As filed with the Securities and Exchange Commission on July 17, 2024
Registration No. 333-280670
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 1 to
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
Incannex Healthcare Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 2834 | 93-2403210 | ||
| (State or other jurisdiction of incorporation or organization) | (Primary Standard Industrial Classification Code Number) | (I.R.S. Employer Identification Number) |
Suite 105, 8 Century Circuit
Norwest, NSW 2153
Australia
+61 409 840 786
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
George Anastassov
Director
Incannex Healthcare Inc.
221 Dosoris Lane
Glen Cove, NY 11542
+1 917 607 2057
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Andrew Reilly
Rimôn Law
Level 2, 50 Bridge Street
Sydney, NSW 2000
Australia
+61 2 9055 6965
andrew.reilly@rimonlaw.com
Approximate date of commencement of proposed sale to the public: As soon as practicable after this Registration Statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act, check the following box: ☐
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ | Non-accelerated filer ☒ | Smaller reporting company ☒ |
Emerging growth company ☒
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☐
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and we are not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to completion, dated July 17, 2024
PRELIMINARY PROSPECTUS

Up to 4,000,000 Shares of Common Stock
Pre-Funded Warrants to Purchase up to 4,000,000 Shares of Common Stock
Common Warrants to Purchase up to 4,000,000 Shares of Common Stock
Up to 8,000,000 Shares of Common Stock Underlying the Pre-Funded Warrants and Common Warrants
We are offering 4,000,000 shares of common stock together with common warrants to purchase up to 4,000,000 shares of common stock at an assumed public offering price for each share of common stock and accompanying common warrant of $2.76, the last reported sale price of our common stock on the Nasdaq Global Market on July 11, 2024. Each share of common stock, or pre-funded warrant in lieu thereof, is being sold together with a common warrant to purchase one share of common stock. The shares of common stock, or pre-funded warrants in lieu thereof, and common warrants are immediately separable and will be issued separately in this offering, but must be purchased together in this offering. Each common warrant will have an exercise price of $ , will be immediately exercisable and will expire on the fifth anniversary of the original issuance date.
We are also offering to certain purchasers whose purchase of shares of common stock in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock immediately following the consummation of this offering, the opportunity to purchase, if any such purchaser so chooses, pre-funded warrants, in lieu of shares of common stock that would otherwise result in such purchaser’s beneficial ownership exceeding 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock. The public offering price of each pre-funded warrant to purchase one share of common stock and accompanying common warrant to purchase one share of common stock will be equal to the price at which one share of common stock and accompanying common warrant to purchase one share of common stock is sold to the public in this offering, minus $0.001, and the exercise price of each pre-funded warrant will be $0.001 per share. The pre-funded warrants will be immediately exercisable and may be exercised at any time until all of the pre-funded warrants are exercised in full.
This offering will terminate on September 15, 2024, unless we decide to terminate the offering (which we may do at any time in our discretion) prior to that date. We will have one closing for all the securities purchased in this offering.
Our Common Stock is listed on the Nasdaq Global Market, or Nasdaq, under the symbol “IXHL”. On July 11, 2024, the last reported sale price of our common stock on Nasdaq was $2.76 per share of common stock. The public offering price per share of common stock and per common warrant and per pre-funded warrant will be determined between us and investors based on market conditions at the time of pricing, and may be at a discount to the then current market price of our common stock. The recent market price used throughout this prospectus may not be indicative of the actual offering price. The actual public offering price may be based upon a number of factors, including our history and our prospects, the industry in which we operate, our past and present operating results, the previous experience of our executive officers and the general condition of the securities markets at the time of this offering.
There is no established public trading market for the pre-funded warrants or common warrants and we do not expect a market to develop. Without an active trading market, the liquidity of the common warrants will be limited. In addition, we do not intend to list the pre-funded warrants or common warrants on the Nasdaq Global Market, any other national securities exchange or any other trading system.
We have engaged JonesTrading Institutional Services LLC and Lake Street Capital Markets, LLC (the “joint placement agents”) to act as our exclusive joint placement agents in connection with this offering. The joint placement agents have agreed to use their reasonable best efforts to arrange for the sale of the securities offered by this prospectus. The joint placement agents are not purchasing or selling any of the securities we are offering and the joint placement agents are not required to arrange the purchase or sale of any specific number of securities or dollar amount. We have agreed to pay to the joint placement agents the joint placement agents fees set forth in the table below, which assumes that we sell all of the securities offered by this prospectus. Since we will deliver the securities to be issued in this offering upon our receipt of investor funds, there is no arrangement for funds to be received in escrow, trust or similar arrangement.
There is no minimum offering requirement as a condition of closing of this offering. Because there is no minimum offering amount required as a condition to closing this offering, we may sell fewer than all of the securities offered hereby, which may significantly reduce the amount of proceeds received by us, and investors in this offering will not receive a refund in the event that we do not sell an amount of securities sufficient to pursue our business goals described in this prospectus. In addition, because there is no escrow account and no minimum offering amount, investors could be in a position where they have invested in our company, but we are unable to fulfill all of our contemplated objectives due to a lack of interest in this offering.
Further, any proceeds from the sale of securities offered by us will be available for our immediate use, despite uncertainty about whether we would be able to use such funds to effectively implement our business plan. See the section entitled “Risk Factors” for more information. We will bear all costs associated with the offering. See “Plan of Distribution” on page 126 of this prospectus for more information regarding these arrangements.
| Per Share and Common Warrant | Per Pre-Funded Warrant and Warrant | Total | ||||||||||
| Public offering price | $ | $ | $ | |||||||||
| Placement agent fees | $ | $ | $ | |||||||||
| Proceeds, before expenses, to us | $ | $ | $ | |||||||||
Delivery of the shares of common stock, pre-funded warrants and common warrants offered hereby is expected to be made on or about ____, 2024, subject to the satisfaction of customary closing conditions.
Investing in our securities involves a high degree of risk. See the section entitled “Risk Factors” appearing on pages 8 of this prospectus and elsewhere in this prospectus and the accompanying base prospectus for a discussion of information that should be considered in connection with an investment in our securities.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
| Jones | Lake Street |
The date of this prospectus is , 2024
TABLE OF CONTENTS
You should rely only on the information contained in this prospectus and any related free-writing prospectus that we authorize to be distributed to you. We have not authorized any person to provide you with information different from that contained in this prospectus or any related free-writing prospectus that we authorize to be distributed to you. This prospectus is not an offer to sell, nor is it seeking an offer to buy, these securities in any state where the offer or sale is not permitted. The information in this prospectus speaks only as of the date of this prospectus unless the information specifically indicates that another date applies, regardless of the time of delivery of this prospectus.
No action is being taken in any jurisdiction outside the United States to permit a public offering of the securities or possession or distribution of this prospectus in that jurisdiction. Persons who come into possession of this prospectus in jurisdictions outside the United States are required to inform themselves about and to observe any restrictions as to this offering and the distribution of the prospectus applicable to that jurisdiction.
i
Trademarks
We own or have rights to trademarks and trade names that we use in connection with the operation of our business, including our corporate name, logos, product names and website names. Solely for your convenience, some of the trademarks and trade names referred to in this prospectus are listed without the ® and TM symbols, but we will assert, to the fullest extent under applicable law, our rights to our trademarks and trade names.
Statistical and Other Industry and Market Data
This prospectus includes statistical and other industry and market data and contains estimates and information concerning our industry and our business, including estimated market size and projected growth rates of the markets for our product candidates. Unless otherwise expressly stated, we obtained this industry, business, market, medical and other information from reports, research surveys, studies and similar data prepared by third parties, industry, medical and general publications, government data and similar sources.
This information involves a number of assumptions and limitations. Although we are responsible for all of the disclosure contained in this prospectus and we believe the third-party market position, market opportunity and market size data included in this prospectus are reliable, we have not independently verified the accuracy or completeness of this third-party data. In addition, projections, assumptions and estimates of our future performance and the future performance of the industry in which we operate are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in “Risk Factors.” These and other factors could cause results to differ materially from those expressed in these publications and reports.
Special Note Regarding Forward-Looking Statements
This prospectus contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, adopted pursuant to the Private Securities Litigation Reform Act of 1995. All statements other than statements of historical facts contained in this prospectus, including statements regarding our future results of operations, financial condition, business strategy and plans and objectives of management for future operations, are forward-looking statements. In some cases, you can identify forward-looking statements because they contain words such as “anticipate,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “target,” “will,” or “would,” or the negative of these words or other similar terms or expressions.
We have based these forward-looking statements largely on our current expectations and projections about future events and trends that we believe may affect our financial condition, results of operations, business strategy and financial needs. These forward-looking statements are subject to a number of known and unknown risks, uncertainties, other factors and assumptions, including the risks described in “Risk Factors” and elsewhere in this prospectus, regarding, among other things:
| ● | our product development and business strategy, including the potential size of the markets for our products and future development and/or expansion of our products and therapies in our markets; |
| ● | our research and development activities, including clinical testing and manufacturing and the related costs and timing; |
| ● | the impact that a pandemic could have on business operations; |
ii
| ● | the sufficiency of our cash resources; |
| ● | our ability to commercialize products and generate product revenues; |
| ● | our ability to raise additional funding when needed; |
| ● | any statements concerning anticipated regulatory activities or licensing or collaborative arrangements, including our ability to obtain regulatory clearances; |
| ● | our research and development expenses; |
| ● | our intellectual property; and |
| ● | any statement of assumptions underlying any of the foregoing. |
These risks are not exhaustive. Other sections of this prospectus may include additional factors that could harm our business and financial performance.
You should not rely on forward-looking statements as predictions of future events. We have based the forward-looking statements contained in this prospectus primarily on our current expectations and projections about future events and trends that we believe may affect our business, financial condition and operating results. We undertake no obligation to update any forward-looking statements made in this prospectus to reflect events or circumstances after the date of this prospectus or to reflect new information or the occurrence of unanticipated events, except as required by law. We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based on information available to us as of the date of this prospectus. While we believe that information provides a reasonable basis for these statements, that information may be limited or incomplete. Our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all relevant information. These statements are inherently uncertain, and investors are cautioned not to unduly rely on these statements.
We qualify all our forward-looking statements by these cautionary statements.
iii
Prospectus Summary
This summary highlights selected information contained elsewhere in this prospectus and is qualified in its entirety by the more detailed information and financial statements included elsewhere in this prospectus. This summary does not contain all of the information you should consider before investing in our common stock. You should carefully read this entire prospectus, including the information in the sections titled “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Special Note Regarding Forward-Looking Statements,” and our financial statements and related notes included elsewhere in this prospectus, before making an investment decision. Unless the context requires otherwise, references in this prospectus to “Incannex,” the “Company,” “we,” “us,” and “our” refer to Incannex Healthcare Inc.
Overview
Incannex is a biotech company developing cannabinoid and psychedelic compound medicines. Our mission is to create premier ethical pharmaceutical drugs and therapies for patients with unmet or inadequately addressed medical needs, in all instances fulfilling regulatory requirements of the U.S. Food and Drug Administration (“FDA”) and other relevant regulatory agencies. We aim to be recognized as a leading specialty drug development company, committed to restoring health and transforming the lives of patients through the development of novel pharmaceutical products and treatments.
We develop targeted and scientifically validated fixed-dose combinations of cannabinoids with generic partners, novel formulations of cannabinoids, and psychedelic agents, applying proprietary insights in an effort to create long term value for our patients and shareholders. We focus on clinical indications that we believe represent unmet or inadequately addressed medical needs that also represent compelling commercial opportunities.
We are developing three novel pharmaceutical compositions to target five indications: obstructive sleep apnea, traumatic brain injury and concussion, rheumatoid arthritis, inflammatory bowel disease and inflammatory lung conditions. We are also developing a treatment for generalized anxiety disorder utilizing psilocybin combined with innovative psychotherapy methods.
We acquired APIRx Pharmaceutical USA, LLC (“APIRx”) in August 2022 and, since that acquisition, we continued to develop cannabinoid products to target additional indications. Our initial focus will be on opioid addiction, smoking cessation, cannabis use disorder, vitiligo, atopic dermatitis and psoriasis. We expect to pursue FDA registration and marketing approval for each product and therapy under development.
The acquisition of APIRx brings to Incannex a diverse portfolio of therapeutic candidates targeted at treating a broad range of conditions including addiction disorders, skin conditions, Parkinson’s disease, gastrointestinal diseases, periodontitis, and ophthalmic conditions. The acquisition of APIRx strengthens our position in the area of cannabinoid and psychedelic treatment development. In particular, it:
| ● | adds a large portfolio of intellectual property with granted and pending patents; |
| ● | significantly expands Incannex’s addressable markets globally and addressable market sizes; |
| ● | further enhances Incannex’s technical and drug development capability; and |
| ● | expands Incannex’s drug delivery capability to include APIRx’s patented delivery technologies. |
Re-domiciliation
In November 2023, Incannex Healthcare Inc., a Delaware corporation, acquired all the outstanding ordinary shares of Incannex Healthcare Limited, an Australian corporation (“Incannex Australia”), pursuant to a Scheme of Arrangement under Australian law (the “Re-domiciliation”). As a result of the Re-domiciliation, Incannex Australia became a wholly-owned subsidiary of Incannex Healthcare Inc., which is the new ultimate parent company.
Until the Re-domiciliation, Incannex Australia’s ordinary shares were listed on the Australian Securities Exchange (“ASX”) and American Depositary Shares (“ADSs”), each representing 25 ordinary shares of Incannex Australia, traded on Nasdaq. Following completion of the Re-domiciliation, Incannex Australia’s ordinary shares were delisted from the ASX and Incannex Healthcare Inc. assumed Incannex Australia’s listing on Nasdaq.
1
Pursuant to the Re-domiciliation, holders of Incannex Australia’s ordinary shares received one share of common stock in Incannex Healthcare Inc. for every 100 ordinary shares held in Incannex Australia and holders of ADSs in Incannex Australia received one share of common stock of Incannex Healthcare Inc. for every 4 ADSs held in Incannex Australia.
The issued and outstanding shares of our common stock as shown in this prospectus have been adjusted in the consolidated financial statements included in this prospectus to reflect the 100:1 exchange ratio as if it had occurred on July 1, 2022.
Corporate Information
Our legal name is Incannex Healthcare Inc. Incannex was incorporated in Delaware in July 2023. Our principal office is located at Suite 105, 8 Century Circuit, Norwest 2153, New South Wales, Australia, and our telephone number is +61 409 840 786. Our address on the Internet is www.incannex.com. Our website address is provided as an inactive textual reference only, and the information on, or accessible through, our website is not part of this prospectus.
Implications of Being an Emerging Growth Company and a Smaller Reporting Company
We qualify as an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”). An emerging growth company and a smaller reporting company may take advantage of certain reduced disclosures and other requirements that are otherwise applicable to public companies. These provisions include:
| ● | being permitted to provide only two years of audited financial statements, in addition to any required unaudited interim financial statements, with correspondingly reduced “Management’s Discussion and Analysis of Financial Condition and Results of Operations” disclosure; |
| ● | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as amended (the “Sarbanes-Oxley Act”); |
| ● | not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, unless the U.S. Securities and Exchange Commission (“SEC”) determines the new rules are necessary for protecting the public; |
| ● | reduced disclosure obligations regarding executive compensation in our periodic reports, proxy statements and registration statements; and |
| ● | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We may take advantage of these provisions until the last day of our fiscal year following the fifth anniversary of the date of the first sale of our common equity securities pursuant to an effective registration statement under the Securities Act of 1933, as amended (the Securities Act). However, if certain events occur prior to the end of such five-year period, including if we become a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act, our annual gross revenues exceed $1.235 billion or we issue more than $1.0 billion of non-convertible debt in any three-year period, we will cease to be an emerging growth company prior to the end of such five-year period.
We have elected to take advantage of certain of the reduced disclosure obligations in this prospectus and in the registration statement of which this prospectus is a part and may elect to take advantage of other reduced reporting requirements in future filings. As a result, the information in this prospectus and that we provide to our stockholders in the future may be different than what you might receive from other public reporting companies in which you hold equity interests.
In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. We have elected to avail ourselves of this exemption and, therefore, we will not be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. As a result, our financial statements may not be comparable to companies that comply with new or revised accounting pronouncements as of public company effective dates. We intend to rely on other exemptions provided by the JOBS Act, including without limitation, not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act.
2
We are also a “smaller reporting company” as defined in the Exchange Act. We may continue to be a smaller reporting company even after we are no longer an emerging growth company. We may take advantage of certain of the scaled disclosures available to smaller reporting companies and will be able to take advantage of these scaled disclosures for so long as our voting and non-voting common stock held by non-affiliates is less than $250.0 million measured on the last business day of our second fiscal quarter, or our annual revenue is less than $100.0 million during the most recently completed fiscal year and our voting and non-voting common stock held by non-affiliates is less than $700.0 million measured on the last business day of our second fiscal quarter.
Risk Factors Summary
Our ability to execute our business strategy is subject to numerous risks and uncertainties that you should consider before investing in us, as more fully described in the section titled “Risk Factors” immediately following this Prospectus Summary. These risks include, among others:
Risks related to our business
| ● | We have a history of operating losses, we may not achieve or maintain profitability in the future which makes our ability to continue as a going concern at risk. |
| ● | Our research and development activities could be adversely impacted if our funding sources are insufficient. |
| ● | We currently have no source of product revenue, may never become profitable and may encounter difficulties in managing our growth. |
| ● | We will require additional financing and may be unable to raise sufficient capital, which could have a material impact on our research and development programs or commercialization of our drug candidates. |
| ● | We may find it difficult to enroll patients in our clinical trials and patients could discontinue their participation in our clinical trials, which could delay or prevent our current and any future clinical trials of our drug candidates and make those trials more expensive to undertake. |
| ● | Any failure to implement our business strategy could negatively impact our business, financial condition and results of operations. |
| ● | Positive results from preclinical studies of our drug candidates are not necessarily predictive of the results of our planned clinical trials of our drug candidates. |
| ● | Ongoing and future clinical trials of drug candidates may not show sufficient safety and efficacy to obtain requisite regulatory approvals for commercial sale. |
| ● | We may be unable to commercialize our drug candidates. |
3
| ● | Even if our drug candidates receive regulatory approval, we may still face development and regulatory difficulties that may delay or impair future sales of drug candidates, including approvals under the applicable controlled substance laws and regulations, or we may be unable to achieve market acceptance of our products. |
| ● | We have limited manufacturing experience with our drug candidates. |
| ● | To the extent we rely significantly on contractors, we will be exposed to risks related to the business and operational conditions of our contractors. |
| ● | We depend on, and will continue to depend on, collaboration and strategic alliances with third parties. To the extent we are able to enter into collaborative arrangements or strategic alliances, we will be exposed to risks related to those collaborations and alliances. |
| ● | As we rely on third-party manufacturing and supply partners, our supply of research and development, preclinical and clinical development materials may become limited or interrupted or may not be of satisfactory quantity or quality. |
| ● | The integration of APIRx with our business operations could undermine our results of operations and we may be unable to achieve the expected synergies. |
| ● | The outbreak of a pandemic could adversely impact our business, including our non-clinical studies and clinical trials. |
Risks related to intellectual property
| ● | Our success depends on our ability to protect our intellectual property and our proprietary technology. |
| ● | Intellectual property rights of third parties could adversely affect our ability to commercialize our drug candidates. |
| ● | We may be unable to protect our intellectual property rights or gain intellectual property rights that protect our competitive advantage. |
| ● | Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed. |
| ● | Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements. |
4
The Offering
| Common stock offered by us | Up to 4,000,000 shares of common stock.
We are also registering 8,000,000 shares of common stock issuable upon the exercise of the common warrants and pre-funded warrants. | |
| Pre-funded warrants offered by us | Each purchaser whose purchase of shares in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock immediately following the consummation of this offering, has the opportunity to purchase, if the purchaser so chooses, pre-funded warrants (each pre-funded warrant to purchase one share of our common stock) in lieu of shares that would otherwise result in the purchaser’s beneficial ownership exceeding 4.99% of our outstanding common stock (or, at the election of the purchaser, 9.99%). The purchase price of each pre-funded warrant will equal the price at which one share of common stock is being sold to the public in this offering, minus $0.001, and the exercise price of each pre-funded warrant will be $0.001 per share. The pre-funded warrants will be immediately exercisable and may be exercised at any time until all of the pre-funded warrants are exercised in full. For each pre-funded warrant we sell, the number of shares we are offering will be decreased on a one-for-one basis. We are also registering the shares of common stock issuable upon the exercise of the pre-funded warrants. | |
| Common warrants to be Offered | Common warrants to purchase up to 4,000,000 shares of common stock. Each common warrant has an exercise price of $ per share, will be immediately exercisable and will expire on the fifth anniversary of the original issuance date. The exercise price is subject to customary adjustments for stock splits and similar recapitalization transactions. The shares of common stock and the accompanying common warrants can only be purchased together in this offering but will be issued separately and will be immediately separable upon issuance. | |
| Term of the offering | This offering will terminate on September 15, 2024, unless we decide to terminate the offering (which we may do at any time in our discretion) prior to that date. | |
| Common stock to be outstanding after this offering | 19,873,113 shares, assuming no sale of pre-funded warrants, which, if sold, would reduce the number of shares of common stock that we are offering on a one-for-one basis, and further assuming no exercise of any common warrants. | |
| Use of proceeds | We intend to use the net proceeds from this offering, together with our existing cash, to advance our clinical programs, for working capital and other general corporate purposes. See “Use of Proceeds” for additional information. | |
| Risk factors | Investing in our common stock involves a high degree of risk. See the section titled “Risk Factors” and other information included in this prospectus for a discussion of risks you should consider carefully before deciding to invest in our securities. | |
| Nasdaq Global Market symbol for the Common Stock | Our common stock is listed on the Nasdaq Global Market under the symbol “IXHL”. There is no established trading market for the pre-funded warrants and common warrants, and we do not expect a trading market to develop. We do not intend to list the common warrants or the pre-funded warrants on any securities exchange or other trading market. Without a trading market, the liquidity of the common warrants and pre-funded warrants will be extremely limited. |
The number of shares of common stock that will be outstanding after this offering is based on 17,642,832 shares of common stock outstanding as of June 30, 2024, assumes no exercise of pre-funded warrants and common warrants, and excludes:
| ● | 1,978,338 shares of common stock issuable upon the exercise of outstanding warrants that were issued to stockholders in connection with the Re-domiciliation; and |
| ● | 5,243,328 shares of common stock reserved for issuance under the Company’s Registration Statement on Form S-8 filed on December 20, 2023 (“S-8 Registration Statement”). |
In addition, unless we specifically state otherwise, the information in this prospectus assumes (i) no exercise of outstanding options to purchase shares of common stock and (ii) no vesting of the outstanding restricted stock issued to the directors of the Company.
5
Summary Consolidated Financial Data
The following tables set forth a summary of our historical financial data as of, and for the periods ended on, the dates indicated. We have derived the summary Consolidated Statement of Profit or Loss and Other Comprehensive Income data and the Consolidated Statements of Financial Position Data for the years ended June 30, 2022 and 2023 from our audited financial statements included elsewhere in this prospectus. We have derived the summary Consolidated Statement of Profit or Loss and Other Comprehensive Income data for the nine months ended March 31, 2023 and 2024 and the summary balance sheet data as of March 31, 2024 from our unaudited condensed financial statements included elsewhere in this prospectus. The unaudited condensed financial statements have been prepared on a basis consistent with our audited financial statements included in this prospectus and, in the opinion of management, reflect all adjustments, consisting only of normal recurring adjustments, necessary to fairly state the financial information in those statements. You should read this data together with our financial statements and related notes included elsewhere in this prospectus and the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
Consolidated Statement of Profit or Loss and Other Comprehensive Income
| Year ended June 30, | Nine months ended March 31, | |||||||||||||||
| 2023 | 2022 | 2024 | 2023 | |||||||||||||
| (in $’000, except share amounts) | (in $’000, except share amounts) | |||||||||||||||
| R&D tax incentive | 683 | 568 | 8,150 | 684 | ||||||||||||
| Foreign exchange expense | (67 | ) | (48 | ) | (17 | ) | — | |||||||||
| Interest income | 241 | 4 | 166 | 153 | ||||||||||||
| Research and development | (6,309 | ) | (3,899 | ) | 8,520 | 4,597 | ||||||||||
| Acquisition of in-process research and development | (35,347 | ) | — | — | (35,347 | ) | ||||||||||
| General and administrative | (8,012 | ) | (7,443 | ) | (11,777 | ) | (5,530 | ) | ||||||||
| Currency translation adjustment, net of tax | (2,292 | ) | (1,302 | ) | (403 | ) | (2,029 | ) | ||||||||
| Total Comprehensive loss | (51,103 | ) | (12,120 | ) | (12,401 | ) | (46,666 | ) | ||||||||
| Net loss per share– basic and diluted | (3.32 | ) | (1.02 | ) | 0.76 | 2.93 | ||||||||||
| Weighted average number of shares of common stock outstanding – basic and diluted | 15,384,704 | 11,921,292 | 15,873,113 | 15,221,900 | ||||||||||||
| Dividends per share | — | — | — | — | ||||||||||||
6
Consolidated Statements of Financial Position Data ($ in thousands)
| June 30, 2023 | March 31, 2024 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 22,120 | $ | 9,305 | ||||
| Prepaid expenses and other assets | 877 | 7,014 | ||||||
| Total current assets | 22,997 | 16,319 | ||||||
| Property, plant and equipment, net | 294 | 523 | ||||||
| Operating lease right-of-use assets | 492 | 408 | ||||||
| Total assets | $ | 23,783 | $ | 17,250 | ||||
| Liabilities and stockholders’ equity | ||||||||
| Current liabilities: | ||||||||
| Trade and other payables | $ | 1,748 | $ | 1,255 | ||||
| Accrued expenses and other current liabilities | 689 | 1,448 | ||||||
| Operating lease liabilities, current | 113 | 161 | ||||||
| Total current liabilities | 2,550 | 2,864 | ||||||
| Operating lease liabilities, non-current | 408 | 248 | ||||||
| Total liabilities | 2,958 | 3,112 | ||||||
| Stockholders’ equity: | ||||||||
| Common stock, $0.0001 par value – 100,000,000 shares authorized; 15,873,113 and 12,926,349 shares issued and outstanding at June 30, 2023 and 2022, respectively | 2 | 2 | ||||||
| Preferred stock, $0.0001 par value per share, 10,000,000 shares authorized; no shares issued or outstanding at June 30, 2023 and 2022, respectively | - | - | ||||||
| Additional paid-in capital | 116,290 | 122,004 | ||||||
| Accumulated deficit | (92,212 | ) | (104,210 | ) | ||||
| Foreign currency translation reserve | (3,255 | ) | (3,658 | ) | ||||
| Total shareholders’ equity | 20,825 | 14,138 | ||||||
| Total liabilities and stockholders’ equity | $ | 23,783 | $ | 17,250 | ||||
7
Risk Factors
Investing in our common stock involves a high degree of risk. You should consider carefully the risks and uncertainties described below, together with all of the other information in this prospectus, including our financial statements and related notes included elsewhere in this prospectus and in the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” before making an investment decision. If any of the following risks are realized, our business, financial condition, results of operations and prospects could be materially and adversely affected. In that event, the trading price of our common stock could decline and you could lose part or all of your investment. Additional risks and uncertainties not presently known to us or that we currently believe to be immaterial also may impair our business, financial condition, results of operations and prospects.
Risks Related to Our Business
We have a history of operating losses and may not achieve or maintain profitability in the future.
We have experienced significant recurring operating losses and negative cash flows from operating activities since inception. For example, for the nine months ended March 31, 2023 and 2024, we had total comprehensive losses of $46.7 million and $12.4 million, respectively, and we had negative cash flows from operating activities of $8.2 million and $12.2 million, respectively. As of March 31, 2024, we had accumulated losses of $104.2 million.
We are a clinical stage pharmaceutical development company and the success of our drug candidates is therefore uncertain. We focus on medicinal synthetic cannabidiol pharmaceutical products and psychedelic medicine therapies.
We expect to continue to incur losses from operations for the foreseeable future and expect the costs of drug development to increase in the future as more patients are recruited for clinical trials. In particular, we expect to continue to incur significant losses in the development of our drug candidates. Because of the numerous risks and uncertainties associated with the development, manufacturing, sales and marketing of our drug candidates, we may experience larger than expected future losses and may never become profitable.
Moreover, there is a substantial risk that we, or our development partners, may not be able to complete the development of our current drug candidates or develop other pharmaceutical products. It is possible that none of them will be successfully commercialized, which could prevent us from ever achieving profitability.
Our research and development activities could be adversely impacted if our funding sources are insufficient.
We anticipate that the costs related to the development of our clinical trials will increase and we will require additional funds to achieve our long-term goals of commercialization and further development of our drug candidates. In addition, we will require funds to pursue regulatory applications, defend intellectual property rights, contract manufacturing capacity, develop marketing and sales capability and fund operating expenses. We intend to seek such additional funding through public or private financings and/or through licensing of our assets or other arrangements with corporate partners. However, such financing, licensing opportunities or other arrangements may not be available from any sources on acceptable terms, or at all. Any shortfall in funding could result in us having to curtail or cease our research and development activities, thereby adversely affecting our business, financial condition and results of operations.
In addition, because of the numerous risks and uncertainties associated with the development of our drug candidates, we are unable to predict the timing or amount of increased research and development costs, or when, or if, we will be able to achieve or maintain profitability. Our costs could significantly increase beyond current expectations if the applicable regulatory authorities require further studies in addition to those currently anticipated. In any case, even if our drug candidates are approved for commercial sale, we anticipate incurring significant costs associated with the commercial launch of such drug candidates and there can be no guarantee that we will ever generate significant revenues.
8
We currently have no source of product revenue and may never become profitable.
None of our drug candidates has been approved for commercial sale, and we expect it to be several years before any of them are approved, if ever, and we are then able to commence sales of our drug candidates. To date, we have not generated any revenue from the licensing or commercialization of our drug candidates and do not expect to receive revenue from them for a number of years, if ever. We will not be able to generate product revenue unless and until our drug candidates, alone or with future partners, successfully complete clinical trials, receive regulatory approval and are successfully commercialized. Although we may seek to obtain revenue from collaboration or licensing agreements with third parties, we currently have no such agreements that could provide us with material, ongoing future revenue and we may never enter into any such agreements.
We will require additional financing and may be unable to raise sufficient capital, which could have a material impact on our research and development programs or commercialization of our drug candidates.
We have historically devoted most of our financial resources to research and development, including pre-clinical and clinical development activities. To date, we have financed a significant amount of our operations through equity financings. The amount of our future net losses will depend, in part, on the rate of our future expenditures and our ability to obtain funding through equity or debt financings or strategic collaborations. The amount of such future net losses, as well as the possibility of future profitability, will also depend on our success in developing and commercializing products that generate significant revenue. Our failure to become and remain profitable would depress the value of our shares of common stock and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product offerings or even continue our operations.
We anticipate that our expenses will increase substantially for the foreseeable future if, and as, we:
| ● | continue our research and preclinical and clinical development of our drug candidates; |
| ● | expand the scope of our current proposed clinical studies for our drug candidates; |
| ● | initiate additional preclinical, clinical or other studies for our drug candidates; |
| ● | change or add additional manufacturers or suppliers; |
| ● | seek regulatory and marketing approvals for our drug candidates that successfully complete clinical studies; |
| ● | seek to identify and validate additional drug candidates; |
| ● | acquire or in-license other drug candidates and technologies; |
| ● | maintain, protect and expand our intellectual property portfolio; |
| ● | attract and retain skilled personnel; |
| ● | create additional infrastructure to support our operations as a publicly quoted company and our product development and planned future commercialization efforts; |
| ● | add an internal sales force; and |
| ● | experience any delays or encounter issues with any of the above. |
Until our drug candidates become commercially available, we will need to obtain additional funding in connection with the further development of our drug candidates. Our ability to obtain additional financing will be subject to a number of factors, including market conditions, our operating performance and investor sentiment. As such, additional financing may not be available to us when needed, on acceptable terms, or at all. If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research and development programs or any future commercialization efforts or obtain funds by entering agreements on unattractive terms.
9
Furthermore, any additional equity fundraising in the capital markets may be dilutive for shareholders and any debt-based funding may bind us to restrictive covenants and curb our operating activities and ability to pay potential future dividends even when profitable. We cannot guarantee that future financing will be available in sufficient amounts or on acceptable terms, if at all. If we are unable to raise additional capital in sufficient amounts or on acceptable terms, we will be prevented from pursuing research and development efforts. This could harm our business, operating results and financial condition and cause the price of our shares of common stock to fall.
If we are unable to secure sufficient capital to fund our operations, then we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to third parties to develop and market drug candidates that we would otherwise prefer to develop and market ourselves. For example, strategic collaborations could require us to share commercial rights to our drug candidates with third parties in ways that we do not intend currently or on terms that may not be favorable to us. Moreover, we could also have to relinquish valuable rights to our technologies, future revenue streams, research programs or drug candidates or grant licenses on terms that may not be favorable to us.
We may need additional capital, and financing may not be available on terms acceptable to us, or at all.
Although our current cash and cash equivalents, anticipated cash flows from operating activities will be sufficient to meet our anticipated working capital requirements and capital expenditures in the ordinary course of business for at least 12 months following this offering, there is a risk that we may need additional cash resources in the future to fund our growth plans or if we experience adverse changes in business conditions or other developments. We may also need additional cash resources in the future if we find and wish to pursue opportunities for new investments, acquisitions, capital expenditures or similar actions. If we determine that our cash requirements exceed the amount of cash and cash equivalents we have on hand at the time, we may seek to issue equity or debt securities or obtain credit facilities. We cannot assure you that financing will be available in amounts or on terms acceptable to us, if at all. The issuance and sale of additional equity would result in further dilution to our shareholders. One or more new debt financings could subject us to any or all of the following risks:
| ● | default and foreclosure on our assets if our operating revenue is insufficient to repay debt obligations; | |
| ● | acceleration of obligations to repay the indebtedness (or other outstanding indebtedness), even if we make all principal and interest payments when due, if we breach any covenants that require the maintenance of certain financial ratios or reserves without a waiver or renegotiation of that covenant; | |
| ● | our inability to obtain necessary additional financing if the debt security contains covenants restricting our ability to obtain such financing while the debt security is outstanding; | |
| ● | diverting a substantial portion of cash flow to pay principal and interest on such debt, which would reduce the funds available for expenses, capital expenditures, acquisitions and other general corporate purposes; and | |
| ● | creating potential limitations on our flexibility in planning for and reacting to changes in our business and in the industry in which we operate. |
The occurrence of any of these risks could adversely affect our operations or financial condition.
10
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to design and implement an effective system of internal control may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses. Ineffective internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock. Our internal control over financial reporting may not prevent or detect all misstatements because of inherent limitations. Additionally, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with our policies and procedures. A material weakness is defined as a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. Management has concluded that we did not maintain effective disclosure controls and procedures due to the material weakness in internal control over financial reporting which existed as of March 31, 2024, relating to the documentation of accounting policies and procedures, particularly relating to the correct application of complex accounting measures.
The measures that we are undertaking to remediate the material weakness in internal control over financial reporting have and will include: (i) hiring qualified internal control personnel or consultants to manage the implementation of internal control policies, procedures and improvement of the internal audit function, as applicable; (ii) developing and implementing written policies and procedures for accounting and financial reporting that meet the standards applied to public companies listed in the United States; and (iii) conducting internal control training to management, key operations personnel and the accounting department, so that management and relevant personnel understand the requirements and elements of internal control over financial reporting mandated by the U.S. securities laws.
We believe we have made progress in accordance with our remediation plan even though the material weaknesses will not be considered remediated until we have completed implementing the necessary additional applicable controls and operate with them for a sufficient period of time to allow management and our auditors to concluded that these controls are operating effectively.
We cannot determine when our remediation plan will be fully completed and we cannot provide any assurance that these remediation efforts will be successful or that our internal control over financial reporting will be effective as a result of these efforts..
We have a limited operating history and a history of losses and expect future losses. There can be no assurances that we will achieve and sustain profitability, which puts our ability to continue as a going concern at risk. In particular, our independent auditor included in its audit report, which is part of this Registration Statement, a going concern opinion raising substantial doubt about our ability to continue as a going concern.
We have incurred significant net losses and negative cash flow from operations since our inception. We reported a total comprehensive loss of $12.4 million as of March 31, 2024. We will expect to continue to incur losses and will need to continue to raise additional funds through equity offerings for our business to survive.
Our financial statements have been prepared assuming that we will continue as a going concern. The application of the going concern principle is dependent upon us achieving profitable operations to generate sufficient cash flows to fund continued operations, or, in the absence of adequate cash flows from operations, obtaining additional financing. However, there is a material uncertainty as to our ability to continue as a going concern and, therefore, that we may be unable to realize our assets and discharge our liabilities in the normal course of business. In particular, our independent auditor included in its audit report, which is part of this Registration Statement, a going concern opinion raising substantial doubt about our ability to continue as a going concern. Such material uncertainty as to our ability to continue as a going concern may adversely impact our ability to obtain the debt or equity financing we need to continue our business operations. If we are unable to achieve profitable operations or obtain additional debt or equity financing, we may be required to reduce or to limit operations, or cease operations altogether and our securityholders may lose some or all of their investment.
11
We may find it difficult to enroll patients in our clinical trials and patients could discontinue their participation in our clinical trials, which could delay or prevent our current and any future clinical trials of our drug candidates and make those trials more expensive to undertake.
Identifying and qualifying patients to participate in current and any future clinical trials of our drug candidates is critical to our success. The timing of our clinical trials depends on the speed at which we can recruit patients to participate in testing our drug candidates. Patients may be unwilling to participate in any future clinical trials because of negative publicity from adverse events in the biotechnology industry. Patients could be unavailable for other reasons, including competitive clinical trials for similar patient populations, and the timeline for recruiting patients, conducting trials and obtaining regulatory approval of potential products may be delayed. If we have difficulty enrolling a sufficient number of patients to conduct any future clinical trials as planned, we may need to delay, limit or discontinue those clinical trials. Clinical trial delays could result in increased costs, slower product development, setbacks in testing the safety and effectiveness of our technology or discontinuation of the clinical trials altogether.
Any failure to implement our business strategy could negatively impact our business, financial condition and results of operations.
The development and commercialization of our drug candidates is subject to many risks, including:
| ● | additional clinical or pre-clinical trials may be required beyond what we currently expect; |
| ● | regulatory authorities may disagree with our interpretation of data from our preclinical studies and clinical studies or may require that we conduct additional studies; |
| ● | regulatory authorities may disagree with our proposed design of future clinical trials; |
| ● | regulatory authorities may delay approval of our drug candidates, thus preventing milestone payments from our collaboration partners; |
| ● | regulatory authorities may not accept data generated at our clinical study sites; |
| ● | we may be unable to obtain and maintain regulatory approval of our drug candidate in any jurisdiction; |
| ● | the prevalence and severity of any side effects of any drug candidate could delay or prevent commercialization, limit the indications for any approved drug candidate, require the establishment of a risk evaluation and mitigation strategy, or cause an approved drug candidate to be taken off the market; |
| ● | regulatory authorities may identify deficiencies in manufacturing processes; |
| ● | regulatory authorities may change their approval policies or adopt new regulations; |
| ● | the third party manufacturers we expect to depend on to supply or manufacture our drug candidates may not produce adequate supply; |
| ● | we, or our third party manufacturers, may not be able to source or produce materials that meet current Good Manufacturing Practice (“cGMP”) standards for the production of our drug candidates; |
| ● | we may not be able to manufacture our drug candidates at a cost or in quantities necessary to make commercially successful products; |
| ● | we may not be able to obtain adequate supply of our drug candidates for our clinical trials; |
| ● | we may experience delays in the commencement of, enrolment of patients in and timing of our clinical trials; |
12
| ● | we may not be able to demonstrate that our drug candidates are safe and effective as a treatment for its indications to the satisfaction of regulatory authorities, and we may not be able to achieve and maintain compliance with all regulatory requirements applicable to our drug candidates; |
| ● | we may not be able to maintain a continued acceptable safety profile of our products following approval; |
| ● | we may be unable to establish or maintain collaborations, licensing or other arrangements; |
| ● | the market may not accept our drug candidates; |
| ● | we may be unable to establish and maintain an effective sales and marketing infrastructure, either through the creation of a commercial infrastructure or through strategic collaborations, and the effectiveness of our own or any future strategic collaborators’ marketing, sales and distribution strategy and operations will affect our profitability; |
| ● | we may experience competition from existing products or new products that may emerge; |
| ● | we and our licensors may be unable to successfully obtain, maintain, defend and enforce intellectual property rights important to protect our drug candidates; and |
| ● | we may not be able to obtain and maintain coverage and adequate reimbursement from third party payors. |
If any of these risks materialize, we could experience significant delays or an inability to successfully develop and commercialize our drug candidates we or our partners may develop, which would have a material adverse effect on our business, financial condition and results of operations.
Positive results from preclinical studies of our drug candidates are not necessarily predictive of the results of our planned clinical trials of our drug candidates.
Positive results in preclinical proof of concept and animal studies of our drug candidates may not result in positive results in clinical trials in humans. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical trials after achieving positive results in preclinical development or early-stage clinical trials, and we cannot be certain that we will not face similar setbacks. These setbacks can be caused by preclinical findings made while clinical trials were underway or safety or efficacy observations made in clinical trials, including adverse events. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses. Many companies that believed their drug candidates performed satisfactorily in preclinical studies and clinical trials nonetheless failed to obtain U.S. Food and Drug Administration (“FDA”) or other regulatory authority approval. If we fail to produce positive results in our clinical trials of our drug candidates, the development timeline and regulatory approval and commercialization prospects for our drug candidates, and, correspondingly, our business and financial prospects, would be negatively impacted.
Ongoing and future clinical trials of drug candidates may not show sufficient safety and efficacy to obtain requisite regulatory approvals for commercial sale.
Phase I and Phase II clinical trials are not primarily designed to test the efficacy of a drug candidate, but rather to test safety and to understand the drug candidate’s side effects at various doses and schedules. Furthermore, success in preclinical and early clinical trials does not ensure that later large-scale trials will be successful, nor does it predict final results. Acceptable results in early trials may not be repeated in later trials. Further, Phase III clinical trials may not show sufficient safety or efficacy to obtain regulatory approval for marketing. In addition, clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. Negative or inconclusive results or adverse medical events during a clinical trial could require that the clinical trial be redone or terminated. The length of time necessary to complete clinical trials and to submit an application for marketing approval by applicable regulatory authorities may also vary significantly based on the type, complexity and novelty of the drug candidate involved, as well as other factors. If we suffer any significant delays, quality issues, setbacks or negative results in, or termination of, our clinical trials, we may be unable to continue the development of our drug candidates or generate revenue and our business may be severely harmed.
13
If we do not obtain the necessary regulatory approvals, we will be unable to commercialize our drug candidates.
The clinical development, manufacturing, sales and marketing of our drug candidates are subject to extensive regulation by regulatory authorities in the United States, the United Kingdom, the European Union, Australia and elsewhere. Despite the substantial time and expense invested in preparation and submission of a Biologic License Application or equivalents in other jurisdictions, regulatory approval is never guaranteed. The number, size and design of preclinical studies and clinical trials that will be required will vary depending on the product, the disease or condition for which the product is intended to be used and the regulations and guidance documents applicable to any particular product. Additionally, during the review process and prior to approval, the FDA and/or other regulatory bodies may require additional data, including with respect to whether our products have the potential for abuse, which may delay approval and any potential controlled substance scheduling processes. The FDA or other regulators can delay, limit or deny approval of a product for many reasons, including, but not limited to, the fact that regulators may not approve our or a third-party manufacturer’s processes or facilities, or that new laws may be enacted, or regulators may change their approval policies, or adopt new regulations requiring new or different evidence of safety and efficacy for the intended use of a product.
Successful results in clinical trials and in the subsequent application for marketing approval are not guaranteed. If we are unable to obtain regulatory approvals, we will not be able to generate revenue from our drug candidates. Even if we receive regulatory approval for any of our drug candidates, our profitability will depend on our ability to generate revenues from their sale or the licensing of our technology.
Even if our drug candidates receive regulatory approval, we may still face development and regulatory difficulties that may delay or impair future sales of drug candidates.
Even if we or our licensing partners receive regulatory approval to sell any drug candidates, the relevant regulatory authorities may, nevertheless, impose significant restrictions on the indicated uses, manufacturing, labelling, packaging, adverse event reporting, storage, advertising, promotion and record keeping or impose ongoing requirements for post-approval studies. In addition, regulatory agencies subject a marketed product, its manufacturer and the manufacturer’s facilities to continual review and periodic inspections. Previously unknown problems with the drug candidate, including adverse events of unanticipated severity or frequency, may result in restrictions on the marketing of the product, and could include withdrawal of the product from the market. In addition, new statutory requirements or additional regulations may be enacted that could prevent or delay regulatory approval of our drug candidates.
We have limited manufacturing experience with our drug candidates.
We have no manufacturing capabilities and are dependent on third parties for cost effective manufacture and manufacturing process development of our drug candidates. Problems with third-party manufacturers or the manufacturing process, or the scaling up of manufacturing activities as such, may delay clinical trials and commercialization of our drug candidates.
To the extent we rely significantly on contractors, we will be exposed to risks related to the business and operational conditions of our contractors.
We are a small company, with few internal staff and limited facilities. We are and will be required to rely on a variety of contractors to manufacture and transport our drug candidates, to perform clinical testing and to prepare regulatory dossiers. Adverse events that affect one or more of our contractors could adversely affect us, such as:
| ● | a contractor is unable to retain key staff that have been working on our drug candidates; |
| ● | a contractor is unable to sustain operations due to financial or other business issues; |
| ● | a contractor loses their permits or licenses that may be required to manufacture our drug candidates; or |
| ● | errors, negligence or misconduct that occur within a contractor may adversely affect our business. |
14
We depend on, and will continue to depend on, collaboration and strategic alliances with third partners. To the extent we are able to enter into collaborative arrangements or strategic alliances, we will be exposed to risks related to those collaborations and alliances.
An important element of our strategy for developing, manufacturing and commercializing our drug candidates is entering into partnerships and strategic alliances with other pharmaceutical companies or other industry participants.
Any partnerships or alliances we have or may have in the future may be terminated for reasons beyond our control or we may not be able to negotiate future alliances on acceptable terms, if at all. These arrangements may result in us receiving less revenue than if we sold our products directly, may place the development, sales and marketing of our products outside of our control, may require us to relinquish important rights or may otherwise be on unfavorable terms. Collaborative arrangements or strategic alliances will also subject us to a number of risks, including the risk that:
| ● | we may not be able to control the amount and timing of resources that our strategic partner/collaborators may devote to the drug candidates; |
| ● | strategic partner/collaborators may experience financial difficulties; |
| ● | the failure to successfully collaborate with third parties may delay, prevent or otherwise impair the development or commercialization of our drug candidates or revenue expectations; |
| ● | products being developed by partners/collaborators may never reach commercial stage resulting in reduced or even no milestone or royalty payments; |
| ● | business combinations or significant changes in a collaborator’s business strategy may also adversely affect a collaborator’s willingness or ability to complete their obligations under any arrangement; |
| ● | a collaborator could independently move forward with a competing product developed either independently or in collaboration with others, including our competitors; and |
| ● | collaborative arrangements are often terminated or allowed to expire, which would delay the development and may increase the cost of developing drug candidates. |
Because we rely on third-party manufacturing and supply partners, our supply of research and development, preclinical and clinical development materials may become limited or interrupted or may not be of satisfactory quantity or quality.
We rely on third-party supply and manufacturing partners to manufacture and supply the materials for our research and development and preclinical and clinical study supplies. We do not own manufacturing facilities or supply sources for such materials.
There can be no assurance that our supply of research and development, preclinical and clinical development biologics and other materials will not be limited, interrupted or restricted in certain geographic regions, be of satisfactory quality or continue to be available at acceptable prices. Replacement of a third-party manufacturer could require significant effort, cost and expertise because there may be a limited number of qualified replacements.
The manufacturing process for a drug candidate is subject to FDA and foreign regulatory authority review. Suppliers and manufacturers must meet applicable manufacturing requirements and undergo rigorous facility and process validation tests required by regulatory authorities in order to comply with regulatory standards. In the event that any of our suppliers or manufacturers fails to comply with such requirements or to perform its obligations to us in relation to quality, timing or otherwise, or if our supply of components or other materials becomes limited or interrupted for other reasons, we may be forced to manufacture the materials ourselves or enter into an agreement with another third party, which would be costly and delay any future clinical trials.
15
Further, if any third-party provider fails to meet its obligations to manufacture our products, or fails to maintain or achieve satisfactory regulatory compliance, the development of such substances and the commercialization of any therapies, if approved, could be stopped, delayed or made commercially unviable, less profitable or may result in enforcement actions against us.
Our research and development efforts will be jeopardized if we are unable to retain key personnel and cultivate key academic and scientific collaborations.
Changes in our senior management could be disruptive to our business and may adversely affect our operations. For example, when we have changes in senior management positions, we may elect to adopt different business strategies or plans. Any new strategies or plans, if adopted, may not be successful and if any new strategies or plans do not produce the desired results, our business may suffer.
Moreover, competition among biotechnology and pharmaceutical companies for qualified employees is intense and as such we may not be able to attract and retain personnel critical to our success. Our success depends on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel, manufacturing personnel, sales and marketing personnel and on our ability to develop and maintain important relationships with clinicians, scientists and leading academic and health institutions. If we fail to identify, attract, retain and motivate these highly skilled personnel, we may be unable to continue our product development and commercialization activities.
In addition, biotechnology and pharmaceutical industries are subject to rapid and significant technological change. Our drug candidates may be or become uncompetitive. To remain competitive, we must employ and retain suitably qualified staff that are continuously educated to keep pace with changing technology, but may not be in a position to do so.
We may encounter difficulties in managing our growth, which could negatively impact our operations.
As we advance our clinical development programs for drug candidates, seek regulatory approval in the United States and elsewhere and increase the number of ongoing product development programs, we anticipate that we will need to increase our product development, scientific and administrative headcount. We will also need to establish commercial capabilities in order to commercialize any drug candidates that may be approved. Such an evolution may impact our strategic focus and our deployment and allocation of resources.
Our ability to manage our operations and growth effectively depends upon the continual improvement of our procedures, reporting systems and operational, financial and management controls. We may not be able to implement administrative and operational improvements in an efficient or timely manner and may discover deficiencies in existing systems and controls. If we do not meet these challenges, we may be unable to execute our business strategies and may be forced to expend more resources than anticipated addressing these issues.
We may acquire additional technology and complementary businesses in the future. Acquisitions involve many risks, any of which could materially harm our business, including the diversion of management’s attention from core business concerns, failure to effectively exploit acquired technologies, failure to successfully integrate the acquired business or realize expected synergies, or the loss of key employees from either our business or the acquired businesses.
In addition, in order to continue to meet our obligations as a publicly listed company and to support our anticipated long-term growth, we will need to increase our general and administrative capabilities. Our management, personnel and systems may not be adequate to support this future growth.
If we are unable to successfully manage our growth and the increased complexity of our operations, our business, financial position, results of operations and prospects may be harmed.
16
The integration of APIRx with our business operations could undermine our results of operations.
In August 2022, we completed the acquisition of 100% of APIRx Pharmaceutical USA, LLC (the “Acquisition”). However, our ability to successfully integrate APIRx will depend on the timely integration and consolidation of operations, facilities, procedures, policies and technologies, and the harmonization of differences in the business cultures between APIRx and us. Such integration and consolidation could be complex and time consuming, will involve additional expense and could disrupt our business and divert management’s attention from ongoing business concerns and our clinical trials. Any failure to successfully integrate the business, operations and employees of APIRx could undermine our results of operations.
We may be unable to achieve the expected synergies following the Acquisition.
We believe that the Acquisition will provide us with the opportunity to achieve synergies between Incannex’s clinical trials and APIRx’s clinical projects. The synergies we expect to realize from the Acquisition are, necessarily, based on projections and assumptions about the combined businesses and assume the successful integration of APIRx’s operations into our business and operations. Our projections and assumptions concerning the Acquisition could prove to be inaccurate, however, and we may not successfully integrate APIRx and our operations in a timely manner, or at all. We could also be exposed to unexpected contingencies or liabilities of APIRx or litigation regarding APIRx’s intellectual property portfolio. If we do not realize the anticipated synergies from the Acquisition, our growth strategy and future profitability could be adversely affected.
Future potential sales of our drug candidates may suffer if they are not accepted in the marketplace by physicians, patients and the medical community.
There is a risk that our drug candidates may not gain market acceptance among physicians, patients and the medical community, even if they are approved by the regulatory authorities. The degree of market acceptance of any of our approved drug candidates will depend on a variety of factors, including:
| ● | timing of market introduction, number and clinical profile of competitive products; |
| ● | our ability to provide acceptable evidence of safety and efficacy and our ability to secure the support of key clinicians and physicians for our drug candidates; |
| ● | cost-effectiveness compared to existing and new treatments; |
| ● | availability of coverage, reimbursement and adequate payment from health maintenance organizations and other third-party payers; |
| ● | prevalence and severity of adverse side effects; and |
| ● | other advantages over other treatment methods. |
As controlled substances, the products may generate public controversy. Physicians, patients, payers or the medical community may be unwilling to accept, use or recommend our drug candidates which would adversely affect our potential revenues and future profitability. Adverse publicity or public perception regarding cannabis and psilocybin to our investigational therapies using these substances may negatively influence the success of these therapies.
We face competition from entities that may develop drug candidates for our target disease indications, including companies developing novel treatments and technology platforms based on modalities and technology similar to ours.
The development and commercialization of drug candidates is highly competitive. Multinational pharmaceutical companies and specialized biotechnology companies could develop drug candidates and processes competitive with our drug candidates. Competitive therapeutic treatments include those that have already been approved and accepted by the medical community, patients and third-party payers, and any new treatments that enter the market.
17
There may be a significant number of products that are currently under development, and may become commercially available in the future, for the treatment of conditions for which we are developing, and may in the future try to develop, drug candidates.
Multinational pharmaceutical companies and specialized biotechnology companies could have significantly greater financial, technical, manufacturing, marketing, sales and supply resources and experience than we have. If we successfully obtain approval for any drug candidate, we could face competition based on many different factors, including the safety and effectiveness of our drug candidates, the ease with which our drug candidates can be administered and the extent to which patients accept relatively new routes of administration, the timing and scope of regulatory approvals for these drug candidates, the availability and cost of manufacturing, marketing and sales capabilities, price, reimbursement coverage and patent position.
Competing products could present superior treatment alternatives, including by being more effective, safer, less expensive or marketed and sold more effectively than any products we may develop. Competitive products may make any products we develop obsolete or non-competitive before we recover the expense of developing and commercializing our drug candidates. Such competitors could also recruit our employees, which could negatively impact our level of expertise and our ability to execute our business plan.
If healthcare insurers and other organizations do not pay for our drug candidates or impose limits on reimbursement, our future business may suffer.
Our drug candidates may be rejected by the market due to many factors, including cost. The continuing efforts of governments, insurance companies and other payers of healthcare costs to contain or reduce healthcare costs may affect our future revenues and profitability. In Australia and certain foreign markets, the pricing of pharmaceutical products is subject to government control. We expect initiatives for similar government control to continue in the United States and elsewhere. The adoption of any such legislative or regulatory proposals could harm our business and prospects.
Successful commercialization of our drug candidates will depend in part on the extent to which reimbursement for the cost of our products and related treatment will be available from government health administration authorities, private health insurers and other organizations. Our drug candidates may not be considered cost-effective and reimbursement may not be available to consumers or may not be sufficient to allow our products to be marketed on a competitive basis. Third-party payers are increasingly challenging the price of medical products and treatment. If third-party coverage is not available for our drug candidates, the market acceptance of these drug candidates will be reduced. Cost-control initiatives could decrease the price we might establish for drug candidates, which could result in product revenues lower than anticipated. If the price for our drug candidates decreases or if governmental and other third-party payers do not provide adequate coverage and reimbursement levels, our potential revenue and prospects for profitability will suffer.
We could become exposed to product liability claims that could adversely affect our business.
The testing, marketing and sale of therapeutic products entails an inherent risk of product liability. We rely on a number of third-party researchers and contractors to produce, collect, and analyze data regarding the safety and efficacy of our drug candidates. We also have quality control and quality assurance in place to mitigate these risks, as well as professional liability and clinical trial insurance to cover financial damages in the event that human testing is done incorrectly or the data is analyzed incorrectly.
18
Notwithstanding our control procedures, we may face product liability exposure related to the testing of our drug candidates in human clinical trials. If any of our drug candidates are approved for sale, we may face exposure to claims by an even greater number of persons than were involved in the clinical trials once marketing, distribution and sales of our drug candidates begin. Regardless of merit or eventual outcome, liability claims may result in:
| ● | decreased demand for our drug candidates; |
| ● | injury to our reputation; |
| ● | withdrawal of clinical trial participants; |
| ● | costs of related litigation; |
| ● | substantial monetary awards to patients and others; |
| ● | loss of revenues; and |
| ● | the inability to commercialize drug candidates. |
With respect to product liability claims, we could face additional liability beyond insurance limits if testing mistakes were to endanger any human subjects. In addition, if a claim is made against us in conjunction with these research testing activities, the market price of our shares of common stock may be negatively affected.
The outbreak of a pandemic could adversely impact our business, including our non-clinical studies and clinical trials.
Public health crises such as pandemics or similar outbreaks might adversely impact our business. In December 2019, a novel strain of coronavirus (“COVID-19”) surfaced in China and then spread to most countries in the world.
As a result of the COVID-19 outbreak, or any future pandemic, we have and may in the future experience disruptions that could severely impact our business, preclinical studies and clinical trials, including:
| ● | delays or difficulties in enrolling patients in our clinical trials; |
| ● | delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff; |
| ● | delays or disruptions in non-clinical experiments and investigational new drug application-enabling good laboratory practice standard toxicology studies due to unforeseen circumstances at contract research organizations and vendors along their supply chain; |
| ● | increased rates of patients withdrawing from our clinical trials following enrollment as a result of contracting a virus, being forced to quarantine, or not wanting to attend hospital visits; |
| ● | diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials; |
| ● | interruption of key clinical trial activities, such as clinical trial site data monitoring, due to limitations on travel imposed or recommended by national, state or local governments, employers and others or interruption of clinical trial subject visits and study procedures (particularly any procedures that may be deemed non-essential), which may impact the integrity of subject data and clinical study endpoints; |
| ● | interruption or delays in the operations of the FDA, the European Medicines Agency, the Australian Therapeutic Goods Administration or other foreign regulatory agencies, which may impact approval timelines; |
| ● | interruption of, or delays in receiving, supplies of our drug candidates from our contract manufacturing organizations due to staffing shortages, production slowdowns or stoppages and disruptions in our supply chain or distribution vendors’ ability to ship drug candidates; and |
| ● | limitations on employee resources that would otherwise be focused on the conduct of our non-clinical studies and clinical trials, including because of sickness of employees or their families, the desire of employees to avoid contact with large groups of people, an increased reliance on working from home or mass transit disruptions. |
19
Product shipment delays could have a material adverse effect on our business, results of operations and financial condition.
The shipment, import and export of our drug candidates and the Active Pharmaceutical Ingredient (“API”) used to manufacture them, along with the drugs used in our psychedelic-assisted psychotherapy services, will require import and export licenses. In the United States, the FDA, U.S. Customs and Border Protection, and the U.S. Drug Enforcement Administration (the “DEA”); in Canada, the Canada Border Services Agency, Health Canada; in Europe, the European Medicines Agency (the “EMA”) and the European Commission; in Australia and New Zealand, the Australian Customs and Board Protection Service, the Therapeutic Goods Administration (the “TGA”), the New Zealand Medicines and Medical Device Safety Authority and the New Zealand Customs Service; and in other countries, similar regulatory authorities, regulate the import and export of pharmaceutical products that contain controlled substances. Specifically, the import and export processes require the issuance of import and export licenses by the relevant controlled substance authority in both the importing and exporting country.
We may not be granted, or if granted, maintain, such licenses from the authorities in certain countries. Even if we obtain the relevant licenses, shipments of API and our drug candidates may be held up or lost in transit, which could cause significant delays and may lead to product batches being stored outside required temperature ranges. Inappropriate storage may damage the product shipment resulting in delays in clinical trials or, upon commercialization, a partial or total loss of revenue from one or more shipments of API or our drug candidates. A delay in a clinical trial or, upon commercialization, a partial or total loss of revenue from one or more shipments of API or our drug candidates, could have a material adverse effect on our business, results of operations and financial condition.
Our drug candidates will be subject to controlled substance laws and regulations. Failure to receive necessary approvals may delay the launch of our drug candidates and failure to comply with these laws and regulations may adversely affect the results of our business operations.
Our drug candidates contain controlled substances as defined in the Controlled Substance Act (the “CSA”). Controlled substances that are pharmaceutical products are subject to a high degree of regulation under the CSA, which establishes, among other things, certain registration, manufacturing quotas, security, recordkeeping, reporting, import, export and other requirements administered by the DEA. The DEA classifies controlled substances into five schedules: Schedule I, II, III, IV or V substances. By definition, Schedule I substances have a high potential for abuse, have not currently “accepted medical use” in the United States, lack accepted safety for use under medical supervision, and may not be prescribed, marketed or sold in the United States. Pharmaceutical products approved for use in the United States may be listed as Schedule II, III, IV or V, with Schedule II substances considered to present the highest potential for abuse or dependence and Schedule V substances the lowest relative risk of abuse among such substances. Schedule I and II drugs are subject to the strictest controls under the CSA, including manufacturing and procurement quotas, security requirements and criteria for importation. In addition, dispensing of Schedule II drugs is further restricted. For example, they may not be refilled without a new prescription.
As a synthetic cannabinoids pharmaceutical product with psychedelic agents, our drug candidates are likely to be scheduled as Schedule II or III controlled substance. We will need to identify wholesale distributors with the appropriate DEA registrations and authority to distribute the products to pharmacies and other healthcare providers, and these distributors would need to obtain Schedule II or III distribution registrations. The failure to obtain, or delay in obtaining, or the loss of any of those registrations could result in increased costs to us. If any of our drug candidates is a Schedule II drug, pharmacies would have to maintain enhanced security with alarms and monitoring systems, and they must adhere to additional recordkeeping and inventory requirements. This may discourage some pharmacies from carrying the product. Furthermore, state and federal enforcement actions, regulatory requirements, and legislation intended to reduce prescription drug abuse, such as the requirement that physicians consult a state prescription drug monitoring program, may make physicians less willing to prescribe, and pharmacies to dispense, Schedule II products.
20
We intend to manufacture the commercial supply of our drug candidates outside of the United States. If any of our products are approved by the FDA and classified as a Schedule II or III substance, an importer can import for commercial purposes if it obtains from the DEA an importer registration and files an application with the DEA for an import permit for each import. The failure to identify an importer or obtain the necessary import authority could affect the availability of our drug candidates and have a material adverse effect on our business, results of operations and financial condition. In addition, an application for a Schedule II importer registration must be published in the Federal Register, and there is a waiting period for third party comments to be submitted. The failure to maintain the necessary registrations or comply with applicable laws could delay the commercialization of our drug candidates and could delay the completion of the clinical studies. Furthermore, failure to maintain compliance with the CSA, particularly non-compliance resulting in loss or diversion, could result in regulatory action that could have a material adverse effect on our business, financial condition and results of operations. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate proceedings to restrict, suspend or revoke those registrations. In certain circumstances, violations could lead to criminal proceedings. In addition, if the FDA, DEA, or any foreign regulatory authority determines that our drug candidates may have potential for abuse, it may require us to generate more clinical or other data than we currently anticipate to establish whether or to what extent the substance has an abuse potential, which could increase the cost and/or delay the launch of our drug candidates.
