As filed with the Securities and Exchange Commission on November 24, 2021.
Registration No. 333-260923
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
Amendment No. 1 to
FORM F-1
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
Genenta Science S.p.A.
(Exact name of registrant as specified in its charter)
| Republic of Italy | 2836 | Not Applicable | ||
| (State or other jurisdiction of incorporation or organization) | (Primary Standard Industrial Classification Code Number | (I.R.S. Employer Identification Number) |
| Pierluigi Paracchi Chief Executive Officer Via Olgettina No. 58 20132 Milan, Italy Tel: +39-02-2643-4681 | Cogency Global Inc. 122 East 42nd Street, 18th Floor New York, New York 10168 Tel: +1.800.221.0102 | |
| (Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices) | (Name, address, including zip code, and telephone number, including area code, of agent for service) |
Copies to:
Mitchell S. Nussbaum, Esq. Norwood P. Beveridge, Esq. Loeb & Loeb LLP 345 Park Avenue New York, NY 10154 | Thomas S. Levato, Esq. Ettore A. Santucci, Esq. Goodwin Procter LLP 620 Eighth Avenue New York, New York 10018 | ||
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date hereof.
If any of the securities being registered on this form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act, check the following box. [X]
If this form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
If this form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
If this form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933.
Emerging growth company [X]
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 7(a)(2)(B) of the Securities Act. [ ]
†The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
CALCULATION OF REGISTRATION FEE
| Title of each class of securities to be registered | Proposed maximum aggregate offering price (1)(2)(3) | Amount of registration fee (3) | ||||||
| Ordinary shares, with no par value, as represented by American Depositary Shares | $ | 34,500,000 | $ | 3,198.15 | ||||
| Representatives’ warrants to purchase ordinary shares as represented by American Depositary Shares(4) | ||||||||
| Ordinary shares as represented by American Depositary Shares underlying Representatives’ warrants(5) | 460,000 | 42.64 | ||||||
| Total | $ | 34,960,000 | $ | 3,240.79 | ||||
| (1) | The ordinary shares may be represented by American Depositary Shares, or ADSs, each of which represents one ordinary share. A separate Registration Statement on Form F-6 (Registration No. 333-261223) has been filed for the registration of ADSs issuable upon deposit of the ordinary shares. | |
| (2) | Pursuant to Rule 416 under the Securities Act of 1933, as amended, or the Securities Act, the ordinary shares registered hereby also include an indeterminate number of additional ordinary shares as may from time to time become issuable by reason of stock splits, stock dividends, recapitalizations or other similar transactions. | |
| (3) | Estimated solely for purposes of calculating the amount of the registration fee pursuant to Rule 457(o) under the Securities Act. Includes the offering price of ordinary shares that the underwriters have the option to purchase to cover over-allotments, if any. | |
| (4) | In accordance with Rule 457(g) under the Securities Act, because the registrant’s ordinary shares as represented by American Depositary Shares underlying the Representatives’ warrants are being registered hereby, no separate registration fee is required with respect to the warrants registered hereby. | |
| (5) | As estimated solely for the purpose of calculating the registration fee pursuant to Rule 457(g) under the Securities Act based on an estimated 1,000,000 ordinary shares or ADSs reserved for sale outside the U.S., with respect to which the Representatives will not be entitled to compensation equal to 4% warrant coverage. The warrants are exercisable at a per share exercise price equal to 125% of the public offering price, and the proposed maximum aggregate offering price of shares underlying the representative’s warrants is $460,000. |
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED NOVEMBER 24, 2021
PRELIMINARY PROSPECTUS
2,608,695 Ordinary Shares
![]()
(Including 1,608,695 Ordinary Shares in the Form of American Depositary Shares)
This is a public offering of 2,608,695 ordinary shares with no par value, or ordinary shares, of Genenta S.p.A., which consists of (i) an initial public offering of 1,608,695 ordinary shares in the form of American Depositary Shares, or ADSs, each representing one of our ordinary shares, which we refer to as the “ADS offering,” and (ii) a concurrent offering of 1,000,000 ordinary shares exclusively offered to existing shareholders in Italy, as a mitigant for the exclusion of pre-emptive rights as resolved by the general shareholders meeting of the company in accordance with Italian corporate law and our by-laws, which we refer to as the “Reserved Offering”. We refer in this prospectus to the ADS offering and the Reserved Offering collectively as the “offering.” Ordinary shares may be offered directly or in the form of ADSs, which likewise may be evidenced by American Depositary Receipts, or ADRs.
No public market currently exists for the ADSs or ordinary shares. We estimate the initial public offering price of the ADSs and ordinary shares will be between $10.50 and $12.50 per ADS or ordinary share, as the case may be. We refer to the ADSs, including the underlying ordinary shares and the ordinary shares being offered hereby, collectively, as the “securities.”
We have applied to list the ADSs on the Nasdaq Capital Market, or Nasdaq, under the symbol “GNTA.” There can be no assurance that we will be successful in listing the ADSs on Nasdaq.
We are an “emerging growth company” and a “foreign private issuer” under applicable U.S. federal securities laws and are eligible for reduced public company reporting requirements. See “Prospectus Summary—Implications of Being an Emerging Growth Company” and “Prospectus Summary—Implications of Being a Foreign Private Issuer” for additional information.
Investing in our securities involves a high degree of risk, including the risk of losing your entire investment. See “Risk Factors” beginning on page 12 of this prospectus for a discussion of information that should be considered in connection with an investment in our securities.
Neither the Securities and Exchange Commission nor any state or foreign securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
| Per ADS | Total | ||||||
| Initial public offering price | $ | $ | |||||
| Underwriting discounts and commissions (1) | $ | $ | |||||
| Proceeds to us (before expenses) | $ | $ | |||||
| (1) | We have agreed to reimburse the underwriters for certain expenses. Certain of our existing shareholders in Italy are expected to purchase ordinary shares directly from us concurrently with the closing of this offering for which we will not incur any underwriting discount or commission but will owe an advisory fee to the representatives of the underwriters. The amounts reflected as total underwriting discounts and commissions and proceeds to us (before expenses) are reflective of these arrangements. See “Underwriting” beginning on page 191 of this prospectus for additional disclosure regarding underwriter compensation and offering expenses. |
We have granted the underwriters an option for a period of 30 days from the date of this prospectus to purchase up to an additional 241,304 ADSs to cover over-allotments, if any.
Delivery of the ADSs will be made on or about December , 2021, subject to customary closing conditions.
Sole Book-Running Manager
| Roth Capital Partners |
Lead Manager
| Maxim Group LLC |
The date of this prospectus is December , 2021
TABLE OF CONTENTS
You should rely only on the information contained in this prospectus and any free writing prospectus prepared by or on behalf of us or to which we have referred you. We have not authorized anyone to provide you with information that is different. We are offering to sell our securities, and seeking offers to buy our securities, only in jurisdictions where offers and sales are permitted. The information in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or any sale of our securities.
For investors outside of the United States: Neither we nor any of the underwriters have done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. You are required to inform yourselves about and to observe any restrictions relating to this offering and the distribution of this prospectus.
We are organized under the laws of Italy and our registered office and domicile is located in Milan, Italy. Under the rules of the U.S. Securities and Exchange Commission, or the SEC, we are currently eligible for treatment as a “foreign private issuer.” As a foreign private issuer, we will not be required to file periodic reports and financial statements with the SEC as frequently or as promptly as domestic registrants whose securities are registered under the Securities Exchange Act of 1934, as amended, or the Exchange Act. Moreover, the chairman of the Board, the CEO and most of our directors are not residents of the United States, and all or a substantial portion of the assets of such persons are located outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon us or upon such persons or to enforce against them judgments obtained in U.S. courts, including judgments in actions predicated upon the civil liability provisions of the federal securities laws of the United States. We have been advised by our Italian counsel that there is doubt as to the enforceability in Italy of original actions, or in actions for enforcement of judgments of U.S. courts, of civil liabilities to the extent solely predicated upon the federal and state securities laws of the United States. See “Enforceability of Civil Liabilities” in this prospectus for additional information.
In May 2021, we changed the legal form of our company under Italian law (the “Corporate Conversion”) from a limited liability company (società a responsabilità limitata, or S.r.l.) to a joint stock company (società per azioni, or S.p.A.). Unless otherwise indicated or the context otherwise requires, all references in this prospectus to the terms “we,” “us,” “our,” the “Company” and “Genenta” refer to Genenta Science S.p.A. after the transformation of Genenta Science S.r.l. to an Italian joint stock company.
All trademarks or trade names referred to in this prospectus are the property of their respective owners. Solely for convenience, the trademarks and trade names in this prospectus are referred to without the ® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend the use or display of other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
Our reporting currency and functional currency is the Euro. Unless otherwise expressly stated or the context otherwise requires, references in this prospectus to “dollars,” “USD” or “$” mean U.S. dollars, and references to “euros,” “EUR” or “€” are to the European Union euros. U.S. dollar translations of EUR amounts presented in this prospectus were done on different dates in accordance with the date as of such entry in our books and are derived from our audited financial statements included elsewhere in this prospectus. U.S. dollar translations of EUR amounts presented in this prospectus that are not derived from our audited financial statements included elsewhere in this prospectus are translated using the rate of € 1.00 to $1.1271, based on the euro foreign exchange reference rate provided by the European Central Bank on November 19, 2021.
This prospectus includes statistical, market and industry data and forecasts which we obtained from publicly available information and independent industry publications and reports that we believe to be reliable sources. These publicly available industry publications and reports generally state that they obtain their information from sources that they believe to be reliable, but they do not guarantee the accuracy or completeness of the information. Although we believe that these sources are reliable, we have not independently verified the information contained in such publications.
This summary highlights information contained elsewhere in this prospectus. This summary does not contain all of the information you should consider before investing in our securities. Before you decide to invest in our securities, you should read the entire prospectus carefully, including the “Risk Factors” section and the financial statements and related notes appearing at the end of this prospectus.
Our Company
We are a clinical-stage biotechnology company engaged in the development of hematopoietic stem cell gene therapies for the treatment of solid tumors. We have developed a novel biologic platform which involves the ex-vivo gene transfer of a therapeutic candidate into autologous hematopoietic stem/progenitor cells (HSPCs) to deliver immunomodulatory molecules directly to the tumor by infiltrating monocytes/macrophages (Tie2 Expressing Monocytes - TEMs). Our technology is designed to turn TEMs, which normally have an affinity for and travel to tumors, into a “Trojan Horse” to counteract cancer progression and to prevent tumor relapse. Our technology is not target dependent, and therefore we believe it can be used as a treatment for a broad variety of cancers.
Our technology incorporates the use of a lentiviral vector (LVV) that combines a therapeutic transgene sequence, or payload, with our proprietary platform. Our proprietary platform consists of (i) the Tie-2 promoter, that drives transgene sequence transcription specifically in TEMs, and (ii) miRNA-126 target sequences to downregulate transgene expression post-transcription in those cells where the Tie-2 promoter is active and the miRNA-126 is present. We believe there are many advantages to our approach:
| ● | Trojan Horse Mechanism of Action (“MoA”): We use and modify TEMs, a subpopulation of tumor-associated myeloid cells, known to be involved in tumor growth and in the inhibition of immune system response, to allow the immune system to recognize the tumor and to deliver to the cancer site a chosen therapeutic. | |
| ● | Select Regulation of Transgene Expression: Our selected control of the chosen therapeutic gene expression is designed to avoid off-target and systemic toxicity. | |
| ● | Potential Long-Term Effect: Through the use of hematopoietic stem cells, our candidate is designed to be a “living therapy” that has the potential to break the cancer-induced immune tolerance and to establish a competent immune surveillance throughout the life of the patient. | |
| ● | Agnostic Response: In contrast to antigen-restricted chimeric antigen receptor T cells (CAR-T), our platform is not restricted to a pre-selected tumor antigen, nor any one tumor type. As such, it may be applied to a broad number of solid tumors and cancer subtypes, thereby overcoming one of the central unresolved challenges of immune-oncology cancer therapies. |
Our lead product candidate, Temferon, was developed using our platform and carries an interferon-alpha (IFN-α) payload. IFN-α is a well-known therapeutic that was previously administered intravenously for treatment of various cancers but is currently rarely used due to its systemic toxicity. The Temferon-modified TEMs express the transgene payload, IFN-α, in the tumor microenvironment resulting in the breakdown of tumor induced immune-tolerance. As a result, the immune system can recognize the tumor, respond, and inhibit tumor growth. Because Temferon delivers the IFN-α payload directly to the tumor, we believe it will demonstrate clinical activity without the side effect profile of systemic delivery of IFN-α. In preclinical cancer mouse models treated with Temferon, both direct (anti-angiogenic, pro-apoptotic) and indirect (immune response) effects were observed.
We are currently developing Temferon for the treatment of glioblastoma multiforme (GBM) in patients with GBM who have an unmethylated MGMT gene promoter (uMGMT-GBM). GBM is the most common malignant primary brain tumor accounting for more than half of all central nervous system (CNS) cancers, and unmethylated MGMT promoter status is identified in approximately 60% of GBM patients. Patients suffering from GBM have limited, non-curative treatment options. Although these treatments may improve survival, the prognosis for GBM patients remains poor with a median overall survival (mOS) of approximately 13 to 15 months and only 5.5% of patients estimated to be alive five years after diagnosis. With no curative treatments available and such poor prognosis for patients, there remains a large, unmet medical need. We chose GBM among our first targets for clinical development after considering the medical need, the active role that TEMs have in GBM pathology, and the high number of newly diagnosed uMGMT-GBM patients potentially interested in participating in our study. As a result, we believe GBM offers a good profile for our initial proof of concept trial in humans. We are currently conducting a Phase 1/2a dose-escalation clinical trial with Temferon in newly diagnosed uMGMT-GBM patients in Italy. We intend to complete patient enrollment by the second quarter of 2022 and to use the preliminary results of our Phase 1/2a clinical trial to support a Clinical Trial Application to conduct in Europe a multicenter Phase 2 trial in uMGMT-GBM, which we currently intend to conduct primarily in Italy. As of November 15th, 2021, we have administered Temferon to a total of 15 patients in clinical trials. The preliminary results to date show that Temferon was generally well tolerated with no dose limiting toxicities identified so far.
| 1 |
We also intend to develop Temferon for the treatment of other solid tumor indications, and locally advanced hepatocellular carcinoma (HCC) and intra-hepatic cholangiocarcinoma (ICC) are two indications under consideration. HCC and ICC are gastrointestinal (GI) cancers affecting the digestive system. HCC is a primary malignancy of the liver that occurs predominantly in patients with underlying chronic liver disease and cirrhosis. ICC is a biliary tract cancer and represents approximately 3% of all GI malignancies. The prognosis for patients with locally advanced HCC or ICC remains poor with few therapeutic options which have limited clinical benefits. While we are considering development of Temferon for these liver indications for similar reasons as GBM (i.e. the high unmet need, TEMs’ role in HCC and ICC pathology, and the number of newly diagnosed patients potentially eligible for our study), we are also evaluating development of Temferon for other solid tumor indications. If HCC and ICC will be selected, we intend to submit a Clinical Trial Application (CTA) for conducting a Phase 1/2a study of Temferon to the Italian Medicines Agency (Agenzia Italiana del Farmaco, or AIFA), the public institution responsible for the regulatory activity of pharmaceuticals in Italy.
In addition to our Temferon programs being developed in uMGMT-GBM patients, or other solid tumor indications such as HCC and ICC, we have exclusive option rights to license (i) Temferon for the treatment of additional indications, and (ii) other drug candidates that are currently in the preclinical stage of development both as standalone treatments and as combination therapies.
AGC Biologics’ facility in Milan, Italy, will continue manufacturing LVV and Temferon to support Genenta’s trials. For further larger studies we may use AGC’s 60,000 square meter cell and gene therapy manufacturing facility located in Longmont, Colorado (U.S.), which AGC purchased from Novartis in July 2021 or another US-based CMO.
Research and Development Pipeline
Our portfolio of clinical and preclinical ex-vivo autologous gene cancer therapies is based on our technology platform, which was originally developed in our founders’ laboratories at Ospedale San Raffaele, or OSR. Through our collaboration with OSR, we have worldwide commercial rights to Temferon for the treatment of GBM, HCC and ICC, as well as exclusive option rights to license all of our other programs. Specifically, we retain exclusive option rights to license (i) any platform improvements, including our second-generation technology, which includes developments to enable the on/off regulation of the therapeutic transgene, (ii) products for additional indications that utilize our platform technology but use different transgene payloads, and (iii) combinations of our platform with therapies in the immuno-oncology (IO) field, such as immune checkpoint inhibitors (ICI), CAR-T cell therapies and T cell receptor (TCR) therapies.
| 2 |
Our current pipeline, with clinical and preclinical stage programs, is summarized below:
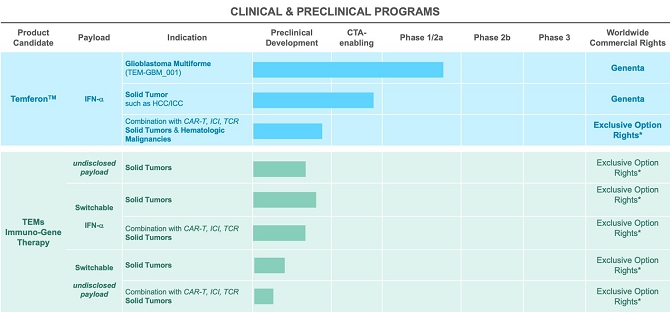
*Genenta has options/rights on IP derived from preclinical data generated at SR-Tiget laboratories.
Strategy
We are developing novel cancer therapeutics using our autologous ex-vivo gene therapy platform, to initially address the unmet medical needs of GBM patients and patients suffering from another solid cancer indication such as HCC and ICC. Ultimately, we hope to broaden our platform to treat a wide variety of cancers by pursuing the following strategies:
Advance development of our leading clinical-stage product candidate, Temferon in the U.S.
We are currently conducting a Phase 1/2a dose-escalation study to primarily evaluate the safety and tolerability of Temferon in up to 21 uMGMT-GBM patients, following radiotherapy treatment. In the fourth quarter of 2022, we intend to file the regulatory package required to initiate in Europe a multicenter Phase 2 trial in uMGMT-GBM, which we currently intend to conduct primarily in Italy, to evaluate the safety and efficacy of Temferon in up to 27 uMGMT-GBM patients (both newly diagnosed and with progressive disease) who have an unmethylated MGMT promoter where we intend to measure progression free survival (PFS) and overall survival (OS) as endpoints. In advance of our CTA submission and although it is not required for our European trial, we submitted a pre-IND meeting request to the FDA and received written responses from the Agency regarding future possible Phase 2 clinical studies and drug product manufacturing as part of our development of long-term strategies following our European trial. There cannot be any assurance that such meetings will lead to clinical and manufacturing strategies in the US in the future, which is dependent on feedback from the FDA and advice from key opinion leaders and US partnering clinical centers, as well as the availability of financial and other resources.
Extend our product pipeline across multiple indications
We intend to extend our product pipeline by:
| ● | Identifying additional indications suitable for Temferon. We are in the planning stages for a second study using Temferon in another solid tumor indication and locally advanced HCC and ICC are under consideration. The Istituto Superiore di Sanità (ISS), an independent committee with oversight from the Italian Ministry of Health, must issue a positive opinion regarding our CTA before AIFA will approve it. In addition to these indications, we believe there may be additional cancer indications which actively recruit TEMs to proliferate for which Temferon may be a suitable therapy. | |
| ● | Using our platform with different transgene payloads. Our platform technology is designed to enable us to use different transgene payloads to potentially achieve therapeutic outcomes in selected cancer indications. We are currently conducting preclinical studies for two therapeutics using our platform with different payloads targeting solid tumors. | |
| ● | Developing a second-generation platform that enables the “on-demand” release of the transgene payload. We intend to develop a second-generation technology platform that allows the drug products to be switched on to exert the therapeutic effects and switched off if they are no longer needed, or to mitigate toxicity. This technology may enable us to expand our treatment options to broader patient populations. |
| 3 |
| ● | Exploring combination therapies. We will seek to enter into collaborations with other companies to explore combination studies of our therapeutics with other cancer therapies, such as ICI, CAR-T cell therapies and TCR therapies. We believe our product candidate, as a result of its MoA, has the potential to enhance the durability and efficacy of the existing therapies, thus abolishing the immune tolerance to the tumor. | |
| ● | Exploiting in-licensing opportunities with OSR. We intend to exploit in-licensing opportunities with OSR, a co-founding shareholder. |
Develop and maintain efficient manufacturing processes to support anticipated growth
Currently, for Temferon manufacturing we rely on AGC Biologics, a contract development and manufacturing organization (CDMO), at their cell and gene therapy facility located in Milan (Italy). After the completion of our planned multicenter Phase 2 uMGMT- GBM study most likely to be conducted in Italy, to meet our drug product supply needs for conducting larger trials we intend to enter into a supply agreement with a US-based CMO for the manufacturing of our products. In July 2021 AGC Biologics purchased a 60,000 square meters cells and gene therapy manufacturing facility located in Longmont, Colorado (U.S.).
Establish a patient-centered infrastructure and strong relationships with key U.S. opinion leaders working in our disease area
Since cell and gene-based therapies are relatively new approaches in oncology, we intend to implement programs to improve patient and physician education regarding the availability of gene therapy-based products for those cancers with a high unmet medical need. To this end, we are discussing with Antonio Chiocca, MD, Professor Neurosurgeon-in-Chief and Chairman, Department of Neurosurgery at Brigham and Women’s Hospital in Boston, MA, Frederick Lang, MD, Professor and Chairman of the Department of Neurosurgery at MD Anderson in Houston, TX, and David A. Reardon, MD, Department of Medical Oncology at Dana-Farber Cancer Institute in Boston, MA.
Develop opportunistic partnership(s) with pharmaceutical company(s)
We may choose to partner with larger pharmaceutical companies whose core competencies and oncology strategies are in line with ours.
Our Strengths
We believe that our growing body of early clinical data supporting the potential of our autologous ex-vivo gene therapy approach, coupled with our founders’ expertise in the development, manufacturing and commercialization of gene and cell therapies, positions us well to provide potentially transformative therapies through a single administration to patients suffering from a broad range of cancers. We believe our key strengths include:
| ● | Unique and valuable expertise. We are conducting our clinical trials at OSR, a leading center for ex-vivo gene therapy for inherited diseases. OSR has treated more than 121 patients worldwide (one of the highest number of patients treated with gene therapy for rare diseases in a research hospital) using an ex-vivo viral vector platform similar to the one we are developing for cancer treatment. Members of our executive leadership team have held senior positions at GSK, Merck, Annapurna-Adverum and other companies specializing in gene and cell therapies and rare diseases. We have partnered with academic institutions that are pioneers in autologous ex-vivo gene therapy and hold exclusive option rights to license additional patents and know-how to build our portfolio. Partnerships with leading academic institutions that are well recognized in the gene therapy field, such as SR-Tiget and OSR, are a core part of our research engine through which we are working to advance the clinical development of our product candidates and to identify new opportunities that we believe have comparably high probabilities of success in a preclinical setting. We believe our expertise, combined with our plan to leverage our relationships with leading academic institutions, will help expedite the commercialization of our lead clinical-stage product candidate and further expand our pipeline. |
| 4 |
| ● | Deep pipeline with broad utility. We believe that the flexibility of our technology platform combined with our exclusive option rights to in-license additional programs, give us the ability to grow our pipeline by targeting a broad set of cancer diseases. | |
| ● | Durable therapeutic potential. Preliminary interim clinical data collected from fifteen uMGMT- GBM patients following a single administration of Temferon displayed modified cells at 18 months, the last measured timepoint to date. | |
| ● | Designed for tumor restricted therapeutic payload delivery and release. The design of our transgene expression cassette is intended to restrict payload expression to the tumor microenvironment. The local and tumor restricted therapeutic gene deployment approach is designed to focus the pleiotropic anti-tumor activities of the selected payload, by limiting the toxic manifestation that results from standard systemic administration of the payload. | |
| ● | Agnostic approach. Our immune-gene therapy approach is designed to be a tumor-agnostic immunotherapy that does not rely on any specific target or tumor type, and we believe it could be successfully applied to a potentially broad range of cancers and immune contexts. | |
| ● | Solid tumors targeting. Our platform has the potential to efficiently target solid tumors. Solid tumors are difficult to treat, even by the most novel and leading-edge technologies such as ICIs and CAR-T cells. Our cellular carrier, TEMs, is spontaneously and actively recruited by growing tumors and is found in several human solid tumors, irrespective of location. | |
| ● | Active and sustained tumor surveillance. Our immune-gene therapy is designed to trigger the patient’s own immune response that establishes an active immune surveillance. Our preclinical work, which used different cancer models (B-cell acute lymphoblastic leukemia - B-ALL and GBM), as well as preliminary data collected from our uMGMT-GBM patients suggests the occurrence of changes in the immune system. | |
| ● | Fine dose tuning. Our platform holds promise to fine tune the dose to be administered based on individual patient characteristics. | |
| ● | Preliminary data indicate that our approach is feasible and well-tolerated. Temferon has been well-tolerated in the limited number of patients treated to date. Our ex-vivo modification of the patient’s own HSPCs and cryopreservation allow us to formulate the patient’s drug product prior to administering the therapy. | |
| ● | LVV as transgene payload delivery vehicles. LVVs are particularly attractive for clinical applications due to their capacity to transfer large genes/payloads and their ability to efficiently transduce non-proliferating or slowly proliferating cells, such as hematopoietic stem and progenitor cells that allow a persistent gene expression in transduced cells. Moreover, LVVs have a potentially reduced risk of genotoxicity compared to gamma-retroviral vectors (gRV). A large number of patients have been treated both with other LVV gene therapy products approved for sale and with clinical-stage product candidates for rare diseases worldwide, and generally these therapies have been well tolerated. We believe that long-term extensive follow-up across multiple diseases, with vectors expressing different genes, demonstrates the potential safety of our LVV-based autologous ex-vivo gene therapy approach. | |
| ● | Applicability to a potentially large number of patients and indications. We believe our autologous ex-vivo gene therapy approach has broad therapeutic potential across a large number of malignancies. The ex-vivo transduction of HSPCs allows for the potentially long-term production of a differentiated cellular carrier loaded with the therapeutic gene and the consequent distribution of the therapeutic payload throughout multiple organs and tissues containing solid tumors. |
| 5 |
Company History and Management Team
We were founded in 2014 by San Raffaele Hospital (OSR) in Milan, a globally recognized premier research hospital for ex-vivo gene therapy, with Pierluigi Paracchi (our CEO), Luigi Naldini (Chairman of our Executive Scientific Board) and Bernhard Gentner (a member of our Executive Scientific Board), to develop potential ground-breaking cell and gene cancer therapies. We leverage the vast experience in LVV technology of the San Raffaele Telethon Institute for Gene Therapy (SR-Tiget). SR-Tiget, a joint venture between OSR and Fondazione Telethon (Telethon), is a world leading cell and gene therapy research institution at the forefront of developing therapies for rare diseases. SR-Tiget has a proven track record for successful collaborative clinical research programs in ex-vivo gene therapy. Its research has resulted in a number of approved products, including Strimvelis, an ex-vivo gammaretroviral vector-based gene therapy for adenosine deaminase severe combined immunodeficiency (ADA-SCID), and Libmeldy, an ex-vivo gene therapy for the treatment of early-onset metachromatic leukodystrophy (MLD) patients, both marketed by Orchard Therapeutics. Our platform was developed in the SR-Tiget laboratories of our founders, Prof. Naldini and Dr. Gentner, and we hold exclusive rights and option rights, to certain intellectual property (IP) originating there.
Since closing our first round of funding in May 2015, we have recruited a leading management team, established a manufacturing process for our drug product candidate, completed preclinical activities (research and Good Laboratory Practice – GLP – grades), engaged with Italian, European and U.S. Key Opinion Leaders (KOLs) to identify our clinical lead indications, and submitted our first CTA (June 2018).
Our leadership team has a proven track record as biotech executives. Their expertise spans from finance and venture capital to medical affairs, from scientific research to clinical drug product development and clinical trial management. For example, members of our management team have been involved in the successful development of Ethical Oncology Science, which was acquired in 2013 for over $400 million, and Strimvelis the first ever ex-vivo approved gene therapy product that was developed under the guidance of Carlo Russo, our Chief Medical Officer and Head of Development (formerly Head of Development of R&D Biopharm and Rare Disease Units at GSK). Our management team members have played important roles in both large pharma companies such as Merck and GSK, and biotech startups, such as Adverum, Annapurna, VaxInnate Corporation, OncoSec Medical, Biological Dynamics and GenMark Diagnostics. We believe this multi-disciplinary competence, provides a unique blend for the development of innovative gene and cell therapy products, and constitutes a fertile ground for alliances with industrial partners that could help us bring new therapies to patients.
Risks Associated with Our Business
Our business, and investing in our securities, are subject to numerous risks and uncertainties, as more fully described in the section “Risk Factors” included elsewhere in this prospectus. You should read these risks before making a decision to invest in our securities. If any of these risks actually occur, our business, financial condition or results of operations would likely be materially adversely affected. In each case, the trading price of our securities would likely decline, and you may lose all or part of your investment. The following is a summary of some of the principal risks we face:
| ● | We have a limited operating history and have incurred significant losses since our inception. We have never generated revenue and will require significant additional funds, which may not be available on acceptable terms or at all. As a result, you could lose your entire investment. | |
| ● | Our lentiviral-based gene therapy product candidates are based on a novel technology that is in preliminary stages of evaluation, which makes it difficult to predict the time and cost of product candidate development or the likelihood of receiving required regulatory approvals. Our rights to the intellectual property underlying our novel technology derive solely from our license agreement with OSR and any failure to comply with the terms of such license agreement could have a material adverse effect on our intellectual property position and our ability to seek approval for and ultimately commercialize such product candidates. | |
| ● | Even if we do receive regulatory approvals for our product candidates, they may face commercialization issues from significantly larger oncology competitors, unfavorable pricing regulations or lack of acceptance by doctors, hospitals, patients and insurers. Our product candidates and the process for administering them may also cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit their commercial potential or result in significant negative consequences following any potential marketing approval. |
| 6 |
| ● | We currently have very few employees and rely almost entirely on the efforts of third parties over which we have limited control and in certain cases are reliant on a sole supplier for our materials. Our contract research organizations, or CROs, may fail to observe the standards to which our studies must be conducted and our product candidates may not be approved as a result. Likewise, our contract manufacturing organizations, or CMOs, may not continue producing the needed materials for preclinical and clinical testing, whether as a result of their commitments to other customers or otherwise. Any failure of these third parties to meet our expectations would have a materially adverse effect on our product development efforts. | |
| ● | Our clinical trials for Temferon must be successful if we are to seek and obtain regulatory marketing application through the submission of a new Biological License Application (BLA) and marketing authorization application (MAA) with the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA), respectively. Advanced clinical trials are often not successful even if prior trials were successful, and even if we are able to conduct advanced clinical trials and those trials are successful, we may not obtain necessary regulatory approvals for Temferon or we may be unable to successfully commercialize our products even if we receive the necessary regulatory approvals. | |
| ● | The ongoing COVID-19 pandemic could adversely impact our ongoing and planned clinical trials, operations and financial condition, and our overall generation of revenues may not succeed on the time frames we expect or at all. | |
| ● | Our Chief Executive Officer, directors and shareholders who own more than 5% of our outstanding ordinary shares before this offering currently own approximately 47.1% of our outstanding ordinary shares and will own approximately 40.2% of our ordinary shares upon the completion of this offering and will therefore be able to exert significant control over matters submitted to our shareholders for approval. | |
| ● | As a public company following the conclusion of this offering, we will need to comply with extensive additional U.S. and Italian governmental and Nasdaq regulations, which will be expensive, and which will require significant management attention. | |
| ● | As a company organized under the laws of Italy and whose shares are represented by ADSs, the rights of investors in the company following this offering will differ in several material respects from the rights of holders of shares of common stock of a US domestic company and may not afford investors the same protections. |
Corporate Information
Genenta was formed as an Italian limited liability company (società a responsabilità limitata, or S.r.l.) in 2014. In May 2021, we changed the legal form of our company under Italian law to a joint stock company (società per azioni, or S.p.A.).
Our principal executive offices are located at Via Olgettina No. 58, 20132 Milan, Italy. Our telephone number in Italy is +39.02.2643.6639. Our website address is www.genenta.com. The information contained on, or that can be accessed through, our website is not part of this prospectus. We have included our website address in this prospectus solely as an inactive textual reference.
The Securities and Exchange Commission, or the SEC, also maintains an Internet website that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC. Our filings with the U.S. Securities and Exchange Commission, or the SEC, will also be available to the public through the SEC’s website at http://www.sec.gov.
| 7 |
Implications of Being an “Emerging Growth Company”
We qualify as an “emerging growth company,” as defined in Section 2(a) of the Securities Act of 1933, as amended, or the Securities Act, as modified by the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. As such, we are eligible to, and intend to, take advantage of certain exemptions from various reporting requirements applicable to other public companies that are otherwise applicable generally to public companies. These provisions include:
| ● | only two years of audited financial statements in addition to any required unaudited interim financial statements with correspondingly reduced “Management’s Discussion and Analysis of Financial Condition and Results of Operations” disclosure; | |
| ● | reduced disclosure about our executive compensation arrangements; | |
| ● | exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting; and | |
| ● | an exemption from new or revised financial accounting standards until they would apply to private companies and from compliance with any new requirements adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotation. |
We could remain an “emerging growth company” for up to five years, or until the earliest of (a) the last day of the first fiscal year in which our annual gross revenue exceeds $1.07 billion, (b) the date that we become a “large accelerated filer” as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, or the Exchange Act, which would occur if the market value of our securities that is held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter, or (c) the date on which we have issued more than $1 billion in nonconvertible debt during the preceding three-year period. We may choose to take advantage of some but not all of these exemptions. We have taken advantage of reduced reporting requirements in this prospectus. Accordingly, the information contained herein may be different from the information you receive from other public companies in which you hold stock. We have elected to avail ourselves of the exemption for the delayed adoption of certain accounting standards and, therefore, will not be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
Implications of being a “Foreign Private Issuer”
Upon consummation of this offering, we will be subject to the information reporting requirements of the Exchange Act that are applicable to “foreign private issuers,” and under those requirements we will file reports with the SEC. As a foreign private issuer, we will not be subject to the same requirements that are imposed upon U.S. domestic issuers by the SEC. Under the Exchange Act, we will be subject to reporting obligations that, in certain respects, are less detailed and less frequent than those of U.S. domestic reporting companies. For example, we will not be required to issue quarterly reports, proxy statements that comply with the requirements applicable to U.S. domestic reporting companies, or individual executive compensation information that is as detailed as that required of U.S. domestic reporting companies. We also will have four months after the end of each fiscal year to file our annual report with the SEC and will not be required to file current reports as frequently or promptly as U.S. domestic reporting companies. Our officers, directors and principal shareholders will be exempt from the requirements to report transactions in our equity securities and from the short-swing profit liability provisions contained in Section 16 of the Exchange Act. As a foreign private issuer, we will not be subject to the requirements of Regulation FD (Fair Disclosure) promulgated under the Exchange Act. In addition, as a foreign private issuer, we will be permitted to follow certain home country corporate governance practices instead of those otherwise required under the Nasdaq Stock Market rules for domestic U.S. issuers and will not be required to be compliant with all Nasdaq Stock Market rules as of the date of our initial listing on Nasdaq as would domestic U.S. issuers (see “Risk Factors—Risks Related to this Offering and Ownership of Our Securities”). These exemptions and leniencies will reduce the frequency and scope of information and protections available to you in comparison to those applicable to a U.S. domestic reporting company. We intend to take advantage of the exemptions available to us as a foreign private issuer during and after the period we qualify as a “foreign private issuer.”
| 8 |
THE OFFERING
| Securities offered by us | 2,608,695, including ADSs representing 1,608,695 ordinary shares |
| Ordinary shares issued and outstanding prior to this offering | 15,000,000 ordinary shares |
| Ordinary shares to be issued and outstanding after this offering | 17,608,695 ordinary shares (or 18,000,000 ordinary shares if the underwriters exercise their option to purchase additional ADSs within 30 days of the date of this prospectus from us in full) (includes ordinary shares represented by the ADSs) |
Reserved Offering | At our request, 1,000,000 ordinary shares covered by the registration statement of which this prospectus is part have been reserved for sale, at the initial public offering price, by the Company, directly or through agents, in the Reserved Offering to existing shareholders of the Company, as a mitigant for the exclusion of pre-emptive rights as resolved by the general shareholders meeting of the Company in accordance with Italian corporate law and our by-laws. The Reserved Offering will be effected in compliance with the laws of Italy, at the same price to investors as the offering of ADSs. We will receive the full proceeds from the Reserved Offering and will not pay any underwriting discounts or commissions with respect to ordinary shares that we sell in the Reserved Offering but have agreed to pay an advisory fee to the representatives with respect to such sales. The offer and sale of ordinary shares in the Reserved Offering (i) is contingent upon the completion of the offering of ADSs and will commence and be consummated concurrently with such offering of ADSs, (ii) has been registered for purposes of the securities laws of the United States under the registration statement of which this prospectus is part, and (iii) is conducted in Italy under applicable exemptions from Italian and European regulations concerning prospectus. |
| The ADSs | Each ADS represents one of our ordinary shares, with no par value. The ADSs may be evidenced by American Depositary Receipts, or ADRs. The depositary will hold in custody the ordinary shares underlying the ADSs and you will have the rights of an ADS holder as provided in the deposit agreement among us, the depositary and owners and holders of ADSs from time to time.
To better understand the terms of the ADSs, you should carefully read the “Description of the Offered Securities” section of this prospectus. We also encourage you to read the deposit agreement, which is incorporated by reference as an exhibit to the registration statement that includes this prospectus. |
| Over-allotment option | We have granted to the underwriters an option exercisable for a period of 30 days after the date of this prospectus to purchase up to 241,304 additional ADSs from us solely to cover over-allotments, if any. If the underwriters exercise all or part of this option, it will purchase securities covered by the option at the public offering price per ADS, less the underwriting discounts and advisory fees. See “Underwriting.” |
| Use of proceeds | We estimate that the net proceeds from our issuance and sale of 2,608,695 securities in this offering will be approximately $26.4 million, assuming an offering price of $11.50 per ADS, the midpoint of the estimated price range set forth on the cover page of this prospectus, and after deducting underwriting discounts and commissions and offering expenses payable by us. If the underwriters exercise the over-allotment option in full, we estimate that the net proceeds from this offering will be approximately $30.6 million, assuming an offering price of $11.50 per ADS, and after deducting underwriting discounts and commissions and offering expenses payable by us. We currently expect to use the net proceeds from this offering:
● to conduct a clinical trial in selected cancer patient populations;
● to support the ongoing Temferon TEM-GBM 001 trial and its long term follow up;
● to start a new Temferon clinical program in a second solid tumor indication;
● to fund further preclinical research for the development of Temferon across broad cancer indications;
● to fund Temferon manufacturing activities including LVV manufacturing, stability and process scalability studies, and tech transfer activities; and
● working capital and general corporate purposes.
See “Use of Proceeds” for additional information. See “Use of Proceeds” for additional information. |
| 9 |
| Depositary | The Bank of New York Mellon |
| Custodian | The Bank of New York Mellon, as custodian, acting through an office located in the United Kingdom |
| Proposed Nasdaq trading symbol and listing | We have applied to list the ADSs on the Nasdaq Capital Market under the symbol “GNTA.” No assurance can be given that such listing will be approved or that a liquid trading market will develop for the ADSs. |
| Lock-up | Our directors, executive officers, and certain shareholders have agreed with the underwriters not to offer for sale, issue, sell, contract to sell, pledge or otherwise dispose of any of our ordinary shares or securities convertible into ordinary shares for a period of 180 days following the date of this prospectus. See “Underwriting.” |
| Risk factors | You should read the “Risk Factors” section starting on page 12 of this prospectus for a discussion of factors to consider carefully before deciding to invest in our securities. |
The number of our ordinary shares that are and will be outstanding immediately before and after this offering as shown above is based on 15,000,000 ordinary shares, with no par value, outstanding as of June 30, 2021, and excludes ordinary shares that are available for future issuance under our 2021-2025 Equity Incentive Plan.
Unless otherwise indicated, this prospectus reflects and assumes no exercise by the underwriters of their over-allotment option or the warrants to purchase ADSs at an exercise price per share equal to 125% of the initial public offering price per ADS or $14.375, that will be issued to the representatives of the underwriters in connection with this offering (the “Representatives’ Warrants”).
| 10 |
SUMMARY FINANCIAL DATA
We have derived the following summary statements of operations data for the years ended December 31, 2020 and 2019, and the six months ended June 30, 2021 and 2020, and summary balance sheet data as of June 30, 2021 from our audited and unaudited financial statements included elsewhere in this prospectus. Our historical results are not necessarily indicative of the results that may be expected in the future. The following summary financial data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and related notes included elsewhere in this prospectus, and are qualified entirely by reference to such financial statements.
Our financial statements included in this prospectus are prepared and presented in accordance with accounting principles generally accepted in the United States, or U.S. GAAP.
| Year Ended | Six Months Ended | |||||||||||||||
| December 31, | June 30, | |||||||||||||||
| in Euros, except share data | 2020 | 2019 | 2021 | 2020 | ||||||||||||
| (Unaudited) | ||||||||||||||||
| Research and development expenses | € | 4,688,461 | € | 3,702,982 | € | 3,199,234 | € | 2,072,129 | ||||||||
| General and administrative expenses | 901,765 | 921,520 | 842,236 | 404,885 | ||||||||||||
| Total operating expenses | 5,590,226 | 4,624,502 | 4,041,470 | 2,477,014 | ||||||||||||
| Loss from operations | (5,590,226 | ) | (4,624,502 | ) | (4,041,470 | ) | (2,477,014 | ) | ||||||||
| Other income | 5,966 | 36,331 | 2,679 | 559 | ||||||||||||
| Foreign exchange loss | (7,754 | ) | (9,552 | ) | (9,111 | ) | (2,849 | ) | ||||||||
| Loss | (5,592,014 | ) | (4,597,723 | ) | (4,047,902 | ) | (2,479,304 | ) | ||||||||
| Comprehensive loss: | — | — | — | — | ||||||||||||
| Total comprehensive loss | € | (5,592,014 | ) | € | (4,597,723 | ) | € | (4,047,902 | ) | € | (2,479,304 | ) | ||||
| Pro forma information (unaudited): | ||||||||||||||||
| Pro forma net loss | € | (5,592,014 | ) | € | (4,597,723 | ) | € | (4,047,902 | ) | € | (2,479,304 | ) | ||||
| Pro forma net loss per share - basic and diluted | € | (0.38 | ) | € | (0.36 | ) | € | (0.27 | ) | € | (0.17 | ) | ||||
| Weighted average pro forma number of shares outstanding - basic and diluted | 14,539,534 | 12,651,158 | 14,772,610 | 14,420,904 | ||||||||||||
| (1) | We have presented pro forma basic and diluted loss per share for the years ended December 31, 2020 and 2019 and the six months ended June 30, 2021 and 2020, which consists of our historical loss attributable to Genenta Science S.r.l., divided by the pro forma basic and diluted weighted average number of shares outstanding after giving effect to the Corporate Conversion. See Notes 2 and 10 to our interim financial statements and our Management Discussion and Analysis included elsewhere in this prospectus for additional information regarding the method used to calculate the pro forma basic and diluted loss per share and the pro forma weighted average number of shares used in the computation of the per share amounts. |
| As of June 30, 2021 (Unaudited) | ||||||||
| in Euros | Actual | Proforma As Adjusted(1) | ||||||
| Balance Sheet Data: | ||||||||
| Cash and cash equivalents | € | 10,553,906 | € | 33,967,971 | ||||
| Total assets | 13,291,009 | 36,705,074 | ||||||
| Total long-term liabilities | 21,645 | 21,645 | ||||||
| Accumulated deficit | (25,538,377 | ) | (25,538,377 | ) | ||||
| Total equity | 11,601,054 | 35,015,119 | ||||||
| (1) | The as adjusted balance sheet data gives effect to the issuance and sale 2,608,695 securities, including ADSs, representing 1,608,695 ordinary shares, in this offering at the assumed initial public offering price of $11.50 per ADS or ordinary share, the midpoint of the price range set forth on the cover page of this prospectus, after deducting the estimated underwriting discounts, estimated advisory fees and estimated offering expenses payable by us. Each $1.00 increase (decrease) in the assumed initial public offering price of $11.50 per ADS would increase (decrease) the pro forma as adjusted amount of each of cash and cash equivalents, total assets and total equity by approximately €2.6 million, assuming that the number of ADSs offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts, estimated advisory fees and estimated offering expenses payable by us. Similarly, each increase (decrease) of 1.0 million in the number of ADSs and ordinary shares offered by us at the assumed initial public offering price would increase (decrease) each of cash and cash equivalents, total assets and total equity by approximately €11.5 million. The as adjusted information discussed above is illustrative only and will be adjusted based on the actual initial public offering price and other terms of our initial public offering determined at pricing. |
| 11 |
An investment in our securities involves a high degree of risk. We operate in a dynamic and rapidly changing industry that involves numerous risks and uncertainties. You should consider carefully the risks described below, as well as the financial or other information included in this prospectus, including our financial statements and the related notes, before you decide to buy our securities. The risks and uncertainties described below are not the only risks facing us. We may face additional risks and uncertainties not currently known to us or that we currently deem to be immaterial. Any of the risks described below, and any such additional risks, could materially adversely affect our business, financial condition or results of operations. In such case, you may lose all or part of your original investment.
Risks Related to Our Financial Position and Capital Requirements
We are a clinical-stage biopharmaceutical company with limited operating history, which may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
We are an emerging biotechnology company with a limited operating history. Investment in biopharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk that any potential product candidate will fail to demonstrate adequate effect or an acceptable safety profile, gain regulatory approval and become commercially viable. All of our product candidates are in early development and none have been approved for commercial sale. We have not demonstrated an ability to successfully complete late-stage clinical trials, obtain regulatory approvals, manufacture our product candidates at commercial scale or arrange for a third-party to do so on our behalf, conduct sales and marketing activities necessary for successful commercialization, or obtain reimbursement in the countries of sale. We may encounter unforeseen expenses, difficulties, complications, and delays in achieving our business objectives. Our short history as an operating company makes any assessment of our future success or viability subject to significant uncertainty. If we do not address these risks successfully or are unable to transition at some point from a company with a research and development focus to a company capable of supporting commercial activities, then our business will be materially harmed.
We have incurred significant losses in every year since our inception. We expect to continue to incur losses over the next several years and may never achieve or maintain profitability.
We have no products approved for commercial sale, have not generated any revenue from commercial sales of our product candidates, and have incurred losses each year since our inception. Our losses for the six months ended June 30, 2021 and 2020 were approximately €4.0 million and €2.5, respectively; and for the years ended December 31, 2020 and 2019 were approximately €5.6 million and €4.6 million, respectively. As of June 30, 2021 and December 31, 2020, we had an accumulated deficit of approximately €(25.5) million and €(21.5) million, respectively. Substantially all of our operating losses resulted from costs incurred in connection with our research and development activities, including pre- and non-clinical development of our gene therapy product candidates, namely our leading product candidate Temferon, and from general and administrative costs associated with our operations.
We expect that it will be several years, if ever, before we have any product approved for commercial sale. We have funded our operations to date primarily through proceeds from the private placement of ordinary shares to our founding shareholders. We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. We anticipate that our expenses will increase substantially if, and as, we
| ● | continue the research and development of our gene therapy product candidates, including continuing and conducting preclinical studies and clinical trials of Temferon and conducting preclinical studies and clinical trials for any additional product candidates that we may pursue in the future; | |
| ● | develop and obtain regulatory approval for registration studies for our current product candidate Temferon and any additional product candidates that we may pursue in the future; |
| 12 |
| ● | seek regulatory and marketing approvals for our product candidates that successfully complete clinical studies, if any, including obtaining orphan drug designation; | |
| ● | establish a sales, marketing, and distribution infrastructure to commercialize any product candidates for which we may obtain marketing approval; | |
| ● | industrialize our lentivirus ex-vivo gene therapy approach into a robust, scalable and, if approved, commercially viable process; | |
| ● | maintain, protect, and expand our intellectual property portfolio; | |
| ● | hire and retain qualified technical personnel, such as clinical, quality control, commercial and scientific personnel; | |
| ● | expand our infrastructure and facilities to support our operations, including adding equipment and physical infrastructure to support our research and development; and | |
| ● | incur additional legal, accounting and other expenses associated with operating as a public company. |
We have not generated revenue from product sales and may never be profitable.
Our ability to generate revenue from product sales and achieve profitability depends on our ability, alone or with partners, to successfully complete the development of, and obtain the regulatory approvals necessary to commercialize, our product candidates. We do not anticipate generating revenues from product sales for the next several years, if ever, and our ability to do so depends heavily on our success in many areas, including but not limited to:
| ● | completing research and pre- and non-clinical development of our products candidates | |
| ● | seeking and obtaining regulatory and marketing approvals for product candidates for which we complete clinical studies, if any; | |
| ● | establishing and maintaining supply and manufacturing processes and relationships with third parties that can provide adequate (in amount and quality) products and services, and at acceptable costs, to support clinical development and market demand for our product candidates, if marketing approval is received; | |
| ● | negotiating favorable terms in any collaboration, licensing or other arrangements into which we may enter; and | |
| ● | obtaining market acceptance of our product candidates, if approved for marketing, as viable treatment options. |
Even if one or more of the product candidates that we develop is approved for commercial sale, we anticipate incurring significant costs associated with commercialization, with all associated risks and uncertainties. Therefore we cannot predict when, or if, we will be able to achieve profitability. Additional clinical trials or delays in the initiation and completion of clinical trials could cause our expenses to increase significantly and profitability to be further delayed.
Our failure to become and remain profitable would decrease the value of our company and could impair our ability to raise capital, maintain our research and development efforts, expand our business or continue our operations. A decline in the value of our company also could cause you to lose all or part of your investment.
| 13 |
We will need additional capital in the future. Raising additional capital by issuing securities may cause dilution to existing shareholders. Financing may not be available on acceptable terms, or at all. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate our product candidate development efforts or other operations.
As of June 30, 2021 and December 31, 2020, our cash and cash equivalents were approximately €10.6 and €15.5 million, respectively. If we continue to use cash at our historical rates of use we will need significant additional financing, which we may seek through a combination of private and public equity offerings, debt financings and collaboration, strategic alliance and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of any such offerings may include liquidation or other preferences that may adversely affect the then existing shareholders rights. Debt financing, if available, would result in increased fixed payment obligations, and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. If we raise additional funds through collaboration, strategic alliance or licensing arrangements with third parties, we may be required to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us. Even if we believe that we have sufficient funds for our current or future operating plans, we may seek additional capital if market conditions are favorable or if we have specific strategic considerations.
Our future funding requirements will depend on many factors, including but not limited to:
| ● | the scope, progress, results and costs of drug discovery, laboratory testing, pre- and non-clinical development and clinical trials for our product candidates, including Temferon; | |
| ● | the cost, timing and outcome of regulatory review of our product candidates; | |
| ● | the costs of future activities, including product sales, marketing, manufacturing and distribution, for any of our product candidates for which we receive marketing approval; | |
| ● | the cost of preparing, filing and prosecuting patent and trademark applications, maintaining and enforcing our intellectual property rights and defending our intellectual property-related claims; | |
| ● | any product liability or other lawsuits related to our products; | |
| ● | the expenses needed to attract and retain skilled personnel; and | |
| ● | the costs associated with being a public company. |
Any additional fundraising efforts may divert our management from their day-to-day activities, which may adversely affect our ability to develop and commercialize our product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of holders of our securities and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our securities to decline.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay or discontinue one or more of our research or development programs or the development or commercialization, if any, of any product candidates or be unable to expand our operations or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition and results of operations.
| 14 |
Risks Related to Product Development, Regulatory Approval and Commercialization
Our lentivirus ex-vivo gene transfer therapy product candidates are based on a novel technology, which makes it difficult to predict the time and cost of product candidate development and likelihood of subsequently obtaining regulatory approval.
We have concentrated our research and development efforts on our lentivirus ex-vivo gene transfer strategy approach, and our future success is highly dependent upon our successful development of commercially viable gene therapy product candidates. There can be no assurance that we will not experience problems or delays in developing new product candidates and that such problems or delays will not cause unanticipated costs, or that any such development problems can be solved. Because lentivirus ex-vivo gene transfer cell therapies represent a relatively new field of cellular immunotherapy and cancer treatment generally, developing and commercializing our product candidates subjects us to a number of risks and challenges, including:
| ● | obtaining regulatory approval for our product candidates, as the FDA, the EMA, the AIFA and other regulatory authorities have limited experience with lentivirus ex-vivo gene transfer therapies for cancer; | |
| ● | developing and deploying consistent and reliable processes for engineering a patient’s HSPCs ex vivo and infusing the engineered HSPCs back into the patient; | |
| ● | sourcing clinical and, if approved, commercial supplies of the materials used to manufacture our product candidates; | |
| ● | developing programming modules with the desired properties, while avoiding adverse reactions; | |
| ● | creating viral vectors capable of delivering multiple programming modules; | |
| ● | developing a reliable and consistent ex vivo gene modification and manufacturing process; | |
| ● | securing manufacturing capacity suitable for the manufacture of our product candidates in line with expanding enrollment in our clinical studies and our projected commercial requirements; | |
| ● | minimizing and avoiding infection and contamination during production of product candidates; | |
| ● | developing protocols for the safe administration of our product candidates; | |
| ● | educating medical personnel regarding our lentivirus ex-vivo gene transfer technologies and the potential side effect profile of each of our product candidates, such as potential adverse effects related to pyrexia and infections; | |
| ● | establishing integrated solutions in collaboration with specialty treatment centers in order to reduce the burdens and complex logistics commonly associated with the administration of lentivirus ex-vivo gene transfer cell therapies; | |
| ● | if and when we obtain any required regulatory approvals, establishing sales and marketing capabilities or partnerships to successfully launch and commercialize our product candidates and gaining market acceptance of a novel therapy; and | |
| ● | the availability of coverage and adequate reimbursement from third-party payors. |
| 15 |
We may not be able to successfully develop our lentivirus ex-vivo gene transfer product candidates or our technology in a manner that will yield products that are safe, effective, scalable or profitable. Additionally, because our technology involves the genetic modification of patient cells ex vivo, we are subject to additional regulatory challenges and risks, including:
| ● | regulatory requirements governing gene and cell therapy products have changed frequently and may continue to change in the future. To date, few CAR-T cell therapy products that involve the genetic modification of patient cells have been approved in the United States and/or the European Union, and no lentivirus ex-vivo gene transfer product candidates have been approved by any regulatory authority. In this regard, the European Commission has granted conditional marketing authorization for ZYNTEGLO; | |
| ● | genetically modified products could lead to lymphoma, leukemia or other cancers, or other aberrantly functioning cells in the event of improper insertion of a gene sequence into a patient’s chromosome, or due to other unknown causes; | |
| ● | although our viral vectors are not able to replicate, there is a risk with the use of lentiviral vectors that they could lead to new or reactivated pathogenic strains of virus or other infectious diseases; and | |
| ● | the FDA recommends a 15-year follow-up observation period for patients who receive treatment using gene therapies and guidance promulgated by the EMA requires a similar follow-up observation period for patients who receive cell therapeutic products, which has to be sufficient to observe the subjects for risks that may be due to the characteristics of the product, the nature and extent of the exposure, and the anticipated time of occurrence of delayed adverse reactions and could be as long as life-time, and we may need to adopt an observation period for our product candidates. |
Moreover, public perception and awareness of cell and gene therapy safety issues may adversely influence the willingness of subjects to participate in clinical trials of our product candidates, or if approved, of physicians to prescribe our products. Physicians, hospitals and third-party payors often are slow to adopt new products, technologies and treatment practices that require additional upfront costs and training. Treatment centers may not be willing or able to devote the personnel and establish other infrastructure required for the administration of lentivirus ex-vivo gene transfer cell therapies. Physicians may not be willing to undergo training to adopt this novel and personalized therapy, may decide the therapy is too complex to adopt without appropriate training and may choose not to administer the therapy. Based on these and other factors, hospitals and payors may decide that the benefits of this new therapy do not or will not outweigh its costs.
Our gene therapy product candidates and the process for administering our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit their commercial potential or result in significant negative consequences following any potential marketing approval.
Following treatment with our gene therapy product candidates, patients may experience changes in their health, including illnesses, injuries, discomforts or a fatal outcome. It is possible that as we study and test Temferon or other product candidates in larger, longer and more extensive clinical programs, or as use of our product candidates becomes more widespread if they receive regulatory approval, illnesses, injuries, discomforts and other adverse events that were observed in earlier clinical trials, as well as conditions that did not occur or went undetected in previous clinical trials, will be reported by patients. Gene therapies are also subject to the potential risk that occurrence of adverse events will be delayed following administration of the gene therapy due to persistent biological activity of the genetic material or other components of the vectors used to carry the genetic material. Many times, additional safety risks, contraindications, drug interactions, adverse events and side effects are only detectable after investigational products are tested in larger scale clinical trials or, in some cases, after they are made available to patients on a commercial scale after approval. Moreover, as noted above the FDA generally requires a long-term follow-up of study subjects for potential gene therapy-related adverse events for a 15-year period, including a minimum of five years of annual examinations followed by ten years of annual queries, either in person or by questionnaire, of study subjects. If additional clinical experience indicates that Temferon or any other product candidates or similar products developed by other companies has side effects or causes serious or life-threatening side effects, the development of the product candidate may fail or be delayed, or, if the product candidate has received regulatory approval, such approval may be revoked or limited.
| 16 |
There have been several significant adverse side effects in gene therapy treatments in the past, including reported cases of leukemia with the use of gammaretrovirus vector and patient deaths in other clinical trials. There have been recent case reports of suspected unexpected serious adverse reactions (SUSARs) involving an ex-vivo transduced lentivirus vector (LVV) gene therapy product, BlueBird Bio’s elivaldogene autotemcel (“Lenti-D”), involving two SUSARs for cases of acute myeloid leukemia (AML), and one case involving myelodysplastic syndrome.
In July 2021, the European Medicines Agency’s (EMA) safety committee (Pharmacovigilance Risk Assessment Committee - PRAC) announced that there is no evidence the LVV used in both Lenti-D and the EU-approved gene therapy Zynteglo spurred the AML cases.
BlueBird Bio announced on August 9, 2021 that the SUSAR involving myelodysplastic syndrome occurred in one patient treated with Lenti-D over a year previously, that this SUSAR “is likely mediated by Lenti-D lentiviral vector (LVV) insertion,” and that “[e]vidence currently available suggests that specific design features of Lenti-D LVV likely contributed to this event.” As a result of this SUSAR, the FDA has placed a clinical hold on BlueBird Bio’s Lenti-D phase 3 trial for cerebral adrenoleukodystrophy (CALD).
Gene therapy is still a relatively new approach to disease treatment and additional adverse side effects could develop. Possible adverse side effects that may occur with treatment with gene therapy products include an immunologic reaction early after administration that could substantially limit the effectiveness of the treatment or represent safety risks for patients. Another safety concern for gene therapies using viral vectors has been the possibility of insertional mutagenesis by the vectors, leading to malignant transformation of transduced cells. While our lentivirus ex-vivo gene transfer therapy approach is designed to avoid immunogenicity after administration, there can be no assurance that patients would not create antibodies that may impair treatment. Our approach involves the use of integrating vectors which have the potential for genomic disruption and therefore could interfere with other genes with adverse clinical effects. If any of our gene therapy product candidates demonstrates adverse side effects, we may decide or be required to halt or delay clinical development of such product candidates.
Potential risks for gene therapy products can be identified, in addition to side effects caused by the product candidate itself, as part of the entire process required for their manufacturing and administration. For Temferon manufacturing, each patient needs to be subjected to a mobilization and harvesting process for hemapoietic stem progenitor cells (HSPCs) collection. This procedure is associated with risks linked to the administration of mobilization agents. The conditioning regimen required for administering our product candidate and the associated procedures can also cause adverse side effects. A gene therapy patient is generally administered with cytotoxic drugs to remove stem cells from the bone marrow to create sufficient space for the modified stem cells to engraft and produce their progeny. This procedure compromises the patient’s immune system, and adverse events related to preconditioning have been observed in our ongoing clinical trial. If in the future we are unable to demonstrate that such adverse events were caused by the conditioning regimens used, or by their administration process or related procedure, the FDA, EMA or other regulatory authorities could order us to cease further development of, or deny the approval of, Temferon or our other product candidates for any or all target indications. Even if we are able to demonstrate that adverse events are not related to our drug product, such occurrences could affect the ability to enroll patients to complete the clinical trial, or the commercial viability of any product candidates that obtain regulatory approval.
To date, Temferon has only been administered to a small number of human subjects in our ongoing Phase 1/2a study. Due to the lack of a broader experience in human subjects, there is limited information available about the relationship of adverse events to administration of Temferon. Adverse events experienced in our clinical trials and attributed to autologous stem cell transplant (ASCT), concomitant medications, and disease progression have included febrile neutropenia and other infectious complications, venous thromboembolism, poor performance status, liver enzyme elevation, brain abscess and hemiparesis. While most of these adverse events were managed with treatment and supportive care, two GBM patients died (day +60 and +122) due to complications following the conditioning regimens.
Patient deaths and severe adverse effects caused by any investigational product candidates could result in the delay, suspension, clinical hold or termination of clinical trials by Sponsors, ethics committees and regulatory authorities. If we elect or are required to delay, suspend or terminate any clinical trial of any product candidates that we develop, the commercial prospects of such product candidates will be harmed and our ability to generate product revenue from any of these product candidates would be delayed or eliminated. Serious adverse events observed in clinical trials could hinder or prevent market acceptance of the product candidate at issue. Any of these occurrences may harm our business, prospects, financial condition and results of operations significantly.
| 17 |
Additionally, if any of our product candidates receives marketing approval, the FDA could require us to adopt a Risk Evaluation and Mitigation Strategy, or REMS, and other non-U.S. regulatory authorities could impose other specific obligations as a condition of approval to ensure that the benefits outweigh its risks, which may include, among other things, a medication guide outlining the risks of the product for distribution to patients, a communication plan to health care practitioners, and restrictions on how or where the product can be distributed, dispensed or used. Furthermore, if we or others later identify undesirable side effects caused by Temferon or any of our other product candidates, several potentially significant negative consequences could result, including:
| ● | regulatory authorities may suspend or withdraw approvals of such a product candidate; | |
| ● | regulatory authorities may require additional warnings or limitations of use in product labeling; | |
| ● | we may be required to change the way a product candidate is distributed, dispensed, or administered or conduct additional clinical trials; | |
| ● | we could be sued and held liable for harm caused to patients; and | |
| ● | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of our product candidates and could significantly harm our business, prospects, financial condition and results of operations.
We are evaluating Temferon in a Phase 1/2a clinical trial in newly diagnosed unmethylated MGMT gene promoter glioblastoma tumor patients (TEM-GBM Study). To date, the TEM-GBM Study is ongoing and not complete.
We are at a very early stage of development for all of our gene therapy product candidates. At this stage, only our lead product candidate Temferon has been authorized by the AIFA to be evaluated in a Phase 1/2a clinical trial in Italy. A study testing Temferon in multiple myeloma study was also approved by AIFA, but we closed the study due to lack of enrollment feasibility, rather than clinical events, as no multiple myeloma patients have been treated with Temferon.
In order to commence a clinical trial in the United States, we will be required to seek FDA acceptance of an IND for each of our product candidates, including Temferon. We cannot be sure any IND we submit to the FDA, or any similar clinical trial application we submit in other countries, will be accepted. If we will be required by regulatory authorities to conduct additional preclinical testing prior to filing an IND or similar application to clinically evaluate any of our product candidates, including Temferon, this may result in delay in our product candidate development. The results of any such preclinical testing may not be positive and may not support an application to study Temferon or any of our other product candidates in additional clinical trials.
It is possible that the FDA or EMA will not view our ongoing or planned trials as providing adequate support for future clinical trials or for an application for marketing approval, for any one or more reasons, including elements of the design or execution of the trials or safety concerns or other trial results. If we are unable to confirm or replicate the results of our trials in larger patient group or if negative results are obtained, we would likely be further delayed or prevented from advancing further clinical development of Temferon or any of our other product candidates.
Additionally, the FDA or EMA may disagree with the sufficiency of our proposed reliance upon the preclinical, manufacturing or clinical data generated by third-party academic-sponsored trials, or our interpretation of preclinical, manufacturing or clinical data from our ongoing trials. If so, the FDA or EMA may require us to obtain and submit additional preclinical, manufacturing or clinical data.
We need to complete our Phase 1/2a clinical trial for Temferon, as well as additional clinical trials in order to obtain regulatory approvals to market Temferon. Carrying out later-stage clinical trials is a complicated process. We are a small organization with limited experience in preparing, submitting and prosecuting regulatory filings, and we have not previously submitted a biologics license application, or BLA, to the FDA for any product candidate.
| 18 |
In addition, we have not yet conducted clinical trials of any our product candidates in the United States, and we cannot be certain how many clinical trials of Temferon or any of our other product candidates will be required or how such trials should be designed. Consequently, we may be unable to successfully and efficiently execute and complete necessary clinical trials in a way that leads to a BLA submission and approval of Temferon or any of our other product candidates. We may require more time and incur greater costs than our competitors and may not succeed in obtaining regulatory approvals of product candidates that we develop. Failure to commence or complete, or delays in, our planned clinical trials, could prevent us from or delay us in commercializing Temferon.
We may encounter substantial delays in commencement and completion of clinical trials.
Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we must conduct extensive clinical studies to demonstrate the safety and efficacy of the product candidates in humans. Clinical development is a long, expensive and uncertain process, and delay or failure can occur at any stage of any of our clinical trials. We cannot guarantee that any clinical studies will be conducted or completed on schedule, if at all. Clinical trials can be delayed or prevented for a number of reasons, including:
| ● | delays in reaching a consensus with regulatory agencies on study design; | |
| ● | changes in regulatory requirements and guidance that require amending or submitting new clinical protocols; | |
| ● | difficulties obtaining regulatory approval to commence a clinical trial or complying with conditions imposed by a regulatory authority regarding the scope or term of a clinical trial; | |
| ● | difficulties obtaining institutional review board, or IRB, approval to conduct a clinical trial at a prospective site; | |
| ● | failure to perform in accordance with the FDA’s good clinical practices, or GCP, or applicable regulatory guidelines in other countries; | |
| ● | delays in reaching or failing to reach agreement on acceptable terms with prospective contract research organizations, or CROs, and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; | |
| ● | failure by CROs, other third parties or us to adhere to clinical trial protocol and record keeping requirements; | |
| ● | trial sites or patients dropping out of a study; | |
| ● | the occurrence of SAEs associated with the product candidate that are viewed to outweigh its potential benefits; | |
| ● | insufficient or inadequate supply or quality of a product candidate or other materials necessary to conduct our clinical trials; | |
| ● | delays in the testing, validation, manufacturing and delivery of our product candidates to the clinical sites; and | |
| ● | if the FDA or the EMA or other regulatory authorities elect to enact policy changes, as a result of the COVID-19 pandemic or otherwise. |
| 19 |
Clinical trials may also be delayed or terminated as a result of ambiguous or negative interim results. In addition, a clinical trial may be suspended or terminated by us, the FDA, the IRBs at the sites where the IRBs are overseeing a trial, a data safety monitoring board overseeing the clinical trial at issue or by other regulatory authorities due to a number of factors, including:
| ● | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; | |
| ● | inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities; | |
| ● | unforeseen safety issues (including those that result from the COVID-19 pandemic) or lack of effectiveness; and | |
| ● | lack of adequate funding to continue the clinical trial. |
Any inability to successfully complete preclinical and clinical development could result in additional costs to us or impair our ability to generate revenues. This could result in increased costs, delays in advancing our product candidates, delays in testing the effectiveness of our product candidates or termination of the clinical trials altogether.
In addition, if we make changes to our product candidates, we may need to conduct additional studies to bridge our modified product candidates to earlier versions, this will increase the costs and could delay our clinical development plan, or marketing approval for our product candidates. For example, among our preclinical candidates we are developing a “switchable” system. This system has the potential to be a “switchable” on/off system that may limit the long-term exposure to any selected therapeutic payloads, but it requires further preclinical testing as well as additional manufacturing validation. Moreover, our platform is designed to allow us to use other therapeutic payloads, other than IFN-α. This has the potential to open a multitude of therapeutic indications but further preclinical testing as well as additional manufacturing validation are required. Any modification of our product candidates will likely require updates to our clinical trial applications and INDs with the relevant regulatory authorities, which may result in delay, suspension or termination of ongoing or future clinical trials pending our submission, and the agencies’ review, of such updates. Clinical study delays could also shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
The results of preclinical studies, early-stage clinical trials, data obtained from real-world use, and published third-party studies may not be indicative of results in future clinical trials and we cannot assure you that any clinical trials will lead to results sufficient for the necessary regulatory approvals.
The results of preclinical studies may not be predictive of the results of clinical trials, and the results of any completed clinical trials, including studies derived from real-world use and studies in published literature, or clinical trials we commence may not be predictive of the results of later-stage clinical trials. Additionally, interim results and analyses from our ongoing clinical trials do not necessarily predict final results. Moreover, preliminary data and analyses from our ongoing clinical trials may change as more patient data become available. In general, we conduct interim analyses at pre-specified times, which do not include data subsequent to the cut-off date and will not be available until the next planned interim analysis. From time to time, preliminary data and analyses might be presented, typically by academic investigators at scientific conferences or in scientific publications. Interim data and analyses are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and/or more patient data become available to us. Interim and preliminary data/analyses also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data available to us or that we previously published. As a result, preliminary and interim data/analyses should be viewed with caution until the final data are available. Material adverse changes in the final data compared to the preliminary or interim data/analyses could significantly harm our business prospects.
Indeed, our product candidates may fail to show the desired safety and efficacy in clinical development despite demonstrating positive results in preclinical studies or having successfully advanced through initial clinical trials. Later clinical trial results may not replicate earlier clinical trials for a variety of reasons, including differences in trial design, different trial endpoints (or lack of trial endpoints in exploratory studies), subject population, number of subjects, subject selection criteria, trial duration, drug dosage and formulation and lack of statistical power in the earlier studies. Our company has limited experience in designing and conducting clinical trials and we may be unable to design and execute a clinical trial to support regulatory approval. There can be no assurance that any of our clinical trials will ultimately be successful or support further clinical development of any of our product candidates. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical development even after achieving promising results in earlier studies, and any such setbacks in our clinical development could have a negative impact on our business. Any of our product candidates, including Temferon, may fail to show the desired safety and efficacy in clinical development despite positive results in preclinical studies. Any such failure would cause us to abandon the product candidate.
| 20 |
Additionally, our ongoing clinical trial utilizes, and our planned clinical trials may utilize, an “open-label” trial design. An “open-label” clinical trial is one where both the patient and investigator know whether the patient is receiving the investigational product candidate or either an existing approved drug or placebo. Most typically, open-label clinical trials test only the investigational product candidate and sometimes may do so at different dose levels. Open-label clinical trials are subject to various limitations that may exaggerate any therapeutic effect as patients in open-label clinical trials are aware when they are receiving treatment. Open-label clinical trials may be subject to a “patient bias” where patients perceive their symptoms to have improved merely due to their awareness of receiving an experimental treatment. In addition, open-label clinical trials may be subject to an “investigator bias” where those assessing and reviewing the physiological outcomes of the clinical trials are aware of which patients have received treatment and may interpret the information of the treated group more favorably given this knowledge. The results from an open-label trial may not be predictive of future clinical trial results with any of our product candidates for which we include an open-label clinical trial when studied in a controlled environment with a placebo or active control.
We may find it difficult to enroll patients in our clinical trials, which could delay or prevent us from proceeding with clinical trials of our product candidates.
Identifying and qualifying patients to participate in clinical trials of our product candidates is critical to our success. The timing of our clinical trials depends on our ability and on the speed at which we can recruit patients to participate in testing our product candidates, as well as the completion of required follow-up periods. We may experience delays in our clinical trials if we encounter difficulties in enrollment. Patients may be unwilling to participate in our gene therapy clinical trials because of negative publicity from adverse events related to the biotechnology or gene therapy fields, the safety profile of our product candidate under study, the perceived risks and benefits of the product candidate under study; the perceived risks and benefits of gene therapy-based approaches to treatment of diseases, including any required pretreatment conditioning regimens, the existence of competitive clinical trials for similar patient populations.
In addition, we may not be able to identify, recruit and enroll a sufficient number of patients due to the existence of efficacious alternative treatments, the size of the patient population and process for identifying subjects, the design of the trial protocol, the exclusion/inclusion criteria that we are currently targeting may limit the pool of patients that may be enrolled in our ongoing or planned clinical trials, the proximity and availability of clinical trial sites for prospective subjects and the patient referral practices of physicians; the ability to obtain and maintain subject consent; the risk that enrolled subjects will drop out before completion of the trial.
In addition, the evolving COVID-19 pandemic may directly or indirectly impact the pace of enrollment in our clinical trials as patients may avoid or may not be able to travel to healthcare facilities and physicians’ offices due to a health emergency and clinical trial staff can no longer get to the clinic. Additionally, such facilities and offices have been and may continue to be required to focus limited resources on non-clinical trial matters, including treatment of COVID-19 patients, thereby decreasing availability, in whole or in part, for clinical trial services. See “Risks Related to Our Business Operations – We face business disruption and related risks resulting from the recent outbreak of COVID-19, which could have a material adverse effect on our business and results of operations” for additional information.
If patients are unwilling to participate in our studies for any reason, the timeline for recruiting patients, conducting studies and obtaining regulatory approval of potential product candidates will be delayed.
If we experience delays in the commencement or completion or termination of any clinical trial of our product candidates, the commercial prospects of our product candidates will be harmed, and our ability to generate product candidate revenue from any of these product candidates could be delayed or prevented. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product candidate sales and generate revenue. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
The results of clinical trials conducted at clinical sites outside the United States may not be accepted by the FDA and the results of clinical trials conducted at clinical sites in the United States may not be accepted by international regulatory authorities.
To date our only ongoing recruiting clinical trial has been conducted in Europe but we are planning to globally develop Temferon, including in the United States. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of this data is subject to certain conditions imposed by the FDA. For example, the clinical trial must be well-designed and conducted and performed by qualified investigators in accordance with, GCPs, ethical principles such as or IRB or ethics committee approval and informed consent. Generally, the subject population for any clinical trials conducted outside of the United States must be representative of the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. In addition, while these clinical trials are subject to the applicable local laws, FDA acceptance of the data will be dependent upon its determination that the trials were conducted consistent with all applicable U.S. laws and regulations. There can be no assurance the FDA or international regulatory authorities will accept data from trials conducted outside of the location in which each regulatory authority is based as adequate support of a marketing application in a given jurisdiction. If the FDA does not accept the data from sites in our globally conducted clinical trials, or if international regulatory authorities do not accept the data from our U.S. clinical trials, it would likely result in the need for additional trials, which would be costly and time-consuming and could delay or permanently halt the development of one or more of our product candidates.
| 21 |
Our ability to successfully initiate, enroll and complete a clinical trial in any foreign country including the United States, is subject to numerous risks unique to conducting business in foreign countries, including:
| ● | difficulty in establishing or managing relationships with contract research organizations, or CROs, and physicians; | |
| ● | different standards for the conduct of clinical trials; | |
| ● | the absence in some countries of established groups with sufficient regulatory expertise for review of gene therapy protocols; | |
| ● | our inability to locate qualified local consultants, physicians and partners; and | |
| ● | the potential burden of complying with a variety of laws, medical standards and regulatory requirements, including the regulation of pharmaceutical and biotechnology products and treatment. |
Changes in methods of product candidate manufacturing or formulation may result in additional costs or delay.
As product candidates proceed through preclinical studies to late-stage clinical trials towards potential approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods and formulation, are altered along the way in an effort to optimize processes and results. Such changes carry the risk that they will not achieve these intended objectives. We may also experience delays in developing a sustainable, reproducible and scalable manufacturing process or delays in transferring that process to commercial partners, which may prevent us from initiating, completing or expanding our clinical trials or commercializing our products, if any, on a timely or profitable basis, if at all. For example, the anticipated transition of our cell processing to a different commercial partner in the U.S., or to a commercial partner(s) relying on automated closed system, if available, using all disposable supplies would require regulatory approvals, may not be successful or may experience unforeseen delays, which may cause shortages or delays in the supply of our products available for clinical trials and future commercial sales, if any. In addition, there is no assurance that products manufactured using a different commercial partner or an automated closed system, if and when available, will achieve the same results observed to date in Temferon clinical and preclinical and non-clinical studies. Any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials conducted with the materials manufactured using altered processes. Such changes may also require additional testing such as comparability studies, FDA or EMA notification or FDA approval. This could delay completion of clinical trials, require the conduct of bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of our product candidates and jeopardize our ability to commence sales and generate revenue.
| 22 |
Even if we complete the necessary preclinical and clinical studies, we cannot predict when or if we will obtain regulatory approval to commercialize a product candidate and the approval may be for a more narrow indication than we seek.
We cannot commercialize a product until the appropriate regulatory authorities have reviewed and approved the product candidate. Even if our product candidates demonstrate safety and efficacy in preclinical and clinical studies, the regulatory agencies may not complete their review processes in a timely manner, or we may not be able to obtain regulatory approval. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in preclinical testing and earlier-stage clinical trials. Additional delays may result if an FDA Advisory Committee or other regulatory authority does not recommend approval or recommends restrictions on approval. In addition, we may experience delays or rejections based upon additional government regulation from future legislation or administrative action, or changes in regulatory agency policy during the period of product development, clinical studies and the review process. Regulatory agencies also may approve a product candidate for fewer or more limited indications than requested or may grant approval subject to the performance of post-marketing studies. In addition, regulatory agencies may not approve the labeling claims that are necessary or desirable for the successful commercialization of our product candidates. If we are unable to obtain necessary regulatory approvals, our business, prospects, financial condition and results of operations may suffer.
We may seek designations for our product candidates with the FDA and other comparable regulatory authorities that are intended to confer benefits such as a faster development process or an accelerated regulatory pathway, but there can be no assurance that we will successfully obtain such designations. In addition, even if one or more of our product candidates are granted such designations, we may not be able to realize the intended benefits of such designations.
The FDA, and other comparable regulatory authorities, offer certain designations for product candidates that are intended to encourage the research and development of pharmaceutical and biotechnology products addressing conditions with significant unmet medical need. These designations may confer benefits such as additional interaction with regulatory authorities, a potentially accelerated regulatory pathway and priority review. There can be no assurance that we will successfully obtain such designation for Temferon. In addition, while such designations could expedite the development or approval process, they do not change the standards for approval. Even if we obtain such designations for one or more of our product candidates, there can be no assurance that we will realize their intended benefits.
For example, we may seek a Breakthrough Therapy Designation from the FDA for one or more of our product candidates. A Breakthrough Therapy Designation is defined as a therapy that is intended, alone or in combination with one or more other therapies, to treat a serious or life-threatening disease or condition, if preliminary clinical evidence indicates that the therapy may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. For therapies that have Breakthrough Therapy Designation, interaction and communication between the FDA and the sponsor of the trial can help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Therapies with Breakthrough Therapy Designation from the FDA are also eligible for accelerated approval. Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if we believe one of our product candidates meets the criteria for Breakthrough Therapy Designation, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of a Breakthrough Therapy Designation for a product candidate may not result in a faster development process, review or approval compared to therapies considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA. In addition, even if one or more of our product candidates qualify for Breakthrough Therapy Designation, the FDA may later decide that such product candidates no longer meet the conditions for qualification.
We may also seek Fast Track Designation from the FDA for some of our product candidates. If a therapy is intended for the treatment of a serious or life-threatening condition and the therapy demonstrates the potential to address unmet medical needs for this condition, the therapy sponsor may apply for Fast Track Designation. The FDA has broad discretion whether or not to grant this designation, so even if we believe a particular product candidate is eligible for this designation, there can be no assurance that the FDA would decide to grant it. Even if we do receive Fast Track Designation, we may not experience a faster development process, review or approval compared to conventional FDA procedures, and receiving a Fast Track Designation does not provide assurance of ultimate FDA approval. The FDA may withdraw Fast Track Designation if it believes that the designation is no longer supported by data from our clinical development program.
| 23 |
In addition, we may seek a regenerative medicine advanced therapy, or RMAT, designation for some of our product candidates. An RMAT is defined as a cell therapy, therapeutic tissue engineering products, human cell and tissue products, or any combination products using any such therapies or products. Gene therapies, including genetically modified cells that lead to a durable modification of cells or tissues may meet the definition of a regenerative medicine therapy. The RMAT program is intended to facilitate efficient development and expedite review of RMATs, which are intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition, and for which preliminary clinical evidence indicates that the candidate has potential to address unmet medical needs for such disease or condition. A new drug application or a BLA for an RMAT may be eligible for priority review or accelerated approval through (1) surrogate or intermediate endpoints reasonably likely to predict long-term clinical benefit or (2) reliance upon data obtained from a meaningful number of sites. Benefits of such designation also include early interactions with FDA to discuss any potential surrogate or intermediate endpoint to be used to support accelerated approval. A regenerative medicine therapy that is granted accelerated approval and is subject to post-approval requirements may fulfill such requirements through the submission of clinical evidence, clinical studies, patient registries, or other sources of real world evidence, such as electronic health records; the collection of larger confirmatory data sets; or post-approval monitoring of all patients treated with such therapy prior to its approval. RMAT designation is within the discretion of the FDA. Accordingly, even if we believe one of our product candidates meets the criteria for designation as a regenerative medicine advanced therapy, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of RMAT designation for a product candidate may not result in a faster development process, review or approval compared to product candidates considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA. In addition, even if one or more of our product candidates qualify as for RMAT designation, the FDA may later decide that the biological products no longer meet the conditions for qualification.
We may seek a conditional marketing authorization in Europe for some or all of our current product candidates, but we may not be able to obtain or maintain such designation.
As part of its marketing authorization process, the EMA may grant marketing authorizations for certain categories of medicinal products on the basis of less complete data than is normally required, where the benefit of immediate availability of the medicine outweighs the risk inherent in the fact that additional data are still required or in the interests of public health. In such cases, it is possible for the Committee for Medicinal Products for Human Use, or CHMP, to recommend the granting of a marketing authorization, subject to certain specific obligations to be reviewed annually, which is referred to as a conditional marketing authorization. This may apply to medicinal products for human use that fall under the jurisdiction of the EMA, including those that aim at the treatment, the prevention, or the medical diagnosis of seriously debilitating or life-threatening diseases and those designated as orphan medicinal products.
A conditional marketing authorization may be granted when the CHMP finds that, although comprehensive clinical data referring to the safety and efficacy of the medicinal product have not been supplied, all the following requirements are met:
| ● | the risk-benefit balance of the medicinal product is positive; | |
| ● | the applicant will provide the comprehensive clinical data post-authorization; | |
| ● | unmet medical needs will be fulfilled; and | |
| ● | the benefit to public health of the immediate availability on the market of the medicinal product concerned outweighs the risk inherent in the fact that additional data is still required. |
The granting of a conditional marketing authorization is restricted to situations in which only the clinical part of the application is not yet fully complete. Incomplete preclinical or quality data may only be accepted if duly justified and only in the case of a product intended to be used in emergency situations in response to public health threats. Conditional marketing authorizations are valid for one year, on a renewable basis. The holder will be required to complete ongoing trials or to conduct new trials with a view to confirming that the benefit-risk balance is positive. In addition, specific obligations may be imposed in relation to the collection of pharmacovigilance data.
| 24 |
Granting a conditional marketing authorization allows medicines to reach patients with unmet medical needs earlier than might otherwise be the case and will ensure that additional data on a product is generated, submitted, assessed and acted upon. Although we may seek a conditional marketing authorization for one or more of our product candidates by the EMA, the CHMP may ultimately not agree that the requirements for such conditional marketing authorization have been satisfied and hence delay the commercialization of our product candidates.
We may be unable to obtain orphan drug designation for our product candidates and, even if we obtain such designation, we may not be able to realize the benefits of such designation, including potential marketing exclusivity of our product candidates, if approved.
Regulatory authorities in some jurisdictions, including the United States and EU, may designate drugs for relatively small patient populations as “orphan drugs.” Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is a drug intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals in the United States or a patient population of 200,000 or more individuals in the United States, but for which there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. In the EU, the European Commission grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 persons in the EU community. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the EU would be sufficient to justify the necessary investment in developing the drug or biologic product. In either case, the applicant for orphan designation must also demonstrate that no satisfactory method of diagnosis, prevention, or treatment for the condition has been authorized (or, if a method exists, the new product would be a significant benefit to those affected compared to the product available).
If we request orphan drug designation (or the international equivalent) for Temferon or any of our other product candidates, there can be no assurances that the FDA or international regulatory authorities will grant any of our product candidates such designation. This designation of a product candidate as an orphan product does not mean that any regulatory agency will accelerate regulatory review of, or ultimately approve, that product candidate, nor does it limit the ability of any regulatory agency to grant orphan drug designation to product candidates of other companies that treat the same indications as our product candidates prior to our product candidates receiving exclusive marketing approval.
Generally, if a product candidate with an orphan drug designation subsequently receives the first marketing approval for the indication for which it has such designation, the drug may be entitled to a period of marketing exclusivity, which precludes the FDA or the EMA from approving another marketing application for the same drug for the same indication for that time period, except in limited circumstances. If another sponsor receives such approval before we do (regardless of our orphan drug designation), we will be precluded from receiving marketing approval for our product for the applicable exclusivity period. The applicable period is seven years in the United States and ten years in the EU. The exclusivity period in the EU can be reduced to six years if a drug no longer meets the criteria for orphan drug designation or if the drug is sufficiently profitable so that market exclusivity is no longer justified. Orphan drug exclusivity may be revoked if any regulatory agency determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition.
Even if we obtain orphan drug exclusivity for a product candidate, that exclusivity may not effectively protect the product candidate from competition because different drugs can be approved for the same indication. Even after an orphan drug is approved, the FDA may subsequently approve another drug for the same condition if the FDA concludes that the latter drug is not the same drug or is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care. In the EU, marketing authorization may be granted to a similar medicinal product for the same orphan indication if:
| ● | the second applicant can establish in its application that its medicinal product, although similar to the orphan medicinal product already authorized, is safer, more effective or otherwise clinically superior; |
| 25 |
| ● | the holder of the marketing authorization for the original orphan medicinal product consents to a second orphan medicinal product application; or | |
| ● | the holder of the marketing authorization for the original orphan medicinal product cannot supply sufficient quantities of orphan medicinal product. |
Even if we obtain and maintain regulatory approval for a product candidate, our products will remain subject to ongoing regulatory oversight.
Even if we obtain any regulatory approval for our product candidates, they will be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping and submission of safety and other post-market information. Any regulatory approvals that we receive for our product candidates also may be subject to a REMS, limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the quality, safety and efficacy of the product. For example, as noted above in the United States, the holder of an approved BLA is obligated to monitor and report adverse events and any failure of a product to meet the specifications in the BLA. FDA guidance advises that patients treated with some types of gene therapy undergo follow-up observations for potential adverse events for as long as 15 years. The holder of an approved marketing application also must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling or manufacturing process.
We must comply with requirements concerning advertising and promotion for any product candidates for which we obtain marketing approval. Promotional communications with respect to therapeutics are subject to a variety of legal and regulatory restrictions and continuing review by the FDA or comparable foreign regulatory authorities, Department of Justice, Department of Health and Human Services’, or HHS, Office of Inspector General, state attorneys general, members of Congress, and the public. When the FDA or comparable foreign regulatory authorities issue regulatory approval for a product candidate, the regulatory approval is limited to those specific uses and indications for which a product is approved. If we are not able to obtain FDA or comparable foreign regulatory authority approval for desired uses or indications for our current product candidates and any future product candidates, we may not market or promote them for those indications and uses, referred to as off-label uses, and our business, financial condition, results of operations, stock price and prospects will be materially harmed. We also must sufficiently substantiate any claims that we make for our products, including claims comparing our products to other companies’ products, and must abide by the FDA or a comparable foreign regulatory authority’s strict requirements regarding the content of promotion and advertising.
While physicians may choose to prescribe products for uses that are not described in the product’s labeling and for uses that differ from those tested in clinical trials and approved by the regulatory authorities, we and any third parties engaged on our behalf are prohibited from marketing and promoting the products for indications and uses that are not specifically approved by the FDA or comparable foreign regulatory authorities. Regulatory authorities in the United States generally do not restrict or regulate the behavior of physicians in their choice of treatment within the practice of medicine. Regulatory authorities do, however, restrict communications by biopharmaceutical companies concerning off-label use.
If we are found to have impermissibly promoted any of our current product candidates and any future product candidates, we may become subject to significant liability and government fines. The FDA and other agencies actively enforce the laws and regulations regarding product promotion, particularly those prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted a product may be subject to significant sanctions. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed.
In the European Union, the advertising and promotion of our products are subject to European Union laws governing promotion of medicinal products, interactions with physicians, misleading and comparative advertising, and unfair commercial practices. In addition, other legislation adopted by individual European Union Member States may apply to the advertising and promotion of medicinal products. These laws require that promotional materials and advertising for medicinal products are consistent with the product’s Summary of Product Characteristics, or SmPC, as approved by the competent authorities. The SmPC is the document that provides information to physicians concerning the safe and effective use of the medicinal product. It forms an intrinsic and integral part of the marketing authorization granted for the medicinal product. Promotion of a medicinal product that does not comply with the SmPC is considered to constitute off-label promotion. The off-label promotion of medicinal products is prohibited in the European Union. The applicable laws at European Union level and in the individual European Union Member States also prohibit the direct-to-consumer advertising of prescription-only medicinal products. Violations of the rules governing the promotion of medicinal products in the European Union could be penalized by administrative measures, fines and imprisonment. These laws may further limit or restrict the advertising and promotion of our products to the general public, and may also impose limitations on our promotional activities with health care professionals.
In addition, product manufacturers and their facilities are subject to payment of user fees and continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current good manufacturing practices, or GMP, requirements and adherence to commitments made in the BLA or foreign marketing application. If we, or a regulatory authority, discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured or disagrees with the promotion, marketing or labeling of that product, a regulatory authority may impose restrictions relative to that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing.
If we fail to comply with applicable regulatory requirements, a regulatory authority may:
| ● | issue an untitled letter or warning letter that we are in violation of the law; | |
| ● | seek an injunction or impose administrative, civil or criminal penalties or monetary fines; | |
| ● | suspend or withdraw regulatory approval; |
| 26 |
| ● | suspend any ongoing clinical trials; | |
| ● | refuse to approve BLA or comparable foreign marketing application (or any supplements thereto) submitted by us or our strategic partners; | |
| ● | restrict the marketing or manufacturing of the product; | |
| ● | seize or detain the products or require the withdrawal of the product from the market; | |
| ● | refuse to permit the import or export of the products; or | |
| ● | refuse to allow us to enter into supply contracts, including government contracts. |
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize our product candidates and adversely affect our business, financial condition, results of operations and prospects.
In addition, the FDA’s policies, and those of the EMA and other regulatory authorities, may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would materially and adversely affect our business, financial condition, results of operations and prospects.
Both marketing authorization holders and manufacturers of medicinal products are subject to comprehensive regulatory oversight by the EMA and the competent authorities of the individual European Union Member States both before and after grant of the manufacturing and marketing authorizations. This includes control of compliance with GMP rules, which govern quality control of the manufacturing process and require documentation policies and procedures. We and our third-party manufacturers would be required to ensure that all of our processes, quality systems, methods, and equipment are compliant with GMP. Failure by us or by any of our third-party partners, including suppliers, manufacturers, and distributors to comply with European Union laws and the related national laws of individual European Union Member States governing the conduct of clinical trials, manufacturing approval, marketing authorization of medicinal products, both before and after grant of marketing authorization, and marketing of such products following grant of authorization may result in administrative, civil, or criminal penalties. These penalties could include delays in or refusal to authorize the conduct of clinical trials or to grant marketing authorization, product withdrawals and recalls, product seizures, suspension, or variation of the marketing authorization, total or partial suspension of production, distribution, manufacturing, or clinical trials, operating restrictions, injunctions, suspension of licenses, fines, and criminal penalties. operating restrictions, injunctions, suspension of licenses, fines, and criminal penalties.
In addition, European Union legislation related to pharmacovigilance, or the assessment and monitoring of the safety of medicinal products, provides that EMA and the competent authorities of the European Union Member States have the authority to require companies to conduct additional post-approval clinical efficacy and safety studies. The legislation also governs the obligations of marketing authorization holders with respect to additional monitoring, adverse event management and reporting. Under the pharmacovigilance legislation and its related regulations and guidelines, we may be required to conduct a burdensome collection of data regarding the risks and benefits of marketed products and may be required to engage in ongoing assessments of those risks and benefits, including the possible requirement to conduct additional clinical trials, which may be time-consuming and expensive and could impact our profitability. Non-compliance with such obligations can lead to the variation, suspension or withdrawal of marketing authorization or imposition of financial penalties or other enforcement measures.
| 27 |
We do not have sales, distribution, and marketing capabilities. If we are unable to develop these capabilities or enter into agreements with third parties to market and sell Temferon and our other product candidates, we will be unable to generate any product revenue.
We currently have no sales, distribution or marketing organization. To successfully commercialize any of our current or future product candidates, if approved, we will need to develop these capabilities, either on our own or with others. The establishment and development of our own commercial team or the establishment of a contract sales force to market any product candidate we may develop will be expensive and time-consuming and could delay any product launch. Moreover, we cannot be certain that we will be able to successfully develop this capability. We may enter into collaborations regarding any approved product candidates with other entities to utilize their established marketing and distribution capabilities, but we may be unable to enter into such agreements on favorable terms, if at all. If any future collaborators do not commit sufficient resources to commercialize our product candidates, or we are unable to develop the necessary capabilities on our own, we will be unable to generate sufficient product revenue to sustain our business. We compete with many companies that currently have extensive, experienced and well-funded sales, distribution and marketing operations to recruit, hire, train and retain marketing and sales personnel. We also face competition in our search for third parties to assist us with the sales and marketing efforts of our product candidates, if approved. Without an internal team or the support of a third-party to perform marketing and sales functions, we may be unable to compete successfully against these more established companies.
