Exhibit 99.1
DESCRIPTION OF BUSINESS OF BAUDAX BIO, INC.
Company Overview
Baudax Bio, Inc. is a biotechnology company focused on developing T cell receptor (“TCR”) therapies utilizing human regulatory T cells (“Tregs”), as well as a portfolio of clinical stage Neuromuscular Blocking Agents (“NMBs”) and an associated reversal agent. Our TCR Treg programs primarily focus on immune modulating therapies for orphan diseases or complications associated with such diseases, as well as the treatment of autoimmune disorders. We believe that our TCR Treg programs have the potential to provide valuable therapeutic options to patients suffering from diseases for which there are limited treatment options and significant unmet need, as well as to prescribers and payers in these markets.
On June 29, 2023, we acquired TeraImmune, Inc. (“TeraImmune”), a Delaware corporation. TeraImmune was a privately-held biotechnology company focused on discovery and development of novel Treg-based cell therapies for autoimmune diseases. TeraImmune’s proprietary and patented technology platforms include a method for expansion of the Treg without losing its function and stability, as well as a method to target specific receptors including TCRs, Chimeric Antigen Receptors (“CARs”) and B cell Antigen Receptors (“BARs”). TerraImmune has also in-licensed through an exclusive, sublicensable, royalty-bearing license, a patent family covering methods of producing T cell populations enriched for regulatory T cells and cell culture compositions from U.S. Department of Health and Human Services, as represented by National Institute of Allergy and Infectious Diseases of the National Institutes of Health. In addition, TeraImmune has developed Treg manufacturing procedures in accordance with regulatory guidance from the U.S. Food and Drug Administration (“FDA”).
In June 2022, TeraImmune’s Investigational New Drug (“IND”) application to commence clinical trials of a Factor VIII (“FVIII”) TCR-Treg treatment for Hemophilia A with inhibitors was cleared by the FDA.
Tregs are designed to recognize and target certain cells through the engagement of target-specific receptors by peptide antigens presented on the surface of the target cell by the major histocompatibility complex. Our proprietary and patented technology platform consists of two approaches: (1) TREGable™, which involves the isolation of natural Tregs, and (2) TREGing™, which involves engineering effector T (“Teff”) cells into antigen-specific Tregs. Each approach is intended to recognize and attack pathogens while avoiding an attack on healthy cells and tissues. The lead product candidate we acquired in the acquisition with TeraImmune, TI-168, is being developed for the treatment of Hemophilia A with inhibitors, which received IND clearance in 2022. We have in-licensed two patent families relating to TI-168, nucleic acids constructs encoding T cell receptors, methods of producing TI-168, immunosuppressive induced regulatory T cells from the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. (“HJF”) under two worldwide, exclusive, sublicensable royalty-bearing licenses. We also exclusively license a family of pending U.S. and foreign patent applications directed to immunosuppressive induced regulatory T cells and methods of producing these cells, which if issued would expire in 2041 subject to any applicable disclaimer or extensions.
We also hold exclusive global rights to two new molecular entities, which are centrally acting NMBs, BX1000, an intermediate duration of action NMB that recently completed a successful Phase II clinical trial, and BX2000, an ultra-short acting NMB currently undergoing a Phase I clinical trial. A proprietary blockade reversal agent, BX3000, is currently being evaluated in preclinical studies intended to support an IND filing in 2023. BX3000 is an agent that is expected to rapidly reverse BX1000 and BX2000 blockade. All three agents are licensed from Cornell University. We believe these agents, when an NMB and BX3000 are administered in succession, allow for a rapid onset of centrally acting neuromuscular blockade, followed by a rapid reversal of the neuromuscular blockade with BX3000. These novel agents have the potential to meaningfully reduce time to onset and reversal of blockade and improve the reliability of onset and offset of neuromuscular blockade. This can potentially reduce time in operating rooms or post operative units, resulting in potential clinical and cost advantages, as well as valuable cost savings for hospitals and ambulatory surgical centers and has the potential for an improved clinical profile in terms of safety.
In mid-2020, we launched our first commercial product, ANJESO, in the United States. ANJESO was the first and only 24-hour, intravenous, analgesia agent. ANJESO is a cyclooxygenase-2 preferential, non-steroidal anti-inflammatory drug (“NSAID”) for the management of moderate to severe pain, which could be administered alone or in combination with other non-NSAID analgesics. We discontinued commercial sales of ANJESO in December 2022 and further withdrew its New Drug Application (“NDA”) related to ANJESO in late March 2023.
Our Strategy
We believe that we can bring valuable therapeutic options for patients suffering from certain orphan diseases and autoimmune disorders for which there are limited treatment options and significant unmet need, and prescribers and payers in these markets, as well as to the acute care and related markets. We believe we can create value for our shareholders through the development, and potential approval and commercialization of TI-168 for treatment of Hemophilia A with inhibitors, as well as our other pipeline product candidates we develop for the treatment of autoimmune disorders utilizing Treg-based therapies. In addition to our Treg pipeline, we continue to modestly progress the NMB and related assets, and will consider select acquisitions, especially those that could contribute revenue and cash flow.
Our near-term goals include:
| • | Leveraging our development experience to progress TI-168, and the research experience of the TeraImmune team to progress additional Treg research product candidates. We intend to leverage our drug development expertise to commence a Phase 1/2a clinical trial of TI-168 for treatment of the Hemophilia A with inhibitors, and continue development of TI-168 and other candidates for the treatment of other orphan or autoimmune disorders. We intend to approach the development of such programs in a cost-effective manner, including by potentially leveraging the data we generate from our TI-168 clinical trials for the further expansion of our Treg-based therapy platform, enabling us to expand our target indications and product candidates. Our overall goal is to utilize our drug development expertise to safely develop these Treg product candidates. |
| • | Continue development of our clinical stage NMB portfolio of product candidates. Our legacy clinical stage product pipeline includes proprietary NMB blockade product candidates for use in anesthesia, BX1000 and BX2000, as well as an NMB reversal agent currently in preclinical studies, BX3000, which is currently being evaluated in preclinical studies intended to support an IND filing in the last quarter of 2023. We believe the concurrent development of a blocking agents and reversal agent used safely in the same patient, once certain stand alone and initial combination information is available, will allow our programs to provide clinical, financial, and temporal advantages to providers and patients. |
Products and Pipeline
Clinical Development
TI-168
Our lead Treg-based product candidate, TI-168, is being developed for the treatment of Hemophilia A with inhibitors. Hemophilia A is significant bleeding disorder characterized by impaired clotting as a result of deficiencies in the production or coagulation FVIII. Hemophilia A is an orphan disease that is lifelong and with limited treatment options. Approximately 30% of Hemophilia A patients develop inhibitors to FVIII, which can add complexity to its treatment regimen, and increase costs significantly. Inhibitors have historically been contraindicated with gene therapy, further limiting treatment options. The current standard of care for patients with FVIII inhibitors includes immune tolerance induction (“ITI”), along with the use of Emicizumab and bypass agents. We believe ITI therapy has important limitations, including that a significant portion of the patient population does not respond to ITI (up to 60%), recurrence can occur in a significant number of patients (up to 29%), the therapy can be expensive with respect to both time and resources, and may render a patient ineligible for gene therapy. We believe that TI-168, subject to FDA approval and commercialization, may provide Hemophilia A patients with the ability to avoid the potentially prohibitive cost, inconvenience, efficacy concerns and other limitations of ITI. Further, we believe that patients with FVIII tolerance that receive TI-168 will be potential candidates for gene therapy.
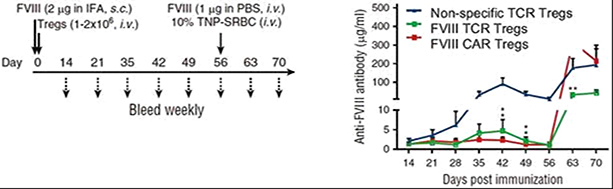
TI-168 is an autologous FVIII TCR-Treg cell therapy for the treatment of Hemophilia A patients with refractory inhibitors, which is designed to replace ITI treatments. Preclinical studies have shown that FVIII TCR-Tregs showed FVIII-specific immunosuppressive efficacy, with TCRs outperforming CAR Tregs. As illustrated in the images below, in a preclinical study, hemophilic mice were subcutaneously immunized for FVIII, and four hours thereafter, infused with either a TCR Treg, CAR Treg or nonspecific Treg. Over the course of the study, the nonspecific Treg could not effectively control the development of anti-FVIII antibodies, while the anti-FVIII antibody response was effectively suppressed by both the TCR and CAR Tregs over a period of approximately 8 weeks. Further, rechallenge with FVIII at day 56 resulted in a much higher loss of tolerance for the CAR Treg group, while the TCR Treg group’s loss of tolerance remained significantly lower. We believe that FVIII TCR Tregs may provide a therapeutic option in controlling anti-FVIII antibody formation in refractory Hemophilia A patients, and have the potential to perform better than CAR Treg therapies.
