Exhibit 99.2
![]()
Management’s Report on Financial Position and Operating Results
For the three-month period ended March 31, 2019
LETTER TO SHAREHOLDERS
Dear Fellow Shareholders,
The DPX-Survivac program continues to be a major value-driver for IMV, with its unique mechanism of action providing significant clinical differentiation and a potential much-needed innovation for hard-to-treat cancer. Highlights of our progress this quarter includes:
Reported promising initial data from the phase 2 cohort of the DeCidE1 clinical study which underscores the potential of DPX-Survivac as monotherapy;
Awarded grant with le Centre de Recherche du CHU de Québec-Université Laval to develop a first in-class dual target T Cell therapy in bladder cancer, and in bladder cancer; and
Completed a $29.46 million financing with Wells Fargo acting as lead underwriter that provided the Company that provided the Corporation with increased financial flexibility and an expansion of our shareholder base.
DPX-Survivac Clinical Program Updates:
Phase 2 Cohort of the DeCedE1 Clinical Study in Ovarian Cancer
We provided a clinical update in March indicating that six patients receiving DPX-Survivac monotherapy with intermittent low-dose cyclophosphamide had reached the first CT scan assessment. Key related findings were as follows:
83% of the participants (5 of 6) showed stable disease, including two tumor regressions; and
80% (4 of 5) of those with stable disease were subjects with a lower baseline tumor burden of less than 5 centimeters, which also included the two tumor regressions.
In earlier stages of this trial, durable clinical responses occurred after 140 days, and at the date of this latest update, they had lasted for 20 months or more. The amended phase 2 cohort of the DeCidE1 trial focuses on patients with low tumor burden (less than 5 centimeters) and is targeting enrollment of at least 16 additional patients at sites in the U.S. and Canada.
IMV will present additional data on DeCidE1 at the 2019 American Society of Clinical Oncology (ASCO) annual meeting.
Phase 2 Study in Combination with KEYTRUDA® in Relapsed/Refractory DLBCL (SPiReL)
As of April 5, 2019, ten patients had been enrolled across four different clinical sites in Canada. Additional patients are being screened and IMV expects to report updated clinical data at the bi-annual International Conference on Malignant Lymphoma, which will be held in Lugano Switzerland in June 2019.
Phase 2 Basket Trial in Combination with KEYTRUDA® in Multiple Solid Tumors
Screening and enrollment of patients is ongoing at multiple clinical sites across the U.S. and Canada for five cohorts of patients with bladder, liver (hepatocellular carcinoma), ovarian, or non-small cell lung (NSCLC) cancers, as well as tumors shown to be positive for the microsatellite instability high (MSI-H) biomarker.
The first patients have been treated in the ovarian, NSCLC and MSI-H cohorts and IMV expects to report preliminary clinical results on several of the solid tumor indications before the end of 2019.
The following table indicates IMV expected milestones between now and the first half of 2020:
| Phase 2 monotherapy clinical results in Ovarian – ASCO | June 2019 |
| Phase 2 clinical results with Merck Keytruda in DLBCL – ICML | June 2019 |
| Preliminary clinical results Basket trial in 5 indications | H2 2019 |
| Topline monotherapy clinical results in Ovarian | H2 2019 |
| Top line clinical results for Basket trial | H2 2020 |
We are pleased at the steady progress we’ve made this far in 2019, and we look forward to leveraging our technology to improve immunotherapy treatment options, particularly in underserved cancers. We are grateful for the continued support of our shareholders and partners and look forward to a very productive remainder of 2019.

Frederic Ors
Chief Executive Officer
3
MANAGEMENT DISCUSSION AND ANALYSIS (“MD&A”)
The following analysis provides a review of the unaudited interim condensed consolidated results of operations, financial condition, and cash flows for the three-month period ended March 31, 2019 (“Q1 2019”), with information compared to the three months ended March 31, 2018 (“Q1 2018”), for IMV Inc. (“IMV” or the “Corporation”). This analysis should also be read in conjunction with the information contained in the annual audited consolidated financial statements and related notes for the years ended December 31, 2018 and December 31, 2017.
The Corporation prepares its unaudited interim condensed consolidated financial statements in accordance with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board (IASB). Management is responsible for the preparation of the consolidated financial statements and other financial information relating to the Corporation included in this report. The Board of Directors is responsible for ensuring that management fulfills its responsibilities for financial reporting. In furtherance of the foregoing, the Board of Directors has appointed an Audit Committee comprised of independent directors. The Audit Committee meets with management and the auditors in order to discuss the results of operations and the financial condition of the Corporation prior to making recommendations and submitting the consolidated financial statements to the Board of Directors for its consideration and approval for issuance to shareholders. The information included in this MD&A is as of May 9, 2019, the date when the Board of Directors approved the Corporation’s unaudited interim condensed consolidated financial statements for the three months ended March 31, 2019, on the recommendation of the Audit Committee.
Amounts presented in this MD&A are approximate and have been rounded to the nearest thousand except for per share data. Unless specified otherwise, all amounts are presented in Canadian dollars.
Additional information regarding the business of the Corporation, including the Annual Information Form of the Corporation for the year ended December 31, 2018 (the “AIF”) and included in the Corporation’s registration statement on Form 40-F filed with the U.S. Securities and Exchange Commission, is available on SEDAR at www.sedar.com and on EDGAR at www.sec.gov/edgar.
FORWARD-LOOKING STATEMENTS
Certain statements in this MD&A may constitute “forward-looking” statements which involve known and unknown risks, uncertainties and other factors that may cause the actual results, performance, or achievements of the Corporation, or industry results, to be materially different from any future results, performance, or achievements expressed or implied by such forward-looking statements. When used in this MD&A, such statements use such words as “will”, “may”, “could”, “intends”, “potential”, “plans”, “believes”, “expects”, “projects”, “estimates”, “anticipates”, “continue”, “potential”, “predicts” or “should” and other similar terminology. These statements reflect current expectations of management regarding future events and operating performance and speak only as of the date of this MD&A. Forward-looking statements include, among others:
The Corporation’s business strategy;
Statements with respect to the sufficiency of the Corporation’s financial resources to support its activities;
Potential sources of funding;
The Corporation’s ability to obtain necessary funding on favorable terms or at all;
The Corporation’s expected expenditures and accumulated deficit level;
The Corporation’s expected outcomes from its ongoing and future research and research collaborations;
The Corporation’s exploration of opportunities to maximize shareholder value as part of the ordinary course of its business through collaborations, strategic partnerships, and other transactions with third parties;
The Corporation’s plans for the research and development of certain product candidates;
The Corporation’s strategy for protecting its intellectual property;
The Corporation’s ability to identify licensable products or research suitable for licensing and commercialization;
The Corporation’s ability to obtain licences on commercially reasonable terms;
The Corporation’s plans for generating revenue;
The Corporation’s plans for future clinical trials; and
The Corporation’s hiring and retention of skilled staff.
Forward-looking statements involve significant risks and uncertainties, should not be read as guarantees of future performance or results, and will not necessarily be accurate indications of whether or not such results will be achieved. A number of factors could cause actual results to differ materially from the results discussed in the forward-looking statements, including, but not limited to, the factors discussed in the AIF, under the heading “Risk Factors and Uncertainties.” Although the forward-looking statements contained in this MD&A are
4
based upon what management of the Corporation believes are reasonable assumptions, the Corporation cannot provide any assurance to investors that actual results will be consistent with these forward-looking statements and should not be unduly relied upon by investors.
Actual results, performance and achievements are likely to differ, and may differ materially, from those expressed or implied by the forward-looking statements contained in this MD&A. Such statements are based on a number of assumptions which may prove to be incorrect, including, but not limited to, assumptions about:
Obtaining additional funding on reasonable terms when necessary;
Positive results of pre-clinical studies and clinical trials;
The Corporation’s ability to successfully develop existing and new products;
The Corporation’s ability to hire and retain skilled staff;
The products and technology offered by the Corporation’s competitors;
General business and economic conditions;
The Corporation’s ability to protect its intellectual property;
The Corporation’s ability to manufacture its products and to meet demand; and
Regulatory approvals.
These statements reflect management’s current views and beliefs and are based on estimates, assumptions, and information currently available to, and considered reasonable by, management. The information contained herein is dated as of May 9, 2019, the date of the Board’s approval of the Q1 2019 unaudited interim condensed consolidated financial statements and of the MD&A. For additional information on risks, uncertainties, and assumptions, including a more detailed assessment of the risks that could cause actual results to materially differ from current expectations, please refer to the AIF of IMV filed on SEDAR at www.sedar.com and included in the registration statement on Form 40-F filed on EDGAR at www.sec.gov/edgar.