Even if any of our product candidates receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success.
The commercial success of Temferon will depend upon the acceptance of each product by the medical community, including physicians, patients and third-party payors. The degree of market acceptance of any approved product will depend on a number of factors, including:
| ● | the efficacy and safety of the product; | |
| ● | the potential advantages of the product compared to available therapies; | |
| ● | the convenience and ease of administration compared to alternative treatments; | |
| ● | limitations or warnings, including use restrictions contained in the product’s approved labeling; | |
| ● | distribution and use restrictions imposed by the FDA, the EMA or other regulatory authority or agreed to by us as part of a mandatory or voluntary risk management plan; | |
| ● | availability of alternative treatments, including competitive products expected to be commercially launched in the near future; | |
| ● | pricing and cost effectiveness in relation to alternative treatments; | |
| ● | if the product is included under physician treatment guidelines as a first-, second,- or third-line therapy; | |
| ● | the strength of sales, marketing and distribution support; | |
| ● | the availability of third-party coverage and adequate reimbursement and the willingness of patients to pay out-of-pocket in the absence of coverage by third-party payors; | |
| ● | the strength of sales, marketing and distribution support; | |
| ● | the willingness of patients to pay for drugs out of pocket in the absence of third-party coverage; and |
| 28 |
| ● | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies. |
If Temferon is approved but does not achieve an adequate level of acceptance by physicians, third party payors and patients, we may not generate sufficient revenue from the product, and we may not become or remain profitable. In addition, our efforts to educate the medical community and third-party payors on the benefits of the product may require significant resources and may never be successful.
In addition, we may choose to collaborate with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems. If we enter into arrangements with third parties to perform sales, marketing and distribution services for our products, the resulting revenues or the profitability from these revenues to us are likely to be lower than if we had sold, marketed and distributed our products ourselves. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to successfully commercialize any of our product candidates that receive regulatory approval. Depending on the nature of the third-party relationship, we may have little control over such third parties, and any of these third parties may fail to devote the necessary resources and attention to sell, market and distribute our products effectively. If we are not successful in commercializing our product candidates, either on our own or through collaborations with one or more third parties, our future product revenue will suffer, and we may incur significant additional losses.
Even if we are able to commercialize any product candidates, the products may become subject to unfavorable pricing regulations or third-party coverage and reimbursement policies, any of which could harm our business.
Our ability to commercialize any product candidates successfully will depend, in part, on the extent to which coverage and reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and impact reimbursement levels.
Obtaining and maintaining adequate reimbursement for our products may be difficult. We cannot be certain if and when we will obtain an adequate level of reimbursement for our products by third party payors. Even if we do obtain adequate levels of reimbursement, third-party payors, such as government or private healthcare insurers, carefully review and increasingly question the coverage of, and challenge the prices charged for, drugs. Reimbursement rates from private health insurance companies vary depending on the company, the insurance plan and other factors. A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for drugs. We may also be required to conduct expensive pharmacoeconomic studies to justify coverage and reimbursement or the level of reimbursement relative to other therapies. If coverage and reimbursement are not available or reimbursement is available only to limited levels, we may not be able to successfully commercialize any product candidate for which we obtain marketing approval, and the royalties resulting from the sales of those products may also be adversely impacted.
There may be significant delays in obtaining reimbursement for newly approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or similar regulatory authorities outside the United States. Moreover, eligibility for reimbursement does not imply that a drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Our inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for any approved products that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
| 29 |
The regulations that govern marketing approvals, pricing, coverage and reimbursement for new drug products vary widely from country to country. Current and future legislation may significantly change the approval requirements in ways that could involve additional costs and cause delays in obtaining approvals. Some countries require approval of the sale price of a drug before it can be reimbursed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription drug pricing remains subject to continuing governmental control, including possible price reductions, even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the product, possibly for lengthy time periods, and negatively impact the revenues we are able to generate from the sale of the product in that country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates, even if our product candidates obtain marketing approval. There can be no assurance that our product candidates, if they are approved for sale in the United States or in other countries, will be considered medically necessary or cost-effective for a specific indication, or that coverage or an adequate level of reimbursement will be available.
Obtaining and maintaining regulatory approval of our product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product candidates in other jurisdictions. Our failure to obtain regulatory approval in international jurisdictions would prevent our product candidates from being marketed abroad, and any approval we are granted for our product candidates in the United States would not assure approval of product candidates in foreign jurisdictions.
In order to market any products outside of the United States, we must establish and comply with numerous and varying regulatory requirements of other countries regarding clinical trial design, safety and efficacy. The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of drugs are subject to extensive regulation by the FDA in the United States and other regulatory authorities in other countries. These regulations differ from country to country. Even if we obtain and maintain regulatory approval of our product candidates in one jurisdiction, such approval does not guarantee that we will be able to obtain or maintain regulatory approval in any other jurisdiction, but a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. For example, even if the FDA grants marketing approval of a product candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion of the product candidate in those countries.
Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from those in the United States, including additional non-clinical studies or clinical trials as investigations conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to approval. These regulatory procedures can result in substantial delays in such countries. In other countries, product approval depends on showing superiority to an approved alternative therapy. This can result in significant expense for conducting complex clinical trials.
Approval of a product candidate in the United States by the FDA does not ensure approval of such product candidate by the EMA or other regulatory authorities in other countries or jurisdictions, and approval by the EMA or another regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. Sales of our product candidates outside of the United States will be subject to foreign regulatory requirements governing clinical trials and marketing approval. Even if the FDA grants marketing approval for a product candidate, comparable regulatory authorities of foreign countries also must approve the manufacturing and marketing of the product candidates in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and more onerous than, those in the United States, including additional preclinical studies or clinical trials. In many countries outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that country. In some cases, the price that we intend to charge for our products, if approved, is also subject to approval. We intend to submit an MAA to the EMA for approval of our product candidates in the European Union but obtaining such approval from the European Commission following the opinion of EMA is a lengthy and expensive process. Even if a product candidate is approved, the FDA or the European Commission may limit the indications for which the product may be marketed, require extensive warnings on the product labeling or require expensive and time consuming additional clinical trials or reporting as conditions of approval. Regulatory authorities in countries outside of the United States and the European Union also have requirements for approval of product candidates with which we must comply prior to marketing in those countries. Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our product candidates in certain countries.
| 30 |
Finally, we do not have any products approved for sale in any jurisdiction, including international markets, and we do not have experience in obtaining regulatory approval. If we, or any third parties with whom we work, fail to comply with regulatory requirements in United States or international markets or to obtain and maintain required approvals or if regulatory approvals in international markets are delayed, our target market may be reduced and our ability to realize the full market potential of our products will likely be harmed. The inability to meet continuously evolving regulatory standards for approval may result in our failing to obtain regulatory approval to market our current product candidates, which could significantly harm our business, results of operations and prospects.
Additionally, on June 23, 2016, the electorate in the United Kingdom voted in favor of leaving the EU, commonly referred to as “Brexit.” On March 29, 2017, the country formally notified the EU of its intention to withdraw pursuant to Article 50 of the Lisbon Treaty and the withdrawal of the United Kingdom from the EU took effect on January 31, 2020. There was a transition period, during which EU pharmaceutical law remained applicable in the United Kingdom, however this ended on December 31, 2020. Since a significant proportion of the regulatory framework governing the development and commercialization of medicinal products in the United Kingdom is derived from EU Directives and Regulations, Brexit, now that the transition period is over, could materially impact the regulatory regime with respect to the approval of our product candidates in the United Kingdom or the EU, as United Kingdom legislation now has the potential to diverge from EU legislation. Any delay in obtaining, or an inability to obtain, any regulatory approvals, as a result of Brexit or otherwise, would prevent us from commercializing our product candidates in the United Kingdom and/or the EU and restrict our ability to generate revenue and achieve and sustain profitability. If any of these outcomes occur, we may be forced to restrict or delay efforts to seek regulatory approval in the United Kingdom and/or EU for our product candidates, which could significantly and materially harm our business.
We operate in a rapidly changing industry and face significant competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
The development and commercialization of new biopharmaceutical products is highly competitive and subject to rapid and significant technological advancements. We face competition from major multi-national pharmaceutical companies, biotechnology companies and specialty pharmaceutical companies with respect to our current and future product candidates that we may develop and commercialize in the future. There are a number of large pharmaceutical and biotechnology companies that currently market and sell products or are pursuing the development of product candidates for the treatment of cancer. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Potential competitors also include academic institutions, government agencies and other public and private research organizations. Our competitors may succeed in developing, acquiring or licensing technologies and products that are more effective, more effectively marketed and sold or less costly than any product candidates that we may develop, which could render our product candidates noncompetitive and obsolete.
Many of our competitors, either alone or with their strategic collaborators, have substantially greater financial, technical and human resources than we do. Accordingly, our competitors may be more successful than we are in obtaining approval for treatments and achieving widespread market acceptance, which may render our treatments obsolete or noncompetitive. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical study sites and patient registration for clinical studies, as well as in acquiring technologies complementary to, or necessary for, our programs.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive or better reimbursed than any products that we may commercialize. Our competitors also may obtain EMA, FDA or other regulatory approval for their products more rapidly than we do, which could result in our competitors establishing a strong market position for either the product or a specific indication before we are able to enter the market.
| 31 |
Our product candidates may face competition sooner than anticipated from biosimilar products.
Even if we are successful in achieving regulatory approval to commercialize a product candidate faster than our competitors, our product candidates may face competition from biosimilar products. In the United States, our product candidates are regulated by the FDA as biologic products and we intend to seek approval for these product candidates pursuant to the BLA pathway. The Biologics Price Competition and Innovation Act of 2009, or BPCIA, created an abbreviated pathway for the approval of biosimilar and interchangeable biologic products. The abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing brand product. Under the BPCIA, an application for a biosimilar product cannot be approved by the FDA until 12 years after the original branded product was approved under a BLA. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation, and meaning are subject to uncertainty.
There is a risk that any exclusivity we may be afforded if any of our product candidates are approved as a biologic product under a BLA could be shortened due to congressional action, the results of recent litigation, or otherwise, or that the FDA will not consider our product candidates to be reference products for competing products, potentially creating the opportunity for generic or biosimilar competition sooner than anticipated. Moreover, the extent to which a biosimilar product, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biologic products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing. In addition, a competitor could decide to forego the biosimilar approval path and submit a full BLA after completing its own preclinical studies and clinical trials. In such cases, any exclusivity to which we may be eligible under the BPCIA would not prevent the competitor from marketing its product as soon as it is approved.
In addition, critics of the 12-year exclusivity period in the biosimilar pathway law will likely continue to seek to shorten the data exclusivity period and/or to encourage the FDA to interpret narrowly the law’s provisions regarding which new products receive data exclusivity. In December 2019, the US agreed to remove from the United States-Mexico-Canada Agreement a requirement for at least 10 years of data exclusivity for biologic products. Also, the FDA is considering whether subsequent changes to a licensed biologic would be protected by the remainder of the reference product’s original 12-year exclusivity period (a concept known in the generic drug context as “umbrella exclusivity”). If the FDA were to decide that umbrella exclusivity does not apply to biological reference products or were to make other changes to the exclusivity period, this could expose us to biosimilar competition at an earlier time. There also have been, and may continue to be, legislative and regulatory efforts to promote competition through policies enabling easier generic and biosimilar approval and commercialization, including efforts to lower standards for demonstrating biosimilarity or interchangeability, limit patents that may be litigated and/or patent settlements and implement preferential reimbursement policies for biosimilars.
In Europe, the European Commission has granted marketing authorizations for several biosimilar products pursuant to a set of general and product class-specific guidelines for biosimilar approvals issued over the past few years. In the EEA, innovative medicinal products generally receive eight years of data exclusivity and an additional two years of marketing exclusivity. Data exclusivity prevents biosimilar applicants from referencing the innovator’s preclinical and clinical trial data when applying for a biosimilar marketing authorization, during a period of eight years from the date on which the reference product was first authorized in the EEA. During the additional two-year period of market exclusivity, biosimilar marketing authorization can be submitted, and the innovator’s data may be referenced, but no biosimilar product can be marketed until the expiration of the market exclusivity period. This 10-year marketing exclusivity period may be extended to 11 years if, during the first eight of those 10 years, the marketing authorization holder obtains an approval for one or more new therapeutic indications that bring significant clinical benefits compared with existing therapies. However, even if an innovative medicinal product gains the prescribed period of data exclusivity, another company may market another version of the product if such company obtained marketing authorization based on an application with a complete independent data package of pharmaceutical tests, preclinical tests and clinical trials. In addition, companies may be developing biosimilar products in other countries that could compete with our products, if approved.
If competitors are able to obtain marketing approval for biosimilars referencing our product candidates, if approved, such products may become subject to competition from such biosimilars, with the attendant competitive pressure and potential adverse consequences. Such competitive products may be able to immediately compete with us in each indication for which our product candidates may have received approval.
| 32 |
If product liability lawsuits are brought against us, we may incur substantial liabilities, even if we have appropriate insurance policies, and we may be required to limit commercialization of our product candidates.
We are exposed to potential product liability and professional indemnity risks that are inherent in the research, development, manufacturing, marketing and use of biopharmaceutical and biotechnology products. Currently, we have no products that have been approved for marketing or commercialization; however, the use of our product candidates in clinical trials, and the sale of these product candidates, if approved, in the future, may expose us to liability claims. Product liability claims may be brought against us or our partners by participants enrolled in our clinical trials, patients, health care providers, biotechnology and pharmaceutical companies, our collaborators or others using, administering or selling any of our future approved products. If we cannot successfully defend ourselves against any such claims, we may incur substantial liabilities, even if we have product liability or such other applicable insurance policies in effect. We may not be able to maintain adequate levels of insurance for these liabilities at reasonable cost and/or reasonable terms. Excessive insurance costs or uninsured claims would add to our future operating expenses and adversely affect our financial condition. As a result of such lawsuits and their potential results, we may be required to limit commercialization of our product candidates. Regardless of the merits or eventual outcome, liability claims may result in:
| ● | decreased demand for our product candidates; | |
| ● | termination of clinical trial sites or entire trial programs; | |
| ● | injury to our reputation and negative media attention; | |
| ● | product recalls or increased warnings on product labels; | |
| ● | withdrawal of clinical trial participants; | |
| ● | costs of to defend the related litigation; | |
| ● | diversion of management and our resources; | |
| ● | substantial monetary awards to, or costly settlements with, clinical trial participants, patients or other claimants; | |
| ● | higher insurance premiums; | |
| ● | loss of initiation of investigations by regulators or other authorities; and | |
| ● | the inability to successfully commercialize our product candidates, if approved. |
Gene therapies are novel, complex and difficult to manufacture. We could experience production problems that result in delays in our development or commercialization programs or otherwise adversely affect our business.
Biological products are inherently difficult to manufacture, and gene therapy products are complex biological products, the development and manufacture of which necessitates substantial expertise and capital investment.
Temferon is individually manufactured for each patient using complex processes in specialized facilities. Our production process requires a variety of raw materials, some of which are highly specialized, including the viral vector that encodes for the therapeutic payload. Some of these raw materials have limited and, in some cases, sole suppliers. Even though we plan to have back-up supplies of raw materials whenever possible, we cannot be certain such supplies will be sufficient if our primary sources are unavailable. A shortage of a critical raw material or a technical issue during manufacturing may lead to delays in clinical development or commercialization of our product candidates.
Our product candidate, Temferon, is being studied for GBM patients with unmethylated MGMT status, as determined by a laboratory test. If approved for use only in uMGMT-GBM patients, use of such a laboratory test would be required for each patient before treatment with Temferon. There are several currently-marketed, CE-marked tests for uMGMT status in the EU, one or more of which may be used in our clinical trials and which we would expect to be used in clinical practice upon approval of Temferon. If a regulatory authority were, however, to deem that no currently-available tests are appropriate for use with Temferon, or if appropriate tests were to become commercially unavailable, we might be required to develop and obtain regulatory approval for our own version of such a companion diagnostic test, or work with another entity to develop such a test, in which case we could experience significant delays in obtaining regulatory approval or interruptions in our ability to market Temferon.
| 33 |
Several factors could cause production interruptions, including equipment malfunctions, facility contamination, raw material shortages or contamination, natural disasters, disruption in utility services, human error or disruptions in the operations of our suppliers. We have limited experience manufacturing our product candidates. We have contracted with a third party CMO for the manufacture of our viral vectors and drug product for clinical trials. We expect this CMO will be capable of providing sufficient quantities of our viral vectors and gene therapy products to meet the anticipated scales for our clinical trials, in due course, and commercial demands, if approved. However, to meet our projected needs for further commercial manufacturing and large scale clinical trials, third parties with whom we currently work might need to increase their scale and frequency of production, and we will likely need to secure alternate suppliers or develop our own capabilities. We believe that there are alternate sources of supply that can satisfy our requirements, although we cannot be certain that identifying and establishing relationships with such sources, if necessary, would not result in significant delay or material additional costs.
In addition, two vaccines for COVID-19 were granted Emergency Use Authorization by the FDA in late 2020, and more are likely to be authorized in the coming months. The resultant demand for vaccines and potential for manufacturing facilities and materials to be commandeered under the Defense Production Act of 1950, or equivalent foreign legislation, may make it more difficult to obtain materials or manufacturing slots for the products needed for our clinical trials, which could lead to delays in these trials.
All manufacturers of pharmaceutical products must comply with strictly enforced requirements and complex regulations. Any failure by our CMO to adhere to or document compliance to such regulatory requirements could lead to a delay or interruption in the availability of our product candidate for clinical trials or result in sanctions, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of raw materials, product candidates or products, operating restrictions and criminal prosecutions, any of which could have significant adverse consequences on us. Our potential future dependence upon others for the manufacture of our gene therapies may also adversely affect our future profit margins and our ability to commercialize any product candidates that receive regulatory approval on a timely and competitive basis.
Delays in obtaining regulatory approval of our or our CMOs’ manufacturing process and facility or disruptions in our manufacturing process may delay or disrupt our commercialization efforts.
Before we can begin to commercially manufacture our viral vector or product candidates in our own facility, or the facility of a CMO, we must obtain regulatory approval from the FDA for our manufacturing processes and for the facility in which manufacturing is performed. A manufacturing authorization must also be obtained from the appropriate European Union regulatory authorities. In addition, we must pass a pre-approval inspection of our or our CMOs manufacturing facility by the FDA and other relevant regulatory authorities before any of our gene therapy product candidates can obtain marketing approval. Since March 2020, foreign and domestic inspections by FDA have largely been on hold with FDA announcing plans in July 2020 to resume prioritized domestic inspections. Should FDA determine that an inspection is necessary for approval of a marketing application and an inspection cannot be completed during the review cycle due to restrictions on travel, FDA has stated that it generally intends to issue a complete response letter. Further, if there is inadequate information to make a determination on the acceptability of a facility, FDA may defer action on the application until an inspection can be completed. In 2020, several companies announced receipt of complete response letters due to the FDA’s inability to complete required inspections for their applications. Regulatory authorities outside the U.S. may adopt similar restrictions or other policy measures in response to the COVID-19 pandemic and may experience delays in their regulatory activities. In order to obtain approval, we will need to ensure that all of our processes, quality systems, methods, equipment policies and procedures are compliant with GMP, and perform extensive audits of vendors, contract laboratories, CMOs and suppliers. If any of our vendors, contract laboratories, CMOs or suppliers is found to be out of compliance with GMP, we may experience delays or disruptions in manufacturing while we work with these third parties to remedy the violation or while we work to identify suitable replacement vendors. The GMP requirements govern quality control of the manufacturing process and documentation policies and procedures. In complying with GMP, we will be obligated to expend time, money and effort in production, record keeping and quality control to assure that the product meets applicable specifications and other requirements. If we fail to comply with these requirements, we would be subject to possible regulatory action and may not be permitted to sell any products that we may develop.
| 34 |
Failure to comply with ongoing regulatory requirements could cause us to suspend production or put in place costly or time-consuming remedial measures.
The regulatory authorities may, at any time following approval of a product for sale, audit the manufacturing facilities for such product. If any such inspection or audit identifies a failure to comply with applicable regulations, or if a violation of product specifications or applicable regulations occurs independent of such an inspection or audit, the relevant regulatory authority may require remedial measures that may be costly or time-consuming to implement and that may include the temporary or permanent suspension of a clinical trial or commercial sales or the temporary or permanent closure of a manufacturing facility. Any such remedial measures imposed upon our CMO or us could harm our business, financial condition, results of operations and prospects.
If our CMOs or we fail to comply with applicable GMP regulations, FDA and foreign regulatory authorities can impose regulatory sanctions including, among other things, refusal to approve a pending application for a new product candidate or suspension or revocation of a pre-existing approval. Such an occurrence may cause our business, financial condition, results of operations and prospects to be harmed.
Additionally, if supply from any CMO or us is delayed or interrupted, there could be a significant disruption in the clinical or commercial supply of our product candidates. We have agreements in place with our CMO pursuant to which we are collaborating on GMP manufacturing processes and analytical methods for the manufacture and release of our viral vectors and drug product. Therefore, if we are unable to enter into an agreement with our CMO to manufacture clinical or commercial material for our product programs, or if our agreement with our CMOs were terminated, we would have to find suitable alternative manufacturers. This could delay our or our collaborators’ ability to conduct clinical trials or commercialize our current and future product candidates. The regulatory authorities also may require additional clinical trials and other nonclinical and or analytical evaluations if a new manufacturer is relied upon for clinical or commercial production. Switching manufacturers may involve substantial costs, require significant comparability studies and could result in a delay in our desired clinical and commercial timelines.
Any contamination in our manufacturing process, shortages of materials or failure of any of our key suppliers to deliver necessary components could result in interruption in the supply of our product candidates and delays in our clinical development or commercialization schedules.
Given the nature of biologics manufacturing, there is a risk of contamination in our manufacturing processes. Any contamination could materially adversely affect our ability to produce product candidates on schedule and could, therefore, harm our results of operations and cause reputational damage.
Some of the materials required in our manufacturing process are derived from biologic sources. Such materials are difficult to procure and may be subject to contamination or recall. A material shortage, contamination, recall or restriction on the use of biologically derived substances in the manufacture of our product candidates could adversely impact or disrupt the commercial manufacturing or the production of clinical material, which could materially and adversely affect our development timelines and our business, financial condition, results of operations and prospects.
| 35 |
Patients’ cellular source material must be transported from the clinical collection site to the manufacturing facility and the cryopreserved drug product must be returned to the clinical site for administration into the patient using controlled temperature shipping containers.
Once collected from the patient, the cellular source material must be transported to the manufacturing facility using a shipping container that maintains the material at a required temperature and be delivered typically within three days of collection. While we intend to use reputable couriers and agents for the transport of such materials, if the shipping container is opened or damaged such that the required temperature is not maintained, the cellular source material may be adversely impacted and it may not be feasible to manufacture a drug product for the patient. Similarly, if a shipment is delayed due to adverse weather, misrouting, other events or held up at a customs point, the cellular source material may not be delivered within a time window that will allow for its use for the successful manufacture of a drug product. Similarly, the patient’s autologous drug product must be returned to the clinical site for administration into the patient using a specialized shipping container that maintains the material at a very low temperature for a period of typically up to ten days. While we intend to use reputable couriers and agents for the transport of our drug products, if the shipping container is opened or damaged such that the very low temperature is not maintained, the drug product may be adversely impacted and it may be unsuitable for administration to the patient or harmful. Similarly, if a shipment is delayed due to adverse weather, misrouting, held up at a customs point or other events, and is not delivered to the clinical site within the time period that the very low temperature is maintained, the drug product may be unsuitable for administration to the patient or harmful.
Our gene therapies are for autologous use only. Therefore, if a drug product is administered to the wrong patient, the patient could suffer harm.
Our gene therapies are autologous, so they must be administered back only to the patient from which the cellular source material was collected. While we implement specific identifiers, lot numbers and labels with cross checks for our products and operations from collection of cellular source material, through manufacture of drug product, transport of product to the clinical site up to thawing and administration of the product, it is possible that a product may be administered into the wrong patient. If an autologous gene therapies were to be administered into the wrong patient, the patient could suffer harm, including experiencing a severe adverse immune reaction and this event, should it happen, could adversely affect our business, financial condition, results of operations and prospects.
Our focus on developing our current product candidates may not yield any commercially viable products, and our failure to successfully identify and develop additional product candidates could impair our ability to grow.
As part of our growth strategy, we intend to identify, develop and market additional product candidates beyond our existing product candidate, namely Temferon. We may spend several years completing our development of any particular current or future product candidates, and failure can occur at any stage. The product candidates to which we allocate our resources may not end up being successful. Because we have limited resources, we may forego or delay pursuit of opportunities with certain programs or product candidates or for indications that later prove to have greater commercial potential than Temferon or our other product candidates. Our spending on current and future research and development programs may not yield any commercially viable product candidates. If we do not accurately evaluate the commercial potential for a particular product candidate, we may relinquish valuable rights to that product candidate through strategic collaborations, licensing or other arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate. If any of these events occur, we may be forced to abandon our development efforts with respect to a particular product candidate or fail to develop a potentially successful product candidate.
In addition, certain of our current or future product candidates may not demonstrate in patients any or all of the pharmacological benefits we believe they may possess or compare favorably to existing, approved therapies. We have not yet succeeded and may never succeed in demonstrating efficacy and safety of our product candidates or any future product candidates in preclinical studies, clinical trials or in obtaining marketing approval thereafter and, therefore, may not result in the discovery and development of commercially viable products.
If we are unsuccessful in our development efforts, we may not be able to advance the development of our product candidates, commercialize products, raise capital, expand our business or continue our operations.
| 36 |
Risks Related to Our Reliance on Third Parties
We utilize, and expect to continue to utilize, third parties to conduct some or all aspects of our vector production and product manufacturing for the foreseeable future, and these third parties may not perform satisfactorily.
We currently rely on our CMO for the production of our viral vectors and product candidate for our ongoing clinical trials and preclinical studies. For future clinical trials we intend to utilize materials manufactured by GMP-compliant CMOs. If our partners do not successfully carry out their contractual duties, meet expected deadlines or manufacture our viral vector and product candidates in accordance with regulatory requirements or if there are disagreements between us and our CMO, we will not be able to complete, or may be delayed in completing, the clinical trials required to support approval of our product candidates or the FDA, EMA or other regulatory agencies may refuse to accept our clinical or preclinical data. In such instances, we may need to enter into an appropriate replacement third-party relationship, which may not be readily available or available on acceptable terms, which would cause additional delay or increased expense prior to the approval of our product candidates and would thereby have a negative impact on our business, financial condition, results of operations and prospects.
We have partnered with a commercial GMP-compliant CMO and intend to utilize viral vectors and gene therapy products manufactured by such CMO for our future clinical trials and products for which we obtain marketing approval. There is no assurance that our CMO, or any other future third-party manufacturer that we engage, will be successful in producing any or all of our viral vector or product candidates, that any such product will, if required, pass the required comparability testing, or that any materials produced by any other third-party manufacturer that we engage will have the same effect in patients that we have observed to date. We believe that our manufacturing network will have sufficient capacity to meet demand for our clinical and existing and expected initial commercial needs, but there is a risk that if supplies are interrupted or result in poor yield or quality, it would materially harm our business. Additionally, if the gene therapy industry were to grow, we may encounter increasing competition for the raw materials and consumables necessary for the production of our product candidates. Furthermore, demand for CMO GMP manufacturing capabilities may grow at a faster rate than existing manufacturing capacity, which could disrupt our ability to find and retain third-party manufacturers capable of producing sufficient quantities of our viral vectors or product candidates for future clinical trials or to meet expected initial commercial demand.
Under certain circumstances, our current CMO is entitled to terminate their engagements with us. If we need to enter into alternative arrangements, it could delay our development activities. Our reliance on our CMO for certain manufacturing activities will reduce our control over these activities but will not relieve us of our responsibility to ensure compliance with all required regulations. In addition to our current CMO, we may rely on additional third parties to manufacture ingredients of our viral vectors and or drug product in the future and to perform quality testing, and reliance on these third parties entails risks including:
| ● | reduced control for certain aspects of manufacturing activities; | |
| ● | termination or nonrenewal of manufacturing and service agreements with third parties in a manner or at a time that is costly or damaging to us; and | |
| ● | disruptions to the operations of our third-party manufacturers and service providers caused by conditions unrelated to our business or operations, including the bankruptcy of the manufacturer or service provider. |
Any of these events could lead to clinical trial delays or failure to obtain regulatory approval, or impact our ability to successfully commercialize any of our product candidates. Some of these events could be the basis for FDA, EMA or other regulatory authority action, including injunction, recall, seizure or total or partial suspension of product manufacture.
| 37 |
We rely on third parties to conduct our preclinical and clinical studies and perform other tasks for us. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or comply with regulatory requirements, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be substantially harmed.
We do not expect to independently conduct all aspects of our lentiviral vector protocol development, research and preclinical and clinical testing. We currently rely, and plan to continue to rely, upon third-party CROs to monitor and manage data for our ongoing preclinical and clinical programs. Pursuant to the OSR License Agreement, we agreed to use OSR as the primary site in any preclinical study or clinical trial (including all phases thereof) relating to any licensed products in the field of use, subject to OSR maintaining any required quality standards and providing its services on customary and reasonable terms and consistent with then-applicable market standards. We rely on these parties, including OSR, for execution of our preclinical and clinical studies, but we can only control limited aspects of their activities. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards and our reliance on the CROs does not relieve us of our regulatory responsibilities. We and our CROs and other vendors are required to comply with current GMP, Good Clinical Practices, or GCP, and Good Laboratory Practices, or GLP, which are regulations and guidelines enforced by the FDA, the Competent Authorities of the Member States of the EEA, and comparable foreign regulatory authorities for all of our product candidates in clinical development. Regulatory authorities enforce these regulations through periodic inspections of study sponsors, principal investigators, study sites and other contractors. If we or any of our CROs or vendors fail to comply with applicable regulations, the clinical data generated in our clinical studies may be deemed unreliable and the FDA, the EMA or other comparable regulatory authorities may require us to perform additional clinical studies before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical studies comply with GCP regulations. In addition, our clinical studies must be conducted with product candidates which are produced under GMP regulations. These risks may be heightened as a result of the evolving COVID-19 pandemic due to difficulties in recruiting study subjects during times of travel restriction, delays in obtaining required regulatory inspections, and potential unavailability of our CROs due to their involvement with COVID-related development activities. Our failure to comply with these regulations may require us to repeat clinical studies, which would delay the regulatory approval process.
We and our collaborators and contract manufacturers are subject to significant regulation with respect to manufacturing our product candidates. The manufacturing facilities on which we rely may not meet regulatory requirements and have limited capacity.
Contract manufacturers and their facilities are required to comply with extensive regulatory requirements, including ensuring that quality control and manufacturing procedures conform to GMPs. These GMP regulations cover all aspects of manufacturing relating to our product candidates and components used in clinical studies. These regulations govern manufacturing processes and procedures (including record keeping) and the implementation and operation of quality systems to control and assure the quality of investigational product candidates and products approved for sale. Poor control of production processes can lead to the introduction of contaminants or to inadvertent changes in the properties or stability of our product candidates that may not be detectable in final product testing. We, our collaborators or our contract manufacturers must supply all necessary documentation in support of an BLA or MAA on a timely basis and must adhere to GLP and GMP regulations enforced by the FDA and other regulatory authorities through their facilities inspection program. The facilities and quality systems of some or all of our collaborators and third-party contractors must pass a pre-approval inspection for compliance with the applicable regulations as a condition of regulatory approval of our product candidates or any of our other potential product candidates. In addition, the regulatory authorities may, at any time, audit or inspect a manufacturing facility involved with the preparation of our product candidates or our other potential product candidates or the associated quality systems for compliance with the regulations applicable to the activities being conducted. We do not control the manufacturing process of, and are completely dependent on, our contract manufacturing partners for compliance with the regulatory requirements. If these facilities do not pass a pre-approval plant inspection, regulatory approval of the product candidates may not be granted or may be substantially delayed until any violations are corrected to the satisfaction of the regulatory authority, if ever. Moreover, if our contract manufacturers fail to achieve and maintain high manufacturing standards, in accordance with applicable regulatory requirements, or there are substantial manufacturing errors, this could result in patient injury or death, product shortages, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could seriously harm our business.
| 38 |
We are dependent on a limited number of suppliers and, in some instances, a sole supplier, for some of our components and materials used in our product candidates.
We currently depend on a limited number of suppliers and, in some instances, a sole supplier, for some of the components and equipment necessary for the production of our viral vectors and drug product. We cannot be sure that these suppliers will remain in business, or that they will not be purchased by one of our competitors or another company that is not interested in continuing to produce these materials for our intended purpose. Our use of a sole or a limited number of suppliers of raw materials, components and finished goods exposes us to several risks, including disruptions in supply, price increases, late deliveries and an inability to meet customer demand. There are, in general, relatively few alternative sources of supply for these components, and in some cases, no alternatives. These vendors may be unable or unwilling to meet our future demands for our clinical trials or commercial sale. Establishing additional or replacement suppliers for these components could take a substantial amount of time and it may be difficult to establish replacement suppliers who meet regulatory requirements. Any disruption in supply from any supplier or manufacturing location could lead to supply delays or interruptions which would damage our business, financial condition, results of operations and prospects.
If we are required to switch to a replacement supplier, the manufacture and delivery of our viral vectors and product candidates could be interrupted for an extended period, adversely affecting our business. Establishing additional or replacement suppliers may not be accomplished quickly. If we are able to find a replacement supplier, the replacement supplier would need to be qualified and may require additional regulatory authority approval, which could result in further delay. For example, the FDA or EMA could require additional supplemental data, manufacturing data and comparability data up to and including clinical trial data if we rely upon a new supplier. We may be unsuccessful in demonstrating the comparability of clinical supplies which could require the conduct of additional clinical trials. While we seek to maintain adequate inventory of the components and materials used in our product candidates, any interruption or delay in the supply of components or materials, or our inability to obtain components or materials from alternate sources at acceptable prices in a timely manner, could impair our ability to conduct our clinical trials and, if our product candidates are approved, to meet the demand of our customers and cause them to cancel orders.
In addition, as part of the FDA’s approval of our product candidates, the FDA must review and approve the individual components of our production process, which includes raw materials, the manufacturing processes and facilities of our suppliers. Some of our current suppliers have not undergone this process nor have they had any components included in any product approved by the FDA.
Our reliance on these suppliers subjects us to a number of risks that could harm our reputation, business, and financial condition, including, among other things:
| ● | the interruption of supply resulting from modifications to or discontinuation of a supplier’s operations; | |
| ● | delays in product shipments resulting from uncorrected defects, reliability issues, or a supplier’s variation in a component; | |
| ● | a lack of long-term supply arrangements for key components with our suppliers; | |
| ● | the inability to obtain adequate supply in a timely manner, or to obtain adequate supply on commercially reasonable terms; | |
| ● | difficulty and cost associated with locating and qualifying alternative suppliers for our components in a timely manner; | |
| ● | production delays related to the evaluation and testing of products from alternative suppliers, and corresponding regulatory qualifications; | |
| ● | a delay in delivery due to our suppliers prioritizing other customer orders over ours; |
| 39 |
| ● | damage to our reputation caused by defective components produced by our suppliers; | |
| ● | increased cost of our warranty program due to product repair or replacement based upon defects in components produced by our suppliers; and | |
| ● | fluctuation in delivery by our suppliers due to changes in demand from us or their other customers. |
If any of these risks materialize, costs could significantly increase and our ability to conduct our clinical trials and, if our product candidates are approved, to meet demand for our products could be impacted.
Any collaboration arrangements that we may enter into in the future may not be successful, which could adversely affect our ability to develop and commercialize our current and potential future product candidates.
We may seek collaboration arrangements with biotechnology or pharmaceutical companies for the development or commercialization of our current and potential future product candidates. We may enter into these arrangements on a selective basis depending on the merits of retaining commercialization rights for ourselves as compared to entering into selective collaboration arrangements with other biotechnology or pharmaceutical companies for each product candidate, both in the United States and internationally. We will face, to the extent that we decide to enter into collaboration agreements, significant competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time consuming to negotiate, document and implement. We may not be successful in our efforts to establish and implement collaborations or other alternative arrangements should we so choose to enter into such arrangements. The terms of any collaborations or other arrangements that we may establish may not be favorable to us.
Disagreements between parties to a collaboration arrangement regarding clinical development and commercialization matters can lead to delays in the development process or commercializing the applicable product candidate and, in some cases, termination of the collaboration arrangement. These disagreements can be difficult to resolve if neither of the parties has final decision-making authority.
Collaborations with biotechnology or pharmaceutical companies and other third parties often are terminated or allowed to expire by the other party. Any such termination or expiration could adversely affect us financially and could harm our business reputation.
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or otherwise disclosed.
Because we rely on third parties to develop and manufacture our product candidates, we must, at times, share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, such as trade secrets. Despite the contractual provisions employed when working with third parties, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, a competitor’s discovery of our trade secrets or other unauthorized use or disclosure would impair our competitive position and may have a material adverse effect on our business.
In addition, these agreements typically restrict the ability of our collaborators, advisors, employees and consultants to publish data potentially relating to our trade secrets. Our principal investigators, physicians and academic collaborators typically have rights to publish data, provided that we are notified in advance and may delay publication for a specified time in order to secure our intellectual property rights arising from the collaboration. Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of these agreements, independent development or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication. A competitor’s discovery of our trade secrets would impair our competitive position and have an adverse impact on our business.
| 40 |
Risks Related to Intellectual Property
We depend on license agreements with OSR to permit us to use patents and patent applications, as well as to exploit specific OSR know-how. Termination of these rights or the failure to comply with obligations under these agreements could materially harm our business and prevent us from developing or commercializing our product candidates (Temferon in particular).
We are party to a license agreement with OSR under which we were granted rights to patents and patent applications, as well as proprietary technologies, that are important and necessary to our business, including our Temferon based product candidates. Our rights to use these patents and patent applications and employ the inventions claimed in these licensed patents, as well as the exploitation of OSR proprietary technology, are subject to the continuation of, and our compliance with, the terms of our license agreement.
Our license agreement with OSR imposes upon us various diligence, payment and other obligations, including the following:
| ● | our obligation to pay OSR various milestone payments in the aggregate amount of up to €10 million related to the Lympho-Hematopoietic Indication of each Licensed Product and up to €53 million related to each Solid Cancer indication, contingent upon our achievement of certain late-stage regulatory and sales milestones with respect to use of Temferon for GBM, and additional amounts for milestones with other solid cancer indications upon exercising those rights. However, starting with the fifth Solid Cancer indication, the first two related milestone payments totaling €7.0 million are reduced to €3.5 million. | |
| ● | our obligation to pay OSR royalties based on net sales of each licensed product that we commercialize under the agreement. | |
| ● | our obligation to pay a percentage of income derived from sublicensees for each licensed product sublicensed under the agreement. | |
| ● | our obligation to pay fees associated with the prosecution, maintenance, or filing of the patents and patent applications we have licensed. |
If we fail to comply with any of our obligations under the OSR license agreement, or we are subject to a bankruptcy or dissolution, OSR may have the right to terminate the license agreement, in which event we would not be able to market any product candidates covered by the license.
We do not currently own any patents, and we are heavily reliant upon license from OSR to certain patent rights that are important or necessary to the development of our technology and product candidates, including the patents relating to Temferon. Our license is exclusive only to specific fields of use, namely: GBM, solid liver cancer and any lympho-hematopoietic indication. Although we have exclusive option rights to license additional fields of use, or indications, upon the payment of additional fees to OSR, there is no guarantee that we will be in a position to do so within the time period specified to exercise such right. As a result, we may not be able to prevent competitors from developing and commercializing competitive products.
We do not control the prosecution, maintenance, or filing of the patents and patent applications that are licensed to us under the OSR license agreement (unless OSR chooses i) not to file and/or prosecute certain patent applications, or ii) to abandon such patent application and issued patents, in which cases we have the right to – at our expense – file, prosecute and/or maintain such patent applications), or the enforcement of these patents and patent applications against infringement by third parties. Thus, these patents and patent applications were not drafted by us or our attorneys, and we do not control or have any input into the prosecution of these patents and patent applications. We cannot be certain that drafting or prosecution of the patents and patent applications licensed to us has been conducted in compliance with applicable laws and regulations or will result in valid and enforceable patents. OSR controls the preparation, filing and prosecution of patent applications, and is responsible for maintaining the patents, covering technology that we license.
Pursuant to our license, we are required to file an IND regarding Temferon for GBM prior to February 2022. If we fail to comply with the obligations under our license agreement, including as a result of COVID-19 impacting our operations or due to lack of funds, or if we use the licensed intellectual property in an unauthorized manner, we may be required to pay damages and our licensors may have the right to terminate the license. If our license agreement is terminated, we may not be able to develop, manufacture, market or sell the product candidates covered by our agreement and those being tested or approved in combination with such products. Such an occurrence could materially adversely affect the value of the product candidates being developed under any such agreement.
| 41 |
Disputes may arise regarding intellectual property subject to, and any of our rights and obligations under, any license or other strategic agreement, including:
| ● | the scope of rights granted under the license agreement and other interpretation-related issues; | |
| ● | the extent to which our technology and processes infringe, misappropriate or violate the intellectual property of the licensor that is not subject to the license agreement; | |
| ● | our diligence obligations under the license agreement and what activities satisfy those diligence obligations; | |
| ● | the sublicensing of patent and other rights to third parties under any such agreement or collaborative relationships; | |
| ● | the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and | |
| ● | the priority of invention of patented technology. |
In addition, the agreements under which we license intellectual property or technology to or from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates.
Our business also would suffer if any current or future licensors fail to abide by the terms of the license, if the licensors fail to enforce licensed patents against infringing third parties, if the licensed patents or other rights are found to be invalid or unenforceable, or if we are unable to enter into necessary licenses on acceptable terms. Moreover, our licensors may own or control intellectual property that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are infringing, misappropriating or otherwise violating the licensor’s rights.
In addition, if we are unable to successfully obtain rights to required third-party intellectual property rights or maintain the existing intellectual property rights we have, we may have to seek alternative options, such as developing new product candidates with design-around technologies, which may require more time and investment, or abandon development of the relevant research programs or product candidates and our business, financial condition, results of operations and prospects could suffer.
We have been granted licenses in certain fields of use to patent applications. There can be no assurance that any of the patent applications that we have licenses to will result in issued patents. As a result, our ability to protect our proprietary technology in the marketplace may be limited.
We have been granted licenses in certain fields of use to patent applications in many countries worldwide. These applications cover a range of areas including: applications relating, in general terms, to the use of gene vectors comprising a miRNA target sequence, and the use of gene vectors comprising an interferon-alpha transgene operably linked to a miRNA-130a or miRNA-126 target sequence. Unless and until the pending patent applications are issued, their protective scope is impossible to determine. It is also impossible to predict whether or how many of the patent applications will result in issued patents. Even if pending applications are issued, they may be issued with coverage significantly narrower than what is currently sought.
| 42 |
Our proprietary position for our product candidates currently depends in part upon licenses to patents protecting methods of use, which may not prevent a competitor or other third party from using the same product candidate for another use.
Composition of matter patent claims on the active pharmaceutical ingredient, or API, in pharmaceutical drug products are generally considered to be the favored form of intellectual property protection for pharmaceutical products, as such patents generally provide protection without regard to any particular method of use, manufacture or formulation of the API used. Method of use patent claims protect the use of a product for the specified method and dosing. These types of patent claims do not prevent a competitor or other third party from making and marketing an identical API for an indication that is outside the scope of the method claims or from developing a different dosing regimen. Moreover, even if competitors or other third parties do not actively promote their product for our targeted indications or uses for which we may obtain patents, physicians may recommend that patients use these products off-label, or patients may do so themselves. Although off-label use may infringe or contribute to the infringement of method of use patents, the practice is common and such infringement is difficult to prevent or prosecute.
Even if patents are issued based on patent applications to which we have been granted a license, because the patent positions of pharmaceutical and biotechnology products are complex and uncertain, we cannot predict the scope and extent of patent protection for our product candidates.
Any patents that may be issued based on patent applications that we have been granted licenses to will not ensure sufficient protection with respect to our activities for a number of reasons, including without limitation the following:
| ● | any issued patents may not be broad or strong enough to prevent competition from other gene therapy products including identical or similar products; | |
| ● | if patents are not issued or if issued patents expire, there would be no protections against competitors making generic equivalents; | |
| ● | there may be prior art of which we are not aware that may affect the validity or enforceability of a patent claim; | |
| ● | there may be other patents existing, now or in the future, in the patent landscape for Temferon, or any other product candidates that we seek to commercialize or develop, if any, that will affect our freedom to operate; | |
| ● | if patents that we have been granted licenses to are challenged, a court could determine that they are not valid or enforceable; | |
| ● | a court could determine that a competitor’s technology or product does not infringe patents that we have been granted licenses to; | |
| ● | patents to which we have been granted licenses could irretrievably lapse due to failure to pay fees or otherwise comply with regulations, or could be subject to compulsory licensing; and | |
| ● | if we encounter delays in our development or clinical trials, the period of time during which we could market our products under patent protection would be reduced. |
| 43 |
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and patent protection could be reduced or eliminated for noncompliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the United States Patent and Trademark Office (USPTO) and foreign Intellectual Property Offices in several stages over the term of the patent. Maintenance fees are also due for pending patent applications in some countries. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Noncompliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to office actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. In such an event, our competitors might be able to enter the market, which would have a material adverse effect on our business.
The life of patent protection is limited, and third parties could develop and commercialize products and technologies similar or identical to ours and compete directly with us after the patent licensed to us expires, which could materially and adversely affect our ability to commercialize our products and technologies.
The life of a patent and the protection it affords is limited. For example, in the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. In Europe, the expiration of an invention patent is 20 years from its filing date. Even if we successfully obtain patent protection for an approved drug candidate, it may face competition from generic or biosimilar medications. Manufacturers of generic or biosimilar drugs may challenge the scope, validity or enforceability of the patents underlying our technology in court or before a patent office, and the patent holder may not be successful in enforcing or defending those intellectual property rights and, as a result, we may not be able to develop or market the relevant product candidate exclusively, which would materially adversely affect any potential sales of that product.
Given the amount of time required for the development, testing and regulatory review of new drug candidates, patents protecting such drug candidates might expire before or shortly after such drug candidates are commercialized. As a result, the patents and patent applications licensed to us may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours. Even if we believe that the patents involved are eligible for certain (and time-limited) patent term extensions, there can be no assurance that the applicable authorities, including the FDA and the USPTO, and any equivalent regulatory authority in other countries, will agree with our assessment of whether such extensions are available, and such authorities may refuse to grant extensions to such patents, or may grant more limited extensions than requested. For example, depending upon the timing, duration and specifics of any FDA marketing approval of any product candidates we may develop, one or more of the U.S. patents licensed to us may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Action of 1984, or Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent extension term of up to five years as compensation for patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. However, we may not be granted an extension because of, for example, failing to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents, or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than requested. If we are unable to obtain patent term extension or term of any such extension is less than requested, our competitors may obtain approval of competing products following our patent expiration, and our business could be harmed. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
The patents and pending patent applications licensed to us for our product candidates are expected to expire on various dates as described in “Business—Intellectual Property.” Upon the expiration, we will not be able to assert such licensed patent rights against potential competitors, which would materially adversely affect our business, financial condition, results of operations and prospects.
| 44 |
We may need to license intellectual property from third parties, and such licenses may not be available or may not be available on commercially reasonable terms or at all.
There may be intellectual property rights existing now, or in the future, relevant to Temferon, or any other product candidates that we seek to commercialize or develop, if any, that may affect our ability to commercialize such product candidates. Although the Company is not aware of any such intellectual property rights, a third-party may hold intellectual property rights, including patent rights, that are important or necessary to the development or manufacture of our product candidates. Even if all our main product candidates are covered by patents, it may be necessary for us to use the patented or proprietary technology of third parties to commercialize our product candidates, in which case we would be required to obtain a license from these third parties. Such a license may not be available on commercially reasonable terms, or at all, and we could be forced to accept unfavorable contractual terms. In that event, we may be required to expend significant time and resources to redesign our technology, product candidates, or the methods for manufacturing them or to develop or license replacement technology, all of which may not be feasible on a technical or commercial basis. If we are unable to do so, our business could be harmed.
The licensing or acquisition of third-party intellectual property rights is a competitive area, and several more established companies may pursue strategies to license or acquire third party intellectual property rights that we may consider attractive or necessary. These established companies may have a competitive advantage over us due to their size, capital resources and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third party intellectual property rights on terms that would allow us to make an appropriate return on our investment or at all. If we are unable to successfully obtain rights to required third party intellectual property rights or maintain the existing intellectual property rights we have, we may have to abandon development of the relevant program or product candidate, which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
We may become involved in lawsuits to protect or enforce our intellectual property, which could be expensive, time consuming and unsuccessful.
In addition to the possibility of litigation relating to infringement claims asserted against it, we may become a party to other patent litigation and other proceedings, including inter partes review proceedings, post-grant review proceedings, derivation proceedings declared by the USPTO and similar proceedings in foreign countries, regarding intellectual property rights with respect to our current or future technologies or product candidates or products. Pursuant to the OSR License Agreement, OSR has the right to enforce the patents at its own expense. However, if OSR fails to do so, we have the right to enforce the licensed patents in the field of use, at our expense. The cost to us of any patent litigation or other proceeding, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. Patent litigation and other proceedings may also absorb significant management time. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could impair our ability to compete in the marketplace.
Competitors may infringe or otherwise violate our intellectual property, including patents that may issue to or be licensed by us. As a result, we may be required to file claims in an effort to stop third-party infringement or unauthorized use. Any such claims could provoke these parties to assert counterclaims against us, including claims alleging that we infringe their patents or other intellectual property rights, and/or that any of our IP, including licensed IP, is invalid and/or unenforceable. This can be prohibitively expensive, particularly for a company of our size, and time-consuming, and even if we are successful, any award of monetary damages or other remedy we may receive may not be commercially valuable. In addition, in an infringement proceeding, a court may decide that our asserted intellectual property is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our intellectual property does not cover its technology. An adverse determination in any litigation or defense proceedings could put our intellectual property at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
If the breadth or strength of our patent or other intellectual property rights is compromised or threatened, it could allow third parties to exploit and, in particular, commercialize our technology or products or result in our inability to exploit and/or commercialize our technology and products without infringing third-party intellectual property rights. Further, third parties may be dissuaded from collaborating with us.
Interference or derivation proceedings brought by the USPTO or its foreign counterparts may be necessary to determine the priority of inventions with respect to our patent applications, and we may also become involved in other proceedings, such as re-examination proceedings, before the USPTO or its foreign counterparts. Due to the substantial competition in the pharmaceutical space, the number of such proceedings may increase. This could delay the prosecution of our pending patent applications or impact the validity and enforceability of any future patents that we may obtain. In addition, any such litigation, submission or proceeding may be resolved adversely to us and, even if successful, may result in substantial costs and distraction to our management.
| 45 |
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation, even if some jurisdictions have specific rules so as to maintain confidentiality during the proceedings. Moreover, intellectual property law relating to the fields in which we operate is still evolving and, consequently, patent and other intellectual property rights in our industry are subject to change and are often uncertain. We may not prevail in any of these suits or other efforts to protect our technology, and the damages or other remedies awarded, if any, may not be commercially valuable. During the course of this type of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, the market price for our common stock could be significantly harmed.
Intellectual property litigation could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Litigation or other legal proceedings relating to intellectual property claims, with or without merit, is unpredictable and generally expensive and time consuming and is likely to divert significant resources from our core business, including distracting our technical and management personnel from their normal responsibilities. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock and negatively impact our ability to raise additional funds. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities.
We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon or misappropriating or from successfully challenging our intellectual property rights. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
Any trademarks we may obtain may be infringed or successfully challenged, resulting in harm to our business.
We expect to rely on trademarks, including Temferon, as one means to distinguish any of our product candidates that are approved for marketing from the products of our competitors. Other than Temferon, which we have registered in the EU and the U.S., we have not yet selected trademarks for our product candidates and have not yet begun the process of applying to register trademarks for any other of our product candidates. Once we select trademarks and apply to register them, our trademark applications may not be approved. Third parties may oppose our trademark applications, or otherwise challenge our use of the trademarks. In the event that our trademarks are successfully challenged, we could be forced to rebrand our products, which could result in loss of brand recognition and could require us to devote resources to advertising and marketing new brands. Our competitors may infringe our trademarks and we may not have adequate resources to enforce our trademarks.
In addition, any proprietary name we propose to use with our clinical-stage product candidates or any other product candidate in the United States must be approved by the FDA, regardless of whether we have registered it, or applied to register it, as a trademark. The FDA typically conducts a review of proposed product names, including an evaluation of the potential for confusion with other product names. If the FDA objects to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable proprietary product name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA. The EMA may also object to our proposed proprietary product name that infringes the existing rights of third parties.
| 46 |
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively and our business may be adversely affected. Our efforts to enforce or protect our proprietary rights related to trademarks, trade secrets, domain names, copyrights or other intellectual property may be ineffective and could result in substantial costs and diversion of resources and could adversely affect our business, financial condition, results of operations and growth prospects.
We may not be able to enforce intellectual property rights throughout the world.
Filing, prosecuting, and defending patent applications and issued patents on product candidates in all countries throughout the world would be prohibitively expensive, and intellectual property rights that we have been granted licenses to in some countries outside the United States and Italy can be less extensive than those in the United States and Italy. In addition, the laws of some foreign countries do not protect intellectual property to the same extent as laws in the United States and Italy. Consequently, we may not be able to seek to prevent third parties from practicing inventions that are the subject of patents that we have been granted licenses to in all countries outside the United States and Italy, or from selling or importing products made using inventions that are the subject of patents that we have been granted licenses to in and into the United States or other jurisdictions. Competitors, for example, may use technologies that are the subject of patents that we have been granted licenses to in jurisdictions where we have not licensed patents to develop their own products and further, may export otherwise infringing products to territories where we have been granted licenses to patents, but enforcement is not as strong as that in the United States and Italy.
Many companies have encountered significant problems in protecting and defending intellectual property in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets and other intellectual property, particularly those relating to pharmaceutical and biotechnology products, which could make it difficult for us to stop the infringement of patents that we have been granted licenses to or marketing of competing products in violation of our proprietary rights generally. To date, we have not sought to enforce any issued patents in these foreign jurisdictions. Proceedings to enforce patent rights that we have been granted licenses to in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put patents that we have been granted licenses to at risk of being invalidated or interpreted narrowly and patent applications that we have been granted licenses to at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. The requirements for patentability may differ in certain countries, particularly developing countries. Certain countries in Europe and developing countries, including China and India, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In those countries, we and our licensors may have limited remedies if patents are infringed or if we or our licensors are compelled to grant a license to a third party, which could materially diminish the value of those patents. This could limit our potential revenue opportunities. Accordingly, our efforts to enforce intellectual property rights that we have been granted licenses to around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
If we are unable to maintain effective proprietary rights for our product candidates, we may not be able to compete effectively in our markets.
In addition to the protection afforded by any issued patents to which we have been granted licenses and future patents that may be granted, our license agreement with OSR provides rights to access know-how, or trade secrets. We seek to preserve the integrity and confidentiality of our data, trade secrets and intellectual property by maintaining physical security of our premises and physical and electronic security of our information technology systems, as well as by entering into confidentiality agreements. Agreements or security measures may be breached or could expire, and we may not have adequate remedies for any breach and/or expiration. In addition, our trade secrets may otherwise become known or be independently discovered by competitors.
We cannot provide any assurances that trade secrets and other confidential proprietary information will not be disclosed in violation of confidentiality agreements or that competitors will not otherwise gain access to trade secrets or independently develop substantially equivalent information and techniques. Also, misappropriation or unauthorized and unavoidable disclosure of trade secrets could impair our competitive position and may have a material adverse effect on our business. Additionally, if the steps taken to maintain trade secrets and intellectual property are deemed inadequate, we may have insufficient recourse against third parties for misappropriating any trade secret.
| 47 |
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends upon our ability to develop, manufacture, market and sell our platform technology without infringing the proprietary rights of third parties. There is considerable intellectual property litigation in the biotechnology and pharmaceutical industries. While no such judicial litigation has been brought against us and we have not been held by any court to have infringed a third party’s intellectual property rights, we cannot guarantee that our technology or use of our technology does not infringe third-party patents. It is also possible that we have failed to identify relevant third-party patents or applications. For example, applications filed before November 29, 2000 and certain applications filed after that date that will not be filed outside the United States remain confidential until patents issue. Patent applications in the United States and elsewhere are published approximately 18 months after the earliest filing, which is referred to as the priority date in cases where priority is claimed. Therefore, patent applications covering our technology could have been filed by others without our knowledge. Additionally, pending patent applications which have been published can, subject to certain limitations, be later amended in a manner that could cover our technology.
In March 2013, the United States transitioned to a ‘first to file’ system in which the first inventor to file a patent application will be entitled to the patent. Third parties are allowed to submit prior art prior to the issuance of a patent by the USPTO and may become involved in post-grant review or derivation proceedings for applications filed on or after March 16, 2013, interference proceedings for applications filed before March 16, 2013, ex parte reexamination, or inter partes review challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, which could adversely affect our competitive position with respect to third parties. We may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our technology, including inter partes review, interference, or derivation proceedings before the USPTO and similar bodies in other countries. Third parties may assert infringement claims against us based on existing intellectual property rights and intellectual property rights that may be granted in the future.
We are aware of issued patents in the United States that cover the lentiviral vectors used in the manufacture of our product candidates.
If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue developing and marketing our technology. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Any inability to secure licenses or alternative technology could result in delays in the introduction of our products or lead to prohibition of the manufacture or sale of products by us. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. We could be forced, including by court order, to cease commercializing the infringing technology. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our technology or force us to cease some of our business operations, which could materially harm our business. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business.
We may be subject to claims asserting that our employees, consultants or advisors have wrongfully used or disclosed alleged trade secrets, inventions or intellectual property rights of their current or former employers or claims asserting ownership of what we regard as intellectual property that we have been granted licenses to.
Certain of our employees, consultants or advisors are currently, or were previously, employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees, consultants and advisors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that these individuals or we have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such individual’s current or former employer. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management. Our licensors may face similar risks, which could have an adverse impact on intellectual property that is licensed to us.
In this respect, our key people Mr. Luigi Naldini and Mr. Bernhard Gentner are also employees of Ospedale San Raffaele, and have been appointed, according to a consultancy agreement, as directors of our scientific committee for the purpose of designing and developing the preclinic research and clinic experimentation program in the area of cancers (Prof. Naldini) and mieloma (Dr. Gentner) gene therapy. The relevant consultancy agreements do not set forth any specific representation and warranty in our favor that their activities do not infringe any third-party’s intellectual property rights (in particular, of OSR). In this respect, Mr. Naldini and Mr. Gentner have executed a statement whereby they have declared that their consultancy activities in our favor have been carried out by the same without infringing upon the intellectual property rights of OSR. OSR is not part of this statement and, therefore, OSR could in any case address claims against us with respect to an infringement of its intellectual property right by Mr. Naldini and Mr. Gentner in relation to their activity in our favor.
| 48 |
There may be claims challenging the inventorship of patents and other intellectual property that we have been granted licenses to.
There may be claims that former employees, collaborators or other third parties have an interest in patents or other intellectual property that we have been granted licenses to as an inventor or co-inventor. For example, we may have inventorship disputes arise from conflicting obligations of consultants or others who are involved in developing our product candidates. Litigation may be necessary to defend against these and other claims challenging inventorship. If we fail in defending any such claims, in addition to paying monetary damages, there may be a loss of valuable intellectual property rights to us or our licensors, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. In addition, we may receive less revenue from future products if any of our employees successfully claim for compensation for their work in developing intellectual property, which in turn could impact our future profitability.
Under applicable employment laws, we may not be able to prevent our employees or key consultants, after the termination of their relationship with us or - with reference to key consultants - during the same, to perform competitive activity in favor of other companies nor to enforce covenants not to compete and therefore may be unable to prevent our competitors from benefiting from the expertise of such employees or consultants. In addition, employees and consultants may be entitled to seek compensation for their inventions irrespective of their agreements with us.
To date, we have not entered into non-competition agreements with our current employees in order to prevent them, after the termination of their employment, to perform competitive activity in favor of other employers. Therefore, we cannot exclude that, after the termination of the employment, such employers may benefit from the expertise of our current employees developed while working for us. We sometimes enter into non-competition agreements with certain key consultants. These agreements prohibit key consultants, if they cease working for us, from competing directly with us or working for our competitors or clients for a limited period of time. We may be unable to enforce these agreements under the laws of the jurisdictions in which our consultants work and it may be difficult for us to restrict our competitors from benefitting from the expertise our former consultants developed while working for us. Under Italian law, a non-competition agreement could be invalidated if, for example, the geographic scope of the non-competition agreement is too broad, or, alternatively, such an agreement could be deemed by an Italian court to be an occupation ban. Such actions would make enforcing our non-competition agreements more challenging and could make it easier for our competitors to employ or benefit from the expertise of our key consultants. In addition, we cannot exclude that our current independent consultants may perform activities –during their relationship with us- which could result in competition / conflict with our activity (e.g., in case they perform their activity for the benefit of other employers or companies). Lastly, with reference to the key consultants with whom no non-competition agreement has been entered into, we cannot exclude that, after the termination of their relationship with us or during the same, other employers or companies may benefit from the expertise of such consultants developed while working for us.
In addition, under Italian law, in case of inventions developed by our employees, which were developed while performing their employment activities, but outside the performance of their contractual duties, the rights to the inventions belong to us but we are required to compensate the employees for the rights to their respective inventions. With regard to independent consultants, Italian law provides that, save for the case in which the inventive activity of the same has been set forth as the subject of the consulting agreement and compensated for this purpose, the rights to economically exploit the original contributions and inventions realized in the execution of the consulting agreement will belong to consultant.
To date, both the employment agreements and consultancy agreements do not provide any specific compensation related to the inventive activity.
As a consequence, employees and independent consultants may ask for a fair compensation due to such inventions and, with regard to independent consultants, the failure to pay a fair compensation could prevent us from obtaining rights on their inventions, and this could have a material adverse effect on our operations and ability to effectively compete.
Intellectual property rights do not necessarily address all potential threats.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
| ● | any product candidates we may develop will eventually become commercially available in generic or biosimilar product forms; | |
| ● | others may be able to make products that are similar to any product candidates we may develop or utilize similar technology but that are not covered by the claims of the patents that we may own or license now or in the future; | |
| ● | we, or any future license partners or collaborators, might not have been the first to make the inventions covered by the issued patent or pending patent application that we own or license now or in the future; | |
| ● | we, or any future license partners or collaborators, might not have been the first to file patent applications covering certain of our or their inventions; |
| 49 |
| ● | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; | |
| ● | it is possible that pending patent applications currently licensed or those to which we may enter into a license regarding in the future will not lead to issued patents; | |
| ● | it is possible that there are prior public disclosures that could invalidate the issued patents that have been licensed to us, or parts of such issued patents; | |
| ● | it is possible that there are unpublished applications or patent applications maintained in secrecy that may later issue with claims covering our product candidates or technology similar to ours; | |
| ● | issued patents to which we hold rights to may be held invalid or unenforceable, including as a result of legal challenges by our competitors; | |
| ● | the claims of patent applications, if and when issued, may not cover our product candidates; | |
| ● | our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; | |
| ● | the laws of foreign countries may not protect our proprietary rights or the proprietary rights of license partners or current or future collaborators to the same extent as the laws of the United States; | |
| ● | the inventors of our patent applications may become involved with competitors, develop products or processes that design around our patents, or become hostile to us or the patents or patent applications on which they are named as inventors; | |
| ● | we engage in scientific collaborations and will continue to do so in the future, and our collaborators may develop adjacent or competing products that are outside the scope of our patents; | |
| ● | any product candidates we develop may be covered by third parties’ patents or other exclusive rights; | |
| ● | the patents of others may harm our business; | |
| ● | we may not develop additional proprietary technologies that are patentable; and | |
| ● | we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third-party may subsequently file a patent covering such intellectual property. |
Should any of these events occur, they could have a material adverse effect on our business, financial condition, results of operations, and prospects. The patent positions of biotechnology and pharmaceutical companies generally are highly uncertain, involve complex legal, technical and factual questions and have in recent years been the subject of much litigation. As a result, the issuance, scope, validity, enforceability and commercial value of our patents, including those patent rights licensed to us by third parties, are highly uncertain.
Risks Related to Our Business Operations
As a company currently with substantial operations outside of the United States, our business is subject to economic, political, regulatory and other risks associated with international operations.
As a company with substantial operations in Italy, our business is subject to risks associated with conducting business outside the United States. Many of our suppliers and clinical trial relationships are located outside the United States. Accordingly, our future results could be harmed by a variety of factors, including:
| ● | economic weakness, including inflation, or political instability in particular non-U.S. economies and markets; | |
| ● | differing and changing regulatory requirements for product approvals; | |
| ● | differing jurisdictions could present different issues for securing, maintaining or obtaining freedom to operate in such jurisdictions; | |
| ● | potentially reduced protection for intellectual property rights; |
| 50 |
| ● | difficulties in compliance with different, complex and changing laws, regulations and court systems of multiple jurisdictions and compliance with a wide variety of foreign laws, treaties and regulations; | |
| ● | changes in non-U.S. regulations and customs, tariffs and trade barriers; | |
| ● | foreign exchange risks and currency controls; | |
| ● | changes in a specific country’s or region’s political or economic environment; | |
| ● | trade protection measures, import or export licensing requirements or other restrictive actions by governments; | |
| ● | differing reimbursement regimes and price controls in certain non-U.S. markets; | |
| ● | negative consequences from changes in tax laws; | |
| ● | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad, including, for example, the variable tax treatment in different jurisdictions of options granted under our share incentive plans; | |
| ● | workforce uncertainty in countries where labor unrest is more common than in the United States; | |
| ● | litigation or administrative actions resulting from claims against us by current or former employees or by employees of third party contractors or consultants, individually or as part of class actions, including: (i) claims of wrongful terminations and payment of the related damages, (ii) discrimination, (iii) misclassification, (iv) claims for salary differences or for a different classification according to national collective bargaining agreement, (v) claims for the payment of social security charges or severance benefits, (vi) claims from suppliers’ employees or external consultants such as, by way of example, claims for reclassification as employees, rather than independent contractors, or, as indicated above, requests for payment of salary / social security charges, (vii) any sanctions due to the above-mentioned obligations, (viii) or other violations of labor law or other alleged conduct; | |
| ● | difficulties associated with staffing and managing international operations, including differing labor relations; | |
| ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and | |
| ● | business interruptions resulting from geo-political actions, including war and terrorism, health epidemics, or natural disasters including earthquakes, typhoons, floods and fires. |
We manage our business through a small number of employees, key consultants and third-party contractors.
Our key people include our Chief Executive Officer and current Vice Chairman, Pierluigi Paracchi, who co-founded our company in 2014 along with Luigi Naldini, our Executive Scientific Board Chairman, and Bernhard Gentner, a member of our Executive Scientific Board. Our other key people include Carlo Russo, our Chief Medical Officer & Head of Development, Richard Slansky, our Chief Financial Officer, and Stefania Mazzoleni, our Scientific Project Manager and Communications Officer. With the exception of Ms. Mazzoleni, our key personnel are not (currently) engaged as full-time employees. Dr. Russo shall become a full-time employee after this offering and Mr. Slansky has signed an agreement to become a full-time employee at the earlier of the conclusion of this offering or November 1, 2021, whichever is earlier. We expect that some of our key personnel and other personnel that join us after the completion of this offering will be engaged as full-time employees in the immediate future. Our future growth and success depend on our ability to recruit, retain, manage, and motivate our employees and key consultants. The loss of the services of our Chief Executive Officer or any of our key personnel or the inability to hire or retain experienced management personnel could adversely affect our ability to execute our business plan and harm our operating results. Although we expect to enter into employment agreements with management, these agreements will likely be terminable at will with notice.
In addition, laws and regulations on executive compensation, including legislation in our home country, Italy, may restrict our ability to attract, motivate and retain the required level of qualified personnel.
Because of the specialized scientific and managerial nature of our business, we rely heavily on our ability to attract and retain qualified scientific and technical consultants. In particular, the loss of one or more of our key personnel could be detrimental to us if we cannot recruit suitable replacements in a timely manner. Recruiting and retaining other qualified employees, consultants and advisors for our business, including scientific and technical personnel, will also be critical to our success. There is currently a shortage of skilled executives in our industry, which is likely to continue. As a result, competition for skilled personnel, including in gene therapy research and vector manufacturing, is intense and the turnover rate can be high. We may not be able to attract and retain personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for individuals with similar skill sets. In addition, failure to succeed in preclinical or clinical trials may make it more challenging to recruit and retain qualified personnel. The inability to recruit or the loss of the services of any executive, key employee, consultant or advisor may impede the progress of our research, development and commercialization objectives. We do not currently carry “key person” insurance on the lives of members of senior management. The competition for qualified personnel in the biotechnology and pharmaceutical fields is intense. Due to this intense competition, we may be unable to attract and retain qualified personnel necessary for the development of our business or to recruit suitable replacement personnel.
| 51 |
We face business disruption and related risks resulting from the recent outbreak of the COVID-19 pandemic, which could have a material adverse effect on our business and results of operations.
In December 2019, a novel strain of coronavirus, COVID-19, was identified in Wuhan, China. This virus continues to spread globally and, as of February 2021, has spread to over 200 countries and territories, including Italy and the United States. The spread of COVID-19 from China to other countries has resulted in the World Health Organization declaring the outbreak of COVID-19 as a “pandemic,” or a worldwide spread of a new disease. Many countries around the world have imposed quarantines and restrictions on travel and mass gatherings to slow the spread of the virus. Authorities around the world have and may continue implementing similar restrictions on businesses and individuals in their jurisdictions. While COVID-19 is still spreading and the final implications of the pandemic are difficult to estimate at this stage, it is clear that it has affected the lives of a large portion of the global population, including significant infections in Italy and the United States.
Our operations and business have experienced disruption due to the unprecedented conditions surrounding the COVID-19 pandemic spreading throughout Italy and the world. There can be no assurance we will be able to enact any remedial measures that will enable us to avoid part or all of any impact from the spread of COVID-19 or its consequences, including downturns in business sentiment generally or in our sector in particular. In addition, the impact of COVID-19 may cause delays to our preclinical studies and future clinical trials, and may make it difficult for us to enroll patients to clinical trials.
It is not possible at this time to estimate the full impact that the COVID-19 pandemic, the continued spread of COVID-19, and any additional measures taken by governments, health officials or by us in response to such spread, could have on our business, including the timing of our future clinical trials, results of operations and financial condition. The COVID-19 pandemic and mitigation measures have also negatively impacted global economic conditions, which, in turn, could adversely affect our business, including the timing of our future clinical trials, results of operations and financial condition. We continue to monitor our operations and government recommendations. A significant reduction in our workforce and our compliance with instructions imposed by Italian and other European authorities may harm our ability to continue operating our business and materially and adversely affect our operations and financial condition. Therefore, there can be no assurances that we will be able to immediately comply with all government regulations and we may be subject to authorities’ inspections which may result, in case of non-compliance, in the application of sanctions (in the worst case, even the suspension of work activity). Moreover, we cannot foresee whether the Italian or other European authorities or the U.S. federal government will impose further restrictive instructions, which if implemented may lead to significant changes. The spread of COVID-19 may also result in the inability of our suppliers to deliver components or raw materials on a timely basis. The extent to which COVID-19 impacts our business, results of operations and financial condition will depend on future developments, which are highly uncertain and cannot be predicted, including new information which may emerge concerning the severity of COVID-19 and the actions to contain COVID-19 or treat its impact, among others. See “Risks Related to Product Development, Regulatory Approval and Commercialization – We may find it difficult to enroll patients in our clinical trials. Difficulty in enrolling patients could delay or prevent clinical trials of our product candidates” for additional information.
| 52 |
We will need to significantly increase the size of our organization, and we may experience difficulties in managing growth.
We currently have a very limited number of employees. If we are successful in executing our business strategy and in order to commercialize our products, if approved, we will need to substantially increase our operations, including expanding our employee base of managerial, operational and financial personnel. Any future growth will impose significant added responsibilities on members of management, including the need to identify, recruit, maintain and integrate additional employees. To that end, we must be able to:
| ● | manage our clinical trials and the regulatory process effectively; | |
| ● | develop our administrative, accounting and management information systems and internal controls; | |
| ● | hire and train additional qualified personnel; and | |
| ● | integrate current and additional management, administrative, financial and sales and marketing personnel. |
Our employees, principal investigators, consultants and commercial partners may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of fraud or other misconduct by our employees, principal investigators, consultants and commercial partners. Misconduct by these parties could include intentional failures to comply with the regulations of the FDA, EMA or of other foreign regulatory authorities, provide accurate information to the FDA, EMA and other foreign regulatory authorities, comply with healthcare fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. in particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Such misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and cause serious harm to our reputation. We plan to adopt a code of conduct applicable to all of our employees, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws and health information privacy and security laws. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our operations may be directly or indirectly through our customers, subject to various federal and state fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute, the federal False Claims Act and physician sunshine laws and regulations. These laws may impact, among other things, our proposed sales, marketing and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; | |
| ● | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent; | |
| ● | the Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
| 53 |
| ● | HIPAA, as amended by the Health Information Technology and Clinical Health Act, and its implementing regulations, which imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; | |
| ● | the federal physician sunshine requirements under the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or the PPACA, requires manufacturers of drugs, devices and medical supplies to report annually to the U.S. Department of Health and Human Services information related to payments and other transfers of value to physicians, other healthcare providers and teaching hospitals and ownership and investment interests held by physicians and other healthcare providers and their immediate family members and applicable group purchasing organizations; | |
| ● | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require biotechnology and pharmaceutical companies to comply with the biotechnology and pharmaceutical industries’ voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; and | |
| ● | European and other foreign law equivalents of each of the laws, including reporting requirements detailing interactions with and payments to healthcare providers, and the European General Data Protection Regulation, or GDPR, which became effective in May 2018 and contains new provisions specifically directed at the processing of health information, higher sanctions and extra-territoriality measures intended to bring non-EU companies under the regulation, including companies like us that conduct clinical trials in the EU; we anticipate that over time we may expand our business operations to include additional operations in the EU and with such expansion, we would be subject to increased governmental regulation in the EU countries in which we might operate, including the GDPR and all relevant data protection rulings and further legislation. |
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other related governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, including, without limitation, damages, fines, imprisonment, disgorgement, exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid, reputational harm, additional oversight and reporting obligations if we become subject to a corporate integrity agreement or similar settlement to resolve allegations of non-compliance with these laws and the curtailment or restructuring of our operations. If any of the physicians or other healthcare providers or entities with whom we expect to do business is found to be not in compliance with applicable laws, they may be subject to similar actions, penalties and sanctions. Efforts to ensure that our business arrangements comply with applicable healthcare laws and regulations, as well as responding to possible investigations by government authorities, can be time- and resource-consuming and can divert a company’s attention from the business.
Healthcare legislative and regulatory reform measures may have a material adverse effect on our business and results of operations.
Our industry is highly regulated and changes in law may adversely impact our business, operations or financial results. The PPACA is a sweeping measure intended to, among other things, expand healthcare coverage within the United States, primarily through the imposition of health insurance mandates on employers and individuals and expansion of the Medicaid program. Several provisions of the law may affect us and increase certain of our costs.
| 54 |
In addition, other legislative changes have been adopted since the PPACA was enacted. These changes include aggregate reductions in Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013 and, following passage of the Bipartisan Budget Act of 2018, will remain in effect through 2027 unless additional Congressional action is taken. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our customers and, accordingly, our financial operations.
We anticipate that the PPACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and an additional downward pressure on the reimbursement our customers may receive for our products. Further, there have been, and there may continue to be, judicial and Congressional challenges to certain aspects of the PPACA. For example, the U.S. Tax Cuts and Jobs Act of 2017, or TCJA, includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the PPACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” Additional legislative and regulatory changes to the PPACA, its implementing regulations and guidance and its policies, remain possible. However, it remains unclear how any new legislation or regulation might affect the prices we may obtain for any of our product candidates for which regulatory approval is obtained. Any reduction in reimbursement from Medicare and other government programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our products.
In addition, the delivery of healthcare in the European Union, including the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively a matter for national, rather than EU, law and policy. National governments and health service providers have different priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context. In general, however, the healthcare budgetary constraints in most EU member states have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing EU and national regulatory burdens on those wishing to develop and market products, this could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to commercialize any products for which we obtain marketing approval.
We are currently unable to predict what additional legislation or regulation, if any, relating to the health care industry may be enacted in the future or what effect recently enacted federal legislation or any such additional legislation or regulation would have on our business. The pendency or approval of such proposals or reforms could result in a decrease in the price of our securities or limit our ability to raise capital or to enter into collaboration agreements for the further development and potential commercialization of our products.
The use of any of our product candidates could result in product liability or similar claims that could be expensive, damage our reputation and harm our business.
Our business exposes us to an inherent risk of potential product liability or similar claims. The biotechnology and pharmaceutical industries has historically been litigious, and we face financial exposure to product liability or similar claims if the use of any of our products were to cause or contribute to injury or death. There is also the possibility that defects in the design or manufacture of any of our products might necessitate a product recall. Although we plan to maintain product liability insurance, the coverage limits of these policies may not be adequate to cover future claims. In the future, we may be unable to maintain product liability insurance on acceptable terms or at reasonable costs and such insurance may not provide us with adequate coverage against potential liabilities. A product liability claim, regardless of merit or ultimate outcome, or any product recall could result in substantial costs to us, damage to our reputation, customer dissatisfaction and frustration and a substantial diversion of management attention. A successful claim brought against us in excess of, or outside of, our insurance coverage could have a material adverse effect on our business, financial condition and results of operations.
| 55 |
Our internal information technology systems, or those of our third-party vendors, collaborators or other contractors or consultants, may fail or suffer security breaches or other unauthorized or improper access, which could result in a significant disruption of our product development programs, give rise to significant liability, subject us to costly and protracted litigation, cause significant reputational harm and impact our ability to operate our business effectively.
We are increasingly dependent upon information technology systems, infrastructure, and data to operate our business. In the ordinary course of business, we collect, store, and transmit confidential information (including but not limited to intellectual property, proprietary business information, and personal information). It is critical that we do so in a secure manner to maintain the confidentiality and integrity of such information. We also have outsourced elements of our operations to third parties, and as a result we manage a number of third-party vendors and other contractors and consultants who have access to our confidential information.
Our internal information technology systems and those of our current and any future third-party vendors, collaborators and other contractors or consultants may be vulnerable to a variety of disruptive elements, including data breaches, cyber-attacks by malicious third parties (including the deployment of computer viruses, harmful malware, ransomware, denial-of-service attacks, social engineering, and other means to affect service reliability and threaten the confidentiality, integrity, and availability of information), unauthorized access, natural disasters, terrorism, war, telecommunication and electrical failures and persons with access to systems inside our organization. In particular, the risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers and cyber terrorists, has generally increased as the number, intensity, and sophistication of attempted attacks and intrusions from around the world have increased. We may not be able to anticipate all types of security threats, and we may not be able to implement preventive measures effective against all such security threats. Because the techniques used by cyber criminals change frequently, may not be recognized until launched, and can originate from a wide variety of sources, including outside groups such as external service providers, organized crime affiliates or terrorist organizations, we and our partners may be unable to anticipate these techniques or implement adequate preventative measures. Further, we do not have any control over the operations of the facilities or technology of third parties that collect, process and store personal data on our behalf.
While we have not experienced any significant system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations or a loss of, or damage to, our data or applications, or those of our third-party vendors and other collaborators, contractors and consultants, it could result in a disruption of our development programs and our business operations, whether due to a loss of our trade secrets or other confidential, personal or proprietary information, significant delays or setbacks in our research, or other similar disruptions. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential, personal or proprietary information, we could incur significant liability, our competitive position could be harmed, our reputation could be damaged, and the further development and commercialization of our product candidates could be delayed.
Unauthorized disclosure of sensitive or confidential data, including personal information, whether through a breach of computer systems, systems failure, employee negligence, fraud or misappropriation, or otherwise, or unauthorized access to or through our information systems and networks, whether by our employees or third parties, could result in negative publicity, damage to our reputation and/or compel us to comply with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action, and otherwise subject us to liability under laws and regulations that protect the privacy and security of personal information. The costs related to significant security breaches or disruptions could be material. If the information technology systems of our third-party vendors and other collaborators, contractors and consultants become subject to disruptions or security breaches, we may be exposed to material liability and have insufficient recourse against such third parties and we may have to expend significant resources to mitigate the impact of such an event, and to develop and implement protections to prevent future events of this nature from occurring. Any of the foregoing could adversely affect our business, financial condition, results of operations or prospects.
| 56 |
We or the third parties upon whom we depend may be adversely affected by earthquakes or other natural disasters and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
Earthquakes or other natural disasters could severely disrupt our operations, and have a material adverse effect on our business, results of operations, financial condition and prospects. If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as the manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place currently are limited and are unlikely to prove adequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which, particularly when taken together with our lack of earthquake insurance, could have a material adverse effect on our business, financial condition, results of operations and prospects.
Unsuccessful compliance with certain European privacy regulations could have an adverse effect on our business and reputation.
The collection and use of personal health data in the EU is governed, as of May 2018, by the General Data Protection Regulation 2016/679 (GDPR) as implemented by European Data Protection Board (EDPB) guidelines and EU Member States national legislations. General EU data protection rules impose several requirements relating to the consent of the individuals to whom the personal data relates, the information provided to the individuals, notification of data processing obligations to the competent national data protection authorities and the security and confidentiality of the personal data. The GDPR also extends the geographical scope of EU data protection law to non-EU entities under certain conditions, tightens existing EU data protection principles and creates new obligations for companies and new rights for individuals. Failure to comply with the requirements of the GDPR, the EDPB guidelines and the related national data protection laws of the EU Member States may result in fines and other administrative penalties. The GDPR introduces new data protection requirements in the EU and substantial fines for breaches of the data protection rules, including violation of articles 44 to 49 GDPR related to transfer of personal data to a recipient in a non-EU country. The GDPR regulations impose additional responsibility and liability in relation to personal data that we process, and we intend to put in place additional mechanisms ensuring compliance with these and/or new data protection rules. In addition, other jurisdictions, including Italy, have implemented regulations similar to GDPR. With regard to Italian legislation, the national Privacy and Data Protection Code has been amended according to GDPR provisions (Legislative Decree n. 196/2003 as amended and updated by Legislative Decree n. 101/2018) and imposes additional fines and administrative penalties in relation to the processing of health data and processing of data for scientific research purposes. Moreover, European data protection background is constantly changing under the drive of the European Data Protection Board (EDPB) on the correct interpretation and application of GDPR and the ruling activity of the Court of Justice of the European Union (see, for instance, the recent CJEU case C-3111/18, also known as Schremes II which invalidated the EU-US Privacy Shield Framework for transfer of data to United States).
The Company is compliant with most recent legislative changes in European data protection rules, adopting Data Processing Agreements containing Standard Contractual Clauses with all partners based in the United States and (for the transition period until June 2021) in the United Kingdom. However, changes to these European privacy regulations (and similar regulations in other jurisdictions) and unsuccessful compliance may be onerous and adversely affect our business, financial condition, prospects, results of operations and reputation.
Risks Related to this Offering and Ownership of Our Securities
An active trading market for the ADSs may not develop and you may not be able to resell the ADSs at or above the initial offering price, or at all.
This offering constitutes the initial public offering of the ADSs, and no public market has previously existed for the ADSs. Any delay in the commencement of trading of the ADSs on Nasdaq would impair the liquidity of the market for the ADSs and make it more difficult for holders to sell the ADSs. There can be no assurance that an active trading market for the ADSs will develop or be sustained after this offering is completed. The lack of an active trading market may also reduce the fair market value of the ADSs. The initial offering price was determined by negotiations among the lead underwriters and us. Among the factors considered in determining the initial public offering price were our future prospects and the prospects of our industry in general, our revenue, net income and certain other financial and operating information in recent periods, and the market prices of securities and certain financial and operating information of companies engaged in activities similar to ours. However, there can be no assurance that, following the completion of this offering, the ADSs will trade at a price equal to or greater than the initial public offering price.
Our management will have broad discretion as to the use of the net proceeds from this offering and may not use the proceeds effectively.
We currently intend to use the net proceeds of this offering for working capital and general corporate purposes, possible in licensing of additional intellectual property and product candidates, and next generation product development. See “Use of Proceeds.” However, our management will have broad discretion in the application of the net proceeds. Our shareholders may not agree with the manner in which our management chooses to allocate the net proceeds from this offering. The failure by our management to apply these funds effectively could have a material adverse effect on our business, financial condition and results of operation. Pending their use, we may invest the net proceeds from this offering in a manner that does not produce income. The decisions made by our management may not result in positive returns on your investment and you will not have an opportunity to evaluate the economic, financial or other information upon which our management bases its decisions.
| 57 |
If you purchase the securities in this offering, you will incur immediate and substantial dilution in the book value of the securities.
Because the price per security being offered is substantially higher than our net tangible book value per ADS or ordinary share, you will suffer substantial dilution in the net tangible book value of any ADSs or ordinary shares you purchase in this offering. After giving effect to the sale by us of securities in this offering, based on an assumed public offering price of $11.50 per security, the midpoint of the estimated price range set forth on the cover page of this prospectus, and after deducting underwriting discounts, estimated advisory fees and offering expenses payable by us, our as adjusted net tangible book value of the securities would be approximately $35.0 million, or approximately $1.76 per ADS or ordinary share, as of June 30, 2021. If you purchase ADSs or ordinary shares in this offering, you will suffer immediate and substantial dilution of our as adjusted net tangible book value of approximately $9.74 per ADS or ordinary share. As a result of this dilution, investors purchasing shares in this offering may receive significantly less than the purchase price paid in this offering in the event of liquidation. See “Dilution” for additional information.
ADS representing a substantial percentage of our outstanding ordinary shares may be sold in this offering, which could cause the price of the ADSs to decline.
We may sell in this offering securities representing approximately 1,608,695 ADSs and approximately 1,000,000 ordinary shares, or approximately 17%, of our outstanding securities as of June 30, 2021. This sale and any future sales of a substantial number of ADSs in the public market, or the perception that such sales may occur, could materially adversely affect the price of the ADSs. We cannot predict the effect, if any, that market sales of those ADSs or the availability of those ADSs for sale will have on the market price of the ADSs. While the securities sold to non-affiliates in this offering will be freely tradable in the public market, substantially all of the securities owned prior to this offering are expected to be subject to lock-up agreements with the underwriters that restrict the ability of these shareholders to transfer our securities held by them for at least six months from the date of this prospectus. These outstanding securities that are subject to lock-up agreements are expected to become eligible for unrestricted sale upon expiration of the lock-up period. In addition, securities issued or issuable upon exercise of options and warrants vested as of the expiration of the lock-up period will be eligible for sale at that time. Sales of securities by these shareholders could have a material adverse effect on the trading price of the ADSs.
We do not know what the trading price of the ADSs will be following this offering and as a result it may be difficult for you to sell the ADSs.
The trading price of the ADSs is likely to be volatile. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of the ADSs:
| ● | adverse results or delays in pre- and non-clinical studies or clinical trials; | |
| ● | reports of adverse events in other gene therapy products or clinical studies of such products; | |
| ● | inability to obtain additional funding; | |
| ● | inability to obtain the approvals necessary to commence clinical trials; | |
| ● | unsatisfactory results of clinical trials; | |
| ● | announcements of regulatory approval or the failure to obtain it, or specific label indications or patient populations for its use, or changes or delays in the regulatory review process; |
| 58 |
| ● | announcements of therapeutic innovations or new products by us or our competitors; | |
| ● | adverse actions taken by regulatory authorities with respect to our clinical trials, manufacturing supply chain or sales and marketing activities; | |
| ● | changes or developments in laws or regulations applicable to the treatment of cancer tumors, or any other indication that we may seek to develop; | |
| ● | any adverse changes to our relationship with manufacturers or suppliers; | |
| ● | any intellectual property infringement actions in which we may become involved; | |
| ● | announcements concerning our competitors or the biotechnology and pharmaceutical industries in general; | |
| ● | achievement of expected product sales and profitability or our failure to meet expectations; | |
| ● | our commencement of, or involvement in, litigation; | |
| ● | any major changes in our board of directors or management; | |
| ● | our ability to recruit and retain qualified regulatory, research and development personnel; | |
| ● | legislation in the United States relating to the sale or pricing of biotechnology or gene therapy products; | |
| ● | the depth of the trading market in the ADSs; | |
| ● | termination of the lock-up agreements or other restrictions limiting our ability or that of any of our existing shareholders to sell our securities (or any other securities that we may issue, if any) after this offering; | |
| ● | economic weakness, including inflation, or political instability in particular foreign economies and markets; | |
| ● | business interruptions resulting from a local or worldwide pandemic, such as COVID-19, geopolitical actions, including war and terrorism, or natural disasters; | |
| ● | the granting or exercise of employee stock options or other equity awards; | |
| ● | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; | |
| ● | additions or departures of key scientific or management personnel; | |
| ● | significant lawsuits, including patent or stockholder litigation; and | |
| ● | changes in investors’ and securities analysts’ perception of the business risks and conditions of our business. |
In addition, the stock market in general, and the Nasdaq Stock Market in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of small companies. Broad market and industry factors may negatively affect the market price of the ADSs, regardless of our actual operating performance. Further, a systemic decline in the financial markets and related factors beyond our control may cause our ADS price to decline rapidly and unexpectedly.
| 59 |
Holders of ADSs may not receive the same distributions or dividends as those we make to the holders of our ordinary shares, and, in some limited circumstances, you may not receive dividends or other distributions on our ordinary shares and you may not receive any value for them, if it is illegal or impractical to make them available to you.
The Depositary for the ADSs has agreed to pay to you the cash dividends or other distributions it or the custodian receives on ordinary shares or other deposited securities underlying the ADSs, after deducting its fees and expenses and subject to the terms of the deposit agreement. You will receive these distributions in proportion to the number of ordinary shares the ADSs represent. However, in accordance with the limitations set forth in the deposit agreement the Depositary is not responsible if it decides that it is unlawful or impractical to make a distribution available to any holders of ADSs. For example, it would be unlawful to make a distribution to a holder of ADSs if it consists of securities that require registration under the Securities Act, but that are not properly registered or distributed under an applicable exemption from registration. In addition, conversion into U.S. dollars from foreign currency that was part of a dividend made in respect of deposited ordinary shares may require the approval or license of, or a filing with, any government or agency thereof, which may be unobtainable. In these cases, the Depositary may determine not to distribute such property and hold it as “deposited securities” or may distribute the net cash proceeds from the sale of the dividends. We have no obligation to register under U.S. securities laws any ADSs, ordinary shares, rights or other securities received through such distributions. We also have no obligation to take any other action to permit the distribution of ADSs, ordinary shares, rights or anything else to holders of ADSs. In addition, the Depositary may withhold from such dividends or distributions its fees and an amount on account of taxes or other governmental charges to the extent the Depositary believes it is required to make such withholding. This means that you may not receive the same distributions or dividends as those we make to the holders of our ordinary shares, and, in some limited circumstances, you may not receive any value for such distributions or dividends if it is illegal or impractical for us to make them available to you. These restrictions may cause a material decline in the value of the ADSs.
Holders of ADSs must act through the Depositary to exercise voting rights relating to the ordinary shares.
Holders of the ADSs do not have the same rights of our shareholders and may only exercise the voting rights with respect to the underlying ordinary shares in accordance with the provisions of the Deposit Agreement. When a shareholder meeting is convened, holders of ADSs may not receive sufficient notice of a shareholder meeting to permit them to cancel their ADSs and withdraw ordinary shares to allow them to directly cast their vote with respect to any specific matter. In addition, the Depositary and its agents may not be able to send voting instructions to holders of ADSs or carry out their voting instructions in a timely manner. We will make all reasonable efforts to cause the Depositary to extend voting rights to holders of the ADSs in a timely manner, but we cannot assure holders that they will receive the voting materials in time to ensure that they can instruct the Depositary to vote the ordinary shares underlying their ADSs. Furthermore, the Depositary and its agents will not be responsible for any failure to carry out any instructions to vote, for the manner in which any vote is cast or for the effect of any such vote. As a result, holders of the ADSs may not be able to exercise their right to vote and they may lack recourse if the ordinary shares underlying their ADSs are not voted as they requested. In addition, in the capacity as a holder of ADSs, they will not be able to call a shareholder meeting.
ADSs holders may not be entitled to a jury trial with respect to claims arising under the deposit agreement, which could augur less favorable results to the plaintiff(s) in any such action.
The deposit agreement governing the ADSs representing our shares provides that owners and holders of ADSs, including those who purchase the ADSs in a secondary transaction, irrevocably waive the right to a trial by jury in any legal proceeding arising out of or relating to the deposit agreement, our shares or the ADSs or the transactions contemplated thereby, including claims under federal securities laws, against us or the depositary to the fullest extent permitted by applicable law. If this jury trial waiver provision is prohibited by applicable law, an action could nevertheless proceed under the terms of the deposit agreement with a jury trial. To our knowledge, the enforceability of a jury trial waiver under the federal securities laws has not been finally adjudicated by a federal court. However, we believe that a jury trial waiver provision is generally enforceable under the laws of the State of New York, which govern the deposit agreement, by a court of the State of New York or a federal court in New York, which have non-exclusive jurisdiction over matters arising under the deposit agreement, applying such law. In determining whether to enforce a jury trial waiver provision, New York courts and federal courts will consider whether the visibility of the jury trial waiver provision within the agreement is sufficiently prominent such that a party has knowingly waived any right to trial by jury. We believe that this is the case with respect to the deposit agreement, our shares and the ADSs and the transactions contemplated thereby. In addition, New York courts will not enforce a jury trial waiver provision in order to bar a viable setoff or counterclaim sounding in fraud or one which is based upon a creditor’s negligence in failing to liquidate collateral upon a guarantor’s demand, or in the case of an intentional tort claim (as opposed to a contract dispute), none of which we believe are applicable in the case of the deposit agreement, our shares or the ADSs or the transactions contemplated thereby. No condition, stipulation or provision of the deposit agreement or ADSs serves as a waiver by any owner or holder of ADSs or by us or the depositary of compliance with any provision of the federal securities laws. If you or any other owner or holder of ADSs brings a claim against us or the depositary in connection with matters arising under the deposit agreement, our shares or the ADSs or the transactions contemplated thereby, you or such other owner or holder may not be entitled to a jury trial with respect to such claims, which may have the effect of limiting and discouraging lawsuits against us and/or the depositary, lead to increased costs to bring a claim, limited access to information and other imbalances of resources between such owner or holder and us, or limit such holder’s ability to bring a claim in a judicial forum that such holder finds favorable. If a lawsuit is brought against us and/or the depositary under the deposit agreement, it may be heard only by a judge or justice of the applicable trial court, which would be conducted according to different civil procedures and may augur different results than a trial by jury would have had, including results that could be less favorable to the plaintiff(s) in any such action, depending on, among other things, the nature of the claims, the judge or justice hearing such claims, and the venue of the hearing.
| 60 |
We have identified material weaknesses in our internal control over financial reporting and, if our remediation of the material weaknesses is not effective or if we identify additional material weaknesses in the future, we may not be able to accurately or timely report our financial results, or prevent fraud, and investor confidence in our Company and the market price of our shares may be adversely affected.
