UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| | | | | |
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2024
OR
| | | | | |
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _______________ to _______________
Commission file number 001-38485
Amneal Pharmaceuticals, Inc.
(Exact name of registrant as specified in its charter)
Delaware
(State or other jurisdiction of incorporation or organization)
400 Crossing Boulevard, Bridgewater, NJ
(Address of principal executive offices)
93-4225266
(I.R.S. Employer Identification No.)
08807
(Zip Code)
(908) 947-3120
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| | | | | | | | |
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
| Class A Common Stock, par value $0.01 per share | AMRX | The Nasdaq Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
| | | | | | | | | | | |
| Large accelerated filer | ☒ | Accelerated filer | ☐ |
| Non-accelerated filer | ☐ | Smaller reporting company | ☐ |
| | | Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management's assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☒
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the registrant’s outstanding shares of common stock, other than shares held by persons who may be deemed affiliates of the registrant, computed by reference to the price at which the registrant’s common stock was last sold on The Nasdaq Stock Market LLC as of the last business day of the registrant’s most recently completed second fiscal quarter (June 30, 2024), was approximately $1.6 billion.
As of February 14, 2025, there were 309,966,341 shares of the registrant’s Class A common stock outstanding, with a par value of $0.01.
DOCUMENTS INCORPORATED BY REFERENCE
Certain information required to be furnished pursuant to Part III of this Form 10-K will be set forth in, and is hereby incorporated by reference herein from, the registrant’s definitive proxy statement for its 2025 Annual Meeting of Stockholders, to be filed by the registrant with the Securities and Exchange Commission pursuant to Regulation 14A no later than 120 days after December 31, 2024 (the “2025 Proxy Statement”).
Amneal Pharmaceuticals, Inc.
Table of Contents
Cautionary Note Regarding Forward-Looking Statements
This Annual Report on Form 10-K and other publicly available documents of Amneal Pharmaceuticals, Inc. contain “forward-looking statements” within the meaning of the safe harbor provisions of the United States (“U.S.”) Private Securities Litigation Reform Act of 1995. Management and representatives of Amneal Pharmaceuticals, Inc. and its subsidiaries (“the Company”, “we”, “us” or “our”) also may from time to time make forward-looking statements. Forward-looking statements do not relate strictly to historical or current facts and reflect management’s assumptions, views, plans, objectives and projections about the future. Forward-looking statements may be identified by the use of words such as “plans,” “expects,” “will,” “anticipates,” “targets,” “estimates,” and other words of similar meaning in conjunction with, among other things: discussions of future operations; expected operating results and financial performance; impact of planned acquisitions and dispositions; our strategy for growth; product development; regulatory approvals; market position and expenditures.
Because forward-looking statements are based on current beliefs, expectations and assumptions regarding future events, they are subject to uncertainties, risks and changes that are difficult to predict and many of which are outside of our control. Investors should realize that if underlying assumptions prove inaccurate, known or unknown risks or uncertainties materialize, or other factors or circumstances change, our actual results and financial condition could vary materially from expectations and projections expressed or implied in our forward-looking statements. Investors are therefore cautioned not to rely on these forward-looking statements.
Summary of Material Risks
Risks and uncertainties that make an investment in the Company speculative or risky or that could cause our actual results to differ materially from the forward-looking statements contained in this Annual Report on Form 10-K, include, but are not limited to:
•our ability to successfully develop, license, acquire and commercialize new products on a timely basis;
•the competition we face in the pharmaceutical industry from brand and generic drug product companies, and the impact of that competition on our ability to set prices;
•our ability to obtain exclusive marketing rights for our products;
•the impact of illegal distribution and sale by third parties of counterfeit versions of our products or stolen products;
•the impact of negative market perceptions of us and the safety and quality of our products;
•our revenues are derived from the sales of a limited number of products, a substantial portion of which are through a limited number of customers;
•the continuing trend of consolidation of certain customer groups;
•our dependence on third-party suppliers and distributors for raw materials for our products and certain finished goods;
•the imposition of tariffs may adversely affect our business, results of operations and financial condition
•legal, regulatory and legislative efforts by our brand competitors to deter competition from our generic alternatives;
•our dependence on information technology systems and infrastructure and the potential for cybersecurity incidents, and risks associated with artificial intelligence;
•the impact of a prolonged business interruption within our supply chain;
•our ability to attract, hire and retain highly skilled personnel;
•risks related to federal regulation of arrangements between manufacturers of branded and generic products;
•our reliance on certain licenses to proprietary technologies from time to time;
•the significant amount of resources we expend on research and development (“R&D”);
•the risk of claims brought against us by third parties such as those described in Note 20. Commitments and Contingencies - Other Litigation Related to the Company’s Business;
•risks related to changes in the regulatory environment, including U.S. federal and state laws related to government contracting, healthcare fraud abuse and health information privacy and security and changes in such laws;
•changes to Food and Drug Administration (“FDA”) product approval requirements;
•the impact of healthcare reform and changes in coverage and reimbursement levels by governmental authorities and other third-party payers;
•our dependence on third-party agreements for a portion of our product offerings;
•our substantial amount of indebtedness and our ability to generate sufficient cash to service our indebtedness in the future, and the impact of interest rate fluctuations on such indebtedness;
•our potential expansion into additional international markets subjecting us to increased regulatory, economic, social and political uncertainties;
•our ability to identify, make and integrate acquisitions or investments in complementary businesses and products on advantageous terms;
•the impact of global economic, political or other catastrophic events;
•our obligations under a tax receivable agreement may be significant;
•the high concentration of ownership of our Class A common stock and the fact that we are controlled by the Amneal Group (as defined below in Item 1. Business); and
•such other factors as may be set forth elsewhere in this Annual Report on Form 10-K, particularly in the section entitled 1A. Risk Factors and our public filings with the SEC.
Investors also should carefully read the Risk Factors described in Item 1A. Risk Factors for a description of certain risks that could, among other things, cause our actual results to differ materially from those expressed in our forward-looking statements. Investors should understand that it is not possible to predict or identify all such factors and should not consider the risks described above and in Item 1A. Risk Factors to be a complete statement of all potential risks and uncertainties. The Company does not undertake to publicly update any forward-looking statement that may be made from time to time, whether as a result of new information or future events or developments.
PART I.
Item 1. Business
Overview
Amneal Pharmaceuticals, Inc. (the “Company”, “we,” “us,” or “our”) is a global biopharmaceutical company that develops, manufactures, markets, and distributes a diverse portfolio of essential medicines. Our Affordable Medicines segment includes retail generics, injectables, and biosimilars. In our Specialty segment, we offer a portfolio of branded pharmaceuticals focused primarily on central nervous system and endocrine disorders. Through our AvKARE segment, we are a distributor of pharmaceuticals and other products for the U.S. federal government, retail, and institutional markets. We operate principally in the United States (“U.S.”), India, and Ireland. Refer to the section “Segments of the Business” below for an overview of our segments, including the change in name of the Affordable Medicines segment.
Corporate Structure
We are a holding company, whose principal assets are common units (the “Amneal Common Units”) of Amneal Pharmaceuticals, LLC (“Amneal”). Immediately prior to the Reorganization (as defined herein), we held 50.4% of the Amneal Common Units and the group, together with their affiliates and certain assignees, who owned Amneal when it was a private company (the “Members” or the “Amneal Group”) held the remaining 49.6%. On November 7, 2023, we implemented a plan pursuant to which the Company and Amneal reorganized and simplified our corporate structure by eliminating our umbrella partnership-C-corporation structure and converting to a more traditional C-corporation structure whereby all stockholders hold their voting and economic interests directly through the public company (the “Reorganization”). Effective with the Reorganization, we hold 100% of the Amneal Common Units and continue to consolidate the financial statements of Amneal and its subsidiaries. Refer to Note 1. Nature of Operations in our consolidated financial statements for additional information about the Reorganization.
Although we had a minority economic interest in Amneal prior to March 2023, we were Amneal’s sole managing member (and we continue to be the sole managing member), having the sole voting power to make all of Amneal’s business decisions and control its management. Therefore, we consolidated the financial statements of Amneal and its subsidiaries for all periods prior to the Reorganization. We recorded non-controlling interests for the portion of Amneal’s economic interests that we did not hold prior to the Reorganization.
Alliance and Collaboration
Collaboration to Develop and Supply Medicines for Obesity and Metabolic Diseases
On September 30, 2024, we entered into a collaboration agreement to develop and supply a new portfolio of weight loss medicines globally with Metsera, Inc. (“Metsera”), a clinical stage biopharmaceutical company (the “Metsera Agreement”). We will serve as Metsera’s preferred supply partner for developed markets, including the United States and Europe. In addition, we have been granted an exclusive license to commercialize Metsera products covered under the agreement in selected emerging markets, including India and certain countries in Southeast Asia, Africa and the Middle East.
Under the terms of the Metsera Agreement, we will be responsible for performing certain development activities on behalf of Metsera. Upon Metsera obtaining regulatory approval for any or all the weight loss medicines referred to above, we will manufacture commercial products on behalf of Metsera.
We plan to construct two new greenfield manufacturing facilities in India; one for peptide synthesis and one for sterile fill-finish manufacturing. Metsera will contribute an agreed percentage of the construction costs, up to $100 million, subject to annual maximums, as defined in the Metsera agreement.
The initial term of the Metsera Agreement is seven years from the first commercial sale. Metsera has the sole right to renew the agreement for an additional five-year period. Following this initial renewal, the agreement may be extended by mutual written consent.
License Agreement with Zambon Biotech
On February 23, 2024, we entered into a license, distribution and supply agreement with Zambon Biotech S.A. (“Zambon”) granting Zambon the exclusive rights to seek regulatory approval and commercialize IPX203 in Europe (the “Zambon License Agreement”). The term for the Zambon License Agreement is 15 years commencing from the commercial launch of the product, which can automatically renew for successive two-year periods unless either party provides notice declining such
renewal at least one year in advance. Zambon will be responsible for the performance of all R&D activities, regulatory approval, commercialization, and marketing activities for the territories in the agreement to be conducted to obtain regulatory approval for each product. Upon achieving regulatory approval for products, we will be responsible for manufacturing and supplying products to Zambon.
As of December 31, 2024, IPX203 had not been approved for sale outside of the United States, where it is marketed as CREXONT®. CREXONT® (combination of carbidopa and levodopa extended release capsules) is indicated in the United States for the treatment of Parkinson’s disease.
Knight Therapeutics International S.A. License Agreement
On January 24, 2024, we entered into a 15-year license, distribution and supply agreement with Knight Therapeutics International S.A. (“Knight”) granting Knight the exclusive rights to seek regulatory approval and commercialize IPX203 in Canada and Latin America (the “Knight License Agreement”). The Knight License Agreement will automatically renew for successive two-year periods unless either party provides notice declining such renewal at least one year in advance.
Knight will be responsible for the performance of all R&D activities, regulatory approval, commercialization, and marketing activities for the territories in the agreement to be conducted to obtain regulatory approval for each product. Upon achieving regulatory approval for products, Amneal will be responsible for manufacturing and supplying products to Knight.
As of December 31, 2024, IPX203 had not been approved for sale outside of the United States, where it is marketed as CREXONT®.
ONGENTYS® License and Supply Agreement
On December 5, 2023, we entered into a license agreement with BIAL-Portela & Ca., S.A. (“BIAL”) for the exclusive rights to market and distribute ONGENTYS® (opicapone) in the U.S. starting on December 18, 2023 and ending at such time when generic opicapone sales reach certain predetermined thresholds (the “BIAL Agreement”). ONGENTYS® is BIAL’s proprietary, once-daily, peripherally-acting, highly-selective catechol-O-methyltransferase inhibitor approved by the U.S. Food and Drug Administration (the “FDA”) in 2020 as an add-on treatment to carbidopa/levodopa in patients with Parkinson’s disease experiencing “off” episodes. Under the BIAL Agreement, we are responsible for commercialization and marketing of ONGENTYS® in the U.S. and BIAL is responsible for manufacturing and supply. We commenced distribution of ONGENTYS® in early 2024.
Orion Corporation License Agreement
On December 28, 2022, we signed a long-term license agreement with Orion Corporation (“Orion”), a globally operating Finnish pharmaceutical company, to commercialize a number of our complex generic products in most parts of Europe, Australia and New Zealand (the “Orion Agreement”). The initial term of the Orion Agreement commences upon commercial launch of the products and will continue for eight years. The Orion Agreement will automatically renew for successive two-year terms unless either party declines such renewal in writing at least one year in advance. As of December 31, 2024, the initial term has not commenced.
Licensing and Supply Agreement with mAbxience S.L.
On May 7, 2018, we entered into a licensing and supply agreement with mAbxience S.L. (“mAbxience”), for its biosimilar candidate for Avastin® (bevacizumab). We are the exclusive partner in the U.S. market. On April 13, 2022, the FDA approved our biologics license application for bevacizumab-maly, a biosimilar referencing Avastin®.
License and Supply Agreement with Kashiv Biosciences LLC
In December 2022, we entered into a development and supply agreement specific to four generic product candidates with Kashiv Biosciences LLC (“Kashiv”). Pursuant to the development supply agreement, we maintained a right of first offer and negotiation to the licensing of each generic product candidate. In March 2024, we entered into a license and supply agreement with Kashiv for the development and commercialization of a long-acting injectable (the “Injectable License and Supply Agreement"). The existing development supply agreement remains effective for the remaining three generic product candidates.
Subject to the terms of the Injectable License and Supply Agreement, we are responsible for development, regulatory approval, and commercialization of the product candidate in the U.S., whereas Kashiv is responsible for development and regulatory approval of the product candidate for all other territories outside the U.S. Contingent upon Kashiv obtaining regulatory approval outside the U.S., we shall manufacture the commercial supply for Kashiv at a stated price. The term of the agreement is 10 years from the respective product’s launch date in the U.S. As of December 31, 2024, the term has not commenced.
For additional information about our alliance and collaboration agreements, refer to Note 5. Alliance and Collaboration and Note 23. Related Party Transactions in our consolidated financial statements.
Acquisition
Baclofen Franchise
On December 30, 2021, we entered into an asset purchase agreement with certain entities affiliated with Saol International Limited (collectively, “Saol”), a private specialty pharmaceutical company, pursuant to which we agreed to acquire Saol’s baclofen franchise, including Lioresal®, LYVISPAH™, and a pipeline product under development (the “Saol Acquisition”). The Saol Acquisition expanded our commercial institutional and specialty portfolio in neurology and added commercial infrastructure in advance of our entry into the biosimilar institutional market during October 2022. Consideration for the Saol Acquisition included approximately $84.7 million, paid at closing with cash on hand, and contingent royalty payments based on annual net sales for certain acquired assets, beginning in 2023. The transaction closed on February 9, 2022.
For additional information about our acquisition, refer to Note 3. Acquisition in our consolidated financial statements.
Segments of the Business
We have three reportable segments: Affordable Medicines (formerly known as Generics), Specialty, and AvKARE.
During the fourth quarter of 2024, we changed the name of our Generics segment to “Affordable Medicines” to reflect the full product offering of the segment. The segment name change did not result in any change to the composition of our reportable segments and, therefore, did not result in any changes to our historical segment results.
Affordable Medicines
Prescription pharmaceutical products are sold either as branded or generic products. Generic pharmaceutical products have the same active product ingredient (“API”), dosage form, strength, route of administration, and conditions of use as patented branded pharmaceutical products, are bioequivalent to the brand it copies, and are usually marketed under their chemical (generic) names rather than brand names. Generic pharmaceutical products are intended to provide a cost-effective alternative for consumers while maintaining the safety, efficacy, quality and stability of the branded product, and as such are generally sold at prices below their branded equivalents. Typically, a generic pharmaceutical may not be marketed until the expiration of applicable patent(s) on the corresponding branded product, unless the resolution of patent litigation results in an earlier opportunity to enter the market. Generic manufacturers are required to file and receive approval for an Abbreviated New Drug Application (“ANDA”) to market a generic pharmaceutical product. Manufacturers of biosimilars are required to file a Biologics License Application (“BLA”) to introduce, or deliver for introduction, a biologic product into interstate commerce and market the product for one or more indications. Refer to Item 1. Business - Pharmaceutical Approval Process in the United States - New Drug Application and Biologics License Application for additional information.
Our Affordable Medicines segment includes approximately 270 product families covering an extensive range of dosage forms and delivery systems, including both immediate and extended-release oral solids, powders, liquids, sterile injectables, nasal sprays, inhalation and respiratory products, biosimilar products, ophthalmics, films, transdermal patches and topicals. We focus on developing products that have substantial barriers-to-entry due to complex drug formulations or manufacturing, or legal or regulatory challenges. Focusing on these products allows us the opportunity to offer first-to-file (“FTF”), first-to-market (“FTM”) and other high-value products to customers. A generic pharmaceutical product is considered an FTF product if the ANDA filed with respect to such product is the first to be filed for such product. Pursuant to the Hatch-Waxman Amendments, FTF products may receive a statutory 180-day exclusivity period, subject to certain conditions. A generic product that does not qualify as an FTF may still be an FTM product. A generic product is considered an FTM product if it is the first marketed generic version of a branded pharmaceutical. FTF, FTM and high-value products tend to be more profitable and often have longer life cycles than other generic pharmaceuticals. As such, the timing of new product introductions can have a significant impact on our financial results. Market entry by additional competitors generally has a negative impact on the volume and pricing of the affected products. Additionally, pricing is often affected by factors outside of our control. Refer to “Pharmaceutical Approval Process in the United States,” below, for more information.
As of December 31, 2024, our Affordable Medicines segment had 76 products with a pending ANDA and another 56 products in various stages of development in our pipeline, 74% of which are non-oral solid products. We have an integrated, team-based approach to product development that combines our formulation, regulatory, legal, manufacturing and commercial capabilities.
Our Affordable Medicines segment has a growing portfolio of institutional injectable products primarily for the U.S. hospital market. Our R&D pipeline has prioritized new product innovations in injectables, such as drug/device combinations, peptides, long-acting injectables and large volume parenteral bags. We have expanded our manufacturing capabilities and infrastructure to support the needs of this expanding business with a focus on development, commercialization and scaling a differentiated injectables portfolio. During 2023, we launched 39 new products, of which 14 were injectables. In May 2023, the FDA approved for manufacturing our fourth and largest injectable site. During 2024, we launched 22 new products, of which 12 were injectables.
In 2022 we began to commercialize an initial portfolio of oncology biosimilars in the U.S. Alymsys®, a biosimilar referencing Avastin®, launched in October 2022, followed by Releuko®, a biosimilar referencing Neupogen®, in November 2022 and Flynetra™, a biosimilar referencing Neulasta®, in May 2023. On October 12, 2023, we announced the addition of two denosumab biosimilars referencing both Prolia® and XGEVA® to our biosimilar pipeline. The two denosumab products are being developed by mAbxience S.L., a global biotech company with over a decade of experience in the development, manufacture, and commercialization of biopharmaceuticals. To further grow our oncology biosimilars sales, we are focused on serving oncology clinics, integrated health systems and specialty pharmacies. We are focused on expanding our oncology and biosimilar portfolio with additional molecules, and we seek to vertically integrate by expanding our biosimilar capabilities.
In March 2024, we amended the Kashiv Biosimilar Agreement (as defined in Note 23. Related Party Transactions) to include two additional in-development products, a pre-filled auto-injector delivery system for peg-filgrastim and a pre-filled on-body injector (OBI) delivery system for peg-filgrastim. In July 2024, we entered into an exclusive license and commercialization agreement with Kashiv Biosciences LLC to distribute and sell Omalizumab, a biosimilar to XOLAIR®, in the U.S. and India (refer to Note 23. Related Party Transactions.)
Our Affordable Medicines segment had net revenue of $1.69 billion, $1.47 billion and $1.43 billion and operating income of $270.1 million, $276.2 million and $224.2 million for the years ended December 31, 2024, 2023 and 2022, respectively.
Specialty
Our Specialty segment is engaged in the development, promotion, sale and distribution of proprietary branded pharmaceutical products, with a focus on products addressing central nervous system (“CNS”) disorders, including Parkinson’s disease, and endocrine disorders. Our portfolio of products includes CREXONT® (combination of carbidopa and levodopa extended release capsules), RYTARY® (extended release oral capsule formulation of carbidopa-levodopa), UNITHROID® (levothyroxine sodium), and ONGENTYS® (opicapone). On August 7, 2024, the FDA approved our new drug application (“NDA”) for CREXONT®, previously referred to as IPX203. In September 2024, we began selling CREXONT®, which is indicated for the treatment of Parkinson’s disease, Parkinson’s disease caused by infection or inflammation of the brain, or Parkinson’s disease-like symptoms that may result from carbon monoxide or manganese poisoning in adults. RYTARY® is indicated for the treatment of Parkinson’s disease, post-encephalitic parkinsonism, and parkinsonism that may follow carbon monoxide intoxication or manganese intoxication. ONGENTYS® is an add-on treatment to carbidopa/levodopa in patients with Parkinson’s disease experiencing “off” episodes, which we commenced selling in early 2024 under a license agreement with BIAL-Portela & Ca., S.A. UNITHROID®, indicated for the treatment of hypothyroidism, is sold under a license and distribution agreement with Jerome Stevens Pharmaceuticals, Inc.
Our Specialty products are marketed through skilled specialty sales and marketing teams, who call on neurologists, movement disorder specialists, endocrinologists and primary care physicians throughout the U.S. Our Specialty segment also has other product candidates that are in varying stages of development.
For Specialty products, the majority of such products’ commercial value is usually realized during the period in which the product has market exclusivity. In the U.S., when market exclusivity expires and generic versions of a product are approved and marketed, there can often be substantial and rapid declines in the branded product’s sales. In 2025, we expect to lose exclusivity for RYTARY®, an extended-release oral capsule formulation of carbidopa-levodopa for the treatment of Parkinson’s disease.
Our Specialty segment had net revenue of $445.7 million, $390.5 million and $374.1 million and operating income of $113.6 million, $65.3 million and $72.6 million, for the years ended December 31, 2024, 2023 and 2022, respectively.
AvKARE
Our AvKARE segment provides pharmaceuticals, medical and surgical products, and services primarily to governmental agencies, predominantly focused on serving the U.S. Department of Defense and the U.S. Department of Veterans Affairs. AvKARE is also a re-packager of bottle and unit dose pharmaceuticals and vitamins under the registered names of AvKARE and AvPAK. AvKARE is also a wholesale distributor of pharmaceuticals, over the counter drugs and medical supplies to its retail and institutional customers that are located throughout the U.S. focused primarily on entities that provide care to low-income and uninsured patients. Operating results for the sale of Amneal products by AvKARE are included in our Affordable Medicines reportable segment.
Our AvKARE segment had net revenue of $662.9 million, $531.7 million and $406.1 million and operating income of $42.9 million, $31.5 million and $3.3 million for the years ended December 31, 2024, 2023 and 2022, respectively.
Geographic Areas
We operate in the U.S., India, and Ireland. Investments and activities in some countries outside the U.S. are subject to higher risks than comparable U.S. activities because the investment and commercial climate may be influenced by financial instability in international economies, more restrictive economic policies and higher political and legal system uncertainties. See further discussion of this risk in Item 1A. Risk Factors.
Sales & Marketing and Customers
In the U.S. and the Commonwealth of Puerto Rico, we market our Affordable Medicines and Specialty products primarily through major wholesalers and distributors, retail pharmacies, mail-order pharmacies and directly into hospitals and institutions. The majority of our Affordable Medicines pharmaceutical products are marketed to large group purchasing organizations (“GPOs”) and sold through wholesalers, directly to large chain retailers or to mail order customers. Our sterile injectable products and biosimilars utilize a dedicated field-based sales force and are generally marketed to GPOs and specialty distributors, and sold through wholesalers, and occasionally directly to large hospitals and institutions. All of our wholesalers purchase products and warehouse them for retail drug stores, independent pharmacies and managed care organizations, such as hospitals, nursing homes, health maintenance organizations (“HMOs”), clinics, pharmacy benefit management companies and mail-order customers. Our Specialty segment, which promotes branded pharmaceutical products, employs a team of dedicated field-based sales representatives to engage in the direct marketing and promotion of our branded products to physicians and healthcare providers.
Refer to “AvKARE” in the section “Segments of the Business” above for a description of our AvKARE segment’s customers.
For the year ended December 31, 2024, on a consolidated basis, our four largest customers, Cencora, Inc., McKesson Drug Co., Cardinal Health, Inc., and CVS Health Corporation, accounted for approximately 70% of our net revenue. In total, we currently have approximately 1,300 customers (including over 1,100 customers specific to our AvKARE segment), some of which are part of large purchasing groups.
We have no long-term agreements that guarantee future business with any of our major customers and the loss of or substantial reduction in orders from any one or more of these customers could have a material adverse effect on our operating results, prospects and financial condition.
Competition
The pharmaceutical industry is highly competitive and is affected by new technologies, new developments, government regulations, health care legislation, availability of financing, and other factors. Many of our competitors have longer operating histories and substantially greater financial, R&D, marketing, and other resources than we do. Competing manufacturers of generic pharmaceutical products create value for our customers by offering substitutes for branded pharmaceutical products at significantly lower prices, and at times we may not be able to differentiate our product offerings from those of our competitors, successfully formulate and bring to market new products that are less expensive than those of our competitors or offer commercial terms as favorable as those of our competitors. We compete with numerous other companies that currently operate, or intend to operate, in the pharmaceutical industry, including companies that are engaged in the development of controlled-release drug delivery technologies and products, and other manufacturers that may decide to undertake development of such products. Our principal competitors in the generic/biosimilar pharmaceutical products market include Teva Pharmaceutical Industries Ltd., Viatris Inc., Sandoz Group, Pfizer Inc., Fresenius Kabi KGaA, Hikma Pharmaceuticals PLC, Dr. Reddy's Laboratories Ltd., Amphastar Pharmaceuticals, Inc., Sun Pharmaceutical Industries Ltd., Lupin Pharmaceuticals, Inc., Zydus
Pharmaceuticals USA Inc., and Aurobindo Pharma Limited. Our principal competitors in the specialty pharmaceutical products market include Supernus Pharmaceuticals, Inc., Jazz Pharmaceuticals PLC, AbbVie Inc., and Alkermes PLC.
Our AvKARE segment is also highly competitive, with new smaller competitors entering the space regularly. Our competitors are other wholesalers, including Cardinal Health, Inc., Cencora, Inc., McKesson Drug Co., and manufacturers / re-packagers such as Golden State Medical Supply.
By focusing on our high-value products with complex dosage forms and high barriers-to-entry, as well as taking advantage of our vertically integrated supply chain and selective use of internal API, we aim to manufacture more profitable products relative to our competition.
The Hatch-Waxman Amendments amended the Federal Food, Drug and Cosmetic Act (“FDCA”) and provided for a period of 180 days of generic marketing exclusivity for each applicant that is FTF an ANDA with a Paragraph IV certification, which signifies a challenge to at least one brand patent. The holder of an approved FTF ANDA that successfully challenges the relevant innovator drug patent(s) usually enjoys higher market share and sales during the 180-day period of exclusivity. When the exclusivity period concludes, other generic competitors may launch their versions of the product, which may cause significant price erosion and loss of market share. In cases where we are the holder of an ANDA for a FTF product, upon the expiration of the 180-day exclusivity period, we may adjust the price of such product and provide price adjustments to our customers for the difference between the lower price and the price at which we previously sold the product then held in inventory by our customers. These adjustments are commonly known as shelf stock adjustments. In certain circumstances, we may decide not to provide price adjustments to certain customers and, as a result, we may receive returns of unsold product from these customers and forego future sales volume as opposed to reducing pricing.
Authorized generic pharmaceutical products, which are generic labeled versions of pharmaceutical products introduced by brand companies (directly or through a third-party) under the brand’s NDA, have also increased competition in the generic pharmaceutical industry. Authorized generic pharmaceutical products may be sold prior to, during and subsequent to the 180-day exclusivity period and are a significant source of competition, because brand companies do not face any regulatory barriers to rapidly introducing generic versions of their pharmaceutical products.
Additionally, consolidation among wholesalers and retailers and the formation of GPOs has caused increased price competition in the generic pharmaceutical market. The downward price adjustments demanded by distributors of generic pharmaceutical products have reduced revenue and average product gross margin across the industry. Should these price reductions continue or even increase, it could have a material adverse effect on our revenue and gross margin. Further, even if we reduce the prices we charge our customers, that does not ensure that the prices consumers pay for those drugs will be similarly reduced.
The main competitive factors in the generic pharmaceutical market include:
•a generic pharmaceutical products manufacturer’s ability to rapidly develop and obtain regulatory approval for and supply commercial quantities of generic pharmaceutical products;
•the introduction of other generic pharmaceutical manufacturers’ products in direct competition with our products;
•the introduction of authorized generic pharmaceutical products in direct competition with our products;
•consolidation among our customers and the formation of buyer consortia;
•pricing pressures by competitors and customers, even if similar price savings are not passed on to consumers;
•product quality of our generic pharmaceutical competitors;
•our and our competitors’ breadth of product offerings across its portfolio;
•our ability and the ability of our generic pharmaceutical competitors to quickly enter the market after the expiration of patents or statutory exclusivity periods, limiting the extent and duration of profitability for our products;
•the willingness of our customers to switch their source of supply of products among various generic pharmaceutical competitors;
•the ability of our generic pharmaceutical competitors to identify and market niche products;
•our and our competitors’ level of service (including maintenance of inventories for timely delivery) and reputation as a reliable developer and manufacturer of generic pharmaceutical products; and
•product appearance and labeling for our products and those of our competitors.
In the brand-name pharmaceutical market, our principal competitors are pharmaceutical companies that are focused on Parkinson’s disease and other CNS disorders. In addition, with respect to products that we are developing internally and/or any additional products we may in-license from third parties, we expect that we will face increased competition from large
pharmaceutical companies, drug delivery companies and other specialty pharmaceutical companies that have focused on the same disorders as our branded products.
Research and Development
R&D activities represent a significant part of our business. R&D expenditures relate to the processes of discovering, testing and developing new products, upfront payments and milestones, improving existing products, as well as demonstrating product efficacy, if applicable, and regulatory compliance prior to launch. We are committed to investing in R&D with the aim of delivering high quality and innovative products. For the years ended December 31, 2024, 2023 and 2022, our R&D expense was $190.7 million, $164.0 million and $195.7 million, respectively.
Raw Materials
Raw materials, including APIs, essential to our business are generally readily available from various suppliers/sources. We purchase raw materials from manufacturers/distributors of bulk pharmaceutical chemicals and we also manufacture certain APIs at our facilities in India. In some cases, however, the raw materials used to manufacture our products are available only from a single supplier. Further, even if more than one supplier exists, we may choose, and have done so in the case of our API suppliers for a majority of our products, to list only one supplier in our product applications submitted to the FDA. Generally, we would need as long as 18 months to find and qualify a new sole-source supplier. If we receive less than one year’s termination notice from a sole-source supplier that it intends to cease supplying raw materials, it could result in disruption of our ability to produce the drug involved. Although to date we have only experienced occasional interruptions in supplies, no assurance can be given that we will continue to receive uninterrupted or adequate supplies of such raw materials. Any inability to obtain raw materials on a timely basis, or any significant price increases not passed on to customers, could have a material adverse effect on our business.
Because legal and regulatory requirements mandate that our product marketing authorizations specify API and raw material suppliers, if a specified supplier were for any reason unable to continue to supply us, we would need to seek FDA approval of a new supplier. The resulting delay in the manufacture and marketing of the impacted pharmaceutical product during the FDA process to qualify and approve the new supplier could, depending on the product, have a material adverse effect on our results of operations and financial condition. We protect against the risk of such an event by generally providing for, where feasible, two or more suppliers of raw materials for the pharmaceutical products we manufacture, including those for which we manufacture API in-house. Additionally, we may enter into a contract with a raw material distributor in order to secure adequate supply for specific products.
For a discussion of the risks relating to tariffs on APIs, refer to the risk factor “Changes in trade policy, including the imposition of tariffs may adversely affect our business, results of operations and financial condition” in Part 1. Item 1A. Risk Factors.
Manufacturing and Distribution
We have a network of manufacturing sites and co-located R&D centers within the U.S., India and Ireland, with broad dosage capabilities. We also have a distribution center for our Affordable Medicines and Specialty products in Glasgow, Kentucky. We manufacture the majority of our Affordable Medicines products internally; of these products, for the year ended December 31, 2024, those manufactured in our U.S. facilities contributed 44% of Affordable Medicines product net revenue compared to 28% for those manufactured in India. We rely on third-party manufacturers to supply products in our Affordable Medicines portfolio representing approximately 28% of our Affordable Medicines net revenue for the year ended December 31, 2024. Most of our Specialty products are manufactured by third-party manufacturers. In addition, we selectively manufacture API for a subset of our products, which helps to reduce the cost of manufacturing for our products and gives us greater control over our supply chain.
Our AvKARE segment’s distribution centers are located in Fountain Run, Kentucky and Philadelphia, Pennsylvania.
Government Regulation
The business of developing, manufacturing, selling, distributing, and marketing generic, biosimilar, and branded products is subject to significant health, safety, and environmental laws and regulations, including those governing the approval and pricing of products, clinical trials, laboratory procedures, privacy and security of health and other sensitive information and the handling, use, storage, treatment and disposal of hazardous materials and wastes. These regulatory regimes are overseen by
governmental bodies, principally the FDA and, as applicable, the Drug Enforcement Administration (the “DEA”), the Department of Health and Human Services, the Federal Trade Commission (the “FTC”) and several state and local government or other agencies, including individual data protection authorities, in the U.S. and abroad. Failure to comply with the laws and regulations of these governmental agencies may result in legal or other enforcement actions, including suspension of regulatory approval, delays in regulatory approval, clinical holds, orders to cease non-compliant activities and potential civil and criminal actions against us. The regulatory environment, particularly enforcement positions, statutes and legal interpretations applicable to the pharmaceutical industry are constantly in flux and not always clear. Significant changes in this environment could have a material adverse effect on our financial condition and results of operations.
The FDCA, the Public Health Service Act (the “PHSA”), the Controlled Substances Act, the regulations that implement these laws and other statutes and regulations govern the development, testing, manufacture, packaging, use, distribution, safety, effectiveness, labeling, storage, record keeping, approval, marketing, sale, and promotion of our products, as well as post-marketing requirements for safety surveillance and reporting. Failure to comply with these laws and regulations can result in judicial and/or administrative sanctions, such as warning letters, recalls, product seizures, injunctions, fines, total or partial suspension of distribution or production, exclusion or debarment from government programs and contracts, restitution, disgorgement and criminal prosecutions. The FDA has the authority to withdraw its approval of pharmaceuticals at any time, in accordance with its regulatory due process procedures, and can enforce the recall of products.
Pharmaceutical Approval Process in the United States
In the U.S., the FDA regulates pharmaceuticals and biologics under the FDCA and the PHSA, and their implementing regulations. To market a new drug or biologic, considerable data must be submitted to the FDA for review and approval. In addition to approval, the FDA also regulates research, development, preclinical and clinical testing, manufacturing, packaging, storage, distribution, recordkeeping, labeling, advertising, promotion, marketing, post-approval monitoring and reporting, and import and export of drugs and biologics. If we fail to comply with the applicable U.S. regulatory requirements at any time during the product development process, approval process or after approval, we may be subject to a variety of administrative or judicial sanctions, which could include, among other actions, the FDA’s refusal to approve pending applications, delays in approval, suspension or withdrawal of an approval, imposition of a clinical hold, orders to cease non-compliant activities, criminal charges, issuance of warning letters and other types of enforcement-related letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, exclusion from participation in government programs and contracts, restitution, disgorgement of profits, or civil or criminal investigations and penalties brought by the FDA, the U.S. Department of Justice (“DOJ”) or other governmental entities.
Generally, the following types of applications are used to obtain FDA approval.
New Drug Application and Biologics License Application
For a drug product containing an active ingredient not previously approved by the FDA, a prospective manufacturer must submit a complete NDA containing the results of clinical studies supporting the drug product’s safety and efficacy in addition to data and information related to drug product quality and manufacturing. An NDA is also required for a drug with a previously approved active ingredient if the drug will be used to treat an indication for which the drug was not previously approved or if the dosage form, strength or method of delivery is changed and requires clinical studies to support the change. A BLA is required to introduce, or deliver for introduction, a biologic product into interstate commerce and market the product for one or more indications. The process required by the FDA before a new pharmaceutical or biological product may be approved for marketing in the U.S. generally involves the steps listed below.
•Laboratory and clinical tests;
•Submission to the FDA of an Investigational New Drug (“IND”) application;
•Adequate and well-controlled human clinical studies conducted according to the FDA’s requirements for good clinical practice (“GCP”) and additional requirements for the protection of human research subjects and their health information, to establish the safety and efficacy of the proposed product for its proposed conditions of use;
•For pharmaceutical products, submission of an NDA containing the results of the preclinical tests and clinical studies establishing the safety and efficacy of the proposed product for its proposed conditions of use, proposed labeling, extensive data addressing such matters such as drug product quality and manufacturing, and certain information with regards to patents related to the proposed drug product;
•For biological products, submission of a BLA that includes substantive evidence of safety, purity, and potency from results of nonclinical testing and clinical studies, as well as information on the chemistry, manufacturing and controls to ensure product identity and quality, and proposed labeling;
•For certain products, development and implementation of a Risk Evaluation and Mitigation Strategy (“REMS”);
•Scale-up to commercial manufacturing;
•Satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the product is produced to assess compliance with current good manufacturing practices (“cGMP”), and, if applicable, the FDA’s current good tissue practice;
•Potential FDA inspection of the nonclinical and clinical study sites and the clinical study sponsor that generated the data in support of the NDA or BLA; and
•FDA review and approval or licensure of an NDA or of the BLA.
Prior to beginning the first clinical trial with a product candidate, the sponsor must submit an IND to the FDA and the IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises safety concerns or questions about the proposed clinical trial and places the IND on clinical hold within that 30-day time period. The FDA may also impose clinical holds on a product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, studies may not recommence without FDA authorization and then only under terms authorized by the FDA.
The FDA or the sponsor may suspend a clinical trial at any time on various grounds. Similarly, an independent institutional review board (“IRB”) can suspend or terminate approval of a clinical trial at its site if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the product candidate has been associated with unexpected serious harm to patients.
Human clinical studies are typically conducted in three sequential phases that may overlap or be combined. Phase 1 studies are designed to assess safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. If favorable, Phase 2 studies are initiated to evaluate the efficacy of the product for specific targeted diseases or conditions and to determine dosage tolerance, optimal dosage and dosing schedule, as well as identify any adverse effects that could limit the product’s usefulness. If data from the Phase 2 trials are favorable, large-scale Phase 3 trials are undertaken to confirm the product’s efficacy and safety.
During all phases of clinical development, FDA and others require extensive monitoring and auditing of all clinical activities, clinical data, and clinical study investigators, and certain progress and safety reports must be submitted to the FDA. Phase 1, Phase 2 and Phase 3 clinical studies may not be completed successfully within any specified period, if at all.
Assuming successful completion of all required testing in accordance with regulatory requirements, the submission of an NDA or BLA requesting approval to market the product is subject to a substantial application user fee, which may be reduced or waived if the FDA finds that certain criteria are met, and there are certain exemptions for products designated for rare diseases or conditions. The submission of an NDA or BLA is not a guarantee that the FDA will find it complete and accept it for filing. After the application is deemed filed by the FDA, FDA staff will review an NDA or BLA to determine, among other things, whether a product is safe and efficacious for its proposed conditions of use. There can be no assurance that a product will obtain the regulatory approvals necessary for it to be marketed.
If, after reviewing the NDA or BLA, the FDA determines that the application cannot be approved in its current form, the FDA sends the applicant a complete response letter (“CRL”) identifying all outstanding deficiencies that preclude final approval. The FDA then halts its review until the applicant resubmits the NDA or BLA with new information designed to address the deficiencies. An applicant receiving a CRL may resubmit the application with data and information addressing all of the FDA’s concerns or requirements set forth in the CRL, withdraw the application without prejudice to a subsequent submission of a related application or request a hearing on whether there are grounds for denying approval of the application. If a product receives regulatory approval, the approval is limited to specific diseases and dosages or the indications for use for which approval had been sought. In addition, the FDA may require an applicant to conduct Phase 4 testing which involves clinical trials designed to further assess a drug’s safety and effectiveness after approval, and may require surveillance programs to monitor the safety of approved products which have been commercialized. Once issued, the FDA may withdraw product approval if ongoing regulatory requirements are not met or if safety or efficacy questions are raised after the product reaches the market. The agency may also impose requirements that the NDA or BLA holder conduct new studies, make labeling changes, implement a REMS, and take other corrective measures.
The FDA has a number of programs, including fast track, breakthrough therapy, priority review and accelerated approval, intended to expedite the development or review of products that meet certain criteria, and applicants may explore some of these opportunities for their product candidates, if appropriate. These programs do not change the standards for approval, but may expedite the development or approval process. Even if a product qualifies for one or more of these programs, the FDA may
later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened.
Additionally, in the context of public health emergencies, applicants may seek an Emergency Use Authorization (“EUA”) from the FDA, which if granted, allows for the distribution and use of unapproved products during the declared public health emergency or to allow a medical countermeasure to be used in an emergency caused by a chemical, biological, radiological and nuclear agent, in accordance with the conditions set forth in the EUA, unless the EUA is otherwise terminated.
NDA User Fee Program
On September 30, 2022, President Biden signed into law the FDA User Fee Reauthorization Act of 2022, which includes the reauthorization of the Prescription Drug User Fee Act (“PDUFA VII”) from fiscal year 2023 through 2027. The program provides for the continued timely review of new NDAs and BLAs. PDUFA VII enhancements include modernizing the user fee structure, a focus on human resource and financial management improvement including a significant increase in staff capacity and capabilities to support the review of cell and gene therapy products; the creation of capacity planning capability; enhancing use of regulatory tools via benefit-risk, patient-focused drug development, complex innovative trial designs, and model informed drug development; enhancing staffing for breakthrough therapy reviews; focusing on communication with industry; and exploring real world evidence in regulatory decision-making.
Biosimilar and Interchangeable Biologics License Application
A biosimilar product is a biologic product that is highly similar to an existing FDA-approved biological product (which is referred to as the “reference product”) and has no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency. The FDA approves biosimilars through an abbreviated review process, with the goal of demonstrating biosimilarity between the proposed biosimilar and its reference product. Ultimately, the process entails less expensive and fewer clinical trials than a “full” BLA.
Biosimilar license application submissions typically included analytical studies that provide comparative analytical data to demonstrate the structural and functional similarity of the proposed biosimilar product to the reference product and evaluate the impact of any differences identified. The application must also include an assessment of toxicity and a clinical study or studies sufficient to demonstrate safety, purity, and potency in one or more appropriate conditions of use for which the reference product is licensed and for which licensure is sought for the proposed biological product. Pharmacology studies may demonstrate that the proposed biosimilar is processed by the human body in the same way and with the same effects as the reference product. The application may also include an immunogenicity assessment, which evaluates a patient’s immune response to the proposed biosimilar. An applicant may need to conduct additional comparative clinical studies to demonstrate whether there are any clinically meaningful differences between the proposed biosimilar and the reference product.
An interchangeable biosimilar is a biosimilar that may be substituted for the reference product without the intervention of the prescribing health care provider that prescribed the reference product, depending on state pharmacy laws. In addition to establishing biosimilarity to the reference product per the process described above, a manufacturer of an interchangeable biosimilar must also submit information to the FDA to show that the proposed interchangeable product can be expected to produce the same clinical result as the reference product in any given patient. If the biological product is to be administered more than once to an individual, then the manufacturer in certain circumstances also submits information in the application that demonstrates the risk in terms of safety or diminished efficacy of alternating between the biological product and the reference product is not greater than the risk of using the reference product without switching. All biosimilars are approved only after they meet the FDA’s rigorous approval standards.
Biosimilar User Fee Program
The Biosimilar User Fee Act was reauthorized for the second time on September 30, 2022 (“BsUFA III”). In the reauthorization of BsUFA III, enhancements include new supplement types and expedited review timelines; enhancing communication and feedback during the biosimilar biological development process; enhancing pre-licensure inspection communication; introducing a new pilot program to enhance regulatory decision-making and facilitate science-based recommendation; and enhancing financial management, transparency, and strategic hiring.
Abbreviated New Drug Application
For a generic version of an approved drug product approved under an NDA, an ANDA relies on the FDA’s previous finding of safety and effectiveness for a reference listed drug (“RLD”) and, as a result, may be approved without submission of the same
type and extent of information that is requested for a stand-alone NDA to establish the safety and effectiveness of the proposed product. Instead, an ANDA must submit data and information demonstrating the proposed product has the same active ingredient, dosage form, route of administration, and strength, and is bioequivalent to the previously approved RLD indicating that the rate of absorption and levels of concentration of the generic drug in the body do not show a significant difference from those of the reference listed drug. For most orally administered pharmaceutical products, bioequivalence between brand and generic is established when there is no statistically significant difference in the rate and extent to which the API in the product is absorbed into the bloodstream. For certain pharmaceutical products, such as topical, locally acting pharmaceutical products, other means of establishing bioequivalence may be required by the FDA. The proposed product also must have the same labeling as the RLD with certain limited exceptions, include certain information regarding patents for the RLD, and must meet the same legal and regulatory standards for drug product quality and manufacturing as NDA products.
ANDA User Fee Program
On September 30, 2022, the Generic Drug User Fee Amendments (“GDUFA”) program was reauthorized for a second time with provisions that are in effect through September 30, 2027 (“GDUFA III”). The FDA’s commitment letter for GDUFA III, which sets forth performance goals and program enhancements for the reauthorization of GDUFA for fiscal years 2023-2027, sets goals for FDA’s assessment and review of different ANDA submissions, drug master files, and generic-related inspections, and includes enhancements designed to increase pre-submission assessment activities, reduce the number of assessment cycles for ANDAs and facilitate access to generic drugs, in particular for complex products such as those with complex active ingredients or dosage forms that generally are more difficult to genericize and have increased numbers of assessment cycles. The reauthorization also established capacity planning capability.
Current Good Manufacturing Practices
In order to obtain FDA approval for its products, a pharmaceutical manufacturer must demonstrate that its facilities comply with cGMP regulations. The manufacturer is required to comply with cGMP standards at all times during the production and processing of pharmaceuticals, and the FDA may inspect the manufacturer’s sites at any time to ensure compliance.
Patent Provisions
A branded pharmaceutical product is usually protected under patents granted by the U.S. Patent and Trademark Office that allow only the pharmaceutical company that developed the pharmaceutical product to market and sell such product. The sponsor of a branded product must “list” certain patents with the FDA, which in turns publishes that patent information publicly. For a generic pharmaceutical manufacturer to introduce a generic version of an RLD, it must submit to the FDA an ANDA with a certification for each patent listed by the RLD sponsor stating one of the following:
•Paragraph I: That the required patent information relating to the patent for the RLD has not been filed;
•Paragraph II: That the patent for the RLD has expired;
•Paragraph III: That the patent for the RLD will expire on a particular date; or
•Paragraph IV: That the patent for the RLD is invalid, unenforceable and/or will not be infringed by the pharmaceutical product for which approval is being sought.
The FDCA describes only one circumstance in which an ANDA applicant need not certify to a listed patent. Specifically, when a patent is listed only for a method of use, an ANDA applicant seeking to omit that approved method of use from the generic drug’s labeling can submit a “section viii statement,” acknowledging that a given method-of-use patent has been listed, but stating that the patent at issue does not claim a use for which the applicant seeks approval. Filing an ANDA with certifications under Paragraph I or II, referenced above, permits the ANDA to be approved immediately, if it is otherwise eligible. Filing an ANDA with certifications under Paragraph III, referenced above, indicates that the applicant agrees to wait until the relevant patent has expired before seeking final approval of its ANDA. Under Paragraph IV, referenced above, a generic pharmaceutical manufacturer can challenge the patent of the RLD prior to ANDA approval.
If the ANDA for a generic pharmaceutical product has a Paragraph IV certification, the filer must also notify the NDA and patent holders of the ANDA submission and all Paragraph IV certifications upon acceptance of the ANDA filing by the FDA (the “PIV Notice”). If in response to notice of a Paragraph IV certification, the NDA holder or patent owner initiates a patent infringement action against the ANDA applicant within 45 days of receiving the required notice, approval of the ANDA generally will be stayed for 30 months following receipt of the notice or such shorter or longer time as the court might order.
Generic Pharmaceutical Pricing
The pricing of a generic pharmaceutical product generally correlates to the number of companies manufacturing generic versions of such pharmaceutical product. A generic pharmaceutical product is usually at its highest price immediately after the first generic launch of the product, either because qualifying manufacturers have been granted 180-day exclusivity, restricting approval of non-exclusivity eligible ANDAs from marketing, or because only a few manufacturers have entered the market due to other technical or operational obstacles to bringing such product to market, such as raw materials shortages or complex formulation. As additional generic manufacturers enter the market, the price of a generic pharmaceutical product typically falls as manufacturers compete on price to capture market share. Even if we reduce the prices we charge our customers, the prices consumers pay for those drugs may not be similarly reduced. Additionally, consolidation among pharmacy benefit managers, wholesalers and retailers and the formation of GPOs has caused increased price competition in the generic pharmaceutical market and more generally, a deflationary market.
Healthcare Reform
Pricing and reimbursement for our products depend in part on government regulation. In the U.S., there are multiple federal and state proposals, laws and actions related to the pricing of pharmaceuticals and other changes to the healthcare system, including the enactment of the Inflation Reduction Act (“IRA”). The IRA requires: (i) the government to negotiate prices for select high expenditure Medicare Part D drugs (prices effective beginning in 2026) and Part B drugs (prices effective beginning in 2028), (ii) manufacturers to pay a rebate for Medicare Part B and Part D drugs when prices for those drugs increase faster than inflation, and (iii) a Medicare Part D redesign that: (a) establishes a new manufacturing discount program whereby participating manufacturers are required to provide discounts on their applicable drugs in the initial coverage and catastrophic coverage phases of the Part D benefit and (b) replaces the current coverage gap provisions and establishes a $2,000 cap for out-of-pocket costs for Medicare beneficiaries beginning in 2025, with manufacturers being responsible for 10% of costs up to the $2,000 cap and 20% after that cap is reached.
The IRA will permit the Centers for Medicare and Medicaid Services (“CMS”) to negotiate maximum fair prices on up to 60 drugs by 2029, starting with 10 drugs in 2026. The drugs must be approved by the FDA for at least seven years (small molecule drugs) or at least eleven years (biologics). The federal government must consider manufacturer-submitted data including R&D costs, production and distribution costs, market data, and revenue and sales volume data. The government also must consider evidence regarding alternative treatments, including whether a drug represents a therapeutic advance compared to existing therapies and the comparative effectiveness of the drug and therapeutic alternatives, including effects on specific populations (e.g., children, elderly, terminally ill). The IRA also requires Medicare Part D plans to limit beneficiaries’ cost-sharing for insulin products, but does not include an insulin cost-sharing cap for commercial plans.
Any significant efforts at the federal or state level to reform the healthcare system by changing the way healthcare is provided or funded or by more directly imposing controls on drug pricing, government reimbursement, and access to products could have a material impact on us. Efforts by states and the federal government to regulate prices or payment for pharmaceutical products, including proposed actions to facilitate drug importation, limit reimbursement to lower international reference prices, and require deep discounts, could adversely affect our business if implemented. States may also advance drug-pricing initiatives with a focus on affordability review boards, financial penalties related to pricing practices, manufacturer pricing and reporting requirements, as well as regulation of prescription drug assistance or copay accumulator programs in the commercial market. In addition, changes to the Medicaid program or the federal 340B drug pricing program, which imposes ceilings on prices that drug manufacturers can charge for medications sold to certain health care facilities, could have a material impact on our business. It is unclear what, if any, legislative proposals may be adopted or how governmental bodies and private payors will respond to such healthcare reform. As such, we cannot predict the impact of potential legislation on our business and cannot guarantee that such legislation will not have a material adverse effect on our financial condition and results of operations.
Pharmaceutical Pedigree Laws
Various pharmaceutical pedigree laws, such as the Drug Supply Chain Security Act enacted in 2014, require the tracking of all transactions involving prescription pharmaceutical products from the manufacturer to the dispensary (e.g., pharmacy). Compliance with such laws requires extensive tracking systems and tight coordination with customers and manufacturers. While we believe that we comply with these laws and we intend to do so in the future, such legislation and government enforcement regarding these laws is constantly evolving. Failure to comply could result in fines, penalties or loss of business that could have a material adverse effect on our financial results.
Sales and Marketing Regulations
Our marketing practices are subject to state laws, as well as federal laws, such as the Anti-Kickback Statute and False Claims Act, intended to prevent fraud and abuse in the healthcare industry. The Anti-Kickback Statute generally prohibits corruptly soliciting, offering, receiving, or paying anything of value to generate business. The False Claims Act generally prohibits anyone from knowingly and willingly presenting, or causing to be presented, any claims for payment for goods or services, including to government payers, such as Medicare and Medicaid, that are false or fraudulent and generally treat claims generated through kickbacks as false or fraudulent. For example, the DOJ has entered into settlements with pharmaceutical manufacturers that allegedly caused the submission of false claims to Medicare for drugs that were no longer eligible for Medicare coverage because the FDA approved a prescription drug’s conversion to over-the-counter status and the drug was no longer considered a prescription product. The federal government and states also regulate sales and marketing activities and financial interactions between manufacturers and healthcare providers, requiring disclosure to government authorities and the public of such interactions, disclosure of transfers of value from pharmaceutical companies to healthcare providers and healthcare organizations, such as academic teaching hospitals, and the adoption of compliance standards or programs. State attorneys general have also taken action to regulate the marketing of prescription drugs under state consumer protection and false advertising laws.
Federal Regulation of Patent Litigation Settlements and Authorized Generic Arrangements
Pursuant to the Medicare Prescription Drug Improvement and Modernization Act of 2003, generic and brand pharmaceutical companies must file with the DOJ and FTC certain agreements entered into between other brand and/or generic pharmaceutical companies in regards to the settlement of patent litigation and/or the manufacture and marketing of generic versions of branded pharmaceutical products. This requirement impacts the ways in which generic pharmaceutical companies resolve intellectual property (“IP”) litigation and may result in an increase in private-party litigation against pharmaceutical companies and/or additional investigations by the FTC or other governmental organizations.
Pursuant to the Patient Right to Know Drug Prices Act of 2018, the FTC also obtains and reviews patent settlement agreements between reference product and biosimilar manufacturers. This notification allows the FTC to evaluate whether these agreements include, among other things, anticompetitive reverse payments that slow or defeat the introduction of lower-priced medicines, including biosimilars. Such review occurs in the same manner that the FTC reviews patent settlement agreements between branded and generic drug manufacturers.
Other Regulatory Requirements
We are subject to the Maximum Allowable Cost Regulations, which limit reimbursements for certain generic prescription drugs under Medicare, Medicaid, and other programs to the lowest price at which these drugs are generally available. In many instances, only generic prescription drugs fall within the regulations’ limits. Generally, the pricing and promotion of, method of reimbursement and fixing of reimbursement levels for, and the reporting to federal and state agencies relating to drug products is under active review by federal, state and local governmental entities, as well as by private third-party reimbursors and individuals under whistleblower statutes. At present, the DOJ and U.S. Attorneys Offices and State Attorneys General have initiated investigations, reviews, and litigation into industry-wide pharmaceutical pricing and promotional practices, and whistleblowers have filed qui tam suits. We cannot predict the results of those reviews, investigations, and litigation, or their impact on our business. For further detail, see Note 20. Commitments and Contingencies to our consolidated financial statements.
Furthermore, the Company relies on global supply chains and production and manufacturing processes that are complex and subject to increasing regulatory scrutiny and enforcement, including the Trade Agreements Act and Buy American Act, that may affect the sourcing, supply, manufacturing and pricing of materials used in the Company’s products sold to the Federal government. Additionally, the Cybersecurity Maturity Model Certification 2.0 Requirements for healthcare contractors who do business with the Department of Defense will necessitate enhanced oversight including an annual self-assessment and annual affirmation to ensure compliance with 15 security requirements in Federal Acquisition Regulation clause 52.204-21.
Virtually every state, as well as the District of Columbia, has enacted legislation permitting the substitution of equivalent generic and certain biosimilar prescription drugs for brand-name drugs where authorized or not prohibited by the prescribing physician, and some states mandate generic substitution in Medicaid programs.
In addition, numerous state and federal requirements exist for a variety of controlled substances, such as narcotics, that may be part of our product formulations. We must meet the requirements of controlled substances laws, such as the Controlled Substances Act, as amended, DEA regulations for certain of our products and activities, and related state requirements. These laws and regulations relate to the manufacture, shipment, storage, security, inventory, recordkeeping, distribution, sale, dispensing, and use of controlled substances. The DEA and other regulatory agencies limit the availability of the controlled
substances used in certain of our current products and products in development. We must annually, as well as quarterly, apply to the DEA and similar governmental and regulatory authorities for procurement quotas in order to obtain these substances. The DEA may also pursue monetary penalties, administrative penalties such as revocation of a registration to manufacture controlled substances and criminal penalties for controlled substances violations.
Other federal and state regulatory agencies have far reaching authority. For example, the State of California requires that any manufacturer, wholesaler, retailer or other entity in California that sells, transfers, or otherwise furnishes certain so-called precursor substances must have a permit issued by the California Department of Justice, Bureau of Narcotic Enforcement. The substances covered by this requirement include ephedrine, pseudoephedrine, norpseudoephedrine, and phenylpropanolamine, among others. The Bureau has authority to issue, suspend and revoke precursor permits, and a permit may be denied, revoked or suspended for various reasons, including (i) failure to maintain effective controls against diversion of precursors to unauthorized persons or entities; (ii) failure to comply with the Health and Safety Code provisions relating to precursor substances, or any regulations adopted thereunder; (iii) commission of any act which would demonstrate actual or potential unfitness to hold a permit in light of the public safety and welfare, which act is substantially related to the qualifications, functions or duties of the permit holder; or (iv) if any individual owner, manager, agent, representative or employee of the permit applicant/permit holder willfully violates any federal, state or local criminal statute, rule, or ordinance relating to the manufacture, maintenance, disposal, sale, transfer or furnishing of any precursor substances.
