Amryt is a global commercial-stage biopharmaceutical company focused on acquiring, developing and commercializing innovative treatments to help improve the lives of patients with rare and orphan diseases. Amryt comprises a strong and growing portfolio of commercial and development assets.
As used herein, references to “we,” “us,” “Amryt” or the “Group” in this annual report on Form 20-F shall mean Amryt Pharma plc and its global subsidiaries, collectively. References to the “Company” in this annual report shall mean Amryt Pharma plc.
This annual report includes the following historical financial information, presented in US Dollars:
All references in this annual report to “€” mean Euros, all references to “£” mean pounds sterling and all references to “$” mean U.S. dollars. Except as otherwise stated herein, all monetary amounts in this annual report have been presented in US Dollars. For presentation purposes all financial information, including comparative figures from prior periods, have been stated in round thousands, unless otherwise indicated.
This annual report contains forward-looking statements concerning our business, operations and financial performance and condition, as well as our plans, objectives and expectations for our business operations and financial performance and condition. Any statements contained herein that are not statements of historical facts may be deemed to be forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as “aim,” “anticipate,” “assume,” “believe,” “contemplate,” “continue,” “could,” “due,” “estimate,” “expect,” “goal,” “intend,” “may,” “might,” “objective,” “plan,” “predict,” “potential,” “positioned,” “seek,” “should,” “target,” “will,” “would,” and other similar expressions that are predictions of or indicate future events and future trends, or the negative of these terms or other comparable terminology, although not all forward-looking statements contain these identifying words.
By their nature, forward-looking statements involve risk and uncertainty because they relate to future events and circumstances. Forward-looking statements are not guarantees of future performance and have not been reviewed by our auditors. Actual results and developments could differ materially from those expressed or implied by the forward-looking statements due to a variety of factors, including, but not limited to, those identified under the section titled “Risk Factors” in this annual report.
The preceding list is not intended to be an exhaustive list of all of our risks and uncertainties. As a result of these factors, we cannot assure you that the forward-looking statements in this annual report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this annual report, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all relevant information. These statements are inherently uncertain and investors are cautioned not to unduly rely upon these statements.
The forward-looking statements and opinions contained in this annual report are based upon information available to us as of the date of this annual report and, while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. The forward-looking statements contained in this annual report speak only as of the date of this annual report, and we do not undertake any obligation to update them in light of new information or future developments or to release publicly any revisions to these statements in order to reflect later events or circumstances or to reflect the occurrence of unanticipated events.
You should read this annual report and the documents referenced herein and have filed as exhibits to the registration statement, of which this annual report forms a part, completely and with the understanding that our actual future results may be materially different from expectations. We qualify all forward-looking statements in this annual report by these cautionary statements.
This annual report contains estimates, projections and other information concerning our industry, our business and the markets for our products and product candidates. Information that is based on estimates, forecasts, projections, market research or similar methodologies is inherently subject to uncertainties, and actual events or circumstances may differ materially from events and circumstances that are assumed in this information. Unless otherwise expressly stated, we obtained this industry, business, market and other data from our own internal estimates and research as well as from reports, research surveys, studies and similar data prepared by market research firms and other third parties, industry, medical and general publications, government data and similar sources. Management estimates are derived from publicly available information, our knowledge of our industry and assumptions based on such information and knowledge, which we believe to be reasonable.
In addition, assumptions and estimates of our and our industry’s future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in “Risk Factors.” These and other factors could cause our future performance to differ materially from our assumptions and estimates. See “Special Note Regarding Forward-Looking Statements.”
The Amryt logo, Myalept®, Myalepta®, Juxtapid®, Lojuxta®, Episalvan®, Filsuvez®, Oleogel-S-10, Mycapssa®, TPE® and other trademarks or service marks of Amryt appearing in this annual report are the property of Amryt. This annual report includes trademarks, tradenames and service marks, certain of which belong to us and others that are the property of other organizations. Solely for convenience, trademarks, tradenames and service marks referred to in this annual report appear without the ®, TM and SM symbols, but the absence of those symbols is not intended to indicate, in any way, that we will not assert our rights or that the applicable owner will not assert its rights to these trademarks, tradenames and service marks to the fullest extent under applicable law. We do not intend our use or display of other parties’ trademarks, trade names or service marks to imply, and such use or display should not be construed to imply a relationship with, or endorsement or sponsorship of us by, these other parties.
Not applicable.
Not applicable.
The following selected historical consolidated financial data of Amryt Pharma as at December 31, 2021 and 2020 and for each of the years ended December 31, 2021, 2020 and 2019 have been derived from, and should be read in conjunction with, the audited consolidated financial statements and notes thereto set forth in Item 18 of this annual report. The selected historical consolidated financial data as at December 31, 2019 is derived from the audited consolidated financial statements not appearing in this annual report. This data should be read in conjunction with the financial statements, related notes and other financial information included elsewhere herein.
We have not included selected historical consolidated financial data for the years ended December 31, 2018, and 2017 in the table below as we qualify as an emerging growth company (an “Emerging Growth Company”) as defined in Section 2(a)(19) of the Securities Act and we make use of an accommodation for reduced reporting.
A. Selected Consolidated Financial Data of Amryt
B. Capitalization and Indebtedness
Not applicable.
C. Reasons for the Offer and Use of Proceeds
Not applicable.
D. Risk Factors
Our business is subject to a number of risks. You should carefully consider the risks and uncertainties described below and the other information in this annual report, including our consolidated financial statements and related notes. Our business, financial condition, results of operations or prospects could be materially and adversely affected if any of these risks occur, and as a result, the market price of our ADSs could decline.
We have incurred significant operating losses since our inception and we may not achieve or maintain profitability in the future.
To date, we have financed our operations primarily through a combination of revenues from sales of our commercialized products and the sale of our equity securities and convertible bonds. We have incurred operating losses since our inception, including net losses of $50.5 million, $46.5 million and $38.6 million for the years ended December 31, 2019, 2020 and 2021, respectively. Aegerion incurred losses since its inception, including net losses of $96.5 million and $30.9 million for the year ended December 31, 2018, and the six-month period ended June 30, 2019. Chiasma incurred losses since its inception, including net losses of $74,7 million and $50.4 million for the year ended December 31, 2020, and the six-month period ended June 30, 2021. We have devoted most of our financial resources to the acquisition of attractive commercial and near-commercial rare disease assets and research and development. We anticipate that we will continue to incur significant costs associated with the continued commercialization of lomitapide, Mycapssa® and metreleptin, and in connection with ongoing clinical development efforts and post-marketing commitments for these products as well as the continued development of our product candidates. The amount of our future net losses will depend, in part, on the rate of our future expenditures, our ability to continue generating adequate revenues from sales of lomitapide, Mycapssa® and metreleptin and from sales of Oleogel-S10 if approved, and our ability to obtain funding through equity or debt offerings, grant funding, collaborations, strategic partnerships and/or licensing arrangements. If we do become profitable, we may not be able to sustain or increase our profitability on a quarterly or annual basis.
Our future performance depends, in part, on our ability to successfully implement our strategy.
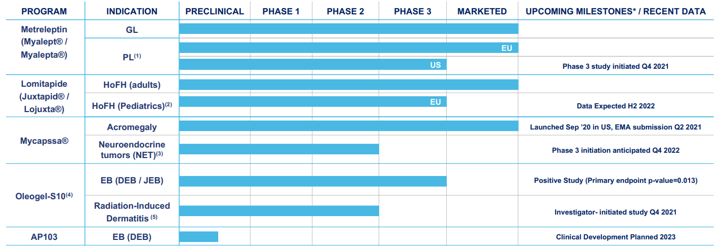
Our future success will depend on our ability to implement our strategy to develop and expand our existing portfolio of drugs to treat patients with rare diseases and to create a rare disease company with a diversified offering of multiple development stage and commercial assets that can provide us with scale to support future growth. The success of our strategy depends, in part, on our ability to drive growth in existing territories and access and drive growth in new territories and indications with existing commercial assets, build a franchise in EB through our lead product candidate, Oleogel-S10 (Filsuvez), and our product candidate AP103, based on our novel polymer-based topical gene therapy delivery platform, expand the indication of Mycapssa® (octreotide capsules) beyond acromegaly into carcinoid syndrome associated with neuroendocrine tumors and expand and diversify our product portfolio through acquisition and in-licensing opportunities. This strategy will require us to, among other things, identify suitable targets, conduct and complete clinical trials, obtain the necessary consents and authorizations for our products and any product candidates, conduct diligence, carry out any acquisition or in-licensing transaction and obtain suitable financing.
Implementing our strategy requires substantial time and resources from our management team. Our Board and management may not be able to successfully implement our strategy or other strategies to be developed by management, and implementing these strategies may not sustain or improve, and could even harm, our business, financial condition, results of operations and prospects. We may be unable to realize the anticipated benefits and synergies of our business strategies, which are based on assumptions about future demand for our current products and product candidates, as well as on our continuing ability to produce our products profitably. Consequently, any predictions made about our future success or viability may not be as accurate as they could be if we had a longer operating history or additional products on the market.
We are dependent primarily on three products, lomitapide, Mycapssa® and metreleptin, to generate revenue and these products may not be successful and may not generate sales at anticipated levels.
Our ability to meet expectations with respect to sales of lomitapide, Mycapssa® and metreleptin, and to generate revenues from such sales, and attain and maintain positive cash flow from operations, in the time periods anticipated, or at all, will depend on a number of factors, including, among others:
If we are unable to continue to generate revenue from our current commercial products, our business, financial condition, results of operations and prospects will be adversely affected.
We may not be successful in our efforts to build a pipeline of product candidates and develop additional marketable products.
We operate in the biopharmaceutical sector and have product candidates in various stages of clinical and preclinical development. In addition, we may continue to explore other opportunities within the sector in order to expand our present development pipeline. Industry experience indicates that there may be a very high incidence of delay or failure to produce valuable scientific results in relation to our present development pipeline. We will also face the risk that in developing new products we may spend substantial sums of money and the new products developed may not effectively meet the perceived need or may not be successfully commercialized. Our ability to develop new products relies on, among other things, the recruitment of sufficiently qualified research and development partners with expertise in the biopharmaceutical sector. We may not be able to develop relationships or recruit research partners of a sufficient caliber to satisfy the rate of growth and develop our future pipeline.
We may need to raise additional funding, which may not be available on acceptable terms, or at all, and failure to obtain this capital when needed may force us to delay, limit or terminate our product development efforts or other operations or to delay payment on future obligations.
On September 24, 2019, we entered into a five-year term loan facility (“Secured Credit Facility”) and issued 5% senior unsecured convertible notes (“Convertible Notes”). As of December 31, 2021, we had borrowings of $93.4 million under our Secured Credit Facility and $125.0 million under our Convertible Notes, for total debt of $218.4 million. The Secured Credit Facility includes an option to prepay the loan in whole or in part at any time subject to payment of an exit fee, which ranged from 0-5% of the principal amount then outstanding on the Secured Credit Facility. On February 18, 2022, the Secured Credit Facility was repaid in full and the Group secured a $125 million senior credit facility (“Senior Credit Facility”) from funds managed by the Credit Group of Ares Management Corporation (“Ares”) of which US$105 million was drawn down to facilitate the prepayment of the existing Secured Credit Facility. In repaying the Secured Credit Facility, Amryt incurred an exit fee of 5.00% of the outstanding principal amount as at the prepayment date. Furthermore, we may be required to satisfy payment obligations of up to $85 million to holders of contingent value rights (“CVRs”) issued to shareholders and option holders in connection with specified milestones, which we may elect to pay in ordinary shares or notes. If we elect to issue notes, we will settle such notes in cash 120 days after their issue. The specified milestones are related to the success of Oleogel-S10 and $50 million of the payment obligations are dependent on FDA and EMA approval. $35 million is due in respect of FDA approval and $15 million is due in respect of EMA approval if approval is obtained before December 31, 2021, with a sliding scale on a linear basis to zero if before July 1, 2022.
We have significant payment obligations in connection with our Senior Credit Facility, the Convertible Notes, the CVRs and royalty obligations under certain of our license agreements.
We have significant milestone and royalty payments payable under our material contracts if/when certain milestones are met, the terms of which are outlined in the “Material Contracts” section.
In addition, while we are currently commercializing our approved products, we are also advancing our product candidates through preclinical and clinical development. Developing product candidates is expensive, lengthy and risky, and we expect our research and development expenses to increase in connection with our ongoing activities, particularly as we advance our product candidates toward commercialization.
As of December 31, 2021, our unrestricted cash and cash equivalents were $113.0 million. We expect that our existing cash and cash equivalents will be sufficient to fund our current operations and our payment obligations to third parties for at least the next 12 months. However, these resources might be insufficient to conduct research and development programs, the cost of product in-taking and possible acquisitions, fully commercialize products and operate our business to the full extent currently planned, and our operating plan may change as a result of many factors currently unknown to us. As a result, we may need to seek additional funds sooner than planned, through public or private equity or debt financings, government or other third-party funding, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements or a combination of these approaches. Even if we believe we have sufficient funds for our current or future operating plans as well as to satisfy our payment obligations to third parties, we may seek additional capital if market conditions are favorable or if we have specific strategic considerations. Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates.
If we cannot obtain adequate funds, we may be required to significantly curtail our commercial plans or one or more of our research and development programs or obtain funds through additional arrangements with corporate collaborators or others that may require us to relinquish rights to some of our technologies or product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of our shareholders and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our ADSs or ordinary shares to decline. The sale of additional equity or convertible securities may be dilutive to our shareholders. We may also enter into additional credit facilities from time to time, which may be secured, to fund certain of our operations. If we raise additional capital through debt financing, we would be subject to payment obligations and may be subject to security interests in our assets and covenants restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional capital through marketing and distribution arrangements, sales of assets or other collaborations, or licensing arrangements with third parties, we may have to relinquish certain valuable rights to our product candidates, technologies, future revenue streams or research programs. We also could be required to seek collaborators for one or more of our current or future product candidates at an earlier stage than otherwise would be desirable or relinquish our rights to product candidates or intellectual property that we otherwise would seek to develop or commercialize ourselves. If we are unable to raise additional capital in sufficient amounts, at the right time, on favorable terms, or at all, we may have to significantly delay, scale back or discontinue the development or commercialization of one or more of our products or product candidates, or one or more of our other research and development initiatives. Any of the above events could significantly harm our business, prospects, financial condition and results of operations, cause the price of our ADSs and ordinary shares to decline, and negatively impact our ability to fund operations.
The terms of our debt and any requirements to incur further indebtedness or refinance our indebtedness in the future could have a material adverse effect on our business and results of operations.
Our level of indebtedness, together with any additional indebtedness we may incur in the future, could adversely affect our business, financial condition, results of operations and prospects. For example, our anticipated level of indebtedness or any additional financing may:
Any of the foregoing, alone or in combination, could have a material adverse effect on our business, financial condition, results of operations and prospects. A breach of, or the inability to comply with, the covenants in the Senior Credit Facility and the Convertible Notes could result in an event of default, in which case the lenders will have the right to declare all borrowings to be immediately due and payable, which would have a material adverse effect on our business, financial condition, results of operations and prospects and could lead to foreclosure on our assets.
In the future, we may need to refinance our indebtedness. However, additional financing may not be available on favorable commercial terms to us, or at all. If, at such time, market conditions are materially different or our credit profile has deteriorated, the cost of refinancing such debt may be significantly higher than our indebtedness existing at that time. Furthermore, we may not be able to procure refinancing at all. Any failure to meet any future debt service obligations through use of cash flow, refinancing or otherwise, could have a material adverse effect on our business, financial condition, results of operations and prospects.
Restrictive covenants in certain of the agreements and instruments governing our indebtedness may adversely affect our financial and operational flexibility.
The terms of our indebtedness as of the date of this annual report include covenants that, among other things, restrict our ability to: incur liens; dispose of our assets (both material assets and exclusive licensing transactions in material territories); consolidate and merge with other entities; make loans and investments; incur indebtedness; engage in transactions with affiliates; make specified payments; and engage in other customary business activities.
If we breach a restrictive covenant under the Senior Credit Facility or the Convertible Notes, or an event of default occurs with respect to such indebtedness, all amounts owing in respect of such indebtedness may be able to be declared due and payable immediately, which could have a material adverse effect on our business, prospects, liquidity, financial condition and results of operations. In addition, both our Senior Credit Facility and the Convertible Notes include cross-default provisions, which generally provide that a default under one facility also results in a default under the other. See “Item 7. Major Shareholders and Related Party Transactions—B. Related Party Transactions—Agreements with Principal Shareholders— Senior Credit Facility” and “Item 7. Major Shareholders and Related Party Transactions—B. Related Party Transactions—Agreements with Principal Shareholders—Convertible Notes.”
If we enter into future debt agreements, they may include similar or more restrictive provisions. These restrictions may also make more difficult or discourage a takeover of our company, whether favored or opposed by management or the Board. Our ability to comply with these covenants may be affected by events beyond our control, and any material deviations from our forecasts could require us to seek waivers or amendments of covenants or alternative sources of financing, or to reduce expenditures. We cannot guarantee that such waivers, amendments or alternative financing could be obtained or, if obtained, would be on terms acceptable to us. A breach of any of the covenants or restrictions in our debt agreements or instruments could result in an event of default. Such a default could allow our debt holders to accelerate repayment of the related debt, as well as any other debt to which a cross-acceleration or cross-default provision applies, or to declare all borrowings outstanding under these agreements to be due and payable. If our debt is accelerated, our assets may not be sufficient to repay such debt.
Holders of the Convertible Notes have the right to require the repurchase of their notes for cash upon the occurrence of a fundamental change, such as a change of control of our company, at a repurchase price equal to 100% of the respective principal amount, plus accrued and unpaid interest, if any. If a fundamental change occurs and the Convertible Notes are converted during the related fundamental change conversion period, then the conversion rate may be increased to a corresponding number of shares and share price set forth in the Convertible Notes. Subject to certain exceptions as provided in the indenture governing the Convertible Notes, a fundamental change includes (a) the acquisition of 50% or more of the voting interests in our company, (b) an event in which we merge or consolidate with another entity, (c) an event in which we convey, sell, transfer or lease all or substantially all of our assets to another entity, (d) our liquidation or (e) delisting of our ordinary shares. Among the exceptions provided in the indenture are for transactions described in (a), (b) or (c) in which (i) our stockholders immediately prior to the transaction have the right to exercise, directly or indirectly, 50% or more of the total voting power of the capital stock of the continuing or surviving entity or transferee or parent thereof following the transaction or (ii) 90% of the consideration paid for our ordinary shares in a transaction consists of stock that is or will be quoted on AIM, the New York Stock Exchange or the Nasdaq. Further, unless we elect to deliver our ordinary shares to settle any conversions of Convertible Notes, we would be required to settle a portion or all of the conversion obligation through the payment of cash. We may not have enough available cash or be able to obtain financing at the time it is required to make repurchases of Convertible Notes surrendered therefor or Convertible Notes being converted. The failure to repurchase Convertible Notes at a time when the repurchase is required by the indenture or to pay any cash payable on future conversions of the Convertible Notes as required by the indenture would constitute a default under the indenture. A default under the indenture or a fundamental change itself could also lead to a default under agreements governing our current and future indebtedness. If the repayment of such indebtedness were to be accelerated after any applicable notice or grace periods, we are unlikely to have sufficient funds to repay such indebtedness and repurchase the Convertible Notes or make cash payments upon conversions thereof or other required payments on its other indebtedness, which would materially and adversely affect our business, financial condition and results of operations on a consolidated basis.
We face potential product liability exposure, and if claims are brought against us, we may incur substantial liability.
The use of any product in clinical trials or early access programs, and the sale and use of any product for which we have obtained marketing approval exposes us to the risk of product liability claims. Product liability claims might be brought against us by consumers, healthcare providers or others selling or otherwise coming into contact with our products. If we cannot successfully defend against product liability claims, we could incur substantial liabilities. In addition, regardless of merit or eventual outcome, product liability claims may result in:
Although we have obtained product liability insurance coverage for both our clinical trials and our commercial exposure, this insurance coverage may not be sufficient to reimburse us for expenses or losses that we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against potential liability. If a claim is brought against us, we might be required to pay legal and other expenses to defend the claim, as well as pay uncovered damages awards resulting from a claim brought successfully against us and these damages could be significant and have a material adverse effect on our financial condition. On occasion, large judgments have been awarded in class action lawsuits relating to drugs that had unanticipated side effects or warnings found to be inadequate. The cost of any product liability litigation or other proceedings, even if resolved in our favor, could be substantial. A product liability claim or series of claims brought against us could harm our reputation and cause the price of our securities to decline and, if the claim is successful and judgments exceed our insurance coverage, it could have a material adverse impact on our business, financial condition, results of operations and prospects.
Adverse events involving any of our products and product candidates may lead the U.S. Food & Drug Administration (“FDA”), the European Medicines Agency (“EMA”) or other regulatory authorities to delay or deny clearance for our products or result in product recalls that could harm our reputation, business and financial results.
The FDA and the EMA, as well as similar governmental authorities in other jurisdictions, have the authority to require the recall of certain commercialized products in the event of adverse side effects, material deficiencies or defects in design or manufacture. Manufacturers may, under their own initiative, recall a product if any material deficiency in a product is found. A government-mandated recall or voluntary recall by us or one of our distributors could occur as a result of adverse side effects, impurities or other product contamination, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our products or product candidates would divert managerial and financial resources and have an adverse effect on our financial condition and results of operations. A recall announcement could harm our reputation with customers and negatively affect our sales, if any.
Our future success depends on our ability to hire and retain key executives and to attract, retain and motivate qualified personnel.
Our future success depends on our ability to attract and retain key management personnel, scientific and technical personnel, particularly in the biopharmaceutical industry. Our ability to continue our operations and implement our strategy depends upon retaining, recruiting and motivating employees, especially with respect to our management team and research personnel. Experienced employees in the biopharmaceutical and biotechnology industries are in high demand and competition for their talents can be intense, especially in Germany, Ireland, and Boston, Massachusetts, where we maintain our principal operations. We have entered into employment agreements with executive officers and other key employees, but any employee may terminate his or her employment at any time or may be unable to continue in his or her role. The loss of any executive or key employee, or an inability to recruit desirable candidates or find adequate third parties to perform such services on reasonable terms and on a timely basis, could have a material adverse effect on our business, financial condition, results of operations and prospects. If we are not able to attract, retain and motivate necessary personnel to accomplish our business objectives, we may experience constraints that could significantly impede our ability to achieve our development and commercial objectives, our ability to raise additional capital and our ability to implement our business strategy.
For U.S. federal income tax purposes, Amryt is treated as a surrogate foreign corporation, and there is a risk that Amryt may be treated as a U.S. corporation under certain circumstances, including as a result a change in law.
Section 7874 of the U.S. Internal Revenue Code of 1986, as amended (the “Code”) and the Treasury regulations promulgated thereunder contain two alternative sets of rules under which a U.S. target corporation may be subjected to certain additional U.S. federal income taxes or a non-U.S. acquiring corporation (such as Amryt) may be treated as a U.S. corporation for U.S. federal income tax purposes as a result of a transaction. Which set of rules applies depends on what percentage of the non-U.S. acquiring corporation’s stock the historic stockholders of the U.S. target corporation own or are treated as owning, under certain counting conventions, by reason of holding shares of the U.S. target corporation following the transaction (the “Section 7874 Percentage”). One set of rules imposes a tax on certain gain and income of the U.S. target corporation, and potentially certain other taxes, if (in addition to other requirements) the Section 7874 Percentage is at least 60 percent (by vote or value). The other set of rules under Section 7874 of the Code treats the non-U.S. acquiring corporation as a U.S. corporation for U.S. federal income tax purposes if (in addition to other requirements) the Section 7874 Percentage is at least 80 percent (by vote or value). If the Section 7874 Percentage is at least 60 percent (by vote or value), the non-U.S. acquiring corporation is considered a “surrogate foreign corporation,” and the U.S. target corporation is considered an “expatriated entity” with respect to the non-U.S. acquiring corporation.
Amryt believes that, as a result of Amryt’s acquisition of Aegerion in 2019 (the “Prior Acquisition”), Amryt is treated as a surrogate foreign corporation (the 60 percent test), but not as a U.S. corporation (the 80 percent test). As a result of Amryt’s status as a surrogate foreign corporation, dividends paid in respect of the Amryt ADSs or Ordinary Shares are not expected to be eligible to be taxed at favorable rates that otherwise are applicable to “qualified dividend income” received by non-corporate U.S. holders if certain additional conditions are satisfied.
Amryt believes that both Aegerion and Chiasma meet the definition of “expatriated entity” within the meaning of section 7874 of the Code. As expatriated entities, several limitations apply to both Aegerion and Chiasma, including, but not limited to, the prohibition, for a period of ten years from the closing date of the Prior Acquisition, of the use of net operating losses, foreign tax credits and other tax attributes to offset the income or gain recognized by reason of transfer of any property to a foreign related person or to offset any income received or accrued during such period by reason of Amryt’s license of any property to a foreign related person. Moreover, in such case, an additional minimum tax under Section 59A of the Code on certain “base eroding” payments to certain affiliates that are foreign corporations may be imposed.
Although Amryt does not expect to be treated as a U.S. corporation under currently applicable law and regulations, it is possible that a future change in law could expand the scope of Section 7874 of the Code on a retroactive basis. If Amryt were treated as a U.S. corporation, its entire net income would be subject to U.S. federal income tax on a net income basis and would be determined under U.S. federal income tax principles. Further, Amryt’s treatment as a U.S. corporation may have material adverse effects on the business, financial condition, results of operations and prospects of Amryt and its subsidiaries.
The application of Section 7874 of the Code is complex and subject to detailed regulations (the application of which is uncertain in various respects and could be impacted by changes in U.S. Treasury regulations with possible retroactive effect). Accordingly, there can be no assurance that the IRS will not challenge the status of Amryt as a non-U.S. corporation for U.S. federal income tax purposes under Section 7874 of the Code or that such challenge would not be sustained by a court. If the IRS were to successfully challenge under Section 7874 of the Code Amryt’s status as a non-U.S. corporation for U.S. federal income tax purposes, Amryt and certain Amryt Shareholders may be subject to significant adverse tax consequences, including a higher effective corporate income tax rate on Amryt, the recognition of gain by certain U.S. Amryt Shareholders upon the deemed conversion of Amryt from a non-U.S. corporation to a U.S. corporation, and future withholding taxes on certain Amryt Shareholders, depending on the application of any applicable income tax treaty that may apply to reduce such withholding taxes.
Our global operations subject us to significant tax risks.
We are subject to tax rules in the jurisdictions in which we operate. Changes in tax rates, tax relief and tax laws, changes in practice or interpretation of the law by the relevant tax authorities, increasing challenges by relevant tax authorities or any failure to manage tax risks adequately could result in increased charges, financial loss, penalties and reputational damage. Tax authorities may actively pursue additional taxes based on retroactive changes to tax laws which could result in a material restatement to its tax position. Any of these factors could have a negative impact on our business, financial condition, results of operations and prospects.
Fluctuations in currency exchange rates and increased inflation could materially adversely affect our financial condition and results of operations.
We have operations in Ireland, the United Kingdom, the United States, Germany, Switzerland, Brazil, France, Italy, Spain, Israel and other select markets throughout the world. As a result of the international scope of our operations, fluctuations in exchange rates, particularly between the U.S. dollar, our reporting currency, and the Euro, may adversely affect us. In the year ended December 31, 2021, 2.6% of our sales were denominated in pound sterling (£), 72.6% of our sales were denominated in U.S. dollars, 24.0% were denominated in Euros and the balance was denominated in other currencies. As a result, weakening of the Euro or pound sterling relative to the U.S. dollar presents the most significant risk to us. Any significant fluctuations in currency exchange rates may have a material impact on our business.
In addition, economies in Central European and Latin American countries have periodically experienced high rates of inflation. Periods of higher inflation may slow economic growth in those countries. As a substantial portion of our expenses (excluding currency losses and changes in deferred tax) is denominated in U.S. dollars or Euros, the relative movement of inflation significantly affects our results of operations. Inflation also is likely to increase some of our costs and expenses, including wages, rents, leases and employee benefit payments, which we may not be able to pass on to our customers and, as a result, may reduce our profitability. To the extent inflation causes these costs to increase, such inflation may materially adversely affect our business. Inflationary pressures could also affect our ability to access financial markets and lead to counter-inflationary measures that may harm our financial condition, or results of operations or materially adversely affect the market price of our securities.
The occurrence of cyber incidents, or a deficiency in cybersecurity, could negatively impact our business by causing a disruption to its operations, a compromise or corruption of confidential information, exposure to legal and regulatory action, or damage to our patient, partner or employee relationships, any of which could subject us to loss and reputational harm.
A cyber incident is considered to be any event that threatens the confidentiality, integrity or availability of information resources. More specifically, a cyber incident is an intentional attack or an unintentional event that can include gaining unauthorized access to systems to disrupt operations, corrupt data or steal confidential information about patients, suppliers, partners or employees. A number of companies have recently experienced serious cyber incidents and breaches of their information technology systems. Cyber incidents pose risks both to our internal systems and to those we have outsourced operations to, including the risk of operational interruption, damage to our reputation and relationships with patients, partners and employees, and private data exposure. We have implemented processes, procedures and controls to help mitigate these risks. However, these measures, as well as an increased awareness of the risk of a cyber incident, do not guarantee that our reputation, operations and financial results will not be adversely affected by such an incident.
In the ordinary course of business, we collect and store sensitive data, including intellectual property, proprietary business information and patient data. This data includes, where required or permitted by applicable laws, personally identifiable information. Certain third parties with whom we contract also collect and store data related to clinical trial subjects and patients. The secure maintenance of this information is critical to our operations and business strategy. Despite our current security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breaches due to employee error, malfeasance or other disruptions. Any such breach could compromise information stored on networks or those of our partners. Any access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, recovery costs, disruption of operations, including delays in our regulatory approval efforts, and damage our reputation, which could adversely affect our business.
Risks Related to Acquisitions, Including our Acquisitions of Aegerion and Chiasma
We have faced, and we may continue to face, challenges integrating the businesses and operations of our company and Chiasma and Aegerion and, as a result, may not realize the expected benefits of the acquisitions.
Integrating the businesses of our company and the businesses of Aegerion and Chiasma is a complex process.. We cannot guarantee that the integration of our business with Aegerion and Chiasma’s will be completed successfully or that we will be able to achieve any of the anticipated benefits. In order for the integration to be successful, we must maintain prior regulatory relationships, clinical trials, product initiatives, license, service DOJ and Corporate Integrity agreement(s) and their related obligations, with violations of these agreements resulting in potential financial penalties, additional time added on to the current terms of the agreements and additional costs for the company. We must also undertake certain actions required to maintain manufacturing, supply and distribution processes. While we have made significant progress, we may not be able to successfully integrate the businesses of our company and Aegerion and Chiasma, and even if successful, the integration may be costly and time consuming, and the anticipated synergies may not be realized, either of which may have a material and adverse effect on our business, financial condition, results of operations and prospects.
Any future acquisitions we make may expose us, to risks that could adversely affect our business, and we may not achieve the anticipated benefits of acquisitions of businesses or technologies.
As a part of our growth strategy, we may make additional acquisitions of complementary businesses. Any future acquisition will involve numerous risks and operational, financial and managerial challenges, including the following, any of which could adversely affect our business, financial condition or results of operations:
| • | limited support and user knowledge for legacy systems of acquired companies; |
| • | problems maintaining uniform procedures, controls and policies with respect to our financial accounting systems; |
| • | difficulties in managing geographically dispersed operations, including risks associated with entering foreign markets in which we have no or limited prior experience; |
| • | underperformance of any acquired technology, product or business relative to our expectations and the price we paid; |
| • | negative near-term impacts on financial results after an acquisition, including acquisition-related earnings charges; |
| • | the potential loss of key employees, customers and strategic partners of acquired companies; |
| • | claims by terminated employees and shareholders of acquired companies or other third parties related to the transaction; |
| • | the assumption or incurrence of additional debt obligations or expenses, or use of substantial portions of our cash; |
| • | the issuance of equity securities to finance or as consideration for any acquisitions that dilute the ownership of our shareholders; |
| • | any collaboration, strategic alliance and licensing arrangement may require us to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us; |
| • | risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing products or product candidates and regulatory approvals; |
| • | diversion of management’s attention and company resources from existing operations of the business; |
| • | inconsistencies in standards, controls, procedures and policies; |
| • | the impairment of intangible assets as a result of technological advancements, or worse-than-expected performance of acquired companies; |
| • | assumption of, or exposure to, historical liabilities of the acquired business, including unknown contingent or similar liabilities that are difficult to identify or accurately quantify; |
| • | our inability to generate revenue from acquired technology or products sufficient to meet our objectives in undertaking the acquisition or even to offset the associated acquisition and maintenance costs; |
| • | risks associated with acquiring intellectual property, including potential disputes regarding acquired companies’ intellectual property; and |
| • | future acquired products, employees policies and operations (including pricing/reimbursement) could be subject to Corporate Integrity Agreement compliance and review. |
There can be no assurance that any of the acquisitions we may make will be successful or will be, or will remain, profitable. Our failure to successfully address the foregoing risks may prevent us from achieving the anticipated benefits from any acquisition in a reasonable time frame, or at all.
If intangible assets and goodwill that we record in connection with our acquisitions become impaired, we may have to take significant charges against earnings.
In connection with the accounting for our acquisitions, a significant value may be recognized in respect of intangible assets, including developed technology and customer relationships relating to the acquired product lines, and goodwill. Under IFRS, we must assess, at least annually and potentially more frequently, whether the value of intangible assets and goodwill has been impaired. Intangible assets and goodwill will be assessed for impairment in the event of an impairment indicator. Any reduction or impairment of the value of intangible assets and goodwill will result in a charge against earnings, which could materially adversely affect our results of operations and shareholders’ equity in future periods.
Risks Related to the Commercialization of our Products
Our assumptions and estimates regarding prevalence and the addressable markets for our products and product candidates may be inaccurate. If there are fewer actual patients than estimated, or if any product approval is based on narrower definitions of patient populations, our revenues and cash position could be materially and adversely affected.
The patient population for the diseases that our products treat is very small, and networking, data gathering and support channels are not as established as those for more prevalent and researched disease indications. There are limited patient registries or methods of establishing with precision the actual number of Homozygous familial Hypercholesteraemia (HoFH), General Lipodystrophy (GL) and Partial Lipodystrophy (PL), acromegaly, Neuroendocrine Tumour (NET) or Epidermolysis Bullosa (EB) patients in any geography. Estimating the prevalence of a rare disease is difficult and we therefore must rely on assumptions, beliefs and an amalgam of information from multiple sources, resulting in potential under or over-reporting. There is no guarantee that our assumptions and beliefs are correct, or that the methodologies used and data collected have generated or will continue to generate accurate estimates. There is therefore uncertainty around the estimated total potential addressable patient population for treatment with lomitapide, Mycapssa, metreleptin or Oleogel-S10 worldwide.
In addition, the potential market opportunity for our product candidates that we may develop is difficult to estimate precisely, particularly given that the orphan drug markets which are targeted are, by their nature, relatively unknown. Our estimates of the potential market opportunity for each of these product candidates are predicated on several key assumptions, such as industry knowledge and publications, third-party research reports and other surveys. If any of our assumptions prove to be inaccurate, then the actual market for lomitapide, Mycapssa, metreleptin, Oleogel-S10 or AP103, or our other or future product candidates, could be smaller than our estimates of the potential market opportunity. If that turns out to be the case, our product revenue may be limited, and we may be unable to achieve or maintain profitability, which could have a material adverse effect on our business, financial condition, results of operations and prospects.
Our products may not gain market acceptance, in which case we may not be able to generate product revenues.
Physicians, healthcare providers, patients, payers or the medical community may not accept or use our approved products. Efforts to educate the medical community and third-party payers on the benefits of the products may require significant resources and may not be successful. Notwithstanding the level of revenues historically generated from the sale of lomitapide, Mycapssa® and metreleptin, if any of our existing marketed products or product candidates do not achieve an adequate level of acceptance, we may struggle to continue to generate significant product revenues and may not in the future generate any profits from operations. The degree of market acceptance will depend on a variety of factors, including, but not limited to:
| • | whether clinicians and potential patients perceive product candidates to have better efficacy, safety, tolerability profile and ease of use, when compared with the products marketed by our competitors and the prevailing standard of care (“SOC”); |
| • | the timing and location of market introduction of any approved products; |
| • | our ability to provide acceptable evidence of safety and efficacy; |
| • | the frequency and severity and causal relationships of any side effects and a continued acceptable safety profile following approval; |
| • | relative convenience and ease of administration; |
| • | patient diagnostics and screening infrastructure in each market; |
| • | marketing and distribution support; |
| • | the availability of healthcare coverage, reimbursement and adequate payment from health maintenance organizations and other third-party payers, both public and private; and |
| • | competition from other therapies. |
We face significant competition from other biotechnology and pharmaceutical companies.
The specific markets in which we operate are highly competitive and this competition could harm our results of operations, cash flows and financial condition. Our competitors include major international pharmaceutical companies as well as smaller or regional specialty pharmaceutical and biotechnology companies. We may be forced to either lower the selling prices of our products in response to competitor pricing or lose patients who choose lower-priced products. Many of our competitors are larger, have greater financial resources and a lower cost structure. As a result, our competitors may be better equipped to withstand changes in economic and industry conditions. These competitors currently engage in, have engaged in or may in the future engage in the development, manufacturing, marketing and commercialization of new pharmaceuticals, some of which may compete with our products. Competition may also arise from, among other things, other drug development technologies, methods of preventing or reducing the incidence of disease, including vaccines and new small molecule or other classes of therapeutic agents. Smaller or early-stage companies may also be significant competitors, particularly through collaborative arrangements with large, established companies. Key competitive factors affecting the commercial success of our products and any other products that we develop or acquire are likely to be safety, efficacy, tolerability profile, reliability, convenience of dosing, price and reimbursement. We may also face future competition from companies selling generic alternatives to our products in countries where we do not have patent coverage, Orphan Drug status or another form of data or marketing exclusivity or where patent coverage or data or marketing exclusivity has expired, is not enforced, or may, in the future, be challenged.
A significant competitor to our lomitapide product is a class of drugs known as PCSK9 inhibitors. Two main brands dominate the marketplace – Praluent and Repatha which are both approved in the European Union and the United States. Sales of PCSK9 inhibitors compete with sales of lomitapide and we expect that this product will continue to compete with lomitapide. In addition, one of our competitors, Regeneron Pharmaceuticals Inc., is developing Evinacumab, a human monoclonal antibody directed against the activity of angiopoietin-like 3 (“ANGPTL3”) for the treatment of HoFH. In August 2019, Regeneron announced positive topline data from its ongoing Phase 3 trial in HoFH and the FDA approved Evinacumab on February 11, 2021, for adults and pediatric patients 12 years and older for treatment of HoFH. EMA approved Evinacumab in June 2021 as an adjunct to diet and other low-density lipoprotein-cholesterol (LDL-C) lowering therapies for the treatment of adult and adolescent patients aged 12 years and older with homozygous familial hypercholesterolaemia (HoFH). Regeneron launched Evinacumab in the United States in March 2021. Evinacumab was licensed to Ultragenyx Pharmaceutical Inc in countries outside the US in January 2022. Although administered through intravenous infusion, physicians may now consider this product for HoFH patients as an alternative to lomitapide. Other competitors may succeed in developing, acquiring or licensing additional pharmaceutical products that are introduced into the market and that are more effective, have a more favorable safety profile or are less costly than our products.
Although Myalept is the first and only product approved in the United States for the treatment of complications of leptin deficiency in patients with GL, there are a number of therapies approved to treat these complications independently that are not specific to GL. Myalepta also faces competition in the European Union, both for the treatment of GL and PL. Our competitors are also developing products, which, if approved, and depending on the labelled indication, could potentially compete with metreleptin.
The acromegaly market has many established competitors, dominated by Novartis and Ipsen, who like the rest of the competition offer injectable SSA’s. Mycapssa® is the only oral SSA available on the market. Crinetics Pharmaceuticals are developing paltusotine, which if approved would be the second oral SSA to enter the market and compete directly with Mycapssa. Crinetics also have a Ph3 study to evaluate effect in treatment-naïve patients. If studies are successful, filings could take place at end of 2023 / early 2024.
Although there are no approved products in the United States and the European Union for the treatment of EB, our competitors are developing products, which, if approved, and depending on the labelled indication, could potentially compete with Oleogel-S10. One such competitor on the near-term horizon is a potential competitor from Krystal Biotech – B-VEC. B-VEC is gene therapy delivered through a herpes simplex viral vector with studies focused on the narrower population of DEB patients. In November 2021 Kyrstal announced that its Ph3 GEM-3 study of B-VEC, known as Vyjuvek™, met its primary endpoint of complete wound healing at six-month timepoints and its secondary endpoint of complete wound healing at three-month timepoints. Additionally, Vyjuvek™ was shown to be well-tolerated, with no drug-related serious adverse events or discontinuations. They could file a Biologics License Application in Q2 2022, with potential approval in the second half of 2022 and file for MAA in EU in the second half of 2022. Although there is overlap with Oleogel-S10, Amryt believe that this lifelong condition will require multiple therapeutic options.
Other competitors may succeed in developing, acquiring or licensing additional pharmaceutical products that are introduced into the market and that are more effective, have a more favorable safety profile, or are less costly than our products. If we do not compete successfully, our operating margins, financial condition and cash flows could be adversely affected.
We commercialize our products through both a direct sales force and through strategic relationships with third parties for commercialization, distribution, sales and marketing in certain jurisdictions. If we are unable to adequately develop and maintain our sales, marketing and distribution capabilities or enter into business arrangements, we may not be successful in commercializing our product candidates.
We sell lomitapide, Mycapssa® and metreleptin directly in the United States using our own marketing and sales resources and also market and sell, or plan to market and sell, our products directly in certain key countries outside the United States where such products are, or may be, approved using country managers or local distributors to the extent rights to commercialize such products are not out-licensed. We also make our products available on a named patient basis as a result of the approval of our products in the United States or the European Union. We use third parties to provide sales, warehousing, shipping, third-party logistics, invoicing, collections and other distribution services on our behalf in connection with the sale of products globally. Metreleptin is also available in: Italy, Greece, France, Germany, Austria, Switzerland, United Kingdom, Denmark, Spain, Portugal, Serbia, Russia, Kazakhstan, Saudi Arabia, Israel, Turkey, Oman, Qatar, Bahrain, UAE, Colombia, Brazil and Argentina. Metreleptin was out-licensed to Shionogi & Co. Ltd. (“Shionogi”) in Japan. Lomitapide is also available in: the Netherlands, Germany, Austria, Spain, Greece, Italy, United Kingdom, Sweden, Norway, Denmark, France, Hungary, Czech Republic, Russia, Qatar, Kuwait, Saudi Arabia, Canada, Brazil, Colombia and Argentina. We also out-licensed lomitapide for sale by Recordati Rare Diseases Inc. (“Recordati”) in Japan. Mycapssa® is currently only sold in the United States.
For example, there is currently a contract with a single specialty pharmacy distributor in the United States for the distribution of lomitapide, Mycapssa® and metreleptin, a single distributor in Brazil for lomitapide and metreleptin, and single distributors, third-party logistics providers, importers and/or specialty pharmacies in certain other countries, including the European Union for lomitapide and metreleptin. We have entered into or may selectively seek to establish sales, distribution and similar forms of arrangements to grow revenue in existing territories and gain access to new territories and to reach patients in certain geographies that we do not believe can be cost-effectively addressed with our own sales and marketing capabilities. If we are unable to establish, maintain and finance the capabilities to sell, market and distribute our products, either through our own capabilities or through arrangements with third parties, and to effectively manage those third parties, or if we are unable to enter into distribution agreements in countries that we do not believe can be cost-effectively addressed with our own sales and marketing capabilities, we may not be able to successfully sell our products. We cannot guarantee that we will be able to establish, maintain and finance our own capabilities or to enter into and maintain favorable distribution agreements with third parties on acceptable terms, if at all.
To the extent that we enter into arrangements with third parties to perform sales, marketing or distribution services, the product revenues or the profitability of these product revenues may be lower than if we were to commercialize the products on our own. We will have limited control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market the products effectively, and may also, despite our compliance diligence reviews, audits and training, engage in non-compliant / illegal activities including U.S. Foreign Corrupt Practices Act of 1977 (“FCPA”) violative activity that, directly or indirectly, impact the use or sales of the products or damage our relationships with relevant stakeholders. Any performance failure, inability or refusal of our specialty pharmacy distributors, or third-party service providers to perform, or any failure to renew existing agreements on favorable terms, or at all, could cause serious disruption and impair our commercial or named patient sales of the products, which may have a material adverse effect on our business, financial condition and results of operations. Adverse actions with third parties and contract manufacturing organizations we enter into arrangements with including adverse regulatory actions could negatively impact the company. Furthermore, the expenses associated with maintaining sales force and distribution capabilities may continue to be substantial compared to the revenues we generate. If we are unable to establish and effectively maintain adequate sales, marketing and distribution capabilities, whether independently or with third parties, we may not be able to generate product revenues, which may have a material adverse effect on our business, financial condition, results of operations and prospects. See “—Risks Related to our Reliance on Third Parties.”
The successful commercialization of our product candidates will depend in part on the extent to which governmental authorities and health insurers establish adequate coverage, reimbursement levels and pricing policies. Failure to obtain or maintain coverage and adequate reimbursement for our product candidates, if approved, could limit our ability to market those products and decrease revenue generating ability.
The availability and adequacy of coverage and reimbursement by governmental healthcare programs such as Medicare and Medicaid, private health insurers and other third-party payers is essential for many patients to be able to afford prescription medications such as our products and potential product candidates, assuming regulatory approval is obtained. Our ability to achieve acceptable levels of coverage and reimbursement for products by governmental authorities, private health insurers and other organizations will affect the success of our approved products and product candidates. Assuming we obtain coverage for our product candidates by third-party payers, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. We cannot be sure that coverage and reimbursement in the United States, the EU Member States, or elsewhere will be available for the product candidates or any product that we may develop, and any reimbursement that may become available may be decreased or eliminated in the future.
Further, it is possible that a third-party payer may consider our product candidates as substitutes and only offer to reimburse patients for a less expensive product. Even if we show improved efficiency or convenience of administration with our product candidates compared to products marketed by our competitors and the prevailing SOC, the pricing of existing therapies may still limit the amount we could charge. Third-party payers may deny or revoke the reimbursement status of any given product or establish new prices for existing marketed products that inhibit us from realizing an appropriate return on our investment in the product candidates. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our product candidates and may not be able to obtain a satisfactory financial return on them.
Outside the United States, the success of our products and operations is subject to extensive governmental price controls and other market regulations which may materially and adversely affect our ability to generate commercially reasonable revenue and profits.
Our operations are subject to extensive governmental price controls and other market regulations in the United Kingdom and other countries outside of the United States. The increasing emphasis on cost-containment initiatives in the various EU Member States and other countries can put pressure on the pricing and usage of currently marketed products and product candidates in the future. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. Some EU Member States have established free-pricing systems, but regulate the pricing for drugs, inter alia, through profit control schemes. However, the UK, which has implemented the most vigorous scheme, has officially left the European Union on January 31, 2020. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside the United States, the reimbursement for our currently marketed products and our product candidates in the future may be reduced and may be insufficient to generate sufficient revenues and profits. Moreover, increasing efforts by governmental and third-party payers in the United States and abroad to control healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for newly approved products and, as a result, they may not cover or provide adequate payment for our products, or any other product candidates we may develop in the future.
A number of adverse effects have been reported in clinical trials for metreleptin, lomitapide, oral octreotide and Oleogel-S10, and the prescribing information for each of lomitapide, metreleptin and oral octreotide contains significant limitations on use and other important warnings and precautions, any of which could negatively affect the market acceptance, dropout rates and marketing approval for these products, and post-marketing commitments could identify additional adverse events and safety or efficacy risks, which could further harm our business.
The prescribing information for lomitapide in the United States and the European Union and in other countries in which lomitapide is approved contains significant limitations on use and other important warnings and precautions, including, but not limited to, a boxed warning in the Juxtapid U.S. labeling, additional monitoring which is identified by a black inverted triangle in the product information for Lojuxta in the European Union, warnings in the prescribing information for Lojuxta citing concerns over liver toxicity associated with use of lomitapide, European Risk Management Plan (RMP) materials for physicians and patients, and a U.S. Risk Evaluation Mitigation Strategy (“REMS”) program. The prescribing information for metreleptin in the United States and European Union contains important warnings and precautions, including but not limited to, a boxed warning on the Myalept label in the United States, citing the risk of anti-metreleptin antibodies with neutralizing activity, risk of lymphoma and a U.S. REMS program. As with the U.S. label, the Myalepta Summary of Product Characteristics in the European Union notes that the consequences of neutralizing antibodies with respect to the loss of efficacy or serious or severe infections is not well characterized but could reduce how well the leptin found naturally in the body works or how well Myalepta works.
Patients reported various adverse reactions in the Phase 3 study of lomitapide, including reports of gastrointestinal events by 93% of patients, and in the HoFH clinical trial, including diarrhea, nausea, vomiting, dyspepsia, abdominal pain, weight loss, abdominal discomfort, abdominal distension, constipation, flatulence, increased alanine aminotransferase, chest pain, influenza, nasopharyngitis, and fatigue. Elevations in liver enzymes and hepatic (liver) fat were also observed.
GL patients in the Phase 3 study of metreleptin reported various adverse drug reactions, including weight loss, hypoglycemia, decreased appetite, fatigue, neutralizing antibodies and alopecia. Additionally, although none were assessed as drug related, there were four reported treatment-emergent deaths over the course of the 14-year study duration. Upon further investigation, these reports were consistent with the underlying morbidity of lipodystrophy and included renal failure, cardiac arrest (with pancreatitis and septic shock), progressive end-stage liver disease (chronic hepatic failure), and hypoxic-ischemic encephalopathy. In the open-label, long-term, investigator-sponsored study of metreleptin for the treatment of metabolic disorders associated with lipodystrophy syndromes (initiated in 2000 and conducted at the National Institutes of Health (“NIH”)), there were two cases of peripheral T-cell lymphoma and one case of a localized anaplastic lymphoma (kinase-positive anaplastic large cell lymphoma, which is a type of T-cell lymphoma). Both of the cases of peripheral T-cell lymphoma were reported in patients with acquired GL, and both had evidence of pre-existing lymphoma and/or bone marrow/hematologic abnormalities before metreleptin therapy. A third case of anaplastic large cell lymphoma occurred in the context of a specific chromosomal translocation.
These adverse events, coupled with the boxed warnings and other label restrictions, could cause healthcare providers, regulators and patients or potential patients to view the risks associated with our products as outweighing the benefits. This could cause patients to discontinue use and limit the number of new patients, thereby negatively affecting our business, financial condition, results of operations and prospects. In addition, as part of the post-marketing commitment to the FDA for both lomitapide and metreleptin, we are conducting post-marketing registries to better understand their long-term safety and effectiveness. For lomitapide, we are conducting a long-term observational cohort study (product exposure registry) to better understand the long-term safety, patterns of use, compliance and long-term effectiveness of controlling LDL levels. For metreleptin, we are conducting a long-term, prospective, observational study (product exposure registry) to identify and better understand any serious risks related to the use of the product. Finally, we are conducting sequential programs to expand the understanding of metreleptin immunogenicity and manufacturing. The final program regarding the immunogenicity of metreleptin was initiated in 2018, and the final post-marketing study related to the manufacturing of metreleptin is expected to be completed by the deadlines agreed with the FDA. In addition, we are working to implement post-marketing commitments in the European Union for metreleptin, including a pediatric study in GL patients, an immunogenicity program, a post-approval study in PL and to enroll European patients in the product exposure registry. A failure to meet post-marketing commitments to the FDA, the EMA or other regulatory authorities could impact the ability to continue to market lomitapide or metreleptin, respectively, in countries where we are unable to meet such commitments. A Phase 3, randomized, multicenter, double-blind, placebo-controlled study of metreleptin in patients with partial lipodystrophy (PL) was initiated at the end of Q4 2021 with registration intent to include this population in the US label. In general, we may not be able to meet our post marketing commitments for our current approved products and those in development which could materially impact our ability to commercialize our products and therefore have a material impact on the success of our business.
In the course of conducting observational cohort studies, additional clinical studies (such as pursuant to a pediatric investigation plan (“PIP”) in the European Union), post-marketing surveillance, or re-evaluation of any completed clinical study data could identify, additional safety information on known or unknown side effects or new undesirable side effects caused by our products or product candidates, or the data may raise other issues with respect to the products. In such instances, a number of potentially significant negative consequences could result, including:
| • | we may experience a negative impact on market acceptance and increased dropout rates; |
| • | regulatory authorities may suspend, withdraw or alter their approval of the relevant product; |
| • | regulatory authorities may require the addition of labeling statements, such as warnings or contraindications or distribution and use restrictions such as, for example, the modifications to the Juxtapid label to include language instructing patients to cease therapy upon the occurrence of severe diarrhea; |
| • | regulatory authorities may issue, or require us to issue additional specific communications such as safety alerts, field alerts, or “Dear Doctor” letters to healthcare professionals; |
| • | regulatory authorities may require us to recall, withdraw, or stop selling a product or take other enforcement action; |
| • | we may receive negative publicity; |
| • | we may be required to change the way the relevant product is administered, conduct additional preclinical studies or clinical trials or restrict the distribution or use of the relevant product; |
| • | patients could suffer harm, and we could be sued and held liable for harm caused to patients; |
| • | the regulatory authorities may require us to amend the relevant REMS program, Risk Management Plan or comparable equivalent; and |
| • | our reputation may suffer. |
Mycapssa® (oral octreotide capsules) is approved in the US for use in patients who have responded to and tolerated octreotide or lanreotide, injectable somatostatin analogues (SSAs). Approval in the European Union is under review by the EMA with an anticipated approval date of Q3 2022. In the US, the prescribing information does not include any boxed warnings. The safety profile of Mycapssa® is consistent with that seen with injectable octreotide with the exception of the absence of injection site reactions. Warnings and precautions include the risks of cholelithiasis and complications of cholelithiasis, hypoglycemia or hyperglycemia, hypothyroidism bradycardia, arrhythmia, or cardiac conduction abnormalities, decreased Vitamin B12 Levels and an abnormal Schilling's Tests. There are no FDA post approval commitments for Mycapssa® in the treatment of Acromegaly patients but the company is conducting a 2 year observation registry study in patients with acromegaly receiving treatment with Mycapssa® as well as other drug treatments for acromegaly.
Oleogel-S10 is under review by the EMA. A phase 3 study (EASE) has completed its double-blind phase and the open label extension is on-going. On April 22, 2022, the European Medicines Agency's (“EMA”) Committee for Medicinal Products for Human Use (“CHMP”) has adopted a positive opinion, recommending the approval of Filsuvez® in the European Union (EU) for the treatment of partial thickness wounds associated with dystrophic and junctional Epidermolysis Bullosa (EB) in patients six months and older. Based on this CHMP recommendation a decision by the European Commission (EC) is expected on the Filsuvez® application within 67 days. The proposed label include warnings and precaution statements on hypersensitivity reactions, use in patients with wound infections, squamous cell carcinoma, use in dominant dystrophic EB (DDEB) and junctional EB (JEB), birch pollen allergy and accidental eye exposure.
Any known safety concerns for our products or product candidates, or any unknown safety issues that may develop or be discovered, including drug interaction problems or an increase in the severity or frequency of known adverse events or the discovery of previously unknown adverse events, or the evaluation or reevaluation of study data, could prevent us from achieving or maintaining market acceptance of the respective product, affect our ability to obtain or retain regulatory marketing approval of the respective product in one or more countries, result in onerous restrictions on such approval or the implementation or modification of the REMS programs (if applicable) or risk management plans for our products, or any other enforcement actions, result in claims, lawsuits and increased regulatory scrutiny, and affect our ability to achieve our financial goals.
Enacted and future legislation and related regulations may increase the difficulty and cost for us to commercialize metreleptin, lomitapide, Mycapssa® or Oleogel-S10 or development candidates and may affect the prices we are able to obtain for our products, if and where approved.
In the United States, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that restrict or regulate post-approval activities, which may affect our ability to profitably sell our products. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We cannot predict whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes for the products may be. In addition, increased scrutiny by Congress of the FDA’s approval process may subject us to more stringent product labeling, post-marketing testing and other requirements. In May 2019, the Centers for Medicare and Medicaid Services adopted a final rule permitting Medicaid Part D plans to apply certain utilization controls on new starts of protected class drugs.
In the United States, most outpatient prescription drugs, including Myalept, Mycapssa, Juxtapid and Oleogel-S10, if approved, may be covered under Medicare Part D for beneficiaries over the age of 65 or with certain disabilities. Medicare Part D prescription drug plans use formularies to limit the number of drugs that will be covered in any therapeutic class and impose differential cost sharing or other utilization management techniques. The possibility that our products and product candidates will be subject to such formularies places pressure on them to contain and reduce costs, which could negatively impact our commercialization efforts. Changes to Medicare Part D, which give plans more freedom to limit coverage or manage utilization, and other cost reduction initiatives in the program could decrease patient access to any approved products, and could be detrimental to our business.
The Patient Protection and Affordable Care Act (“PPACA”) (as amended by the Health Care and Education Reconciliation Act of 2010) substantially changed the way healthcare is financed by both governmental and private insurers. The law contains a number of provisions that affect coverage and reimbursement of drug products and that could potentially reduce the demand for our products, such as:
| • | increasing drug rebates under state Medicaid programs for brand name prescription drugs and extending those rebates to Medicaid managed care; and |
| • | requiring drug manufacturers to provide a 70% discount on Medicare Part D brand name prescription drugs sold to Medicare beneficiaries whose prescription drug costs cause the beneficiaries to be subject to the Medicare Part D coverage cap (i.e., the so-called donut hole). |
We cannot predict the ultimate content, timing or effect of any changes to the PPACA or other federal and state reform efforts. There is no assurance that federal or state healthcare reform will not adversely affect our business and financial results, and we cannot predict how future federal or state legislative, judicial or administrative changes relating to healthcare reform will affect our business.
Multiple rules issued by HHS under the Trump administration could impact access to and reimbursement for prescription drugs if they become effective. A final rule that eliminated the safe harbor shielding Medicare Part D rebates to pharmacy benefit managers from the Anti-Kickback statute was finalized in November 2020. However, The Infrastructure Investment and Jobs Act (Public Law No: 117-58), signed into law on November 15 2021, established a moratorium on implementation of rule relating to eliminating the anti-kickback statute safe harbor protection for prescription drug rebates until January 1, 2026. In addition, the Build Back Better Act (HR 5376), as passed by the House of Representatives and currently under consideration in the Senate, would permanently prevent implementation of the rule so it remains unclear whether this will take effect.
In addition, the Medicaid Drug Rebate Program final rule, issued in December of 2020, finalized policies to change the way in which Medicaid rebates are calculated. This policy would require that copay assistance provided to patients would be applied as a discount to a payer for the purposes and could set a new best price for the purposes of the Medicaid drug rebate calculation unless a manufacturer can “ensure” that the full value of the assistance is passed through to the patient. This is problematic given the rising number of copay accumulator plans in which pharmacy benefit managers block patients from taking full advantage of patient assistance programs without the knowledge of the manufacturer. The Pharmaceutical Researchers and Manufacturers of America (PhRMA), sued to invalidate this rule in May of 2021. The outcome of this litigation remains uncertain and could require changes to patient assistance programs.
In September of 2021, the Build Back Better Act was introduced in the House of Representatives and contained a number of provisions that could impact the biopharmaceutical industry. These provisions include measures aimed at reducing patient out of pocket costs through a redesign of the Medicare Part D benefit, provisions aimed at allowing Medicare to negotiate drug prices for a subset of drugs in Medicare Part B and Part D, and provisions meant to limit drug price increases to the rate of inflation, requiring rebates to the government for price increases taken beyond inflation. The House of Representatives passed the Build Back Better Act in November of 2021, and it has not been considered by the Senate. If any of these measures are adopted into law, they could impact future business actions and access to medicines.
Any significant spending reductions affecting Medicare, Medicaid or other publicly funded or subsidized health programs that may be implemented and/or any significant taxes or fees that may be imposed, as part of any broader deficit reduction effort or legislative replacement to current laws or regulations, could have an adverse impact on our results of operations. In addition, countries outside the United States may make changes to their healthcare systems, which may in the future affect the revenue generated from sales of lomitapide, metreleptin and Oleogel-S10, if approved, or any of our future commercial products.
Existing legislation and federal regulations may permit reimportation of drugs from Canada into the United States where the drugs are sold at lower prices and this may adversely affect our operating results and overall financial condition.
The U.S. Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (“MMA”) contains provisions that may change importation laws and expand pharmacists’ and wholesalers’ abilities to import lower-priced versions of an approved drug and competing products from Canada, where there are government price controls.
In October 2020, the U.S. Department of Health and Human Services and the FDA issued a final rule and guidance concerning two new pathways for importing lower-cost drugs into the United States. The final rule allows States to set up importation schemes after receiving approval of their importation program by the Department of Health and Human Services. These state programs could allow medicines to be imported from Canada, but would not permit the import of certain medicines, including biologics or products subject to REMS programs. The FDA guidance describes procedures for drug manufacturers to facilitate the importation of FDA approved drugs and biologics manufactured abroad and originally intended for sale in a foreign country in the United States. Ongoing litigation on this regulation makes the outcome of any importation schemes uncertain.
If purchasers of Myalept, Mycapssa® or Juxtapid in the United States are able to import lower-priced products from countries outside the United States that place price controls on pharmaceutical products, this may result in a negative impact on the revenues of our products.
The reimportation of metreleptin, Mycapssa® or lomitapide into the U.S. market from a foreign market may negatively impact our revenues and anticipated financial results.
Although the European Union does not permit the re-importation of medicinal products from outside the European Union, parallel trade between EU Member States is possible and can result in third party imports from EU Member States offering lower prices for a product into those reimbursing products at higher costs.
We rely on named patient sales of our products in certain territories, but there are no assurances that named patient sales of our products will continue at current levels, or at all.
In a number of countries, where permitted based on U.S. or EU approval, metreleptin and lomitapide are available on a named patient sales or similar basis. Named patient basis means physician-requested treatment for patients in territories where marketing authorization has not yet occurred. There is no assurance that named patient sales will continue to be authorized in any particular country. Even if they are authorized, we will likely not be permitted to promote, market or otherwise engage in proactive selling activities for products sold on a named patient basis, which makes named patient sales much less predictable, and susceptible to unexpected decreases. If violations of any laws or governmental regulations are found to have occurred in connection with our products significant criminal or civil lawsuits may be filed, or investigations may be commenced. For example, in Brazil, under certain circumstances, we could be barred from sales of products to federal or state governments in Brazil due to penalties imposed by Brazilian regulatory authorities or through civil actions initiated by federal or state public prosecutors, or we could face administrative penalties imposed by Brazilian regulatory authorities and additional damages and fines. It is believed that the investigations in respect of Aegerion in Brazil have contributed to a slower turnaround between price quotation and orders, including reorders, from the federal government, and, in some cases, delays in orders and reorders from the government of the State of São Paulo after a patient has obtained access to lomitapide through the judicial process. These delays may continue, and we may experience other delays or suspensions of the ordering process. Similarly, there has been, and may continue to be, some reluctance by physicians to prescribe lomitapide, and some patients to take or stay on lomitapide, while the investigations are ongoing, particularly given that some of the investigators in Brazil made formal inquiries of certain prescribers of lomitapide, and there has been significant local media coverage of such inquiries and Aegerion’s past activities in Brazil. Further, in October 2017, a new set of regulatory requirements governing use of product candidates was published in Brazil which has added complexity to the process for the purchase, on a named patient basis, of drugs which have not received regulatory and/or pricing and reimbursement approval in Brazil, such as metreleptin and lomitapide, which has, along with the ongoing court proceeding, resulted in delays in the receipt of orders from Brazil for existing metreleptin and lomitapide patients. We believe that this led certain patients to discontinue therapy with metreleptin and lomitapide. In December 2020, we received marketing authorization approval from Agencia Nacional De Vigilancia Sanitaria, the Brazilian Health Regulatory Agency, for lomitapide.
We do not know the full extent of the impact that the approval of PCSK9 inhibitor products or Evinacumab in the United States, or the approval of a PCSK9 inhibitor product, will have on the named patient sales of lomitapide in other countries. We also do not know whether we will be permitted to sell metreleptin, Mycapssa® or lomitapide on a named patient basis in any additional countries. In certain countries, we may decide not to pursue named patient sales even if permitted. Even if named patient sales (or equivalent sales) are permitted in a certain country, and we elect to make metreleptin, Mycapssa® or lomitapide available on such basis, there is no guarantee that physicians in such country will prescribe the product, which they can only do if they proactively reach out to us or our distributors and also undertake the effort, time and cost of following the stringent local requirements to get their patient on therapy on a named patient basis, and that patients will be willing to start and adhere to therapy, or that the country will pay for the product at all, or at a level that is acceptable to us, without delay or imposing other hurdles on payment.