We plan to initiate a Phase 1/2a clinical trial of TI-168 for the treatment of Hemophilia A with inhibitors, with proof of concept data expected on the first 3 patients within the next 12-15 months. We are actively engaging in the Institutional Review Board (“IRB”) clinical trial site process. We intend to seek orphan drug designation in the United States for TI-168 for the treatment of Hemophilia A.
BX1000
We completed a Phase I study in 2021 for BX1000 which evaluated its safety profile when administered with Total Intravenous Anesthesia, as well as the dose response of neuromuscular blockade. We completed a dose-escalation study evaluating BX1000 in a total of 58 healthy volunteers who had already undergone endotracheal intubation while under general anesthesia. After intubation, subjects received a single IV bolus dose of BX1000 and were monitored for neuromuscular blockade and for any changes in vital signs or the presence of adverse events. BX1000 dose-escalations were continued until prespecified effects were observed. Doses of BX1000, up to 0.4 mg/kg, were well tolerated in this study of healthy volunteer subjects. Muscle paralysis was rapidly achieved along with complete spontaneous recovery. Neuromuscular blocking parameters were observed to increase in depth and duration of blockade while the time to onset of blockade was reduced with increasing doses of BX1000. Pharmacokinetic exposures increased with increasing study doses while elimination of the study compound remained rapid. Evaluation of electrocardiogram data using concentration-QTc modeling did not identify a risk of QTc prolongation within the studied dosing range. We engaged with the FDA regarding the design for the Phase II study in patients undergoing elective hernia and similar abdominal surgical procedures utilizing total intravenous anesthesia, in the fall of 2022, and initiated enrollment in the study in the fourth quarter of 2022. In January 2023, we announced the positive outcome of the interim analysis of the randomized, double blind, active controlled clinical Phase II trial, which compared three doses of BX1000 to a standard dose of rocuronium. The interim analysis was performed without breaking the study blind and was based on the first 20 of the 80 total patients being enrolled to the 4 study arms. The primary efficacy endpoint was the proportion of patients meeting criteria for Good or Excellent intubating conditions using a standardized scale. Additionally, the study is evaluating the safety and tolerability of BX1000 as compared to rocuronium in this patient population.
The top-line results for the Phase II clinical trial for BX1000 showed that BX1000 met the primary endpoint of readiness for intubation (evaluated as “Good” or “Excellent” - Viby-Mogensen 1996) at 60 seconds for all three dose levels of BX1000 compared to the active-control, rocuronium. Study treatments were generally well tolerated, with no occurrence of severe or serious adverse events. The frequency and severity of adverse events was similar across all four dose groups, and no notable events were aggregated in any one dose group.
BX2000
We filed an IND for BX2000 in 2020 in order to conduct a first-in-human clinical trial. We conducted an additional toxicology study requested by the FDA in 2021 and in March 2022, FDA notified us that we could proceed with initiation of a first in human, Phase I dose-escalation study in healthy volunteers.
In June of 2022, we announced the completion of dosing of the first cohort of the Phase I dose escalation study for BX2000, which we believe to be a rapid onset, ultra-short acting NMB agent, in healthy volunteers. The study is investigating single, ascending doses of BX2000 administered in a single, intravenous bolus injection compared to placebo. The study is comprised of up to 10 dosing cohorts and each cohort will enroll 8 patients. The study will evaluate the effect of BX2000 on safety, including heart rate, blood pressure, corrected QT interval, pharmacokinetics, and the time course of the neuromuscular blocking profile. Subjects will be monitored at an inpatient facility for 24 hours following administration of BX2000. There are also follow up visits on Day 8 and additional follow ups will take place approximately 2 and 4 weeks after dosing to evaluate the continued safety of study participants. Enrollment began in the second quarter of 2022 and cohort 2 completed, as planned, in the fourth quarter of 2022. Enrollment in cohort 3 was completed in 2023 and despite the challenges of enrollment to this type of protocol and operating on a limited budget, we remain optimistic that we will be close to reaching maximum dosage in 2024.
BX3000
BX3000 is a small molecule that was designed to induce chemical cleaving of BX1000 and BX2000, resulting in the rapid inactivation of those molecules and thus quickly reversing neuromuscular blockade. We are currently engaged with the pre-clinical toxicity studies needed to support an IND filing for BX3000 in the last quarter of 2023. We expect to begin the clinical program for BX3000 in 2023 within a limited budget.
Discovery platform
In addition to our named product candidates, we are actively engaged in a number of earlier stage discovery programs where we believe our Treg platform may provide therapeutic benefits. These discovery stage initiatives are focused on indications with pathological autoantibodies, including, among others, myasthenia gravis and multiple sclerosis. For these and other indications, we plan to use our Treg platform to develop therapeutics that could be used in the treatment of these, and other diseases involving pathological autoantibodies.
Licenses and Agreements
HA FVIII TCR Agreement
On August 5, 2019, TeraImmune entered into an exclusive worldwide license agreement (the “HA FVIII TCR Agreement”) with HJF to utilize the licensed patent rights granted thereunder to research, design, develop, make, use, sell, distribute, exploit, improve and import the licensed products and processes covered thereby. The patent rights covered by the HA FVIII TCR Agreement include certain technologies relating to coagulation FVIII specific T cell receptors (“TCRs”) or BAR expressing Tregs, methods of producing and stabilizing FVIII specific TCR or BAR expressing Tregs, and their use in humans. HJF retains the right to grant non-exclusive licenses to the patent rights covered under the HA FVIII TCR Agreement for non-commercial and research purposes. In addition, HJF retains the right to request that TeraImmune relinquish its exclusive rights under the HA FVIII TCR Agreement if it has not obtained FDA or other regulatory approval to a licensed product within ten years of the effective date of the HA FVIII TCR Agreement.
Pursuant to the HA FVIII TCR Agreement, TeraImmune has agreed to pay mid-single digit percent royalties on net sales (as defined therein) in jurisdictions where a valid claim with respect to the patent rights exist, and low-single digit percent royalties on net sales where no valid claim exists or where valid claims have expired. Additionally, TeraImmune agreed to pay a high-teens percentage of its non-royalty sublicense income received prior to regulatory approval of licensed product and a low-teens percentage of its non-royalty sublicense income received after regulatory approval of a licensed product, as well a minimal annual maintenance fee, which shall be credited against any royalty fees due and payable in for the calendar year relating to such maintenance fee. Further, TeraImmune is obligated to pay an aggregate of $1.3 million in milestone fees in the event such milestones are met. As of March 31, 2023, TeraImmune has paid a license royalty fee and annual royalties of $50,000 to HJF.
The HA FVIII TCR Agreement will remain in effect until the later of (a) the full end of the term or terms of certain patent rights as defined therein on a country-by-country basis or (b) 15 years from the first sale of the licensed product in a given country, whichever is longer, provided, however, that HJF may terminate the HA FVIII TCR Agreement in the event certain milestones are not met within the timeframe required by the HA FVIII TCR Agreement.
BML Agreement
On August 26, 2019, TeraImmune entered into the non-exclusive Biological Materials License Agreement (the “BMLA”) with the National Cancer Institute (“NCI”), a part of the National Institutes of Health (“NIH”), which is part of the U.S. Government Department of Health and Human Services. Pursuant to the BMLA, TeraImmune was granted a world-wide, non-exclusive license to utilize the licensed patent rights granted thereunder to make, have made, use, sell and import autologous T cell therapy products for the treatment of Hemophilia A utilizing the pMSGV1 vector.
Pursuant to the BMLA, TeraImmune agreed to pay minimal non-refundable license, initial royalty, and annual royalty fees. Further, TeraImmune is required to pay a less than 1.0% royalty on net sales of any licensed products under the BMLA. As of March 31, 2023, TeraImmune has paid a license execution fee and annual royalties of $11,000 to NIH. The BMLA shall terminate in accordance with its terms ten years after the effective date thereof, unless extended by us and NCI.