Privacy, Security and Data Standards Regulations
Numerous federal, state, and foreign laws and regulations govern the creation, collection, dissemination, receipt, maintenance, protection, use, transmission, disclosure, privacy, confidentiality, security, availability, integrity, creation, processing, and disposal (collectively, “Processing”) of protected health information (“PHI”) and other personal, sensitive, regulated or confidential data, including personally identifiable information (“PII”). Some of our activities may involve the Processing of PHI and PII.
On the federal level we are subject to a number of sector specific regulations. The federal Health Insurance Portability and Accountability Act of 1996, the Health Information Technology for Economic and Clinical Health Act, the 21st Century Cures Act, Public Law 116-321, and the regulations that implement these laws (collectively, “HIPAA Law”) impose requirements on covered entities and business associates that address the privacy and security of PHI. In the conduct of our business, we may be either a covered entity or business associate, and we may also be held liable for HIPAA Law violations by our vendors that are business associates. HIPAA Law imposes contracting requirements, requires breach notifications, and establishes rules that standardize the format and content of certain electronic transactions, including eligibility and claims. Violations of HIPAA Law may result in enforcement actions, civil and criminal penalties, and settlement, resolution, and monitoring agreements. Further, state attorneys general may bring civil actions seeking either injunctions or damages in response to violations of HIPAA Law that threaten the privacy of state residents and may negotiate settlements for related cases on behalf of their respective residents. There can be no assurance that we will not be the subject of an investigation, audit or compliance review regarding our compliance with HIPAA Law. While HIPAA Law does not create a private right of action, its standards have been used as a basis for the duty of care in state civil suits, such as those for negligence or recklessness in the handling, misuse or breach of PHI. HIPAA Law does not preempt more stringent state health privacy laws and regulations, which may protect the health information of certain individuals, such as minors, and certain types of sensitive health information, such as transgender care, HIV/AIDS status, reproductive health information, genetic information, and mental and behavioral health. Recently, several states have enacted broadly applicable laws to protect the privacy of personal health information. These laws generally require consent for the collection, use or sharing of any “consumer health data”, which is defined as personal information that is linked or reasonably linkable to a consumer and that identifies a consumer’s past, present, or future physical or mental health.
Additionally, under Section 5 of the Federal Trade Commission Act (“FTC Act”), the FTC has jurisdiction over certain privacy and security practices that are deemed unfair and deceptive acts and practices in or affecting commerce. The FTC has charged companies with violating this act based on failures to appropriately and transparently safeguard personal information, respect consumers’ privacy rights, based on disclosures of health and personal information to third parties, the failure to limit third-party use of health information, the failure to implement policies and procedures to prevent the improper or unauthorized disclosure of health information, and the failure to provide notice and obtain consent before the use and disclosure of health information for advertising. In addition to the FTC Act, the FTC also enforces other federal laws and regulations relating to consumers’ privacy and security. For information that is not subject to HIPAA and deemed to be “personal health records”, the FTC may also impose penalties for violations of the Health Breach Notification Rule to the extent we are considered a “personal health record-related entity” or “third party service provider.” Data privacy and security laws and regulations continue to evolve and, as a result, we expect scrutiny by federal and state regulators and others of our collection, use and disclosure of health information.
Over the past several years, the federal government has increasingly focused on the cybersecurity requirements applicable to government contractors, including enhanced guidance and regulation. These include compliance with the Privacy Act of 1974, the Defense Federal Acquisition Regulation Supplement cybersecurity requirements, the Cybersecurity Maturity Model Certification (going into effect over the next several years and based on National Institutes of Standards and Technology Cybersecurity (“NIST”) standards), the Federal Information Security Modernization Act, and the White House’s 2021 Executive Order on Improving the Nation’s Cybersecurity.
State and local authorities are increasingly focused on protecting individuals from identity theft and a number of states have adopted comprehensive data security laws and regulations requiring, among other things, certain minimum data security standards and security breach notifications that may apply to us in certain circumstances, as well as certain limitations on access to and use of PII. These laws and regulations include state general data breach laws, which exist in all fifty states and protect PII generally. Many states also have their own sector-specific laws regarding the Processing of PII which may apply to us as well.
In the past few years, several states have adopted their own comprehensive consumer privacy statutes and many more states are considering doing so. Generally, these statutes exempt data and/or entities regulated by HIPAA Law but are, in varying respects, applicable to other data we collect, such as PII provided by website visitors, and in California, employees and business partners. Additionally, we anticipate federal and state legislators and regulators will continue to enact legislation related to privacy and cybersecurity, including with respect to ransomware incidents.
In addition, international laws, rules and regulations governing the use and disclosure of PII can be more stringent than those in the U.S., and they vary from jurisdiction to jurisdiction. The European Union’s General Data Protection Regulation (“GDPR”), which became effective May 2018, enhanced or created obligations regarding the handling of PII relating to European residents (such as regarding notices, data protection impact assessments and individual rights) and provides for greater penalties for noncompliance than the previous European Directive or laws. Under GDPR, fines of up to €20 million or up to 4% of the annual global revenues, whichever is greater, can be imposed for violations. In addition, many countries outside of Europe where we conduct business have implemented or may implement data protection laws and regulations, some of which include requirements modeled after those in the GDPR. Some non-U.S. jurisdictions are also instituting data residency regulations requiring that data be maintained within the respective jurisdiction or otherwise restricting transfer of personal data across borders unless specified regulatory requirements are met.
Data privacy laws and regulations are constantly evolving and can be subject to significant change or interpretive application. Varying jurisdictional requirements could increase the costs and complexity of our compliance efforts and violations of applicable data privacy laws can result in significant penalties. Any failure, or perceived failure, by us to comply with applicable data protection laws could result in proceedings or actions against us by governmental entities or others, subject us to significant fines, penalties, judgments and negative publicity, require us to change our business practices, increase the costs and complexity of compliance and adversely affect our business.
Environmental Laws
We are subject to comprehensive federal, state and local environmental laws and regulations that govern, among other things, air polluting emissions, wastewater discharges, solid and hazardous waste disposal, and the remediation of contamination associated with current or past generation handling and disposal activities. We are subject periodically to environmental compliance reviews by various environmental regulatory agencies. While it is impossible to predict accurately the future costs associated with environmental compliance and potential remediation activities, compliance with environmental laws is not expected to require significant capital expenditures and has not had, and is not expected to have, a material adverse effect on our business, operations or financial condition.
Patents, Trademarks and Licenses
We own or license a number of patents in the U.S. and other countries covering certain products and product candidates and have also developed brand names and trademarks for other products and product candidates.
Generally, the branded pharmaceutical business relies upon patent protection to ensure market exclusivity for the life of the patent. We consider the overall protection of our patents, trademarks and license rights to be of material value and act to protect these rights from infringement. However, our business is not dependent upon any single patent, trademark or license.
In the branded pharmaceutical industry, the majority of an innovative product’s commercial value is usually realized during the period in which the product has market exclusivity. In the U.S. and some other countries, when market exclusivity expires and generic or biosimilar versions of a product are approved and marketed, there can often be very substantial and rapid declines in
the branded product’s sales, more so for introduction of a generic product as compared to a biosimilar. The rate of this decline also varies by country and by therapeutic category; however, following patent expiration, branded products often continue to have market viability based upon the goodwill of the product name, which typically benefits from trademark protection.
An innovator product’s market exclusivity is generally determined by two forms of IP: patent rights held by the innovator company and any regulatory forms of exclusivity to which the innovator is entitled.
Patents are a key determinant of market exclusivity for most branded pharmaceuticals. Patents provide the innovator with the right to exclude others from practicing an invention related to the medicine. Patents may cover, among other things, the active ingredient(s), various uses of a drug product, pharmaceutical formulations, drug delivery mechanisms and processes for (or intermediates useful in) the manufacture of products. Protection for individual products extends for varying periods in accordance with the expiration dates of patents in the various countries. The protection afforded, which may also vary from country to country, depends upon the type of patent, its scope of coverage and the availability of meaningful legal remedies in the country.
Market exclusivity is also sometimes influenced by regulatory exclusivity rights. Many developed countries provide certain non-patent incentives for the development of medicines. For example, the U.S., the European Union and Japan each provide for a minimum period of time after the approval of a new drug during which the regulatory agency may not rely upon the innovator’s data to approve a competitor’s generic copy or biosimilar. Regulatory exclusivity rights are also available in certain markets as incentives for research on new indications, on orphan drugs and on medicines useful in treating pediatric patients. Regulatory exclusivity rights are independent of any patent rights and can be particularly important when a drug lacks broad patent protection. However, most regulatory forms of exclusivity do not prevent a competitor from gaining regulatory approval prior to the expiration of regulatory data exclusivity on the basis of the competitor’s own safety and efficacy data on its drug, even when that drug is identical to that marketed by the innovator.
We estimate the likely market exclusivity period for each of our branded products on a case-by-case basis. It is not possible to predict the length of market exclusivity for any of our branded products with certainty because of the complex interaction between patent and regulatory forms of exclusivity, and inherent uncertainties concerning patent litigation. We cannot assure that a particular product will enjoy market exclusivity for the full period of time that we currently estimate or that the exclusivity will be limited to the estimate.
In addition to patents and regulatory forms of exclusivity, we also market products with trademarks. Trademarks have no effect on market exclusivity for a product, but are considered to have marketing value. Trademark protection continues in some countries as long as used; in other countries, as long as registered. Registration is for fixed terms and may be renewed indefinitely.
Seasonality
Consistent with the U.S. pharmaceutical industry trends, the first quarter of each year is typically our lowest revenue quarter in the year. Certain products within our portfolio are specifically affected by seasonality. For example, sales of Adrenaclick® (epinephrine injection, USP auto-injector) correlate with allergy seasonality. The seasonal impact of these particular products may affect a quarterly comparison within any fiscal year.
Human Capital
Workforce Demographics
As of December 31, 2024, we had over 8,100 employees (“Amneal Employees”), excluding approximately 200 employees in our AvKARE segment. Of the Amneal Employees, nearly 2,500 employees were in the U.S. and over 5,600 employees were located outside of the U.S., primarily in India and Ireland. Globally, we hired approximately 1,900 Amneal Employees in 2024, and turnover was approximately 19.1%. We monitor our turnover rate and continuously evolve our human capital management strategies to meet our needs while also navigating the dynamic labor market and increasing competition for talent.
Being one of the leading companies in healthcare requires dynamic skill sets, different perspectives, and a continuous pipeline of new ideas and innovation. We expanded our Amneal Women employee resource group in India, and in 2024 our India-based
team successfully trained approximately 90 female new hires through our “AmNeev” initiative - a three-month program designed to help early career individuals bridge the gap between academic learning and pharmaceutical industry requirements.
As of December 31, 2024, six out of ten of our executives identified as diverse by race, ethnicity, or gender. More broadly, as of December 31, 2024, approximately 74% of our U.S. workforce of Amneal Employees identified as diverse by race or ethnicity and women represented approximately 21% of our global workforce of Amneal Employees. In India, women comprise approximately 11% of our workforce, and in the U.S., women represented approximately 40% of our workforce and held approximately 34% of leadership roles at the level of Director and above as of December 31, 2024.
Culture
Our success is driven by an inclusive employee culture that encourages colleagues to bring their best selves to work and be actively engaged, offer new ideas, and deliver real results. Our “Rise, Lead, Succeed” behaviors unite our global teams and foster an environment of open communication (town halls, company updates, video storytelling, access to leaders, etc.), collaboration (digital and traditional), and ownership. They also serve as a dynamic framework for celebrating and rewarding individual and team performance.
Other powerful hallmarks of Amneal culture are a teamwide commitment to quality and an unwavering focus on doing the right thing. Our steadfast commitment to quality is driven by a robust global quality policy and ethics and compliance is guided by our Code of Conduct. Both are championed by our executive management team and every colleague. We also foster a speak up culture where colleagues are encouraged to report potential misconduct or a violation of the Code of Conduct with their managers or via our Amtegrity ethics website/hotline, which is independently monitored 24 hours a day, 7 days a week.
Employee Well-Being
We foster holistic well-being for our employees and their families through our AmWell program. In the United States, this initiative has been powered by Personify Health since 2021, providing an extensive range of mental, physical, and financial resources, content and services. In 2024, we expanded the AmWell platform to our Ireland colleagues while our India-based teams benefited from AmWell wellness solutions tailored to local needs. Our India-based AmWell programming includes health screenings for employees, with a majority of our employees based in India participating in our annual employee health check-up offering in 2024. During 2024, we also saw impressive engagement with our expanding India-based mental health, physical fitness and financial literacy programs.
To deepen value and encourage active participation, we enhanced AmWell with applications focused on mindfulness, meditation, nutrition, fitness, mobility, and financial acumen. We also host wellness challenges throughout the year that spotlight different pillars of well-being and offer meaningful incentives to encourage personal achievements. By listening to employee feedback and regularly updating our offerings, we create tailored programming that empower individuals to thrive in every aspect of their well-being.
Alongside personal wellness, we care deeply about workplace safety and bolstered our efforts via monthly safety education campaigns and continued deployment of the IndustrySafe application, supporting enhanced incident reporting and analysis of safety metrics across all geographies. Colleagues demonstrated our continuous safety focus by participating in the World Health and Safety Day across our global sites.
Total Rewards
We attract and retain talent and reward performance through our robust Total Rewards program, which includes industry competitive benefits, compensation and recognition offerings. Our compensation program includes competitive base salaries, annual cash performance-based incentives, equity-based long-term incentive awards and cash-based long-term incentive awards for eligible employees. We also offer a broad, flexible and competitive benefits program that enables employees to choose the plans and coverage that meet their personal needs. These robust programs, which vary by country, include basic and supplemental health and insurance benefits, health savings and flexible spending accounts, access to a personal health advocate, paid parental leave for birth, adoption or foster placement, family leave, employee assistance programs, travel assistance, tuition reimbursement assistance and retirement savings plans. In addition, we offer a remote work policy that enables eligible U.S.-based employees the flexibility of a hybrid work schedule of three days onsite and two days remote per week.
Recognition is a core element of our culture and in early 2024, we reimagined our global employee recognition program with the launch of a new Amneal Achievers digital platform supported by learning sessions on how gratitude can create connections, convey impact, and foster belonging. By the end of 2024, impressive engagement resulted in nearly 55,000 unique recognitions
shared between colleagues. These recognitions were further amplified over 50,000 times by other peers boosting, liking, and commenting within the platform.
Talent Development
We develop next-level individual and organizational capability through our annual performance management process as well as a continuously evolving learning culture. In 2024, we strengthened management and leadership skills via continued investment in professional development.
We expanded the Amneal Leadership Lab (“all”) curriculum leveraging recognized external experts who collaborated with senior leaders on Leadership Practices including Recognition, Inclusion & Belonging, Managing Energy to Sustain Momentum, and Listening for Collaborative Dialogue. The all core program was further cascaded across the organization and covered psychological safety and direct conversations, building trust, effective decisions, and leading and embracing change. Additionally, we expanded offerings of our all Management Standards curriculum - a series of eight workshops designed for new people leaders covering best practices for elevating manager effectiveness and creating positive employee experiences.
In India, our Young Women Leadership Development Program successfully prepared 23 female employees for their next role and our Frontline Development Program trained more than 100 frontline employees on essential leadership skills. We also launched our Leader Speak and Amneal Accelerate bite size learning initiatives, which reinforced our culture of continuous learning.
To help cascade Amneal leadership and management principles, strengthen a learning culture, and accelerate development across the organization, we also offer access to LinkedIn Learning which also hosts Amneal’s growing library of custom-curated learning options supporting individual and organizational leadership growth.
Employee Engagement
Listening and engagement are important aspects of our culture and colleagues are encouraged to share their opinions, insights, and ideas. We collect and measure that feedback through various channels including town halls, leadership retreats, company email boxes and our Amneal Listens program. In 2024, we leveraged Amneal Listens, powered by a reputable third-party tool, to measure employee engagement. We recorded an 88% overall employee engagement index (11% above the pharmaceutical industry benchmark) as well as very high scores on confidence in the Company (91% favorable on efforts to advance strategy) and retention (81% plan to stay with Amneal for the next 3 or more years). Even with these results, we are taking additional actions to further strengthen engagement, including expanding opportunities to grow via a robust career development program (design initiated in 2024) and supporting functional leadership in addressing key survey themes across the business. We continue to expand our Amneal Listens strategy via our annual employee engagement survey as well as various organizationally aligned ad hoc surveys throughout the year.
Environmental, Social, and Governance (“ESG”) Initiatives
We are a purpose-driven company, and we continue strengthening our longstanding commitment to corporate responsibility.
In 2024, we made several enhancements to our ESG efforts to keep pace with our desire to be an environmentally and socially responsible company while also meeting rapidly evolving global regulatory requirements. We catalyzed our global data collection efforts, including several new metrics to help the company continue to improve our sustainability performance. We also expanded our materiality work to align with global ESG disclosure frameworks. Additionally, our cross-functional ESG task force (including members from finance, internal audit, legal, information technology and ESG) kicked off several projects to enhance our ESG programs and related governance. In social responsibility, we deepened our 2024 philanthropic partnerships with non-profit organizations in support of response efforts from the devastating hurricanes that hit the Southeastern U.S. In addition, we furthered relationships with product donation partners to help address the growing need for critical medicines.
These are just some of our many human capital and ESG initiatives. Every year, we aim to review and enhance these and other programs to ensure that we are improving, staying competitive and putting our people at the center of our success.
Further information on our Responsibility program is available at https://www.amneal.com/about/responsibility. The information on our website is not, and will not be deemed, a part of this Annual Report on Form 10-K or incorporated into any other filings we make with the SEC.
For discussion of the risks relating to the attraction and retention of management and executive management employees, refer to Part 1. Item 1A. Risk Factors.
Available Information
Our main corporate website address is www.amneal.com. The Company files electronically with the Securities and Exchange Commission (“SEC”) required reports on Form 8-K, Form 10-Q, and Form 10-K; proxy materials; ownership reports for insiders required by Section 16 of the Securities Exchange Act of 1934, as amended; registration statements on Forms S-3 and S-8, as necessary; and other forms or reports as required. Copies of our Quarterly Reports on Form 10-Q, Annual Reports on Form 10-K, Current Reports on Form 8-K, proxy statements and any amendments to such reports filed with or furnished to the SEC, are available free of charge on our website as soon as reasonably practicable after having been filed with or furnished to the SEC. All SEC filings are also available at the SEC’s website at www.sec.gov. In addition, the written charters of our Audit Committee, Compensation Committee, Nominating and Governance Committee, and Conflicts Committee of the Board of Directors and our Code of Business Conduct, Corporate Governance Guidelines and other corporate governance materials are available on our website. We may use our website as a distribution channel of material company information. Financial and other important information is routinely posted on and accessible through our website at https://investors.amneal.com. In addition, you may automatically receive email alerts and other information when you enroll your email address by visiting https://investors.amneal.com/investor-resources/email-alerts/default.aspx. The information on our website is not, and will not be deemed, a part of this Annual Report on Form 10-K or incorporated into any other filings we make with the SEC.
Item 1A. Risk Factors
An investment in our common stock involves a high degree of risk. In deciding whether to invest in our common stock, you should consider carefully the following risk factors, as well as the other information included in this Annual Report on Form 10-K. The materialization of any of these risks could have a material adverse effect on our business, results of operations and financial condition.
Operational and Competitive Risks
If we are unable to successfully develop or commercialize new products, our operating results will suffer.
Developing and commercializing a new product is time consuming, costly and subject to numerous factors that may delay or prevent such development and commercialization. Our future results of operations will depend to a significant extent upon our ability to successfully commercialize new products in a timely manner. We face several challenges when developing and commercializing new products, including:
•our ability to develop products in a timely and cost-efficient manner and in compliance with regulatory requirements, including delays associated with the FDA listing and approval process and our ability to obtain required regulatory approvals in a timely manner, or at all, and maintain such approvals if obtained;
•the success of our clinical testing process to ensure that new products are safe and effective or bioequivalent to the reference listed drug;
•the risk that legal action may be brought against our generic drug products by our branded drug product competitors, including patent infringement claims among others;
•the availability, on commercially reasonable terms, of raw materials, including APIs and other key ingredients necessary to the development of our drug products; and
•our ability to scale-up manufacturing methods to successfully manufacture commercial quantities of drug product in compliance with regulatory requirements.
As a result of these and other difficulties, our products in development may or may not receive necessary regulatory approvals on a timely basis or at all, which may result in unsuccessful development or commercialization of new products.
Separately, product licensing involves inherent risks including uncertainties due to matters that may affect the achievement of milestones, as well as the possibility of contractual disagreements with regard to terms such as license scope or termination rights. If any of our products, when acquired or developed and approved, cannot be successfully or timely commercialized, our operating results could be adversely affected. We cannot guarantee that any investment we make in developing, marketing or licensing products will be recouped, even if we are successful in commercializing those products.
We face intense competition in the pharmaceutical industry from both brand and generic drug product companies, which could significantly limit our growth and materially adversely affect our financial results.
The pharmaceutical industry is highly competitive. The principal competitive factors in the pharmaceutical market include:
•introduction of other generic drug manufacturers’ products in direct competition with our generic drug products;
•introduction of authorized generic drug products in direct competition with our generic or branded products, particularly during exclusivity periods;
•the ability of generic drug product competitors to quickly enter the market after the expiration of patents or exclusivity periods of our branded products, diminishing the amount and duration of significant profits;
•consolidation among distribution outlets through mergers and acquisitions and the formation of buying groups;
•the willingness of generic drug customers, including wholesale and retail customers, to switch among products of different pharmaceutical manufacturers;
•pricing pressures by competitors and customers, even if similar price savings are not passed on to consumers;
•a company’s reputation as a manufacturer and distributor of quality products;
•a company’s level of service (including maintaining sufficient inventory levels for timely deliveries);
•a company’s ability to use and integrate artificial intelligence (“AI”);
•product appearance and labeling; and
•a company’s breadth of product offerings.
Many of our competitors have longer operating histories and greater financial, R&D, marketing and other resources than we do. Consequently, some of our competitors may be able to develop products and/or processes competitive with, or superior to, our products and/or processes. Furthermore, we may not be able to (i) differentiate our products from those of our competitors, (ii) successfully develop or introduce new products, on a timely basis or at all, that are less costly than those of our competitors, (iii) integrate new systems or technology, such as AI, as quickly or successfully as our competitors, or (iv) offer customers payment and other commercial terms as favorable as those offered by our competitors. The markets in which we compete and intend to compete are undergoing, and are expected to continue to undergo, rapid and significant change. We expect competition to intensify as technology advances and consolidation continues. New developments by other manufacturers and distributors could render our products uncompetitive or obsolete.
Our principal competitors in the U.S. generic/biosimilar pharmaceutical products market, where we primarily compete, are Teva Pharmaceutical Industries Ltd., Viatris Inc., Sandoz Group, Pfizer Inc., Fresenius Kabi KGaA, Hikma Pharmaceuticals PLC, Dr. Reddy’s Laboratories Ltd., Amphastar Pharmaceuticals, Inc., Sun Pharmaceutical Industries Ltd., Lupin Pharmaceuticals, Inc., Zydus Pharmaceuticals USA Inc., and Aurobindo Pharma Limited. Our principal competitors in the specialty pharmaceutical products market include Supernus Pharmaceuticals, Inc., Jazz Pharmaceuticals PLC, AbbVie Inc. and Alkermes PLC. Our competitors in the AvKARE segment are other wholesalers, including Cardinal Health, Inc., Cencora, Inc., McKesson Drug Co., and manufacturers / re-packagers such as Golden State Medical Supply.
The products produced by these companies, among others, collectively compete with the majority of our products. We also face price competition generally as other generic manufacturers enter the market. Any such price competition may be especially pronounced where our competitors source their products from jurisdictions where production costs may be lower (sometimes significantly) than our production costs. Any of these factors could result in reductions in our sales prices and gross margin. This price competition has led to an increase in demands for downward price adjustments by generic pharmaceutical distributors. Our principal strategy in addressing our competition is to offer customers a consistent supply of our generic drug products, as well as to pursue product opportunities with the potential for limited competition, such as high-barrier-to-entry FTF or FTM products. We cannot provide assurance, however, that this strategy will enable us to compete successfully in the generic drug product industry or that we will be able to develop and implement any new or additional viable strategies.
Competition in the generic drug industry has also increased due to the proliferation of authorized generic pharmaceutical products. Authorized generic drug products are generic drug products that are introduced by brand companies, either directly or through third parties, under the brand’s NDA approval. Authorized generics do not face any regulatory barriers to introduction and are not prohibited from sale during the 180-day marketing exclusivity period granted to the FTF ANDA applicant. The sale of authorized generics adversely impacts the market share of a generic drug product that has been granted 180 days of marketing exclusivity. This is a significant source of competition for us, because an authorized generic drug product can materially decrease the profits that we could receive as an otherwise exclusive marketer of a generic drug product. Such actions have the effect of reducing the potential market share and profitability of our generic drug products and may inhibit us from developing and introducing generic pharmaceutical drug products corresponding to certain branded drugs.
If we fail to obtain exclusive marketing rights for our products or fail to introduce our products to the market on a timely basis, our revenues, gross margin and operating results may decline significantly.
The Hatch-Waxman amendments to the FDCA provide for a period of 180 days of generic marketing exclusivity for any applicant that is first to file an ANDA containing a certification of invalidity, non-infringement or unenforceability related to a patent listed with respect to the corresponding branded drug (commonly referred to as a “Paragraph IV certification”). “First filers” are often able to price the applicable generic drug to yield relatively high gross margins during this 180-day marketing exclusivity period.
With respect to our generic products, ANDAs containing Paragraph IV certifications generally become the subject of patent litigation that can be both lengthy and costly. There is no certainty that we will prevail in any such litigation, that we will be the first to file and thus granted the 180-day marketing exclusivity period, or, if we are granted the 180-day marketing exclusivity period, that we will not forfeit such period. Even where we are awarded marketing exclusivity, we may be required to share our exclusivity period with other first filers. In addition, branded drug product companies often authorize a generic version of the corresponding branded drug product to be sold during any period of marketing exclusivity that is awarded, which reduces gross margins during the marketing exclusivity period. Branded drug product companies may also reduce the price of their branded drug product to compete directly with generic drug products entering the market, which would similarly have the effect of reducing gross margins. Furthermore, timely commencement of the litigation by the patent owner imposes an automatic stay of ANDA approval by the FDA for 30 months, unless the case is decided in the ANDA applicant’s favor during that period. Finally, if the court decision is adverse to the ANDA applicant, the ANDA approval will be delayed until the challenged patent expires, and the applicant forfeits the 180-day marketing exclusivity.
Our future profitability depends, to a significant extent, upon our ability to introduce, on a timely basis, new generic drug products that are either the first-to-market (or among the first-to-market) or that otherwise can gain significant market share. The timeliness of our product introductions is dependent upon, among other things, the timing of regulatory approval of our products, which to a large extent is outside of our control, as well as the timing of the introduction of competing products. As additional distributors introduce comparable generic pharmaceutical products, price competition intensifies, market access narrows, and product sales prices and gross margins decline, often significantly and rapidly, regardless of whether consumers ultimately pay less for the drug. Accordingly, our revenues and future profitability are dependent, in large part, upon our ability or the ability of our development partners to file ANDAs with the FDA in a timely and effective manner or, alternatively, to enter into contractual relationships with other parties that have obtained marketing exclusivity. We cannot provide any assurance that we will be able to develop and introduce successful products in the future within the time constraints necessary to be successful. If we or our development partners are unable to continue to timely and effectively file ANDAs with the FDA or to partner with other parties that have obtained marketing exclusivity, our revenues, gross margin and operating results may decline significantly, and our prospects and business may be materially adversely affected.
With respect to our branded products, generic equivalents for branded pharmaceutical products are typically sold at lower prices than the branded products. The regulatory approval process in the U.S. and European Union exempts generic products from costly and time-consuming clinical trials to demonstrate their safety and efficacy and relies instead on the safety and efficacy of prior products. After the introduction of a competing generic product, a significant percentage of the prescriptions previously written for the branded product are often written for the generic version. In addition, legislation enacted in most U.S. states allows, or in some instances mandates, a pharmacist to dispense an available generic equivalent when filling a prescription for a branded product, in the absence of specific instructions from the prescribing physician. Pursuant to the provisions of the Hatch-Waxman Act, manufacturers of branded products often bring lawsuits to enforce their patent rights against generic products released prior to the expiration of branded products’ patents, but it is possible for generic manufacturers to offer generic products while such litigation is pending. As a result, branded products typically experience a significant loss in revenues and gross profit following the introduction of a competing generic product, even if subject to an existing patent. Our branded pharmaceutical products are or may become subject to competition from generic equivalents because there is no proprietary protection for some of the branded pharmaceutical products we sell, because our patent protection expired or because our patent protection is not sufficiently broad or enforceable. For example, in 2025, we expect to lose exclusivity for RYTARY®, an extended-release oral capsule formulation of carbidopa-levodopa for the treatment of Parkinson’s disease.
As our competitors introduce their own generic equivalents of our generic drug products, our revenues and gross margin from such products generally decline, often rapidly.
Revenues and gross margin derived from generic pharmaceutical products often follow a pattern based on regulatory and competitive factors that we believe are unique to the generic pharmaceutical industry. As the patent(s) for a brand name product
or the statutory marketing exclusivity period (if any) expires, the first generic manufacturer to receive regulatory approval for a generic equivalent of the product is often able to capture a substantial share of the market. However, as other generic manufacturers receive regulatory approvals for their own generic versions, that market share, and the price of that product, will typically decline depending on several factors, including the number of competitors, the price of the branded product and the pricing strategy of the new competitors. We often experience significant competition for many of our generic products, which from time to time, has resulted in a significant decline in our revenue and gross margin. We cannot provide assurance that we will be able to continue to develop such products or that the number of our competitors for any given product will not increase to such an extent that we may stop marketing a generic drug product for which we previously obtained approval, which may have a material adverse impact on our revenues and gross margin.
The illegal distribution and sale by third parties of counterfeit versions of our products or of stolen products could have a negative impact on our reputation and a material adverse effect on our business, results of operations and financial condition.
Third parties could illegally distribute and sell counterfeit versions of our products, which do not meet the rigorous manufacturing and testing standards that our products undergo. Counterfeit products are frequently unsafe or ineffective and can be life-threatening. Counterfeit medicines may contain harmful substances, the wrong dose of the API or no API at all. However, to distributors and users, counterfeit products may be visually indistinguishable from the authentic version.
Reports of adverse reactions to counterfeit drugs or increased levels of counterfeiting could materially affect patient confidence in the authentic product. It is possible that adverse events caused by unsafe counterfeit products will mistakenly be attributed to the authentic product. In addition, thefts of inventory at warehouses, plants or while in-transit, which are not properly stored and which are sold through unauthorized channels, could adversely impact patient safety, our reputation and our business.
Public loss of confidence in the integrity of pharmaceutical products as a result of counterfeiting or theft could have a material adverse effect on our business, results of operations and financial condition.
Our business is highly dependent on market perceptions of us and the safety and quality of our products. Our business, products or product pricing could be subject to negative publicity, which could have a material adverse effect on our business, results of operations and financial condition.
Market perceptions of our business are very important to us, especially market perceptions of the safety and quality of our products. If any of our products or similar products that other companies distribute are subject to market withdrawal or recall or are proven to be, or are claimed to be, harmful to consumers, then this could have a material adverse effect on our business, results of operations and financial condition. Also, because our business is dependent on market perceptions, negative publicity associated with product quality, illness or other adverse effects resulting from, or perceived to be resulting from, our products could have a material adverse impact on our business, results of operations and financial condition.
The pharmaceutical industry has also in recent years been the subject of significant publicity regarding the pricing of pharmaceutical products more generally, including publicity and pressure resulting from prices charged by competitors and peer companies for new products as well as price increases by competitors and peer companies on older products that the public has deemed excessive. Even if we may have reduced the prices we charge our customers for certain products, often consumers do not see similar reductions in the prices they paid. Any downward pricing pressure on the price of certain of our products arising from social or political pressure to lower the cost of pharmaceutical products could have a material adverse impact on our business, results of operations and financial condition.
Accompanying the press and media coverage of pharmaceutical pricing practices and public complaints about the same, has been increasing U.S. federal and state legislative and enforcement interest with respect to drug pricing. For instance, the DOJ issued subpoenas to pharmaceutical companies, including us, seeking information about the sales, marketing and pricing of certain generic drugs. See Note 20. Commitments and Contingencies for additional information on the DOJ investigation. In addition to the effects of any investigations or claims brought against us, our business, results of operations and financial condition could also be adversely affected if any such inquiries, of us or of other pharmaceutical companies or the industry more generally, were to result in legislative or regulatory proposals that limit our ability to increase the prices of our products.
A substantial portion of our total revenues is expected to be derived from sales of a limited number of products.
We expect that we will continue to derive a substantial portion of our revenue from sales of a limited number of products. For the year ended December 31, 2024, our significant product families (defined as our top five products by annual revenue
including both our Affordable Medicines and Specialty segments) accounted for 25% of our consolidated net revenue. The sale of our products may be significantly influenced by market conditions, as well as regulatory actions. We may experience decreases in the sale of our products in the future as a result of actions taken by our competitors, such as price reductions, or as a result of regulatory actions related to our products or to competing products, which could have a material impact on our results of operations. Actions which could be taken by our competitors, which may materially and adversely affect our business, results of operations and financial condition, may include, without limitation, pricing changes and entering or exiting the market for specific products.
Our approved products may not achieve expected levels of market acceptance.
Even if we are able to obtain regulatory approvals for our new products, such as for CREXONT®, the success of those products is dependent upon market acceptance. Levels of market acceptance for our new products could be affected by several factors, including:
•the availability of alternative products from our competitors;
•the prices of our products relative to those of our competitors;
•the timing of our market entry;
•the ability to market our products effectively at the retail level;
•the perception of patients and the healthcare community, including third-party payers, regarding the safety, efficacy and benefits of our drug products compared to those of competing products;
•the healthcare providers’ willingness to prescribe medication based on perceptions, training, personal experiences and guidelines;
•the willingness of payers and insurance companies to cover or add products to their formularies;
•the willingness of hospitals and integrated delivery systems to stock or approve products on their formularies; and
•the acceptance of our products by government and private formularies.
Some of these factors will not be in our control, and our products may not achieve expected levels of market acceptance. Additionally, continuing and increasingly sophisticated studies of the proper utilization, safety and efficacy of pharmaceutical products are being conducted by the industry, government agencies and others which can call into question the utilization, safety and efficacy of products currently or previously marketed by us. In some cases, studies have resulted, and may in the future result, in the discontinuance of product marketing or other risk management programs such as the need for a patient registry.
We may discontinue the manufacture and distribution of certain existing products, which may adversely impact our business, results of operations and financial condition.
We continually evaluate the performance of our products and may determine that it is in our best interest to discontinue the manufacture and distribution of certain of our products. For example, in 2023, there was a reduction in the promotional focus on LYVISPAH™, which resulted in an impairment charge of $34.1 million recorded to cost of goods sold. We cannot guarantee that we have correctly forecasted, or will correctly forecast in the future, the appropriate products to discontinue or that our decision to discontinue various products is prudent if market conditions change. In addition, we cannot assure you that the discontinuance of products will reduce our operating expenses or will not cause us to incur material charges associated with such a decision. Furthermore, the discontinuance of existing products entails various risks, including, in the event that we decide to sell the discontinued product, the risk that we will not be able to find a purchaser for such products or that the purchase price obtained will not be equal to at least the book value of the net assets for such products. Other risks include managing the expectations of, and maintaining good relations with, our customers who previously purchased products from among our discontinued products, which could prevent us from selling other products to them in the future. Moreover, we may incur other significant liabilities and costs associated with our discontinuance of products, which could have a material adverse effect on our business, results of operations and financial condition.
Manufacturing or quality control problems may damage our reputation for quality production, demand costly remedial activities and negatively impact our business, results of operations and financial condition.
As a pharmaceutical company, we are subject to substantial regulation by various governmental authorities. For instance, we must comply with requirements of the FDA, DEA and other healthcare regulators with respect to the manufacture, labeling, sale, distribution, marketing, advertising, promotion and development of pharmaceutical products. We must register our facilities, whether located in the U.S. or elsewhere, with the FDA as well as regulators outside the U.S., and our products must
be made in a manner consistent with cGMP, or similar standards in each territory in which we manufacture. The failure of one of our facilities, or a facility of one of our third-party suppliers, to comply with applicable laws and regulations may lead to breach of representations made to our customers or to regulatory or government action against us related to products made in that facility.
In addition, the FDA, DEA and other agencies periodically inspect our manufacturing facilities. Following an inspection, agencies have in the past issued, and may in the future issue, a notice listing conditions that are believed to violate cGMP or other regulations, or a warning letter for violations of “regulatory significance” that may result in enforcement action if not promptly and adequately corrected. We remain committed to continuing to improve our quality control and manufacturing practices; however, we cannot be assured that the FDA will continue to be satisfied with our corrective actions and with our quality control and manufacturing systems and standards. Failure to comply strictly with these regulations and requirements, or our failure to remedy any deficiencies, may damage our reputation and lead to financial penalties, compliance expenditures, the recall or seizure of products, total or partial suspension of production and/or distribution, withdrawal or suspension of the applicable regulator’s review of our submissions, enforcement actions, injunctions and criminal prosecution. Further, other federal agencies, our customers and partners in our alliance, development, collaboration and other partnership agreements with respect to our products and services may take any such FDA observations or warning letters into account when considering the award of contracts or the continuation or extension of such partnership agreements. Because regulatory approval to manufacture a drug is site-specific, the delay and cost of remedial actions, or obtaining approval to manufacture at a different facility, could negatively impact our business. Any failure by us to comply with applicable laws and regulations and/or any actions by the FDA and other agencies as described above could have a material adverse effect on our business, financial position and results of operations.
Our profitability depends on our major customers. If these relationships do not continue as expected, our business, condition (financial and otherwise), prospects and results of operations could materially suffer.
Our four largest customers, Cencora, Inc., McKesson Drug Co., Cardinal Health, Inc. and CVS Health Corporation, accounted for approximately 70%, 70% and 71% of total net sales of products for the years ended December 31, 2024, 2023 and 2022, respectively. The loss of any one or more of these or any other major customer or the substantial reduction in orders from any one or more of our major customers could have a material impact on our future operating results and financial condition. In total, we currently have over 1,300 customers (including over 1,100 customers specific to our AvKARE segment), some of which are part of large purchasing groups.
We may experience declines in the sales volume and prices of our products as a result of the continuing trend of consolidation of certain customer groups, which could have a material adverse effect on our business, financial position and results of operations.
Our ability to successfully commercialize any generic or branded pharmaceutical product depends in large part upon the acceptance of the product by third parties, including pharmacies, government formularies, other retailers, physicians and patients. Therefore, our success will depend in large part on market acceptance of our products. We make a significant amount of our sales to a relatively small number of drug wholesalers and retail drug chains. These customers represent an essential part of the distribution chain of our pharmaceutical products. Drug wholesalers and retail drug chains have undergone, and are continuing to undergo, significant consolidation. This consolidation may result in these groups gaining additional purchasing leverage and, consequently, increasing the product pricing pressures facing our business. Additionally, the emergence of large buying groups representing independent retail pharmacies and other drug distributors, and the prevalence and influence of managed care organizations and similar institutions, potentially enable such groups to demand larger price discounts on our products. For example, large wholesalers and retailer customers have formed alliances, such as Walgreens and Cencora, Inc., Rite Aid and McKesson Drug Company, and CVS Caremark and Cardinal Health. The result of these developments may have a material adverse effect on our business, financial position and results of operations.
We depend to a large extent on third-party suppliers and distributors for the raw materials for our products, particularly the chemical compounds comprising the APIs that we use to manufacture our products, as well as for certain finished goods. A prolonged interruption in the supply of such products could have a material adverse effect on our business, financial position and results of operations.
We purchase the bulk of the raw materials essential to our manufacturing business from third parties. If we experience supply interruptions or delays, or if a supplier discontinues the sale of certain products, we may have to obtain substitute materials or products, which in turn would require us to obtain amended or additional regulatory approvals, subjecting us to additional expenditures of significant time and resources. In addition, changes in our raw material suppliers could result in significant delays in production, higher raw material costs and loss of sales and customers, because regulatory authorities must generally approve raw material sources for pharmaceutical products, which may be time consuming. For example, we may need as long as 18 months to find and qualify a new sole-source supplier. If we receive less than one year’s termination notice from a sole-source supplier that intends to cease supplying raw materials, it could result in disruption of our ability to produce the drug involved. Any significant supply interruption could have a material adverse effect on our business, condition (financial and otherwise), prospects and results of operations. To date, although we have experienced occasional interruptions in supplies, we have experienced no significant difficulties in obtaining raw materials. However, because the federal drug application process requires specification of raw material suppliers, if raw materials from a specified supplier were to become unavailable, FDA approval of a new supplier would be required. The amount of time required for the FDA to qualify a new supplier and confirm that our manufacturing processes meet the necessary standards could cause delays in the manufacturing and marketing of one or more of our products and could, depending on the particular product, have a material adverse effect on our results of operations and financial condition.
Changes in trade policy, including the imposition of tariffs may adversely affect our business, results of operations and financial condition.
The U.S. and various foreign governments have established certain trade and tariff requirements. From time to time, the U.S. government has indicated a willingness to revise or renegotiate tariffs on certain goods imported into the U.S. Since we rely on APIs, packaging and other components and finished pharmaceutical products from China, India, and certain countries in the European Union, such steps, if adopted, could adversely impact our business, increase our costs, and make our products less competitive.
On February 1, 2025, President Trump announced a 10% additional tariff on imports from China and has expressed the possibility of imposing tariffs on imports from the European Union and India. In addition, on February 13, 2025, President Trump announced a plan to establish reciprocal tariffs with nations that impose tariffs on U.S. products, and directed relevant segments of the U.S. government to assess harm of non-reciprocal trade arrangements and to generate a report of that assessment within 180 days. Reciprocal tariffs may be imposed prior to completion of the report. There also have been statements by the Trump Administration regarding a 25% tariff on pharmaceutical products. For the year ending December 31, 2025, we estimate the impact of tariffs currently imposed on our imports from China will not be material. We are unable to estimate the impacts of any tariffs that have not yet been imposed by the U.S. government.
The time necessary to develop generic drugs may adversely affect whether, and the extent to which, we receive a return on our capital.
We generally begin our development activities for a new generic drug product several years in advance of the patent expiration date of the brand-name drug equivalent. The development process, including drug formulation, testing, and FDA review and approval, often takes three or more years, and is informed by factors outside of our control, including but not limited to, FDA staffing and policy changes. This process requires that we expend considerable capital to pursue activities that do not yield an immediate or near-term return. Also, because of the significant time necessary to develop a product, the actual market for a product at the time it is available for sale may be significantly less than the originally projected market for the product. If this were to occur, our potential return on our investment in developing the product, if approved for marketing by the FDA, would be adversely affected and we may never receive a return on our investment in the product. It is also possible for the manufacturer of the brand-name product for which we are developing a generic drug to obtain approvals from the FDA to switch the brand-name drug from the prescription market to the over-the counter (“OTC”) market. If this were to occur, we would be prohibited from marketing our product other than as an OTC drug, in which case revenues could be substantially less than we anticipated.
The risks and uncertainties inherent in conducting clinical trials could delay or prevent the development and commercialization of our own branded products, which could have a material adverse effect on our business, results of operations and financial condition.
With respect to our branded products which do not qualify for the FDA’s abbreviated application procedures, we must demonstrate through clinical trials that these products are safe and effective for use. We have only limited experience in conducting and supervising clinical trials. The process of completing clinical trials and preparing an NDA may take several years and requires substantial resources. Our studies and filings may not result in FDA approval to market our new drug products and, if the FDA grants approval, we cannot predict the timing of any approval, which is informed by factors outside of our control, including but not limited to, FDA staffing and policy changes. There are substantial filing fees for NDAs that are not refundable if FDA approval is not obtained.
There are a number of risks and uncertainties associated with clinical trials. The results of clinical trials may not be indicative of results that would be obtained from large scale testing. Clinical trials are often conducted with patients having advanced stages of disease and, as a result, during the course of treatment these patients can die or suffer adverse medical effects for reasons that may not be related to the pharmaceutical agents being tested, but which nevertheless affect the clinical trial results. In addition, side effects experienced by the patients may cause delay of approval or limit the profile of an approved product. Moreover, our clinical trials may not demonstrate sufficient safety and efficacy to obtain approval from the FDA or foreign regulatory authorities. The FDA or foreign regulatory authorities may not agree with our assessment of the clinical data or they may interpret it differently. Such regulatory authorities may require additional or expanded clinical trials. Even if the FDA or foreign regulatory authorities approve certain products developed by us, we cannot provide assurance that such regulatory authorities will not subject marketing of such products to certain limits on indicated use.
Developing and commercializing branded pharmaceutical products is generally more costly than developing and commercializing generic products. To grow and achieve success in our branded product business, we must continually identify, develop, acquire and license new products that we can ultimately market. There are many difficulties and uncertainties inherent in pharmaceutical R&D, and there is a high rate of failure inherent in new drug discovery and development. Failure can occur at any point in the process, including late in the process after substantial investment. New product candidates that appear promising in development may fail to reach the market or may have only limited commercial success because of efficacy or safety concerns, inability to obtain necessary regulatory approvals and payer reimbursement, limited scope of approved uses, difficulty or excessive costs to manufacture, or infringement of the patents or IP rights of others. Products that do reach the market may ultimately be subject to recalls or other suspensions in sales. Delays and uncertainties in the FDA approval process and the approval processes in other countries can result in delays in product launches and lost market opportunity. Because there is a high rate of failure inherent in the R&D process of new products, there is a significant risk that funds invested in R&D will not generate financial returns. We cannot be certain when or whether any of our products currently under development will be approved or launched or whether, once launched, such products will be commercially successful. We may be required to spend several years and incur substantial expense in completing certain clinical trials. The length of time, number of trial sites and patients required for clinical trials vary substantially, and we may have difficulty finding a sufficient number of sites and subjects to participate in our trials. Delays in planned clinical trials can result in increased development costs, delays in regulatory approvals and delays in product candidates reaching the market. We rely on independent third-party clinical investigators to recruit subjects and conduct clinical trials in accordance with applicable study protocols and laws and regulations. If regulatory authorities determine that we have not complied with regulations in the development of a product candidate, they may refuse to accept trial data from the site and/or not approve the product candidate, and we would not be able to market and sell that product. If we are not able to market and sell our products after significant expenditures to develop and test them, our business and results of operations could be materially and adversely affected.
The results from early clinical trials may not be predictive of results obtained in later and larger clinical trials, and product candidates in later clinical trials may fail to show the desired safety or efficacy despite having progressed successfully through earlier clinical testing. A number of companies in the pharmaceutical industry, including us, have suffered significant setbacks in clinical trials, even in advanced clinical trials after showing positive results in earlier clinical trials. The completion of clinical trials for our product candidates may be delayed or halted for a variety of reasons in addition to the reasons noted above.
In addition, our product candidates could be subject to competition for clinical study sites and patients from other therapies under development which may delay the enrollment in or initiation of our clinical trials.
The FDA or foreign regulatory authorities may require us to conduct unanticipated additional clinical trials, which could result in additional expense and delays in bringing our product candidates to market. Any failure or delay in completing clinical trials
for our product candidates would prevent or delay the commercialization of our product candidates. We cannot assure you that our expenses related to clinical trials will lead to the development of brand-name drugs that will generate revenues in the near future. Delays or failure in the development and commercialization of our own branded products could have a material adverse effect on our business, results of operations and financial condition.
If we are unable to execute acquisitions or other strategic transactions, or successfully integrate such acquisitions or manage our growth therefrom, it could have a material adverse effect on our business.
We may seek to expand our business through complementary or strategic acquisitions of other businesses, products or assets, or through joint ventures, strategic agreements or other arrangements. Any such acquisitions, joint ventures or other business combinations may involve significant integration challenges, operational complexities and time consumption, adversely affect liquidity and require substantial resources and effort. It may also disrupt our ongoing businesses, which may adversely affect our relationships with customers, employees, regulators and others with whom we conduct business. Further, if we are unable to realize synergies or other benefits expected to result from any acquisitions, joint ventures or other business combinations, or to generate additional revenue to offset any unanticipated inability to realize these expected synergies or benefits, our growth and ability to compete may be impaired, which would require us to focus additional resources on the integration of operations rather than other profitable areas of our business, and may otherwise cause a material adverse effect on our business, results of operations and financial condition. Acquisitions may also have hidden costs, including unforeseen pre-acquisition liabilities or the impairment of customer relationships or certain acquired assets such as goodwill. We may also incur costs and inefficiencies to the extent an acquisition expands the industries, markets or geographies in which we operate due to our limited exposure to and experience in a given industry, market or region. Finally, acquisitions can also involve litigation and/or post-transaction disputes, including with the counterparty regarding purchase price or other working capital adjustment or liabilities for which we believe we were indemnified under the relevant transaction agreements, among other matters.
The use of legal, regulatory and legislative strategies by brand competitors, including authorized generics and citizen’s petitions, as well as the potential impact of proposed legislation, may have an adverse effect on our business.
Brand drug companies often pursue strategies that may serve to prevent or delay competition from our generic alternatives to their branded products. These strategies may include, but are not limited to, using certain tactics with our regulators that could delay our product approvals, restricting our access to samples needed for our testing or using the legal system or other IP-related or regulatory mechanisms to severely delay or disrupt our process. These and other strategies by brand competitors, as well as the potential impact of proposed legislation, may increase our costs associated with the introduction or marketing of our generic products, delay or prevent such introduction and/or significantly reduce the profit potential of our products.
We are increasingly dependent on information technology, and our systems and infrastructure face certain risks, including cybersecurity and data leakage risks.
Significant disruptions to our IT systems or breaches of information security could adversely affect our business. In the ordinary course of business, we collect, store and transmit large amounts of confidential information, and it is critical that we do so in a secure manner to maintain the confidentiality and integrity of such information. Additionally, our IT systems are critical to our ability to store electronic and financial information and to manage a variety of business processes and activities, including manufacturing, financial, logistics, sales, marketing and administrative functions. We depend on our IT infrastructure to communicate internally and externally with employees, customers, suppliers and others. We also use IT networks and systems to comply with regulatory, legal and tax requirements. We have outsourced significant elements of our IT infrastructure; as a result we manage independent vendor relationships with third-parties who are responsible for maintaining significant elements of our IT systems and infrastructure and who may or could have access to our confidential information. The size and complexity of our IT systems, and those of our third-party vendors, make such systems potentially vulnerable to service interruptions and security breaches from inadvertent or intentional actions by our employees, partners or vendors. These systems are also vulnerable to attacks by malicious third parties, such as phishing or ransomware attacks, and may be susceptible to intentional or accidental physical damage to the infrastructure maintained by us or by third parties, including as a result of extreme weather events, such as fires, floods, hurricanes, or tornadoes or as the result of the use of AI or other new technologies. For example, in 2024, CrowdStrike Holdings, Inc., a cybersecurity vendor, distributed a faulty software update that caused widespread problems with computers running Microsoft Windows as their operating system, which impacted individual, business and government users globally. While the Company was impacted by this faulty update, affected systems were remediated within seven business days, with minimal disruption and no material impact to the Company.
Maintaining the secrecy of confidential, proprietary, and/or trade secret information is important to our competitive business position. We continually assess these threats and make investments to increase internal protection, detection, and response
capabilities, as well as ensure our third-party providers have required capabilities and controls, to mitigate these risks. Like other public companies, our computer systems and those of our third-party vendors and service providers are regularly subject to, and will continue to be the target of, computer viruses, malware or other malicious code (including ransomware), unauthorized access, cyber-attacks or other computer-related penetrations, which have caused, and may continue to cause, disruptions to our operations. For example, we have been the victim of phishing attempts, some of which have been successful in evading detection and blocking. While we have experienced threats to our data and systems, to date, we are not aware that we have experienced a material cyber-security breach. Over time, however, the sophistication of these threats continues to increase. Our reliance on unsupported and vulnerable operating systems and other software in certain cases may increase both the likelihood and potential severity of cyber incidents. The preventative actions we take to reduce the risk of cyber incidents and protect our information may be insufficient. Our efforts may not prevent service interruptions or security breaches in our systems or the unauthorized or inadvertent wrongful use or disclosure of confidential information that could adversely affect our business operations or result in the loss, dissemination, or misuse of critical or sensitive information. A breach of our security measures or the accidental loss, inadvertent disclosure, unapproved dissemination, misappropriation or misuse of trade secrets, proprietary information, or other confidential information, whether as a result of theft, hacking, fraud, trickery or other forms of deception, or for any other cause, could enable others to produce competing products, use our proprietary technology or information, and/or adversely affect our business position. Further, any such interruption, security breach, loss or disclosure of confidential information could result in financial, legal, business, and reputational harm to us and could have a material adverse effect on our business, financial position, results of operations and/or cash flow.
Artificial intelligence-based platforms may present new risks and challenges to our business.
AI technologies may exacerbate existing risks, including risks associated with data privacy, cybersecurity, IP, healthcare fraud and abuse, drug development and manufacturing, and risks to patients or human subjects in clinical trials. AI also introduces new risks, due to the autonomous nature of the technology, which, in some cases, may be deployed to perform tasks, inform decisions, automate decisions, and make predictions, sometimes using unverified or false information. AI may amplify biased and discriminatory decision making, perform unreliably and malfunction, generate insights which are difficult to interpret and explain, and cause direct harm to individuals or groups.
Regulators are proposing, adopting, and implementing new AI laws and regulations. We may be required to change our business practices and policies as a result of such laws and regulations and may incur substantial compliance-related costs. Regulators are also using existing laws and regulations to take enforcement actions related to the deployment of AI in ways that result in non-compliance with current laws and regulations. If we fail to comply with AI laws and regulations, we may be subject to sanctions, fines, and reputational damage, orders to stop certain processing of personal data, orders to delete certain data or destroy AI algorithms derived from data collects, legal action on behalf of impacted individuals or other enforcement or other actions. If we or our vendors using AI technologies fail to take steps to protect our confidential data, trade secrets, IP and personal data, we may be subject to legal, regulatory, financial, and reputational risks.
AI technologies present significant opportunities and risks to our business. Harnessing AI’s transformative potential may enable us to speed up the discovery and development of new drugs, optimize our manufacturing processes, and drive efficiencies. Our failure to use AI technologies in a way that maintains trust, quality and control in our business activities and to capitalize on opportunities presented by AI may also place us at a competitive disadvantage. Failure to address AI risks will reduce our ability to deliver strategic objectives. Also, investments in AI may not realize the benefits that were anticipated.
The majority of our products are produced at a limited number of locations, and a business interruption at one or more of these locations or within our supply chain could have a material adverse effect on our business, financial position and results of operations.
We produce the majority of the products that we manufacture at our manufacturing facilities in New York, New Jersey and India, as well as at certain third-party suppliers, one of which is located in Taiwan. Disruptions at these facilities or within our supply chain can occur for many reasons, including events unrelated to us or beyond our control, such as fires and other industrial accidents, floods and other severe weather events, natural disasters, environmental incidents or other catastrophes, utility and transportation infrastructure disruptions, shortages of raw materials, pandemic diseases or viral contagions, and acts of war or terrorism. For example, in November 2023, the Houthi movement, which controls parts of Yemen, began attacking merchant ships in the Red Sea disrupting global supply chains; while the attacks have largely abated since late 2024, it is possible they will resume. Natural disasters and adverse weather conditions can be caused or exacerbated by climate change, and the spate of extreme weather events experienced over the past several years presents an alarming trend. Extreme weather events have compromised our facilities in the past and may do so in the future. Furthermore, work stoppages, whether union-organized or not, can also disrupt operations. Business interruption could also be caused by compliance failures. A significant
disruption at any of these facilities or otherwise within our supply chain, even on a short-term basis, could impair our ability to produce and ship products to the market on a timely basis or at all, which could have a material adverse effect on our business, financial position and results of operations.
Catastrophic events, including severe weather events, war and terrorist attacks, may negatively affect our business and results of operations.
We rely on our network infrastructure and enterprise applications, internal technology systems and websites to run our business as well as our or our third-party partners’ physical facilities, such as our R&D or manufacturing premises. In addition, we rely on third-party hosted services. A disruption, infiltration or failure of these systems, facilities or third-party hosted services in the event of a hurricane, tsunami, tornado, earthquake, wildfire or flooding or other weather event, power loss, telecommunications failure, software or hardware malfunctions, pandemics, cyber-attack, war, terrorist attack or other catastrophic event that our disaster recovery plans do not adequately address, could cause system interruptions, reputational harm, loss of IP, delays in our product development, lengthy interruptions in our services, breaches of data security and loss of critical data. Any of these events could prevent us from conducting our day-to-day activities and could disrupt the operation of our supply chain. For example, we source some of our APIs from the Middle East region, and the armed conflicts that have escalated in the area since October 2023 could threaten our ability to obtain these important inputs. Separately, certain of our products utilize a contract manufacturing company in Taiwan, and an escalation of tensions between China and Taiwan could impair or prevent altogether our ability to source these products. A catastrophic event that results in the destruction or disruption of any of our or our third-party partners’ business centers, manufacturing facilities, data centers, R&D or manufacturing facilities, or our critical business or IT systems could severely affect our ability to conduct normal business operations and, as a result, our future operating results could be adversely affected. The adverse effects of any such catastrophic event would be exacerbated if experienced at the same time as another unexpected and adverse event. Additionally, the impacts of the changing weather on water resources may result in water scarcity, limiting our ability to access sufficient high-quality water in certain locations, which may increase operational costs. Our business interruption plans may be insufficient to mitigate these, and any other, catastrophic events.
Our business is subject to evolving corporate governance and public disclosure regulations and expectations, including with respect to environmental, social and governance matters, that could expose us to numerous risks.