Further, there are countries where we choose to or are required to make our products available under an expanded access program at no cost prior to approval in such countries. There is no assurance that we will be able to obtain marketing approval or reimbursement at all or at acceptable levels or to maintain reimbursement for our products in any country where we have expanded access programs or that patients on such programs will convert to commercial product even if we do obtain requisite approvals. In certain countries where we seek reimbursement for the product during the pre-approval phase, we are able to establish the price for the product, while in other countries we need to negotiate the price. Such negotiations may not result in a price acceptable to us, in which case we may elect not to distribute the products in such country prior to approval or it may curtail distribution. Our expanded access program may result in significant expenses and may not result in expected future sales at desired levels or at all, and could negatively impact our financial results.
Risks Related to Clinical Development
If we are unable to commercialize or receive regulatory approval for Oleogel-S10, or experience significant delays in doing so, or are not granted a Priority Review Voucher, our business could be materially harmed.
Our Phase 3 EASE randomized double-blind placebo control study achieved its primary endpoint and forms the basis of application for regulatory approval. An NDA was submitted to FDA on March 30, 2021, and a Marketing Authorization Application (“MMA”) was submitted to the EMA on March 8, 2021, with a procedure start date of March 25, 2021. On February 28, 2022, we received a Complete Response Letter (“CRL”) from the FDA which asked Amryt to submit additional confirmatory evidence of effectiveness for Oleogel-S10 in EB. Amryt intends to discuss with the FDA the nature of the data required to address the Agency’s concerns. Our inability to obtain approval for and commercialize Oleogel-S10 would materially adversely affect our business, results of operations and prospects. On April 22, 2022, the European Medicines Agency's (“EMA”) Committee for Medicinal Products for Human Use (“CHMP”) has adopted a positive opinion, recommending the approval of Filsuvez® in the European Union (EU) for the treatment of partial thickness wounds associated with dystrophic and junctional Epidermolysis Bullosa (EB) in patients six months and older. Based on this CHMP recommendation a decision by the European Commission (EC) is expected on the Filsuvez® application within 67 days.
Amryt will seek a Priority Review Voucher (“PRV”) as part of the Oleogel-S10 NDA submission which if granted, we can sell, transfer or use to accelerate the approval of a future Amryt NDA. However, to be eligible for a PRV, Oleogel-S10 must have a Pediatric Rare Disease Designation from the FDA, be granted a priority review by FDA, and ultimately the NDA must be approved by the FDA. Amryt was granted a Pediatric Rare Disease Designation by the FDA in August 2018. When the NDA was submitted to the FDA on March 30, 2021, Amryt requested priority review. In June 2021, Amryt received confirmation from the FDA that its NDA for Oleogel-S10 had been accepted and granted priority review. Having received a CRL from the FDA we may no longer be eligible for a PRV and we intend to discuss and clarify this with the FDA.
Clinical trials are expensive, time consuming and difficult to design and implement and involve uncertain outcomes and, furthermore, results of earlier preclinical studies and clinical trials may not be predictive of results of future preclinical studies or clinical trials.
To obtain the requisite regulatory approvals to market and sell any of our product candidates, or to obtain regulatory approvals to market and sell any of our commercial products for new indications, we must demonstrate, through extensive preclinical studies and clinical trials, that our product candidates are safe and effective in humans. Clinical testing is expensive and can take many years to complete and has inherently uncertain outcomes. Failure can occur at any time during the clinical trial process and in addition regulatory authorities may require further studies at additional cost. Furthermore, regulatory authorities may not agree on the same trial design for pivotal studies. The results of preclinical studies and earlier clinical trials, or the results from earlier stages of preclinical studies or clinical trials, may not be predictive of the results of later-stage clinical trials. For example, the results generated to date in preclinical studies or Phase 1 or Phase 2 clinical trials for product candidates do not ensure that later clinical trials will demonstrate similar results. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy outcomes despite having progressed through preclinical studies and initial clinical trials. We may suffer setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding any promising results in earlier clinical trials. As product candidates are developed from preclinical through early to late-stage clinical trials towards approval and commercialization, it is customary that various aspects of the development program, such as manufacturing and methods of administration, are altered along the way in an effort to optimize processes and results. While these types of changes are common and are intended to optimize the product candidates for late-stage clinical trials, approval and commercialization, such changes carry the risk that they will not achieve these intended objectives. In addition, we may experience delays in ongoing or future preclinical studies or clinical trials and we have no certainty as to whether future preclinical studies or clinical trials will begin on time, will need to be redesigned, will enroll an adequate number of subjects or patients on time, if at all, or will be completed on schedule, if at all. Such factors may have a material adverse effect on our business, financial condition, results of operations and prospects.
Our product candidates may not work as intended, may cause undesirable side effects or may have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
Use of our product candidates could be associated with side effects or adverse events which can vary in severity from minor reactions to serious and/or severe adverse events, and in frequency from infrequent to prevalent. Undesirable side effects or unacceptable toxicities caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA, the EMA or comparable regulatory authorities. Results of our trials could reveal a high and unacceptable severity and prevalence of side effects.
If unacceptable side effects arise in the development of our product candidates, we, the FDA, competent authorities of EU Member States, ethics committees, the IRBs, the institutions in which our studies are conducted, or the DSMB could suspend or terminate our clinical trials. The FDA or comparable regulatory authorities could also order us to cease clinical trials or deny approval of our product candidates for any or all targeted indications. Treatment-related side effects could also affect patient recruitment or the ability of enrolled patients to complete any of our clinical trials or result in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff. We expect to have to train medical personnel using our product candidates to understand the side effect profiles of our product candidates in our clinical trials and upon any commercialization of any of our product candidates. Inadequate training in recognizing or managing the potential side effects of our product candidates could result in patient injury or death. Any of these occurrences may harm our business, financial condition, results of operations and prospects significantly.
The regulatory approval processes of the EMA, the FDA and other comparable regulatory agencies may be lengthy and time consuming and the outcome is unpredictable.
Our future success is partly dependent upon our ability to successfully develop, obtain regulatory approval for, and commercialize one or more of our product candidates. There can be no assurance that any development product candidates will be successful in clinical trials or receive regulatory approval. We cannot predict with certainty if or when we might submit for regulatory approval of any of our product candidates currently under development. Any approvals we may obtain may not cover all of the clinical indications for which we are seeking approval. Also, an approval might contain significant limitations in the form of narrow indications, warnings, precautions, or contra-indications with respect to conditions of use. Applications for any of our product candidates could fail to receive regulatory approval for many reasons, including, but not limited to, the following:
| • | the EMA, the FDA or any other comparable regulatory agency may disagree with the design or implementation of clinical trials or interpretation of data from non-clinical trials or clinical trials; |
| • | the population studied in the clinical program may not be sufficiently broad or representative to ensure that the clinical data can be relied on safely in the full population for which we are seeking approval; |
| • | the data collected from clinical trials of our product candidates may not be sufficient to support a finding that has statistically significant clinical meaningfulness or support the submission of a new drug application or other submission, or to obtain regulatory approval in relevant jurisdictions, such as the European Union and the United States; |
| • | we may be unable to demonstrate to the EMA, the FDA or any other comparable regulatory agency that a product candidate’s risk-benefit ratio for its proposed indication is acceptable; |
| • | the EMA, the FDA or any other comparable regulatory agency may fail to approve the manufacturing processes, test procedures and specifications or facilities of Amryt or third-party manufacturers with which we contract for clinical and commercial supplies; |
| • | a positive opinion from the CHMP of the EMA may not be ratified by the European Commission; and |
| | • | the approval policies or regulations of the EMA, the FDA or any other comparable regulatory agency may significantly change in a manner rendering clinical data insufficient for approval. |
Disruptions at the FDA, the EMA and other regulatory agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, in recent years, including in 2018 and 2019, the U.S. government shut down several times and certain regulatory agencies, such as the FDA, had to furlough critical employees and stop critical activities. Separately, in response to the COVID-19 pandemic, on March 10, 2020, the FDA announced its intention to postpone most inspections of foreign manufacturing facilities and products except for those deemed to be “mission critical.” On March 18, 2020, the FDA announced its intention to temporarily postpone routine surveillance inspections of domestic manufacturing facilities and provided guidance regarding the conduct of clinical trials. In July 2020, FDA resumed “prioritized” domestic inspections, which generally include pre-approval and surveillance inspections based on the COVID-19 Advisory Rating System, which it followed up on with an August 2020 guidance document on inspections. Following the August 2020 guidance, FDA resumed domestic inspections, while foreign inspections remained limited to those deemed “mission critical.” In March 2021, the U.S. Government Accountability Office issued a report indicating that FDA faces an inspections backlog that may take years to clear, which could delay drug approvals. In April 2021, the FDA issued guidance related to remote inspections to further elaborate on the oversight tools which may be considered in relation to mission critical inspections. Regulatory authorities outside the United States may adopt similar restrictions or other policy measures in response to the COVID-19 pandemic. If a prolonged government shutdown occurs, or if global health concerns continue to prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities, it could significantly impact the ability of such regulatory authorities to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, in our operations as a public company, future government shutdowns or delays could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.
Any of our current or future product candidates could take a significantly longer time to gain regulatory approval than expected or may never gain regulatory approval. This could delay or eliminate any potential product revenue by delaying or terminating the potential commercialization of product candidates. For example, any centralized marketing authorization application made to the EMA involving ‘Advanced Therapy Medicinal Products’ (such as AP103) will be subject to scientific evaluation by the Committee for Advanced Therapies, in addition to the CHMP.
We intend to seek regulatory approvals to commercialize the product candidates in the United States and the European Union. To obtain regulatory approval in other countries, we must comply with numerous and varying regulatory requirements of such other jurisdictions, which may include (without limitation) safety, efficacy, chemistry, manufacturing and controls, clinical trials, commercial sales, pricing and distribution of product candidates. Even if we are successful in obtaining approval in one jurisdiction, there can be no guarantee that it will obtain approval in other jurisdictions. Failure to obtain any marketing authorizations for the product candidates will result in us being unable to market and sell such products. If we fail to obtain approval in any jurisdiction, the geographic market for the product candidates could be limited. Similarly, regulatory agencies may not approve the labelling claims that are necessary or desirable for the successful commercialization of the product candidates.
We may fail to obtain or maintain Orphan Drug marketing exclusivity for our products and product candidates and may face significant competitive threats to the commercialization of these compounds from other manufacturers.
Obtaining Orphan Drug Designation does not guarantee that we will be the first to obtain marketing approval for any particular orphan indication due to the uncertainties associated with developing pharmaceutical products, which could prevent us from marketing the product candidates if another company is able to obtain Orphan Drug exclusivity before we do. Exclusive marketing rights in the United States may be unavailable or lost if: (i) an approval is sought for an indication broader than the orphan-designated indication; (ii) the FDA later determines that the request for designation was materially defective; or (iii) the applicant is unable to secure sufficient quantities of the drug to meet the needs of patients with the rare disease or condition following approval. In the European Union, a marketing authorization granted for an Orphan Drug must only cover the therapeutic indications that meet the Orphan Drug Designation criteria. Further, even if Orphan Drug exclusivity has been obtained, and maintained that exclusivity may not effectively protect the product candidates from competition because different drugs with different active moieties may be approved for the same condition. In addition, the FDA and the EMA may subsequently approve products with the same active moiety for the same condition if the FDA or the EMA concludes that the later drug is safer, more effective, or makes a major contribution to patient care. Orphan Drug Designation neither shortens the development time or regulatory review time of a drug nor gives the drug any advantage in the regulatory review or approval process.
We intend to seek Orphan Drug Designation for existing and future product candidates and we may never receive such designations. Despite exclusivity protections, another company nevertheless could market another version of the product if such company submits a full New Drug Application (“NDA”) in the United States or a full application for marketing authorization in the European Union with a complete human clinical trial program and obtains marketing approval of its product. Failure by us to obtain or maintain Orphan Drug marketing exclusivity for our products and product candidates may have a material and adverse effect on our business, financial condition, results of operations and prospects.
The FDA’s Orphan Drug exclusivity regulations may face legal challenges that could lead to changes which might adversely affect our business.
There have been legal challenges to aspects of the FDA’s regulations and policies concerning the exclusivity provisions of the U.S. Orphan Drug Act of 1983, and future challenges could lead to changes that affect the protections afforded to our product candidates in ways that are difficult to predict and which may have a material adverse effect on their business, financial condition, results of operations and prospects. In 2014, a U.S. district court invalidated the FDA’s denial of orphan exclusivity to an orphan designated drug, which the FDA had based on its determination that the drug was not proven to be clinically superior to a previously approved “same drug.” In response to the decision, the FDA released a policy statement stating that the court’s decision is limited to the facts of that particular case and that the FDA will continue to deny Orphan Drug exclusivity to a designated drug upon approval if the drug is the “same” as a previously approved drug, unless the drug is demonstrated to be clinically superior to that previously approved drug. Since then, similar legal challenges have been initiated against the FDA for its denial of Orphan Drug exclusivity to other designated drugs, and in 2017, Congress amended the U.S. Orphan Drug Act of 1983 to require a demonstration of clinical superiority upon approval as a condition to receiving Orphan Drug exclusivity when another “same drug” has already been approved for the same indication. In the future, there is the potential for additional legal challenges to the FDA’s Orphan Drug regulations and policies, and it is uncertain how ongoing and future challenges might affect our business.
We may seek and fail to obtain Fast Track or breakthrough therapy designations for our current or future product candidates. If we are successful, these programs may not lead to a faster development or regulatory review process, and they do not guarantee we will receive approval for any product candidate.
If a product is intended for the treatment of a serious or life-threatening condition and preclinical or clinical data demonstrate the potential to address an unmet medical need for this condition, the product sponsor may apply for Fast Track Designation. The FDA has broad discretion whether or not to grant this designation, so even if we believe a particular product candidate is eligible for this designation, we cannot assure you that the FDA would decide to grant it. Fast Track Designation does not mean we will necessarily experience a faster development process, review or approval compared to conventional FDA procedures. The FDA may rescind a Fast Track Designation if it believes that the designation is no longer supported by data from our clinical development program.
Risks Related to Government Regulation and Compliance
The laws and regulations in the areas of sales and marketing of pharmaceutical products and interacting with healthcare professionals and patients are very complex and onerous and require a robust compliance program. Failure to comply with these laws and regulations could have a material adverse effect on our business, financial condition and results of operations.
Failure to comply with certain laws and regulations could lead to governmental investigations and result in financial penalties and remedial and compliance measures. For example, compliance failures by Aegerion led to a DOJ investigation and ultimately resulted in three separate settlements (Corporate Integrity Agreement, Consent Decree and Deferred Prosecution Agreement) with multiple government agencies (Office of Inspector General (“OIG”), the FDA and DOJ) and aggregate penalties of approximately $40.1 million, which include $7.2 million in restitution payable over three years beginning January 2018, a civil penalty of $4.1 million to be paid to the SEC pursuant to the SEC judgment, and $28.8 million to be paid pursuant to the settlement agreement with the DOJ. Aegerion had been making the required payments and following the acquisition we assumed responsibility for payment obligations, which were fully satisfied in the first quarter of 2021. Pursuant to the settlement, we are also required to maintain various remedial and compliance measures, which were implemented as required by the settlement. We may be unsuccessful in implementing and complying with all of the elements of the settlement in a timely or satisfactory manner, or at all. Failure to comply with any provisions of these settlements, or if we became subject to new allegations or whistleblower complaints, could result in the imposition of additional fines, penalties and obligations by the applicable government agency, and could subject us to prosecution.
Furthermore, the investigation by the Brazilian authorities of Aegerion’s activities could result in the commencement of formal proceedings, and if the investigation finds any violation of any laws or governmental regulations, then our Brazilian subsidiary may be subject to civil lawsuits and administrative penalties and other potential damages and fines. Under certain circumstances, the Brazilian subsidiary and our company could be barred from further sales to federal or state governments in Brazil, including sales of Juxtapid or Myalepta, due to penalties imposed by Brazilian regulatory authorities or through civil actions initiated by federal or state public prosecutors.
We are subject to extensive legal and compliance obligations as a pharmaceutical company that commercializes products, as well as under Aegerion’s settlements with the DOJ, OIG, FDA, SEC and other federal and state government agencies.
As a pharmaceutical company that develops and commercializes pharmaceutical products, we are subject to an extensive array of broad and complex laws and regulations. These include, without limitation, regulations and laws in the United States and outside the United States related to manufacturing, clinical, quality, drug safety, commercialization, payments to and interactions with healthcare professionals and healthcare organizations, anti-kickbacks, fraud and abuse, the requirement to report payments and other transfers of value to healthcare professionals and healthcare organizations, data protection and privacy, pricing, reimbursement, price reporting, anti-corruption and anti-bribery, and a myriad of other areas and levels of regulation. Any failure by us or our key vendors, contractors, distributors, licensors or other key third-party vendors or service providers to comply with such laws and regulations could have a material adverse effect on our results of operations and financial condition, could result in product approvals being suspended, withdrawn, delayed or denied, could result in litigation or investigations which could be costly and be a significant distraction to executive management and other employees, and could result in damages or prosecution.
In September 2017, Aegerion entered into various settlements with the DOJ, OIG, FDA and SEC (“Aegerion Settlements”) of investigations regarding Aegerion’s U.S. commercial activities and disclosures related to Juxtapid. Under the terms of the Aegerion Settlements, Aegerion was required to pay approximately $40.1 million in penalties, which include $7.2 million in restitution payable over three years beginning January 2018, a civil penalty of $4.1 million to be paid to the SEC pursuant to the SEC judgment, and $28.8 million to be paid pursuant to the settlement agreement with the DOJ. We assumed these obligations in connection with the acquisition and have been making these penalty payments in accordance with the terms of the Aegerion Settlements with all of these payment obligations having been fully satisfied as of the end of the first fiscal quarter 2021. The Aegerion Settlements also mandates extensive remedial and compliance measures. The failure to comply with any provisions of the Aegerion Settlements, including the financial, remedial and compliance measures, or if we became subject to new allegations or whistleblower complaints, could result in the imposition of additional fines, penalties and obligations, and could subject us to prosecution or exclusion from federal healthcare programs in the United States.
The FDA, the EU Member States and other regulatory agencies outside the United States and the European Union enforce laws and regulations governing drug marketing and promotional activities, including prohibiting the promotion of off-label uses. Violations of these laws and regulations, and the resulting enforcement actions by these agencies, can result in significant liability.
The FDA, the regulatory authorities of the EU Member States and other comparable regulatory agencies outside the United States and the European Union strictly regulate the promotional claims that may be made about prescription drug products. In particular, a drug product may not be promoted in a jurisdiction prior to obtaining regulatory marketing approval or for uses that are not approved by the FDA, the European Commission (“EC”), the regulatory authorities of the EU Member States or such other regulatory agencies, as applicable, as reflected in the product’s approved prescribing information or summary of product characteristics. The European Union and the United Kingdom concluded a trade and cooperation agreement (“TCA”) that entered into force on May 1, 2021. Among other areas, the TCA includes provisions affecting the life sciences sector (including on customs and tariffs) but areas for further discussion between the EU and UK remain. In addition, there are some specific provisions concerning pharmaceuticals. These include the mutual recognition of GMP, inspections of manufacturing facilities for medicinal products and GMP documents issued. The TCA does not, however, contain wholesale mutual recognition of UK and EU pharmaceutical regulations and product standards. In the United States, marketing and promotion of drug products is regulated by the FDA and the Federal Trade Commission (“FTC”) to ensure that any claims about such products are consistent with regulatory approvals, not misleading or false in any particular way, and adequately substantiated by clinical data. In the United States and the European Union, the promotions of a drug product in a manner that is false, misleading, unsubstantiated, or for unapproved (or off-label) uses may result in enforcement letters or notices, inquiries and investigations, and civil and criminal sanctions by the FDA, FTC and regulatory authorities of the EU Member States (as applicable). Promotion of products for off-label uses in the United States can also result in false claims litigation under federal and state statutes, which can lead to consent decrees, civil monetary penalties, restitution, criminal fines and imprisonment, and exclusion from participation in Medicare, Medicaid and other federal and state healthcare programs. Aegerion was the subject of such investigations and enforcement in connection with the investigations into Juxtapid and the related Aegerion Settlements. Cooperation with such investigations and negotiation of, and compliance with, the settlement has been and will continue to be costly and burdensome on our management and other resources.
Any future investigations and litigation involving us could have a material adverse effect on our business, reputation, financial condition, results of operations and prospects. It could also distract our management from operating the business and may be disruptive to employees, leading to further employee attrition. In addition, the Aegerion Settlements have impacted, and may continue to impact, our reputation and the willingness of some physicians to prescribe our licensed products.
If we fail to comply with UK, EU or U.S. privacy and data security laws and regulations, we may be subject to civil and criminal penalties and other liability.
We are also subject to laws and regulations covering data privacy and the protection of health-related and other personal information. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues which may affect our business, including recently enacted laws in many jurisdictions where we operate. The collection and use of personal health data in the European Union is governed by the provisions of the General Data Protection Regulation (EU) 2016/679 (“GDPR”). The GDPR, which is wide-ranging in scope and includes extraterritoriality provisions that apply to certain entities located outside of the European Union, imposes several requirements relating to the consent of the individuals to whom the personal data relates, the information provided to the individuals, notification of data processing obligations to the competent national data protection authorities and the security and confidentiality of the personal data, and substantial fines for breaches of the data protection rules.
The GDPR also imposes strict rules on the transfer of personal data out of the European Union to other countries (including the United States). Failure to comply with the requirements of the GDPR and the related national data protection laws of the EU Member States may result in large fines and other administrative penalties: a failure to comply could result in fines up to the greater of 4% of annual worldwide turnover for the preceding financial year or €20 million, whichever is greater, with infringements being grouped into tiers which attract different maximum fine levels. Turnover in this context may include not only the entity in breach but also other group entities. Recent enforcement actions against multinational companies have resulted in significant fines.
Following the UK's withdrawal from the European Union and the EEA, companies have to comply also with the UK’s data protection laws (the UK GDPR, which is based on the EU GDPR), which has the ability to separately fine up to the greater of £17.5 million or 4% of global turnover.
While we have taken steps to comply with the EU GDPR and the UK GDPR, we cannot assure you that our efforts to achieve and remain in compliance have been or will continue to be fully successful. The GDPR regulations may impose additional responsibility and liability in relation to personal data that we process and we may be required to put in place additional mechanisms ensuring compliance with these or new data protection rules. This may be onerous and adversely affect our business, financial condition, results of operations and prospects.
In addition, we obtain patient health information from most healthcare providers that prescribe our products and research institutions with which we collaborate, and they are subject to privacy and security requirements under the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) in the United States. Although we are not directly subject to HIPAA other than with respect to providing certain employee benefits, we could potentially be subject to criminal penalties if we knowingly obtain or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA.
Failure to comply with healthcare laws and laws and regulations covering data privacy and the protection of health-related and other personal information could result in government enforcement actions, which could include civil or criminal penalties, private litigation and adverse publicity and could negatively affect our business, financial condition, results of operations and prospects.
Our relationships with customers and payers in the United States are subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, any breaches of which could expose us to criminal sanctions, civil penalties, contractual damages and reputational harm, could diminish future earnings and could prevent us from achieving our expected financial results.
Our arrangements with third-party payers and customers in the United States expose us to broadly applicable fraud and abuse and other healthcare laws and regulations, including the federal healthcare Anti-Kickback Statute, the False Claims Act, HIPAA and the Physician Payment Sunshine Act, and similar state and foreign laws and regulations that may regulate the business or financial arrangements and relationships through which we market, sell and distribute our products. The number and complexity of both federal and state laws continue to increase, and additional governmental resources are being used to enforce these laws and to prosecute companies and individuals who are believed to be violating them.
In March 2010, the PPACA was passed, which substantially changes the way healthcare is financed by both the government and private insurers, and significantly impacts the U.S. pharmaceutical industry. The PPACA, among other things: (i) addresses a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; (ii) increases the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extends the rebate program to individuals enrolled in Medicaid managed care organizations; (iii) establishes annual fees and taxes on manufacturers of certain branded prescription drugs; (iv) expands the availability of lower pricing under the 340B drug pricing program by adding new entities to the program; and (v) establishes a new Medicare Part D coverage gap discount program effective 2019, in which manufacturers must agree to offer 70% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D. The PPACA also includes a number of provisions aimed at strengthening the government’s ability to pursue anti-kickback and false claims cases against pharmaceutical manufacturers and other healthcare entities, including substantially increased funding for healthcare fraud enforcement activities, enhanced investigative powers, and amendments to the False Claims Act that make it easier for the government and whistleblowers to pursue alleged violations of the Anti-Kickback Statute, the Federal Food, Drug, and Cosmetic Act (“FDCA”), the False Claims Act and other relevant laws. While the evolving nature of this regulatory framework makes it difficult to predict what effect the framework and any recent or future changes will have on our business, we anticipate that government scrutiny of pharmaceutical sales and marketing practices will continue for the foreseeable future, and the risk of government investigations and enforcement actions will continue. For example, federal enforcement agencies recently have shown interest in, and engaged in enforcement actions against, pharmaceutical companies’ product and patient assistance programs, including manufacturer reimbursement support services and relationships with specialty pharmacies, as well as contributions by companies to third-party 501(c)(3) charitable organizations that assist patients in accessing treatment for certain diseases and conditions. This was also a part of the Juxtapid investigations, including certain contributions that were not resolved in the Aegerion Settlements. Some of these investigations have resulted in significant civil and criminal settlements. Responding to a government investigation or enforcement action would be expensive and time consuming and could have a material adverse effect on our reputation, business, financial condition, results of operations and prospects.
Anti-bribery rules in many jurisdictions also prohibit the offer of kick-backs and other inappropriate inducements to prescribe.
We are subject to the UK Bribery Act, the U.S. Foreign Corrupt Practices Act, and other anti-corruption laws, export control laws, import and customs laws, trade and economic sanctions laws and other laws which govern our operations.
Our operations are subject to anti-corruption laws, including the UK Bribery Act, the U.S. Foreign Corrupt Practices Act of 1977 (“FCPA”), the U.S. domestic bribery statute, the U.S. Travel Act, and other anti-corruption laws that apply in countries where we conduct business. The UK Bribery Act, the FCPA and other anti-corruption laws generally prohibit us and our employees and intermediaries from authorizing, promising, offering or providing, directly or indirectly, improper or prohibited payments, or anything else of value, to government officials or other persons to obtain or retain business or gain some other business advantage. Under the UK Bribery Act, we may also be liable for failing to prevent a person associated with us from committing a bribery offense. We and our commercial partners operate in a number of jurisdictions that pose a high risk of potential UK Bribery Act or FCPA violations, and we also participate in collaborations and relationships with third parties whose corrupt or illegal activities could potentially subject us to liability under the UK Bribery Act, FCPA or local anti-corruption laws, even if we did not explicitly authorize or have actual knowledge of such activities. In addition, we cannot predict the nature, scope or effect of future regulatory requirements on our international operations or the manner in which existing laws might be administered or interpreted.
We are also subject to other laws and regulations governing our international operations, including regulations administered by the governments of the United Kingdom and the United States, and authorities in the European Union, including applicable export control regulations, economic sanctions and embargoes on certain countries and persons, anti-money laundering laws, import and customs requirements and currency exchange regulations (collectively, “Trade Control Laws”).
There is no assurance that we will be completely effective in ensuring compliance with all applicable anti-corruption laws, including the UK Bribery Act and the FCPA, or other legal requirements, including Trade Control Laws. If we are not in compliance with the UK Bribery Act, the FCPA and other anti-corruption laws or Trade Control Laws, we may be subject to criminal and civil penalties, disgorgement and other sanctions and remedial measures, and legal expenses, which could have an adverse effect on our business, financial condition, results of operations and liquidity. Likewise, any investigation of any potential violations of the UK Bribery Act, the FCPA, other anti-corruption laws or Trade Control Laws by the United Kingdom, United States, or other authorities could also have an adverse impact on our reputation, business, financial condition, results of operations and prospects.
If we fail to comply with our reporting and payment obligations under the Medicaid Drug Rebate Program or other governmental pricing programs in the United States, we could be subject to additional reimbursement requirements, penalties, sanctions and fines which could materially and adversely affect our business, financial condition, results of operations and prospects.
We participate in various government programs and contracts that require us to calculate and report certain prices for our products to government agencies or provide rebates or discounted pricing on products purchased to certain purchasers or government payers. The requirements for calculating prices and rebates are complex and subject to change. Changes to such requirements may affect our business and operations. We may also have reimbursement obligations or be subject to penalties if we fail to provide timely and accurate information to the government, pay the correct rebates or offer the correct discounted pricing.
To maintain coverage of products under the Medicaid Drug Rebate Program and Medicare Part B, we will be required to extend significant discounts to certain “covered entities” (defined by statute to include certain types of hospitals and other healthcare providers that receive federal grants) that purchase products under the 340B Program. The 340B Program requires participating manufacturers to agree to charge such covered entities no more than the 340B “ceiling price” for the manufacturers’ covered outpatient drugs. The 340B ceiling price is calculated using a statutory formula, which is based on the average manufacturer price and rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate Program. Orphan drugs, such as Myalept and Juxtapid, are exempt from the ceiling price requirements for drugs purchased by certain covered entities (i.e., rural referral centers, sole community hospitals, critical access hospitals and freestanding cancer hospitals). Because of our participation in the Medicaid Drug Rebate Program, we are required to pay a rebate for each unit of metreleptin or lomitapide reimbursed by a state Medicaid program as a condition to avail us of federal funds to the states for our drugs under Medicaid and Medicare Part D.
Pricing and rebate calculations vary among products and programs. The calculations are complex and often subject to interpretation by governmental or regulatory agencies and the courts. For example, the Medicaid rebate amount is computed each quarter based on our submission to the Centers for Medicare & Medicaid Services (“CMS”) of our average manufacturer price and best price for the quarter. If we become aware that reporting for prior quarters was incorrect or has changed as a result of recalculation of pricing data, we must resubmit the corrected data for a period not exceeding 12 quarters from the quarter in which the data was originally due. Such restatements and recalculations would serve to increase our costs for complying with the laws and regulations governing the Medicaid Drug Rebate Program. Any corrections to our rebate calculations could result in an overage or underage in our rebate liability for past quarters, depending on the nature of the correction. Price recalculations also may affect the price that we will be required to charge certain safety net providers under the Public Health Service 340B drug discount program.
If we fail to comply with our reporting and payment obligations under the Federal Supply Schedule pricing program, we could be subject to penalties.
Federal Supply Schedule (“FSS”) contracts are federal procurement contracts that include standard government terms and conditions, separate pricing for each product, and extensive disclosure and certification requirements. All federal agencies and some non-federal entities are authorized to access FSS contracts. FSS contractors are permitted to charge FSS purchasers other than the four federal agencies “negotiated pricing” for covered drugs that is not capped by the statutory Federal Ceiling Price (“FCP”); instead, such pricing is negotiated based on a mandatory disclosure of the contractor’s commercial “most favored customer” pricing. Moreover, all items on FSS contracts are subject to a standard FSS contract clause that requires FSS contract price reductions under certain circumstances where pricing to an agreed “tracking” customer is reduced.
In July 2016, Aegerion concluded negotiations with the Department of Veterans Affairs (“VA”), and on August 15, 2016, Aegerion secured an FSS contract for both Myalept and Juxtapid. Under this program, we are obligated to make Myalept and Juxtapid available for procurement on an FSS contract at a negotiated price and also charge a price to four federal agencies (the VA, Department of Defense, Public Health Service and Coast Guard) that is no higher than the statutory FCP, which we calculate and report to the VA on a quarterly and annual basis. If we fail to comply with our reporting and payment obligations under the FSS pricing program, we could be subject to penalties in the future, which may have a material and adverse effect on our business, financial condition and results of operations.
If we overcharge U.S. government payers for our products, or if we fail to correctly and timely report state drug pricing changes, we may be found liable and could be subject to penalties for errors associated with the submission of pricing data.
To maintain coverage of products under the Medicaid Drug Rebate Program and Medicare Part D and when purchased by the Department of Veterans Affairs, Department of Defense, Public Health Service and Coast Guard, typically a pharmaceutical company must participate in the FSS.
In addition to the FSS contract with the VA, we also participate in the Tricare Retail Pharmacy program, under which we pay quarterly rebates on utilization of Myalept and Juxtapid when the products are dispensed through the Tricare Retail Pharmacy network to Tricare beneficiaries.
If we are found to have knowingly submitted false average manufacturer price or best price information to the government, we may be liable for civil monetary penalties in the amount of $100,000 per item of false information. Failure by us to submit monthly or quarterly average manufacturer price and best price data on a timely basis could result in a civil monetary penalty of $10,000 per day for each day the submission is late beyond the due date. In the event that CMS were to terminate the rebate agreement, no federal payments would be available under Medicaid or Medicare Part D for our products. In addition, if we overcharge the government in connection with the FSS contract or under any other government program, we will be required to refund the difference to the government. Failure to make necessary disclosures or to identify contract overcharges could result in allegations against us under the federal civil False Claims Act and other laws and regulations.
If we fail to correctly and timely report state drug pricing changes on a State by State basis as applicable, we may be liable to pay fines which may have a material and adverse effect on our business, financial condition and results of operations.
Risks Related to our Reliance on Third Parties
We rely on third parties to conduct clinical trials and registry studies and perform related services, and those third parties may not perform satisfactorily, including by failing to meet established deadlines for the completion of such clinical trials and compliance with post-marketing requirements.
We do not have the resources to independently conduct clinical trials or registry studies or perform pharmacovigilance and REMS program and other risk management plan monitoring and reporting, and we rely on third parties, such as contract research organizations, medical institutions, academic institutions, clinical investigators, specialty pharmacies and other third-party service providers, to perform these functions. Reliance on third parties for these functions reduces our control over such functions. However, if we sponsor clinical trials, we are responsible for ensuring that each of the sponsored clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Our reliance on third parties does not relieve us of these responsibilities and requirements. Furthermore, these third parties may have relationships with other entities, some of which may be our competitors.
If the third parties we rely upon fail to successfully carry out their contractual duties or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the data they provide is compromised or delayed due to the failure to adhere to regulatory requirements or clinical trial protocols, or for other reasons, our current marketing authorizations may be revoked, suspended, or revised to be more stringent. Further, our development programs, including any potential clinical studies, may be extended, delayed or terminated. If we were to experience an unexpected loss of supply of any of our product candidates or any of our future product candidates for any reason, whether as a result of manufacturing, supply or storage issues or otherwise, we could experience delays, disruptions, suspensions or terminations of, or be required to restart or repeat, any pending or ongoing clinical trials. Additional marketing approvals for metreleptin, Mycapssa® or lomitapide may be delayed or denied in the targeted indication or jurisdiction, and efforts to successfully commercialize Oleogel-S10 if approved, metreleptin, Mycapssa, lomitapide, or any other product for targeted indications or in the targeted jurisdiction may be delayed or unsuccessful. Should this occur, any existing approvals could be negatively impacted, which could materially and adversely affect our commercialization efforts.
We depend on third-party manufacturers to produce the drug substance and the drug product for lomitapide, Mycapssa® and metreleptin sold globally, as well as the drug product for commercial supply and clinical trials. We also depend on third-party manufacturers to produce the drug product for Oleogel-S10. Even though we have reserve stock, interruption in supply could materially and adversely affect sales.
We have limited internal manufacturing facilities for the production of the active pharmaceutical ingredient in Oleogel-S10. We employ a small number of personnel with manufacturing experience but we are currently dependent upon contract manufacturers to produce metreleptin, Mycapssa® and lomitapide and the drug product for commercial supplies and clinical trials, including for Oleogel-S10, if it is approved.
Aegerion entered into long-term supply agreements with a manufacturer of metreleptin drug substance and a manufacturer of metreleptin drug product. In February 2017, the original contract manufacturer for metreleptin drug product received a warning letter from the FDA citing significant violations of current Good Manufacturing Practice (“cGMP”) regulations at the manufacturing facility where metreleptin drug product was manufactured. We do not have any other agreements in place for redundant supply or a second source for drug substance or drug product for our products. Any interruption, delay or failure to supply our drug product or drug substance for our products from any of our third-party manufacturers could have a material and adverse impact on our sales, depending upon the length of interruption, which in turn may have a material adverse effect on our business, financial condition, results of operations and prospects.
If we are unable to maintain arrangements for third-party manufacturing, are unable to do so on commercially reasonable terms, or are unable to obtain timely regulatory approvals in connection with contract manufacturers, we may not be able to complete development of our product candidates or successfully commercialize our products. We may incur significant added costs and substantial delays in identifying and qualifying any replacement manufacturers, and in obtaining regulatory approval to use such replacement manufacturer in the manufacture of the products. Any such delays could result in significant delay in the supply of drug product for an ongoing clinical trial due to the need to replace a third-party manufacturer and could delay completion of the trial. If for any reason we are unable to obtain adequate supplies of lomitapide, Mycapssa® or metreleptin, or the drug substances used to manufacture them, it will be more difficult or impossible for us to compete effectively, generate revenues, meet expectations for financial performance and further develop our products. In addition, if we are unable to assure a sufficient quantity of the drug for patients with rare diseases or conditions, we may lose any Orphan Drug exclusivity to which our product otherwise would be entitled.
We are subject to minimum production levels with some of these manufacturing companies. If we cannot sell sufficient levels of metreleptin, Mycapssa® or lomitapide, this will result in excess levels of inventory being manufactured and resulting obsolescence.
If our third-party manufacturers are unable to comply with applicable regulatory requirements, unable to source sufficient raw materials, experience manufacturing or distribution difficulties, or are otherwise unable to manufacture and distribute sufficient quantities to meet demand, our commercialization efforts may be materially harmed.
While we manufacture the drug substance for Oleogel-S10 on our own, we do not own or operate manufacturing facilities for the production of clinical or commercial supplies of our products. We have limited personnel with experience in drug manufacturing and lack the resources and the capabilities to manufacture any of our product candidates on a clinical or commercial scale. We currently rely on third parties for supply of drug substances and drug products, and our strategy is to outsource all manufacturing of our product candidates and products to third parties.
We rely on contract manufacturers to consistently produce drug substances and drug products to required specifications, including those imposed by the FDA, the EMA and other regulatory authorities. There can be no assurance that our contractors will consistently be able to produce commercial supplies of drug substance or drug product meeting the approved specifications. A number of factors could cause production interruptions at the facilities of the contract manufacturers, including equipment malfunctions, facility contamination, labor problems, raw material shortages or contamination, the impact of the COVID-19 pandemic, natural disasters, disruption in utility services, terrorist activities, regulatory actions resulting from failure to comply with cGMP, human error or disruptions in the operations of the suppliers. Aegerion experienced failures by third-party manufacturers to produce products that meet specifications in the past, and any future failure by third-party manufacturers to produce products that meet specifications could lead to a shortage of lomitapide, Mycapssa® or metreleptin which could have a material and adverse impact on our sales, depending upon the length of interruption delay or failure, which in turn may have a material adverse effect on our business, financial condition, results of operations and prospects.
The FDA, the EMA and other regulatory authorities require that drug products be manufactured according to cGMPs relating to methods, facilities and controls used in the manufacturing, processing and packaging of the product, which are intended to ensure that drug products are safe and that they consistently meet applicable requirements and specifications. Our contract manufacturers are subject to periodic announced and unannounced inspections by the FDA to assess compliance with cGMP requirements. If an FDA inspection on a manufacturer’s facilities reveals conditions that the FDA determines do not comply with applicable regulatory requirements, the FDA may issue observations through a Notice of Inspectional Observations, commonly referred to as a “Form FDA 483” report. If observations in the Form FDA 483 Report are not addressed in a timely manner and to the FDA’s satisfaction, the FDA may issue a Warning Letter or proceed directly to other forms of enforcement action. Any failure by third-party manufacturers to comply with cGMP or to provide adequate and timely corrective actions in response to deficiencies identified in a regulatory inspection could result in further enforcement action that could lead to a shortage of products and harm our business, including to withdrawal of approvals previously granted, seizure, injunction or other civil or criminal penalties. The failure of any third-party manufacturer to address any concerns raised by the FDA or foreign regulators could also lead to plant shutdown or the delay or withholding of product approval by the FDA in additional indications, or by foreign regulators in any indication. Certain countries may impose additional requirements on the manufacturing of drug products or drug substances, and on third-party manufacturers, as part of the regulatory approval process for products in such countries. The failure by us or our third-party manufacturers to satisfy such requirements could impact our ability to obtain or maintain approval of our products in such countries.
The manufacture of biologic pharmaceuticals, such as metreleptin, is more difficult and riskier than the manufacture of small molecule pharmaceuticals, such as lomitapide and Mycapssa. The process of manufacturing biologics is highly susceptible to product loss due to contamination, oxidation, equipment failure or improper installation or operation of equipment, or vendor or operator error. Even minor deviations from normal manufacturing processes could result in reduced production yields, product defects and other supply disruptions. If microbial, viral or other contaminations are discovered in metreleptin or the facilities of the contract manufacturer, we may need to cease the manufacture of metreleptin for an extended period of time to investigate and remediate the contaminant. A contamination, recall, raw material shortage, or other supply disruption could adversely impact or disrupt commercial manufacturing of metreleptin or could result in a withdrawal of metreleptin from the market. Any such disruption, in turn, could adversely affect our ability to satisfy demand for metreleptin, which could materially and adversely affect our business, financial condition, results of operations and prospects.
We may be unable to scale up our manufacturing capability or successfully outsource manufacturing to third-party contractors.
We may underestimate the cost, time or complexity of installing or operating the manufacturing equipment required for our business. There also is a risk that any new equipment may not function as expected once installed. We may decide to outsource future manufacturing needs to third parties but may be unable to find sufficient supply for our manufacturing needs. Any inability to successfully scale up manufacturing capability, including that of Oleogel-S10, or meet manufacturing need through third-party contractors may materially and adversely affect our business, financial condition, results of operations and prospects.
We rely on third parties for distribution services around the world, and a failure to manage these third parties could harm our business.
We use, and plan to use, third parties to provide warehousing, shipping, third-party logistics, invoicing, collections and other distribution services on our behalf in the United States and in other countries throughout the world. Our failure to establish, maintain and finance the capabilities to sell, market or distribute our products, either through our own commercial infrastructure or through arrangements with third parties, and to effectively manage such third parties, could result in us not being able to successfully sell our products and could, as a result, have a material adverse effect on business, financial condition, results of operations and prospects.
If our licensees do not successfully commercialize the licensed products, we will not receive anticipated royalties and other payments and our results of operations will suffer.
The product sales, milestone payments and royalties that we may be entitled to receive under our license arrangements with Shionogi and Recordati are dependent on such licensees’ ability to successfully develop and commercialize the licensed products for their respective indications. Our licensees may not succeed in their product development efforts. It is possible that our licensees may be unable to obtain regulatory approval of product candidates using our technologies or successfully market and commercialize any such products for which regulatory approval is obtained. We have no control over these licensees and the contractual provisions may not provide adequate remedies in the event that a licensee fails to satisfy its obligations and commitments under the licensing arrangements. In addition, we have limited experience in maintaining multiple licensing arrangements and may be unable to adequately administer or monitor, or comply with, these arrangements, especially in light of limited personnel and cash resources, and these or future license arrangements may require us to provide services, supply or other efforts to the licensee. For example, under the license agreement with Recordati, we are required to provide supply product to Recordati. Moreover, the licensees have the ability to terminate or elect not to renew their respective license agreement in certain circumstances, including if we materially breach the agreement. A decision by a licensee to terminate its relationship, or a failure by a licensee to successfully develop or commercialize the licensed products or indications, could materially and adversely affect our business, financial condition, results of operations and prospects.
Risks Related to our Intellectual Property
It may be challenging or costly for us to obtain, maintain, enforce and defend our intellectual property rights. Failure to obtain or protect these rights could adversely affect our business and our ability to compete.
Our success and ability to compete effectively are in large part dependent upon exploitation of proprietary technologies and product candidates that have been developed internally or have been acquired or in-licensed, our ability to protect and enforce our intellectual property rights so as to preserve our exclusive rights in respect of our technologies and product candidates, and our ability to preserve the confidentiality of our know-how. We rely primarily on exclusivity provided by a combination of Orphan Drug approval, data exclusivity, patent rights, trade secrets and confidentiality to protect our intellectual property rights. There can be no assurance that patents pending or future patent applications will be issued, or that the lack of any such patents will not have a material adverse effect on our ability to develop and market our proposed candidates, or that, if issued, we would have the resources to protect or enforce any such issued patent. Also, no assurance can be given that we will develop technologies or candidates that are patentable or that patents will be sufficient in their scope to provide protection for our products or intellectual property rights against third parties. Nor can there be any assurance as to the ownership, validity, patentability, enforceability or scope of any patents that have been, or may in the future be, issued to us or that claims with respect thereto will not be asserted by third parties. Furthermore, we may develop technology important to our businesses that we cannot successfully patent due to the existence of prior art. Lojuxta did not receive Orphan Drug approval in Europe and relies on data exclusivity and patent protection. This may make our reimbursement discussions more difficult. In addition, there can be no assurance that we will be able to obtain and/or maintain Orphan Drug Designation or Orphan Drug approval for our product candidates. For example, the EMA’s COMP reviews an orphan design of a product if it is approved for a marketing authorization; for a product to benefit from market exclusivity, it must maintain its orphan designation at the time of marketing authorization review by the EMA and approval by the EC.
The pharmaceutical industry is characterized by extensive litigation regarding patents and other intellectual property rights. For example, on August 28, 2015, the Coalition for Affordable Drugs VIII L.L.C. filed two separate inter partes review (“IPR”) petitions with the Patent Trial and Appeal Board (“PTAB”) of the U.S. Patent and Trademark Office, challenging the validity of U.S. Patent Nos. 7,932,268 and 8,618,135, which are directed to methods-of-use for lomitapide. Although the PTAB ruled in our favor, third parties may obtain patents in the future and allege that our products or the use of our technologies infringe their patent claims or that we are employing their proprietary technology and other intellectual property without authorization. Likewise, third parties may infringe upon our existing or future patents.
We also rely on copyright, trademark and trade secret laws, as well as confidentiality procedures and agreements, non-compete and work for hire invention assignment agreements and licensing arrangements with our employees, consultants, contractors, customers and vendors, to establish and protect our rights to our technology and other developments and, to the best extent possible, control the access to and distribution of our technology, software, documentation and proprietary information. We also have procedures and policies in place against the misuse of third party rights.
While we employ physical and electronic protective measures to safeguard our technology and other proprietary information, it may be possible for a third party to copy or otherwise obtain and use our technology without authorization. Once granted, a patent can be challenged both in the patent office and in the courts by third parties. Third parties can bring material and arguments, for example, that the patent office granting the patent may not have been aware of. Therefore, issued patents may be found by a court of law or by the patent office to be invalid or unenforceable or in need of further limitation.
In particular, our commercial success depends on our ability to obtain and maintain the following intellectual property protections:
| • | product, composition of matter, formulation and method of use patents in Europe, the United States and other key global markets for existing and future products; |
| • | Orphan Drug exclusivity granted to our products because they aim to treat rare diseases and conditions, which entitles us to: exclusivity protections for a period of up to seven years after approval in the United States (although metreleptin should also qualify for a 12-year period of exclusivity from biosimilar or interchangeable products) and up to ten years in the European Union and Japan; as well as certain financial incentives. The ten-year Orphan Drug exclusivity period in the European Union can be extended a further two years upon successful completion of a PIP. Conversely, the ten-year exclusivity period may be reduced to six years, if at the end of the fifth year, it is established that a product no longer fulfills the criteria for Orphan Drug Designation; |
| • | medicinal products granted a marketing authorization in the European Union entitles us to eight years’ data exclusivity after approval, and up to ten years’ market exclusivity protection which can be extended for a further year if a new indication is granted; and |
| • | available extensions to the terms of our Orphan Drug exclusivity, product and methods of use patents in Europe and United States. |
If we lose the competitive advantage provided by these intellectual property and other protections, we will not be able to generate sustainable revenues or profits from our product portfolio. If we do not adequately protect and enforce our intellectual property, competitors may erode or negate any competitive advantage we may have, which could materially harm our business and ability to achieve expected financial results.
We may infringe or be alleged to infringe the intellectual property rights of others, which may prevent or delay product development and commercialization efforts, requiring us to expend resources on litigation or other resolutions, which may materially and adversely affect our business.
Our success will depend in part on our ability to operate without infringing the intellectual property and other proprietary rights of third parties. Identification of third-party patent rights that may be relevant to our products and proprietary technology is difficult due to differences in terminology among patents, incomplete databases and the difficulty and uncertainty in assessing the meaning of patent claims. There could be issued patents of which we are or were not aware that our products infringe. There also could be patents that we believe our products do not infringe, but that our products may ultimately be found to infringe. Moreover, a patent application may be maintained in secrecy until a patent on the application is issued. The publication of discoveries in the scientific or patent literature frequently occurs later, often substantially later, than the date on which the underlying discoveries were made and patent applications were filed. Because patents can take many years to issue, there may be currently pending applications of which we are unaware that may later result in issued patents that our products will be found to infringe. For example, there may exist pending applications that provide support or can be amended to recite a claim that is granted and which our products are later found to infringe. Proceedings involving our patents or patent applications or those of others could:
| • | put one or more of our patents at risk of being invalidated, rendered unenforceable or interpreted narrowly; |
| • | adversely impact our ability to obtain patent protection for our inventions relating to our products; |
| • | result in monetary damages, injunctive relief or other harm to our competitive position, including by limiting marketing and selling activities, increasing the risk for generic competition, limiting development and commercialization activities or requiring us to obtain licenses to use the relevant technology (which licenses may not be available on commercially reasonable terms, if at all); and |
otherwise negatively impact the enforceability, validity or scope of protection provided by the patents relating to the products.
We may not have the resources to adequately defend claims by third parties in one or more such proceedings, and even if successful in any such proceedings, we would incur substantial costs and divert management’s time and attention in pursuing these proceedings, putting further strain on our resources, which could have a material adverse effect on our business, financial condition, results of operations and prospects. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, defend an infringement action or challenge the validity of the patents in court or another venue. Patent litigation is costly and time consuming. We may not have sufficient resources to bring these actions to a successful conclusion.
In addition, if we do not obtain a license, develop or obtain non-infringing technology, fail to defend an infringement action successfully or have infringed patents declared invalid, we may:
| • | incur substantial monetary damages; |
| • | encounter significant delays in expanding the market of our products; and |
| • | be precluded from manufacturing or selling any products; |
which, in each case, could have a material adverse effect on our business, financial condition, results of operations and prospects.
Our existing patent protections will expire and protection for these rights may not be extended.
We depend on patent protection to provide exclusive marketing rights for our products. Loss of patent protection for a product typically leads to a significant and rapid loss of sales for that product as lower-priced generic versions of that drug become available. The expiration of patent protection for any product that contributes significantly to our sales can have a material and adverse effect on our business, cash flow, results of operations, financial position and prospects.
The Oleogel-S10 patent portfolio includes two U.S. patents covering oleogel compositions and three U.S. patents covering treatment of EB. One U.S. patent covering oleogel compositions provides exclusivity in the United States into 2025 (compositions) and one U.S. patent covering oleogel compositions provides U.S. exclusivity until 2039. Two U.S. patents covering the treatment of EB provide exclusivity in the U.S. until 2030 and one U.S. patent covering the treatment of EB provides U.S. exclusivity until 2039. The existing Orphan Drug designation for Oleogel-S10 would extend Orphan Drug exclusivity for Oleogel-S10 seven years from the approval date in the United States.
The non-U.S. patents for Oleogel-S10 include European patents covering the Oleogel-S10 composition and methods of healing wounds with Oleogel-S10. Supplementary protection certificates have been obtained in various EU countries, extending the expiration of the composition patents in the European Union from 2025 to 2030. The European method of use patents expire in 2030. There are pending applications covering the Oleogel S10 composition and its use in the treatment of EB in Australia, Brazil, Canada, China, Colombia, Europe, India, Israel, Japan, South Korea, Mexico, Russia, Singapore and South Africa, which if granted would expire in 2039.
The AP103 patent portfolio includes one granted European patent (validated in various European Patent Organisation member states including the United Kingdom, Germany, France, Italy and Spain) claiming highly branched poly β-amino ester (“HPAE”) polymers and polyplexes comprising HPAE polymers, and transfection methods using such polymers, as well as pending U.S. and European applications. These patents and applications have been in-licensed from the University College Dublin and are expected to provide exclusivity into 2035. We have filed U.S. and European applications covering a proprietary genus of HPAEs and novel HPAE polyplexes, as well as U.S. and European applications claiming a proprietary and scalable HPAE polyplex manufacturing methods, which if granted would expire in 2039.
Our lomitapide patent portfolio includes eight Approved Drug Products with Therapeutic Equivalence Evaluations (“Orange Book”) listed patents licensed from the University of Pennsylvania (“UPenn”) claiming methods of treatment that provide protection from 2025 to 2027 in the United States and into 2025 in Europe, Australia, Japan, South Korea, Canada and New Zealand. Applications licensed from UPenn covering lomitapide methods of treatment are pending in the U.S. and India, which if granted would expire in 2025. Supplementary protection certificates (“SPCs”) have been granted in 26 European countries (including the United Kingdom, Germany, France, Italy and Spain) extending patent protection into 2028. Two additional SPCs applications are pending.
The metreleptin patent portfolio includes two issued U.S. patents claiming methods of treating lipoatrophy, two granted European patents (“EP patents”) claiming methods of treatment (validated in numerous European countries, including the United Kingdom, Germany, France, Italy and Spain), granted patents claiming methods of treatment in other jurisdictions including Australia, Canada, and Japan, all of which have been licensed to Aegerion or are owned by Aegerion. The granted patents provide protection from 2022 to 2027 in the United States and into 2022 in Europe, Aegerion received approval for marketing authorization by the EC for metreleptin in the European Union, under the brand name Myalepta, as replacement therapy to treat complications of leptin deficiency in patients with GL and PL. Metreleptin has orphan exclusivity in the European Union until July 2028. The ten-year Orphan Drug exclusivity period in the European Union can be extended a further two years upon successful completion of a PIP.
Our Mycapssa® patent portfolio includes eight Approved Drug Products with Therapeutic Equivalence Evaluations (“Orange Book”) listed patents, including three formulation and one acromegaly use patents which expire in 2029, three acromegaly use patents which expire in 2036, one octreotide method of use patent expiring 2040, as well as one pending U.S. application which covers NET/VIP use which if allowed would expire in 2037. In Europe, there are two granted composition patents which expire in 2029, one pending acromegaly use application, which if allowed would expire 2036 and one NET/VIP use application which if allowed would expire in 2037.
The loss of patent protection could have a material adverse effect on our business, financial condition, results of operations and prospects.
Previously granted regulatory exclusivity will expire and we may not be entitled to such exclusivity in the future.
Orphan Drug Designation can confer various benefits, depending on the jurisdiction, including market exclusivity and regulatory and fiscal incentives. Because our products and product candidates treat rare diseases and conditions that affect relatively small patient populations, we depend on the financial incentives and market exclusivity provided by Orphan Drug Designation to generate revenue from our product portfolio and protect our products from competition from lower-priced generic versions of such drugs. The Orphan Drug Designation in the United States for Juxtapid for the treatment of HoFH expired on December 21, 2019, and Orphan Drug Designation for Myalept for treatment of GL expired in 2021. Aegerion also obtained Orphan Drug Designation for metreleptin for the treatment of PL in the United States, which provides seven years exclusivity after approval for this indication in the United States. In 2012, metreleptin was granted Orphan Drug Designation by the EC for the treatment of Barraquer-Simons syndrome, Berardinelli-Seip syndrome, Lawrence syndrome and familial PL, which is expected to expire ten years from approval of any marketing authorization(s) for such indication(s) by the EMA. The EC provided that at the end of the fifth year into the Orphan Drug exclusivity period it is established that metreleptin continues to fulfill the criteria for Orphan Drug Designation. If not, such exclusivity period is reduced to six years. Following approval by the EMA in July 2018, metreleptin was entitled to ten years of market and Orphan Drug exclusivity in the European Union. In the U.S. Mycapssa® may benefit from Orphan Drug exclusivity for seven years from approval in June 2020. The grant remains pending from FDA. Mycapssa® has been granted Orphan Designation in Europe for the treatment in acromegaly. Maintenance of this orphan designation according to the relevant requirements will be assessed by COMP during the ongoing MAA procedure. Upon the expiration of the Orphan Drug exclusivity periods for our products, we may face substantial competition from generic drugs which could materially and adversely affect the price we are able to receive for the products and its business. Furthermore, the Orphan Drug Designation may not be available in the future by virtue of a number of factors, such as changes in law or regulation, increased competition in the Orphan Drug market, clinical and technological developments, among others. The designation could also result in significant payments to meet ongoing compliance obligations. The expiration or loss of Orphan Drug Designation in respect of one of our products or future products means that financial incentives by governments and non-government entities may not be available, which may have a material adverse effect on our business, financial condition, results of operations and prospects.
We also rely on the data and market exclusivity granted to innovative medicinal products in the European Union to protect our products from competition from lower-priced generic versions of the drugs. Innovative medicinal products authorized in the European Union on the basis of a full marketing authorization application (as opposed to an application for marketing authorization that relies on data in the marketing authorization application for another, previously approved medicinal product) are entitled to eight years’ data exclusivity from the date of notification of the marketing authorization. During this period, applicants for approval of generics of these innovative products cannot rely on data contained in the marketing authorization application submitted for the innovative medicinal product. Innovative medicinal products are also entitled to ten years’ market exclusivity. During this ten-year period, no generic medicinal product can be placed on the EU market. The ten-year period of market exclusivity can be extended to a maximum of 11 years if, during the first eight of those ten years, the marketing authorization holder for the innovative product obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies.
We currently benefit from marketing and data exclusivity periods in the European Union. Metreleptin is entitled to up to ten years of market exclusivity in the European Union from its approval in July 2018, subject to possible reductions to six years when the marketing authorization is renewed if the product no longer meets the criteria for orphan designation. The product is also entitled to eight years' data exclusivity and ten years’ marketing exclusivity in the European Union from its date of authorization. Lomitapide is entitled to eight years’ data exclusivity and ten years’ marketing exclusivity in the European Union from July 31, 2013, the date of the EC’s approval of lomitapide. If the Committee for Orphan Medicinal Products (“COMP”) adopt a positive opinion of the maintenance of the Orphan designation Mycapssa® will be entitled to up to ten years of market exclusivity in the European Union from the date of EC decision. Upon the expiration of these data and market exclusivity periods, in relation to existing and new products we may face substantial competition from generic drugs which could have a material adverse effect on the price we are able to receive for the products and business, which may have a material and adverse effect on our business, financial condition, results of operations and prospects.
Our patents may be challenged, deemed unenforceable, invalidated or circumvented, and if we do not obtain or maintain patent protection for the products, our business may be materially harmed.
The patent positions of biotechnology and pharmaceutical companies involve complex legal and factual questions and, therefore, validity and enforceability cannot be predicted with certainty. U.S. patents and patent applications also may be subject to interference or derivation proceedings, ex parte re-examination, IPR and post-grant review proceedings and supplemental examination and may be challenged in district courts. Patents granted in certain other countries may be subjected to opposition, revocation, or the like before various authorities. These proceedings could result in either loss of a patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, derivation, re-examination, post-grant review, IPR, supplemental examination, opposition or revocation proceedings may be costly. We will be able to protect our proprietary rights against third parties only to the extent that our proprietary technologies are protected by valid and enforceable patents or are effectively maintained as trade secrets.
The degree of future protection for our products and proprietary rights is uncertain, and it cannot be guaranteed that:
| • | we will be able to successfully develop or commercialize our product in some countries before some or all of the relevant patents or regulatory exclusivity expire, or successfully develop or commercialize our product in countries where we do not have any such patent protection or exclusivity; |
| • | we or our licensors were the first to make the inventions claimed by each of the pending patent applications and patents; |
| • | we or our licensors were the first to file, or the first inventors to file, patent applications for these inventions; |
| • | others will not independently develop similar or alternative technologies or duplicate any of our technologies; |
| • | any of our pending patent applications or those that we have licensed will result in issued patents; |
| • | any of our patents or those we have licensed will be valid or enforceable; |
| • | we will be able to license the patents or pending patent applications necessary or desirable to enforce or protect our patent rights on commercially reasonable terms or at all; |
| • | any patents issued to us or to our licensors or collaborators will provide a basis for any additional commercially viable products, will provide us with any competitive advantages or will not be challenged by third parties; |
| • | we will be able to develop additional proprietary technologies that are patent eligible or patentable; |
| • | Orphan Drug exclusivity marketing rights for our products in the United States will be maintained, if, for example, the FDA determines in the future that the request for Orphan Drug Designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition. In addition, the FDA, and the EMA for the European Union, may subsequently approve products with the same active moiety for the same condition if the FDA or the EMA concludes that the later drug is safer, more effective, or makes a major contribution to patient care. Orphan Drug Designation neither shortens the development time nor regulatory review time of a drug nor gives the drug any advantage in the regulatory review or approval process; or |
| • | the patents of others will not have an adverse effect on our business. |
We enjoy only limited geographical protection with respect to certain patents.
Filing and prosecuting patent applications and defending patents covering product candidates in all countries throughout the world would be prohibitively expensive. Competitors may use our and our licensors’ technologies in jurisdictions where patent protection has not yet been obtained to develop their own products or may export infringing products to territories where enforcement rights are not as strong as in the United States or Europe. These products may compete with our product candidates, and our intellectual property rights may not be effective or sufficient to prevent such products from competing with our products. Patent applications may be issued in some non-U.S. jurisdictions with different scope as compared with the corresponding U.S. patent, or they may not be granted at all in view of differences in patent law and practice in jurisdictions around the world. Proceedings to enforce our patent rights in other jurisdictions, whether or not successful, could result in substantial costs and divert efforts and attention from other aspects of the business. They could also put our patents and patent applications at risk of being invalidated, denied or interpreted narrowly, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits or have damages or other remedies awarded to us, and any such remedies, if obtained, they may not be commercially meaningful. Accordingly, our intellectual property rights as enforced may be inadequate to obtain a significant commercial advantage and our efforts to protect our intellectual property rights may be unsuccessful or inadequate, which may adversely affect our ability to successfully commercialize our product candidates, which may have a material adverse effect on our business, financial condition, results of operations and prospects. Furthermore, while we intend to protect our intellectual property rights in our expected significant markets, we cannot ensure that we will be able to initiate or maintain similar efforts in all jurisdictions in which we may wish to market our product candidates. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate, which may have an adverse effect on our ability to successfully commercialize our drug candidates in all of our expected, significant international markets.
Some countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties or laws limiting the enforceability of patents against government agencies or government contractors. In those countries, the value of such patents may be materially diminished at least in those contexts. If we or any of our licensors are forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be adversely affected.
If we do not obtain protection under the Hatch-Waxman Amendments and similar non-U.S. legislation for extending the term of patents covering our product candidates, our ability to compete effectively could be impaired.
Depending upon the timing, duration and conditions of FDA marketing approval of our product candidates, one or more of our U.S. patents may be eligible for patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984 (“Hatch-Waxman Amendments”). The Hatch-Waxman Amendments permit a patent term extension of up to five years for a patent covering an approved product or method of use as compensation for patent term lost during product development and the FDA regulatory review process. A maximum of one patent may be extended per FDA approved product as compensation for the patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only those claims covering such approved drug product, a method for using it or a method for manufacturing it may be extended. Similar patent term extensions may be available in other jurisdictions. In the European Union, a Supplementary Protection Certificate may be applied for approval to recover some of the time lost between the patent application filing date and the date of first marketing authorization. However, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents, or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be less than requested. If we are unable to obtain patent term extensions or the term of any such extension is less than requested, the period during which we can enforce our patent rights for that product will be shortened and competitors may obtain approval to market competing products sooner. As a result, our revenue from applicable products could be reduced materially.
We applied for supplemental protection in the countries in which Lojuxta is approved, on a country-by-country basis, and have been granted SPCs in 26 European countries (including the United Kingdom, Germany, France, Italy and Spain) extending patent protection into 2028. Two additional SPCs applications are also pending.
If we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology and product portfolio could be significantly diminished.
We rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is adequate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to protect trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information, and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. For example, the FDA has periodically considered whether to make additional information, such as clinical information, publicly available on a routine basis, and the EMA has considered amplifying its disclosure rules as well. Changes in this regard could mean that information that we may consider to be a trade secret or otherwise confidential and proprietary may have to be publicly disclosed, and it is currently unclear how FDA and the EMA disclosure policies may change in the future. Costly and time consuming litigation could be necessary to enforce and determine the scope of proprietary rights and we may not have adequate resources at the time to enforce our rights. Failure to obtain or maintain trade secret protection could adversely affect our competitive business position and have a material adverse effect on our business, financial condition, results of operations and prospects.
If we fail to comply with our obligations in the license agreements for our products or are alleged to have breached such agreements, we could lose license rights that are important to our business, have to make additional payments to our licensors or become involved in costly litigation, which would further reduce cash resources.
Our existing license agreements impose various diligence, milestone payment, royalty, insurance, reporting, audit and other obligations on us. If we fail to comply with such obligations or encounters disagreements with its license partners, we could lose license rights that are important to the business. Further, if the license partners allege that a breach of any such license agreements has occurred, defense of such allegations, even if untrue, can be costly to pursue and could lead to litigation or costly settlements, which would be burdensome on our resources. Even if we were able to successfully settle any disagreements or disputes under the license agreements, the settlement arrangements are unlikely to foreclose additional claims from the licensing party or other third parties, and the terms of such settlement could have a material adverse effect on our business, financial condition, results of operations and prospects.
