
July 23, 2020
Division of Corporation Finance
Securities and Exchange Commission
100 F Street, N.E.
Washington, D.C. 20549
| Re: | NuGenerex Immuno-Oncology, Inc. |
| Amendment No. 1 to Registration Statement on Form 10-12G | |
| Filed June 12, 2020 | |
| File No. 000-56153 |
Dear Staff:
On behalf of NuGenerex Immuno-Oncology, Inc. (the “Company”), we have set forth below responses to the comments of the staff (the “Staff”) of the Securities and Exchange Commission (the “SEC”) contained in its letter of June 7, 2020 with respect to Amendment No.1 to Registration Statement on Form 10-12G (the “Form 10”) filed by the Company on June 12, 2020 (File No. 000-56153). For your convenience, the text of the Staff’s comments is set forth below in bold, followed in each case by the Company’s responses. Please note that all references to page numbers in the responses are references to the page numbers in Amendment No. 2 to the Form 10 (the “Form 10/A2”) submitted concurrently with the submission of this letter in response to the Staff’s comments.
Amendment No. 1 to Registration Statement on Form 10-12G
Introductory Comment, page 3
1. Please revise to clarify that the registration statement is effective and clarify your reporting status.
The Company has made the requested revisions to its disclosure in the Introductory Comment on page 3 of the Form 10 A/2.
Item 1. Business
AE37 – Ii-Key/HER2/neu Hybrid Immunotherapeutic Vaccine, page 4
2. We note your disclosure in this section that you are currently developing AE37 for the treatment of cancer, including breast, bladder, prostate, and potentially other indications yet you state on page 13 that your Phase II clinical study using AE37 in combination with Keytruda for treatment of metastatic triple negative breast cancer is your only ongoing research and development project. Please revise or advise. In that regard, we note your response to our prior comment 4 that you are waiting to see whether Shenzhen is successful in achieving positive results in the prostate cancer development program before determining how to proceed. Accordingly, please revise to disclose, if true, that you are not in the process of developing AE37 for prostate cancer as stated on page 4.
The Company has made the requested revisions to its disclosure in the first paragraph of “Business Overview—AE37–Ii-Key/HER/2/neu Hybrid Immunotherapeutic Vaccine” in “Item 1. Business” on page 4 of the Form 10/A2.
3. We note your response to our prior comments 2 and 3. Please revise to disclose the term and the termination provisions for each of the Merck, NSABP and Shenzhen agreements. Please also indicate whether the royalty term under the Shenzhen agreement is the same as the term of the agreement.
The Company has included the following paragraphs on page 5 of the Form 10/A2 with respect to the Merck and NSABP agreements:
“The Collaboration Agreement terminates after the completion of the related phase II clinical trial. The Collaboration Agreement may also be terminated by any party for material breaches of the agreement by the other party, patient safety concerns, regulatory reasons or by Merck if it believes KEYTRUDA is being used in an unsafe manner.
The Clinical Trial Agreement terminates upon the completion of the obligations under such agreement. The Clinical Trial Agreement may be terminated by (i) any party if the authorization to conduct the phase II clinical trial is revoked by the FDA; if the human and/or toxicology results support termination; safety concerns, if the manufacture of a drug used in the phase II clinical trial has been exhausted or (ii) by NSABP if we fail to pay NSABP an undisputed amount under the Clinical Trial Agreement.”
The Company has included the following paragraphs on page 5 of the Form 10/A2 with respect to the Shenzhen agreement:
“The License Agreement terminates upon the later to occur of (x) the expiration of the last to expire licensed patent under the License Agreement and (y) the fifteenth (15th) anniversary of the first approved sale of a licensed product by Shenzhen in China (including Taiwan, Hong Kong and Macau). The License Agreement may also be terminated (i) by any party to the agreement generally upon the bankruptcy or insolvency of the other party or if the other party materially breaches the License Agreement or (ii) by Shenzhen any time after eighteen (18) months from the License Agreement’s effective date upon sixty (60) days’ notice to NGIO. Unless Shenzhen terminates the License Agreement pursuant to clause (y)(ii) above, or we terminate the agreement pursuant to clause (y)(i) above, Shenzhen may sell any licensed products existing at the time of termination for twelve (12) months after such termination; provided that such salse are subject to royalty payments under the License Agreement. No other royalties shall be payable after the termination of the License Agreement other than royalties that have accrued and are unpaid as od the date of termination.”
4. We note your response to our prior comment 8, but we do not see any disclosure concerning the failure to achieve the primary endpoint which prevented moving to a Phase III trial of AE37 in breast cancer. Please revise or advise.
The Company has made the requested revision by adding the following sentences at the end of the third paragraph under “Business Overview—AE37–Ii-Key/HER/2/neu Hybrid Immunotherapeutic Vaccine” in “Item 1. Business” on page 4 of the Form 10/A2:
“The Phase IIb Trial did not achieve the primary endpoint in the entire intent to treat study population, however, the Phase IIb Trial did provide evidence of a positive effect in the triple negative and low HER2 populations. Based upon those results we decided to pursue the further development of AE37 in combination with the newly approved immune checkpoint inhibitors, which enables us to move earlier in the treatment paradigm to evaluate the effect of AE37 on tumors rather than wait another 10 years for the completion of a Phase III registration trial.”