We are subject to changing rules and regulations promulgated by a number of governmental and self-regulatory organizations, including the SEC, the Nasdaq Stock Market LLC (“Nasdaq”) and the Financial Accounting Standards Board. These rules and regulations continue to evolve in scope and complexity and many new requirements have been created in response to laws enacted by Congress, making compliance more difficult and uncertain. In addition, increasingly regulators, customers, investors, employees and other stakeholders are focusing on ESG matters and related disclosures. Concern over severe weather may also result in new or additional legal or regulatory requirements designed to mitigate the effects of severe weather on the environment and businesses. If such laws or regulations are more stringent than current legal or regulatory obligations, we may experience disruption in, or an increase in the costs associated with sourcing, manufacturing and distribution of our products, as well as an increase in costs associated with monitoring, tracking and reporting ESG related information to regulatory bodies, which may adversely affect our business, results of operations or financial condition. At the same time, regulators and legislators have increasingly expressed or pursued opposing views, legislation and investment expectations with respect to sustainability initiatives, including the enactment or proposal of “anti-ESG” legislation or policies. These opposing views may also be adopted by our investors.
These changing rules, regulations and stakeholder expectations have resulted in, and are likely to continue to result in, increased general and administrative expenses and increased management time and attention spent complying with or meeting such regulations and expectations. For example, the State of California recently passed the Climate Corporate Data Accountability Act and the Climate-Related Financial Risk Act that will impose broad climate-related disclosure obligations on certain companies doing business in California, including us. Other U.S. states’ legislatures are considering enactment of similar rules and regulations. In addition, the European Union (“EU”) enacted the Corporate Sustainability Reporting Directive (“CSRD”) legislation in January 2023 which requires certain reporting and disclosure starting during the first quarter of 2026 of calendar year 2025 data relating to ESG matters for companies whose business and assets exceed certain thresholds within EU countries. Due to our subsidiaries in Ireland, the CSRD requirements will apply to us for 2025 reporting, which will require significant preparatory work to comply with the reporting rules. Developing and acting on initiatives within the scope of ESG, and collecting, measuring and reporting ESG related information and metrics can be costly, difficult and time consuming and is subject to evolving reporting standards and similar proposals by other international regulatory bodies. We may also communicate certain initiatives and goals, regarding environmental matters, diversity, responsible sourcing and social investments and other ESG related matters, in our public disclosures. These initiatives and goals within the scope of ESG could be difficult and expensive to implement, the technologies needed to implement them may not be cost effective and may not advance at a sufficient pace, and we could be criticized for the accuracy, adequacy or completeness of the disclosure. Further, statements about our ESG-related initiatives and goals, and progress against those goals, may be based on standards for measuring progress that are still developing, internal controls and processes that continue to evolve, and assumptions that are
subject to change in the future. In addition, we could be criticized for the scope or nature of such initiatives or goals, or for any revisions to these goals. If our ESG-related data, processes and reporting are incomplete or inaccurate, or if we fail to achieve progress with respect to our goals within the scope of ESG on a timely basis, or at all, our reputation, business, financial performance and growth could be adversely affected.
Our future success depends on our ability to attract and retain talented employees and consultants.
Our future success depends, to a substantial degree, upon the continued service of the members of our management team. The loss of the services of members of our management team, or their inability to perform services on our behalf, could have a material adverse effect on our business, condition (financial and otherwise), prospects and results of operations. Our success also depends, to a large extent, upon the contributions of our sales, marketing, scientific and quality assurance staff. We compete with brand and generic pharmaceutical manufacturers for qualified personnel, and our competitors may offer more favorable employment opportunities than we do. If we are not able to attract and retain the necessary personnel to accomplish our business objectives, we could experience constraints that would adversely affect our ability to sell and market our products effectively, to meet the demands of our strategic partners in a timely fashion, and to support our R&D programs. In particular, our sales and marketing efforts depend on the ability to attract and retain skilled and experienced sales, marketing and quality assurance representatives. Although we believe that we have been successful in attracting and retaining skilled personnel in all areas of our business, we cannot provide assurance that we can continue to attract, train and retain such personnel. Any failure in this regard could limit the rates at which we generate sales and develop or acquire new products.
IP and Licensing Risks
Federal regulation of arrangements between manufacturers of branded and generic products could adversely affect our business.
We are involved in numerous patent litigations in which we challenge the validity or enforceability of innovator companies’ listed patents and/or their applicability to our generic and/or biosimilar pharmaceutical products, as well as patent infringement litigation in which other generic/biosimilar companies challenge the validity or enforceability of our patents and/or their applicability to their generic/biosimilar pharmaceutical products, and therefore settling patent litigations has been and is likely to continue to be an important part of our business. As part of the Medicare Prescription Drug and Modernization Act of 2003, companies, including us, are required to file with the FTC and the DOJ agreements entered into between branded and generic and/or biosimilar pharmaceutical companies related to the manufacture, marketing and sale of generic/biosimilar versions of branded drugs for their review. In June 2013, the U.S. Supreme Court in its decision in FTC v. Actavis determined that “reverse payment” patent settlement agreements between brand and generic/biosimilar companies could violate the antitrust laws. The Supreme Court held that such settlement agreements are neither immune from antitrust attack nor presumptively illegal but rather should be analyzed under the “Rule of Reason” test to determine whether they violate the federal antitrust laws. This holding has resulted in heightened scrutiny of such settlement agreements by the FTC and state and local authorities, and has increased the risk of liability in pending antitrust litigation brought by private plaintiffs. The FTC has brought actions against parties to such settlement agreements, including us, and we have become subject to increased FTC investigations or enforcement actions arising from such settlement agreements. Further, private plaintiffs, including direct and indirect purchasers of our products, have also become more active in bringing private litigation claims against us and other brand and generic/biosimilar pharmaceutical companies alleging that such settlement agreements violate the antitrust laws. Accordingly, we have in the past received and may receive formal or informal requests from the FTC for information about a particular settlement agreement, and there is a risk that the FTC, state and local authorities, or private plaintiffs, may commence an action against us alleging violations of the antitrust laws. We have been and are currently involved in private antitrust actions involving certain settlement agreements as described in Note 20. Commitments and Contingencies — Other Litigation Related to the Company’s Business.
Antitrust investigations and claims are generally expensive and time consuming, and we can give no assurance as to the timing or outcome of such investigations or claims or of any future private litigation or government action alleging that one of our settlement agreements violates antitrust laws. The impact of federal regulation of arrangements between manufacturers of brand and generic/biosimilar products, further legislation and the potential for private-party lawsuits associated with such arrangements could adversely affect our business.
From time to time we may need to rely on licenses to proprietary technologies, which may be difficult or expensive to obtain.
We may need to obtain licenses to patents and other proprietary rights held by third parties to develop, manufacture and market products. If we are unable to timely obtain these licenses on commercially reasonable terms, our ability to commercially market
our products may be inhibited or prevented, which could have a material adverse effect on our business, results of operations and financial condition.
Our competitors or other third parties may allege that we are infringing upon their IP, forcing us to expend substantial resources in litigation, the outcome of which is uncertain. Any unfavorable outcome of such litigation, including losses related to “at-risk” product launches, could have a material adverse effect on our business, financial position and results of operations.
Companies that produce branded pharmaceutical products routinely bring litigation against ANDA filers or similar applicants that seek regulatory approval to manufacture and market generic forms of their branded products alleging patent infringement or other violations of IP rights. Patent holders may also bring patent infringement suits against companies that are currently marketing and selling approved generic products. Similarly, companies that produce biologics may bring litigation against abbreviated Biologics License Application (“aBLA”) filers that seek regulatory approval to manufacture and market biosimilars. Litigation often involves significant expense and can delay or prevent introduction or sale of our generic and/or biosimilar products. If valid and enforceable patents are infringed by our products, we would need to delay selling the infringing generic product unless we could obtain a license from the patent holder, and, if we were already selling the infringing product, cease selling and potentially destroy existing product stock.
There may be situations in which we may make business and legal judgments to market and sell products that are subject to claims of alleged patent infringement prior to final resolution of those claims by the courts, based upon our belief that such patents are invalid, unenforceable, or are not infringed by our marketing and sale of such products. This is referred to in the pharmaceutical industry as an “at-risk” launch. The risk involved in an at-risk launch can be substantial because, if a patent holder ultimately prevails against us, the remedies available to such holder may include, among other things, damages measured by the profits lost by the patent holder or treble damages, which can be significantly higher than the profits we make from selling the generic or biosimilar version of the product. We may also be harmed by the loss of any value of such inventory that we are unable to market or sell.
We expend a significant amount of resources on research and development, including milestones on in-licensed products, which may not lead to successful product introductions.
Much of our development effort is focused on technically difficult-to-formulate products and/or products that require advanced manufacturing technology. We expend significant resources on R&D primarily to enable us to manufacture and market FDA-approved pharmaceuticals in accordance with FDA regulations. We have entered into, and may in the future enter into, agreements that require us to make significant milestone payments upon achievement of various R&D events and regulatory approvals. As we continue to develop and in-license new products, we will likely incur increased research and licensing expenses. Because of the inherent risk associated with R&D efforts in the industry, particularly with respect to new drugs, our R&D expenditures may not result in the successful introduction of FDA-approved pharmaceutical products. Additionally, after we or our development partners submit an ANDA or aBLA, the FDA may request that additional studies be conducted. As a result, we may be unable to reasonably determine the total R&D costs required to develop a particular product. Finally, we cannot be certain that any investment made in developing products will be recovered, even if we are successful in commercialization. To the extent that we expend significant resources on R&D efforts and are not ultimately able to successfully introduce new products as a result of those efforts, our business, financial position and results of operations may be materially adversely affected.
We depend on our ability to protect our IP and proprietary rights.
Our success depends on our ability to protect and defend the IP rights associated with our current and future products. If we fail to protect our IP adequately, competitors may manufacture and market products similar to, or that may be confused with, our products, and our generic competitors may obtain regulatory approval to make and distribute generic versions of our branded products. Some patent applications in the U.S. are maintained in secrecy or are not published until the resulting patents issue. We also cannot be certain that patents will be issued with respect to any of our patent applications or that any existing or future patents issued to or licensed by us will provide competitive advantages for our products or will not be challenged, invalidated, circumvented or held unenforceable in proceedings commenced by our competitors or other third parties. Furthermore, our patent rights may not prevent or limit our present and future competitors from developing, making, importing, using or commercializing products that are functionally similar to our products. We rely particularly on trade secrets, trademarks, unpatented proprietary expertise and continuing innovation that we seek to protect, in part, by registering and using marks; and by entering into confidentiality agreements with licensees, suppliers, employees, consultants and other parties. We use this
approach to protecting our IP in large part because few of our products are protected by patents. We cannot provide assurance that these agreements will not be breached or circumvented. We also cannot be certain that we will have recourse to adequate remedies in the event of a breach of such agreements. Disputes may arise concerning the ownership of IP or the applicability of confidentiality agreements. We cannot be sure that our trade secrets and proprietary technology will not be independently developed or otherwise become known by our competitors or, if patents are not issued with respect to our internally developed products, that we will be able to maintain the confidentiality of information relating to these products. In addition, efforts to ensure our IP rights may be costly, time-consuming and/or ultimately unsuccessful. We cannot be sure that we will have the resources to protect our own rights against infringement by third parties. Our inability to protect our IP and proprietary rights could have a material adverse effect on our business, results of operations, financial condition and cash flows.
Legal and Regulatory Risks
We are involved in various legal proceedings and may be involved in future legal proceedings, all of which are uncertain, and existing and future proceedings may require us to incur substantial expense to defend and/or expose us to substantial liability.
The development, manufacture and sale of our drug products involves an inherent risk of product liability and other claims and the associated adverse publicity, and insurance against such potential claims is expensive and may be difficult to obtain. Litigation is inherently subject to uncertainties and we may be required to expend substantial amounts in the defense or resolution of this and similar matters. We regularly monitor the use of our products for trends or increases in reports of adverse events or product complaints, and regularly report such matters to the FDA. In some cases, an increase in adverse event reports may be an indication that there has been a change in a product’s specifications or efficacy. Such changes could lead to a recall of the product in question or, in some cases, increases in product liability claims related to the product in question. If the coverage limits for product liability and other insurance policies are not adequate, or if certain of our products are excluded from coverage, a claim brought against us, whether covered by insurance or not, could have a material adverse effect on our business, results of operations, financial condition and cash flows.
In the ordinary course of our business, we may also be subject to a variety of other types of claims, proceedings, investigations and litigation initiated by government agencies or third parties. These matters may include compliance matters, product regulation or safety, taxes, employee benefit plans, employment discrimination, health and safety, environmental, antitrust, securities law, customs, import/export, government contract compliance, financial controls or reporting, IP, allegations of misrepresentation, false claims or false statements, commercial claims, claims regarding promotion of our products and services, or other similar matters. In addition, government investigations related to the use of our generic drug products may cause reputational harm to us. Negative publicity, whether accurate or inaccurate, about the efficacy, safety or side effects of our generic drug products or product categories, whether involving us or a competitor, could materially reduce market acceptance of our products, cause consumers to seek alternatives to our products, result in product withdrawals and cause our stock price to decline. Negative publicity could also result in an increased number of product liability claims, whether or not these claims have a basis in scientific fact. Any such claims, proceedings, investigations or litigation, regardless of the merits, might result in substantial costs to defend or settle, restrictions on product use or sales, or otherwise injure our business.
We manufacture and derive a portion of our revenue from the sale of pharmaceutical products in the opioid class of drugs. The U.S. Department of Health and Human Services has declared the widespread addiction to and abuse of such products a public health emergency, and the federal government has also announced plans to increase federal oversight on opioid sale and consumption. These plans, along with changing public and clinical perceptions of opioid products and the risks relating to their use may result in the imposition of even stricter regulation of such products and further restrictions on their sale and use. For instance, the DEA has recently increased its scrutiny and regulation over the manufacture, distribution and sale of opioid products, which may require us to incur significant expenses to comply with such regulations. We derive substantial revenues from the sale of certain controlled drug substances that are subject to specific aggregate production quotas established and administered by the DEA in accordance with governing laws and regulations. Our inability to secure our quota allocation, the DEA’s decision to allocate quota in an amount less than the amount we requested, or a delay by the government in the issuance of the quota for these substances can result in a substantial impact to our revenues.
State governments have also taken steps to impose surcharges or taxes on opioid manufacturers or distributors. Any new or stricter regulations imposed by governmental authorities such as the DEA related to opioid products, as well as a potential increase in opioid-related litigation involving us, could result in material adverse effects on our business and results of operations. Refer to Note 20. Commitments and Contingencies — Civil Prescription Opioid Litigation for more information regarding opioid-related litigation, including a nationwide settlement in principle, involving us.
We are subject to United States federal and state laws related to healthcare fraud and abuse and health information privacy and security, and the failure to comply with such laws may adversely affect our business.
In the U.S., many of our products are eligible for reimbursement under federal and state health care programs such as Medicaid, Medicare, TRICARE, and/or state pharmaceutical assistance programs, and as a result, certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are, and will be, applicable to our business. We could be subject to healthcare fraud and abuse and patient privacy regulation by both the federal government and the states in which we conduct our business.
The domestic and foreign laws that may affect our ability to operate include, but are not limited to: (i) the U.S. Anti-Kickback Statute, which applies to our marketing and research practices, educational programs, pricing policies and relationships with healthcare providers or other entities, by prohibiting, among other things, soliciting, receiving, offering or paying remuneration, directly or indirectly, as a means of inducing, or in exchange for, either the referral of an individual or the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; (ii) U.S. federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payers that are false or fraudulent; (iii) the U.S. Health Insurance Portability and Accountability Act of 1996, (“HIPAA”), which among other things created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters, and HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and our implementing regulations, which impose certain requirements relating to the privacy, security and transmission of individually identifiable health information and place restrictions on the use of such information for marketing communications; (iv) the U.S. Physician Payments Sunshine Act, which among other things, requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available under a federal healthcare program to report annually information related to “payments or other transfers of value” made to physicians, physician assistants, advanced practice nurses and teaching hospitals, and ownership and investment interests held by certain healthcare professionals and their immediate family members, and similar state laws; (v) the government pricing rules applicable to the Medicaid, Medicare Part B, 340B Drug Pricing Program, the U.S. Department of Veterans Affairs program, the TRICARE program, and state price reporting laws; and (vi) state and foreign law equivalents of each of the above U.S. laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payer, including commercial insurers, and state and foreign laws governing the privacy and security of health and other sensitive information in certain circumstances, such as the requirements under the European Union’s General Data Protection Regulation and certain U.S. state privacy laws, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. Violations of certain of these laws, including the fraud and abuse laws may result in severe penalties against us and/or our responsible employees, including jail sentences, large fines, and the exclusion of our products from reimbursement under federal and state programs. Additionally, these risks may be compounded by our rapid international expansion, as each international jurisdiction has its own legal and regulatory requirements. Defense of litigation claims and government investigations can be costly, time-consuming, and distract management, and it is possible that we could incur judgments or enter into settlements that would require us to change the way we operate our business. We are committed to conducting the sales and marketing of our products in compliance with the healthcare fraud and abuse laws, but certain applicable laws may impose liability even in the absence of specific intent to defraud. Furthermore, should there be ambiguity, a governmental authority may take a position contrary to a position we have taken, or should an employee violate these laws without our knowledge, a governmental authority may impose civil and/or criminal sanctions.
Any adverse outcome in these types of actions, or the imposition of penalties or sanctions for failing to comply with fraud and abuse laws, could adversely affect us and may have a material adverse effect on our business, results of operations, financial condition and cash flows. Some of the statutes and regulations that govern our activities, such as federal and state anti-kickback and false claims laws, are broad in scope, and while exemptions and safe harbors protecting certain common activities exist, they are often narrowly drawn. While we manage our business activities to comply with these statutory provisions, due to their breadth, complexity and, in certain cases, uncertainty of application, it is possible that our activities could be subject to challenge by various government agencies. In particular, the FDA, the DOJ and other agencies have increased their enforcement activities with respect to the sales, marketing, research and similar activities of pharmaceutical companies in recent years, and many pharmaceutical companies have been subject to government investigations related to these practices. A determination that we are in violation of these and/or other government regulations and legal requirements may result in civil damages and penalties, criminal fines and prosecution, administrative remedies, the recall of products, the total or partial suspension of manufacturing and/or distribution activities, seizure of products, injunctions, whistleblower lawsuits, failure to obtain approval of pending product applications, withdrawal of existing product approvals, exclusion from participation in government healthcare programs and other sanctions.
Any of these types of investigations or enforcement actions could affect our ability to commercially distribute our products and could materially and adversely affect our business, financial condition, results of operations and cash flows.
Approvals for our new generic drug products may be delayed or become more difficult to obtain if the FDA institutes changes to its approval requirements. Similarly, the FDA could change the approval or post-approval regulatory requirements for new drug applications.
Congress may institute changes to the FDA’s user fee structures, such as implementing new or additional fees similar to the fees imposed under GDUFA III, which may make it more difficult or expensive for us to obtain approval for our new generic products. The FDA may also implement changes to the ANDA approval or post-approval regulatory requirements that may directly affect some of our ANDA filings pending approval from the FDA or our already-approved products, such as changes to guidance from the FDA regarding bioequivalency requirements for particular drugs. Such changes may cause our development of such generic drugs to be significantly more difficult or result in delays in FDA approval or result in our decision to abandon or terminate certain projects or the marketing of certain approved products. Any changes in FDA requirements may make it more difficult for us to file ANDAs or obtain approval of our ANDAs and generate revenues and thus have a material adverse effect on our business, results of operations and financial condition.
Healthcare reform and a reduction in the coverage and reimbursement levels by governmental authorities, HMOs, MCOs or other third-party payers may adversely affect our business.
As part of commercializing our products, we have obtained authorization to receive reimbursement at varying levels for the cost of certain products and related treatments from governmental authorities and private health insurers and other organizations, such as HMOs and managed care organizations (“MCOs”). The drug pricing reforms in the IRA have impacted, and may impact in the future, the prices of certain of our products. For example, rebates related to the IRA reduced our net revenue for the years ended December 31, 2024 and 2023 by $8.0 million and $7.9 million, respectively. The trend toward managed healthcare in the U.S., the growth of organizations such as HMOs and MCOs, and legislative proposals to reform healthcare and government insurance programs could significantly influence the purchase of pharmaceutical products, resulting in lower prices and a reduction in product demand. The Patient Protection and Affordable Care Act and the Health Care and Education Reconciliation Act of 2010 were signed into law on March 23, 2010 and March 30, 2010, respectively. These laws are referred to herein as “healthcare reform.” A number of provisions of the healthcare reform laws continue to have a negative impact on the price of our products sold to U.S. government entities. For example, the legislation includes measures that (i) significantly increase Medicaid rebates through the expansion of the program; (ii) substantially expand the Public Health System (340B) program to allow other entities to purchase prescription drugs at substantial discounts; (iii) extend the Medicaid rebate rate to a significant portion of Managed Medicaid enrollees; (iv) apply a 75% discount to Medicare Part D beneficiary spending in the coverage gap for branded and authorized generic prescription drugs; and (v) levy a significant excise tax on the industry to fund healthcare reform. Such cost containment measures and healthcare reform affect our ability to sell our products and have a material adverse effect on our business, results of operations and financial condition. Additionally, the Medicare Part D Prescription Drug Benefit established a voluntary outpatient prescription drug benefit for Medicare beneficiaries (primarily the elderly over 65 and the disabled). These beneficiaries may enroll in private drug plans. There are multiple types of Part D plans and numerous plan sponsors, each with its own formulary and product access requirements. The plans have considerable discretion in establishing formularies and tiered co-pay structures and in placing prior authorization and other restrictions on the utilization of specific products. In addition, Part D plan sponsors are permitted and encouraged to negotiate rebates with manufacturers. The Medicare Part D program, which went into effect January 1, 2006, is administered by the CMS within the Department of Health and Human Services.
The CMS has issued extensive regulations and other sub-regulatory guidance documents implementing the Medicare Part D benefit, and the Office of Inspector General has issued regulations and other guidance in connection with the Medicare Part D program. The federal government can be expected to continue to issue guidance and regulations regarding the obligations of Part D sponsors and their subcontractors. Participating drug plans may establish drug formularies that exclude coverage of specific drugs and payment levels for drugs negotiated with Part D drug plans may be lower than reimbursement levels available through private health plans or other payers. Moreover, beneficiary co-insurance requirements could influence which products are recommended by physicians and selected by patients. There is no guarantee that any drug that we market will be offered by drug plans participating under the Medicare Part D program or of the terms of any such coverage, or that covered drugs will be reimbursed at amounts that reflect current or historical levels. Additionally, any reimbursement granted may not be maintained, or limits on reimbursement available from third-party payers may reduce the demand for, or negatively affect the price of those products, which could significantly harm our business, results of operations, financial condition and cash flows. We may also be subject to lawsuits relating to reimbursement programs that could be costly to defend, divert management’s attention and adversely affect our operating results. Most state Medicaid programs have established preferred
drug lists, and the process, criteria and timeframe for obtaining placement on the preferred drug list varies from state to state. Under the Medicaid drug rebate program, a manufacturer must pay a rebate for Medicaid utilization of a product. The rebate for single source products (including authorized generics) is based on the greater of (i) a specified percentage of the product’s average manufacturer price or (ii) the difference between the product’s average manufacturer price and the best price offered by the manufacturer. The rebate for multiple source products is a specified percentage of the product’s average manufacturer price. In addition, many states have established supplemental rebate programs as a condition for including a drug product on a preferred drug list. The profitability of our products may depend on the extent to which they appear on the preferred drug lists of a significant number of state Medicaid programs and the amount of the rebates that must be paid to such states. In addition, there is significant fiscal pressure on the Medicaid program, and amendments to lower the pharmaceutical costs of the program are possible. Such amendments could materially adversely affect our anticipated revenues and results of operations. Due to the uncertainties regarding the outcome of future healthcare reform initiatives and their enactment and implementation, we cannot predict which, if any, of the future reform proposals will be adopted or the effect such adoption may have on our business. Future rulemaking and reform, including repeal of existing laws, including healthcare reform laws, with respect to the healthcare and pharmaceutical industries, could increase rebates, reduce prices or the rate of price increases for healthcare products and services, or require additional reporting and disclosure. We cannot predict the timing or impact of any future rule making, reform or repeal of healthcare laws.
We depend on third-party agreements for a portion of our product offerings and any failure to maintain these arrangements or enter into similar arrangements with new partners could result in a material adverse effect.
We have broadened our product offering by entering into a variety of third-party agreements covering any combination of joint development, supply, marketing and/or distribution of products. We cannot provide assurance that the development, supply, marketing and/or distribution efforts of our contractual partners will continue to be successful, that we will be able to renew such agreements or that we will be able to enter into new agreements with favorable terms for additional products. Any alteration to, or termination of, our current distribution and marketing agreements, failure to enter into new and similar agreements, or interruption of our product supply under such agreements, could have a material adverse effect on our business, condition (financial and otherwise), prospects or results of operations.
The testing required for the regulatory approval of our products is conducted primarily by independent third parties. Any failure by any of these third parties to perform this testing properly and in a timely manner may have an adverse effect upon our ability to obtain regulatory approvals.
Our applications for regulatory approval of our products, including both internally developed and in-licensed products, incorporate the results of testing and other information that is conducted or gathered primarily by independent third parties (including, for example, manufacturers of raw materials, testing laboratories, contract research organizations or independent research facilities). Our ability to obtain and maintain regulatory approval of the products being tested is dependent upon the quality of the work performed by these third parties, the quality of the third parties’ facilities, and the accuracy of the information provided by third parties. We have little or no control over any of these factors. If this testing is not performed properly, our ability to obtain or maintain regulatory approvals, and to launch or continue selling products, could be restricted or delayed.
Our reporting and payment obligations under the Medicaid rebate program and other governmental purchasing and rebate programs are complex and may involve subjective decisions. Any determination that we have failed to comply with those obligations could subject us to penalties and sanctions, which could have a material adverse effect on our business.
The regulations applicable to us regarding reporting and payment obligations with respect to Medicaid reimbursement and rebates and other governmental programs are complex. Our calculations and methodologies are subject to review and challenge by the applicable governmental agencies, and it is possible that such reviews could adversely affect us and our business. In addition, because our processes for these calculations and the judgments involved in making these calculations involve, and will continue to involve, subjective decisions and complex methodologies, these calculations are subject to the risk of error and misjudgment. Any governmental agencies that have commenced (or that may commence) an investigation of us could impose, based on a claim of violation of anti-fraud and false claims laws or otherwise, civil and/or criminal sanctions, including fines, penalties and possible exclusion from federal health care programs (including Medicaid and Medicare). Some of the applicable laws may impose liability even in the absence of specific intent to defraud. Furthermore, should there be ambiguity with respect to how to properly calculate and report payments, and even in the absence of any such ambiguity, a governmental authority may take a position contrary to a position that we have taken and may impose civil and/or criminal sanctions on us. Any such penalties, sanctions, or exclusion from federal health care programs could have a material adverse effect on our business, financial position and results of operations. From time to time we conduct routine reviews of our government pricing
calculations. These reviews may have an impact on government price reporting and rebate calculations used to comply with various government regulations regarding reporting and payment obligations.
Investigations and litigation concerning the calculation of average wholesale prices may adversely affect our business.
Many government and third-party payers, including Medicare, Medicaid, HMOs and others, reimburse doctors and others for the purchase of certain prescription drugs based on a drug’s average wholesale price (“AWP”). In the past several years, state and federal government agencies have conducted ongoing investigations of manufacturers’ reporting practices with respect to AWP, as a result of which certain agencies have suggested that reporting of inflated AWPs by manufacturers has led to excessive payments for prescription drugs. Numerous pharmaceutical companies have been named as defendants in actions brought by various State Attorneys General and have faced state law qui tam actions brought on behalf of various states, alleging generally that the defendants defrauded state Medicaid systems by purportedly reporting or causing the reporting of AWP and/or “Wholesale Acquisition Costs” that exceeded the actual selling price of the defendants’ prescription drugs. These cases generally seek some combination of actual damages, and/or double damages, treble damages, compensatory damages, statutory damages, civil penalties, disgorgement of excessive profits, restitution, disbursements, counsel fees and costs, litigation expenses, investigative costs, injunctive relief, punitive damages, imposition of a constructive trust, accounting of profits or gains derived through the alleged conduct, expert fees, interest and other relief that the court may have deemed proper.
We can give no assurance that we will be able to settle current or future actions on terms that we deem reasonable, or that such settlements or adverse judgments, if entered, will not exceed the amount of any liability we have recorded. Accordingly, such actions could adversely affect us and may have a material adverse effect on our business, results of operations, financial condition and cash flows.
Failure to comply with our government contracting regulations could adversely affect our business and results of operations.
Our AvKARE segment generates a substantial amount of its net revenue from government contracts. Contracts with federal, state, and local governmental customers are subject to various procurement regulations, contract provisions and other requirements relating to their formation, administration and performance, and are subject to regular audits and investigations. Any failure by us to comply with the government contracting regulations could result in the imposition of various civil and criminal penalties, which may include termination of contracts, forfeiture of profits, suspension of payments, fines and suspension or debarment from future government business. Such failures could also cause reputational damage to our business. In addition, some of AvKARE’s contracts provide for termination by the government, without cause. If one or more of our government contracts is suspended or terminated, or if there are executive or congressional actions that block, freeze or otherwise impact federal government spending, including government shutdowns, or if we are suspended, debarred or otherwise restricted from future government work, our business, results of operations and financial condition could suffer.
Risks Relating to Our Indebtedness
We have a substantial amount of indebtedness, which could adversely affect our financial health.
As of December 31, 2024, we had $2.6 billion of total indebtedness, comprised of $2.3 billion, $192.0 million and $100.0 million in borrowings outstanding on the Term Loan Due 2028, Term Loan Due 2025 and Amended New Revolving Credit Facility, respectively. As of December 31, 2024, we had an ability to borrow up to an additional $523.2 million under our revolving credit facilities, comprised of $495.2 million and $28.0 million of available capacity under the Amended New Revolving Credit Facility and the Amended Rondo Revolving Credit Facility, respectively. In January 2025, the Company used $190.0 million of available funds under the Amended New Revolving Credit Facility to pay the entire remaining principal balance of $192.0 million outstanding on its Term Loan Due 2025, plus accrued interest thereon of $0.7 million, along with cash on hand.
Our substantial level of indebtedness could have important consequences. For example, it could:
•increase our vulnerability to adverse economic and industry conditions;
•limit our ability to obtain additional financing for future working capital, capital expenditures, raw materials, strategic acquisitions and other general corporate requirements;
•expose us to unhedged interest rate fluctuations (such as recent increases in interest rates from 2022 through 2023) because the interest on certain debt under the credit facilities is imposed at variable rates;
•require us to dedicate a substantial portion of our cash flow from operations to payments on our debt (including interest payments), thereby reducing the availability of cash flow for operations and other purposes;
•make it more difficult for us to satisfy our obligations to our lenders, resulting in possible defaults on and acceleration of such indebtedness;
•limit our ability to refinance indebtedness or increase the associated costs;
•require us to sell assets to reduce debt or influence the decision about whether to do so;
•limit our flexibility in planning for, or reacting to, changes in our business and the industry in which we operate or prevent us from carrying out capital spending that is necessary or important to our growth strategy and efforts to improve operating margins or our business; and
•place us at a competitive disadvantage compared to any competitors that have less debt or comparable debt at more favorable interest rates and that, as a result, may be better positioned to withstand economic downturn.
We may not be able to generate sufficient cash to service all of our indebtedness and may be forced to take other actions to satisfy our obligations under our indebtedness, which may not be successful.
Our ability to make scheduled payments on or refinance our debt obligations depends on our financial condition and operating performance, which are subject to prevailing economic and competitive conditions and to certain financial, business, legislative, regulatory and other factors which may be beyond our control. We may be unable to maintain a level of cash flows from operating activities sufficient to permit us to pay the principal, premium, if any, and interest on our indebtedness. As of December 31, 2024, we had approximately $2.6 billion of total indebtedness. In January 2025, the Company paid the entire remaining principal balance of $192.0 million outstanding on its Term Loan Due 2025, plus accrued interest thereon of $0.7 million, with $190.0 million of new borrowings under the Amended New Revolving Credit Facility and cash on hand. During 2025, we expect to make $58.8 million in principal payments on the Term Loan Due 2028. We expect to make interest payments totaling $226.9 million, excluding the impact of our interest rate swap and borrowings under our Amended New Revolving Credit Facility, during 2025 related to the Term Loan Due 2028. Refer to Note 15. Debt and “Commitments and Contractual Obligations” under Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations for additional information.
If our cash flows and capital resources are insufficient to fund our debt service obligations, we could face substantial liquidity problems and could be forced to reduce or delay investments and capital expenditures or to dispose of material assets or operations, seek additional debt or equity capital or restructure or refinance our indebtedness. We may not be able to effect any such alternative measures on commercially reasonable terms or at all and, even if successful, those alternative actions may not allow us to meet our scheduled debt service obligations. Our credit agreements restrict our ability to dispose of assets and use the proceeds from those dispositions and also restrict our ability to raise debt or equity capital to be used to repay other indebtedness when it becomes due. We may not be able to consummate those dispositions or obtain proceeds in an amount sufficient to meet any debt service obligations when due. Any of these circumstances, would materially and adversely affect our financial position and results of operations and our ability to satisfy our obligations, including our indebtedness.
If we cannot make scheduled payments on our debt, we will be in default and, as a result:
•our debt holders could declare all outstanding principal and interest to be due and payable;
•the lenders under our credit agreements could terminate their commitments to lend us capital; and
•we could be forced into bankruptcy or liquidation.
The terms of our credit agreements restrict our operations, particularly our ability to respond to changes or to take certain actions.
Our credit agreements contain a number of restrictive covenants that impose operating and financial restrictions on us and may limit our ability to, among other things: incur additional indebtedness; pay dividends or make other distributions or repurchase or redeem capital stock; prepay, redeem or repurchase certain debt; make loans and investments or sell assets.
A breach of the covenants under such credit agreements could result in an event of default under the applicable indebtedness. Such a default may allow the creditors to accelerate the related debt and may result in the acceleration of any other debt to which a cross-acceleration or cross-default provision applies which could have a material adverse effect on our business, operations and financial results. In the event our lenders accelerated the repayment of the borrowings, we and our subsidiaries may not have sufficient assets to repay that indebtedness. Any acceleration of amounts due under the credit agreements would
likely have a material adverse effect on us. As a result of these restrictions, we may be limited in how we conduct business, unable to raise additional financing and unable to compete effectively. These restrictions may affect our ability to grow in accordance with our strategy.
Economic, Political and Financial Risks
Our current operations in, and potential expansion into additional international markets subjects us to increased regulatory oversight both in those international markets and domestically as well as regulatory, economic, social and political uncertainties, which could cause a material adverse effect on our business, financial position and results of operations.
We are subject to certain risks associated with having substantial assets and operations located in foreign jurisdictions, including our operations in India and Ireland, as well as related to our distribution activities being initiated in new geographies outside the U.S. and India. Over the past several years, we have significantly expanded our Indian operations, and we may in the future expand our international business and operations in these jurisdictions or into jurisdictions in which we have limited operating experience, including with respect to seeking regulatory approvals, marketing or selling products.
Our international operations may be adversely affected by general economic conditions (including inflation, expropriation and other government actions), economic and fiscal policy (including changes in exchange rates and controls, interest rates and taxation policies), changes in IP protections and remedies, trade regulations, tax laws, and increased government regulation (including those affecting approval, production, pricing, and marketing of, reimbursement for and access to our products). With respect to India, our operations could also be adversely affected by any reversal of India’s recent economic liberalization and deregulation policies, as well as social instability and other political, economic or diplomatic developments in the future. Certain jurisdictions have, from time to time, experienced instances of civil unrest and hostilities, both internally and with neighboring countries. Rioting, military activity, terrorist attacks, armed hostilities or unstable government and legal systems could cause our operations in such jurisdictions to be adversely affected or suspended. We generally do not have insurance for losses and interruptions caused by terrorist attacks, military conflicts and wars. In addition, our international operations may subject us to heightened scrutiny under the Foreign Corrupt Practices Act or similar anti-bribery laws, and could subject us to liability under such laws despite our efforts to comply. Some emerging market countries may be particularly vulnerable to periods of financial or political instability or significant currency fluctuations or may have limited resources for healthcare spending. As a result of these and other factors, our strategy to grow in emerging markets may not be successful, and growth rates in these markets may not be sustainable. Government financing and economic pressures can lead to negative pricing pressure in various markets where governments take an active role in setting prices, access criteria (e.g., through health technology assessments) or other means of cost control. Further, notwithstanding our compliance programs, there can be no assurances that our policies will prevent our employees or agents from violating any applicable laws or protect us from any such violations. Additionally, we cannot predict the nature, scope or impact of any future regulatory requirements that may apply to our expanding international operations or how foreign governments will interpret existing or new laws.
We may make acquisitions of, or investments in, complementary businesses or products, which may be on terms that may not turn out to be commercially advantageous, or may require additional debt or equity financing, which could increase our leverage and/or dilute equity holders.
While we regularly review the potential acquisition of technologies, products, product rights and complementary businesses and are currently evaluating, and intend to continue to evaluate, potential product and/or company acquisitions and other business development opportunities, we may not be able to identify suitable acquisition or investment candidates. In addition, to the extent that we do identify candidates that we believe to be suitable, we cannot provide any assurance that we will be able to reach an agreement with the selling party or parties or consummate the transaction on terms that are commercially advantageous to us or at all. If we make any acquisitions or investments, we may finance such acquisitions or investments through our cash reserves, debt financing, which may increase our leverage, or by issuing additional equity interests, which could dilute the holdings of our then-existing shareholders. If, due to capital constraints, we require financing, we cannot provide any assurance that we will be able to obtain such financing when needed on acceptable terms or at all.
Global economic conditions could harm us.
Global efforts to contain health care costs continue to exert pressure on product pricing and market access to pharmaceutical products. In many international markets, government-mandated pricing actions have reduced prices of patented drugs, and it is possible that the U.S. may adopt similar measures to reduce drug prices to consumers. Some countries may be subject to periods of financial instability, may have reduced resources to spend on healthcare or may be subject to economic sanctions, and our business in these countries may be disproportionately affected by these changes. Continued concerns about the systemic
impact of potential geopolitical issues and economic policy uncertainty, particularly in areas in which we operate, could potentially cause economic and market instability in the future and could adversely affect our business, including our financial performance. These conditions may also result in decreased consumer spending, including spending on our products.
Challenging economic conditions have resulted, and may continue to result, in tighter credit conditions. The cost and availability of credit may be adversely affected by illiquid credit markets and wider credit spreads, which could adversely affect the ability of our third-party distributors, partners, manufacturers and suppliers to buy inventory or raw materials and to perform their obligations under agreements with us, which could disrupt our operations and adversely affect our financial performance.
We have increased exposure to tax liabilities, including foreign tax liabilities.
As a U.S. company with subsidiaries in, among other countries, India, Switzerland, Ireland and the United Kingdom, we are subject to, or potentially subject to, income and other taxes in these jurisdictions as well as the U.S. Significant judgment is required in determining our worldwide provision for income taxes and other tax liabilities. Changes in tax laws or tax rulings may have a significant adverse impact on our effective tax rate. In addition, we have potential tax exposures resulting from the varying application of statutes, regulations and interpretations, which include exposures on intercompany terms of cross-border arrangements among foreign subsidiaries in relation to various aspects of our business, including R&D activities and manufacturing. Tax authorities in various jurisdictions may disagree with, and subsequently challenge, the amount of profits taxed in such jurisdictions. Any such challenges may result in increased tax liability, including accrued interest and penalties, which would cause our tax expense to increase and may have a material adverse effect on our business, financial position and results of operations and our ability to satisfy our debt obligations.
In addition, many countries are implementing legislation and other guidance to align their international tax rules with the Organization for Economic Co-operation and Development’s (“OECD”) Base Erosion and Profit Shifting recommendations and action plan that aim to standardize and modernize global corporate tax policy, including changes to cross-border tax, transfer pricing documentation rules, and nexus-based tax incentive practices. The OECD has issued a two-pillar approach to global taxation, focusing on global profit allocation and a global minimum tax rate. The “Pillar One” global profit allocation proposal would not apply to us, because it generally applies to companies with global revenues exceeding €20 billion (approximately $21 billion using the exchange rate as of December 31, 2024). The “Pillar Two” proposal focuses on a global minimum tax of at least 15%. Legislation for the “Pillar Two” proposal, applying to us, has been enacted in Ireland, and was effective with the financial year beginning on January 1, 2024. As the tax rates of the other jurisdictions in which we operate exceed 15%, we do not believe there is any potential additional exposure besides in Ireland.
We assessed that no top-up tax under Pillar 2 of the OECD Inclusive Framework on Base Erosion and Profit Shifting is expected to be due for the year ended December 31, 2024. This assessment is based on the application of safe harbor provisions available in all relevant jurisdictions including UK, Germany, Ireland, Switzerland, U.S. and India.
In certain circumstances, we issue price adjustments and other sales allowances to our customers. Although we may establish liabilities based on our estimates of these amounts, if estimates are incorrect and the liabilities are inadequate, it may result in adjustments to these liabilities that may have a material adverse effect on our financial position and results of operations.
As described above, the first company to file an ANDA containing a Paragraph IV certification that successfully challenges the patent(s) on a branded product may be granted 180 days of generic market exclusivity by the FDA for such generic product. At the expiration of such exclusivity period, other generic distributors may enter the market, resulting in a significant price decline for the drug (in some instances, price declines have exceeded 90%). When we experience price declines following a period of generic marketing exclusivity, or at any time when a competitor enters the market or offers a lower price with respect to a product we are selling, we may, at our discretion, decide to lower the price of our product to retain market share and provide price adjustments to our customers for the difference between our new (lower) price and the price at which we previously sold the product which is still held in inventory by such customers. We accrue for these adjustments when the expected value of an adjustment is greater than zero, based on contractual pricing, actual net sales, accrual rates based on historical average rates, and estimates of the level of inventory of its products in the distribution channel that remain subject to these adjustments. There are also circumstances under which we may decide not to provide price adjustments to certain customers, and consequently, as a matter of business strategy, we may risk a greater level of sale returns of products in a customer’s existing inventory and lose future sales volume to competitors rather than reduce our pricing.
Based on estimates, we establish liabilities for sales allowances including, but not limited to: sales discounts and returns, chargebacks, sales volume rebates, shelf stocks, cash discounts, and Medicaid rebate obligations at the time of sale. Although
we believe our liabilities are adequate as of the date of this report, we cannot provide assurances that our reserves will ultimately prove to be adequate. Increases in sales allowances may exceed our estimates for a variety of reasons, including unanticipated competition or an unexpected change in one or more of our contractual relationships. We will continue to evaluate the effects of competition and will record a price adjustment liability if and when we deem it necessary. Any failure to establish adequate liabilities with respect to sales allowances may result in a material adverse effect on our financial position and results of operations.
If we determine that our goodwill and other intangible assets have become impaired, we may record significant impairment charges, which would adversely affect our results of operations.
Goodwill and other intangible assets represent a significant portion of our assets. Goodwill is the excess of cost over the fair market value of net assets acquired in business combinations. In the future, goodwill and intangible assets may increase as a result of future acquisitions. We review our goodwill and indefinite lived intangible assets at least annually for impairment. We review our intangible assets with finite lives for recoverability whenever events or changes in circumstances indicate that the carrying amount of the assets may not be fully recoverable. Impairment may result from, among other things, deterioration in the performance of acquired businesses, adverse market conditions and adverse changes in applicable laws or regulations, including changes that restrict the activities of an acquired business.
Generic pharmaceuticals have faced regular and increasing price erosion each year, placing even greater importance on our ability to continually introduce new products. If these trends continue or worsen, or if we experience further difficulty in this market or the specialty market, our revenues and profits in our Affordable Medicines and Specialty segments may continue to be affected adversely. A decline in our market capitalization, even if otherwise due to macroeconomic or industry-wide factors, could put pressure on the carrying value of our goodwill in both our Affordable Medicines and Specialty segments and cause us to conduct an interim impairment test. A determination that all or a portion of our goodwill or other intangible assets is impaired, although a non-cash charge against earnings, could have a material adverse effect on our results of operations and financial condition.
Risks Related to Our Tax Receivable Agreement
We are required under a tax receivable agreement to make cash payments in respect of certain tax benefits to which we may become entitled, and we expect that the payments we will be required to make will be substantial.
We are a party to a tax receivable agreement (“TRA”) with each of the members of the group, together with their affiliates and certain assignees, who owned Amneal when it was a private company (“Members” or the “Amneal Group”), dated May 4, 2018. On November 7, 2023, the TRA was amended as part of the Reorganization, and it may be further amended or supplemented from time to time. Under the November 7, 2023 amendment, the parties agreed to reduce our future obligation to pay 85% of the realized tax benefits subject to the TRA to 75% of such realized tax benefits. Therefore, under the TRA, we will be required to make cash payments to the Members and their permitted transferees equal to 75% of certain attributed tax benefits, if any, that we actually realize. The amount of cash payments that we will be required to make under the TRA may be significant.
For the years ended December 31, 2024, 2023 and 2022, we recorded expenses associated with the TRA in total other expense, net of $50.7 million, $3.1 million and $0.6 million, respectively, as a result of the realization of cash tax savings from the TRA attributes for those years. As of December 31, 2024 and 2023, we had a TRA liability of $53.9 million and $3.7 million, respectively. Should we determine that deferred tax assets (“DTAs”) subject to the TRA with a valuation allowance are realizable in a future period, the related valuation allowance will be reversed and if a resulting TRA payment is determined to be probable, a corresponding liability will be recorded. As a result, our future results of operations and earnings could be significantly impacted by these matters. However, if the tax attributes are not utilized in future years, it is reasonably possible no amounts would be paid under the TRA. If utilization of our DTAs becomes more-likely-than-not in the future, at such time, we could incur obligations approximating the $133.8 million unrecorded contingent TRA liability as of December 31, 2024.
The timing and amount of any payments under the TRA may vary, depending upon a number of factors including the timing and amount of our taxable income and the tax rate in effect at the time of realization of the taxable income (the TRA liability is determined based on a percentage of the corporate tax savings from the use of the TRA’s attributes). Because the Amneal Group has sold or exchanged all of their common units, as of the Reorganization, there is no longer the associated risk of increased future obligations under the TRA (i.e., there cannot be further sales or exchanges giving rise to increased TRA liability occurring subsequent to December 31, 2023).
In certain cases, payments under the TRA to the Members or their permitted transferees may be accelerated or significantly exceed the actual benefits we realize in respect of the tax attributes subject to the TRA.
The TRA continues to provide that upon certain mergers, asset sales, other forms of business combinations or other changes of control or if, at any time, we elect an early termination of the TRA, then our obligations under the TRA to make payments would be based on certain assumptions, including an assumption that we would have sufficient taxable income to fully utilize all potential future tax benefits that are subject to the TRA. The parties agreed that there was no change of control from the Reorganization.
As a result of the foregoing, we could be required to make payments under the TRA that (i) are greater than the actual benefits we ultimately realize in respect of the tax benefits that are subject to the TRA and (ii) are based on the present value of the anticipated future tax benefits that are the subject of the TRA, which payment may be required to be made significantly in advance of the actual realization, if any, of such future tax benefits. In these situations, our obligations under the TRA could have a substantial negative impact on our liquidity and could have the effect of delaying or preventing certain mergers, asset sales, other forms of business combinations or other changes of control. There can be no assurance that we will be able to fund or finance our obligations under the TRA.
We will not be reimbursed for any payments made to the Members or their permitted transferees under the TRA in the event that any tax benefits are disallowed.
Payments under the TRA will be based on the tax reporting positions that we determine, and the Internal Revenue Service or another tax authority may challenge all or part of the tax benefits we claim, as well as other related tax positions we take, and a court could sustain such challenge. If the outcome of any such challenge would reasonably be expected to materially adversely affect a recipient’s rights or obligations (including the amount or timing of payments) under the TRA, then we will not be permitted to settle or fail to contest such challenge without the consent of the Members. We will not be reimbursed for any cash payments previously made to the Members or their permitted transferees under the TRA in the event that any tax benefits initially claimed by us and for which payment has been made to the Members or their permitted transferees are subsequently challenged by a taxing authority and are ultimately disallowed. Instead, any excess cash payments made by us to the Members or their permitted transferees will be netted against any future cash payments that we might otherwise be required to make to Members or their permitted transferees under the terms of the TRA. However, we might not determine that we have effectively made an excess cash payment to the Members or their permitted transferees for a number of years following the initial time of such payment. As a result, payments could be made under the TRA in excess of the tax savings that we ultimately realize in respect of the tax attributes with respect to the Members or their permitted transferees.
Risks Related to Our Class A Common Stock
The Amneal Group owns a majority of our outstanding Class A Common Stock. The interests of the Amneal Group may differ from the interests of our other stockholders.
As of December 31, 2024, the Amneal Group controlled the majority of the voting power of all of our outstanding shares of common stock. Accordingly, the Amneal Group has substantial influence over the outcome of corporate actions requiring stockholder approval, including the election of directors, any merger, consolidation, or sale of all or substantially all of our assets or any other significant corporate transactions. These stockholders may also delay or prevent a change of control of the Company, even if such a change of control would benefit our other stockholders. This concentrated control could discourage a potential investor from seeking to acquire Class A common stock and, as a result, might harm the market price of that Class A common stock.
Through its control of a majority of our voting power and the provisions set forth in our charter, bylaws and our Third Amended and Restated Stockholders Agreement, dated November 7, 2023 (as amended to date, the “Stockholders Agreement”), the Amneal Group has the ability to designate and elect a majority of our board of directors. As of December 31, 2024, six out of eleven members of our board of directors (the “Board of Directors”), have been designated by the Amneal Group. The Amneal Group has control over all matters submitted to our stockholders for approval, including changes in capital structure, transactions requiring stockholder approval under Delaware law and corporate governance, subject to the terms of the Stockholders Agreement relating to the Amneal Group's agreement to vote in favor of directors not designated by the Amneal Group and such other matters that are set forth in the Stockholders Agreement. The Amneal Group may have different interests than our other stockholders and may make decisions adverse to such interests.
In the ordinary course of their business activities, the Amneal Group may engage in activities where their interests conflict with our interests or those of our other stockholders. Our certificate of incorporation provides that the Amneal Group have no duty to refrain from engaging in the same business activities or similar business activities or lines of business in which we operate. The Amneal Group also may pursue business opportunities with any of our clients, customers or vendors that may be complementary to our business and, as a result, those acquisition opportunities may not be available to us.
The Amneal Group could also transfer control of us to a third-party by transferring its shares. In addition, members of the Amneal Group have pledged shares of our Class A common stock to secure borrowings, and other members of the Amneal Group could enter into similar arrangements. In connection with these arrangements, we have entered into agreements with certain Amneal Group members and the lending institutions to whom their securities may be pledged. The voluntary or forced sale of some or all of these shares pursuant to a margin call or otherwise could cause our stock price to decline and negatively impact our business. Similarly, a voluntary or forced sale could cause us to lose our “controlled company” status under the Nasdaq listing requirements, which would require us to comply over a transition period with certain corporate governance requirements from which we are currently exempt, including having a fully independent compensation committee. If all of our shares of Class A common stock were pledged to secure borrowings by members of the Amneal Group, a complete foreclosure could result in a change of control.
Future sales of shares by the Amneal Group could cause our Class A Common Stock price to decline.
The majority of our Class A common stock is held by the Amneal Group and is eligible for sale or transfer (subject to certain continuing restrictions). The Amneal Group may elect to sell their shares. If some or all of these shares are sold, or if it is perceived that they will be sold, the trading price of our Class A common stock could decline.
We are a holding company with nominal net worth and depend on dividends and distributions from our subsidiaries.
We are a holding company with nominal net worth and will not have any material assets or conduct any business operations other than our investments in our subsidiaries. Our business operations are conducted primarily out of our direct operating subsidiary, Amneal, and its subsidiaries. As a result, our ability to satisfy our financial obligations and, notwithstanding any restrictions on payment of dividends under our existing indebtedness, our ability to pay dividends, if any, is dependent upon cash dividends and distributions or other transfers from our subsidiaries, including from Amneal.
We do not anticipate that we will pay any cash dividends in the foreseeable future.
We expect that we will retain our future earnings to fund the development and growth of our business. As a result, capital appreciation, if any, of our Class A common stock will be the sole source of gain for our stockholders for the foreseeable future. The payment of future cash dividends, if any, will be at the discretion of our Board of Directors and will be dependent upon our earnings, financial condition, capital requirements and other factors as our Board of Directors may deem relevant.
Item 1B. Unresolved Staff Comments
None.
Item 1C. Cybersecurity
Risk Management & Strategy
Our cybersecurity program includes policies and procedures designed to protect our systems and operations as well as sensitive information and data from anticipated cybersecurity threats. This program is a key component of our approach to enterprise risk management.
Key Processes
Our cybersecurity defenses include multiple layers of processes and technologies that can help prevent, detect and respond to cybersecurity threats. We administer multiple cybersecurity-related training and awareness events annually. These include baseline cybersecurity training for all new employees and an annual, mandatory, interactive cybersecurity training for all who are assigned our user accounts, and frequent cybersecurity topic awareness broadcast emails and targeted training to specific user groups. Additionally, we use a variety of detective and preventive technologies, including:
•email threat detection;
•endpoint detection and response;
•a 24 hours a day, 7 days a week security operations center that monitors system log telemetry and threat intelligence via a security incident and event management platform;
•vulnerability scanning on both internal and externally-facing infrastructure;
•next-generation firewalls with geographic access restriction for sensitive externally-facing systems;
•multi-factor authentication for remote access and certain internal systems;
•data loss prevention for email traffic containing sensitive information;
•domain name service threat detection; and
•internal incident response procedures based on NIST Special Publication 800-61.
We also conduct periodic phishing attack simulations to evaluate users’ vulnerability to emerging email threats. We conduct remediation training where failures occur. On a quarterly basis, our cybersecurity team also conducts scenario-based tabletop exercises with critical business teams to simulate disasters and cyberattacks. The tabletop exercises test and fine-tune our business continuity plans and incident response procedures.
Program Assessments
Our cybersecurity processes are evaluated as part of an ongoing assessment of our internal control environment, which are informed by the five pillars of the NIST Cybersecurity Framework (“NIST CSF”). We employ a third-party service provider to conduct periodic penetration tests and scan different parts of our IT environment for potential vulnerabilities. We prioritize critical or high vulnerabilities for swift remediation. Additionally, we employ a third-party service provider for continuous cybersecurity risk and vulnerability monitoring. We make continuous adjustments to system and network configurations to mitigate or remediate identified vulnerabilities.
Incident Response
Cybersecurity incident response procedures are informed by NIST Special Publication 800-61, and continuously improved following periodic exercises and live incidents. Incident response emphasizes rapid containment following detection of a range of threats including:
•suspicious repeated login failures;
•suspicious network traffic;
•malware detection; and
•other threats as prioritized through a combination of industry threat intelligence via the Healthcare Information Sharing and Analysis Center and the Company’s security operation center.
Third-party Risk Management
We focus on further building cybersecurity resiliency throughout our value chain. We perform risk management via an industry third-party risk management service provider for all critical vendors, partners, and systems (including third-party hosted information systems) meeting our risk management policy criteria, to minimize the likelihood and impact of malicious cybersecurity incidents. During the onboarding phase, our cybersecurity team under the direction of the Sr. Director Information Security, Compliance, and Privacy, performs a technological risk assessment on, and utilizes certain tools to detect external risk posed by, the vendor, partner and/or system. Vendors identified as posing elevated risk are escalated to senior management for informed risk tolerance determination. Following the onboarding phase, the cybersecurity team continuously monitors risks related to the vendor, partner and/or system. All third-party cybersecurity incidents are tracked though the third-party service provider and are communicated to our cybersecurity team upon discovery. We prioritize our mitigation of cybersecurity risks based on relative likelihood and severity (e.g., critical risk, high risk, low risk) and document a mitigation plan that details a resolution timeline.
Risks
Identified cybersecurity risks, including third-party risks and internal risks, are documented and managed in a risk register, which is reviewed regularly with leaders of our internal audit, compliance, and IT departments to ensure visibility and consensus in a separation-of-duties structure. Risks are stratified according to a standard calculus of probability, severity and materiality.
As of the date of this report, we do not believe that any risks from any cybersecurity threats, including as a result of any previous cybersecurity incidents, have materially affected or are reasonably likely to materially affect us, including our business strategy, results of operations or financial condition. However, a significant cybersecurity incident may materially impact our
business strategy, results of operations and financial condition. Such significant cybersecurity incidents include, but are not limited to:
•ransomware infiltrating our critical systems resulting in production delays and/or loss of critical information;
•cyber theft of our IP;
•cyber theft of employee and family member, customer and/or patient information;
•cyberattack on a critical partner that disrupts our supply chain and/or services; and,
•cyberattacks that significantly impact our brand perception.
As discussed more fully under Part 1, Item 1A, Risk Factors, “We are increasingly dependent on information technology, and our systems and infrastructure face certain risks, including cybersecurity and data leakage risks,” the sophistication of cybersecurity threats continues to increase, and the preventative actions we take to reduce the risk of cybersecurity incidents and protect our systems and information may be insufficient. Accordingly, no matter how well designed or implemented our controls are, we will not be able to anticipate all security breaches of these types, and we may not be able to implement effective preventive measures against such security breaches in a timely manner.
Board Oversight and Management’s Responsibilities
Board Oversight
While our Board of Directors is ultimately responsible for risk oversight, committees of the Board of Directors assist in fulfilling oversight responsibilities in certain areas of risk. The Audit Committee of the Board of Directors (the “Audit Committee”) is responsible for overseeing risks from cybersecurity threats. The Audit Committee receives cybersecurity updates from IT leadership at least twice a year. When meeting with the Audit Committee, the IT leadership team highlights significant accomplishments and issues related to our IT infrastructure, including cybersecurity incidents, risks, industry trends, notable incidents facing other companies, incident preparedness and other developments. The Audit Committee also receives updates regarding progress on initiatives to further align with the five pillars of the NIST CSF. These briefings are designed to provide visibility to the Audit Committee about the identification, assessment, and management of critical risks, audit findings, and management’s risk mitigation strategies.
Management Oversight
Our IT department, in conjunction with the compliance department, assess and manage risks related to cybersecurity. The Senior Director of Global Information Security, Compliance and Privacy, is the primary management personnel responsible for our cybersecurity program. He has more than twenty years of experience as an information security specialist and holds various cybersecurity professional certifications, including as a Certified Information Security Manager by the Information Systems Audit and Control Association and a Certified Information Systems Security Professional by the International Information System Security Certification Consortium. In addition, the department heads for IT and internal audit have industry recognized credentials and extensive experience in the area of cybersecurity. Specific cybersecurity incidents are tracked by a third-party service provider through a ticketing system.