If we fail to comply with the obligations and restrictions under the license agreements, the applicable licensor may have the right to seek financial payments or damages and terminate the license, in which case we may not be able to market the products that are covered by the license, or the licensor might require amendments to the license agreement that are overly burdensome. Further, the terms of such license agreements are intensely negotiated, and therefore tend to be complex, which could lead to differing interpretations by the parties. Any breach or alleged breach (whether intentional or unintentional), or termination, of the license agreements applicable to our products, or any disagreements as to the application of the terms of the agreements with licensing partners, could expose us to considerable costs and expenses as such allegations, terminations or disputes can be costly to remedy or resolve (regardless of the merits), and may not be remedied or resolved in a manner favorable to us, or at all. Any breach or alleged breach of the quantitative or qualitative diligence provisions of these agreements could result in termination of the co-development agreement and loss of the program and also expose us to considerable costs and expenses and any disputes may not be remedied or resolved in a manner favorable to us, or at all. Any such difficulties with the license agreements could have a significant adverse effect on our business.
Risks Related to Ownership of our ADSs and our Nasdaq Listing
An active and liquid market for our securities may fail to develop, which could harm the market price of our ADSs.
Although our ADSs are listed on the Nasdaq, an active trading market for our ADSs may never be sustained. In the absence of an active trading market for our ADSs, investors may not be able to sell their ADSs at the time that they would like to sell.
The price and trading volume of our ADSs may be volatile, and purchasers of our ADSs could incur substantial losses.
The market price of our ADSs is likely to be volatile and could decline significantly. The stock market in general, and the market for biotechnology and emerging pharmaceutical companies in particular, have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. In addition, our securities have a relatively small public float and may be less liquid and more volatile than securities of companies with broader public ownership. The market price for our ADSs may be influenced by a variety of factors, including:
| • | actual or anticipated variations in our financial condition and operating results; |
| • | actual or anticipated changes in our growth rate relative to our competitors; |
| • | announcements of technological partnerships, innovations or new products by us or our competitors; |
| • | the success of competitive products or technologies; |
| • | changes in management and members of our Board; |
| • | changes in financial estimates or recommendations by securities analysts; |
| • | changes in the trading volume of our ADSs on the Nasdaq; |
| • | sales of our ADSs or ordinary shares by executive officers or future holders of our equity securities; |
| • | announcements or expectations of additional debt or equity financing efforts; |
| • | unanticipated losses or gains due to unexpected events, including events related to the success of our clinical trials or regulatory approvals; |
| • | significant acquisitions or business combinations, strategic partnerships, joint ventures or capital commitments by or involving us or our competitors; |
| • | changes in our accounting policies or practices; |
| • | disputes or other developments related to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| • | failure to integrate successfully the Aegerion or Chiasma businesses with ours or to realize anticipated benefits from the integration; |
| • | changes in government regulations, including any changes that may affect pricing or reimbursement; and |
| • | conditions in the financial markets or changes in general economic conditions. |
These and other market and industry factors may cause the market price and demand for our ADSs to fluctuate substantially.
In addition, the trading prices for common stock of other biopharmaceutical companies have been highly volatile as a result of the COVID-19 pandemic and the extent to which the continuing effects of the pandemic may impact our business and the prices of our ADSs in the future will depend on future developments, which are highly uncertain and cannot be predicted with confidence.
The Athyrium Funds hold a significant interest in our company and may be able to influence matters relating to our business.
The Athyrium Funds hold approximately 14% of our share capital. The Athyrium Funds are in a position to exert influence over matters requiring approval of the holders of our equity securities and will have the ability to block certain actions requiring shareholder approval, including (but not limited to) disapplication of preemption rights. We are not required to enter and have not entered into a relationship agreement with Athyrium or the Athyrium Funds to manage the relationship between them.
The interests of the Athyrium Funds may be different from the interests of other shareholders and, as a result, the Athyrium Funds’ interests in our ADSs, if of sufficient individual or aggregate size, or if aggregated in any circumstances, or should the Athyrium Funds convert their convertible debentures into ADSs, may permit them to effect certain transactions without the support of other holders of our equity securities, or delay or prevent certain transactions that are in the interests of other holders of our equity securities. Decisions made by Athyrium (on behalf of the Athyrium Funds) may therefore influence our business, financial condition, results of operations and prospects. These decisions may also conflict with the interest of other holders of our equity securities and could prevent other holders of our equity securities from receiving a premium on their securities. The market price of our ADSs may decline if Athyrium uses its influence in ways that are or may be adverse to the interests of other holders of our equity securities.
We will incur significant increased costs as a result of operating as a U.S. listed public company, and our management will be required to devote substantial time to new compliance initiatives.
As a U.S. publicly traded company, and particularly after we cease to be an “emerging growth company” as defined in the JOBS Act, we will incur significant legal, accounting and other expenses that we did not incur prior to the listing of our ADSs in the United States as a result of reporting requirements under the Exchange Act. In addition, the federal securities laws, including the Sarbanes-Oxley Act of 2002 (“Sarbanes-Oxley Act”), and rules subsequently implemented by the SEC and the Nasdaq Stock Market LLC have imposed various requirements on public companies, including requirements to file annual and event-driven reports with respect to our business and financial condition, and to establish and maintain effective disclosure and financial controls and corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time consuming and costly. For example, these regulations may make it more difficult to attract and retain qualified members of the Board and various corporate committees and obtaining director and officer liability insurance will be more expensive. We may not be able to produce reliable financial statements or file these financial statements as part of a periodic report in a timely manner with the SEC or comply with the Nasdaq listing requirements. In addition, we could make errors in our financial statements that could require us to restate our financial statements.
Shareholder activism, the current political environment, and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact the manner in which we operate our business in ways we cannot currently anticipate. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives.
We must maintain effective internal control over financial reporting, and if we are unable to do so, the accuracy and timeliness of our financial reporting may be adversely affected, which could have a material adverse effect on our business, investor confidence and market price.
As a U.S. listed public company, we are required to maintain effective internal control over financial reporting in order to accurately and timely report our results of operations and financial condition. As required by Section 404 of the Sarbanes-Oxley Act, we will engage in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants, and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented, and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that neither we nor our independent registered public accounting firm will be able to conclude within the prescribed timeframe that our internal control over financial reporting is effective as required by Section 404. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
We disclose changes made in our internal controls and procedures and our management is required to assess the effectiveness of these controls annually. However, for as long as we are an emerging growth company, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404. We could be an emerging growth company for up to five years. An independent assessment of the effectiveness of our internal controls over financial reporting could detect problems that our management’s assessment might not. Undetected material weaknesses in our internal controls over financial reporting could lead to restatements of our financial statements and require us to incur the expense of remediation.
If we fail to staff our accounting and finance function adequately or maintain internal control over financial reporting adequate to meet the requirements of the Sarbanes-Oxley Act, our business and reputation may be harmed. Moreover, if we are not able to comply with the applicable requirements of Section 404 in a timely manner, we may be subject to sanctions or investigations by regulatory authorities, including the SEC and the Nasdaq. Furthermore, if we are unable to conclude that our internal control over financial reporting is effective or if our independent registered public accounting firm identifies deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our ADSs and ordinary shares could decline, and we could be subject to sanctions or investigations by the SEC, the Nasdaq or other regulatory authorities. Failure to implement or maintain effective internal control systems as required of public companies could also restrict our access to the capital markets. The occurrence of any of the foregoing would also require additional financial and management resources.
We are an “emerging growth company” and the reduced disclosure requirements applicable to emerging growth companies may make our ADSs less attractive to investors and, as a result, adversely affect the price of the ADSs and result in a less active trading market for the ADSs.
We are an emerging growth company as defined in the JOBS Act, and we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include:
| • | only being required to present three years of audited financials and related discussion in Management’s Discussion & Analysis; |
| • | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act; |
| • | not being required to comply with any requirement that may be adopted by the PCAOB regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; |
| • | reduced disclosure obligations regarding executive compensation; and |
| • | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. |
We may choose to take advantage of some, but not all, of the available exemptions. For example, in this annual report, we have elected to rely on the exemption from the auditor attestation requirement and the exemption that permits only two years of financial statements to be presented. We may avail ourselves of these disclosure exemptions until we are no longer an emerging growth company. We cannot predict whether investors will find our ADSs less attractive because of our reliance on some or all of these exemptions. If investors find our ADSs less attractive, it may adversely affect the price of the ADSs and there may be a less active trading market for the ADSs.
We will cease to be an emerging growth company upon the earliest of:
| • | the last day of the fiscal year during which we have total annual gross revenues of $1.07 billion (as such amount is indexed for inflation every five years by the SEC) or more; |
| • | the date on which we have, during the previous three-year period, issued more than $1.0 billion in non-convertible debt; |
| • | the date on which we are deemed to be a “large-accelerated filer,” as defined in Rule 12b-2 of the Exchange Act, which would occur if the market value of our ADSs that are held by non-affiliates exceeds $700 million as of the last day of our most recently completed second fiscal quarter; and |
In addition, under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. If we are unable to comply timely with these accounting standards, we may be delayed in providing the disclosures required by the Exchange Act.
As a “foreign private issuer,” we are exempt from a number of rules under the U.S. securities laws and the Nasdaq rules, and we are permitted to file less information with the SEC than are U.S. companies. In addition, we are permitted and expect to follow certain home country corporate governance practices in lieu of certain Nasdaq requirements applicable to domestic issuers. This may make our ADSs and ordinary shares less attractive to investors.
We are a “foreign private issuer,” as defined in the rules and regulations of the SEC, and, consequently, we are not subject to all of the disclosure requirements applicable to companies organized within the United States For example, we are exempt from certain rules under the Exchange Act that regulate disclosure obligations and procedural requirements related to the solicitation of proxies, consents or authorizations applicable to a security registered under the Exchange Act. In addition, our officers and directors are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and related rules with respect to their purchases and sales of our securities. Moreover, we are not required to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. public companies. Accordingly, there may be less publicly available information concerning our company than there is for U.S. public companies.
As a foreign private issuer we are permitted to follow certain home country corporate governance practices in lieu of certain Nasdaq requirements. The rights of holders of ordinary shares and, therefore, certain of the rights of holders of the ADSs, are governed by English law, including the provisions of the Companies Act, and by our Articles of Association, which may provide less protection than is afforded to investors under Nasdaq rules applicable to domestic issuers.
In particular, we expect to follow English law instead of Nasdaq practice in the following ways:
| • | We do not intend to follow Nasdaq Rule 5620(c) regarding quorum requirements applicable to meetings of shareholders. Such quorum requirements are not required under English law. In accordance with generally accepted business practice, our Articles of Association provide alternative quorum requirements that are generally applicable to meetings of shareholders. |
| • | We do not intend to follow Nasdaq Rule 5605(b)(2), which requires that independent directors regularly meet in an executive session, where only independent directors are present. The independent directors may choose to meet in an executive session at their discretion. |
Although we may rely on certain home country corporate governance practices, we must comply with Nasdaq Rule 5625, Notification of Noncompliance and Rule 5640, Voting Rights. Further, we must have an audit committee that satisfies Rule 5605(c)(3), which addresses audit committee responsibilities and authority, and that consists of committee members who meet the independence requirements of Rule 5605(c)(2)(A)(ii).
We intend to take all actions necessary to maintain compliance as a foreign private issuer under the applicable corporate governance requirements of the Sarbanes-Oxley Act, the rules adopted by the SEC and the Nasdaq corporate governance rules and listing standards.
It is possible that we will be a passive foreign investment company for U.S. federal income tax purposes.
A non-U.S. corporation will be classified as a passive foreign investment company (a “PFIC”) for U.S. federal income tax purposes for any taxable year if either:
| • | at least 75% of its gross income is “passive income,” or |
| • | at least 50% of the value, determined on the basis of a quarterly average, of its gross assets is attributable to assets that produce or are held for the production of “passive income.” |
Passive income generally includes dividends, interest, royalties, rents (other than certain rents and royalties derived in the active conduct of a trade or business), annuities and gains from assets that produce passive income. Cash is a passive asset for PFIC purposes, even if held as working capital. In addition, a non-U.S. corporation will be treated as owning its proportionate share of the assets and earning its proportionate share of the income of any other corporation in which it owns, directly or indirectly, no more than 25% (by value) of the stock.
Based on the composition of our income and assets, we do not believe that we were a PFIC in 2021 and do not expect to be a PFIC for U.S. federal income tax purposes for the current taxable year or any future taxable year. This conclusion is a factual determination, however, that is made annually and thus may be subject to change. Because PFIC status is determined based on the composition of our income and assets annually and generally cannot be determined until the end of the taxable year, there can be no assurance that we will not be a PFIC for the current taxable year or any future taxable year.
If we are a PFIC for any taxable year during which you own ADSs and ordinary shares, and you are a U.S. Holder (as defined in this annual report in the section titled “Income Tax Considerations”), you will generally be subject to additional taxes and interest charges on the gain from a sale of ADSs or ordinary shares, and upon receipt of an “excess distribution” with respect to the ADSs or ordinary shares. In general, a U.S. Holder would receive an “excess distribution” if the amount of any distribution for U.S. federal income tax purposes in respect of the ADSs and ordinary shares were more than 125% of the average distributions made with respect to the ADSs and ordinary shares within the three preceding taxable years (or shorter period if such U.S. Holder held the ADSs or ordinary shares for a shorter period). A U.S. Holder of a PFIC generally may mitigate these adverse U.S. federal income tax consequences by making a “qualified electing fund” election or a “mark-to-market” election. However, there is no assurance that we would provide information that would enable a U.S. Holder to make a qualified electing fund election. In addition, as a PFIC, dividends in respect of the ADSs or ordinary shares would not be eligible for the special tax rate available to non-corporate U.S. Holders applicable to “qualified dividend income.” U.S. owners of ADSs or ordinary shares should consult their own U.S. tax advisors regarding the potential application of the PFIC rules.
Future sales, or the possibility of future sales, of a substantial number of our ADSs could adversely affect the price of such securities.
Future sales of a substantial number of our ADSs or the perception that such sales will occur, could cause a decline in the market price of ADSs. As of December 31, 2021, we had 319,814,747 ordinary shares issued and outstanding including 219,206,010 ordinary shares represented by ADSs. If holders sell substantial amounts of shares in the public markets, or if the market perceives that such sales may occur, the market price of our ADSs and our ability to raise capital through an issue of equity securities in the future could be adversely affected.
Holders of our ADSs have fewer rights than our ordinary shareholders and are subject to certain additional risks.
ADS holders do not hold ordinary shares directly and, as such, are subject to, among others, the following additional risks:
| • | As an ADS holder, we will not treat you as one of our shareholders and you will not be able to exercise shareholder rights, except through the depositary as permitted by the deposit agreement. |
| • | Distributions on the ordinary shares represented by your ADSs will be paid to the depositary, and before the depositary makes a distribution to you on behalf of your ADSs, any withholding taxes that must be paid will be deducted. Additionally, if the exchange rate fluctuates during a time when the depositary cannot convert the foreign currency, you may lose some or all of the value of the distribution. |
| • | We and the depositary may amend or terminate the deposit agreement without the ADS holders’ consent in a manner that could prejudice ADS holders. |
ADS holders must act through the depositary to exercise their voting rights and, as a result, may be unable to exercise their voting rights on a timely basis.
We will not treat holders of our ADSs (rather than the ordinary shares underlying the ADSs) as shareholders, and they will not be able to exercise shareholder rights. The depositary will be the holder of the ordinary shares underlying the ADSs, and ADS holders will be able to exercise voting rights with respect to the ordinary shares represented by the ADSs only in accordance with the deposit agreement relating to the ADSs. There are practical limitations on the ability of ADS holders to exercise their voting rights due to the additional procedural steps involved in communicating with these holders. For example, holders of our ordinary shares will receive notice of shareholders’ meetings by mail and will be able to exercise their voting rights by either attending the shareholders meeting in person or voting by proxy. ADS holders, by comparison, will not receive notice directly from us. Instead, in accordance with the deposit agreement, we will use commercially reasonable endeavors to provide at least 30 days’ notice to the depositary of any such shareholders’ meeting and details concerning the matters to be voted on in advance of the meeting date. If we so instruct, the depositary will mail to holders of ADSs the notice of the meeting and a statement as to the manner in which voting instructions may be given by holders as soon as practicable after receiving notice from us of any such meeting. To exercise their voting rights, ADS holders must then instruct the depositary as to voting the ordinary shares represented by their ADSs. Due to these procedural steps involving the depositary, the process for exercising voting rights may take longer for ADS holders than for holders of ordinary shares. The ordinary shares represented by ADSs for which the depositary fails to receive timely voting instructions will not be voted.
ADS holders may have difficulty in effecting service of process on our company and certain directors or officers in the United States in enforcing U.S. judgements in the United Kingdom or in enforcing U.S. securities laws in UK courts.
We are incorporated and located outside the United States and certain of our directors and officers are located outside of the United States. As a result, it may not be possible for ADS holders to effect service of process within the United States upon all such persons or our company, or to obtain discovery of relevant documents and/or the testimony of witnesses. ADS holders based in the United States may also have difficulty enforcing in courts outside the United States judgments obtained in U.S. courts against our company or our directors (including actions under the civil liability provisions of the U.S. securities laws). ADS holders may also have difficulty enforcing liabilities under the U.S. securities laws in legal actions originally brought in jurisdictions located outside the United States.
Currency fluctuations may adversely affect the value of our ADSs.
Our ADSs are quoted in U.S. dollars on the Nasdaq. Movements in the U.S. dollar exchange rate may adversely affect the value of the ADSs.
We have never declared or paid dividends and we do not anticipate paying dividends in the foreseeable future.
We have never declared or paid cash dividends. For the foreseeable future, we intend to retain all available funds and any future earnings to support our operations and to finance the growth and development of our business. Any future determination to declare cash dividends will be made at the discretion of the Board, subject to compliance with applicable laws and covenants under current or future debt facilities, which may restrict or limit our ability to pay dividends, and will depend on our financial condition, operating results, capital requirements, general business conditions and other factors that the Board may deem relevant. We do not anticipate paying any cash dividends in the foreseeable future. As a result, a return on any investment will only occur if the price of our ADSs increases.
ADS holders may not receive distributions on the ordinary shares represented by our ADSs or any value for such distribution if it is illegal or impractical to make them available to holders of ADSs.
While we do not anticipate paying any dividends in the foreseeable future, if such a dividend is declared, the depositary for the ADSs has agreed to pay to you the cash dividends or other distributions it or the custodian receives after deducting its fees and expenses. You will receive these distributions in proportion to the number of ordinary shares your ADSs represent. However, in accordance with the limitations set forth in the deposit agreement, it may be unlawful or impractical to make a distribution available to holders of ADSs. We have no obligation to take any other action to permit the distribution of the ADSs, ordinary shares, rights or anything else to holders of our ADSs. This means that you may not receive distributions we make if it is unlawful or impractical to make them available to you. These restrictions may have a material adverse effect on the value of your ADSs.
ADS holders may not be able to participate in rights offerings and may experience dilution of their holdings as a result.
We may from time to time distribute rights to our shareholders, including rights to acquire our securities. However, we may not, and under the deposit agreement for the ADSs, the depositary will not, offer those rights to ADS holders unless both the rights and the underlying securities to be distributed to ADS holders are registered under the Securities Act or the distribution of them to ADS holders is exempt from registration under the Securities Act with respect to all holders of ADSs. We are under no obligation to file a registration statement with respect to any such rights or underlying securities or to endeavor to cause such a registration statement to be declared effective. In addition, we may not be able to rely on an exemption from registration under the Securities Act to distribute such rights and securities. Accordingly, holders of the ADSs may be unable to participate in our rights offerings and may experience dilution in their holdings as a result.
ADS holders may be subject to limitations on transfer of the ADSs.
The ADSs are only transferable on the books of the depositary. However, the depositary may close its transfer books at any time or from time to time when it deems expedient in connection with the performance of its duties. In addition, the depositary may refuse to deliver, transfer or register transfers of ADSs generally when our books or the books of the depositary are closed, or at any time if we or the depositary deem it advisable to do so because of any requirement of law or of any government or governmental body, or under any provision of the deposit agreement, or for any other reason.
| Item 4. | Information on the Company |
A. History and Development of the Company
Our predecessor company was incorporated under the Companies Act 1985 and registered in England and Wales on December 20, 2004, under the name Elm Partners plc. We changed our name to Hamilton Partners plc on December 4, 2006, to Sterling Green Group plc on April 26, 2007, and to Fastnet Oil & Gas plc on June 8, 2012.
On April 18, 2016, we completed a reverse merger into AIM-quoted Fastnet Equity plc. Concurrently with the completion of this reverse merger, we also acquired Birken AG, which offered a range of derma-cosmetic products, including treatments for sensitive, allergy-prone and dry skin. We subsequently changed our name to Amryt Pharma plc.
We were incorporated under the Companies Act 2006 and registered in England and Wales on July 17, 2019, as a private company limited by shares under the name Amryt Pharma Holdings Limited. Amryt was re-registered as a public limited company, Amryt Pharma Holdings plc, on September 24, 2019.
On September 24, 2019, Amryt Pharma Holdings plc became the new parent company of Amryt Pharma plc pursuant to a scheme of arrangement between Amryt Pharma plc and its shareholders under Part 26 of the Companies Act 2006 (“Companies Act”). Amryt Pharma Holdings plc changed its name to Amryt Pharma plc.
On May 5, 2021, Amryt announced that it had signed a definitive agreement to acquire Chiasma, Inc. in an all-stock combination. Under the terms of the transaction, each share of Chiasma common stock issued and outstanding prior to the consummation of the transaction was exchanged for 0.396 Amryt American Depositary Shares (“ADSs”), each representing five Amryt ordinary shares. The merger agreement provided that, upon the terms and subject to the conditions set forth in the merger agreement, at the effective time, Amryt Merger Sub, an indirect wholly owned subsidiary of Amryt, merged with and into Chiasma with Chiasma surviving as an indirect wholly owned subsidiary of Amryt subsequently renamed Amryt Endo, Inc. which then ceased to be a publicly traded company.
On August 5, 2021, Amryt completed the acquisition of Chiasma, Inc. and, in conjunction with the completion, Amryt allotted and issued a total of 127,733,680 ordinary shares as consideration for the acquisition. Following the completion, shareholdings in Chiasma were rounded in being converted to Amryt shares using the exchange ratio of 0.396. Roundings in converting Chiasma shareholdings to Amryt shares were finalized in August 2021 and resulted in an additional 7,015 ordinary shares being allotted and issued by Amryt as consideration for the acquisition. In total, these ordinary shares were issued to the former Chiasma Shareholders in the form of 25,548,139 ADSs at US$10.19 per share, to acquire Chiasma for a value of US$260,336,000. On August 5, 2021, the Group repaid US$116,629,000 of Chiasma long term debt.
Through the acquisition of Chiasma, Inc, we acquired our third commercial product, Mycapssa® (octreotide capsules) which is approved in the US for long-term maintenance therapy in acromegaly patients who have responded to and tolerated treatment with octreotide or lanreotide. Mycapssa® is the first and only oral somatostatin analog approved by the FDA. Mycapssa® has also been submitted to the EMA and is not yet approved in Europe. We believe that following the acquisition the combined company will be a global leader in rare and orphan diseases with three on-market commercial products, a global commercial and operational footprint and a significant development pipeline of therapies with the financial flexibility to execute its growth plans.
Amryt has traded on AIM under the symbol “AMYT” from April 2016 up to January 11, 2022, at which point the Company cancelled the admission to trading of its ordinary shares on AIM. Amryt ADSs representing our ordinary shares have traded on the Nasdaq under the symbol “AMYT” since July 8, 2020. Our registered office is located at Dept 920a 196 High Road, Wood Green, London N22 8HH, United Kingdom, and our principal office is located at 45 Mespil Road, Dublin 4, Ireland. We maintain a website at www.amrytpharma.com. The reference to our website is an inactive textual reference only and the information contained in, or that can be accessed through, our website is not a part of this annual report.
The SEC maintains a website at www.sec.gov that contains reports, proxy and information statements, and other information that we file electronically with the SEC.
B. Business Overview
We are a global, commercial-stage biopharmaceutical company dedicated to acquiring, developing and commercializing novel treatments for rare diseases. Our diversified portfolio is comprised of three commercial rare disease products, as well as a development-stage pipeline focused on rare skin diseases. Two of our three commercial products, lomitapide for the treatment of homozygous familial hypercholesterolemia (“HoFH”), and metreleptin for the treatment of generalized lipodystrophy (“GL”) and partial lipodystrophy (“PL”), are sold globally through our commercial infrastructure. Our third commercial product, Mycapssa® (octreotide capsules) is approved in the US for long-term maintenance therapy in acromegaly patients who have responded to and tolerated treatment with octreotide or lanreotide. Mycapssa® is the first and only oral somatostatin analog approved by the FDA. Mycapssa® has also been submitted to the EMA and is not yet approved in Europe. We acquired this product through the acquisition of Chiasma, Inc. (“Chiasma”) in August 2021. We are also developing our lead product candidate, Oleogel-S10, for the treatment of partial thickness wounds of severe Epidermolysis Bullosa (“EB”), a rare and devastating genetic skin disease for which there is currently no approved therapy. Filsuvez is the approved brand name for Oleogel-S10, which has not received regulatory approval by the Food and Drug Administration (“FDA”) or the European Medicines Agency (“EMA”) for the treatment of EB. We received positive topline data from the pivotal Phase 3 EASE trial of Oleogel-S10 on September 9, 2020. On February 28, 2022, we received a CRL from the FDA which asked Amryt to submit additional confirmatory evidence of effectiveness for Oleogel-S10 in EB. Amryt intends to discuss with the FDA the nature of the data required to address the Agency’s concerns. A MAA for Oleogel-S10 for the treatment of Dystrophic and Junctional EB was validated by EMA March 25, 2021, the assessment process by EMA was completed on April 22, 2022, when the CHMP adopted a positive opinion. The positive opinion recommends the approval of Filsuvez® in the EU for the treatment of partial thickness wounds associated with dystrophic and junctional EB in patients six months and older. Based on this CHMP recommendation a decision by the EC is expected on the Filsuvez® application within 67 days. Further details are discussed in “Business—Our Product Candidates—Oleogel-S10 for the Treatment of Severe EB—Topline Results for Oleogel-S10.” We are also developing Mycapssa® to expand the indication of Mycapssa® (octreotide capsules) beyond acromegaly into carcinoid syndrome associated with neuroendocrine tumors, further details of which are discussed in “Business—Our Product Candidates— Mycapssa® for the Treatment of Neuroendocrine Tumors (NET)—.”
Our next product candidate, AP103, is currently in preclinical development for the treatment of patients with Dystrophic EB (“DEB”), a subset of EB. AP103 is the first gene therapy product candidate based on our novel polymer-based topical gene therapy delivery platform, which also has potential use for the treatment of other rare genetic diseases. In September 2020, the EMA’s Committee for Orphan Medicinal Products (“COMP”) adopted a positive opinion for orphan designation for the use of AP103 in EB. In December 2020, the FDA granted orphan drug designation for AP103 for the treatment of Dystrophic Epidermolysis Bullosa (“DEB”), a subset of EB. We intend to initiate clinical development of AP103 in 2023. To support this milestone, the team intend to qualify and validate the necessary analytical methods required to advance the manufacturing and characterization of our drug product and critical starting materials. We have a proven track record of obtaining rare disease assets, either through acquisition or in-license, and we intend to continue building our portfolio of rare disease programs with the goal of bringing effective treatments to patients in need.
The following table summarizes key information about our product portfolio and clinical development pipeline. We have retained worldwide development and commercial rights to all of our programs, excluding Japan for lomitapide and Japan, South Korea and Taiwan for metreleptin.
Definitions: Dystrophic EB (“DEB”); Junctional EB (‘‘JEB’’)
* Upcoming clinical milestones are subject to the impact of COVID-19 on our business.
(1) Global Phase 3 study to support US label expansion for metreleptin in the treatment of partial lipodystrophy (PL).
(2) We are conducting a Phase 3 study of homozygous familial hypercholesterolemia ("HoFH") in children and adolescents in Europe, the Middle East and Africa ("EMEA") as part of our European Medicines Agency (‘‘EMA’’) pediatric investigation plan (PIP) commitment.
(3) 505(b)(2) pathway Phase 2 not required, Phase 3 initiation anticipated in Q4 2022 for the treatment of carcinoid symptoms in NET.
(4) Oleogel-S10 was approved in 2016 by the EMA for the treatment of partial thickness wounds in adults but has not been commercially launched.
(5) This radiation-induced dermatitis Phase 2 trial is an investigator-initiated study
Lomitapide is an oral therapy approved as an adjunct to a low-fat diet and other lipid-lowering treatments for adults with HoFH. It is marketed in the United States under the trade name Juxtapid and in the European Union under the trade name Lojuxta. HoFH is a rare and serious genetic condition that leads to aggressive and premature heart disease, heart attacks and strokes in patients as young as teenagers. HoFH patients are at a high risk of experiencing life-threatening cardiovascular events as a result of extremely elevated cholesterol levels in the blood and have a substantially reduced life expectancy. HoFH impairs the liver’s ability to remove low density lipoprotein (“LDL”) cholesterol, or “bad” cholesterol, from the blood, which if left untreated can cause aggressive narrowing and blocking of the blood vessels. According to a 2013 European Heart Journal article, the prevalence of HoFH is one person per million. However, according to a 2016 article published in Atherosclerosis, the number may be as high as 6.25 persons per million. Lomitapide is a small molecule microsomal triglyceride transfer protein (“MTP”) inhibitor. MTP exists in both the liver and intestines where it plays a role in the formation of cholesterol-carrying lipoproteins. As a result, inhibition of MTP is an effective cholesterol-lowering therapy in HoFH patients with limited or non-functional LDL receptors. We are conducting a Phase 3 pediatric study, in the EMA pediatric investigation plan (PIP) for the use of lomitapide in children and adolescents with HoFH, for which we expect to report data in the second half of 2022.
In December 2020, we received a marketing authorization approval from Agencia Nacional De Vigilancia Sanitaria, the Brazilian Health Regulatory Agency for Lojuxta.
In December 2021, we received a marketing authorization approval for Lojuxta from the Saudi Food and Drug Authority (SFDA).
Since the completion of the Brexit transition period, a separate national Marketing Authorization (MA) is required for UK and Great Britain (GB). In 2021, we received marketing authorization approval for each of Lojuxta and Myalepta from the UK Medicines and Healthcare products Regulatory Agency (MHRA).
Metreleptin for injection is approved in the United States under the trade name Myalept as an adjunct to diet as replacement therapy to treat the complications of leptin deficiency in patients with congenital or acquired GL. It is approved in the European Union under the trade name Myalepta for the treatment of leptin deficiency in patients with congenital or acquired GL and familial or acquired PL for whom standard treatments have failed to achieve adequate metabolic control. GL and PL are rare diseases characterized by loss or lack of adipose tissues (fat cells), resulting in the deficiency of the hormone leptin. GL and PL patients experience severe metabolic abnormalities including severe insulin resistance, diabetes, hypertriglyceridemia and fatty liver disease. We estimate that the prevalence of GL is approximately one person per million and that of PL is approximately three persons per million. Metreleptin is a recombinant human leptin analog that binds to and activates the human leptin receptor. Metreleptin acts to stimulate fatty acid oxidation throughout the body and lower plasma, hepatic and myocellular triglyceride levels. Amryt received positive feedback from the FDA on the potential for label expansion of metreleptin in the United States to include the treatment of PL. Prior to potential approval, a pivotal Phase 3 randomized double-blind placebo controlled trial to evaluate the safety and efficacy of daily subcutaneous metreleptin treatment in patients with PL is required, which will enroll approximately 51-63 patients globally. This study was initiated in Q4 2021.
Mycapssa® (octreotide capsules) for long-term maintenance treatment in acromegaly patients who have responded to and tolerated treatment with octreotide or lanreotide. U.S. commercial launch of Mycapssa® commenced in September 2020. Mycapssa® is the first and only oral somatostatin analog, or SSA, approved by the FDA and the first product approved by the FDA utilizing Amryt’s proprietary Transient Permeability Enhancer (‘‘TPE®’’) technology. The FDA approval of Mycapssa® was based on the positive results of a randomized, double-blind, placebo-controlled, nine-month Phase 3 Optimal clinical trial of octreotide capsules, which met the primary endpoint and all four secondary endpoints, as well as safety data from all of its Phase 3 clinical trials of Mycapssa. Amryt is focused on the commercialization of Mycapssa® for the treatment of patients with acromegaly in the United States. An MAA for Mycapssa® has been submitted on June 28, 2021, to EMA for long-term maintenance treatment in patients with acromegaly who have responded to and tolerated treatment with somatostatin analogues. The assessment process is ongoing.
Our lead development candidate, Oleogel-S10, is being developed as a potential topical treatment for the partial thickness wounds of severe EB, a rare and devastating genetic skin disease affecting young children and adults for which there is currently no approved treatment. EB is a group of diseases of the skin, mucous membranes and internal epithelial linings characterized by extreme skin fragility that blisters and tears from minor friction or trauma. Patients with severe forms of EB, including Dystrophic EB (“DEB”) and Junctional EB (“JEB”), suffer from severe and chronic blistering, ulceration, scarring, mutilating scarring of the hands and feet, joint contractures, strictures of the esophagus and mucous membranes, a high risk of developing aggressive squamous cell carcinomas, infections and risk of premature death. Market research indicates an incidence among live births of one in 20,000, and, when accounting for life expectancy per EB sub-type, there are an estimated 30 patients per million (total EB prevalence in the general population), of which approximately 31% are DEB & JEB patients. We have completed the Double Blind Phase (DBP) of a pivotal Phase 3 clinical trial of Oleogel-S10 in severe EB, including DEB and JEB. The Open Label Phase (OLP) is ongoing. In January 2019, we reported that the independent Data and Safety Monitoring Board (“DSMB”) performed a pre-specified unblinded interim efficacy analysis and recommended the continuation of the study with an increase in enrollment from 182 to 230 evaluable patients to maintain 80% conditional statistical power. We received positive topline results from the EASE trial on September 9, 2020. These results are outlined in more detail in “Business—Our Product Candidates—Oleogel-S10 for the Treatment of Severe EB—Topline Results for Oleogel-S10.”
On February 28, 2022, we received a CRL from the FDA which asked Amryt to submit additional confirmatory evidence of effectiveness for Oleogel-S10 in EB. Amryt intends to discuss with the FDA the nature of the data required to address the Agency’s concerns. A MAA for Oleogel-S10 for the treatment of Dystrophic and Junctional EB was validated by EMA March 25, 2021, the assessment process by EMA was completed on April 22, 2022, when the CHMP adopted a positive opinion. The positive opinion recommends the approval of Filsuvez® in the EU for the treatment of partial thickness wounds associated with dystrophic and junctional EB in patients six months and older. Based on this CHMP recommendation a decision by the EC is expected on the Filsuvez® application within 67 days. We are also supporting an investigator-led Phase 2 study of Oleogel-S10 for the treatment of severe radiation-induced dermatitis. This trial commenced in the Q4 2021.
We also have a novel polymer-based topical gene therapy delivery platform for potential use in the treatment of rare genetic diseases. Our first product candidate utilizing this platform, AP103, is currently in preclinical development for the treatment of DEB. We intend to initiate clinical development for AP103 in 2023. In September 2020, the EMA’s COMP adopted a positive opinion for orphan designation for the use of AP103 in EB, and on December 23, 2020, the FDA granted orphan designation for AP103 in the treatment of DEB. During 2021, the team engaged with the EMA (Protocol Assistance) and FDA (INTERACT Meeting) to seek scientific guidance on the proposed CMC and non-clinical study plans which has been incorporated into the program.
We sell Mycapssa® in the US and lomitapide and metreleptin in the Americas, Europe and the Middle East through our existing rare disease commercial infrastructure. Our commercial expertise includes market access, marketing, sales managers and sales representatives and is supported by our experienced medical affairs team with medical science liaisons, patient advocates and dieticians in the field. We also have a network of third-party distributors in other key markets throughout the world.
We are led by a management team and board of directors with deep experience at leading biotech companies, large pharmaceutical companies and academic institutions. Our company headquarters are in Dublin, Ireland, and we have over 289 employees throughout the world. We believe the expertise of our leadership team and the strength of our relationships within the industry and medical community are critical to our strategy as we commercialize our existing products, progress our development pipeline and explore new business development opportunities.
Our Commercial Products
Lomitapide for the Treatment of HoFH
Overview
HoFH is a rare genetic disease, which impairs the body’s ability to remove LDL cholesterol, or “bad” cholesterol, typically leading to abnormally high LDL cholesterol levels in the blood. HoFH patients are at a high risk of experiencing life-threatening cardiovascular events at an early age as a result of extremely elevated cholesterol levels in the blood and have a substantially reduced life expectancy relative to unaffected individuals. According to a 2013 European Health Journal article, the prevalence of HoFH is one person per million. However, according to a 2016 article published in Atherosclerosis, the number may be as high as 6.25 persons per million. Aggressive treatment, including dietary modifications plus combination therapy with currently approved lipid lowering drugs at maximum tolerated doses, often fails to reduce LDL cholesterol levels to their recommended targets in these patients. Lomitapide is a small molecule MTP inhibitor with the potential to provide significant reductions in LDL cholesterol levels in this high-risk patient population. Lomitapide, which is marketed as Juxtapid in the United States and as Lojuxta in the EMEA, is an oral, once-a-day treatment for adult patients with HoFH, as an adjunct to a low-fat diet and other lipid-lowering medicinal products, with or without LDL apheresis.
In October 2007, the FDA granted lomitapide Orphan Drug Designation for the treatment of HoFH. The FDA approved Juxtapid in December 2012 and the EMA granted marketing authorization for Lojuxta in July 2013. In addition to the United States and the European Union, lomitapide has received regulatory approval in Japan, Canada, Saudi Arabia and certain Latin American countries. Lomitapide has also been made available upon physician request on a named patient sales basis in certain other jurisdictions. In 2019, we out-licensed to Recordati the rights to sell lomitapide in Japan.
HoFH – Background and Current Treatments
HoFH is a serious, rare genetic disease usually caused by defects in both alleles of the low-density lipoprotein receptor (“LDL-R”) gene, resulting in the impairment of the function of the LDL-R. The LDL-R is a protein on the surface of cells that is responsible for binding and removing LDL from the blood. Malfunction of the LDL-R results in significant elevation of blood cholesterol levels. Cholesterol naturally occurs in the body, synthesized in the liver, and also enters the bloodstream through absorption from food. Cholesterol is transported in the blood for use as a source of energy and cell structure. Excess levels of cholesterol in the blood, also known as hypercholesterolemia, can cause significant complications. HoFH is most commonly caused by genetic mutations in both alleles of the primary gene responsible for LDL-R production, the LDL-R gene, but can also be caused by mutations in other genes. To date, more than 1,600 mutations have been identified that can impair the function of the LDL-R gene, with some mutations leading to a significant reduction or a total lack of LDL-R activity. As a result of elevated levels of LDL, HoFH patients often develop premature and progressive atherosclerosis, a narrowing or blocking of the arteries, usually in combination with arterial thrombosis, and are at high risk of experiencing premature cardiovascular events, such as heart attack or stroke, often experiencing their first cardiovascular event in early adulthood, and have a significantly shortened life expectancy.
Physicians in the United States and in many other countries often use clinical findings and family history to make a clinical diagnosis of HoFH. Clinical diagnosis is typically made using the following criteria: significantly elevated LDL cholesterol levels; physical signs, which may include the presence of cutaneous xanthomas, Achilles tendon thickening, xanthelasma and/or corneal arcus; limited response to statins that is not attributed to statin intolerance or to another identifiable cause (usually dependent on functional LDL receptors); evidence of premature cardiovascular disease (often in the second and third decade of life); and a positive history of high cholesterol and/or premature cardiovascular disease, consistent with having familial hypercholesterolemia on both sides of the family.
The clinical approach taken with HoFH patients typically involves an aggressive treatment plan to reduce lipid levels as much as possible through dietary modifications and a combination of lipid-lowering drug therapies.
Current drug therapies for reducing LDL levels in HoFH patients include statins, cholesterol absorption inhibitors, PCSK9 inhibitors, Evinacumab and lomitapide. Patients are also managed through LDL apheresis (lipid dialysis). Since the introduction of PCSK9 inhibitors in the United States in 2015, healthcare professionals have started most new adult HoFH patients on a PCSK9 inhibitor product before trying lomitapide and have switched some existing lomitapide patients to a PCSK9 inhibitor product because such products may have fewer side effects, are significantly less expensive and do not require that patients follow a special low-fat diet. However, because many therapies, including statins and PCSK9 inhibitor products, act by increasing the activity of LDL-R, HoFH patients often have an inadequate response due to their impaired LDL-R function. For example, high dose statin therapies that typically produce 35% to 47% reductions in LDL levels in the broad hypercholesterolemic patient population, on average, produce only a 10% to 25% reduction in HoFH patients. Also, a study published in Lancet in 2014 showed that although PCSK9 inhibitor evolocumab produced a mean LDL reduction of 23% in HoFH patients at 12 weeks, a subset of HoFH patients with very low or no LDL-R function did not respond to treatment. HoFH patients who are unable to reach their recommended target LDL levels on drug therapy are sometimes treated using LDL apheresis. However, apheresis provides only temporary reductions in LDL levels, so it must be repeated frequently to avoid rapid rebound that typically occurs within approximately four days. In addition, except in certain countries in the European Union, apheresis is often not readily available, due to the limited number of treatment centers that perform this procedure.
Recently Regeneron Pharmaceuticals Inc. launched Evinacumab for the treatment of HoFH in the US. In August 2019, Regeneron announced positive topline data from its ongoing Phase 3 trial in HoFH and the FDA approved Evinacumab on February 11, 2021, for adults and pediatric patients 12 years and older for treatment of HoFH. EMA approved Evinacumab in June 2021 as an adjunct to diet and other low-density lipoprotein-cholesterol (LDL-C) lowering therapies for the treatment of adult and adolescent patients aged 12 years and older with homozygous familial hypercholesterolaemia (HoFH). Regeneron launched Evinacumab in the United States in March 2021. Evinacumab was licensed to Ultragenyx Pharmaceutical Inc in countries outside the US in January 2022. Although the drug is administered by intravenous infusion, physicians may consider use of this product in HoFH patients before initiation of lomitapide treatment or may switch patients currently treated with lomitapide to Evinacumab treatment.
Our Solution: Lomitapide for the Treatment of HoFH
We believe lomitapide has the potential to address and mitigate the root cause of HoFH. Lomitapide is a small molecule MTP inhibitor. MTP exists in both the liver and intestines where it plays a role in the formation of cholesterol-carrying lipoproteins. As a result, the inhibition of MTP is an effective cholesterol-lowering therapy in HoFH patients with limited or non-functional LDL receptors. We believe lomitapide has the potential to become the standard of care (“SOC”) for the reduction of LDL levels in HoFH patients who have not responded to other therapies.
Treatment and Adverse Reaction Rates
The recommended starting dosage of lomitapide is 5 mg once daily (up to a maximum of 60 mg, depending on patient response to treatment and goal of therapy), and the dose should be escalated gradually based on acceptable safety and tolerability. Lomitapide should be taken once daily with a glass of water, without food, at least two hours after the evening meal. Lomitapide is contraindicated in the following conditions: pregnancy, concomitant administration of lomitapide with moderate or strong CYP3A4 inhibitors, patients with moderate or severe hepatic impairment and patients with active liver disease.
One single-arm, open-label, 78-week trial has been conducted treating 29 patients with lomitapide for HoFH, 23 of whom completed at least one year of treatment. The initial dosage of lomitapide in such trial was 5 mg daily, with titration up to 60 mg daily during an 18-week period based on safety and tolerability. In this trial, the mean age was 30.7 years (range, 18 to 55 years), 16 (55%) patients were men, 25 (86%) patients were Caucasian, two (7%) were Asian, one (3%) was African American, and one (3%) was multi-racial. The most common adverse reactions were gastrointestinal, reported by 27 (93%) of 29 patients. Adverse reactions reported by ≥ 8 (28%) patients in the HoFH clinical trial included diarrhea, nausea, vomiting, dyspepsia and abdominal pain. Other common adverse reactions, reported by five to seven (17-24%) patients, included weight loss, abdominal discomfort, abdominal distension, constipation, flatulence, increased alanine aminotransferase (“ALT”), chest pain, influenza, nasopharyngitis and fatigue. Adverse reactions of severe intensity were reported by eight (28%) of 29 patients, with the most common being diarrhea, vomiting, increased ALT or hepatotoxicity and abdominal pain, distension, and/or discomfort.
Expansion Opportunities in Pediatric HoFH and Post-Approval Obligations
We are conducting a Phase 3 pediatric study in the European Union for the use of lomitapide in children and adolescents with HoFH. We expect to report data from this trial in the second half of 2022.
Clinical Development and Post-Approval Obligations
In 2014, we initiated an observational cohort study in the United States and European Union to generate additional data on the long-term safety profile of lomitapide, the patterns of use and compliance and the long-term effectiveness of lomitapide in controlling LDL levels. The FDA has required target enrollment of 300 HoFH patients worldwide, and has mandated study of enrolled patients for a period of ten years. The EMA has required that all patients taking lomitapide in the European Union be encouraged to participate in the study, and that the study period be open-ended. In the study, investigators will follow each patient to track malignancies, tumors, teratogenicity, hepatic effects and gastrointestinal adverse reactions, events associated with coagulopathy, major adverse cardiovascular events and death. The EMA also required that a study be conducted to determine the impact of lomitapide on vascular endpoints. In addition, certain drug-drug interaction studies have been completed with results submitted to the EMA.
Metreleptin for the Treatment of GL and PL
Overview
Metreleptin is a recombinant analog of human leptin. It is marketed as Myalept in the United States as an adjunct to diet as a replacement therapy to treat the complications of leptin deficiency in patients with congenital or acquired GL. It is marketed as Myalepta in the European Union as an adjunct to diet as a replacement therapy to treat the complications of leptin deficiency in adults and children two years of age and above with congenital or acquired GL. Myalepta is also approved in the European Union for adults and children 12 years of age and above with familial or acquired PL for whom standard treatments have failed to achieve adequate metabolic control with congenital or acquired GL and also congenital or acquired PL. Leptin, which is deficient in patients with GL, is the key hormone responsible for regulating appetite and also has an important regulatory effect on energy expenditure. Leptin is a naturally occurring hormone derived from fat cells and an important regulator of energy, fat and glucose metabolism, reproductive capacity and other physiological functions. The predominant cause of metabolic complications in GL is excess triglyceride accumulation in the liver and skeletal muscle due to the inability to store triglycerides in fat cells. As a result of the deficiency of leptin associated with GL, patients experience significant fatigue as well as hyperphagia, or unregulated appetite. The loss of fat tissue caused by this disease often leads to severe metabolic abnormalities that contribute to increased morbidity and mortality.
Lipodystrophy – GL, PL and Current Treatments
Lipodystrophy is a heterogeneous group of rare syndromes characterized by selective but variable loss of fat tissue. Both the total amount and the appropriate distribution of fat deposits contribute to patients’ metabolic state. Because of the loss of fat tissue and fat cells, secreted hormone leptin is very low. Leptin circulates in blood and acts on the brain to regulate food intake and energy expenditure. When fat mass falls, plasma leptin levels fall, stimulating appetite and suppressing energy expenditure until fat mass is restored. When fat mass increases, leptin levels increase, suppressing appetite until weight is lost. This system maintains homeostatic control of fat cell mass.
Due to the lack of fat cells in individuals with lipodystrophy, energy can no longer be stored as fat in these cells and fat accumulates in the muscles and organs such as the heart, liver and pancreas, causing lipotoxicity and organ damage. In addition, deposition of fat in these unusual locations leads to extreme insulin resistance and its associated complications such as diabetes mellitus, hypertriglyceridemia, hepatic steatosis, polycystic ovary syndrome and high blood pressure. These severe metabolic abnormalities are typically resistant to conventional therapies. As a result of the deficiency of leptin associated with lipodystrophy, patients experience significant fatigue as well as unregulated appetite. The voracious appetite itself significantly aggravates the metabolic abnormalities and further reduces the ability to successfully treat these metabolic abnormalities with conventional therapies.
GL is characterized by a near complete lack of fat cells and, consequently, leads to early and significant morbidity and mortality. Classification of GL (versus PL) is made based on the anatomical distribution of fat loss, which is widespread in GL patients, as well as the younger age and greater rapidity of onset and severity of the metabolic abnormalities. The severe metabolic abnormalities associated with GL may result in premature diabetic nephropathy, retinopathy, cardiomyopathy, recurrent attacks of acute pancreatitis, enlarged liver and organ failure. These complications themselves increase morbidity and mortality due to their negative long-term impacts. The key diagnostic indicators of GL are most commonly a combination of physical appearance and severe metabolic abnormalities.
PL is characterized by a less uniform loss of fat cells and with a later age of onset. There can be considerable heterogeneity in the extent of fat cell loss, levels of leptin and degree of metabolic abnormalities. In PL patients with relative or near complete leptin deficiency, the metabolic abnormalities and longer impact on disease progression can closely mirror that of patients with GL.
Clinical diagnosis of lipodystrophy is largely based on history and physical exam but may be informed by genetic markers and blood tests, including adiponectin and leptin levels. Patients may be diagnosed well into adulthood even after seeing multiple doctors. Treatment is usually initiated by specialists, including adult and pediatric endocrinologists and lipidologists. A multi-disciplinary approach is required for ongoing care, which can also include hepatologists, cardiologists, nephrologists and rheumatologists.
Just under one person per million suffers from GL and approximately three persons per million for PL. We believe that the prevalence rate of GL in countries outside the United States is likely to be consistent with the prevalence rate in the United States. However, we anticipate that physicians will use metreleptin to treat the approximately one in three PL patients who suffer from severe PL.
Our Solution: Metreleptin for the Treatment of GL and PL
Metreleptin is a recombinant human leptin analog that binds to and activates the human leptin receptor. Clinical studies show that metreleptin stimulates fatty acid oxidation throughout the body and lowers plasma, hepatic and myocellular lipid levels (especially triglycerides), resulting in increased insulin sensitivity; improves insulin suppression of glucose production by the liver and increases insulin-stimulated peripheral glucose uptake into muscle; and corrects hyperphagia secondary to leptin deficiency with concomitant reduction in caloric and fat intake. Clinical studies have demonstrated the potential for metreleptin to effectively and consistently target leptin deficiencies and related health complications and improve patient outcomes.
Treatment and Adverse Reaction Rates
Metreleptin should be injected subcutaneously once daily at the same time every day, and may be administered any time of day without regard to the timing of meals. Metreleptin should be administered to patients as prescribed by their physician. Recommended dosage is as follows: Persons with body weight 40 kg or less: starting dose 0.06 mg/kg/day, increase or decrease by 0.02 mg/kg to a maximum daily dose of 0.13 mg/kg; Men greater than 40 kg body weight: starting dose 2.5 mg/day, increase or decrease by 1.25 mg to 2.5 mg/day to a maximum dose of 10 mg/day; and women greater than 40 kg body weight: starting dose 5 mg/day, increase or decrease by 1.25 mg to 2.5 mg/day to a maximum dose of 10 mg/day. Metreleptin is contraindicated in patients with general obesity not associated with congenital leptin deficiency. The safety of metreleptin was evaluated in 48 patients with GL in a single-arm, open-label study. The median duration of exposure in this trial was 2.7 years with a range of 3.6 months to 10.9 years. Five to six patients (10-13%) experienced the most common adverse reactions: headache, hypoglycemia, decreased weight and abdominal pain.
Expansion Opportunities and Post-Approval Obligations
Amryt received positive feedback from the FDA on the potential for label expansion of metreleptin in the United States to include the treatment of PL. Prior to potential approval, a pivotal Phase 3 randomized double-blind placebo controlled trial to evaluate the safety and efficacy of daily subcutaneous metreleptin treatment in patients with PL will be required, which will enroll approximately 51-63 patients globally. This Phase 3 study was initiated in Q4 2021.
Clinical Development and Post-Marketing Commitments
A long-term, prospective, non-interventional, observational study (product exposure registry) in patients to evaluate serious risks related to the use of the product is under way. In the US, the study plans to enroll at least 100 patients treated with metreleptin and will be followed for a minimum of ten years unless they withdraw consent to participate in the registry or die. In the EEA, the registry will offer enrolment to all patients treated with metreleptin. Patients will be followed for the duration of the lifecycle of the product unless they withdraw consent to participate in the registry or die.
The US registry will attempt to enroll at least 100 new patients treated with metreleptin. Enrollment will close after five years or after 100 new patients have been enrolled, whichever occurs first. The registry will continue for ten years from the date of last patient’s enrollment.
We set forth below the additional post-approval obligations required by the FDA for metreleptin:
| • | the development, validation and implementation of a ligand binding assay to supplement the neutralizing bioassay that tests for the presence of neutralizing antibodies in serum samples from patients with GL. An update has been provided to FDA and written feedback received in November 2021. |
| • | testing all banked clinical samples from the GL clinical program for the presence of neutralizing antibodies against leptin using the ligand binding assay and to correlate neutralizing antibodies with clinical events (completed); and |
| • | a prospective study to assess the immunogenicity of metreleptin in patients receiving metreleptin. |
The presence of neutralizing antibodies will be assessed using both a validated cell-based assay and a validated ligand-binding assay in samples that are confirmed positive for binding antibodies to leptin. In addition, we are required to conduct certain studies related to the manufacture of metreleptin, including in order to validate new test methods, implement a risk-based reference standard program approach, and reassess product acceptance criteria with a larger data set from more manufactured batches. Post-approval obligations related to manufacturing metreleptin are completed or are on track for completion by their respective deadlines. Finally, we have ongoing obligations to assess spontaneous reports of serious risks related to the use of metreleptin, including the risk to exposed pregnancies and pregnancy outcomes, regardless of indication, for ten years from the date of approval of metreleptin in the United States.
In connection with specific obligations to the EMA to complete post-marketing measures, it has been our intention to conduct the following studies:
| • | Enroll patients in the non-interventional, prospective, observational study (product exposure registry) of GL and PL patients initiating treatment with metreleptin. As mentioned above, for the patients from the EEA, this study will be open to all patients treated with metreleptin and will continue to the life-span of the product; |
| • | a post-approval efficacy study in PL patients; and |
| • | an integrated analysis of immunogenicity that includes testing, using validated assays, on samples from historical studies, as well as the registry, the pediatric investigation plan (“PIP”) and the post-approval efficacy study in PL patients. |
We have also committed to the EMA, as part of our PIP, to conduct a study in GL patients under six years old to further evaluate the pharmacokinetics, activity and safety of metreleptin in this pediatric sub-population. The required in vitro study to assess the binding of metreleptin to proteins in serum, and the non-clinical tissue distribution study have been completed and were approved by EMA in September 2021.
Mycapssa® for the Treatment of Acromegaly
Mycapssa® (octreotide capsules) is a combination of octreotide acetate and excipients collectively called Transient Permeability Enhancer (TPE®). TPE improves the oral bioavailability of poorly absorbed drugs such as octreotide by increasing the permeability of the intestine. The mode of action of TPE is thought to involve a transient opening of the tight junctions between epithelial cells lining the intestine.
Acromegaly is a rare disease most often caused by a benign pituitary tumor and characterized by an excess of growth hormone and insulin-like growth factor-1 hormone. Treatment options include surgery, medication and radiation or a combination of these. Mycapssa® (octreotide capsules) is approved in the US for long-term maintenance therapy in acromegaly patients who have responded to and tolerated treatment with octreotide or lanreotide. Mycapssa® is the first and only oral somatostatin analog approved by the FDA. Mycapssa® has also been submitted to the EMA and is not yet approved in Europe.
Imlan: Therapeutic Topical Cream
We acquired Imlan, a derma-cosmetic product, as part of our acquisition of Birken AG in 2016. Imlan is a topical skin cream marketed solely in Germany for the daily care of highly sensitive skin.
Our Product Candidates
Oleogel-S10 for the Treatment of Severe EB
Overview
Oleogel-S10 is an oil-based gel that we are developing as a potential treatment for severe EB, a rare and devastating genetic skin disease that causes the skin layers and internal body linings to separate, affecting infants, children and adults, for which there is currently no approved treatment. Patients with EB suffer from a genetic disease resulting in the inability to produce a certain type of collagen, which plays an important role in anchoring the dermal and epidermal layers of the skin. Patients with severe forms of EB, including DEB and JEB, suffer from severe and chronic blistering, ulceration, scarring, mutilating scarring of the hands and feet, joint contractures, strictures of the esophagus and mucous membranes, a high risk of developing aggressive squamous cell carcinomas, infections and risk of premature death.
We commenced our Phase 3 Efficacy and Safety Study of Oleogel-S10 in EB (“EASE”), a pivotal study of Oleogel-S10 in patients with severe EB, in March 2017 and enrolled the first patient in April 2017. This study is the largest Phase 3 study conducted in patients with EB. In January 2019, we reported that the DSMB performed a pre-specified unblinded interim efficacy analysis and recommended the continuation of the study with an increase in enrollment from 182 to 230 evaluable patients to maintain 80% conditional statistical power. On September 9, 2020, we received positive topline data of the global pivotal Phase 3 trial. Further information in relation to the topline data can be found in “Business—Our Product Candidates—Oleogel-S10 for the Treatment of Severe EB—Topline Results for Oleogel-S10.”
Oleogel-S10 has been granted Orphan Drug Designation for the treatment of EB by both the FDA and the EMA. Oleogel-S10 was granted Fast Track Designation by the FDA in September 2019. Oleogel-S10 was also granted Rare Pediatric Disease Designation by the FDA. On February 28, 2022, we received a CRL from the FDA which asked Amryt to submit additional confirmatory evidence of effectiveness for Oleogel-S10 in EB. Amryt intends to discuss with the FDA the nature of the data required to address the Agency’s concerns. A MAA for Oleogel-S10 for the treatment of Dystrophic and Junctional EB was validated by EMA March 25, 2021, the assessment process by EMA was completed on April 22, 2022, when the CHMP adopted a positive opinion. The positive opinion recommends the approval of Filsuvez® in the EU for the treatment of partial thickness wounds associated with dystrophic and junctional EB in patients six months and older. Based on this CHMP recommendation a decision by the EC is expected on the Filsuvez® application within 67 days.
About EB
EB is a heterogeneous group of diseases of the skin, mucous membranes and internal epithelial linings characterized by extreme skin fragility that blisters and tears from minor friction or trauma. The severity and symptoms of EB can range from blistering of the hands, feet, knees and elbows in mild cases to widespread blistering that can lead to vision loss, disfigurement and serious medical problems in severe cases. There are currently no approved treatments for EB.
EB results from mutations in the genes responsible for the production of structural proteins essential for healthy skin function. Such genetic mutations cause impaired or absent function of the proteins that normally give the skin its mechanical strength. Market research indicates an incidence among live births of one in 20,000, and, when accounting for life expectancy per EB sub-type, there are an estimated 30 patients per million (total EB prevalence in the general population), of which approximately 31% are DEB & JEB patients. EB has been categorized as encompassing five major types (EB Simplex (“EBS”), DEB, JEB, Kindler Syndrome and Aquisita) and 31 subtypes. EBS is the most common and least severe form of EB. Approximately 70% of people with EB have EBS. Patients with EBS often heal quickly with minimal or no scarring. We are developing Oleogel-S10 to treat the cutaneous manifestations of the approximately 30% of EB patients that have either JEB, DEB and Kindler Syndrome. Patients with these more severe forms of EB may suffer from severe and chronic blistering, ulceration, scarring, mutilating scarring of the hands and feet, joint contractures, strictures of the esophagus and mucous membranes, a high risk of developing aggressive squamous cell carcinomas, infections and risk of premature death. Other manifestations of these more severe types of EB may include anemia, cardiomyopathy, syndactyly (fusion of the fingers and toes), renal insufficiency, dysphagia (difficulty swallowing), malnourishment, cancer, constipation, osteoporosis, muscular dystrophy and pyloric atresia.
Background on Dystrophic Epidermolysis Bullosa
DEB is the second most common variation of EB, occurring in approximately 25% of people with EB. DEB derives its name from the tendency of healing blisters to scar, which leads to contraction of the joints, fusion of the fingers and toes and contraction of the mouth membranes and narrowing of the esophagus. The intensity of DEB varies significantly, but the severity of this disease increases with age due to complications driven by scarring, the fusion of digits and wastage of skin tissue.
DEB is sub-classified into RDEB and Dominant Dystrophic EB (“DDEB”). RDEB is generally a more severe form of DEB. Infants affected by RDEB are typically born with widespread blistering and areas of missing skin, caused by trauma during birth. Most frequently, blistering occurs over the entire body and affects mucous membranes such as the lining of the mouth and digestive tract. Healing blisters result in severe scarring. Scarring in the mouth and esophagus can make it difficult to chew and swallow food, leading to chronic malnutrition and slow growth. Additional complications of progressive scarring can include fusion of the fingers and toes, loss of fingernails and toenails, joint deformities (contractures) that restrict movement and eye inflammation leading to vision loss. Persons with RDEB have reduced life expectancy and are at increased risk of developing aggressive types of skin cancer before the age of 35. DDEB is a less common and less severe form of DEB.
Background on Junctional Epidermolysis Bullosa
Approximately 5% of people living with EB have JEB, which is sub-classified into Herlitz JEB and non-Herlitz JEB. Herlitz JEB is a more severe form of JEB. From birth or early infancy, affected individuals have blistering over large regions of the body. Blistering also affects the mucous membranes, such as the moist lining of the mouth and digestive tract, which often makes consumption and digestion difficult. From birth or early infancy, affected individuals have blistering over large regions of their body. Children with Herlitz JEB often die in infancy. Non-Herlitz JEB is a less severe form of JEB. The blistering associated with non-Herlitz JEB may be limited to the hands, feet, knees and elbows, and it often improves after the conclusion of the newborn period. Persons living with non-Herlitz JEB intermediate typically experience a normal lifespan.
Our Solution: Oleogel-S10 for the Treatment of Severe EB
Oleogel-S10 is a topical product incorporating betulin and other related triterpene compounds. In in vitro studies using normal cultured keratinocytes, Oleogel-S10 has been observed to stimulate keratinocyte migration and promote differentiation into mature-epithelial skin cells, thereby ensuring more rapid wound healing. We conducted a pivotal Phase 3 clinical trial of Oleogel-S10 in severe EB, including DEB and JEB, and have communicated efficacy and safety data from the 90-day double-blind treatment period of the trial.
Clinical Development of Oleogel-S10
We have set forth below the clinical trials conducted to date for Oleogel-S10 for the treatment of severe EB.
Phase 2a Study
Our Phase 2a study of Oleogel-S10 was a prospective, case-controlled study to compare the efficacy and tolerance of Oleogel-S10 to SOC in EB wounds. Half of each wound was treated with Oleogel-S10, together with a non-adhesive wound dressing, and the other half was treated with wound dressing alone. The primary endpoint of the study was the rate of progress of re-epithelialization from baseline to either day 14 or day 28 (depending on whether the wound was acute or chronic). A blinded assessment of efficacy was conducted by two independent experts based on a chronological series of photographs taken before start of treatment, during wound dressing changes and at the end of treatment on day 14 and day 28. Safety variables of the study were incidence, severity and causality of adverse events and assessment of tolerability by the investigators and patients and/or his/her legal representative.
Ten patients were enrolled and 12 wounds were treated with study medication. Reviewers were asked to assess which half of the wound was healing faster. Results of this trial are set forth in the table below.
Results of Blinded Efficacy Evaluation by Patient and Wound
Patient (wound #) | | | Blinded Efficacy Assessment Experts | | | Result |
| | Reviewer 1 | | | Reviewer 2 | |
| 1 | | | Non-adhesive wound dressing | | | Oleogel-S10 | | | Undecided |
| 2 | | | Oleogel-S10 | | | Equal | | | Oleogel-S10 |
| 3 | | | Oleogel-S10 | | | Oleogel-S10 | | | Oleogel-S10 |
| 4 | | | Oleogel-S10 | | | Equal | | | Oleogel-S10 |
| 5 | | | Equal | | | Equal | | | Undecided |
| 6 | | | Oleogel-S10 | | | Oleogel-S10 | | | Oleogel-S10 |
| 7 | | | Equal | | | Equal | | | Undecided |
| 8 | | | Oleogel-S10 | | | Non-adhesive wound dressing | | | Undecided |
| 9 (1) | | | Oleogel-S10 | | | Oleogel-S10 | | | Oleogel-S10 |
| 9 (2) | | | Equal | | | Oleogel-S10 | | | Oleogel-S10 |
| 10 (1) | | | Oleogel-S10 | | | Oleogel-S10 | | | Oleogel-S10 |
| 10 (2) | | | Oleogel-S10 | | | Oleogel-S10 | | | Oleogel-S10 |
We observed that Oleogel-S10 together with the non-adhesive wound dressing significantly accelerated healing (eight of eight decided cases; p = 0.0078, binomial test) compared to non-adhesive wound dressing only (zero of eight decided cases). Of the four undecided cases, two consisted of opposite conclusions by the two reviewers, and the other two wounds were considered to have equal healing in both halves by the two reviewers.