CORPORATE OVERVIEW
IMV is a clinical-stage biopharmaceutical company dedicated to making immunotherapy more effective, more broadly applicable, and more widely available to people facing cancer and other serious diseases. IMV is pioneering a new class of immunotherapies based on the Corporation’s proprietary drug delivery platform (“DPX”). This patented technology leverages a novel mechanism of action (“MOA”) discovered by the Corporation. This MOA does not release the active ingredients at the site of injection but forces an active uptake and delivery of active ingredients into immune cells and lymph nodes. It enables the programming of immune cellsin vivo, which are aimed at generating powerful new synthetic therapeutic capabilities. DPX’s no-release MOA can be leveraged to generate “first-in-class” T cell therapies with the potential, in the opinion of IMV, to be transformative in the treatment of cancer.
The Corporation’s first cancer immunotherapy uses survivin-based peptides licensed from Merck KGaA, on a world-wide exclusive basis, formulated in DPX (“DPX-Survivac”). Survivin is a well characterized and tumour associated antigen known to be overexpressed in more than 20 different cancers. DPX-Survivac leverages the MOA of DPX to generate a constant flow of T cells in the blood that are targeted against survivin expressed on cancer cells. It is comprised of five minimal MHC class I peptides to activate naïve T cells against survivin.
DPX-Survivac is currently being tested in:
A phase 2 clinical trial that evaluates DPX-Survivac in an open label safety and efficacy study in ovarian cancer patients with advanced platinum-sensitive and resistant ovarian cancer;
Two investigator-sponsored phase 2 clinical trials in combination with the checkpoint inhibitor Keytruda® (pembrolizumab) of Merck & Co Inc. (“Merck”) in patients with recurrent, platinum-resistant, and sensitive ovarian cancer and in patients with measurable or recurrent diffuse large B cell lymphoma (“DLBCL”); and
A phase 2 basket trial in combination with Merck’s Keytruda® (pembrolizumab) in patients with select advanced or recurrent solid tumours in bladder, liver (hepatocellular carcinoma), ovarian, or non-small-cell lung (NSCLC) cancers, as well as tumours shown to be positive for the microsatellite instability high (MSI-H) biomarker.
In infectious disease vaccine applications, the Corporation has completed a demonstration phase 1 clinical trial with a target against the respiratory syncytial virus (“RSV”). The Corporation also has a commercial licensing agreement with Zoetis for the development of two cattle vaccines and is also conducting several research and clinical collaborations, including a collaboration with the Dana-Farber Cancer Institute (“Dana-Farber”) for Human Papillomavirus (“HPV”) related cancers and with Leidos, Inc. (“Leidos”) in the United States for the development of vaccine candidates for malaria and the Zika virus.
5
The common shares of the Corporation are listed on the Nasdaq Stock Market LLC and on the Toronto Stock Exchange under the symbol “IMV.”
BUSINESS MODEL AND STRATEGY
IMV is dedicated to making immunotherapy more effective, more broadly applicable, and more widely available to people facing cancer. The Corporation’s lead product candidate, DPX-Survivac, has demonstrated the ability to induce prolonged T cell activation leading to tumour regressions in advanced ovarian cancer and is currently being used in clinical trials as a monotherapy and in combination with Merck’s KEYTRUDA® checkpoint inhibitor.
Foremost, the Corporation’s clinical strategy is to establish monotherapy activity of DPX-Survivac in order to increase value, de-risk clinical development, and to target late stage unmet medical needs for a shorter path to clinical demonstration and first regulatory approval and to establish strategic partnerships to support further development and commercialization. In addition, the Corporation is evaluating DPX Survivac in combination with Merck’s KEYTRUDA® checkpoint inhibitor in multiple oncology targets.
The Corporation is focusing on a fast path to market in ovarian and diffuse large DLBCL cancers and on repeating its clinical demonstrations of activity in other indications.
In collaboration with commercial and academic partners, the Corporation is also expanding the application of DPX as a delivery platform for other applications. Pre-clinical and clinical studies have indicated to date that the Corporation’s delivery platform may allow for the development of enhanced vaccines for a wide range of infectious diseases by generating a stronger and more durable immune response than is possible with existing delivery methods.
The Corporation intends to be opportunistic in the development of products by exploring a variety of avenues, including co-development through potential collaborations, strategic partnerships or other transactions with third parties. The Corporation may seek additional equity and non-dilutive funding and partnerships to advance the development of its product candidates.
PLATFORM AND PRODUCTS IN DEVELOPMENT
Delivery Platform
The DPX platform is a unique and patented formulation discovered by the Corporation that provides a new way to deliver active ingredients to the immune system using a novel MOA. This MOA does not release the active ingredients at the site of injection but forces an active uptake and delivery of active ingredients into immune cells and lymph nodes. IMV is exploiting this MOA to pioneer a new class of immunotherapies that represents a paradigm shift from current approaches. By not releasing the active ingredients at the site of injection, it bypasses the steps involved in conventional immune “native responses,” such as vaccines, and enables access and programming of immune cellsin-vivoto generate new “synthetic” therapeutic capabilities. The DPX no-release MOA can be leveraged to generate “first-in-class” T cell therapies with the potential to be transformative in the treatment of cancer. The Corporation believes that the novel MOA of DPX makes the platform uniquely suitable for cancer immunotherapies, which are designed to target tumour cells. DPX can induce prolonged, target-specific, and polyfunctional T cell activation, which are postulated to be required for effective tumour control.
Figure 1: Illustrative representation of IMV’s DPX new MOA
The DPX platform is based on active ingredients formulated in lipid nanoparticles and, after freeze drying, suspended directly into oil. DPX-based products are stored in a dry format, which provides the added benefit of an extended shelf life. The formulation is designed to be easy to re-suspend and administer to patients.
6
DPX also has multiple manufacturing advantages: it is fully synthetic; can accommodate hydrophilic and hydrophobic compounds; is amenable to a wide-range of applications (for example, peptides, small-molecules, RNA/DNA, or antibodies); and provides long term stability as well as low cost of goods.
The DPX platform forms the basis of all of IMV’s product development programs.
DPX-Survivac
Product Candidate Overview
DPX-Survivac, the Corporation’s first cancer immunotherapy candidate, uses survivin-based peptides licensed from Merck KGaA on a world-wide exclusive basis that are formulated in DPX. DPX-Survivac leverages the MOA of DPX to generate a constant flow of T cells in the blood that are targeted against survivin expressed on cancer cells, and is comprised of five minimal MHC class I peptides to activate naïve T cells against survivin.
Survivin is a well characterized and recognized tumour associated antigen known to be expressed during fetal development and across most tumour cell types, but it is rarely present in normal non-malignant adult cells. Survivin controls key cancer processes (apoptosis, cell division, and metastasis) and has been associated with chemoresistance and cancer progression. It has been shown that survivin was expressed in all 60 different human tumour lines used in the National Cancer Institute’s cancer drug
screening program and is documented in the literature to be overexpressed in more than 20 indications.

Figure 2: Examples of % of patients with survivin expression in different indications
IMMUNO-ONCOLOGY
DPX-Survivac is being tested in 6 different cancer indications through multiple phase 2 clinical trials.
Ongoing Clinical Trials

DPX- Survivac – Ongoing Clinical Trials
Monotherapy
7
Ovarian subpopulation – DeCidE1 phase 2
The DeCidE1 (DPX-Survivac with low dose intermittent cyclophosphamide) phase 2 study is an open label safety and efficacy study for individuals with advanced platinum-sensitive and resistant ovarian cancer. Primary and secondary end points include:
Safety profile;
Objective Response Rate (ORR) and Duration of Response (DOR) using Recist 1.1 criteria;
Induction of systemic survivin-specific T-cells in the blood; and
Induction of T-cell infiltration into tumours.
As of the March 25, 2019, the data cut-off date, 13 patients had been enrolled in the phase 2 portion of this trial. Five patients were randomized into the DPX-Survivac monotherapy cohort. Seven patients had been randomized into DPX-Survivac/mCPA in combination with epacadostat before the phase 2 protocol was amended to stop enrolment in the combination arm. One of the patients in the combination arm elected to switch to the monotherapy arm of the trial.
