effectiveness of the investigational drug for the proposed indication. Once Phase III trials are completed, drug developers submit the results of preclinical studies and clinical trials to the FDA in the form of an NDA or a BLA for approval to commence commercial sales. In most cases, the submission of an NDA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act (“PDUFA”) guidelines that are currently in effect, the FDA has a goal of ten months from the date of filing of a standard NDA for a new molecular entity to review and act on the submission. This review typically takes twelve months from the date the NDA is submitted to FDA because the FDA has approximately two months to make a “filing” decision.
In addition, under the Pediatric Research Equity Act of 2003 as amended and reauthorized, certain NDAs or supplements to an NDA must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults or full or partial waivers from the pediatric data requirements.
| 26 |
The FDA also may require submission of a “risk evaluation and mitigation strategy” (“REMS”) plan to ensure that the benefits of the drug outweigh its risks. The REMS plan could include medication guides, physician communication plans, assessment plans, and/or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality and purity.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with current GMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical trial sites to assure compliance with “Good Clinical Practice” requirements.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA and may require additional clinical or preclinical testing in order for FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements and FDA review and approval.
| 27 |
We will also have to complete an approval process similar to that in the United States in virtually every foreign target market for our products in order to commercialize our product candidates in those countries. The approval procedure and the time required for approval vary from country to country and may involve additional testing. Foreign approvals may not be granted on a timely basis, or at all. In addition, regulatory approval of prices is required in most countries other than the United States. The resulting prices may not be sufficient to generate an acceptable return to us or our corporate collaborators.
Special FDA Expedited Review and Approval Programs
The FDA has various programs, including “fast track” designation, accelerated approval, priority review, and breakthrough therapy designation, which are intended to expedite or simplify the process for the development and FDA review of drugs that are intended for the treatment of serious or life threatening diseases or conditions and demonstrate the potential to address unmet medical needs. The purpose of these programs is to provide important new drugs to patients earlier than under standard FDA review procedures.
To be eligible for a “fast track” designation, the FDA must determine, based on the request of a sponsor, that a product is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address an unmet medical need. The FDA will determine that a product will fill an unmet medical need if it will provide a therapy where none exists or provide a therapy that may be potentially superior to existing therapy based on efficacy or safety factors. The FDA may review sections of the NDA for a “fast track” product on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA, the FDA agrees to accept sections of the NDA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA.
The FDA may give a priority review designation to drugs that offer major advances in treatment, or provide a treatment where no adequate therapy exists. A priority review means that the goal for the FDA to review an application is six months, rather than the standard review of ten months under current PDUFA guidelines. These six and ten-month review periods are measured from the filing date rather than the receipt date for NDAs for new molecular entities, which typically adds approximately two months to the timeline for review and decision from the date of submission. Most products that are eligible for “fast track” designation are also likely to be considered appropriate to receive a priority review.
In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may be eligible for accelerated approval and may be approved on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require a sponsor of a drug receiving accelerated approval to perform post-marketing studies to verify and describe the predicted effect on irreversible morbidity or mortality or other clinical endpoint, and the drug may be subject to accelerated withdrawal procedures.
A “breakthrough therapy” is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs designated as “breakthrough therapies” are also eligible for accelerated approval. The FDA must take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a “breakthrough therapy”.
| 28 |
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened.
FDA Post-Approval Requirements
Following the approval of a BLA or NDA, the FDA continues to require adverse event reporting and submission of periodic reports. The FDA also may require post-marketing testing, known as Phase IV testing, REMS, and surveillance to monitor the effects of an approved product, or the FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control, drug manufacture, packaging, and labeling procedures must continue to conform to GMP after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing facilities to assess compliance with GMP. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain compliance with GMP. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA or BLA. For example, the FDA may require post-marketing testing, including Phase IV clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with current GMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from GMP requirements and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain GMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
| 29 |
| ● | Restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| ● | Fines, warning letters or holds on post-approval clinical trials; |
| ● | Refusal of the FDA to approve pending NDAs/BLAs or supplements to approved NDAs/BLAs, or suspension or revocation of product approvals; |
| ● | Product seizure or detention, or refusal to permit the import or export of products; or |
| ● | Injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
Insurance Coverage & Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any product candidates for which we obtain regulatory approval. In the United States and markets in other countries, sales of any products for which we receive regulatory approval will depend, in part, on the extent to which third-party payors provide coverage and establish adequate reimbursement levels for such drug products.
In the United States, third-party payors include federal and state healthcare programs, government authorities, private managed care providers, private health insurers and other organizations. Third-party payors are increasingly challenging the price, examining the medical necessity and reviewing the cost-effectiveness of medical drug products and medical services, in addition to questioning their safety and efficacy. Such payors may limit coverage to specific drug products on an approved list, also known as a formulary, which might not include all of the FDA-approved drugs for a particular indication. We may need to conduct expensive pharmaco-economic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain the FDA approvals. Nonetheless, our product candidates may not be considered medically necessary or cost-effective.
Moreover, the process for determining whether a third-party payor will provide coverage for a drug product may be separate from the process for setting the price of a drug product or for establishing the reimbursement rate that such a payor will pay for the drug product. A payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a drug product does not assure that other payors will also provide coverage for the drug product. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
| 30 |
The marketability of any product candidates for which we or our collaborators receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, emphasis on managed care in the United States has increased and we expect will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Human Health Product Regulation in the U.S.
In the U.S., the FDA regulates pharmaceuticals under the Federal Food, Drug, and Cosmetic Act (“FDCA”) and related regulations. Pharmaceuticals are also subject to other federal, state, and local statutes and regulations. Failure to comply with applicable U.S. regulatory requirements at any time during the product development process, approval process or after approval may subject an applicant to administrative or judicial sanctions. These sanctions could include the imposition by the FDA of an Institutional Review Board (“IRB”), a clinical hold on trials, a refusal to approve pending applications, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.
The FDA and comparable regulatory agencies in state and local jurisdictions impose substantial requirements upon the clinical development, manufacture and marketing of pharmaceutical products. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, distribution, record keeping, approval, advertising and promotion of our products.
The FDA’s policies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of new disease indications or label changes. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or elsewhere.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of certain FDA-regulated products, including prescription drugs, are required to register and disclose certain clinical trial information on a public website maintained by the U.S. National Institutes of Health. Information related to the product, patient population, phase of investigation, study sites and investigator, and other aspects of the clinical trial is made public as part of the registration. Sponsors are also obligated to disclose the results of these trials after completion. Disclosure of the results of these trials can be delayed until the product or new indication being studied has been approved. Competitors may use this publicly-available information to gain knowledge regarding the design and progress of our development programs.
The Drug Price Competition and Patent Term Restoration Act
The Drug Price Competition and Patent Term Restoration Act, also known as the Hatch-Waxman Act, requires pharmaceutical companies to divulge certain information regarding their products which have the effect of making it easier for other companies to manufacture generic drugs to compete with those products.
The U.S. generally requires an environmental assessment, which discusses a company’s proposed action, possible alternatives to the action, and whether the further analysis of an environmental impact statement is necessary. Certain exemptions are available from the requirement to perform an environmental assessment and an environmental impact statement. Once an exemption is claimed, a company must state to the FDA that no extraordinary circumstances exist that may significantly affect the environment.
| 31 |
Patient Protection and Affordable Care Act
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, the “ACA”), which includes measures that have or will significantly change the way health care is financed by both governmental and private insurers, became law in the U.S. The ACA is a sweeping measure intended to expand health care coverage within the U.S., primarily through the imposition of health insurance mandates on employers and individuals and expansion of the Medicaid program. The ACA has significantly impacted the pharmaceutical industry. The ACA will require discounts under the Medicare drug benefit program and increased rebates on drugs covered by Medicaid. In addition, the ACA imposes an annual fee, which will increase annually, on sales by branded pharmaceutical manufacturers. At this time, the financial impact of these discounts, increased rebates and fees and the other provisions of the ACA on our business are unclear. However, the fees, discounts and other provisions of this law are expected to have a significant negative effect on the profitability of pharmaceuticals.
Human Health Product Regulation in the European Union
In addition to regulations in the U.S., we may eventually be subject, either directly or through our distribution partners, to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products, if approved.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in non-U.S. countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the U.S. have a process that requires the submission of a clinical trial application prior to the commencement of human clinical trials. In Europe, for example, a Clinical Trial Application (“CTA”) must be submitted to the competent national health authority and to independent ethics committees in each country in which a company intends to conduct clinical trials. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed in that country.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country, even though there is already some degree of legal harmonization in the European Union (“EU”) Member States resulting from the national implementation of underlying EU legislation. In all cases, the clinical trials are conducted in accordance with GCP and other applicable regulatory requirements.
To obtain regulatory approval of an investigational drug under EU regulatory systems, we must submit a marketing authorization application. This application is similar to the BLA in the U.S., with the exception of, among other things, country-specific document requirements. Drugs can be authorized in the EU by using (i) the centralized authorization procedure, (ii) the mutual recognition procedure, (iii) the decentralized procedure or (iv) national authorization procedures.
