Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 1 to
FORM 20-F
| ☑ | REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
| ☐ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
OR
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
OR
| ☐ | SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Date of event requiring this shell company report
Commission file number
ACCUSTEM SCIENCES LIMITED
(Exact name of Registrant as specified in its charter)
Not Applicable
(Translation of Registrant’s name into English)
England and Wales
(Jurisdiction of incorporation or organization)
107 Cheapside, 9th Floor
London EC2V 6DN United Kingdom
(Address of principal executive offices)
Keeren Shah
55 Park Lane
London W1K 1NA United Kingdom
Telephone: +44 (0) 20 7066 1000
(Name, telephone, e-mail and/or facsimile number and address of company contact person)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
Not Applicable
(Title of Class)
Securities registered or to be registered pursuant to Section 12(g) of the Act.
Title of each class |
| American Depositary Shares, each representing two ordinary shares, nominal value £0.01 per share |
| Ordinary shares, nominal value £0.01 per share* |
| * Not for trading, but only in connection with the registration of American Depositary Shares. |
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act.
Not Applicable
(Title of Class)
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report.
Ordinary shares, nominal value £0.01 per share: 204,193,543 as of March 9, 2021
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☐ Yes ☑ No
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. Yes ☐ No ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☐ No ☑
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 229.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☐ No ☑
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated file, a non-accelerated filer, or an emerging growth company. See the definitions of “large accelerated filer”, “accelerated filer”, and “emerging growth company” in Rule 12b-2 of the Exchange Act.:
Large accelerated filer | ☐ | Accelerated filer | ☐ | |||
Non-accelerated filer | ☐ | Emerging growth company | ☑ |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
† The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark which basis for accounting the registrant has used to prepare the financing statements included in this filing:
| U.S. GAAP ☐ | International Financial Reporting Standards as issued | Other ☐ | ||||
| by the International Accounting Standards Board ☑ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow. ☐ Item 17 ☐ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☐
Table of Contents
ACCUSTEM SCIENCES LIMITED
Amendment No. 1 to
FORM 20-F
1
Table of Contents
On October 30, 2020, Tiziana Life Sciences plc, a London-listed biotechnology company focused on innovative therapeutics for oncology, inflammation and infectious diseases (“Tiziana”), completed the demerger and spin-off (which we refer to as the “Demerger”, as further discussed below) of its StemPrintER and SPARE projects, a novel biology-based genomic predictor of distant recurrence in breast cancer, into a separate business, StemPrintER Sciences Limited, together with £1 million in cash, and then contributed StemPrintER Sciences Limited to AccuStem Sciences Limited (the “Company”) in exchange for rights to receive a new issuance of the Company’s Ordinary Shares. Next, Tiziana declared a dividend in specie (a distribution to shareholders of the shares of another company) to Tiziana’s shareholders of the right to receive the Company’s Ordinary Shares, when issued. The Company is filing this registration statement to register the distribution of those Ordinary Shares under the Exchange Act, which is a requirement for Tiziana to complete the distribution of the Company’s Ordinary Shares to Tiziana’s shareholders by issuing them to Tiziana’s shareholders.
Unless otherwise indicated, “AccuStem”, “the Company”, “we”, “us” and “our” refer to AccuStem Sciences Limited. References to “U.K. Pounds Sterling”, “pence”, “£” or “p” are to the lawful currency of the United Kingdom, references to “€” are to the common currency of the European Monetary Union, and references to “U.S. dollars”, “$” or “cents” are to the lawful currency of the United States. Reference to “the Ordinary Shares” are references to AccuStem’s ordinary shares, nominal value £0.01 per share, and references to “the ADSs” are to AccuStem’s American Depositary Shares (each representing two Ordinary Shares), which are evidenced by American Depositary Receipts (“ADRs”).
The following definitions apply throughout this registration statement:
| “Admission” | admission of the Ordinary Shares to listing on the standard segment of the Official List and to trading on the Main Market; | |
| “ADSs” | American Depositary Shares; | |
| “affiliate” or “affiliates” | an affiliate of, or person affiliated with, a person; a person that, directly or indirectly, or indirectly through one or more intermediaries, controls or is controlled by, or is under common control with, the person specified; | |
| “AGM” | an annual general meeting of the Company; | |
| “Articles” or “Articles of Association” | the articles of association of the Company in force from time to time; | |
| “Audit Committee” | the audit committee of the Board; | |
| “Brexit” | the formal exit from the EU by the U.K. on January 31, 2020; | |
| “CE marking” or “CE mark” | European conformity marking which is a mandatory conformity mark for products placed on the market in the EEA which ensures that the products conform with the essential requirements of the applicable European regulations and directives; | |
| “CLIA” | U.S. Clinical Laboratory Improvement Amendments of 1988; | |
| “Change of Control” | following any acquisition, the acquisition of Control of the Company by any person or party (or by any group of persons or parties who are acting in concert); | |
| “Companies Act” | the U.K. Companies Act 2006; | |
| “Company” or “AccuStem” | AccuStem Sciences Limited, a company incorporated in England and Wales with company number 12647178; | |
1
Table of Contents
| “Control” | (i) the power (whether by way of ownership of shares, proxy, contract, agency or otherwise) to: (a) cast, or control the casting of, more than 50% of the maximum number of votes that might be cast at a general meeting of the Company; or (b) appoint or remove all, or the majority, of the Directors or other equivalent officers of the Company; or (c) give directions with respect to the operating and financial policies of the Company with which the Directors or other equivalent officers of the Company are obliged to comply; and/or (ii) the holding beneficially of more than 50% of the issued shares of the Company (excluding any issued shares that carry no right to participate beyond a distribution of either profits or capital), but excluding in the case of each of (i) and (ii) above any such power or holding that arises as a result of the issue of Ordinary Shares by the Company in connection with the acquisition; | |
| “CREST” or “CREST System” | the system for the paperless settlement of trades in securities and the holding of uncertificated securities operated by Euroclear in the system for the paperless settlement of trades in securities and the holding of uncertificated securities operated by Euroclear in accordance with the CREST Regulations; | |
| “CREST Regulation” | the Uncertificated Securities Regulations 2001 (SI 2001 No 3755); | |
| “Demerger” | the demerger of the Company from Tiziana Life Sciences plc on October 30, 2020; | |
| “Demerger Agreement” | the agreement between the Company and Tiziana, dated October 5, 2020, pursuant to which Tiziana declared a dividend in specie on the Ordinary Shares equal to the book value (of approximately £3.07 million) of Tiziana’s shareholding in StemPrintER Sciences Limited, the entity within the Group which holds all of the assets and intellectual property relating to the StemPrintER and SPARE projects and £1 million in cash, as amended on October 30, 2021; | |
| “Demerger Record Time” | 7:00 a.m. on October 30, 2020; | |
| “Depositary” | JPMorgan Chase Bank, N.A. | |
| “Directors”, “Board” or “Board of Directors” | the directors of the Company, whose names appear in “Item 1.A.—Directors and Senior Management” of this registration statement, or the board of directors from time to time of the Company, as the context requires, and “Director” is to be construed accordingly; | |
| “EEA” | the European Economic Area, comprising the EU, Iceland, Norway and Liechtenstein; | |
| “EMA” | the European Medicines Agency; | |
| “EN ISO 13485” | specifies requirements for a quality management system where an organisation needs to demonstrate its ability to provide medical devices and related services that consistently meet customer requirements and regulatory requirements applicable to medical devices and related services; | |
| “ER+” | oestrogen receptor positive; | |
| “Exchange Act” | U.S. Securities Exchange Act of 1934, as amended; | |
| “EU” or “European Union” | the European Union first established by the treaty made at Maastricht on February 7, 1992; | |
| “Euroclear” | Euroclear U.K. & Ireland Limited (a company incorporated in England and Wales with company number 02878738, being the operator of CREST); | |
| “EUWA” | European Union (Withdrawal) Act 2018; | |
| “FCA” | the U.K. Financial Conduct Authority or any successor thereof, the single U.K. statutory regulator under FSMA; | |
| “FDA” | the U.S. Food and Drug Administration; | |
| “FSMA” | the U.K. Financial Services and Markets Act 2000; | |
| “FTC” | U.S. Federal Trade Commission; | |
| “GDPR” | General Data Protection Regulation (EU) 2016/679; | |
| “general meeting” | a general meeting of the shareholders of the Company or a class of shareholders of the Company, as the context requires; | |
| “Group” | AccuStem and its wholly-owned subsidiary, StemPrintER Sciences Limited; | |
| “HER2-” | human epidermal growth factor receptor 2 negative; | |
2
Table of Contents
| “HIPAA” | U.S. Health Insurance Portability and Accountability Act of 1996; | |
| “HMRC” | Her Majesty’s Revenue and Customs; | |
| “IEO/University of Milan” | Istituto Europeo di Oncologia, Fondazione FIRC per l’Oncologia Molecolare and the University of Milan; | |
| “IFRS” | International Financial Reporting Standards, as issued by the International Accounting Standards Board; | |
| “IVDs” | In vitro medical devices; | |
| “IVD Directive” | In Vitro Diagnostic Medical Device Directive 98/79/EC; | |
| “IVDR” | In Vitro Diagnostic Medical Device Regulation (EU) 2017/746; | |
| “LDT” | lab developed test; | |
| “License” | the exclusive license agreement dated June 24, 2014 between Tiziana and IEO/University of Milan, which was assigned to the Company on October 30, 2020, pursuant to which the Company holds a worldwide, royalty-bearing, exclusive licence under certain patents and a worldwide, royalty-bearing, non-exclusive license under certain know-how, respectively, of IEO/University of Milan to develop and commercialize licensed products in connection with a multi-gene prognostic tool; | |
| “Listing Rules” | the listing rules made by the FCA under section 73A of FSMA; | |
| “LN-” | lymph node negative; | |
| “London Stock Exchange” | London Stock Exchange plc (a company registered in England and Wales with company number 2075721); | |
| “Main Market” | the main market for listed securities of the London Stock Exchange; | |
| “Market Abuse Regulation” | Market Abuse Regulation (EU) 596/2014; | |
| “Medicaid” | a national health insurance program administered by the Centers for Medicaid and Medicare Services of the U.S. Federal Government which assists with healthcare costs of Americans with limited income and/or resources; | |
| “Medicare” | a national health insurance program administered by the Centers for Medicaid and Medicare Services of the U.S. Federal Government which provides health care insurance for Americans aged 65 and older who have contributed to the fund via the payroll tax during the course of their working lives; | |
| “U.K. Market Abuse Regulation” | the Market Abuse Regulation, which forms part of retained law by virtue of the EUWA; | |
| “Medical Device Directive” | Medical Device Directive 93/42/EEC; | |
| “Medical Device Regulation” or “MDR” | EU Medical Device Regulation (EU) 2017/745; | |
| “MHRA” | U.K. Medicines and Healthcare products Regulatory Agency; | |
| “mRNA” | messenger ribonucleic acid; | |
| “Nasdaq” | the Nasdaq Global Market operated by NASDAQ, Inc.; | |
| “Nomination Committee” | the nomination committee of the Board; | |
| “Official List” | the official list maintained by the FCA pursuant to Part VI of FSMA; | |
| “Order” | the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005; | |
| “Ordinary Resolution” | resolution of shareholders requiring a simple majority of not less than 50%; | |
| “Ordinary Shares” | ordinary shares of nominal value 1 pence each in the capital of the Company; | |
| “R&D” | research and development; | |
3
Table of Contents
| “Registrar” | Link Market Services, a trading name of Link Market Services Limited, of The Registry, 34 Beckenham Road, Beckenham, Kent BR3 4TU, or any other registrar appointed by the Company from time to time; | |
| “Regulation S” | Regulation S under the Securities Act; | |
| “Remuneration Committee” | the remuneration committee of the Board; | |
| “SaMD” | software as a medical device; | |
| “SEC” | U.S. Securities and Exchange Commission; | |
| “Securities Act” | U.S. Securities Act of 1933, as amended; | |
| “Senior Managers” | the members of the senior management team of the Company, the name(s) of which are appear in Item 1.A.—“Directors and Senior Management”; | |
| “Shareholder” | a holder of Ordinary Shares; | |
| “Special Resolution” | a resolution of shareholders requiring a majority of not less than 75%; | |
| “Standard Listing” | a standard listing under Chapter 14 of the Listing Rules; | |
| “Takeover Code” | the City Code on Takeovers and Mergers; | |
| “Tiziana” | Tiziana Life Sciences plc (Nasdaq: TLSA / LSE: TILS), a public limited company incorporated in England and Wales with company number 03508592; | |
| “Transition Period” | the transition period following Brexit during which the U.K. continued to follow all EU rules between January 31, 2020 and December 31, 2020; | |
| “U.K. Corporate Governance Code” | the U.K. Corporate Governance Code issued by the Financial Reporting Council (or successor bodies thereto) in the U.K. from time to time; | |
| “U.K. MDR 2002” | the U.K. Medical Devices Regulations 2002, as amended by 2019 No. 791; | |
| “uncertificated” or “uncertificated form” | in relation to a share or other security, a share or other security, title to which is recorded in the relevant register of the share or other security concerned as being held in uncertificated form (i.e., in CREST) and title to which may be transferred by using CREST; | |
| “United Kingdom” or “U.K.” | the United Kingdom of Great Britain and Northern Ireland; | |
| “United States” or “U.S.” | the United States of America, its possessions or territories, any State of the United States of America and the district of Columbia or any area subject to its jurisdiction or any political subdivision thereof; and | |
| “VAT” | (i) within the EU, any tax imposed by any Member State in conformity with the Directive of the Council of the European Union on the common system of value added tax (2006/112/EC), and (ii) outside the EU, any tax corresponding to, or substantially similar to, the common system of value added tax referred to in paragraph (i) of this definition. | |
References to a “company” in this registration statement shall be construed so as to include any company, corporation or other body corporate, wherever and however incorporated or established.
All references to legislation or regulation in this registration statement are to the legislation of England and Wales unless the contrary is indicated. Any reference to any provision of any legislation or regulation shall include any amendment, modification, supplement, re-enactment or extension thereof. Words importing the singular shall include the plural and vice versa, and words importing the masculine gender shall include the feminine or neutral gender.
For the purpose of this registration statement, “subsidiary” and “subsidiary undertaking” have the meanings given by the Companies Act.
In this registration statement any reference to any EU directive, EU regulation, EU decision, EU tertiary legislation or provision of the EEA agreement (an “EU Matter”) which forms part of domestic law by application of the EUWA shall be read as a reference to that EU Matter as it forms (by virtue of the EUWA) part of retained EU law and as modified by domestic law from time to time. For the purposes of this paragraph, (i) “domestic law” shall have the meaning given in the EUWA; and (ii) any other words and expressions shall, unless the context otherwise provides, have the meanings given in the EUWA.
4
Table of Contents
Solely for convenience, the trademarks, service marks and trade names in this registration statement may be referred to without the ® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. This registration statement contains additional trademarks, service marks and trade names of others, which are the property of their respective owners. We do not intend to use or display other companies’ trademarks, service marks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This registration statement contains forward-looking statements that involve substantial risks and uncertainties. All statements contained in this registration statement, other than statements of historical fact, including statements regarding our strategy, future operations, future financial position, future revenues, projected costs, prospects, plans and objectives of management, are forward-looking statements. The words “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “plan,” “predict,” “project,” “target,” “potential,” “would,” “could,” “should,” “continue” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. The forward-looking statements in this registration statement include, among other things, statements about:
| • | the success, cost and timing of our clinical development of our products, including the progress of, and results from, our preclinical and clinical trials of StemPrintER and SPARE products, our discovery programs and other potential product candidates; |
| • | our ability to obtain and maintain regulatory approval of our product candidates, and any related restrictions, limitations or warnings in the label of any of our product candidates, if approved; |
| • | our ability to compete with companies currently marketing or engaged in the development of treatments for indications that our product candidates are designed to target; |
| • | our plans to pursue research and development of other future product candidates; |
| • | the potential advantages of our product candidates and those being developed; |
| • | the rate and degree of market acceptance and clinical utility of our product candidates; |
| • | the success of our collaborations and partnerships with third parties; |
| • | our estimates regarding the potential market opportunity for our product candidates; |
| • | our sales, marketing and distribution capabilities and strategy; |
| • | our ability to establish and maintain arrangements for manufacture of our product candidates; |
| • | our intellectual property position; |
| • | our expectations related to the use of capital; |
| • | the effect of the COVID-19 pandemic, including mitigation efforts and economic effects, on any of the foregoing or other aspects of our business operations, including but not limited to our preclinical studies and future clinical trials; |
| • | our estimates regarding expenses, future revenues, capital requirements and needs for additional financing; |
| • | the impact of government laws and regulations; and |
| • | our competitive position. |
Forward-looking statements reflect our current views with respect to future events, are based on assumptions and are subject to risks, uncertainties and other important factors. We operate in a very competitive and rapidly changing environment. New risks emerge from time to time. We cannot assure you that our plans, intentions or expectations will be achieved. Our actual results, performance or achievements could differ materially from those contemplated, expressed or implied by the forward-looking statements contained in this registration statement as described in “Item 3.D.—Risk Factors”, “Item 4—Information
5
Table of Contents
on the Company” and “Item 5—Operating and Financial Review and Prospects”. Given these risks, uncertainties and other important factors, you should not place undue reliance on these forward-looking statements. All forward-looking statements attributable to us or persons acting on our behalf are expressly qualified in their entirety by the cautionary statements set forth in this registration statement. Also, these forward-looking statements represent our estimates and assumptions only as of the date such forward-looking statements are made. Except as required by law, we assume no obligation to update any forward-looking statements publicly, whether as a result of new information, future events or otherwise.
| ITEM 1. | IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISERS |
A. Directors and Senior Management
The following table sets forth the names and positions of our senior management and Directors, all of whom can be reached at our business address of 107 Cheapside, 9th Floor, London EC2V 6DN United Kingdom:
| NAME | POSITION | |
Senior Management | ||
Keeren Shah | Finance Director* | |
* Not a statutory Director of the Company | ||
Non-Employee Directors | ||
Gabriele Cerrone (3) | Chairman | |
Dr. Kunwar Shailubhai, Ph.D., M.B.A. | Director | |
Willy Simon (1) (2) (3) | Director | |
John Brancaccio (1) (2) | Director | |
| (1) | Member of the Audit Committee. |
| (2) | Member of the Remuneration Committee. |
| (3) | Member of the Nomination Committee. |
Executive Officers
Keeren Shah serves as our Finance Director. Ms. Shah currently also serves as the Finance Director of Tiziana and OKYO Pharma Limited and Rasna Therapeutics Inc., having previously served as the Group Financial Controller for both businesses from June 2016 to July 2020. Prior to joining the Company, Ms. Shah spent 10 years at Visa, Inc. as a Senior Leader in its finance team where she was responsible for key financial controller activities, financial planning and analysis, and core processes as well as leading and participating in key transformation programmes and Visa Inc.’s initial public offering. Before joining Visa, Ms. Shah has also held a variety of finance positions at other leading companies including Arthur Andersen and BBC Worldwide. She holds a Bachelor of arts with honours in Economics and is a member of the Chartered Institute of Management Accountants.
Non-Employee Directors
Gabriele Cerrone has a successful track record and extensive experience in the financing and restructuring of micro-cap biotechnology companies. He has founded ten biotechnology companies in oncology, infectious diseases and molecular diagnostics, and has taken seven of these companies to the Nasdaq Market and two to the Main Market and AIM Market in London. Mr. Cerrone co-founded Cardiff Oncology, Inc. (Nasdaq: CRDF), an oncology company and served as its Co-Chairman; he was a co-founder and served as Chairman of both Synergy Pharmaceuticals, Inc. (Nasdaq: SGYP) and Callisto Pharmaceuticals, Inc. (OTCMKTS: CLSP), and was a Director of and led the restructuring of Siga Technologies, Inc. (Nasdaq: SIGA). Mr. Cerrone also co-founded FermaVir Pharmaceuticals, Inc. and served as Chairman of the Board until its merger in September 2007 with Inhibitex, Inc. Mr. Cerrone served as a director of Inhibitex, Inc. until its $2.5bn sale to Bristol Myers Squibb Co in 2012. Mr. Cerrone is the Executive Chairman and Founder of dual-listed Tiziana Life Sciences plc (Nasdaq:TLSA, LSE: TILS) an oncology focused therapeutics company; Co-Founder of Rasna Therapeutics Inc. (OTCMKTS: RASP), a company focused on the development of therapeutics for leukaemias; Co-Founder of Hepion Pharmaceuticals, Inc. (Nasdaq: HEPA); Executive Chairman and Co-Founder of Gensignia Life Sciences, Inc., a molecular diagnostics company focused on oncology using microRNA technology; Non-Executive Chairman and Founder of OKYO Pharma Limited; and founder of BioVitas Capital Ltd.
6
Table of Contents
Kunwar Shailubhai, Ph.D., M.B.A. serves as Chief Executive Officer and Chief Scientific Officer of Tiziana and is also an Executive Director of the Company. Dr. Shailubhai brings more than 25 years of experience within the life science industry, combined with a distinguished track record of success in translating drugs from concept through commercialisation to market. He also currently serves as CEO of Rasna Therapeutics, Inc., a developer of therapeutics to address the high unmet need that exists for AML and other forms of leukaemia. As co-founder, EVP and CSO of Synergy Pharmaceuticals, Inc. (Nasdaq: SGYP) he led the non-clinical, CMC and clinical development of Trulance from inception to approval by the FDA. Earlier, from 2003 until 2008, Dr. Shailubhai served as Senior Vice President, Drug Discovery and from 2001 to 2003, he held the position of Vice President, Drug Discovery at Synergy, where he pioneered therapeutic applications of GC-C agonists in a variety of human diseases such as Asthma, COPD and cholesterol lowering. Prior to Synergy, he was with Monsanto Company, serving as Group Leader, Cancer Prevention and previously served as a Senior Staff Fellow at the National Institutes of Health, and as an Assistant Professor at the University of Maryland. Dr. Shailubhai received his Ph.D. in microbiology from the University of Baroda, India, and his MBA from the University of Missouri, St. Louis.
Willy Jules Simon is a banker and worked at Kredietbank N.V. and Citibank London before serving as an executive member of the Board of Generale Bank NL from 1997 to 1999 and as the chief executive of Fortis Investment Management from 1999 to 2002. He acted as chairman of Bank Oyens & van Eeghen from 2002 to 2004. From 2004 until 2012, he served as a non-executive director of Redi & Partners Ltd., a fund of funds. He was previously chairman of AIM-traded Velox3 plc (formerly 24/7 Gaming Group Holdings plc) until 2015 and had been a director of Playlogic Entertainment Inc., a Nasdaq OTC listed company, and Frasia Holdings S.A. Mr. Simon is also currently a director of Rasna Therapeutics, Inc., OKYO Pharma Limited, African Metals Limited, Bever Holding N.V. and Ducat Maritime Limited.
Mr. Brancaccio, retired CPA, is a financial executive with extensive international and domestic experience in pharmaceutical and biotechnology for privately and publicly held companies. From 2000 to 2002, Mr. Brancaccio was the Chief Financial Officer/Chief Operating Officer of Eline Group, an entertainment and media company. From May 2002 until March 2004, Mr. Brancaccio was the Chief Financial Officer of Memory Pharmaceuticals Corp., a biotechnology company. From April 2004 until May 2017, Mr. Brancaccio was the Chief Financial Officer of Accelerated Technologies, Inc., an incubator for medical device companies. Mr. Brancaccio is also currently a director of Cardiff Oncology, Inc., Rasna Therapeutics, Inc., OKYO Pharma Limited and Hepion Pharmaceuticals, Inc.
B. Advisers
Our principal United States legal advisers are Orrick, Herrington & Sutcliffe LLP, located at 51 West 52nd Street, New York, NY 10166 and our principal United Kingdom legal advisers are Orrick, Herrington & Sutcliffe (UK) LLP, located at 107 Cheapside, London EC2V 6DN, United Kingdom.
C. Auditors
Mazars LLP has been our auditor since our incorporation on June 5, 2020; its address is Tower Bridge House, St Katharine’s Way, London E1W 1DD, United Kingdom. Mazars is registered to perform audits in the U.K. by the Institute of Chartered Accountants in England and Wales and is a registered auditor with the PCAOB.
| ITEM 2. | OFFER STATISTICS AND EXPECTED TIMETABLE |
Not applicable.
| ITEM 3. | KEY INFORMATION |
A. SELECTED FINANCIAL DATA
We derived the selected consolidated financial data as of and for the period from incorporation on June 5, 2020 to December 31, 2020 from our audited financial statements included elsewhere in this registration statement. We present our consolidated financial statements in accordance with International Financial Reporting Standards, as issued by the International Accounting Standards Board (“IFRS”).
The summary financial data below should be read together with those financial statements as well as “Item 5.—Operating and Financial Review and Prospects.” Our historical results for any prior period are not necessarily indicative of results to be expected in any future period, and our interim period results are not necessarily indicative of results to be expected for a full year or any other interim period.
7
Table of Contents
Consolidated Income Statement Data
| December 31, 2020 | ||||
Revenue | £ | — | ||
Research and development expenses | (4,347 | ) | ||
Administrative expenses | (36,342 | ) | ||
Operating result | (40,689 | ) | ||
Finance income/(expense) | — | |||
Profit before taxation | (40,689 | ) | ||
Income tax | — | |||
Profit for the period and total comprehensive income for the period | (40,689 | ) | ||
Consolidated Balance Sheet Data
| As at December 31, 2020 | ||||
ASSETS | ||||
Current Assets | ||||
Cash receivable | £ | 1,151,383 | ||
Non-Current Assets | ||||
Intangible asset in process research & development | 2,073,930 | |||
Total assets | 3,225,313 | |||
LIABILITIES | ||||
Related party payable | 9,892 | |||
Other payable | 30,797 | |||
Total liabilities | 40,689 | |||
EQUITY AND LIABILITIES | ||||
Equity attributable to owners | ||||
Ordinary Share capital | 2,041,935 | |||
Share premium | 55,571 | |||
Merger reserve | 1,127,807 | |||
Retained earnings | (40,689 | ) | ||
Total equity attributable to shareholders | 3,184,624 | |||
Total equity and liabilities | 3,225,313 | |||
B. CAPITALIZATION AND INDEBTEDNESS
The table below sets forth our cash and cash equivalents and shows our capitalization as of February 28, 2021. You should read this table in conjunction with our audited consolidated financial statements included in this registration statement, together with the accompanying notes and the other information appearing under the heading “Item 5.— Operating and Financial Review and Prospects”.
| As of December 31, 2020 | ||||
Cash and cash equivalents | £ | 1,151,383 | ||
Shareholders’ equity: | ||||
Share capital, nominal value £0.01 par value per ordinary share; 204,193,543 shares issued or allotted and outstanding (1) | 2,041,935 | |||
Share premium | 55,571 | |||
Merger reserve | 1,127,807 | |||
Retained earnings | (40,689 | ) | ||
Total Capitalization | 3,184,624 | |||
(1) Of these, 1 ordinary share has been issued and 204,193,543 have been allotted but not yet issued. | ||||
8
Table of Contents
C. REASONS FOR THE OFFER AND USE OF PROCEEDS
Not applicable.
D. RISK FACTORS
Holding Ordinary Shares and ADSs carries a significant degree of risk, including risks in relation to our business strategy, risks relating to taxation and risks relating to the ADSs. However, as the risks which we face relate to events and depend on circumstances that may or may not occur in the future, you should consider the risks and uncertainties described below.
The risks referred to below are those risks which we believe to be the material risks relating to our company. However, there may be additional risks that we do not currently consider to be material or of which we are not currently aware that may adversely affect our business, financial condition, results of operations or prospects. If any of the risks referred to in this registration statement were to occur, our results of operations, financial condition and prospects could be materially adversely affected. If that were to be the case, the trading price of the Ordinary Shares and/or the level of dividends or distributions (if any) received from the Ordinary Shares could decline significantly.
Risks Specific to the Development of the Business
We do not have collaborations in place with institutions for utility studies and there is no guarantee that we will be able to demonstrate prospective clinical utility of the StemPrintER and SPARE products
Following the completion of the initial retrospective validation studies in two spatial and temporal independent cohorts in respect of the StemPrinter and SPARE products, we are likely to run clinical utility studies to support applications for reimbursement, which are necessary for successful commercialization and to provide further evidence to support marketing claims. We have not yet identified which institutions will carry out the utility studies and have not yet entered into the relevant agreements with these institutions. There is a risk that we will not be able to secure these collaborations, which would impact our ability to proceed to the utility study stage. Whilst the utility studies are not a source of continuing revenue, such studies do provide a short-term revenue stream from sales of the tests run on the StemPrintER and SPARE products.
Furthermore, we may not be able to demonstrate the clinical utility of the StemPrintER and SPARE products in a real-world setting, which would impact our ability to secure reimbursement. If such reimbursement is not achieved, it will make commercialization of the StemPrintER and SPARE products significantly more challenging and would impact our ability to generate revenue and, accordingly, result in a material adverse impact on our business, financial condition and results of operations and those of our wholly-owned subsidiary, StemPrintER Sciences Limited (which we refer to collectively as the “Group”).
There are risks associated with the process of establishing a U.S. Clinical Laboratory Improvement Amendments of 1988 (“CLIA”) certified laboratory and in offering StemPrintER and SPARE products as laboratory developed tests (“LDTs”) which are outside our control
The StemPrintER and SPARE products do not as yet have status as LDTs and we do not yet have a CLIA-certified laboratory.
Even if we eventually obtain the required FDA clearance and certification as LDTs for our products and proceed to commercialization, there are inherent risks associated with offering the StemPrintER and SPARE products as LDTs that are outside our control, including test uptake, which would have an impact on the amount of revenue we could generate. Further, we may not be able to generate the revenue amounts that we anticipate, if any, from offering the StemPrintER and SPARE products as LDTs.
We are dependent on other third parties who provide certain resources and services to us, as we have limited resources in the short-term
We rely in part on external resources to conduct the research, development, supply of supplies and clinical testing of our StemPrintER and SPARE products, including in relation to our laboratory systems which rely on software developed by external manufacturers. The future development of the StemPrintER and SPARE products and other products will partly depend upon the performance of these third parties. We cannot guarantee that the relevant third parties will be able to carry out their obligations under the relevant arrangements. In the future, we may depend on external resources in marketing, sales and distribution of its products. We cannot guarantee that we will be able to assign competent partners to conduct these tasks or that these tasks can be completed on the basis of terms which are beneficial to us. Additionally, whilst the Directors are responsible for making decisions on our behalf, the Directors will rely to a certain extent on the advice of external professional advisors. There is no guarantee that we will receive the correct advice from such advisors.
9
Table of Contents
Disagreements between us and any third parties could lead to delays in our research and development (“R&D”) program and/or commercialization plans. If any third parties were to terminate their relationships with us, we would be required to obtain development and/or commercialization services from other third parties or develop the relevant functions internally, which could have an adverse effect on our business, results of operations and financial condition.
We are subject to research and product development risk
We may not be able to develop new products or to identify specific market needs that can be addressed by tests or solutions developed us. Product development will be a key ongoing activity for us. However, there can be no guarantee that further products will be developed, successfully launched, or accepted by the market. All new product development has an inherent level of risk and can be a lengthy process and suffer unforeseen delays, cost overruns and setbacks, such as difficultly recruiting patients into clinical trials. The nature of the medical device industry may mean new products may become obsolete as a result of competition or regulatory changes which could have a material adverse effect on our business, results of operations and financial condition.
In addition, R&D may be subject to various requirements, such as research subject protection for individuals participating in clinical evaluations of new products, institutional review board oversight, regulatory authorizations, and design control requirements. Failure to comply with requirements could result in penalties, delay, or prevent commercialization of products.
We are subject to risks associated with medical and technological change and obsolescence
Demand for our products could be adversely impacted by the development of alternative technology and alternative medicines. There can be no assurance that the technology and products currently being developed by us will not be rendered obsolete. As a result, there is the possibility that new technology or products may be superior to, or render obsolete, the technology and products that we are currently developing. Any failure of ours to ensure that our products remain up to date with the latest advances may have a material adverse impact on our competitiveness and financial performance. Our success will depend, in part, on our ability to develop and adapt to these technological changes and industry trends and failure to do so could have a material adverse effect on our business, results of operations and financial condition.
Risks Relating to Intellectual Property
Our rights to develop and commercialize our product candidates are subject to the terms and conditions of licenses granted to us by others. If we fail to comply with our obligations under our existing and any future intellectual property licenses with third parties, we could lose license rights that are important to our business
We are reliant upon licenses and sublicenses from Istituto Europeo di Oncologia, Fondazione FIRC per l’Oncologia Molecolare and the University of Milan (“IEO/University of Milan”) to certain patent rights and proprietary technology that are important or necessary to the development of our technology and product candidates, including the patents and know-how relating to manufacture.
On June 24, 2014, Tiziana entered into an exclusive licence agreement with IEO/University of Milan (the “License”), pursuant to which it obtained a worldwide, royalty-bearing, exclusive licence under certain patents and a worldwide, royalty-bearing, non-exclusive licence under certain know-how, respectively, of IEO/University of Milan to develop and commercialize licensed products in connection with a multi-gene prognostic tool. The License was assigned to us as part of the arrangements contained in the Demerger Agreement on October 30, 2020. Pursuant to the terms of the License, we are obliged to use commercially reasonable efforts in connection with the development and commercialization of the licensed products, including in accordance with specified diligence milestones. If the Company fails to meet its obligations under the License or if the License is terminated for any reason, it could negatively impact the Company’s business and strategic goals. The License may also be terminated for other reasons including breach and insolvency.
IEO/University of Milan have not provided the level of assurances and representations that are usually expected of similar options or licences, and generally the rights are granted on an ‘as is’ basis. Although it is appreciated that as an academic institution IEO/University of Milan is not in the habit of providing warranties, but generally it does leave us commercially exposed. Furthermore, our liability under the License is not capped.