Should cybersecurity issues arise throughout the quarter, management would determine whether a cybersecurity incident was material and decide on appropriate reporting and mitigation measures. Following this determination, management would promptly schedule a meeting with the Audit Committee.
Item 2. Properties
Amneal owns or leases numerous properties in domestic and foreign locations. Amneal’s principal properties include manufacturing facilities, R&D laboratories, warehouses, and corporate offices. Our properties are generally used to support the operations of our Affordable Medicines, Specialty and AvKARE segments.
Our significant properties are as follows:
| | | | | | | | | | | | | | |
| Property Address | | Legal
Status | | Purpose |
| Bridgewater, New Jersey | | Leased | | Executive Office |
| Glasgow, Kentucky | | Leased | | Administrative, Distribution and Warehouse |
| Glasgow, Kentucky | | Leased | | Warehouse |
| Yaphank, New York | | Leased | | Warehouse |
| Glasgow, Kentucky | | Owned | | Warehouse |
| Glasgow, Kentucky | | Owned | | Warehouse |
| Glasgow, Kentucky | | Leased | | Warehouse |
| Piscataway, New Jersey | | Leased | | Warehouse |
| Piscataway, New Jersey | | Leased | | Manufacturing |
| Piscataway, New Jersey | | Leased | | R&D, Manufacturing |
| Branchburg, New Jersey | | Leased | | Manufacturing |
| Branchburg, New Jersey | | Leased | | Manufacturing |
| Piscataway, New Jersey | | Leased | | Manufacturing |
| Branchburg, New Jersey | | Leased | | Warehouse |
| East Hanover, New Jersey | | Leased | | Warehouse |
| Bridgewater, New Jersey | | Leased | | R&D |
| Yaphank, New York | | Leased | | Manufacturing, R&D, Quality and Regulatory |
| Pulaski, Tennessee | | Leased | | Warehouse and office space |
| Philadelphia, Pennsylvania | | Leased | | Warehouse and office space |
| Fountain Run, Kentucky | | Leased | | Warehouse and office space |
| Cashel Co, Tipperary, Ireland | | Owned | | R&D, Manufacturing |
| Ahmedabad, Gujarat, India | | Owned | | Oral Solids Manufacturing and R&D |
| Matoda, Gujarat, India | | Leased | | Oral Solids and Injectables Manufacturing and R&D |
| Ahmedabad, Gujarat, India | | Leased | | R&D (Injectables), Corporate Office |
| Ahmedabad, Gujarat, India | | Leased | | Corporate Office |
| Mahabubnagar, Telangana, India | | Leased | | Oncology R&D and Manufacturing |
| Visakhapatam, Apandhra Pradesh, India | | Owned | | API Manufacturing and R&D |
| Bharuch, Gujarat, India | | Leased | | API Manufacturing |
| Ahmedabad, Gujarat, India | | Leased | | R&D |
| Mehsana, Gujarat, India | | Owned | | Injectables Manufacturing |
| Ahmedabad, Gujarat, India | | Leased | | Office space |
| Ahmedabad, Gujarat, India | | Owned | | Injectables Manufacturing and R&D |
| Mumbai, Maharashtra, India | | Leased | | Office Space |
| Ahmedabad, Gujarat, India | | Leased | | Warehouse |
Item 3. Legal Proceedings
Information pertaining to legal proceedings can be found in Note 20. Commitments and Contingencies and is incorporated by reference herein.
Item 4. Mine Safety Disclosures
Not applicable.
PART II.
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information and Holders
Our Class A common stock trades under the symbol “AMRX” on the Nasdaq. According to the records of our transfer agent, we had 154 holders of record of our Class A common stock as of February 14, 2025. A substantially greater number of holders of our Class A common stock are “street name” or beneficial holders, whose shares of record are held by banks, brokers, and other financial institutions.
Performance Graph
Set forth below is a line graph comparing the change in the cumulative total shareholder return on our Class A common stock with the cumulative total returns of the Nasdaq Composite Total Return Index, the Russell 2000 Index and the Dow Jones U.S. Select Pharmaceuticals Index for the period from December 31, 2019 to December 31, 2024, assuming the investment of $100 on December 31, 2019, and the reinvestment of dividends. For the years ended December 31, 2024 and December 31, 2023, we utilized the Russell 2000 Index as a broad equity market index for purposes of meeting the disclosure requirements of Regulation S-K Item 201(e)(1)(i) and the Dow Jones U.S. Select Pharmaceuticals Index as a published industry index for purposes of meeting the disclosure requirements of Regulation S-K Item 201(e)(1)(ii)(A). The Class A common stock price performance shown on the graph only reflects the change in our Class A common stock price relative to the noted indices and is not necessarily indicative of future price performance.
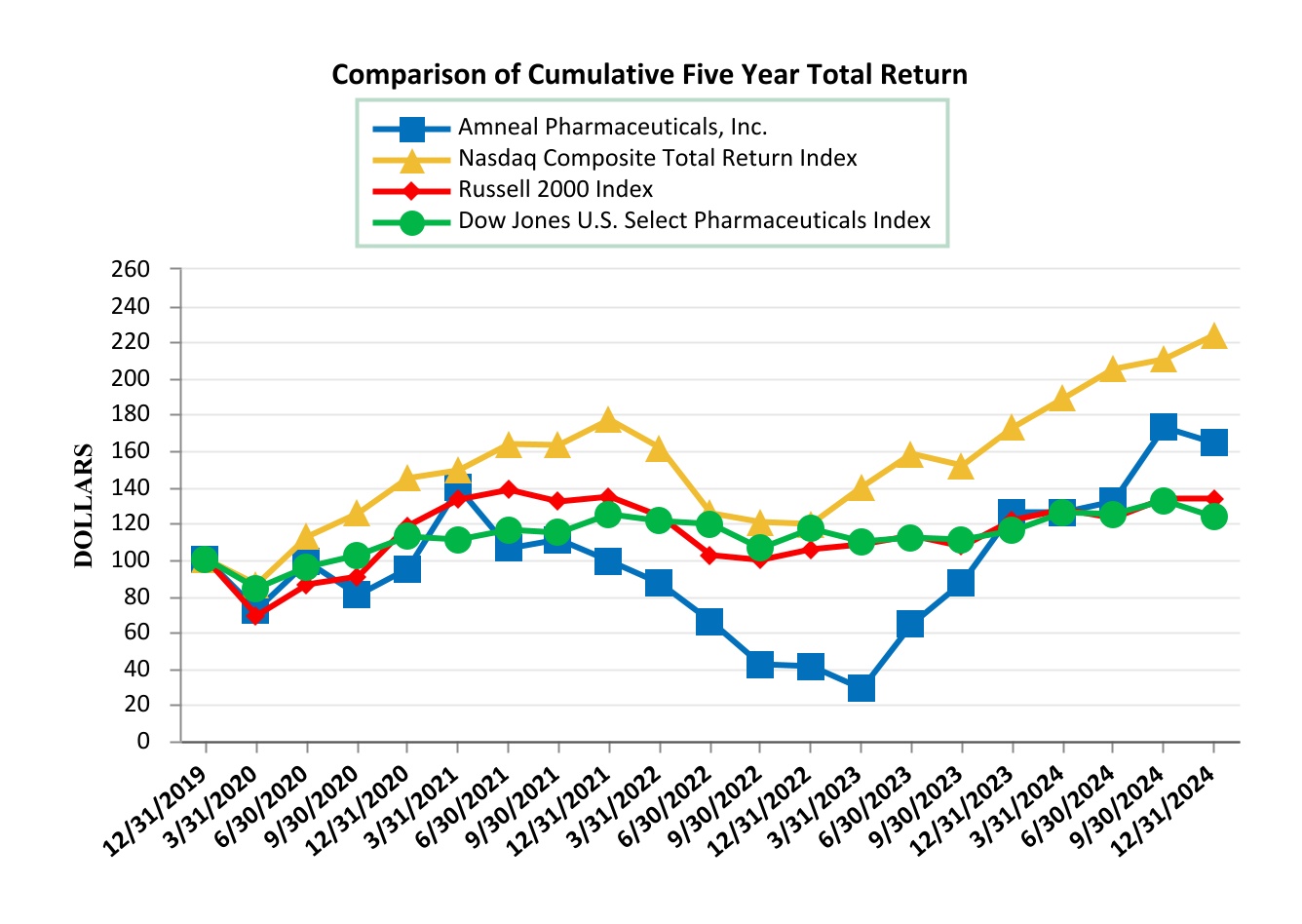
Dividends
We have never paid cash dividends on any class of our common stock and have no present plans to do so. Our current policy is to retain all earnings, if any, for use in the operation of our business or to reduce our debt.
Issuer Purchases of Equity Securities
We did not purchase any shares of our Class A common stock during the three months ended December 31, 2024.
Item 6. Reserved
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
Amneal Pharmaceuticals, Inc. (the “Company”, “we,” “us,” or “our”) is a global pharmaceutical company that develops, manufactures, markets, and distributes a diverse portfolio of essential medicines. Our Affordable Medicines segment includes retail generics, injectables, and biosimilars. In our Specialty segment, we offer a portfolio of branded pharmaceuticals focused primarily on central nervous system and endocrine disorders. Through our AvKARE segment, we are a distributor of pharmaceuticals and other products for the U.S. federal government, retail, and institutional markets. We operate principally in the United States (“U.S.”), India, and Ireland. Refer to the section “Segments” below for an overview of our segments, including the change in name of the Affordable Medicines segment.
Prior to the Reorganization (as defined herein), we were a holding company, whose principal assets were common units (the “Amneal Common Units”) of Amneal Pharmaceuticals, LLC (“Amneal”). As of September 30, 2023, we held 50.4% of the Amneal Common Units and the group, together with their affiliates and certain assignees, who owned Amneal when it was a private company (the “Members” or the “Amneal Group”) held the remaining 49.6%. On November 7, 2023, we implemented a plan pursuant to which we and Amneal reorganized and simplified our corporate structure by eliminating our umbrella partnership-C-corporation structure and converting to a more traditional structure whereby all stockholders hold their voting and economic interests directly through the public company (the “Reorganization”). Effective with the Reorganization, we hold 100% of the Amneal Common Units and consolidate the financial statements of Amneal and its subsidiaries. Refer to Note 1. Nature of Operations in our consolidated financial statements for additional information about the Reorganization.
Although we had a minority economic interest in Amneal prior to March 31, 2023, we were Amneal’s sole managing member, having the sole voting power to make all of Amneal’s business decisions and control its management. Therefore, we consolidated the financial statements of Amneal and its subsidiaries prior to the Reorganization. We recorded non-controlling interests for the portion of Amneal’s economic interests that we did not hold prior to the Reorganization.
The following discussion and analysis contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of certain factors, including those set forth under Item 1A. Risk Factors and under the heading Forward-Looking Statements in this Annual Report on Form 10-K. The following discussion and analysis, as well as other sections in this report, should be read in conjunction with the consolidated financial statements and related notes to consolidated financial statements included elsewhere herein.
For a discussion of our financial condition and results of operations for the year ended December 31, 2023 compared to the year ended December 31, 2022, see “Results of Operations” and “Liquidity and Capital Resources” under Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations in our 2023 Annual Report on Form 10-K, filed with the U.S. Securities and Exchange Commission on March 14, 2024.
Overview
Segments
We have three reportable segments: Affordable Medicines (formerly known as Generics), Specialty, and AvKARE.
During the fourth quarter of 2024, we changed the name of our Generics segment to “Affordable Medicines” to reflect the full product offering of the segment. The segment name change did not result in any change to the composition of our reportable segments and, therefore, did not result in any changes to our historical segment results.
Affordable Medicines
Our Affordable Medicines segment includes approximately 270 product families covering an extensive range of dosage forms and delivery systems, including both immediate and extended-release oral solids, powders, liquids, sterile injectables, nasal sprays, inhalation and respiratory products, biosimilar products, ophthalmics, films, transdermal patches and topicals. We focus on developing products that have substantial barriers-to-entry due to complex drug formulations or manufacturing, or legal or regulatory challenges.
Generic products, particularly in the U.S., generally contribute most significantly to revenues and gross margins at the time of their launch, and even more so in periods of market exclusivity, or in periods of limited generic competition. As such, the timing of new product introductions can have a significant impact on our financial results. The entrance into the market of additional competition generally has a negative impact on the volume and/or pricing of the affected products. Additionally, pricing is determined by market place dynamics and is often affected by factors outside of our control.
Specialty
Our Specialty segment is engaged in the development, promotion, sale and distribution of proprietary branded pharmaceutical products, with a focus on products addressing central nervous system disorders, including Parkinson’s disease, and endocrine disorders. Our portfolio of products includes CREXONT® (combination of carbidopa and levodopa extended release capsules), RYTARY® (extended release oral capsule formulation of carbidopa-levodopa), UNITHROID® (levothyroxine sodium), and ONGENTYS® (opicapone). On August 7, 2024, the FDA approved our new drug application (“NDA”) for CREXONT®, previously referred to as IPX203. In September 2024, we began selling CREXONT®, which is indicated for the treatment of Parkinson’s disease, Parkinson’s disease caused by infection or inflammation of the brain, or Parkinson’s disease-like symptoms that may result from carbon monoxide or manganese poisoning in adults. RYTARY® is indicated for the treatment of Parkinson’s disease, post-encephalitic parkinsonism, and parkinsonism that may follow carbon monoxide intoxication or manganese intoxication. ONGENTYS® is an add-on treatment to carbidopa/levodopa in patients with Parkinson’s disease experiencing “Off” episodes, which we commenced selling in early 2024 under a license agreement with BIAL-Portela & Ca., S.A. UNITHROID®, indicated for the treatment of hypothyroidism, is sold under a license and distribution agreement with Jerome Stevens Pharmaceuticals, Inc.
Our Specialty products are marketed through skilled specialty sales and marketing teams, who call on neurologists, movement disorder specialists, endocrinologists and primary care physicians in key markets throughout the U.S. Our Specialty segment also has a number of product candidates that are in varying stages of development.
For Specialty products, the majority of the product’s commercial value is usually realized during the period in which the product has market exclusivity. In the U.S., when market exclusivity expires and generic versions of a product are approved and marketed, there can often be very substantial and rapid declines in the branded product’s sales.
AvKARE
Our AvKARE segment provides pharmaceuticals, medical and surgical products and services primarily to governmental agencies, predominantly focused on the U.S. Department of Defense and the U.S. Department of Veterans Affairs. AvKARE is a re-packager of bottle and unit dose pharmaceuticals under the registered names of AvKARE and AvPAK. AvKARE is also a wholesale distributor of pharmaceuticals, over the counter drugs and medical supplies to its retail and institutional customers that are located throughout the U.S. focused primarily on entities that provide care to low-income and uninsured patients. Operating results for the sale of Amneal products by AvKARE are included in our Affordable Medicines reportable segment.
The Pharmaceutical Industry
The pharmaceutical industry is highly competitive and highly regulated. As a result, we face a number of industry-specific factors and challenges, which can significantly impact our results. For a more detailed explanation of our business and its risks, refer to Item 1. Business and Item 1A. Risk Factors in this Form 10-K.
Inflation
While it is difficult to accurately measure the impact of inflation, we estimate our business did not experience a material increase in costs due to inflation for the year ended December 31, 2024. We do not expect a material impact related to inflation for the year ending December 31, 2025. Notwithstanding our estimates, rising inflationary pressures due to higher input costs, including higher material, transportation, labor and other costs, could exceed our expectations and may adversely impact our operating results in future periods.
Results of Operations
Consolidated Results
The following table sets forth our summarized, consolidated results of operations (dollars in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, | | Change |
| | 2024 | | 2023 | | $ | | % |
| Net revenue | $ | 2,793,957 | | | $ | 2,393,607 | | | $ | 400,350 | | | 16.7% |
| Cost of goods sold | 1,773,519 | | | 1,573,042 | | | 200,477 | | | 12.7% |
| Gross profit | 1,020,438 | | | 820,565 | | | 199,873 | | | 24.4% |
| Selling, general and administrative | 476,436 | | | 429,675 | | | 46,761 | | | 10.9% |
| Research and development | 190,714 | | | 163,950 | | | 26,764 | | | 16.3% |
| In-process research and development impairment charges | — | | | 30,800 | | | (30,800) | | | (100.0)% |
| Intellectual property legal development expenses | 5,845 | | | 3,828 | | | 2,017 | | | 52.7% |
| Restructuring and other charges | 2,355 | | | 1,749 | | | 606 | | | 34.6% |
| Change in fair value of contingent consideration | (930) | | | (14,497) | | | 13,567 | | | (93.6)% |
| Charges related to legal matters, net | 96,692 | | | 1,824 | | | 94,868 | | | nm |
| Other operating income | — | | | (1,138) | | | 1,138 | | | (100.0)% |
| Operating income | 249,326 | | | 204,374 | | | 44,952 | | | 22.0% |
| Total other expense, net | (304,339) | | | (244,644) | | | (59,695) | | | 24.4% |
| Loss before income taxes | (55,013) | | | (40,270) | | | (14,743) | | | 36.6% |
| Provision for income taxes | 18,863 | | | 8,452 | | | 10,411 | | | 123.2% |
| Net loss | $ | (73,876) | | | $ | (48,722) | | | $ | (25,154) | | | 51.6% |
nm - not meaningful
Net Revenue
Net revenue for the year ended December 31, 2024 increased 16.7% from the prior year primarily due to:
•Growth in our Affordable Medicines segment of $213.9 million, primarily due to new products launched in 2024 and 2023, which included biosimilars that contributed $59.7 million of year-over-year growth and other new products that contributed $144.1 million of year-over-year growth, and strong volume growth, partially offset by price erosion. Net revenue for the year ended December 31, 2023 included a non-recurring customer order of $21.0 million.
•Growth in our AvKARE segment of $131.2 million primarily driven by growth in our distribution and government channels resulting from new product introductions.
•Growth in our Specialty segment of $55.3 million primarily driven by a $44.7 million increase in our promoted Parkinson’s franchise, of which $16.6 million was comprised of sales of ONGENTYS®, which launched in January 2024, and initial sales of CREXONT®, which launched in September 2024. Additionally, growth in our promoted endocrinology portfolio of $20.8 million was partially offset by declines in our non-promoted products.
Cost of Goods Sold and Gross Profit
Cost of goods sold increased 12.7% for the year ended December 31, 2024 as compared to the prior year. The increase in cost of goods sold was primarily due to increased AvKARE and Affordable Medicines volume, increased plant and freight costs, and an increased inventory provision, partially offset by efficiencies in our supply costs. Cost of goods sold for the year ended December 31, 2023 included $11.0 million associated with the non-recurring customer order in our Affordable Medicines segment discussed above and a marketed product intangible asset impairment charge of $34.1 million in our Specialty segment related to a reduction in the promotional focus on LYVISPAH®.
Gross profit as a percentage of net revenue increased to 36.5% for the year ended December 31, 2024 from 34.3% in the prior year, primarily as a result of the factors noted above.
Selling, General and Administrative
Selling, general and administrative (“SG&A”) expenses for the year ended December 31, 2024 increased 10.9% as compared to the prior year primarily due to increases in employee compensation, promotion associated with ONGENTYS® and CREXONT®, increased expenses associated with our growing biosimilars, and annual fees assessed on branded prescription drug manufacturers, which are also applicable to certain of our Affordable Medicines products.
Research and Development
Research and development (“R&D”) expenses for the year ended December 31, 2024 increased 16.3% from the prior year primarily due to an increase in in-licensing and upfront milestone payments of $30.0 million, including $20.0 million associated with our exclusive license of Omalizumab (refer to Note 23. Related Party Transactions for additional information), partially offset by operating efficiencies in our infrastructure.
In-Process Research and Development Impairment Charges
In process research and development (“IPR&D”) impairment charges of $30.8 million for the year ended December 31, 2023 were related to one Affordable Medicines asset and one Specialty asset, both of which experienced adverse clinical trials results in the fourth quarter of 2023 and resulted in significantly lower than expected future cash flows.
Change in Fair Value of Contingent Consideration
The year-over-year variance of $13.6 million in change in fair value of contingent consideration for the year ended December 31, 2024 as compared to the prior year was primarily related to a reduction in promotional focus on LYVISPAHTM during the year ended December 31, 2023. Refer to Note 18. Fair Value Measurements for additional information.
Charges Related to Legal Matters, Net
For the year ended December 31, 2024, charges related to legal matters, net of $96.7 million were primarily associated with an Affordable Medicines settlement in principle on the primary financial terms for a nationwide resolution to the opioids cases that have been filed and that might have been filed against us by political subdivisions and Native American tribes across the U.S.
For the year ended December 31, 2023, charges related to legal matters, net of $1.8 million were comprised of $3.9 million in charges associated with Affordable Medicines civil prescription opioid litigation, a $3.0 million charge for the settlement of an Affordable Medicines customer claim, a $3.0 million charge for the settlement of Affordable Medicines commercial antitrust litigation, and a $1.9 million charge for the settlement of a corporate stockholder derivative lawsuit, partially offset by $10.0 million from the settlement of Affordable Medicines patent infringement matters.
For additional information, refer to Note 20. Commitments and Contingencies.
Total Other Expense, Net
Total other expense, net increased 24.4% for the year ended December 31, 2024. The increase was primarily driven by a $48.0 million increase in net interest expense as a result of higher rates on our variable rate debt and an increase in the average amount outstanding on our revolving credit facility throughout 2024, and a $47.6 million increase in our tax receivable agreement liability (refer to Note 6. Income Taxes for additional information), partially offset by a $40.8 million loss on refinancing the Term Loan Due 2025 and amending the New Revolving Credit Facility in 2023 (refer to Note 15. Debt for additional information).
Provision For Income Taxes
The provision for income taxes was $18.9 million and $8.5 million for the years ended December 31, 2024 and 2023, respectively. The effective tax rates for the years ended December 31, 2024 and 2023 were (34.3)% and (21.0)%, respectively. The change in the effective income tax rate for the year ended December 31, 2024 as compared to the prior year was primarily due to the timing and jurisdictional mix of income and the Reorganization, which resulted in allocating all of Amneal’s income to the Company. Refer to Note 6. Income Taxes for additional information.
Affordable Medicines
The following table sets forth the results of operations for our Affordable Medicines segment (dollars in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, | | Change |
| | 2024 | | 2023 | | $ | | % |
| Net revenue | $ | 1,685,263 | | | $ | 1,471,401 | | | $ | 213,862 | | | 14.5% |
| Cost of goods sold | 1,011,363 | | | 913,869 | | | 97,494 | | | 10.7% |
| Gross profit | 673,900 | | | 557,532 | | | 116,368 | | | 20.9% |
| Selling, general and administrative | 129,578 | | | 119,912 | | | 9,666 | | | 8.1% |
| Research and development | 171,771 | | | 132,233 | | | 39,538 | | | 29.9% |
| In-process research and development impairment charges | — | | | 26,500 | | | (26,500) | | | (100.0)% |
| Intellectual property legal development expenses | 5,685 | | | 3,708 | | | 1,977 | | | 53.3% |
| Restructuring and other charges | 70 | | | 211 | | | (141) | | | (66.8)% |
| Charges (credit) related to legal matters, net | 96,692 | | | (64) | | | 96,756 | | | nm |
| Other operating income | — | | | (1,138) | | | 1,138 | | | (100.0)% |
| Operating income | $ | 270,104 | | | $ | 276,170 | | | $ | (6,066) | | | (2.2)% |
nm - not meaningful
Net Revenue
Affordable Medicines net revenue for the year ended December 31, 2024 increased 14.5% as compared to the prior year, primarily due to new products launched in 2024 and 2023, which included biosimilars that contributed $59.7 million of year-over-year growth and other new products that contributed $144.1 million of year-over-year growth, and strong volume growth, partially offset by price erosion. Net revenue for the year ended December 31, 2023 included a non-recurring customer order of $21.0 million.
Cost of Goods Sold and Gross Profit
Affordable Medicines cost of goods sold for the year ended December 31, 2024 increased 10.7% compared to the prior year primarily due to costs associated with increased sales volume and increased plant and freight costs and an increased inventory provision, partially offset by efficiencies in our supply costs. Cost of goods sold for the year ended December 31, 2023 included $11.0 million associated with the non-recurring customer order discussed above.
Affordable Medicines gross profit as a percentage of net revenue increased to 40.0% for the year ended December 31, 2024 from 37.9% in the prior year as a result of the factors described above.
Selling, General, and Administrative
Affordable Medicines SG&A for the year ended December 31, 2024 increased by 8.1% compared to the prior year primarily due to increases in employee compensation driven by infrastructure expansion and promotion associated with our biosimilar launches and the annual fees assessed on branded prescription drug manufacturers, which are also applicable to certain of our affordable medicine products, partially offset by reduced legal fees.
Research and Development
Affordable Medicines R&D expense for the year ended December 31, 2024 increased 29.9% as compared to the prior year primarily due to an increase in in-licensing and upfront milestone payments of $30.0 million, including $20.0 million associated with our exclusive license of Omalizumab (refer to Note 23. Related Party Transactions for additional information), partially offset by operating efficiencies.
In-Process Research and Development Impairment Charges
Affordable Medicines IPR&D impairment charges for the year ended December 31, 2023 were related to one asset that experienced adverse clinical trials results in the fourth quarter of 2023 and resulted in significantly lower than expected future cash flows.
Charges (Credit) Related to Legal Matters, Net
For the year ended December 31, 2024, Affordable Medicines charges related to legal matters, net of $96.7 million were primarily associated with a settlement in principle on the primary financial terms for a nationwide resolution to the opioids cases that have been filed and that might have been filed against us by political subdivisions and Native American tribes across the U.S. Refer to Note 20. Commitments and Contingencies for additional information.
For the year ended December 31, 2023, the Affordable Medicines credit related to legal matters, net was $(0.1) million, comprised of $10.0 million received from the settlement of patent infringement matters, net of $3.9 million in charges associated with civil prescription opioid litigation, a $3.0 million charge for the settlement of a customer claim, and a $3.0 million charge for the settlement of commercial antitrust litigation.
Specialty
The following table sets forth the results of operations for our Specialty segment (dollars in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| Years Ended December 31, | | Change |
| 2024 | | 2023 | | $ | | % |
| Net revenue | $ | 445,749 | | | $ | 390,457 | | | $ | 55,292 | | | 14.2% |
| Cost of goods sold | 202,821 | | | 214,277 | | | (11,456) | | | (5.3)% |
| Gross profit | 242,928 | | | 176,180 | | | 66,748 | | | 37.9% |
| Selling, general and administrative | 109,658 | | | 88,137 | | | 21,521 | | | 24.4% |
| Research and development | 18,943 | | | 31,717 | | | (12,774) | | | (40.3)% |
| In-process research and development impairment charges | — | | | 4,300 | | | (4,300) | | | (100.0)% |
| Intellectual property legal development expenses | 160 | | | 120 | | | 40 | | | 33.3% |
| Restructuring and other charges | 1,517 | | | 1,105 | | | 412 | | | 37.3% |
| Change in fair value of contingent consideration | (930) | | | (14,497) | | | 13,567 | | | (93.6)% |
| Operating income | $ | 113,580 | | | $ | 65,298 | | | $ | 48,282 | | | 73.9% |
Net Revenue
Specialty net revenue for the year ended December 31, 2024 increased 14.2% as compared to the prior year, primarily driven by a $44.7 million increase in our promoted Parkinson’s franchise, of which $16.6 million was comprised of sales of ONGENTYS®, which launched in January 2024, and initial sales of CREXONT®, which launched in September 2024. Additionally, growth in our promoted endocrinology portfolio of $20.8 million was partially offset by declines in our non-promoted products.
Cost of Goods Sold and Gross Profit
Specialty cost of goods sold for the year ended December 31, 2024 decreased 5.3% as compared to the prior year due to a marketed product intangible asset impairment charge of $34.1 million in 2023 related to reduced promotional focus on LYVISPAH®, partially offset by increased sales in our promoted products. Specialty gross profit as a percentage of net revenue increased to 54.5% for the year ended December 31, 2024 as compared to 45.1% in the prior year as a result of the factors described above.
Selling, General, and Administrative
Specialty SG&A expense for the year ended December 31, 2024 increased 24.4% as compared to the prior year primarily due to increases in promotional costs associated with ONGENTYS® and CREXONT®.
Research and Development
Specialty R&D expense for the year ended December 31, 2024 decreased 40.3% as compared to the prior year primarily due to reduced project spend of $8.6 million and reduced infrastructure costs.
Change in Fair Value of Contingent Consideration
The year-over-year variance of $13.6 million in change in fair value of contingent consideration for the year ended December 31, 2024 as compared to the prior year was primarily related to a reduction in promotional focus on LYVISPAHTM during the year ended December 31, 2023. Refer to Note 18. Fair Value Measurements for additional information.
AvKARE
The following table sets forth the results of operations for our AvKARE segment (dollars in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| Years Ended December, 31 | | Change |
| 2024 | | 2023 | | $ | | % |
| Net revenue | $ | 662,945 | | | $ | 531,749 | | | $ | 131,196 | | | 24.7% |
| Cost of goods sold | 559,335 | | | 444,896 | | | 114,439 | | | 25.7% |
| Gross profit | 103,610 | | | 86,853 | | | 16,757 | | | 19.3% |
| Selling, general and administrative | 60,709 | | | 55,341 | | | 5,368 | | | 9.7% |
| Operating income | $ | 42,901 | | | $ | 31,512 | | | $ | 11,389 | | | 36.1% |
Net Revenue
AvKARE net revenue for the year ended December 31, 2024 increased 24.7% as compared to the prior year primarily driven by growth in our distribution and government channels resulting from new product introductions.
Cost of Goods Sold and Gross Profit
AvKARE cost of goods sold for the year ended December 31, 2024 increased 25.7% as compared to the prior year, and gross profit as a percentage of net revenue decreased to 15.6% for the year ended December 31, 2024 from 16.3% in the prior year primarily due to the increase in sales through our lower margin distribution channel and an increased inventory provision.
Selling, General, and Administrative
AvKARE SG&A expense for the year ended December 31, 2024 increased 9.7% as compared to the prior year primarily due to higher sales-related expenses, increases in employee compensation and higher professional fees.
Liquidity and Capital Resources
Our primary source of liquidity is cash generated from operations, available cash and borrowings under debt financing arrangements (as defined and described in Note 15. Debt), including $495.2 million of available capacity on our Amended New Revolving Credit Facility and $28.0 million of available capacity under the Amended Rondo Revolving Credit Facility as of December 31, 2024. We believe these sources are sufficient to fund our planned operations, meet our interest and contractual obligations and provide sufficient liquidity over the next 12 months. However, our ability to satisfy our working capital requirements and debt obligations will depend upon economic conditions, our ability to negotiate and maintain satisfactory terms under our borrowing and debt facilities in the future, and demand for our products, which are factors that may be out of our control. Our primary uses of capital resources are to fund operating activities, including R&D expenses associated with new product filings, and pharmaceutical product manufacturing expenses, license payments, spending on production facility expansions, capital equipment, acquisitions, and legal settlements.
We estimate that we will invest approximately $120.0 million during 2025 for capital expenditures to support and grow our existing operations, primarily related to investments in manufacturing equipment, IT and facilities. Our 2025 estimate includes capital expenditures for our collaboration and supply agreement with Metsera, of which we expect Metsera to reimburse us approximately $20.0 million. We expect such reimbursements to primarily be included in our financing cash flows.
Debt Instruments
Over the next 12 months, we expect to make substantial payments for monthly interest and quarterly principal amounts due for our Term Loan Due 2028, monthly interest on our Amended New Credit Facility, and contractual payments for leased premises. In January 2025, the Company paid the entire remaining principal balance of $192.0 million outstanding on its Term
Loan Due 2025, plus accrued interest thereon of $0.7 million, with $190.0 million of new borrowings under the Amended New Revolving Credit Facility and cash on hand.
Annually, we are also required to calculate the amount of excess cash flow payments, as defined in our term loan agreements. Based on the results of the excess cash flows calculation for the years ended December 31, 2024, 2023 and 2022, no excess cash flows principal payments were required.
Settlement in Principle on Nationwide Civil Prescription Opioid Litigation
In late April 2024, we reached a nationwide settlement in principle on the primary financial terms, with no admission of wrongdoing, for a nationwide resolution to the opioids cases that have been filed and that might have been filed by Attorneys General, political subdivisions and Native American tribes. Refer to Note 20. Commitments and Contingencies for additional information. Refer to Note 20. Commitments and Contingencies for additional information.
Tax Receivable Agreement
As part of the Reorganization, our existing tax receivable agreement (“TRA”) was amended to reduce our future obligation to pay 85% of the realized tax benefits subject to the TRA to 75% of such realized benefits. As of December 31, 2024, the unrecorded contingent TRA liability, including the impact of the amendment, was $133.8 million.
The timing and amount of any payments under the TRA may vary, depending upon a number of factors including the timing and amount of our taxable income, and the corporate tax rate in effect at the time of realization of our taxable income. The timing and amount of payments may also be accelerated under certain conditions, such as a change of control or other early termination event, which could give rise to our obligation to make TRA payments in advance of tax benefits being realized.
For further information, refer to Item 1A. Risk Factors and Note 6. Income Taxes.
Tax-related Distributions
In 2020, we acquired a 65.1% controlling interest in both AvKARE Inc., a Tennessee corporation, now a limited liability company, and R&S. The sellers of AvKARE, LLC and R&S (the “AvKARE Sellers”) hold the remaining 34.9% interest in the holding company that directly owns the acquired companies (“Rondo”). We attribute 34.9% of the net income or loss associated with Rondo to redeemable non-controlling interests. During the year ended December 31, 2024, 2023 and 2022, we made cash tax distributions of $19.8 million, $14.2 million and $6.9 million, respectively, to the AvKARE Sellers. There was no liability for tax distributions payable to the AvKARE Sellers as of December 31, 2024 or 2023.
Development and Supply Agreement
Pursuant to a development and supply agreement with Kashiv Biosciences LLC (“Kashiv”) for a long-acting injectable, we paid Kashiv $10.0 million in February 2025 for the achievement of a regulatory milestone, which was accrued as of December 31, 2024. Refer to Note 23. Related Party Transactions for additional information.
Cash Balances
At December 31, 2024, our cash and cash equivalents consist of cash on deposit and highly liquid investments. A portion of our cash flows are derived outside the U.S. As a result, we are subject to market risk associated with changes in foreign exchange rates. We maintain cash balances at both U.S. based and foreign country based commercial banks. At various times during the year, our cash balances held in the U.S. may exceed amounts that are insured by the Federal Deposit Insurance Corporation. We make our investments in accordance with our investment policy. The primary objectives of our investment policy are liquidity and safety of principal.
Cash Flows
For a discussion comparing of our cash flows for the fiscal years 2023 to 2022, see Cash Flows under Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations in our 2023 Annual Report on Form 10-K.
The following table sets forth our summarized, consolidated cash flows for the years ended December 31, 2024 and 2023 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, | | Change |
| | 2024 | | 2023 | | $ | | % |
| Cash provided by (used in): | | | | | | | |
| Operating activities | $ | 295,099 | | | $ | 345,577 | | | $ | (50,478) | | | (14.6) | % |
| Investing activities | (62,996) | | | (69,189) | | | 6,193 | | | (9.0) | % |
| Financing activities | (211,791) | | | (212,573) | | | 782 | | | (0.4) | % |
| Effect of exchange rate changes on cash | (999) | | | 65 | | | (1,064) | | | nm |
| Net increase (decrease) in cash, cash equivalents, and restricted cash | $ | 19,313 | | | $ | 63,880 | | | $ | (44,567) | | | (69.8) | % |
nm - not meaningful
Cash Flows from Operating Activities
Net cash provided by operating activities was $295.1 million for the year ended December 31, 2024 as compared to $345.6 million for the prior year. The year-over-year decrease in net operating cash flows for the year ended December 31, 2024 as compared to the prior year period was primarily driven by (i) lower collections of outstanding accounts receivable due to timing of sales in the quarter ended December 31, 2022, which benefited the year ended December 31, 2023 and (ii) receipt of a $21.4 million upfront payment associated with the license agreement with Orion Corporation during the year ended December 31, 2023, partially offset by (i) increased profitability adjusted for non-cash items, (ii) lower period over period payments associated with the Opana ER® antitrust litigation settlement and (iii) favorable working capital movements, most notably an increase in days payables outstanding.
Cash Flows from Investing Activities
Net cash used in investing activities was $63.0 million for the year ended December 31, 2024 as compared to $69.2 million for the prior year. The year-over-year decrease in net cash used in investing activities was primarily due to $12.0 million in proceeds from the sale of a subsidiary in India to a subsidiary of Kashiv in April 2024 (refer to Note 23. Related Party Transactions for additional information) and a year-over-year decrease in cash paid for intangible assets associated with marketed product licenses, partially offset by higher capital expenditures.
Cash Flows from Financing Activities
Net cash used in financing activities of $211.8 million for the year ended December 31, 2024 was relatively flat compared to the prior year as a decrease in payments associated with refinancing our Term Loan Due 2025 in the prior year of $162.3 million and a reduction in tax distributions paid to non-controlling interests of $51.1 million were offset by a year-over-year decrease in net debt and financing leases of $207.8 million and an increase in employee payroll tax withholding on restricted stock unit vesting of $5.6 million. Refer to Note 15. Debt for additional information about the refinancing our Term Loan Due 2025. The decrease in tax distributions was the result of the Reorganization (refer to Note 21. Stockholders’ (Deficiency) Equity for additional information).
Commitments and Contractual Obligations
Our contractual obligations as of December 31, 2024 were as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Payments Due by Period |
| | Total | | Less
Than 1
Year | | 1-3
Years | | 3-5
Years | | More
Than 5
Years |
Term Loan Due 2025 (1) | | $ | 191,979 | | | $ | 191,979 | | | $ | — | | | $ | — | | | $ | — | |
Interest payments on Term Loan Due 2025 (1) | | 5,271 | | | 5,271 | | | — | | | — | | | — | |
Term Loan Due 2028 (1) | | 2,292,856 | | | 58,791 | | | 117,582 | | | 2,116,483 | | | — | |
Interest payments on Term Loan Due 2028 (1) | | 735,460 | | | 226,929 | | | 436,230 | | | 72,301 | | | — | |
Amended New Revolving Credit Facility (2) | | 100,000 | | | 100,000 | | | — | | | — | | | — | |
Operating lease obligations (3) | | 58,754 | | | 16,914 | | | 24,059 | | | 14,762 | | | 3,019 | |
Financing lease obligation (4) | | 105,632 | | | 7,508 | | | 12,897 | | | 11,320 | | | 73,907 | |
Tax receivable agreement liability (5) | | 53,885 | | | 2,985 | | | 50,900 | | | — | | | — | |
Non-cancelable marketing and royalty obligations (6) | | 17,068 | | | 10,159 | | | 6,909 | | | — | | | — | |
| Total | | $ | 3,560,905 | | | $ | 620,536 | | | $ | 648,577 | | | $ | 2,214,866 | | | $ | 76,926 | |
(1)A description of our Term Loan Due 2025 and Term Loan Due 2028, and related debt service and interest requirements is contained in Note 15. Debt. Interest on our Term Loan Due 2025 and Term Loan Due 2028 was calculated based on applicable rates at December 31, 2024, excluding the impact of our interest rate swap. In January 2025, the Company paid the entire remaining principal balance of $192.0 million outstanding on its Term Loan Due 2025, plus accrued interest thereon of $0.7 million, with $190.0 million of new borrowings under the Amended New Revolving Credit Facility and cash on hand.
(2)A description of our Amended New Revolving Credit Facility is contained in Note 15. Debt. The table assumes the balance outstanding as of December 31, 2024 will be repaid or refinanced by December 31, 2025. The actual balance outstanding may fluctuate significantly in future periods. The interest rate on borrowings under the Amended New Revolving Credit Facility resets every 30, 90 or 180 days based on the term that we select. In January 2025, the Company paid the entire remaining principal balance of $192.0 million outstanding on its Term Loan Due 2025, plus accrued interest thereon of $0.7 million, with $190.0 million of new borrowings under the Amended New Revolving Credit Facility and cash on hand.
(3)Amounts represent future minimum rental payments under non-cancelable facility leases. A discussion of our operating lease obligations is contained in Note 17. Leases.
(4)Amounts primarily represent future minimum rental payments under a non-cancelable financing lease obligation for a production facility in New York. A discussion of our financing lease obligations is contained in Note 17. Leases.
(5)Represents the tax receivable agreement liability as of December 31, 2024. A discussion on our tax receivable agreement is contained in Note 6. Income Taxes.
(6)Represents minimum sales and marketing spending obligations and a minimum royalty obligation.
The foregoing table does not include milestone payments potentially payable by us under our collaboration agreements. Such payments are dependent upon the occurrence of specific and contingent events, and not the passage of time. A discussion of our significant contingent milestones is contained in Note 5. Alliance and Collaboration and Note 23. Related Party Transactions.
Off-Balance Sheet Arrangements
We did not have any off-balance sheet arrangements as of December 31, 2024.
Critical Accounting Policies
Our significant accounting policies are described in Note 2. Summary of Significant Accounting Policies.
Included within these policies are certain policies which contain critical accounting estimates and, therefore, have been deemed to be “critical accounting policies.” Critical accounting estimates are those which require management to make assumptions about matters that were uncertain at the time the estimate was made and for which the use of different estimates, which
reasonably could have been used, or changes in the accounting estimates that are reasonably likely to occur from period to period could have a material impact on our financial condition or results of operations. We have identified the following to be our critical accounting policies: certain sales-related deductions, business combinations, impairment of goodwill and intangible assets, income taxes and contingencies.
Certain Sales-Related Deductions
Our gross product revenue is subject to a variety of deductions, which are estimated and recorded in the same period that the revenue is recognized. Certain deductions represent estimates of rebates related to gross sales for the reporting period and, as such, knowledge and judgment of market conditions and practice are required when estimating the impact of these revenue deductions on gross sales for a reporting period.
Historically, our changes of estimates reflecting actual results or updated expectations have not been material to our overall business. If any of our ratios, factors, assessments, experiences or judgments are not indicative or accurate predictors of our future experience, our results could be materially affected. The sensitivity of our estimates can vary by program, type of customer and geographic location. However, estimates associated with Medicaid rebates and sales returns are most at risk for material adjustment because of the extensive time delay between the recording of the accrual and its ultimate settlement, an interval that can generally range up to one year. Because of this time lag, in any given quarter, our adjustments to actual can incorporate revisions of several prior quarters.
Business Combinations
We account for acquired businesses using the acquisition method of accounting, which requires that assets acquired and liabilities assumed be recorded at the date of acquisition at their respective fair values. The consolidated financial statements and results of operations reflect an acquired business after the completion of the acquisition. The fair value of the consideration paid is assigned to the underlying net assets of the acquired business based on their respective fair values as determined using a market participant concept. Any excess of the purchase price over the fair value of net assets and other identifiable intangible assets acquired is recorded as goodwill.
Intangible assets are amortized over the estimated useful life of the asset. Significant judgments are used in determining the estimated fair values assigned to the assets acquired and liabilities assumed and in determining estimates of useful lives of long-lived assets. Fair value determinations and useful life estimates are based on, among other factors, estimates of expected future net cash flows, estimates of appropriate discount rates used to present value expected future net cash flow streams, the assessment of each asset’s life cycle, competitive trends impacting the asset and each cash flow stream, as well as other factors. These judgments can materially impact the estimates used to allocate acquisition date fair values to assets acquired and liabilities assumed and the future useful lives. For these and other reasons, actual results may vary significantly from estimated results.
Impairment of Goodwill and Intangible Assets
Goodwill
Goodwill, which represents the excess of purchase price over the fair value of net assets acquired, is carried at cost. Goodwill is not amortized; rather, it is subject to a periodic assessment for impairment by applying a fair value based test. We test goodwill for possible impairment annually during the fourth quarter, or whenever events or circumstances indicate that the carrying amount may not be recoverable.
In order to test goodwill for impairment, an entity is permitted to first assess qualitative factors to determine whether a quantitative assessment of goodwill is necessary. The qualitative factors that we consider may include, but are not limited to, general economic conditions, our outlook, market performance of our industry and recent and forecasted financial performance. Further testing is only required if the entity determines, based on the qualitative assessment, that it is more likely than not that a reporting unit’s fair value is less than its carrying amount. Otherwise, no further impairment testing is required. If a quantitative assessment is required, we determine the fair value of our reporting unit using a combination of the income and market approaches. If the net book value of the reporting unit exceeds its fair value, we recognize a goodwill impairment charge for the reporting unit equal to the lesser of (i) the total goodwill allocated to that reporting unit and (ii) the amount by which that reporting unit’s carrying amount exceeds its fair value.
Goodwill is allocated and evaluated for impairment at the reporting unit level, which is defined as an operating segment or one level below an operating segment. Our reportable segments are the same as the respective operating segments and reporting
units. As of December 31, 2024, $366.3 million, $161.7 million, and $69.5 million of goodwill was allocated to our Specialty, Affordable Medicines, and AvKARE segments, respectively. During the fourth quarter of 2024, we tested each of our reporting units for impairment using a quantitative assessment. The determination of fair value in the quantitative assessment required us to make significant estimates and assumptions. These estimates and assumptions primarily included, but were not limited to: the selection of appropriate peer group companies, the discount rate, terminal growth rates, forecasts of revenue, operating income, depreciation and amortization, restructuring charges and capital expenditures. For more information about goodwill, including our interim impairment test, see Note 12. Goodwill and Other Intangible Assets. There was no impairment of goodwill in any reporting unit for the year ended December 31, 2024.
Significant judgment is used in determining the assumptions utilized in our quantitative assessment. Accordingly, any changes in assumptions described above could have a material impact on our consolidated results of operations. Additionally, for each of our reporting units, there are a number of future events and factors that may impact future results and the outcome of subsequent goodwill impairment testing. For a list of these factors, see Item 1A. Risk Factors.
Intangible Assets
We review our long-lived assets, including intangible assets with finite lives, for recoverability whenever events or changes in circumstances indicate that the carrying amount of the assets may not be fully recoverable. We evaluate assets for potential impairment by comparing estimated future undiscounted net cash flows to the carrying amount of the asset. If the carrying amount of the assets exceeds the estimated future undiscounted cash flows, impairment is measured based on the difference between the carrying amount of the assets and fair value which is generally an expected present value cash flow technique. Our policy in determining whether an impairment indicator exists comprises measurable operating performance criteria as well as other qualitative measures. Events giving rise to impairment are an inherent risk in the pharmaceutical industry and cannot be predicted. Factors that we consider in deciding when to perform an impairment review include significant under-performance of a product in relation to expectations, significant negative industry or economic trends, and significant changes or planned changes in our use of the assets. If our assumptions are not correct, there could be an impairment loss in subsequent periods or, in the case of a change in the estimated useful life of the asset, a change in amortization expense.
Intangible assets with indefinite lives, including IPR&D, are tested for impairment if impairment indicators arise and, at a minimum, annually. However, an entity is permitted to first assess qualitative factors to determine if a quantitative impairment test is necessary. Further testing is only required if the entity determines, based on the qualitative assessment, that it is more likely than not that an indefinite-lived intangible asset’s fair value is less than its carrying amount. Otherwise, no further impairment testing is required. The indefinite-lived intangible asset impairment test consists of a one-step analysis that compares the fair value of the intangible asset with its carrying amount. If the carrying amount of an intangible asset exceeds its fair value, an impairment loss is recognized in an amount equal to that excess. We consider many factors in evaluating whether the value of its intangible assets with indefinite lives may not be recoverable, including, but not limited to the discount rate, terminal growth rates, general economic conditions, our outlook and market performance of our industry and recent and forecasted financial performance.
For the year ended December 31, 2024, intangible asset impairment charges were not material. For the year ended December 31, 2023, we recognized $66.9 million of intangible asset impairment charges, of which $36.1 million was recognized in cost of goods sold and $30.8 million was recognized in in-process research and development impairment charges. Cost of sales impairment charges for the year ended December 31, 2023 of $36.1 million primarily related to a reduction in promotional focus on LYVISPAH™ in our Specialty segment, resulting in significantly lower than expected future cash flows. IPR&D impairment charges for the year ended December 31, 2023 of $30.8 million were related to one Affordable Medicines asset and one Specialty asset, both of which experienced adverse clinical trials results in the fourth quarter of 2023 and resulted in significantly lower than expected future cash flows.
Income Taxes
We record valuation allowances against our DTAs when it is more likely than not that all or a portion of a DTA will not be realized. We routinely evaluate the realizability of our DTAs by assessing the likelihood that our DTAs will be recovered based on all available positive and negative evidence, including scheduled reversals of deferred tax liabilities, estimates of future taxable income, tax planning strategies and results of operations. Estimating future taxable income is inherently uncertain and requires judgment. In projecting future taxable income, we consider our historical results and incorporate certain assumptions, including projected new product launches, revenue growth, and operating margins, among others.
A valuation allowance, if needed, reduces DTAs to the amount expected to be realized. When determining the amount of net DTAs that are more likely than not to be realized, we assess all available positive and negative evidence. This evidence
includes, but is not limited to, prior earnings history, projected future earnings, carryback and carry-forward periods and the feasibility of ongoing tax strategies that could potentially enhance the likelihood of the realization of a DTA. The weight given to the positive and negative evidence is commensurate with the extent the evidence may be objectively verified. As such, it is generally difficult for positive evidence regarding projected future taxable income to outweigh objective negative evidence of recent financial reporting losses.
As of December 31, 2024, based upon all available objective and verifiable evidence both positive and negative, including historical levels of pre-tax loss and income both on a consolidated basis and tax reporting entity basis, legislative developments, expectations and risks associated with estimates of future pre-tax income, and prudent and feasible tax planning strategies, we determined that it is more likely than not that we will not realize the benefits of our gross DTAs. Accordingly, as of December 31, 2024, this valuation allowance was $590.3 million and reduced the carrying value of these gross DTAs, net of the impact of the reversal of taxable temporary differences, to zero.
As described in Item 1A. Risk Factors and Note 6. Income Taxes in our consolidated financial statements, we are a party to a TRA under which we are generally required to pay to the Amneal Group 75% of the applicable tax savings, if any, in U.S. federal and state income tax that we are deemed to realize and that are created as a result of tax benefits attributable to payments made under the TRA.
The timing and amount of any payments under the TRA may vary, depending upon a number of factors including the timing and amount of our taxable income, and the tax rate in effect at the time of realization of the our taxable income. Because the Amneal Group has sold or exchanged all of their Amneal Common Units, effective with the Reorganization, there is no longer the associated risk of increased future obligations under the TRA (i.e., there cannot be further sales or exchanges giving rise to increased TRA liability occurring subsequent to December 31, 2023).
The projection of future taxable income involves significant judgment. Actual taxable income may differ materially from our estimates, which could significantly impact the timing and payment of the TRA. As noted above, we have determined it is more-likely-than-not we will be unable to utilize all of our DTAs subject to the TRA; and, as of December 31, 2024 and 2023, we had not recognized the entire contingent liability under the TRA related to the tax savings we may realize from Amneal Common Units sold or exchanged. If utilization of these DTAs becomes more-likely-than-not in the future, at such time, these TRA liabilities (which amount to approximately $133.8 million as of December 31, 2024, as a result of basis adjustments under Internal Revenue Code Section 754) will be recorded through charges to our statements of operations. However, if the tax attributes are not utilized in future years, it is reasonably possible no amounts would be paid under the TRA in excess of the $53.9 million accrued as of December 31, 2024. Should we determine that a DTA with a valuation allowance is realizable in a subsequent period, the related valuation allowance will be reversed and if a resulting TRA payment is determined to be probable, a corresponding TRA liability will be recorded.
Contingencies
We are involved in various litigation, government investigations and other legal proceedings that arise from time to time in the ordinary course of business. Our legal proceedings are complex, constantly evolving and subject to uncertainty. As such, we cannot predict the outcome or impact of our legal proceedings.
While we believe we have valid claims and/or defenses for the matters described in Note 20. Commitments and Contingencies, the nature of litigation is unpredictable and the outcome of the proceedings could include damages, fines, penalties and injunctive or administrative remedies. For any proceedings where losses are probable and reasonably capable of estimation, we accrue for a potential loss. When we have a probable loss for which a reasonable estimate of the liability is a range of losses and no amount within that range is a better estimate than any other amount, we record the loss at the low end of the range. While these accruals have been deemed reasonable by our management, the assessment process relies heavily on estimates and assumptions that may ultimately prove inaccurate or incomplete. Additionally, unforeseen circumstances or events may lead us to subsequently change our estimates and assumptions. The process of analyzing, assessing and establishing reserve estimates relative to legal proceedings involves a high degree of judgment.
The ultimate resolution of any or all claims, legal proceedings or investigations are inherently uncertain and difficult to predict, could differ materially from our estimates and could have a material adverse effect on our results of operations and/or cash flows in any given accounting period, or on our overall financial condition.
For further details, refer to Note 20. Commitments and Contingencies.
Recently Issued Accounting Standards
Recently issued accounting standards are discussed in Note 2. Summary of Significant Accounting Policies.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Our cash is held on deposit in demand accounts at large financial institutions in amounts in excess of the FDIC insurance coverage limit of $250,000 per depositor, per FDIC-insured bank, per ownership category. Our cash equivalents are comprised of highly rated money market funds. We had no short-term investments as of December 31, 2024 and December 31, 2023.
Financial instruments that potentially subject us to concentrations of credit risk consist principally of cash equivalents and accounts receivable. We limit our credit risk associated with cash equivalents by placing investments with high credit quality securities, including U.S. government securities, treasury bills, corporate debt, short-term commercial paper and highly rated money market funds. As discussed in Note 15. Debt, we are party to term loans with an aggregate principal amount of $2.5 billion, the Amended New Revolving Credit Facility and the Amended Rondo Revolving Credit Facility under which loans and letters of credit up to a principal amount of $495.2 million and $28.0 million are available as of December 31, 2024, respectively (principal amount of up to $20.2 million and $18.0 million remain available for letters of credit, respectively). The proceeds for any loans made under our asset backed revolving credit facility are available for capital expenditures, acquisitions, working capital needs and other general corporate purposes. In January 2025, the Company paid the entire remaining principal balance of $192.0 million outstanding on its Term Loan Due 2025, plus accrued interest thereon of $0.7 million, with $190.0 million of new borrowings under the Amended New Revolving Credit Facility and cash on hand.
We limit our credit risk with respect to accounts receivable by performing credit evaluations when deemed necessary. We do not require collateral to secure amounts owed to us by our customers.
By the nature of our global operations, we are exposed to cash flow and earnings fluctuations resulting from foreign exchange rate variation. These exposures are transactional and translational in nature. Since we manufacture and sell our products throughout the world, we believe our foreign currency risk is diversified. Principal drivers of this diversified foreign exchange exposure include the European Euro and the Indian Rupee. Our transactional exposure arises from the purchase and sale of goods and services in currencies other than the functional currency of our operational units. We also have exposure related to the translation of financial statements of our foreign divisions into U.S. dollars, our functional currency. The financial statements of our operations outside the U.S. are measured using the local currency as the functional currency. Adjustments to translate the assets and liabilities of these foreign operations into U.S. dollars are accumulated as a component of other comprehensive loss. Transaction gains and losses are included in the determination of our net loss in our statements of operations. Such foreign currency transaction gains and losses include fluctuations related to long term intercompany loans that are payable in the foreseeable future.
While it is difficult to accurately measure the impact of inflation, we estimate our business did not experience a material increase in costs due to inflation for the year ended December 31, 2024. We do not expect a material impact related to inflation for the year ending December 31, 2025. However, rising inflationary pressures due to higher input costs, including higher material, transportation, labor and other costs, could exceed our expectations and may adversely impact our operating results in future periods.
In the normal course of operations, we are exposed to market risks relating to our long-term debt arising from adverse changes in interest rates. Market risk is defined for these purposes as the potential change in the fair value of a financial asset or liability resulting from an adverse movement in interest rates. Changes in interest rates impact fixed and variable rate debt differently. For fixed rate debt, a change in interest rates will impact only the fair value of the debt, whereas for variable rate debt, a change in the interest rates will impact interest expense and cash flows.
At December 31, 2024 and 2023, we had $2.48 billion and $2.54 billion, respectively, of variable rate debt. Our debt as of December 31, 2024 comprised of our Term Loan Due 2028 with principal outstanding of $2.29 billion and our Term Loan Due 2025 with principal outstanding of $192.0 million. We estimated the fair values of the Term Loan Due 2028 and Term Loan Due 2025 using quoted prices in active markets and yields for the same or similar types of borrowings, taking into account the underlying terms of the debt instruments. At December 31, 2024 and 2023, we estimated the fair value of the Term Loan Due 2025 to be $192.6 million and $190.8 million, respectively. At December 31, 2024 and 2023, we estimated the fair value of the Term Loan Due 2028 to be $2.36 billion and $2.33 billion, respectively.
In October 2019, we entered into an interest rate lock agreement for a total notional amount of $1.3 billion whereby we exchanged floating for fixed rate interest payments for our LIBOR based borrowing under our Term Loan Due 2025 (the
“October 2019 Swap”). On May 31, 2023, we executed an amendment to the interest rate swap that changed the reference rate from LIBOR to the one-month secured overnight financing rate (“SOFR”). On November 14, 2023, in connection with our refinancing of the Term Loan Due 2025 and the New Credit Facility, as defined in Note 15. Debt, we novated the October 2019 Swap to another counterparty and, in connection with such novation, amended the October 2019 Swap. Specifically, the amendments modified (i) the fixed rate payable by the counterparty from 1.3660% to a new fixed rate of 2.7877% and (ii) extended the termination date through May 4, 2027 (i.e., one year before the Term Loan Due 2028 matures). The amendments did not change the notional amount of $1.3 billion. Refer to Note 19. Financial Instruments for additional information. At inception and at year end, we assessed hedge effectiveness and determined it to continue to be highly effective. We also reviewed the credit standing of the counterparty at year end and deemed the counterparties to have the ability to honor their obligations. The fair value of the variable-to-fixed interest rate swap was an asset of $35.9 million as of December 31, 2024. We estimated that a hypothetical 100 basis point increase in the forward one-month SOFR curve would potentially increase the fair value of the variable-to-fixed interest rate swap asset by $27.0 million as of December 31, 2024. We estimated that a hypothetical 100 basis point decrease in the forward one-month SOFR curve would potentially decrease the fair value of the variable-to fixed interest rate swap asset by $28.2 million as of December 31, 2024.