The amended phase 2 cohort of the DeCidE1 trial is targeting an enrolment of at least additional 16 patients in the population with a lower tumour burden. Enrolment is continuing at multiple sites in the United States and Canada.
On March 26, 2019, IMV provided preliminary data from the phase 2 cohort of the DeCidE1 clinical study. Six patients receiving DPX-Survivac monotherapy with intermittent low-dose cyclophosphamide (“mCPA”) have reached the first CT scan assessment with key related findings as follows:
83 per cent of the subjects (five of six) show stable disease (“SD”), including two tumour regressions; and
80 per cent (four of five) with stable disease are in subjects with a lower baseline tumour burden (BTB), which also includes the two tumour regressions.
This initial phase 2 data confirms the earlier trends we saw in the phase 1b portion of the study and it supports the potential of DPX-Survivac as a monotherapy and the use of IMV’s patient selection strategy. The Corporation is encouraged by these early initial results and is committed to advancing this program quickly with the goal of providing an additional treatment option to patients with advanced ovarian cancer.
Importantly, in earlier stages of this trial, durable clinical responses occurred after 140 days, and have now lasted for 20 months or more. Additional data at the 140-day mark of this cohort will be available by the end of the first half of 2019.
In December, 2018, IMV met with the U.S. Food and Drug Administration (“FDA”) in a Type B meeting to discuss the results to date of its DeCidE1 clinical trial and continuing development plan, as well as to obtain agency guidance on a potential accelerated regulatory pathway for DPX-Survivac as a T cell immunotherapy for the treatment of advanced ovarian cancer in patients with progressing disease.
The purpose of IMV's Type B meeting with the FDA was to request feedback on the design of the clinical program for DPX-Survivac. This program includes the continuing DeCidE1 phase 2 clinical study and a potential future registration trial for accelerated approval in a subset of ovarian cancer patients.
The FDA reviewed the Corporation’s proposed clinical development plan and acknowledged the potential for accelerated approval in advanced ovarian cancer based on ORR according to Recist 1.1 criteria with reported median DOR. In addition, the FDA provided important guidance on clinical design considerations for different lines of therapy and platinum-sensitive and resistant patient populations.

8
Figure 3: Examples of previous US FDA accelerated approvals in ovarian cancer (source: FDA website)
In addition, IMV submitted a protocol amendment for a predictive enrichment approach to the phase 2 DeCidE1 trial, and further discussed those details with the FDA during the Type B meeting. The phase 2 primary end point, based on ORR per Recist 1.1 criteria, is intended to confirm the high response rate and duration of clinical benefits observed in previously announced results in a patient population defined by a clinical biomarker based on BTB.
The Corporation believes that there is still an urgent medical need in advanced recurrent ovarian cancer (Sources: 1. NCCN Guidelines Ovarian Cancer V2.2018; SEER Ovarian Cancer; JCO, vol 33; 32 Nov 2015, Gyn Onc 133(2014) 624-631):
Nearly 70% of ovarian cancers are diagnosed in advanced stage;
The overall 5-year survival rate is 46.5%, and only 29% for advanced disease;
Most patients develop advanced, platinum-resistant, poor prognosis disease; and
Limited options exist with current single-agents at 6-30% response rates and median progression free survival (“mPFS”) of 2.1 - 4.2 months.
The Corporation believes that it has the potential to be “best-in-class” in the competitive landscape of recurrent ovarian cancer as other immunotherapeutic treatments tested in this patient population (Incyte Corporation’s epacadostat, Merck’s Keytruda, and Pfizer/Merck KGaA’s Bavencio) are unlikely to proceed into registration trials based on the published results available:
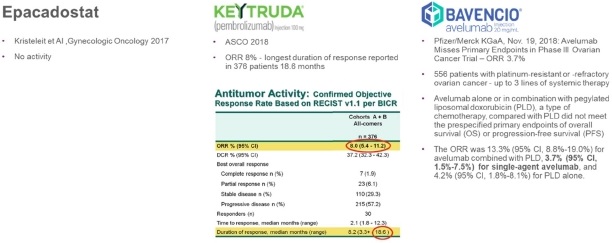
Figure 4: Recurrent ovarian cancer immunotherapy competitive landscape
Subject to phase 2 results, IMV plans to schedule a follow-up meeting with FDA to finalize the design of a potential pivotal trial based on ORR and DOR.
IMV expects to provide a clinical update at the 2019 annual meeting of the American Society of Clinical Oncology (“ASCO”) and investigators are also planning to submit the study findings for scientific publication.
The Corporation’s clinical strategy with this trial is to establish monotherapy activity in order to increase value and de-risk clinical development, and to target late stage unmet medical needs for a shorter path to clinical demonstration and first regulatory approval.
The Corporation currently anticipates that, in addition to general clinical expenses, which are distributed amongst the various clinical projects, the costs to complete this phase 2 clinical trial are estimated at $2,500,000 of which $1,000,000 is expected to occur in 2019.
9
Combinations
Phase 2 clinical trial in Diffuse large B-cell lymphoma (“DLBCL”) with Merck (investigator-sponsored)
This phase 2 study is a triple-combination immunotherapy in patients with measurable or recurrent DLBCL led by Sunnybrook Research Institute. This investigator sponsored trial, announced initially in May 2017, is designed to evaluate the safety and efficacy of DPX-Survivac, Merck’s pembrolizumab, and low-dose cyclophosphamide. Primary and secondary end points include:
Safety profile; and
ORR and DOR using Recist 1.1 criteria.
The non-randomized, open label study is expected to enroll 25 evaluable participants at five centers in Canada. As of April 5, 2019, 10 patients have been enrolled across four different clinical sites in Canada. Additional patients are being screened and IMV expects to be in a position to report updated clinical data at about the same time as the biennial International Conference on Malignant Lymphoma, which will be held in Lugano, Switzerland, in June.
Researchers conducting the investigator sponsored study are testing the novel immunotherapy combination in patients whose DLBCL expresses survivin, a tumour antigen highly expressed in 60 percent of DLBCL patients. DPX Survivac stimulates the immune system to produce T cell responses targeting survivin.
On November 8, 2017, the Corporation announced that Health Canada had granted Sunnybrook Research Institute regulatory clearance to begin recruiting patients. On March 28, 2018, the Corporation announced that the first patient had been treated.
On September 18, 2018, IMV announced details of the initial data from this clinical trial. The preliminary data included assessments of safety and clinical activity (based on modified Cheson criteriai) for the first four evaluable patients who have completed their first CT scan after the start of treatment. The data showed that:
Two of the first four evaluable participants showed tumour regressions at the first on-treatment CT scan:g
The first enrolled participant demonstrated a tumour regression of 48% at first on-treatment scan; and
The second participant demonstrated a partial response (PR) via a tumour regression of 66% at first on-treatment scan.
Preliminary data from the third participant demonstrated stable disease.
The other participant had early disease progression less than two months following treatment initiation and was discontinued from the study.
The combination therapy appears to demonstrate an acceptable safety profile, with no serious adverse events reported to date.
iCheson, B.D., Pfistner, B., Juweid, M.E., Gascoyne, R.D., Specht, L., Horning, S.J. and Diehl, V. (2007). Revised Response Criteria for Malignant Lymphoma. Journal of Clinical Oncology, 25(5) DOI: 10.1200/JCO.2006.09.2403
The Corporation currently anticipates that, in addition to general clinical expenses, which are distributed amongst the various clinical projects, its share of the cost to complete this study will be approximately $1,500,000, of which $1,000,000 is expected to be spent in 2019.
Phase 2 basket trial in 5 indications with Merck
On September 11, 2018, the Corporation announced the expansion of its clinical program with a phase 2 basket trial in collaboration with Merck evaluating its lead candidate, DPX-Survivac, in combination with low dose cyclophosphamide, and Merck’s anti-PD-1 therapy, KEYTRUDA® (pembrolizumab), in patients with select advanced or recurrent solid tumours.
The open-label, multicenter, phase 2 basket study will evaluate the safety and efficacy of the immunotherapeutic combination in patients with bladder, liver (hepatocellular carcinoma), ovarian, or non-small cell lung (NSCLC) cancers, as well as tumours shown to be positive for the microsatellite instability high (MSI-H) biomarker. Investigators plan to enroll more than 200 patients across five indications at multiple medical centers in Canada and the United States.
10
The ASCO defines a basket clinical study as a trial that investigates the effects of a drug regimen in multiple tumour types that share a common molecular target, regardless of where the disease originated.