10
Table of Contents
IEO/University of Milan may terminate the License immediately if we de-list from a public stock exchange, among other things, meaning that the IEO/University of Milan may trigger this termination right if we were to be taken private following admission of our Ordinary Shares to the London Stock Exchange, which could impact our value.
If we are unable to obtain and maintain patent protection for our product candidates and technology, or if the scope of our patent protection is not sufficiently broad, our competitors could develop and commercialize similar products and technology
Our success depends, in large part, on our ability to seek, obtain and maintain patent protection in the United States, U.K. and other countries with respect to our product candidates and technology. Our licensors have sought, and we intend to seek, to protect our proprietary position by filing patent applications in the United States, the U.K. and elsewhere, related to certain technologies and our product candidates, StemPrintER and SPARE, that are important to our business.
Our current patent portfolio contains a limited number of patent applications, which are in-licensed from third parties. If we are unable to assert any such patents to prevent others from reproducing our technology and product candidates, or are unable to identify patentable aspects of our R&D output before it is too late to obtain patent protection, failure to do so could have a material adverse effect on our business, results of operations and financial condition.
Our intellectual property is open to challenge
No assurance can be given that any current or future trademark, design right or patent applications will result in registered trademarks, design rights or patents, that the scope of any patent, design or trademark protection or the protection provided by copyright or database rights or the right to bring actions for breach of confidentiality will exclude competitors or provide competitive advantages to us, that any of our owned or licensed-in patents, design rights or trademarks will be held valid if challenged or that third parties will not claim rights or ownership of the patents, design rights, trademarks or other intellectual property rights held by us.
If we cannot successfully enforce our intellectual property rights, this could have a material adverse effect on our business, financial condition and prospects. We may be subject to claims in relation to the infringement of patents, design rights, trademarks or other intellectual property rights owned by third parties. Adverse judgments against us may give rise to significant liabilities in monetary damages, legal fees and/or an inability to manufacture, market or sell products either at all or in particular territories.
Our strategy involves generating commercially valuable intellectual property that can be protected
We intend to augment our intellectual property portfolio. No assurance can be given that any future patent applications will result in granted patents, that the scope of any patent protection will exclude competitors or provide competitive advantages to us, that any of our patents will be held valid if challenged or that third parties will not claim rights in or ownership of the patents and other proprietary rights held by us. Should we fail to successfully obtain additional patent protection in respect its technology and products could have a material adverse effect on our business, results of operations and financial condition.
Market and Competitive Risks
We operate in a competitive market and may face competition from competitors involved in multi-gene prognostic assay for the prediction of risk of recurrence in luminal ER+/HER2- breast cancer patients
We may face competition from competitors involved in developing a multi-gene prognostic assay for the prediction of risk of recurrence in luminal ER+/HER2- breast cancer patients. Some of our competitors may have access to greater research, development, marketing, financial and personnel resources which may provide commercial advantages to those competitors. New products may be more effective, cheaper or more effectively marketed than StemPrintER and SPARE. A substantial increase in competition for any of these reasons could require us to, for example, increase our marketing or capital expenditure or require us to change our business model to remain competitive, which may have an adverse impact on our business including our profitability and/or financial condition.
The market opportunities for our product candidates may be smaller than we anticipate
We are focusing our R&D efforts on a multi-gene prognostic tool for predicting the recurrence of certain breast cancers. Our understanding of both the number of people who have these diseases, as well as the subset of people with these diseases who have the potential to benefit from our prognostic assay, is based on estimates. These estimates may prove to be incorrect and
11
Table of Contents
new studies may reduce the estimated incidence or prevalence of these diseases. The number of patients in the United States, the U.K., the EU and elsewhere may turn out to be lower than expected, may not be otherwise amenable to assessment with our product candidates or patients may become increasingly difficult to identify and access, all of which would adversely affect our business, financial condition, results of operations and prospects.
Further, there are several factors that could contribute to making the actual number of patients who receive our potential products, if and when approved, less than the potentially addressable market, such as the lack of widespread availability of, and limited reimbursement for, new therapies in many underdeveloped markets.
The future commercial success of our product candidates will depend upon the degree of each product candidates’ market acceptance by physicians, patients, third-party payors and others in the medical community
We have no product authorized for marketing; our product candidates are at the validation study stage of development, and we may never have a product that is commercially successful. The commercial success of our product candidates will depend, in part, on their acceptance by physicians, patients and third-party payors as medically necessary, cost-effective and safe. If these products do not achieve an adequate level of acceptance, we may not generate significant product revenue and may not become profitable. Even if some product candidates achieve market acceptance, the market may not prove to be large enough to generate significant revenues. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on several factors, including, but not limited to:
| • | the effectiveness and safety of our product candidates as demonstrated in clinical trials; |
| • | the potential and perceived advantages of our product candidates over alternative prognostic tools; |
| • | the availability and cost of use relative to alternative prognostic tools; |
| • | changes in the standard of care for the targeted indications for any product candidate; |
| • | the willingness of physicians to use, and the target patient population to try, new prognostic tools; |
| • | product labelling or product insert requirements of the FDA, the U.K. Medicines and Healthcare products Regulatory Agency (“MHRA”), the European Medicines Agency (“EMA”) or other regulatory authorities, including any limitations or warnings contained in a product’s approved labelling; |
| • | the timing of market introduction of competitive products; |
| • | sales, distribution and marketing support; |
| • | publicity concerning our product candidates or competing products and treatments; |
| • | potential product liability claims; |
| • | any restrictions on the use of our products together with other medications; and |
| • | favourable third-party payor coverage and adequate reimbursement. |
Even if a potential product displays favourable clinical properties and safety profile in preclinical studies and clinical trials, market acceptance of the product will not be fully known until after it is launched.
The insurance coverage and reimbursement status of newly approved products is uncertain. Failure to obtain or maintain adequate coverage and reimbursement for our approved product candidates could limit our ability to market those products
We expect that coverage and adequate reimbursement by government and private payors will be essential for most patients to be able to afford our approved product candidates. Accordingly, sales of our product candidates will depend substantially, both domestically and abroad, on the extent to which the costs of our product candidates will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organisations, or will be reimbursed by government authorities, private health coverage insurers and other third-party payors. Coverage and reimbursement by a third-party payor may depend upon several factors, including the third-party payor’s determination that use of a product is:
| • | a covered benefit under our health plan; |
| • | safe, effective and medically necessary; |
| • | appropriate for the specific patient; |
| • | cost-effective; and |
12
Table of Contents
| • | neither experimental nor investigational. |
Obtaining coverage and reimbursement for a product from third-party payors is a time-consuming and costly process that could require us to provide to the payor supporting scientific, clinical and cost-effectiveness data. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. If coverage and reimbursement are not available, or are available only at limited levels, we may not be able to successfully commercialize our product candidates. Even if coverage is provided, the approved reimbursement amount may not be adequate to realise a sufficient return on our investment.
Market acceptance and sales of our products will depend significantly on the availability of adequate coverage and reimbursement from third-party payors and may be affected by existing and future healthcare reform measures.
Regulatory Risks
Our failure to maintain compliance of its future clinical laboratory operations with applicable laws could result in substantial civil or criminal penalties
The operation of a clinical laboratory by us will be in a highly regulated environment which, among other things, will require maintaining compliance with CLIA certification and state clinical laboratory licensing requirements. Failure to maintain compliance with these requirements may result in a range of enforcement actions, including certificate or license suspension, limitation, or revocation, directed plan of action, onsite monitoring, civil monetary penalties and criminal sanctions. Such failure may also result in significant adverse publicity. Any of these consequences could limit or entirely prevent our continued operation and therefore impact our financial performance.
We are subject to various health regulatory laws pertaining to fraud and abuse and related matters, and any failure to comply with such laws could result in substantial civil or criminal penalties
Our employees, independent contractors, consultants, and collaborators may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements, which could cause significant liability for us and harm our operations and reputation. We are exposed to the risk that our employees, independent contractors, consultants, and collaborators may engage in fraud or other misconduct to comply with manufacturing standards we have established, to comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable non-U.S. regulatory authorities, to report financial information or data accurately or to disclose unauthorized activities to us. Such misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter misconduct, and the precautions we will take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with such laws, standards or regulations. If any such actions are instituted against us, or our key employees, independent contractors, consultants, or collaborators, and we are not successful in defending ourself or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant criminal, civil and administrative sanctions including monetary penalties, damages, fines, disgorgement, individual imprisonment, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, reputational harm, and we may be required to curtail or restructure our operations.
Our failure to prevent a data breach would result in serious reputational damage to us and may result in civil or criminal lawsuits and associated penalties
We take our responsibility to maintain patient confidentiality and protect patient data extremely seriously. By its nature, the de-identified data that is being processed is highly sensitive and includes genetic and demographic information, the processing of which is subject to the most onerous obligations of applicable data protection legislation. If, due to a technical oversight, human error or malicious action by an employee or third party, the privacy, security or integrity of the data were compromised, we may be obliged to report such breach once we became aware of it under applicable laws and regulations such as HIPAA, GDPR, DPA 2018 or other U.S. state, U.K. or EU member state specific laws as well as the data privacy laws of other countries such as Japan, Singapore, Hong Kong and China. Depending on the nature and extent of the breach, we may become subject to a regulatory investigation, which would divert time and financial resources from the day-to-day operation of the business and may result in civil or criminal lawsuits and financial fines and penalties as well as adverse publicity. If third parties and/or customers of ours become aware of such breaches, they may opt to cancel existing contracts or not enter new contracts with us, reducing revenue. We may also be required to personally inform the patients whose data
13
Table of Contents
was released or accessed as a result of a data breach, which may increase the severity of the reputational damage and may lead to patients revoking their consent for the data to be used by us. In addition, patients may have the right to bring claims for compensation for such breaches which might be brought by way of class or representative actions and claim significant sums as damages. To mitigate the risk of a data breach or related issue, we will employ technical security measures to protect data and work closely with our data providers to ensure that each party understands its obligations to protect personal data.
Risks Specific to our Current Size and Headcount
We are reliant upon the expertise and continued service of a small number of key individuals of our management, Board of Directors and scientific advisors
We rely on the expertise and experience of a small number of key individuals of its management, Directors and scientific advisors to continue to develop and manage our business. The retention of their services cannot be guaranteed. Accordingly, the departure of these key individuals could have a negative impact on our operations, financial condition, our ability to execute our business strategy and future prospects.
Going forwards, we will rely, in part, on the recruitment of appropriately qualified personnel, including personnel with a high level of scientific and technical expertise in the industry. We may be unable to find a sufficient number of appropriately highly trained individuals to satisfy its growth rate which could affects its ability to develop products as planned.
In addition, if we fail to succeed in pre-clinical or clinical studies, it may make it more challenging to recruit and retain appropriately qualified personnel. Our inability to recruit key personnel or the loss of the services of key personnel or consultants may impede the progress of our R&D objectives as well as the commercialization of our lead and other products, which could have a material adverse effect on our business, results of operations and financial condition.
We will need to expand our organization and may experience difficulties in managing this growth, which could disrupt our operations
As we mature, we expect to expand our full-time employee base and to hire more scientists, technicians and other skilled or experienced personnel. The management may need to divert a disproportionate amount of its attention away from the day-to-day activities and devote a substantial amount of time toward managing these growth activities. We may not be able to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Our expected growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of additional products or technologies. If the management is unable to effectively manage our growth, our expenses may increase more than expected, the ability to generate and/or grow revenues could be reduced, and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize products and compete effectively will depend, in part, on our ability to effectively manage any future growth.
Risks Specific to the Future Financing of the Business
We need substantial additional funding to complete the development of its product candidates, which may not be available on acceptable terms, if at all. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate certain of our product development, research operations or future commercialization efforts, if any
Our operations have consumed substantial amounts of cash since inception, and we expect our expenses to increase in connection with our ongoing activities, particularly as we continue the R&D of, initiate further clinical trials of and seek marketing approval for, our product candidates. In addition, if we obtain marketing approval for our product candidates, we expect to incur significant expenses related to product sales, marketing, manufacturing and distribution.
Furthermore, we expect to incur additional costs associated with operating as a public reporting company in the United States.
If we are unable to obtain adequate funding on a timely basis, we may be required to significantly curtail, delay or discontinue our R&D programmes of our product candidates or any future commercialization efforts, be unable to expand our operations or be unable to otherwise capitalize on our business opportunities, as desired, which could harm our business and potentially cause a discontinuation of operations.
14
Table of Contents
We may need to raise additional funding to take advantage of future opportunities
We may need to raise additional funding to take advantage of future opportunities. Such additional funding may not be available or, if available, may not be on terms that are favorable to us or our shareholders. If we are unable to obtain additional funding as required, we may be required to reduce the scope of our operations or anticipated expansion.
Risks Related to Being a Newly Independent, Public Company
We have no history of operating as a separate, U.S. public company.
Our historical financial information is not necessarily representative of the results that we would have achieved as a separate, U.S. public company and may not be a reliable indicator of our future results. Prior to the Demerger, in which Tiziana contributed its StemPrintER project to us to operate as a separate, independent business, our operations were conducted by Tiziana as part of its broader corporate organization, rather than as a separate company. The corporate functions that Tiziana performed, such as paying filing fees, accounting, payroll, auditing and legal fees, are now our responsibility to manage . Our future financial results will reflect corporate expenses for such functions that we have not yet had to manage as an independent company. We will also incur additional expenses to comply with our obligations as a public company in the U.S.
Risks Related to Legal, Political and Economic Uncertainty
The relationship of the U.K. with the EU could impact our ability to operate efficiently in certain jurisdictions or in certain markets
The U.K. formally exited the EU on January 31, 2020 (“Brexit”). Under the terms of its departure, the U.K. entered a transition period during which it continued to follow all EU rules until December 31, 2020 (the “Transition Period”). On December 30, 2020, the U.K. and EU signed the Trade and Cooperation Agreement, which includes an agreement on free trade between the two parties.
There is considerable uncertainty resulting from a lack of precedent and the complexity of the U.K. and EU’s intertwined legal regimes as to how Brexit (following the Transition Period) will impact the medical devices industry in Europe. Since a significant proportion of the regulatory framework in the U.K. applicable to our business and product candidates is derived from EU directives and regulations, Brexit could materially impact the regulatory regime with respect to the development, manufacture, importation, approval and commercialization of our product candidates in the U.K. or the EU. The impact will largely depend on the model and means by which the U.K.’s relationship with the EU is governed post-Brexit and the extent to which the U.K. chooses to diverge from the EU regulatory framework. For example, following the Transition Period, the U.K. will no longer be covered by the centralized procedures for obtaining EU-wide marketing authorizations and our product candidates will therefore require a separate marketing authorization for such products to be marketed in the U.K.. It is also unclear as to whether the relevant authorities in the EU and the U.K. are adequately prepared for the additional administrative burden caused by Brexit. Any delay in obtaining, or an inability to obtain, any marketing approvals, as a result of Brexit or otherwise, would prevent us from, or delay commercialization of, product candidates in the U.K. and/or the EEA and restrict our ability to generate revenue and achieve and sustain profitability.
If any of these outcomes occur, we may be forced to restrict or delay efforts to seek regulatory approval in the U.K. for our product candidates, which could significantly and materially harm our business. There is a degree of uncertainty regarding the overall impact that Brexit will have on process to obtain regulatory approval in the U.K. for product candidates.
Further, the U.K.’s withdrawal from the EU has resulted in the relocation of the EMA from the U.K. to the Netherlands. This relocation has caused, and may continue to cause, disruption in the administrative and medical scientific links between the EMA and the MHRA, including delays in granting clinical trial authorisation or marketing authorization, disruption of importation and export of medical devices. The cumulative effects of the disruption to the regulatory framework may add considerably to the development lead time to marketing authorization and commercialization of product candidates in the EU and/or the U.K. Brexit may also result in a reduction of funding to the EMA once the U.K. no longer makes financial contributions to EU institutions, such as the EMA. If funding to the EMA is so reduced, it could create delays in the EMA issuing regulatory approvals for the Company’s product candidates and, accordingly, have a material adverse effect on our business, financial condition, results of operations or prospects.
15
Table of Contents
Risks Related to our Business Operations
Risks relating to managing growth, employee matters and other risks relating to our business
Growth may place significant demands on our management and resources. We expect to experience growth in the number of our employees and the scope of our operations in connection with the continued development and, in due course, the potential commercialisation of our products.
This potential growth will place a significant strain on our management and operations, and we may have difficulty managing this future potential growth.
We are highly dependent on our current Directors and the Senior Manager (together, the “Persons Discharging Managerial Responsibility” (“PDMR”)) and their services are critical to the successful implementation of our product development and regulatory strategies. Whilst suitable contracts of employment or engagement are in place including six to 12 months’ notice periods for all PDMRs, they may give notice to terminate their employment with us at any time. The loss of the services of any of the PDMRs and our inability to find suitable replacements could harm our business, prospects, financial condition, results of operations and ability to achieve the successful development or commercialisation of our products.
Challenges in identifying and retaining key personnel could impair our ability to conduct and grow our operations effectively
Our ability to compete in the highly competitive medical device industry depends upon our ability to attract and retain highly qualified management and sales teams. We are intending to recruit our own commercial team and expand our existing central infrastructure team. Many of the other pharmaceutical companies and academic institutions that we compete against for qualified personnel have greater financial and other resources, different risk profiles and a longer history in the industry than it does. We might not be able to attract or retain these key persons on conditions that are economically acceptable. Our inability to attract and retain these key persons could have a material adverse effect on our business, prospects, financial conditions and results of operation.
We may become subject to product liability claims
We face an inherent risk of product liability and associated adverse publicity as a result of the clinical testing of our products and sales of our products once marketing approval is received from relevant regulatory authorities.
Criminal or civil proceedings might be filed against us by study subjects, patients, relevant regulatory authorities, pharmaceutical companies, and any other third party using or marketing our products. Any such product liability claims may include allegations of defects in manufacturing or design, negligence, strict liability, a breach of warranties and a failure to warn of dangers inherent in the product.
If we cannot successfully defend ourself against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products, if approved. Even if we successfully defends ourself against such product liability claims it could require significant financial and management resources. Regardless of the merits or eventual outcome, product liability claims may result in:
| • | decreased demand for our products due to negative public perception; |
| • | injury to our reputation; |
| • | withdrawal of clinical study participants or difficulties in recruiting new study participants; |
| • | initiation of investigations by regulators; |
| • | costs to defend or settle the related litigation; |
| • | diversion of management’s time and our resources; |
| • | substantial monetary awards to patients, study participants or subjects; |
| • | product recalls, withdrawals or labelling, marketing or promotional restrictions; |
| • | loss of revenues from product sales; or |
| • | the inability to commercialize any our products, if approved. |
16
Table of Contents
Although we will maintain levels of insurance customary for our sector to cover our current and future business operations, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. In such cases, we would have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
If we or our partners, licensees and subcontractors were unable to obtain and maintain appropriate insurance coverage at an acceptable cost, or to protect ourself or themselves in any way against actions for damages, this would seriously affect the marketing of our products and, more generally, be detrimental to our business, prospects, results of operations or financial condition.
Risks Relating to Taxation
We may be classified as a passive foreign investment company for United States federal income tax purposes
We may be classified as a passive foreign investment company for U.S. federal income tax purposes. If we are so classified, we may, but are not obliged to, provide to U.S. holders of Ordinary Shares the information that would be necessary in order for such persons to make a qualified electing fund election with respect to the Ordinary Shares for any year in which we are a passive foreign investment company.
Changes in tax law and practice may impact shareholders and the Group
The tax treatment of shareholders and the Group are subject to changes in tax laws or tax authority practices in the United Kingdom or any other relevant jurisdiction. Any change may reduce any net return derived by investors from a shareholding in us.
You should not rely on the general guide to taxation set out in this registration statement and should seek their own specialist advice. The tax rates referred to in this registration statement are those currently applicable and they are subject to change.
We may not be able to make returns for shareholders in a tax efficient manner
We intend to structure the Group, including any company or business acquired, to maximise returns for shareholders in as fiscally efficient a manner as is practicable. We have made certain assumptions regarding taxation. However, if these assumptions are not borne out in practice, taxes may be imposed with respect to any of the Group’s assets, or the members of the Group may be subject to tax on income, profits, gains or distributions in a particular jurisdiction or jurisdictions in excess of taxes that were anticipated. This could alter the post-tax returns for shareholders (or shareholders in certain jurisdictions). The level of return for shareholders may also be adversely affected. Any change in laws or tax authority practices could also adversely affect any post-tax returns of capital to shareholders or payments of dividends (if any, of which we do not envisage the payment, at least in the short to medium term). In addition, we may incur costs in taking steps to mitigate any such adverse effect on the post-tax returns for Shareholders.
Risks Related to Our Ordinary Shares and ADSs
We do not expect that an active trading market will develop for our ADSs, which will make it difficult for you to sell your ADSs
While we intend for our Ordinary Shares to be admitted to a Standard Listing and to trade on the Main Market of the London Stock Exchange later in 2021, the ADSs registered hereunder constitute the first opportunity to receive our ADSs in the United States. No public market has previously existed for our ADSs or our Ordinary Shares, and we do not anticipate a public market for the ADSs or the Ordinary Shares developing in the near term. We do not intend to apply to have our ADSs listed on any stock exchange or over-the-counter trading market in the United States, which will make it difficult for holders to sell their ADSs; instead, holders may withdraw the underlying Ordinary Shares and trade those Ordinary Shares on the Main Market of the London Stock Exchange, when and if such shares are admitted to trading later in 2021.
17
Table of Contents
There is currently no market for our Ordinary Shares, and a market for our Ordinary Shares may not develop, which would adversely affect the liquidity and price of our Ordinary Shares
There is currently no market for our Ordinary Shares. Although our current intention is that our Ordinary Shares should trade on the Main Market of the London Stock Exchange, we cannot assure you that we will do so. If we do proceed with a listing on the Main Market of the London Stock Exchange, an active trading market for our Ordinary Shares may not develop or, if developed, may not be maintained. You may be unable to sell the Ordinary Shares underlying your ADSs unless a market can be established and maintained, and if we subsequently obtain a listing on an exchange in addition to, or in lieu of, the Main Market of the London Stock Exchange, the level of liquidity of the Ordinary Shares may decline.
The market price of our Ordinary Shares, when and if they are admitted to a Standard Listing and to trading of the Main Market of the London Stock Exchange, may be highly volatile
The market price of our Ordinary Shares, when and if they are admitted for trading on the Main Market of the London Stock Exchange, is likely to be volatile. Stock markets in general, and for biopharmaceutical companies in particular, have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, you may not be able to sell the Ordinary Shares underlying your ADSs.
Further issuances of Ordinary Shares may be dilutive
We may decide to offer additional shares in the future for capital raising or other purposes. Shareholders who do not take up or who are not eligible to take such an offer will find their proportionate ownership and voting interests in us to be reduced. An additional offering could also have a material adverse effect on the market price of the Ordinary Shares as a whole.
Economic conditions and current economic weakness
Any economic downturn either globally or locally in any area in which we operate may have an adverse effect on the demand for our services. A more prolonged economic downturn may restrict our ability to generate a profit.
In addition, although signs of economic recovery have been perceptible in certain countries, the sustainability of a global economic upturn is not yet assured. If economic conditions remain uncertain this might have an adverse impact on our operations and business results.
Our ability to pay dividends in the future is not certain
We cannot guarantee that we will have sufficient cash resources to pay dividends in the future. The declaration, payment and amount of any future dividends are subject to the shareholders’ discretion, or in the case of interim dividends, the Board’s discretion, and will depend upon our earnings, financial position, cash requirements, availability or profits, any dividends and profits that we receive from our subsidiary companies, as well as provisions for relevant laws or generally accepted accounting principles from time to time.
We are likely to have a number of outstanding options which, if exercised, could have a material dilutive effect on existing shareholders
Whilst we currently have no outstanding options over Ordinary Shares, options may be issued in connection with our proposed admission to a Standard Listing and to trading of the Main Market of the London Stock Exchange. Such convertible instruments would have a material dilutive effect on shareholders when and if they are exercised.
Pre-emption rights for U.S. and other non-U.K. holders of Ordinary Shares may be unavailable
In the case of certain increases in the Company’s issued share capital, existing holders of Ordinary Shares are generally entitled to pre-emption rights to subscribe for such shares, unless shareholders waive such rights by a resolution at a shareholders’ meeting. U.S. holders of ordinary shares in U.K. companies are customarily excluded from exercising any such pre-emption rights they may have, unless a registration statement under the Securities Act is effective with respect to those rights, or an exemption from the registration requirements thereunder is available.
Any exemption from the registration requirements of the Securities Act or applicable non-U.S. securities law may not be available to enable U.S. or other non-U.K. holders to exercise such pre-emption rights or, if available, the Company may not utilize any such exemption.
18
Table of Contents
Shareholders may be unable to enforce judgments obtained in U.S. courts
The Company is incorporated and registered in England and Wales under the Companies Act. Service of process upon our Directors and officers, all of whom reside outside the United States, may be difficult to obtain within the United States. Furthermore, since all of our directly owned assets and those of our Directors are located outside the United States, any judgment obtained in the United States against it or them may not be enforceable outside of the United States, including without limitation judgments based upon the civil liability provisions of the U.S. federal securities laws or the laws of any state or territory within the U.S. In addition, an award or awards of punitive damages in actions brought in the U.S. or elsewhere may be unenforceable in the U.K. Shareholders may also have difficulties enforcing, in original actions brought in courts in jurisdictions outside the U.S., liabilities under U.S. securities laws.
Holders of ADSs are not treated as holders of our Ordinary Shares
After purchasing an ADS, you will become a holder of ADSs with rights to the underlying shares in a company incorporated under English law. Holders of ADSs are not treated as holders of our Ordinary Shares, unless they withdraw the Ordinary Shares underlying their ADSs in accordance with the deposit agreement and applicable laws and regulations. The Depositary is the holder of the Ordinary Shares underlying the ADSs. Holders of ADSs therefore do not have any rights as holders of our Ordinary Shares, other than the rights that they have pursuant to the deposit agreement. See “Item 12.D.—American Depositary Shares.”
Holders of ADSs may be subject to limitations on the transfer of their ADSs and the withdrawal of the underlying ordinary shares
ADSs are transferable on the books of the Depositary. However, the Depositary may close its books at any time or from time to time when it deems expedient in connection with the performance of its duties. The Depositary may refuse to deliver, transfer or register transfers of ADSs generally when our books or the books of the Depositary are closed, or at any time if we or the Depositary think it is advisable to do so because of any requirement of law, government or governmental body, or under any provision of the deposit agreement, or for any other reason, subject to the right of ADS holders to cancel their ADSs and withdraw the underlying ordinary shares. Temporary delays in the cancellation of your ADSs and withdrawal of the underlying ordinary shares may arise because the depositary has closed its transfer books or we have closed our transfer books, the transfer of ordinary shares is blocked to permit voting at a shareholders’ meeting or we are paying a dividend on our ordinary shares. In addition, ADS holders may not be able to cancel their ADSs and withdraw the underlying ordinary shares when they owe money for fees, taxes and similar charges and when it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of ordinary shares or other deposited securities. See “Item 12.D.—American Depositary Shares.”
19
Table of Contents
You will not have the same voting rights as the holders of our Ordinary Shares and may not receive voting materials in time to be able to exercise your right to vote
Except as described in this registration statement and the deposit agreement, holders of the ADSs will not be able to exercise voting rights attaching to the Ordinary Shares represented by the ADSs. Under the terms of the deposit agreement, holders of the ADSs may instruct the Depositary to vote the Ordinary Shares underlying their ADSs. Otherwise, holders of ADSs will not be able to exercise their right to vote unless they withdraw the Ordinary Shares underlying their ADSs to vote them in person or by proxy in accordance with applicable laws and regulations and our Articles of Association. Even so, ADS holders may not know about a meeting far enough in advance to withdraw those Ordinary Shares. If we ask for the instructions of holders of the ADSs, the Depositary, upon timely notice from us, will notify ADS holders of the upcoming vote and arrange to deliver our voting materials to them. Upon our request, the Depositary will mail to holders a shareholder meeting notice that contains, among other things, a statement as to the manner in which voting instructions may be given. We cannot guarantee that ADS holders will receive the voting materials in time to ensure that they can instruct the depositary to vote the ordinary shares underlying their ADSs. A shareholder is only entitled to participate in, and vote at, the meeting of shareholders, provided that it holds our Ordinary Shares as of the record date set for such meeting and otherwise complies with our Articles of Association. In addition, the Depositary’s liability to ADS holders for failing to execute voting instructions or for the manner of executing voting instructions is limited by the deposit agreement. As a result, holders of ADSs may not be able to exercise their right to give voting instructions or to vote in person or by proxy and they may not have any recourse against the Depositary or us if their Ordinary Shares are not voted as they have requested or if their Ordinary Shares cannot be voted.
You may not receive distributions on our Ordinary Shares represented by the ADSs or any value for them if it is illegal or impractical to make them available to holders of ADSs
The Depositary has agreed to pay to you any cash dividends or other distributions it or the custodian receives on our Ordinary Shares or other deposited securities after deducting its fees and expenses. You will receive these distributions in proportion to the number of our Ordinary Shares your ADSs represent. However, in accordance with the limitations set forth in the deposit agreement, it may be unlawful or impractical to make a distribution available to holders of ADSs. We have no obligation to take any other action to permit distribution on the ADSs, ordinary shares, rights or anything else to holders of the ADSs. This means that you may not receive the distributions we make on our ordinary shares or any value from them if it is unlawful or impractical to make them available to you. These restrictions may have an adverse effect on the value of your ADSs.
ADSs holders may not be entitled to a jury trial with respect to claims arising under the deposit agreement, which could result in less favorable outcomes to the plaintiff(s) in any such action
The deposit agreement provides that, to the fullest extent permitted by law, holders and beneficial owners of ADSs irrevocably waive the right to a jury trial of any claim they may have against us or the depositary arising out of or relating to the ADSs or the deposit agreement.
If this jury trial waiver provision is not permitted by applicable law, an action could proceed under the terms of the deposit agreement with a jury trial. If we or the depositary opposed a jury trial demand based on the waiver, the court would determine whether the waiver was enforceable based on the facts and circumstances of that case in accordance with the applicable state and federal law. To our knowledge, the enforceability of a contractual pre-dispute jury trial waiver in connection with claims arising under the federal securities laws has not been finally adjudicated by the U.S. Supreme Court. However, we believe that a contractual pre-dispute jury trial waiver provision is generally enforceable, including under the laws of the State of New York, which govern the deposit agreement, by a federal or state court in the City of New York, which has non-exclusive jurisdiction over matters arising under the deposit agreement. In determining whether to enforce a contractual pre-dispute jury trial waiver provision, courts will generally consider whether a party knowingly, intelligently and voluntarily waived the right to a jury trial. We believe that this is the case with respect to the deposit agreement and the ADSs. It is advisable that you consult legal counsel regarding the jury waiver provision before entering into the deposit agreement.
If you or any other holders or beneficial owners of ADSs bring a claim against us or the Depositary in connection with matters arising under the deposit agreement or the ADSs, including claims under federal securities laws, you or such other holder or beneficial owner may not be entitled to a jury trial with respect to such claims, which may have the effect of limiting and discouraging lawsuits against us and/or the depositary. If a lawsuit is brought against us and/or the depositary under the deposit agreement, it may be heard only by a judge or justice of the applicable trial court, which would be conducted according to different civil procedures and may result in different outcomes than a trial by jury would have had, including results that could be less favorable to the plaintiff(s) in any such action, depending on, among other things, the nature of the claims, the judge or justice hearing such claims, and the venue of the hearing.
20
Table of Contents
No condition, stipulation or provision of the deposit agreement or ADSs serves as a waiver by any holder or beneficial owner of ADSs or by us or the Depositary of compliance with the U.S. federal securities laws and the rules and regulations promulgated thereunder.
Our principal shareholder has a significant holding in the company which may give them influence in certain matters requiring approval by shareholders, including approval of significant corporate transactions in certain circumstances
As of December 31, 2020, Gabriele Cerrone and Planwise Group Limited, a company in which Mr. Cerrone is the sole beneficial owner, held a beneficial ownership right (through his allotment of our shares as a Tiziana shareholder upon their issue and distribution following the effective date of this registration statement) in aggregate of approximately 37.12 per cent. of our Ordinary Shares. Accordingly, Mr. Cerrone and Planwise Group Limited may, as a practical matter, be able to influence certain matters requiring approval by shareholders, including approval of significant corporate transactions in certain circumstances. Such concentration of ownership may also have the effect of delaying or preventing any future proposed change in control of the Company. The trading price of the Ordinary Shares, when and if admitted to a Standard Listing and to trading of the Main Market of the London Stock Exchange, could be adversely affected if potential new investors are disinclined to invest in us because they perceive disadvantages to a large shareholding being concentrated in the hands of a single shareholder. The interests of Tiziana and holders that acquire ADSs may not be aligned. Tiziana may make acquisitions of, or investments in, other businesses in the same sectors as us. These businesses may be, or may become, competitors of us. In addition, funds or other entities managed or advised by Tiziana may be in direct competition with us on potential acquisitions of, or investments in, certain businesses.
We are an “emerging growth company”, and there are reduced disclosure requirements applicable to emerging growth companies
We are an “emerging growth company” as defined in the SEC’s rules and regulations and we will remain an emerging growth company until the earlier to occur of (1) the last day of 2025, (2) the last day of the fiscal year in which we have total annual gross revenues of at least $1.07 billion, (3) the last day of the fiscal year in which we are deemed to be a “large accelerated filer”, under the SEC’s rules, which means the market value of our equity securities that is held by non-affiliates exceeds $700 million as of the prior June 30th, and (4) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period. For so long as we remain an emerging growth company, we are permitted and intend to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include:
| • | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act; |
| • | not being required to comply with any requirement that has or may be adopted by the Public Company Accounting Oversight Board (PCAOB) regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; |
| • | being permitted to provide only two years of audited financial statements in this initial registration statement, in addition to any required unaudited interim financial statements, with correspondingly reduced “Management’s Discussion and Analysis of Financial Condition and Results of Operations” disclosure; |
| • | reduced disclosure obligations regarding executive compensation; and |
| • | an exemption from the requirement to seek nonbinding advisory votes on executive compensation or golden parachute arrangements. |
We may choose to take advantage of some, but not all, of the available exemptions. We have taken advantage of reduced reporting burdens in this registration statement. In particular, we have not included all of the executive compensation information that would be required if we were not an emerging growth company. We cannot predict whether you will find our ADSs less attractive if we rely on certain or all of these exemptions.