The European Medicines Agency (“EMA”) implemented the centralized procedure for the approval of human drugs to facilitate marketing authorizations that are valid throughout the EU. This procedure results in a single marketing authorization granted by the European Commission that is valid across the EU, as well as in Iceland, Liechtenstein and Norway (the “European Community”). The centralized procedure is compulsory for human drugs that are: (i) derived from biotechnology processes, such as genetic engineering, (ii) contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative diseases, autoimmune and other immune dysfunctions and viral diseases, (iii) officially designated orphan drugs, and (iv) advanced-therapy medicines, such as gene-therapy, somatic cell-therapy or tissue-engineered medicines. The centralized procedure may at the request of the applicant also be used for human drugs which do not fall within the above mentioned categories if the human drug (a) contains a new active substance which, on the date of entry into force of Regulation (EC) No. 726/2004, was not authorized in the European Community; or (b) the applicant shows that the medicinal product constitutes a significant therapeutic, scientific or technical innovation or that the granting of authorization in the centralized procedure is in the interests of patients at European Community level.
| 32 |
Under the centralized procedure in the EU, the maximum timeframe for the evaluation of a Marketing Authorization Application (“MAA”) by the EMA is two hundred ten (210) days, though the date count stops whenever the Committee for Medicinal Products for Human Use (“CHMP”) asks the applicant for additional written or oral information, with adoption of the actual marketing authorization by the European Commission thereafter. Accelerated evaluation might be granted by the CHMP in exceptional cases, as when a medicinal product is expected to be of a major public health interest from the point of view of therapeutic innovation, defined by three cumulative criteria: (i) the seriousness of the disease to be treated; (ii) the absence of an appropriate alternative therapeutic approach; and (iii) anticipation of exceptional high therapeutic benefit. In this circumstance, EMA ensures that the evaluation for the opinion of the CHMP is completed within 150 days and the opinion issued thereafter.
The Mutual Recognition Procedure (“MRP”), for the approval of human drugs is an alternative approach to facilitate individual national marketing authorizations within the EU. Basically, the MRP may be applied for all human drugs for which the centralized procedure is not obligatory. The MRP is applicable to the majority of conventional medicinal products, and is based on the principle of recognition of an already existing national marketing authorization by one or more Member States.
The characteristic of the MRP is that the procedure builds on an already existing marketing authorization in a Member State of the EU that is used as reference in order to obtain marketing authorizations in other EU Member States. In the MRP, a marketing authorization for a drug already exists in one or more Member States of the EU and subsequently marketing authorization applications are made in other EU Member States by referring to the initial marketing authorization. The Member State in which the marketing authorization was first granted will then act as the reference Member State. The Member States where the marketing authorization is subsequently applied for act as concerned Member States.
The MRP is based on the principle of the mutual recognition by EU Member States of their respective national marketing authorizations. Based on a marketing authorization in the reference Member State, the applicant may apply for marketing authorizations in other Member States. In such case, the reference Member State shall update its existing assessment report about the drug in 90 days. After the assessment is completed, copies of the report are sent to all Member States, together with the approved summary of product characteristics, labeling and package leaflet. The concerned Member States then have 90 days to recognize the decision of the reference Member State and the summary of product characteristics, labeling and package leaflet. National marketing authorizations shall be granted within 30 days after acknowledgement of the agreement.
Should any Member State refuse to recognize the marketing authorization by the reference Member State, on the grounds of potential serious risk to public health, the issue will be referred to a coordination group. Within a timeframe of 60 days, Member States shall, within the coordination group, make all efforts to reach a consensus. If this fails, the procedure is submitted to an EMA scientific committee for arbitration. The opinion of this EMA Committee is then forwarded to the Commission, for the start of the decision-making process. As in the centralized procedure, this process entails consulting various European Commission Directorates General and the Standing Committee on Human Medicinal Products.
| 33 |
Human Health Product Regulation in the Rest of World
For other countries outside of the EU, such as countries in Eastern Europe or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCPs and the other applicable regulatory requirements. If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension of clinical trials, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Other Regulatory Considerations
Once a BLA or NDA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of pharmaceuticals, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities on the internet and elsewhere. While doctors are free to prescribe any pharmaceutical approved by the FDA for any use, a company can only make claims relating to the safety and efficacy of a pharmaceutical that are consistent with the FDA approval, and is only allowed to actively market a pharmaceutical for the particular indication approved by the FDA. Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new BLA or BLA supplement before the change can be implemented. A BLA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing BLA supplements as it does in reviewing NDAs.
In addition, any claims we make for our products in advertising or promotion must be appropriately balanced with important safety information and otherwise be adequately substantiated. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising, injunctions and potential civil and criminal penalties. Government regulators recently have increased their scrutiny of the promotion and marketing of pharmaceuticals.
Other Health Care Laws and Compliance Requirements
In the U.S., our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services (formerly the Health Care Financing Administration), other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice and individual U.S. Attorney offices within the Department of Justice, and state and local governments. For example, sales, marketing and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, the privacy provisions of the Health Insurance Portability and Accountability Act, and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veterans Health Care Act of 1992 (“VHCA”), each as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements will apply. Under the VHCA, drug companies are required to offer certain drugs at a reduced price to a number of federal agencies including U.S. Department of Veteran Affairs and U.S. Department of Defense, the Public Health Service and certain private Public Health Service designated entities in order to participate in other federal funding programs including Medicare and Medicaid. Recent legislative changes require that discounted prices be offered for certain U.S. Department of Defense purchases for its TRICARE program via a rebate system. Participation under the VHCA requires submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulations.
| 34 |
In order to distribute products commercially, we must comply with state laws that require the registration of manufacturers and wholesale distributors of pharmaceutical products in a state, including, in certain states, manufacturers and distributors that ship products into the state even if such manufacturers or distributors have no place of business within the state. Some states also impose requirements on manufacturers and distributors to establish the pedigree of product in the chain of distribution, including some states that require manufacturers and others to adopt new technology capable of tracking and tracing product as it moves through the distribution chain. Several states have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities or register their sales representatives. Other legislation has been enacted in certain states prohibiting pharmacies and other health care entities from providing certain physician prescribing data to pharmaceutical companies for use in sales and marketing, and prohibiting certain other sales and marketing practices. All of our activities are potentially subject to federal and state consumer protection and unfair competition laws.
Healthcare Reform
The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
By way of example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the “ACA”), was signed into law, which intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add transparency requirements for the healthcare and health insurance industries, impose taxes and fees on the health industry and impose additional health policy reforms. Among the provisions of the ACA of importance to our potential drug candidates are:
| ● | an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs; |
| ● | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13.0% of the average manufacturer price for branded and generic drugs, respectively; |
| ● | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; |
| ● | extension of a manufacturer’s Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| ● | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a manufacturer’s Medicaid rebate liability; |
| 35 |
| ● | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for a manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| ● | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; and |
| ● | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
We expect that the ACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and lower reimbursement, and in additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payor.
Competition
The biotechnology and pharmaceutical industries are subject to rapid and intense technological change. We face, and will continue to face, competition in the development and marketing of our product candidates from biotechnology and pharmaceutical companies, research institutions, government agencies and academic institutions. Competition may also arise from, among other things:
| ● | other drug development technologies; |
| ● | methods of preventing or reducing the incidence of disease, including vaccines; and |
| ● | new small molecule or other classes of therapeutic agents. |
Developments by others may render our product candidates or technologies obsolete or noncompetitive. We are performing research on or developing products for the treatment of several disorders including Parkinson’s disease (L-dopa Induced Dyskinesia), Alzheimer’s disease, Huntington’s disease, Multiple Sclerosis, Encephalitis, Post-Operative Cognitive Dysfunction (also known as “POCD”), Migraine, Optic Neuritis, Neuromyelitis Optica, Inclusion-Body Myositis (also known as “IBM”), Inflammatory Myopathy, Glaucoma, Ulcerative Colitis, Uveitis, Rheumatoid Arthritis, Lupus, Amyotrophic Lateral Sclerosis (also known as “ALS” or “Lou Gehrig’s Disease”), Diabetes, and Chronic Obstructive Pulmonary Disease (also known as “COPD”), Cystic Fibrosis (also known as “CF”), and other neurological-related diseases and disorders.
| 36 |
Item 2. Management's Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of our operations together with our interim financial statements and the notes thereto appearing elsewhere in this Form 1-K. This discussion contains forward-looking statements reflecting our current expectations, whose actual outcomes involve risks and uncertainties. Actual results and the timing of events may differ materially from those stated in or implied by these forward-looking statements. These factors include our research and development activities, distributor channel; compliance with regulatory impositions; and our capital needs. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. These forward looking statements, are subject to risks, uncertainties and assumptions, including those discussed in the sections entitled “Risk Factors,” "Cautionary Statement regarding Forward-Looking Statements" in the Offering Circular.
