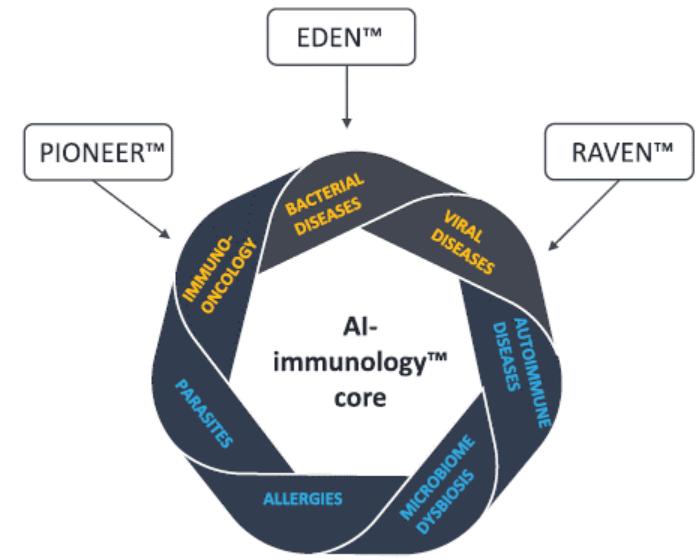
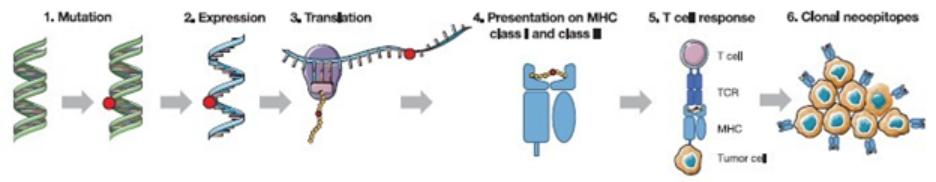
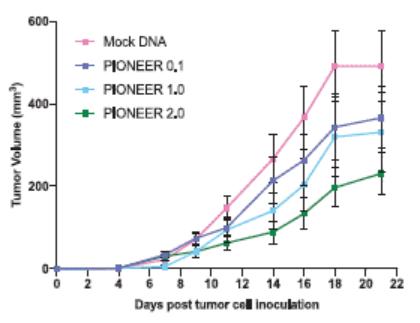
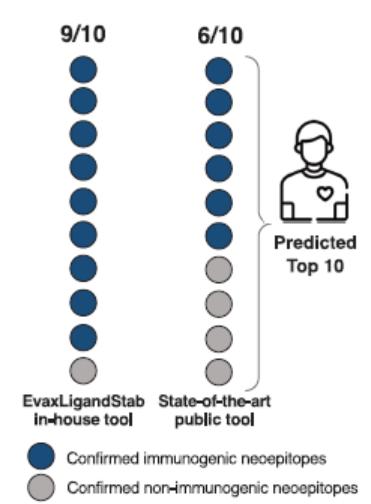
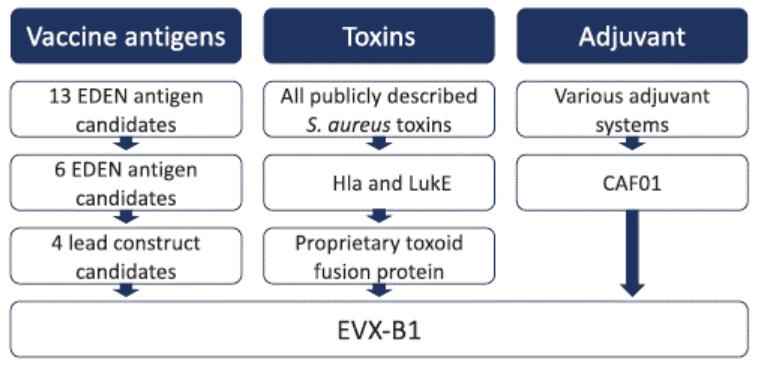
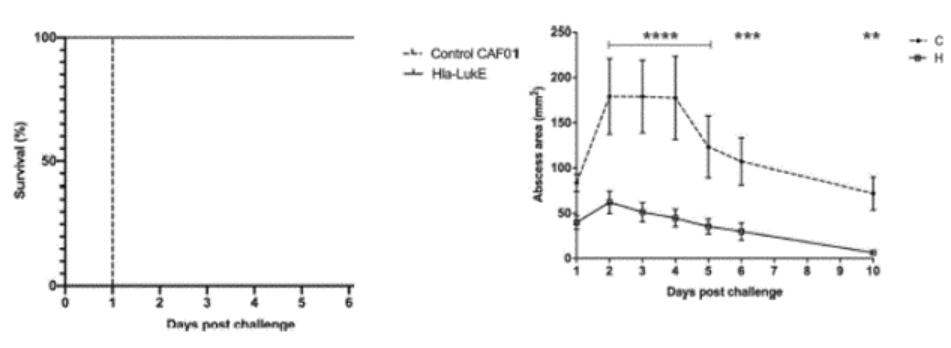
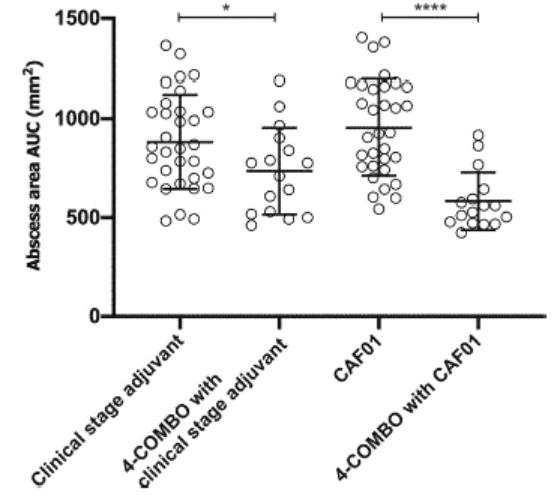
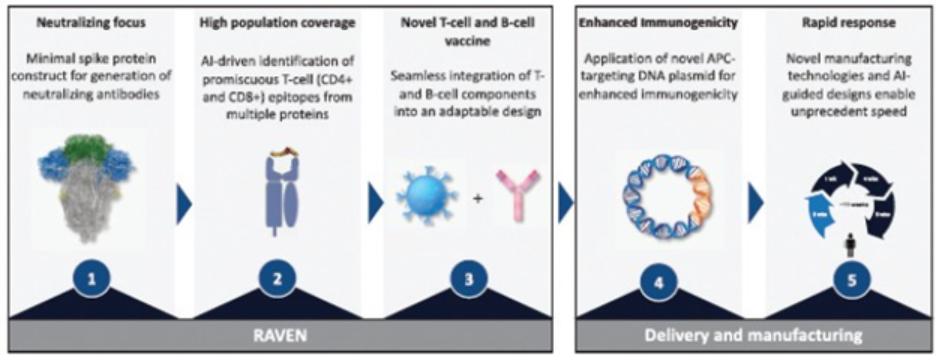
European Union member states. Failure to comply with these requirements could result in reputational risk, public reprimands, administrative penalties, fines or imprisonment.
We are subject to certain anti-corruption, anti-money laundering, export control, sanctions, and other trade laws and regulations. We can face serious consequences for violations.
Among other matters, anti-corruption, anti-money laundering, export control, sanctions, and other trade laws and regulations, which are collectively referred to as “trade laws”, prohibit companies and their employees, agents, CROs, CMOs, legal counsel, accountants, consultants, contractors and other future partners from authorizing, promising, offering, providing, soliciting, or receiving directly or indirectly, corrupt or improper payments or anything else of value to or from recipients in the public or private sector. Violations of trade laws can result in substantial criminal fines and civil penalties, imprisonment, the loss of trade privileges, debarment, tax reassessments, breach of contract and fraud litigation, reputational harm, and other consequences. We have direct or indirect interactions with officials and employees of government agencies or government-affiliated hospitals, universities and other organizations. We plan to engage third parties for clinical trials and/or to obtain necessary permits, licenses, intellectual property (including patents) and other regulatory approvals, and we can be held liable for the corrupt or other illegal activities of our personnel, agents or future partners, even if we do not explicitly authorize or have prior knowledge of such activities.
We are subject to stringent privacy laws, information security policies and contractual obligations governing the use, processing, and cross-border transfer of personal information and our data privacy and security practices.
We receive, generate and store significant and increasing volumes of sensitive information, such as employee, personal and patient data. We are subject to a variety of local, state, national and international laws, directives and regulations that apply to the collection, use, storage, retention, protection, disclosure, transfer and other processing of personal data, collectively referred to as “data processing”, in the different jurisdictions in which we operate, including comprehensive regulatory systems in the United States and Europe. Legal requirements relating to data processing continue to evolve and may result in ever-increasing public scrutiny and escalating levels of enforcement, sanctions and increased costs of compliance.