In addition, the JOBS Act provides that an emerging growth company may take advantage of an extended transition period for complying with new or revised accounting standards. This allows an emerging growth company to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We are considering whether we will take advantage of the extended transition period for complying with new or revised accounting standards. Since IFRS makes no distinction between public and private companies for purposes of compliance with new or revised accounting standards, the requirements for our compliance as a private company and as a public company are the same.
21
Table of Contents
We are not, and do not intend to become, regulated as an “investment company” under the Investment Company Act and if we were deemed an “investment company” under the Investment Company Act, applicable restrictions could make it impractical for us to continue our business as contemplated and could have a material adverse effect on our business
The Investment Company Act and the rules thereunder contain detailed parameters for the organization and operation of investment companies. Among other things, the Investment Company Act and the rules thereunder limit or prohibit transactions with affiliates, impose limitations on the issuance of debt and equity securities and impose certain governance requirements. We have not been and do not intend to become regulated as an investment company, and we intend to conduct our activities so that we will not be deemed to be an investment company under the Investment Company Act. In order to ensure that we are not deemed to be an investment company, we may be limited in the assets that we may continue to own and, further, may need to dispose of or acquire certain assets at such times or on such terms as may be less favorable to us than in the absence of such requirement. If anything were to happen which would cause us to be deemed to be an investment company under the Investment Company Act (such as significant changes in the value of our Founded Entities or a change in circumstance that results in a reclassification of our interests in our Founded Entities for purposes of the Investment Company Act), the requirements imposed by the Investment Company Act could make it impractical for us to continue our business as currently conducted, which would materially adversely affect our business, results of operations and financial condition. In addition, if we were to become inadvertently subject to the Investment Company Act, any violation of the Investment Company Act could subject us to material adverse consequences, including potentially significant regulatory penalties and the possibility that certain of our contracts could be deemed unenforceable.
As a foreign private issuer, we are exempt from a number of rules under the U.S. securities laws and are permitted to file less information with the SEC than a U.S. company. This may limit the information available to holders of ADSs or our ordinary shares
We are a “foreign private issuer”, as defined in the SEC’s rules and regulations and, consequently, we are not subject to all of the disclosure requirements applicable to public companies organized within the United States. For example, we are exempt from certain rules under the Exchange Act, that regulate disclosure obligations and procedural requirements related to the solicitation of proxies, consents or authorizations applicable to a security registered under the Exchange Act, including the U.S. proxy rules under Section 14 of the Exchange Act. In addition, our officers and Directors are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and related rules with respect to their purchases and sales of our securities. Moreover, while we currently make annual and semi-annual filings with respect to our listing on the LSE, we will not be required to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. domestic issuers and will not be required to file quarterly reports on Form 10-Q or current reports on Form 8-K under the Exchange Act. Accordingly, there will be less publicly available information concerning our company than there would be if we were not a foreign private issuer.
We may lose our foreign private issuer status in the future, which could result in significant additional cost and expense
While we currently qualify as a foreign private issuer, the determination of foreign private issuer status is made annually on the last business day of an issuer’s most recently completed second fiscal quarter and, accordingly, the next determination will be made with respect to us on June 30, 2021.
In the future, we would lose our foreign private issuer status if we to fail to meet the requirements necessary to maintain our foreign private issuer status as of the relevant determination date. For example, if more than 50 per cent. of our securities are held by U.S. residents and more than 50 per cent. of the members of our executive committee or members of our Board of Directors are residents or citizens of the United States, we could lose our foreign private issuer status.
22
Table of Contents
The regulatory and compliance costs to us under U.S. securities laws as a U.S. domestic issuer may be significantly more than costs we incur as a foreign private issuer. If we are not a foreign private issuer, we will be required to file periodic reports and registration statements on U.S. domestic issuer forms with the SEC, which are more detailed and extensive in certain respects than the forms available to a foreign private issuer. We would be required under current SEC rules to prepare our financial statements in accordance with U.S. GAAP, rather than IFRS, and modify certain of our policies to comply with corporate governance practices associated with U.S. domestic issuers. Such conversion of our financial statements to U.S. GAAP will involve significant time and cost. In addition, we may lose our ability to rely upon exemptions from certain corporate governance requirements on U.S. stock exchanges that are available to foreign private issuers such as the ones described above and exemptions from procedural requirements related to the solicitation of proxies.
We will incur increased costs as a result of operating as a U.S. public company, and our management will be required to devote substantial time to new compliance initiatives
As a U.S. public company, and particularly after we are no longer an emerging growth company, we will incur significant legal, accounting and other expenses that we will not incur as a public company listed on the LSE. In addition, the Sarbanes-Oxley Act of 2002, and rules subsequently implemented by the SEC have imposed various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly. For example, we expect that these rules and regulations may make it more difficult and more expensive for us to obtain director and officer liability insurance.
Pursuant to Section 404, we will be required to furnish a report by our management on our internal control over financial reporting, including an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. However, while we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk we will not be able to conclude within the prescribed timeframe that our internal control over financial reporting is effective as required by Section 404. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
If we are unable to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, shareholders could lose confidence in our financial and other public reporting, which would harm our business
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. In addition, any testing by us conducted in connection with Section 404, or any subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our financial statements or identify other areas for further attention or improvement. Inferior internal controls could also cause you to lose confidence in our reported financial information.
Our management will be required to assess the effectiveness of these controls annually. However, for as long as we are an emerging growth company, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404. We could be an emerging growth company for up to five years. An independent assessment of the effectiveness of our internal controls over financial reporting could detect problems that our management’s assessment might not. Undetected material weaknesses in our internal controls over financial reporting could lead to financial statement restatements and require us to incur the expense of remediation.
23
Table of Contents
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud
Upon completion of this registration, we will become subject to certain reporting requirements of the Exchange Act. Our disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by us in reports we file or submit under the Exchange Act is accumulated and communicated to management, recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements or insufficient disclosures due to error or fraud may occur and not be detected.
We may be unable to use net operating loss and tax credit carryforwards and certain built-in losses to reduce future U.K. tax liabilities
As a U.K. incorporated and tax resident entity, we are subject to U.K. corporate taxation on its tax-adjusted trading profits. Due to the nature of our business, we have generated losses since inception and therefore we have not paid any U.K. corporation tax. Subject to numerous utilization criteria and restrictions (including those that limit the percentage of profits that can be reduced by carried forward losses and those that can restrict the use of carried forward losses where there is a change of ownership of more than half the ordinary shares of the company and a major change in the nature, conduct or scale of the trade), we expect these to be eligible for carry forward and utilization against future U.K. operating profits.
Future changes to tax laws could materially adversely affect our company and reduce net returns to our shareholders
The tax treatment of the company is subject to changes in tax laws, regulations and treaties, or the interpretation thereof, tax policy initiatives and reforms under consideration and the practices of tax authorities in jurisdictions in which we operate, as well as tax policy initiatives and reforms related to the Organisation for Economic Co-Operation and Development, Base Erosion and Profit Shifting Project, the European Commission’s state aid investigations and other initiatives. Such changes may include (but are not limited to) the taxation of operating income, investment income, dividends received or (in the specific context of withholding tax) dividends paid. We are unable to predict what tax reform may be proposed or enacted in the future or what effect such changes would have on our business, but such changes, to the extent they are brought into tax legislation, regulations, policies or practices, could affect our financial position and overall or effective tax rates in the future in countries where we have operations, reduce post-tax returns to our shareholders, and increase the complexity, burden and cost of tax compliance.
Tax authorities may disagree with our positions and conclusions regarding certain tax positions, resulting in unanticipated costs, taxes or non-realization of expected benefits
A tax authority may disagree with tax positions that we have taken, which could result in increased tax liabilities. For example, HM Revenue & Customs (“HMRC”), the Internal Revenue Service or another tax authority could challenge our allocation of income by tax jurisdiction and the amounts paid between certain of our Founded Entities pursuant to our intercompany arrangements and transfer pricing policies, including amounts paid with respect to our intellectual property development. Similarly, a tax authority could assert that we are subject to tax in a jurisdiction where we believe we have not established a taxable connection, often referred to as a “permanent establishment” under international tax treaties, and such an assertion, if successful, could increase our expected tax liability in one or more jurisdictions. A tax authority may take the position that material income tax liabilities, interest and penalties are payable by us, in which case, we expect that we might contest such assessment. Contesting such an assessment may be lengthy and costly and if we were unsuccessful in disputing the assessment, the implications could increase our anticipated effective tax rate, where applicable.
Shareholder protections found in provisions under the U.K. City Code on Takeovers and Mergers (the “Takeover Code”), will not apply if our securities are no longer admitted to trading on a regulated market or a multilateral trading facility in the United Kingdom or on any stock exchange in the Channel Islands or the Isle of Man and our place of management and control is considered to change to outside the United Kingdom
We intend to re-register as a public limited company incorporated in England and Wales in the near future and have our Ordinary Shares admitted to trading on the Main Market of the London Stock Exchange. Accordingly, if and when we proceed with an admission to trading on the Main Market of the London Stock Exchange, we will become subject to the Takeover Code and, as a result, our shareholders are entitled to the benefit of certain takeover offer protections provided under the Takeover Code. The Takeover Code provides a framework within which takeovers of companies are regulated and conducted. If, at the time of a takeover offer, we have de-listed from the Main Market of the London Stock Exchange (and do not maintain a listing of securities on any other regulated market or a multilateral trading facility in the United Kingdom or on any stock exchange in the Channel Islands or the Isle of Man) and the Panel on Takeovers and Mergers determine that we
24
Table of Contents
do not have our place of central management and control in the United Kingdom, then the Takeover Code may not apply to us and our shareholders would not be entitled to the benefit of the various protections that the Takeover Code affords. In particular, we would not be subject to the rules regarding mandatory takeover bids. The following is a brief summary of some of the most important rules of the Takeover Code:
| • | when any person acquires, whether by a series of transactions over a period of time or not, an interest in shares which (taken together with shares already held by that person and an interest in shares held or acquired by persons acting in concert with him or her) carry 30 per cent. or more of the voting rights of a company that is subject to the Takeover Code, that person is generally required to make a mandatory offer to all the holders of any class of equity share capital or other class of transferable securities carrying voting rights in that company to acquire the balance of their interests in the company; |
| • | when any person who, together with persons acting in concert with him or her, is interested in shares representing not less than 30 per cent. but does not hold more than 50 per cent. of the voting rights of a company that is subject to the Takeover Code, and such person, or any person acting in concert with him or her, acquires an additional interest in shares which increases the percentage of shares carrying voting rights in which he or she is interested, then such person is generally required to make a mandatory offer to all the holders of any class of equity share capital or other class of transferable securities carrying voting rights of that company to acquire the balance of their interests in the company; |
| • | a mandatory offer triggered in the circumstances described in the two paragraphs above must be in cash (or be accompanied by a cash alternative) and at not less than the highest price paid within the preceding 12 months to acquire any interest in shares in the company by the person required to make the offer or any person acting in concert with him or her; |
| • | in relation to a voluntary offer (i.e. any offer which is not a mandatory offer), when interests in shares representing 10 per cent. or more of the shares of a class have been acquired for cash by an offeror (i.e., a bidder) and any person acting in concert with it in the offer period and the previous 12 months, the offer must be in cash or include a cash alternative for all shareholders of that class at not less than the highest price paid for any interest in shares of that class by the offeror and by any person acting in concert with it in that period. Further, if an offeror acquires for cash any interest in shares during the offer period, a cash alternative must be made available at not less than the highest price paid for any interest in the shares of that class; |
| • | if the offeror acquires an interest in shares in an offeree company (i.e., a target) at a price higher than the value of the offer, the offer must be increased to not less than the highest price paid for the interest in shares so acquired; |
| • | the offeree company must obtain competent advice as to whether the terms of any offer are fair and reasonable and the substance of such advice must be made known to all the shareholders, together with the opinion of the board of directors of the offeree company; |
| • | special or favorable deals for selected shareholders are not permitted, except in certain circumstances where independent shareholder approval is given and the arrangements are regarded as fair and reasonable in the opinion of the financial adviser to the offeree; |
| • | all shareholders must be given the same information; |
| • | each document published in connection with an offer by or on behalf of the offeror or offeree must state that the directors of the offeror or the offeree, as the case may be, accept responsibility for the information contained therein; |
| • | profit forecasts, quantified financial benefits statements and asset valuations must be made to specified standards and must be reported on by professional advisers; |
| • | misleading, inaccurate or unsubstantiated statements made in documents or to the media must be publicly corrected immediately; |
| • | actions during the course of an offer by the offeree company, which might frustrate the offer are generally prohibited unless shareholders approve these plans. Frustrating actions would include, for example, lengthening the notice period for directors under their service contract or agreeing to sell off material parts of the target group; |
| • | stringent and detailed requirements are laid down for the disclosure of dealings in relevant securities during an offer, including the prompt disclosure of positions and dealing in relevant securities by the parties to an offer and any person who is interested (directly or indirectly) in 1 per cent. or more of any class of relevant securities; and |
25
Table of Contents
| • | employees of both the offeror and the offeree company and the trustees of the offeree company’s pension scheme must be informed about an offer. In addition, the offeree company’s employee representatives and pension scheme trustees have the right to have a separate opinion on the effects of the offer on employment appended to the offeree board of directors’ circular or published on a website. |
The rights of our shareholders may differ from the rights typically offered to shareholders of a U.S. corporation
The Company is incorporated under the laws of England and Wales. The rights of holders of ordinary shares and, therefore, certain of the rights of holders of ADSs, are governed by English law, including the provisions of the Companies Act, and by our Articles of Association. These rights differ in certain respects from the rights of shareholders in typical U.S. corporations. See “Articles of Association—Differences in Corporate Law” in this registration statement for a description of the principal differences between the provisions of the Companies Act applicable to us and, for example, the Delaware General Corporation Law relating to shareholders’ rights and protections.
The principal differences include the following:
| • | under English law and our Articles of Association, each shareholder present at a meeting has only one vote when voting by a show of hands unless demand is made for a vote on a poll, in which case each holder gets one vote per share owned. The Company usually conducts all of its shareholder votes on a poll. Under U.S. law, each shareholder typically is entitled to one vote per share at all meetings; |
| • | under English law, it is only on a poll that the number of shares determines the number of votes a holder may cast. You should be aware, however, that the voting rights of ADSs are also governed by the provisions of a deposit agreement with our depositary bank; |
| • | under English law, subject to certain exceptions and disapplications, each shareholder generally has preemptive rights to subscribe on a proportionate basis to any issuance of ordinary shares or rights to subscribe for, or to convert securities into, ordinary shares for cash. Under U.S. law, shareholders generally do not have preemptive rights unless specifically granted in the certificate of incorporation or otherwise; |
| • | under English law and our Articles of Association, certain matters require the approval of 75 per cent. of the shareholders who vote (in person or by proxy) on the relevant resolution (or on a poll of shareholders representing 75 per cent. of the ordinary shares voting (in person or by proxy)), including amendments to the Articles of Association. This may make it more difficult for us to complete certain corporate transactions deemed advisable by our Board of Directors. Under U.S. law, generally only majority shareholder approval is required to amend the certificate of incorporation or to approve other significant transactions; |
| • | in the U.K., takeovers may be structured as takeover offers or as schemes of arrangement. Under English law, for so long as we continue to be subject to the Takeover Code, a bidder seeking to acquire us by means of a takeover offer would need to make an offer for all of our outstanding ordinary shares/ADSs. If acceptances are not received for 90 per cent. or more of the ordinary shares/ADSs under the offer, under English law, the bidder cannot complete a “squeeze out” to obtain 100 per cent. control of us. Accordingly, acceptances of 90 per cent. of our outstanding ordinary shares/ADSs will likely be a condition in any takeover offer to acquire us, not 50 per cent. as is more common in tender offers for corporations organized under Delaware law. By contrast, a scheme of arrangement, the successful completion of which would result in a bidder obtaining 100 per cent. control of us, requires the approval of a majority of shareholders voting at the meeting and representing 75 per cent. of the ordinary shares voting for approval; |
| • | under English law and our Articles of Association, shareholders and other persons whom we know or have reasonable cause to believe are, or have been, interested in our shares may be required to disclose information regarding their interests in our shares upon our request, and the failure to provide the required information could result in the loss or restriction of rights attaching to the shares, including prohibitions on certain transfers of the shares, withholding of dividends and loss of voting rights. Comparable provisions generally do not exist under U.S. law; and |
| • | the quorum requirement for a shareholders’ meeting is a minimum of two shareholders entitled to vote at the meeting and present in person or by proxy or, in the case of a shareholder which is a corporation, represented by a duly authorized officer. Under U.S. law, a majority of the shares eligible to vote must generally be present (in person or by proxy) at a shareholders’ meeting in order to constitute a quorum. The minimum number of shares required for a quorum can be reduced pursuant to a provision in a company’s certificate of incorporation or bylaws, but typically not below one-third of the shares entitled to vote at the meeting. |
26
Table of Contents
| ITEM 4. | INFORMATION ON THE COMPANY |
A. HISTORY AND DEVELOPMENT OF THE COMPANY
We operate a genomics-based personalized medicine business, particularly focused on breast cancer patients. Our pre-clinical product candidate is StemPrintER, a multi-gene prognostic assay intended for the prediction of the risk of distant recurrence (“DR”) in luminal, ER+/HER2-negative breast cancer patients, based on the detection of 20 stem cell markers normalized to four housekeeping genes. StemPrintER has been evaluated in an initial retrospective validation study using a consecutive cohort of approximately 2,400 patients with breast cancer. In that validation study, StemPrintER outperformed Oncotype DX risk score (“RS”) in 10-year DR risk prediction in all patients, as well as in N0 and N1-3 patients, and outperformed RS in adding prognostic information to the Clinical Treatment Score (“CTS”) over the current market leader Oncotype DX in delivering prognostic information as part of the therapeutic decision-making process in ER+/HER2- breast cancer patients. Our next generation derivative risk model, SPARE, combines StemPrintER with two clinical markers, namely lymph nodal status and tumor size, in a more refined risk model.
We aim to commercialize the StemPrintER and SPARE platform technology. The Company was incorporated in England and Wales on June 5, 2020 as a private company with limited liability under the Companies Act with indefinite life and company number 12647178, we intend to re-register as a public limited company if and when we may apply to have our shares traded on the London Stock Exchange. The Company was created in connection with its Demerger (spin-off) from Tiziana Life Sciences plc, a company admitted to a Standard Listing and to trading on both the Main Market of the London Stock Exchange and Nasdaq.
On October 5, 2020, we entered into a Demerger Agreement with Tiziana. The Demerger was conditional upon, among other things, Court approval of a Tiziana capital reduction, which was approved by special resolution of Tiziana’s shareholders on October 2, 2020. The Court sanctioned the related Tiziana capital reduction on October 27, 2020, and the Demerger became effective on October 30, 2020.
The Demerger Agreement provides for the transfer by Tiziana to us of the entire issued share capital of StemPrintER Sciences Limited, the Tiziana entity to which Tiziana contributed all of the assets and intellectual property relating to the StemPrintER and SPARE projects and £1.0 million in cash. The Demerger consideration will be satisfied by the Company’s allotment and issue of the new Ordinary Shares directly to Tiziana’s shareholders. For the purposes of English company law and U.K. tax principles, Tiziana directed that the consideration shares in the Company that would have been due to it would instead be allotted and issued to Tiziana’s shareholders directly by the Company. Tiziana’s act of “directing” the issue of the Company consideration shares to its shareholders constitutes a distribution (for legal and tax purposes) by Tiziana to its own shareholders (for which it required the sanction of the High Court in England). Tiziana’s making the in-specie distribution of the Company consideration shares to its shareholders implements the final step of the Court-sanctioned demerger or “spin-off”. The Company has allotted the consideration shares to Tiziana’s shareholders (and accordingly, appear in the Company’s share capital); however, completing the actual distribution of those shares (i.e., their physical issuance) to Tiziana’s shareholders cannot be completed until Tiziana’s U.S. shareholders (holding their shares through Tiziana ADSs) can lawfully be issued and receive the Company’s shares in a transaction that complies with the registration requirements of the Securities Act, which is dependent upon this registration statement becoming or being declared effective.
For the purposes of the Demerger, Tiziana first transferred the assets relating to the StemPrintER project and the SPARE project (primarily the benefit of the License from IEO/University of Milan and an outsourced research program) to a separate company, StemPrintER Sciences Limited, together with £1 million in cash. As a result of this step, StemPrintER Sciences Limited became an operating business. In the next step, Tiziana transferred StemPrintER Sciences Limited’s shares to us in return for an allotment of Ordinary Shares to Tiziana’s shareholders, on a one for one basis, and Tiziana declared a dividend in specie to its shareholders of those Ordinary Shares. While the Ordinary Shares were allotted to the Tiziana shareholders on completion of the Demerger, as noted above, no Ordinary Shares or ADSs have actually been issued or distributed. Tiziana shareholders will continue to have an entitlement to our Ordinary Shares until this registration statement becomes or is declared effective, and Tiziana is able to complete the distribution by directing us to issue Ordinary Shares and distribute the Ordinary Shares to Tiziana’s shareholders that are not U.S. persons and, with respect to Tiziana shareholders that are U.S. persons, deposit the corresponding Ordinary Shares with the Depositary so that they receive ADSs representing the Ordinary Shares.
27
Table of Contents
The objective of the Demerger was to maximize value to Tiziana’s shareholders through the further commercialization of the StemPrintER project by transferring Tiziana’s interest in the StemPrintER project assets and intellectual property. The separation of Tiziana’s existing life sciences research and development related to the StemPrintER project into the Company will enable us and Tiziana to separately direct our respective business strategies to maximize value for our respective shareholders and other stakeholders. In addition, the separation of the StemPrintER project into a separate company is intended to enhance the visibility and transparency of our and Tiziana’s respective businesses, provide choice and liquidity for all investors to choose to invest, or not, in the bioscience and diagnostics businesses, and provide clear accountability, together with targeted incentive arrangements, for management and employees. The demerger will allow the Company to focus these commercialization efforts as a separate company with cash reserves of approximately £1,000,000.
Tiziana will continue to provide certain limited management and administrative services to the Company for a 12-month period following the completion of the demerger. Tiziana and the Company have entered into a Demerger Agreement which sets out certain agreements that govern certain aspects of the relationship between Tiziana and the Company and their respective subsidiaries following the Demerger, details of the Demerger Agreement are more fully outlined in “Item 5.A.—Material Contracts”.
Our principal capital expenditures are devoted to conducting research and development of our product candidate, as further discussed in “Item 5.A.—Operating Results”.
The Company’s legal name is AccuStem Sciences Limited. Our registered office is situated at 107 Cheapside, 9th floor, London EC2V 6DN, United Kingdom, and our telephone number is +44 (0) 20 7066 1000. We have one wholly owned subsidiary: StemPrintER Sciences Limited, a private company incorporated in England and Wales with limited liability under the Companies Act. Our website address is www.accustem.com. The reference to our website is an inactive textual reference only and information contained in, or that can be accessed through, our website or any other website cited in this registration statement is not part of hereof.
B. BUSINESS OVERVIEW
Overview and Market Opportunity
Endocrine receptor positive (ER+) breast cancers constitute the majority of breast cancer cases (~75-80%) and display remarkable variability in clinical behavior. This heterogeneity makes prognosis and therapy response often challenging to predict using the standard clinicopathological features of the tumor. Although the overall prognosis for this group of patients is good, a significant proportion (>20%) of these patients will experience distant recurrence in the first 10 years post-surgery. For ER+ patients who also have a negative HER2 status (HER2-), the standard of care is endocrine therapy with the addition of adjuvant chemotherapy in those patients considered to be at risk of recurrence according to clinicopathological parameters.
However, it has become apparent that these parameters are often insufficient to predict risk of recurrence in ER+/HER2- breast cancer patients, and, as a consequence, a significant proportion of these patients are either over- or under-treated. Multigene prognostic tests can assist decision making on treatments, differentiating low risk patients who could be safely be spared chemotherapy from higher risk patients who might benefit from chemotherapy or prolonged endocrine treatments. We anticipate this in vitro prognostic test to be used in conjunction with clinical evaluation to identify those patients at increased risk for early and/or late metastasis.
StemPrintER is designed to help physicians distinguish ER+/HER2- patients. StemPrintER has a novel biological basis in the stem cell biology and interrogates the intrinsic content and aggressiveness of cancer stem cells of the primary tumor. The assay uses a reliable real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR), formalin-fixed, paraffin-embedded (FFPE) platform for testing.
StemPrintER has been initially developed and clinically evaluated in a retrospective validation study using a consecutive cohort of approximately 2,400 patients with breast cancer. Subsequently, StemPrintER has been further evaluated in an independent retrospective cohort of more than 800 ER+/HER2- postmenopausal patients from the TransATAC trial.
Scientific Background
The identification and development of multi-gene assays for accurate prognostication of individual breast cancer patients has represented an expanding area of research for more than a decade. In this context, it has become progressively clear that biomarkers for the prediction of clinical outcome should be able to interrogate the underlying biology of the tumors of individual breast cancer patients. The increasingly recognized relevance of cancer stem cells to breast cancer heterogeneity and disease course suggests that the knowledge of the “degree of stemness” of a breast cancer might substantially advance individualised patient management.
28
Table of Contents
StemPrintER was developed to be a novel genomic predictor based on a cluster of 20 stem cell genes whose high expression levels were capable of identifying a homogeneous group of patients with adverse clinical outcome in the meta-analysis of four distinct public breast cancer datasets.
Based on existing published research, several of the 20 stem cell genes display evident connection to metastatic dissemination through their role in matrix degradation, migration, invasion and engraftment (e.g., MMP1, SNF, MIEN1, PHLDA2, EPB41L5).
For other genes (RACGAP1, H2AFZ, H2AFJ, APOBEC3B, CENPW, TOP2A CDK1), it was considered possible, or even probable, that they were significant in the establishment of cancer stem cells phenotypes and might be linked to involvement in genomic instability, which could be reasonably hypothesized based on their role in processes, such as DNA replication and repair, chromatin remodelling and mitotic control of chromosome segregation.
A final set of genes, whose putative role in metastasis is less obvious, might be linked – directly or indirectly—to the development of adaptive plastic responses required for cancer stem cells to withstand and survive in hostile environmental conditions, such as hypoxia and nutrient deprivation, both at the primary tumor and metastatic site level, and/or to resist hormonal or chemotherapy treatments, often in the broader context of the activation of an epithelial-to-mesenchymal transition (EMT) program. These genes include those involved in: (a) metabolism reprogramming and mitochondrial physiology (MRPS23, NDUFB10, Phb); (b) mRNA ribonucleoparticle bioGENEsis, mRNA transcription, splicing and export, and RNA processing and degradation events (ALYREF, EXOSC4); and (c) survival/escape from apoptosis, which is connected to resistance to hormonal and/or chemotherapy through hijacking of signalling pathways, such as TGF-beta and pi3k-AKT-mTOR (NOL3, LY6E, EIF4EBP1). Additional evidence for a mechanistic link between the 20 genes and the cancer stem cell phenotype comes from the observation that these genes are frequently overexpressed in breast cancer, sometimes as a consequence of gene amplification.
Whilst further studies may be needed to establish whether the genes comprised in the signature are causal in the determination and/or maintenance of cancer stem cells in breast cancers, and possibly of their metastatic potential, the ability of the 20 stem cell gene signature to predict distant metastasis in breast cancer patients also support the proposition that the metastatic potential of individual breast cancers can be traced back to the molecular characteristics of a rare subpopulation of tumor cells that display cancer stem cell traits.
Summary of Clinical Research History
Through validation studies in a large prospective-retrospective cohort of breast cancer patients with high-quality follow-up, and functional prospective studies based on the use of fresh tumor samples from an additional consecutive series of breast cancer patients, it has been established that the 20-gene SC-based assay predicts the individual likelihood to develop distant metastasis in breast cancer, in particular in triple negative breast cancer and luminal ER+/HER2- breast cancer cancers and does so, most likely, by interrogating the number and biological characteristics of their cancer stem cells. Of note, our genomic predictor comprises a set of genes that do not belong (with one exception) to any other genomic tool or molecular classifier described for triple negative breast cancer and luminal breast cancers.
We accordingly believe that the result of our research is the development of a unique tool capable of probing into the “degree of stemness”, and hence into the clinical outcome, of breast cancers.
Our initial research focused on genes discriminating mammary stem cells from progenitors in the normal gland. Only those genes that were expressed at higher levels in mammary stem cells versus progenitors were selected. Selection criteria was based on the premise that cancer stem cells might display traits reminiscent of those present in normal mammary stem cells and, since cancer stem cells are rare, the selection of overexpressed genes (mammary stem cells versus progenitors) afforded a higher likelihood of scoring differences, with respect to under-expressed genes.
Accordingly, these findings have important potential implications from both the biological and clinical perspective.
29
Table of Contents
Clinical and Commercial Viability
From a clinical standpoint, although future studies in independent retrospective and/or prospective breast cancer patient cohorts are warranted to increase the level of clinical evidence of the reliability and transportability of the 20 stem cell gene genomic tool, in light of the recent independent validation study in the TransATAC cohort of the Royal Marsden Hospital and Queen Mary University in London, the results of our research have immediate relevance to the clinical management of breast cancer patients, in particular for the subgroup of ER+/HER2- breast cancer patients. These patients represent the majority (~75%) of the cases and display high molecular heterogeneity and variability in their clinical behaviour. Accordingly, ER+/HER2- breast cancer patients can greatly benefit from accurate stratification of their risk of recurrence, for the administration of the optimal therapy, while avoiding under- or over-treatment.
Based on the 20 stem cell -gene signature, StemPrintER is a specific risk model for ER+/HER2- breast cancer patients. StemPrintER is an independent predictor of both early and late metastasis. This places StemPrintER among the more recently developed second generation multi-gene assays for breast cancer, such as Prosigna and EndoPredict.
In particular, StemPrintER predicts early metastasis in lymph node-negative breast cancer patients, and both early and late metastasis in lymph node-positive breast cancer patients. Accordingly, we consider that StemPrintER could have immediate clinical application as a tool to tailor the administration of adjuvant chemotherapy, in addition to the standard endocrine therapy, in those ER+/HER2- breast cancer patients at high risk of early recurrence, while sparing unnecessary chemotherapy to low risk patients.
In addition, StemPrintER could also represent a valuable tool to identify ER+/HER2- breast cancer patients at high risk of late recurrence, who might benefit from prolongation of endocrine therapy beyond the standard 5 years of treatment. This is an important issue in the clinical management of ER+/HER2- breast cancer patients, who remain at persistent risk of recurrence for at least 15–20 years, with >50% of relapses and more than two-thirds of deaths occurring >5 years after the original diagnosis. However, while continuation of endocrine therapy reduces the proclivity to develop late recurrences, its benefits must be weighed against side effects and quality of life, avoiding overtreatment through accurate patient stratification.
Research Results
A team of scientists from IEO/University of Milan conducted an independent validation of StemPrintER and its derivative, SPARE, in the TransATAC cohort of ER+/HER2- postmenopausal breast cancer patients, in collaboration with the Royal Marsden Hospital and Queen Mary University in London, alongside a head-to-head comparison of StemPrintER with Oncotype DX in these patients.
The likelihood c2 (LRc2) and Kaplan-Meier survival analyses were used to assess prognostic information provided by StemPrintER, Oncotype DX RS and the CTS. Comparative analyses were made for DR risk over the 10-year follow-up, as well as in the early (0-5 years) or late (5-10 years) interval, according to nodal status. Our study results showed that StemPrintER outperformed Oncotype DX RS in 10-year DR risk prediction in all patients, as well as in N0 and N1-3 patients and is superior to RS in adding prognostic information to CTS. See the results in the diagram below:
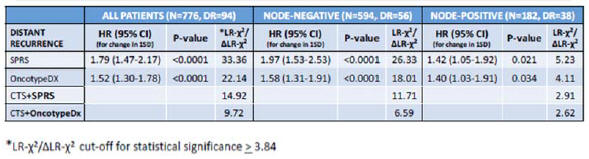
The results also showed that StemPrintER significantly stratifies patients at high versus low recurrence risk among low and intermediate, but not high, pre-specified RS groups.
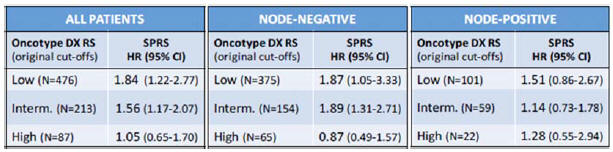
Commercialization
We intend to continue our collaboration strategy to further develop, validate and commercialize the StemPrintER/SPARE project. We intend to conduct further validation and utility studies with the intention of filing for regulatory review under the CLIA system and, ultimately, for reimbursement review. We also expect to pursue a strategy to achieve appropriate regulatory review with European and Asian regulatory agencies to expand the addressable market for its products.
30
Table of Contents
Regulatory Process
For diagnostics in the U.S., all laboratories performing clinical testing must achieve compliance with federal regulations under CLIA. These regulations are administered at the state level through the individual state departments of health where states may add further regulation alongside the federal. There are two states, New York and Washington, where the state level regulations are considered at least or more rigorous than the federal, and in those two states, the state level credentialing is allowed to show compliance at the federal level exempting from the need to achieve direct CLIA certification.