As a public company, we will be required to maintain internal control over financial reporting and to report any material weaknesses in such internal control. Section 404 of the Sarbanes-Oxley Act requires that we evaluate and determine the effectiveness of our internal control over financial reporting. A material weakness is a deficiency or combination of deficiencies in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our financial statements will not be prevented or detected on a timely basis.
To date, we have had limited financial and accounting personnel, which has resulted in a limited segregation of duties to fully execute our accounting processes and address our internal control over financial reporting. In connection with the audits of our financial statements as of and for the years ended December 31, 2020 and 2019, we identified certain material weaknesses in our internal control over financial reporting, including but not limited to our lack of adequate staff to: (i) process financial information in a timely manner; (ii) analyze and account for complex, non-routine transactions - including those subject to our critical accounting policies; and, (iii) maintain adequate segregation of duties; and, the lack of documentation related to our internal control over financial reporting including our policy over related party relationships and transactions.
We plan to take steps to address the internal control deficiencies that contributed to the material weaknesses, including the following:
| ● | hiring of additional finance and accounting personnel with prior experience working for finance departments and technical accounting experience, supplemented by third-party resources; | |
| ● | documenting and formally assessing our accounting and financial reporting policies and procedures; and | |
| ● | increasing the use of third-party consultants in assessing significant accounting transactions and other technical accounting and financial reporting issues, preparing accounting memoranda addressing these issues and maintaining these memoranda in our corporate records. |
While we believe that these efforts will improve our internal control over financial reporting, the implementation of these measures is ongoing and will require validation and testing of the design and operating effectiveness of internal controls over a sustained period of financial reporting cycles. We cannot assure you that the measures we have taken to date, and are continuing to implement, will be sufficient to remediate the material weaknesses we have identified or avoid potential future material weaknesses. If the steps we take do not correct the material weaknesses in a timely manner, we will be unable to conclude that we maintain effective internal control over financial reporting. Accordingly, there could continue to be a reasonable possibility that these deficiencies or others could result in a misstatement of our accounts or disclosures that would result in a material misstatement of our financial statements that would not be prevented or detected on a timely basis.
Our management and independent registered public accounting firm did not perform an evaluation of our internal control over financial reporting during any period in accordance with the provisions of the Sarbanes-Oxley Act. Had we and our independent registered public accounting firm performed an evaluation of our internal control over financial reporting in accordance with the provisions of the Sarbanes-Oxley Act, additional control deficiencies amounting to material weaknesses may have been identified. If we identify new material weaknesses in our internal control over financial reporting, if we are unable to comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner, if we are unable to assert that our internal control over financial reporting is effective, or if our independent registered public accounting firm is unable to express an opinion as to the effectiveness of our internal control over financial reporting, we may be late with the filing of our periodic reports, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our common stock could be negatively affected. As a result of such failures, we could also become subject to investigations by the stock exchange on which our securities are listed, the SEC, or other regulatory authorities, and become subject to litigation from investors and stockholders, which could harm our reputation, financial condition or divert financial and management resources from our core business.
| 61 |
We may be subject to securities litigation, which is expensive and could divert management attention.
In the past, companies that have experienced volatility in the market price of their shares have been subject to securities class action litigation. We may be the target of this type of litigation in the future. Litigation of this type could result in substantial costs and diversion of management’s attention and resources, which could seriously hurt our business. Any adverse determination in litigation could also subject us to significant liabilities.
Our Chief Executive Officer, directors and shareholders who own more than 5% of our outstanding ordinary shares before this offering currently own approximately 47.14% of our outstanding ordinary shares and will own approximately 40.17% of our ordinary shares upon the completion of this offering. They will therefore be able to exert significant control over matters submitted to our shareholders for approval.
After this offering, our Chief Executive Officer and directors, and shareholders who own more than 5% of our outstanding ordinary shares before this offering will, in the aggregate, beneficially own approximately 40.17% of our ordinary shares (assuming no exercise of the underwriters’ over-allotment option). This significant concentration of share ownership may adversely affect the trading price for the ADSs because investors often perceive disadvantages in owning securities in companies with controlling shareholders. As a result, these shareholders, if they acted together, could significantly influence or even unilaterally approve matters requiring approval by our shareholders, including the election of directors and the approval of mergers or other business combination transactions. The interests of these shareholders may not always coincide with our interests or the interests of other shareholders. Also, the concentration of our beneficial ownership may have the effect of delaying, deterring or preventing a change in our control, or may discourage bids for our shares at a premium over the market price of the shares. The significant concentration of share ownership may adversely affect the trading price of our ordinary shares due to investors’ perception that conflicts of interest may exist or arise.
If we were to be characterized as a “passive foreign investment company” for U.S. tax purposes, U.S. holders of the ADSs could have adverse U.S. income tax consequences.
In general, we will be treated as a passive foreign investment company, or a PFIC, for U.S. federal income tax purposes in any taxable year in which either (1) at least 75% of our gross income is “passive income” or (2) on average at least 50% of our assets by value produce passive income or are held for the production of passive income. Passive income for this purpose generally includes, among other things, certain dividends, interest, royalties, rents and gains from commodities and securities transactions and from the sale or exchange of property that gives rise to passive income. Passive income also includes amounts derived by reason of the temporary investment of funds, including those raised in a public offering. In determining whether a non-U.S. corporation is a PFIC, a proportionate share of the income and assets of each corporation in which it owns, directly or indirectly, at least a 25% interest (by value) is taken into account. We have not made the formal analysis necessary to determine whether or not we are currently a PFIC or whether we have ever been a PFIC, although preliminarily it appears we may have been a PFIC at certain points in the past. The tests for determining PFIC status depend, in part, on the application of complex U.S. federal income tax rules, which are subject to differing interpretations. In addition, whether any corporation will be a PFIC for any taxable year depends on the assets and income of such corporation over the course of each such taxable year and, as a result, it is difficult to make accurate projections of future income and assets which are relevant to this determination for the current taxable year or any future period. If we are a PFIC in any taxable year during which a U.S. taxpayer holds the ADSs, such U.S. taxpayer would be subject to certain adverse U.S. federal income tax rules. In particular, if the U.S. taxpayer did not make an election to treat us as a “qualified electing fund,” or QEF, or make a “mark-to- market” election, then “excess distributions” to the U.S. taxpayer, and any gain realized on the sale or other disposition of the ADSs by the U.S. taxpayer: (1) would be allocated ratably over the U.S. taxpayer’s holding period for the ADSs; (2) the amount allocated to the current taxable year and any period prior to the first day of the first taxable year in which we were a PFIC would be taxed as ordinary income; and (3) the amount allocated to each of the other taxable years would be subject to tax at the highest rate of tax in effect for the applicable class of taxpayer for that year, and an interest charge for the deemed deferral benefit would be imposed with respect to the resulting tax attributable to each such other taxable year. In addition, if the U.S. Internal Revenue Service, or the IRS, determines that we are a PFIC for a year with respect to which we have determined that we were not a PFIC, it may be too late for a U.S. taxpayer to make a timely QEF or mark-to-market election. U.S. taxpayers that have held the ADSs during a period when we were a PFIC will be subject to the foregoing rules, even if we cease to be a PFIC in subsequent years, subject to exceptions for U.S. taxpayer who made a timely QEF or mark-to-market election. A U.S. taxpayer can make a QEF election by completing the relevant portions of and filing IRS Form 8621 in accordance with the instructions thereto. U.S. taxpayers that hold the ADSs are strongly urged to consult their tax advisors about the PFIC rules, including tax return filing requirements and the eligibility, manner, and consequences to them of making a QEF or mark-to-market election with respect to the ADSs in the event that we are a PFIC. See “Taxation —U.S. Federal Income Tax Consequences—Passive Foreign Investment Companies” for additional information.
| 62 |
If securities or industry analysts do not publish or cease publishing research or reports about us, our business or our market, or if they adversely change their recommendations or publish negative reports regarding our business or our securities, our ADS price and trading volume could decline.
The trading market for the ADSs will be influenced by the research and reports that industry or securities analysts may publish about us, our business, our market or our competitors. We do not have any control over these analysts and we cannot provide any assurance that analysts will cover us or provide favorable coverage. If any of the analysts who may cover us adversely change their recommendation regarding our securities, or provide more favorable relative recommendations about our competitors, our ADS price would likely decline. If any analyst who may cover us were to cease coverage of our Company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our ADS price or trading volume to decline.
We have not paid, and do not intend to pay, dividends on our ordinary shares and, therefore, unless our traded securities appreciate in value, our investors may not benefit from holding our securities.
We have never declared or paid cash dividends on our ordinary shares. We do not anticipate paying any cash dividends on our ordinary shares in the foreseeable future. Consequently, investors may need to rely on sales of their ADSs after price appreciation, which may never occur, as the only way to realize any future gains on their investment. Investors seeking cash dividends should not purchase the ADSs. Moreover, Italian law imposes certain restrictions on our ability to declare and pay dividends. In particular, Italian law prohibits distributing dividends other than from net income or distributable reserves set forth in a company’s statutory accounts approved by a meeting of shareholders and after the establishment of certain compulsory reserves. In addition, if losses from previous fiscal years have reduced a company’s capital, dividends may not be paid until the capital is reconstituted or its stated amount is reduced by the amount of such losses. The application of these restrictions limits our ability to make distributions to holders of our shares See “Dividend Policy” and “Description of Share Capital and Governing Documents—Dividends and Other Distributions” for additional information.
The requirements associated with being a public company will require significant company resources and management attention.
Following this offering, we will become subject to the reporting requirements of the Exchange Act, Nasdaq listing requirements and other applicable securities rules and regulations. The Exchange Act requires that we file periodic reports with respect to our business and financial condition and maintain effective disclosure controls and procedures and internal control over financial reporting. In addition, subsequent rules implemented by the SEC and the Nasdaq Stock Market may also impose various additional requirements on public companies. As a result, we will incur additional legal, accounting and other expenses that we did not incur as a nonpublic company, particularly after we are no longer an “emerging growth company” as defined in the JOBS Act. In the period following this offering, we estimate that these expenses will be at least several hundred thousand dollars annually. Further, the need to establish the corporate infrastructure demanded of a public company may divert management’s attention from implementing our development plans. We have made changes to our corporate governance standards, disclosure controls and financial reporting and accounting systems to meet our reporting obligations. The measures we take, however, may not be sufficient to satisfy our obligations as a public company, which could subject us to delisting of our securities, fines, sanctions and other regulatory action and potentially civil litigation.
| 63 |
The JOBS Act allows us to postpone the date by which we must comply with some of the laws and regulations intended to protect investors and to reduce the amount of information we provide in our reports filed with the SEC, which could undermine investor confidence in our Company and adversely affect the market price of the ADSs.
For so long as we remain an “emerging growth company” as defined in the JOBS Act, we intend to take advantage of certain exemptions from various requirements that are applicable to public companies that are not “emerging growth companies” including:
| ● | the provisions of the Sarbanes-Oxley Act requiring that our independent registered public accounting firm provide an attestation report on the effectiveness of our internal control over financial reporting; | |
| ● | Section 107 of the JOBS Act, which provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. This means that an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We are electing to delay such adoption of new or revised accounting standards. As a result of this election, our financial statements may not be comparable to companies that comply with the public company effective date; | |
| ● | any rules that may be adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotation or a supplement to the auditor’s report on the financial statements; and | |
| ● | our ability to furnish two rather than three years of income statements and statements of cash flows in various required filings. |
We intend to take advantage of these exemptions until we are no longer an “emerging growth company.” We will remain an emerging growth company until the earlier of (1) the last day of the fiscal year (a) following the fifth anniversary of the date of the completion of this offering, (b) in which we have total annual gross revenue of at least $1.07 billion, or (c) in which we are deemed to be a large accelerated filer, as defined in the rule under the Exchange Act, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
We cannot predict if investors will find our securities less attractive because we may rely on these exemptions. If some investors find our securities less attractive as a result, there may be a less active trading market for the ADSs, and our ADS price may be more volatile and may decline.
As a foreign private issuer, we are permitted, and intend, to follow certain home country corporate governance practices instead of otherwise applicable SEC and Nasdaq requirements, which may result in less protection than is accorded to investors under rules applicable to domestic U.S. issuers.
Our status as a foreign private issuer exempts us from compliance with certain SEC laws and regulations and certain regulations of the Nasdaq Stock Market, including the proxy rules, the short-swing profits recapture rules, and certain governance requirements such as independent director oversight of the nomination of directors and executive compensation. In addition, we will not be required under the Exchange Act to file current reports and financial statements with the SEC as frequently or as promptly as U.S. domestic companies whose securities are registered under the Exchange Act and we will generally be exempt from filing quarterly reports with the SEC. Furthermore, as a foreign private issuer, we are also not subject to the requirements of Regulation FD (Fair Disclosure) promulgated under the Exchange Act. These exemptions and leniencies will reduce the frequency and scope of information and protections to which you are entitled as an investor. See “Management – Differences between Italian Laws and Nasdaq Requirements” for additional information.
As a foreign private issuer, we are permitted, and intend, to phase-in our compliance with certain Nasdaq Listing Rules, as permitted by Nasdaq Listing Rule 5615(b)(1), instead of otherwise having to be in compliance with such rules as of the date of our initial listing on Nasdaq, which may result in less protection than is accorded to investors under rules applicable to domestic U.S. issuers and if we do not obtain compliance within the allotted time, we could become subject to delisting by Nasdaq.
In accordance with Nasdaq Listing Rule 5615(b)(1), as a foreign private issuer, we are permitted, and intend, to phase-in our compliance with certain Nasdaq Listing Rules, instead of otherwise having to be in compliance with such rules as of the date of our initial listing on Nasdaq. For instance, although we are a foreign private issuer and have opted into following our home country rules, which are the laws of the Italy, in lieu of following certain Nasdaq Listing Rules, we are still required to have an audit committee that satisfies Nasdaq Listing Rule 5605(c)(3) and ensure that such audit committee members meet the independence requirement in Nasdaq Listing Rule 5605(c)(2)(A)(ii), provided, however, that in light of Nasdaq Listing Rule 5615(b)(1), we have up to one year from the date of our listing to have an audit committee and members who meet such requirements. This phased-in period of compliance may result in less protection than is accorded to investors under rules applicable to domestic U.S. issuers and if we do not obtain compliance within the allotted time, we could become subject to delisting by Nasdaq.
We may become taxable in a jurisdiction other than Italy and this may increase the aggregate tax burden on us.
Since incorporation, we have, on a continuous basis, had our place of effective management in Italy. We are therefore a tax resident of Italy under Italian tax law. However, we may become subject to limited income tax liability in other countries with respect to our operations in other countries, for example, the United States, due to the existence of a permanent establishment or a permanent representative. The applicable tax laws or interpretations thereof may change. We have our place of effective management in Italy and, as such, we believe we are tax residents in Italy, although that determination is largely a matter of fact and degree based on all the circumstances, rather than a question of law, which facts and degree may also change. Changes to applicable laws or interpretations thereof and changes to applicable facts and circumstances (for example, a change of board members or the place where board meetings take place), may result in us becoming a tax resident of a jurisdiction other than Italy. As a consequence, our overall effective income tax rate and income tax expense could materially increase, which could have a material adverse effect on our business, results of operations, financial condition and prospects. However, if there is a double tax treaty between Italy and the respective other country, double taxation of income may be avoided and the detrimental tax effects mitigated by the application of the treaty.
| 64 |
Risks Related to Italian Law and Our Operations in Italy
We are an Italian corporation. The rights of our shareholders may be different from the rights of shareholders in companies governed by the laws of U.S. jurisdictions.
We are an Italian corporation. Our corporate affairs are governed by our articles of association and by the laws governing companies incorporated in Italy. The rights of our shareholders and the responsibilities of members of our board of directors may be different from the rights and obligations of shareholders and directors of companies governed by the laws of U.S. jurisdictions. While performing its duties, our board of directors is required by Italian law to act with the diligence required by the nature of their assignment and by their specific expertise. Italian corporate law limits the ability of our shareholders to challenge resolutions made or other actions taken by our board of directors in court. Our shareholders generally are not permitted to file a suit to reverse or void a decision or an action taken by our board of directors, except for those decisions that are detrimental to their rights. If a board resolution has not been taken in accordance with the Italian law or the company’s articles of association, only the board of statutory auditors and the absent or dissenting members of the board of directors may challenge it within 90 days of such resolution. However, the shareholders may, where they represent the thresholds provided for by Italian law, bring corporate liability action against our directors where they have acted in violation of their duties of conduct. The individual shareholder may also take action for compensation for the damage directly caused to them by the director’s conduct. Under Italian law, shareholders’ claims against a member of our board of directors for breach of their duties of conduct must be filed in Milan, Italy, as the place where the company was incorporated. See “Share Capital Description and Reference Documents” and “Comparison between Delaware Law and Italian Law”.
Our shares are not listed in Italy, our home jurisdiction. As a result, our shareholders will not benefit from certain provisions of Italian law that are designed to protect shareholders in a public takeover offer or a change-of-control transaction and may not be protected in the same degree in a public takeover offer or a change-of-control transaction as are shareholders of certain U.S. companies or in an Italian company listed in Italy.
Because the ADSs will be listed exclusively on Nasdaq and not in Italy’s stock exchange, our shareholders will not benefit from the protection afforded by certain provisions of Italian law that are designed to protect shareholders in the event of a public takeover offer or a change-of-control transaction. For example, Article 120 of the Italian Financials’ Consolidated Act and its implementing provisions require investors to disclose their interest in the relevant listed company if they reach, exceed or fall below certain ownership thresholds. Similarly, the Italian takeover regime imposes a duty on any person or group of persons who acquires more than the 30% of a company’s voting rights (or the 25% if such company is not a small-medium enterprise, where there is no other shareholder holding a higher stake) to make a mandatory offer for all of the company’s outstanding listed equity securities. In addition, the Italian takeover regime imposes certain restrictions and obligations on bidders in a voluntary public takeover offer that are designed to protect shareholders. However, these protections are applicable only to issuers that list their equity securities in Italy and, because the ADSs will be listed exclusively on Nasdaq, will not be applicable to us. Furthermore, since Italian law restricts our ability to implement rights plans or U.S.-style “poison pills,” our ability to resist an unsolicited takeover attempt or to protect minority shareholders in the event of a change of control transaction may be limited. Therefore, our shareholders may not be protected in the same degree in a public takeover offer or a change-of-control transaction as are shareholders in certain U.S. companies or in an Italian company listed in Italy.
| 65 |
The process of seeking to raise additional funds is cumbersome, subject to the verification of an Italian notary public in compliance with our bylaws and applicable law and may require prior approval of our shareholders at an extraordinary shareholder meeting.
We are incorporated under the laws of the Republic of Italy. The principal laws and regulations that apply to our operations, those of Italy and the European Union, are different from those of the United States. With some exceptions, in order to issue new equity or debt securities convertible into equity, we must increase our authorized capital. In order to do so, our board of directors must meet and resolve to recommend that our shareholders approve an amendment to our bylaws increasing our capital. The holders of the majority of our outstanding shares must then approve that amendment at an extraordinary shareholder meeting duly called. These meetings take time to call and it might be very difficult to get a majority of the holders of all outstanding shares to vote in favor of the proposed resolution. In addition, an Italian notary public must verify that the capital increase is in compliance with our bylaws and with applicable Italian law. Further, under Italian law, our existing shareholders and any holders of convertible securities have preemptive rights (except in specific cases) to acquire any such shares pro-rated on their percentage interest in our company, and on the same terms as approved for such capital increase. Alternatively, our shareholders can delegate the power to increase our capital to the board of directors, but the board’s right to exercise such power, if delegated, will expire after five years. If the board does not approve a capital increase by the end of those five years, our board and shareholders would need to meet again to re-delegate this authority.
With respect to shareholder resolutions approving a capital increase, Italian law provides that in the absence of meeting minutes, or in the event of the impossibility or illegality of the resolution, any interested person may, for a period of 180 days following the filing of the shareholder resolution with the competent Register of Companies, challenge such resolution. If a shareholder meeting was not called to approve the capital increase, the relevant resolution should be considered invalid and, any interested person may challenge the capital increase for a period of 90 days following the approval of the financial statements referring to the year during which the shareholder resolution has been, also partially, executed. In addition, once our shareholders authorize a capital increase, all those authorized shares that have been subscribed need to be entirely paid-up before the shareholders may perform/execute a new capital increase. These restrictions could limit our ability to issue new equity or convertible debt securities on a timely basis.
Italian law places restrictions on the amount of debt securities that we may issue relative to our equity to the extent that such debt securities are not listed on regulated markets or do not otherwise provide the holder of such securities the right to purchase or convert the same into our shares.
Under Italian law, we may issue debt securities in an amount not to exceed twice the sum of our capital, our legal reserve and any other disposable reserves appearing on our latest Italian GAAP balance sheet approved by our shareholders, unless the debt securities are listed on regulated markets or provide the holder of such securities the right to purchase or convert the same into our shares, in which case such restrictions do not apply. The legal reserve is a reserve to which we allocate 5% of our Italian GAAP net income each year until it equals at least 20% of our capital. One of the other reserves that we maintain on our balance sheet is a “share premium reserve,” meaning amounts paid for our ordinary shares in excess of the amount of such ordinary shares that is allocated to the capital. At December 31, 2020, the sum of our capital, legal reserves and other reserves on our unaudited Italian GAAP financial statements was € 18.3 million. If, in the future, we issue debt securities that are not listed on regulated markets or do not provide the holder of the securities the right to purchase or convert the same into our shares, until such debt securities are repaid in full, we may not voluntarily reduce our capital or allocate our reserves (such as by declaring dividends) if it results in the aggregate of the capital and reserves being less than half of the outstanding amount of the debt. In such a case, if our equity is reduced by losses or otherwise such that the amount of the outstanding debt securities is more than twice the amount of our equity, some legal scholars are of the opinion that the ratio must be restored through a recapitalization of our company. If our equity is reduced, we could recapitalize by issuing new shares or having our shareholders contribute additional capital to us, although there can be no assurance that we would be able to find purchasers of new shares or that any of our current shareholders would be willing to contribute additional capital.
| 66 |
If we suffer losses that reduce our capital to less than €50,000, we would need to recapitalize, change our form of entity or be liquidated.
Italian law requires us to reduce our shareholders’ equity and, in particular, our capital, to reflect on-going losses, in certain cases of losses exceeding 1/3 of the capital of the company. Also, as an S.p.A., we are also required to maintain a minimum capital of €50,000. At December 31, 2020, our Italian GAAP capital was €37,056; however, in May 2021, it was increased to €50,000 due to the exercise of options on €715 Quota B plus a quotaholder action at a special meeting which converted the Company from an S.r.l. to an S.p.A. and increased the share capital. If we suffer losses from operations that reduce our capital to less than €50,000, then we must either increase our capital (which we could do by issuing new shares or having our shareholders contribute additional capital to our company) to at least €50,000 (or convert the form of our company into an S.r.l. but such conversion would not be applicable since the S.r.l. form is not consistent with being listed pursuant to Italian law). If we do not take these steps, our company could be liquidated.
We apply our operational losses against our legal reserves and capital. If our capital is reduced more than one-third as a result of losses, our board of directors must call a shareholder meeting as soon as possible. The shareholders should take appropriate measures, which may include, inter alia, reducing the legal reserves and capital by the amount of the remaining losses, or carrying the losses forward for up to one year. If the shareholders vote to carry the losses forward up to one year, and the losses are still more than one-third of the amount of the capital at the end of the year, then we must reduce our capital by the amount of the losses.
Italian labor laws could impair our flexibility to restructure our business.
In Italy, our employees are protected by various laws which afford them consultation rights with respect to specific matters regarding their employers’ business and operations, including the downsizing or closure of facilities and employee terminations. In particular, among other applicable Italian laws: (i) Laws no. 604/1966, 300/1970 and 92/2012 regulate the individual dismissals; (ii) Law no. 223/1991, concerns the collective dismissal procedure; (iii) Law no. 428/1990 as amended by legislative decree no. 18/2001, provides for the information and consultation procedure in case of a transfer of the undertaking or a part thereof; (iv) Legislative decree no. 25/2007, introduces a general right to information and consultation for employees and (v) Legislative Decree no. 23/2015 regulates the consequences of individual dismissals with specific reference to the employees hired starting from March 7, 2015. In addition, due to COVID-19 emergency, various government decrees have introduced a specific ban on dismissals for objective reasons and collective dismissal procedures. These laws and the collective bargaining agreements to which we are subject could impair our flexibility if we need to restructure our business.
Purchasers of our Ordinary Shares and ADSs may be exposed to increased transaction costs as a result of the Italian financial transaction tax or the proposed European financial transaction tax.
On February 14, 2013, the European Commission adopted a proposal for a directive on the financial transaction tax (“EU FTT”) to be implemented under the enhanced cooperation procedure by eleven Member States initially (Austria, Belgium, Estonia, France, Germany, Greece, Italy, Portugal, Slovenia, Slovakia and Spain). Following Estonia’s formal withdrawal on March 16, 2016, ten Member States are currently participating in the negotiations on the proposed directive. Member States may join or leave the group of participating Member States at later stages and, subject to an agreement being reached by the participating Member States, a final directive will be enacted. The participating Member States will then implement the directive in local legislation. If the proposed directive is adopted and implemented in local legislation, investors in Ordinary Shares and ADSs may be exposed to increased transaction costs.
The Italian financial transaction tax (the “IFTT”) applies with respect to trades entailing the transfer of (i) shares or equity-like financial instruments issued by companies resident in Italy, such as the Ordinary Shares; and (ii) securities representing the shares and financial instruments under (i) above (including depositary receipts such as the ADSs), regardless of the residence of the issuer. The IFTT may also apply to the transfer of Ordinary Shares and ADSs by a U.S. resident. The IFTT does not apply to companies having an average market capitalization lower than €500 million in the month of November of the year preceding the year in which the trade takes place. In order to benefit from this exemption, companies whose securities are listed on a foreign regulated market, such as the Company, need to be included on a list published annually by the Italian Ministry of Economy and Finance. The Company is in the process of starting the relevant procedures to be included in such list by the end of 2020. For so long as the Company is not included in such list, investors in the Ordinary Shares and ADSs may be exposed to increased transaction costs. See “Taxation.”
| 67 |
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
Some of the statements made under “Prospectus Summary,” “Risk Factors,” “Use of Proceeds,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” “Business” and elsewhere in this prospectus constitute forward- looking statements. In some cases, you can identify forward-looking statements by terminology such as “may,” “will,” “should,” “expects,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “potential” “intends” or “continue,” or the negative of these terms or other comparable terminology.
Forward-looking statements include, but are not limited to, statements about:
| ● | the timing, progress and results of preclinical studies and clinical trials for our programs and product candidates, including statements regarding the timing of initiation and completion of studies or trials and related preparatory work, the period during which the results of the trials will become available and our research and development programs; | |
| ● | the interactions with the regulatory bodies, key opinion leaders and partnering clinical centers that may result in changes to clinical trials and manufacturing programs; | |
| ● | the timing, scope or likelihood of regulatory filings and approvals; | |
| ● | our ability to develop and advance product candidates into, and successfully complete, clinical studies; | |
| ● | our expectations regarding the size of the patient populations for our product candidates, if approved for commercial use; | |
| ● | the implementation of our business model and our strategic plans for our business, product candidates and technology; | |
| ● | our commercialization, marketing and manufacturing capabilities and strategy; | |
| ● | the pricing and reimbursement of our product candidates, if approved; | |
| ● | the scalability and commercial viability of our manufacturing methods and processes; | |
| ● | the rate and degree of market acceptance and clinical utility of our product candidates, in particular, and gene therapy, in general; | |
| ● | our ability to establish or maintain collaborations or strategic relationships or obtain additional funding; | |
| ● | our competitive position; | |
| ● | the scope of protection we and/or our licensors are able to establish and maintain for intellectual property rights covering our product candidates; | |
| ● | developments and projections relating to our competitors and our industry; | |
| ● | our expectations related to the use of proceeds from this offering; | |
| ● | our estimates regarding expenses, future revenue, capital requirements and needs for additional financing; | |
| ● | our ability to remediate the material weaknesses that we and our independent registered public accounting firm identified and avoid any findings of material weaknesses or significant deficiencies in the future; |
| 68 |
| ● | the impact of laws and regulations; | |
| ● | the ability of our management team to lead the development of our product candidates; | |
| ● | our expectations regarding the time during which we will be an emerging growth company under the JOBS Act; and | |
| ● | our expectations regarding the impact of the COVID-19 pandemic, including on our planned clinical trials, operations and financial position. |
Forward-looking statements are not guarantees of future performance and are subject to risks and uncertainties. We have based these forward-looking statements on assumptions and assessments made by our management in light of their experience and their perception of historical trends, current conditions, expected future developments and other factors they believe to be appropriate.
These statements are only current predictions and are subject to known and unknown risks, uncertainties and other factors that may cause our or our industry’s actual results, levels of activity, performance or achievements to be materially different from those anticipated by the forward-looking statements. We discuss many of these risks in this prospectus in greater detail under the heading “Risk Factors” and elsewhere in this prospectus. You should not rely upon forward-looking statements as predictions of future events.
Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance, or achievements. Except as required by law, we are under no duty to update or revise any of the forward-looking statements, whether as a result of new information, future events or otherwise, after the date of this prospectus.
| 69 |
We estimate that the net proceeds to us in this offering will be approximately $26.4 million, after deducting the estimated underwriting discounts, estimated advisory fees and estimated offering expenses payable by us, based on an assumed public offering price of $11.50 per security, which is the mid-point of the price range set forth on the cover page of this prospectus, and assuming approximately 1.0 million ordinary shares are sold in the Reserved Offering and approximately 1.6 million ADS are sold in the public offering. If the underwriters exercise their option to purchase additional ADSs in full, we estimate that the net proceeds to us from this offering will be approximately $30.6 million, after deducting the estimated underwriting discounts and estimated offering expenses payable by us.
A $1.00 increase (decrease) in the assumed initial public offering price of $11.50 per security (ADS or ordinary shares) would increase (decrease) the gross proceeds to us from this offering by approximately $2.6 million.
We currently expect that we will use the net proceeds from this offering, together with our existing cash and cash equivalents, as follows:
| ● | approximately $5.3 million to complete our current Temferon TEM-GBM 001 trial, which is expected to enroll its last patient in the second quarter of 2022, and has been designed to establish clinical proof of concept and determine a therapeutic dosage of Temferon that is safe and well tolerated for patients in order to advance our GBM clinical program to Phase 2; | |
| ● | approximately $0.6 million to support long-term monitoring of the patients in the Temferon TEM-GBM 001 trial required by applicable regulations, which is expected to run through 2029; | |
| ● | approximately $1.0 million to fund preclinical research performed in the laboratories of our founders, Professor Naldini and Dr. Gentner at OSR. The aims of the research programs include the development of Temferon across a broad range of cancer indications, second generation switchable Temferon, additional payloads and combination therapies. We have exclusive licensing options extending over the scope of supported research pursuant to existing arrangements; | |
| ● | approximately $11.2 million to conduct an Italian clinical Phase 2 synthetic controlled study of Temferon in approximately thirty (30) uMGMT-GBM patients expected to take place from early 2023 to late 2025, with the objectives of safety, tolerability and efficacy of a single dose compared to matched patients in the synthetic arm receiving standard of care. The results of this study could help us develop an adaptive study design for the registration of Temferon in newly diagnosed GBM patients; | |
| ● | approximately $7.2 million to fund Temferon manufacturing activities for the Phase 2 trial, including LVV manufacturing, stability and process scalability studies and technology transfer activities; and, | |
| ● | the remainder to fund ongoing business development activities, general and administrative expenses, the costs of operating as a public company, working capital, and other general corporate purposes. |
We envision the future development of Temferon for the treatment of other solid tumor indications and we have already identified locally advanced hepatocellular carcinoma (HCC) and intra-hepatic cholangiocarcinoma (ICC) as appropriate additional target indications; however, the funding of this future development is not expected to be covered by the net proceeds we will receive from this offering. We expect additional capital would need to be raised, either through additional equity financings through future offerings of the Company’s ADSs, a strategic funding event or an alternative funding strategy to begin work on these other solid tumor indications.
| 70 |
It is difficult to predict the cost and timing required to complete such trials due to, among other factors; the final study design, the timeline for regulatory agency approval, the rate of enrollment of patients, and/or the timing and costs of manufacturing and supplying our product candidates for our planned trials. Furthermore, we estimated our budget for European clinical trial activities currently planned by referencing the rates we are paying to sites and third-party vendors involved in our ongoing programs in Italy, and subsequently applying an adjustment to those rates. Our estimated rates may change as a result of discussions with regulatory bodies, key opinion leaders and partnering clinical centers. We may also use a portion of the net proceeds to in-license, acquire or invest in additional businesses, technologies, products and/or assets. Therefore, we cannot predict with certainty all of the particular uses of the net proceeds from the offering or the actual amounts that we will spend on the uses set forth above.
As a result, we believe that the expected net proceeds from this offering, together with our existing cash and cash equivalents, will be sufficient to enable us to fund the development of Temferon through Phase 2 and our other operating expenses until 2025. While we have based this estimate on assumptions that we believe to be reasonable, there can be no assurance that those assumptions will not be proven wrong by future events, including the ongoing effects of the COVID 19 pandemic and related issues. Therefore we could deplete our available capital resources more quickly than we currently expect.
This expected use of net proceeds from this offering represents our intentions based upon our current plans and business conditions, which could change in the future as our plans and business conditions evolve. We may also use a portion of the net proceeds to in-license, acquire, or invest in additional businesses, technologies, products, or assets. We may invest or raise additional capital to begin a new Temferon clinical program in a second solid tumor indication. We cannot predict with certainty all of the particular uses for the net proceeds to be received upon the closing of this offering or the amounts that we will actually spend on the uses set forth above. Predicting the cost necessary to develop product candidates can be difficult and the amounts and timing of our actual expenditures may vary significantly depending on numerous factors, many of which are beyond our control, including the progress of our development, the status of and results from clinical trials, the availability of manufacturing capabilities, any collaborations that we may enter into with third parties for our product candidates and any unforeseen cash needs. As a result, although we currently intend to focus on the development of Temferon in uMGMT-GBM in a clinical Phase 2 trial to be performed in Italy, we will retain broad discretion over the allocation of the net proceeds from this offering.
Pending our use of the net proceeds from this offering, we may invest the net proceeds in a variety of capital preservation investments, including short-term, investment grade, and interest-bearing instruments and government securities.
| 71 |
We have never declared or paid any cash dividends on our ordinary shares and do not anticipate paying any cash dividends in the foreseeable future. Payment of cash dividends, if any, in the future will be at the discretion of our shareholders, upon proposal by our board of directors, and will depend on then-existing conditions, including our financial condition, operating results, contractual restrictions, capital requirements, business prospects and other factors our board of directors may deem relevant.
Under Italian law, Italian companies are required to furnish certain information to the Italian tax authorities regarding the identity of non-resident shareholders in connection with the payment of dividends. Shareholders are required to provide their Italian tax identification number, if any, or alternatively, in the case of legal entities, their name, country of establishment and address, or in the case of individuals, their name, address and place and date of birth, or in the case of partnerships, the information required for individuals with respect to one of their representatives. Payment of dividends may be subject to Italian withholding taxes. See “Taxation—Italy Tax Considerations” for additional information. However, beneficial U.S. holders are entitled to a reduction of the withholding taxes applicable to dividends paid to them under the income tax convention for the avoidance of double taxation between the United States and Italy, which was signed on August 25, 1999 and went into effect on December 16, 2009 (the “Income Tax Convention”); provided, however, that conditions set out in the Income Tax Convention are met and subject to the applicable anti-avoidance provisions contained therein. In order for you to benefit from that reduction, we are required to furnish certain information about you to the Italian tax authorities and, therefore, any claim by you for those benefits would need to be accompanied by the required information.
| 72 |
The following table sets forth our cash and cash equivalents and our capitalization as of June 30, 2021:
| ● | on an actual basis; | |
| ● | on an as adjusted basis to give effect to the issuance and sale of 2,608,695 securities (approximately 1,608,695 ordinary shares and approximately 1,000,000 ADSs) in this offering at an assumed public offering price of $11.50 per ADS or ordinary share, the midpoint of the estimated price range set forth on the cover page of this prospectus, after deducting estimated underwriting discounts, estimated advisory fees and estimated offering expenses payable by us, and to the application of the net proceeds as described in “Use of Proceeds,” as if the sale of the securities had occurred on June 30, 2021. |
The as adjusted information set forth in the table below is illustrative only and will be adjusted based on the actual public offering price and other terms of this offering determined at pricing.
You should read this table in conjunction with the sections titled “Selected Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and related notes included elsewhere in this prospectus.
As of June 30, 2021 | ||||||||
| in Euros, except share data | Actual (1) | Proforma As Adjusted (1) | ||||||
| (Unaudited) | ||||||||
| Cash and cash equivalents | € | 10,553,906 | € | 33,967,971 | ||||
| Shareholders’ deficit: | ||||||||
| Ordinary shares, with no par value, 59,700,000 shares authorized: 15,000,000 shares issued and outstanding; 17,608,695 shares, pro forma | 37,139,431 | 60,553,496 | ||||||
| Accumulated deficit | (25,538,377 | ) | (25,538,377 | ) | ||||
| Total (deficit) equity | 11,601,054 | 35,015,119 | ||||||
| Total capitalization | € | 11,601,054 | € | 35,015,119 | ||||
| (1) | A $1.00 increase or decrease in the assumed public offering price of $11.50 per security would increase or decrease the amount of each of cash and cash equivalents and total shareholders’ deficit by approximately $2.6 million, assuming that the number of securities offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts, estimated advisory fees and estimated offering expenses payable by us. An increase or decrease of 1,000,000 in the number of securities offered by us would increase or decrease each of cash and cash equivalents and total shareholders’ deficit by approximately €11.5 million, assuming the assumed public offering price remains the same, after deducting estimated underwriting discounts, estimated advisory fees and any estimated offering expenses payable by us. |
The number of our securities to be outstanding immediately after this offering is 17,608,695 based on 15,000,000 ordinary shares outstanding as of June 30, 2021, after the Corporate Conversion, plus 1,000,000 ordinary shares and 1,608,695 ADSs sold in this offering and excludes:
| ● | ordinary shares that are available for future issuance under our 2021-2025 Equity Incentive Plan; | |
| ● | ordinary shares issuable upon exercise of the Representatives’ Warrants; and | |
| ● | ordinary shares and/or debentures convertible into ordinary shares issuable by the board of directors in connection with certain events such as the acquisition of stocks, assets or convertible notes and certain private placements and/or public offerings. |
| 73 |
If you invest in our securities, your interest will be diluted immediately to the extent of the difference between the public offering price per security you will pay in this offering and the pro forma net tangible book value per security after the offering. At June 30, 2021, we had net tangible book value of approximately €11.6 million, corresponding to a net tangible book value of approximately €0.77 per ordinary share or $0.69 per ordinary share (using the ratio of one ordinary share to one ADS). Net tangible book value per ordinary share or per ADS represents the amount of our total tangible assets less our total liabilities, divided by 15.0 million, the total number of ordinary shares outstanding at June 30, 2021, or 15.0 million, the total number of ADSs that would represent such total number of shares based on an ordinary share-to-ADS ratio of one-to-one.
After giving pro forma effect to the Corporate Conversion and the sale of the securities offered by us in this offering and after deducting the estimated underwriting discounts, estimated advisory fees and estimated offering expenses payable by us, our pro forma estimated net tangible book value at June 30, 2021 would have been approximately €35.0 million, representing €1.99 per security or $1.76 per security. At the assumed public offering price for this offering of $11.50 per security, the midpoint of the estimated price range set forth on the cover page of this prospectus, this represents an immediate increase in pro forma net tangible book value of €1.22 per security or $1.08 per security to existing shareholders and an immediate dilution in pro forma net tangible book value of €8.64 per security or $9.74 per security to purchasers of ADSs or ordinary shares in this offering. Dilution for this purpose represents the difference between the price per security paid by these purchasers and pro forma net tangible book value per security immediately after the completion of this offering.
The following table illustrates this dilution on a per security basis to purchasers of securities in this offering:
| Assumed public offering price per security, the midpoint of the estimated price range set forth on the cover page of this prospectus | $ | 11.50 | ||||||
| Historical net tangible book value (deficit) per security as of June 30, 2021 | $ | 0.68 | ||||||
| Increase in net tangible book value per security attributable to new investors in this offering | $ | 1.08 | ||||||
| Pro forma as adjusted net tangible book value per security after offering | $ | 1.76 | ||||||
| Dilution in tangible book value per security to new investors | $ | 9.74 | ||||||
| Percentage of dilution in net tangible book value per security for new investors | 85 | % |
The dilution information set forth in the table above is illustrative only and will be adjusted based on the actual public offering price and other terms of this offering determined at pricing.
A $1.00 increase or decrease in the assumed initial public offering price of $11.50 per security would increase or decrease our as adjusted net tangible book value per security after this offering by approximately $0.11 and the dilution per security to new investors by approximately $0.11, assuming the number of securities offered by us, as set forth on the cover page of this prospectus, remains the same, after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
We may also increase or decrease the number of securities we are offering. An increase or decrease of 1,000,000 in the number of securities offered by us would increase or decrease our as adjusted net tangible book value after this offering by approximately $10.2 million and the pro forma net tangible book value per security after this offering by $0.51 per security and would increase or decrease the dilution per security to new investors by $0.51, assuming the assumed public offering price remains the same, after deducting estimated underwriting discounts and estimated offering expenses payable by us.
If the underwriters exercise their over-allotment option in this offering in full, the pro forma net tangible book value per security after this offering would be $1.86 per security, and the dilution in pro forma as adjusted net tangible book value per security to new investors purchasing securities in this offering would be $9.64 per security, after deducting estimated underwriting discounts, estimated advisory fees and estimated offering expenses payable by us.
| 74 |
The following table summarizes, on an as adjusted basis as of June 30, 2021, the differences between the number of securities acquired from us, the total amount paid and the average price per security paid by the existing holders of our ordinary shares and by investors in this offering and based upon an assumed public offering price of $11.50 per security, the midpoint of the estimated price range set forth on the cover page of this prospectus.
| Shares | Total Consideration | Average Price Per Ordinary | |||||||||||||||||
| Number | Percent | Amount | Percent | Share | |||||||||||||||
| Existing shareholders | 15,000,000 | 85 | % | $ | 37,862,540 | 56 | % | $ | 2.52 | ||||||||||
| New investors | 2,608,695 | 15 | % | $ | 29,999,993 | 44 | % | $ | 11.50 | ||||||||||
| Total | 17,608,695 | 100 | % | $ | 67,862,533 | 100 | % | ||||||||||||
The number of our securities to be outstanding immediately after this offering is 17,608,695 based on 15.0 million ordinary shares outstanding as June 30, 2021, approximately 1.0 million ordinary shares and approximately 1.6 million ADSs sold in this offering and excludes:
| ● | ordinary shares that are available for future issuance under our 2021-2024 Equity Incentive Plan; | |
| ● | ordinary shares issuable upon exercise of the Representatives’ Warrants; and | |
| ● | ordinary shares and/or debentures convertible into ordinary shares issuable by the board of directors in connection with certain events such as the acquisition of stocks, assets or convertible notes and certain private placements and/or public offerings. |
If the representatives of the underwriters exercises the over-allotment option to purchase additional ADSs in full in this offering, the number of securities represented by ADSs held by new investors will increase from 1,608,695, or 9% to 1,849,999, or 10% of the total number of securities outstanding after this offering and the percentage of ordinary shares held by existing shareholders will decrease from 91% to 90% of the total securities outstanding.
| 75 |
MANAGEMENT’S DISCUSSION AND ANALYSIS
OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis should be read in conjunction with our financial statements and related notes included elsewhere in this prospectus. This discussion and other parts of the prospectus contain forward-looking statements based upon current expectations that involve risks and uncertainties. Our actual results and the timing of selected events could differ materially from those anticipated in these forward-looking statements as a result of several factors, including those set forth under “Risk Factors” and elsewhere in this prospectus.
Prior to this offering, we were a limited liability company, but the change in incorporation did not affect the financial information herein presented, except for the transformation of our quotas into ordinary shares. As such the historical financial statements will not be retrospectively adjusted.
Overview
We are a clinical-stage biotechnology company engaged in the development of hematopoietic stem cell gene therapies for the treatment of solid tumors. We have developed a novel biologic platform which involves the ex-vivo gene transfer of a therapeutic candidate into autologous hematopoietic stem/progenitor cells (HSPCs) to deliver immunomodulatory molecules directly to the tumor by infiltrating monocytes/macrophages (Tie2 Expressing Monocytes - TEMs). Our technology is designed to turn TEMs, which normally have an affinity for and travel to tumors, into a “Trojan Horse” to counteract cancer progression and prevent tumor relapse. Because our technology is not target dependent, we believe it can be used for treatment across a broad variety of cancers.
Since our inception in 2014, we have devoted substantially all of our resources to organizing and staffing our company, business planning, raising capital, acquiring or discovering product candidates and securing related intellectual property rights, conducting discovery, research and development activities for our programs and planning for eventual commercialization. We do not have any products approved for sale and have not generated any revenue from product sales. To date, we have funded our operations with proceeds from the sales of equity securities, which through December 31, 2020, aggregated gross cash proceeds of approximately €35.1 million.
We do not have any products approved for sale, have not generated any revenue from commercial sales of our product candidates, and have incurred net losses each year since our inception. Our ability to generate product revenue sufficient to achieve profitability will depend heavily on the successful development and eventual commercialization of one or more of our current or future product candidates and programs. Our net losses for the six months ended June 30, 2021 and 2020 were approximately €4.0 million and approximately €2.5 million, respectively. Our net losses for the years ended December 31, 2020 and 2019 were approximately €5.6 million and approximately €4.6 million, respectively. At June 30, 2021 and December 31, 2020, we had an accumulated deficit of approximately €25.5 million and €21.5 million. Substantially all of our operating losses resulted from costs incurred in connection with our research and development activities, including preclinical and clinical development of our gene therapy product candidates, namely our leading product candidate Temferon, and from general and administrative costs associated with our operations.
We expect to continue to incur significant expenses for at least the next several years as we advance our product candidates from discovery through preclinical development and clinical trials and seek regulatory approval of our product candidates. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant commercialization expenses related to product manufacturing, marketing, sales and distribution. We may also incur expenses in connection with the in-licensing or acquisition of additional product candidates. Furthermore, we expect to continue incurring additional costs associated with operating as a public company, including significant legal, accounting, investor relations and other expenses.
| 76 |
As a result, for our long-term strategy, we will need substantial additional funding to support our continuing operations and pursue our growth strategy. Until such time as we can generate significant revenue from product sales, if ever, we expect to finance our operations with proceeds from outside sources, with most of such proceeds to be derived from sales of equity securities, including the net proceeds from our IPO and follow-on offerings. We also plan to pursue additional funding from outside sources, including but not limited to our entry into or expansion of new borrowing arrangements; research and development incentive payments, government grants, pharmaceutical companies and other corporate sources; and our entry into potential future collaboration agreements with pharmaceutical companies or other third parties for one or more of our programs. We may be unable to raise additional funds or enter into such other agreements or arrangements when needed on favorable terms, or at all. If we fail to raise capital or enter into such agreements as, and when, needed, we may have to significantly delay, scale back or discontinue the development and eventual commercialization of one or more of our product candidates or delay our pursuit of potential in-licenses or acquisitions.
We are unable to predict the timing or amount of increased expenses or when or if we will be able to achieve or maintain profitability, mainly due to the numerous risks and uncertainties associated with product development and related regulatory filings, which we expect to make in multiple jurisdictions. When we are eventually able to generate product sales, those sales may not be sufficient to become profitable. If we fail to become profitable or are unable to sustain profitability on a continuing basis, we may be unable to continue our operations at planned levels and be forced to reduce or terminate our operations.
As of June 30, 2021, we had cash and cash equivalents of approximately €10.6 million. We believe that our existing cash and cash equivalents as of June 30, 2021 will enable us to fund our operating expenses and capital expenditure requirements for at least the next twelve months. We have based this estimate on assumptions that may prove to be wrong, and we could exhaust our available capital resources sooner than we expect. See “—Liquidity and Capital Resources.” To finance our continuing operations, we will need to raise additional capital, which cannot be assured.
COVID-19 Update
As of the date of this filing, the global healthcare community continues to respond to the COVID-19 pandemic, including the recent emergence of the “delta variant.” In February 2020, the COVID-19 pandemic commenced in Italy. Regulatory guidance was issued in March and updated in April 2020 relating to the management of clinical trials during the pandemic. As the global healthcare community continues to respond to the COVID-19 pandemic, many hospitals, including our clinical sites, temporarily paused elective medical procedures, including dosing of new patients in clinical trials of our investigational gene therapies. While dosing of new patients and data collection from enrolled patients has resumed at clinical sites, the extent to which clinical activities continue to be delayed or interrupted will depend on future developments that are highly uncertain. We have not experienced significant interruptions related to COVID-19, although one patient tested positive for COVID-19 and had to delay treatment with Temferon. We may find it difficult to enroll patients in our clinical trials, which could delay or prevent us from proceeding with clinical trials of our product candidates. We continue to closely monitor this rapidly evolving situation and the potential impact on us.
Components of Operating Results
Revenue
We have not generated any revenue since inception and do not expect to generate any revenue from the sale of products in the near future until we obtain regulatory approval of, and commercialize, our product candidates.
Operating Expenses
Our current operating expenses consist of two components – research and development expenses, and general and administrative expenses.
| 77 |
Research and Development Expenses
We expense research and development costs as incurred. These expenses consist of costs incurred in connection with the development of our product candidates, including:
| ● | license fees and milestone payments incurred in connection with our license agreements; | |
| ● | expenses incurred under agreements with contract research organizations, or CROs, contract manufacturing organizations, or CMOs, as well as investigative sites and consultants that conduct our clinical trials, preclinical studies and other scientific development services; | |
| ● | manufacturing scale-up expenses and the cost of acquiring and manufacturing preclinical and, in due course, clinical trial materials and commercial materials, including manufacturing validation batches; | |
| ● | employee-related expenses, including salaries, social security charges, related benefits, severance indemnity in case of termination of employees’ relationships, travel and stock-based compensation expense for employees engaged in research and development functions and consulting fees; | |
| ● | costs related to compliance with regulatory requirements; and | |
| ● | facilities costs, depreciation and other expenses, which include rent and utilities. |
Our research and development expenses are tracked on a program-by-program basis for our product candidates and consist primarily of external costs, such as fees paid to outside consultants, CROs, CMOs, and central laboratories in connection with our preclinical development, process development, manufacturing and clinical development activities. Our research and development expenses by program also include fees incurred under license agreements, as well as option agreements with respect to licensing rights. We do not allocate employee costs or facility expenses, including depreciation or other indirect costs, to specific programs because these costs are deployed across multiple programs and, as such, are not separately classified. We primarily use internal resources to oversee research and discovery activities as well as for managing our preclinical development, process development, manufacturing, and clinical development activities. These employees work across programs, and therefore, we do not track their costs by program. We elected to present the research and development credit net of the related research and development expenditure on the statements of operations and comprehensive loss. However, not all of our research and development expenses are allocated by program:
| 78 |
| For the Six Months Ended | ||||||||
| June 30, | June 30, | |||||||
| 2021 | 2020 | |||||||
(Unaudited) | ||||||||
| Direct research and development expenses by program: | ||||||||
| TEM-GBM (including TEM-LT) | € | 1,692,458 | € | 948,019 | ||||
| TEM-MM | 18,609 | 173,680 | ||||||
| TEM-HC | 667 | - | ||||||
| Unallocated costs: | ||||||||
| Personnel (including share-based compensation) | 401,380 | 318,833 | ||||||
| Consultants and other third parties | 760,831 | 354,599 | ||||||
| Materials & supplies | 312,844 | 243,892 | ||||||
| Travel & entertainment | 10,445 | 31,106 | ||||||
| Other | 2,000 | 2,000 | ||||||
| Total research and development expenses | € | 3,199,234 | € | 2,072,129 | ||||
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Direct research and development expenses by program: | ||||||||
| TEM-GBM | € | 3,353,369 | € | 1,853,929 | ||||
| TEM-MM | 190,764 | 769,146 | ||||||
| Option fee – second indication 1 | 500,000 | — | ||||||
| Unallocated costs: | ||||||||
| Personnel (including share-based compensation) | 472,100 | 753,238 | ||||||
| Consultants and other third party | 70,035 | 73,736 | ||||||
| Materials & supplies | 62,600 | 64,151 | ||||||
| Travel & entertainment | 34,466 | 133,573 | ||||||
| Other | 5,127 | 55,209 | ||||||
| Total research and development expenses | € | 4,688,461 | € | 3,702,982 | ||||
1 Although the second indication is currently liver cancer, the Company has the right to change the indication.
Research and development activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We expect that our research and development expenses will increase substantially over the next several years, particularly as we increase personnel costs, including stock-based compensation, contractor costs and facilities costs, as we continue to advance the development of our product candidates. We also expect to incur additional expenses related to milestone and royalty payments payable to third parties with whom we have entered into license agreements to acquire the rights to our product candidates.
| 79 |
The successful development and commercialization of our product candidates is highly uncertain. At this time, we cannot reasonably estimate or know the nature, timing and costs of the efforts that will be necessary to complete the preclinical and clinical development of any of our product candidates or when, if ever, material net cash inflows may commence from any of our product candidates. This uncertainty is due to the numerous risks and uncertainties associated with product development and commercialization, including the uncertainty of:
| ● | the scope, progress, outcome and costs of our preclinical development activities, clinical trials and other research and development activities; | |
| ● | the impact of the COVID-19 pandemic on our preclinical development activities, clinical trials and other research and development activities; | |
| ● | establishing an appropriate safety profile with IND-enabling studies; | |
| ● | successful patient enrollment in, and the design, initiation and completion of, clinical trials; | |
| ● | the timing, receipt and terms of any marketing approvals from applicable regulatory authorities; | |
| ● | establishing and maintaining clinical and commercial manufacturing capabilities or making arrangements with third-party manufacturers; | |
| ● | development and timely delivery of commercial-grade drug formulations that can be used in our clinical trials and for commercial launch; | |
| ● | obtaining, maintaining, defending and enforcing patent claims and other intellectual property rights; | |
| ● | significant and changing government regulation; | |
| ● | qualifying for, and maintaining, adequate coverage and reimbursement by the government and other payors for any product candidate for which we obtain marketing approval; | |
| ● | launching commercial sales of our product candidates, if and when approved, whether alone or in collaboration with others; | |
| ● | addressing any competing technological and market developments; and | |
| ● | maintaining a continued acceptable safety profile of the product candidates following approval. |
We may never succeed in achieving regulatory approval for any of our product candidates. We may obtain unexpected results from our clinical trials. We may elect, or be forced by regulatory authorities, to discontinue, delay or modify clinical trials of some product candidates or focus on others. Any changes in the outcome of any of these variables with respect to the development of our product candidates in preclinical and clinical development could mean a significant change in the costs and timing associated with the development of these product candidates. For example, if the EMA, FDA or another regulatory authority were to delay our planned start of clinical trials or require us to conduct clinical trials or other testing beyond those that we currently expect, or if we experience significant delays in enrollment in or treatment as part of any of our ongoing and planned clinical trials for any reason, including as a result of the ongoing COVID-19 pandemic, we could be required to expend significant additional financial resources and time on the completion of clinical development of that product candidate. Identifying potential product candidates and conducting preclinical testing and clinical trials is a time-consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain marketing approval and achieve product sales. In addition, our product candidates, if approved, may not achieve commercial success.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and consulting fees, related benefits, travel and stock-based compensation expense for personnel in executive, finance and administrative functions. General and administrative expenses also include professional fees for legal, consulting, accounting, and audit services.
| 80 |
We anticipate that our general and administrative expenses will increase in the future as we increase our headcount to support our continued research activities and development of our product candidates. We also anticipate that we will continue to incur additional accounting, audit, legal, regulatory, compliance, director and officer insurance costs as well as investor and public relations expenses associated with being a public company. Additionally, if and when we believe a regulatory approval of a product candidate appears likely, we anticipate an increase in payroll and other expense as a result of our preparation for commercial operations, especially as it relates to the sales and marketing of our product candidate.
Other Income (Expense)
Other income (expense) consists primarily of interest income/(expense), foreign exchange income/(loss) and, for the year ended December 31, 2019, an award given to the Company by international institutions for the successful results achieved in clinical trials.
Income taxes
We are subject to taxation in Italy. Taxes are recorded on an accrual basis. They therefore represent the allowances for taxes paid or to be paid for the year, calculated according to the current enacted rates and applicable laws. Due to the tax loss position reported, no income taxes were due for the six months ended June 30, 2021 and 2020 and the years ended December 31, 2020 and 2019 or the interim periods.
As of each reporting date, we consider existing evidence, both positive and negative, that could impact our view regarding to future realization of deferred tax assets. We believe that it is more likely than not that the benefit for deferred tax assets will not be realized. In recognition of this uncertainty, a full valuation allowance was applied to the deferred tax assets. Future realization depends on our future earnings, if any, the timing and amount of which are uncertain as of June 30, 2021. In the future, should management conclude that it is more likely than not that the deferred tax assets are partially or fully realizable, the valuation allowance would be reduced to the extent of such expected realization and the amount would be recognized as a deferred income tax benefit in our statements of operations.
There are open statutes of limitations for Italian tax authorities to audit our tax returns. There have been no material income tax-related interests or penalties assessed or recorded.
There is no liability related to uncertain tax positions reported in our financial statements.
In line with the legislation in force until December 31, 2019, companies in Italy that invested in eligible research and development activities, regardless of the legal form and economic sector in which they operate, could benefit from a tax credit up to 50% of the increase of annual research and development expenses compared to the median expense for the years 2012-2014, which could be used as compensation in order to reduce most taxes payable, including income tax or regional tax on productive activities, as well as of social security contributions.
The 2020 Italian Budget Law established that: (i) the tax credit due is up to 12% of the research and development costs incurred (up to a maximum of € 3.0 million); (ii) the actual support of eligible expenditure and its correspondence with the accounting documents must result from a specific certification issued by the person responsible for the legal audit; (iii) the tax credit due can only be used as compensation in three equal annual instalments. The 2021 Italian Budget Law established that: (i) the tax credit due is up to 20% of the costs incurred (up to a maximum of € 4.0 million); (ii) the tax credit can be used for 2021 and 2022 fiscal years; (iii) it is necessary to have, besides the audit report, a technical report.
| 81 |
Results of Operations
Comparison of the Six Months Ended June 30, 2021 to the Six Months Ended June 2020
The following table summarizes our results of operations for the six months ended June 30, 2021 and 2020:
| Six-Months Ended June 30, | ||||||||
| 2021 | 2020 | |||||||
| (Unaudited) | ||||||||
| Operating expenses | ||||||||
| Research and development | € | 3,199,234 | € | 2,072,129 | ||||
| General and administrative | 842,236 | 404,885 | ||||||
| Total operating expenses | 4,041,470 | 2,477,014 | ||||||
| Loss from operations | (4,041,470 | ) | (2,477,014 | ) | ||||
| Other income (expense) | ||||||||
| Other income | 2,679 | 559 | ||||||
| Foreign exchange loss | (9,111 | ) | (2,849 | ) | ||||
| Total other income (expense), net | (6,432 | ) | (2,290 | ) | ||||
| Net loss before income taxes | (4,047,902 | ) | (2,479,304 | ) | ||||
| Income taxes (Note 5) | - | - | ||||||
| Loss | € | (4,047,902 | ) | € | (2,479,304 | ) | ||
| Comprehensive loss | ||||||||
| Total comprehensive loss | (4,047,902 | ) | (2,479,304 | ) | ||||
| Loss per share and pro forma information (unaudited): | ||||||||
| Loss and pro forma loss | € | (4,047,902 | ) | € | (2,479,304 | ) | ||
| Loss per share and pro forma loss per share - basic and diluted | € | (0.27 | ) | € | (0.17 | ) | ||
| Weighted average and pro forma weighted average number of shares outstanding - basic and diluted | 14,772,610 | 14,420,904 | ||||||
We have presented actual and pro forma basic and diluted loss per share at June 30, 2021 and 2020, respectively, which consists of our historical loss attributable to Genenta Science S.r.l. (and subsequently as of June 18, 2021 Genenta Science S.p.A.), divided by the weighted average and pro forma basic and diluted weighted average number of ordinary shares outstanding, on an “as converted” basis (similar to a stock split transaction) using the same conversion rate of 300 common shares per weighted average quota percentage of ownership during the six months ended June 30, 2021 and 2020, respectively, after giving effect to the Corporate Conversion. See our disclosures in this prospectus for additional information regarding the method used to calculate the pro forma basic and diluted loss per ordinary share and the pro forma weighted average number of ordinary shares used in the computation of the per share amounts.
| 82 |
Research and Development Expenses
Research and development expenses were approximately €3.2 million for the six months ended June 30, 2021, as compared to approximately €2.1 million for the six months ended June 30, 2020. The increase of approximately €1.1 million was primarily due to the increase of approximately €1.3 million related the Company’s GMB research and clinical activities (including personnel costs and related stock option exercise expenses), partially offset by €0.2 million related to the decrease of MM research and clinical related expenses.
General and Administrative Expenses
General and administrative expenses were approximately €0.8 million for the six months ended June 30, 2021, as compared to approximately €0.4 million for the six months ended June 30, 2020. The increase of approximately €0.4 million of general and administrative expenses was primarily due to €0.2 million of accounting, legal and consulting fees and €0.2 million of compensation expense (including stock-based compensation and social costs).
Other Income
Other income was approximately €2,700 for the six months ended June 30, 2021, as compared to approximately €600 for the six months ended June 30, 2020. The increase of approximately €2,100 was primarily due to the number of awards granted to the Company in 2020.
Foreign Exchange Loss
Foreign exchange loss was approximately €9,100 for the six months ended June 30, 2021, as compared to approximately €2,800 for the six months ended June 30, 2020. The increase of approximately €6,300 was primarily due to the net of exchange rate gains and losses.
Loss
Our loss was approximately €4.0 million for the year six months June 30, 2021, as compared to approximately €2.5 million for the six months ended June 30, 2020. The increase of approximately €1.5 million was primarily due to the increased spending related to our overall research and clinical activities and to the increased spending related to advisory and consulting expenses related to our fundraising activities.
Comparison of the Year Ended December 31, 2020 to the Year Ended December 31, 2019
The following table summarizes our results of operations for the years ended December 31, 2020 and 2019:
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| Operating expenses | ||||||||
| Research and development | € | 4,688,461 | € | 3,702,982 | ||||
| General and administrative | 901,765 | 921,520 | ||||||
| Total operating expenses | 5,590,226 | 4,624,502 | ||||||
| Loss from operations | (5,590,226 | ) | (4,624,502 | ) | ||||
| Other income (expense) | ||||||||
| Other income | 5,966 | 36,331 | ||||||
| Finance expense | (7,754 | ) | (9,552 | ) | ||||
| Total other income (expense), net | (1,788 | ) | 26,779 | |||||
| Loss before income taxes | (5,592,014 | ) | (4,597,723 | ) | ||||
| Income tax benefit (expense) | - | - | ||||||
| Net loss | (5,592,014 | ) | (4,597,723 | ) | ||||
| Comprehensive loss | - | - | ||||||
| Total comprehensive loss | € | (5,592,014 | ) | € | (4,597,723 | ) | ||
| Pro forma information (unaudited): | ||||||||
| Pro forma net loss | € | (5,592,014 | ) | € | (4,597,723 | ) | ||
| Pro forma net loss per share - basic and diluted | € | (0.38 | ) | € | (0.36 | ) | ||
| Weighted average pro forma number of shares outstanding - basic and diluted | 14,539,534 | 12,651,158 | ||||||
| 83 |
We have presented pro forma basic and diluted loss per share at December 31, 2020 and 2019, respectively, which consists of our historical loss attributable to Genenta Science S.r.l., divided by the pro forma basic and diluted weighted average number of ordinary shares outstanding, on an “as converted” basis, after giving effect to the Corporate Conversion, which occurred on June 18, 2021. See our disclosures in this prospectus for additional information regarding the method used to calculate the pro forma basic and diluted loss per ordinary share and the pro forma weighted average number of ordinary shares used in the computation of the per share amounts. We did not present basic and diluted loss per share for the years ended December 31, 2020 and 2019 in our audited Statements of Operations, since we were an S.r.l. at the time and maintained classes of quota (similar to membership interests in a limited liability company in the United States) rather than shares, which it now has post-conversion to an S.p.A.,(similar to a C-corporation in the United States). The quotas represented percentage ownership in us and not actual shares. We did not believe that representing loss per quota was meaningful.
Research and Development Expenses
Research and development expenses were approximately €4.7 million for the year ended December 31, 2020, as compared to approximately €3.7 million for the year ended December 31, 2019. The increase of approximately €1.0 million was primarily due to the increase of approximately €1.6 million related the Company’s GBM research and clinical activities partially offset by €0.6 million related to the decrease of MM related expenses. In addition, we exercised our option with OSR for our second cancer indication, which triggered an option fee of €0.5 million; however, this was offset by a decrease of €0.3 million of personnel costs and €0.2 million of other research, development, and clinical related expenses.
General and Administrative Expenses
General and administrative expenses were approximately €0.9 million for the year ended December 31, 2020, as compared to approximately €0.9 million for the year ended December 31, 2019. The general and administrative expenses remained essentially flat due to our ability to control expenses and focus our resources on research, development, and clinical activities.
Other Income
Other income was approximately €6,000 for the year ended December 31, 2020, as compared to approximately €36,000 for the year ended December 31, 2019. The decrease of approximately €30,000 was primarily due to the awards granted to the Company in 2019, i.e., the Company won two international prizes for its results on research programs in 2019 without a corresponding award in 2020.
Finance Expense
Finance expense was approximately €7,800 for the year ended December 31, 2020, as compared to approximately €9,600 for the year ended December 31, 2019. The increase of approximately €1,800 was primarily due to small exchange rate gains and losses.
| 84 |
Loss
Our loss was approximately €5.6 million for the year ended December 31, 2020, as compared to approximately €4.6 million for the year ended December 31, 2019. The increase of approximately €1.0 million was primarily due to the increased spending related to our GBM research and clinical activities.
Liquidity and Capital Resources
Overview
Since our inception, we have not generated any revenue and have incurred significant operating losses and negative cash flows from our operations. We have funded our operations to date primarily with proceeds from the sales of quotas. Through June 30, 2021, we had received gross cash proceeds of approximately €35.1 million from sales of our quotas. As of June 30, 2021, we had approximately €10.6 million in cash and cash equivalents.
The table below presents our cash flows for the periods indicated:
| (in Euros) | For the Six Months Ended June 30, | For the Year Ended December 31, | ||||||||||||||
(Unaudited) | ||||||||||||||||
| 2021 | 2020 | 2020 | 2019 | |||||||||||||
| Net cash used in operating activities | € | (4,690,869 | ) | € | (1,943,466 | ) | € | (6,044,581 | ) | € | (2,490,187 | ) | ||||
| Net cash used in investing activities | (3,727 | ) | (13,194 | ) | (20,871 | ) | - | |||||||||
| Net cash provided by (used in) financing activities | (216,741 | ) | 136 | 1,389,316 | 14,768,202 | |||||||||||
| Net (decrease) increase in cash and cash equivalents | € | (4,911,337 | ) | € | (1,956,524 | ) | € | (4,676,136 | ) | € | 12,278,015 | |||||
| Cash and cash equivalents at beginning of year | 15,465,243 | 20,141,379 | 20,141,379 | 7,863,364 | ||||||||||||
| Cash and cash equivalents at end of year | € | 10,553,906 | € | 18,184,855 | € | 15,465,243 | € | 20,141,379 | ||||||||
Operating Activities
During the six months ended June 30, 2021 and 2020, operating activities used approximately €4.7 million and €1.9 million, respectively of cash and cash equivalents, resulting from our loss of approximately €4.0 million and by cash used for changes in our operating assets and liabilities of approximately €1.4 million and partially offset non-cash charges of approximately €0.5 million. The net changes in our operating assets and liabilities were primarily due to an increase in payment of related party research and clinical accrued expenses as well as an increase of prepaid and other current assets mainly related to a VAT (value added tax) receivable. The non-cash charges primarily included approximately €0.5 million of stock-based compensation expense and a deminimus amount of depreciation and retirement benefit obligation expense.
During the year ended December 31, 2020, operating activities used approximately €6.0 million of cash and cash equivalents, resulting from our net loss of approximately €5.6 million and by cash used for changes in our operating assets and liabilities of approximately €0.9 million and partially offset non-cash charges of approximately €0.5 million. The net changes in our operating assets and liabilities were primarily due to an increase in payment of related party research and clinical accrued expenses as well as an increase of prepaid and other current assets mainly related to a VAT (value added tax) receivable. The non-cash charges primarily included approximately €0.5 million of stock-based compensation expense and a deminimus amount of depreciation and retirement benefit obligation expense.
During the year ended December 31, 2019, operating activities used approximately €2.5 million of cash and cash equivalents, resulting from our net loss of approximately €4.6 million partially offset by cash provided by changes in our operating assets and liabilities of approximately €1.4 million and non-cash charges of approximately €0.7 million. The net changes in our operating assets and liabilities were primarily due to an increase in accruals and liabilities linked to the clinical trials made in cooperation with Ospedale San Raffaele and the related clinical manufacturers. The non-cash charges relate primarily to approximately €0.7 million of share-based compensation expense.
| 85 |
Financing Activities
During the six months ended June 30, 2021 and 2020, cash flow from financing activities were €(216,741) and €136, respectively, consisting of cash proceeds from the exercise of options on our class B quota. €172 class B quota was repurchased from Drs. Naldini and Gentner at nominal value, cancelled, and allocated to the option plan as available for grant.
During the year ended December 31, 2020, net cash provided by financing activities was approximately €1.4 million, primarily consisting of net cash proceeds from the sale of our class E quota, which raised net proceeds of approximately €1.4 million.
During the year ended December 31, 2019, net cash provided by financing activities was approximately €14.8 million, consisting of net cash proceeds received from the rounds of equity fundraising that occurred in 2019.
Current Outlook
We have financed our operations to date primarily through proceeds from sales of our quotas to our founding quota holders and other third-party investors. We have incurred losses and generated negative cash flows from operations since inception in 2014. To date we have not generated revenue, and we do not expect to generate significant revenues from the sale of our product candidates in the near future.
As of June 30, 2021, our cash and cash equivalents were approximately €10.6 million. Our primary cash obligations relate to payments to OSR pursuant to our license agreement and other providers of clinical trial related services. We believe that our existing cash and cash equivalents will be sufficient to fund our projected cash requirements through the fourth quarter of 2022. Therefore, we will require significant additional financing in the future to fund our operations. As we continue to assess the effects of the COVID-19 pandemic, we do believe that it is possible that the COVID- 19 pandemic may make financing opportunities scarcer or more difficult or, if such funds are available to us, that such additional financing may not be available in an amount that is sufficient to meet our needs. Our failure to raise capital or enter into such other arrangements when needed would have a negative impact on our financial condition and could force us to delay, reduce or terminate our planned clinical trials or other operations, or grant rights to develop and commercialize product candidates that we would otherwise prefer to develop and commercialize ourselves.
We currently anticipate that we will require approximately €5.2 million for research and development activities over the course of the next twelve months. We also anticipate that we will require approximately €3.1 million for operational expenditures over such twelve-month period, which consists primarily of expenditures for supporting preclinical studies required for obtaining approval to conduct such clinical studies for our product candidates, general and administration labor costs, and fundraising activities. In light of our financial condition, we reduced our operating expenses, including for those purposes listed above, before the start of the COVID-19 pandemic. There can be no assurance that the analysis that we have undertaken or remedial measures that have been enacted will enable us to avoid part or all of any impact from the spread of COVID-19 or its consequences.
This expected use of net proceeds from this offering represents our intentions based upon our current plans and business conditions, which could change in the future as our plans and business conditions evolve. We may also use a portion of the net proceeds to in-license, acquire, or invest in additional businesses, technologies, products or assets. We cannot predict with certainty all of the particular uses for the net proceeds to be received upon the closing of this offering or the amounts that we will actually spend on the uses set forth above. Predicting the cost necessary to commercialize approved products and develop product candidates can be difficult and the amounts and timing of our actual expenditures may vary significantly depending on numerous factors, including the progress of our development, our plans to develop our in-house drug product and vector manufacturing capabilities, the status of and results from clinical trials, any collaborations that we may enter into with third parties for our product candidates and any unforeseen cash needs. As a result, our management will retain broad discretion over the allocation of the net proceeds from this offering.
| 86 |
Based on our planned use of the net proceeds from this offering and our existing cash, we estimate that such funds will be sufficient to fund our operations and capital expenditure requirements into 2024. We have based this estimate on assumptions that may prove to be wrong, and we could use our available capital resources sooner than we currently expect.
In addition, our operating plans may change as a result of many factors that may currently be unknown to us, and we may need to seek additional funds sooner than planned. Our future capital requirements will depend on many factors, including:
| ● | the length of the COVID-19 pandemic and its impact on our planned clinical trials, operations and financial condition; | |
| ● | the progress and costs of our preclinical studies, clinical trials and other research and development activities; | |
| ● | the scope, prioritization and number of our clinical trials and other research and development programs; | |
| ● | any cost that we may incur under in- and out-licensing arrangements relating to our product candidate that we may enter into in the future; | |
| ● | the costs and timing of obtaining regulatory approval for our product candidates; | |
| ● | the costs of filing, prosecuting, enforcing and defending patent claims and other intellectual property rights; | |
| ● | the costs of, and timing for, amending current manufacturing agreements for production of sufficient clinical and commercial quantities of our product candidates, or entering into new agreement with existing or new CMOs; | |
| ● | the potential costs of contracting with third parties to provide marketing and distribution services for us or for building such capacities internally; and | |
| ● | the costs of acquiring or undertaking the development and commercialization efforts for additional, future therapeutic applications of our product candidates and the magnitude of our general and administrative expenses. |
Until we can generate significant revenues, if ever, we expect to satisfy our future cash needs through our existing cash, cash equivalents and short-term deposits, the net proceeds from this current offering, loans, debt or equity financings. We cannot be certain that additional funding will be available to us on acceptable terms, if at all. If funds are not available, we may be required to delay, reduce the scope of, or eliminate research or development plans for, or commercialization efforts with respect to, one or more applications of our product candidates.
Critical Accounting Policies
Our financial statements are prepared in accordance with generally accepted accounting principles in the United States. The preparation of our financial statements and related disclosures requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, costs and expenses, and the disclosure of contingent assets and liabilities in our financial statements. We base our estimates on historical experience, known trends and events and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. We evaluate our estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates under different assumptions or conditions.
| 87 |
We believe that the accounting policies described below are critical in order to understand the judgements and estimates used in the financial statements and to fully understand and evaluate our financial condition and results of operations.
Accrued Research and Development Expenses
As part of the process of preparing our financial statements, we are required to estimate our accrued research and development expenses. This process involves reviewing open contracts and purchase orders, communicating with our personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual costs. The majority of our service providers invoice us in arrears for services performed, on a pre-determined schedule or when contractual milestones are met; however, some require advanced payments. We make estimates of our accrued expenses as of each balance sheet date in the financial statements based on facts and circumstances known to us at that time. We periodically confirm the accuracy of these estimates with the service providers and make adjustments, if necessary. Examples of estimated accrued research and development expenses include fees paid to:
| ● | vendors, including central laboratories, in connection with preclinical development activities, especially, OSR, a co-founding quotaholder, significant related party vendor and a leading center for ex-vivo gene therapy for inherited diseases; | |
| ● | CROs and investigative sites in connection with preclinical and clinical studies; and | |
| ● | CMOs in connection with drug substance and drug product formulation of preclinical and clinical trial materials. |
We base our expenses related to preclinical studies and clinical trials on our estimates of the services received and efforts expended pursuant to quotes and contracts with multiple research institutions and CROs that conduct and manage preclinical studies and clinical trials on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. There may be instances in which payments made to our vendors will exceed the level of services provided and result in a prepayment of the expense. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from the estimate, we adjust the accrual or the amount of prepaid expenses accordingly. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in reporting amounts that are too high or too low in any particular period. To date, there have not been any material adjustments to our prior estimates of accrued research and development expenses.
Share-based compensation
To reward the efforts of employees, directors, and certain consultants to promote our growth, our Board of Directors has historically approved, during its existence, various share-based awards, although no share-based awards were granted during the year ended December 31, 2020; however, during the six months ended June 30, 2021, the Board granted fully vested options on €169 quota B. In addition, the Board accelerated the vesting of other stock options on €546 quota B that were previously granted. This amounted to a total of €715 quota B that were exercised prior to the Corporate Conversion. All options were awarded with an exercise price of €1 per quota and, when exercised, were first converted to Quota B of Genenta Science S.r.l. and then to ordinary shares giving effect to our Corporate Conversion to Genenta Science S.p.A. All options have now been granted and there were no outstanding options as of June 30, 2021. The Board is implementing a new stock option plan (the Company’s “Equity Incentive Plan 2021–2025”) based on a maximum of 2.7 million of new ordinary shares (i.e., common stock) or 10% of the number of shares outstanding after the next round of financing. The new stock option plan will be implemented by the Compensation and Nominating Committee of the Board of Directors after the next round of financing.
| 88 |
We measure share-based awards granted to employees and directors based on the fair value on the date of the grant and recognize compensation expense for those awards over the requisite service period, which is the vesting period of the respective award. Forfeitures are accounted for as they occur. The measurement date for option awards is the date of the grant. We classify share-based compensation expense in our statements of operations and comprehensive loss in the same manner in which the award recipient’s payroll costs are classified or in which the award recipient’s service payments are classified.
With the adoption of Accounting Standards Update (“ASU”) No. 2018-07, Compensation—Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting (“ASU 2018-07”) on January 1, 2019, the measurement date for non-employee awards is the date of the grant. The compensation expense for non-employees is recognized, without changes in the fair value of the award, over the requisite service period, which is the vesting period of the respective award.
Quota B valuations (Pre-Conversion to S.p.A.)
The fair value of the Quota B of Genenta Science S.r.l. underlying our stock-based compensation grants prior to our Corporate Conversion has historically been determined by our board of directors, with input from management and third-party valuations. We believe that the board of directors has the relevant experience and expertise to determine the fair value of the Quota B, when also securing third-party assistance. Given the absence of a public trading market of our equity, and in accordance with the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held Company Equity Securities Issued as Compensation, the board of directors exercised reasonable judgment and considered numerous objective and subjective factors to determine the best estimate of the fair value of our equity at each grant date. These factors include:
| ● | valuations of the Quota B equity performed by third-party specialists; | |
| ● | the price of our equity to third-party, arms-length, sophisticated, and qualified investors, which was used in the OPM backsolve model; | |
| ● | the prices, rights, preferences, and privileges of our Quota C, D, and E preferred equity classes relative to those of our equity; | |
| ● | lack of marketability of the Quota B; | |
| ● | lack of voting rights of the Quota B; | |
| ● | current business conditions and projections; | |
| ● | hiring of key personnel and the experience of management; | |
| ● | our stage of development; | |
| ● | the timing, progress and results of our preclinical studies and clinical trials for our programs and product candidates; including statements regarding the timing of initiation and completion of trials or studies and related preparatory work, the period during which the results of the trials will become available and our research and development programs; | |
| ● | likelihood of achieving a liquidity event, such as an initial public offering, a merger or acquisition given prevailing market conditions, or other liquidation events for us; | |
| ● | the market performance of comparable publicly traded companies; and | |
| ● | the European, U.S. and global capital market conditions. |
| 89 |
In valuing the Quota B, the board of directors determined the equity value of our business using various valuation methods. The board of directors engaged a third-party valuation firm who performed analyses in accordance with the guidance outlined in the American Institute of Certified Public Accountants’ Accounting and Valuation Guide, Valuation of Privately-Held Company Equity Securities Issued as Compensation. Our option valuations were prepared using an option pricing method (“OPM”), which used market approaches to estimate our enterprise value.
The OPM treats each equity class as a call options on the total equity value of a company, with exercise prices (i.e., breakpoints) based on the value thresholds at which the allocation among the various holders of a company’s securities changes. A discount was considered for Lack of Marketability (“DLOM”), which is an amount or percentage that is deducted from the value in order to reflect the absence of a viable market. The DLOM was then applied to arrive at an indication of value for the option. Also, considered in the valuation was volatility and the fact that the Quota B did not carry voting rights. The expected volatility used in the OPM is based upon the historical volatility of a number of publicly traded companies in similar stages of clinical development.
Application of our approach involved the use of estimates, judgment, and assumptions that are highly complex and subjective, such as those regarding the selection of comparable companies, and the expected timing of an initial public offering (“IPO”) or other liquidity event. Changes in any or all of these estimates and assumptions or the relationships between those assumptions impact the valuations at each valuation date and may have a material impact on the valuation of the Quota B, and consequently, our share-based compensation expense could be materially different. For valuations after the completion of an initial public offering, the board of directors will determine the fair value of each share of underlying equity shares based on the closing price of the shares as reported on the date of grant.
On June 18, 2021, all Quota B were converted, with all other classes of Quota, into ordinary common stock based essentially on their ownership percentages. All previous rights and privileges of the classes of quotaholders were cancelled and all owners received their pro rata ownership in the same class of ordinary common stock.
Research and development tax credit receivables
We account for our research and development tax credit receivable in accordance with IAS 20 Accounting for Government Grants and Disclosure of Government Assistance. The receivable is recognized when there is reasonable assurance that: (1) the recipient will comply with the relevant conditions and (2) the grant will be received. We elected to present the credit net of the related expenditure on the statements of operations and comprehensive loss. While these tax credits can be carried forward indefinitely, we recognized an amount which reflects management’s best estimate of the amount that is reasonably assured to be realized or utilized in the foreseeable future based on historical benefits realized, adjusted for expected changes, as applicable.
Emerging Growth Company Status
We are an “emerging growth company.” Under the JOBS Act, an emerging growth company can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies. We have irrevocably elected to avail ourselves of this exemption from new or revised accounting standards, and, therefore, will not be subject to the same new or revised accounting standards as public companies that are not emerging growth companies.
Off-Balance Sheet Arrangements
We have not engaged in any off-balance sheet arrangements, such as the use of unconsolidated subsidiaries, structured finance, special purpose entities or variable interest entities.
| 90 |
We do not believe that our off-balance sheet arrangements and commitments have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that is material to investors.
Quantitative and Qualitative Disclosure About Market Risk
We are exposed to market risks in the ordinary course of our business. Market risk represents the risk of loss that may impact our financial position due to adverse changes in financial market prices and rates. Our current investment policy is to invest available cash in bank deposits with banks that have a credit rating of at least A-. Accordingly, a substantial majority of our cash and cash equivalents is held in deposits that bear interest. Given the current low rates of interest we receive, we will not be adversely affected if such rates are reduced. Our market risk exposure is primarily a result of foreign currency exchange rates, which is discussed in detail in the following paragraph.
Foreign Currency Exchange Risk
Our results of operations and cash flow are not subject to significant fluctuations due to changes in foreign currency exchange rates. As discussed above, most of our liquid assets and our expenses are denominated in EUR. Changes of 5% and 10% in the USD/EUR exchange rate would not have significantly increased/decreased our operating expenses. As we continue to grow our business, our results of operations and cash flows might be subject to fluctuations due to changes in foreign currency exchange rates, which could adversely impact our results of operations.
We do not hedge our foreign currency exchange risk. In the future, we may enter into formal currency hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange rates of our principal operating currencies. These measures, however, may not adequately protect us from the material adverse effects of such fluctuations.
| 91 |
Overview
We are a clinical-stage biotechnology company engaged in the development of hematopoietic stem cell gene therapies for the treatment of solid tumors. We have developed a novel biologic platform that involves the ex-vivo gene transfer of a therapeutic candidate into autologous hematopoietic stem/progenitor cells (HSPCs) to deliver immunomodulatory molecules directly to the tumor by infiltrating monocytes/macrophages (Tie2 Expressing Monocytes - TEMs). Our technology is designed to turn TEMs, which normally have an affinity for and travel to tumors, into a “Trojan Horse” to counteract cancer progression and to prevent tumor relapse. Our technology is not target dependent, and therefore we believe it can be used as a treatment for a broad variety of cancers.
Our technology incorporates the use of a lentiviral vector (LVV) that combines a therapeutic transgene sequence, or payload, with our proprietary platform. Our proprietary platform consists of (i) the Tie-2 promoter, that drives transgene sequence transcription specifically in TEMs, and (ii) miRNA-126 target sequences to downregulate transgene expression post-transcription in those cells where the Tie-2 promoter is active and the miRNA-126 is present. We believe there are many advantages to our approach:
| ● | Trojan Horse Mechanism of Action (“MoA”): We use and modify TEMs, a subpopulation of tumor-associated myeloid cells, known to be involved in tumor growth and in the inhibition of immune system response, to allow the immune system to recognize the tumor and to deliver to the cancer site a chosen therapeutic. | |
| ● | Select Regulation of Transgene Expression: Our selected control of the chosen therapeutic gene expression is designed to avoid off-target and systemic toxicity. | |
| ● | Potential Long-Term Effect: Through the use of hematopoietic stem cells, our therapeutic candidate is designed as a “living therapy” intended to break the cancer-induced immune tolerance and to establish a competent immune surveillance throughout the life of the patient. | |
| ● | Agnostic Response: In contrast to antigen-restricted CAR-T cells, our platform is not restricted to a pre-selected tumor antigen, nor any one tumor type. As such, it may be applied to a broad range of solid tumors and cancer subtypes, which would overcome one of the central unresolved challenges of immune-oncology cancer therapies. |
Our lead product candidate, Temferon, was developed using our platform and carries an interferon-alpha (IFN-α) payload. IFN-α is a well-known therapeutic that was previously administered intravenously for treatment of various cancers, but it is currently rarely used because of its systemic toxicity. The Temferon-modified TEMs express the transgene payload, IFN-α, in the tumor microenvironment resulting in the breakdown of tumor induced immune-tolerance. As a result, the immune system can recognize the tumor, respond, and inhibit tumor growth. Because Temferon is designed to deliver the IFN-α payload directly to the tumor, we believe it will demonstrate clinical activity without the side effect profile of systemic delivery of IFN-α. In preclinical mouse cancer models treated with Temferon both direct (anti-angiogenic, pro-apoptotic) and indirect (immune response) effects were observed.
We are currently developing Temferon for the treatment of glioblastoma multiforme (GBM) in patients who have an unmethylated MGMT gene promoter (uMGMT-GBM). GBM is the most common malignant primary brain tumor, accounting for more than half of all central nervous system (CNS) cancers. Patients suffering from GBM have limited, non-curative treatment options. Although these treatments may improve survival, the prognosis for GBM patients remains poor, with a median overall survival (mOS) of approximately 13 to 15 months and only 5.5% of patients estimated to be alive 5 years after diagnosis. With no curative treatments available and such poor prognosis for patients, there remains a large, unmet medical need. We chose uMGMT-GBM among our first targets for clinical development after considering the medical need, the active role that TEMs have in GBM pathology, and the high number of newly diagnosed uMGMT-GBM patients potentially interested in participating in our study. As a result, we believe uMGMT-GBM offers a good profile for our initial proof of concept trial in humans. We are currently conducting a Phase 1/2a clinical trial with Temferon in newly diagnosed uMGMT-GBM patients in Italy. We anticipate completing enrollment and patient dosing by the second quarter of 2022. We intend to use the preliminary results of our Phase 1/2a clinical trial to support a Clinical Trial Application to conduct in EU a multicenter Phase 2 trial in uMGMT-GBM, which we currently intend to conduct primarily in Italy. As of November 15th, 2021, we have dosed a total of 15 patients. The preliminary results show that Temferon has been generally well tolerated, with no dose limiting toxicities identified so far.
| 92 |
We also intend to develop Temferon for the treatment of other solid tumor indications, and locally advanced hepatocellular carcinoma (HCC) and intra-hepatic cholangiocarcinoma (ICC) are under consideration. HCC and ICC are gastrointestinal (GI) cancers affecting the digestive system. HCC is a primary malignancy of the liver that occurs predominantly in patients with underlying chronic liver disease and cirrhosis. ICC is a biliary tract cancer and represents approximately 3% of all GI malignancies. The prognosis for patients with locally advanced HCC or ICC remains poor, with few therapeutic options, having limited clinical benefits. While we are considering development of Temferon for these liver indications for similar reasons as GBM (i.e. the high unmet need, TEMs’ role in HCC and ICC pathology, and the number of newly diagnosed patients potentially eligible for our study), we are also evaluating development of Temferon for other solid tumor indications.
In addition to our Temferon programs that target uMGMT-GBM or other solid tumor indications such as HCC and ICC, we have exclusive option rights to license (i) Temferon for the treatment of additional indications, and (ii) other drug candidates that are currently in the preclinical stage of development both as standalone treatments and as combination therapies.
AGC Biologics facility in Milan, Italy, will continue manufacturing LVV and Temferon to support Genenta’s trials. For further larger studies we may use AGC’s 60,000 square meter cell and gene therapy manufacturing facility located in Longmont, Colorado (U.S.), which AGC purchased from Novartis in July 2021 or another US-based CMO.
We were founded in 2014 by San Raffaele Hospital (Ospedale San Raffaele or OSR) in Milan, a globally recognized premier research hospital for ex-vivo gene therapy, with Pierluigi Paracchi (our CEO), Luigi Naldini (Chairman of our Executive Scientific Board) and Bernhard Gentner (a member of our Executive Scientific Board), to develop potential ground-breaking cell and gene cancer therapies. We leverage the vast experience in LVV technology of the San Raffaele Telethon Institute for Gene Therapy (SR-Tiget), a world leading cell and gene therapy research institution on the forefront of developing therapies for rare diseases that is a joint venture between OSR and Fondazione Telethon (Telethon). SR-Tiget has a proven track record for successful collaborative clinical research programs in ex-vivo gene therapy that give rise to approved products, including Strimvelis, an ex-vivo gammaretroviral vector-based gene therapy for adenosine deaminase severe combined immunodeficiency (ADA-SCID), and Libmeldy, an ex-vivo gene therapy for the treatment of early-onset metachromatic leukodystrophy (MLD) patients, both marketed by Orchard Therapeutics. Our platform was developed in the SR-Tiget laboratories of our founders, Prof. Naldini and Dr. Gentner and we hold exclusive rights, and exclusive option rights, to certain intellectual property originating there.
Since closing our first round of funding in May 2015, we have recruited a leading management team, established a manufacturing process for our drug product candidate, completed preclinical activities (research and Good Laboratory Practice – GLP – grades), engaged with Italian, European and U.S. Key Opinion Leaders (KOLs) to identify our clinical lead indications and submitted our first CTA.
Our leadership team has a proven track record as biotech executives. Their expertise span from finance and venture capital to medical affairs, scientific research, clinical drug product development and clinical trial management and conduction. For example, members of our management team have been involved in the successful development of Ethical Oncology Science, which was acquired in 2013 for over $400 million, and Strimvelis the first ever approved ex-vivo gene therapy product that was developed under the guide of Carlo Russo, our Chief Medical Officer and Head of Development (formerly Head of Development of the Biopharm Unit and Head of Research & Development of the Rare Disease Unit at GSK). Our management team members have played important roles in both large pharma companies such as Merck and GSK, and life science startups, such as Adverum, Annapurna, VaxInnate Corporation, OncoSec Medical, Biological Dynamics and GenMark Diagnostics. We believe this multi-disciplinary competence, provides a unique blend for the development of innovative gene and cell therapy products, and constitutes a fertile ground for alliances with industrial partners that could help us bring new therapies to patients.
| 93 |
Research and Development Pipeline
Our portfolio of clinical and preclinical ex-vivo autologous gene cancer therapies is based on our technology platform, which was originally developed in our founders’ laboratories at OSR. Through our collaboration with OSR, we have worldwide commercial rights to Temferon (though our current trademark rights to Temferon are limited to the US and Europe) for the treatment of GBM, HCC and ICC, as well as exclusive option rights to license all of our other programs. Specifically, we retain exclusive option rights to license (i) any platform improvements, including our second-generation technology, which includes developments to enable the on/off regulation of the therapeutic transgene, (ii) products for additional indications that utilize our platform technology but use different transgene payloads, and (iii) combinations of our platform with therapies in the immuno-oncology (IO) field, such as ICI, CAR-T cell therapies and TCR therapies.
Our current pipeline, with clinical and preclinical stage programs, is summarized below:
*Genenta has options/rights on IP derived from preclinical data generated at SR-Tiget laboratories.
Strategy
We are developing novel cancer therapeutics using our autologous ex-vivo gene therapy platform, to initially address the unmet medical needs of uMGMT-GBM patients and patients suffering from another solid cancer indication such as HCC and ICC, but ultimately, we hope to broaden our platform to treat a wide variety of cancers by pursuing the following strategies:
Advance development of our leading clinical-stage product candidate, Temferon in the U.S.
