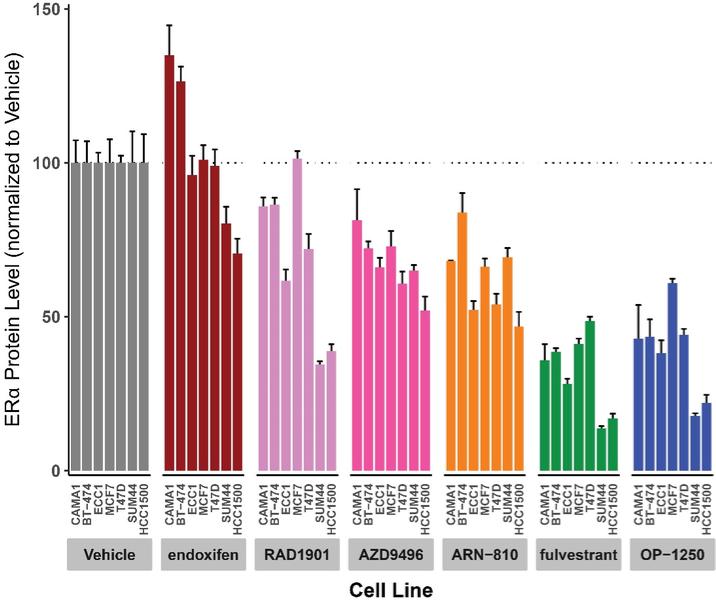
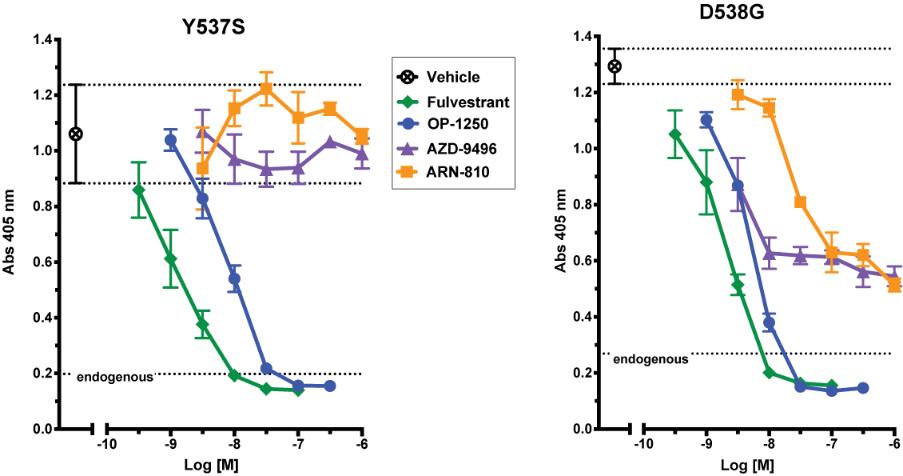
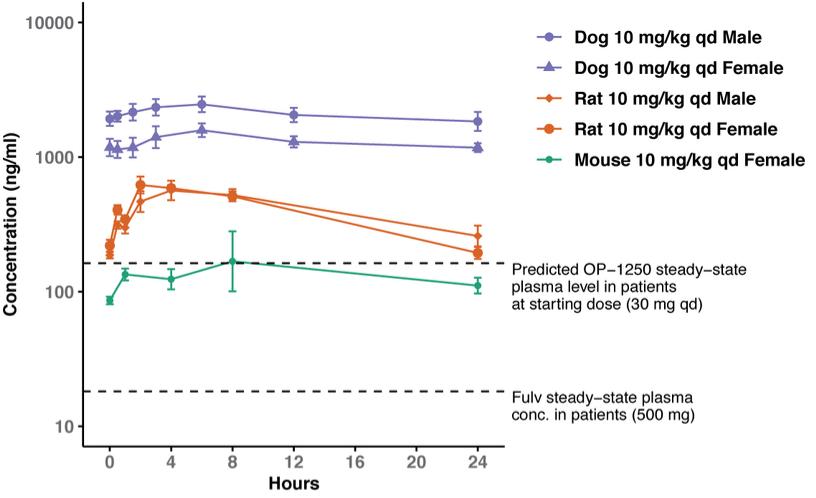
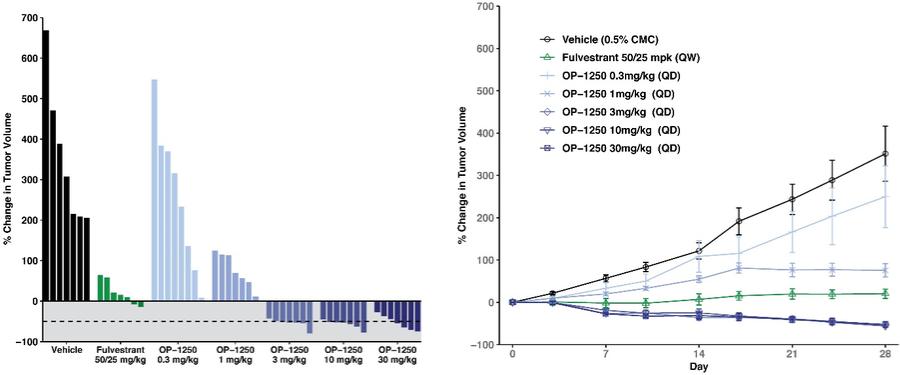
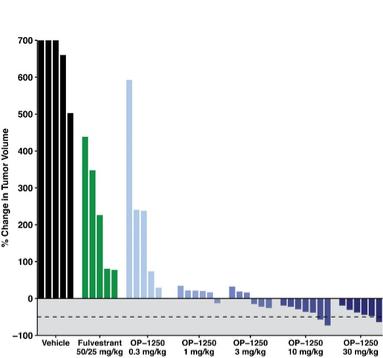
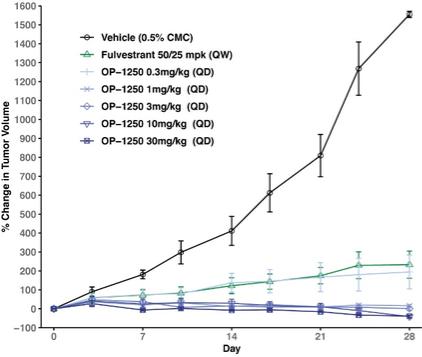
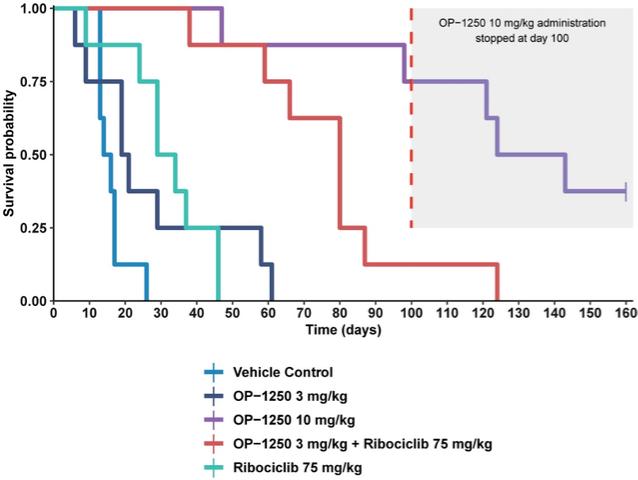
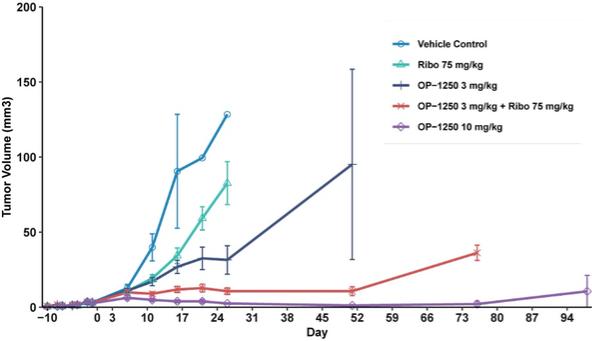
were unsecured and were due and payable, including accrued interest, on May 2, 2020 (“maturity date”). The Company was not permitted to prepay the outstanding principal and interest without the consent of the note holders. In the event of a default, all unpaid principal and accrued interest would become immediately due.