HA ODN Agreement
On June 18, 2020, TeraImmune entered into an exclusive license agreement (the “HA ODN Agreement”) with the National Institute of Allergy and Infectious Diseases (“NIAID”), a part of NIH. Pursuant to the HA ODN Agreement, TeraImmune was granted a non-exclusive license to utilize the licensed patent rights granted thereunder to make, have made, use, have used, sell and have sold, offer to sell and import certain autologous T cell therapy products for the treatment of Hemophilia A for patients who have inhibitory anti-FVIII auto-antibodies in the United States.
Pursuant to the HA ODN Agreement, TeraImmune agreed to pay mid-single digit percent royalties on net sales (as defined therein) of any licensed products covered by the HA ODN Agreement. TeraImmune also agreed to a minimal non-refundable license, initial royalty, and annual royalty fees. Additionally, TeraImmune agreed to reimburse NIAID for certain patent expenses on a payment schedule that may amount in total reimbursements of up to $45,000, including expenses incurred in maintaining the patent. As of March 31, 2023, TeraImmune has reimbursed an aggregate expense of $22,000 for patent prosecution fees and paid a license royalty fee and annual royalties of $33,000 to NIAID. The HA ODN Agreement also requires the payment of up to $1.1 million in milestone payments upon the achievement of certain regulatory and commercialization milestones. The HA ODN Agreement will remain in effect until the expiration of the last to expire of certain licensed patent rights or U.S. orphan drug exclusivity, each as defined therein, provided, however, that NIAID may terminate the HA ODN Agreement in the event that certain milestones are not met within the timeframe required by the HA ODN Agreement. iTreg Agreement
On November 11, 2020, TeraImmune entered into an exclusive worldwide license agreement (the “iTreg Agreement”) with HJF to utilize the licensed patent rights granted thereunder to practice, research, design, develop, make, use, sell, distribute, exploit, improve and import the licensed products and processes covered thereby. The patents rights covered by the iTreg Agreement include technology related to inducable regulatory T (“iTreg”) cells, methods for producing iTreg cells and their use in humans. TeraImmune agreed to take responsibility for the maintenance and prosecution of the Patent Rights in consultation with HJF on all strategic global filing and prosecution decisions. HJF retains the right to grant non-exclusive licenses to the patent rights covered by the iTreg Agreement for non-commercial and research purposes. In addition, HJF retains the right to request that TeraImmune relinquish its exclusive rights under the iTreg Agreement if it has not obtained FDA or other regulatory approval to a licensed product within six years of the effective date of the iTreg Agreement.
Pursuant to the iTreg Agreement, TeraImmune has agreed to pay low-single digit percent royalties to HJF on net sales (as defined therein) of any licensed product covered thereby and a low-teens percentage on its non-royalty sublicense income. TeraImmune paid a non-refundable license fee of $25,000 to HJF in December 2020. The iTreg Agreement will remain in effect until the full end of the term or terms of certain patent rights as defined therein on a country-by-country basis, provided, however, that HJF may terminate the iTreg Agreement in the event that certain milestones are not met within the timeframe required by the iTreg Agreement.
Intellectual Property
We license the patents and other intellectual property covering the NMBs and the related reversal agent and related methods of use under a worldwide, exclusive, sublicensable, royalty-bearing license from Cornell University. We exclusively license issued patents in the U.S. and other major foreign markets directed to BX1000 that expire in 2027, subject to any applicable disclaimer or extension, along with a pending PCT application directed to certain methods of using BX1000 that, if issued, would expire in 2041, subject to any applicable disclaimer or extension. We exclusively license issued patents in the U.S. and other major foreign markets directed to BX2000 that expire in 2033, subject to any applicable disclaimer or extension, along with a pending PCT application directed to certain methods of using BX-2000 that, if issued, would expire in 2041, subject to any applicable disclaimer or extension. Under the license agreement, we are obligated to pay Cornell University (i) an annual license maintenance fee payment which ranges from $15,000 to $125,000 until the first commercial sale of a licensed compound; (ii) milestone payments upon the achievement of certain milestones, up to a maximum, for each NMB, of $5 million for U.S. regulatory approval and commercialization milestones and $3 million for European regulatory approval and commercialization milestones; and (iii) royalties on net sales of the NMBs and the related reversal agent at rates ranging from low to mid-single digits, depending on the applicable licensed compound and whether there is a valid patent claim in the applicable country, subject to an annual minimum royalty amount of that increases after the fourth year of sales. In addition, we will reimburse Cornell University for past and ongoing patent costs related to prosecution and maintenance of the patents related to the licensed compounds. The license agreement is terminable by us at any time upon 90 days’ written notice and by Cornell University upon our material breach, subject to a cure period, and upon our filing any claim asserting the invalidity of any of Cornell University’s licensed patent rights. The royalty term for each licensed compound expires, on a country-by-country basis, on the later of (i) the expiration date of the longest-lived licensed patent, (ii) the expiration of any granted statutory period of marketing exclusivity, or (iii) the first commercial sale of a generic equivalent of the applicable licensed compound. On the last to expire royalty term the license agreement will automatically convert to a royalty-free nonexclusive license.
We have in-licensed a patent family covering methods of producing T cell populations enriched for regulatory T cells and cell culture compositions from U.S. Department of Health and Human Services, as represented by National Institute of Allergy and Infectious Diseases of the NIH under an exclusive, sublicensable royalty-bearing license. We exclusively license 2 issued U.S. patents covering methods of producing a population of cells having stable, regulatory T cells and cell culture compositions containing isolated human regulatory T cells, antibodies and an oligonucleotide that expire in 2033 subject to any applicable disclaimer or extensions. Under the license agreement with the NIH, we are obligated to pay the NIH (i) single-digit royalties on net sales subject a minimum annual royalty, which may be credited against royalties due for sales made in a particular year; and (ii) development milestone payments. Upon expiration of the licensed patent rights, a reduced royalty rate will be applied to net sales for the duration of the U.S. Orphan Drug Exclusivity period. The NIH license agreement is terminable by us at any time upon 60 days written notice and by the NIH if we fail in the performance of any material obligations under the license agreement subject to a cure period.
We have also in-licensed 2 patent families relating to TI-168, nucleic acids constructs encoding T cell receptors, methods of producing TI-168, immunosuppressive induced regulatory T cells and methods of producing these cells from HJF under two worldwide, exclusive, sublicensable royalty-bearing licenses. We exclusively license 2 issued patents in the U.S. and a pending European patent application relating to methods of producing and stabilizing T cell populations enriched for regulatory T cells and cell culture compositions that expire in 2034 subject to any applicable disclaimer or extension. We also exclusively license a family of pending U.S. and foreign patent applications directed to immunosuppressive induced regulatory T cells and methods of producing these cells which if issued would expire in 2041 subject to any applicable disclaimer or extensions. Under the license agreement with HJF, covering the 2 issued U.S. patents and pending European patent application, we are obligated to pay HJF (i) an annual license maintenance fee creditable against royalty payments due in the same year; (ii) single-digit royalties on net sales until expiration of the licensed patent rights, after which a reduced royalty rate will be applied to net sales for 15 years from the first sale of a licensed product; and (iii) development and funding milestone payments. Under the license agreement with HJF covering the pending U.S. and foreign patent applications directed to immunosuppressive induced regulatory T cells and methods of producing these cells, we are obligated to pay HJF single-digit royalties on net sales until expiration of the patent rights on a country-by-country basis. The HJF license agreements are terminable by us at any time upon 90 days written notice and by HJF if we fail in the performance of any obligations under the license agreement which in some instances are subject to a cure period.
We own patents and patent applications directed to the analgesia indication, formulations and intranasal methods of use of dexmedetomidine in the United States and certain major foreign markets. Several patents have issued outside the United States for transmucosal methods, and the resulting patent protection will last into 2030, subject to any disclaimers or extensions. In addition, patents related to intranasal methods has issued in the United States and certain major foreign markets, and the resulting patent protection will last into 2032, subject to any disclaimers or extensions.
We intend to rely on a combination of patents and trade secrets, as well as confidentiality agreements and license agreements, to protect our product candidates. Our patent strategy is designed to facilitate commercialization of our current product candidates and future product candidates, as well as create barriers to entry for third parties. One focus of our claim strategy is on formulation claims and other related claims.