Increases or decreases in interest rates would affect our annual interest expense. Based on the principal amount of the Term Loan Due 2028 outstanding as of December 31, 2024, a hypothetical 100 basis point increase or decrease in interest rates would have affected our annual interest expense by approximately $22.9 million, before the impact of the interest rate lock agreement discussed above. Based on the principal amount of the Term Loan Due 2025 outstanding as of December 31, 2024, a hypothetical 100 basis point increase or decrease in interest rates would have affected our annual interest expense by approximately $1.9 million.
In January 2025, the Company paid the entire remaining principal balance of $192.0 million outstanding on its Term Loan Due 2025, plus accrued interest thereon of $0.7 million, with $190.0 million of new borrowings under the Amended New Revolving Credit Facility and cash on hand.
Item 8. Financial Statements and Supplementary Data
The consolidated financial statements listed in Item 15. Exhibits, Financial Statement Schedules are filed as part of this Annual Report on Form 10-K and incorporated by reference herein.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
Not applicable.
Item 9A. Controls and Procedures
Disclosure Controls and Procedures
We maintain disclosure controls and procedures (as defined in Rule 13a-15(e) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”)) that are designed to ensure information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Co-Chief Executive Officers and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
Our management, with the participation of our Co-Chief Executive Officers and Chief Financial Officer, evaluated the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based upon that evaluation, our Co-Chief Executive Officers and Chief Financial Officer concluded that our disclosure controls and procedures were effective as of December 31, 2024.
Management's Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act). Management conducted an assessment of the effectiveness of our internal control over financial reporting based on the criteria set forth in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework). Based on the assessment, management has concluded that our internal control over financial reporting was effective as of December 31, 2024. Ernst & Young LLP has independently assessed the effectiveness of our internal control over financial reporting and its report is included below.
Changes in Internal Control over Financial Reporting
During the quarter ended December 31, 2024, there were no changes in internal control over financial reporting which materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Limitation on Effective Controls
Management, including our Co-Chief Executive Officers and Chief Financial Officer, does not expect that our disclosure controls and procedures or our system of internal control over financial reporting will prevent or detect all errors and all fraud. A control system, no matter how well designed or operated, can provide only reasonable, but not absolute, assurance that the objectives of the system of internal control are met. The design of our control system reflects the fact that there are resource constraints, and that the benefits of such control system must be considered relative to their costs. Further, because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control failures and instances of fraud, if any, have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the intentional acts of individuals, by collusion of two or more people, or by management override of the controls. The design of any system of controls is also based in part on certain assumptions about the likelihood of future events, and there can be no assurance that the design of any particular control will always succeed in achieving its objective under all potential future conditions.
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Amneal Pharmaceuticals, Inc.
Opinion on Internal Control Over Financial Reporting
We have audited Amneal Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2024, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Amneal Pharmaceuticals, Inc. (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2024, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 2024 and 2023, the related consolidated statements of operations, comprehensive loss, changes in stockholders’ equity (deficiency) and cash flows for each of the three years in the period ended December 31, 2024, and the related notes and our report dated February 28, 2025 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Iselin, New Jersey
February 28, 2025
Item 9B. Other Information
Not applicable.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
PART III.
Item 10. Directors, Executive Officers and Corporate Governance
The information required in this Item 10 will be included in the following sections in the 2025 Proxy Statement, which sections are incorporated in this Item 10 by reference: “Proposal No. 1-Election of Directors”, “Our Management”, “Committees of the Board of Directors”, “Audit Committee”, “Corporate Governance” and, if included in the 2025 Proxy Statement, “Delinquent Section 16(a) Reports”.
Code of Business Conduct for Principal Executive Officer, Principal Financial Officer and Principal Accounting Officer. We have adopted a Code of Business Conduct that applies to all of our employees, officers and directors. The full text of our Code of Business Conduct is available at the investors section of our website, http://investors.amneal.com. We intend to disclose any amendment to, or waiver from, a provision of the Code of Business Conduct that applies to our principal executive officer, principal financial officer or principal accounting officer in the investors section of our website.
Item 11. Executive Compensation
The information required in this Item 11 will be included in the following sections in the 2025 Proxy Statement, which sections are incorporated in this Item 11 by reference to the extent required by this Item 11: “Compensation Discussion and Analysis,” “Executive Compensation,” “Director Compensation,” “The Board’s Role in Risk Oversight,” “Compensation Committee Interlocks and Insider Participation” and “Report of the Compensation Committee.” Notwithstanding the foregoing, the information in the section entitled “Report of the Compensation Committee” and “Pay Versus Performance” is only “furnished” herein and shall not be deemed “filed” for purposes of Section 18 of the Exchange Act.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Except as set forth below, the information required in this Item 12 will be included in the section entitled “Beneficial Ownership” in the 2025 Proxy Statement, which section is incorporated in this Item 12 by reference.
Securities Authorized for Issuance Under Equity Compensation Plans. The following table summarizes information, as of December 31, 2024, relating to the Amneal Pharmaceuticals, Inc. 2018 Incentive Award Plan, which was approved by the Company’s stockholders and which authorizes the grant of stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, other stock or cash-based awards and dividend equivalent awards to employees, non-employee directors and consultants.
Equity Compensation Plan Information
| | | | | | | | | | | | | | | | | | | | |
| Plan Category | | Number of securities to
be issued upon exercise
of outstanding options,
warrants and rights
(a) | | Weighted-average
exercise price of
outstanding options,
warrants and rights
(b) | | Number of securities
remaining available for
future issuance under
equity compensation
plans (excluding
securities reflected in
column (a))
(c) |
| Equity compensation plans approved by security holders | | 19,595,634 | | (1) | $ | 4.86 | | (2) | 23,723,461 | |
| Equity compensation plans not approved by security holders | | — | | | — | | | — | |
| Total | | 19,595,634 | | | $ | 4.86 | | | 23,723,461 | |
(1)Equity compensation plans approved by security holders which are included in column (a) of the table are the amended and restated 2018 Incentive Award Plan (including 1,991,075 shares of Class A Common Stock to be issued upon exercise of outstanding options and 17,576,183 shares of Class A Common Stock to be issued upon vesting and settlement of outstanding restricted stock units (“RSUs”) and market performance-based restricted stock units (“MPRSUs”) subject to continued employment) and 28,376 of options remaining from the Impax Laboratories, Inc (“Impax”) option conversion associated with the acquisition of Impax on May 4, 2018. RSUs and MPRSUs included
in column (a) of the table represent the full number of RSUs and MPRSUs awarded and outstanding whereas the number of shares of Class A Common Stock to be issued upon vesting will be lower than what is reflected on the table because the value of shares required to meet employee tax withholding requirements are not issued.
(2)Column (b) relates to stock options and does not include any exercise price for RSUs and MPRSUs because their value is dependent upon attainment of continued employment or service and they are settled for shares of Class A Common Stock on a one-for-one basis.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required in this Item 13 will be included in the following sections in the 2025 Proxy Statement, which sections are incorporated in this Item 13 by reference: “Certain Related Parties and Related Party Transactions,” “Controlled Company Status” and “Committees of the Board of Directors.”
Item 14. Principal Accounting Fees and Services
The independent registered public accounting firm is Ernst & Young LLP, Iselin, NJ, PCAOB ID 42.
The information required in this Item 14 will be included in the section entitled “Independent Registered Public Accounting Firm Fees” in the 2025 Proxy Statement, which section is incorporated in this Item 14 by reference.
PART IV.
Item 15. Exhibits, Financial Statement Schedules
(a)(1) Consolidated Financial Statements
Index to financial statements and supplementary data filed as part of this Report.
(a)(2) Financial Statement Schedules
All schedules are omitted because they are not required or because the required information is included in the Consolidated Financial Statements or notes thereto.
(a)(3) Exhibits
See the “Exhibit Index” prior to the signature page of this Annual Report on Form 10-K.
Item 16. Form 10-K Summary
None.
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Amneal Pharmaceuticals, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Amneal Pharmaceuticals, Inc. (the Company) as of December 31, 2024 and 2023, the related consolidated statements of operations, comprehensive loss, changes in stockholders’ equity (deficiency) and cash flows for each of the three years in the period ended December 31, 2024, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2024 and 2023, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2024, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2024, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 28, 2025 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
| | | | | | | | |
| | Medicaid Rebates |
| | |
| Description of the Matter | | As discussed in Note 4 to the consolidated financial statements, the Company recognizes revenue from product sales based on amounts due from customers net of allowances for variable consideration, which include, among others, rebates mandated by law under Medicaid and other government pricing programs. The Company includes an estimate of variable consideration in its transaction price at the time of sale, when control of the product transfers to the customer. The Company estimates its Medicaid and other government pricing accruals based on monthly sales, historical experience of claims submitted by the various states and jurisdictions, historical rebate rates and estimated lag time of the rebate invoices. At December 31, 2024, the Company had $135 million in accrued Medicaid and commercial rebates, which are presented within accounts payable and accrued expenses on the consolidated balance sheet. Auditing the allowances for Medicaid rebates was complex and challenging due to the significant estimation involved in management’s assumptions to calculate expected future claims and the amount of projected shipments from wholesalers that will be dispensed to eligible benefit plan participants, as well as the complexity of governmental pricing calculations. The allowances for Medicaid rebates are sensitive to these significant assumptions and calculations. |
| | |
| How We Addressed the Matter in Our Audit | | We obtained an understanding, evaluated the design and tested the operating effectiveness of controls over management’s review of the allowances for Medicaid rebates. For example, we tested controls over management’s review of the significant assumptions including the completeness and accuracy of inputs utilized in significant assumptions as well as controls over management’s review of the application of the government pricing regulations. To test the allowances for Medicaid rebates, we performed audit procedures that included, among others, evaluating the methodologies used and testing the significant assumptions discussed above. We compared the significant assumptions used by management to historical trends, evaluated the change in the accruals from prior periods, and assessed the historical accuracy of management’s estimates against actual results. We also tested the completeness and accuracy of the underlying data used in the Company’s calculations through third-party invoices, claims data and actual cash payments. In addition, we involved our government pricing subject matter professionals to assist in evaluating management’s methodology and calculations used to measure certain estimated rebates. |
| | |
| | | | | | | | |
| | Sales Returns |
| | |
| Description of the Matter | | As discussed in Note 4 of the consolidated financial statements, the Company permits the return of product under certain circumstances, including product expiration, shipping errors, damaged product, and product recalls. The Company accrues for the customer’s right to return as part of its variable consideration at the time of sale, when control of the product transfers to the customer. The Company’s product returns accrual is primarily based on estimates of future product returns, estimates of the level of inventory of its products in the distribution channel that remain subject to returns, estimated lag time of returns and historical return rates. At December 31, 2024, the Company had $160 million in accrued returns allowance, which are presented within accounts payable and accrued expenses on the consolidated balance sheet. Auditing the allowance for sales returns was complex due to the significant estimation required in determining inventory in the distribution channel that will not ultimately be sold to the end user and returned. The allowances for sales returns is sensitive to the level of inventory and turnover of inventory in the distribution channel, which could exceed future market demand and be subject to return. |
| | |
| How We Addressed the Matter in Our Audit | | We obtained an understanding, evaluated the design and tested the operating effectiveness of the Company's controls over the estimation of sales returns. For example, we tested controls over management’s review of the significant assumptions including review of the inventory on hand in the distribution channel, estimated lag time of returns, and the completeness and accuracy of inputs utilized in the estimate of sales returns. To test the estimated sales return reserve, we performed audit procedures that included, among others, testing the historical return rate and estimated lag time of returns and verifying the completeness and accuracy of sales data and sales returns data used in calculating the historical return rate and lag time. In addition, we tested the Company’s quarterly analysis of inventory in the distribution channel and analytically reviewed daily sales at period end for unusual activity. We also performed direct inquiries with management including the Sales and Legal departments, obtained representations confirming key contract terms at period end from the executive sales representatives, and agreed representations obtained to executed contracts and reserve calculations. |
| | |
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2008.
Iselin, New Jersey
February 28, 2025
Amneal Pharmaceuticals, Inc.
Consolidated Statements of Operations
(in thousands, except per share amounts)
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Net revenue | $ | 2,793,957 | | | $ | 2,393,607 | | | $ | 2,212,304 | |
| Cost of goods sold | 1,773,519 | | | 1,573,042 | | | 1,427,596 | |
| Gross profit | 1,020,438 | | | 820,565 | | | 784,708 | |
| Selling, general and administrative | 476,436 | | | 429,675 | | | 399,700 | |
| Research and development | 190,714 | | | 163,950 | | | 195,688 | |
| In-process research and development impairment charges | — | | | 30,800 | | | 12,970 | |
| Intellectual property legal development expenses | 5,845 | | | 3,828 | | | 4,358 | |
| Acquisition, transaction-related and integration expenses | — | | | — | | | 709 | |
| Restructuring and other charges | 2,355 | | | 1,749 | | | 1,421 | |
| Change in fair value of contingent consideration | (930) | | | (14,497) | | | 731 | |
| Insurance recoveries for property losses and associated expenses, net | — | | | — | | | (1,911) | |
| Charges related to legal matters, net | 96,692 | | | 1,824 | | | 269,930 | |
| Other operating income | — | | | (1,138) | | | (3,960) | |
| Operating income (loss) | 249,326 | | | 204,374 | | | (94,928) | |
| Other (expense) income: | | | | | |
| Interest expense, net | (258,595) | | | (210,629) | | | (158,377) | |
| Foreign exchange (loss) gain, net | (6,846) | | | 1,671 | | | (12,364) | |
| Loss on refinancing | — | | | (40,805) | | | (291) | |
| Increase in tax receivable agreement liability | (50,680) | | | (3,124) | | | (631) | |
| Other income, net | 11,782 | | | 8,243 | | | 18,464 | |
| Total other expense, net | (304,339) | | | (244,644) | | | (153,199) | |
| Loss before income taxes | (55,013) | | | (40,270) | | | (248,127) | |
| Provision for income taxes | 18,863 | | | 8,452 | | | 6,662 | |
| Net loss | (73,876) | | | (48,722) | | | (254,789) | |
| Less: Net (income) loss attributable to non-controlling interests | (43,010) | | | (35,271) | | | 125,241 | |
| Net loss attributable to Amneal Pharmaceuticals, Inc. before accretion of redeemable non-controlling interest | (116,886) | | | (83,993) | | | (129,548) | |
| Accretion of redeemable non-controlling interest | — | | | — | | | (438) | |
| Net loss attributable to Amneal Pharmaceuticals, Inc. | $ | (116,886) | | | $ | (83,993) | | | $ | (129,986) | |
| | | | | |
| Net loss per share attributable to Amneal Pharmaceuticals, Inc.'s Class A common stockholders: | | | | | |
| Basic and diluted | $ | (0.38) | | | $ | (0.48) | | | $ | (0.86) | |
| Weighted-average common shares outstanding: | | | | | |
| Basic and diluted | 308,978 | | | 176,136 | | | 150,944 | |
The accompanying notes are an integral part of these consolidated financial statements.
Amneal Pharmaceuticals, Inc.
Consolidated Statements of Comprehensive Loss
(in thousands)
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Net loss | $ | (73,876) | | | $ | (48,722) | | | $ | (254,789) | |
| Less: Net (income) loss attributable to non-controlling interests | (43,010) | | | (35,271) | | | 125,241 | |
| Net loss attributable to Amneal Pharmaceuticals, Inc. before accretion of redeemable non-controlling interest | (116,886) | | | (83,993) | | | (129,548) | |
| Accretion of redeemable non-controlling interest | — | | | — | | | (438) | |
| Net loss attributable to Amneal Pharmaceuticals, Inc. | (116,886) | | | (83,993) | | | (129,986) | |
| Other comprehensive (loss) income: | | | | | |
| Foreign currency translation adjustments arising during the period | (5,788) | | | (1,059) | | | (26,891) | |
| Unrealized (loss) gain on cash flow hedge, net of tax of $0 | (1,168) | | | (48,497) | | | 97,059 | |
| Reclassification of cash flow hedge to earnings, net of tax of $0 | (26,205) | | | (3,366) | | | — | |
| Less: Other comprehensive loss (income) attributable to non-controlling interests | — | | | 9,875 | | | (35,292) | |
| Other comprehensive (loss) income attributable to Amneal Pharmaceuticals, Inc. | (33,161) | | | (43,047) | | | 34,876 | |
| Comprehensive loss attributable to Amneal Pharmaceuticals, Inc. | $ | (150,047) | | | $ | (127,040) | | | $ | (95,110) | |
The accompanying notes are an integral part of these consolidated financial statements.
Amneal Pharmaceuticals, Inc.
Consolidated Balance Sheets
(in thousands) | | | | | | | | | | | |
| December 31,
2024 | | December 31,
2023 |
| Assets | | | |
| Current assets: | | | |
| Cash and cash equivalents | $ | 110,552 | | | $ | 91,542 | |
| Restricted cash | 7,868 | | | 7,565 | |
| Trade accounts receivable, net | 775,731 | | | 613,732 | |
| Inventories | 612,454 | | | 581,384 | |
| Prepaid expenses and other current assets | 80,717 | | | 82,685 | |
| Related party receivables | 484 | | | 955 | |
| Total current assets | 1,587,806 | | | 1,377,863 | |
| Property, plant and equipment, net | 424,908 | | | 447,574 | |
| Goodwill | 597,436 | | | 598,629 | |
| Intangible assets, net | 732,377 | | | 890,423 | |
| Operating lease right-of-use assets | 31,388 | | | 30,329 | |
| Operating lease right-of-use assets - related party | 10,964 | | | 12,954 | |
| Financing lease right-of-use assets | 56,433 | | | 59,280 | |
| Other assets | 60,133 | | | 55,517 | |
| Total assets | $ | 3,501,445 | | | $ | 3,472,569 | |
| Liabilities and Stockholders’ (Deficiency) Equity | | | |
| Current liabilities: | | | |
| Accounts payable and accrued expenses | $ | 735,450 | | | $ | 534,662 | |
| Current portion of liabilities for legal matters | 31,755 | | | 76,988 | |
| Revolving credit facility | 100,000 | | | 179,000 | |
| Current portion of long-term debt, net | 224,213 | | | 34,125 | |
| Current portion of operating lease liabilities | 9,435 | | | 9,207 | |
| Current portion of operating lease liabilities - related party | 3,396 | | | 2,825 | |
| Current portion of financing lease liabilities | 3,211 | | | 2,467 | |
| Related party payables - short term | 22,311 | | | 7,321 | |
| Total current liabilities | 1,129,771 | | | 846,595 | |
| Long-term debt, net | 2,161,790 | | | 2,386,004 | |
| Note payable - related party | — | | | 41,447 | |
| Operating lease liabilities | 24,814 | | | 24,095 | |
| Operating lease liabilities - related party | 9,391 | | | 12,787 | |
| Financing lease liabilities | 56,889 | | | 58,566 | |
| Related party payable - long term | 50,900 | | | 11,776 | |
| Liabilities for legal matters - long term | 85,479 | | | 316 | |
| Other long-term liabilities | 26,949 | | | 29,679 | |
| Total long-term liabilities | 2,416,212 | | | 2,564,670 | |
| Commitments and contingencies (Notes 5, 20 and 23) | | | |
| Redeemable non-controlling interests | 64,974 | | | 41,293 | |
| Stockholders’ (Deficiency) Equity: | | | |
| Preferred stock, $0.01 par value, 2,000 shares authorized at December 31, 2024 and 2023; none issued at both December 31, 2024 and 2023 | — | | — |
| Class A common stock, $0.01 par value, 900,000 shares authorized at both December 31, 2024 and 2023; 309,881 and 306,565 shares issued at December 31, 2024 and 2023, respectively | 3,099 | | | 3,066 | |
| Class B common stock, $0.01 par value, 300,000 shares authorized at December 31, 2024 and 2023; none issued at December 31, 2024 and 2023 | — | | | — | |
| Additional paid-in capital | 560,206 | | | 539,240 | |
| Stockholders' accumulated deficit | (607,062) | | | (490,176) | |
| Accumulated other comprehensive loss | (65,510) | | | (32,349) | |
| Total Amneal Pharmaceuticals, Inc. stockholders' (deficiency) equity | (109,267) | | | 19,781 | |
| Non-controlling interests | (245) | | | 230 | |
| Total stockholders' (deficiency) equity | (109,512) | | | 20,011 | |
| Total liabilities and stockholders' (deficiency) equity | $ | 3,501,445 | | | $ | 3,472,569 | |
The accompanying notes are an integral part of these consolidated financial statements.
Amneal Pharmaceuticals, Inc.
Consolidated Statement of Changes in Stockholders’ Equity (Deficiency)
(in thousands)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| New PubCo | | | | | | | | | | | | | |
| Class A Common Stock | | Additional
Paid-in
Capital | | Stockholders’
Accumulated
Deficit | | Accumulated Other Comprehensive Loss | | Non-
Controlling
Interests | | Total Equity (Deficiency) | | | Redeemable
Non-
Controlling Interests |
| Shares | | Amount | | | | | | | |
| Balance at December 31, 2023 | 306,565 | | | $ | 3,066 | | | $ | 539,240 | | | $ | (490,176) | | | $ | (32,349) | | | $ | 230 | | | $ | 20,011 | | | | $ | 41,293 | |
| Net (loss) income | — | | | — | | | — | | | (116,886) | | | — | | | (475) | | | (117,361) | | | | 43,485 | |
| Foreign currency translation adjustments | — | | | — | | | — | | | — | | | (5,788) | | | — | | | (5,788) | | | | — | |
| Stock-based compensation | — | | | — | | | 27,768 | | | — | | | — | | | — | | | 27,768 | | | | — | |
| Exercise of stock options | 397 | | | 4 | | | 1,150 | | | — | | | — | | | — | | | 1,154 | | | | — | |
| Restricted stock unit vesting, net of shares withheld to cover payroll taxes | 2,919 | | | 29 | | | (7,952) | | | — | | | — | | | — | | | (7,923) | | | | — | |
| Unrealized loss on cash flow hedge, net of tax of $0 | — | | | — | | | — | | | — | | | (1,168) | | | — | | | (1,168) | | | | — | |
| Tax distributions, net | — | | | — | | | — | | | — | | | — | | | — | | | — | | | | (19,804) | |
| Reclassification of cash flow hedge to earnings, net of tax of $0 | — | | | — | | | — | | | — | | | (26,205) | | | — | | | (26,205) | | | | — | |
| Balance at December 31, 2024 | 309,881 | | | $ | 3,099 | | | $ | 560,206 | | | $ | (607,062) | | | $ | (65,510) | | | $ | (245) | | | $ | (109,512) | | | | $ | 64,974 | |
The accompanying notes are an integral part of these consolidated financial statements.
Amneal Pharmaceuticals, Inc.
Consolidated Statement of Changes in Stockholders’ Equity
(in thousands)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Old PubCo | | | New PubCo | | | | | | | | | | | | | |
| Class A Common Stock | | Class B Common Stock | | | Class A Common Stock | | Additional
Paid-in
Capital | | Stockholders’
Accumulated
Deficit | | Accumulated Other Comprehensive Income (Loss) | | Non-
Controlling
Interests | | Total
Equity | | | Redeemable
Non-
Controlling Interests |
| Shares | | Amount | | Shares | | Amount | | | Shares | | Amount | | | | | | | |
| Balance at December 31, 2022 | 151,490 | | | $ | 1,514 | | | 152,117 | | | $ | 1,522 | | | | — | | | $ | — | | | $ | 691,629 | | | $ | (406,183) | | | $ | 9,939 | | | $ | (114,442) | | | $ | 183,979 | | | | $ | 24,949 | |
| Net (loss) income | — | | | — | | | — | | | — | | | | — | | | — | | | — | | | (83,993) | | | — | | | 4,728 | | | (79,265) | | | | 30,543 | |
| Foreign currency translation adjustments | — | | | — | | | — | | | — | | | | — | | | — | | | — | | | — | | | (433) | | | (626) | | | (1,059) | | | | — | |
| Stock-based compensation | — | | | — | | | — | | | — | | | | — | | | — | | | 26,822 | | | — | | | — | | | — | | | 26,822 | | | | — | |
| Exercise of stock options | 148 | | | 1 | | | — | | | — | | | | 15 | | | 1 | | | 447 | | | — | | | 4 | | | (2) | | | 451 | | | | — | |
| Restricted stock unit vesting, net of shares withheld to cover payroll taxes | 2,789 | | | 28 | | | — | | | — | | | | 6 | | | — | | | 2,594 | | | — | | | 77 | | | (5,069) | | | (2,370) | | | | — | |
| Unrealized loss on cash flow hedge, net of tax of $0 | — | | | — | | | — | | | — | | | | — | | | — | | | — | | | — | | | (39,248) | | | (9,249) | | | (48,497) | | | | — | |
| Tax distributions, net | — | | | — | | | — | | | — | | | | — | | | — | | | — | | | — | | | — | | | (56,684) | | | (56,684) | | | | (14,199) | |
| Reclassification of cash flow hedge to earnings, net of tax of $0 | — | | | — | | | — | | | — | | | | — | | | — | | | — | | | — | | | (3,366) | | | — | | | (3,366) | | | | — | |
| Effect of the Reorganization | (154,427) | | | (1,543) | | | (152,117) | | | (1,522) | | | | 306,544 | | | 3,065 | | | (182,252) | | | — | | | 678 | | | 181,574 | | | — | | | | — | |
| Balance at December 31, 2023 | — | | | $ | — | | | — | | | $ | — | | | | 306,565 | | | $ | 3,066 | | | $ | 539,240 | | | $ | (490,176) | | | $ | (32,349) | | | $ | 230 | | | $ | 20,011 | | | | $ | 41,293 | |
The accompanying notes are an integral part of these consolidated financial statements.
Amneal Pharmaceuticals, Inc.
Consolidated Statement of Changes in Stockholders’ Equity
(in thousands)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Old PubCo | | | | | | | | | | | | | |
| Class A Common Stock | | Class B Common Stock | | Additional
Paid-in
Capital | | Stockholders’
Accumulated
Deficit | | Accumulated Other Comprehensive (Loss) Income | | Non-
Controlling
Interests | | Total
Equity | | | Redeemable Non-Controlling Interests |
| Shares | | Amount | | Shares | | Amount | | | | | | | |
| Balance at December 31, 2021 | 149,413 | | | $ | 1,492 | | | 152,117 | | | $ | 1,522 | | | $ | 658,350 | | | $ | (276,197) | | | $ | (24,827) | | | $ | 6,633 | | | $ | 366,973 | | | | $ | 16,907 | |
| Net (loss) income | — | | | — | | | — | | | — | | | — | | | (129,548) | | | — | | | (141,036) | | | (270,584) | | | | 15,795 | |
| Foreign currency translation adjustments | — | | | — | | | — | | | — | | | — | | | — | | | (13,394) | | | (13,497) | | | (26,891) | | | | — | |
| Stock-based compensation | — | | | — | | | — | | | — | | | 31,847 | | | — | | | — | | | — | | | 31,847 | | | | — | |
| Exercise of stock options | 207 | | | 2 | | | — | | | — | | | 615 | | | — | | | — | | | 45 | | | 662 | | | | — | |
| Restricted stock unit vesting, net of shares withheld to cover payroll taxes | 1,870 | | | 20 | | | — | | | — | | | 817 | | | — | | | (110) | | | (4,289) | | | (3,562) | | | | — | |
| Unrealized gain on cash flow hedge, net of tax of $0 | — | | | — | | | — | | | — | | | — | | | — | | | 48,270 | | | 48,789 | | | 97,059 | | | | — | |
| Tax distributions, net | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (10,642) | | | (10,642) | | | | (6,914) | |
| Reclassification of redeemable non-controlling interests | — | | | — | | | — | | | — | | | — | | | (438) | | | — | | | (445) | | | (883) | | | | 883 | |
| Acquisition of non-controlling interest from Puniska Acquisition | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | | (1,722) | |
| Balance at December 31, 2022 | 151,490 | | | $ | 1,514 | | | 152,117 | | | $ | 1,522 | | | $ | 691,629 | | | $ | (406,183) | | | $ | 9,939 | | | $ | (114,442) | | | $ | 183,979 | | | | $ | 24,949 | |
The accompanying notes are an integral part of these consolidated financial statements.
Amneal Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
(in thousands)
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Cash flows from operating activities: | | | | | |
| Net loss | $ | (73,876) | | | $ | (48,722) | | | $ | (254,789) | |
| Adjustments to reconcile net loss to net cash provided by operating activities: | | | | | |
| Depreciation and amortization | 236,191 | | | 229,400 | | | 240,175 | |
| Unrealized foreign currency loss (gain) | 7,191 | | | (768) | | | 15,190 | |
| Amortization of debt issuance costs and discount | 29,097 | | | 11,548 | | | 8,595 | |
| Reclassification of cash flow hedge | (26,205) | | | (3,366) | | | — | |
| Loss on refinancing | — | | | 40,805 | | | 291 | |
| Intangible asset impairment charges | 920 | | | 66,932 | | | 24,081 | |
| Change in fair value of contingent consideration | (930) | | | (14,497) | | | 731 | |
| Stock-based compensation | 27,768 | | | 26,822 | | | 31,847 | |
| Inventory provision | 96,558 | | | 74,686 | | | 51,096 | |
| Insurance recoveries for property and equipment losses | — | | | — | | | (1,000) | |
| Other operating charges and credits, net | 2,453 | | | 9,923 | | | 8,828 | |
| Changes in assets and liabilities: | | | | | |
| Trade accounts receivable, net | (162,637) | | | 126,289 | | | (79,717) | |
| Inventories | (130,530) | | | (126,182) | | | (102,396) | |
| Prepaid expenses, other current assets and other assets | (959) | | | 37,814 | | | 9,882 | |
| Related party receivables | 482 | | | (490) | | | 646 | |
| Accounts payable, accrued expenses and other liabilities | 235,135 | | | (94,446) | | | 109,568 | |
| Related party payables | 54,441 | | | 9,829 | | | 2,072 | |
| Net cash provided by operating activities | 295,099 | | | 345,577 | | | 65,100 | |
| Cash flows from investing activities: | | | | | |
| Purchases of property, plant and equipment | (51,924) | | | (43,216) | | | (46,407) | |
| Acquisition of intangible assets | (14,650) | | | (22,388) | | | (41,800) | |
| Deposits for future acquisition of property, plant, and equipment | (8,416) | | | (3,585) | | | (2,388) | |
| Acquisitions of business | — | | | — | | | (84,714) | |
| Proceeds from insurance recoveries for property and equipment losses | — | | | — | | | 1,000 | |
| Proceeds from sale of subsidiary | 11,994 | | | — | | | — | |
| Net cash used in investing activities | (62,996) | | | (69,189) | | | (174,309) | |
| Cash flows from financing activities: | | | | | |
| Payments of deferred financing, refinancing costs and debt extinguishment costs | (71) | | | (162,415) | | | (1,663) | |
| Payments of principal on debt, revolving credit facility, financing leases and other | (188,918) | | | (414,080) | | | (123,272) | |
| Proceeds from issuance of debt | — | | | 217,732 | | | — | |
| Borrowings on revolving credit facility | 48,000 | | | 219,000 | | | 85,000 | |
| Proceeds from exercise of stock options | 1,154 | | | 451 | | | 662 | |
| Employee payroll tax withholding on restricted stock unit vesting | (7,952) | | | (2,378) | | | (3,571) | |
| Payments of deferred consideration for acquisitions - related party | — | | | — | | | (44,498) | |
| Acquisition of redeemable non-controlling interests | — | | | — | | | (1,722) | |
| Tax distributions to non-controlling interest | (19,804) | | | (70,883) | | | (17,556) | |
| Repayment of related party note | (44,200) | | | — | | | — | |
| Net cash used in financing activities | (211,791) | | | (212,573) | | | (106,620) | |
| Effect of foreign exchange rate on cash | (999) | | | 65 | | | (5,683) | |
| Net increase (decrease) in cash, cash equivalents, and restricted cash | 19,313 | | | 63,880 | | | (221,512) | |
| Cash, cash equivalents, and restricted cash - beginning of period | 99,107 | | | 35,227 | | | 256,739 | |
| Cash, cash equivalents, and restricted cash - end of period | $ | 118,420 | | | $ | 99,107 | | | $ | 35,227 | |
| Cash and cash equivalents - end of period | $ | 110,552 | | | $ | 91,542 | | | $ | 25,976 | |
| Restricted cash - end of period | 7,868 | | | 7,565 | | | 9,251 | |
| Cash, cash equivalents, and restricted cash - end of period | $ | 118,420 | | | $ | 99,107 | | | $ | 35,227 | |
The accompanying notes are an integral part of these consolidated financial statements.
Amneal Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows (continued)
(in thousands)
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Supplemental disclosure of cash flow information: | | | | | |
| Cash paid for interest | $ | 263,518 | | | $ | 192,806 | | | $ | 142,722 | |
| Cash paid, net for income taxes | $ | 15,220 | | | $ | 2,496 | | | $ | 12,649 | |
| | | | | |
| Supplemental disclosure of non-cash investing and financing activity: | | | | | |
| Contingent consideration for acquisition | $ | — | | | $ | — | | | $ | 8,796 | |
| Payable for acquisition of product rights and licenses | $ | 3,900 | | | $ | 2,100 | | | $ | — | |
The accompanying notes are an integral part of these consolidated financial statements.
Amneal Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
1. Nature of Operations
Amneal Pharmaceuticals, Inc. (the “Company”) is a global biopharmaceutical company that develops, manufactures, markets, and distributes a diverse portfolio of essential medicines. The Company’s Affordable Medicines segment includes retail generics, injectables, and biosimilars. In its Specialty segment, the Company offers a portfolio of branded pharmaceuticals focused primarily on central nervous system and endocrine disorders. Through the Company’s AvKARE segment, it is a distributor of pharmaceuticals and other products for the U.S. federal government, retail, and institutional markets. The Company operates principally in the United States (“U.S.”), India, and Ireland. Refer to Note 25. Segment Information for an overview of our segments, including the change in segment name from “Generics” to “Affordable Medicines” during the fourth quarter of 2024.
The Company is a holding company, whose principal assets are common units (“Amneal Common Units”) of Amneal Pharmaceuticals, LLC (“Amneal”). Immediately prior to the Reorganization (as defined herein), the Company held 50.4% of the Amneal Common Units and the group, together with their affiliates and certain assignees, who owned Amneal when it was a private company (the “Members” or the “Amneal Group”) held the remaining 49.6%. On November 7, 2023, the Company implemented a plan pursuant to which the Company and Amneal reorganized and simplified the Company’s corporate structure by eliminating its umbrella partnership-C-corporation structure and converting to a more traditional C-corporation structure whereby all stockholders hold their voting and economic interests directly through the public company (the “Reorganization”). Effective with the Reorganization, the Company holds 100% of the Amneal Common Units.
Following the implementation of the Reorganization, Amneal Pharmaceuticals, Inc. (“Old PubCo”) became a wholly owned subsidiary of a new holding company, Amneal NewCo Inc. (“New PubCo”), which replaced Old PubCo as the public company trading on the New York Stock Exchange under Old PubCo’s ticker symbol “AMRX.” In addition, New PubCo changed its name to “Amneal Pharmaceuticals, Inc.” and Old PubCo changed its name to “Amneal Intermediate, Inc.” In connection with the Reorganization, holders of shares of Class A common stock, par value $0.01 per share, of Old PubCo (“Old PubCo Class A Common Stock”) ceased to hold such shares and received an equivalent number of shares of Class A common stock, par value $0.01 per share, of New PubCo that have the same voting and economic rights as Old PubCo Class A Common Stock. Additionally, holders of shares of Class B common stock, par value $0.01 per share, of Old PubCo (“Old PubCo Class B Common Stock”), ceased to hold such shares and received an equivalent number of shares of Class A common stock, par value $0.01 per share, of New PubCo that have the same voting and economic rights as Old PubCo Class A Common Stock. All outstanding shares of Old PubCo Class B Common Stock were surrendered and canceled. Accordingly, upon consummation of the Reorganization, Old PubCo stockholders automatically became stockholders of New PubCo, on a one-for-one basis, with the same number and ownership percentage of shares they held in Old PubCo immediately prior to the effective time of the Reorganization. On December 27, 2023, the Company voluntarily withdrew the listing of its Class A common stock from the New York Stock Exchange and transferred the listing to the Nasdaq Stock Market LLC under the same name and ticker symbol.
2. Summary of Significant Accounting Policies
Accounting Principles
The financial statements and accompanying notes are prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”). All intercompany accounts and transactions have been eliminated.
Principles of Consolidation
The consolidated financial statements include the accounts of all entities controlled by the Company, including Amneal and its subsidiaries, through the Company’s direct or indirect ownership of a majority voting interest. The Company records non-controlling interests for the portion of its subsidiaries’ economic interests that it does not hold.
Although the Company had a minority economic interest in Amneal prior to March 31, 2023, it was Amneal’s sole managing member (and it continues to be the sole managing member), having the sole voting power to make all of Amneal’s business decisions and control its management. Therefore, the Company also consolidated the financial statements of Amneal and its subsidiaries for all periods prior to the Reorganization.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires the Company's management to make estimates and assumptions that affect the reported financial position at the date of the financial statements and the reported results of operations during the reporting period. Such estimates and assumptions affect the reported amounts of assets, liabilities, revenues and expenses, and disclosure of contingent assets and liabilities in the consolidated financial statements and accompanying notes. The following are some, but not all, of such estimates: the determination of chargebacks, sales returns, rebates, billbacks, valuation of intangible and other assets acquired in business combinations, allowances for accounts receivable, accrued liabilities, liabilities for legal matters, contingent liabilities, initial and subsequent valuation of contingent consideration recognized in business combinations, stock-based compensation, valuation of inventory balances, the determination of useful lives for product rights and the assessment of expected cash flows used in evaluating goodwill and other long-lived assets for impairment. Actual results could differ from those estimates.
Revenue Recognition
When assessing its revenue recognition, the Company performs the following five steps: (i) identify the contract with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the entity satisfies the performance obligation. The Company recognizes revenue when it transfers control of its products to customers, in an amount that reflects the consideration to which the Company expects to be entitled to receive in exchange for those products.
From time to time, the Company may enter into arrangements where it licenses certain products to a third-party distributor. Licensing arrangement performance obligations generally include intellectual property (“IP”) rights and research and development (“R&D”) and contract manufacturing services. The Company accounts for IP rights and services separately if they are distinct. The consideration is allocated between IP rights and services based on their relative stand-alone selling prices.
Revenue for distinct IP rights is accounted for based on the nature of the promise to grant the license. In determining whether the Company’s promise is to provide a right to access its IP or a right to use its IP, the Company considers the nature of the IP to which the customer will have rights. IP is either functional IP which has significant standalone functionality or symbolic IP which does not have significant standalone functionality. Revenue from functional IP is recognized at the point in time when control of the distinct license is transferred to the customer. Revenue from symbolic IP is recognized over the access period to the Company’s IP.
Revenue from sales-based milestones and royalties promised in exchange for a license of IP is recognized only when, or as, the later of subsequent sale or the performance obligation to which some or all of the sales-based royalty has been allocated, is satisfied.
For further details on the Company’s revenue recognition policies, refer to Note 4. Revenue Recognition.
Stock-Based Compensation
The Company’s stock-based compensation consists of stock options, restricted stock units (“RSUs”) and market performance-based restricted stock units (“MPRSUs”) awarded to employees and non-employee directors. Stock options are measured at their fair value on the grant date or date of modification, as applicable. RSUs, including MPRSUs, are measured at the stock price on the grant date or date of modification, as applicable. The Company recognizes compensation expense on a straight-line basis over the requisite service and/or performance period, as applicable. Forfeitures of awards are accounted for as a reduction in stock-based compensation expense in the period such awards are forfeited. The Company's policy is to issue new shares upon option exercises and the vesting of RSUs and MPRSUs.
Contingent consideration
Business acquisitions may include future payments that are contingent upon the occurrence of certain pharmaceutical regulatory milestones or net sales of pharmaceutical products. For acquisitions that are accounted for as a business combination, the obligations for such contingent consideration payments are recorded at fair value on the acquisition date. For contingent milestone payments, the Company uses a probability-weighted income approach utilizing an appropriate discount rate. For contingent tiered royalties on net sales, the Company uses a Monte Carlo simulation model. Contingent consideration liabilities are revalued to fair value at the end of each reporting period. Changes in the fair value of contingent consideration, other than changes due to payments, are recognized as a gain or loss and recorded within change in fair value of contingent consideration
in the consolidated statements of operations. Refer to Note 3. Acquisition and Note 18. Fair Value Measurements for additional information.
Foreign Currencies
The Company has operations in the U.S., India, Ireland, and other foreign jurisdictions. Generally, the Company’s foreign operating subsidiaries’ functional currency is the local currency. The results of its non-U.S. dollar based operations are translated to U.S. dollars at the average exchange rates during the period. Assets and liabilities are translated at the rate of exchange prevailing on the balance sheet date. Translation adjustments are included in accumulated other comprehensive (loss) income and non-controlling interests in the consolidated balance sheets and are included in comprehensive loss. Transaction gains and losses are included in net loss in the Company’s consolidated statements of operations as a component of foreign exchange (loss) gain, net. Such foreign currency transaction gains and losses include fluctuations related to long-term intercompany loans that are payable in the foreseeable future. Translation gains and losses on intercompany balances of a long-term investment nature are included in foreign currency translation adjustments in accumulated other comprehensive (loss) income and non-controlling interests, and comprehensive loss.
Business Combinations
Business combinations are accounted for using the acquisition method of accounting. Under the acquisition method, the acquiring entity in a business combination records the assets acquired and liabilities assumed at the date of acquisition at their fair values. Any excess of the purchase price over the fair value of net assets and other identifiable intangible assets acquired is recorded as goodwill. Acquisition-related costs, primarily professional fees, are expensed as incurred.
Cash and Cash Equivalents
Cash and cash equivalents consist of cash on deposit and highly liquid investments with original maturities of three months or less. A portion of the Company’s cash flows are derived outside the U.S. As a result, the Company is subject to market risk associated with changes in foreign exchange rates. The Company maintains cash balances at both U.S.-based and international-based commercial banks. At various times during the year, cash balances in the U.S. may exceed amounts that are insured by the Federal Deposit Insurance Corporation.
Restricted Cash
At December 31, 2024 and 2023, the Company had restricted cash balances of $7.9 million and $7.6 million, respectively, in its bank accounts primarily related to the purchase of certain land and equipment in India.
Accounts Receivable and Allowance for Credit Losses
Trade accounts receivable are recorded at the invoiced amount and do not bear interest. The Company limits its credit risk with respect to accounts receivable by performing credit evaluations when deemed necessary. The Company does not require collateral to secure amounts owed to it by its customers.
Trade accounts receivable are stated at their net realizable value. The allowance for credit losses reflects the best estimate of expected credit losses of the accounts receivable portfolio determined on the basis of historical experience, current information, and forecasts of future economic conditions. The Company determines its allowance methodology by pooling receivable balances at the customer level. The Company considers various factors, including its previous loss history, individual credit risk associated to each customer, and the current and future condition of the general economy. These credit risk factors are monitored on a quarterly basis and updated as necessary. To the extent that any individual debtor is identified whose credit quality has deteriorated, the Company establishes allowances based on the individual risk characteristics of such customer. The Company makes concerted efforts to collect all outstanding balances due from customers; however, account balances are charged off against the allowance when management believes it is probable the receivable will not be recovered. The Company does not have any off-balance-sheet credit exposure related to customers.
Chargebacks Received from Manufacturers
When a sale occurs on a contracted item, the difference between the cost the Company pays to the manufacturer of that item and the contract price that the end customer has with the manufacturer is rebated to the Company by the manufacturer as a chargeback. Chargebacks are recorded as a reduction to cost of sales and either a reduction in the amount due to the manufacturer (if there is a right of offset) or as a receivable from the manufacturer.
Inventories
Inventories consist of finished goods held for sale, raw materials, and work in process. Inventories are stated at net realizable value, with cost determined using the first-in, first-out method. Adjustments for excess and obsolete inventories are established based upon historical experience and management’s assessment of current product demand. These assessments include inventory obsolescence based on its expiration date, damaged or rejected product, and slow-moving products.
Property, Plant, and Equipment
Property, plant, and equipment are stated at historical cost less accumulated depreciation. Depreciation expense is computed primarily using the straight-line method over the estimated useful lives of the assets, which are as follows:
| | | | | | | | |
| Asset Classification | | Estimated Useful Life |
| Buildings | | 30 years |
| Computer equipment | | 5 years |
| Furniture and fixtures | | 7 years |
| Leasehold improvements | | Shorter of asset's useful life or remaining life of lease |
| Machinery and equipment | | 5 - 10 years |
| Vehicles | | 5 years |
Upon retirement or disposal, the cost of the asset disposed and the accumulated depreciation are removed from the accounts, and any gain or loss is reflected as part of operating income (loss) in the period of disposal. Expenditures that significantly increase value or extend useful lives of property, plant, and equipment are capitalized, whereas those for normal maintenance and repairs are expensed. The Company capitalizes interest on borrowings during the construction period of major capital projects as part of the related asset and amortizes the capitalized interest into earnings over the related asset’s remaining useful life.
Leases
All significant lease arrangements are recognized as right-of-use (“ROU”) assets and lease liabilities at lease commencement. ROU assets represent the Company’s right to use an underlying asset for the lease term, and lease liabilities represent its obligation to make lease payments arising from the lease. ROU assets and liabilities are recognized at the commencement date based on the present value of the future lease payments using the Company's incremental borrowing rate.
Operating lease expense is recognized on a straight-line basis over the lease term. At each balance sheet date, operating and financing lease liabilities continue to represent the present value of the future payments. Financing lease ROU assets are expensed using the straight-line method, unless another basis is more representative of the pattern of economic benefit, to lease expense. Interest on financing lease liabilities is recognized in interest expense.
Leases with an initial term of 12 months or less (short-term leases) are not recognized in the balance sheet and the related lease payments are recognized as incurred over the lease term. The Company separates lease and non-lease components. A portion of the Company's real estate leases are subject to periodic changes in the Consumer Price Index (“CPI”). The changes to the CPI are treated as variable lease payments and recognized in the period in which the obligation for those payments was incurred.
For further details regarding the Company's leases, refer to Note 17. Leases.
In-Process Research and Development
The fair value of in-process research and development (“IPR&D”) acquired in a business combination is determined based on the present value of each research project’s projected cash flows using an income approach. Revenues are estimated based on relevant market size and growth factors, expected industry trends, individual project life cycles and the life of each research project’s underlying marketability. In determining the fair value of each research project, expected cash flows are adjusted for certain risks of completion, including technical and regulatory risk.
The value attributable to IPR&D projects at the time of acquisition is capitalized as an indefinite-lived intangible asset and tested for impairment until the project is completed or abandoned. Upon completion of the project, the indefinite-lived intangible asset is then accounted for as a finite-lived intangible asset and amortized over the estimated useful life of the asset based on the pattern in which the economic benefits are expected to be consumed or otherwise used up or, if that pattern is not
readily determinable, on a straight-line basis. If the project is abandoned, the indefinite-lived intangible asset is charged to expense.
Intangible assets with indefinite lives, including IPR&D, are tested for impairment if impairment indicators arise and, at a minimum, annually. However, an entity is permitted to first assess qualitative factors to determine if a quantitative impairment test is necessary. Further testing is only required if the entity determines, based on the qualitative assessment, that it is more likely than not that an indefinite-lived intangible asset’s fair value is less than its carrying amount. Otherwise, no further impairment testing is required. The indefinite-lived intangible asset impairment test consists of a one-step analysis that compares the fair value of the intangible asset with its carrying amount. If the carrying amount of an intangible asset exceeds its fair value, an impairment loss is recognized in an amount equal to that excess. The Company considers many factors in evaluating whether the value of its intangible assets with indefinite lives may not be recoverable, including, but not limited to, expected growth rates, the cost of equity and debt capital, general economic conditions, the Company’s outlook and market performance of the Company’s industry and recent and forecasted financial performance.
Goodwill
Goodwill, which represents the excess of purchase price over the fair value of net assets acquired, is carried at cost. Goodwill is not amortized; rather, it is subject to a periodic assessment for impairment by applying a fair value based test. The Company reviews goodwill for possible impairment annually during the fourth quarter, or whenever events or circumstances indicate that the carrying amount may not be recoverable.
In order to test goodwill for impairment, an entity is permitted to first assess qualitative factors to determine whether a quantitative assessment of goodwill is necessary. The qualitative factors considered by the Company may include, but are not limited to, general economic conditions, the Company’s outlook, market performance of the Company’s industry and recent and forecasted financial performance. Further testing is only required if the entity determines, based on the qualitative assessment, that it is more likely than not that a reporting unit’s fair value is less than its carrying amount. Otherwise, no further impairment testing is required. If a quantitative assessment is required, the Company determines the fair value of its reporting unit using a combination of the income and market approaches. If the net book value of the reporting unit exceeds its fair value, the Company recognizes a goodwill impairment charge for the reporting unit equal to the lesser of (i) the total goodwill allocated to that reporting unit and (ii) the amount by which that reporting unit’s carrying amount exceeds its fair value. See Note 12. Goodwill and Other Intangible Assets, for further discussion of the Company's qualitative and quantitative assessments of goodwill.
Assumptions and estimates used in the evaluation of impairment may affect the carrying value of long-lived assets, which could result in impairment charges in future periods. Such assumptions include projections of future cash flows and the current fair value of the asset.
Amortization of Intangible Assets with Finite Lives
Intangible assets, other than indefinite-lived intangible assets, are amortized over the estimated useful life of the asset based on the pattern in which the economic benefits are expected to be consumed or otherwise used up or, if that pattern is not readily determinable, on a straight-line basis. The useful life is the period over which the assets are expected to contribute directly or indirectly to future cash flows. Intangible assets are not written-off in the period of acquisition unless they become impaired during that period.
The Company regularly evaluates the remaining useful life of each intangible asset that is being amortized to determine whether events and circumstances warrant a revision to the remaining period of amortization. If the estimate of the intangible asset’s remaining useful life is changed, the remaining carrying amount of the intangible asset is amortized prospectively over that revised remaining useful life. See Note 12. Goodwill and Other Intangible Assets for further discussion of the Company’s intangible assets.
Impairment of Long-Lived Assets (Including Intangible Assets with Finite Lives)
The Company reviews its long-lived assets, including intangible assets with finite lives, for recoverability whenever events or changes in circumstances indicate that the carrying amount of the assets may not be fully recoverable. The Company evaluates assets for potential impairment by comparing estimated future undiscounted net cash flows to the carrying amount of the asset. If the carrying amount of the assets exceeds the estimated future undiscounted cash flows, impairment is measured based on the difference between the carrying amount of the assets and fair value, which is generally an expected present value cash flow technique. Management’s policy in determining whether an impairment indicator exists comprises measurable operating
performance criteria as well as other qualitative measures. See Note 12. Goodwill and Other Intangible Assets for further discussion of the Company’s assessment of intangible asset impairments.
Financial Instruments
The Company minimizes its risks from interest fluctuations through its normal operating and financing activities and, when deemed appropriate through the use of derivative financial instruments. Derivative financial instruments are used to manage risk and are not used for trading or other speculative purposes. The Company does not use leveraged derivative financial instruments. Derivative financial instruments that qualify for hedge accounting must be designated and effective as a hedge of the identified risk exposure at the inception of the contract. Accordingly, changes in fair value of the derivative contract must be highly correlated with changes in fair value of the underlying hedged item at inception of the hedge and over the life of the hedge contract.
All derivatives are recorded on the balance sheet as assets or liabilities and measured at fair value. For derivatives designated as cash flow hedges, the effective portion of the changes in fair value of the derivatives are recorded in accumulated other comprehensive (loss) income net of income taxes and subsequently amortized as an adjustment to interest expense over the period during which the hedged forecasted transaction affects earnings, which is when the Company recognizes interest expense on the hedged cash flows. Cash flows of such derivative financial instruments are classified consistent with the underlying hedged item.
Highly effective hedging relationships that use interest rate swaps as the hedging instrument and that meet criteria under ASC 815, Derivatives and Hedging (“ASC 815”), may qualify for the “short-cut method” of assessing effectiveness. The short-cut method allows the Company to make the assumption of no ineffectiveness, which means that the change in fair value of the hedged item can be assumed to be equal to the change in fair value of the derivative. Unless critical terms change, no further evaluation of effectiveness is performed for these hedging relationships unless a critical term is changed.
For a hedging relationship that does not qualify for the short-cut method, the Company measures its effectiveness using the “hypothetical derivative method”, in which the change in fair value of the hedged item must be measured separately from the change in fair value of the derivative. At inception and quarterly thereafter, the Company formally assesses whether the derivatives that are used in hedging transactions are highly effective in offsetting changes in the fair value or cash flows of the hedged item. The Company compares the change in the fair value of the actual interest rate derivative to the change in the fair value of a hypothetical interest rate derivative with critical terms that match the hedged interest rate payments. After the initial quantitative assessment, this analysis is performed on a qualitative basis and, if it is determined that the hedging relationship was and continues to be highly effective, no further analysis is required.
All components of each derivative financial instrument's gain or loss are included in the assessment of hedge effectiveness. If it is determined that a derivative ceases to be a highly effective hedge, the Company discontinues hedge accounting and any deferred gains or losses related to a discontinued cash flow hedge shall continue to be reported in accumulated other comprehensive (loss) income net of income taxes, unless it is probable that the forecasted transaction will not occur. If it is probable that the forecasted transaction will not occur by the originally specified time period, the Company discontinues hedge accounting, and any deferred gains or losses reported in accumulated other comprehensive (loss) income are reclassified into earnings immediately.
The Company is subject to credit risk as a result of nonperformance by counterparties to the derivative agreements. Upon inception and quarterly thereafter, the Company makes judgments on each counterparty’s creditworthiness for nonperformance by counterparties.
Income Taxes
The Company accounts for income taxes in accordance with ASC 740, Accounting for Income Taxes (“ASC 740”), which requires the recognition of tax benefits or expenses on temporary differences between the financial reporting and tax bases of its assets and liabilities by applying the enacted tax rates in effect for the year in which the differences are expected to reverse. Such net tax effects on temporary differences are reflected on the Company’s consolidated balance sheets as deferred tax assets and liabilities. Deferred tax assets are reduced by a valuation allowance when the Company believes that it is more-likely-than-not that some portion or all of the deferred tax assets will not be realized.
ASC 740-10 prescribes a two-step approach for the recognition and measurement of tax benefits associated with the positions taken or expected to be taken in a tax return that affect amounts reported in the financial statements. The Company has reviewed and will continue to review the conclusions reached regarding uncertain tax positions, which may be subject to review
and adjustment at a later date based on ongoing analyses of tax laws, regulations and interpretations thereof. To the extent that the Company’s assessment of the conclusions reached regarding uncertain tax positions changes as a result of the evaluation of new information, such change in estimate will be recorded in the period in which such determination is made. The Company reports income tax-related interest and penalties relating to uncertain tax positions, if applicable, as a component of income tax expense.
Comprehensive Loss
Comprehensive loss includes net loss and all changes in stockholders’ equity (except those arising from transactions with stockholders) including foreign currency translation adjustments resulting from the consolidation of foreign subsidiaries’ financial statements and unrealized (losses) gains on cash flows hedges, net of income taxes.
Research and Development
R&D activities are expensed as incurred. R&D expenses primarily consist of direct and allocated expenses incurred with the process of formulation, clinical research, and validation associated with new product development. Upfront and milestone payments made to third parties in connection with R&D collaborations are expensed as incurred up to the point of regulatory approval or when there is no alternative future use.
Intellectual Property Legal Development Expenses
The Company expenses external IP legal development expenses as incurred. These costs relate to legal challenges of innovator’s patents for invalidity or non-infringement, which are customary in the generic pharmaceutical industry, and are incurred predominately during development of a product and prior to regulatory approval. Associated costs include, but are not limited to, formulation assessments, patent challenge opinions and strategy, and litigation expenses to defend the IP supporting the Company's regulatory filings.
Shipping Costs
The Company records the costs of shipping product to its customers as a component of selling, general, and administrative expenses as incurred. Shipping costs were $18.9 million, $21.7 million and $18.7 million for the years ended December 31, 2024, 2023 and 2022, respectively.
Advertising Costs
Advertising costs are expensed as incurred. Advertising costs are included in selling, general and administrative expenses and were $34.4 million, $12.4 million, and $16.8 million for the years ended December 31, 2024, 2023 and 2022, respectively.
Recently Adopted Accounting Pronouncements
In November 2023, the Financial Accounting Standards Board (“FASB”) issued ASU 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures (“ASU 2023-07”), which provides improvements to reportable segment disclosure requirements, primarily through enhanced disclosures about significant segment expenses. ASU 2023-07 requires disclosures to include the title and position of the chief operating decision maker (“CODM”), significant segment expenses that are regularly provided to the CODM, a description of other segment items by reportable segment, and any additional measures of a segment’s profit or loss used by the CODM when deciding how to allocate resources. ASU 2023-07 also requires all annual disclosures currently required by Topic 280 to be included in interim periods. The Company adopted the annual and interim disclosure requirements of ASU 2023-07 effective December 31, 2024 (refer to Note 25. Segment Information). The adoption of this ASU only affects the Company’s disclosures, with no impact to its financial condition and results of operations.
Recently Issued Accounting Pronouncements
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures (“ASU 2023-09”), which enhances the transparency and usefulness of income tax disclosures. ASU 2023-09 requires that public business entities on an annual basis disclose specific categories in the rate reconciliation and provide additional information for reconciling items that meet a quantitative threshold. The update is effective for fiscal years beginning after December 15, 2024, with early adoption permitted for annual financial statements that have not yet been issued or made available for issuance. The Company is currently evaluating the impact this guidance will have on its consolidated financial statements.
In November 2024, the FASB issued ASU 2024-03, Income Statement - Reporting Comprehensive Income - Expense Disaggregation Disclosures (Subtopic 220-40): Disaggregation of Income Statement Expenses (“ASU 2024-03”), which requires a public business entity to provide disaggregated disclosures, in the notes to the financial statements, of certain categories of expenses that are included in expense line items on the face of the income statement. ASU 2024-03 is effective for fiscal years beginning after December 15, 2026, and interim reporting periods beginning December 15, 2027, with early adoption permitted. Upon adoption, ASU 2024-03 may be applied prospectively for reporting periods after the effective date or retrospectively to any or all prior periods presented in the financial statements. The Company is currently evaluating the impact this guidance will have on its consolidated financial statements.
