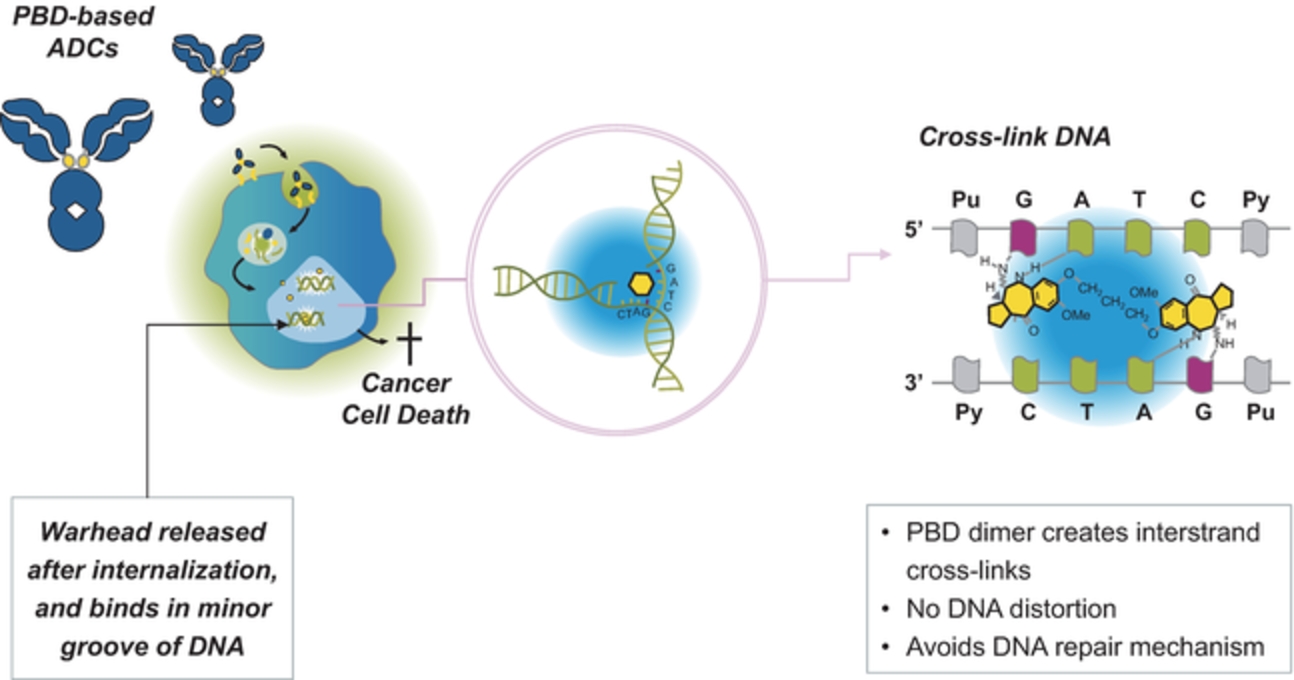
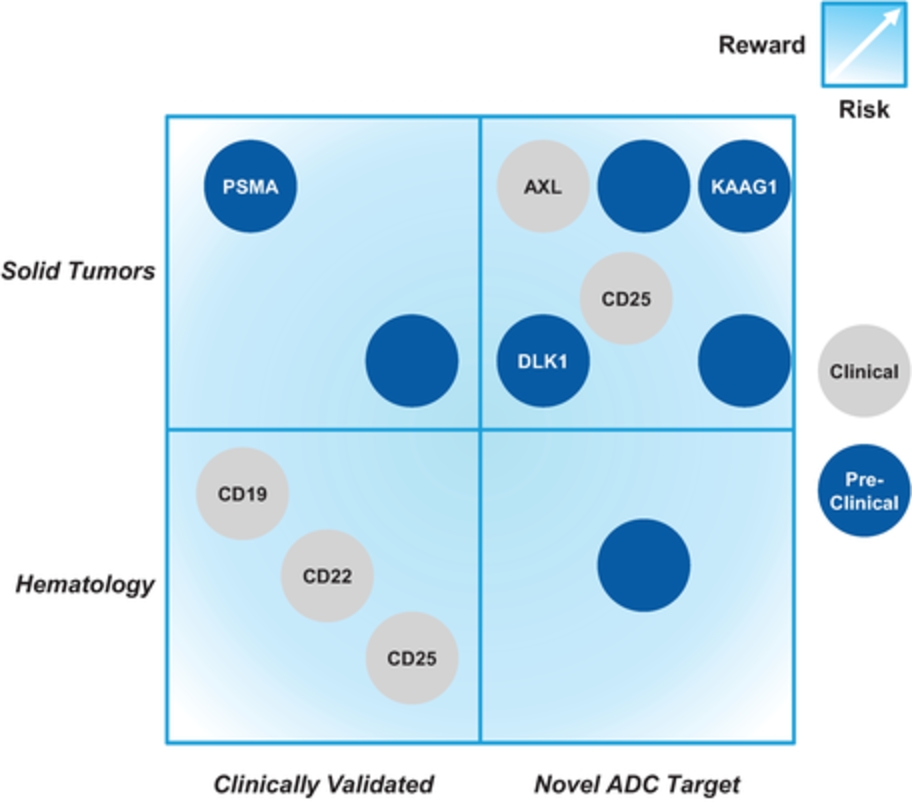
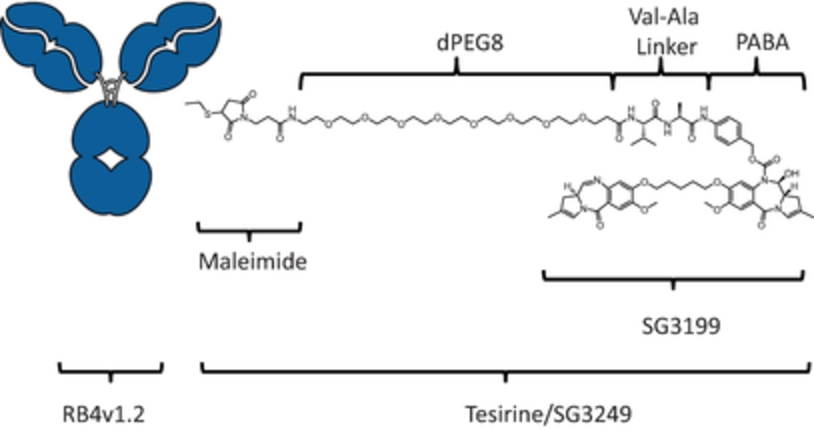
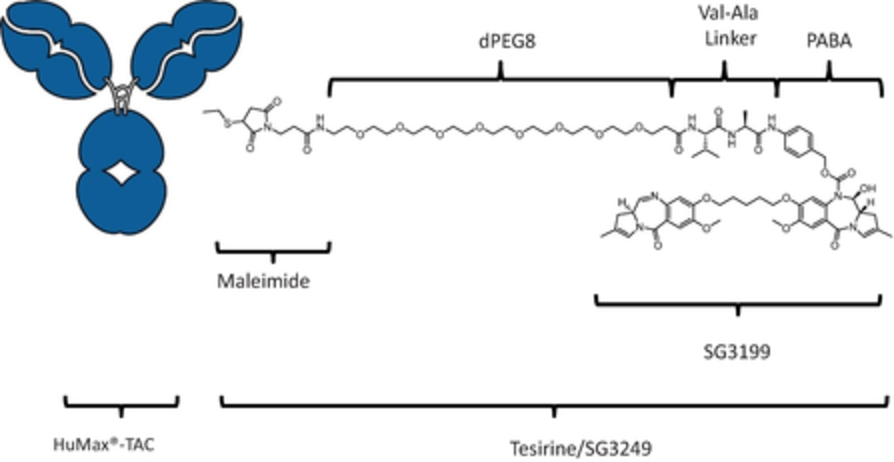
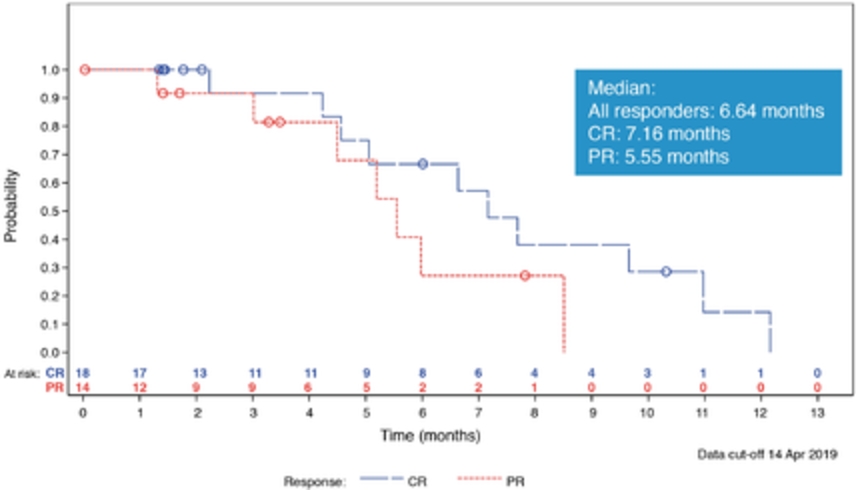
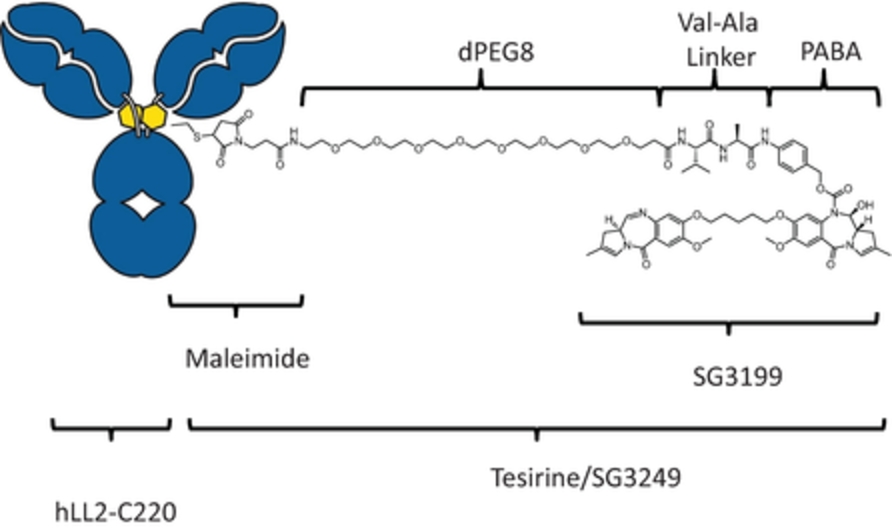
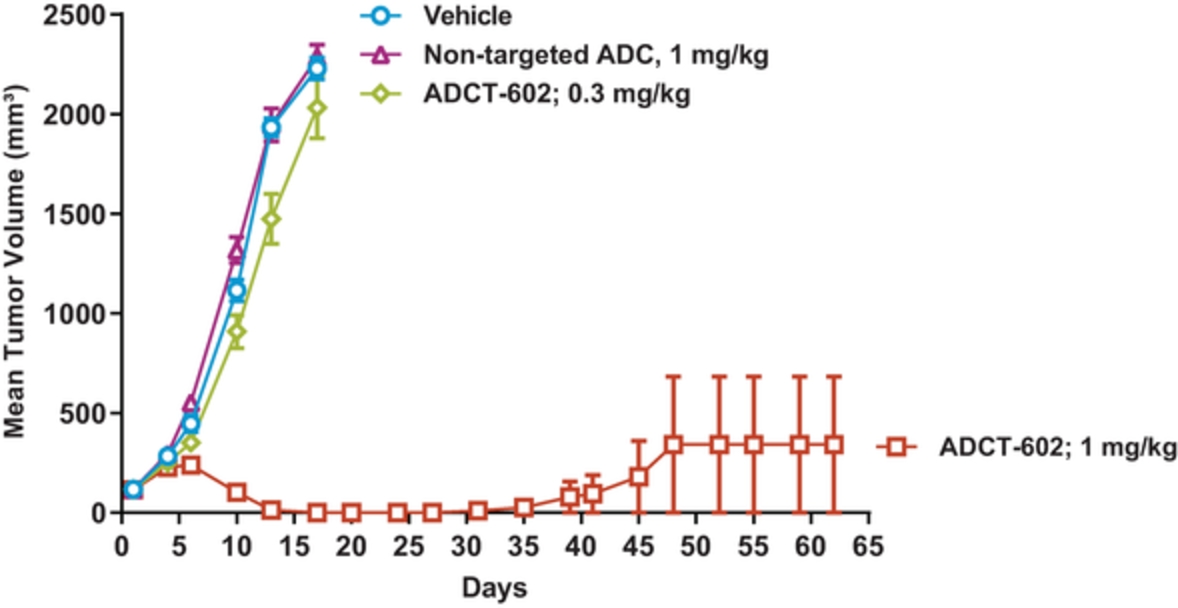
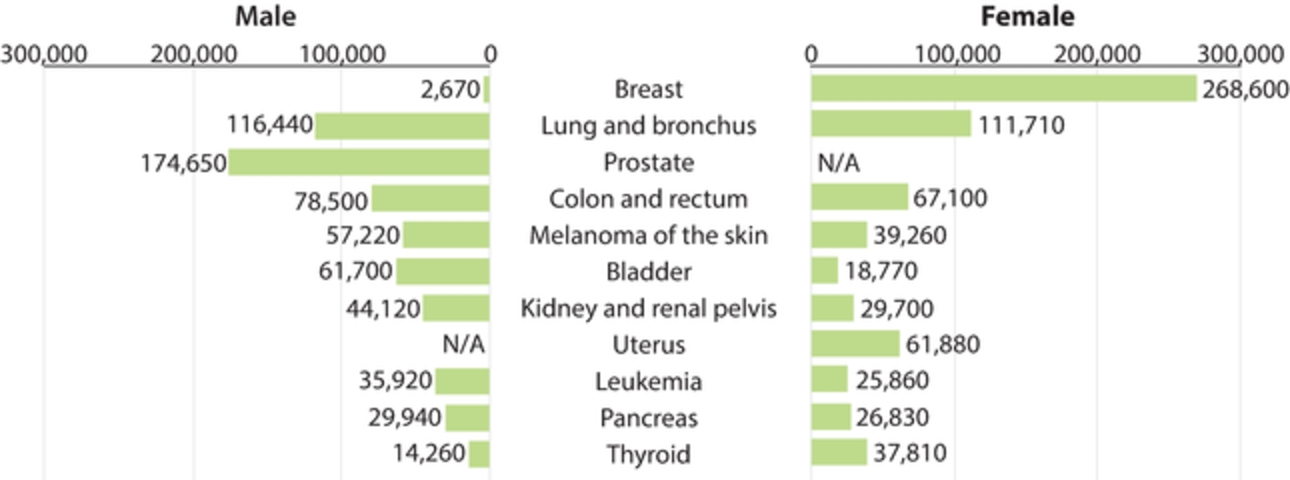
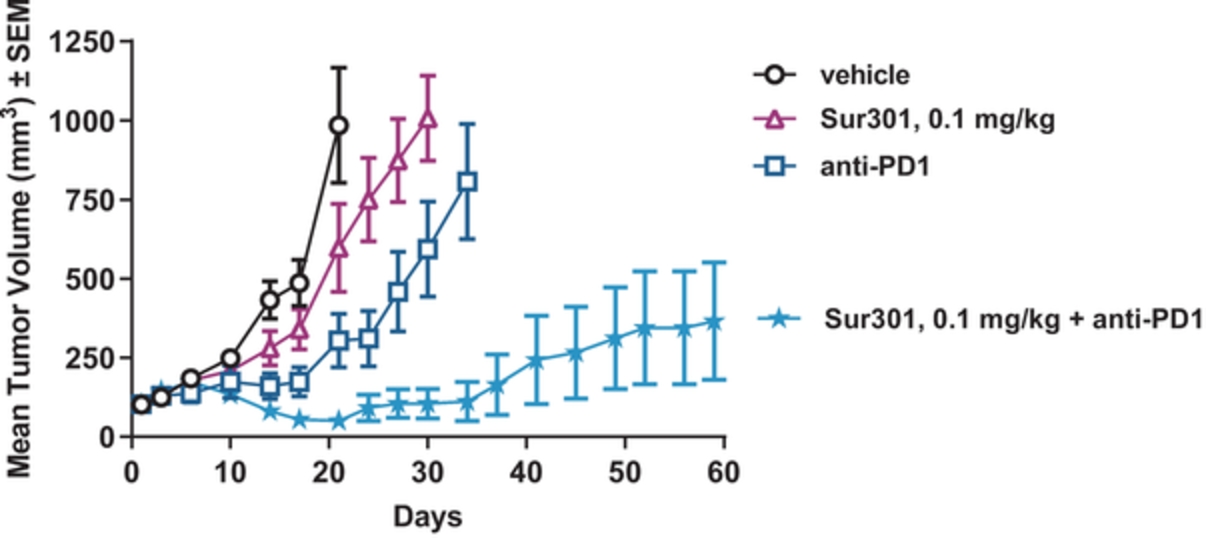
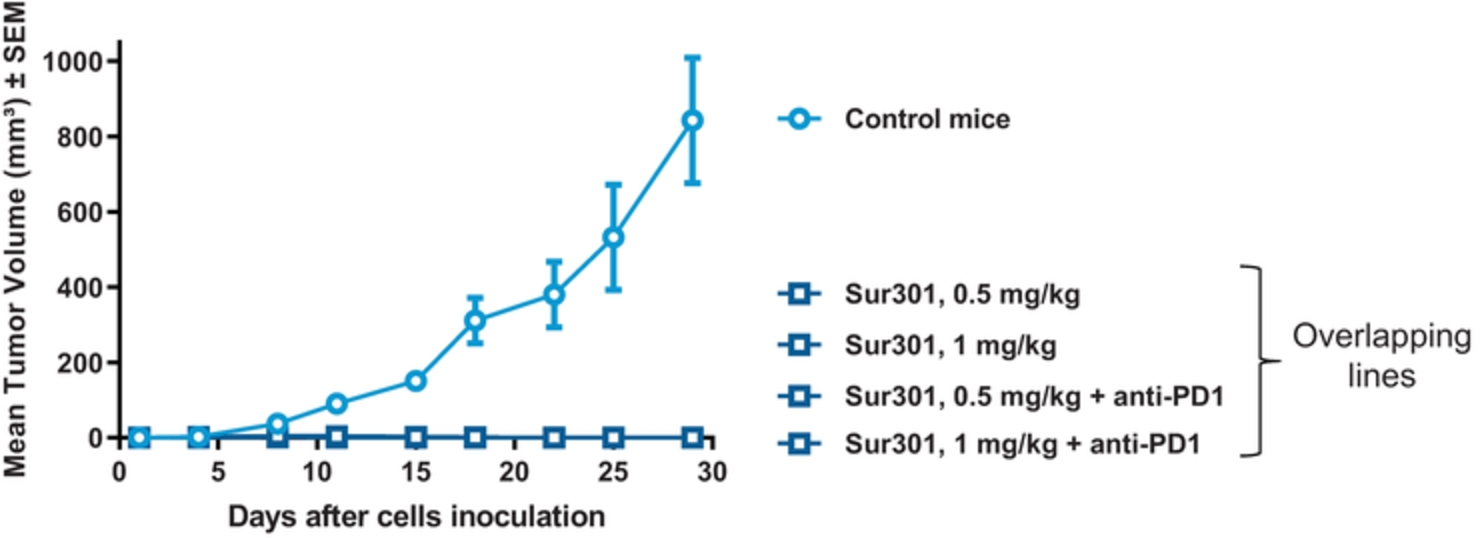
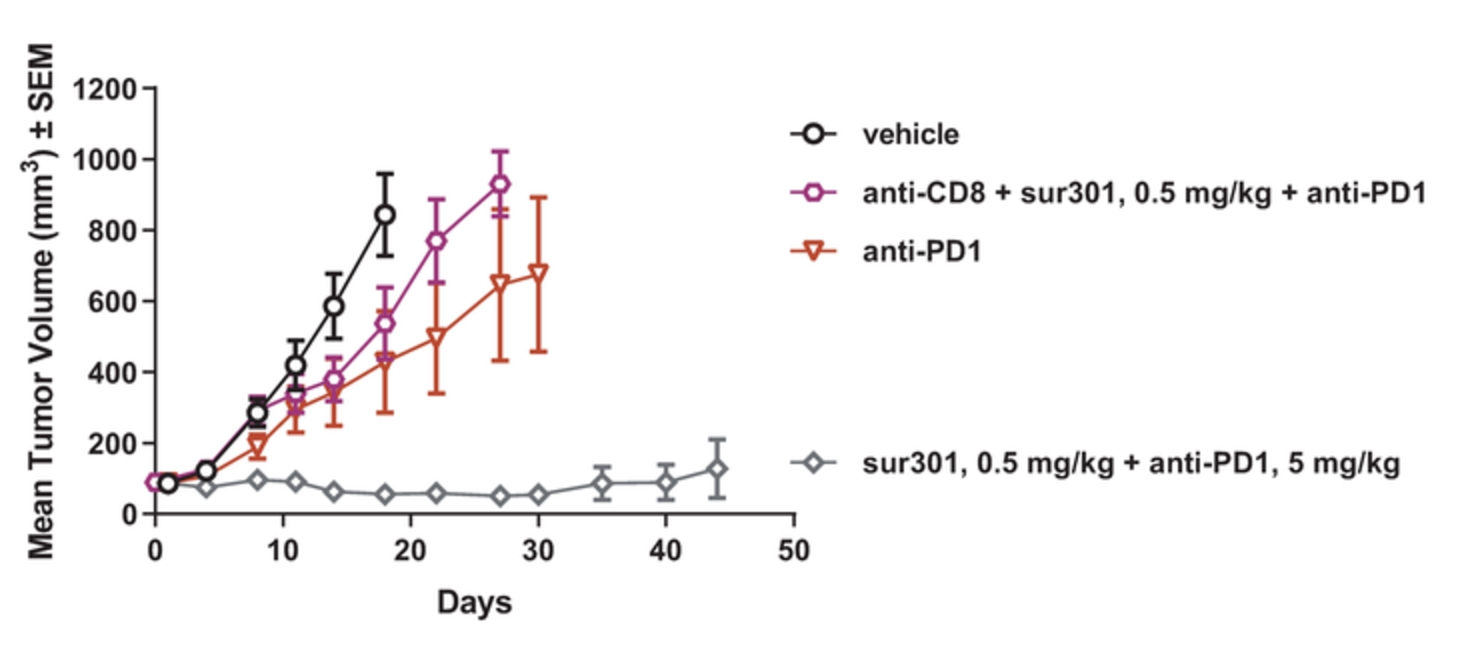
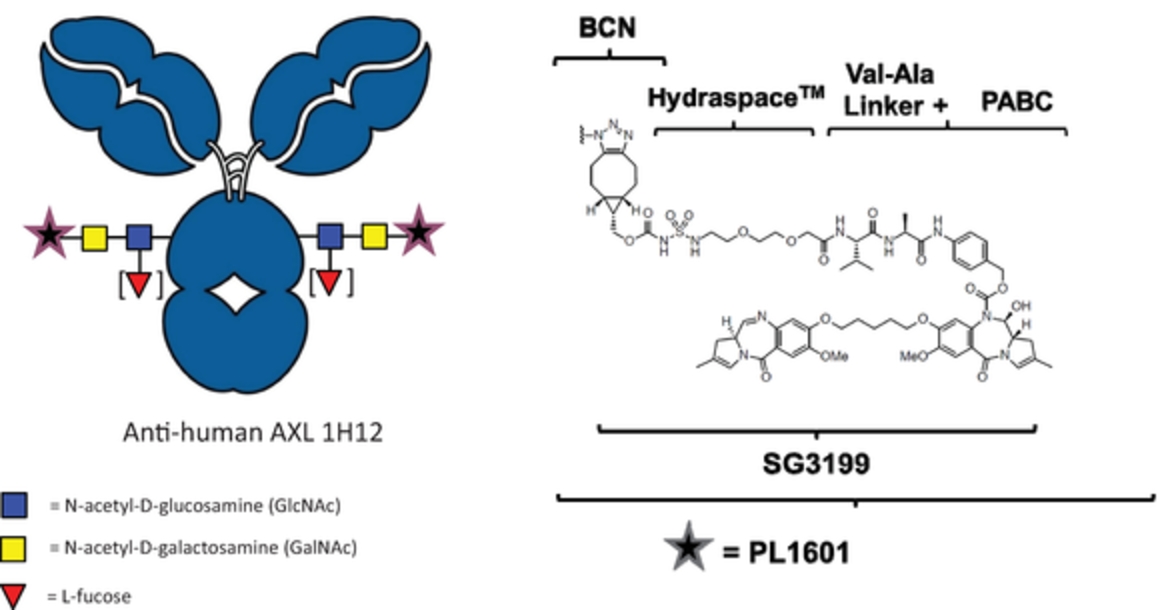
field including new payload classes, new conjugation approaches and new targeting moieties. Specifically, we are aware of multiple companies