The following table summarizes the results of our operations for the year ended December 31, 2018 and 2017:
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Operating expenses: | ||||||||
| Research and development | $ | (9,563 | ) | $ | (53,426 | ) | ||
| General and administrative | (741,193 | ) | (753,558 | ) | ||||
| Total operating expenses | (750,756 | ) | (806,984 | ) | ||||
| Net loss | $ | (750,756 | ) | $ | (806,984 | ) | ||
| Weighted-average common shares outstanding, basic and diluted | 43,334,000 | 43,334,000 | ||||||
| Net loss per share, basic and diluted | $ | (0.02 | ) | $ | (0.02 | ) | ||
Comparison of year ended December 31, 2018 and 2017.
Net Loss
The net loss for the year ended December 31, 2018 was $751,000 compared to $807,000 for the year ended December 31, 2017. The decline was primarily attributed to a decrease in research and development expense.
Revenues
NeurMedix, Inc., is a pre-revenue development stage biopharmaceutical company that engages in developing products for the treatment of neurological and neuro-degenerative disorders. We have no products approved for commercial sale and have not generated any revenues from product sales since our inception in 2014.
Research and Development expenses
Our research and development expenses were approximately $10,000 and $53,000 for the year ended December 31, 2018 and 2017, respectively. In 2018 the Company’s focus was on capital raising efforts and activity in research and development has been minimal to nil. The research and development expense in 2017 was primarily related to clinical programs to further the Orphan Drug Development and filings with the FDA during 2017. All research and development expenses were funded by the Company’s sole shareholder.
| 37 |
General and Administrative expenses
Our general and administrative expenses of $741,000 for the year ended December 31, 2108 was comparable to $754,000 for the year ended December 31, 2017. The primary components of the net decline consisted of the following: a decline in legal and professional fees of approximately $151,000 to $302,000 in 2018 from $453,000 in 2017 due to the original preparation and filings the Regulation A which occurred in 2017, offset by approximately $166,000 of expenses for the continued development of the market campaign and materials for capital raising efforts. All general and administrative expenses are funded by the Company’s sole shareholder.
Liquidity and Capital Resources
The accompanying interim financial statements were prepared assuming the Company will continue as a going concern, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The Company expects substantial losses in future periods. As of December 31, 2018, the Company has an accumulated deficit of approximately $6.3 million, a stockholder’s deficit of approximately $624,000 and a working capital deficit of $659,000. The Company’s future operations are dependent on raising additional capital to continue its development and commercialization efforts. The Company has funded its operations exclusively in its shareholder’s contributions, and there is no formal agreement for such arrangement to continue. Although we are in the process of raising additional interim capital from other private sources there can be no assurance that such needed capital will be available or adequate to fund future operations.
There is no assurance that we will successfully obtain the required capital or revenues, or, if obtained, that the amounts will be sufficient to fund our ongoing operations. The inability to secure additional capital would have a material adverse effect on us, including the possibility that we would have to sell or forego a portion or all of our assets or cease operations. If we discontinue our operations, we will not have sufficient funds to pay any amounts to our stockholders and their vendors.
Because our working capital requirements depend upon numerous factors there can be no assurance that our current cash resources will be sufficient to fund our operations. At present, we have no committed external sources of capital, and do not expect any significant product revenues for the foreseeable future. Thus, we will require immediate additional financing to fund future operations. There can be no assurance, however, that we will be able to obtain funds on acceptable terms, if at all.
These circumstances raise substantial doubt on our ability to continue as a going concern. These financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or amounts and classification of liabilities that might result from this uncertainty.
Credit Facilities
We do not have any credit facilities or other access to bank credit.
Capital Expenditures
We do not have any contractual obligations for ongoing capital expenditures at this time. We may, however, purchase equipment, supplies and software necessary to conduct our operations on an as needed basis.
| 38 |
Contractual Obligations, Commitments and Contingencies
Contingencies
Certain conditions may exist as of the date the financial statements are issued, which may result in a loss to the Company, but which will only be resolved when one or more future events occur or fail to occur. The Company's management, in consultation with its legal counsel as appropriate, assesses such contingent liabilities, and such assessment inherently involves an exercise of judgment. In assessing loss contingencies related to legal proceedings that are pending against the Company or unasserted claims that may result in such proceedings, the Company, in consultation with legal counsel, evaluates the perceived merits of any legal proceedings or unasserted claims, as well as the perceived merits of the amount of relief sought or expected to be sought therein. If the assessment of a contingency indicates it is probable that a material loss has been incurred and the amount of the liability can be estimated, then the estimated liability would be accrued in the Company's financial statements. If the assessment indicates a potentially material loss contingency is not probable, but is reasonably possible, or is probable, but cannot be estimated, then the nature of the contingent liability, together with an estimate of the range of possible loss, if determinable and material, would be disclosed. Loss contingencies considered remote are generally not disclosed unless they involve guarantees, in which case the guarantees would be disclosed. We are not aware of any matters which result in a loss contingency. At December 31, 2018, a payment of past warehousing fees to a certain vendor is contingent upon obtaining funding through our capital raise efforts. Although the Company is undertaking a “best efforts” offering of its common stock to raise additional capital, there is no assurance that such an offering will be successful. Upon successful completion of such offering, the contingent liability would become due and payable. The estimated possible maximum amount that potentially may be due upon securing funding was estimated at approximately $310,000. This amount has not been recognized in the accompanying financial statements as of December 31, 2018 as the funding event is not deemed probable at the date of this filing.
Off-Balance Sheet Arrangements
There were no off-balance sheet arrangements during the periods presented, or as of December 31, 2018 the Company did not have any off-balance sheet arrangements.
Quantitative and Qualitative Disclosures about Market Risk
In the ordinary course of our business, we are not exposed to market risk of the sort that may arise from changes in interest rates or foreign currency exchange rates, or that may otherwise arise from transactions in derivatives.
| 39 |
| Item 3. | Directors and Officers |
As of the date of this Annual Report, Terren S. Peizer, is the Chief Executive Officer, Treasurer, Principal Financial Officer, Secretary and sole Director. However, below is a list of the executive officers and directors and their respective positions which will become effective as of the date this Offering Circular is qualified by the Commission, unless otherwise indicated:
| Name | Position | Age | Term of Office (*) | |||
| Executive Officers: | ||||||
| Terren S. Peizer | Chief Executive Officer, Treasurer & Secretary | 59 | Since November 2014 | |||
| Joanne Wendy Kim | Chief Financial Officer | 64 | Since November 2018 (**) | |||
| Clarence Ahlem | Chief Operating Officer | 63 | Since November 2014 | |||
| Christopher L. Reading | Chief Scientific Officer | 72 | Since November 2014 | |||
| Robert J. Hariri | Chairman of Executive Committee | 60 | Since October 2017 (**) | |||
| Directors: | ||||||
| Terren S. Peizer | Director | 59 | Since November 2014 | |||
| Robert J. Hariri | Director & Chairman of the Executive Committee | 60 | Since October 2017 (**) | |||
| Richard A. Berman | Director | 74 | Since October 2017 (**) | |||
| Steve Gorlin | Director & Vice Chairman | 81 | Since October 2017 (**) | |||
| Richard J. Berman | Director | 76 | Since October 2017 (**) |
(*) With the exception of Terren S. Peizer, the other persons designated above as officers of the Company have served as independent contractors on a consulting basis and are responsible to pay their own taxes. Upon closing of the Offering, such persons will become full time employees, officers and directors of the Company.
(**) These individuals will become board members upon the closing of the Offering.
During the past five years, none of the persons identified above has been involved in any bankruptcy or insolvency proceeding or convicted in a criminal proceeding, excluding traffic violations and other minor offenses. There is no arrangement or understanding between the persons described above and any other person pursuant to which the person was selected to his or her office or position.
| 40 |
Executive Officers and Directors
Terren S. Peizer, Chief Executive Officer, Treasurer, Secretary & Director. Mr. Peizer is an entrepreneur, investor, and financier with a particular interest in healthcare having founded and successfully commercialized several healthcare companies. Most recently, Mr. Peizer is the founder of Catasys, Inc., a leader in behavioral and mental health management services. He has also served as its CEO and Chairman of the Board of Directors since the Company’s inception in 2004. Mr. Peizer also is the Founder, Chairman and CEO NeurMedix, Inc., a biotechnology company with a focus on inflammatory, neurological and neuro-degenerative diseases. NeurMedix’s lead compound NE3107 has shown efficacy in 11 pre-clinical and 6 human studies. These studies resulted in publications in peer-reviewed journals. Mr. Peizer is Chairman of Acuitas Group Holdings, LLC, his personal holding company that is the owner of all of his portfolio company interests. Through Acuitas, Mr. Peizer owns Crede Capital Group, LLC an industry leader in investing in micro and small capitalization equities, having invested over $1.2 Billion directly into portfolio companies. Mr. Peizer has been the largest beneficial shareholder, and has held various senior executive positions with several other publicly-traded growth companies, including Chairman of internationally renowned Cray, Inc., the industry’s leading supercomputer company. Mr. Peizer has a background in venture capital, investing, mergers and acquisitions, corporate finance, and previously held senior executive positions with the investment banking firms Goldman Sachs, First Boston, and Drexel Burnham Lambert. He received his B.S.E. in finance from The Wharton School of Finance and Commerce.