We are seeking patent protection in the United States and internationally for our product candidates. Our policy is to pursue, maintain and defend patent rights and to protect the technology, inventions and improvements that are commercially important to the development of our business. We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents granted to us in the future will be commercially useful in protecting our technology. We also intend to rely on trade secrets to protect our product candidates. Our commercial success also depends in part on our non-infringement of the patents or proprietary rights of third parties.
Our success will depend significantly on our ability to:
| • | obtain and maintain patent and other proprietary protection for our product candidates, as well as obtaining and maintaining biologic product regulatory exclusivity for our products; |
| • | develop trade secrets as needed and preserve the confidentiality of our trade secrets; and |
| • | operate our business without infringing the patents and proprietary rights of third parties. |
We have taken steps to build and will continue to build proprietary positions for our product candidates and related technology in the United States and abroad. We note that the patent laws of foreign countries differ from those in the United States, and the degree of protection afforded by foreign patents may be different from the protection offered by United States patents.
Government Regulation
Governmental authorities in the United States at the federal, state and local level, and the equivalent regulatory authorities in other countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, marketing, export and import of products such as those we are developing. Our product candidates must be approved by the FDA before they may legally be marketed in the United States. In addition, to the extent we choose to clinically evaluate or market any products in other countries or develop these products for future licensing to third parties, we are subject to a variety of regulatory requirements and to the authority of the competent regulatory authorities of those other countries.
U.S. Drug Development Process
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act (“FDCA”) and implementing regulations. The process of obtaining regulatory approvals and ensuring compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process, or after approval, may subject an applicant to administrative enforcement or judicial sanctions. This enforcement could include, without limitation, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, corrective actions, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| • | completion of preclinical laboratory tests, animal studies and formulation studies, some of which must be conducted according to good laboratory practices regulations; |
| • | submission to the FDA of an IND, which must become effective before human clinical trials may begin; |
| • | performance of adequate and well-controlled human clinical trials according to the FDA’s good clinical practices (“GCPs”) to establish the safety and efficacy of the proposed drug for its intended use; |
| • | submission to the FDA of an NDA for a new drug or Biologics License Application (“BLA”) for a new biologic product; |
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities identified in the NDA or BLA; |
| • | review and approval of proposed proprietary name; and |
| • | FDA review and approval of the NDA or BLA. |
The testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all.
Once a pharmaceutical product candidate is identified for development, it enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity, formulation and stability, as well as animal studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data and any available clinical data or literature, to the FDA as part of the IND. The sponsor must also include a protocol detailing, among other things, the objectives of the initial clinical trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated if the initial clinical trial lends itself to an efficacy evaluation. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the clinical trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Clinical holds also may be imposed by the FDA at any time before or during trials due to safety concerns regarding the product candidate or non-compliance with applicable requirements.
All clinical trials of a product candidate must be conducted under the supervision of one or more qualified investigators, in accordance with GCP regulations. These regulations include the requirement that all research subjects provide informed consent. Further, an IRB must review and approve the plan for any clinical trial before it commences at any institution. The IRB’s role is to protect the rights and welfare of human subjects involved in clinical studies by evaluating, among other things, the potential risks and benefits to subjects, processes for obtaining informed consent, monitoring of data to ensure subject safety, and provisions to protect the subjects’ privacy. The IRB approves the information regarding the clinical trial and the consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
Once an IND is in effect, each new clinical protocol, and any amendments to the protocol, must be submitted to the IND for FDA review and to the IRBs for approval. Protocols detail, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters to be used to monitor subject safety.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| • | Phase I. The product is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing may be conducted in patients. |
| • | Phase II. Phase II trials involve investigations in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted indications and to determine dosage tolerance and optimal dosage and schedule. |
| • | Phase III. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for regulatory approval and product labeling. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA, and safety reports must be submitted to the FDA and the investigators for serious and unexpected side effects. Phase I, Phase II and Phase III testing may not be completed successfully within any specified period, if at all. Results from earlier trials are not necessarily predictive of results from later trials. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the product and finalize a process for manufacturing the product in commercial quantities in accordance with current good manufacturing practice (“cGMP”) requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
U.S. Review and Approval Processes
The results of product development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the drug, proposed labeling and other relevant information, are submitted to the FDA as part of an NDA for a new drug or BLA for a new biologic agent, requesting approval to market the product.
The submission of an NDA generally is subject to the payment of a substantial user fee for a human drug application. In addition, under the Pediatric Research Equity Act of 2003, an NDA or supplement to an NDA for a new indication, dosage form, dosing regimen, route of administration, or active ingredient, must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may waive or defer pediatric studies under certain circumstances.
Section 505(b)(2) New Drug Applications. As an alternate path to FDA approval, particularly for modifications to drug products previously approved by the FDA, an applicant may submit an NDA under Section 505(b)(2) of the FDCA (“Section 505(b)(2) NDA”). Section 505(b)(2) was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments, and it permits approval of applications other than those for duplicate products and permits reliance for such approvals on literature or on the FDA’s findings of safety and effectiveness of an approved drug product. A Section 505(b)(2) NDA is an application where at least some of the information required for approval comes from clinical trials not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The FDA requires submission of information needed to support any changes relative to a previously approved drug, known as the reference product, such as published data or new studies conducted by the applicant, including bioavailability or bioequivalence studies, or clinical trials demonstrating safety and effectiveness. The FDA may then approve the Section 505(b)(2) NDA for all or some of the labeled indications for which the reference product has been approved, as well as for any new indication sought by the applicant, unless such indications or uses are protected by patent or exclusivity provisions covering the reference product. To the extent that a Section 505(b)(2) NDA relies on clinical trials conducted for a previously approved drug product or the FDA’s prior findings of safety and effectiveness for a previously approved drug product, the Section 505(b)(2) applicant must submit patent certifications in its application with respect to any patents for the reference product that are listed in the FDA’s publication, Approved Drug Products with Therapeutic Equivalence Evaluations, commonly referred to as the Orange Book. Specifically, the applicant must certify for each listed patent that, in relevant part, (1) the required patent information has not been filed; (2) the listed patent has expired; (3) the listed patent has not expired but will expire on a particular date and approval is not sought until after patent expiration; or (4) the listed patent is invalid, unenforceable or will not be infringed by the proposed new product. A certification that the new product will not infringe the previously approved product’s listed patent or that such patent is invalid or unenforceable is known as a Paragraph IV certification. If the applicant does not challenge one or more listed patents through a Paragraph IV certification, the FDA will not approve the Section 505(b)(2) NDA until all the listed patents claiming the referenced product have expired.
Further, the FDA will also not approve a Section 505(b)(2) NDA until any non-patent exclusivity, such as, for example, five-year exclusivity for obtaining approval of a new chemical entity, three-year exclusivity for an approval based on new clinical trials, or pediatric exclusivity, listed in the Orange Book for the reference product, has expired.
If the Section 505(b)(2) NDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the owner of the reference product and relevant patent holders within 20 days after the Section 505(b)(2) NDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement suit against the Section 505(b)(2) applicant. Under the FDCA, the filing of a patent infringement lawsuit within 45 days of receipt of the notification regarding a Paragraph IV certification automatically prevents the FDA from approving the Section 505(b)(2) NDA for 30 months, beginning on the date the patent holder receives notice, or until the patent expires or a court deems the patent unenforceable, invalid or not infringed, whichever is earlier. Even if a patent infringement claim is not brought within the 45-day period, a patent infringement claim may be brought under traditional patent law, but it does not invoke the 30-month stay. Moreover, in cases where a Section 505(b)(2) application containing a Paragraph IV certification is submitted after the fourth year of a previously approved drug’s five-year exclusivity period, and the patent holder brings suit within 45 days of notice of certification, the 30-month period is automatically extended to prevent approval of the Section 505(b)(2) application until the date that is seven and one-half years after approval of the previously approved reference product. The court also has the ability to shorten or lengthen either the 30-month or the seven and one-half year period if either party is found not to be reasonably cooperating in expediting the litigation. Thus, the Section 505(b)(2) applicant may invest a significant amount of time and expense in the development of its product only to be subject to significant delay and patent litigation before its product may be commercialized. Alternatively, if the NDA applicant or relevant patent holder does not file a patent infringement lawsuit within the specified 45-day period, the FDA may approve the Section 505(b)(2) application at any time, assuming the application is otherwise approvable.