5. We note your disclosure that the primary and secondary endpoints were met in the 2006 and 2007 trials for AE37. Please provide the data from the trials that support these statements and disclose how many subjects were in the 2006 trial.
The Company has included in the Form 10/A2 (i) the peer reviewed publication that provides the data for the 2006 Phase I breast cancer trial as Exhibit 99.1; (ii) the peer reviewed publication that provides the data for the 2007 Phase I prostate cancer trial as Exhibit 99.2; (iii) the peer reviewed publication that provides the preliminary data for the Phase IIb Trial (as defined in the Form 10/A2) as Exhibit 99.3 and (iv) and the peer reviewed publication that provides the final data for the Phase IIb Trial, as Exhibit 99.4.
The number of patients enrolled in the 2006 trial was 16, which was included in the second sentence of the second paragraph under “Business Overview—AE37–Ii-Key/HER/2/neu Hybrid Immunotherapeutic Vaccine” in “Item 1. Business” on page 4 of the Form 10/A.
6. We note your response to our prior comment 5 and reissue in part. Please revise to clarify the next steps for each of your product candidates, including how much funding you will need, what steps remain to achieve regulatory approval and the planned timeline.
The company has included the following paragraphs on page 6 of the Form 10/A2 with respect to the Shenzhen agreement:
“According to a published report in the Journal of Health Economics form Tufts Centy for the Study of Drug Development, developinga new prescription medicine that gains marketing approval is estimated to cost drug makers $1.4 billion in out of pocket costs. Furthermore, the estimated cost of post-approval research and development is $312 million. Research and development costs include studies to test new indications, new formulations, new dosage strength and regimens, and to monitor safety and long-term side effects in patients as required by the FDA as a condition of approval.
Given this market environment, and the early stage of development for our Ii-Key immunotherapeutic peptide vaccines, we are focused on developing our product candidates through Phase II proof of concept trials, with the expectation that positive Phase II data will lead to a partnership or joint venture agreement with a major pharmaceutical company that is positioned, financially, operationally, and commercially to bring a drug through the regulatory process and into the market with a successful drug launch. To that end, we do not plan to bring the products to market, but rather to a partnership opportunity with development milestones, licensing fees, and royalties, as is common throughout the biotechnology industry. Each product candidate will require between $15 and $50 million to complete the clinical development and manufacturing process that will be needed to obtain such partnerships and we cannot predict when, if at all, we will obtain such funding and even if such funding is obtained we cannot guarantee that we will be able to enter into the partnerships or joint ventures discussed above.”
7. Please revise the disclosure on page 5 to explain your basis for the statement that "funding for the development of a vaccine of SARS-CoV-2 virus is in advanced discussions and currently expected that the development costs will be borne by the U.S. and foreign government agencies.
The Company has deleted the sentence referred to by the Staff.
Clinical Development Plans for Ii-Key Immunotherapeutic Peptides, page 6
8. We note your response to our prior comment 9. Please revise to provide the detail from your response that the discussions with the major oncology research centers are currently confidential, and the clinical trials are in the planning stages.
In bladder/urothelial cancer, HER2/neu is over-expressed in 30% of patients. There is a significant unmet need for new bladder cancer treatments. Additionally, a number of immune checkpoint inhibitors like Keytruda are approved by FDA in bladder cancer, as illustrated in the Table below:
FDA Approved Immune Checkpoint Inhibitors in Bladder Cancer
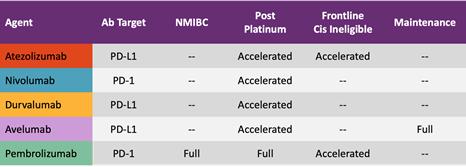
As NGIO is now focused on the development of cancer immunotherapeutics using Ii-Key peptide vaccines in combination with checkpoint inhibitors, we have approached a leading oncology research institution that specializes in immunotherapy of bladder cancer. We are working with clinicians at the institution to develop a protocol for a Phase II trial.
In addition to the bladder cancer trial, NGIO plans to initiate clinical trials with the Ii-Key-GP100 and Ii-Key-Tyrosinase peptide vaccines for melanoma. The company has conducted extensive pre-clinical work with Ii-Key-GP100 that demonstrated positive immune responses in vaccinated transgenic mouse models (J Immunotherapy, Volume 28, Number 4, July/August 2005). We have initiated discussions with a major oncology research hospital in Florida that specializes in the immunotherapy of melanoma. There are 6 checkpoint inhibitors approved as first line therapy in melanoma, so the planned Phase II trial will be a combination study of Ii-Key-GP100 and Ii-Key-Tyrosinase peptides with a checkpoint inhibitor. We will advance protocol development upon funding.
Competition, page 7
9. We note your response to our prior comment 11. Please revise to disclose, if true, that NGIO has never received regulatory approval for a product candidate or had commercial sales.
The Company has included the requested disclosure as the last sentence of the third paragraph under the subheading “—Competition on page 7 of the Form 10/A2.”