Compliance with United States and international data protection laws and regulations could cause us to incur substantial costs or require us to change our business practices and compliance procedures in a manner adverse to our business. Moreover, complying with these various laws could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. Failure to comply with United States, European and other international data protection laws and regulations could result in government enforcement actions (which could include civil or criminal penalties), private litigation and/or adverse publicity and could negatively affect our operating results and business. Claims that we have violated individuals’ privacy rights, failed to comply with data protection laws, or breached our contractual obligations, even if we are not found liable, could be expensive and time consuming to defend, could result in adverse publicity and could have a material adverse effect on our business, financial condition and results of operations.
Various U.S. states, such as California and Massachusetts, have implemented similar privacy laws and regulations, such as the California Confidentiality of Medical Information Act, that impose restrictive requirements regulating the use and disclosure of patient health information and other personal information. In addition to fines and penalties imposed upon violators, some of these state laws also afford private rights of action to individuals who believe their personal information has been misused. California’s patient privacy laws, for example, provide for penalties of up to $250,000 and permit injured parties to sue for damages. In addition to the California Confidentiality of Medical Information Act, California also recently enacted the California Consumer Privacy Act of 2018, or CCPA, which became effective on January 1, 2020. The CCPA has been characterized as the first “GDPR-like” privacy statute to be enacted in the United States because it mirrors a number of the key provisions of the EU General Data Protection Regulation (described below). The CCPA establishes a new privacy framework for covered businesses in the State of California, by creating an expanded definition of personal information, establishing new data privacy rights for consumers imposing special rules on the collection of consumer data from minors, and creating a new and potentially severe statutory damages framework for violations of the CCPA and for businesses that fail to implement reasonable security procedures and practices to prevent data breaches. An initiative called the California Privacy Rights Act, or CCPA 2.0, passed in November 2020