Notwithstanding the approval of many products by the FDA pursuant to Section 505(b)(2), over the last few years, some pharmaceutical companies and other stakeholders have objected to the FDA’s interpretation of Section 505(b)(2). If the FDA changes its interpretation of Section 505(b)(2), or if the FDA’s interpretation is successfully challenged in court, this could delay or even prevent the FDA from approving any Section 505(b)(2) NDA that we submit.
FDA Review of New Drug Applications. The FDA reviews all NDAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. If the FDA does not find an NDA to be sufficiently complete for filing, it may request additional information rather than accepting the NDA for filing. In this event, the sponsor must resubmit the NDA with the additional information. The re-submitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether clinical data demonstrates that a product is safe and effective for its intended use and whether its manufacturing process can assure the product’s identity, strength, quality and purity. Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. The FDA may refer the NDA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions. An advisory committee is a panel of independent experts who provide advice and recommendations when requested by the FDA. The FDA is not bound by the recommendation of an advisory committee.
The approval process is lengthy and difficult, and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter (“CRL”) if the agency decides not to approve the NDA in its present form. The CRL usually describes all the specific deficiencies that the FDA identified in the NDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the CRL may include recommended actions that the applicant might take to place the application in a condition for approval. If a CRL is issued, the applicant may either resubmit the NDA, addressing all the deficiencies identified in the letter, withdraw the application or request an opportunity for a hearing.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages, or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling, and the agency also may require a risk evaluation and mitigation strategy (“REMS”) if it determines that a REMS is necessary to assure that the benefits of a drug outweigh its risks. In addition, the FDA may require Phase IV testing, which involves clinical trials designed to further assess a drug’s safety and effectiveness after NDA approval, and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.
U.S. Biologic nonclinical and clinical development
Prior to beginning the first clinical trial with a biologic product candidate in the U.S., we must submit an IND to the FDA. An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The IND includes the clinical protocols and general development plan, as well as results of animal and in vitro studies assessing the toxicology, pharmacokinetic (“PK”), pharmacology and PD characteristics of the product candidate; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions. In such a case, the IND may be placed on clinical hold until the IND sponsor and the FDA resolve the outstanding concerns or questions. The FDA also may impose clinical holds at any time before or during clinical studies due to safety concerns or non-compliance. If the FDA imposes a clinical hold, studies may not recommence without FDA authorization and then only under terms authorized by the FDA. Submission of an IND therefore does not guarantee that FDA authorization to begin a clinical trial will be granted or that, once begun, issues will not arise that adversely impact, suspend or terminate such studies.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments and additional information such as toxicology or Chemistry, Manufacturing and Controls data in support of the investigational product(s). For new indications, a separate new IND is usually required. Outside of the U.S., clinical trial applications are generally required to conduct clinical studies in each country. Furthermore, an independent IRB for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at that site, and must monitor the study until completed to ensure that the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits.
Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives. Some studies also include oversight by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board or independent data monitoring committee, which provides direction for whether or not a study may move forward at designated check points based on access to certain data from the study and may halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical studies and clinical study results to public registries.
Information about most clinical trials must be submitted within specific timeframes for publication on thewww.clinicaltrials.gov website. For purposes of BLA/market authorization application approval, human clinical trials are typically conducted in three sequential phases that may overlap or be combined.
| • | Phase 1 — The investigational product is initially introduced into healthy human subjects or patients with the target disease or condition. These studies are designed to test the safety, tolerability, absorption, metabolism, distribution and elimination of the investigational product in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on pharmacodynamics and effectiveness. |
| • | Phase 2 — The investigational product is administered to a limited patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. |
| • | Phase 3 — The investigational product is administered to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple global clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the investigational product and to provide an adequate basis for product approval. These trials may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to support chronic use of a product during marketing. |
In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product. These so-called Phase 4 studies may be made a condition to approval of the BLA or, in certain circumstances, mandated after approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication and are commonly intended to generate additional safety data regarding use of the product in a clinical setting or in some cases to support full approval for products that are approved via an accelerated pathway as described below. During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data, and clinical study investigators. Annual progress reports detailing the results of the clinical studies must be submitted to the FDA, and IND safety reports must be submitted to the FDA, other regulators, and investigators within a regulated timeframe for serious and unexpected adverse events, any findings from other studies, tests in laboratory animals or in vitro testing that suggest a significant risk for human subjects, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure or adverse events reported by anti-FcRn product candidates developed by others.
Concurrent with clinical trials, companies may complete additional animal studies and develop additional information about the biological characteristics of the product candidate and must finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final product, or for biologics, the safety, purity and potency. The FDA may require such testing to occur on a lot-by-lot basis in order to release product for clinical use. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
BLA Submission, Review and Approval
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, nonclinical studies and clinical trials are submitted to the FDA as part of a BLA requesting approval to market the product for one or more indications. The BLA must include all relevant data available from pertinent preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other information. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. The submission of a BLA requires payment of a substantial application user fee to FDA, unless a waiver or exemption applies. There can be no assurance that the FDA will accept the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all.
Once a BLA has been submitted, the FDA reviews the BLA within 60 days to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Under the goals and policies agreed to by the FDA under the Prescription Drug User Fee Act (“PDUFA”), the FDA’s goal is to review standard applications within ten months after it accepts the application for filing, or, if the application qualifies for priority review, six months after the FDA accepts the application for filing. In both standard and priority reviews, the FDA does not always meet PDUFA goal dates, and the review process can be significantly extended by FDA requests for additional information or clarification or company submissions of substantial data during the review. The FDA reviews a BLA to determine, among other things, whether a product is safe, pure and potent and the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the product’s continued safety, purity and potency. The FDA may convene an advisory committee to provide clinical insight on application review questions with emphasis on risk and benefit of the molecule and proposed indications, and provide a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. Before approving a BLA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites, preclinical studies, and/or the sponsor to assure compliance with GCP. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
After the FDA evaluates a BLA and conducts inspections of manufacturing facilities where the investigational product and/or its drug substance will be produced, the FDA may issue an approval letter or a Complete Response letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response letter will describe all of the deficiencies that the FDA has identified in the BLA, and where the FDA determines that the data supporting the application are inadequate to support approval, the FDA may issue the Complete Response letter prior to inspections, testing submitted product lots, and/or reviewing proposed labeling. In issuing the Complete Response letter, the FDA may recommend actions that the applicant might take to place the BLA in condition for approval, including requests for additional information or clarification, completion of other significant and time-consuming requirements related to clinical trials, and/or conduct of additional preclinical studies or manufacturing activities. Even if such data and information are submitted, the FDA may determine that the BLA does not satisfy the criteria for approval. FDA approval of a BLA must be obtained before a biologic may be marketed in the U.S. The FDA may delay or refuse approval of a BLA if applicable regulatory criteria are not satisfied and may require additional clinical testing or safety information.
If regulatory approval of a product is granted, such approval will be granted for particular indications and may entail limitations on the indicated uses for which such product may be marketed, which could limit the commercial value of the product. For example, the FDA may approve the BLA with a REMS, to ensure the benefits of the product outweigh its risks. A REMS is a safety strategy to manage a known or potential serious risk associated with a product, and could include medication guides, healthcare professional and/or patient communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA will evaluate if any labeling or risk management plans are necessary to ensure safe use of the product in the targeted patient population and indication. Once approved, the FDA has the authority to withdraw the product approval if compliance with pre-and post-marketing requirements is not maintained or if problems occur after the product reaches the marketplace. The FDA may impose post-marketing requirements and commitments such as additional manufacturing data or testing; additional preclinical data or evaluation; additional clinical data from Phase 3 studies (e.g. long-term extension data); and may require one or more Phase 4 post-market studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization and may limit further marketing of the product based on the results of these post-marketing studies.
Expedited Development and Review Programs and other Marketing Authorization Procedures
Any marketing application for a biologic submitted to the FDA for approval may be eligible for FDA programs intended to expedite the FDA review and approval process, such as priority review, fast track designation, breakthrough therapy and accelerated approval.