Reclassification
The prior period balances related to the TRA (as defined in Note 6. Income Taxes) of $3.1 million and $0.6 million, formerly included in “Other income, net”, for the year ended December 31, 2023 and 2022, respectively, have been reclassified to the income statement caption “Increase in tax receivable agreement liability” to conform to the current period presentation in the consolidated statements of operations. This reclassification did not impact total other expense, net or net income.
The prior period balance related to long-term liabilities for legal matters of $0.3 million, formerly included in “Other long-term liabilities” as of December 31, 2023, has been reclassified to the balance sheet caption “Liabilities for legal matters - long term” to conform to the current period presentation in the consolidated balance sheets. This reclassification did not impact total long-term liabilities or total liabilities.
3. Acquisition
Saol Baclofen Franchise Acquisition
On December 30, 2021, the Company entered into an asset purchase agreement with certain entities affiliated with Saol International Limited (collectively, “Saol”), a private specialty pharmaceutical company, pursuant to which it agreed to acquire Saol’s baclofen franchise, including Lioresal®, LYVISPAH™, and a pipeline product under development (the “Saol Acquisition”). The Saol Acquisition expanded the Company’s commercial institutional and specialty portfolio in neurology while adding commercial infrastructure in advance of its entry into the biosimilar institutional market. The transaction closed on February 9, 2022.
Consideration for the Saol Acquisition included $84.7 million, paid at closing with cash on hand, and contingent royalty payments based on annual net sales for certain acquired assets, beginning in June 2023. Cash paid at closing included $1.1 million for inventory acquired in excess of the normalized level, as defined in the asset purchase agreement (working capital adjustment).
For the year ended December 31, 2022, the Company incurred $0.1 million in transaction costs associated with the Saol Acquisition, which was recorded in acquisition, transaction-related and integration expenses.
The Saol Acquisition was accounted for under the acquisition method of accounting, with Amneal as the accounting acquirer. The purchase price was calculated as follows (in thousands):
| | | | | |
| Cash | $ | 84,714 | |
Contingent consideration (royalties) (1) | 8,796 | |
| Fair value of consideration transferred | $ | 93,510 | |
(1)The estimated fair value of contingent consideration on the acquisition date of $8.8 million was based on significant Level 3 inputs that were not observable in the market. Key assumptions included the discount rate, projected year of payments and expected net product sales. Refer to Note 18. Fair Value Measurements for additional information on the methodology and determination of this liability.
From the acquisition date of February 9, 2022 to December 31, 2022, the Saol Acquisition contributed net revenue and an operating loss of $19.8 million and $7.1 million, respectively.
Acquisition, Transaction-Related and Integration Expenses
For the year ended December 31, 2022, acquisition, transaction-related and integration expenses of $0.7 million primarily consisted of professional services fees associated with the Saol Acquisition.
4. Revenue Recognition
Pharmaceutical Product Sales
Performance Obligations
The Company’s performance obligation is the supply of finished pharmaceutical products to its customers. The Company’s customers consist primarily of major wholesalers, distributors, retail pharmacies, managed care organizations, purchasing co-ops, hospitals, government agencies, institutions and pharmaceutical companies. The Company’s customer contracts generally consist of both a master agreement, which is signed by the Company and its customer, and a customer submitted purchase order, which is governed by the terms and conditions of the master agreement. Customers purchase product by direct channel sales from the Company or by indirect channel sales through various distribution channels.
Revenue is recognized when the Company transfers control of its products to the customer, which typically occurs at a point-in-time, upon shipment or delivery. Substantially all of the Company’s net revenues relate to products which are transferred to the customer at a point-in-time.
The Company offers standard payment terms to its customers and has elected the practical expedient to not adjust the promised amount of consideration for the effects of a significant financing, since the period between when the Company transfers the product to the customer and when the customer pays for that product is one year or less. Taxes collected from customers relating to product sales and remitted to governmental authorities are excluded from revenues. The consideration amounts due from customers as a result of product sales are subject to variable consideration, as described further below.
The Company offers standard product warranties which provide assurance that the product will function as expected and in accordance with specifications. Customers cannot purchase warranties separately and these warranties do not give rise to a separate performance obligation.
The Company permits the return of product under certain circumstances, mainly upon product expiration, instances of shipping errors or where product is damaged in transit. The Company accrues for the customer’s right to return as part of its variable consideration. See below for further details.
Variable Consideration
The Company includes an estimate of variable consideration in its transaction price at the time of sale, when control of the product transfers to the customer. Variable consideration includes but is not limited to: chargebacks, distribution fees, rebates, group purchasing organization (“GPO”) fees, prompt payment (cash) discounts, consideration payable to the customer, billbacks, Medicaid and other government pricing programs, price protection and shelf stock adjustments, sales returns, and profit shares.
The Company assesses whether or not an estimate of its variable consideration is constrained and has determined that the constraint does not apply, since it is probable that a significant reversal in the amount of cumulative revenue will not occur in the future when the uncertainty associated with the variable consideration is subsequently resolved. The Company’s estimates for variable consideration are adjusted as required at each reporting period for specific known developments that may result in a change in the amount of total consideration it expects to receive.
Chargebacks
In the case an indirect customer purchases product from their preferred wholesaler instead of directly from the Company, and the contract price charged to the indirect customer is lower than the wholesaler pricing, the Company pays the direct customer (wholesaler) a chargeback for the price differential. The Company estimates its chargeback accrual based on its estimates of the level of inventory of its products in the distribution channel that remain subject to chargebacks, current contract terms and historical experience. The estimate of the level of products in the distribution channel is based primarily on data provided by key customers.
Rebates
The Company pays fixed or volume-based rebates to its customers based on a fixed amount, fixed percentage of product sales or based on the achievement of a specified level of purchases. The Company’s rebate accruals are based on actual net sales, contractual rebate rates negotiated with customers, and expected purchase volumes / corresponding tiers based on actual sales to date and forecasted amounts.
Group Purchasing Organization Fees
The Company pays fees to GPOs for administrative services that the GPOs perform in connection with the purchases of product by the GPO participants who are the Company’s customers. The Company’s GPO fee accruals are based on actual net sales, contractual fee rates negotiated with GPOs and the mix of the products in the distribution channel that remain subject to GPO fees.
Prompt Payment (Cash) Discounts
The Company provides customers with prompt payment discounts which may result in adjustments to the price that is invoiced for the product transferred, in the case that payments are made within a defined period. The Company’s prompt payment discount accruals are based on actual net sales and contractual discount rates.
Consideration Payable to the Customer
The Company pays administrative and service fees to its customers based on a fixed percentage of the product price. These fees are not in exchange for a distinct good or service and therefore are recognized as a reduction of the transaction price. The Company accrues for these fees based on actual net sales, contractual fee rates negotiated with the customer and the mix of the products in the distribution channel that remain subject to fees.
Billbacks
In the case an indirect customer purchases product from their preferred wholesaler instead of directly from the Company, and the contract price charged to the indirect customer is higher than contractual pricing, the Company pays the indirect customer a billback for the price differential. The Company estimates its billback accrual based on its estimates of the level of inventory of its products in the distribution channel that remain subject to billbacks, current contract terms and historical experience. The estimate of the level of products in the distribution channel is based primarily on data provided by key customers.
Medicaid and Other Government Pricing Programs
The Company complies with required rebates mandated by law under Medicaid and other government pricing programs. The Company estimates its government pricing accruals based on monthly sales, historical experience of claims submitted by the various states and jurisdictions, historical rates and estimated lag time of the rebate invoices.
Price Protection and Shelf Stock Adjustments
The Company provides customers with price protection and shelf stock adjustments which may result in an adjustment to the price charged for the product transferred, based on differences between old and new prices which may be applied to the customer’s on-hand inventory at the time of the price change. The Company accrues for these adjustments when its expected value of an adjustment is greater than zero, based on contractual pricing, actual net sales, accrual rates based on historical average rates, and estimates of the level of inventory of its products in the distribution channel that remain subject to these adjustments. The estimate of the level of products in the distribution channel is based primarily on data provided by key customers.
Sales Returns
The Company permits the return of product under certain circumstances, mainly due to product expiration, instances of shipping errors or where product is damaged in transit, and occurrences of product recalls. The Company’s product returns accrual is primarily based on estimates of future product returns based generally on actual net sales, estimates of the level of inventory of its products in the distribution and retail channels that remain subject to returns, estimated lag time of returns and historical return rates. The estimate of the level of products in the distribution channel is based primarily on data provided by key customers.
Profit Shares
For certain product sale arrangements, the Company earns a profit share upon the customer’s sell-through of the product purchased from the Company. The Company estimates its profit shares based on actual net sales by third parties, estimated inventory sold to our partners not yet sold by them, and historical rates of profit shares earned. The estimate of the level of products in the distribution channel is based primarily on data provided by key customers.
License Agreements
Refer to Note 5. Alliance and Collaboration for further information related to revenue recognition associated with a license agreement with multiple performance obligations.
Concentration of Revenue
The following table summarizes the percentages of net revenues from each of the Company's customers that individually accounted for 10% or more of its net revenues:
| | | | | | | | | | | | | | | | | | | | |
| | For the year ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Customer A | | 23 | % | | 24 | % | | 21 | % |
| Customer B | | 15 | % | | 16 | % | | 18 | % |
| Customer C | | 23 | % | | 21 | % | | 22 | % |
| Customer D | | 9 | % | | 9 | % | | 10 | % |
Disaggregated Revenue
During the fourth quarter of 2024, the Company changed the presentation of disaggregated net revenue in its Affordable Medicines segment from a classification primarily based on significant therapeutic classes to a classification primarily based on significant dosage forms to reflect the full product offering of the segment. The new presentation did not change the composition of the Company’s reportable segments and, therefore, did not change historical total net revenue in any segment. All prior periods were changed to conform to the current period’s presentation.
The Company’s significant dosage forms for its Affordable Medicines segment, therapeutic classes for its Specialty segment and sales channels for its AvKARE segment, as determined based on net revenue for the years ended December 31, 2024, 2023 and 2022, are set forth below (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | Year ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Affordable Medicines | | | | | |
| Oral solid | $ | 689,436 | | | $ | 649,998 | | | $ | 652,743 | |
| Auto-injector | 209,497 | | | 160,853 | | | 140,560 | |
| Transdermal | 180,917 | | | 159,084 | | | 119,294 | |
| Injectable | 165,430 | | | 141,854 | | | 188,938 | |
| Biosimilar | 125,422 | | | 65,923 | | | 3,241 | |
| Oral liquid | 103,421 | | | 111,045 | | | 108,405 | |
| Other dosage forms (1) | 202,188 | | | 180,479 | | | 209,406 | |
| Subtotal dosage forms | 1,676,311 | | | 1,469,236 | | | 1,422,587 | |
| International | 8,952 | | | 2,165 | | | 1,468 | |
| License agreement (2) | — | | | — | | | 8,018 | |
| Total Affordable Medicines net revenue | 1,685,263 | | | 1,471,401 | | | 1,432,073 | |
| Specialty | | | | | | |
| Hormonal / allergy | 130,426 | | | 110,486 | | | 91,465 | |
| Central nervous system | 280,174 | | | 249,981 | | | 255,656 | |
| Other therapeutic classes | 28,622 | | | 29,990 | | | 27,000 | |
| Subtotal therapeutic classes | 439,222 | | | 390,457 | | | 374,121 | |
| License agreement (2) | 6,527 | | | — | | | — | |
| Total Specialty net revenue | 445,749 | | | 390,457 | | | 374,121 | |
| AvKARE | | | | | | |
| Distribution | 433,981 | | | 347,406 | | | 260,560 | |
| Government | 159,957 | | | 121,829 | | | 98,234 | |
| Institutional | 42,750 | | | 38,016 | | | 27,742 | |
| Other | 26,257 | | | 24,498 | | | 19,574 | |
| Total AvKARE net revenue | 662,945 | | | 531,749 | | | 406,110 | |
| Total net revenue | $ | 2,793,957 | | | $ | 2,393,607 | | | $ | 2,212,304 | |
(1)Includes net revenue from sales of transmucosal, ophthalmic, topical, nasal and inhalation dosage forms.
(2)Refer to Note 5. Alliance and Collaboration for information on revenue recognized under license agreements.
A rollforward of the major categories of sales-related deductions for the years ended December 31, 2024, 2023 and 2022 is as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| | Contract Charge-
backs and Sales
Volume
Allowances | | Cash
Discount
Allowances | | Accrued
Returns
Allowance | | Accrued
Medicaid and
Commercial
Rebates |
| Balance at December 31, 2021 | $ | 503,902 | | | $ | 23,642 | | | $ | 161,978 | | | $ | 85,737 | |
| Provision related to sales recorded in the period | 3,416,149 | | | 112,609 | | | 84,306 | | | 129,203 | |
| Credits/payments issued during the period | (3,346,459) | | | (108,797) | | | (101,224) | | | (128,910) | |
| Balance at December 31, 2022 | 573,592 | | | 27,454 | | | 145,060 | | | 86,030 | |
| Provision related to sales recorded in the period | 3,384,360 | | | 113,396 | | | 73,172 | | | 246,608 | |
| Credits/payments issued during the period | (3,398,618) | | | (116,958) | | | (81,746) | | | (241,948) | |
| Balance at December 31, 2023 | 559,334 | | | 23,892 | | | 136,486 | | | 90,690 | |
| Provision related to sales recorded in the period | 3,684,804 | | | 126,035 | | | 98,118 | | | 274,534 | |
| Credits/payments issued during the period | (3,745,601) | | | (123,959) | | | (74,114) | | | (229,736) | |
| Balance at December 31, 2024 | $ | 498,537 | | | $ | 25,968 | | | $ | 160,490 | | | $ | 135,488 | |
5. Alliance and Collaboration
The Company has entered into several alliance, collaboration, license, distribution and similar agreements with respect to certain of its products and services with third-party pharmaceutical companies. The consolidated statements of operations include revenue recognized under agreements the Company has entered into to develop marketing and/or distribution relationships with its partners to fully leverage the technology platform and revenue recognized under development agreements which generally obligate the Company to provide R&D services over multiple periods. The Company's significant arrangements are discussed below.
Orion Corporation License Agreement
On December 28, 2022, Amneal signed a long-term license agreement with Orion Corporation (“Orion”), a globally operating Finnish pharmaceutical company, to commercialize a number of our complex generic products in most parts of Europe, Australia and New Zealand (the “Orion Agreement”). The initial term of the Orion Agreement commences upon commercial launch of the products and will continue for eight years. The Orion Agreement will automatically renew for successive two-year terms unless either party declines such renewal in writing at least one year in advance.
Under the terms of the Orion Agreement, Amneal granted Orion licenses to certain generic products commercially available in the U.S. today and select high-value pipeline products currently under development. In addition, Amneal will be responsible for the performance of all R&D activities to be conducted to obtain regulatory approval for each product. Amneal is entitled to be reimbursed for a percentage of mutually agreed upon R&D expenses from Orion. Orion will be responsible for preparing and filing regulatory documentation, along with paying any application fees seeking regulatory approval for the products.
Upon achieving regulatory approval for products, Amneal will be responsible for manufacturing and supplying products to Orion. Orion will be responsible for all commercialization and marketing activities for the territories described above. Amneal will earn revenue for supplying products to Orion at the greater of: (i) cost plus a stated margin, or (ii) a fixed percentage of the net selling price, as defined in the Orion Agreement.
Upon signing of the Orion Agreement, Amneal was entitled to an upfront, non-refundable payment of €20.0 million, or $21.4 million (based on the exchange rate at that date). Amneal is eligible to receive certain one-time sales-based milestones in the aggregate of €45.0 million, or $46.7 million (based on the exchange rate as of December 31, 2024) contingent upon whether Orion achieves certain annual sales targets.
The Orion Agreement is within the scope of ASC Topic 808, Collaborative Arrangements (“ASC 808”). The Company identified performance obligations related to: (1) the grant of a license of functional IP, (2) the performance of R&D activities, and (3) the supply of products. The Company evaluated that the grant of licenses is in the scope of ASC 606, whereas the performance of R&D activities is in the scope of ASC 730-20, Research and Development Arrangements, because the Company determined that performing R&D activities on behalf of other parties is not part of the ordinary activities of its business. The Company will record reimbursement received from Orion for R&D activities as a reduction of R&D expense. The Company concluded each future purchase order from Orion represents a separate contract. Amneal will record revenue related to each purchase order when it transfers control of the products to Orion. For the years ended December 31, 2024 and 2023, Amneal recognized $0.9 million and $0.5 million, respectively, as a reduction of R&D expense for reimbursable R&D activities under the Orion Agreement.
The Company determined that the transaction price under the arrangement was the upfront payment of $21.4 million, which was allocated to the performance obligations based on their relative standalone selling prices. The remaining sales-based milestones payments of $46.7 million are variable consideration and were not included in the transaction price because they were fully constrained under ASC 606.
As of December 31, 2022, the Company had recorded a $21.4 million receivable in prepaid expenses and other current assets for the upfront payment due from Orion, which was received in January 2023. For the year ended December 31, 2022, the Company recognized $8.0 million in license revenue related to the delivery of functional IP, which was recorded in net revenues. The remaining $13.4 million of the transaction price was allocated to the R&D activities performance obligation and was recorded as deferred income, of which $6.7 million was recorded in accounts payable and accrued expenses and $6.7 million was recorded in other long-term liabilities as of December 31, 2022.
During the year ended December 31, 2023, the Company recognized $0.9 million as a reduction to R&D expense related to services performed under the Orion Agreement. As of December 31, 2023, deferred income of $7.8 million and $4.7 million was recorded in accounts payable and accrued expenses and other long-term liabilities, respectively.
During the year ended December 31, 2024, the Company recognized $4.0 million as a reduction to R&D expense related to services performed under the Orion Agreement. As of December 31, 2024, deferred income of $5.0 million and $3.5 million was recorded in accounts payable and accrued expenses and other long-term liabilities, respectively. As of December 31, 2024, no products have been supplied by Amneal under the Orion Agreement.
ONGENTYS® License Agreement
On December 5, 2023, the Company entered into a license agreement with BIAL-Portela & Ca., S.A. (“BIAL”) for the exclusive royalty-free right to market and distribute ONGENTYS® (opicapone) in the U.S. starting on December 18, 2023 and ending at such time when generic opicapone sales reach certain predetermined thresholds (the “BIAL License Agreement”). ONGENTYS® is BIAL’s proprietary, once-daily, peripherally-acting, highly-selective catechol-O-methyltransferase inhibitor approved by the FDA in 2020 as an add-on treatment to carbidopa/levodopa in patients with Parkinson’s disease experiencing “Off” episodes. Under the BIAL Agreement, the Company is responsible for commercialization and marketing of ONGENTYS® in the U.S., and BIAL is responsible for manufacturing and supply. The BIAL Agreement also requires the Company to spend a minimum of $6.0 million in medical and marketing activities directly related to ONGENTYS® of which $5.7 million was expensed through December 31, 2024. The Company commenced distribution of ONGENTYS® in January 2024.
During December 2023, the Company paid a nonrefundable license fee of $12.5 million to BIAL, which was capitalized as an intangible asset and will be amortized to cost of sales over a period of eight years. The BIAL License Agreement provides for potential future milestone payments totaling $22.5 million, depending on cumulative net sales of ONGENTYS®.
Knight Therapeutics International S.A. License Agreement
On January 24, 2024, the Company entered into a 15-year license, distribution and supply agreement with Knight Therapeutics International S.A. (“Knight”) granting Knight the exclusive rights to seek regulatory approval for and commercialize IPX203 in Canada and Latin America (the “Knight License Agreement”). The Knight License Agreement will automatically renew for successive two-year periods unless either party provides notice declining such renewal at least one year in advance.
Knight will be responsible for the performance of all R&D activities, regulatory approval, commercialization, and marketing activities for the territories in the agreement to be conducted to obtain regulatory approval for each product. Upon achieving regulatory approval for products, Amneal will be responsible for manufacturing and supplying products to Knight.
During the year ended December 31, 2024, the Company recorded net revenue of $2.0 million for payments received for a nonrefundable license fee and a regulatory milestone. The Knight License Agreement provides for potential future milestone payments totaling $9.5 million, contingent upon regulatory approval, launch dates and cumulative net sales targets by Knight. The agreement also includes low-double digit royalty payments based on net sales of IPX203.
License Agreement with Zambon Biotech
On February 23, 2024, the Company entered into a license, distribution and supply agreement with Zambon Biotech S.A. (“Zambon”) granting Zambon the exclusive rights to seek regulatory approval for and commercialize IPX203 in Europe (the “Zambon License Agreement”). The term for the Zambon License Agreement is 15 years commencing from the commercial launch of the product, which can automatically renew for successive two-year periods unless either party provides notice declining such renewal at least one year in advance. Zambon will be responsible for the performance of all R&D activities, regulatory approval, commercialization, and marketing activities for the territories in the agreement to be conducted to obtain regulatory approval for each product. Upon achieving regulatory approval for products, Amneal will be responsible for manufacturing and supplying products to Zambon.
In connection with the execution of the agreement, the Company was entitled to a nonrefundable license fee of €5.0 million, or $5.4 million, which was received in April 2024. Of the license fee, the Company allocated €3.2 million, or $3.5 million, to the delivery of a functional license, which was recorded as net revenue during the year ended December 31, 2024. In September 2024, the Company received €1.5 million, or $1.6 million, for a regulatory milestone. Of the regulatory milestone, the Company allocated €1.0 million, or $1.0 million, to the delivery of a functional license, which was recorded as net revenue during the year ended December 31, 2024. In addition, the Company is eligible to receive future milestone payments totaling €70.0 million, or $72.7 million, as of December 31, 2024, from Zambon, contingent upon regulatory approval of the product, and achievement of certain annual net sales targets by Zambon. The Zambon License Agreement also includes single-digit to low-double digit royalty payments based on net sales of IPX203.
Biosimilar Licensing and Supply Agreements
Bevacizumab
On May 7, 2018, the Company entered into a licensing and supply agreement with mAbxience S.L. (“mAbxience”), for its biosimilar candidate for Avastin® (bevacizumab). The supply agreement was subsequently amended on March 2, 2021, and the licensing agreement was amended on March 4, 2021. The Company is the exclusive partner in the U.S. market.
On April 13, 2022, the Food and Drug Administration (“FDA”) approved the Company’s biologics license application for bevacizumab-maly, a biosimilar referencing Avastin®. In connection with this regulatory approval and associated activity, the Company paid milestones of $26.5 million during the year ended December 31, 2022, which were capitalized as product rights intangible assets and are being amortized to cost of sales over their estimated useful lives of seven years. On March 29, 2024, the Company paid a sales-based milestone of $9.5 million, which was capitalized as a product rights intangible asset and is being amortized to cost of sales. The agreement provides for potential future milestone payments to mAbxience of up to $14.0 million for commercial milestones.
Denosumab
On October 12, 2023, the Company entered into a licensing and supply agreement with mAbxience to be the exclusive U.S. partner for two denosumab biosimilars referencing both Prolia® and XGEVA®. Denosumab is a monoclonal antibody drug that inhibits bone reabsorption. It is indicated for two major categories of therapy: bone metastasis from various forms of cancer and prevention of bone pain and fractures, including osteoporosis-related injuries. mAbxience is responsible for the clinical and regulatory approval for the two products and regulatory fees will be shared by the parties. Upon approval of each product, mAbxience will be responsible for supply and the Company will be responsible for commercialization.
During the year ended December 31, 2023, the Company recorded R&D expense for a $2.5 million payment made upon execution of the agreement and an additional $2.5 million for a developmental milestone. During the year ended December 31, 2024, the Company recorded R&D expense of $6.5 million for clinical, development and regulatory milestones. The agreement provides for potential additional future milestone payments to mAbxience of up to $62.5 million as follows: up to $15.0 million for regulatory approval and initial commercial launch milestones; and up to $47.5 million for the achievement of annual commercial milestones.
Collaboration to Develop and Supply Medicines for Obesity and Metabolic Diseases
On September 30, 2024, the Company and Metsera, Inc. (“Metsera”), a clinical stage biopharmaceutical company, entered into a collaboration agreement to develop and supply a new portfolio of weight loss medicines globally (the “Metsera Agreement”). The Company will serve as Metsera’s preferred supply partner for developed markets, including the United States and Europe. In addition, the Company has been granted an exclusive license to commercialize Metsera products covered under the agreement in selected emerging markets, including India and certain countries in Southeast Asia, Africa and the Middle East.
Under the terms of the Metsera Agreement, the Company will be responsible for performing certain development activities on behalf of Metsera and will receive cost plus a margin, as defined. Upon Metsera obtaining regulatory approval for any or all the weight loss medicines referred to above, the Company will manufacture commercial products on behalf of Metsera for cost plus a margin, as defined. The Company is also entitled to a tiered quarterly earn-out calculated as a low-single digit percentage of Metsera’s gross profit, as defined in the Metsera Agreement.
The Company plans to construct two new greenfield manufacturing facilities (the “Manufacturing Facilities”) in India: one for peptide synthesis and one for sterile fill-finish manufacturing. Metsera will contribute an agreed percentage of the construction costs, up to $100 million, subject to annual maximums, as defined. In consideration for the funding by Metsera, the Company will (i) provide a rebate on the price of each unit of commercial injectable product produced by the Company and purchased by Metsera and (ii) provide a payment to Metsera for each unit of commercial product manufactured on behalf of itself or third parties using the Manufacturing Facilities, in aggregate, up to the amount funded by Metsera for construction costs.
The initial term of the Metsera Agreement is seven years from the first commercial sale. Metsera has the sole right to renew the agreement for an additional five-year period. Following this initial renewal, the agreement may be extended by mutual written consent.
The Metsera Agreement did not have a material impact on the Company’s consolidated financial statements as of and for the year ended December 31, 2024.
Agreements with Kashiv Biosciences, LLC
For detail on the Company’s related party agreements with Kashiv Biosciences, LLC, refer to Note 23. Related Party Transactions.
6. Income Taxes
Amneal is a limited liability company that is treated as a partnership for U.S. federal and most applicable state and local income tax purposes. As a partnership, Amneal is not subject to U.S. federal and certain state and local income taxes. Any taxable income or loss generated by Amneal is passed through to and included in the taxable income or loss of its members, including the Company, on a pro rata basis subject to applicable tax regulations. The Company is subject to U.S. federal income taxes, in addition to state and local income taxes with respect to its allocable share of any taxable income or loss of Amneal, as well as any stand-alone income or loss generated by the Company. Amneal provides for income taxes in the various foreign jurisdictions in which it operates. Effective with the Reorganization on November 7, 2023, the Company and a wholly-owned subsidiary are the only members of Amneal.
The Company records its valuation allowances against its deferred tax assets (“DTAs”) when it is more likely than not that all or a portion of a DTA will not be realized. The Company routinely evaluates the realizability of its DTAs by assessing the likelihood that its DTAs will be recovered based on all available positive and negative evidence, including scheduled reversals of deferred tax liabilities, estimates of future taxable income, tax planning strategies and results of operations.
The Company established a valuation allowance based upon all available objective and verifiable evidence both positive and negative, including historical levels of pre-tax income (loss) both on a consolidated basis and tax reporting entity basis, legislative developments, expectations and risks associated with estimates of future pre-tax income, and prudent and feasible tax planning strategies. Since first establishing a valuation allowance, the Company has generated cumulative consolidated three-year pre-tax losses through December 31, 2024. As a result of the losses through December 31, 2024, the Company determined that it is more likely than not that it will not realize the benefits of its gross DTAs and therefore maintained its valuation allowance. As of December 31, 2024, this valuation allowance was $590.3 million, and it reduced the carrying value of these gross DTAs, net of the impact of the reversal of taxable temporary differences, to zero.
In 2018, the Company entered into a tax receivable agreement (“TRA”) with the Members pursuant to which it was generally required to pay the Members, on a one-to-one basis, 85% of the applicable tax savings, if any, in U.S. federal and state income tax that it is deemed to realize as a result of certain tax attributes of their Amneal common units sold to the Company (or exchanged in a taxable sale) and that were created as a result of (i) the sales of their Amneal common units for shares of Class A common stock of the Company prior to the Reorganization (as defined in Note 1. Nature of Operations) and (ii) tax benefits attributable to payments made under the TRA. As part of the Reorganization, the TRA was amended to reduce the Company’s future obligation to pay 85% of the tax benefits subject to the TRA to 75% of such realized benefits. The Reorganization’s amendment to the agreement will not cause the acceleration of TRA payments.
In conjunction with the valuation allowance recorded on the DTAs, the Company reversed the entire accrued TRA liability of $192.8 million during 2019. The Company did not record a TRA liability as of December 31, 2021, 2020, and 2019 because future TRA payments were not probable and estimable. Payments made under the TRA represent amounts that otherwise would have been due to taxing authorities in the absence of attributes obtained by the Company as a result of the sales or exchanges of Amneal common units discussed above. Such amounts will be paid after cash tax savings are realized from the TRA attributes. Payments under the TRA are only expected to be made in periods following the filing of a tax return in which the Company is able to utilize certain tax benefits to reduce its cash taxes paid to a taxing authority.
For the years ended December 31, 2024, 2023, and 2022, the Company recorded expenses associated with the TRA in total other expense, net of $50.7 million, $3.1 million, and $0.6 million, respectively, as a result of the realization of cash tax savings from the TRA attributes for those years. As of December 31, 2024 and 2023, the Company had TRA liabilities of $53.9 million and $3.7 million, respectively (refer to Note 23. Related Party Transactions for the current and long-term portions of the TRA liability). The Company’s cumulative cash tax benefit recorded through December 31, 2024 associated with the TRA was approximately $72.3 million, of which the Company recognized cumulative expenses under the TRA of $54.4 million.
As noted above, the Company has determined it is more-likely-than-not it will be unable to utilize its DTAs subject to the TRA; therefore, as of December 31, 2024, the Company had not recognized the contingent liability under the TRA related to the tax savings it may realize in future years from Amneal common units sold or exchanged. If utilization of these DTAs becomes more-likely-than-not in the future, at such time, the unrecorded contingent TRA liability (which amounted to $133.8 million as of December 31, 2024) will be recorded through charges in the Company’s consolidated statements of operations. If the TRA
attributes are not utilized in future years, it is reasonably possible no amounts would be paid under the TRA in excess of the $53.9 million accrued as of December 31, 2024.
The timing and amount of any payments under the TRA may vary, depending upon a number of factors including the timing and amount of the Company’s taxable income, and the corporate tax rate in effect at the time of realization of the Company’s taxable income. The timing and amount of payments may also be accelerated under certain conditions, such as a change of control or other early termination event, which could give rise to the Company being obligated to make TRA payments in advance of tax benefits being realized.
For the years ended December 31, 2024, 2023 and 2022 the Company's provision for income taxes and effective tax rates were $18.9 million and (34.3)%, $8.5 million and (21.0)%, and $6.7 million and (2.7)%, respectively.
The Company and its subsidiaries file income tax returns in the U.S. federal, and various state, local and foreign jurisdictions. In the U.S., income tax returns are generally subject to examination for a period of three years. The majority of states in which the Company files income tax returns follow the three-year U.S. federal statute of limitations, and a few states have a four-year statute of limitations in which to assess state taxes four years after the return is filed. Because the Company has unused state NOL carryovers generated more than three or four years ago, the relevant state taxing authorities may audit and adjust otherwise closed carryover tax years to the extent such NOL carryover is utilized in an open year. Neither the Company nor any of its other affiliates is currently under audit by the Internal Revenue Service. The Amneal partnership is currently under examination in certain states and the Company does not expect any material adjustments as of December 31, 2024.
The components of the Company's (loss) income before income taxes were as follows (in thousands):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| United States | $ | (69,020) | | | $ | (59,781) | | | $ | (260,616) | |
| International | 14,007 | | | 19,511 | | | 12,489 | |
| Total loss before income taxes | $ | (55,013) | | | $ | (40,270) | | | $ | (248,127) | |
The provision for (benefit from) income taxes was comprised of the following (in thousands):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Current: | | | | | |
| Domestic | $ | 8,314 | | | $ | 2,470 | | | $ | (1,073) | |
| Foreign | 10,549 | | | 5,982 | | | 7,735 | |
| Total current income tax | $ | 18,863 | | | $ | 8,452 | | | $ | 6,662 | |
For the years ended December 31, 2024, 2023, and 2022, the Company did not record a provision for deferred income taxes as a result of recording a full valuation allowance on its DTAs.
The Company’s effective tax rates were as follows:
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Federal income tax at the statutory rate | 21.0 | % | | 21.0 | % | | 21.0 | % |
| State income tax, net of federal benefit | (7.2) | | | (5.5) | | | (0.8) | |
| Income not subject to tax | 16.3 | | | 14.5 | | | (10.7) | |
| Foreign rate differential | (17.0) | | | (19.5) | | | (3.4) | |
| Permanent book/tax differences | (30.2) | | | (2.1) | | | (0.3) | |
| Change in prior year estimates | (4.8) | | | 7.7 | | | 0.7 | |
| Deferred tax adjustment | — | | | (5.7) | | | — | |
| Valuation allowance | (15.5) | | | (32.3) | | | (10.3) | |
| Other | 3.1 | | | 0.9 | | | 1.1 | |
| Effective income tax rate | (34.3) | % | | (21.0) | % | | (2.7) | % |
The change in effective income tax rate for the year ended December 31, 2024 compared to the year ended December 31, 2023 was primarily due to the timing and jurisdictional mix of income and the exit of the umbrella partnership-C-corporation structure as a result of the Reorganization (refer to Note 1. Nature of Operations), which has the effect of allocating all of the operating company’s income to the corporate parent.
The change in effective income tax rate for the year ended December 31, 2023 compared to the year ended December 31, 2022 was primarily due to the timing and mix of income and the reversal of liabilities for uncertain tax positions in 2022.
The following table summarizes the changes in the Company’s valuation allowance on deferred tax assets (in thousands):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Balance at the beginning of the period | $ | 566,544 | | | $ | 434,895 | | | $ | 416,588 | |
| Increase due to net operating losses and temporary differences | 15,139 | | | 23,078 | | | 25,589 | |
| Increase due to stock-based compensation | 2,665 | | | 1,652 | | | 224 | |
| Decrease recorded against goodwill | — | | | — | | | (1,590) | |
| Increase recorded against additional paid-in capital | (1,794) | | | 96,316 | | | 2,720 | |
| Increase (decrease) recorded against other comprehensive income | 7,779 | | | 10,603 | | | (8,636) | |
| Balance at the end of the period | $ | 590,333 | | | $ | 566,544 | | | $ | 434,895 | |
At December 31, 2024, the Company had approximately $167.4 million of foreign net operating loss carryforwards. These net operating loss carryforwards will partially expire, if unused, between 2029 and 2033. At December 31, 2024, the Company had approximately $19.0 million of federal and $147.3 million of state net operating loss carry forwards. The federal net operating losses are generally allowed to be carried forward indefinitely, and the majority of the state net operating losses will expire, if unused, between 2032 and 2042. At December 31, 2024, the Company had approximately $7.1 million of federal R&D credit carry forwards and $12.3 million of state R&D credit carry forwards. The majority of the federal R&D credit carry forwards will expire if unused, between 2038 and 2044, and the majority of state credits will expire if unused by 2037. At December 31, 2024, the Company had approximately $5.0 million of federal capital loss carry forwards, which will expire, if unused, in 2028.
The tax effects of temporary differences that give rise to deferred taxes were as follows (in thousands):
| | | | | | | | | | | |
| | December 31,
2024 | | December 31,
2023 |
| Deferred tax assets: | | | |
| Partnership interest in Amneal | $ | 360,055 | | | $ | 318,140 | |
| Projected imputed interest on TRA | 18,169 | | | 22,730 | |
| Net operating loss carryforward | 33,403 | | | 74,340 | |
| IRC Section 163(j) interest carryforward | 105,621 | | | 72,513 | |
| Capitalized costs | 2,048 | | | 2,537 | |
| Accrued expenses | — | | | 648 | |
| Stock-based compensation | 17,337 | | | 14,672 | |
| Intangible assets | 19,701 | | | 21,901 | |
| Tax credits and other | 33,999 | | | 39,063 | |
| Total deferred tax assets | 590,333 | | | 566,544 | |
| Valuation allowance | (590,333) | | | (566,544) | |
| Net deferred tax assets | $ | — | | | $ | — | |
| | | |
| | | |
| | | |
| | | |
The Company's Indian subsidiaries are primarily export-oriented, and the tax holiday benefits provided by the Indian government for export activities within Special Economic Zones (“SEZ”) expired in March 2023. Without availing the SEZ benefit in India, the Company is eligible to claim a reduced tax rate of approximately 25.17%.
The Company accounts for income tax contingencies using the benefit recognition model. The Company will recognize a benefit if a tax position is more likely than not to be sustained upon audit, based solely on the technical merits. The benefit is measured by determining the amount that is greater than 50% likely of being realized upon settlement, presuming that the tax position is examined by the appropriate taxing authority that has full knowledge of all relevant information. The amount of
unrecognized tax benefits at December 31, 2024, 2023, and 2022, was $3.9 million, $3.7 million and $3.6 million, respectively, of which $3.9 million, $3.6 million and $3.5 million, respectively, would impact the Company’s effective tax rate if recognized. The Company currently does not believe that the total amount of unrecognized tax benefits will increase or decrease significantly over the next 12 months. Interest expense related to income taxes is included in provision for income taxes. Net interest expense (benefit) related to unrecognized tax benefits for the years ended December 31, 2024 and 2022 was $0.8 million and $(0.7) million, respectively (minimal for the year ended December 31, 2023). Accrued interest expense as of December 31, 2024, 2023, and 2022 was $0.9 million, $0.1 million, and $0.1 million, respectively. Income tax penalties are included in provision for income taxes. Accrued tax penalties as of December 31, 2024, 2023 and 2022 were immaterial.
A rollforward of unrecognized tax benefits for the years ended December 31, 2024, 2023 and 2022 is as follows (in thousands):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Unrecognized tax benefits at the beginning of the period | $ | 3,735 | | | $ | 3,616 | | | $ | 5,489 | |
| Gross change for current period positions | 210 | | | 170 | | | 110 | |
| Gross change for prior period positions | (1) | | | (51) | | | (1,983) | |
| Unrecognized tax benefits at the end of the period | $ | 3,944 | | | $ | 3,735 | | | $ | 3,616 | |
In India, the income tax returns for the fiscal years ending March 31, 2022 and 2023 are currently being reviewed by tax authorities as part of the normal procedures, and the Company is not expecting any material adjustments. There are no other income tax returns in the process of examination, administrative appeal, or litigation. Income tax returns are generally subject to examination for a period of three years, five years, and four years after the tax year in India, Switzerland, and Ireland, respectively.
Applicable foreign taxes (including withholding taxes) have not been provided on the approximately $137.8 million of undistributed earnings of foreign subsidiaries as of December 31, 2024. These earnings have been and currently are considered to be indefinitely reinvested. Quantification of additional taxes that may be payable on distribution is not practicable.
The Company continuously monitors government proposals to make changes to tax laws, including comprehensive tax reform in the U.S. and proposed legislation in certain foreign jurisdictions resulting from the adoption of the Organization for Economic Cooperation and Development (“OECD”) policies. If legislative changes are enacted in other countries, any of these proposals may include increasing or decreasing existing statutory tax rates. A change in statutory tax rates in any country would result in the revaluation of Amneal’s deferred tax assets and liabilities related to that particular jurisdiction in the period in which the new tax law is enacted.
As a U.S. company with subsidiaries in, among other countries, India, Switzerland, Ireland and the U.K, we carefully evaluate how many of these many countries are implementing legislation and other guidance to align their international tax rules with the OECD Base Erosion and Profit Shifting recommendations and action plan that aim to standardize and modernize global corporate tax policy, including changes to cross-border tax, transfer pricing documentation rules, and nexus-based tax incentive practices. The OECD has issued a two-pillar approach to global taxation, focusing on global profit allocation and a global minimum tax rate. The “Pillar One” global profit allocation proposal would not apply to the Company, since it generally applies to companies with global revenues exceeding €20 billion (approximately $21 billion using the exchange rate as of December 31, 2024). The “Pillar Two” proposal focuses on a global minimum tax of at least 15%. Legislation for the “Pillar Two” proposal, applying to the Company, has been enacted in Ireland, and became effective with the financial year beginning on January 1, 2024. As the tax rates of the other jurisdictions in which the Company operates exceed 15%, the Company does not believe there is any potential additional exposure besides in Ireland.
The Company assessed that no top-up tax under Pillar 2 of the OECD Inclusive Framework on Base Erosion and Profit Shifting is expected to be due for the year ended December 31, 2024. This assessment is based on the application of safe harbor provisions available in all relevant jurisdictions including UK, Germany, Ireland, Switzerland, U.S. and India.
Since Pillar Two taxes are an alternative minimum tax, deferred taxes will not need to be recorded or remeasured. Instead, Pillar Two taxes will be expensed as incurred.
7. Loss per Share
Following the implementation of the Reorganization on November 7, 2023 (refer to Note 1. Nature of Operations for additional information), all outstanding shares of Old PubCo Class A Common Stock and Old PubCo Class B Common Stock were exchanged for an equivalent number of shares of Class A common stock of the Company.
Basic loss per share of Class A common stock was computed by dividing net loss attributable to Amneal Pharmaceuticals, Inc. by the weighted-average number of shares of Class A common stock outstanding during the period. Diluted loss per share of Class A common stock was computed by dividing net loss attributable to Amneal Pharmaceuticals, Inc. by the weighted-average number of shares of Class A common stock outstanding during the period, adjusted to give effect to potentially dilutive securities. The weighted-average number of shares of Class A common stock for all periods prior to the Reorganization includes shares of Old PubCo Class A Common Stock.
The following table sets forth reconciliations of the numerators and denominators used to compute basic and diluted loss per share of Class A common stock (in thousands, except per share amounts):
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Numerator: | | | | | |
| Net loss attributable to Amneal Pharmaceuticals, Inc. | $ | (116,886) | | | $ | (83,993) | | | $ | (129,986) | |
| Denominator: | | | | | |
| Weighted-average shares outstanding - basic and diluted | 308,978 | | | 176,136 | | | 150,944 | |
| Net loss per share attributable to Amneal Pharmaceuticals, Inc.'s Class A common stockholders: | | | | | |
| Basic and diluted | $ | (0.38) | | | $ | (0.48) | | | $ | (0.86) | |
Prior to the Reorganization, shares of Old PubCo Class B Common Stock did not share in the earnings or losses of the Company and, therefore, were not participating securities. As such, separate presentation of basic and diluted loss per share of Old PubCo Class B Common Stock under the two-class method was not presented. Effective with the Reorganization, all outstanding shares of Old PubCo Class B Common Stock were surrendered and canceled.
The following table presents potentially dilutive securities excluded from the computations of diluted loss per share of Class A common stock (in thousands):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| | 2024 | | 2023 | | 2022 |
Stock options (1) | 2,019 | | | 2,416 | | | 2,648 | |
Restricted stock units (1) | 9,967 | | | 10,511 | | | 10,755 | |
Performance stock units (1) | 7,609 | | | 6,944 | | | 7,174 | |
Shares of Old PubCo Class B Common Stock (2) | — | | | — | | | 152,117 | |
(1)Excluded from the computation of diluted loss per share of Class A common stock because the effect of their inclusion would have been anti-dilutive since there was a net loss attributable to the Company during the period.
(2)Shares of Old PubCo Class B Common Stock were considered potentially dilutive shares of Class A common stock. Shares of Old PubCo Class B Common Stock were excluded from the computations of diluted loss per share of Class A common stock for the year ended December 31, 2022 because the effect of their inclusion would have been anti-dilutive under the if-converted method.
8. Trade Accounts Receivable, Net
Trade accounts receivable, net is comprised of the following (in thousands):
| | | | | | | | | | | |
| | December 31,
2024 | | December 31,
2023 |
| Gross accounts receivable | $ | 1,303,788 | | | $ | 1,199,980 | |
| Allowance for credit losses | (3,552) | | | (3,022) | |
Contract charge-backs and sales volume allowances (1) | (498,537) | | | (559,334) | |
Cash discount allowances (1) | (25,968) | | | (23,892) | |
| Subtotal | (528,057) | | | (586,248) | |
| Trade accounts receivable, net | $ | 775,731 | | | $ | 613,732 | |
(1) Refer to Note 4. Revenue Recognition for additional information.
Concentration of Receivables
Trade accounts receivables from customers representing 10% or more of the Company’s total trade accounts receivable were as follows:
| | | | | | | | | | | |
| December 31, 2024 | | December 31, 2023 |
| Customer A | 37 | % | | 40 | % |
| Customer B | 21 | % | | 24 | % |
| Customer C | 29 | % | | 22 | % |
9. Inventories
Inventories are comprised of the following (in thousands):
| | | | | | | | | | | |
| | December 31,
2024 | | December 31,
2023 |
| Raw materials | $ | 207,697 | | | $ | 217,744 | |
| Work in process | 52,835 | | | 59,563 | |
| Finished goods | 351,922 | | | 304,077 | |
| Total inventories | $ | 612,454 | | | $ | 581,384 | |
10. Prepaid Expenses and Other Current Assets
Prepaid expenses and other current assets are comprised of the following (in thousands):
| | | | | | | | | | | |
| | December 31,
2024 | | December 31,
2023 |
| Deposits and advances | $ | 1,868 | | | $ | 2,200 | |
| Prepaid insurance | 8,264 | | | 8,334 | |
| Prepaid regulatory fees | 6,958 | | | 6,331 | |
| Income and other tax receivables | 16,829 | | | 13,168 | |
| Prepaid taxes | 7,516 | | | 11,899 | |
| Other current receivables | 9,142 | | | 9,929 | |
Chargebacks receivable (1) | 6,378 | | | 7,876 | |
| Other prepaid assets | 23,762 | | | 22,948 | |
| Total prepaid expenses and other current assets | $ | 80,717 | | | $ | 82,685 | |
(1)When a sale occurs on a contract item, the difference between the cost paid to the manufacturer by the Company and the contract cost that the end customer has with the manufacturer is rebated back to the Company by the manufacturer. The Company establishes a chargeback (rebate) receivable and a reduction to cost of goods sold in the same period as the related sale.
11. Property, Plant, and Equipment, Net
Property, plant, and equipment, net was comprised of the following (in thousands):
| | | | | | | | | | | |
| December 31,
2024 | | December 31,
2023 |
| Land | $ | 8,112 | | | $ | 9,024 | |
| Buildings | 224,655 | | | 227,837 | |
| Leasehold improvements | 130,905 | | | 126,461 | |
| Machinery and equipment | 459,026 | | | 443,532 | |
| Furniture and fixtures | 15,003 | | | 14,757 | |
| Vehicles | 2,034 | | | 2,098 | |
| Computer equipment | 70,495 | | | 64,227 | |
| Construction-in-progress | 78,198 | | | 67,665 | |
| Total property, plant, and equipment | 988,428 | | | 955,601 | |
| Less: Accumulated depreciation | (563,520) | | | (508,027) | |
| Property, plant, and equipment, net | $ | 424,908 | | | $ | 447,574 | |
Depreciation expense for the years ended December 31, 2024, 2023 and 2022 was $62.6 million, $66.2 million and $68.1 million, respectively.
12. Goodwill and Other Intangible Assets
The changes in goodwill by segment were as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| Affordable Medicines | | Specialty | | AvKARE | | Total |
| Balance as of December 31, 2022 | $ | 163,076 | | | $ | 366,312 | | | $ | 69,465 | | | $ | 598,853 | |
| Currency translation | (224) | | | — | | | — | | | (224) | |
| Balance as of December 31, 2023 | 162,852 | | | 366,312 | | | 69,465 | | | 598,629 | |
| Currency translation | (1,193) | | | — | | | — | | | (1,193) | |
| Balance as of December 31, 2024 | $ | 161,659 | | | $ | 366,312 | | | $ | 69,465 | | | $ | 597,436 | |
Annual Goodwill Impairment Test
The Company performed a quantitative annual goodwill impairment test for each reporting unit on October 1, 2024, the measurement date. The analysis performed included estimating the fair value of each reporting unit using both the income and market approaches. Based on the results of the annual impairment test, the Company determined that the estimated fair values of the Affordable Medicines, Specialty and AvKARE reporting units exceeded their respective carrying amounts as of the measurement date; therefore, the Company did not record an impairment charge for the year ended December 31, 2024. There were no indicators of goodwill impairment during the year ended December 31, 2024, including the period subsequent to the measurement date.
In performing the quantitative annual goodwill impairment test, the Company utilized long-term growth rates for its reporting units ranging from no growth to 1.0% and a discount rate ranging from 10.5% to 14.0% in its estimation of fair value. As of October 1, 2024, the estimated fair value of the Affordable Medicines reporting unit was in excess of its carrying value by approximately 112%, the estimated fair value of the Specialty reporting unit was in excess of its carrying value by approximately 113%, and the estimated fair value of the AvKARE reporting unit was in excess of its carrying value by approximately 728%. A 500-basis point increase in the assumed discount rates utilized in each test would not have resulted in a goodwill impairment charge in any of the Company's reporting units.
While management believes the assumptions used were reasonable and commensurate with the views of a market participant, changes in key assumptions for these reporting units, including increasing the discount rate, lowering forecasts for revenue and operating margin or lowering the long-term growth rate, could result in a future goodwill impairment.
Intangible assets were comprised of the following (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | December 31, 2024 | | December 31, 2023 |
| | Weighted-
Average
Amortization
Period
(in years) | | Cost | | Accumulated
Amortization | | Net | | Cost | | Accumulated Amortization | | Net |
| Amortizing intangible assets: | | | | | | | | | | | | | |
| Product rights | 6.9 | | $ | 1,550,469 | | | $ | (856,914) | | | $ | 693,555 | | | $ | 1,198,971 | | | $ | (703,297) | | | $ | 495,674 | |
| | | | | | | | | | | | | |
| Other intangible assets | 2.6 | | 83,200 | | | (58,678) | | | 24,522 | | | 111,800 | | | (72,896) | | | 38,904 | |
| Total | | | 1,633,669 | | | (915,592) | | | 718,077 | | | 1,310,771 | | | (776,193) | | | 534,578 | |
| In-process research and development | | | 14,300 | | | — | | | 14,300 | | | 355,845 | | | — | | | 355,845 | |
| Total intangible assets | | | $ | 1,647,969 | | | $ | (915,592) | | | $ | 732,377 | | | $ | 1,666,616 | | | $ | (776,193) | | | $ | 890,423 | |
For the year ended December 31, 2024, intangible asset impairment charges were immaterial.
For the year ended December 31, 2023, the Company recognized a total of $66.9 million of intangible asset impairment charges, of which $36.1 million was recognized in cost of goods sold and $30.8 million was recognized in in-process research and development impairment charges. Cost of sales impairment charges for the year ended December 31, 2023 of $36.1 million primarily related to a reduction in promotional focus on LYVISPAH™, resulting in significantly lower than forecasted future cash flows. IPR&D impairment charges for the year ended December 31, 2023 of $30.8 million were related to one Affordable Medicines asset and one Specialty asset, both of which experienced adverse clinical trials results in the fourth quarter of 2023 and resulted in significantly lower than expected future cash flows.
For the year ended December 31, 2022, the Company recognized a total of $24.1 million of intangible asset impairment charges, of which $11.1 million was recognized in cost of goods sold and $13.0 million was recognized in in-process research and development impairment charges. Cost of sales impairment charges for the year ended December 31, 2022 of $11.1 million related to currently marketed products of which (i) one product experienced significant price erosion during 2022, resulting in significantly lower than expected future cash flows and negative margins, (ii) the supply agreement of one product was terminated during 2022 and therefore the asset was not recoverable and (iii) one product was no longer expected to be sold to a key customer, and therefore the asset was not recoverable. IPR&D impairment charges for the year ended December 31, 2022 of $13.0 million related to (i) one asset that experienced a delay in its expected launch date and (ii) one asset that experienced significant expected price erosion, both of which resulted in significantly lower than expected future cash flows.
Amortization expense related to intangible assets for the years ended December 31, 2024, 2023 and 2022 was $173.6 million, $163.2 million and $172.1 million, respectively.
The following table presents future amortization expense for the next five years and thereafter, excluding $14.3 million of IPR&D intangible assets (in thousands):
| | | | | |
| | Future
Amortization |
| 2025 | $ | 163,086 | |
| 2026 | 115,497 | |
| 2027 | 95,063 | |
| 2028 | 75,677 | |
| 2029 | 68,909 | |
| Thereafter | 199,845 | |
| Total | $ | 718,077 | |
13. Other Assets
Other assets were comprised of the following (in thousands):
| | | | | | | | | | | |
| | December 31,
2024 | | December 31,
2023 |
Interest rate swap (1) | $ | 35,921 | | | $ | 37,089 | |
| Security deposits | 3,752 | | | 3,602 | |
| Long-term prepaid expenses | 12,362 | | | 3,273 | |
| Deferred revolving credit facility costs | 2,820 | | | 4,427 | |
| Other long-term assets | 5,278 | | | 7,126 | |
| Total | $ | 60,133 | | | $ | 55,517 | |
(1)Refer to Note 18. Fair Value Measurements and Note 19. Financial Instruments for information about the Company’s interest rate swap.
14. Accounts Payable and Accrued Expenses
Accounts payable and accrued expenses were comprised of the following (in thousands):
| | | | | | | | | | | |
| | December 31,
2024 | | December 31,
2023 |
| Accounts payable | $ | 258,691 | | | $ | 143,572 | |
Accrued returns allowance (1) | 160,490 | | | 136,486 | |
| Accrued compensation | 72,959 | | | 71,122 | |
Accrued Medicaid and commercial rebates (1) | 135,488 | | | 90,690 | |
| Accrued royalties | 23,687 | | | 23,342 | |
| Commercial chargebacks and rebates | 10,226 | | | 10,226 | |
| Accrued professional fees | 17,339 | | | 11,005 | |
| Accrued other | 56,570 | | | 48,219 | |
| Total accounts payable and accrued expenses | $ | 735,450 | | | $ | 534,662 | |
(1)Refer to Note 4. Revenue Recognition for additional information.
15. Debt
The following is a summary of the Company’s term loan indebtedness (in thousands):
| | | | | | | | | | | |
| | December 31,
2024 | | December 31,
2023 |
| Term Loan Due 2025 | $ | 191,979 | | | $ | 191,979 | |
| Term Loan Due 2028 | 2,292,856 | | | 2,351,647 | |
| Total debt | 2,484,835 | | | 2,543,626 | |
| Less: debt issuance costs | (98,832) | | | (123,497) | |
| Total debt, net of debt issuance costs | 2,386,003 | | | 2,420,129 | |
| Less: current portion of long-term debt | (224,213) | | | (34,125) | |
| Total long-term debt, net | $ | 2,161,790 | | | $ | 2,386,004 | |
Overview of Amneal Credit Facilities
On May 4, 2018 the Company entered into a Term Loan Credit Agreement (the “Term Loan Credit Agreement”) that provided a term loan (“Term Loan Due 2025”) with a principal amount of $2.7 billion and an asset backed revolving credit facility (“Revolving Credit Facility”) under which loans and letters of credit up to a principal amount of $500.0 million were available (principal amount of up to $25.0 million was available for letters of credit).
On June 2, 2022, the Company entered into a revolving credit agreement (the “New Credit Agreement”) that terminated the lender commitments under the Revolving Credit Facility, and replaced them with a new $350.0 million senior secured
revolving credit facility that matures on June 2, 2027 (the “New Revolving Credit Facility”). In addition, the New Credit Agreement (i) provided for up to $25.0 million for the purpose of issuing letters of credit, (ii) provided for up to $35.0 million for the purpose of issuing swingline loans, and (iii) allowed the Company to request an incremental increase in the revolving facility commitments by up to $150.0 million.
On November 14, 2023, the Company and certain existing consenting lenders under the Term Loan Credit Agreement entered into an amendment to the Term Loan Due 2025 (the “New Term Loan Credit Agreement”). As a result of this transaction (the “Refinancing”), the Company exchanged and refinanced $2.35 billion of the $2.54 billion of principal then outstanding under the Term Loan Due 2025, at par, for new term loans that mature on May 4, 2028 (the “Term Loan Due 2028”). After the Refinancing, the principal remaining on the Term Loan Due 2025 was $192.0 million.
In October 2019, the Company entered into an interest rate lock agreement for a total notional amount of $1.3 billion to hedge part of the Company’s interest rate exposure associated with the variability in future cash flows with its Term Loan Due 2025. In connection with the Refinancing, the Company amended this interest rate agreement. For further details on this, refer to Note 19. Financial Instruments.
Additionally on November 14, 2023, the Company entered into an amendment to the New Revolving Credit Facility (the “Amended New Revolving Credit Facility”), pursuant to which certain lenders agreed to increase their commitments such that the aggregate revolving commitments increased to up to $600.0 million.
The Term Loan Due 2028, Term Loan Due 2025, Amended New Revolving Credit Facility, New Revolving Credit Facility and Revolving Credit Facility are collectively referred to as the “Senior Secured Credit Facilities”. The proceeds of any loans made under the Senior Secured Credit Facilities can be used for capital expenditures, acquisitions, working capital needs and other general purposes, subject to covenants as described below.
Amneal Term Loans
Term Loan Due 2025
The Term Loan Due 2025 required principal repayments in equal quarterly installments at a rate of 1.00% of the original principal amount annually, with the balance payable at maturity on May 4, 2025. Subject to the refinancing on November 14, 2023, the Company is no longer required to repay quarterly principal installments, and as a result, the Company was required to repay the remaining principal balance of $192.0 million at maturity on May 4, 2025. In January 2025, the Company paid the entire remaining principal balance of $192.0 million outstanding on its Term Loan Due 2025, plus accrued interest thereon of $0.7 million, with $190.0 million of new borrowings under the Amended New Revolving Credit Facility and cash on hand.