A total of 11 adverse events (“AEs”) in six patients were reported; nine of 11 AEs (81.8%) involved the skin, one AE (9.1%) was classified as a flu-like syndrome, and one AE (9.1%) was an infection of a wound. All AEs affecting the skin were erosions within or next to the study and control wounds, respectively. None of the AEs was assessed as being related to study treatment (n = 6 not related, 54.5%; n = 5 probably not related, 45.5%). All AEs were mild to moderate in severity: nine AEs affecting the skin were evaluated as being Common Terminology Criteria for Adverse Events (“CTCAE”) grade 1 (81.8%), only the flu-like syndrome and the infection of a wound were rated as CTCAE grade 2 (18.2%). Treatment with Oleogel-S10 was not adjusted or interrupted due to AEs. No serious adverse event (“SAE”) or significant AE occurred throughout the study; none of the patients died or were lost to follow-up.
Phase 3 Trials in Partial Thickness Wounds
Before we acquired Oleogel-S10, three Phase 3 studies were conducted looking at the safety and efficacy of Oleogel-S10 in other partial thickness wounds (“PTWs”). This led to the approval of Oleogel-S10 for the treatment of partial thickness wounds by adults by the EMA in 2016. Although we do not intend to commercialize Oleogel-S10 for partial thickness wounds, we do intend to rely on these Phase 3 clinical trials in our new drug application and marketing authorization application because we consider the data from these trials supportive in the assessment of safety and efficacy of Oleogel-S10 for the treatment of EB. Patients with EB also suffer from PTWs and the process that leads to wound healing involves the same cellular mechanisms as those seen in other examples of PTWs such as burns and split-thickness skin graft donor sites.
The observations of accelerated wound healing in the Phase 3 trials of PTWs contribute key clinical evidence of the biological activity and pharmacodynamic effect of Oleogel-S10. In addition, we expect that the FDA, EMA and other health authorities will evaluate safety data to supplement the EASE trial data due to the small size of the study, even though the data must be interpreted with caution due to differences in study design and patient population disease characteristics. Specifically, the Phase 3 trials in PTWs enrolled adult patients who were generally healthy. Each patient had a single wound that was treated for a maximum period of 28 days. Each wound was divided into two halves and each half received topical treatment with Oleogel-S10 or standard care. Their wounds healed rapidly and without long term complications. In contrast, EB patients studied in the EASE trial are predominantly adolescents, children or infants with multiple complications of their disease including anemia, malnutrition, and impaired kidney function. These disease-related complications are expected to slow the wound healing process. Furthermore, due to the underlying skin fragility caused by EB, patients may develop new wounds during the course of the study and may experience re-opening of previously healed wounds. Patients in the EASE trial are required to apply Oleogel-S10 or the control gel to multiple wounds over a significant surface area of their skin for the duration of the double-blind phase of the trial (three months), and then Oleogel-S10 during the open label phase for a further two years. As a result, the efficacy and safety data from the EASE trial will provide the primary (pivotal) evidence required for benefit-risk assessment of Oleogel-S10 in EB. Data from the Phase 3 PTW trials are expected to provide supportive evidence only.
We have presented pooled safety data for all three PTW Phase 3 trials. The rationale is based on the fact that the trials were almost identical in design. The enrolled patient population characteristics were very similar, and pooling safety data is always performed when possible to increase the number of exposed patients for a more precise estimation of the true incidence of any adverse events that might be expected when much larger numbers of patients are exposed. This approach has been endorsed by the EMA.
Phase 3 Trial in Grade 2a Burns
One of the Phase 3 trials conducted for Oleogel-S10 (known as BBW-11) was a blinded, prospective, randomized, intra-individually controlled, multicenter, Phase 3 clinical trial comparing the efficacy and safety of Oleogel-S10 with wound dressing to SOC treatment (an antiseptic gel plus wound dressing) in accelerating the healing of Grade 2a burns. The primary endpoint was the percentage of patients with earlier healing (at least 95% epithelialization) of the wound half treated with Oleogel-S10 compared to SOC, as evaluated by the majority decision of three independent, blinded experts. Assessment of efficacy was primarily based on blinded photo evaluation.
66 patients were enrolled at ten centers in Germany, Sweden, Switzerland and the United Kingdom. Of the 66 patients, 61 patients received treatment with both Oleogel-S10and SOC. Of the 61 patients that received treatment, 50 patients completed treatment and 11 discontinued prematurely. The primary analysis evaluated only those patients for whom a difference was observed between treated wound halves (35 patients).
As set forth in the table below, the percentage of patients showing earlier healing of Oleogel-S10 treated wound half compared with SOC treated half was higher than the percentage of patients showing the opposite result (85.7% versus 14.3%, intention-to-treat population).
Primary Endpoint: Earlier Healing of Wound Half - Majority Decision of Blinded Expert Evaluation
| | n (%) | | 95% CI | | p value(1) |
| Patients with earlier healing of SOC treated wound half | 5 (14.3) | | 4.8, 30.3 | | |
| Patients with earlier healing of Oleogel-S10 treated wound half | 30 (85.7) | | 69.7, 95.2 | | < 0.0001 |
| (1) | Based on one-sided, exact binomial test evaluating the rate of superiority of Oleogel-S10 being > 0.5. |
CI = confidence interval, n = number of patients with the indicated difference in wound healing.
A representative series of burn wound photographs from one of the studied patients is shown below:
Secondary endpoints supported the primary finding indicating faster healing and epithelialization of the wound with Oleogel-S10 compared to standard of care.
Two Phase 3 Trials in Patients with Split-Thickness Skin Graft Donor Sites
Oleogel-S10 was also studied in two blinded, prospective, randomized, intra-individually controlled, multicenter Phase 3 clinical trials (known as BSH-12 and BSG-12) comparing the efficacy and safety of Oleogel-S10 plus SOC (non-adhesive wound dressing) versus SOC alone in accelerating the wound healing of split-thickness skin graft (“STSG”) donor sites. Since the design and conduct of these studies was identical the pooled results are presented here.
After the STSG surgery, the wound was split into halves and the two wound halves were randomized to treatment with either Oleogel-S10 plus SOC or SOC alone. Treatment was applied until complete wound closure of both halves occurred but no longer than 28 days. Wound dressing change and medication application were performed at least every three or four days.
The primary endpoint of both trials was the intra-individual difference in time to wound closure (at least 95% epithelialization) between wound halves based on photo evaluation by three independent, blinded experts. 107 and 112 patients were enrolled and received treatment in the two trials respectively. However, two patients in the BSG-12 study did not provide written informed consent and are therefore removed from the analysis below.
As set forth in the table below, wound halves treated with Oleogel-S10 healed faster than the wound halves treated with SOC (mean 1.1 days, p < 0.0001, two-sided paired t-test, based on blinded reader evaluation).
Difference in Time (Days) to Wound Closure(1) (total n = 217)
| Study | | n | | Median | | Min, Max | | Mean | | 95% CI | | p value(2) |
| BSH-12 | | 107 | | -0.3 | | -10.0, 2.3 | | -1.4 | | -1.8, -0.9 | | < 0.001 |
| BSG-12 | | 110 | | 0.0 | | -18.3, 12.3 | | -0.8 | | -1.5, -0.1 | | 0.0232 |
| Pooled | | 217 | | -0.3 | | -18.3, 12.3 | | -1.1 | | -1.5, -0.7 | | < 0.0001 |
| (1) | Difference in time to wound closure was set to zero for photos rated as not evaluable. If wound closure was not observed, wound closure was set to one day after last photograph of the series (= “conservative +1 day approach”). |
| (2) | Two-sided paired t-test evaluating the mean difference as different from zero. |
CI = confidence interval, n = number of patients in analysis, Max = maximum, Min = minimum.
A representative photographic series from one of the studied patients of a split-thickness skin graft donor site is shown below:
Pooled Safety Data From All Three Phase 3 Studies in Partial Thickness Wounds Including EBW-11, BSH-12 and BSG-12
Safety data from the three Phase 3 studies in PTWs were pooled and included 280 patients. One or more AEs were reported for a total of 35% of patients. The system organ classes with the highest incidence were infections and infestations (11%), injury, poisoning and procedural complications (8%) and skin and subcutaneous tissue disorders (10%). Most frequent AEs (by preferred term) comprised pyrexia and wound infection, skin infection, pain of skin, and pruritus, each reported at a rate of 4% to 6%.
Treatment-related AEs were reported by 25 patients (9%), including pain of skin (2.9%), pruritus (1.4%) and a few patients with other AEs, e.g., wound complication and post procedural complications. Application-site AEs were reported for the Oleogel-S10 treatment half for four patients (1.4%), for the SOC treatment half for 15 patients (5.4%) and for both treatment halves (meaning that the location of the AE could not be further differentiated) for 36 patients (12.9%). In cases where the AE could be clearly ascribed to one wound half, the number of local AEs was lower in the Oleogel-S10 treated wound half compared with the SOC treated wound half.
The number of patients in the pooled analysis (study BSH 12, BSG 12 and BBW 11) who experienced one or more serious SAEs was low (15 or 5.4% of the patients). The most frequently reported SAEs were wound infection (four or 1.4% of patients) and condition aggravated (two or 0.7% of patients). All other SAEs occurred in only one (0.4%) patient per event. Only one SAE was considered possibly related to treatment (wound necrosis) for which causality between treatment and event was reported as “yes: unknown” by the investigator. In this patient both wound halves had changed from Grade 2a to Grade 2b and required skin grafting resulting in prolonged hospital stay. Based on clinical experience a change in burn wound depth from Grade 2a to Grade 2b was expected for up to 30% of patients in the study. Skin grafting is the standard treatment for Grade 2b burn wounds.
The number of patients who discontinued the study due to AEs was low (10 or 3.6% of patients) and none were considered related to treatment except for the case of wound necrosis described above.
No deaths were reported in the treatment phase of the studies. Two deaths in the 12 months follow-up period were considered not related to the treatment.
Pivotal Phase 3 Trial of Oleogel-S10 in EB (EASE)
In 2017, we commenced a double-blind, randomized, vehicle-controlled, Phase 3 study with a 24-month open-label period follow-up comparing the safety and efficacy of Oleogel-S10 versus a gel vehicle in the promotion of healing of PTWs in patients with severe EB (JEB, DEB and Kindler Syndrome). Qualifying target wounds were between 10 cm2 and 50 cm2 in surface area and at least 21 days old, but less than nine months old.
The primary endpoint is the proportion of patients with first complete closure of the EB target wound by day 45±7 days compared to control gel based on clinical assessment by the investigator (the wound will be rated as “closed” at first appearance of complete re-epithelialization without drainage).
Key secondary endpoints are:
| • | Time to first complete closure of the EB target wound as evidenced by clinical assessment until the end of the double-blind phase (day 90±7 days); |
| • | Proportion of patients with first complete closure of the EB target wound at day 90±7 days based on clinical assessment by the investigator; |
| • | The incidence of target wound infection between baseline and day 90±7 days as evidenced by adverse events and/or use of topical and/or systemic antibiotics (related to wound infection); |
| • | The maximum severity of target wound infection between baseline and day 90±7 days as evidenced by adverse events and/or use of topical and/or systemic antibiotics (related to wound infection); |
| • | Change from baseline in total body wound burden as evidenced by clinical assessment using Section I of the EB Disease Activity and Scarring Index at day 90±7 days; and |
| • | Change from baseline in itching using the Itch Man Scale in patients ≥ 4 years and up to 13 years of age and the Leuven Itch Scale in patients ≥ 14 years of age before wound dressing changes at day 90±7 days. |
Safety endpoints include:
| • | Incidence, severity and relatedness of adverse events; |
| • | Local tolerability as judged by the investigator; |
| • | Safety laboratory data; and |
| • | Systemic exposure to betulin. |
Following completion of the 90 day double-blind period all patients randomized to the control gel switched to open label treatment with Oleogel-S10. Patients who had been randomized to Oleogel-S10 will continue on open label treatment until the end of the study.
During the open label safety follow up period, patients will be monitored for safety evaluation by collection of adverse event information. The efficacy of Oleogel-S10 during this phase of the study will be evaluated by assessment of total wound burden, impact on symptoms of disease and additional endpoints reflecting severity of disease and quality of life.
Unblinded Interim Efficacy Analysis and Safety Analysis
In January 2019, we reported that the DSMB performed a pre-specified unblinded interim efficacy analysis and recommended the continuation of the study with an increase in enrollment from 182 to 230 evaluable patients to maintain 80% conditional statistical power. This increase in sample size was based upon maintenance of 80% conditional statistical power to detect a 20% difference between treatment groups with respect to the primary efficacy endpoint.
In February 2019, the independent DSMB performed a pre-specified unblinded interim safety analysis which included a review of safety and PK data from children enrolled in the trial. Following this review, the DSMB recommended expansion of the trial to include neonates, infants and children from ≥ 21 days of age. On September 9, 2020, we received positive topline data of the global pivotal Phase 3 trial. Further information in relation to the topline data can be found in “—Topline Results for Oleogel-S10.”
Topline Results for Oleogel-S10
Further to our announcement of September 9, 2020, we released additional results from the pivotal Phase 3 EASE trial of Oleogel-S10 on October 29, 2020.
Highlights of the results are:
| • | 223 patients were randomized and included in the full analysis set (“FAS”). Of these, 109 patients were randomized to receive Oleogel S-10, and 114 patients were randomized to receive the control gel; |
| • | 156 (70%) of the FAS were children (under 18 years old), and 175 patients (78.5%) had RDEB, of which 124 (55.6%) had RDEB generalized severe sub-type. The remaining 48 patients were distributed between sub-types as follows: DDEB 20 patients, JEB 26 patients, EB simplex two patients; |
| • | At baseline the median target wound size was 15.6 cm2, and the median wound age was 35.5 days. The baseline characteristics of the two treatment groups was similar; |
| • | The primary endpoint (the proportion of patients with first complete closure of EB target wound within 45 days) was 41.3% in the Oleogel-S10 group and 28.9% in the control group (p value = 0.013); |
| • | This equates to a 44% increase in the probability of target wound closure with Oleogel-S10 compared to the control gel; |
| • | The proportion of RDEB patients with first complete closure of the EB target wound within 45 days was 44.0% in the Oleogel-S10 group and 26.2% in the control group (nominal p = 0.008); |
| • | This represents a 72% increase in the probability of target wound closure with Oleogel-S10 compared to the control gel in RDEB patients; |
| • | The proportion of DDEB patients with first complete closure of the EB target within 45 days was 50% in the Oleogel-S10 group and 50% in the control group (nominal p = 0.844). The proportion of JEB patients with first complete closure of the EB target within 45 days was 18.2% in the Oleogel-S10 group and 26.7% in the control group (nominal p = 0.522); |
| • | A greater reduction in pain associated with dressing changes was observed in the Oleogel-S10 treatment group at each time point compared with the control group. At Day 14, this difference was nominally significant (p = 0.02); |
| • | There was a greater reduction in total body wound burden as measured by EB Disease Activity and Scarring Index (“EBDASI”) and total body surface area of EB partial thickness wounds with Oleogel-S10 although the differences were not statistically significant; and |
| • | Oleogel-S10 had an acceptable safety profile and was well tolerated when compared with the control gel. |
Primary Efficacy Endpoint
The proportion of patients with first complete closure of EB target wound within 45 days was 41.3% in the Oleogel-S10 group and 28.9% in the control group. This difference reached statistical significance (p value = 0.013). This translates to a 44% increase in the probability of target wound closure with Oleogel-S10 compared to the control gel.
Proportion of patients with first complete closure of EB target wound within 45 days
| | | | Oleogel-S10 Group N = 104 | | | Control Group N = 114 |
| Primary Endpoint | | | 41.3% | | | 28.9% |
Relative Risk (95% CI) | | | 1.44 (1.01, 2.05) |
| p value* | | | 0.013 |
| * | after pre-specified adjustment to account for IDMC (Independent Data Monitoring Committee) interim sample size re-estimation |
The difference observed in the proportion of target wounds healed within 45 days between treatment groups for the primary endpoint was driven by the RDEB subgroup. The proportion of RDEB patients with first complete closure of EB target wound within 45 days was 44.0% in the Oleogel-S10 group and 26.2% in the control group (nominal p value = 0.008). This translates to a 72% increase in the probability of target wound closure with Oleogel-S10 compared to the control gel in RDEB patients.
Proportion of patients with first complete closure of target wound within 45 days by EB subtype
| Subtype | | | Oleogel-S10 Group | | | Control Group | | | Relative Risk | | | p value* |
| | | | n | | | Closure Rate | | | n | | | Closure Rate | | | | | | |
| RDEB** | | | 91 | | | 44.0% | | | 84 | | | 26.2% | | | 1.72 | | | 0.008 |
| DDEB** | | | 6 | | | 50.0% | | | 14 | | | 50.0% | | | 1.10 | | | 0.844 |
| JEB** | | | 11 | | | 18.2% | | | 15 | | | 26.7% | | | 0.61 | | | 0.522 |
| ** | Recessive Dystrophic EB (RDEB) / Dominant Dystrophic EB (DDEB) / Junctional EB (JEB) |
Secondary Efficacy Endpoints
In wounds that achieved complete closure over the 90-day treatment period, the mean time to first closure in the Oleogel-S10 group was 37.7 days compared to 44.5 days in the control group. During the entire 90-day double-blind treatment period separation in target wound closure occurred around Day 30 with the difference narrowing around Day 90. The proportion of first completely closed target wounds within the 90-day treatment period was 50.5% in the Oleogel-S10 group compared with 43.9% in the control group (p value = 0.296). The difference in the time to wound healing between the two treatment groups over the 90 days was not statistically significantly different (p = 0.302).
A greater reduction in pain associated with dressing changes was observed in the Oleogel-S10 treatment group at each time point compared with the control group. At Day 14, this difference was nominally significant (p = 0.022). At Day 90, the difference just missed nominal significance (p value = 0.051).
Procedural pain mean change from baseline (Wong-Baker FACES pain rating scale)
Patients ≥4 years of age
| | | | Day 14 | | | Day 30 | | | Day 45 | | | Day 60 | | | Day 90 |
| | | | n | | | | | | n | | | | | | n | | | | | | n | | | | | | n | | | |
| Oleogel-S10 Group | | | 90 | | | -1.44 | | | 90 | | | -1.04 | | | 84 | | | -0.93 | | | 84 | | | -1.29 | | | 76 | | | -1.32 |
| Control Group | | | 95 | | | -0.78 | | | 90 | | | -0.27 | | | 85 | | | -0.78 | | | 86 | | | -0.56 | | | 78 | | | -0.18 |
| Nominal p value | | | | | | 0.022 | | | | | | 0.152 | | | | | | 0.805 | | | | | | 0.095 | | | | | | 0.051 |
There was a greater reduction in total body wound burden as measured by EBDASI and total body surface area of EB partial thickness wounds with Oleogel-S10 although the differences were not statistically significant.
Reduction in total body wound burden (EBDASI)
| | | | Day 30 | | | Day 60 | | | Day 90 |
| | | | n | | | Mean change from baseline | | | n | | | Mean change from baseline | | | n | | | Mean change from baseline |
| Oleogel-S10 Group | | | 99 | | | -2.3 | | | 91 | | | -3.1 | | | 84 | | | -3.4 |
| Control Group | | | 99 | | | -2.2 | | | 96 | | | -2.0 | | | 85 | | | -2.8 |
Reduction in total body surface area of EB Partial Thickness Wounds
(Body Surface Area Percentage)
| | | | Day 30 | | | Day 60 | | | Day 90 |
| | | | n | | | Mean change from baseline | | | n | | | Mean change from baseline | | | n | | | Mean change from baseline |
| Oleogel-S10 Group | | | 98 | | | -2.56 | | | 92 | | | -2.92 | | | 86 | | | -4.32 |
| Control Group | | | 98 | | | -2.64 | | | 96 | | | -1.69 | | | 85 | | | -2.53 |
Target wound infections occurred infrequently (seven patients in total, with two patients in the Oleogel-S10 group and five in the control group). Five patients had a target wound infection reported as an AE. Four of these occurred in the control group of which three were classified as “moderate” and one “severe.” The single target wound infection reported in the Oleogel-S10 group was classified as “mild.”
In patients ≥14 years of age, both treatment groups experienced an improvement in itch, however the pattern across the six assessment domains did not suggest a consistent effect in favor of either treatment group. In patients between four and 13 years of age, there was a modest improvement in itch in both treatment groups with a small difference favoring the control group (not statistically significant).
Safety Profile
The incidence of patients with AEs was similar in the treatment groups. As expected in this patient population, approximately 80% of patients experienced at least one AE. The majority of these AEs were classed as mild or moderate in severity. There were 13 patients with severe (grade 3/4) AEs in the Oleogel-S10 group compared to six in the control group. However, this imbalance was mainly due to reports of “anemia,” a very common baseline finding in this population, and one that frequently requires treatment. The incidence of patients with related AEs and patients with AEs leading to study withdrawal was similar in the two treatment groups.
Summary of Adverse Events during 90 day treatment period
| Adverse Event Category | | | Oleogel-S10 (N = 109) n (%) | | | Control (N = 114) n (%) | | | All Patients (N = 223) n (%) |
Patients with any adverse event | | | 89 (81.7) | | | 92 (80.7) | | | 181 (81.2) |
| Mild AEs (grade 1) | | | 46 (42.2) | | | 41 (36.0) | | | 87 (39.0) |
| Moderate AEs (grade 2) | | | 30 (27.5) | | | 45 (39.5) | | | 75 (33.6) |
| Severe AEs (grade 3/4) | | | 13 (11.9) | | | 6 (5.3) | | | 19 (8.5) |
| Any Related AEs | | | 27 (24.8) | | | 26 (22.8) | | | 53 (23.8) |
Any AE leading to study withdrawal | | | 3 (2.8) | | | 2 (1.8) | | | 5 (2.2) |
The most frequently reported AEs (Oleogel-S10 vs Control) were wound complication (61.5% vs 53.5%), pyrexia (8.3% vs 13.2%), wound infection (7.3% vs 8.8%), pruritus (7.3% vs 5.3%) and anemia (7.3% vs 3.5%).
Oleogel-S10 previously received Fast Track Designation and Rare Pediatric Disease Designation from the US Food and Drug Administration (FDA).
As the final module in a Rolling Review a New Drug Application (NDA) was submitted to the FDA for Oloegel-S10 on March 31, 2021, for the treatment of Dystrophic and Junctional EB. The MAA is supported by data from the EASE pivotal phase 3 trial in EB.
In June 2021, the NDA was accepted by the FDA and was granted Priority Review. Priority Review is granted by the FDA to applications for medicines that, if approved, would provide significant improvements in the effectiveness or safety of the treatment, diagnosis, or prevention of serious conditions when compared to standard applications. In general, the FDA’s Priority Review designation accelerates the review time from ten months to a goal of six months from the date of acceptance of the filing. FDA set a Prescription Drug User Fee Act (“PDUFA”) target action date for the Oleogel-S10 NDA of November 30, 2021. We received and responded to Information Requests (IR) from FDA during the assessment period and met with FDA at the scheduled Mid Cycle and Late Cycle Meeting.
In November 2021, the FDA extended the PDUFA goal date to allow time to review additional analyses of data previously submitted by Amryt. The submission of this additional information was determined by the FDA to constitute a Major Amendment to the NDA, resulting in an extension of the PDUFA goal date by three months to February 28, 2022. Three months is a standard review extension period to allow the FDA additional time to review information already submitted.
On February 28, 2022, we received a CRL from the FDA which asked Amryt to submit additional confirmatory evidence of effectiveness for Oleogel-S10 in EB. Amryt intends to discuss with the FDA the nature of the data required to address the Agency’s concerns.
A MAA for Oleogel-S10 for the treatment of Dystrophic and Junctional EB was validated by EMA March 25, 2021, to be reviewed according to standard timelines. The MAA is supported by Phase 3 data from the EASE trial which was the largest ever global trial conducted in patients with EB, performed across 58 sites in 28 countries.. Questions and review comments were received from the CHMP according to the procedure timetable, responses and updated documents have been submitted by us according to the required deadlines. Given the rarity of the disease without any approved therapies, the EMA proposed that an Ad-Hoc Expert Group (AHEG), comprised of both EB clinical experts and patients with EB, be consulted to provide external and independent EB specific advice. Recommendations from the AHEG were provided directly to CHMP ahead of an Oral Explanation (OE) between us and the CHMP. On April 22, 2022, the CHMP adopted a positive opinion, recommending the approval of Filsuvez® in the EU for the treatment of partial thickness wounds associated with dystrophic and junctional EB in patients six months and older. Based on this CHMP recommendation a decision by the EC is expected on the Filsuvez® application within 67 days.
Additional Opportunity for Oleogel-S10
We are also supporting an investigator-led Phase 2 study of Oleogel-S10 for the treatment of severe radiation-induced dermatitis. This study commenced in Q4 2021.
AP103 for the Treatment of DEB
In March 2018, we acquired the rights to a novel polymer-based topical gene therapy delivery platform for potential use in the treatment of rare genetic diseases. The technology involves the use of highly branched poly β-amino ester (“HPAE”) polymers as the topical delivery vehicle for gene therapy. Our first product candidate utilizing this platform, AP103, is currently in preclinical development for the treatment of patients with DEB. Patients with DEB have a defect in the COL7A1 gene resulting in the inability to produce collagen VII, which plays an important role in anchoring the dermal and epidermal layers of the skin. AP103 is the combination of this polymer technology and the COL7A1 gene. If successful, we believe this could eliminate the requirement for viruses as topical delivery vectors.
In preclinical studies in a human mouse xenograph model of EB, we observed that topical application of AP103 restored production of collagen VII. In separate preclinical studies, AP103 was observed to restore collagen VII to levels exceeding those produced by healthy human keratinocytes (cells that regenerate the outer layer of the skin). In addition, we did not observe evidence of cellular toxicity after repeated administration in these studies. Our preclinical development of AP103 is ongoing. To support this critical milestone, significant effort has been invested in 2021 to advance the manufacturing and characterization of the AP103 drug product and constituent components. To date, the team have generated the initial qualified batches of the HPAE polymer and DNA required to generate AP103. This advancement will facilitate the initiation of key aspects of the non-clinical safety program in 2022, which need to be completed prior to launch of first in human (FIH) studies. We intend to initiate clinical development of AP103 in 2023. In September 2020, the EMA’s COMP adopted a positive opinion for orphan designation for the use of AP103 in EB and on December 23, 2020, the FDA granted orphan designation for AP103 in the treatment of DEB.
In December 2018, a consortium comprised of us (as lead partner), University College Dublin, National University of Ireland (“University College Dublin”), Curran Scientific Limited and DEBRA Ireland was awarded (subject to contract negotiation) grant funding totaling €8.4 million over three years from the Disruptive Technologies Innovation Fund, part of the Irish Government’s Department of Business, Enterprise and Innovation, to develop the AP103 gene therapy platform. The grant funding will be matched by the consortium partners at various funding levels over the three-year term of the project. The grant will fund further development of the AP103 gene therapy platform from preclinical testing to proof of concept in humans. The initial funds will be used for research and development and staff costs associated with the project and, if preclinical work is successful the funds will also support research into the development of the HPAE polymer technology for the potential treatment of other genetic diseases. In 2022, the consortium successfully designed, engineered and delivered an advanced working protype for the manufacturing of the AP103 product. This complexing system which generates the DNA and Polymer nanoparticles at the center of AP103, will be used to supply material for the non-clinical safety and FIH clinical studies. The complexing system delivers scalable and consistent AP103 product that has demonstrated to induce expression of the therapeutic gene, COL7A1, in pre-clinical models of DEB.
Mycapssa® for the Treatment of Neuroendocrine Tumors (NET)
Neuroendocrine tumors (NETs) are a heterogeneous group of cancer subtypes that arise in endocrine cells that exist in different organ systems throughout the body. Most NETs (approximately 70%) occur in the gastrointestinal (GI) tract or pancreas. Tumors arising from the GI tract are termed carcinoid tumors. NET may also occur in the respiratory tract, central nervous system, thyroid, skin, breast, and urogenital system. Up to 20% of carcinoid tumors are estimated to have carcinoid syndrome (CS). CS are mainly associated with midgut metastatic carcinoid tumors. Most commonly, CS presents with diarrhea and flushing episodes due to excessive secretion of serotonin. Injectable SRLs, such as octreotide long-acting release, subcutaneous octreotide immediate release, and lanreotide, are the first-line treatment for CS associated symptoms as they significantly improve flushing episodes and diarrhea symptoms by inhibiting the secretion of serotonin among other hormones and vasoactive substances.
Pharmacokinetic studies have been completed and the data supports the higher doses of Mycapssa® (octreotide capsules) required in the planned Phase 3 study in patients with carcinoid symptoms due to NET.
The FDA has confirmed that a single positive Phase 3 study would be sufficient for approval consistent with the 505(b)(2) regulatory pathway previously agreed. Amryt is currently finalizing the study protocol with the FDA and plans to initiate the Phase 3 study in Q4 2022.
Medical Affairs
We have a medical affairs team in the United States, the EMEA and Latin America. The medical affairs team maintains product registries, performs retrospective reviews of clinical data, and conducts post-marketing clinical trials as needed, to support the value propositions of our products. The medical affairs team also supports independent medical education programs and investigator-initiated studies, by providing financial grants in a number of medical and disease-related areas. The responsibilities of medical affairs personnel also include providing training and education to physicians through the dissemination of medical information, industry research and publications, providing support in connection with our post-approval clinical commitments, as well as assembling and managing scientific and medical advisory boards to obtain valuable input from experts and practitioners on a variety of medical topics relevant to our products and the diseases our products treat.
Sales, Marketing and Reimbursement
In the United States, our sales team promotes Myalept to healthcare professionals for the treatment of GL patients, Juxtapid for the treatment of adult HoFH patients and Mycapssa® for appropriate patients with acromegaly. The most frequent physician call points for our products are endocrinologists (including pediatric endocrinologists) with respect to Myalept and Mycapssa, and lipidologists and cardiologists with respect to Juxtapid.
Our sales and marketing efforts are also focused on obtaining market access for our products, which primarily represents securing pricing and reimbursement approvals at acceptable levels. In the United States, we mobilize a team supporting market access initiatives with the primary responsibility of collaborating with insurance plans, health maintenance organizations and other payers on securing reimbursement and formulary status for Myalept, Mycapssa® and Juxtapid. This team also anticipates authorization and reauthorization requirements and educating physicians and office staff regarding criteria to continue coverage for our products.
Outside the United States, we support this effort primarily through the work of Country Managers and local consultants. We have successfully secured market access for both lomitapide and metreleptin in certain key EU and LATAM countries. We also continue to submit market access dossiers in other EU and LATAM countries where reimbursement has not occurred to date. We intend to enter into price negotiations in such countries following approval of the relevant Health Technology Assessment (“HTA”) body. One of our primary objectives is to strengthen the value proposition for metreleptin and lomitapide for payers through the generation of market access studies to enhance patient, physician and payer knowledge of GL, HoFH, and, in the European Union, PL and the real-world burden of these diseases. We have successfully achieved coverage in the United States and reimbursement in Japan, Turkey, Saudi Arabia, Germany, Italy, the United Kingdom, France, Austria, the Netherlands and Slovenia. Individual patient funding has been obtained in Czech Republic and Hungary. Pricing and reimbursement processes are ongoing in Brazil, Spain, Denmark, Norway, Portugal , Israel, Kuwait, Belgium, Poland, Romania and Latvia. Mycapssa® has been submitted to the EMA in 2021 and is not yet approved in Europe.
Manufacturing Supply and Distribution
We and our contract manufacturers are subject to the FDA’s Current Good Manufacturing Practice (“cGMP”) regulations in the United States and other rules and regulations prescribed by regulatory authorities outside the United States.
Lomitapide is a small molecule drug that is synthesized with readily available raw materials using conventional chemical processes. Hard gelatin capsules are prepared in 5 mg, 10 mg, 20 mg and 30 mg strengths. Metreleptin is a recombinant protein biologic that is produced using conventional fermentation, isolation and purification processing techniques. The drug product is provided globally in nominal 10 mg vials that are reconstituted prior to injection. We developed and received approval from the EMA for new presentations in nominal 2.5 mg and 5 mg vials for commercial distribution in the European Union. Mycapssa is an oral presentation of octreotide acetate for acromegaly. It contains 20mg active in enteric coated hard gelatin capsules. Octreotide acetate is manufactured by solid state peptide synthesis and is compendial material. It is marketed in the US in a 28-count child-resistant DosePack wallet.
We rely on contract manufacturers to produce the drug substance for lomitapide, Mycapssa® and metreleptin and to produce the drug product for commercial supplies and for clinical trials. We manufacture the drug substance for Oleogel-S10 at our facility in Germany. We have long-term supply agreements with the manufacturers of lomitapide, Mycapssa® and metreleptin. We may encounter any issue with our suppliers, for example, in the event a manufacturer notifies us that it intends to terminate its arrangement or if the manufacturer is operating in violation of the cGMP regulations. Prior to appointing a new supplier, we will undertake a rigorous validation and approval process of that manufacturer.
We require supplies of bacteriostatic water for injection (“BWFI”), an approved diluent for reconstitution of metreleptin that allows for use of a reconstituted vial of metreleptin for up to three days when stored appropriately. BWFI is only available for sale and approved for use in the United States, and we or our contract manufacturers will purchase it for supply with named patient sales outside the United States and other expanded access distribution to reduce the number of metreleptin vials provided. BWFI is not required in Europe following the launch of appropriate vial sizes to accommodate the dosing needs of the patient population.
In the United States, lomitapide, metreleptin and Mycapssa® are distributed exclusively through a specialty pharmacy network. Lomitapide and metreleptin are distributed through Accredo Health Group, Inc. (“Accredo”) and Mycapssa® is distributed through AcariaHealth, Inc (“AcariaHealth”). Accredo Specialty and AcariaHealth take title of products upon delivery and have specialty pharmacy networks that provide the products directly to patients and, under limited circumstances, to other U.S. purchasers. The specialty pharmacy takes title upon delivery of products. These specialty pharmacies provide contractual patient service support on behalf of Amryt. For commercial sales outside the United States, including sales of Myalepta and Lojuxta, named patient sales and other expanded access distribution, we use our existing infrastructure and third party providers to distribute products either directly to the purchaser in the applicable country or to our local third party distributor or service provider for such country.
Competition
The industry in which we operate is highly competitive and subject to rapid and significant technological change. Our competitors and potential competitors include large pharmaceutical and biotechnology companies, specialty pharmaceutical and generic drug companies, academic institutions, government agencies and research institutions.
All of these competitors currently engage in, have engaged in or may engage in the future in the development, manufacturing, marketing and commercialization of pharmaceuticals that compete with or may in the future compete with metreleptin, lomitapide or other products or product candidates we may develop or acquire. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Academic institutions, government agencies and other public and private research organizations also are conducting research activities, seeking patent protection and may commercialize products on their own or through joint ventures. The existence of these products, or other products or treatments of which we are not aware, or products or treatments that may be developed in the future, may adversely affect the marketability of products we develop.
| • | Lomitapide: Competition to lomitapide is a class of drugs known as PCSK9 inhibitors, including alirocumab and Evolocumab. These are approved both in the United States and the European Union. Sales of these PCSK9 inhibitors compete with sales of the lomitapide product and we expect they will continue to compete with lomitapide. However, because many therapies, including PCSK9 inhibition products, act by increasing the activity of LDL-R, HoFH patients often have an inadequate response due to their impaired LDL-R function. |
Recently Regeneron Pharmaceuticals Inc. launched Evinacumab for the treatment of HoFH in the US. In August 2019, Regeneron announced positive topline data from its ongoing Phase 3 trial in HoFH and the FDA approved Evinacumab on February 11, 2021, for adults and pediatric patients 12 years and older for treatment of HoFH. EMA approved Evinacumab in June 2021 as an adjunct to diet and other low-density lipoprotein-cholesterol (LDL-C) lowering therapies for the treatment of adult and adolescent patients aged 12 years and older with homozygous familial hypercholesterolaemia (HoFH). Regeneron launched Evinacumab in the United States in March 2021. Evinacumab was licensed to Ultragenyx Pharmaceutical Inc in countries outside the US in January 2022. Although the drug is administered by intravenous infusion, physicians may consider use of this product in HoFH patients before initiation of lomitapide treatment or may switch patients currently treated with lomitapide to Evinacumab treatment.
In addition, other companies may succeed in developing, acquiring or licensing additional pharmaceutical products that are introduced into the market and that are more effective or less costly than our products.
| • | Metreleptin: Our competitors are also developing products, which, if approved and depending on the labelled indication, could potentially compete with metreleptin, including Regeneron Pharmaceuticals, Inc. and Akcea Therapeutics, Inc. Although Myalept is the first and only product approved in the United States for the treatment of complications of leptin deficiency in patients with GL, there are a number of therapies approved to treat these complications independently that are not specific to GL. Myalepta also faces competition in the European Union, both for the treatment of GL and PL. |
| • | Mycapssa: The acromegaly market has many established competitors, dominated in by Novartis and Ipsen, who like the rest of the competition offer injectable SSA’s. Mycapssa® is the only oral SSA available on the market. Crinetics Pharmaceuticals are developing paltusotine, which if approved would be the second oral SSA to enter the market and compete directly with Mycapssa. Crinetics also have a Ph3 study to evaluate effect in treatment-naïve patients. If studies are successful, filings could take place at end of 2023 / early 2024. |
| • | Oleogel-S10: Although there are no approved products in the United States or the European Union for the treatment of EB, some of our competitors are developing products which, if approved, and depending on the labelled indication, could potentially compete with Oleogel-S10. Some companies, including Krystal Biotech, Inc. and Abeona Therapeutics Inc., are developing gene therapy treatments, while other companies, including Castle Creek Pharmaceuticals Holdings, Inc. and Wings Therapeutics, Inc., are developing non-gene therapy treatments. In November 2021 Kyrstal announced that its Ph3 GEM-3 study of B-VEC, known as Vyjuvek™, met its primary endpoint of complete wound healing at six-month timepoints and its secondary endpoint of complete wound healing at three-month timepoints. Additionally, Vyjuvek™ was shown to be well-tolerated, with no drug-related serious adverse events or discontinuations. They could file a Biologics License Application in Q2 2022, with potential approval in the second half of 2022 and file for MAA in EU in the second half of 2022. |
Intellectual Property
We rely on our own patents licensed from third parties, and on other means to protect our technology, inventions and improvements that are commercially important to our business.
Our policy is to file patent applications on a worldwide basis in those jurisdictions where it is considered beneficial to our business, depending on the subject matter and the commercialization strategy.
We have a portfolio of patents and pending patent applications covering various aspects of our lomitapide, metreleptin, Mycapssa, Oleogel-S10 and AP103 assets in a variety of jurisdictions. Our lomitapide patent portfolio includes eight Approved Drug Products with Therapeutic Equivalence Evaluations (“Orange Book”) listed patents licensed from the University of Pennsylvania (“UPenn”) claiming methods of treatment that provide protection from 2025 to 2027 in the United States and into 2025 in Europe, Australia, Japan, South Korea, Canada and New Zealand. Applications are pending in the U.S. and India. Supplementary protection certificates (“SPCs”) have been granted in 26 European countries (including the United Kingdom, Germany, France, Italy and Spain) extending patent protection into 2028. Two additional SPCs applications are pending.
The metreleptin patent portfolio includes two issued U.S. patents claiming methods of treating lipoatrophy, two granted European patents (“EP patents”) claiming methods of treatment (validated in numerous European countries, including the United Kingdom, Germany, France, Italy and Spain), granted patents claiming methods of treatment in other jurisdictions including Australia, Canada, and Japan as well a pending applications in the United States, all of which have been licensed to Amryt or are owned by Amryt. The granted patents provide protection from 2022 to 2027 in the United States and into 2022 in Europe and other jurisdictions. A US patent claiming methods of detecting anti-leptin neutralizing antibodies provides protection until 2038. Additional applications claiming methods of detecting anti-leptin neutralizing antibodies are pending in the United States, Brazil, Canada, China, Eurasia, India, Hong Kong, Japan and Mexico; if granted, patent protection would expire in 2037.
Our Mycapssa® patent portfolio includes eight Approved Drug Products with Therapeutic Equivalence Evaluations (“Orange Book”) listed patents, including three formulation and one acromegaly use patents which expire in 2029, three acromegaly use patents which expire in 2036, one octreotide method of use patent expiring 2040, as well as one pending U.S. application which covers NET/VIP use which if allowed would expire in 2037. In Europe, there are two granted composition patents which expire in 2029, one pending Acromegaly use Application, which if allowed would expire 2036 and one NET Use Application which if allowed would expire in 2037.
We were first granted patent protection for Oleogel-S10 in Japan in 2010. Key patent grants for Oleogel-S10 in Europe and the United States followed in 2013. If Oleogel-S10 is approved in the U.S. our Oleogel-S10 patent portfolio includes one U.S. patent that will be listable on the Orange Book covering oleogel compositions providing exclusivity in the United States to 2025, two U.S. patents that will be listable on the Orange Book covering treatment of EB that provide exclusivity in the United States into 2030 and one U.S. patent that will be listable on the Orange Book covering treatment of EB into 2039. Our non-U.S. patents for Oleogel-S10, include EP patents (validated in various European countries including the United Kingdom, Germany, France, Italy and Spain) covering the Oleogel-S10 composition and methods of healing wounds with Oleogel-S10, expiring in 2025 and 2030, respectively. Supplementary protection certificates have been obtained in various EU countries, extending the expiration of the composition patents in Europe from 2025 to 2030. We have filed non U.S. patent applications directed to the clinical formulation and methods of manufacturing and treatment therewith; the term of any patents that are derived from this application would expire in 2039.
The AP103 patent portfolio includes one granted EP patent (validated in various countries including the United Kingdom, Germany, France, Italy and Spain) claiming HPAE polymers and polyplexes comprising HPAE polymers, and transfection methods using such polymers, as well as pending U.S. and European applications. These patents and applications have been in-licensed from the University College Dublin and are expected to provide exclusivity into 2035. We have also filed U.S. and European applications claiming a proprietary genus of HPAEs and novel HPAE polyplexes, as well as claiming a proprietary and scalable HPAE polyplex manufacturing methods; the term of any patents that are derived from these applications would expire in 2039. See “—Material Contracts—University College Dublin In-License Agreement.”
Juxtapid was granted Orphan Drug status in the United States for the treatment of HoFH and for the treatment of FCS. This designation provided exclusivity to December 2019 for the approved HoFH indication and can provide potentially seven years of exclusivity for the FCS indication, if that indication is approved. Lojuxta obtained Orphan Drug status in the European Union for the treatment of FCS. Upon approval of an FCS indication in the European Union, 10 years of exclusivity (plus two years on successful completion of a PIP) could be obtained.
Following approval by the EMA in July 2018, metreleptin is entitled to ten years of market and Orphan Drug exclusivity in the European Union (to July 2028 which can be extended a further two years upon successful completion of a PIP) for the treatment of familial partial lipodystrophy, Barraquer-Simons syndrome, Lawrence syndrome, and Berardinelli-Seip syndrome. In the United States Myalept should also qualify for a 12-year period of exclusivity from biosimilar or interchangeable products, which will expire in 2026, under the Biologics Price Competition and Innovation Act. Myalept could also receive orphan designation upon submission and approval of a PL indication in the United States which would provide seven years of exclusivity for this indication following approval in the United States.
In January 2020, an exclusivity justification orphan drug exclusivity for Mycapssa, for the treatment of Acromegaly in the U.S. was submitted to the Office of Orphan Products Development (OOPD) at FDA. If awarded Mycapssa® will have 7 years orphan drug exclusivity for the treatment of Acromegaly in the U.S. If Mycapssa® is approved by EMA in the European Union and, subject a positive opinion from COMP regarding maintenance of the Orphan Designation (OD), Mycapssa® will have 10 years market exclusivity from the date of approval.
Oleogel-S10 has been granted Orphan Drug status for the treatment of EB in the European Union and the United States. Should Oleogel-S10 be granted approval it will be entitled to Orphan Drug exclusivity for the treatment of EB, extending protections for seven years in the United States and, subject to a positive opinion from COMP regarding maintenance of the Orphan Designation (OD), ten years in the European Union from the date of approval in the respective jurisdictions. The ten-year Orphan Drug exclusivity period in the European Union can be extended a further two years upon successful completion of a PIP.
Material Contracts
The following summary of our material contracts is not intended to be a complete description of these contracts, and it is qualified in its entirety by reference to the full text of such contracts, which are filed as exhibits to the registration statement of which this annual report forms a part. We urge you to review these exhibits in their entirety.
The following summary excludes contracts with our principal shareholders that we entered in connection with the acquisition of Aegerion, which are summarized under “Related Party Transactions.”
Agreement and Plan of Merger Between Amryt Pharma Plc. Amryt Merger Sub and Chiasma, Inc.
On May 5, 2021, Amryt announced that it had signed a definitive agreement to acquire Chiasma, Inc. in an all-stock combination. Under the terms of the transaction, each share of Chiasma common stock issued and outstanding prior to the consummation of the transaction was exchanged for 0.396 Amryt American Depositary Shares (“ADSs”), each representing five Amryt ordinary shares. The merger agreement provided that, upon the terms and subject to the conditions set forth in the merger agreement, at the effective time, Amryt Merger Sub, an indirect wholly owned subsidiary of Amryt, merged with and into Chiasma with Chiasma surviving as an indirect wholly owned subsidiary of Amryt subsequently renamed Amryt Endo, Inc. which then ceased to be a publicly traded company.
On August 5, 2021, Amryt completed the acquisition of Chiasma, Inc. and, in conjunction with the completion, Amryt allotted and issued a total of 127,733,680 ordinary shares as consideration for the acquisition. Following the completion, shareholdings in Chiasma were rounded in being converted to Amryt shares using the exchange ratio of 0.396. Roundings in converting Chiasma shareholdings to Amryt shares were finalized in August 2021 and resulted in an additional 7,015 ordinary shares being allotted and issued by Amryt as consideration for the acquisition. In total, these ordinary shares were issued to the former Chiasma Shareholders in the form of 25,548,139 ADSs at US$10.19 per share, to acquire Chiasma for a value of US$260,336,000. On August 5, 2021, the Group repaid US$116,629,000 of Chiasma long term debt.
Amryt’s $125m Debt-Refinancing
On February 22, 2022, Amryt announced that it secured US$125 million of senior credit facilities from funds managed by the Credit Group of Ares Management Corporation (“Ares”). Amryt used a portion of the proceeds to refinance its previous secured term debt facility, which had an outstanding balance of US$94.0, million as at February 22, 2022, an interest rate of 13.00% and a maturity date of September 2024. The new facilities will generate significant annual interest cost savings as well as provide for important strategic flexibility as Amryt looks to continue to grow its global rare disease presence.
Key features of the facilities include:
| • | Total new facilities of $125 million, consisting of: |
| o | $85 million Term Loan Facility with interest rate of Secured Overnight Financing Rate (“SOFR”)+6.75%, subject to a 0.90% SOFR floor |
| o | $40 million Revolving Credit Facility with $20 million drawn at close and interest rate of SOFR+4.00%, subject to a 0.90% SOFR floor |
| o | Quarterly blended cash interest rate of SOFR+5.87% (assuming fully drawn), subject to a 0.90% SOFR floor, substantially lower than Amryt’s current secured term debt facility at 13.00% interest |
| • | Requires interest-only payments until facility matures in February 2027 |
| • | There are no warrants or any equity conversion features associated with the new facilities |
| • | The proceeds will be used to refinance existing debt, for general corporate and product development purposes; and potentially for shareholder approved share repurchase programs. |
Charges were taken over certain assets of the company and its material entities as guarantee and collateral for the provision of the debt.
Share Purchase and Transfer Agreement between Software AG – Stiftung and Dr. Armin Scheffler and Amryt Pharmaceuticals DAC
On October 16, 2015, Amryt Pharmaceuticals DAC entered into a share purchase and transfer agreement (“Birken SPA”) with Software AG – Stiftung and Dr. Armin Scheffler in their capacity as sellers (together, “Birken Sellers”) relating to the purchase of the entire issued share capital of Birken AG (now Amryt GmbH). Under the Birken SPA, the consideration for the shares of Birken AG comprised the allotment and issue of new ordinary shares in Amryt Pharmaceuticals DAC, the sum of €1,000,000 plus additional milestone and royalty payments if/when certain milestones are met. As of the date of this annual report, we have made milestone payments of €12 million under this agreement. Remaining milestone payments are as follows:
| • | €10 million once net ex-factory sales/net revenue of Oleogel-S10 first exceed €50 million in any calendar year; |
| • | €15 million once net ex-factory sales/net revenue of Oleogel-S10 first exceed €100 million in any calendar year; and |
| • | €10 million on receipt of marketing approval by the EMA or the FDA for the treatment of EB. |
The royalty payments payable to the Birken Sellers are as follows: (a) 9% of (i) net ex-factory sales, and (ii) net revenues in either case relating to Oleogel-S10; and (b) 6% of: (i) net ex-factory sales; and (ii) net revenues relating to other betulin products, with the relevant royalty periods essentially being ten years from first commercial sale of the relevant product (other than in respect of Imlan).
University College Dublin In-License Agreement
On March 14, 2018, Amryt Genetics Limited entered into an agreement with University College Dublin relating to the patent rights for AP103. Under this agreement, University College Dublin granted an exclusive, worldwide license to the patent rights for the platform technology to develop one or more gene therapy products for therapeutic use in any disorder in humans and animals. University College Dublin retains an irrevocable, perpetual, royalty-free, worldwide right to use the licensed intellectual property for the purposes of academic teaching, publication and non-commercial research. The subject platform technology relates to a non-viral gene therapy, which offers a potential treatment for patients with EB. Preliminary data available to us suggests that the treatment could be potentially disease-modifying for patients with RDEB. We intend to conduct various preclinical studies in connection with the agreement.
In consideration of the agreement, Amryt Genetics Limited made an upfront payment of €40,000 and agreed to the following milestone payments:
| • | €100,000 on the successful completion of a Phase 2a proof of concept study; |
| • | €100,000 on the successful completion of a Phase 2b study; |
| • | €200,000 upon the first commercial sale of a gene therapy product incorporating or utilizing the licensed technology; and |
| • | €200,000 upon the first commercial sale of each subsequent product requiring a separate marketing authorization. |
Upon the sale of product, we are obligated to make royalty payments to University College Dublin as follows: (a) 2% on net sales in a calendar year up to €100 million, and (b) 3% on net sales in a calendar year in excess of €100 million. Under the agreement, we must also pay to University College Dublin a portion of the royalties we receive from our affiliates in connection with the product: (a) 30% of net royalties prior to the initiation of a Phase 1 study, (b) 20% of net royalties commencing upon the initiation of a Phase 1 study and before the completion of a Phase 2a proof of concept study, and (c) 10% of net royalties commencing upon the completion of a Phase 2a proof of concept study.
The agreement continues in full force on a product-by-product and country-by-country basis until the later of (a) the expiration of the last valid claim of patent rights, (b) the expiration of orphan drug exclusivity, or (c) 15 years after the first commercial sale of the first product by us or our affiliates or sub-licensees. University College Dublin may terminate the agreement with 30 business days’ prior written notice under the following circumstances:
| • | if we are in material breach of any provision of the agreement and, after receiving notice from University College Dublin identifying a material breach, we fail to cure said material breach within 60 business days, University College Dublin may issue a notice of default. If we do not cure the material breach within 60 business days from receipt of the notice of default, then University College Dublin may terminate the agreement; |
| • | if we become insolvent, or if an interim order is applied for or made, or a voluntary arrangement approved, or a voluntary arrangement is proposed or approved or an administration order is made, or a receiver or administrative receiver is appointed for any of our assets or undertaking or a winding-up resolution or petition is passed or presented (otherwise than for the purposes of reconstruction or amalgamation), or if any circumstances arise which entitle the court or a creditor to appoint a receiver, administrative receiver or administrator or to prevent a winding-up petition or make a winding-up order, or other similar or equivalent action is taken against or by us by reason of our insolvency or in consequence of debt, or if we make any arrangement with our creditors; |
| • | if we or our affiliates challenge the validity of the patent applications when granted, or assist any third party to commence legal proceedings to challenge such validity; |
| • | if, according to an independent expert’s judgment, we have failed to use commercially reasonable efforts to develop, use and exploit the intellectual property licensed under the agreement, and six months after the independent expert’s judgment we have still failed to take the specific actions to develop, use and exploit the intellectual property licensed, then University College Dublin may at any time within three months after the end of that six-month period, provide at least three months’ notice to us to terminate the agreement; |
| • | if we fail to pay any amount due under the agreement within 30 business days having received written notice from University College Dublin of our failure to pay, and such failure to pay is not subject to a good faith dispute between the parties; and |
| • | if we dispose of all, or a substantial part of our business involving the licensing of the intellectual property under the agreement in circumstances where we do not enter into a novation agreement pursuant to us becoming insolvent or any other circumstance described above. |
We may terminate the agreement at any time by giving University College Dublin at least 60 business days’ prior written notice. Upon expiration or termination of the agreement, all rights and licenses granted to us under the agreement also terminate.
University of Pennsylvania Lomitapide License Agreement
On May 19, 2006, Aegerion entered into an agreement with UPenn for the in-license of lomitapide. Pursuant to the terms of the agreement Aegerion was granted an exclusive and worldwide license (including right to sublicense) to certain patents of UPenn and certain patents assigned to UPenn by Bristol-Myers Squibb (“Bristol-Myers”). The license is provided for the following field of use: (a) monotherapy or with other dyslipidemic therapies to treat patients with HoFH; and (b) monotherapy or in combination with other dyslipidemic therapies for treatment of patients with other severe forms of hypercholesterolemia unable to come within 15% of National Cholesterol Education Program’s (“NCEP”) goal on maximal tolerated oral therapy, as determined by the patient’s prescribing physician, with severe combined hyperlipidemia unable to come within 15% of NCEP’s non-high-density lipoprotein-cholesterol goal on maximal tolerated oral therapy, as determined by the patient’s prescribing physician, or with severe hypertriglyceridemia unable to reduce triglycerides less than 1,000 on maximal tolerated therapy.
In partial consideration of the in-license of lomitapide, we are required to pay UPenn a royalty of 2% based on patent coverage of the product and annual net sales on a country-by-country basis made pursuant to the terms of the agreement. The sub-license fees are as follows: (i) 25% of sub-licensing fees received for net sales in a country that is covered by a valid claim of the licensed patent rights and (ii) 15% of all other sub-license fees.
If the indication is not HoFH or severe refractory hypercholesterolemia, then additional payments are required to be made upon the initiation of a Phase 3 trial ($300,000), the filing of a U.S. NDA ($750,000) and the grant by the FDA of approval of an NDA ($1,500,000).
The agreement expires on a country-by-country basis upon there no longer being a valid claim of licensed patent rights. Upon the expiration in a country, the license becomes irrevocable and fully paid-up. We can also terminate the agreement at any time.
Japan Lomitapide License Agreement
On February 5, 2019, Aegerion entered into a license agreement with Recordati for the commercialization of Juxtapid in Japan. Under the terms of the License Agreement, and subject to the conditions set forth therein, Aegerion granted to Recordati an exclusive license in Japan, for the current marketed indication for HoFH. During the term of the License Agreement, Recordati also has an exclusive right of first negotiation to any new indications for Juxtapid in Japan that may be developed and the right to grant sub-licenses and to manufacture and commercialize Juxtapid, under specific circumstances.
Pursuant to the terms of the License Agreement, and subject to the conditions set forth therein, Recordati is required to make the following payments: (i) $25 million as a one-time upfront payment on the effective date of the agreement (which was paid in the first quarter of 2019), (ii) $5 million as a one-time payment within 45 days following the date on which the Japan marketing authorization for Juxtapid is successfully transferred to Recordati (“Completion Date”) (which was paid in the second quarter of 2019), (iii) quarterly royalty payments, during the term of the License Agreement, equal to 22.5% of all net sales of Juxtapid in Japan, and (iv) 20% of all other sublicense revenues received by Recordati or any of its affiliates.
In addition, pursuant to the terms of the License Agreement, we may receive from Recordati commercial milestone payments (up to a total of $80 million) for net sales in Japan, conditioned and based upon the achievement of certain net sales levels in Japan, the first $12.5 million installment of which becomes payable at the end of the first quarter in which cumulative net sales in Japan reach $70 million, and which are payable in incremental installments thereafter at the end of each quarter in which cumulative net sales in Japan increase by $70 million (in incremental payments of $12.5 million until cumulative net sales reach $280 million and then in incremental installments of $5 million for each incremental $70 million of revenues until cumulative net sales reach $700 million).
Further, pursuant to the current license agreement with UPenn, UPenn is entitled to receive 15% of the $25 million upfront payment, 15% of the marketing authorization transfer milestone and any subsequent sales milestone payments received from Recordati and 25% of royalty payments that we receive from Recordati under the License Agreement.
We have also entered into a customary supply agreement with Recordati under which we will supply Juxtapid to Recordati (or its affiliate) at cost plus an agreed upon markup for an initial term of two years with automatic renewal for successive two year terms.
The initial term of the License Agreement continues until the latest of: (i) expiration of the last valid claim of the licensed patents covering Juxtapid in Japan, (ii) expiration of data or regulatory exclusivity in relation to Juxtapid in Japan, or (c) ten years from the Completion Date. Thereafter the term of the License Agreement will automatically renew for a single five-year term, and then thereafter for successive five-year terms unless either party provides written notice at least 18 months prior to the end of the then current renewal term. Either party may terminate the License Agreement for cause if the other party materially breaches or defaults in the performance of its obligations, and, if curable, such material breach remains uncured for 90 days (15 days for non-payment).
Metreleptin Asset Purchase Agreements
In November 2014, Aegerion entered into an asset purchase agreement relating to the acquisition from Amylin Pharmaceuticals, LLC (“Amylin”) and AstraZeneca Pharmaceuticals LP (“AstraZeneca”), an affiliate of Amylin, of certain assets and rights associated with the biological product metreleptin for injection. Under the terms of the agreement, Aegerion paid AstraZeneca $325 million upfront to acquire the global rights to develop, manufacture and commercialize metreleptin, subject to an existing distributor license with Shionogi covering Japan, South Korea and Taiwan, which Aegerion assumed upon closing. The transaction did not include the transfer of any AstraZeneca employees or facilities.
On January 9, 2015, Aegerion entered into a letter agreement with AstraZeneca and Bristol-Myers, which was subsequently amended on April 30, 2015. This agreement was entered into pursuant to Aegerion’s acquisition of metreleptin from AstraZeneca. Under the asset purchase agreement and letter agreement, Aegerion agreed to fulfill certain of AstraZeneca’s original obligations to Bristol-Myers including reporting, milestone and royalty payments and diligence obligations in relation to metreleptin.
Pursuant to the terms of the agreements we are required to pay 5-10% royalties to Bristol-Myers, as previously paid by AstraZeneca, on the net U.S. sales of metreleptin.
Amgen License Agreement
In connection with Aegerion’s acquisition of metreleptin in January 2015, Aegerion acquired a license agreement between Amgen and Amylin, dated February 7, 2006 (“Amgen License”) pursuant to which Aegerion obtained an exclusive worldwide license from Amgen to certain know-how and patents and patent applications covering the composition of matter and methods of use of metreleptin to develop, manufacture and commercialize a preparation containing metreleptin (“Amgen Licensed Products”).
As part of the Amgen License, Aegerion also obtained an exclusive sublicense of Amgen’s exclusive rights to certain metreleptin-related patents and patent applications owned by the Rockefeller University and exclusively licensed to Amgen under a license agreement dated April 14, 1995, as amended (“Rockefeller License”) and an exclusive sublicense of Amgen’s non-exclusive rights to certain metreleptin-related patents and patent applications owned by The Regents of the University of California and non-exclusively licensed to Amgen under a license agreement dated July 13, 2005 (“UCSF License”). Amgen retains rights to conduct research, development, manufacturing and commercialization activities with respect to products other than the Amgen Licensed Products.
We may grant sublicenses under the licenses and sublicenses granted by Amgen, subject to certain limitations, including Amgen’s right of first offer for any out-license, partnership, co-development, commercialization, co-promotion or similar agreement related to metreleptin or the Amgen Licensed Products, which expired in February 2021.
We are required to make royalty payments to Amgen and Rockefeller University on net sales of each Amgen Licensed Product on a country-by-country basis (i) at a royalty rate in the low double digits where the Amgen Licensed Product has patent protection or market exclusivity granted by a regulatory authority at the time of regulatory approval in the applicable country during the applicable royalty term, which runs on a country-by-country basis until the later of (a) the expiration of the last-to-expire valid claim covering an Amgen Licensed Product in the applicable country, (b) the expiration of any market exclusivity granted by a regulatory authority, and (c) ten years from the date on which an Amgen Licensed Product is first sold to a third party in a country after regulatory approval for the Amgen Licensed Product has been granted in such country (“Amgen Royalty Term”) or (ii) at a royalty rate in the mid-single digits to low double digits where the Amgen Licensed Product receives patent protection or market exclusivity following the time of regulatory approval in the applicable country, in either case subject to a variety of customary reductions.
Under the Amgen License, we are also required to directly meet certain payment obligations under the Rockefeller License and UCSF License. We are required to make royalty payments to Rockefeller University on net sales of each product with patent rights or know-how in the field of obesity genes, obesity gene products, and molecules that modulate or mediate their action and/or regulation on a country-by-country basis at a range of royalty rates in the low single digits depending on whether the product has an orphan product designation or not until the later to occur of expiration of (i) patent protection, (ii) any market exclusivity period granted in the applicable country, or (iii) any data exclusivity period in the applicable country (with certain limitations related to the number of units sold). Since acquiring this license agreement in January 2015, a one-time $5 million milestone payment was paid to Rockefeller University in February 2015, which was due 12 months following the receipt of marketing approval for Myalept in the United States. We will also be required to pay to Rockefeller University a percentage in the low double digits of any upfront license fees or one-time fees it receives in consideration for a sublicense of the licensed rights. There are no material payment obligations outstanding under the UCSF License.
Current patent protection exists from 2022 to 2027 in the United States and into 2022 in the European Union, as well as orphan exclusivity in the European Union into 2028. A US patent application claiming methods of detecting anti-leptin neutralizing antibodies provides protection until 2038. Additional applications claiming methods of detecting anti-leptin neutralizing antibodies are pending in the United States, Brazil, Canada, China, Eurasia, India, Japan and Mexico; if granted, patent protection would expire in 2037.and 2039, if extended upon successful completion of a PIP, respectively
We assumed this agreement upon the acquisition of Aegerion. We have the right to terminate the agreement at any time.
National Institutes of Health (“NIH”) License Agreement
In February 2017, Aegerion entered into an in-license agreement in respect of patent rights for metreleptin with the NIH. Pursuant to the terms of the agreement Aegerion was granted an exclusive and worldwide license to (including the right to sub-license) the NIH patent rights for the use of leptin, leptin analogs and derivatives for the diagnosis, prevention and treatment of human lipodystrophy.
Upon the date of the first commercial sale of a licensed product in a country other than the United States, we have agreed to pay NIH a royalty percentage of 0.4% of net sales, on a country-by-country basis, in such countries in which a valid patent claim exists. No U.S. royalty is due on the sales.
The agreement expires upon the expiration of the last to expire licensed patent rights. We can terminate the agreement at any time and NIH has the following termination and modification rights:
Other than NIH having the right to immediately terminate the agreement should we become insolvent, file for bankruptcy or receive notice of a third party’s intention to file an involuntary petition in bankruptcy, NIH may terminate the agreement if we default on any material obligation under the agreement which has not been remedied within 90 days of the notice of default being provided, including the following:
| • | we have not achieved certain agreed performance milestones; |
| • | we have willfully made a false statement of, or willfully omitted, a material fact in the license application or in any report required by the agreement; |
| • | we have committed a material breach of a covenant or agreement contained in the agreement that has not been remedied within 90 days; |
| • | we have not been keeping the licensed products or the licensed processes reasonably available to the public after commercial use commences; or |
| • | we cannot reasonably satisfy unmet health and safety needs. |
Accredo Agreements
On December 6, 2013, Aegerion entered into a distribution agreement in respect of lomitapide with Accredo.
Pursuant to the agreement Accredo is appointed as an authorized non-exclusive distributor of record of lomitapide. Amryt may appoint other distributors upon 60 days’ notice to Accredo (such notice must also be provided by Amryt when other specialty pharmacies are added to the distribution network). If Amryt exercises this right and such an appointment causes Accredo to no longer be one of three (or less) specialty pharmacies, Accredo may terminate the agreement. In addition, either party may terminate the agreement at any time upon 180 days’ written notice to the other party.
On December 6, 2013, Aegerion entered into a master services agreement in respect of metreleptin with Accredo. The agreement sets the framework of non-exclusive commercial relationship between the parties. Pursuant to the agreement, Accredo agreed to perform services on our behalf, related to metreleptin as described in one or more statements of work provided to us. Such services include, but are not limited to case management and ongoing clinical support services. In consideration for services provided, we are required to pay Accredo the fees set forth in the applicable statement of work. We have the right to terminate at any time upon 120 days’ notice and Accredo may terminate on one year’s notice.
The distribution agreement expires on June 30, 2022, and the Accredo master services agreement expires in December 2023.
Metreleptin Contract Manufacturing Agreement
We currently have in place a contract manufacturing agreement with Sandoz GmbH in respect of metreleptin, effective as of September 30, 2010. Pursuant to the terms of the agreement, Sandoz GmbH is appointed as non-exclusive manufacturer of metreleptin. Sandoz GmbH is obligated to produce a majority of our metreleptin product demand and Amryt has committed to purchase such amount and is committed to a contract value for 2022 in the amount of €9.9 million. The agreement expires on December 31, 2023. Either party may terminate the agreement for any scientific, regulatory, safety or economic reason to the end of each calendar year by giving 24 months’ prior written notice.