This is the third clinical trial evaluating the combination of DPX-Survivac, low dose cyclophosphamide, and pembrolizumab in advanced recurrent cancers.
The first patients have been treated in the ovarian, NSCLC and MSI-H cohorts and the Corporation expects to disclose preliminary data in the second half of 2019 and currently anticipates that, in addition to general clinical expenses, which are distributed amongst the various clinical projects, $12,600,000 is estimated to be spent for the safety lead-in for this trial, with $5,000,000 is estimated to be spent in 2019.
Phase 2 clinical trial in ovarian cancer with Merck (investigator-sponsored)
In February 2017, the Corporation announced an investigator-sponsored phase 2 clinical trial in ovarian cancer in combination with Merck’s checkpoint inhibitor pembrolizumab in patients with recurrent, platinum-resistant ovarian cancer. University Health Network’s (“UHN”) Princess Margaret Cancer Centre will conduct the phase 2 non-randomized, open-label trial designed to evaluate the potential anti-tumour activity of the combination of pembrolizumab, DPX-Survivac, and low-dose cyclophosphamide. It is expected to enroll 42 subjects with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer. The study’s primary objective is to assess overall response rate. Secondary study objectives include progression free survival rate, overall survival rate, and potential side effects, over a five-year period. At this stage, the Corporation has no specific plan on the next steps after this trial as it will have to be assessed with its partner based on the clinical trial results.
As of May 1, 2019, 12 patients were enrolled in the trial and the Corporation will disclose results once provided by the UHN Princess Margaret Cancer Centre and currently anticipates that, in addition to general clinical expenses, which are distributed amongst the various clinical projects, its share of the costs to complete this study, expected to be spent in 2019, are estimated at $400,000.
Orphan Drug Status and Fast Track Designation
The Corporation announced, in November 2016, that the European Medicines Agency (EMA) had granted orphan drug designation status to IMV’s DPX-Survivac in ovarian cancer. In July 2015, the FDA also granted orphan drug status to DPX-Survivac for the treatment of ovarian cancer. This designation is valid for all applications of DPX-Survivac in ovarian cancer without restriction to a specific stage of disease.
IMV had previously received FDA fast track designation for DPX-Survivac. The designation is intended for patients with no measurable disease after their initial surgery and chemotherapy.
Other Programs
Oncology
DPX-NEO
On January 17, 2019, treatment of the first patient occurred in the phase 1 trial evaluating neoepitopes formulated in the Corporation's proprietary DPX delivery platform in patients with ovarian cancer. The study is part of the Corporation's DPX-NEO program, which is a continuing collaboration between UConn Health and IMV to develop neoepitope-based anti-cancer therapies.
Investigators will assess the safety and efficacy of using patient-specific neoepitopes discovered at UConn Health and formulated in IMV's proprietary DPX-based delivery technology in women with ovarian cancer. Investigators plan to enroll up to 15 patients in the phase 1 study. UConn Health is financing the trial with IMV providing materials and advice.
The Corporation expects to disclose results when provided by UConn Health.
DPX-E7
On April 17, 2017, the Corporation announced that the first study participant has been treated in a phase 1b/2 clinical study evaluating an investigational cancer target for HPV (E7) formulated in DPX and in combination with low-dose cyclophosphamide in patients with incurable oropharyngeal, cervical, and anal cancers related to HPV.
Dana-Farber is leading the DPX -E7 study through a $1.5 million research grant from Stand Up To Cancer and the Farrah Fawcett Foundation to clinically evaluate collaborative translational research that addresses critical problems in HPV-related cancers. The Dana-
11
Farber study is a single center, open label, non-randomized clinical trial that will investigate the safety and clinical efficacy in a total of 44 treated participants. Its primary objectives are to evaluate changes in CD8+ T cells in peripheral blood and tumour tissue, and to evaluate the safety in HLA-A2 positive patients with incurable HPV-related head and neck, cervical, or anal cancers. IMV has the option to produce the DPX -E7 vaccine if it proves successful in the clinical trials.
The Corporation expects to disclose results when provided by Dana-Farber.
Other Applications
Product Overview
A component of the Corporation’s business strategy is partnering the DPX platform within infectious and other diseases. The DPX platform has the potential to generate a rapid and robust immune response, often in a single dose. The unique single-dose capability could prove to be beneficial in targeting difficult infectious and other disease candidates.
RSV
The Corporation has performed preclinical research activities for a vaccine targeting RSV, which is the second leading cause of respiratory illness in infants, the elderly, and the immunosuppressed. Currently, there is no vaccine available for this virus and IMV is seeking to develop a novel vaccine formulation to be used in elderly and healthy adults, including women of child-bearing age. IMV has in-licensed the RSV antigen exclusively from VIB VZW (“VIB”), a non-profit life sciences research institute funded by the Flemish government, to expand its pipeline of vaccine candidates. The novel RSV antigen being evaluated in DPX is based on the short hydrophobic protein present at low levels on the surface of the RSV virion. But, more importantly, it is also present on the surface of RSV-infected cells. This vaccine has a unique mechanism of action in which the resultant antibodies bind to and destroy infected cells rather than directly bind to and neutralize the free virus.
Phase 1 clinical trial in RSV
A phase 1 clinical study has been conducted in Canada with the Corporation’s RSV vaccine candidate in healthy adults. The RSV vaccine is formulated in IMV’s proprietary DPX platform and is initially being developed to protect the elderly population from infection. The phase 1 study, which was the first clinical trial of a DPX-based vaccine in an infectious disease indication, evaluated the safety and immune response profile of the RSV vaccine candidate in 40 healthy older adult volunteers (age 50-64 years) and two dose cohorts, with 20 subjects in each cohort.
In July 2016, the Corporation announced positive interim results from this trial. Investigators analyzed the safety and immune response data of all participants up to study day 84. The safety analysis indicates that DPX-RSV was well tolerated among all study participants, with no SAEs recorded. Furthermore, immunogenicity data supported DPX-RSV’s ability to generate a relevant immune response: the vaccine candidate obtained antigen-specific antibody responses in 75 percent of subjects vaccinated with the lower dose and 100 percent of those vaccinated with the higher dose.
In October 2016, the Corporation announced positive topline results from this trial. The report outlined that more than nine months after the last vaccination, 15 of 16 participants (93%) who received DPX-RSV demonstrated antigen-specific immune responses. The vaccine candidate also continued to have a positive safety profile and was well tolerated with no SAEs among all study participants.
On April 12, 2017, the Corporation announced additional positive data from an extended evaluation of patients in this trial. An amendment had been submitted to Health Canada to test subjects who received the higher dose of vaccine out to one year after the booster vaccination. In the 25µg dose cohort, which was the only dose tested out to one year, 100 percent of older adults (7/7 immune responders) vaccinated with DPX-RSV maintained the antigen-specific immune responses one year after receiving the booster dose. At one year, the antibody levels measured were still at peak with no sign of decrease.
On September 27, 2018, IMV announced results of ongoing research to further explore the novel MOA of its vaccine candidate. New data from a preclinical study highlighted the effects of two potential approaches to preventing RSV, comparing a single dose bovine version of DPX-RSV to a two-dose conventional investigational bovine RSV vaccine. Researchers found that IMV’s vaccine candidate yielded strong antigen-specific immune responses and a protective effect on disease pathology. The degree of protection was comparable between the two vaccine candidates.
In this study, researchers compared the effects of both the IMV and conventional RSV vaccine approaches among bovines with known RSV infections (the bovine animal model is considered an optimal model of RSV infection). Researchers administered one dose of DPX-
12
bRSV to one cohort; the second received two doses of a subunit RSV bovine vaccine. Researchers measured immune response with an antibody titer test and assessed disease pathology with a lung lesion score and other clinical parameters (such as body temperature changes).
They found SH antibodies in 14 of the 15 subjects that received DPX-bRSV, and the improvements observed in disease pathology were comparable between the two cohorts. These were the first bovine animal health data to directly correlate the vaccine-induced immune response against IMV’s novel RSV target - the SH viral protein– with measures of disease protection.