Joanne Wendy Kim, CPA, Chief Financial Officer. Wendy Kim has more than 30 years’ experience in accounting and finance. Serving as a CFO of private and public entities where she restructured and oversaw Corporate Finance/Operational Groups, closed M&A transactions, secured bank financings, developed and implemented new business strategies, managed risk and implemented new policies, procedures and investor reporting including managing investor relations. As a certified public accountant, she served eight years in BDO USA LLP’s National Office in the SEC Department and provided accounting, SEC filings reviews and other business consultative services to companies and engagement teams. Wendy Kim also served eleven years at KPMG from 1981 to 1992. She is a current member of the American Institute of Certified Public Accountants and California Society of CPA’s. Wendy received her Bachelor’s degree in Business Administration – Accounting and Finance from California State University, Long Beach.
Clarence Ahlem, M.S., Chief Operating Officer.Clarence Ahlem has more than 30 years of pharmaceutical development experience. His scientific breadth includes the enzymology of DNA replication, synthetic bifunctional antibodies, hematopoietic stem cell purification, and pharmacological characterization of the dehydroepiandrosterone metabolome and associated pharmaceutical science. Clarence worked for four years at UCSD prior to joining the Therapeutics Division at Hybritech in San Diego, CA, where he worked for six years developing synthetic bifunctional antibodies and their clinical applications. Mr. Ahlem left Hybritech to initiate product development at Systemix, Inc. in Palo Alto, CA, where he was manager of bio-organic chemistry for 4 years. He left Systemix to join Prolinx Biochemistry in Bothell, WA, and moved to San Diego in 1997 to join Hollis-Eden Pharmaceuticals (subsequently Harbor BioSciences and Harbor Therapeutics), where he was Vice President of Product Development until 2014.
Christopher L. Reading, Ph.D., Chief Scientific Officer. Chris Reading received his Ph.D. in Biochemistry from UC Berkeley, performed post-doctoral studies in cancer biology at UC Irvine, and joined MD Anderson Cancer Center and the University of Texas, Graduate School of Biomedical Sciences in Houston for 13 years, where he became Associate Professor of Medicine in the Department of Developmental Therapeutics with a joint appointment in the Department of Tumor Biology. He then accepted a position at SyStemix/Novartis in Palo Alto, where he became Vice President of Product and Process Development. From there, he moved to San Diego where he has spent over 15 years on the NE3107 platform development at Harbor Therapeutics. He has over 35 years of research and drug development experience, and over 130 peer-reviewed scientific publication, and has authored numerous patents in the areas of monoclonal antibodies, cell separation technologies, stem cell transplantation, and sterol drug development.
| 41 |
Robert J. Hariri, M.D., Ph.D., Chairman of the Executive Committee & Director. Dr. Hariri a surgeon, biomedical scientist and highly successful serial entrepreneur in two technology sectors: biomedicine and aerospace. The former Chairman, Founder, and Chief Executive Officer of Celgene Cellular Therapeutics, one of the world’s largest human cellular therapeutics companies, Dr. Hariri has pioneered the use of stem cells to treat a range of life threatening diseases and has made transformative contributions in the field of tissue engineering. He co-founded Human Longevity, Inc., the world’s largest gene sequencing operation with genomics legend, J. Craig Venter and serves as Vice Chairman. His activities and experience includes academic neurosurgeon at Cornell, biotechnology executive, military and defense scientist, surgeon, aviator and aerospace innovator. Dr. Hariri has 150 issued and pending patents, has authored over 100 published chapters, articles and abstracts and is most recognized for his discovery of pluripotent stem cells from the placenta and as a member of the team which discovered TNF (tumor necrosis factor). Dr. Hariri was recipient of the Thomas Alva Edison Award in 2007 and 2011, The Fred J. Epstein Lifetime Achievement Award and has received numerous other honors for his many contributions to biomedicine and aviation.
Dr. Hariri also serves on numerous Boards of Directors including Bionik Laboratories and Provista Diagnostics. Dr. Hariri is an Adjunct Associate Professor of Pathology at the Mount Sinai School of Medicine and a member of the Board of Visitors of the Columbia University School of Engineering & Applied Sciences and the Science & Technology Council of the College of Physicians and Surgeons, and is a member of the scientific advisory board for the Archon X PRIZE for Genomics, which is awarded by the X PRIZE Foundation. Dr. Hariri is also a Trustee of the Liberty Science Center and has been appointed to the New Jersey Commission on Cancer Research by Governor Chris Christie. Dr. Hariri received his undergraduate training at Columbia College and Columbia University School of Engineering and Applied Sciences and was awarded his M.D. and Ph.D. degrees from Cornell University Medical College. Dr. Hariri received his surgical training at The New York Hospital-Cornell Medical Center where he also directed the Aitken Neurosurgery Laboratory and the Center for Trauma Research.
Richard A. Berman, Director. Mr. Berman is currently the President and Chief Executive Officer of LICAS, a K-12, College and University, Health Care consulting firm. In addition, he currently serves as Chairman of the board of directors of Emblem Health's Quality of Care Committee and a member of its Audit Committee. Mr. Berman is also an Entrepreneur in Residence at GaTech’s ATDC and visiting professor at USF MUMA College of Business. Mr. Berman has held healthcare, educational, housing and community development positions around the world. He has worked with several foreign governments, the United Nations, the U.S. Department of Health, the FDA, and as a cabinet level official for the state of New York. He has also worked with Manhattanville College, McKinsey & Co, NYU Medical Center, Westchester Medical, EmblemHealth, and numerous startups. Mr. Berman has a Bachelor of Business Administration, an MBA and Masters, in Public Health. We believe Mr. Berman’s qualifications to serve on our Board of Directors include his extensive experience as an executive in several healthcare firms. In addition, as a board member of a health plan we believe he has an understanding of our customer base and current developments and strategies in the health insurance industry.
| 42 |
Steve Gorlin, Director & Vice Chairman. Mr. Gorlin is an entrepreneur who has founded numerous successful biotechnology and pharmaceutical companies over the last 40 years, including Medivation and Entremed. He currently serves as Executive Chairman to Conkwest, Inc. and served as Chairman of the Board of MiMedx, Inc., a wound care Company, from November 2006 to June 2013. Mr. Gorlin served many years on the Business Advisory Council to the Johns Hopkins School of Medicine as well as on the advisory board of the Johns Hopkins BioMedical Engineering Advisory Board. We believe Mr. Gorlin’s qualifications to serve on our Board of Directors include his experience in the healthcare industry, his extensive business development experience, and his current and past executive experience in numerous private and publicly traded companies.
Richard J. Berman, Director. Richard Berman’s business career spans over 35 years of venture capital, senior management and merger and acquisitions experience. In the past 5 years, Mr. Berman has served as a director and/or officer of over a dozen public and private companies. In November 2014, he was named Chairman of MetaStat, Inc. a public company in the cancer diagnostic field. From 2006 to 2011, he was Chairman of National Investment Managers, a company with $12 Billion in pension administration assets. In 2012, he became vice chairman of Energy Smart Resources, Inc. From 2002 to 2010, he was director of Nexmed Inc. where he also served as Chairman and CEO in 2008 and 2009. From 1998 to 2012, Mr. Berman served as a Director of Easy Link International and served as the Chairman and CEO from 1998 to 2000. Most recently, Mr. Berman was appointed a partner, chairman and director at Scylax Aviation. In addition, Mr. Berman is currently a director of four (4) public companies: Advaxis, Inc., Calandrius Biosciences, Inc., Cryoport, Inc., and MetaStat, Inc. Previously, Mr. Berman worked at Goldman Sachs; was Senior Vice President of Bankers Trust Company, where he started the M&A and Leveraged Buyout Departments; created the largest battery company in the world in the 1980s by merging Prestolite, General Battery and Exide to form Exide Technologies (XIDE); helped to create what is now Soho (NYC) by developing five buildings; and advised on over $4 Billion of M&A transactions (completed over 300 deals). He is a past Director of the Stern School of Business of NYU where he obtained his B.S. and M.B.A. He also has US and foreign law degrees from Boston College and The Hague Academy of International Law, respectively. Mr. Berman’s extensive knowledge of our industry, his role in the governance of publicly held companies and his directorships in other life science companies qualify him to serve as our director.
Board Leadership Structure and Risk Oversight
The Board oversees our business and considers the risks associated with our business strategy and decisions. The Board currently implements its risk oversight function as a whole. Each of the Board committees, when established, will also provide risk oversight in respect of its areas of concentration and reports material risks to the board for further consideration.
Term of Office
Directors hold office until his or her successor is elected and qualified, unless sooner displaced. Officers are appointed to serve for one year until the meeting of the Board following the annual meeting of shareholders and until their successors have been elected and qualified.