Individual states have also established controlled substance laws and regulations. Though state-controlled substance laws often mirror federal law, because the states are separate jurisdictions, they may separately schedule our drug candidates as well. While some states automatically schedule a drug based on federal action, other states schedule drugs through rulemaking or a legislative action. State scheduling may delay commercial sale of any product for which we obtain federal regulatory approval and adverse scheduling could have a material adverse effect on the commercial attractiveness of such product. We or our partners must also obtain separate state registrations, permits or licenses in order to be able to obtain, handle, and distribute controlled substances for clinical trials or commercial sale, and failure to meet applicable regulatory requirements could lead to enforcement and sanctions by the states in addition to those from the DEA or otherwise arising under federal law.
We intend to contract manufacturers in Australia to produce the drug product for our clinical trials and the API for our drug candidates. In addition, we may decide to develop, manufacture or commercialize our drug candidates in additional countries. As a result, we will also be subject to controlled substance laws and regulations from the TGA in Australia and from other regulatory agencies in other countries where we develop, manufacture or commercialize our drug candidates in the future. We plan to submit New Drug Applications (“NDAs”) for our drug candidates to the FDA upon completion of all requisite clinical trials and may require additional DEA scheduling decisions at such time as well.
Changes in U.S. healthcare law and implementing regulations, as well as changes in healthcare policy, may impact our business in ways that we cannot currently predict and may harm our business and results of operations.
There have been, and likely will continue to be, several executive, legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of drug candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any drug candidates for which we obtain marketing approval. Among policy makers and payors in the United States, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. The pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, collectively referred to as the Affordable Care Act, substantially changed the way healthcare is financed by both the government and private insurers, and significantly impacts the U.S. pharmaceutical industry. The Affordable Care Act, among other things: (1) introduced a new average manufacturer price definition for drugs and biologics that are inhaled, infused, instilled, implanted or injected and not generally dispensed through retail community pharmacies; (2) increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and expanded rebate liability from fee-for-service Medicaid utilization to include the utilization of Medicaid managed care organizations as well; (3) established a branded prescription drug fee that pharmaceutical manufacturers of branded prescription drugs must pay to the federal government; (4) expanded the list of covered entities eligible to participate in the 340B drug pricing program by adding new entities eligible for the program; (5) established a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% (increased to 70% pursuant to the Bipartisan Budget Act of 2018, or BBA, effective as of January 2019) point-of-sale discounts off negotiated prices of applicable branded drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; (6) extended manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; (7) expanded eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals, including individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability; (8) created a licensure framework for follow-on biologic products; and (9) established a Center for Medicare and Medicaid Innovation at the CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending. We continue to monitor any changes or challenges to the Affordable Care Act that, in turn, may potentially impact our business in the future.
21
Other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. These changes include aggregate reductions to Medicare payments to providers of 2% per fiscal year pursuant to the Budget Control Act of 2011 and subsequent laws, which began in 2013 and will remain in effect through 2030, unless additional Congressional action is taken. Subsequently, the American Taxpayer Relief Act of 2012 further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. New laws could result in additional reductions in Medicare and other healthcare funding, which may materially negatively affect customer demand and affordability for our drug candidates and, accordingly, the results of our financial operations. Also, there has been heightened governmental scrutiny recently over the manner in which pharmaceutical companies set prices for their marketed products, which have resulted in several Congressional inquiries and proposed federal legislation, as well as state efforts, designed to, among other things, bring more transparency to product pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. At the state level, individual states in the United States are increasingly active in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions on coverage or access could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce the ultimate demand for our drug candidates that we successfully commercialize or put pressure on our product pricing.
In addition, proposed federal and state legislation may increase competition as it relates to cannabis derived products. Under the Cannabis Administration and Opportunity Act, the U.S. Senate proposed legalizing the use of hemp-derived CBD in dietary supplements by amending the Federal Food, Drug, and Cosmetic Act (the “FDCA”). The Hemp Access and Consumer Safety Act of 2021 (SB 1698) also permits hemp-derived CBD to be used in dietary supplements. States are considering the reimbursement of medical marijuana. As the availability and reimbursement of cannabis-derived products potentially expand, the pharmaceutical industry may directly compete with state-regulated cannabis businesses for market share.
We expect that these and other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and lower reimbursement, and put additional downward pressure on the price that we receive for any approved product. It is possible that additional governmental action is taken in response to the COVID-19 pandemic. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action in the United States. If we or any third parties we may engage are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or such third parties are not able to maintain regulatory compliance, our drug candidates may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability.
22
Risks Related to Intellectual Property
Our success depends on our ability to protect our intellectual property and our proprietary technology.
Our success is to a certain degree also dependent on our ability to obtain and maintain protection of our intellectual property portfolio, including the assets acquired through the Acquisition or, where applicable, to receive/maintain orphan drug designation/status and resulting marketing exclusivity for our drug candidates.
We may be materially adversely affected by our failure or inability to protect our intellectual property rights. Without the granting of these rights, the ability to pursue damages for infringement would be limited. Similarly, any know-how that is proprietary or particular to our technologies may be subject to risk of disclosure by employees or consultants despite having confidentiality agreements in place.
Any future success will depend in part on whether we can obtain and maintain patents to protect our own products and technologies; obtain licenses to the patented technologies of third parties; and operate without infringing on the proprietary rights of third parties. Biotechnology patent matters can involve complex legal and scientific questions, and it is impossible to predict the outcome of biotechnology and pharmaceutical patent claims. Any of our future patent applications may not be approved, or we may not develop additional products or processes that are patentable. Some countries in which we may sell our drug candidate or license our intellectual property may fail to protect our intellectual property rights to the same extent as the protection that may be afforded in the United States or Australia. Some legal principles remain unresolved and there has not been a consistent policy regarding the breadth or interpretation of claims allowed in patents in the United States, the United Kingdom, the European Union, Australia or elsewhere. In addition, the specific content of patents and patent applications that are necessary to support and interpret patent claims is highly uncertain due to the complex nature of the relevant legal, scientific and factual issues. Changes in either patent laws or in interpretations of patent laws in Australia, the United States, the United Kingdom, the European Union or elsewhere may diminish the value of our intellectual property or narrow the scope of our patent protection. Even if we are able to obtain patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner. We may also fail to take the required actions or pay the necessary fees to maintain our patents.
Moreover, any of our pending applications may be subject to a third-party pre-issuance submission of prior art to the U.S. Patent and Trademark Office, or USPTO, the European Patent Office, or EPO, the Intellectual Property Office, or IPO, in the United Kingdom, the Australian Patent and Trademark Office and/or any patents issuing thereon may become involved in opposition, derivation, reexamination, post grant review, interference proceedings or other patent office proceedings or litigation, in the United States or elsewhere, challenging our patent rights. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, and allow third parties to commercialize our technology or products and compete directly with us, without payment to us. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to exploit our intellectual property or develop and commercialize drug candidates.
The issuance of a patent is not conclusive as to the inventorship, scope, validity or enforceability, and our patents may be challenged in the courts or patent offices in the United States, the European Union, Australia and elsewhere. Such challenges may result in loss of ownership or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit the duration of the patent protection of our technology and products. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
In addition, other companies may attempt to circumvent any regulatory data protection or market exclusivity that we obtain under applicable legislation, which may require us to allocate significant resources to preventing such circumvention. Such developments could enable other companies to circumvent our intellectual property rights and use our clinical trial data to obtain marketing authorizations in the European Union, Australia and in other jurisdictions. Such developments may also require us to allocate significant resources to prevent other companies from circumventing or violating our intellectual property rights.
23
Intellectual property rights of third parties could adversely affect our ability to commercialize our drug candidates, such that we could be required to litigate with or obtain licenses from third parties in order to develop or market our drug candidates.
Our commercial success may depend upon our future ability and the ability of our potential collaborators to develop, manufacture, market and sell our drug candidates without infringing valid intellectual property rights of third parties. If a third-party intellectual property right exists it may require the pursuit of litigation or administrative proceedings to nullify or invalidate the third-party intellectual property right concerned, or entry into a license agreement with the intellectual property right holder, which may not be available on commercially reasonable terms, if at all.
Third-party intellectual property right holders, including our competitors, may bring infringement claims against us. We may not be able to successfully settle or otherwise resolve such infringement claims. If we are unable to successfully settle future claims or otherwise resolve such claims on terms acceptable to us, we may be required to engage in or continue costly, unpredictable and time-consuming litigation and may be prevented from, or experience substantial delays in, marketing our drug candidate.
If we fail to settle or otherwise resolve any such dispute, in addition to being forced to pay damages, we or our potential collaborators may be prohibited from commercializing any drug candidates we may develop that are held to be infringing, for the duration of the patent term. We might, if possible, also be forced to redesign our formulations so that we no longer infringe such third-party intellectual property rights. Any of these events, even if we were ultimately to prevail, could result in injury to our reputation or require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
Because we collaborate with various organizations and academic institutions on the advancement of our technology and drug candidates, we may, at times, share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements or other similar agreements with our collaborators, advisors, employees and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, such as trade secrets. Despite these contractual provisions, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known by potential competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, discovery by a third party of our trade secrets or other unauthorized use or disclosure would impair our intellectual property rights and protections in our drug candidates.
In addition, these agreements typically restrict the ability of our collaborators, advisors, employees and consultants to publish data potentially relating to our trade secrets. Our academic collaborators typically have rights to publish data, provided that we are notified in advance and may delay publication for a specified time in order to secure our intellectual property rights arising from the collaboration. In other cases, publication rights are controlled exclusively by us. In other cases, we may share these rights with other parties. Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of these agreements, independent development or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and applications are required to be paid to the United State Patent and Trademark Office and other governmental patent agencies outside of the United States in several stages over the lifetime of the patents and applications. The USPTO and various corresponding governmental patent agencies outside of the United States require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process and after a patent has issued. There are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction.
24
We may become involved in lawsuits to protect and defend our patents or other intellectual property, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or other intellectual property and we may inadvertently infringe the patent or intellectual property of others. To counter infringement or unauthorized use, we may be required to file claims, and any related litigation and/or prosecution of such claims can be expensive and time consuming. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringe their intellectual property. In addition, in a patent infringement proceeding, a court may decide that a patent of ours is invalid in whole or in part, unenforceable, or construe the patent’s claims narrowly allowing the other party to commercialize competing products on the grounds that our patents do not cover such products.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our technical and management personnel from their normal responsibilities. Such litigation or proceedings could substantially increase our operating losses and reduce our resources available for development activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. The effects of patent litigation or other proceedings could therefore have a material adverse effect on our ability to compete in the marketplace.
Confidentiality and invention assignment agreements with our employees, advisors and consultants may not adequately prevent disclosure of trade secrets and protect other proprietary information.
We consider proprietary trade secrets and/or confidential know-how and unpatented know-how to be important to our business. We may rely on trade secrets and/or confidential know-how to protect our technology, especially where patent protection is believed by us to be of limited value. However, trade secrets and/or confidential know-how can be difficult to maintain as confidential.
To protect this type of information against disclosure or appropriation by competitors, our policy is to require our employees, advisors and consultants to enter into confidentiality and invention assignment agreements with us. However, current or former employees, advisors and consultants may unintentionally or willfully disclose our confidential information to competitors, and confidentiality and invention assignment agreements may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. Enforcing a claim that a third party obtained illegally and is using trade secrets and/or confidential know-how is expensive, time consuming and unpredictable. The enforceability of confidentiality and invention assignment agreements may vary from jurisdiction to jurisdiction.
Failure to obtain or maintain trade secrets and/or confidential know-how trade protection could adversely affect our competitive position. Moreover, our competitors may independently develop substantially equivalent proprietary information and may even apply for patent protection in respect of the same. If successful in obtaining such patent protection, our competitors could limit our use of our trade secrets and/or confidential know-how, our competitive position or commercial advantage may be impaired and our business and results of operations may be adversely affected.
25
Intellectual property rights do not address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
| ● | Others may be able to make products that are similar to ours but that are not covered by our intellectual property rights. |
| ● | Others may independently develop similar or alternative technologies or otherwise circumvent any of our technologies without infringing our intellectual property rights. |
| ● | We or any of our collaboration partners might not have been the first to conceive and reduce to practice the inventions covered by the patents or patent applications that we own, license or will own or license. |
| ● | We or any of our collaboration partners might not have been the first to file patent applications covering certain of the patents or patent applications that we or they own or have obtained a license, or will own or will have obtained a license. |
| ● | It is possible that any pending patent applications that we have filed, or will file, will not lead to issued patents. |
| ● | Issued patents that we own may not provide us with any competitive advantage, or may be held invalid or unenforceable, as a result of legal challenges by our competitors. |
| ● | Our competitors might conduct research and development activities in countries where we do not have patent rights, or in countries where research and development safe harbor laws exist, and then use the information learned from such activities to develop competitive products for sale in our major commercial markets. |
| ● | Ownership of our patents or patent applications may be challenged by third parties. |
| ● | The patents of third parties or pending or future applications of third parties, if issued, may have an adverse effect on our business. |
We may face difficulties with protecting our intellectual property in certain jurisdictions, which may diminish the value of our intellectual property rights in those jurisdictions.
The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws in the United States and the European Union, and many companies have encountered significant difficulties in protecting and defending such rights in such jurisdictions. If we or our collaboration partners encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished and we may face additional competition from others in those jurisdictions.
Some countries in Europe and China have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we are, or any of our licensors is, forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position or commercial advantage may be impaired and our business and results of operations may be adversely affected.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect our drug candidates and any future drug candidates.
As is the case with other biotechnology and pharmaceutical companies, our success is heavily dependent on intellectual property rights, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves technological and legal complexity, and obtaining and enforcing biopharmaceutical patents is costly, time-consuming and inherently uncertain. The U.S. Supreme Court in recent years has issued rulings either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations or ruling that certain subject matter is not eligible for patent protection. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by Congress, the federal courts, the USPTO and equivalent bodies in non-U.S. jurisdictions, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce existing patents and patents we may obtain in the future.
26
Patent reform laws, such as the Leahy-Smith America Invents Act, or the Leahy-Smith Act, as well as changes in how patent laws are interpreted, could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. The Leahy-Smith Act made a number of significant changes to U.S. patent law. These include provisions that affect the filing and prosecution strategies associated with patent applications, including a change from a “first-to-invent” to a “first-inventor-to-file” patent system, and a change allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO-administered post-grant proceedings, including post-grant review, inter partes review and derivation proceedings. The USPTO has developed regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act and, in particular, the “first-inventor-to-file” provisions. The Leahy-Smith Act and its implementation may increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could harm our business, financial condition and results of operations.
Price controls may be imposed in non-U.S. markets, which may negatively affect our future profitability.
In some countries, particularly EU member states, Japan, Australia and Canada, the pricing of prescription drugs is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after receipt of marketing approval for a product. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various EU member states and parallel distribution, or arbitrage between low-priced and high-priced member states, can further reduce prices. In some countries, we or our collaborators may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our drug candidates to other available therapies in order to obtain or maintain reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If reimbursement of our drug candidates is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business, revenues or profitability could be harmed.
Risks Related to Investing in our Securities
The price of our common stock has been and may continue to be highly volatile, which may make it difficult for stockholders to sell our common stock when desired or at attractive prices.
Following our Re-domiciliation, the trading price of our common stock has been volatile and could be subject to fluctuations in response to various factors, some of which are beyond our control. Factors such as announcements of variations in our quarterly financial results and fluctuations in revenue could also cause the market price of our common stock to fluctuate. Additionally, the stock markets have at times experienced price and volume fluctuations that have affected and might in the future affect the market prices of equity securities of many companies. These fluctuations have, in some cases, been unrelated or disproportionate to the operating performance of these companies. Further, the trading prices of publicly traded shares of companies in our industry have been particularly volatile and may be very volatile in the future. These broad market and industry fluctuations, as well as general economic, political and market conditions such as recessions, interest rate changes, international currency fluctuations or political unrest, may negatively impact the market price of our common stock. If the price of our common stock declines, our ability to raise funds through the issuance of equity or otherwise use our common stock as consideration will be reduced. A low price for our equity may negatively impact our ability to access additional debt capital. These factors may limit our ability to implement our operating and growth plans.
27
U.S. investors may have difficulty enforcing civil liabilities against our directors or members of senior management.
Several of our officers and directors are non-residents of the United States, and a substantial portion of the assets of such persons are located outside the United States. As a result, it may be difficult to serve process on such persons in the United States or to enforce judgments obtained in U.S. courts against them based on civil liability provisions of the securities laws of the United States. Even if you are successful in bringing such an action, there is doubt as to whether Australian courts would enforce certain civil liabilities under U.S. securities laws in original actions or judgments of U.S. courts based upon these civil liability provisions.
Certain provisions of our amended and restated certificate of incorporation may discourage, delay or prevent a change in control of our company and, therefore, depress the trading price of our securities.
Our amended and restated certificate of incorporation provides that our board of directors is classified into three classes of directors, each with staggered three-year terms. Such provision may discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for our securities.
In addition, as a Delaware corporation, we would also generally be subject to provisions of Delaware law, including Section 203 of the Delaware General Corporation Law, which limits the ability of stockholders holding shares representing more than 15% of the voting power of our outstanding voting stock from engaging in certain business combinations with us. However, our amended and restated certificate of incorporation provides that we are not subject to Section 203 of Delaware General Corporation Law.
The existence of the foregoing provisions and anti-takeover measures could limit the price that investors might be willing to pay in the future for shares of our common stock. They could also deter potential acquirers of our company, thereby reducing the likelihood that you could receive a premium for your common stock in an acquisition.
There is no public market for common warrants or the pre-funded warrants to purchase shares of our common stock being offered by us in this offering.
There is no established public trading market for the common warrants or the pre-funded warrants to purchase shares of our common stock that are being offered as part of this offering, and we do not expect a market to develop. In addition, we do not intend to apply to list the common warrants or the pre-funded warrants on any national securities exchange or other nationally recognized trading system, including the Nasdaq Global Market. Without an active market, the liquidity of the common warrants and the pre-funded warrants will be limited.
The common warrants and the pre-funded warrants are speculative in nature.
The common warrants and the pre-funded warrants do not confer any rights of common stock ownership on their holders, such as voting rights or the right to receive dividends, but rather merely represent the right to acquire shares of common stock at a fixed price. Specifically, commencing on the date of issuance, holders of the common warrants may exercise their right to acquire common stock and pay an exercise price of $ per share, subject to certain adjustments, prior to five years from the date on which such common warrants were issued, after which date any unexercised common warrants will expire and have no further value. Holders of pre-funded warrants have identical rights, except that the pre-funded warrants have an exercise price of $0.001 and do not expire until exercised in full. Moreover, following this offering, the market value of the common warrants and pre-funded warrants, if any, is uncertain and there can be no assurance that the market value of the common warrants or the pre-funded warrants will equal or exceed their imputed offering price.
Holders of the common warrants and pre-funded warrants offered hereby will have no rights as common stockholders with respect to the shares our common stock underlying the common warrants and pre-funded warrants until such holders exercise their common warrants and pre-funded warrants and acquire our common stock, except as otherwise provided in the common warrants and pre-funded warrants.
Until holders of the common warrants and pre-funded warrants acquire shares of our common stock upon exercise thereof, such holders will have no rights with respect to the shares of our common stock underlying such warrants. Upon exercise of the common warrants and pre-funded warrants, the holders will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs after the exercise date.
28
This is a best efforts offering, no minimum amount of securities is required to be sold, and we may not raise the amount of capital we believe is required for our business plan.
The joint placement agents have agreed to use their reasonable best efforts to solicit offers to purchase the securities in this offering. The joint placement agents have no obligation to buy any of the securities from us or to arrange for the purchase or sale of any specific number or dollar amount of the securities. There is no required minimum number of securities that must be sold as a condition to completion of this offering. Because there is no minimum offering amount required as a condition to the closing of this offering, the actual offering amount, joint placement agents fees and proceeds to us are not presently determinable and may be substantially less than the maximum amounts contemplated in this prospectus. We may sell fewer than all of the securities offered hereby, which may significantly reduce the amount of proceeds received by us, and investors in this offering will not receive a refund in the event that we do not sell an amount of securities sufficient to support our continued operations, including our near-term continued operations.
Our management has broad discretion as to the use of the net proceeds from this offering.
Our management will have broad discretion in the application of the net proceeds from this offering, including for any of the purposes described in the section titled “Use of Proceeds.” Because of the number and variability of factors that will determine our use of the net proceeds from this offering, their ultimate use may vary substantially from their currently intended use. Our management might not apply our net proceeds in ways that ultimately increase the value of your investment, and the failure by our management to apply these funds effectively could harm our business. If we do not invest or apply the net proceeds from this offering in ways that enhance stockholder value, we may fail to achieve expected results, which could cause our common stock price to decline.
If you purchase securities in this offering, you will experience immediate and substantial dilution and may experience additional dilution in the future.
Investors purchasing shares of our common stock and accompanying common warrants in this offering will pay a price per share that substantially exceeds the as adjusted net tangible book value per share. As a result, investors purchasing our common stock and accompanying common warrants in this offering will incur immediate dilution of $0. per share, representing the difference between the assumed public offering price of $2.76 per share, which is the last reported sale price of our common stock on the Nasdaq Global Market on July 11, 2024, and our as adjusted net tangible book value as of March 31, 2024. For more information on the dilution you may suffer as a result of investing in this offering, see “Dilution.”
Because there is no minimum required for the offering to close, investors in this offering will not receive a refund in the event that we do not sell an amount of securities sufficient to pursue the business goals outlined in this prospectus.
We have not specified a minimum offering amount nor have or will we establish an escrow account in connection with this offering. Because there is no escrow account and no minimum offering amount, investors could be in a position where they have invested in our company, but we are unable to fulfill our objectives due to a lack of interest in this offering. Further, because there is no escrow account in operation and no minimum investment amount, any proceeds from the sale of securities offered by us will be available for our immediate use, despite uncertainty about whether we would be able to use such funds to effectively implement our business plan. Investor funds will not be returned under any circumstances whether during or after the offering.
Future sales of shares of our common stock in the public market, or the perception that such sales could occur, could cause our stock price to fall.
Sales of a substantial number of shares of our common stock in the public market, or the perception that these sales could occur, following this offering could cause the market price of our common stock to decline. A substantial majority of the outstanding shares of our common stock are, and the shares of common stock sold in this offering upon issuance will be, freely tradable without restriction or further registration under the Securities Act. We cannot predict the effect that future sales of common stock or other equity-related securities would have on the market price of our common stock.
29
Use of Proceeds
We estimate that we will receive net proceeds of approximately $ million, after deducting the joint placement agents fees and estimated offering expenses payable by us (based on an assumed public offering price of $2.76 per share, which was the last reported sales price of our common stock on the Nasdaq Global Market on July 11, 2024) and assuming no sale of any pre-funded warrants and no exercise of common warrants.
We currently intend to use the net proceeds from this offering, together with our existing cash resources, toward the following activities in their order of importance:
| ● | advancement of our clinical programs; and |
| ● | fund working capital and other general corporate purposes. |
This expected use of net proceeds from this offering represents our intentions based upon our current plans and business conditions. We have not determined the exact amounts we may spend on any of the items listed above or the timing of these expenditures.
Our management will retain broad discretion over the allocation of the net proceeds from this offering. We may find it necessary or advisable to use the net proceeds from this offering for other purposes, and we will have broad discretion in the application of net proceeds. Although we may use a portion of the net proceeds of this offering for the acquisition or licensing, as the case may be, of additional technologies, other assets or businesses, we have no current understandings, agreements or commitments to do so.
Dividend Policy
We have never declared or paid any dividends on our shares of common stock. We intend to retain any earnings for use in our business and do not currently intend to pay cash dividends on our shares of common stock. Dividends, if any, on our outstanding shares of common stock will be declared by and subject to the discretion of our board of directors, and subject to Delaware law.
30
Capitalization
The following table sets forth our cash and our capitalization as of March 31, 2024, on:
| ● | an actual basis; and |
| ● | on an as adjusted basis, giving effect to the issuance and sale of 4,000,000 shares of common stock in this offering at the assumed public offering price of $2.76 per share, which is the last reported sale price of our common stock on the Nasdaq Global Market on July 11, 2024, and after deducting joint placement agents fees and estimated offering expenses payable by us and excludes the proceeds, if any, from the sale of pre-funded warrants and the exercise of any common warrants issued in this offering. |
You should read this information in conjunction with our consolidated financial statements and the related notes included elsewhere in this prospectus, the information set forth in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and other financial information contained elsewhere in this prospectus.
| Actual | As Adjusted | |||||||
| (in $’000, except share data) | (in $’000) | |||||||
| Total cash and cash equivalents | 9,305 | 19,245 | ||||||
| Borrowing | - | - | ||||||
| Equity | ||||||||
| Additional paid-in capital: 15,873,113 common stock, outstanding, actual; 19,873,113 common stock, outstanding, as adjusted | 122,006 | 131,946 | ||||||
| Accumulated deficit | (104,210 | ) | (104,210 | ) | ||||
| Foreign Currency Translation Reserves | (3,658 | ) | (3,658 | ) | ||||
| Total equity | 14,138 | 24,078 | ||||||
| Total capitalization | 14,138 | 24,078 | ||||||
The number of shares of common stock that will be outstanding after this offering is based on 17,642,832 shares of common stock outstanding as of June 30, 2024, and excludes:
| ● | 1,978,338 shares of common stock issuable upon the exercise of outstanding warrants that were issued to stockholders in connection with the Re-domiciliation; and |
| ● | 5,243,328 shares of common stock reserved for issuance under the Company’s S-8 Registration Statement. |
31
Dilution
If you invest in the Common Stock in this offering, your ownership interest will be immediately diluted to the extent of the difference between the public offering price per share of common stock and the as adjusted net tangible book value per share of common stock immediately after this offering.
As of March 31, 2024, our historical net tangible book value was $14.1 million or $0.89 per share of common stock. Historical net tangible book value per share of common stock represents our total tangible assets less total liabilities, divided by the number of shares of common stock outstanding as March 31, 2024.
After giving effect to the receipt of the net proceeds from our sale of Common Stock in this offering at an assumed public offering price of $2.76 per share of common stock, assuming no sale of pre-funded warrants in this offering and no exercise of common warrants and after deducting estimated joint placement agent commissions and offering expenses payable by us, our as adjusted net tangible book value as of March 31, 2024, was $1.21 per share of common stock. This represents an immediate increase in net tangible book value of $0.32 per share of common stock to our existing shareholders and immediate dilution of $1.55 per share of common stock to investors purchasing Common Stock in this offering.
The following table illustrates this dilution on a per share of common stock basis, assuming all shares of common stock outstanding as of March 31, 2024:
| Assumed public offering price per share of common stock | $ | 2.76 | ||||||
| Historical net tangible book value per share of common stock as of March 31, 2024 | $ | 0.89 | ||||||
| Increase in net tangible book value per share of common stock attributed to this offering | 0.32 | |||||||
| As adjusted net tangible book value per share of common stock after this offering | 1.21 | |||||||
| Dilution in net tangible book value per share of common stock to investors in this offering | $ | 1.55 |
A $0.50 increase or decrease in the assumed public offering price of $2.76 per share of common stock, based on the last reported sale price for our common stock as reported on the Nasdaq Global Market on July 11, 2024, would decrease the number of shares of our common stock offered in this offering by approximately 613,497 shares or increase the number of shares of our common stock offered in this offering by approximately 884,956 shares.
We may also increase or decrease the number of shares of common stock we are offering. An increase of 100,000 in the number of shares of common stock offered by us would increase our as adjusted net tangible book value by approximately $0.2 million, or $0.003 per share, and decrease the dilution per share to investors participating in this offering by $0.003 per share, assuming the assumed offering price per share remains the same and after deducting the estimated joint placement agents commissions and estimated offering expenses payable by us. Similarly, a decrease of 100,000 in the number of shares of common stock offered by us would decrease our as adjusted net tangible book value by approximately $0.3 million or $0.01 per share, and increase the dilution per share to investors participating in this offering by $0.01 per share, assuming the assumed offering price per share remains the same and after deducting the estimated joint placement agents commissions and estimated offering expenses payable by us.
The dilution information above is for illustration purposes only. Our as adjusted net tangible book value following the closing of this offering will depend on the actual public offering price and other terms of this offering determined at pricing.
The number of shares of common stock that will be outstanding after this offering is based on 17,642,832 shares of common stock outstanding as of June 30, 2024, and excludes:
| ● | 1,978,338 shares of common stock issuable upon the exercise of outstanding warrants that were issued to stockholders in connection with the Re-domiciliation; and |
| ● | 5,243,328 shares of common stock reserved for issuance under the Company’s S-8 Registration Statement. |
To the extent any outstanding options are exercised, new options or other equity awards are issued under our equity incentive plans, or we issue additional equity or convertible securities in the future, there will be further dilution to new investors participating in this offering.
32
Management’s Discussion and Analysis of
Financial Condition and Results of Operations
You should read the following discussion and analysis of our audited and unaudited financial condition and results of operations together with our financial statements and the related notes included elsewhere in this prospectus. Some of the information contained in this discussion and analysis or set forth elsewhere in this prospectus, including information with respect to our plans and strategy for our business and related financing, and includes forward-looking statements that involve risks and uncertainties. As a result of many factors, including those factors set forth in the section titled “Risk Factors” our actual results could differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis. See also the section titled “Special Note Regarding Forward-Looking Statements.
We are a development stage enterprise at an early stage in the development of our drug candidates. We have incurred net losses since inception and expect to incur substantial and increasing losses for the next several years as we expand our research and development activities (“R&D”) and move our drug candidates into later stages of development. The process of carrying out the development of our drug candidates to later stages of development may require significant additional research and development expenditures, including pre-clinical testing and clinical trials, as well as for obtaining regulatory approval. To date, we have funded our operations primarily through the sale of equity securities, proceeds from the exercise of options, tax grants from R&D activities and interest income.
Our mission is to create premier ethical pharmaceutical drugs and therapies for patients with unmet or under met medical needs, in all instances fulfilling regulatory requirements of the FDA and other relevant regulatory agencies. We aim to be recognized as a leading specialty drug development company, committed to restoring health and transforming the lives of patients through the development of novel pharmaceutical products and treatments.
We incurred total comprehensive losses of $51.1 million and $12.1 million for fiscal 2023 and 2022, respectively. We incurred net losses of $12.0 million and $44.6 million for the nine months ended March 31, 2024 and 2023, respectively. As of March 31, 2024, we had accumulated losses of $104.2 million.
Components of Our Results of Operations
Other income
We receive tax incentives from the Australian government for R&D activities. Subject to certain exclusions, the Australian Government tax incentives provide benefits for eligible R&D activities. Entities are entitled to either (i) a 43.5% refundable tax offset for eligible companies with an aggregated turnover of less than A$20 million per annum or (ii) a non-refundable 38.5% tax offset for all other eligible companies. Our aggregated turnover is less than A$20 million and not be controlled by one or more income tax exempt entities, we anticipate being entitled to a claim of 43.5% refundable tax offset for costs relating to eligible R&D activities during the year.
We have not generated any revenue from the sale of products, however, and do not expect to generate any revenue from the sale of products in the foreseeable future, if at all. If our development efforts and any future product candidates are successful and result in regulatory approval, we may generate revenue in the future from product sales, payments from existing or potential future collaboration or license agreements with third parties, or any combination thereof.
Operating Expenses
Our operating expenses consist of (i) research and development expenses and (ii) general and administrative expenses.
Research and Development Expenses
Research and development (R&D) expenses consist primarily of external and internal costs incurred in performing clinical and preclinical development activities.
33
Our R&D expenses consist of:
| ● | external costs incurred under agreements with CROs, contract manufacturers, consultants and other third parties to conduct and support our clinical trials and preclinical studies; and |
| ● | internal costs, including R&D personnel-related expenses such as salaries, and benefits, as well as allocated facilities costs and dues and subscriptions. |
We expense R&D costs as incurred. We track R&D expenses on clinical trial basis.
Although R&D activities are central to our business model, the successful development of our product candidates is highly uncertain. There are numerous factors associated with the successful development of our product candidates, including future trial design and various regulatory requirements, many of which cannot be determined with accuracy at this time based on our stage of development. In addition, future regulatory factors beyond our control may impact our clinical development programs. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. As a result, we expect our R&D expenses will increase substantially in connection with our ongoing and planned clinical and preclinical development activities in the near term and in the future. At this time, we cannot accurately estimate or know the nature, timing and costs of the efforts that will be necessary to complete the preclinical and clinical development of our product candidates. Our future R&D expenses may vary significantly based on a wide variety of factors such as:
| ● | the number and scope, rate of progress, expense and results of our clinical trials and preclinical studies, including any modifications to clinical development plans based on feedback that we may receive from regulatory authorities; |
| ● | per patient trial costs; |
| ● | the number of trials required for approval; |
| ● | the number of sites included in the trials; |
| ● | the countries in which the trials are conducted; |
| ● | the length of time required to enroll eligible patients; |
| ● | the number of patients that participate in the trials; |
| ● | the number of doses that patients receive; |
| ● | the drop-out or discontinuation rates of patients; |
| ● | the potential additional safety monitoring requested by regulatory agencies; |
| ● | the duration of patient participation in the trials and follow-up; |
| ● | the cost and timing of manufacturing of our product candidates; |
| ● | the costs, if any, of obtaining third-party drugs for use in our combination trials; |
| ● | the extent of changes in government regulation and regulatory guidance; |
| ● | the efficacy and safety profile of our product candidates; |
| ● | the timing, receipt, and terms of any approvals from applicable regulatory authorities; and |
| ● | the extent to which we establish additional collaboration, license, or other arrangements. |
A change in the outcome of any of these variables with respect to the development of our product candidates could significantly change the costs and timing associated with the development of that product candidate. We may never succeed in obtaining regulatory approval for any product candidate.
34
Other Operating Expenses
Other Operating expenses consist primarily of general and administrative expenses which relate to personnel-related expenses finance and accounting, human resources and other administrative functions. In addition, other operating expenses also concern salaries, stock-based compensation and benefits, legal fees, expenses relating to patent and corporate matters and professional fees paid for accounting, auditing, consulting and tax services, as well as facilities-related costs not otherwise included in R&D expenses and other costs such as insurance costs and travel expenses.
We anticipate our operating expenses will increase substantially in the future as we expand our operations, including increasing our headcount to support our continued R&D activities and preparing for potential commercialization of our product candidates. We also anticipate we will incur increased accounting, audit, legal, regulatory, compliance, director and officer insurance, and investor and public relations expenses associated with operating as a public company.
Results of Operations
Comparison of the Nine Months Ended March 31, 2024 and 2023
The following tables summarize our results of operations for the periods presented (in thousands):
| For the Nine Months Ended March 31 | ||||||||||||||||
| 2024 | 2023 | $ Change | % Change | |||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | (8,520 | ) | $ | (4,597 | ) | $ | (3,923 | ) | (85 | )% | |||||
| Acquisition of in-process research and development | $ | - | $ | (35,347 | ) | $ | 35,347 | (100 | )% | |||||||
| General and administrative | (11,777 | ) | (5,530 | ) | (6,247 | ) | (113 | )% | ||||||||
| Total operating expenses | (20,297 | ) | (45,474 | ) | 25,177 | 55 | % | |||||||||
| Loss from operations | (20,297 | ) | (45,474 | ) | 25,177 | 55 | % | |||||||||
| Other income/(expense): | - | - | - | - | ||||||||||||
| Benefit from R&D tax credit | 8,150 | 684 | 7,466 | 1,092 | % | |||||||||||
| Foreign exchange gains (losses) | (17 | ) | - | (17 | ) | - | ||||||||||
| Interest income | 166 | 153 | 13 | 8 | % | |||||||||||
| Total other income/(expense), net | 8,299 | 837 | 7,462 | 892 | % | |||||||||||
| Currency translation adjustment, net of tax | (403 | ) | (2,029 | ) | 1,626 | 80 | % | |||||||||
| Comprehensive loss | $ | (12,401 | ) | $ | (46,666 | ) | $ | 34,265 | 73 | % | ||||||
Operating Expenses
Research and development
Research and development expenses increased by $3.9 million for the nine months ended March 31, 2024 compared to the nine months ended March 31, 2023. The increase was primarily due to due to the commencement of a Bioequivalence/Bioavailability clinical trial investigating IHL-42X in healthy volunteers, a Phase 2 clinical trial investigating IHL-675A in patients with Rheumatoid arthritis, and a Phase 2/3 clinical trial investigating IHL-42X in patients with obstructive sleep apnea.
35
Acquisition of in-process research and development
Acquisition of in-process research and development expense was recorded exclusively in the nine months ended March 31, 2023, because such expenses related to the acquisition of APIRx Pharmaceutical USA, LLC, which was recorded as an asset acquisition during such period. The acquisition of APIRx was completed in August 2022. We concluded that the acquisition of APIRx did not meet the definition of business under ASC 805, Business Combinations as APIRx did not have outputs present and a substantive process was not acquired.
General and Administrative
General and administrative expenses increased by $6.2 million for the nine months ended March 31, 2024, compared to the nine months ended March 31, 2023. The increase was due to increases of $4.6 million in salaries and benefits for employees and directors (mostly due to the value of stock awards), $1.2 million in compliance, legal and regulatory as a result of increased legal and accounting costs due to the increase expenses of our reporting obligations following our re-domiciliation (from $1.2 million to $2.4 million), $0.1 million in occupancy expenses, $0.04 million in advertising and investor relations, $0.3 million in other administration expenses, and $0.02 million in depreciation expense.
Other Income (Expense)
Benefit from R&D tax credit
Benefit from R&D tax credit increased by $7.5 million for the nine months ended March 31, 2024 compared to the nine months ended March 31, 2023. The increase was due to an increase in the research and development tax credit received from the Australian Taxation Office for our research and clinical trials activities in Australia following finalization of the fiscal 2023 Australian tax return.
Foreign exchange losses and Interest Income
Foreign exchange losses increased by $17,000 for the nine months ended March 31, 2024 compared to the nine months ended March 31, 2023, due to unfavorable currency exchange rates, partially offset by an increase in interest income received from cash deposited in our bank accounts by $13,000 during the same period.
Currency translation adjustment, net of tax
Currency translation adjustment, net of tax decreased $1.8 million for the nine months ended March 31, 2024 compared to the nine months ended March 31, 2023. The decrease was due to currency translation of the financial statements from the Australian dollar to the U.S. dollar. We maintain our consolidated financial statements in Australian dollar, which is our functional currency. However, our financial statements are translated into U.S. dollars for reporting purposes. See our note 2 “Foreign Currency Translation” to our unaudited financial statements included in this Registration Statement for further information.
Comparison of Fiscal Year Ended June 30, 2023 to June 30, 2022
The following tables set forth our results of operations in for the years ended June 30, 2023 and 2022.
Year ended June 30 | ||||||||||||||||
| 2023 | 2022 | $ Change | % Change | |||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | (6,309 | ) | $ | (3,899 | ) | $ | (2,410 | ) | (62 | )% | |||||
| Acquisition of in-process research and development | $ | (35,347 | ) | $ | - | $ | (35,347 | ) | - | |||||||
| General and administrative | (8,012 | ) | (7,443 | ) | (569 | ) | (8 | )% | ||||||||
| Total operating expenses | (49,668 | ) | (11,342 | ) | (38,326 | ) | (338 | )% | ||||||||
| Loss from operations | (49,668 | ) | (11,342 | ) | (38,326 | ) | 338 | % | ||||||||
| Other income/(expense): | ||||||||||||||||
| R&D tax incentive | 683 | 568 | 115 | 20 | % | |||||||||||
| Foreign exchange income (expense) | (67 | ) | (48 | ) | (19 | ) | (40 | )% | ||||||||
| Interest income | 241 | 4 | 237 | 5,925 | % | |||||||||||
| Total other income/(expense), net | 857 | 524 | 333 | 64 | % | |||||||||||
| Currency translation adjustment, net of tax | (2,292 | ) | (1,302 | ) | (990 | ) | (76 | )% | ||||||||
| Total comprehensive loss | $ | (51,103 | ) | $ | (12,120 | ) | $ | (38,983 | ) | (322 | )% | |||||
36
Operating Expenses
Research and Development (in thousands):
Research and development expenses increased by $2.4 million in fiscal 2023 compared to fiscal 2022. The increase was primarily due to increases in development costs related to our clinical trials, particularly with respect to Psi-GAD, IHL-675A and IHL-42X.
Acquisition of in-process research and development
Acquisition of in-process research and development expense was recorded exclusively in the fiscal year 2023, because such expenses related to the acquisition of APIRx Pharmaceutical USA, LLC, which was recorded as an asset acquisition during such period. The acquisition of APIRx was completed in August 2022. We concluded that the acquisition of APIRx did not meet the definition of business under ASC 805, Business Combinations as APIRx did not have outputs present and a substantive process was not acquired.
General and Administrative
General and administrative expenses increased $569,000 in fiscal 2023 compared to fiscal 2022. The increase was due to increases of $0.9 million in salaries, and other employee benefits (from $1.5 million to $2.4 million), $88,000 in depreciation expense (from nil to $88,000), $2,000 in occupancy expenses (from $82,000 to $84,000), $76,000 in other administration expenses (from $258,000 to $316,000), $1.1 million in share based payment expenses (from 1.0 million to 2.1 million) partially offset by a decrease of $0.8 million in compliance, legal and regulatory (from $2.6 million to $1.8 million), $0.8 million in advertising and investor relations (from $2.0 million to $1.2 million).
Other Income (Expense)
R&D tax incentive
R&D tax incentive increased by $115,000 in fiscal 2023 compared to fiscal 2022. The increase was due to an increase in the research and development tax credit received from the Australian Taxation Office as a result of our research and clinical trials activities in Australia.
Foreign exchange losses and Interest Income
Foreign exchange losses increased by $19,000 for fiscal 2023 compared to fiscal 2022. The increase was due to an unfavorable exchange rate between the Australian dollar and the US dollar. Instead, interest income increased by $237,000 for fiscal 2023 compared to fiscal 2022 due to an increase in interest rates.
Currency translation adjustment, net of tax
Currency translation adjustment, net of tax increased $990,000 for fiscal 2023 compared to fiscal 2022. The increase was due to currency translation of the financial statements from the Australian dollar to the US dollar. We maintain our consolidated financial statements in Australian dollar, which is our functional currency. However, our financial statements are translated into US dollars for reporting purposes. See our note 2 “Foreign Currency Translation” to our financial statements included in this Registration Statement for further information.
37
Liquidity and Capital Resources
Sources of Liquidity
Since our inception, our operations have mainly been financed through the issuance of equity securities. Additional funding has come through interest earned from cash on term deposit.
As of March 31, 2024, we had cash of $9.3 million. We anticipate that our current cash will be sufficient for the current fiscal year and to fund our operations at least until December 2024. However, our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement that involves risks and uncertainties, and actual results could vary materially.
For the fiscal year ended June 30, 2023, we incurred a net loss after tax of $48.8 million and experienced net cash outflows from operating activities of $13.0 million. In addition, as of March 31, 2024, we had cash assets of $9.3 million and current assets exceeded our current liabilities by $13.5 million.
Our ability to continue as going concern and to pay our debts as and when they fall due is dependent on the following:
| ● | the ability to raise additional funding; | |
| ● | managing costs in line with management’s forecasts; and | |
| ● | receipt of Australian R&D tax incentives in line with management’s estimates for the amount and expected timing. |
We believe that we will be able to raise additional funding, continue to receive R&D tax incentives and that we will manage efficiently our costs in the foreseeable future. Thus, we believe that we will continue to operate as going concern in the foreseeable future.
Due to our focus on research and development activities, we do not have ready access to credit facilities and, therefore, are not subject to externally imposed capital requirements. Our objective in relation to capital risk management is to balance our current working capital position against the requirements to meet research and development programs and corporate overheads.
We anticipate that we will require substantial additional funds in order to achieve our long-term goals and complete the research and development of our current drug candidates. We do not expect to generate significant revenue until we obtain regulatory approval to market and sell our drug candidate and sales of our drug candidate have commenced. We therefore expect to continue to incur substantial losses in the near future.
Our future capital requirements are difficult to forecast and will depend on many factors, including:
| ● | the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
| ● | the scope, results and timing of preclinical studies and clinical trials; |
| ● | the costs and timing of regulatory approvals; and |
| ● | the costs of establishing sales, marketing and distribution capabilities. |
38
Off-Balance Sheet Arrangements
We did not have, during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined in the rules and regulations of the SEC.
Contractual obligations
Excluding accounts payable and lease obligations, which are reflected in our balance sheet, we did not have any other contractual obligations during the periods presented.
Contingent liabilities
We did not have any material contingent liabilities outstanding during the periods presented.
Capital commitments
We did not have any material capital expenditure commitments outstanding during the periods presented.
Cash Flows
| For the Nine Months Ended March 31, 2024 | For the Nine Months Ended March 31, 2023 | |||||||
| Net cash used in operating activities | $ | (12,203 | ) | $ | (8,167 | ) | ||
| Net cash used in investing activities | (274 | ) | (145 | ) | ||||
| Net cash provided by financing activities | - | 8,207 | ||||||
| Net decrease in cash | $ | (12,477 | ) | $ | (105 | ) | ||
Cash flows from operating activities
Cash used in operating activities increased by $4.0 million for the nine months ended March 31, 2024, compared to the nine months ended March 31, 2023. The increase was due to an increase in prepaid expenses and other current assets (from $264,000 to $6.2 million), partially offset by positive cash flow regarding trade and other payables by $0.8 million (from cash used equal to $504,000 to cash received equal to $302,000).
Cash flows from investing activities
Cash used in financing activities increased by $129,000 for the nine months ended March 31, 2024, compared to the nine months ended March 31, 2023. The increase was due to an increase in cash used to purchase equipment to conduct our clinical trials.
Cash flows from financing activities
Cash provided by financing activities decreased to nil for the nine months ended March 31, 2024, compared to the nine months ended March 31, 2023. The decrease was due to a decrease in the proceeds from issuances of shares of common stock.
39
Comparison of cash flows for the fiscal year ended June 30, 2022, with June 30, 2023
The following table summarizes our cash flows for the periods presented:
| Year Ended June 30, | ||||||||
| 2023 | 2022 | |||||||
| Net cash used in operating activities | $ | (13,041 | ) | $ | (10,218 | ) | ||
| Net cash used in investing activities | (316 | ) | - | |||||
| Net cash provided by financing activities | 8,175 | 29,566 | ||||||
| Net (decrease)/increase in cash, cash equivalents and restricted cash | $ | (5,182 | ) | $ | 19,348 | |||
Cash flows from operating activities
Cash used in operating activities increased by $2.8 million in fiscal 2023 compared to fiscal 2022. The increase was due to an increase in cash used to conduct our research and development activities and for general and administrative expenses, and an increase in prepaid expenses and other current assets (from $27,000 to $626,000), partially offset by positive cash flow regarding trade and other payables by $0.3 million.
Cash flows from investing activities
Cash used in investing activities increased by $0.3 million in fiscal 2023 compared to fiscal 2022. The increase was due to payments for the addition of property, plant and equipment.
Cash flows from financing activities
Cash provided by financing activities decreased by $21.4 million in fiscal 2023 compared to fiscal 2022. The decrease was due to a decrease in the proceeds from issuances of shares of common stock.
Critical Accounting Policies and Significant Judgments and Estimates
Our financial statements are prepared in accordance with generally accepted accounting principles in the United States. The preparation of our financial statements and related disclosures requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, costs and expenses, and the disclosure of contingent assets and liabilities in our financial statements. We base our estimates on historical experience, known trends and events, and various other factors we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. We evaluate our estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are described in more detail in Note 2 to our financial statements included elsewhere in this prospectus, we believe the following accounting policies are those most critical to the judgments and estimates used in the preparation of our financial statements.
Acquisitions
The Company evaluate acquisitions under the accounting framework in ASC 805, Business Combinations, to determine whether the transaction is a business combination or an asset acquisition. In determining whether an acquisition should be accounted for as a business combination or an asset acquisition, the Company first perform a screen test to determine whether substantially all of the fair value of the gross assets acquired is concentrated in a single identifiable asset or a group of similar identifiable assets. If this is the case, the acquired set is not deemed to be a business and is instead accounted for as an asset acquisition. If this is not the case, the Company further evaluate whether the acquired set includes, at a minimum, an input and a substantive process that together significantly contribute to the ability to create outputs. If so, the Company conclude that the acquired set is a business.
The Company measures and recognizes asset acquisitions that are not deemed to be business combinations based on the cost to acquire the assets, which includes pre-acquisition direct costs recorded in accrued professional and consulting fees. Goodwill is not recognized in asset acquisitions.
40
During the year ended June 30, 2023 the Company acquired APIRx Pharmaceutical USA, LLC (“APIRx”). The Company concluded that the acquisition of APIRx did not meet the definition of business under ASC 805, Business Combinations as the acquired set did not have outputs present and a substantive process was not acquired. Therefore, the Company accounted for the transaction as an asset acquisition rather than a business combination.
In accordance with ASC 730-10-25-2(c), intangible assets used in research and developmental activities acquired in an asset acquisition should be expensed at the acquisition date if there is no alternative future use in other R&D projects or otherwise (i.e., if they have no economic value). Additionally, in an asset acquisition, direct transaction costs are accumulated as a component of the consideration transferred and expensed with the acquired IPR&D that has no alternative use.
The Company determined that product candidates pertaining to APIRx had no alternative future use at the time of acquisition and charged $35.3 million including transaction costs of $2.43 million, to the acquisition of in-process research and development (IPR&D) expense as of the date of acquisition.
Share-based compensation
The Company accounts for share-based compensation arrangements with employees and non-employees using a fair value method which requires the recognition of compensation expense for costs related to all share-based payments including share options. The fair value method requires the Company to estimate the fair value of share-based payment awards on the date of grant using an option-pricing model. The Company uses either the trinomial pricing or Black-Scholes option-pricing model to estimate the fair value of options granted. Share-based compensation awards are expensed using the graded vesting method over the requisite service period, which is generally the vesting period, for each separately-vesting tranche. The Company has elected a policy of estimating forfeitures at grant date. Option valuation models, including the trinomial pricing and Black-Scholes option-pricing model, require the input of several assumptions. These inputs are subjective and generally require significant analysis and judgment to develop.
Benefit from Research and Development Tax Incentive
Benefit from R&D tax credit consists of the R&D tax credit received in Australia, which is recorded within other income (expense), net. The Company recognizes grants once both of the following conditions are met: (i) the Company is able to comply with the relevant conditions of the grant and (ii) the grant is received.
Emerging Growth Company Status and Smaller Reporting Company Status
We are an emerging growth company, as defined in the JOBS Act. The JOBS Act permits an emerging growth company such as us to take advantage of an extended transition period to comply with new or revised accounting standards. We have elected to avail ourselves of such extended transition period, which means that when a standard is issued or revised and it has different application dates for public or private companies, we can adopt the new or revised standard at the time private companies adopt the new or revised standard and may do so until such time that we either (i) irrevocably elect to opt out of such extended transition period or (ii) no longer qualify as an emerging growth company. We may choose to early adopt any new or revised accounting standards whenever such early adoption is permitted for private companies.
We will continue to remain an emerging growth company until the earliest of the following:
| ● | the last day of the fiscal year following the fifth anniversary of the date of the completion of this offering; |
| ● | the last day of the fiscal year in which our total annual gross revenue is equal to or more than $1.235 billion; |
| ● | the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years; or |
| ● | the date on which we are deemed to be a large accelerated filer under the rules of the SEC. |
We are also a smaller reporting company as defined in the Exchange Act. We may continue to be a smaller reporting company even after we are no longer an emerging growth company. We may take advantage of certain of the scaled disclosures available to smaller reporting companies and will be able to take advantage of these scaled disclosures for so long as our voting and non-voting common stock held by non-affiliates is less than $250.0 million measured on the last business day of our second fiscal quarter, or our annual revenue is less than $100.0 million during the most recently completed fiscal year and our voting and non-voting common stock held by non-affiliates is less than $700.0 million measured on the last business day of our second fiscal quarter.
41
Business
History and Development of the Company
Our legal name is Incannex Healthcare Inc., which was incorporated in Delaware in July 2023.
Since 2019, we have been conducting research and development for medicinal cannabis pharmaceutical products and psychedelic medicine therapies for treatment of a range of indications.
In January 2019, the Department of Health of Victoria granted us licenses to sell or supply cannabinoid substances, and in particular cannabis, cannabidiol (“CBD”), tetrahydrocannabinols (“THC”) and dronabinol.
In June 2020, we discontinued the sale of mouthguards for sports activities to focus our resources on cannabinoid sales and development activities. As a result, on June 30, 2020, we sold our wholly-owned subsidiary Gameday International Pty Ltd.
In June 2021, in order to focus on the development of our drug candidates, we terminated our distribution agreement for the sale of cannabinoid products and, as a result, have not had any sales of such products since then.
In August 2022, we acquired APIRx Pharmaceutical USA, LLC, which focuses on the research and development of prescription pharmaceutical cannabinoid medicines. We issued 218,169,497 ordinary shares, at a price of A$0.225 per share, in exchange for 100% of the equity interests in APIRx. Upon completion of the acquisition, the Founders of APIRx, Dr. George Anastassov and Mr Lekhram Changoer joined Incannex as a Director and our Chief Technology Officer, respectively. While APIRx owned intellectual property at the time of the acquisition, it did not have any other material assets or liabilities. The acquisition of APIRx presents Incannex with both long and short-term opportunities for significant value growth. APIRx has active clinical and pre-clinical research and development projects underpinned by an intellectual property portfolio that includes 28 granted patents and 11 pending patents. It holds a diverse portfolio of promising therapeutic candidates targeted at treating an extensive range of conditions including pain disorders, addiction disorders, mental illnesses, gastrointestinal diseases, gum disease, skin conditions and ophthalmic conditions.
On November 28, 2023, the redomiciliation of Incannex Healthcare Limited, an Australian corporation was implemented under Australian law in accordance with the Scheme Implementation Deed, as amended and restated on September 13, 2023, between Incannex Australia and the Company. As a result of the redomiciliation, Incannex Australia became a wholly-owned subsidiary of the Company, which is the new ultimate parent company.
Our principal office is located at Suite 105, 8 Century Circuit, Norwest 2153, NSW Australia and our telephone number is +61 409 840 786. Our address on the Internet is www. incannex.com. The information on, or accessible through, our website is not part of this Registration Statement. All information we file with the U.S. Securities and Exchange Commission (“SEC”) is available through the SEC’s Electronic Data Gathering, Analysis and Retrieval system, which may be accessed through the SEC’s website at www.sec.gov.
Business Overview
Incannex is a biotech company developing cannabinoid and psychedelic compound medicines.
The recent acquisition of APIRx brings to Incannex a diverse portfolio of therapeutic candidates targeted at treating a broad range of conditions including addiction disorders, skin conditions, gastrointestinal diseases, periodontitis, and ophthalmic conditions.
The acquisition of APIRx strengthens our position in the area of cannabinoid and psychedelic treatment development. In particular, it:
| ● | adds a large portfolio of intellectual property with granted and pending patents; |
| ● | significantly expands Incannex’s addressable markets globally and addressable market sizes; |
| ● | further enhances Incannex’s technical and drug development capability; and |
| ● | expands Incannex’s drug delivery capability to include APIRx’s patented delivery technologies. |
42
Strategy
Our mission is to create premier ethical pharmaceutical drugs and therapies for patients with unmet or under met medical needs, in all instances fulfilling regulatory requirements of the FDA and other relevant regulatory agencies. We aim to be recognized as a leading specialty drug development company, committed to restoring health and transforming the lives of patients through the development of novel pharmaceutical products and treatments.
We develop targeted and scientifically validated fixed-dose combinations of cannabinoids with generic partners, novel formulations of cannabinoids, and psychedelic agents, applying proprietary insights in an effort to create long term value for our patients and shareholders. We focus on clinical indications that we believe represent unmet or inadequately addressed medical needs that also represent compelling commercial opportunities. In particular, we are developing three novel pharmaceutical compositions to target five indications: obstructive sleep apnea (“OSA”), traumatic brain injury and concussion (“TBI”), rheumatoid arthritis (“RA”), inflammatory bowel disease (“IBD”) and inflammatory lung conditions (“ARDS”, “COPD”, Asthma, Bronchitis). We are also developing a treatment for generalized anxiety disorder (“GAD”) utilizing psilocybin combined with innovative psychotherapy methods. With the acquisition of APIRx we are also developing cannabinoid products to target additional indications. Our initial focus will be on opioid addiction, smoking cessation, cannabis use disorder, vitiligo, atopic dermatitis and psoriasis.
We are pursuing FDA registration and marketing approval for each product and therapy under development.
Additionally, we seek to secure patents on our drug candidates in conjunction with our medical and scientific staff, advisors and the investigators of our research studies that constitute our advisory board. Our advisory board is comprised of industry and academic experts familiar with our business, and we meet with the advisory board regularly. The current members of our advisory board are Dr. Mark Bleackley (our Chief Scientific Officer), Dr George Anastassov (Non-executive director), Lekhram Changoer (our Chief Technical Officer), and Dr Paul Liknaitzky (psychedelic principal investigator from Monash University).
To achieve our goals, we intend to:
| ● | Advance our novel investigational drug candidates towards approval in the United States and elsewhere. We are pursuing FDA approval of all our drug candidates currently in development. All preclinical and clinical trials are structured to ensure that each program is FDA compliant. We will be pursuing a New Drug Application (“NDA”) with the FDA with respect to each of our drug candidates. If the NDA is approved, the product may be marketed in the United States. Once an NDA for one of our drug candidates is approved in the United States, we plan to pursue marketing approval of our drug candidates in other regions including the Europe Union, Japan, Australia and Israel. | |
| ● | Take advantage of accelerated commercialization pathway options for our drug candidates. We and our regulatory consultants believe that each of our drug candidates will qualify for one or more FDA expedited review programs (breakthrough designation, accelerated approval, priority review and/or fast track), as there are a limited amount of pharmaceutical drug treatments approved in the U.S. to treat the indications that we are targeting with our drug candidates, and the pharmaceutical treatments that do exist provide limited treatment and are costly. These expedited review programs often result in accelerated and less-costly regulatory pathways to approval compared with traditional regulatory pathways. We have not yet approached the FDA about the suitability of our products for these accelerated approval pathways and such designations do not guarantee accelerated review by the FDA. | |
| ● | Develop future drug candidates targeting unmet medical needs. We intend to develop drug candidates that treat unmet medical or inadequately addressed conditions. As a result, we may have opportunities to accelerate commercialization of such products. | |
43
| ● | Maintain a strong intellectual property portfolio. We have developed a global intellectual property strategy to support our commercial objectives. We are monitoring the results of our research and development programs to identify new intellectual property that aligns with those commercial objectives. We intend to take a global approach to our intellectual property strategy and we intend to pursue patent protection in key global markets, including the United States, Europe, Japan and Israel. We have one granted patent for IHL-216A and multiple pending patent applications relating to our drug candidates IHL-42X, IHL-216A and IHL-675A and we own a further 28 granted and 11 pending patents resulting from the APIRx acquisition. Our patents approach aligns with our regulatory strategy, including the proposed submission of Pre-Investigational New Drug Application (“pre-IND”) meeting requests and Investigational New Drug (IND) applications to the FDA for our clinical programs. |
Clinical Approach
We are pursuing FDA approval for all our drug candidates currently being developed. We will continue to work with FDA to ensure each clinical program is structured to meet regulatory requirements. FDA approval will be sought following the completion of successful phase 3 studies. Once we receive FDA approval for our drug candidates, we will be able to commercialize our drug candidates in the United States and pursue regulatory approval for the drug to be made available in other jurisdictions, including the Europe, Japan, Australia and Israel.
Market Opportunity
The combined annual global market size of the indications we are targeting is over US$249 billion, which is derived from the total addressable market for the treatment of all indications over which we are developing drug candidates. The indications being pursued include: OSA, TBI, concussions, rheumatoid arthritis, inflammatory bowel disease, inflammatory lung conditions (ARDS, COPD, Asthma, Bronchitis), GAD, addiction disorders, skin conditions, gastrointestinal diseases, periodontitis and ophthalmic conditions. Thus, there is significant economic potential to shareholders, as well as benefit to patients suffering from these medical conditions.
Our Drug Candidates
IHL-42X
Obstructive Sleep Apnea
Obstructive sleep apnea (“OSA”) is characterized by a narrowing or obstruction of the upper airway in sleep, interfering with breathing and interrupting sleep. OSA is a disease of sleep disordered breathing where the upper airway repeatedly completely or partially collapses during sleep. This disrupts airflow, reduces oxygen uptake and leads to poor sleep quality. Presentation of OSA often includes snoring and waking up gasping for air. This relatively common and chronic disorder is underdiagnosed and inadequately treated. It is understood to contribute to a wide range of serious long-term outcomes, including cardiovascular disease, cognitive impairments such as memory loss, poor concentration and judgment, depression and death or injury due to traffic accidents resulting from excessive daytime sleepiness. The costs associated with OSA are substantial, relating to healthcare utilization lost productivity, workplace and motor vehicle accidents.
A 2019 article published by the Lancet premised on literature-based analysis of 17 studies across 16 countries, estimated that OSA affects some 936 million adults worldwide. This alarming statistic is also thought to be increasing due to growing prevalence of obesity and an ageing global population. Many people with OSA develop high blood pressure (hypertension), which can increase the risk of cardiovascular disease. The more severe the OSA, the greater the risk of coronary artery disease, heart attack, heart failure and stroke.
There are no registered drugs for OSA. Current treatment options include: continuous positive airway pressure (“CPAP”) in which an external device pneumatically splints the airway open to prevent disruptions in breathing; oral appliances to advance the mandible or to retain the tongue, putting the mouth in a position more conducive to breathing; surgery to remove physical obstructions to air flow; and implantable electronic stimulators to activate muscles at the base of the tongue, opening the airway in synchrony with respiration. However, all of these therapies are poorly tolerated, inadequate, expensive, and for implantable stimulators and surgery, invasive.
44
The standard treatment option is the mechanical CPAP device, however, patient compliance to CPAP devices is low due to discomfort and claustrophobia resulting from pressurized air being pumped into the patient’s nose and/or mouth during sleep. Despite these discomforts, the global annual market for sleep apnea devices is over US$8 billion and growing. The estimated compound annual growth rate (“CAGR”) for OSA detection and treatment using devices from 2024 to 2029 is 7.33%.
IHL-42X in Obstructive Sleep Apnea
IHL-42X is a fixed-dose combination of acetazolamide, a registered pharmaceutical, and dronabinol, a synthetic form of delta-9-tetrahydrocannabinol (“THC”); both agents have been shown to reduce the apnea hypopnea index (“AHI”). We believe that the activity of dronabinol on cannabinoid receptors causes dilation of the airway, and acetazolamide induces modest metabolic acidosis, signaling to the body that there is excess CO2 in the blood, thus increasing respiration. By exploiting two mechanisms that both reduce AHI in one pharmaceutical formulation, we believe that IHL-42X has a therapeutic benefit at doses of each constituent drug that are safe and tolerable.
Phase 2 Clinical Trial for IHL-42X for Obstructive Sleep Apnea (“OSA”)
We completed a proof-of-concept Phase 2 clinical trial in Australia to establish the safety and efficacy of IHL-42X for treatment of obstructive sleep apnea support our Investigational New Drug (“IND”) application with FDA and to inform the design of the pivotal Phase 2/3 clinical trial. The IND was cleared in August 2023.
The primary endpoint of the trial was the change in AHI relative to baseline and the secondary endpoints included change in oxygen desaturation index (“ODI”), daytime somnolence measured by the Epworth Sleepiness Scale, improvement in mood as measured by the Profile of Moods State (“POMS”), and well-being as measured by the Short Form 36 and the safety of the IHL-42X combination was assessed through adverse event monitoring.
The study was conducted at the Alfred Hospital in Melbourne Australia and the University of Western Australia Centre for Sleep Science in Perth. Novotech, a global contract research organization, was engaged to manage and monitor the study. In July 2021, a confidential interim analysis of the data from the phase 2 double blind randomized placebo-controlled clinical trial was performed, and these results were utilized to support a patent application regarding compositions and methods of use for the treatment of obstructive sleep apnea. In December 2021, we completed the dosing of participants in the phase 2 clinical trial.
A total of eleven participants were recruited to the study and ten participants competed treatment periods. The cross over design of the trial, which assessed low, medium and high doses of IHL-42X and the placebo in all ten trial participants, increased the power of the study compared to a traditional parallel arm design.
In June 2022, we announced the full and complete analysis of data from the phase 2 proof-of-concept clinical trial investigating IHL-42X for treatment of OSA:
| ● | The following table presents the average AHI data for baseline and each treatment period. All doses of IHL-42X reduced AHI in patients with sleep apnea compared to baseline. This reduction was substantially greater than observed for placebo. |
45
Average AHI data for baseline and each treatment period
| Baseline | Placebo | Low | Medium | High | ||||||||||||||||
| Average AHI | 42.84 | 40.08 | 21.13 | 22.22 | 27.78 | |||||||||||||||
| Standard deviation | 20.33 | 18.16 | 15.92 | 15.52 | 17.61 | |||||||||||||||
| % Reduction relative to baseline | N/A | 6.44 | 50.69 | 48.13 | 35.16 | |||||||||||||||
| p value compared to baseline | N/A | 0.76 | 0.029 | 0.031 | 0.12 | |||||||||||||||