TEVA Manufacture & Supply Agreement
On December 31, 2015, Chiasma entered into a commercial manufacture, purchase and supply agreement with TEVA API Inc for the manufacture, purchase and supply of R&D and registration quantities of API (active pharmaceutical ingredient) for further processing into Mycapssa.
The terms of the agreement require Chiasma to purchase minimum percentages of Chiasma’s total annual requirements of Octreotide acetate in the manufacturing of Mycapssa. The consideration payable by Chiasma to TEVA API Inc is calculated based on the annual consumption by Chiasma of the API set forth in binding purchase orders issued in a calendar year. The agreement expires on the seventh anniversary of the first commercial sale of Mycapssa® to an independent third-party in a region or any country in a region outlined in the supply agreement.
AcariaHealth, Inc Specialty Pharmacy Provider Dispensing and Services Master Agreement
On August 21, 2020, Chiasma entered into a product purchase and pharmacy services master agreement with AcariaHealth whereby Chiasma agreed to sell Mycapssa® to AcariaHealth in the United States and Puerto Rico on a non-exclusive basis. Additionally, the parties agree that AcariaHealth will perform certain services performed in the normal course of business for similarly situated pharmacies dispensing products including enhanced services for which a fair market value fee is payable by Chiasma. The agreement has an initial term of two years and automatically renews for successive one year renewal terms and can be terminated on ninety days’ notice along with other customary termination provisions.
AssistRx Master Services Agreement
On May 8, 2020, Chiasma entered into a master services agreement with AssistRx, Inc. to provide certain specialty pharmacy services for Mycapssa. The agreement is for a period of three years with automatic one year renewals unless either party terminates within ninety days of the expiration of the initial term or renewal term (as applicable). The agreement may also be terminated by either party for a material and continuing breach that remains uncured beyond thirty days following receipt of a written notice of breach from the non-breaching party along with other customary termination provisions.
Cardinal Health Exclusive Distribution Agreement
On June 23, 2020, entered into an exclusive distribution agreement with Cardinal Health 105, Inc., as its exclusive third-party logistics distribution agent for commercial sales in the United States of all pharmaceutical products manufactured and/or marketed by Chiasma as well as traditional 3PL Operating Guidelines including storage, distribution, returns, customer support, financial support, EDI and system access support.
The term of the agreement is for an initial period of three years following the first shipment of FDA-approved product to a commercial customer unless terminated earlier with one year automatic renewals unless terminated by either party at least ninety days prior to the end of the initial term or any renewal term. Either party has the right to immediately terminate the agreement upon other customary material termination provisions.
Government Regulation and Product Approval
Government authorities in the United States, at the federal, state and local level, the European Union, EU Member States, and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labelling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of the products. In the future, if we were to develop or acquire any other products, or any product candidates, they would also be subject to such regulations and oversight. Our products must be approved by the FDA through the NDA or the BLA process before they may be legally marketed in the United States and must be approved by foreign regulatory authorities via various procedures before they can be marketed in the applicable country, including the EMA or the regulatory authorities of the EU Member States before they can be placed on the market in the European Union.
U.S. Regulatory Matters
Drug and Biologic Development Process
In the United States, the FDA regulates drugs under the FDCA, and regulates biologics under both the FDCA and the Public Health Service Act of 1944 (“PHSA”). The process of obtaining marketing approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject a company to administrative or judicial sanctions. These sanctions could include, among other things, the FDA’s refusal to approve pending applications, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters and other types of enforcement-related letters, product recalls, product seizures, changes to the conditions surrounding marketing approval such as labelling changes or changes to a REMS program, total or partial suspension of production or distribution, injunctions, civil money penalties, fines, refusals of government contracts, debarment, restitution, disgorgement of profits, or civil or criminal investigations and penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
The process required by the FDA before a drug or biologic may be marketed in the United States is extensive and generally involves the following:
| • | completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices and other applicable regulations; |
| • | submission to the FDA of an investigational new drug (“IND”) application, which must become effective before human clinical trials may begin; |
| • | performance of human clinical trials, including adequate and well-controlled trials, according to Good Clinical Practice to establish the safety and efficacy of the proposed drug for its intended use, or the safety, purity and potency of a biological product; |
| • | approval by an independent IRB, representing each clinical site before each clinical trial may be initiated; |
| • | submission to the FDA of an NDA or BLA; |
| • | completion of registration batches and validation of the manufacturing process to show that the producer is capable of consistently producing quality batches of product; |
| • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with cGMP to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
| • | FDA review and approval of the NDA or BLA. |
Once a pharmaceutical candidate is identified for development it enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies, to assess the safety and quality of the product. Animal studies must be performed in compliance with the FDA regulations and the U.S. Department of Agriculture’s Animal Welfare Act of 1966. Human clinical trials cannot commence until an IND application is submitted and becomes effective. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. The sponsor also will include a protocol detailing, among other things, the objectives of the first phase of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated, if the first phase lends itself to an efficacy evaluation. Preclinical testing may continue after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30 day time period, places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Clinical holds also may be imposed by the FDA at any time before or during studies due to safety concerns or non-compliance, or other reasons.
All clinical trials must be conducted under the supervision of one or more qualified investigators. The conduct of clinical trials is subject to extensive regulation, including the FDA’s bioresearch monitoring regulations and Good Clinical Practice (“GCP”) requirements, which establish standards for conducting, recording data from, and reporting the results of, clinical trials, and are intended to assure that the data and reported results are credible and accurate, and that the rights, safety, and well-being of study participants are protected. These regulations include the requirement that all research subjects provide informed consent. Further, an IRB must review and approve the plan for any clinical trial before it commences at any institution. An IRB considers, among other things, whether the risks to individuals participating in the trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the information regarding the trial and the consent form that must be provided to each trial subject or his or her legal representative and continues to provide oversight of the study until it is completed. In addition, companies sponsoring the clinical trials, investigators, and IRBs also must comply with regulations and guidelines for complying with the protocol and investigational plan, adequately monitoring the clinical trial, and timely reporting of adverse events. Foreign studies conducted under an IND must meet the same requirements that apply to studies being conducted in the United States. Data from a foreign study not conducted under an IND may be submitted in support of an NDA or BLA if the study was conducted in accordance with GCP and the FDA is able to validate the data through an onsite inspection, if necessary.
Each new clinical protocol must be submitted for FDA review, and to the IRBs for approval. Protocols detail, among other things, the objectives of the study, the primary and secondary endpoints of the study, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| • | Phase 1. The investigational drug is initially introduced into healthy human subjects, and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing may be conducted in patients with the target diseases. |
| • | Phase 2. This phase involves trials in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| • | Phase 3. This phase involves trials undertaken after preliminary evidence of effectiveness has been obtained and is intended to further evaluate dosage and clinical efficacy and safety of the drug, or the safety, purity, and potency of a biological product, in an expanded patient population, often at geographically dispersed clinical trial sites. These trials are intended to establish the overall risk/benefit ratio of the product, and to provide an adequate basis for product approval and product labelling. |
Progress reports detailing developments associated with the clinical testing program must be submitted at least annually to the FDA, and safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events or animal test results that suggest a significant risk to human subjects. Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within any specified period, if at all. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Further, success in either preclinical studies or early-stage clinical trials does not assure success in later-stage clinical trials. Frequently, product candidates that have shown promising results in early clinical trials have subsequently suffered significant setbacks in later clinical trials. In addition, the design of a clinical trial can determine whether its results will support approval of a product, and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. Sponsors of certain interventional clinical trials, except for Phase 1 trials, are required to submit certain clinical trial information for inclusion in the public clinical trial registry and results data bank maintained by the NIH, which are publicly available at http://clinicaltrials.gov.
Concurrent with clinical trials, companies usually complete additional studies in non-human models, and must also develop additional information about the chemistry and physical characteristics of the product, and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements.
The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. In addition, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
U.S. Review and Approval Processes
The results of product development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the product, proposed labelling, and other relevant information are submitted to the FDA as part of an NDA or BLA requesting approval to market the product. The submission of an NDA or BLA generally is subject to the payment of a user fee, which is currently ca. $3.1 million for FY 2022 for applications requiring clinical data, although NDAs or BLAs for designated Orphan Drugs are exempt from this fee. An approved NDA or BLA is generally subject to an annual program fee, currently $369,413, per strength up to a maximum of five user fees per NDA or BLA, for FY 2022.
In addition, under the U.S. Pediatric Research Equity Act of 2007, as amended, an application or supplement to an application for a drug with certain novel features (e.g., new active ingredient, new indication, new dosage form) must contain data to assess the safety and effectiveness of the drug or biologic for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant deferrals or full or partial waivers for submission of this data. Unless otherwise required by regulation, the act does not apply to any drug or biologic for an indication for which orphan designation has been granted.
The FDA conducts a preliminary review of all NDAs and BLAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. The FDA may request additional information rather than accept an application for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. Under the Food and Drug Administration Amendments Act of 2007, the FDA is required to refer an NDA for a new chemical entity to an advisory committee prior to approval or explain why such review is not necessary. The FDA is not bound by the recommendation of an advisory committee, but it frequently follows such recommendations. The approval or licensure process is lengthy and difficult, and the FDA may refuse to approve an NDA or BLA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data and information. The FDA reviews an application to determine, among other things, whether a drug is safe and effective for its intended use, or whether a biologic is safe, pure, and potent, and whether its manufacturing is cGMP-compliant to assure and preserve the product’s identity, strength, quality and purity. Before approving an NDA or BLA, the FDA will typically inspect the facility or facilities where the product is manufactured. In addition, the FDA often will conduct a bioresearch monitoring inspection of the clinical trial sites involved in conducting pivotal studies to ensure data integrity and compliance with applicable GCP requirements.
Applications receive either standard review or priority review, which is reserved for a product that represents a major advance in treatment or a treatment where no adequate therapy exists. Under the U.S. Prescription Drug User Fee Act of 1992 (“PDUFA”), the FDA has ten months in which to complete its initial review of a standard new molecular entity NDA or original BLA and six months for a priority review new molecular entity NDA, BLA, or efficacy supplement. The FDA does not always meet its PDUFA goal dates and in certain circumstances the PDUFA goal date may be extended. In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and which provide meaningful therapeutic benefit over existing treatments, may receive accelerated approval. In that situation, the product may be approved on the basis of adequate and well-controlled clinical trials establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, a sponsor of a drug or biologic receiving accelerated approval must perform post-marketing studies to validate the surrogate endpoint or otherwise confirm the effect of the product on a clinical endpoint, and the product may be subject to accelerated withdrawal procedures.
If a product receives marketing approval, the approval may be significantly limited to specific diseases, dosages or patient populations, or the indications for use may otherwise be limited, which could restrict the commercial value of the product. In addition, the FDA may impose distribution and use restrictions and other limitations on labelling and communication activities with respect to an approved product via a REMS program, which could include medication guides, patient package inserts, physician communication plans, restricted distribution methods, and patient registries.
The goal of the Juxtapid REMS is to mitigate the risk of hepatotoxicity associated with the use of Juxtapid by ensuring that:
| • | prescribers are educated about the approved indication for Juxtapid, the risk of hepatotoxicity associated with the use of Juxtapid; and the need to monitor patients during treatment with Juxtapid as per product labeling, |
| • | Juxtapid is dispensed only to patients with a clinical or laboratory diagnosis consistent with homozygous familial hypercholesterolemia (HoFH), and |
| • | patients are informed about the risk of hepatotoxicity associated with the use of Juxtapid and the need for baseline and periodic monitoring. |
The Juxtapid REMS includes the following Elements to Assure Safe Use: (i) certification of Healthcare Providers who prescribe Juxtapid, (ii) certification of the pharmacies that dispense Juxtapid, and (iii) Juxtapid must be dispensed only to patients with evidence or other documentation of safe-use conditions. Further information can be found at http://www.juxtapidremsprogram.com.
The goal of the Myalept REMS is to mitigate:
| • | the risks of serious adverse sequelae (such as severe infections, excessive weight gain, glucose intolerance, diabetes mellitus) due to the development of anti-metreleptin antibodies that neutralize endogenous leptin and/or Myalept, and |
| • | the risk of lymphoma by: |
| • | Educating prescribers about the development of neutralizing anti-metreleptin antibodies, the serious adverse sequelae that may result from these antibodies, and the risk for lymphoma associated with Myalept. |
| • | Limiting the population exposed to Myalept by requiring prescriber certification, pharmacy certification, and prescriber attestation that each patient has a diagnosis consistent with the approved indication. |
The Myalept REMS includes the following Elements to Assure Safe Use: (i) certification of Healthcare Providers who prescribe Myalept, (ii) certification of the pharmacies that dispense Myalept, and (iii) Myalept must be dispensed only to patients with evidence or other documentation of safe-use conditions as well as a Letter for Healthcare Providers (risk of severe infections) communication.
Further information can be found at http://myaleptrems.com.
The Hatch-Waxman Act, Marketing Exclusivity and Patent Term Restoration
The Hatch-Waxman Amendments established two abbreviated approval pathways for drug products that are in some way follow-on versions of already approved NDA products.
Generic Drugs: a generic drug is approved by means of an abbreviated NDA (“ANDA”) when its sponsor demonstrates that the proposed product is identical or bioequivalent to the approved, brand-name drug, referred to as the Reference Listed Drug (“RLD”). Generic drug applications are “abbreviated” because they are generally not required to include preclinical (animal) and clinical (human) data to establish safety and effectiveness. Instead, the applicant must show that the generic product performs in the same way as the RLD. Generally, an ANDA must contain data and information showing that the proposed generic product and RLD have the same active ingredients, in the same strength and dosage form, delivered via the same route of administration; are intended for the same uses, have the same labelling and, if applicable, are bioequivalent. An ANDA need not independently demonstrate the proposed product’s safety and effectiveness because the proposed product’s safety and effectiveness are inferred from the fact that the product is demonstrated to be the same as, and bioequivalent to, the RLD. These drugs are commonly referred to as “generic equivalents,” and, under state law, can typically be substituted by pharmacists under prescriptions written for the RLD. 505(b)(2) NDAs: if a product is similar to, but not the same as, an already approved product and thus is not eligible for submission of an ANDA, it may be submitted for approval via an NDA under section 505(b)(2) of the FDCA. Like an ANDA, a 505(b)(2) application is permitted to rely on the FDA’s finding that the RLD is safe and effective to the extent the products share similar features, but the sponsor must submit its own product-specific safety and effectiveness data to support any differences between the proposed and reference products.
RLD Patents: an NDA sponsor must identify to the FDA any patents that claim the drug substance, drug product or method of using the drug. These patents are among the information about the product that is listed in the FDA publication, Approved Drug Products with Therapeutic Equivalence Evaluations, also known as the “Orange Book.” The sponsor of an ANDA or 505(b)(2) application seeking to rely on an RLD must make one of several certifications regarding each listed patent, which in turn affect the timing of approval of the application.
Marketing Exclusivities in the United States
The Hatch-Waxman Act provides periods of regulatory exclusivity for products that would serve as RLDs. If a drug is a new chemical entity (“NCE”), generally meaning that the FDA has not previously approved any other drug containing the same active moiety (the molecule or ion responsible for the action of the drug substance) there is a period of up to five years from the product’s approval during which the FDA may not accept for filing any ANDA or 505(b)(2) application for a drug with the same active moiety. Juxtapid’s NCE exclusivity expired on December 21, 2017, which means that an ANDA or 505(b)(2) NDA may be submitted for Juxtapid.
A product that is not an NCE may qualify for three years of marketing exclusivity following approval when the application contains new clinical investigations, other than bioavailability studies, conducted or sponsored by the applicant and deemed by the FDA to be essential to the approval of the application. Three-year exclusivity is often available for changes to a previously approved drug product, such as new indications, strengths or dosage forms. Although this exclusivity period does not preclude filing or review of an ANDA or 505(b)(2) application, the FDA cannot grant final approval to the ANDA or 505(b)(2) application until three years after approval of the RLD. In addition, this three-year exclusivity covers only the conditions of use associated with the new clinical investigations and, as a general matter, does not prohibit the FDA from approving ANDAs or 505(b)(2) NDAs for generic versions of the original, unmodified drug product.
Pediatric exclusivity is available in the United States under section 505A of the FDCA based on the voluntary completion of pediatric trials and submission of pediatric data in response to an FDA written request. If reports are submitted to and accepted by the FDA within statutory time limits, any periods of regulatory exclusivity or Orange Book-listed patent protections that cover the drug (other than a 30 month stay) are extended by six months. Pediatric exclusivity does not extend a patent term but instead effectively extends the regulatory period during which the FDA cannot approve an ANDA or 505(b)(2) application. Pediatric exclusivity can only extend any regulatory exclusivity or patent protection if the FDA makes its determination that the pediatric studies fairly respond to the written request prior to nine months before the expiration of the exclusivity or patent protection period.
Patent Term Restoration
The Hatch-Waxman Act established a patent restoration term of up to five years as compensation for patent term lost during product development and regulatory review. The maximum period of restoration is five years and cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension, and the extension must be applied for prior to expiration of the patent and within 60 days of the NDA approval. The United States Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. A five-year patent term extension has been granted for the U.S. patent covering the composition of matter of lomitapide, extending the patent term to 2020 from the originally scheduled expiration of early 2015. With respect to metreleptin, the U.S. method-of-use patent directed to treating lipoatrophy has been elected for a 1,445-day patent term extension that will extend its expiration date to 2027.
The Biologics Price Competition and Innovation Act
The BPCI Act, enacted in 2010 as part of the Patient Protection and Affordable Care Act (“PPACA”) authorizes the FDA to license a biological product that is highly similar (“biosimilar”) to, and possibly interchangeable with, a PHSA-licensed reference biological product through an abbreviated pathway. The objectives of the BPCI Act are conceptually similar to those of the Hatch-Waxman Act, which established abbreviated pathways for the approval of small molecule drug products. The BPCI Act establishes criteria for determining that a product is biosimilar to an already-licensed biologic (a reference product), and establishes a process by which an abbreviated BLA for a biosimilar product is submitted, reviewed and approved. The BPCI Act provides periods of exclusivity that protect a reference product from biosimilar competition. Under the BPCI Act, innovator manufacturers of original reference products are granted 12 years of exclusive use before biosimilar versions of such products can be licensed for marketing in the United States. This means that the FDA may not approve an application for a biosimilar version of a reference product until 12 years after the date of approval of the reference product, although a biosimilar application may be submitted 4 years after the date of licensure of the reference product. In addition, the BPCI Act establishes procedures that require the biosimilar applicant to provide information about its application and product to the reference product sponsor, allow information about potentially relevant patents to be shared and permit litigation over patents in advance of approval. The BPCI Act also provides a period of exclusivity for the first biosimilar to be determined by the FDA to be interchangeable with the reference product. The Directors believe that under the BPCI Act, metreleptin has 12 years of exclusivity in the United States until February 24, 2026.
The FDA has issued guidance documents related to BPCI Act implementation concerning biosimilarity and interchangeability, BLA submission requirements, and exclusivity. In February 2020, the FDA issued a final rule to amend its definition of “biological product” to align with the statutory definition in the BPCIA, as amended by the Further Consolidated Appropriations Act, and to provide regulatory clarity around the term. The Directors anticipate that contours of the BPCI Act will be further defined through issuance of additional guidance documents by the FDA, proposed regulations, and decisions in the course of considering specific applications.
Like pediatric exclusivity for drug products approved under the FDCA, pediatric exclusivity for products approved under the BPCI Act is only available if the FDA determines the submitted pediatric study met the terms of its written request prior to 9 months before the expiration of either the 12-year reference product or the seven-year Orphan Drug exclusivity periods. The BPCI Act sets forth a complex mechanism for resolving patent disputes that involves a step-wise exchange of information prior to the initiation of a patent infringement lawsuit against a biosimilar or interchangeable product sponsor. Unlike the Hatch-Waxman Act, the BPCI Act provides no automatic stay on approval of a biosimilar or interchangeable product application based on patents.
Orphan Drug Designation
Under the U.S. Orphan Drug Act of 1983, the FDA may grant Orphan Drug Designation to a drug or biological product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or for which there is no reasonable expectation that development and production costs will be recovered from sales of the drug for such disease or condition in the U.S. Orphan Drug Designation must be requested before submitting an NDA or BLA. After the FDA grants Orphan Drug Designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. If a product that has Orphan Drug Designation subsequently receives FDA approval and is the first drug approved for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication for seven years, except in limited circumstances such as a demonstration that the subsequent drug is clinically superior or the inability of the existing manufacturer to supply the market. See “—Intellectual Property” for more information regarding the Orphan Drug status of our products.
Expedited Development and Review Programs
The FDA’s Fast Track program facilitates the development and review of drugs that are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs. Under the program, the sponsor of a new drug may request that the FDA designate the drug for a specific indication as a Fast Track product concurrent with or after the filing of the IND for the product candidate. A drug that receives Fast Track Designation may be eligible for more frequent meetings with the FDA to discuss the development; more frequent written correspondence from the FDA about the design of the proposed clinical trials; and rolling review of its NDA. Drugs with Fast Track Designation may also become eligible for Priority Review within a six month time frame from the time the complete NDA is accepted, as opposed to the ten month time frame for a standard review. The FDA grants priority review to products that demonstrate the potential to significantly improve safety or effectiveness of treatment, prevention, or diagnosis of a serious condition. We were granted Fast Track Designation by the FDA for Oleogel-S10 in September 2019 following the opening of the IND in the United States.
Rare Pediatric Disease Priority Review Voucher
Certain drugs may also be candidates for Rare Pediatric Disease designation by the FDA. In order to qualify for such designation, a sponsor must demonstrate that the proposed indication is for the treatment or prevention of a rare disease or condition that affects fewer than 200,000 individuals in the United States, or that affects more than 200,000 individuals in the United States and there is no reasonable expectation that the cost of developing and making available in the United States a drug for this type of disease or condition will be recovered from sales in the United States for that product, and is a serious or life-threatening disease in which the serious or life-threatening manifestations primarily affect individuals aged from birth to 18 years. Under the FDCA, a sponsor who is granted a priority review, receives approval of an NDA for a product for the prevention or treatment of a rare pediatric disease and meets certain additional criteria, may qualify for a Rare Pediatric Disease PRV. A PRV can be redeemed to receive priority review under an expedited timeframe for a subsequent marketing application for a different product. A PRV may also be sold or transferred from the initial sponsor to another sponsor and may be further transferred any number of times before it is used. In December 2020, Congress extended the FDA’s authority to award rare pediatric disease vouchers until September 30, 2024, and until September 30, 2026, for products that receive Rare Pediatric Disease designation by September 30, 2024. We were granted a Rare Pediatric Disease Designation in August 2018. It is our intent to discuss the status our PRV is to be discussed with the FDA when we discuss the nature of the data required to address the FDA’s concerns in the CRL we received which asked Amryt to submit additional confirmatory evidence of effectiveness for Oleogel-S10 in EB.
Post-Approval Requirements
Following FDA approval of a drug product, our activities, as well as those of our third-party manufacturers, remain subject to ongoing regulation and close monitoring by the FDA. The FDA may withdraw an approval, following notice and an opportunity for a hearing, if, among other things, compliance with certain regulatory standards is not maintained or if safety or efficacy problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. If new safety issues are identified following approval, the FDA may require the NDA sponsor to take certain measures, such as revising the approved labelling to reflect the new safety information, conducting post-market studies or clinical trials to assess the new safety information, and/or implementing or changing a REMS program to mitigate newly-identified risks. These are often referred to as Phase 4 or post-marketing studies, and may involve additional clinical trials, nonclinical testing and surveillance programs to monitor the safety of approved products which have been commercialized. After approval, most changes to the approved product, such as adding new indications, manufacturing changes and additional labelling claims, require supplemental applications for FDA review and approval. Manufacturers and other entities involved in the manufacture and distribution of approved drugs or licensed biologics are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP requirements and other laws. In its approvals of Juxtapid and Myalept, the FDA required extensive post-marketing requirements and commitments. The post-marketing requirements (“PMR”) for Juxtapid and Myalept are listed below:
| • | PMR1 - Juvenile Tox Study (completed in 2014); |
| • | PMR2 - Enhanced Pharmacovigilance (ongoing); and |
| • | PMR3 - Observational Registry (ongoing); and |
| • | PMR1 - Observational Registry (ongoing); |
| • | PMR2 - Immunogenicity Assay (completed in 2016); |
| • | PMR3 - Immunogenicity Analysis (completed in 2017); |
| • | PMR4 - Immunogenicity study in GL patients (ongoing); and |
| • | PMR5 - Enhanced Pharmacovigilance (ongoing). |
Manufacturing, Packaging and Distribution
Companies engaged in manufacturing drug products or their components must comply with applicable cGMP requirements and product-specific regulations enforced by the FDA and other regulatory agencies. The FDA’s cGMPs are intended to ensure that pharmaceutical products are safe and that they consistently meet applicable requirements and specifications. Compliance with cGMP requires adherence to requirements relating to methods, facilities and controls used in the manufacturing, processing and packaging of a pharmaceutical product, including with respect to personnel, facilities, equipment, control of components and drug product containers and closures, production and process controls, packaging and labelling controls, holding and distribution, laboratory controls, and records and reporting requirements. If, after receiving approval, a company makes a material change in manufacturing equipment, location, or process (all of which are, to some degree, incorporated in the NDA or BLA) additional regulatory review and approval may be required.
The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
In addition, companies manufacturing or distributing drug products pursuant to FDA approvals are subject to record-keeping requirements; requirements on reporting of adverse experiences with the drug, and providing the FDA with updated safety and efficacy or safety, purity, and potency information for drugs and biologics, respectively; compliance within REMS program reporting obligations; drug and biologic sampling and distribution requirements; compliance with certain electronic records and signature requirements, and compliance with the FDA promotion and advertising requirements. As discussed more fully below, the FDA strictly regulates labelling, advertising, promotion and other types of information regarding marketed products that are placed on the market. Drugs and biologics may be promoted only for their approved indications and in accordance with the provisions of the approved labelling.
The FDA also conducts regular, periodic visits to inspect facilities following the initial approval of a product. Accordingly, manufacturers must continue to spend time, money and effort to maintain cGMP compliance. The Directors cannot be certain that our future third party manufacturers will consistently comply with cGMP and other applicable FDA regulatory requirements.
If an FDA investigator identifies conditions in a manufacturer’s facilities that do not comply with applicable regulatory requirements, the FDA investigator may issue observations through a Notice of Inspectional Observations, commonly referred to as a “Form FDA 483” report. If observations in the Form FDA 483 report are not addressed in a timely manner and to the FDA’s satisfaction, the FDA may issue a Warning Letter or proceed directly to other forms of enforcement action. The failure to provide adequate and timely corrective actions can result in further enforcement action, as described below.
Title II of the Drug Quality and Security Act (“DQSA”), known as the Drug Supply Chain Security Act, calls for the establishment of a nationwide electronic system that tracks certain prescription drugs at each point in the supply chain in order to prevent the introduction of counterfeit, adulterated, or mislabeled drugs into the market. Implementation began in 2015 and is scheduled to be completed by 2023. The FDA has issued regulations and guidance implementing the DQSA, which require manufacturers, distributors and dispensers to comply with various regulatory requirements related to product identification, product tracing, product verification, detection and response, notification and wholesaler licensing.
If there are problems with our products, such as adverse events of unanticipated severity or frequency, problems with the facility where the product is manufactured, or non-compliance with the FDCA or comparable laws, then there may be restrictions imposed on the product, the manufacturing facility or them, or we may be required to take other action such as recall or withdrawal of the product from the market or suspension of manufacturing. If our, our respective product candidates, or the manufacturing facilities for our respective product candidates fail to comply with applicable regulatory requirements, or undesirable side effects caused by such products are identified, a governmental authority may:
| • | issue safety alerts, “Dear Doctor” letters, press releases or other communications containing warnings about such product; |
| • | mandate modifications to promotional materials or require us to provide corrective information to healthcare practitioners; |
| • | require that we conduct post-marketing studies; |
| • | require us to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; |
| • | seek an injunction or impose civil or criminal penalties or monetary fines; |
| • | suspend marketing of, withdraw regulatory approval of or recall such product; |
| • | suspend any ongoing clinical studies; |
| • | refuse to approve pending applications or supplements to applications filed by us; |
| • | suspend or impose restrictions on operations, including costly new manufacturing requirements; or |
| • | seize or detain products, refuse to permit the import or export of products or require us to initiate a product recall. |
The occurrence of any event or penalty described above may inhibit our ability to commercialize its products and generate revenue.
Promotional Activities and Interactions with Healthcare Providers and Patients
The FDA and other regulatory agencies strictly regulate promotional claims about prescription drug and biological products through, among other things, standards and regulations for direct-to-consumer advertising, communications regarding unapproved uses, industry-sponsored scientific and educational activities, and promotional activities involving the internet and social media. A product generally cannot be commercially promoted before it is approved. In general, after approval, a drug product may not be promoted for uses that are not approved by the FDA, the EC, and the regulatory authorities of the EU Member States or such other regulatory agencies as reflected in the product’s prescribing information. Moreover, the promotion of prescription-only medicinal products to non-healthcare professionals is prohibited in the European Union. In the United States, healthcare professionals are generally permitted to prescribe drugs and biologics for “off-label” uses (i.e., uses not described in the drug’s labelling) because the FDA does not regulate the practice of medicine. However, the FDA regulates the advertising and promotion of prescription drug and biological products to ensure that claims made with respect to such products are consistent with regulatory approvals, are adequately substantiated by reasonable data, and are neither false nor misleading in any respect. With respect to off-label uses, FDA regulations impose stringent restrictions on manufacturers’ communications regarding off-label uses. A manufacturer may not promote a drug or biologic for off-label use, but under very specific conditions, it may be permissible for a manufacturer to engage in non-promotional, truthful, non-misleading communication regarding off-label information. If a company is found to have made product claims that are false, misleading, unsubstantial or that promote off-label uses, it may become subject to adverse publicity and enforcement action by the FDA, the DOJ, or the Office of the Inspector General of the U.S. Department of Health & Human Services, as well as state authorities. This could subject a company to a range of penalties that could have a significant commercial impact, including civil and criminal fines and agreements that materially restrict the manner in which a company promotes or distributes drug or biological products. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed. For example, to resolve investigations conducted by the DOJ and SEC regarding Aegerion’s U.S. commercial activities and disclosures related to Juxtapid, in September 2017 Aegerion was required, among other things, to pay approximately $40.1 million in aggregate penalties, which includes $7.2 million in restitution payable over three years beginning January 2018, a civil penalty of $4.1 million to be paid to the SEC pursuant to the SEC judgment, and $28.8 million to be paid pursuant to the settlement agreement with the DOJ.
Other Laws Regulating Prescription Drug and Biological Products
From time to time, legislation is drafted, introduced and passed in U.S. Congress that could significantly change the statutory provisions governing the testing, approval, manufacturing and marketing of products regulated by the FDA. In addition to new legislation, FDA regulations and guidance are often revised or interpreted by the agency in ways that may significantly affect our business and products. Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (a) changes to its manufacturing arrangements; (b) additions or modifications to product labelling; (c) the recall or discontinuation of its products; or (d) additional record-keeping requirements.
Manufacturing, sales, promotion and other activities following product approval are also subject to regulation by numerous regulatory authorities in the United States in addition to the FDA, including the CMS, other divisions of the U.S. Department of Health & Human Services, the DOJ, the Drug Enforcement Administration (“DEA”), the Consumer Product Safety Commission, the FTC, the Occupational Safety & Health Administration, the Environmental Protection Agency and state and local governments.
Under the Controlled Substances Act, manufacturers and distributors of controlled substances must maintain registration with the DEA and comply with various regulatory requirements, including with respect to maintaining records and inventory, reporting to the DEA, and meeting certain security and operational safeguards.
U.S. Pricing and Reimbursement Environment
Significant uncertainty exists regarding the coverage and reimbursement status of products approved by the FDA and other government authorities. Sales of pharmaceutical products depend in significant part on the availability and adequacy of third party reimbursement. Third party payers include government health administrative authorities, managed care providers, private health insurers and other organizations. Third party payers are increasingly challenging the prices charged for, examining the medical necessity of, and assessing the cost-effectiveness of medicinal products and services. Coverage may also be more limited than the purposes for which the product is approved by the FDA or comparable foreign regulatory authorities. Further, one payer’s determination to provide coverage for a product does not assure that other payers will also provide coverage for the product.
Government and private third party payers have a variety of methodologies for paying for drugs and biologics. Payers are increasingly considering new metrics as the basis for reimbursement rates, such as average sales price, average manufacturer price or actual acquisition cost.
Private Payers
Coverage of drugs and biologics by private health insurance varies. Private payers may use a variety of utilization management techniques designed to control costs, including requiring pre-approval of coverage for drug therapies before reimbursing healthcare providers or patients that use such therapies. In addition, a payer’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be provided. Coverage may not be sufficient to maintain price levels high enough to realize an appropriate return on investment in product development.
Medicare
In the United States, the Medicare program provides health insurance for people who are 65 or older, as well as certain people with disabilities and other conditions irrespective of their age. The Medicare program is funded by the federal government and administered by the CMS. The U.S. Medicare Prescription Drug, Improvement, and Modernization Act of 2003, established the Medicare Part D program to provide a voluntary prescription drug benefit to Medicare beneficiaries. Medicare Part D is a voluntary prescription drug benefit through which beneficiaries may enroll in prescription drug plans offered by private entities under contract with CMS for coverage of certain outpatient prescription drugs. Medicare Part D generally provides coverage for medically necessary self-administered drugs. Coverage and reimbursement for outpatient drugs under Part D is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs the plan will cover and to what extent so long as the plan includes drugs from each therapeutic category and class of covered drugs, though not necessarily all the drugs in each category or class.
Although the availability of coverage under Medicare Part D may increase demand for Myalept, Juxtapid and Mycapssa®, any negotiated prices for our products covered by a Part D prescription drug plan likely will be lower than the prices it might otherwise obtain. In order for Myalept, Juxtapid and Mycapssa® to remain on or be included on the formularies of Part D prescription drug plans, we may have to offer discounts on the price of those products. In addition, manufacturers, are required to provide a 70% discount on brand name prescription drugs utilized by Medicare Part D beneficiaries when those beneficiaries reach the so-called “donut hole” in their drug benefits in a particular year (i.e., a coverage gap between initial coverage and catastrophic coverage). Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payers because private payers use Medicare coverage policies and payment limitations in setting their own rates.
The Directors believe that investigations and enforcement actions by certain government agencies have caused a reduction in contributions to these third party patient organizations, which may prevent these organizations from providing adequate financial assistance, including assistance with co-payment obligations, to individuals who would otherwise be unable to afford our products. As a result, Medicare Part D patients may not be able to afford their out-of-pocket co-payments for our products. In 2017, for example, the Directors believe that a considerable number of Juxtapid patients who were covered by Medicare Part D ceased treatment with Juxtapid, due to reductions in contributions to patient assistance programs operated by independent charitable 501(c)(3) organizations, which resulted in prior sources of financial support for these patients being less available.
We may develop products that, once approved, may be administered by a physician. Under currently applicable U.S. law, certain products not usually self-administered (including injectable drugs) may be eligible for coverage under Medicare through Medicare Part B. Medicare Part B is the part of Medicare that covers outpatient services and supplies, including certain pharmaceutical products that are medically necessary to treat a beneficiary’s health condition. As a condition of receiving Medicare Part B reimbursement for a manufacturer’s eligible drugs, the manufacturer is required to participate in other government healthcare programs, including the Medicaid Drug Rebate Program and the Public Health Service 340B Drug Pricing Program, discussed below.
Medicaid
Medicaid is a health insurance program with mandatory coverage for certain low-income children, families, pregnant women, and people with disabilities. States also have the option of expanding Medicaid coverage to low-income individuals generally and many states have done so. Medicaid is jointly funded by the federal and state governments, but administered by the states.
We participate in the Medicaid Drug Rebate Program. The Medicaid Drug Rebate Program requires pharmaceutical manufacturers to enter into and have in effect a national rebate agreement with the Secretary of the Department of Health and Human Services as a condition for states to receive federal matching funds for the manufacturer’s outpatient drugs furnished to Medicaid patients. Under the Medicaid Drug Rebate Program, we are required to pay a rebate for each unit of our product reimbursed by a state Medicaid program as a condition of having federal funds made available to the states for our drugs under Medicaid and Medicare Part B. Those rebates are based on pricing data that we report on a monthly and quarterly basis to CMS, the federal agency that administers the Medicaid Drug Rebate Program. We may also participate in state Medicaid supplemental rebate programs which require payment of an incremental rebate to state Medicaid programs for covered utilization of our products. Price reductions as well as price increases that exceed the rate of inflation for our products may result in increasing the rebates we are required to pay under the Medicaid Drug Rebate Program or state Medicaid supplemental rebate programs and the discounts we will be required to offer under the Public Health Service 340B drug pricing discount program, known as the “340 B Program.” The American Rescue Plan Act of 2021 (Public Law No: 117-2), signed into law on March 11, 2021, ended the cap on Medicaid rebates, effective January 1, 2024. Beginning in 2024, manufacturers may have Medicaid rebate liability greater than 100% dependent upon past price increases. As a result of the substantial price increase for Myalept in February 2015, the Directors expect a significant gross-to-net adjustment for Medicaid rebates which will offset the majority of revenues from Medicaid and negatively impact net product sales in future quarters, since Medicaid rebates directly reduce our net product sales. The degree of such impact on our overall financial performance will depend on the percentage of Myalept patients that have Medicaid as their primary insurance coverage and the quantity of units ordered per patient. To date, approximately 35% of patients prescribed Myalept have been Medicaid beneficiaries. The number of patients prescribed Myalept in the future who are Medicaid beneficiaries could be higher than historical rates.
To maintain coverage of our products under the Medicaid Drug Rebate Program and Medicare Part B, it will be required to extend significant discounts to certain “covered entities” (defined by statute to include certain types of hospitals and other healthcare providers that receive federal grants) that purchase products under the 340B Program. The 340B Program requires participating manufacturers to agree to charge such covered entities no more than the 340B “ceiling price” for the manufacturers’ covered outpatient drugs. The 340B ceiling price is calculated using a statutory formula, which is based on the average manufacturer price and rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate Program. Orphan drugs (those designated under section 526 of the FDCA, such as Myalept, Juxtapid and Mycapssa) are exempt from the ceiling price requirements with respect to drugs purchased by certain covered entities (i.e., rural referral centers, sole community hospitals, critical access hospitals and free-standing cancer hospitals). The PPACA also obliges the Health Resources and Services Administration, the agency which administers the 340B Program, to promulgate various regulations and implement processes to improve the integrity of the 340B Program. The status of new and pending regulations and guidance is uncertain under the new presidential administration.
Recent regulations have granted Medicare Part D plans authority to apply new cost control measures to steer patients toward lower-priced drug products prior to covering non-preferred, more expensive products. This could potentially have the result of reducing access to our products under Medicare Part D.
Drug products are subject to discounted pricing when purchased by federal agencies via the Federal Supply Schedule (“FSS”). FSS participation is required for a drug product to be covered and reimbursed by certain federal agencies and for coverage under Medicaid, Medicare Part B and the 340B Program. FSS pricing is negotiated periodically with the Department of Veterans Affairs. FSS pricing is intended not to exceed the price that a manufacturer charges its most-favored non-federal customer for its product. In addition, prices for drugs purchased by the Veterans Administration, Department of Defense (including drugs purchased by military personnel and dependents through the Tricare Retail Pharmacy program), Coast Guard and PHS are subject to a cap on pricing (known as the federal ceiling price) and may be subject to an additional discount if pricing increases more than the rate of inflation. We have had an FSS contract in place for its products since 2016. We also participate in the Tricare Retail Pharmacy program, under which we pay quarterly rebates on utilization of Myalept and Juxtapid when the products are dispensed through the Tricare Retail Pharmacy network to Tricare beneficiaries.
The marketability of any products for which we will receive regulatory approval for commercial sale may suffer if the government and third party payers fail to provide adequate coverage and reimbursement. An increasing emphasis on cost containment measures in the United States has increased and the Directors expect will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
In recent years, additional laws have resulted in direct or indirect reimbursement reductions for certain Medicare providers. These laws, and future state and federal healthcare reform measures may be adopted in the future, any of which may result in additional reductions in Medicare and other healthcare funding and otherwise affect the prices we may obtain for any product candidates for which we may obtain regulatory approval or the frequency with which any such product candidate is prescribed or used.
Further, in the United States, the cost of pharmaceuticals continues to generate substantial governmental and third party payer interest. There have been several recent U.S. congressional inquiries and proposed federal and proposed and enacted state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the costs of drugs under Medicare and reform government program reimbursement methodologies for drug products. Individual state legislatures have become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing. Some of these measures include price or patient reimbursement constraints, discounts, restrictions on certain product access, marketing cost disclosure and transparency measures, and in some cases measures designed to encourage importation from other countries and bulk purchasing. In addition, regional health care authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other health care programs. These measures could reduce the ultimate demand for our products, once approved, or put pressure on our product pricing.
Other Regulatory and Liability Matters
We may be subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. From time to time and in the future, our operations may involve the use of hazardous and flammable materials, including chemicals and biological materials, and may also produce hazardous waste products. Even if we contract with third parties for the disposal of these materials and waste products, we cannot completely eliminate the risk of contamination or injury resulting from these materials.
We will maintain insurance to cover us for costs and expenses we may incur due to injuries to its employees, but this insurance may not provide adequate coverage against potential liabilities.
U.S. Healthcare Reform
Our revenues and operations could be affected by changes in healthcare spending and policy in the United States. We operate in, a highly regulated industry and new laws, regulations or judicial decisions, or new interpretations of existing laws, regulations or decisions, related to health care availability, the method of delivery or payment for health care products and services could negatively impact our business, operations and financial condition. As noted above, the U.S. Congress, federal agencies and state legislatures from time to time propose and adopt initiatives aimed at cost containment, which could impact our ability to sell our products profitably. For example, the PPACA substantially changed the way healthcare is financed by both governmental and private insurers. The law contains a number of provisions that affect coverage and reimbursement of drug products and/or that could potentially reduce the demand for our products such as:
| • | increasing drug rebates under state Medicaid programs for brand name prescription drugs and extending those rebates to Medicaid managed care; |
| • | requiring drug manufacturers to provide a 70% discount on Medicare Part D brand name prescription drugs sold to Medicare beneficiaries whose prescription drug costs cause the beneficiaries to be subject to the Medicare Part D coverage cap (i.e., the so-called donut hole); and |
| • | expanding programs designed to test innovative payment models, service delivery models, or value-based arrangements. |
There have been continuous legal and political challenges to certain aspects of the Affordable Care Act. Although the Supreme Court has upheld several key provisions of the Affordable Care Act against legal challenges, including in 2021, opponents of the law may continue to challenge aspects of the law and could be successful in the future.
The Affordable Care Act, if substantially maintained in its current form, will continue to result in downward pressure on coverage and the price that we may receive for any approved product. Any reduction in reimbursement from Medicare and other government programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our products.
Other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. For example, in August 2011, President Barack H. Obama signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. This committee did not achieve a targeted deficit reduction of at least $1.2 trillion for fiscal years 2012 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, which went into effect beginning on April 1, 2013. Section 4408 of the CARES Act temporarily suspended Medicare sequestration during the period of May 1, 2020 through December 31, 2020, while extending the Medicare sequestration sunset date through 2030. The Protecting Medicare and American Farmers from Sequester Cuts Act (Public Law No: 117-71), signed into law on December 10, 2021, extended the moratorium on the 2% Medicare sequester payment reductions through March 31, 2022. After March, the law phases in the Medicare sequester cuts by implementing a 1% cut from April 1 through June 30, 2022, and then allowing the full 2% cut to go into effect on July 1, 2022. In order to pay for this grace period, the sequester cuts are increased in 2030 to 2.25% for the first six months and 3% for the next six months of that year.
Multiple rules issued by HHS under the Trump administration could impact access to and reimbursement for prescription drugs if they become effective. A final rule that eliminated the safe harbor shielding Medicare Part D rebates to pharmacy benefit managers from the Anti-Kickback statute was finalized in November 2020. However, The Infrastructure Investment and Jobs Act (Public Law No: 117-58), signed into law on November 15, 2021, established a moratorium until January 1, 2026, on implementation of that rule as it relates to eliminating the anti-kickback statute safe harbor protection for prescription drug rebates. In addition, the Build Back Better Act (HR 5376), as passed by the House of Representatives and currently under consideration in the Senate, would permanently prevent implementation of the rule so it remains unclear whether this will take effect.
In addition, the Medicaid Drug Rebate Program final rule, issued in December of 2020, finalized policies to change the way in which Medicaid rebates are calculated. This policy would require that copay assistance provided to patients would be applied as a discount to a payer for the purposes and could set a new best price for the purposes of the Medicaid drug rebate calculation unless a manufacturer can “ensure” that the full value of the assistance is passed through to the patient. This is problematic given the rising number of copay accumulator plans in which pharmacy benefit managers block patients from taking full advantage of patient assistance programs without the knowledge of the manufacturer. The Pharmaceutical Researchers and Manufacturers of America (PhRMA) sued to invalidate this rule in May of 2021. The outcome of this litigation remains uncertain and could require changes to patient assistance programs.
In September of 2021, the Build Back Better Act was introduced in the House of Representatives and contained a number of provisions that could impact the biopharmaceutical industry. These provisions include measures aimed at reducing patient out of pocket costs through a redesign of the Medicare Part D benefit, provisions aimed at allowing Medicare to negotiate drug prices for a subset of drugs in Medicare Part B and Part D, and provisions meant to limit drug price increases to the rate of inflation, requiring rebates to the government for price increases taken beyond inflation. The House of Representatives passed the Build Back Better Act in November of 2021 and it has not been considered by the Senate. If any of these measures are adopted into law, they could impact future business actions and access to medicines.
Other Laws Affecting Us in the United States
Healthcare providers, physicians and third party payers will play a primary role in the recommendation and prescription of any products for which we obtain marketing approval. Our future arrangements with third party payers, healthcare providers, patients and patient advocacy organizations, teaching hospitals, other licensed prescribers and physicians may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute any drugs for which we obtain marketing approval. In the United States, these laws include, without limitation, state and federal anti-kickback, false claims, physician transparency, and patient data privacy and security laws and regulations, including but not limited to those described below.
The U.S. Anti-Kickback Statute makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf), to knowingly and willfully solicit, receive, offer or pay any remuneration, directly or indirectly, in cash or in kind, that is intended to induce or reward referrals, including the purchase, recommendation, order or prescription of a particular drug, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. Violations of this law are punishable by up to five years in prison, criminal fines, administrative civil money penalties and exclusion from participation in federal healthcare programs. The term remuneration has been interpreted broadly to include anything of value.
There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution. The exceptions and safe harbors are drawn narrowly and practices that involve remuneration that may be alleged to be intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances. Our practices may not in all cases meet all of the criteria for protection under a statutory exception or regulatory safe harbor. The statutory exceptions and regulatory safe harbors are also subject to change and some safe harbors, such as the safe harbor for certain rebate arrangements, are currently under review for amendments or reversal.
The intent standard under the federal Anti-Kickback Statute was amended by the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, to a stricter standard such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. Several courts have also interpreted the Anti-Kickback Statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated, even if there are additional, legitimate purposes for the remuneration. In addition, the Affordable Care Act also codified case law that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act (“FCA”).
The Federal False Claims Act imposes civil penalties, including through civil whistle-blower or qui tam actions, against individuals or entities (including manufacturers) for, among other things, knowingly presenting, or causing to be presented false or fraudulent claims for payment by a federal healthcare program or making a false statement or record material to payment of a false claim or avoiding, decreasing or concealing an obligation to pay money to the federal government. The government may deem manufacturers to have “caused” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers or promoting a product off-label. Claims which include items or services resulting from a violation of the federal Anti-Kickback Statute are false or fraudulent claims for purposes of the False Claims Act. Our future marketing and activities relating to the reporting of wholesaler or estimated retail prices for our products, the reporting of prices used to calculate Medicaid rebate information and other information affecting federal, state and third party reimbursement for our products, and the sale and marketing of its products and any future product candidates, are subject to scrutiny under this law.
HIPAA imposes criminal and civil liability for knowingly and willfully executing a scheme, or attempting to execute a scheme, to defraud any healthcare benefit program, including private payers, or falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment for healthcare benefits, items or services.
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 (“HITECH”), and their respective implementing regulations impose, among other things, specified requirements on covered entities and their business associates relating to the privacy and security of individually identifiable health information including mandatory contractual terms and required implementation of technical safeguards of such information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. Like the Anti-Kickback Statute, the Affordable Care Act amended the intent standard for certain healthcare fraud statutes under HIPAA such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
The Physician Payments Sunshine Act, enacted as part of the PPACA, imposes new annual reporting requirements for certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, for certain payments and “transfers of value” provided to physicians, patients, patient advocacy organization, teaching hospitals and other licensed prescribers, as well as ownership and investment interests held by physicians and their immediate family members. This information is made publicly available on a CMS website, and failure to report accurately could result in penalties.
Analogous state and foreign fraud and abuse laws and regulations, such as state anti-kickback and false claims laws, may be broader in scope and apply regardless of payer. Such laws are enforced by various state agencies and through private actions. Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant federal government compliance guidance, require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers, and restrict marketing practices or require disclosure of marketing expenditures. State and foreign laws also govern the privacy and security of health information in some circumstances. Such data privacy and security laws may differ from each other in significant ways and often are not pre-empted by HIPAA, thus complicating compliance efforts.
In order to distribute products commercially companies must comply with state laws that require the registration of manufacturers and wholesale distributors of drug and biological products in a state, including, in certain states, manufacturers and distributors who ship products into the state even if such manufacturers or distributors have no place of business within the state. Some states also impose requirements on manufacturers and distributors to establish the pedigree of product in the chain of distribution, including some states that require manufacturers and others to adopt new technology capable of tracking and tracing product as it moves through the distribution chain. Several states have enacted legislation requiring pharmaceutical and biotechnology companies to establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities, and/or register their sales representatives, as well as to prohibit pharmacies and other healthcare entities from providing certain physician prescribing data to pharmaceutical and biotechnology companies for use in sales and marketing, and to prohibit certain other sales and marketing practices. All of our activities are potentially subject to federal and state consumer protection and unfair competition laws.
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. If our operations are found to be in violation of any of these laws or any other related governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, disgorgement, exclusion of drugs from government funded healthcare programs, such as Medicare and Medicaid, reputational harm, additional oversight and reporting obligations and the curtailment or restructuring of our operations. For example, in September 2017, Aegerion entered into a settlement agreement with the DOJ (as further described under “—Legal Proceedings” in this section) to resolve investigations conducted by the DOJ and the SEC regarding Aegerion’s U.S. commercial activities and disclosures related to Juxtapid, and on January 30, 2018, a U.S. District Court judge sentenced Aegerion after the judge accepted Aegerion’s guilty criminal plea. Aegerion was required, among other things, to pay approximately $40.1 million in aggregate penalties over three years and was put on probation for three years. We are also subject to a CIA and a civil consent decree (which took effect in March 2019) with the FDA and the DOJ relating to the Juxtapid REMS program.
EU Regulatory Matters
The United Kingdom UK formally left the EU on January 31, 2020 and the transition period, during which EU laws continued to apply to the UK, expired on December 31, 2020. This means EU laws now only apply to the UK in respect of Northern Ireland as laid out in the Protocol on Ireland and Northern Ireland. Following the end of the transition period, the EU and the UK concluded a trade and cooperation agreement (“TCA”), which applied provisionally from January 1, 2021 and entered into force on May 1, 2021.
The TCA includes provisions affecting the life sciences sector (including on customs and tariffs) but areas for further discussion between the EU and the UK remain. In addition, there are some specific provisions concerning pharmaceuticals. These include the mutual recognition of Good Manufacturing Practice (“GMP”) and issued GMP documents. The TCA does not, however, contain wholesale mutual recognition of UK and EU pharmaceutical regulations and product standards.
Since January 1, 2021, the EU laws which have been transposed into UK law through secondary legislation continue to be applicable in the UK as “retained EU law”. As there is no general power to amend these regulations, the UK government has enacted the Medicines and Medical Devices Act 2021. The purpose of the act is to enable the existing regulatory frameworks in relation to human medicines, clinical trials of human medicines, veterinary medicines and medical devices to be updated. The powers under the act may only be exercised in relation to specified matters and must safeguard public health.
Specified provisions of the Medicines and Medical Devices Act 2021 entered into force on February 11, 2021. The remaining provisions came into effect within two months of February 11, 2021 or will otherwise come into effect as stipulated in subsequent statutory instruments. The Medicines and Medical Devices Act 2021 supplements the UK Medical Devices Regulations 2002 (“UK Regulations”), which are based on the EU Medical Devices Directive as amended to reflect the UK’s post-Brexit regulatory regime. Notably, the UK Regulations do not include any of the revisions that have been made by the EU Medical Devices Regulation (EU) 2017/745, which, since May 26, 2021, now applies in all EU Member States.
The UK’s Medicines and Healthcare products Regulatory Agency (“MHRA”) conducted a comprehensive consultation between September and November 2021 on proposals to develop a new UK regime for medical devices in the UK. The proposals include more closely aligning definitions for medical devices and in vitro medical devices with internationally recognised definitions and changing the classification of medical devices according to levels or risk. The proposals are intended to improve patient and public safety and increase the appeal of the UK market. The new regime is planned to come into force on July 1, 2023, which will align with the date from which the UK is due to stop accepting CE marked medical devices and require UKCA (“UK Conformity Assessed”) marking. It is envisaged that, in Northern Ireland, the amended regime could run in parallel with any existing or future EU rules in accordance with the Protocol on Ireland and Northern Ireland.
Drug and Biologic Development Process
The conduct of clinical trials in the European Union is currently governed by the EU Clinical Trials Directive 2001/20/EC, or Clinical Trials Directive, and will be replaced by the EU Clinical Trials Regulation (EU) No. 536/2014 (“CTR”) once the latter comes into effect. The CTR introduces a complete overhaul of the existing regulation of clinical trials for medicinal products in the EU. It entered into force on January 31, 2022.
Under the current regime, which will expire after a transition period of one or three years, respectively, as outlined below in more detail, before a clinical trial can be initiated it must be approved in each EU Member State where there is a site at which the trial is to be conducted. The approval must be obtained from two separate entities: the National Competent Authority (“NCA”) and one or more Ethics Committees. The NCA of the EU Member States in which the clinical trial will be conducted must authorize the conduct of the trial, and the independent Ethics Committee must grant a positive opinion in relation to the conduct of the clinical trial in the relevant EU Member State before the commencement of the trial. Any substantial changes to the trial protocol or other information submitted with the clinical trial applications must be submitted to or approved by the relevant NCA and Ethics Committees. Under the current regime all suspected unexpected serious adverse reactions to the investigated drug that occur during the clinical trial must be reported to the NCA and to the Ethics Committees of the EU Member State where they occur.
A more unified procedure will apply under the new CTR. A sponsor will be able to submit a single application for approval of a clinical trial through a centralized EU clinical trials portal. One national regulatory authority (the reporting EU Member State proposed by the applicant) will take the lead in validating and evaluating the application consult and coordinate with the other concerned Member States. If an application is rejected, it may be amended and resubmitted through the EU clinical trials portal. If an approval is issued, the sponsor may start the clinical trial in all concerned Member States. However, a concerned EU Member State may in limited circumstances declare an “opt-out” from an approval and prevent the clinical trial from being conducted in such Member State. The CTR also aims to streamline and simplify the rules on safety reporting, and introduces enhanced transparency requirements such as mandatory submission of a summary of the clinical trial results to the EU Database. The CTR foresees a three-year transition period. Member States will work in CTIS immediately after the system has gone live. For one year, until 31 January 2023, clinical trial sponsors can still choose whether to submit an initial clinical trial application in line with the current system (Clinical Trials Directive) or via CTIS. From 31 January 2023, submission of initial clinical trial applications via CTIS becomes mandatory, and by 31 January 2025, all ongoing trials approved under the current Clinical Trials Directive will be governed by the new Regulation and have to be transitioned to CTIS.
Under both the current regime and the new CTR, national laws, regulations, and the applicable Good Clinical Practice and Good Laboratory Practice standards must also be respected during the conduct of the trials, including the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (“ICH”) guidelines on Good Clinical Practice (“GCP)” and the ethical principles that have their origin in the Declaration of Helsinki.
During the development of a medicinal product, the European Medical Agency (“EMA”) and national regulators within the EU provide the opportunity for dialogue and guidance on the development program. At the EMA level, this is usually done in the form of scientific advice, which is given by the Committee for Medicinal Products for Human Use (“CHMP”) on the recommendation of the Scientific Advice Working Party (“SAWP”). A fee is incurred with each scientific advice procedure, but is significantly reduced for designated orphan medicines. Advice from the EMA is typically provided based on questions concerning, for example, quality (chemistry, manufacturing and controls testing), nonclinical testing and clinical studies, and pharmacovigilance plans and risk-management programs. Advice is not legally binding with regard to any future Marketing Authorization Application (“MAA”) of the product concerned.The approval process for clinical trials in other countries outside the United States and the European Union varies from country to country, and the time may be longer or shorter than that required for FDA approval.
Drug Marketing Authorization
In the EU and in Iceland, Norway and Liechtenstein (together the European Economic Area or “EEA”), after completion of all required clinical testing, medicinal products may only be placed on the market after obtaining a Marketing Authorization (“MA”). To obtain an MA of a drug under European Union regulatory systems, an applicant can submit an Marketing Authorization Application (“MAA”) through, amongst others, a centralized or decentralized procedure.
The centralized procedure provides for the grant of a single MA by the European Commission (EC) that is valid for all EU Member States and, after respective national implementing decisions, in the three additional EEA Member States. The centralized procedure is compulsory for specific medicinal products, including for medicines developed by means of certain biotechnological processes, products designated as orphan medicinal products, advanced therapy medicinal products (“ATMP”) and medicinal products with a new active substance indicated for the treatment of certain diseases (AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases). For medicinal products containing a new active substance not yet authorized in the EEA before May 20, 2004 and indicated for the treatment of other diseases, medicinal products that constitute significant therapeutic, scientific or technical innovations or for which the grant of a MA through the centralized procedure would be in the interest of public health at EU level, an applicant may voluntarily submit an application for a marketing authorization through the centralized procedure.
Under the centralized procedure, the Committee for Medicinal Products for Human Use (“CHMP”), established at the EMA, is responsible for conducting the initial assessment of a drug. The CHMP is also responsible for several post-authorization and maintenance activities, such as the assessment of modifications or extensions to an existing marketing authorization. Under the centralized procedure, the timeframe for the evaluation of an MAA by the EMA’s CHMP is, in principle, 210 days from receipt of a valid MAA. However, this timeline excludes clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP, so the overall process typically takes a year or more, unless the application is eligible for an accelerated assessment. Accelerated assessment might be granted by the CHMP in exceptional cases when a medicinal product is of major interest from the point of view of public health and in particular from the viewpoint of therapeutic innovation. On request, the CHMP can reduce the time frame to 150 days if the applicant provides sufficient justification for an accelerated assessment. The CHMP will provide a positive opinion regarding the application only if it meets certain quality, safety and efficacy requirements. However, the EC has final authority for granting the MA within 67 days after receipt of the CHMP opinion.
The decentralized procedure permits companies to file identical MA applications for a medicinal product to the competent authorities in various EU Member States simultaneously if such medicinal product has not received marketing approval in any EU Member State before. This procedure is available for medicinal products not falling within the mandatory scope of the centralized procedure. The competent authority of a single EU Member State, known as the reference EU Member State, is appointed to review the application and provide an assessment report. Under this procedure, an applicant submits an application based on identical dossiers and related materials, including a draft summary of product characteristics, and draft labeling and package leaflet, to the reference EU Member State and concerned EU Member States. The reference EU Member State prepares a draft assessment report and drafts of the related materials within 120 days after receipt of a valid application. Subsequently each concerned EU Member State must decide whether to approve the assessment report and related materials. If an EU Member State cannot approve the assessment report and related materials on the grounds of potential serious risk to public health, the disputed points are subject to a dispute resolution mechanism and may eventually be referred to the EC, whose decision is binding for all EU Member States.
All new MAAs must include a Risk Management Plan (“RMP”), describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the MA. RMPs and Periodic Safety Update Reports (“PSURs”) are routinely available to third parties requesting access, subject to limited redactions.
Marketing authorizations have an initial duration of five years. After these five years, the authorization may subsequently be renewed on the basis of a re-evaluation of the risk-benefit balance. Once renewed, the marketing authorization is valid for an unlimited period unless the EC or the national competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal. Applications for renewal must be made to the EMA at least nine months before the five-year period expires.
Episalvan was granted a marketing authorization under the centralized procedure for the treatment of partial-thickness skin wounds in adults. The outcome of the Episalvan five year renewal procedure recommended that one additional five-year renewal be required on the basis of limited safety information due to limited marketing of Episalvan. Lojuxta and Myalepta were each granted a marketing authorization under exceptional circumstances by the EC under the centralized procedure and are subject to additional monitoring (which aims to enhance reporting of suspected adverse drug reactions for drugs for which the clinical evidence base is less well developed). So-called “marketing authorizations under exceptional circumstances” are intended for medicinal products where the applicant can demonstrate that it is not possible to provide comprehensive clinical data on efficacy and safety under normal conditions of use. For instance, because the indications for which the product in question is intended are encountered so rarely that the applicant cannot reasonably be expected to provide comprehensive evidence, or in the present state of scientific knowledge, comprehensive information cannot be provided, or it would be contrary to generally accepted principles of medical ethics to collect such information. Consequently, marketing authorizations under exceptional circumstances may be granted subject to certain specific obligations.
In the case of Lojuxta and Myalepta:
The Lojuxta marketing authorization under exceptional circumstances includes a commitment to conduct a long-term observational study to collect information on the safety and effectiveness outcomes of patients treated with Lojuxta. The EMA has required that all patients taking Lojuxta in the European Union be encouraged to participate in the study, and that the study period be open-ended. In the study, physicians will follow each patient to evaluate serum lipid control, hepatic effects, tumor occurrence, teratogenicity, gastrointestinal adverse reactions, events associated with coagulopathy, major adverse cardiovascular events and death. The EMA also required that a study be conducted to determine the impact of lomitapide on vascular endpoints. In addition, an appropriate risk-management educational program to inform healthcare professionals and patients must be established prior to the product being launched in each country.
The Myalepta authorization under exceptional circumstances includes a commitment to establish a registry including all patients with GL or PL treated with Myalepta to further evaluate the long-term safety and effectiveness of Myalepta under normal conditions of clinical practice, along with a commitment to conduct an efficacy and safety study to further investigate the effect of Myalepta on poor metabolic control once background therapy is maximized in patients with familial or acquired PL. In order to address the potential safety concerns and/or lack of efficacy related to immunogenicity of Myalepta, the EMA also requires an integrated analysis of immunogenicity measured accordingly to validated assays, including samples from all available historical samples from previous studies with patients with GL or PL and samples obtained from patients that will be included in the efficacy and safety study in PL patients, the pediatric investigation plan (“PIP”) study and the patients registry. There have been delays in compliance and completion of some of the conditions to the Lojuxta and Myalepta marketing authorizations, but the Company is engaging with the CHMP to amend the relevant conditions, including where appropriate amendments to study protocols and extensions to deadlines.
The Lojuxta and Myalepta exceptional circumstances marketing authorizations both require an annual reassessment of the risk/benefit balance for each of Lojuxta and Myalepta by the CHMP. A negative assessment could potentially result in the relevant marketing authorization being suspended or revoked. The renewal of a marketing authorization of a medicinal product under exceptional circumstances, however, follows the same rules as a “normal” marketing authorization. Thus, a marketing authorization under exceptional circumstances is granted for an initial five years, after which the authorization will become valid indefinitely, unless the EMA decides that safety grounds merit one additional five-year renewal.
It is anticipated that the AP103 product candidate will be regulated as an advanced therapy medicinal product (“ATMP”) under the EU Regulation (EC) No. 1394/2007 on advanced therapy medicinal products, or the ATMP Regulation. ATMPs comprise gene therapy medicinal products, somatic cell therapy medicinal products and tissue engineered products, which are cells or tissues that have undergone substantial manipulation and that are administered to human beings in order cure, diagnose or prevent diseases or to regenerate, repair or replace a human tissue. Pursuant to the ATMP Regulation, the Committee on Advanced Therapies (“CAT”) is responsible in conjunction with the CHMP for the evaluation of ATMPs. The CAT is primarily responsible for the scientific evaluation of ATMPs and prepares a draft opinion on the quality, safety and efficacy of each ATMP for which a marketing authorization application is submitted. The CAT’s opinion is then taken into account by the CHMP when giving its final recommendation regarding the authorization of a product in view of the balance of benefits and risks identified. Although the CAT’s draft opinion is submitted to the CHMP for final approval, the CHMP may depart from the draft opinion, if it provides detailed scientific justification. The CHMP and CAT are also responsible for providing guidelines on ATMPs and have published numerous guidelines, including specific guidelines on gene therapies and cell therapies. These guidelines provide additional guidance on the factors that the EMA will consider in relation to the development and evaluation of ATMPs and include, among other things, the pre-clinical studies required to characterize ATMPs; the manufacturing and control information that should be submitted in a marketing authorization application; and post-approval measures required to monitor patients and evaluate the long term efficacy and potential adverse reactions of ATMPs. Although such guidelines are not legally binding, compliance with them is often necessary to gain and maintain approval for product candidates. In addition to the mandatory RMP, the holder of a MA for an ATMP must put in place and maintain a system to ensure that each individual product and its starting and raw materials, including all substances coming into contact with the cells or tissues it may contain, can be traced through the sourcing, manufacturing, packaging, storage, transport and delivery to the relevant healthcare institution where the product is used.