A product is eligible for priority review if it is intended to treat a serious condition and, if approved, would provide a significant improvement in safety or effectiveness. Priority review designation means the FDA’s goal under PDUFA is to take action on the marketing application within six months of the 60-day filing date (compared with ten months under standard review). To be eligible for a fast track designation, the FDA must determine, based on the request of a sponsor, that a product is intended to treat a serious or life threatening disease or condition and demonstrates the potential to address an unmet medical need by providing a therapy where none exists or a therapy that may be potentially superior to existing therapy based on efficacy or safety factors. Fast track designation is intended to facilitate development and expedite review of a product, and also provides opportunities for frequent interactions with the FDA review team. The FDA may also review complete sections of the BLA for a fast track product on a rolling basis before the entire application is submitted, if the sponsor and FDA agree on a schedule for the submission of the application sections, and the sponsor pays any required user fees upon submission of the first section of the BLA. The review clock generally does not begin until the final section of the BLA is submitted.
In addition, a sponsor can request designation of a product candidate as a “breakthrough therapy.” A breakthrough therapy is defined as a drug or biologic that is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug or biologic may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The FDA will take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a breakthrough therapy.
Additionally, products studied for their safety and effectiveness in treating serious or life-threatening diseases or conditions and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical studies establishing that the product has an effect on a validated surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint that can be measured earlier than survival or irreversible morbidity and is reasonably likely to predict an effect on survival, irreversible morbidity or another clinical benefit. As a condition of accelerated approval, the FDA requires the sponsor to perform adequate and well-controlled post-marketing confirmatory clinical studies to verify and describe the anticipated effect on irreversible morbidity or mortality or other clinical benefit. Approval may be withdrawn if the confirmatory study does not verify the anticipated clinical benefit. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which the sponsor must plan to provide all commercial materials and seek approval prior to the launch of the product.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review and approval will not be shortened. Furthermore, priority review, fast track designation, breakthrough therapy designation, and accelerated approval do not change the standards for approval and may not ultimately expedite the development or approval process.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biologic intended to treat a rare disease or condition, which is a disease or condition that affects fewer than 200,000 individuals in the U.S., or more than 200,000 individuals in the U.S. for which there is no reasonable expectation that the cost of developing and making available in the U.S. a drug or biologic for this type of disease or condition will be recovered from sales in the U.S. for that drug or biologic. Orphan designation must be requested before submitting a BLA. After the FDA grants orphan designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. The orphan drug designation does not convey any advantage in, or automatically shorten the duration of, the regulatory review or approval process. If a product that has orphan designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan exclusivity, which means that the FDA may not approve any other applications, including a full BLA, to market the same product for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity by means of greater effectiveness, greater safety or providing a major contribution to patient care or in instances of drug supply issues. Orphan exclusivity does not prevent FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the BLA application fee. A designated orphan product may not receive orphan exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. In addition, exclusive marketing rights in the U.S. may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Biosimilars and Reference Product Exclusivity
The Patient Protection and Affordable Care Act (“Affordable Care Act”) signed into law in 2010 includes a subtitle called the Biologics Price Competition and Innovation Act of 2009 (“BPCIA”), which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-approved reference biological product. To date, a number of biosimilars have been licensed under the BPCIA, but no interchangeable biologic has been approved in the U.S. The FDA has issued several guidance documents outlining its approach to the review and approval of biosimilars. Biosimilarity, which requires that the biological product be highly similar to the reference product notwithstanding minor differences in clinically inactive components and that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, can be shown through analytical studies, animal studies, and a clinical study or studies. Interchangeability requires that a product is biosimilar to the reference product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product in any given patient and, for products that are administered multiple times to an individual, the biologic and the reference biologic may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing that applicant’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of its product. The BPCIA also created certain exclusivity periods for biosimilars approved as interchangeable products.
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specific circumstances of FDA marketing approval of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. Subject to certain limitations, the patent term restoration period is generally equal to one-half of the time between the effective date of an IND and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application. However, each phase of the regulatory review period may be reduced by any time that the FDA finds the applicant did act not act with due diligence. Only one patent applicable to an approved drug is eligible for the extension, it must be the first approval of the active ingredient of the product, and the application for the extension must be submitted prior to the expiration of the patent and within sixty days of approval of the drug. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent term for patents that issue from some of our currently owned or licensed patents or patent applications to add patent life beyond their current expiration dates, depending on the expected length of the clinical trials, the eligibility of the product and other factors involved in the filing of the relevant NDA.
Market exclusivity provisions under the FDCA can also delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to NDAs for products containing chemical entities never previously approved by the FDA alone or in combination. A new chemical entity means a drug that contains no active moiety that has been approved by the FDA in any application submitted under Section 505(b) of the FDCA. An active moiety is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application (“ANDA”), or a Section 505(b) (2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. This exclusivity provision does not prevent the submission or approval of another full Section 505(b)(1) NDA, but such an NDA applicant would be required to conduct its own preclinical and adequate, well-controlled clinical trials to demonstrate safety and effectiveness. The FDCA also provides three years of marketing exclusivity for an NDA, Section 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application. Such clinical trials may, for example, support new indications, dosages, routes of administration or strengths of an existing drug, or for a new use. This exclusivity, which is sometimes referred to as clinical investigation exclusivity, prevents the FDA from approving an application under a Section 505(b)(2) NDA or an ANDA for the same conditions of use associated with the new clinical investigations before the expiration of three years from the date of approval. Such three-year exclusivity, however, would not prevent the approval of another application if the applicant submits a Section 505(b)(1) NDA and has conducted its own adequate, well-controlled clinical trials demonstrating safety and efficacy, nor would it prevent approval of an ANDA or a Section 505(b)(2) NDA product that did not incorporate the exclusivity-protected aspects of the approved drug product. Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing that applicant’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of its product. The BPCIA also created certain exclusivity periods for biosimilars approved as interchangeable products.
Pediatric exclusivity is another type of exclusivity in the United States. Pediatric exclusivity, if granted, provides an additional six months of exclusivity to any existing exclusivity (e.g., three- or five-year exclusivity) or patent protection for a drug. This six-month exclusivity, which runs from the end of other exclusivity or patent protection, may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial.
Orange Book Listing
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent whose claims cover the applicant’s product or a method of using the product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an ANDA or an application covered by Section 505(b)(2) of the FDCA. An ANDA provides for marketing of a drug product that has the same active ingredients, generally in the same strengths and dosage form, as the listed drug and has been shown through PK testing to be bioequivalent to the listed drug. Drugs approved in this way are commonly referred to as “generic equivalents” to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug. Other than the requirement for bioequivalence testing, ANDA applicants are generally not required to conduct, or submit results of, preclinical studies or clinical tests to prove the safety or effectiveness of their drug product. Section 505(b)(2) applications provide for marketing of a drug product that may have the same active ingredients as the listed drug and contains full safety and effectiveness data as an NDA, but at least some of this information comes from studies not conducted by or for the applicant. This alternate regulatory pathway enables the applicant to rely, in part, on the FDA’s findings of safety and efficacy for an existing product, or published literature, in support of its application. The FDA may then approve the new drug candidate for all or some of the labeled indications for which the referenced product has been approved, as well as for any new indication sought by the 505(b)(2) applicant.
The ANDA or Section 505(b)(2) applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. The ANDA or Section 505(b)(2) applicant may also elect to submit a statement certifying that its proposed ANDA label does not contain, or carves out, any language regarding a patented method of use rather than certify to such listed method of use patent. If the applicant does not challenge the listed patents by filing a certification that the listed patent is invalid or will not be infringed by the new product, the ANDA or Section 505(b)(2) application will not be approved until all the listed patents claiming the referenced product have expired.
A certification that the new product will not infringe the already approved product’s listed patents, or that such patents are invalid, is called a Paragraph IV certification. If the ANDA or Section 505(b)(2) applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA or Section 505(b)(2) application has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA or Section 505(b)(2) application until the earliest of 30 months, expiration of the patent, settlement of the lawsuit, and a decision in the infringement case that is favorable to the ANDA or Section 505(b)(2) applicant. This prohibition is generally referred to as the 30-month stay. Thus, approval of an ANDA or 505(b)(2) NDA could be delayed for a significant period of time depending on the patent certification the applicant makes and the reference drug sponsor’s decision to initiate patent litigation.
The ANDA or Section 505(b)(2) application also will not be approved until any applicable non-patent exclusivity listed in the Orange Book for the referenced product has expired.
Post-Approval Requirements
Any drugs for which we receive FDA approval will be subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, and complying with FDA promotion and advertising requirements.
