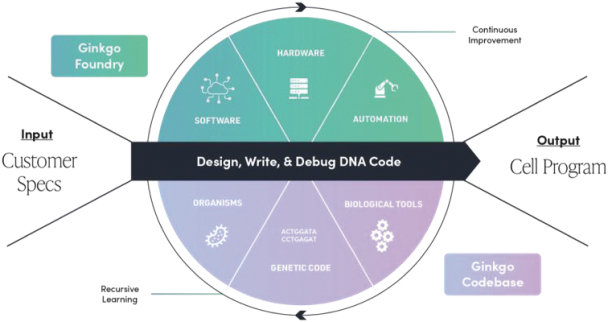
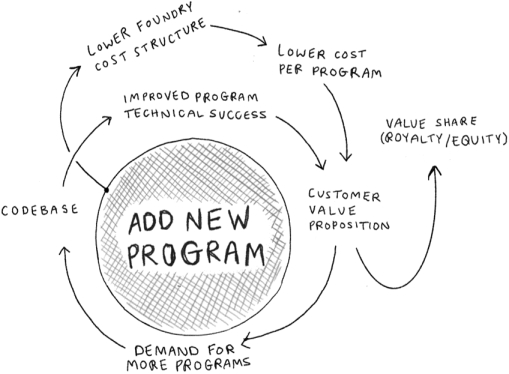
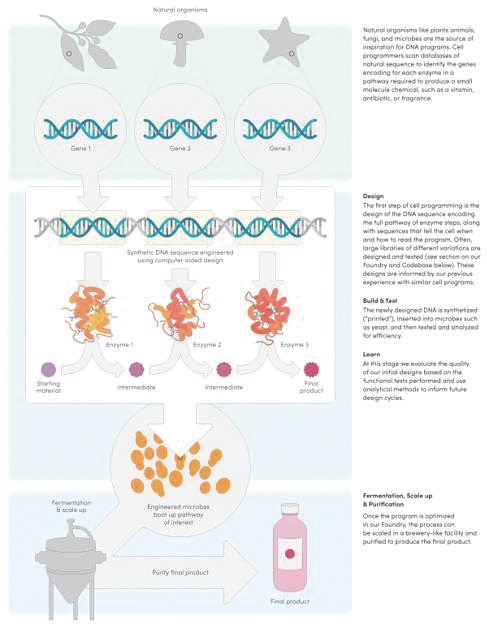
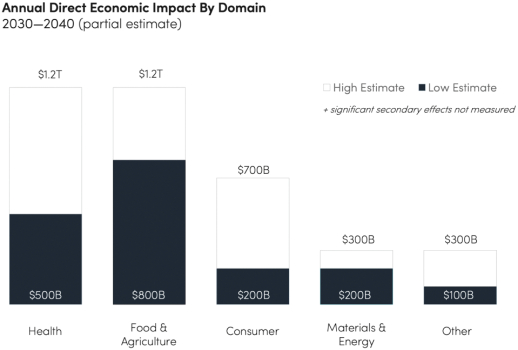
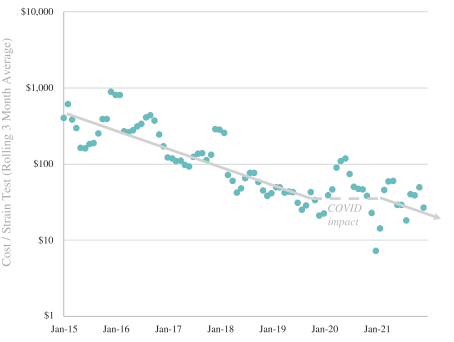
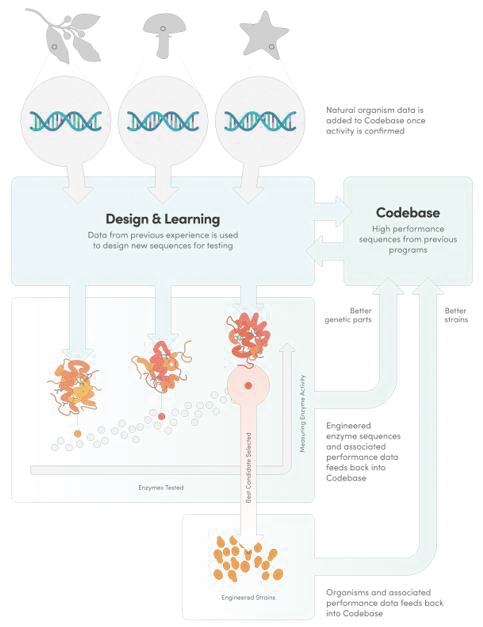
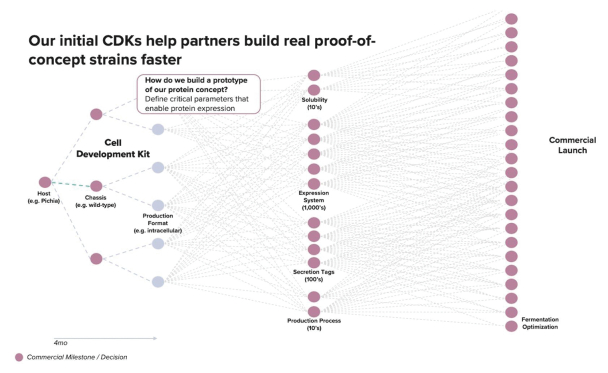
We provide cell engineering and product discovery services to customers engaged in the manufacture of foods, cosmetics and pharmaceutical products. The FDA regulates the research, development, testing, quality control, import, export, safety, effectiveness, storage, recordkeeping, premarket review, approval or licensure, processing, formulation, manufacturing, packaging, labeling, advertising, promotion, marketing, distribution, sale, post-market monitoring and reporting of our customers’ pharmaceuticals, cosmetics and food products, and the FTC also regulates the advertising and promotion of these products.
We also act as a systems integrator and authorized distributor of certain
COVID-19
diagnostic test and collection kits manufactured by independent third parties, and we work with laboratory partners that provide clinical laboratory testing services as part of the
COVID-19
testing services we offer, and these tests and test kits may be subject to regulation by the FDA. In particular, the tests and test kits used in our
COVID-19
testing services may be subject to regulation by the FDA as medical devices, and may be required to comply with the requirement that such products have obtained clearance, approval, or other marketing authorizations, such as an EUA, before they can be commercialized, as well as post-market requirements such as adverse event reporting and restrictions on labeling, marketing, and distribution.
The HHS and FDA issued several policy statements in November 2021 governing the regulation of
COVID-19
Laboratory Developed Tests (“LDTs”) that resume FDA premarket review of
COVID-19
LDTs that HHS halted in August 2020. Pursuant to these new policy statements, FDA expects laboratories to seek FDA marketing authorization and otherwise comply with FDA device regulations when marketing
COVID-19
LDTs. An LDT is an in vitro diagnostic test that is intended for clinical use and is designed, manufactured, and used within a single laboratory. LDTs are classified as medical devices, but the FDA has historically exercised enforcement discretion and has generally not enforced FDA requirements, including premarket review, with respect to laboratories that offer LDTs. While HHS and FDA have announced their intention to require premarket review of