Conventional RSV vaccine candidates target either the F or G proteins of the virus and provide protection by neutralizing the RSV virus. Clinical measures of efficacy focus on the amount of neutralizing antibodies in the bloodstream. DPX-RSV works differently; it targets the SH viral ectodomain of the RSV virus and, instead of neutralizing the virus, it enables the immune system to recognize and destroy infected cells. Because there are no neutralizing antibodies resulting from the DPX-RSV MOA, a different clinical assessment is required to determine the vaccine candidate’s protective effect. IMV has exclusive worldwide licenses on applications that target the SH ectodomain antigen in RSV. The Corporation intends to explore opportunities to out-license this product to potential partners.
Malaria
In 2016, IMV was awarded a subcontract by Leidos, a health, national security, and infrastructure solutions company, to evaluate IMV’s DPX platform for the development of peptide-based malaria vaccine targets. The subcontract is funded through Leidos’ prime contract from the U.S. Agency for International Development (“USAID”) to provide vaccine evaluations in the preclinical, clinical, and field stages of malaria vaccine development. Leidos and IMV are working together to identify adjuvant and antigen combinations that can be used to protect against malaria and, with the DPX delivery system, formulate promising vaccine candidates for potential clinical testing.
In November 2017, an expansion of this collaboration was announced. Following the achievement of several preclinical milestones in the collaboration with USAID, Leidos and USAID selected the DPX-based platform as one of the preferred formulations for further development under a new contract extension. Under the new subcontract, the collaborators will conduct additional research that focuses on identifying the most promising target-formulation combinations.
Zoetis Collaboration
On August 31, 2017, the Corporation announced the achievement of several milestones in its ongoing collaboration with global animal health company Zoetis to develop cattle vaccines. In recent controlled studies, the IMV formulations met efficacy and duration of immunity endpoints against two disease targets. These results will enable Zoetis to advance two IMV-formulated vaccine candidates into late-stage testing.
Licensing Agreements
While the Corporation is focused on developing a pipeline of cancer immunotherapies, it is also pursuing opportunities to license its platform technology to other parties interested in creating enhanced vaccines on an application-by-application basis.
In April 2018, IMV signed a licensing agreement and granted SpayVac-for-Wildlife (SFW Inc.) a license to two of its proprietary delivery platforms. SFW Inc. has global exclusive rights to use both of these platforms to develop humane, immuno-contraceptive vaccines for control of overabundant, feral and invasive wildlife populations against royalties on sales.
MARKET OVERVIEW
Cancer Immunotherapies
Cancer is considered one of the most widespread and prevalent diseases globally. According to Global Cancer Facts & Figures, 4th edition (released in 2018 by the American Cancer Society), it is predicted that new cancer cases will rise to 27.5 million and the number of cancer deaths to 16.3 million by 2040 simply due to the growth of the aging population. Conventional cancer treatment involves surgery to remove the tumour whenever possible, as well as chemotherapy and radiation. Chemotherapies are widely used, despite their associated toxicities, because they interfere with the ability of cancer cells to grow and spread. However, tumours often develop resistance to chemotherapies, thus limiting their efficacy in preventing tumour recurrence. Despite recent advances, independent sources note a high unmet medical need in cancer therapy, noting the median survival rate remains poor. Cancer immunotherapies may provide new and effective treatments. According to a Market & Markets report released in January 2017, the global immunotherapy drug market is projected to reach USD$201.52 billion by 2021 from USD$108.41 billion in 2016, growing at a compound annual growth rate (“CAGR”) of 13.5% during the forecast period of 2016 to 2021. The major players operating in the immunotherapy drug market include F. Hoffmann-La Roche AG
13
(Switzerland), GlaxoSmithKline (U.K.), AbbVie, Inc. (U.S.), Amgen, Inc. (U.S.), Merck & Co., Inc. (U.S.), Bristol-Myers Squibb (U.S.), Novartis International AG (Switzerland), Eli Lilly and Corporation (U.S.), Johnson & Johnson (U.S.), and AstraZeneca plc (U.K.).
Cancer immunotherapy seeks to harness the immune system to assist in the destruction of tumours and to prevent their recurrence. There has been significant interest in the field of cancer immunotherapy stemming from recent clinical success in prolonging patient survival with novel compounds. The ability to apply these appropriately has resulted from a greater understanding of the immune dysfunction that is characteristic of cancer. One area in which there have been breakthroughs has been in the area of checkpoint inhibitors, which are compounds that target key regulatory molecules of the immune system. Yervoy® (anti CTLA 4, or ipilumumab, developed by Bristol Myers Squibb) was the first compound in this class to be approved for use in advanced metastatic melanoma. In cancer, these regulators (CTLA 4, PD 1 and its ligand PD L1) act to inhibit CD8 T cell-mediated anti-tumour immune responses that are crucial for tumour control. Monoclonal antibodies that target PD 1 and PD L1 have shown unusual efficacy in cancer patients, with a significant percentage of patients experiencing durable response to these therapies. Several of these compounds have been approved in multiple indications. Merck’s KEYTRUDA® (pembrolizumab) and Bristol Myers Squibb’s OPDIVO® (nivolumab) received FDA approval in 2014 for advanced melanoma patients who have stopped responding to other therapies. These therapies have subsequently been approved for use in other advanced cancers including bladder cancer, non-small cell lung cancer, Hodgkin’s Lymphoma, squamous cell carcinoma of the head and neck and stomach cancer. In addition, KEYTRUDA® in particular has been approved for use in cancers with a specific molecular indication irrelevant of cancer type, having been approved in May 2017 for use to treat solid tumours having a biomarker for microsatellite instability (MSI-H), which is a defect in the DNA repair pathway. This represents about 5% of a number of different tumour types, including colorectal, breast, prostate, and thyroid cancers.
These drugs have been shown to be helpful in treating several types of cancer but with success only in a limited percentage of patients. It is not yet known exactly why, though researchers have noticed that these drugs seem to work especially well for patients whose cancer cells have a higher number of mutations.
Key opinion leaders in the field have indicated that that the solution lies in combining checkpoint inhibitors with other cancer treatments and that the ideal combination is likely to be a therapy that drives tumour specific immune responses. These include novel T cell-based therapies. These therapies fit well with checkpoint inhibition therapy because they simultaneously activate strong tumour-specific T cell activation, while also releasing the brakes on immune suppression. The success of such combinations should allow pharmaceutical companies to significantly expand the market of their checkpoint inhibitors.
The Corporation believes that T cell therapies will become an important component of these novel combination immunotherapies, with the potential of synergistic benefits to become an essential part of a multi-pronged approach for the treatment of cancer.
INTELLECTUAL PROPERTY
The Corporation strives to protect its intellectual property in established, as well as emerging, markets around the world. The Corporation’s intellectual property portfolio relating to its platform technology includes 17 patent families, the first of which contains eight patents issued in five jurisdictions (United States, Europe, Canada, Japan, and Australia). The 16 other families collectively contain 36 patents issued in 10 jurisdictions (United States, Europe, Canada, Australia, Japan, India, Israel, Singapore, China, and, separately. Hong Kong) and 47 pending patent applications in 9 jurisdictions. Taking into account the validations of the European patents, the Corporation’s intellectual property portfolio includes 89 patents. More details on the Corporation’s intellectual property strategy and patents can be found in the AIF filed on SEDAR at www.sedar.com.
The Corporation owns registered trademarks in the United States, Canada, and Europe.
RECENT AND QUARTERLY DEVELOPMENTS
Key developments and achievements
The Corporation announced:
On April 3, 2019, that it presented preclinical research at the American Association for Cancer Research (AACR) Annual Meeting 2019 that demonstrated how the MOA of IMV’s proprietary DPX technology can enhance a broad spectrum of immune cell infiltration into tumours, which included T cells, Natural Killer (NK) cells, and macrophages. Analysis also revealed the differentiated characteristics of the immune cell responses and the potential implications for enhanced anti-tumour activity. In the poster titled, T-distributed stochastic neighbor embedding (t-SNE) analysis of tumour infiltrating lymphocytes after treatment with a T cell activating therapy identifies a unique population of recruited CD8+ T cells and novel options for combination immunotherapy, IMV researchers used specialized data analytics to examine how DPX-based agents, when combined with cyclophosphamide (CPA), induced T cells to infiltrate tumours and attack cancerous cells. The study closely examined the types
14
On March 26, 2019, preliminary data from the phase 2 cohort of the DeCidE1 clinical study. Six patients receiving DPX-Survivac monotherapy with intermittent low-dose cyclophosphamide (mCPA) have reached the first CT scan assessment with key related findings as follows:
83 per cent of the subjects (five of six) show stable disease, including two tumour regressions; and
80 per cent (four of five) with stable disease are in subjects with a lower BTB, which also includes the two tumour regressions.
of immune cell responses and how and why they were able to affect disease. The data indicated that this approach stimulated the infiltration of a broad base of immune cells into tumours, including T cells, NK cells, and macrophages. The specific T cell population that moved into tumours could be grouped based on the co-expression of different checkpoint molecules such as PD-1 and Tim-3. However, those stimulated to infiltrate tumours generally did not express CTLA-4 (a protein found on T cells that inhibits the immune response).
