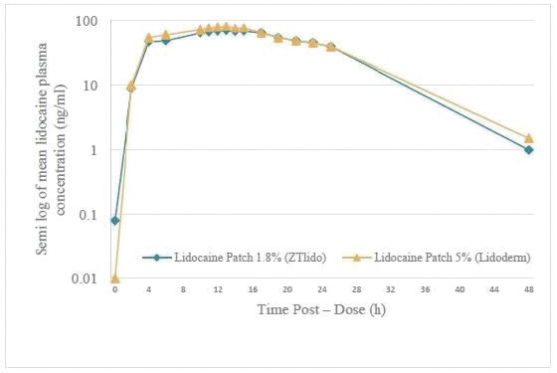
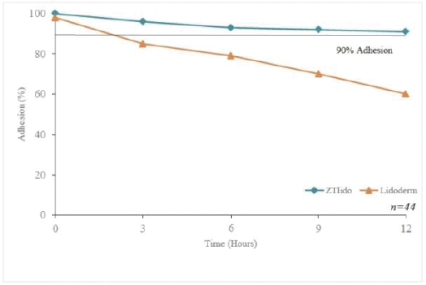
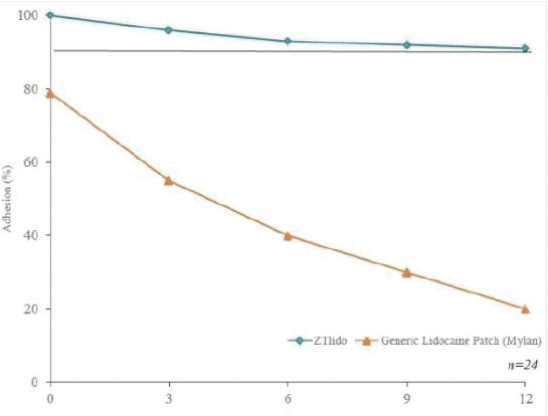
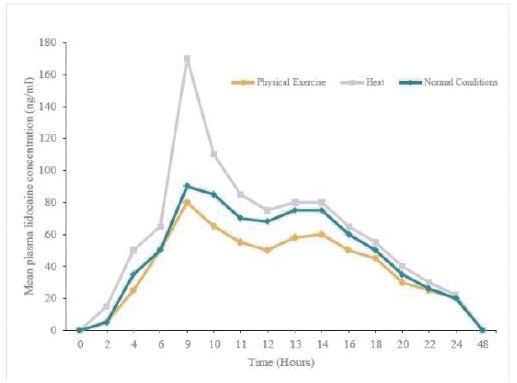
2022/076470 (and all patents and patent applications that claim priority rights thereto), which covers oral delayed burst formulations of low-dose naloxone or naltrexone and related methods of treatment. We continue to seek to maximize the scope of our patent protection for all our programs. We have six issued U.S. patents and two pending U.S. patent applications relating to lidocaine topical system compositions and methods of treating pain with lidocaine topical system compositions. The patents are U.S. Pat. Nos. 9,283,174, 9,925,264, 9,931,403, 10,765,749, 10,765,640 and 11,278,623 all of which expire in 2031. Related to GLOPERBA, our licensed patents include U.S. Pat. Nos. 9,907,751, 10,226,423, 10,383,820, and 10,383,821, which relate to liquid colchicine formulations for oral administration and associated methods of use. U.S. Pat. No. 10,226,423 expires in 2037. The remaining patents related to GLOPERBA expire in 2036. We believe that we have certain know-how and trade secrets relating to our technology and product candidates. We rely on trade secrets to protect certain aspects of our technology related to our current and future product candidates. However, trade secrets can be difficult to protect. We seek to protect our trade secrets, in part, by entering into confidentiality agreements with our employees, consultants, scientific advisors, service providers, and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems.
Trademarks, Trade Secrets and Other Proprietary Information
We file trademark applications and pursue registrations in the United States and abroad when appropriate. We own registered U.S. trademarks for the marks “SCILEX,” “ZTlido” and “RESPONSIBLE BY DESIGN.” We also own pending trademark applications for “SEMNUR PHARMACEUTICALS” and “SEMDEXA” in the United States. We also license the GLOPERBA trademark as discussed in relation to the Romeg Agreement described above.
We also depend upon the skills, knowledge and experience of our scientific and technical personnel, as well as that of our advisors, consultants and other contractors. To help protect our proprietary know-how, we rely on trade secret protection and confidentiality agreements to protect our interests. Our policy is to require each of our employees, consultants and advisors to execute a confidentiality and inventions assignment agreement before beginning their employment, consulting or advisory relationship with us. The agreements generally provide that the individual must keep confidential and not disclose to other parties any confidential information developed or learned by the individual during the course of the individual’s relationship with us except in limited circumstances. These agreements generally also provide that we will own all inventions conceived by the individual in the course of rendering services to us.