Companies which create a test and offer the diagnostic product as a service (returning results to individuals/clinicians) as a LDT under the CLIA process should obtain clearance from the FDA for that LDT. Performing the LDT in a CLIA compliant laboratory without first clearing the test though FDA is more simple and faster but does necessitate both the laboratory and the product being certificated and require specialist staff and approval processes.
CLIA requires that laboratories demonstrate or verify the analytical validity of all tests they perform. A clinical laboratory analyzing specimens based on a proprietary test method (i.e., an LDT) must, among other things, document the accuracy, precision, specificity, sensitivity of, and establish a reference range for, such test.
Revenue Strategy
Our revenue is expected to be derived from different sources including standard private third-party and government medical insurance coverage and reimbursement models. Prior to full commercial scaling, we expect to focus our first revenues from a small number of early adopting sites, where we may also subsidize the full cost of the test to encourage use and create further validation data. This is expected to be within 24 months of Admission, subject to successful validation trials and approvals under the CLIA certification or upon the obtaining of a CE mark for the test.
Competition
The current market leader in the field is the Oncotype DX assay, a 21-gene assay that predicts the likelihood of chemotherapy benefit and 10-year risk of distant recurrence to inform adjuvant treatment decisions in certain women with early-stage invasive breast cancer. Oncotype DX is owned by Exact Sciences Corp., a leading provider of cancer screening and diagnostic tests.
Whilst Exact Sciences Corp. is market leader, as noted above, a team of scientists from IEO/University of Milan conducted a head-to-head comparison of StemPrintERwith Oncotype DX in collaboration with the Royal Marsden Hospital and Queen Mary University in London. The results of the study and the performance of StemPrintER are more fully described in “Research Results” above.
EndoPredict is a CE-marked assay that is designed to predict the likelihood of metastases developing within 10 years of an initial breast cancer diagnosis. The test is for pre- and post-menopausal people with early breast cancer with ER+, HER2- and LN- or LN-positive disease (up to 3 positive nodes).
MammaPrint is a CE-marked assay that is designed to assess the risk of distant recurrence within 5 and 10 years and whether a person would benefit from chemotherapy. The test is for pre- and post-menopausal people with stage 1 or 2 breast cancer, with a tumor size of 5 centimeters or less, and LN-negative or LN-positive disease (up to 3 positive nodes). The test can be used irrespective of ER and HER2 status.
Prosigna is a CE-marked assay designed to provide information on breast cancer subtype and to predict distant recurrence-free survival at 10 years. The test is for postmenopausal people with early breast cancer that is ER+/HER2- and LN-negative or LN-positive (up to 3 positive nodes).
As was noted by the U.K. National Institute for Health and Care Excellence (NICE) in its review of available prognostic testing, there are few available studies of the market leaders on a direct comparison basis focused on head-to-head utility.
Government Regulation
EU and U.K. regulatory overview
The following provides an overview of key aspects of medical device regulation within the EU and U.K. It should be noted this overview does not address every facet of regulation but only those that would generally be most relevant to the activities discussed in this registration statement.
31
Table of Contents
We operate and intend to operate in a highly regulated industry that is subject to a changing political, economic and regulatory landscape across many countries. Our products will be subject to national and supra-national EU laws and regulations. Our products will also be subject to U.K. laws and regulations.
Current EU and U.K. regulatory framework
Software applications (whether stand-alone or components of a larger system) qualify as medical devices or medical device accessories under EU rules if they meet the relevant definition under the EU Medical Device Directive 93/42/EEC (the “Medical Device Directive”) or they may be classified as in vitro medical 38 devices and governed by the In Vitro Diagnostic Medical Device Directive 98/79/EC (the “IVD Directive”) as the regulatory route to bear CE marking.
Which directive is applicable to our products will depend on a number of factors, including whether or not our software interprets data derived from human tissue or blood samples, or data which itself has been derived from an in vitro medical device. Given that the data our software may analyze may come from multiple sources and may change over time, we have set out below an overview of both the Medical Device Directive and the IVD Directive. Included in discussion below is the upcoming change to meet EU Medical Device Regulation (EU) 2017/745) (the “MDR”) and EU In Vitro Diagnostic Regulation (EU) 2017/746 (the “IVDR”).
As published on September 1, 2020 guidance document, the MHRA will continue to recognize CE marks and certificates issued by EEA-based notified bodies until June 30, 2023 and then medical devices and IVDs distributed in Great Britain will be required to bear the new U.K.CA mark from July 1, 2023. Commercialization of products in Northern Ireland will continue to require a CE Mark as achieved through the MDD or IVDD regime (or MDR by May 26, 2021 and IVDR by May 26, 2022).
Due to the delay with MDR and IVDR taking effect after Brexit, IVDR and MDR will not automatically apply in Great Britain. It is anticipated that additional changes to requirements for commercializing medical devices and IVDs in Great Britain may be forthcoming as suggested in MHRA’s guidance document. Until then, under U.K. law, Medical Devices Regulations 2002 (“U.K. MDR 2002”, and as amended by 2019 No. 791), as transposed from MDD and IVDD into U.K. law will continue to apply post-Brexit.
The Medical Device Directive
Under the Medical Device Directive, a software medical device may be placed on the EU market only if it conforms with the “essential requirements” set out in Annex I to the Medical Device Directive. To assist manufacturers in satisfying the essential requirements of the Medical Device Directive, the European standards organizations have prepared European standards applicable to medical devices. These include harmonized international quality standards (including International Organization for Standardization Standards and International Electrotechnical Commission standards) aimed at ensuring that medical devices are correctly designed and manufactured. While not mandatory, compliance with these standards entitles the manufacturer to a presumption of conformity with the essential requirement that is covered by the standards concerned.
The manufacturer is obliged to demonstrate that the device conforms to the relevant essential requirements through a conformity assessment procedure. For Class I non-sterile and non-measuring medical devices, the manufacturer is responsible for performing the conformity assessment procedure. For Class II and III devices, as well as the sterility/measuring aspects of Class I devices, the manufacturer declares conformity with the essential requirements, but this must be backed up with a conformity assessment by a notified body resulting in a CE certificate. Depending on the conformity assessment route agreed with the notified body, separate certificates may be issued for the device and the underlying quality assurance system against harmonized standard EN ISO 13485.
EU government regulatory bodies are not involved in the pre-market approval of medical devices. The onus of ensuring a device is safe enough to be placed on the market is ultimately the responsibility of the manufacturer and, where relevant, the notified body. Notified bodies are entities licensed by the individual member states to provide independent certification of certain classes of medical device. They apply for and are designated to carry out this function by the relevant national competent authorities, which carry out periodic assessment audits to determine whether the notified bodies continue to satisfy the requirements set out in the Medical Device Directive and the guidance developed by the Notified Body Oversight Group. A notified body must possess the resources (e.g., facilities and staff) for the conformity assessment of medical devices for which it is designated and must conduct such assessments in a competent, transparent, independent and impartial manner.
32
Table of Contents
Once the appropriate conformity assessment procedure for a medical device has been completed, the manufacturer must draw up a written declaration of conformity and affix the CE mark to the device. The device can then be marketed throughout the EEA. Notified bodies perform surveillance and unannounced audits at the manufacturer and critical suppliers with respect to the devices covered by the certificates issued by them. If non-conformities raised during the audits are not timely remedied by the manufacturer, the notified body may (partially or wholly) suspend or withdraw the certificate concerned.
Manufacturers of medical devices are subject to post-market requirements, notably device vigilance and safety reporting obligations. EU member states are responsible for enforcing the EU’s medical device rules and for ensuring that only compliant medical devices are placed on the market or put into service in their jurisdictions. They have powers to suspend the marketing and use, or demand the recall, of unsafe or noncompliant devices. They also have the power to bring enforcement action against companies or individuals for breaches of the device rules, but this is extremely rare absent a public health risk. Non-compliance may also result in notified bodies revoking any certificate of conformity that they have issued for a device or the manufacturer’s quality system.
The IVD Directive
The EU regulates in vitro medical devices (“IVDs”) as a specific category of medical devices with particular differences, which means they are regulated under a separate regime. The Company’s software application is envisaged to process input data from EHRs and from ‘wet’ samples and is envisaged to be installed as a local software layer in clinical institutions that will communicate with the cloud. The EU Guidance document “Qualification and Classification of standalone software” provides that stand alone software fulfilling the definition of medical device and intended to be used for the purpose of providing information derived from in vitro examination of a specimen derived from the human body falls under the IVD Directive. The software may therefore qualify as ‘expert function software’ for the purposes of the applicable guidance, in which case it would therefore be regulated under the IVD Directive.
The IVD Directive sets out certain “essential requirements” set out in Annex I of the IVD Directive and with which IVDs must comply before being placed on the market in the EU. Not all the essential requirements will apply to all devices and it is up to the manufacturer of the device to assess which are appropriate for that particular product. As for the Medical Device Directive, one way in which manufacturers can demonstrate that they have met the essential requirements is to comply with the relevant national standards that transpose harmonized standards.
There are four categories of IVDs, reflecting the perceived risk. Annex II of the IVD Directive sets out specific device types that are categorized as either high risk (List A) or moderate risk (List B). There are also self-test IVDs, which are those devices intended by the manufacturer to be able to be used by lay persons in a home environment, and then the final category covers all IVDs which are not classified as List A, List B or Self-test IVDs, known as general IVDs. As for other medical devices, pre-market approval is delegated to notified bodies. For List A, List B and Self-Test IVDs this means that the manufacturer must gain independent certification by a notified body in order to complete the conformity route process, apply for CE marking and be able to place the device on the European market.
Manufacturers of IVDs are subject to post-market requirements, including setting up an on-going systematic process to review experience gained for their device on the market and to have a vigilance procedure to immediately inform relevant competent authorities. Each competent authority has the right to remove a device that they believe is unsafe from their national market.
The EU Medical Device Regulation
In May 2017, the European Commission finalized and adopted the text of the Medical Device Regulation, which will repeal and replace the Medical Device Directive. Due to COVID-19 pandemic, implementation of the Medical Device Regulation will take effect May 26, 2021. The Company will need to ensure compliance with the Medical Device Regulation in the future if it is to place software that is a medical device on the EU market.
The Medical Device Regulation contains a new classification rule specific to software in Annex VIII (rule 11). All software intended to provide information used to make diagnostic or therapeutic decisions will be in Class IIa, except if the decisions may cause death or an irreversible deterioration in health, in which case it will be in Class III. Where decisions could result in a serious deterioration in a person’s state of health or a surgical intervention, they will be in Class IIb. Software intended to monitor physiological processes will be in Class IIa, except if it is intended to monitor vital physiological parameters and variations in those parameters could result in immediate danger to the patient, in which case it is classified as Class IIb. All other software will be in Class I. Software that currently qualifies as a Class I device under the Medical Device Directive may therefore need to be reclassified as Class IIa or higher once the Medical Device Regulation becomes applicable. This will require a notified body conformity assessment in accordance with the requirements of the Medical Device Regulation.
33
Table of Contents
The Medical Device Regulation will require significantly more clinical data for CE marking than is currently required by the Medical Device Directive. It also promulgates new design requirements for software and will not grandfather any previous CE mark under the Medical Device Directive. We will need to obtain timely CE marking under the new regulation.
The EU In Vitro Diagnostic Medical Device Regulation
The EU In Vitro Diagnostic Medical Device Regulation (“IVDR”) was adopted in May 2017 and will repeal the existing In Vitro Diagnostic Medical Devices Directive (98/79/EC). The majority of the provisions of the IVDR apply from May 26, 2022 and will harmonize the law on in vitro medical devices across the EU. We will need to ensure compliance with the IVDR in the future if it is to place software that is a medical device which is used as an in vitro diagnostic on the EU market after this regulation comes into force. The IVDR will require significantly more clinical data for CE marking than is currently required by the IVD Directive. It also promulgates new design requirements for software and will not grandfather any previous CE mark under the IVD Directive.
As a result, should the IVDR apply to our products, we will need to obtain timely CE marking under the new regulation.
The IVDR contains a new classification regime for all IVDs, including software that qualifies as an IVD, in Annex VIII. The new classification regime groups all IVDs in four risk classes A, B, C, and D, of which only risk class A remains subject to self-declaration for CE marking. Because the software is intended to work with blood-based biomarkers it is likely that, if it is regulated by the IVDR, it will be classified in the highest risk class (D) and will be subject to notified body conformity assessment. Depending on the intended use and inherent risks of the devices, it may alternatively be classified in risk Class C per Rule 2 or Rule 3 as defined in Annex VIII of the IVDR. Class C devices per IVDR are also subject to notified body conformity assessment.
With implementation of IVDR and MDR, Medical Device Coordination Group (MDCG) have established a set of guidelines. The MDCG guidelines are specific to IVDR and MDR and therefore replaces prior MEDEV guidelines that are specific to IVDD and MDD.
U.K. MDR 2002 (as amended by 2019 No.791, MDR 2019)
At this time, essential requirements and conformity assessments follow similarly to those described per MDD and IVDD with specific modifications as transposed into U.K. MDR 2002 and their respective amendments. Process by which requirements for conformity assessments as specified in U.K. MDR 2002 (and amendments) are supplemented by guidance issued by the MHRA. With respect to software as the medical device defined by U.K. MDR 2002, similar definitions to MDD and IVDD applied. Guidance to medical device stand-alone software including apps and in vitro diagnostic medical device provides an interim guide until such time as the MHRA publishes a new set of guidance.
Other Healthcare Standards
Development, clinical evaluation and marketing of digital health software products are subject to significant global regulation by governments and global regulatory agencies. Many approvals require clinical evaluation data relating to safety, quality and efficacy of a product. Many countries, including the U.S. (510(k) clearance), Europe (CE marking), China (CFDA), and Japan (PMDA) have high standards of technical appraisal and have a risk of delays in the approval process.
Data Protection Legislation
On May 25, 2018, the GDPR came into effect in the European Union which regulation continued to apply in the U.K. for the Brexit transition period with effect from January 31, 2020 to December 31, 2020 alongside, the Data Protection Act 2018 (“DPA 2018”) which is and will, post Brexit, be the domestic law governing the processing of personal data in the U.K.. The obligations in the DPA 2018 are for all practical purposes the same as those set out in the GDPR. The GDPR imposes an enhanced data protection regulatory regime with potentially significant sanctions for non-compliance. To the extent we process personal data that is subject to the GDPR, in the future, we will need to comply with the GDPR and the DPA 2018, and any other applicable laws with respect to the handling of personal data respectively for EU and U.K.. U.K. and European hospitals which may, in the future, provide patient data to the us for the us to analyze using its artificial intelligence technology will also be responsible for ensuring that there is a legal basis on which they may collect the data and anonymize it effectively so that it can be transferred to us as anonymous data to which GDPR and the DPA 2018 do not apply (because once anonymized effectively it will no long be personal data). In vitro diagnostic kit development will include, at a minimum, compliance to ISO 13485:2016 – Medical devices – Quality management systems – Requirements for regulatory purposes, 21CFR820 (U.S. Quality Systems Regulations) and other product design and manufacturing standards as identified to support our commercial plan.
34
Table of Contents
U.S. health regulatory overview
The following provides an overview of key aspects of laboratory service and medical device regulation within the U.S. It should be noted this overview does not address every facet of regulation at the federal and state level, but only those that would generally be most relevant to the activities described in this registration statement.
Federal and state clinical laboratory licensing requirements
The CLIA, governs the operations of all clinical laboratories operating in or returning results to individuals in the U.S. CLIA is administered by CMS, in partnership with state health departments. A clinical laboratory is defined as a laboratory that performs testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease, or the assessment of health. Clinical laboratories must hold a certificate applicable to the type of laboratory examinations they perform and must demonstrate compliance with regulations addressing, among other things, personnel qualification and training, record keeping, quality control, and proficiency testing, all of which are intended to ensure the timeliness, reliability, and accuracy of clinical laboratory testing services. CLIA requires that laboratories demonstrate or verify the analytical validity of all tests they perform. Where a clinical laboratory analyses specimens based on a proprietary test method (i.e., an LDT), the laboratory must, among other things, document the accuracy, precision, specificity, sensitivity of, and establish a reference range for, such test.
CMS provides for exemption from CLIA for states that develop clinical laboratory standards that are at least as stringent as federal requirements. Both New York and Washington State are exempt from CLIA. The NYS Clinical Laboratory Evaluation Program requires all independent clinical laboratories operating in, or testing specimens from, NYS to obtain a laboratory permit prior to commencing operations, and all clinical laboratories performing LDTs to submit test validation documentation demonstrating the tests’ analytical and clinical validity.
Failure to comply with CLIA certification and state clinical laboratory licensure requirements may result in a range of enforcement actions, including certificate or license suspension, limitation, or revocation, directed plan of action, onsite monitoring, civil monetary penalties, criminal sanctions, and revocation of the laboratory’s approval to receive Medicare and Medicaid payment for its services, as well as significant adverse publicity.
FDA
The FDA regulates, among other medical products, “medical devices” which include certain articles intended for use in the diagnosis, prevention, cure, mitigation, or treatment of disease or intended to effect the structure or function of the body. Whether a product is intended for use as a medical device is generally determined, in the first instance, based on the manufacturer’s product labelling, which includes the label affixed to the product, materials distributed with the product, and promotional communications concerning the product.
Devices classified as Class I (low risk), generally may be marketed without FDA pre-market review, but are subject to “general controls”, including establishment registration, device listing, record keeping, medical device reporting, and quality system regulations, including design controls. Devices classified as Class II (moderate risk), may, in addition to general controls, also be subject to “special controls” (e.g., performance standards / manufacturing standards, post-market surveillance, patient registries, special labelling requirements, pre-market data requirements and guidelines), and also generally must obtain 510(k) premarket clearance or DeNovo authorization from FDA. Class III (high risk) devices must, in addition to general controls, obtain FDA pre-market approval through the submission of a pre- market approval application that contains evidence, including data from adequate and well- controlled clinical studies, demonstrating that the device is safe and effective for its intended use. In general, devices that require FDA pre-market clearance or DeNovo authorization may not be commercially distributed or promoted prior to obtaining such authorization, although they may be distributed and used for the purpose of developing the clinical data necessary to support FDA marketing applications, subject to certain limitations. Post-market changes to a cleared / authorized or approved device also may be subject to prior review by FDA, depending on the scope of the change and its potential impact on device safety and effectiveness.
It should also be emphasized that this pre-market review process is only one facet of FDA’s regulation. For example, FDA regulates product labelling, including promotional claims; the manufacturing of medical devices, including their design, under FDA quality system requirements; clinical trials with new or modified products; and post-market monitoring for, reporting of, and action related to, safety concerns. Failure to comply with applicable pre- and post-market device
35
Table of Contents
requirements can result in a determination by FDA that a device is “adulterated” (Section 501) or “misbranded” (Section 502) in violation of the U.S. Federal Food, Drug, and Cosmetics Act. The statute provides for a number of penalties, including seizure, injunction, criminal, and civil monetary penalties, for the sale or distribution of adulterated or misbranded devices. In general, prior to undertaking enforcement action, FDA will notify a regulated entity of a violation or suspected 36 violation through a communication, such as a “Warning Letter” or “Untitled Letter”. If FDA identifies violations during inspection of a manufacturer’s facility, the agency will issue a Form 483 listing the identified violation and directing the manufacturer to make the necessary corrections.
FDA regulation of software
Commercially distributed software applications that meet the definition of a medical device may be subject to FDA pre-market authorization, depending on their classification and software function. These include both applications that are components of a hardware medical device and certain “stand-alone” software. In 2017, FDA issued final guidance adopting international principles established by the International Medical Device Regulators Forum for the clinical evaluation of software as a medical device (“SaMD”), which refers to software that is intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device. In 2019, FDA issued a guidance that provides guidance on FDA’s oversight of device software functions including mobile medical apps that meet the definition of a device. While the guidance is not binding on either FDA or regulated industry, FDA intends to consider the principles in developing regulatory approaches for SaMD as well as for digital health technologies.
FDA regulation of LDTs
FDA regulates a category of medical devices, called in vitro diagnostic medical devices, or IVDs, that are used in the collection, preparation, and examination of specimens from the human body. IVDs include reagents, instruments, and systems that are intended for use in diagnosis of disease or other conditions, including the state of health, in order to cure, mitigate, treat, or prevention disease or its sequelae. FDA historically has taken the position that tests developed in-house by a clinical laboratory and used to analyze patient specimens meet the definition of an IVD and fall within the agency’s regulatory jurisdiction. At the same time, FDA historically has for the most part exercised “enforcement discretion,” i.e., has not required clinical laboratories performing LDTs to comply with IVD device requirements. In the past, FDA has signaled intent to modify its enforcement discretion policy with regard to LDT regulation, and in 2014 proposed a regulatory framework for LDTs, which it abandoned before implementation in 2016. As of August 19, 2020, the U.S. Department of Health and Human Services (HHS) determined that LDTs will not require a premarket review with FDA, but rather an applicant may voluntarily submit a premarket notification or premarket approval (or an Emergency Use Authorization in the case of COVID-19 tests) for their LDT. It is possible that Congress will enact legislation directing FDA to regulate LDTs.
The U.S. Federal Trade Commission and Consumer Protection Laws
Within the U.S., the U.S. Federal Trade Commission (“FTC”), has authority to regulate advertising for most medical devices and for laboratory services. In addition, various state consumer protection laws exist which can similarly regulate claims that are being made by entities with respect to what benefits their products or services can provide to consumers. In some instances, FTC or U.S. states have taken action with respect to medical products based on claims being made with respect to, e.g., their benefits to patients, seeking various penalties, such as injunctions and substantial fines. Activities have focused more, to date, on products that are sold directly to consumers, such as dietary supplements, as opposed to prescription products ordered by physicians, although the possibility exists that FTC or other consumer protection bodies could take steps to regulate claims with respect to IVDs or LDTs.
Fraud and Abuse
The significant U.S. fraud and abuse laws include the:
| • | Anti-Kickback Statute: the federal U.S. Anti-Kickback Statute (42 U.S. Code § 1320a–7b(b) imposes criminal penalties on persons and entities for, among other things, knowingly and willfully soliciting, offering, receiving or providing remuneration (including any kickback, bribe or rebate), directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, lease or order of a good, facility, item or service for which payment may be made under a government healthcare program such as Medicare and Medicaid. |
| • | False Claims Act: the U.S. federal false claims and civil monetary penalties laws, including the federal civil U.S. False Claims Act (31 USC. §§ 3729 – 3733), impose criminal and civil penalties, including through civil whistleblower or qui tam actions against individuals or entities for, among other things knowingly presenting or |
36
Table of Contents
causing to be presented false or fraudulent claims for payment by a federal healthcare program or making a false statement or record material to payment of a false claim or avoiding, decreasing, or concealing an obligation to pay money to the federal government, with potential liability including mandatory treble damages, significant per-claim penalties, and administrative penalties. |
Transparency requirements
The U.S. Physician Payments Sunshine Act (known as Affordable Care Act Section 6002: Transparency Reports and Reporting of Physician Ownership or Investment Interests) requires certain manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, with specific exceptions, to report annually to the CMS information related to payments or transfers of value made to physicians and teaching hospitals, as well as information regarding ownership and investment interests held by physicians and their immediate family members. Any failure to report or providing incomplete or misleading information may subject the Company to penalties. Analogous state laws. Analogous state fraud and abuse laws and regulations, such as U.S. state antikickback and false claims laws, can apply to sales or marketing arrangements, and claims involving healthcare items or services reimbursed by governmental or non-governmental third-party payors. These laws are generally broad and are enforced by many different U.S. federal and state agencies as well as through private actions. Some state laws require adherence to compliance guidelines promulgated by the U.S. federal government and require device and drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures.
Data privacy and security
HIPAA
The HIPAA imposes criminal and civil liability for, among other things, failing to protect the privacy of patient and security of patient data. Additionally, the HIPAA by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, also imposes obligations on covered entities and their business associates that perform certain functions or activities that involve the use or disclosure of protected health information on their behalf, including mandatory contractual terms as well as implementing reasonable and appropriate administrative, physical and technical safeguards with respect to maintaining the privacy, security and transmission of protected health information.
FTC
The FTC has taken an active role with regard to protection of personal information, relying on its broad consumer protection powers to seek substantial penalties where companies that have made deceptive or misleading statements regarding practices of collecting and safeguarding data or did not have adequate safeguards to protect information consistent with their claims regarding data security.
State laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not pre-empted by HIPAA, thus complicating compliance efforts.
37
Table of Contents
Intellectual Property
Our intellectual property portfolio comprises the following patents:
Family | Subject | Priority | Status | Expires | Jurisdiction | |||||
StemPrintER | Methods and kits comprising gene signature for stratifying breast cancer patients | 2017 | Pending | 2037 | United States, Canada | |||||
StemPrintER | Methods and kits comprising gene signature for stratifying breast cancer patients | 2017 | Allowed | 2037 | Europe | |||||
StemPrintER | Methods and kits for determining the risk of breast cancer recurrence | 2020 | Pending | 2041 | Europe |
We have rights to a patent family that discloses methods and kits for stratifying risk in breast cancer patients, licensed from IEO/University of Milan. This patent family includes pending applications in Canada and the United States and an allowed application in Europe. Patents issued in family will expire in June 2037, excluding any patent term extensions available in several jurisdictions.
We have rights to a second patent family that discloses methods and kits for determining the risk of breast cancer recurrence, licensed from IEO/University of Milan. This patent family includes one pending application in Europe. Patents issued in this family will expire in May 2041, excluding any patent term extensions available in several jurisdictions.
We are not aware of any third-party claims or contested proceedings in relation to our intellectual property portfolio.
Legal Proceedings
We are not party to any material legal matters or claims. In the future, we may become party to legal matters and claims arising in the ordinary course of business, the resolution of which we do not anticipate would have a material adverse impact on our financial position, results of operations or cash flows.
C. ORGANIZATIONAL STRUCTURE
We have one wholly owned subsidiary: StemPrintER Sciences Limited, a private company incorporated in England and Wales with limited liability under the Companies Act.
D. PROPERTY, PLANTS AND EQUIPMENT
We do not own any property but sublease approximately 75 square feet of office space in London from Tiziana for which we reimburse Tiziana at cost. We believe that our existing facilities are adequate to meet our current needs for the foreseeable future, and that suitable additional alternative spaces will be available in the future on commercially reasonable terms if needed.
| ITEM 4A. | UNRESOLVED STAFF COMMENTS |
Not Applicable.
| ITEM 5. | OPERATING AND FINANCIAL REVIEW AND PROSPECTS |
You should read the following discussion and analysis together with “Selected Financial Data” and our consolidated financial statements, including the notes thereto, included elsewhere in this registration statement. Some of the information contained in this discussion and analysis or set forth elsewhere in this registration statement, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. As a result of many factors, including those factors set forth in the “Risk Factors” section of this registration statement, our actual results could differ materially from the results described in or implied by these forward-looking statements. Our audited consolidated financial statements as of and for the year ended December 31, 2020 have been prepared in accordance with IFRS.
38
Table of Contents
A. OPERATING RESULTS
Overview
We operate a genomics-based personalized medicine business, particularly focused on breast cancer patients. Our pre-clinical product candidate is StemPrintER, a multi-gene prognostic assay intended for the prediction of the risk of distant recurrence (“DR”) in luminal, ER+/HER2-negative breast cancer patients, based on the detection of 20 stem cell markers normalized to four housekeeping genes. StemPrintER has been evaluated in an initial retrospective validation study using a consecutive cohort of approximately 2,400 patients with breast cancer. In that validation study, StemPrintER outperformed Oncotype DX risk score (“RS”) in 10-year DR risk prediction in all patients, as well as in N0 and N1-3 patients, and outperformed RS in adding prognostic information to the Clinical Treatment Score (“CTS”) over the current market leader Oncotype DX in delivering prognostic information as part of the therapeutic decision-making process in ER+/HER2- breast cancer patients. Our next generation derivative risk model, SPARE, combines StemPrintER with two clinical markers, namely lymph nodal status and tumor size, in a more refined risk model.
StemPrintER Sciences Limited was transferred to the Company on October 30, 2020 pursuant to the Demerger of the StemPrintER and SPARE projects from Tiziana. The objective of the Demerger was to maximize value to the shareholders of Tiziana through the further commercialization of the StemPrintER project and its assets and intellectual property. The Demerger will allow us to continue its collaboration strategy to further develop, validate and commercialize the StemPrintER/SPARE platform as a separate listed company with cash reserves of approximately £1.095 million.
Since our inception, we have devoted substantially all of our resources to conducting research and development of our product candidate. Our revenue is expected to be derived from different sources including standard private third-party and government medical insurance coverage and reimbursement models. To date, Tiziana, the former parent of our subsidiary StemPrintER Sciences Limited, raised a total of £89.9 million from external funding sources, such as major investment funds and other leading investors. On completion of the Demerger, Tiziana transferred £1.0 million in cash to StemPrintER Sciences Limited and the entire issued share capital of StemPrintER Sciences Limited to the Company and has agreed to invest a further £2.0 million in our equity securities if and when we list on the London Stock Exchange. As of December 31, 2020, we had cash, cash equivalents and short-term investments of £1,151,383.
We expect our expenses to increase substantially in connection with our ongoing development activities related to our preclinical and clinical programs. We intend to conduct further validation and utility studies with the intention of filing for regulatory review under the CLIA system and, ultimately, for reimbursement review. We also expect to pursue a strategy to achieve appropriate regulatory review with European and Asian regulatory agencies to expand the addressable market for its products. In addition, we plan to build out our management team and infrastructure.
In addition, upon the effectiveness of this registration statement, we expect to incur additional costs associated with operating as a public company in the United States. We expect that our expenses and capital requirements will increase substantially in the near to mid-term as we:
| • | continue our research and development efforts; |
| • | seek regulatory approvals for any product candidates that successfully complete clinical trials; |
| • | add clinical, scientific, operational financial and management information systems and personnel, including personnel to support our product development and potential future commercialization claims; and |
| • | operate as a U.S. public company. |
As a result, we may need substantial additional funding to support our continuing operations and pursue our growth strategy. Until such time as we can generate significant revenue from product sales, if ever, we expect to finance our operations through a combination of public or private equity or debt financings or other sources, which may include collaborations with third parties. We may be unable to raise additional funds or enter into such other agreements or arrangements when needed on favorable terms, or at all. If we fail to raise capital or enter into such agreements as, and when needed, we may have to significantly delay, scale back or discontinue the development and commercialization of our product candidates.
Material Contracts
The following are all of the contracts (not being contracts entered into in the ordinary course of business) that have been entered into by us (including our wholly-owned subsidiary StemPrintER Sciences Limited) since our incorporation which: (i) are, or may be, material to us (or our wholly-owned subsidiary StemPrintER Sciences Limited); or (ii) contain obligations or entitlements which are, or may be, material to us (or our wholly-owned subsidiary StemPrintER Sciences Limited) as at the date of this registration statement.
39
Table of Contents
Demerger Agreement
On October 5, 2020, we entered into an agreement with Tiziana pursuant to which Tiziana declared a dividend in specie on its ordinary shares equal to the book value (of approximately £3.07 million) of Tiziana’s shareholding in StemPrintER Sciences Limited, the entity within the Tiziana Group which held all of the assets and intellectual property relating to the StemPrintER and SPARE projects (primarily the benefit of the License from IEO/University of Milan and an outsourced research program) and £1.0 million in cash.
We determined the fair value of Tiziana’s intellectual property contribution following principles and guidance in IFRS 13 “Fair value measurement”. Depending on the type of transaction, fair value is normally determined on a market, income or cost approach. In this instance, as no market value could be determined as no market transactions involving identical or comparable assets were identified, and no revenue streams were as yet associated with the StemPrintER project and its intellectual property, the Company applied a cost approach by considering the total allocated expenses of the project when held by Tiziana. This expenditure consisted of patent maintenance fees, Contract Research Organisation (“CRO”) costs and other StemPrintER project specific costs. It did not include any apportionment of general R&D expenses, because Tiziana allocated all costs to projects specifically. In the absence of a comparable market transaction for the diagnostic tool, the valuation of the asset was also based on Tiziana’s expenditure from 2014 to the disposition date in September 2020.
The dividend in specie was satisfied by the transfer by Tiziana to us of the shares in StemPrintER Sciences Limited. In return for this transfer, we allotted Ordinary Shares to Tiziana shareholders who were registered on the Tiziana Share Register at the Demerger Record Time (being 7:00 a.m. on October 30, 2020), on the basis of one Ordinary Share for each Tiziana ordinary share held by them at that time, save that the number of Ordinary Shares allotted to our initial subscriber (a Tiziana shareholder) was reduced by the number of Ordinary Shares already held by it so that, on the Demerger becoming effective, each Tiziana Shareholder (including our initial subscriber) held one Ordinary Share for each Tiziana ordinary share held at the Demerger Record Time.
The Demerger was conditional, among other things, upon Court approval of a capital reduction which was approved by special resolution of shareholders on October 2, 2020. The Court sanctioned the related capital reduction on October 27, 2020; the Demerger became effective on October 30, 2020; and we allotted 194,612,288 Ordinary Shares to the Tiziana shareholders.
Supplemental Demerger Agreement
On October 30, 2020, we entered into a supplementary agreement to the Demerger Agreement with Tiziana, pursuant to which we allotted and agreed to issue 9,581,254 additional Ordinary Shares at a price of £0.01 per share to reflect various rights which existed in Tiziana prior to the Demerger, and Tiziana agreed to subscribe for £2.0 million in additional Ordinary Shares upon completion of the London listing.
Asset Transfer Agreement
On October 5, 2020, StemPrintER Sciences Limited entered into an agreement with Tiziana pursuant to which Tiziana transferred all of the StemPrintER project intellectual property and other business assets, together with £1 million in cash to StemPrintER Sciences Limited in exchange for the issue by StemPrintER Sciences Limited of 3,070,000 Ordinary Shares, each credited as fully paid.