On March 18, 2019, that Canadian bioresearch consortium, CQDM, awarded a grant to a collaboration among IMV, Centre de recherche du CHU de Quebec-Universite Laval (“CHU”), and La Fondation du CHU de Quebec (“FCHUQc”). Under the leadership of Yves Fradet, M.D., professor of surgery and researcher in cancer immunotherapy, this project will receive a grant of up to $1.2M from CQDM and $300,000 from the FCHUQc, to develop a novel dual target T cell therapy for an initial clinical application in bladder cancer. The work will target immunogenic peptides identified by Dr. Fradet’s team from the MAGE protein family member A9 (MAGE-A9). These peptides will be combined with selected immunogenic peptides from the survivin protein composing DPX-Survivac. The collaborators will combine these peptides with IMV’s proprietary DPX technology to develop a first-in-class dual target T cell therapy (DPX-SurMAGE).
This initial phase 2 data confirms the earlier trends observed in the phase 1b portion of the study. The Corporation believes it supports the potential of DPX-Survivac as a monotherapy and the use of its patient selection strategy.
Importantly, in earlier stages of this trial, durable clinical responses occurred after 140 days, and have now lasted for 20 months or more. Additional data at the 140-day mark of this cohort will be available by the end of the first half of 2019.
On March 6, 2019, that it completed a public offering of common shares of the Corporation. An aggregate of 4,900,000 common shares was issued at a price of $5.45 per common share, raising gross proceeds of $26.7 million (the “March 2019 Public Offering”) and on March 11, 2019, that the underwriters partially exercised their over-allotment option to purchase additional common shares, resulting in the issuance of an additional 504,855 common shares of the Corporation at a price of $5.45 per share for additional gross proceeds of approximately $2.75 million, increasing the gross proceeds to approximately $29.46 million, before deducting the underwriting commissions and offering expenses. The Corporation intends to use the net proceeds of the Offering to accelerate the development of DPX-Survivac in combination with Keytruda as part of the phase 2 basket trial with Merck in patients with select advanced or recurrent solid tumours in bladder, liver (hepatocellular carcinoma), ovarian, or non- small-cell lung cancers, as well as tumours shown to be positive for the microsatellite instability high biomarker and for general corporate purposes.
On January 30, 2019, an update on its clinical program for its lead investigational treatment, DPX-Survivac, as a potential monotherapy in advanced recurrent ovarian cancer. In December, 2018, IMV met with the FDA in a Type B meeting to discuss the results to date of its DeCidE1 clinical trial and continuing development plan, as well as to obtain agency guidance on a potential accelerated regulatory pathway for DPX-Survivac as a T-cell immunotherapy for the treatment of advanced ovarian cancer in patients with progressing disease.
FDA meeting highlights include:
The purpose of IMV's Type B meeting with the FDA was to request feedback on the design of the clinical program for DPX-Survivac. This program includes the continuing DeCidE1 phase 2 clinical study and a potential future registration trial for accelerated approval in a subset of ovarian cancer patients.
The FDA reviewed the Corporation's proposed clinical development plan and acknowledged the potential for accelerated approvals in advanced ovarian cancer based on ORR according to Recist 1.1 criteria with reported median DOR. In addition, the FDA provided important guidance on clinical design considerations for different lines of therapy and platinum-sensitive and resistant patient populations.
In addition, IMV submitted a protocol amendment for a predictive enrichment approach to the phase 2 DeCidE1 trial, and further discussed those details with the FDA during the Type B meeting. The phase 2 primary endpoint, based on ORR per Recist 1.1 criteria, is intended to confirm the high response rate and duration of clinical benefits observed in previously announced results in a patient population defined by a clinical biomarker based on BTB.
15
On January 17, 2019, treatment of the first patient in its phase 1 trial evaluating neoepitopes formulated in the Corporation's proprietary DPX delivery platform in patients with ovarian cancer. The study is part of the Corporation's DPX-NEO program, which is a continuing collaboration between UConn Health and IMV to develop neoepitope-based anti-cancer therapies.
Investigators will assess the safety and efficacy of using patient-specific neoepitopes discovered at UConn Health and formulated in IMV's proprietary DPX-based delivery technology in women with ovarian cancer. Investigators plan to enroll up to 15 patients in the phase 1 study. UConn Health is financing the trial with IMV providing materials and advice.
Multiple clinical sites are now open for enrolment in the DeCidE1 phase 2 trial. Subject to phase 2 results, IMV plans to schedule a follow-up meeting with the FDA to finalize the design of a potential pivotal trial based on ORR and DOR.
SELECTED FINANCIAL INFORMATION
| Three months ended | Three months ended | |
| March 31, 2019 | March 31, 2018 | |
| $ | $ | |
| Net loss and comprehensive loss for the period | 5,943,000 | 3,067,000 |
| Basic and diluted loss per share | 0.13 | 0.07 |
| As at | As at | |
| March 31, 2019 | December 31, 2018 | |
| $ | $ | |
| Cash and cash equivalents | 34,207,000 | 14,895,000 |
| Total assets | 42,395,000 | 22,925,000 |
| Long term debt | 8,444,000 | 8,069,000 |
RESULTS FOR THE THREE MONTHS ENDED MARCH 31, 2019, COMPARED TO THE THREE MONTHS ENDEDMARCH 31, 2018
| Three months ended | Three months ended | |
| March 31, 2019 | March 31, 2018 | |
| $ | $ | |
| Revenue | (82,000) | (96,000) |
| Research and development | 4,013,000 | 1,882,000 |
| General and administrative | 1,960,000 | 1,290,000 |
| Government assistance | (346,000) | (275,000) |
| Accreted interest | 398,000 | 266,000 |
| Net loss and comprehensive loss for the period | 5,943,000 | 3,067,000 |
Revenue
Revenue decreased by $14,000 in Q1 2019 in comparison with Q1 2018. This is attributable to a decrease in subcontract revenue of $19,000 offset by a slight increase in interest income of $5,000.
16
Research and development expenses
R&D expenses include salaries and benefits, expenses associated with the clinical trials of DPX-Survivac, clinical research and manufacturing of DPX-Survivac and DPX-SurMAGE, consulting fees paid to various independent contractors with specific expertise required by the Corporation, the cost of animal care facilities, laboratory supplies, peptides and other raw materials, insurance, as well as other R&D related expenses.
The Corporation’s R&D efforts and related expenses included costs surrounding the Corporation’s clinical trials of DPX-Survivac, namely the phase 2 DeCidE1 clinical trial in ovarian cancer, phase 2 clinical trial collaboration with Merck in ovarian cancer, phase 2 clinical trial collaboration with Merck in DLBCL, phase 2 basket trial in 5 indications with Merck and costs related to the Corporation’s ongoing R&D activities associated with the investigation, and analysis and evaluation of other potential product candidates and technologies.
Research and development expenses consist of the following:
| Three months | Three months | |
| ended March | ended March | |
| 31, 2019 | 31, 2018 | |
| $ | $ | |
| General R&D expenses | 553,000 | 427,000 |
| DPX-Survivac preclinical and clinical expenses | 2,339,000 | 715,000 |
| Salaries and benefits | 992,000 | 647,000 |
| Stock-based compensation | 100,000 | 69,000 |
| Depreciation of equipment and amortization of intangible | 29,000 | 24,000 |
| Total | 4,013,000 | 1,882,000 |
The increase in general R&D expenses from $427,000 in Q1 2018 to $553,000 in Q1 2019 is mainly attributable to $182,000 in expenses relating to the DPX-SurMAGE collaboration with CQDM, CHU, and FCHUQc, a $62,000 increase in raw materials and supplies offset by a $62,000 decrease in professional fees and outsourced testing, a $43,000 decrease in rent expense as a result of rent now being allocated entirely to G&A and a $15,000 decrease in regulatory consulting.
