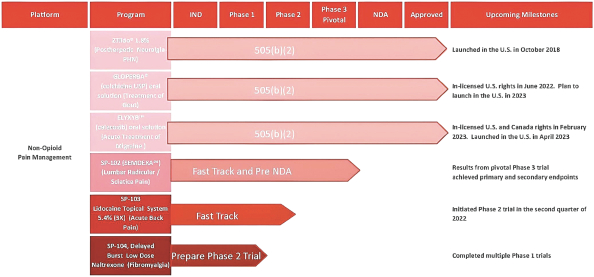
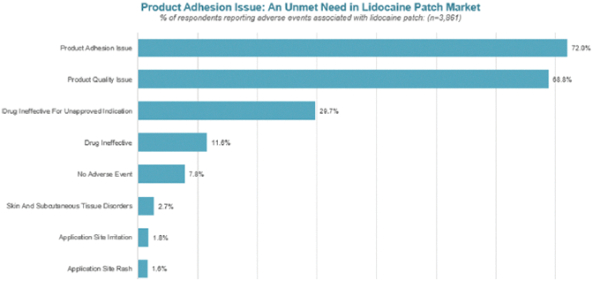
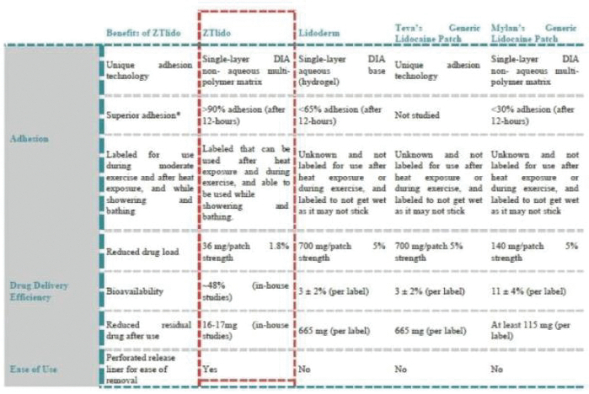
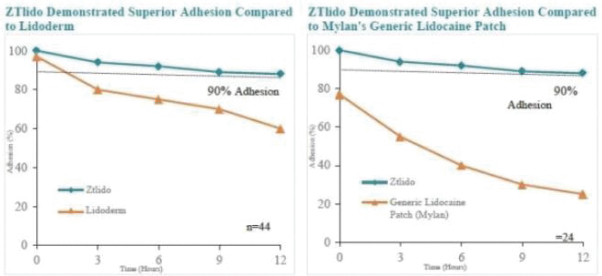
The foregoing is a summary of the material terms of the Romeg Agreement, which is in the form filed as an exhibit to the registration statement of which this prospectus forms a part. You should read the form of the agreement for a complete understanding of all of its terms.
Intellectual Property
Our success depends in part on our ability to obtain and maintain proprietary protection for our product candidates, technology and know-how, to operate without infringing the proprietary rights of others and to prevent others from infringing our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, pursuing and obtaining patent protection in the United States and in jurisdictions outside of the United States related to our novel adhesion and delivery technology, inventions, improvements and product candidates that are important to the development and implementation of our business. Our patent portfolio is intended to cover our product candidates, their methods of use and processes for their manufacture, and any other inventions that are commercially important to our business. We also rely on trade secret protection of our confidential information and know-how relating to our novel adhesion and delivery technology, platforms and product candidates.
Generally, patents are granted a term of 20 years from the earliest claimed non-provisional filing date. In certain instances, patent term can be adjusted to recapture a portion of delay by the PTO in examining the patent application or extended to account for term effectively lost as a result of the FDA regulatory review period, or both. We cannot provide any assurance that any patents will be issued from our pending or future applications or that any patents will adequately protect our product or product candidates.
Our patent portfolio, consisting of owned and/or licensed IP as of March 31, 2023 contains approximately sixteen issued and unexpired U.S. patents, six pending U.S. patent applications, and one pending Patent Cooperation Treaty (PCT) application. Our portfolio also includes certain foreign counterparts of these patents and patent applications including Australia, Brazil, Canada, China, Hong Kong, India, Indonesia, Israel, Japan, Korea, Malaysia, Mexico, New Zealand, Panama, Peru, Philippines, Russian Federation, Singapore, South Africa, Taiwan, Ukraine, and certain countries within the European Patent Convention.
With respect to ZTlido and SP-103, our patents and patent applications cover compositions and methods of treatment. With respect to our product candidate SEMDEXA, our patents and patent applications include formulations and methods of treatment. The patents are U.S. Pat. Nos. 10,500,284, 10,117,938, and 11,020,485, all of which expire in 2036. With respect to SP-104, our pending Patent Cooperation Treaty application is WO 2022/076470 (and all patents and patent applications that claim priority rights thereto), which covers oral delayed burst formulations of low-dose naloxone or naltrexone and related methods of treatment. We continue to seek to maximize the scope of our patent protection for all our programs. We have six issued U.S. patents and two pending U.S. patent applications relating to lidocaine topical system compositions and methods of treating pain with lidocaine topical system compositions. The patents are U.S. Pat. Nos. 9,283,174, 9,925,264, 9,931,403, 10,765,749, 10,765,640 and 11,278,623 all of which expire in 2031. Related to GLOPERBA, our licensed patents include U.S. Pat. Nos. 9,907,751, 10,226,423, 10,383,820, and 10,383,821, which relate to liquid colchicine formulations for oral administration and associated methods of use. U.S. Pat. No. 10,226,423 expires in 2037. The remaining patents related to GLOPERBA expire in 2036. We believe that we have certain know-how and trade secrets relating to our technology and product candidates. We rely on trade secrets to protect certain aspects of our technology related to our current and future product candidates. However, trade secrets can be difficult to protect. We seek to protect our trade secrets, in part, by entering into confidentiality agreements with our employees, consultants, scientific advisors, service providers, and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems.
173