License
On June 24, 2014, Tiziana entered into an exclusive license agreement with IEO/University of Milan, pursuant to which it obtained a worldwide, royalty-bearing, exclusive license under certain patents and a worldwide, royalty-bearing, non-exclusive license under certain know-how, respectively, of IEO/University of Milan to develop and commercialize licensed products in connection with a multi-gene prognostic tool. This license was assigned to us pursuant to the terms of the Asset Transfer Agreement.
40
Table of Contents
Under the License, we have full control and authority over the research, development and commercialization of licensed products and are required to use commercially reasonable efforts in connection with the development and commercialization of the licensed products. Following completion of the research plan, which is still in process, we must meet various diligence requirements.
For the term of the License, we are required to make certain milestone payments:
| • | €50,000 within 30 days of completion of development of a commercial test; |
| • | €100,000 within 30 days of the first commercial sale of a licensed product; and |
| • | €150,000 within 30 days of first regulatory approval in the U.S. or any other major market. |
Tiziana was also required, as licensee prior to the assignment to us of the License, to fund €50,000 per year for sponsored research for up to four years from the effective date of the License, subject to certain conditions. We are also required to pay all ongoing patent prosecution and maintenance costs and for the royalty term (until the expiration of the last claim in an issued, unexpired patent within the licensed patents or a claim that has not been pending more than four years which covers the sale of such licensed product or service in such country) a royalty of 1.5% on net sales of licensed products and services (and a 15% royalty of sub-license revenues for each country for the term of the License).
We may terminate the License at any time on 30 days’ notice and either party may terminate the License by written notice for a material payment breach or any other material breach, subject to 45-day and 120-day cure periods, respectively. Absent early termination, the License will remain in effect, on a product by product and country by country basis, until the date on which the patents and patent applications expire. The License may also be terminated in the case of insolvency.
If we fail to meet our obligations under the License or if the License is terminated for any reason, it could negatively impact our business and strategic goals.
Results of Operations
We derived the selected financial data as of and for the period from incorporation on June 5, 2020 to December 31, 2020 from our audited financial statements included elsewhere in this registration statement. We present our financial statements in accordance with IFRS. We have not prepared ‘carve-out’ predecessor financial statements related to the StemPrintER business for the two-year period prior to its transfer to us, because we do not consider that these historical cost amounts would be useful information to a user of our financial statements and would require us to incur unnecessary cost and third party expense. As discussed below, Tiziana incurred minimal historical research and development costs in the two-year period before the StemPrintER project intellectual property was formally transferred to StemPrintER Sciences Limited in 2020, and prior to it transfer, the StemPrintER project essentially in ‘maintenance’ mode, and was not operated as a stand-alone business, asset or project.
The summary financial data below should be read together with those financial statements. Our historical results for any prior period are not necessarily indicative of results to be expected in any future period, and our interim period results are not necessarily indicative of results to be expected for a full year or any other interim period.
Consolidated Income Statement Data
| December 31, 2020 | ||||
Revenue | £ | — | ||
Research and development expenses | (4,347 | ) | ||
Administrative expenses | (36,342 | ) | ||
Operating result | (40,689 | ) | ||
Finance income/(expense) | — | |||
Profit before taxation | (40,689 | ) | ||
Income tax | — | |||
Profit for the period and total comprehensive income for the period | (40,689 | ) | ||
Basic and diluted earnings per Ordinary Share (pence) | — | |||
41
Table of Contents
Components of Our Results of Operations
Revenue
We have not yet generated any revenue from product sales, and we do not expect to generate any revenue from product sales for the foreseeable future.
Operating Expenses
Research and Development Expenses
Research and development expenses consist primarily of costs incurred for our research activities, including our discovery efforts, and the development of our product candidates, which include:
| • | employee-related expenses, including salaries, related benefits and equity-based compensation; |
| • | expenses incurred in connection with the preclinical and clinical development of our product candidates, including our agreements with contract research organizations, or CROs; |
| • | expenses incurred under agreements with consultants who supplement our internal capabilities; |
| • | the cost of lab supplies and acquiring, developing and manufacturing preclinical study materials and clinical trial materials; |
| • | costs related to compliance with regulatory requirements; and |
| • | facilities, depreciation and other expenses, which include direct and allocated expenses for rent and maintenance of facilities, insurance and other operating costs. |
We expense all research costs in the periods in which they are incurred and development costs are capitalized only if certain criteria are met. For the periods presented, we have not capitalized any development costs since we have not met the necessary criteria required for capitalization. Costs for certain development activities are recognized based on an evaluation of the progress to completion of specific tasks using information and data provided to us by our vendors and third-party service providers.
The table below summarizes our research and development expenses incurred by project:
Direct Research and Development Expenses Incurred by Project
| Period from June 5, 2020 to December 31, 2020 | ||||
StemPrintER | £ | 4,347 | ||
SPARE | — | |||
Total direct research and development expense | 4,347 | |||
Indirect research and development expense | — | |||
Total research and development expense | 4,347 | |||
Tiziana incurred historical research and development costs before the intellectual property was formally transferred to StemPrintER Sciences Limited in 2020. Tiziana’s expenditure in respect of the StemPrintER project for the nine month period ended September 30, 2020 was $55,000, for the year ended December 31, 2019 was $9,000; for the year ended December 31, 2018 was $166,000; for the year ended December 31, 2017 was $1,201,000, and for the year ended December 31, 2016 was $529,000.
In the two years prior to the Demerger, Tiziana’s expenditure on the StemPrintER project was limited and primarily related to maintenance and expenditure to retain rights to the intellectual property. Tiziana did not attribute any significant time or resources to the StemPrintER project during 2019 or 2020.
Since the transfer of the StemPrintER project to StemPrintER Sciences Limited and then to us, we now have the cash resources, business plans and people with the specific skills and knowledge to move the project forward in 2021. Specifically, we continue to maintain the StemPrintER patents, and we are engaged in discussions with a CRO to finalize an agreement and budget for the commercial development of the asset.
42
Table of Contents
Research and development activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We expect that our research and development expenses will continue to increase for the foreseeable future in connection with our planned preclinical and clinical development activities in the near term and in the future. The successful development of our product candidates is highly uncertain. As such, at this time, we cannot reasonably estimate or know the nature, timing and estimated costs of the efforts that will be necessary to complete the remainder of the development of these product candidates. We are also unable to predict when, if ever, material net cash inflows will commence from our product candidates. This is due to the numerous risks and uncertainties associated with developing products.
Comparison of Results of Operations
Not applicable.
Accounting Policies
Our consolidated financial statements are prepared in accordance with IFRS. The preparation of our consolidated financial statements and related disclosures requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, costs and expenses, and the disclosure of contingent assets and liabilities in our consolidated financial statements. We base our estimates on historical experience, known trends and events and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. We evaluate our estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are described in more detail in our consolidated financial statements, we believe that the following accounting policies are those most critical to the judgments and estimates used in the preparation of our consolidated financial statements.
Demerger Transaction Accounting
Tiziana’s capitalization of the Company and creation of the separate StemPrintER business by way of a demerger under English law happened in two distinct steps. In the first step, in September 2020, Tiziana transferred all ownership rights and intellectual property relating to the StemPrintER project, in the form of patents and a license, to a Tiziana subsidiary, StemPrintER Sciences Limited. In the second step, on October 5, 2020, Tiziana transferred StemPrintER Sciences Limited to the Company.
In the first step, the transfer of all the ownership rights and intellectual property was treated as an asset transfer. The treatment as a separate asset acquisition at this stage reflected the fact that, immediately prior to transfer, Tiziana carried out only limited maintenance type activity on the StemPrintER project and the concentration of fair value was in the StemPrintER intellectual property asset. Tiziana tracked the expenses incurred in maintaining the StemPrintER project in the form of patent maintenance fees, CRO fees and project consultancy fees, the total amounts for which between 2014 and the transfer date were £2.07 million. Tiziana used the aggregate amount of these expenses as its determination of fair value to credit an account in the books of StemPrintER Sciences Limited by £2.07m. Tiziana received 3,070,000 shares in StemPrintER Sciences Limited as consideration for the asset transfer. Tiziana also contributed capital and resources, consisting of £1.0 million in cash.
In the second step of the transaction, on October 5, 2020, we entered into an agreement with Tiziana to acquire StemPrintER Sciences Limited, including the ownership rights and intellectual property relating to the StemPrintER project, the SPARE project and cash of £1.0 million. In exchange for the transfer of ownership (shares in StemPrintER Sciences Limited), the Company allotted shares to Tiziana shareholders on a one for one basis based on Tiziana’s ownership on October 30, 2020 (i.e., the Demerger). At this step, Tiziana was deemed to have undertaken a business combination under common control, because Tiziana’s management considered StemPrintER Sciences Limited a “business” under IFRS, as opposed to a “moth-balled” concentrated asset. As a business, Tiziana’s management considered StemPrintER Sciences Limited to have inputs and the ability to contribute to the creation of outputs, using an outsourced pharmaceutical research service organization (a CRO) to develop the asset, which is market practice once there is sufficient financial resource.
43
Table of Contents
Recent Accounting Pronouncements
For information on recent accounting pronouncements, see our consolidated financial statements and the related notes found elsewhere in this registration statement.
Internal Control over Financial Reporting
We are a private company which intends to seek a listing on the London Stock Exchange with limited requirements to implement and test internal controls under a U.K. framework. As such, we have not been subject to the internal control over financial reporting requirements of the Sarbanes-Oxley Act of 2002 and the standards of the PCAOB and our independent registered public accounting firm has not conducted an audit of our internal control over financial reporting in accordance with such rules. As a U.S. public company, Section 404 of the Sarbanes-Oxley Act will require that our management assess our internal control over financial reporting and include a report of management on our internal control over financial reporting in our annual report on Form 20-F beginning with our second annual report. Although we will adhere to all internal control requirements made relevant by the governance of the LSE, the requirements pertaining to the design and implementation of internal controls over financial reporting as contemplated under the Sarbanes-Oxley Act had not been considered in the production of financial statements included elsewhere in this registration statement.
B. LIQUIDITY AND CAPITAL RESOURCES
Sources of Liquidity
Since our inception, we have not generated any revenue and have incurred significant operating losses. Our potential products are at various phases of preclinical and clinical development. We do not expect to generate significant revenue from product sales for several years, if at all. To date, Tiziana, our former parent, raised a total of £89.9 million from external funding sources, such as major investment funds and other leading investors. Pursuant to the Demerger, Tiziana committed to transfer £1.0 million in cash to StemPrintER Sciences Limited and the entire issued share capital of StemPrintER Sciences Limited to the Company. For a description of our structure see “Item 4.B.—Business Overview”.
Our cash flows may fluctuate and are difficult to forecast and will depend on many factors. As of February 28, 2021, our cash receivable balance was approximately £1.09m.
Cash Flows
The following table summarizes our cash flows for from the date of incorporation on June 5, 2020 to December 31, 2020:
Period Ended £ | ||||
Cash flows from operating activities | ||||
Loss before income tax | (40,689 | ) | ||
Related party and other payables | 40, 689 | |||
Net cash from operating activities | — | |||
Net increase in cash and cash equivalents | — | |||
Cash and cash equivalents at beginning of period | — | |||
Cash and cash equivalents at end of period | — | |||
Operating Activities
Related party payables were due to Tiziana of £9.892 for expenses it had paid on our behalf. Other trade payables of £30,797 consisted of the audit fee due for this period.
Investing Activities
There was no net cash used in or provided by investing activities for the period to December 31, 2020.
Financing Activities
There was no net cash received in financing investing activities for the period to December 31, 2020. There is a cash receivable balance due from Tiziana of £1,151,383 for shares issued but not yet paid.
44
Table of Contents
Material Capital Expenditure Commitments
We have no material commitment for capital expenditures.
Funding Requirements
We expect that our expenses will increase and operating losses will be generated, and we have no retained earnings as at December 31, 2020. Based on our current plans, we believe our existing cash and cash equivalents will be sufficient to fund our operations and capital expenditure requirements into 2022. We expect to incur substantial additional expenditures in the near term to support our ongoing activities. Additionally, if we elect to proceed, we expect to incur additional costs as a result of operating as a public company whose Ordinary Shares are admitted to a Standard Listing on the Official List and to trading on the Main Market of the London Stock Exchange. We expect to incur net losses for the foreseeable future. Our ability to fund our product development and clinical operations as well as commercialization of our product candidates, will depend on the amount and timing of cash received from planned financings. Our future capital requirements will depend on many factors, including:
| • | the costs, timing and outcomes of clinical trials and regulatory reviews associated with our product candidates; |
| • | the costs of commercialization activities, including product marketing, sales and distribution; |
| • | the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property-related claims; |
| • | the emergence of competing technologies and products and other adverse marketing developments; |
| • | the effect on our product development activities of actions taken by the FDA, EMA or other regulatory authorities; |
| • | our degree of success in commercializing our product candidates, if and when approved; and |
| • | the number and types of future products we develop and commercialize. |
A change in the outcome of any of these or other variables with respect to the development of any of our product candidates could significantly change the costs and timing associated with the development of that product candidate. Further, our operating plans may change in the future, and we may need additional funds to meet operational needs and capital requirements associated with such operating plans.
Until such time, if ever, as we can generate substantial product revenue, we expect to finance our operations through a combination of equity financings, debt financings, collaborations with other companies or other strategic transactions. We do not currently have any committed external source of funds. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making acquisitions or capital expenditures or declaring dividends. If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings or other arrangements when needed, we may be required to delay, limit, reduce or terminate our research, product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Further, our operating plans may change, and we may need additional funds to meet operational needs and capital requirements for clinical trials and other research and development activities. We currently have no credit facility or committed sources of capital. Because of the numerous risks and uncertainties associated with the development and commercialization of our product candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures associated with our current and anticipated product development programs.
Quantitative and Qualitative Disclosures about Financial Risks
Interest Rate Sensitivity
As at December 31, 2020, we had cash and cash equivalents of £1,151,383. Our exposure to interest rate sensitivity is the interest received on the cash held which is immaterial.
45
Table of Contents
Foreign Currency Exchange Risk
As all monetary assets and liabilities and all our transactions are denominated in our functional currency, we are not exposed to any foreign currency risk.
We do not currently engage in currency hedging activities in order to reduce our currency exposure, but we may begin to do so in the future. Instruments that may be used to hedge future risks include foreign currency forward and swap contracts. These instruments may be used to selectively manage risks, but there can be no assurance that we will be fully protected against material foreign currency fluctuations.
JOBS Act Exemptions and Foreign Private Issuer Status
We qualify as an “emerging growth company” as defined in the U.S. Jumpstart Our Business Startups Act of 2012. An emerging growth company may take advantage of specified reduced reporting and other requirements that are otherwise applicable generally to public companies. This includes an exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting pursuant to the Sarbanes-Oxley Act. We may take advantage of this exemption for up to five years or such earlier time that we are no longer an emerging growth company. We will cease to be an emerging growth company if we have more than $1.07 billion in total annual gross revenue, have more than $700.0 million in market value of our ordinary shares held by non-affiliates or issue more than $1.0 billion of non-convertible debt over a three-year period. We may choose to take advantage of some but not all of these provisions that allow for reduced reporting and other requirements.
We are considering whether we will take advantage of the extended transition period provided under Section 7(a)(2)(B) of the Securities Act, for complying with new or revised accounting standards. Since IFRS makes no distinction between public and private companies for purposes of compliance with new or revised accounting standards, the requirements for our compliance as a private company and as a public company are the same.
C. RESEARCH AND DEVELOPMENT, PATENTS AND LICENSES
For a discussion of our research and development activities, see “Item 4.B.—Business Overview” and “Item 5.A.—Operating Results.”
D. TREND INFORMATION
For a discussion of trends and uncertainties relating to our business, see “Item 5.A.—Operating Results.”
E. OFF-BALANCE SHEET ARRANGEMENTS
We have no other off-balance sheet arrangements that have had, or are reasonably likely to have, a material current or future effect on our consolidated financial statements or changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources.
F. TABULAR DISCLOSURE OF CONTRACTUAL OBLIGATIONS
Contractual Obligations and Commitments
The following table summarizes our contractual commitments and obligations as of as of December 31, 2020:
| Payments Due By Period | ||||||||||
| Total | Less Than 1 Year | 1-3 Years | 3-5 Years | More Than 5 Years | ||||||
| None | ||||||||||
We have no current payment obligations under a license agreement with IEO/University of Milan. Under this agreement we are required to make milestone payments upon successful completion and achievement of certain sales milestones. The payment obligations under the License are contingent upon future events such as our achievement of specified commercial milestones, and we will be required to make royalty payments in connection with the sale of products developed under these agreements. As the achievement and timing of these future milestone payments are not probable or estimable, such amounts have not been included in our unaudited condensed consolidated financial statements or in the contractual obligations table above. For additional information regarding our License, see “Item 5.A.—Material Contracts” above.
46
Table of Contents
G. SAFE HARBOR
This registration statement contains forward-looking statements within the meaning of Section 27A of the Securities Act and Section 21E of the Exchange Act and as defined in the Private Securities Litigation Reform Act of 1995. See “Special Note Regarding Forward-Looking Statements” included elsewhere in this registration statement.
| ITEM 6. | DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES |
A. DIRECTORS AND SENIOR MANAGEMENT
For information about our Directors and senior management, see “Item 1.A.—Identity of Directors, Senior Management and Advisers – Directors and Senior Management.”
B. COMPENSATION
Executive Compensation
For the period from June 5, 2020 to December 31, 2020, we did not pay any cash compensation or benefits, such as pension, retirement or similar benefits, to our executive officer.
Outstanding Equity Awards
We currently have no outstanding option awards although we anticipate that certain option awards will be made in conjunction with the intended listing of our Ordinary Shares on the London Stock Exchange.
Employment Arrangements
We have not entered into any employment agreements.
Consultancy Agreements
Keeren Shah
We entered into a consultancy agreement with Ms. Shah on March 1, 2021 to provide finance director services. Compensation under the consultancy agreement becomes payable from the date on which our Ordinary Shares are listed on the London Stock Exchange. This agreement entitles Ms. Shah to receive a base fee of £10,000 per annum. Ms. Shah may also be eligible to receive a bonus in an amount to be determined in our sole discretion. Ms. Shah is not entitled to any fringe benefits. If Ms. Shah’s consultancy with the Company is terminated without cause, Ms. Shah will be entitled to a payment in lieu of notice to the equal to her basic salary for all or any remaining part of the relevant period of notice. The payment in lieu shall consist solely of her base fee for the relevant period and shall exclude any other entitlements or benefits.
Ms. Shah is also subject to a 6-month non-competition and non-solicitation covenant.
Share Option and Other Compensation Plans
The Company currently has no share option or other compensation plans.
Annual Bonus
The Company currently has no annual bonus program.
Retirement Plan
The Company currently has no retirement plans.
Limitations on Liability and Indemnification
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our Directors, officers or controlling persons, we have been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
47
Table of Contents
NON-EMPLOYEE DIRECTOR COMPENSATION
The following table sets forth information regarding compensation earned by our non-employee Directors during the period from June 5, 2020 to December 31, 2020. We have agreed that, from the date on which our Ordinary Shares are admitted to a Standard Listing and to trading on the Main Market of the London Stock Exchange, our Chairman, Gabriele Cerrone, will receive a fee of £50,000 per annum and our Non-Executive Directors will receive fees of £20,000 per annum. We also intend to reimburse non-employee members of our Board of Directors for reasonable travel expenses with effect from the date on which are Ordinary Shares are admitted to a Standard Listing and to trading on the Main Market of the London Stock Exchange. The compensation of our Finance Director is discussed above in “Executive Compensation.”
NAME | FEES EARNED OR PAID IN CASH(£) | TOTAL(£) | ||||||
Dr. Kunwar Shailubhai | — | — | ||||||
Gabriele Cerrone | — | — | ||||||
Willy Simon | — | — | ||||||
John Brancaccio | — | — | ||||||
Non-Employee Director Compensation Policy
We do not currently pay our Directors. With effect from the date on which our Ordinary Shares are admitted to a Standard Listing and to trading on the Main Market of the London Stock Exchange, we intend to pay our Directors in accordance with the following non-employee Director compensation policy. Such amounts are subject to annual adjustment, as determined by the Remuneration Committee. Each non-employee Director will be paid a fee per annum of £20,000 for their services on our Board of Directors with any additional advisory services provided to be agreed on a case by case basis.
C. BOARD PRACTICES
Board Composition
Our Board of Directors currently consists of four members, the Chairman and three Non-Executive Directors. Each Directors’ term expires at the next annual general meeting of shareholders in 2021.
As a foreign private issuer that is not listed on any stock market, we are not required to have independent directors on our Board of Directors, except that our audit committee is required to consist fully of independent Directors, subject to certain phase-in schedules.
Corporate Governance and Committees of the Board
Corporate Governance
The Directors are responsible for carrying out our objectives, implementing our business strategy and conducting our overall supervision. The Board will provide leadership within a framework of prudent and effective controls. The Board will establish our corporate governance values and will have overall responsibility for setting our strategic aims, defining the business plan and strategy and managing our financial and operational resources. The Board will schedule quarterly meetings and hold additional meetings as and when required. The expectation is that this will not result in more than four meetings of the Board each year.
While we are not under an obligation to adopt a governance code on a ‘comply or explain’ basis given our intention to admit our Ordinary Shares to a Standard Listing and to trading on the Main Market of the London Stock Exchange, the Directors have opted to observe the requirements of the U.K. Corporate Governance Code to the extent they consider appropriate in light of the Company’s size, stage of development and resources. The Directors will take into account the Corporate Governance Guidelines for Smaller Quoted Companies published by the Quoted Companies Alliance so far as it is practicable and appropriate.
We will hold Board meetings periodically as issues arise which require the Board’s attention. The Board will be responsible for the management of our business, setting our strategic direction, establishing our policies and appraising the making of all material investments. It will be the Board’s responsibility to oversee our financial position and monitor our business and affairs on behalf of shareholders, to whom the Directors are accountable. The primary duty of the Board will be to act in the best interests of the Company at all times. The Board will also address issues relating to internal control and our approach to risk management.
48
Table of Contents
Other Corporate Governance Matters
The Sarbanes-Oxley Act of 2002, as well as related rules subsequently implemented by the SEC, requires foreign private issuers, including our company, to comply with various corporate governance practices. We intend to take all actions necessary for us to maintain compliance as a foreign private issuer under the applicable corporate governance requirements of the Sarbanes-Oxley Act of 2002 and the rules adopted by the SEC.
Because we are a foreign private issuer, our Directors and senior management are not subject to short-swing profit and insider trading reporting obligations under Section 16 of the Exchange Act. They will, however, be subject to the obligations to report changes in share ownership under Section 13 of the Exchange Act and related SEC rules.
Committees of the Board
Our Board of Directors has established an audit committee, a nomination committee and a remuneration committee. Each of these committees operates under formally delegated duties and responsibilities.
Audit Committee
The audit committee of the Board comprises John Brancaccio and Willy Simon. It is chaired by John Brancaccio, and is responsible for:
| • | monitoring the quality of internal controls and ensuring our financial performance is properly measured and reported on; |
| • | consideration of the Directors’ risk assessment and suggesting items for discussion at the full Board; |
| • | receipt and review of reports from our management and auditors relating to the interim and annual accounts, including a review of accounting policies, accounting treatment and disclosures in the financial reports; |
| • | consideration of the accounting and internal control systems in use throughout the Company and its subsidiaries; and |
| • | overseeing our relationship with external auditors, including making recommendations to the Board as to the appointment or re-appointment of the external auditors, reviewing their terms of engagement, and monitoring the external auditors’ independence, objectivity and effectiveness. |
The Audit Committee meets not less than twice in each financial year and has unrestricted access to our auditors.
Our Board of Directors has determined that John Brancaccio is an “audit committee financial expert” as defined in Item 16.A. of Form 20-F.
In order to satisfy the independence criteria for audit committee members set forth in Rule 10A-3 under the Exchange Act, each member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the Board of Directors, or any other board committee, accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries or otherwise be an affiliated person of the listed company or any of its subsidiaries. We believe that the composition of our audit committee will meet the requirements for independence under current SEC rules and regulations.
Remuneration Committee
The remuneration committee of the Board comprises Willy Simon and John Brancaccio. It is chaired by Willy Simon, and is responsible for:
| • | the review of the performance of the Executive Directors; |
| • | recommendations to the Board on matters relating to the remuneration and terms of service of the Executive Directors; and |
| • | recommendations to the Board on proposals for the granting of share options and other equity incentives pursuant to any share option scheme or equity incentive scheme in operation from time to time. |
49
Table of Contents
In making their recommendations, the Remuneration Committee will have due regard to our shareholders’ interests and our performance.
In order to satisfy the independence criteria for Remuneration Committee members set forth in Rule 10C-1 under the Exchange Act, all factors specifically relevant to determining whether a director has a relationship to such company which is material to that director’s ability to be independent from management in connection with the duties of a remuneration committee member must be considered, including, but not limited to: (1) the source of compensation of the director, including any consulting advisory or other compensatory fee paid by such company to the director; and (2) whether the director is affiliated with the company or any of its subsidiaries or affiliates. We believe the composition of our remuneration committee will meet the requirements for independence under current SEC rules and regulations.
Nomination Committee
The Nomination Committee of the Board comprises Gabriele Cerrone and Willy Simon. It is chaired by Gabriele Cerrone, and is responsible for:
| • | drawing up selection criteria and appointment procedures for Directors; |
| • | recommending nominees for election to our Board of Directors and its corresponding committees; |
| • | assessing the functioning of individual members of our Board of Directors and executive officers and reporting the results of such assessment to our Board of Directors; and |
| • | developing corporate governance guidelines. |
None of the members of our compensation committee is, or has ever been, an officer or employee of our company.
D. EMPLOYEES
As of December 31, 2020, we had no employees.
E. SHARE OWNERSHIP
For information regarding the share ownership of our Directors and management, see “Item 6.B.—Compensation” and “Item 7.A.—Major Shareholders”.
| ITEM 7. | MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS |
A. MAJOR SHAREHOLDERS
The following table sets forth information with respect to the beneficial ownership of our Ordinary Shares as of December 31, 2020by:
| • | each of our Directors; |
| • | each of our executive officers; and |
| • | each person, or group of affiliated persons, who is known by us to beneficially own more than 5 per cent. of our outstanding Ordinary Shares. |
| Number of Ordinary Shares Beneficially Owned(1) | ||||||||
Name and address of beneficial owner | Shares | % | ||||||
Planwise Group Limited (2) | 65,886,784 | 32.27 | % | |||||
Executive Officers and Directors (3) | ||||||||
Gabriele Cerrone (4) | 75,806,147 | 37.12 | % | |||||
Dr. Kunwar Shailubhai | * | |||||||
Willy Simon | * | |||||||
50
Table of Contents
| Number of Ordinary Shares Beneficially Owned(1) | ||||||
Name and address of beneficial owner | Shares | % | ||||
Keeren Shah | * | |||||
* Represents beneficial ownership of less than 1 per cent. of our outstanding ordinary shares. |
| (1) | “Percentage of Shares Beneficially Owned” is based on 204,193,543 Ordinary Shares allotted as at December 31, 2020, as for UK companies law and tax purposes, allotted shares are part of a company’s share capital. |
| (2) | Gabriele Cerrone, Chairman, is the beneficial owner of the entire issued share capital of Planwise Group Limited. Planwise Group Limited is incorporated in the British Virgin Islands with a registered address at Vistra Corporate Services Centre, Wickhams Cay II, Road Town, Tortola, VG1110, British Virgin Islands. Planwise is the registered owner of our initial subscriber share and will remain so for Companies Act purposes until the Demerger is completed; however, Planwise has neither voting power not disposal authority for the subscriber share. |
| (3) | The address of our executive officers and Directors is 107 Cheapside, 9th Floor, London EC2V 6DN, United Kingdom. |
| (4) | This includes shares held by Mr. Cerrone personally and shares held by Planwise Group Limited and Panetta Partners Limited (being entities in which Mr. Cerrone is considered to have a beneficial interest). |
Beneficial ownership is determined in accordance with the rules and regulations of the SEC and includes voting or investment power with respect to our ordinary shares. Ordinary shares subject to options that are currently exercisable or exercisable within 60 days after December 31, 2020 are considered outstanding and beneficially owned by the person holding the options for the purpose of calculating the percentage ownership of that person but not for the purpose of calculating the percentage ownership of any other person. Except as otherwise noted, the persons and entities in this table have sole voting and investment power with respect to all of the ordinary shares beneficially owned by them, subject to community property laws, where applicable. The information in the table below is based on information known to us or ascertained by us from public filings made by the shareholders. We have also set forth below information known to us regarding any significant change in the percentage ownership of our ordinary shares by any major shareholders during the past three years. The major shareholders listed below do not have voting rights with respect to their Ordinary Shares that are different from the voting rights of other holders of our Ordinary Shares.
To our knowledge there has been no significant change in the percentage ownership held by the major shareholders listed above in the last three years, except as described in “Item 7.B.—Related Party Transactions” included in this registration statement.
We are not aware that the Company is directly owned or controlled by another corporation, any foreign government or any other natural or legal person(s) severally or jointly. We are not aware of any arrangement, the operation of which may result in a Change of Control of the Company.
B. RELATED PARTY TRANSACTIONS
The following is a description of related party transactions we have entered into with the beneficial owners of 10 per cent. or more of our ordinary shares, which are our only voting securities, senior management and members of our Board of Directors, since June 5, 2020.
Agreements with Our Executive Officers and Directors
We have entered into an employment agreement with our Finance Director and Director compensation agreements with our Non-Executive Directors. See “Item 6.B.—Compensation” These agreements contain customary provisions and representations, including confidentiality, non-competition and non-solicitation undertakings by the executive officer. However, the enforceability of the non-competition provisions may be limited under applicable law.
Demerger Agreement
We entered into a Demerger Agreement with Tiziana on October 5, 2020 pursuant to which Tiziana declared a dividend in specie on its ordinary shares equal to the book value (of approximately £3.07 million) of Tiziana’s shareholding in StemPrintER Sciences Limited, the entity within the Tiziana Group which holds all of the assets and intellectual property relating to the StemPrintER and SPARE projects (primarily the benefit of the License from IEO/University of Milan and an outsources research program) and £1.0 million in cash.
51
Table of Contents
The dividend in specie was satisfied by the transfer by Tiziana to us of the shares in StemPrintER Sciences Limited. In return for this transfer, we allotted Ordinary Shares to the Tiziana shareholders who were registered on the Tiziana Share Register at the Demerger Record Time (being 7:00 a.m. on October 30, 2020), on the basis of one Ordinary Share for each Tiziana ordinary share held by them at that time, save that the number of Ordinary Shares allotted to our initial subscriber (a Tiziana shareholder) was reduced by the number of Ordinary Shares already held by it so that, on the Demerger becoming effective, each Tiziana shareholder (including our initial subscriber) held one Ordinary Share for each Tiziana ordinary share held at the Demerger Record Time.
The Demerger was conditional, among other things, upon Court approval of a capital reduction which was approved by special resolution of shareholders on October 2, 2020. The Court sanctioned the related capital reduction on October 27, 2020; the Demerger became effective on October 30, 2020; at which time we allotted 194,612,288 Ordinary Shares to the Tiziana shareholders.
Supplemental Demerger Agreement
On October 30, 2020, we entered into a supplementary agreement to the Demerger Agreement with Tiziana, pursuant to which we allotted and agreed to issue 9,581,254 additional Ordinary Shares at a price of £0.01 per share to reflect various rights which existed in Tiziana prior to the Demerger, and Tiziana agreed to subscribe for £2.0 million in additional Ordinary Shares upon completion of the London listing.
Family Relationships
None.
C. INTERESTS OF EXPERTS AND COUNSEL
Not applicable.
| ITEM 8. | FINANCIAL INFORMATION |
A. CONSOLIDATED STATEMENTS AND OTHER FINANCIAL INFORMATION
Consolidated Financial Statements
Our audited consolidated financial statements for the fiscal year December 31, 2020 are included in Item 18 of this registration statement.
Legal proceedings
From time to time, we may become involved in legal, governmental or arbitration proceedings or be subject to claims arising in the ordinary course of our business. We are not presently a party to any legal, governmental or arbitration proceeding. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Dividend Distribution Policy
We have never declared or paid any dividends on our ordinary shares, and we currently do not plan to declare or pay dividends on our ordinary shares in the foreseeable future. Under English law, we may only pay dividends if our accumulated realized profits, which have not been previously distributed or capitalized, exceed our accumulated realized losses, so far as such losses have not been previously written off in a reduction or reorganization of capital. Therefore, we must have sufficient distributable profits before issuing a dividend. Distributable profits are determined at the holding company level and not on a consolidated basis. Subject to such restrictions and to any restrictions set out in the Articles of Association, declaration and payment of cash dividends in the future, if any, will be at the discretion of our Board of Directors (and in the case of final dividends, must be approved by our shareholders), and will depend upon such factors as results of operations, capital requirements, contractual restrictions, our overall financial condition or applicable laws and any other factors deemed relevant by our Board of Directors.
52
Table of Contents
B. SIGNIFICANT CHANGES
Except as otherwise disclosed in this registration statement, no significant change has occurred since the date of the most recent financial statements included in this registration statement.
| ITEM 9. | THE OFFER AND LISTING |
A. OFFER AND LISTING DETAILS
Not applicable.
For a description of the rights of our ADSs, see “Item 12.D. —American Depositary Shares.”
B. PLAN OF DISTRIBUTION
Not applicable.
C. MARKETS
We intend to re-register as a public limited company incorporated in England and Wales in the near future and have our Ordinary Shares admitted to trading on a regulated market in the United Kingdom (being the Main Market of the London Stock Exchange). We presently have no intention to list the ADSs on any stock exchange.
D. SELLING SHAREHOLDERS
Not applicable.
E. DILUTION
Not applicable.
F. EXPENSES OF THE ISSUE
Not applicable.
| ITEM 10. | ADDITIONAL INFORMATION |
A. SHARE CAPITAL
Issued Share Capital
Our share capital as of March 9, 2021 is 204,193,543 Ordinary Shares with a par value of £0.01 per Ordinary Share. 1 Ordinary Share has been issued and fully paid. 204,193,543 Ordinary Shares have been allotted but not yet issued. We currently have no deferred shares in our issued share capital.