We are currently conducting a Phase 1/2a dose-escalation study in Italy to primarily evaluate the safety and tolerability of Temferon in up to 21 uMGMT-GBM patients who have an unmethylated MGMT promoter, following radiotherapy treatment. We plan to initiate in Europe a multicenter Phase 2 trial in uMGMT-GBM, which we currently intend to conduct primarily in Italy to evaluate the safety and efficacy of Temferon in up to 27 uMGMT-GBM patients who have an unmethylated MGMT promoter where we intend to measure progression free survival (PFS) and overall survival (OS) as endpoints. In advance of our CTA submission and not required for our European trial, we have submitted a pre-IND meeting request to the FDA and received written responses from the Agency in the third quarter of 2021 regarding the proposed Phase 2 clinical study design and drug product manufacturing strategy. The plans for the Phase 2 clinical study and drug product manufacturing strategy will be informed by the Agency comments.
| 94 |
Extend our product pipeline across multiple indications
We intend to expand our product pipeline by:
| ● | Identifying additional indications suitable for Temferon. We are in the planning stages for a second study using Temferon in a second solid tumor indication and locally advanced HCC and ICC are under consideration. The Istituto Superiore di Sanità (ISS), an independent committee with oversight from the Italian Ministry of Health, must issue a positive opinion regarding our CTA before AIFA will approve it and we may start patient’s recruitment. In addition to HCC and ICC, we believe there may be additional cancer indications which actively recruit TEMs to proliferate for which Temferon may be a suitable therapy. | |
| ● | Using our platform with different transgene payloads. Our platform technology is designed to enable us to use different transgene payloads to potentially achieve therapeutic outcomes in selected cancer indications. We are currently conducting preclinical studies for two therapeutics using our platform with different payloads targeting solid tumors. | |
| ● | Developing a second-generation platform that enables the “on-demand” release of the transgene payload. We intend to develop a second-generation technology platform that allows the drug products to be switched on to exert the therapeutic effects and switched off if they are no longer needed, or to mitigate toxicity. This technology may enable us to expand our treatment options to broader patient populations. | |
| ● | Exploring combination therapies. We will seek to enter into collaborations with other companies to explore combination studies of our therapeutics with other cancer therapies, such as ICI, CAR-T cell therapies and TCR therapies. We believe our product, as a result of its MoA, has the potential to enhance the durability and efficacy of the existing therapies, thus abolishing the immune tolerance to the tumor. | |
| ● | Exploiting in-licensing opportunities with OSR. We intend to exploit in-licensing opportunities with OSR, a co-founding shareholder. |
Develop and maintain efficient manufacturing processes to support anticipated growth
To meet our drug product supply needs for conducting larger trials after the completion of our planned Phase 2 uMGMT-GBM study in Europe, we intend to enter into a supply agreement with a US-based CMO for the manufacturing of our products. Currently, Temferon, is manufactured by AGC Biologics, a leading global contract development and manufacturing organization (CDMO), which is headquartered in Italy and specializes in the manufacturing of viral vectors and genetically engineered cells. Their facility is certified by AIFA. AGC Biologics facility in Milan, Italy, will continue manufacturing LVV and Temferon to support Genenta’s trials. For further larger studies we may use AGC’s 60,000 square meter cell and gene therapy manufacturing facility located in Longmont, Colorado (U.S.), which AGC purchased from Novartis in July 2021 or another US-based CMO.
Establish a patient-centered infrastructure and strong relationships with key U.S. opinion leaders working in our disease area
Since cell and gene-based therapies are relatively new approaches in oncology, we intend to implement programs to improve patient and physician education regarding the availability of gene therapy-based products for those cancers with a high unmet medical need. To this end, we are discussing with Antonio Chiocca, MD, Professor Neurosurgeon-in-Chief and Chairman, Department of Neurosurgery at Brigham and Women’s Hospital in Boston, MA, Frederick Lang, MD, Professor and Chairman of the Department of Neurosurgery at MD Anderson in Houston, TX, and David A. Reardon, MD, Department of Medical Oncology at Dana-Farber Cancer Institute in Boston, MA.
| 95 |
Develop opportunistic partnership(s) with pharmaceutical company(s)
We may choose to partner with larger pharmaceutical companies whose core competencies and oncology strategies are in line with ours.
Our Strengths
We believe that our growing body of early clinical data evidencing the potential of our autologous ex-vivo gene therapy approach, coupled with our founders’ expertise in the development, manufacturing and commercialization of gene and cell therapies, positions us well to provide potentially transformative therapies through a single administration to patients suffering from a broad range of cancers. We believe our key strengths include:
| ● | Unique and valuable expertise. We are conducting our clinical trials at OSR, a leading center for ex-vivo gene therapy for inherited diseases. OSR has treated more than 121 patients worldwide (one of the highest number of patients treated with gene therapy for rare diseases in a research hospital), using an ex-vivo viral vector platform similar to the one we are developing for cancer treatment. Members of our executive leadership team have held senior positions at GSK, Merck, Annapurna-Adverum and other companies specializing in gene and cell therapies and rare diseases. We have partnered with academic institutions that are pioneers in autologous ex-vivo gene therapy and hold exclusive option rights to license additional patents and know-how to build our portfolio. Partnerships with leading academic institutions well recognized in the gene therapy field, such as SR-Tiget and OSR, are a core part of our research engine through which we are working to advance the clinical development of our product candidates and to identify new opportunities that we believe have comparably high probabilities of success in a preclinical setting. We believe our expertise, combined with our plan to leverage our relationships with leading academic institutions, will help expedite the commercialization of our lead clinical-stage product candidate and further expand our pipeline. | |
| ● | Deep pipeline with broad utility. We believe that the flexibility of our technology platform combined with our exclusive option rights to in-license additional programs, gives us the ability to grow our pipeline by targeting a broad set of cancer diseases. | |
| ● | Durable therapeutic potential. Preliminary interim clinical data collected from twelve treated uMGMT-GBM patients following a single administration of Temferon displayed modified cells at 18 months, the last measured timepoint to date. | |
| ● | Designed for tumor restricted therapeutic payload delivery and release. The design of our transgene expression cassette is intended to restrict payload expression to the tumor microenvironment. The local and tumor restricted therapeutic gene deployment approach is designed to focus the pleiotropic anti-tumor activities of the selected payload, by limiting the toxic manifestation that results from standard systemic administration of the payload. | |
| ● | Agnostic approach. Our immune-gene therapy approach is designed to be a tumor-agnostic immunotherapy that does not rely on any specific target or tumor type, and we believe it could be successfully applied to a potentially broad range of cancers and immune contexts. | |
| ● | Solid tumors targeting. Our platform has the potential to efficiently target solid tumors. Solid tumors are difficult to treat, even by the most novel and leading-edge technologies such as ICIs and CAR-T cells. Our cellular carrier, TEMs, is spontaneously and actively recruited by growing tumors and is found in several human solid tumors, irrespective of location. | |
| ● | Active and sustained tumor surveillance. Our immune-gene investigational therapy is designed to trigger the patient’s own immune response and establish an active immune surveillance. Our preclinical work, which used different cancer models (B-cell acute lymphoblastic leukemia - B-ALL and GBM) as well as preliminary data collected from our uMGMT-GBM patients, suggests the occurrence of changes in the immune system. | |
| ● | Fine dose tuning. Our platform holds promise to fine tune the dose to be administered based on individual patient characteristics. |
| 96 |
| ● | Preliminary data indicate that our approach is feasible and well-tolerated. Temferon has been well-tolerated in the limited number of patients treated to date. Our ex-vivo modification of the patient’s own HSPCs and cryopreservation allow us to formulate the patient’s drug product prior to administering the therapy. | |
| ● | LVV as transgene payload delivery vehicles. LVVs are particularly attractive for clinical applications due to their capacity to transfer large genes/payloads and their ability to efficiently transduce non-proliferating or slowly proliferating cells, such as hematopoietic stem and progenitor cells that allow a persistent gene expression in transduced cells. Moreover, LVVs have a potentially reduced risk of genotoxicity compared to gamma-retroviral vectors (gRV). A large number of patients have been treated both with other LVV gene therapy products approved for sale and with clinical-stage LVV gene therapy product candidates for rare diseases worldwide, and generally these therapies have been well tolerated. We believe that long-term extensive follow-up across multiple diseases, with vectors expressing different genes, demonstrates the potential safety of our LVV-based autologous ex-vivo gene therapy approach. | |
| ● | Applicability to a potentially large number of patients and indications. We believe our autologous ex-vivo gene therapy approach has broad therapeutic potential across a large number of malignancies. The ex-vivo transduction of HSPCs allows for the potentially long-term production of a differentiated cellular carrier loaded with the therapeutic gene and the consequent distribution of the therapeutic payload throughout multiple organs and tissues containing solid tumors. |
Status of Current IO Treatments
Despite new therapeutic approaches and new drugs having been developed or approved, substantial unmet need remains for many of the most common cancers. Immuno-oncology therapies seek to work in conjunction with the patient’s own immune system to recognize and attack cancer cells selectively, without affecting normal cells, or to deliver immune system components that prevent the spread of cancer. Immuno-oncology therapy is recognized as an important type of cancer treatment in addition to more established options such as surgery, chemotherapy, targeted therapy and radiation therapy. Indeed, the number of IO therapeutics in development worldwide grew 233% from 2017 to 2020. IO therapeutics, which rely on the natural activity of the immune system to fight cancers in various ways, are grouped, by the Cancer Research Institute, in five main classes reported in the tables below.
| 97 |


Current Limitations of IO Approaches
Despite significant advances, the clinical application of immunotherapy for cancer patients still faces challenges, including:
| ● | Development of tumor resistance (positive selection of tumor cells bearing advantageous mutations); | |
| ● | Dependence on specific targets; | |
| ● | Poor response for many patients; | |
| ● | Lack of a durable response; | |
| ● | Side effects; | |
| ● | Need for multiple dosing for most IO classes; and | |
| ● | Inability to efficiently target many solid tumors. |
| 98 |
In addition, manufacturing scalability of some IO approaches remains a challenge and significantly limits market penetration. The table below reports some of the main limitations for each IO class.
| IO Class | Limitations | ||
| Cell-based immunotherapies | ● | Graft versus host disease (GVHD) | |
| ● | Only a limited number of antigens may be targeted currently | ||
| ● | Inability to target multiple antigens at the same time | ||
| ● | Some antigens targeting may be ineffective | ||
| ● | The majority of TILs within the tumor microenvironment are exhausted so even tumor-specific T cells are hypo-responsive | ||
| ● | CAR-T cells can only recognize antigens that are naturally expressed on the cell surface, so the range of potential antigen targets is smaller than with TCRs | ||
| ● | Presence of target-negative tumor cells cause relapse in the long-term | ||
| ● | Limited penetration and distribution into solid tumor tissues | ||
| Immunomodulators | ● | Loss of self-tolerance (establishment of autoimmunity associated with failure of tumor rejection) | |
| ● | Absence of reliable biomarkers and thresholds to identify the most likely responsive population | ||
| ● | Induction of overactive immune responses as well off-target responses against healthy cells | ||
| ● | Limited penetration and distribution into solid tumor tissues | ||
| Vaccines | ● | Absence of universal antigens (each individual’s tumor is unique and has its own distinguishing antigens) | |
| ● | Presence of a compromised/weak/immunosuppressed immune system | ||
| Antibody-based targeted therapies | ● | Limited penetration and distribution into solid tumor tissues (mAbs directed against tumor-specific antigens largely remain in the blood and no more than 20% of the administered dose typically interacts with the tumor) | |
| ● | Low binding affinity between the antibody and its receptor | ||
| ● | Antibody uptake limit | ||
| ● | Competition for target binding between the therapeutic antibodies and a patient’s antibodies | ||
| ● | Speed of diffusion through tumors is mAb size dependent (large tumor masses may be more difficult to treat by mAb therapy) | ||
| Oncolytic viruses | ● | Development of neutralizing antibodies by the host, which limits the viral delivery to cancer sites and the therapeutic effect | |
| ● | Limited penetration and distribution of the virus into tumor tissues | ||
| ● | Limited viral tropism and oncolysis capacity | ||
| ● | Tricky dosing strategies | ||
| ● | Induction of overactive immune responses as well off-target responses against healthy cells | ||
| ● | Risk of infection | ||
IO for Solid Tumors
Despite the success of IO therapies for some hematological cancers, significant gaps remain in the development of efficacious IO therapies for solid tumors. There are still a number of challenges that IO therapies need to resolve to treat solid tumors including the ability to target delivery of a therapeutic to the solid tumor and identification of suitable prominent cell surface targets. Cell therapies have not been as successful in solid tumors in comparison to blood cancers mainly because of the absence of a suitable prominent cell surface target and the high risk of toxicity when a potential solid tumor target is expressed, even at a low level, on normal tissue. Even if targets for solid tumors with a suitable tumor-selectivity profile can be identified, other factors may limit the activity of cell therapies, including limited cell-therapy penetration and distribution, low oxygen concentration (hypoxia) barriers around cancers that may prevent T cell access to the tumor, expression by tumor cells of certain checkpoint genes and an inability to target multiple antigens at the same time.
| 99 |
To overcome the current limitations of IO therapies in solid tumors, a new, effective tumor therapeutic must:
| 1. | achieve a local and tumor-targeted delivery; | |
| 2. | maximize on-target effects; | |
| 3. | reach the desired therapeutic index; | |
| 4. | minimize the off-target side effects; and | |
| 5. | potentially provide long term results. |
Our Platform
Our platform technology utilizes a novel mechanism of action that we believe has the potential to address the limitations and challenges of current IO technologies. Through a single administration, our platform is designed to provide a broadly applicable treatment to deliver a tumor-targeted therapeutic, including to solid tumors. It does so by exploiting a naturally occurring cancer-induced biological process, allowing for the local delivery of the payload with a potentially durable response, in a manner that we believe will limit systemic toxicity. The ability to deliver localized and tumor-targeted payloads, by avoiding systemic or off-target toxicity, may also allow for the use of well-established immunotherapies, such as the immunomodulator IFN-α, that has shown efficacy but has had limited therapeutic applications due to side effects associated with its intravenous delivery.
Specifically, we adapted an autologous ex-vivo gene therapy method to direct the patient’s own hematopoietic stem and progenitor cells (HSPCs) by loading them with an immunotherapeutic transgene sequence, or payload, that is able to counteract cancer progression and prevent tumor relapse. We believe that by delivering a targeted therapeutic specific to cancer cells, we can reach the desired on-target anti-tumor effect while reducing off-target side effects.
Our platform technology employs the following key components:
| a) | use of the patient’s own autologous HSPCs; | |
| b) | use of LVVs for ex vivo HSPCs transduction; and | |
| c) | payload delivery within the tumor microenvironment (TME) using specific tumor-associated myeloid cell (Tie2-expressing monocytes – TEMs). This “cell-confined” transgene expression is ensured by the selected promoter (Tie-2 promoter) and the imposed post-transcriptional regulation layer represented by a miRNA target sequence (miRNA-126 target sequences). |
The image below illustrates the steps of our ex-vivo approach to transform patient’s autologous HSPCs into a therapeutic product.
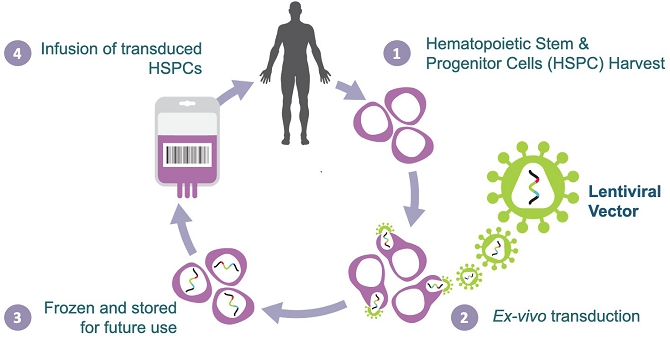
Illustration of our ex-vivo approach (steps 1-2-3) and treatment process (step 4)
(1) Patient’s HSPCs are harvested by means of an apheresis process, and (2) ex-vivo modified by an LVV. The obtained drug product is frozen and stored for clinical use (3). When needed, the therapeutic product may be thawed and infused back in patient’s blood stream (4). The engineered HSPCs will repopulate the entire hematopoietic system, giving rise to differentiated progeny bearing the introduced modification.
| 100 |
a) HSPCs are the Source of the Delivery Vehicle for Our Gene Therapy Approach
By re-introducing gene-modified HSPCs into the patient, we seek to take advantage of the self-renewing and multi-differentiation capability of HSPCs to enable durable and potentially long-term effects following a single treatment. HSPCs are self-renewing cells that can differentiate into all types of blood cells, including white blood cells, red blood cells and platelets. HSPCs can be obtained directly from the bone marrow or from the patient’s peripheral blood with the use of a mobilizing agent that induces HSPCs to relocate from the bone marrow into the peripheral blood where they may be collected by apheresis. The advantages of using a patient’s own HSPCs include the absence of graft versus host disease (GVHD) that could occur using allogeneic cells, and the potential long-term treatment durability of this approach.
b) Ex-vivo LVV based Transduction
After collection, a functional copy of the therapeutic gene is inserted into the patient’s own HSPCs using a non-replicating LVV. This is an ex-vivo process called transduction. We have chosen an ex-vivo gene therapy approach because it enables us to optimize the quantity, or dose, of modified cells to be infused in each patient since we know, ahead of the administration, the drug product characteristics.
We believe that LVVs are the first choice for ex-vivo gene therapy in humans because they can (i) carry large transgenes that will allow us to expand the therapeutic options to a multitude of payloads without “size” limits and (ii) efficiently transduce non-proliferating, or slowly proliferating cells, such as hematopoietic stem and progenitor cells. Most importantly, there is already an abundance of safety data generated using these vectors to develop investigational products currently under clinical testing, including CARs, TCRs, as well as commercial products such as Kymriah® (CD-19 CAR-T, Novartis Pharma) and Zynteglo® (β-Thal, BlueBird Bio). With more than 100 clinical trials either completed or in progress using LVVs worldwide, this delivery method accounts for more than a third of ex-vivo modified gene therapy clinical trials.
Accordingly, extensive clinical ex-vivo gene therapy studies, based on LVV gene transfer, have been performed in recent years by SR-Tiget for the prevention and treatment of some severe inherited disorders, resulting in approved drugs, such as LibmeldyTM. These studies have shown that LVVs constitute a valuable and safer alternative to gamma-retroviral vectors (gRV), enabling a more efficient gene transfer into HSPCs and resulting in a robust and long-term transgene expression in their progeny. The studies also have demonstrated an alleviated risk of genotoxicity because of the vector design.
Differences exist between LVVs used for ex-vivo transduction that could, in theory, lead to differences in the long-term safety profile of products, particularly in terms of genotoxic potential. Use of strong promoters in conditions where a high pre-existing risk for hematologic malignancies exists, such as sickle cell disease (SCD), could in the long-term (i.e. 5 years or more) contribute to the development of leukemia. There have been several significant adverse side effects in gene therapy treatments involving an ex-vivo transduced lentivirus vector (LVV) gene therapy product, BlueBird Bio’s elivaldogene autotemcel (“Lenti-D”), involving two SUSARs for cases of acute myeloid leukemia (AML), and one case involving myelodysplastic syndrome.
In February 2021, BlueBird Bio temporary suspended its gene therapy clinical trials for SCD (HGB-206 and HGB-210) and the marketing of Zynteglo® due to a suspected unexpected serious adverse reaction (SUSAR) of acute myeloid leukemia (AML) in a SCD patient who received the product more than five years ago. In July 2021, the European Medicines Agency’s (EMA) safety committee (Pharmacovigilance Risk Assessment Committee - PRAC) announced that there is no evidence the LVV used in both Lenti-D and the EU-approved gene therapy Zynteglo spurred the AML cases.
BlueBird Bio announced on August 9, 2021 that the SUSAR involving myelodysplastic syndrome occurred in one patient treated with Lenti-D over a year previously, that this SUSAR “is likely mediated by Lenti-D lentiviral vector (LVV) insertion,” and that “[e]vidence currently available suggests that specific design features of Lenti-D LVV likely contributed to this event.” As a result of this SUSAR, the FDA has placed a clinical hold on BlueBird Bio’s Lenti-D phase 3 trial for cerebral adrenoleukodystrophy (CALD).
We believe that the intrinsic characteristics of the LVV we have selected as well as the properties of the promoter and control mechanisms, combined with HSPCs’ ability to self-renew, allow for a stable integration of the modified gene into the HSPCs and their related differentiated progeny, potentially achieving long-term safety and protection after only a single treatment.
c) Tumor-Targeted Payload Delivery
Our platform technology, used by all of our product candidates including Temferon, is designed to turn TEMs, which normally have an affinity for and travel to tumors, into a “Trojan Horse” to deliver a tumor-targeted payload. The technology ensures that the payload is only expressed in TEMs and not in other types of cells. The following key components make up our platform technology: (i) a Tie-2 promoter that drives transgene sequence transcription specifically in TEMs, and (ii) a post-transcriptional regulation layer represented by miRNA-126 target sequences that induces the downregulation of the transgene expression in those cells where the Tie2 promoter is active and the miRNA-126 target sequence is present. This transcriptional / post-transcriptional regulatory mechanism prevents off-target effects and allows the expression of the payload by the selected cellular carrier (TEMs).
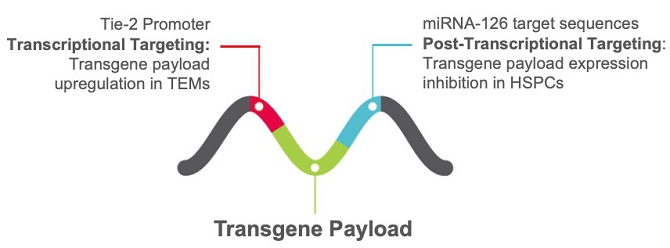
Our transgene payload expression cassette consists of two key components: the Tie-2 promoter (RED) and miRNA-126 target sequences (LIGHT BLUE)
| 101 |
| (i) | Tie-2 promoter. The Tie-2 promoter enables the transformation of TEMs into a “Trojan Horse”, to deliver the therapeutic payload within the tumor microenvironment. Tumor development and progression is a multi-step process leading to cancer growth. The so called “angiogenic switch” is one of the required steps and refers to a time-restricted event during tumor progression where the balance between pro- and anti-angiogenic factors tilts towards a pro-angiogenic outcome, resulting in the transition from a “dormant” avascularized tumor to an outgrowing vascularized cancer. It is well recognized that TEMs play an active role in this regard. Indeed, TEMs are actively recruited by proliferating tumors, through signals produced by the cancer cells or stromal/endothelial components, to promote the neo-vascularization and to contribute to the establishment of an immunosuppressive tumor microenvironment that leads to the failure in tumor eradication by the immune system. Amongst chemoattractant factors of monocytes, angiopoietins (Ang) play a crucial role. These are adhesion molecules and known vascular growth factors expressed by peritumoral blood vessels. One Ang in particular, Ang-2, attracts TEMs, which binds to the Tie-2 receptor. Expression of Ang-2 is upregulated by tumor hypoxia and may function as a chemoattractant for Tie2-expressing monocytes. Moreover, TEMs’ penetration into the tumor microenvironment in response to these stimuli cause Tie-2 receptor upregulation, which enhances the delivery of the payload to the tumor. Since TEMs recruitment is a naturally occurring event in the tumor development process and is a key aspect shared by several different cancers, we believe that our platform which enables the tumor targeted delivery of therapeutics represents a unique approach that may have broad applicability. | |
| (ii) | miRNA-126 target sequences. The miRNA-126 target sequence serves as a post-transcriptional regulation layer that allows the expression of the transgene payload only in cells where miRNA-126 is not expressed. In our case, because miRNA-126 is highly expressed in HSPCs but down-regulated in the differentiated progeny, it switches off transgene expression in the stem and progenitor cell compartment. Indeed, Tie2 is a weak promoter expressed, in the hematopoietic compartment, by Tie2-expressing monocytes and by hematopoietic stem cells (HSC). In HSC, it works as a membrane-bound receptor that keeps HSC cell-to-cell interaction and adhesion with the bone marrow niche and preserves the HSC quiescent/low proliferating state. |
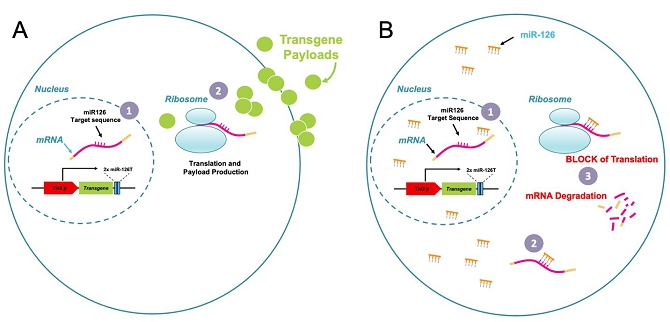
Post-transcriptional control mechanism of transgene expression
A) Transgene expression is allowed only in cells where miRNA-126 is not expressed; (1) mRNA is transcribed into the nucleus (2) the transgene is then translated in the cytoplasm and released.
B) In those cells expressing miRNA-126 the payload production is prohibited; (1) mRNA is transcribed into the nucleus (2) miRNA-126 recognizes its target sequences on the mRNA and forms double strands of RNA (3) that are degraded or block the translation process.
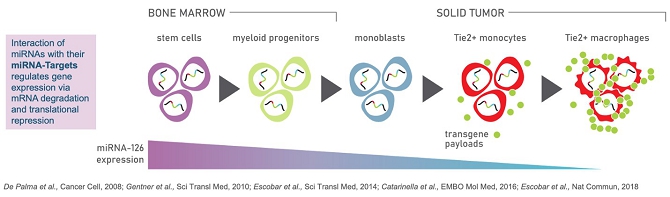
Transgene payload expression as the result of the transcriptional (promoter) and post-transcriptional regulation (miRNA-126 target sequence) imposed by our expression cassette.
We believe that combining our built-in post-transcriptional control mechanism (miRNA-126 target sequences), with TEMs designed as a “Trojan Horse” allows our platform to restrict transgene expression and avoid systemic toxicity while reaching the therapeutic index to drive efficacy.
| 102 |
Our Leading Product Candidate: Temferon
Our lead product candidate, Temferon, consists of genetically modified HSPCs which use our platform to deliver interferon-alpha (IFN-α), within the tumor microenvironment via TEMs (HSPCs differentiated myeloid progeny). The IFN-α reduces angiogenesis, counteracts tumor cells proliferation and breaks the established immune-tolerance, enabling the immune system to recognize the tumor. IFN-α is a proven and known immunomodulatory molecule, or cytokine, that has limited clinical use due to the systemic toxicity associated with its intravenous administration. Our technology is designed to protect the HSPCs from IFN-α mediated activation that could negatively impact their repopulation capacity as reported in some studies of repeated systemic administration of high doses of IFN-α. We believe that this protection technology, whereby we restrict payload expression to TEMs, and the release of IFN-α within the TME, has the potential to provide efficacy without inducing systemic toxicity.
Because TEMs are associated with the growth of numerous cancer types, including solid tumors, we believe that Temferon is tumor type and tumor target agnostic and therefore may be used across a large variety of cancers. Currently, we are developing Temferon for uMGMT-GBM.
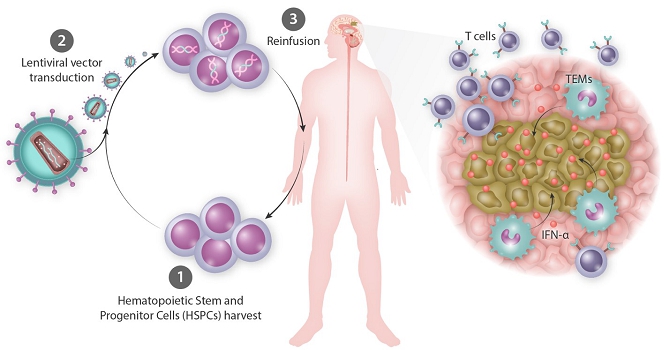
Overview of Temferon manufacturing process and mechanism of action.
Patient’s HSPCs are harvested, (1) ex-vivo modified by LVVs (2) and re-introduced back in patient’s blood stream (3). Once recruited within the tumor microenvironment, TEMs release IFN-α that reduces angiogenesis, counteracts tumor cells proliferation and enables the immune system to recognize the tumor.
GBM is a solid tumor affecting the brain. We have chosen this indication due to the following factors:
| ● | High unmet medical need. The prognosis for GBM patients remains poor with few therapeutic options having limited clinical benefits. | |
| ● | Temferon’s MoA targets TEMs which have an active role in uMGMT-GBM pathology. GBMs are highly vascularized tumors that critically depend on the generation of tumor-associated blood vessels. Several studies demonstrate that infiltrating myeloid cells, including Tie2-expressing monocytes, contribute significantly to tumor angiogenesis, presumably by secreting pro-angiogenic factors and promoting malignant glioma growth by creating a local immunosuppressive microenvironment. Moreover, TEMs have been identified in the normal/tumor boundary from human biopsy samples of GBM patients who received treatment to reduce angiogenesis using the anti-VEGF treatment bevacizumab, and the Tie2 pathway has been implicated in the triggering of a bevacizumab-mediated VEGF-independent angiogenesis that explains the long-term refractoriness of GBMs to anti-VEGF treatment. | |
| ● | Immunosuppressive tumor microenvironment. GBM is characterized by an immunosuppressive microenvironment that is mediated by tumor associated myeloid cells (including TEMs) that prevent the immune system from recognizing and rejecting the tumor. Our treatment approach is designed to exploit TEMS to deliver IFN-α to the tumor so that the immune system recognizes the tumor and halts tumor cell proliferation and recurrence. |
| 103 |
| ● | Availability of a “competent” immune system. Our approach relies on a patient’s immune system being capable of providing an immune response upon recognition of the tumor. Therefore, we believe newly diagnosed uMGMT-GBM patients who have relatively “competent” immune systems, not yet damaged by repeated cycles of chemotherapies, are strong candidates for our candidate. | |
| ● | Compelling preclinical data. Our preclinical studies, published in peer-reviewed papers, suggest that TEMs play an active role in uMGMT-GBM, and when used as a “Trojan Horse,” significantly shrink the tumor and to control disease progression. In more recent unpublished studies, we have also demonstrated, in a preclinical immunocompetent GBM mouse model, that treatment by Temferon resulted in a long-lasting immune response in surviving mice, even after repeated tumor challenge intended to replicate possible tumor recurrences. | |
| ● | Market Opportunity. Based on currently available treatments, the global market size for all GBM is projected to grow to over $1.5 billion by 2026. We believe a novel therapeutic which demonstrates improvement over existing therapies would greatly increase the market size. |
We are discussing a second study using Temferon in a second solid tumor indication. Locally advanced HCC and ICC are the leading indications under consideration and, as with GBM, these liver cancer indications have been selected for similar reasons as described above.
Temferon for uMGMT-GBM
Disease Overview
GBM is the most common malignant primary brain tumor accounting for more than half of all central nervous system (CNS) cancers and for which there is a high unmet medical need. The incidence rate is 3.20 per 100,000 persons with over 13,000 deaths per year in the United States. This disease is lethal and left untreated, the median survival is three (3) months. The current standard of care includes using a combination of surgery, radiation therapy, and chemotherapy for treatment. Although these treatments may improve survival, the prognosis for GBM patients remains poor with a median overall survival (mOS) of approximately 13 to 15 months and only 5.5% of patients estimated to be alive 5 years after diagnosis. GBM may occur at any age, but 70% of cases are seen in patients between 45 and 70 years of age (median 64 years). The disease often progresses rapidly (over 2 to 3 months). Neurological signs are nonspecific as they result from intracranial hypertension and include headaches and vomiting, often associated with behavioral changes or focal neurological deficits. Variants of GBM include secondary glioblastoma (20% of total diagnosed GBM), gliosarcoma (2%) and giant cell glioblastoma (1%). We are not including these variants in our studies because they do not fully meet our selection criteria discussed above.
Current Treatment Landscape and Limitations
The current standard of care for GBM includes surgery to remove the accessible tumor followed by radiation therapy (RTx), chemotherapy with temozolomide (TMZ) and/or tumor treating fields (TTFields).
| ● | Surgery remains the mainstay of initial treatment. If the tumor is located in a resectable region of the brain, it is used to histologically confirm the diagnosis and level of tumor burden. For many patients, removal of the tumor also results in a decrease of tumor mass-associated symptoms. Although the extent of the surgical removal of the tumor is linked to longer survival, due to the invariably infiltrative nature of the disease, even the complete removal of the accessible tumor is not curative and most people with GBM later develop recurrent tumors either near the original site or at more distant locations within the brain. Additionally, as a possible consequence of surgical procedures, permanent brain damage may occur. | |
| ● | Radiation therapy improves survival and is typically started approximately 3 to 4 weeks after surgery. RTx is performed daily for approximately 6 weeks. RTx induces the formation of neo-antigens and a pro-inflammatory response that are key aspects for immune system mediated disease control. However, the efficacy of RTx is impaired by hypoxia and by the negative effects of RTx on tumor infiltrating immune cells. |
| 104 |
| ● | Chemotherapy. Temozolomide, the current chemotherapeutic standard of care, is a DNA-alkylating agent that can cross the blood-brain barrier to reach therapeutic concentrations in the brain. The drug is administered every day during radiation therapy and then for six to 12 cycles after radiation as a maintenance therapy. Each cycle lasts 28 days, with TMZ given the first five days of each cycle, followed by 23 days of rest. TMZ adds a methyl group to DNA that, if unrepaired, leads to DNA strand breaks and cytotoxicity. More than one-third of glioblastomas are deficient in methylguanine methyltransferase (MGMT), a repair protein that removes the methyl group. This MGMT deficiency occurs through the methylation (silencing) of the MGMT gene promoter. Glioblastoma patients with a silenced MGMT gene who are treated with TMZ have a longer survival than those with an unmethylated MGMT. TMZ has several adverse side effects, including a cumulative bone marrow toxicity. | |
| ● | Tumor-Treating Fields (TTFields). The use of TTFields to extend temozolomide maintenance chemotherapy for newly diagnosed glioblastoma patients has recently been incorporated as a new standard of care. TTFields are applied via multiple electrodes that are directly fixed to the scalp. These low-intensity, alternating electrical fields interfere with cell division ultimately leading to cell cycle arrest, aneuploidy, and apoptosis. The most common TTFields-associated adverse events (AEs) are mild-to-moderate array-associated contact dermatitis. | |
| ● | Experimental Treatments. Along with the above-mentioned treatments, the addition of the antiangiogenic agent bevacizumab (BEV) to RTx and TMZ has been explored with mixed clinical results BEV was tested both a first-line treatment together with RTx and concomitant TMZ administration in newly diagnosed glioblastoma patients, as well as in combination with RTx in recurrent GBM patients. BEV was approved by the FDA as monotherapy for recurrent glioblastoma in 2009 under the name Avastin®. The EMA declined to approve BEV for recurrent glioblastoma due to the absence of a non-bevacizumab control arm, a modest overall survival increment versus historic controls, inadequate elucidation of true antitumor effect, and challenges with radiographic response assessment. More recently, an immune-checkpoint blocker nivolumab was tested in combination with TMZ and RTx in a Phase 3 trial in recurrent glioblastoma patients but showed minimal activity and no benefit in terms of mOS, resulting in failure to meet one of its primary endpoints, progression free survival (PFS). |
Currently available GBM surgical treatments have not been able to prevent GBM recurrence because of the infiltrative nature of this disease and the absence of an effective immune system. A therapeutic able to cross the blood-brain barrier and selectively impact proliferation of cancer cells independently from the region of the brain where the tumor resides would be a significant advancement.
Our Solution
We believe that our investigational product, Temferon, has the potential to address the world recognized GBM unmet need. Through a single administration, we believe Temferon may be able to provide a long-lasting immune response, minimize systemic toxicity, counteract cancer progression and prevent tumor relapse.
Temferon utilizes our platform and ex-vivo gene therapy approach to introduce a functional copy of IFN-α which is Temferon’s transgene payload, into the patient’s autologous HSPCs, resulting in a drug product that can then be reintroduced into the patient as outlined above (see “Our Leading Product Candidate: Temferon” and “Our Platform”). Temferon is designed to colonize the patient’s bone marrow with the genetically modified HSPCs to continuously generate TEMs containing the IFN-α payload. Since TEMs are recruited within the tumor microenvironment, IFN-α is released solely at the targeted tumor, which may result in clinical activity without inducing systemic toxicity.
| 105 |
Once within the tumor microenvironment, IFN-α is expected to act both directly by promoting cancer cell apoptosis and inhibiting vascularization and indirectly by restoring the body’s anti-tumor immune response, as follows:
| ● | Direct Effects. IFN-α suppresses tumor cell proliferation and promotes the apoptosis of tumor and stromal cells by induction of proapoptotic genes or repression of anti-apoptotic genes. Moreover, IFN-α inhibits angiogenesis by downregulating the expression of proangiogenic factors. | |
| ● | Indirect Effects. IFN-α stimulates early innate immune responses and the subsequent adaptive immune response via multiple pathways and mechanisms, including: maturation and cross-priming capacity of dendritic cells (DCs); upregulation of the expression of tumor-associated surface antigens and MHC class I molecules on tumor cells and of MHC class I and II molecules on DCs; enhanced priming and survival of T cells; enhanced humoral immunity; increased cytotoxic activity of NK cells and macrophages; control of helper T cell population balance (Th1=Th2); immunoglobulin class switching of B cells; and the regulation of CD8+ cytotoxic T-lymphocyte (CTL) responses. |
We believe that through these immunomodulatory functions and based on our preclinical data, IFN-α increases tumor immunogenicity, recruits and activates immune cells within the tumor milieu, breaks established tumor-induced immunotolerance and may induce tumor rejection.
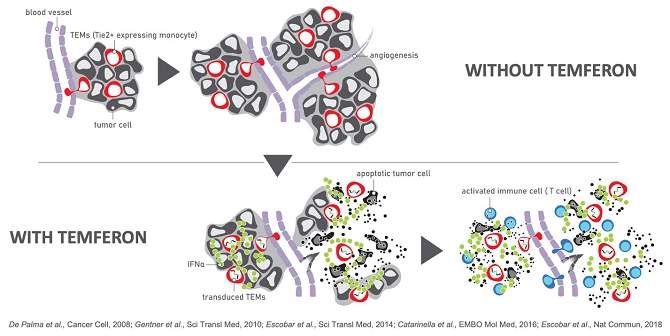
Overview of Temferon antitumor activity
Once recruited at the tumor site, instead of fostering tumor growth and inhibiting the immune systems, TEMs start to release IFN-α that triggers cancer cell apoptosis and arms the immune cells to fight the cancer.
Clinical Development of Temferon in uMGMT-GBM
Preclinical Data
We have preclinical data, published in 2018, suggesting that TEMs play an active role in GBM disease. Further studies published in 2008 showed that when TEMs were used as a “Trojan Horse,” as utilized by Temferon, the GBM tumor volume decreased and the disease progression was controlled. In more recent, unpublished preclinical studies, we have also demonstrated, in an immunocompetent GBM mouse model, that treatment with Temferon resulted in a long-term immune response in surviving mice, even after repeated tumor challenge intended to replicate possible tumor recurrences.
| 106 |
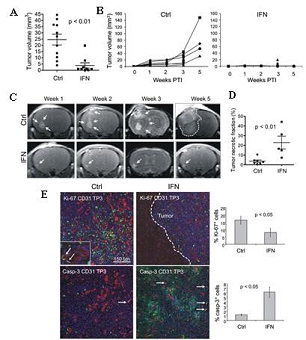
Inhibition of Human Gliomas in IFN-α Gene Therapy Treated Athymic Mice (De Palma et al., 2008).
A-B) Glioma growth (mean tumor volume ± SEM, measured by MRI) in individual control and IFN-α gene therapy treated mice (Tie2-IFN) at 3 weeks post tumor injection (PTI). Tumor volume and progression decreased in IFN-α gene therapy treated mice. C) MRI images showing brain tumor growth at the indicated time points PTI in representative control (Ctrl) and Tie2-IFN mice. Intracranial gliomas are indicated by arrows and dashed line. The tumor did not progress in Tie2-IFN mice. D) Measurement of tumor necrotic fraction (mean necrotic fraction, % ± SEM) by MRI in individual Ctrl and Tie2-IFN mice at 3 weeks PTI, evidencing cancer cell death. E) The Tie2-IFN tumors that grew sufficiently to be analyzed displayed decreased cell proliferation and greater apoptosis (assessed by Ki-67 and cleaved caspase-3 immunostaining, respectively a proliferation and an apoptosis marker) as compared to the control tumors. (Caspase-3 = Casp-3; green) and CD31 (marker of blood vessels; red) TO-PRO-3 (TP3; nuclear staining, blue). Arrows show Ki-67+CD31+ or caspase-3+CD31+ (ECs; dashed line indicates tumor margin).
Phase 1/2a Clinical Trial
In the second quarter of 2019, we enrolled our first patient in a single-arm, open label, dose escalation, Phase 1/2a clinical trial, in adult patients aged 18 to 70 years (“TEM-GBM 001 study”). The trial is being conducted at two clinical centers of excellence located in Milan, Italy: (i) Istituto Nazionale Neurologico “Carlo Besta”, an internationally recognized leading center in neuroscience, specializing in the diagnosis and treatment of neurological diseases in adults and children, and (ii) OSR, which has a recognized expertise in complex and innovative diagnostic and therapeutic approaches in onco-hematological patients and in gene therapy treatments.
| 107 |
The primary objective of the TEM-GBM 001 study is to evaluate the safety, tolerability, feasibility and biological activity of Temferon in uMGMT-GBM patients with an unmethylated MGMT gene, who as a result of the gene, have a poor prognosis and are expected to not respond to TMZ treatment. These patients are identified immediately after surgery, upon confirmation of the diagnosis and MGMT methylation status. After enrollment, each patient is screened for eligibility, and if eligible subjected to the mobilization procedure to induce HSPCs to exit from the bone marrow niche and to migrate into the peripheral blood where they are collected by leukapheresis. Immediately after collection, each patient’s HSPCs are sent to our CMO to be genetically modified and become Temferon. After Temferon administration, each patient is followed for two years. The figure below depicts the different stages of TEM-GBM 001 study enrollment and treatment.
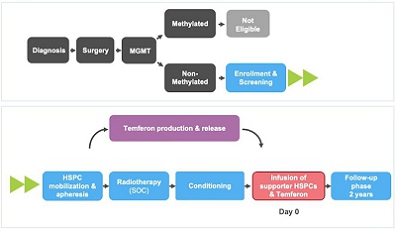
TEM-GBM 001 Study Design.
Our study aims to evaluate the short-term (up to 90 Days) and long-term (up to 2 years) tolerability and safety of five escalating doses of Temferon in up to 21 patients with uMGMT-GBM and an unmethylated MGMT promoter, following first line radiotherapy.
Initially the study was designed with a dose escalation phase in 15 patients assigned to 5 cohorts (Part A) to test three different escalating Temferon doses and two different conditioning regimens, with the aim of determining the best Temferon engraftment associated with fewer side effects to be then applied to an expansion phase (Part B) in six additional patients.
Patients in cohorts 1, 2 and 3 received Temferon dose escalation up to 2 x 106 CD34+ cells/kg (Dose Level 3) and 3 x 106 CD34+ cells/kg as unmanipulated supporter HSPCs. Patients in Cohorts 4 and 5 were infused with Temferon Dose Level 3 but received 2 x 106 CD34+ cells/kg and were conditioned with 2 different conditioning regimens (BCNU & thiotepa for Cohort 4, also utilized in Cohort 1-3 and Busulfan & thiothepa for Cohort 5).
Based on the results collected in the Part A of the study, we have amended the study to add two additional dose escalation cohorts (numbers 6 and 7). The Clinical Protocol amendment was submitted on July 20th, 2021 to the Italian Competent Authorities, and approved by AIFA in September 2021.
As per the submitted clinical protocol amendment, Cohorts 6 and 7 will recruit three patients each who will be conditioned with BCNU and thiotepa and will receive 2 x 106/kg supporter HSPCs and a Temferon dose of 3 x 106 cells/kg and (Cohort 6, Dose Level 4), and 4 x 106 cells/kg (Cohort 7, Dose Level 5), respectively.
The figure below represents the amended TEM-GBM 001 study scheme with interim and anticipated milestones. The study EudraCT Number is 2018-001404-11 and can be found at clinicaltrialsregister.eu/ctr-search/trial/2018-001404-11/IT.
| 108 |
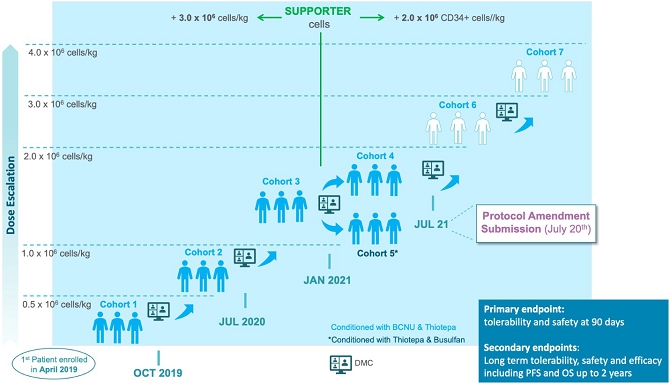
TEM-GBM 001 Amended Study Design.
Preliminary Interim Results.
We designed the TEM-GBM 001 study to: (i) obtain rapid accrual of patients (ii) assess the safety profile of Temferon, (iii) identify the optimal dose, and (iv) measure the biological activity of Temferon on those patients who progress to the point of requiring a second surgery.
We designed our study anticipating that patients receiving lower doses of Temferon could progress to the point of needing a second surgery, which is often necessary in GBM patients. A second surgery provides the only source of GBM specimens post treatment to evaluate the biological activity of Temferon and to evaluate its mechanism of action in patients. We enrolled our first patient in April 2019 and as of November 15th, 2021, we had dosed 15 patients, from cohort 1 to 5. We expect to complete the enrollment and the dosing of patients to be assigned to cohorts 6 and 7 by the end of the second quarter of 2022.
Overview of the current status of TEM-GBM 001 clinical development program
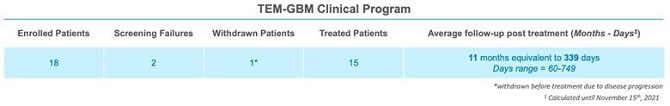
| 109 |
There have been no dose limiting toxicities identified in our clinical study to date. All patients showed rapid engraftment and hematological recovery after administering the sub-myeloablative conditioning regimens (Busulfan BCNU + Thiotepa or Busulfan and Thiothepa). Evidence of presence and persistence of Temferon-derived differentiated cells is assessed by measuring the vector genomes in the DNA of peripheral blood and bone marrow cells (absolute number quantified by ddPCR). The vector copy number (VCN) positive cells were present within 14 days post treatment and were detectable, albeit at lower levels, in the long-term (up to 18 months, the last measured timepoint to date).
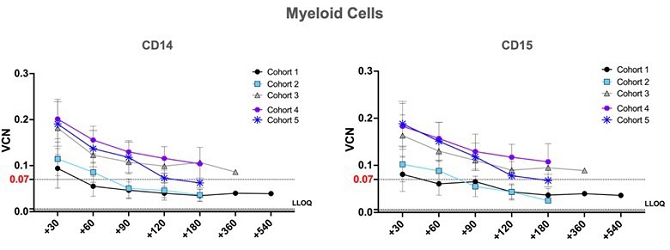
Engineered myeloid cells persist in peripheral blood for up to 18 months
We also detected very low concentrations of IFN-α in the plasma and cerebrospinal fluid, suggesting the transgene expression control mechanism may be working as intended by limiting systemic IFN-a exposure.
Serious adverse events have mainly been attributed by the Investigator to the effects of the conditioning regimen (pneumonia, pulmonary embolism, febrile neutropenia, fatigue, C.diff infection, CMV reactivation, sepsis) or disease progression (worsening left hemiparesis, seizure, brain abscess, sudden death). Three instances of elevated gamma glutamyl transferase (GGT) have been observed: two mild cases attributed to chemotherapy and HSPC harvesting, each of which has resolved; and one case occurring at D+78 following Temferon administration, which was classified as a suspected unexpected serious adverse reaction (SUSAR) possibly related to Temferon, and which has also resolved. Nine deaths have been reported to date: seven at +241, +322, +340, +402, +478, +646 and +749 days after Temferon administration due to disease progression, and two deaths at +60 and +122 days which the Investigator considered not related to Temferon but possibly related to complications following the chronic steroid treatment and the conditioning regimens prior to Temferon administration.
The last evaluable disease status as assessed by immunotherapy response assessment for neuro-oncology (iRANO) criteria as of November 15th, 2021, the last evaluable time point for all 15 treated patients (prior to death, where applicable), was as follows: six patients had shown stable disease (SD), two patients had demonstrated partial response, seven patients had shown progressive disease (PD). For reference, The iRANO guidelines specifically address interpretation of initial progressive imaging findings in the context of neuro-oncology patients with a goal of decreasing the likelihood of premature discontinuation of potentially beneficial therapies while ensuring maximum patient safety. iRANO empirically stipulates a three-month window for confirmation of progression on follow-up imaging, and further advises that progressive imaging changes beyond six months after immunotherapy initiation are more likely true tumor progression.
So far, eight out of the nine patients from the first three cohorts and one patient in cohort 4 progressed. The PD occurred after a median of + 123 days (range 83–239 days) following administration of Temferon. Two patients of cohort 3 progressed before Temferon administration (11 and 12 days before Temferon administration). Preliminary analyses performed on the tumor specimens belonging to patients with PD who underwent a second surgery (four patients) confirmed the presence of TEMs within the tumor, as assessed by flow cytometry, and an increased expression of IFN-responsive gene signatures compared to diagnosis, as assessed by quantitative polymerase chain reaction (PCR) tests. We believe that these findings suggest intra-tumor IFN-α release.
Of the four patients who underwent the second surgery, one patient had a prior lesion, which was not removed during the first surgery. When this patient underwent the second surgery following treatment with Temferon, it was observed that this lesion was stable and had not grown. In addition, this patient presented a relapsing progressing lesion that had developed at the first surgery site. Both lesions were removed and biopsied at the second surgery. Notably, as assessed by flow cytometry, the stable lesion had a higher proportion of T cells and TEMs within the myeloid infiltrate and as detected by quantitative PCR a markedly increased IFN-response signature as compared to the relapse-progressing lesion.
We also analyzed the peripheral blood of the patients from cohorts 1 to 3. In these patients, the peripheral blood showed, on two of the four subjects with samples at the first and second surgery, a change in the T cell immune repertoire post treatment, revealing expansion of tumor-associated clones, which suggested that changes in the immune system are occurring.
We believe these results align with our preclinical data and suggest the immune activation effects of Temferon. Specifically, the preliminary interim results, including the tumor analysis conducted after second surgery described above, provide biological evidence to support the hypothesis that cellular and molecular changes were triggered by Temferon in uMGMT-GBM patients despite the low dose administered. We have yet to reach the maximum tolerated dose of Temferon and as outlined in our study protocol, we will be increasing the dose in cohorts 6 and 7.
TEM-GBM Clinical Development Plan
We plan to use the results of our TEM-GBM Phase 1/2a study (NCT03866109) to support a Clinical Trial Application to be conducted in Europe, a multicenter Phase 2 study in uMGMT-GBM, which we currently intend to conduct primarily in Italy. In advance of our CTA submission and not required for our European trial, we have submitted a pre-IND meeting request to the FDA and received written responses from the Agency in the third quarter of 2021 regarding the proposed Phase 2 clinical study design and drug product manufacturing strategy. The plans for the Phase 2 clinical study and drug product manufacturing strategy will be informed by the Agency comments. We intend to submit the CTA for a Temferon Phase 2 study for uMGMT-GBM patients by the fourth quarter of 2022. The Phase 2 study will evaluate the safety, tolerability, biological reprogramming of the tumor microenvironment in Temferon treated patients compared to the current standard of care, with primary endpoints, of PFS and mOS. We are also working with our current CMO and potentially other CMOs based in the U.S. to manufacture and to supply Temferon in the U.S. for our larger trials to be conducted after the completion of our Phase 2 study.
Key components of the anticipated clinical trial are as follows:
| ● | Multicenter study evaluating Temferon for safety, tolerability and efficacy compared to the current standard of care; | |
| ● | Enrollment and treatment of up to 27 patients with newly diagnosed and recurrent uMGMT-GBM; |
| 110 |
| ● | PFS and mOS as co-primary study endpoints |
The study design as discussed with FDA at the Pre-IND meeting may be subject to modifications.
Second Solid Tumor Indication
We are also evaluating liver cancers as a potential second solid tumor to be investigated. HCC and ICC are gastrointestinal cancers affecting the digestive system. We chose to pursue these indications for similar reasons as the uMGMT-GBM indication, namely:
| ● | High unmet medical need and market opportunity. The prognosis for patients with locally advanced HCC and ICC remains poor with few therapeutic options having limited clinical benefits. | |
| ● | TEMs have an active role in HCC and ICC pathology. |
| ○ | Several third-party studies have shown that tumor infiltration by tumor associated myeloid cells, including TEMs, is a negative prognostic factor in HCC and ICC. | |
| ○ | In HCC, TEMs are the most abundant proportion of tumor associated myeloid cells. | |
| ○ | Hepatitis B (HBV) infections and hepatitis C (HCV) infections, both of which predispose individuals to the development of chronic liver disease and the subsequent liver cancers, upregulate Ang-2 expression, further contributing to tumor angiogenesis and TEMs migration. | |
| ○ | In HCC, Ang-2 mRNA expression is significantly increased when compared to adjacent liver tissue and angiopoietins and tumor infiltrating TEMs appear to be associated with metastasis and recurrent disease. |
All the above evidence highlights the role of TEMs in fostering liver cancer growth, by promoting angiogenesis.
| ● | Pro-inflammatory and immunosuppressive tumor microenvironment. Both locally advanced HCC and ICC are characterized by a pro-inflammatory and immunosuppressive tumor microenvironment with TEMs playing a key role. | |
| ● | IFN-α and liver tumors. Chronic inflammation resulting from HBV and HCV infection are key contributors for the development of HCC or ICC. Before the introduction of antiviral agents in 2011, parenterally administered interferons, including IFN-α, were utilized as the standard of care in order to achieve viral clearance and/or suppression. The use of IFN-based regimens for HCV and HBV infections has been associated with a reduction in HCC risk. Evidence suggests that use of IFN-α as an adjuvant therapy in HCC patients after curative therapy may reduce the recurrence rates for up to 5 years, particularly in patients with concurrent HCV infection. | |
| ● | Available preclinical data. We have preclinical data, published in peer reviewed papers suggesting that TEMs used as a “Trojan Horse” for cancer that has metastasized to the liver were able to induce a statistically significant tumor shrinkage and control disease progression. | |
| ● | Market Opportunity. Based on currently available treatments, the HCC and ICC combined global market value is projected to grow to over $2.6 billion by 2026. We believe a novel therapeutic which demonstrates improvement over existing therapies would greatly grow the market size. |
| 111 |
Disease Overview
Liver and intrahepatic bile duct cancers are upper gastrointestinal (GI) cancers that affect the digestive system. They are the fifth most common cause of cancer deaths in men in the U.S., and the seventh most common cause of death in women. The American Cancer Society (ACS) estimates that 42,230 new cases of liver cancer (including intrahepatic bile duct cancers) will be diagnosed in 2021. Of these cases, approximately three-quarters were HCC, while less than one-quarter were bile duct cancers (both intrahepatic and extrahepatic). We are developing Temferon to treat the two tumor types listed below.
| ● | Hepatocellular carcinoma (HCC) is a primary malignancy of the liver that occurs predominantly in patients with underlying chronic liver disease and cirrhosis. The incidence of HCC has been rising worldwide over the last 20 years, with about eight new cases per 100,000 adults per year. The highest incidence of HCC occurs in Asia and Africa, where the endemically high prevalence of hepatitis B (HBV) and hepatitis C (HCV) strongly predisposes individuals to the development of chronic liver disease and the subsequent development of HCC. Patients with HCC have, when treated with the current standard of care, an expected median OS of only 11 months with approximately 10-40% of patients surviving 3 years. Up to 25% of HCC patients have no history of cirrhosis and do not present any risk factors for chronic liver disease, such as HBV and HCV. | |
| ● | Cholangiocarcinoma is a biliary tract cancer and represents approximately 3% of all GI malignancies. Of this 3%, ICC accounts for 5% to 10% of those cases. The incidence is 2.1 per 100,000 person years. Despite surgery, the prognosis remains poor with disease recurrence and progression occurring in approximately two-thirds of patients and a 5-year survival rate of only 30%. |
HCC Current Treatment Landscape and Limitations
In general, HCC patients are asymptomatic in the early stages of the disease, and as a result, the diagnosis is usually obtained in later stages of the disease when there are no curative therapies available. The implementation of surveillance, and the improvements in imaging techniques, have enabled the diagnosis at earlier stages of HCC. In terms of first line treatment approaches, patients with very early HCC are optimal candidates for surgical resection and liver transplantation but when these options are not suitable due to tumor localization or the absence of a matched liver donor, local ablation through percutaneous ethanol injection or radiofrequency are the available alternative treatments. For late-stage HCC, the transcatheter arterial chemoembolization (TACE) is the standard of care.
| ● | Surgical Resection is the recommended first line therapy for patients with a single HCC nodule, achieving the highest effectiveness in terms of disease control and overall survival. However, despite the accurate selection of patients with early tumors and the complete tumor removal, HCC patients are at high risk of tumor recurrence. | |
| ● | Percutaneous ethanol injection (PEI) is an effective technique used for early-stage HCC with a low complication rate that induces the complete necrosis of small HCC lesions. However, PEI has limitations including the need for multiple treatment sessions (4-6) and a prolonged treatment time. Although generally well tolerated, PEI can result in death and rare instances of tumor seeding. PEI-treated lesions have a high rate of local recurrence (33% - 43%). | |
| ● | Radiofrequency ablation (RFA) has advantages compared to PEI including ease of performance, effectiveness similar to that of surgical resection, high safety, and low invasiveness. RFA is the first-choice post-surgery procedure for patients with early-stage HCC. However, despite the advantages of RFA, complete tumor ablation remains difficult to achieve in some specific liver sites. Many patients treated with RFA develop atrial fibrillation on long-term follow-up. | |
| ● | Transarterial chemoembolization (TACE) is the current standard of care for patients with intermediate/late-stage HCC and who have relatively preserved liver function. TACE combines the “tumor embolization”, meaning the treatment blocks the vascular supply to a tumor accompanied by a local administration of chemotherapy. This permits high concentration of drugs in the tumor area, while simultaneously reducing systemic exposure. However, the technique is not standardized and there is no universal consensus as to TACE application in the clinical setting thereby limiting reliable comparison of results. |
| 112 |
| ● | Sorafenib has been the first line chemotherapy standard of care for patients with advanced unresectable hepatocellular carcinoma (HCC) since 2007 and has also been found to be useful in association with TACE as an effective chemotherapeutic agent to prolong survival in inoperable HCCs. However, Sorafenib is associated with adverse events (AEs) such as hand-foot skin reaction, rash, upper and lower gastrointestinal distress (i.e. diarrhea), fatigue, and hypertension. Furthermore, a proportion of treated patients show no response to the drug. |
ICC Current Treatment Landscape and Limitations
Surgery (radical excision) is the only treatment available for ICC. However, the disease will recur in 40–85% of patients who have a median survival of 36 months. Neo-adjuvant approaches have largely been unsuccessful and adjuvant therapy (radiotherapy, chemoradiotherapy or chemotherapy alone) is offered to patients only as a palliative solution. Based on a single, positive Phase 3 study, chemotherapy with gemcitabine and cisplatin is considered the standard of care and plays an established role as a palliative care solution.
Our Solution
Similar to Temferon for GBM, we believe that Temferon may overcome the current treatment limitations of HCC and ICC that are not curative/resolutive by stopping/delaying disease progression.
Clinical Development of Temferon in HCC and ICC
Preclinical Data
As described in the study published by Giovanni Sitia’s research group at OSR in collaboration with Luigi Naldini, our IFN-α gene delivery platform was tested in a mouse model of liver metastases from colorectal cancer (CRC) resulting in a substantially delayed or complete prevention of tumor growth with prolonged long-term survival. Our approach was also effective on pre-established liver metastases that more closely mimics tumor treatment in human.
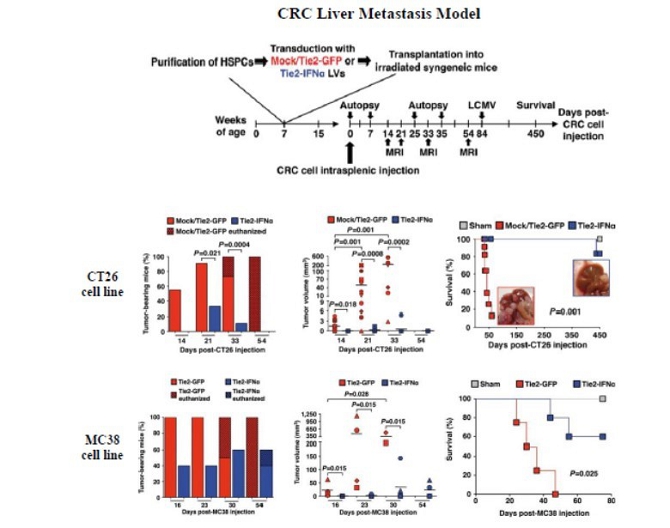
In vivo data on Tie2-IFN gene therapy platform in a mouse model of colorectal cancer (CRC) liver metastasis (Catarinella et. al; 2016)
IFN-α gene therapy treatment impairs the metastatic growth in the liver CRC liver metastasis.
CT26 and MC38 cell line panels: Percentage and tumor volume quantification measured by MRI analysis of mice injected with CT26 cells bearing at least one CRC liver metastasis estimated by MRI analysis and Kaplan–Meier survival curves of the indicated groups. The inset images show representative macroscopic photographs of the metastatic progression in the liver. Sham i.e., mice transplanted with non-transduced HSPCs and intrasplenically injected with saline thereafter.
Second Solid Tumor Study Design
As with the TEM-GBM 001 study, we plan to propose to the Italian regulatory bodies a non-randomized, open label, single center, Phase 1/2a, therapeutic-exploratory, prospective study, involving a single injection of Temferon administration in up to 9 patients. The study will recruit and track patients at OSR.
Patients will be administered Temferon following the same process as our TEM-GBM trial, namely: (i) HSPCs harvesting, (ii) standard of care chemotherapy concomitant to Temferon manufacturing, (iii) administration of the conditioning regimen followed by Temferon infusion, and (iv) 2-years of follow-up.
Additional Pipeline Pre-Clinical Programs
Our platform technology was designed to be flexible so that it can be potentially adapted to treat a variety of cancers. We believe that Temferon lends itself to be used in combination with a variety of current existing therapeutics to potentially enhance overall efficacy because its mechanism of action is intended to abrogate tumor induced tolerance. Additionally, we are developing a second-generation platform which is designed to release our therapeutic payload “on demand”. Finally, our proprietary platform is designed to control the expression of a potentially wide variety of therapeutic payloads we may choose for different targets.
Combination Treatment
While therapies to treat several types of cancers, such as ICI, CAR-T and TCR, are rapidly transforming the practice of medical oncology, clinical data point to the risk of late relapses after treatment with these therapeutics. Thus, the data suggest that the durability of the response to these therapies remains a significant challenge. We believe that due the potentially agnostic nature of Temferon, its potential activity, which includes the abrogation of tumor induced tolerance, and its potentially synergistic mechanism of action, makes it an ideal candidate to be considered for combination treatments. Specifically, Temferon may be a good candidate to be used in combination, for very aggressive tumors, with other immune-oncology drugs, such as CAR-T and ICI, to extend the durability of the response in very aggressive tumors. We believe that this additional Temferon application is supported by the promising results coming from the combination studies performed using Temferon with CAR-T, TCR and ICI in our pre-clinical programs as discussed below.
In preclinical studies conducted in the laboratories of our founders, Professor Luigi Naldini and Dr. Bernhard Gentner, Temferon was evaluated in combination with CAR T, TCR-edited T cells directed against tumor-associated antigens and immune checkpoint blockers. The results showed promising additive-to-synergistic anti-tumor activity in leukemia experimental models (Escobar et al., Nature Communication 2018), glioblastoma models, and multiple myeloma mouse models (manuscripts in preparation). These results lead us to believe that IFN gene therapy might also boost the efficacy of other immunotherapies.
Specifically, in a leukemia mouse model, a CD19 CAR-T approach had detectable, but not significant effect on the tumor burden. However, when used in combination with Temferon, the combination treatment resulted in a significant inhibition of the hematological malignancy with a significant fraction of CAR-T/Temferon treated mice surviving at the latest timepoint of analysis. Similarly, the combination of IFN gene therapy to α-CTLA4, an immune check point blocker, or adoptive T cell therapy, significantly improved the survival of the mice (Escobar et al., Nature Communication 2018).
In a multiple myeloma mouse model, Temferon was administered in combination with human TCR-edited T cells directed against NY-ESO1 and anti-myeloma drugs. The combination approach showed promising additive-to-synergistic effects leading to more pronounced disease control compared to most single-agent regimens, and without exacerbating hematologic or systemic toxicities (manuscript in preparation).
Switchable Platform
Our founders are developing a second-generation platform designed to release the therapeutic payload “on demand” to allow in vivo control of its potential efficacy. Potential advantages of this application include (i) broadening the clinical application to patient populations with more favorable pre-treatment prognoses; (ii) control of long-term side effects that may arise from the chronic exposure to immunostimulatory molecules and (iii) the ability to activate the immune system on demand to recognize tumors based on clinical need.
An inducible version of the IFN-a payload has been generated by fusing the protein with a destabilizing domain (DD), which targets the protein to proteasomal degradation, unless a small molecule ligand binding to the DD and stabilizing it, is administered. The optimized fusion construct is delivered by the TEM platform and the exogenous administration of the ligand switches on its secretion within the tumor. The results from experiments performed in the laboratory of Professor Naldini with a glioblastoma mouse model showed similar anti-tumor activity of the inducible and wild-type IFN payload. Moreover, the inducible construct allows switching of IFN release upon tumor clearance (manuscript to be submitted for publication). We plan to use our second generation platform carrying an IFN-a payload in combination with CAR T cells to target glioblastoma-associated antigens or immune checkpoint blockers in an experimental tumor model in mice.
Other Payloads
Our platform is designed to allow the control of the expression of any payload we use. Similar to IFN-a, there are several alternative payloads with immunotherapeutic properties that were previously systemically delivered to patients, but were discontinued due to significant toxicity that prevented the drug from reaching therapeutic dosages (e.g. TNF-a). Because we believe that our first- and second-generation platforms may overcome the limitations associated with systemic administration, we are testing them with additional payloads such as IFN-g, IL-12 and TNF-a. Because each payload triggers a unique biological response, we believe our platform may enable a personalized treatment approach unique amongst the current treatment paradigms.
Additional immune activating cytokines have been tested for TEM-mediated gene-based delivery to tumor models in mice. Current pre-clinical results suggest the feasibility and specificity of tumor-targeted delivery of IFN-g and TNF-a and further support our hypothesis that the specific transcriptional and microRNA regulated expression of the payload prevents toxicity. Data generated in the laboratory of our founders in a leukemia model showed that IFN-g but not TNF-a mediate anti-tumor activity when delivered ex vivo. Further ex vivo studies showed enhanced anti-tumor activity upon combined delivery of two cytokines by the TEM-based platform.
Moreover, experiments conducted with our second-generation inducible platform expressing IL-12 supported the hypothesis that our proprietary transcriptional and microRNA regulated expression of the payload to may prevent toxicity. Indeed, IL-12 is a potent cytokine that must be kept within a therapeutic dose range to prevent toxicity. Targeted delivery and anti-tumor activity of the new inducible payload are being investigated (abstracts to be presented at EACR in June 2021; at ASGCT 2021 in May 2021; and manuscript to be submitted). The laboratory of Professor Naldini plans to test the combination of TEM-mediated gene-based delivery of inducible Il-12 or additional inducible payloads with CAR-T, TCR and ICI in experimental murine tumor models.
Competition
Biotechnology and pharmaceutical industries put significant resources in developing novel and proprietary therapies for the treatment of cancer. For the cell therapy field in particular, this results in rapidly advancing and changing technologies, intense competition and a strong emphasis on intellectual property. While we believe that our leading product candidate, Temferon and our scientific expertise in the field of cell and gene therapy provide us with competitive advantages, we face potential competition from various sources, including larger and better-funded pharmaceutical, specialty pharmaceutical and biotechnology companies, as well as from academic institutions, governmental agencies and public and private research institutions.
| 113 |
Manufacturing
The manufacturing process for our autologous cell and gene therapy approach requires the following steps:
| 1. | HSPCs harvesting in a specialized clinical center (leukapheresis) | |
| 2. | Shipping of apheresis bag/s to the selected contract manufacturing organization (CMO) | |
| 3. | CD34+ cells enrichment | |
| 4. | Ex-vivo transduction of CD34+ cells with our LVV, | |
| 5. | Cryopreservation, characterization and release by a Qualified Person of the obtained drug product. |
The LVV manufacturing needed for the ex-vivo transduction process, as well as steps from 3 to 5, are conducted by the CMO. The figure below delineates the steps and the timeline for manufacturing Temferon.
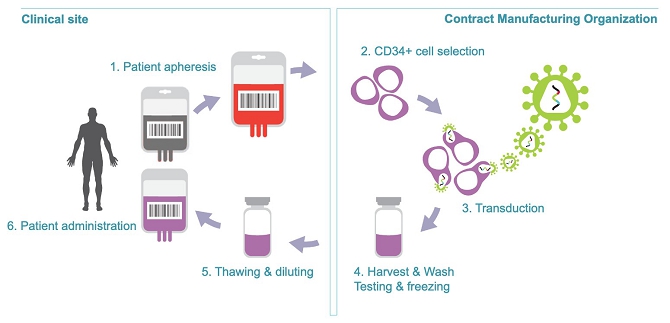
Overview of Temferon manufacturing Process
We have entered into agreements with AGC Biologics to manufacture our LVV and drug product for our ongoing clinical programs in Italy. AGC Biologics is a leading global contract development and manufacturing organization (CDMO), providing world-class development and manufacture of mammalian and microbial-based therapeutic proteins of plasmid DNA (pDNA) and recently with the acquisition of Molecular Medicine S.p.A. (“MolMed”), of viral vectors and genetically engineered cells. MolMed S.p.A. is recognized as the leading cell and gene therapy CDMO focused on research, development, production and clinical validation of cell and gene therapies for the treatment of cancer and rare diseases. Indeed, Strimvelis, the first ever market approved ex-vivo gene therapy for children, was developed and is still currently manufactured by AGC Biologics (former MolMed). Accordingly, Orchard Therapeutics (NasdaqGS: ORTX) in July 2020 renewed their collaboration agreement with AGC which will continue to support activities related to the development and manufacturing of vectors and drug products for several of Orchard’s investigational ex-vivo hematopoietic stem cell (HSC) gene therapies in the upcoming years, including the recent EU market authorized gene therapy drug Libmeldy.
| 114 |
AGC Biologics Headquarters and Capabilities
Our agreement with AGC (MolMed) establishes agreed-upon timelines for purchase order submissions and manufacturing date changes/cancellation. The supply agreement also sets milestones both during the clinical phase and any future commercial phase of our product candidates and for technology transfer if required, as well as customary termination provisions, allowing for termination by a party upon the other party’s uncured material breach or upon the other party’s insolvency. AGC Biologics facility in Milan, Italy, will continue manufacturing LVV and Temferon to support Genenta’s trials. For further larger studies we may use AGC’s 60,000 square meter cell and gene therapy manufacturing facility located in Longmont, Colorado (U.S.), which AGC purchased from Novartis in July 2021 or another US-based CMO.
Intellectual Property
Our goal is to obtain, maintain and enforce patent protection for our product candidates, formulations, processes, methods and any other proprietary technologies, preserve our trade secrets, and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. Our policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for our current product candidates and any future product candidates, proprietary information and proprietary technology through a combination of contractual arrangements and patents, both in the United States and abroad. However, patent protection may not afford us with complete protection against competitors who seek to circumvent our patents.
We also depend upon the skills, knowledge, experience and know-how of our management and research and development personnel, as well as that of our advisors, consultants and other contractors. To help protect our proprietary know-how, which is not patentable, and for inventions for which patents may be difficult to enforce, we currently rely and will in the future rely on trade secret protection and confidentiality agreements to protect our interests. To this end, we require all of our employees, consultants, advisors and other contractors to enter into confidentiality agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business.
| 115 |
Temferon
Temferon is protected by the following patents families that contain both issued and/or pending patent applications. While the following patent families are jointly owned by OSR and Fondazione Telethon (Telethon), as set forth in our December 15, 2014 license agreement with OSR (described below), Telethon granted OSR a worldwide exclusive license, with the right to sublicense, its rights in the patent families pursuant to a separate cooperation agreement between OSR and Telethon. As described below, we have a worldwide exclusive license, from OSR, to the following patent families (including the U.S. and European family members indicated) in the fields of: GBM, solid liver cancer (LC) and any lympho-hematopoietic indication:
| Focus / Family | U.S. | E.U. | Expiration | |||
Gene Vector comprising mi-RNA (composition and method of treatment claims) PCT/IB2006/002266 (WO 2007/000668). | USP 10,000,757* USP 9,556,438 USSN 16/004,394 (pending) | EP 2002003 B1 | 5/26/2026* | |||
Gene Vector comprising mi-RNA (composition and method of treatment claims) PCT/IB2010/001166 (WO / 2010/125471) | USP 10,287,579 USP 9,951,328 USSN 16/384,571 (pending) | EP 2424571 B1 | 4/30/2030 | |||
Monocyte Cell (Tie-2) activation process (composition claims) USP 7,833,789 | USP 7,833,789 | - | 10/5/2027 | |||
Methods for Genetic Modification of Stem Cells (method of treatment claims) PCT/IB2014/065594 (WO 2015/059674) | USP 10,617,721* USSN 16/827,708 (pending) | EP 3060670 B1 EP 19185334 (pending) | 10/24/2034* | |||
Vector Production (composition and method of treatment claims) PCT/IB2015/055286 (WO 2016/009326) | USP 10,912,824 17/143,953 (pending) | EP 3169788 (pending) | 7/13/2035 | |||
Type 1 Interferon Gene Therapy (method of treatment claims) PCT/EP2018/060238 (WO 2018/193119) | USSN 16/604,484 (pending) | EP 18724470.2 (pending) | 4/20/2038** |
* Later expiration for certain U.S. patents pursuant to patent term adjustment (35 U.S.C. §154(b)).
** Application pending, anticipated expiration based on 20 year patent term.
Our technology incorporates the use of a lentiviral vector (LVV) that combines a therapeutic transgene sequence, or payload, with our proprietary platform. Our proprietary platform consists of (i) the Tie-2 promoter, that drives transgene sequence transcription specifically in TEMs, and (ii) miRNA-126 target sequences to downregulate transgene expression post-transcription in those cells where the Tie-2 promoter is active and the miRNA-126 is present. Intellectual property protection for our proprietary platform includes an exclusive license to all issued patents and pending applications (if any) in the PCT/IB2006/002266 (WO 2007/000668) and PCT/IB2010/001166 (WO / 2010/125471) families, as well as trade secrets. As discussed below, we have the option to license exclusively USP 7,833,789 and issued patents and pending applications (in the PCT/IB2014/065594 and PCT/IB2015/055286 patent families we have not yet exercised the option rights), and improvements, for other indications (fields of use) and other product candidates.
In addition to patents and patent applications that we have been granted licenses to, we may also rely on unpatented trade secrets, know-how, and continuing technological innovation to develop and maintain our competitive position. We seek to protect know-how and trade secrets through an active program of legal mechanism including invention assignments, confidentiality agreements, material transfer agreements, research collaborations and licenses to protect our product candidates. These agreements may be breached, and we may not have adequate remedies for any breach. In addition, trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our employees, consultants, scientific advisors or other contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions. For a more comprehensive discussion of the risks related to our intellectual property, please see “Risk Factors — Risks Related to Our Intellectual Property.”
Collaboration / Licensing
License Agreement with Ospedale San Raffaele
Effective December 15, 2014, we entered into a license agreement with OSR (OSR License Agreement) pursuant to which OSR granted us an exclusive, royalty-bearing, non-transferrable (except with the prior written consent of OSR) worldwide license, subject to certain retained rights, to certain patents, patent applications and existing know-how for (1) the use in the field(s) of IFN gene therapy by lentiviral based-HSPC gene transfer with respect to (a) any Lympho-Hematopoietic Indication and/or (b) any Solid Cancer Indication that we exercise a future option (discussed below); and (2) certain products developed during the license term for use in the aforementioned field(s) consisting of any lentivirals vector regulated by miR126 and/or miR130 and/or other miRs with the same expression pattern as miR126 and miR130 in hematopoietic cells for the expression of IFN under the control of a Tie2 promoter. Lympho-Hematopoietic Indications mean any indication related to lympho-hematopoietic malignancies and Solid Cancer Indication means any solid cancer indication (e.g., without limitation, breast, pancreas, colon cancer), wherein each affected human organ being considered as a specific Solid Cancer Indication.
| 116 |
The rights retained by OSR, and extending to its affiliates, include the right to use the licensed technology for internal research within the field(s) of use, and the right to use the licensed technology for any use outside the field(s) of use, but subject to the options described below. In addition, we granted OSR a perpetual, worldwide, royalty-free, non-exclusive license to any improvement generated by us with respect to the licensed technology, to conduct internal research within the field of use directly, or in or with the collaboration third parties; and, for any use outside the field of use, in which case the license is sublicenseable by OSR. Finally, the world-wide rights for the field of use granted to us regarding the Lentigen know-how are non-exclusive and cannot be sublicensed due to a pre-existing nonexclusive sublicense to these rights between OSR and GlaxoSmithKline Intellectual Property Development Limited.
As stated, we have an exclusive option to (i) certain additional patents and patent applications, and OSR improvements at no additional cost, which could be useful for the development and/or commercialization of licensed products in the field of use; and (ii) any Solid Cancer Indication to be included as part of the field of use, on an indication-by-indication basis, subject to the payment of specified option fees and milestone payments.
As consideration, we paid OSR an upfront fee of €250,000, and we agreed to pay OSR royalties on a single digit percentage of the net sales of each licensed product. The royalty may be reduced upon the introduction of generic competition or patent stacking, but in no event would the royalty be less than half of what it would have otherwise been, but for the generic competition or patent stacking. We also agreed to pay OSR a royalty of our net sublicensing income for each licensed product and to pay OSR certain milestone payments upon the achievement of certain milestone events, such as the initiation of different phases of clinical trials of a licensed product, MAA approval by a major EU country, BLA acceptance by the FDA, the first commercial sale of a licensed product in the United States and certain EU countries, and achievement of certain net sales levels.
As part of the license, we agreed to use OSR as the primary site in any preclinical study or clinical trial (including all phases thereof) relating to any licensed products in the field of use, subject to OSR maintaining any required quality standards and providing its services on customary and reasonable terms and consistent with then-applicable market standards. We are also obligated to carry out our development activities using highly skilled professionals and sufficient level of resources and, specifically, to invest (a) at least €5,425,000 with respect to the development of the licensed products, and (b) at least €2,420,000 with respect to the manufacturing of such licensed products (subject to certain adjustments).
OSR maintains control of the preparation, prosecution and maintenance of the patents licensed. We are obligated to pay those costs unless additional licensees benefit from these rights, in which case the cost will be shared pro rata. OSR controls enforcement of the patents and know-how rights, at its own expense. In the event that OSR fails to file suit to enforce such rights after notice from Genenta, we have the right to enforce the licensed technology within the field of use. Both us and OSR must consent to settlement of any such litigation, and all monies recovered will be shared equally between the parties after reimbursement for costs, or failing a bona fide agreement between us and OSR, on a 50% - 50% basis.
The OSR License expires upon the expiry of the “Royalty Term” for all licensed products in all countries, unless terminated earlier. The Royalty Term begins on the first commercial sale of a licensed product and ends upon the later of the (a) expiration of the commercial exclusivity for such product (wherein the commercial exclusivity refers to any remaining valid licensed patent claims covering such licensed product, or any remaining regulatory data exclusivity for such licensed product), and (b) 10 years from the first commercial sale of such licensed product. The parties may terminate the agreement in the event the other party breaches its obligations therein, which termination shall become effective 60 business days following written notice thereof to the breaching party. The breaching party shall have the right to cure such breach or default during such 60 business days. OSR may terminate the agreement for failure to pay in the event that we fail to pay any of the upfront payment, sublicensing income or milestone payments within 30 days of due dates for each. In addition, OSR may terminate our rights as to certain fields of use for our failure to develop (a) with respect to a solid cancer indication, upon third anniversary of the date we exercised such option, if we have not filed an IND with respect to such optioned solid cancer indication specifically, as to GBM, we are required to file an IND regarding Temferon for GBM prior to February 2022, or (b) with respect to a lympho-hematopoietic indication, on the earlier of (i) the fifth anniversary of the initiation (first patient dosed) of the first human clinical trial for a licensed product in any lympho hematopoietic indication or solid cancer indication if a patient has not been dosed with a licensed product in a Phase 3 clinical trial and (ii) September 1, 2025.
In March 2017, the OSR License Agreement was amended to provide us with additional exclusive option to expand the license to include the use of lentiviral based-HSPC gene therapy platform to use tumor necrosis factor (TNF) as an alternative payload (rather than IFN) and for the use of that product alone or in combination with other products and committed to spend €500,000 on such product should the option be exercised. We have until September 30, 2022 to exercise this option, upon the payment of specified option fees, as further amended by the third amendment to the OSR License Agreement described below.
| 117 |
In February 2019, we entered into a second amendment to the OSR License Agreement, whereby we exercised an option with respect to GBM as the first solid cancer indication. We agreed to pay OSR a GBM option fee of €1.0 million upon the dosing of the tenth patient in a specified clinical trial of GBM. We also agreed to extend the period to exercise the option rights set forth by the license agreement until September 30, 2021. This date has been extended to September 30, 2022 pursuant to the fourth amendment to the to the OSR License Agreement described below.
In December 2020, we entered into a third amendment to the OSR License Agreement, whereby the GBM option fee included in the second amendment was reduced to €500,000 pursuant to an agreement to enter into a sponsored research agreement (SRA) within forty-five (45) days (and later amended to seventy (70) days) from the effective date of the third amendment (discussed below) in relation to research programs aimed at further studies regarding Temferon, with all intellectual property generated by such SRA being owned by OSR/Telethon subject to the grant by OSR to us of exclusive option rights with respect to such intellectual property. With the third amendment we also exercised an option with respect to a second solid cancer indication, namely solid liver cancer (LC). We agreed to pay OSR an LC option fee of €500,000 upon the earlier of (i) June 30, 2021 and (ii) the enrollment of the first patient within the Phase I clinical study for an LC licensed product. Under the terms of the third amendment, if we are unable to obtain regulatory approval to initiate human clinical trial with respect to solid liver cancer within nine (9) months from the third amendment effective date, we have the right, at no additional cost, to convert the option exercise for the second solid liver cancer indication to an alternative indication. In addition, pursuant to the third amendment, the option period for us to exercise an option with respect to any other solid cancer indication is extended until the second anniversary of the third amendment effective date, or December 23, 2022. The aggregate amount paid to date under the OSR License Agreement is €0.75 million. In addition, €0.5 million is currently due under the OSR License Agreement. Future potential payments that are not yet considered probable under this agreement include €53 million relating to GBM, €47.5 million relating to LC and €0.3 million relating to the license fee option for the third indication, if exercised. In addition, the OSR agreement was amended to extend the exclusive option as to Alternative Payload/Competing Product to September 30, 2022, as noted above.
In February 2021, we entered into a Sponsored Research Agreement (“SRA”) with OSR to conduct certain research projects related to Temferon. Unless terminated earlier or extended by mutual agreement, the SRA ends upon the earlier of (a) the date of completion of all activities relating to the sponsored research and (b) December 31, 2022. The total consideration to be paid by the Company under the SRA will be €1.0 million with payments scheduled quarterly over 2021 and 2022. The aggregate amount paid in 2021 to date under the SRA is €0.25 million.
In September 2021, the OSR License Agreement was amended to extend the exclusive option. Specifically, if Genenta is not able to obtain approval of the Regulatory Authorities to initiate a human clinical trial in any country with respect to solid liver cancer on or before September 30, 2022, then Genenta has the right, at no additional cost, to convert the option exercise for the second Solid Cancer Indication to an indication other than solid liver cancer. We now have until September 30, 2022 to exercise this option.
Know-How License Agreement with Fondazione Telethon
In February 2016, we entered into a Know-How License Agreement with Telethon (Telethon License Agreement). Telethon granted us a non-exclusive, perpetual, sublicensable (through multiple tiers), royalty-bearing, worldwide license to use its manufacturing know-how in the research and development, sale and export of any product, which is defined therein as any lentiviral vector regulated by miRNA 126 and/or miRNA 130 and/or other miRNAs with the same expression pattern as miRNA 126 and/or miRNA 130 in hematopoietic cells for the expression of any anticancer protein under the control of a Tie2 promoter or INF under the control of any promoter other than Tie2 for any cancer indication. As consideration for the license, we agreed to pay Telethon a royalty equal to a low single digit percentage of any actual payments (excluding taxes) to any CMO for the manufacturing of any product using the licensed know-how. The royalty payments must be made for eight (8) years from the effective date, or until February 2, 2024. The parties may terminate the agreement in the event the other party breaches its obligations therein, which termination shall become effective sixty (60) business days following written notice thereof to the breaching party.
Government Regulation; Coverage, Reimbursement, Pricing
Government authorities in the United States, at the federal, state and local levels, and in other countries and jurisdictions, including the EU, extensively regulate, among other things, the research, development, testing, manufacture, sales, pricing, reimbursement, quality control, approval, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, post-approval monitoring and reporting, and import and export of biopharmaceutical products. The processes for obtaining marketing approvals in the United States and in foreign countries and jurisdictions, along with compliance with applicable statutes and regulations and other regulatory authorities, require the expenditure of substantial time and financial resources.
Approval and Regulation of Drugs in the United States
In the United States, drug products are regulated by the FDA under the Federal Food, Drug, and Cosmetic Act (“FDCA”) and other laws, including, in the case of biologics, the Public Health Service Act (“PHSA”). Drug products are also subject to other federal, state and local statutes and regulations. A failure to comply with any applicable requirements during the product development, approval, or post-approval periods may lead to administrative or judicial sanctions, including, among other things, the imposition by the FDA or an institutional review board (“IRB”) of a hold on clinical trials, refusal to approve pending marketing applications or supplements, withdrawal of approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, or administrative, civil and/or criminal investigation, penalties or prosecution.
| 118 |
In the United States, all of our product candidates are regulated by the FDA as biologics. Biologics require the submission of a BLA and approval by the FDA prior to being marketed in the United States. Manufacturers of biologics may also be subject to state and local regulation.
The steps required before a biologic may be marketed in the United States generally include:
| ● | completion of preclinical studies, animal studies and formulation studies, performed in accordance with the FDA’s good laboratory practices (“GLP”) requirements, and applicable requirements for the humane use of laboratory animals or other applicable regulations; | |
| ● | submission to the FDA of an application for an investigational new drug application (“IND”), which must become effective before human clinical trials may commence; | |
| ● | approval of the protocol and related documentation by an independent IRB or ethics committee at each clinical trial site before each trial may be initiated; | |
| ● | performance of adequate and well-controlled clinical trials under protocols submitted to the FDA and reviewed and approved by each IRB, conducted in accordance with the FDA’s good clinical practices (“GCPs”) requirements and any additional requirements for the protection of human research subjects and their health information to establish the safety, purity and potency of the biologic for each targeted indication; | |
| ● | preparation of and submission to FDA of a biologics license application (“BLA”) for marketing approval that includes sufficient evidence of establishing the safety, purity, and potency of the proposed biological product for its intended indication, including from results of nonclinical testing and clinical trials; | |
| ● | a determination by the FDA within 60 days of its receipt of a BLA to accept and file the application; | |
| ● | satisfactory completion of an FDA pre-license inspection of the manufacturing facility or facilities where the biological product is produced to assess compliance with current good manufacturing practices (“cGMPs”) to assure that the facilities, methods and controls are adequate to preserve the biological product’s identity, strength, quality and purity; | |
| ● | satisfactory completion of an FDA Advisory Committee review, if applicable; | |
| ● | potential FDA audit of the preclinical study and clinical trial sites that generated the data in support of the BLA in accordance with any applicable expedited programs or designations; | |
| ● | payment of user fees for FDA review of the BLA (unless a fee waiver applies); | |
| ● | FDA review and approval, or licensure, of the BLA to permit commercial marketing of the product for particular indications for use in the U.S.; | |
| ● | satisfactory completion of an FDA inspection of the manufacturing facilities at which the biologic is produced to assess compliance with current Good Manufacturing Practices (“cGMP”) and to assure that the facilities, methods and controls are adequate; and | |
| ● | FDA review and approval of the BLA. |
| 119 |
Preclinical Studies and the IND Process
Before testing any biological product candidate in humans, the product candidate enters the preclinical testing stage. Preclinical studies include laboratory evaluation of a product’s biological characteristics, chemistry, formulation and toxicity, as well as animal studies to assess the characteristics and potential safety and efficacy of the product candidate. The conduct of the preclinical studies must comply with federal regulations and requirements, including, as applicable, GLP and the Animal Welfare Act.
Prior to commencing an initial clinical trial in humans with a product candidate in the U.S., an IND must be submitted to the FDA and the FDA must allow the IND to proceed. An IND is an exemption from the FD&C Act that allows an unapproved product candidate to be shipped in interstate commerce for use in an investigational clinical trial and a request for FDA allowance that such investigational product may be administered to humans in connection with such trial. Such authorization must be secured prior to interstate shipment and administration. In support of a request for an IND, the clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. An IND must become effective before human clinical trials may begin. Once submitted, the IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the IND on a full or partial clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial or part of the study can begin. Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial. The FDA also may impose clinical holds on a sponsor’s IND at any time before or during clinical trials due to, among other considerations, unreasonable or significant safety concerns, inability to assess safety concerns, lack of qualified investigators, a misleading or materially incomplete investigator brochure, study design deficiencies, interference with the conduct or completion of a study designed to be adequate and well-controlled for the same or another investigational product, insufficient quantities of investigational product, lack of effectiveness, or non-compliance. If the FDA imposes a clinical hold, studies may not recommence without FDA authorization and then only under terms authorized by the FDA.
Clinical Trials
Clinical trials involve the administration of the product candidate to healthy volunteers or patients under the supervision of qualified investigators. Clinical trials are subject to extensive regulation and must be conducted in compliance with (i) federal regulations, (ii) GCP standards, which set safeguards to protect the rights and health of patients and establish standards for conducting, recording data from, and reporting results of clinical trials, and (iii) protocols detailing, among other things, the objectives of the trial, subject selection and exclusion criteria, the parameters and criteria to be used in monitoring safety, and the effectiveness criteria to be evaluated, if any. Foreign studies conducted under an IND generally must meet the same requirements that apply to studies being conducted in the United States. The informed written consent of each study patient must be obtained before the patient may begin participation in the clinical trial. The study protocol, study plan, and informed consent forms for each clinical trial must be reviewed and approved by an IRB for each clinical site, and the study must be conducted under the auspices of an IRB for each trial site. Investigators and IRBs must also comply with FDA regulations and guidelines, including those regarding oversight of study patient informed consent, complying with the study protocol and investigational plan, adequately monitoring the clinical trial, and timely reporting of adverse events. An IRB can suspend or terminate approval of a clinical trial at its institution, or an institution it represents, if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the product candidate has been associated with unexpected serious harm to patients.
The clinical trial program for a product candidate is generally divided into three phases. Although the phases are usually conducted sequentially, they may overlap or be combined. The three phases are as follows:
| ● | Phase 1. Phase 1 involves the initial introduction of a product candidate into humans. Phase 1 clinical trials are typically conducted in healthy human subjects, but in some situations are conducted in patients with the target disease or condition. These clinical trials are generally designed to evaluate the safety, metabolism, pharmacokinetic (“PK”) properties and pharmacologic actions of the product candidate in humans, the side effects associated with increasing doses and, if possible, to gain early evidence of effectiveness. During Phase 1 clinical trials, sufficient information about the product candidate’s PK properties and pharmacological effects may be obtained to inform and support the design of Phase 2 clinical trials. |
| 120 |
| ● | Phase 2. Phase 2 includes the controlled clinical trials conducted to obtain initial evidence of effectiveness of the product candidate for a particular indication(s) in patients with the target disease or condition, to determine dosage tolerance and optimal dosage, and to gather additional information on possible adverse side effects and safety risks associated with the product candidate. Phase 2 clinical trials are typically well-controlled, closely monitored, and conducted in a limited patient population; and | |
| ● | Phase 3. Phase 3 clinical trials are clinical trials conducted in an expanded patient population at geographically dispersed clinical trial sites. They are performed after preliminary evidence suggesting effectiveness of the product candidate has been obtained and are intended to further evaluate dosage, clinical effectiveness and safety, to establish the overall benefit-risk relationship of the product candidate and to provide an adequate basis for regulatory approval. In most cases, the FDA requires two adequate and well controlled Phase 3 clinical trials to demonstrate the efficacy of the product candidate, although a single Phase 3 clinical trial with other confirmatory evidence may be sufficient in certain instances. |
In August 2018, the FDA released a draft guidance entitled “Expansion Cohorts: Use in First-In-Human Clinical Trials to Expedite Development of Oncology Drugs and Biologics,” which outlines how developers can utilize an adaptive trial design commonly referred to as a seamless trial design in early stages of oncology biological product development (i.e., the first-in-human clinical trial) to compress the traditional three phases of trials into one continuous trial called an expansion cohort trial. Information to support the design of individual expansion cohorts are included in IND applications and assessed by FDA. Expansion cohort trials can potentially bring efficiency to biological product development and reduce developmental costs and time.
In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product. These post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may also be made a condition to approval of the BLA. Failure to exhibit due diligence regarding conducting required Phase 4 clinical trials could result in withdrawal of approval for products.
Concurrent with clinical trials, companies usually complete additional animal studies and also must develop additional information about the chemistry and physical characteristics of the biological product as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. To help reduce the risk of the introduction of adventitious agents with use of biological products, the PHSA, emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final biological product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the biological product candidate does not undergo unacceptable deterioration over its shelf life.