with ADC technologies that may be competitive to our product candidates, including, but not limited to, AbbVie, Inc., Astellas Pharma Inc., AstraZeneca plc, BioAtla, LLC, Bristol-Myers Squibb Company, CytomX Therapeutics, Daiichi Sankyo Company, Eli Lilly and Company, Genentech, Inc., Genmab A/S, ImmunoGen, Inc., Immunomedics, Inc., Mersana Therapeutics Inc., Millennium Pharmaceuticals, Inc., MorphoSys AG, Novartis International AG, Pfizer Inc., F. Hoffmann-La Roche AG, Sanofi S.A., Seattle Genetics, Inc., Sutro Biopharma, Inc., Takeda Pharmaceutical Company Ltd and Wyeth Pharmaceuticals, Inc. Currently, there are five approved ADCs: (i) brentuximab vedotin (Adcetris®), marketed by Seattle Genetics, Inc. and Takeda Pharmaceutical Company Ltd, (ii) ado-trastuzumab emtansine (Kadcyla®), marketed by Genentech, Inc., (iii) inotuzumab ozogamicin (Besponsa®), marketed by Pfizer Inc., (iv) gemtuzumab ozogamicin (Mylotarg®), also marketed by Pfizer Inc. and (v) polatuzumab vedotin (Polivy®), marketed by Genentech, Inc. In addition, as of 2018, there are approximately 175 ADCs in development, the vast majority of which are being developed for the treatment of cancer.
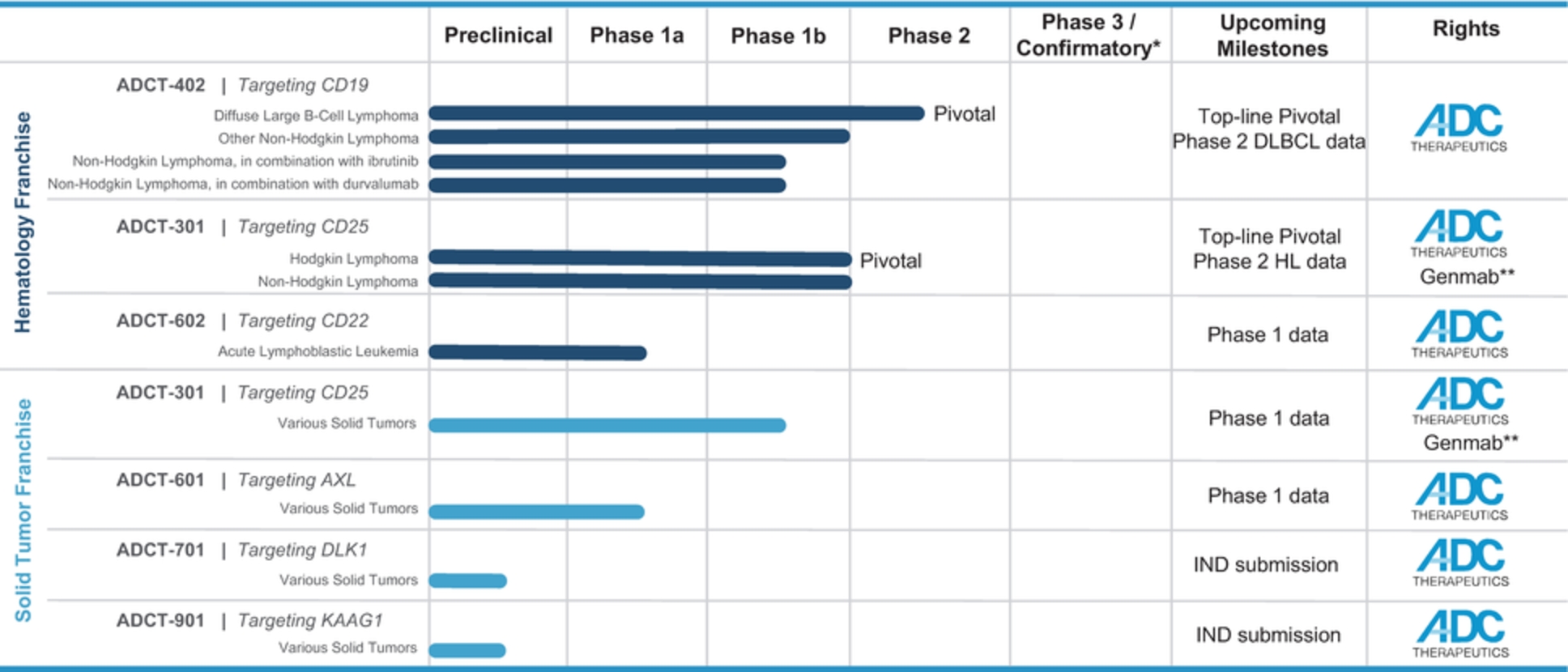
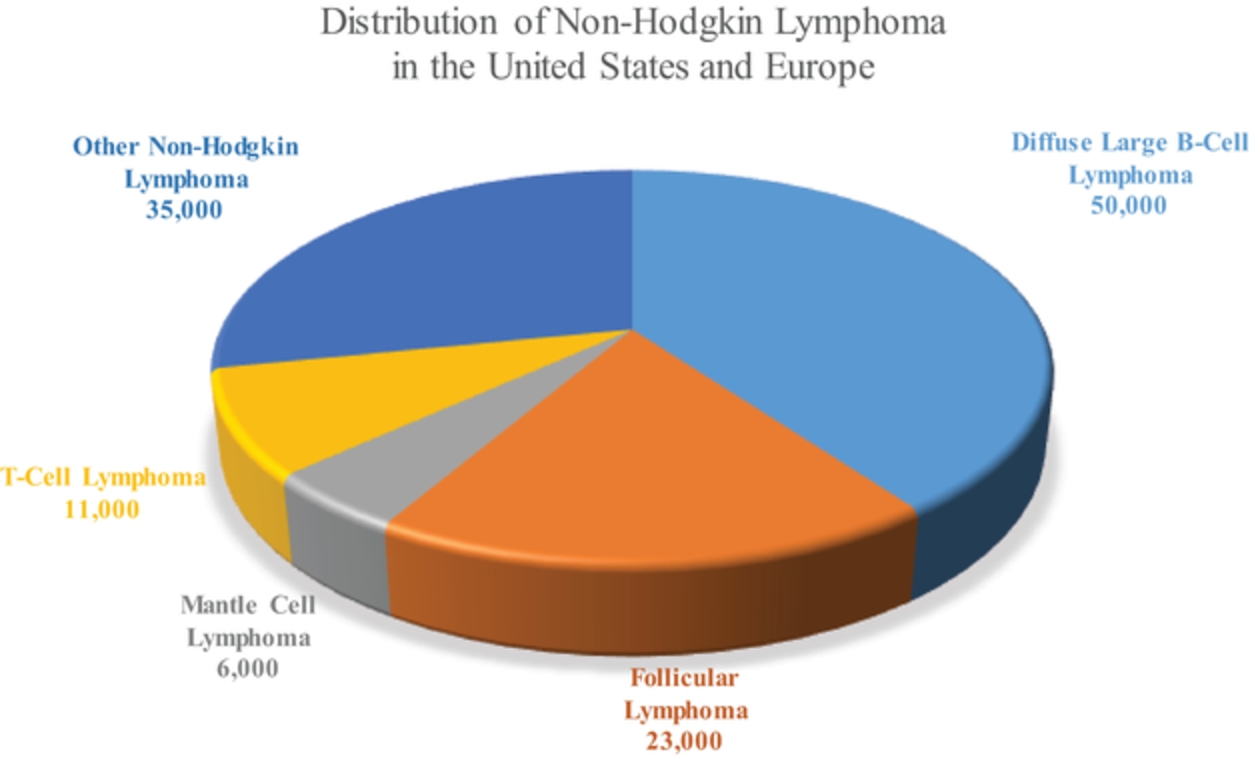