Ordinary Shares
The holders of Ordinary Shares are entitled to receive dividends in proportion to the number of Ordinary Shares held by them and according to the amount paid up on such Ordinary Shares during any portion or portions of the period in respect of which the dividend is paid. Holders of Ordinary Shares are entitled, in proportion to the number of Ordinary Shares held by them and to the amounts paid up thereon, to share in any surplus in the event of our winding up. The holders of Ordinary Shares are entitled to receive notice of, attend either in person or by proxy or, being a corporation, by a duly authorized representative, and vote at general meetings of shareholders.
53
Table of Contents
Share Register
We are required by the Companies Act 2006 to keep a register of our shareholders. Under English law, the Ordinary Shares are deemed to be issued when the name of the shareholder is entered in our share register. The share register therefore is prima facie evidence of the identity of our shareholders, and the shares that they hold. The share register generally provides limited, or no, information regarding the ultimate beneficial owners of our Ordinary Shares. Our share register is maintained by our registrar, Link Market Services.
As an ADS holder, we will not treat you as one of our shareholders and your name will therefore not be entered into our share register. The Depositary will be the holder of the shares underlying our ADSs. For discussion on
our ADSs and ADS holder rights see “Item 12.D.—American Depository Shares” in this registration statement. As an ADS holder, you have a right to receive the ordinary shares underlying your ADSs as discussed at “Item 12.D.—American Depository Shares” in this registration statement.
Under the Companies Act 2006, we must enter an allotment of shares in our share register as soon as practicable and in any event within two months of the allotment. We are also required by the Companies Act 2006 to register a transfer of shares (or give the transferee notice of and reasons for refusal) as soon as practicable and in any event within two months of receiving notice of the transfer.
We, any of our shareholders or any other affected person may apply to the court for rectification of the share register if:
| • | the name of any person is wrongly entered in or omitted from our register of members; or |
| • | there is a failure or unnecessary delay in amending the register of members to show the date a member ceased to be a member. |
History of Share Capital
For the dates described below, the Company allotted Ordinary Shares as follows:
Allotment Date | Action | Number of Ordinary Shares Allotted | Number of Shares after Allotment | Consideration Received | ||||||||||
| June 5, 2020 | Subscriber share issued on incorporation | 1 | 1 | (1) | ||||||||||
| October 30, 2020 | Allotment pursuant to the Demerger | 194,612,288 | 194,612,289 | (2) | ||||||||||
| October 30, 2020 | Allotment pursuant to the terms of the Supplemental Demerger Agreement | 9,581,254 | 204,193,543 | (1)(3) | ||||||||||
| (1) | We received cash consideration for this issuance. |
| (2) | This allotment was pursuant to the Demerger Agreement, as described in “Item 5.A.—Material Contracts”, without consideration. The Ordinary Shares were allotted on a one for one basis except that the number of Ordinary Shares allotted to our initial subscriber (Planwise Group Limited, a Tiziana shareholder) was reduced by the number of Ordinary Shares already held by it so that, upon the Demerger becoming effective, each Tiziana shareholder (including our initial subscriber) holds the entitlement to one of our Ordinary Shares for each Tiziana share held by such shareholder at the Demerger Record Time. |
| (3) | This allotment was pursuant to the Supplemental Demerger Agreement, as described in “Item 5.A.—Material Contracts” for the future cash consideration described on satisfaction of the conditions specified above. |
B. ARTICLES OF ASSOCIATION
Articles of Association
Objects
Section 31 of the Companies Act provides that the objects of a company are unrestricted unless any restrictions are set out in the articles. There are no such restrictions in the Articles and our objects are therefore unrestricted.
Voting Rights
The shareholders have the right to receive notice of, and to vote at, our general meetings. Each shareholder who is present in person (or, being a corporation, by representative) at a general meeting on a show of hands has one vote and, on a poll, every such holder who is present in person (or, being a corporation, by representative) or by proxy has one vote in respect of every share held by him.
54
Table of Contents
Dividends
We may, subject to the provisions of the Companies Act and the Articles, by ordinary resolution from time to time declare dividends to be paid to members not exceeding the amount recommended by the Directors. Subject to the provisions of the Companies Act in so far as, in the Directors’ opinions our profits justify such payments, the Directors may pay interim dividends on any class of shares.
Any dividend unclaimed after a period of 12 years from the date such dividend was declared or became payable shall, if the Directors resolve, be forfeited and shall revert to us. No dividend or other moneys payable on or in respect of a share shall bear interest as against us.
Allotment of Shares and Pre-Emption Rights
Subject to the Companies Act and to any rights attached to existing shares, any share may be issued with or have attached to it such rights and restrictions as we may by ordinary resolution determine, or if no ordinary resolution has been passed or so far as the resolution does not make specific provision, as the Directors may determine (including shares which are to be redeemed, or are liable to be redeemed at our option or the holder of such shares).
In accordance with section 551 of the Companies Act, the Directors may be generally and unconditionally authorised to exercise all our powers to allot shares up to an aggregate nominal amount equal to the amount stated in the relevant ordinary resolution authorising such allotment.
The provisions of section 561 of the Companies Act (which confer on shareholders rights of pre-emption in respect of the allotment of equity securities which are paid up in cash) apply to us except to the extent disapplied by special resolution of the Company.
Distribution of Assets on a Winding-Up
If the Company is wound up (whether the liquidation is voluntary, under supervision of the court or by the court), the liquidator may, with the sanction of a special resolution and any other sanction required by Act, divide among the members in specie the whole or any part of our assets. For this purpose, the liquidator may set such value as he considers fair on any one or more class or classes of property and may determine how such division shall be carried out as between members or classes of members. The liquidator may, with the same authority, transfer the whole or any part of the assets to trustees on such trusts for the benefit of members as he thinks fit.
Transfer of Shares
Every transfer of shares which are in certificated form must be in writing in any usual form or in any form approved by the Board and shall be executed by or on behalf of the transferor and (in the case of a transfer of a share which is not fully paid up) by or on behalf of the transferee.
Every transfer of shares which are in uncertificated form must be made by means of a relevant system (such as CREST).
The Board may, in its absolute discretion and without giving reason, refuse to register any transfer of certificated shares unless: (a) it is in respect of a share which is fully paid up; (b) it is in respect of a share upon which we have no lien; (c) it is only for one class of share; (d) it is duly stamped or is duly certificated or otherwise shown to the satisfaction of the Board to be exempt from stamp duty; and (e) it is delivered for registration to our registered office (or such other place as the Board may determine), accompanied (except in the case of a transfer by a person to whom we are not required by law to issue a certificate and to whom a certificate has not been issued or in the case of a renunciation) by the certificate for the shares to which it relates and such other evidence as the Board may reasonably require to prove the title of the transferor (or person renouncing) and the due execution of the transfer or renunciation by him or, if the transfer or renunciation is executed by some other person on his behalf, the authority of that person to do so.
The Directors may refuse to register a transfer of uncertificated shares in any circumstances that are allowed or required by the CREST Regulations and the CREST System.
55
Table of Contents
Restrictions on Voting Rights
If a member or any person appearing to be interested in shares held by such a member has been duly issued with a notice under section 793 of the Companies Act, a failure in relation to any shares to provide the required information could result in the loss or restriction of rights attaching to the shares, including prohibitions on certain transfers of the shares, withholding of dividends and loss of voting rights.
Variation of Class Rights
Whenever our share capital is divided into different classes of shares, the special rights attached to any class may be varied or abrogated either with the consent in writing of the holders of three-fourths in nominal value of the issued shares of that class or with the sanction of a special resolution passed at a general meeting of the holders of the shares of that class and may be so varied and abrogated whilst the Company is a going concern or during or in contemplation of a winding up.
Share Capital, Changes in Capital and Purchase of Own Shares
We may issue shares with such rights or restrictions as may be determined by ordinary resolution, including shares which are to be redeemed, or are liable to be redeemed at our option or the holder of such shares.
We may by ordinary resolution consolidate or divide all of its share capital into shares of larger nominal value than its existing shares, or cancel any shares which, at the date of the ordinary resolution, have not been taken or agreed to be taken by any person and diminish the amount of its share capital by the nominal amount of shares so cancelled or sub-divide its shares, or any of them, into shares of smaller nominal value.
We may, in accordance with the Companies Act, reduce or cancel its share capital or any capital redemption reserve or share premium account in any manner and with and subject to any conditions, authorities and consents required by law.
General Meetings
We must convene and hold AGMs in accordance with the Companies Act.
No business shall be transacted at any general meeting unless a quorum is present when the meeting proceeds to business, but the absence of a quorum shall not preclude the choice or appointment of a Chair of the meeting which shall not be treated as part of the business of the meeting. Save as otherwise provided by the articles, two shareholders present in person or by proxy and entitled to vote shall be a quorum for all purposes.
Borrowing Powers
Subject to the Articles and the Companies Act, the Board may exercise all of our powers to:
| • | borrow money; |
| • | indemnify and guarantee; |
| • | mortgage or charge; |
| • | create and issue debentures and other securities; and |
| • | give security either outright or as collateral security for any debt, liability or obligation of the Company or of any third party. |
Capitalization of profits
The Directors may, if they are so authorised by an ordinary resolution of the shareholders, decide to capitalize any of our undivided profits (whether or not they are available for distribution), or any sum standing to the credit of our share premium account or capital redemption reserve. The Directors may also, subject to the aforementioned ordinary resolution, appropriate any sum which they so decide to capitalize to the persons who would have been entitled to it if it were distributed by way of dividend and in the same proportions.
56
Table of Contents
Uncertificated shares
Subject to the Companies Act, the Directors may permit title to shares of any class to be issued or held otherwise than by a certificate and to be transferred by means of a ‘relevant system’ (i.e., the CREST System) without a certificate.
The Directors may take such steps as it sees fit in relation to the evidencing of and transfer of title to uncertificated shares, any records relating to the holding of uncertificated shares and the conversion of uncertificated shares to certificated shares, or vice-versa.
We may by notice to the holder of an uncertificated share, require that share to be converted into certificated form.
The Board may take such other action that the Board considers appropriate to achieve the sale, transfer, disposal, forfeiture, re-allotment or surrender of an uncertified share or otherwise to enforce a lien in respect of it.
Directors
Unless otherwise determined by us by ordinary resolution, the number of Directors (other than any alternate Directors) shall not be less than two and shall not be more than fifteen.
Subject to the Articles and the Companies Act, we may by ordinary resolution appoint a person who is willing to act as a Director and the Board shall have power at any time to appoint any person who is willing to act as a Director, in both cases either to fill a vacancy or as an addition to the existing Board.
At each AGM all Directors shall retire from office except any Director appointed by the Board after the notice of that AGM has been given and before that AGM has been held.
Subject to the provisions of the Articles, the Board may regulate their proceedings as they think fit. A Director may, and the secretary at the request of a Director shall, call a meeting of the Directors.
The quorum for a Directors’ meeting shall be fixed from time to time by a decision of the Directors, and unless otherwise fixed, it is two.
Questions and matters requiring resolution arising at a meeting shall be decided by a majority of votes of the participating Directors. In the case of an equality of votes the chairman of the meeting shall have a second or casting vote (unless he is not entitled to vote on the resolution in question).
Each of the Directors may be paid a fee at such rate as may from time to time be determined by the Board. However, the aggregate of all fees payable to the Directors (other than amounts payable under any other provision of the Articles) must not exceed £2,000,000 a year or such higher amount as may from time to time be decided by us by ordinary resolution. The salary or remuneration of any Director appointed to hold any employment or executive office may be either a fixed sum of money, or may altogether or in part be governed by business done or profits made or otherwise determined by the Board, and may be in addition to or instead of any fee payable to him for his services as Director.
The Directors shall also be entitled to be paid all reasonable expenses properly incurred by them in connection with their attendance at meetings of shareholders or class meetings, board or committee meetings or otherwise in connection with the exercise of their powers and the discharge of their responsibilities in relation to us.
Directors’ Interests
The Board may, in accordance with the requirements in the Articles, authorise any matter proposed to them by any Director which would, if not authorised, involve a Director breaching his duty under the Companies Act to avoid conflicts of interests.
A Director seeking authorisation in respect of such conflict shall declare to the Board the nature and extent of their interest in a conflict as soon as is reasonably practicable.
The Director shall provide the Board with such details of the matter as are necessary for the Board to decide how to address the conflict together with such additional information as may be requested by the Board.
57
Table of Contents
Any authorisation by the Board will be effective only if:
| • | to the extent permitted by the Companies Act, the matter in question shall have been proposed by any Director for consideration in the same way that any other matter may be proposed to the Directors under the provisions of the Articles; |
| • | any requirement as to the quorum for consideration of the relevant matter is met without counting the conflicted Director and any other conflicted Director; and |
| • | the matter is agreed to without the conflicted Director voting or would be agreed to if the conflicted Director’s and any other interested Director’s vote is not counted. |
Subject to the provisions of the Companies Act, every Director, secretary or other officer of the Company (other than an auditor) is entitled to be indemnified against all costs, charges, losses, damages and liabilities incurred by him in the actual purported exercise or discharge of his duties or exercise of his powers or otherwise in relation to them.
Differences in Corporate Law
The applicable provisions of the Companies Act differ from laws applicable to U.S. corporations and their shareholders. Set forth below is a summary of certain differences between the provisions of the Companies Act applicable to us and the Delaware General Corporation Law relating to shareholders’ rights and protections. This summary is not intended to be a complete discussion of the respective rights and it is qualified in its entirety by reference to Delaware law and English law.
England and Wales | Delaware | |||
| Number of Directors | Under the Companies Act, a public limited company must have at least two directors and the number of directors may be fixed by or in the manner provided in a company’s articles of association. | Under Delaware law, a corporation must have at least one director and the number of directors shall be fixed by or in the manner provided in the bylaws. | ||
| Removal of Directors | Under the Companies Act, shareholders may remove a director without cause by an ordinary resolution (which is passed by a simple majority of those voting in person or by proxy at a general | Under Delaware law, any director or the entire board of directors may be removed, with or without cause, by the holders of a majority of the shares then entitled to vote at an election of directors, except (i) unless the | ||
| meeting) irrespective of any provisions of any service contract the director has with the company, provided 28 clear days’ notice of the resolution has been given to the company and its shareholders. On receipt of notice of an intended resolution to remove a director, the company must forthwith send a copy of the notice to the director concerned. Certain other procedural requirements under the Companies Act must also be followed, such as allowing the director to make representations against his or her removal either at the meeting or in writing. | certificate of incorporation provides otherwise, in the case of a corporation whose board of directors is classified, stockholders may effect such removal only for cause, or (ii) in the case of a corporation having cumulative voting, if less than the entire board of directors is to be removed, no director may be removed without cause if the votes cast against his removal would be sufficient to elect him if then cumulatively voted at an election of the entire board of directors, or, if there are classes of directors, at an election of the class of directors of which he is a part. | |||
58
Table of Contents
England and Wales | Delaware | |||
| Vacancies on the Board of Directors | Under English law, the procedure by which directors, other than a company’s initial directors, are appointed is generally set out in a company’s articles of association, provided that where two or more persons are appointed as directors of a public limited company by a resolution of the shareholders, resolutions appointing each director must be voted on individually. | Under Delaware law, vacancies and newly created directorships may be filled by a majority of the directors then in office (even though less than a quorum) or by a sole remaining director unless (i) otherwise provided in the certificate of incorporation or bylaws of the corporation or (ii) the certificate of incorporation directs that a particular class of stock is to elect such director, in which case a majority of the other directors elected by such class, or a sole remaining director elected by such class, will fill such vacancy. | ||
| Annual General Meeting | Under the Companies Act, a public limited company must hold an annual general meeting in each six-month period following its annual accounting reference date. | Under Delaware law, the annual meeting of stockholders shall be held at such place, on such date and at such time as may be designated from time to time by the board of directors or as provided in the certificate of incorporation or by the bylaws. | ||
| General Meeting | Under the Companies Act, a general meeting of the shareholders of a public limited company may be called by the directors. | Under Delaware law, special meetings of the stockholders may be called by the board of directors or by such person or persons as may be authorized by the certificate of incorporation or by the bylaws. | ||
| Shareholders holding at least 5% of the paid-up capital of the company carrying voting rights at general meetings (excluding any paid up capital held as treasury shares) can require the directors to call a general meeting and, if the directors fail to do so within a certain period, may themselves (or any of them representing more than one half of the total voting rights of all of them) convene a general meeting. | ||||
59
Table of Contents
England and Wales | Delaware | |||
| Notice of General Meetings | Subject to a company’s articles of association providing for a longer period, under the Companies Act, 21 clear days’ notice must be given for an annual general meeting and any resolutions to be proposed at the meeting, Subject to a company’s articles of association providing for a longer period, at least 14 clear days’ notice is required for any other general meetings. In addition, certain matters, such as the removal of directors or auditors, require special notice, which is 28 clear days’ notice. The shareholders of a company may in all cases consent to a shorter notice period, the proportion of shareholders’ consent required being 100% of those entitled to attend and vote in the case of an annual general meeting and, in the case of any other general meeting, a majority in number of the members having a right to attend and vote at the meeting, being a majority who together hold not less than 95% in nominal value of the shares giving a right to attend and vote at the meeting. | Under Delaware law, unless otherwise provided in the certificate of incorporation or bylaws, written notice of any meeting of the stockholders must be given to each stockholder entitled to vote at the meeting not less than 10 nor more than 60 days before the date of the meeting and shall specify the place, date, hour and purpose or purposes of the meeting. | ||
| Quorum | Subject to the provisions of a company’s articles of association, the Companies Act provides that two shareholders present at a meeting (in person, by proxy or authorised under the Companies Act) shall constitute a quorum for companies with more than one shareholder. | The certificate of incorporation or bylaws may specify the number of shares, the holders of which shall be present or represented by proxy at any meeting in order to constitute a quorum, but in no event shall a quorum consist of less than one third of the shares entitled to vote at the meeting. In the absence of such specification in the certificate of incorporation or bylaws, a majority of the shares entitled to vote, present in person or represented by proxy, shall constitute a quorum at a meeting of stockholders. | ||
| Proxy | Under the Companies Act, at any meeting of shareholders, a shareholder may designate another person to attend, speak and vote at the meeting on their behalf by proxy. | Under Delaware law, at any meeting of stockholders, a stockholder may designate another person to act for such stockholder by proxy, but no such proxy shall be voted or acted upon after three years from its date, unless the proxy provides for a longer period. A director of a Delaware corporation may not issue a proxy representing the director’s voting rights as a director. | ||
60
Table of Contents
England and Wales | Delaware | |||
| Issue of New Shares | Under the Companies Act, the directors of a company must not exercise any power to allot shares or grant rights to subscribe for, or to convert any security into, shares unless they are authorized to do so by the company’s Articles or by an ordinary resolution of the shareholders. Any authorization given must state the maximum amount of shares that may be allotted under it and specify the date on which it will expire, which must be not more than five years from the date the authorization was given. The authority can be renewed by a further resolution of the shareholders. | Under Delaware law, if the corporation’s certificate of incorporation so provides, the directors have the power to authorize additional stock. The directors may authorize capital stock to be issued for consideration consisting of cash, any tangible or intangible property or any benefit to the corporation or any combination thereof. In the absence of actual fraud in the transaction, the judgment of the directors as to the value of such consideration is conclusive. | ||
| Pre-emptive Rights | Under the Companies Act, “equity securities,” being (i) shares in the company other than shares that, with respect to dividends and capital, carry a right to participate only up to a specified amount in a distribution, referred to as “ordinary shares,” or (ii) rights to subscribe for, or to convert securities into, ordinary shares, proposed to be allotted for cash must be offered first to the existing equity shareholders in the company in proportion to the respective nominal value of their holdings, unless an exception applies or a special resolution to the contrary has been passed by shareholders in a general meeting or the articles of association provide otherwise in each case in accordance with the provisions of the Companies Act. | Under Delaware law, stockholders have no pre-emptive rights to subscribe to additional issues of stock or to any security convertible into such stock unless, and except to the extent that, such rights are expressly provided for in the certificate of incorporation. | ||
| Authority to Allot | Under the Companies Act, the directors of a company must not allot shares or grant of rights to subscribe for or to convert any security into shares unless an exception applies or an ordinary resolution to the contrary has been passed by shareholders in a general meeting or the articles of association provide otherwise, in each case in accordance with the provisions of the Companies Act. | Under Delaware law, if the corporation’s charter or certificate of incorporation so provides, the board of directors has the power to authorize the issuance of stock. The board of directors may authorize capital stock to be issued for consideration consisting of cash, any tangible or intangible property or any benefit to the corporation or any combination thereof. In the absence of actual fraud in the transaction, the judgment of the directors as to the value of such consideration is conclusive. | ||
61
Table of Contents
England and Wales | Delaware | |||
| Liability of Directors and Officers | Under the Companies Act, any provision, whether contained in a company’s Articles or any contract or otherwise, that purports to exempt a director of a company, to any extent, from any liability that would otherwise attach to him in connection with any negligence, default, breach of duty or breach of trust in relation to the company is void. Any provision by which a company directly or indirectly provides an indemnity, to any extent, for a director of the company or of an associated company against any liability attaching to him in connection with any negligence, default, breach of duty or breach of trust in relation to the company of which he is a director is also void except as permitted by the Companies Act, which provides exceptions for the company to: (i) purchase and maintain insurance against such liability; (ii) provide a “qualifying third party indemnity” (being an indemnity against liability incurred by the director to a person other than the company or an associated company or criminal proceeding in which such director is convicted); and (iii) provide a “qualifying pension scheme indemnity” (being an indemnity against liability incurred in connection with the company’s activities as trustee of an occupational pension plan). | Under Delaware law, a corporation’s certificate of incorporation may include a provision eliminating or limiting the personal liability of a director to the corporation and its stockholders for damages arising from a breach of fiduciary duty as a director. However, no provision can limit the liability of a director for:
• any breach of the director’s duty of loyalty to the corporation or its stockholders;
• acts or omissions not in good faith or that involve intentional misconduct or a knowing violation of law;
• intentional or negligent payment of unlawful dividends or stock purchases or redemptions; or
• any transaction from which the director derives an improper personal benefit. | ||
| Voting Rights | Under English law, unless a poll is demanded by the shareholders of a company or is required by the chairman of the meeting or the company’s articles of association, shareholders shall vote on all resolutions on a show of hands. Under the Companies Act, a poll may be demanded by: (i) not fewer than five shareholders having the right to vote on the resolution; (ii) any | Delaware law provides that, unless otherwise provided in the certificate of incorporation, each stockholder is entitled to one vote for each share of capital stock held by such stockholder. | ||
62
Table of Contents
England and Wales | Delaware | |||
shareholder(s) representing not less than 10% of the total voting rights of all the shareholders having the right to vote on the resolution (excluding any voting rights attaching to treasury shares); or (iii) any shareholder(s) holding shares in the company conferring a right to vote on the resolution (excluding any voting rights attaching to treasury shares) being shares on which an aggregate sum has been paid up equal to not less than 10% of the total sum paid up on all the shares conferring that right. A company’s articles of association may provide more extensive rights for shareholders to call a poll.
Under English law, an ordinary resolution is passed on a show of hands if it is approved by a simple majority (more than 50%) of the votes cast by shareholders present (in person or by proxy) and entitled to vote. If a poll is demanded, an ordinary resolution is passed if it is approved by holders representing a simple majority of the total voting rights of shareholders present, in person or by proxy, who, being entitled to vote, vote on the resolution. Special resolutions require the affirmative vote of not less than 75% of the votes cast by shareholders present, in person or by proxy, at the meeting. If a poll is demanded, a special resolution is passed if it is approved by holders representing not less than 75% of the total voting rights of shareholders in person or by proxy who, being entitled to vote, vote on the resolution. | ||||
| Shareholder Vote on Certain Transactions | The Companies Act provides for schemes of arrangement, which are arrangements or compromises between a company and any class of shareholders or creditors and used in certain types of reconstructions, amalgamations, capital reorganizations or takeovers. These arrangements require: | Generally, under Delaware law, unless the certificate of incorporation provides for the vote of a larger portion of the stock, completion of a merger, consolidation, sale, lease or exchange of all or substantially all of a corporation’s assets or dissolution requires: | ||
• the approval at a shareholders’ or creditors’ meeting convened by order of the court, of a majority in number of shareholders or creditors
or class thereof representing 75% in value, the class of shareholders or creditors, or class thereof present and voting, either in person or by proxy; and
• the approval of the court. | • the approval of the board of directors; and
• the approval by the vote of the holders of a majority of the outstanding stock or, if the certificate of incorporation provides for more or less than one vote per share, a majority of the votes of the outstanding stock of the corporation entitled to vote on the matter. |
63
Table of Contents
| Standard of Conduct for Directors | Under English law, a director owes various statutory and fiduciary duties to the company, including:
• to act in the way he considers, in good faith, would be most likely to promote the success of the company for the benefit of its members as a whole;
• to avoid a situation in which he has, or can have, a direct or indirect interest that conflicts, or possibly conflicts, with the interests of the company; | Delaware law does not contain specific provisions setting forth the standard of conduct of a director. The scope of the fiduciary duties of directors is generally determined by the courts of the State of Delaware. In general, directors have a duty to act without self-interest, on a well-informed basis and in a manner they reasonably believe to be in the best interest of the stockholders. | ||
• to act in accordance with the company’s constitution and only exercise his powers for the purposes for which they are conferred;
• to exercise independent judgment;
• to exercise reasonable care, skill and diligence;
• not to accept benefits from a third party conferred by reason of his being a director or doing, or not doing, anything as a director; and
• to declare any interest that he has, whether directly or indirectly, in a proposed or existing transaction or arrangement with the company. | Directors of a Delaware corporation owe fiduciary duties of care and loyalty to the corporation and to its stockholders. The duty of care generally requires that a director act in good faith, with the care that an ordinarily prudent person would exercise under similar circumstances. Under this duty, a director must inform himself of all material information reasonably available regarding a significant transaction. The duty of loyalty requires that a director act in a manner he reasonably believes to be in the best interests of the corporation. He must not use his corporate position for personal gain or advantage. In general, but subject to certain exceptions, actions of a director are presumed to have been made on an informed basis, in good faith and in the honest belief that the action taken was in the best interests of the corporation. However, this presumption may be rebutted by evidence of a breach of one of the fiduciary duties. Delaware courts have also imposed a heightened standard of conduct upon directors of a Delaware corporation who take any action designed to defeat a threatened change in control of the corporation. | |||
| In addition, under Delaware law, when the board of directors of a Delaware corporation approves the sale or break-up of a corporation, the board of directors may, in certain circumstances, have a duty to obtain the highest value reasonably available to the stockholders. |
64
Table of Contents
| Shareholder Litigation | Under English law, generally, the company, rather than its shareholders, is the proper claimant in an action in respect of a wrong done to the company or where there is an irregularity in the company’s internal management. Notwithstanding this general position, the Companies Act provides that (i) a court may allow a shareholder to bring a derivative claim (that is, an action in respect of and on behalf of the company) in respect of a cause of action arising from a director’s negligence, default, breach of duty or breach of trust and (ii) a shareholder may bring a claim for a court order where the company’s affairs have been or are being conducted in a manner that is unfairly prejudicial to some of its shareholders. | Under Delaware law, a stockholder may initiate a derivative action to enforce a right of a corporation if the corporation fails to enforce the right itself. The complaint must:
• state that the plaintiff was a stockholder at the time of the transaction of which the plaintiff complains or that the plaintiffs shares thereafter devolved on the plaintiff by operation of law; and
• allege with particularity the efforts made by the plaintiff to obtain the action the plaintiff desires from the directors and the reasons for the plaintiff’s failure to obtain the action; or
• state the reasons for not making the effort.
Additionally, the plaintiff must remain a stockholder through the duration of the derivative suit. The action will not be dismissed or compromised without the approval of the Delaware Court of Chancery. |
C. MATERIAL CONTRACTS
Except as otherwise set forth below or as otherwise disclosed in this registration statement, we are not currently, and have not been in the last two years, party to any material contract, other than contracts entered into in the ordinary course of business. See the section titled “Item 5.A.—Material Contracts”.
D. EXCHANGE CONTROLS
Other than certain economic sanctions which may be in place from time to time, there are currently no U.K. laws, decrees or regulations restricting the import or export of capital or affecting the remittance of dividends or other payment to holders of ordinary shares who are non-residents of the United Kingdom. Similarly, other than certain economic sanctions which may be in force from time to time, there are no limitations relating only to nonresidents of the United Kingdom under English law or the Articles of Association on the right to be a holder of, and to vote in respect of, the Ordinary Shares.
E. TAXATION
Certain United Kingdom Tax Considerations
The following is a general summary of certain U.K. tax considerations relating to the ownership and disposal of an ordinary share or ADS and does not address all possible tax consequences relating to holding an ordinary share or ADS. It is based on U.K. tax law and generally published HMRC, practice (which may not be binding on HMRC) as of the date of this registration statement, both of which are subject to change, possibly with retrospective effect.
Save as provided otherwise, this summary applies only to a person who is the absolute beneficial owner of an ordinary share or ADS and who is resident (and, in the case of an individual, domiciled) in the United Kingdom for tax purposes and who is not resident for tax purposes in any other jurisdiction and does not have a permanent establishment or fixed base in any other jurisdiction with which the holding of an ordinary share or ADS is connected (“U.K. Holders”). A person (a) who is not resident (or, if resident, is not domiciled) in the United Kingdom for tax purposes, including an individual and company who trades in the United Kingdom through a branch, agency or permanent establishment in the United Kingdom to which an ordinary share or ADS is attributable, or (b) who is resident or otherwise subject to tax in a jurisdiction outside the United Kingdom, is recommended to seek the advice of professional advisors in relation to their taxation obligations.
65
Table of Contents
This summary is for general information only and is not intended to be, nor should it be considered to be, legal or tax advice to any particular shareholder. It does not address all of the tax considerations that may be relevant to you in light of your particular circumstances or to those of you subject to special treatment under U.K. tax law. In particular:
| • | this summary only applies to an absolute beneficial owner of an ordinary share or ADS and any dividend paid in respect of the ordinary share where the dividend is regarded for U.K. tax purposes as that person’s own income (and not the income of some other person); |
| • | this summary: (a) only addresses the principal U.K. tax consequences for a holder of an ordinary share or ADS as a capital asset, (b) does not address the tax consequences that may be relevant to certain special classes of holders such as a dealer, broker or trader in shares or securities and any other person who otherwise holds an ordinary share or ADS, (c) does not address the tax consequences for a holder that is a financial institution, insurance company, collective investment scheme, pension scheme, charity or tax-exempt organization, (d) assumes that a holder is not an officer or employee of the company (nor of any related company) and has not (and is not deemed to have) acquired the an ordinary share or ADS by virtue of an office or employment, and (e) assumes that a holder does not control or hold (and is not deemed to control or hold), either alone or together with one or more associated or connected persons, directly or indirectly (including through the holding of an ordinary share or ADS), an interest of 10 per cent. or more in the issued share capital (or in any class thereof), voting power, rights to profits or capital of the company, and is not otherwise connected with the company. |
This summary further assumes that a holder of an ordinary share or ADS is the beneficial owner of the underlying ordinary share for U.K. direct tax purposes.
YOU SHOULD SATISFY YOURSELF AS TO THE OVERALL TAX CONSEQUENCES, INCLUDING, SPECIFICALLY, THE CONSEQUENCES UNDER U.K. TAX LAW AND HMRC PRACTICE OF THE ACQUISITION, OWNERSHIP AND DISPOSAL OF THE ORDINARY SHARES OR ADSs, IN YOUR OWN PARTICULAR CIRCUMSTANCES BY CONSULTING YOUR TAX ADVISERS.
Taxation of Dividends
Withholding Tax
A dividend payment in respect of an ordinary share may be made without withholding or deduction for or on account of U.K. tax.
Income Tax
A dividend received by individual U.K. Holders may, depending on his or her particular circumstances, be subject to U.K. income tax on the gross amount of the dividend paid.
An individual holder of an ordinary share or ADS who is not a U.K. Holder will not be chargeable to U.K. income tax on a dividend paid by the company, unless such holder carries on (whether solely or in partnership) a trade, profession or vocation in the United Kingdom through a permanent establishment in the United Kingdom to which the ordinary share or ADS is attributable. In these circumstances, such holder may, depending on his or her individual circumstances, be chargeable to U.K. income tax on a dividend received from the company.
All dividends received by a U.K. Holder from the Company or from other sources will form part of the U.K. Holder’s total income for U.K. income tax purposes and will constitute the top slice of that income. The rate of U.K. income tax that is chargeable on dividends received in the tax year 2020/2021 by (i) an additional rate taxpayer is 38.1 percent, (ii) a higher rate taxpayer is 32.5 percent, and (iii) a basic rate taxpayer is 7.5 percent. A nil rate of income tax will apply to the first £2,000 of taxable dividend income received by an individual U.K. Holder in a tax year.
Corporation Tax
A U.K. Holder within the charge to U.K. corporation tax may be entitled to exemption from U.K. corporation tax in respect of dividend payments, provided the dividends qualify for exemption (which is likely) and certain conditions are met (including anti-avoidance conditions). If the conditions for the exemption are not satisfied, or such U.K. Holder elects for an otherwise exempt dividend to be taxable, U.K. corporation tax will be chargeable on the gross amount of a dividend. If you are in any doubt as to your position, you should consult your own professional advisers.
66
Table of Contents
A corporate holder of an ordinary share or ADS that is not a U.K. Holder will not be subject to U.K. corporation tax on a dividend received from the company, unless it carries on a trade in the United Kingdom through a permanent establishment to which the ordinary share or ADS is attributable. In these circumstances, such holder may, depending on its individual circumstances and if the exemption from U.K. corporation tax discussed above does not apply, be chargeable to U.K. corporation tax on dividends received from the company.