On the issuance date the Company determined that the conversion option associated with the 2020 Notes met the definition of a beneficial conversion feature (“BCF”) as the fair value of the underlying instrument at the time of issuance exceeded the contractual conversion price. The BCF was recognized at its aggregate intrinsic value of $1.1 million as a debt discount with a corresponding credit to additional paid-in capital in the Company’s balance sheet. The debt discount was amortized over the term of the 2020 Notes through the recognition of interest expense via the effective interest method.
On March 17, 2020 (the “settlement date”), the Company issued and sold 2,545,277 shares of Series B convertible preferred stock at $4.712 per share for gross proceeds of approximately $12.0 million (see Note 7, “Convertible Preferred Stock”). On the settlement date, the principal and accrued interest then outstanding under the 2020 Notes of $3.0 million were converted into 638,270 shares of Series B convertible preferred stock (“March 2020 conversion”).
On the settlement date, the unamortized debt discount on the 2020 Notes was $0.4 million and the intrinsic value of the BCF was $2.6 million representing an increase of $1.5 million from the issuance date of the 2020 Notes. The March 2020 conversion was accounted for as a debt extinguishment. However, as the note holders were previous investors of the Company, the increase in the intrinsic value of the BCF was deemed to be a capital contribution and therefore not income attributable to common stockholders, and accordingly, the Company recorded the $1.5 million gain on extinguishment of the debt within additional paid-in capital.
6. Convertible Preferred Stock
Upon the closing of the Company’s IPO, each then outstanding share of convertible preferred stock was converted into one share of common stock.
As of December 31, 2019, the Company’s certificate of incorporation, as amended and restated, authorized the Company to issue 4,640,126 shares of Series A convertible preferred stock at par value of $0.0001 per share. In September 2020, the Company purchased and retired 181,503 shares of Series A convertible preferred stock from investors at a price of $11.063 per share, or approximately $2.0 million.
In September 2020, the Company purchased and retired 25,319 shares of Series A-1 convertible preferred stock at a price of $11.063 per share or approximately $0.3 million.
Series B Convertible Preferred Stock
On March 13, 2020, the Company filed its fourth amended and restated certificate of incorporation, which authorized the Company to sell and issue up to 26,627,219 shares of Series B convertible preferred stock with a par value of $0.0001 per share. On March 17, 2020, the Company issued and sold 2,545,277 shares of Series B convertible preferred stock at $4.712 per share for gross proceeds of approximately $12.0 million. At the same time, the Company issued an additional 638,270 shares of Series B convertible preferred stock in conjunction with its conversion of the 2020 Notes (see Note 6, “Convertible Notes” “2020 Convertible Notes”), for a total of 3,183,547 shares of Series B convertible preferred stock issued on March 17, 2020. On March 20, 2020, the Company issued and sold 3,183,550 shares of Series B convertible preferred stock at $4.712 per share for gross proceeds of approximately $15.0 million. On May 28, 2020, the Company filed an amendment to its fourth amended and restated certificate of incorporation, which authorized the Company to sell and issue up to 32,781,066 shares of Series B convertible preferred stock. On June 1, 2020, the Company issued and sold 5,072,450 shares of Series B convertible preferred stock at $4.712 per share for gross proceeds of approximately $23.9 million (collectively, “Series B convertible preferred stock issuances”). In total, 11,439,547 shares of Series B convertible preferred stock were issued for gross cash proceeds of approximately $50.9 million.