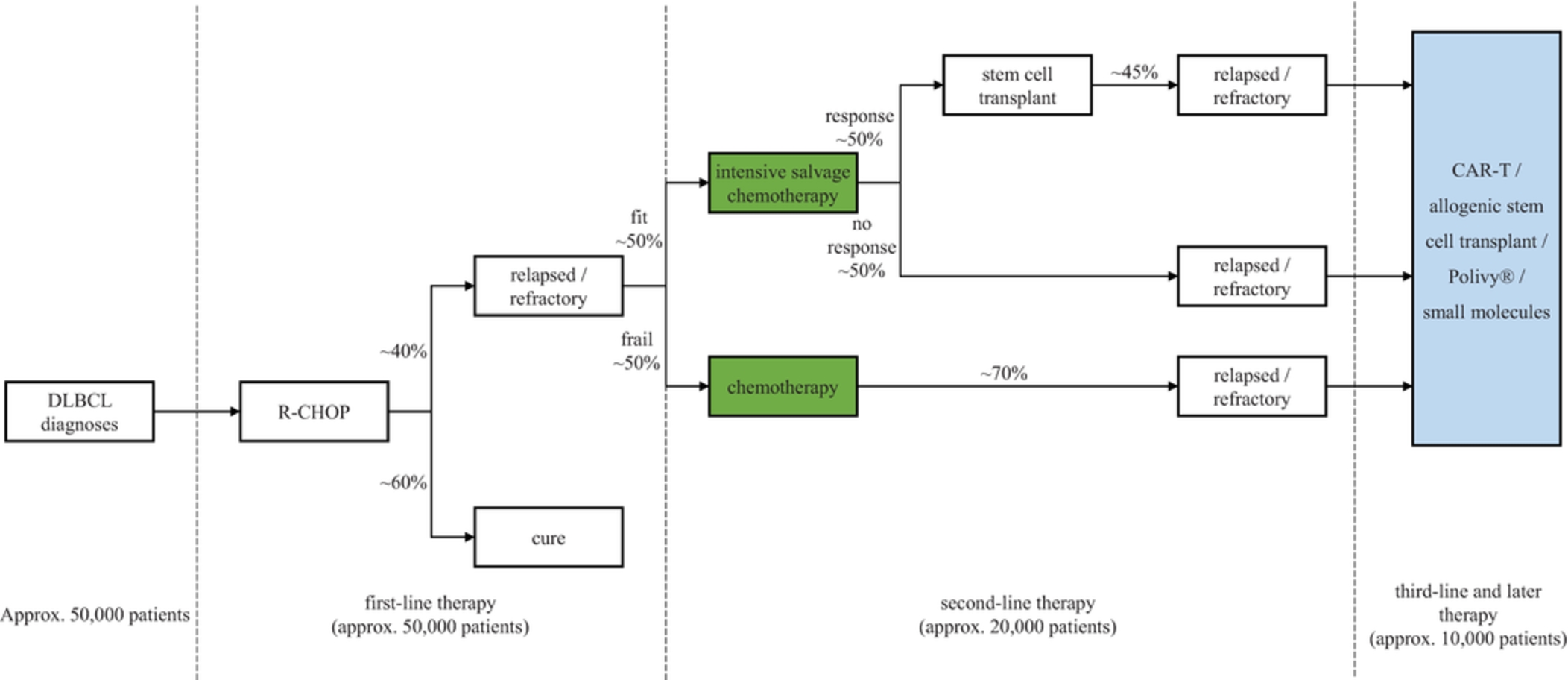
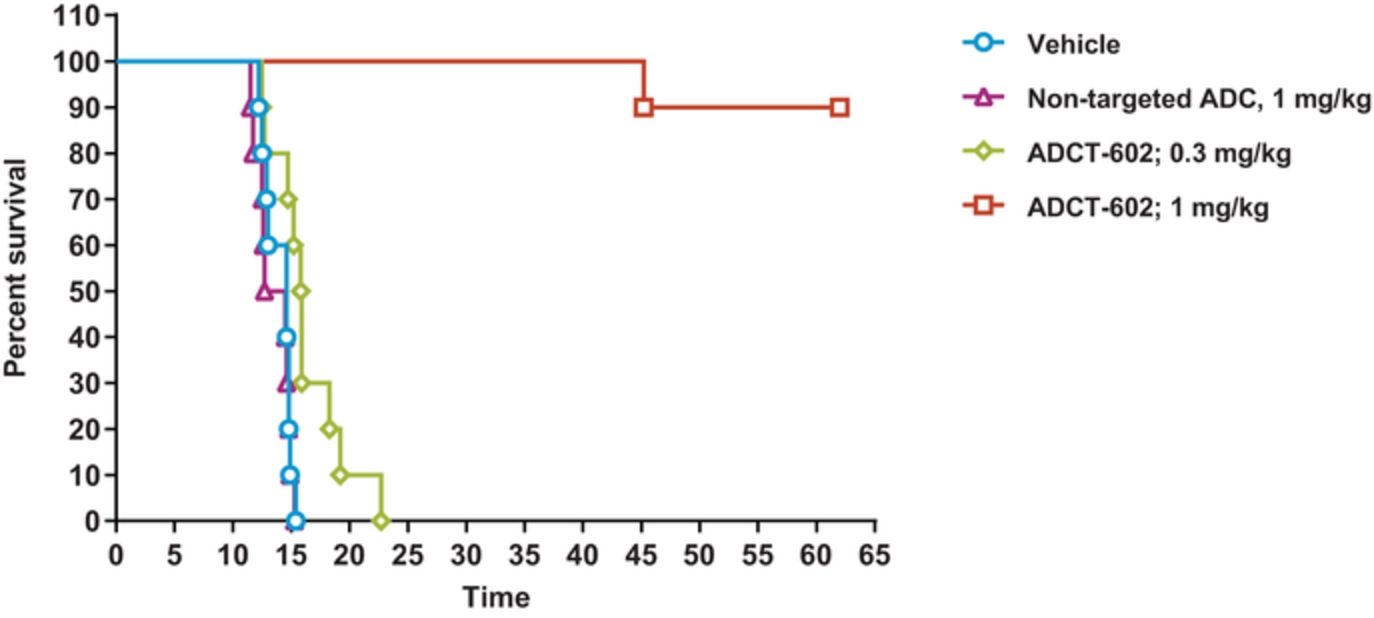
In the relapsed or refractory DLBCL setting, for which we are developing ADCT-402, current third-line treatment options include CAR-T, allogeneic stem cell transplant and chemotherapy using small molecules. Recently, Polivy® was approved for third-line use in combination with bendamustine and a rituximab product. In addition, we expect new competitors, including MOR208 and bispecific antibodies, to enter the market as treatment options for such patients.
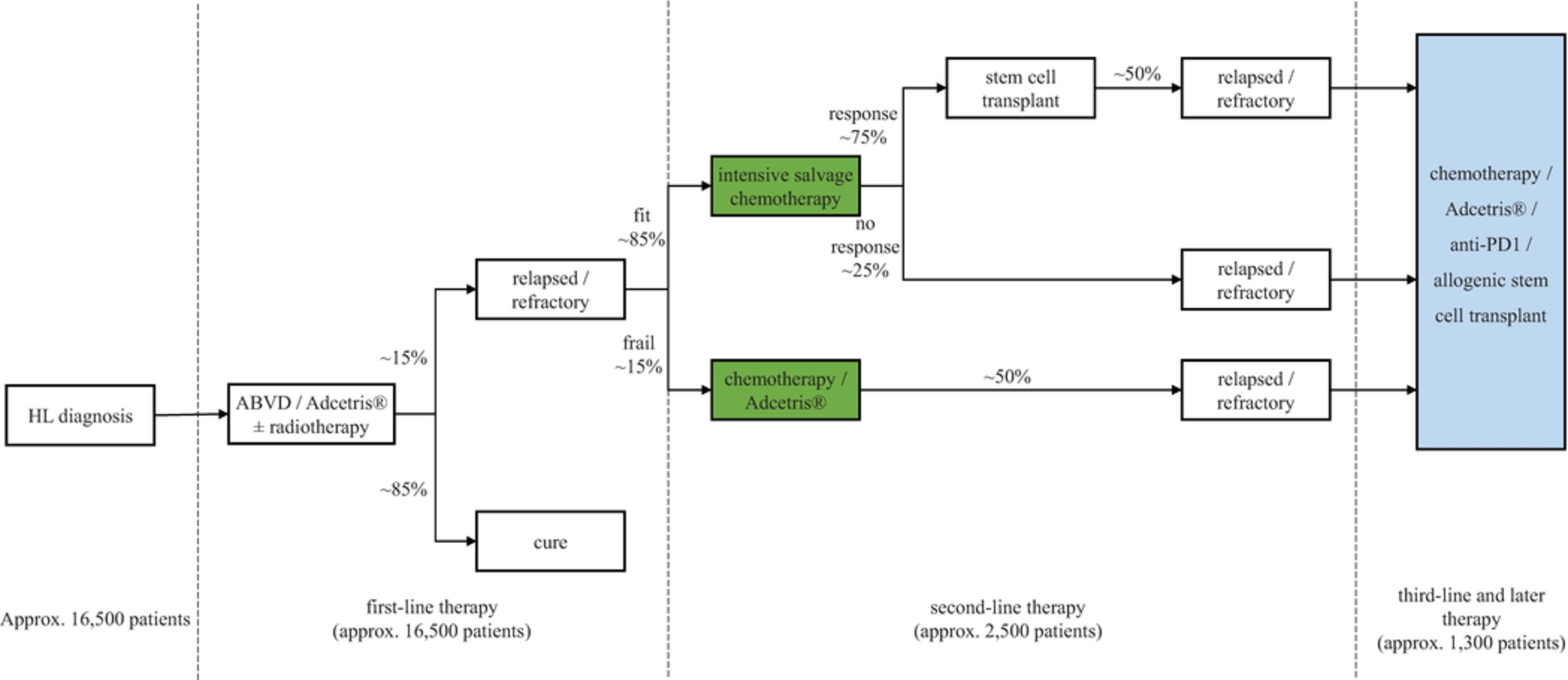
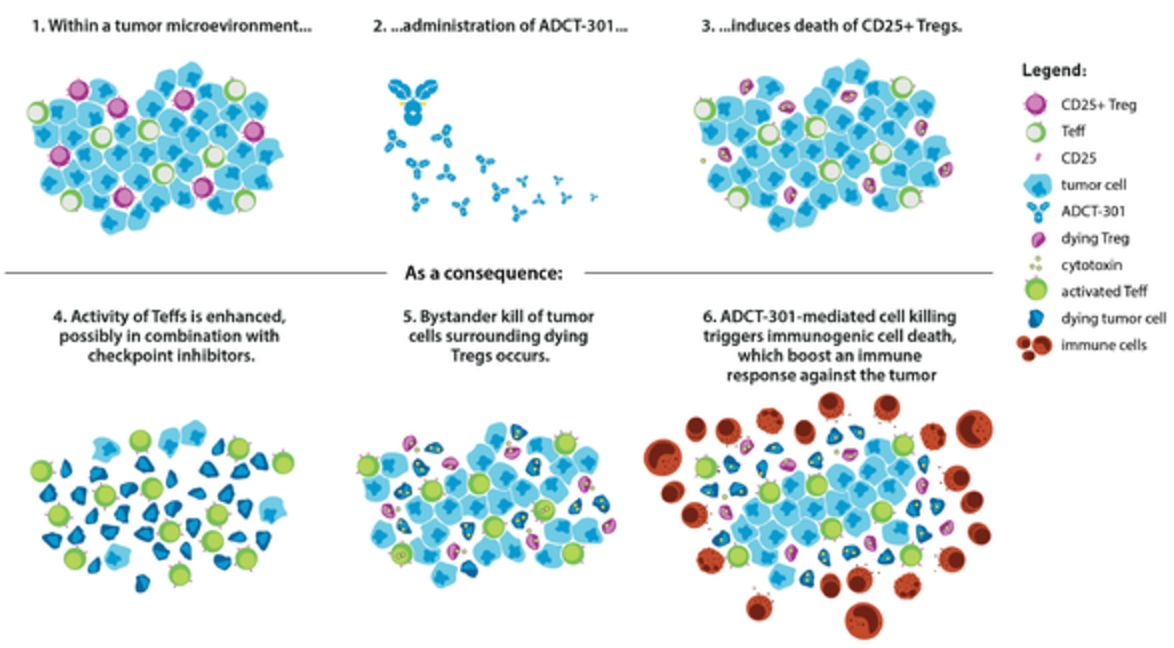
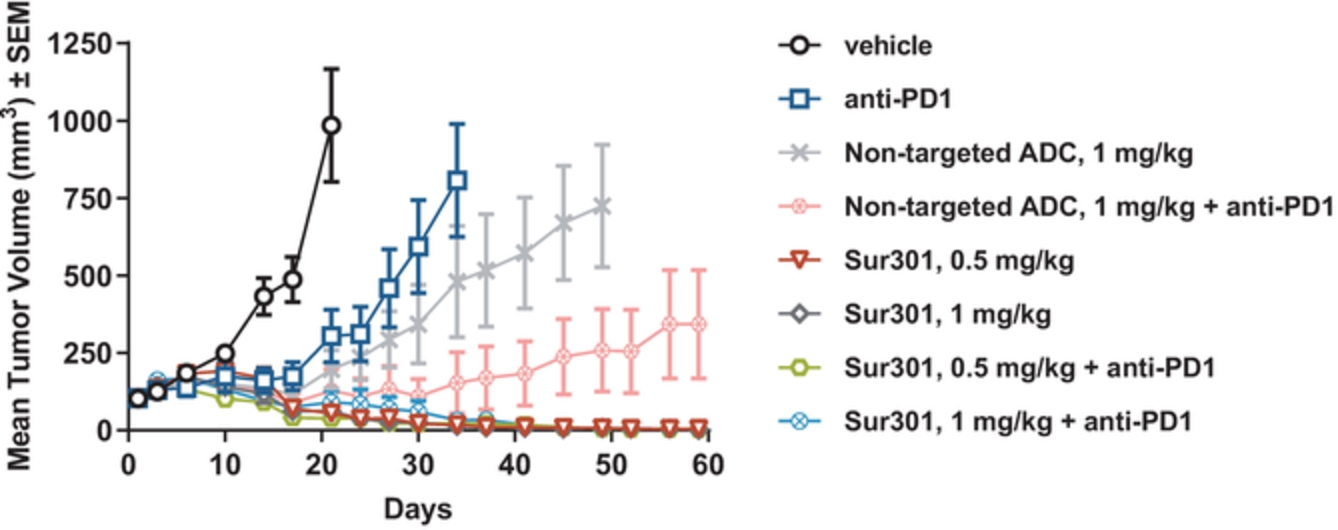