Director Independence
We use the definition of “independence” of The NASDAQ Stock Market to make this determination. NASDAQ Listing Rule 5605(a)(2) provides that an “independent director” is a person other than an officer or employee of the company or any other individual having a relationship which, in the opinion of the Company’s Board, would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. The NASDAQ listing rules provide that a director cannot be considered independent if:
| • | the director is, or at any time during the past three years was, an employee of the company; |
| 43 |
| • | the director or a family member of the director accepted any compensation from the company in excess of $120,000 during any period of twelve consecutive months within the three years preceding the independence determination (subject to certain exemptions, including, among other things, compensation for board or board committee service); |
| • | the director or a family member of the director is a partner in, controlling shareholder of, or an executive officer of an entity to which the company made, or from which the company received, payments in the current or any of the past three fiscal years that exceed 5% of the recipient’s consolidated gross revenue for that year or $200,000, whichever is greater (subject to certain exemptions; |
| • | the director or a family member of the director is employed as an executive officer of an entity where, at any time during the past three years, any of the executive officers of the company served on the compensation committee of such other entity; or |
| • | the director or a family member of the director is a current partner of the company’s outside auditor, or at any time during the past three years was a partner or employee of the company’s outside auditor, and who worked on the company’s audit. |
Under such definitions, we have no independent directors. However, our Common Stock is not currently quoted or listed on any national exchange or interdealer quotation system with a requirement that a majority of our Board be independent and, therefore, the Company is not subject to any director independence requirements.
Family Relationships
There are no family relationships among any of our officers or directors.
Involvement in Certain Legal Proceedings
Except as disclosed below, to our knowledge, none of our current directors or executive officers has, during the past ten years:
| ¨ | been convicted in a criminal proceeding or been subject to a pending criminal proceeding (excluding traffic violations and other minor offenses); |
| ¨ | had any bankruptcy petition filed by or against the business or property of the person, or of any partnership, corporation or business association of which he was a general partner or executive officer, either at the time of the bankruptcy filing or within two years prior to that time; |
| ¨ | been subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction or federal or state authority, permanently or temporarily enjoining, barring, suspending or otherwise limiting, his involvement in any type of business, securities, futures, commodities, investment, banking, savings and loan, or insurance activities, or to be associated with persons engaged in any such activity; |
| ¨ | been found by a court of competent jurisdiction in a civil action or by the SEC or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated; |
| 44 |
| ¨ | been the subject of, or a party to, any federal or state judicial or administrative order, judgment, decree, or finding, not subsequently reversed, suspended or vacated (not including any settlement of a civil proceeding among private litigants), relating to an alleged violation of any federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies including, but not limited to, a temporary or permanent injunction, order of disgorgement or restitution, civil money penalty or temporary or permanent cease-and-desist order, or removal or prohibition order, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or |
| ¨ | been the subject of, or a party to, any sanction or order, not subsequently reversed, suspended or vacated, of any self-regulatory organization (as defined in Section 3(a)(26) of the Exchange Act), any registered entity (as defined in Section 1(a)(29) of the Commodity Exchange Act), or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
Except as set forth above and in our discussion below in “Certain Relationships and Related Transactions,” none of our directors or executive officers has been involved in any transactions with us or any of our directors, executive officers, affiliates or associates which are required to be disclosed pursuant to the rules and regulations of the SEC.
We are not currently a party to any legal proceedings, the adverse outcome of which, individually or in the aggregate, we believe will have a material adverse effect on our business, financial condition or operating results.
Code of Business Conduct and Ethics
Our Board plans to adopt a written code of business conduct and ethics (“Code”) that applies to our directors, officers and employees, including our principal executive officer, principal financial officer and principal accounting officer or controller, or persons performing similar functions. We intend to post on our website a current copy of the Code and all disclosures that are required by law in regard to any amendments to, or waivers from, any provision of the Code.
Executive Compensation
The following table sets forth information regarding the total compensation received by, or earned by, our Chief Executive Officer, our President and Chief Operating Officer and our Chief Financial Officer (collective, the “named executive officers”) during the year ended December 31, 2018.
| Name and Principal Position | Year | Salary ($) | Option Awards ($) | Total ($) | ||||||||||||
| Terren S. Peizer,Chief Executive Officer, Treasurer, Secretary & Director | 2018 | -0- | -0- | -0- | ||||||||||||
| Joanne Wendy Kim,CPA, Chief Financial Officer | 2018 | -0- | -0- | -0- | ||||||||||||
| Clarence Ahlem,Chief Operating Officer | 2018 | -0- | -0- | -0- | ||||||||||||
| Christopher L. Reading,Chief Scientific Officer | 2018 | -0- | -0- | -0- | ||||||||||||
| 45 |
The following table represents information regarding thetotal planned compensation for our executive officers and directors of the Company to be paid following completion of the Offering and assuming we sell a minimum of 833,333 Shares for gross proceeds of $5,000,000 receive(1):
| Cash | Other | Total | ||||||||||
| Compensation | Compensation | Compensation | ||||||||||
| Name and Principal Position | ($)(1) | ($)(2)(3) | ($)(2)(3) | |||||||||
| Terren S. Peizer,Chief Executive Officer, Treasurer, Secretary & Director | $ | 450,000 | $ | 0.00 | $ | 450,000 | ||||||
| Joanne Wendy Kim, CPA, Chief Financial Officer | $ | 200,000 | $ | 0.00 | $ | 200,000 | ||||||
| Clarence Ahlem,Chief Operating Officer | $ | 220,000 | (4) | $ | 0.00 | $ | 220,000 | |||||
| Christopher L. Reading,Chief Scientific Officer | $ | 220,000 | (4) | $ | 0.00 | $ | 220,000 | |||||
| Robert J. Hariri,Chairman of the Executive Committee & Director | $ | 0.00 | $ | 0.00 | $ | 0.00 | ||||||
| Steve Gorlin,Director & Vice Chairman | $ | 0.00 | $ | 0.00 | $ | 0.00 | ||||||
| Richard A. Berman,Director | $ | 0.00 | $ | 0.00 | $ | 0.00 | ||||||
| Richard J. Berman,Director | $ | 0.00 | $ | 0.00 | $ | 0.00 | ||||||
| (1) | The cash compensation amounts reflect the full calendar year compensation that the Company intends to pay to its officers and directors from the proceeds of the Offering, following the completion of this Offering and assuming the Company is successful in raising at least $5,000,000 in net proceeds in the Offering. |
| (2) | Any values reported in the “Other Compensation”, if applicable, column represents the aggregate grant date fair value, computed in accordance with Accounting Standards Codification ("ASC") 718Share Based Payments, of grants of stock options to each of our named executive officers and directors. |
| (3) | The Company intends to grant equity compensation (in the form of stock options, warrants, and/or stock grants) in an aggregate amount up to 10% of the Company’s issued and outstanding Common Stock to its officers and directors following the completion of the Offering. The actual type and amounts of equity compensation to be paid to the Company’s officers and directors has not yet been determined. |
| (4) | Mr. Ahlem and Mr. Reading have historically served as independent contractors on a consulting basis and are responsible to pay their own taxes. Upon closing of the Offering, such persons will become full time employees, officers and directors of the Company. During the Company’s the last fiscal year, the Company paid consulting fees totaling $90,000 to Mr. Ahlem and paid consulting fees totaling $78,000 to Mr. Reading as compensation for their services as independent contractors. |
| 46 |
Employment Agreements
We do not currently have employment agreements with any of our officers or employees.
Item 4. Security Ownership of Management and Certain Securityholders
The following table shows the beneficial ownership of our Common Stock as of the date of this Annual Report held by (i) each person known to us to be the beneficial owner of more than 5% of any class of our shares; (ii) each director; (iii) each executive officer; and (iv) all directors and executive officers as a group. As of the date of this Annual Report, there were 43,334,000 shares of our Common Stock issued and outstanding.
Beneficial ownership is determined in accordance with the rules of the Commission, and generally includes voting power and/or investment power with respect to the securities held. Shares of Common Stock subject to options and warrants currently exercisable or which may become exercisable within 60 days of the date of this Annual Report, are deemed outstanding and beneficially owned by the person holding such options or warrants for purposes of computing the number of shares and percentage beneficially owned by such person, but are not deemed outstanding for purposes of computing the percentage beneficially owned by any other person. Except as indicated in the footnotes to this table, the persons or entities named have sole voting and investment power with respect to all shares of Common Stock shown as beneficially owned by them.