PRIME Designation
In March 2016, the EMA launched an initiative to facilitate development of product candidates in indications, often rare, for which few or no therapies currently exist. The PRIority MEdicines, or PRIME, scheme is intended to encourage drug development in areas of unmet medical need and provides accelerated assessment of products representing substantial innovation reviewed under the centralized procedure. Products from small- and medium-sized enterprises may qualify for earlier entry into the PRIME scheme than larger companies on the basis of compelling non-clinical data and tolerability data from initial clinical trials. Many benefits accrue to sponsors of product candidates with PRIME designation, including but not limited to, early and proactive regulatory dialogue with the EMA, frequent discussions on clinical trial designs and other development program elements, and potentially accelerated marketing authorization application assessment once a dossier has been submitted. Importantly, once a candidate medicine has been selected for the PRIME scheme, a dedicated contact point and rapporteur from the CHMP or from CAT are appointed facilitating increased understanding of the product at EMA’s Committee level. A kick-off meeting with the CHMP/CAT rapporteur initiates these relationships and includes a team of multidisciplinary experts to provide guidance on the overall development plan and regulatory strategy. PRIME eligibility does not change the standards for product approval, and there is no assurance that any such designation or eligibility will result in expedited review or approval.
New Chemical Entity Exclusivity
As in the United States, it may be possible to obtain a period of market and / or data exclusivity in the EU that would have the effect of postponing the entry into the marketplace of a competitor’s generic, hybrid or biosimilar product (even if the pharmaceutical product has already received a MA) and prohibiting another applicant from relying on the MA holder’s pharmacological, toxicological and clinical data in support of another MA for the purposes of submitting an application, obtaining MA or placing the product on the market.
New Chemical Entities (“NCE”) approved in the EU qualify for eight years of data exclusivity and 10 years of marketing exclusivity. An additional non-cumulative one-year period of marketing exclusivity is possible if during the data exclusivity period (the first eight years of the 10-year marketing exclusivity period), the MA holder obtains an authorization for one or more new therapeutic indications that are deemed to bring a significant clinical benefit compared to existing therapies.
The data exclusivity period begins on the date of the product’s first MA in the EU. After eight years, a generic product application may be submitted and generic companies may rely on the MA holder’s data. However, a generic product cannot launch until two years later (or a total of 10 years after the first MA in the EU of the innovator product), or three years later (or a total of 11 years after the first MA in the EU of the innovator product) if the MA holder obtains MA for a new indication with significant clinical benefit within the eight-year data exclusivity period. Additionally, another noncumulative one -year period of data exclusivity can be added to the eight years of data exclusivity where an application is made for a new indication for a well-established substance, provided that significant pre-clinical or clinical studies were carried out in relation to the new indication. Another year of data exclusivity may be added to the eight years, where a change of classification of a pharmaceutical product has been authorized on the basis of significant pre-trial tests or clinical trials (when examining an application by another applicant for or holder of market authorization for a change of classification of the same substance the competent authority will not refer to the results of those tests or trials for one year after the initial chance was authorized).
Products may not be granted data exclusivity since there is no guarantee that a product will be considered by the European Union’s regulatory authorities to include a NCE. Even if a compound is considered to be a NCE and the MA applicant is able to gain the prescribed period of data exclusivity, another company nevertheless could also market another version of the medicinal product if such company can complete a full MAA with their own complete database of pharmaceutical tests, preclinical studies and clinical trials and obtain MA of its product.
Myalepta is entitled to 10 years of market exclusivity by the European Union from its approval in July 2018. On July 31, 2013 lomitapide was granted a centralized marketing authorization from the European Commission. Lomitapide has eight years’ data exclusivity and 10 years’ marketing exclusivity in the European Union from July 31, 2013.
Orphan Designation and Exclusivity
The criteria for designating an orphan medicinal product in the European Union are similar in principle to those in the United States. The EMA grants orphan drug designation if the medicinal product is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting no more than five in 10,000 persons in the European Union (prevalence criterion). In addition, Orphan Drug Designation can be granted if, for economic reasons, the medicinal product would be unlikely to be developed without incentives and if there is no other satisfactory method approved in the European Union of diagnosing, preventing, or treating the condition, or if such a method exists, the proposed medicinal product is a significant benefit to patients affected by the condition. An application for orphan drug designation (which is not a marketing authorization, as not all orphan-designated medicines reach the authorization application stage) must be submitted first before an application for marketing authorization of the medicinal product is submitted. The applicant will receive a fee reduction for the marketing authorization application if the orphan drug designation has been granted, but not if the designation is still pending at the time the marketing authorization is submitted, and sponsors must submit an annual report to EMA summarizing the status of development of the medicine. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. Designated orphan medicines are eligible for conditional marketing authorization.
The EMA’s Committee for Orphan Medicinal Products (“COMP”) reassesses the orphan drug designation of a product in parallel with the review for a marketing authorization; for a product to benefit from market exclusivity it must maintain its orphan drug designation at the time of marketing authorization review by the EMA and approval by the EC. Additionally, any marketing authorization granted for an orphan medicinal product must only cover the therapeutic indication(s) that are covered by the orphan drug designation. Upon the grant of a marketing authorization, orphan drug designation provides up to ten years of market exclusivity in the orphan indication.
During the 10-year period of market exclusivity, with a limited number of exceptions, the regulatory authorities of the EU Member States and the EMA may not accept applications for marketing authorization, accept an application to extend an existing marketing authorization or grant marketing authorization for other similar medicinal products for the same therapeutic indication. A similar medicinal product is defined as a medicinal product containing a similar active substance or substances as contained in a currently authorized orphan medicinal product, and which is intended for the same therapeutic indication. An orphan medicinal product can also obtain an additional two years of market exclusivity for an orphan-designated condition when the results of specific studies are reflected in the Summary of Product Characteristics (“SmPC”), addressing the pediatric population and completed in accordance with a fully compliant Pediatric Investigation Plan (“PIP”). No extension to any supplementary protection certificate can be granted on the basis of pediatric studies for orphan indications.
The 10-year market exclusivity may be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria for orphan designation, i.e. the condition prevalence or financial returns criteria under Article 3 of Regulation (EC) No. 141/2000 on orphan medicinal products. When the period of orphan market exclusivity for an indication ends, the orphan drug designation for that indication expires as well. Orphan exclusivity runs in parallel with normal rules on data exclusivity and market protection. Additionally, a marketing authorization may be granted to a similar medicinal product (orphan or not) for the same or overlapping indication subject to certain requirements.
In 2012, metreleptin was granted four Orphan Drug Designations by the European Commission for the treatment of acquired and congenital GL (Lawrence syndrome and Berardinelli-Seip syndrome respectively), and acquired and familial PL (Barraquer-Simons syndrome). Metreleptin is entitled to ten years of market exclusivity by the European Union from its approval as Myalepta in July 2018. Despite the prevalence rate, lomitapide does not have Orphan Drug exclusivity in the European Union for the treatment of HoFH because the EMA views the relevant condition, for Orphan Drug purposes, to include both HoFH and the more prevalent HeFH. In August 2013 orphan designation was granted by the European Commission for octreotide acetate (oral use) for the treatment of acromegaly. Mycapssa® is not approved in the EU. Maintenance of the orphan designation will be determined by COMP in parallel to the evaluation of the MAA by CHMP.
In the European Union, certain patents relating to Lojuxta may qualify for a supplemental protection certificate that would extend patent protection for up to five years after patent expiration upon marketing authorization in the European Union. Grant of such supplemental protection certificate is, however, subject to strict conditions and it is not automatic. Amryt has applied for such protection in the countries in which Lojuxta is approved, on a country-by-country basis, and in some countries, supplemental protection has been granted to extend patent protection to July or August of 2028, while in other countries, the applications are still pending.
Pediatric Development
In the European Union, companies developing a new medicinal product are obligated to study their product in children and must therefore submit a PIP together with a request for agreement to the EMA. The EMA issues a decision on the PIP based on an opinion of the EMA’s Pediatric Committee (“PDCO”). Companies must conduct pediatric clinical trials in accordance with the PIP approved by the EMA, unless a deferral (e.g. until enough information to demonstrate its effectiveness and safety in adults is available) or a waiver (e.g. because the relevant disease or condition occurs only in adults) has been granted by the EMA. The marketing authorization application for the medicinal product must include the results of all pediatric clinical trials performed and details of all information collected in compliance with the approved PIP, unless a waiver or a deferral has been granted, in which case the pediatric clinical trials may be completed at a later date. Medicinal products that are granted a marketing authorization on the basis of the pediatric clinical trials conducted in accordance with the approved PIP are eligible for a six month extension of the protection under a supplementary protection certificate (if any is in effect at the time of approval) or, in the case of orphan medicinal products, a two year extension of the orphan market exclusivity. This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the approved PIP are developed and submitted. An approved PIP is also required when a marketing-authorization holder wants to add a new indication, pharmaceutical form or route of administration for a medicine that is already authorized and covered by intellectual property rights.
Post-Approval Regulation
Similar to the United States, both MA holders and manufacturers of medicinal products are subject to comprehensive regulatory oversight by the EMA, the EC and/or the competent regulatory authorities of the EU Member States. This oversight applies both before and after grant of manufacturing licenses and marketing authorizations. It includes control of compliance with EU good manufacturing practices rules, manufacturing authorizations, pharmacovigilance rules and requirements governing advertising, promotion, sale, and distribution, recordkeeping, importing and exporting of medicinal products.
Failure by us or by any of our third-party partners, including suppliers, manufacturers and distributors to comply with EU laws and the related national laws of individual EU Member States governing the conduct of clinical trials, manufacturing approval, MA of medicinal products and marketing of such products, both before and after grant of MA, statutory health insurance, bribery and anti-corruption or other applicable regulatory requirements may result in administrative, civil or criminal penalties. These penalties could include delays or refusal to authorize the conduct of clinical trials or to grant MA, product withdrawals and recalls, product seizures, suspension, withdrawal or variation of the MA, total or partial suspension of production, distribution, manufacturing or clinical trials, operating restrictions, injunctions, suspension of licenses, fines and criminal penalties.
The holder of a MA for a medicinal product must also comply with EU pharmacovigilance legislation and its related regulations and guidelines, which entail many requirements for conducting pharmacovigilance, or the assessment and monitoring of the safety of medicinal products.
These pharmacovigilance rules can impose on holders of MAs the obligation to conduct a labor intensive collection of data regarding the risks and benefits of marketed medicinal products and to engage in ongoing assessments of those risks and benefits, including the possible requirement to conduct additional clinical studies or post-authorization safety studies to obtain further information on a medicine’s safety, or to measure the effectiveness of risk-management measures, which may be time consuming and expensive and could impact our profitability. MA holders must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance, who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of Periodic Safety Update Reports (“PSURs”) in relation to medicinal products for which they hold MAs. The EMA reviews PSURs for medicinal products authorized through the centralized procedure. If the EMA has concerns that the risk benefit profile of a product has varied, it can adopt an opinion advising that the existing MA for the product be suspended, withdrawn or varied. The agency can advise that the MA holder be obliged to conduct post-authorization Phase IV safety studies. If the EC agrees with the opinion, it can adopt a decision varying the existing MA. Failure by the MA holder to fulfill the obligations for which the EC’s decision provides can undermine the ongoing validity of the MA.
More generally, non-compliance with pharmacovigilance obligations can lead to the variation, suspension or withdrawal of the MA for the product or imposition of financial penalties or other enforcement measures.
The manufacturing process for medicinal products in the European Union is highly regulated and regulators may shut down manufacturing facilities that they believe do not comply with regulations. Manufacturing requires a manufacturing authorization, and the manufacturing authorization holder must comply with various requirements set out in the applicable EU laws, regulations and guidance, including Directive 2001/83/EC, Directive 2003/94/EC, Regulation (EC) No 726/2004 and the European Commission Guidelines for Good Manufacturing Practice (“GMP”). These requirements include compliance with EU GMP standards when manufacturing medicinal products and active pharmaceutical ingredients, including the manufacture of active pharmaceutical ingredients outside of the European Union with the intention to import the active pharmaceutical ingredients into the European Union. Similarly, the distribution of medicinal products into and within the European Union is subject to compliance with the applicable EU laws, regulations and guidelines, including the requirement to hold appropriate authorizations for distribution granted by the competent authorities of the EU Member States. The manufacturer or importer must have a qualified person who is responsible for certifying that each batch of product has been manufactured in accordance with GMP, before releasing the product for commercial distribution in the European Union or for use in a clinical trial. Manufacturing facilities are subject to periodic inspections by the competent authorities for compliance with GMP.
We and our third-party manufacturers are subject to GMP, which are extensive regulations governing manufacturing processes, stability testing, record keeping and quality standards as defined by the EMA, the European Commission, the competent authorities of EU Member States and other regulatory authorities. Companies may be subject to civil, criminal or administrative sanctions. These include suspension of manufacturing authorization in case of non-compliance with the European Union or EU Member States’ requirements governing the manufacturing of medicinal products.
Advertising and Promotion
The advertising and promotion of our products is also subject to EU laws concerning promotion of medicinal products, interactions with physicians, misleading and comparative advertising and unfair commercial practices. In addition, other national legislation of individual EU Member States may apply to the advertising and promotion of medicinal products and may differ from one country to another. These laws require that promotional materials and advertising in relation to medicinal products comply with the product’s SmPC as approved by the competent regulatory authorities. The SmPC is the document that provides information to physicians concerning the safe and effective use of the medicinal product. It forms an intrinsic and integral part of the marketing authorization granted for the medicinal product. Promotion of a medicinal product that does not comply with the SmPC is considered to constitute off-label promotion. All advertising and promotional activities for the product must be consistent with the approved SmPC and therefore all off-label promotion is prohibited. Direct-to-consumer advertising of prescription-only medicines is also prohibited in the EU. Violations of the rules governing the promotion of medicinal products in the European Union could be penalized by administrative measures, fines and imprisonment. These laws may further limit or restrict the advertising and promotion of our products to the general public and may also impose limitations on its promotional activities with healthcare professionals.
Pricing and Reimbursement Environment
Even if a medicinal product obtains a marketing authorization in the European Union, there can be no assurance that reimbursement for such product will be secured on a timely basis or at all. The EU Member States are free to restrict the range of medicinal products for which their national health insurance systems provide reimbursement, and to control the prices and reimbursement levels of medicinal products for human use. An EU Member State may approve a specific price or level of reimbursement for the medicinal product, or alternatively adopt a system of direct or indirect controls on the profitability of the company responsible for placing the medicinal product on the market, including volume-based arrangements, caps and reference pricing mechanisms.
Reference pricing used by various EU Member States and parallel distribution, or arbitrage between low-priced and high-priced member states, can further reduce prices. In some countries, we may be required to conduct a clinical study or other studies that compare the cost-effectiveness of our product candidates, if any, to other available therapies in order to obtain or maintain reimbursement or pricing approval. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, medicinal products launched in the European Union do not follow price structures of the United States and generally published and actual prices tend to be significantly lower. Publication of discounts by third party payers or authorities and public tenders may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries.
The so-called Health Technology Assessment (“HTA”) of medicinal products is becoming an increasingly common part of the pricing and reimbursement procedures in some EU Member States, including France, Germany, Ireland, Italy and Sweden. The HTA process, which is governed by the national laws of these countries, is the procedure according to which the assessment of the public health impact, therapeutic impact, and the economic and societal impact of use of a given medicinal product in the national healthcare systems of the individual country is conducted. HTA generally focuses on the clinical efficacy and effectiveness, safety, cost, and cost-effectiveness of individual medicinal products as well as their potential implications for the healthcare system. Those elements of medicinal products are compared with other treatment options available on the market. The outcome of HTA regarding specific medicinal products will often influence the pricing and reimbursement status granted to medicinal products by the regulatory authorities of individual EU Member States. A negative HTA of one of our products by a leading and recognized HTA body could not only undermine our ability to obtain reimbursement for such product in the EU Member State in which such negative assessment was issued, but also in other EU Member States. For example, EU Member States that have not yet developed HTA mechanisms could rely to some extent on the HTA performed in other countries with a developed HTA framework, when adopting decisions concerning the pricing and reimbursement of a specific medicinal product.
On January 31, 2018, the European Commission adopted a proposal for a regulation on health technology assessment. This legislative proposal is intended to boost EU level cooperation among EU Member States in assessing health technologies, including new medicinal products, and providing the basis for cooperation at the EU level for joint clinical assessments in these areas. The proposal provides that EU Member States will be able to use common HTA tools, methodologies and procedures across the European Union, working together in four main areas, including joint clinical assessment of the innovative health technologies with the most potential impact for patients, joint scientific consultations whereby developers can seek advice from HTA authorities, identification of emerging health technologies to identify promising technologies early, and continuing voluntary cooperation in other areas. Individual EU Member States will continue to be responsible for assessing non-clinical (e.g., While EU Member States could choose to delay participation in the joint work until three years after the rules enter into force, it will become mandatory after six years. The European Commission has stated that the role of the HTA regulation is not to influence pricing and reimbursement decisions in the individual EU Member States, but there can be no assurance that the HTA regulation will not have effects on pricing and reimbursement decisions. The HTA entered into force on January 11, 2022 and applies as of January 2025 followed by a further three-year transitional period during which EU member states must fully adapt to the new system.
To obtain reimbursement or pricing approval in some countries, including the EU Member States, we may be required to conduct studies that compare the cost-effectiveness of our product candidates to other therapies that are considered the local standard of care. There can be no assurance that any country will allow favorable pricing, reimbursement and market access conditions for any of our products, or that we will be feasible to conduct additional cost-effectiveness studies, if required.
In certain of the EU Member States, medicinal products that are designated as orphan medicinal products may be exempted or waived from having to provide certain clinical, cost-effectiveness and other economic data in connection with their filings for pricing/reimbursement approval. As noted above, Lojuxta was not granted an Orphan Drug Designation by the EMA for the treatment of HoFH. As such, it is not eligible for benefits related to Orphan Drug Designation. As a result, we may not be able to provide all of the data required to obtain pricing/reimbursement approvals in certain EU Member States, which has and could, in the future, result in delays of pricing/reimbursement approvals for Lojuxta, Lojuxta not obtaining pricing/reimbursement approval at all, or Lojuxta obtaining approvals at less than acceptable levels or with significant restrictions on use or reimbursement.
Many countries outside the United States and the European Union have pricing and reimbursement approvals and regimes that are comparable to those in the key EU markets. We have successfully achieved coverage in the United States and reimbursement in Japan, Turkey, Saudi Arabia, Germany, Italy, the United Kingdom, France, Austria, the Netherlands and Slovenia. Individual patient funding has been obtained in Czech Republic and Hungary. Pricing and reimbursement processes are ongoing in Brazil, Spain, Denmark, Norway, Portugal, Israel, Kuwait, Belgium, Poland, Romania and Latvia. Mycapssa® has been submitted to the EMA in 2021 and is not yet approved in Europe.
European Data Laws
The collection and use of personal health data and other personal information in the European Union is governed by the provisions of the European General Data Protection Regulation (EU) 2016/697 (“GDPR”), which came into force in May 2018 and related implementing laws in individual EU Member States.
. The GDPR imposes a number of strict obligations and restrictions on the ability to process (processing includes collection, analysis and transfer of) personal data of individuals within the European Union and in the EEA, including health data from clinical trials and adverse event reporting. The GDPR also includes requirements relating to the consent of the individuals to whom the personal data relates, the information provided to the individuals prior to processing their personal data or personal health data, notification of data processing obligations to the national data protection authorities and the security and confidentiality of the personal data. EU Member States may also impose additional requirements in relation to health, genetic and biometric data through their national implementing legislation.
Under the GDPR, personal data can only be transferred within the EU Member States and the three additional European Economic Area countries (Norway, Iceland and Liechtenstein) that have adopted a national law implementing the GDPR. Appropriate safeguards are required to enable cross-border transfers of personal data from the EU and EEA Member States to a “third country” (a country outside the EU or EEA). This status has a number of significant practical consequences, in particular for international data transfers, competent supervisory authorities and enforcement of the GDPR.
In conclusion, he GDPR also prohibits the transfer of personal data to countries outside of the European Union/EEA that are not considered by the European Commission to provide an adequate level of data protection (including the United States), except if the data controller meets very specific requirements such as the use of standard contractual clauses, (“SCCs”), issued by the European Commission. In this respect recent legal developments in Europe have created complexity and compliance uncertainty regarding certain transfers of personal data from the EU/EEA. For example, following the Schrems II decision of the Court of Justice of the European Union on July 16, 2020, in which the Court invalidated the Privacy Shield under which personal data could be transferred from the EU/EEA to United States entities who had self-certified under the Privacy Shield scheme, there is uncertainty as to the general permissibility of international data transfers under the GDPR. The Court did not invalidate the then current SCCs, but ruled that data exporters relying on these SCCs are required to verify, on a case-by-case basis, if the law of the third country ensures an adequate level of data protection that is essentially equivalent to that guaranteed in the EU/EEA. In light of the implications of this decision we may face difficulties regarding the transfer of personal data from the European Union/EEA to third countries. However, on June 4, 2021 the EU Commission issued a new set of SCCs for data transfers from controllers or processors in the EU/EEA to controllers or processors established outside the EU/EEA. These SCCs replace the old sets of SCCs that were adopted under the previous European Data Protection Directive 95/46. Since September 27, 2021, it is no longer possible to conclude contracts incorporating these previous versions of the SCCs. In addition, for contracts concluded before September 27, 2021, it is still possible to rely on the previous SCCs until the end of an additional 15 months’ transitional period (until December 27, 2022), provided that the processing operations which are the subject matter of the contract remain unchanged and reliance on previous SCCs ensures that the transfer is subject to appropriate safeguards. On November 11, 2021, the European Data Protection Board has adopted recommendations on such appropriate safeguards that supplement transfer mechanisms. These recommendations aim to assist data exporters with their duty to identify and implement appropriate supplementary measures where they are needed to ensure an essentially equivalent level of protection to the personal data they transfer to third countries.
On March 25, 2022, the United States and the European Commission published a joint statement on a new Trans-Atlantic Data Privacy Framework. While such an agreement may facilitate data transfers to the United States in the future, the mere announcement has no impact and so far does not provide the legal basis for transferring personal data to the United States.
Failure to comply with the requirements of the GDPR and the related national data protection laws of the EU Member States may result in significant monetary fines, for noncompliance of up to €20 million or 4% of the annual global revenues of the noncompliant company, whichever is greater, other administrative penalties and a number of criminal offenses (punishable by uncapped fines) for organizations and in certain cases their directors and officers as well as civil liability claims from individuals whose personal data was processed. Data protection authorities from the different EU Member States may still implement certain variations, enforce the GDPR and national data protection laws differently, and introduce additional national regulations and guidelines, which adds to the complexity of processing personal data in the European Union. Guidance developed at both EU level and at the national level in individual EU Member States concerning implementation and compliance practices are often updated or otherwise revised.
There is, moreover, a growing trend towards required public disclosure of clinical trial data in the European Union which adds to the complexity of obligations relating to processing health data from clinical trials. Such public disclosure obligations are provided in the new EU CTR, EMA disclosure initiatives and voluntary commitments by industry. Failing to comply with these obligations could lead to government enforcement actions and significant penalties against us, harm to our reputation, and adversely impact our business and operating results. The uncertainty regarding the interplay between different regulatory frameworks, such as the Clinical Trials Regulation and the GDPR, further adds to the complexity that we face with regard to data protection regulation.
On June 28, 2021, the European Commission adopted two adequacy decisions for the United Kingdom – one under the GDPR and the other for the Law Enforcement Directive. Personal data may now freely flow from the European Union to the United Kingdom since the United Kingdom is deemed to have an adequate data protection level. Additionally, following the UK's withdrawal from the European Union and the EEA, companies have to comply also with the UK’s data protection laws (including the GDPR as incorporated into UK national law), the latter regime having the ability to separately fine up to the greater of £17.5 million or 4% of global turnover.
Promotional Activities
In the European Union, interactions between pharmaceutical companies and physicians are also governed by strict laws, regulations, industry self-regulation codes of conduct and physicians’ codes of professional conduct both at EU level and in the individual EU Member States. The provision of benefits or advantages to physicians to induce or encourage the prescription, recommendation, endorsement, purchase, supply, order or use of medicinal products is prohibited in the European Union. The provision of benefits or advantages to physicians is also governed by the national anti-bribery laws of the EU Member States. Violation of these laws could result in substantial fines and imprisonment.
Payments made to physicians in certain EU Member States must be publicly disclosed. Moreover, agreements with physicians must often be the subject of prior notification and approval by the physician’s employer, his/her regulatory professional organization, and/or the competent authorities of the individual EU Member States. These requirements are provided in the national laws, industry codes, or professional codes of conduct, applicable in the individual EU Member States. Failure to comply with these requirements could result in reputational risk, public reprimands, administrative penalties, fines or imprisonment.
While the UK has left the EU, as mentioned above, it should be noted that the UK still has the strictest anti-bribery regime in Europe, the UK Bribery Act 2010. The Act is applicable English law and continues to apply to any company incorporated in or “carrying on business” in the United Kingdom, irrespective of where in the world the alleged bribery activity occurs.
Expanded Access Outside the United States and the European Union
In certain countries, drug products approved in the United States or the European Union can be accessed by patients before the drug has obtained marketing approval in such country. Various forms of this access include sale of product, often to the government, on a named patient basis and provision of the product free of charge on a named patient basis or a compassionate use basis. Each country has its own laws and regulations that apply to these forms of access. Amryt has made lomitapide available in Brazil, Canada, Czech Republic, France, Germany, Greece, Israel, Italy, Turkey, Poland, Spain, South Africa, Switzerland, Taiwan and USA and plan to continue to consider access to additional countries. When Aegerion acquired metreleptin from AstraZeneca in January 2015, there were a number of patients receiving metreleptin therapy free of charge in certain countries outside the United States that allow use of a drug before marketing approval has been obtained in such country. Where permitted in accordance with applicable requirements, we have continued to make metreleptin available free of charge under such program, which has resulted in significant costs to the Company. In 2016, we began generating revenues from named patient sales of metreleptin in certain markets where named patient sales of metreleptin are possible and to the extent permitted by applicable law and local regulatory authorities. Where appropriate we have supported the transfer of patients from free of charge supply to locally approved pre-authorization funding programs.
Additional Laws and Regulations Governing International Operations
For other countries outside of the European Union and the United States, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In addition, the clinical trials must be conducted in accordance with GCP requirements and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
The UK Bribery Act, the FCPA and other anti-corruption laws generally prohibit us and our employees and intermediaries from authorizing, promising, offering or providing, directly or indirectly, improper or prohibited payments, or anything else of value, to government officials or other persons to obtain or retain business or gain some other business advantage. Under the UK Bribery Act, we may also be liable for failing to prevent a person associated with us from committing a bribery offense. We and our commercial partners operate in a number of jurisdictions that pose a high risk of potential UK Bribery Act or FCPA violations, and we also participate in collaborations and relationships with third parties whose corrupt or illegal activities could potentially subject us to liability under the UK Bribery Act, FCPA or local anti-corruption laws, even if we did not explicitly authorize or have actual knowledge of such activities. The FCPA also requires us, as a public company, to make and keep books and records that accurately and fairly reflect all of our transactions and to devise and maintain an adequate system of internal accounting controls.
Compliance with the FCPA is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, the FCPA presents particular challenges in the pharmaceutical industry, because, in many countries, hospitals are operated by the government, and doctors and other hospital employees are considered foreign officials. Certain payments to hospitals in connection with clinical trials and other work have been deemed to be improper payments to government officials and have led to FCPA enforcement actions against other companies.
Various laws, regulations and executive orders also restrict the use and dissemination outside of the United States, or the sharing with certain non-U.S. nationals, of information classified for national security purposes, as well as certain products and technical data relating to those products. If we are successful in expanding our presence outside of the United States, such expansion may require us to dedicate additional resources to comply with these laws, and these laws may preclude us from developing, manufacturing, or selling certain products and product candidates outside of the United States, which could limit our growth potential and increase our development costs.
We will be subject to compliance with the anti-bribery laws of other countries, including Brazil. Our activities outside the United States or those of its employees, licensees, distributors, manufacturers, clinical research organizations, or other third parties who act on its behalf or with whom we do business could subject us to investigation or prosecution under foreign or U.S. laws. For example, federal, Brasilia and São Paulo authorities in Brazil are each conducting an investigation to determine whether there have been any violations of Brazilian laws related to the sales of Juxtapid in Brazil.
The failure to comply with laws governing international business practices may result in substantial civil and criminal penalties and suspension or debarment from government contracting. The SEC also may suspend or bar issuers from trading securities on U.S. exchanges for violations of the FCPA’s accounting provisions.
Our present and future business has been and will continue to be subject to various other laws and regulations. Various laws, regulations and recommendations relating to safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import and export and use and disposal of hazardous or potentially hazardous substances, including radioactive compounds and infectious disease agents, used or that we may use in the future in connection with our development work are or may be applicable to our activities. Certain agreements we enter into involving exclusive license rights or acquisitions may be subject to national or supranational antitrust regulatory control, the effect of which cannot be predicted. The extent of government regulation, which might result from future legislation or administrative action, cannot accurately be predicted.
C. Organizational Structure
Amryt Pharma plc is a public limited company organized under the laws of the England and Wales.
Group Structure as of December 31, 2021
| Subsidiary | Owner | Ownership Percentage |
| Amryt Pharma Holdings Limited | Amryt Pharma Plc | 100% |
| Amryt Pharmaceuticals Designated Activity Company | Amryt Pharma Holdings Limited | 100% |
| Amryt Pharmaceuticals Inc. | Amryt Pharma Holdings Limited | 100% |
| Amryt Endo Inc. | Amryt Pharmaceuticals Inc. | 100% |
| Chiasma Securities Corp | Amryt Endo Inc. | 100% |
| Chiasma (Israel) Limited | Amryt Endo Inc. | 100% |
| SomPharmaceuticals SA | Amryt Pharmaceuticals Designated Activity Company | 100% |
| Amryt Endocrinology Limited | Amryt Pharmaceuticals Designated Activity Company | 100% |
| Amryt GmbH | Amryt Pharmaceuticals Designated Activity Company | 100% |
| Amryt Research Limited | Amryt Pharmaceuticals Designated Activity Company | 100% |
| Amryt Lipidology Limited | Amryt Pharmaceuticals Designated Activity Company | 100% |
| Amryt Distribution Limited | Amryt Pharmaceuticals Designated Activity Company | 100% |
| Amryt Pharma Italy SRL | Amryt Pharmaceuticals Designated Activity Company | 100% |
| Amryt Pharma (UK) Limited | Amryt Pharmaceuticals Designated Activity Company | 100% |
| Amryt Pharma Spain SL | Amryt Pharmaceuticals Designated Activity Company | 100% |
| Amryt Genetics Limited | Amryt Pharmaceuticals Designated Activity Company | 100% |
| Cala Medical Limited. | Amryt Pharmaceuticals Designated Activity Company | 100% |
| Aegerion Pharmaceuticals Holdings, Inc. (DE) | Amryt Pharmaceuticals Inc. (DE) | 100% |
| Aegerion Argentina S.R.L. (Argentina) | Amryt Pharmaceuticals Inc. (DE) | 99.31% |
| Aegerion Pharmaceuticals Holdings, Inc. (DE) | 0.69% |
| Aegerion International Limited (Bermuda) | Amryt Pharmaceuticals Inc. (DE) | 100% |
| Aegerion Pharmaceuticals Limited (Bermuda) | Aegerion International Limited (Bermuda) | 100% |
| Aegerion Pharmaceuticals (Canada) Limited (BC) | Aegerion Pharmaceuticals Holdings Inc. (DE) | 100% |
| Amryt Colombia S.A.S. (Colombia) | Amryt Pharmaceuticals Inc. (DE) | 100% |
| Aegerion Pharmaceuticals Limited (England & Wales) | Aegerion Pharmaceuticals Limited (Bermuda) | 100% |
| Amryt Pharmaceuticals SAS (France) | Aegerion Pharmaceuticals Limited (England & Wales) | 100% |
| Amryt Pharma GmbH (Germany) | Aegerion Pharmaceuticals Limited (England & Wales) | 100% |
| Aegerion Pharmaceuticals Srl (Italy) | Aegerion Pharmaceuticals Limited (England & Wales) | 100% |
| Aegerion Pharmaceuticals B.V. (Netherlands) | Aegerion Pharmaceuticals Limited (England & Wales) | 100% |
| Aegerion Pharmaceuticals Spain, S.L. (Spain) | Aegerion Pharmaceuticals Limited (England & Wales) | 100% |
| Aegerion Pharmaceuticals SARL (Switzerland) | Aegerion Pharmaceuticals Limited (England & Wales) | 100% |
| Amryt Turkey İlaç Ticaret Limited Şirketi (Turkey) | Aegerion Pharmaceuticals Limited (England & Wales) | 100% |
| Amryt Brasil Comercio E Importacao De Medicamentos LTDA (Brazil) | Amryt Pharmaceuticals Inc. (DE) | 100% |
D. Property, Plants and Equipment
Our principal executive office is in Dublin, Ireland, where we lease office space under leases that expire in 2040. Our U.S. headquarters are in Boston, Massachusetts, where we lease office space under a lease that expires in 2027. We also have additional offices, research facilities and manufacturing facilities in Germany. We believe these facilities are adequate for our current needs and that additional or replacement space will be available on commercially reasonable terms as needed.
| Item 4A. | Unresolved Staff Comments |
Not applicable.
| Item 5. | Operating and Financial Review and Prospects |
A. Operating Results
You should read the following discussion and analysis of our financial condition and results of operations together with the information in and the Consolidated Financial Statements of Amryt, including the notes thereto. The following discussion is based on financial information prepared in accordance with IFRS as issued by the IASB. The following discussion includes forward-looking statements that involve risks, uncertainties and assumptions. Actual results may differ materially from those anticipated in these forward-looking statements as a result of many factors, including but not limited to those described under “Risk Factors” and elsewhere in this annual report.
Overview
We are a global, commercial-stage biopharmaceutical company dedicated to acquiring, developing and commercializing novel treatments for rare diseases. We have built a diverse portfolio of commercial and development stage assets including:
| • | lomitapide, an approved treatment in the United States and the European Union for adult patients with HoFH; |
| • | metreleptin, an approved treatment in the United States for GL and in the European Union for GL and PL; |
| • | oral octreotide, an approved treatment in the United States for long-term maintenance therapy in acromegaly; |
| • | Oleogel-S10, our lead development asset, which has completed its pivotal Phase 3 trial as a potential treatment for severe EB and received positive topline data on September 9, 2020. On February 28, 2022, we received a CRL from the FDA which asked Amryt to submit additional confirmatory evidence of effectiveness for Oleogel-S10 in EB. Amryt intends to discuss with the FDA the nature of the data required to address the Agency’s concerns. A MAA for Oleogel-S10 for the treatment of Dystrophic and Junctional EB was validated by the EMA on March 25, 2021, the assessment process by EMA was completed on April 22, 2022, when the CHMP adopted a positive opinion. The positive opinion recommends the approval of Filsuvez® in the EU for the treatment of partial thickness wounds associated with dystrophic and junctional EB in patients six months and older. Based on this CHMP recommendation a decision by the European Commission (“EC”) is expected on the Filsuvez® application within 67 days; |
| • | We are also developing Mycapssa® to expand the indication of Mycapssa® (octreotide capsules) beyond acromegaly into carcinoid syndrome associated with neuroendocrine tumors; |
| • | AP103, our first product candidate utilizing our novel polymer-based topical gene therapy delivery platform, which is in preclinical development as a potential treatment for patients with EB and other topical indications; and |
| • | Imlan, a range of derma-cosmetic products marketed solely in Germany as a treatment for sensitive, allergy-prone and dry skin. |
On September 24, 2019, we completed the acquisition of Aegerion, a wholly-owned operating subsidiary of Novelion Therapeutics Inc. We believe that this acquisition has enabled the Group to advance our ambition to create a global leader in rare and orphan diseases. Prior to the Acquisition, we licensed the rights from Aegerion to sell lomitapide in Europe and the Middle East, while Aegerion retained the rights to this product in the United States and elsewhere. Following the acquisition, we sell lomitapide and metreleptin globally, excluding Japan for lomitapide and Japan, South Korea and Taiwan for metreleptin.
On August 5, 2021, we completed the acquisition of Chiasma, Inc. We believe that following the acquisition the combined company will be a global leader in rare and orphan diseases with three on-market commercial products, a global commercial and operational footprint and a significant development pipeline of therapies with the financial flexibility to execute its growth plans.
Key Factors Affecting Our Business
Our ability to expand approved indications and markets for lomitapide, metreleptin and Mycapssa
We intend to take steps necessary to seek approval for the use of lomitapide to treat pediatric HoFH and for the use of metreleptin to treat PL in the United States. We are also developing Mycapssa® to expand the indication of Mycapssa® (octreotide capsules) beyond acromegaly into carcinoid syndrome associated with neuroendocrine tumors. In addition, we intend to seek marketing approval for metreleptin and lomitapide in certain South American countries as well as Mycapssa® in the European Union. Our ability to generate revenue from these potential new indications and markets depends on our success in completing development of and commercializing such products, which will require a significant investment of time and resources and may not ultimately prove successful.
Our ability to commercialize Oleogel-S10, if approved
We received positive top line data from the pivotal Phase 3 EASE Trial of Oleogel-S10 in September 2020, details of which are discussed further in “Business—Our Product Candidates—Oleogel-S10 for the Treatment of Severe EB—Topline Results for Oleogel-S10,” and in 2021, we submitted applications for approval of Oleogel-S10 and, if approved, commercialize it under the name Oleogel-S10 in the United States and Europe through our existing commercial infrastructure. On February 28, 2022, we received a CRL from the FDA which asked Amryt to submit additional confirmatory evidence of effectiveness for Oleogel-S10 in EB. Amryt intends to discuss with the FDA the nature of the data required to address the Agency’s concerns. A MAA for Oleogel-S10 for the treatment of Dystrophic and Junctional EB was validated by EMA March 25, 2021, the assessment process by EMA was completed on April 22, 2022, when the CHMP adopted a positive opinion. The positive opinion recommends the approval of Filsuvez® in the EU for the treatment of partial thickness wounds associated with dystrophic and junctional EB in patients six months and older. Based on this CHMP recommendation a decision by the EC is expected on the Filsuvez® application within 67 days. If the commercialization effort requires more than minimal additions, our financial results could suffer.
International operations and foreign currency exchange
We operate on a global basis with offices, sales and activities throughout the world. Our worldwide operations and sales have increased as a result of the acquisition of Aegerion and Chiasma and the continued expansion of our commercial organization. Our global operations subject our financial results to fluctuations in foreign currency exchange rates, changes in general economic and political conditions in countries where we operate, particularly as a result of ongoing economic instability within foreign jurisdictions, sensitivity to governmental actions relating to tariffs or trade agreements, complex and restrictive employment and labor laws and regulations, as well as union and works council restrictions, sensitivity to changes in tax laws or rulings in jurisdictions across the world, longer payment cycles from customers in certain geographies, and legal compliance costs and risks.
Our ability to add rare disease assets to our portfolio
Identifying, acquiring and developing new products and product candidates to build shareholder value is key to our goal of becoming a global leader in the treatment of rare diseases. Our future profitability and growth will depend on our ability to acquire and in-license product candidates on favorable terms. Moreover, competitors may succeed in developing, acquiring, licensing or introducing new pharmaceutical products that are more effective, have a more favorable safety profile or are less costly than our products. See “—Risk Factors” for additional information.
Key Components of Our Results of Operations
Revenues
We generated revenues from the sale of lomitapide globally (marketed as Juxtapid in the U.S. and as Lojuxta in the EU), metreleptin globally (marketed as Myalept in the U.S. and as Myalepta in the EU), royalties paid to us from our Juxtapid and Myalept licensees in Japan, and from the sale of Imlan. Following the acquisition of Chiasma, we also generated revenues from the sale of Mycapssa® in the U.S. (the non-EU marketing name for octreotide).
Cost of Sales
Cost of sales is comprised of the cost of producing products, royalties on sales and the cost of delivery of goods sold to customers, including the costs associated with the services provided by the distributors to import and deliver the goods. Cost of sales also includes amortization of intangible assets that we acquired as part of the acquisitions of Aegerion and Chiasma, as well as amortization of the step-up in the fair value of the inventory acquired in both acquisitions and any write-down of inventories to fair value less costs to sell recognized as an expense.
Selling, General and Administrative (“SG&A”) expenses
SG&A expenses includes payroll and payroll-related costs, insurance costs, fees payable to auditors, pension costs, marketing expenses and general corporate expenses. Following the acquisition of Aegerion and Chiasma, our SG&A expenses increased significantly with the expansion of the selling, regulatory, quality and compliance infrastructure in place for a company with a global commercial footprint.
We expect SG&A expenses in 2022 to increase compared to 2021, due to an increase in compensation-related expenses driven by higher headcount and other expenses related to the expansion and support of our business. We also expect an increase in expenses related to the preparation for the potential commercial launch of Oleogel-S10 and the continuation of the commercial launch of metreleptin in the European Union following its approval in July 2018.
Research and Development (“R&D”) expenses
R&D expenses include employee-related expenses, such as salaries and other benefits, for our R&D personnel; costs for production of drug product used in our clinical trials and development of our manufacturing processes; fees and other costs paid to third parties in connection with R&D, consultants and other suppliers to conduct our clinical trials, preclinical and non-clinical studies, and post approval commitment studies; and costs of facilities, materials and equipment related to our clinical trials and preclinical and non-clinical studies. We expense R&D costs as incurred, because they do not qualify to be capitalized as intangible assets under IFRS since the technical feasibility of the materials is not proven and no alternative use for them exists in the absence of marketing approval.
For 2022 and beyond, we expect that our R&D expenses will continue to increase from previous levels. We also anticipate that costs will increase as we prepare for anticipated regulatory submissions and data read-outs from clinical trials, initiate and undertake additional clinical trials and related development work, and potentially acquire rights to additional product candidates. Specific anticipated drivers of increases in R&D expenses are our efforts to complete clinical development and seek marketing approval for NET, initiate clinical development of AP103, and continue with life cycle development opportunities and post approval commitments for lomitapide and metreleptin. R&D expenses may vary substantially from period to period based on the timing of our research and development activities, including timing due to regulatory approval and moving from preclinical to the clinic and enrolling patients into clinical trials.
If Oleogel-S10 is approved within certain time frames, we will incur both significant milestone payment obligations and significant financial obligations (under our CVRs and other milestones).
Finance Items
Finance income (expense) includes interest on our realized and unrealized exchange rate gains (losses), as well as interest and fees paid on our outstanding indebtedness.
As part of the acquisition of Aegerion, we entered into a U.S. dollar denominated $81,021,000 Secured Credit Facility with various lenders. The Secured Credit Facility was due to mature on September 24, 2024. Interest was payable on a quarterly basis at the rate of (i) 11% per annum paid in cash on a quarterly basis or, at our option, (ii) at the rate of 6.5% paid in cash plus 6.5% paid in kind. The Secured Credit Facility could be prepaid, in whole or in part, at any time subject to payment of an exit fee, which depending on the stage of the loan term, ranges from 5.0% to 0% of the principal then outstanding. On February 18, 2022, the Secured Credit Facility was repaid in full and the Group secured a $125 million Senior Credit Facility from funds managed by Ares of which US$105 million was drawn down to facilitate the prepayment of the existing Secured Credit Facility. In repaying the Secured Credit Facility Amryt incurred an exit fee of 5.00% of the outstanding principal as at the prepayment date.
As part of the acquisition of Aegerion, we also issued Convertible Notes with an aggregate principal amount of $125 million to certain of Aegerion’s pre-bankruptcy creditors. The Convertible Notes are senior unsecured obligations and bear interest at a rate of 5.0% per year, payable semi-annually in arrears on April 1 and October 1 of each year. The Convertible Notes will mature on April 1, 2025, unless earlier repurchased or converted. As a result of the conversion feature in the Convertible Notes, the notes were assessed to have both a debt and an equity component. The two components were assessed separately and classified as a financial liability and equity instrument, respectively. The financial liability component was measured at fair value based on the discounted cash flows expected over the estimated term of the notes using a discount rate based on a market interest rate that a similar debt instrument without a conversion feature would be subject to. The liability component is recorded on our Consolidated Statement of Financial Position at amortized cost, and interest is calculated by applying the estimated prevailing market interest rate at the time of issue. The equity component is recognized in equity and is not subsequently remeasured.
All interest-bearing loans and borrowings are recognized initially at fair value less attributable transaction costs which are subsequently carried at amortized cost using the effective interest method. Interest is charged to the Consolidated Statement of Comprehensive Income/(Loss).
Exchange rate adjustments recognized in the financial income or expenses reflect adjustments to the value of assets and liabilities denominated in foreign currencies, other than our functional currency, which is U.S. dollars for the year ended December 31, 2021.
We include the non-cash discount component of contingent consideration which is being unwound to the Consolidated Statement of Comprehensive Income/(Loss) in Finance expenses. The contingent consideration arose as part of the acquisition of Birken AG (now Amryt GmbH) in 2016 through which we acquired Oleogel-S10. The fair value of contingent consideration is determined by discounting the contingent amounts payable to their present value as of each reporting date. The discount component is being unwound as a non-cash financing charge/gain in the Consolidated Statement of Comprehensive Income/(Loss) over the life of the obligation. We review the fair value of the contingent consideration at each reporting date.
We include the non-cash CVR gain / (loss) which is recognized in the Consolidated Statement of Comprehensive Income/(Loss) in Finance expenses. The CVR finance gain / (loss) arises from the issuance of CVRs to the shareholders and option holders of Amryt as at September 20, 2019, prior to the acquisition of Aegerion. The value of the potential payout of the CVRs was calculated using the probability-weighted expected returns method. Using this method, the potential payment amounts were multiplied by the probability of achievement and discounted to present value. The discount component is being unwound as a non-cash financing charge in the Consolidated Statement of Comprehensive Income/(Loss) over the life of the obligation. We review the estimates and where necessary, will adjust the carrying amount of CVR to reflect the actual and revised estimated contractual cash flows. At each reporting date, we recalculate the amortized cost of the CVR as the present value of the re-estimated future contractual cash flows at the original effective interest rate.
Tax on ordinary activities
Our corporate tax is comprised of current tax and the adjustment of deferred taxes during the period. In any given period, the adjustment to our deferred tax position, including the reversal of valuation allowances, may partially or wholly offset current tax expense.
Results of Operations of Amryt
Financial results of Amryt for the Year Ended December 31, 2021, compared to the Year Ended December 31, 2020.
The following table sets forth certain consolidated statement of operations data for Amryt:
| | | Year ended December 31, | |
| | | 2021 | | | 2020 | |
| | | (US$’000, except per share data) | |
| Revenue | | | 222,543 | | | | 182,607 | |
| Cost of sales | | | (106,119 | ) | | | (119,029 | ) |
| Gross profit | | | 116,424 | | | | 63,578 | |
| Research and development expenses | | | (37,729 | ) | | | (27,618 | ) |
| Selling, general and administrative expenses | | | (91,995 | ) | | | (76,673 | ) |
| Restructuring and acquisition costs | | | (16,947 | ) | | | (1,017 | ) |
| Share based payment expenses | | | (8,341 | ) | | | (4,729 | ) |
| Operating loss before finance expense | | | (38,588 | ) | | | (46,459 | ) |
| Non-cash change in fair value of contingent consideration | | | 18,407 | | | | (27,827 | ) |
| Non-cash contingent value rights gain/(loss) | | | 41,525 | | | | (12,004 | ) |
| Net finance expense - other | | | (27,906 | ) | | | (19,569 | ) |
| Loss on ordinary activities before taxation | | | (6,562 | ) | | | (105,859 | ) |
| Tax credit on loss on ordinary activities | | | 7,562 | | | | 1,332 | |
| Profit/(loss) for the year attributable to the equity holders of the Company | | | 1,000 | | | | (104,527 | ) |
| Total other comprehensive income/(loss) | | | 4,423 | | | | (2,164 | ) |
| Total comprehensive income/(loss) for the year attributable to the equity holders of the Company | | | 5,423 | | | | (106,691 | ) |
| Basic earnings/(loss) per share attributable to ordinary equity holders of the parent | | | 0.00 | | | | (0.66 | ) |
| Diluted earnings/(loss) per share attributable to ordinary equity holders of the parent | | | 0.00 | | | | (0.66 | ) |
Revenues
The revenues for each of our significant products were as follows:
| | | Year ended December 31, | | | | | | | |
| Revenues: | | 2021 | | | 2020 | | | Increase / (Decrease) | |
| | | US$’000 | | | US$’000 | | | US$’000 | | | % | |
Metreleptin | | | 141,242 | | | | 106,872 | | | | 34,370 | | | | 32.2 | % |
Lomitapide | | | 73,867 | | | | 74,750 | | | | (883 | ) | | | (1.2 | %) |
Mycapssa® | | | 6,407 | | | | — | | | | 6,407 | | | | — | |
Other | | | 1,027 | | | | 985 | | | | 42 | | | | 4.3 | % |
| Total revenues | | | 222,543 | | | | 182,607 | | | | 39,936 | | | | 21.9 | % |
Total product sales were $222.5 million for the year ended December 31, 2021, compared to $182.6 million for the year ended December 31, 2020. The increase in revenues was due to increased sales of metreleptin as well as our acquisition of Chiasma in August 2021. Sales of metreleptin lomitapide, and Mycapssa® comprise product sales and royalties on sales, respectively, made by our licensees.
Metreleptin
We generated revenues from product sales of metreleptin of $141.1 million and royalties of $0.1 million from Shionogi for the year ended December 31, 2021, compared to $106.8 million and $0.1 million for the year ended December 31, 2020, respectively. The increase is driven by continued EMEA launch success, regular tender orders in Brazil and US patient growth. 49.7% of product sales for metreleptin were in the United States, with the remaining 50.3% in the European Union and other international markets.
Future net revenues of metreleptin are highly dependent on our ability to:
| • | maintain existing patients on therapy; |
| • | continue to build market acceptance for metreleptin in the United States; |
| • | build market acceptance in the European Union following the approval by the EMA in July 2018, and continue to obtain pricing and reimbursement approvals in key markets in the European Union and other territories; |
| • | secure regulatory approval from the FDA for the treatment of PL in the United States; and |
| • | obtain regulatory approvals for metreleptin in new markets for the treatment of GL and PL based on the data package which secured approval in Europe. |
In addition, we expect to continue to pay significant Medicaid rebates for metreleptin in the United States. An increase in the relative mix of patients that have Medicaid as their primary insurance coverage would have an adverse impact on our ability to increase revenues. We have successfully achieved coverage in the United States and reimbursement in Japan, Turkey, Germany, Italy, the United Kingdom, France and Austria. Price and reimbursement processes are ongoing in Spain, Denmark, Norway, Portugal, the Netherlands, Belgium, Poland, Slovenia, Romania and Latvia.
Lomitapide
We generated revenues from product sales of lomitapide of $70.5 million and Recordati royalties of $3.4 million for the year ended December 31, 2021, compared to $71.8 million and $3.0 million for the year ended December 31, 2020, respectively. The decrease is primarily due to the impact of competition in the US offsetting underlying continued growth in Europe and other territories. 44.5% of product sales for lomitapide were in the United States, with the remaining 55.5% in the European Union and other international markets.
Future net revenues of lomitapide are highly dependent on our ability to:
| • | maintain existing patients on therapy even in the face of competing pressure following the recent launch of Evinacumab in the US; |
| • | continue to build market acceptance for lomitapide in existing markets; |
| • | continue to support patient access programs in all territories; |
| • | obtain pricing and reimbursement approvals in new markets in the European Union and other territories; and |
| • | secure regulatory approval for lomitapide in key markets. |
In addition, we expect to continue to pay significant Medicaid rebates for lomitapide in the United States. An increase in the relative mix of patients that have Medicaid as their primary insurance coverage would have an adverse impact on our ability to increase revenues. We have successfully achieved coverage in the United States and reimbursement in Japan, Turkey, Saudi Arabia, , Italy, the United Kingdom, France, Netherlands, Austria and Slovenia. Individual patient funding has been obtained in Czech Republic and Hungary. Price and reimbursement processes are ongoing in Brazil, Norway, Israel and Kuwait.
Mycapssa®
We generated revenues from product sales of Mycapssa® of $6.4 million for the period from the date of the acquisition of Chiasma on August 5, 2021, to December 31, 2021.
Future net revenues of Mycapssa® are highly dependent on our ability to:
| • | maintain existing patients on therapy even in the face of competing pressure from other products; |
| • | continue to build market acceptance by the medical community and patients of Mycapssa® as a safe and effective product; |
| • | continue to support patient access programs; |
| • | secure regulatory approval for octreotide in key markets. |
Other
Other revenues relate to sales from our in-house derma-cosmetic range of products, Imlan, and our early access program for Oleogel-S10. Imlan is marketed solely in Germany as a treatment for sensitive, allergy-prone skin. The increase in revenues in the year ended December 31, 2021, was mainly due to higher sales from our early access program product, Oleogel-S10. We intend to market Oleogel-S10 under the brand name of Filsuvez if it is approved for the treatment of EB.
Cost of Sales
| | | Year ended December 31, | | | | | | | |
| Cost of Sales: | | 2021 | | | 2020 | | | Increase / (Decrease) | |
| | | US$’000 | | | US$’000 | | | US$’000 | | | % | |
| Cost of product sales | | | 22,029 | | | | 21,796 | | | | 233 | | | | 1.1 | % |
| Write-down of inventories | | | 5,688 | | | | 4,058 | | | | 1,630 | | | | 40.2 | % |
| Reversal of write-down of inventories | | | (932 | ) | | | — | | | | (932 | ) | | | (100.0 | %) |
| Amortization of acquired intangibles | | | 48,945 | | | | 42,966 | | | | 5,979 | | | | 13.9 | % |
| Amortization of inventory fair value step-up | | | 4,417 | | | | 27,617 | | | | (23,199 | ) | | | (84.0 | %) |
| Royalty expenses | | | 25,973 | | | | 22,592 | | | | 3,381 | | | | 15.0 | % |
| Total cost of sales | | | 106,119 | | | | 119,029 | | | | (12,910 | ) | | | (10.8 | %) |
Total cost of sales was $106.1 million for the year ended December 31, 2021, representing the cost, including royalties, of selling metreleptin, lomitapide and Mycapssa®, the cost of delivery of goods sold to customers, including the costs associated with the services provided by the distributors to import and deliver the goods, the non-cash intangible amortization, and the non-cash inventory fair value step-up expenses and write-down of inventories to fair value less costs to sell recognized as an expense. Total cost of sales was $119.0 million for the year ended December 31, 2020. The decrease is driven by a reduction in the non-cash inventory fair value step-up expenses which reduced due to the full amortization of the Aegerion related inventory fair value step-up in early 2021 and a lower amortization from the Chiasma related inventory fair value step-up from August 5, 2021. This decrease is offset by additional costs related to the cost, including royalties, of selling metreleptin, lomitapide and Mycapssa®, and non-cash intangible amortization.
The cost of product sales in the year ended December 31, 2021, increased by $0.9 million, and royalty expenses increased by $3.4 million in 2021 compared to the year ended December 31, 2020. The acquisition of Mycapssa® as well as increased costs for lomitapide for markets outside the EMEA and metreleptin for all markets largely drove this increase in costs. Following the acquisition of Chiasma, we are now selling three commercial products with two being sold on a global basis and one commercial product being solely sold in the United States. This results in a higher cost of producing our commercial products, higher royalties on sales, and higher costs of delivery of goods sold to customers, including the costs associated with the services provided by our distributors to import and deliver the goods.
Amortization of acquired intangible assets was $48.9 million in 2021 compared to $43.0 million in 2020. This relates to the amortization charge on the three commercial assets purchased as part of the Aegerion and Chiasma acquisitions. The increase is driven by amortization related to the period from the date of the Chiasma acquisition on August 5, 2021, to December 31, 2021.
The non-cash inventory fair value step-up expense was $4.4 million in 2021, compared to $27.6 million in 2020. This relates to the difference between the estimated fair value and the book value of inventory acquired from as part of the acquisitions of Aegerion and Chiasma which is being amortized over the estimated period that we expect to sell this inventory. The decrease in the non-cash inventory fair value step-up expense is due to the inventory step-up recognized as part of the Aegerion acquisition being fully amortized at the beginning of 2021 and the inventory fair value step-up from the Chiasma acquisition, which was lower than that from the Aegerion acquisition, being amortized from August 5, 2021.
Research and Development Expenses
Research and development expenses consist primarily of costs related to clinical studies and outside services, post-approval commitment studies, personnel expenses and other research and development costs. Study costs and outside services costs relate primarily to services performed by clinical research organizations, materials and supplies, and other third-party fees. Research and development expenses for the year ended December 31, 2021, were $37.7 million, representing 24.3% of our total operating expenses, compared to $27.6 million, or 25.1% of total operating expenses, for the year ended December 31, 2020. Research and development expenses in both years were primarily driven by the clinical advancement of Oleogel-S10 as we continued our global EASE study. Research expenses in 2021 comprised $15.9 million in employee compensation, $14.9 million of amounts paid to clinical research organizations, and $6.9 million of other outsourced services. Research expenses in 2020 comprised $11.7 million in employee compensation, $11.3 million of amounts paid to clinical research organizations, and $4.6 million of other outsourced services.
Selling, General and Administrative Expenses
Selling, general and administrative expenses were $92.0 million for the year ended December 31, 2021, representing 59.3% of our total operating expenses, compared to $76.7 million for the year ended December 31, 2020, representing 69.7% of our total operating expenses. The increase in selling, general and administrative expenses was primarily due to an increase in compensation-related expenses, primarily driven by higher headcount following the acquisition of Chiasma, and an increase in other expenses related to the expansion and support of our business.
Restructuring and Acquisition Costs
Restructuring and acquisition costs for the year ended December 31, 2021, were $16.9 million compared to $1.0 million for the year ended December 31, 2020. These costs primarily relate to professional fees associated with the acquisition of Chiasma, which was predominantly completed during 2021. The expenses also include severance costs following the completion of the Chiasma acquisition.
Share-Based Payment Expenses
Non-cash share-based payment expenses for the year ended December 31, 2021, were $8.3 million, compared to $4.7 million in the year ended December 31, 2020. We issue share options and restricted share units as an incentive to senior management and employees. The fair value is measured at the grant date using the Black-Scholes model and amortized over the period during which the awards vest.
Impairment charge
There was no impairment charge recorded for the years ended December 31, 2021 and December 31, 2020.
Non-Cash Change in Fair Value of Contingent Consideration
We compute the fair value of the contingent consideration arising from the acquisition of Birken AG (now Amryt GmbH).
The Amryt GmbH consideration relates to milestone payments of up to $35 million and royalty payments that are payable to the previous owners of Amryt GmbH, which are triggered by regulatory approvals of Oleogel-S10 for the treatment of EB from the FDA or the EMA, as well as future sales-driven milestones.
The finance gain for the year ended December 31, 2021, was $18.4 million compared to a charge of $27.8 million for the year ended December 31, 2020. The gain in 2021 is driven by a change in the probabilities and a decrease in discount rates used in calculating the fair value of the contingent consideration. The probability chance of success, based on management’s expertise and experience for orphan drugs and taking into account the unique circumstances applying to approval process of this product, was revised for the financial year ended December 31, 2021. The probability chance of success was updated following the receipt of a CRL from the FDA which asked Amryt to submit additional confirmatory evidence of effectiveness for Oleogel-S10 in EB, and following the positive opinion adopted by the CHMP, recommending the approval of Filsuvez® in the EU for the treatment of partial thickness wounds associated with dystrophic and junctional EB in patients six months and older. Based on this CHMP recommendation a decision by the EC is expected on the Filsuvez® application within 67 days. Additionally, the discount rate used in the calculation of the fair value of the contingent consideration was decreased which was due to the significant risk reduction in the Group over the last 12 months following the growth in commercial revenues, the positive top-line data on the Phase 3 EASE trial of Oleogel-S10, the positive CHMP opinion recommending the approval of Filsuvez in the EU, the addition of a third commercial product in Mycapssa, and the recent refinancing of our term debt facilities with a significant reduction in the interest rate.
Non-Cash Contingent Value Rights Finance Expense
We issued CVRs pursuant to which up to $85 million may become payable to Amryt shareholders and option holders who were shareholders prior to completion of the Aegerion acquisition, if certain regulatory approval and revenue milestones are met in relation to Oleogel-S10.
The $41.5 million non-cash CVR gain for the year ended December 31, 2021, represents the revised estimated expected cash flows and the effective interest rate unwind on amortized cost between the carrying value of the CVRs from the initial recognition date to the reporting date of December 31, 2021. The non-cash CVR loss for the year ended December 31, 2020, was $12.0 million. The gain recognized in the 2021 Consolidated Statement of Comprehensive Income/(Loss) is mainly driven by changes in the expected timing of milestones being met and the related expected amount due as well as a change in the probability chance of success. Milestone payments related to the regulatory approval from the FDA and EMA have been updated to reflect the expecting timing of achieving regulatory approval and, in turn, the related amount due, which is based on a sliding scale on a linear basis from December 31, 2021 to zero if approved before July 1, 2022. The probability chance of success was updated based on management’s expertise and experience for orphan drugs and taking into account the unique circumstances applying to approval process of this product, following the receipt of a CRL from the FDA, which asked Amryt to submit additional confirmatory evidence of effectiveness for Oleogel-S10 in EB, and following the positive opinion adopted by the CHMP, recommending the approval of Filsuvez® in the EU for the treatment of partial thickness wounds associated with dystrophic and junctional EB in patients six months and older. Based on this CHMP recommendation a decision by the EC is expected on the Filsuvez® application within 67 days.
Net Finance Expense - Other
Other net finance expense was $27.9 million for the year ended December 31, 2021, compared to $19.6 million for the year ended December 31, 2020. Other net finance expense mainly relates to interest on loans and foreign exchange losses, which amounted to $23.2 million and $4.1 million, respectively, for the year ended December 31, 2021. Interest on loans was $22.0 million for the year ended December 31, 2020. The increase in 2021 is mainly due to the compounding of interest on the Secured Credit Facility, where interest at 6.5% is added to the principal loan balance outstanding at each quarter. In 2020 the foreign exchange gain amounted to $2.7 million and in both years the foreign exchange gain/(loss) primarily relates to the translation of euro and sterling-denominated net monetary amounts held by subsidiaries with a non-U.S. dollar functional currency.
Financial results of Amryt for the Year Ended December 31, 2020, compared to the Year Ended December 31, 2019
For information relating to the financial results of Amryt for the year ended December 31, 2020, compared to the year ended December 31, 2019, see “Item 5. Operating and Financial Review and Prospects” in our annual report on Form 20-F for the fiscal year ended December 31, 2020, filed with the SEC on April 30, 2021.
B. Liquidity and Capital Resources
We had unrestricted cash and cash equivalents of $113.0 million and $118.6 million as at December 31, 2021, and December 31, 2020, respectively. We have financed our operations primarily through sales of our commercial products, sales of our ordinary shares and debt financing. We expect to incur significant expenses for the foreseeable future as we continue commercializing our approved products and advancing the clinical development of our product candidates. We expect that our R&D and SG&A costs will increase in connection with conducting clinical trials for our product candidates and any new product candidates we acquire or develop and due to the costs of seeking marketing approval for our product candidates in Europe, the United States and other jurisdictions.
Financing
The principal debt obligations related to our $81 million Secured Credit Facility and our Convertible Notes with an aggregate principal amount of $125 million and the interest associated with these facilities. The Secured Credit Facility had a five-year term from date of draw down and matures in 2024. Interest was payable at our option at the rate of 11% per annum paid in cash on a quarterly basis or at a rate of 6.5% paid in cash plus 6.5% paid in kind that would be paid when the principal was repaid, which rolls up and is included in the principal balance outstanding, on a quarterly basis. For the purposes of the contractual obligations table above, we assume that we choose to pay interest at a rate of 6.5% paid in cash plus 6.5% paid in kind that will be paid when the principal is repaid. For more information on this indebtedness, see “Item 7. Major Shareholders and Related Party Transactions—B. Related Party Transactions.” On February 18, 2022, the Secured Credit Facility was repaid in full and the Group secured a $125 million Senior Credit Facility from funds managed by the Ares of which US$105 million was drawn down to facilitate the prepayment of the existing Secured Credit Facility. In repaying the Secured Credit Facility, Amryt incurred an exit fee of 5.00% of the outstanding principal amount as at the prepayment date.
The Convertible Notes bear interest at a rate of 5.0% per year, payable semi-annually in arrears on April 1 and October 1 of each year, beginning on April 1, 2020. The Convertible Notes will mature on April 1, 2025, unless earlier repurchased or converted. For further detail on our principal debt, see “Note 19: Long term loan” and “Note 20: Convertible notes” in our Consolidated Financial Statements.
Contingent consideration and contingent value rights
Contingent consideration and contingent value rights arose as part of (i) the acquisition of Amryt GmbH in 2016, through which we acquired Oleogel-S10, and (ii) the issuance of CVRs to Amryt shareholders and option holders prior to the acquisition of Aegerion. The contingent consideration and contingent value rights arising on these transactions are payable on achieving various milestones and sales royalties. For further detail, see “Note 6: Business combinations and asset acquisitions” in our Consolidated Financial Statements.