Figure 1. Average reduction in apnea hypopnea index (AHI) for each treatment period, relative to baseline, in the IHL-42X proof of concept phase 2 clinical trial.

Figure 2. Proportion of patients in each IHL-42X proof of concept treatment period who experienced a reduction in AHI of >50% and >80% relative to baseline.
46
| ● | At the group level the difference relative to baseline with low dose and medium dose was statistically significant (p<0.05). |
| ● | When comparing directly to baseline within patients the difference in AHI compared to baseline between all three doses and placebo was statistically significant (p<0.001), as shown in the table below: |
Change in AHI from baseline within subject (least square mean)
| Average change in AHI from baseline | p-value relative to placebo (Bonferroni adjusted) | Proportion of subjects with AHI reduction >50% relative to baseline (%) | Proportion of subjects with AHI reduction >80% relative to baseline (%) | |||||||||||||
| Placebo | 1.95 | N/A | 10 | 0 | ||||||||||||
| Low | 17.51 | <0.001 | 62.5 | 25 | ||||||||||||
| Medium | 14.86 | <0.001 | 33.3 | 11.1 | ||||||||||||
| High | 16.18 | <0.001 | 22.2 | 11.1 | ||||||||||||
| ● | Low dose IHL-42X reduced AHI by >50% relative to baseline in 62.5% of patients and by >80% in 25% of patients. |
| ● | Low dose IHL-42X reduced AHI to the greatest extent at both the group level and when comparing the within patient reduction relative to baseline. |
| ● | Low dose IHL-42X reduced AHI to a greater extent than predicted based on published data for dronabinol and acetazolamide alone (Table 3). |
Comparison of reduction in AHI relative to baseline with low dose IHL-42X and the predicted reduction with component drugs as monotherapies at equivalent doses based on reported data.
| Reduction in AHI compared to baseline (%) | ||||
| 2.5 mg dronabinol (1) | 23.4 | |||
| 125 mg acetazolamide (2) | 23.4 | |||
| Low dose IHL-42X | 50.7 | |||
The reduction in AHI observed during IHL-42X treatment periods means that when treated with our proprietary drug, the patient’s breathing was interrupted less frequently during sleep. This supports our hypothesis that IHL-42X is an effective treatment for OSA. Furthermore, greater reduction in AHI with low dose IHL-42X compared to dronabinol and acetazolamide at equivalent doses supports our hypothesis that the two drugs are acting synergistically to produce a superior outcome than would be expected from dronabinol and acetazolamide as monotherapies.
The ODI is a measure similar to AHI, but instead measures the number of times there is insufficient blood oxygen levels or desaturation events. With respect to the oxygen desaturation index (“ODI”), the data from the phase 2 proof-of-concept clinical trial supported the following:
| ● | all three doses of IHL-42X reduced ODI compared to baseline to a greater extent than placebo. |
| ● | For low and medium dose IHL-42X the difference in reduction in ODI relative to baseline compared to placebo was statistically significant (p<0.05). |
47
Reduction in ODI compared to baseline during each treatment period.
| Reduction in ODI relative to baseline (least squares mean) | Reduction in ODI relative to baseline (%) | p value compared to placebo (Bonferroni adjusted) | ||||||||||
| Placebo | 1.8 | 18.3 | N/A | |||||||||
| Low | 11.7 | 59.7 | 0.021 | |||||||||
| Medium | 12 | 59.0 | 0.012 | |||||||||
| High | 8.32 | 28.5 | 0.162 | |||||||||

Figure 3. Average reduction in oxygen desaturation index (ODI) for each treatment period, relative to baseline, in the IHL-42X proof of concept phase 2 clinical trial.
The study also measured the Plasma THC levels in patients’ blood. Plasma samples were collected 2 hours post dose 1 and the morning after dose 7 for each treatment period. The morning after dose 7, THC levels in the low dose IHL-42X samples had an average of 0.20 ng/ml and a maximum of 0.45 ng/ml, both of which are below the thresholds for impaired driving imposed in countries that have set limits for THC. With medium and high dose IHL-42X, the average THC concentrations the morning after dose 7 were 0.86 and 0.52 ng/mL respectively. The following diagram presents the average THC concentrations in plasma samples collected during each of the treatment periods of the IHL-42X proof of concept clinical trial. The average is calculated for samples for which there was THC detected; in the placebo treatment period this was a single sample.
48

Figure 4. Average THC concentrations in plasma samples collected
During the IHL-42X treatment periods, patients more frequently reported that their sleep quality was good or very good when compared to placebo. The highest level of patient reported sleep quality was observed with low and high dose IHL-42X.
Patient reported sleep quality during each treatment period
| Proportion of subjects reporting good or very good sleep quality | ||||
| Placebo | 26.50 | % | ||
| Low | 49.49 | % | ||
| Medium | 38.47 | % | ||
| High | 50.13 | % | ||

Figure 5. Proportion of patients in each IHL-42X proof of concept treatment period who reported good or very good sleep quality.
49
For the duration of the clinical trial, patients wore an Actiwatch, a watch-like device that uses actigraphy to capture data on activity and sleep. IHL-42X at all doses improved sleep efficiency (the percentage of time in bed a patient is asleep), the number of awakenings per night, and the total minutes every patient was awake during the night (WASO) compared to placebo (Table 6). These improvements were greatest for low and high dose IHL-42X. This means that while taking IHL-42X trial participants were asleep for a greater proportion of time they were in bed and woke up less often.
Sleep metrics captured by actigraphy
| Placebo | Low | Medium | High | |||||||||||||||
| Sleep efficiency | average | 76.83 | 84.81 | 81.34 | 84.17 | |||||||||||||
| p value compared to placebo | N/A | 0.0048 | 0.058 | 0.0078 | ||||||||||||||
| Awakenings per night | average | 49.31 | 35.8 | 41.44 | 37.33 | |||||||||||||
| p value compared to placebo | N/A | 0.0053 | 0.055 | 0.012 | ||||||||||||||
| WASO (min) | average | 62.59 | 37.55 | 47.22 | 38.55 | |||||||||||||
| p value compared to placebo | NA | 0.00011 | 0.0031 | 0.0010 | ||||||||||||||
Adverse events were recorded from the time the patients enrolled in the trial until their end of study visit. After recording treatment emergent adverse events (“TEAE”), the study team, including investigators and medical monitors, reviewed the TEAEs to determine whether they were likely related to the investigational product. The TEAEs were consistent with what has been reported for dronabinol and acetazolamide alone. For each treatment period the proportion of patients reporting one or more TEAEs (Table 7) as well as the total number of TEAEs (Table 8) were extracted from the clinical study report. Low dose IHL-42X had a similar proportion of patients reporting TEAEs and a lower number of total TEAEs than placebo. This indicated that low dose IHL-42X is well tolerated.
Proportion subjects of TEAEs reported for each treatment period
| Placebo | Low | Medium | High | |||||||||||||
| Total TEAE (%) | 81.8 | 33.3 | 55.6 | 66.7 | ||||||||||||
| Related TEAE (%) | 27.3 | 22.2 | 44.4 | 55.6 | ||||||||||||
Total number of TEAEs reported during each treatment period
| Placebo | Low | Medium | High | |||||||||||||
| Total TEAE | 15 | 6 | 22 | 16 | ||||||||||||
| Related TEAE | 7 | 4 | 16 | 12 | ||||||||||||
50

Figure 6. Total number of treatment emergent adverse events (TEAE), and TEAE that were probably or possibly related to the treatment, reported during each IHL-42X treatment period.
Formulation Development and Manufacturing of IHL-42X
In October 2021 Incannex engaged Procaps for development and manufacturing of the IHL-42X fixed dose combination product. Procaps is able to provide an end-to-end solution from formulation development through to GMP manufacturing at commercial scale.
Bioavailability/bioequivalence clinical trial
In November 2022, Incannex announced that it had engaged CMAX Clinical Research and Novotech CRO to undertake a bioequivalence/bioavailability (BA/BE) clinical trial for IHL-42X. The BA/BE study focuses on assessing the pharmacokinetics and tolerability of IHL-42X’s active pharmaceutical ingredients (“APIs”), dronabinol (THC) and acetazolamide, in comparison to FDA reference listed drugs Marinol and Taro acetazolamide tablets respectively. The study will also investigate the effect of food on IHL-42X tolerability and pharmacokinetics. The BA/BE study involves at least 116 participants and will evaluate the concentrations of APIs and metabolites in blood samples over 48 hours. This study design adheres to FDA recommendations for bioequivalence studies. The outcomes of the BA/BE trial will be a crucial component of a forthcoming NDA, serving as a bridging mechanism to the reference listed drugs, thereby facilitating regulatory approval via the FDA505(b)2 regulatory pathway.
Approval was received in July 2023 from Bellberry Human Research Ethics Committee (“HREC”) for the conduct of the BA/BE clinical trial. As of April 2024 participant recruitment and dosing is ongoing.
Phase 2/3 clinical trial investigating IHL-42X in patients with OSA (“RePOSA study”)
The next step in the development of IHL-42X is a global Phase 2/3 clinical trial, the RePOSA study, investigating the effect of the drug product in patients with OSA who are non-compliant, intolerant or naïve to positive airway pressure devices, such as CPAP. The Phase 2/3 clinical trial is randomized, double-blind clinical trial to determine the safety and efficacy of IHL-42X in subjects with OSA who are intolerant, non-compliant, or naïve to positive airway pressure (PAP), such as that administered via a continuous positive airway pressure (CPAP) machine..
51
The RePOSA study consists of two component studies. A four-week Phase 2 dose ranging trial that will determine the optimal dose of IHL-42X based on safety and efficacy in OSA patients, and a 52-week Phase 3 factorial trial that will compare the optimal dose of IHL-42X to the component APIs, dronabinol and acetazolamide, at equivalent doses, as well as placebo. The study is designed to facilitate a seamless transition between Phase 2 and Phase 3, reducing downtime and accelerating development timelines.
The endpoints, inclusion criteria and study procedures are similar across both component studies, which streamlines the transition process from Phase 2 to Phase 3. The target patient population is individuals aged 18 years or older with OSA who are intolerant, non-compliant or naïve to Positive Airway Pressure. Approximately 560 patients will be recruited, with a total of 355 patients receiving IHL-42X and 205 patients receiving placebo over the course of the study.
Start-up for the Phase 2/3 trial is in progress with all 55 sites selected. This includes 25 in the United States, 16 in Germany, 7 in Spain, 2 in Finland and 5 in the United Kingdom. IRB approval of the protocol allows the U.S.-based sites to proceed with site-specific approval, which is a critical step in site activation.
Appointment of lead principal investigators for Phase 2/3 trial
Dr. John Douglas Hudson, MD, of FutureSearch Trials of Neurology, Austin, Texas and Dr. Dennis Lacey, MD, of NeuroTrials Research Inc, Atlanta, Georgia, had been recruited as co-Lead Principal Investigators for the IHL-42X Phase 2/3 Study.
Dr. John Douglas Hudson, MD, is board certified in Neurology and Sleep Medicine. He serves as the Principal Investigator for FutureSearch Trials of Neurology, Austin, Texas. Dr. Hudson has supervised over 300 clinical trials over the past 20 years mostly related to neurological and sleep disorders and has been a national and international speaker for these disorders. Dr. Hudson completed his neurology residency at the University of Iowa and was Austin’s first board certified sleep specialist. Past activities include founding the Austin Neurological Clinic and Sleep Medicine Consultants. He held the position of President of the Texas Neurological Society, with a Lifetime Achievement Award and was President of the Capital Area American Heart Association.
FutureSearch Trials consists of two clinical research facilities in Austin and Dallas, Texas which have been in operation for over 15 years. The Austin site where Dr. Hudson is the Principal Investigator focuses on clinical research studies for treatment of neurological, pain and sleep disorders and features an on-site sleep lab.
Dr. Dennis Lacey, MD, holds certifications from the American Board of Psychiatry and Neurology, Specialty in Sleep Medicine. He has over 30 years of clinical research experience, acting as investigator or sub-investigator in more than 200 trials in Phase I through Phase IV.
NeuroTrials Research Inc is a clinical research facility in Atlanta, Georgia that has been in operation for over 25 years. NeuroTrials Research is focused on delivery of trials in neurology/CNS and sleep indications.
Investigational New Drug Application
On July 20, 2023, Incannex submitted an IND application to the FDA for review. The IND dossier compiled by the Incannex team included comprehensive modules on the safety and efficacy of IHL-42X and its component active pharmaceutical ingredients. It also included detailed information on the development, manufacturing, quality and stability of the IHL-42X drug product, as well as the clinical protocol and investigator information for the Phase 2/3 IND opening clinical trial.
The modules of the IND were:
| ● | Module 1 – Administrative Information and Prescribing Information |
| ● | Module 2 – Nonclinical/Clinical Overviews and Summaries |
| ● | Module 3 – Quality data |
52
| ● | Module 4 – Nonclinical Study Reports and Key Literature References |
| ● | Module 5 – Clinical Study Reports, Clinical Protocol and Investigator Information |
Submitting and clearing an IND with the FDA is crucial for companies to gain regulatory approval, conduct clinical trials, and engage in scientific dialogue with FDA whilst they progress investigational drugs through the stages of development in the United States. The FDA review process for an IND application involves evaluation of the scientific, clinical, and safety aspects to ensure that the proposed clinical trial meets regulatory requirements.
In August 2023, FDA completed their review of the IND application within the allocated 30-day period, and Incannex received confirmation from the FDA that the IND application has cleared and the IND opening study may proceed based on the agency’s assessment of the trial protocol, lead trial investigators, and a risk benefit analysis of the trial and prospective product. Clearance of the IND application is a critical milestone that is required to conduct clinical trials in the United States. The IND opening trial is the RePOSA trial described above that will assess the effect of IHL-42X in obstructive sleep apnea patients who are non-compliant, intolerant, or naïve to positive airway pressure treatment, such as that administered by CPAP devices.
Incannex is now working with Fortrea, the CRO engaged to manage the RePOSA study, following the commencement of patient dosing for the Phase 2/3 clinical trial in May 2024.
IHL-216A
IHL-216A for Concussion/Traumatic Brain Injury and Chronic traumatic encephalopathy
Concussion/Traumatic Brain Injuries (“TBIs”) are caused by a rapid acceleration/deceleration of the brain caused by a direct blow to the head or sudden impact to the body that jolts the skull. This causes the brain to compress against the skull. The impact of the brain against the skull causes both macro and micro scale damage to the brain which sets off a series of physiological events called secondary injury cascades. These secondary injury cascades are what cause many of the neurocognitive deficits seen in TBI patients.
Falls, vehicle collisions, violence, sports and combat injuries are the main activities leading to TBI and concussion. The signs and symptoms of a concussion can be subtle and may not show up immediately. Symptoms can last for days, weeks or even longer. Common symptoms after a concussive traumatic brain injury include headaches, loss of memory (amnesia) and confusion. Amnesia usually involves forgetting the event that caused the concussion. Other symptoms include nausea, vomiting, fatigue, blurry vision and ringing in the ears.
Complications can occur immediately or soon after a traumatic brain injury. Severe injuries increase the risk of a greater number of and more severe complications. Moderate to severe traumatic brain injury can result in prolonged or permanent changes in a person’s state of consciousness, awareness or responsiveness. Many people who have had a significant brain injury will experience changes in their cognitive ability, have executive functioning problems and or communication, emotional and behavioral problems. Some research suggests that repeated or severe traumatic brain injuries might increase the risk of degenerative brain diseases, but this risk cannot be predicted for an individual.
Chronic traumatic encephalopathy (“CTE”) is the term used to describe brain degeneration likely caused by repeated head traumas. CTE is a diagnosis made only at autopsy by studying sections of the brain. CTE is a rare disorder that is not yet well understood. CTE is not related to the immediate consequences of a late-life episode of head trauma. CTE has a complex relationship with head traumas such as persistent post-concussive symptoms and second impact syndrome that occur earlier in life.
Experts are still trying to understand how repeated head traumas, including how many head injuries and the severity of those injuries, and other factors might contribute to the changes in the brain that result in CTE.
CTE has been found in the brains of football players, boxers and other athletes that play contact sports, along with military personnel who were exposed to explosive blasts. Some signs and symptoms of CTE are thought to include difficulties with thinking (cognition) and emotions, physical problems and other behaviors. Symptoms of CTE often manifest decades after head trauma occurs.
53
CTE cannot be made as a diagnosis during life except in those rare individuals with high-risk exposures. Researchers do not yet know the frequency of CTE in the population and do not understand the causes. There is no cure for CTE. Researchers are currently developing diagnostic biomarkers for CTE, but none have been validated yet.
The total global addressable market for TBI was estimated to be US$3.46 billion in 2022 and the anticipated market in 2030 is US$5.53 billion. There are currently no pharmacological treatments for the secondary neurological effects of TBI.
IHL-216A Formulation development for clinical trials
IHL-216A is a fixed dose combination of isoflurane, a registered pharmaceutical, and CBD, intended for administration in the immediate period after primary blunt head injury to prevent development of brain injuries. Isoflurane is approved in the United States for induction and maintenance of anaesthesia. CBD is approved for use in seizure disorders and has shown effects on neuroinflammatory responses to brain injury. Isoflurane is a registered pharmaceutical, and has also demonstrated neuroprotective activity (neuroprotective activity, or neuroprotection, is defined as reduced neuronal cell death or disruption) in animal studies of TBI and is thought to act by modulating glutamate release and calcium uptake as well as via effects on mitochondrial membrane depolarization and excitatory neurotransmission. Thus, we believe that IHL-216A may affect neuroexcitation, neuro-inflammation, cerebral blood flow and cerebral oxygen consumption resulting in overall neuroprotection. We are also assessing its ability to protect the brain against secondary injury mechanisms that cause neuronal cell death and raised intracranial pressure in the days and weeks following head trauma in sports, and all other applicable scenarios resulting in head trauma (falls, vehicle collisions, violence, combat, among other causes). Reducing secondary brain injury may improve positive outcomes for long term neurological sequelae, including CTE, a major health risk associated with contact sports.
The formulation of IHL-216A presents a novel research and development opportunity. We have formulated IHL-216A as a combined inhalational product with nebulized drug delivery that involves using air pressure or ultrasonic vibrations to turn a liquid drug solution into an aerosol. We engaged Vectura, a UK based contract development and manufacturing organization, to develop the nebulized CBD formulation and device for delivery of the CBD to the isoflurane anaesthetic circuit. Vectura specializes in the development of inhaled drugs and has an excellent track record of bringing products to market and have formulated pharmaceutical drugs for multinational pharmaceutical companies including Bayer, Sandoz and Novartis. Development of the nebulized formulation was an iterative process starting with three steps of refinement based on properties of the solution, generated aerosol and dose delivery.
In August 2022, we engaged Curia Global, Inc. (“Curia”) to further develop and manufacture GMP-grade IHL-216A. The scalable manufacturing process has been developed at Curia. Experimental batch manufacturing has been completed and samples set down for stability analysis. However, GMP manufacturing has been put on hold due changes in company priorities as a result of shifts in global economic environment and market conditions.
In January 2023, via written pre-IND meeting correspondence, the FDA provided valuable, multidisciplinary feedback on the proposed clinical development of IHL-216A. The FDA also confirmed that FDA505(b)2 may be the appropriate regulatory pathway for IHL-216A, whereby some of the information required for marketing approval may derive from studies already completed on the drug components of IHL-216A and in the public domain.
FDA provided critical guidance on the data requirements for opening an IND for IHL-216A, particularly related to the intricacies of developing an inhaled drug product and conducting clinical trials that involve an anaesthetic. Incannex is drafting a follow-up request for additional information on the FDA’s recommendations and will provide an update to ASX and Nasdaq investor platforms when it has been received.
Due to the product’s potential therapeutic utility in contact sports, IHL-216A has been developed to satisfy the World Anti-doping Authority (“WADA”) specifications for use by elite and amateur athletes at risk of TBI and CTE.
54
Stage 1 pre-clinical study for IHL-216A for TBI and CTE
In December 2020, we completed an animal study to formally assess the neuroprotective capability of IHL-216A. The study introduced rodents to head trauma in a highly controlled manner to inflict a reproducible injury. Various doses of IHL-216A or its active pharmaceutical ingredients were administered to eight cohorts of rodents soon after traumatic head injury. Behavioral tests were used to assess the neurocognitive and motor function over time. We also monitored secondary injury cascades and performed micro-scale cellular analysis post-mortem to discern and compare neuronal damage across the cohorts.
As detailed below, we found that the IHL-216A components, CBD and isoflurane, act synergistically to reduce indicators of neuronal damage, neuroinflammation and behavioral deficits that are consequences of TBI, as IHL-216A had a greater effect than the predicted effect of CBD and isoflurane combined. The predicted result is determined by analyzing the results of isoflurane and CBD independently, and then based on those results predicting how well the drugs would do in combination; to the extent IHL-216A exceeds the predicted result, we can conclude that the drugs strengthen the effectiveness of one another and synergy exists. The study also found that IHL-216A reduced neuronal damage, neuroinflammation and cognitive deficits in a rodent model of TBI to a greater extent than either CBD or isoflurane applied on a standalone basis. These results have not been assessed for statistical significance.
Post-mortem analysis of rat brains also detected synergy between CBD and isoflurane. Brains were fixed and sectioned prior to Nissl staining to identify neuronal damage. Nissl staining is a microscopy technique to visualize Nissl bodies. Healthy neurons typically have more Nissl bodies than damaged ones. Neuronal damage is indicated by the ratio of Nissl bodies to neurons across different sections of the hippocampus with a lower Nissl/neuron ratio indicative of increased neuronal damage. Synergy between CBD and isoflurane was detected in hippocampal regions cornu ammonis 1 (CA1) and cornu ammonis 2 (CA2). These regions of the brain are known to be important in the formation and storage of memories. In the study, the improvement in Nissl/Neuron ratio observed for IHL-216A treated animals was increased by 53% for CA1 and 60% for CA2 relative to CBD alone, 28% for CA1 and 145% for CA2 relative to isoflurane alone, and by 20% for CA1 and 53% for CA2 relative to the predicted effect of CBD and isoflurane combined. These results demonstrated that less neuronal damage was observed in the rats treated with IHL-216A relative to the predicted value.

Figure 7. Synergistic activity of CBD and isoflurane (IHL-216A) in neuronal damage as assessed by Nissl staining. Rodents that had undergone TBI and treated with CBD and/or isoflurane or vehicle were assessed for neuronal damage by post-mortem analysis of fixed brain sections by Nissl staining. Nissl staining permits the quantitation of the ratio of Nissl bodies to total neurons, a lower ratio being indicative of increased neuronal damage. The Nissl/neuron ratio observed in hippocampal regions (A) CA1 and (B) CA2 contralateral to the site of injury in the group treated with IHL-216A was greater than that predicted based on the groups treated with each drug alone, which is indicative of synergy. Values are average of the respective treatment group. Group sizes: Uninjured n=6, vehicle n=6, CBD n=6, isoflurane n=5, IHL-216A n=6. Neuroinflammation Marker — Iba1.
A post-mortem analysis of the rat brains also determined that CBD and isoflurane were synergistic in reducing levels of the neuroinflammation marker Iba1 as detected using immunofluorescence. Iba1 is a protein expressed in microglia, a type of innate immune cell in the brain, that is an established marker of microglial activation and neuroinflammation. The levels of Iba1 in the brain are detected using immunofluorescence, which is a microscopy technique that employs antibodies specific to Iba1 which are detected using a fluorescent tag. Increased levels of Iba1 are indicative of increased neuroinflammation. in groups treated with IHL-216A, levels of the Iba1 neuroinflammation marker were reduced by 35% more relative to CBD alone and 123% more relative to isoflurane administered alone. IHL-216A also reduced the Iba1 neuroinflammation marker by 10% more than the predicted value of the combined CBD and isoflurane treatments.
55

Figure 8. Synergistic activity of CBD and isoflurane (IHL-216A) in reducing levels of the neuroinflammatory marker Iba1. Rodents that had undergone TBI and treated with CBD and/or isoflurane or vehicle were assessed for neuroinflammation through immunofluorescence analysis of the neuroinflammatory marker Iba1. Iba1 levels increase after TBI and a reduction in Iba1 is indicative of a reduction in neuroinflammation. Iba1 levels in brain sections ipsilateral to the site of injury in the group treated with IHL-216A were reduced more than would be predicted based on the reduction observed in groups treated with each drug alone, which is indicative of synergy. Values are average of the respective treatment group. Group sizes: Uninjured n=6, vehicle n=5, CBD n=6, isoflurane n=3, IHL-216A n=5.
Synergy between CBD and isoflurane was detected in the behavioral outcomes assessed using the Morris Water Maze. In the Morris Water Maze animals are trained to find a platform in a pool of water. After a number of training sessions, the platform is removed and the mice are monitored to determine whether they return to the location of the platform, which is a measure of spatial learning and memory. The number of animals treated with IHL-216A that returned to the location of the platform per group and the proportion of rats in the group that returned to the location of the platform was greater than that predicted based on the effect of CBD and isoflurane by 87 % and 24 % respectively. The improved performance of IHL-216A treated rats compared to the predicted effect demonstrated the synergistic effect of CBD and isoflurane.

Figure 9. Synergistic activity of CBD and isoflurane (IHL-216A) in the Morris Water Maze assessment. Rodents that had undergone TBI and treated with CBD and/or isoflurane or vehicle were assessed for spatial learning and memory using the Morris Water Maze. The observed performance with respect to both (A) relative instances of animal in platform location and (B) proportion of animals in that reached the platform location was better in the group treated with the CBD isoflurane combination (IHL-216A) than what was predicted based on the performance of the groups treated with each drug alone, which is indicative of synergy. Values are average of the respective treatment group. Group sizes: Uninjured n=6, vehicle n=6, CBD n=5, isoflurane n=6, IHL-216A n=6.
56
Stage 2 pre-clinical study for IHL-216A
In May 2022, we announced that the stage 2 study had been completed and that IHL-216A was observed to have a strong neuroprotective effect in a widely, known model of sports concussion developed in collaboration with the NFL to accurately represent the type of brain injury that occurs in sports-related concussion. This study compared six groups of twenty-four Sprague Dawley rats. When animals were tested in a Y-maze task, which assesses spatial memory by determining the animal’s ability to discriminate between a novel (new) and familiar arm, twenty-four hours after injury, animals treated with IHL-216A were found to have no difference in discrimination index compared sham (uninjured) animals (mean difference= 0.0598, p=0.5855) (Figure 10).
In contrast, injured animals treated with either vehicle or isoflurane alone after injury, the discrimination index was significantly reduced compared to sham animals (mean diff=0.2704, p=0.0498 and mean diff=0.3095, p=0.0245 respectively). The group treated with CBD alone had intermediate performance in the Y-maze between sham and vehicle treated animals (mean diff.0.1745, p==0.2933). These findings indicate that the defect in spatial memory observed at 1 day post injury is restored in animals treated with IHL-216A.

Figure 10. IHL-216A restores the deficit in Y-maze novel/familiar arm discrimination index assessment 24 h post TBI. A Y-maze was used to assess spatial memory 24 h after induction of TBI. Sham + Vehicle treated animals displayed a clear preference for the novel arm. This preference was reduced in TBI + vehicle animals, indicating that there is a deficit in novel arm discrimination associated with TBI. Each group consisted of 24 rodents.
57
IHL-675A
IHL-675A for treatment of inflammatory conditions
We are developing IHL-675A, a proprietary fixed dose combination product that contains CBD and hydroxychloroquine sulphate (“HCQ”) for the treatment of inflammatory conditions. Inflammatory conditions occur when the body’s immune system attacks its own tissues and organs causing inflammation, pain, discomfort, and damage to the affected tissues. IHL-675A is a multi-use anti-inflammatory drug targeting rheumatoid arthritis, inflammatory bowel disease (colitis and Crohn’s disease) and lung inflammation (COPD, asthma, bronchitis, and ARDS). IHL-675A comprises a combination of hydroxychloroquine, a registered pharmaceutical, and CBD. HCQ is a disease modifying anti-rheumatic drug that regulates the activity of the immune system, which may be overactive in some conditions. HCQ can modify the underlying disease process, rather than simply treating the symptoms. We have demonstrated that IHL-675A components, CBD and HCQ, act synergistically to inhibit production of key inflammatory cytokines in an in vitro study and in 4 distinct successful in vivo experiments using established models of inflammation. We were able to determine whether synergies exist in IHL-675A studies by comparing the predicted result of CBD and HCQ acting together to the actual IHL-675A results. The predicted result is determined by analyzing the results of HCQ and CBD independently in the study, and then based on those results predicting how well the drugs would do on a combined basis; to the extent IHL-675A exceeds the predicted result, we can conclude that the drugs strengthen the effectiveness of one another and synergies exist.
We have evaluated the results of these experiments and believe IHL-675A to be a multi-use drug candidate for the prevention and treatment of inflammatory lung conditions (ARDS, COPD, asthma, and bronchitis), rheumatoid arthritis and inflammatory bowel diseases (colitis and Crohn’s disease). Potentially, this could mean that IHL-675A is a better alternative to CBD or HCQ based products for certain inflammatory diseases, subject to further examination.
The hypothesis of synergistic anti-inflammatory activity was confirmed in a series of preclinical studies using human peripheral blood mononuclear cells and animal models of inflammatory diseases including arthritis, inflammatory bowel disease and inflammatory lung disease. Following these results, we developed a novel fixed dose combination product for assessment in clinical trials with the goal of regulatory approval by bodies including the FDA and TGA.
In July 2021 we engaged Procaps SA for development and production of the IHL-675A drug product using Procap’s proprietary patented Unigel technology. Procaps provides a complete supply chain solution from development to GMP manufacture at commercial scale. Their manufacturing facilities have been inspected and approved by multiple global regulatory agencies including the FDA.
We completed a phase 1 clinical trial investigating safety and pharmacokinetics of IHL-675A. The trial measured the safety, tolerability, and pharmacokinetic profiles of IHL-675A compared to the reference listed drugs, Epidiolex (CBD) and Plaquenil (HCQ). The key endpoints of the trial were the adverse events reported and the plasma levels of the active pharmaceutical ingredients (APIs), CBD and HCQ, and their major metabolites over a 28-day period. Three cohorts of 12 participants (n = 36) received either IHL-675A, CBD or HCQ and the assessments were identical across the three arms of the trial. Patient recruitment commenced in August 2022 and dosing was completed in September 2022. The final clinical study report was delivered to us in July 2023. IHL-675A was well tolerated and both the active pharmaceutical ingredients were bioavailable.
We have completed pre-IND meetings with the FDA to discuss the regulatory pathway for the development of IHL-675A for lung inflammation and rheumatoid arthritis in the United States and plan to open INDs for each of the three indications. The FDA provided feedback that marketing applications for IHL-675A could be appropriate as 505(b)(2) applications due to the existence of certain safety and efficacy information on the active ingredients of IHL-675A originating from historical studies that we are entitled to use in a new drug application. In July 2023, following the pre-IND meeting for use of IHL-675A for treatment of rheumatoid arthritis, the FDA confirmed that no further non-clinical studies are needed to open an IND and provided guidance on the proposed clinical development plan for IHL-675A in RA. The IND application will define the company’s strategy to demonstrate IHL-675A is safe and effective in the target indications via a series of randomized, controlled clinical trials.
58
Rheumatoid Arthritis
Rheumatoid arthritis is a chronic inflammatory disorder that can affect joints, skin, eyes, lungs, heart and blood vessels. As an autoimmune disorder, rheumatoid arthritis is caused by attacks to body tissues by one’s immune system. Unlike the wear-and-tear damage caused by osteoarthritis, rheumatoid arthritis causes a painful swelling that can eventually result in bone erosion and joint deformity. The total global addressable market for the pharmaceutical treatment of rheumatoid arthritis was estimated at US$60.1 billion in 2021 with a CAGR of 1.75% for the period of 2022-2030.
HCQ is approved for treatment of rheumatoid arthritis in the form of hydroxychloroquine sulphate and marketed as Plaquenil and generic equivalents.
Lung Inflammation (COPD, Asthma, ARDS and Bronchitis)
Chronic obstructive pulmonary disease (“COPD”) is a chronic inflammatory lung disease that causes obstructed airflow from the lungs. Symptoms include breathing difficulty, cough, mucus (sputum) production and wheezing. It is typically caused by long-term exposure to irritating gases or particulate matter, most often from cigarette smoke. People with COPD are at increased risk of developing heart disease, lung cancer and a variety of other conditions.
Asthma is a condition in which inflammation causes the airways to narrow and swell and which may cause the patient to produce extra mucus. This can make breathing difficult and trigger coughing, a whistling sound (wheezing) during breathing and shortness of breath. For some people, asthma is a minor nuisance. For others, it can be a major problem that interferes with daily activities and may lead to a life-threatening asthma attack. According to Acumen Research and Consulting, the Global COPD and asthma drug market was US$36.7 billion in 2022, growing at a CAGR of 5.2% from 2023 to 2032.
Acute respiratory distress syndrome (“ARDS”) occurs when fluid builds up in the air sacs (alveoli) located in the lungs. The fluid prevents oxygen from reaching the bloodstream. This deprives organs of the oxygen they need to function. ARDS typically occurs in people who are already critically ill or who have significant injuries. Severe shortness of breath (the main symptom of ARDS) usually develops within a few hours to a few days after the primary injury or infection. It is the one of the main causes of death resulting from COVID-19 and many people who develop ARDS do not survive. The risk of death increases with age and severity of illness. People who survive ARDS may experience lasting damage to their lungs.
Bronchitis is an inflammation of the lining of the bronchial tubes of the lungs. Bronchitis may be either acute or chronic. While acute bronchitis is common, chronic bronchitis, a more serious condition, is a constant irritation or inflammation of the lining of the bronchial tubes.
Inflammatory Bowel Disease
Inflammatory Bowel Disease (“IBD”) is an umbrella term used to describe disorders that involve chronic inflammation of the digestive tract. Significant types of IBD include:
| ● | Ulcerative colitis. This condition involves inflammation and sores (ulcers) along the superficial lining of the large intestine (colon) and rectum. |
| ● | Crohn’s disease. This type of IBD is characterized by inflammation of the lining of the digestive tract, which often can involve the deeper layers of the digestive tract. |
Both ulcerative colitis and Crohn’s disease are usually characterized by diarrhea, rectal bleeding, abdominal pain, fatigue and weight loss. IBD can be debilitating and sometimes leads to life-threatening complications.
The precise cause of inflammatory bowel disease remains unknown. Previously, diet and stress were suspected. However, currently medical practitioners acknowledge that these factors may aggravate, but are not the cause, of IBD. One possible cause is an immune system malfunction. When the immune system attempts to defeat an invading virus or bacterium, an abnormal immune response can cause the immune system to attack the cells in the digestive tract. The total global addressable market for IBD is estimated at US$21 billion in 2021 and the IBD global market is anticipated to grow at a CAGR of 5.1% from 2022 to 2031.
59
In vitro study of IHL-675A anti-inflammatory activity
On November 5, 2020, we released the results of our first in vitro study to investigate the synergistic activity of IHL-675A to inhibit inflammation. To test the anti-inflammatory potential of IHL-675A, human peripheral blood mononuclear cells (“PBMCs”) were stimulated with bacterial lipopolysaccharide (“LPS”). PBMCs were incubated with a range of concentrations of CBD and HCQ in combination or each drug alone and then stimulated with LPS to induce an inflammatory response. The inflammatory response was assessed by measuring cytokine levels in the culture medium after 24 hours. A reduction in cytokine levels in response to drug treatment is indicative of anti-inflammatory activity.
Cytokine levels were averaged across three replicates from two donors and normalized to maximum values to yield a relative inhibition value. A relative inhibition of 1 is complete inhibition of cytokine release whereas a value of 0 is no inhibition of cytokine release. Anti-inflammatory synergy was determined using the standard scientific “Excess over Bliss” (“EOB”) method where the predicted inhibition, as calculated using the formula Epred A+B=(EA+EB)-(EAEB), is subtracted from the observed inhibition to yield an EOB score. An EOB score of greater than zero indicates that the combination is synergistic. None of the below data has been analyzed for statistical significance.
The study demonstrated that CBD and HCQ act synergistically to inhibit production of the assessed inflammatory cytokines IL-1β, IL-6, TNF-α, IL-1aα, and MIP-1α by PBMCs from the donors. The average EOB scores ranged from 0.32-0.57. The reduction in levels of the five cytokines (relative to vehicle treated PBMCs) observed in PBMCs treated with IHL-675A was 436% to 1320% greater relative to those treated with HCQ alone, 109% to 767% greater relative to those treated with CBD alone and 87% to 767% greater relative to the predicted combinatorial effect of CBD and HCQ.
The results in Figure 11 A, B, C, D and E presented below, display the optimal fixed dose IHL-675A combination assessed for each cytokine. The bars noted as ‘Predicted CBD+HCQ’ represent what our expectation was based on the activity of each drug individually. The observed inhibition of cytokine release upon treatment with the CBD HCQ combination was greater than predicted based on the activity of each drug alone for each cytokine analyzed.

60


Figure 11. Inhibition of LPS-induced cytokine release from human PBMCs by CBD and HCQ. Data is presented is the average relative inhibition for the PBMC donors. Predicted inhibition by CBD+HCQ was calculated using the formula Epred A+B=(EA+EB)-(EAEB). Observed CBD+HCQ is the level of inhibition observed in the experiment. (A) IL-1b, (B) IL-1a, (C) IL-6, (D) MIP-1a, and (E) TNF-a. Error bars are standard error of the mean of the donors.
Preclinical in vivo study of IHL-675A against inflammation
In November of 2020, we announced the results of an in vivo study assessing IHL-675A in a mouse model of sepsis. To determine whether CBD and HCQ synergize in vivo, mice from 11 groups of 10 mice, weighing 18-20g were injected with CBD and HCQ both alone and in combination. After one hour, the mice were injected with LPS to induce an inflammatory response. Each mouse in every cohort was assessed for each of the 5 inflammatory cytokines. Two hours after LPS injection, blood was collected from the mice by cardiac puncture. Sera were processed and analyzed for cytokine levels using a Luminex based assay. For synergy analysis, data was baseline subtracted using sham treated (no LPS injection) cytokine levels and then the values for each cytokine were normalized relative to maximum values across the groups. The normalized values were used to calculate the relative inhibition where a value of 1 is complete inhibition and a value of 0 is no inhibition. Synergy was calculated using the EOB method, or the difference between the observed and predicted inhibition between the combination of drug concentrations where the predicted inhibition is determined using the equation Epred A+B=(EA+EB)-(EAEB). An EOB score of greater than 0 is indicative of synergy.
The results of the in vivo study are presented in Figure 12, showing the optimal fixed dose IHL-675A combination assessed for each cytokine in 11 groups of 10 mice. The bars noted as ‘Predicted CBD + HCQ’ represent IHL’s expectation based on the activity of each drug alone. The observed results from the study significantly exceeded the predicted results across the inflammatory cytokines analyzed. CBD and HCQ synergize to inhibit the production of inflammatory cytokines IL-1β, IL-6, TNF- α, IL12(p70), and IFN-γ in a mouse model of LPS induced sepsis.
61
The average EOB scores ranged from 0.15-0.30. Levels of the five inflammatory cytokines were reduced compared to animals treated with vehicle to a greater extent in animals treated with IHL-675A than in those treated with CBD alone. Reduction in cytokine levels compared to vehicle treated group in the group treated with IHL-675A was 26% to 81% greater relative to the predicted effect of the CBD HCQ combination across the five analyzed cytokines after 2 hours.


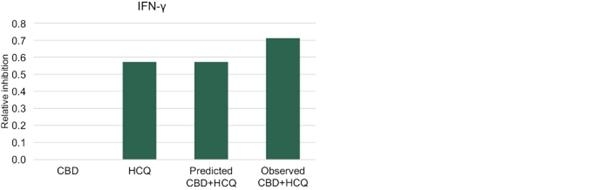
Figure 12. Synergistic anti-inflammatory activity of CBD and HCQ in a mouse sepsis model. The anti-inflammatory activity of the combination of CBD and HCQ was greater than that predicted using the Excess over Bliss method. The CBD+HCQ combination was synergistic at inhibiting release of IL-1β, IL-6, TNF-α, IL12(p70), and IFN-γ.
Preclinical in vivo study of IHL-675A against Pulmonary Inflammation (ARDS, COPD, Asthma and Bronchitis)
In February 2021, we announced the results of an in vivo study assessing IHL-675A anti-inflammatory capabilities regarding chronic obstructive pulmonary disease, asthma, bronchitis, and other inflammatory respiratory conditions. We also assessed the anti-inflammatory effect of our proprietary IHL-675A formulation on Pulmonary Neutrophilia, which is a primary underlying cause of COPD, asthma, bronchitis, and other inflammatory respiratory conditions. We reported encouraging results, as discussed below, which facilitate a substantial expansion of the potential uses for IHL-675A and represent new patient treatment opportunities.
62
A rodent model of pulmonary inflammation was used to assess the anti-inflammatory efficacy of IHL-675A in lungs. In this study, ten groups of six mice each were pre-treated with either CBD, HCQ or IHL-675A prior to intratracheal administration of bacterial lipopolysaccharide (“LPS”), which was then inhaled and acts as an inflammatory stimulus in the lungs. A sham group where LPS was not administered to the mice was also included as a control. The lungs were flushed with a saline solution 24 hours after LPS administration and bronchoalveolar lavage fluid (“BALF”) was analyzed for cytokine levels using a Luminex based assay. Cytokines are proteins that mediate the inflammatory response and a reduction in cytokine levels is indicative of reduced inflammation. A white blood cell (“WBC”) count was also performed on the BALF. When inflammation occurs in the lungs, WBCs are recruited as part of the inflammatory response. A reduction in WBC count is also indicative of reduced inflammation.
Cytokine levels were normalized to those detected in vehicle treated mice and then the relative inhibition was calculated. IHL-675A reduced levels of all assessed inflammatory cytokines IL-1β, IL-6, TNF-α, CXCL1 and MCP-1 to a greater extent than either CBD or HCQ alone. WBC counts were normalized using the same method used for cytokines and IHL-675A reduced WBC counts to a greater extent than CBD or HCQ alone. These results indicate that IHL-675A has superior anti-inflammatory activity compared to CBD and HCQ in a mouse pulmonary inflammation model. Based on these results IHL-675A will be assessed for efficacy in the treatment of pulmonary inflammation in humans. These results have not been analyzed for statistical significance.