Note 2- Summary of Significant Accounting Policies
Research and Development Costs, page F-8
10. We note from your response and revised disclosures to comment 21 that you completed a Phase IIb trial of AE37 in combination with GM-CSF in November of 2019. Please address the following:
| • | Clarify in the filing if the trial which was completed, as discussed on page 4, is the same trial in which costs were incurred for AE37 in combination with Keytruda as disclosed on page 13 in which you incurred $251,459 of costs. If not, please tell us where the costs for AE37 in combination with GM-CSF were recorded in your financial statements and revise your MD&A as necessary to discuss the costs incurred. |
The Phase IIb Trial, which is discussed on page 4, is for AE37 in combination GM-CSF and is not the same trial as the Phase II trial for AE7 in combination with Keytruda. There were no costs incurred by the Company related to the Phase IIb Trial in fiscal years 2018, 2019 or in the nine months ended April 30, 2020.
| • | You state on page 5 that "based on the results from the Phase II trial described above, NGIO entered into a Clinical Trial Collaboration and Supply Agreement" on June 28, 2017 with Merck. Since the Phase IIb trial was not completed until November of 2019. Please clarify in the filing what results you are referring to which resulted in the collaboration agreement with Merck in 2017. |
The Company has revised the beginning of the third paragraph under “Business Overview—AE37–Ii-Key/HER/2/neu Hybrid Immunotherapeutic Vaccine” in “Item 1. Business” on page 4 of the Form 10/A2 to state:
“In April of 2007 we commenced patient enrollment for a Phase IIb trial of AE37 in combination with GM-CSF on 300 patients with respect to the prevention of cancer recurrence in women who were at high risk of recurrence after undergoing successful primary standard of care breast cancer therapies and were disease free at time of enrollment (the “Phase IIb Trial”). The Phase IIb Trial was completed in 2016 and preliminary results of the Phase IIb Trial were published in 2016 by Elizabeth A. Mittendorf, MD, F.A.C.S. (Annals of Oncology 27: Published online 30 March 2016). The Phase IIb Trial was officially concluded with site close-out and database lock in November of 2019.”
The Company also revised the first sentence of the immediately following paragraph on page 5 of the Form 10/A2 to state:
“Based on the preliminary results from the Phase IIb Trial, NGIO entered into a Clinical Trial Collaboration and Supply Agreement (the “Collaboration Agreement”) on June 28, 2017 with Merck Sharpe & Dohme B.V. (“Merck”) to evaluate the safety and efficacy of AE37 in combination with the anti-PD-1 therapy, KEYTRUDA (pembrolizumab) in patients with metastatic triple-negative breast cancer.”
11. We acknowledge your response and revised disclosures to comment 20. We believe the significant terms of each material agreement are required to be presented in the filing such as the rights and obligations of each party, including any significant milestone payments paid/received to date and aggregate potential milestones and the triggering factors thereof, the royalty percentages or a range, profit sharing, and termination clauses. For instance, it does not appear sufficient to omit disclosure of the termination provision in the agreement with Merck because you do not believe an accrual for a liability is warranted. Please revise your disclosure accordingly.
The Company has included the termination clauses for the Merck agreement the NSABP agreement and the Shenzhen agreement as decribed in our response to the Staff’s comment 3, included above. All of the other material terms of these agreements were previously provided, except the Company included a description of a provision under the Shenzhen agreement that provides for the reduction in the royaly rate in certain circumstatnces on page 5 of the Form 10/A2.
Note 3 - Commitments and Contingencies
Payable to Foundation, page F-10
12. We note from your response and revised disclosures to comment 22 that effective August 1, 2015, you capitalized all outstanding unpaid interest on the outstanding balance. Please explain to us your basis for capitalizing the unpaid interest on this payable.
We added the accrued interest to the loan balance(‘Capitalized the interest) because as stated Accounting Standards Codification (“ASC”) 835-30-25-7b the entity shall apply
“an interest factor to compensate the supplier over the life of the note for the use if the funds that would have been received in a cash transaction at the time of the exchange”
In the Forbearance Agreement effective September 9, 2013 (“Forebearance Agreement”) among the Company,its parent Generex Biotechnology Corp, and the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. (the “Foundation”). The Company and the Foundation acknowledged that the Company was $1,315,817.38 in arrears in its payment and interest obligations. The Forebearance Agreement further states in the preamble, and the definition of “Forebearance Amount” that interest is required on over due payments. Consequently we believe that interest should be accrued on any unpaid balances including any unpaid interest. The Forebearance Agreement provides that the rate of interest to be applied to such overdue payments is one and one half percent (1 ½%) per month. We have continued to accrue interest at this rate.
Exhibits
13. Please file an executed copy of Exhibit 10.3. In this regard, we note that it is unclear whether the counterparty signed the agreement.
The Company has filed the requested exhibit with the Form 10/A2.
Should you have any questions relating to the foregoing or wish to discuss any aspect of the Company’s filing, please contact me at 212-658-0458.
Jeffrey P. Wofford, Esq.
Carmel, Milazzo & Feil LLP