The increase of $1,624,000 in Q1 2019 in DPX-Survivac preclinical and clinical expenses is mainly attributable to increased clinical activity including: milestone payments for phase 2 study in DLBCL ($123,000) and expenses related to the basket trial ($1,246,000). The increase is also attributable to manufacturing activities to support the increased clinical activity including purchasing of raw materials and contract manufacturing organization costs ($293,000).
The increase in R&D salaries in 2019 is mainly attributable to the hiring of eleven new R&D positions (two at a director level) since the beginning of 2018.
General and administrative expenses
G&A expenses consist of the following:
| Three months | Three months | |
| ended March | ended March | |
| 31, 2019 | 31, 2018 | |
| $ | $ | |
| General and administrative expenses, excluding salaries | 1,150,000 | 850,000 |
| Salaries and benefits | 645,000 | 458,000 |
| Stock-based and deferred share unit compensation | 87,000 | (37,000) |
| Depreciation of furniture, leaseholds and equipment | 78,000 | 19,000 |
| Total | 1,960,000 | 1,290,000 |
17
For Q1 2019, G&A expenses, excluding salaries, increased by $300,000. The increase is mainly explained by an $82,000 increase relating to the new facility opened in Q2 2018, an increase of $162,000 in insurance premiums and $64,000 in regulatory fees following the NASDAQ listing in Q2 2018, and $45,000 in directors’ fees. These increases are partly offset by a decrease of $48,000 in travel and a $72,000 foreign exchange gain.
Salaries and benefits increased by $187,000 in Q1 2019 due to two new positions since Q1 2018, including a senior director of investor relations and communications, as well as an overall increase in compensation for the senior executive team.
The increase in stock-based and deferred share unit compensation in 2019 is explained by an increase of $217,000 in stock-based compensation related to new positions as well as the 2018 option grant for executives occurring in late March, partly offset by a decrease in the fair value of DSUs.
The increase in depreciation compared to Q1 2018 is attributable to the leasehold improvements, equipment and leased space and associated with the new facility occupied since Q2 2018.
Government assistance
Government assistance consists of the following:
| Three Months Ended | Three Months Ended | |
| March 31, | March 31, | |
| 2019 | 2018 | |
| $ | $ | |
| Investment tax credits (“ITC”) | 346,000 | 259,000 |
| Government loans and assistance | - | 16,000 |
| Total | 346,000 | 275,000 |
The increase in investment tax credit in 2019 is explained by the increase in R&D salaries and raw materials as well as increased clinical trial activity being performed in Canada.
Accreted Interest
Accreted interest relates entirely to the valuation of low-interest bearing government loans which are repayable based on a percentage of future gross revenue and is comparable to 2018.
Net loss and comprehensive loss
The net loss and comprehensive loss was $5,943,000 or $0.13 per basic and diluted share compared to $3,067,000 or $0.07 per basic and diluted share for three months ended March 31, 2018.
CASH FLOWS, LIQUIDITY AND CAPITAL RESOURCES
At March 31, 2019, the Corporation had cash and cash equivalents of $34,207,000 and working capital of $33,893,000, compared to $14,895,000 and $12,247,000, respectively as at December 31, 2018.
Since the Corporation’s inception, operations have been financed through the issuance of equity securities, debt, revenue from licenses, cost recoveries from collaborations, interest income on funds available for investment, government assistance and tax credits.
During Q1 2019, $8,206,000 was used in operating activities. This included the reported net loss of $5,943,000 prior to being decreased for non-cash expenses including DSU compensation, depreciation, accretion of long-term debt and lease obligations, loss on disposal of assets and stock-based compensation. The Corporation had a net decrease of cash of $3,002,000 as a result of changes in working capital balances.
Sources of cash included: $29,456,000 raised through financing activities less cash issuance costs of $1,862,000; and $116,000 through the exercise of stock options and warrants. The Corporation used $80,000 to repay long-term debt and lease obligations during the period.
18
During Q1 2019, the Corporation purchased furniture and equipment for ongoing research and operating activities for an aggregate amount of $112,000.
The Corporation aims to maintain adequate cash and cash resources to support planned activities which include: phase 2 DeCidE1 clinical trial in ovarian cancer with DPX-Survivac; the two phase 2 investigator-sponsored combination trials with DPX-Survivac and Merck’s checkpoint inhibitor, pembrolizumab in ovarian cancer and DLBCL; the basket trial in 5 indications with DPX-Survivac and Merck’s checkpoint inhibitor, pembrolizumab; and other research and development activities, business development efforts, administration costs, and intellectual property maintenance and expansion.
At March 31, 2019, the Corporation had approximately $36.5 million of existing and identified potential sources of cash including:
cash and equivalents of $34.2 million; and
amounts receivable and investment tax credits receivable of $2.3 million.
For Q1 2019, the Corporation’s “cash burn rate” (defined as net loss for the period adjusted for operations not involving cash - interest on lease obligation, depreciation, accretion of long-term debt, stock-based compensation and DSU compensation) was $5.2 million. Based on the current business plan and depending on the timing of certain clinical expenses, the Corporation forecasts the cash burn rate to be between $5 million to $6 million per quarter for 2019, as it continues to execute its clinical plan.
It is common for early-stage biotechnology companies to require additional funding to further develop product-candidates until successful commercialization of at least one product candidate. IMV’s product candidates are still in the early-development stage of the product cycle and therefore are not generating revenue to fund operations. The Corporation continuously monitors its liquidity position, the status of its development programs including those of its partners, cash forecasts for completing various stages of development, the potential to license or co-develop each vaccine candidate, and continues to actively pursue alternatives to raise capital, including the sale of its equity securities, debt and non-dilutive funding.
Management believes that its cash resources of $34.2 million and its additional potential cash resources of $2.3 million, as at March 31, 2019, will be sufficient to fund operations for the next twelve months while maintaining adequate working capital well into 2020. The Corporation continually reassesses the adequacy of its cash resources, evaluating existing clinical trials, research projects and/or potential collaboration opportunities, to determine when and how much additional funding is required.
JUNE 2017 EQUITY OFFERING AND USE OF PROCEEDS
On June 21, 2017, the Corporation completed a public offering, issuing 7,692,308 common shares common shares pre-consolidation (2,403,846 post-consolidation) at a price of $1.30 per share pre-consolidation ($4.16 post-consolidation) for aggregate proceeds of $10,000,000. The Corporation intends to use the net proceeds of this offering for the research and development and clinical advancement of its cancer and infectious disease vaccine candidates and for working capital and general corporate purposes. The table below provides the amount used to date and any variances (except for working capital and general corporate purposes).
| Intended Use of Proceeds | Estimated amount | Amount | Variances |
| $ | to date | ||
| $ | |||
| phase 2 clinical trial in DLBCL with Merck | 2,400,000 | 1,359,000 | No variances anticipated |
| phase 1 clinical trial for multiple indications | 4,200,000 | 3,080,000 | No variances anticipated |
FEBRUARY 2018 EQUITY OFFERING AND USE OF PROCEEDS
On February 15, 2018, the Corporation completed a public offering, issuing 7,187,500 common shares pre-consolidation (2,246,094 post-consolidation) at a price of $2.00 per share pre-consolidation ($6.40 post-consolidation) for aggregate proceeds of $14,375,000. The Corporation intends to use the net proceeds of this offering to continue to advance the Corporation’s pipeline and conduct a phase 1 basket
19
trial in up to five indications to be identified, for research and development, working capital, and for general corporate purposes. The table below provides the amount used to date and any variances (except for working capital and general corporate purposes).
| Intended Use of Proceeds | Estimated amount | Amount | Variances |
| $ | to date | ||
| $ | |||
| Clinical trials in 2019 | 4,800,000 | 393,000 | No variances anticipated |
| Research & development in 2019 | 5,300,000 | 2,104,000 | No variances anticipated |
MARCH 2019 EQUITY OFFERING AND USE OF PROCEEDS
On March 6, 2019, the Corporation completed a public offering, issuing 5,404,855 common shares pre-consolidation (including 504,855 common shares upon the exercise of the underwriters’ over-allotment option on March 11, 2019) at a price of $5.45 per share for aggregate proceeds of $29.5M. The Corporation intends to use the net proceeds of this offering to accelerate the development of DPX-Survivac in combination with Keytruda as part of the basket trial in selected advanced or recurrent solid tumours in bladder, liver (hepatocellular carcinoma), ovarian or non-small-cell lung cancers, as well as tumours shown to be positive for the microsatellite instability high biomarker and for general corporate purposes. The table below provides the amount used to date and any variances (except for working capital and general corporate purposes).