Figure 13. Reduction in cytokine levels and white blood cell count in BALF resulting from treatment with by IHL-675A, CBD or HCQ in a mouse model of pulmonary inflammation. Mice were treated with CBD, HCQ or a combination of CBD and HCQ (IHL-675A) and then LPS was administered intratracheally. Twenty-four hours after LPS administration bronchioalveolar lavage fluid (BALF) was analyzed for cytokine levels and white blood cell count. The reduction in cytokine levels by IHL-675A was greater than that for either drug alone. Drug concentrations were 1 mg/kg CBD and 25 mg/kg HCQ for (A) IL-1β, (B) IL-6, (C) MCP1 and (E) TNF-α, 10 mg/kg CBD and 2.5 mg/kg HCQ for CXCL-1 and WBC (white blood cell count).
63
Preclinical study of IHL-675A in a model of Rheumatoid Arthritis
In March 2021, we announced the results of an in vivo study assessing IHL-675A’s anti-inflammatory capabilities in a rheumatoid arthritis model. Results indicate that a low dose of IHL-675A was 1.06 to 3.52 times more effective at reducing disease severity scores across multiple assessments including clinical score, paw volume, pannus score, total histology score and serum cytokine levels compared to a standard dose of HCQ only. HCQ is approved and widely used for the treatment of rheumatoid arthritis in the form of hydroxychloroquine sulfate, which is marketed as Plaquenil.
In this model of rheumatoid arthritis, female Lewis rats were challenged with porcine type-II collagen with Freund’s adjuvant on Day 1 (0.2 mg/0.2 mL/rat) by subcutaneous injection at the base of the tail to induce arthritis. A booster injection at 0.1 mg/0.1 mL/rat was injected on day 7. On day 16, rats were allocated into groups of six. There were ten groups of modelled rats and one sham injected group. CBD, HCQ or IHL-675A were injected intraperitoneally once per day from day 17 to 30 (total of 14 days). Drug doses were 1 and 10 mg/kg CBD and 2.5 and 25 mg/kg HCQ. The 10 mg/kg CBD and 25 mg/kg HCQ doses were selected as they are representative of standard doses in humans based on the FDA body surface area dose equivalence estimation for rats to humans of 6/37. For a 60 kg person, the 10 mg/kg CBD dose in rats is equivalent to 97 mg and the 25 mg/kg HCQ dose in rats is equivalent to 243 mg. The maintenance dose range recommended for rheumatoid arthritis in the Plaquenil prescribing information is 200-400 mg daily.
Disease severity was assessed by measuring hind paw volume with a plethysmometer and using a qualitative severity score system on days 1, 7 ,10, 14, 16, 18, 20, 22, 24, 26, 28 and 30. Post termination on day 30, blood was collected from all rats and analyzed for levels of the inflammatory cytokines IL-1β and IL-6 using commercially available ELISA kits. These two cytokines were selected as they are known to be involved in the pathophysiology of rheumatoid arthritis. Both hind paws were harvested, weighed and formalin-fixed for histopathology. Histopathological evaluation consisted of an evaluation of cartilage and bone destruction by pannus formation (an abnormal layer of fibrovascular or granulated tissue) and mononuclear cell infiltration in synovial joint tissues. A total histology score, which is a sum of the pannus formation and mononuclear cell infiltration scores, was also calculated. For all assessments, the score was sham subtracted and then the reduction relative to the vehicle group was calculated.
In the rat model of arthritis, IHL-675A treated animals had a greater reduction (relative to vehicle treated animals) in clinical score and paw volume at days 24 and 30, pannus formation, total histology score, IL-1β and IL-6 than animals treated with HCQ alone or CBD alone (at equivalent doses). The reduction in disease assessments by IHL-675A was 1.07-8.72 times that observed for HCQ alone at an equivalent dose, which indicates that IHL-675A has a benefit in a rat model of arthritis greater than that of HCQ alone and demonstrates that IHL-675A has potential as a treatment for rheumatoid arthritis in humans.
64

Figure 14. Comparison of IHL-675A to its component drugs CBD and HCQ in reduction of disease assessments in a rat model of rheumatoid arthritis. Groups of rats that had undergone collagen-induced arthritis modelling were treated with IHL-675A, CBD or HCQ at equivalent doses (1 mg/kg CBD, 2.5 mg/kg HCQ). The reduction in arthritis disease severity in IHL-675A treated rats was greater than for either CBD or HCQ treated rats with respect to (A) clinical score at day 24, (B) paw volume at day 24, (C) pannus formation, (D) total histology score, (E) serum IL-1b levels and (F) serum IL-6 levels.
Preclinical studies of IHL-675A in models of inflammatory bowel disease
In February 2021, we announced the results of an in vivo study assessing IHL-675A’s anti-inflammatory capabilities regarding inflammatory bowel disease. IHL-675A demonstrated a reduction in the colitis index of 46%, while CBD only and HCQ only treatment achieved a reduction of 25% and 27% respectively, demonstrating that IHL-675A has superior anti-inflammatory activity compared to CBD only and HCQ only, which indicates that IHL-675A has the potential to be a treatment for inflammatory bowel disease in humans.
65
This study used eleven groups of six mice. Mice were treated with IHL-675A, CBD or HCQ for four consecutive days after administration of TNBS/ethanol to induce ulcerative colitis. A vehicle treated group and sham group were included in the study. Stool consistency was monitored over the course of the experiment. On Day 5 mice were sacrificed, blood collected for cytokine analysis and the colon removed for analysis.
Endpoint measurements include stool consistency score (an ordinal scale that measures stool consistency with a higher number indicative of looser stools), colon weight, colon macroscopic damage score (an ordinal scale that combines adhesions, strictures, ulcers/inflammations and instances of wall thickening), colitis index (a composite scale from the histological examination of colon sections) and myeloperoxidase (an enzyme abundantly expressed in neutrophil granulocytes that contributes to inflammatory damage in IBD) levels in the colon tissue at day 5. The results from each of these endpoints were sham subtracted and the relative reduction was calculated. The data was not analyzed for statistical significance.
Animals treated with IHL-675A displayed a greater reduction (relative to vehicle treated animals) in colitis index, macroscopic damage score, stool consistency score, colon to body weight ratio and myeloperoxidase (MPO) levels than animals treated with either CBD or HCQ alone. These results indicate that IHL-675A has a benefit in a mouse model of ulcerative colitis greater than that of CBD or HCQ alone, which indicates that IHL-675A is a potential treatment for inflammatory bowel disease in humans.


Figure 15. Reduction in colitis score assessments by CBD and HCQ (IHL-675A) in a mouse model of colitis. Colitis was induced in mice by intracolonic installation of TNBS/ethanol and then treated with CBD, HCQ or CBD and HCQ (IHL-675A). After 4 days, mice were sacrifice and the colons extracted for macro and microscopic analysis. The reduction in colitis severity was greater in mice treated with IHL-675A than for either CBD or HCQ alone for (A) colitis index, (B) macroscopic damage score, (C) relative colon weight, (D) stool consistency and (E) MPO levels. Drug dose in all assessments was 1 mg/kg CBD and 2.5 mg/kg HCQ.
66
Phase 1 clinical trial for IHL-675A
A Phase 1 clinical trial to assess the safety and pharmacokinetics of IHL-675A in healthy volunteers was conducted in Australia, the results of which will form part of our FDA IND submissions across the indications of lung inflammation, rheumatoid arthritis and inflammatory bowel disease. The aims of this study were to demonstrate that there are no, or minimal, additional risks/side effects associated with the combination of CBD and HCQ compared to each drug alone and that the uptake and metabolism (pharmacokinetics) of the two drugs do not interfere with one another. A total of 36 subjects participated in the trial, evenly divided across three arms. The three arms of 12 subjects each received one of IHL-675A, Epidiolex (CBD), or Plaquenil (HCQ). The safety and pharmacokinetic assessments were identical across the three arms.
CBD and HCQ both have been used historically as treatments for our targeted indications when used independently. However, as with any pharmaceuticals there are risks involved. Part of the strategy in the design of IHL-675A is that the combination of CBD with HCQ permits a reduction in HCQ, which reduces the known risks associated with cumulative HCQ dose, without sacrificing efficacy. Results from the preclinical studies we have conducted to-date have led to the hypothesis that a lower cumulative dose of HCQ, when combined with CBD, will also reduce disease severity scores in IHL-675A’s target indications in humans. Nonetheless, there is always potential for two drugs to interact and exacerbate minor concerns that exist when used alone or lead to new safety concerns. Demonstrating that a combination drug containing CBD and HCQ has a similar safety profile to the component drugs is an important step in the development program and is a requirement set out by regulatory agencies. Safety assessments included cardiac monitoring via 24-hour holter monitor and ECG, and blood biomarkers, serum liver enzyme levels, blood cell counts and biochemistry, monitoring of vital signs and mental health questionnaires.
The other component of this study was monitoring the pharmacokinetics of the two active pharmaceutical ingredients (“API”) of IHL-675A, CBD and HCQ, and comparing them to their respective reference listed drugs Epidiolex and Plaquenil. Study participants were dosed with either IHL-675A, Epidiolex or Plaquenil with equivalent amounts of the respective API. Blood samples were drawn at predetermined intervals over a 72-hour period and analyzed for levels of CBD and HCQ as well as their major metabolites. For each molecule the maximum concentration (“Cmax”), time to maximum concentration (“Tmax”) and total exposure (“AUC”) were determined. The pharmacokinetic parameters for IHL-675A, Epidiolex and Plaquenil were compared to determine whether the APIs in IHL-675A are bioequivalent to the reference listed drugs. Bioequivalence is an important component of the FDA 505(b)2 approval pathway that IHL is targeting with IHL-675A.
Approval from the Human Research Ethics Committee to conduct the phase 1 study was received in July 2022. Participant recruitment commenced in August 2022 and dosing was completed in September 2022.
Participants were monitored until the end of October 2022, after which blood samples collected during the study were assessed for levels of CBD, HCQ and major metabolites to characterize the pharmacokinetics of each active pharmaceutical ingredient.
IHL-675A was well tolerated, with no adverse events of concern and no serious adverse events reported, as shown in Figure 16 below. The same number of treatment related TEAEs were reported for IHL-675A as for Epidiolex. Treatment-related TEAEs included abdominal pain, dizziness, fatigue, frequent bowel movements, headache and somnolence. All TEAEs were minor, with the exception of one incidence of moderate severity abdominal cramps, which was resolved soon after onset.
67

Figure 16. Percentage of subjects who reported a treatment related treatment emergent adverse event in each of the treatment groups of the IHL-675A Phase 1 clinical trial.
CBD Pharmacokinetic Results
Comparison of the average pharmacokinetics of CBD in participants administered IHL-675A compared to those administered Epidiolex revealed that the CBD was taken up from IHL-675A more quickly and reached a higher maximum concentration than from Epidiolex, as shown in Figure 17 below. The average maximum concentration (Cmax) of CBD from IHL-675A was 1.57 times higher than for Epidiolex. The time to reach the maximum concentration (Tmax) was 26% faster for IHL-675A than Epidiolex. CBD administered in IHL-675A was also cleared more quickly than Epidiolex. The half-life (t1/2) of CBD from IHL-675A was 13% faster than Epidiolex. The total exposure (AUCinf) was similar for CBD administered as IHL-675A and Epidiolex. These patterns are trends at this point (p >0.05). Similar results were observed for CBD metabolites 7-COOH-CBD and 7-OH-CBD.

Figure 17. Average plasma concentrations of CBD over time for the IHL-675A and Epidiolex treatment groups in the IHL-675A Phase 1 clinical trial.
68
The following table presents the pharmacokinetic parameters, namely the CBD and metabolite PK parameters, from the IHL-675A Phase 1 study:
| IHL-675A | Epidiolex | |||||||||||||||||||||||||||||||||
| Cmax | Tmax | AUCinf | T1/2 | Cmax | Tmax | AUCinf | T1/2 | |||||||||||||||||||||||||||
| (ng/mL) | (hr) | (hr*ng/mL) | (hr) | (ng/mL) | (hr) | (hr*ng/mL) | (hr) | |||||||||||||||||||||||||||
| CBD | Mean | 207.04 | 2.13 | 841.08 | 220.17 | 131.89 | 2.88 | 725.8 | 231.22 | |||||||||||||||||||||||||
| SD | 117.44 | 0.91 | 358.63 | 53.85 | 61.92 | 1.21 | 223.98 | 56.45 | ||||||||||||||||||||||||||
| Min | 72.6 | 1.02 | 391 | 113.84 | 45.6 | 1.5 | 355 | 144.41 | ||||||||||||||||||||||||||
| Max | 472 | 4 | 1699 | 301.17 | 241 | 6 | 1121 | 305.88 | ||||||||||||||||||||||||||
| 7-OH-CBD | Mean | 55.24 | 2.17 | 389.18 | 40.54 | 21.06 | 3 | 262.24 | 21.15 | |||||||||||||||||||||||||
| SD | 34.58 | 0.94 | 214.49 | 52.79 | 9.15 | 1.22 | 103.95 | 10.05 | ||||||||||||||||||||||||||
| Min | 14.9 | 1.02 | 220 | 10.78 | 7.7 | 1.5 | 149 | 10.54 | ||||||||||||||||||||||||||
| Max | 116 | 4 | 950 | 202.58 | 38.4 | 6 | 448 | 49.36 | ||||||||||||||||||||||||||
| 7-COOH-CBD | Mean | 479.75 | 2.83 | 18753.9 | 167.87 | 362.17 | 4.97 | 16267.9 | 153.68 | |||||||||||||||||||||||||
| SD | 218.74 | 1.2 | 8979.02 | 95.47 | 299.63 | 1.3 | 11069.2 | 92.41 | ||||||||||||||||||||||||||
| Min | 209 | 1.5 | 11445 | 46.03 | 116 | 2.5 | 4475 | 18.47 | ||||||||||||||||||||||||||
| Max | 921 | 6 | 43714 | 332.65 | 1180 | 6.05 | 42018 | 317.68 | ||||||||||||||||||||||||||
Hydroxychloroquine Pharmacokinetic Results
A comparison of the average pharmacokinetics of hydroxychloroquine in participants administered IHL-675A compared to those administered Plaquenil revealed that hydroxychloroquine was taken up more slowly from IHL-675A than from Plaquenil, but the two drugs had a similar maximum plasma concentration, as shown in Figure 18 below. The time to reach the maximum concentration (Tmax) for HCQ administered as IHL-675A was 46% slower than for Plaquenil. The hydroxychloroquine clearance and total exposure were similar for the two drugs. These patterns are trends at this point (p >0.05). Plasma concentrations of hydroxychloroquine metabolites desethylhydroxychloroquine, bisdesethylhydroxychloroquine and desethylchloroquine were detected only at low levels (<2 ng/mL) at all points in the study.