Prior to May 31, 2023, the variable annual interest rate of the Term Loan Due 2025 was a LIBOR-designated rate plus 3.5%. On May 31, 2023, the Company executed an amendment to the Term Loan Due 2025, which changed the variable reference rate from LIBOR to the one-month adjusted term secured overnight financing rate (“SOFR”), subject to a floor of (0.11448%) plus 3.5%. After adopting ASC 848, Reference Rate Reform and electing certain applicable practical expedients, this amendment did not have a material impact on the Company’s consolidated financial statements for the year ended December 31, 2023. As of December 31, 2024, the interest rate for borrowings under the Term Loan Due 2025 was approximately 8.0%.
The Term Loan Due 2025 was recorded in the balance sheet net of issuance costs. In 2018, the Company incurred costs associated with the Term Loan Due 2025 of $38.1 million, which were capitalized and amortized over the life of the Term Loan Due 2025 to interest expense using the effective interest method. Subject to the amendment to the Term Loan Due 2025, unamortized debt issuance costs of $0.6 million and $7.3 million were allocated on a pro rata basis to the Term Loan Due 2025 and Term Loan Due 2028, respectively. The remaining unamortized debt issuance costs will be amortized over the life of the Term Loan Due 2025 to interest expense using the effective interest method. Amortization of debt issuance costs related to the Term Loan Due 2025 was not material for the year ended December 31, 2024. For the years ended December 31, 2023 and 2022, amortization of debt issuance costs related to the Term Loan Due 2025 were $4.7 million and $5.4 million, respectively.
Refinancing and Term Loan Due 2028
The Term Loan Due 2028 is repayable in equal quarterly installments in an amount equal to 2.50% per annum of the original principal amount thereof, with the remaining balance due at final maturity on May 4, 2028. Interest is payable on borrowings under the Term Loan Due 2028 at a rate equal to the term SOFR benchmark rate or the base rate, plus an applicable margin, in each case, subject to a term SOFR benchmark rate floor of 0.00% or a base rate floor of 1.00%, as applicable. The applicable margin for borrowings under the Term Loan Due 2028 is 5.50% per annum for term SOFR benchmark rate loans and 4.50% per
annum for base rate loans. As of December 31, 2024, the interest rate for borrowings under the Term Loan Due 2028 was approximately 9.9%.
The Refinancing involved multiple lenders that were considered members of a loan syndicate. In determining whether the refinancing of the Term Loan Due 2025 was to be accounted for as a debt extinguishment or a debt modification, the Company considered whether creditors remained the same or changed and whether the changes in debt terms were substantial, on a lender-by-lender basis, in accordance with the guidance in ASC 470, Debt. As a result of this analysis, the Company legally has separate loans from each lender in the syndicate of the Term Loan Due 2028 and each lender has a contractual right to payments from the Company. The Company concluded that, on a lender-by-lender basis, debt held by 99% of the lenders included in the Refinancing is considered modified, with the remaining debt held by lenders considered to be extinguished. In accordance with ASC 470, Debt, the Company capitalized costs of $118.6 million associated with the Term Loan Due 2028, primarily comprised of lender fees, which were combined with $7.3 million of unamortized debt issuance costs associated with the Term Loan Due 2025 (as discussed above), both to be amortized to interest expense over the life of the Term Loan Due 2028 using the effective interest method. In connection with the Refinancing, the Company recognized a loss of $40.8 million for the year ended December 31, 2023, which was primarily comprised of debt issuance costs associated with the portion of the Term Loan Due 2025 that was modified. For the years ended December 31, 2024 and 2023, amortization of debt issuance costs related to the Term Loan Due 2028 was $24.3 million and $2.9 million, respectively.
The borrowings under the Term Loan Due 2028 are guaranteed by certain wholly-owned subsidiaries of the Company that also guarantee the Term Loan Due 2025 (together with the Company, the “Loan Parties”).
The Loan Parties’ obligations under the New Term Loan Credit Agreement are secured, subject to customary permitted liens and other agreed upon exceptions, by a perfected security interest in (i) all tangible and intangible assets of the Loan Parties, except for certain excluded assets, and (ii) all of the equity interests of the subsidiaries of the Loan Parties (except for certain excluded subsidiaries and excluded assets and limited, in the case of the voting equity interests of certain foreign subsidiaries and certain domestic subsidiaries that hold no assets other than equity interests of foreign subsidiaries, to 65% of the voting equity interests of such subsidiaries).
New Credit Agreement, New Revolving Credit Facility and Amended New Credit Facility
The New Revolving Credit Facility bears an interest rate equal to the alternate base rate (“ABR”) or SOFR, plus an applicable margin, in each case, subject to an ABR floor of 1.00% or a SOFR floor of 0.00%, as applicable. The applicable margin for the New Revolving Credit Facility was initially 0.25% per annum for ABR loans and 1.25% per annum for SOFR loans. The applicable margin on borrowings under the New Revolving Credit Facility thereafter adjusts, ranging from 0.25% to 0.50% per annum for ABR loans and from 1.25% to 1.50% per annum for SOFR loans determined by the average historical excess availability. The Company paid a commitment fee based on the average daily unused amount of the New Revolving Credit Facility at a rate of 0.25% per annum.
The Amended New Revolving Credit Facility bears interest rate consistent with the New Revolving Credit Facility. The maturity date of the Amended New Revolving Credit Facility is June 2, 2027, subject to a springing maturity in certain circumstances set forth in the Amended New Revolving Credit Facility.
The Company incurred costs associated with the Revolving Credit Facility of $4.6 million, which were capitalized and are being amortized over the life of the agreement. Subject to the June 2, 2022 refinancing, there was a decrease in the borrowing capacity of certain lenders between the New Revolving Credit Facility and the Revolving Credit Facility. As a result, the Company recorded a $0.3 million charge for the year ended December 31, 2022 in loss on refinancing. Additionally, the Company incurred costs of $1.6 million associated with the New Credit Agreement, which were capitalized as deferred financing costs with the remaining unamortized costs associated with the Revolving Credit Facility, and were amortized over the life of the New Credit Agreement.
Subject to the November 14, 2023 amendment, there was an increase in the borrowing capacity of all lenders between the Amended New Revolving Credit Facility and the New Revolving Credit Facility. The Company incurred costs of $2.4 million associated with the Amended New Revolving Credit Facility, which were capitalized as deferred financing costs with the remaining unamortized costs associated with the New Revolving Credit Facility, and will be amortized over the life of the Amended New Credit Agreement.
Costs associated with the Amended New Revolving Credit Facility and the New Revolving Credit Facility have been recorded in other assets. For the years ended December 31, 2024, 2023 and 2022, amortization of deferred financing costs were $1.1 million, $0.5 million and $0.7 million, respectively.
During the year ended December 31, 2024, the Company borrowed $20.0 million and repaid $99.0 million under the Amended New Revolving Credit Facility. As of December 31, 2024, the Company had $100.0 million in borrowings and $495.2 million of available capacity under the Amended New Revolving Credit Facility (principal amount of up to $20.2 million remained available for letters of credit). As discussed above, in January 2025, the Company paid the entire remaining principal balance of $192.0 million outstanding on its Term Loan Due 2025, plus accrued interest thereon of $0.7 million, with $190.0 million of new borrowings under the Amended New Revolving Credit Facility and cash on hand.
As of December 31, 2023, the Company had $179.0 million in borrowings and $225.2 million of available capacity under the Amended New Revolving Credit Facility (principal amount of up to $20.9 million remained available for letters of credit).
Covenants to the Senior Secured Credit Facilities
The Senior Secured Credit Facilities contain a number of covenants that, among other things, create liens on Amneal’s and its subsidiaries’ assets. The Senior Secured Credit Facilities contain certain negative covenants that, among other things and subject to certain exceptions, restrict Amneal’s and its subsidiaries’ ability to incur additional debt or guarantees, grant liens, make loans, acquisitions or other investments, dispose of assets, merge, dissolve, liquidate or consolidate, pay dividends or other payments on capital stock, make optional payments or modify certain debt instruments, modify certain organizational documents, enter into arrangements that restrict the ability to pay dividends or grant liens, or enter into or consummate transactions with affiliates. The Senior Secured Credit Facilities contain customary events of default, subject to certain exceptions. Upon the occurrence of certain events of default, the obligations under the Senior Secured Credit Facilities may be accelerated and the commitments may be terminated. In addition, the Amended New Revolving Credit Facility also included a financial covenant whereby the Company was required to maintain a minimum fixed-charge coverage ratio if certain borrowing conditions were met. At December 31, 2024, Amneal was in compliance with all covenants associated with the Senior Secured Credit Facilities.
Annually, the Company is also required to calculate the amount of excess cash flows, as defined in the Term Loan Credit Agreement and New Term Loan Credit Agreement. Based on the results of the excess cash flows calculation for the years ended December 31, 2024, 2023 and 2022, no additional principal payments were due.
Rondo Credit Facilities and Note Payable - Related Party
Rondo Acquisitions Financing - Revolving Credit and Term Loan Agreement
On January 31, 2020, in connection with the Rondo Acquisitions, Rondo Intermediate Holdings, LLC (“Rondo Holdings”), a wholly-owned subsidiary of Rondo Holdings, LLC, entered into a revolving credit and term loan agreement (“Rondo Credit Facility”) that provided a term loan (“Rondo Term Loan”) with a principal amount of $180.0 million and a revolving credit facility (“Rondo Revolving Credit Facility”), which loaned up to a principal amount of $30.0 million. During the year ended December 31, 2023, the Company paid the remaining outstanding principal under the Rondo Term Loan from cash on hand. The Rondo Credit Facility bore a variable annual interest rate, which originated as one-month LIBOR plus 3.0%.
On April 30, 2023, the Company executed an amendment to the Rondo Revolving Credit Facility, which changed the variable reference rate in the Rondo Term Loan from LIBOR to the one-month adjusted term SOFR, subject to a floor of 0.1% plus 2.25%. This amendment to the Rondo Revolving Credit Facility did not have a material impact on the Company’s consolidated financial statements for the year ended December 31, 2023. On September 21, 2023, the Company executed an amendment to the Rondo Revolving Credit Facility (the “Amended Rondo Revolving Credit Facility”) that, among other things, (i) increased the aggregate revolving commitment from $30.0 million to $70.0 million, and (ii) increased the letter of credit commitment from $10.0 million to $60.0 million. The Amended Rondo Revolving Credit Facility bears a variable annual interest rate, which did not change as a result of this amendment, of one-month adjusted term SOFR, subject to a floor of 0.1% plus 2.25%. On December 16, 2024, the Company executed an amendment to the Amended Rondo Revolving Credit Facility to extend the maturity from January 31, 2025 to April 30, 2025. No other material changes were made to the terms of the Revolving Credit facility. At December 31, 2024, the variable annual interest rate was one-month SOFR plus 2.5%. Additionally, the annual interest rate for borrowings under the Amended Rondo Revolving Credit Facility may be reduced or increased by 0.25% based on step-downs and step-ups determined by the total net leverage ratio, as defined in that agreement.
A commitment fee based on the average daily unused amount of the Amended Rondo Revolving Credit Facility is assessed at a rate based on total net leverage ratio, between 0.25% and 0.50% per annum. At December 31, 2024, the Amended Rondo Revolving Credit Facility commitment fee rate was 0.25% per annum.
Costs associated with the Amended Rondo Revolving Credit Facility of $0.6 million were capitalized and amortized over the life of the facility to interest expense using the effective interest method. Costs associated with the Amended Rondo Revolving Credit Facility were recorded in other assets. For the years ended December 31, 2024 and 2023, amortization of deferred financing costs associated with the Amended Rondo Revolving Credit Facility were $0.6 million and $0.2 million, respectively.
The Amended Rondo Revolving Credit Facility contains a number of covenants that, among other things, create liens on the equity securities and assets of Rondo Holdings, Rondo, AvKARE, LLC and Dixon-Shane, LLC d/b/a R&S Northeast LLC (“R&S”). The Amended Rondo Revolving Credit Facility contains certain negative, affirmative and financial covenants that, among other things, restrict the ability to incur additional debt, grant liens, transact in mergers and acquisitions, make certain investments and payments or engage in certain transactions with affiliates. The Amended Rondo Revolving Credit Facility also contains customary events of default. Upon the occurrence of certain events of default, the obligations under the Amended Rondo Revolving Credit Facility may be accelerated and/or the interest rate may be increased. At December 31, 2024, Rondo was in compliance with all covenants. The Company is not party to the Amended Rondo Revolving Credit Facility and is not a guarantor of any debt incurred thereunder.
During the year ended December 31, 2024, the Company borrowed $28.0 million under the Amended Rondo Revolving Credit Facility for working capital purposes. The Company repaid $28.0 million of these borrowings during the year ended December 31, 2024 from cash on hand.
On September 26, 2023, Rondo entered into a standby letter of credit guarantee arrangement under the Amended Rondo Revolving Credit Facility in the amount of $42.0 million for purposes of securing inventory from a certain supplier. As of December 31, 2024, the Company had no outstanding borrowings and outstanding letters of credit of $42.0 million under the Amended Rondo Revolving Credit Facility and unused borrowing capacity of $28.0 million.
Rondo Acquisitions Financing – Notes Payable-Related Party
On January 31, 2020, the closing date of the Rondo Acquisitions, Rondo Partners, LLC or its subsidiary, Rondo Top Holdings, LLC, issued notes to the sellers (the “Sellers Notes”) with a stated aggregate principal amount of $44.2 million and a short-term note to a seller (the “Short-Term Seller Note”) with a stated principal amount of $1.0 million. The Sellers Notes were unsecured and accrued interest at a rate of 5% per annum, not compounded, until June 30, 2025. The Sellers Notes were subject to prepayment at the option of Rondo, as the obligor, without premium or penalty. Mandatory payment of the outstanding principal and interest was due on June 30, 2025 if certain financial targets were achieved, the borrowers’ cash flows were sufficient (as defined in the Sellers Notes) and repayment was not prohibited by senior debt. If repayment of all outstanding principal and accrued interest on the Sellers Notes was not made on June 30, 2025, the requirements for repayment were to be revisited on June 30 of each subsequent year until all principal and accrued interest were satisfied no later than January 31, 2030 or earlier, upon a change in control, as defined. The Short-Term Sellers Note was also unsecured, accrued interest at a rate of 1.6%, and was paid during February 2021.
In accordance with ASC 805, Business Combinations, all consideration transferred was measured at its acquisition-date fair value. The Sellers Notes were stated at the fair value estimate of $35.0 million, which was estimated using the Monte-Carlo simulation approach under the option pricing framework. The Short-Term Sellers Note of $1.0 million was recorded at the stated principal amount of $1.0 million, which approximated fair value. The $9.2 million discount on the Sellers Notes was to be amortized to interest expense using the effective interest method from January 31, 2020 to June 30, 2025 and the carrying value of the Sellers Notes was to accrete to the stated principal amount of $44.2 million. During the year ended December 31, 2024, the Company repaid principal of $44.2 million and interest of $10.0 million associated with the Sellers Notes from cash on hand. As of December 31, 2024, the Sellers Notes and accrued interest had been fully repaid. The Sellers Notes, net of unamortized discount, were included in notes payable-related party and accrued interest was included in related party payables-short term and long-term as of December 31, 2023. During the year ended December 31, 2024, 2023 and 2022, amortization of the discount related to the Sellers Notes was $1.1 million, $1.7 million, and $1.7 million, respectively.
The Company was not party to or a guarantor of the Sellers Notes.
16. Other Long-Term Liabilities
Other long-term liabilities were comprised of the following (in thousands):
| | | | | | | | | | | |
| December 31, 2024 | | December 31, 2023 |
| Uncertain tax positions | $ | 1,252 | | | $ | 497 | |
Long-term compensation (1) | 17,125 | | | 21,283 | |
| Contingent consideration | — | | | 433 | |
| Other long-term liabilities | 8,572 | | | 7,466 | |
| Total other long-term liabilities | $ | 26,949 | | | $ | 29,679 | |
(1) Includes $8.5 million and $11.1 million of long-term liabilities under deferred compensation plans (refer to Note 18. Fair Value Measurements for certain deferred compensation plan liabilities measured at fair value) as of December 31, 2024 and 2023, respectively, and $8.6 million and $10.2 million of long-term employee benefits for the Company’s international employees as of December 31, 2024 and 2023, respectively.
17. Leases
The majority of the Company's operating and financing lease portfolio consists of corporate offices, manufacturing sites, warehouse space, R&D facilities, and land. The Company's leases have remaining lease terms of 1 year to 20 years (excluding international land easements with remaining terms of approximately 19-94 years). Rent expense for the years ended December 31, 2024, 2023 and 2022 was $21.0 million, $21.7 million, and $22.6 million, respectively.
The components of total lease costs were as follows (in thousands):
| | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| 2024 | | 2023 | | 2022 |
Operating lease cost (1) | $ | 16,429 | | | $ | 16,734 | | | $ | 17,800 | |
| Finance lease cost: | | | | | |
| Amortization of right-of-use assets | 4,583 | | | 4,972 | | | 4,808 | |
| Interest on lease liabilities | 4,542 | | | 4,583 | | | 4,508 | |
| Total finance lease cost | 9,125 | | | 9,555 | | | 9,316 | |
| Total lease cost | $ | 25,554 | | | $ | 26,289 | | | $ | 27,116 | |
(1)Includes variable and short-term lease costs.
Supplemental balance sheet information related to the Company's leases was as follows (in thousands):
| | | | | | | | | | | |
| Operating leases | December 31, 2024 | | December 31, 2023 |
| Operating lease right-of-use assets | $ | 31,388 | | | $ | 30,329 | |
Operating lease right-of-use assets - related party (1) | 10,964 | | | 12,954 | |
| Total operating lease right-of-use assets | $ | 42,352 | | | $ | 43,283 | |
| | | | |
| Operating lease liabilities | $ | 24,814 | | | $ | 24,095 | |
Operating lease liabilities - related party (1) | 9,391 | | | 12,787 | |
| Current portion of operating lease liabilities | 9,435 | | | 9,207 | |
Current portion of operating lease liabilities - related party (1) | 3,396 | | | 2,825 | |
| Total operating lease liabilities | $ | 47,036 | | | $ | 48,914 | |
| | | | |
| Financing leases | | | |
| Financing lease right of use assets | $ | 56,433 | | | $ | 59,280 | |
| | | | |
| Financing lease liabilities | $ | 56,889 | | | $ | 58,566 | |
| Current portion of financing lease liabilities | 3,211 | | | 2,467 | |
| Total financing lease liabilities | $ | 60,100 | | | $ | 61,033 | |
(1) Refer to Note 23. Related Party Transactions for information about related party leases.
Supplemental cash flow information related to leases was as follows (in thousands):
| | | | | | | | | | | |
| Years Ended December 31, |
| | 2024 | | 2023 |
| Cash paid for amounts included in the measurement of lease liabilities: | | | |
| Operating cash flows from finance leases | $ | 4,542 | | | $ | 4,583 | |
| Operating cash flows from operating leases | $ | 17,117 | | | $ | 16,036 | |
| Financing cash flows from finance leases | $ | 3,251 | | | $ | 3,588 | |
| Non-cash activity: | | | |
| Right-of-use assets obtained in exchange for new operating lease liabilities | $ | 9,981 | | | $ | 773 | |
| Right-of-use assets obtained in exchange for new financing lease liabilities | $ | 1,889 | | | $ | 856 | |
The table below reflects the weighted average remaining lease term and weighted average discount rate for the Company's operating and finance leases:
| | | | | | | | | | | |
| | December 31, 2024 | | December 31, 2023 |
| Weighted average remaining lease term - operating leases | 4 years | | 5 years |
| Weighted average remaining lease term - finance leases | 17 years | | 18 years |
| Weighted average discount rate - operating leases | 9.6% | | 8.9% |
| Weighted average discount rate - finance leases | 7.3% | | 7.3% |
Maturities of lease liabilities as of December 31, 2024 were as follow (in thousands):
| | | | | | | | | | | |
| | Operating
Leases | | Financing
Leases |
| 2025 | $ | 16,914 | | | $ | 7,508 | |
| 2026 | 13,496 | | | 7,007 | |
| 2027 | 10,563 | | | 5,890 | |
| 2028 | 8,195 | | | 5,667 | |
| 2029 | 6,567 | | | 5,653 | |
| Thereafter | 3,019 | | | 73,907 | |
| Total lease payments | 58,754 | | | 105,632 | |
| Less: Imputed interest | (11,718) | | | (45,532) | |
| Total | $ | 47,036 | | | $ | 60,100 | |
Maturities of lease liabilities as of December 31, 2023 were as follows (in thousands):
| | | | | | | | | | | |
| | Operating
Leases | | Financing
Leases |
| 2024 | $ | 15,978 | | | $ | 6,856 | |
| 2025 | 14,544 | | | 6,874 | |
| 2026 | 10,693 | | | 6,140 | |
| 2027 | 7,742 | | | 5,647 | |
| 2028 | 5,467 | | | 5,647 | |
| Thereafter | 6,916 | | | 79,573 | |
| Total lease payments | 61,340 | | | 110,737 | |
| Less: Imputed interest | (12,426) | | | (49,704) | |
| Total | $ | 48,914 | | | $ | 61,033 | |
18. Fair Value Measurements
Fair value is the exit price that would be received to sell an asset or paid to transfer a liability. Fair value is a market-based measurement that should be determined using assumptions that market participants would use in pricing an asset or liability. Valuation techniques used to measure fair value should maximize the use of observable inputs and minimize the use of unobservable inputs. To measure fair value, the Company uses the following fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable:
| | | | | | | | |
| Level 1 – | Quoted prices in active markets for identical assets or liabilities. |
| | |
| Level 2 – | Inputs other than Level 1 that are observable for the asset or liability, either directly or indirectly, such as quoted prices for similar assets and liabilities in active markets; quoted prices for identical or similar assets or liabilities in markets that are not active; or other inputs that are observable or can be corroborated by observable market data by correlation or other means. |
| | |
| Level 3 – | Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. Value is determined using pricing models, discounted cash flow methodologies, or similar techniques and also includes instruments for which the determination of fair value requires significant judgment or estimation. |
Assets and Liabilities Measured at Fair Value on a Recurring Basis
The Company evaluates its financial assets and liabilities subject to fair value measurements on a recurring basis to determine the appropriate level of classification for each reporting period. The following table sets forth the Company’s financial assets and liabilities that were measured at fair value on a recurring basis (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Fair Value Measurement Based on |
| December 31, 2024 | | Total | | Quoted
Prices in
Active
Markets
(Level 1) | | Significant
Other
Observable
Inputs
(Level 2) | | Significant
Unobservable
Inputs
(Level 3) |
| Assets | | | | | | | | |
Interest Rate Swap (1) | | $ | 35,921 | | | $ | — | | | $ | 35,921 | | | $ | — | |
| Liabilities | | | | | | | | |
Deferred compensation plan liabilities (2) | | $ | 8,224 | | | $ | — | | | $ | 8,224 | | | $ | — | |
| | | | | | | | |
| December 31, 2023 | | | | | | | | |
| Assets | | | | | | | | |
Interest Rate Swap (1) | | $ | 37,089 | | | $ | — | | | $ | 37,089 | | | $ | — | |
| Liabilities | | | | | | | | |
Deferred compensation plan liabilities (2) | | $ | 9,100 | | | $ | — | | | $ | 9,100 | | | $ | — | |
Contingent consideration liability (3) | | $ | 921 | | | $ | — | | | $ | — | | | $ | 921 | |
(1)The fair value measurement of the Company’s interest rate swap classified within Level 2 of the fair value hierarchy is a model-derived valuation as of a given date in which all significant inputs are observable in active markets including certain financial information and certain assumptions regarding past, present, and future market conditions. Refer to Note 19. Financial Instruments for information about the Company’s interest rate swap.
(2)These liabilities are recorded at the value of the amount owed to the plan participants, with changes in value recognized as compensation expense. The calculation of the deferred compensation plan obligation is derived from observable market data by reference to hypothetical investments selected by the participants.
(3)The fair value measurement of contingent consideration liability has been classified as a Level 3 recurring liability as its valuation requires judgment and estimation of factors that are not currently observable in the market. If different assumptions were used for various inputs, the estimated fair value could be higher or lower than what the Company determined. As of December 31, 2023, the contingent consideration liability associated with the Saol Acquisition included $0.1 million recorded in accounts payable and accrued expenses and $0.4 million recorded in other-longer term liabilities. As of December 31, 2023, the contingent consideration liability associated with the acquisition of Kashiv Specialty Pharmaceuticals, LLC was $0.4 million and was recorded within related party payables - long term. Contingent consideration liability was $0 as of December 31, 2024.
There were no transfers between levels in the fair value hierarchy during the year ended December 31, 2024.
Contingent Consideration
On February 9, 2022, the Company completed the Saol Acquisition (refer to Note 3. Acquisition), which provides for contingent royalty payments that are tiered depending on the aggregate annual net sales for certain pharmaceutical products, beginning in 2023.
Contingent royalty payments for the years ended December 31, 2024 and 2023 were not material.
The following table provides a reconciliation of the contingent consideration liability measured at fair value on a recurring basis using significant unobservable inputs (Level 3) for the year ended December 31, 2023 (in thousands):
| | | | | |
| Year Ended December 31, 2023 |
| Balance, beginning of period | $ | 15,427 | |
| Net change in fair value during period | (14,491) | |
| Payments | (15) | |
| Balance, end of period | $ | 921 | |
The fair value measurement of the contingent consideration liabilities was determined based on significant unobservable inputs, including the discount rate, estimated probabilities of success, timing of achieving specified regulatory milestones and the estimated amount of future sales of the acquired products. The contingent consideration liability is estimated by applying a probability-weighted expected payment model for contingent milestone payments and a Monte Carlo simulation model for contingent royalty payments, which are then discounted to present value. Changes to fair value of the contingent consideration liabilities can result from changes to one or a number of the aforementioned inputs. If different assumptions were used for various inputs, the estimated fair value could be higher or lower than what the Company determined. The change in the fair value of the contingent consideration liability for the year ended December 31, 2024 and the contingent consideration liability as of December 31, 2024 was not material.
Assets and Liabilities Not Measured at Fair Value on a Recurring Basis
The carrying amounts of cash, accounts receivable and accounts payable approximate their fair values due to the short-term maturity of these instruments.
The following is a summary of the Company’s indebtedness at fair value (in thousands):
| | | | | | | | | | | | | | |
| | December 31, 2024 | | December 31, 2023 |
| Term Loan Due 2025 | | $ | 192,579 | | | $ | 190,779 | |
| Term Loan Due 2028 | | $ | 2,364,508 | | | $ | 2,328,130 | |
| Sellers Notes | | $ | — | | | $ | 41,033 | |
The Term Loan Due 2025 and Term Loan Due 2028 are in the Level 2 category within the fair value level hierarchy. The fair values were determined using market data for valuation. The Sellers Notes were in the Level 2 category within the fair value level hierarchy. During the year ended December 31, 2024, the Company fully repaid amounts outstanding under the Sellers Notes. Refer to Note 15. Debt for additional information about its indebtedness, including definitions of terms.
Assets and Liabilities Measured at Fair Value on a Non-Recurring Basis
There were no non-recurring fair value measurements during the years ended December 31, 2024 and 2023.
19. Financial Instruments
The Company uses an interest rate swap to manage its exposure to market risks for changes in interest rates.
Interest Rate Risk
Interest income earned on cash and cash equivalents may fluctuate as interest rates change; however, due to their relatively short maturities, the Company does not hedge these assets or their investment cash flows and the impact of interest rate risk is not material. The Company is exposed to interest rate risk on its debt obligations. The Company's debt obligations consist of variable-rate and fixed-rate debt instruments (for further details, refer to Note 15. Debt). The Company's primary objective is to achieve the lowest overall cost of funding while managing the variability in cash outflows within an acceptable range. To achieve this objective, the Company initially entered into an interest rate swap on the Term Loan Due 2025. On November 14, 2023, in connection with the refinancing of the Term Loan Due 2025 and the New Credit Facility, the Company novated its swap agreement to another counterparty and, in connection with such novation, amended the interest rate swap agreement. Refer to section “Interest Rate Derivative - Cash Flow Hedge” below for additional information.
Interest Rate Derivative – Cash Flow Hedge
The interest rate swap involves the periodic exchange of payments without the exchange of underlying principal or notional amounts. In October 2019, the Company entered into an interest rate lock agreement for a total notional amount of $1.3 billion to hedge part of the Company's interest rate exposure associated with the variability in future cash flows from changes in the one-month LIBOR associated with its Term Loan Due 2025 (the “October 2019 Swap”). On May 31, 2023, the Company executed an amendment to the October 2019 Swap that, among other things, changed the variable reference rate from LIBOR to the one-month SOFR (the “Amended October 2019 Swap”).
On November 14, 2023, in connection with the Company’s refinancing of the Term Loan Due 2025 and the New Credit Facility (refer to Note 15. Debt), the Company novated its Amended October 2019 Swap to another counterparty and subsequently amended the interest rate agreement. Specifically, the amendments modified (i) the fixed rate payable by the counterparty from 1.3660% to a new fixed rate of 2.7877% and (ii) extended the termination date through May 4, 2027 (i.e., one year before the Term Loan Due 2028 matures) (the “November 2023 Swap”). The amendments did not change the notional amount of $1.3 billion. The purpose of the November 2023 Swap is to hedge part of the Company's interest rate exposure associated with the variability in future cash flows from changes in the one-month SOFR associated with the Term Loan Due 2028.
The Company used a strategy commonly referred to as “blend and extend,” which allows the existing asset position of the swap agreement to be effectively blended into the new interest rate swap agreement. As a result of this transaction, on November 14, 2023, the Amended October 2019 Swap was de-designated and the unrealized gain of $66.7 million was recorded within accumulated other comprehensive loss and will be amortized as a reduction of interest expense, net, over the original term of the of the Amended October 2019 Swap (until May 2025), as the hedged transactions affect earnings. Additionally, the November 2023 Swap had a fair value of $66.7 million at inception, and will be ratably recorded to accumulated other comprehensive loss and reclassified to interest expense, net, over the term of the November 2023 Swap (until May 2027), as the hedged transactions affect earnings.
At the inception of the November 2023 Swap, the Company determined that the swap qualified for cash flow hedge accounting under ASC 815. Therefore, changes in the fair value of the swap, net of taxes, will be recognized in other comprehensive loss each period, then reclassified into the consolidated statements of operations as a component of interest expense, net in the period in which the hedged transaction affects earnings. The November 2023 Swap is the only swap agreement outstanding as of December 31, 2024.
The effectiveness of the outstanding November 2023 Swap will be assessed qualitatively by the Company during the life of the hedge by (i) comparing the current terms of the hedge with the related hedged debt to assure they continue to coincide based upon initial quantitative assessment of the amended swap and (ii) through an evaluation of the ability of the counterparty to the hedge to honor its obligations under the hedge.
During the year ended December 31, 2024, the Company reclassified a net gain of $26.2 million from accumulated other comprehensive loss to a reduction of interest expense, net. Approximately $3.8 million of net losses included in accumulated other comprehensive loss as of December 31, 2024 are expected to be reclassified into earnings as interest expense within the next 12 months as interest payments are made on the Company’s Term Loan Due 2028 and amortization of the amounts included in accumulated other comprehensive loss occurs.
As of December 31, 2024, the total gain, net of income taxes, related to the Company’s cash flow hedge of $6.4 million was recognized in accumulated other comprehensive loss. As of December 31, 2023, the total gain, net of income taxes, related to the Company’s cash flow hedge of $33.7 million was recognized in accumulated other comprehensive loss.
A summary of the fair values of derivative instruments in the consolidated balance sheets was as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| | December 31, 2024 | | December 31, 2023 |
| Derivatives Designated as Hedging Instruments | Balance Sheet
Classification | | Fair Value | | Balance Sheet
Classification | | Fair Value |
| Variable-to-fixed interest rate swap | Other Assets | | $ | 35,921 | | | Other Assets | | $ | 37,089 | |
20. Commitments and Contingencies
Commitments
Commercial Manufacturing, Collaboration, License, and Distribution Agreements
The Company continues to seek to enhance its product line and develop a balanced portfolio of differentiated products through product acquisitions and in-licensing. Accordingly, the Company, in certain instances, may be contractually obligated to make potential future development, regulatory, and commercial milestone, royalty and/or profit-sharing payments in conjunction with collaborative agreements or acquisitions that the Company has entered with third parties. The Company has also licensed certain technologies or IP from various third parties. The Company is generally required to make upfront payments and other payments upon successful completion of regulatory or sales milestones. The agreements generally permit the Company to terminate the agreement with no significant continuing obligation. The Company could be required to make significant payments pursuant to these arrangements. These payments are contingent upon the occurrence of certain future events and, given the nature of these events, it is unclear when, if ever, the Company may be required to pay such amounts. Further, the timing of any future payment is not reasonably estimable. Refer to Note 5. Alliance and Collaboration for additional information. Certain of these arrangements are with related parties. Refer to Note 23. Related Party Transactions for additional information.
Contingencies
Legal Proceedings
The Company's legal proceedings are complex, constantly evolving, and subject to uncertainty. As such, the Company cannot predict the outcome or impact of its significant legal proceedings which are set forth below. Additionally, the Company manufactures and derives a portion of its revenue from the sale of pharmaceutical products in the opioid class of drugs and may therefore face claims arising from the regulation and/or consumption of such products. While the Company believes it has meritorious claims and/or defenses to the matters described below (and intends to vigorously prosecute and defend them), the nature and cost of litigation is unpredictable, and an unfavorable outcome of such proceedings could include damages, fines, penalties and injunctive or administrative remedies.
For any proceedings where losses are probable and reasonably capable of estimation, the Company accrues a potential loss. When the Company has a probable loss for which a reasonable estimate of the liability is a range of losses and no amount within that range is a better estimate than any other amount, the Company records the loss at the low end of the range. While these accruals have been deemed reasonable by the Company’s management, the assessment process relies heavily on estimates and assumptions that may ultimately prove inaccurate or incomplete. Additionally, unforeseen circumstances or events may lead the Company to subsequently change its estimates and assumptions. Unless otherwise indicated below, the Company is unable at this time to estimate the possible loss or the range of loss, if any, associated with such legal proceedings and claims. Any such claims, proceedings, investigations or litigation, regardless of the merits, might result in substantial costs to defend or settle, borrowings under the Company’s debt agreements, restrictions on product use or sales, or otherwise harm the Company’s business. The ultimate resolution of any or all claims, legal proceedings or investigations are inherently uncertain and difficult to predict, could differ materially from the Company’s estimates and could have a material adverse effect on its results of operations and/or cash flows in any given accounting period, or on its overall financial condition. The Company currently intends to vigorously prosecute and/or defend these proceedings as appropriate. From time to time, however, the Company may settle or otherwise resolve these matters on terms and conditions that it believes to be in its best interest. An insurance recovery, if any, is recorded in the period in which it is probable the recovery will be realized.
For the year ended December 31, 2024, charges related to legal matters, net of $96.7 million were primarily associated with the Affordable Medicines segment’s settlement in principle on the primary financial terms for a nationwide resolution to the opioids cases that have been filed and that might have been filed against the Company by political subdivisions and Native American tribes across the U.S. (refer to the section Civil Prescription Opioid Litigation below). For the year ended December 31, 2023, charges related to legal matters, net of $1.8 million were comprised of $3.9 million in charges associated with Affordable Medicines civil prescription opioid litigation, a $3.0 million charge for the settlement of an Affordable Medicines customer claim, a $3.0 million charge for the settlement of Affordable Medicines commercial antitrust litigation, and a $1.9 million charge for the settlement of a corporate stockholder derivative lawsuit, partially offset by a $10.0 million credit from the settlement of Affordable Medicines patent infringement matters. For the year ended December 31, 2022, the Company recorded charges related to legal matters, net of $269.9 million, primarily for corporate Opana® ER antitrust litigation of $262.8 million and Affordable Medicines civil prescription opioid litigation of $18.0 million, partially offset by corporate insurance recoveries associated with securities class actions of $15.5 million.
Liabilities for legal matters were comprised of the following (in thousands):
| | | | | | | | | | | |
| December 31, |
| Matter | 2024 | | 2023 |
Opana ER® antitrust litigation | $ | — | | | $ | 50,000 | |
Opana ER® antitrust litigation-accrued interest | — | | | 2,347 | |
| Civil prescription opioid litigation | 29,671 | | | 21,189 | |
Other | 2,084 | | | 3,452 | |
| Current portion of liabilities for legal matters | $ | 31,755 | | | $ | 76,988 | |
| | | |
| Civil prescription opioid litigation (Liabilities for legal matters - long term) | $ | 85,479 | | | $ | 316 | |
Refer to the respective discussions below for information about the significant matters summarized above.
Medicaid Reimbursement and Price Reporting Matters
The Company is required to provide pricing information to state agencies, including agencies that administer federal Medicaid programs. Certain state agencies have alleged that manufacturers have reported improper pricing information, which allegedly caused them to overpay reimbursement costs. Other agencies have alleged that manufacturers have failed to timely file required reports concerning pricing information. Liabilities are periodically established by the Company for any potential claims or settlements of overpayment. The Company intends to vigorously defend against any such claims. The ultimate settlement of any potential liability for such claims may be higher or lower than estimated.
Patent Litigation
There is substantial litigation in the pharmaceutical, biological, and biotechnology industries with respect to the manufacture, use, and sale of new products which are the subject of conflicting patent and IP claims. One or more patents often cover the brand name products for which the Company is developing generic versions, and the Company typically has patent rights covering the Company’s branded products.
Under federal law, when a drug developer files an Abbreviated New Drug Application (“ANDA”) for a generic drug seeking approval before expiration of a patent which has been listed with the FDA as covering the brand name product, the developer must certify its product will not infringe the listed patent(s) and/or the listed patent is invalid or unenforceable (commonly referred to as a “Paragraph IV” certification). Notices of such certification must be provided to the patent holder, who may file a suit for patent infringement within 45 days of the patent holder’s receipt of such notice. If the patent holder files suit within the 45-day period, the FDA can review and tentatively approve the ANDA, but generally is prevented from granting final marketing approval of the product until a final judgment in the action has been rendered in favor of the generic drug developer, or 30 months from the date the notice was received, whichever is sooner. The Company’s Affordable Medicines segment is typically subject to patent infringement litigation brought by branded pharmaceutical manufacturers in connection with the Company’s Paragraph IV certifications seeking an order delaying the approval of the Company’s ANDA until expiration of the patent(s) at issue in the litigation.
The uncertainties inherent in patent litigation make the outcome of such litigation difficult to predict. For the Company’s Affordable Medicines segment, the potential consequences in the event of an unfavorable outcome in such litigation include
delaying launch of its generic products until patent expiration. If the Company were to launch its generic product prior to successful resolution of a patent litigation, the Company could be liable for potential damages measured by the profits lost by the branded product manufacturer rather than the profits earned by the Company if it is found to infringe a valid, enforceable patent, or enhanced treble damages in cases of willful infringement. For the Company’s Specialty segment, an unfavorable outcome may significantly accelerate generic competition ahead of expiration of the patents covering the Company’s branded products. All such litigation typically involves significant expense.
The Company is generally responsible for all of the patent litigation fees and costs associated with current and future products not covered by its alliance and collaboration agreements. The Company has agreed to share legal expenses with respect to third-party and Company products under the terms of certain of the alliance and collaboration agreements. The Company records the costs of patent litigation as expense in the period when incurred for products it has developed, as well as for products which are the subject of an alliance or collaboration agreement with a third party.
Other Litigation Related to the Company’s Business
Opana ER® Antitrust Litigation
From June 2014 to April 2015, a number of complaints styled as class actions on behalf of direct purchasers and indirect purchasers (or end-payors) and several separate individual complaints on behalf of certain direct purchasers (the “opt-out plaintiffs”) of Opana ER® were filed against Endo Pharmaceuticals Inc. and Impax Laboratories, Inc. (“Impax”) and consolidated into multi-district litigation (“MDL”) in the U.S. District Court for the Northern District of Illinois.
Impax subsequently entered into settlement agreements with all of the plaintiffs that were subsequently approved by the court. Pursuant to the settlement agreements, the Company agreed to pay a total of $265.0 million between 2022 and mid-January 2024 to resolve substantially all of the plaintiffs’ claims. As of December 31, 2023, the liability for the final settlement payment of $50.0 million, plus 3% stated interest thereon, was included in the current portion of liabilities for legal matters and was paid in January 2024 with cash on hand. The settlement agreements are not an admission of liability or fault by Impax, the Company or its subsidiaries. Upon court approval of the final settlement agreements as discussed above, substantially all the claims and lawsuits in the litigation were resolved.
United States Department of Justice Investigations
On May 15, 2023, Amneal received a Civil Investigative Demand (“CID”) from the Civil Division of the United States Department of Justice (the “Civil Division”) requesting information and documents related to the manufacturing and shipping of diclofenac sodium 1% gel labeled as “prescription only” after the reference listed drug’s label was converted to over-the-counter. In October 2024, the Company received supplemental CIDs seeking additional information related to the same subject matter. The Company is continuing to cooperate with the Civil Division’s investigation. However, no assurance can be given as to the timing or outcome of the investigation.
In Re Generic Pharmaceuticals Pricing Antitrust Litigation
Beginning in March 2016, various purchasers of generic drugs filed multiple putative antitrust class action complaints against a substantial number of generic pharmaceutical manufacturers, including the Company and Impax, alleging an illegal conspiracy to fix, maintain, stabilize, and/or raise prices, rig bids, and allocate markets or customers. They seek unspecified monetary damages and equitable relief, including disgorgement and restitution. The lawsuits were consolidated in the United States District Court for the Eastern District of Pennsylvania (See In re Generic Pharmaceuticals Pricing Antitrust Litigation, No. 2724 (E.D. Pa.)) (“MDL No. 2724”).
In 2019 and 2020, Attorneys General of 43 States and the Commonwealth of Puerto Rico named the Company in two complaints alleging a similar conspiracy and seeking similar damages. These cases are pending in the District of Connecticut. See Connecticut, et al. v. Teva Pharmaceuticals USA, Inc., et al., 3:19-cv-00710-MPS and Connecticut, et al. v. Sandoz, Inc. et al., 3:20-cv-00802-MPS.
Fact discovery is underway in MDL No. 2724 and in the State Attorneys General cases naming the Company as a defendant. Expert discovery is complete in Connecticut, et al. v. Sandoz, Inc. et al., 3:20-cv-00802-MPS. In Connecticut, et al. v. Sandoz, Inc. et al., 3:20-cv-00802-MSP, defendants’ joint motions for summary judgment were filed in November 2024 and defendant-specific motions for summary judgment are due in July 2025. In Connecticut, et al. v. Teva Pharmaceuticals USA, Inc., et al., 3:19-cv-00710-MPS, defendants jointly moved to dismiss the complaint and Amneal individually moved to dismiss the states’ Ranitidine, Bethanechol, and overarching conspiracy claims. These motions were fully briefed on February 14, 2025. In the
MDL, defendants including the Company and Impax jointly moved to dismiss certain complaints in December 2024. Amneal individually moved to dismiss plaintiffs’ Bethanechol Chloride claims in American Airlines, Inc., et al v. Actavis Holdco U.S., Inc., et al, 2:24-cv-01430. These motions were fully briefed on February 20, 2025. The MDL Court ordered that trials for the first MDL cases chosen for bellwether treatment, none of which name the Company or Impax as defendants, will begin August 8, 2025. The MDL Court has identified the second round of MDL cases chosen for bellwether treatment, one of which names Impax as a defendant. No scheduling orders have been set.
Civil Prescription Opioid Litigation
The Company is named in over 900 state and federal cases relating to the sale of prescription opioid pain relievers. Plaintiffs are political subdivisions, schools, hospitals, Native American tribes, pension funds, third-party payors, and individuals. Nearly all federal court cases are consolidated for pre-trial proceedings in Case No. 17-mdl-2804, USDC N.D. OH. The Company also is named in state court cases pending in seven states. There are no firm trial dates in those state-court cases except in Texas, where the trial date in the Dallas County case is September 29, 2025, and the trial-ready date in the Bexar County case is March 16, 2026.
The Company has received a subpoena from the New York Attorney General, a subpoena from the Maryland Attorney General, and a CID issued by the Alaska Attorney General all seeking information regarding its business concerning opioid-containing products. The Company has cooperated and continues to cooperate with these requests.
In 2023, the Company reached settlements with the New Mexico Attorney General and West Virginia political subdivisions and a settlement in principle with a group of private hospitals in Alabama. In late April 2024, the Company reached a nationwide settlement in principle on the primary financial terms, with no admission of wrongdoing, for a nationwide resolution to the opioids cases filed and that might have been filed by state Attorneys General, political subdivisions and Native American tribes. The settlement in principle is subject to execution of a definitive settlement agreement. The settlement would be payable over ten years. Under the settlement in principle, the Company would agree to pay $92.5 million in cash and provide $180.0 million (valued at $125/twin pack) in naloxone nasal spray to help treat opioid overdoses. In lieu of receiving product, the settling parties can opt to receive 25% of the naloxone nasal spray’s value (up to $45.0 million) in cash during the last four years of the ten years payment term, which could increase the total amount of cash the Company would agree to pay up to $137.5 million.
As of March 31, 2024, the Company concluded the loss related to the opioid litigation was probable, and the related loss was reasonably estimable considering the settlement in principle. As a result, the Company recorded a charge of $94.4 million associated with the settlement in principle during the three months ended March 31, 2024, to increase the liability as of March 31, 2024 to $115.6 million. The liability as of December 31, 2024 was $115.2 million, of which $85.5 million was classified as long-term. While this liability has been deemed reasonable by the Company’s management, it could significantly change as the definitive settlement agreement is finalized. As of December 31, 2023, the Company had a liability of $21.5 million related to its prescription opioid litigation, of which $0.3 million was classified as long-term. For the remaining cases not covered by the settlement in principle, primarily brought by other hospitals, schools and individuals, the Company has not recorded a liability as of December 31, 2024 or 2023, because it concluded that a loss was not probable and estimable.
United States Department of Justice / Drug Enforcement Administration Subpoenas
On July 7, 2017, Amneal Pharmaceuticals of New York, LLC received an administrative subpoena issued by the Long Island, NY District Office of the Drug Enforcement Administration (the “DEA”) requesting information related to compliance with certain recordkeeping and reporting requirements. On or about April 12, 2019 and May 28, 2019, the Company received grand jury subpoenas from the U.S. Attorney’s Office for the Eastern District of New York (the “USAO”) relating to similar topics concerning the Company’s suspicious order monitoring program and its compliance with the Controlled Substances Act. The Company is cooperating with the USAO in responding to the subpoenas. The Company has entered into a tolling agreement with respect to potential criminal charges through May 15, 2025. The Company entered into a tolling agreement with the USAO that tolled the statute of limitations for potential civil claims through November 15, 2024. It is not possible to determine the exact outcome of these investigations.
On March 14, 2019, Amneal received a subpoena from an Assistant U.S. Attorney for the Southern District of Florida (the “AUSA”). The subpoena requested information and documents generally related to the marketing, sale, and distribution of oxymorphone. The Company is cooperating with the AUSA regarding the subpoena. However, no assurance can be given as to the timing or outcome of its underlying investigation.
On October 7, 2019, Amneal received a subpoena from the New York State Department of Financial Services seeking documents and information related to sales of opioid products in the state of New York. The Company is cooperating with the request and providing responsive information. It is not possible to determine the exact outcome of this investigation.
Ranitidine Litigation
The Company was named, along with numerous other brand and generic pharmaceutical manufacturers, wholesale distributors, retail pharmacy chains, and repackagers of ranitidine-containing products in a federal MDL (In re Zantac/Ranitidine NDMA Litigation (MDL No. 2924), Southern District of Florida). Plaintiffs alleged defendants failed to disclose and/or concealed the alleged inherent presence of N-Nitrosodimethylamine (or “NDMA”) in ranitidine products and the alleged associated risk of cancer. The MDL Court’s dismissal of claims by all plaintiffs against the Company and other generic drug manufacturers on preemption grounds is on appeal in the 11th Circuit. Plaintiffs filed their merits brief on April 10, 2024. The generic drug manufacturers, including the Company, filed their briefs on July 25, 2024. Plaintiffs’ reply brief was filed November 8, 2024. The briefing also addresses the MDL Court’s December 6, 2022 exclusion of plaintiff’s general causation experts. The 11th Circuit has not set an oral argument date.
The Company has also been named in state court cases in four states. The Company has filed motions to dismiss those cases. On August 17, 2023, the judge in the consolidated Illinois state court cases granted a motion to dismiss all such cases in which the Company had been named, holding all claims preempted. On December 10, 2024, plaintiffs filed a motion in the Illinois state court cases seeking entry of partial final judgment as to the Company and other generic drug manufacturer defendants to allow plaintiffs to appeal the dismissals of those defendants. The Company has reached an agreement in principle, which is not material, to settle the 95 cases pending against it in California state court. Currently, there is a September 15, 2025 trial date in the one case pending in New Mexico brought by the Attorney General, but the court recently indicated that date is likely to be continued. There are no other trial dates involving the Company in any of the state court cases.
Metformin Litigation
Beginning in 2020, Amneal was named as a defendant in several putative class action lawsuits filed and consolidated in the United States District Court for the District of New Jersey, seeking compensation for economic loss allegedly incurred in connection with their purchase of generic metformin allegedly contaminated with NDMA. See In Re Metformin Marketing and Sales Practices Litigation (No. 2:20-cv-02324-MCA-MAH) (“In re Metformin”), Marcia E. Brice v. Amneal Pharmaceuticals, Inc., No. 2:20-cv-13728 (D.N.J.), and Michael Hann v. Amneal Pharmaceuticals of New York, LLC et al., No. 2:23-cv-22902 (D.N.J.). On January 7, 2025, the court dismissed the Third Amended Complaint in In Re Metformin without prejudice and granted plaintiffs the opportunity to amend their complaint. On February 20, 2025, plaintiffs filed a Fourth Amended Complaint in In Re Metformin, which incorporated the allegations of plaintiff Brice and plaintiff Hann, and then filed notices of voluntary dismissal of Marcia E. Brice v. Amneal Pharmaceuticals, Inc., No. 2:20-cv-13728 (D.N.J.) and Michael Hann v. Amneal Pharmaceuticals of New York, LLC et al., No. 2:23-cv-22902 (D.N.J.) as standalone actions. Defendants will file a motion to dismiss the Fourth Amended Complaint by March 7, 2025. Plaintiffs’ response in opposition is due on April 7, 2025 and defendants’ reply is due on April 22, 2025.
On March 29, 2021, a plaintiff filed a complaint in the United States District Court for the Middle District of Alabama asserting claims against manufacturers of valsartan, losartan, and metformin based on the alleged presence of nitrosamines in those products. The only allegations against the Company concern metformin (See Davis v. Camber Pharmaceuticals, Inc., et al., C.A. No. 2:21-00254 (M.D. Ala.) (the “Davis Action”)). On May 5, 2021, the United States Judicial Panel on Multidistrict Litigation transferred the Davis Action into the In re: Valsartan, Losartan, and Irbesartan Products Liability Litigation MDL for pretrial proceedings.
Xyrem® (Sodium Oxybate) Antitrust Litigation
Amneal was named as a defendant, along with Jazz Pharmaceuticals, Inc. (“Jazz”) and numerous other manufacturers of generic versions of Jazz’s Xyrem® (sodium oxybate), in several class action lawsuits filed in the United States District Court for the Northern District of California and the United States District Court for the Southern District of New York, alleging that the generic manufacturers entered into anticompetitive agreements with Jazz in connection with the settlement of patent litigation related to Xyrem®. The actions were consolidated in the United States District Court for the Northern District of California for pretrial proceedings (In re Xyrem (Sodium Oxybate) Antitrust Litigation, No. 5:20-md-02966-LHK (N.D. Cal.)).
Amneal was also named as a defendant in a similar action filed by Aetna Inc. (“Aetna”) in California state court (Aetna Inc. v. Jazz Pharms., Inc. et. al, No. 22CV010951 (Cal. Super. Ct.)). The California state court held that it lacks jurisdiction over several defendants, including Amneal, on December 27, 2022, and later issued an order dismissing Amneal without prejudice. On August 25, 2023, Aetna filed a motion seeking leave to file a second amended complaint adding Amneal as a defendant, which the Court tentatively granted on October 20, 2023. Aetna filed a second amended complaint naming Amneal on November 17, 2023.
On February 28, 2023, Amneal executed a $1.9 million settlement agreement with class plaintiffs in the federal litigation. Class plaintiffs filed a motion for final approval of the settlement on November 10, 2023, and entered an order granting final approval, certifying settlement class, and dismissing class plaintiffs’ against Amneal with prejudice on April 17, 2024. On December 18, 2023, Amneal executed a $4.0 million settlement with Aetna, United Healthcare Services, Inc. (“United”), Humana Inc. (“Humana”), Molina Healthcare Inc. (“Molina”), and Health Care Service Corporation (“HCSC”). Pursuant to that settlement, the federal court dismissed United, Humana, Molina and HCSC’s claims against Amneal, with prejudice, on February 26, 2024, and the California state court dismissed Aetna’s claims against Amneal, with prejudice, on February 29, 2024. Thus, all claims against Amneal in the federal and state court have been voluntarily dismissed with prejudice pursuant to settlements. In December 2023, the Company recorded $3.0 million for the settlement of claims associated with Xyrem® antitrust litigation. As of December 31, 2023, the Company had a liability of $2.0 million associated with this settlement, which was paid in January 2024.
UFCW Local 1500 Welfare Fund v. Takeda Pharmaceuticals U.S.A., Inc.
On November 14, 2023, UFCW Local 1500 Welfare Fund and other health plans filed a purported class action lawsuit in the United States District Court for the Southern District of New York against multiple manufacturers, including the Company, alleging an illegal conspiracy to restrict output of generic COLCRYS®. See UFCW Local 1500 Welfare Fund et al. v. Takeda Pharma. U.S.A., Inc. et al, No. 1:23-cv-10030 (S.D.N.Y.). On February 28, 2024, Takeda Pharmaceuticals U.S.A., Inc. filed a motion to transfer the case to the United States District Court for the Eastern District of Pennsylvania. On March 13, 2024 and March 27, 2024, Amneal submitted a letter and brief, respectively, informing the Court of its position that the Eastern District of Pennsylvania lacks personal jurisdiction over Amneal. That motion remains pending, and the deadline to respond to the complaint is set at 45 days after the court resolves the motion to transfer.
Indian Tax Authority Matters
Amneal Pharmaceuticals Pvt. Ltd., RAKS Pharmaceuticals Pvt. Ltd., and Puniska Healthcare Pvt. Ltd., which are subsidiaries of the Company, are currently involved in litigations with Indian tax authorities concerning Central Excise Tax, Service Tax, Goods & Services Tax, and Value Added Tax for various periods of time between 2014 and 2017. These subsidiaries have contested certain of these assessments, which are at various stages of the administrative process. The Company strongly believes its Indian subsidiaries have meritorious defenses in the matter.
Guaifenesin Litigation
On September 5, 2024, Amneal was named as a defendant along with CVS Pharmacy, Inc. (“CVS”) in a putative consumer class action lawsuit in the United States District Court for the Northern District of California alleging that generic guaifenesin products manufactured by Amneal contain benzene through the use of carbomer, an inactive ingredient. See Leonard v. CVS Pharmacy, Inc., No. 5:24-cv-06280 (N.D. Cal.). The complaint purports to plead, on behalf of a nationwide class and California subclass, the following counts: breach of warranty; unjust enrichment; fraud; and violation of California’s Unfair Competition Law. The complaint seeks damages, including punitive damages, restitution, other equitable monetary relief, injunctive relief, prejudgment interest and attorneys’ fees and costs. On December 30, 2024, the Company and CVS jointly filed a motion to dismiss. On January 21, 2025, in lieu of filing a response to defendants’ motion to dismiss, plaintiff filed an amended complaint. Defendants’ motion to dismiss the amended complaint was filed on February 20, 2025. Plaintiff’s response to the motion to dismiss is due March 24, 2025, and defendants’ reply is due April 14, 2025.
Amneal Pharmaceuticals LLC et al. v. Sandoz Inc., D.N.J. 3:25-cv-00181-GC-TJB
On November 25, 2024, the Company and Impax received a notice letter from Sandoz Inc. (“Sandoz”) stating that it had filed an ANDA with the FDA seeking approval to market generic versions of CREXONT®, an extended-release oral capsule formulation of carbidopa and levodopa for the treatment of Parkinson’s disease. The notice letter included a Paragraph IV certification alleging that certain patents covering CREXONT® are invalid, unenforceable, or will not be infringed by the manufacture, use, or sale of Sandoz’s generic product.
In response to this notice letter, on January 7, 2025, the Company and Impax filed a patent infringement lawsuit against Sandoz in the U.S. District Court for the District of New Jersey, alleging infringement of U.S. Patent Nos. 10,098,845, 10,292,935,
10,688,058, 10,973,769, 10,987,313, 11,357,733, 11,622,941, 11,666,538, 11,986,449, 12,064,521, 12,109,185, and 12,128,141. The current deadline for Sandoz to respond to the complaint is March 11, 2025. The filing of this lawsuit triggered a 30-month stay of FDA approval of the Sandoz ANDA from the date of receipt of the notice letter. CREXONT® is also subject to a regulatory exclusivity until August 7, 2027.
21. Stockholders’ (Deficiency) Equity
On November 7, 2023, the Company implemented the Reorganization, a plan pursuant to which the Company and Amneal reorganized and simplified the Company’s corporate structure by eliminating its umbrella partnership-C-corporation structure and converting to a more traditional C-corporation structure whereby all stockholders hold their voting and economic interests directly through the public company. Effective with the Reorganization, the Company holds 100% of the Amneal Common Units. Refer to Note 1. Nature of Operations for additional information on the Reorganization.
In connection with the Reorganization, the Company amended and restated its certificate of incorporation (“Charter”). The voting rights, dividend rights and participation rights of holders of Class A common stock of the Company did not materially change as a result of the amendment. There were no shares of Class B common stock of the Company outstanding as of December 31, 2024 and 2023.
Voting Rights
Holders of Class A common stock and Class B common stock are entitled to one vote for each share of stock held, except as required by law. Holders of Class A common stock and Class B common stock vote together as a single class on each matter submitted to a stockholder vote, including to elect, remove or replace all other directors to the Board subject to rights of holders of any preferred stock. Holders of Class A common stock and Class B common stock are not entitled to vote on any amendment to the Company’s Charter that relates solely to the terms of one or more outstanding series of preferred stock if the holders of such affected series are entitled, either separately or together with the holders of one or more other such series, to vote on such terms pursuant to the Company’s Charter or law. Holders of Class A common stock do not have cumulative voting rights.
