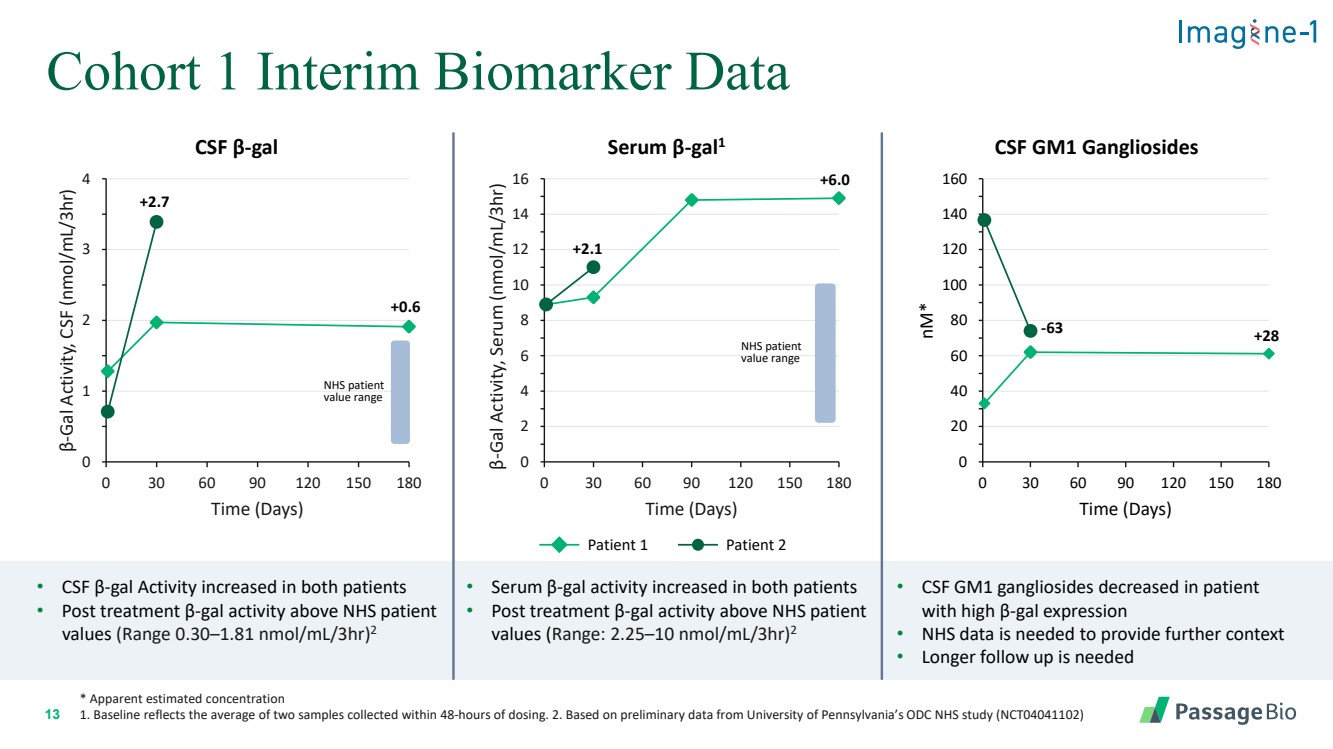
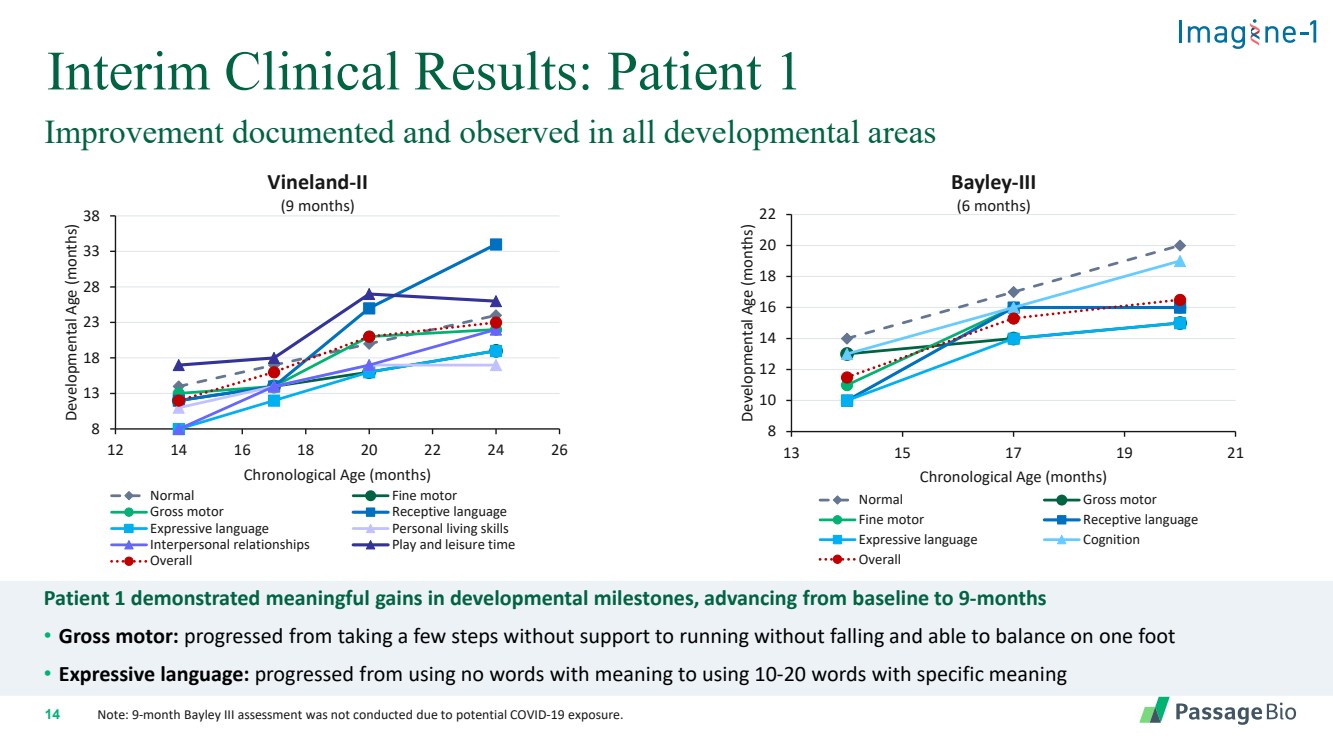
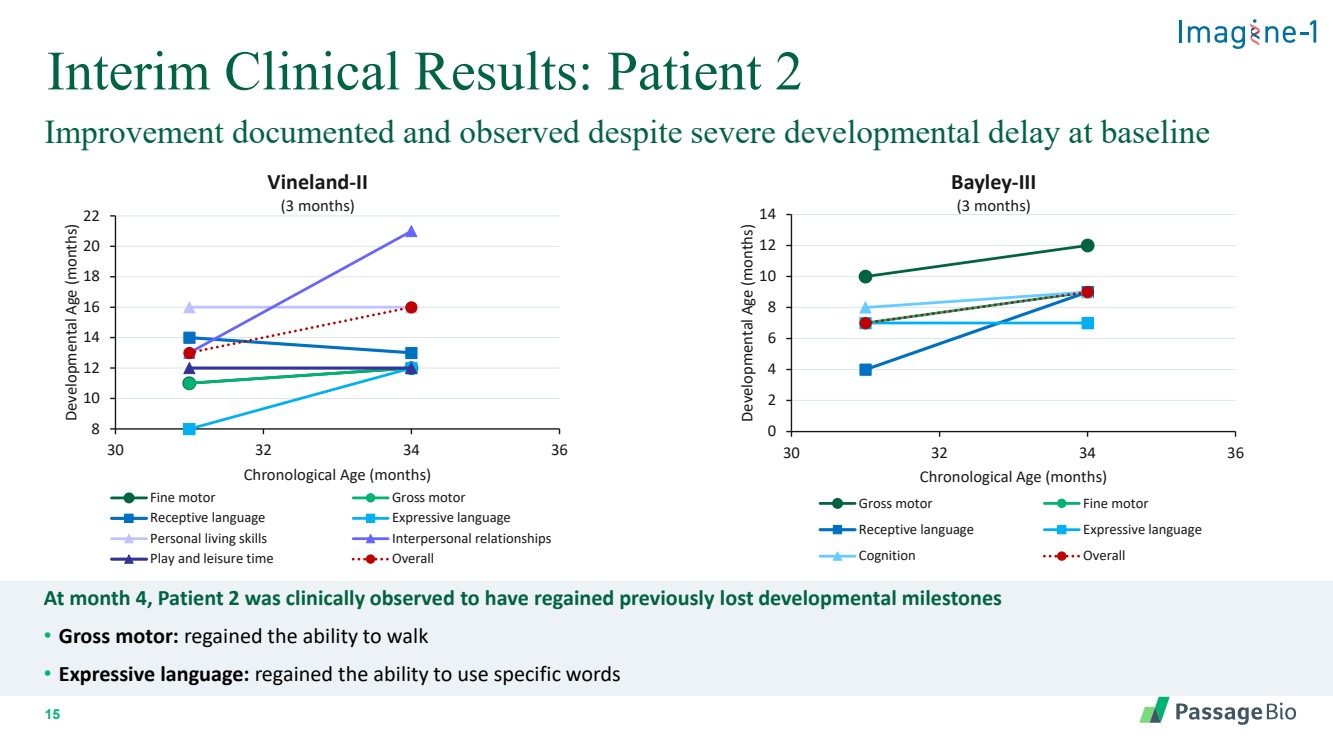
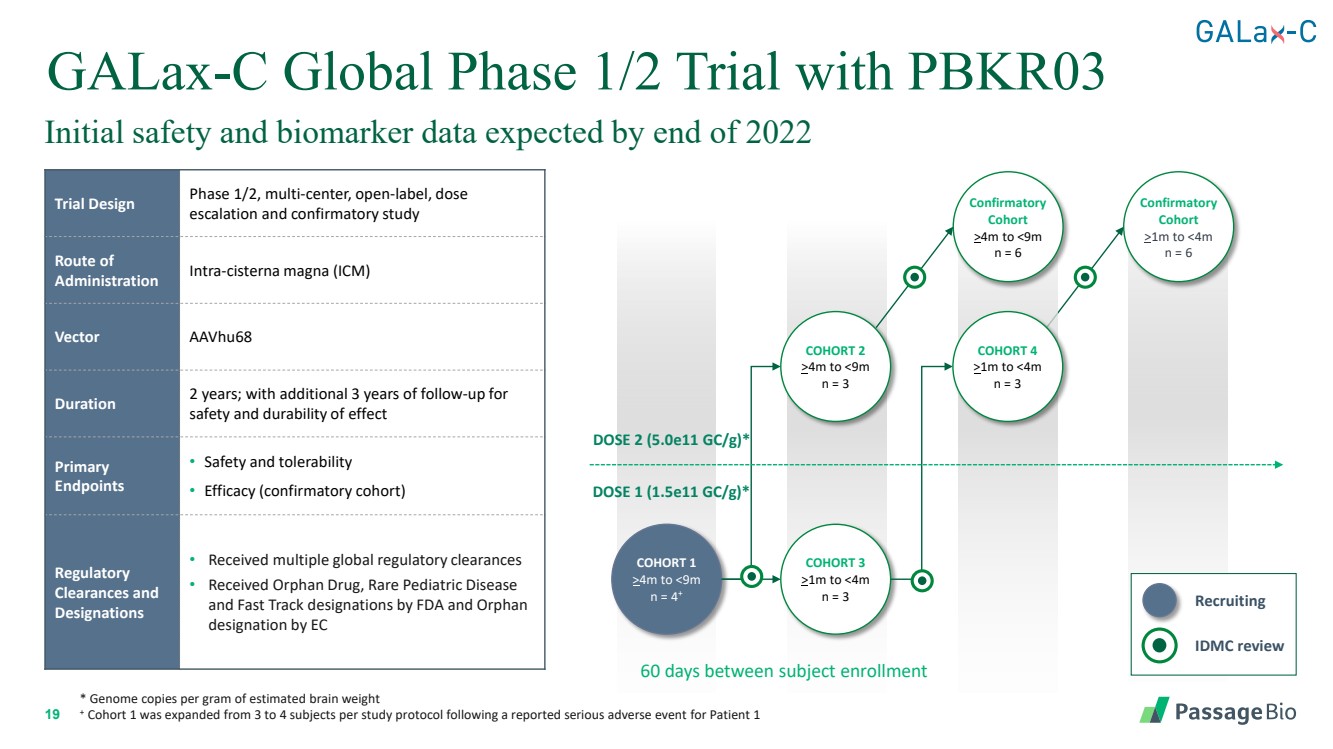
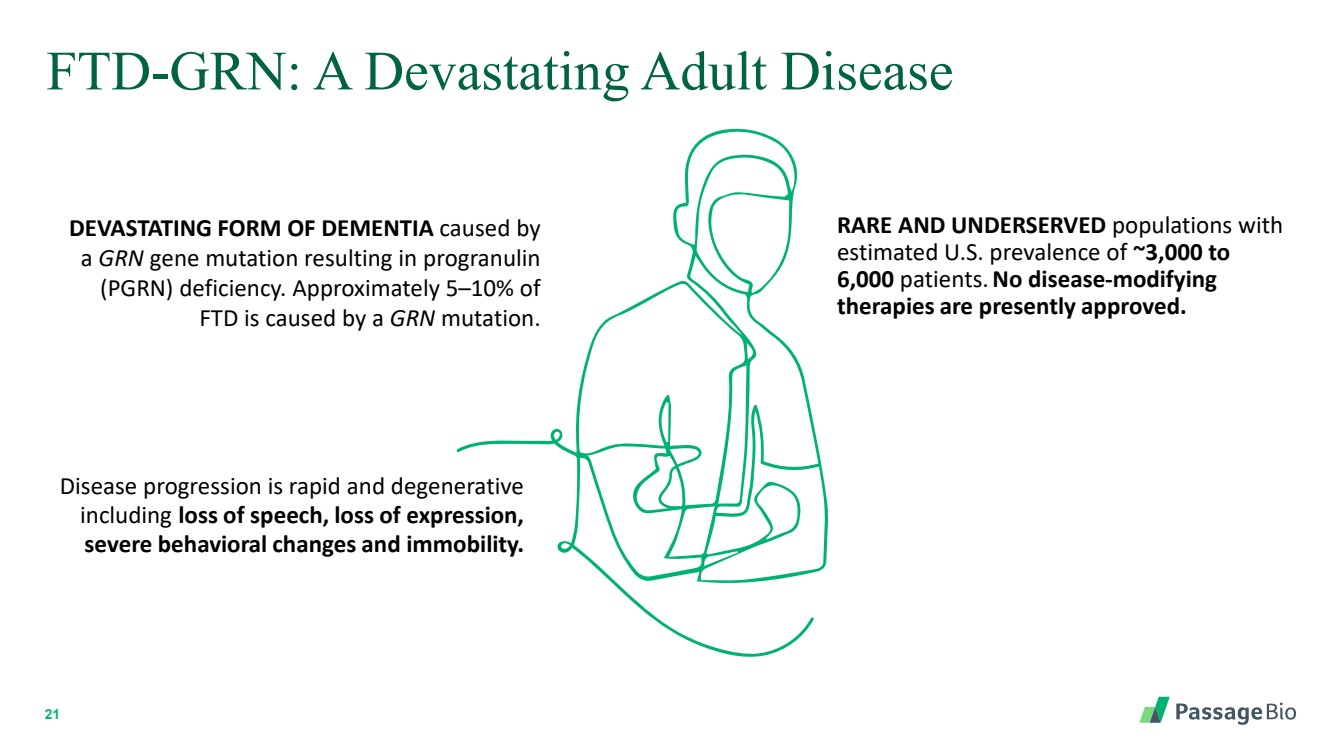
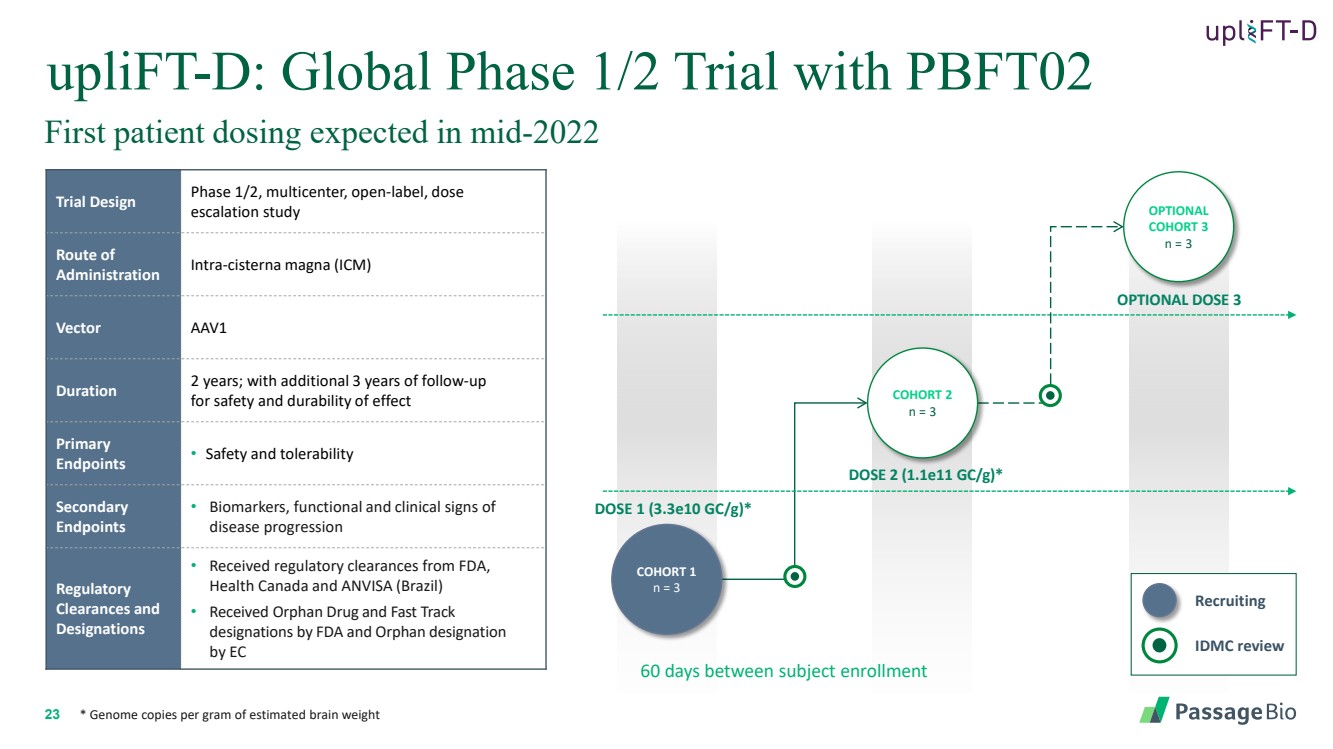
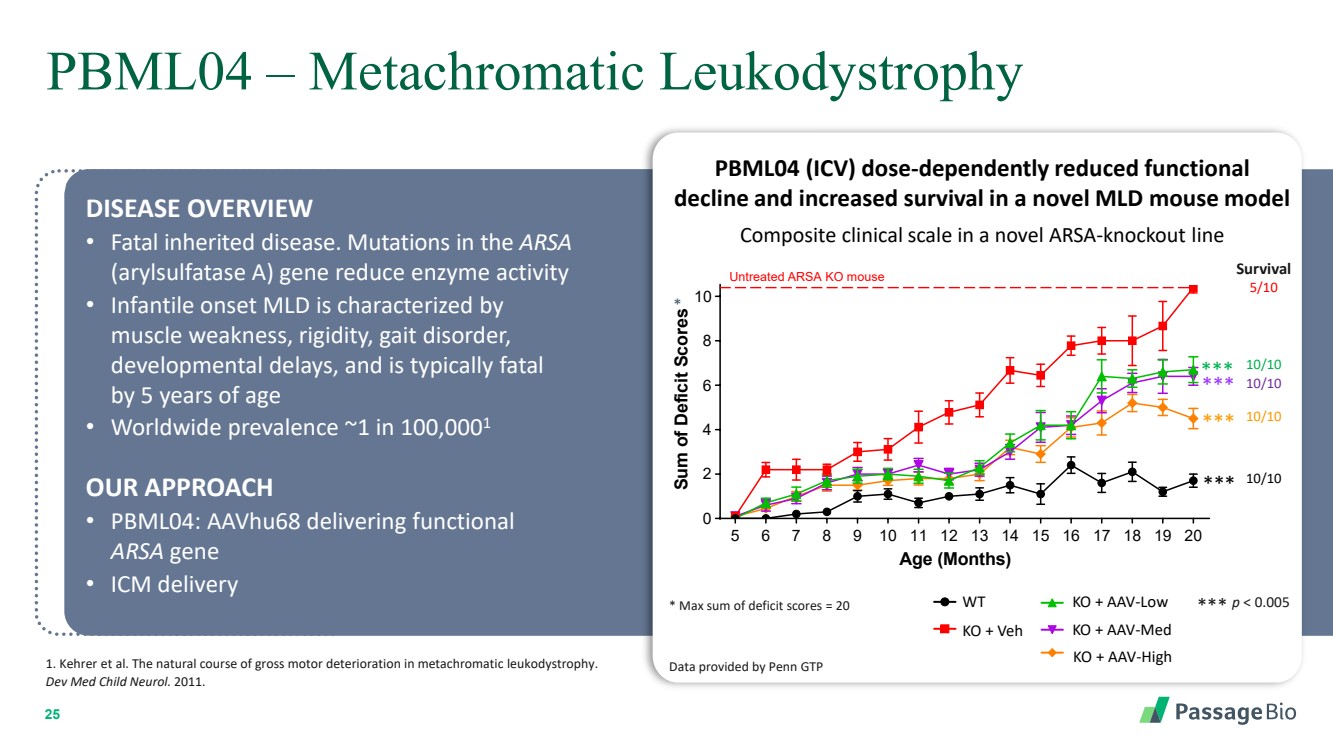
| 2 Forward-Looking StatementThis presentation includes “forward-looking statements” within the meaning of, and made pursuant to the safe harbor provisions of, the Private Securities Litigation Reform Act of 1995, including, but not limited to: our expectation about timing and execution of anticipated milestones, including our planned initiation of clinical