Taxation of Disposals
U.K. Holders
A disposal or deemed disposal of an ordinary share or ADS by an individual U.K. Holder may, depending on his or her individual circumstances, give rise to a chargeable gain or to an allowable loss for the purpose of U.K. capital gains tax. The principal factors that will determine the capital gains tax position on a disposal of an ordinary share or ADS are the extent to which the holder realizes any other capital gains in the tax year in which the disposal is made, the extent to which the holder has incurred capital losses in that or any earlier tax year and the level of the annual exemption for tax-free gains in that tax year (the “annual exemption”). The annual exemption for the 2020/2021 tax year is £12,500. If, after all allowable deductions, an individual U.K. Holder’s total taxable income for the year exceeds the basic rate income tax limit, a taxable capital gain accruing on a disposal of an ordinary share or an ADS is taxed at the rate of 20 percent. In other cases, a taxable capital gain accruing on a disposal of an ordinary share or ADS may be taxed at the rate of 10 per cent. save to the extent that any capital gains exceed the unused basic rate tax band. In that case, the rate currently applicable to the excess would be 20 percent.
An individual U.K. Holder who ceases to be resident in the United Kingdom (or who fails to be regarded as resident in a territory outside the United Kingdom for the purposes of double taxation relief) for a period of five tax years or less than five years and who disposes of an ordinary share or ADS during that period of temporary non-residence may be liable to U.K. capital gains tax on a chargeable gain accruing on such disposal on his or her return to the United Kingdom (or upon ceasing to be regarded as resident outside the United Kingdom for the purposes of double taxation relief) (subject to available exemptions or reliefs).
A disposal (or deemed disposal) of an ordinary share or ADS by a corporate U.K. Holder may give rise to a chargeable gain or an allowable loss for the purpose of U.K. corporation tax. Any gain or loss in respect of currency fluctuations over the period of holding an ordinary share or an ADS are also brought into account on a disposal.
Non-U.K. Holders
An individual holder who is not a U.K. Holder should not normally be liable to U.K. capital gains tax on capital gains realized on the disposal of an ordinary share or ADS unless such holder carries on (whether solely or in partnership) a trade, profession or vocation in the United Kingdom through a permanent establishment in the United Kingdom to which the ordinary share or ADS is attributable. In these circumstances, such holder may, depending on his or her individual circumstances, be chargeable to U.K. capital gains tax on chargeable gains arising from a disposal of his or her ordinary share or ADS.
A corporate holder of an ordinary share or ADS that is not a U.K. Holder will not be liable for U.K. corporation tax on chargeable gains realized on the disposal of an ordinary share or ADS unless: (i) it carries on a trade in the United Kingdom through a permanent establishment to which the ordinary share or ADS is attributable; or (ii) the corporate holder is disposing of an interest in a company and that disposal is of an asset that derives 75 per cent. or more of its gross asset value from U.K. land and that holder has a substantial indirect interest in U.K. land (broadly at least 25 per cent. at any time during the previous two years). In these circumstances, a disposal (or deemed disposal) of an ordinary share or ADS by such holder may give rise to a chargeable gain or an allowable loss for the purposes of U.K. corporation tax.
Inheritance Tax
If, for the purposes of the Double Taxation Relief (Taxes on Estates of Deceased Persons and on Gifts) Treaty United States of America Order 1979 (S1 1979/1454) between the United States and the United Kingdom, an individual holder is domiciled at the time of their death or at the time of a transfer made during their lifetime in the United States and is not a
67
Table of Contents
national of the United Kingdom, any ordinary share or ADS beneficially owned by that holder should not generally be subject to U.K. inheritance tax, provided that any applicable U.S. federal gift or estate tax liability is paid, except where (i) the ordinary share or ADS is part of the business property of a U.K. permanent establishment or pertain to a U.K. fixed base used for the performance of independent personal services; or (ii) the ordinary share or ADS is comprised in a settlement unless, at the time the settlement was made, the settlor was domiciled in the United States and not a national of the U.K. (in which case no charge to U.K. inheritance tax should apply).
Stamp Duty and Stamp Duty Reserve Tax
The stamp duty and stamp duty reserve tax, or SDRT, treatment of the issue, transfer and agreement to transfer an ordinary share outside a depositary receipt system or a clearance service are discussed in the paragraphs under “General” below. The stamp duty and SDRT treatment of such transactions in relation to such systems are discussed in the paragraphs under “Depositary Receipt Systems and Clearance Services” below.
General
The issue of an ordinary share or ADS does not give rise to a SDRT liability, according to the HM Revenue & Customs practice and recent case law and is not subject to stamp duty.
A transfer of an ordinary share or ADS will generally be subject to stamp duty at the rate of 0.5 per cent. of the consideration given for the transfer (rounded up to the next £5). An exemption from stamp duty is available on an instrument transferring an ordinary share or ADSs where the amount or value of the consideration is £1,000 or less, and it is certified on the instrument that the transaction effected does not form part of a larger transaction or series of transactions in respect of which the aggregate amount or value of the consideration exceeds £1,000. The purchaser normally pays the stamp duty.
An unconditional agreement to transfer an ordinary share or ADS will normally give rise to a charge to SDRT at the rate of 0.5 per cent. of the amount or value of the consideration payable for the transfer. SDRT is, in general, payable by the purchaser.
If a duly stamped transfer completing an agreement to transfer is produced within six years of the date on which the agreement is made (or, if the agreement is conditional, the date on which the agreement becomes unconditional) any SDRT already paid is generally repayable, normally with interest, and any SDRT charge yet to be paid is cancelled.
No stamp duty will, in practice, be payable on a written instrument transferring an ordinary share or an ADS or on an unconditional agreement to transfer an ordinary share or an ADS if the instrument of transfer or the unconditional agreement to transfer is executed and remains at all times outside the U.K..
Depositary Receipt Systems and Clearance Services
Based on current HMRC practice and recent case law in respect of the European Council Directives 69/335/EC and 2009/7/EC, on the Capital Duties Directive, no SDRT is generally payable when shares are issued or transferred to a clearance service or depositary receipt system as an integral part of a raising of capital.
Where an ordinary share or ADS is otherwise transferred (i) to, or to a nominee or an agent for, a person whose business is or includes the provision of clearance services or (ii) to, or to a nominee or an agent for a person whose business is or includes issuing depositary receipts, stamp duty or SDRT will generally be payable at the higher rate of 1.5 per cent. of the amount or value of the consideration given or, in certain circumstances, the value of the shares.
There is an exception from the 1.5 per cent. charge on the transfer to, or to a nominee or agent for, a clearance service where the clearance service has made and maintained an election under section 97A(1) of the Finance Act 1986, which has been approved by HM Revenue & Customs. In these circumstances, SDRT at the rate of 0.5 per cent. of the amount or value of the consideration payable for the transfer will arise on any transfer of ordinary share into such an account and on subsequent agreements to transfer such shares within such account.
Any liability for stamp duty or SDRT in respect of a transfer into a clearance service or depositary receipt system, or in respect of a transfer within such a service, which does arise will strictly be accountable by the clearance service or depositary receipt system operator or their nominee, as the case may be, but will, in practice, be borne by the participants in the clearance service or depositary receipt system.
68
Table of Contents
Taxation in the United States
The following summary of the material U.S. federal income tax consequences of the acquisition, ownership and disposition of our ordinary shares or ADSs is based upon current law and does not purport to be a comprehensive discussion of all the tax considerations that may be relevant to a decision to purchase our ordinary shares or ADSs. This summary is based on current provisions of the Internal Revenue Code of 1986, as amended (the “Code”), existing, final, temporary and proposed U.S. Treasury Regulations, administrative rulings and judicial decisions, in each case as available on the date of this registration statement. All of the foregoing are subject to change, which change could apply retroactively and could affect the tax consequences described below.
This section summarizes the material U.S. federal income tax consequences to U.S. holders and certain non-U.S. holders, each as defined below, of our ordinary shares or ADSs. This summary addresses only the U.S. federal income tax considerations for holders that acquire our ordinary shares or ADSs at their original issuance and hold our ordinary shares or ADSs as capital assets. This summary does not address all U.S. federal income tax matters that may be relevant to a particular holder. You should consult a professional tax advisor with respect to the tax consequences of the acquisition, ownership or disposition of our ordinary shares or ADSs. This summary does not address tax considerations applicable to a holder of our ordinary shares or ADSs that may be subject to special tax rules including, without limitation, the following:
| • | banks or other financial institutions; |
| • | insurance companies; |
| • | dealers or traders in securities, currencies, or notional principal contracts; |
| • | tax-exempt entities, including an “individual retirement account” or “Roth IRA” retirement plan; |
| • | regulated investment companies or real estate investment trusts; |
| • | “qualified foreign pension funds,” or entities wholly owned by a “qualified foreign pension fund”; persons who have elected to mark securities to market |
| • | persons that hold the ordinary shares as part of a hedge, straddle, conversion, constructive sale or similar transaction involving more than one position; |
| • | holders (whether individuals, corporations or partnerships) that are treated as expatriates for some or all U.S. federal income tax purposes; |
| • | persons who acquired the ADSs as compensation for the performance of services; |
| • | persons holding the ADSs in connection with a trade or business conducted outside of the United States; |
| • | holders that own (or are deemed to own) 10 per cent. or more of our ordinary shares or ADSs, by vote or value; and |
| • | holders that have a “functional currency” other than the U.S. dollar. |
Further, this summary does not address any aspects of any U.S. state, local or non-U.S. tax law, alternative minimum tax, gift or estate consequences, the rules regarding qualified small business stock within the meaning of Section 1202 of the Code, any election to apply Section 1400Z-2 of the Code to gains recognized with respect to our ordinary shares, any other U.S. federal tax other than the income tax or the indirect effects on the holders of equity interests in entities that own our ordinary shares or ADSs.
For the purposes of this summary, a “U.S. holder” is a beneficial owner of ordinary shares or ADSs that is (or is treated as), for U.S. federal income tax purposes:
| • | an individual who is either a citizen or resident of the United States; |
| • | a corporation, or other entity that is treated as a corporation for U.S. federal income tax purposes, created or organized in or under the laws of the United States or any state of the United States or the District of Columbia; |
| • | an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or |
| • | a trust, if a court within the United States is able to exercise primary supervision over its administration and one or more U.S. persons have the authority to control all of the substantial decisions of such trust or has a valid election in effect under applicable U.S. Treasury Regulations to be treated as a U.S. person. |
69
Table of Contents
If a partnership holds ordinary shares or ADSs, the tax treatment of a partner will generally depend upon the status of the partner and upon the activities of the partnership. This discussion does not address the tax treatment of partnerships or other entities that are pass-through entities for U.S. federal income tax purposes or persons that hold their ordinary shares or ADSs through partnerships or other pass-through entities. A partner in a partnership or other pass-through entity that will hold our ordinary shares or ADSs should consult his, her or its tax advisor regarding the tax consequences of acquiring, holding and disposing of our ordinary shares or ADSs through a partnership or other pass-through entity, as applicable.
We will not seek a ruling from the U.S. Internal Revenue Service (the “IRS”), with regard to the U.S. federal income tax treatment of holding our ordinary shares or ADSs, and we cannot assure you that the IRS will agree with the conclusions set forth below.
YOU SHOULD CONSULT YOUR TAX ADVISORS AS TO THE PARTICULAR TAX CONSEQUENCES APPLICABLE TO YOU RELATING TO THE ACQUISITION, OWNERSHIP AND DISPOSITION OF THE ORDINARY SHARES OR ADSs, INCLUDING THE APPLICABILITY OF U.S. FEDERAL, STATE AND LOCAL TAX LAWS.
Ownership of ADSs
For U.S. federal income tax purposes, a holder of ADSs generally will be treated as the owner of the ordinary shares represented by such ADSs. Gain or loss will generally not be recognized on account of exchanges of ordinary shares for ADSs, or of ADSs for ordinary shares. References to ordinary shares in the discussion below are deemed to include ADSs, unless context otherwise requires.
F. DIVIDENDS AND PAYING AGENTS
Not applicable.
G. STATEMENT BY EXPERTS
The Company’s consolidated financial statements of as of and for the year ended December 31, 2020, have been included herein and in the registration statement in reliance upon the report of Mazars LLP, independent registered public accounting firm, appearing elsewhere herein, and upon the authority of said firm as experts in accounting and auditing.
H. DOCUMENTS ON DISPLAY
When this registration statement on Form 20-F becomes effective, we will be subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers, and under those requirements will file reports with the SEC. The SEC also maintains a website at http://www.sec.gov from which filings may be accessed.
As a foreign private issuer, we will be exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, Directors and principal shareholders will be exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we will not be required under the Exchange Act to file annual, quarterly and current reports and financial statements with the SEC as frequently or as promptly as U.S. companies whose securities are registered under the Exchange Act. However, we will file with the SEC, within 120 days after the end of each fiscal year, or such applicable time as required by the SEC, an annual report on Form 20-F containing financial statements audited by an independent registered public accounting firm.
I. SUBSIDIARY INFORMATION
Not applicable.
| ITEM 11. | QUANTITIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
For information about our quantitative and qualitative disclosures about market risk, see “Item 5.B.—Operating and Financial Review and Prospects—Liquidity and Capital Resources” under the sub-heading “Quantitative and Qualitative Disclosures about Financial Risks.”
70
Table of Contents
| ITEM 12. | DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES |
A. DEBT SECURITIES
Not applicable.
B. WARRANTS AND RIGHTS
Not applicable.
C. OTHER SECURITIES
Not applicable.
D. AMERICAN DEPOSITARY SHARES
JPMorgan Chase Bank, N.A., as depositary will issue the ADSs which you will be entitled to receive in this offering. Each ADS will represent an ownership interest in a designated number of ordinary shares which we will deposit with the custodian, as agent of the depositary, under the deposit agreement among ourselves, the depositary and yourself as an ADR holder. In the future, each ADS will also represent any securities, cash or other property deposited with the depositary but which they have not distributed directly to you. Unless certificated ADRs are specifically requested by you, all ADSs will be issued on the books of our depositary in book-entry form and periodic statements will be mailed to you which reflect your ownership interest in such ADSs. In our description, references to American depositary receipts, or ADRs, shall include the statements you will receive which reflect your ownership of ADSs.
The depositary’s office is located at 383 Madison Avenue, Floor 11, New York, NY, 10179.
You may hold ADSs either directly or indirectly through your broker or other financial institution. If you hold ADSs directly, by having an ADS registered in your name on the books of the depositary, you are an ADR holder. This description assumes you hold your ADSs directly. If you hold the ADSs through your broker or financial institution nominee, you must rely on the procedures of such broker or financial institution to assert the rights of an ADR holder described in this section. You should consult with your broker or financial institution to find out what those procedures are.
As an ADR holder, we will not treat you as a shareholder of ours and you will not have any shareholder rights. The law of England and Wales governs shareholder rights. Because the depositary or its nominee will be the shareholder of record for the ordinary shares represented by all outstanding ADSs, shareholder rights rest with such record holder. Your rights are those of an ADR holder. Such rights derive from the terms of the deposit agreement to be entered into among us, the depositary and all registered holders from time to time of ADRs issued under the deposit agreement. The obligations of our company, the depositary and its agents are also set out in the deposit agreement. Because the depositary or its nominee will actually be the registered owner of the ordinary shares, you must rely on it to exercise the rights of a shareholder on your behalf. The deposit agreement and the ADSs are governed by New York law. Under the deposit agreement, as an ADR holder, you agree that any legal suit, action or proceeding against or involving us or the depositary, arising out of or based upon the deposit agreement, the ADSs or the transactions contemplated thereby, may only be instituted in a state or federal court in New York, New York, and you irrevocably waive any objection which you may have to the laying of venue of any such proceeding and irrevocably submit to the exclusive jurisdiction of such courts in any such suit, action or proceeding.
The following is a summary of what we believe to be the material terms of the deposit agreement. Notwithstanding this, because it is a summary, it may not contain all the information that you may otherwise deem important. For more complete information, you should read the entire deposit agreement and the form of ADR which contains the terms of your ADSs. You can read a copy of the deposit agreement which is filed as an exhibit to the registration statement of which this prospectus forms a part. You may also obtain a copy of the deposit agreement at the SEC’s Public Reference Room which is located at 100 F Street, NE, Washington, DC 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-732-0330. You may also find the registration statement and the attached deposit agreement on the SEC’s website at www.sec.gov.
71
Table of Contents
Share Dividends and Other Distributions
How will I receive dividends and other distributions on the ordinary shares underlying my ADSs?
We may make various types of distributions with respect to our securities. The depositary has agreed that, to the extent practicable, it will pay to you the cash dividends or other distributions it or the custodian receives on ordinary shares or other deposited securities, after converting any cash received into U.S. dollars (if it determines such conversion may be made on a reasonable basis) and, in all cases, making any necessary deductions provided for in the deposit agreement. The depositary may utilize a division, branch or affiliate of JPMorgan Chase Bank, N.A. to direct, manage and/or execute any public and/or private sale of securities under the deposit agreement. Such division, branch and/or affiliate may charge the depositary a fee in connection with such sales, which fee is considered an expense of the depositary. You will receive these distributions in proportion to the number of underlying securities that your ADSs represent.
Except as stated below, the depositary will deliver such distributions to ADR holders in proportion to their interests in the following manner:
| • | Cash. The depositary will distribute any U.S. dollars available to it resulting from a cash dividend or other cash distribution or the net proceeds of sales of any other distribution or portion thereof (to the extent applicable), on an averaged or other practicable basis, subject to (i) appropriate adjustments for taxes withheld, (ii) such distribution being impermissible or impracticable with respect to certain registered ADR holders, and (iii) deduction of the depositary’s and/or its agents’ expenses in (1) converting any foreign currency to U.S. dollars to the extent that it determines that such conversion may be made on a reasonable basis, (2) transferring foreign currency or U.S. dollars to the United States by such means as the depositary may determine to the extent that it determines that such transfer may be made on a reasonable basis, (3) obtaining any approval or license of any governmental authority required for such conversion or transfer, which is obtainable at a reasonable cost and within a reasonable time and (4) making any sale by public or private means in any commercially reasonable manner. If exchange rates fluctuate during a time when the depositary cannot convert a foreign currency, you may lose some or all of the value of the distribution. |
| • | Ordinary Shares. In the case of a distribution in ordinary shares, the depositary will issue additional ADRs to evidence the number of ADSs representing such ordinary shares. Only whole ADSs will be issued. Any ordinary shares which would result in fractional ADSs will be sold and the net proceeds will be distributed in the same manner as cash to the ADR holders entitled thereto. |
| • | Rights to receive additional ordinary shares. In the case of a distribution of rights to subscribe for additional ordinary shares or other rights, if we timely provide evidence satisfactory to the depositary that it may lawfully distribute such rights, the depositary will distribute warrants or other instruments in the discretion of the depositary representing rights to acquire additional ADRs. However, if we do not timely furnish such evidence, the depositary may: (i) sell such rights if practicable and distribute the net proceeds in the same manner as cash to the ADR holders entitled thereto; or (ii) if it is not practicable to sell such rights by reason of the non-transferability of the rights, limited markets therefor, their short duration or otherwise, do nothing and allow such rights to lapse, in which case ADR holders will receive nothing and the rights may lapse. |
| • | Other Distributions. In the case of a distribution of securities or property other than those described above, the depositary may either (i) distribute such securities or property in any manner it deems equitable and practicable or (ii) to the extent the depositary deems distribution of such securities or property not to be equitable and practicable, sell such securities or property and distribute any net proceeds in the same way it distributes cash. |
| • | Elective Distributions. In the case of a dividend payable at the election of our shareholders in cash or in additional ordinary shares, we will notify the depositary at least 30 days prior to the proposed distribution stating whether or not we wish such elective distribution to be made available to ADR holders. The depositary shall make such elective distribution available to ADR holders only if (i) we shall have timely requested that the elective distribution is available to ADR holders, (ii) the depositary shall have determined that such distribution is reasonably practicable and (iii) the depositary shall have received satisfactory documentation within the terms of the deposit agreement including any legal opinions of counsel that the depositary in its reasonable discretion may request. If the above conditions are not satisfied, the depositary shall, to the extent permitted by law, distribute to the ADR holders, on the basis of the same determination as is made in the local market in respect of the ordinary shares for which no election is made, either (x) cash or (y) additional ADSs representing such additional ordinary shares. If the above conditions are satisfied, the depositary shall establish procedures to enable ADR holders to elect the receipt of the proposed dividend in cash or in additional ADSs. There can be no assurance that ADR holders generally, or any ADR holder in particular, will be given the opportunity to receive elective distributions on the same terms and conditions as the holders of ordinary shares. |
72
Table of Contents
If the depositary determines in its discretion that any distribution described above is not practicable with respect to any specific registered ADR holder, the depositary may choose any method of distribution that it deems practicable for such ADR holder, including the distribution of foreign currency, securities or property, or it may retain such items, without paying interest on or investing them, on behalf of the ADR holder as deposited securities, in which case the ADSs will also represent the retained items.
Any U.S. dollars will be distributed by checks drawn on a bank in the United States for whole dollars and cents. Fractional cents will be withheld without liability and dealt with by the depositary in accordance with its then current practices.
The depositary is not responsible if it fails to determine that any distribution or action is lawful or reasonably practicable.
There can be no assurance that the depositary will be able to convert any currency at a specified exchange rate or sell any property, rights, shares or other securities at a specified price, nor that any of such transactions can be completed within a specified time period. All purchases and sales of securities will be handled by the Depositary in accordance with its then current policies, which are currently set forth in the “Depositary Receipt Sale and Purchase of Security” section of www.adr.com/Investors/FindOutAboutDRs, the location and contents of which the Depositary shall be solely responsible for.
Deposit, withdrawal and Cancellation
How does the depositary issue ADSs?
The depositary will issue ADSs if you or your broker deposit ordinary shares or evidence of rights to receive ordinary shares with the custodian and pay the fees and expenses owing to the depositary in connection with such issuance. In the case of the ADSs to be issued under this prospectus, we will arrange with the underwriters named herein to deposit such ordinary shares.
Ordinary shares deposited in the future with the custodian must be accompanied by certain delivery documentation and shall, at the time of such deposit, be registered in the name of the depositary, the custodian or a nominee of either.
The custodian will hold all deposited ordinary shares (including those being deposited by or on our behalf in connection with this offering to which this prospectus relates) for the account and to the order of the depositary for the benefit of registered holders of ADRs, to the extent not prohibited by law. ADR holders thus have no direct ownership interest in the ordinary shares and only have such rights as are contained in the deposit agreement. The custodian will also hold any additional securities, property and cash received on or in substitution for the deposited ordinary shares. The deposited ordinary shares and any such additional items are referred to as “deposited securities.”
Upon each deposit of ordinary shares, receipt of related delivery documentation and compliance with the other provisions of the deposit agreement, including the payment of the fees and charges of the depositary and any taxes or other fees or charges owing, the depositary will issue an ADR or ADRs in the name or upon the order of the person entitled thereto evidencing the number of ADSs to which such person is entitled. All of the ADSs issued will, unless specifically requested to the contrary, be part of the depositary’s direct registration system, and a registered holder will receive periodic statements from the depositary which will show the number of ADSs registered in such holder’s name. An ADR holder can request that the ADSs not be held through the depositary’s direct registration system and that a certificated ADR be issued.
How do ADR holders cancel an ADS and obtain deposited securities?
When you turn in your ADR certificate at the depositary’s office, or when you provide proper instructions and documentation in the case of direct registration ADSs, the depositary will, upon payment of certain applicable fees, charges and taxes, deliver the underlying ordinary shares to you or upon your written order. Delivery of deposited securities in certificated form will be made at the custodian’s office. At your risk, expense and request, the depositary may deliver deposited securities at such other place as you may request.
The depositary may only restrict the withdrawal of deposited securities in connection with:
| • | temporary delays caused by closing our transfer books or those of the depositary or the deposit of ordinary shares in connection with voting at a shareholders meeting, or the payment of dividends; |
| • | the payment of fees, taxes and similar charges; or |
| • | compliance with any U.S. or foreign laws or governmental regulations relating to the ADRs or to the withdrawal of deposited securities. |
73
Table of Contents
This right of withdrawal may not be limited by any other provision of the deposit agreement.
Record Dates
The depositary may, after consultation with us if practicable, fix record dates (which, to the extent applicable, shall be as near as practicable to any corresponding record dates set by us) for the determination of the registered ADR holders who will be entitled (or obligated, as the case may be):
| • | to receive any distribution on or in respect of deposited securities; |
| • | to give instructions for the exercise of voting rights; |
| • | to pay the fee assessed by the depositary for administration of the ADR program and for any expenses as provided for in the ADR; or |
| • | to receive any notice or to act in respect of other matters, |
| • | all subject to the provisions of the deposit agreement. |
Voting Rights
How do I vote?
If you are an ADR holder and the depositary asks you to provide it with voting instructions, you may instruct the depositary how to exercise the voting rights for the ordinary shares which underlie your ADSs. Subject to the next sentence, as soon as practicable after receipt from us of notice of any meeting at which the holders of ordinary shares are entitled to vote, or of our solicitation of consents or proxies from holders of ordinary shares, the depositary shall fix the ADS record date in accordance with the provisions of the deposit agreement in respect of such meeting or solicitation of consent or proxy. The depositary shall, if we request in writing in a timely manner (the depositary having no obligation to take any further action if our request shall not have been received by the depositary at least 30 days prior to the date of such vote or meeting) and at our expense and provided no legal prohibitions exist, distribute to the registered ADR holders a notice stating such information as is contained in the voting materials received by the depositary, stating that that each registered holder of ADRs on the ADS record date will, subject to any applicable provisions of the law of England and Wales, be entitled to instruct the depositary as to the exercise of any voting rights pertaining to ordinary shares underlying such holder’s ADSs, and describing how you may instruct the depositary to exercise the voting rights for the ordinary shares which underlie your ADSs, including instructions for giving a discretionary proxy to a person designated by us. For instructions to be valid, the depositary must receive them in the manner and on or before the date specified. The depositary will try, as far as is practical, subject to the provisions of or governing the underlying ordinary shares or other deposited securities, to vote or cause to be voted the ordinary shares or other deposited securities as you instruct. The depositary will only vote or attempt to vote as you instruct. Holders are strongly encouraged to forward their voting instructions to the depositary as soon as possible. Voting instructions will not be deemed to be received until such time as the ADR department responsible for proxies and voting has received such instructions notwithstanding that such instructions may have been physically received by the depositary prior to such time. The depositary will not itself exercise any voting discretion. Furthermore, neither the depositary nor its agents are responsible for any failure to carry out any voting instructions, for the manner in which any vote is cast or for the effect of any vote. Notwithstanding anything contained in the deposit agreement or any ADR, the depositary may, to the extent not prohibited by law or regulations, or by the requirements of the stock exchange on which the ADSs are listed, in lieu of distribution of the materials provided to the depositary in connection with any meeting of, or solicitation of consents or proxies from, holders of deposited securities, distribute to the registered holders of ADRs a notice that provides such holders with, or otherwise publicizes to such holders, instructions on how to retrieve such materials or receive such materials upon request (i.e., by reference to a website containing the materials for retrieval or a contact for requesting copies of the materials).
There is no guarantee that you will receive voting materials in time to instruct the depositary to vote and it is possible that you, or persons who hold their ADSs through brokers, dealers or other third parties, will not have the opportunity to exercise a right to vote.
Reports and Other Communications
Will ADR holders be able to view our reports?
The depositary will make available for inspection by ADR holders at the offices of the depositary and the custodian the deposit agreement, the provisions of or governing deposited securities, and any written communications from us which are both received by the custodian or its nominee as a holder of deposited securities and made generally available to the holders of deposited securities.
74
Table of Contents
Additionally, if we make any written communications generally available to holders of our ordinary shares, and we furnish copies thereof (or English translations or summaries) to the depositary, it will distribute the same to registered ADR holders.
Fees and Expenses
What fees and expenses will I be responsible for paying?
The depositary may charge each person to whom ADSs are issued, including, without limitation, issuances against deposits of ordinary shares, issuances in respect of share distributions, rights and other distributions, issuances pursuant to a stock dividend or stock split declared by us or issuances pursuant to a merger, exchange of securities or any other transaction or event affecting the ADSs or deposited securities, and each person surrendering ADSs for withdrawal of deposited securities or whose ADSs are cancelled or reduced for any other reason, $5.00 for each 100 ADSs (or any portion thereof) issued, delivered, reduced, cancelled or surrendered, as the case may be. The depositary may sell (by public or private sale) sufficient securities and property received in respect of a share distribution, rights and/or other distribution prior to such deposit to pay such charge.
The following additional charges shall be incurred by the ADR holders, by any party depositing or withdrawing ordinary shares or by any party surrendering ADSs and/or to whom ADSs are issued (including, without limitation, issuance pursuant to a stock dividend or stock split declared by us or an exchange of stock regarding the ADSs or the deposited securities or a distribution of ADSs), whichever is applicable:
| • | a fee of U.S.$1.50 per ADR or ADRs for transfers of certificated or direct registration ADRs; |
| • | a fee of up to U.S.$0.05 per ADS for any cash distribution made pursuant to the deposit agreement; |
| • | an aggregate fee of up to U.S.$0.05 per ADS per calendar year (or portion thereof) for services performed by the depositary in administering the ADRs (which fee may be charged on a periodic basis during each calendar year and shall be assessed against holders of ADRs as of the record date or record dates set by the depositary during each calendar year and shall be payable in the manner described in the next succeeding provision); |
| • | a fee for the reimbursement of such fees, charges and expenses as are incurred by the depositary and/or any of its agents (including, without limitation, the custodian and expenses incurred on behalf of holders in connection with compliance with foreign exchange control regulations or any law or regulation relating to foreign investment) in connection with the servicing of the ordinary shares or other deposited securities, the sale of securities (including, without limitation, deposited securities), the delivery of deposited securities or otherwise in connection with the depositary’s or its custodian’s compliance with applicable law, rule or regulation (which fees and charges shall be assessed on a proportionate basis against holders as of the record date or dates set by the depositary and shall be payable at the sole discretion of the depositary by billing such holders or by deducting such charge from one or more cash dividends or other cash distributions); |
| • | a fee for the distribution of securities (or the sale of securities in connection with a distribution), such fee being in an amount equal to the $0.05 per ADS issuance fee for the execution and delivery of ADSs which would have been charged as a result of the deposit of such securities (treating all such securities as if they were ordinary shares) but which securities or the net cash proceeds from the sale thereof are instead distributed by the depositary to those holders entitled thereto; |
| • | stock transfer or other taxes and other governmental charges; |
| • | SWIFT, cable, telex and facsimile transmission and delivery charges incurred at your request in connection with the deposit or delivery of ordinary shares, ADRs or deposited securities; |
| • | transfer or registration fees for the registration or transfer of deposited securities on any applicable register in connection with the deposit or withdrawal of deposited securities; |
| • | in connection with the conversion of foreign currency into U.S. dollars, JPMorgan Chase Bank, N.A. shall deduct out of such foreign currency the fees, expenses and other charges charged by it and/or its agent (which may be a division, branch or affiliate) so appointed in connection with such conversion; and |
| • | fees of any division, branch or affiliate of the depositary utilized by the depositary to direct, manage and/or execute any public and/or private sale of securities under the deposit agreement. |
75
Table of Contents
JPMorgan Chase Bank, N.A. and/or its agent may act as principal for such conversion of foreign currency. For further details see www.adr.com.
We will pay all other charges and expenses of the depositary and any agent of the depositary (except the custodian) pursuant to agreements from time to time between us and the depositary. The charges described above may be amended from time to time by agreement between us and the depositary. The right of the depositary to receive payment of fees, charges and expenses as provided above shall survive the termination of the deposit agreement.
The depositary may make available to us a set amount or a portion of the depositary fees charged in respect of the ADR program or otherwise upon such terms and conditions as we and the depositary may agree from time to time. The depositary collects its fees for issuance and cancellation of ADSs directly from anyone depositing ordinary shares or surrendering ADSs for the purpose of withdrawal or from intermediaries acting for them. The depositary collects fees for making distributions to you by deducting those fees from the amounts distributed or by selling a portion of distributable property to pay the fees. The depositary may collect its annual fee for depositary services by deduction from cash distributions, or by directly billing you, or by charging the book-entry system accounts of participants acting for them. The depositary will generally set off the amounts owing from distributions made to holders of ADSs. If, however, no distribution exists and payment owing is not timely received by the depositary, the depositary may refuse to provide any further services to holders that have not paid those fees and expenses owing until such fees and expenses have been paid. At the discretion of the depositary, all fees and charges owing under the deposit agreement are due in advance and/or when declared owing by the depositary.
Payment of Taxes
If any taxes or other governmental charges (including any penalties and/or interest) shall become payable by or on behalf of the custodian or the depositary with respect to any ADR, any deposited securities represented by the ADSs evidenced thereby or any distribution thereon, such tax or other governmental charge shall be paid by the holder thereof to the depositary and by holding or having held an ADR the holder and all prior holders thereof, jointly and severally, agree to indemnify, defend and save harmless each of the depositary and its agents in respect thereof. If an ADR holder owes any tax or other governmental charge, the depositary may (i) deduct the amount thereof from any cash distributions, or (ii) sell deposited securities by public or private sale (after attempting by reasonable means to notify the ADR holder hereof prior to such sale) and deduct the amount owing from the net proceeds of such sale. In either case the ADR holder remains liable for any shortfall. If any tax or governmental charge is unpaid, the depositary may also refuse to effect any registration, registration of transfer, split-up or combination of deposited securities or withdrawal of deposited securities until such payment is made. If any tax or governmental charge is required to be withheld on any cash distribution, the depositary may deduct the amount required to be withheld from any cash distribution or, in the case of a non-cash distribution, sell the distributed property or securities (by public or private sale) in such amounts and in such manner as the depositary deems necessary and practicable to pay such taxes and distribute any remaining net proceeds or the balance of any such property after deduction of such taxes to the ADR holders entitled thereto.