Other short-term obligations
The Group has a liability for revenue rebates due on Myalepta sales in a country in the EMEA region from agreeing a reimbursement price with the government authorities resulting in a one-off payment related to sales up to the date of approval, which occurred in March 2021. For further detail, see “Note 21: Trade and other payables” in our Consolidated Financial Statements.
Cash Flows
The table below provides selected cash flow information for the periods indicated (in thousands):
| | | Year ended December 31, | |
| | | 2021 | | | 2020 | |
| | | US$’000 | |
| Net cash flow from operating activities | | | 15,540 | | | | 26,891 | |
| Net cash flow from / (used in) investing activities | | | 106,402 | | | | (2,379 | ) |
| Net cash flow (used in) / from financing activities | | | (125,426 | ) | | | 26,028 | |
| Exchange and other movements | | | (2,282 | ) | | | 1,029 | |
| Net change in cash and cash equivalents | | | (5,766 | ) | | | 51,569 | |
Net Cash Flow From / (Used in) Operating Activities
Net cash from operating activities was $15.5 million for the year ended December 31, 2021, compared to net cash from operating activities of $26.9 million for the year ended December 31, 2020. The decrease of $11.4 million was primarily driven by the increased restructuring and acquisition costs related to the acquisition of Chiasma along with the increased scale of our business and working capital fluctuations.
Net Cash Flow From / (Used in) Investing Activities
Net cash from investing activities was $106.4 million for the year ended December 31, 2021, and primarily related to the Chiasma cash balance of $107.9 million, which we received in acquiring Chiasma.
Net cash used in investing activities was $2.4 million for the year ended December 31, 2020, and primarily related to payments for property, plant and equipment and payments for intangible assets.
Net Cash Flow (Used in) / From Financing Activities
Net cash flow used in financing activities was $125.4 million for the year ended December 31, 2021. In conjunction with the acquisition of Chiasma, we repaid US$116.6 million of debt that was outstanding on August 5, 2021. The remaining cash outflows mainly consisted of interest paid on our Secured Credit Facility of $5.8 million and on the Convertible Notes of $6.3 million.
Net cash flow from financing activities was $26.0 million for the year ended December 31, 2020. On December 8, 2020, we entered into a securities purchase agreement with several institutional accredited investors for the private placement of 3,200,000 ADSs, at a purchase price of $12.50 per ADS, yielding gross proceeds of $40 million and net proceeds of $37.9 million. The private placement included new and existing investors including Stonepine Capital, LP, Aquilo Capital Management, LLC, Amati Global Investors, Athyrium Capital Management, LP and Highbridge Capital Management, among others. These cash inflows were partially offset by interest paid on our Secured Credit Facility of $4.1 million and on the Convertible Notes of $6.4 million.
C. Research and Development, Patents and Licenses, etc.
Amryt has established a strong, positive reputation as a highly-transactional biopharmaceutical company, forging creative deals that benefit all parties. We continue to seek to identify additional products to build-out our portfolio through either in-licensing or acquiring innovative assets which improve the lives of people living with rare and orphan diseases. We are currently commercializing our approved products and progressing our product candidates through preclinical and clinical development. For further information on research and development see “Item 4. Information on the Company—Item 4.B Business overview.” For a description of the Company’s research and development expenses see “Item 5. Operating and Financial Review and Prospects—A. Operating Results—Financial Overview—Research and Development Expenses.”
D. Trend Information
Other than as disclosed in this annual report, we are not aware of any trends, uncertainties, demands, commitments or events for the current financial year that are reasonably likely to have a material effect on our net sales or revenues, income from continuing operations, profitability, liquidity or capital resources, or that would cause reported financial information not necessarily to be indicative of future operating results or financial conditions.
E. Critical Accounting Judgments and Estimates
Our financial statements have been prepared in accordance with IFRS as issued by the IASB. In the application of accounting policies, certain judgments, estimates and assumptions about the value of assets and liabilities for which there is no definitive third-party reference were required. The estimates and associated assumptions are based on historical experience and other factors that were considered to be relevant. Actual results may differ from these estimates. These estimates and assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimate is revised if the revision affects only that period or in the period of the revisions and future periods if the revision affects both current and future periods. For a discussion of the estimates and assumptions used by us in the preparation of our financial statements, see “Note 2: Accounting policies” in our Consolidated Financial Statements for the year ended December 31, 2021.
| Item 6. | Directors, Senior Management and Employees |
A. Directors and Senior Management
The following table presents information about our executive officers and directors:
| Name | Age | Position |
| Executive Officers | | |
| | | |
| Dr. Joseph A. Wiley | 51 | Chief Executive Officer |
| | | |
| Rory P. Nealon | 54 | Chief Financial Officer and Chief Operating Officer |
| | | |
| Non-Executive Directors | | |
| | | |
| Raymond T. Stafford | 75 | Chairman of the Board |
| | | |
| George P. Hampton, Jr. | 52 | Non-executive Director |
| | | |
| Raj Kannan | 58 | Non-executive Director |
| | | |
| Dr. Roni Mamluk | 55 | Non-executive Director |
| | | |
| Dr. Alain H. Munoz | 73 | Non-executive Director |
| | | |
| Donald K. Stern | 76 | Non-executive Director |
| | | |
| Dr. Patrick V.J.J. Vink | 58 | Non-executive Director |
| | | |
| Stephen T. Wills | 65 | Non-executive Director |
The business address for each of our executive officers and directors is c/o Amryt Pharma plc, 45 Mespil Road, Dublin 4, Ireland.
The following are biographies of our executive officers and directors:
Dr. Joseph A. Wiley, Dr. Wiley founded Amryt and has served as Chief Executive Officer since 2015. He has over 20 years of experience in the pharmaceutical, medical and venture capital industries. Prior to Amryt, Dr. Wiley opened and led the European office of Sofinnova Ventures Inc. He was previously a medical director at Astellas Pharma Limited. Prior to joining Astellas, he held investment roles at Spirit Capital SA, Inventages Venture Capital Investment Inc. and Aberdeen Asset Managers Private Equity Limited. Dr. Wiley trained in general medicine at Trinity College Dublin, specializing in neurology. He holds a Masters of Business Administration from INSEAD and is also a Member of the Royal College of Physicians in Ireland.
Rory P. Nealon, Mr. Nealon founded Amryt and has served as Chief Financial Officer and Chief Operating Officer since 2015. He was previously a board member of Trinity Biotech Plc where he joined as Chief Financial Officer in January 2003 and subsequently was appointed as Chief Operations Officer in November 2007, a position he held until leaving Trinity Biotech Plc in 2014. Prior to joining Trinity Biotech Plc, Mr. Nealon served as Chief Financial Officer of Conduit Plc, an Irish directory services provider with operations in Ireland, the UK, Austria and Switzerland. Prior to joining Conduit Plc, Mr. Nealon was an associate director for AIB Capital Markets, a subsidiary of AIB Group plc, the Irish banking group. Mr. Nealon holds a Bachelor of Commerce degree from University College Dublin, is a Fellow of the Institute of Chartered Accountants in Ireland, a member of the Institute of Taxation in Ireland, and a member of the Institute of Corporate Treasurers in the UK.
Raymond T. Stafford, Mr. Stafford has been a director of Amryt since 2016. He has worked in the pharmaceutical industry for more than 30 years. He has served as Chairman, Chief Executive Officer and majority shareholder of the Tosara Group which owned, manufactured and marketed the successful international brand Sudocrem, and was ultimately integrated into the U.S.-based, NYSE-listed company Forest Laboratories, Inc. in 1988. Mr. Stafford held numerous senior positions within such corporations, including Chief Executive Officer of Forest UK and Ireland as well as Chief Executive Officer of Forest Laboratories Europe since 1999. Mr. Stafford retired in 2014 following the sale of Forest Laboratories, Inc. to Actavis Plc (now Allergan plc) in a $28 billion transaction where Mr. Stafford was Executive Vice President of Global Marketing. Separately, Mr. Stafford also founded one of Ireland’s current leading multi-channel sales, marketing and distribution service providers approved by the Irish Medicines Board (now, The Health Products Regulatory Authority) to service the wholesale and retail trade.
George P. Hampton, Jr., Mr. Hampton has been a director of Amryt since 2019. He joined Currax Pharmaceuticals in April of 2019 as Chief Executive Officer and serves on its board of directors. Prior to joining Currax, Mr. Hampton served as executive vice president, primary care business unit for Horizon Pharmaceuticals (HZNP), a public biopharmaceutical company. In this role he was tasked with leading the company’s forward-looking strategy, as well as establishing operational goals for the business. Previously, Mr. Hampton served as executive vice president, global orphan business unit and international operations for Horizon Pharmaceuticals. He has more than 25 years of experience as a successful executive in the pharmaceutical and biotechnology field on both a national and international scale including specific expertise in rare disease (ACTIMMUNE, RAVICTI, PROCYSBI), autoimmune (HUMIRA), primary care, orthopedic (CELEBREX), diabetes (BYETTA), anti-infectives and cardiovascular spaces. This includes roles of increasing responsibility in sales, marketing and operations at G.D. Searle, Abbott (now AbbVie), Amylin and Horizon Pharmaceuticals. Mr. Hampton earned his Bachelor of Science from Miami University in Oxford, Ohio. He previously served on the board of IMAC (Nasdaq: IMAC) regeneration medical centers.
Raj Kannan, Mr. Kannan was appointed Chief Executive Officer of Chiasma, Inc. in June 2019. On August 6, 2021, Mr. Kannan resigned as Chief Executive Officer of Chiasma and joined the board of Amryt as a non-executive director. Mr. Kannan is the CEO of Aerie Pharmaceuticals. He has over 25 years of experience leading and developing companies. He has effectively led and grown organizations and supported multiple successful launches across therapeutic areas in the U.S. and globally. Prior to joining Aerie, Mr. Kannan was Chief Executive Officer and President of Chiasma, Inc., where he led the organization through the approval and the launch of the first oral therapy in over a decade for patients with acromegaly and subsequently through the acquisition by Amryt Pharma Plc. Before that, Mr. Kannan was Chief Commercial Officer at Kiniksa Pharmaceuticals, Ltd. (“Kiniksa”), where he built the commercial operations, including sales, marketing, and business analytics functions. Prior to Kiniksa, he served as the Global Head of the Neurology and Immunology business franchise at Merck KGaA, where he was responsible for transforming the largest franchise into a growth franchise with $2 billion in annual revenues through significant strategic shifts in investment to support new product introductions and through recalibration of pipeline investments. Before that, Mr. Kannan spent 10 years at Boehringer Ingelheim International GmbH in the U.S., Canada, and in Germany, including as Global Marketing Head of the Cardiovascular Franchise, where he was responsible for more than $3.5 billion in annual revenues.
Dr. Roni Mamluk, Dr. Mamluk, Ph.D. joined the Board of Directors of Chiasma, Inc. in June 2017. On August 6, 2021, Dr. Mamluk resigned as a non-executive director of Chiasma and joined the board of Amryt as a non-executive director. Dr. Mamluk currently serves as President and Chief Executive Officer of Ayala Pharmaceuticals, Inc., a clinical-stage biopharmaceutical company dedicated to developing targeted cancer therapies for people living with genetically defined cancers, and serves on its board of directors. She joined Chiasma in 2006 and led the creation of its TPE technology and subsequently Mycapssa® development. Dr. Mamluk fulfilled multiple roles at Chiasma including Chief Development Officer from March 2015 to March 2017, Chief Executive Officer from April 2013 to March 2015 and held various roles in the Company from 2006 to April 2013, including Chief Operating Officer and Vice President, Research and Development. Prior to joining Chiasma, Dr. Mamluk led nonclinical research and development at Adnexus Therapeutics, Inc. Dr. Mamluk received her B.A. and Ph.D. from the Hebrew University. She completed her post-doctoral fellowship at Children’s Hospital/Harvard Medical School in the field of angiogenesis.
Dr. Alain H. Munoz, Dr. Munoz has been a director of Amryt since 2019. He is an entrepreneur and independent management consultant in the pharmaceutical and biotechnology industry and has over 30 years of experience in the industry at the executive level. Dr. Munoz worked with the Fournier Group as Research and Development director and thereafter as Senior Vice President of the Pharmaceutical Division. Prior to serving at Fournier, he served at Sanofi Group, first as director in the cardiovascular and anti-thrombotic products department, and thereafter as Vice President of international development. Dr. Munoz is qualified in cardiology and anesthesiology from the University Hospital of Montpellier, France where he was head of the clinical cardiology department. He has been a member of the Scientific Committee of the French drug agency and Chairman of the Board of Hybrigenics SA and Novagali Pharma acquired by Santen Pharmaceuticals. He presently is an independent board member of Auris Medical Holding AG (Nasdaq: EARS), Zealand Pharma A/S (Nasdaq: ZEAL) and Chairman of Acticor-biotech (Euronext: ALACT.PA). Mr. Munoz received an undergraduate degree from International Institute for Management Development, a doctorate from the University of Montpellier and a graduate degree from Centre Hospitalier Universitaire Pitie-Salpetriere.Donald K. Stern, Mr. Stern has been a director of Amryt since 2019. He was previously a director of Novelion, Aegerion’s former parent company, and was a member of Aegerion’s board of directors from September 2015 to October 2016. Mr. Stern serves as Managing Director of Corporate Monitoring & Consulting Services at Affiliated Monitors, Inc., a consulting firm providing independent integrity monitoring services and compliance services across a wide range of regulated industries and professions. He is also Of Counsel to the Boston law firm of Yurko Partners, P.C.. He has had a diverse and distinguished legal career, split between private practice and public service. Prior to joining Affiliated Monitors, Inc., Mr. Stern was a partner at three major law firms: Cooley LLP, Bingham McCutchen LLP and Hale & Dorr LLP (now Wilmer Cutler Pickering Hale and Dorr LLP). Mr. Stern also served as the U.S. Attorney for the District of Massachusetts, the Chief Legal Counsel to Governor Michael S. Dukakis and the Chief of the Government Bureau in the Massachusetts Attorney General’s office. Mr. Stern holds a Masters in Laws from University of Pennsylvania Law School, a Juris Doctor degree from Georgetown University Law Center and a Bachelor of Arts from Hobart College.
Dr. Patrick V.J.J. Vink, Dr. Vink has been a director of Amryt since 2019. He has significant experience as a senior executive, having worked in the pharmaceutical industry for more than 30 years. Dr. Vink serves as Chairman at BiognoSys AG, a privately held proteomics company in Switzerland. Dr. Vink also serves as Chairman of venture capital-backed NMD Pharma, a neurology biopharmaceutical company in Denmark and F2G Ltd, a rare fungal disease UK and Austria based company. In addition, Dr. Vink is a board member at Santhera AG and Spero Therapeutics, Inc. and in 2019 began working with Athyrium as a Senior Advisor. While serving in these capacities, Dr. Vink has been involved in initial public listings and geographic expansions and has contributed to the achievement of significant development and commercial milestones. Earlier in his career he held several leadership positions across the industry, including Head of Global Biopharmaceuticals for the Sandoz division of the Novartis Group, Vice President International Business for Biogen Inc., and Head of Worldwide Marketing, Cardiovascular and Thrombosis at Sanofi-Synthelabo Ltd. Dr. Vink also served as a member of the Executive Committee of the European Federation of Pharmaceutical Industries and Associations from 2013 to 2015. Dr. Vink graduated as a medical doctor from the University of Leiden, Netherlands in 1988 and obtained his Masters of Business Administration in 1992 at the University of Rochester.
Stephen T. Wills, Mr. Wills became a director of Amryt in 2019. He currently serves as the Chief Financial Officer (since 1997), and Chief Operating Officer (since 2011) of Palatin Technologies, Inc. (NYSE: PTN), a biopharmaceutical company developing targeted, receptor-specific peptide therapeutics for the treatment of diseases with significant unmet medical need and commercial potential. Mr. Wills serves as Chief Financial Officer of Cactus Acquisition Corp (Nasdaq: CCTS), a Special Purpose Acquisition Company (SPAC). Mr. Wills serves on the boards of directors of MediWound Ltd. (Nasdaq: MDWD), a biopharmaceutical company focused on treatment in the fields of severe burns, chronic and other hard to heal wounds, since April 2017, and as Chairman since January 2018, and of Gamida Cell Ltd. (Nasdaq: GMDA), a leading cellular and immune therapeutics company, since March 2019 (audit chair and compensation and finance committee member). Mr. Wills also has served on the board of trustees and executive committee of The Hun School of Princeton, a college preparatory day and boarding school, since 2013, and its Chairman since June 2018. Mr. Wills served on the board of directors of Caliper Corporation, a psychological assessment and talent development company, since March 2016, and as Chairman from December 2016 to December 2019, when Caliper was acquired by PSI. Mr. Wills served as Executive Chairman and Interim Principal Executive Officer of Derma Sciences, Inc., a provider of advanced wound care products, from December 2015 to February 2017, when Derma Sciences was acquired by Integra Lifesciences (Nasdaq: IART). Previously, Mr. Wills served on the board of directors of Derma Sciences as the lead director and chairman of the audit committee from June 2000 to December 2015. Mr. Wills served as the Chief Financial Officer of Derma Sciences from 1997 to 2000. Mr. Wills served as the President and Chief Operating Officer of Wills, Owens & Baker, P.C., a public accounting firm, from 1991 to 2000. Mr. Wills, a certified public accountant, earned his Bachelor of Science in accounting from West Chester University, and a Master of Science in taxation from Temple University.
Family Relationships
There are no family relationships among any of the directors or senior management.
B. Compensation
2021 Director Remuneration
For the fiscal year ended December 31, 2021, the aggregate compensation accrued or paid to the members of our Board and our executive officers for services in all capacities was $3.1 million. The following table sets forth the approximate remuneration paid during the year ended December 31, 2021, to our directors.
| | | Base Salary and Fees | | | Bonus | | | Pension Contributions | | | Other Benefits | | | 2021 Total | |
| | | US$’000 | |
| Raymond T. Stafford | | | 88 | | | | — | | | | — | | | | — | | | | 88 | |
| Dr. Joseph A. Wiley | | | 747 | | | | 728 | | | | 71 | | | | 174 | | | | 1,720 | |
| George P. Hampton, Jr. | | | 65 | | | | — | | | | — | | | | — | | | | 65 | |
| Raj Kannan | | | 20 | | | | — | | | | — | | | | — | | | | 20 | |
| Dr. Roni Mamluk | | | 20 | | | | — | | | | — | | | | — | | | | 20 | |
| Dr. Alain H. Munoz | | | 58 | | | | — | | | | — | | | | — | | | | 58 | |
| Donald K. Stern | | | 80 | | | | — | | | | — | | | | — | | | | 80 | |
| Dr. Patrick V.J.J. Vink | | | 60 | | | | — | | | | — | | | | — | | | | 60 | |
| Stephen T. Wills | | | 88 | | | | — | | | | — | | | | — | | | | 88 | |
| Rory P. Nealon | | | 505 | | | | 378 | | | | 49 | | | | 7 | | | | 939 | |
| Total | | | 1,731 | | | | 1,106 | | | | 120 | | | | 181 | | | | 3,138 | |
Directors’ Service Agreements and Letters of Appointment
Raymond T. Stafford
We entered into a letter of appointment with Raymond Stafford on August 27, 2019, under which he was appointed a non-executive director with effect from September 24, 2019. Mr. Stafford was also appointed as a member of the Audit Committee from that date. The appointment is subject to our Articles of Association, and pursuant to terms of the letter of appointment may be terminated at any time upon three months’ written notice served by either party. Mr. Stafford is entitled to an annual fee of $77,500 for his role as non-executive chairman, a fee of $10,000 for his role as member of the Audit Committee, and reimbursement of reasonable expenses. Mr. Stafford is also subject to a six-month non-compete restrictive covenant.
George P. Hampton, Jr.
We entered into a letter of appointment with George Hampton on August 27, 2019, under which he was appointed a non-executive director with effect from September 24, 2019. Mr. Hampton was also appointed as a member and chair of the Remuneration Committee from that date. The appointment is subject to our Articles of Association, and pursuant to terms of the letter of appointment may be terminated at any time upon three months’ written notice served by either party. Mr. Hampton is entitled to an annual fee of $50,000 for his role as non-executive director, a fee of $15,000 for his role as chair and member of the Remuneration Committee, and reimbursement of reasonable expenses. Mr. Hampton is also subject to a six-month non-compete restrictive covenant.
Raj Kannan
We entered into a letter of appointment with Raj Kannan on August 6, 2021, under which he was appointed a non-executive director with effect from August 6, 2021. The appointment is subject to our Articles of Association, and pursuant to terms of the letter of appointment may be terminated at any time upon three months’ written notice served by either party. Mr. Kannan is entitled to an annual fee of $50,000 for his role as non-executive director and reimbursement of reasonable expenses. Mr. Kannan is also subject to a six-month non-compete restrictive covenant.
Dr. Roni Mamluk
We entered into a letter of appointment with Dr. Mamluk on August 6, 2021, under which she was appointed a non-executive director with effect from August 6, 2021. The appointment is subject to our Articles of Association, and pursuant to terms of the letter of appointment may be terminated at any time upon three months’ written notice served by either party. Dr. Mamluk is entitled to an annual fee of $50,000 for her role as non-executive director and reimbursement of reasonable expenses. Dr. Mamluk is also subject to a six-month non-compete restrictive covenant.
Dr. Alain H. Munoz
We entered into a letter of appointment with Alain Munoz on August 27, 2019, under which he was appointed a non-executive director with effect from September 24, 2019. Dr. Munoz was also appointed as a member of the Remuneration Committee from that date. The appointment is subject to our Articles of Association, and pursuant to terms of the letter of appointment may be terminated at any time upon three months’ written notice served by either party. Dr. Munoz is entitled to an annual fee of $50,000 for his role as non-executive director, a fee of $7,500 for his role as member of the Remuneration Committee and reimbursement of reasonable expenses. Dr. Munoz is also subject to a six-month non-compete restrictive covenant.
Donald K. Stern
We entered into a letter of appointment with Donald Stern on August 27, 2019, under which he was appointed a non-executive director with effect from September 24, 2019. Mr. Stern was also appointed as a member of the Audit Committee and a member and chair of the Compliance Committee from that date. The appointment is subject to our Articles of Association, and pursuant to terms of the letter of appointment may be terminated at any time upon three months’ written notice served by either party. Mr. Stern is entitled to an annual fee of $50,000 for his role as non-executive director, a fee of $10,000 for his role as member of the Audit Committee, a fee of $20,000 for his role as the chair of the Compliance Committee and reimbursement of reasonable expenses. Mr. Stern is also subject to a six-month non-compete restrictive covenant.
Dr. Patrick V.J.J. Vink
We entered into a letter of appointment with Patrick Vink on August 27, 2019, under which he was appointed a non-executive director with effect from September 24, 2019. Dr. Vink was also appointed as a member the Compliance Committee from that date. The appointment is subject to our Articles of Association, and pursuant to terms of the letter of appointment may be terminated at any time upon three months’ written notice served by either party. Dr. Vink is entitled to an annual fee of $50,000 for his role as non-executive director, a fee of $10,000 for his role as member of the Compliance Committee and reimbursement of reasonable expenses. Dr. Vink is also subject to a six-month non-compete restrictive covenant.
Stephen T. Wills
We entered into a letter of appointment with Stephen Wills on August 27, 2019, under which he was appointed a non-executive director with effect from September 24, 2019. Mr. Wills was also appointed as a member and chair of the Audit Committee, a member of the Remuneration Committee and a member of the Compliance Committee from that date. The appointment is subject to our Articles of Association, and pursuant to terms of the letter of appointment may be terminated at any time upon three months’ written notice served by either party. Mr. Wills is entitled to an annual fee of $50,000 for his role as non-executive director, a fee of $20,000 for his role as chair of the Audit Committee, a fee of $7,500 for his role as a member of the Remuneration Committee, a fee of $10,000 for his role as member of the Compliance Committee and reimbursement of reasonable expenses. Mr. Wills is also subject to a six-month non-compete restrictive covenant.
A registration rights agreement to which we are a party contains provisions regarding the terms of appointment of our Board. See “Item 7. Major Shareholders and Related Party Transactions—B. Related Party Transactions—Agreements with Principal Shareholders—Registration Rights Agreement” for further information.
C. Board Practices
Composition of Board of Directors
Our Board consists of nine members. The Board has determined that eight of its nine directors, Raymond Stafford, Stephen Wills, Donald Stern, George Hampton, Patrick Vink, Alain Munoz, Raj Kannan and Dr. Roni Mamluk do not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of director and that each of these directors is “independent” as that term is defined under the rules of the Nasdaq. As a foreign private issuer, we are not required to meet the Nasdaq rule that our Board be comprised of a majority of independent directors. However, we voluntarily comply and intend to continue to comply with this requirement.
As required under the Nasdaq listing standards, a majority of the members of a listed company’s board of directors must qualify as “independent,” as affirmatively determined by the board of directors. Our board of directors consults with counsel to ensure that the board’s determinations are consistent with relevant securities and other laws and regulations regarding the definition of “independent,” including those set forth in the applicable Nasdaq listing standards, as in effect from time to time. Consistent with these considerations, after review of all relevant transactions or relationships between each director, or any of his or her family members, and our company, our senior management and our independent registered public accounting firm, the board of directors affirmatively determined that all of our current directors are independent directors within the meaning of the applicable Nasdaq listing standards, except that Dr. Joe Wiley, our Chief Executive Officer, is not independent by virtue of his employment with our company. In addition, our board of directors has determined that each member of the audit committee, compensation committee and nominating and corporate governance committee meets the applicable Nasdaq and SEC rules and regulations regarding “independence” and that each member is free of any relationship that would impair his or her individual exercise of independent judgment with regard to the company.
In accordance with our Articles of Association and with the Companies Act, the directors shall not be fewer than two and, unless otherwise determined by special resolution of our shareholders, shall not be more than nine in number. Each of the directors is subject to compulsory retirement and at each annual general meeting which occurs after September 25, 2021, one-third of the current directors (or, if their number is not a multiple of three, the number nearest to but not greater than one-third, subject to a minimum of one) shall retire from office by rotation. A director retiring by rotation shall be eligible for re-election.
A registration rights agreement to which we are a party contains provisions regarding the composition of our Board. See “Item 7. Major Shareholders and Related Party Transactions—B. Related Party Transactions—Agreements with Principal Shareholders—Registration Rights Agreement” for further information.
Role of the Board in Risk Oversight
Our Board is primarily responsible for the oversight of our risk management activities and has delegated to the audit committee the responsibility to assist our Board in this task. While our Board oversees our risk management, our management is responsible for day-to-day risk management processes. We believe this division of responsibilities is the most effective approach for addressing the risks we face. Our Board expects our management to consider risk and risk management in each business decision, to proactively develop and monitor risk management strategies and processes for day-to-day activities and to effectively implement risk management strategies adopted by the Board.
Diversity
On August 6, 2021, the SEC approved the Nasdaq Stock Market’s proposal to amend its listing standards to encourage greater board diversity and to require board diversity disclosures for Nasdaq-listed companies. Pursuant to the amended listing standards, a Board diversity matrix is required to be included in the Annual Report on Form 20-F, containing certain demographic and other information regarding members of the Board. The Board diversity matrix is set out below.
| Board Diversity Matrix for (as of March 31, 2021) |
| Country of Principal Executive Offices | Ireland | | | |
| Foreign Private Issuer | Yes | | | |
| Disclosure Prohibited Under Home Country Law | No | | | |
| Total Number of Directors | 9 | | | |
| | Female | Male | Non-Binary | Did not Disclose Gender |
| Part I: Gender Identity |
| Directors | 1 | 8 | - | - |
| Part II: Demographic Background |
| Underrepresented Individual in Home Country Jurisdiction | - | 1 | - | - |
| LGBTQ+ | - | - | - | - |
| Did Not Disclose Demographic Background | - | - | - | - |
Corporate Governance Practices
As a “foreign private issuer,” as defined by the SEC, although we are permitted to follow certain governance practices of the United Kingdom instead of those otherwise required under the Nasdaq rules for domestic issuers, we intend to follow the Nasdaq corporate governance rules applicable to foreign private issuers, though we may in the future decide to use the foreign private issuer exemption with respect to some or all of the other Nasdaq corporate governance rules. While we voluntarily follow most Nasdaq corporate governance rules, we intend to take advantage of the following limited exemptions:
| • | Exemption from filing quarterly reports on Form 10-Q and current reports on Form 8-K disclosing significant events within four days of their occurrence. |
| • | Exemption from Section 16 rules regarding sales of ordinary shares by insiders, which will provide less data in this regard than to shareholders of U.S. companies that are subject to the Exchange Act. |
| • | Exemption from the Nasdaq rules applicable to domestic issuers requiring disclosure within four business days of any determination to grant a waiver of the code of business conduct and ethics to directors and officers. Although we will require approval from our Board of any such waiver, we may choose not to disclose the waiver in the manner set forth in the Nasdaq rules, as permitted by the foreign private issuer exemption. |
| • | Exemption from the requirements that director nominees are selected, or recommended for selection by our Board, either by (1) independent directors constituting a majority of our Board’s independent directors in a vote in which only independent directors participate, or (2) a committee comprised solely of independent directors, and that a formal written charter or board resolution, as applicable, addressing the nominations process is adopted. |
Furthermore, Nasdaq Rule 5615(a)(3) provides that a foreign private issuer, like our company, may rely on home country corporate governance practices in lieu of certain of the rules in the Nasdaq Rule 5600 Series and Rule 5250(d), provided that we nevertheless comply with the Nasdaq’s Notification of Noncompliance requirement (Rule 5625), the Voting Rights requirement (Rule 5640) and that we have an audit committee that satisfies Rule 5605(c)(3), consisting of committee members who meet the independence requirements of Rule 5605(c)(2)(A)(ii). Although we generally intend to follow Nasdaq corporate governance rules, we do not intend to follow Nasdaq Rule 5620(c) regarding quorum requirements applicable to meetings of shareholders. Such quorum requirements are not required under English law. In accordance with generally accepted business practice, our Articles of Association provide alternative quorum requirements that are generally applicable to meetings of shareholders.
We intend to take all actions necessary to maintain compliance as a foreign private issuer under the applicable corporate governance requirements of the Sarbanes-Oxley Act, the rules adopted by the SEC and the Nasdaq corporate governance rules and listing standards.
Committees of Board of Directors
The Board has three standing committees: an Audit Committee, a Remuneration Committee and a Compliance Committee.
Audit Committee
The Audit Committee has responsibility for, among other things, the monitoring of the financial integrity of our financial statements and the involvement of our auditors in that process. The Committee focuses in particular on compliance with accounting policies and ensuring that an effective system of internal and external audit and financial control is maintained, including oversight of the annual audit and the extent of the non-audit work undertaken by our external auditors, as well as advising on the appointment of external auditors. The Audit Committee comprises three members, who are independent non-executive directors who are financially sophisticated and experienced: Stephen Wills, Raymond Stafford and Donald Stern. The Committee is chaired by Stephen Wills. Our Board has determined that Mr. Wills qualifies as an “audit committee financial expert” as defined by applicable SEC rules and has the requisite financial sophistication as defined under applicable Nasdaq rules and regulations. The Board has determined that all of the members of the Audit Committee satisfy the “independence” requirements set forth in Rule 10A-3 under the Exchange Act. The Audit Committee is governed by terms of reference that comply with the Nasdaq rules for a charter of such body.
Among the responsibilities of the Audit Committee are:
| • | monitoring the integrity of our financial statements, including our annual and half-yearly reports, interim management statements, preliminary results announcements and any other formal announcement relating to our financial performance; |
| • | advising on and recommending the appointment of the independent auditors; |
| • | evaluating our independent auditors’ qualifications, performance and independence, and presenting its conclusions to the full Board on at least an annual basis; |
| • | reviewing and challenging where necessary our accounting policies, methods and applications of accounting standards; |
| • | ensuring that we maintain an effective system of internal and external audit and financial control; |
| • | reviewing and considering the scope of the annual audit and the extent of the non-audit work undertaken by our independent auditors. |
The Audit Committee will meet as often as one or more members of the Audit Committee deem necessary, but in any event will meet at least four times a year at the appropriate times in the financial reporting and audit cycle.
Remuneration Committee
The Remuneration Committee has responsibility for determining senior management compensation. The Remuneration Committee comprises three members, who are independent non-executive directors: George Hampton, Alain Munoz and Stephen Wills. The Committee is chaired by George Hampton. Under the SEC and Nasdaq rules, there are heightened independence standards for members of the Remuneration Committee, including a prohibition against the receipt of any compensation from us other than as a standard board member. Although foreign private issuers are not required to meet this heightened standard, we voluntarily comply and intend to continue to comply with this heightened standard. The Remuneration Committee is governed by terms of reference that comply with the Nasdaq rules for a charter of such body.
The responsibilities of the Remuneration Committee include:
| • | proposing and recommending specific remuneration packages for each of the executive directors, including pension rights and any compensation payments; |
| • | recommending and monitoring the level and structure of remuneration for senior management, and the implementation of share option, restricted share units or other performance-related schemes; |
| • | determining and monitoring policy on and setting levels of remuneration for executive directors and senior management; |
| • | determining policy on termination of senior management; |
| • | determining performance-related pay, pension arrangements, share incentive plans and reporting and disclosure; |
| • | reviewing the Company’s arrangements for its employees to raise concerns, in confidence, about possible wrongdoing in financial reporting or other matters; and |
| • | establishing the selection criteria for, selecting, appointing and setting the terms of reference for, any remuneration consultants who advise the Remuneration Committee. |
The Remuneration Committee will meet as often as one or more members of the Remuneration Committee deem necessary, but in any event will meet at least two times a year at the appropriate times in the financial reporting and audit cycle.
Compliance Committee
The Compliance Committee has responsibility for overseeing our compliance with laws, regulations, internal procedures and industry standards the violation of which may cause us significant business, regulatory or reputational damage, as well as legal and business trends and public policy issues. The Compliance Committee comprises three members, who are independent non-executive directors: Donald Stern, Patrick Vink and Stephen Wills. The Committee is chaired by Donald Stern.
The responsibilities of the Compliance Committee include oversight of the development and implementation of compliance and ethics policies and practices.
The Compliance Committee will meet as often as one or more members of the Compliance Committee deem necessary, but in any event will meet at least two times a year at the appropriate times in the financial reporting and audit cycle.
Code of Ethics
We have adopted a Code of Ethics that is applicable to all of our employees, executive officers and directors. A committee of the Board will be responsible for overseeing the Code of Ethics and will be required to approve any waivers of the Code of Ethics for employees, executive officers and directors. We intend to disclose any amendments to the Code of Ethics or any waivers of its requirements on our website.
Conflicts of Interest
Our Board, in the manner set out in the Companies Act, may authorize any interest of a director that conflicts, or may conflict, with our interests as a company, provided that the conflict is not a direct transaction or arrangement between the director and us as a company. Subject to specified exceptions in our Articles of Association, a director shall not vote nor be counted in the quorum on any resolution of the Board in which that director is materially interested.
D. Employees
As of December 31, 2021, we had 289 employees, excluding temporary workers, 29% of whom are based in Dublin, Ireland, 32% in the United States, and 39% elsewhere globally. None of our employees has engaged in any labor strikes. We have no collective bargaining agreements with our employees, but we maintain company agreements (Betriebsvereinbarungen) with respect to certain topics at our Niefern site in Germany. Our German entity, Amryt GmbH, has a works counsel that works with the Amryt GmbH management team.
E. Share Ownership
Share Ownership
The total number of ordinary shares of the Company beneficially owned by our directors and executive officers including in the form of ADSs, as of the date of this annual report, was 18,316,612 (3,663,322 ADSs) (which includes shares subject to options that are currently exercisable or will be exercisable within 60 days of March 31, 2022) which represents 5.66% of the total number of ordinary shares outstanding as of March 31, 2022. See Item 7.A. below.
Equity Compensation Arrangements
The Equity Incentive Plan
Our Employee Equity Incentive Plan (“Equity Incentive Plan”) was adopted on September 23, 2019, and effective from September 24, 2019. Prior to such date, we granted options under a prior employee share option plan, which had the same terms and conditions as the Equity Incentive Plan. On September 24, 2019, all options held under our prior share option plan were rolled over into options to subscribe for our ordinary shares with the key terms including strike price, vesting and the expiration date of such rolled over options remaining the same as they were on the issue date of the options under the prior share option plan. The Equity Incentive Plan was approved for amendment by the Board on May 18, 2020, and provides for the granting of share options, Restricted Share Units and Performance Share Units (as defined below) (together “Equity Incentives”) to directors, employees and consultants of the Company and associated companies. The Equity Incentive Plan was approved for amendment by the Board on August 3, 2021, and November 2, 2021, to provide for a number of changes including the addition of an Israel sub-plan for Israeli employees, to address the behavior of Amryt’s Equity’s in the instance of a merger or acquisition as well as the ability to issue ADS’s under the Plan.
On May 18, 2020, the Board also approved the adoption of a U.S. Sub-Plan under the terms of the existing Equity Incentive Plan (“U.S. Sub-Plan”) in order to facilitate the grant of Restricted Share Units (as defined below), Incentive Stock Options (as defined below) and non-statutory stock options under the U.S. Sub-Plan (i.e., a share option granted under the U.S. Sub-Plan that does not qualify as an Incentive Stock Option and which does not benefit from any tax favored treatment (“Non-statutory Stock Option” and, together with Incentive Stock Options, “U.S. Options”)) to U.S. Participants. “U.S. Participants” are certain of our employees, directors and consultants who are either U.S. residents or U.S. taxpayers, and who shall have been nominated to participate in the U.S. Sub-Plan by the Board in compliance with U.S. tax, securities and other applicable laws.
All equity incentives granted are in the form of ordinary shares. Share option exercise prices below are the exercise price per ordinary share. The ADS equivalent exercise price will be the ordinary share exercise price multiplied by five and the number of ADSs will be the number of ordinary shares divided by five.
On November 28, 2017, a total of 343,521 share options were granted to Dr. Wiley at an exercise price of £1.2072 per share, on May 21, 2019, a total of 316,039 share options were granted to Dr. Wiley at an exercise price of £0.7584 per share, on November 5, 2019, a total of 5,777,900 share options were granted to Dr. Wiley at an exercise price of £1.215 per share, on March 8, 2021, a total of 2,031,350 share options were granted to Dr. Wiley at an exercise price of $2.804 per share, and on March 11, 2022, a total of 3,401,100 share options at an exercise price of $1.418 per share and 347,700 performance stock units were granted to Dr. Wiley. Performance conditions determine how many of these performance stock units will vest and, if performance targets are exceeded, additional performance stock units will be issued and vest in accordance with the terms of the relevant performance stock units award. The PSUs vest based on the Total Shareholder Return (TSR) of Amryt’s NASDAQ traded common stock relative to the TSRs of the constituents that comprise the NASDAQ Biotechnology Index (the Peer Group) as of January 1, 2022. TSR for Amryt and each peer company will be measured over the period from January 1, 2022, to December 31, 2024. The payout schedule can produce payout percentages ranging from 0% to 150%. The maximum amount of performance stock units that could be issued is 521,600 and the minimum amount is nil.
On November 28, 2017, a total of 137,408 share options were granted to Mr. Nealon at an exercise price of £1.2072 per share, on May 21, 2019, a total of 251,915 share options were granted to Mr. Nealon at an exercise price of £0.7584 per share, on November 5, 2019, a total of 4,437,500 share options were granted to Mr. Nealon at an exercise price of £1.215 per share, on March 8, 2021, a total of 1,400,000 share options were granted to Mr. Nealon at an exercise price of $2.804 per share and on March 11, 2022, a total of 2,346,800 share options at an exercise price of $1.418 per share and 274,700 performance stock units were granted to Mr Rory Nealon. Performance conditions determine how many of these performance stock units will vest and, if performance targets are exceeded, additional performance stock units will be issued and vest in accordance with the terms of the relevant performance stock units award. The PSUs vest based on the Total Shareholder Return (TSR) of Amryt’s NASDAQ traded common stock relative to the TSRs of the constituents that comprise the NASDAQ Biotechnology Index (the Peer Group) as of January 1, 2022. TSR for Amryt and each peer company will be measured over the period from January 1, 2022, to December 31, 2024. The payout schedule can produce payout percentages ranging from 0% to 150%. The maximum amount of performance stock units that could be issued is 359,900 and the minimum amount is nil.
On July 9, 2020, a total of 220,000 share options were granted to each of the six non-executive directors at an exercise price of $2.25 per share. On August 9, 2021, a total of 110,000 share options were granted to each of the six non-executive directors and a total of 220,000 share options were granted to Raj Kannan, a new non-executive director, at an exercise price of $2.04 per share. On September 14, 2021, 2021 a total of 220,000 share options were granted to Dr. Roni Mamluk, a new non-executive director, at an exercise price of $2.14 per share.
The U.S. Sub-Plan
The U.S. Sub-Plan permits the Board, as approved by the Company shareholders at our annual general meeting on July 29, 2020, to grant tax-qualified incentive stock options (i.e., a share options granted under the U.S. Sub-Plan that is intended to be, and qualifies as, within the meaning of section 422 of the Code, an incentive stock option (“Incentive Stock Options”)) to certain U.S. Participants.
The Israel Sub-Plan
The Israel Sub-Plan, as approved by the Board on August 3, 2021, permits the Board to grant Equity Incentives, including share options, Restricted Share Units and other equity-based awards, granted under the Equity Incentive Plan to certain Israeli Participants.
Description of Restricted Share Units
A Restricted Share Unit is an unfunded, unsecured right to receive, at a future date, one ordinary share (or an amount in cash or other consideration determined by the Board to be of equal value of such shares at such future date) granted to a Participant pursuant to the terms of the Equity Incentive Plan (including, for the avoidance of doubt, restricted share units granted to U.S. Participants pursuant to the terms of the U.S. Sub-Plan (“U.S. Restricted Share Units”)) (together, “Restricted Share Units”). The Equity Incentive Plan allows the Board to grant Restricted Share Units which shall vest at such time or times, whether or not in instalments, as shall be determined by the Board at or after the date of grant. At the time of grant of a Restricted Share Unit, the Board may in its discretion attach any condition to the vesting or forfeiture of that Restricted Share Unit, including any condition relating to the future performance of the Participant, which must be satisfied before that Restricted Share Unit vests. The Board may also provide that a Participant is entitled to receive a dividend equivalent in respect of a Restricted Share Unit.
Eligibility, Awards and Administration of the Equity Incentive Plan
The Equity Incentive Plan provides for the grant of Equity Incentives (including U.S. Options and U.S. Restricted Share Units) to our directors, employees and consultants eligible to receive awards. The Board may grant Equity Incentives to our directors, employees or consultants or the directors, employees or consultants of any associated company (companies in which we hold a 20% or greater interest). The maximum number of shares over which Equity Incentives (including U.S. Options and U.S. Restricted Share Units) may be in issue at any one time pursuant to the Equity Incentive Plan cannot currently exceed 15% of our share capital from time to time (“Equity Incentives Limit”). On January 1 in each calendar year, the then Equity Incentive Limit will automatically increase by 5% of our issued share capital from time to time. The Equity Incentive Limit from time to time shall decrease by the number of our ordinary shares in relation to which Equity Incentives are exercised (such decrease to be calculated by reference to the percentage that the number of ordinary shares in relation to which Equity Incentives are exercised bears to our issued share capital immediately prior to such exercise).
Options granted under the Equity Incentive Plan typically contain a three-year vesting period and typically have seven years within which they must be exercised. When options are granted, the Board may at its discretion attach conditions to the exercise of the options, which must be satisfied before an option may be exercised.
Before the date of adoption of the Equity Incentive Plan, we had 4,308,805 options in issue to purchase ordinary shares granted to employees and consultants of which 778,363 have been exercised and 129,599 have lapsed as at March 31, 2022. On September 24, 2019, these options were rolled over into new options to subscribe for our ordinary shares. Since then, we have issued 40,834,234 additional share options as at March 31, 2022, of which 87,542 share options have been exercised and 1,524,465 share options have lapsed. Of such outstanding options of 42,623,202, as at March 31, 2022, 11,435,228 had fully vested and were exercisable and 31,187,974 were not yet exercisable.
Details of Restricted Share Units granted to employees are as follows:
| | Date |
| Number of restricted share units granted |
| | August 8, 2020 | | 1,477,775 |
| | September 16, 2020 | | 79,185 |
| | March 8, 2021 | | 293,180 |
| | April 5, 2021 | | 162,690 |
| | August 9, 2021 | | 86,525 |
| | September 14, 2021 | | 9,010 |
| | November 9, 2021 | | 64,090 |
| | December 20, 2021 | | 9,710 |
Of the total Restricted Share units granted in 2020 and 2021, 250,555 had lapsed at December 31, 2021 and 362,855 have vested at December 31, 2021. On March 11, 2022, the Board granted 2,388,365 Restricted Share Units and 1,851,222 Performance Share units to employees. A further 118,145 of the total Restricted Share units were exercised and 28,315 Restricted Share units lapsed in the three month period to March 31, 2022. The total Restricted Share Units outstanding at March 31, 2022 was 3,810,661.
Lapse of Options (including U.S. Options)
Options and U.S. Options lapse on the last date upon which any part thereof may be exercised under the terms of the Equity Incentive Plan, which date shall be determined by the Board and specified in the option certificate issued to a participant in connection with the grant of an option, but in no event shall such date be later than the date preceding the tenth anniversary of the date of grant of an option (“Final Option Date”).
To the extent then exercisable, options and U.S. Options also lapse on the occurrence of certain events unless exercised within certain specified periods (or, if earlier, by no later than the Final Option Date), subject to the exercise by the Board of its discretion, as follows:
| • | on death, one year from the date of death; |
| • | on the option holder ceasing to meet the requirements of a participant in the Equity Incentive Plan due to retirement or resignation or early retirement due to disability or ill health, on the expiration of one year after having resigned or retired; or |
| • | on resignation, retirement or dismissal for reasons other than termination summarily for serious misconduct, 90 days after such event. On termination summarily for serious misconduct, options lapse immediately on such termination. |
If options were not already exercisable on the occurrence of the events referred to above, the options will lapse (subject to the discretion of the Board). The Board may at its discretion extend the periods set out above.
Options and U.S. Options shall also lapse in the event of a liquidator being appointed.
Certain additional restrictions apply under the U.S. Sub-Plan and the Code in relation to post-termination exercise periods relating to Incentive Stock Options.
Certain Transactions
Under the Equity Incentive Plan, on a merger, takeover or other re-organization resulting in a change of control of us (or the Board considers this is about to occur), each Equity Incentive (including U.S. Options and U.S. Restricted Share Units) issued prior to November 2, 2021 and to the extent covered by contractual commitments shall accelerate and become exercisable in full as of a date specified by the Board and subject to such conditions as the Board may at its discretion determine. A reverse takeover by us of another company will not result in an acceleration of the Equity Incentives (including U.S. Options and U.S. Restricted Share Units) vesting. If the Equity Incentives are substituted with new Equity Incentives of a public company successor entity or parent thereof in a a merger, takeover or other reorganization, or change of control subsequently occurs of the successor entity or the Equity Incentive Plan participant is terminated or resigns for good cause either immediately or within twelve (12) months from the occurrence of the change of control of Amryt or within three (3) years of a change of control of the public company successor entity, each substituted Equity Incentive shall automatically accelerate and vest and Equity Incentive Plan participant shall be entitled to exercise any or all their Equity Incentives immediately. The Remuneration Committee may also make such other comparable arrangements to replace any Equity Incentives (including U.S. Options and U.S. Restricted Share Units) (whether exercisable or not) as it determines in its discretion.
Adjustments
Under the Equity Incentive Plan, Israel Sub-Plan and the U.S. Sub-Plan, in the event of a consolidation, sub-division or reduction of our share capital, or any other variation in our share capital, the Board may make such adjustment to the relevant exercise price, the number, and/or the class of shares subject to each option or U.S. Option (as applicable) as it deems appropriate. If shareholders are granted rights to subscribe for further ordinary shares (such rights being related to the number of ordinary shares held by them respectively) the Board shall at its absolute discretion decide whether the granting of such rights and the subscriptions made thereunder shall result in the depletion in the value of each ordinary share and the Board may make such adjustment to the exercise price, the number and/or class of shares subject to each option as it deems appropriate.
Amendment and Termination
The Equity Incentive Plan may be terminated at any time by resolution of our Board. Subsequent to any termination of the Equity Incentive Plan, we shall not grant any further options, but no such termination shall affect or modify any subsisting rights or obligations of participants in respect of any options, and notwithstanding such termination, we shall continue to administer and manage the Equity Incentive Plan in accordance with the rules of the Equity Incentive Plan.
We may at any time by resolution of the Board vary, amend or revoke any of the provisions of the Equity Incentive Plan in such manner as the Board sees fit.
Under the U.S. Sub-Plan, the Board may grant U.S. Equity Incentives under the terms of the U.S. Sub-Plan until the earlier of: (a) ten years from May 18, 2020, being the date that the Board agreed to adopt the U.S. Sub-Plan; and (b) the termination of the Equity Incentive Plan. No U.S. Equity Incentive may be granted under the U.S. Sub-Plan later than the date of termination or expiration of the Equity Incentive Plan.
The Board may amend, suspend or terminate any provision of the U.S. Sub-Plan at any time and may amend, modify or terminate any U.S. Equity Incentive. The approval of shareholders is required for any amendment to the U.S. Sub-Plan that would require such approval in order to satisfy the rules relating to Incentive Stock Options under the Code or any other applicable law.
Chiasma Equity Incentive Plan
When Amryt acquired Chiasma in August 2021, the Chiasma Stock Option and Incentive Plan transferred across to Amryt. Each outstanding and unexercised Chiasma Stock Option or RSU, whether vested or not vested, ceased to represent a right to acquire shares of Chiasma common stock and were converted into an option to purchase Amryt ADSs on the same terms and conditions as were applicable under such Chiasma Stock Option and Incentive Plan immediately prior to the acquisition.
On August 5, 2021, a total of 18,139,060 stock options were issued by Amryt to replace Chiasma stock options. Of the total amount of Chiasma stock options, 3,320,515 stock options were exercised and 4,098,275 stock options had lapsed by December 31, 2021. A further 226,690 stock options were exercised in and a further 2,171,425 lapsed in the three month period to March 31, 2022. The number of legacy Chiasma stock options outstanding at March 31, 2022 is 8,322,005.
On August 5, 2021, a total of 202,145 RSUs were transferred across to Amryt. Of the total amount of Chiasma RSUs transferred across to Amryt, 39,180 RSUs were exercised, and 56,405 RSUs had lapsed by December 31, 2021. A further 49,300 RSUs were exercised and a further 4,605 lapsed in the three month period to March 31, 2022. The number of legacy Chiasma RSUs outstanding at March 31, 2022 is 52,655.
No new stock option or RSUs will be granted under the Chiasma stock option and incentive plan.
Terms and Conditions of New Grants and Grants under the Chiasma Equity Incentive Plan
The terms and conditions of new grants are as follows, whereby all options are settled by physical delivery of shares:
Vesting conditions
The employee share options vest following a period of service by the officer or employee. The required period of service is determined by the Remuneration Committee at the date of grant of the options (usually the date of approval by the Remuneration Committee). There are no market conditions associated with the share option vesting periods.
Contractual life
The term of an option is determined by the Remuneration Committee provided that the term may not exceed a period of seven to ten years from the date of grant. All options will terminate 90 days after termination of the option holder’s employment, service or consultancy with the Group except where a longer period is approved by the Board of Directors. Under certain circumstances involving a change in control of the Group, some options will automatically accelerate and become exercisable in full as of a date specified by the Board of Directors.
Bonus Payments
All executive directors and senior management are eligible for a discretionary annual bonus. Annual cash bonuses are paid on the achievement of pre-set strategic objectives. The Remuneration Committee in conjunction with the Board reviews and sets the objectives at the start of each calendar year.
Pension, Retirement or Similar Benefits
We operate defined contribution schemes in various locations where employees are based. Contributions to the defined contribution schemes are recognized in our statement of comprehensive income in the period in which the related services are received from the employee. Under these schemes we have no obligation, either legal or constructive, to pay further contributions in the event that the fund does not hold sufficient assets to meet our benefit commitments.
As of December 31, 2021, we had accrued $463,000 in pension benefits which was paid to the pension administrators in early 2022. With the exception of Dr. Wiley and Mr. Nealon, no director or secretary has any pension or retirement benefits. In addition, no director or senior manager is entitled to any pension, retirement or related benefit upon termination of his employment.
Insurance and Indemnification
To the extent permitted by law, each of our directors, alternate directors, officers or employees is entitled to be indemnified out of our assets against all losses or liabilities which he or she may sustain or incur in or about the execution of the duties of his or her office or otherwise in relation thereto. We maintain directors’ and officers’ insurance to insure such persons against certain liabilities.
To the extent permitted by law, the directors have the power to purchase and maintain insurance for or for the benefit of any persons who are or were at any time:
| • | directors, alternate directors, officers or employees of a group company; or |
| • | trustees of any pension fund in which our employees or employees of any other group company are interested, including in each case insurance against any liability incurred by such persons in respect of any act or omission in the actual or purported execution and/or discharge of their duties and/or in the exercise or purported exercise of their powers and/or otherwise in relation to their duties, powers or offices in relation to us or any other group company or pension fund. |
Insofar as indemnification of liabilities arising under the Securities Act may be permitted to the Board, executive officers, or persons controlling us pursuant to the foregoing provisions, we are informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Options and Warrants
Directors may issue options or warrants if authorized to do so by our Articles of Association or an ordinary resolution of the shareholders. We may issue shares upon the exercise of options or warrants without further shareholder authorization, up to the relevant authorized limit.
Options
As of December 31, 2021, there were options to purchase 28,502,942 ordinary shares outstanding under the Equity Incentive Plan with a weighted average exercise price of $1.994 per ordinary share (based on an exchange rate of £1.00 to $1.34898 as of December 31, 2021). The options generally lapse after seven years from the date of the grant.
As of December 31, 2021, there were options to purchase 10,720,120 ordinary shares outstanding under the Chiasma Stock Option and Incentive Plan with a weighted average exercise price of $2.663 per ordinary share (based on an exchange rate of £1.00 to $1.34898 as of December 31, 2021). The options generally lapse after seven years from the date of the grant. The outstanding options were issued under the Equity Incentive Plan.
Warrants
As at December 31, 2021, there was no category of warrants to purchase our ordinary shares outstanding.
As of December 31, 2020, there were two categories of warrants to purchase our ordinary shares outstanding:
| • | Warrants issued on September 24, 2019, to purchase 345,542 ordinary shares with a weighted average exercise price of £1.44 per ordinary share. These warrants generally lapse after five years from the date of the grant. These warrants were issued to Shore Capital and Davy in exchange for warrants granted to such parties on April 19, 2016, in connection with the placement undertaken by us on that date. In March 2021, 283,389 ordinary shares were issued from treasury following the exercise of these warrants and the remaining warrants lapsed in April 2021. |
| • | Warrants to purchase 8,966,520 ordinary shares, with an exercise price of zero. 13,976,722 warrants were issued to certain Aegerion creditors who expressed a wish to restrict their percentage share interest in our issued share capital and who elected to take warrants in lieu of ordinary shares. In addition 4,864,656 warrants were issued to certain investors in exchange for the repurchase of 4,864,656 ordinary shares by us from these investors on November 14, 2019. On December 19, 2019, certain investors elected to exercise 1,645,105 warrants in exchange for 1,645,105 ordinary shares. On July 9, 2020, certain investors elected to exercise 4,000,000 warrants in exchange for 4,000,000 ordinary shares and on September 22, 2020, Nineteen 77 Global Multi Strategy Alpha Master Limited elected to exercise 4,229,753 warrants in exchange for 4,229,753 ordinary shares. On August 5, 2021, Highbridge Tactical Credit Master Fund, L.P. elected to exercise 8,966,520 warrants in exchange for 8,966,520 ordinary shares. |
| Item 7. | Major Shareholders and Related Party Transactions |
A. Major Shareholders
The following table sets forth information relating to the beneficial ownership of our ordinary shares as of March 31, 2022, by:
| • | each person, or group of affiliated persons, known to us to beneficially own 5% or more of our outstanding ordinary shares; |
| • | each member of our Board and each of our executive officers; and |
| • | all Board members and executive officers as a group. |
Beneficial ownership is determined in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities as well as any ordinary shares that the individual has the right to acquire within 60 days of March 31, 2022, through the exercise of any option, warrant or other right. The percentage ownership information shown in the table below is based upon 320,699,807 ordinary shares, which are outstanding as of March 31, 2022.
Except as otherwise indicated, all of the shares reflected in the table are ordinary shares and all persons listed below have sole voting and investment power with respect to the ordinary shares beneficially owned by them, subject to applicable community property laws. The information is not necessarily indicative of beneficial ownership for any other purpose.
In computing the number of ordinary shares beneficially owned by a person and the percentage ownership of that person, we deemed outstanding ordinary shares subject to options and warrants held by that person that are immediately exercisable or exercisable within 60 days of March 31, 2022. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person.
| | | Beneficially Owned | |
| | | ADS | | | Ordinary Shares | | | | |
| Name of beneficial owner | | Number | | | Number | | | Percentage | |
| 5% or greater shareholders: | | | | | | | | | |
Athyrium Capital Management, LP(1) | | | 13,159,248 | | | | | | | | 19.22% |
|
| Highbridge Capital Management, LLC(2) | | | 9,838,220 | | | | 49,191,100 | | | | 14.50% | |
Stonepine Capital Management, LLC(3) | | | 6,069,775 | | | | 30,348,875 | | | | 9.46% |
|
Rubric Capital Management, L.P.(4) | | | 3,544,468 | | | | 17,722,340 | | | | 5.53% |
|
| (1) | Based on information known to Amryt and information contained in a Statement on Schedule 13F filed by Athyrium Capital Management, L.P. in February, 2022 and 21,509,901 ordinary shares issuable upon conversion of Convertible Notes held by the following affiliates of Athyrium Capital Management, LP: Athyrium Opportunities II Acquisition 2 LP, Athyrium Opportunities III Acquisition 2 LP, Athyrium Opportunities II Acquisition LP and Athyrium Opportunities III Acquisition LP. |
| (2) | Based on information known to Amryt and information contained in a Statement on Schedule 13F filed by Highbridge Capital Management, LLC in February, 2022 and 18,520,040 ordinary shares issuable upon conversion of Convertible Notes held by the following affiliates of Highbridge: Highbridge MSF International Ltd., Highbridge SCF Special Situations SPV, L.P. and Highbridge Tactical Credit Master Fund, L.P. Pursuant to the terms of the Convertible Notes, Highbridge may not convert any Convertible Notes that it beneficially owns to the extent that such conversions and exercises would result in Highbridge beneficially owning in excess of 9.99% of our ordinary shares. After giving effect to these blockers, as of November 30, 2020, Highbridge’s beneficial ownership is limited to 9.99%. |
| (3) | Based on information known to Amryt and information contained in a Statement on Schedule 13F filed by Stonepine Capital Management, LLC in February, 2022 |
| (4) | Based on information known to Amryt and information contained in a Statement on Schedule 13F filed by Rubric Capital Management, L.P. in February, 2022. |
B. Related Party Transactions
The following is a summary of related party transactions, since January 1, 2018 to date, in which we have been a participant and in which the amount involved exceeded or will exceed $120,000, and in which any of our then directors, executive officers or holders of more than 5% of any class of our voting securities at the time of such transaction, or any members of their immediate family, had or will have a direct or indirect material interest.
Agreements with Principal Shareholders
We entered into a number of agreements with our principal shareholders in connection with the acquisition of Aegerion. The following summary of these agreements is not intended to be a complete description of these agreements, and it is qualified in its entirety by reference to the full text of such agreements, which are filed as exhibits to the registration statement of which this annual report forms a part. We urge you to review these exhibits in their entirety.
The Restructuring Support Agreement
On May 20, 2019, we entered into a Restructuring Support Agreement (as subsequently amended on June 12, 2019, “Restructuring Support Agreement”) with (i) Aegerion and Aegerion Pharmaceuticals Holdings, Inc. (together, “Debtors”), (ii) Novelion, as holder of 100% of the outstanding equity interests of Aegerion and holder of 100% by principal amount of claims under Aegerion’s Amended and Restated Loan Credit Agreement dated March 15, 2018, (iii) the holders of 100% in principal amount under Aegerion’s Bridge Loan Credit Agreement dated November 8, 2018, and (iv) holders of in excess of 67% in principal amount of Aegerion’s 2% Senior Unsecured Convertible Notes due 2019. The holders of Aegerion debt party to the Restructuring Support Agreement are collectively referred to herein as the “Consenting Lenders.”
Under the Restructuring Support Agreement, we shall, among other things, negotiate in good faith the documents necessary to complete the transactions in connection with the Acquisition. The Debtors agreed to, among other things, (a) use reasonable best efforts to take any actions necessary in connection with the transaction; (b) commence the Debtors’ bankruptcy cases and file and seek approval of “first day” motions; and (c) file and prosecute confirmation of the Plan of Reorganization.
Each of the Consenting Lenders agreed to, among other things, (a) vote to accept the Plan of Reorganization and otherwise support the transactions; (b) not object or impede confirmation or implementation of the Plan of Reorganization or the transactions, including by soliciting an alternative transaction; (c) not support any other Chapter 11 plan or other restructuring or reorganization of the Debtors other than the Plan of Reorganization; and (d) not take any action that would interfere with consummation of the transactions and take any and all necessary, appropriate, or advisable action in furtherance of the transactions.
Each Consenting Lender also agreed that, if the Restructuring Support Agreement was terminated pursuant to the specified provisions contained therein on or after the date Aegerion received a proposal for an alternative transaction that is still outstanding at the time of such termination, it would vote against, and use its reasonable best efforts to oppose, approval of an alternative transaction unless, following a written inquiry by the Consenting Lenders regarding whether we would be willing to complete the transactions in accordance with the definitive documents, we did not confirm within five business days following such inquiry (subject to withdrawal at any time upon five business days’ notice) that we would be willing to consummate the transactions if the documents are re-executed by the other parties thereto.
Plan Funding Agreement
On May 20, 2019, we entered into the Plan Funding Agreement with Aegerion setting forth the terms and conditions of the acquisition of Aegerion.
In consideration of the acquisition, Aegerion’s former creditors received our ordinary shares (including shares represented by ADSs and zero cost warrants that may be exercised at any time), constituting approximately 61.6% of our outstanding share capital prior to our $60 million dollar equity issuance in September 2019 (“September 2019 Equity Raise”). The number of our ordinary shares that were issued as consideration was calculated by multiplying the aggregate amount of all issued and outstanding ordinary shares of Amryt immediately prior to the closing of the Acquisition by 1.59, after giving effect to the transactions, but before giving effect to equity underlying the issuance of the Convertible Notes, the September 2019 Equity Raise, our ordinary shares that may be issuable in satisfaction of the CVRs if the relevant milestones are achieved on the terms of the CVRs, and equity that was reserved for issuance under any management equity compensation plan adopted by us.
The Plan Funding Agreement contains reciprocal representations and warranties made by us and Aegerion relating to organization, qualification, due authorization of the parties and authority, consents and approvals, capitalization, financial statements, no undisclosed liabilities, recent events, contracts and commitments, real property, intellectual property, privacy and data security, legal compliance, permits, environmental compliance and conditions, litigation, tax matters, insurance, illegal or improper payments, related party transactions, brokers’ fees, employees and healthcare compliance matters.
Under the terms of the Plan Funding Agreement:
| • | We were not required to take any action which is prohibited or restricted by the Takeover Code in relation to any alternative transaction. Similarly, we were not required to refrain from any action which is required by the Takeover Code in relation to any alternative transaction. |
| • | Following the approval by the U.S. Bankruptcy Court of the Plan Funding Agreement Approval Motion on June 28, 2019, Aegerion had a 55-day period to solicit an alternative transaction. Subject to the limitations of the Plan Funding Agreement¸ Aegerion was also entitled to respond to unsolicited proposals that Aegerion determined reasonably likely to result in a superior transaction. |
| • | At any time prior to and after the completion of the 55-day period, Aegerion was entitled to receive offers from competing bidders and was able to respond to any such offer if its board deemed that the offer constituted or was likely to constitute a superior proposal. |
The Plan Funding Agreement included termination rights for both us and Aegerion and related provisions that allocated fees and expenses among the parties if the Plan Funding Agreement was terminated prior to closing.
Upon termination of the Restructuring Support Agreement, we had the right to terminate the Plan Funding Agreement for any reason, so long as the termination of the Restructuring Support Agreement was not principally due to our material uncured breach. Aegerion also had a right to terminate the Plan Funding Agreement upon termination of the Restructuring Support Agreement, so long as the termination of the Restructuring Support Agreement was not principally due to Aegerion’s or the Consenting Lenders’ material uncured breach.
In specified circumstances, upon termination, we had the obligation to pay Aegerion a termination fee of $2,050,000. Additionally, upon a termination in other specified circumstances, Aegerion was obligated to pay us an expense reimbursement amount equal to all of our reasonable and documented fees and expenses incurred in connection with the negotiation, preparation and implementation of the documents for the transactions, up to $4,000,000. In addition, if Aegerion disclosed an alternative transaction and did not withdraw it and Aegerion entered into an agreement for an alternative transaction with a third party within one year of the date of the Plan Funding Agreement, Aegerion was liable to pay us a termination fee of $11,850,000 upon consummation of such transaction.