trials and the availability of clinical data from such trials; our expectations about our collaborators’ and partners’ ability to execute key initiatives; our expectations about our manufacturing plans and strategies; estimates regarding our cash forecasts; the expected impact of the COVID-19 pandemic on our operations; and the ability of our lead product candidates to treat their respective target CNS disorders. These forward-lookingstatements may be accompanied by such words as “aim,” “anticipate,” “believe,” “could,” “estimate,” “expect,” “forecast,” “goal,” “intend,” “may,” “might,”“plan,” “potential,” “possible,” “will,” “would,” and other words and terms of similar meaning. These statements involve risks and uncertainties that could cause actual results to differ materially from those reflected in such statements, including: our ability to develop, obtain regulatory approval for and commercialize PBGM01, PBFT02, PBKR03 and future product candidates; the timing and results of preclinical studies and clinical trials; the risk that positive results in a preclinical study or clinical trial may not be replicated in subsequent trials or success in early stage clinical trials may not be predictive of results in later stage clinical trials; risks associated with clinical trials, including our ability to adequately manage clinical activities, unexpected concerns that may arise from additional data or analysis obtained during clinical trials, regulatory authorities may require additional information or further studies, or may fail to approve or may delay approval ofour drug candidates; the occurrence of adverse safety events; failure to protect and enforce our intellectual property, and other proprietary rights; failure to successfully execute or realize the anticipated benefits of our strategic and growth initiatives; risks relating to technology failures or breaches; our dependence on collaborators and other third parties for the development of product candidates and other aspects of our business, which are outside of our full control; risks associated with current and potential delays, work stoppages, or supply chain disruptions caused by the coronavirus pandemic; risks associated with current and potential future healthcare reforms; risks relating to attracting and retaining key personnel; failure to comply with legal and regulatory requirements; risks relating to access to capital and credit markets; and the other risks and uncertainties that are described in the Risk Factors section of our most recent filings with the U.S. Securities and Exchange Commission. These statements are based on our current beliefs and expectations and speak only as of the date of this presentation. We do not undertake any obligation to publicly update any forward-looking statements except as required by law. By attending or receiving this presentation you acknowledge that you are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date such statements are made; you will be solely responsible for your own assessment of the market and our market position; and that you will conduct your own analysis and besolely responsible for forming your own view of the potential future performance of Passage Bio. |