The FDA strictly regulates marketing, labeling, advertising, and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other government agencies enforce the laws and regulations prohibiting the false or misleading promotion of drugs. The FDA also limits the promotion of product candidates prior to their approval. With limited exceptions, pre-approval promotion is prohibited under the FDA’s regulations.
Further, manufacturers of drugs must continue to comply with cGMP requirements, which are extensive and require considerable time, resources and ongoing investment to ensure compliance. In addition, changes to the manufacturing process may require prior FDA approval before being implemented, and other types of changes to the approved product, such as adding new indications and additional labeling claims, are subject to further FDA review and approval. Drug manufacturers and other entities involved in the manufacturing and distribution of approved drugs, and those supplying products, ingredients, and components of them, are required to list their products and to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP, applicable product tracking and tracing requirements, and other laws. The cGMP requirements apply to all stages of the manufacturing process, including the production, processing, sterilization, packaging, labeling, storage and shipment of the drug. Manufacturers must establish validated systems to ensure that products meet specifications and regulatory standards and test each product batch or lot prior to its release. We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our product candidates. FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution or may require substantial resources to correct.
The FDA may withdraw a product approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Further, the failure to maintain compliance with regulatory requirements may result in administrative or judicial actions, such as fines, untitled and warning letters, holds on clinical trials, product recalls or seizures, product detention or refusal to permit the import or export of products, refusal to approve pending applications or supplements, restrictions on marketing or manufacturing, consent decrees, injunctions or the imposition of civil or criminal penalties.
From time to time, legislation is drafted, introduced and passed in the U.S. Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. In addition to new legislation, the FDA regulations and policies are often revised or reinterpreted by the agency in ways that may significantly affect our business and our product candidates. It is impossible to predict whether further legislative or FDA regulation or policy changes will be enacted or implemented and what the impact of such changes, if any, may be.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials of our product candidates and commercial sales and distribution of any product for which we obtain regulatory approval outside of the United States. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. As in the United States, post-approval regulatory requirements, such as those regarding product manufacture, marketing or distribution, would apply to any product that is approved outside the United States.
For example, in the European Union, we may submit applications for marketing authorizations either under a centralized, decentralized, or mutual recognition marketing authorization procedure. The centralized procedure provides for the grant of a single marketing authorization for a medicinal product by the European Commission on the basis of a positive opinion by the European Medicines Agency. A centralized marketing authorization is valid for all European Union member states and three of the four European Free Trade Association States (Iceland, Liechtenstein and Norway). The decentralized procedure and the mutual recognition procedure apply between European Union member states. The decentralized marketing authorization procedure involves the submission of an application for marketing authorization to the competent authority of all European Union member states in which the product is to be marketed. One national competent authority, selected by the applicant, assesses the application for marketing authorization. The competent authorities of the other European Union member states are subsequently required to grant marketing authorization for their territory on the basis of this assessment, except where grounds of potential serious risk to public health require this authorization to be refused. The mutual recognition procedure provides for mutual recognition of marketing authorizations delivered by the national competent authorities of European Union member states by the competent authorities of other European Union member states. The holder of a national marketing authorization may submit an application to the competent authority of a European Union member state requesting that this authority recognize the marketing authorization delivered by the competent authority of another European Union member state for the same medicinal product.
We are also subject to the U.K. Bribery Act, and other third country anti-corruption laws and regulations pertaining to our financial relationships with foreign government officials. The U.K. Bribery Act, which applies to any company incorporated or doing business in the UK, prohibits giving, offering, or promising bribes in the public and private sectors, bribing a foreign public official or private person, and failing to have adequate procedures to prevent bribery amongst employees and other agents. Penalties under the Bribery Act include potentially unlimited fines for companies and criminal sanctions for corporate officers under certain circumstances. Liability in relation to breaches of the U.K. Bribery Act is strict. This means that it is not necessary to demonstrate elements of a corrupt state of mind. However, a defense of having in place adequate procedures designed to prevent bribery is available.
Formulary Approvals and Third-Party Payer Coverage and Reimbursement
In both the United States and foreign markets, our ability to successfully commercialize our product candidates for which we receive regulatory approval, and to attract commercialization partners for our product candidates, depends in significant part on the availability of institutional formulary approvals and on adequate financial coverage and reimbursement from third-party payers, including, in the United States. These payers include the Centers for Medicare & Medicaid Services (“CMS”), the federal program that runs the Medicare program, and monitors the Medicaid programs offered by each state, as well as national and regional commercial plans. Medicare is a federally funded program managed by CMS through local Medicare Administrative Contractors that administer coverage and reimbursement for certain healthcare items and services furnished to the elderly, disabled and other individuals with certain conditions. Medicaid is an insurance program for certain categories of patients whose income and assets fall below state defined levels that is both federally and state funded and managed by each state. The federal government sets general guidelines for Medicaid and each state creates specific regulations that govern its individual program. Each government or commercial plan has its own process and standards for determining whether it will cover and reimburse a procedure or particular product and how much it will pay for that procedure or product. Commercial plans often rely on the lead of the governmental payers in rendering coverage and reimbursement determinations. Therefore, achieving favorable Medicare coverage and reimbursement is usually an essential component of successfully launching a new product. The competitive position of some of our products will depend, in part, upon the extent of coverage and adequate reimbursement for such products and for the procedures in which such products are used. Reimbursement can be subject to challenge, reduction or denial by government and other commercial plans.
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government healthcare programs and other third-party payers are increasingly challenging the prices charged for medical products and services and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy, and have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payers are challenging the prices charged for medical products and requiring that drug companies provide them with predetermined discounts from list prices.
The Inflation Reduction Act of 2022 (the “IRA”) contains substantial drug pricing reforms, including the establishment of a drug price negotiation program within the U.S. Department of Health and Human Services that would require manufacturers to charge a negotiated “maximum fair price” for certain selected drugs or pay an excise tax for noncompliance, the establishment of rebate payment requirements on manufacturers of certain drugs payable under Medicare Parts B and D to penalize price increases that outpace inflation, and requires manufacturers to provide discounts on Part D drugs. Substantial penalties can be assessed for noncompliance with the drug pricing provisions in the IRA. The IRA could have the effect of reducing the prices we can charge and reimbursement we receive for our products, if approved, thereby reducing our profitability, and could have a material adverse effect on our financial condition, results of operations, and growth prospects. The effects of the IRA on our business and the pharmaceutical industry in general is not yet known.
Payers also are increasingly changing the metrics for reimbursement rates, such as basing payment on average sales price, average manufacturer price, and wholesale acquisition cost. The existing data for reimbursement based on these metrics is relatively limited, although certain states have begun to survey acquisition cost data for the purpose of setting Medicaid reimbursement rates. CMS surveys and publishes retail community pharmacy acquisition cost information in the form of National Average Drug Acquisition Cost files to provide state Medicaid agencies with a basis of comparison for their own reimbursement and pricing methodologies and rates. It may be difficult to project the impact of these evolving reimbursement mechanics on the willingness of payers to cover any products.
If we successfully commercialize any of our products, we may participate in the Medicaid Drug Rebate Program. Participation is required for federal funds to be available for our products under Medicaid and Medicare Part B. Under the Medicaid Drug Rebate Program, we would be required to pay a quarterly rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for our drugs under Medicaid and Part B of the Medicare program.
Federal law requires that any company that participates in the Medicaid Drug Rebate Program also participate in the Public Health Service’s 340B drug pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B drug pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. These 340B covered entities include a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share of low-income patients.
Additionally, in order to be eligible to have its products paid for with federal funds under the Medicaid and Medicare Part B programs and purchased by certain federal agencies and grantees, a manufacturer also must participate in the Department of Veterans Affairs, Federal Supply Schedule (“FSS”), pricing program, established by Section 603 of the Veterans Health Care Act of 1992 (“VHCA”). Under this program, the manufacturer is obligated to make its innovator and single source products available for procurement on an FSS contract and charge a price to four federal agencies, Department of Veterans Affairs, Department of Defense (“DoD”), Public Health Service, and Coast Guard, that is no higher than the statutory Federal Ceiling Price. Moreover, pursuant to regulations issued by the DoD’s TRICARE Management Activity, now the Defense Health Agency, to implement Section 703 of the National Defense Authorization Act for Fiscal Year 2008, manufacturers are required to provide rebates on utilization of their innovator and single source products that are dispensed to TRICARE beneficiaries by TRICARE network retail pharmacies. The formula for determining the rebate is established in the regulations and is based on the difference between the annual non-federal average manufacturer price and the Federal Ceiling Price (these price points are required to be calculated by us under the VHCA). The requirements under the 340B, FSS, and TRICARE programs could reduce the revenue we may generate from any products that are commercialized in the future.