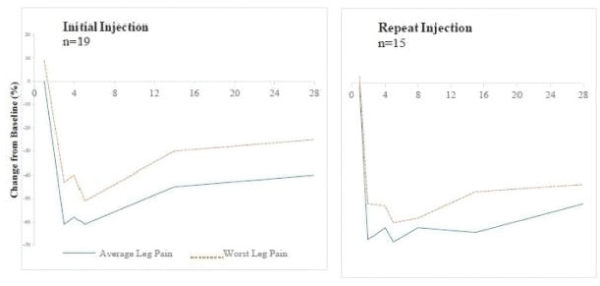
SEMDEXA benefits from our substantial intellectual property portfolio and other technical barriers to entry for potential competitors. Further, we are a party to an exclusive supply agreement for a proprietary biocompatible excipient with no generic equivalent. Our complex manufacturing process, specialized equipment and know-how for sterile viscous product candidates are also key to our competitive edge. We believe that our competitors will be required to conduct lengthy and costly preclinical and clinical trials to establish products with comparable safety and efficacy to SEMDEXA.
Government Regulation and Product Approval
Government authorities in the United States at the federal, state and local levels, and in other countries and jurisdictions, extensively regulate, among other things, the research, development, testing, manufacture, pricing, reimbursement, sales, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products such as those we are marketing and developing. SP-102, SP-103, SP-104 and any other product candidate that we develop must be approved by the FDA or otherwise authorized for marketing before they may be legally marketed in the United States and by the corresponding foreign regulatory agencies before they may be legally marketed in foreign countries. The processes for obtaining marketing approvals, along with subsequent compliance with applicable statutes and regulations and other regulatory authorities, require the expenditure of substantial time and financial resources.
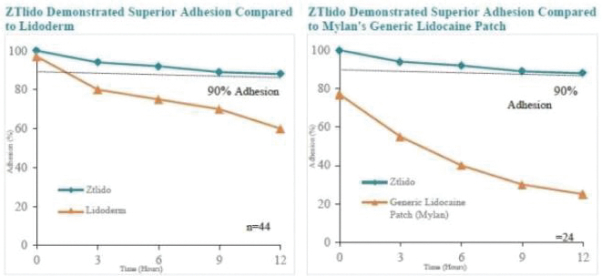
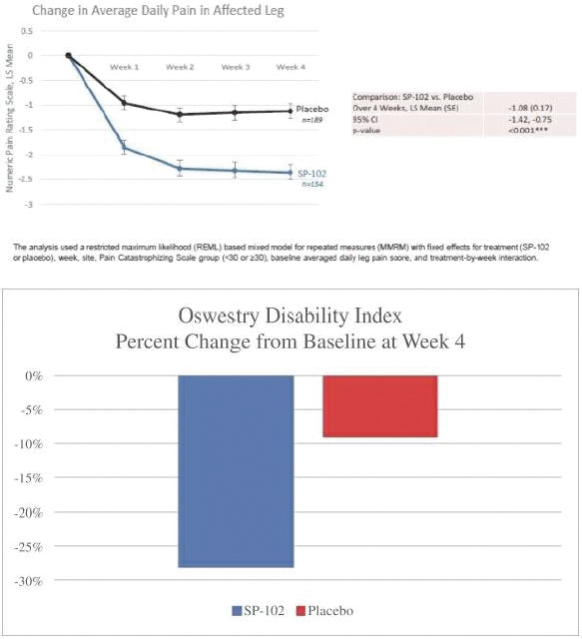
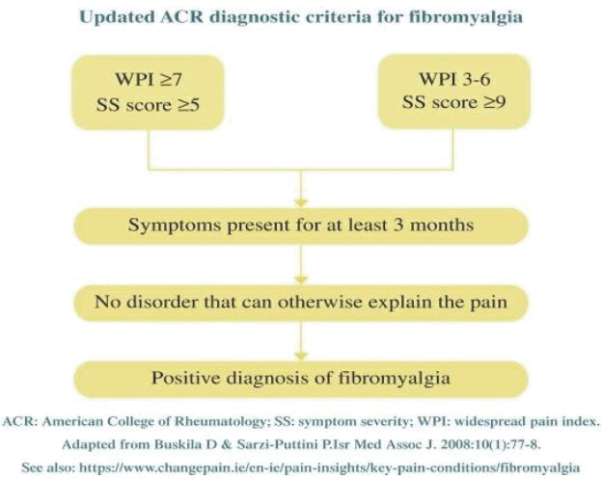
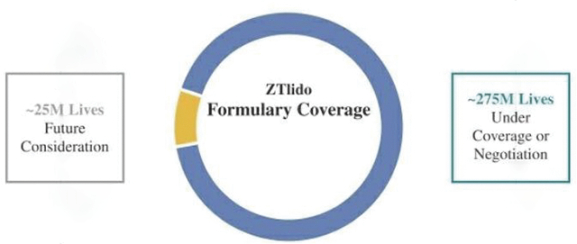
167