Dividend Rights
The holders of Class A common stock are entitled to receive dividends, if any, payable in cash, property, or securities of the Company, as may be declared by the Company's board of directors, out of funds legally available for the payment of dividends, subject to any preferential or other rights of the holders of any outstanding shares of preferred stock. The holders of Class B common stock will not be entitled to receive any dividends.
Participation Rights
The holders of Class A common stock and Class B common stock have no participation rights.
Issuance and Restrictions on Company Common Stock
No shares of Class B common stock may be issued except to a holder of Common Units or its affiliates.
Liquidation Rights
On the liquidation, dissolution or winding-up of the Company, whether voluntary or involuntary, the holders of Class A common stock are entitled to share equally in all assets of the Company available for distribution among the stockholders of the Company after payment to all creditors and subject to any preferential or other rights of the holders of any outstanding shares of preferred stock. The holders of Class B common stock are not entitled to share in such net assets.
Preferred Stock
Under the Company’s Charter, the Company’s Board of Directors has the authority to issue preferred stock and set its rights and preferences. As of December 31, 2024 and 2023, no preferred stock had been issued.
Non-Controlling Interests
As discussed in Note 2. Summary of Significant Accounting Policies, the consolidated financial statements of the Company include the accounts of all entities controlled by the Company, including Amneal and its subsidiaries, through the Company’s direct or indirect ownership of a majority voting interest. The Company records non-controlling interests for the portion of its
subsidiaries’ economic interests that it does not hold. Prior to the Reorganization, non-controlling interests were adjusted for capital transactions that impacted the non-publicly held economic interests in Amneal.
Prior to the Reorganization, Amneal was obligated to make tax distributions to the Members. For the years ended December 31, 2023 and 2022, the Company recorded net tax distributions of $56.7 million and $10.6 million, respectively, as a reduction of non-controlling interests. Subsequent to the Reorganization, the Company is no longer obligated to make tax distributions to the Members. There was no liability for tax distributions payable to Members as of December 31, 2024 and 2023.
The Company acquired a 98% interest in Kashiv Specialty Pharmaceuticals, LLC (“KSP”) on April 2, 2021. The sellers of KSP, a related party, hold the remaining interest. The Company attributes 2% of the net income or loss of KSP to these non-controlling interests.
Redeemable Non-Controlling Interests - AvKARE, LLC and R&S
The Company acquired a 65.1% interest in both AvKARE, LLC and R&S on January 31, 2020. The sellers hold the remaining 34.9% interest (“Rondo Class B Units”) in the holding company that directly owns the acquired companies (“Rondo”). Beginning on January 1, 2026, the holders of the Rondo Class B Units have the right (“Put Right”) to require the Company to acquire the Rondo Class B Units for a purchase price that is based on a multiple of Rondo’s earnings before income taxes, depreciation, and amortization (EBITDA) if certain financial targets and other conditions are met. Additionally, beginning on January 31, 2020, the Company has the right to acquire the Rondo Class B Units based on the same value and conditions as the Put Right (the “Call Option”). The sellers holding the Rondo Class B Units have the one-time right to defer the exercise of the Call Option until the subsequent calendar year. The Rondo Class B Units are also redeemable by the holders upon a change in control. Since the redemption of the Rondo Class B Units is outside of the Company’s control, the units have been presented outside of stockholders’ equity as redeemable non-controlling interests. The Company attributes 34.9% of the net income of Rondo to the redeemable non-controlling interests. The Company will also accrete the redeemable non-controlling interests to redemption value upon an event that makes redemption certain. For the years ended December 31, 2024, 2023 and 2022, tax distributions of $19.8 million, $14.2 million and $6.9 million, respectively, were recorded as reductions of redeemable non-controlling interests. As of December 31, 2024 and 2023, there were no amounts due for tax distributions related to these redeemable non-controlling interests.
Redeemable Non-Controlling Interests - Puniska Healthcare Pvt. Ltd.
The Company acquired a 74% interest in Puniska Healthcare Pvt. Ltd. (“Puniska”) on November 2, 2021. Amneal was required pursuant to the purchase agreement to acquire the remaining 26% of Puniska upon approval of the transaction by the government of India. Since approval of the government of India was outside of the Company’s control, upon closing of the Puniska Acquisition, the equity interests of Puniska that the Company did not own were presented outside of stockholders’ equity as redeemable non-controlling interests. The Company attributed 26% of the net losses of Puniska to the redeemable non-controlling interests.
Upon approval of the transaction by the government of India in March 2022, the Company paid the $1.7 million redemption value for the remaining 26% of the equity interests of Puniska. For the year ended December 31, 2022, the Company recorded accretion of $0.9 million to increase the redeemable non-controlling interests to redemption value.
Changes in Accumulated Other Comprehensive Income (Loss) by Component (in thousands):
| | | | | | | | | | | | | | | | | |
| | Foreign
currency
translation
adjustment | | Unrealized gain (loss) on cash flow hedge, net of tax | | Accumulated other comprehensive income (loss) |
| Balance December 31, 2022 | $ | (32,382) | | | $ | 42,321 | | | $ | 9,939 | |
| Other comprehensive income before reclassification | (433) | | | (39,248) | | | (39,681) | |
| Reallocation of ownership interests | (33,257) | | | 34,016 | | | 759 | |
| Reclassification of cash flow hedge to earnings, net of tax of $0 | — | | | (3,366) | | | (3,366) | |
| Balance December 31, 2023 | (66,072) | | | 33,723 | | | (32,349) | |
| Other comprehensive income before reclassification | (5,788) | | | (1,168) | | | (6,956) | |
| Reclassification of cash flow hedge to earnings, net of tax of $0 | — | | | (26,205) | | | (26,205) | |
| Balance December 31, 2024 | $ | (71,860) | | | $ | 6,350 | | | $ | (65,510) | |
22. Stock-Based Compensation
Amneal Pharmaceuticals, Inc. 2018 Incentive Award Plan
In May 2018, the Company adopted the Amneal Pharmaceuticals, Inc. 2018 Incentive Award Plan (“2018 Plan”) under which the Company may grant stock options, restricted stock units and other equity-based awards to employees and non-employee directors providing services to the Company and its subsidiaries. The stock option, RSU and MPRSU award grants are made in accordance with the Company’s 2018 Plan and are subject to forfeiture if the vesting conditions are not met. On May 5, 2020, the stockholders of the Company approved an amendment to the 2018 Plan, which authorized an additional 14 million shares of Class A common stock available for issuance under the 2018 Plan. On May 9, 2023, the stockholders of the Company approved an amendment and restatement of the 2018 Plan, which authorized an additional 20 million shares of Class A common stock available for issuance under the 2018 Plan, resulting in a total shares reserved under the Stock Plan of 57 million shares, and extends the term of the 2018 Plan until May 9, 2033. As of December 31, 2024, the Company had 23,723,461 shares available for issuance under the 2018 Plan.
The Company recognizes the grant date fair value of each option and share of restricted stock unit over its vesting period. Stock options and RSU awards are granted under the Company’s 2018 Plan and generally vest over a 4 year period and, in the case of stock options, have a term of 10 years.
The following table summarizes all of the Company’s stock option activity for the years ended December 31, 2024, 2023, and 2022:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Stock Options | | Number of
Shares
Under Option | | Weighted-
Average
Exercise
Price
per Share | | Weighted-
Average
Remaining
Contractual
Life | | Aggregate
Intrinsic
Value
(in millions) |
| Outstanding at December 31, 2021 | | 3,051,500 | | | $ | 4.17 | | | | | |
| Options exercised | | (207,452) | | | 2.75 | | | | | |
| Options forfeited | | (195,607) | | | 2.77 | | | | | |
| Outstanding at December 31, 2022 | | 2,648,441 | | | $ | 4.38 | | | 6.0 | | $ | — | |
| Options exercised | | (163,824) | | | 2.75 | | | | | |
| Options forfeited | | (68,252) | | | 2.75 | | | | | |
| Outstanding at December 31, 2023 | | 2,416,365 | | | $ | 4.54 | | | 5.0 | | $ | 6.6 | |
| Options exercised | | (396,914) | | | 2.91 | | | | | |
| Outstanding at December 31, 2024 | | 2,019,451 | | | $ | 4.86 | | | 4.0 | | $ | 8.5 | |
| Options exercisable at December 31, 2024 | | 2,019,451 | | | $ | 4.86 | | | 4.0 | | $ | 8.5 | |
The intrinsic value of options exercised during the year ended December 31, 2024 was approximately $1.8 million. There were no options granted in the years ended December 31, 2024, 2023 and 2022.
The following table summarizes all of the Company's restricted stock unit activity for the years ended December 31, 2024, 2023, and 2022:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Restricted Stock Units | | Number of
Restricted
Stock Units | | Weighted-
Average
Grant Date
Fair Value | | Weighted-
Average
Remaining
Years | | Aggregate
Intrinsic
Value
(in millions) |
| Non-vested at December 31, 2021 | | 13,183,600 | | | $ | 5.25 | | | | | |
| Granted | | 10,117,037 | | | 4.54 | | | | | |
| Vested | | (2,697,134) | | | 5.95 | | | | | |
| Forfeited | | (2,674,890) | | | 5.07 | | | | | |
| Non-vested at December 31, 2022 | | 17,928,613 | | | $ | 4.77 | | | 1.3 | | $ | 35.7 | |
| Granted | | 7,519,565 | | | 1.91 | | | | | |
| Vested | | (3,888,602) | | | 4.53 | | | | | |
| Forfeited | | (4,104,873) | | | 3.41 | | | | | |
| Non-vested at December 31, 2023 | | 17,454,703 | | | $ | 3.92 | | | 1.2 | | $ | 105.4 | |
| Granted | | 7,268,315 | | | 5.40 | | | | | |
| Vested | | (4,325,941) | | | 3.33 | | | | | |
| Forfeited | | (2,820,894) | | | 6.12 | | | | | |
| Non-vested at December 31, 2024 | | 17,576,183 | | | $ | 4.32 | | | 1.2 | | $ | 139.2 | |
The table above includes 2,893,669 MPRSUs granted to executives during 2024. Vesting of these awards is contingent upon the Company’s achievement of stock price hurdles over the performance period starting March 4, 2024 and requires the employee’s continued employment or service through February 28, 2027. The MPRSUs cliff vest at the end of the three-year period and have a maximum potential to vest at 200% (5,787,338 shares) based on the Company's stock price performance. The related share-based compensation expense is determined based on the estimated fair value of the underlying shares on the date of grant and is recognized straight-line over the vesting term. The estimated fair value per share of the MPRSUs was $5.21 and was calculated using a Monte Carlo simulation model. 2,857,002 of these MPRSUs remained outstanding and unvested at December 31, 2024.
The table above includes 2,431,521 MPRSUs granted to executives during March and April 2023. Vesting of the March 2023 awards is contingent upon the Company’s achievement of stock price hurdles over the performance period starting March 3, 2023 and requires the employee’s continued employment or service through February 28, 2026. Vesting of the April 2023 awards is contingent upon the Company’s achievement of stock price hurdles over the performance period starting April 28, 2023 and requires the employee’s continued employment or service through February 28, 2026. The MPRSUs cliff vest at the end of the three-year period and have a maximum potential to vest at 200% (4,863,042 shares) based on the Company's stock price performance. The related share-based compensation expense is determined based on the estimated fair value of the underlying shares on the date of grant and is recognized straight-line over the vesting term. The estimated fair value per share of the MPRSUs ranged from $1.81 to $2.17 and was calculated using a Monte Carlo simulation model. 2,291,995 of these MPRSUs remained outstanding and unvested at December 31, 2024.
The table above includes 3,053,738 MPRSUs granted to executives during 2022. Vesting of these awards is contingent upon the Company’s achievement of stock price hurdles over the performance period starting March 1, 2022 and requires the employee’s continued employment or service through February 28, 2025. The MPRSUs cliff vest at the end of the three-year period and have a maximum potential to vest at 200% (6,107,476 shares) based on the Company's stock price performance. The related share-based compensation expense is determined based on the estimated fair value of the underlying shares on the date of grant and is recognized straight-line over the vesting term. The estimated fair value per share of the MPRSUs was $6.22 and was calculated using a Monte Carlo simulation model. 2,460,107 of these MPRSUs remained outstanding and unvested at December 31, 2024.
As of December 31, 2024, the Company had total unrecognized stock-based compensation expense of $40.7 million related to all of its stock-based awards, which is expected to be recognized over a weighted average period of 1.8 years.
The amount of stock-based compensation expense recognized by the Company was as follows (in thousands):
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Cost of goods sold | $ | 3,564 | | | $ | 3,561 | | | $ | 4,811 | |
| Selling, general and administrative | 20,343 | | | 18,922 | | | 20,746 | |
| Research and development | 3,645 | | | 4,339 | | | 6,290 | |
| Restructuring and other charges | 216 | | | — | | | — | |
| Total | $ | 27,768 | | | $ | 26,822 | | | $ | 31,847 | |
23. Related Party Transactions
The Company has various business agreements with certain third-party companies in which there is some common ownership and/or management between those entities, on the one hand, and the Company, on the other hand. The Company has no direct ownership or management in any of such related party companies. The following table summarizes the Company’s related party transactions (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | Year ended December 31, |
| Related Party and Nature of Transaction | Caption in Balance Sheet and Statement of Operations | | 2024 | | 2023 | | 2022 |
| A. | Kashiv Biosciences LLC | | | | | | | |
| i. | Development and commercialization agreement - Omaluzimab | Research and development | | $ | 20,000 | | | $ | — | | | $ | — | |
| ii. | Development and commercialization agreement - Filgrastim and Pegfilgrastim - Royalty expense (Releuko and Fylnetra) | Cost of goods sold | | $ | 16,124 | | | $ | 5,114 | | | $ | — | |
| iii. | Development and commercialization agreement - Long-acting injectable | Research and development | | $ | 10,500 | | | $ | — | | | $ | — | |
| ii. | Inventory purchases under development and commercialization agreement - Filgrastim and Pegfilgrastim (Releuko and Fylnetra) | Inventory and cost of goods sold | | $ | 9,210 | | | $ | 1,022 | | | $ | 260 | |
| iv. | Sale of subsidiary - gain on sale | Other income, net | | $ | (3,760) | | | $ | — | | | $ | — | |
| iii. | Generic development supply agreement - research and development material | Research and development | | $ | (681) | | | $ | (2,809) | | | $ | — | |
| iv. | Sale of subsidiary - interest income on loan receivable | Interest expense, net | | $ | (515) | | | $ | — | | | $ | — | |
| v. | Storage agreement | Research and development | | $ | (223) | | | $ | (107) | | | $ | (126) | |
| vi. | Parking space lease | Research and development | | $ | 100 | | | $ | 100 | | | $ | 100 | |
| ii. | License and commercialization agreement - Filgrastim and Pegfilgrastim - regulatory approval milestone for Pegfilgrastim-pbbk | Intangible asset | | $ | — | | | $ | — | | | $ | 15,000 | |
| ii. | License and commercialization agreement - Filgrastim and Pegfilgrastim - regulatory approval milestone for Filgrastim | Selling, general and administrative | | $ | — | | | $ | — | | | $ | 5,000 | |
| vii. | Development and commercialization agreement - Ganirelix Acetate and Cetrorelix Acetate | Research and development | | $ | — | | | $ | (25) | | | $ | 1,761 | |
| | | | | | | | |
| B. | Members - tax receivable agreement | Increase in tax receivable agreement liability | | $ | 50,680 | | | $ | 3,124 | | | $ | 631 | |
| C. | Apace KY, LLC d/b/a Apace Packaging LLC - packaging agreement | Inventory and cost of goods sold | | $ | 19,574 | | | $ | 15,873 | | | $ | 2,742 | |
| D. | AzaTech Pharma LLC - supply agreement | Inventory and cost of goods sold | | $ | 11,587 | | | $ | 8,746 | | | $ | 4,963 | |
| E. | Kanan, LLC - operating lease | Inventory and cost of goods sold | | $ | 2,368 | | | $ | 2,540 | | | $ | 2,104 | |
| F. | Sellers Notes - interest | Interest expense, net | | $ | 1,325 | | | $ | 2,210 | | | $ | 2,210 | |
| G. | Sutaria Family Realty, LLC - operating lease | Inventory and cost of goods sold | | $ | 1,286 | | | $ | 1,352 | | | $ | 1,211 | |
| H. | Tracy Properties LLC - operating lease | Selling, general and administrative | | $ | 609 | | | $ | 625 | | | $ | 565 | |
| I. | Avtar Investments, LLC - consulting services | Research and development | | $ | 251 | | | $ | 321 | | | $ | 216 | |
| J. | Alkermes Plc | Inventory and cost of goods sold | | $ | 229 | | | $ | 464 | | | $ | 235 | |
| K. | AvPROP, LLC - operating lease | Selling, general and administrative | | $ | 184 | | | $ | 167 | | | $ | 178 | |
| L. | Fosun International Limited - license and supply agreement | Net revenue | | $ | — | | | $ | (80) | | | $ | — | |
| M. | TPG Capital BD, LLC | Loss on refinancing | | $ | — | | | $ | 3,000 | | | $ | — | |
| N. | R&S Solutions LLC - logistics services | Selling, general and administrative | | $ | — | | | $ | 102 | | | $ | 85 | |
| O. | PharmaSophia, LLC - research and development services income | Research and development | | $ | — | | | $ | — | | | $ | (45) | |
| O. | PharmaSophia, LLC - license and commercialization agreement | Research and development | | $ | — | | | $ | — | | | $ | 1,093 | |
| P. | TPG Operations, LLC - consulting services | Selling, general and administrative | | $ | — | | | $ | — | | | $ | 19 | |
| Q. | Asana Biosciences, LLC | Research and development | | $ | — | | | $ | — | | | $ | (5) | |
The following table summarizes the amounts due to or from the Company for related party transactions (in thousands):
| | | | | | | | | | | | | | |
| | December 31, 2024 | | December 31, 2023 |
| A. | Kashiv - various agreements | $ | 447 | | | $ | 954 | |
| D. | AzaTech Pharma LLC | 21 | | | — | |
| J. | Alkermes Plc | 16 | | | 1 | |
| Related party receivables - short term | $ | 484 | | | $ | 955 | |
| | | | |
| A. | Kashiv - various agreements | $ | 16,908 | | | $ | 3,179 | |
| B. | Members - tax receivable agreement | 2,985 | | | 549 | |
| C. | Apace Packaging, LLC - packaging agreement | 1,205 | | | 1,091 | |
| D. | AzaTech Pharma LLC - supply agreement | 1,151 | | | 1,958 | |
| I. | Avtar Investments LLC - consulting services | 60 | | | 100 | |
| J. | Alkermes Plc | 2 | | | 2 | |
| F. | Sellers of AvKARE LLC and R&S - accrued interest on Sellers Notes | — | | | 442 | |
| Related party payables - short term | $ | 22,311 | | | $ | 7,321 | |
| | | | |
| B. | Members - tax receivable agreement | $ | 50,900 | | | $ | 3,207 | |
| A. | Kashiv - contingent consideration | — | | | 430 | |
| F. | Sellers of AvKARE LLC and R&S - accrued interest on Sellers Notes | — | | | 8,139 | |
| Related party payables - long term | $ | 50,900 | | | $ | 11,776 | |
Related Party Transaction Descriptions
A. Kashiv Biosciences LLC
Kashiv Biosciences LLC (“Kashiv”) is a vertically integrated biopharmaceutical company with a diverse portfolio of commercial and clinical-stage assets. Amneal has various business agreements with Kashiv. Certain executive officers of the Company beneficially own, directly and through certain revocable or irrevocable trusts for the benefit of their immediate families, outstanding equity securities of Kashiv. In addition, they serve on the Board of Managers of Kashiv.
On January 11, 2021, the Company and Kashiv entered into a definitive agreement for Amneal to acquire a 98% interest in KSP, which was a subsidiary of Kashiv and focuses on the development of complex generics, innovative drug delivery platforms and novel 505(b)(2) drugs. The acquisition closed on April 2, 2021 and included contingent payments for the achievement of certain regulatory milestones and potential royalty payments based on annual net sales for certain future pharmaceutical products. As of December 31, 2023, the contingent consideration liability associated with the acquisition of KSP of $0.4 million was recorded within related party payables - long term. Contingent consideration liability was $0 as of December 31, 2024.
Below is a summary of the related party arrangements held between the Company and Kashiv that were not impacted by the KSP Acquisition:
i.On July 1, 2024, Kashiv and Amneal entered into an exclusive license and commercialization agreement to distribute and sell Omalizumab, a biosimilar to XOLAIR®, in the U.S. and India. Kashiv is responsible for development, regulatory filings, obtaining FDA approval, and manufacturing for the product, and Amneal is responsible for marketing, selling, and pricing activities. The term of the agreement is 10 years from the respective product’s launch date and automatically renews for terms of three years unless either party provides written notification of termination.
The agreement requires the Company to pay $10.0 million as an up-front amount to Kashiv and potential future milestone payments of up to $75.0 million, upon achieving certain developmental and regulatory achievements within agreed-upon timelines. The milestones include: (i) up to $32.5 million in developmental milestone payments, (ii) $22.5 million in regulatory approval and commercial launch milestone payments, and (iii) a $20.0 million sales-based milestone payment, which is contingent upon reaching a defined annual commercial sales volume for the product. In addition, the agreement provides for Amneal to pay a profit share up to 45% of net profits, after considering manufacturing, marketing, royalty and shipping costs.
During the year ended December 31, 2024, the Company expensed amounts paid to Kashiv for: (i) the upfront amount of $10.0 million in connection with the execution of the agreement and (ii) an additional $10.0 million related to the first developmental milestone.
ii.In 2017, Kashiv and Amneal entered into an exclusive license and commercialization agreement (the “Kashiv Biosimilar Agreement”) to distribute and sell two biosimilar products, Filgrastim and Pegfilgrastim, in the U.S. Kashiv is responsible for development, regulatory filings, obtaining FDA approval, and manufacturing, and Amneal is responsible for marketing, selling, and pricing activities. The term of the agreement is 10 years from the respective product’s launch date.
The Kashiv Biosimilar Agreement provided for potential future milestone payments to Kashiv of up to $183.0 million, as follows: (i) up to $22.5 million relating to regulatory approval and execution, (ii) up to $43.0 million for successful delivery of commercial launch inventory, (iii) up to $50.0 million depending on the number of competitors at launch for one product, and (iv) between $15.0 million and $67.5 million for the achievement of cumulative net sales for both products.
In July 2022, the Company and Kashiv amended the Kashiv Biosimilar Agreement to, among other things, (i) eliminate milestones related to the manufacturing and delivery of the Kashiv products, (ii) revise the net sales milestones to provide for future milestone payments by the Company to Kashiv of up to $37.5 million for the achievement of cumulative combined net sales goals for both products, and (iii) adjust the supply price of product that Kashiv manufacturers and supplies to the Company, which will lower the cost per unit of both products.
The remaining milestones are subject to reaching certain commercial sales volume objectives. In addition, the agreement provides for Amneal to pay a profit share equal to 50% of net profits, after considering manufacturing and marketing costs.
On May 27, 2022, the FDA approved the Company’s biologic license application, associated with the amended Kashiv Biosimilar Agreement, for Pegfilgrastim-pbbk. In connection with this regulatory approval and associated activity, the Company incurred and paid a milestone of $15.0 million during the year ended December 31, 2022, payable to Kashiv. The milestone was capitalized as an intangible asset and will be amortized to cost of sales over an estimated useful life of 8.3 years.
The Company recognized a $5.0 million milestone in selling, general and administrative expense upon FDA approval of Filgrastim in February 2022.
In March 2024, the Company amended the Kashiv Biosimilar Agreement to include two additional in-development products, a pre-filled auto-injector delivery system for peg-filgrastim and a pre-filled on-body injector (OBI) delivery system for peg-filgrastim. Consistent with the existing terms, Kashiv is responsible for development, regulatory filings, obtaining FDA approval, and manufacturing, and Amneal is responsible for marketing, selling, and pricing activities of these product candidates. The amendment did not change the contractual terms related to existing commercialized biosimilar products.
The amendment provides an incremental $14.5 million in potential future milestone payments specific to these in-development products, including $7.0 million for clinical and developmental milestones and $7.5 million for regulatory approval and first commercial-sales milestones. In addition, the amendment clarifies that future net sales milestones payments of up to $37.5 million, which did not change, shall be contingent upon reaching certain commercial sales volume objectives for the aggregate of all products under the amended agreement. The agreement provides for Amneal to pay a profit share equal to 50% of net profits, after considering manufacturing and marketing costs.
No amounts were paid or recognized during the year ended December 31, 2024, pursuant to this amendment.
iii.In December 2022, Amneal and Kashiv entered into a development and supply agreement specific to four generic product candidates. Amneal is responsible for manufacturing batch products and performing certain developmental activities on behalf of Kashiv. Kashiv, as owner of the intellectual property, is responsible for regulatory filings, obtaining FDA approval, marketing, selling, and pricing activities. Pursuant to the terms of the development supply agreement, Amneal is eligible to earn up to $2.4 million related to the aforementioned services. As of December 31, 2024 and 2023, deferred revenue related to this arrangement was $0.2 within accounts payable and accrued expenses on the consolidated balance sheet.
Pursuant to the development supply agreement, Amneal maintained a right of first offer and negotiation to the licensing of each generic product candidate. In March 2024, Amneal and Kashiv entered into a license and supply agreement for the development and commercialization of a long-acting injectable (the “Injectable License and Supply Agreement”). The existing development supply agreement remains effective for the remaining three generic product candidates.
Subject to the terms of the Injectable License and Supply Agreement, Amneal is responsible for development, regulatory approval, and commercialization of the product candidate in the U.S., whereas Kashiv is responsible for development and regulatory approval of the product candidate for all other territories outside the U.S. Contingent upon Kashiv obtaining regulatory approval outside the U.S., Amneal shall manufacture the commercial supply for Kashiv at a stated price. The term of the agreement is 10 years from the respective product’s launch date in the U.S.
During the year ended December 31, 2024, the Company recorded R&D expense of $10.5 million comprised of (i) $0.5 million for payment made upon execution of the license and supply agreement and (ii) $10.0 million for the achievement of a regulatory milestone upon the FDA’s written acceptance of a drug approval application filing. The agreement provides for potential future milestone payments to Kashiv of up to $25.0 million as follows: (i) up to $10.0 million relating to developmental milestones; (ii) up to $10.0 million for U.S. regulatory approval and initial commercial launch milestones; and (iii) up to $5.0 million for the achievement of annual commercial milestones. In addition, the agreement provides for Amneal to pay a profit share equal to 50% of net profits, after considering manufacturing and marketing costs. As of December 31, 2024, the Company had a liability of $10.0 million related to the achievement of a regulatory milestone, which was paid by the Company in February 2025.
iv.On April 30, 2024, Amneal closed on the sale of a wholly owned subsidiary in India to a subsidiary of Kashiv for total consideration of ₹1.0 billion, or $12.2 million. Total consideration consisted of a ₹416.2 million, or $5.0 million, cash payment at closing and the assumption of a loan payable of ₹598.6 million, or $7.2 million, payable to another subsidiary of Amneal in India. The loan payable bore interest of 11% on the unpaid principal and was due on or before December 31, 2024. The Company was permitted to offset royalties or other amounts payable to Kashiv with any overdue principal and accrued interest on the loan payable. On December 27, 2024, Kashiv paid the loan of ₹598.6 million, or $7.0 million (based on foreign exchange rates at that date) and accrued interest in full. The subsidiary’s assets and liabilities were primarily comprised of a building under construction and a note payable, respectively. The subsidiary had no business activity, other than the construction of the building. As a result of the sale, the Company recognized a pre-tax gain of $3.8 million in other income, net in its Affordable Medicines segment for the year ended December 31, 2024.
v.The parties entered into a pallet storage agreement for Amneal to store materials for Kashiv.
vi.The parties entered into a lease for parking spaces in Piscataway, NJ. The annual lease cost is $0.1 million per year for the lease agreement.
vii.Amneal and Kashiv entered into a product development agreement for the development and commercialization of two generic peptide products, Ganirelix Acetate and Cetrorelix Acetate. Under the agreement, the IP and abbreviated new drug application for these products are owned by Amneal, and Kashiv will receive a profit share for all sales of the products made by Amneal. In connection with the agreement, Amneal made an upfront payment of $1.1 million in August 2020. The agreement also provides for potential future milestone payments to Kashiv of (i) up to $2.1 million relating to development milestones, and (ii) up to $0.3 million relating to regulatory filings. The milestones are subject to certain performance conditions which may or may not be achieved, including FDA filings. In addition, Amneal agreed to pay $2.6 million of development fees to Kashiv as the development work is completed.
B. Tax Receivable Agreement
In 2018, the Company entered into the TRA pursuant to which it was generally required to pay the holders of Amneal Common Units, on a one-to-one basis, 85% of the applicable tax savings, if any, in U.S. federal and state income tax that it is deemed to realize as a result of certain tax attributes of their Amneal common units sold to the Company (or exchanged in a taxable sale) and that were created as a result of (i) the sales of their Amneal common units for shares of Class A common stock of the Company prior to the Reorganization (as defined in Note 1. Nature of Operations) and (ii) tax benefits attributable to payments made under the TRA. As part of the Reorganization, the TRA was amended to reduce the Company’s future obligation to pay 85% of the tax benefits subject to the TRA to 75% of such realized benefits. Refer to Note 6. Income Taxes for additional information.
C. Apace KY, LLC d/b/a Apace Packaging LLC
Apace KY, LLC d/b/a Apace Packaging LLC (“Apace”) provides packaging solutions pursuant to packaging agreements for Amneal and R&S. Apace markets its services which include bottling and blistering for the pharmaceutical industry. A member of Company management beneficially owns outstanding equity securities of Apace.
D. AzaTech Pharma, LLC
R&S purchases inventory from AzaTech Pharma LLC (“AzaTech”) for resale. A member of Company management beneficially owns outstanding equity securities of AzaTech.
E. Kanan, LLC
Kanan, LLC (“Kanan”) is a real estate company that owns Amneal’s manufacturing facilities located at 65 Readington Road, Branchburg, New Jersey, 131 Chambers Brook Road, Branchburg, New Jersey and 1 New England Avenue, Piscataway, New Jersey. Certain executive officers of the Company beneficially own, through certain revocable trusts, equity securities of Kanan. In addition, they serve on the Board of Managers of Kanan. Amneal leases these facilities from Kanan under two separate triple-net lease agreements that expire in 2027 and 2031, respectively, at an annual rental cost of approximately $2.0 million combined, subject to CPI rent escalation adjustments as provided in the lease agreements.
F. Sellers of AvKARE LLC and R&S
Notes Payable – Related Party
Certain holders of the Rondo Class B Units were also holders of the Sellers Notes. During the year ended December 31, 2024, the Company repaid principal of $44.2 million and interest of $10.0 million associated with the Sellers Notes from cash on hand. As of December 31, 2024, the Sellers Notes and accrued interest had been fully repaid. The Sellers Notes, net of unamortized discount, were included in notes payable-related party and accrued interest was included in related party payables-short term and long-term as of December 31, 2023. For additional information, refer to Note 15. Debt.
Refer to Note 21. Stockholders’ (Deficiency) Equity for related party transactions associated with Rondo.
Tax Distributions
Under the terms of the limited liability company agreement between the Company and the holders of the Rondo Class B Units, Rondo is obligated to make tax distributions to those holders, subject to certain limitations as defined in the Rondo Credit Facility. For the years ended December 31, 2024, 2023 and 2022, tax distributions of $19.8 million, $14.2 million and $6.9 million, respectively, were recorded as reductions of redeemable non-controlling interests. As of December 31, 2024 and 2023, there were no amounts due for tax distributions related to these redeemable non-controlling interests. For further details, refer to Note 21. Stockholders’ (Deficiency) Equity.
G. Sutaria Family Realty, LLC
Industrial Real Estate Holdings NY, LLC (“IRE”) is a real estate management entity, which was the sub-landlord of Amneal’s leased manufacturing facility located at 75 Adams Avenue, Hauppauge, New York. IRE is controlled by a member of the Amneal Group who also serves as an observer on our Board of Directors. Effective June 1, 2020, the lease was assigned to the Company with the consent of the landlord, Sutaria Family Realty, LLC, which is also a related party because a member of Company management is a beneficial owner. Concurrently with the assignment of the lease, the Company exercised a renewal option for $0.1 million to extend the lease by five years until March 31, 2026. Monthly rent payments are $0.1 million and increase by 3% annually.
H. Tracy Properties LLC
R&S leases operating facilities, office and warehouse space from Tracy Properties LLC (“Tracy”). A member of Company management beneficially owns outstanding equity securities of Tracy.
I. Avtar Investments, LLC
Avtar Investments, LLC (“Avtar”) is a private investment firm. Certain executive officers of the Company beneficially own, directly and through certain revocable or irrevocable trusts for the benefit of their immediate families, outstanding equity securities of Avtar. During April 2020, the Company entered into an agreement under which Avtar will provide R&D consulting services.
J. Alkermes Plc
Rondo Partners LLC purchases inventory from Alkermes Plc for resale. A member of the Board of Directors of the Company is also a member of the Board of Directors of Alkermes Plc.
K. AvPROP, LLC
AvKARE LLC leases its operating facilities from AvPROP, LLC (“AvPROP”). A member of Company management beneficially owns outstanding equity securities of AvPROP.
L. Fosun International Limited
Fosun International Limited (“Fosun”) is a Chinese international conglomerate and investment company that was a significant shareholder of the Company. On June 6, 2019, the Company entered into a license and supply agreement with a subsidiary of Fosun, which is a Chinese pharmaceutical company. Under the terms of the agreement, the Company will hold the imported drug license required for pharmaceutical products manufactured outside of China and will supply Fosun with finished, packaged products for Fosun to then sell in the China market. Fosun will be responsible for obtaining regulatory approval in China and for shipping the product from Amneal’s facility to Fosun’s customers in China. In consideration for access to the Company’s U.S. regulatory filings to support its China regulatory filings and for the supply of product, Fosun paid the Company a $1.0 million non-refundable fee, net of tax, in July 2019 and was required to pay the Company $0.3 million for each of eight products upon the first commercial sale of each in China in addition to a supply price and a profit share. On August 11, 2023, the Company and Fosun amended the license and supply agreement to, among other things, (i) increase the products in the agreement from eight to ten, (ii) eliminate the first commercial sales milestone of $0.3 million for each product and (iii) decrease the profit share percentage applicable to all products.
On August 12, 2021, the Company entered into an active pharmaceutical ingredient (“API”) co-development agreement with a subsidiary of Fosun. Under the terms of the agreement, the Company provided Fosun a license to manufacture and sell two pharmaceutical products outside of the U.S. Fosun will be responsible for obtaining regulatory approval outside the U.S. Fosun paid the Company a $0.2 million non-refundable fee in 2021 and will be required to pay the Company $0.1 million for each of the two products upon the first commercial sale of each in China in addition to a profit share.
Effective March 31, 2024, Fosun was no longer a significant shareholder of the Company; therefore, it was no longer a related party as of that date.
M. TPG Capital BD, LLC
TPG Capital BD, LLC (“TPG Capital”), provided the Company with advice and assistance with respect to the refinancing of the Term Loan Due 2025 and the Amended New Revolving Credit Facility for which the Company paid TPG Capital $3.0 million. An observer of our Board is a partner in TPG Capital. Refer to Note 15. Debt for additional information on the refinancing of the Company’s debt.
N. R&S Solutions LLC
R&S Solutions LLC provides logistic services to the Company. A member of Company management beneficially owns outstanding equity securities of R&S Solutions LLC.
O. PharmaSophia, LLC
PharmaSophia, LLC (“PharmaSophia”) is a joint venture formed by Nava Pharma, LLC (“Nava”) and Oakwood Laboratories, LLC for the purpose of developing certain products. Certain executive officers of the Company beneficially own, directly and through certain revocable or irrevocable trusts for the benefit of their immediate families, outstanding equity securities of Nava. Nava beneficially owns 50% of the outstanding equity securities of PharmaSophia. In addition, these executive officers also serve on the Board of Managers of PharmaSophia.
PharmaSophia and Nava are parties to an R&D agreement pursuant to which Nava provides R&D services to PharmaSophia (the “Nava Agreement”). Nava subcontracted this obligation to Amneal under a subcontract R&D services agreement pursuant to which Amneal provides R&D services to Nava in connection with the products being developed by PharmaSophia. On August 28, 2023, Amneal and Nava terminated the subcontract R&D services agreement by mutual consent.
In October 2022, PharmaSophia and Amneal entered into an exclusive license and commercialization agreement (the “PharmaSophia Agreement”) to develop, manufacture, and sell one injectable product. Under the terms of the agreement, Amneal committed to spend up to $6.0 million to further develop the product, including all related expenses up to submission of the ANDA, which will be owned by Amneal. Also under the terms of the PharmaSophia Agreement, PharmaSophia settled a liability of $1.1 million payable to Amneal under the terms of the Nava Agreement by reducing the amount of Amneal’s committed spending under the terms of the PharmaSophia Agreement to $4.9 million. Amneal recorded $1.1 million of research and development expense for the year ended December 31, 2022 as a result of the settlement. PharmaSophia will receive a 50% profit share for all sales of product made by Amneal under the PharmaSophia Agreement.
P. TPG Operations, LLC
TPG Operations LLC (“TPG Operations”) is a private investment firm that provides financial services. An observer of our Board is a partner in TPG Capital. In March 2020, the Company entered into an agreement in which TPG Operations provided financial consulting services for a period of seven months. The agreement was subsequently extended until March 2022.
Q. Asana Biosciences, LLC
Asana Biosciences, LLC (“Asana”) is an early-stage drug discovery and R&D company focusing on several therapeutic areas, including oncology, pain and inflammation. Certain executive officers of the Company beneficially own, directly and through certain revocable or irrevocable trusts for the benefit of their immediate families, outstanding equity securities of Asana. In addition, they serve on the Board of Managers of Asana. From time to time, Amneal provides R&D services to Asana under a development and manufacturing agreement and storage services under a storage agreement.
Other Agreements
Tarsadia Investments, LLC
Tarsadia Investments, LLC (“Tarsadia”) is a private investment firm that provides financial services. Tarsadia is a significant shareholder of the Company. A member of Amneal Group, and an observer to the Board, is the Chairman and Founder of Tarsadia. Another member of the Amneal Group, and a member of the Board, is a Managing Director of Tarsadia. Tarsadia offers capital and strategic support for companies with substantial growth potential primarily in the healthcare, financial services, real estate, and clean technology sectors. The Company entered into an agreement in which Tarsadia will provide financial consulting services. The services are not expected to have a material impact to the Company’s financial statements. There was no activity for the years ended December 31, 2024, 2023 and 2022.
24. Employee Benefit Plans
The Company has voluntary defined contribution plans covering eligible employees in the U.S. which provide for a Company match. For the years ended December 31, 2024, 2023 and 2022, the Company made matching contributions of $9.2 million, $9.9 million and $9.5 million, respectively.
The Company also has a deferred compensation plan for certain former executives and employees of Impax, which the Company acquired in 2018, some of whom are currently employed by the Company. In January 2019, the Company announced that it will no longer accept contributions from employees or make matching contributions for the deferred compensation plan. Deferred compensation liabilities are recorded at the value of the amount owed to the plan participants, with changes in value recognized as compensation expense. The calculation of the deferred compensation plan obligation is derived by reference to hypothetical investments selected by the participants and is included in accounts payable and accrued expenses and other long-term liabilities. Refer to Note 16. Other Long-Term Liabilities and Note 18. Fair Value Measurements for additional information.
25. Segment Information
The Company has three reportable segments: Affordable Medicines (formerly known as Generics), Specialty, and AvKARE.
During the fourth quarter of 2024, the Company changed the name of its Generics segment to “Affordable Medicines” to reflect the full product offering of the segment. The name change did not result in any change to the composition of the Company’s reportable segments and, therefore, did not result in any change to its historical results.
Affordable Medicines
The Company’s Affordable Medicines segment includes approximately 270 product families covering an extensive range of dosage forms and delivery systems, including both immediate and extended-release oral solids, powders, liquids, sterile injectables, nasal sprays, inhalation and respiratory products, biosimilar products, ophthalmics, films, transdermal patches and topicals.
Specialty
The Company’s Specialty segment includes branded products, primarily focused on central nervous system disorders, including Parkinson’s disease, and endocrine disorders.
AvKARE
The Company’s AvKARE segment provides pharmaceuticals, medical and surgical products and services primarily to governmental agencies, predominantly focused on the U.S. Department of Defense and the U.S. Department of Veterans Affairs. AvKARE is a re-packager of bottle and unit dose pharmaceuticals under the registered names of AvKARE and AvPAK. AvKARE is also a wholesale distributor of pharmaceuticals, over the counter drugs and medical supplies to its retail and institutional customers that are located throughout the U.S. focused primarily on entities that provide care to low-income and uninsured patients. Operating results for the sale of Amneal products by AvKARE are included in the Company’s Affordable Medicines reportable segment.
Chief Operating Decision Makers
The Company’s Co-Chief Executive Officers are the Company’s chief operating decision makers (“CODMs”). The CODMs evaluate the financial performance of the Company based upon segment operating income (loss). Items below operating income (loss) are not reported by segment, since they are excluded from the measure of segment profitability reviewed by the Company’s CODMs. Additionally, general and administrative expenses, certain selling expenses, certain litigation settlements, and non-operating income and expenses are included in “Corporate and Other.” The Company does not report balance sheet information by segment since it is not reviewed by the Company’s CODMs.
The tables below present segment information reconciled to total Company financial results, with segment operating income or loss, including gross profit less direct selling expenses, R&D expenses, and other operating expenses to the extent specifically identified by segment (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, 2024 | | Affordable Medicines (1) | | Specialty | | AvKARE | | Corporate
and Other | | Total
Company |
Net revenue (2) | | $ | 1,685,263 | | | $ | 445,749 | | | $ | 662,945 | | | $ | — | | | $ | 2,793,957 | |
| Cost of goods sold | | 1,011,363 | | | 202,821 | | | 559,335 | | | — | | | 1,773,519 | |
| Gross profit | | 673,900 | | | 242,928 | | | 103,610 | | | — | | | 1,020,438 | |
| Selling, general and administrative | | 129,578 | | | 109,658 | | A | 60,709 | | | 176,491 | | | 476,436 | |
| Research and development | | 171,771 | | B | 18,943 | | B | — | | | — | | | 190,714 | |
| Intellectual property legal development expenses | | 5,685 | | | 160 | | | — | | | — | | | 5,845 | |
| Restructuring and other charges | | 70 | | | 1,517 | | | — | | | 768 | | | 2,355 | |
| Change in fair value of contingent consideration | | — | | | (930) | | | — | | | — | | | (930) | |
| Charges related to legal matters, net | | 96,692 | | | — | | | — | | | — | | | 96,692 | |
| Operating income (loss) | | $ | 270,104 | | | $ | 113,580 | | | $ | 42,901 | | | $ | (177,259) | | | $ | 249,326 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, 2023 | | Affordable Medicines (1) | | Specialty | | AvKARE | | Corporate
and Other | | Total
Company |
Net revenue (2) | | $ | 1,471,401 | | | $ | 390,457 | | | $ | 531,749 | | | $ | — | | | $ | 2,393,607 | |
| Cost of goods sold | | 913,869 | | | 214,277 | | | 444,896 | | | — | | | 1,573,042 | |
| Gross profit | | 557,532 | | | 176,180 | | | 86,853 | | | — | | | 820,565 | |
| Selling, general and administrative | | 119,912 | | | 88,137 | | A | 55,341 | | | 166,285 | | | 429,675 | |
| Research and development | | 132,233 | | B | 31,717 | | B | — | | | — | | | 163,950 | |
| In-process research and development impairment charges | | 26,500 | | | 4,300 | | | — | | | — | | | 30,800 | |
| Intellectual property legal development expenses | | 3,708 | | | 120 | | | — | | | — | | | 3,828 | |
| Restructuring and other charges | | 211 | | | 1,105 | | | — | | | 433 | | | 1,749 | |
| Change in fair value of contingent consideration | | — | | | (14,497) | | | — | | | — | | | (14,497) | |
| (Credit) charges related to legal matters, net | | (64) | | | — | | | — | | | 1,888 | | | 1,824 | |
| Other operating income | | (1,138) | | | — | | | — | | | — | | | (1,138) | |
| Operating income (loss) | | $ | 276,170 | | | $ | 65,298 | | | $ | 31,512 | | | $ | (168,606) | | | $ | 204,374 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Year Ended December 31, 2022 | | Affordable Medicines (1) | | Specialty | | AvKARE | | Corporate
and Other | | Total
Company |
Net revenue (2) | | $ | 1,432,073 | | | $ | 374,121 | | | $ | 406,110 | | | $ | — | | | $ | 2,212,304 | |
| Cost of goods sold | | 896,031 | | | 182,432 | | | 349,133 | | | — | | | 1,427,596 | |
| Gross profit | | 536,042 | | | 191,689 | | | 56,977 | | | — | | | 784,708 | |
| Selling, general and administrative | | 109,781 | | | 90,031 | | A | 53,659 | | | 146,229 | | | 399,700 | |
| Research and development | | 167,509 | | B | 28,179 | | B | — | | | — | | | 195,688 | |
| In-process research and development impairment charges | | 12,970 | | | — | | | — | | | — | | | 12,970 | |
| Intellectual property legal development expenses | | 4,251 | | | 107 | | | — | | | — | | | 4,358 | |
| Acquisition, transaction-related and integration expenses | | 25 | | | 49 | | | — | | | 635 | | | 709 | |
| Restructuring and other charges | | 821 | | | — | | | — | | | 600 | | | 1,421 | |
| Change in fair value of contingent consideration | | — | | | 731 | | | — | | | — | | | 731 | |
| Insurance recoveries for property losses and associated expenses, net | | (1,911) | | | — | | | — | | | — | | | (1,911) | |
| Charges related to legal matters, net | | 22,400 | | | — | | | — | | | 247,530 | | | 269,930 | |
| Other operating income | | (3,960) | | | — | | | — | | | — | | | (3,960) | |
| Operating income (loss) | | $ | 224,156 | | | $ | 72,592 | | | $ | 3,318 | | | $ | (394,994) | | | $ | (94,928) | |
(1)Operating results for the sale of Amneal products by AvKARE are included in Affordable Medicines.
(2)Net revenue from external customers is attributed to countries based on the location of the product shipment. For the years ended December 31, 2024, 2023, and 2022, net revenue from external customers attributed to foreign countries was immaterial.
Significant Expense Categories Provided to the Chief Operating Decision Makers
Selling, General and Administrative Expenses - Specialty Segment
A The CODMs review certain selling, general and administrative expenses (“SG&A”) for the Specialty segment and, separately, on a departmental basis. The CODMs do not review SG&A for the Affordable Medicines and AvKARE segments. SG&A for the Specialty segment was comprised of the following (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | For the Year Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Employee compensation and benefits | | $ | 34,908 | | | $ | 36,352 | | | $ | 34,893 | |
| Product marketing | | 44,179 | | | 27,431 | | | 31,481 | |
| Commercial operations and salesforce | | 25,567 | | | 20,748 | | | 20,256 | |
Other (1) | | 5,004 | | | 3,606 | | | 3,401 | |
| Total | | $ | 109,658 | | | $ | 88,137 | | | $ | 90,031 | |
(1)Other includes professional fees and other expenses not presented to the CODMs.
Research and Development Expenses - Affordable Medicines and Specialty Segments
B Research and development expenses for the Affordable Medicines and Specialty segments were comprised of the following (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | For the Year Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| | Affordable Medicines | | Specialty | | Affordable Medicines | | Specialty | | Affordable Medicines | | Specialty |
| Employee compensation and benefits | | $ | 46,551 | | | $ | 7,863 | | | $ | 47,185 | | | $ | 10,120 | | | $ | 51,945 | | | $ | 7,987 | |
| Materials and supplies | | 40,585 | | | 1,221 | | | 21,382 | | | 5,107 | | | 32,912 | | | 1,305 | |
| Product development and studies | | 3,707 | | | 1,408 | | | 8,434 | | | 2,967 | | | 23,203 | | | 2,182 | |
| Regulatory fees | | 5,439 | | | — | | | 8,913 | | | 2,024 | | | 5,540 | | | 3,117 | |
| Milestones | | 38,475 | | | — | | | 8,425 | | | — | | | 6,414 | | | 4,500 | |
| Facilities costs | | 6,714 | | | 5,858 | | | 7,485 | | | 5,655 | | | 9,399 | | | 3,680 | |
Other (1) | | 30,300 | | | 2,593 | | | 30,409 | | | 5,844 | | | 38,096 | | | 5,408 | |
| Total | | $ | 171,771 | | | $ | 18,943 | | | $ | 132,233 | | | $ | 31,717 | | | $ | 167,509 | | | $ | 28,179 | |
(1)For the Affordable Medicines segment, other includes repairs and maintenance, outside testing, equipment calibration and other expenses not presented to the CODMs. For the Specialty segment, other includes repairs and maintenance, outside testing, professional fees and other expenses not presented to the CODMs.
Long-Lived Assets
Long-lived assets, which are comprised of property, plant and equipment, net and operating and financing lease right-of-use assets, are attributed based on physical location. Long-lived assets by country were as follows (in thousands):
| | | | | | | | | | | | | | |
| | December 31, 2024 | | December 31, 2023 |
| United States | | $ | 310,702 | | | $ | 316,947 | |
| India | | 159,650 | | | 179,401 | |
| Ireland | | 53,341 | | | 53,789 | |
| | $ | 523,693 | | | $ | 550,137 | |
26. Subsequent Events
Term Loan Due 2025
In January 2025, the Company paid the entire remaining principal balance of $192.0 million outstanding on its Term Loan Due 2025, plus accrued interest thereon of $0.7 million, with $190.0 million of new borrowings under the Amended New Revolving Credit Facility and cash on hand.
Securities Purchase Agreement and License and Collaboration Agreement - Related Party
On January 3, 2025, the Company entered into a securities purchase agreement and a license and collaboration agreement with Ellodi Pharmaceuticals, L.P. (“Ellodi”) and certain entities affiliated with TPG for $3 million. An observer of our Board is a partner in TPG Capital and a board director of Ellodi. Ellodi is a gastroenterology-focused specialty pharmaceutical company.
Amneal has the option to obtain, under certain conditions, an exclusive royalty bearing and sub-licensable world-wide license to a late-stage gastroenterology-focused pipeline product under development. If exercised, the Company will be responsible for remaining development activities and obtaining regulatory approval of the product. The license and collaboration agreement provides for potential future milestone payments to Ellodi for regulatory and commercial milestones of up to $48.5 million and royalties on commercial sales.
EXHIBIT INDEX
| | | | | | | | |
| Exhibit No. | | Description of Document |
| | |
| | Equity Purchase Agreement, dated December 10, 2019, by and among the Jerry Brian Shirley Business Trust, the Darren Thomas Shirley Business Trust, the Steve Shirley Business Trust, the Jerry Shirley Business Trust, Troy Mizell, Darrell Calvert, AvKARE, Dixon-Shane, LLC d/b/a R&S Northeast LLC and Rondo Acquisition LLC. In accordance with the instructions to Item 601(b)(2) of Regulation S-K, the schedules and exhibits to the Equity Purchase Agreement are not filed herewith. The Equity Purchase Agreement identifies such schedules and exhibits, including the general nature of their content. The Company undertakes to provide such schedules and exhibits to the SEC upon request (incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed on December 10, 2019). |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | | | | | | | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | Second Amended and Restated Stockholders Agreement, dated as of December 16, 2017, among Atlas Holdings, Inc., Amneal Pharmaceuticals Holdings Company LLC, AP Class D Member, LLC, AP Class E Member, LLC and AH PPU Management, LLC (incorporated by reference to Exhibit 10.4 to the Company's Registration Statement on Form S-1, filed on May 7, 2018). |
| | |
| | Amendment No. 1, dated as of August 2, 2019, to Second Amended and Restated Stockholders Agreement, by and among Amneal Pharmaceuticals Holding Company, LLC, a Delaware limited liability company, AP Class D Member, LLC, a Delaware limited liability company, AP Class E Member, LLC, a Delaware limited liability company, AH PPU Management, LLC, a Delaware limited liability company, and Amneal Pharmaceuticals, Inc. (incorporated by reference to Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2019, filed on August 5, 2019). |
| | |
| | |
| | |
| | | | | | | | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | Guaranty and Security Agreement, dated as of January 31, 2020, by and among Rondo Intermediate Holdings, LLC, and Rondo Holdings, LLC, AvKARE, R&S Northeast, and the Administrative Agent (incorporated by reference to Exhibit 2.3 to the Company's Current Report on Form 8-K, filed on February 3, 2020). |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | Amendment No. 1 to Revolving Credit and Term Loan Agreement, dated as of April 20, 2023, by and among Rondo Holdings, LLC, Rondo Intermediate Holdings, LLC, the Subsidiary Loan Parties party hereto, the Lenders party hereto and Truist Bank as Administrative Agent (incorporated by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2023, filed on November 9, 2023). |
| | |
| | Amendment No. 2 Revolving Credit and Term Loan Agreement, dated as of September 21, 2023, by and among Rondo Holdings, LLC, Rondo Intermediate Holdings, LLC, the Subsidiary Loan Parties party hereto, the Lenders party hereto and Truist Bank as Administrative Agent (incorporated by reference to Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2023, filed on November 9, 2023). |
| | |
| | |
| | |
| | | | | | | | |
| | |
| | |
| | |
| | |
| | Amendment to No. 1, dated as of November 7, 2023, to the Employment Agreement, dated as of January 24, 2018, by and among Amneal Pharmaceuticals LLC, Amneal Holdings, LLC and Andrew Boyer, as modified (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on November 8, 2023).† |
| | |
| | |
| | |
| | |
| | |
| | Amendment No. 1, dated as of November 7, 2023, to the Tax Receivable Agreement, dated as of May 4, 2018, by and among Amneal Intermediate Inc. (formerly Amneal Pharmaceuticals, Inc.), Amneal Pharmaceuticals LLC and the certain former Members of Amneal Pharmaceuticals LLC from time to time party thereto among Amneal Intermediate Inc. (formerly Amneal Pharmaceuticals, Inc.), Amneal Pharmaceuticals, Inc. (formerly Amneal NewCo Inc.), Amneal Pharmaceuticals LLC and Padmesh Patel, solely in his capacity as the Member Representative (incorporated by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K filed on November 8, 2023). |
| | |
| | Third Amended and Restated Stockholders Agreement, dated as of November 7, 2023, by and among AP Class D Member, LLC, AP Class E Member, LLC, AH PPU Management, LLC, Amneal Intermediate (formerly Amneal Pharmaceuticals, Inc. and Atlas Holdings, Inc.) and Amneal Pharmaceuticals, Inc. (formerly Amneal NewCo Inc.) (incorporated by reference to Exhibit 10.5 to the Company’s Current Report on Form 8-K filed on November 8, 2023). |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | | | | | | | |
| 101 | | The following materials from the Company’s Annual Report on Form 10-K for the year ended December 31, 2024, formatted in inline XBRL (eXtensible Business Reporting Language): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Operations, (iii) Consolidated Statements of Comprehensive Loss, (iv) Consolidated Statements of Changes in Stockholders’ Equity (Deficiency), (v) Consolidated Statements of Cash Flows, and (vi) Notes to Consolidated Financial Statements. |
| | |
| 104 | | Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101) |
* Filed herewith
** This certificate is being furnished solely to accompany the report pursuant to 18 U.S.C. 1350 and is not being filed for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, and is not to be incorporated by reference into any filing of the Company, whether made before or after the date hereof, regardless of any general incorporation language in such filing.
*** Certain portions of this exhibit have been omitted pursuant to Item 601(b)(10)(iv) of Regulation S-K.
† Denotes management compensatory plan or arrangement.
The agreements and other documents filed as exhibits to this report are not intended to provide factual information or other disclosure other than the terms of the agreements or other documents themselves, and you should not rely on them for that purpose. In particular, any representations and warranties made by the Company in these agreements or other documents were made solely within the specific context of the relevant agreement or document and may not describe the actual state of affairs at the date they were made or at any other time.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| | | | | | | | |
| Date: February 28, 2025 | Amneal Pharmaceuticals, Inc. |
| | | |
| | By: | /s/ Anastasios Konidaris |
| | | Anastasios Konidaris |
| | | Executive Vice President, Chief Financial Officer
(Principal Financial and Accounting Officer) |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | | | | | | | | | | | |
| Signature | | Title | | Date |
| /s/ Chirag Patel | | President, Co-Chief Executive Officer and Director | | February 28, 2025 |
| Chirag Patel | | (Co-Principal Executive Officer) | | |
| | | | |
| /s/ Chintu Patel | | Co-Chief Executive Officer and Director | | February 28, 2025 |
| Chintu Patel | | (Co-Principal Executive Officer) | | |
| | | | |
| /s/ Anastasios Konidaris | | Executive Vice President, Chief Financial Officer | | February 28, 2025 |
| Anastasios Konidaris | | (Principal Financial and Accounting Officer) | | |
| | | | |
| /s/ Paul M. Meister | | | | February 28, 2025 |
| Paul M. Meister | | Chairman of the Board and Director | | |
| | | | |
| /s/ Emily Peterson Alva | | | | February 28, 2025 |
| Emily Peterson Alva | | Director | | |
| | | | |
| /s/ Deb Autor | | | | February 28, 2025 |
| Deb Autor | | Director | | |
| | | | |
| /s/ J. Kevin Buchi | | | | February 28, 2025 |
| J. Kevin Buchi | | Director | | |
| | | | |
| /s/ Jeffrey P. George | | | | February 28, 2025 |
| Jeffrey P. George | | Director | | |
| | | | |
| /s/ John J. Kiely, Jr. | | | | February 28, 2025 |
| John J. Kiely, Jr. | | Director | | |
| | | | |
| /s/ Ted Nark | | | | February 28, 2025 |
| Ted Nark | | Director | | |
| | | | |
| /s/ Gautam Patel | | | | February 28, 2025 |
| Gautam Patel | | Director | | |
| | | | |
| /s/ Shlomo Yanai | | | | February 28, 2025 |
| Shlomo Yanai | | Director | | |