There may be significant delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that a drug will be paid for in all cases or at a rate that covers costs, including research, development, manufacturing, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover costs and may only be temporary. Reimbursement rates vary according to the use of the drug and the clinical setting in which it is used. Product reimbursement may also be incorporated into existing bundled payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or commercial payers and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Limited coverage may impact the demand for, or the price of, any product candidate for which marketing approval is obtained. Third-party payers also may seek additional clinical evidence, including expensive pharmacoeconomic studies, beyond the data required to obtain marketing approval, demonstrating clinical benefits and value in specific patient populations, before covering our products for those patients. If reimbursement is available only for limited indications, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval. Our inability to promptly obtain coverage and profitable reimbursement rates from both government-funded and commercial payers for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
United States Healthcare Reform
The United States and many foreign jurisdictions have enacted or proposed legislative and regulatory changes affecting the healthcare system, including implementing cost-containment programs to limit the growth of government-paid healthcare costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs.
In the United States, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, the “Affordable Care Act”), was intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry, and impose additional health policy reforms.
Among the provisions of the Affordable Care Act that have been implemented since enactment and are of importance to the commercialization of our product and product candidates, if approved, are the following:
| • | an annual, nondeductible fee on any entity that manufactures, or imports specified branded prescription drugs or biologic agents; |
| • | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program; |
| • | expansion of healthcare fraud and abuse laws, including the U.S. civil False Claims Act and the Anti-Kickback Statute, new government investigative powers, and enhanced penalties for noncompliance; |
| • | a Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for a manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| • | extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| • | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted, or injected; |
| • | expansion of eligibility criteria for Medicaid programs; |
| • | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; |
| • | requirements to report certain financial arrangements with physicians and teaching hospitals; |
| • | a requirement to annually report certain information regarding drug samples that manufacturers and distributors provide to physicians; and |
| • | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
There have been significant ongoing judicial, administrative, executive and legislative efforts to modify or eliminate the Affordable Care Act. For example, the Tax Act enacted on December 22, 2017, repealed the shared responsibility payment for individuals who fail to maintain minimum essential coverage under section 5000A of the Internal Revenue Code, commonly referred to as the individual mandate. Other legislative changes have been proposed and adopted since passage of the Affordable Care Act. The Budget Control Act of 2011, among other things, created the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee did not achieve its targeted deficit reduction of an amount greater than $1.2 trillion for the fiscal years 2012 through 2021, triggering the legislation’s automatic reductions to several government programs. These reductions included aggregate reductions to Medicare payments to healthcare providers of up to 2.0% per fiscal year, which went into effect in April 2013. Subsequent litigation extended the 2% reduction, on average, to 2030 unless additional Congressional action is taken. The Coronavirus Aid, Relief and Economic Security Act, which was designed to provide financial support and resources to individuals and businesses affected by the COVID-19 pandemic, suspended the 2% Medicare sequester from May 1, 2020 to March 31, 2022. From April through June 2022, a 1% reduction was in effect. As of July 2, 2022, the 2% cut resumed. The sequester will remain in place through 2030. On January 2, 2013, the American Taxpayer Relief Act was signed into law, which, among other things, reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
The Affordable Care Act has also been subject to challenges in the courts. On December 14, 2018, a Texas U.S. District Court Judge ruled that the Affordable Care Act is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. On December 18, 2019, the Fifth Circuit U.S. Court of Appeals held that the individual mandate is unconstitutional and remanded the case to the Texas District Court to reconsider its earlier invalidation of the entire Affordable Care Act. An appeal was taken to the U.S. Supreme Court. On June 17, 2021, the Supreme Court ruled that the plaintiffs lacked standing to challenge the law as they had not alleged personal injury traceable to the allegedly unlawful conduct. As a result, the Supreme Court did not rule on the constitutionality of the Affordable Care Act or any of its provisions.
Further changes to and under the Affordable Care Act remain possible but it is unknown what form any such changes or any law proposed to replace or revise the Affordable Care Act would take, and how or whether it may affect our business in the future. We expect that changes to the Affordable Care Act, the Medicare and Medicaid programs, changes allowing the federal government to directly negotiate drug prices and changes stemming from other healthcare reform measures, especially with regard to healthcare access, financing or other legislation in individual states, could have a material adverse effect on the healthcare industry. We also expect that the Affordable Care Act, as well as other healthcare reform measures that have and may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for our product and product candidates, if approved. Any reduction in reimbursement from Medicare, Medicaid, or other government programs may result in a similar reduction in payments from private payers.
Other Healthcare Laws and Compliance Requirements
If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our activities will be subject to various federal and state fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute, the federal civil False Claims Act, and laws and regulations pertaining to limitations on and reporting of healthcare provider payments (physician sunshine laws). These laws and regulations are interpreted and enforced by various federal, state and local authorities including CMS, the Office of Inspector General for the U.S. Department of Health and Human Services, the U.S. Department of Justice, individual U.S. Attorney offices within the Department of Justice, and state and local governments. These laws include:
| • | the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, offering, receiving or paying any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, lease, order, or arranging for or recommending the purchase, lease or order of, any good or service, for which payment may be made, in whole or in part, under federal healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
| • | the U.S. civil False Claims Act (which can be enforced through “qui tam,” or whistleblower actions, by private citizens on behalf of the federal government), prohibits any person from, among other things, knowingly presenting, or causing to be presented false or fraudulent claims for payment of government funds or knowingly making, using or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly and improperly avoiding, decreasing or concealing an obligation to pay money to the U.S. federal government; |
| • | U.S. federal Health Insurance Portability and Accountability Act of 1996, which imposes criminal liability and amends provisions on the reporting, investigating, enforcement, and penalizing of civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement, in connection with the delivery of, or payment for healthcare benefits, items or services by a healthcare benefit program, which includes both government and privately funded benefits programs; similar to the U.S. federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
| • | state laws and regulations, including state anti-kickback and false claims laws, that may apply to our business practices, including but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by any third-party payer, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the U.S. federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; and state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information, which requires tracking gifts and other remuneration and items of value provided to healthcare professionals and entities; and |
| • | the Physician Payments Sunshine Act, implemented as the Open Payments program, and its implementing regulations, requires certain manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid, or the Children’s Health Insurance Program to report annually to CMS information related to certain payments made in the preceding calendar year |
and other transfers of value to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; beginning in 2022, applicable manufacturers were required to report such information regarding payments and transfers of value provided, as well as ownership and investment interests held, during the previous year to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, and certified nurse-midwives.
Violations of any of these laws or any other governmental regulations that may apply to us, may subject us to significant civil, criminal and administrative sanctions including penalties, damages, fines, imprisonment, and exclusion from government funded healthcare programs, such as Medicare and Medicaid, and/or adverse publicity.
Moreover, government entities and private litigants have asserted claims under state consumer protection statutes against pharmaceutical and medical device companies for alleged false or misleading statements in connection with the marketing, promotion and/or sale of pharmaceutical and medical device products, including state investigations and litigation by certain government entities regarding the marketing of opioid products.
Foreign Corrupt Practices Act
The Foreign Corrupt Practices Act (“FCPA”) generally prohibits offering, promising, giving, or authorizing others to give anything of value, either directly or indirectly, to a non-U.S. government official in order to influence official action, or otherwise obtain or retain business. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect the transactions of the corporation and to devise and maintain an adequate system of internal accounting controls. Our industry is heavily regulated and therefore involves significant interaction with public officials, including officials of non-U.S. governments. Additionally, in many other countries, the health care providers who prescribe pharmaceuticals are employed by their government, and the purchasers of pharmaceuticals are government entities; therefore, our dealings with these prescribers and purchasers are subject to regulation under the FCPA. Recently, the SEC and Department of Justice have increased their FCPA enforcement activities with respect to pharmaceutical companies. Violations could result in fines, criminal sanctions against us, our officers, or our employees, the closing down of our facilities, requirements to obtain export licenses, cessation of business activities in sanctioned countries, implementation of compliance programs, and prohibitions on the conduct of our business.
Corporate Information
We were incorporated in Pennsylvania in 2019 and our office headquarters is located at 490 Lapp Road, Malvern, Pennsylvania 19355.