Figure 18. Average plasma concentrations of hydroxychloroquine over time for the IHL-675A and Plaquenil treatment groups in the IHL-675A Phase 1 clinical trial.
69
The following table presents the pharmacokinetic parameters, namely the Hydroxychloroquine and metabolite PK parameters, from the IHL-675A Phase 1 study:
| IHL-675A | Plaquenil | |||||||||||||||||||||||||||||||||
| Cmax | Tmax | AUCinf | T1/2 | Cmax | Tmax | AUCinf | T1/2 | |||||||||||||||||||||||||||
| (ng/mL) | (hr) | (hr*ng/mL) | (hr) | (ng/mL) | (hr) | (hr*ng/mL) | (hr) | |||||||||||||||||||||||||||
| HCQ | Mean | 54.71 | 5.59 | 2986 | 182.62 | 55.52 | 3.46 | 3430.9 | 251.6 | |||||||||||||||||||||||||
| SD | 23.85 | 2.51 | 1244.46 | 93.7 | 24.81 | 1.94 | 1104.38 | 73.65 | ||||||||||||||||||||||||||
| Min | 22 | 2 | 800 | 35.68 | 26.1 | 1 | 2073 | 163.92 | ||||||||||||||||||||||||||
| Max | 105 | 12.03 | 4217 | 311.57 | 124 | 6 | 5888 | 421.51 | ||||||||||||||||||||||||||
| Desethyl-hydroxy-chloroquine | Mean | 1.38 | 81.08 | NA | * | NA | * | 1.29 | 17.46 | NA | * | NA | * | |||||||||||||||||||||
| SD | 1.24 | 183.01 | NA | * | NA | * | 1.04 | 35.04 | NA | * | NA | * | ||||||||||||||||||||||
| Min | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||||||||||||||||||||||||
| Max | 4.4 | 673.83 | 0 | 0 | 3.3 | 123.93 | 0 | 0 | ||||||||||||||||||||||||||
| Desethyl-chloroquine | Mean | 0.79 | 7.77 | NA | * | NA | * | 0.41 | 5.59 | NA | * | NA | * | |||||||||||||||||||||
| SD | 0.72 | 13.03 | NA | * | NA | * | 0.84 | 13.58 | NA | * | NA | * | ||||||||||||||||||||||
| Min | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||||||||||||||||||||||||
| Max | 2 | 49.05 | 0 | 0 | 2.9 | 49.07 | 0 | 0 | ||||||||||||||||||||||||||
| Bisdesethyl-hydroxy-chloroquine | Mean | 0 | 0 | NA | * | NA | * | 0 | 0 | NA | * | NA | * | |||||||||||||||||||||
| SD | 0 | 0 | NA | * | NA | * | 0 | 0 | NA | * | NA | * | ||||||||||||||||||||||
| Min | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||||||||||||||||||||||||
| Max | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||||||||||||||||||||||||
| * | NA: metabolite not detected at levels sufficient to calculate PK parameter |
Interpretation of the results from the phase 1 clinical trial
IHL-675A is well tolerated in healthy volunteers. Adverse events for IHL-675A were consistent with what was observed, and has been publicly reported, for Epidiolex and Plaquenil. Both active pharmaceutical ingredients, CBD and HCQ, are absorbed from IHL-675A. Trends in PK profiles indicate that the uptake of CBD may be more rapid for IHL-675A than Epidiolex and uptake of HCQ may be slower for IHL-675A than Plaquenil. This could be advantageous for IHL-675A. CBD provides immediate relief for inflammation and pain, whereas HCQ is a slower acting molecule and provides extended relief.
Phase 2 clinical trial assessing the effects of IHL-675A on pain and function in patients with rheumatoid arthritis
In February 2023, we commenced a Phase 2 clinical trial to assess the safety and efficacy of IHL-675A on pain and function in patients with rheumatoid arthritis. The patients will be randomized according to one of four arms: either IHL-675A, CBD alone, HCQ alone or placebo. The treatments will be double blinded, meaning neither the investigators nor patients will know which treatment an individual is receiving. The study is managed by Avance Clinical, an Australian and US CRO, who identified clinical trial sites with expertise in rheumatoid arthritis to conduct patient recruitment and assessments. Avance Clinical will manage the sites and study conduct, ensure that the data is of the necessary quality, and conduct the analysis of data collected across all the trial sites.
The trial will include 128 participants who meet the eligibility criteria. Participants are randomized to one of 4 arms: either IHL-675A, CBD alone, HCQ alone or placebo. The primary endpoint for the study is pain and function relative to baseline determined via the score on the RAPID3 assessment at 24 weeks. Participants will also record their pain and function outcomes daily, by completing questionnaires on pain, fatigue, joint stiffness and quality of life, using an electronic Patient Reported Outcomes device (similar to completing a questionnaire on an electronic tablet). The participants will attend monthly visits at the clinical trial site, where blood tests, and physical examinations will monitor additional safety and efficacy outcomes, including inflammatory biomarkers. In July 2023, we received approval from HREC for a phase 2 clinical trial investigating IHL-675A in patients with rheumatoid arthritis. In January 2024, we commenced patient recruitment and dosing commenced.
70
Psilocybin-assisted Psychotherapy for General Anxiety Disorder (Psi-GAD)
Generalized Anxiety Disorder
Generalized Anxiety Disorder (“GAD”) is characterized by diffuse, excessive, uncontrollable anxiety that frequently occurs and is not restricted to any particular environmental circumstances. Symptoms are variable, including feelings of persistent and excessive worry, nervousness, restlessness, difficulty in concentrating fatigue, irregular sleeping patterns, muscle tension, irritability, and nausea.
Generalized anxiety disorder is a relatively common and serious psychiatric condition affecting around 4-6% of the population during their lifetime. GAD can severely affect quality of life and professional career prospects. It is a highly comorbid disorder, with estimations of lifetime mental disorder comorbidity as high as 90%. It is most comorbid with major depression, and also commonly comorbid with other anxiety disorders, other mood disorders, and non-psychiatric disorders such as chronic pain and irritable bowel syndrome. An estimated 8 million people in Australia and the United States have moderate to severe GAD at any point in time, of which, 1 million people reside in Australia and 7 million people reside in the United States.
Existing treatments
International guidelines for GAD treatment recommend selective serotonin reuptake inhibitors (“SSRIs”), serotonin and noradrenaline reuptake inhibitors (“SNRIs”), and pregabalin as first-line options, with benzodiazepines such as diazepam as second-line options. GAD is also treated with psychotherapy alone or in combination with pharmacotherapies. However, these treatments show limited efficacy, with less than half of patients achieving remission following these treatments and substantial treatment side-effects and cost. In particular, the side effects associated with long term use of these pharmacotherapies include emotional numbness, reduced positivity, weight gain, sexual disfunctions, and suicidal thoughts. Due to the limitations of existing treatments, we believe there is a significant unmet need for new therapies to improve quality of life outcomes for patients diagnosed with GAD.
Psilocybin as a treatment for generalized anxiety disorder
Psychedelic-assisted psychotherapy may provide rapid, significant, and lasting benefits in treating unipolar depression, depression and anxiety symptoms associated with a terminal illness, and substance misuse. Psilocybin is a psychoactive molecule that occurs naturally in several genera of mushrooms, which primarily acts on the serotonin receptor system, and can modulate states of consciousness, cognition, perception, and mood.
When combined with specialized forms of psychotherapeutic support, psilocybin does not lead to clinically significant adverse events and can reduce scores on mental health severity assessments. Through the 1950s and 1960s, tens of thousands of individuals participated in psychedelic research. While methodologically limited by modern standards, the findings from many of these studies showed substantial improvements in anxiety, depression and addiction levels, and quality of life.
Following decades of socio-political obstruction to psychedelic treatments, an increasing number of clinical psychedelic trials are now being conducted at highly esteemed institutions around the world, including Imperial College London, Johns Hopkins University, University of California, and now Monash University, Melbourne, in partnership with us.
Over the past decade, the therapeutic potential of psilocybin in anxiety, depression and addiction has been demonstrated in various academic-sponsored studies. In these studies, psilocybin-assisted psychotherapy, provided a rapid reduction in anxiety and depression symptoms on the day of administration with generally maintained treatment effects at follow-up assessments many months later. These studies have shown psilocybin to be generally well-tolerated, with low toxicity and no serious adverse events reported.
71
Two psilocybin research programs for depression have received breakthrough designation from the FDA. A small number of other psilocybin treatment development programs are underway globally. Should the results from any of these research programs be positive, approval of psilocybin-assisted psychotherapy as a prescription treatment could occur within the next five years.
Incannex’s investigational psilocybin therapy for Generalized Anxiety Disorder
Our psilocybin therapy combines psilocybin with psychological therapy that has been specifically designed for patients diagnosed with generalized anxiety disorder by a multidisciplinary team of experts led by Principal Investigator Dr Paul Liknaitzky, along with Co-Investigators Professor Suresh Sundram and Professor Murat Yucel. The wider research team includes experts in psychedelic-assisted therapies, psychometric evaluation, qualitative research, therapist training, and risk management.
Psilocybin therapy protocol
Our psilocybin therapy comprises administration of medication with psychotherapy by mental health professionals who have undergone our specialized therapist training program. The therapy is designed to optimize patient safety and therapeutic outcomes in GAD with specific support before, during and after psilocybin dosing sessions.
Each participant receives two therapeutic doses of our investigational product, which will be composed of a specified dosage of psilocybin, with psychotherapy before, during and after each dose session. The psychotherapy comprises four distinct phases:
| ● | Preliminary psychotherapy: conducted during the screening stage with key focus on clinical formulation, therapeutic alliance, psychedelic treatment psychoeducation and practical preparation for dosing. |
| ● | Preparation psychotherapy: conducted following full enrollment and prior to the first dosing session with a key focus on extending preliminary psychotherapy work and covering more targeted and GAD-specific psychological and practical preparation for dosing. |
| ● | After dosing support: conducted within a week following the preparation session with a key focus on trust, suitable mindset, conducive physical setting, and participant-led support. Dosing support is the psychotherapy session. |
| ● | Integration psychotherapy: conducted following the dosing sessions, including the day directly following each dosing session, with key focus on sustaining benefits through specific mindful, emotion and somatic-focused therapy, meaning-centered support, and facilitating contextual changes that support outcomes. |
FDA development plan and pre-IND meeting
In October 2021, we conducted a pre-IND meeting with the FDA on the psilocybin-assisted psychotherapy for GAD program. The pre-IND meeting package was prepared with the assistance of Camargo Pharmaceuticals LLC, who also attended the meeting with us. The FDA provided guidance on Incannex’s proposed long-term development strategy with regards to what will be required for a successful NDA (FDA approval) and marketing authorization.
Phase 2 exploratory clinical trial
Our Phase 2 Australian exploratory clinical trial was approved by the Human Research Ethics Committee (“HREC”) in late 2021 and this approval from an independent board of examiners permitted us to recruit trial participants in Australia. Participant screening and recruitment commenced in February 2022 and the first participants in the trial commenced treatment in March 2022.
72
The study was a Phase 2 randomized triple-blind active-placebo-controlled trial to assess the safety and efficacy of psilocybin-assisted psychotherapy for GAD. Participants experienced two psilocybin or active-placebo dosing sessions and up to 11 non-drug, specialist psychotherapy sessions over a period of 10 weeks.
Primary outcomes were safety, efficacy and tolerability, and secondary outcomes were quality of life, functional impairment, and comorbidities. Safety is assessed by monitoring adverse events including but not limited to liver function tests and scores on the Ultra Brief Checklist of Suicidality. Efficacy is assessed by comparing the change in Hamilton Anxiety Rating Scale from baseline between the placebo and treatment group. Tolerability is assessed by comparing the proportion of participants who complete both dosing sessions in the placebo and treatment groups. Secondary endpoints will be assessed by monitoring disability, comorbidity, productivity and quality of life using patient reported outcome measures.
In January 2024, the dosing and treatment protocols for all 72 participants in the Phase 2 study were completed. In February 2024, top-line results from the Phase 2 study were received. The trial met its primary endpoint, supporting a large clinical effect in the psilocybin treatment group compared to the placebo group.
The reduction in HAM-A score from baseline in the psilocybin group was 12.8 points, from 29.5 at baseline to 16.8 at week 11 (6 weeks following the final dosing session). This reduction in Hamilton Anxiety Ratings Scale score observed in the psilocybin group was 9.2 points greater than the reduction observed in the placebo group (-12.8 psilocybin vs. -3.6 placebo; p<0.0001). In the study, 44% of patients in the psilocybin group were observed to show a clinically meaningful improvement of at least 50% reduction in anxiety score from baseline; a ‘response rate’ more than four times higher than that of the placebo group. 27% of patients in the psilocybin group achieved full disease remission; a rate five times higher than that of psychotherapy with placebo.
Psilocybin within the context of PsiGAD psychotherapy was observed to be well-tolerated, with only mild and moderate adverse events (AEs) reported. The reported AEs were consistent with the known effects of the drug. No serious or severe adverse events were observed. Only one person of the 73 participants withdrew from the trial during the 7-week treatment program.
Next steps in PsiGAD clinical development
In August 2023, our subsidiary Psychennex Pty Ltd commenced preparing an IND application for the PsiGAD program. The results of the PsiGAD 1 study are being incorporated into the IND dossier along with the finalization of the other modules in preparation for submission to the FDA.
We have designed the follow-up Phase 2B clinical trial, PsiGAD2, with the assistance of Clerkenwell Health, a UK based contract research organization specializing in psychiatry and central nervous system treatments This trial is expected to be conducted at multiple sites in the United States, once the IND application is cleared by the FDA, and the United Kingdom.
Development and manufacture of cGMP psilocybin drug product
Based on the promising outcome of the interim analysis from PsiGAD1, we engaged Catalent for the development and cGMP manufacture of our own psilocybin drug product in February 2023. This drug product will be used in future clinical trials and potential wider commercial use. This development project is ongoing.
We finalized the development of the formulation of our psilocybin drug product, PSX-001. Final preparations for the manufacture of the cGMP clinical trial supply of PSX-001 are currently ongoing. Documentation on the formulation development and cGMP manufacture will support our FDA IND application that we commenced preparations of in August 2023.
Monash University
In December 2020, we entered into a partnership agreement with Monash University (“Monash”) in Australia to conduct a psilocybin-assisted psychotherapy trial to treat GAD. Monash sponsored our initial Phase 2 exploratory clinical trial, ensuring rigorous scientific independence and the highest standards in ethical and safe research. We are funding and supporting this investigator-initiated trial, and retain all intellectual property created by the trial. We are also investigating the commencement of other psychedelic medicine research projects that would offer an opportunity to address what we believe is an unmet need in patients diagnosed with other mental illnesses.
73
Monash is one of Australia’s leading universities and consistently ranks among the world’s top 100. Psychedelic treatments for our exploratory trials are delivered within BrainPark, a state-of-the-art research platform at Monash’s Turner Institute for Brain and Mental Health and Biomedical Imaging Facility, that provides a highly conducive environment for psychedelic treatments in a research context. Both the School of Psychological Sciences within the Turner Institute for Brain and Mental Health, and the Department of Psychiatry within the School of Clinical Sciences, have combined forces to conduct psychedelic research and the team comprises leading researchers and clinicians in relevant fields of psychiatry, psychotherapy, and mental health treatment development.
Virtual Reality (“VR”) Exposure Response Therapy (“ERP”) and psychedelics
In March 2022, we entered into a license agreement with Monash to develop a novel treatment that combines Virtual Reality and psychedelics. The license agreement provides an exclusive and perpetual license over an immersive therapeutic Virtual Reality environment developed by BrainPark. The license allows Incannex to investigate the use of the Virtual Reality therapy tool in combination with a psychedelic drug to develop a new treatment for severe forms of one or more anxiety disorders.
The associated research and development will be led by Dr. Paul Liknaitzky at Monash, a highly reputable, globally recognized, and innovative university that ranked #40 in the world in the US News and World Report 2022. Incannex and Monash are in advanced stages of discussion in relation to a research agreement for the clinical trials required to develop the new treatment form. The initial clinical trial will assess the efficacy, safety, tolerability, and optimal dose of the treatment method.
Clinical trial investigators
The Principal Investigator is Dr. Paul Liknaitzky, with Co-Investigators Professor Murat Yucel and Professor Suresh Sundram.
Dr. Liknaitzky is Head of the Clinical Psychedelic Research Lab within the Turner Institute and the Department of Psychiatry, at Monash. He is a Chief Principal Investigator and Research Fellow at Monash University, and has Adjunct or Honorary appointments at St Vincent’s Hospital, Macquarie University, Deakin University, and the University of Melbourne. He earned an Honors in Neuroscience and a PhD in Psychology from the University of Melbourne. His work examines mechanisms of mental illness and treatment development primarily within mood, anxiety and addiction research. Liknaitzky is an investigator across a number of Australia’s first clinical psychedelic trials. He has been invited to deliver numerous academic, professional, and public talks on psychedelic-assisted psychotherapy, and has been interviewed on the topic for print media, radio, and podcasts. Liknaitzky leads Australia’s first clinical psychedelic lab, coordinates Australia’s first applied psychedelic therapist training program, and is establishing Australia’s largest psychedelic trial (Psi-GAD). His work is focused on developing a rigorous program of research in psychedelic medicine at Monash University that seeks to evaluate therapeutic effects, innovate on treatment design, mitigate known risks, explore potential drawbacks, and understand therapeutic mechanisms.
Professor Murat Yucel gained a PhD combined with specialist clinical training in Clinical Neuropsychology in 2001 at La Trobe University. He then worked across as numerous mental health research centers at the University of Melbourne and was promoted to professor in 2012. He now works within the Monash School of Psychological Sciences, where he heads the mental health and addiction research programs. He is a director of BrainPark — a world-first neuroscience research clinic designed to bring the latest neuroscience with diagnostic or therapeutic benefit to the community in an accessible way.
Professor Suresh Sundram is the Head of the Department of Psychiatry, School of Clinical Sciences, Monash University and Director of Research, Mental Health Program, Monash Health. He has been investigating the molecular pathology of schizophrenia and related psychotic disorders using pharmacological, neurochemical and neuropathological approaches. These inter-related methods have been applied to parse components of the disorder such as treatment resistance and suicide to better understand their neurobiological substrates. He undertook his doctoral and post-doctoral studies at the Mental Health Research Institute in Melbourne before establishing his laboratory there and subsequently at the Florey Institute and concurrently establishing a clinical research laboratory undertaking clinical trials and biomarker research in psychotic disorders. He then transferred to and integrated his research program at Monash University and Monash Medical Centre.
74
Intellectual Property Strategy
We strategically protect our innovations with a harmonized IP strategy, combining patent protection with regulatory and market exclusivity. We are pursuing patent protection for aspects of our psilocybin therapy program. The patent position that will be available to us is unlikely to cover psilocybin alone as a clinical entity. However, we are pursuing a patent position in relation to methods of treatment using psilocybin including combination therapies (e.g., formulations, actives plus psychotherapeutic modalities) and other therapeutic methods (e.g., specific dosage regimens).
Cannabinoid Chewing Gums and chewable tablets
Medicated chewing gum and chewable tablets (“MCGT”) are drug delivery systems growing in favor in the medical community due to their application as an extended-release dosage form that supports continuous, ongoing release of the medicine contained. MCGTs are fast acting as they deliver the active ingredients into the oral mucosa, reducing the potential for gastric intolerance amongst patients. These qualities make MCGTs an excellent delivery system for medicinal combinations designed to treat sustaining pain and addiction disorders. MCGTs are also well tolerated by patients as there are no capsules to swallow or liquids to administer. The benefits of mastication, otherwise known as chewing, are well documented and include improved cerebral circulation, an anti-anxiety effect, memory improvement, neuroprotection, and an analgesic effect. These qualities make MCGTs an excellent delivery system for medicinal combinations designed to treat sustaining pain and addiction disorders.
Our subsidiary APIRx has multiple patents for cannabinoid-based drug candidates designed for the treatment of addiction to different drug classes (including marijuana addiction and opioid addiction) as well as sustaining pain (including for pain and spasticity in Multiple Sclerosis).
MedChew Dronabinol for chemotherapy induced nausea and vomiting
According to the WHO, cancer is one of the leading causes of death and chemotherapy is utilized by approximately ten (10) million cancer patients annually, and this statistic is expected to grow by 53% by 2040. Nausea and vomiting are two of the most dreaded cancer treatment-related side effects. Dronabinol, which is synthetic Tetrahydrocannabinol (“THC”), is an approved treatment for chemotherapy-associated nausea and vomiting as well as anorexia associated with HIV/AIDS. Oral dronabinol is taken up slowly, however, taking 1-2.5 hours to reach peak plasma concentration, and is also subject to first pass metabolism, which means that only 10-20% of the dose reaches the circulation.
MedChew Dronabinol is a chewable variant of Dronabinol that has been developed and patented by APIRx. In a phase 1a study of MedChew Dronabinol, THC appears in circulation within 10 minutes and a sustained release profile of 4 to 8 hours was observed in most study subjects so that the product is more useful in the time in which it is required. The next developmental step for the product is to conduct a bioavailability/bioequivalence clinical study to support the application for approval by bridging to publicly available data on Marinol, the marketing name of generic dronabinol. The economic size of the global drug market for chemotherapy induced nausea and vomiting was estimated to be US$6.04 billion in 2023 and is expected to expand at a CAGR of 6% through 2030.
MedChew Rx for pain and spasticity in multiple sclerosis (‘MS’)
Up to 84% of people suffering from MS also experience spasticity, which causes involuntary muscle stiffness and spasms. Pain is also a common symptom in MS, with up to two-thirds of people with MS reporting pain in worldwide studies. MedChew™ Rx is designed to be absorbed through the oral mucosal membrane and bypasses the liver, and first pass metabolism. MedChewTM Rx contains the same constituent formulation of CBD and THC as the product Sativex, which was initially approved in Canada in 2005 and is now available in 25 countries, including 18 countries in Europe, and Australia. MedChew Rx, however, facilitates extended dosing and reduces the need to readminister, which for Sativex is up to 12 times per day. It does not contain alcohol, which Sativex does, and will not exacerbate the dry mouth that is often associated with MS pharmacotherapy. MedChew Rx has underlying patent protection via granted patents related to chewing gums comprising cannabinoids. APIRx staff have completed regulatory meetings with Swiss-Medic (Switzerland) and CBG-MEG (Netherlands). We believe there could be potential to fast track to drug approval in some countries in Europe with a bioequivalence phase 1 study to bridge to Sativex CBD/THC oral spray safety and efficacy data.
75
Medicated Chewing Gum and Chewable Tablets for Treatment of Addiction
Medicated chewing gums deliver their active ingredients directly into the circulation of the oral mucosa, ensuring that the effects of the ingredients are delivered rapidly, but also in a sustained manner to reduce cravings for longer than other delivery methods. Rapid onset and sustained effect are both qualities desirable for the treatment of addiction disorders. Furthermore, the act of chewing, known as mastication, also has a multi-action, anti-anxiety effect that has been demonstrated in other scientific assessments.
Incannex has chosen Quest Pharmaceutical Services as its partner for regulatory guidance and clinical trial management for the advancement of the CannQuit™ and Renecann™ product lines designed for addiction and immune-disordered skin diseases.
CheWell for Cannabis Dependence
CheWell is a CBD chewable tablet with high bioavailability that can be used in the treatment of people with marijuana addiction. Cannabis dependence is predicted to be the fastest growing segment of drug dependence market and preliminary data observed by APIRx suggest a possible beneficial impact of CBD on mitigating the craving effect of cannabis. A case report has shown positive outcomes for one patient treated with CBD during the withdrawal and relapse phases of cannabis dependence. A pre-IND for the use of CheWell in patients with cannabis dependence with the FDA is currently in preparation.
We have data for CheWell as a high bioavailability product. A Phase 1 pharmacokinetic (PK) study demonstrated that the CheWell formulation led to a >10x increase maximum CBD levels in the blood compared to the standard CBD chewing gum delivery mechanisms. An international regulatory analysis is being undertaken to identify what is required for commercial launch in different jurisdictions. Improved bioavailability means that even small doses of CBD within MCGTs could be highly effective even without a prescription from a doctor, thus meeting the TGA requirements for an OTC product. Increased bioavailability also reduces the cost of goods, which increases margins.
CannQuitN for Smoking Cessation
CannQuitN is a medicated chewing gum that combines cannabinoids and nicotine to reduce addiction to cigarettes. CannQuitN is designed to better assist addicted smokers to quit smoking and we intend to trial our product for effectiveness against existing nicotine chewing gums. A more effective and cost-effective cannabinoid/nicotine combination medicated gum may have the potential to disrupt the incumbent global nicotine gum market, which was valued at US$1.3 billion in 2023 with an estimated CAGR of 5.6% through 2030.
In November 2022 Eurofins Scientific was engaged to develop and manufacture CannQuitN. Data collected on the quality and stability of CannQuitN during its development and manufacturing at Eurofins will be key components of future regulatory packages. These data packages include IND applications and NDA filings with the FDA.
A pre-IND meeting was conducted with the FDA in February 2024. During the meeting the agency provided feedback on the proposed development strategy for CannQuitN. This feedback is currently being incorporated into the development pipeline for CannQuitN.
76
CannQuit O for Opioid Addiction
CanQuit O is a medicated chewing gum that combines cannabinoids with opioid agonists and/or antagonists, which is designed to suppress opioid-based drug addiction in people addicted to opioids. We intend for CanQuit O to be a prescription product to help combat the ongoing opioid addiction crisis in the United States and elsewhere. We believe CanQuit O has the potential to be a simple solution to a complex addiction disorder and nationwide problem with far reaching consequences. Opioid use disorder has an annual addressable market size estimated to be US$3.5 billion in 2022 with a CAGR of 11.3% from 2023-2028, and with many people being addicted but untreated.
In November 2022 Eurofins Scientific was engaged to develop and manufacture CannQuit O. Data collected on the quality and stability of CannQuit O during its development and manufacturing at Eurofins will be key components of future regulatory packages. These data packages include IND applications and NDA filings with the FDA.
A pre-IND meeting was conducted with the FDA in April 2024. During the meeting the agency provided feedback on the proposed development strategy for CannQuit O This feedback is currently being incorporated into the development pipeline for CannQuit O.
CanChew Rx and SuppoCan for Irritable Bowel Syndrome and Inflammatory Bowel Disease
APIRx has developed a CBD-containing controlled-release functional chewing gum called CanChew Rx and a cannabinoid containing suppository called SuppoCan to be used independently or in conjunction with one another to treat bowel diseases.
Irritable bowel syndrome (IBS) is a condition that affects up to 11% of the population globally. IBS is characterized by abdominal pain, altered bowel habits, as well as diarrhea, constipation or both. Although the first marketing claim will be for IBS, we believe that the CanChew product could potentially provide a therapeutic benefit for a range of indications where CBD may assist patients. Data from 36 patient phase 2 proof of concept trial observed a 50% reduction in abdominal pain in CanChew treated IBS patients, supporting a therapeutic effect in IBS.
In 2017, it was estimated that there were 6.8 million people worldwide who suffered from IBD. Signs and symptoms of IBD, which encompass both Crohn’s disease and ulcerative colitis, include diarrhea, fatigue, abdominal pain and cramping, reduced appetite, and unintended weight loss. The main medications currently available for IBD are anti-inflammatory medications and analgesics. Anti-inflammatories include courses of corticosteroids which are used to induce remission but are immunosuppressing. CBD has shown efficacy in treating IBD in animals and we intend to undertake a phase 1 clinical trial to assess CanChew Rx and SuppoCan.
ReneCann topical CBD/CBG product for treatment of dermatological conditions.
ReneCann™ is Incannex’s proprietary topical cannabinoid formulation for the treatment of dermatological conditions caused by disorders of the immune system, including vitiligo, psoriasis, and atopic dermatitis, otherwise known as eczema. The ReneCann™ formulation is commercially protected by granted and pending patents acquired by Incannex as part of the APIRx acquisition that was finalised in August 2022.
The formulation combines cannabigerol (“CBG”) and CBD. CBG is a non- psychoactive cannabinoid with potent anti-inflammatory properties. A previous formulation of hempseed oil CBG/CBD combined as it will be used for ReneCann™ was used in an in-human proof of concept study with dosing over a 6-week period. The study was conducted at the Maurits Clinic, The Netherlands, and led by a world-renowned dermatologist Dr. Marcus Meinardi, MD, PhD.
In the study, the hempseed oil CBG/CBD formulation reduced disease scores in patients with each of the target skin diseases. Patients with vitiligo, psoriasis and atopic dermatitis were observed to experience improvements in symptoms of 10%, 33% and 22% respectively. While the 3% CBG/CBD oil treatment showed no improvement, the 15% CBG/CBD oil showed 16% and 33% improvement on the two psoriasis vulgaris patients respectively, 11% and 22% improvement on two atopic dermatitis patients with only CBG/CBD oil treatment respectively, and 10% improvement in the vitiligo patient with the 15% CBG/CBD oil treatment.
77
Vitiligo is observed when pigment-producing cells (melanocytes) stop producing melanin, causing the loss of skin colour in patches and the discoloured areas generally become larger over time. ReneCann™ was associated with diffuse re-pigmentation (usually perifollicular or from the borders of the lesion) and efficacy lasted for weeks eventually before depigmentation recurred.
In November 2022, Incannex engaged Eurofins Scientific to develop and manufacture ReneCann. The ReneCann™ drug product that is produced by Eurofins will be used in clinical trials to confirm the safety and therapeutic effect of ReneCann™ in vitiligo, psoriasis, and atopic dermatitis. Data on the quality and stability of ReneCann™ generated during development and manufacturing at Eurofins will be used in the chemistry and manufacturing control modules of future regulatory packages with the FDA. ReneCann™ also has the potential to be assessed for efficacy in other diseases where topical application may provide a benefit over conventional orally dosed cannabinoid formulations.
A pre-IND meeting was conducted with the FDA in October 2023. During the meeting the agency provided feedback on the proposed development strategy for ReneCann™. This feedback is currently being incorporated into the development pipeline for ReneCann™.
OraxiMax for Periodontal Disease and Gingivitis
Up to 50% of adults worldwide suffer from moderate to severe periodontitis and/or gingivitis. Periodontal disease treatment has been limited to professional dental cleaning and the use of systemic antibiotics. We are developing OraxiMax Toothpaste and Mouthwash contain CBD and Cannabigerol (CBG) which can prevent dental plaque formation, and thus gingivitis and periodontitis. Due to their proprietary formulations, the local availability of APIs is increased while systemic absorption is kept to a minimum.
Benefits of CBD in dental protection include:
| ● | Reduction in inflammation that can lead to gum diseases |
| ● | Reduction of bacteria associated with tooth decay, reducing the risk of cavities. |
| ● | Relieves dental and gum sensitivity |
| ● | Encourages tooth remineralization, and |
| ● | Restores pH balance. |
Cannabinoids for Ophthalmic Conditions
Through the Acquisition, we have two granted patents for ophthalmic formulations of cannabinoids. Anecdotal evidence supports therapeutic benefit for cannabis and cannabinoids drug products in the treatment of glaucoma and conjunctivitis. We believe that a therapeutic effect in these eye conditions is derived from the neuroprotective, anti-inflammatory, and anti-microbial activities of cannabinoids.
Clarion Clinics
In March 2023, we announced our intention to open multiple psychedelic-assisted psychotherapy clinics in Australia and overseas. We had been developing the commercialization plans for psychedelic clinics for some time, well before the TGA decision to down-schedule psilocybin for treatment-resistant depression (“TRD”) and MDMA for Post-Traumatic Stress Disorder (“PTSD”) was announced. The announcement from TGA led to the expansion and announcement of these plans.
78
We have entered a partnership with Australia’s leading clinical psychedelic professionals, all of whom have extensive experience within clinical psychedelic research, treatment, and training, including the following individuals:
| ● | Dr Paul Liknaitzky: Co-Founder, Director, Chief Strategy Officer, and Chief Scientific Officer |
Dr. Paul Liknaitzky has played a central role in establishing the clinical psychedelic field in Australia and leads the largest group of psychedelic researchers and clinicians in the country. Dr. Paul Liknaitzky is the Chief Principal Investigator on a program of psychedelic trials and collaborates on numerous others nationally. He has led the development of psychedelic trial protocols, treatment design, trial coordination, therapist selection and training, and has established active collaborations with an extensive network of international experts and organizations in the field. Dr. Paul Liknaitzky’s work is focused on developing innovative psychedelic therapies, evaluating benefits, exploring potential drawbacks, predicting treatment response, mitigating risks, understanding therapeutic mechanisms, and translating research into practice.
| ● | Professor Suresh Sundram: Co-Founder, Director, Chief Medical Officer, and Head of Psychiatry |
Professor Suresh Sundram is a Fellow of the Royal Australian and New Zealand College of Psychiatrists and a consultant psychiatrist. He holds senior leadership positions in academic and clinical psychiatry and has published more than 150 scientific articles, books, book chapters, and conference abstracts. He has presented as plenary and invited speaker at international and national conferences, served as Deputy Editor for the Asian Journal of Psychiatry, and as an advisor to the United Nations, and to national and state governments. Professor Suresh Sundram has led over 50 clinical trials and studies in psychiatric disorders. He has extensive experience with the use of psychedelics within psychotherapy and has overseen multiple research projects in this field.
| ● | Sean O’Carroll: Co-Founder, Director, and Head of Psychotherapy |
Mr. Sean O’Carroll is an integrative psychotherapist and academic specializing in experiential, relational, and transpersonal psychotherapy. Since 2019, he has developed and delivered psychedelic-assisted psychotherapy training for several clinical psychedelic research teams. He has served as lead psychotherapist on two clinical research trials, continues to supervise one of these teams, and works as a psychedelic-assisted psychotherapy consultant within industry, with an emphasis on psychotherapy training and protocol development. Mr. Sean O’Carroll began lecturing in transpersonal psychology in 2011 and has over ten years’ experience working with what he calls “psychedelic casualties”. Through the Wild Mind Institute, he offers training for mental health practitioners in psychedelic-assisted psychotherapy, “bad trip” integration, and eco-psychotherapy.
In May 2023, we announced that we signed a lease for the first clinic in Abbottsford, a suburb of Melbourne, Victoria. The clinic is designed as a commercial scale prototype, which can be scaled up and replicated to other locations. It is estimated to have the capacity to treat over 600 patients per year in normal working hours and substantially more in extended hours of operation. The company also announced that it had secured an initial supply of psilocybin and MDMA to facilitate the commencement of clinical operations.
The Clarion Clinics Advisory board is made up of world leading clinical psychedelics experts, including the following individuals:
| ● | Dr. Bill Richards is among the world’s best known psychedelic researchers and practitioners. He has had a multi-decade career at the forefront of psychedelic research, therapy, and training, and is a mentor and trainer to numerous research groups around the world. He co-founded the psychedelic research group at Johns Hopkins University and is the Director of Therapy at Sunstone Therapies in Maryland, US. |
| ● | Dr. Andrea Jungaberle is the Chief Medical Officer of Ovid Clinics in Berlin and co-founder of the MIND Foundation, Europe’s leading psychedelic research and education group. She has conducted and/or supervised psychedelic-assisted psychotherapy for hundreds of patients and works both within Germany’s largest clinical psilocybin trial and within clinical service delivery. |
79
| ● | Professor Matthew Johnson is one of the world’s most published psychedelic scientists. He has been central in the establishment and leading track record of the Johns Hopkins Center for Psychedelic & Consciousness Research, and his work has contributed to standards in practice within clinical psychedelic science. As a high-profile scientist in his field, he is frequently interviewed by national and international media outlets. |
In August 2023, Clarion Clinics announced that it would begin accepting registrations for psychedelic treatment interest by potential patients as part of pre-screening in readiness for opening. In December 2023, the Company received HREC endorsement for psychedelic assisted therapy. The Melbourne Clarion Clinic will be the first dedicated Psychedelic-assisted therapy clinic in Australia. In February 2024, Clarion Clinic’s Co-Founder and Head of Psychiatry, Professor Suresh Sundram, received approval from the TGA to prescribe MDMA in the treatment of Post-traumatic Stress Disorder and psilocybin for Treatment-resistant Depression.
Intellectual Property
We have implemented a patent filing strategy as we develop our products and therapies in conjunction with our medical advisory board. As of March 31, 2024, we own pending patent applications relating to our cannabinoid drug candidates. A summary of the number of patents, patent types and jurisdictions are listed in the table below. Once converted to the complete/PCT stage, the provisional patents will also be applicable to all PCT contracting states. International search reports and written opinions of the International Search Authority have confirmed that the key claims in our filed Patent Cooperation Treaty applications are novel and inventive and that the invention meets the requirements of industrial applicability. The preparation of the International Search Report (ISR) and International Search Opinion (ISO) for PCT applications is one of the main procedural steps of the international phase of the Patent Cooperation Treaty (PCT). The purpose of conducting the searches at the international phase is to identify the relevant prior art and for the International Searching Authority to establish a preliminary opinion as to whether the claims are novel, involve an inventive step and are industrially applicable. While the ISR and the ISO are non-binding, in the sense that national patent offices are not obliged to accept any finding of the International Searching Authority, these reports often represent a useful guide in relation to the patentability of the subject matter claimed in the PCT application.
In the context of the PCT applications that cover the cannabinoid drug candidates, IHL-216A, IHL-675A and IHL-42X, the International Searching Authority is the Australian Patent Office. Accordingly, the opinion expressed in the ISR / ISO for each of these PCT applications is based on searches that have been conducted by Australian Patent Examiners.
| Product/technology | Number of applications | Type of patent protection | Applicable jurisdictions | |||
| IHL-42X/Compositions and methods for the treatment of obstructive sleep apnoea (OSA) | 10 | Standard/utility | AU, CA, CO, EP, IL, JP, NZ, US | |||
| IHL-675A/Compositions and methods for the treatment of an inflammatory conditions | 16 | Standard/utility | AU, CA, CO, EP, IL, JP, NZ, US | |||
| IHL-216A/Compositions and methods for the treatment or prevention of traumatic brain injury (TBI) | 6 | Standard/utility | AU, CA, EP, IL, NZ, US | |||
| PsiGAD | 1 | Standard/utility | Provisional |
The cannabinoid drug candidate, IHL-675A, is a combination of CBD and hydroxychloroquine, which is specifically defined by claim 16 of International (PCT) Application No. PCT/AU2021/050226. The International Searching Authority considers claim 16 to be both novel and inventive.
The cannabinoid drug candidate, IHL-42X, is a combination of THC and acetazolamide for use in the treatment of obstructive sleep apnea (OSA), which is specifically defined by claim 3 of International (PCT) Application No. PCT/AU2021/050734. The International Searching Authority considers claim 3 to be both novel and inventive.
80
The cannabinoid drug candidate, IHL-216A, is a combination of CBD and isoflurane, which is specifically defined by claim 23 of International (PCT) Application No. PCT/AU2020/051056. The International Searching Authority considers claim 23 to be both novel and inventive.
In addition to pursuing patent protection for all our assets, we rely on unpatented trade secrets, know-how and other confidential information as well as proprietary technological innovation and expertise that are protected in part by confidentiality and invention assignment agreements with our employees, advisors and consultants.
Patent matters in biotechnology are highly uncertain and involve complex legal and factual questions. The availability and breadth of claims allowed in biotechnology and pharmaceutical patents cannot be predicted. Statutory differences in patentable subject matter may limit the scope of protection we can obtain on some or all of our licensed inventions or prevent us from obtaining patent protection either of which could harm our business, financial condition and results of operations. Since patent applications are not published until at least 18 months from their first filing date and the publication of discoveries in the scientific literature often lags behind actual discoveries, we cannot be certain that we, or any of our licensors, were the first creator of inventions covered by pending patent applications, or that we or our licensors, were the first to file patent applications for such inventions. Additionally, the grant and enforceability of a patent is dependent on a number of factors that may vary between jurisdictions. These factors may include the novelty of the invention, the requirement that the invention not be obvious in the light of prior art (including prior use or publication of the invention), the utility of the invention and the extent to which the patent clearly describes the best method of working the invention. In short, this means that claims granted in various territories may vary and thereby influence commercial outcomes.
While we have applied for and will continue to file for protection as appropriate for our therapeutic products and technologies, we cannot be certain that any future patent applications we file, or licensed to us, will be granted, or that we will develop additional proprietary products or processes that are patentable or that we will be able to license any other patentable products or processes. We cannot be certain that others will not independently develop similar products or processes, duplicate any of the products or processes developed or being developed by the company or licensed to us, or design around the patents owned or licensed by us, or that any patents owned or licensed by us will provide us with competitive advantages.
Furthermore, we cannot be certain that patents held by third parties will not prevent the commercialization of products incorporating the technology developed by us or licensed to us, or that third parties will not challenge or seek to narrow, invalidate or circumvent any of the issued, pending or future patents owned or licensed by us.
Our commercial success will also depend, in part, on our ability to avoid infringement of patents issued to others. If a court determines that we were infringing any third-party patents, we could be required to pay damages, alter our products or processes, obtain licenses or cease certain activities. We cannot be certain that the licenses required under patents held by third parties would be made available on terms acceptable to us or at all. To the extent that we are unable to obtain such licenses, we could be foreclosed from the development, export, manufacture or commercialization of the product requiring such license or encounter delays in product introductions while we attempt to design around such patents, and any of these circumstances could have a material adverse effect on our business, financial condition and results of operations. We may have to resort to litigation to enforce any patents issued or licensed to us or to determine the scope and validity of third-party proprietary rights. Such litigation could result in substantial costs and diversion of effort by us. We may have to participate in opposition proceedings before the Australian Patent and Trademark Office or another foreign patent office, or in interference proceedings declared by the United States Patent and Trademark Office, to determine the priority of invention for patent applications filed by competitors. Any such litigation interference or opposition proceeding, regardless of outcome, could be expensive and time consuming, and adverse determinations in any such proceedings could prevent us from developing, manufacturing or commercializing our products and could have a material adverse effect on our business, financial condition and results of operations.
As of March 31, 2024, the Company also owns trademark registrations in Australia and the United States.
81
Patent Portfolio
The following table presents our portfolio of patents and patent applications filed by Incannex, including their status (as at March 31 2024) and title. Patents can be valid for approximately 20 years from their effective filing date if maintained.
The following table presents our portfolio of patents and patent applications filed by Incannex, including their status (as at March 31, 2024) and title.
| Patent Family | Title | Status | Expires | |||
| AU 2019903734 | Inhalable compositions and uses thereof | Continued as PCT application | NA | |||
| PCT/AU2020/051056 | Compositions for the treatment or prevention of traumatic brain injury | National / regional phase entered | NA | |||
| AU 2020359294 | Compositions for the treatment or prevention of traumatic brain injury | Granted | 10/02/2040 | |||
| CA 3152806 | Compositions for the treatment or prevention of traumatic brain injury | Pending | 10/02/2040* | |||
| EP 20870484.1 | Compositions for the treatment or prevention of traumatic brain injury | Pending | 10/02/2040* | |||
| IL 291874 | Compositions for the treatment or prevention of traumatic brain injury | Pending | 10/02/2040* | |||
| NZ 786889 | Compositions for the treatment or prevention of traumatic brain injury | Pending | 10/02/2040* | |||
| US 17/638264 | Compositions for the treatment or prevention of traumatic brain injury | Pending | 10/02/2040* | |||
| AU 2020901030 | A method of treatment | Continued as PCT application | NA | |||
| AU 2020902432 | A method of treatment | Continued as PCT application | NA | |||
| AU 2020903985 | A method of treatment | Continued as PCT application | NA | |||
| AU 2020904264 | A method of treatment | Continued as PCT application | NA | |||
| AU 2021900241 | A method of treatment | Continued as PCT application | NA | |||
| AU 2021900324 | A method of treatment | Continued as PCT application | NA | |||
| PCT/AU2021/050226 | Methods and compositions for treating or preventing an inflammatory condition | National / regional phase entered | NA | |||
| AU 2021250462 | Methods and compositions for treating or preventing an inflammatory condition | Pending | 03/15/2041* | |||
| CA 3169702 | Methods and compositions for treating or preventing an inflammatory condition | Pending | 03/15/2041* | |||
| CO NC2022/0015048 | Methods and compositions for treating or preventing an inflammatory condition | Pending | 03/15/2041* | |||
| EP 21781628.9 | Methods and compositions for treating or preventing an inflammatory condition | Pending | 03/15/2041* | |||
| IL 296943 | Methods and compositions for treating or preventing an inflammatory condition | Pending | 03/15/2041* | |||
| JP 2022-559967 | Methods and compositions for treating or preventing an inflammatory condition | Pending | 03/15/2041* | |||
| NZ 793870 | Methods and compositions for treating or preventing an inflammatory condition | Pending | 03/15/2041* | |||
| US 17/907322 | Methods and compositions for treating or preventing an inflammatory condition | Pending | 03/15/2041* | |||
| AU 2020902368 | A method of treatment | Continued as PCT application | NA |
82
| PCT/AU2021/050734 | Compositions and methods for the treatment of obstructive sleep apnoea (OSA) | National / regional phase entered | NA | |||
| AU 2021306424 | Compositions and methods for the treatment of obstructive sleep apnoea (OSA) | Pending | 07/09/2041* | |||
| CA 3182528 | Compositions and methods for the treatment of obstructive sleep apnoea (OSA) | Pending | 07/09/2041* | |||
| CO NC2023/0000118 | Compositions and methods for the treatment of obstructive sleep apnoea (OSA) | Pending | 07/09/2041* | |||
| EP 21837113.6 | Compositions and methods for the treatment of obstructive sleep apnoea (OSA) | Pending | 07/09/2041* | |||
| IL 299212 | Compositions and methods for the treatment of obstructive sleep apnoea (OSA) | Pending | 07/09/2041* | |||
| JP 2023-501090 | Compositions and methods for the treatment of obstructive sleep apnoea (OSA) | Pending | 07/09/2041* | |||
| NZ 795230 | Compositions and methods for the treatment of obstructive sleep apnoea (OSA) | Pending | 07/09/2041* | |||
| US 18/000,880 | Compositions and methods for the treatment of obstructive sleep apnoea (OSA) | Pending | 07/09/2041* | |||
| AU 2021902170 | A composition and uses thereof | Continued as PCT application | NA | |||
| PCT/AU2022/050731 | Composition comprising cannabidiol and hydroxychloroquine in a fixed dose combination capsule | National Regional Phase entered | NA | |||
| AU 2022309612 | Composition comprising cannabidiol and hydroxychloroquine in a fixed dose combination capsule | Pending | 07/13/2042* | |||
| CA 3224079 | Composition comprising cannabidiol and hydroxychloroquine in a fixed dose combination capsule | Pending | 07/13/2042* | |||
| CO NC2024/0000294 | Composition comprising cannabidiol and hydroxychloroquine in a fixed dose combination capsule | Pending | 07/13/2042* | |||
| EP 22840862.1 | Composition comprising cannabidiol and hydroxychloroquine in a fixed dose combination capsule | Pending | 07/13/2042* | |||
| IS 309886 | Composition comprising cannabidiol and hydroxychloroquine in a fixed dose combination capsule | Pending | 07/13/2042* | |||
| JP 2024-501866 | Composition comprising cannabidiol and hydroxychloroquine in a fixed dose combination capsule | Pending | 07/13/2042* | |||
| US18/576218 | Composition comprising cannabidiol and hydroxychloroquine in a fixed dose combination capsule | Pending | 07/13/2042* | |||
| AU 2021903210 | A composition and uses thereof | Continued as PCT application | NA | |||
| PCT/AU2022/051200 | Oil-in-water emulsion for inhalation administration comprising cannabidiol | Pending | 10/07/2042^ | |||
| AU 2022903614 | A method of treatment | Continued as PCT application | NA | |||
| PCT/AU2023/051226 | A method of treating obstructive sleep apnoea | Pending | 11/29/2043^ | |||
| AU 2023903778 | A composition and uses thereof | Pending | 11/23/2043# | |||
| AU 2024900439 | A method of treatment | Pending | 02/23/2045$ |
| * | Expiry date may be subject to any patent term extensions or adjustments that may be available. |
| ^ | Assumes that national / regional phase will be entered in key jurisdictions |
| # | Assumes completion as a PCT application on the completion deadline of November 23, 2024 |
| $ | Assumes completion as a PCT application on the completion deadline of February 23, 2025 |
83
The acquisition of APIRx has added 28 granted patents and 11 pending patent applications to the Incannex patent portfolio. These patents cover all aspects of cannabinoid drug development including extraction, formulation and methods of use.
Competition
We are targeting indications that have no registered, limited or costly pharmacological solutions. Thus, competitor drugs for the indications we are assessing with our drug candidates either do not exist or are limited in efficacy or have unpleasant side effect profiles for certain cohorts of patients. The table below outlines existing drugs and therapies used to treat the illnesses we aim to treat with our drug candidates and their associated pitfalls for patients.
| IHL Drug Candidate | Indication | Existing Products | Existing Product Pitfalls | |||
| IHL-42X | Obstructive Sleep Apnea | - CPAP device, dental device | - Noisy mechanical device worn during sleep; - potential poor patient compliance due to discomfort. | |||
| IHL-216A | Traumatic Brain Injury/Concussion | None | N/A | |||
| IHL-675A | Lung Inflammation | - Corticosteroids - Ventilator | - Corticosteroids reduce immune system activity; - ventilators are associated with a high rate of mortality. | |||
| IHL-675A | Rheumatoid Arthritis | - Corticosteroids - DMARDS - Biologic agents | - High expense, significant side effect profiles; - lack of efficacy or tolerability in certain patient cohorts. | |||
| IHL-675A | Inflammatory Bowel Disease | - Corticosteroids - Immune system suppressors (ISSs) - Biologic agents | - Corticosteroids can reduce immune system activity; - ISSs can damage the digestive tract lining; | |||
| PSI-GAD | Generalized Anxiety Disorder | - Antidepressants (SSRI/SNRI classes) | - Non-curative, poor side effect profile; - some patients become treatment resistant. | |||
| ReneCann | Vitiligo | Topical corticosteroids, calcineurin inhibitors, phototherapy, systemic immunosuppressants | Poor efficacy, unpleasant side effects with long term use. Multiple applications required, lag before response is seen | |||
| ReneCann | Atopic dermatitis | Topical corticosteroids, calcineurin inhibitors, phototherapy, systemic immunosuppressants, Crisaborole, Dupilumab | Poor treatment adherence, safety concerns, limited efficacy | |||
| ReneCann | Psoriasis | Topical corticosteroids, calcineurin inhibitors, keratinolytics, biologics | Need to inject treatment, limited efficacy | |||
| CannQuitN | Smoking cessation | Nicotine replacement therapy, bupropion, varenicline | Limited efficacy | |||
| CannQuitO | Opioid use disorder | Buprenorphine, methadone | Limited efficacy, abuse potential, side effects |
84
Material Contracts
Share Sale and Purchase Agreement between Incannex and the sellers of APIRx Pharmaceutical USA, LLC
In May 2022, we entered into a Share Sale and Purchase Agreement to acquire 100% equity interests in APIRx. As consideration, we issued 218,169,506 ordinary shares in Incannex to the sellers of APIRx, at A$0.225 per share. Under the terms of the agreement, we acquired all assets and intellectual property rights of APIRx. We completed the acquisition in August 2022.
Master Consultancy Agreement with Clinical Network Services (CNS) Pty Ltd
On June 29, 2020, we entered into a Master Consultancy Agreement with Clinical Network Services (CNS) Pty Ltd (“Clinical Network”). Under the terms of the agreement, Clinical Network is to act as Australian and New Zealand consultant to product development and management of clinical research programs. Incannex will pay market-standard fees to Clinical Network. The agreement may be terminated by either party for cause, for the reasons set forth in the agreement.
Research Services Agreement with Monash University, dated November 27, 2020
On November 27, 2020, we entered into a Research Services Agreement with Monash University. Under the terms of the agreement, Monash University is to conduct research services with respect to Psi-GAD. Research activities are to be conducted with respect to a phase 2A randomized double-blind active-placebo-controlled trial to assess the safety and efficacy of Psi-GAD. Incannex will provide all the information required to conduct the study and will pay market-standard fees to Monash University. The agreement may be terminated by either party for cause, for the reasons set forth in the agreement.
Master Service Agreement with Avance Clinical Pty Limited
On July 12, 2021, we entered into a Master Service Agreement with Avance Clinical Pty Limited (“Avance”). Under the terms of the agreement, Avance will perform services to support Incannex’s clinical trials and studies, as requested by Incannex. The agreement has an initial term of five years. Each party may terminate the agreement by delivering a written notice three months prior to the expiration of the term of the contract.
Regulatory Authorities
The ongoing research and development, clinical, regulatory, commercial and manufacturing activities of our drug candidates are subject to extensive regulation by numerous governmental authorities, including (i) in Australia, principally the Therapeutic Goods Administration, or TGA; (ii) in the United States, principally the Food and Drug Administration, or FDA, as well as the Drug Enforcement Agency (DEA); and (iii) in Europe, principally the European Medicines Agency, or EMA and local competent authorities, ethics committees (ECs), institutional research boards (IRBs) and other regulatory authorities at federal, state or local levels. We, along with our third-party contractors, will be required to navigate the various preclinical, clinical and commercial approval and post-approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of our drug candidates.
85
United States
FDA process
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and its implementing regulations. Regulations that govern the pharmaceutical quality, safety, efficacy, labeling, storage, record keeping, advertising and promotion of such products under the Federal Food, Drug and Cosmetic Act, the Public Health Service Act, and their implementing regulations. In particular, controlled substances, like synthetic cannabidiol, THC, and psilocybin are regulated by the U.S. Drug Enforcement Administration, or DEA.
The process of obtaining FDA and DEA approval and achieving and maintaining compliance with applicable laws and regulations requires the expenditure of substantial time and financial resources. Failure to comply with applicable FDA or other requirements may result in refusal to approve pending applications, a clinical hold, warning letters, civil or criminal penalties, recall or seizure of products, partial or total suspension of production or withdrawal of the product from the market. FDA approval is required before any new drug or biologic, including a new use of a previously approved drug for a new indication, can be marketed in the United States. All applications for FDA approval must contain, among other things, information relating to safety and efficacy, pharmaceutical quality, packaging, labeling and quality control.
Government oversight of the pharmaceutical industry is usually classified into pre-approval and post-approval categories. Most of the therapeutically significant innovative products marketed today are the subject of New Drug Applications, or NDAs, or Biologics License Applications, or BLAs. Pre-approval activities are used to assure the product is safe and effective before marketing.
Drug Approval Process — FDA
None of our drug candidates may be marketed in the United States until the drug has received FDA approval. The steps required before a drug may be marketed in the United States generally include the following:
| ● | completion of extensive pre-clinical laboratory tests, animal studies, and formulation studies in accordance with the FDA’s GLP and GMP regulations; |
| ● | submission to the FDA of an Investigational New Drug, or IND, application for human clinical testing, which must become effective before human clinical trials may begin; |
| ● | receive approval from the DEA prior to commencement of any clinical trials in the United States that involve the use of Schedule I controlled substances; |
| ● | performance of adequate and well-controlled human clinical trials in accordance with GCP requirements to establish the safety and efficacy of the drug for each proposed indication; |
| ● | submission to the FDA of an NDA/BLA after completion of all pivotal clinical trials; |
| ● | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the active pharmaceutical ingredient, or API, and finished drug product are produced and tested to assess compliance with current Good Manufacturing Practices, or cGMPs; and |
| ● | FDA review and approval of the NDA/BLA and DEA scheduling (for a controlled substance) prior to any commercial marketing or sale of the drug in the United States. |
The manufacturing and quality, as well as preclinical and clinical testing and approval process requires substantial time, effort and financial resources, and we cannot guarantee approval of our drug candidates will be granted on a timely basis, if at all. Notably, the FDA may reach different conclusions than we have after analyzing the same data, or there may be differences of opinion amongst members of FDA’s review team.
86
The FDA may inspect and audit domestic and foreign development facilities, planned production facilities, clinical trial sites and laboratory facilities. There is a pre-approval inspection after submission to market a new product, routine inspection of a regulated facility and a “for-cause” inspection to investigate a specific problem that has come to the FDA’s attention. After the product is approved and marketed, the FDA uses different mechanisms for assuring that firms adhere to the terms and conditions of approval described in the application and that the product is manufactured in a consistent and controlled manner. This is done by periodic unannounced inspections of production and quality control facilities by the FDA’s field investigators and analysts.
Preclinical tests include laboratory evaluation of toxicity in animals and in vitro (laboratory tests). The results of preclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND application to the FDA. The IND application is based on the results of initial testing done on animals for pharmacology and toxicity, which is used to develop a plan for testing the drug on humans. Only after preclinical testing, the FDA determines whether the drug should be tested in people.
Further, an independent institutional review board, or IRB, covering each medical center proposing to conduct clinical trials must review and approve the plan for any clinical trial before it commences at that center and it must monitor the study until completed. The FDA, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive Good Clinical Practice, or GCP, regulations, which include requirements that all research subjects provide informed consent and that all clinical studies be conducted under the supervision of one or more qualified investigators.
Clinical trials (under an IND) involve administration of the investigational drug to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol must be provided to the FDA as part of a separate submission to the IND. Further, an Institutional Review Board, or IRB, for each medical center proposing to conduct the clinical trial must review and approve the study protocol and informed consent information for study subjects for any clinical trial before it commences at that center, and the IRB must monitor the study until it is completed. There are also requirements governing reporting of ongoing clinical trials and clinical trial results to public registries. Study subjects must sign an informed consent form before participating in a clinical trial.
Progress reports detailing the results of the clinical studies must be submitted at least annually to the FDA and safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse reactions in case of an open IND. For purposes of an NDA or BLA submission and approval, human clinical trials are typically conducted in the following sequential phases, which may overlap:
| ● | Phase I: Trials are initially conducted in a limited population of healthy human (in oncology Phase I trials are often conducted in patients) subjects or patients to test the drug candidate for safety and dose tolerance. These studies are designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. |
| ● | Phase II: The investigational product is administered to a limited patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. |
| ● | Phase III: The investigational product is administered to an expanded patient population in adequate and well-controlled studies to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit relationship of the investigational product and to provide an adequate basis for product approval. |
| ● | Phase IV: In some cases, the FDA may condition approval of an NDA or BLA on the sponsor’s agreement to conduct additional clinical trials to further assess the drug candidate’s safety, purity and potency after NDA or BLA approval. Such post-approval trials are typically referred to as Phase IV clinical trials. |
87
Concurrent with clinical studies, sponsors usually complete additional animal studies and must also develop additional information about the product and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, the manufacturer must develop and validate methods for testing the identity, strength, quality and purity of the final product. Moreover, appropriate packaging must be selected and tested and stability studies must be conducted to assure product integrity and demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
The results of product development, preclinical studies and clinical trials, along with the aforementioned manufacturing information, are submitted to the FDA as part of a BLA/NDA. Under the Prescription Drug User Fee Act, or PDUFA, the FDA agrees to specific goals for NDA/BLA review time. The testing and approval process requires substantial time, effort and financial resources. The FDA will review the BLA/NDA and may deem it to be inadequate to support approval, and we cannot be sure that any approval will be granted on a timely basis, if at all. The FDA may also refer the application to the appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendations of the advisory committee, but it typically follows such recommendations.
The FDA may deny approval of a BLA/NDA if the applicable regulatory criteria are not satisfied, or it may require additional clinical data or additional pivotal Phase III clinical trials. Even if such data are submitted, the FDA may ultimately decide that the NDA/BLA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than the sponsor does. Once issued, product approval may be withdrawn by the FDA if ongoing regulatory requirements are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing, including Phase IV clinical trials, and surveillance programs to monitor the effect of approved products that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs which may include pediatric assessment, and potentially studies required for an application for a new indication, new dosage form, a new dosing regimen, a new route of administration or a new active ingredient. Products may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the drug, including changes in indications, labeling or manufacturing processes or facilities, approval of a new or supplemental BLA may be required, which may involve conducting additional preclinical studies and clinical trials.
Expedited Review and Approval
The FDA has various programs, including fast track designation, priority review, accelerated approval, and breakthrough therapy designation, which are intended to expedite or simplify the process for reviewing drugs and/or provide for approval on the basis of surrogate endpoints. Even if a drug qualifies for one or more of these programs, the FDA may later decide that the drug no longer meets the conditions for qualification or that the time period for FDA review or approval will not be shortened. In particular, if accelerated approval is granted for any particular drug candidate, the FDA can subsequently revoke the marketing authorization for such product if post-market clinical trial results are unsuccessful. Generally, drugs that may be eligible for these programs are those for serious or life-threatening diseases or conditions, those with the potential to address unmet medical needs, and those that offer meaningful benefits over existing treatments.
Other U.S. Regulatory Requirements
After approval, products are subject to extensive continuing regulation by the FDA, which include company obligations to manufacture products in accordance with GMP, maintain and provide to the FDA updated safety and efficacy information, report adverse experiences with the product, keep certain records and submit periodic reports, obtain FDA approval of certain manufacturing or labelling changes and comply with FDA promotion and advertising requirements and restrictions. Failure to meet these obligations can result in various adverse consequences, both voluntary and FDA-imposed, including product recalls, withdrawal of approval, restrictions on marketing, and the imposition of civil fines and criminal penalties against the BLA holder — all of which may become public. In addition, later discovery of previously unknown safety or efficacy issues may result in restrictions on the product, manufacturer or application holder.
88
We, and any manufacturers of our drug candidates, are required to comply with applicable FDA manufacturing requirements contained in the FDA’s GMP regulations. GMP regulations require, among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation. The manufacturing facilities for our drug candidates must meet GMP requirements. We, and any third-party manufacturers, are also subject to periodic inspections of facilities by the FDA and other authorities, including procedures and operations used in the testing and manufacture of our drug candidates to assess our compliance with applicable regulations.
With respect to post-market product advertising and promotion, the FDA imposes a number of complex regulations on entities that advertise and promote pharmaceuticals, which include, among others, standards for direct-to-consumer advertising, promoting products for uses or in patient populations that are not described in the product’s approved labelling (known as “off-label use”), industry-sponsored scientific and educational activities, and promotional activities involving the Internet. Failure to comply with FDA requirements can have negative consequences, including adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors and civil or criminal penalties. Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such off-label uses.
Changes to some of the conditions established in an approved application, including changes in indications, labelling, or manufacturing processes or facilities, require submission and FDA approval of a new BLA or BLA supplement before the change can be implemented. A BLA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing BLA supplements as it does in reviewing BLAs.
Adverse event reporting and submission of periodic reports are required following FDA approval of a BLA. The FDA also may require post-marketing testing, known as Phase IV testing, risk mitigation strategies, and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product.
Controlled Substances
The Controlled Substances Act (CSA) and its implementing regulations establish a “closed system” of distribution for controlled substances. The CSA imposes registration, security, recordkeeping and reporting, storage, manufacturing, distribution, labeling, importation, exportation, disposal and other requirements under the oversight of the DEA. The DEA is the federal agency responsible for regulating controlled substances, and requires those individuals or entities that manufacture, import, export, distribute, research, or dispense controlled substances to comply with the regulatory requirements to prevent the diversion of controlled substances to illicit channels of commerce.
Facilities that research, manufacture, distribute, import or export any controlled substance must register annually with the DEA. The DEA registration is specific to the particular location, activity(ies) and controlled substances utilized. For example, separate registrations are required for importation and manufacturing activities, and each registration authorizes which schedules of controlled substances the registrant may handle. However, certain coincident activities are permitted without obtaining a separate DEA registration, such as distribution of controlled substances by the manufacturer that produces them.
The DEA categorizes controlled substances into one of five schedules — Schedule I, II, III, IV, or V — with varying qualifications for listing in each schedule. Scheduling determination by the DEA are dependent on approval of a substance or a specific formulation of a substance. Schedule I substances by definition have a high potential for abuse, have no currently “accepted medical use” in treatment in the United States and lack accepted safety for use under medical supervision. They may be used only in federally-approved research programs and may not be marketed or sold for dispensing to patients in the United States. Pharmaceutical products having a currently accepted medical use may be listed as Schedule II, III, IV or V substances, with Schedule II substances presenting the highest potential for abuse and physical or psychological dependence, and Schedule V substances presenting the lowest relative potential for abuse and dependence. The regulatory requirements are more restrictive for Schedule II substances than Schedule III-V substances. For example, all Schedule II drug prescriptions must be signed by a physician, physically presented to a pharmacist in most situations, and cannot be refilled. Marijuana and THC are Schedule I controlled substances under the CSA. Products approved for medical use in the United States that contain marijuana, THC or marijuana/THC extracts, must be placed in Schedules II-V, since approval by the FDA satisfies the “acceptable medical use” requirement. While marijuana and THC are controlled substances, the Agricultural Improvement Act of 2018 amended the CSA to exclude Cannabis meeting the statutory definition of hemp from the definition of marijuana. As a result, Cannabis that contains 0.3 percent or less of delta-9 THC on a dry weight basis is no longer considered a controlled substance. By extension, Cannabis-derived cannabidiol that satisfies the same limitation concerning delta-9 THC is also excluded from CSA regulatory controls. Because the definition of hemp does not expressly include synthetic equivalents of Cannabis or its derivatives, however, there is a lack of clarity about the CSA control status of pharmaceutically manufactured cannabidiol. Absent guidance to the contrary from the DEA, Cannabis and those products which contain Cannabis, that do not meet the definition of hemp remain in Schedule I of the CSA for purposes of development and research activities.
89
The DEA inspects all manufacturing facilities to review security, record keeping, reporting and compliance with other DEA regulatory requirements prior to issuing a controlled substance registration. The specific security requirements vary by the type of business activity and the schedule and quantity of controlled substances handled. The most stringent requirements apply to manufacturers of Schedule I and Schedule II substances. Required security measures commonly include background checks on employees and physical control of controlled substances through storage in approved vaults, safes and cages, and through use of alarm systems and surveillance cameras. An application for a manufacturing registration as a bulk manufacturer (not a dosage form manufacturer or a repacker/relabeler) for a Schedule I or II substance must be published in the Federal Register and is open for 30 days to permit interested persons to submit comments, objections, or requests for a hearing. A copy of the notice of the Federal Register publication is forwarded by the DEA to all those registered, or applicants for registration, as bulk manufacturers of that substance. Once registered, manufacturing facilities must maintain records documenting the manufacture, receipt and distribution of all controlled substances. Manufacturers must submit periodic reports to the DEA of the distribution of Schedule I and II controlled substances, Schedule III narcotic substances, and other designated substances. Registrants must also report any controlled substance thefts or significant losses and must adhere to certain requirements to dispose of controlled substances. As with applications for registration as a bulk manufacturer, an application for an importer registration for a Schedule I or II substance must also be published in the Federal Register, which remains open for 30 days for comments. Imports of Schedule I and II controlled substances for commercial purposes are generally restricted to substances not already available from a domestic supplier or where there is not adequate competition among domestic suppliers. In addition to an importer or exporter registration, importers and exporters must obtain a permit for every import or export of a Schedule I and II substance, Schedule III, IV and V narcotic, specially designated Schedule III non-narcotics, or Schedule IV or V narcotic controlled in Schedule I or II by the Convention on Psychotropic Substances and submit import or export declarations for Schedule III, IV and V non-narcotics.
For drugs manufactured in the United States, the DEA establishes annually an aggregate quota for the amount of substances within Schedules I and II that may be manufactured or produced in the United States based on the DEA’s estimate of the quantity needed to meet legitimate medical, scientific, research and industrial needs. This limited aggregate amount of Cannabis that the DEA allows to be produced in the United States each year is allocated among individual companies, which, in turn, must annually apply to the DEA for individual manufacturing and procurement quotas. The quotas apply equally to the manufacturing of the active pharmaceutical ingredient and the production of dosage forms. The DEA may adjust aggregate production quotas a few times per year, and individual manufacturing or procurement quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments for individual companies.
The states also maintain separate controlled substance laws and regulations, including licensing, recordkeeping, security, distribution, and dispensing requirements. State Authorities, including Boards of Pharmacy, regulate use of controlled substances in each state. Failure to maintain compliance with applicable requirements, particularly as manifested in the loss or diversion of controlled substances, can result in enforcement action that could have a material adverse effect on our business, operations and financial condition. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate proceedings to revoke those registrations. In certain circumstances, violations could lead to criminal prosecution.