COVID-19
LDTs, either agency may change its position in the future.
Medical products, including
COVID-19
tests, that are granted an EUA or other marketing authorization must comply fully with the terms and conditions provided in the EUA or other marketing authorization. For example, EUAs for
COVID-19
tests may include conditions of authorization applicable to the EUA holder, authorized distributors and authorized laboratories. Noncompliance with applicable requirements could result in negative consequences, including adverse publicity, judicial or administrative enforcement, warning letters or untitled letters from the FDA, mandated corrective promotional materials, advertising or communications with doctors, and civil or criminal penalties, among others. The FDA can also withdraw marketing authorization for the applicable product, and in the case of a product subject to an EUA, the authorization to market the product under the EUA lasts only as long as the declared public health emergency.
We are engaged in the research, development, and export of certain products that may be regulated as controlled substances, including microbes designed to generate precursors to cannabinoids or other chemical intermediates. The CSA, as amended from time to time, establishes registration, security, recordkeeping, reporting, storage, distribution and other requirements administered by the DEA. The DEA is concerned with the control of handlers of controlled substances, and with the equipment and raw materials used in their manufacture and packaging, in order to prevent loss and diversion into illicit channels of commerce. The DEA regulates controlled substances as Schedule I, II, III, IV or V substances. Schedule I substances by definition have no established medicinal use, and may not be marketed or sold in the United States. Schedule I substances are considered to present the highest risk of abuse, and Schedule V substances the lowest relative risk of abuse among controlled substances. Marijuana is classified as a Schedule I controlled substance. However, the term does not include “hemp,” which means the cannabis plant and any part of that plant, including the seeds and all derivatives, extracts, cannabinoids, isomers, acids, salts, and salts of isomers, whether growing or not, with a THC concentration of not more than 0.3% on a dry weight basis.