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data, and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA and the investigators for serious and unexpected adverse events, any findings from other studies, tests in laboratory animals or in vitro testing that suggest a significant risk for human subjects, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information.
| 121 |
The decision to suspend or terminate development of a product candidate may be made by either a health authority body, such as the FDA, by an IRB, or by a company for various reasons and during any phase of clinical trials. The FDA may order the temporary or permanent discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. In some cases, clinical trials are overseen by a data safety monitoring board (“DSMB”), which is an independent group of qualified experts organized by the trial sponsor to evaluate at designated points in time whether or not a trial may move forward and/or should be modified. These decisions are based on unblinded access to data from the ongoing trial and generally involve determinations regarding the benefit-risk ratio for study patients and the scientific integrity and validity of the clinical trial.
In addition, there are requirements for the registration of certain clinical trials of product candidates on public registries, such as www.clinicaltrials.gov, and the submission of certain information pertaining to these trials, including clinical trial results, after trial completion.
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, a sponsor submits extensive information about the product candidate to the FDA in the form of a BLA to request marketing approval for the product candidate in specified indications.
Biologics License Applications
In order to obtain approval to market a biologic in the United States, a BLA must be submitted to the FDA that provides data establishing the safety and effectiveness of the product candidate for the proposed indication. The BLA includes all relevant data available from pertinent preclinical studies and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls and proposed labeling, among other things. Data can come from company-sponsored clinical trials intended to test the safety and effectiveness of a product candidate, or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and effectiveness of the product candidate to the satisfaction of the FDA.
Under the Prescription Drug User Fee Act (“PDUFA”), as amended, the fees payable to the FDA for reviewing an original BLA, as well as annual program fees for approved products, can be substantial, subject to certain limited deferrals, waivers and reductions that may be available. The FDA has 60 days from receipt of a BLA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. The FDA may refuse to accept for filing any BLA that it deems incomplete or not properly reviewable at the time of submission, in which case the BLA will have to be updated and resubmitted. The FDA’s PDUFA review goal (which is not a legal requirement) is to review 90% of priority BLA applications within six months of filing and 90% of standard applications within 10 months of filing, but the FDA can and frequently does extend this review timeline to consider certain later-submitted information or information intended to clarify or supplement information provided in the initial submission. The FDA may not complete its review or approve a BLA within these established goal review times. The FDA reviews the BLA to determine, among other things, whether the proposed product is safe, pure, and potent for its intended use, and whether the product is being manufactured in compliance with cGMP to ensure its continued safety, purity and potency. The FDA may also refer applications for novel product candidates which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving a BLA, the FDA will inspect the facilities at which the product is manufactured or the facilities that are significantly involved in the product development and distribution process, and will not approve the product candidate unless cGMP compliance is satisfactory and the manufacturing processes and facilities are adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP requirements.
Under the Pediatric Research Equity Act, certain BLAs must include an assessment, generally based on clinical trial data, of the safety and effectiveness of the biological product in relevant pediatric populations. The FDA may waive or defer the requirement for a pediatric assessment, either at a company’s request or by its own initiative, including waivers for certain products not likely to be used in a substantial number of pediatric patients. Unless otherwise required by regulation, products with orphan drug designation are exempt from these requirements for orphan-designated indications with no formal waiver process required.
| 122 |
After the FDA evaluates the BLA and the manufacturing facilities, the FDA may issue either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the BLA, the FDA may issue an approval letter. The FDA’s PDUFA review goal is to review such resubmissions within two or six months of receipt, depending on the type of information included. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval and deny approval of a resubmitted BLA. FDA approval of any application may include many delays or never be granted. An approval letter authorizes commercial marketing of the product candidate with specific prescribing information for specific indications. As a condition of BLA approval, the FDA may require a risk evaluation and mitigation strategy (“REMS”) to help ensure that the benefits of the product outweigh the potential risks. REMS can include Medication Guides, communication plans for healthcare professionals, and also may include elements to assure safe use (“ETASU”). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the biologic. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the biologic’s safety, purity, or potency, which can be costly.
Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new BLA or a supplemental BLA before the change can be implemented. A supplemental BLA for a new indication typically requires clinical data similar to that in the original application, and the FDA generally uses the same procedures and actions in reviewing a supplemental BLA as it does in reviewing a new BLA.
Product approvals may be withdrawn if compliance with regulatory standards is not maintained or if safety or manufacturing problems occur following initial marketing. For example, quality control and manufacturing procedures must conform, on an ongoing basis, to cGMP requirements, and the FDA periodically inspects manufacturing facilities to assess compliance with cGMP. Accordingly, manufacturers must continue to spend time, money and effort to maintain cGMP compliance. In addition, new or modified government requirements, including from new legislation, may be established that could delay or prevent regulatory approval of our product candidates under development or affect our ability to maintain product approvals we have obtained.
Regulation of Companion Diagnostics.
For drugs and therapeutic biologics where the use of a specific diagnostic test is essential for the safe and effective use of the therapeutic product, such as when the use of a product is limited to a specific patient subpopulation that can be identified by using the test, regulatory authorities may require, as a condition of approval, that a relevant “companion diagnostic” test also be approved or cleared for the appropriate indication. This general policy approach may be inapplicable in cases where the drug or therapeutic biologic is intended to treat a serious or life-threatening condition for which no satisfactory alternative treatment exists and the benefits from the use of a product with an unapproved companion diagnostic device are so pronounced as to outweigh the risks from the lack of an approved companion diagnostic. Companion diagnostics are generally regulated as medical devices by regulatory authorities and relevant statutes and regulations govern, among other things, medical device design and development, preclinical and clinical testing, premarket regulatory review, manufacturing, labeling, storage, advertising and promotion, sales and distribution, export and import, and post-market surveillance.
Orphan Drug Designation
The FDA may grant orphan drug designation to biologics intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the United States or a disease or condition that affects 200,000 or more individuals in the United States but there is no reasonable expectation that the cost of developing and making the biologic would be recovered from sales in the United States. Orphan drug designation must be requested before submitting a BLA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
In the United States, orphan drug designation entitles a party to financial incentives, such as opportunities for grant funding towards clinical trial costs, tax credits for certain research and user fee waivers under certain circumstances. In addition, if a company receives the first FDA approval of a drug or biologic for the indication for which it has orphan drug designation, the product is entitled to seven years of orphan exclusivity, which means the FDA may not approve any other application for the “same” drug or biologic for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority over the product with orphan exclusivity. The FDA can revoke a product’s orphan drug exclusivity under certain circumstances, including when the product sponsor is unable to assure the availability of sufficient quantities of the product to meet patient needs. Orphan drug exclusivity does not prevent the FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition.
| 123 |
In the EEA, the criteria for designating an “orphan medicinal product” are similar in principle to those in the United States. Under Article 3 of Regulation (EC) 141/2000, a medicinal product may be designated as an orphan medicinal product if it meets the following criteria: (a) it is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating conditions; (b) either such condition that affects no more than five in 10,000 people in the E.U.; or without the benefits derived from orphan status, it must be unlikely that the marketing of the medicine would generate sufficient return in the E.U. to justify the investment needed for its development; and, (c) there exists no satisfactory method of diagnosis, prevention or treatment of the condition concerned, or, if such a method exists, the medicinal product would be of significant benefit to those affected by the condition. The application for orphan designation must be submitted to the EMA and approved by the European Commission before an application is made for marketing authorization for the product. Once designated, orphan medicinal product designation also entitles a party to financial incentives such as reduction of fees or fee waivers. Moreover, ten years of market exclusivity is granted, if the product continues to be designated as an orphan medicinal product upon grant of the marketing authorization. During this ten-year period, with a limited number of exceptions, neither the competent authorities of the E.U. Member States, the EMA, or the European Commission are permitted to accept applications or grant marketing authorization for other similar medicinal products with the same therapeutic indication. A “similar medicinal product” is defined as a medicinal product containing a similar active substance or substances as contained in an authorized orphan medicinal product, and which is intended for the same therapeutic indication. However, marketing authorization may be granted to a similar medicinal product with the same orphan indication during the ten-year period with the consent of the marketing authorization holder for the original orphan medicinal product or if the manufacturer of the original orphan medicinal product is unable to supply sufficient quantities. Marketing authorization may also be granted to a similar medicinal product with the same orphan indication if this latter product is demonstrated to be safer, more effective or otherwise clinically superior to the original orphan medicinal product. This period of market exclusivity may be reduced to six years if, at the end of the fifth year, it is established that the orphan designation criteria are no longer met, including where it can be demonstrated on the basis of available evidence that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Expedited Programs in the United States and Other Jurisdictions
In the United States, the FDA has various programs, including fast track designation, breakthrough therapy designation, accelerated approval and priority review, that are intended to expedite or simplify the process for the development and FDA review of drugs and biologics that are intended for the treatment of serious or life-threatening diseases or conditions. A product may be granted fast track designation if it is intended for the treatment of a serious or life-threatening condition and demonstrates the potential to address unmet medical needs for such condition. Fast track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug or biologic may request the FDA to designate the drug or biologic as a fast track product at any time during the clinical development of the product. With fast track designation, the sponsor may be eligible for more frequent opportunities to obtain the FDA’s feedback. Another benefit of fast track designation, for example, is that the FDA may consider for review sections of the marketing application on a rolling basis before the complete application is submitted if certain conditions are satisfied, including an agreement with the FDA on the proposed schedule for submission of portions of the application and the payment of applicable user fees before the FDA may initiate a review. Even if a product candidate receives fast track designation, the designation can be rescinded and provides no assurance that a product will be reviewed or approved more expeditiously than would otherwise have been the case, or that the product will be approved at all.
Under the FDA’s breakthrough therapy program, a sponsor may seek FDA designation of its product candidate as a breakthrough therapy if the product candidate is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that it may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Breakthrough therapy designation comes with all of the benefits of fast-track designation. The FDA may take other actions appropriate to expedite the development and review of the product candidate, including holding meetings with the sponsor and providing timely advice to, and interactive communication with, the sponsor regarding the development program. Even if one or more of our product candidates receives breakthrough therapy designation, the designation can be rescinded and provides no assurance that a product will be reviewed or approved more expeditiously than would otherwise have been the case, or that the product will be approved at all.
A product candidate may be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical trials establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on an intermediate clinical endpoint other than survival or irreversible morbidity or mortality, that is reasonably likely to predict irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA generally requires that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials to verify the clinical benefit in relationship to the surrogate endpoint or ultimate outcome in relationship to the clinical benefit. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. The FDA may withdraw approval of a drug or indication approved under accelerated approval if, for example, the confirmatory trial fails to verify the predicted clinical benefit of the product.
| 124 |
The FDA may grant priority review designation to a product candidate, which sets the user fee target date for FDA action on the application at six months from FDA filing. Priority review may be granted where a product is intended to treat a serious or life-threatening disease or condition and, if approved, has the potential to provide a safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in safety or efficacy compared to available therapy. If criteria are not met for priority review, the standard FDA review period is ten months from FDA filing. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Under the 21st Century Cures Act, a drug is eligible for regenerative medicine advanced therapy (“RMAT”) designation if (i) the drug is a regenerative medicine therapy, which is defined by FDA to include cell therapy, therapeutic tissue engineering product, human cell and tissue product, any combination product using such therapies or products, and certain human gene therapies and xenogeneic cell products, except for human cells, tissues, and cellular and tissue-based products (“HCT/P’s”) that are regulated solely under Section 361 of the Public Health Service Act and part 1271 of Title 21, Code of Federal Regulations; (ii) the drug is intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition; and (iii) preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such disease or condition. An RMAT designation include all the benefits of the fast track and breakthrough therapy designation programs, including early interactions with FDA, and the potential to support accelerated approval and address post-approval requirements. An RMAT designation request should be submitted with the IND or after and, ideally, no later than the end-of-phase 2 meeting. Even if a product candidate receives RMAT designation, the designation can be rescinded and provides no assurance that a product will be reviewed or approved more expeditiously than would otherwise have been the case, or that the product will be approved at all.
Under the centralized procedure in the EEA, the maximum timeframe for the evaluation of a marketing authorization application is 210 days (excluding ‘‘clock stops,’’ when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP). Clock stops may extend the timeframe of evaluation of a marketing authorization application considerably beyond 210 days. Accelerated evaluation might be granted by CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest, which should be justified and assessed on a case-by-case basis. In this circumstance, EMA ensures that the opinion of CHMP is given within 150 days (excluding clock stops).
| 125 |
Expanded Access to an Investigational Drug for Treatment Use
Expanded access, also called “compassionate use,” is the use of investigational new drug products outside of clinical trials to treat patients with serious or immediately life-threatening diseases or conditions when there are no comparable or satisfactory alternative treatment options. The FDA regulations allow access to investigational drugs under an IND by the sponsor or the treating physician for treatment purposes on a case-by-case basis for individual patients, intermediate-size patient populations, and larger populations for use of the drug under a treatment protocol or Treatment IND Application.
The suitability of treating a patient or a group of patients under expanded access is determined by the following: if patient(s) have a serious or immediately life-threatening disease or condition, there is no comparable or satisfactory alternative therapy to diagnose, monitor, or treat the disease or condition, the potential patient benefit justifies the potential risks of the treatment and the potential risks are not unreasonable in the context or condition to be treated, and the expanded use of the investigational drug for the requested treatment will not interfere with the initiation, conduct or completion of clinical investigations that could support marketing approval of the product candidate or otherwise compromise the potential development of the product candidate.
Sponsors of one or more investigational drugs for the treatment of a serious disease(s) or condition(s) make publicly available their policies for evaluating and responding to requests for expanded access for individual patients. This provision requires drug companies to make publicly available their policies for expanded access for individual patient access to products intended for serious diseases. Sponsors are required to make such policies publicly available upon the earlier of initiation of a Phase 2 or Phase 3 study or 15 days after the drug receives breakthrough therapy, fast track, or regenerative medicine advanced therapy designation. Additionally, in 2018 the Right to Try Act, was signed into law. The law, among other things, provides a federal framework for certain patients to access certain investigational new drug products that have completed a Phase 1 clinical trial and that are undergoing investigation for FDA approval. Under certain circumstances, eligible patients can seek treatment without enrolling in clinical trials and without obtaining FDA permission under the FDA expanded access program. There is no obligation for a drug manufacturer to make its drug products available to eligible patients as a result of the Right to Try Act.
U.S. market exclusivity
A biological product can obtain pediatric market exclusivity in the U.S. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods, including some regulatory exclusivity periods tied to patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study.
The Biologics Price Competition and Innovation Act of 2009 (“BPCIA”), created an abbreviated approval pathway for biological products shown to be biosimilar to, or interchangeable with, an FDA-licensed reference biological product. This amendment to the PHSA attempts to minimize duplicative testing. Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, can be shown through analytical studies, animal studies, and a clinical trial or trials. Interchangeability requires that a product is biosimilar to the reference product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product and, for products administered multiple times, the biologic and the reference biologic may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. However, complexities associated with the larger, and often more complex, structure of biological products, as well as the process by which such products are manufactured, pose significant hurdles to implementation that are still being worked out by the FDA.
| 126 |
FDA will not accept an application for a biosimilar or interchangeable product based on the reference biological product until four years after the date of first licensure of the reference product, and FDA will not approve an application for a biosimilar or interchangeable product based on the reference biological product until 12 years after the date of first licensure of the reference product. “First licensure” typically means the initial date the particular product at issue was licensed in the U.S. Date of first licensure does not include the date of licensure of (and a new period of exclusivity is not available for) a biological product if the licensure is for a supplement for the biological product or for a subsequent application by the same sponsor or manufacturer of the biological product (or licensor, predecessor in interest, or other related entity) for a change (not including a modification to the structure of the biological product) that results in a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device or strength, or for a modification to the structure of the biological product that does not result in a change in safety, purity, or potency. The BPCIA is complex and continues to be interpreted and implemented by the FDA. In addition, government proposals have sought to reduce the 12-year reference product exclusivity period. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. As a result, the ultimate implementation and impact of the BPCIA is subject to significant uncertainty.
Post-approval requirements
Rigorous and extensive FDA regulation of biological products continues after approval, particularly with respect to cGMP requirements, as well as requirements relating to record keeping, reporting of adverse experiences, periodic reporting, product sampling and distribution, and advertising and promotion of the product. Manufacturers of products are required to comply with applicable requirements in the cGMP regulations, including quality control and quality assurance and maintenance of records and documentation. Other post-approval requirements applicable to biological products, include reporting of cGMP deviations that may affect the identity, potency, purity and overall safety of a distributed product, record keeping requirements, reporting of adverse effects, reporting updated safety and efficacy information, and complying with electronic record and signature requirements. After a BLA is approved, the product also may be subject to official lot release. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release by the FDA, the manufacturer submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot. The FDA also may perform certain confirmatory tests on lots of some products before releasing the lots for distribution by the manufacturer. In addition, the FDA conducts laboratory research related to the regulatory standards on the safety, purity, potency, and effectiveness of biological products.
As a condition of BLA approval, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization. Regulatory approval of oncology products often requires that patients in clinical trials be followed for long periods to determine the overall survival benefit of the product. In addition, as a holder of an approved BLA, a company would be required to report adverse reactions and production problems to the FDA, to provide updated safety and efficacy information, and to comply with requirements concerning advertising and promotional labeling for any of its products.
Manufacturers must comply with the FDA’s advertising and promotion requirements, such as those related to direct-to-consumer advertising, the prohibition on promoting products for uses or in patient populations that are not described in the product’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities, and promotional activities involving the internet. Discovery of previously unknown problems with a product or failure to comply with applicable regulatory requirements after approval may result in serious and extensive restrictions on a product or the manufacturer or holder of an approved BLA, as well as lead to potential market disruptions. These restrictions may include suspension of product manufacturing until the FDA is assured that quality standards can be met, continuing oversight of manufacturing by the FDA under a ‘‘consent decree,’’ which frequently includes the imposition of costs and continuing inspections over a period of many years, as well as possible withdrawal of the product from the market. Other potential consequences include interruption of production, issuance of warning letters or other enforcement letters, refusal to approve pending BLAs or supplements to approved BLAs, product seizure or detention, product recalls, fines, and injunctions or imposition of civil and/or criminal penalties.
In addition, changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation, correction, and reporting of any deviations from cGMP and impose reporting and documentation requirements upon a company and any third-party manufacturers that a company may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
Biological product manufacturers and other entities involved in the manufacture and distribution of approved biological products are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance.
Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures, such as additional post-market clinical trials to assess new safety risks or distribution-related or other restrictions under a REMS. In addition, changes to the manufacturing process or facility generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
| 127 |
Patent Term Restoration and Extension
Depending upon the timing, duration and specifics of the FDA approval of our product candidates, some of our U.S. patents may be eligible for limited patent term extension. The provisions of the Drug Price Competition and Patent Term Restoration Act, informally known as the Hatch-Waxman Act, permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of a BLA plus the time between the submission date of a BLA and the approval of that application. Only one patent applicable to an approved product is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The United States Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we may apply for restoration of patent term for one of our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant BLA.
Many other countries also provide for patent term extensions or similar extensions of patent protection for biologic products. For example, in Japan, it may be possible to extend the patent term for up to five years and in Europe, it may be possible to obtain a supplementary patent certificate that would effectively extend patent protection for up to five years.
Health Care Laws and Regulations
Health care providers and third-party payors play a primary role in the recommendation and prescription of drug products that are granted marketing approval. Arrangements with providers, consultants, third-party payors and customers are subject to broadly applicable fraud and abuse, anti-kickback, false claims laws, patient privacy laws and regulations and other health care laws and regulations that may constrain business and/or financial arrangements. Restrictions under applicable federal and state health care laws and regulations include the following:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, paying, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made, in whole or in part, under a federal health care program such as Medicare and Medicaid; | |
| ● | the federal civil and criminal false claims laws, including the civil False Claims Act, and civil monetary penalties laws, which prohibit individuals or entities from, among other things, knowingly presenting, or causing to be presented, directly or indirectly, to the federal government, claims for payment that are false, fictitious or fraudulent or knowingly making, using or causing to made or used a false record or statement to avoid, decrease or conceal an obligation to pay money to the federal government; | |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal laws that prohibit, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any health care benefit program or making false statements relating to health care matters; | |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, and their respective implementing regulations, including the Final Omnibus Rule published in January 2013, which impose obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; | |
| ● | the federal false statements statute, which prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for health care benefits, items or services; |
| 128 |
| ● | the FCPA which prohibits companies and their intermediaries from making, or offering or promising to make improper payments to non-U.S. officials for the purpose of obtaining or retaining business or otherwise seeking favorable treatment; | |
| ● | the federal transparency requirements known as the federal Physician Payments Sunshine Act, under the PPACA, which requires certain manufacturers of drugs, devices, biologics and medical supplies to report annually to the Centers for Medicare & Medicaid Services within the United States Department of Health and Human Services, information related to payments and other transfers of value made by that entity to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; and | |
| ● | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to health care items or services that are reimbursed by non-government third-party payors, including private insurers. |
Further, some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring manufacturers to report information related to payments to physicians and other health care providers or marketing expenditures. Additionally, some state and local laws require the registration of pharmaceutical sales representatives in the jurisdiction. State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Pharmaceutical Insurance Coverage and Health Care Reform
In the United States and markets in other countries, patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated health care costs. Significant uncertainty exists as to the coverage and reimbursement status of drug products approved by the FDA and other government authorities. Thus, even if a product candidate is approved, sales of such product will depend, in part, on the extent to which third-party payors, including government health programs in the United States such as Medicare and Medicaid, commercial health insurers and managed care organizations, provide coverage and establish adequate reimbursement levels for, the product. The process for determining whether a payor will provide coverage for a drug product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the drug product once coverage is approved. Third-party payors are increasingly challenging the prices charged, examining the medical necessity and reviewing the cost-effectiveness of medical products and services and imposing controls to manage costs. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the approved products for a particular indication.
In order to secure coverage and reimbursement for any product candidate that might be approved for sale, a company may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product, in addition to the costs required to obtain FDA or other comparable marketing approvals. Nonetheless, product candidates may not be considered medically necessary or cost effective. A decision by a third-party payor not to cover a product candidate could reduce physician utilization once the product candidate is approved. Additionally, a payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage and reimbursement for the product, and the level of coverage and reimbursement can differ significantly from payor to payor.
The containment of health care costs also has become a priority of federal, state and foreign governments and the prices of products have been a focus in this effort. Governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic drug products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit a company’s revenue generated from the sale of any approved drug products. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more drug products for which a company or its collaborators receive marketing approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
| 129 |
There have been a number of federal and state proposals during the last few years regarding the pricing of pharmaceutical and biopharmaceutical products, limiting coverage and reimbursement for drugs and biologics and other medical products, government control and other changes to the health care system in the United States.
In March 2010, the United States Congress enacted the PPACA, which, among other things, included changes to the coverage and payment for drug products under government health care programs. The PPACA effected the following changes of importance to our potential product candidates:
| ● | established an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs; | |
| ● | expanded eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a manufacturer’s Medicaid rebate liability; | |
| ● | expanded manufacturers’ rebate liability under the Medicaid Drug Rebate Program by increasing the minimum rebate for both branded and generic drugs and revising the definition of “average manufacturer price” for calculating and reporting Medicaid drug rebates on outpatient prescription drug prices; | |
| ● | addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; | |
| ● | expanded the types of entities eligible for the 340B drug discount program; | |
| ● | established the Medicare Part D coverage gap discount program by requiring manufacturers to provide a 70% point-of-sale discount off of the negotiated price of applicable brand drugs to eligible beneficiaries during their coverage gap period as a condition for the manufacturers’ outpatient drugs to be covered under Medicare Part D; and | |
| ● | established a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
Other legislative changes have been proposed and adopted in the United States since the PPACA was enacted. In August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This included aggregate reductions of Medicare payments to providers up to 2% per fiscal year, which went into effect in April 2013 and, due to subsequent legislative amendments, will remain in effect through 2027 unless additional congressional action is taken. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Since enactment of the PPACA, there have been numerous legal challenges and congressional actions to repeal and replace provisions of the law. For example, with enactment of the TCJA, which was signed by the President on December 22, 2017, Congress repealed the “individual mandate.” The repeal of this provision, which requires most Americans to carry a minimal level of health insurance, will become effective in 2019. According to the Congressional Budget Office, the repeal of the individual mandate will cause 13 million fewer Americans to be insured in 2027 and premiums in insurance markets may rise. Additionally, on January 22, 2018, the President signed a continuing resolution on appropriations for fiscal year 2018 that delayed the implementation of certain PPACA-mandated fees, including the so-called “Cadillac” tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share, and the medical device excise tax on non-exempt medical devices. Further, the Bipartisan Budget Act of 2018, among other things, amended the PPACA, effective January 1, 2019, to increase from 50% to 70% the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D and to close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole”. Congress will likely consider other legislation to replace elements of the PPACA during the next congressional session.
| 130 |
The presidential administration has also taken executive actions to undermine or delay implementation of the PPACA. In January 2017, the President signed an Executive Order directing federal agencies with authorities and responsibilities under the PPACA to waive, defer, grant exemptions from, or delay the implementation of any provision of the PPACA that would impose a fiscal or regulatory burden on states, individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. In October 2017, the President signed a second Executive Order allowing for the use of association health plans and short-term health insurance, which may provide fewer health benefits than the plans sold through the PPACA exchanges. At the same time, the presidential administration announced that it will discontinue the payment of cost-sharing reduction, or CSR, payments to insurance companies until Congress approves the appropriation of funds for such CSR payments. The loss of CSR payments is expected to increase premiums on certain policies issued by qualified health plans under the PPACA. A bipartisan bill to appropriate funds for CSR payments was introduced in the Senate, but the future of that bill is uncertain. In addition, the Centers for Medicare & Medicaid Services has recently proposed regulations that would give states greater flexibility in setting benchmarks for insurers in the individual and small group marketplaces, which may have the effect of relaxing the essential health benefits required under the PPACA for plans sold through such marketplaces.
Further, there have been several recent U.S. congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the costs of drugs under Medicare and reform government program reimbursement methodologies for drug products. At the federal level, the presidential administration’s budget proposal for fiscal year 2019 contained further drug price control measures that could be enacted during the 2019 budget process or in other future legislation, including, for example, measures to permit Medicare Part D plans to negotiate the price of certain drugs under Medicare Part B, to allow some states to negotiate drug prices under Medicaid, and to eliminate cost sharing for generic drugs for low-income patients. While any proposed measures will require authorization through additional legislation to become effective, Congress and the presidential administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs.
At the state level, individual states are increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition, regional health care authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other health care programs.
Review and Approval of Medicinal Products in Europe
In order to market any medicinal product outside of the United States, a company must also comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of medicinal products. Whether or not it obtains FDA approval for a product candidate, an applicant will need to obtain the necessary approvals by the comparable non-U.S. regulatory authorities before it can commence clinical trials or marketing of the product in those countries or jurisdictions. Some countries outside of the United States have a similar process that requires the submission of a CTA much like the IND prior to the commencement of human clinical trials. In the E.U., for example, a CTA must be submitted to the national competent authorities of the E.U. Member States where the clinical trial is conducted and to an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed.
In April 2014, the E.U. adopted a new Clinical Trials Regulation (EU) No 536/2014, which is set to replace the current Clinical Trials Directive 2001/20/EC. It will overhaul the current system of approvals for clinical trials in the E.U. Specifically, the new regulation, which will be directly applicable in all Member States (meaning that no national implementing legislation in each E.U. Member State is required), aims at simplifying and streamlining the approval of clinical trials in the E.U. For instance, the new Clinical Trials Regulation provides for a streamlined application procedure via a single entry point and strictly defined deadlines for the assessment of clinical trial applications. It is expected that the new Clinical Trials Regulation will come into effect following confirmation of full functionality of the Clinical Trials Information System, the centralized EU portal and database for clinical trials foreseen by the new Clinical Trials regulation, through an independent audit, which is currently expected to occur in December 2021.
| 131 |
Clinical Trial Regulations in Italy
Under the E.U. and EU-member country legislation, any application for marketing authorization must be accompanied by the results of clinical trials conducted in accordance with applicable regulations. A unified regulation on clinical trial procedures has been approved (EU Reg. 536/2014), but is not yet effective. The currently applicable rule is EU Directive 2001/20, as implemented in the various EU member countries from time to time through national laws and regulations.
We are currently conducting or planning Phase I/II clinical trials on Temferon in Italy, in accordance with the specific regulations applicable to such early-phase trials. As discussed elsewhere in this prospectus, we are currently conducting our TEM-GBM 001 study on UMGMT-GBM patients and plan to start new ones on hepatic tumor (HCC and IHC) in the near future by means of appropriate application to the Italian Regulatory Authority (“AIFA”).
The applicable Italian regulation is the Decree of April 27, 2015 of the Ministry of Health, providing a precise sequence of approvals for the start of Phase I studies and subsequent amendments to the related protocols. According to such Decree, an initial request must be submitted to AIFA seeking a technical-scientific opinion of Istituto Superiore di Sanità (ISS), acting on behalf of AIFA, on the admissibility of the request. Upon the favorable opinion of ISS, AIFA issues an authorization to proceed with the planned study, and the rules generally governing the conduct of clinical trials (Legislative Decree 211 of June 24, 2003, implementing in Italy EU Directive 2001/20, Decree of December 17, 2004 of the Ministry of Health for non-profit studies, plus procedural rules such as the Decree of December 21, 2007, so called “CTA decree”, for the prescribed formats), are of application.
Based on the AIFA approval, the Independent Ethics Committees (“IECs”) of the research centers participating in the trial issue their opinions on the conduct of the study, having evaluated the study protocol and all other relevant documentation such as the informed consent form (“ICF”), the insurance policy underwritten by the sponsor, the information and consent form for data protection purposes. The IEC of the Coordinating Center issues first its opinion – the so-called Parere Unico, lit. “sole opinion” (“PU”) - and then the IECs of the other participating centers accept or refuse the PU in its entirety (they may seek amendment to the ICF on the basis of local operating circumstances).
All documents pertaining to each specific step of the procedure, in the right sequence, must be loaded on the online database of Aifa (“Osservatorio sulle Sperimentazioni Cliniche”, or OsSC); the OsSC system provides certain controls to make sure that e.g. no IEC opinion can be loaded before the pertinent AIFA approval, or that the opinions of the participating sites cannot be loaded before the PU is loaded. It may occur however that, due to calendar mismatches in the calendars of IEC meeting (usually held on a monthly basis), an approval may precede by a few days a “prior” one (typically, the PU or the AIFA approval): in such cases the IEC approval is issued under reservation (“con riserva”) and can be loaded in advance accordingly, under the assumption that the documents subjected to evaluation - protocol (updated) version, ICF and the rest – coincide exactly.
Marketing Authorization Application for Biologic Medicinal Products
To obtain regulatory approval to commercialize a new drug in the EEA (comprising the E.U. Member States plus Iceland, Liechtenstein and Norway, we must submit a marketing authorization application.
In the E.U., a marketing authorization for a medicinal product can be obtained through a centralized, mutual recognition, decentralized procedure, or national procedure (single country). The centralized procedure is mandatory for certain medicinal products, including orphan medicinal products, those produced by biotechnology, advanced therapy medicinal products (gene therapy, somatic cell therapy and tissue-engineered products) and those with a new active substance indicated for the treatment of HIV, AIDS, cancer, neurodegenerative disorders, autoimmune and other immune dysfunctions, viral diseases or diabetes, and is optional for certain other products, including medicinal products with a new active substance for other indications, and products that are a significant therapeutic, scientific or technical innovation, or whose authorization would be in the interest of public health.
| 132 |
Under the centralized procedure, the applicant can submit a single application for marketing authorization to the EMA which will provide a positive opinion regarding the application if it meets certain quality, safety, and efficacy requirements. Based on the opinion of the EMA, the European Commission takes a final decision to grant a centralized marketing authorization which permits the marketing of a product throughout the EEA. Under the centralized procedure, the maximum timeframe for the evaluation of a marketing authorization application is 210 days (excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the EMA Committee for Medicinal Products for Human Use (“CHMP”)). Clock stops may extend the timeframe of evaluation of a marketing authorization application considerably beyond 210 days. Where the CHMP gives a positive opinion, it provides the opinion together with supporting documentation to the European Commission, who make the final decision to grant a marketing authorization, which is issued within 67 days of receipt of the EMA’s recommendation. Accelerated assessment might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of major public health interest, particularly from the point of view of therapeutic innovation. The timeframe for the evaluation of a marketing authorization application under the accelerated assessment procedure is 150 days, excluding clock stops, but it is possible that the CHMP may revert to the standard time limit for the centralized procedure if it determines that it is no longer appropriate to conduct an accelerated assessment.
Now that the United Kingdom (which comprises Great Britain and Northern Ireland) has left the E.U., Great Britain will no longer be covered by centralized marketing authorizations (under the Northern Irish Protocol, centralized marketing authorizations will continue to be recognized in Northern Ireland). All medicinal products with a current centralized marketing authorization were automatically converted to Great Britain marketing authorizations on January, 1 2021. For a period of two years from January 1, 2021, the Medicines and Healthcare products Regulatory Agency, or MHRA, the United Kingdom medicines regulator, may rely on a decision taken by the European Commission on the approval of a new marketing authorization in the centralized procedure, in order to more quickly grant a new Great Britain marketing authorization. A separate application will, however, still be required.
For other countries outside of the E.U., such as the United Kingdom and countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. Internationally, clinical trials are generally required to be conducted in accordance with cGCPs, applicable regulatory requirements of each jurisdiction and the medical ethics principles that have their origin in the Declaration of Helsinki. If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Advertising, Promotion and Compliance
In the E.U., the advertising and promotion of our products will also be subject to E.U. laws and E.U. Member States’ national laws governing promotion of medicinal products, interactions with physicians, misleading and comparative advertising and unfair commercial practices. These laws require that promotional materials and advertising in relation to medicinal products comply with the product’s Summary of Product Characteristics (“SmPC”), as approved by the competent authorities. The SmPC is the document that provides information to physicians concerning the safe and effective use of the medicinal product. The SmPC forms an intrinsic and integral part of the marketing authorization granted for the medicinal product. Promotion of a medicinal product that does not comply with the SmPC is considered to constitute off-label promotion and is prohibited in the E.U. The applicable laws at the E.U. level and in the individual E.U. Member States also prohibit the direct-to-consumer advertising of prescription-only medicinal products. Violations of the rules governing the promotion of medicinal products in the E.U. could be penalized by administrative measures, fines and imprisonment. As the United Kingdom medicinal products legislation is still largely based on EU legislation, the promotion of prescription-only medicines to the public and promotion of medicinal products not in compliance with the SmPC are both also prohibited under United Kingdom law.
During all phases of development (pre- and post-marketing), failure to comply with applicable regulatory requirements may result in administrative or judicial sanctions. These penalties could include the imposition of a clinical hold on trials, refusal to approve pending applications, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, product detention or refusal to permit the import or export of products, injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.
Regulatory Data Protection in the EEA
In the EEA, innovative medicinal products approved on the basis of a complete independent data package qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity pursuant to Directive 2001/83/EC. Regulation (EC) No 726/2004 repeats this entitlement for medicinal products authorized in accordance the centralized authorization procedure. Data exclusivity prevents applicants for authorization of generics or biosimilars of these innovative products from referencing the innovator’s data when applying for a generic or biosimilar marketing authorization for a period of eight years from the date on which the innovator’s product was first authorized in the EEA. During an additional two-year period of market exclusivity, a generic or biosimilar marketing authorization can be submitted and authorized, and the innovator’s data may be referenced, but no generic or biosimilar medicinal product can be placed on the EU market until the expiration of the market exclusivity. The overall 10-year period will be extended to a maximum of 11 years if, during the first eight years of those 10 years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. Even if an innovative medicinal product gains the prescribed period of data exclusivity, another company may market another version of the product if such company obtained a marketing authorization based on a marketing authorization application with a complete independent data package of pharmaceutical tests, preclinical tests and clinical trials.
| 133 |
Periods of Authorization and Renewals
A marketing authorization has an initial validity for five years in principle. The marketing authorization may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance by the EMA or by the competent authority of the relevant Member State. To this end, the marketing authorization holder must provide the EMA or the competent authority with a consolidated version of the file in respect of quality, safety and efficacy, including all variations introduced since the marketing authorization was granted, at least six months before the marketing authorization ceases to be valid. The European Commission or the competent authorities of the EEA Member States may decide, on justified grounds relating to pharmacovigilance, to proceed with one further five-year period of marketing authorization. Once subsequently definitively renewed, the marketing authorization shall be valid for an unlimited period. Any authorization which is not followed by the actual placing of the medicinal product on the EEA market (in case of centralized procedure) or on the market of the authorizing EEA Member State within three years after authorization ceases to be valid (the so-called sunset clause).
Pediatric Studies and Exclusivity
Prior to obtaining a marketing authorization in the EEA, applicants must demonstrate compliance with all measures included in an EMA-approved PIP covering all subsets of the pediatric population, unless the EMA has granted a product-specific waiver, a class waiver, or a deferral for one or more of the measures included in the PIP. The respective requirements for all marketing authorization procedures are laid down in Regulation (EC) No 1901/2006, commonly referred to as the Pediatric Regulation. This requirement also applies when a company wants to add a new indication, pharmaceutical form or route of administration for a medicine that is already authorized. The Pediatric Committee of the EMA, or PDCO, may grant deferrals for some medicines, allowing a company to delay development of the medicine for children until there is enough information to demonstrate its effectiveness and safety in adults. The PDCO may also grant waivers when development of a medicine for children is not needed or is not appropriate, such as for diseases that only affect an adult population. Before a marketing authorization application can be filed, or an existing marketing authorization can be amended, the EMA must determine that a company actually complied with the agreed studies and measures listed in each relevant PIP, unless the EMA has granted: (i) a product-specific waiver, (ii) a class waiver or (iii) a deferral for one or more of the measures included in the PIP. If an applicant obtains a marketing authorization in all EEA Member States, or a marketing authorization granted in the centralized procedure by the European Commission, and the study results of the pediatric clinical trials conducted in accordance with the PIP are included in the drug product information, even when negative, the medicine is then eligible for an additional six-month period of qualifying patent protection through extension of the term of the Supplementary Protection Certificate. In the case of orphan medicinal products, a two year extension of the orphan market exclusivity may be available. This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the PIP are developed and submitted.
Regulatory Requirements after a Marketing Authorization has been Obtained
In case an authorization for a medicinal product in the EEA is obtained, the holder of the marketing authorization is required to comply with a range of requirements applicable to the manufacturing, marketing, promotion and sale of medicinal products. These include:
| ● | Compliance with the EU’s stringent pharmacovigilance or safety reporting rules must be ensured. These rules can impose post-authorization studies and additional monitoring obligations. | |
| ● | The manufacturing of authorized medicinal products, for which a separate manufacturer’s license is mandatory, must also be conducted in strict compliance with the applicable EU laws, regulations and guidance, including Directive 2001/83/EC, Directive 2003/94/EC, Regulation (EC) No 726/2004 and the European Commission Guidelines for Good Manufacturing Practice, or EU cGMP. These requirements include compliance with EU cGMP standards when manufacturing medicinal products and APIs, including the manufacture of API outside of the EU with the intention to import the API into the EU. | |
| ● | The marketing and promotion of authorized drugs, including industry-sponsored continuing medical education and advertising directed toward the prescribers of drugs and/or the general public, are strictly regulated in the EU notably under Directive 2001/83EC, as amended, and E. Member State laws. Direct-to-consumer advertising of prescription medicines is prohibited across the EU. |
| 134 |
Brexit and the Regulatory Framework in the United Kingdom
On June 23, 2016, the electorate in the United Kingdom voted in favor of Brexit. Thereafter, on March 29, 2017, the country formally notified the EU of its intention to withdraw pursuant to Article 50 of the Lisbon Treaty. The withdrawal of the United Kingdom from the EU took effect on January 31, 2020, however, there was a transition period until December 31, 2020 during which EU laws, including in respect of medicinal products, continued to be applicable in the United Kingdom. Since the regulatory framework for pharmaceutical products in the United Kingdom covering quality, safety and efficacy of pharmaceutical products, clinical trials, marketing authorization, commercial sales and distribution of pharmaceutical products is derived from EU Directives and Regulations, Brexit could materially impact the future regulatory regime which applies to drug products and the approval of product candidates in the United Kingdom, now that the United Kingdom legislation has the potential to diverge from EU legislation. It remains to be seen how Brexit will impact regulatory requirements for product candidates and approved drug products in the United Kingdom in the long term. The MHRA, the United Kingdom’s medicines and medical devices regulator, has recently published detailed guidance for industry and organizations to follow from January 1, 2021 now the transition period is over, which will be updated as the United Kingdom’s regulatory position on medicinal products evolves over time.
General Data Protection Regulation
The collection, use, disclosure, transfer or other processing of personal data of individuals in the EU, including personal health data, is governed by the GDPR, which became effective on May 25, 2018. After effectiveness of GDPR, the European data protection law background has been constantly implemented through the activity of the European Data Protection Board (EDPB) concerning the correct interpretation and application of GDPR, as well as through the ruling of the Court of Justice of the European Union (CJEU). The GDPR and EU Member States national data protection legislation, including Italy, are wide-ranging in scope and impose numerous requirements on companies that process personal data, including requirements relating to processing health and other sensitive data, obtaining consent of the individuals to whom the personal data relates, providing notice to individuals regarding data processing activities, implementing safeguards to protect the security and confidentiality of personal data, providing notification of data breaches, and taking certain measures when engaging third party processors. The GDPR also imposes strict rules on the transfer of personal data to countries outside the EU, including the United States and now – after Brexit – the United Kingdom, and permits data protection authorities to impose large penalties for violations of the GDPR, including potential fines of up to €20 million or 4% of annual global revenues, whichever is greater. Concerning the transfer of (pseudonymized) personal data to the United States, the recent CJEU case C-3111/18, also known as Schremes II, invalidated the European Commission’s adequacy decision for the EU-U.S. Privacy Shield Framework, on which the majority of U.S. companies relied to conduct trans-Atlantic trade in compliance with EU data protection rules. The decision reinforced the importance of data protection to global commerce and imposed EU companies trading with US companies or organizations to rely the transfer of personal data on other legal basis or appropriate safeguards provided for in the GDPR, such as Standard Contractual Clauses (SCC), Binding Corporate Rules (BCR) or derogations for specific situations. Regarding the transfer of personal data to United Kingdom, the EU / UK agreement regulating the transition period after Brexit (“Statement on the end of the Brexit transition period”) provides that, until June 2021, “all data flows of personal data between stakeholders subject to GDPR and UK organizations will not be considered as transfers to a third country”, thus implying the adoption of appropriate legal basis and safeguards provided for in the GDPR for any communication of personal data to a third party, such as Data Processing Agreement containing Standard Contractual Clauses.
The GDPR also confers a private right of action on data subjects and consumer associations to lodge complaints with supervisory authorities, seek judicial remedies and obtain compensation for damages.
Compliance with the GDPR and all relevant EU data protection rules will be a rigorous and time-intensive process that may increase our cost of doing business.
Pricing Decisions for Approved Drug Products
In the EU, pricing and reimbursement schemes vary widely from country to country. Some countries provide that drug products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular product candidate to currently available therapies or so-called health technology assessments, in order to obtain reimbursement or pricing approval. For example, the EU provides options for the EU Member States to restrict the range of drug products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. EU Member States may approve a specific price for a product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the product on the market. Other EU Member States allow companies to fix their own prices for drug products, but monitor and control prescription volumes and issue guidance to physicians to limit prescriptions. Recently, many countries in the EU have increased the amount of discounts required on pharmaceuticals and these efforts could continue as countries attempt to manage health care expenditures, especially in light of the severe fiscal and debt crises experienced by many countries in the EU. The downward pressure on health care costs in general, particularly with respect to prescription products, has become intense. As a result, increasingly high barriers are being erected to the entry of new drug products. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various EU Member States, and parallel trade, i.e., arbitrage between low-priced and high-priced EU Member States, can further reduce prices. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any drug products, if approved in those countries.
Employees and consultants
As of September 15, 2021, we had four full-time employees, all located in Milan, Italy and we rely on consultants and a large number of collaborators at SR-TIGET and OSR. Our full-time employees and consultants are engaged in clinical, research and development, product development and quality assurance activities. We consider our relationship with our employees to be good.
| 135 |
Property and Facilities
Our corporate headquarters is located in Milan, Via Olgettina 58 within San Raffaele Hospital, Italy, where we lease approximately 51 square meters of office space (3 offices). The lease commenced in January 2020 and has a 6-year initial term. It will expire on December 1, 2025 and may be renewed for additional 6 years. We also have an office in a co-working space located in Alexandria Center - LaunchLabs, 430 East 29th Street, New York, NY. We believe that our existing facilities are adequate for our near-term needs, and we believe that suitable additional or alternative office will be available as required in the future on commercially reasonable terms.
Legal Proceedings
From time to time, we may be involved in various claims and legal proceedings relating to claims arising out of our operations. We are not currently a party to any legal proceedings that, in the opinion of our management, are likely to have a material adverse effect on our business and have not been notified of any claims in respect thereof, other than as set forth below:
By letter dated February 25, 2019, Theravectys notified the Company of the possible infringement by Genenta of Theravectys’ exclusive license to patents no. EP 1071804 (and of the corresponding US patent US 6,682,907), EP 1224314, and EP 1222300 (and of the corresponding US patent US 7,968,332) granted from the owner of the patents Institut Pasteur. Theravectys requested Genenta engage in discussions as to possible contractual arrangements including the opportunity to either enter into (i) a manufacturing and supply agreement; or (ii) a non-exclusive license for Genenta’s use of the technologies allegedly protected under the patent(s). Each of these patents is now expired, having each reached the end of it its patent term on April 23, 2019 for EP1071804 and October 10, 2020 for EP 1224314, and EP 1222300.
To date, Genenta has not engaged in any such discussions with Theravectys nor has Genenta received any further claim/request from Theravectys in relation to the above.
| 136 |
Executive Officers and Directors
The following table sets forth the name, age as of the date of this prospectus, and position of the individuals who will serve as directors and executive officers of Genenta Science S.p.A. following the Corporate Conversion in May 2021 and the closing of this offering. The following also includes certain information regarding the individual experience, qualifications, attributes and skills of our directors and executive officers as well as brief statements of those aspects of our directors’ backgrounds that led us to conclude that they are qualified to serve as directors:
| Name | Age | Position | Year elected or appointed | |||
| Stephen Squinto, Ph.D. | 65 | Chairman of the Board of Directors(1)(2) | 2021 | |||
| Pierluigi Paracchi | 48 | Chief Executive Officer, Vice Chairman of the Board of Directors and General Manager * | 2014 | |||
| Richard B. Slansky | 64 | Chief Financial Officer | 2021 | |||
| Carlo Russo, M.D. | 68 | Chief Medical Officer, Head of Development | 2021 | |||
| Roger Abravanel | 75 | Director(1)(2) | 2017 | |||
| Daniela Bellomo, Ph.D. | 53 | Director | 2019 | |||
| Guido Guidi | 68 | Director(1)(2) | 2017 | |||
| Luca Guidotti | 60 | Director | 2018 | |||
| Anthony Marucci | 59 | Director(1) | 2021 | |||
| Stefania Mazzoleni, Ph.D. | 40 | Scientific Project Manager and Communications Officer | 2016 |
* Role of General Manager effective upon listing
| (1) | Independent Director (as defined under Nasdaq Stock Market rules) | |
| (2) | Member of the Compensation, Nomination and Governance Committee |
The directors above were elected at the Company’s general shareholders’ meeting held on May 20, 2021, for a three-year term. The board of directors consists of 7 (seven) members.
The board of directors’ term will expire with the Shareholders’ Meeting called to approve the financial statements for the year ending December 31, 2023.
Following the expiration of the above board of directors, future members will be appointed by means of a slate voting mechanism: slates may be submitted by the shareholders representing, individually or collectively with other shareholders joining in the submission of the slate, at least 6% of share capital eligible to vote at the shareholders’ meeting at which directors are to be elected, such eligibility to be established by filing an appropriate certification to that effect. The board of directors will be appointed as follows: (a) candidates for election as directors equal to the number of seats on the board of directors minus 1 (one) will be drawn from the slate that has obtained the highest number of votes cast on the shareholders’ meeting (the Majority Slate), based on the progressive order in which they are listed on the slate, while (b) the first candidate listed on the slate that receives the second greatest number of votes cast (the Minority Slate) will be elected as director so long as the Minority Slate has no connection in any way, whether directly or indirectly, with the shareholders who have submitted or voted the Majority Slate.
Each shareholder and shareholders belonging to the same group shall not submit, or contribute to submit, or cast their vote for more than one slate, including through a nominee. Each candidate may only be listed on a single slate or, otherwise, will be ineligible for election if named on multiple slates.
| 137 |
For additional information concerning the slate voting mechanism, see “Description of Share Capital and Governing Documents” – “Board of Directors”.
Management
Pierluigi Paracchi, Chief Executive Officer, Vice Chairman of the Board of Directors and General Manager
Mr. Paracchi has over 15 years of combined experience as an investor and director of life science companies, including as Founder and CEO of Quantica SGR and in senior roles at Axòn Capital, Sofinnova Partners and AurorA Science. He was also a board member and investor in Ethical Oncology Science, which was acquired in 2013 for a total deal of $470 million. Pierluigi Paracchi is a member of the Assobiotec Steering Committee, the Italian Association for the development of biotechnology. He also serves on the Board of Directors of the autoimmune disease and cancer company Altheia Science, as non-executive Chairman at medical device company Lipogems International and is a venture partner with AurorA Science, an independent biotech investment vehicle.
Richard B. Slansky, Chief Financial Officer
Mr. Slansky is a senior financial executive with more than 30 years of experience as Chief Financial Officer in various biopharmaceutical, diagnostic and life science companies, including OncoSec Medical, Biological Dynamics and GenMark Diagnostics. His experience spans across public and private healthcare and technology companies at various stages of growth, pre-revenue to commercial. He has been responsible for strategic vision and oversight of financial and operational teams, organizational leadership and creating maximum stakeholder value. He also serves on the Board of Directors of several private companies, including Nuclear RNA Networks, an early-stage RNA gene transcription therapeutics company.
Carlo Russo, M.D., Chief Medical Officer & Head of Development
Dr. Russo has extensive experience as a biotech executive focused on medical affairs and research and development. He has served as Head of Development of GSK’s R&D Biopharm and Rare Disease Units and the Cardiovascular Metabolic Center. Previously, Dr. Russo served as an Executive VP and CMO of Adverum, CMO & Head of Research & Development of Annapurna and President and CEO of VaxInnate Corporation, among other senior roles. Dr Russo holds a number of senior positions at research institutions, including Cornell University Medical College, Columbia University and Scripps Research Institute. He holds his MD and Board Certification in Hematology from the University of Genoa Medical School and is the author of more than 70 scientific publications.
Stefania Mazzoleni, Ph.D., Scientific Project Manager and Communications Officer
Dr. Mazzoleni manages and oversees the scientific development of parallel immuno-gene therapy studies in oncology indications and provides scientific support for investor interactions. Dr. Mazzoleni has more than 15 years’ experience in life science research and development, oncology and project management, including over 4 years of drug development and cell and gene therapy experience acquired while working at various academic institutions (San Raffaele Hospital, National Institute of Molecular Genetics) and pharmaceutics (Nerviano Medical Sciences). Dr. Mazzoleni received a MSc in Medical Biotechnology in 2005, holds a PhD in Molecular and Cellular Biology from San Raffaele Vita-Salute University, has a second level vocational Master’s in Pharmacy and Pharmaceutical Oncology and is a member of the European Academy of Tumor Immunology.
| 138 |
Board of Directors
Stephen Squinto, Ph.D., Chairman of the Board of Directors
Dr. Squinto has more than 25 years’ experience in the biotech industry and is an Executive Partner of the healthcare investment company OrbiMed Advisors. He was previously CEO of the gene therapy company, Passage Bio, and co-founded Alexion Pharmaceuticals, where he served as Chief Global Operations Officer and Global Head of Research, and held several senior leadership positions at Regeneron Pharmaceuticals. Dr Squinto currently serves on the Board of Directors of several biotech and healthcare companies and has received numerous honors and awards from academic and professional organizations for his scientific work.
Roger Abravanel, Director
Mr. Abravanel worked at McKinsey & Company for 34 years as a consultant for Italian and multinational corporations in Europe, the United States and the Far East, and is now an emeritus director. He is a former board member of Luxottica, COFIDE, Teva and Admiral, is currently Chairman of the INSEAD’s advisory group in Italy and is the author of several best-selling business books.
Daniela Bellomo, Ph.D., Director
Dr. Bellomo is Head of Business Development at San Raffaele Hospital, where she oversees technology transfer, development and value generation of research in biotech, medical technology and digital health. Dr. Bellomo is a board member of San Raffaele’s spin off Genespire and has previously been on the boards of Parco Tecnologico Padano and BiovelocITA, as well as an advisor to several life science start-ups, venture capital and incubators in the biotech and medical technology field.
Guido Guidi, Director
Mr. Guidi has 35 years of experience in top global roles in large pharmaceuticals companies, managing up to 7,000 employees and a turnover of more than €7 billion. He was previously Head of Pharma EU at Novartis, Head of Oncology at Novartis EU, overseeing major products including Cosentyx, Entresto, Lucentis, Gilenya, Xolair, Ultibro, Seebri, Galvus and Exforge.
Luca Guidotti, MD, PhD., Director
Dr. Guidotti is an experimental pathologist renowned internationally in the field of viral hepatitis. Dr. Guidotti spent more than 20 years as a Faculty of the Scripps Research Institute in La Jolla, California and he currently serves as Deputy Scientific Director of San Raffaele Hospital, Milan. He has published works in prestigious scientific journals including Cell, Nature, Science, Nature Medicine, Journal of Clinical Investigation and Journal of Experimental Medicine.
Anthony Marucci, Director
Mr. Marucci is a seasoned life sciences and public company leader who has raised $1.7 billion in capital in multiple organizations over his 30 years’ experience. He is currently President and CEO of Celldex Therapeutics, the company he co-founded in 2004 and which develops targeted therapeutics, including immunotherapies and other targeted biologics. Prior to founding Celldex, he was Treasurer at Medarex, from which Celldex was spun out, and he holds an MBA from Columbia University and a MHL from Brown University.
Executive Scientific Board
Luigi Naldini, M.D., Ph.D., Chairman of the Executive Scientific Board
Professor Naldini is a deeply experienced scientist and academic, considered by many to be the father of lentiviral gene therapy. Dr. Naldini is Professor of Cell and Tissue Biology and Cell and Gene Therapy at the Vita-Salute San Raffaele University School of Medicine in Milan, and Director of the San Raffaele-Telethon Institute for Gene Therapy and of the Division of Regenerative Medicine, Stem Cells & Gene Therapy at the San Raffaele Scientific Institute. He has previously served as President of the European Society of Gene and Cell Therapy and a member of the Board of Directors and Advisory Council of the American Society of Gene and Cell Therapy. Dr. Naldini is also a scientific advisor on EMEA and WHO committees for the evaluation of novel gene transfer medicines and has authored more than 250 scientific publications.
| 139 |
Bernhard Gentner, M.D., Member of the Executive Scientific Board
Dr. Gentner is a physician scientist, serving as Group Leader of the Translational Stem Cell and Leukemia Research Unit at the San Raffaele-Telethon Institute for Gene Therapy in Milan and Staff Hematologist in the Hematology and Bone Marrow Transplantation Unit of San Raffaele Hospital. Dr. Gentner completed his MD studies at the University of Heidelberg, Germany, the MD Anderson Cancer Center and Baylor College of Medicine, Houston, USA. He completed his internal medicine training at Erlangen University Hospital, Germany and his hematology training at San Raffaele Vita-Salute University and has authored more than 30 scientific publications.
Strategic Advisors
Advisors to the Company include:
| ● | Gaurav Shah, M.D.: co-founder of Rocket Pharma (NASDAQ: RCKT) and serves as its Chief Executive Officer and Corporate Board Member. Prior to this role Dr. Shah was a Global Program Head in the Cell & Gene Therapies Unit at Novartis, where he had strategic oversight of 12 functions and helped spearhead pivotal trials with CART-19 for patients with leukemia and lymphoma. | |
| ● | Brad Loncar: Founder and Chief Executive Officer at Loncar Investments. Mr. Loncar is a biotech investor, index provider, and creator of two Nasdaq-listed exchange traded funds. | |
| ● | Alexander Ross: Distinguished Visiting Professor at The University of Bologna Business School and a Board Partner at Amplo, a global venture capital firm. He is the author of New York Times–bestselling The Industries of the Future and Former Senior Advisor for Innovation to the US Secretary of State (2009-2013). |
Family Relationships
There are no family relationships among our executive officers and directors.
Arrangements Concerning Election of Directors and Members of Management
There are no arrangements or understandings with major shareholders, customers, suppliers or others pursuant to which any of our directors or members of senior management were selected as such.
Compensation
The following table presents in the aggregate all compensation we paid to all of our directors and senior management as a group for the year ended December 31, 2020. The table does not include any amounts we paid to reimburse any of such persons for costs incurred in providing us with services during this period. We are not required to provide the compensation, on an individual basis, of our executive officers and directors under Italian law. As a matter of Italian law, the compensation of directors is established at the time of their appointment or by the shareholders’ meeting. The compensation of the managing directors shall be established by the board of directors, with the opinion of the board of statutory auditors. Our bylaws provides that the shareholders’ meeting may determine a total amount for the compensation of the directors, including managing directors.
All amounts reported in the table below reflect the cost to the Company, in thousands of Euros, for the year ended December 31, 2020.
Salary, Bonuses and Related Benefits | Pension, Retirement and Other Similar Benefits | Share Based Compensation | ||||||||||
| All directors and senior management as a group, consisting of 9 persons | € | 617,904 | € | - | € | 460,194 | ||||||
| 140 |
Differences between Italian Laws and Nasdaq Requirements
The Sarbanes-Oxley Act, as well as related rules subsequently implemented by the SEC, requires foreign private issuers, such as us, to comply with various corporate governance practices. In addition, following the listing of the ADSs on Nasdaq, we will be required to comply with the Nasdaq Stock Market Rules. Under those rules, we may elect to follow certain corporate governance practices permitted under Italian law in lieu of compliance with corresponding corporate governance requirements otherwise imposed by the Nasdaq Stock Market Rules for U.S. domestic registrants.
In accordance with Italian law and practice and subject to the exemption set forth in Rule 5615 of the Nasdaq Stock Market Rules, as a foreign private issuer, we have elected to rely on home country governance requirements and certain exemptions thereunder rather than the Nasdaq Stock Market Rules, with respect to the following requirements:
| ● | Composition of the board of directors. Italian law does not require that the majority of our board of directors consist of independent directors. Our board of directors therefore may include fewer independent directors than would be required if we were subject to Nasdaq Listing Rule 5605(b)(1). In addition, we will not be subject to Nasdaq Listing Rule 5605(b)(2), which requires that independent directors must regularly have scheduled meetings at which only independent directors are present. | |
| ● | Quorum. In accordance with Italian law quorum requirements generally applicable to general meetings of shareholders are set forth in the Italian Civil Code (see “Description of Share Capital and Governing Documents”—“Meeting of shareholders”) therefore our bylaws may not provide a specific regulation of them. Our practice thus varies from the requirement of Nasdaq Listing Rule 5620(c), which requires an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting stock. | |
| According to Italian law, the management report and the annual financial statements shall be communicated to Company’s auditor and to the board of statutory auditors at least 30 days prior to the general meeting of shareholders convened for its approval. The board of statutory auditors must report to the shareholders’ meeting on the results of the financial year and on the activities carried out in the performance of its duties, and make observations and proposals regarding the financial statements and their approval. The financial statements, together with the reports of the directors, statutory auditors and Company’s auditors, must remain deposited at the Company’s registered office for the 15 days preceding the shareholders’ meeting called to approve them. | ||
| ● | Proxy Solicitations. Under Italian law shareholders may appoint attorneys-in-fact by delivering in writing appropriate power of attorney to represent them in an ordinary or extraordinary shareholders’ meeting of the Company. Our directors, auditors and employees may not be proxies. Italian law does not have a specific regulatory regime for the solicitation of proxies in private companies; thus our practice will vary from the requirement of Nasdaq Listing Rule 5620(b), which sets forth certain requirements regarding the solicitation of proxies. | |
| ● | Share Issuances. Pursuant to Italian law, prior to this offering, we will opt out of shareholder approval requirements by way of including authorized and conditional share capital (see “Description of Share Capital and Governing Documents—General —Authorization of shares”) for the issuance of securities in connection with certain events such as the acquisition of stock, assets or convertible notes, certain private placements and/or public offering. To this extent, our practice varies from the requirements of Nasdaq Listing Rule 5635, which generally requires an issuer to obtain shareholder approval for the issuance of securities in connection with such events. | |
| ● | Audit Committee. US companies listed on Nasdaq are required to have an audit committee that satisfies the requirements of Rule 10A-3 under the Exchange Act and certain additional requirements set by the Nasdaq. In particular, all members of this committee must be independent and the committee must adopt a written charter. The committee’s prescribed responsibilities include (i) the appointment, compensation, retention and oversight of the external auditors; (ii) establishing procedures for handling “whistle blower” complaints regarding accounting, internal accounting controls, or auditing matters; (iii) engaging independent counsel and other advisers, as it determines necessary to carry out its duties and (iv) determine appropriate funding for payments to the external auditor, advisors employed by the audit committee and other necessary administrative expenses of the audit committee. A company must also have an internal audit function, which may be outsourced, except to the independent auditor. We follow the “traditional” model of corporate governance for Italian companies and accordingly have established a board of statutory auditors established in accordance with Italian law which performs substantially the same functions, (see “Description of Share Capital and Governing Documents” – Statutory auditors”) and is accordingly exempt from the audit committee requirements established by Rule 10A-3 and Nasdaq rules. The Company’s reliance on such exemption is based on the circumstance that the Company’s board of statutory auditors meets the following requirements set forth in Exchange Act Rule 10A-3(c)(3): |
| 141 |
| (i) | the board of statutory auditors is established and selected pursuant to Italian law expressly permitting such a board; | |
| (ii) | the board of statutory auditors is required under Italian law to be separate from the Company’s board of directors; | |
| (iii) | the board of statutory auditors is not elected by management of the Company and no executive officer of the Company is a member of the board of statutory auditors; | |
| (iv) | Italian law provides for standards for the independence of the board of statutory auditors from the Company and its management; | |
| (v) | the board of statutory auditors, in accordance with applicable Italian law and the Company’s governing documents, is responsible, to the extent permitted by Italian law, for the appointment, retention and oversight of the work (including, to the extent permitted by law, the resolution of disagreements between management and the auditor regarding financial reporting) of any registered public accounting firm engaged for the purpose of preparing or issuing an audit report or performing other audit, review or attest services for the Company. |
Our reliance on Rule 10A-3(c)(3) does not, in our opinion, materially adversely affect the ability of its board of statutory auditors to act independently and to satisfy the other requirements of Rule 10A-3.
| ● | Compensation, Nomination and Governance Committee. Italian law does not require the appointment of a Compensation, Nomination and Governance Committee as required by the Nasdaq Listing Rules. As a matter of Italian law applicable to Italian stock corporations whose shares are not listed on a regulated market in the European Union and under our bylaws, the compensation of executive directors, including the CEO, is determined by the board of directors, after consultation with the board of statutory auditors, while the Company’s shareholders, according to Italian law and our bylaws, may determine a total amount for the compensation of the directors, including managing directors. Compensation of the Company’s executive officers is determined by board of directors or by the CEO, if duly empowered. Nevertheless, although not required under Italian law, the Company intend to establish a Compensation, Nomination and Governance Committee.
| |
| ● | Code of Business. Conduct and Ethics. Pursuant to Italian law, prior to this offering, we will adopt an “Organization and Operational Model” as required by Italian Legislative Decree of June 8, 2001, No. 231 (relating to administrative responsibility) that we expect will consist of: (i) a Code of Etichs; (ii) operating procedures and reporting systems applicable to all of our directors, officers and employees, which may not comply with the requirements of Nasdaq Listing Rule 5610. |
| 142 |
Committees of the Board of Directors
We currently follow the historical Italian corporate governance system, with a board of directors (consiglio di amministrazione) and a separate board of statutory auditors (collegio sindacale) with supervisory functions. The two boards are separate and no individual may be a member of both corporate bodies. Both the members of the board of directors and the members of the board of statutory auditors owe duties of loyalty and care to the Company.
Statutory Auditors
During 2020, the Company’s statutory auditors received approximately €35,000 in compensation in the aggregate for their services to the Company.
At the Company’s annual general shareholders’ meeting held on May 20, 2021, the following individuals were elected or re-appointed to the Company’s board of statutory auditors for a three-year term. The board consists of three members, one of which is the chairman, and two alternates. The board of statutory auditors’ term will therefore expire with the Shareholders’ Meeting called to approve the financial statements for the year ending December 31, 2023.
| Name | Age | Position | Year elected or re-appointed | |||
| Carlo-Alberto Nicchio | 46 | Chairman of the Board of Statutory Auditors | 2021 | |||
| Cesare Lazzaroni | 70 | Statutory auditor | 2021 | |||
| Jacopo Doveri | 48 | Statutory auditor | 2021 | |||
| Lorenzo Gianluigi Grossi | 55 | Alternate auditor | 2021 | |||
| Alessandro Arpiani | 37 | Alternate auditor | 2021 |
Following the expiration of the above board of statutory auditors, future members will be appointed by means of a slate voting mechanism: slates may be submitted by the shareholders representing, individually or collectively with other shareholders joining in the submission of the slate, at least 6% of share capital eligible to vote at the shareholders’ meeting at which auditors are to be elected, such eligibility to be established by filing an appropriate certification to that effect. According to the Company’s bylaws, the board of statutory auditors will be appointed as follows: (a) candidates for election as auditors equal to two statutory auditors and one alternate auditor will be drawn from the slate that has obtained the highest number of votes on the shareholders meeting (the Majority Slate), based on the progressive order in which they are listed, while (b) the remaining statutory auditor (who will act as President of the board of statutory auditors) and alternate auditor will be drawn from the slate that has obtained the second greatest number of votes cast (the Minority Slate) so long as the Minority Slate has no connection in any way, whether directly or indirectly, with the shareholders who have submitted or voted the Majority Slate.
Each shareholder and shareholders belonging to the same group shall not submit, or contribute to submit, or to cast their vote for more than one slate, including through a nominee. Each candidate may only be listed on single slate or, otherwise, will be ineligible for election if named in multiple slates.
For additional information concerning the slate voting mechanism, see “Description of Share Capital and Governing Documents” – “Board of Statutory auditors”.
The Company relies on an exemption from the Rule 10A-3 requirements provided by Rule 10A-3(c)(3) of the Exchange Act for foreign private issuers with a board of statutory auditors established in accordance with local law or listing requirements and subject to independence requirements under local law or listing requirements. See “— Differences between Italian Laws and Nasdaq Requirements” for more information.
| 143 |
Additional Board Committees
Although Italian law does not require that we adopt a Compensation, Nomination and Governance Committee, in connection with the Corporate Conversion, we have established a Compensation, Nomination and Governance Committee according to Nasdaq Listing Rule 5615(a)(3). The members of our compensation, nomination and governance committee include Stephen Squinto, Roger Abravanel and Guido Guidi. The compensation, nomination and governance committee will assist our board of directors in overseeing our cash compensation and equity award recommendations for our executive officers along with the rationale for such recommendations, as well as summary information regarding the aggregate compensation provided to our executive officers.
Equity Incentive Plan
2021 – 2025 Equity Incentive Plan
On May 20, 2021, our shareholders meeting approved a capital increase to allow for issuance of up to 2,700,000 ordinary shares to the service of a four-year employees’ share option plan (the “2021-2025 Equity Incentive Plan”) to be adopted by the board of directors. The purpose of the 2021-2025 Equity Incentive Plan is to motivate and reward performance of our employees, directors, non-employee directors and consultants, and in the best interests of the Company and our shareholders.
On May 20, 2021 our board of directors approved the specific terms (e.g., regulation) of our 2021 – 2025 Equity Incentive Plan which will become effective upon the effectiveness of the registration statement of which this prospectus is part. Under Italian law, we do not need to obtain the approval of the specific terms of our equity incentive plans by our shareholders. Except where the context indicates otherwise, reference hereunder to our ordinary shares shall be deemed to include a number of ADSs equal to the number of ordinary shares.
Plan Administration. Our 2021 – 2025 Equity Incentive Plan is administered by our board of directors in consultation with the Compensation, Nomination and Governance Committee, unless and until the board delegates administration to this latter. The board of directors has the authority to take all actions and make all determinations under the 2021 – 2025 Equity Incentive Plan, to interpret the 2021 – 2025 Equity Incentive Plan and award agreements and to adopt, amend and repeal rules for the administration of the 2021 – 2025 Equity Incentive Plan as it deems advisable, subject to certain limitations imposed under the 2021 – 2025 Equity Incentive Plan, and other applicable laws and stock exchange rules.
Eligible Participants. The board of directors jointly with the Compensation, Nomination and Governance Committee will be able to offer equity awards at its discretion under the 2021 – 2025 Equity Incentive Plan to:
| ● | any employees of us or any of our subsidiaries; | |
| ● | any non-employee directors serving on our board of directors; | |
| ● | any consultants to us or any of our subsidiaries (the “Eligible Participants”). |
Grant of Awards; Shares Available for Awards. The 2021 – 2025 Equity Incentive Plan provides for the grant of options to purchase up to a maximum of 2.7 million ordinary shares (i.e. approximately 10% of the issued share capital of the Company upon this offering on a fully diluted basis) in the future upon written exercise notice. This number is subject to adjustment in the event of a split-up, share dividend or other change in our capitalization. If any award expires, is cancelled, or is terminated, unexercised or is forfeited, the number of shares subject thereto is again available for grant under the 2021 – 2025 Equity Incentive Plan.
| 144 |
The number of stock options to be granted to executives and directors cannot be determined at this time as the grant of stock options and/or restricted shares is dependent upon various factors such as hiring requirements and job performance.
Terms of exercise – Stock Options.
Options may be granted to selected Eligible Participants (the “Optionee”) on such terms and conditions as the board of directors may determine; provided, however, that the exercise price of an option may not be less than the fair market value of a Company share as of the grant date as determined by the board of directors, or the Compensation, Nomination and Governance Committee, in its reasonable discretion and the term of the option my not exceed three years from the grant date.
Vesting. The vesting conditions for stock options granted under the 2021 – 2025 Equity Incentive Plan are set forth in the applicable award documentation.
Termination of Service and Change in Control. In the event of termination of an Optionee’s employment or service without cause or an Optionee’s resignation for just cause (as defined in the 2021 – 2025 Equity Incentive Plan) following a change in control of the company (as defined in the 2021 – 2025 Equity Incentive Plan), any awards outstanding to the Optionee (unless otherwise provided in the award agreement) will immediately vest, and options will become fully exercisable during the twelve (12) months following the Change in Control of the Company, or until the date of expiration of the applicable option, if earlier. In the event of a change in control that involves a merger, acquisition or other corporate transaction, any outstanding award not assumed, substituted, replaced or continued in connection with the transaction will immediately vest, and options will become fully exercisable and upon the effectiveness of such corporate transaction, the 2021 – 2025 Equity Incentive Plan and all awards will automatically terminate.
Amendment and Termination. Within the limitation of the 2021 – 2025 Equity Incentive Plan, the board of directors or the Compensation, Nomination and Governance Committee may amend, suspend or terminate the 2021 – 2025 Equity Incentive Plan, at any time and for any reason. The suspension or termination of the 2021 – 2025 Equity Incentive Plan, or any amendment thereof, shall not affect any option previously granted under the 2021 – 2025 Equity Incentive Plan. No option shall be granted under the 2021 – 2025 Equity Incentive Plan after its suspension or termination. An amendment of the 2021 – 2025 Equity Incentive Plan shall be subject to the approval of the Company’s shareholders only to the extent required by applicable laws or regulations.
Right to amend options. The board of directors at any time, and from time to time, may amend the terms of any one or more options; provided, however, that the rights under any option shall not be impaired by any such amendment unless (i) the Company requests the consent of the optionee and (ii) the optionee consents in writing.
2021- 2025 Chairman Sub-Plan
The 2021 – 2025 Chairman Sub-Plan allows for the grant of options to our chairman of the board of directors, Mr. Squinto. In particular, Mr. Squinto is entitled to receive options to subscribe: (i) no. 147,783 shares at a price equal to €6.38 each upon listing; and (ii) no. 73,892 shares at a price equal to €6.38 each, during the year starting from the first anniversary of the listing.
Except for the above, the 2021 – 2025 Chairman Sub-Plan provides for identical terms and conditions under our 2021 – 2025 Equity Incentive Plan.
Accelerated Vesting of Existing Options Represented by Class B Quotas
The following table summarizes, as of the date of this prospectus, previously outstanding options subject to accelerated vesting upon the occurrence of a liquidity event. Prior to the consummation of the Corporation Conversion all such options were converted into Quota B and then subsequently converted into our ordinary shares.
| Name | Class B quotas Underlying Options Awarded | Exercise Price (€/Share) | ||||||
| Roger Abravanel | 82 | 1 | ||||||
| Guido Guidi | 82 | 1 | ||||||
| Luca Guidotti | 82 | 1 | ||||||
| Carlo Russo | 300 | 1 | ||||||
| Total | 546 | 1 | ||||||
| 145 |
BENEFICIAL OWNERSHIP OF PRINCIPAL SHAREHOLDERS AND MANAGEMENT
The following table sets forth information regarding beneficial ownership of our ordinary shares as of November 15, 2021 by:
| ● | each person, or group of affiliated persons, known to us to be the beneficial owner of more than 5% of our outstanding ordinary shares; | |
| ● | each of our directors and executive officers; and | |
| ● | all of our directors and executive officers as a group. |
Beneficial ownership is determined in accordance with the rules of the SEC, and includes voting or investment power with respect to ordinary shares. Percentage of shares beneficially owned before this offering is based on 15,000,000 shares outstanding on November 15, 2021. The number of ordinary shares deemed outstanding after this offering includes the ordinary shares being offered for sale in this offering but assumes no exercise by the representative of the underwriters of the over-allotment option.
Except as indicated in footnotes to this table, we believe that the shareholders named in this table have sole voting and investment power with respect to all shares shown to be beneficially owned by them, based on information provided to us by such shareholders. Unless otherwise noted below, each beneficial owner’s address is: c/o Genenta Science S.p.A., Via Olgettina no. 58, 20132 Milan, Italy.
No. of Shares Beneficially Owned Prior to this Offering | Percentage Owned Before this Offering(1) | Percentage Owned After this Offering(3) | ||||||||||
| Directors and executive officers | ||||||||||||
| Stephen Squinto | — | — | — | |||||||||
| Pierluigi Paracchi | 2,275,516 | 15.17 | % | 12.92 | % | |||||||
| Roger Abravanel | 73,729 | * | % | * | %(4) | |||||||
| Daniela Bellomo | 16,357 | * | % | * | % | |||||||
| Guido Guidi | 61,553 | * | % | * | % | |||||||
| Luca Guidotti | 49,469 | * | % | * | % | |||||||
| Anthony Marucci | — | — | — | |||||||||
| Richard B. Slansky | 19,947 | * | % | * | % | |||||||
| Carlo-Russo | 598,417 | 3.99 | % | 3.40 | % | |||||||
| Luigi Naldini | 1,386,145 | 9.24 | % | 7.87 | % | |||||||
| Bernard Gentner | 692,871 | 4.62 | % | 3.93 | % | |||||||
| All directors and executive officers as a group (11 persons) | 5,174,004 | 34.49 | % | 29.40 | % | |||||||
| 5% Shareholders | ||||||||||||
| Ospedale San Raffaele | 1,896,730 | 12.64 | % | 10.77 | % | |||||||
| Spafid(2) | 3,404,688 | 22.70 | % | 19.34 | % | |||||||
* Less than 1%.
| (1) | The percentages shown are based on 15,000,000 ordinary shares issued and outstanding as of November 15, 2021. |
| (2) | Spafid is a public limited liability company established under the laws of Italy, having its registered office at Via Filodrammatici no. 10, 20121 Milan, Italy, and having its principal place of business at Foro Buonaparte no. 10, 20121 Milan, Italy, registered under number 00717010151, that acts pursuant to Italian Law 23 November 1939 no. 1966 on “fiduciary Activity”. Spafid is indicated as Owner Entity because identified as formal owner of the shares. As Fiduciary Company, according to Italian Rules, Spafid operates on behalf of Beneficial Owner of Fiduciary Mandate. Shares of Spafid are wholly owned by Mediobanca – Banca di Credito Finanziario S.p.A., a leading Italian investment bank. In such a capacity of Fiduciary company, Spafid owns no. 3,404,688 ordinary shares of the Company on behalf of no. 43 beneficial owners (“fiducianti”). |
| (3) | The percentages shown are based on 17,608,695 securities issued and outstanding after this offering. |
| (4) | Includes 2,450 ordinary shares that Mr. Abravanel intends to purchase in the Reserved Offering. |
Although the shares of the Company owned by Spafid represent 22.59% of the Company’s share capital: (i) the average shareholding owned through Spafid in the Company’s share capital by each beneficial owner is equal to 0.53%; while (ii) the maximum shareholding owned through Spafid in the Company’s share capital by a single beneficial owner is equal to 2.30%. Spafid exercises - and will exercise - the relevant voting rights in the shareholders’ meeting of the Company according to voting instructions provided for by each beneficial owner.
Record Holders
As of November 15, 2021, there was a total of 68 holders of record of our ordinary shares, none of which had, to the best of our knowledge, a registered address in the United States.
We are not controlled by another corporation, by any foreign government or by any natural or legal persons except as set forth herein, and there are no arrangements known to us which would result in a change in control of the Company at a subsequent date.
| 146 |
Agreements with OSR
We have a longstanding relationship with OSR. Dr. Guidotti, a member of our Board of Directors, currently serves as Deputy Scientific Director of OSR. On June 4, 2015, we entered into a service agreement with OSR to provide certain services (accounting/bookkeeping and rent of spaces, the latter with an addendum effective from January 1, 2016) free of charge. Beginning in January 2020, we engaged a third-party provider to perform these services. We determined that the value of these services provided in 2019 and in prior years were not material to our financial statements. Beginning January 1, 2020, we entered into a six-year lease agreement for the use of office space in the OSR building. We paid OSR annual rent of €13,400 in 2020 with a security deposit of €3,350.
We entered into a license agreement with OSR effective December 15, 2014, pursuant to which OSR granted us an exclusive, royalty-bearing, non-transferable, worldwide license, subject to certain retained rights, to certain patents, patent applications and existing know-how in exchange for certain ongoing payment obligations. See “Business — Collaboration/Licensing.” In February 2021, we entered into a Sponsored Research Agreement (“SRA”) with OSR to conduct certain research projects related to Temferon. The total consideration to be paid by the Company under the SRA will be €1.0 million with payments scheduled quarterly during 2021 and 2022.
Employment, Consulting and Services Agreements
Dr. Russo is currently a party to a Service Agreement dated July 2017 through an affiliated company, XDG BioMed LLC, which, as amended, provides for fixed annual fees of €300,000 gross and a discretionary annual variable gross remuneration up to a minimum amount of €50,000, tax included. We entered into consulting agreements in October 2015 and April 2016 with Prof. Naldini and Dr. Gentner, which, as amended, provided for gross annual fees of €50,000 and €30,000, respectively. These agreements renew automatically each year.
We entered into a directorship agreement with Mr. Paracchi in December 2019, which provides for a gross annual salary of €250,000 and a discretionary €50,000 gross performance bonus payment to be approved by the Board of Directors. Such agreement will be terminated by the parties for mutual consent upon signature of the below employment agreement.
Proposed New Employment Agreements
We intend to enter into new employment agreements with each of Mr. Paracchi, Dr. Russo and Mr. Slansky effective upon the consummation of this offering, and in the case of Mr. Slansky, the earlier of the consummation of this offering or November 1, 2021, whichever is earlier. The forms of such proposed employment agreements have been filed as exhibits to the registration statement of which this prospectus forms a part. Pursuant to such proposed employment agreements, Mr. Paracchi, Dr. Russo and Mr. Slansky are entitled to gross annual base salaries of €420,000, $500,000 and $300,000, respectively, which is subject to annual review by and at the sole discretion of the Compensation, Nomination and Governance Committee of our board of directors. Mr. Paracchi and Dr. Russo are also eligible to receive an annual cash bonus equal to or exceeding 20% of base salary, and Mr. Slansky is eligible to receive an annual cash bonus equal to or exceeding 30% of base salary, provided that such individual achieves performance targets determined by the Compensation, Nomination and Governance Committee of the board of directors.
The proposed employment agreements of Dr. Russo and Mr. Slansky, are governed by US Law and include the following terms and conditions, among others:
| (a) | each proposed employment agreement has a term commencing on the date of consummation of this offering and continuing until terminated (i) upon death of the employee, (ii) upon disability, (iii) for cause or good reason, (iv) without cause, or (v) voluntarily. The employment agreement also contains, among other things, the following material provisions: (i) reimbursement for all reasonable travel and other out-of-pocket expenses incurred in connection with such individual’s employment; (ii) paid vacation leave; (iii) health benefits; and (iv) a severance payment equal to twelve (12) months of base salary and a prorated portion of the applicable cash bonus upon termination by such individual for just cause or by the Company without cause (each as defined in the relevant agreement), with restrictive covenants applicable for a corresponding period after termination. |
| (b) | in the event such individual is terminated six months prior to or two years after a Change of Control (as defined in the agreement) by the Company for any reason other than cause or by such individual for good reason, then the executive shall be entitled to receive a cash payment equal to times such individual’s then-current annual base salary, plus 1.0 times his cash bonus for the year in which the termination occurs. Such payment shall be in lieu of the severance payment described above. |
The proposed employment agreement of Mr. Paracchi is governed by Italian Law and include the following terms and conditions, among others:
| (a) | the duties of general manager (direttore generale) with direct report to the Board of Directors of the Company;
|
| (b) | reimbursement of reasonable expenses incurred in the performance of work duties, health benefits and, subject to the approval of the board of directors, a grant of an equity award under an equity incentive plan to be adopted after the offering;
|
| (c) | in case of Change of Control (as defined in the relevant agreement), in the event of termination not for “cause” by the Company or of resignation for “cause” by the executive (such terms as understood in accordance with Article 2119 of the Italian Civil Code), the executive shall be entitled to receive a cash payment equal to three times such individual’s then-current annual base salary (such indemnity will replace any indemnity provided for by the applicable National Collective Labour Agreement in case of termination); |
| (d) | non-competition and non-solicitation obligations of the executive for a 12 months period after the termination of the employment in consideration for compensation equal to 12 months after termination at the executive’s then-current monthly base salary for each obligation;
Such agreement has a term commencing on the date of consummation of this offering and continuing until terminated, among other things, (a) upon death of the executive, (b) for just cause, (c) with objective or subjective reason, (d) by resignation of the executive, or (e) voluntarily by mutual agreement between the parties.
Proposed New Consultancy Agreements
Each of Messrs. Squinto and Marucci have entered into consultancy agreements in connection with their appointment to the board of directors in order to support the development and growth of the Company. The agreements provide for gross compensations of $25,000 (Mr. Squinto) and $11,875 (Mr. Marucci) per quarter (pro rated for partial quarters). The consultant will be fully and solely responsible for the payment of all tax and/or social security charges and will not be entitled to expenses reimbursement. These agreements have a term of three years, but are subject to termination at the option of either party by written notice given at least 15 days prior to each anniversary of the date of effectiveness of the agreement).
In addition, Messrs. Alexander Ross, Brad Loncar and Gaurav Shah have entered into consultancy agreements for the activity of support the development and the growth of the Company in the US market. All agreements provide for a gross compensation of USD 30,000 for the 12-months’ period of duration of the agreements and a specific no-conflict clause towards the Company. |
| 147 |
Indemnification Agreements
On or before the time of effectiveness of this registration statement, an indemnification agreement with our directors and executive officers will be entered into. The indemnification agreement requires us to indemnify our directors and executive officers to the fullest extent permitted by law, save for a limited number of instances, including when (i) officers and directors’ acts or omissions constituted willful misconduct or gross negligence, (ii) officers and directors did not act in good faith, for a purpose which they reasonably believed to be in, or not opposed to, the best interests of the Company and (iii) officers and directors are held liable towards the Company.
Insofar as indemnification of liabilities arising under the Securities Act may be permitted to executive officers and board members or persons controlling us pursuant to the foregoing provisions, we have been informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable
Issuances of Share Capital
Since our founding, we have obtained equity financing in an aggregate amount of approximately €35.1 million in four separate rounds based on the credentials and reputation of our founders and management team and our relationship with SR-TIGET, OSR, and Telethon Institute for Gene Therapy.
Upon its organization in July 2014, the Company was capitalized with €11,650 through the issuance of the following quotas:
| Name | Quotas (€) | Aggregate Purchase Price (€) | ||||||
| Ospedale San Raffaele | 4,754.37 | 1,500 | ||||||
| Pierluigi Paracchi | 5,703.84 | 10,000 | ||||||
| Luigi Naldini | 794.53 | 100 | ||||||
| Bernhard Gentner | 397.26 | 50 | ||||||
The quotas held by Mr. Paracchi and OSR, on the one hand, and by Prof. Naldini and Dr. Gentner, on the other hand, were converted into class A and B quotas, respectively, in connection with our first round of equity financing approved by shareholders on December 23, 2014 (which resolution has been subsequently amended and restated), as follows:
| ● | issuance of a maximum of €6,237.50 of class B quotas to Prof. Naldini and Dr. Gentner, directors, employees and other individuals in recognition of their contributions to the Company’s growth; and | |
| ● | issuance of class C quotas (including class C quotas issuable upon conversion of convertible notes) for an aggregate amount equal to €10.0 million. |
The class C quotas were offered on the basis of a pre-money valuation of the Company of €20.0 million with assistance from Banca Esperia, the formerly Private Bank of Mediobanca, and purchased by accredited investors, including entrepreneurs, managers, and family offices, including affiliates of the Ferrari family, which controls FIS Holding, one of the leading manufacturers of pharmaceuticals in Europe, and an early investor in Advanced Accelerator Application (NASDAQ: AAAP) (“AAAP”), a biotech company later acquired by Novartis (NYSE: NVS) for $3.9 billion.
In connection with the class C quota offering, Roger Abravanel (former McKinsey & Company director and board member of TEVA) joined the Company’s board of directors.
On June 27, 2017, our shareholders approved the issuance of class D quotas for an aggregate amount equal to €7.0 million on the basis of a pre-money valuation of the Company of €45.0 million. The class D quotas were purchased by Italian, British, and Swiss private investors, family offices, and angel investors, including FIDIM S.r.l., the holding company for the Rovati family, former owner of Rottapharm, which was acquired in 2014 by Meda/Mylan for $2.2 billion) (“FIDIM”), some other early investors in AAAP, and Mr. Giuseppe Vita, former Chairman of Schering-Plough.
In connection with the class D quota offering, Guido Guidi, then Head of Novartis in Europe joined the Company’s board of directors. During his tenure as Head of Novartis’ Oncology Europe division, Mr. Guidi led the development and launch of a number of highly successful products, such as Cosentyx, Entresto, Lucentis, Gilenya, Xolair, Ultibro, Seebri, Galvus, Exforge, Zometa, Femara and Glivec. At that time, Kenneth C. Anderson, Kraft Family Professor of Medicine at Harvard Medical School, President Elect of the American Society of Hematology, and Director of the Lebow Institute for Myeloma Therapeutics and the Jerome Lipper Myeloma Center at DanaFaber Cancer Institute, joined the Company’s Scientific Advisory Board.
| 148 |
In August 2019, our shareholders approved the issuance of class E quotas for an aggregate amount equal €17.1 million, subscribed for €15.1 million, on the basis of a pre-money valuation of the Company of €70.0 million. The lead investor in the class E quota offering was Qianzhan Investment Management, a Shanghai based private company active in private equity and venture capital investments, and FIDIM. Qianzhan was an early investor in Tencent Music (NASDAQ: TME) and in pharmaceutical and biotech companies in China and in the U.S.
In July 2020, our shareholders approved the issuance of an additional of class E quotas for an aggregate amount equal to € 1.5 million on the basis of a pre-money valuation of the Company of €90.0 million. The lead investor was GM Investimenti, a private company controlled by Giuseppe Miroglio, former CEO and current Chairman of Miroglio Group.
The tables below set forth the aggregate number of class B and D quotas issued and subscribed by related parties as part of the offerings described above. No related parties purchased class C or E quotas.
| Name | Class B Quotas (€) | Aggregate Purchase Price (€) | ||||||
| Luigi Naldini | 2,795 | 2,795 | ||||||
| Bernhard Gentner | 1,396.5 | 1,396.5 | ||||||
| Carlo Russo | 1,200 | 1,200 | ||||||
| Roger Abravanel | 74 | 74 | ||||||
| Guido Guidi | 55 | 55 | ||||||
| Luca Guidotti | 42 | 42 | ||||||
| Stefania Mazzoleni | 30 | 30 | ||||||
| Name | Class D Quotas (€) | Aggregate Purchase Price (€) | ||||||
| Roger Abravanel | 28.81 | 50,000 | ||||||
| Guido Guidi | 17.29 | 30,000 | ||||||
| 149 |
DESCRIPTION OF SHARE CAPITAL AND GOVERNING DOCUMENTS
General
The following description summarizes important terms of our capital stock and certain provisions of our articles of association and by-laws, each of which will be in effect upon the closing of this offering. Copies of these documents will be filed with the SEC as exhibits to our registration statement, of which this prospectus forms a part. The description of our capital stock reflects the completion of the Corporate Conversion that occurred in May 2021.
As of the date of filing of this prospectus our authorized capital stock is equal to Euro 50,000 represented by 15,000,000 ordinary shares with no par value.
No preference shares are designated, issued or outstanding.
The following is a summary of certain information concerning our ordinary shares and bylaws (Statuto), as well as Italian law provisions applicable to companies like ours whose shares are not listed in a “regulated market” within the European Union, as in effect at the date of this prospectus. The summary contains such information as we consider material regarding the ordinary shares but does not purport to be complete and is qualified in its entirety by reference to our bylaws or Italian law, as the case may be.
Under Italian law, most of the procedures regulating our Company, including certain rights of shareholders, are contained in our bylaws. Amendments to our bylaws must be approved at an extraordinary meeting of shareholders, as described below.
Authorization of shares
Our shareholders may authorize the issuance of additional shares at any time at an extraordinary shareholders’ meeting. However, the newly issued shares may not be purchased before all the outstanding shares (i.e., the shares already subscribed) are entirely paid for. Moreover, although Italian law generally provides shareholders with preemptive rights when new shares are issued for cash, it is possible, in certain case, for general meeting to exclude or limit preemptive rights. Such an exclusion of preemptive rights must be illustrated by the directors with a specific report which sets out the reasons for the exclusion or limitation of the pre-emptive right, or, if the exclusion derives from a contribution in kind, the reasons for such contribution in kind, and in any case the criteria adopted for determining the issue price. On May 20, 2021, our shareholders approved or delegated our board of directors to approve — several capital increases. In particular, the shareholders’ meeting unanimously resolved, inter alia
| ● | to increase the share capital against payment in one or more instalments on a divisible basis, with the exclusion of the pre-emptive rights under Article 2441, paragraph 5, of Italian Civil Code, up to a maximum amount of € 115 million (including share premium), with allocation to capital of € 0.10 per share, by issuance of a maximum of 11.5 million new ordinary shares with no par value and with regular dividend entitlement, to the service of our initial public offering, granting authority to the board of directors to implement the proposed capital increase in one or more tranches, in the amounts quoted above, within the time limit of December 31, 2021, of which a maximum number of 10 million ordinary shares against a countervalue of € 100 million (including share premium), shall be issued to the service of the initial public offering; while a maximum number of 1.5 million ordinary shares (subject to the maximum limit of 15% of the number of shares actually used to the service of our initial public offering) against a countervalue of € 15 million (including share premium), shall be issued to the service of potential exercise of the over-allotment option (greenshoe); | |
| ● | to grant to the board of directors the authority to approve the issuance of a maximum number of 0.5 million warrants to be granted free of charge to the Underwriters and to increase the share capital, on a divisible basis, to the service of warrants exercised by the Underwriters, by maximum par values of €5,000,000 (including share premium), with allocation to capital of €0.10 per share, by issuance of a further maximum number of 500,000 ordinary shares (subject to the maximum limit of 4% of the ADSs to service our initial public offering as well as criteria set forth in agreements executed by the Company in relation to our initial public offering), all represented by ADSs, to be issued by no later than December 31, 2021, being it specified that, if such increase is not fully subscribed within that time limit, the increase shall remain valid within the limits of the subscriptions collected; being it further specified that such capital increase shall be implemented, pursuant to the warrants regulation, by the board of directors duly authorized to set the issue price and the portions to be allocated to capital and to share premium; and | |
| ● | to grant to the board of directors the authority, in accordance with Article 2443 of Italian Civil Code, to further increase the share capital against payment, in one or more instalments on a divisible basis, up to a maximum amount of €300,000,000 (including share premium), by issuance of a maximum number of 30,000,000 new ordinary shares with no par value and with regular dividend entitlement, also with the exclusion of pre-emptive rights or free of charge, in accordance with Article 2441, paragraphs 4, 5 and 8, of Italian Civil Code for a five-year period, also in support of third-party grants of participating interests and/or industrial and intellectual property rights and similar intangible assets (such as patents, marks and know-how) which can be granted and held by the board of directors itself in accordance to the scope of the corporate purpose, in addition to the authority, pursuant to Article 2420-ter of Italian Civil Code, to issue convertible debentures in one or more instalments, convertible into ordinary shares, within the same aggregate maximum amount of €300,000,000 (including share premium), with consequent capital increase to the service of the conversion, also with the exclusion of pre-emptive rights in accordance with Article 2441, paragraphs 4, 5 and 8 of Italian Civil Code, likewise for a period of five years. |
Form and transfer of shares
Our ordinary shares are not represented by share certificates (certificati azionari) as they are dematerialised (azioni dematerializzate). The ownership of the shares, their transfer, the related rights and restrictions on the shares (if any) results from the electronical register managed by an intermediary (banks and other financial institutions). The entitlement to exercise the rights attached to the shares is then proven by the exhibition of certifications or communications to the issuer made by the intermediary, pursuant to its own accounting records, in favor of the subject entitled to the right.
| 150 |
There are no limitations on the right to own or vote our ordinary shares, which applies to non-Italian residents and foreign residents except for Golden Power’s rules and Antitrust rule (see Section “Notification of Acquisition of Shares”). There are no provisions in our articles of association or bylaws that would have the effect of delaying, deferring or preventing a change of control of our Company and that would operate only with respect to a merger, acquisition or corporate restructuring involving our Company. There are no provisions in our bylaws governing the ownership threshold which shareholder ownership must be disclosed. There are no provisions discriminating against any existing or prospective holder of our ordinary shares as a result of such shareholder owning a substantial number of our shares. There are no sinking fund provisions or provisions providing for liability for further capital calls by our Company.