| Intended Use of Proceeds | Estimated amount | Amount | Variances |
| $ | to date | ||
| $ | |||
| Phase 2 clinical trial for multiple indications | 16,000,000 | Nil | No variances anticipated |
SUMMARY OF QUARTERLY RESULTS
The following consolidated quarterly data was drawn from the annual audited consolidated financial statements and the unaudited interim condensed consolidated financial statements. All values discussed below are rounded to the nearest thousand. The information is reported on an IFRS basis.
| Quarter Ended In | Total Revenue | Total Expenses | Loss | Basic and Diluted Loss Per Share |
| $ | $ | $ | $ | |
| Q1- March 31, 2019 | 82,000 | 6,025,000 | (5,943,000) | (0.13) |
| Q4- December 31, 2018 | 133,000 | 7,818,000 | (7,685,000) | (0.17) |
| Q3- September 30, 2018 | 125,000 | 6,112,000 | (5,987,000) | (0.14) |
| Q2– June 30, 2018 | 129,000 | 5,325,000 | (5,196,000) | (0.12) |
| Q1– March 31, 2018 | 96,000 | 3,163,000 | (3,067,000) | (0.07) |
| Q4 -December 31, 2017 | 66,000 | 4,997,000 | (4,931,000) | (0.13) |
| Q3- September 30, 2017 | 53,000 | 2,175,000 | (2,122,000) | (0.06) |
| Q2– June 30, 2017 | 36,000 | 2,641,000 | (2,605,000) | (0.06) |
Revenues from quarter to quarter may vary significantly. Revenues are non-recurring by nature and are generated by license agreements as well as contract research agreements. It is also important to note that historical patterns of expenses cannot be taken as an indication of future expenses. The amount and timing of expenses and availability of capital resources vary substantially from quarter to quarter,
20
depending on the level of R&D activities being undertaken at any time and the availability of funding from investors or collaboration partners.
OUTLOOK FOR 2019
The Corporation has many clinical studies ongoing and expects the following timing to disclose results for the following studies:
![]()
The exact timing of disclosure of the above results could differ from our expectations but are currently management’s best estimate.
RELATED PARTY TRANSACTIONS
During Q1 2019, there were no related party transactions (Q1 2018 - $nil).
CONTRACTUAL OBLIGATIONS
There is no material change in the contractual obligations of the Corporation since the beginning of the 2019 fiscal year. Details on the contractual obligations of the Corporation can be found in the in the annual audited consolidated financial statements and related notes for the year ended December 31, 2018.
OFF-BALANCE SHEET ARRANGEMENTS
The Corporation was not party to any off-balance sheet arrangements as of March 31, 2019.
OUTSTANDING SECURITIES
As of May 9, 2019, the number of issued and outstanding common shares was 50,597,306 and a total of 2,030,471 stock options, warrants, and deferred share units were outstanding.
RISKS AND UNCERTAINTIES
The Corporation is a clinical-stage company that operates in an industry that is dependent on a number of factors that include the capacity to raise additional capital on reasonable terms, obtain positive results of clinical trials - including clinical trials on DPX-Survivac, obtain positive results of clinical trials without serious adverse or inappropriate side effects, and obtain market acceptance of its product by physicians, patients, healthcare payers and others in the medical community for commercial success, etc. An investment in the Corporation’s common shares is subject to a number of risks and uncertainties. An investor should carefully consider the risks described in the Corporation’s AIF and the registration statement on Form 40-F filed with the U.S. Securities and Exchange Commission, as well as the other information filed with the securities regulators before investing in the Corporation’s common shares. If any of the such described risks occur, or if others occur, the Corporation’s business, operating results and financial condition could be seriously harmed and investors may lose a significant proportion of their investment.
There are important risks which management believes could impact the Corporation’s business. For information on risks and uncertainties, please also refer to the “Risk Factors” section of our most recent AIF filed on SEDAR atwww.sedar.comand included in the registration statement on Form 40-F filed on EDGAR at www.sec.gov/edgar.
21
DISCLOSURE CONTROLS AND PROCEDURES AND INTERNAL CONTROLS OVER FINANCIAL REPORTING
Disclosure Controls and Procedures
The Chief Executive Officer (the “CEO”) and the Chief Financial Officer (the “CFO”) of the Corporation are responsible for establishing and maintaining the Corporation’s disclosure controls and procedures (“DCP”) including adherence to the Disclosure Policy adopted by the Corporation. The Disclosure Policy requires all staff to keep senior management fully apprised of all material information affecting the Corporation so that they may evaluate and discuss this information and determine the appropriateness and timing for public disclosure.
The Corporation maintains DCP designed to ensure that information required to be disclosed in reports filed under applicable securities laws, is recorded, processed, summarized and reported within the appropriate time periods and that such information is accumulated and communicated to the Corporation’s management, including the CEO and CFO, to allow for timely decisions regarding required disclosure.
The CEO and CFO have evaluated whether there were changes to the DCP during the year ended December 31, 2018 that have materially affected, or are reasonably likely to materially affect, the DCP. No such changes were identified through their evaluation.
In designing and evaluating DCP, the Corporation recognizes that any disclosure controls and procedures, no matter how well conceived or operated, can only provide reasonable, not absolute, assurance that the objectives of the control system are met, and management is required to exercise its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Internal Control over Financial Reporting
The Corporation’s management, including the CEO and the CFO, are responsible for establishing and maintaining adequate internal control over financial reporting (“ICFR”) for the Corporation to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with IFRS. The fundamental issue is ensuring all transactions are properly authorized and identified and entered into a well-designed, robust and clearly understood accounting system on a timely basis to minimize risk of inaccuracy, failure to fairly reflect transactions, failure to fairly record transactions necessary to present financial statements in accordance with IFRS, unauthorized receipts and expenditures, or the inability to provide assurance that unauthorized acquisitions or dispositions of assets can be detected.
The CEO and CFO have evaluated whether there were changes to the ICFR during the year ended December 31, 2018 that have materially affected, or are reasonably likely to materially affect, the ICFR. No such changes were identified through their evaluation.
The Corporation’s ICFR may not prevent or detect all misstatements because of inherent limitations. Additionally, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because changes in conditions or deterioration in the degree of compliance with the Corporation’s policies and procedures.
BASIS OF PRESENTATION OF CONSOLIDATED FINANCIAL STATEMENTS AND SIGNIFICANT ACCOUNTING POLICIES
The consolidated financial statements have been prepared in accordance with the IFRS as issued by the IASB. The accounting policies, methods of computation and presentation applied in the consolidated financial statements are consistent with those of previous financial year except for business development and investor relations expenses are now presented in general and administrative expenses on the consolidated statements of loss and comprehensive loss. Certain comparative figures have been reclassified to conform the presentation adopted in the current year for general and administrative expenses.
The significant accounting policies of IMV are detailed in the notes to the annual audited consolidated financial statements for the year ended December 31, 2018 filed on SEDAR www.sedar.com and included in the registration statement on Form 40-F filed on EDGAR at www.sec.gov/edgar.
CRITICAL ACCOUNTING ESTIMATES AND JUDGEMENTS
Estimates and assumptions are continually evaluated and are based on historical experience and other factors, including expectations of future events that are believed to be reasonable under the circumstances. The determination of estimates requires the exercise of judgement based on various assumptions and other factors such as historical experience and current and expected economic conditions. Actual results could differ from those estimates.
Critical judgements in applying the Corporation’s accounting policies are detailed in the annual audited consolidated financial statements for the year ended December 31, 2018 filed on SEDAR www.sedar.com and included in the registration statement on Form 40-F filed on EDGAR at www.sec.gov/edgar.
22
FINANCIAL INSTRUMENTS
Financial instruments are defined as a contractual right or obligation to receive or deliver cash on another financial asset. The Corporation recognizes financial instruments based on their classification. Depending on the financial instrument’s classification, changes in subsequent measurements are recognized in net loss or other comprehensive loss.
A description of the financial instruments, their fair value and risk management is included in the Corporation’s annual audited consolidated financial statements for the year ended December 31, 2018 filed on SEDAR www.sedar.com and included in the registration statement on Form 40-F filed on EDGAR at www.sec.gov/edgar.
| (Signed) Frédéric Ors | (Signed) Pierre Labbé |
| Frédéric Ors | Pierre Labbé |
| Chief Executive Officer | Chief Financial Officer |
May 9, 2019
23