The percentages below are based on fully diluted shares of our Common Stock as of the date of this Annual Report.
| Number of | ||||||||||||
| shares of | ||||||||||||
Common Stock | ||||||||||||
| Beneficially | ||||||||||||
Owned as of | Percentage | |||||||||||
December 31, 2018 | Before Offering(2) | Beneficially Owned After Maximum Offering(2), (3) | ||||||||||
| Directors and Officers: | ||||||||||||
| Terren S. Peizer(1) | 43,334,000 | 100 | % | 84 | % | |||||||
| All directors and named executive officers as a group (1 person) | 43,334,000 | 100 | % | 84 | % | |||||||
| Greater than 5% Beneficial Owners: | ||||||||||||
| Terren S. Peizer(1) | 43,334,000 | 100 | % | 84 | % | |||||||
| (1) | Represents Common Stock beneficially owned and controlled by NeurMedix, LLC (formerly, Reserva, LLC), an entity wholly-owned and controlled by Terren S. Peizer, our founder, Director, CEO, Treasurer and Secretary. |
| (2) | Figures exclude the following: (a) any underlying shares of Common Stock issuable upon the exercise of any Company stock option grants, warrants or equity grants issued under the Company’s equity incentive plan, that are not currently exercisable or exercisable within 60 day of the date of this Annual Report. There have not been any such instruments issued to date. |
| (3) | Assumes that upon the sale of all common stock shares being offered by the Company in the Offering, the Company will have 51,667,333 Common Stock shares issued and outstanding. |
| 47 |
Item 5. Interest of Management and Others in Certain Transactions
Transactions with Related Persons
On December 9, 2014, Reserva, LLC, the sole shareholder of the Company, bought all of the assets related to NE3107 from Harbor Therapeutics, Inc., a wholly owned subsidiary of Harbor Diversified, Inc., and an unrelated party of the Company, for cash consideration of $2.5 Million on behalf of the Company. Pursuant to the Asset Purchase Agreement, Reserva, LLC acquired NE3107, which was in pre-clinical testing state, including inventory of NE3107 products and their components, trade secrets and proprietary information, licenses and other contract rights, patents and patent applications related to NE3107 use, production and manufacturing. The purchase of NE3107 was executed on behalf of the Company and thus has been recorded as a research and development license expense on the Company’s financial statements and related statements of operations. Reserva, LLC is wholly owned and controlled by Terren S. Peizer, our CEO, Treasurer, Secretary & Director. On February 16, 2015, Reserva, LLC assigned its right, title and interest to the patents, patent applications and trademarks acquired from Harbor Therapeutics, Inc., to the Company. On March 9, 2016, Reserva, LLC changed its name to NeurMedix, LLC. All of the activities related to NE3107 funded by NeurMedix, LLC (formerly Reserva, LLC) on behalf of the Company, were treated as a capital contribution by the sole shareholder through March 31, 2018.
Our mailing address is NeurMedix, Inc., 6165 Greenwich Drive, Suite 150, San Diego, California 92122, however, we also operate our business from offices maintained by Terren S. Peizer, our President, CEO, Director, founder and sole shareholder, and Mr. Peizer’s unrelated businesses, which are located at 11601 Wilshire Boulevard, Suite 1100, Los Angeles, California 90025.
We entered into an equity contribution agreement (“Contribution Agreement”), effective as of June 1, 2014, with NeurMedix, LLC (formerly, Reserva, LLC) and our CEO, Treasurer, Secretary, Director, and founder, Terren S. Peizer, pursuant to which the parties agreed that all activities of NeurMedix, LLC were made on behalf of the Company since all expenses related the Company’s business (since the Company’s inception) were funded and recorded by NeurMedix, LLC in 2014 and 2015. Pursuant to the terms of the Contribution Agreement, the parties also agreed that all capital expenditures and expenses paid for by NeurMedix, LLC on behalf or for the benefit of the Company (the “Company Payments”): (a) are comprehensive and represent a full and complete record of all such Company Payments; (b) pertain solely to the business and operations of the Company, (c) are in fact “equity contributions” to the Company by NeurMedix, LLC and/or Terren S. Peizer and are not intended to be repaid by the Company at any time, (d) shall not be reimbursable to NeurMedix, LLC or Mr. Peizer and the Company shall have no obligation to repay such Company Payments; and (e) the Company Payments shall not be deemed liabilities of the Company. The cumulative amount of such equity contributions was approximately $5.3 million and $5.5 million as of December 31, 2017 and March 31, 2018, respectively. Subsequent to March 31, 2018, there was no further equity contributions. Commencing April 1, 2018, Terren S. Peizer, the Parent’s sole stockholder, CEO and founder, has paid or advanced monies on behalf of the Company to pay certain operating expenses of the Company and expenses related to this Regulation A offering. From April 1, 2018 through December 31, 2018, the amounts advanced totaled approximately $423,000 and were recorded as liabilities in the accompanying balance sheet. The amounts advanced to the Company by its Parent and Terren S. Peizer may be reimbursed to him out of the net proceeds received by the Company from the Regulation A+ Offering or its capital raising efforts.
| 48 |
To the best of our knowledge, since the period from inception (November 12, 2014) to the date of this Annual Report, other than as set forth above, there were no material transactions, or series of similar transactions, or any currently proposed transactions, or series of similar transactions, to which we were or are to be a party, in which any director or executive officer, or any security holder who is known by us to own of record or beneficially own more than 5% of any class of our Common Stock, or any member of the immediate family of any of the foregoing persons, has an interest (other than compensation to our officers and directors in the ordinary course of business).
Review, Approval and Ratification of Related Party Transactions
Given our small size and limited financial resources, we have not adopted formal policies and procedures for the review, approval or ratification of transactions, such as those described above, with our executive officer(s), Director(s) and significant stockholders. We intend to establish formal policies and procedures in the future, once we have sufficient resources and have appointed additional Directors, so that such transactions will be subject to the review, approval or ratification of our Board of Directors, or an appropriate committee thereof. On a moving forward basis, our Directors will continue to approve any related party transaction.
Item 6. Other Information
Change of Selling Agent
In January 2019, NeurMedix Inc., a Delaware corporation (“NeurMedix” or the “Company”), engaged NMS Capital Advisors, LLC, a registered broker/dealer, as its new managing selling agent (the “Selling Agent”), in place of its previous selling agent, Westpark Capital, Inc. Under the terms of its engagement agreement with the Company, the Selling Agent may engage one or more sub-selling agents. However, neither the Selling Agent nor any sub-selling agent shall have any marketing or sales obligations other than to process indications of interest forwarded to the Selling Agent or sub-selling agent by the Company or its management. As of the date of this Annual Report, Selling Agent is conducting its due diligence on the Company, finalizing a formal Selling Agent Agreement with the Company and obtaining approval of the Financial Industry Regulatory Authority (“FINRA”) of the compensation terms to be included in the long-form Selling Agent Agreement.
Under the terms of its engagement agreement with NMS Capital Advisors, LLC, the Company has agreed to pay the Selling Agent a commission and fee equal to five and a half percent (5.5%) of all gross proceeds received by the Company in the Offering in consideration for the Selling Agent’s commitment to process orders on behalf of the Company and its management.
A copy of the engagement agreement is filed asExhibit A to this Annual Report on Form 1-K and any summary of the terms of such documents are subject to, and qualified in their entirety by, the full text of such documents, which are incorporated herein by reference.
| 49 |
Item 7. Financial Statements
NEURMEDIX, INC.
TABLE OF CONTENTS
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
NeurMedix, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of NeurMedix, Inc. (the “Company") as December 31, 2018 and 2017, and the related statements of operations, changes in stockholder’s deficit, and cash flows for each of the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2018 and 2017, and the results of its operations and its cash flows for each of the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has experienced significant losses and negative cash flows from operations since inception and is wholly dependent upon funding from its sole shareholder, these factors raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
| /s/ EisnerAmper LLP | |
| We have served as the Company’s auditor since 2017. | |
| EISNERAMPER LLP | |
| Iselin, New Jersey | |
| April 30, 2019 | |
| F-1 |
BALANCE SHEETS
| December 31, | December 31, | |||||||
| 2018 | 2017 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash | $ | 5,376 | $ | 13,591 | ||||
| Prepaid expenses and other current assets | 189,395 | 62,646 | ||||||
| Total current assets | 194,771 | 76,237 | ||||||
| Furniture and fixtures, net | 2,337 | 7,256 | ||||||
| Deposit | 32,442 | 32,442 | ||||||
| Total assets | $ | 229,550 | $ | 115,935 | ||||
| LIABILITIES AND STOCKHOLDER'S DEFICIT | ||||||||
| Current liabilities: | ||||||||
| Accrued expenses | $ | 49,750 | $ | 10,000 | ||||
| Accounts payable | 375,259 | 396,515 | ||||||
| Advances from shareholder (Note 1) | 422,809 | - | ||||||
| Deferred rent payable | 829 | 7,049 | ||||||
| Other liabilities | 5,376 | - | ||||||
| Total current liabilities | 854,023 | 413,564 | ||||||
| Deferred rent payable | - | 829 | ||||||
| Total liabilities | 854,023 | 414,393 | ||||||
| Commitments and contingencies (Note 5) | ||||||||
| Stockholder's deficit: | ||||||||
| Preferred stock, par value $.000001, authorized 40,000,000 shares, no shares issued or outstanding at December 31, 2018 and December 31, 2017, respectively | - | - | ||||||
| Common stock, par value $0.000001 per share - 200,000,000 shares authorized; 43,334,000 shares issued and outstanding at December 31, 2018 and December 31, 2017, respectively | 43 | 43 | ||||||
| Additional paid-in capital | 5,723,148 | 5,298,407 | ||||||
| Accumulated deficit | (6,347,664 | ) | (5,596,908 | ) | ||||
| Total stockholder's deficit | (624,473 | ) | (298,458 | ) | ||||
| Total liabilities and stockholder's deficit | $ | 229,550 | $ | 115,935 | ||||
See accompanying notes to financial statements.
