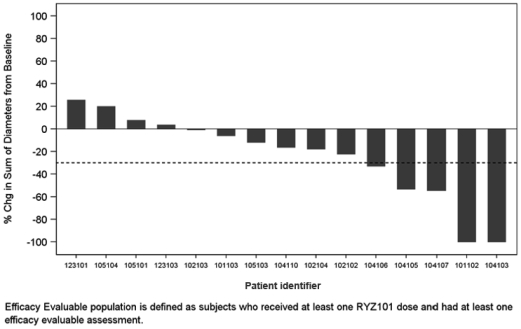
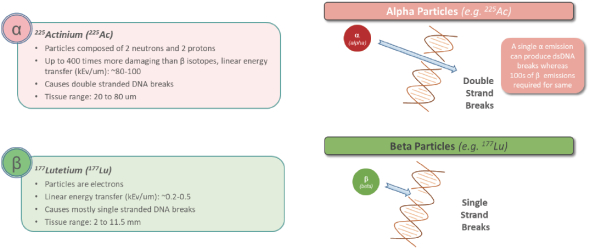
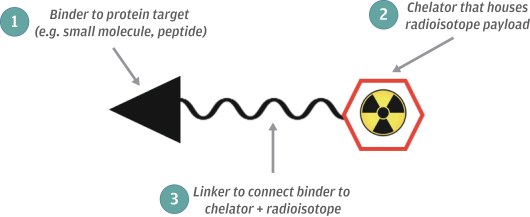
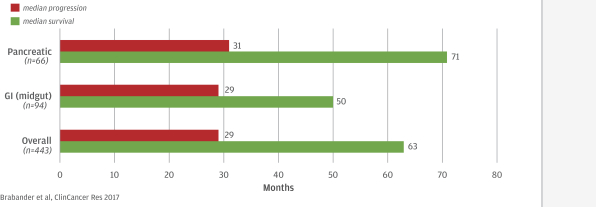
In addition, at the state level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. As a result, we may be unable to use all or a material portion of our NOL carryforwards and other tax attributes, which could adversely affect our future cash flows.
Changes in tax laws or regulations that are applied adversely to us or our customers may have a material adverse effect on our business, cash flow, financial condition or results of operations.
New income, sales, use or other tax laws, statutes, rules, regulations, or ordinances could be enacted at any time, which could adversely affect our business operations and financial performance. Further, existing tax laws, statutes, rules, regulations, or ordinances could be interpreted, changed, modified or applied adversely to us. For example, the Tax Cuts and Jobs Act, the Coronavirus Aid, Relief, and Economic Security Act, and the Inflation Reduction Act have all made many significant changes to the U.S. tax laws. Future guidance from the Internal Revenue Service and other tax authorities with respect to such legislation may affect us, and certain aspects of such legislation could be repealed or modified in future legislation. It is also possible that future legislation could have an adverse effect on our operations, cash flows and results of operations and contribute to overall market volatility. In addition, it is uncertain if and to what extent various states will conform to recent or newly enacted federal tax legislation. Changes in corporate tax rates, the realization of net deferred tax assets relating to our operations, the taxation of foreign earnings, and the deductibility of expenses could have a material impact on the value of our deferred tax assets, could result in significant one-time charges, and could increase our future U.S. tax expense.
If our information technology systems or data, or those of third parties upon which we rely, are or were compromised, we could experience adverse consequences resulting from such compromise, including but not limited to regulatory investigations or actions, litigation, fines and penalties, disruptions of our business operations, reputational harm, loss of revenue or profits, and other adverse consequences. Our internal computer systems, or those of our third-party CROs, manufacturers, contractors or consultants, or current or future collaborators or other third parties, may fail or suffer security breaches or other unauthorized or improper access, which could result in a material disruption of our development programs.
We are increasingly dependent upon information technology systems, infrastructure and data to operate our business. In the ordinary course of business, we collect, store, handle, share, use, retain, safeguard, transmit, analyze and otherwise process large amounts of data, including, without limitation, confidential information, such as proprietary business information, health information, and personal information. It is critical that we do so in a manner to maintain the confidentiality and integrity of such confidential information.
Despite the implementation of security measures, given the size and complexity of our internal information technology systems and those of our third-party vendors and other contractors and consultants, and the increasing amounts of confidential information that they maintain, such internal computer and technology systems and those of our current and any future third-party CROs, manufacturers, and other contractors, consultants and collaborators are potentially vulnerable to breakdown or other damage or interruption from service interruptions, system malfunction, information security threats, such as data breaches, damage from computer viruses, cyber-attacks (such as the deployment of social engineering malware, denial-of-service attacks, ransomware attacks, and supply chain attacks), unauthorized access, intentional or accidental actions or inaction by our employees, third-party vendors, contractors, consultants, business partners or other third parties that introduce vulnerabilities, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any material system failures, accidents, security breaches or intrusions to date, if such an event were to occur and cause interruptions in our operations or result in any inadvertent or unauthorized disclosure of or access to personal information, protected health information, or other sensitive, confidential or proprietary information, it could result in a material disruption of our programs and/or material liability and
88