In the relapsed or refractory HL setting, for which we are developing ADCT-301, current third-line treatment options include chemotherapy, immunotherapy and Adcetris®. In addition, we expect changes to the treatment paradigm, such as the movement of Adcetris® to earlier lines of therapy and the potential expanded use of checkpoint inhibitors, and new entrants such as bispecific antibodies.
Any product candidates that we successfully develop and commercialize may compete directly with approved therapies and any new therapies that may be approved in the future. Competition will be based on their safety and effectiveness, the timing and scope of marketing approvals, the availability and cost of supply, marketing and sales capabilities, reimbursement coverage, price levels and discounts offered, patent position and other factors. Our competitors may succeed in developing competing products before we do, obtaining marketing approval for products and gaining acceptance for such products in the same markets that we are targeting.
Government Regulation
Government authorities in the United States at the federal, state and local level and in other countries and jurisdictions, including the European Union, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug and biological products, such as our investigational medicines and any future investigational medicines. Generally, before a new drug or biologic can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific for each regulatory authority, submitted for review and approved by the regulatory authority.
Regulatory Approval in the United States
In the United States, pharmaceutical products are subject to extensive regulation by the FDA. The FDCA, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Biological products used for the prevention, treatment or cure of a disease or condition of a human being are subject to regulation under the FDCA, except the section of the FDCA that governs the approval of new drug applications (“NDAs”). Biological products, such as our ADC product candidates, are approved for marketing under provisions of the Public Health Service Act (the “PHSA”), via a BLA. However, the application process and requirements for approval of BLAs are very similar to those for NDAs, and biologics are associated with similar approval risks and costs as drugs. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as clinical hold, FDA refusal to approve pending NDAs or BLAs, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.