| F-2 |
STATEMENTS OF OPERATIONS
| Year ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Operating expenses: | ||||||||
| Research and development | $ | (9,563 | ) | $ | (53,426 | ) | ||
| General and administrative | (741,193 | ) | (753,558 | ) | ||||
| Total operating expenses | (750,756 | ) | (806,984 | ) | ||||
| Net loss | $ | (750,756 | ) | $ | (806,984 | ) | ||
| Weighted-average common shares outstanding, basic and diluted | 43,334,000 | 43,334,000 | ||||||
| Net loss per share, basic and diluted | $ | (0.02 | ) | $ | (0.02 | ) | ||
See accompanying notes to financial statements.
| F-3 |
STATEMENTS OF CHANGES IN STOCKHOLDER’S DEFICIT FOR THE
YEARS ENDED DECEMBER 31, 2018 AND 2017
| Total | ||||||||||||||||||||
| Common Stock | Additional | Accumulated | Stockholder's (Deficit) | |||||||||||||||||
| Shares | Amount | Paid-In Capital | Deficit | /Equity | ||||||||||||||||
| Balance at December 31, 2016 | 43,334,000 | $ | 43 | $ | 4,852,844 | $ | (4,789,924 | ) | $ | 62,963 | ||||||||||
| Capital contributions | - | - | 445,563 | - | 445,563 | |||||||||||||||
| Net loss | - | - | - | (806,984 | ) | (806,984 | ) | |||||||||||||
| Balance at December 31, 2017 | 43,334,000 | 43 | 5,298,407 | (5,596,908 | ) | (298,458 | ) | |||||||||||||
| Capital contributions | - | - | 424,741 | - | 424,741 | |||||||||||||||
| Net loss | - | - | - | (750,756 | ) | (750,756 | ) | |||||||||||||
| Balance at December 31, 2018 | 43,334,000 | $ | 43 | $ | 5,723,148 | $ | (6,347,664 | ) | $ | (624,473 | ) | |||||||||
See accompanying notes to financial statements.
| F-4 |
STATEMENTS OF CASH FLOWS
| Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||
| Net loss | $ | (750,756 | ) | $ | (806,984 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 4,919 | 4,920 | ||||||
| Deferred rent | (7,049 | ) | (5,277 | ) | ||||
| Write-off of deferred financing costs | - | 47,500 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | (126,749 | ) | (52,201 | ) | ||||
| Accrued expenses | 39,750 | (9,525 | ) | |||||
| Accounts payable | (21,256 | ) | 389,595 | |||||
| Other liabilities | 5,376 | - | ||||||
| Net cash used in operating activities | (855,765 | ) | (431,972 | ) | ||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||
| Advances from shareholder | 422,809 | - | ||||||
| Proceeds from shareholder contributions | 424,741 | 445,563 | ||||||
| Net cash provided by financing activities | 847,550 | 445,563 | ||||||
| Net change in cash | (8,215 | ) | 13,591 | |||||
| Cash, beginning of period | 13,591 | - | ||||||
| Cash, end of period | $ | 5,376 | $ | 13,591 | ||||
See accompanying notes to financial statements.
| F-5 |
NOTES TO FINANCIAL STATEMENTS
| 1. | Description of Business and Going Concern Uncertainty |
Neurmedix, Inc. (the “Company”) was incorporated in the state of Delaware on November 12, 2014 and is headquartered in San Diego, California. The Company was formed to discover and develop transformative therapeutics for neurological diseases.
The Company amended its articles of incorporation in February 2017, increasing the number of shares authorized and converted the 1,000 common shares previously issued to 71,428,571 shares. On June 29, 2017, the Company reduced the shares outstanding to 50,000,000 shares held by the sole shareholder without any change in the par value per share. On September 27, 2017, the Company reduced the shares outstanding to 43,334,000 shares held by the sole shareholder without any change in the par value per share. For financial reporting purposes, this has been reflected as if it were a stock split and all share and per share amounts have been retroactively adjusted. Since the par value of the common stock remained at $0.000001 per share, the value of the “Common Stock” retroactively decreased to reflect the par value of the restated outstanding shares, with a corresponding increase to “Additional Paid in Capital.” The amended articles of incorporation permit the issuance of two classes of stock – common and preferred, and permits the Board of Directors to set the rights and privileges of any class of preferred stock. No preferred shares have been issued through the date of this report. All share information in this report has been restated to reflect this recapitalization.
In December 2014, Reserva, LLC,wholly owned and controlled by Terren S. Peizer, our CEO, Treasurer, Secretary & Director, bought all of the assets related to NE-3107, a pre-clinical novel immune-modulatory therapeutic for use in neurological diseases for cash consideration of $2.5 million on behalf of the Company from Harbor Therapeutics, Inc. (an unrelated party).
On February 16, 2015, Reserva, LLC assigned its right, title and interest to the patents, patent applications and trademarks acquired from Harbor Therapeutics, Inc. to the Company. On March 9, 2016, Reserva, LLC changed its name to NeurMedix, LLC (“Parent”).
All activities related to NE-3107 and related molecules are funded by Parent on behalf of the Company, and are treated as a capital contribution by the sole shareholder.
The Company has entered into an equity contribution agreement (“Contribution Agreement”), effective as of June 1, 2014, with Parent and the CEO, Treasurer, Secretary, Director, and founder, Terren S. Peizer, pursuant to which the parties agreed that all activities of Parent were made on behalf of the Company since all expenses related to the Company’s business (since the Company’s inception) were funded and recorded by the Parent in 2014 and 2015 . Pursuant to the terms of the Contribution Agreement, the parties also agreed that all capital expenditures and expenses paid for by Parent on behalf of or for the benefit of the Company (the “Company Payments”): (a) are comprehensive and represent a full and complete record of all such Company Payments, (b) pertain solely to the business and operations of the Company, (c) are in fact “equity contributions” to the Company by Parent and/or Terren S. Peizer and are not intended to be repaid by the Company at any time, (d) shall not be reimbursable to parent or Mr. Peizer and the Company shall have no obligation to repay such Company Payments; and (e) the Company Payments shall not be deemed liabilities of the Company. The cumulative amount of such equity contributions was approximately $5.3 million and $5.5 million as of December 31, 2017 and March 31, 2018, respectively. Subsequent to March 31, 2018, there was no further equity contributions. Commencing April 1, 2018, Terren S. Peizer, the Parent’s sole stockholder, CEO and founder, has paid or advanced monies on behalf of the Company to pay certain operating expenses of the Company and expenses related to the Regulation A offering. From April 1, 2018 through December 31, 2018, the amounts advanced totaled approximately $423,000 and were recorded as liabilities in the accompanying balance sheet. The amounts advanced to the Company by its Parent and Terren S. Peizer may be reimbursed to him out of the net proceeds received by the Company from the Regulation A+ Offering or its capital raising efforts.
| F-6 |
Going Concern Uncertainty
The Company’s operations are subject to a number of factors that can affect its operating results and financial conditions. Such factors include, but are not limited to: the results of clinical testing and trial activities of the Company’s products, the Company’s ability to obtain regulatory approval to market its products, competition from products manufactured and sold or being developed by other companies, the price of, and demand for, Company products, the Company’s ability to negotiate favorable licensing or other manufacturing and marketing agreements for its products, and the Company’s ability to raise capital. The Company’s financial statements have been prepared assuming the Company will continue as a going concern, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The Company has experienced losses since inception and has an accumulated deficit of approximately $6.3 million at December 31, 2018. The Company has limited cash resources of its own. The Company has funded operations exclusively with the proceeds from capital contributions and advances in the form of expenditures paid by Parent on behalf of the Company, and there is no formal agreement for such arrangement to continue.
The future viability of the Company is largely dependent upon its ability to raise additional capital to finance its operations. Management expects that future sources of funding may include sales of equity, obtaining loans, or other strategic transactions. Although management continues to pursue these plans, there is no assurance that the Company will be successful in obtaining sufficient financing on terms acceptable to the Company to fund continuing operations, if at all. These circumstances raise substantial doubt on the Company’s ability to continue as a going concern. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
| F-7 |
The Company is undertaking a “best efforts” offering of its common stock to raise additional capital. There is no assurance that such an offering will be successful. Upon successful completion of such
offering, the Company plans to institute a stock option plan, hire employees and enter into employment agreements, engage members of the Board of Directors and enter into agreements with organizations to conduct clinical trials and other research and development processes.
| 2. | Significant Accounting Policies |
Basis of Presentation
The accompanying financial statements and related notes have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”).