Backstop Agreement
On July 10, 2019, we entered into the Backstop Agreement with Highbridge MSF International Ltd., Highbridge SCF Special Situations SPV, L.P., 1992 Tactical Credit Master Fund, L.P., Athyrium Opportunities II Acquisition 2 LP, Athyrium Opportunities III Acquisition 2 LP, Whitebox Relative Value Partners, LP, Whitebox GT Fund, LP; Whitebox Multi-Strategy Partners, LP, Pandora Select Partners, LP, Nineteen77 Global Multi-Strategy Alpha Master Limited and Nineteen77 Global Convertible Bond Master Limited (each a “Backstop Party” and collectively, “Backstop Parties”), whereby each Backstop Party agreed to subscribe for a specified percentage of any unsubscribed shares in the September 2019 Equity Raise. In consideration for the Backstop Parties’ undertaking to subscribe for our ordinary shares, we agreed to pay the Backstop Parties the sum of $3 million in cash as commission.
The obligations of each Backstop Party were conditional on the satisfaction of, among others, the following conditions: (a) the rights attaching to our ordinary shares not having been altered at the date of execution of the Restructuring Support Agreement; (b) the approval of specified resolutions by our shareholders; and (c) a final order confirming the Plan of Reorganization having been entered (in form and substance reasonably satisfactory to the Backstop Parties and to us), the Effective Date, having occurred, and all conditions precedent to the occurrence of the Effective Date having been satisfied or waived by the Backstop Parties. The Backstop Parties, acting unanimously, had the ability to waive in whole or in part all or any of the conditions.
DIP Funding Agreement
On June 28, 2019, Aegerion (as borrower) entered into a $20 million aggregate maximum principal amount post-petition super-priority secured debtor-in-possession credit facility (“DIP Funding Agreement”) with the Athyrium Funds and certain affiliates of Highbridge, as lenders, and Cantor Fitzgerald Securities, as administrative agent. The DIP Funding Agreement was fully and unconditionally guaranteed on a super-priority senior secured basis by Aegerion’s U.S. subsidiaries (other than Aegerion Securities Corporation) (“DIP Subsidiary Guarantors”).
The loans under the DIP Funding Agreement bore interest at a rate of 12.5% per annum, to be paid in cash on a monthly basis. The DIP Funding Agreement matured on the earliest to occur of: (a) the Effective Date; (b) the date that is 150 days after May 20, 2019 (being October 17, 2019), subject to a 60-day extension in accordance with the Plan Funding Agreement; and (c) the date on which the obligations under the DIP Funding Agreement are accelerated and became due and payable following the occurrence of an event of default.
The DIP Funding Agreement contained negative covenants restricting Aegerion and its subsidiaries, including limitations on, or related to, liens, investments, the incurrence of indebtedness, fundamental changes, dispositions, restricted payments, changes in business, transactions with affiliates, prepayments and modifications of debt, negative pledges, changes to organizational documents, use of proceeds, compliance with laws and orders of the bankruptcy court, ownership of subsidiaries, Chapter 11 claims, revisions of orders and applications to the bankruptcy court, and adequate protection.
The DIP Funding Agreement contained events of default, including failure to make payments when due, breach of covenants, breach of representations or warranties, cross-defaults, judgments, ERISA, invalidity of loan documents, change of control, failure of liens to be perfected, dissolution or liquidation, discontinuation of business, maintenance of independent directors, and bankruptcy matters including failure to confirm the approved Plan, non-compliance or revocation or modification of bankruptcy orders, and non-compliance with the Restructuring Support Agreement.
The DIP Funding Agreement was repayable from time to time and contained mandatory repayments from the proceeds of certain dispositions of assets, casualty events, and the incurrence of indebtedness. However, the DIP Funding Agreement facility was never utilized by Aegerion, therefore no repayments were required to be made.
Registration Rights Agreement
On the closing of the acquisition of Aegerion, we entered into a Registration Rights Agreement with affiliates of Athyrium (“Athyrium Parties”) and affiliates of Highbridge (“Highbridge Parties”). Pursuant to the agreement, we were to use commercially reasonable efforts to list our ordinary shares, or ADSs representing such shares, on the Nasdaq within 90 days of the closing of the Acquisition. The agreement also provided the Athyrium Parties and the Highbridge Parties with the right to demand share registration up to two times in any 12-month period and a right to participate in future registration statements filed by us. We are obligated to pay any expenses relating to such registrations and indemnify the Athyrium Parties and the Highbridge Parties (and their respective partners, members, directors, offices and professional advisers, among others) participating in such registration against specified liabilities that may arise in connection with such registration. Furthermore, each of the Athyrium Parties and the Highbridge Parties participating in such registration will indemnify us and certain other persons against specified limited liabilities that may arise in connection with such registration.
The agreement provided the Athyrium Parties and the Highbridge Parties with specified rights to nominate directors and designate observers to the Board. The agreement specified that, on closing of the Acquisition, the initial Board would be made up of seven members. The Board was to initially consist of Joseph Wiley, our Chief Executive Officer, one independent chairman and one independent director proposed by Amryt, one independent director proposed by the Athyrium Parties, one non-independent director proposed by the Athyrium Parties and two independent directors proposed by the Highbridge Parties. The Highbridge Parties have the right to subsequently select a non-independent director to replace one of its nominated independent directors, subject to the shareholding requirements set out below. The agreement also provided that Joseph Wiley would continue as Chief Executive Officer for no less than two years after the closing of the Acquisition, unless he: (a) resigns; (b) is removed from the position by a vote of a majority of the Board, which majority must include the vote of at least one of the directors nominated by us; or (c) is removed for cause under the terms of his employment agreement.
The initial Board is subject to compulsory retirement and will be put up for re-election at our first annual general meeting to be held at least 24 months after the closing of the Acquisition. For so long as each of the Athyrium Parties or the Highbridge Parties (or their respective affiliates) respectively hold at least 10% of our issued share capital, the Athyrium Parties and the Highbridge Parties (as applicable) are each entitled to nominate a replacement of the non-independent director (as applicable) selected by them on his or her resignation or retirement. Any such director shall serve on the Board until our next annual general meeting, where such director’s appointment will be subject to approval by an ordinary resolution of our shareholders.
The agreement will also grant the Athyrium Parties or the Highbridge Parties the right to designate a board observer to attend Board meetings under the Secured Credit Facility (as described below), subject to restrictions on attendance when specified matters are discussed. Each of the Athyrium Parties and the Highbridge Parties have the ability to terminate this right, and this right will be terminated when the Secured Credit Facility is either refinanced or paid off in full. To date, only Athyrium has exercised this right.
Secured Credit Facility
On September 24, 2019, we entered into the Secured Credit Facility. The Secured Credit Facility is fully and unconditionally guaranteed on a senior secured basis by us and, to the extent permitted by law, each of our and Aegerion’s existing and subsequently acquired or formed direct and indirect subsidiaries (other than certain immaterial non-U.S. subsidiaries and Aegerion Securities Corporation) (“Facility Guarantors”). Proceeds from the Secured Credit Facility were used to refinance Aegerion’s existing secured bridge loan in the principal amount of approximately $50 million held by certain funds managed by Athyrium and Highbridge, respectively, and our existing €20 million (in principal) secured loan facility with the EIB.
The loans under the Secured Credit Facility bear interest at a rate of: (a) 11% per annum paid in cash on a quarterly basis; or (b) 6.5% per annum paid in cash plus 6.5% per annum paid in kind on a quarterly basis. The Secured Credit Facility will mature five years after issuance, unless earlier repaid.
The Secured Credit Facility contains the following negative covenants: (a) limitations on liens; (b) limitations on disposition of assets (including restrictions on dispositions of material assets and exclusive licensing transactions in material territories but permitting, among other things, (i) dispositions consisting of licenses of Oleogel-S10 and AP103 assets outside of the United States and European territories, (ii) dispositions of obsolete and surplus property that are immaterial, and (iii) a general basket for dispositions less than $250,000 individually and $2,500,000 in the aggregate); (c) limitations on consolidations and mergers; (d) limitations on loans and investments (with restrictions on investments and indebtedness to non-Facility Guarantor subsidiaries to be agreed); (e) limitations on indebtedness (with exceptions permitting, among other things, the indebtedness under the CVR deed poll); (f) limitations on transactions with affiliates (with exceptions permitting, among other things, certain de minimis transactions, certain transactions for the provision of inter-company services among Facility Guarantors and other transactions with non-Facility Guarantor affiliates in the ordinary course of business on an arm’s-length basis not to exceed an amount to be agreed upon, including Transactions with non-Facility Guarantor affiliates in respect of intercompany services charged on a cost plus 10% basis); (g) compliance with ERISA; (h) limitations on restricted payments (including restrictions on dividends (stock or cash), share repurchases, spin-off and split-off transactions and payments to non- Facility Guarantors); (i) limitations on changes in business, changes in structure, changes in accounting, name and jurisdiction of organization, amendments to organizational documents that are adverse to the lenders, any amendments, modifications or waivers to the CVR deed poll; (j) limitations on material changes to other indebtedness documents; (k) limitations on restricted debt payments; (l) no negative pledges or burdensome agreements; and (m) certain other customary restrictions.
The Secured Credit Facility contains the following events of default under the Secured Credit Facility: (a) failure to pay principal, interest or any other amount when due; (b) representations and warranties incorrect in any material respect when made or deemed made; (c) failure to comply with covenants; (d) cross-default to other indebtedness; (e) failure to satisfy or stay execution of judgments; (f) bankruptcy or insolvency; (g) actual or asserted invalidity or impairment of any part of the credit documentation (including the failure of any lien to remain perfected); (h) ERISA default; (i) failure to conduct business; (j) change of ownership or control; and (k) liquidation or dissolution.
Repayments of the Secured Credit Facility are subject to a prepayment premium as follows: (a) 105% of the amount repaid plus interest that would have accrued on such amount repaid through the second anniversary of the closing of the Acquisition discounted at a rate equal to the yield on U.S. Treasury notes with a maturity closest to the second anniversary of the closing of the Acquisition plus 50 basis points in years one and two; (b) 105% of the amount repaid in year three; (c) 101% of the amount repaid in year four; and (d) thereafter, 100%. Such premium will apply to repayments after acceleration, but will not apply to prepayments from insurance and condemnation proceeds.
The Secured Credit Facility also contains a covenant requiring the maintenance of minimum liquidity of $10 million at all times, subject to an add back with respect to cash expenditures made to French pricing authorities related to the Myalepta product. The Secured Credit Facility requires mandatory prepayments for certain specified debt issuances, asset sales or other dispositions of assets (including exclusive licensing transactions and insurance and condemnation proceeds), subject to certain exceptions and thresholds.
On February 18, 2022, the Secured Credit Facility was repaid in full and the Group secured a $125 million Senior Credit Facility from funds managed by Ares of which US$105 million was drawn down to facilitate the prepayment of the existing Secured Credit Facility. In repaying the Secured Credit Facility, Amryt incurred an exit fee of 5.00% of the outstanding principal amount as at the prepayment date.
Convertible Notes
On September 24, 2019, we issued $125 million aggregate principal amount of Convertible Notes due 2025 to certain creditors of Aegerion. The Convertible Notes are fully and unconditionally guaranteed on a senior unsecured basis by us and, to the extent permitted by law, each of our and Aegerion’s existing and subsequently acquired or formed direct and indirect subsidiaries (other than certain immaterial non-U.S. subsidiaries and Aegerion Securities Corporation) (“Notes Guarantors”). The Convertible Notes are issued pursuant to an indenture by and among Amryt Pharmaceuticals Inc and the Notes Guarantors and a trustee for the Convertible Notes.
The Convertible Notes bear interest at a rate of 5% per annum, payable in cash semi-annually. The Convertible Notes will mature approximately five and a half years after issuance, unless earlier repurchased, redeemed or converted.
The Convertible Notes and guarantees are the general senior unsecured obligations of Amryt Pharmaceuticals Inc and the Notes Guarantors, respectively, ranking equally in right of payment with all of the existing and future liabilities of such entities that are not so subordinated.
Convertible Note holders are entitled to convert their Convertible Notes, at their option, at any time prior to the close of business on the second scheduled trading day immediately prior to the maturity date, unless the Convertible Notes have been previously repurchased or redeemed. The Convertible Notes are initially convertible into 386.75 ordinary shares for each $1,000 principal amount of Convertible Notes, which is equivalent to an initial conversion price of approximately $2.59 per ordinary share or $12.95 per ADS. The conversion rate is subject to customary anti-dilution adjustments as well as adjustments following certain fundamental changes (as set out below) and adjustments to account for the final conversion share price. Upon conversion, Amryt Pharmaceuticals Inc may choose to pay or deliver, as the case may be, cash, ordinary shares or a combination of cash and our ordinary shares.
The Convertible Notes have the following events of default: (a) a default in the payment when due of the principal of, or the redemption price or repurchase price for, any Convertible Note; (b) a default for 30 days in the payment when due of interest on any Convertible Note; (c) the failure to deliver, when required, a repurchase notice; (d) a default in the obligation to convert a Convertible Note upon the exercise of the conversion right with respect thereto and such failure continues for five business days; (e) a default in obligations with respect to successors to us or Amryt Pharmaceuticals Inc; (f) a default in other obligations or agreements under the Convertible Notes or the indenture where such default is not cured or waived within 60 days after notice; (g) certain defaults with respect to any one or more mortgages, agreements or other instruments under which there is outstanding, or by which there is secured or evidenced, any indebtedness for money borrowed of at least $12 million (or its foreign currency equivalent) subject to a 60-day grace period; (h) one or more final judgments for the payment of at least $12 million in the aggregate, where such judgment is not timely discharged or stayed, subject to a 60-day grace period; and (i) certain events of bankruptcy, insolvency and reorganization with respect to Amryt Pharmaceuticals Inc, us or any of our or their significant subsidiaries.
Amryt Pharmaceuticals Inc has the right to redeem all or any portion of the Convertible Notes on or after the third anniversary of issuance, provided the per share volume-weighted average price of our ordinary shares exceeds 150% of the conversion price on: (a) each of at least 20 trading days (whether or not consecutive) during the 30 consecutive trading days ending on, and including, the trading day immediately before the date Amryt Pharmaceuticals Inc sends the notice of such redemption; and (b) the trading day immediately before the date Amryt Pharmaceuticals Inc sends such notice. Amryt Pharmaceuticals Inc may also redeem the Convertible Notes upon certain changes in tax law or regulation. The redemption price will be an amount in cash equal to 100% of the principal amount of the Convertible Notes redeemed, plus any accrued and unpaid interest.
Upon the occurrence of events deemed a fundamental change, Convertible Note holders may require Amryt Pharmaceuticals Inc to repurchase all or a portion of their Convertible Notes at a repurchase price of 100% of the principal amount of the Convertible Notes, plus any accrued and unpaid interest. With certain exceptions, a fundamental change will occur if: (a) a “person” or “group” (within the meaning of Section 13(d)(3) of the Exchange Act), other than us, any of our wholly owned subsidiaries or their respective employee benefit plans, has filed any report with the SEC indicating that such person or group has become the direct or indirect beneficial owner of our ordinary shares representing more than 50% of the voting power of all of our then-outstanding ordinary shares; (b) the consummation of (i) any sale, lease or other transfer, in one transaction or a series of Transactions, of all or substantially all of the assets of us and our subsidiaries, taken as a whole, to any person, other than one or more of our wholly owned subsidiaries; or (ii) any transaction or series of related transactions in connection with which (whether by means of merger, consolidation, share exchange, combination, reclassification, recapitalization, acquisition, liquidation or otherwise) all of our ordinary shares are exchanged for, converted into, acquired for, or constitute solely the right to receive, other securities, cash or other property (other than changes resulting solely from a subdivision or combination, or a change in par value, of our ordinary shares); (c) our shareholders approve any plan or proposal for the liquidation or dissolution of our company; or (d) our ordinary shares cease to be traded on AIM or another permitted exchange.
If a fundamental change occurs or if Amryt Pharmaceuticals Inc initiates a redemption of the Convertible Notes, the conversion rate will increase for any conversion occurring in connection with such fundamental change or redemption. The increase in the conversion rate will be determined based on a schedule set forth in the indenture and is subject to adjustment in accordance with the final conversion share price.
Zero Cost Warrant
We agreed, for certain Aegerion creditors who wished to restrict their percentage share interest in our issued share capital, to issue to the relevant Aegerion creditor, as an alternative to our ordinary shares, an equivalent number of new zero cost warrants to subscribe for our ordinary shares to be constituted on the terms of the zero cost warrant. The relevant Aegerion creditor is entitled at any time to exercise the zero cost warrants, at which point in time we would issue to that Aegerion creditor the relevant number of fully paid ordinary shares in return for the exercise of the zero cost warrants.
In accordance with the terms of the zero cost warrant, each zero cost warrant entitles the holder thereof to subscribe for one ordinary share at any time after our admission to trade on AIM. In no event shall any holder of zero cost warrants have the right to subscribe for zero additional consideration for, nor shall we issue to such holder of zero cost warrants, ordinary shares to the extent that such subscription would result in the holder of zero cost warrants and its affiliates, including groups that include the holder of zero cost warrants and its affiliates, together beneficially owning more than their prescribed maximum percentage to be set in the zero cost warrant. Any issue of our ordinary shares in contravention of this condition will be null and void.
The zero cost warrants constitute our direct and unsecured obligations and rank pari passu and without any preference among themselves (save for any obligations to be preferred by law) at least equally with our other present and future unsecured and unsubordinated obligations. The zero cost warrants are not transferrable except with our prior written consent.
On September 24, 2019, certain of Aegerion’s creditors elected to receive 13,976,722 warrants to subscribe for our ordinary shares as consideration for the Acquisition.
On November 14, 2019, we repurchased a combined 4,864,656 ordinary shares from Highbridge Tactical Credit Master Fund L.P., Highbridge SCF Special Situations SPV, L.P. and Nineteen77 Global Multi Strategy Alpha Master Limited. In exchange for the ordinary shares, these institutions were issued an equivalent number of zero cost warrants. Each warrant entitles the holder to subscribe for one ordinary share at zero cost.
On December 19, 2019, Highbridge MSF International Ltd exercised 1,645,105 zero cost warrants in exchange for 1,645,105 ordinary shares. On July 9, 2020, Highbridge Tactical Credit Master Fund, L.P. exercised 4,000,000 zero cost warrants in exchange for 4,000,000 ordinary shares. On September 22, 2020, Nineteen 77 Global Multi Strategy Alpha Master Limited exercised 4,229,753 zero cost warrants in exchange for 4,229,753 ordinary shares. On August 5, 2021, Highbridge Tactical Credit Master Fund, L.P. exercised 8,966,520 warrants in exchange for 8,966,520 ordinary shares, 4,208,314 of which were issued from treasury shares.
There were no remaining warrants outstanding as at December 31, 2021.
Interim Subscription Agreements
On August 27, 2019, we entered into subscription agreements in the total amount of $8 million which included:
| • | a subscription agreement between Amryt, Highbridge MSF International Ltd. and Highbridge SCF Special Situations SPV, L.P. (together, “Highbridge Subscribers”) pursuant to which the Highbridge Subscribers agreed to subscribe for 907,193 ordinary shares for an aggregate subscription price of $1 million and, subject to the GM Resolutions being passed, a further 2,765,901 ordinary shares for an aggregate subscription price of $3 million; |
| • | a subscription agreement between Amryt and Raymond Stafford pursuant to which Raymond Stafford agreed to subscribe for 918,273 Amryt ordinary shares for an aggregate subscription price of $1 million; and |
| • | a subscription agreement between Amryt and Stonepine Capital, LP pursuant to which Stonepine Capital, LP agreed to subscribe for 459,136 Amryt ordinary shares for an aggregate subscription price of $500,000. |
Private Placement
| • | On December 8, 2020, we entered into securities purchase agreements with several institutional accredited investors for the private placement of 3,200,000 ADSs, at a purchase price of $12.50 per ADS, yielding gross proceeds of $40 million. |
| • | The private placement included new and existing investors including Stonepine Capital, LP, Aquilo Capital Management, LLC, Amati Global Investors, Athyrium Capital Management, LP and Highbridge Capital Management, among others. |
C. Interests of Experts and Counsel
Not applicable.
| Item 8. | Financial information |
| | A. | Consolidated Statements and Other Financial Information |
Please refer to “Item 18. Financial statements.”
We do not anticipate paying any dividends in the foreseeable future.
Legal Proceedings
Other than as outlined below, we are not subject to any material legal proceedings.
Investigations in Brazil
Federal prosecutors in Brazil are conducting an investigation to determine whether there have been violations of Brazilian laws related to the sales of Juxtapid in Brazil. In July 2016, the Ethics Council of Interfarma fined Aegerion’s subsidiary in Brazil (“Aegerion Brazil”) approximately $0.5 million for violations to its Code of Conduct, to which Aegerion Brazil is bound due to its affiliation with Interfarma. Also, the Board of Directors of Interfarma imposed an additional penalty of suspension of Aegerion Brazil’s membership, without suspension of Aegerion Brazil’s membership contribution, for a period of 180 days for Aegerion Brazil to demonstrate the implementation of effective measures to cease alleged irregular conduct, or exclusion of Aegerion Brazil’s membership in Interfarma if such measures are not implemented. Aegerion Brazil paid the fine of approximately $0.5 million during the third quarter of 2016. In March 2017, after the suspension period ended, Interfarma’s Board of Directors decided to reintegrate Aegerion Brazil, enabling it to participate regularly in Interfarma activities, subject to meeting certain obligations. In March 2019, the Board of Directors of Interfarma agreed that Aegerion Brazil had successfully met all of the requirements imposed by the association, and the investigation was closed.
In June 2017, the Federal Public Prosecutor of the City of São José dos Campos, State of São Paulo, in connection with its criminal investigation into former employees of Aegerion Brazil, requested that a Brazilian federal court provide federal investigators with access to the bank records of certain individuals and entities, including Aegerion Brazil, certain former Aegerion Brazil employees, a Brazilian patient association, and 462 Brazilian physicians. The Federal Trial Court Judge issued a decision on July 12, 2018 authorizing access to the banking records on the terms that the Federal Public Prosecutor of the City of São José dos Campos had requested. On July 16, 2018 Aegerion Brazil filed a motion for clarification of the decision that authorized the breach of the banking secrecy, which was denied by the Federal Court Judge. In 2018, a related investigation was opened by the federal investigators in the state of Brasília which focused on a number of cases that occurred in Brasília with alleged signs of irregularities. The Public Prosecutor in São José dos Campos continues to gather information in connection with this investigation.
At this time, the Company does not know whether the inquiry of the Public Prosecutor in São José dos Campos will result in the commencement of any formal proceeding, but if Aegerion’s activities in Brazil are found to violate any laws or governmental regulations, we may be subject to significant civil lawsuits to be filed by the Public Prosecution office, and administrative penalties imposed by Brazilian regulatory authorities and additional damages and fines. Under certain circumstances, we could be barred from further sales to federal and/or state governments in Brazil, including sales of Juxtapid and/or Myalept, due to penalties imposed by Brazilian regulatory authorities or through civil actions initiated by federal or state public prosecutors. Additionally, the SEC conducted inquiries with Aegerion concerning the investigations by Brazilian authorities and, in July 2019, the SEC concluded the investigation and no enforcement action was made against Aegerion. The Company cannot determine if a loss is probable as a result of the investigations and inquiry in Brazil and whether the outcome will have a material adverse effect on the Company’s business.
Claim for Refund in Italy
The Agenzia Italiana del Farmaco (“AIFA”), also known as the Italian Medicines Agency, is the public institution responsible for the regulation of pharmaceuticals in Italy. AIFA has claimed that Amryt Pharma Italy S.r.l. refund approximately €1.9 million related to sales of Lojuxta in Italy during 2018. AIFA alleges that it classified Lojuxta incorrectly as an “Orphan” product and thus claims the repayment of funds paid to Amryt Pharma Italy. AIFA have also made a claim regarding a refund due to them for sales of Lojuxta in 2019 of approximately €700,000 based on the same underlying misclassification of Lojuxta as an “Orphan” product as the 2018 claim. We adhere to the view that Lojuxta has all the characteristics to be considered an “Orphan” product, and we intend to defend against AIFA’s claims for a refund.
In April 2021, the Italian courts passed their first judgment on the issue of the 2018 refund with the courts deciding that no further payments are due by the companies involved in this judgment to AIFA. We expect that the dispute regarding Amryt will be reviewed by the Italian Courts in due course with the outcome likely to be the same as the judgment that was recently issued and that no refund for 2018 will be due by Amryt. Regarding the 2019 refund, we appealed this to the Italian courts and an initial determination to reject our appeal was made on April 22, 2022. A final court hearing is now set for October 2022 at which point a final decision will be issued.
B. Significant Changes
No significant change, other than as otherwise described in this annual report on Form 20-F, has occurred in our operations since the date of our consolidated financial statements included in this annual report on Form 20-F.
| Item 9. | The Offer and Listing |
A. Offer and Listing Details
Our ADSs have been listed on the Nasdaq Global Select Market (“Nasdaq”) under the symbol “AMYT” since July 8, 2020, where each ADS represents five Ordinary Shares.
B. Plan of Distribution
Not applicable.
C. Markets
See “—A. Offer and Listing Details.”
D. Selling Shareholders
Not applicable.
E. Dilution
Not applicable.
F. Expenses of the Issue
Not applicable.
| Item 10. | Additional information |
A. Share Capital
Not applicable.
B. Memorandum and Articles of Association
Pursuant to the instructions to Form 20-F, the information called for by this section of Item 10 is contained in our Registration Statement on Form F-1, as originally filed with the SEC on June 23, 2020, as subsequently amended, under the heading “Description of Share Capital and Certain Corporate Matters,” and is hereby incorporated by reference.
C. Material Contracts
For a description of our material contracts, please see “Item 4. Information on the Company— B. Business Overview—Material Contracts.”
D. Exchange Controls
There are no governmental laws, decrees, regulations or other legislation in the United Kingdom that may affect the import or export of capital, including the availability of cash and cash equivalents for use by us, or that may affect the remittance of dividends, interest, or other payments by us to non-resident holders of our ordinary shares or ADSs, other than withholding tax requirements. There is no limitation imposed by English law or in the Articles on the right of non-residents to hold or vote shares. However, where a holder has a registered address within the United Kingdom and has not notified us of an address within the United Kingdom at which notices may be given to him or her, such holder shall not be entitled to receive any notice.
E. Taxation
Material U.S. Federal Income Tax Considerations
The following describes the material U.S. federal income tax consequences relating to the acquisition, ownership and disposition of the ADSs. This section does not purport to be a comprehensive description of all of the U.S. federal income tax considerations that may be relevant to a particular person’s decision to acquire the ADSs. This discussion is based on the U.S. Internal Revenue Code of 1986, as amended (“Code”), and U.S. Treasury regulations promulgated thereunder (“Treasury Regulations”), as well as judicial and administrative interpretations thereof as in effect as of the date of this annual report. All of the foregoing authorities are subject to change or differing interpretations that could apply retroactively and could affect the tax consequences described below, and there can be no assurance that the U.S. Internal Revenue Service (“IRS”), or U.S. courts will agree with the tax consequences described in this section.
We undertake no obligation to publicly update or otherwise revise this section whether as a result of new Treasury Regulations, Code sections, judicial and administrative interpretations or otherwise. This section is also based in part upon the assumption that each obligation in the Deposit Agreement and any related agreement will be performed in accordance with its terms.
This description applies only to U.S. Holders (as defined below) that purchase ADSs representing our ordinary shares and hold such ADSs as capital assets within the meaning of Section 1221 of the Code (generally, property held for investment). This section does not address any U.S. federal estate and gift tax, alternative minimum tax or Medicare tax on net investment income consequences, or any U.S. state or local or non-U.S. tax consequences. This section also does not address the tax considerations that may be relevant to certain types of investors subject to special treatment under U.S. federal income tax laws, such as:
| • | banks and other financial institutions; |
| • | regulated investment companies or real estate investment trusts; |
| • | dealers or traders in securities or currencies that use a mark-to-market method of accounting; |
| • | tax exempt organizations, retirement plans, individual retirement accounts and other tax deferred accounts; |
| • | persons holding the ADSs as part of a straddle, hedging, conversion or integrated transaction for U.S. federal income tax purposes; |
| • | U.S. Holders whose functional currency is not the U.S. dollar; |
| • | any entity or arrangement classified as partnership for U.S. federal income tax purposes or investors therein; |
| • | persons who own or are deemed to own, directly or indirectly, 10% or more of the total combined voting power of all classes of our voting stock or 10% or more of the total value of shares of all classes of our stock; |
| • | persons subject to special tax accounting rules as a result of any item of gross income with respect to the ADSs being taken into account in an applicable financial statement; and |
| • | persons that held, directly, indirectly or by attributions, ownership interest in us prior to the effectiveness of the registration statement of which this annual report forms a part. |
THE DESCRIPTION OF U.S. FEDERAL INCOME TAX CONSEQUENCES SET OUT BELOW IS FOR GENERAL INFORMATION ONLY. PROSPECTIVE INVESTORS SHOULD CONSULT THEIR TAX ADVISORS REGARDING THE APPLICATION OF THE U.S. FEDERAL TAX RULES TO THEIR PARTICULAR CIRCUMSTANCES AS WELL AS THE STATE, LOCAL, NON-U.S. AND OTHER TAX CONSEQUENCES TO THEM OF THE ACQUISITION, OWNERSHIP AND DISPOSITION OF THE ADSs.
As used in this discussion, the term “U.S. Holder” means a beneficial owner of the ADSs that is for U.S. federal income tax purposes:
| • | a citizen or individual resident of the United States; |
| • | a corporation (or other entity treated as a corporation) created or organized in or under the laws of the United States, any state thereof or the District of Columbia; |
| • | an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or |
| • | trust that (1) is subject to the primary supervision of a court within the United States and the control of one or more U.S. persons for all substantial decisions of the trust or (2) has a valid election in effect under applicable Treasury Regulations to be treated as a U.S. person. |
The U.S. federal income tax treatment of a partner in an entity or arrangement treated as a partnership for U.S. federal income tax purposes that holds ADSs generally will depend on the status of the partner and the activities of the partnership. Partnerships considering an investment in the ADSs and partners in such partnerships should consult their tax advisors regarding the specific U.S. federal income tax consequences to them of the acquisition, ownership and disposition of the ADSs.
The discussion below assumes that the representations contained in the deposit agreement and any related agreement are true and that the obligations in such agreements will be complied with in accordance with their terms. The discussion below also assumes that we are not treated as a U.S. domestic corporation under Section 7874 of the Code, but are treated as a surrogate foreign corporation, as a result of the acquisition of Aegerion. See “Item 3. Key Information—D. Risk Factors—Risks Related to our Business, Financial Condition and Capital Requirements.”
ADSs
Taking into account the earlier assumptions, for U.S. federal income tax purposes, U.S. Holders of ADSs generally will be treated as the beneficial owners of the underlying shares represented by the ADSs and an exchange of ADSs for our shares generally will not be subject to U.S. federal income tax.
The U.S. Treasury Department and the IRS have expressed concerns that U.S. Holders of ADSs may be claiming foreign tax credits in situations where an intermediary in the chain of ownership between the holder of an ADS and the issuer of the security underlying the ADS has taken actions that are inconsistent with the U.S. Holder of the ADS being treated as the beneficial owner of the underlying security. Such actions (for example, a pre-release of an ADS by a depositary) also may be inconsistent with the claiming of the reduced rate of tax applicable to certain dividends received by non-corporate U.S. Holders of ADSs, including individual U.S. Holders. Accordingly, the availability of foreign tax credits or the reduced U.S. federal income tax rate for “qualified dividend income,” each discussed below, could be affected by actions taken by intermediaries in the chain of ownership between the holder of an ADS and us, if as a result of such actions the U.S. Holder of an ADS is not properly treated as the beneficial owner of the underlying share.
U.S. Holders should consult their own tax advisers concerning the U.S. federal, state, local and non-U.S. tax consequences of acquiring, owning and disposing of ADSs in their particular circumstances.
Dividends and Other Distributions
Subject to the passive foreign investment company (“PFIC”), rules discussed below, the gross amount of any distribution made by us to a U.S. Holder with respect to the ADSs (including the amount of any taxes withheld therefrom) generally will be included in such holder’s gross income as non-U.S. source dividend income in the year actually or constructively received by the depositary, but only to the extent that the distribution is paid out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). As a non-U.S. company, we do not maintain calculations of our earnings and profits under U.S. federal income tax principles. Therefore, it is expected that any distributions generally will be reported to U.S. Holders as dividends. Any dividends that we pay will not be eligible for the dividends-received deduction allowed to qualifying corporations under Section 243 of the Code.
We believe that we are treated as a surrogate foreign corporation. Accordingly, we do not believe that dividends paid in respect of the ADSs will be eligible to be taxed at favorable rates that otherwise are applicable to “qualified dividend income” received by non-corporate U.S. Holders if certain additional conditions are satisfied.
The amount of any distribution paid in a currency other than U.S. dollars will be included in a U.S. Holder’s income in an amount equal to the U.S. dollar value of such currency calculated by reference to the exchange rate in effect on the date the distribution is actually or constructively received by the depositary, regardless of whether the payment is in fact converted into U.S. dollars at that time. If the distribution is converted into U.S. dollars on the date of receipt, a U.S. Holder generally should not be required to recognize foreign currency gain or loss in respect of the distribution. A U.S. Holder may have foreign currency gain or loss if the distribution is converted into, or exchanged for, U.S. dollars after the date of receipt.
Any dividends we pay to U.S. Holders generally will constitute non-U.S. source “passive category” income for U.S. foreign tax credit limitation purposes. However, if 50% or more of our stock is treated as held by U.S. persons, we will be treated as a “U.S.-owned foreign corporation.” In that case, dividends may be treated, for foreign tax credit limitation purposes, as income from sources outside the United States to the extent attributable to our non-U.S. source earnings and profits, and as income from sources within the United States to the extent attributable to our U.S. source earnings and profits. If any UK taxes are withheld with respect to dividends paid to a U.S. Holder with respect to the ADSs, subject to certain conditions and limitations provided in the Code and the applicable Treasury Regulations (including a minimum holding period requirement), such taxes may be treated as non-U.S. taxes eligible for credit against such U.S. holder’s U.S. federal income tax liability (to the extent not exceeding the withholding rate applicable to the U.S. Holder). In lieu of claiming a foreign tax credit, U.S. Holders may, at their election, deduct non-U.S. taxes, including any UK taxes withheld from dividends on the ADSs, in computing their taxable income, subject to generally applicable limitations under U.S. federal income tax law. An election to deduct non-U.S. taxes instead of claiming foreign tax credits applies to all non-U.S. taxes paid or accrued in the taxable year.
If a refund of the tax withheld is available under the laws of the United Kingdom or under an applicable income tax treaty, the amount of tax withheld that is refundable will not be eligible for such credit against a U.S. Holder’s U.S. federal income tax liability (and will not be eligible for the deduction against U.S. federal taxable income).
The rules relating to the determination of the U.S. foreign tax credit and the deduction of non-U.S. taxes are complex, and U.S. Holders should consult their tax advisors to determine whether and to what extent a credit or deduction may be available in their particular circumstances.
Taxable Dispositions of the ADSs
Subject to the PFIC rules discussed below, a U.S. Holder generally will recognize taxable gain or loss on any sale, exchange or other taxable disposition of an ADS in an amount equal to the difference between the sum of the fair market value of any property and the amount of cash received in such disposition and the holder’s tax basis in the ADS. The U.S. Holder’s tax basis in the ADSs generally will equal the cost of the ADSs to the U.S. Holder. The gain or loss generally will be capital gain or loss, and generally will be a long term capital gain or loss if the U.S. Holder has held the ADS for more than one year at the time of disposition. For certain non-corporate taxpayers (including individuals), long term capital gains are subject to tax at reduced rates. The deductibility of capital losses is subject to limitations.
Any gain or loss that a U.S. Holder recognizes on a sale or other taxable disposition of an ADS generally will be treated as U.S. source income or loss for U.S. foreign tax credit limitation purposes. U.S. Holders should consult their tax advisors regarding the proper treatment of any gain or loss in their particular circumstances, including the effects of any applicable income tax treaties.
Passive Foreign Investment Company Considerations
Based on the current and anticipated value of our assets and the nature and composition of our income and assets, we do not believe we were a PFIC for the taxable year ending December 31, 2021, and we do not expect to be a PFIC in the current taxable year ending December 31, 2022, or in the foreseeable future. However, the determination of PFIC status is based on an annual determination that cannot be made until the close of a taxable year, involves extensive factual investigation, including ascertaining the fair market value of all of our assets on a quarterly basis and the active or passive character of each item of income that we earn, and is subject to uncertainty in several respects. Changes in the nature or composition of our income or assets, the structure of our operation or the value of our assets may cause us to become a PFIC. The determination of the value of our assets may depend in part upon the value of our goodwill not reflected on our balance sheet (which may depend upon the market value of the ADSs from time to time, which may be volatile). Accordingly, we cannot assure you that we will not be a PFIC for our current taxable year ending December 31, 2022, or for any future taxable year. If we are a PFIC for any year during which a U.S. Holder holds the ADSs, we generally will continue to be treated as a PFIC with respect to that U.S. Holder for all succeeding years during which the U.S. Holder holds the ADSs, even if we cease to meet the threshold requirements for PFIC status in any particular year, unless the U.S. Holder has made a “deemed sale” election under the PFIC Rules when we cease to be a PFIC.
A non-U.S. corporation such as us will be treated as a PFIC for U.S. federal income tax purposes for any taxable year if, applying applicable look-through rules, either:
| • | at least 75% of its gross income for such year is “passive income” for purposes of the PFIC rules; or |
| • | at least 50% of the value of its assets (determined based on a quarterly average) during such year is attributable to assets that produce or are held for the production of passive income. |
For this purpose, passive income generally includes dividends, interest, royalties and rents other than certain royalties and rents derived in the active conduct of a trade or business and not derived from a related person. We will be treated as owning a proportionate share of the assets and earning a proportionate share of the income of any other corporation in which we own, directly or indirectly, more than 25% by value of the stock.
If we were a PFIC for any taxable year during which a U.S. Holder holds ADSs, then, unless such U.S. Holder makes a “mark-to-market” election or a “qualified electing fund” election (each as discussed below), such U.S. Holder generally would be subject to special adverse tax rules with respect to any “excess distribution” that it receives from us and any gain that it recognizes from a sale or other disposition, including, in certain circumstances, a pledge, of ADSs. For this purpose, distributions that a U.S. Holder receives in a taxable year that are greater than 125% of the average annual distributions that it received during the shorter of the three preceding taxable years or your holding period for the ADSs will be treated as an excess distribution. Under these rules:
| • | the excess distribution or recognized gain would be allocated ratably over the U.S. Holder’s holding period for the ADSs; |
| • | the amount of the excess distribution or recognized gain allocated to the taxable year of distribution or gain, and to any taxable years in the U.S. Holder’s holding period prior to the first taxable year in which we were treated as a PFIC, would be treated as ordinary income; and |
| • | the amount of the excess distribution or recognized gain allocated to each other taxable year would be subject to the highest tax rate in effect for individuals or corporations, as applicable, for each such year and the resulting tax will be subject to the interest charge generally applicable to underpayments of tax. |
If we were a PFIC for any taxable year during which a U.S. Holder holds ADSs and any of our non-U.S. subsidiaries or other corporate entities in which we own equity interests is also a PFIC, the U.S. Holder would be treated as owning a proportionate amount (by value) of the shares of each such non-U.S. entity classified as a PFIC, each such entity referred to as a lower-tier PFIC, for purposes of the application of these rules. U.S. Holders should consult their own tax advisor regarding the application of the PFIC rules to any of our lower-tier PFICs.
If we were a PFIC for any taxable year during which a U.S. Holder holds ADSs, then in lieu of being subject to the tax and interest-charge rules discussed above, the U.S. Holder may make an election to include gain on the ADSs as ordinary income under a mark-to-market method, provided that our ADSs constitute “marketable stock.” Marketable stock is stock that is regularly traded on a qualified exchange or other market, as defined in applicable Treasury Regulations. Shares or ADSs generally will be considered regularly traded during any calendar year during which they are traded, other than in de minimis quantities, on at least 15 days during each calendar quarter. Any trades that have as their principal purpose meeting this requirement will be disregarded. Our ADSs, but not our shares, are listed on the Nasdaq, which is a qualified exchange or other market for these purposes.
Consequently, if the ADSs are regularly traded, we expect that the mark-to-market election would be available to U.S. Holders of ADSs if we were to become a PFIC, but no assurances are given in this regard.
Because a mark-to-market election cannot be made for any lower-tier PFICs that we may own (unless the shares in such lower-tier PFIC are themselves treated as marketable stock), if we were a PFIC for any taxable year, a U.S. Holder that makes the mark-to-market election may continue to be subject to the tax and interest charges under the general PFIC rules with respect to such U.S. Holder’s indirect interest in any investments held by us that are treated as an equity interest in a PFIC for U.S. federal income tax purposes.
In certain circumstances, a shareholder in a PFIC may avoid the adverse tax and interest-charge regime described above by making a “qualified electing fund” election to include in income its share of the corporation’s income on a current basis. However, a U.S. Holder may make a qualified electing fund election with respect to the ADSs only if we agree to furnish such U.S. Holder annually with a PFIC annual information statement as specified in the applicable Treasury Regulations. We do not intend to provide such information, and thus we do not expect that a U.S. Holder will be able to make a qualified electing fund election.
If a U.S. Holder owns ADSs during any year in which we are a PFIC, such U.S. Holder (including, potentially, indirect holders) generally will be required to file an IRS Form 8621 with such holder’s U.S. federal income tax return for that year.
U.S. Holders should consult their own tax advisors regarding the application of the PFIC rules to their ownership of the ADSs.
Information Reporting and Backup Withholding
Dividend payments with respect to the ADSs and proceeds from a sale, exchange, redemption or other taxable disposition of the ADSs made within the United States or through certain U.S. related financial intermediaries may be subject to information reporting to the IRS and possible U.S. backup withholding. Backup withholding will not apply, however, to a U.S. Holder that furnishes a correct taxpayer identification number and makes any other required certification on IRS Form W-9 or that is otherwise exempt from backup withholding. U.S. Holders of the ADSs should consult their tax advisors regarding the application of the U.S. information reporting and backup withholding rules.
Backup withholding is not an additional tax. Amounts withheld as backup withholding may be credited against such U.S. Holder’s U.S. federal income tax liability, and such holder may obtain a refund of any excess amounts withheld under the backup withholding rules by filing an appropriate claim for refund with the IRS and furnishing any required information in a timely manner.
Certain U.S. Holders may be required to comply with certain reporting requirements relating to the ADSs, including filing IRS Form 8938, with respect to the holding of certain foreign financial assets, including stock of foreign issuers (such as us), either directly or through certain foreign financial institutions, if the aggregate value of all such assets exceeds $50,000 on the last day of the tax year or $75,000 at any time during the tax year. U.S. Holders who fail to report the required information could be subject to substantial penalties. U.S. Holders should consult their own tax advisors regarding the application of these rules to their ownership of the ADSs.
THE DISCUSSION ABOVE IS A GENERAL SUMMARY. IT DOES NOT COVER ALL TAX MATTERS THAT MAY BE IMPORTANT TO YOU. PROSPECTIVE INVESTORS SHOULD CONSULT THEIR TAX ADVISORS REGARDING THE APPLICATION OF THE U.S. FEDERAL TAX RULES TO THEIR PARTICULAR CIRCUMSTANCES AS WELL AS THE STATE, LOCAL, NON-U.S. AND OTHER TAX CONSEQUENCES TO THEM OF THE ACQUISITION, OWNERSHIP AND DISPOSITION OF THE ADSs.
UK Tax Considerations
The following is a general summary of certain UK tax considerations relating to the ownership and disposal of the ADSs and does not address all possible tax consequences relating to an investment in the ADSs. It is based on current UK tax law and generally published HM Revenue & Customs (“HMRC”) practice as at the date of this annual report supplement, both of which are subject to change, possibly with retrospective effect.
Save as provided otherwise, this summary applies only to persons who are resident (and, in the case of individuals, domiciled) in the United Kingdom for tax purposes and who are not resident for tax purposes in any other jurisdiction and do not have a permanent establishment or fixed base in any other jurisdiction with which the holding of ADSs is connected (“UK Holders”). Persons (a) who are not resident (or, if resident are not domiciled) in the United Kingdom for tax purposes, including those individuals and companies who trade in the United Kingdom through a branch, agency or permanent establishment in the United Kingdom to which the ADSs are attributable, or (b) who are resident or otherwise subject to tax in a jurisdiction outside the United Kingdom, are recommended to seek the advice of professional advisers in relation to their taxation obligations.
This summary is for general information only and is not intended to be, nor should it be considered to be, legal or tax advice to any investor. It does not address all of the tax considerations that may be relevant to specific investors in light of their particular circumstances or to investors subject to special treatment under UK tax law. In particular, this summary:
| • | only applies to the absolute beneficial owners of the ADSs and any dividends paid in respect of the ordinary shares represented by the ADSs where the dividends are regarded for UK tax purposes as that person’s own income (and not the income of some other person); and |
| • | (a) only addresses the principal UK tax consequences for investors who hold the ADSs as capital assets/investments, (b) does not address the tax consequences that may be relevant to certain special classes of investor such as dealers, brokers or traders in shares or securities and other persons who hold the ADSs otherwise than as an investment, (c) does not address the tax consequences for holders that are financial institutions, insurance companies, collective investment schemes, pension schemes, charities or tax-exempt organizations, (d) assumes that the holder is not an officer or employee of the Company (or of any related company) and has not (and is not deemed to have) acquired the ADSs by reason of an office or employment, and (e) assumes that the holder does not control or hold (and is not deemed to control or hold), either alone or together with one or more associated or connected persons, directly or indirectly (including through the holding of the ADSs), an interest of 10% or more in the issued share capital (or in any class thereof), voting power, rights to profits or capital of the Company, and is not otherwise connected with the Company. |
This summary further assumes that a holder of ADSs will be treated as the beneficial owner of the underlying ordinary shares for UK direct tax purposes.
POTENTIAL INVESTORS IN THE ADSs SHOULD SATISFY THEMSELVES PRIOR TO INVESTING AS TO THE OVERALL TAX CONSEQUENCES, INCLUDING, SPECIFICALLY, THE CONSEQUENCES UNDER UK TAX LAW AND HMRC PRACTICE OF THE ACQUISITION, OWNERSHIP AND DISPOSAL OF THE ADSs (OR UNDERLYING ORDINARY SHARES), IN THEIR OWN PARTICULAR CIRCUMSTANCES BY CONSULTING THEIR OWN TAX ADVISERS.
Taxation of dividends
Withholding Tax
Dividend payments in respect of the ordinary shares represented by the ADSs may be made without withholding or deduction for or on account of UK tax.
Income Tax
Dividends received by individual UK Holders will be subject to UK income tax on the amount of the dividend paid.
The first £2,000 of dividend income received by an individual UK Holder in a tax year will be subject to a nil rate of tax regardless of the amount of the individual’s other taxable income.
For the UK tax year 2021/2022, dividend income in excess of the £2,000 to which the nil rate of tax applies will be taxed at the rate of 7.5% to the extent that the dividend, when treated as the top slice of the relevant UK Holder’s income, does not exceed the basic rate income tax limit, at the rate of 32.5% to the extent that the dividend, when treated as the top slice of the relevant UK Holder’s income, exceeds the basic rate income tax limit but does not exceed the higher rate income tax limit, and at the rate of 38.1% to the extent that the dividend, when treated as the top slice of the relevant UK Holder’s income, exceeds the higher rate income tax limit.
An individual holder of ADSs who is not a UK Holder will not be chargeable to UK income tax on dividends paid by the Company, unless such holder carries on (whether solely or in partnership) a trade, profession or vocation in the United Kingdom through a branch or agency in the United Kingdom to which the ADSs (or underlying ordinary shares) are attributable. In these circumstances, such holder may, depending on his or her individual circumstances, be chargeable to UK income tax on dividends received from the Company.
Corporation Tax
A UK Holder within the charge to UK corporation tax may be entitled to exemption from UK corporation tax in respect of dividend payments. If the conditions for the exemption are not satisfied, or such UK Holder elects for an otherwise exempt dividend to be taxable, UK corporation tax will be chargeable on the gross amount of any dividends (the current rate of which is 19%). If potential investors are in any doubt as to their position, they should consult their own professional advisers.
A corporate holder of ADSs that is not a UK Holder will not be subject to UK corporation tax on dividends received from the Company, unless it carries on a trade in the United Kingdom through a permanent establishment to which the ADSs (or underlying ordinary shares) are attributable. In these circumstances, such holder may, depending on its individual circumstances and if the exemption from UK corporation tax discussed above does not apply, be chargeable to UK corporation tax on dividends received from the company.
Taxation of disposals
UK Holders
A disposal or deemed disposal of ADSs by an individual UK Holder may, depending on his or her individual circumstances, give rise to a chargeable gain or to an allowable loss for the purpose of UK capital gains tax. The principal factors that will determine the capital gains tax position on a disposal of ADSs are the extent to which the holder realizes any other capital gains in the tax year in which the disposal is made, the extent to which the holder has incurred capital losses in that or any earlier tax year and the level of the annual allowance of tax-free gains in that tax year (“annual exemption”). The annual exemption for individuals for the 2021/2022 tax year is £12,300. If, after all allowable deductions, an individual UK Holder’s total taxable income (which, for the avoidance of doubt, includes any dividend income within the £2,000 nil rate band described above) for the year exceeds the basic rate income tax limit, a taxable capital gain accruing on a disposal of ADSs will be taxed at 20%. If, after all allowable deductions, an individual UK Holder’s total taxable income for the year does not exceed the basic rate income tax limit, and assuming the individual does not have any other taxable capital gains in the tax year that would use up the remaining basic rate allowance, a taxable capital gain accruing on a disposal of ADSs will be taxed at 10% on an amount that, when treated as the top slice of the relevant UK Holder’s income/gains, does not exceed the basic rate income tax and at 20% on the remainder.
A disposal of ADSs by a corporate UK Holder may give rise to a chargeable gain or an allowable loss for the purposes of UK corporation tax.
Any gains or losses in respect of currency fluctuations over the period of holding the ADSs would also be brought into account on the disposal.
Non-UK Holders
An individual holder who is not a UK Holder will not be liable to UK capital gains tax on capital gains realized on the disposal of his or her ADSs unless such holder carries on (whether solely or in partnership) a trade, profession or vocation in the United Kingdom through a branch or agency in the United Kingdom to which the ADSs (or underlying ordinary shares) are attributable. In these circumstances, such holder may, depending on his or her individual circumstances, be chargeable to UK capital gains tax on chargeable gains arising from a disposal of his or her ADSs. Furthermore, an individual holder of ADSs who has ceased to be resident for tax purposes in the United Kingdom for a period of less than five years and who disposes of ADSs during that period may be liable on his or her return to the United Kingdom to UK tax on any capital gain realized (subject to any available exemption or relief).
A corporate holder of ADSs that is not a UK Holder will not be liable for UK corporation tax on chargeable gains realized on the disposal of its ADSs unless it carries on a trade in the United Kingdom through a permanent establishment to which the ADSs (or underlying ordinary shares) are attributable. In these circumstances, a disposal of ADSs by such holder may give rise to a chargeable gain or an allowable loss for the purposes of UK corporation tax.
Stamp Duty and Stamp Duty Reserve Tax
Issue of Ordinary Shares
No UK stamp duty or stamp duty reserve tax (“SDRT”) is payable on the issue of the underlying ordinary shares in the company.
Transfer of Ordinary Shares
Stamp duty or SDRT will generally apply to transfers of, or agreements to transfer, ordinary shares. Where applicable, the purchaser normally pays the stamp duty or SDRT.
Shareholders who elect to deposit holdings of Ordinary Shares for delivery of Nasdaq-listed ADSs will generally incur a stamp duty, or SDRT, charge at the rate of 1.5 per cent. of the market value of the Ordinary Shares being deposited.
Issue and Transfer of ADSs
No UK stamp duty or SDRT is payable on the issuance of the ADSs. No UK stamp duty should be required to be paid on any written instrument transferring an ADS or on any written agreement to transfer an ADS, also noting that based on current HMRC published practice, a depositary receipt is not regarded as “stock” or a “marketable security” for UK stamp duty purposes.
No SDRT will be payable on an agreement to transfer an ADS, as a depositary receipt (and any interest in a depositary receipt) is not treated as a “chargeable security” for the purposes of SDRT.
F. Dividends and Paying Agents
Not applicable.
G. Statement by Experts
Not applicable.
H. Documents on Display
Documents concerning us that are referred to herein may be inspected at our principal executive headquarters at 45 Mespil Road, Dublin 4, Ireland. Electronic copies of these documents, including the related exhibits and schedules, and any documents we file with the SEC without charge at the SEC’s website, www.sec.gov.
Our internet address is www.amrytpharma.com. Investors and others should note that we announce material information to investors through the investor relations page on our website, SEC filings, press releases, public conference calls and webcasts. We encourage investors, the media and others to review the information we post from time to time on our website. The information contained on or connected to our website is not incorporated by reference into this Annual Report on Form 20-F and should not be considered part of this or any other report filed with the SEC.
I. Subsidiary Information
Not applicable.
| Item 11. | Quantitative and Qualitative Disclosures About Market Risk |
Our operations expose us to some financial risks arising from our use of financial instruments, the most significant ones being liquidity, market risk and credit risk. The Board is responsible for our risk management policies and while retaining responsibility for them it has delegated the authority for designing and operating processes that ensure the effective implementation of the objectives and policies to our finance function. See “Note 24: Fair value measurement and financial risk management” in our Consolidated Financial Statements for the year ended December 31, 2021.
| Item 12. | Description of Securities Other than Equity Securities |
A. Debt Securities
Not applicable.
B. Warrants and Rights
Not applicable.
C. Other Securities
Not applicable.
D. American Depositary Shares
Citibank, N.A. (“Citibank”) acts as the depositary for our American Depository Shares (“ADSs”), each of which represents five ordinary shares, pursuant to a deposit agreement.
A summary description of our ADSs is contained in our Registration Statement on F-1, as originally filed with the SEC on June 23, 2020, as subsequently amended, under the heading “Description of American Depositary Shares”.
Fees and Charges
ADS holders are required to pay the following fees under the terms of the deposit agreement:
| Service | | Fees |
| Issuance of ADSs (e.g., an issuance of ADS upon a deposit of ordinary shares, upon a change in the ADS(s)-to-ordinary shares ratio or for any other reason), excluding ADS issuances as a result of distributions of ordinary shares | | Up to U.S. 5¢ per ADS issued |
| | | |
| Cancellation of ADSs (e.g., a cancellation of ADSs for delivery of deposited property, upon a change in the ADS(s)-to-ordinary shares ratio or for any other reason) | | Up to U.S. 5¢ per ADS cancelled |
| | | |
| Distribution of cash dividends or other cash distributions (e.g., upon a sale of rights and other entitlements) | | Up to U.S. 5¢ per ADS held |
| | | |
| Distribution of ADSs pursuant to (i) stock dividends or other free stock distributions, or (ii) exercise of rights to purchase additional ADSs | | Up to U.S. 5¢ per ADS held |
| | | |
| Distribution of securities other than ADSs or rights to purchase additional ADSs (e.g., upon a spin-off) | | Up to U.S. 5¢ per ADS held |
| | | |
| ADS services | | Up to U.S. 5¢ per ADS held on the applicable record date(s) established by the depositary bank |
| | | |
Registration of ADS transfers (e.g., upon a registration of the transfer of registered ownership of ADSs, upon a transfer of ADSs into DTC and vice versa, or for any other reason) | | Up to U.S. 5¢ per ADS (or fraction thereof) transferred |
| | | |
Conversion of ADSs of one series for ADSs of another series (e.g., upon conversion of Partial Entitlement ADSs for Full Entitlement ADSs, or upon conversion of Restricted ADSs (each as defined in the deposit agreement) into freely transferable ADSs, and vice versa). | | Up to U.S. 5¢ per ADS (or fraction thereof) converted |
ADS holders are also responsible to pay certain charges such as:
| • | taxes (including applicable interest and penalties) and other governmental charges; |
| • | the registration fees as may from time to time be in effect for the registration of ordinary shares on the share register and applicable to transfers of ordinary shares to or from the name of the custodian, the depositary bank or any nominees upon the making of deposits and withdrawals, respectively; |
| • | certain cable, telex and facsimile transmission and delivery expenses; |
| • | the fees, expenses, spreads, taxes and other charges of the depositary bank and/or service providers (which may be a division, branch or affiliate of the depositary bank) in the conversion of foreign currency; |
| • | the reasonable and customary out-of-pocket expenses incurred by the depositary bank in connection with compliance with exchange control regulations and other regulatory requirements applicable to ordinary shares, ADSs and ADRs; and |
| • | the fees, charges, costs and expenses incurred by the depositary bank, the custodian, or any nominee in connection with the ADR program. |
ADS fees and charges for (i) the issuance of ADSs and (ii) the cancellation of ADSs are charged to the person for whom the ADSs are issued (in the case of ADS issuances) and to the person for whom ADSs are cancelled (in the case of ADS cancellations). In the case of ADSs issued by the depositary into DTC, the ADS issuance and cancellation fees and charges may be deducted from distributions made through DTC, and may be charged to the DTC participant(s) receiving the ADSs being issued or the DTC participant(s) holding the ADSs being cancelled, as the case may be, on behalf of the beneficial owner(s) and will be charged by the DTC participant(s) to the account of the applicable beneficial owner(s) in accordance with the procedures and practices of the DTC participants as in effect at the time. ADS fees and charges in respect of distributions and the ADS service fee are charged to the holders as of the applicable ADS record date. In the case of distributions of cash, the amount of the applicable ADS fees and charges is deducted from the funds being distributed. In the case of (i) distributions other than cash and (ii) the ADS service fee, holders as of the ADS record date will be invoiced for the amount of the ADS fees and charges and such ADS fees and charges may be deducted from distributions made to holders of ADSs. For ADSs held through DTC, the ADS fees and charges for distributions other than cash and the ADS service fee may be deducted from distributions made through DTC, and may be charged to the DTC participants in accordance with the procedures and practices prescribed by DTC and the DTC participants in turn charge the amount of such ADS fees and charges to the beneficial owners for whom they hold ADSs. In the case of (i) registration of ADS transfers, the ADS transfer fee will be payable by the ADS Holder whose ADSs are being transferred or by the person to whom the ADSs are transferred, and (ii) conversion of ADSs of one series for ADSs of another series, the ADS conversion fee will be payable by the Holder whose ADSs are converted or by the person to whom the converted ADSs are delivered.
In the event of refusal to pay the depositary fees, the depositary may, under the terms of the deposit agreement, refuse the requested service until payment is received or may set off the amount of the depositary fees from any distribution to be made to the ADS holder. Note that the fees and charges you may be required to pay may vary over time and may be changed by us and by the depositary. You will receive prior notice of such changes. The depositary may reimburse us for certain expenses we incur in respect of the ADR program, by making available a portion of the ADS fees charged in respect of the ADR program or otherwise, upon such terms and conditions as we and the depositary agree from time to time.
PART II
| Item 13. | Defaults, Dividend Arrearages and Delinquencies |
None.
| Item 14. | Material Modifications to the Rights of Security Holders and Use of Proceeds |
Not applicable.
| Item 15. | Controls and Procedures |
A. Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures pursuant to Rules 13a-15(d) of the Exchange Act as of December 31, 2021. Based on such evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that, as of December 31, 2021, our disclosure controls and procedures were effective in recording, processing, summarizing and reporting, on a timely basis, information required to be included in periodic filings under the Exchange Act and that such information is accumulated and communicated to management, including our Chief Executive Officer and Chief Financial Officer, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure.
In designing and evaluating our disclosure controls and procedures, our management, with the participation of the Chief Executive Officer and Chief Financial Officer, recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and our management necessarily was required to apply its judgement in evaluating the cost-benefit relationship of possible controls and procedures. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the Company have been detected.
B. Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rule 13a-15(f) under the Exchange Act.
Our internal control over financial reporting includes policies and procedures that pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect transactions and dispositions of assets; provide reasonable assurances that transactions are recorded as necessary to permit preparation of the financial statements in accordance with IFRS and that receipts and expenditures are being made only in accordance with the authorization of management and the directors of Amryt; and provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of Amryt’s assets that could have a material effect on our financial statements. Because of its inherent limitations, internal control over financial reporting may not prevent or detect all misstatements.
Management has assessed the effectiveness of internal control over financial reporting based on criteria established in the 2013 Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). Based on this assessment, management has concluded that the Group’s internal control over financial reporting was effective as of December 31, 2021.
C. Attestation Report of the Registered Public Accounting Firm
Since Amryt Pharma is an emerging growth company as defined in the JOBS Act, our auditor, Grant Thornton, an independent registered public accounting firm, is not required to issue an attestation report on our internal control over financial reporting as of December 31, 2021.
D. Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the period covered by this annual report that have materially affected, or that are reasonably likely to materially affect, our internal control over financial reporting.
| Item 16A. | Audit Committee Financial Expert |
Our Board has determined that Mr. Stephen Wills qualifies as an “audit committee financial expert” as defined by applicable SEC rules and has the requisite financial sophistication as defined under applicable Nasdaq rules and regulations. The Board has determined that Mr. Stephen Wills satisfies the “independence” requirements under the corporate governance standards of the Nasdaq listing and the audit committee independence requirements set forth in Rule 10A-3 under the Exchange Act. The Audit Committee is governed by terms of reference that comply with the Nasdaq rules for a charter of such body. For more information see “Item 6. Directors, Senior Management and Employees—C. Board Practices—Audit Committee.”
We have adopted a Code of Ethics that is applicable to all of our employees, executive officers and directors. A Committee of the Board will be responsible for overseeing the Code of Ethics and will be required to approve any waivers of the Code of Ethics for employees, executive officers and directors. The Code of Ethics can be found on our website at www.amrytpharma.com.
| Item 16C. | Principal Accountant Fees and Services |
Fees Billed by Independent Public Accountants
The following table provides information regarding fees paid by us to Grant Thornton for all services, for the years ended December 31, 2021 and 2020:
| | | December 31, | |
| | | 2021 | | | 2020 | |
| | | US$’000 | | | US$’000 | |
| Audit fees | | | 893 | | | | 814 | |
| Audit-related fees | | | 92 | | | | 44 | |
| Tax fees | | | — | | | | — | |
| All other fees | | | — | | | | — | |
| Total fees | | | 985 | | | | 858 | |
Audit services include audit of our consolidated financial statements, review of our interim financial statements and statutory audits. Audit related services are for assurance and related services performed by the independent auditor, including due diligence services and grant claim reviews.
Policy on Pre-approval of Audit and Non-Audit Services of Independent Auditors
Our Audit Committee has adopted policies and procedures for the pre-approval of audit and non-audit services rendered by our independent public accountants, Grant Thornton. The policy generally pre-approves certain specific services in the categories of audit services, audit-related services, and tax services up to specified amounts, and sets requirements for specific case-by-case pre-approval of discrete projects, those which may have a material effect on our operations or services over certain amounts.
Pre-approval may be given as part of the Audit Committee’s approval of the scope of the engagement of our independent auditor or on an individual basis. The pre-approval of services may be delegated to one or more of the Audit Committee’s members, but the decision must be presented to the full Audit Committee at its next scheduled meeting. The policy prohibits retention of the independent public accountants to perform the prohibited non-audit functions defined in Section 201 of the Sarbanes-Oxley Act or the rules of the SEC, and also considers whether proposed services are compatible with the independence of the public accountants.
During the year ended December 31, 2021, all audit and non-audit services provided by our independent public accountants were pre-approved in accordance with such policies and procedures.
| Item 16D. | Exemptions from the Listing Standards for Audit Committees |
Not applicable.
| Item 16E. | Purchases of Equity Securities by the Issuer and Affiliated Purchasers |
Following the Company’s delisting from the AIM market of the London Stock Exchange, which became effective on January 11, 2022, the Company may only repurchase its Ordinary Shares (including Ordinary Shares represented by ADSs) in accordance with the Companies Act procedures for “off-market purchases”. As such, these repurchases may only be made pursuant to a form of share repurchase contract that was approved by Shareholders on March 2, 2022. In addition, the Company may only conduct share repurchases with counterparties which were approved by Shareholders on March 2, 2022. These approvals are valid for five years. Approval of the forms of contract and counterparties is not an approval of the amount or timing of any repurchase activity. There can be no assurance as to whether the Company will repurchase any of its Ordinary Shares or as to the amount of any such repurchases or the prices at which such repurchases may be made.
No such repurchases have been made to date.
| Item 16F. | Change in Registrant’s Certifying Accountant |
Not applicable.
| Item 16G. | Corporate Governance |
As Amryt Pharma is a foreign private issuer, it is not required to comply with all of the corporate governance requirements set forth in NASDAQ Rule 5600 as they apply to U.S. domestic companies. Our corporate governance measures differ in the following significant way: we have not appointed an independent nominations committee or adopted a board resolution addressing the nominations process. At present, the Board as a whole address the nominations process.
| Item 16H. | Mine Safety Disclosure |
Not applicable.
| Item 16I. | Disclosure Regarding Foreign Jurisdictions that Prevent Inspections |
Not applicable.