Dividend rights
Our payment of any annual dividend must be proposed by our board of directors to the shareholders and is subject to the approval of our shareholders at the annual ordinary shareholders’ meeting. Before dividends may be paid out of our unconsolidated net income in any year, we must allocate an amount equal to 5% of the Italian GAAP net income to our legal reserve until such reserve is at least equal to 20% of our corporate capital. If a loss in our corporate capital occurs, we may not pay dividends until the capital is reconstituted or reduced by the amount of such losses. We may pay dividends out of available retained earnings from prior years, provided that after such payment, we will have a legal reserve at least equal to the legally required minimum of 20% of the corporate capital. We may not approve or pay dividends until this minimum (i.e., 20% of the capital) is met. If the minimum is met, the board of directors could propose the issuance of a dividend to the shareholders and the shareholders’ resolution might approve that issuance. The shareholders’ resolution will specify the manner and the date for dividend payment. Any dividends which shareholders do not collect within five years of the date on which they become payable will be forfeited by those shareholders and come back to us. The board of directors may not approve interim dividends at times between our annual ordinary shareholders’ meetings.
Governance
Under Nasdaq rules, the Company is permitted, as a listed foreign private issuer, to adhere to the corporate governance rules of our home country in lieu of certain Nasdaq corporate governance rules.
Corporate governance rules for Italian corporations (società per azioni) like the Company provide that the Company can be managed by a board of directors (consiglio di amministrazione) and a separate board of statutory auditors (collegio sindacale) with supervisory functions concerning the compliance of the by-laws and Italian laws. The two boards are separate and no individual may be a member of both boards. Both the members of the board of directors and the members of the board of statutory auditors owe duties of loyalty and care to the Company. As required by Italian law, an external auditing firm (società di revisione) is in charge of auditing the Company’s financial statements. The members of the Company’s board of directors and board of statutory auditors, as well as the external auditor, are directly and separately appointed by shareholders’ resolution at the shareholders’ meetings.
Board of directors
Our board of director consists of seven members (see “Management” for additional information). Our board of directors is elected at an ordinary shareholders’ meeting for the period established at the time of election but in any case, for no longer than three fiscal years. Our directors, who may but are not required to be shareholders, may be reappointed for successive terms.
Following the expiration of the term of directors currently in office, future members of the board of directors will be elected by means of a slate voting mechanism. Slates may be submitted by shareholders representing, individually or collectively with other shareholders joining in the submission of the slate, at least 6% of the share capital eligible to vote at the shareholders’ meeting at which directors are to be elected, such eligibility to be established by filing with the Company a certification to that effect. The board of directors shall be elected as follows: (a) candidates for election as directors equal to the total number of seats on the board of directors minus one shall be drawn from the slate that has obtained the highest number of votes cast at the shareholders meeting (the Majority Slate), in the order in which their name is listed on the Majority Slate, (b) the first candidate listed on the slate that receives the second greatest number of votes cast (the Minority Slate) shall be elected as director so long as none of the shareholders joining in the submission of the Minority Slate has any connection whatsoever, whether directly or indirectly, with the shareholders who have submitted or voted the Majority Slate.
| 151 |
Each shareholder and shareholders belonging to the same group shall not submit, or join in submitting, or cast their vote for more than one slate, including through a nominee or agent. Each candidate for election as director may only be listed on single slate and will be ineligible for election if named on multiple slates.
In the event that the Majority Slate does not contain a sufficient number of candidates to fill the number of vacancies on the board of directors to be filled as provided under clause (a) above, all candidates listed in the Majority Slate shall be elected as directors and the remaining directors shall be elected from the Minority Slate according to the order in which they are listed in such slate. The voting procedure according to slates described above shall be applicable only in case of election of the entire board of directors at the same shareholders’ meeting. In the event of a tie between slates, a new vote shall be taken and the candidates obtaining the largest number of votes shall be elected without regard to the slate on which they are listed or application of the slate voting mechanism. Should a single slate be submitted, the shareholders eligible to vote at the meeting shall cast their vote on such slate and, so long as more votes are cast for such slate than votes cast against such slate, all the members of the board of directors shall be elected from that slate in accordance with applicable law at the time. If no slates are submitted, or a single slate is submitted and such slate does not obtain the requisite number of votes, or the number of directors to be elected on the basis of the slates submitted is less than the full number of directors to be elected, or the entire board of directors is not to be elected at the same shareholders’ meeting, or in case it is otherwise not possible for any reason to elect the board of directors in accordance with the provisions set out in our by-laws, the members of the board of directors shall be elected at the shareholders’ meeting in accordance with generally applicable procedures and required majorities under applicable law, without application of the slate voting mechanism.
Our board of directors has all ordinary and extraordinary powers to manage our affairs as it deems advisable for the achievement of our corporate purposes, except for the actions reserved, by applicable law or the bylaws, to a vote of the shareholders at an ordinary or extraordinary shareholders’ meeting.
If we cannot repay our creditors, and a court determines that our directors did not perform their duties regarding the preservation of our assets, the court may find our directors liable to our creditors. However, the judgement on the director’s diligence in carrying out his or her mandate should never cover the management choices or the manner and circumstances of such choices, even if the same entail significant economic risks, but should cover only the diligence shown by the director in appreciating in advance the risk margins connected with the transaction to be undertaken, and therefore, the possible omission of those precautions, assessments and information normally required for a decision of that type, implemented in those circumstances and with those methods. Directors are liable to the company’s creditors when their improper management conduct impairs the company’s assets in such a way that the same may not be sufficient to satisfy creditors’ claims.
If a chairman (presidente) has not already been elected by the shareholders, the board of directors must appoint one. The board of directors may appoint a vice-chairman. The chairman of the board of directors is the legal representative of the Company. Our board of directors may delegate certain powers to one or more managing directors (amministratori delegati), determine the nature and scope of the powers delegated to each such managing director and revoke such delegation at any time. Any managing director shall report to the board of directors and the board of statutory auditors at least every 180 days on the Company’s business and the principal business carried out by the Company or by its subsidiaries. Our board of directors may also appoint one or more general managers (direttori generali) who must report directly to the board and may delegate authority for specific matters or categories of matters to employees of the Company or persons unaffiliated with the Company. The general managers may be employees, and the board of directors may delegate certain powers to general managers that the board has not already delegated to managing directors or an executive committee, subject to the following limitations: under Italian law, the board of directors may not delegate to officers or employees of the Company certain responsibilities, including the preparation and approval of draft financial statements, the approval of merger and de-merger plans to be presented to shareholders’ meetings, increases in the amount of our share capital or the issuance of convertible debentures (if any of such powers has been delegated to our board of directors by our shareholders at an extraordinary shareholders’ meeting).
The board of directors may establish one or more committees with advisory, deliberative or oversight functions in accordance with applicable laws and regulations in whatsoever applicable jurisdiction, as well as with codes of conduct and corporate governance best practices. If one or more committees are established, their composition, powers and operation shall be as determined by the board of directors.
Under Italian law, the members of the board of directors must perform the duties imposed on them by law and company’s bylaws with the degree of diligence that is required by the nature of their office and pursuant to their specific level of competence.
Meetings of our board of directors are called with no less than four days’ notice or, in case of urgency, at least one day’s notice given by registered letter or e-mail sent by the chairman, the deputy chairman or a managing director. Statutory auditors are normally required to attend meetings of the board of directors, but if a meeting has been duly called, the board of directors may validly take action at the meeting even if the board of statutory auditors does not participate. If the meeting has not been duly called, the meeting is nevertheless validly constituted if all of the directors in office and all of the statutory auditors are in attendance.
Meetings of our board of directors may be held in person, or by audio-conference or video-conference, in Italy and in any member state of the European Union or the United States. The quorum for meetings of our board of directors is a majority of the directors in office. Resolutions are adopted by the vote of the majority of the directors in attendance at a meeting at which a quorum is met.
| 152 |
Our directors are not subject to the non-compete obligations provided by Italian law. As a result, our directors may (i) be equity holders in competing companies, including entities where they have unlimited liability, (ii) exercise in person or on behalf of third parties activities in competition with that of the Company, and (iii) be elected as directors of, or act as general managers (direttori generali) in, competing companies.
Under Italian law, directors having any interest in a proposed transaction must disclose their interest to the board of directors and to the board of statutory auditors, even if such interest is not in conflict with the Company’s interest in the same transaction. The interested director is not required to abstain from voting on the resolution approving the transaction, but the resolution must state explicitly the reasons for, and the benefit to us of, the approved transaction. If these provisions are not complied with, or if the transaction would not have been approved without the vote of the interested director, the resolution may be challenged by a director or by our board of statutory auditors if the approved transaction may be prejudicial to us. A director having any interest in a proposed transaction that he or she has authority to approve must solicit prior board approval of such transaction. The interested director may be held liable for damages to us resulting from a resolution adopted in breach of the above rules. Finally, directors may be held liable for damages to us if they illicitly profit from insider information or corporate opportunities.
Our board of directors may transfer the Company’s registered office within Italy, set up and eliminate secondary offices and, if the company’s bylaws (as in our case) so provide, approve mergers by absorption into the Company of any subsidiary in which the Company holds at least 90% of the issued share capital. Our board of directors may also approve the issuance of shares or convertible debentures and reductions of the Company’s share capital in the case of withdrawal of a shareholder if so authorized by the Company’s extraordinary shareholders’ meeting.
Under Italian law, directors may be removed from office at any time by the vote of shareholders at any shareholders’ meeting. Whilst the removal is immediately effective within the company, it is effective vis-à-vis third parties only upon entry of the relevant resolution in the companies register. The resolution of the shareholders’ meeting in favor of a liability suit against a director and the occurrence of causes of ineligibility constitute just cause for removal. If the removal of a director occurs without just cause, such director may have a claim for damages against us. These damages may include, but are not limited to, compensation that would otherwise have been paid to the director for the remainder of his or her term and damage to his or her reputation. Directors may resign at any time by written notice to the board of directors and to the chairman of the board of statutory auditors. The board of directors must appoint substitute directors to fill vacancies arising from the removal or resignation of any director, subject to the approval of the board of statutory auditors, to serve until the next shareholders’ meeting. If, at any time, more than half of the members of our board of directors appointed by the shareholders’ meeting of the Company resign, such resignation is ineffective until the majority of the new board of directors has been appointed. In such a case, the remaining members of the board of directors (or the board of statutory auditors if all members of the board of directors have resigned or cease to be directors) must promptly call a shareholders’ meeting to elect new directors.
Our Compensation, Nomination and Governance Committee will recommend the compensation of our directors to the board of directors, which in turn will make recommendations to our shareholders. Under Italian law, our shareholders determine the compensation of our directors relating to basic board service, such as annual fees for serving on the board and/or fees for attending board meetings. The board of directors, after consultation with the board of statutory auditors, may determine the remuneration of directors that serve on the various board committees and/or perform management or other special services for the Company, such as managing directors and general directors. Our directors are entitled to reimbursement for expenses incurred in connection with their service as directors, such as travel expenses to attend board meetings.
Statutory auditors
The board of statutory auditors (Collegio Sindacale) is elected by the shareholders and must be comprised of individuals qualified to act in such capacity under Italian law. Statutory auditors are elected for a term of three fiscal years, they may be re-elected for successive terms and may be removed only for cause and with the approval of a competent court. Each member of the board of statutory auditors must provide evidence that such individual is qualified to act in such capacity under Italian law and meets certain professional standards.
| 153 |
Under Italian law, at least one standing statutory auditor and one substitute statutory auditor must be chosen among persons registered with the Register of Auditors established with the Italian Ministry of Justice. The other statutory auditors shall be chosen among those registered with any register established by decree of the Ministry of Justice or among University professors in economics or law, if they are not registered with the Register of Auditors. The following persons may not serve as statutory auditors:
| ● | a person legally incapacitated, bankrupt, or disqualified from holding public or an executive office under Italian law; | |
| ● | the spouse, parent or relative-in-law of a director of the company or an affiliate of the company; and | |
| ● | a person whose independence may be impaired due to an employment or consulting relationship or any other economic relationship with the company or an affiliate of the company. |
Our bylaws currently provides that the board of statutory auditors shall consist of three standing statutory auditors and two alternate auditors (who will automatically replace a statutory auditor who resigns or is otherwise unable to serve).
Following the expiration of the members of the board of statutory auditors currently in office, future members will be elected by means of a slate voting mechanism: slates may be submitted by shareholders representing, individually or collectively with other shareholders joining in the submission of the slate, at least 6% of share capital eligible to vote at the shareholders’ meeting at which auditors are to be elected, such eligibility to be established by filing with the Company an appropriate certification to that effect. The slates of candidates shall be split into two sections, one for candidates for the position of statutory auditor and one for candidates for the position of alternate auditor. According to the Company’s bylaws, the board of statutory auditors will be elected as follows: (a) two statutory auditors and one alternate auditor shall be drawn from the slate that has obtained the highest number of votes on the shareholders meeting (the Majority Slate), in the order in which their name is listed in the Majority Slate, and(b) the remaining statutory auditor (who will act as President of the board of statutory auditors) and alternate auditor shall be drawn from the slate that receives the second greatest number of votes cast (the Minority Slate) so long as none of the shareholders joining in the submission of the Minority Slate has any connection whatsoever, whether directly or indirectly, with the shareholders who have submitted or voted the Majority Slate.
Each shareholder and shareholders belonging to the same group shall not submit, or contribute join in submitting, or cast their vote for more than one slate, including through a nominee or agent. Each candidate for election as statutory auditor or alternate auditor may only be listed on single slate and will be ineligible for election if named on multiple slates.
In the event that the Majority Slate does not contain a sufficient number of candidates to fill the number of vacancies on the board of statutory auditors to be filled as provided under paragraph (a) above, all candidates listed in the Majority Slate shall be elected statutory auditors and the remaining statutory auditors shall be drawn from the Minority Slate according to the order in which they are listed in such slate. The voting procedure according to slates provided above shall be applicable only in case of election of the entire board of statutory auditors. In the event of a tie between slates, a new vote shall be taken and the candidates obtaining the largest number of votes shall be elected without regard to the slate on which they are listed or application of the slate voting mechanism. Should a single slate be submitted, the shareholders eligible to vote at the meeting shall cast their vote on such slate and, so long as more votes are cast for such slate than votes cast against such slate, all the members of the board of statutory auditors shall be elected from that slate in accordance with applicable law at the time. If no slates are submitted, or a single slate is submitted and such slate does not obtain the requisite number of votes, or the number of statutory auditors to be elected on the basis of the slates submitted is less than the full number of statutory auditors to be elected, or the board of statutory auditors is not to be entirely elected, or it is otherwise not possible for any reason to elect the board of statutory auditors in accordance with the provisions of this title, the members of the board of statutory auditors shall be elected at the shareholders’ meeting in accordance with generally applicable procedures and required majorities under applicable law, without application of the slate voting mechanism.
In the event of the resignation or removal termination of a member of the board of statutory auditors who was drawn from the Majority Slate or from the Minority Slate, as the case may be, alternate statutory auditors drawn from the same slate shall fill the vacancy, with older alternates being seated before younger alternates, subject to compliance with the requirements of our bylaws regarding the composition of the board of statutory auditors
The board of statutory auditors is required, among other things, to verify that we:
| ● | comply with applicable laws and our bylaws; | |
| ● | respect principles of good governance; and | |
| ● | maintain adequate organizational structure, internal controls and administrative and accounting system. |
The board of statutory auditors is required to meet at least once every ninety days and is expected to attend meetings of our board of directors and our shareholders. In case a statutory auditor, without just cause, does not attend the shareholders’ meetings or does not attend two consecutive meetings of the board of directors or of the board of statutory auditors during the same fiscal year, such statutory auditor shall automatically be removed from office. Our statutory auditors may decide to call a shareholders’ meeting, ask for information about our management from our directors, carry out inspections and verifications at our offices and exchange information with our external auditors. Any shareholder may submit a complaint to our board of statutory auditors regarding facts that the shareholder believes should be investigated and if shareholders collectively representing 5% of our corporate capital, or 2% of the corporate capital should our company be classified as an Open Company (as defined in Section “Applicable Laws”), submit such a complaint, our board of statutory auditors must promptly undertake an investigation and present its findings and any recommendations to a shareholders’ meeting (which must be convened immediately if the complaint appears to have a reasonable basis and there is an urgent need to take action). Our board of statutory auditors may report serious breaches of directors’ duties to a competent court which then may take such actions as inspecting the company’s operations, removing directors, appointing temporary administrators to manage the company or any other actions that the court feels is necessary to preserve the value of our company for our creditors and shareholders.
| 154 |
External auditor
Mayer Hoffman McCann, P.C. remains the PCAOB-registered, independent registered public accounting firm for the purposes of all of our filings in the United States; however, Italian law requires us to appoint an external auditor or a firm of external auditors (revisore legale dei conti), each of them qualified to act in such capacity under Italian law, to verify during the fiscal year that our accounting records are correctly kept and accurately reflect our activities, and to audit our financial statements for statutory filings.This is separate and apart from the work of Mayer Hoffman McCann, P.C. The Italian external auditor’s opinion on the financial statements must be published in Italy.
The Italian external auditor is appointed for a three-year term by the vote of our shareholders at an ordinary shareholders’ meeting. Once appointed, the shareholders may remove the auditors only for cause and with the approval of the board of statutory auditors and of a competent court. Following removal, the Company and the external auditor or auditing firm must timely inform the Italian Ministry of Economics and Finance, explaining the reasons justifying the removal. In case we are considered an Open Company, such communication must be notified also to Consob.
On May 20, 2021, our shareholders appointed Kreston GV Italy – Audit S.r.l., subject to the effective listing of the Company on the Nasdaq Capital Market, as our Italian external auditors for three-year term expiring at the time of the annual shareholders meeting to approve the financial statement for the financial year ending on December 31, 2023.
Meetings of shareholders
Shareholders are entitled to attend and vote at ordinary and extraordinary shareholders’ meetings. Votes may be cast personally or by proxy. Shareholders’ meetings may be called by the board of directors (or by the board of statutory auditors) and must be called if requested by holders of at least 10% of the outstanding shares. Shareholders are not entitled to request that a meeting of shareholders be convened to vote on issues which as a matter of law fall within the authority of the board of directors. If the shareholders’ meeting is not called despite a shareholders’ request and such refusal is unjustified, a competent court may call the meeting.
We may hold meetings of shareholders at our registered office, or elsewhere within Italy and any member state of the European Union or in the United States. Our bylaws provides that shareholders’ meetings, both ordinary and extraordinary, shall be called by notice published in the Italian daily newspaper “Il Sole 24 Ore” at least 15 days prior to the date of the meeting or otherwise given by our Company, provided that so long as the Company is allowed by applicable Italian law, by sending, either in the alternative or in addition to the foregoing published notice, a notice to the shareholders, to the members of the board of directors and to the members of the board of statutory auditors, by: (a) registered mail with return receipt requested; (b) e-mail with electronic acknowledgment of receipt; as long as the notice is received by the addressee at least eight calendar days before the date set for the meeting with evidence thereof as provided above. The notice shall also be posted on our Company’s web site. However, if the above procedures are not complied with, the shareholders’ meeting will still be deemed validly held if all outstanding shares are represented, all other holders having the right to vote are present and a majority of the board of directors and the board of statutory auditors are in attendance; provided that in such event, any shareholder or other participant present may object to the discussion of items with respect to which he/she does not deem to have been adequately informed.
We must convene an ordinary shareholders’ meeting at least once a year. Our annual stand-alone financial statements are prepared by the board of directors and submitted for approval to the ordinary shareholders’ meeting, which must be convened within 120 days after the end of the fiscal year to which such financial statements relate. This term may be extended by up to 180 days after the end of the fiscal year, as long as the Company continues to be bound by law to draw up financial statements or if particular circumstances concerning our structure or our purposes so require. At ordinary shareholders’ meetings, our shareholders also appoint the external auditors, approve any distribution of dividends that have been proposed by our board of directors, elect our board of directors and statutory auditors, determine their remuneration and vote on any business matter for which resolution or authorization is entrusted to the shareholders by law.
| 155 |
We may call extraordinary shareholders’ meetings to vote upon spin-offs, split-ups, dissolutions, appointment of receivers and similar extraordinary actions. We may also call extraordinary shareholders’ meetings to vote upon proposed amendments to our bylaws, issuance of convertible debentures, mergers and de-mergers and capital increases and reductions, if such actions may not be authorized by the board of directors without shareholder approval. The board of directors has the authority to transfer our registered office within Italy, authorize, on a non-exclusive basis, amendments to our bylaws that are required by law, authorize mergers by absorption to our subsidiaries in which we hold all or at least 90% of the outstanding shares, if the Company’s bylaws (as in our case) so provide, authorize reductions of our share capital in case of withdrawal of a shareholder and indicate who among the directors is our legal representative. If the shareholders authorize the issuance of shares or other securities at an extraordinary meeting, they may delegate the power to make specific issuances to the board of directors.
Once our shareholders have authorized the issuance of securities, the securities that have been subscribed must be fully paid for before the shareholders may authorize the issuance of additional securities, unless the shareholders meet and vote to cancel those authorized but unsubscribed securities.
The notice of our shareholders’ meeting may specify two or more meeting dates for an ordinary or extraordinary shareholders’ meeting.
The quorum for an ordinary meeting of our shareholders on the first call, also in the event we are considered an Open Company, is at least 50% of the outstanding ordinary shares, while on second call there is no quorum requirement. In either case, resolutions may be adopted with the majority of ordinary shares in attendance or represented at the meeting. The quorum for an extraordinary shareholders’ meeting is more than half of the outstanding ordinary shares on the first call and more than one-third of the outstanding shares on second call. Extraordinary shareholders’ meeting resolutions may be adopted with the majority of the outstanding ordinary shares on first call and at least two-thirds of the holders of shares in attendance or represented at the meeting on second call. In the event we are considered an Open Company, the quorum for an extraordinary shareholders’ meeting is, on first call, the majority of at least two-thirds of the shares present or represented at such meeting, provided that a quorum of at least half of the share capital is attending; on second call, the majority of at least two-thirds of the shares present or represented at such meeting, provided that a quorum of more than one-third of the share capital is attending. In addition, certain matters (such as, for example, a change in our corporate purpose, the transfer of our registered office outside Italy or our liquidation prior to the date set forth in our bylaws) must be adopted by shareholders representing more than one-third of the outstanding ordinary shares (not just the ordinary shares in attendance or represented at the meeting).
Shareholders are entitled to one vote per ordinary share. Neither Italian law nor our bylaws limit the right of non-resident or foreign owners to hold or vote their shares. Shareholders do not need to present their share certificates (due to the fact that our shares are not represented by share certificates) but the entitlement to attend the meetings is then proven by exhibition of the certificate or communication to the Company made by the intermediary (banks and other financial institutions) which manages the electronical register whereby ownership of the shares, their transfer, the related rights and restrictions on shares (if any) are recorded.
Shareholders may appoint proxies or attorneys-in-fact by delivering in writing the proxies to represent them in an ordinary or extraordinary shareholders’ meeting. Our directors, auditors and employees may not be proxies. Italian law provides that no proxy may represent more than twenty shareholders prior to the company “seeking access to the risk capital market.” It is unsettled whether listing shares on a stock exchange outside of the European Union constitutes “seeking access to the risk capital market” for this purpose.
Preemptive rights
Pursuant to Italian law, holders of outstanding ordinary shares and convertible debentures are entitled to subscribe for newly issued ordinary shares or convertible debentures in proportion to their holdings at the time that the shareholders authorize the capital increase for those issuances, unless those issuances are for non-cash considerations. Those who exercise their preemptive rights, provided they make such request simultaneously, have a pre-emption right on the purchase of shares and debentures convertible into shares that have not been subscribed. Preemptive rights may be excluded or limited by resolution of the shareholders at an extraordinary shareholders’ meeting, or by the board of directors if the bylaws delegate such power to the board of directors (including the power to exclude or limit the preemptive right), and provided that such exclusion or limitation is in the interest of the company, or if the shares are to be paid by means of contributions in kind. According to Italian law proposals to increase share capital with exclusion or limitation of preemptive rights must be accompanied by a report of the board of directors setting forth the reasons for the exclusion or limitation of pre-emptive rights, or, if the exclusion derives from a contribution in kind, the reasons for such contribution in kind, and the report must in all cases set forth the criteria adopted for determining the issue price. The report must be communicated by the board of directors to the board of statutory auditors and to the external auditor at least 30 days prior to the date set for the shareholders’ meeting. Within 15 days, the board of statutory auditors must express its opinion on the fairness of the issue price of the shares. The opinion of the board of statutory auditors and, only in the case of contributions in kind, the sworn report of an expert appointed by a competent court or documentation provided by Italian law, must remain deposited at the Company’s registered office during the 15 days prior to the shareholders’ meeting and until the latter has passed a resolution. The resolution shall determine the issue price of the shares on the basis of of shareholders’ equity, taking into account, in the case of shares listed on regulated markets, also the trend in prices over the last six months. The foregoing procedure shall apply also in case of capital increase delegated to the board of directors.
| 156 |
Preference shares; other securities
Italian law permits us to issue preference shares with limited voting rights, other classes of equity securities with different economic and voting rights, shares with economic rights related to the results of the corporate activity in a specific sector, “participation instruments” with limited economic and voting rights against the contribution, by shareholders or third-parties, of work or services, as well as “participation instruments” in favor of employees.
Our bylaws allow us to issue shares without voting rights, with voting rights limited to particular subjects, with voting rights subject to the occurrence of particular conditions not merely arbitrary or with multiple voting rights (each multiple voting right share may have a maximum of three votes). According to Italian law, the total value of such shares may not exceed half of the share capital. Our bylaws also provides that, in relation to the quantity of shares held by the same party, the voting right may be limited to a maximum extent or may be staggered.
“Participation instruments” may include convertible equity and/or “hybrid” instruments that confer on the holder the right to vote on specific matters and/or grants economic rights, to be determined at the time of the issuance of such instruments, or management rights, such as the right to appoint, in accordance with the procedures established by the bylaws, an independent member of the board of directors or a statutory auditor.
Our bylaws currently allow us to issue these securities. We may also issue convertible and non-convertible debt securities. In order to issue convertible debt securities, our board of directors would need to recommend to our shareholders that they approve the issuance of particular securities in connection with a capital increase, and the shareholders would need to vote to approve such an issuance and capital increase at an extraordinary meeting. The board of directors would also need to recommend, and the shareholders would need to approve by vote at the extraordinary meeting, specific terms of the securities. The shareholders may vote at the extraordinary shareholders’ meeting to delegate authority to the board of directors to issue those securities from time to time, but not for more than five years from the date of the extraordinary shareholders’ meeting.
Debt-equity ratio
Italian law provides that we may not issue debt securities for an amount exceeding twice the value of the sum of our equity capital, our legal reserve and any other disposable reserves appearing on the latest balance sheet approved by our shareholders. The board of statutory auditors must certify compliance with such limitation. This limitation may be exceeded if the debt securities issued in excess are intended for subscription by professional investors subject to prudential supervision pursuant to special laws. In the event of subsequent circulation of the debt security, whoever transfers them is liable for the solvency of the company vis-à-vis buyers who are not professional investors. The rules indicated above do not apply in case we intend to issue debt securities to be listed on regulated markets or multilateral trading systems or which have attached the right to purchase or subscribe shares. The legal reserve is a reserve to which we are required to allocate 5% of our Italian GAAP net income each year until it equals at least 20% of our equity capital. One of the other reserves that we maintain on our balance sheet is a “share premium reserve”, meaning amounts paid for our ordinary shares in excess of the amount of such ordinary shares that is allocated to stated capital (valore nominale). Until our outstanding debt securities are repaid in full, we may not voluntarily reduce our equity capital or distribute our reserves (such as by declaring dividends) in the event the aggregate of the capital plus reserves, after giving effect to such reduction, is less than half of the outstanding amount of the debt securities. If our equity capital is reduced by losses or otherwise such that the amount of the outstanding debt securities is more than twice the amount of our equity capital, we cannot distribute profits to our shareholders until the ratio between the amount of our debt securities and our equity capital plus reserves is restored. If our equity capital is reduced, we could recapitalize by means of issuing new shares or having our current shareholders contribute additional capital to our company, although there can be no assurance that we would be able to find purchasers for new shares or that any of our current shareholders would be willing to contribute additional capital. The legal requirements regarding the ratio of debt securities to equity capital plus reserves do not apply to issuances of debt securities to professional investors (as defined by Italian law). However, in such a case, should the professional investors transfer such debt securities to third parties not qualified as professional investors, the former remains liable for the payment of such securities.
| 157 |
Reduction of equity by losses
Italian law requires us to reduce our shareholders’ equity in certain situations. Our shareholders’ equity has three main components: capital, legal reserves and other shareholders’ equity (such as share premium and retained earnings). We first apply our losses from operations against our shareholders’ equity other than legal reserves and capital. If additional losses remain and, after the legal reserves, our corporate capital is reduced by more than one-third, our board of directors must call a shareholders’ meeting as soon as possible. The shareholders should take appropriate measures, which may include, among others, either reducing the legal reserves and capital by the amount of the remaining losses, or carrying the losses forward for up to one year. If the shareholders vote to elect to carry the losses forward for up to one year, and the losses are still more than one-third of the amount of the capital at the end of the year, then we must reduce our capital by the amount of the losses.
We have no present intention to enter into any such transaction and no such transaction is currently in effect.
Liquidation rights
Pursuant to Italian law and subject to the satisfaction of the claims of all creditors, our shareholders are entitled to a distribution in liquidation that is equal to an amount resulting from the division of the positive liquidation balance by the number of shares (to the extent available out of our net assets). Preferred shareholders and holders of “participating certificates” typically do not participate in the distribution of assets of a dissolved corporation beyond their established contractual preferences. Once the rights of preferred shareholders and holders of participating certificates and the claims of all creditors have been fully satisfied, holders of ordinary shares are entitled to the distribution of any remaining assets.
Purchase of shares by us (Treasury Stocks)
We are permitted to purchase our outstanding shares, subject to certain conditions and limitations provided for by Italian law. We may only purchase the shares out of profits available for dividends or out of distributable reserves, in each case as appearing on the latest shareholder-approved financial statements; in lack, the shares in excess must be cancelled and the corporate capital must be reduced accordingly. Further, we may only repurchase fully paid-in shares. Such purchases and the conditions thereto must be authorized by our shareholders by vote at an ordinary shareholders’ meeting and the authorization may be issued for a period not exceeding the term of eighteen (18) months.
A corresponding reserve equal to the purchase price of such shares must be created in the balance sheet, and such reserve is not available for distribution, unless such shares are sold or cancelled. Shares purchased and held by us may be resold only pursuant to a resolution of our shareholders adopted at an ordinary shareholders’ meeting. The voting rights attaching to the shares held by us or our subsidiaries cannot be exercised, but the shares can be counted for quorum purposes in shareholders’ meetings. Dividends and other rights, including pre-emptive rights, attaching to such shares will accrue to the benefit of other shareholders.
The foregoing limitations do not apply in case we purchase our shares: (i) by giving execution to shareholders’ meeting resolution authorizing capital reduction through repurchase or cancellation; (ii) for free, to the extent they are fully paid-in; (iii) as a consequence of universal succession, merger or demerger; (iv) on occasion of foreclosures authorized to satisfy a credit of our company, to the extent they are fully paid-in.
As long as such shares remain the property of the company, the right to profits and the right of option are attributed proportionally to the other shares. The right to vote is suspended, but such shares are nevertheless taken into account for the purposes of calculating the majority and the quorum required for the constitution and for the resolutions of the shareholders’ meetings.
The Company does not hold any of its shares.
| 158 |
Notification of the acquisition of shares
In accordance with Italian antitrust laws, the Italian Antitrust Authority could prohibit, if certain threshold requirements are met, the acquisition of control in a company which would thereby create or strengthen a dominant position in the domestic market or a significant part thereof and which would result in the elimination or substantial reduction, on a lasting basis, of competition, provided that certain turnover thresholds are exceeded. However, if the turnover of the acquiring party and the company to be acquired exceed certain other monetary thresholds, the antitrust review of the acquisition falls within the exclusive jurisdiction of the European Commission.
In addition, if we fall under the scope of the Law Decree No. 21 of March 15, 2012 (so-called Italian “Golden Power” regulations), as subsequently amended and supplemented, (i) certain resolutions of the Company and, if specific thresholds requirements are met, (ii) certain third-party investors’ purchases of our shares may be subject to ad hoc notifications to the Italian Government and this latter may object to the transaction thereof.
In particular, in such cases the Government would have, among others:
(i) the power to veto or to impose specific conditions with respect to the acquisition of certain shareholdings by any foreign entity outside the European Union with respect to companies having assets and business in sectors of strategic importance; and
(ii) the power to veto or impose specific conditions with regard to the adoption of specific corporate resolutions, acts or transactions by the same companies.
Minority shareholders’ rights; withdrawal rights
Shareholders’ resolutions which are not adopted in conformity with applicable law or our bylaws may be challenged (with certain limitations and exceptions) within 90 days of such resolution (or, if such resolution is subject to registration or filing with the Italian Company Register, within ninety days of its registration or filing) by absent, dissenting or abstaining shareholders representing individually or in the aggregate at least 5% of our share capital (as well as by our board of directors or our board of statutory auditors). Shareholders not reaching this threshold or shareholders not entitled to vote at our meetings may only claim damages arising from the challenged resolution.
Dissenting or absent shareholders may withdraw from the company as a result of shareholders’ resolutions approving, among other things, material modifications of our corporate purpose or of the voting rights of our ordinary shares, our transformation from a share corporation into a different legal entity or the transfer of our registered seat outside Italy. In such a case, our other shareholders would have a pre-emptive right to purchase the shares of the withdrawing shareholder. Should no shareholder exercise that pre-emptive right, the shares must be offered to third parties or, in the absence of any third party wishing to buy them, they will be purchased by us by using the available reserves. In the event that no reserve is available, our equity capital must be reduced accordingly. Any repurchase of such shares by us must be on terms authorized by our board of directors, upon consultation with our board of statutory auditors and our external auditor, having regard to our net asset value, our prospective earnings and the market value of our ordinary shares, if any. Under Italian law, we may set forth different criteria in our bylaws for the consideration to be paid to withdrawing shareholders. We have not done so as of the date of this prospectus.
Any shareholder may bring to the attention of the board of statutory auditors facts or acts which such shareholder deems wrongful. If such shareholders represent at least 5% of our share capital, or in case we are considered an Open Company 2% of the same, our board of statutory auditors must investigate without delay and report its findings and recommendations at our shareholders’ meeting. Shareholders representing more than 10% of our share capital, or, in case we are considered an Open Company one-twentieth, have the right to report to the competent court serious breaches of the duties of the directors which may be prejudicial to us or to our subsidiaries. In addition, shareholders representing at least 20% of our share capital may commence derivative suits before the competent court against our directors, statutory auditors and general managers. We may waive or settle the suit unless shareholders holding at least 20% of the shares vote against such waiver or settlement. We will reimburse the legal costs of such action in the event that the claim of such shareholder is successful and the court does not award such costs against the relevant directors, statutory auditors or general managers.
| 159 |
Applicable Laws
As of the date of this prospectus and upon completion of this offering, the Company is and will be governed by the corporate laws of Italy, and is and will be legally considered and treated, according to the Italian Civil Code, as a private company being our shares not listed on a regulated market in Italy or within the European Union.
It should be noted that under Italian corporate law, while joint-stock companies are all the same type of legal entity, there is a distinction between those companies that do not have access to the capital markets (Private Companies) and companies that have such access. This latter category comprises both companies listed on European regulated markets (Public Companies) and companies whose securities are not listed on such markets, insofar as they have completed a significant distribution of their securities among the public (so-called “emittenti aventi strumenti finanziari diffusi tra il pubblico in maniera rilevante”), according to the relevant provisions set forth in Italian Financials’ Consolidated Act and its implementing provisions (Open Companies).
Pursuant to Article 2-bis of the Issuer’s Regulation implemented by Consob (Regolamento emittenti), Open Companies must meet the following requirements:
| a) | having at least 500 shareholders in addition to the majority shareholders who hold at least 5% of the outstanding share capital; and | |
| b) | exceeding at least two out of the following three thresholds: |
| - | total assets side of €4.4 million; | |
| - | total revenues of €8.8 million; and, | |
| - | an average of 50 employees during the year. |
If, following completion of this offering, we issue financial instruments widely distributed among the public, the regulation relating to Open Companies could apply to us.
Stock Exchange Listing
We have reserved the symbol “GNTA” for purposes of listing the ADSs on the Nasdaq Capital Market and have applied to list the ADSs on the Nasdaq Capital Market.
Registrar of Shares
Our share register is currently kept by Spafid S.p.A., which acts as registrar. The share register reflects only record owners of our ordinary shares.
| 160 |
DESCRIPTION OF AMERICAN DEPOSITARY SHARES
The Bank of New York Mellon, as depositary, will register and deliver American Depositary Shares, also referred to as ADSs. Each ADS will represent one share (or a right to receive one share) deposited with The Bank of New York Mellon, as custodian, acting through an office located in the United Kingdom. Each ADS will also represent any other securities, cash or other property that may be held by the depositary. The deposited shares together with any other securities, cash or other property held by the depositary are referred to as the deposited securities. The depositary’s office at which the ADSs will be administered and its principal executive office are located at 240 Greenwich Street, New York, New York 10286.
You may hold ADSs either (A) directly (i) by having an American Depositary Receipt, also referred to as an ADR, which is a certificate evidencing a specific number of ADSs, registered in your name, or (ii) by having uncertificated ADSs registered in your name, or (B) indirectly by holding a security entitlement in ADSs through your broker or other financial institution that is a direct or indirect participant in The Depository Trust Company, also called DTC. If you hold ADSs directly, you are a registered ADS holder, also referred to as an ADS holder. This description assumes you are an ADS holder. If you hold the ADSs indirectly, you must rely on the procedures of your broker or other financial institution to assert the rights of ADS holders described in this section. You should consult with your broker or financial institution to find out what those procedures are.
Registered holders of uncertificated ADSs will receive statements from the depositary confirming their holdings.
As an ADS holder, we will not treat you as one of our shareholders and you will not have shareholder rights. Italian law governs shareholder rights. The depositary will be the holder of the shares underlying the ADSs. As a registered holder of ADSs, you will have ADS holder rights. A deposit agreement among us, the depositary, ADS holders and all other persons indirectly or beneficially holding ADSs sets out ADS holder rights as well as the rights and obligations of the depositary. New York law governs the deposit agreement and the ADSs.
The following is a summary of the material provisions of the deposit agreement. For more complete information, you should read the entire deposit agreement and the form of ADR. Directions on how to obtain copies of those documents are provided under the heading “Where You Can Find More Information.”
Dividends and Other Distributions
How will you receive dividends and other distributions on the shares?
The depositary has agreed to pay or distribute to ADS holders the cash dividends or other distributions it or the custodian receives on shares or other deposited securities, upon payment or deduction of its fees and expenses. You will receive these distributions in proportion to the number of shares the ADSs represent.
Cash. The depositary will convert any cash dividend or other cash distribution we pay on the shares into U.S. dollars, if it can do so on a reasonable basis and can transfer the U.S. dollars to the United States. If that is not possible or if any government approval is needed and cannot be obtained, the deposit agreement allows the depositary to distribute the foreign currency only to those ADS holders to whom it is possible to do so. It will hold the foreign currency it cannot convert for the account of the ADS holders who have not been paid. It will not invest the foreign currency and it will not be liable for any interest.
Before making a distribution, any withholding taxes, or other governmental charges that must be paid will be deducted. See “Taxation.” The depositary will distribute only whole U.S. dollars and cents and will round fractional cents to the nearest whole cent. If the exchange rates fluctuate during a time when the depositary cannot convert the foreign currency, you may lose some of the value of the distribution.
Shares. The depositary may distribute additional ADSs representing any shares we distribute as a dividend or free distribution. The depositary will only distribute whole ADSs. It will sell shares which would require it to deliver a fraction of an ADS (or ADSs representing those shares) and distribute the net proceeds in the same way as it does with cash. If the depositary does not distribute additional ADSs, the outstanding ADSs will also represent the new shares. The depositary may sell a portion of the distributed shares (or ADSs representing those shares) sufficient to pay its fees and expenses in connection with that distribution.
| 161 |
Rights to purchase additional shares. If we offer holders of our securities any rights to subscribe for additional shares or any other rights, the depositary may (i) exercise those rights on behalf of ADS holders, (ii) distribute those rights to ADS holders or (iii) sell those rights and distribute the net proceeds to ADS holders, in each case after deduction or upon payment of its fees and expenses. To the extent the depositary does not do any of those things, it will allow the rights to lapse. In that case, you will receive no value for them. The depositary will exercise or distribute rights only if we ask it to and provide satisfactory assurances to the depositary that it is legal to do so. If the depositary will exercise rights, it will purchase the securities to which the rights relate and distribute those securities or, in the case of shares, new ADSs representing the new shares, to subscribing ADS holders, but only if ADS holders have paid the exercise price to the depositary. U.S. securities laws may restrict the ability of the depositary to distribute rights or ADSs or other securities issued on exercise of rights to all or certain ADS holders, and the securities distributed may be subject to restrictions on transfer.
Other Distributions. The depositary will send to ADS holders anything else we distribute on deposited securities by any means it thinks is legal, fair and practical. If it cannot make the distribution in that way, the depositary has a choice. It may decide to sell what we distributed and distribute the net proceeds, in the same way as it does with cash. Or, it may decide to hold what we distributed, in which case ADSs will also represent the newly distributed property. However, the depositary is not required to distribute any securities (other than ADSs) to ADS holders unless it receives satisfactory evidence from us that it is legal to make that distribution. The depositary may sell a portion of the distributed securities or property sufficient to pay its fees and expenses in connection with that distribution. U.S. securities laws may restrict the ability of the depositary to distribute securities to all or certain ADS holders, and the securities distributed may be subject to restrictions on transfer.
The depositary is not responsible if it decides that it is unlawful or impractical to make a distribution available to any ADS holders. We have no obligation to register ADSs, shares, rights or other securities under the Securities Act. We also have no obligation to take any other action to permit the distribution of ADSs, shares, rights or anything else to ADS holders. This means that you may not receive the distributions we make on our shares or any value for them if it is illegal or impractical for us to make them available to you.
Deposit, Withdrawal and Cancellation
How are ADSs issued?
The depositary will deliver ADSs if you or your broker deposits shares or evidence of rights to receive shares with the custodian. Upon payment of its fees and expenses and of any taxes or charges, such as stamp taxes or stock transfer taxes or fees, the depositary will register the appropriate number of ADSs in the names you request and will deliver the ADSs to or upon the order of the person or persons that made the deposit.
How can ADS holders withdraw the deposited securities?
You may surrender the ADSs to the depositary for the purpose of withdrawal. Upon payment of its fees and expenses and of any taxes or charges, such as stamp taxes or stock transfer taxes or fees, the depositary will deliver the shares and any other deposited securities underlying the ADSs to the ADS holder or a person the ADS holder designates at the office of the custodian. Or, at your request, risk and expense, the depositary will deliver the deposited securities at its office, if feasible. However, the depositary is not required to accept surrender of ADSs to the extent it would require delivery of a fraction of a deposited share or other security. The depositary may charge you a fee and its expenses for instructing the custodian regarding delivery of deposited securities.
How do ADS holders interchange between certificated ADSs and uncertificated ADSs?
You may surrender your ADR to the depositary for the purpose of exchanging your ADR for uncertificated ADSs. The depositary will cancel that ADR and will send to the ADS holder a statement confirming that the ADS holder is the registered holder of uncertificated ADSs. Upon receipt by the depositary of a proper instruction from a registered holder of uncertificated ADSs requesting the exchange of uncertificated ADSs for certificated ADSs, the depositary will execute and deliver to the ADS holder an ADR evidencing those ADSs.
| 162 |
Voting Rights
How do you vote?
ADS holders may instruct the depositary how to vote the number of deposited shares their ADSs represent. If we request the depositary to solicit your voting instructions (and we are not required to do so), the depositary will notify you of a shareholders’ meeting and send or make voting materials available to you. Those materials will describe the matters to be voted on and explain how ADS holders may instruct the depositary how to vote. For instructions to be valid, they must reach the depositary by a date set by the depositary. The depositary will try, as far as practical, subject to the laws of Italy and the provisions of our articles of association or similar documents, to vote or to have its agents vote the shares or other deposited securities as instructed by ADS holders. If we do not request the depositary to solicit your voting instructions, you can still send voting instructions, and, in that case, the depositary may try to vote as you instruct, but it is not required to do so.
Except by instructing the depositary as described above, you will not be able to exercise voting rights unless you surrender the ADSs and withdraw the shares. However, you may not know about the meeting enough in advance to withdraw the shares. In any event, the depositary will not exercise any discretion in voting deposited securities and it will only vote or attempt to vote as instructed.
We cannot assure you that you will receive the voting materials in time to ensure that you can instruct the depositary to vote your shares. In addition, the depositary and its agents are not responsible for failing to carry out voting instructions or for the manner of carrying out voting instructions. This means that you may not be able to exercise voting rights and there may be nothing you can do if your shares are not voted as you requested.
In order to give you a reasonable opportunity to instruct the depositary as to the exercise of voting rights relating to Deposited Securities, if we request the Depositary to act, we agree to give the depositary notice of any such meeting and details concerning the matters to be voted upon at least 45 days in advance of the meeting date.
Fees and Expenses
| Persons depositing or withdrawing shares or ADS holders must pay: | For: | |
| $5.00 (or less) per 100 ADSs (or portion of 100 ADSs) | Issuance of ADSs, including issuances resulting from a distribution of shares or rights or other property. Cancellation of ADSs for the purpose of withdrawal, including if the deposit agreement terminates | |
| $.05 (or less) per ADS | Any cash distribution to ADS holders | |
| A fee equivalent to the fee that would be payable if securities distributed to you had been shares and the shares had been deposited for issuance of ADSs | Distribution of securities distributed to holders of deposited securities (including rights) that are distributed by the depositary to ADS holders | |
| $.05 (or less) per ADS per calendar year | Depositary services | |
| Registration or transfer fees | Transfer and registration of shares on our share register to or from the name of the depositary or its agent when you deposit or withdraw shares | |
| Expenses of the depositary | Cable (including SWIFT) and facsimile transmissions (when expressly provided in the deposit agreement)Converting foreign currency to U.S. dollars | |
| Taxes and other governmental charges the depositary or the custodian has to pay on any ADSs or shares underlying ADSs, such as stock transfer taxes, stamp duty or withholding taxes | As necessary | |
| Any charges incurred by the depositary or its agents for servicing the deposited securities | As necessary |
| 163 |
The depositary collects its fees for delivery and surrender of ADSs directly from investors depositing shares or surrendering ADSs for the purpose of withdrawal or from intermediaries acting for them. The depositary collects fees for making distributions to investors by deducting those fees from the amounts distributed or by selling a portion of distributable property to pay the fees. The depositary may collect its annual fee for depositary services by deduction from cash distributions or by directly billing investors or by charging the book-entry system accounts of participants acting for them. The depositary may collect any of its fees by deduction from any cash distribution payable (or by selling a portion of securities or other property distributable) to ADS holders that are obligated to pay those fees. The depositary may generally refuse to provide fee-attracting services until its fees for those services are paid.
From time to time, the depositary may make payments to us to reimburse us for costs and expenses generally arising out of establishment and maintenance of the ADS program, waive fees and expenses for services provided to us by the depositary or share revenue from the fees collected from ADS holders. In performing its duties under the deposit agreement, the depositary may use brokers, dealers, foreign currency dealers or other service providers that are owned by or affiliated with the depositary and that may earn or share fees, spreads or commissions.
The depositary may convert currency itself or through any of its affiliates, or the custodian or we may convert currency and pay U.S. dollars to the depositary. Where the depositary converts currency itself or through any of its affiliates, the depositary acts as principal for its own account and not as agent, advisor, broker or fiduciary on behalf of any other person and earns revenue, including, without limitation, transaction spreads, that it will retain for its own account. The revenue is based on, among other things, the difference between the exchange rate assigned to the currency conversion made under the deposit agreement and the rate that the depositary or its affiliate receives when buying or selling foreign currency for its own account. The depositary makes no representation that the exchange rate used or obtained by it or its affiliate in any currency conversion under the deposit agreement will be the most favorable rate that could be obtained at the time or that the method by which that rate will be determined will be the most favorable to ADS holders, subject to the depositary’s obligation to act without negligence or bad faith. The methodology used to determine exchange rates used in currency conversions made by the depositary is available upon request. Where the custodian converts currency, the custodian has no obligation to obtain the most favorable rate that could be obtained at the time or to ensure that the method by which that rate will be determined will be the most favorable to ADS holders, and the depositary makes no representation that the rate is the most favorable rate and will not be liable for any direct or indirect losses associated with the rate. In certain instances, the depositary may receive dividends or other distributions from the us in U.S. dollars that represent the proceeds of a conversion of foreign currency or translation from foreign currency at a rate that was obtained or determined by us and, in such cases, the depositary will not engage in, or be responsible for, any foreign currency transactions and neither it nor we make any representation that the rate obtained or determined by us is the most favorable rate and neither it nor we will be liable for any direct or indirect losses associated with the rate.
Payment of Taxes
You will be responsible for any taxes or other governmental charges payable on the ADSs or on the deposited securities represented by any of the ADSs. The depositary may refuse to register any transfer of the ADSs or allow you to withdraw the deposited securities represented by the ADSs until those taxes or other charges are paid. It may apply payments owed to you or sell deposited securities represented by the ADSs to pay any taxes owed and you will remain liable for any deficiency. If the depositary sells deposited securities, it will, if appropriate, reduce the number of ADSs to reflect the sale and pay to ADS holders any proceeds, or send to ADS holders any property, remaining after it has paid the taxes.
Tender and Exchange Offers; Redemption, Replacement or Cancellation of Deposited Securities
The depositary will not tender deposited securities in any voluntary tender or exchange offer unless instructed to do so by an ADS holder surrendering ADSs and subject to any conditions or procedures the depositary may establish.
| 164 |
If deposited securities are redeemed for cash in a transaction that is mandatory for the depositary as a holder of deposited securities, the depositary will call for surrender of a corresponding number of ADSs and distribute the net redemption money to the holders of called ADSs upon surrender of those ADSs.
If there is any change in the deposited securities such as a sub-division, combination or other reclassification, or any merger, consolidation, recapitalization or reorganization affecting the issuer of deposited securities in which the depositary receives new securities in exchange for or in lieu of the old deposited securities, the depositary will hold those replacement securities as deposited securities under the deposit agreement. However, if the depositary decides it would not be lawful and practical to hold the replacement securities because those securities could not be distributed to ADS holders or for any other reason, the depositary may instead sell the replacement securities and distribute the net proceeds upon surrender of the ADSs.
If there is a replacement of the deposited securities and the depositary will continue to hold the replacement securities, the depositary may distribute new ADSs representing the new deposited securities or ask you to surrender your outstanding ADRs in exchange for new ADRs identifying the new deposited securities.
If there are no deposited securities underlying ADSs, including if the deposited securities are cancelled, or if the deposited securities underlying ADSs have become apparently worthless, the depositary may call for surrender of those ADSs or cancel those ADSs upon notice to the ADS holders.
Amendment and Termination
How may the deposit agreement be amended?
We may agree with the depositary to amend the deposit agreement and the ADRs without your consent for any reason. If an amendment adds or increases fees or charges, except for taxes and other governmental charges or expenses of the depositary for registration fees, facsimile costs, delivery charges or similar items, or prejudices a substantial right of ADS holders, it will not become effective for outstanding ADSs until 30 days after the depositary notifies ADS holders of the amendment. At the time an amendment becomes effective, you are considered, by continuing to hold the ADSs, to agree to the amendment and to be bound by the ADRs and the deposit agreement as amended.
How may the deposit agreement be terminated?
The depositary will initiate termination of the deposit agreement if we instruct it to do so. The depositary may initiate termination of the deposit agreement if
| ● | 60 days have passed since the depositary told us it wants to resign but a successor depositary has not been appointed and accepted its appointment; |
| ● | we delist the ADSs from an exchange in the United States on which they were listed and do not list the ADSs on another exchange in the United States or make arrangements for trading of ADSs on the U.S. over-the-counter market; |
| ● | we delist our shares from an exchange outside the United States on which they were listed and do not list the shares on another exchange outside the United States; |
| ● | the depositary has reason to believe the ADSs have become, or will become, ineligible for registration on Form F-6 under the Securities Act of 1933; |
| ● | we appear to be insolvent or enter insolvency proceedings; | |
| ● | all or substantially all the value of the deposited securities has been distributed either in cash or in the form of securities; | |
| ● | there are no deposited securities underlying the ADSs or the underlying deposited securities have become apparently worthless; or | |
| ● | there has been a replacement of deposited securities. |
| 165 |
If the deposit agreement will terminate, the depositary will notify ADS holders at least 90 days before the termination date. At any time after the termination date, the depositary may sell the deposited securities. After that, the depositary will hold the money it received on the sale, as well as any other cash it is holding under the deposit agreement, unsegregated and without liability for interest, for the pro rata benefit of the ADS holders that have not surrendered their ADSs. Normally, the depositary will sell as soon as practicable after the termination date.
After the termination date and before the depositary sells, ADS holders can still surrender their ADSs and receive delivery of deposited securities, except that the depositary may refuse to accept a surrender for the purpose of withdrawing deposited securities or reverse previously accepted surrenders of that kind that have not settled if it would interfere with the selling process. The depositary may refuse to accept a surrender for the purpose of withdrawing sale proceeds until all the deposited securities have been sold. The depositary will continue to collect distributions on deposited securities, but, after the termination date, the depositary is not required to register any transfer of ADSs or distribute any dividends or other distributions on deposited securities to the ADSs holder (until they surrender their ADSs) or give any notices or perform any other duties under the deposit agreement except as described in this paragraph.
Limitations on Obligations and Liability
Limits on our Obligations and the Obligations of the Depositary; Limits on Liability to Holders of ADSs
The deposit agreement expressly limits our obligations and the obligations of the depositary. It also limits our liability and the liability of the depositary. We and the depositary:
| ● | are only obligated to take the actions specifically set forth in the deposit agreement without negligence or bad faith, and the depositary will not be a fiduciary or have any fiduciary duty to holders of ADSs; | |
| ● | are not liable if we are or it is prevented or delayed by law or by events or circumstances beyond our or its ability to prevent or counteract with reasonable care or effort from performing our or its obligations under the deposit agreement; | |
| ● | are not liable if we or it exercises discretion permitted under the deposit agreement; | |
| ● | are not liable for the inability of any holder of ADSs to benefit from any distribution on deposited securities that is not made available to holders of ADSs under the terms of the deposit agreement, or for any special, consequential or punitive damages for any breach of the terms of the deposit agreement; | |
| ● | have no obligation to become involved in a lawsuit or other proceeding related to the ADSs or the deposit agreement on your behalf or on behalf of any other person; | |
| ● | may rely upon any documents we believe or it believes in good faith to be genuine and to have been signed or presented by the proper person; |
are not liable for the acts or omissions of any securities depository, clearing agency or settlement system; and
| ● | the depositary has no duty to make any determination or provide any information as to our tax status, or any liability for any tax consequences that may be incurred by ADS holders as a result of owning or holding ADSs or be liable for the inability or failure of an ADS holder to obtain the benefit of a foreign tax credit, reduced rate of withholding or refund of amounts withheld in respect of tax or any other tax benefit. |
In the deposit agreement, we and the depositary agree to indemnify each other under certain circumstances.
| 166 |
Requirements for Depositary Actions
Before the depositary will deliver or register a transfer of ADSs, make a distribution on ADSs, or permit withdrawal of shares, the depositary may require:
| ● | payment of stock transfer or other taxes or other governmental charges and transfer or registration fees charged by third parties for the transfer of any shares or other deposited securities; | |
| ● | satisfactory proof of the identity and genuineness of any signature or other information it deems necessary; and | |
| ● | compliance with regulations it may establish, from time to time, consistent with the deposit agreement, including presentation of transfer documents. |
The depositary may refuse to deliver ADSs or register transfers of ADSs when the transfer books of the depositary or our transfer books are closed or at any time if the depositary or we think it advisable to do so.
Your Right to Receive the Shares Underlying the ADSs
ADS holders have the right to cancel their ADSs and withdraw the underlying shares at any time except:
| ● | when temporary delays arise because: (i) the depositary has closed its transfer books or we have closed our transfer books; (ii) the transfer of shares is blocked to permit voting at a shareholders’ meeting; or (iii) we are paying a dividend on our shares; | |
| ● | when you owe money to pay fees, taxes and similar charges; or | |
| ● | when it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of shares or other deposited securities. |
This right of withdrawal may not be limited by any other provision of the deposit agreement.
Direct Registration System
In the deposit agreement, all parties to the deposit agreement acknowledge that the Direct Registration System, also referred to as DRS, and Profile Modification System, also referred to as Profile, will apply to the ADSs. DRS is a system administered by DTC that facilitates interchange between registered holding of uncertificated ADSs and holding of security entitlements in ADSs through DTC and a DTC participant. Profile is a feature of DRS that allows a DTC participant, claiming to act on behalf of a registered holder of uncertificated ADSs, to direct the depositary to register a transfer of those ADSs to DTC or its nominee and to deliver those ADSs to the DTC account of that DTC participant without receipt by the depositary of prior authorization from the ADS holder to register that transfer.
In connection with and in accordance with the arrangements and procedures relating to DRS/Profile, the parties to the deposit agreement understand that the depositary will not determine whether the DTC participant that is claiming to be acting on behalf of an ADS holder in requesting registration of transfer and delivery as described in the paragraph above has the actual authority to act on behalf of the ADS holder (notwithstanding any requirements under the Uniform Commercial Code). In the deposit agreement, the parties agree that the depositary’s reliance on and compliance with instructions received by the depositary through the DRS/Profile system and in accordance with the deposit agreement will not constitute negligence or bad faith on the part of the depositary.
| 167 |
Shareholder communications; inspection of register of holders of ADSs
The depositary will make available for your inspection at its office all communications that it receives from us as a holder of deposited securities that we make generally available to holders of deposited securities. The depositary will send you copies of those communications or otherwise make those communications available to you if we ask it to. You have a right to inspect the register of holders of ADSs, but not for the purpose of contacting those holders about a matter unrelated to our business or the ADSs.
Jury Trial Waiver
The deposit agreement provides that, to the extent permitted by law, ADS holders waive the right to a jury trial of any claim they may have against us or the depositary arising out of or relating to our shares, the ADSs or the deposit agreement, including any claim under the U.S. federal securities laws. If we or the depositary opposed a jury trial demand based on the waiver, the court would determine whether the waiver was enforceable in the facts and circumstances of that case in accordance with applicable case law.
You will not, by agreeing to the terms of the deposit agreement, be deemed to have waived our or the depositary’s compliance with U.S. federal securities laws or the rules and regulations promulgated thereunder.
| 168 |
COMPARISON OF ITALIAN LAW AND DELAWARE LAW
The following comparison table summarizing the most significant differences in shareholder rights between the applicable provisions of the Delaware General Corporation Law and the corporate laws of Italy, also taking into account those main differences existing between the Private Companies and Open Companies.
It should be considered that this is only a general summary of certain provisions applicable to companies in Delaware and Italy. Certain Delaware and Italian companies may be permitted to exclude certain of the provisions summarized below in their charter documents.
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Mergers and similar arrangements | ||
| Under the Delaware General Corporation Law, with certain exceptions, a merger, consolidation, sale, lease or transfer of all or substantially all of the assets of a corporation must be approved by the board of directors and a majority of the outstanding shares entitled to vote thereon. A shareholder of a Delaware corporation participating in certain major corporate transactions may, under certain circumstances, be entitled to appraisal rights pursuant to which such shareholder may receive cash in the amount of the fair value of the shares held by such shareholder (as determined by a court) in lieu of the consideration such shareholder would otherwise receive in the transaction. The Delaware General Corporation Law also provides that a parent corporation, by resolution of its board of directors, may merge with any subsidiary of which it owns at least 90.0% of each class of capital stock without a vote by the shareholders of such subsidiary. Upon any such merger, dissenting shareholders of the subsidiary would have appraisal rights. | Under Italian law, a merger requires the approval of more than half of the share capital at an extraordinary shareholders’ meeting of each company which is part to the merger. The merger may only be adopted after the expiry of a period of sixty (60) days from the filing of the merger resolution and the related documents with the companies’ register. This time limit, in particular, is intended to allow the company’s creditors, subject to limited exceptions, to file an opposition to the merger, provided however that if such an opposition is deemed groundless and the company has given adequate financial assurances, the competent court may nevertheless decide that the merger shall take place notwithstanding the opposition.
Italian law also provides for simplified forms of merger (with less documentation needed) in cases where the merger is carried out by combining wholly-owned subsidiaries and subsidiaries which are at least 90% owned. In both cases the decision to merge is delegated to the board of directors, provided that the company’s bylaws (as in our case) so provide. The delegation of powers to the board of directors does not, in any event, release it from the obligation to file and/or publish, as the case may be, the merger plan in order to inform shareholders of the merger and to postpone consummation for 30 days from the filing/publication of the merger plan, unless the shareholders waive their right. Shareholders of the “acquiring” company representing at least 5% of the share capital may request the decision on the merger to be taken by the extraordinary shareholders’ meeting. In such a case the authority to approve the merger shifts to the extraordinary shareholders’ meeting, as provided for the non-simplified procedure. | |
| 169 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Shareholders’ suits | ||
| Class actions and derivative actions generally are available to shareholders of a Delaware corporation for, among other things, breach of fiduciary duty, corporate waste and actions not taken in accordance with applicable law. In such actions, the court has discretion to permit the winning party to recover attorneys’ fees incurred in connection with such action. | Shareholders may bring to the attention of the board of statutory auditors facts or acts which such shareholder deems wrongful. If such shareholders represent 5% of our share capital, or 2% should the company be classified as an Open Company, the board of statutory auditors must investigate without delay and report its findings and recommendations at a shareholders’ meeting.
If there is a well-founded suspicion that the directors, in breach of their duties, have committed serious irregularities in the management of the company’s business that may cause damage to the company, shareholders representing, in Private Companies, one-tenth of the share capital or, in Open Companies, one-twentieth of the share capital can report the facts to a competent court. The court, having heard the directors and the statutory auditors, may order an investigation of the matter. The court will not order an investigation and will delay legal proceedings for a fixed period if the shareholders replace the directors and the statutory auditors and the new directors and auditors take action without delay to ascertain whether wrongdoing has been committed and, if so, to remedy it, report to the court their findings and the actions undertaken by them. If the reported wrongdoing did occur or if the investigations fail to address the matters at issue, the court may order appropriate provisional measures and call a shareholders’ meeting to adopt appropriate resolutions. In the most serious cases, the court can replace directors and the statutory auditors and appoint a receiver, determining his or her powers and duration of office.
Liability claims against directors and general managers may be brought either by the shareholders through resolutions approved at a shareholders’ meeting, by the board of statutory auditors (following a resolution passed with the favorable vote of two thirds of its members) or directly by shareholders representing a specified percentage of the share capital.
With reference to resolutions to be adopted at a shareholders’ meeting, a resolution concerning the liability of directors may be approved at a meeting for that purpose or at the meeting called to approve the financial statements. Unless otherwise provided for in the bylaws, resolution shall require approval by standard majority. Claims against directors are subject to a five year statute of limitations from the date of termination of the director’s office and can be waived or settled upon resolution approved by the shareholder, unless shareholders representing at least one-fifth of the share capital, in Private Companies such as ours, or, in Open Companies, one-twentieth of the share capital, vote against the resolution. The legal costs of such action shall be borne by the company unless the court orders the directors, statutory auditors or general managers to pay them.
Shareholders representing a certain percentage of the share capital may bring claims against directors. In case of a Private Company such as ours, the percentage required is one-fifth of the share capital, while for an Open Company the percentage is one-fortieth of the share capital. | |
| 170 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Dissenters’ rights | ||
| Any shareholder of a Delaware corporation has the right to dissent from any plan of merger or consolidation to which the corporation is a party, except that, unless the certificate of incorporation provides otherwise, a shareholder shall not have the right to dissent from any plan of merger or consolidation, with respect to shares of a class or series that are listed on a national securities exchange or held of record, by not less than 2,000 holders on the record date fixed to determine the shareholders entitled to vote upon the plan of merger or consolidation. A dissenting shareholder has a right to appraisal of its shares. | Under Italian law, shareholders’ resolutions which are not adopted in compliance with applicable law or the bylaws may be challenged (with certain limitations and exceptions) within 90 days of such resolution (or, if such resolution is subject to registration or filing with the Italian Company Register, within 90 days of its registration or filing) by shareholders who did not attend the meeting, dissented or abstained representing, individually or in the aggregate, at least 5% of the share capital in case of a Private Company such as ours, or for an Open Company, at least one per thousand of the share capital, as well as by the board of directors or the board of statutory auditors. Shareholders who do not meet the threshold or are not otherwise entitled to vote at our meetings may only claim damages arising from the resolution. Alongside the challenge to the resolution, an urgent application may also be filed to suspend the implementation of the resolution being challenged, in order to preserve the rights of challengers. Dissenting or absent shareholders may withdraw from the company as a result of shareholders’ resolutions approving, among other things, material modifications to the corporate purpose, the rights attached to ordinary shares (e.g., voting rights and/or economic rights related to ordinary shares), conversion from a share corporation into a different legal entity or a transfer of the registered office outside of Italy. Under any such circumstances, non-withdrawing shareholders would have pre-emptive rights to purchase the shares of the withdrawing shareholders. Should no shareholder exercise its pre-emptive rights, the shares must be offered to third parties. If no third-party desires to purchase the shares, the Company must purchase them out of available reserves. In the event that there are no reserves available, the company must reduce its capital accordingly. According to Italian law, the liquidation value of any such shares must be on terms determined by the board of directors, after consultation with the board of statutory auditors and the external auditor, after careful consideration of the company’s net asset value, prospective earnings and the market value of the ordinary shares. Under Italian law, a company may include provisions in its bylaws governing the criteria for determining the liquidation value of shares in the event of withdrawal. We have not done so as of the date of this offering. | |
| 171 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Shareholder vote on board and management compensation | ||
| Under the Delaware General Corporation Law, the board of directors has the authority to fix the compensation of directors, unless otherwise restricted by the certificate of incorporation or bylaws. | Under Italian Law, the compensation of directors is established at the time of appointment or by resolution adopted at as shareholders’ meeting. The compensation of the chief executive officers shall be established by the board of directors, with input from the board of statutory auditors. If the bylaws so provide, the shareholders’ meeting may determine a total amount for the compensation of all directors, including the chief executive officer. | |
| Annual vote on board renewal | ||
| Unless otherwise specified in the certificate of incorporation or bylaws of the corporation, directors shall be elected by a plurality of votes of the shareholders on a date and at a time designated by or in the manner provided in the bylaws. Re-election is possible. | The board of directors is elected by resolution adopted at the annual shareholders’ meeting of the Company for the period established at the time of election but in no case for longer than three fiscal years. Directors may, but are not required to be, shareholders and may be reappointed for successive terms. | |
| Indemnification of directors and executive management and limitation of liability | ||
The Delaware General Corporation Law provides that a certificate of incorporation may contain a provision eliminating or limiting the personal liability of directors, officers, employees or agents of the corporation for monetary damages for breach of a fiduciary duty as a director, except no provision in the certificate of incorporation may eliminate or limit the liability of a director for:
● any breach of a director’s duty of loyalty to the corporation or its shareholders;
● acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law;
● statutory liability for unlawful payment of dividends or unlawful share purchase or redemption; or
● any transaction from which the director derived an improper personal benefit.
A Delaware corporation may indemnify any person who was or is a party or is threatened to be made a party to any proceeding, other than an action by or on behalf of the corporation, because the person is or was a director or officer, against liability incurred in connection with the proceeding if the director or officer acted in good faith and in a manner reasonably believed to be in, or not opposed to, the best interests of the corporation; and the director or officer, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful.
Unless ordered by a court, any foregoing indemnification is subject to a determination that the director or officer has met the applicable standard of conduct:
● by a majority vote of the directors who are not parties to the proceeding, even though less than a quorum;
● by a committee of directors designated by a majority vote of the eligible directors, even though less than a quorum;
● by independent legal counsel in a written opinion if there are no eligible directors, or if the eligible directors so direct; or
● by the shareholders.
Moreover, a Delaware corporation may not indemnify a director or officer in connection with any proceeding in which the director or officer has been adjudged to be liable to the corporation unless and only to the extent that the court determines that, despite the adjudication of liability but in view of all the circumstances of the case, the director or officer is fairly and reasonably entitled to indemnity for those expenses which the court deems proper. | Italian law requires directors and members of any committee designated by the board of directors to perform their duties with the degree of diligence required by the nature of their office and according to their specific level of competence. Liability should never arise from business judgments under the circumstances, even if the decisions made entail significant economic risks, but be attributable only to lack of diligence by the director in appreciating in advance the risk involved in the transaction to be undertaken, and therefore, the omission of possible precautions, assessments or the lack of information normally required for a decision of that type, taken under those circumstances and in that manner. Directors are liable to the company’s creditors when their improper management conduct impairs the company’s assets and prevents the company from satisfying creditors’ claims. If the company cannot repay its creditors, and a court determines that the directors did not adequately perform their duties relating to the preservation of assets, the court may find directors liable.
| |
| 172 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Directors’ fiduciary duties | ||
A director of a Delaware corporation has a fiduciary duty to the corporation and its shareholders. This duty has two components: the duty of care and the duty of loyalty.
The duty of care requires that a director act in good faith, with the care that an ordinarily prudent person would exercise under similar circumstances. Under this duty, a director must inform himself of, and disclose to shareholders, all material information reasonably available regarding a significant transaction. The duty of loyalty requires that a director act in a manner he reasonably believes to be in the best interests of the corporation. He must not use his corporate position for personal gain or advantage. This duty prohibits self-dealing by a director and mandates that the best interest of the corporation and its shareholders take precedence over any interest possessed by a director, officer or controlling shareholder and not shared by the shareholders generally. In general, actions of a director are presumed to have been made on an informed basis, in good faith and in the honest belief that the action taken was in the best interests of the corporation. However, this presumption may be rebutted by evidence of a breach of one of the fiduciary duties.
Should such evidence be presented concerning a transaction by a director, a director must prove the procedural fairness of the transaction, and that the transaction was of fair value to the corporation. | Under Italian law, directors must perform their duties and responsibilities with the degree of diligence required by the nature of their tasks and according to their level of competence. | |
| 173 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Shareholder action by written consent | ||
| A Delaware corporation may, in its certificate of incorporation, eliminate the right of shareholders to act by written consent. | Under Italian law, shareholders may not act without a meeting. | |
| Special/extraordinary meetings of shareholders | ||
| Under Delaware law, special meetings of shareholders may be called by the board of directors or by such person or persons as may be authorized by the certificate of incorporation or the bylaws. | Under Italian law, a shareholders’ meeting, both ordinary and extraordinary, may be called by the board of directors and must be called if requested by holders of at least 10% of the share capital in the case of a Private Company, as is our case, or for an Open Company, at least 5% of the share capital, unless in either case the bylaws establish a lower threshold. Shareholders are not entitled to request that a meeting of shareholders be convened to vote on issues which as a matter of law are within the authority of the board of directors. If a request by the shareholders for a meeting is rejected by the board of directors, and such refusal is unjustified, the meeting may be called by a competent court. | |
| Shareholder proposals | ||
| A shareholder of a Delaware corporation has the right to put any proposal before the annual meeting of shareholders, provided it complies with the notice provisions in the governing documents. A special meeting may be called by the board of directors or any other person authorized to do so in the governing documents, but shareholders may be precluded from calling special meetings. | Registered holders of the Company’s ordinary shares are entitled to attend and vote at all ordinary and extraordinary shareholders’ meetings.
When shareholders representing at least 10% of the share capital in the case of a Private Company, as is our case, or for an Open Company, at least 5% of the share capital, unless in either case the bylaws establish lower threshold, request the board of directors to call a meeting of shareholders, they must set out in their request the proposals to be discussed. | |
| Cumulative voting | ||
| Under the Delaware General Corporation Law, cumulative voting for elections of directors is not permitted unless the corporation’s certificate of incorporation provides for it. | Under Italian law, shareholders are not entitled to elect directors through cumulative voting. Directors are elected by a majority of the votes cast by the shareholders entitled to vote in person or by proxy at an ordinary meeting of shareholders at which the relevant quorum is met. | |
| Board action by written consent | ||
| Under Delaware law, any action required or permitted to be taken at any meeting of the board of directors may be taken without a meeting if all members of the board consent to the action in writing or by electronic transmission, and the writing or electronic transmission is filed with the minutes of proceedings of the board, unless otherwise restricted by the certificate of incorporation or bylaws. | Under Italian law, directors of a corporation may not act without a meeting. | |
| 174 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Removal of directors | ||
| Unless there is cumulative voting or there is a classified board, generally a director or the entire board of directors may be removed, with or without cause, by the holders of a majority of the shares then entitled to vote at an election of directors. | Under Italian law, a director may be removed from office at any time by the vote of shareholders at an ordinary shareholders’ meeting. The removal is immediately effective against third parties only upon entry of the relevant resolution in the Companies Register.
A resolution approved at a shareholders’ meeting of claim for liability against a director or the occurrence of causes of ineligibility to serve under applicable law constitute just cause for removal.
If the removal of a director is without just cause, the director may have a claim for damages against the company. These damages may include, but are not limited to, compensation that would otherwise have been paid to the director for the remainder of his or her term and damage to his or her reputation.
Directors may resign at any time by written notice to the board of directors and to the chairman of the board of statutory auditors. The board of directors, subject to the approval of the board of statutory auditors, must appoint substitute directors to fill any vacancies caused by removal or resignation of directors, and the substitute will serve until the next ordinary shareholders’ meeting. If, at any time, more than half of the members of the board of directors elected by shareholders resign, such resignation is ineffective until a majority of the new board of directors has been elected. In such a case, the remaining members of the board of directors (or the board of statutory auditors if all the members of the board of directors have resigned or ceased to be directors) must promptly call an ordinary shareholders’ meeting to elect new directors. | |
| Transactions with interested shareholders or directors | ||
| The Delaware General Corporation Law generally prohibits a Delaware corporation from engaging in certain business combinations with an “interested shareholder” for three years following the date that such person becomes an interested shareholder. An interested shareholder generally is a person or group who or which owns or owned 15.0% or more of the corporation’s outstanding voting shares within the past three years. | Italian law provides that a resolution approved at a shareholders’ meeting with the vote of shareholders who have, directly or indirectly, a conflict of interest with the company may be challenged.
Under Italian law, a director having a personal interest in a proposed transaction must disclose his or her interest to the board of directors and to the board of statutory auditors, even if such interest does not conflict with the company’s interest in the transaction. The interested director is not required to abstain from voting on the resolution approving the transaction, but the resolution must explicitly state the reasons for the approved transaction and the benefit of the transaction to the company. If these provisions are not complied with, or if the transaction would not have been approved without the vote of the interested director, the resolution may be challenged within 90 days by any director or by the board of statutory auditors on grounds that the approved transaction would be prejudicial to the company. An authorized representative of the company having any interest in a proposed transaction that he or she has authority to approve must solicit prior board approval before consenting to such transaction. An interested director may be held liable for damages to the company resulting from a resolution adopted in breach of the above requirements. Finally, a director may be held liable for illicitly profiting from insider information or a corporate opportunity. | |
| 175 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Dissolution; winding up | ||
| Unless the board of directors of a Delaware corporation approves the proposal to dissolve, dissolution must be approved by shareholders holding 100.0% of the total voting power of the corporation. Only if the dissolution is initiated by the board of directors may it be approved by a simple majority of the corporation’s outstanding shares. Delaware law allows a Delaware corporation to include in its certificate of incorporation a supermajority voting requirement in connection with dissolutions initiated by the board. | Under Italian law, and subject to the satisfaction of the claims of all creditors, upon liquidation shareholders are entitled to a distribution that is equal to a per share amount equal to positive liquidation balance divided by the number of shares outstanding. Asset distribution to preferred shareholders and holders of “participating certificates” upon corporate dissolution is typically provided by established contractual preferences. Once the rights of preferred shareholders and holders of participating certificates have been fully satisfied, holders of ordinary shares are entitled to any remaining assets. | |
| Variation of rights of shares | ||
| A Delaware corporation may vary the rights of a class of shares with the approval of a majority of the outstanding shares of such class, unless the certificate of incorporation provides otherwise. | Italian law allows the Company to issue preference shares with limited voting rights, other classes of equity securities with different economic and voting rights, shares with economic rights related to the results of the corporate activity in a specific sector, “participation instruments” (strumenti finanziari partecipativi) with limited economic and voting rights against the contribution, by shareholders or third parties, of work or services, as well as “participation instruments” in favor of employees. | |
| 176 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Amendment of governing documents | ||
| A Delaware corporation’s governing documents may be amended with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. | Under Italian law, the charter documents consist of articles of association and bylaws. An amendment to these documents requires the approval of shareholders at an extraordinary shareholders’ meeting with the following majorities for a Private Company, such as ours: on the first call, more than half of the share capital, and on the second call, at least two-thirds of the shares present or represented at the meeting, provided that a quorum of more than one-third of the share capital is present or represented. Certain matters (such as a change in business purpose or corporate form, demergers, mergers, the transfer of the registered office outside Italy, liquidation prior to the term set forth in its bylaws, extension of the company’s term, the revocation of liquidation and the issuance of special categories of shares) must be approved by the extraordinary shareholders’ meeting pursuant to the same requirements.
For an Open Company, according to Italian law, such resolutions are adopted, on the first call, by a majority of at least two-thirds of the shares present or represented at such meeting, provided that a quorum of at least half of the share capital is present or represented; on the second call, by a majority of at least two-thirds of the shares present or represented at such meeting, provided that a quorum of more than one-third of the share capital is present or represented; on the third and subsequent calls, by a majority of at least two-thirds of the shares present or represented at such meeting, provided that a quorum of at least one-fifth of the share capital is present or represented.
Under Italian law, a shareholder who has not participated in a resolution amending the bylaws that involves a change in the rights attached to its shares has the right to withdraw from the company. | |
| 177 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Inspection of Books and Records | ||
| Shareholders of a Delaware corporation, upon written demand under oath stating the purpose thereof, have the right during the usual hours for business to inspect for any proper purpose, and to obtain copies of list(s) of shareholders and other books and records of the corporation and its subsidiaries, if any, to the extent the books and records of such subsidiaries are available to the corporation. | Under Italian law, our shareholders may review the report of the board of directors on the management of the company and the report of the statutory auditors and external auditor on the financial statements during the 15 days prior to the ordinary shareholders’ meeting called to approve those financial statements. The report remains on file at the company’s office and is also filed with the Companies’ Register of Milan and made publicly available. Moreover, any shareholder is entitled to examine the ledger of the minutes of the shareholders’ meeting at any time.
For an Open Company, insofar as the ownership of the share capital may vary on a daily basis and transfers are electronically registered by intermediaries (banks and financial institutions), the record holder shall be determined according to certifications or communications to the company from intermediaries, based to their own accounting records. | |
| Payment of dividends | ||
The board of directors may approve a dividend without shareholder approval. Subject to any restrictions contained in its certificate of incorporation, the board may declare and pay dividends upon the shares of its capital stock either:
● out of its surplus, or
● in case there is no such surplus, out of its net profits for the fiscal year in which the dividend is declared and/or the preceding fiscal year. | Under Italian law, any payment of dividends must be proposed by the board of directors to the shareholders and is subject to shareholder approval at the annual ordinary shareholders’ meeting. Before dividends may be paid out of profits in any year, the company must allocate an amount equal to 5% of the net profit to a legal reserve until such reserve is at least equal to 20% of the corporate capital. If the corporate capital is reduced as a result of accumulated losses, the company may not pay dividends until the capital is reconstituted or reduced by the amount of such losses. The company may distribute reserves constituting earnings retained from prior years, provided that after such payment the company must have a legal reserve at least equal to the legally required minimum of 20% of corporate capital. If the minimum requirement of a reserve equal to 20% of corporate capital is met, the board of directors could propose the payment of a dividend and the shareholders approve it, the shareholders’ resolution must specify the manner and date of dividend payment. Any dividends which shareholders do not collect within five years of the payment date will be forfeited and the money returned to the company. The board of directors may not approve dividends during the period between annual ordinary shareholders’ meetings. | |
| 178 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Return of Capital | ||
| Delaware law provides that corporations may return capital by dividend, redemption or repurchase subject to certain solvency tests. Shareholder approval is not required for these transactions so long as the corporation meets the solvency tests. | Under Italian law, a company are permitted to purchase outstanding shares, subject to certain conditions and limitations (so-called “azioni proprie”). A company may only purchase shares out of profits available for dividends or out of distributable reserves, in each case as appearing on the latest financial statements approved by shareholders. Further, the company may only repurchase fully paid-in shares. Such purchases and the conditions thereto must be authorized by the shareholders at an ordinary shareholders’ meeting and the approval may not be for a period exceeding 18 months. Shares purchases in excess of the amount approved by shareholders must be sold within one year of the date of purchase and, if not sold, must be cancelled and the corporate capital must be reduced accordingly. Similar limitations apply with respect to purchases of shares by subsidiaries.
A reserve equal to the purchase price of such shares must be created on the balance sheet, and such reserve is not available for dividends, unless the shares are sold or cancelled. Shares purchased and held by the company may be resold only pursuant to a resolution adopted at an ordinary shareholders’ meeting. The voting rights attaching to the shares held by the company or its subsidiaries cannot be exercised, but the shares can be counted for quorum purposes. Dividends and other rights, including pre-emptive rights, attaching to such shares, will accrue to the benefit of other shareholders.
The foregoing limitations do not apply in case the company purchases shares: (i) pursuant to a shareholder resolution approved at a shareholders’ meeting authorizing a capital reduction through repurchase or cancellation; (ii) without consideration, to the extent the shares are fully paid-in; (iii) as a consequence of merger or demerger; (iv) by foreclosure to satisfy a debt owed to the company, to the extent the shares are fully paid-in.
As long as such shares remain the property of the company, the right to profits and the right of option are attributed proportionally to the other shares. The right to vote is suspended, but such shares are nevertheless taken into account for the purposes of calculating the majority and the quora required for the constitution and for the resolutions of the shareholders’ meetings. | |
| 179 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Creation and issuance of new shares | ||
Shareholder approval is required to authorize capital stock in excess of that provided in the charter. The corporation must file a certificate of amendment to its certificate of incorporation before the creation of additional authorized shares may become effective.
The board of directors may, without shareholder consent, authorize capital stock to be issued for consideration consisting of cash, any tangible or intangible property or any benefit to the corporation, or any combination thereof. | Under Italian law, the issuance of any shares, ordinary or otherwise, requires an amendment to the bylaws to increase the share capital, which must be recommended to the shareholders by the board of directors and approved by the shareholders at an extraordinary shareholders’ meeting. The board of directors shall then be authorized to issue the securities according to the terms and conditions approved by shareholders. Alternatively, the shareholders can delegate the power to increase the share capital to the board of directors, but the board’s right to exercise such power for up to five years. If the board does not approve a capital increase within five years, the authority expires automatically. With respect to shareholders’ resolutions approving capital increases, Italian law provides that in the absence of meeting minutes or in the event of the impossibility or illegality of the resolution, any interested person may challenge such resolution for a period of 180 days following the filing of the shareholders’ resolution with the Register of Companies. If a shareholders’ meeting was not properly held to approve the capital increase, the relevant resolution should be considered invalid, and any interested person may challenge the capital increase for a period of 90 days following the approval of the financial statements for the year during which the capital increase was executed in whole or in part. Finally, once shareholders authorize a capital increase, all the new authorized shares that have been subscribed must be paid-up in full before a new capital increase may be executed.
By means of shareholders’ extraordinary resolution, the company can also increase its corporate capital (without requesting contributions from its shareholders) by allocating to capital its available reserves and other funds registered in its financial statements (so-called “aumento di capitale gratuito”). In this case, the newly issued shares must have the same characteristics as those previously issued, and they must be placed to the shareholders proportionally to their shareholdings in the company. | |
| 180 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Option Rights | ||
| Under Delaware law, shareholders do not possess option rights with respect to the issuance of additional securities by the corporation, unless the certificate of incorporation provides otherwise. | Under Italian law, shareholders and holders of convertible debentures are entitled to subscribe for newly issued ordinary shares or convertible debentures in proportion to their holdings at the time of authorization of the issuance, except in the case of contributions in kind. Those who exercise such option right, provided they make such request at the same time, have a further pre-emption right with respect to shares and debentures convertible into shares that have not been subscribed. The option rights may be excluded or limited by a shareholders’ resolution approved at an extraordinary meeting of the shareholders, if such exclusion or limitation is in the interest of the company, or if the shares are to be paid for by means of a contribution in kind.
According to Italian law proposals to increase the share capital with the exclusion or limitation of pre-emptive rights must be accompanied by a report of the board of directors setting forth the reasons for the exclusion or limitation, or, if the exclusion derives from a contribution in kind, the reasons for such contribution in kind, and in all cases the criteria adopted for determining the issue price. The report must be communicated by the directors to the board of statutory auditors and to the external auditor at least 30 days prior to the date of the shareholders’ meeting at which the capital increase is to be approved. Within 15 days, the board of statutory auditors must express its opinion on the fairness of the issue price of the new shares. The opinion of the board of statutory auditors and, only in the case of contribution in kind, the sworn report of the expert appointed by a competent court or the documentation provided by Italian law, must be available at the company’s registered office during the 15 days prior to the shareholders’ meeting. Such terms may be waived by unanimous consent of the shareholders. The shareholders’ resolution shall determine the issue price of the shares on the basis of shareholders’ equity, taking into account, in the case of shares listed on a regulated market, also the trend in prices over the last six months. The foregoing procedure shall apply also in case of capital increase delegated to the board of directors. | |
| 181 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Debt-equity ratio | ||
| Under Delaware law, there are no restrictions on the amount of debt securities that a corporation may issue. | Under Italian law, a company may issue debt securities in an amount not to exceed twice the sum of its corporate capital, plus legal reserves and any other disposable reserves appearing on the latest balance sheet approved by the shareholders. The board of statutory auditors must certify compliance with such limitation. This limitation may be exceeded if the debt securities issued in excess are intended for subscription by professional investors subject to regulatory supervision pursuant to special laws. In the event of subsequent circulation of the debt security, whoever transfers them is liable for the solvency of the company vis-à-vis buyers who are not professional investors. The rules indicated above do not apply in case we intend to issue debt securities to be listed on regulated markets or multilateral trading systems or which have attached the right to purchase or subscribe shares.
The legal reserve is a reserve to which a company must allocate 5% of its net income each year until it equals at least 20% of the corporate capital. Another reserve is a “share premium reserve,” which reflects the amount paid for ordinary shares in excess of the amount allocated to corporate capital. Until its outstanding debt securities are repaid in full, a company may not voluntarily reduce its corporate capital or distribute reserves (such as by declaring dividends) if, by doing so, it fails to meet the limitation indicated above. In the event of mandatory capital reduction, or if reserves reduce as a consequence of losses, the company may not distribute dividends until the aggregate of corporate capital and reserves, following such reduction of capital and/or distribution of reserves, is equal or exceeding half of the outstanding principal amount of debt securities. If our equity is reduced by losses or otherwise, such that the principal amount of outstanding debt securities is more than twice the amount of the company’s equity, the company cannot pay dividends to shareholders until the minimum ratio between the amount of debt securities and our capital and reserves is restored. Some legal scholars are of the opinion that the ratio must be restored by a recapitalization of the company. If equity is reduced, a company could recapitalize by means of issuing new shares or having current shareholders contribute additional capital, although there can be no assurance that we would be able to find purchasers for new shares or that any of our current shareholders would be willing to contribute additional capital. These provisions regarding the ratio of debt securities to corporate capital and reserves do not apply to the issuance of debt securities to professional investors (as defined by Italian law). However, professional investors who transfer such debt securities issued by us to third parties not qualified as professional investors would remain liable to our third-party investors for the payment of such securities. | |
| 182 |
| DELAWARE CORPORATE LAW | ITALIAN CORPORATE LAW | |
| Reduction of equity by losses | ||
| Under Delaware law, shareholder equity in a corporation is reduced by losses and may become negative. | Italian law requires a company to reduce its shareholders’ equity in certain situations. Shareholders’ equity has three main components: capital, legal reserves and other shareholders’ equity (such as any premium paid for the shares over the nominal value and any retained earnings). Losses from operations are first offset against shareholders’ equity other than legal reserves and corporate capital. If additional losses remain and, after setting aside legal reserves, corporate capital is reduced by more than one-third as a result of losses, the board of directors must call a shareholders’ meeting as soon as possible. The shareholders, having received an updated report on the company’s financial situation accompanied by a report of the board of statutory auditors, should take appropriate measures, which may include, among others, reducing the legal reserves and corporate capital by the amount of the remaining losses, or carrying the losses forward up to one year. If the shareholders vote to carry the losses forward up to one year, and the losses are still more than one-third of the amount of corporate capital at the end of that year, then the company must reduce its corporate capital by the amount of the losses suffered. An S.p.A. like us must have a corporate capital of at least €50,000. If the amount of the losses would reduce our corporate capital to less than €50,000, then:
● we would need to increase our capital, which we could do by issuing new shares or having our shareholders contribute additional capital to our company, although there can be no assurance that we would be able to find purchasers for new shares or that any of our current shareholders would be willing to contribute additional capital;
● our shareholders would need to convert our company to an “S.r.l.”, a private limited liability company, which has a lower capital requirement of €10,000. Such arrangement would, however, prevent the Company to remain listed on Nasdaq since the S.r.l. form is not consistent with being listed pursuant to Italian law; or
● if neither of these options are pursued, our shareholders or, if the directors do not convene a shareholders’ meeting, a court of competent jurisdiction, at the instance of individuals shareholders, could appoint a liquidator, who need not be an Italian citizen, to liquidate our company. | |
EXCHANGE CONTROLS
No exchange control consent is required in Italy for the transfer of dividends or other distributions with respect to shares of an Italian company or proceeds from the sale thereof to persons outside of Italy.
| 183 |
The following description is not intended to constitute a complete analysis of all tax consequences relating to the ownership or disposition of our ordinary shares. You should consult your own tax advisor concerning the tax consequences of your particular situation, as well as any tax consequences that may arise under the laws of any state, local, foreign, including Italian, or other taxing jurisdiction.
Italy Tax Considerations
General. Under Italian law, financial instruments issued by an Italian company are subject to the same tax regime as are shares, provided that their remuneration is entirely represented by participation in the economic results of the issuer. Pursuant to Article 10(3) of the Income Tax Convention, the tax regime of dividends applies to income from corporate rights of an Italian company, which is subject to the same taxation treatment as income from shares under the laws of Italy.
Income Tax Withholding on Dividends. We do not anticipate making any distributions on our ordinary shares in the foreseeable future. However, if we were to make distributions on our ordinary shares, we would generally be required under Italian law, except as otherwise discussed below, to apply a definitive withholding tax on payments made to holders of our ordinary shares who are not residents of Italy for tax purposes.
Notably, dividends paid to beneficial owners who are not Italian residents and do not have a permanent establishment in Italy are generally subject to a 26 percent substitute tax rate. Therefore, the amount of the dividends that the holders of ADRs or holders of equity shares not residing in Italy will initially receive will be net of such Italian substitute tax.
All non-Italian resident owners of equity shares or ADRs may benefit from reduced withholding tax settled in the relevant anti-double tax treaty undersigned between Italy and the country of residence for tax purposes of the owners of equity shares or ADRs. The reduced withholding tax rate under the relevant anti-double tax treaty will be applicable provided that the non-resident owners of the equity shares or ADRs are able to produce the documentation attesting the requirements to be eligible for the application of the relevant anti-double tax treaty.
Under Italian law, US owners can claim, in accordance with Presidential Decree No. 600 of October 16, 1973, Article 27(3), a refund of up to eleven-twenty-sixths (i.e. 11/26) of the Italian withholding tax withheld on dividends upon presenting evidence to the Italian tax authorities that income taxes have been fully paid on the dividends in the country of residence of the US owners in an amount at least equal to the total refund claimed. US holders should consult an independent tax advisor concerning the availability of this refund, which has traditionally become payable only after extensive delays.
Under the double tax treaty in force between Italy and the United States of America (“US/Italy Income Tax Treaty”), if the payee is the beneficial owner of the payment, dividends paid to US owners will be subject to Italian withholding tax at a reduced rate of: 1) 5%, if the beneficiary is a company owning at least 25% of the payer’s voting shares (for at least 12 months preceding the dividend distribution); 2) 15% in any other case. The aforementioned regime (both 1 and 2) is applicable only if the payee does not carry out an entrepreneurial activity in Italy through a permanent establishment or performs independent personal services through a fixed place situated therein.
Companies or entities subject to corporation tax and resident in States that are European Union Member States or participants in the EEA (included in the list provided for by Italian Ministerial Decree, September 4, 1996, amended and supplemented by Ministerial Decree March 23, 2017) may be entitled to a reduced tax rate of 1.2% on dividends distributed. The pensions funds established in an EU Member State or EEA country may be entitled to a reduced tax rate of 11% or, under certain conditions, to exemption from Italian taxation on dividends.
Income Tax on Capital Gains.
As a general rule, gains from shares in Italian companies, under custody in Italy, could give rise to a taxable income for the non-resident transferor.
| 184 |
Capital Gains exempt from taxation in Italy - “Non-qualified shareholdings” are those which are below 2% of the voting rights and 5% of the capital of an exchange-listed company. Gains from the disposal of non-qualified share investments in Italian listed companies by non-Italian residents are not subject to Italian income tax under domestic rule.