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts expensed during the reporting period. Management bases its estimates on historical experience and on various other assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results could differ from those estimates.
Prepaid Expenses
Prepaid expenses represent costs incurred that benefit future periods. These costs are amortized over a specific time-period based on the specific agreements.
Property and Equipment
Property and equipment are recorded at cost, net of accumulated depreciation. Depreciation is recorded using the straight-line method over the estimated useful lives of the assets, three to five years. Major replacements and improvements are capitalized, while general repairs and maintenance are expensed as incurred.
Long-Lived Assets
The Company evaluates the carrying amount of its long-lived assets whenever events or changes in circumstances indicate that the assets may not be recoverable. An impairment loss is recognized when estimated future cash flows expected to result from the use of the asset and its eventual disposition is less than the carrying amount of the asset. To date, there have been no such impairment losses.
| F-8 |
Research and development
Research and development expenses are charged to expense as incurred. Research and development expenses include laboratory supplies, consulting costs, external contract research and development expenses, and allocated overhead, including rent, equipment depreciation and utilities. Advance payments for goods or services for future research and development activities are deferred and expensed as the goods are delivered or the related services are performed.
These estimates are based on communications with the third-party service providers and the Company’s estimates of accrued expenses and on information available at each balance sheet date. If the actual timing of the performance of services or the level of effort varies from the estimate, the Company will adjust the accrual accordingly.
Income taxes
The Company uses the asset-and-liability method for deferred income taxes. Deferred tax assets and liabilities are recognized for future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on the deferred tax assets and liabilities of a change in tax rate is recognized in the period that includes the enactment date. A valuation allowance is recorded if it is more likely than not that some portion or all the deferred tax assets will not be recovered in future periods.
The Company assesses uncertain tax positions using a two-step approach of recognition and measurement. The first step involves assessing whether the tax position is more likely than not to be sustained upon examination based upon its technical merits. The second step involves measurement of the amount to recognize.
As of December 31, 2018 and 2017, the Company had no uncertain tax positions. There was no interest or penalties related to income taxes for the year ended December 31, 2018 and 2017.
Net Loss Per Share
Basic net loss per common share is calculated by dividing the net loss attributable to common stockholders by the weighted-average number of common shares outstanding during the period, without consideration for potentially dilutive securities. Diluted net loss per share is computed by dividing the net loss attributable to common stockholders by the weighted-average number of common shares and potentially dilutive securities outstanding for the period determined using the treasury-stock and if-converted methods. Under this method, options and warrants are assumed to be exercised at the beginning of the period (or at the time of issuance, if later), and as if funds obtained thereby were used to purchase common stock at the average market price during the period. There were no potentially dilutive securities outstanding during the years ended December 31, 2018 and 2017.
| F-9 |
Recent accounting pronouncements
The Company considered the applicability and impact of recent accounting pronouncements and determined them to be either not applicable or expected to have minimal impact on our balance sheets or statements of operations, except for the following.
In February 2016, the Financial Accounting Standards Board issued Accounting Standards Update (“ASU”) 2016-02,Leases (Topic 842). This ASU requires a lessee to recognize in the statement of financial position a liability to make lease payments (the lease liability) and a right-to-use asset representing its right to use the underlying asset for the lease term. This ASU is effective for the Company’s interim periods within fiscal years beginning after December 15, 2019, with early adoption permitted. The Company is currently evaluating the impact of adoption of this update on its financial statements.
| 3. | Significant contracts |
The Company entered into an agreement on June 29, 2017 for marketing efforts associated with Company’s capital raising efforts for total cash consideration of $250,000. In 2017, $100,000 was advanced at signing and $50,000 of that amount expensed during the fiscal year ended December 31, 2017 proportionate to the services provided for the pre-development of the marketing campaign and materials.
In February 2018, the second payment of $100,000 was made and the funds were used in July 2018 to further develop, update and revise the market campaign and materials. In December 2018, an additional amount of $200,000 was paid for further market campaign and materials for capital raising efforts to be launched in 2019. The total amount recognized in general and administrative expenses in the accompanying statementsof operations for the fiscal year ended December 31, 2018 was $216,000 and the remaining $133,000 is reported as prepaid expenses and other current assets in the accompanying balance sheetsat December 31, 2018. The prepaid amounts are being amortized over a three-month period in which the services and materials are developed.
| F-10 |
| 4. | Furniture and fixtures, net |
Furniture and fixtures, net, consists of the following:
| December 31, | December 31, | |||||||
| 2018 | 2017 | |||||||
| Furniture and fixtures | $ | 19,130 | 19,130 | |||||
| Less: accumulated depreciation | (16,793 | ) | (11,874 | ) | ||||
| Furniture and fixtures, net | $ | 2,337 | $ | 7,256 | ||||
Depreciation expense was approximately $4,900 for the years ended December 31, 2018 and 2017, which is included in the general and administrative expense in the accompanying statements of operations.
| 5. | Commitments and Contingencies |
Facility Lease Agreement
The Company entered into an agreement to lease office space for its headquarters in San Diego, California on October 20, 2015. This operating lease agreement expired February 1, 2019 and was extended for six months to August 2019. This lease agreement provides for escalation of rent payments each year. The Company records rent expense on a straight-line basis over the term of the lease. As of December 31, 2018, future minimum commitments under facility operating leases were approximately $66,000.
The Company has provided a security deposit in the amount of $32,442, which is held by the lessor in connection with the Company’s facility lease agreement.
Rent expense was approximately $87,000 for each of the years ended December 31, 2018 and 2017, respectively, which is included in general and administrative expense in the accompanying statements of operations.
Contingencies
The Company may be subject to a variety of claims and lawsuits in the ordinary course of business. As of December 31, 2018, management believes there are no such outstanding claims or lawsuits that, individually or in the aggregate, would have a material adverse effect on the Company’s financial position, the results of its operations, or its cash flows.
At December 31, 2018, a payment of past warehousing fees to a certain vendor is contingent upon obtaining funding through our capital raise efforts. Although the Company is undertaking a “best efforts” offering of its common stock to raise additional capital, there is no assurance that such an offering will be successful. Upon successful completion of such offering, the contingent liability would become due and payable. At December 31, 2018, the estimated possible amount that potentially may be due upon securing funding was estimated at approximately $310,000. This amount has not been recognized in the accompanying financial statements at December 31, 2018 as the funding event was not deemed probable at date of this reporting.
| F-11 |
Indemnifications
In the normal course of business, the Company enters into contracts and agreements that contain a variety of representations and warranties and provide for general indemnifications. The Company’s exposure under these agreements is unknown because it involves claims that may be made against the Company in the future, but have not yet been made. To date, the Company has not paid any claims or been required to defend any action related to its indemnification obligations. However, the Company may record charges in the future as a result of these indemnification obligations.
| 6. | Income Taxes |
The Company is a C-corporation and has not filed a tax return in 2014, 2015, 2016, 2017 or 2018. All activities related to the Company’s business were recorded by the Parent in 2014, 2015, 2016, 2017 and 2018 and any tax benefits related to such activities were recognized by Parent. Deferred income taxes resulting from book versus tax basis differences would not have been significant and would have a full valuation allowance against such deferred tax assets. Accordingly, there are no current or deferred tax expenses on the Company’s financial statements for the year ended December 31, 2018 and 2017, and there are no operating loss carryforwards at December 31, 2018.
As of December 31, 2018, the Company did not have a liability related to unrecognized tax benefits.
| 7. | Subsequent Events |
The Company evaluated subsequent events through April 30, 2019, the date that the accompanying financial statements were available to be issued. There were no material subsequent events that required disclosure in these financial statements, except that advances from shareholder made by the Parent for the period from January 1, 2019 to April 30, 2019 were approximately $80,000 .
The Company received subscriptions to purchase common stock in 2018 of $5,376, which is included in the accompanying balance sheet at December 31, 2018. The total amount related to subscriptions to purchase common stock received at April 30, 2019 was $90,000. The subscriptions to purchase common stock have not yet been accepted by the Company.
| F-12 |
Item 7. Exhibits
The exhibits listed in the following Exhibit Index are filed as part of this Registration Statement.
| Exhibit No. | Description | |
| EX1A-1A | Regulation A Offering Engagement Agreement with Selling Agent |
SIGNATURES
Pursuant to the requirements of Regulation A, the issuer has duly caused this annual report on Form 1-K to be signed on its behalf by the undersigned, thereunto duly authorized, in Los Angeles California on April 30, 2019.
| NEURMEDIX, INC.. | ||
| By: | /s/ Terren S. Peizer | |
| Name: | Terren S. Peizer | |
| Title: | Chief Executive Officer, Treasurer & Secretary | |
Pursuant to the requirements of Regulation A, this report has been signed below by the following persons on behalf of the issuer in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Joanne Wendy Kim | Chief Executive Officer and Principal Accounting Officer | April 30, 2019 | ||
| Joanne Wendy Kim | ||||
| /s/ Terren S. Peizer | April 30, 2019 | |||
| Terren S. Peizer | Sole Director |