We will be subject to DEA approval to conduct our clinical trials and manufacturing activities in the United States. All parties engaged for Incannex projects, including but not limited to formulation development, manufacturing, preclinical and clinical research, involving controlled substances in the United States will have the appropriate licenses and permits from the DEA. We may also decide to develop, manufacture or commercialize our drug candidates in additional countries. As a result, we will be subject to controlled substance laws and regulations from the TGA in Australia, Health Canada’s Office of Controlled Substances in Canada, the Drugs & Firearms Unit (Home Office) of the National Drug Control System in the United Kingdom, and from other regulatory agencies in other countries where we develop, manufacture or commercialize each drug asset in the future.
90
Foreign Regulation
In addition to regulations in the United States, we are subject to a variety of foreign regulations governing clinical trials and the commercial sales and distribution of our drug candidates. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials vary greatly from country to country.
European Union and United Kingdom
In the European Economic Area, or EEA, which is comprised of the Member States of the European Union plus Norway, Iceland and Liechtenstein, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA by the EMA. Under European Union regulatory systems, we must submit and obtain authorization for a clinical trial application in each member state in which we intend to conduct a clinical trial. When conducting clinical trials in the European Union, we must adhere to the provisions of the European Union Clinical Trials Directive (Directive 2001/20/EC) and the laws and regulations of the EU Member States implementing them. These provisions require, among other things, that the prior authorization of an Ethics Committee and the competent Member State authority is obtained before commencing the clinical trial. In April 2014, the European Union passed the Clinical Trials Regulation (Regulation 536/2014), which will replace the current Clinical Trials Directive. To ensure that the rules for clinical trials are identical throughout the European Union, the EU Clinical Trials Regulation was passed as a regulation that is directly applicable in all EU member states. All clinical trials performed in the European Union are required to be conducted in accordance with the Clinical Trials Directive until the Clinical Trials Regulation becomes applicable. According to the current plans of the EMA, the Clinical Trials Regulation is expected to become applicable.
After we have completed our clinical trials, we must obtain marketing authorization before we can market our product. We may submit applications for marketing authorizations under a centralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states.
The European Medicines Agency, or EMA, is a body of the European Union located in Amsterdam. The EMA is responsible for the scientific evaluation of medicines developed by pharmaceutical companies for use in the European Union. The EMA is involved in the scientific evaluation of medicines that fall within the scope of the centralized procedure. Like the FDA, there is a harmonization between regulators and the EMA may inspect and audit the development facilities, planned production facilities, clinical trial sites and laboratory facilities. Additionally, after the product is approved and marketed, the EMA uses different mechanisms for assuring that firms adhere to the terms and conditions of approval described in the application and that the product is manufactured in a consistent and controlled manner. This is done by periodic unannounced inspections of production and quality control facilities.
Regulation and Procedures Governing Approval of Medicinal Products in the European Union
In order to market any product outside of the United States, a company must also comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of products. Whether or not it obtains FDA approval for a product, an applicant will need to obtain the necessary approvals by the comparable foreign regulatory authorities before it can initiate clinical trials or marketing of the product in those countries or jurisdictions. Specifically, the process governing approval of medicinal products in the European Union generally follows the same lines as in the United States, although the approval of a medicinal product in the United States is no guarantee of approval of the same product in the European Union, either at all or within the same timescale as approval may be granted in the United States. It entails satisfactory completion of pharmaceutical development, non-clinical studies and adequate and well-controlled clinical trials to establish the safety and efficacy of the medicinal product for each proposed indication. It also requires the submission to relevant competent authorities for clinical trials authorization for a marketing authorization application, or MAA, and the granting of a marketing authorization by these authorities before the product can be marketed and sold in the European Union or its member states (as well as Iceland, Norway and Liechtenstein). If we fail to comply with applicable requirements, we may be subject to, among other things, fines, suspension of clinical trials, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
91
Clinical Trial Approval
Pursuant to the currently applicable Clinical Trials Directive 2001/20/EC and the Directive 2005/28/EC on GCP, a system for the approval of clinical trials in the European Union has been implemented through national legislation of the member states. Under this system, an applicant must obtain approval from the national competent authority, or NCA, of a European Union member state in which the clinical trial is to be conducted or in multiple member states if the clinical trial is to be conducted in a number of member states. Furthermore, the applicant may only start a clinical trial at a specific study site after the independent ethics committee, or EC, has issued a favorable opinion in relation to the clinical trial. The clinical trial application must be accompanied by an investigational medicinal product dossier with supporting information prescribed by Directive 2001/20/EC and Directive 2005/28/EC and corresponding national laws of the member states and further detailed in applicable guidance documents. Under the current regime all suspected unexpected serious adverse reactions to the investigated drug that occur during the clinical trial have to be reported to the NCA and ECs of the member state where they occurred.
In April 2014, the European Union adopted a new Clinical Trials Regulation (EU) No 536/2014, which is set to replace the current Clinical Trials Directive 2001/20/EC. It will overhaul the current system of approvals for clinical trials in the European Union. Specifically, the new legislation, which will be directly applicable in all EU member states (meaning that no national implementing legislation in each European Union member state is required), aims at simplifying and streamlining the approval of clinical trials in the European Union. For instance, the new Clinical Trials Regulation provides for a streamlined application procedure via a single-entry point and strictly defined deadlines for the assessment of clinical trial applications. The new Clinical Trials Regulation became effective on January 31, 2022.
Marketing Authorization
To obtain a marketing authorization for a product in the European Economic Area (comprised of the EU member states plus Norway, Iceland and Liechtenstein), or EEA, an applicant must submit a MAA, either under a centralized procedure administered by the EMA or one of the procedures administered by competent authorities in the EEA member states (decentralized procedure, national procedure, or mutual recognition procedure). A marketing authorization may be granted only to an applicant established in the EEA.
The centralized procedure provides for the grant of a single marketing authorization by the European Commission that is valid throughout the EEA. For those products for which the use of the centralized procedure is not mandatory, applicants may elect to use the centralized procedure where either the product contains a new active substance indicated for the treatment of diseases other than those on the mandatory list, or where the applicant can show that the product constitutes a significant therapeutic, scientific or technical innovation, or for which a centralized process is in the interest of public health.
Under the centralized procedure, the Committee for Medicinal Products for Human use, or the CHMP, which is the EMA’s committee that is responsible for human medicines, established at the EMA is responsible for conducting the assessment of whether a medicine meets the required quality, safety and efficacy requirements, and whether it has a positive benefit/risk profile. Under the centralized procedure, the maximum timeframe for the evaluation of a MAA is 210 days from the receipt of a valid MAA, excluding clock stops when additional information or written or oral explanation is to be provided by the applicant in response to questions of the CHMP. Clock stops may extend the timeframe of evaluation of a MAA considerably beyond 210 days. Where the CHMP gives a positive opinion, it provides the opinion together with supporting documentation to the European Commission, who make the final decision to grant a marketing authorization. Accelerated evaluation may be granted by the CHMP in exceptional cases, when a medicinal product is of major interest from the point of view of public health and, in particular, from the viewpoint of therapeutic innovation. If the CHMP accepts such a request, the timeframe of 210 days for assessment will be reduced to 150 days (excluding clock stops), but it is possible that the CHMP may revert to the standard time limit for the centralized procedure if it determines that the application is no longer appropriate to conduct an accelerated assessment.
92
PRIME Scheme
EMA now offers a scheme that is intended to reinforce early dialogue with, and regulatory support from, EMA in order to stimulate innovation, optimize development and enable accelerated assessment of PRIority MEdicines, or PRIME. It is intended to build upon the scientific advice scheme and accelerated assessment procedure offered by EMA. The scheme is voluntary and eligibility criteria must be met for a medicine to qualify for PRIME.
The PRIME scheme is open to medicines under development and for which the applicant intends to apply for an initial marketing authorization application through the centralized procedure. Eligible products must target conditions for which there is an unmet medical need (there is no satisfactory method of diagnosis, prevention or treatment in the European Union or, if there is, the new medicine will bring a major therapeutic advantage) and they must demonstrate the potential to address the unmet medical need by introducing new methods or therapy or improving existing ones. Applicants will typically be at the exploratory clinical trial phase of development and will have preliminary clinical evidence in patients to demonstrate the promising activity of the medicine and its potential to address to a significant extent an unmet medical need. In exceptional cases, applicants from the academic sector or SMEs (small and medium sized enterprises) may submit an eligibility request at an earlier stage of development if compelling non-clinical data in a relevant model provide early evidence of promising activity, and first in man studies indicate adequate exposure for the desired pharmacotherapeutic effects and tolerability.
If a medicine is selected for the PRIME scheme, EMA:
| ● | appoints a rapporteur from the CHMP or from the Committee for Advanced Therapies (CAT) to provide continuous support and to build up knowledge of the medicine in advance of the filing of a marketing authorization application; |
| ● | issues guidance on the applicant’s overall development plan and regulatory strategy; |
| ● | organizes a kick-off meeting with the rapporteur and experts from relevant EMA committees and working groups; |
| ● | provides a dedicated EMA contact person; and |
| ● | provides scientific advice at key development milestones, involving additional stakeholders, such as health technology assessment bodies and patients, as needed. |
Medicines that are selected for the PRIME scheme are also expected to benefit from EMA’s accelerated assessment procedure at the time of application for marketing authorization. Where, during the course of development, a medicine no longer meets the eligibility criteria, support under the PRIME scheme may be withdrawn.
Regulatory Data Protection in the European Union
In the EEA, innovative medicinal products approved on the basis of a complete independent data package qualify for eight years of data exclusivity upon grant of a marketing authorization and an additional two years of market exclusivity pursuant to Regulation (EC) No. 726/2004, as amended, and Directive 2001/83/EC, as amended. Data exclusivity prevents generic and biosimilar applicants from referencing the innovator’s preclinical and clinical trial data contained in the dossier of the reference product when applying for a marketing authorization for a period of eight years from the date on which the reference product was first authorized in the EEA. During the additional two-year period of market exclusivity, a generic or biosimilar marketing authorization application can be submitted, and the innovator’s data may be referenced, but no generic or biosimilar medicinal product can be marketed until the expiration of the market exclusivity period. The overall 10-year period will be extended to a maximum of 11 years if, during the first eight years of those 10 years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to authorization, is held to bring a significant clinical benefit in comparison with existing therapies. Even if a compound is considered to be an innovative medicinal product so that the innovator gains the prescribed period of data exclusivity, another company may market another version of the product if such company obtained marketing authorization based on a MAA with a completely independent data package of pharmaceutical tests, preclinical tests and clinical trials.
93
Periods of Authorization and Renewals
A marketing authorization is valid for five years, in principle, and it may be renewed after five years on the basis of a re-evaluation of the risk benefit balance by the EMA or by the competent authority of the authorizing member state. To that end, the marketing authorization holder must provide the EMA or the competent authority with a consolidated version of the file in respect of quality, safety and efficacy, including all variations introduced since the marketing authorization was granted, at least nine months before the marketing authorization ceases to be valid. Once renewed, the marketing authorization is valid for an unlimited period, unless the European Commission or the competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal period. Any authorization that is not followed by the placement of the product on the EEA market (in the case of the centralized procedure) or on the market of the authorizing member state within three years after authorization ceases to be valid.
Controlled Drugs Classification
The position in the member states of the European Union is not harmonized. Member states have implemented the relevant U.N. Conventions (the Single Convention on Narcotic Drugs 1961 and the Convention on Psychotropic Substances 1971) into their national legislation, which has led to differences in how controlled substances are regulated in different countries of the European Union. It is therefore important to determine at a national level whether a substance is controlled and to comply with the applicable legal requirements.
Regulatory Requirements After Marketing Authorization
Following approval, the holder of the marketing authorization is required to comply with a range of requirements applicable to the manufacturing, marketing, promotion and sale of the medicinal product.
These include compliance with the European Union’s stringent pharmacovigilance or safety reporting rules, pursuant to which post-authorization studies and additional monitoring obligations can be imposed. The holder of a marketing authorization must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance, who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports, or PSURs.
In addition, all new MAAs must include a risk management plan, or RMP, describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the marketing authorization. Such risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of additional clinical trials or post-authorization safety studies. RMPs and PSURs are routinely available to third parties requesting access, subject to limited redactions.
Furthermore, the manufacturing of authorized products, for which a separate manufacturer’s license is mandatory, must also be conducted in strict compliance with the EMA’s cGMP requirements and comparable requirements of other regulatory bodies in the European Union, which mandate the methods, facilities and controls used in manufacturing, processing and packing of products to assure their safety and identity.
Finally, the marketing and promotion of authorized products, including industry-sponsored continuing medical education and advertising directed toward the prescribers of products, are strictly regulated in the European Union under Directive 2001/83/EC, as amended. The advertising of prescription-only medicines to the general public is not permitted in the European Union, or in the UK under the Human Medicines Regulations 2021. Although general requirements for advertising and promotion of medicinal products are established under EU Directive 2001/83/EC as amended, the details are governed by regulations in each European Union member state (as well as Iceland, Norway and Liechtenstein) and can differ from one country to another.
94
United Kingdom
The United Kingdom (UK) has left the European Union and will declare its independent processes to approve clinical research and marketing authorizations. Since the regulatory framework in the UK covering quality, safety and efficacy of pharmaceutical products, clinical trials, marketing authorization, commercial sales and distribution of pharmaceutical products is derived from EU Directives and Regulations, Brexit could materially impact the future regulatory regime which applies to products and the approval of drug candidates in the UK, as UK legislation now has the potential to diverge from EU legislation. It remains to be seen how Brexit will impact regulatory requirements for drug candidates and products in the UK in the long-term. The MHRA has published detailed guidance for industry and organizations to follow from January 1, 2021, which will be updated as the UK’s regulatory position on medicinal products evolves over time. How precisely clinical research within the UK will be performed and how approval for drugs will be organized is subject to ongoing discussions.
The UK will no longer be covered by centralized marketing authorizations (under the Northern Irish Protocol, centralized marketing authorizations will continue to be recognized in Northern Ireland). All medicinal products with a current centralized marketing authorization were automatically converted to Great Britain marketing authorizations on January 1, 2021. From January 1, 2021 to December 31, 2023, the MHRA may rely on a decision taken by the European Commission on the approval of a new marketing authorization in the centralized procedure, in order to more quickly grant a new Great Britain marketing authorization. A separate application will, however, still be required. As of January 1, 2024, a new marketing recognition framework will apply.
Australia
In Australia, the relevant regulatory body responsible for the pharmaceutical industry is the Therapeutics Goods Administration, or TGA. As with the EMA and FDA there is a harmonization and collaboration between regulatory authorities. The TGA requires notification of all clinical trials via an electronic submission of a Clinical Trial Notification (CTN) prior to commencing the clinical trial.
Third-Party Payer Coverage and Reimbursement
Although our drug candidates have not been commercialized for any indication, if they are approved for marketing, the commercial success of our drug candidates will depend, in part, upon the availability of coverage and reimbursement from third party payers at the federal, state and private levels.
In the United States and internationally, sales of any product that we market in the future, and our ability to generate revenues from such sales, are dependent, in significant part, on the availability of adequate coverage and reimbursement from third party payors, such as state and federal governments, managed care providers and private insurance plans.
Private insurers, such as health maintenance organizations and managed care providers, have implemented cost-cutting and reimbursement initiatives and likely will continue to do so in the future. These include establishing formularies that govern the drugs and biologics that will be offered and the out-of-pocket obligations of member patients for such products. We may need to conduct pharmacoeconomic studies to demonstrate the cost-effectiveness of our drug candidates for formulary coverage and reimbursement. Even with such studies, our drug candidates may be considered less safe, less effective or less cost-effective than existing products, and third-party payors may not provide coverage and reimbursement for our drug candidates, in whole or in part. In addition, particularly in the United States and increasingly in other countries, we are required to provide discounts and pay rebates to state and federal governments and agencies in connection with purchases of our drug candidates that are reimbursed by such entities. It is possible that future legislation in the United States and other jurisdictions could be enacted to potentially impact reimbursement rates for the drug candidates we are developing and may develop in the future and could further impact the levels of discounts and rebates paid to federal and state government entities. Any legislation that impacts these areas could impact, in a significant way, our ability to generate revenues from sales of drug candidates that, if successfully developed, we bring to market. Political, economic and regulatory influences are subjecting the healthcare industry in the United States to fundamental changes. There have been, and we expect there will continue to be, legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our future business.
95
Similar political, economic and regulatory developments are occurring in the European Union and may affect the ability of pharmaceutical companies to profitably commercialize their products. In addition to continuing pressure on prices and cost containment measures, legislative developments at the European Union or member state level may result in significant additional requirements or obstacles. The delivery of healthcare in the European Union, including the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively a matter for national, rather than European Union, law and policy. National governments and health service providers have different priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context. In general, however, the healthcare budgetary constraints in most EU member states have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing EU and national regulatory burdens on those wishing to develop and market products, this could restrict or regulate post-approval activities and affect the ability of pharmaceutical companies to commercialize their products. In international markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. In the future, there may continue to be additional proposals relating to the reform of the healthcare system in the United States and international healthcare systems. Future legislation, or regulatory actions implementing recent or future legislation may have a significant effect on our business. Our ability to successfully commercialize products depends in part on the extent to which reimbursement for the costs of our drug candidates and related treatments will be available in the United States and worldwide from government and health administration authorities, private health insurers and other organizations. The adoption of certain proposals could limit the prices we are able to charge for our drug candidates, the amounts of reimbursement available for our drug candidates, and limit the acceptance and availability of our drug candidates. Therefore, substantial uncertainty exists as to the reimbursement status of newly approved health care products by third party payors.
Data Privacy and Security Laws
Numerous state, federal, and foreign laws, regulations and standards govern the collection, use, access to, confidentiality, and security of health-related and other personal information, and could apply now or in the future to our operations or the operations of our partners. In the United States, numerous federal and state laws and regulations, including data breach notification laws, health information privacy and security laws, and consumer protection laws and regulations govern the collection, use, disclosure, and protection of health-related and other personal information. In addition, certain foreign laws govern the privacy and security of personal data, including health-related data. Privacy and security laws, regulations, and other obligations are constantly evolving, may conflict with each other to complicate compliance efforts, and can result in investigations, proceedings, or actions that lead to significant civil and/or criminal penalties and restrictions on data processing.
Legal Proceedings
We are not involved in any legal or arbitration proceedings that could have a material adverse impact on our financial position or profitability. We are not involved in any governmental proceedings.
Inflation and Seasonality
Management believes inflation has not had a material impact on our operations or financial condition. Management further believes that our operations are not currently subject to seasonal influences due to our current lack of marketed products. Moreover, the targets of our drug candidates, are not seasonal diseases. Accordingly, once we have marketed products, management does not expect that our business will be subject to seasonal influences.
96
Manufacturing and Raw Materials
We have no manufacturing capabilities and are dependent on third parties for cost effective manufacture and manufacturing process development of our drug candidates. Problems with third party manufacturers or the manufacturing process as such may delay or jeopardize clinical trials and commercialization of our drug candidates.
Human Capital Resources
As of June 30, 2024, we had 10 employees. Of these employees, 6 were employed in research and development and 4 were employed in general management and administration. As of the end of fiscal year 2023, we had 10 employees.
Each of our full-time employees has entered into an agreement with an unlimited term. We may only terminate the employment of any of our employees in accordance with the relevant employee’s contract of employment.
Our standard contract of employment for full time employees provides that we can terminate the employment of an employee without notice for serious misconduct or with between one to six months’ notice without cause (as set out in the relevant employee’s contract of employment).
Organizational Structure
Below is a list of our significant subsidiaries, including our ownership percentage, its date of formation and its jurisdiction. These subsidiaries were established to allow us to conduct commercial and clinical operations in Europe and the United States and expand our operations in Australia.
| Subsidiary | Ownership | Date of Formation/Acquisition | Jurisdiction | |||||
| Incannex Healthcare Limited | 100 | % | November 30, 2023 | Victoria, Australia | ||||
| Incannex Pty Ltd | 100 | % | November 30, 2018 | Victoria, Australia | ||||
| Psychennex Pty Ltd | 100 | % | November 20, 2020 | Victoria, Australia | ||||
| APIRx Pharmaceutical USA, LLC | 100 | % | August 5, 2022 | Delaware | ||||
Property, Plants and Equipment
We own computer equipment, office furniture and laboratory equipment, which is primarily placed at our own offices and laboratories.
| Office Location | Lease expiry date | |
| 1/8 Century Circuit, Norwest, NSW 2153 | July 2026 | |
| Suite 909, 401 Docklands Drive, Docklands, VIC 3008 | March 2028 | |
| Ground floor, 18-62 Trenerry Crescent, Abbotsford VIC 3067 | April 2026 | |
| Suite 2, 3 Statesman Parade, Baldivis WA | July 2024 | |
| 221 Dosoris Lane, Glen Cove, NY 11542 | - |
97
Management
Executive Officers and Directors
The following table sets forth the name, age and position of each of our executive officers and directors as of June 30, 2024.
| Name | Age | Position(s) | ||
| Executive Officers and Employee Directors | ||||
| Joel Latham | 35 | Chief Executive Officer, President and Director | ||
| Lekhram Changoer | 58 | Chief Technology Officer | ||
| Joseph Swan | 33 | Chief Financial Officer, Treasurer and Secretary | ||
| Non-Employee Directors | ||||
| Troy Valentine(1)(2) | 50 | Chairman | ||
| Peter Widdows(1)(2) | 58 | Director | ||
| Dr. George Anastassov | 60 | Director | ||
| Robert Clark(1)(2) | 64 | Director |
| (1) | Member of the compensation committee. |
| (2) | Member of the audit committee. |
Executive Officers and Employee Directors
Joel Latham. Joel Latham has been the Chief Executive Officer, President and Director of Incannex since July 2023, and he was the Chief Executive Officer and Managing Director of Incannex Australia from July 2018 to November 2023. Mr. Latham is responsible for the Company’s commercial operations, strategic decision-making, and oversight of all clinical development assets for Incannex. Prior to his appointment as Chief Executive Officer, Mr. Latham had been a key member of our senior leadership team acting as General Manager since 2016. During this time, he was instrumental in the marketing and procurement of multiple revenue-generating opportunities and partnerships, including with Pacific Smiles (ASX:PSQ), 1300 Smiles (ASX: ONT), the National Rugby League, the Australian Football League, ONE Fighting Championship, FIT Technologies and Cannvalate. During his time at the Company, Mr. Latham has been pivotal in the development and execution of Incannex’s drug development and regulatory strategy. Prior to joining Incannex, Mr. Latham had over 14 years’ experience, with major firms such as Mars Foods, Tabcorp and Philip Morris International in management and commercial operational roles.
Lekhram Changoer. Mr. Changoer has been Chief Technology Officer of Incannex since July 2023 and he was the Chief Technology Officer of Incannex Australia from June 2022 to November 2023. He is responsible for the development and implementation of science and technical strategies for clinical and commercial manufacturing of pharmacotherapies. He has been a director of APIRx Pharmaceuticals B.V. since September 2018. Prior to joining Incannex, Mr. Changoer was director at APIRx from January 2019 to August 2022. Previously, Mr. Changoer was CTO and Co-founder of AXIM Biotechnologies since 2014 to 2020.
Joseph Swan. Joseph Swan has been Chief Financial Officer and Secretary since February 2024, Treasurer since July 2023 and was Controller of the Company from July 2023 to February 2024. In addition, Mr. Swan was Head of Finance of Incannex Australia from November 2021 until November 2023, when Incannex Australian re-domiciled to Delaware with the Company becoming the parent entity and Incannex Australia becoming a subsidiary. Prior to joining Incannex Australia, Mr. Swan had been an Audit Supervisor at HLB Mann Judd, an Australian accounting and advisory firm, from May 2020 to November 2021, an internal auditor an INPEX Australia, an energy company, from July 2017 to July 2018 and an analyst at Deloitte Australia from February 2015 to June 2017. Mr. Swan is a chartered accountant in Australia and New Zealand and holds a university degree in commerce and accounting from the University of Western Australia.
Non-Employee Directors
Troy Valentine. Troy Valentine has been Chairman of the Board of Directors since November 2023 and he was Chairman of the Borad of Directors of Incannex Australia from December 2017 to November 2023. Mr. Valentine is a finance professional with managerial and Board experience spanning over 27 years. He commenced his career with Australian brokerage firm Hartley Poynton (now Euroz Hartleys Limited) in 1994 before moving to Patersons Securities (now Canaccord Genuity) in 2000 where he subsequently became an Associate Director. During his time at Patersons, he was responsible for managing both retail and institutional accounts. Mr. Valentine has significant corporate and capital raising experience, especially with start-ups and small to mid-cap size companies. He is currently also a director of Australian boutique corporate advisory firm Alignment Capital Pty Ltd, which he co-founded in 2014.
98
Peter Widdows. Peter Widdows has been a Director since November 2023 and he was a Director of Incannex Australia from 2018 to November 2023. He is a Fellow Chartered Accountant with experience across various functions of business. He has extensive experience in Australian and international consumer goods markets and has worked as a senior executive in numerous geographies, including Europe, the United States and Asia Pacific. In particular, Mr. Widdows served as the Regional Chief Executive Officer — Australasia and Greater China at the H. J. Heinz Company from 2008 to 2010 and as the Chief Executive Officer and Managing Director — Australia at the H. J. Heinz Company from 2002 to 2008 and as the General Manager Strategy & Planning at Starkist Foods Inc. in Cincinnati from 1998 to 2000. Since September 2018, Mr. Widdows has been Chairman of Sunny Queen Australia Ltd, Australia’s largest shell egg and egg-based meal producer and is also a Non-Executive Director of Youi Insurance Holdings Ltd, an Australian general insurance company.
Dr. George Anastassov. Dr. George Anastassov has been a Director since November 2023 and he was a Director of Incannex Australia from June 2022 to November 2023. Dr Anastassov has developed substantial experience regarding liaising and negotiating with FDA and the EMA, due to the fact that he has presented numerous regulatory submissions, including IND meeting packages and IND applications, to regulatory agencies over many years. Dr Anastassov is one of the developers of the first-in-the world cannabinoid-containing chewing gum-based delivery system. Prior to his appointment as a Director, Dr. Anastassov had been the founding managing director of APIRx Pharmaceuticals LLC since 2017 to 2022. whilst also being a key member of the medical and scientific advisory team, assisting with the development of the Combination Compounds. Previously, Dr Anastassov had been CEO and co-founder of AXIM Biotechnologies, which achieved an all-time-high market capitalization of approximately US$1.2 billion, since 2014 to 2018.
Robert Clark. Robert Clark has been a Director since November 2023 and he was a Director of Incannex Australia from August 2022 to November 2023. Mr. Clark is a senior-level strategic regulatory affairs expert with over 38 years of U.S. and international regulatory experience, including more than 20 years with Pfizer Inc. and more than 10 years with Novo Nordisk A/S. He is an expert on FDA and EMA matters, U.S. pharmaceutical advertising practices and regulatory aspects related to healthcare professionals and sales force activities, having contributed to the FDA approval of a notable twelve significant new drugs since 2012. Since May 2012, Mr. Clark has been Vice President, U.S. Regulatory Affairs for Novo Nordisk, where he provides strategic leadership to a team of more than 50 regulatory staff and scientists in the development of new medicines. Prior to his appointment as a Director, Mr. Clark had been Vice President of Worldwide Regulatory Strategy and U.S. Regulatory Affairs at Pfizer from 1992 to 2021, where he led a team of up to 150 regional regulatory professionals supporting the drug development and approval processes.
Board Composition and Election of Directors
Director Independence
Our board of directors currently consists of five members. Our board of directors has determined that Peter Widdows, Robert Clark and George Anastassov are independent directors in accordance with the listing requirements of the Nasdaq Stock Market (Nasdaq). The Nasdaq independence definition includes a series of objective tests, including that the director is not, and has not been for at least three years, one of our employees and that neither the director nor any of his or her family members has engaged in various types of business dealings with us. In addition, as required by Nasdaq rules, our board of directors has made a subjective determination as to each independent director that no relationships exist, which, in the opinion of our board of directors, would interfere with the exercise of independent judgment in carrying out the responsibilities of the director. In making these determinations, our board of directors reviewed and discussed information provided by the directors and us with regard to each director’s business and personal activities and relationships as they may relate to us and our management.
99
Family Relationships
There are no family relationships among any of our executive officers or directors.
Classified Board of Directors
In accordance with the terms of our amended and restated certificate of incorporation, our board of directors will be divided into three classes with staggered, three-year terms. At each annual meeting of stockholders, the directors whose terms then expire will be eligible for reelection until the third annual meeting following reelection. Effective upon the closing of this offering, our directors will be divided among the three classes as follows:
| ● | the Class I directors will be Mr. Peter Widdows, and his term will expire at our first annual meeting of stockholders; |
| ● | the Class II directors will be Dr. George Anastassov and Mr. Robert Clark, and their terms will expire at our second annual meeting of stockholders; and |
| ● | the Class III directors will be Mr. Joel Latham and Mr. Troy Valentine, and their terms will expire at our third annual meeting of stockholders. |
Our amended and restated certificate of incorporation provides that the authorized number of directors may be changed only by resolution of the board of directors. The division of our board of directors into three classes with staggered three-year terms may delay or prevent a change of our board of directors or a change in control of our company. Our directors may be removed only for cause by the affirmative vote of the holders of at least two-thirds of our outstanding voting stock then entitled to vote in an election of directors.
Role of Board in Risk Oversight Process
Our board of directors has responsibility for the oversight of our risk management processes and, either as a whole or through its committees, regularly discusses with management our major risk exposures, their potential impact on our business and the steps we take to manage them. The risk oversight process includes receiving regular reports from board committees and members of senior management to enable our board of directors to understand our risk identification, risk management and risk mitigation strategies with respect to areas of potential material risk, including operations, finance, legal, regulatory, strategic and reputational risk.
The audit committee reviews information regarding liquidity and operations as well as oversees our management of financial risks. Periodically, the audit committee reviews our policies with respect to risk assessment, risk management, loss prevention and regulatory compliance. Oversight by the audit committee includes direct communication with our external auditors, and discussions with management regarding significant risk exposures and the actions management has taken to limit, monitor or control such exposures. The compensation committee is responsible for assessing whether any of our compensation policies or programs have the potential to encourage excessive risk-taking. While each committee is responsible for evaluating certain risks and overseeing the management of such risks, the entire board of directors is regularly informed through committee reports about such risks. Matters of significant strategic risk are considered by our board of directors as a whole.
Board Committees and Independence
Our board of directors has established two standing committees – audit and compensation – each of which operates under a charter that has been approved by our board of directors.
Audit Committee
The audit committee’s main function is to oversee our accounting and financial reporting processes and the audits of our financial statements. This committee’s responsibilities include, among other things:
| ● | appointing our independent registered public accounting firm; |
| ● | evaluating the qualifications, independence and performance of our independent registered public accounting firm; |
| ● | approving the audit and non-audit services to be performed by our independent registered public accounting firm; |
| ● | reviewing the design, implementation, adequacy and effectiveness of our internal accounting controls and our critical accounting policies; |
100
| ● | discussing with management and the independent registered public accounting firm the results of our annual audit and the review of our quarterly unaudited financial statements; |
| ● | reviewing, overseeing and monitoring the integrity of our financial statements and our compliance with legal and regulatory requirements as they relate to financial statements or accounting matters; |
| ● | reviewing with management and our auditors any earnings announcements and other public announcements regarding our results of operations; |
| ● | preparing the report that the SEC requires in our annual proxy statement; |
| ● | reviewing and approving any related party transactions and reviewing and monitoring compliance with our code of conduct and ethics; and |
| ● | reviewing and evaluating, at least annually, the performance of the audit committee and its members including compliance of the audit committee with its charter. |
The members of our audit committee are Mr. Peter Widdows, Mr. Robert Clark and Mr. Troy Valentine. Mr. Widdows serves as the chairperson of the committee. All members of our audit committee meet the requirements for financial literacy under the applicable rules and regulations of the SEC and Nasdaq. Our board of directors has determined that Mr. Widdows is an “audit committee financial expert” as defined by applicable SEC rules and has the requisite financial sophistication as defined under the applicable Nasdaq listing standards. Our board of directors has determined that each of Mr. Widdows and Mr. Clark is independent under the applicable rules of the SEC and Nasdaq. Mr. Valentine is not independent under the applicable rules of the SEC and Nasdaq, however the board of directors has determined that Mr. Valentine’s appointment as a member of the audit committee is in the best interest of the Company and its shareholders, according to Nasdaq Listing Rule 5605(c)(2)(B). Mr. Valentine was appointed on October 5, 2023, and will be able to serve as a member of the audit committee until October 5, 2025, according to Nasdaq Listing Rule 5605(c)(2)(B). The audit committee will operate under a written charter that satisfies the applicable standards of the SEC and Nasdaq.
Compensation Committee
Our compensation committee approves policies relating to compensation and benefits of our officers and employees. The compensation committee approves corporate goals and objectives relevant to the compensation of our Chief Executive Officer and other executive officers, evaluates the performance of these officers in light of those goals and objectives and approves the compensation of these officers based on such evaluations. The compensation committee also approves the issuance of stock options and other awards under our equity plans. The compensation committee will review and evaluate, at least annually, the performance of the compensation committee and its members, including compliance by the compensation committee with its charter.
The members of our compensation committee are Mr. Peter Widdows, Mr. Robert Clark and Mr. Troy Valentine. Mr. Widdows serves as the chairperson of the committee. Our board of directors has determined that each of Mr. Widdows and Mr. Clark is independent under the applicable Nasdaq listing standards and is a ”non-employee director” as defined in Rule 16b-3 promulgated under the Exchange Act. Mr. Valentine is not independent under the applicable rules of the SEC and Nasdaq, however the board of directors has determined that Mr. Valentine’s appointment as a member of the audit committee is in the best interest of the Company and its shareholders, according to Nasdaq Listing Rule 5605(d)(2)(B). Mr. Valentine was appointed on October 5, 2023, and will be able to serve as a member of the audit committee until October 5, 2025, according to Nasdaq Listing Rule 5605(d)(2)(B). The compensation committee will operate under a written charter, which the compensation committee will review and evaluate at least annually.
Prior to the Re-domiciliation, Incannex Australia did not have a compensation committee. Instead, compensation its executive officers was determined by the board of directors. Compensation packages were based on fixed and variable components, determined by the executives’ position, experience and performance. The compensation was not directly based on financial performance, but rather on industry practice, given we operate in the biotechnology sector and our primary focus is research activities with a long-term objective of developing and commercializing the research and development results. During fiscal 2023, there were no interlocking directorships (i.e., no executive officer of Incannex Australia served on a compensation committee or board of directors of another company where an executive officer served as a director of Incannex Australia).
101
Board Diversity
Our board of directors is responsible for reviewing the appropriate characteristics, skills and experience required for the board of directors as a whole and its individual members. In evaluating the suitability of individual candidates (both new candidates and current members) for election or appointment, the board of directors takes into account many factors, including the following:
| ● | personal and professional integrity, ethics and values; |
| ● | experience in corporate management, such as serving as an officer or former officer of a publicly-held company; |
| ● | experience as a board member or executive officer of another publicly-held company; |
| ● | strong finance experience; |
| ● | diversity of expertise and experience in substantive matters pertaining to our business relative to other board members; |
| ● | diversity of background and perspective, including, but not limited to, with respect to age, gender, race, place of residence and specialized experience; |
| ● | experience relevant to our business industry and with relevant social policy concerns; and |
| ● | relevant academic expertise or other proficiency in an area of our business operations. |
Code of Business Conduct and Ethics
We have adopted a written code of business conduct and ethics that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions, which will be effective upon the closing of this offering. Our code of business conduct and ethics is available under the Corporate Governance section of our website at www.incannex.com. In addition, we intend to post on our website all disclosures that are required by law or the listing standards of Nasdaq concerning any amendments to, or waivers from, any provision of the code. We have included our website address in this prospectus solely as an inactive textual reference. The reference to our website address does not constitute incorporation by reference of the information contained at or available through our website, and you should not consider it to be a part of this prospectus.
102
Executive and Director Compensation
Overview
Our named executive officers for fiscal year 2024 are:
| ● | Joel Latham, Chief Executive Officer, President and Director; |
| ● | Lekhram Changoer, Chief Technical Officer; and |
| ● | Joseph Swan, Chief Financial Officer, Treasurer and Secretary. |
This discussion may contain forward-looking statements that are based on our current plans, considerations, expectations, and determinations regarding future compensation programs. Actual compensation programs that we adopt following the closing of this offering may differ materially from the currently planned programs summarized in this discussion.
The following table sets forth information regarding compensation earned with respect to the fiscal year ended June 30, 2024 by our named executive officers.
2024 Summary Compensation Table
| Name and Principal Position | Year | Salary ($) | Bonus ($) | Stock Awards ($)(1) | Option Awards ($)(1) | Non-Equity Incentive Plan Compensation ($) | Nonqualified deferred compensation earnings ($) | All Other Compensation ($) | Total ($) | |||||||||||||||||||||||||
| Joel Latham | 2024 | 941,695 | - | 4,955,360 | 189,429 | - | - | - | 6,086,484 | |||||||||||||||||||||||||
| 2023 | 552,206 | 286,878 | 713,604 | 471,838 | - | - | - | 2,024,526 | ||||||||||||||||||||||||||
| Madhukar Bhalla(2) | 2024 | 41,293 | - | - | - | - | - | - | 41,293 | |||||||||||||||||||||||||
| 2023 | 56,567 | - | - | - | - | - | - | 56,567 | ||||||||||||||||||||||||||
| Lekhram Changoer | 2024 | 137,645 | - | - | - | - | - | - | 137,645 | |||||||||||||||||||||||||
| 2023 | 106,061 | - | - | - | - | - | - | 106,061 | ||||||||||||||||||||||||||
| Joseph Swan | 2024 | 150,214 | 26,873 | 64,663 | 8,417 | - | - | - | 250,167 | |||||||||||||||||||||||||
| 2023 | 85,211 | 10,943 | 35,965 | 24,199 | - | - | - | 158,652 | ||||||||||||||||||||||||||
| (1) | The amounts reported in the “Stock Awards” and “Option Awards” column represent the aggregate grant date fair value of the stock options awarded to our named executive officers during the applicable fiscal year, calculated in accordance with Financial Accounting Standards Board (FASB), Accounting Standards Codification (ASC) Topic 718. The assumptions used in calculating the grant date fair value of the awards reported in this column are set forth in our financial statements included elsewhere in this prospectus. The amounts reported in these columns reflect the accounting cost for the stock awards and stock options and do not reflect the actual economic value that will be realized by the individual upon the vesting of the stock awards and stock options, the vesting of the stock awards and the exercise of the stock options or the sale of the common stock underlying such awards. |
| (2) | Madhukar Bhalla resigned effective February 29, 2024. |
Narrative to Summary Compensation Table
Annual Base Salary
The compensation of our named executive officers is generally determined and approved by our board of directors. The 2024 base salaries of each of our named executive officers are described under the subsection titled “Employment and Service Agreements with our Named Executive Officers” below.
Annual Bonus
In addition to base salaries, our named executive officers are eligible to receive annual performance-based cash bonuses, which are designed to provide appropriate incentives to our executives to achieve annual corporate goals and to reward our executives for individual achievement towards these goals. The annual performance-based bonus each named executive officer is eligible to receive is based on the extent to which we achieve the corporate goals that our board of directors establishes each year. At the end of the year, our board of directors reviews our performance against each corporate goal and determines the extent to which we achieved each of our corporate goals.
103
The corporate goals the board of directors established for 2023 related to regulatory, clinical and development goals, as well as operational objectives. Bonuses are usually determined at the end of the fiscal year and paid in the first quarter of the following fiscal year.
Outstanding Equity Awards
The following table presents information regarding the outstanding stock options held by each of our named executive officers as of June 30, 2024.
| Option Awards | ||||||||||||||||||||
| Name | Grant Date | Number of Securities Underlying Unexercised Options Exercisable | Number of Securities Underlying Unexercised Options Unexercisable | Equity incentive plan awards: Number of securities underlying unexercised unearned options | Option Exercise Price ($) | Option Expiration Date | ||||||||||||||
| Joel Latham | 11/28/23 | 7,500 | — | — | $ | 3.29 | 6/30/2025 | |||||||||||||
| 11/28/23 | 7,500 | — | ) | — | $ | 3.29 | 6/30/2026 | |||||||||||||
| 11/28/23 | 7,500 | — | — | $ | 3.29 | 6/30/2027 | ||||||||||||||
| 11/28/23 | 7,500 | — | — | $ | 3.29 | 6/30/2026 | ||||||||||||||
| 11/28/23 | 7,500 | — | — | $ | 3.29 | 6/30/2027 | ||||||||||||||
| 11/28/23 | 7,500 | — | — | $ | 3.29 | 6/30/2028 | ||||||||||||||
| 11/28/23 | 9,334 | — | — | $ | 17.10 | 7/1/2025 | ||||||||||||||
| 11/28/23 | 9,334 | — | — | $ | 20.39 | 7/1/2026 | ||||||||||||||
| 11/28/23 | 9,334 | ) | — | $ | 23.02 | 7/1/2027 | ||||||||||||||
| 11/28/23 | 9,334 | — | $ | 17.10 | 7/1/2026 | |||||||||||||||
| 11/28/23 | 9,334 | — | $ | 20.39 | 7/1/2027 | |||||||||||||||
| 11/28/23 | 9,334 | — | $ | 23.02 | 7/1/2028 | |||||||||||||||
| 11/28/23 | 15,833 | — | $ | 16.44 | 4/30/2026 | |||||||||||||||
| Madhukar Bhalla(1) | — | — | — | ) | — | — | — | |||||||||||||
| Lekhram Changoer | — | — | — | — | $ | — | — | |||||||||||||
| Joseph Swan | 11/28/23 | 1,000 | — | $ | 17.10 | 7/1/2025 | ||||||||||||||
| 11/28/23 | 1,000 | — | $ | 20.39 | 7/1/2026 | |||||||||||||||
| 11/28/23 | — | 1,000 | — | $ | 23.02 | 7/1/2027 | ||||||||||||||
The following table presents information regarding the outstanding stock awards held by each of our named executive officers as of June 30, 2024.
| Stock Awards | ||||||||||||||||
| Name | Number of shares or units of stock that have not vested | Market Value of shares or units of stock that have not vested ($) | Equity incentive plan awards: number of unearned shares, units or rights that have not vested | Equity incentive plan awards: Market or payout value of unearned shares, units or other rights that have not vested ($) | ||||||||||||
| Joel Latham | 335,000 | 1,373,500 | — | — | ||||||||||||
| Madhukar Bhalla(1) | — | — | — | ) | — | |||||||||||
| Lekhram Changoer | — | — | — | — | ||||||||||||
| Joseph Swan | 33,862 | 133,332 | — | — | ||||||||||||
| (1) | Madhukar Bhalla resigned effective February 29, 2024. |
104
Employment and Service Agreements with Our Executive Officers
We have entered into employment and service agreements with certain of our executive officers, including our named executive officers, which govern the terms of their employment and service with us. Regardless of the manner in which our named executive officers’ employment or service terminates, they are entitled to receive certain accrued amounts previously earned during their term of employment or service, including unpaid salary, reimbursement of expenses owed, and accrued but unpaid paid time off and any continuation of benefits required by applicable law.
Joel Latham
We entered into an employment agreement with Mr. Joel Latham on July 1, 2020. The employment agreement has no fixed term. Each party can terminate at will by giving three months’ notice. However, if the termination is for cause, no notice is required.
Mr. Latham’s compensation consists of a base salary of US$588,000 per year, cash bonus of US$275,000 plus superannuation payments under Australian law for his role as executive officer and US$95,000 as fees for his role as director. Mr. Latham is also entitled to a vehicle allowance of A$20,000 per year.
Lekhram Changoer
We entered into a service agreement with Mr. Lekhram Changoer on July 5, 2022. This service agreement has a 1 year term as of October 10, 2022. Following such date, the agreement continues to be effective unless terminated by either party. Mr. Changoer may terminate the contract by giving a 21-day notice to the Company. We can terminate the contract for cause.
Mr. Changoer is paid a base salary of A$210,000 per year for services as Chief Technical Officer.
Joseph Swan
We entered into an employment agreement with Mr. Joseph Swan on February 27, 2024. Under the terms of his employment contract, Mr. Swan will receive an annual base salary of A$205,000. Mr. Swan will also be eligible to receive up to 20% of his fixed salary, payable in cash, if certain short-term targets are met and up to a total value of A$200,000 of shares of common stock of the Company if certain long-term targets are met. Mr. Swan’s employment contract has no fixed term and can be terminated at will by either party with three months’ notice or for cause by the Company.
401(k) Plan
The Company does not sponsor, nor intend to sponsor in the foreseeable future, the participation of its employees in a plan established under subsection 401(k) of the U.S. Internal Revenue.
Recovery Policy
On October 5, 2023, we adopted a policy on the recovery of erroneously awarded incentive compensation that is compliant with the Nasdaq Listing Rules. This policy is available on our website www.incannex.com under the “Corporate Governance” section of our website.
Equity Incentive Plans
The principal features of Incannex’s equity incentive plan are summarized below. This summary is qualified in its by reference to the actual text of the applicable plan, which is or will be filed as an exhibit to the registration statement of which this prospectus is a part.
105
2023 Equity Incentive Plan
On November 20, 2023, our Board adopted our 2023 Equity Incentive Plan (“Incentive Plan”). The material terms of the Incentive Plan are summarized below.
Purpose. The purpose of the Incentive Plan is to provide a means through which we and our affiliates may attract and retain key personnel and to provide a means whereby our and our affiliate’s directors, employees, and consultants (and prospective directors, employees, and consultants) can acquire and maintain an equity interest in the Company, or be paid incentive compensation, which may, but need not, be measured by reference to the value of our shares of common stock (“Shares”), thereby strengthening their commitment to the success of the Company and its affiliates and aligning their interests with those of our stockholders.
Eligibility and administration. Employees, consultants, and directors of Incannex and its affiliates, as well as prospective employees, consultants, and directors who have accepted offers of employment or consultancy from Incannex or its affiliates are eligible to receive one or more types of Awards under the Incentive Plan.
The Incentive Plan is administered by a committee of two directors (“Committee”). Currently, Messrs. Widdows and Clark are members of the Committee. The Committee has complete authority to determine which employees, consultants, and/or non-employee directors will be granted Awards under the Incentive Plan. The Committee members must qualify as “non-employee directors” under Rule 16b-3 of the Securities Exchange Act of 1934, as amended.
Subject to the terms of the Incentive Plan, the Committee has all discretion and authority to administer the Incentive Plan and to control its operation, in accordance with the Incentive Plan’s provisions, including, but not limited to, the power to (a) determine which employees, consultants, and non-employee directors will be granted Awards, (b) prescribe the terms and conditions of the Awards (which need not be the same), (c) interpret the Incentive Plan and the Awards, (d) adopt such procedures and/or subplans deemed necessary or appropriate for the purpose of satisfying applicable foreign laws or for qualifying for favorable tax treatment under applicable foreign laws, (e) to institute and determine the terms and conditions of an award exchange program; provided, however, that the Committee shall not implement an award exchange program without the approval of the majority of the Company’s stockholders entitled to vote at any annual or special meeting of Company’s stockholders, and (f) make whatever rules it considers appropriate for the administration and interpretation of the Incentive Plan.
The Committee may delegate any of its authority and powers under the Incentive Plan to one or more officers of Incannex. However, the Committee may not delegate its authority and powers with respect to any Awards that are granted to Incannex’s executive officers or directors who are subject to Section 16(b) of the Securities Exchange Act. All interpretations, determinations and decisions made by the Committee, the Board, and any delegate of the Committee will be final and binding on all persons and will be given the maximum possible deference permitted by law.
Limitation on Awards and Shares Available. The maximum number of Shares available for issuance under the Incentive Plan is 5,000,000 Shares (the “Share Reserve”). In no event shall the maximum aggregate number of Shares that may be issued under the Incentive Plan pursuant to incentive stock options exceed the Share Reserve. The Share Reserve is subject to further adjustment as provided in the Incentive Plan. In no event shall fractional Shares be issued under the Incentive Plan. The maximum number of Shares that may be granted under the Incentive Plan during any single fiscal year to a non-employee director, when taken together with any cash fees paid to such non-employee director during such year in respect of his or her service as a non-employee director (including service as a member or chair of any committee of the Board), shall not exceed US$750,000 in total value (calculating the value of any such Awards based on the grant date fair value of such Awards for financial reporting purposes).
In the event there is a specified type of change in our capital structure, such as a stock split, reverse stock split, or recapitalization, appropriate adjustments will be made to (i) the class and maximum number of Shares reserved for issuance under the Incentive Plan and (ii) the class and maximum number of Shares that may be issued on the exercise of ISOs.
Awards. The Incentive Plan permits Incannex to grant various types of discretionary equity compensation awards under the Incentive Plan (“Awards”), including:
| ● | Incentive stock options or ISOs |
| ● | Nonqualified stock options or NSOs |
106
| ● | Stock appreciation rights or SARs |
| ● | Restricted stock |
| ● | Restricted stock units or RSUs |
| ● | Stock bonus awards, and |
| ● | Performance awards. An individual who has received one or more Awards under the Incentive Plan is referred to in this summary as a “participant”. |
A brief description of each award type follows.
| ● | ISOs and NSOs. Stock options provide for the purchase of Shares in the future at an exercise price set by the Committee on the grant date. ISOs are stock options that by their terms qualify for, and are intended to qualify for, favorable U.S. federal tax treatment. NSOs are stock options that by their terms either do not qualify for or are not intended to qualify as ISOs. Incannex may grant ISOs only to employees of Incannex or a subsidiary at the time of grant. The exercise price of each NSO will be determined by the Committee in its discretion but must be at least one hundred percent (100%) of the fair market value of the Shares on the grant date or otherwise compliant with Section 409A of the Internal Revenue Code of 1986, as amended (the “Code”). The exercise price of an ISO must be at least one hundred percent (100%) of the fair market value of the Shares on the grant date (although in rare circumstances, the exercise price must be at least 110% of the fair market value of the Shares on the grant date), except with respect to certain substitute options granted in connection with a corporate transaction. Stock options will not be exercisable after the expiration of 10 years from the date of grant (or 5 years, in the case of an ISO issued to a 10% stockholder). | |
| ● | SARs. SARs entitle the participant, upon exercise, to receive an amount equal to the appreciation of the Shares subject to the Award between the grant date and the exercise date. The exercise price of a SAR will not be less than 100% of the fair market value of the underlying Share on the grant date (except with respect to certain substitute SARs granted in connection with a corporate transaction). SARs will not be exercisable after the expiration of 10 years from the grant date. | |
| ● | Restricted stock and RSUs. Restricted stock is an award of nontransferable Shares that remain forfeitable unless and until specified conditions are met, and which may be subject to a purchase price. RSUs are contractual promises to pay cash or deliver Shares in the future, which may also remain forfeitable unless and until specified conditions are met. Delivery of the shares underlying RSUs may be deferred under the terms of the Award or at the election of the participant, if the Committee permits such a deferral. | |
| ● | Stock bonuses. A stock bonus is the issuance of Shares to a participant. The Shares issued pursuant to a stock bonus typically are unrestricted, meaning that they are not subject to vesting requirements. | |
| ● | Performance awards. Performance awards include any of the foregoing Awards that are granted subject to vesting and/or payment based on the attainment of specified performance goals or other criteria the Committee may determine, which may or may not be objectively determinable. Such performance goals may be based solely by reference to our performance or the performance of a subsidiary, division, business segment or business unit, or based upon performance relative to performance of other companies or upon comparisons of any of the indicators of performance relative to performance of other companies. |
Vesting. The Committee may determine the time and conditions under which the Award will vest and may specify partial vesting in one or more vesting tranches, which may be based solely upon continued employment or service for a specified period of time or may be based upon the achievement of specific performance goals established by the Committee in its discretion.
For all purposes of the Incentive Plan, “vesting” of an Award shall mean:
| ● | For an ISO, NSO, or SAR, the time at which the participant has the right to exercise the Award. |
| ● | For restricted stock or RSUs, the time at which all conditions for vesting, as stated in the applicable award agreement or the Incentive Plan, are satisfied. |
| ● | For performance shares, the time at which the participant has satisfied the requirements to receive payment on such performance shares, as stated in the applicable award agreement or the Incentive Plan. |
107
Vesting need not be uniform among Awards granted at the same time or to persons similarly situated. Vesting requirements shall be set forth in the applicable award agreement.
If the date of the vesting of any Award, other than an ISO, NSO, or SAR, held by participant who is subject to Incannex’s policy regarding trading of its Shares by its officers and directors and the Shares are not within a “window period” applicable to the participant, as determined by Incannex in accordance with such policy, then the vesting of such Award shall not occur on such original vesting date and shall instead occur on the first day of the next “window period” applicable to the participant pursuant to such policy.
Certain transactions; Adjustments. In the event of (i) any dividend (other than ordinary cash dividends) or other distribution (whether in the form of cash, Shares, other securities or other property), recapitalization, stock split, reverse stock split, reorganization, merger, amalgamation, consolidation, spin-off, split-up, split-off, combination, or other similar corporate transaction or event that affects the Shares, or (ii) unusual or infrequently occurring events affecting the Company, any affiliate, or the financial statements of the Company or any affiliate, or changes in applicable rules, rulings, regulations or other requirements of any governmental body or securities exchange or inter-dealer quotation system, accounting principles or law, such that in either case the Committee in its sole discretion may adjust any or all of (A) the number of Shares or other securities of the Company (or number and kind of other securities or other property) that may be delivered in respect of Awards or with respect to which Awards may be granted under the Incentive Plan and (B) the terms of any outstanding Award, including, without limitation, (1) the number of Shares or other securities of the Company (or number and kind of other securities or other property) subject to outstanding Awards or to which outstanding Awards relate, (2) the exercise price with respect to any Award, or (3) any applicable performance measures.
Treatment of Awards Upon a Change in Control. In the event of a “change in control” of the Company, as defined in the Incentive Plan, then unless otherwise provided in an award agreement, the Committee may, in its sole discretion: (i) cancel awards for a cash payment equal to their fair value (as determined in the sole discretion of the Committee), (ii) provide for the issuance of replacement awards, (iii) terminate stock options without providing accelerated vesting, (iv) immediately vest the unvested portion of any Award or (v) take any other action with respect to the awards the Committee deems appropriate. The treatment of awards upon a change in control may vary among participants and types of awards in the Committee’s sole discretion. Awards subject to performance goals shall be settled upon a “change in control” of the Company based upon the extent to which the performance goals underlying such awards have been achieved as determined in the sole discretion of the Committee.
Clawback provisions, transferability, and participant payments. All Awards will be subject to the provisions of any clawback policy implemented by Incannex and to the extent set forth in such clawback policy or in the applicable award agreement. With limited exceptions according to the laws of descent and distribution, Awards under the Incentive Plan are generally nontransferable prior to vesting and are exercisable only by the participant. With regard to tax withholding obligations arising in connection with Awards under the Incentive Plan and exercise price obligations arising in connection with the exercise of stock options under the Incentive Plan, the Committee may, in its discretion, accept cash, wire transfer, or check, shares of our common stock that meet specified conditions (a market sell order) or such other consideration as it deems suitable or any combination of the foregoing.
Incentive Plan amendment and termination. The Board may amend, suspend, or terminate the Incentive Plan at any time; however, Incannex will obtain stockholder approval of any material amendment to the Incentive Plan. No amendment, suspension or termination of the Incentive Plan can, without the consent of the participant, alter or impair any rights or obligations under his or her outstanding Award(s). No award may be granted pursuant to the Incentive Plan after the tenth anniversary of the date on which our Board of directors adopted Incentive Plan.
2023 Australian Incentive Sub-Plan
Our Board and our stockholders have adopted and approved the 2023 Australian Incentive Sub-Plan (“Australian Subplan”), effective as of November 20, 2023.
The Australian Subplan for Australian Participants applies to employees, directors, and consultants of the Company and of its subsidiaries and affiliates, who are either Australian residents or Australian taxpayers, and who shall have been nominated to participate in this Australian Subplan by the Committee (each such person, an “Australian Participant”). Pursuant to Section 15(d) the Incentive Plan, the Committee has the authority to adopt an addendum or subplan for the benefit of Australian Participants. The purpose of this Australian Subplan is to facilitate compliance with Australian tax, securities, and other applicable laws, and to permit the Company to issue Awards to eligible Participants who are Australian residents.
108
All rights granted to Australian Participants will be governed by the terms of the Incentive Plan, when read together with the Australian Subplan (on the basis that, for these purposes, when reading the Incentive Plan, the “Incentive Plan” shall include this Australian Subplan) In the case of an irreconcilable contradiction (as determined by the Committee) between the provisions of this Australian Subplan and the Incentive Plan, the provisions of this Australian Subplan will govern.
Non-Employee Director Compensation
We pay directors’ fees to our non-employee directors for their service on our board of directors. We also have a policy of reimbursing all of our non-employee directors for their reasonable out-of-pocket expenses in connection with attending board of directors and committee meetings.
The following table sets forth information regarding compensation earned with respect to the fiscal year ended June 30, 2024 by each individual who served as a non-employee director during such fiscal year.
| Name | Fees Earned or Paid in Cash ($) | Stock Awards ($) | Option Awards ($) | Non-equity incentive plan compensation ($) | Nonqualified deferred compensation earnings ($) | All Other Compensation ($) | Total ($) | |||||||||||||||||||||
| Troy Valentine | 312,976 | 2,477,681 | 94,715 | - | - | - | 2,885,372 | |||||||||||||||||||||
| Peter Widdows | 283,770 | 343,849 | - | - | - | - | 627,619 | |||||||||||||||||||||
| Dr. George Anastassov | 132,000 | 343,849 | - | - | - | - | 475,849 | |||||||||||||||||||||
| Robert Clark | 75,000 | 343,849 | - | - | - | - | 418,849 | |||||||||||||||||||||
| 1) | The amounts reported in the “Stock Awards” and “Option Awards” column represent the aggregate grant date fair value of the stock options awarded to our named executive officers during the applicable fiscal year, calculated in accordance with Financial Accounting Standards Board (FASB), Accounting Standards Codification (ASC) Topic 718. The assumptions used in calculating the grant date fair value of the awards reported in this column are set forth in our financial statements included elsewhere in this prospectus. The amounts reported in these columns reflect the accounting cost for the stock awards and stock options and do not reflect the actual economic value that will be realized by the individual upon the vesting of the stock awards and stock options, the vesting of the stock awards and the exercise of the stock options or the sale of the common stock underlying such awards. |
Limitations of Liability and Indemnification Matters
Our amended and restated bylaws provide that we will indemnify our directors and officers to the fullest extent permitted by the Delaware General Corporation Law, which prohibits from limiting the liability of our directors for the following:
| ● | any breach of the director’s duty of loyalty to us or our stockholders; |
| ● | acts or omissions not in good faith or that involve intentional misconduct or a knowing violation of law; |
| ● | unlawful payment of dividends or unlawful stock repurchases or redemptions; or |
| ● | any transaction from which the director derived an improper personal benefit. |
Our amended and restated bylaws also provide that the personal liability of a director is limited to the fullest extent permitted by Delaware law. This limitation of liability does not apply to liabilities arising under the federal securities laws and does not affect the availability of equitable remedies such as injunctive relief or rescission.
Our amended and restated bylaws also permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in this capacity, regardless of whether our amended and restated bylaws would permit indemnification. We have obtained directors’ and officers’ liability insurance.
109
We have entered into separate indemnification agreements with our directors and executive officers, in addition to indemnification provided for in our amended and restated certificate of incorporation and amended and restated bylaws. These agreements, among other things, provide for indemnification of our directors and executive officers for expenses, judgments, fines and settlement amounts incurred by this person in any action or proceeding arising out of this person’s services as a director or executive officer or at our request. We believe that these provisions in our amended and restated bylaws and indemnification agreements are necessary to attract and retain qualified persons as directors and executive officers.
The above description of our amended and restated bylaws and our indemnification agreements is not complete and is qualified in its entirety by reference to these documents, each of which is filed as an exhibit to the registration statement of which this prospectus is a part.
The limitation of liability and indemnification provisions may discourage stockholders from bringing a lawsuit against directors for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against directors and officers, even though an action, if successful, might benefit us and our stockholders. A stockholder’s investment may be harmed to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions. Insofar as indemnification for liabilities under the Securities Act may be permitted to directors, officers or persons controlling us pursuant to the foregoing provisions, we have been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable. There is no pending litigation or proceeding naming any of our directors or officers as to which indemnification is being sought, nor are we aware of any pending or threatened litigation that may result in claims for indemnification by any director or officer.
110
Certain Relationships and Related Party Transactions
The following is a description of our related party transactions since July 1, 2021 and we note that all of them were negotiated at arm’s length.
No related party transactions occurred in fiscal year 2024.
During fiscal year 2023, A$247,122 in fees was paid to Cannvalate, an entity in which Dr. Sud Agarwal (resigned June 28, 2022 as director of Incannex Australia) is a director. The fees accrued and were payable in fiscal 2022 with respect to patient research activities conducted by Cannvalate.
During fiscal years 2023 and 2022, respectively, Troy Valentine was paid A$254,000 and A$240,000 for consulting fees invoiced to Incannex Australia, outside of his directors’ fees for his service as director of Incannex Australia. Peter Widdows was also paid A$160,000 in fiscal year 2023 for consulting fees invoiced to Incannex Australia, outside of his directors’ fees for his service as director of Incannex Australia.
In fiscal year 2022, A$407,824 in fees were paid to Alignment Capital Pty Ltd (“Alignment”), an entity controlled by our Chairman Troy Valentine, as consideration for its services as lead manager with respect to Incannex Australia’s ASX-listed options program.
Director and Officer Indemnification
We have entered into indemnification agreements with each of our directors and executive officers. These agreements, among other things, require us or will require us to indemnify each director (and executive officer to the fullest extent permitted by Delaware law, including indemnification of expenses such as attorneys’ fees, judgments, fines and settlement amounts incurred by the director or executive officer in any action or proceeding, including any action or proceeding by or in right of us, arising out of the person’s services as a director or executive officer.
Our amended and restated bylaws provide that we will indemnify each of our directors and officers to the fullest extent permitted by the Delaware General Corporation Law. Further, we have purchased a policy of directors’ and officers’ liability insurance that insures our directors and officers against the cost of defense, settlement or payment of a judgment under certain circumstances. For further information, see the section titled “Executive and Director Compensation—Limitations of Liability and Indemnification Matters.”
Policies and Procedures for Related Person Transactions
Our board of directors has adopted a written related person transaction policy setting forth the policies and procedures for the review and approval or ratification of related person transactions. This policy covers, with certain exceptions set forth in Item 404 of Regulation S-K under the Securities Act, any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships in which we were or are to be a participant, where the amount involved exceeds the lesser of $120,000 or one percent of the average of our total assets at year-end for the last two completed fiscal years, and a related person had or will have a direct or indirect material interest, including, without limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness and employment by us of a related person. In reviewing and approving any such transactions, our audit committee is tasked to consider all relevant facts and circumstances, including, but not limited to, whether the transaction is on terms comparable to those that could be obtained in an arm’s length transaction and the extent of the related person’s interest in the transaction. All of the transactions described above occurred prior to the adoption of this policy.
111
Principal Shareholders
The following table sets forth information with respect to the beneficial ownership of our common stock as of June 30, 2024.
The number of shares beneficially owned by each stockholder is determined under rules issued by the SEC. Under these rules, beneficial ownership includes any shares as to which a person has sole or shared voting power or investment power. Applicable percentage ownership is based on 17,642,832 shares of common stock outstanding on June 30, 2024. In computing the number of shares beneficially owned by a person and the percentage ownership of that person, shares of common stock subject to options or other rights held by such person that are currently exercisable or that will become exercisable or otherwise vest within 60 days of June 30, 2024, are considered outstanding, although these shares are not considered outstanding for purposes of computing the percentage ownership of any other person. The table below excludes any potential purchases in this offering by the beneficial owners identified in the table below.
Unless otherwise indicated, the address of Joel Latham, Troy Valentine, Peter Widdows and Joseph Swan is 105/8 Century Circuit, Norwest, New South Wales, Australia 2153, and the address of Dr. George Anastassov, Robert Clark and Lekhram Changoer is 221 Dosoris Lane, Glen Cove, NY 11542. We believe, based on information provided to us, that each of the stockholders listed below has sole voting and investment power with respect to the shares beneficially owned by the stockholder unless noted otherwise, subject to community property laws where applicable.
| Amount and Nature of | Percentage of Shares Beneficially Owned | |||||||||||
| Name of Beneficial Owner | Beneficial Ownership | Before Offering | After Offering | |||||||||