Capital Gains subject to tax in Italy - “Qualified shareholdings” in a listed company are those representing more than the 2% of the voting rights or more than the 5% of the capital of an exchange-listed company. Capital gains from the disposal of a qualified shareholding in a listed company are subject to a withholding tax of 26% under the domestic rule.
The “qualified shareholding” thresholds must be verified over a 12-month monitoring period, starting from the day on which the investor has held at least a qualifying stake, either actual or potential (this rule aimed at preventing that a buy/sale kind of trading resulting in an overall disposition of over 2% in 12 months may result in a qualifying gain having to be declared even when the investor has never owned an actual or potential qualifying stake). As a consequence, all trades cumulatively carried out in a 12-month period should be considered. More in details: (i) until the investor holds a qualifying shareholding at any point in time, trades are not relevant for capital gain purposes, even if the overall amount disposed in a 12-month period exceeds the relevant thresholds; and (ii) starting from the day when the taxpayer holds a qualifying shareholding, all the trades carried out in any consecutive 12 months give rise to qualified capital gains if the overall amount disposed of exceeds one of the relevant thresholds.
However please be informed that in accordance with rules stated in the anti-double tax treaty, in force between Italy and the country of residence for tax purposes of the transferor, is possible to claim the benefit of exemption of the 26% taxation on capital gains. In principle, and more in details, the art. 13 of the OECD model convention basically states that the capital gain is only taxed in the transferor’s country of tax residence. The Italy – U.S.A. anti-double treaty tax convention states a taxation criterion in line with the above. In the light of the above and upon conditions that all the requirements relevant for the application of the Italy – U.S.A. anti-double treaty tax convention are met, an US investor may benefit from the fully exemption of taxation in Italy.
Furthermore, save for any applicable anti-avoidance provision, pursuant to the Income Tax Convention, a US owner will not be subject to Italian capital gain tax or to Italian individual or corporate income tax unless such US owner has a permanent establishment or fixed base in Italy to which the owner’s ordinary shares is effectively connected. To this end, US owners selling ordinary shares and claiming benefits under the Income Tax Convention may be required to produce appropriate documentation establishing that the above-mentioned conditions have been met.
Estate and Gift Tax. Inheritance and gift taxes, which were abolished in 2001, have been re-introduced in the Italian system by Law Decree No. 262 of October 3, 2006 (converted into law, with amendments, by Law Decree No. 286 of November 24, 2006), as amended. Such taxes will apply to the overall net value of the relevant assets, at the following rates, depending on the relationship between the testate (or donor) and the beneficiary (or donee): (a) 4%, if the beneficiary (or donee) is the spouse or a direct ascendant or descendant (such rate only applying on the net asset value exceeding, for each person, €1.0 million); (b) 6%, if the beneficiary (or donee) is a brother or sister (such rate only applying on the net asset value exceeding, for each person, €0.1 million); (c) 6% if the beneficiary (or donee) is another relative within the fourth degree or a direct relative-in-law as well as an indirect relative-in-law within the third degree; and (d) 8% if the beneficiary is a person other than those mentioned under (a), (b) and (c), above. If the beneficiary has a serious disability recognized under applicable law, inheritance and gift taxes will apply to its portion of the net asset value exceeding €1.5 million.
Transfer tax. In connection with the Italian stamp duty tax on the transfer of shares, according to article 37 of Law No. 248 of December 31, 2007, converted with amendments into Law No. 31 of February 28, 2008, the stamp duty has been abolished with regard to contracts having as their object the transfer of shares. In certain cases the relevant transfer acts would be subject to the registration tax at a flat amount equal to €200.
Communications Stamp Duty. A stamp duty has been introduced under article 19 of Law Decree No. 201 of December 6, 2011, converted into Law No. 214 of December 22, 2011, to be imposed on communications (issued by banks and financial intermediaries) to clients relating to securities, even where the deposit of such securities is not mandatory (although certain entities are excluded). The amount of the stamp duty is based on the market value of the securities or, in the absence of a market value, on the nominal amount or the amount payable on redemption. As a general comment, the stamp duty rate is 0.2% on a yearly pro-rata temporis basis (from January 1 up to December 31). The minimum amount is fixed of € 34,20 up to a maximum amount of €14,000. The communication is deemed to be sent to clients at least once a year, even where there is no obligation to issue any such communication.
| 185 |
Financial Transaction Tax. Law 228 of December 24, 2012, Article 1(491 – 500) introduced the Italian Financial Transaction Tax applicable (i) to the transfer of shares and other participative financial instruments issued by companies resident in Italy (“Italian Equity”) and securities representing Italian Equity, regardless of the country where the issuer has its residence (together with Italian Equity are referred to as “Qualifying Equity”); (ii) on the basis of the “value of the transaction”; (iii) regardless of the place where the transaction is concluded and of the State where the parties have their residence; (iv) to transactions on “regulated markets and on multilateral trading facilities” with a reduced rate; (v) to over-the-counter transactions with a full rate.
The taxable event, triggering Italian Financial Transaction Tax, is the transfer of ownership of Qualifying Equity. Securities representing Italian Equity are in scope of the Italian Financial Transaction Tax, regardless of the State where the issuer has its residence. This provision is aimed at including in the scope of the Italian Financial Transaction Tax, American Depository Receipts (“ADRs”), Global Depository Receipts (“GDRs”) and any other certificate of deposit, where the underlying securities are Italian Equity.
The value of the transaction is determined on the basis of the net balance of the transactions settled daily, calculated for each taxpayer with reference to the number of securities traded under the transactions settled in the same day and relating to the same financial instrument.
The calculation is made by the financial intermediary responsible for the payment of the tax, i.e., the one receiving the order to execute the transaction directly from the purchaser or final counterparty.
The Italian Financial Transaction Tax is due by the person in whose favor the transfer of ownership of the Qualifying Equities occurs.
The tax rate applicable is 0.20% while the reduced rate for transactions on “regulated markets and on multilateral trading facilities” is 0.10%.
The tax shall be paid by the 16th day of the month following the one in which the relevant triggering event occurs.
The Italian Financial Transaction Tax does not apply to the transfer of ownership of Italian Equity where the issuing companies are listed in regulated markets and have a market capitalization below 500 million Euros. Such exclusion also applies to the transfer of ownership of securities representing Italian Equity.
U.S. Federal Income Tax Consequences
THE FOLLOWING SUMMARY IS INCLUDED HEREIN FOR GENERAL INFORMATION AND IS NOT INTENDED TO BE, AND SHOULD NOT BE CONSIDERED TO BE, LEGAL OR TAX ADVICE. EACH U.S. HOLDER SHOULD CONSULT WITH HIS OR HER OWN TAX ADVISOR AS TO THE PARTICULAR U.S. FEDERAL INCOME TAX CONSEQUENCES OF THE PURCHASE, OWNERSHIP AND SALE OF ADSS, INCLUDING THE EFFECTS OF APPLICABLE STATE, LOCAL, FOREIGN OR OTHER TAX LAWS AND POSSIBLE CHANGES IN THE TAX LAWS.
Subject to the limitations described in the next paragraph, the following discussion summarizes the material U.S. federal income tax consequences to a “U.S. Holder” arising from the purchase, ownership and sale of the ADSs. For this purpose, a “U.S. Holder” is a holder of ADSs that is: (1) an individual citizen or resident of the United States, including an alien individual who is a lawful permanent resident of the United States or meets the substantial presence residency test under U.S. federal income tax laws; (2) a corporation (or entity treated as a corporation for U.S. federal income tax purposes) or a partnership (other than a partnership that is not treated as a U.S. person under any applicable U.S. Treasury regulations) created or organized under the laws of the United States or the District of Columbia or any political subdivision thereof; (3) an estate, the income of which is includable in gross income for U.S. federal income tax purposes regardless of source; (4) a trust if a court within the United States is able to exercise primary supervision over the administration of the trust and one or more U.S. persons have authority to control all substantial decisions of the trust; or (5) a trust that has a valid election in effect to be treated as a U.S. person to the extent provided in U.S. Treasury regulations.
| 186 |
This summary is for general information purposes only and does not purport to be a comprehensive description of all of the U.S. federal income tax considerations that may be relevant to a decision to purchase the ADSs. This summary generally considers only U.S. Holders that will own the ADSs as capital assets. This summary does not consider the U.S. federal tax consequences to a person that is not a U.S. Holder, nor does it describe the rules applicable to determine a taxpayer’s status as a U.S. Holder. This summary is based on the provisions of the Internal Revenue Code of 1986, as amended, or the Code, final, temporary and proposed U.S. Treasury regulations promulgated thereunder, administrative and judicial interpretations thereof, and the U.S./Italy Income Tax Treaty, all as in effect as of the date hereof and all of which are subject to change, possibly on a retroactive basis, and all of which are open to differing interpretations. We will not seek a ruling from the IRS with regard to the U.S. federal income tax treatment of an investment in the ADSs by U.S. Holders and, therefore, can provide no assurances that the IRS will agree with the conclusions set forth below.
This discussion does not address all of the aspects of U.S. federal income taxation that may be relevant to a particular U.S. holder based on such holder’s particular circumstances and in particular does not discuss any estate, gift, generation-skipping, transfer, state, local, excise or foreign tax considerations. In addition, this discussion does not address the U.S. federal income tax treatment of a U.S. Holder who is: (1) a bank, life insurance company, regulated investment company, or other financial institution or “financial services entity;” (2) a broker or dealer in securities or foreign currency; (3) a person who acquired our securities in connection with employment or other performance of services; (4) a U.S. Holder that is subject to the U.S. alternative minimum tax; (5) a U.S. Holder that holds our securities as a hedge or as part of a hedging, straddle, conversion or constructive sale transaction or other risk-reduction transaction for U.S. federal income tax purposes; (6) a tax-exempt entity; (7) real estate investment trusts or grantor trusts; (8) an expatriate or a former long-term resident of the United States; or (9) a U.S. Holder having a functional currency other than the U.S. dollar. This discussion does not address the U.S. federal income tax treatment of a U.S. Holder that owns, directly or constructively, at any time, securities representing 10% or more of the voting power or value of our shares. Additionally, the U.S. federal income tax treatment of partnerships (or other pass-through entities) or persons who hold securities through a partnership or other pass-through entity are not addressed.
Each prospective investor is advised to consult his or her own tax adviser for the specific tax consequences to that investor of purchasing, holding or disposing of our securities, including the effects of applicable state, local, foreign or other tax laws and possible changes in the tax laws.
Taxation of Dividends Paid on Ordinary Shares
We do not intend to pay dividends in the foreseeable future. In the event that we do pay dividends, and subject to the discussion under the heading “Passive Foreign Investment Companies” below and the discussion of “qualified dividend income” below, a U.S. Holder, other than certain U.S. Holders that are U.S. corporations, will be required to include in gross income as ordinary income the amount of any distribution paid on ordinary shares (including the amount of any Italy tax withheld on the date of the distribution), to the extent that such distribution does not exceed our current and accumulated earnings and profits, as determined for U.S. federal income tax purposes. The amount of a distribution which exceeds our earnings and profits will be treated first as a non-taxable return of capital, reducing the U.S. Holder’s tax basis for the ordinary shares to the extent thereof, and then capital gain. We do not expect to maintain calculations of our earnings and profits under U.S. federal income tax principles and, therefore, U.S. Holders should expect that the entire amount of any distribution generally will be reported as dividend income.
In general, preferential tax rates for “qualified dividend income” and long-term capital gains are applicable for U.S. Holders that are individuals, estates or trusts. For this purpose, “qualified dividend income” means, inter alia, dividends received from a “qualified foreign corporation.” A “qualified foreign corporation” is a corporation that is entitled to the benefits of a comprehensive tax treaty with the United States which includes an exchange of information program. The IRS has stated that the Italy/U.S. Tax Treaty satisfies this requirement and we believe we are eligible for the benefits of that treaty.
| 187 |
In addition, our dividends will be qualified dividend income if our ordinary shares are readily tradable on Nasdaq or another established securities market in the United States. Dividends will not qualify for the preferential rates if we are treated, in the year the dividend is paid or in the prior year, as a PFIC, as described below under “Passive Foreign Investment Companies.” A U.S. Holder will not be entitled to the preferential rate: (1) if the U.S. Holder has not held our ordinary shares for at least 61 days of the 121 day period beginning on the date which is 60 days before the ex-dividend date, or (2) to the extent the U.S. Holder is under an obligation to make related payments on substantially similar property. Any days during which the U.S. Holder has diminished its risk of loss on our ordinary shares are not counted towards meeting the 61-day holding period. Finally, U.S. Holders who elect to treat the dividend income as “investment income” pursuant to Code section 163(d)(4) will not be eligible for the preferential rate of taxation.
The amount of a distribution with respect to our ordinary shares will be measured by the amount of the fair market value of any property distributed, and for U.S. federal income tax purposes, the amount of any Italian taxes withheld therefrom. Cash distributions paid by us in Euros will be included in the income of U.S. Holders at a U.S. dollar amount based upon the spot rate of exchange in effect on the date the dividend is includible in the income of the U.S. Holder, and U.S. Holders will have a tax basis in such Euros for U.S. federal income tax purposes equal to such U.S. dollar value. If the U.S. Holder subsequently converts the Euros into U.S. dollars or otherwise disposes of it, any subsequent gain or loss in respect of such Euros arising from exchange rate fluctuations will be U.S. source ordinary exchange gain or loss.
Taxation of the Disposition of Ordinary Shares
Except as provided under the PFIC rules described below under “Passive Foreign Investment Companies,” upon the sale, exchange or other disposition of our ordinary shares, a U.S. Holder will recognize capital gain or loss in an amount equal to the difference between such U.S. Holder’s tax basis for the ordinary shares in U.S. dollars and the amount realized on the disposition in U.S. dollar (or its U.S. dollar equivalent determined by reference to the spot rate of exchange on the date of disposition, if the amount realized is denominated in a foreign currency). The gain or loss realized on the sale, exchange or other disposition of ordinary shares will be long-term capital gain or loss if the U.S. Holder has a holding period of more than one year at the time of the disposition. Individuals who recognize long-term capital gains may be taxed on such gains at reduced rates of tax. The deduction of capital losses is subject to various limitations.
Passive Foreign Investment Companies
Special U.S. federal income tax laws apply to U.S. taxpayers who own shares of a corporation that is a PFIC. We will be treated as a PFIC for U.S. federal income tax purposes for any taxable year that either:
| ● | 75% or more of our gross income (including our pro rata share of gross income for any company, in which we are considered to own 25% or more of the shares by value), in a taxable year is passive; or | |
| ● | At least 50% of our assets, averaged over the year and generally determined based upon fair market value (including our pro rata share of the assets of any company in which we are considered to own 25% or more of the shares by value) are held for the production of, or produce, passive income. |
For this purpose, passive income generally consists of dividends, interest, rents, royalties, annuities and income from certain commodities transactions and from notional principal contracts. Cash is treated as generating passive income.
We have not made the formal analysis necessary to determine whether or not we are currently a PFIC or whether we have ever been a PFIC, although preliminarily it appears we may have been a PFIC at certain points in the past. The tests for determining PFIC status depend, in part, on the application of complex US federal income tax rules, which are subject to differing interpretations. In addition, whether any corporation will be a PFIC for any taxable year depends on the assets and income of such corporation over the course of each such taxable year and, as a result, it is difficult to make accurate projections of future income and assets which are relevant to this determination for the current taxable year or any future period. Accordingly, there can be no assurance that we currently are not or will not become a PFIC.
| 188 |
If we currently are or become a PFIC, each U.S. Holder who has not elected to mark the shares to market (as discussed below), would, upon receipt of certain distributions by us and upon disposition of our ordinary shares at a gain: (1) have such distribution or gain allocated ratably over the U.S. Holder’s holding period for the ordinary shares, as the case may be; (2) the amount allocated to the current taxable year and any period prior to the first day of the first taxable year in which we were a PFIC would be taxed as ordinary income; and (3) the amount allocated to each of the other taxable years would be subject to tax at the highest rate of tax in effect for the applicable class of taxpayer for that year, and an interest charge for the deemed deferral benefit would be imposed with respect to the resulting tax attributable to each such other taxable year. In addition, when shares of a PFIC are acquired by reason of death from a decedent that was a U.S. Holder, the tax basis of such shares would not receive a step-up to fair market value as of the date of the decedent’s death, but instead would be equal to the decedent’s basis if lower, unless all gain were recognized by the decedent. Indirect investments in a PFIC may also be subject to these special U.S. federal income tax rules.
The PFIC rules described above would not apply to a U.S. Holder who makes a QEF election for all taxable years that such U.S. Holder has held the ordinary shares while we are a PFIC, provided that we comply with specified reporting requirements. Instead, each U.S. Holder who has made such a QEF election is required for each taxable year that we are a PFIC to include in income such U.S. Holder’s pro rata share of our ordinary earnings as ordinary income and such U.S. Holder’s pro rata share of our net capital gains as long-term capital gain, regardless of whether we make any distributions of such earnings or gain. In general, a QEF election is effective only if we make available certain required information. The QEF election is made on a shareholder-by-shareholder basis and generally may be revoked only with the consent of the IRS.
In addition, the PFIC rules described above would not apply if we were a PFIC and a U.S. Holder made a mark-to-market election. A U.S. Holder of our ordinary shares which are regularly traded on a qualifying exchange, including the Nasdaq Capital Market, can elect to mark the ordinary shares to market annually, recognizing as ordinary income or loss each year an amount equal to the difference as of the close of the taxable year between the fair market value of the ordinary shares and the U.S. Holder’s adjusted tax basis in the ordinary shares. Losses are allowed only to the extent of net mark-to-market gain previously included income by the U.S. Holder under the election for prior taxable years.
U.S. Holders who hold our ordinary shares during a period when we are a PFIC will be subject to the foregoing rules, even if we cease to be a PFIC. U.S. Holders are strongly urged to consult their tax advisors about the PFIC rules.
Tax on Net Investment Income
U.S. Holders who are individuals, estates or trusts will generally be required to pay a 3.8% Medicare tax on their net investment income (including dividends on and gains from the sale or other disposition of our ordinary shares), or in the case of estates and trusts on their net investment income that is not distributed. In each case, the 3.8% Medicare tax applies only to the extent the U.S. Holder’s total adjusted income exceeds applicable thresholds.
| 189 |
Information Reporting and Withholding
A U.S. Holder may be subject to backup withholding at a rate of 24% with respect to cash dividends and proceeds from a disposition of ordinary shares. In general, backup withholding will apply only if a U.S. Holder fails to comply with specified identification procedures. Backup withholding will not apply with respect to payments made to designated exempt recipients, such as corporations and tax-exempt organizations. Backup withholding is not an additional tax and may be claimed as a credit against the U.S. federal income tax liability of a U.S. Holder, provided that the required information is timely furnished to the IRS.
A U.S. Holder with interests in “specified foreign financial assets” (including, among other assets, our ordinary shares, unless such ordinary shares are held on such U.S. Holder’s behalf through a financial institution) may be required to file an information report with the IRS if the aggregate value of all such assets exceeds $50,000 on the last day of the taxable year or $75,000 at any time during the taxable year (or such higher dollar amount as may be prescribed by applicable IRS guidance); and may be required to file a Report of Foreign Bank and Financial Accounts if the aggregate value of the foreign financial accounts exceeds $10,000 at any time during the calendar year. You should consult your own tax advisor as to the possible obligation to file such information report.
| 190 |
Roth Capital Partners, LLC and Maxim Group LLC are acting as representatives of the underwriters named below. We have entered into an underwriting agreement dated , 2021 with the representatives. Subject to the terms and conditions of the underwriting agreement, we have agreed to sell to each underwriter named below, and each underwriter named below has severally agreed to purchase, at the public offering price less the underwriting discount set forth on the cover page of this prospectus supplement, the number of ADSs listed next to its name in the following table:
| Underwriter | Number of ADSs | |||
| Roth Capital Partners, LLC | ||||
| Maxim Group LLC | ||||
| Total | ||||
The underwriters are committed to purchase all the ADSs offered by the Company, other than those covered by the over-allotment option to purchase additional ADSs as described below. The obligations of the underwriters may be terminated upon the occurrence of certain events specified in the underwriting agreement. Furthermore, the underwriting agreement provides that the obligations of the underwriters to pay for and accept delivery of the ADSs offered by us in this prospectus are subject to various representations and warranties and other customary conditions specified in the underwriting agreement, such as receipt by the underwriters of officers’ certificates and legal opinions.
We have agreed to indemnify the underwriters against specified liabilities, including liabilities under the Securities Act, and to contribute to payments the underwriters may be required to make in respect thereof.
The underwriters are offering the ADSs subject to prior sale, when, as and if issued to and accepted by them, subject to approval of legal matters by their counsel and other conditions specified in the underwriting agreement. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part.
We have granted the underwriters an over-allotment option. This option, which is exercisable for up to 30 days after the date of this prospectus, permits the underwriters to purchase up to an aggregate of 241,304 additional ADSs (equal to 15% of the total number of ADSs sold in this offering) at the public offering price per share, less underwriting discounts and commissions, solely to cover over-allotments, if any. If the underwriters exercise this option in whole or in part, then the underwriters will be severally committed, subject to the conditions described in the underwriting agreement, to purchase the additional ADSs in proportion to their respective commitments set forth in the prior table.
Discounts, Commissions and Reimbursement
The representatives have advised us that the underwriters propose to offer the ADSs to the public at the initial public offering price per share set forth on the cover page of this prospectus. The underwriters may offer ADSs to securities dealers at that price less a concession of not more than $ per ADS of which up to $ per ADS may be reallowed to other dealers. After the initial offering to the public, the public offering price and other selling terms may be changed by the representatives.
The following table summarizes the underwriting discounts and commissions, advisory fees payable to the underwriters and proceeds, before expenses, to us assuming both no exercise and full exercise by the underwriters of their over-allotment option:
| Total | |||||||||||
| Per ADS | Without Option | With Option | |||||||||
| Public offering price | $ | $ | $ | ||||||||
| Underwriting discounts and commissions (public investors, 7%) | |||||||||||
| Advisory fee (Reserved Offering, 4.5%) | $ | $ | $ | ||||||||
| Proceeds, before expenses, to us | $ | $ | $ | ||||||||
| 191 |
The underwriters have agreed that the maximum aggregate underwriting discounts and commissions shall not exceed 6.5% of the total gross proceeds from the offering. In addition, we have also agreed to pay the reasonable and documents expenses of the underwriters relating to the offering (including the costs of document preparation, production and distribution (such as printing and binding, and photocopies), and third-party research and database services, and the reasonable fees and disbursements of independent counsel), up to an aggregate of $400,000.
We estimate the expenses of this offering payable by us, not including underwriting discounts and commissions, will be approximately $ .
Representative Warrants
Upon the closing of this offering, we have agreed to issue to Roth Capital Partners LLC and Maxim Partners LLC warrants, or the Warrants, to purchase a number of ADSs equal to 4% of the total number of ADSs sold in this public offering to investors other than (i) non-US retail, family office or institutional investors identified by the Company and mutually agreed with Roth Capital Partners LLC and (ii) existing investors in the Company. The Warrants will be exercisable at a per share exercise price equal to 125% of the public offering price per ADS sold in this offering. The Warrants are exercisable at any time and from time to time, in whole or in part, during the four and one-half year period commencing six months from the deemed effective date of the registration statement related to this offering. The Warrants also provide for one demand registration right of the ADSs underlying the Warrants, and unlimited “piggyback” registration rights with respect to the registration of the ADSs underlying the Warrants and customary antidilution provisions. The demand registration right provided will not be greater than five years from the date of the underwriting agreement related to this offering in compliance with FINRA Rule 5110(g)(8)(D). The piggyback registration right provided will not be greater than seven years from the date of the underwriting agreement related to this offering in compliance with FINRA Rule 5110(g)(8)(D).
The Warrants and the ADSs underlying the Warrants have been deemed compensation by the Financial Industry Regulatory Authority, or FINRA, and are therefore subject to a 180-day lock-up pursuant to Rule 5110(g)(1) of FINRA. The holder, or permitted assignees under such rule, may not sell, transfer, assign, pledge, or hypothecate the Warrants or the ADSs underlying the Warrants, nor will the holder engage in any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of the Warrants or the underlying ADSs for a period of 180 days from the deemed effective date of the registration statement. Additionally, the Warrants may not be sold transferred, assigned, pledged or hypothecated for a 180-day period following the deemed effective date of the registration statement except to any underwriter and selected dealer participating in the offering and their bona fide officers or partners. The Warrants will provide for adjustment in the number and price of the Warrants and the ADSs underlying such Warrants in the event of recapitalization, merger, stock split or other structural transaction, or a future financing undertaken by us.
Reserved Offering/Directed Share Program
At our request, 1,000,000 ordinary shares covered by the registration statement of which this prospectus is part have been reserved for sale, at the initial public offering price, by the Company, directly or through agents, in the Reserved Offering to existing shareholders of the Company, as a mitigant for the exclusion of pre-emptive rights as resolved by the general shareholders meeting of the Company in accordance with Italian corporate law and our by-laws. The Reserved Offering will be effected in compliance with the laws of Italy, at the same price to investors as the offering of ADSs. We will receive the proceeds from the Reserved Offering minus an advisory fee payable to the underwriters equal to 4.5% of the offering price of ordinary shares sold to existing shareholders. The offer and sale of ordinary shares in the Reserved Offering (i) is contingent upon the completion of the offering of ADSs and will commence and be consummated concurrently with such offering of ADSs, (ii) has been registered for purposes of the securities laws of the United States under the registration statement of which this prospectus is part, and (iii) is conducted in Italy under applicable exemptions from Italian and European regulations concerning prospectus.
In addition, at our request, the underwriters have reserved up to ordinary shares, representing % of the ADSs offered by this prospectus, for sale to investors identified by our directors and management through a directed share program to be administered at our direction by Roth Capital Partners, LLC. If and to the extent that these persons purchase ADSs, those purchases will reduce the number of ADSs available for sale to the general public Ordinary shares committed to be purchased by such persons that are not so purchased will be reallocated for sale as ADSs to the general public in the offering. All such sales of reserved ordinary shares will be made at the initial public offering price per ADS set forth on the cover page of this prospectus, less an underwriting discount of %.
| 192 |
Lock-Up Agreements
We have agreed with the underwriters that we will not, without the prior consent of the representatives, for a period of 180 days following the date of this prospectus, offer, sell, contract to sell, pledge, grant any option to purchase, purchase any option or contract to sell, right or warrant to purchase, make any short sale, file a registration statement with respect to any ordinary shares or ADSs or any securities that are convertible into or exercisable or exchangeable for ordinary shares or ADSs, or otherwise transfer or dispose of (including entering into any swap or other agreement that transfers to any other entity, in whole or in part, any of the economic consequences of ownership interest): (1) the ADSs and depositary shares representing our ordinary shares; (2) shares of our controlled affiliates and depositary shares representing those shares; and (3) securities that are substantially similar to such ADSs or depositary shares. We have also agreed to cause affiliates to abide by the restrictions of the lock-up agreement. In addition, each of our directors and executive officers and each beneficial owner of 10% or more of the ADSs or ordinary shares will abide by similar 180-day lock-up agreement with respect to their ordinary shares, ADSs, depositary shares representing the ADSs and securities that are substantially similar to the ADSs or depositary shares representing our ordinary shares, subject to customary exceptions for transfers among affiliates. The restrictions of our lock-up agreement do not apply to: (1) the issuance of securities pursuant to our employee share incentive plan which is described in this prospectus, and (2) a transfer by us to our affiliate, provided that such transfer is not a disposition for value and that such affiliate agrees to be bound in writing by the restrictions set forth in the lock-up agreement to which we are subject. The exceptions also permit our executive officers and directors and other existing security holders, subject to certain restrictions, to transfer ADSs, depositary shares representing the ADSs and securities that are substantially similar to the ADSs or our ordinary shares: (i) as a bona fide gift or gifts, (ii) to the person’s immediate family or any trust, partnership or similar entity for the direct or indirect benefit of the person or their immediate family, (iii) by operation of law, such as pursuant to a qualified domestic order or as required by a divorce settlement, (iv) as a distribution to the person’s limited partners or stockholders, (v) to the person’s affiliates or any investment fund or other entity controlled or managed by the person, or (vi) by will, other testamentary document or intestate succession upon death, including to the transferee’s nominee or custodian. Furthermore, during the lock-up period, our executive officers, directors, and other existing security holders may sell ordinary shares purchased from us in the Reserved Offering and ADSs purchased from the underwriters or in the open market following this offering, provided that such sales are not required to be reported in any public report or filing with the SEC or otherwise, and the seller does not voluntarily effect any public filing or report regarding such sales during the lock-up period.
Indemnification
We have agreed to indemnify the several underwriters against certain liabilities, including certain liabilities under the Securities Act. If we are unable to provide this indemnification, we have agreed to contribute to payments the underwriters may be required to make in respect of those liabilities.
Other Relationships
Some of the underwriters and their affiliates have engaged in, and may in the future engage in, investment banking and other commercial dealings in the ordinary course of business with us or our affiliates. They have received, or may in the future receive, customary fees and commissions for these transactions.
No Public Market
Prior to this offering, there has not been a public market for our securities in the U.S. and the public offering price for the ADSs will be determined through negotiations between us and the underwriters. Among the factors to be considered in these negotiations will be prevailing market conditions, our financial information, market valuations of other companies that we and the underwriters believe to be comparable to us, estimates of our business potential, the present state of our development and other factors deemed relevant.
We offer no assurances that the initial public offering price will correspond to the price at which the ADSs will trade in the public market subsequent to this offering or that an active trading market for the ADSs will develop and continue after this offering.
| 193 |
Stock Exchange
We have applied to list the ADSs on the Nasdaq Capital Market under the symbol “GNTA.” There can be no assurance that we will be successful in listing the ADSs on the Nasdaq Capital Market.
Electronic Distribution
A prospectus in electronic format may be made available on websites or through other online services maintained by one or more of the underwriters of this offering, or by their affiliates. Other than the prospectus in electronic format, the information on any underwriter’s website and any information contained in any other website maintained by an underwriter is not part of this prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or any underwriter in its capacity as underwriter, and should not be relied upon by investors.
Stabilization
In connection with this offering, the underwriters may engage in stabilizing transactions, over-allotment transactions, syndicate-covering transactions, penalty bids and purchases to cover positions created by short sales.
Stabilizing transactions permit bids to purchase ADSs so long as the stabilizing bids do not exceed a specified maximum, and are engaged in for the purpose of preventing or retarding a decline in the market price of the ADSs while the offering is in progress.
Over-allotment transactions involve sales by the underwriters of ADSs in excess of the number of ADSs the underwriters are obligated to purchase. This creates a syndicate short position which may be either a covered short position or a naked short position. In a covered short position, the number of ADSs over-allotted by the underwriters is not greater than the number of ADSs that they may purchase in the over-allotment option. In a naked short position, the number of ADSs involved is greater than the number of ADSs in the over-allotment option. The underwriters may close out any short position by exercising their over-allotment option and/or purchasing ADSs in the open market.
Syndicate covering transactions involve purchases of ADSs in the open market after the distribution has been completed in order to cover syndicate short positions. In determining the source of ADSs to close out the short position, the underwriters will consider, among other things, the price of ADSs available for purchase in the open market as compared with the price at which they may purchase ADSs through exercise of the over-allotment option. If the underwriters sell more shares than could be covered by exercise of the over-allotment option and, therefore, have a naked short position, the position can be closed out only by buying ADSs in the open market. A naked short position is more likely to be created if the underwriters are concerned that after pricing there could be downward pressure on the price of the ADSs in the open market that could adversely affect investors who purchase in the offering.
Penalty bids permit the representatives to reclaim a selling concession from a syndicate member when the ADSs originally sold by that syndicate member are purchased in stabilizing or syndicate covering transactions to cover syndicate short positions.
These stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market price of the ADSs or preventing or retarding a decline in the market price of the ADSs. As a result, the price of the ADSs in the open market may be higher than it would otherwise be in the absence of these transactions. Neither we nor the underwriters make any representation or prediction as to the effect that the transactions described above may have on the price of the ADSs. These transactions may be effected on Nasdaq in the over-the-counter market or otherwise and, if commenced, may be discontinued at any time.
| 194 |
Other Relationships
Certain of the underwriters and their affiliates may in the future provide various investment banking, commercial banking and other financial services for us and our affiliates for which they may in the future receive customary fees.
Offer restrictions outside the United States
Other than in the United States, no action has been taken by us or the underwriters that would permit a public offering of the securities offered by this prospectus in any jurisdiction where action for that purpose is required. The securities offered by this prospectus may not be offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with the offer and sale of any such securities be distributed or published in any jurisdiction, except under circumstances that will result in compliance with the applicable rules and regulations of that jurisdiction. Persons into whose possession this prospectus comes are advised to inform themselves about and to observe any restrictions relating to the offering and the distribution of this prospectus. This prospectus does not constitute an offer to sell or a solicitation of an offer to buy any securities offered by this prospectus in any jurisdiction in which such an offer or a solicitation is unlawful.
Australia
This prospectus is not a disclosure document under Chapter 6D of the Australian Corporations Act, has not been lodged with the Australian Securities and Investments Commission and does not purport to include the information required of a disclosure document under Chapter 6D of the Australian Corporations Act. Accordingly, (i) the offer of the securities under this prospectus is only made to persons to whom it is lawful to offer the securities without disclosure under Chapter 6D of the Australian Corporations Act under one or more exemptions set out in section 708 of the Australian Corporations Act, (ii) this prospectus is made available in Australia only to those persons as set forth in clause (i) above, and (iii) the offeree must be sent a notice stating in substance that by accepting this offer, the offeree represents that the offeree is such a person as set forth in clause (i) above, and, unless permitted under the Australian Corporations Act, agrees not to sell or offer for sale within Australia any of the securities sold to the offeree within 12 months after its transfer to the offeree under this prospectus.
China
The information in this document does not constitute a public offer of the securities, whether by way of sale or subscription, in the People’s Republic of China (excluding, for purposes of this paragraph, Hong Kong Special Administrative Region, Macau Special Administrative Region and Taiwan). The securities may not be offered or sold directly or indirectly in the PRC to legal or natural persons other than directly to “qualified domestic institutional investors.”
European Economic Area
In relation to each member state of the European Economic Area (each, a relevant member state), no securities have been offered or will be offered pursuant to the offering described in this prospectus to the public in that relevant member state prior to the publication of a prospectus in relation to the securities that has been approved by the competent authority in that relevant member state, all in accordance with the Prospectus Regulation, except that offers of securities may be made to the public in that relevant member state at any time:
• to qualified investors as defined under article 2(e) of the Prospectus Regulation;
• to fewer than 150 natural or legal persons (other than qualified investors as defined under article 2(e) of the Prospectus Regulation); or
• in any other circumstances falling within article 1(4) of the Prospectus Regulation,
provided that no such offer of securities shall require us or any underwriter to publish a prospectus pursuant to article 3 of the Prospectus Regulation or supplement a prospectus pursuant to article 23 of the Prospectus Regulation.
For purposes of this provision, the expression an “offer to the public” in relation to any securities in any relevant member state means the communication in any form and by any means of sufficient information on the terms of the offer and any securities to be offered so as to enable an investor to decide to purchase or subscribe for any securities, and the expression “Prospectus Regulation” means Regulation (EU) 2017/1129.
Israel
The securities have not been approved or disapproved by the Israeli Securities Authority (the ISA), or ISA, nor have such securities been registered for sale in Israel. The shares may not be offered or sold, directly or indirectly, to the public in Israel, absent the publication of a prospectus. The ISA has not issued permits, approvals or licenses in connection with the offering or publishing the prospectus; nor has it authenticated the details included herein, confirmed their reliability or completeness, or rendered an opinion as to the quality of the securities being offered. Any resale in Israel, directly or indirectly, to the public of the securities offered by this prospectus is subject to restrictions on transferability and must be effected only in compliance with the Israeli securities laws and regulations.
Japan
The securities have not been and will not be registered under Article 4, paragraph 1 of the Financial Instruments and Exchange Law of Japan (Law No. 25 of 1948), as amended (the “FIEL”) pursuant to an exemption from the registration requirements applicable to a private placement of securities to Qualified Institutional Investors (as defined in and in accordance with Article 2, paragraph 3 of the FIEL and the regulations promulgated thereunder). Accordingly, the securities may not be offered or sold, directly or indirectly, in Japan or to, or for the benefit of, any resident of Japan other than Qualified Institutional Investors. Any Qualified Institutional Investor who acquires securities may not resell them to any person in Japan that is not a Qualified Institutional Investor, and acquisition by any such person of securities is conditional upon the execution of an agreement to that effect.
| 195 |
Switzerland
The securities may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (“SIX”) or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering material relating to the securities may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering material relating to the securities have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of securities will not be supervised by, the Swiss Financial Market Supervisory Authority (FINMA).
This document is personal to the recipient only and not for general circulation in Switzerland.
United Arab Emirates
Neither this document nor the securities have been approved, disapproved or passed on in any way by the Central Bank of the United Arab Emirates or any other governmental authority in the United Arab Emirates, nor has the Company received authorization or licensing from the Central Bank of the United Arab Emirates or any other governmental authority in the United Arab Emirates to market or sell the securities within the United Arab Emirates. This document does not constitute and may not be used for the purpose of an offer or invitation. No services relating to the securities, including the receipt of applications and/or the allotment or redemption of such shares, may be rendered within the United Arab Emirates by the Company.
No offer or invitation to subscribe for securities is valid or permitted in the Dubai International Financial Centre.
United Kingdom
Neither the information in this document nor any other document relating to the offer has been delivered for approval to the Financial Services Authority in the United Kingdom and no prospectus (within the meaning of section 85 of the Financial Services and Markets Act 2000, as amended (“FSMA”) has been published or is intended to be published in respect of the securities. This document is issued on a confidential basis to “qualified investors” (within the meaning of section 86(7) of FSMA) in the United Kingdom, and the securities may not be offered or sold in the United Kingdom by means of this document, any accompanying letter or any other document, except in circumstances which do not require the publication of a prospectus pursuant to section 86(1) FSMA. This document should not be distributed, published or reproduced, in whole or in part, nor may its contents be disclosed by recipients to any other person in the United Kingdom.
Any invitation or inducement to engage in investment activity (within the meaning of section 21 of FSMA) received in connection with the issue or sale of the securities has only been communicated or caused to be communicated and will only be communicated or caused to be communicated in the United Kingdom in circumstances in which section 21(1) of FSMA does not apply to the Company.
In the United Kingdom, this document is being distributed only to, and is directed at, persons (i) who have professional experience in matters relating to investments falling within Article 19(5) (investment professionals) of the Financial Services and Markets Act 2000 (Financial Promotions) Order 2005 (“FPO”), (ii) who fall within the categories of persons referred to in Article 49(2)(a) to (d) (high net worth companies, unincorporated associations, etc.) of the FPO or (iii) to whom it may otherwise be lawfully communicated (together “relevant persons”). The investments to which this document relates are available only to, and any invitation, offer or agreement to purchase will be engaged in only with, relevant persons. Any person who is not a relevant person should not act or rely on this document or any of its contents.
Canada
The securities may be sold in Canada only to purchasers purchasing, or deemed to be purchasing, as principal that are accredited investors, as defined in National Instrument 45-106 Prospectus Exemptions or subsection 73.3(1) of the Securities Act (Ontario), and are permitted clients, as defined in National Instrument 31-103 Registration Requirements, Exemptions and Ongoing Registrant Obligations. Any resale of the securities must be made in accordance with an exemption from, or in a transaction not subject to, the prospectus requirements of applicable securities laws. Securities legislation in certain provinces or territories of Canada may provide a purchaser with remedies for rescission or damages if this prospectus (including any amendment thereto) contains a misrepresentation, provided that the remedies for rescission or damages are exercised by the purchaser within the time limit prescribed by the securities legislation of the purchaser’s province or territory. The purchaser should refer to any applicable provisions of the securities legislation of the purchaser’s province or territory for particulars of these rights or consult with a legal advisor. Pursuant to section 3A.3 of National Instrument 33-105 Underwriting Conflicts (NI 33-105), the underwriters are not required to comply with the disclosure requirements of NI33-105 regarding underwriter conflicts of interest in connection with this offering.
| 196 |
Set forth below is an itemization of the total expenses, excluding underwriting discounts, expected to be incurred in connection with the offer and sale of the ordinary shares by us. With the exception of the SEC registration fee and the FINRA filing fee, all amounts are estimates:
| SEC registration fee | $ | 3,241 | ||
| Nasdaq listing fee | $ | 4,740 | ||
| FINRA filing fee | $ | 5,744 | ||
| Printer fees and expenses | $ | 15,000 | ||
| Legal fees and expenses | $ | 875,000 | ||
| Accounting fees and expenses | $ | 280,000 | ||
| Miscellaneous | $ | 25,000 | ||
| Total | $ | 1,208,725 |
Certain legal matters concerning this offering will be passed upon for us by Loeb & Loeb LLP, New York, New York. Certain legal matters with respect to the legality of the issuance of the securities offered by this prospectus will be passed upon for us by Giovannelli e Associati, Studio Legale, Italy. Certain legal matters related to the offering will be passed upon for the underwriters by Goodwin Procter LLP, New York, New York. LCA Studio Legale, Italy is representing the underwriters with respect to certain matters of Italian law.
The financial statements of Genenta for its fiscal years ended December 31, 2020 and 2019 included herein have been audited by Mayer Hoffman McCann P.C., independent registered public accounting firm, as set forth in their report thereon. Such financial statements are included in this prospectus and registration statement in reliance upon such report given on the authority of such firm as experts in accounting and auditing. The address of Mayer Hoffman McCann P.C. is 13500 Evening Creek Drive North #450, San Diego, California, United States.
ENFORCEABILITY OF CIVIL LIABILITIES
We are incorporated under the laws of Italy and our registered office and domicile is located in Milan, Italy. Moreover, a majority of our directors and executive officers are not residents of the United States, and all or a substantial portion of our assets are located outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon us or upon such persons or to enforce against them judgments obtained in U.S. courts, including judgments in actions predicated upon the civil liability provisions of the federal securities laws of the United States.
We have been advised by our Italian counsel that the recognition and enforcement of foreign judgements in Italy is regulated either by (i) treaties or conventions, bilateral or multilateral, between Italy and the foreign country, whose court issued the judgement, or (ii) Italian Law no. 218 of May 31, 1995 (the “International Private Law Act”). In this regard, the provisions of the applicable treaties and conventions, if any, prevail on the provisions of the International Private Law Act. Indeed, Section 2 of the International Private Law Act states that the provisions of the International Private Law Act are “without prejudice to the application of the international conventions binding on Italy”.
That said, Italian counsels advise us that there exist no treaties or other conventions in existence between the Republic of Italy and the United States, or between the Republic of Italy and the US laws, relating to the recognition and enforcement of civil judgments. There follows that a civil judgment of a court of New York or of a United States federal court applying New York law will be recognized in Italy under the general provisions of the International Private Law Act.
| 197 |
Section 64 of the International Private Law Act provides that a judgment issued in a foreign country is recognized in Italy, without any proceedings (i.e., without a rehearing on the merits) being necessary, if all of the following conditions are met:
a) the judge who issued the judgment had the power to decide the case pursuant to the principles on jurisdiction provided by Italian law;
b) the writ of summons (or equivalent pleading) was duly served upon the defendant in compliance with the applicable provisions of the lex fori (i.e., the application of the rules of the legal system to which the judge belongs);
c) the parties entered an appearance, or their default was duly declared in compliance with the applicable provisions of the lex fori;
d) the judgment to be recognized is a final judgment subject to no further appeal;
e) the judgment to be recognized does not contrast with a final judgment issued by an Italian Court;
f) there are no pending proceedings between the same parties and in relation to the same matter, which proceedings were commenced prior to the commencement of the foreign proceedings; and
g) the judgment to be recognized does not conflict with Italian public order.
When the foreign civil judgment must be enforced in Italy, the above-mentioned conditions must be verified by the Italian Court of Appeal based in the area where the judgment must be executed, as indicated in Article 67 of the International Private Law Act. The subsequent decision of the competent Court of Appeal constitutes the title to enforce the foreign decision. Whether these requirements are met in respect of a judgment based upon the civil liability provisions of the United States securities laws, including whether the award of monetary damages under such laws would constitute a penalty, is an issue for the court making such decision.
Subject to the foregoing, investors may be able to enforce in Italy judgments in civil and commercial matters that have been obtained from U.S. federal or state courts. Nevertheless, we cannot assure you that those judgments will be recognized or enforceable in Italy.
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We have filed with the SEC a registration statement on Form F-1 under the Securities Act relating to this offering of our ordinary shares. This prospectus does not contain all of the information contained in the registration statement. The rules and regulations of the SEC allow us to omit certain information from this prospectus that is included in the registration statement. Statements made in this prospectus concerning the contents of any contract, agreement or other document are summaries of all material information about the documents summarized, but are not complete descriptions of all terms of these documents. If we filed any of these documents as an exhibit to the registration statement, you may read the document itself for a complete description of its terms.
The SEC maintains an Internet website that contains reports and other information regarding issuers that file electronically with the SEC. Our filings with the SEC are also available to the public through the SEC’s website at www.sec.gov.
Upon completion of this offering, we will be subject to the information reporting requirements of the Exchange Act that are applicable to foreign private issuers, and under those requirements will file reports with the SEC. Those other reports or other information may be inspected without charge at the locations described above. As a foreign private issuer, we will be exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we will not be required under the Exchange Act to file annual, quarterly and current reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act. However, we will file with the SEC, within 120 days after the end of each fiscal year, or such applicable time as required by the SEC, an annual report on Form 20-F containing financial statements audited by an independent registered public accounting firm, and will submit to the SEC, on Form 6-K, unaudited quarterly financial information.
We maintain a corporate website at www.genenta.com. Information contained on, or that can be accessed through, our website does not constitute a part of this prospectus. We have included our website address in this prospectus solely as an inactive textual reference.
| 198 |
YEARS ENDED DECEMBER 31, 2020 AND 2019
SIX MONTHS ENDED JUNE 30, 2021 AND 2020
| Statements of Operations and Comprehensive Loss | F-28 |
| Balance Sheets | F-29 |
| Statements of Changes in Quotaholders’ Equity | F-30 |
| Statements of Cash Flows | F-31 |
| Notes to the Financial Statements | F-32 |
| F-1 |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Quotaholders of Genenta Science S.r.l.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Genenta Science S.r.l. (the “Company”) as of December 31, 2020 and 2019, and the related statements of operations and comprehensive loss, changes in quotaholders’ equity, and cash flows for each of the two years in the period ended December 31, 2020, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2020 and 2019, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2020, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
We have served as the Company’s auditor since 2020.
/s/ Mayer Hoffman McCann P.C.
San Diego, California
April 22, 2021
| F-2 |
Statements of Operations and Comprehensive Loss
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| Operating expenses | ||||||||
| Research and development | € | 4,688,461 | € | 3,702,982 | ||||
| General and administrative | 901,765 | 921,520 | ||||||
| Total operating expenses | 5,590,226 | 4,624,502 | ||||||
| Loss from operations | (5,590,226 | ) | (4,624,502 | ) | ||||
| Other income (expense) | ||||||||
| Other income | 5,966 | 36,331 | ||||||
| Finance expense | (7,754 | ) | (9,552 | ) | ||||
| Total other income (expense), net | (1,788 | ) | 26,779 | |||||
| Loss before income taxes | (5,592,014 | ) | (4,597,723 | ) | ||||
| Income tax benefit (expense) | - | - | ||||||
| Net loss | (5,592,014 | ) | (4,597,723 | ) | ||||
| Comprehensive loss | - | - | ||||||
| Total comprehensive loss | € | (5,592,014 | ) | € | (4,597,723 | ) | ||
The accompanying notes are an integral part of these financial statements.
| F-3 |
Balance Sheets
| At December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | € | 15,465,243 | € | 20,141,379 | ||||
| Prepaid expenses and other current assets | 947,501 | 458,118 | ||||||
| Prepaid expenses and other current assets - related party | 121,432 | - | ||||||
| Total current assets | 16,534,176 | 20,599,497 | ||||||
| Non-current assets | ||||||||
| Property and equipment, net | 18,971 | - | ||||||
| Other asset- related party | 3,350 | - | ||||||
| Other non-current assets | 945,618 | 748,681 | ||||||
| Total non-current assets | 967,939 | 748,681 | ||||||
| Total assets | € | 17,502,115 | € | 21,348,178 | ||||
| Liabilities and quotaholders’ equity | ||||||||
| Current liabilities | ||||||||
| Accounts payable | € | 544,988 | € | 279,699 | ||||
| Accounts payable - related party | 10,027 | 292,741 | ||||||
| Accrued expenses | 365,969 | 213,973 | ||||||
| Accrued expenses - related party | 1,359,191 | 1,651,620 | ||||||
| Other current liabilities | 53,243 | 48,000 | ||||||
| Total current liabilities | 2,333,418 | 2,486,033 | ||||||
| Long-term liabilities | ||||||||
| Retirement benefit obligation | 17,388 | 11,332 | ||||||
| Total long-term liabilities | 17,388 | 11,332 | ||||||
| Commitments and contingencies (Note 13) | ||||||||
| Quotaholders’ equity | ||||||||
| Corporate capital | 37,056 | 36,049 | ||||||
| Additional paid-in capital | 36,604,728 | 34,713,225 | ||||||
| Accumulated deficit | (21,490,475 | ) | (15,898,461 | ) | ||||
| Total quotaholders’ equity | 15,151,309 | 18,850,813 | ||||||
| Total liabilities and quotaholders’ equity | € | 17,502,115 | € | 21,348,178 | ||||
The accompanying notes are an integral part of these financial statements.
| F-4 |
Statement of Changes in Quotaholders’ Equity
| (in Euros, except quotas) | Corporate capital | Additional paid-in capital | Accumulated deficit | Total | ||||||||||||
| Balance at January 1, 2019 | € | 29,179 | € | 19,202,381 | € | (11,300,738 | ) | € | 7,930,822 | |||||||
| Capital increase, net of issuance costs | 6,870 | 14,761,332 | - | 14,768,202 | ||||||||||||
| Share-based compensation | - | 749,512 | - | 749,512 | ||||||||||||
Net loss | - | - | (4,597,723 | ) | (4,597,723 | ) | ||||||||||
| Balance at December 31, 2019 | € | 36,049 | € | 34,713,225 | € | (15,898,461 | ) | € | 18,850,813 | |||||||
| Capital increase, net of issuance costs | 1,007 | 1,431,309 | - | 1,432,316 | ||||||||||||
| Share-based compensation | - | 460,194 | - | 460,194 | ||||||||||||
Net loss | - | - | (5,592,014 | ) | (5,592,014 | ) | ||||||||||
| Balance at December 31, 2020 | € | 37,056 | € | 36,604,728 | € | (21,490,475 | ) | € | 15,151,309 | |||||||
The accompanying notes are an integral part of these financial statements.
| F-5 |
Statements of Cash Flows
| Year-Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| Cash flows from operating activities | ||||||||
| Net loss | € | (5,592,014 | ) | € | (4,597,723 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation expense | 1,900 | - | ||||||
| Retirement benefit obligation | 6,056 | 4,867 | ||||||
| Share-based compensation | 460,194 | 749,512 | ||||||
| Changes in operating assets and liabilities | ||||||||
| Prepaid expenses and other current assets | (454,082 | ) | 151,091 | |||||
| Other non-current assets | (200,287 | ) | (588,681 | ) | ||||
| Accounts payable | 265,289 | 190,088 | ||||||
| Accounts payable - related party | (282,714 | ) | (217,670 | ) | ||||
| Accrued expenses | 38,263 | 133,797 | ||||||
| Accrued expenses - related party | (292,429 | ) | 1,666,295 | |||||
| Other current liabilities | 5,243 | 18,237 | ||||||
| Net cash used in operating activities | (6,044,581 | ) | (2,490,187 | ) | ||||
| Cash flows from investing activities | ||||||||
| Purchases of property and equipment | (20,871 | ) | - | |||||
| Net cash used in investing activities | (20,871 | ) | - | |||||
| Cash flows from financing activities | ||||||||
| Proceeds from the issuance of quotas | 1,500,436 | 15,086,708 | ||||||
| Issuance costs | (68,120 | ) | (318,506 | ) | ||||
| Prepaid offering costs | (43,000 | ) | - | |||||
| Net cash provided by financing activities | 1,389,316 | 14,768,202 | ||||||
| Net (decrease) increase in cash and cash equivalents | (4,676,136 | ) | 12,278,015 | |||||
| Cash and cash equivalents at beginning of year | 20,141,379 | 7,863,364 | ||||||
| Cash and cash equivalents at end of year | € | 15,465,243 | € | 20,141,379 | ||||
| Non-cash financing activities: deferred offering costs accrued at year end | € | 113,733 | € | - | ||||
The accompanying notes are an integral part of these financial statements.
| F-6 |
Notes to the Financial Statements
1. Nature of business
Genenta Science S.r.l. (the “Company”) is an early-stage company developing potential ground-breaking cell and gene cancer therapies. The Company is initially developing its clinical leading product, Temferon, to treat glioblastoma multiforme (“GBM”), a solid tumor affecting the brain. The Company intends to start a second clinical trial to study Temferon in liver cancers and is planning to expand its clinical trial on UMGMT-GBM indications in the United States.
Genenta Science S.r.l. is an Italian limited liability company (società a responsabilità limitata, or S.r.l.), which is similar to a limited liability company in the United States. The Company was founded by San Raffaele Hospital (“OSR”), in Milan, Italy, Pierluigi Paracchi, Luigi Naldini and Bernhard Gentner and was incorporated in July 2014. The Company is organized under the laws of Italy, with registered office in Milan. The corporate capital of an S.r.l. is called a “quota.” A quota is a varying ownership or portion of subscribed capital in the S.r.l. Each quota has specific rights and value. (See Note 10. Quotaholders’ Equity.) The Company’s reporting currency is Euros.
The Company is subject to risks and uncertainties common to early-stage clinical companies in the life-science and biotechnology industries, including but not limited to, risks associated with completing preclinical studies and clinical trials, receiving regulatory approvals for product candidates, development by competitors of new competing products, dependence on key personnel, protection of proprietary technology, compliance with government regulations and the ability to secure additional capital to fund operations. The clinical product candidates currently under development will require significant additional research and development efforts, including regulatory approval and clinical testing prior to commercialization. These efforts require significant amounts of additional capital, adequate personnel and infrastructure and extensive compliance-reporting capabilities. Even if the Company’s product development efforts are successful, it is uncertain when, if ever, the Company will realize revenue from product sales.
Liquidity and Risks
The Company has incurred recurring losses since its inception, including a net loss of €5,592,014 and €4,597,723 for the years ended December 31, 2020 and 2019. In addition, as of December 31, 2020, the Company had an accumulated deficit of €21,490,475. The Company has primarily funded these losses through the proceeds from sales of convertible debt and quotas. Although the Company has incurred recurring losses and expects to continue to incur losses for the foreseeable future, the Company expects that its existing cash and cash equivalents on hand as of December 31, 2020 of €15,465,243 will be more than sufficient to fund current planned operations and capital expenditure requirements for at least the next twelve months from the filing date of these financial statements. However, the future viability of the Company is dependent on its ability to raise additional capital to finance its operations. The Company’s inability to raise capital as and when needed could have a negative impact on its financial condition and ability to pursue its business strategies. There can be no assurance that the current operating plan will be achieved or that additional funding will be available on terms acceptable to the Company, or at all.
The Company has evaluated whether there are conditions and events considered in the aggregate that raise substantial doubt about the Company’s ability to continue as a going concern. The Company’s business model, typical of biotechnology companies developing new therapeutic products that have not reached a balanced income and financial position, features negative cash flows. This is due to the fact that, at this stage, costs must be borne in relation to services and personnel, directly connected to research and development activities, and return for these activities is not certain and, in any case, it is expected in future years. Based on the accounting policies adopted, requiring full recognition of research and development costs in the statement of operations and comprehensive loss in the year they are incurred, the Company has reported a loss since its inception, and expects to continue to incur significant costs for research and development in the foreseeable future. There is no certainty that the Company will become profitable in the future.
| F-7 |
The Company has primarily funded its operations with proceeds from several capital increases and at March 31, 2021, had approximately €13.1 million of cash and cash equivalents. Therefore, management and the Board of Directors believe that the Company has adequate financial resources to support business operations in the foreseeable future and for at least 12 months from the date of this report. Accordingly, the accompanying financial statements have been prepared assuming that the Company will continue as a going concern.
The Company will require additional capital to meet its long-term operating requirements. It expects to raise additional capital through, among other things, the sale of equity or debt securities. If adequate funds are not available in the future, the Company may be forced to delay, reorganize, or cancel research and development programs, or to enter into financing, licensing or collaboration agreements with unfavorable conditions or waive rights to certain products which otherwise it would not have waived, resulting in negative effects on the activity and on the economic, patrimonial and /or financial situation of the Company.
In February 2020, the COVID-19 pandemic commenced in Italy. Regulatory guidance was issued in March and updated in April 2020 relating to the management of clinical trials during the pandemic. As the global healthcare community continues to respond to the COVID-19 pandemic, many hospitals, including the Company’s clinical sites, temporarily paused elective medical procedures, including dosing of new patients in clinical trials of our investigational gene therapies. While dosing of new patients and data collection from enrolled patients has resumed at clinical sites, the extent to which clinical activities continue to be delayed or interrupted will depend on future developments that are highly uncertain. The Company has not experienced significant interruptions related to COVID-19, although one patient tested positive for COVID-19 and had to delay treatment with Temferon™. The Company may find it difficult to enroll patients in its clinical trials, which could delay or prevent the Company from proceeding with clinical trials of its product candidates. The Company continues to closely monitor this rapidly evolving situation and the potential impact on the Company.
Basis of presentation
The accompanying financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”). Any reference in these notes to applicable guidance is meant to refer to the authoritative United States generally accepted accounting principles as found in the Accounting Standards Codification (“ASC”) and Accounting Standards Update (“ASU”) of the Financial Accounting Standards Board (“FASB”), unless otherwise stated.
A summary of the significant accounting policies applied in the preparation of these financial statements is presented below, only for the categories and headings now applicable and that might be applicable in the future based on the Company’s business. These policies have been consistently applied, unless otherwise stated.
2. Summary of significant accounting policies
Use of estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts reported in the financial statements and the disclosures made in the accompanying notes. Estimates and assumptions reflected in these financial statements include, but are not limited to, the accrual for research and development expenses and related milestone payments, share-based compensation expense, valuation of Research & Development (R&D) tax credits, the valuation of equity and the recoverability of the Company’s net deferred tax assets and related valuation allowance. Estimates are periodically reviewed considering changes in circumstances, facts and experience. Actual results may differ from these estimates under different assumptions or conditions. Changes in estimates are recorded in the period in which they become known. The areas involving a higher degree of judgment or complexity, or areas where assumptions and estimates are significant to the financial statements are disclosed below.
Cash and cash equivalents
The Company considers all highly liquid investments purchased with original maturities of 90 days or less at acquisition to be cash equivalents. In the cash flow statement cash and cash equivalents includes cash on hand, deposits held with banks, and other short-term highly liquid investments. In the balance sheet, bank overdrafts, if any, are shown in current liabilities. Cash and cash equivalents are detailed as follows:
| At December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| Cash in bank | € | 15,462,805 | € | 20,137,033 | ||||
| Cash in hand & prepaid cards | 2,438 | 4,346 | ||||||
| Total | € | 15,465,243 | € | 20,141,379 | ||||
| F-8 |
Net loss per share
Net loss per share (“EPS”) is computed in accordance with U.S. GAAP. Basic EPS is computed by dividing net loss by the weighted average number of common shares outstanding during the period. Diluted EPS reflects potential dilution and is computed by dividing net loss by the weighted average number of common shares outstanding during the period increased by the number of additional common shares that would have been outstanding if all potential common shares had been issued and were dilutive. Historical EPS or QPS (quota per share) has not been included in these financial statements because the Company has determined it is not a meaningful or material disclosure due to the Company’s current capital structure. Net loss per share has not been presented at December 31, 2020 and 2019, since the Company was an S.r.l. at the time and maintained classes of quota (similar to membership interests in a limited liability company in the United States) rather than shares, which it now has post-conversion to an S.p.A.,(similar to a C-corporation in the United States). The quotas represented percentage ownership in the Company and not actual shares. The Company did not believe that representing loss per quota was a meaningful measurement.
Foreign currency translation
The reporting and functional currency of the Company is Euros. All amounts are presented in Euros unless otherwise stated. All amounts disclosed in the financial statements and notes have been rounded to the nearest Euro unless otherwise stated. Foreign currency transactions, if any, are translated into Euros using the exchange rates prevailing at the dates of the transactions or valuation where items are re-measured. Foreign exchange gains and losses resulting from the settlement of such transactions and from the translation at year-end exchange rates of monetary assets and liabilities denominated in foreign currencies are recognized in the statement of operations. During the years ended December 31, 2020 and 2019, foreign exchange gains and losses were insignificant.
Emerging growth company status
The Company is an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act, or JOBS Act, and may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies. The Company may take advantage of these exemptions until the Company is no longer an “emerging growth company.” Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards. The Company has elected to use the extended transition period for complying with new or revised accounting standards and as a result of this election, its financial statements may not be comparable to companies that comply with public company effective dates. The Company may take advantage of these exemptions up until the last day of the fiscal year following the fifth anniversary of its initial public offering (“IPO”) or such earlier time that it is no longer an “emerging growth company.”
| F-9 |
Fair value measurements
Certain assets and liabilities of the Company are carried at fair value under U.S. GAAP. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. Financial assets and liabilities carried at fair value are to be classified and disclosed in one of the following three levels of the fair value hierarchy, of which the first two are considered observable and the last is considered unobservable:
| ● | Level 1 — Quoted prices in active markets for identical assets or liabilities. | |
| ● | Level 2 — Observable inputs (other than Level 1 quoted prices), such as quoted prices in active markets for similar assets or liabilities, quoted prices in markets that are not active for identical or similar assets or liabilities, or other inputs that are observable or can be corroborated by observable market data. | |
| ● | Level 3 — Unobservable inputs that are supported by little or no market activity that are significant to determining the fair value of the assets or liabilities, including pricing models, discounted cash flow methodologies and similar techniques. |
To the extent that the valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. Accordingly, the degree of judgment exercised by the Company in determining fair value is greatest for instruments categorized in Level 3. A financial instrument’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement.
The carrying values of the Company’s research and development tax credits, VAT credits, accounts payable, accrued expenses and other current liabilities approximate their fair values due to the short-term nature of these assets and liabilities.
Segment information
Operating segments are identified as components of an enterprise for which separate discrete financial information is available for evaluation by the chief operating decision-maker in making decisions regarding resource allocation and assessing performance. The Company and its chief operating decision-maker view the Company’s operations and manages its business in one operating segment, which is the research and development in the pharmaceutical sector with a focus on developing novel therapeutics to treat cancer.
Tax credit on investments in research and development
In line with the legislation in force until December 31, 2020, companies in Italy that invest in eligible research and development activities, regardless of the legal form and economic sector in which they operate, can benefit from a tax credit which can be used in order to reduce most taxes payable, including income tax or regional tax on productive activities, as well as of social security contributions and withholding taxes. The tax credit calculation methodology changed in 2020, and the credit was determined as 12% of eligible expenses up to €3.0 million. Up until December 31, 2019, the tax credit was up to 50% of the increase of annual research and development expenses compared to the median expense for the years 2012-2014.
The eligible activities consist of fundamental research, industrial research, and experimental development as defined respectively of the letters m), q) and j) of point 15, par. 1.3 of the Communication no. 198/2014 of the European Commission.
| F-10 |
To determine the cost basis of the benefit, the following expenses are eligible:
| ● | Personnel costs. | |
| ● | Depreciation charges, costs of the financial or simple lease and other expenses related to movable tangible assets and software used in research and development projects. | |
| ● | Expenses for extra-euro research contracts concerning the direct execution of eligible research and development activities by the provider. | |
| ● | Depreciation charges related to industrial privatives. | |
| ● | Expenses for consultancy services and equivalent services related to research and development eligible activities. | |
| ● | Expenses for materials, supplies, and other similar products used in the research and development projects. |
The Company accounts for this receivable in accordance with International Accounting Standards (IAS) 20, Accounting for Government Grants and Disclosure of Government Assistance. The receivable is recognized when there is reasonable assurance that: (1) the recipient will comply with the relevant conditions and (2) the grant will be received. The Company elected to present it net of the related expenditure on the statement of operations and comprehensive loss.
While these tax credits can be carried forward indefinitely, the Company recognized an amount which reflects management’s best estimate of the amount that is reasonably assured to be realized or utilized in the foreseeable future based on historical benefits realized, adjusted for expected changes, as applicable. The tax credits are recorded as an offset to research and development expenses in the Company’s statement of operations and comprehensive loss.
Share-based compensation
To reward the efforts of employees, directors, and certain consultants to promote the growth of the Company, the Company’s Board of Directors has approved, during its existence, various share-based awards. All options have been awarded with an exercise price of €1 per quota and, when exercised, all options have been converted to Quota B. The options granted have the vesting condition that the individual must remain in his/her role at least one year or as otherwise specified for each person.
The Company measures share-based awards granted to employees and directors based on the fair value on the date of the grant and recognizes compensation expense for those awards over the requisite service period, which is the vesting period of the respective award. Forfeitures are accounted for as they occur. The measurement date for option awards is the date of the grant. The Company classifies share-based compensation expense in its statements of operations and comprehensive loss in the same manner in which the award recipient’s payroll costs are classified or in which the award recipient’s service payments are classified.
With the adoption of Accounting Standards Update (“ASU”) No. 2018-07, Compensation—Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting (“ASU 2018-07”) on January 1, 2019, the measurement date for non-employee awards is the date of the grant. The compensation expense for non-employees is recognized, without changes in the fair value of the award, over the requisite service period, which is the vesting period of the respective award.
| F-11 |
Property and equipment
Property and equipment are stated at cost, including any accessory and direct costs that are necessary to make the assets fit for use, and adjusted by the corresponding accumulated depreciation. The depreciation recorded in the financial statements of operations has been calculated on a straight line basis by taking into consideration the use, purpose and financial-technical duration of the assets, on the basis of their estimated useful economic lives. The Company believes the above criteria to be represented by the following estimated useful lives:
- Equipment & Furniture: 15 years
- Electronic office equipment: 10 years
- Leasehold improvements: based on the length of the lease
Ordinary maintenance costs are entirely attributed to the statements of operations in the year in which they are incurred. Extraordinary maintenance costs, the purpose of which is to extend the useful economic life of the asset, to technologically upgrade it and/or to increase its productivity or safety for the purposes of the economic productivity of the Company, are attributed to the asset to which they refer and depreciated on the basis of its estimated useful economic life. Amortization of leasehold improvements is computed using the straight-line method based upon the terms of the applicable lease or estimated useful life of the improvements, whichever is less.
Impairment of long-lived assets
In accordance with ASC Topic 360-10-20, ‘‘Property, Plant and Equipment,’’ the Company performs an impairment test whenever events or circumstances indicate that the carrying value of long-lived assets with finite lives may be impaired. Impairment is measured by comparing the carrying value of the long-lived assets to the estimated undiscounted pre-tax cash flows expected to result from the use of such assets and their ultimate disposition. In circumstances where impairment is determined to exist, the Company will write down the asset to its fair value based on the present value of estimated cash flows. To date, no impairments have been identified by management for the years ended December 31, 2020 and 2019.
Deferred Offering Costs
Deferred public offering costs, which primarily consist of direct, incremental legal and accounting fees relating to the initial public offering (IPO), are capitalized within prepaid expenses and other current assets. The deferred offering costs will be offset against IPO proceeds upon the consummation of the offering. In the event the offering is terminated, deferred offering costs will be expensed. The Company has incurred €156,000 in IPO offering costs as of December 31, 2020.
Recently issued accounting pronouncements
In December 2019, the FASB issued ASU 2019-12, Income Taxes: Simplifying the Accounting for Income Taxes. The new standard intended to simplify the accounting for income taxes by eliminating certain exceptions related to the approach for intra-period tax allocation, the methodology for calculating income taxes in an interim period and the recognition of deferred tax liabilities for outside basis differences. The new guidance also simplifies aspects of the accounting for franchise taxes and enacted changes in tax laws or rates and clarifies the accounting for transactions that result in a step-up in the tax basis of goodwill. For non-public entities, the standard is effective for annual periods beginning after December 15, 2021, with early adoption permitted. Adoption of the standard requires certain changes to primarily be made prospectively, with some changes to be made retrospectively. The Company is currently evaluating the impact that the adoption of ASU 2019-12 will have on its financial statements.
In July 2017, the FASB issued ASU No. 2017-11, Earnings Per Share (Topic 260), Distinguishing Liabilities from Equity (Topic 480), Derivatives and Hedging (Topic 815) (Part I) Accounting for Certain Financial Instruments with Down Round Features (Part II) Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Noncontrolling Interests with a Scope Exception (‘‘ASU 2017-11’’). Part I applies to entities that issue financial instruments such as warrants, convertible debt or convertible preferred stock that contain down-round features. Part II replaces the indefinite deferral for certain mandatorily redeemable noncontrolling interests and mandatorily redeemable financial instruments of non-public entities contained within ASC Topic 480 with a scope exception and does not impact the accounting for these mandatorily redeemable instruments. For non-public entities, ASU 2017-11 is effective for annual reporting periods beginning after December 15, 2019, including interim periods within those fiscal years. The Company does not expect the adoption of ASU 2017-11 to have a significant impact on its financial position, results of operations, or cash flows.
In August 2018, the FASB issued No. ASU 2018-13, Fair Value Measurement (Topic 820)—Disclosure Framework (‘‘ASU 2018-13’’), which improves the disclosure requirements for fair value measurements. For non-public entities, ASU 2018-13 is effective for annual reporting periods beginning after December 15, 2019, including interim periods within those fiscal years. Early adoption is permitted for any removed or modified disclosures. The Company is currently evaluating the impact that the adoption of ASU 2018-13 will have on its financial statements. The FASB issued authoritative guidance that amends guidance on reporting credit losses for financial assets, including available-for-sale marketable securities and any other financial assets not excluded from the scope that result in a contractual right to receive cash. For available-for-sale marketable securities, credit losses should be measured in a manner similar to current generally accepted accounting standards; however, ASU 2016-13, Financial Instruments—Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments, will require that credit losses be presented as an allowance rather than as a write-down. For non-public entities, the standard is effective for annual periods beginning after December 15, 2022, with early adoption permitted. The Company is currently evaluating the impact that the adoption of ASU 2016-13 will have on its financial statements.
| F-12 |
3. Research and development
Research and development costs are expensed as incurred. Research and development expenses consist of costs incurred in performing research and development activities, including salaries, share-based compensation and benefits, facilities costs, third-party license fees, certain milestone payments, and external costs of outside vendors and consultants engaged to conduct clinical development activities and clinical trials, (e.g., contract research organizations or “CROs”), as well as costs to develop a manufacturing processes, perform analytical testing and manufacture clinical trial materials, (e.g., contract manufacturing organizations or “CMOs”). Non-refundable prepayments for goods or services that will be used or rendered for future research and development activities are recorded as prepaid expenses. Such amounts are recognized as an expense as the goods are delivered or the related services are performed, or until it is no longer expected that the goods will be delivered, or the services rendered. In addition, funding from research grants, if any, is recognized as an offset to research and development expense based on costs incurred on the research program.
The Company yearly sustains a significant amount of research costs to meet its business objectives. The Company has various research and development contracts, and the related costs are recorded as research and development expenses as incurred. When billing terms under these contracts do not coincide with the timing of when the work is performed, the Company is required to make estimates of outstanding obligations as of period end to those third parties. Any accrual estimates are based on several factors, including the Company’s knowledge of the progress towards completion of the research and development activities, invoicing to date under the contracts, communication from the research institution or other companies of any actual costs incurred during the period that have not yet been invoiced, and the costs included in the contracts. Significant judgments and estimates may be made in determining the accrued balances at the end of any reporting period. Actual results could differ from the estimates made by the Company. The historical accrual estimates made by the Company have not been materially different from the actual costs. (For further details please refer to the Related Parties disclosure in Note 12 below.)
4. General and administrative
General and administrative costs consist primarily of salaries, share-based compensation, benefits and other related costs for personnel and consultants in the Company’s executive and finance functions, professional fees for legal, finance, accounting, auditing, tax and consulting services, travel expenses and facility-related expenses, which include rent and maintenance of facilities and other operating costs not otherwise included in research and development expense.
5. Income taxes
The Company is subject to taxation in Italy, including the standard corporate income tax (“IRES”) and a regional business tax (“IRAP”). Taxes are recorded on an accrual basis. They therefore represent the allowances for taxes paid or to be paid for the year, calculated according to the current enacted rates and applicable laws. Due to the tax loss position reported, no income taxes were due for the years ending December 31, 2020 and 2019. The Company is taxed in various countries where it has permanent establishment, as applicable.
The Company uses the asset and liability method of accounting for deferred income taxes. Under this method, deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the carrying amounts and the tax basis of assets and liabilities, measured at tax rates expected to be enacted at the time of their reversals. These temporary differences primarily relate to net operating losses carried forward available to offset future taxable income.
| F-13 |
As of each reporting date, the Company considers existing evidence, both positive and negative, that could impact its view with regards to future realization of deferred tax assets. In consideration of the start-up status of the Company, a full valuation allowance has been established to offset the deferred tax assets, as the related realization is currently uncertain. In the future, should management conclude that it is more likely than not that the deferred tax assets are partially or fully realizable, the valuation allowance will be reduced to the extent of such expected realization, and the corresponding amount will be recognized as income tax benefit in the Company’s statement of operations and comprehensive loss.
The Company recognizes tax liabilities from an uncertain tax position if it is more likely than not that the tax position will not be sustained upon examination by the taxing authorities, based on the technical merits of the tax position. There are no uncertain tax positions that have been recognized in the accompanying financial statements. The prior five (5) years’ tax returns are potentially subject to audit.
A reconciliation of the Company’s effective tax rate is summarized as follows:
| At December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| Income taxes at Italy statutory rate | € | (1,342,083 | ) | € | (1,103,454 | ) | ||
| Permanent differences | 3,750 | 11,370 | ||||||
| Change in valuation allowance | 1,338,333 | 1,092,084 | ||||||
| Total provision expense for income taxes | € | - | € | - | ||||
Significant components of the Company’s net deferred tax assets are summarized as follows:
| At December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| Deferred tax assets | ||||||||
| Net operating loss carryforwards | € | 4,692,844 | € | 3,192,643 | ||||
| Other temporary differences | - | 144,000 | ||||||
| Allowance for corporate equity | 210,620 | 178,972 | ||||||
| Total deferred tax assets | 4,903,464 | 3,515,615 | ||||||
| Valuation allowance | (4,903,464 | ) | (3,515,615 | ) | ||||
| Net deferred tax assets | € | - | € | - | ||||
Tax loss carryforwards expire as follows:
| At December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| No expiration date | € | 5,487,085 | € | 5,487,085 | ||||
| No expiration date - DL 98/2011 | 14,066,434 | 7,815,593 | ||||||
| Total | € | 19,553,519 | € | 13,302,678 | ||||
In 2011, the Italian tax authorities issued a set of rules that modified the previous treatment of tax loss carryforwards. According to the DL 98/2011, at the end of 2011, all existing tax loss carryforwards will never expire but they can off-set only 80% of the taxable income of the year. The rules do not affect the tax loss carryforwards that refer to the start-up period, defined as the first three (3) years of operations starting from the inception of the Company.
| F-14 |
6. Prepaid expenses and other current assets
Prepaid expenses and other current assets consist of the following:
| At December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| Value Added Tax (VAT) | € | 480,000 | € | 237,569 | ||||
| Research and Development Tax Credit | 280,631 | 192,020 | ||||||
Deferred offering costs | 156,261 | - | ||||||
Advances to suppliers – related party | 121,432 | - | ||||||
| Other | 30,609 | 28,529 | ||||||
| Total | € | 1,068,933 | € | 458,118 | ||||
Value Added Tax (VAT) receivables are linked to purchases. Italian VAT (Imposta sul Valore Aggiunto) applies to the supply of goods and services carried out in Italy by entrepreneurs, professionals, or artists and on imports carried out by anyone. Intra-Community acquisitions are also subject to VAT under certain situations. The Italian standard VAT rate for 2020 and 2019 is 22%. Reduced rates are provided for specifically listed supplies of goods and services. It is carried forward indefinitely and does not expire. The Company reclassified to other non-current assets a portion of the receivable which is expected to be realized beyond 12 months.
Tax credits on research and development represent a special tax relief offered to Italian companies operating in the research and development sector and can be used to offset most taxes payable. Tax credits on research and development available were €3.9 million at December 31, 2020 and €3.5 million at December 31, 2019, which can be carried forward indefinitely and does not expire. However, given the start-up status of the Company and the fact that it will not be profitable in the foreseeable future which limits the utilization of the credit, the Company recognized a receivable balance at December 31, 2020, that represents the Company’s best estimate of the amount of tax credit that can be used in offsetting taxes payable for 2021 and 2022, which represents the current projected available cash to support operations. In 2019, the limitation on the Company’s tax credit was based on three years of cash available to support operations. In 2020 and 2019, the Company utilized €280,631 and €192,020 to offset certain taxes payable. The benefit recorded in 2020 and 2019 to offset research and development expenses was €265,834 and €448,079, respectively. The Company reclassified to other non-current assets a portion of the receivable which is expected to be realized beyond 12 months.
The remaining balance is comprised of miscellaneous minor prepaid expenses and deferred offering costs.
| F-15 |
7. Property and equipment, net
Property and equipment consist of the following:
| At December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| Computer | € | 16,196 | € | - | ||||
| Furniture and fixtures | 4,675 | - | ||||||
| Total property and equipment | 20,871 | - | ||||||
| Less: accumulated depreciation | (1,900 | ) | - | |||||
| Property and equipment, net | € | 18,971 | € | - | ||||
Property and equipment consist of computers and furniture and fixtures of our office space in Milan. No disposals, nor impairments occurred during the periods. Depreciation has been calculated by taking into consideration the use, purpose, and financial-technical duration of the assets, based on their estimated economic lives. Property and equipment were entirely purchased in 2020.
Depreciation expense for the year ended December 31, 2020 was €1,900. There was no depreciation expense for the year ended December 31, 2019.
8. Other non-current assets
Other non-current assets consist of the long-term portion of the VAT receivable and R&D tax credit, as follows:
| At December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| Value Added Tax (VAT) | € | 664,987 | € | 364,641 | ||||
| Research and Development Tax Credit | 280,631 | 384,040 | ||||||
| Total | € | 945,618 | € | 748,681 | ||||
9. Retirement benefit obligation
Employees in Italy are entitled to Trattamento di Fine Rapporto (“TFR”), commonly referred to as an employee leaving indemnity, which represents deferred compensation for employees in the private sector. Under Italian law, an entity is obligated to accrue for TFR on an individual employee basis payable to each individual upon termination of employment (including both voluntary and involuntary dismissal). The annual accrual is approximately 7% of total pay, with no ceiling, and is revalued each year by applying a pre-established rate of return of 1.50%, plus 75% of the Consumer Price Index, and is recorded by a book reserve. TFR is an unfunded plan. The costs of the retirement benefit obligation is accounted for under the provisions of ASC 715, Compensation – Retirement Benefits. The amount of the obligation at December 31, 2020 and 2019 was €17,388 and €11,332, respectively.
10. Quotaholders’ equity
The Company is an S.r.l., which is an Italian limited liability company similar to a limited liability company in the United States. The Articles of Incorporation, Shareholders’ Agreement and the By-laws of the Company provide for different quotas, which represent the Company’s corporate capital, rather than shares of stock as ownership.
| F-16 |
Corporate capital
As an S.r.l., the Company’s ownership is called “corporate capital” and “quotas” rather than shares, stock or units.
The Company’s capital is divided between the five quotas as summarized below at December 31, 2020 and 2019:
| At December 31, | At December 31, | |||||||||||||||
| Quota | 2019 | Ownership % | 2020 | Ownership % | ||||||||||||
| A | € | 10,458 | 29 | % | € | 10,458 | 28 | % | ||||||||
| B | 6,450 | 18 | % | 6,886 | 19 | % | ||||||||||
| C | 8,645 | 24 | % | 8,645 | 23 | % | ||||||||||
| D | 4,034 | 11 | % | 4,034 | 11 | % | ||||||||||
| E | 6,462 | 18 | % | 7,033 | 19 | % | ||||||||||
| Total | € | 36,049 | 100 | % | € | 37,056 | 100 | % | ||||||||
The Company’s corporate capital at January 1, 2019 was €29,179, consisting of quota A from founders, options converted to quota B, and quotas C & D from investment capital. Additional capital was raised in 2019 by the sale of quota E, along with options converted to quota B, so corporate capital at December 31, 2019 was €36,049. Additional capital was raised in 2020 by the sale of quota E, so corporate capital at December 31, 2020 was €37,056.
The Company currently has five (5) quotas:
| ● | Quota A. Quota A was reserved for certain founders. One of the founders has the right to appoint three (3) board members out of five (5), appoint the Chair from these three (3) persons and appoint one (1) member of the Board of Statutory Auditors. One other founder has the right to appoint two (2) board members out of five (5), appoint two (2) statutory auditors and appoint the Chair of the statutory auditors from the two (2) appointees. Quota A has voting rights. | |
| ● | Quota B. Quota B has no voting rights, the same profit-sharing rights as Quota A and is priced at a nominal amount of €1.00. The Company has historically utilized Quota B for its share-based compensation program offered to board members, employees, and consultants. Quota B is also held by certain co-founders. The Company’s stock options are exercisable into Quota B for past and present board members, employees, and consultants. | |
| ● | Quota C. Quota C has the right to appoint one (1) member of the Board of Statutory Auditors; specifically, the one (1) that a founder had the right to appoint. Investors received Quota C in the Company’s first funding round (2014/2015) where approximately €10 million was raised. | |
| ● | Quota D. Investors received Quota D in the Company’s second funding round (2017) where approximately €7 million was raised. | |
| ● | Quota E. Investors received Quota E in the Company’s third funding round (through December 31, 2019) where approximately €14.8 million was raised approximately (€15.1 million gross, net of approximately €0.3 million of financing fees). Investors received Quota E in the Company’s second tranche of the third funding round (through December 31, 2020) where approximately €1.4 million was raised (approximately €1.5 million gross, net of approximately €0.1 million of financing fees). |
| F-17 |
| ● | Quotas A, C, D & E. During a divestiture proceeding (meaning Quotas representing 100% of the corporate capital of the Company) or a dissolution of the Company, Quotas C, D & E all have the same rights with respect to the proceeds of a divestiture, i.e., all three (3) quotas share the divestiture consideration equally (on a pari passu basis) up to the amount of their investment. If there is any consideration remaining after payment to quotas C, D & E, then quota A shall be entitled to the amount remaining up to the amount of their investment. If proceeds of a divestiture are less than or equal to €50 million, then any proceeds remaining after payment of quotas A, C, D & E, shall be shared equally among quotas A, C, D & E; however, if proceeds of a divestiture are greater than €50 million, then any proceeds remaining after payment of quotas A, C, D & E, shall be distributed to each quota separately according to a detailed formula specified in the Company’s By-Laws, including quota B. Similar to a divestiture, net profits, if any, shall be distributed in the same manner to quotas A, B, C, D & E, after deducting not less than five (5) percent for a legal reserve (up to where this reserve equals one-fifth of the quota capital). A, C, D, E have equal voting rights and the Company By-laws specify protective provisions for each class of quota for A, C, D & E. |
11. Share-based compensation
The Company has granted options on its corporate capital to certain directors, officers, employees, and consultants, as an incentive and as additional compensation. All options convert into Quota B when vested and exercised. All options have an exercise price of €1.00 per quota. Options generally vest over a one-to-three-year period and are exercised when vested. All options expire on December 31, 2021, if not exercised. The Board of Directors approves any options granted and has authorized and granted 4,835 options to date. At December 31, 2019, there were no options available for grant, as all remaining authorized options were granted in 2019; therefore, no options were granted in 2020.
The Company’s quota option activity for the years ended December 31, 2020 and 2019 is represented by the following:
| Number of Options | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value | |||||||||||||
| (in €’s) | (in €’s) | |||||||||||||||
| Outstanding, vested and expected to vest as of December 31, 2019 | 982 | 1.00 | 2.00 | 1,056,316 | ||||||||||||
| Granted | - | - | - | - | ||||||||||||
| Vested and exercised | 436 | 1.00 | 1.00 | 472,184 | ||||||||||||
| Cancelled or forfeited | - | - | - | - | ||||||||||||
| Outstanding, vested and expected to vest as of December 31, 2020 | 546 | 1.00 | 1.00 | 584,132 | ||||||||||||
| Exerciseable as of December 31, 2020 | - | - | - | - | ||||||||||||
The Company’s share-based compensation expense for the years ended December 31, 2020 and 2019 is represented by the following table:
| Year ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Research & development expense | € | - | € | 21,804 | ||||
| Research & development expense - related party | 326,400 | 581,949 | ||||||
| General & administrative expense | 133,794 | 137,057 | ||||||
| Total | 460,194 | 740,810 | ||||||
| Unrecognized expense at December 31, | € | 313,273 | € | 769,779 | ||||
| F-18 |
The Company awarded eight options to a consultant on September 30, 2019 for assistance in raising additional Quota E capital for the Company in 2019. The Company recorded the €8,702, which was the fair value of the option as a charge to additional paid-in capital. The weighted average grant date fair value of the options granted during 2019 was €1,088. The Company’s unrecognized expense was €313,273 and €769,779 at December 31, 2020 and 2019, respectively, and will be expensed as the options vest.
Quota B Valuations
The fair value of the Quota B underlying the Company’s stock-based compensation grants has historically been determined by the Company’s board of directors, with input from management and third-party valuations. The Company believes that the board of directors has the relevant experience and expertise to determine the fair value of its Quota B, when also securing third-party assistance. Given the absence of a public trading market of the Company’s equity, and in accordance with the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately- Held Company Equity Securities Issued as Compensation, the board of directors exercised reasonable judgment and considered numerous objective and subjective factors to determine the best estimate of the fair value of the Company’s equity at each grant date. These factors include:
| ● | valuations of the Quota B equity performed by third-party specialists; | |
| ● | the price of the Company’s equity to third-party, arms-length, sophisticated, and qualified investors, which was used in the OPM Backsolve Model; | |
| ● | the prices, rights, preferences, and privileges of the Company’s Quota C, D, and E preferred equity classes relative to those of the Company’s equity; | |
| ● | lack of marketability of the Quota B; | |
| ● | lack of voting rights of the Quota B; | |
| ● | current business conditions and projections; | |
| ● | hiring of key personnel and the experience of management; | |
| ● | the Company’s stage of development; | |
| ● | the timing, progress and results of the Company’s pre-clinical studies and clinical trials for the Company’s programs and product candidates; including statements regarding the timing of initiation and completion of trials or studies and related preparatory work, the period during which the results of the trials will become available and the Company’s research and development programs; | |
| ● | likelihood of achieving a liquidity event, such as an initial public offering, a merger or acquisition of the Company given prevailing market conditions, or other liquidation events; | |
| ● | the market performance of comparable publicly traded companies; and | |
| ● | the European, U.S. and global capital market conditions. |
In valuing the Company’s Quota B class of options, the board of directors determined the equity value of the Company’s business using various valuation methods. The board of directors engaged a third-party valuation firm who performed analyses in accordance with the guidance outlined in the American Institute of Certified Public Accountants’ Accounting and Valuation Guide, Valuation of Privately-Held-Company Equity Securities Issued as Compensation. The Company’s option valuations were prepared using an option pricing method (“OPM”), which used market approaches to estimate the Company’s enterprise value.
| F-19 |
The OPM treats each equity class as a call options on the total equity value of a company, with exercise prices (i.e., breakpoints) based on the value thresholds at which the allocation among the various holders of a company’s securities changes. A discount was considered for Lack of Marketability (“DLOM”), which is an amount or percentage that is deducted from the value in order to reflect the absence of a viable market. The DLOM was then applied to arrive at an indication of value for the option. Also, considered in the valuation was volatility and the fact that the Quota B class of equity did not carry voting rights. The expected volatility used in the OPM is based upon the historical volatility of a number of publicly traded companies in similar stages of clinical development.
Application of the Company’s approach involved the use of estimates, judgment, and assumptions that are highly complex and subjective, such as those regarding the selection of comparable companies, and the expected timing of an initial public offering (“IPO”) or other liquidity event. Changes in any or all of these estimates and assumptions or the relationships between those assumptions impact the valuations at each valuation date and may have a material impact on the valuation of the Company’s Quota B equity class, and consequently, the Company’s share-based compensation expense could be materially different.
12. Related parties
The Company’s research and development expenses are a combination of third-party expenses and related party expenses, as detailed below:
| For the Year Ended December 31, 2020 | ||||||||||||
| (In Euros) | Third Parties | Related Parties | Total | |||||||||
| Consultants & other third parties | € | 1,454,576 | € | 2,009,884 | € | 3,464,460 | ||||||
| Materials & supplies | 709,183 | 3,124 | 712,307 | |||||||||
| Compensation (including share-based) | 145,700 | 326,400 | 472,100 | |||||||||
| Travel & entertainment | 16,742 | 17,724 | 34,466 | |||||||||
| Other | 5,128 | - | 5,128 | |||||||||
| Total | € | 2,331,329 | € | 2,357,132 | € | 4,688,461 | ||||||
| For the Year Ended December 31, 2019 | ||||||||||||
| (In Euros) | Third Parties | Related Parties | Total | |||||||||
| Consultants & other third parties | € | 74,518 | € | 2,036,870 | € | 2,111,388 | ||||||
| Compensation (including share-based) | 171,289 | 581,949 | 753,238 | |||||||||
| Materials & supplies | 649,574 | - | 649,574 | |||||||||
| Travel & entertainment | 97,063 | 36,510 | 133,573 | |||||||||
| Other | 55,209 | - | 55,209 | |||||||||
| Total | € | 1,047,653 | € | 2,655,329 | € | 3,702,982 | ||||||
The Company recorded a research and development tax credit in the amount of €265,834 and €448,079 for the years ended December 31, 2020 and 2019, respectively, to offset Consultants and other third parties related research and development expenses.
| F-20 |
The Company’s general and administrative expenses are also a combination of third-party and related party expenses, as detailed below:
| For the Year Ended December 31, 2020 | ||||||||||||
| (In Euros) | Third Parties | Related Parties | Total | |||||||||
| Compensation (including share-based) | € | 191,998 | € | 298,628 | € | 490,626 | ||||||
| Accounting, legal & other professional | 239,861 | - | 239,861 | |||||||||
| Communication & IT related | 64,430 | - | 64,430 | |||||||||
| Facility & insurance related | 24,685 | 14,402 | 39,087 | |||||||||
| Consultants & other third parties | 59,648 | 1,495 | 61,143 | |||||||||
| Other | 6,618 | - | 6,618 | |||||||||
| Total | € | 587,240 | € | 314,525 | € | 901,765 | ||||||
| For the Year Ended December 31, 2019 | ||||||||||||
| (In Euros) | Third Parties | Related Parties | Total | |||||||||
| Consultants & other third parties | € | 375,201 | € | - | € | 375,201 | ||||||
| Compensation (including share-based) | 171,698 | 277,849 | 449,547 | |||||||||
| Accounting, legal & other professional | 40,745 | - | 40,745 | |||||||||
| Communication & IT related | 14,759 | - | 14,759 | |||||||||
| Facility & insurance related | 13,630 | - | 13,630 | |||||||||
| Other | 22,336 | 5,302 | 27,638 | |||||||||
| Total | € | 638,369 | € | 283,151 | € | 921,520 | ||||||
The Company’s accounts payable to related parties are comprised as follows:
| At December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| San Raffaele Hospital | € | 4,085 | € | 283,115 | ||||
| XDG Biomed | 5,942 | 9,626 | ||||||
| Total | € | 10,027 | € | 292,741 | ||||
The Company’s accrued expenses to related parties are comprised as follows:
| At December 31, | ||||||||
| 2020 | 2019 | |||||||
| (in Euros) | ||||||||
| San Raffaele Hospital | € | 1,309,191 | € | 1,454,430 | ||||
| Pierluigi Paracchi | 25,000 | 50,000 | ||||||
| XDG Biomed | 25,000 | 147,190 | ||||||
| Total | € | 1,359,191 | € | 1,651,620 | ||||
The Company has identified the following related parties:
| ● | Pierluigi Paracchi (director and co-founder of the Company); | |
| ● | Luigi Naldini (co-founder of the Company and executive scientific advisory board chairman); | |
| ● | Carlo Russo (through XDG Biomed LLC- Chief Medical Officer); | |
| ● | Bernard Rudolph Gentner (co-founder of the Company and member of scientific advisory board); and, | |
| ● | San Raffaele Hospital (co-founder and main supplier of services). |
| F-21 |
The following is a description of the nature of the transactions between the Company and these related parties:
Pierluigi Paracchi
He is the Chief Executive Officer and the Company’s Vice-Chairman of the Board of Directors as well as co-founder. In addition to his annual remuneration of €250,000 for the year 2020 and €201,980 for the year 2019, Mr. Paracchi also obtained a funding fee of €185,510 in 2019 for the funding activity of the first financing as approved by the Board of Directors and by quotaholders at the Quotaholders’ Meeting. This was recorded in additional paid-in capital as a direct reduction of proceeds. Mr. Paracchi was awarded a bonus of €50,000 by the Board of Directors in July 2020. The bonus was accrued in the 2019 financial statements and paid in July 2020. At December 31, 2020, the Company also accrued a bonus of €25,000 for the period July to December 2020: as per his Directorship agreement, Mr. Paracchi is entitled to receive a bonus of up to €50,000 to be approved by the Board of Directors each year to cover the period August 1st to July 31st of each year. Such Directorship agreement will be terminated by the parties for mutual consent upon signature of the employment agreement.
Luigi Naldini/Bernard Rudolph Gentner
They are co-founders of Genenta and part of the SAB – Scientific Advisory Board, with Dr. Naldini as Chairman, and Dr. Gentner as a member. Dr. Naldini has an advisory agreement approved by the Board of Directors and performs the pre-clinical studies for Genenta. In particular, the pre-clinical experiments are in solid tumor indications. The last agreement with Dr. Naldini is still in effect and was signed on December 12, 2019. In 2020 and 2019, Dr. Naldini’s compensation amounted to €56,250 and €55,500, respectively. Dr. Gentner, like Dr. Naldini, oversees pre-clinical research related to the platform technology. In addition, he analyzes clinical biological data. The last agreement with Dr. Gentner, which is still in force, was signed on October 26, 2017. In 2020 and 2019, Dr. Gentner’s compensation was €18,750 and €44,350, respectively.