By holding an ADR or an interest therein, you will be agreeing to indemnify us, the depositary, its custodian and any of our or their respective officers, Directors, employees, agents and affiliates against, and hold each of them harmless from, any claims by any governmental authority with respect to taxes, additions to tax, penalties or interest arising out of any refund of taxes, reduced rate of withholding at source or other tax benefit obtained.
Reclassifications, Recapitalizations and Mergers
If we take certain actions that affect the deposited securities, including (i) any change in nominal value, split-up, consolidation, cancellation or other reclassification of deposited securities or (ii) any distributions of ordinary shares or other property not made to holders of ADRs or (iii) any recapitalization, reorganization, merger, consolidation, liquidation, receivership, bankruptcy or sale of all or substantially all of our assets, then the depositary may choose to, and shall if reasonably requested by us:
| (1) | amend the form of ADR; |
| (2) | distribute additional or amended ADRs; |
| (3) | distribute cash, securities or other property it has received in connection with such actions; |
| (4) | sell any securities or property received and distribute the proceeds as cash; or |
| (5) | none of the above. |
76
Table of Contents
If the depositary does not choose any of the above options, any of the cash, securities or other property it receives will constitute part of the deposited securities and each ADS will then represent a proportionate interest in such property.
Amendment and Termination
How may the deposit agreement be amended?
We may agree with the depositary to amend the deposit agreement and the ADSs without your consent for any reason. ADR holders must be given at least 30 days’ notice of any amendment that imposes or increases any fees or charges (other than stock transfer or other taxes and other governmental charges, transfer or registration fees, SWIFT, cable, telex or facsimile transmission costs, delivery costs or other such expenses), or otherwise prejudices any substantial existing right of ADR holders. Such notice need not describe in detail the specific amendments effectuated thereby, but must identify to ADR holders a means to access the text of such amendment. If an ADR holder continues to hold an ADR or ADRs after being so notified, such ADR holder is deemed to agree to such amendment and to be bound by the deposit agreement as so amended. Any amendments or supplements which (i) are reasonably necessary (as agreed by us and the depositary) in order for (a) the ADSs to be registered on Form F-6 under the Securities Act or (b) the ADSs or ordinary shares to be traded solely in electronic book-entry form and (ii) do not in either such case impose or increase any fees or charges to be borne by ADR holders, shall be deemed not to prejudice any substantial rights of ADR holders. Notwithstanding the foregoing, if any governmental body or regulatory body should adopt new laws, rules or regulations which would require amendment or supplement of the deposit agreement or the form of ADR to ensure compliance therewith, we and the depositary may amend or supplement the deposit agreement and the ADR at any time in accordance with such changed laws, rules or regulations, which amendment or supplement may take effect before a notice is given or within any other period of time as required for compliance. No amendment, however, will impair your right to surrender your ADSs and receive the underlying securities, except in order to comply with mandatory provisions of applicable law.
How may the deposit agreement be terminated?
The depositary may, and shall at our written direction, terminate the deposit agreement and the ADRs by mailing notice of such termination to the registered holders of ADRs at least 30 days prior to the date fixed in such notice for such termination; provided, however, if the depositary shall have (i) resigned as depositary under the deposit agreement, notice of such termination by the depositary shall not be provided to registered holders unless a successor depositary shall not be operating under the deposit agreement within 60 days of the date of such resignation, and (ii) been removed as depositary under the deposit agreement, notice of such termination by the depositary shall not be provided to registered holders of ADRs unless a successor depositary shall not be operating under the deposit agreement on the 120th day after our notice of removal was first provided to the depositary. After termination, the depositary’s only responsibility will be (i) to deliver deposited securities to ADR holders who surrender their ADRs, and (ii) to hold or sell distributions received on deposited securities. As soon as practicable after the expiration of six months from the termination date, the depositary will sell the deposited securities which remain and hold the net proceeds of such sales (as long as it may lawfully do so), without liability for interest, in trust for the ADR holders who have not yet surrendered their ADRs. After making such sale, the depositary shall have no obligations except to account for such proceeds and other cash.
Limitations on Obligations and Liability to ADR holders
Limits on our obligations and the obligations of the depositary; limits on liability to ADR holders and holders of ADSs
Prior to the issue, registration, registration of transfer, split-up, combination, or cancellation of any ADRs, or the delivery of any distribution in respect thereof, and from time to time in the case of the production of proofs as described below, we or the depositary or its custodian may require:
| • | payment with respect thereto of (i) any stock transfer or other tax or other governmental charge, (ii) any stock transfer or registration fees in effect for the registration of transfers of ordinary shares or other deposited securities upon any applicable register and (iii) any applicable fees and expenses described in the deposit agreement; |
| • | the production of proof satisfactory to it of (i) the identity of any signatory and genuineness of any signature and (ii) such other information, including without limitation, information as to citizenship, residence, exchange control approval, beneficial ownership of any securities, compliance with applicable law, regulations, provisions of or governing deposited securities and terms of the deposit agreement and the ADRs, as it may deem necessary or proper; and |
| • | compliance with such regulations as the depositary may establish consistent with the deposit agreement. |
77
Table of Contents
The issuance of ADRs, the acceptance of deposits of ordinary shares, the registration, registration of transfer, split-up or combination of ADRs or the withdrawal of ordinary shares, may be suspended, generally or in particular instances, when the ADR register or any register for deposited securities is closed or when any such action is deemed advisable by the depositary; provided that the ability to withdraw ordinary shares may only be limited under the following circumstances: (i) temporary delays caused by closing transfer books of the depositary or our transfer books or the deposit of ordinary shares in connection with voting at a shareholders meeting, or the payment of dividends, (ii) the payment of fees, taxes, and similar charges, and (iii) compliance with any laws or governmental regulations relating to ADRs or to the withdrawal of deposited securities.
The deposit agreement expressly limits the obligations and liability of the depositary, ourselves and each of our and the depositary’s respective agents, provided, however, that no disclaimer of liability under the Securities Act or the Exchange Act, to the extent applicable, is intended by any provision of the deposit agreement. In the deposit agreement it provides that neither we nor the depositary nor any such agent will be liable to registered holders or beneficial owners of ADSs if:
| • | any present or future law, rule, regulation, fiat, order or decree of the United States, England and Wales or any other country or jurisdiction, or of any governmental or regulatory authority or securities exchange or market or automated quotation system, the provisions of or governing any deposited securities, any present or future provision of our charter, any act of God, war, terrorism, nationalization, expropriation, currency restrictions, work stoppage, strike, civil unrest, revolutions, rebellions, explosions, computer failure or circumstance beyond our, the depositary’s or our respective agents’ direct and immediate control shall prevent or delay, or shall cause any of them to be subject to any civil or criminal penalty in connection with, any act which the deposit agreement or the ADRs provide shall be done or performed by us, the depositary or our respective agents (including, without limitation, voting); |
| • | it exercises or fails to exercise discretion under the deposit agreement or the ADRs including, without limitation, any failure to determine that any distribution or action may be lawful or reasonably practicable; |
| • | it performs its obligations under the deposit agreement and ADRs without gross negligence or willful misconduct; or |
| • | it takes any action or refrains from taking any action in reliance upon the advice of or information from legal counsel, accountants, any person presenting ordinary shares for deposit, any registered holder of ADRs, or any other person believed by it to be competent to give such advice or information. |
We, the depositary and its agents may rely and shall be protected in acting upon any written notice, request, direction, instruction or document believed by them to be genuine and to have been signed, presented or given by the proper party or parties.
Neither the depositary nor its agents have any obligation to appear in, prosecute or defend any action, suit or other proceeding in respect of any deposited securities or the ADRs. We and our agents shall only be obligated to appear in, prosecute or defend any action, suit or other proceeding in respect of any deposited securities or the ADRs, which in our opinion may involve us in expense or liability, if indemnity satisfactory to us against all expense (including fees and disbursements of counsel) and liability is furnished as often as may be required. The depositary and its agents may fully respond to any and all demands or requests for information maintained by or on its behalf in connection with the deposit agreement, any registered holder or holders of ADRs, any ADRs or otherwise related to the deposit agreement or ADRs to the extent such information is requested or required by or pursuant to any lawful authority, including without limitation laws, rules, regulations, administrative or judicial process, banking, securities or other regulators. The depositary shall not be liable for the acts or omissions made by, or the insolvency of, any securities depository, clearing agency or settlement system. Furthermore, the depositary shall not be responsible for, and shall incur no liability in connection with or arising from, the insolvency of any custodian that is not a branch or affiliate of JPMorgan Chase Bank, N.A. Notwithstanding anything to the contrary contained in the deposit agreement or any ADRs, the depositary shall not be responsible for, and shall incur no liability in connection with or arising from, any act or omission to act on the part of the custodian except to the extent that any registered holder of ADRs has incurred liability directly as a result of the custodian having (i) committed fraud or willful misconduct in the provision of custodial services to the depositary or (ii) failed to use reasonable care in the provision of custodial services to the depositary as determined in accordance with the standards prevailing in the jurisdiction in which the custodian is located. The depositary shall not have any liability for the price received in connection with any sale of securities, the timing thereof or any delay in action or omission to act nor shall it be responsible for any error or delay in action, omission to act, default or negligence on the part of the party so retained in connection with any such sale or proposed sale.
The depositary has no obligation to inform ADR holders or other holders of an interest in any ADSs about the requirements of the law of England and Wales, rules or regulations or any changes therein or thereto.
78
Table of Contents
Neither the depositary nor its agents will be responsible for any failure to carry out any instructions to vote any of the deposited securities, for the manner in which any such vote is cast or for the effect of any such vote. The depositary may rely upon instructions from us or our counsel in respect of any approval or license required for any currency conversion, transfer or distribution. The depositary shall not incur any liability for the content of any information submitted to it by us or on our behalf for distribution to ADR holders or for any inaccuracy of any translation thereof, for any investment risk associated with acquiring an interest in the deposited securities, for the validity or worth of the deposited securities, for the credit-worthiness of any third party, for allowing any rights to lapse upon the terms of the deposit agreement or for the failure or timeliness of any notice from us. The depositary shall not be liable for any acts or omissions made by a successor depositary whether in connection with a previous act or omission of the depositary or in connection with any matter arising wholly after the removal or resignation of the depositary. Neither the depositary nor any of its agents shall be liable to registered holders or beneficial owners of interests in ADSs for any indirect, special, punitive or consequential damages (including, without limitation, legal fees and expenses) or lost profits, in each case of any form incurred by any person or entity, whether or not foreseeable and regardless of the type of action in which such a claim may be brought.
In the deposit agreement each party thereto (including, for avoidance of doubt, each holder and beneficial owner and/or holder of interests in ADRs) irrevocably waives, to the fullest extent permitted by applicable law, any right it may have to a trial by jury in any suit, action or proceeding against the depositary and/or us directly or indirectly arising out of or relating to the ordinary shares or other deposited securities, the ADSs or the ADRs, the deposit agreement or any transaction contemplated therein, or the breach thereof (whether based on contract, tort, common law or any other theory).
The depositary and its agents may own and deal in any class of securities of our company and our affiliates and in ADSs.
Disclosure of Interest in ADSs
To the extent that the provisions of or governing any deposited securities may require disclosure of or impose limits on beneficial or other ownership of deposited securities, other ordinary shares and other securities and may provide for blocking transfer, voting or other rights to enforce such disclosure or limits, you agree to comply with all such disclosure requirements and ownership limitations and to comply with any reasonable instructions we may provide in respect thereof. We reserve the right to instruct you to deliver your ADSs for cancellation and withdrawal of the deposited securities so as to permit us to deal with you directly as a holder of ordinary shares and, by holding an ADS or an interest therein, you will be agreeing to comply with such instructions.
Books of Depositary
The depositary or its agent will maintain a register for the registration, registration of transfer, combination and split-up of ADRs, which register shall include the depositary’s direct registration system. Registered holders of ADRs may inspect such records at the depositary’s office at all reasonable times, but solely for the purpose of communicating with other holders in the interest of the business of our company or a matter relating to the deposit agreement. Such register may be closed at any time or from time to time, when deemed expedient by the depositary.
The depositary will maintain facilities for the delivery and receipt of ADRs.
Appointment
In the deposit agreement, each registered holder of ADRs and each person holding an interest in ADSs, upon acceptance of any ADSs (or any interest therein) issued in accordance with the terms and conditions of the deposit agreement will be deemed for all purposes to:
| • | be a party to and bound by the terms of the deposit agreement and the applicable ADR or ADRs; and |
| • | appoint the depositary its attorney-in-fact, with full power to delegate, to act on its behalf and to take any and all actions contemplated in |
the deposit agreement and the applicable ADR or ADRs, to adopt any and all procedures necessary to comply with applicable laws and to take such action as the depositary in its sole discretion may deem necessary or appropriate to carry out the purposes of the deposit agreement and the applicable ADR and ADRs, the taking of such actions to be the conclusive determinant of the necessity and appropriateness thereof.
79
Table of Contents
Governing Law
The deposit agreement and the ADRs shall be governed by and construed in accordance with the laws of the State of New York. In the deposit agreement, we have submitted to the jurisdiction of the courts of the State of New York and appointed an agent for service of process on our behalf. Notwithstanding the foregoing, any action based on the deposit agreement or the transactions contemplated thereby may be instituted by the depositary in any competent court in England and Wales.
By holding an ADS or an interest therein, registered holders of ADRs and owners of ADSs each irrevocably agree that any legal suit, action or proceeding against or involving us or the depositary, arising out of or based upon the deposit agreement, the ADSs or the transactions contemplated thereby, may only be instituted in a state or federal court in New York, New York, and each irrevocably waives any objection which it may have to the laying of venue of any such proceeding, and irrevocably submits to the exclusive jurisdiction of such courts in any such suit, action or proceeding.
| ITEM 13. | DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES |
Not applicable.
| ITEM 14. | MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS |
Not applicable.
| ITEM 15. | CONTROLS AND PROCEDURES |
Not applicable.
| ITEM 16. | RESERVED |
| ITEM 16A. | AUDIT COMMITTEE FINANCIAL EXPERT |
Our Board of Directors has determined that John Brancaccio is an “audit committee financial expert” as defined in Item 16.A. of Form 20-F.
| ITEM 16B. | CODE OF ETHICS |
Not applicable
| ITEM 16C. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
Not applicable.
| ITEM 16D. | EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES |
Not applicable.
| ITEM 16E. | PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS |
Not applicable.
| ITEM 16F. | CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT |
Not applicable.
| ITEM 16G. | CORPORATE GOVERNANCE |
Not applicable.
80
Table of Contents
| ITEM 16H. | MINE SAFETY DISCLOSURE |
Not applicable.
| ITEM 17. | FINANCIAL STATEMENTS |
We have elected to furnish financial statements and related information specified in Item 18.
| ITEM 18. | FINANCIAL STATEMENTS |
See the financial statements beginning on page F-1 of this registration statement.
| ITEM 19. | EXHIBITS |
The Exhibits listed in the Exhibit Index at the end of this registration statement are filed as Exhibits to this registration statement.
81
Table of Contents
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Consolidated Financial Statements as of and for the Year Ended December 31, 2020
Report of Mazars LLP, Independent Registered Public Accounting Firm | F-2 | |||
| F-3 | ||||
Consolidated Statements of Operations and Comprehensive Loss | F-4 | |||
| F-5 | ||||
| F-6 | ||||
| F-7 |
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Shareholders of AccuStem Sciences Ltd.
Opinion on the special period Financial Statements
We have audited the accompanying balance sheet of AccuStem Sciences Ltd. as of December 31, 2020, together with the related statements of operations and comprehensive loss, changes in shareholders’ equity, and cash flows for the period from June 5, 2020 to December 31, 2020, including the related notes (collectively, the financial statements).
In our opinion, the financial statements present fairly, in all material respects, the financial position of the Group as of December 31, 2020, and the results of its operations and its cash flows for each of the period June 5, 2020 to December 31, 2020, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Basis for Opinion
These financial statements are the responsibility of the company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Group in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Mazars LLP
London, England
March 10, 2021
F-2
Table of Contents
| As at December 31, 2020 | ||||
ASSETS | ||||
Non-Current Assets | ||||
Intangible asset In Process Research & Development | £ | 2,073,930 | ||
Current Assets | ||||
Related party receivable | 1,151,383 | |||
Total assets | 3,225,313 | |||
LIABILITIES | ||||
Related party payable | 9,892 | |||
Other payable | 30,797 | |||
Total liabilities | 40,689 | |||
EQUITY AND LIABILITIES | ||||
Equity attributable to owners | ||||
Ordinary Share capital | 2,041,935 | |||
Share premium | 55,571 | |||
Merger reserve | 1,127,807 | |||
Retained earnings | (40,689 | ) | ||
Total equity attributable to Shareholders | 3,184,624 | |||
Total equity and liabilities | 3,225,313 | |||
F-3
Table of Contents
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
| Period from June 5, 2020 to December 31, 2020 | ||||
Revenue | £ | — | ||
Research and development expenses | (4,347 | ) | ||
Administrative expenses | (36,342 | ) | ||
Operating result | — | |||
Finance income/(expense) | — | |||
Profit before taxation | (40,689 | ) | ||
Income tax | — | |||
Profit for the period and total comprehensive income for the period | (40,689 | ) | ||
Total basic and diluted loss per share (£ per share) | (0.00 | ) | ||
F-4
Table of Contents
CONSOLIDATED STATEMENT OF SHAREHOLDERS’ EQUITY
Share £ | Share £ | Merger £ | Retained £ | Total £ | ||||||||||||||||
Balance at 5 June 2020 | — | — | — | — | — | |||||||||||||||
Transactions with owners | ||||||||||||||||||||
Issue of share capital | 2,041,935 | 55,571 | 1,127,807 | 3,225,313 | ||||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total transactions with owners | 2,041,935 | 55,571 | 1,127,807 | 3,225,313 | ||||||||||||||||
Comprehensive income | ||||||||||||||||||||
Loss for the period | — | — | (40,689 | ) | (40,689 | ) | ||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total comprehensive income | — | — | (40,689 | ) | (40,689 | ) | ||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Balance at 31 December 2020 | 2,041,935 | 55,571 | 1,127,807 | (40,689 | ) | 3,184,624 | ||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
F-5
Table of Contents
CONSOLIDATED STATEMENT OF CASH FLOWS
Period from £ | ||||
CASH FLOWS FROM OPERATING ACTIVITIES: | ||||
Loss from operations before income taxes | (40,689 | ) | ||
Increase in related party payable | 9,892 | |||
Increase in trade payable | 30,797 | |||
Net cash used in operating activities | — | |||
Net increase in cash and cash equivalents | — | |||
|
| |||
Cash and cash equivalent, beginning of period | — | |||
|
| |||
Cash and cash equivalent, end of period | — | |||
|
| |||
Non cash items: | ||||
Cash receivable from related party | 1,000,000 | |||
Shares allotted but not yet been paid | 151,383 | |||
F-6
Table of Contents
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| 1. | General Information |
AccuStem Sciences Limited is a private limited company incorporated on June 5, 2020 in the United Kingdom under the Companies Act. The principal activities of the Company and its subsidiaries (the “Group”) are that of a genomics-based personalized medicine business, particularly focused on breast cancer patients.
The ultimate parent of the group is AccuStem Sciences Limited.
These financial statements are have been prepared for the period from June 5, 2020 to December 31, 2020 and are presented in thousands UK Pounds Sterling (£) which is the presentational and functional currency of the Company, indicative of the primary economic environment in which the Company operates.
2. Accounting Policies
The principal accounting policies applied in the preparation of these consolidated financial statements are set out below. These policies have been applied to the period presented unless otherwise stated.
Basis of preparation
The consolidated financial statements of the Group have been prepared in accordance with International Financial Reporting Standards (IFRS) as issued by the International Accounting Standards Board (IASB), and IFRIC interpretations as applicable to companies reporting under IFRS.
New and Revised Standards
Standards in effect in 2020
An amendment to IFRS 3 ‘Definition of a business’ has come into effect from January 1, 2020. The adoption of the amendment has had no impact on the company.
IFRS in issue but not applied in the current financial statements
The Directors do not expect that the adoption of new IFRS Standards, Interpretations and Amendments that have been issued but are not yet effective will have a material impact on the financial statements of the Group in future periods.
A number of IFRS and IFRIC interpretations are also currently in issue which are not relevant for the Group’s activities and which have not therefore been adopted in preparing these financial statements.
Going Concern
The financial statements have been prepared on the going concern basis, which contemplates continuity of normal business activities and the realization of assets and discharge of liabilities in the normal course of business.
The directors believe that there are reasonable grounds to believe that the company and consolidated entity will be able to continue as going concerns, after consideration of the following factors;
| • | Cash receivable totaling £1.2 million at 31 December 2020 |
| • | Tiziana Life Sciences plc (Tiziana) has agreed to invest £2m in the Company upon the Company’s listing. Tiziana has sufficient funds to meet this obligation. |
| • | The Directors have prepared cash flow projections that include the costs associated with development of the StemPrintER asset. On the basis of those projections, the directors conclude that the company will be able to meet its liabilities as they fall due for the next 12 months from the date when these financial statements are issued. |
Accordingly, the directors believe that the Company and consolidated entities will be able to continue as going concerns and that it is appropriate to adopt the going concern basis in the preparation of these financial statements. The financial statements do not include any adjustment relating to the amounts or classification of recorded assets or liabilities that might be necessary if the Company and consolidated entities do not continue as going concerns.
F-7
Table of Contents
Basis of consolidation
Subsidiary undertakings are all entities over which the Group has the power to govern the financial and operating policies of the subsidiary and therefore exercises control. The existence and effect of both current voting rights and potential voting rights that are currently exercisable or convertible are considered when assessing whether control of an entity is exercised. Subsidiaries are consolidated from the date at which the Group obtains control and are de-consolidated from the date at which control ceases.
Inter-company transactions, balances, and unrealized gains on transactions between group companies are eliminated upon consolidation. Unrealized losses are also eliminated. Accounting policies of subsidiaries have been changed where necessary to ensure consistency with the policies adopted by the group.
Common control combinations
Merger accounting is used by the Group for common control combinations, which are transactions between entities that are ultimately controlled by the same party or parties. This method treats the merged entities as if they had been combined throughout the current and comparative accounting periods.
There are however no comparatives presented in these financial statements as the business asset did not exist until the statutory demerger of StemPrintER Sciences Ltd.
Merger accounting principles for these combinations result in the recognition of a merger reserve in the consolidated statement of financial position, being the difference between the nominal value of any new shares issued or allotted by the parent company for the acquisition of the shares of the subsidiary and the subsidiary’s Net Asset Value. See note 4.
Financial instruments
The Group classifies a financial instrument, or its component parts, as a financial liability, a financial asset or an equity instrument in accordance with the substance of the contractual arrangement and the definitions of a financial liability, a financial asset and an equity instrument.
The Group evaluates the terms of the financial instrument to determine whether it contains an asset, a liability or an equity component. Such components shall be classified separately as financial assets, financial liabilities or equity instruments.
The Group’s financial liabilities include trade and other payables.
The Group does not hold any financial assets or liabilities at fair value through profit or loss or fair value through other comprehensive income.
Intangible Assets
The StemPrintER asset has been recognized as intangible in-process research & development (“IPR&D”) with an indefinite useful life. This asset arose through the transaction described in note 4.
IPR&D assets are considered to have an indefinite useful life until the completion or abandonment of the associated research and development project. Management evaluates indefinite life intangible assets for impairment on an annual basis and on an interim basis if events or changes in circumstances between annual impairment tests indicate that the asset might be impaired. The ongoing evaluation for impairment of its indefinite life intangible assets requires significant management estimates and judgment.
Management has prepared a budget for the ongoing development of the asset and concludes that as at December 31, 2020 there has been no impairment trigger.
F-8
Table of Contents
Impairment
Impairment of financial assets measured at amortized cost
At each reporting date the Group recognizes a loss allowance for expected credit losses on financial assets measured at amortized cost.
In establishing the appropriate amount of loss allowance to be recognized, the Group applies either the general approach or the simplified approach, depending on the nature of the underlying group of financial assets.
General approach
The general approach is applied to the impairment assessment of cash and cash and cash equivalents.
Under the general approach the Group recognizes a loss allowance for a financial asset at an amount equal to the 12-month expected credit losses, unless the credit risk on the financial asset has increased significantly since initial recognition, in which case a loss allowance is recognized at an amount equal to the lifetime expected credit losses.
Simplified approach
Under the simplified approach the Group always recognizes a loss allowance for a financial asset at an amount equal to the lifetime expected credit losses.
Non-financial assets are tested for impairment whenever events or changes in circumstances indicate that the carrying amount may not be recoverable.
Non-financial assets are impaired when its carrying amount exceed its recoverable amount. The recoverable amount is measured as the higher of fair value less cost of disposal and value in use. The value in use is calculated as being net projected cash flows based on financial forecasts discounted back to present value.
Share capital
Ordinary shares of the Parent Company are classified as equity.
| 3. | CRITICAL ACCOUTING AND JUDGEMENTS |
The preparation of financial information in accordance with generally accepted accounting practice, in the case of the Group being International Financial Reporting Standards as issued by the International Accounting Standards Board, requires the directors to make estimates and judgements that affect the reported amount of assets, liabilities, income and expenditure and the disclosures made in the financial statements. Such estimates and judgements must be continually evaluated based on historical experience and other factors, including expectations of future events.
The Group has been involved in a demerger transaction during the period and management has used its judgement to determine whether the transaction was one of common control, or an acquisition through a business combination.
The Group has also made a judgement on the impact of Brexit and COVID during the preparation of the financial statements and considered it to not be significant. Indeed, the Company is planning to file a registration statement form 20-F due to a biotech boom in US, caused in part by the importance of this industry driven by the COVID pandemic.
The Group has also made a judgment regarding the recoverability of the related party receivable due. The Group does not deem there to be any risk involved with recoverability of the receivable.
F-9
Table of Contents
| 4. | ACQUISITION OF SUBSIDIARY |
The consolidated position of the Group is a result of the demerger of the legal entity StemPrintER Sciences Limited from Tiziana Life Sciences plc (Tiziana) on October 30, 2020. The transaction is as detailed below:
In September 2020, Tiziana Life Sciences plc transferred all the ownership rights and intellectual property relating to StemPrintER along with £1.0 million in cash to its wholly owned subsidiary, StemPrinter Sciences Limited in exchange for shares in the subsidiary.
On October 5, 2020, the Company entered into an agreement with Tiziana Life Sciences plc to acquire its subsidiary StemPrintER Sciences Limited, including the ownership rights and intellectual property relating to StemPrintER and cash of |£1.0 million contained within the entity. In exchange for the transfer of ownership, the Company allotted 194,612,288 ordinary shares of £0.01 par value to Tiziana shareholders on a one for one basis based on the Tiziana ownership as at October 30, 2020.
The initial distribution by Tiziana was recorded at fair value, and that is what drives the fair value of the intangible asset in StemPrintER Sciences Limited with a fair value of £2,073,930. The fair value of the asset was approximated by its cost to recreate, using historical cost, as this was identified as the best valuation method for determining fair value.
As the transaction was between entities that were ultimately controlled by the same parties, the acquisition has been treated as a common control combination. No comparatives have been presented, however as the StemPrintER asset did not exist in the prior year.
The net assets acquired within StemPrintER Sciences Limited are therefore as follows:
| £ | ||||
Intangible asset | 2,073,930 | |||
Cash receivable (Note 7) | 1,000,000 | |||
|
| |||
Net Assets | 3,073,930 | |||
|
| |||
| 5. | INTANGIBLE ASSETS |
The IPR&D intangible asset has an indefinite useful life.
Management evaluates indefinite life intangible assets for impairment on an annual basis and on an interim basis if events or changes in circumstances between annual impairment tests indicate that the asset might be impaired. The ongoing evaluation for impairment of its indefinite life intangible assets requires significant management estimates and judgment.
Management has prepared a budget for the ongoing development of the asset and concludes that as at December 31, 2020 there has been no impairment trigger. The long term intention by management is to invest more than £2m in the asset.
| Company and Group | IPR&D £ | |||
Cost | ||||
At June 5, 2020 | — | |||
Acquired through common control combination | 2,073,930 | |||
|
| |||
At December 31, 2020 | 2,073,930 | |||
|
| |||
F-10
Table of Contents
| 6. | SHARE CAPITAL AND MERGER RESERVE |
Share Capital
| Company and Group | Ordinary Number | Share £ | Share Premium £ | Merger £ | Total £ | |||||||||||||||
In issue at June 5, 2020 | 1 | — | — | — | ||||||||||||||||
Allotted in exchange for acquisition of StemPrintER Sciences Ltd | 194,612,288 | 1,946,123 | — | 1,127,807 | 3,073,930 | |||||||||||||||
Allotment of additional shares | 9,581,254 | 95,812 | 55,571 | — | 151,383 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Allotted or issued at December 31, 2020 | 204,193,543 | 2,041,935 | 55,571 | 1,127,807 | 3,225,313 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Ordinary Shares
Ordinary shares have a par value of £0.01. Every holder of ordinary shares is entitled to one vote, to participate in dividends, and to share in the proceeds of winding up the company in proportion to the number of and amounts paid on the shares held. On a show of hands every holder of ordinary shares present at a meeting in person or by proxy, is entitled to one vote, and upon a poll each share is entitled to one vote. The Company does not have a limited amount of authorized capital.
The additional shares were allotted to option and warrant holders who were identified as eligible to receive shares in the Company but were not on the Tiziana register as at 7.00am on October 30, 2020 due to constraints on the timing of the delivery of Tiziana shares under Note 11 to Rule 9 of the Takeover code.
Merger reserve
On October 30, 2020, the Group acquired StemPrinter Sciences Ltd from its then parent company Tiziana Life Sciences plc. StemPrinter Sciences Ltd had a net asset value of £3,073,930. The transfer was effected by the allotment of 194,612,288 ordinary shares to Tiziana shareholders on a one for one basis based on the Tiziana ownership as at October 30, 2020. giving rise to a merger reserve of £1,127,807 in the consolidated statement of financial position, being the difference between the nominal value of the shares issued or allotted by the Company for the acquisition of the shares of the subsidiary and the subsidiary’s net asset value.
| 7. | LOSS PER SHARE |
Basic loss per share is calculated by dividing the loss attributable to equity holders of the company by the weighted average number of ordinary shares in issue during the year.
| December 31, 2020 | ||||
(Loss) attributable to equity holders of the Company (£) | (40,689 | ) | ||
Weighted average number of ordinary shares in issue | 204,193,543 | |||
|
| |||
Basic loss per share (£ per share) | (0.00 | ) | ||
|
| |||
The basic and diluted earnings per share as presented on the face of the Income Statement are identical. All earnings per share figures presented above arise from continuing and total operations and therefore no earnings per share for discontinued operations are presented.
| 8. | RELATED PARTY TRANSACTIONS |
Tiziana Life Sciences PLC (“Tiziana”)
Tiziana Life Sciences plc is a related party as it is under common control, it shares directors, officers and shareholders. The Group has also been formed due to an acquisition of a subsidiary company of Tiziana, see Note 4 for further details.
As at December 31, 2020, £1,151,383 was due from Tiziana, made up of the cash due to StemPrintER Sciences Limited of £1,000,000 plus an additional share subscription of 9,581,254 shares allotted at £0.0158 of £151,383.
As at December 31, 2020, £9,892 was also due to Tiziana, as Tiziana had paid for expenses on behalf of the Company.
F-11
Table of Contents
| 9. | FINANCIAL INSTRUMENTS |
The main risks arising from the Group’s financial instruments are liquidity risk, foreign currency risk and credit risk. The directors regularly review and agree policies for managing each of these risks which are summarized below.
Credit risk
The Group has exposure to credit risk arising from its outstanding related party receivables balance. The Group reviews its related party arrangements carefully to minimize such risks and as Tiziana is a related party with common directors, officers and shareholders, this risk is viewed as minimal, especially as the liquidity of Tiziana is sufficient to cover this receivable.
Liquidity risk
The Group’s policy is to regularly monitor current and expected liquidity requirements to ensure that it maintains sufficient reserves of cash to meet its liquidity requirements in the short and long term. The Group finances its activities through cash generated from by private offerings of equity and debt securities.
The table below summarizes the maturity profile of the Group’s financial liabilities based on contractual undiscounted payments:
| December 31, 2020 | ||||||||||||
| £000 | Less than 1 year | Above 1 year | Total | |||||||||
Trade and other payables | 30,797 | — | 30,797 | |||||||||
Related party payables | 9,892 | — | 9.892 | |||||||||
|
|
|
|
|
| |||||||
| 40,689 | — | 40,689 | ||||||||||
|
|
|
|
|
| |||||||
| 10. | CAPITAL RISK MANAGEMENT |
For the purpose of the Group’s capital management, capital includes called up share capital share premium and merger reserve attributable to the equity holders of the parent as reflected in the statement of financial position.
The Company’s objectives when managing capital are to safeguard the Company’s ability to continue as a going concern and to maximize shareholder value through the optimization of the equity balance.
The Group adjusts its capital structure in light of changes in economic conditions and expected business demands on capital. In order to maintain or adjust its capital structure, the Group considers whether or not to pay dividends and adjusts the amount of any dividend payments to shareholders. The Group may also return capital to shareholders or issue additional shares.
| 11. | FINANCIAL COMMITMENTS |
The Group’s main financial commitment relates to the contractual payments in respect of its licensing agreements. Due to the uncertain nature of scientific research and development and the length of time required to reach product commercialization, the Group does not detail pre-clinical, clinical and commercial payments until there is a reasonable certainty that the obligation will become due. The Group details contractual commitments when the amounts are known and certain.
F-12
Table of Contents
EXHIBIT INDEX
| * | Previously filed. |
Table of Contents
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this registration statement on its behalf.
Date: May 6, 2021
ACCUSTEM SCIENCES LIMITED | ||
| By: | /S/ KEEREN SHAH | |
Name: Keeren Shah | ||
Title: Finance Director | ||