| Named Executive Officers and Directors | ||||||||||||
| Joel Latham(1) | 1,684,988 | 9.31 | % | % | ||||||||
| Troy Valentine(2) | 1,084,284 | 5.96 | % | % | ||||||||
| Peter Widdows(3) | 281,897 | 1.60 | % | % | ||||||||
| Dr. George Anastassov(4) | 769,721 | 4.36 | % | % | ||||||||
| Robert Clark(5) | 150,000 | * | % | |||||||||
| Lekhram Changoer(6) | 639,549 | 3.62 | % | % | ||||||||
| Joseph Swan(7) | 70,724 | * | % | |||||||||
| All executive officers and directors as a group (7 persons) | 4,681,163 | 26.01 | % | % | ||||||||
| * | Less than 1%. |
| (1) | Joel Latham owns 1,242,485 shares of common stock, 335,000 restricted shares and 107,503 shares of common stock underlying warrants. |
| (2) | Troy Valentine owns 531,750 shares of common stock in his own name. Troy Valentine also owns a 50% equity interest in Alignment Capital Pty Ltd, which owns 242,862 shares of common stock of Incannex. Troy Valentine is a director of Tranaj Nominees Pty Ltd, which owns 10,000 shares of common stock in Incannex. Troy Valentine is a director of Valplan Pty Ltd, which owns 30,000 shares of common stock in Incannex. Troy Valentine is a director and the sole shareholder of Cityside Pty Ltd, which owns 44,400 shares of common stock of Incannex. Troy Valentine is the beneficiary of the GFCR Investments Trust managed by Ekirtson Nominees Pty Ltd as trustee, which owns 10,000 shares of common stock in Incannex. Troy Valentine also owns 167,500 restricted shares and 47,772 shares of common stock underlying warrants. |
| (3) | Peter Widdows owns 245,847 shares of common stock, 25,000 restricted shares and 11,050 shares of common stock underlying warrants. |
| (4) | Dr George Anastassov owns 744,721 shares of common stock and 25,000 restricted shares. |
| (5) | Robert Clark owns 75,000 shares of common stock, 25,000 restricted shares and 50,000 shares of common stock underlying warrants. |
| (6) | Lekhram Changoer owns 639,549 shares of common stock held by Prash BV, a company controlled by Mr. Changoer. |
| (7) | Joseph Swan owns 1,000 shares of common stock, 50,793 restricted shares and 2,000 shares of common stock underlying warrants. |
112
Description of Capital STOCK
The following description summarizes some of the terms of our amended and restated certificate of incorporation and amended and restated bylaws that will be in effect upon the closing of this offering, our investors’ rights agreement and of the Delaware General Corporation Law. Because it is only a summary, it does not contain all the information that may be important to you. For a complete description you should refer to our amended and restated certificate of incorporation, amended and restated bylaws and our investors’ rights agreement, copies of which have been filed as exhibits to the registration statement of which this prospectus is a part.
As of June 30, 2024, there were 17,642,832 shares of our common stock outstanding and held of record by 4,991 stockholders. Our authorized capital stock consists of 100,000,000 shares of common stock, $0.0001 par value per share, and 10,000,000 shares of preferred stock, $0.0001 par value per share.
General
Under the Certificate of Incorporation, the Company is authorized to issue up to 100,000,000 shares of common stock and 10,000,000 shares of preferred stock, par value $0.0001 per share.
Common Stock
Voting Rights. The holders of our common stock are entitled to one vote per share on all matters on which stockholders are generally entitled to vote; provided, however, that, except as otherwise required by law, holders of common stock, as such, are not entitled to vote on any amendment to the Certificate of Incorporation that relates solely to the terms of one or more outstanding series of preferred stock if the holders of such affected series are entitled, either separately or together with the holders of one or more other such series, to vote thereon pursuant to the Certificate of Incorporation. Holders of our common stock do not have cumulative voting rights in the election of directors. Accordingly, the holders of a majority of the combined voting power of our common stock could, if they so choose, elect all the directors.
Dividends. Subject to the rights of the holders of any outstanding series of preferred stock, holders of common stock are entitled to receive any dividends to the extent permitted by law when, as and if declared by our board of directors.
Liquidation. Upon our dissolution, liquidation or winding up of the Company, subject to the rights of the holders of any outstanding series of preferred stock, the holders of shares of common stock are entitled to receive the assets of the Company available for distribution to its stockholders ratably in proportion to the number of shares held by them.
Other Matters. The Certificate of Incorporation does not entitle holders of our common stock to preemptive or conversion rights or other subscription rights. There are no redemption or sinking fund provisions applicable to our common stock. The common stock may be subdivided or combined in any manner unless the other class is subdivided or combined in the same proportion. All outstanding shares of our common stock are fully paid and non-assessable.
Authorized but unissued Preferred Stock
Unless required by law or by any stock exchange on which our common stock may be listed, the authorized shares of preferred stock will be available for issuance without further action by our stockholders. Delaware law does not require stockholder approval for any issuance of authorized shares. However, the listing requirements of Nasdaq, which apply as long as our common stock is listed on Nasdaq, require stockholder approval of certain issuances equal to or exceeding 20% of the combined voting power of our common stock. These additional shares may be used for a variety of corporate purposes, including future public offerings to raise additional capital, acquisitions and employee benefit plans.
Our Certificate of Incorporation authorizes our board of directors to establish from time to time the number of shares to be included in each series of preferred stock, and to fix the designation, powers, preferences, and relative, participating, optional or other rights, if any, and the qualifications, limitations or restrictions, if any, of the shares of each series of preferred stock. Our board of directors is also able to increase or decrease the number of authorized shares of any series of preferred stock (but not below the number of shares of that series of preferred stock then outstanding) without any further vote or action by the stockholders.
113
The existence of unissued and unreserved common stock or preferred stock may enable our board of directors to issue shares to persons friendly to current management, which could render more difficult or discourage an attempt to obtain control of the Company by means of a merger, tender offer, proxy contest or otherwise, and could thereby protect the continuity of our management and possibly deprive stockholders of opportunities to sell their shares of common stock at prices higher than prevailing market prices.
Anti-Takeover Effects of Delaware Law, the Certificate of Incorporation and the Bylaws
Certain provisions of Delaware law, the Certificate of Incorporation and the Bylaws could make the acquisition of the Company more difficult and could delay, defer or prevent a tender offer or other takeover attempt that a stockholder might consider to be in its best interest, including takeover attempts that might result in the payment of a premium to stockholders over the market price for their shares. These provisions also may promote the continuity of our management by making it more difficult for a person to remove or change the incumbent members of our board of directors.
Authorized but Unissued Shares; Undesignated Preferred Stock. The authorized but unissued shares of our common stock are available for future issuance without stockholder approval except as required by law or by any stock exchange on which our common stock may be listed. These additional shares may be utilized for a variety of corporate purposes, including future public offerings to raise additional capital, acquisitions and employee benefit plans. In addition, our board of directors may authorize, without stockholder approval, the issuance of undesignated preferred stock with voting rights or other rights or preferences designated from time to time by our board of directors. The existence of authorized but unissued shares of common stock or preferred stock may enable our board of directors to render more difficult or to discourage an attempt to obtain control of us by means of a merger, tender offer, proxy contest or otherwise.
Board Classification. The Certificate of Incorporation provides that our board of directors is divided into three classes of directors, with the classes to be as nearly equal in number as possible, and with the directors serving three-year terms. As a result, approximately one-third of our board of directors is elected each year. The classification of directors has the effect of making it more difficult for stockholders to change the composition of our board of directors. The Certificate of Incorporation and the Bylaws provide that, subject to any rights of holders of preferred stock to elect additional directors under specified circumstances, the number of directors may be fixed from time to time exclusively pursuant to a resolution adopted by our board of directors.
No Cumulative Voting. Holders of our common stock do not have cumulative voting rights in the election of directors.
Special Meetings of Stockholders. The Certificate of Incorporation and the Bylaws provide that special meetings of our stockholders may be called only by our board of directors. Only such business shall be conducted at a special meeting of stockholders as shall have been brought before the meeting by or at the direction of our board of directors.
Stockholder Action by Written Consent. Pursuant to Section 228 of the DGCL, any action required to be taken at any annual or special meeting of the stockholders may be taken without a meeting, without prior notice and without a vote if a consent or consents in writing, setting forth the action so taken, is signed by the holders of outstanding stock having not less than the minimum number of votes that would be necessary to authorize or take such action at a meeting at which all shares of our stock entitled to vote thereon were present and voted, unless our certificate of incorporation provides otherwise. The Certificate of Incorporation precludes stockholder action by written consent.
Advance Notice Requirements for Stockholder Proposals and Nomination of Directors. The Bylaws require stockholders seeking to bring business before an annual meeting of stockholders, or to nominate individuals for election as directors at an annual or special meeting of stockholders, to provide timely notice in writing. To be timely, a stockholder’s notice must be delivered to the secretary at our principal executive offices not later than the close of business on the 90th day nor earlier than the close of business on the 120th day, prior to the anniversary of the preceding year’s annual meeting. However, in the event that the date of the annual meeting is more than 30 days before or more than 60 days after such anniversary date, such notice will be timely only if delivered not earlier than the close of business on the 120th day prior to such annual meeting and not later than the close of business on the later of the 90th day prior to such annual meeting or the tenth day following the date on which a public announcement of the date of such annual meeting is first made by us. The Bylaws also specify requirements as to the form and content of a stockholder’s notice. These provisions may preclude our stockholders from bringing matters before our annual meeting of stockholders or from making nominations for directors at our meetings of stockholders. These provisions may also discourage or deter a potential acquiror from conducting a solicitation of proxies to elect the potential acquiror’s own slate of directors or otherwise attempting to obtain control of the Company.
114
Removal of Directors; Vacancies. Under the DGCL, unless otherwise provided in the Certificate of Incorporation, directors serving on a classified board may be removed by the stockholders only for cause. The Certificate of Incorporation provides that directors may only be removed for cause and only by the affirmative vote of holders of at least 66 2/3% in the voting power of the stock outstanding and entitled to vote thereon. In addition, the Certificate of Incorporation also provides that any newly created directorship on our board of directors resulting from any increase in the authorized number of directors and any vacancies in our board of directors may be filled solely by the affirmative vote of a majority of the remaining directors then in office, even though less than a quorum, or by the sole remaining director (and not by the stockholders).
Supermajority Provisions. The Certificate of Incorporation and the Bylaws provide that our board of directors is expressly authorized to adopt, amend or repeal the Bylaws without a stockholder vote. Any adoption, amendment or repeal of the Bylaws by our stockholders requires the affirmative vote of the holders of at least 66 2/3% of the voting power of the stock outstanding and entitled to vote thereon, voting together as a single class.
The DGCL provides generally that the affirmative vote of a majority of the outstanding shares entitled to vote thereon, voting together as a single class, is required to amend a corporation’s certificate of incorporation, unless the certificate of incorporation requires a greater percentage. The Certificate of Incorporation provides that the affirmative vote of at least 66 2/3% of the voting power of the stock outstanding and entitled to vote thereon, voting together as a single class, is required to amend or repeal, or adopt any provision inconsistent with, the following provisions in the Certificate of Incorporation, among others:
| ● | the provisions providing for a classified board of directors (the election and term of our directors); | |
| ● | the provisions regarding removal of directors; | |
| ● | the provisions regarding filling vacancies on our board of directors and newly created directorships; | |
| ● | the provisions regarding calling special meetings of stockholders; | |
| ● | the provision requiring a 66 2/3% supermajority vote for stockholders to amend the Bylaws; | |
| ● | the provisions eliminating monetary damages for breaches of fiduciary duty by a director; and | |
| ● | the amendment provision requiring that the above provisions be amended only with a 66 2/3% supermajority vote. |
Section 203 of the Delaware General Corporation Law. The Certificate of Incorporation provides that we are not governed by, or otherwise subject to, Section 203 of the DGCL.
Transfer Agent and Registrar
The transfer agent and registrar for the Company’s common stock is Computershare Trust Company, N.A. The transfer agent and registrar’s address is 250 Royall St., Canton, Massachusetts 02021.
115
DESCRIPTION OF SECURITIES WE ARE OFFERING
We are offering up to shares of our common stock or pre-funded warrants to purchase up to shares of our common stock together with common warrants to purchase up to shares of common stock. For each pre-funded warrant we sell, the number of shares of common stock we are offering will be decreased on a one-for-one basis. Each share of common stock or pre-funded warrant is being sold together with a common warrant to purchase one share of common stock. The shares of common stock or pre-funded warrants and accompanying common warrants will be issued separately and will be immediately separable upon issuance but must be purchased together in this offering. We are also registering the shares of common stock issuable from time to time upon exercise of the common warrants and pre-funded warrants offered hereby.
Common Stock
The material terms and provisions of our common stock are described under the caption “Description of Capital Stock” in this prospectus and are incorporated herein by reference.
Pre-Funded Warrants
The following summary of certain terms and provisions of the pre-funded warrants that are being offered hereby is not complete and is subject to, and qualified in its entirety by, the provisions of the pre-funded warrant, the form of which is filed as an exhibit to the registration statement of which this prospectus forms a part. Prospective investors should carefully review the terms and provisions of the form of pre-funded warrant for a complete description of the terms and conditions of the pre-funded warrants.
Duration and Exercise Price
Each pre-funded warrant offered hereby will have an initial exercise price per share equal to $0.001. The pre-funded warrants will be immediately exercisable and may be exercised at any time until the pre-funded warrants are exercised in full. The exercise price and number of shares of common stock issuable upon exercise is subject to appropriate adjustment in the event of stock dividends, stock splits, reorganizations or similar events affecting our common stock and the exercise price.
Exercisability
Each pre-funded warrant may be exercised, in cash or by a cashless exercise at the election of the holder at any time following the date of issuance and from time to time thereafter until the pre-funded warrants are exercised in full. The pre-funded warrants will be exercisable in whole or in part by delivering to the Company a completed instruction form for exercise and complying with the requirements for exercise set forth in the pre-funded warrant. Payment of the exercise price may be made in cash or pursuant to a cashless exercise, in which case the holder would receive upon such exercise the net number of shares of common stock determined according to the formula set forth in the pre-funded warrant.
Cashless Exercise
At the time a holder exercises its pre-funded warrants, in lieu of making the cash payment otherwise contemplated to be made to us upon such exercise in payment of the aggregate exercise price, the holder may elect instead to receive upon such exercise (either in whole or in part) the net number of shares of common stock determined according to a formula set forth in the pre-funded warrants.
Exercise Limitation
In general, a holder will not have the right to exercise any portion of a pre-funded warrant if the holder (together with its Attribution Parties (as defined in the pre-funded warrant)) would beneficially own in excess of 4.99% or 9.99%, at the election of the holder, of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the pre-funded warrant. However, any holder may increase or decrease such percentage to any other percentage not in excess of 9.99% upon notice to us, provided, that any increase in this limitation will not be effective until 61 days after such notice from the holder to us and such increase or decrease will apply only to the holder providing such notice.
116
Transferability
Subject to applicable laws, a pre-funded warrant may be transferred at the option of the holder upon surrender of the pre-funded warrant to us together with the appropriate instruments of transfer.
Fractional Shares
No fractional shares of common stock will be issued upon the exercise of the pre-funded warrants. Rather, the number of shares of common stock to be issued will, at our election, either be rounded up to the nearest whole number or we will pay a cash adjustment in respect of such final fraction in an amount equal to such fraction multiplied by the exercise price.
Trading Market
There is no trading market available for the pre-funded warrants on any securities exchange or nationally recognized trading system.
Right as a Stockholder
Except as otherwise provided in the pre-funded warrants or by virtue of such holder’s ownership of shares of our common stock, the holders of the pre-funded warrants do not have the rights or privileges of holders of our common stock, including any voting rights, until they exercise their pre-funded warrants.
Warrant Agent
The pre-funded warrants are expected to be issued in registered form under a warrant agreement between Computershare Inc. and its affiliate, Computershare Trust Company, N.A., collectively, as warrant agent, and us.
Common Warrants
The following summary of certain terms and provisions of the common warrants that are being offered hereby is not complete and is subject to, and qualified in its entirety by, the provisions of the common warrant, the form of which is filed as an exhibit to the registration statement of which this prospectus forms a part. Prospective investors should carefully review the terms and provisions of the form of common warrant for a complete description of the terms and conditions of the common warrants.
We are offering common warrants to purchase up to an aggregate of shares of our common stock.
Each common warrant issued in this offering represents the right to purchase one share of common stock at an initial exercise price of $ per share. Each common warrant may be exercised, in cash or by a cashless exercise at the election of the holder immediately upon issuance and from time to time thereafter through and including the fifth anniversary of the initial exercise date.
The common warrants will be exercisable in whole or in part by delivering to the Company a completed instruction form for exercise and complying with the requirements for exercise set forth in the common warrant. Payment of the exercise price may be made in cash or pursuant to a cashless exercise, in which case the holder would receive upon such exercise the net number of shares of common stock determined according to the formula set forth in the common warrants.
117
No Fractional Shares
No fractional shares or scrip representing fractional shares shall be issued upon the exercise of the common warrants. As to any fraction of a share which the holder would otherwise be entitled to purchase upon such exercise, the number of shares of common stock to be issued shall be rounded up to the nearest whole number.
Exercise Limitation
In general, a holder will not have the right to exercise any portion of a common warrant if the holder (together with its Attribution Parties (as defined in the common warrant)) would beneficially own in excess of 4.99% or 9.99%, at the election of the holder, of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the common warrant. However, any holder may increase or decrease such percentage to any other percentage not in excess of 9.99% upon notice to us, provided that any increase in this limitation will not be effective until 61 days after such notice from the holder to us and such increase or decrease will apply only to the holder providing such notice.
Cashless Exercise
If, at the time a holder exercises its common warrants, a registration statement registering the issuance of the shares of common stock underlying the common warrants under the Securities Act, is not then effective or available for the issuance of such shares, then in lieu of making the cash payment otherwise contemplated to be made to us upon such exercise in payment of the aggregate exercise price, the holder may elect instead to receive upon such exercise (either in whole or in part) the net number of shares of common stock determined according to a formula set forth in the common warrant.
Adjustment for Stock Splits
The exercise price and the number of shares of common stock purchasable upon the exercise of the common warrants are subject to adjustment upon the occurrence of specific events, including sales of additional shares of common stock, stock dividends, stock splits, and combinations of our common stock.
Dividends or Distributions
If we declare or make any dividend or other distribution of its assets (or rights to acquire its assets) to holders of shares of our common stock, by way of return of capital or otherwise (including, without limitation, any distribution of cash, stock or other securities, property, options, evidence of indebtedness or any other assets by way of a dividend, spin off, reclassification, corporate rearrangement, scheme of arrangement or other similar transaction) at any time after the issuance of the common warrants, then, in each such case, the holders of the common warrants shall be entitled to participate in such distribution to the same extent that the holders would have participated therein if the holders had held the number of shares of common stock acquirable upon complete exercise of the common warrants.
Purchase Rights
If we grant, issue or sell any shares of our common stock or securities exercisable for, exchangeable for or convertible into our common stock, or rights to purchase stock, common warrants, securities or other property pro rata to the record holders of any class of shares of our common stock, referred to as Purchase Rights, then each holder of the common warrants will be entitled to acquire, upon the terms applicable to such Purchase Rights, the aggregate Purchase Rights which the holder could have acquired if the holder had held the number of shares of common stock acquirable upon complete exercise of the common warrants immediately before the record date, or, if no such record is taken, the date as of which the record holders of shares of common stock are to be determined, for the grant, issue or sale of such Purchase Rights.
118
Fundamental Transactions
In the event of a fundamental transaction, as described in the common warrants and generally including any reorganization, recapitalization or reclassification of our common stock, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, the consummation of a business combination with another person or group of persons whereby such other person or group acquires greater than 67% of the voting power of the outstanding common stock and preferred stock, the holders of the common warrants will be entitled to receive upon exercise of the common warrants the kind and amount of securities, cash or other property that the holders would have received had they exercised the common warrants immediately prior to such fundamental transaction.
Transferability
Subject to applicable laws, the common warrants may be offered for sale, sold, transferred or assigned. There is currently no trading market for the common warrants and a trading market is not expected to develop.
Rights as a Stockholder
Except as otherwise provided in the common warrants or by virtue of a holder’s ownership of shares of our common stock, the holders of the common warrants do not have the rights or privileges of holders of our common stock, including any voting rights, unless and until they exercise their common warrants.
Amendments
The common warrants may be amended with the written consent of the holder of such common warrant and us.
Listing
There is no established public trading market for the common warrants, and we do not expect a market to develop. In addition, we do not intend to apply for listing of the common warrants on any national securities exchange.
Warrant Agent
The common warrants are expected to be issued in registered form under a warrant agreement between Computershare Inc. and its affiliate, Computershare Trust Company, N.A., collectively, as warrant agent, and us.
119
MATERIAL UNITED STATES FEDERAL INCOME TAX CONSEQUENCES
The following discussion is a summary of material U.S. federal income tax considerations generally applicable to the purchase, ownership and disposition of our common stock, pre-funded warrants and common warrants. Throughout this summary, all references to our common stock are meant to include our pre-funded warrants and common warrants. The common stock, pre-funded warrants and common warrants are collectively referred to herein as our securities. All prospective holders of our securities should consult their tax advisors with respect to the U.S. federal, state, local, and non-U.S. tax consequences of the purchase, ownership, and disposition of our securities. This discussion is not a complete analysis of all potential U.S. federal income tax consequences relating to the purchase, ownership, and disposition of our securities. This summary is based upon current provisions of the Code, existing U.S. Treasury Regulations promulgated thereunder, published administrative pronouncements, and rulings of the U.S. Internal Revenue Service (the “IRS”), and judicial decisions, all as in effect as of the date of this prospectus. These authorities are subject to change and differing interpretation, possibly with retroactive effect. Any change or differing interpretation could alter the tax consequences to holders described in this discussion. There can be no assurance that a court or the IRS will not challenge one or more of the tax consequences described herein, and we have not obtained, nor do we intend to obtain, a ruling with respect to the U.S. federal income tax consequences to a holder of the purchase, ownership, or disposition of our securities. We assume in this discussion that a holder holds our securities as a “capital asset” within the meaning of Section 1221 of the Code (generally, property held for investment). This discussion does not address all aspects of U.S. federal income taxation that may be relevant to a particular holder in light of that holder’s individual circumstances, nor does it address the special tax accounting rules under Section 451(b) of the Code, any alternative minimum, Medicare contribution, estate or gift tax consequences, or any aspects of U.S. state, local or non-U.S. taxes, or any other U.S. federal tax laws. This discussion also does not address consequences relevant to holders subject to special tax rules, such as holders that own, or are deemed to own, more than 5% of our capital stock (except to the extent specifically set forth below), corporations that accumulate earnings to avoid U.S. federal income tax, tax-exempt organizations, governmental organizations, banks, financial institutions, investment funds, insurance companies, brokers, dealers or traders in securities, commodities or currencies, regulated investment companies or real estate investment trusts, persons that have a “functional currency” other than the U.S. dollar, tax-qualified retirement plans, holders who hold or receive our securities pursuant to the exercise of employee stock options or otherwise as compensation, holders holding our securities as part of a hedge, straddle, or other risk reduction strategy, conversion transaction or other integrated investment, holders deemed to sell our securities under the constructive sale provisions of the Code, passive foreign investment companies, controlled foreign corporations, and certain former U.S. citizens or long-term residents.
In addition, this discussion does not address the tax treatment of partnerships (or entities or arrangements that are treated as partnerships for U.S. federal income tax purposes) or persons that hold our securities through such partnerships. If a partnership, including any entity or arrangement treated as a partnership for U.S. federal income tax purposes, holds our securities, the U.S. federal income tax treatment of a partner in such partnership will generally depend upon the status of the partner and the activities of the partnership. Such partners and partnerships should consult their tax advisors regarding the tax consequences of the purchase, ownership and disposition of our securities.
For purposes of this discussion, a “U.S. Holder” means a beneficial owner of our securities (other than a partnership or an entity or arrangement treated as a partnership for U.S. federal income tax purposes) that is, for U.S. federal income tax purposes:
| ● | an individual who is a citizen or resident of the United States; | |
| ● | a corporation, or an entity treated as a corporation for U.S. federal income tax purposes, created or organized in the United States or under the laws of the United States or of any state thereof or the District of Columbia; | |
| ● | an estate, the income of which is subject to U.S. federal income tax regardless of its source; or | |
| ● | a trust if (a) a U.S. court can exercise primary supervision over the trust’s administration and one or more U.S. persons have the authority to control all of the trust’s substantial decisions or (b) the trust has a valid election in effect under applicable U.S. Treasury Regulations to be treated as a U.S. person. |
For purposes of this discussion, a “non-U.S. Holder” is a beneficial owner of our securities that is neither a U.S. Holder nor a partnership or an entity or arrangement treated as a partnership for U.S. federal income tax purposes.
120
Tax Considerations Applicable to U.S. Holders
Taxation of Distributions
If we pay distributions or make constructive distributions (other than certain distributions of our stock or rights to acquire our stock) to U.S. Holders of shares of our Common Stock, such distributions generally will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. Distributions in excess of our current and accumulated earnings and profits will constitute a return of capital that will be applied against and reduce (but not below zero) the U.S. Holder’s adjusted tax basis in our Common Stock. Any remaining excess will be treated as gain realized on the sale or other disposition of the Common Stock and will be treated as described under “Tax Considerations Applicable to U.S. Holders—U.S. Holders—Gain or Loss on Sale, Taxable Exchange, or Other Taxable Disposition of Common Stock” below.
Dividends we pay to a U.S. Holder that is a taxable corporation will generally qualify for the dividends received deduction (or a partial deduction for dividends received) if the requisite holding period is satisfied. With certain exceptions (including dividends treated as investment income for purposes of investment interest deduction limitations), and provided certain holding period requirements are met, dividends we pay to a non-corporate U.S. Holder will generally constitute “qualified dividends” that will be subject to tax at the maximum tax rate accorded to long-term capital gains. If the holding period requirements are not satisfied, a corporation may not be able to qualify for the dividends received deduction and would have taxable income equal to the entire dividend amount, and non-corporate holders may be subject to tax on such dividend at ordinary income tax rates instead of the preferential rates that apply to qualified dividend income.
Gain or Loss on Sale, Taxable Exchange, or Other Taxable Disposition of Common Stock
A U.S. Holder generally will recognize gain or loss on the sale, taxable exchange, or other taxable disposition of our Common Stock. Any such gain or loss will be capital gain or loss and will be long-term capital gain or loss if the U.S. Holder’s holding period for the Common Stock so disposed of exceeds one year. The amount of gain or loss recognized will generally be equal to the difference between (1) the sum of the amount of cash and the fair market value of any property received in such disposition and (2) the U.S. Holder’s adjusted tax basis in its Common Stock so disposed of. A U.S. Holder’s adjusted tax basis in its Common Stock will generally equal the U.S. Holder’s acquisition cost for such Common Stock, less any prior distributions treated as a return of capital. Long-term capital gains recognized by non-corporate U.S. Holders are generally eligible for reduced rates of tax. If the U.S. Holder’s holding period for the Common Stock so disposed of is one year or less, any gain on a sale or other taxable disposition of the shares would be subject to short-term capital gain treatment and would be taxed at ordinary income tax rates. The deductibility of capital losses is subject to limitations.
Information Reporting and Backup Withholding.
In general, information reporting requirements may apply to dividends paid to a U.S. Holder and to the proceeds of the sale or other disposition of our shares of Common Stock, unless the U.S. Holder is an exempt recipient. Backup withholding may apply to such payments if the U.S. Holder fails to provide a taxpayer identification number (or furnishes an incorrect taxpayer identification number) or a certification of exempt status or has been notified by the IRS that it is subject to backup withholding (and such notification has not been withdrawn).
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules will be allowed as a credit against a U.S. Holder’s U.S. federal income tax liability and may entitle such holder to a refund, provided the required information is timely furnished to the IRS. Taxpayers should consult their tax advisors regarding their qualification for an exemption from backup withholding and the procedures for obtaining such an exemption.
121
Sale or Other Disposition, Exercise or Expiration of Common Warrants
A U.S. Holder will recognize gain or loss on the sale or other taxable disposition of common warrants in an amount equal to the difference, if any, between the amount of cash plus the fair market value of any property received and such U.S. Holder’s tax basis in common warrants sold or otherwise disposed of, in each case as determined in U.S. dollars. Gain or loss recognized on such sale or other taxable disposition generally will be a capital gain or loss, which will be long-term capital gain or loss if the common warrant is held for more than one year. The gain or loss will generally be U.S.-source gain or loss for foreign tax credit purposes. Deductions for capital losses are subject to complex limitations under the Code.
A U.S. Holder should not recognize gain or loss on the exercise of common warrants and related receipt of common stock. A U.S. Holder’s initial tax basis in the common stock received on the exercise of a common warrant should be equal to the sum of (i) such U.S. Holder’s initial tax basis in such common warrant plus (ii) the exercise price paid by such U.S. Holder on the exercise of such common warrant.
In certain limited circumstances, a U.S. Holder may be permitted to undertake a cashless exercise of the common warrants into shares of common stock. The U.S. federal income tax treatment of a cashless exercise of common warrants into shares of common stock is unclear, and the tax consequences of a cashless exercise could differ from the consequences upon the exercise of a common warrant described in the preceding paragraph. U.S. Holders should consult their own tax advisors regarding the U.S. federal income tax consequences of a cashless exercise of common warrants.
Upon the lapse or expiration of a common warrant, a U.S. Holder will recognize a loss in an amount equal to such U.S. Holder’s tax basis in the common warrant. Any such loss generally will be a capital loss and will be long-term capital loss if the common warrants are held for more than one year. Deductions for capital losses are subject to complex limitations under the Code.Certain Adjustments to the Common Warrants Under Section 305 of the Code, an adjustment to the number of shares of common stock that will be issued on the exercise of the common warrants, or an adjustment to the exercise price of the common warrants, may be treated as a constructive distribution to a U.S. Holder of the common warrants if, and to the extent that, such adjustment has the effect of increasing such U.S. Holder’s proportionate interest in our earnings and profits or our assets, depending on the circumstances of such adjustment (for example, if such adjustment is to compensate for a distribution of cash or property to the shareholders). Adjustments to the exercise price of the common warrants made pursuant to a bona fide reasonable adjustment formula that has the effect of preventing dilution of the interest of the holders of the common warrants should generally not be considered to result in a constructive distribution. Any such constructive distribution would be taxable whether or not there is an actual distribution of cash or other property (see more detailed discussion of the rules applicable to distributions we make at “Taxation of Distributions” above).
Certain Adjustments to the Common Warrants and Pre-funded Warrants
Under Section 305 of the Code, an adjustment to the number of shares of common stock that will be issued on the exercise of the common warrants or pre-funded warrants, or an adjustment to the exercise price of the common warrants or pre-funded warrants, may be treated as a constructive distribution to a U.S. Holder of the common warrants or pre-funded warrants if, and to the extent that, such adjustment has the effect of increasing such U.S. Holder’s proportionate interest in our earnings and profits or our assets, depending on the circumstances of such adjustment (for example, if such adjustment is to compensate for a distribution of cash or property to the shareholders). Adjustments to the exercise price of the common warrants or pre-funded warrants made pursuant to a bona fide reasonable adjustment formula that has the effect of preventing dilution of the interest of the holders of the common warrants or pre-funded warrants should generally not be considered to result in a constructive distribution. Any such constructive distribution would be taxable whether or not there is an actual distribution of cash or other property (see more detailed discussion of the rules applicable to distributions we make at “Taxation of Distributions” above).
122
Treatment of Pre-Funded Warrants
Although it is not entirely free from doubt, we believe a pre-funded warrant should be treated as common stock for U.S. federal income tax purposes and a holder of pre-funded warrants should generally be taxed in the same manner as a holder of our common stock, as described below. Accordingly, no gain or loss should be recognized upon the exercise of a pre-funded warrant and, upon exercise, the holding period of a pre-funded warrant should carry over to the common stock received. Similarly, the tax basis of the pre-funded warrant should carry over to the common stock received upon exercise, increased by the exercise price of $0.001 per share. However, our characterization of a pre-funded warrant is not binding on the IRS, and the IRS may treat our pre-funded warrants as warrants to acquire our common stock. If so, the amount and character of your gain with respect to an investment in our pre-funded warrants could change. Accordingly, each holder should consult his, her or its own tax advisor regarding the risks associated with the acquisition of pre-funded warrants pursuant to this offering (including potential alternative characterizations). The balance of this discussion generally assumes that our characterization described above is respected for U.S. federal income tax purposes.
Tax Considerations Applicable to Non-U.S. Holders
Taxation of Distributions
In general, any distributions (including constructive distributions) we make to a non-U.S. Holder of shares on our Common Stock, to the extent paid out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles), will constitute dividends for U.S. federal income tax purposes and, provided such dividends are not effectively connected with the non-U.S. Holder’s conduct of a trade or business within the United States or, if an applicable tax treaty so requires, are not attributable to a U.S. permanent establishment or fixed base maintained by the non-U.S. Holder, we will be required to withhold tax from the gross amount of the dividend at a rate of 30%, unless such non-U.S. Holder is eligible for a reduced rate of withholding tax under an applicable income tax treaty and provides proper certification of its eligibility for such reduced rate (usually on an IRS Form W-8BEN or W-8BEN-E, as applicable). In the case of any constructive dividend (as described below under “Tax Considerations Applicable to Non-U.S. Holders—Possible Constructive Distributions”), it is possible that this tax would be withheld from any amount owed to a non-U.S. Holder by the applicable withholding agent, or other property subsequently paid or credited to such holder. Any distribution not constituting a dividend will be treated first as reducing (but not below zero) the non-U.S. Holder’s adjusted tax basis in its shares of our Common Stock and, to the extent such distribution exceeds the non-U.S. Holder’s adjusted tax basis, as gain realized from the sale or other disposition of the Common Stock, which will be treated as described under “Tax Considerations Applicable to Non-U.S. Holders—Gain on Sale, Taxable Exchange or Other Taxable Disposition of Common Stock” below. In addition, if we determine that we are likely to be classified as a “United States real property holding corporation” (see the section titled “Tax Considerations Applicable to Non-U.S. Holders—Gain on Sale, Exchange, or Other Taxable Disposition of Common Stock” below), we will withhold 15% of any distribution that exceeds our current and accumulated earnings and profits.
Distributions that constitute dividends as described in the prior paragraph that we pay to a non-U.S. Holder and that are effectively connected with such non-U.S. Holder’s conduct of a trade or business within the United States (and if a tax treaty applies are attributable to a U.S. permanent establishment or fixed base maintained by the non-U.S. Holder) will generally not be subject to U.S. withholding tax, provided such non-U.S. Holder complies with certain certification and disclosure requirements (generally by providing an IRS Form W-8ECI). Instead, such dividends generally will be subject to U.S. federal income tax, net of certain deductions, at the same individual or corporate rates applicable to U.S. Holders. If the non-U.S. Holder is a corporation, dividends that are effectively connected income may also be subject to a “branch profits tax” at a rate of 30% (or such lower rate as may be specified by an applicable income tax treaty).
Gain on Sale or Other Disposition of Common Stock, Pre-Funded Warrants and Common Warrants
Subject to the discussion below regarding backup withholding and FATCA, a Non-U.S. Holder generally will not be required to pay U.S. federal income tax on any gain realized upon the sale or other disposition of our common stock, pre-funded warrant or common warrants unless:
| ● | the gain is effectively connected with the Non-U.S. Holder’s conduct of a trade or business within the United States and not eligible for relief under an applicable income tax treaty, in which case the Non-U.S. Holder will be required to pay tax on the net gain derived from the sale under regular graduated U.S. federal income tax rates, and for a Non-U.S. Holder that is a corporation, such Non-U.S. Holder may be subject to the branch profits tax at a 30% rate (or such lower rate as may be specified by an applicable income tax treaty) on such effectively connected gain, as adjusted for certain items; |
123
| ● | the Non-U.S. Holder is an individual who is present in the United States for a period or periods aggregating 183 days or more during the calendar year in which the sale or disposition occurs and certain other conditions are met, in which case the Non-U.S. Holder will be required to pay a flat 30% tax on the gain derived from the sale, which tax may be offset by U.S. source capital losses (even though the Non-U.S. Holder is not considered a resident of the United States) (subject to applicable income tax or other treaties); or |
| ● | we are a “U.S. real property holding corporation” for U.S. federal income tax purposes, or a USRPHC, at any time within the shorter of the five-year period preceding the disposition or the Non-U.S. Holder’s holding period for our common stock. We believe we are not currently and do not anticipate becoming a USRPHC. However, because the determination of whether we are a USRPHC depends on the fair market value of our United States real property interests relative to the fair market value of our other business assets, there can be no assurance that we will not become a USRPHC in the future. Even if we become a USRPHC, however, gain arising from the sale or other taxable disposition by a Non-U.S. Holder of our common stock will not be subject to United States federal income tax if (a) shares of our common stock are “regularly traded,” as defined by applicable Treasury Regulations, on an established securities market, such as Nasdaq, and (b) the Non-U.S. Holder owns or owned, actually and constructively, 5% or less of the shares of our common stock throughout the five-year period ending on the date of the sale or exchange. If the foregoing exception does not apply, such Non-U.S. Holder’s proceeds received on the disposition of shares will generally be subject to withholding at a rate of 15% and such Non-U.S. Holder will generally be taxed on any gain in the same manner as gain that is effectively connected with the conduct of a U.S. trade or business, except that the branch profits tax generally will not apply. |
Exercise or Expiration of Pre-funded Warrant and Common Warrants
In general, a Non-U.S. Holder will not be required to recognize income, gain or loss upon the exercise of a pre-funded warrant or common warrant by payment of the exercise price, except possibly to the extent of cash paid in lieu of a fractional share. However, no discussion is provided herein regarding the U.S. federal income tax treatment on the exercise of a pre-funded warrant or a common warrant on a cashless basis, and Non-U.S. Holders are urged to consult their tax advisors as to the exercise of a pre-funded warrant or a warrant on a cashless basis.
If a pre-funded warrant or a common warrant expires without being exercised, a Non-U.S. Holder that is engaged in a U.S. trade or business to which any income from the pre-funded warrant or the common warrant would be effectively connected or who is present in the United States for a period or periods aggregating 183 days or more during the calendar year in which the expiration occurs (and certain other conditions are met) will recognize a capital loss in an amount equal to such Non-U.S. Holder’s tax basis in the pre-funded warrant or the common warrant. The amount paid to purchase our common stock, pre-funded warrants and common warrants will be apportioned between them in proportion to the respective fair market values of the common stock, pre-funded warrants and common warrants, and the apportioned amount will be the tax basis of the common stock, pre-funded warrants and common warrants respectively. The fair market value of our common stock for this purpose will generally be its trading value immediately after issuance.
Information Reporting and Backup Withholding
Information returns may be filed with the IRS in connection with distributions on common stock, and the proceeds of a sale or other disposition of common stock, pre-funded warrants or common warrants. A non-exempt U.S. Holder may be subject to U.S. backup withholding on these payments if it fails to provide its taxpayer identification number to the withholding agent and comply with certification procedures or otherwise establish an exemption from backup withholding.
124
A Non-U.S. Holder may be subject to U.S. information reporting and backup withholding on these payments unless the Non-U.S. Holder complies with certification procedures to establish that it is not a U.S. person (within the meaning of the Code). The certification requirements generally will be satisfied if the Non-U.S. Holder provides the applicable withholding agent with a statement on the applicable IRS Form (or a suitable substitute or successor form), together with all appropriate attachments, signed under penalties of perjury, stating, among other things, that such Non-U.S. Holder is not a U.S. Person. Applicable Treasury Regulations provide alternative methods for satisfying this requirement. In addition, the amount of distributions on common stock paid to a Non-U.S. Holder, and the amount of any U.S. federal tax withheld therefrom, must be reported annually to the IRS and the holder. This information may be made available by the IRS under the provisions of an applicable tax treaty or agreement to the tax authorities of the country in which the Non-U.S. Holder resides.
Payment of the proceeds of the sale or other disposition of common stock, pre-funded warrants or common warrants to or through a non-U.S. office of a U.S. broker or of a non-U.S. broker with certain specified U.S. connections generally will be subject to information reporting requirements, but not backup withholding, unless the Non-U.S. Holder certifies under penalties of perjury that it is not a U.S. person or an exemption otherwise applies. Payments of the proceeds of a sale or other disposition of common stock, pre-funded warrants or common warrants to or through a U.S. office of a broker generally will be subject to information reporting and backup withholding, unless the Non-U.S. Holder certifies under penalties of perjury that it is not a U.S. person or otherwise establishes an exemption.
Backup withholding is not an additional tax. The amount of any backup withholding from a payment generally will be allowed as a credit against the holder’s U.S. federal income tax liability and may entitle the holder to a refund, provided that the required information is timely furnished to the IRS.
125
PLAN OF DISTRIBUTION
We have engaged JonesTrading Institutional Services LLC and Lake Street Capital Markets, LLC, or the joint placement agents, to act as our exclusive joint placement agents to solicit offers to purchase the shares of our common stock, pre-funded warrants and common warrants offered by this prospectus. The joint placement agents are not purchasing or selling any such securities, nor are they required to arrange for the purchase and sale of any specific number or dollar amount of such securities, other than to use their “reasonable best efforts” to arrange for the sale of such securities by us. Therefore, we may not sell all of the shares of common stock, pre-funded warrants and common warrants being offered. The terms of this offering were subject to market conditions and negotiations between us, the joint placement agents and prospective investors. The joint placement agents will have no authority to bind us. This is a best efforts offering and there is no minimum offering amount required as a condition to the closing of this offering. The joint placement agents may retain sub-agents and selected dealers in connection with this offering. Investors purchasing securities offered hereby will have the option to execute a securities purchase agreement with us. In addition to rights and remedies available to all purchasers in this offering under federal securities and state law, the purchasers who enter into a securities purchase agreement will also be able to bring claims of breach of contract against us. The ability to pursue a claim for breach of contract is material to larger purchasers in this offering as a means to enforce the following covenants uniquely available to them under the securities purchase agreement: (i) a covenant to not enter into variable rate financings for a period of 90 days following the closing of the offering, subject to certain exceptions (provided that an issuance or sale of securities pursuant in this offering shall not be deemed a variable rate financing); and (ii) a covenant to not enter into any equity financings for 45 days from closing of the offering, subject to certain exceptions.
The nature of the representations, warranties and covenants in the securities purchase agreements shall include:
| ● | standard issuer representations and warranties on matters such as organization, qualification, authorization, no conflict, no governmental filings required, current in SEC filings, no litigation, labor or other compliance issues, environmental, intellectual property and title matters and compliance with various laws such as the Foreign Corrupt Practices Act; and |
| ● | covenants regarding matters such as registration of warrant shares, no integration with other offerings, filing of a Current Report on Form 8-K to disclose entering into these securities purchase agreements, no shareholder rights plans, no material nonpublic information, use of proceeds, indemnification of purchasers and reservation and listing of common stock. |
Delivery of the shares of common shares, pre-funded warrants and common warrants offered hereby is expected to occur on or about, 2024, subject to satisfaction of certain customary closing conditions.
We have agreed to pay the joint placement agents an aggregate fee equal to 7% of the gross proceeds received in the offering. In addition, we have agreed to reimburse the joint placement agents for its legal fees and expenses and other out-of-pocket expenses in an amount up to $100,000.
We estimate the total expenses of this offering paid or payable by us, exclusive of the joint placement agents’ cash fee of 7% of the gross proceeds and expenses, will be approximately $772,800. After deducting the fees due to the joint placement agents and our estimated expenses in connection with this offering, we expect the net proceeds from this offering will be approximately $ million, based on an assumed public offering price per share of $2.76, which was the last reported sales price of our common stock on the Nasdaq Global Market on July 11, 2024.
The following table shows the per share and total cash fees we will pay to the joint placement agents in connection with the sale of the common stock, pre-funded warrants, common warrants and shares of common stock underlying the pre-funded warrants and common warrants pursuant to this prospectus.
| Per Share and Common Warrant | Per Pre-Funded Warrant and Common Warrant | Total | ||||||||||
| Public offering price | $ | $ | $ | |||||||||
| Placement agent fees | $ | $ | $ | |||||||||
| Proceeds, before expenses, to us | $ | $ | $ | |||||||||
126
Indemnification
We have agreed to indemnify the joint placement agents against certain liabilities, including liabilities under the Securities Act and liabilities arising from breaches of representations and warranties contained in our engagement letter with the joint placement agents. We have also agreed to contribute to payments the joint placement agents may be required to make in respect of such liabilities.
Lock-up Agreements
We and each of our executive officers and directors have agreed with the joint placement agents to be subject to a lock-up period of days following the date of closing of the offering pursuant to this prospectus. This means that, during the applicable lock-up period, we and such persons may not offer for sale, contract to sell, sell, distribute, grant any option, right or warrant to purchase, pledge, hypothecate or otherwise dispose of, directly or indirectly, any of our shares of common stock or any securities convertible into, or exercisable or exchangeable for, shares of common stock, subject to customary exceptions. The joint placement agents may waive the terms of these lock-up agreements in its sole discretion and without notice.
Other Relationships
From time to time, the joint placement agents may provide in the future various advisory, investment and commercial banking and other services to us in the ordinary course of business, for which they have received and may continue to receive customary fees and commissions.
Regulation M Compliance
The joint placement agents may be deemed to be an underwriter within the meaning of Section 2(a)(11) of the Securities Act, and any commissions received by it and any profit realized on the sale of our securities offered hereby by it while acting as principal might be deemed to be underwriting discounts or commissions under the Securities Act. The joint placement agents will be required to comply with the requirements of the Securities Act and the Exchange Act, including, without limitation, Rule 10b-5 and Regulation M under the Exchange Act. These rules and regulations may limit the timing of purchases and sales of our securities by the joint placement agents. Under these rules and regulations, the joint placement agents may not (i) engage in any stabilization activity in connection with our securities; and (ii) bid for or purchase any of our securities or attempt to induce any person to purchase any of our securities, other than as permitted under the Exchange Act, until they have completed their participation in the distribution.
Trading Market
Our common stock is listed on the Nasdaq Global Market under the symbol “IXHL.” There is no established public trading market for the pre-funded warrants or common warrants to be sold in this offering, and we do not expect a market to develop. In addition, we do not intend to apply for listing of the pre-funded warrants or common warrants on any national securities exchange.
127
Legal Matters
The validity of the shares of common stock offered hereby will be passed upon for us by Rimôn. Certain legal matters will be passed upon for the joint placement agents by Duane Morris LLP.
Experts
The consolidated financial statements of Incannex Healthcare Inc. as of June 30, 2022, and 2023 have been audited by Grant Thornton Audit Pty Ltd (“Grant Thornton”), independent external auditor, as set forth in their report thereon relating to the consolidated financial statements of the Company, appearing elsewhere in the prospectus, and are included in reliance on the report of such firm given upon their authority as experts in accounting and auditing.
The offices of Grant Thornton are located at Level 43, 152 - 158 St Georges Terrace, Perth, WA 6000.
Where You Can Find More Information
We have filed with the SEC a registration statement on Form S-1 under the Securities Act with respect to the shares of our common stock offered hereby. This prospectus, which constitutes a part of the registration statement, does not contain all of the information set forth in the registration statement or the exhibits and schedules filed therewith. For further information about us and the common stock offered hereby, we refer you to the registration statement and the exhibits and schedules filed therewith. Statements contained in this prospectus regarding the contents of any contract or any other document that is filed as an exhibit to the registration statement are not necessarily complete, and each such statement is qualified in all respects by reference to the full text of such contract or other document filed as an exhibit to the registration statement.
The SEC maintains a website at www.sec.gov that contains reports, proxy statements and other information regarding companies that file electronically with it. Our periodic and current reports, proxy statements and other information are available at www.sec.gov.
We also maintain a website at www.incannex.com. You may access our proxy statements, annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act with the SEC free of charge at our website as soon as reasonably practicable after such material is electronically filed with, or furnished to, the SEC. The reference to our website address does not constitute incorporation by reference of the information contained on our website, and you should not consider the contents of our website in making an investment decision with respect to our common stock. We have included our website address in this prospectus solely as an inactive textual reference.
128
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Page | ||
| Consolidated Financial Statements for the years ended June 30, 2022 and 2023 | ||
| Report of Independent External Auditor | F-2 | |
| Consolidated Statements of Financial Position | F-3 | |
| Consolidated Statements of Comprehensive Income/(Loss) | F-4 | |
| Consolidated Statements of Changes in Equity | F-5 | |
| Consolidated Statements of Cash Flows | F-6 | |
| Notes to the Consolidated Financial Statements | F-7 |
INDEX TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS
| Page | ||
| Consolidated Financial Statements for the nine months ended March 31, 2023 and 2024 | ||
| Consolidated Statements of Financial Position | F-21 | |
| Consolidated Statements of Comprehensive Income/(Loss) | F-22 | |
| Consolidated Statements of Changes in Equity | F-23 | |
| Consolidated Statements of Cash Flows | F-25 | |
| Notes to the Consolidated Financial Statements | F-26 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Shareholders
Incannex Healthcare Inc.
Opinion on the financial statements
We have audited the accompanying consolidated balance sheets of Incannex Healthcare Inc. (a Delaware corporation) and subsidiaries (the “Company”) as of June 30, 2023 and 2022, the related consolidated statements of comprehensive income, changes in shareholders’ equity, and cash flows for each of the two years in the period ended June 30, 2023, and the related notes (collectively referred to as the “financial statements”).
In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of June 30, 2023 and 2022, and the results of its operations and its cash flows for each of the two years in the period ended June 30, 2023 and 2022, in conformity with accounting principles generally accepted in the United States of America.
Going concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, The Company has incurred a net loss after tax of $48,811,000 (2022: $10,818,000) and experienced net cash outflows from operating activities of $13,041,000 (2022: $10,218,000) for the year ended June 30, 2023. As at June 30, 2023, the group had cash assets of $22,120,000 (2022: $25,834,000) and reported current assets exceed its current liabilities by $20,447,000 (2022: $24,710,000). These conditions, along with other matters as set forth in Note 2, raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ GRANT THORNTON AUDIT PTY LTD
We have served as the Company’s auditor since 2023.
Perth, Australia
July 3, 2024
F-2
INCANNEX HEALTHCARE INC.
Consolidated Balance Sheets
(in thousands, except share and per share amounts)
(expressed in U.S. Dollars, unless otherwise stated)
| June 30, 2023 | June 30, 2022 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 22,120 | $ | 25,834 | ||||
| Prepaid expenses and other assets | 877 | 261 | ||||||
| Total current assets | 22,997 | 26,095 | ||||||
| Property, plant and equipment, net | 294 | - | ||||||
| Operating lease right-of-use assets | 492 | - | ||||||
| Total assets | $23,783 | $ | 26,095 | |||||
| Liabilities and stockholders’ equity | ||||||||
| Current liabilities: | ||||||||
| Trade and other payables | $ | 1,748 | $ | 896 | ||||
| Accrued expenses and other current liabilities | 689 | 489 | ||||||
| Operating lease liabilities, current | 113 | - | ||||||
| Total current liabilities | 2,550 | 1,385 | ||||||
| Operating lease liabilities, non-current | 408 | - | ||||||
| Total liabilities | 2,958 | 1,385 | ||||||
| Commitments and contingencies (Note 7) | ||||||||
| Stockholders’ equity: | ||||||||
| Common stock, $0.0001 par value – 100,000,000 shares authorized; 15,873,113 and 12,926,349 shares issued and outstanding at June 30, 2023 and 2022, respectively | 2 | 1 | ||||||
| Preferred stock, $0.0001 par value per share, 10,000,000 shares authorized; no shares issued or outstanding at June 30, 2023 and 2022, respectively | - | - | ||||||
| Additional paid-in capital | 116,290 | 69,074 | ||||||
| Accumulated deficit | (92,212 | ) | (43,401 | ) | ||||
| Foreign currency translation reserve | (3,255 | ) | (963 | ) | ||||
| Total shareholders’ equity | 20,825 | 24,711 | ||||||
| Total liabilities and stockholders’ equity | $ | 23,783 | $ | 26,096 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
INCANNEX HEALTHCARE INC.
Consolidated Statements of Operations and Comprehensive Loss
(in thousands, except share and per share amounts)
(expressed in U.S. Dollars, unless otherwise stated)
| For the year ended June 30, | ||||||||
| 2023 | 2022 | |||||||
| Operating expenses: | ||||||||
| Research and development | (6,309 | ) | (3,899 | ) | ||||
| Acquisition of in-process research and development | (35,347 | ) | - | |||||
| General and administrative | (8,012 | ) | (7,443 | ) | ||||
| Total operating expenses | (49,668 | ) | (11,342 | ) | ||||
| Loss from operations | (49,668 | ) | (11,342 | ) | ||||
| Other income, net: | ||||||||
| R&D tax incentive | 683 | 568 | ||||||
| Foreign exchange expense | (67 | ) | (48 | ) | ||||
| Interest income | 241 | 4 | ||||||
| Total other income, net | 857 | 524 | ||||||
| Loss before income tax expense | (48,811 | ) | (10,818 | ) | ||||
| Income tax expense | - | - | ||||||
| Net loss | $ | (48,811 | ) | $ | (10,818 | ) | ||
| Other comprehensive loss: | ||||||||
| Currency translation adjustment, net of tax | (2,292 | ) | (1,302 | ) | ||||
| Total comprehensive loss | $ | (51,103 | ) | $ | (12,120 | ) | ||
| Net loss per share: Basic and diluted | $ | (3.32 | ) | (1.02 | ) | |||
| Weighted average number of shares outstanding, basic and diluted | 15,384,704 | 11,921,292 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
INCANNEX HEALTHCARE INC.
Consolidated Statements of Shareholders’ Equity (Deficit)
(in thousands, except share amounts)
(expressed in U.S. Dollars, unless otherwise stated)
| Common stock | Additional paid-in capital | Accumulated deficit | Foreign currency translation reserve | Total stockholders’ equity (deficit) | ||||||||||||||||||||
| Share | Amount | Amount | Amount | Amount | Amount | |||||||||||||||||||
| # | $ | $ | $ | $ | $ | |||||||||||||||||||
| Balance at June 30, 2021 | 10,687,120 | 1 | 38,689 | (32,583 | ) | 339 | 6,446 | |||||||||||||||||
| Options exercised | 2,076,507 | - | 29,045 | - | - | 29,045 | ||||||||||||||||||
| Share-based compensation | 100,000 | - | 1,009 | - | - | 1,009 | ||||||||||||||||||
| Share placements | 50,000 | - | 290 | - | - | 290 | ||||||||||||||||||
| Share issued to advisors | 12,722 | - | 320 | - | - | 320 | ||||||||||||||||||
| Issuance costs | - | - | (279 | ) | - | - | (279 | ) | ||||||||||||||||
| Net loss | - | - | - | (10,818 | ) | - | (10,818 | ) | ||||||||||||||||
| Currency translation adjustment, net of tax | - | - | - | - | (1,302 | ) | (1,302 | ) | ||||||||||||||||
| Balance at June 30, 2022 | 12,926,349 | 1 | 69,074 | (43,401 | ) | (963 | ) | 24,711 | ||||||||||||||||
| Options issued to advisors | - | - | 476 | - | - | 476 | ||||||||||||||||||
| Option placements | 21 | - | 71 | - | - | 71 | ||||||||||||||||||
| Share-based compensation | - | - | 2,149 | - | - | 2,149 | ||||||||||||||||||
| Share placements | 634,146 | - | 8,829 | - | - | 8,829 | ||||||||||||||||||
| Share issued to advisors | 130,902 | - | 2,050 | - | - | 2,050 | ||||||||||||||||||
| Asset acquisition shares issued | 2,181,695 | 1 | 34,170 | - | - | 34,171 | ||||||||||||||||||
| Issuance costs | - | - | (529 | ) | - | - | (529 | ) | ||||||||||||||||
| Net loss | - | - | - | (48,811 | ) | - | (48,811 | ) | ||||||||||||||||
| Currency translation adjustment, net of tax | - | - | - | - | (2,292 | ) | (2,292 | ) | ||||||||||||||||
| Balance at June 30, 2023 | 15,873,113 | 2 | 116,290 | (92,212 | ) | (3,255 | ) | 20,825 | ||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
INCANNEX HEALTHCARE INC.
Consolidated Statements of Cash Flows
(in thousands, except share and per share amounts)
(expressed in U.S. Dollars, unless otherwise stated)
| For the year ended June 30, | ||||||||
| 2023 | 2022 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (48,811 | ) | $ | (10,818 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | 88 | - | ||||||
| Share-based compensation expense | 2,149 | 1,063 | ||||||
| Change in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | (626 | ) | (27 | ) | ||||
| Trade and other payables | 1,104 | 866 | ||||||
| Acquisition of in-process research and development | 35,347 | - | ||||||
| Net cash used in operating activities | (13,041 | ) | (10,218 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Purchase of property, plant and equipment | (316 | ) | - | |||||
| Net cash used in investing activities | (316 | ) | - | |||||
| Cash flows from financing activities: | ||||||||
| Proceeds from issuance of common stocks, net of issuance costs | 8,175 | 29,566 | ||||||
| Net cash provided by financing activities | 8,175 | 29,566 | ||||||
| Effect of exchange rate changes on cash and cash equivalents | 1,468 | (373 | ) | |||||
| Net (decrease)/increase in cash and cash equivalents | (5,182 | ) | 19,348 | |||||
| Cash and cash equivalents at beginning of period | 25,834 | 6,859 | ||||||
| Cash and cash equivalents at end of period | $ | 22,120 | $ | 25,834 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
INCANNEX HEALTHCARE INC.
Notes To Audited Consolidated Financial Statements
June 30, 2023 and 2022
Note 1 – Re-domiciliation and Business
Incannex Healthcare Inc. is a corporation formed under the laws of Delaware, United States in July 2023. In November 2023, Incannex Healthcare Inc. acquired all the outstanding ordinary shares of Incannex Healthcare Limited, an Australian corporation (“Incannex Australia”), pursuant to a scheme of arrangement under Australian law (the “Re-domiciliation”). As a result of the Re-domiciliation, Incannex Australia became a wholly-owned subsidiary of Incannex Healthcare Inc., which is the new ultimate parent company.
Until the Re-domiciliation, Incannex Australia’s ordinary shares were listed on the Australian Securities Exchange (“ASX”) and American Depositary Shares (“ADSs”), each representing 25 ordinary shares of Incannex Australia, traded on Nasdaq. Following completion of the Re-domiciliation, Incannex Australia’s ordinary shares were delisted from the ASX and Incannex Healthcare Inc. assumed Incannex Australia’s listing on Nasdaq.
Pursuant to the Re-domiciliation, holders of Incannex Australia’s ordinary shares received one share of common stock in Incannex Healthcare Inc. for every 100 ordinary shares held in Incannex Australia and holders of ADSs in Incannex Australia received one share of common stock of Incannex Healthcare Inc. for every 4 ADSs held in Incannex Australia.
The issued and outstanding shares of our common stock as shown in this report have been adjusted in the consolidated financial statements to reflect the 100:1 exchange ratio as if it had occurred on July 1, 2022.
Incannex Healthcare Inc. and its subsidiaries are referred to as “the Company” unless the text otherwise requires.
The Company’s fiscal year end is June 30. References to a particular “fiscal year” are to our fiscal year ended June 30 of that calendar year.
The consolidated financial statements of the Company are presented in United States dollars and consist of Incannex Healthcare Inc. and the following wholly-owned subsidiaries:
| Subsidiary | Jurisdiction | |
| Incannex Healthcare Limited | Victoria, Australia | |
| Incannex Pty Ltd | Victoria, Australia | |
| Psychennex Pty Ltd | Victoria, Australia | |
| APIRx Pharmaceutical USA, LLC | Delaware, United States | |
| APIRx Pharmaceuticals Holding BV | IJsselstein, Netherlands | |
| Clarion Clinics Group Pty Ltd | Victoria, Australia | |
| Clarion Model Clinic Pty Ltd | Victoria, Australia | |
| Psychennex Licensing and Franchising Pty Ltd | Victoria, Australia |
F-7
Note 2 – Basis of Presentation and Summary of Significant Accounting Policies
Basis of Presentation
On November 28, 2023, the Company implemented the transaction to redomicile from Australia to United States and became the parent of Incannex Australia and the wholly owned subsidiaries listed in Note 1. The historical financial statements of Incannex Australia became the historical financial statements of the combined company upon consummation of the Re-domiciliation. As a result, the financial statements included in this report reflect (i) the historical operating results of Incannex Australia and subsidiaries prior to the Re-domiciliation; (ii) the combined results of the Company, Incannex Australia, and subsidiaries following the completion of the Re-domiciliation; and (iii) the Company’s equity structure for all periods presented, including adjusting the issued and outstanding shares of common stock to reflect the 100:1 exchange ratio as if it had occurred on July 1, 2021.
The Company’s consolidated financial statements included in this report have been prepared in accordance with accounting principles generally accepted in the United States of America (“US GAAP”) and pursuant to the rules and regulations of the SEC. Prior to the Re-domiciliation, Incannex Australia reported its consolidated financial statements in accordance with International Financial Reporting Standards (“IFRS”). Following the Re-domiciliation, the Company transitioned to US GAAP and applied US GAAP retrospectively for all prior periods presented.
Reference is frequently made herein to the Financial Accounting Standards Board (the “FASB”) Accounting Standards Codification (“ASC”). This is the source of authoritative US GAAP recognized by the FASB to be applied to non-governmental entities.
Going concern basis
The financial report has been prepared on the going concern basis, which assumes continuity of normal business activities and the realization of assets and the settlement of liabilities in the ordinary course of business.
The group has incurred a net loss after tax of $48,811,000 (2022: $10,818,000) and experienced net cash outflows from operating activities of $13,041,000 (2022: $10,218,000) for the year ended June 30, 2023.
As at June 30, 2023, the group had cash assets of $22,120,000 (2022: $25,834,000) and reported current assets exceed its current liabilities by $20,447,000 (2022: $24,710,000).
The ability of the Consolidated Entity and Company to continue as going concern and to pay their debts as and when they fall due is dependent on the following:
| ● | the ability to raise additional funding; | |
| ● | managing all costs in line with management’s forecasts; and | |
| ● | receipt of Research and Development tax incentive in line with management’s estimates for the amount and expected timing. |
The Directors have prepared a cash flow forecast which indicates that the group will have sufficient cash flows to meet minimum operating overheads and committed expenditure requirements for the 12 month period from the date of signing the financial report if they are successful in relation to matters referred to above.
The Directors are confident that they will achieve the matters set out above and therefore the going concern basis of preparation is appropriate. The financial report has therefore been prepared on the going concern basis.
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries (the “Group”). Details of all controlled entities are set out in Note 1. All intercompany balances and transactions have been eliminated on consolidation.
F-8
Use of Estimates
The preparation of financial statements in conformity with US GAAP requires management to make estimates and assumptions that impact the reported amounts of assets, liabilities and expenses and the disclosure of contingent assets and liabilities in the Company’s consolidated financial statements and accompanying notes.
The most significant estimates and assumptions in the Company’s consolidated financial statements include the valuation of acquisitions, equity-based instruments issued for other than cash, accrued research and development expense, and research and development tax credit. Estimates are periodically reviewed in light of changes in circumstances, facts and experience. Changes in estimates are recorded in the period in which they become known. Actual results could differ materially from those estimates.
Risks and Uncertainties
The Company is subject to risks and uncertainties common to companies in the biopharmaceutical industry. The Company believes that changes in any of the following areas could have a material adverse effect on future financial position or results of operations: ability to obtain future financing; regulatory approval and market acceptance of, and reimbursement for, product candidates; performance of third-party clinical research organizations and manufacturers upon which the Company relies; protection of the Company’s intellectual property; litigation or claims against the Company based on intellectual property, patent, product, regulatory or other factors; the Company’s ability to attract and retain employees.
There can be no assurance that the Company’s research and development will be successfully completed, that adequate protection for the Company’s intellectual property will be obtained or maintained, that any products developed will obtain necessary government regulatory approval or that any approved products will be commercially viable. Even if the Company’s product development efforts are successful, it is uncertain when, if ever, the Company will generate significant revenue from product sales. The Company operates in an environment of rapid technological change and substantial competition from other pharmaceutical and biotechnology companies. In addition, the Company is dependent upon the services of its employees, consultants and other third parties.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentration of credit risk consist primarily of cash and cash equivalents. The Company has not experienced any losses in such accounts, and management believes that the Company is not exposed to significant credit risk due to the financial position of the depository institutions in which those deposits are held. As of June 30, 2023 and 2022, all deposit in banks outside of the United States.
Cash and Cash Equivalents
Cash and cash equivalents, which includes cash and deposits held at call with financial institutions with original maturities of three months or less that are readily convertible to known amounts of cash, are carried at cost, which approximates fair value.
Property, Plant and Equipment, Net
Recognition and Measurement
All property, plant and equipment is recognised at historical cost less depreciation.
Depreciation
Depreciation is calculated using the straight-line method to allocate their cost, net of their residual values, over their estimated useful lives or, in the case of leasehold improvements and certain leased plant and equipment, the shorter lease term as follows:
| ● | Machinery 10-15 years | |
| ● | Vehicles 3-5 years | |
| ● | Furniture, fittings and equipment 3-8 years |
Furniture, fittings and equipment include assets in the form of office fit outs. These assets and other leasehold improvements are recognised at their fair value and depreciated over the shorter of their useful life or the lease term, unless the entity expects to use the assets beyond the lease term.
F-9
Impairment of Long-Lived Assets
Long-lived assets consist primarily of property, plant and equipment, net, and are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. If circumstances require that a long-lived asset be tested for possible impairment, the Company compares the undiscounted cash flows expected to be generated by the asset group to the carrying amount of the asset group. If the carrying amount of the long-lived asset is not recoverable on an undiscounted cash flow basis, an impairment is recognized to the extent that the carrying amount exceeds its fair value. Fair value is generally determined using the asset’s expected future discounted cash flows or market value, if readily determinable.
During the years ended June 30, 2023 and 2022, the Company has not recorded impairment charges on its long-lived assets.
Leases
The Company determines if an arrangement is, or contains, a lease at inception and then classifies the lease as operating or financing based on the underlying terms and conditions of the contract. Leases with terms greater than one year are initially recognized on the consolidated balance sheets as right-of-use assets and lease liabilities based on the present value of lease payments over the expected lease term. The Company has also elected to not apply the recognition requirement to any leases within its existing classes of assets with a term of 12 months or less and does not include any options to purchase the underlying asset that the Company is reasonably certain to exercise.