XDG Biomed LLC
This is the LLC of Dr. Carlo Russo. Dr. Russo has a single contract signed by XDG and the Company that has been approved by the Board of Directors and was subject to multiple amendments. In particular, Dr. Russo, via XDG, serves as the Company’s Chief Medical Officer and Head of Development. Dr. Russo is responsible for the clinical development of Temferon™, the Company’s gene therapy platform. The applicable recurring fees are €300,000 per year effective from August 1, 2019, (€200,000 prior to that date) plus a variable compensation of €50,000 based on the results obtained and depending on the Company’s Board of Directors approval. In 2020, the Company incurred €342,872. The Company accrued €25,000 for a potential bonus relating to the period of July to December 2020. The amounts owed at December 31, 2020 and 2019 are €30,942 and €156,816, respectively. In 2019, the Company granted options on 1,000 class B quotas to Dr. Russo, of which 400 were exercised and converted to class B quotas in 2019 and 300 were exercised and converted to class B quotas in 2020; the remaining 300 are outstanding and will vest by the earlier of July 31, 2021 or the effectiveness of the Company’s registration statement. The fair value of the options based on the Company’s valuation was €1,084 per quota B. There were no options granted in 2020.
San Raffaele Hospital - OSR
San Raffaele Hospital (“OSR”) is a co-founder of the Company and the Company is a corporate and research spin-off of OSR. OSR owns approximately 13% of the Company, as of December 31, 2020. The Hospital is one of the leading biomedical research institutions in Italy and Europe, with a 45-year history of developing innovative therapies and procedures. The Company has several contractual arrangements with OSR. The Company has agreements to license technology, to perform research, pre-clinical and clinical activities, as well as to lease facilities and obtain certain other support functions. The Company’s headquarters is currently in an OSR facility.
| F-22 |
License Agreement
The Company has a License Agreement with OSR entered in December 2014, for the exclusive use of different patents. In particular, OSR granted the Company an exclusive, world-wide, royalty bearing license under certain technology to conduct research and develop, make, use, import, and sell licensed products. The License Agreement covers patents and patent applications, as well as proprietary technologies. The Company’s rights to use these patents and patent applications and to utilize the inventions claimed in these licensed patents are subject to the continuation of, and the Company’s compliance with, the terms of the License Agreement.
Based on the preclinical studies carried out by OSR, in particular by its SR-TIGET Institute (San Raffaele Telethon Institute for Gene Therapy), on a specific gene therapy strategy with respect to lympho-hematopoietic indication and/or solid cancer indication, the Company decided to develop a new therapy to treat cancer through a cell and gene therapy strategy. The “Field of Use” as defined in the License Agreement is:
| a) | Lympho-Hematopoietic Indication; and, | |
| b) | Solid Cancer Indication. |
The agreement provided for an upfront fee of €250,000 (which was paid by the Company in 2015), plus future option fees are as follows:
| ● | option fee on the first indication = €1.0 million (subsequently reduced to €0.5 million); | |
| ● | option fee on the second indication = €0.5 million; | |
| ● | option fee on the third indication = €0.3 million; and, | |
| ● | option fee on any additional indications = no license fee. |
In addition, the Company would be obligated make payments on milestones depending on the Field of Use (as defined in the agreement) and pay royalties of 4% of net sales of each Licensed Product (as defined in the agreement). For information relating to the contingent milestones for these indications, please refer to Note 13 - Commitments and Contingencies.
In February 2019, the Company and OSR entered into Amendment #2 of the License Agreement to conduct a clinical trial according to the protocol TEM-GBM_001 and EudraCT 2018-001404-11 entitled: “A phase I/IIa dose escalation study evaluating the safety and efficacy of autologous CD34+ enriched hematopoietic progenitor cells genetically modified with a lentiviral vector encoding for the human interferon-α2 in patients with glioblastoma multiforme who have an unmethylated O-6-methylguanine-DNA methyltransferase gene promoter.” In Amendment #2, the Company and OSR also revised the license fee requirement for the first Solid Cancer indication (GBM). In relation to the GBM trial, the Company and OSR agreed that the Company would be obligated to pay OSR the €1.0 million Option Fee only in the event that the Company was able to dose its tenth patient. Under this Amendment, the Company is also obligated to pay for the costs of the study-related procedures performed on the patients recruited in the trial, according to periodic study reports delivered by OSR. The first GBM patient was recruited in April 2019 and related clinical activity costs were recorded by the Company in the amount of €1,145,202 in 2019. In 2020, the comparable costs incurred and expensed for the glioblastoma program were €989,556.
In December 2020, the Company and OSR entered into Amendment #3 of the License Agreement. The initial €1.0 million license fee for the first indication (i.e., GBM) would have become due when the tenth patient was dosed in the GBM trial; however, the license fee was reduced to €0.5 million, in exchange for the Company’s agreement to exercise a second option for an additional Solid Cancer indication (i.e., liver cancer) and an agreement to execute a Sponsored Research Agreement in February 2021. If the Company is not be able to obtain approval from the competent authorities to initiate a human clinical trial in liver cancer on or before the expiration of nine months (from December 2020), the Company has the right, at no additional costs, to convert the second solid cancer indication (i.e., liver cancer) to an “Alternate Indication,” as defined in the agreement (i.e., to an indication other than liver cancer.)
| F-23 |
In summary, Amendment #3 to the license agreement amended the existing agreement as follows:
| ● | reduction of option fee on the first solid cancer indication = €0.5 million (accrued at December 31, 2019, since it was considered probable and paid in December 2020); plus, | |
| ● | commitment to enter into a Sponsored Research Agreement, which was executed in February 2021; and, | |
| ● | exercise of option fee on the second indication = €0.5 million (accrued at December 31, 2020, to be paid by June 30, 2021. |
The Company has paid €0.75 million to OSR, since inception under the license agreement. No events have occurred or have been achieved (and none are considered probable) to trigger any contingent payments under the license agreement as of December 31, 2020. For information relating to the contingency payments or future milestones for these indications, please refer to Note 13 - Commitments and Contingencies.
OSR may terminate the Company’s rights as to certain fields of use for the Company’s failure to develop (a) with respect to a solid cancer indication, upon third anniversary of the date the Company exercised such option, if the Company has not filed an IND with respect to such optioned solid cancer indication specifically, as to GBM, the Company is required to file an IND regarding Temferon for GBM prior to February 2022, or (b) with respect to a lympho-hematopoietic indication, on the earlier of (i) the fifth anniversary of the initiation (first patient dosed) of the first human clinical trial for a licensed product in any lympho hematopoietic indication or solid cancer indication if a patient has not been dosed with a licensed product in a Phase 3 clinical trial and (ii) September 1, 2025.
Research Funding Agreement
In March 2019, the Company and OSR entered into a Research Funding Agreement to conduct a clinical trial according to the multiple myeloma protocol, TEM-MM-101 and EudraCT 2018-001741-14, entitled “A Phase I/II dose escalation study evaluating safety and activity of autologous CD34+ enriched hematopoietic progenitor cells genetically modified with a lentiviral vector encoding for the human interferon-α2 in multiple myeloma patients with early relapse after intensive front-line therapy.” This agreement required OSR to perform certain clinical procedures and exploratory analyses on the study population, as per the protocol approved by the relevant competent authorities. The Company was required to fund the costs of the study-related procedures performed on patients recruited in the Trial, according to periodic study reports delivered by OSR. The first TEM-MM-101 trial patient was enrolled in August 2019, after the approval received from national competent authorities.
In 2019, the Company expensed €130,589 for the analysis performed by OSR for multiple myeloma and €364,042 for the clinical procedures performed by OSR’s Hematology and Bone Transplant Unit for multiple myeloma. For the multiple myeloma program, the Company recorded expenses in 2020 of €104,626; however, the discussion to discontinue the program began in late 2020. The final decision to discontinue the program was reached in early 2021, mainly due to the small number of eligible patients, the increasing competitive landscape, and the faster progress and promising result on the concurrent solid tumor glioblastoma trial (TEM-GBM_001). No product indication was exercised, and no milestones were achieved related to the multiple myeloma indication, and so no option fee or contingent payments were due to OSR for this indication.
Sponsor Research Agreement (SRA)
As stated above, in exchange for a reduction in the first solid cancer indication (i.e., GBM) option fee from €1.0 million to €0.5 million, the Company agreed to enter into a Sponsored Research Agreement (SRA) in December 2020. Under the SRA, which was executed in February 2021, two sponsored research activities will be conducted for between €0.5 million and €1.0 million. The first SRA payment was made in early 2021 in the amount of €250,000.
Service Agreement
A service agreement to provide certain services (accounting/bookkeeping and rent of spaces, the latter with an addendum effective from January 1, 2016) free of charge was in place between the Company and OSR from 2014 to 2019. Beginning in January 2020, a third-party provider was engaged by the Company to perform these services. The Company determined the value of these services that were provided in 2019 and in prior years were not material to its financial statements.
Beginning January 1, 2020, the Company entered into a six-year lease agreement for the use of office space in the OSR building. The Company paid OSR annual rent of €13,400 in 2020 with a security deposit of €3,350.
| F-24 |
13. Commitments and contingencies
The Company exercises considerable judgment in determining the exposure to risks and recognizing provisions or providing disclosure for contingent liabilities related to pending litigations or other outstanding claims and liabilities. Judgment is necessary in assessing the likelihood that a pending claim will succeed, or a liability will arise and to quantify the possible range of the final settlement. Provisions are recorded for liabilities when losses are considered probable and can be reasonably estimated. Because of the inherent uncertainties in making such judgments, actual losses may be different from the originally estimated provision. Estimates are subject to change as new information becomes available, primarily with the support of internal specialists or outside consultants, such as actuaries or legal counsel. Adjustments to provisions may significantly affect future operating results.
The following table summarizes our obligations by contractual maturity for years following December 31, 2020:
| Payments by period | ||||||||||||||||||||
| (in Euros) | Total | Less than a year | 1 to 3 years | 4 to 5 years | More than 5 years | |||||||||||||||
| Sponsored research agreement (SRA) with OSR | € | 1,000,000 | € | 500,000 | € | 500,000 | € | - | € | - | ||||||||||
| License option fee for liver cancer (or second cancer indication) | 500,000 | 500,000 | - | - | - | |||||||||||||||
| Operating leases | 80,400 | 13,400 | 26,800 | 26,800 | 13,400 | |||||||||||||||
| Total | € | 1,580,400 | € | 1,013,400 | € | 526,800 | € | 26,800 | € | 13,400 | ||||||||||
The above amounts include an option fee for the second cancer indication payable to OSR. Refer to the Related parties - Note 12 for further information.
The Company entered into certain license agreements under which it is obligated to pay option license fees and to make contingent milestone payments. The Company has not included future milestone and royalty payments in the table above because the payment obligations under these agreements are contingent upon future events, such as the Company’s achievement of specified milestones or generating product sales, and the amount, timing and likelihood of such payments are unknown and are not yet considered probable.
The Company enters into contracts in the normal course of business with CMOs, CROs and other third parties for exploratory studies, manufacturing, clinical trials, testing, and services (shipments, travel logistics, etc.). These contracts do not contain minimum purchase commitments and, except as discussed below, are cancelable by the Company upon prior written notice. Payments due upon cancellation consist only of payments for services provided or expenses incurred, including non-cancelable obligations of the Company’s vendors or third-party service providers, up to the date of cancellation. These payments are not included in the table above as the amount and timing of such payments are not known.
OSR - San Raffaele Hospital
The OSR agreements are non-cancelable, except in the case of breach of contract, and include total potential milestone payments of up to €10 million related to the Lympho-Hematopoietic Indication of each Licensed Product, and up to €53 million related to each Solid Cancer indication; however, starting with the fifth Solid Cancer indication, the first two related milestone payments totaling €7.0 million, are reduced to €3.5 million. The milestones relate to certain events such as, dosing of the first patient with a licensed product in Phase II and III of the trial, MAA (marketing authorization application) and NDA (new-drug application) approval of the licensed product, the first commercial sale of the product in the US and major European countries, and annual sales for the licensed product exceeding a certain amount in different territories.
| F-25 |
Multiple myeloma (MM)
As discussed in Note 12, the Company’s MM program was discontinued in early 2021 due to the relatively small number of eligible patients, and the highly competitive MM landscape. No milestones were achieved with respect to the MM program, and as such no contingent payments were due under the agreement.
Glioblastoma multiforme (GBM)
As discussed in Note 12, in December 2020, the Company had one indication ongoing, glioblastoma multiforme. The Company’s contingent liability for this first solid cancer indication potentially payable to OSR was €53 million.
Liver cancer (LC)
In relation to the option exercised by the Company for the second solid cancer indication, the Company and OSR agreed that the payment due in relation to the “First patient dosed with a Licensed Product in Phase I/II Clinical Trial,” as stated in the agreement, was reduced to €0.5 million rather than €1.0 million. The reduction applied to the first license fee payment only. All the additional contingent payments, other than the last contingent payment of €5.0 million, remained a contingent liability of the Company and potentially payable to OSR. Therefore, for the second solid cancer indication (liver cancer), the total potential commitment of possible contingent payments could amount to €47.5 million.
The agreements also include a €7.8 million commitment related to the development and manufacturing of licensed products, of which the Company has incurred €1.5 million of expense in 2020 compared to €2.6 million of cumulative expense for 2019 and prior years.
In February 2021, the Company entered into a Sponsored Research Agreement (“SRA”) with OSR to conduct certain research projects related to Temferon. The total consideration to be paid by the Company under the SRA will be €1.0 million with payments scheduled quarterly over 2021 and 2022. The Company made no payments under the SRA in 2020.
AGC Biologics (formerly MolMed)
The AGC Biologics agreement is non-cancelable, except in the case of breach of contract, and includes a potential milestone of €0.3 million if a phase 3 study is approved by the relevant authority, as well as potential royalty fees between 0.5% and 1.0% depending on the volume of annual net sales of the first commercial and named patient sale of the product. In the AGC Agreement, the Company entrusts AGC with certain development activities that will allow the Company to carry out activities related to its clinical research and manufacturing. The AGC agreement also includes a technology transfer fee of €0.5 million related to the transfer of the manufacturing know-how and €1.0 million related to the marketability approval by regulatory authorities. The agreement is a “pay-as-you-go” type arrangement with all services expensed in the period the services were performed. In February 2020, the Company entered into Amendment 4 to the Framework Service Agreement with AGC Biologics related to production and testing of the Company’s GBM trials, for a total amount of €360,000. In March 2020, the Company entered into Amendment 5 to the Framework Service Agreement with AGC Biologics related to production and testing of the Company’s GBM trials, for a total amount of €259,000. In March 2020, the Company entered into Amendment 6 to the Framework Service Agreement with ACG Biologics related to production and testing of the Company’s GBM trials, for a total amount of €41,000. In August 2020, the Company entered into Amendment 7 to the Framework Service Agreement with ACG Biologics related to production and testing of the Company’s GBM trials, which provides the Company with an option to accelerate GBM production as stated in Amendment 5 at a 20% cost increase. In October 2020, the Company entered into Amendment 8 to the Framework Service Agreement with ACG Biologics related to production and testing of the Company’s GBM trials, for a total amount of €17,000.
Adaptive Biotechnologies
The Adaptive Biotechnologies agreement on exploratory analyses for trial endpoints is non-cancelable, except in the case of breach of contract, and carries a total cost for the entire trial of €0.2 million, of which approximately €3,000 and approximately €50,000 have been incurred as of December 31, 2020 and 2019, respectively.
| F-26 |
Biogazelle NV
The Biogazelle NV agreement on exploratory analyses for trial endpoints is non-cancelable, except in the case of breach of contract, and carries a total cost for the entire trial of €106,000, of which €28 thousand and €6 thousand have been incurred as of December 31, 2020 and 2019, respectively.
Operating leases
The Company entered into a non-cancelable lease agreement for office space in January 2020. (See Footnote 12 – Related parties under OSR for further information.)
Legal proceedings
The Company is not currently party to any material legal proceedings. At each reporting date, the Company evaluates whether or not a potential loss amount or a potential range of loss is probable and reasonably estimable under the provisions of ASC 450, Contingencies. The Company was notified by Theravectys of the possible infringement by the Company of Theravectys’ exclusive license to patents no. EP 1071804, EP 1224314, and EP 1222300 granted from the owner of the patents Institut Pasteur. Each of these patents is now expired, having each reached the end of it its patent term on April 23, 2019 for EP 1071804 and October 10, 2020 for EP 1224314, and EP 1222300. The Company considered the situation and determined that the likelihood of a material adverse effect on its business is remote. To date, the Company has not engaged in any such discussions with Theravectys nor has the Company received any further communication from Theravectys. The Company expenses as incurred the costs related to its legal proceedings, if any.
14. Subsequent events.
In February 2021, the Company entered into a Sponsored Research Agreement (“SRA”) with OSR to conduct certain research projects related to Temferon. The total consideration to be paid by the Company under the SRA will be €1.0 million with payments scheduled quarterly over 2021 and 2022.
| F-27 |

(formerly, Genenta Science S.r.l.)
Consolidated Statements of Operations and Comprehensive Loss
| Six-Months Ended June 30, | ||||||||
| 2021 | 2020 | |||||||
| (unaudited) | ||||||||
| Operating expenses | ||||||||
| Research and development | € | 3,199,234 | € | 2,072,129 | ||||
| General and administrative | 842,236 | 404,885 | ||||||
| Total operating expenses | 4,041,470 | 2,477,014 | ||||||
| Loss from operations | (4,041,470 | ) | (2,477,014 | ) | ||||
| Other income (expense) | ||||||||
| Other income | 2,679 | 559 | ||||||
| Foreign exchange loss | (9,111 | ) | (2,849 | ) | ||||
| Total other income (expense), net | (6,432 | ) | (2,290 | ) | ||||
| Loss before income taxes | (4,047,902 | ) | (2,479,304 | ) | ||||
| Income taxes (Note 5) | - | - | ||||||
| Loss | € | (4,047,902 | ) | € | (2,479,304 | ) | ||
| Comprehensive loss | ||||||||
| Total comprehensive loss | (4,047,902 | ) | (2,479,304 | ) | ||||
| Loss per share information (unaudited): | ||||||||
| Loss | € | (4,047,902 | ) | - | ||||
| Loss per share - basic and diluted | € | (0.27 | ) | - | ||||
| Weighted average number of shares outstanding - basic and diluted | 14,772,610 | - | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-28 |
(formerly, Genenta Science S.r.l.)
Consolidated Balance Sheets
| At June 30, | At December 31, | |||||||
| 2021 | 2020 | |||||||
| (Unaudited) | ||||||||
| Assets | ||||||||
| Current assets | ||||||||
| Cash and cash equivalents | € | 10,553,906 | € | 15,465,243 | ||||
| Prepaid expenses and other current assets (Note 6) | 1,549,766 | 947,501 | ||||||
| Prepaid expenses and other current assets - related party (Note 12) | - | 121,432 | ||||||
| Total current assets | 12,103,672 | 16,534,176 | ||||||
| Non-current assets | ||||||||
| Property and equipment, net (Note 7) | 20,612 | 18,971 | ||||||
| Other asset- related party (Note 12) | 3,350 | 3,350 | ||||||
| Other non-current assets (Note 8) | 1,163,375 | 945,618 | ||||||
| Total non-current assets | 1,187,337 | 967,939 | ||||||
| Total assets | € | 13,291,009 | € | 17,502,115 | ||||
| Liabilities and quotaholders’ and stockholders’ equity | ||||||||
| Current liabilities | ||||||||
| Accounts payable | € | 270,941 | € | 544,988 | ||||
| Accounts payable - related party (Note 12) | 88,392 | 10,027 | ||||||
| Accrued expenses | 418,571 | 365,969 | ||||||
| Accrued expenses - related party (Note 12) | 809,605 | 1,359,191 | ||||||
| Other current liabilities | 80,801 | 53,243 | ||||||
| Total current liabilities | 1,668,310 | 2,333,418 | ||||||
| Long-term liabilities | ||||||||
| Retirement benefit obligation (Note 9) | 21,645 | 17,388 | ||||||
| Total long-term liabilities | 21,645 | 17,388 | ||||||
| Commitments and contingencies (Note 13) | - | - | ||||||
| Quotaholders’ and stockholders’ equity | ||||||||
| Corporate capital | - | 37,056 | ||||||
| Additional paid-in capital | - | 36,604,728 | ||||||
| Common stock, no par value, 59,700,000 shares authorized and 15,000,000 shares issued and outstanding (unaudited) (Note 10) | 37,139,431 | - | ||||||
| Accumulated deficit | (25,538,377 | ) | (21,490,475 | ) | ||||
| Total quotaholders’ and stockholders’ equity | 11,601,054 | 15,151,309 | ||||||
| Total liabilities and quotaholders’ and stockholders’ equity | € | 13,291,009 | € | 17,502,115 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-29 |
(formerly, Genenta Science S.r.l.)
Consolidated Statement of Changes in Quotaholders’ and
Stockholders’ Equity
| Corporate capital | Additional paid-in capital | Common shares outstanding | Common stock, no par | Accumulated deficit | Total | |||||||||||||||||||
| Balance at December 31, 2019 | € | 36,049 | € | 34,713,225 | - | € | - | € | (15,898,461 | ) | € | 18,850,813 | ||||||||||||
Capital increase from exercise of options on Quota B (Unaudited) | 136 | - | - | - | - | 136 | ||||||||||||||||||
Share-based compensation (Unaudited) | 230,097 | - | - | - | 230,097 | |||||||||||||||||||
Accumulated deficit (Unaudited) | - | - | - | - | (2,479,304 | ) | (2,479,304 | ) | ||||||||||||||||
| Balance at June 30, 2020 (Unaudited) | € | 36,185 | € | 34,943,322 | - | € | - | € | (18,377,765 | ) | € | 16,601,742 | ||||||||||||
| Balance at December 31, 2020 | € | 37,056 | € | 36,604,728 | - | € | - | € | (21,490,475 | ) | € | 15,151,309 | ||||||||||||
Capital increase from exercise of options on Quota B (Unaudited) | 715 | - | - | - | - | 715 | ||||||||||||||||||
Share -based compensation (Unaudited) | 497,104 | - | - | - | 497,104 | |||||||||||||||||||
Quota B repurchased (Unaudited) | (172 | ) | - | - | - | - | (172 | ) | ||||||||||||||||
| Corporate capital adjustment from Srl to SpA (Unaudited) | (37,599 | ) | - | 11,279,700 | 37,599 | - | - | |||||||||||||||||
| Capital increase related to SpA (Unaudited) | - | (12,401 | ) | 3,720,300 | 12,401 | - | - | |||||||||||||||||
| Conversion adjustment from Srl to SpA (Unaudited) | - | (37,089,431 | ) | - | 37,089,431 | - | - | |||||||||||||||||
| Accumulated deficit (Unaudited) | - | - | - | - | (4,047,902 | ) | (4,047,902 | ) | ||||||||||||||||
| Balance at June 30, 2021 (Unaudited) | € | - | € | - | 15,000,000 | € | 37,139,431 | € | (25,538,377 | ) | € | 11,601,054 | ||||||||||||
The accompanying notes are an integral part of these financial consolidated statements.
| F-30 |
(formerly, Genenta Science S.r.l.)
Consolidated Statements of Cash Flows
| Six Months Ended June 30, | ||||||||
| 2021 | 2020 | |||||||
| Unaudited | ||||||||
| Cash flows from operating activities | ||||||||
| Net loss | € | (4,047,902 | ) | € | (2,479,304 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation expense | 2,086 | 566 | ||||||
| Retirement benefit obligation | 4,256 | 3,014 | ||||||
| Share-based compensation | 497,104 | 230,097 | ||||||
| Changes in operating assets and liabilities | ||||||||
| Prepaid expenses and other current assets | (263,548 | ) | (61,337 | ) | ||||
| Other non-current assets | (217,757 | ) | 100,229 | |||||
| Accounts payable | (274,047 | ) | 70,769 | |||||
| Accounts payable - related party | 78,365 | 796,753 | ||||||
| Accrued expenses | 52,602 | (30,647 | ) | |||||
| Accrued expenses - related party | (549,586 | ) | (579,138 | ) | ||||
| Other current liabilities | 27,558 | 5,532 | ||||||
| Net cash used in operating activities | (4,690,869 | ) | (1,943,466 | ) | ||||
| Cash flows from investing activities | ||||||||
| Purchases of property and equipment | (3,727 | ) | (13,194 | ) | ||||
| Net cash used in investing activities | (3,727 | ) | (13,194 | ) | ||||
| Cash flows from financing activities | ||||||||
| Proceeds from the exercise of stock options | 715 | 136 | ||||||
| Quota B repurchased | (172 | ) | - | |||||
| Prepaid offering costs | (217,284 | ) | - | |||||
| Net cash provided (used in) by financing activities | (216,741 | ) | 136 | |||||
| Net decrease in cash and cash equivalents | (4,911,337 | ) | (1,956,524 | ) | ||||
| Cash and cash equivalents at beginning of period | 15,465,243 | 20,141,379 | ||||||
| Cash and cash equivalents at end of period | € | 10,553,906 | € | 18,184,855 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-31 |
(formerly Genenta Science S.r.l.)
Notes to the Consolidated Financial Statements
1. Nature of business
Genenta Science S.p.A. (formerly Genenta Science S.r.l.) (the “Company”) is an early-stage company developing first-in-class cell and gene cancer therapies. The Company is initially developing its clinical leading product, Temferon™, to treat glioblastoma multiforme (“GBM”), a solid tumor affecting the brain. The Company intends to continue its clinical trials in Europe and eventually start a second clinical trial to study Temferon™ in liver cancers, possibly in the United States.
Genenta Science S.p.A. (formerly Genenta Science S.r.l. “società a responsabilità limitata,” similar to a Limited Liability Company in the United States) converted to an Italian corporation (“società per azioni”, or “S.p.A.”) in June 2021, which is similar to a C corporation in the United States. The Company was founded by San Raffaele Hospital (“OSR”), in Milan, Italy, Pierluigi Paracchi, Luigi Naldini and Bernhard Gentner and was incorporated in July 2014. On May 20, 2021, the quotaholders resolved that the Company converted from an S.r.l. to an S.p.A. and determined that the outstanding quota be converted to 15 million common shares at no par value. (See Note 10. Quotaholder’s and stockholder’s equity.) New Bylaws have been adopted, two new Board members were appointed and the existing Board of Directors and Board of Statutory Advisors were re-appointed. The registered office remained located in Milan, Italy. The Company’s reporting currency is Euros. In May 2021, the Company formed a wholly owned, Delaware incorporated subsidiary, Genenta Science, Inc., intended for future operations in the United States. The Company’s subsidiary will operate in US Dollars.
The Company is subject to risks and uncertainties common to early-stage clinical companies in the life-science and biotechnology industries, including but not limited to, risks associated with completing preclinical studies and clinical trials, receiving regulatory approvals for product candidates, development by competitors of new competing products, dependence on key personnel, protection of proprietary technology, compliance with government regulations and the ability to secure additional capital to fund operations. The clinical product candidates currently under development will require significant additional research and development efforts, including regulatory approval and clinical testing prior to commercialization. These efforts require significant amounts of additional capital, adequate personnel and infrastructure and extensive compliance-reporting capabilities. Even if the Company’s product development efforts are successful, it is uncertain when, if ever, the Company will realize revenue from product sales and profit from operations.
Liquidity and Risks
The Company has incurred losses since its inception, including a net loss of €4,047,902 and €2,479,304 for the six-month periods ended June 30, 2021 and 2020, respectively. In addition, at June 30, 2021, the Company had an accumulated deficit of €25,538,377. There is no certainty that the Company will become profitable in the future. The Company has primarily funded these losses through the proceeds from sales of convertible debt and equity quotas, prior to the Company’s conversion into an S.p.A.
The Company has evaluated whether there are conditions and events considered in the aggregate that raise substantial doubt about the Company’s ability to continue as a going concern. Although the Company has incurred recurring losses and expects to continue to incur losses for the foreseeable future, the Company expects that its existing consolidated cash and cash equivalents on hand at September 27, 2021 of approximately €8.9 million will be sufficient to fund projected cash requirements through the fourth quarter of 2022. Therefore, the Company will require significant additional financing in the future to fund operations. Accordingly, the accompanying financial statements have been prepared assuming that the Company will continue as a going concern.
| F-32 |
The future viability of the Company is dependent on its ability to raise additional capital to finance its operations and the Company intends to pursue financings in the US public market. The Company’s inability to raise capital as and when needed could have a negative impact on its financial condition and ability to pursue its business strategies. There can be no assurance that the current operating plan will be achieved or that additional funding will be available on terms acceptable to the Company, or at all.
Basis of presentation
The accompanying unaudited interim consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles for interim financial information and with the instructions to Form 20-F and Article 10 of Regulation S-X promulgated by the Securities and Exchange Commission (“SEC”). Accordingly, certain information and footnote disclosures normally included in annual financial statements have been condensed or omitted. In the opinion of management, the accompanying unaudited interim consolidated financial statements reflect all adjustments (including normal recurring adjustments and the elimination of intercompany accounts) considered necessary for a fair statement of all periods presented. The results of operations of the Company for any interim periods are not necessarily indicative of the results of operations for any other interim periods or for a full fiscal year. These unaudited interim condensed consolidated financial statements for the six months ended June 30, 2021 and 2020 should be read in conjunction with the audited consolidated financial statements and footnotes thereto for the years ended December 31, 2020 and 2019, included elsewhere herein.
A summary of the significant accounting policies applied in the preparation of these financial statements is presented below, only for the categories and headings now applicable and that might be applicable in the future based on the Company’s business. These policies have been consistently applied, unless otherwise stated.
2. Summary of significant accounting policies
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of the Company and its wholly owned subsidiary. All significant intercompany accounts and transactions have been eliminated in consolidation.
Use of estimates
The preparation of financial statements in conformity with US GAAP requires management to make estimates and assumptions that affect the reported amounts reported in the financial statements and the disclosures made in the accompanying notes. Estimates and assumptions reflected in these consolidated financial statements include, but are not limited to, the accrual for research and development expenses and related milestone payments, share-based compensation expense, valuation of Research & Development (R&D) tax credits, the valuation of equity and the recoverability of the Company’s net deferred tax assets and related valuation allowance. Estimates are periodically reviewed considering changes in circumstances, facts and experience. Actual results may differ from these estimates under different assumptions or conditions. Changes in estimates are recorded in the period in which they become known. The areas involving a higher degree of judgment or complexity, or areas where assumptions and estimates are significant to the financial statements are disclosed below.
| F-33 |
Cash and cash equivalents
The Company considers all highly liquid investments purchased with original maturities of 90 days or less at acquisition to be cash equivalents. In the consolidated cash flow statements, cash and cash equivalents include: cash on hand, deposits held with banks, and other short-term highly liquid investments. In the consolidated balance sheets, bank overdrafts, if any, are shown in current liabilities. Cash and cash equivalents are detailed as follows:
At June 30, | At December 31, | |||||||
| 2021 | 2020 | |||||||
| Cash in bank | € | 10,552,591 | € | 15,462,805 | ||||
| Cash in hand & prepaid cards | 1,315 | 2,438 | ||||||
| Total | € | 10,553,906 | € | 15,465,243 | ||||
Loss per share
Loss per share (“EPS”) is computed in accordance with US GAAP. Basic EPS is computed by dividing net loss by the weighted average number of common shares outstanding during the period. Diluted EPS reflects potential dilution and is computed by dividing loss by the weighted average number of common shares outstanding during the period increased by the number of additional common shares that would have been outstanding if all potential common shares had been issued and were dilutive. Since the Company was an S.r.l. in previous periods, QPS (quota per share) has not been included in the historical audited financial statements or the interim Statement of Operations and Comprehensive Loss at June 30, 2020 because the Company determined it was not a meaningful or material disclosure due to the Company’s capital structure. This calculation was applied, after the increase in capital to €50,000 required to be an S.p.A. by Italian law. The Company’s stockholders authorized 59.7 million ordinary common shares. The Company has 15.0 million common shares issued and outstanding at June 30, 2021 with no outstanding options or warrants and 2.7 million common shares reserved for the Company’s Equity Incentive Plan 2021–2025. Diluted EPS is not relevant at June 30, 2021, as there were no common share equivalents. (See Note 10. Quotaholders’ and stockholders’ equity and Note 11. Share-based compensation.)
Foreign currency translation
The reporting and functional currency of the Company is Euros. All amounts are presented in Euros unless otherwise stated. All amounts disclosed in the consolidated financial statements and notes have been rounded to the nearest Euro unless otherwise stated. Foreign currency transactions, if any, are translated into Euros using the exchange rates prevailing at the dates of the transactions or valuation where items are re-measured. Foreign exchange gains and losses resulting from the settlement of such transactions and from the translation at period-end exchange rates of monetary assets and liabilities denominated in foreign currencies are recognized in the consolidated statements of operations. During the periods ended June 30, 2021 and 2020, foreign exchange gains and losses were insignificant.
Emerging growth company status
The Company is an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act, or JOBS Act, and may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies. The Company may take advantage of these exemptions until the Company is no longer an “emerging growth company.” Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards. The Company has elected to use the extended transition period for complying with new or revised accounting standards and, because of this election, its consolidated financial statements may not be comparable to companies that comply with public company effective dates. The Company may take advantage of these exemptions up until the last day of the fiscal year following the fifth anniversary of its initial public offering (“IPO”) or such earlier time that it is no longer an “emerging growth company.”
| F-34 |
Fair value measurements
Certain assets and liabilities of the Company are carried at fair value under U.S. GAAP. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. Financial assets and liabilities carried at fair value are to be classified and disclosed in one of the following three levels of the fair value hierarchy, of which the first two are considered observable and the last is considered unobservable:
| ● | Level 1 — Quoted prices in active markets for identical assets or liabilities. |
| ● | Level 2 — Observable inputs (other than Level 1 quoted prices), such as quoted prices in active markets for similar assets or liabilities, quoted prices in markets that are not active for identical or similar assets or liabilities, or other inputs that are observable or can be corroborated by observable market data. |
| ● | Level 3 — Unobservable inputs that are supported by little or no market activity that are significant to determining the fair value of the assets or liabilities, including pricing models, discounted cash flow methodologies and similar techniques. |
To the extent that the valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment. Accordingly, the degree of judgment exercised by the Company in determining fair value is greatest for instruments categorized in Level 3. A financial instrument’s level within the fair value hierarchy is based on the lowest level of any input that is significant to the fair value measurement.
The carrying values of the Company’s research and development tax credits, VAT credits, accounts payable, accrued expenses and other current liabilities approximate their fair values due to the short-term nature of these assets and liabilities.
Segment information
Operating segments are identified as components of an enterprise for which separate discrete financial information is available for evaluation by the chief operating decision-maker in making decisions regarding resource allocation and assessing performance. The Company and its chief operating decision-maker view the Company’s operations and manages its business in one operating segment, which is the research and development in the pharmaceutical sector with a focus on developing novel therapeutics to treat cancer.
Tax credit on investments in research and development
In line with the legislation in force until June 30, 2021, companies in Italy that invest in eligible research and development activities, regardless of the legal form and economic sector in which they operate, can benefit from a tax credit which can be used in order to reduce most taxes payable, including income tax or regional tax on productive activities, as well as social security contributions and payroll withholding taxes. For eligible R&D activities, the tax credit is equal to 20% (12% in FY 2020) of the eligible costs incurred, with a maximum annual amount of €4.0 million (€3.0 million for FY 2020).
| F-35 |
The eligible activities consist of fundamental research, industrial research, and experimental development as defined respectively of the letters m), q) and j) of point 15, par. 1.3 of the Communication no. 198/2014 of the European Commission.
To determine the cost basis of the benefit, the following expenses are eligible:
| ● | Personnel costs; |
| ● | Depreciation charges, costs of the financial or simple lease and other expenses related to movable tangible assets and software used in research and development projects; |
| ● | Expenses for extra-euro research contracts concerning the direct execution of eligible research and development activities by the provider; |
| ● | Depreciation charges; |
| ● | Expenses for consulting services and equivalent services related to eligible research and development activities; and, |
| ● | Expenses for materials, supplies, and other similar products used in research and development projects. |
The Company, by analogy, accounts for this receivable in accordance with International Accounting Standards (IAS) 20, Accounting for Government Grants and Disclosure of Government Assistance. The receivable is recognized when there is reasonable assurance that: (1) the recipient will comply with the relevant conditions; and, (2) the grant will be received. The Company has elected to present it net of the related expenditure on the consolidated statements of operations and comprehensive loss.
While these tax credits can be carried forward indefinitely, the Company recognized an amount which reflects management’s best estimate of the amount that is reasonably assured to be realized or utilized in the foreseeable future based on historical benefits realized, adjusted for expected changes, as applicable. The tax credits are recorded as an offset to research and development expenses in the Company’s consolidated statements of operations and comprehensive loss.
Share-based compensation
To reward the efforts of employees, directors, and certain consultants and to promote the growth of the Company, the Company’s Board of Directors has approved, prior to the Company’s conversion to an S.p.A., various share-based awards. All options have been awarded with an exercise price of €1 per quota and, when exercised, all options have been converted to Quota B. The options granted had the vesting condition that the individual must remain in his/her role at least one year or as otherwise specified for each person. At June 30, 2021, there were no outstanding options, as all options had been vested and/or accelerated and exercised prior to the Company’s conversion to an S.p.A.
The Company measures share-based awards granted to recipients based on the fair value on the date of the grant and recognizes compensation expense for those awards over the requisite service period(s), which is the vesting period of the respective award. Forfeitures were accounted for as they occurred. The measurement date for option awards was the date of the grant. The Company classifies share-based compensation expense in its consolidated statements of operations and comprehensive loss in the same manner in which the award recipient’s payroll costs are classified or in which the award recipient’s service payments are classified.
| F-36 |
With the adoption of ASU No. 2018-07, Compensation— Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting (“ASU 2018-07”) on January 1, 2019, the measurement date for non-employee awards is the date of the grant. The compensation expense for non-employees is recognized, without changes in the fair value of the award, over the requisite service period, which is the vesting period of the respective award.
Property and equipment
Property and equipment are stated at cost, including any accessory and direct costs that are necessary to make the assets fit for use, and adjusted by the corresponding accumulated depreciation. The depreciation rates recorded in the consolidated financial statements have been calculated by taking into consideration the use, purpose, and financial-technical duration of the assets, on the basis of their estimated useful economic lives. The Company believes the above criteria to be represented by the following depreciation rates:
| ● | Equipment & Furniture: 15 years; |
| ● | Electronic office equipment: 10 years; and, |
| ● | Leasehold Improvements: based on the shorter of the life of the leasehold improvement or the remaining term of the lease. |
Ordinary maintenance costs are expensed to the consolidated statements of operations and comprehensive loss in the year in which they are incurred. Extraordinary maintenance costs, the purpose of which is to extend the useful economic life of the asset, to technologically upgrade it and/or to increase its productivity or safety for the purpose of economic productivity of the Company, are attributed to the asset to which they refer and depreciated on the basis of its estimated useful economic life. Amortization of leasehold improvements is computed using the straight-line method based upon the terms of the applicable lease or estimated useful life of the improvements, whichever is less.
Impairment of long-lived assets
In accordance with ASC Topic 360-10-20, ‘‘Property, Plant and Equipment,” the Company performs an impairment test whenever events or circumstances indicate that the carrying value of long- lived assets with finite lives may be impaired. Impairment is measured by comparing the carrying value of the long-lived assets to the estimated undiscounted pre-tax cash flows expected to result from the use of such assets and their ultimate disposition. In circumstances where impairment is determined to exist, the Company will write-down the asset to its fair value based on the present value of estimated cash flows. To date, no impairments have been identified for the periods ended June 30, 2021 and 2020.
Deferred Offering Costs
Deferred public offering costs, which primarily consist of direct, incremental legal and accounting fees relating to fundraising activities (e.g., an initial public offering (IPO)), are capitalized within prepaid expenses and other current assets. The deferred offering costs will be offset against proceeds from fundraising activities (e.g., an IPO) upon the consummation of an offering. In the event the fundraising activities are terminated without raising capital, any deferred offering costs will be expensed. The Company has incurred €373,545 in deferred offering costs for the six months ended June 30, 2021. (Also, see Note 6.)
| F-37 |
Recently issued accounting pronouncements
In December 2019, the FASB issued ASU 2019-12, Income Taxes: Simplifying the Accounting for Income Taxes. The new standard intended to simplify the accounting for income taxes by eliminating certain exceptions related to the approach for intra-period tax allocation, the methodology for calculating income taxes in an interim period and the recognition of deferred tax liabilities for outside basis differences. The new guidance also simplifies aspects of the accounting for franchise taxes and enacted changes in tax laws or rates and clarifies the accounting for transactions that result in a step-up in the tax basis of goodwill. For non-public entities, the standard is effective for annual periods beginning after December 15, 2021, with early adoption permitted. Adoption of the standard requires certain changes to primarily be made prospectively, with some changes to be made retrospectively. The Company is currently evaluating the impact that the adoption of ASU 2019-12 will have on its consolidated financial statements.
3. Research and development
Research and development costs are expensed as incurred. Research and development expenses consist of costs incurred in performing research and development activities, including salaries, share- based compensation and benefits, facilities costs, third-party license fees, and external costs of outside vendors and consultants engaged to conduct clinical development activities and clinical trials, (e.g., contract research organizations [or “CROs”]), as well as costs to develop a manufacturing processes, perform analytical testing and manufacture clinical trial materials, (e.g., contract manufacturing organizations [or “CMOs”]). Non-refundable prepayments for goods or services that will be used or rendered for future research and development activities are recorded as prepaid expenses. Such amounts are recognized as an expense as the goods are delivered or the related services are performed, or until it is no longer expected that the goods will be delivered, or the services rendered. In addition, funding from research grants, if any, is recognized as an offset to research and development expense based on costs incurred on the research program.
The Company yearly sustains a significant amount of research costs to meet its business objectives. The Company has various research and development contracts, and the related costs are recorded as research and development expenses as incurred. When billing terms under these contracts do not coincide with the timing of when the work is performed, the Company is required to make estimates of outstanding obligations at period end to those third parties. Any accrual estimates are based on several factors, including the Company’s knowledge of the progress towards completion of the research and development activities, invoicing to date under the contracts, communication from the research institution or other companies of any actual costs incurred during the period that have not yet been invoiced, and the costs included in the contracts. Significant judgments and estimates may be made in determining the accrued balances at the end of any reporting period. Actual results could differ from the estimates made by the Company. The historical accrual estimates made by the Company have not been materially different from the actual costs. For further details, please refer to the Related Parties disclosures in Note 12 below.
4. General and administrative
General and administrative costs consist primarily of salaries, share-based compensation, benefits and other related costs for personnel and consultants in the Company’s executive and finance functions, professional fees for legal, finance, accounting, auditing, tax and consulting services, travel expenses and facility-related expenses, which include rent and maintenance of facilities and other operating costs not otherwise included in research and development expense.
| F-38 |
5. Income taxes
The Company is subject to taxation in Italy, including the standard corporate income tax (“IRES”) and a regional business tax (“IRAP”). Taxes are recorded on an accrual basis. They therefore represent the allowances for taxes paid or to be paid for the year, calculated according to the current enacted rates and applicable laws. The Company is taxed in various countries where it has permanent establishment, as applicable. Due to the tax loss position reported, no income taxes were due for the periods ending June 30, 2021 and 2020.
The Company uses the asset and liability method of accounting for deferred income taxes. Under this method, deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the carrying amounts and the tax basis of assets and liabilities, measured at tax rates expected to be enacted at the time of their reversals. These temporary differences primarily relate to net operating losses carried forward available to offset future taxable income.
At each reporting date, the Company considers existing evidence, both positive and negative, that could impact its view with regards to future realization of deferred tax assets. In consideration of the start-up status of the Company, a full valuation allowance has been established to offset the deferred tax assets, as the related realization is currently uncertain. In the future, should the Company conclude that it is more likely than not that the deferred tax assets are partially or fully realizable, the valuation allowance will be reduced to the extent of such expected realization, and the corresponding amount will be recognized as income tax benefit in the Company’s consolidated statements of operations and comprehensive loss.
The Company recognizes tax liabilities from an uncertain tax position if it is more likely than not that the tax position will not be sustained upon examination by the taxing authorities, based on the technical merits of the tax position. There are no uncertain tax positions that have been recognized in the accompanying consolidated financial statements. The prior five years of tax returns (2016-2020) are potentially subject to audit.
At June 30, 2021 and 2020, the Company believes there are no significant differences with regards to its deferred tax assets and its relevant components, compared to the computations of the preceding periods.
In 2011, the Italian tax authorities issued a set of rules that modified the previous treatment of tax loss carryforwards. According to the DL 98/2011, at the end of 2011, all existing tax loss carryforwards will never expire but they can off-set only 80% of the taxable income of the year. The rules do not affect the tax loss carryforward that refer to the start-up period, defined as the first three years of operations starting from the inception of the Company. The impact of the updated calculation of tax losses carryforward at June 30, 2021 and 2020 is deemed not significant with respect of the preceding periods.
| F-39 |
6. Prepaid expenses and other current assets
Prepaid expenses and other current assets consist of the following:
At June 30, | At December 31, | |||||||
| 2021 | 2020 | |||||||
| Value Added Tax (VAT) | € | 724,157 | € | 480,000 | ||||
| Research and development tax credit | 374,667 | 280,631 | ||||||
| Deferred offering costs | 373,545 | 156,261 | ||||||
| Advances to suppliers- related party | - | 121,432 | ||||||
| Other prepaids | 77,397 | 30,609 | ||||||
| Total | € | 1,549,766 | € | 1,068,933 | ||||
Value Added Tax (VAT) receivables are linked to purchases. Italian VAT (Imposta sul Valore Aggiunto) applies to the supply of goods and services carried out in Italy by entrepreneurs, professionals, or artists and on imports carried out by anyone. Intra-Community acquisitions are also subject to VAT under certain situations. The Italian standard VAT rate for 2021 and 2020 is 22%. Reduced rates are provided for specifically listed supplies of goods and services. It is carried forward indefinitely and does not expire. The Company reclassified to other non-current assets a portion of the receivable which is expected to be realized beyond 12 months.
Tax credits on research and development represent a special tax relief offered to Italian companies operating in the research and development sector and can be used to offset most taxes payable. The Company has a total research and development tax credit available to be used of approximately €4.2 million at June 30, 2021, and approximately €3.9 million at December 31, 2020, which can be carried forward indefinitely and does not expire. However, given the start-up status of the Company, and the fact that it will not be profitable in the foreseeable future (which limits the utilization of the credit), the Company recognized a receivable balance that represents the Company’s best estimate of the amount of tax credit that can be used in offsetting taxes payable for 2022.
During the six months ended June 30, 2021, the Company utilized approximately €204,000 to offset certain social contributions and taxes payable, while during the financial year 2020, the Company utilized approximately €281,000. The benefit recorded for the six months ended June, 2021 and 2020, to offset research and development expenses was approximately €204,000 and €136,000, respectively. The Company reclassified to other non-current assets as a portion of the receivable, which is expected to be realized beyond 12 months. The utilization rate of research and development credit, in the periods following June 30, 2021, is projected in a consistent manner to previous periods.
The amount of €373,545 in “deferred offering costs” in the chart above, mainly relate to legal fees incurred from fundraising activities (€263,368) and audit expenses (€110,177). Fundraising deferred offering costs will be offset against the funding proceeds and reclassified to common stock upon completion of the funding effort. If the funding is unsuccessful, the Company will expense all the related fundraising costs. (See Note 2.) The amount of €77,397 in “other prepaids” in the chart above, mainly relates to insurance expenses (€30,824) and various advance payments and operational prepaid expenses.
The remaining balance is comprised of miscellaneous minor prepaid expenses.
| F-40 |
7. Property and equipment, net
Property and equipment consist of the following:
At June 30, | At December 31, | |||||||
| 2021 | 2020 | |||||||
| Computer | € | 22,740 | € | 16,196 | ||||
| Furniture and fixtures | 1,858 | 4,675 | ||||||
| Total property and equipment | 24,598 | 20,871 | ||||||
| Less: accumulated depreciation | (3,986 | ) | (1,900 | ) | ||||
| Property and equipment, net | € | 20,612 | € | 18,971 | ||||
Property and equipment consist of computers and furniture and fixtures of our office space in Milan, Italy. There were no disposals, nor impairments during the periods. Depreciation has been calculated by taking into consideration the use, purpose and financial-technical duration of the assets, based on their estimated economic lives. No significant purchases occurred during the six months ended June 30, 2021.
8. Other non-current assets
Other non-current assets consist of the long-term portion of the VAT receivable and R&D tax credit, as follows:
At June 30, | At December 31, | |||||||
| 2021 | 2020 | |||||||
| Value Added Tax (VAT) | € | 975,215 | € | 664,987 | ||||
| Research and development tax credit | 187,333 | 280,631 | ||||||
| Other assets | 827 | - | ||||||
| Other assets- related party | 3,350 | - | ||||||
| Total | € | 1,166,725 | € | 945,618 | ||||
Other assets - related party include a security deposit of €3,350 paid to Ospedale San Raffaele (“OSR”) as security guarantee for the office lease contract. (See Note 13. Commitments and contingencies.)
9. Retirement benefit obligation
Employees in Italy are entitled to Trattamento di Fine Rapporto (“TFR”), commonly referred to as an employee leaving indemnity, which represents deferred compensation for employees in the private sector. Under Italian law, an entity is obligated to accrue for TFR on an individual employee basis payable to each individual upon termination of employment (including both voluntary and involuntary dismissal). The annual accrual is approximately 7% of total pay, with no ceiling, and is revalued each year by applying a pre-established rate of return of 1.50%, plus 75% of the Consumer Price Index, and is recorded by a book reserve. TFR is an unfunded plan. The costs of the retirement benefit obligation are accounted for under the provisions of ASC 715, Compensation – Retirement Benefits. The amount of the obligation at June 30, 2021, and December 31, 2020 was €21,645 and €17,388, respectively.
10. Quotaholders’ and stockholders’ equity
The Company was an S.r.l., which is an Italian limited liability company similar to a limited liability company in the United States. The Articles of Incorporation, Shareholders’ Agreement and the By-laws of the Company provided for different quotas, which represented the Company’s corporate capital, rather than shares of stock as ownership.
| F-41 |
Corporate capital
As an S.r.l., the Company’s ownership was called “corporate capital” and “quotas” rather than shares, stock or units.
The Company’s capital was divided between the five quotas as summarized below at December 31, 2020:
| At December 31, | Ownership | |||||||
| Quota | 2020 | % | ||||||
| A | € | 10,458 | 28 | % | ||||
| B | 6,886 | 19 | % | |||||
| C | 8,645 | 23 | % | |||||
| D | 4,034 | 11 | % | |||||
| E | 7,033 | 19 | % | |||||
| Total | € | 37,056 | 100 | % | ||||
The Company had five (5) quotas:
| ● | Quota A. Quota A was reserved for certain founders. One of the founders had the right to appoint three (3) board members out of five (5), appoint the Chair from these three (3) persons and appoint one (1) member of the Board of Statutory Auditors. One other founder had the right to appoint two (2) board members out of five (5), appoint two (2) statutory auditors and appoint the Chair of the statutory auditors from the two (2) appointees. Quota A had voting rights. | |
| ● | Quota B. Quota B had no voting rights, the same profit-sharing rights as Quota A and was priced at a nominal amount of €1.00. The Company had historically utilized Quota B for its share-based compensation program offered to board members, employees, and consultants. Quota B was also held by certain co-founders. The Company’s stock options were exercisable into Quota B for past and present board members, employees, and consultants. | |
| ● | Quota C. Quota C had the right to appoint one (1) member of the Board of Statutory Auditors; specifically, the one (1) that a founder had the right to appoint. Investors received Quota C in the Company’s first funding round (2014/2015) where approximately €10 million was raised. | |
| ● | Quota D. Investors received Quota D in the Company’s second funding round (2017) where approximately €7 million was raised. | |
| ● | Quota E. Investors received Quota E in the Company’s third funding round (through December 31, 2019) where approximately €14.8 million was raised approximately (€15.1 million gross, net of approximately €0.3 million of financing fees). Investors received Quota E in the Company’s second tranche of the third funding round (through December 31, 2020) where approximately €1.4 million was raised (approximately €1.5 million gross, net of approximately €0.1 million of financing fees). |
| F-42 |
| ● | Quotas A, C, D & E. During a divestiture proceeding (meaning Quotas representing 100% of the corporate capital of the Company) or a dissolution of the Company, Quotas C, D & E all had the same rights with respect to the proceeds of a divestiture, i.e., all three (3) quotas shared the divestiture consideration equally (on a pari passu basis) up to the amount of their investment. If there was any consideration remaining after payment to quotas C, D & E, then quota A was entitled to the amount remaining up to the amount of their investment. If proceeds of a divestiture were less than or equal to €50 million, then any proceeds remaining after payment of quotas A, C, D & E, were to be shared equally among quotas A, C, D & E; however, if proceeds of a divestiture were greater than €50 million, then any proceeds remaining after payment of quotas A, C, D & E, were to be distributed to each quota separately according to a detailed formula specified in the Company’s By-Laws, including quota B. Similar to a divestiture, net profits, if any, were to be distributed in the same manner to quotas A, B, C, D & E, after deducting not less than five (5) percent for a legal reserve (up to where this reserve equals one-fifth of the quota capital). A, C, D, E had equal voting rights and the Company By-laws specify protective provisions for each class of quota for A, C, D & E. |
During the six months ended June 30, 2021, two events occurred which together had a significant impact on the Company’s equity:
On April 1, 2021, the Board of Directors resolved to grant to employees and non-employees stock options and accelerate the vesting of other stock options on €715 quota B and €172 quota B were repurchased at nominal value, cancelled, and allocated to the option plan as available for grant by Drs. Naldini and Gentner, leaving a net equity increase of €543 quota B. All quota B ownership has limited rights and carry a par value of €1 per quota. The Corporate Capital amount was € 37,771 (€37,056 corporate capital at December 31, 2020 + €715 exercise of Quota B options before the conversion).
On May 20, 2021, at a special Quotaholders’ meeting in May 2021, the Quotaholders resolved to convert the Company from an S.r.l. to an S.p.A., which conversion became effective on June 18, 2021. As consequence of the conversion, the Corporate Capital has been converted to ordinary common stock with no par value and it was increased to €50,000 to satisfy the minimum capital requirement to qualify as an S.p.A. This increase was an adjustment from Additional Paid-in Capital to Common stock, no par value.
As a result of the Company conversion, the Corporate Capital was reclassified as ordinary Common Stock no par value, combining the minimum capital amount of €50,000 with the Additional Paid-In Capital for a total of €37,139,431. The outstanding quota of €50,000 before the conversion were all converted into 15 million shares of ordinary common stock no par value after the conversion at the same conversion rate of approximately 300 quota percentage of ownership. All preferences related to the quota classes were terminated and all stockholders now hold ordinary common stock, no par value. All of shares outstanding after the conversion are held in ledger form. The Company adopted a new Article of Association, appointed two new directors (including Mr. Squinto as Chairman) and re-appointed the existing members of the Board of Directors and the existing Board of Statutory Auditors.
| 11. | Share-based compensation |
The Company granted options on its Corporate Capital to certain directors, officers, employees, and consultants, as an incentive and as additional compensation prior to the Company’s conversion to an S.p.A. All options converted into Quota B when vested and exercised. All options had an exercise price of €1.00 per quota. Options generally vested over a one-to-three-year period and have been exercised when vested.
At December 31, 2019, there were no options available for grant, as all remaining authorized options were granted in 2019; therefore, no options were granted in 2020. However, in April 2021, €172 of quota B shares were repurchased, cancelled and allocated to the option plan as available for grant. The Board approved new option grants on €169 of quota B and accelerated the vesting of options on €546 quota B and all options were exercised. This totals €715 quota B that were issued and exercised during the six months ended June 30, 2021.
| F-43 |
Also, in May 2021 as part of the conversion to an S.p.A., the shareholders adopted a medium-to long-term incentive plan, “Equity Incentive Plan 2021–2025,” (the “Plan”) with the purpose of providing incentive and retention for key officers, directors, employees and consultants with a view to developing the Company’s business over the next few years. The Board of Directors and the Board of Statutory Auditors reviewed a draft of the Plan. The Plan is intended to last four years and will have a maximum of 2,700,000 new ordinary common shares, or 10% of the total outstanding common shares of the Company. No grants were made under the Plan and the Compensation Committee of the Board of Directors will administer the Plan.
At June 18, 2021, the date of the Company’s conversion to an S.p.A., all stock options were granted, fully vested, exercised and converted into ordinary common stock with no par value. At June 30, 2021, there were no outstanding stock options.
| Number of Options on Quota | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value | |||||||||||||
| Outstanding, vested and expected to vest as of January 1, 2020 | € | 982 | 1 | 2 | € | 1,056,316 | ||||||||||
| Granted | - | - | - | - | ||||||||||||
| Vested and exercised | 436 | 1 | 1 | 472,184 | ||||||||||||
| Cancelled or forfeited | - | - | - | - | ||||||||||||
| Outstanding, vested and expected to vest as of December 31, 2020 | 546 | 1 | 1 | 584,132 | ||||||||||||
| Exercisable as of December 31, 2020 | - | - | - | - | ||||||||||||
| Granted | 169 | 1 | - | 183,831 | ||||||||||||
| Vested and exercised | 715 | 1 | - | 777,920 | ||||||||||||
| Outstanding, vested and expected to vest as of June 30, 2021 | - | - | - | - | ||||||||||||
| Exercisable as of June 30, 2021 | € | - | - | - | € | - | ||||||||||
The Company’s share-based compensation expense for the six months ended June 30, 2021 and 2020, is represented by the following table:
| Six Months Ended June 30, | ||||||||
| 2021 | 2020 | |||||||
| Research & development expense | € | 82,669 | € | - | ||||
| Research & development expense - related party | 179,480 | 163,200 | ||||||
| General & administrative expense | 234,955 | 66,897 | ||||||
| Total | € | 497,104 | € | 230,097 | ||||
The weighted average fair value of the options granted during 2021 was €1,088.
For the periods ended June 30, 2021 and 2020, the Company recorded €497,104 and €230,097, respectively as the fair value of the stock options granted. There was no amount of unrecognized expense at June 30, 2021, since there were no outstanding stock options and approximately €540,000 of unrecognized expense at June 30, 2020.
| F-44 |
Quota B Valuations
The fair value of the Quota B underlying the Company’s stock-based compensation grants had historically been determined by the Company’s board of directors, with input from management and third-party valuations. The Company believes that the board of directors has the relevant experience and expertise to determine the fair value of its Quota B, when also securing third-party assistance. Given the absence of a public trading market of the Company’s equity, and in accordance with the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately- Held Company Equity Securities Issued as Compensation, the board of directors exercised reasonable judgment and considered numerous objective and subjective factors to determine the best estimate of the fair value of the Company’s equity at each grant date. These factors include:
| ● | valuations of the Quota B equity performed by third-party specialists; | |
| ● | the price of the Company’s equity to third-party, arms-length, sophisticated, and qualified investors; | |
| ● | the prices, rights, preferences, and privileges of the Company’s Quota C, D, and E preferred equity classes relative to those of the Company’s equity; | |
| ● | lack of marketability of the Quota B; | |
| ● | lack of voting rights of the Quota B; | |
| ● | current business conditions and projections; | |
| ● | hiring of key personnel and the experience of management; | |
| ● | the Company’s stage of development; | |
| ● | the timing, progress and results of the Company’s pre-clinical studies and clinical trials for the Company’s programs and product candidates; including statements regarding the timing of initiation and completion of trials or studies and related preparatory work, the period during which the results of the trials will become available and the Company’s research and development programs; | |
| ● | likelihood of achieving a liquidity event, such as an initial public offering, a merger or acquisition of the Company given prevailing market conditions, or other liquidation events; | |
| ● | the market performance of comparable publicly traded companies; and | |
| ● | the European, U.S. and global capital market conditions. |
In valuing the Company’s Quota B class of options, the board of directors determined the equity value of the Company’s business using various valuation methods. The board of directors engaged a third-party valuation firm who performed analyses in accordance with the guidance outlined in the American Institute of Certified Public Accountants’ Accounting and Valuation Guide, Valuation of Privately-Held-Company Equity Securities Issued as Compensation. The Company’s option valuations were prepared using an option pricing method (“OPM”), which used market approaches to estimate the Company’s enterprise value.
The OPM treats each equity class as a call option on the total equity value of a company, with exercise prices (i.e., breakpoints) based on the value thresholds at which the allocation among the various holders of a company’s securities changes. A discount was considered for Lack of Marketability (“DLOM”), which is an amount or percentage that is deducted from the value in order to reflect the absence of a viable market. The DLOM was then applied to arrive at an indication of value for the option. Also, considered in the valuation was volatility and the fact that the Quota B class of equity did not carry voting rights. The expected volatility used in the OPM is based upon the historical volatility of a number of publicly traded companies in similar stages of clinical development.
| F-45 |
Application of the Company’s approach involved the use of estimates, judgment, and assumptions that are highly complex and subjective, such as those regarding the selection of comparable companies, and the expected timing of an initial public offering (“IPO”) or other liquidity event. Changes in any or all of these estimates and assumptions or the relationships between those assumptions impact the valuations at each valuation date and may have a material impact on the valuation of the Company’s Quota B equity class, and consequently, the Company’s share-based compensation expense could be materially different.
Weighted average shares
As a result of the Company’s conversion to an S.P.A., the Company was able to calculate both a weighted average and pro forma weighted average number of shares outstanding. The calculation was performed by taking the number of quota outstanding during a given period and weighting them for the number of days that number of quota were outstanding and then converting the quota to common stock using the same conversion that was used at June 18, 2021. The quota carried different rights and privileges, as indicated above, so the Company was not sure how all quota would be treated in a conversion; however, all classes of quota were treated the same. Therefore, the conversion was straight forward. For the six months ended June 30, 2021 and 2020, there was a weighted average of €37,029.65 and €35, 190.54 quota, respectively, which converted into 14,772,610 and 14,038.916 shares of the Company’s common stock, no par value, after the Corporate Capital adjustment.
| 12. | Related parties |
The Company’s research and development expenses are a combination of third-party expenses, and related party expenses, as detailed below:
| For the Six Months Ended June 30, | ||||||||||||
| 2021 | ||||||||||||
| Third Parties | Related Parties | Total | ||||||||||
| Consultants & other Services | € | 525,978 | € | 1,541,526 | € | 2,067,504 | ||||||
| Materials & supplies | 717,905 | - | 717,905 | |||||||||
| Compensation (including share-based) | 221,900 | 179,480 | 401,380 | |||||||||
| Travel & entertainment | 10,445 | - | 10,445 | |||||||||
| Other | 2,000 | - | 2,000 | |||||||||
| Total | € | 1,478,228 | € | 1,721,006 | € | 3,199,234 | ||||||
| For the Six Months Ended June 30, | ||||||||||||
| 2020 | ||||||||||||
| Third Parties | Related Parties | Total | ||||||||||
| Consultants & other Services | € | 383,901 | € | 727,458 | € | 1,111,359 | ||||||
| Materials & supplies | 608,831 | - | 608,831 | |||||||||
| Compensation (including share-based) | 155,633 | 163,200 | 318,833 | |||||||||
| Travel & entertainment | 31,106 | - | 31,106 | |||||||||
| Other | 2,000 | - | 2,000 | |||||||||
| Total | € | 1,181,471 | € | 890,658 | € | 2,072,129 | ||||||
Research and development (related party) expenses during the six months ended June 30, 2021, mainly relate to the clinical trial activity done as per the agreement with the San Raffaele Hospital. This trial activity increased significantly from the second half of 2020 both for number of patients enrolled in the trial and the number of patients subsequently dosed. (Also see OSR – San Raffaele Hospital section below.)
| F-46 |
The Company’s general and administrative expenses are also a combination of third-party and related party expenses, as detailed below:
| For the Six Months Ended June 30, | ||||||||||||
| 2021 | ||||||||||||
| Third Parties | Related Parties | Total | ||||||||||
| Compensation (including share-based) | € | 258,514 | € | 150,000 | € | 408,514 | ||||||
| Accounting, legal & other professional | 181,354 | - | 181,354 | |||||||||
| Facility & insurance related | 1,712 | 7,197 | 8,909 | |||||||||
| Consultants & other third parties | 127,988 | - | 127,988 | |||||||||
| Other | 115,471 | - | 115,471 | |||||||||
| Total | € | 685,039 | € | 157,197 | € | 842,236 | ||||||
| For the Six Months Ended June 30, | ||||||||||||
| 2020 | ||||||||||||
| Third Parties | Related Parties | Total | ||||||||||
| Compensation (including share-based) | € | 116,329 | € | 150,000 | € | 266,329 | ||||||
| Accounting, legal & other professional | 82,863 | - | 82,863 | |||||||||
| Communication & IT related | - | - | - | |||||||||
| Facility & insurance related | 9,474 | 7,200 | 16,674 | |||||||||
| Consultants & other third parties | - | - | - | |||||||||
| Other | 39,019 | - | 39,019 | |||||||||
| Total | € | 247,685 | € | 157,200 | € | 404,885 | ||||||
The Company’s accounts payable to related parties are comprised as follows:
| At June 30, | At December 31, | |||||||
| 2021 | 2020 | |||||||
| San Raffaele Hospital | € | 88,193 | € | 4,085 | ||||
| XDG Biomed | - | 5,942 | ||||||
| Pierluigi Paracchi | 27 | - | ||||||
| Naldini | 115 | - | ||||||
| Genter | 57 | - | ||||||
| Total | € | 88,392 | € | 10,027 | ||||
Drs. Naldini and Gentner outstanding balances represent the “quotas” reimbursement due to the Company as a result of the quota repurchase.
| F-47 |
The Company’s accrued expenses to related parties are comprised as follows:
| At June 30, | At December 31, | |||||||
| 2021 | 2020 | |||||||
| San Raffaele Hospital | € | 809,605 | € | 1,309,191 | ||||
| Pierluigi Paracchi | - | 25,000 | ||||||
| XDG Biomed | - | 25,000 | ||||||
| Total | € | 809,605 | € | 1,359,191 | ||||
No amounts were due and recorded at June 30, 2021 to Mr. Paracchi and XDG Biomed, since their performance bonuses were paid in June 2021.
The Company has identified the following related parties:
| ● | Pierluigi Paracchi (director and co-founder of the Company); | |
| ● | Luigi Naldini (co-founder of the Company and executive scientific advisory board chairman); | |
| ● | Carlo Russo (through XDG Biomed LLC- Chief Medical Officer); | |
| ● | Bernard Rudolph Gentner (co-founder of the Company and member of scientific advisory board); and, | |
| ● | San Raffaele Hospital (co-founder and main supplier of services). |
The following is a description of the nature of the transactions between the Company and these related parties:
Pierluigi Paracchi
Pierluigi Parcchi, former President and Chairman of the Company prior to the conversion, is the Chief Executive Officer as well as co-founder. His annual compensation amounts to of €250,000 plus an annual performance bonus of €50,000, maturing in the period July-June of each year payable after Board of Directors approval. In 2020, the bonus was paid in July 2020. In 2021, the bonus was paid in June 2021. For the six months ended June 30, 2021 and 2020, the Company expensed €150,000 each period, respectively, related to compensation for Mr. Paracchi. At June 30, 2021 and the year ended December 31, 2020, the Company accrued €0 and €25,000, respectively, related to compensation for Mr. Paracchi.
Luigi Naldini/Bernard Rudolph Gentner
Drs. Naldini and Gentner are co-founders of Genenta and part of the SAB – Scientific Advisory Board, with Dr. Naldini as Chairman, and Dr. Gentner as a member. Dr. Naldini has an advisory agreement approved by the Board of Directors and performs the pre-clinical studies for the Company. In particular, the pre- clinical experiments are in solid tumor indications. The last agreement with Dr. Naldini was signed in 2019 and it is still in force. The annual fee is €50,000. During the six months ended June 30, 2020, he billed additional fees for €6,250. For the six months ended June 30, 2021 and 2020, the Company expensed €25,000 and €31,250, respectively, related to Dr. Naldini. At June 30, 2021 and the year ended December 31, 2020, the Company did not accrue anything related to Dr. Naldini.
Dr. Gentner, like Dr. Naldini, oversees pre-clinical research related to the platform technology. In addition, he analyzes clinical biological data. The last agreement with Dr. Gentner, which is still in force, was signed in 2017. His annual fee is €30,000. For the six months ended June 30, 2021 and 2020, the Company expensed €15,000 and €22,950, respectively, related to Dr. Gentner. At June 30, 2021 and the year ended December 31, 2020, the Company did not accrue anything related to Dr. Gentner.
| F-48 |
XDG Biomed LLC
This is the LLC of Dr. Carlo Russo. Dr. Russo has a single contract signed by XDG and the Company that has been approved by the Board of Directors and was subject to multiple amendments. In particular, Dr. Russo, via XDG, serves as the Company’s Chief Medical Officer and Head of Development. Dr. Russo is responsible for the clinical development of Temferon™, the Company’s gene therapy platform. The applicable recurring fees are €300,000 per year, plus a performance bonus of €50,000 maturing for the period July-June of each year and payable after Board of Directors approval. In 2020, the bonus was paid in July 2020. In 2021, the bonus was paid in June 2021. For the six months ended June 30, 2021 and 2020, the Company expensed €175,000 and €175,000, respectively, related to XDG Biomed. At June 30, 2021 and the year ended December 31, 2020, the Company accrued €0 and €25,000, respectively, related to XDG Biomed.
OSR – San Raffaele Hospital
San Raffaele Hospital (“OSR”) is a co-founder of the Company, and the Company is a corporate and research spin-off of OSR. At June 30, 2021, OSR held 12.64% of the Company’s common shares. OSR is one of the leading biomedical research institutions in Italy and Europe, with a 45-year history of developing innovative therapies and procedures. The Company has agreements to license technology, to perform research, pre-clinical and clinical activities, as well as to lease facilities and obtain certain other support functions. The Company’s headquarters is currently in an OSR facility.
License Agreement
The Company has a License Agreement with OSR entered in December 2014, for the exclusive use of different patents. In particular, OSR granted the Company an exclusive, world-wide, royalty bearing license under certain technology to conduct research and develop, make, use, import and sell licensed products. The license agreement covers patents and patent applications, as well as proprietary technologies. The Company’s rights to use these patents and patent applications and to utilize the inventions claimed in these licensed patents are subject to the continuation of, and the Company’s compliance with, the terms of the license agreement.
Based on the preclinical studies carried out by OSR, in particular by its SR-TIGET Institute (San Raffaele Telethon Institute for Gene Therapy), on a specific gene therapy strategy with respect to lympho-hematopoietic indication and/or solid cancer indication, the Company decided to develop a new therapy to treat cancer through a cell and gene therapy strategy. The “Field of Use” as defined in the License Agreement is:
| a) | Lympho-Hematopoietic Indication1; and, | |
| b) | Solid Cancer Indication. |
The agreement provided for an upfront fee of €250,000 (which was paid in 2015), future option fees are as follows:
| ● | option fee on the first indication = €1.0 million (subsequently reduced to €0.5 million); | |
| ● | option fee on the second indication = €0.5 million; | |
| ● | option fee on the third indication = €0.3 million; and, | |
| ● | option fee on any additional indications = no license fee. |
1 The Company later amended the License agreement focusing on GBM options. The TEM-MM option fee has never been exercised and instead the related research was abandoned in early 2021.
| F-49 |
In addition, the Company would be obligated make payments on milestones depending on the Field of Use (as defined in the agreement) and pay royalties of 4% of net sales of each Licensed Product (as defined in the agreement).
In connection to the License Agreement, the Company engaged OSR to provide certain research activities regarding the Licensed Products in the Field of Use, based on a mutually agreed study plan and utilizing the extensive resources at OSR. (See Note 13. Commitments and contingencies.) In consideration of the research activities provided by OSR, the Company agreed to pay scientific collaboration research fees in advance. In December 2014, the Company and OSR signed a Scientific Collaboration Agreement and subsequently modified the Agreement with Research Addenda 1, 2 and 3 in 2016, 2017 and 2018, respectively. During the six months ended June 30, 2021 and 2020, there were no costs incurred for the above activities.
The protocol TEM-GBM_001received approval by national Competent Authorities in September 2018 and recruited the first patient in April 2019.
License Agreement Amendment #2
In February 2019, the Company and OSR entered into Amendment #2 of the License Agreement to conduct a clinical trial according to the protocol TEM-GBM_001 and EudraCT 2018-001404-11 entitled: “A phase I/IIa dose escalation study evaluating the safety and efficacy of autologous CD34+ enriched hematopoietic progenitor cells genetically modified with a lentiviral vector encoding for the human interferon-α2 in patients with GBMwho have an unmethylated O-6-methylguanine-DNA methyltransferase gene promoter.” In Amendment #2, the Company and OSR also revised the license fee requirement for the first Solid Cancer indication (GBM). In relation to the GBM trial, the Company and OSR agreed that the Company would be obligated to pay OSR the €1.0 million Option Fee only in the event that the Company was able to dose its tenth patient. Under this Amendment, the Company is also obligated to pay for the costs of the study-related procedures performed on the patients recruited in the trial, according to periodic study reports delivered by OSR. The first GBM patient was recruited in April 2019 and related clinical activity costs were recorded by the Company. During the six months ended June 30, 2021, the comparable costs incurred and expensed for the GBM program were approximately €0.8 million. During the six months ended June 30, 2020, the comparable costs incurred and expensed for the GBM program were approximately €0.5 million.
Under this Amendment, the Company is obligated to cover the costs of the study-related procedures performed on the patients recruited in the Trial, according to periodic study reports delivered by OSR.
License Agreement Amendment #3
In December 2020, the Company and OSR entered into Amendment #3 of the License Agreement: The initial €1.0 million payment in the event of the tenth patient dosed in the GBM trial was reduced to €0.5 million, in exchange for the Company’s agreement to exercise a second option for an additional Solid Cancer indication (possibly Liver Cancer) and an agreement to execute a Sponsored Research Agreement in February 2021. Note: If the Company is not be able to obtain approval from the competent authorities to initiate a human clinical trial on or before the expiration of nine months (from December 2020), the Company has the right, at no additional costs, to convert this second solid cancer option to an “Alternate Indication,” i.e., an indication other than liver cancer.
In summary, the Amendment #3 formalized the new arrangement as follows:
| - | exercise of option fee on the first solid cancer indication = €0.5 million (accrued in 2019, since it was considered probable, and paid in December 2020); plus, | |
| - | commitment to enter into a Sponsored Research Agreement by February 2021; and, | |
| - | exercise of option fee on the second indication = €0.5 million (accrued at December 31, 2020 and was paid on June 30, 2021). |
| F-50 |
At June 30, 2021, no milestones were achieved related to any indication, as provided by License Agreement and subsequent amendments, therefore, no payments were due to OSR. The Company has paid €0.75 million to OSR, since inception under the license agreement. No events have occurred or have been achieved (and none are considered probable) to trigger any contingent payments under the license agreement at December 31, 2020. For information relating to the contingency payments or future milestones for these indications, please refer to Note 13 - Commitments and Contingencies.
OSR may terminate the Company’s rights as to certain fields of use for the Company’s failure to develop (a) with respect to a solid cancer indication, upon third anniversary of the date the Company exercised such option, if the Company has not filed an IND with respect to such optioned solid cancer indication specifically, as to GBM, the Company is required to file an IND regarding Temferon for GBM prior to February 2022, or (b) with respect to a lympho-hematopoietic indication, on the earlier of (i) the fifth anniversary of the initiation (first patient dosed) of the first human clinical trial for a licensed product in any lympho hematopoietic indication or solid cancer indication if a patient has not been dosed with a licensed product in a Phase 3 clinical trial and (ii) September 1, 2025.
During the six months ended June 30, 2021, the Company recorded expenses for OSR for clinical trial in the amount of approximately €1.3 million, while during the six months ended June 30, 2020, the expenses for clinical trials was approximately €0.5 million. The difference between the two periods is due to the increase of the trial activity in the second half of 2020, since a greater number of patients were included in the trial and subsequently dosed.
At June 30, 2021, the total amount of expenses for the OSR clinical trial activity amounted to €8.1 million and it includes the cost for the exercise of the first and the second Solid Cancer indication Option fee.
Research Funding Agreement
In March 2019, the Company and OSR entered a Research Funding Agreement to conduct a clinical trial according to the multiple myeloma protocol, TEM-MM-101 and EudraCT 2018-001741-14, entitled “A Phase I/II dose escalation study evaluating safety and activity of autologous CD34+ enriched hematopoietic progenitor cells genetically modified with a lentiviral vector encoding for the human interferon-α2 in multiple myeloma patients with early relapse after intensive front-line therapy.” This agreement required OSR to perform certain clinical procedures and exploratory analyses on the study population, as per the protocol approved by the relevant competent authorities. The Company was required to fund the costs of the study-related procedures performed on patients recruited in the Trial, according to periodic study reports delivered by OSR. TEM-MM-101 received approval by national Competent Authorities in November 2018 and the first TEM-MM-101 trial patient was enrolled in August 2019.
For the six months ended June 30, 2021 and 2020, the Company expensed €0 and approximately €65,000, respectively, for the analysis performed by OSR for multiple myeloma and there were no clinical procedures performed by OSR’s Hematology and Bone Transplant Unit for multiple myeloma. The Company discontinued the multiple myeloma program in early 2021 due to the relatively small number of eligible patients, and the highly competitive MM landscape. (See Note 13.)
Sponsor Research Agreement (SRA)
As stated above, in exchange for a reduction in the first Solid Cancer indication option fee from €1.0 million to €0.5 million, the Company agreed to enter into a Sponsored Research Agreement (SRA). The Company and OSR executed the agreement in February 2021. Under the SRA, sponsored research activities will be conducted for between €0.5 million and €1.0 million (minimum commitment €0.5 million). The activities relate to:
| F-51 |
| ● | Research 1: Additional preclinical mouse model studies directed to identify Temferon effectors cells (transduced Tie2-expressing cells) and to test Temferon in combination with CAR-T in a GBM mouse model; and, | |
| ● | Research 2: Additional exploratory analyses, including single cell sequencing, to be conducted on samples collected from patients belonging to TEM-GBM_001 clinical trial aimed to deepen Temferon mechanism of action and get a broader insight on its biological activity in humans. |
For the six months ended June 30, 2021, the Company paid and expensed €375,000 related to the SRA. At June 30, 2021, the Company accrued €125,000 related to the SRA.
Operating leases
The Company entered into a non-cancelable lease agreement for office space in January 2020.
| 13. | Commitments and contingencies |
The Company exercises considerable judgment in determining the exposure to risks and recognizing provisions or providing disclosure for contingent liabilities related to pending litigations or other outstanding claims and liabilities. Judgment is necessary in assessing the likelihood that a pending claim will succeed, or a liability will arise and to quantify the possible range of the final settlement. Provisions are recorded for liabilities when losses are considered probable and can be reasonably estimated. Because of the inherent uncertainties in making such judgments, actual losses may be different from the originally estimated provision. Estimates are subject to change as new information becomes available, primarily with the support of internal specialists or outside consultants, such as actuaries or legal counsel. Adjustments to provisions may significantly affect future operating results.
The following table summarizes the Company obligations by contractual maturity at June 30, 2021:
| Payments by period | ||||||||||||||||||||
| Total | Less than a year | 1 to 3 years | 4 to 5 years | More than 5 years | ||||||||||||||||
| OSR operating leases and office rent | € | 33,500 | € | 6,700 | € | 26,800 | € | - | € | - | ||||||||||
| AGC | 40,250 | 8,050 | 32,200 | - | - | |||||||||||||||
| Total | € | 73,750 | € | 14,750 | € | 59,000 | € | - | € | - | ||||||||||
The Company has not included future milestone and royalty payments in the table above because the payment obligations under these agreements are contingent upon future events, such as the Company’s achievement of specified milestones or generating product sales, and the amount, timing and likelihood of such payments are unknown and are not yet considered probable.
CMOs and CROs agreements
The Company enters into contracts in the normal course of business with CMOs, CROs and other third parties for exploratory studies, manufacturing, clinical trials, testing, and services (shipments, travel logistics, etc.). These contracts do not contain minimum purchase commitments and, except as discussed below, are cancelable by the Company upon prior written notice. Payments due upon cancellation consist only of payments for services provided or expenses incurred, including non- cancelable obligations of the Company’s vendors or third-party service providers, up to the date of cancellation. These payments are not included in the table above as the amount and timing of such payments are not known.
| F-52 |
OSR - San Raffaele Hospital
The license agreement in place with OSR provides milestone payments and royalties. The OSR agreements are non-cancelable, except in the case of breach of contract, and include total potential milestone payments of up to €10 million related to the Lympho-Hematopoietic Indication of each Licensed Product, and up to €53 million related to each Solid Cancer indication (as defined in the agreement); however, starting with the fifth Solid Cancer indication, the first two related milestone payments totaling €7.0 million, are reduced to €3.5 million. The milestones relate to certain events such as, dosing of the first patient with a licensed product in Phase II and III of the trial, MAA (marketing authorization application) and NDA (new-drug application) approval of the licensed product, the first commercial sale of the product in the US and major European countries, and annual sales for the licensed product exceeding a certain amount in different territories.
Multiple myeloma (MM)
As discussed in Note 12, the Company’s MM program was discontinued in early 2021 due to the relatively small number of eligible patients, and the highly competitive MM landscape. No milestones were achieved with respect to the MM program, and as such no contingent payments were due under the agreement.
Glioblastoma multiforme (GBM)
As discussed in Note 12, in December 2020, the Company had one indication ongoing, GBM. The Company’s contingent liability for this first solid cancer indication potentially payable to OSR was €53 million.
Liver cancer (LC)
In relation to the option exercised by the Company for the second solid cancer indication, the Company and OSR agreed that the payment due in relation to the “First patient dosed with a Licensed Product in Phase I/II Clinical Trial,” as stated in the agreement, was reduced to €0.5 million rather than €1.0 million. The reduction applied to the first license fee payment only. All the additional contingent payments, other than the last contingent payment of €5.0 million, remained a contingent liability of the Company and potentially payable to OSR. Therefore, for the second solid cancer indication (liver cancer), the total potential commitment of possible contingent payments could amount to €47.5 million.
The agreements also include a €7.8 million commitment related to the development and manufacturing of licensed products, of which the Company had incurred €1.5 million of expense during the six months ended June 30, 2020 compared to €2.6 million of cumulative expense for 2019 and prior years. The Company has incurred approximately €682,000 during the six months ended June 30, 2021.
AGC Biologics (formerly MolMed)
The AGC Biologics agreement is non-cancelable, except in the case of breach of contract, and includes a potential milestone of €0.3 million if a phase 3 study is approved by the relevant authority, as well as potential royalty fees between 0.5% and 1.0% depending on the volume of annual net sales of the first commercial and named patient sale of the product. In the AGC Agreement, the Company entrusts AGC with certain development activities that will allow the Company to carry out activities related to its clinical research and manufacturing. The AGC agreement also includes a technology transfer fee of €0.5 million related to the transfer of the manufacturing know-how and €1.0 million related to the marketability approval by regulatory authorities. The agreement is a “pay-as-you-go” type arrangement with all services expensed in the period the services were performed. In February 2020, the Company entered into Amendment 4 to the Framework Service Agreement with AGC Biologics related to production and testing of the Company’s GBM trials, for a total amount of €360,000. In March 2020, the Company entered into Amendment 5 to the Framework Service Agreement with AGC Biologics related to production and testing of the Company’s GBM trials, for a total amount of €259,000. In March 2020, the Company entered into Amendment 6 to the Framework Service Agreement with ACG Biologics related to production and testing of the Company’s GBM trials, for a total amount of €41,000. In August 2020, the Company entered into Amendment 7 to the Framework Service Agreement with ACG Biologics related to production and testing of the Company’s GBM trials, which provides the Company with an option to accelerate GBM production as stated in Amendment 5 at a 20% cost increase. In October 2020, the Company entered into Amendment 8 to the Framework Service Agreement with ACG Biologics related to production and testing of the Company’s GBM trials, for a total amount of €17,000.
| F-53 |
In the early 2020, the Company and AGC amended the Master Service Agreement for the fourth time to regulate some new production activities for which the total estimated budget amounts to €0.3 million. For the six months ended June 30, 2021, the Company is committed to pay a total of €40,250 relating to various stability timepoints, which will be realized and come due at different times through 2025.
Adaptive Biotechnologies
The Adaptive Biotechnologies agreement on exploratory analyses for trial endpoints is non-cancelable, except in the case of breach of contract, and carries a total cost for the entire trial of €0.2 million, of which approximately €20,000 and €20,000 have been incurred for the six months ended June 30, 2021 and 2020, respectively.
Biogazelle NV
The Biogazelle agreement on exploratory analyses for trial endpoints is a five-year agreement that can be withdrawn by each party with a notice period of six months, or in case of breach. The Company shall compensate Biogazelle for any and all work (including but not limited to work-in-progress) performed or commenced prior to the effective date of termination or expiration and reimburse Biogazelle for any and all costs, expenses and consumables (including non-refundable) approved by the Company and incurred by Biogazelle prior to the effective date of termination or expiration of this agreement. For the six months ended June 30, 2021 and 2020, the Company had incurred and paid approximately €14,000 and €12,000, respectively. At June 30, 2021, there were no future commitment to Biogazelle identified by the Company.
Operating leases
On January 1, 2020, the Company began a six-year non-cancelable lease agreement for office space with OSR. Withdrawal is allowed from the fourth year with a notice of 12 months. Since the annual rent amounts to €13,400, at June 30, 2021, outstanding minimum payments amount to €33,500 until January 1, 2023.
Legal proceedings
The Company is not currently party to any material legal proceedings. At each reporting date, the Company evaluates whether or not a potential loss amount or a potential range of loss is probable and reasonably estimable under the provisions of ASC 450, Contingencies. The Company was notified by Theravectys of the possible infringement by the Company of Theravectys’ exclusive license to patents no. EP 1071804, EP 1224314, and EP 1222300 granted from the owner of the patents Institut Pasteur. Each of these patents is now expired, having each reached the end of it its patent term on April 23, 2019 for EP 1071804 and October 10, 2020 for EP 1224314, and EP 1222300. The Company considered the situation and determined that the likelihood of a material adverse effect on its business is remote. To date, the Company has not engaged in any such discussions with Theravectys nor has the Company received any further communication from Theravectys. The Company expenses as incurred the costs related to its legal proceedings, if any.
| F-54 |
Coronavirus Pandemic
On March 11, 2020, the World health Organization declared the outbreak of a coronavirus (COVID-19) pandemic. Significant uncertainties may arise with respect to potential shutdowns of operations or government orders to cease activities due to emergency declarations, inability to operate, or employee shortages, claims for business interruption insurance, etc. Although the Company has experienced minimal disruption to date and still has staff working remotely from home, the Company may find it difficult to enroll patients in its clinical trials, which could delay or prevent the Company from proceeding with the clinical trials of its product candidates; therefore, the coronavirus pandemic may still have a significant impact on the future results of the Company.
14. Subsequent events.
In July 2021, the Company signed a financial advisory agreement with Roth Capital. The minimum commitment relates to some expenses to be reimbursed by the Company up to US$50,000, or €42,373, using the spot exchange rate at the time the agreement was signed. The agreement also calls for the Company to pay Roth variable commission, estimated to average six percent of funds raised, if and when determined.
In September 2021, the Company entered a new service agreement for shipment services with Marken. The budget provided through 2024 amounts to approximately €106,000, but the agreement can be withdrawal at any time with a reasonable but non-specific notice period.
In August and September 2021, the Company entered into two new purchase orders with AGC for a total amount of €155,200 to be delivered by the end of the year. The purchase orders with AGC are non-cancelable, except in the case of breach of contract, consistent with the disclosure in Note 13 above.
In September 2021, the Company signed two purchase order with Cross NT amounting to €141,006 to be executed in three years through January 2024. Based on the Master Service Agreement in place with Cross NT, the Company can withdraw any task order with a notice period of 60 business days.
| F-55 |
American Depositary Shares
Representing Ordinary Shares
![]()
Genenta Science S.p.A.
PROSPECTUS
Sole Book-Running Manager
| Roth Capital Partners |
Lead Manager
| Maxim Group LLC |
, 2021
Through and including , 2021 (25 days after the date of this prospectus), all dealers that effect transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This delivery requirement is in addition to the dealer’s obligation to deliver a prospectus when acting as an underwriter and with respect to unsold allotment or subscription.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 6. Indemnification of Directors, Officers and Employees
Under Italian law, a corporation is not required to indemnify its directors or officers against losses and expenses, including attorney’s fees, judgments, fines and settlement amounts actually and reasonably incurred in a civil or criminal action, suit or proceeding arising from being the representative of, or serving at the request of, the corporation.
Italian law requires directors and members of any committee designated by the board of directors to perform their duties in good faith and with that degree of diligence that is required by the nature of their office and under their specific level of competence. If directors fail to comply with such obligations, they may be liable towards the company, its creditors, individual shareholders and third parties.
Corporate responsibility action may be brought by:
| ● | the shareholders’ meeting of the company; | |
| ● | shareholders representing at least 20% of the share capital. |
The company may waive the liability action and may settle, provided that the waiver and settlement are approved by express resolution of the shareholders’ meeting, and provided that there is no vote against by a minority of shareholders representing at least 20% of the share capital.
Liability action against directors for the non-fulfilment of their duties may also be brought by creditors when the company’s assets are insufficient to satisfy their claims.
The responsibility of directors is imperative and the articles of association cannot, as general rule, exempt directors from the liability provided for by law nor limit it to malicious conduct alone.
We intend to enter into directors’ and officers’ liability insurance to cover certain actions undertaken by our board of directors and executive officers.
Insofar as indemnification for liabilities arising under the Securities Act of 1933, as amended, or the Securities Act, may be permitted to directors, officers and controlling persons of the Company, the Company has been advised that, in the opinion of the Securities and Exchange Commission, such indemnification is against public policy as expressed in the Securities Act, and is, therefore, unenforceable.
Item 7. Recent Sales of Unregistered Securities
Set forth below are the sales of all securities by the Company since November 9, 2021, which were not registered under the Securities Act. The Company believes that each of such issuances was exempt from registration under the Securities Act in reliance on Section 4(a)(2) of the Securities Act, Rule 701 and/or Regulation S under the Securities Act.
In 2014, the Company was organized with a total corporate capital equal to €11,650, comprised of €10,458.21 class A Quotas and €1,191.79 class B Quotas.
In 2015 through 2017, the Company issued 8,644.40 class C Quotas from convertible notes and 2,958.50 class B Quotas.
In 2017, the Company issued an aggregate maximum amount of 4,033.85 of class D Quotas.
In 2018 and 2019 the Company issued 2,300 of class B Quotas and an additional 6,462.12 class E Quotas.
| II-1 |
In 2020, the Company issued 571.34 class E Quotas, from the 2019 offering which was extended in 2020 to provide an additional €1.5 million plus 436.00 class B Quotas from options.
No underwriters were involved in the foregoing sales of securities. The sales of securities described above were deemed to be exempt from registration pursuant to Section 4(a)(2) of the Securities Act, including Regulation D and Rule 506 promulgated thereunder, as transactions by an issuer not involving a public offering. All of the purchasers in these transactions were acquiring the securities for investment and not distribution. Such purchasers were advised the securities had not been registered under the Securities Act and that any resale in the United States must be made pursuant to a registration or an available exemption from such registration. All of the foregoing securities are deemed restricted securities for the purposes of the Securities Act.
Item 8. Exhibits and Financial Statement Schedules
Exhibits:
| * | Filed herewith |
| ** | Previously filed |
# | Portions of this exhibit (indicated by markouts) have been redacted in compliance with Regulation S-K Item 601(b)(10)(iv). |
Financial Statement Schedules:
All financial statement schedules have been omitted because either they are not required, are not applicable or the information required therein is otherwise set forth in the Company’s financial statements and related notes thereto.
Item 9. Undertakings
(a) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the provisions described in Item 6 hereof, or otherwise, the registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
| (b) | The undersigned registrant hereby undertakes that: |
| (1) | That for purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4), or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. | |
| (2) | That for the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| II-2 |
Pursuant to the requirements of the Securities Act of 1933, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form F-1 and has duly caused this registration statement on Form F-1 to be signed on its behalf by the undersigned, thereunto duly authorized, in Milan, Italy on November 24, 2021.
| Genenta Science S.p.A. | ||
| By: | /s/ Pierluigi Paracchi | |
| Name: | Pierluigi Paracchi | |
| Title: | Chief Executive Officer | |
Pursuant to the requirements of the Securities Act of 1933, this registration statement on Form F-1 has been signed by the following persons in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Pierluigi Paracchi | Chief Executive Officer and Vice Chairman | November 24, 2021 | ||
| Pierluigi Paracchi | (Principal Executive Officer) | |||
| /s/ Richard Slansky | Chief Financial Officer | November 24, 2021 | ||
| Richard Slansky | (Principal Financial and Accounting Officer) | |||
| * | Director | November 24, 2021 | ||
| Roger Abravanel | ||||
| * | Director | November 24, 2021 | ||
| Daniela Bellomo | ||||
| * | Director | November 24, 2021 | ||
| Guido Guidi | ||||
| * | Director | November 24, 2021 | ||
| Luca Guidotti | ||||
| * | Chairman of the Board and Director | November 24, 2021 | ||
| Stephen Squinto | ||||
| * | Director | November 24, 2021 | ||
| Anthony Marucci |
| *By | /s/ Pierluigi Paracchi | ||||
Attorney-in-Fact |
| II-3 |
SIGNATURE OF AUTHORIZED REPRESENTATIVE IN THE UNITED STATES
Pursuant to the Securities Act of 1933, as amended, the undersigned, Cogency Global Inc., the duly authorized representative in the United States of Genenta Science S.p.A., has signed this registration statement on November 24, 2021.
| By: | /s/ Colleen A. De Vries | |
| Name: | Colleen A. De Vries | |
| Title: | Senior Vice-President on behalf of Gogency Global Inc. |
| II-4 |