Lease expense for minimum lease payments on operating leases is recognized on a straight-line basis over the lease term. Variable lease payments are excluded from the right-of-use assets and operating lease liabilities and are recognized in the period in which the obligation for those payments is incurred. Operating lease expenses are categorized within research and development and general and administrative expenses in the consolidated statements of operations and comprehensive loss. Operating lease cash flows are categorized under net cash used in operating activities in the consolidated statements of cash flows.
As most of the Company’s leases do not provide an implicit rate, the Company uses its incremental borrowing rate based on the information available at commencement date in determining the present value of future payments.
Trade and other payables
These amounts represent liabilities for goods and services provided to the Company prior to the end of the period and which are unpaid. Due to their short-term nature, they are measured at amortized cost and are not discounted. The amounts are unsecured and are usually paid within 30 days of recognition.
Segment information
The Company operates and manages its business as one reportable and operating segment, which is the research and development of the use of psychedelic medicine and therapies for the treatment of mental health disorders. The Company’s Chief Executive Officer, who is the chief operating decision maker, reviews financial information on an aggregate basis for the purposes of allocating resources and evaluating financial performance. The Company’s long-lived assets are primarily in Australia.
F-10
Research and Development Costs
Research and development costs are expensed as incurred. Research and development consist of salaries, benefits and other personnel related costs including equity-based compensation expense, laboratory supplies, preclinical studies, clinical trials and related clinical manufacturing costs, costs related to manufacturing preparations, fees paid to other entities to conduct certain research and development activities on the Company’s behalf and allocated facility and other related costs.
Nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities are deferred and capitalized as prepaid expenses until the related goods are delivered or services are performed.
The Company records accrued liabilities for estimated costs of research and development activities conducted by third-party service providers, which include the conduct of pre-clinical studies and clinical trials, and contract manufacturing activities. The Company records the estimated costs of research and development activities based upon the estimated amount of services provided but not yet invoiced and includes these costs in trade and other payables on the consolidated balance sheets and within research and development expenses on the consolidated statements of operations and comprehensive loss.
The Company accrues for these costs based on factors such as estimates of the work completed and in accordance with agreements established with its third-party service providers. The Company makes significant judgments and estimates in determining the accrued liabilities balance at the end of each reporting period. As actual costs become known, the Company adjusts its accrued liabilities. The Company has not experienced any material differences between accrued costs and actual costs incurred.
Acquisitions
The Company evaluate acquisitions under the accounting framework in ASC 805, Business Combinations, to determine whether the transaction is a business combination or an asset acquisition. In determining whether an acquisition should be accounted for as a business combination or an asset acquisition, the Company first perform a screen test to determine whether substantially all of the fair value of the gross assets acquired is concentrated in a single identifiable asset or a group of similar identifiable assets. If this is the case, the acquired set is not deemed to be a business and is instead accounted for as an asset acquisition. If this is not the case, the Company further evaluate whether the acquired set includes, at a minimum, an input and a substantive process that together significantly contribute to the ability to create outputs. If so, the Company conclude that the acquired set is a business.
The Company measures and recognizes asset acquisitions that are not deemed to be business combinations based on the cost to acquire the assets, which includes pre-acquisition direct costs recorded in accrued professional and consulting fees. Goodwill is not recognized in asset acquisitions.
During the year ended June 30, 2023 the Company acquired APIRx Pharmaceutical USA, LLC (“APIRx”). The Company concluded that the acquisition of APIRx did not meet the definition of business under ASC 805, Business Combinations as the acquired set did not have outputs present and a substantive process was not acquired. Therefore, the Company accounted for the transaction as an asset acquisition rather than a business combination.
In accordance with ASC 730-10-25-2(c), intangible assets used in research and developmental activities acquired in an asset acquisition should be expensed at the acquisition date if there is no alternative future use in other R&D projects or otherwise (i.e., if they have no economic value). Additionally, in an asset acquisition, direct transaction costs are accumulated as a component of the consideration transferred and expensed with the acquired IPR&D that has no alternative use.
The Company determined that product candidates pertaining to APIRx had no alternative future use at the time of acquisition and charged $35.3 million including transaction costs of $2.43 million, to the acquisition of in-process research and development (IPR&D) expense as of the date of acquisition.
F-11
Share-based compensation
The Company accounts for share-based compensation arrangements with employees and non-employees using a fair value method which requires the recognition of compensation expense for costs related to all share-based payments including share options. The fair value method requires the Company to estimate the fair value of share-based payment awards on the date of grant using an option-pricing model. The Company uses either the trinomial pricing or Black-Scholes option-pricing model to estimate the fair value of options granted. Share-based compensation awards are expensed using the graded vesting method over the requisite service period, which is generally the vesting period, for each separately-vesting tranche. The Company has elected a policy of estimating forfeitures at grant date. Option valuation models, including the trinomial pricing and Black-Scholes option-pricing model, require the input of several assumptions. These inputs are subjective and generally require significant analysis and judgment to develop. Refer to Note 11 for a discussion of the relevant assumptions.
Benefit from Research and Development Tax Incentive
Benefit from R&D tax credit consists of the R&D tax credit received in Australia, which is recorded within other income (expense), net. The Company recognizes grants once both of the following conditions are met: (1) the Company is able to comply with the relevant conditions of the grant and (2) the grant is received.
Interest income
Interest income is recognized as interest accrues using the effective interest method. This is a method of calculating the amortised cost of a financial asset and allocating the interest income over the relevant period using the effective interest rate, which is the rate that exactly discounts estimated future cash receipts through the expected life of the financial asset to the net carrying amount of the financial asset.
Foreign Currency Translation
The Company maintains its consolidated financial statements in its functional currency, which is Australian Dollar. Monetary assets and liabilities denominated in currencies other than the functional currency are translated into the functional currency at rates of exchange prevailing at the balance sheet dates. Non-monetary assets and liabilities denominated in foreign currencies are translated into the functional currency at the exchange rates prevailing at the date of the transaction. Exchange gains or losses arising from foreign currency transactions are included in other income (expense), net in the consolidated statements of operations and comprehensive loss.
For financial reporting purposes, the consolidated financial statements of the Company have been presented in the U.S. dollar, the reporting currency. The financial statements of entities are translated from their functional currency into the reporting currency as follows: assets and liabilities are translated at the exchange rates at the balance sheet dates, expenses and other income (expense), net are translated at the average exchange rates for the periods presented and shareholders’ equity is translated based on historical exchange rates. Translation adjustments are not included in determining net loss but are included as a foreign exchange adjustment to other comprehensive income, a component of shareholders’ equity.
The following table presents data regarding the dollar exchange rate of relevant currencies:
| June 30, 2023 | June 30, 2022 | |||||||
| Exchange rate on balance sheet dates | ||||||||
| USD: AUD Exchange Rate | 0.6630 | 0.6889 | ||||||
| Average exchange rate for the period | ||||||||
| USD: AUD Exchange Rate | 0.6734 | 0.7258 | ||||||
F-12
Income tax
The Company is governed by Australia and United States income tax laws. The Company follows ASC 740, Accounting for Income Taxes, when accounting for income taxes, which requires an asset and liability approach to financial accounting and reporting for income taxes. Deferred income tax assets and liabilities are computed annually for temporary differences between the financial statements and tax bases of assets and liabilities that will result in taxable or deductible amounts in the future based on enacted tax laws and rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amount more likely than not to be realized.
For uncertain tax positions that meet a “more likely than not” threshold, the Company recognizes the benefit of uncertain tax positions in the consolidated financial statements. The Company’s practice is to recognize interest and penalties, if any, related to uncertain tax positions in income tax expense in the consolidated statements of operations.
Net loss per share attributable to ordinary shareholders
The Company has reported losses since inception and has computed basic net loss per share by dividing net loss by the weighted-average number of common stocks outstanding for the period, without consideration for potentially dilutive securities. The Company computes diluted net loss per share after giving consideration to all potentially dilutive shares, including unvested restricted shares and outstanding options. Because the Company has reported net losses since inception, these potential common stocks have been anti-dilutive and basic and diluted loss per share were the same for all periods presented.
Comprehensive Loss
Comprehensive loss includes net loss as well as other changes in shareholders’ equity that result from transactions and economic events other than those with shareholders. For the year ended June 30, 2023 and 2023, the only component of accumulated other comprehensive loss is foreign currency translation adjustment.
Note 3 – Prepaid expenses and other current assets
| June 30, 2023 $ | June 30, 2022 $ | |||||||
| (in thousands) | ||||||||
| Prepayments1 | 686 | 58 | ||||||
| GST recoverable | 191 | 203 | ||||||
| Total other assets | 877 | 261 | ||||||
| 1 | Prepayments consist of prepaid clinical trial insurances, prepaid R&D expenditure relating to PsiGAD and IHL-675A clinical trials and scientific, marketing, and adverting subscription services. |
| 2 | R&D tax incentive receivable for the fiscal year ended June 30, 2023. |
Note 4 – Property, Plant and Equipment, net
| June 30, 2023, $ | June 30, 2022 $ | |||||||
| (in thousands) | ||||||||
| Furniture, fittings and equipment | 157 | - | ||||||
| Assets under construction | 160 | - | ||||||
| Total property, plant and equipment, gross | 317 | - | ||||||
| Accumulated depreciation and amortization | (23 | ) | - | |||||
| Total property, plant and equipment, net | $ | 294 | $ | - | ||||
F-13
Depreciation expense is recorded general and administrative in the Consolidated Statements of Operations and Comprehensive Loss and amounted to $23 thousands and $nil for the year ended June 30, 2023 and 2022, respectively.
Note 5 – Trade and other payables, accrued expenses and other current liabilities
| June 30, 2023 $ | June 30, 2022 $ | |||||||
| (in thousands) | ||||||||
| Current liabilities | ||||||||
| Trade payables | 1,748 | 896 | ||||||
| Accrued expenses | 426 | 286 | ||||||
| Employee leave entitlements | 263 | 203 | ||||||
| Total trade and other payables, accrued expenses and other current liabilities | 2,437 | 1,385 | ||||||
Trade and other payables are unsecured, non-interest bearing and are normally settled within 30 days. The carrying amounts are a reasonable approximation of fair value.
Note 6 – Leases
For fiscal 2023 the Group entered into a three new lease agreement for its corporate head office in Sydney, Melbourne office and Clarion Clinic site. The leases have four, five and three-year terms respectively. These leases require monthly lease payments that may be subject to annual increases throughout the lease term. Certain of these leases also include renewal options at the election of the Company to renew or extend the lease for an additional three to five years. These optional periods have not been considered in the determination of the right-of-use assets or lease liabilities associated with these leases as the Company did not consider it reasonably certain it would exercise the options.
The following table summarizes the weighted-average remaining lease term and discount rates for the Company’s operating leases:
| June 30, 2023 | June 30, 2022 | |||||||
| Lease term (years) | 1.79 | - | ||||||
| - | ||||||||
| Discount rate | 9.18 | % | - | |||||
The following table summarizes the lease costs pertaining to the Company’s operating leases:
| June 30, 2023 $ | June 30, 2022 $ | |||||||
| (in thousands) | ||||||||
| Operating lease cost | 66 | - | ||||||
F-14
Cash paid for amounts included in the measurement of operating lease liabilities during fiscal years June 30, 2023 and 2022 was $nil and $66, respectively, and was included within net cash used in operating activities in the cash flows.
The following table summarizes the future minimum lease payments due under operating leases as of June 30, 2023, (in thousands):
| Operating leases | Amount $ (in thousands) | |||
| June 30, 2024 | 158 | |||
| June 30, 2025 | 189 | |||
| June 30, 2026 | 183 | |||
| June 30, 2027 | 47 | |||
| June 30, 2028 | 32 | |||
| Total minimum lease payments | 609 | |||
| Less amount representing interest | (87 | ) | ||
| Total operating lease liabilities | 522 | |||
As of June 30, 2023, the Company’s operating lease has a weighted-average remaining lease term of 1.98 years and a discount rate of 9.18%.
Note 7 – Commitments and contingencies
The Company records a loss contingency when it is probable that a liability has been incurred and the amount of the loss can be reasonably estimated. The Company also discloses material contingencies when it believes a loss is not probable but reasonably possible. Accounting for contingencies requires us to use judgment related to both the likelihood of a loss and the estimate of the amount or range of loss. Although the Company cannot predict with assurance the outcome of any litigation or tax matters, it does not believe there are currently any such actions that, if resolved unfavorably, would have a material impact on the Company’s operating results, financial position or cash flows.
F-15
Note 8 – Stockholder’s equity/Issued capital
Common stock
The Company has one class of common stock. In connection with the re-domiciliation, the Company’s amended and restated certificate of incorporation became effective, which provides for authorized the issuance of 100,000,000 authorized shares of common stock with a par value of $0.0001 per share, with one vote per share. Holders of common stock are entitled to receive any dividends as may be declared from time to time by the Company’s board of directors.
On November 28, 2023, the Company effected the Re-domiciliation. All references in these consolidated financial statements to the Company’s outstanding common stock, including per share information, have been retrospectively adjusted to reflect this Re-domiciliation.
| For the year ended, June 30, 2023 | ||||||||
| $ | No. of shares | |||||||
| (in thousands, except per share data) | ||||||||
| Opening balance | 2 | 15,873,113 | ||||||
| Closing balance | 2 | 15,873,113 | ||||||
| For the year ended June 30, 2023 | ||||||||
| $ | No. of shares | |||||||
| Opening balance | 1 | 12,926,349 | ||||||
| Issues of new shares – placements1 | - | 634,146 | ||||||
| Issues of new shares – acquisition2 | 1 | 2,181,695 | ||||||
| Issues of new shares – employees and directors | - | - | ||||||
| Exercise of options | - | 21 | ||||||
| Shares in lieu of advisor fees3 | - | 130,902 | ||||||
| Share issue costs | - | - | ||||||
| Closing balance | 2 | 15,873,113 | ||||||
| 1 | In December 2022, Incannex Australia raised $8.83 million from a placement of 634,146 ordinary shares to institutional and professional investors in a private placement. |
| 2 | In August 2022, Incannex Australia completed the acquisition on APIRx Pharmaceuticals via the issuance of 2,181,695 ordinary shares of Incannex Australia to the owners of APIRx in an all–scrip transaction. |
| 3 | In August 2022, Incannex Australia issued 130,902 ordinary shares to Ryba LLC as lead M&A Advisors on the APIRx acquisition. |
F-16
Note 9 – Additional paid-in capital
Additional paid-in capital:
| June 30, 2023 $ | June 30, 2022 $ | |||||||
| (in thousands, expect per share data) | ||||||||
| Opening balance | 69,074 | 38,689 | ||||||
| Options exercised | - | 29,045 | ||||||
| Options issued to advisors1 | 476 | - | ||||||
| Issues of new options – placement | 71 | - | ||||||
| Equity instruments issued to management and directors2 | 2,149 | 1,009 | ||||||
| Share placements3 | 8,829 | 290 | ||||||
| Share issued to advisors5 | 2,050 | 320 | ||||||
| Asset acquisition shares issued4 | 34,170 | - | ||||||
| Issuance costs6 | (529 | ) | (279 | ) | ||||
| At June 30, 2023 | 116,290 | 69,074 | ||||||
| 1 | In August 2022, Incannex Australia issued 9,000,000 options to Ryba LLC pursuant to the mandate executed between the parties in November 2021. As the transaction between the Company and APIRx was deemed complete in August 2022, the options were issued then. |
| 2 | Relates to the amortization of shares and options issued as share-based payments during the current and prior periods. |
| 3 | In December 2022, Incannex Australia raised $8.83 million from a placement of 634,146 ordinary shares to institutional and professional investors in a private placement. |
| 4 | In August 2022, Incannex Australia completed the acquisition on APIRx Pharmaceuticals via the issuance of 2,181,695 ordinary shares of Incannex Australia to the owners of APIRx in an all–scrip transaction. |
| 5 | In August 2022, Incannex Australia issued 130,902 ordinary shares to Ryba LLC as lead M&A Advisors on the APIRx acquisition. |
| 6 | In December 2022, Incannex Australia paid a commission of $530 to Bell Potter, as placement agent, for its services leading the private placement completed that month. |
The equity based premium reserve is used to record the value of equity issued to raise capital, and for share-based payments.
F-17
Note 10 – General and Administration expenses
| June 30, 2023 $ | June 30, 2022 $ | |||||||
| (in thousands) | ||||||||
| Salaries, and other employee benefits | (2,352 | ) | (1,463 | ) | ||||
| Share-based payments expense | (2,149 | ) | (1,063 | ) | ||||
| Depreciation expense | (88 | ) | 0 | |||||
| Compliance, legal and regulatory | (1,774 | ) | (2,584 | ) | ||||
| Occupancy expenses | (84 | ) | (82 | ) | ||||
| Advertising and investor relations | (1,249 | ) | (1,993 | ) | ||||
| Other administration expenses | (316 | ) | (258 | ) | ||||
| Total general and administration expenses | (8,012 | ) | (7,443 | ) | ||||
Note 11 – Share-based payments
| June 30, 2023 $ | June 30, 2022 $ | |||||||
| (in thousands) | ||||||||
| Research and development | - | - | ||||||
| General and administrative | (2,149 | ) | (1,063 | ) | ||||
| Total share-based compensation expense | (2,149 | ) | (1,063 | ) | ||||
Restricted stocks
A summary of the changes in the Company’s restricted stock activity for the year ended June 30, 2023, are as follows:
| Numbers of Shares | Weighted Average Grant Date Fair Value $ | |||||||
| (in thousands, except per share data) | ||||||||
| Unvested and Outstanding as of June 30, 2022 | 109,847 | 24 | ||||||
| Granted | - | - | ||||||
| Vested | 47,333 | 24 | ||||||
| Forfeited | - | - | ||||||
| Unvested and Outstanding as of June 30, 2023 | 62,514 | 149 | ||||||
F-18
Stock Options
A summary of the changes in the Company’s share options activity for the year ended June 30, 2023, are as follows:
| Number of Shares | Weighted Average Exercise Price ($) | Weighted Average Remaining | Aggregate Intrinsic Value (in thousands) ($) | |||||||||||||
| Outstanding as of June 30, 2022 | 545,008 | 16.16 | 2.20 | 567 | ||||||||||||
| Granted | 140,000 | 59.89 | 1.67 | 113 | ||||||||||||
| Exercised | 51,500 | 12.29 | 0.40 | 844 | ||||||||||||
| Cancelled or forfeited | - | - | - | - | ||||||||||||
| Outstanding as of June 30, 2023 | 736,508 | 24.19 | 1.35 | 442 | ||||||||||||
| Unvested as of June 30, 2023 | 110,171 | 49.22 | 2.75 | 70 | ||||||||||||
The aggregate intrinsic value of share options is calculated as the difference between the exercise price of the share options and the fair value of the Company’s shares of common stock for those share options that had exercise prices lower than the fair value of the Company’s shares of common stock.
As of June 30, 2023, there was $1,009 of unrecognized compensation cost related to unvested share options, which is expected to be recognized over a weighted-average period of 0.74 years.
Share Options Valuation
The weighted-average assumptions used in the Black-Scholes option pricing model to determine the fair value of the share options granted to employees and directors during the year ended June 30, 2023 and 2022, respectively, were as follow:
| June 30, 2023 | June 30, 2022 | |||||||
| Expected option life (years) | 1.5 | 3.0 | ||||||
| Expected volatility | 90 | % | 80 | % | ||||
| Risk-free interest rate | 3.18 | % | 3.12 | % | ||||
| Expected dividend yield | - | - | ||||||
| Fair value of underlying shares of common stock | 1.17 | 17.01 | ||||||
Note 12 – Income Tax
The prima facie income tax benefit on pre-tax accounting loss from operations reconciles to the income tax benefit in the financial statements as follows:
| June 30, 2023 $ | June 30, 2022 $ | |||||||
| (in thousands) | ||||||||
| Accounting loss before tax | (48,811 | ) | (10,818 | ) | ||||
| Income tax benefit at the applicable tax rate of 30% | (14,643 | ) | (3,245 | ) | ||||
| Non-deductible expenses | 36,406 | 492 | ||||||
| Non-assessable income | (206 | ) | (170 | ) | ||||
| Deferred tax assets not recognized | 700 | 460 | ||||||
| Income tax benefit | - | - | ||||||
| Unrecognized Deferred Tax Asset | ||||||||
| Deferred tax asset not recognized in the financial statements: | ||||||||
| Unused tax losses | 5,314 | 4,950 | ||||||
| Net unrecognized tax benefit at 30% | 6,014 | 5,410 | ||||||
F-19
ASC 740 requires that the tax benefit of net operating losses, temporary differences and credit carry forwards be recorded as an asset to the extent that management assesses that realization is “more likely than not.” Realization of the future tax benefits is dependent on the Company’s ability to generate sufficient taxable income within the carryforward period. Because of the Company’s recent history of operating losses, management believes that recognition of the deferred tax assets arising from the above-mentioned future tax benefits is currently not likely to be realized and, accordingly, has provided a valuation allowance. As of June 30, 2023 and 2022, the Company established a valuation allowance against its deferred tax assets due to the uncertainty surrounding the realization of such assets.
Note 13 – Loss per share
All share and earnings per share amounts presented below reflect the impact of the Re-domiciliation as if it had taken effect on July 1, 2022.
Basic and diluted net loss per share attributable to shareholders was calculated as follows (in thousands, except share and per share amounts):
| June 30, 2023 $ | June 30, 2022 $ | |||||||
| Basic and diluted loss per share | 332.17 | 101.66 | ||||||
| The loss and weighted average number of common stocks used in the calculation of basic loss per share is as follows: | 3.32 | 1.02 | ||||||
| Total comprehensive loss for the year | 51,103 | 12,120 | ||||||
| - Weighted average number of common stocks (number) | 15,384,704 | 11,921,292 | ||||||
The company notes that the diluted loss per share is the same as basic loss per share.
Note 14 – Related Party Transactions
Transactions between related parties are on commercial terms and conditions, no more favourable than those available to other parties unless otherwise stated.
F-20
INCANNEX HEALTHCARE INC.
Consolidated Balance Sheets
(unaudited)
(in thousands, except share and per share amounts)
(expressed in U.S. Dollars, unless otherwise stated)
| March 31, 2024 | June 30, 2023 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 9,305 | $ | 22,120 | ||||
| Prepaid expenses and other assets | 7,014 | 877 | ||||||
| Total current assets | 16,319 | 22,997 | ||||||
| Property, plant and equipment, net | 523 | 294 | ||||||
| Operating lease right-of-use assets | 408 | 492 | ||||||
| Total assets | $ | 17,250 | $ | 23,783 | ||||
| Liabilities and stockholders’ equity | ||||||||
| Current liabilities: | ||||||||
| Trade and other payables | $ | 1,255 | $ | 1,748 | ||||
| Accrued expenses and other current liabilities | 1,448 | 689 | ||||||
| Operating lease liabilities, current | 161 | 113 | ||||||
| Total current liabilities | 2,864 | 2,550 | ||||||
| Operating lease liabilities, non-current | 248 | 408 | ||||||
| Total liabilities | 3,112 | 2,958 | ||||||
| Commitments and contingencies (Note 8) | ||||||||
| Stockholders’ equity: | ||||||||
| Common stock, $0.0001 par value – 100,000,000 shares authorized; 15,873,113 and 12,926,349 shares issued and outstanding at March 31, 2024 and June 30, 2023, respectively | 2 | 2 | ||||||
| Preferred stock, $0.0001 par value per share, 10,000,000 shares authorized, no shares issued or outstanding at March 31, 2024 and June 30, 2023, respectively | - | - | ||||||
| Additional paid-in capital | 122,004 | 116,290 | ||||||
| Accumulated deficit | (104,210 | ) | (92,212 | ) | ||||
| Foreign currency translation reserve | (3,658 | ) | (3,255 | ) | ||||
| Total stockholders’ equity | 14,138 | 20,825 | ||||||
| Total liabilities and stockholders’ equity | $ | 17,250 | $ | 23,783 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-21
INCANNEX HEALTHCARE INC.
Consolidated Statements of Operations and Comprehensive Loss
(unaudited)
(in thousands, except share and per share amounts)
(expressed in U.S. Dollars, unless otherwise stated)
| For the three months ended March 31, | For the nine months ended March 31, | |||||||||||||||
| 2024 | 2023 | 2024 | 2023 | |||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 3,277 | $ | 1,639 | $ | 8,520 | $ | 4,597 | ||||||||
| Acquisition of in-process research and development | - | - | - | 35,347 | ||||||||||||
| General and administrative | 4,138 | 2,012 | 11,777 | 5,530 | ||||||||||||
| Total operating expenses | $ | 7,415 | $ | 3,651 | $ | 20,297 | $ | 45,474 | ||||||||
| Loss from operations | (7,415 | ) | (3,651 | ) | (20,297 | ) | (45,474 | ) | ||||||||
| Other income/(expense), net: | - | - | - | - | ||||||||||||
| R&D tax incentive | 1,320 | (83 | ) | 8,150 | 684 | |||||||||||
| Foreign exchange expense | (11 | ) | - | (17 | ) | - | ||||||||||
| Interest income | 75 | 163 | 166 | 153 | ||||||||||||
| Total other income, net | $ | 1,384 | $ | 80 | $ | 8,299 | $ | 837 | ||||||||
| Loss before income tax expense | (6,031 | ) | (3,571 | ) | (11,998 | ) | (44,637 | ) | ||||||||
| Income tax expense | - | - | - | - | ||||||||||||
| Net loss | $ | (6,031 | ) | $ | (3,571 | ) | $ | (11,998 | ) | $ | (44,637 | ) | ||||
| Other comprehensive loss: | ||||||||||||||||
| Currency translation adjustment, net of tax | (820 | ) | (202 | ) | (403 | ) | (2,029 | ) | ||||||||
| Total comprehensive loss | $ | (6,851 | ) | $ | (3,773 | ) | $ | (12,401 | ) | $ | (46,666 | ) | ||||
| Net loss per share: Basic and diluted | $ | (0.38 | ) | $ | (0.22 | ) | $ | (0.76 | ) | $ | (2.93 | ) | ||||
| Weighted average number of shares outstanding, basic and diluted | 15,873,113 | 15,873,113 | 15,873,113 | 15,221,900 | ||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-22
INCANNEX HEALTHCARE INC.
Consolidated Statements of Stockholders’ Equity (Deficit)
(unaudited)
(in thousands, except share amounts)
(expressed in U.S. Dollars, unless otherwise stated)
| Common stock | Additional paid-in capital | Accumulated deficit | Foreign currency translation reserve | Total Stockholders’ Equity (Deficit) | ||||||||||||||||||||
| Share | Amount | Amount | Amount | Amount | Amount | |||||||||||||||||||
| # | $ | $ | $ | $ | $ | |||||||||||||||||||
| Balance at June 30, 2023 | 15,873,113 | 2 | 116,290 | (92,212 | ) | (3,255 | ) | 20,825 | ||||||||||||||||
| Share-based compensation | 5,714 | 5,714 | ||||||||||||||||||||||
| Net loss | (11,998 | ) | (11,998 | ) | ||||||||||||||||||||
| Currency translation adjustment, net of tax | (403 | ) | (403 | ) | ||||||||||||||||||||
| Balance at March 31, 2024 | 15,873,113 | 2 | 122,004 | (104,210 | ) | (3,658 | ) | 14,138 | ||||||||||||||||
| Common stock | Additional paid-in capital | Accumulated deficit | Foreign currency translation reserve | Total Stockholders’ Equity (Deficit) | ||||||||||||||||||||
| Share | Amount | Amount | Amount | Amount | Amount | |||||||||||||||||||
| # | $ | $ | $ | $ | $ | |||||||||||||||||||
| Balance at June 30, 2022 | 12,926,349 | 1 | 69,074 | (43,401 | ) | (963 | ) | 24,711 | ||||||||||||||||
| Options exercised | 21 | |||||||||||||||||||||||
| Options issued to advisors | 476 | 476 | ||||||||||||||||||||||
| Share-based compensation | 1,631 | 1,631 | ||||||||||||||||||||||
| Share placements | 634,146 | 8,830 | 8,830 | |||||||||||||||||||||
| Share issued to advisors | 130,902 | 2,050 | 2,050 | |||||||||||||||||||||
| Asset acquisition shares issued | 2,181,695 | 1 | 34,170 | 34,171 | ||||||||||||||||||||
| Issuance costs | (531 | ) | (531 | ) | ||||||||||||||||||||
| Net loss | (44,637 | ) | (44,637 | ) | ||||||||||||||||||||
| Currency translation adjustment, net of tax | (2,029 | ) | (2,029 | ) | ||||||||||||||||||||
| Balance at March 31, 2023 | 15,873,113 | 2 | 115,700 | (88,038 | ) | (2,992 | ) | 24,672 | ||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-23
INCANNEX HEALTHCARE INC.
Consolidated Statements of Stockholders’ Equity (Deficit) (Continued)
(unaudited)
(in thousands, except share amounts)
(expressed in U.S. Dollars, unless otherwise stated)
| Common stock | Additional paid-in capital | Accumulated deficit | Foreign currency translation reserve | Total Stockholders’ Equity (Deficit) | ||||||||||||||||||||
| Share | Amount | Amount | Amount | Amount | Amount | |||||||||||||||||||
| # | $ | $ | $ | $ | $ | |||||||||||||||||||
| Balance at January 1, 2024 | 15,873,113 | 2 | 119,887 | (98,179 | ) | (2,838 | ) | 18,872 | ||||||||||||||||
| Share-based compensation | 2,117 | 2,117 | ||||||||||||||||||||||
| Net loss | (6,031 | ) | (6,031 | ) | ||||||||||||||||||||
| Currency translation adjustment, net of tax | (820 | ) | (820 | ) | ||||||||||||||||||||
| Balance at March 31, 2024 | 15,873,113 | 2 | 122,004 | (104,210 | ) | (3,658 | ) | 14,138 | ||||||||||||||||
| Common stock | Additional paid-in capital | Accumulated deficit | Foreign currency translation reserve | Total Stockholders’ Equity (Deficit) | ||||||||||||||||||||
| Share | Amount | Amount | Amount | Amount | Amount | |||||||||||||||||||
| # | $ | $ | $ | $ | $ | |||||||||||||||||||
| Balance at January 1, 2023 | 15,873,092 | 2 | 115,169 | (84,467 | ) | (2,790 | ) | 27,914 | ||||||||||||||||
| Options exercised | 21 | |||||||||||||||||||||||
| Options issued to advisors | ||||||||||||||||||||||||
| Option placements | ||||||||||||||||||||||||
| Share-based compensation | 531 | 531 | ||||||||||||||||||||||
| Share placements | ||||||||||||||||||||||||
| Share issued to advisors | ||||||||||||||||||||||||
| Asset acquisition shares issued | ||||||||||||||||||||||||
| Issuance costs | ||||||||||||||||||||||||
| Net loss | (3,571 | ) | (3,571 | ) | ||||||||||||||||||||
| Currency translation adjustment, net of tax | (202 | ) | (202 | ) | ||||||||||||||||||||
| Balance at March 31, 2023 | 15,873,113 | 2 | 115,700 | (88,038 | ) | (2,992 | ) | 24,672 | ||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-24
INCANNEX HEALTHCARE INC.
Consolidated Statements of Cash Flows
(unaudited)
(in thousands, except share and per share amounts)
(expressed in U.S. Dollars, unless otherwise stated)
| For the nine months ended March 31, | ||||||||
| 2024 | 2023 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (11,998 | ) | $ | (44,637 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | 41 | 22 | ||||||
| Share-based compensation expense | 5,585 | 1,631 | ||||||
| Unrealized gain on foreign currency remeasurement | 17 | (4 | ) | |||||
| Change in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | (6,150 | ) | (264 | ) | ||||
| Trade and other payables | 302 | (504 | ) | |||||
| Acquisition of in-process research and development | 0 | 35,589 | ||||||
| Net cash used in operating activities | (12,203 | ) | (8,167 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Purchase of property, plant and equipment | (274 | ) | (145 | ) | ||||
| Net cash used in investing activities | (274 | ) | (145 | ) | ||||
| Cash flows from financing activities: | ||||||||
| Proceeds from issuance of common stocks, net of issuance costs | - | 8,207 | ||||||
| Net cash provided by financing activities | - | 8,207 | ||||||
| Effect of exchange rate changes on cash and cash equivalents | (338 | ) | (850 | ) | ||||
| Net decrease in cash and cash equivalents | (12,477 | ) | (105 | ) | ||||
| Cash and cash equivalents at beginning of period | 22,120 | 25,835 | ||||||
| Cash and cash equivalents at end of period | $ | 9,305 | $ | 24,880 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-25
INCANNEX HEALTHCARE INC.
Notes To Unaudited Consolidated Financial Statements
(in thousands, except share and per share amounts)
(expressed in U.S. Dollars, unless otherwise stated)
Note 1 – Re-domiciliation and Business
Incannex Healthcare Inc. is a corporation formed under the laws of Delaware in July 2023. In November 2023, Incannex Healthcare Inc. acquired all the outstanding ordinary shares of Incannex Healthcare Limited, an Australian corporation (“Incannex Australia”), pursuant to a scheme of arrangement under Australian law (the “re-domiciliation”). As a result of the re-domiciliation, Incannex Australia became a wholly-owned subsidiary of Incannex Healthcare Inc., which is the new ultimate parent company.
Until the re-domiciliation, Incannex Australia’s ordinary shares were listed on the Australian Securities Exchange (“ASX”) and American Depositary Shares (“ADSs”), each representing 25 ordinary shares of Incannex Australia, traded on Nasdaq. Following completion of the re-domiciliation, Incannex Australia’s ordinary shares were delisted from the ASX and Incannex Healthcare Inc. assumed Incannex Australia’s listing on Nasdaq.
Pursuant to the re-domiciliation, holders of Incannex Australia’s ordinary shares received one share of common stock in Incannex Healthcare Inc. for every 100 ordinary shares held in Incannex Australia and holders of ADSs in Incannex Australia received one share of common stock of Incannex Healthcare Inc. for every 4 ADSs held in Incannex Australia.
The issued and outstanding shares of our common stock as shown in this report have been adjusted in the consolidated financial statements to reflect the 100:1 exchange ratio as if it had occurred on July 1, 2022.
Incannex Healthcare Inc. and its subsidiaries are referred to as “the Company” unless the text otherwise requires.
The Company’s fiscal year end is June 30. References to a particular “fiscal year” are to our fiscal year ended June 30 of that calendar year.
The consolidated financial statements of the Company are presented in United States dollars and consist of Incannex Healthcare Inc. and the following wholly-owned subsidiaries:
| Subsidiary | Jurisdiction | |
| Incannex Healthcare Limited | Victoria, Australia | |
| Incannex Pty Ltd | Victoria, Australia | |
| Psychennex Pty Ltd | Victoria, Australia | |
| APIRx Pharmaceutical USA, LLC | Delaware | |
| APIRx Pharmaceuticals Holding BV | IJsselstein, Netherlands | |
| Clarion Clinics Group Pty Ltd | Victoria, Australia | |
| Clarion Model Clinic Pty Ltd | Victoria, Australia | |
| Psychennex Licensing and Franchising Pty Ltd | Victoria, Australia |
F-26
Note 2 – Basis of Presentation and Summary of Significant Accounting Policies
Basis of Presentation
On November 28, 2023, the Company implemented the transaction to redomicile from Australia to United States and became the parent of Incannex Australia and the wholly owned subsidiaries listed in Note 1. The historical financial statements of Incannex Australia became the historical financial statements of the combined company upon consummation of the re-domiciliation. As a result, the financial statements included in this report reflect (i) the historical operating results of Incannex Australia and subsidiaries prior to the re-domiciliation; (ii) the combined results of the Company, Incannex Australia, and subsidiaries following the completion of the re-domiciliation; and (iii) the Company’s equity structure for all periods presented, including adjusting the issued and outstanding shares of common stock to reflect the 100:1 exchange ratio as if it had occurred on July 1, 2022.
The Company’s consolidated financial statements included in this report have been prepared in accordance with accounting principles generally accepted in the United States (“US GAAP”) and pursuant to the rules and regulations of the SEC. Prior to the re-domiciliation, Incannex Australia reported its consolidated financial statements in accordance with International Financial Reporting Standards (“IFRS”). Following the re-domiciliation, the Company transitioned to US GAAP and applied US GAAP retrospectively for all prior periods presented.
Reference is frequently made herein to the Financial Accounting Standards Board (the “FASB”) Accounting Standards Codification (“ASC”). This is the source of authoritative US GAAP recognized by the FASB to be applied to non-governmental entities.
Unaudited Interim Financial Information
In the opinion of the Company, the accompanying unaudited consolidated financial statements contain all adjustments, consisting of only normal recurring adjustments, necessary for a fair statement of its financial position as of March 31, 2024, and its results of operations for the three months and nine months ended March 31, 2024, and 2023, and cash flows for the nine months ended March 31, 2024, and 2023.
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries (the “Group”). Details of all controlled entities are set out in Note 1. All intercompany balances and transactions have been eliminated on consolidation.
Use of Estimates
The preparation of financial statements in conformity with US GAAP requires management to make estimates and assumptions that impact the reported amounts of assets, liabilities and expenses and the disclosure of contingent assets and liabilities in the Company’s consolidated financial statements and accompanying notes.
The most significant estimates and assumptions in the Company’s consolidated financial statements include the valuation of equity-based instruments issued for other than cash accrued research and development expense, and the research and development tax credit. Estimates are periodically reviewed in light of changes in circumstances, facts and experience. Changes in estimates are recorded in the period in which they become known. Actual results could differ materially from those estimates.
Risks and Uncertainties
The Company is subject to risks and uncertainties common to companies in the biopharmaceutical industry. The Company believes that changes in any of the following areas could have a material adverse effect on future financial position or results of operations: ability to obtain future financing; regulatory approval and market acceptance of, and reimbursement for, product candidates; performance of third-party clinical research organizations and manufacturers upon which the Company relies; protection of the Company’s intellectual property; litigation or claims against the Company based on intellectual property, patent, product, regulatory or other factors; the Company’s ability to attract and retain employees.
F-27
There can be no assurance that the Company’s research and development will be successfully completed, that adequate protection for the Company’s intellectual property will be obtained or maintained, that any products developed will obtain necessary government regulatory approval or that any approved products will be commercially viable. Even if the Company’s product development efforts are successful, it is uncertain when, if ever, the Company will generate significant revenue from product sales. The Company operates in an environment of rapid technological change and substantial competition from other pharmaceutical and biotechnology companies. In addition, the Company is dependent upon the services of its employees, consultants and other third parties.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentration of credit risk consist primarily of cash and cash equivalents. The Company has not experienced any losses in such accounts, and management believes that the Company is not exposed to significant credit risk due to the financial position of the depository institutions in which those deposits are held. As of March 31, 2024 and June 30, 2023 all deposit in banks of the Company is held outside of the United States.
Cash and Cash Equivalents
Cash and cash equivalents, which includes cash and deposits held at call with financial institutions with original maturities of three months or less that are readily convertible to known amounts of cash, are carried at cost, which approximates fair value.
Property, Plant and Equipment, Net
Recognition and Measurement
All property, plant and equipment are recognised at historical cost less depreciation.
Depreciation
Depreciation is calculated using the straight-line method to allocate their cost, net of their residual values, over their estimated useful lives or, in the case of leasehold improvements and certain leased plant and equipment, the shorter lease term as follows:
| ● | Buildings 25-40 years |
| ● | Machinery 10-15 years |
| ● | Vehicles 3-5 years |
| ● | Furniture, fittings and equipment 2-8 years |
Furniture, fittings and equipment include assets in the form of office fit outs. These assets and other leasehold improvements are recognised at their fair value and depreciated over the shorter of their useful life or the lease term, unless the entity expects to use the assets beyond the lease term.
Impairment of Long-Lived Assets
Long-lived assets consist primarily of property, plant and equipment, net, and are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. If circumstances require that an asset group be tested for possible impairment, the Company compares the undiscounted cash flows expected to be generated by the asset group to the carrying amount of the asset group. If the carrying amount of the long-lived asset is not recoverable on an undiscounted cash flow basis, an impairment is recognized to the extent that the carrying amount exceeds its fair value. Fair value is generally determined using the asset’s expected future discounted cash flows or market value, if readily determinable.
During the periods ended March 31, 2024 and 2023, the Company has not recorded impairment charges on its long-lived assets.
F-28
Leases
The Company determines if an arrangement is, or contains, a lease at inception and then classifies the lease as operating or financing based on the underlying terms and conditions of the contract. Leases with terms greater than one year are initially recognized on the consolidated balance sheets as right-of-use assets and lease liabilities based on the present value of lease payments over the expected lease term. The Company has also elected to not apply the recognition requirement to any leases within its existing classes of assets with a term of 12 months or less and does not include any options to purchase the underlying asset that the Company is reasonably certain to exercise.
Lease expense for minimum lease payments on operating leases is recognized on a straight-line basis over the lease term. Variable lease payments are excluded from the right-of-use assets and operating lease liabilities and are recognized in the period in which the obligation for those payments is incurred. Operating lease expenses are categorized within research and development and general and administrative expenses in the consolidated statements of operations and comprehensive loss. Operating lease cash flows are categorized under net cash used in operating activities in the consolidated statements of cash flows
As most of the Company’s leases do not provide an implicit rate, the Company uses its incremental borrowing rate based on the information available at commencement date in determining the present value of future payments.
Trade and other payables
These amounts represent liabilities for goods and services provided to the Company prior to the end of the period and which are unpaid. Due to their short-term nature, they are measured at amortized cost and are not discounted. The amounts are unsecured and are usually paid within 30 days of recognition.
Segment information
The Company operates and manages its business as one reportable and operating segment, which is the research and development of the use of psychedelic medicine and therapies for the treatment of mental health disorders. The Company’s Chief Executive Officer, who is the chief operating decision maker, reviews financial information on an aggregate basis for the purposes of allocating resources and evaluating financial performance. The Company’s long-lived assets are primarily in Australia.
Research and Development Costs
Research and development costs are expensed as incurred. Research and development consist of salaries, benefits and other personnel related costs including, laboratory supplies, preclinical studies, clinical trials and related clinical manufacturing costs, costs related to manufacturing preparations, fees paid to other entities to conduct certain research and development activities on the Company’s behalf and allocated facility and other related costs.
Nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities are deferred and capitalized as prepaid expenses until the related goods are delivered or services are performed.
The Company records accrued liabilities for estimated costs of research and development activities conducted by third-party service providers, which include the conduct of pre-clinical studies and clinical trials, and contract manufacturing activities. The Company records the estimated costs of research and development activities based upon the estimated amount of services provided but not yet invoiced and includes these costs in trade and other payables on the consolidated balance sheets and within research and development expenses on the consolidated statements of operations and comprehensive loss.
The Company accrues for these costs based on factors such as estimates of the work completed and in accordance with agreements established with its third-party service providers. The Company makes significant judgments and estimates in determining the accrued liabilities balance at the end of each reporting period. As actual costs become known, the Company adjusts its accrued liabilities. The Company has not experienced any material differences between accrued costs and actual costs incurred.
F-29
Acquisitions
The Company evaluate acquisitions under the accounting framework in ASC 805, Business Combinations, to determine whether the transaction is a business combination or an asset acquisition. In determining whether an acquisition should be accounted for as a business combination or an asset acquisition, the Company first performs a screen test to determine whether substantially all of the fair value of the gross assets acquired is concentrated in a single identifiable asset or a group of similar identifiable assets. If this is the case, then the acquisition is not deemed to be a business and is instead accounted for as an asset acquisition. If this is not the case, then the Company further evaluates whether the acquisition includes, at a minimum, an input and a substantive process that together significantly contribute to the ability to create outputs. If so, the acquisition constitutes a business for accounting purposes.
The Company measures and recognizes asset acquisitions that are not deemed to be business combinations based on the cost to acquire the assets, which includes pre-acquisition direct costs recorded in accrued professional and consulting fees. Goodwill is not recognized in asset acquisitions.
During the year ended June 30, 2023, the Company acquired APIRx Pharmaceutical USA, LLC (“APIRx”). The Company concluded that the acquisition of APIRx did not meet the definition of business under ASC 805, Business Combinations as the acquisition did not have outputs present and a substantive process was not acquired. Therefore, the Company accounted for the transaction as an asset acquisition rather than a business combination.
In accordance with ASC 730-10-25-2(c), intangible assets used in research and developmental activities acquired in an asset acquisition should be expensed at the acquisition date if there is no alternative future use in other R&D projects or otherwise (i.e., if they have no economic value). Additionally, in an asset acquisition, direct transaction costs are accumulated as a component of the consideration transferred and expensed with the acquired IPR&D that has no alternative use.
The Company determined that product candidates pertaining to APIRx had no alternative future use at the time of acquisition and charged $35.4 million, including transaction costs, to acquisition of in-process research and development (IPR&D) expense as of the date of acquisition.
Share-based compensation
The Company accounts for share-based compensation arrangements with employees and non-employees using a fair value method which requires the recognition of compensation expense for costs related to all share-based payments including share options. The fair value method requires the Company to estimate the fair value of share-based payment awards on the date of grant using an option-pricing model. The Company uses either the trinomial pricing or Black-Scholes option-pricing model to estimate the fair value of options granted. Share-based compensation awards are expensed using the graded vesting method over the requisite service period, which is generally the vesting period, for each separately-vesting tranche. The Company has elected a policy of estimating forfeitures at grant date. Option valuation models, including the trinomial pricing and Black-Scholes option-pricing model, require the input of several assumptions. These inputs are subjective and generally require significant analysis and judgment to develop. Refer to Note 12 for a discussion of the relevant assumptions.
Benefit from Research and Development Tax Incentive
Benefit from R&D tax credit consists of the R&D tax credit received in Australia, which is recorded within other income (expense), net. The Company recognizes grants once both of the following conditions are met: (1) the Company is able to comply with the relevant conditions of the grant and (2) the grant is received.
In the three months ended December 31, 2023, due to multiple years of tax incentives being granted and successful lodgement of overseas findings on the Company’s lead assets, the Company changed its estimates for the R&D tax incentive receivable, primarily based on historical experience of claims. The Company determined this was a change in accounting estimate in accordance with ASC 250-10. The result of this change in estimate resulted in an increase compared to the fiscal year ended June 30, 2023 and the receivable for R&D tax incentive by approximately $5 million. This change also resulted in an increase to other income of approximately $5 million. based on historical experience of claims.
F-30
Interest income
Interest income is recognized as interest accrues using the effective interest method. This is a method of calculating the amortised cost of a financial asset and allocating the interest income over the relevant period using the effective interest rate, which is the rate that exactly discounts estimated future cash receipts through the expected life of the financial asset to the net carrying amount of the financial asset.
Foreign Currency Translation
The Company maintains its consolidated financial statements in its functional currency, which is the Australian Dollar. Monetary assets and liabilities denominated in currencies other than the functional currency are translated into the functional currency at rates of exchange prevailing at the balance sheet dates. Non-monetary assets and liabilities denominated in foreign currencies are translated into the functional currency at the exchange rates prevailing at the date of the transaction. Exchange gains or losses arising from foreign currency transactions are included in other income (expense), net in the consolidated statements of operations and comprehensive loss.
For financial reporting purposes, the consolidated financial statements of the Company have been presented in the U.S. dollar, the reporting currency. The financial statements of entities are translated from their functional currency into the reporting currency as follows: assets and liabilities are translated at the exchange rates at the balance sheet dates, expenses and other income (expense), net are translated at the average exchange rates for the periods presented and stockholders’ equity is translated based on historical exchange rates. Translation adjustments are not included in determining net loss but are included as a foreign exchange adjustment to other comprehensive income, a component of stockholders’ equity.
The following table presents data regarding the dollar exchange rate of relevant currencies:
| March 31, 2024 | June 30, 2023 | |||||||
| Exchange rate on balance sheet dates | ||||||||
| USD: AUD Exchange Rate | 0.6532 | 0.6630 | ||||||
| Average exchange rate for the period | ||||||||
| USD: AUD Exchange Rate | 0.6544 | 0.6764 | ||||||
Income tax
The Company is subject to Australian and U.S. income tax laws. The Company follows ASC 740, Accounting for Income Taxes, when accounting for income taxes, which requires an asset and liability approach to financial accounting and reporting for income taxes. Deferred income tax assets and liabilities are computed annually for temporary differences between the financial statements and tax bases of assets and liabilities that will result in taxable or deductible amounts in the future based on enacted tax laws and rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amount more likely than not to be realized.
For uncertain tax positions that meet a “more likely than not” threshold, the Company recognizes the benefit of uncertain tax positions in the consolidated financial statements. The Company’s practice is to recognize interest and penalties, if any, related to uncertain tax positions in income tax expense in the consolidated statements of operations.
Net loss per share attributable to stockholders
The Company has reported losses since inception and has computed basic net loss per share by dividing net loss by the weighted-average number of common stocks outstanding for the period, without consideration for potentially dilutive securities. The Company computes diluted net loss per share after giving consideration to all potentially dilutive shares, including unvested restricted shares and outstanding options. Because the Company has reported net losses since inception, these potential common stocks have been anti-dilutive and basic and diluted loss per share were the same for all periods presented.
F-31
Comprehensive Loss
Comprehensive loss includes net loss as well as other changes in stockholders’ equity that result from transactions and economic events other than those with stockholders. For the nine months ended March 31, 2024, and 2023, the only component of accumulated other comprehensive loss is foreign currency translation adjustment.
Note 3 – Prepaid expenses and other current assets
| March 31, 2024 $ | June 30, 2023 $ | |||||||
| (in thousands) | ||||||||
| Prepayments1 | 421 | 686 | ||||||
| R&D tax credit recoverable2 | 6,406 | - | ||||||
| GST recoverable | 187 | 191 | ||||||
| Total other assets | 7,014 | 877 | ||||||
| 1 | Prepayments consist of prepaid clinical trial insurances, prepaid R&D expenditure relating to PsiGAD and IHL-675A clinical trials and scientific, marketing, and adverting subscription services. |
| 2 | R&D tax incentive receivable for quarter ended March 31, 2024 and the fiscal year ended June 30, 2023. |
Note 4 – Property, Plant and Equipment, net
| March 31, 2024 $ | June 30, 2023 $ | |||||||
| (in thousands) | ||||||||
| Furniture, fittings and equipment | 194 | 157 | ||||||
| Assets under construction | 393 | 160 | ||||||
| Total property, plant and equipment, gross | 587 | 317 | ||||||
| Accumulated depreciation and amortization | (64 | ) | (23 | ) | ||||
| Total property, plant and equipment, net | $ | 523 | $ | 294 | ||||
Depreciation expense is recorded within general and administrative in the Consolidated Statements of Operations and Comprehensive Loss and amounted to $18 and $0 for the three months ended March 31, 2024 and 2023, respectively, and $41 and $0 for the nine months ended March 31, 2024 and 2023, respectively.
Note 5 – Trade and other payables, accrued expenses and other current liabilities
| March 31, 2024 $ | June 30, 2023 $ | |||||||
| (in thousands) | ||||||||
| Current liabilities | ||||||||
| Trade payables | 1,255 | 1,748 | ||||||
| Accrued expenses | 1,133 | 426 | ||||||
| Employee leave entitlements | 315 | 263 | ||||||
| Total Trade and other payables, accrued expenses and other current liabilities | 2,703 | 2,437 | ||||||
Trade and other payables are unsecured, non-interest bearing and are normally settled within 30 days. The carrying amounts are a reasonable approximation of fair value.
F-32
Note 6 – Leases
For fiscal 2023, the Group entered into a three new lease agreement for its corporate head office in Sydney, Melbourne office and Clarion Clinic site. The leases have four, five and three-year terms respectively. These leases require monthly lease payments that may be subject to annual increases throughout the lease term. Certain of these leases also include renewal options at the election of the Company to renew or extend the lease for an additional three to five years. These optional periods have not been considered in the determination of the right-of-use assets or lease liabilities associated with these leases as the Company did not consider it reasonably certain it would exercise the options.
The following table summarizes the weighted-average remaining lease term and discount rates for the Company’s operating leases:
| March 31, 2024 | June 30, 2023 | |||||||
| Lease term (years) | 1.26 | 1.79 | ||||||
| Discount rate | 9.18 | % | 9.18 | % | ||||
The following table summarizes the lease costs pertaining to the Company’s operating leases
| March 31, 2024 $ | June 30, 2023 $ | |||||||
| (in thousands) | ||||||||
| Operating lease cost | 119 | 66 | ||||||
Cash paid for amounts included in the measurement of operating lease liabilities during nine months ended March 31, 2024 and fiscal year June 30, 2023 was $122 and $61, respectively, and was included within net cash used in operating activities in the cash flows.
The following table summarizes the future minimum lease payments due under operating leases as of March 31, 2024, (in thousands):
| Operating leases | Amount $ (in thousands) | |||
| June 30, 2024 | 50 | |||
| June 30, 2025 | 205 | |||
| June 30, 2026 | 199 | |||
| June 30, 2027 | 48 | |||
| June 30, 2028 | 32 | |||
| Total minimum lease payments | 534 | |||
| Less amount representing interest | 125 | |||
| Total operating lease liabilities | 409 | |||
As of March 31, 2024, the Company’s operating lease has a weighted-average remaining lease term of 1.26 years and a discount rate of 9.18%.
F-33
Note 7 – Commitments and contingencies
The Company records a loss contingency when it is probable that a liability has been incurred and the amount of the loss can be reasonably estimated. The Company also discloses material contingencies when it believes a loss is not probable but reasonably possible. Accounting for contingencies requires us to use judgment related to both the likelihood of a loss and the estimate of the amount or range of loss. Although the Company cannot predict with assurance the outcome of any litigation or tax matters, it does not believe there are currently any such actions that, if resolved unfavorably, would have a material impact on the Company’s operating results, financial position or cash flows.
Note 8 – Stockholder’s equity/Issued capital
Common stock
The Company has one class of common stock. In connection with the re-domiciliation, the Company’s amended and restated certificate of incorporation became effective, which provides for authorized the issuance of 100,000,000 authorized shares of common stock with a par value of $0.0001 per share, with one vote per share. Holders of common stock are entitled to receive any dividends as may be declared from time to time by the Company’s board of directors.
On November 28, 2023, the Company effected the re-domiciliation. All references in these consolidated financial statements to the Company’s outstanding common stock, including per share information, have been retrospectively adjusted to reflect this re-domiciliation.
| For the nine months ended March 31, | For the three months ended March 31, | |||||||||||||||
| 2024 $ | 2024 No, of shares | 2024 $ | 2024 No, of shares | |||||||||||||
| (in thousands, expect per share data) | ||||||||||||||||
| Opening balance | 2 | 15,873,113 | 2 | 15,873,113 | ||||||||||||
| Closing balance | 2 | 15,873,113 | 2 | 15,873,113 | ||||||||||||
| For the nine months ended March 31, | For the three months ended March 31, | |||||||||||||||
| 2023 $ | 2023 No, of shares | 2023 $ | 2023 No, of shares | |||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Opening balance | 1 | 12,926,349 | 2 | 15,873,092 | ||||||||||||
| Issues of new shares – placements1 | - | 634,146 | - | - | ||||||||||||
| Issues of new shares – acquisition2 | 1 | 2,181,695 | - | - | ||||||||||||
| Issues of new shares – employees and directors | - | - | - | - | ||||||||||||
| Exercise of options | - | 21 | - | 21 | ||||||||||||
| Shares in lieu of advisor fees3 | - | 130,902 | - | - | ||||||||||||
| Share issue costs | - | - | - | - | ||||||||||||
| Closing balance | 2 | 15,873,113 | 2 | 15,873,113 | ||||||||||||
| 1 | In December 2022, Incannex Australia raised $8.83 million from a placement of 634,146 ordinary shares to institutional and professional investors in a private placement. |
| 2 | In August 2022, Incannex Australia completed the acquisition on APIRx Pharmaceuticals via the issuance of 2,181,695 ordinary shares of Incannex Australia to the owners of APIRx in an all–scrip transaction. |
| 3 | In August 2022, Incannex Australia issued 130,902 ordinary shares to Ryba LLC as lead M&A Advisors on the APIRx acquisition. |
F-34
Note 9 – Additional paid-in capital
Additional paid-in capital:
| March 31, 2024 $ | March 31, 2023 $ | |||||||
| (in thousands, expect per share data) | ||||||||
| Opening balance | 116,290 | 69,074 | ||||||
| Options issued to advisors1 | - | 476 | ||||||
| Issues of new options – placement | - | - | ||||||
| Equity instruments issued to management and directors2 | 5,714 | 1,631 | ||||||
| Share placements3 | - | 8,830 | ||||||
| Share issued to advisors5 | - | 2,050 | ||||||
| Asset acquisition shares issued4 | - | 34,170 | ||||||
| Issuance costs6 | - | (531 | ) | |||||
| At March 31, 2024 | 122,004 | 115,700 | ||||||
| 1 | In August 2022, Incannex Australia issued 9,000,000 options to Ryba LLC pursuant to the mandate executed between the parties in November 2021. As the transaction between the Company and APIRx was deemed complete in August 2022, the options were issued then. |
| 2 | Relates to the amortization of shares and options issued as share-based payments during the current and prior periods. |
| 3 | In December 2022, Incannex Australia raised $8.83 million from a placement of 634,146 ordinary shares to institutional and professional investors in a private placement. |
| 4 | In August 2022, Incannex Australia completed the acquisition on APIRx Pharmaceuticals via the issuance of 2,181,695 ordinary shares of Incannex Australia to the owners of APIRx in an all–scrip transaction. |
| 5 | In August 2022, Incannex Australia issued 130,902 ordinary shares to Ryba LLC as lead M&A Advisors on the APIRx acquisition. |
| 6 | In December 2022, Incannex Australia paid a commission of $530 to Bell Potter, as placement agent, for its services leading the private placement completed that month. |
The equity based premium reserve is used to record the value of equity issued to raise capital, and for share-based payments.
Note 10 – General and Administration expenses
| For the three months ended March 31, | For the nine months ended March 31, | |||||||||||||||
| 2024 $ | 2023 $ | 2024 $ | 2023 $ | |||||||||||||
| (in thousands) | (in thousands) | |||||||||||||||
| Salaries, and other employee benefits | 2,873 | 979 | 7,706 | 3,122 | ||||||||||||
| Depreciation expense | 17 | (13 | ) | 41 | 21 | |||||||||||
| Compliance, legal and regulatory | 821 | 517 | 2,393 | 1,210 | ||||||||||||
| Occupancy expenses | 99 | 79 | 251 | 113 | ||||||||||||
| Advertising and investor relations | 149 | 388 | 854 | 813 | ||||||||||||
| Other administration expenses | 179 | 62 | 532 | 251 | ||||||||||||
| Total general and administration expenses | 4,138 | 2,012 | 11,777 | 5,530 | ||||||||||||
F-35
Note 11 – Share-based payments
| For the three months ended March 31, | For the nine months ended March 31, | |||||||||||||||
| 2024 $ | 2023 $ | 2024 $ | 2023 $ | |||||||||||||
| (in thousands) | (in thousands) | |||||||||||||||
| General and administrative | 2,116 | 531 | 5,584 | 2,092 | ||||||||||||
| Total share-based compensation expense | 2,116 | 531 | 5,584 | 2,092 | ||||||||||||
Restricted stocks
A summary of the changes in the Company’s restricted stock activity for the nine months ended March 31, 2024, are as follows:
| Numbers of Shares | Weighted Average Grant Date Fair Value $ | |||||||
| (in thousands, expect per share data) | ||||||||
| Unvested and Outstanding as of June 30, 2023 | 62,514 | 149 | ||||||
| Granted | 2,316,715 | 1,007 | ||||||
| Vested | 639,014 | 961 | ||||||
| Forfeited | 1,000 | 29 | ||||||
| Unvested and Outstanding as of March 31, 2024 | 1,739,215 | 1,006 | ||||||
Stock options
A summary of the changes in the Company’s stock options activity for the nine months ended March 31, 2024, are as follows:
| Number of Shares | Weighted Average Exercise Price ($) | Weighted Average Remaining Contractual Term (Years) | Aggregate Intrinsic Value (in thousands) ($) | |||||||||||||
| Outstanding as of June 30, 2023 | 633,508 | 24.19 | 1.35 | 442 | ||||||||||||
| Granted | - | - | - | - | ||||||||||||
| Exercised | - | - | - | - | ||||||||||||
| Cancelled or forfeited | 349,500 | 14.29 | - | 720 | ||||||||||||
| Outstanding as of March 31, 2024 | 284,008 | 36.37 | 1.79 | 99 | ||||||||||||
| Unvested as of March 31, 2024 | 47,337 | 22.23 | 3.75 | 125 | ||||||||||||
F-36
The aggregate intrinsic value of share options is calculated as the difference between the exercise price of the share options and the fair value of the Company’s shares of common stock for those share options that had exercise prices lower than the fair value of the Company’s shares of common stock.
As of March 31, 2024, there was $288 of unrecognized compensation cost related to unvested share options, which is expected to be recognized over a weighted-average period of 0.55 years.
Share Options Valuation
The weighted-average assumptions used in the Black-Scholes option pricing model to determine the fair value of the share options granted to employees and directors as of March 31, 2024 and June 30, 2023 were as follow:
| March 31, 2024 | June 30, 2023 | |||||||
| Expected option life (years) | - | 1.5 | ||||||
| Expected volatility | - | 90 | % | |||||
| Risk-free interest rate | - | 3.18 | % | |||||
| Expected dividend yield | - | - | ||||||
| Fair value of underlying shares of common stock | - | 1.17 | ||||||
Note 12 – Income Tax
The prima facie income tax benefit on pre-tax accounting loss from operations reconciles to the income tax benefit in the financial statements as follows:
| March 31, 2024 $ | June 30, 2023 $ | |||||||
| (in thousands) | ||||||||
| Accounting loss before tax | (11,998 | ) | (52,766 | ) | ||||
| Income tax benefit at the applicable tax rate of 30% | (3,599 | ) | (15,830 | ) | ||||
| Non-deductible expenses | 8,064 | 36,510 | ||||||
| Non-assessable income | (8,154 | ) | (171 | ) | ||||
| Deferred tax assets not recognized | 875 | 581 | ||||||
| Income tax benefit | ||||||||
| Unrecognized Deferred Tax Asset | ||||||||
| Deferred tax asset not recognized in the financial statements: | ||||||||
| Unused tax losses | 8,202 | 4,340 | ||||||
| Net unrecognized tax benefit at 30% | 9,075 | 4,989 | ||||||
ASC 740 requires that the tax benefit of net operating losses, temporary differences and credit carry forwards be recorded as an asset to the extent that management assesses that realization is “more likely than not.” Realization of the future tax benefits is dependent on the Company’s ability to generate sufficient taxable income within the carry forward period. Because of the Company’s recent history of operating losses, management believes that recognition of the deferred tax assets arising from the above-mentioned future tax benefits is currently not likely to be realized and, accordingly, has provided a valuation allowance. As of March 31, 2024 and 2023, the Company established a valuation allowance against its deferred tax assets due to the uncertainty surrounding the realization of such assets.
F-37
Note 13 – Loss per share
All share and earnings per share amounts presented below reflect the impact of the re-domiciliation as if it had taken effect on July 1, 2022.
Basic and diluted net loss per share attributable to stockholders was calculated as follows (in thousands, except share and per share amounts):
| For the three months ended March 31, | For the nine months ended March 31, | |||||||||||||||
| 2024 $ | 2023 $ | 2024 $ | 2023 $ | |||||||||||||
| Basic loss per share – cents per share | 38.00 | 22.50 | 75.59 | 293.24 | ||||||||||||
| Basic loss per share | 0.38 | 0.22 | 0.76 | 2.93 | ||||||||||||
| The loss and weighted average number of common stocks used in the calculation of basic loss per share is as follows: | ||||||||||||||||
| Total comprehensive loss for the year | 6,031 | 3,571 | 11,998 | 44,637 | ||||||||||||
| - Weighted average number of common stocks (number) | 15,873,113 | 15,873,113 | 15,873,113 | 15,221,900 | ||||||||||||
The Company notes that the diluted loss per share is the same as basic loss per share.
Note 14 – Related Party Transactions
Transactions between related parties are on commercial terms and conditions, no more favorable than those available to other parties unless otherwise stated.
Note 15 – Events occurring after the reporting period
There were no events occurring after the reporting period ended March 31, 2024.
F-38
Up to 4,000,000 Shares of Common Stock
Pre-Funded Warrants to Purchase up to 4,000,000 Shares of Common Stock
Common Warrants to Purchase up to 4,000,000 Shares of Common Stock
Up to 8,000,000 Shares of Common Stock Underlying the Pre-Funded Warrants and Common Warrants
PRELIMINARY PROSPECTUS
| Jones | Lake Street |
, 2024
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution.
The following table indicates the expenses to be incurred in connection with the offering described in this registration statement, all of which will be paid by us. All amounts are estimated except the SEC registration fee and the Financial Industry Regulatory Authority, Inc. (FINRA) filing fee.
| Amount Paid or to Be Paid | ||||
| SEC registration fee | $ | 3,259.01 | ||
| FINRA filing fee | $ | 3,812 | ||
| Accountants’ fees and expenses | $ | 50,000 | ||
| Legal fees and expenses | $ | 200,000 | ||
| Transfer Agent’s fees and expenses | $ | 10,000 | ||
| Printing expenses | $ | 50,000 | ||
| Miscellaneous | $ | 10,000 | ||
| Total expenses | $ | 327,071.01 | ||
Item 14. Indemnification of Directors and Officers.
Section 102 of the General Corporation Law of the State of Delaware permits a corporation to eliminate the personal liability of directors of a corporation to the corporation or its stockholders for monetary damages for a breach of fiduciary duty as a director, except where the director breached his duty of loyalty, failed to act in good faith, engaged in intentional misconduct or knowingly violated a law, authorized the payment of a dividend or approved a stock repurchase in violation of Delaware corporate law or obtained an improper personal benefit. Our certificate of incorporation provides that no director of the Registrant shall be personally liable to it or its stockholders for monetary damages for any breach of fiduciary duty as a director, notwithstanding any provision of law imposing such liability, except to the extent that the General Corporation Law of the State of Delaware prohibits the elimination or limitation of liability of directors for breaches of fiduciary duty.
Section 145 of the General Corporation Law of the State of Delaware provides that a corporation has the power to indemnify a director, officer, employee or agent of the corporation, or a person serving at the request of the corporation for another corporation, partnership, joint venture, trust or other enterprise in related capacities against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with an action, suit or proceeding to which he was or is a party or is threatened to be made a party to any threatened, ending or completed action, suit or proceeding by reason of such position, if such person acted in good faith and in a manner he reasonably believed to be in or not opposed to the best interests of the corporation, and, in any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful, except that, in the case of actions brought by or in the right of the corporation, no indemnification shall be made with respect to any claim, issue or matter as to which such person shall have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery or other adjudicating court determines that, despite the adjudication of liability but in view of all of the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which the Court of Chancery or such other court shall deem proper.
II-1
Our amended and restated bylaws provide that we will indemnify any Indemnitee who was or is party or is threatened to be made a party to, or was or is otherwise involved in, any action, suit, arbitration, alternative dispute resolution mechanism, investigation, inquiry, judicial, administrative or legislative hearing, or any other threatened, pending or completed proceeding, whether brought by or in the right of the Company or otherwise, including any and all appeals, whether of a civil, criminal, administrative, legislative, investigative or other nature, by reason of the fact that he or she is or was a director or an officer of the Company or while a director or officer of the Company is or was serving at the request of the Company as a director, officer, employee, agent or trustee of another corporation or of a partnership, joint venture, trust or other enterprise, including service with respect to an employee benefit or by reason of anything done or not done by him or her in any such capacity, shall be indemnified and held harmless by the Company to the fullest extent authorized by the DGCL, as the same exists or may hereafter be amended, against all expense, liability and loss (including attorneys’ fees, judgments, fines, ERISA excise taxes, penalties and amounts paid in settlement by or on behalf of the indemnitee) actually and reasonably incurred by such indemnitee in connection therewith, all on the terms and conditions set forth in our amended and restated bylaws, if the Indemnitee acted in good faith and in a manner he or she reasonably believed to be in, or not opposed to, our best interests, except that no indemnification shall be made with respect to any claim, issue or matter as to which such person shall have been adjudged to be liable to us, unless a court determines that, despite such adjudication but in view of all of the circumstances, he or she is entitled to indemnification of such expenses. Notwithstanding the foregoing, to the extent that any Indemnitee has been successful, on the merits or otherwise, he or she will be indemnified by us against all expenses (including attorneys’ fees) actually and reasonably incurred in connection therewith. Expenses must be advanced to an Indemnitee under certain circumstances.
We have entered into indemnification agreements with each of our directors and officers. These indemnification agreements may require us, among other things, to indemnify our directors and officers for some expenses, including attorneys’ fees, judgments, fines and settlement amounts incurred by a director or officer in any action or proceeding arising out of his or her service as one of our directors or officers, or any of our subsidiaries or any other company or enterprise to which the person provides services at our request.
We maintain a general liability insurance policy that covers certain liabilities of directors and officers of our corporation arising out of claims based on acts or omissions in their capacities as directors or officers.
Item 15. Recent Sales of Unregistered Securities.
Set forth below is information regarding unregistered securities issued by us since July 5, 2023 to the date of this registration statement. Also included is the consideration received by us for such securities and information relating to the section of the Securities Act, or rule of the SEC, under which exemption from registration was claimed.
| ● | On November 28, 2023, we issued 15,873,113 shares of common stock and 2,076,666 options in exchange for all the outstanding ordinary shares and options in Incannex Australia, pursuant to Section 3(a)(10) of the Securities Act, as a result of the completion of the Re-domiciliation. |
II-2
Item 16. Exhibits and Financial Statement Schedules.
(a)
| # | Indicates management contract or compensatory plan. |
| + | Portions of this exhibit (indicated by asterisks) have been omitted pursuant to Item 601 of Regulation S-K because it is both not material and is the type that the registrant treats as private or confidential |
| (b) | Financial Statement Schedules. No financial statement schedules are provided because the information called for is not required or is shown in the financial statements or the notes thereto. |
II-3
Item 17. Undertakings.
The undersigned Registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
| (i) | To include any prospectus required by Section 10(a)(3) of the Securities Act; |
| (ii) | To reflect in the prospectus any facts or events arising after the effective date of this registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high and of the estimated maximum offering range may be reflected in the form of prospectus filed with the Securities and Exchange Commission (the “Commission”) pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and |
| (iii) | To include any material information with respect to the plan of distribution not previously disclosed in this registration statement or any material change to such information in this registration statement; |
provided, however, that paragraphs (1)(i), (1)(ii) and (1)(iii) above do not apply if the information required to be included in a post-effective amendment by those paragraphs is contained in reports filed with or furnished to the Commission by the registrant pursuant to Section 13 or Section 15(d) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), that are incorporated by reference in the registration statement, or is contained in a form of prospectus filed pursuant to Rule 424(b) that is part of the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability of the registrant under the Securities Act to any purchaser in the initial distribution of the securities, in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
| (i) | any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424; |
| (ii) | any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant; |
| (iii) | the portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and |
| (iv) | any other communication that is an offer in the offering made by the undersigned registrant to the purchaser. |
II-4
The undersigned registrant hereby undertakes:
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Act and is therefore unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer or controlling person of the Registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
The undersigned registrant hereby undertakes that:
For purposes of determining any liability under the Securities Act of 1933, as amended, the information omitted from a form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in the form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act of 1933, as amended, shall be deemed to be part of this registration statement as of the time it was declared effective.
For the purpose of determining any liability under the Securities Act of 1933, as amended, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-5
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this Amendment No. 1 to the registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in Sydney, Australia on July 17, 2024.
| Incannex Healthcare Inc. | ||
| By: | /s/ Joel Latham | |
| Name: | Joel Latham | |
| Title: | Chief Executive Officer, President and Director | |
Pursuant to the requirements of the Securities Act of 1933, as amended, this registration statement has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
| Signature | Title | Date | ||
| /s/ Joel Latham | Chief Executive Officer, President and Director | July 17, 2024 | ||
| Joel Latham | (principal executive officer) | |||
| /s/ Joseph Swan | Chief Financial Officer, Treasurer and Secretary | July 17, 2024 | ||
| Joseph Swan | (principal financial and accounting officer) | |||
| * | Chairman | July 17, 2024 | ||
| Troy Valentine | ||||
| * | Director | July 17, 2024 | ||
| Peter Widdows | ||||
| * | Director | July 17, 2024 | ||
| George Anastassov | ||||
| * | Director | July 17, 2024 | ||
| Robert Clark |
| *By: | /s/ Joel Latham | |
| Joel Latham Attorney-in-fact |
II-6